Изобретение относится к области медицинской химии, иммунологии и нейробиологии, и направлено на разработку нового средства для лечения рассеянного склероза, относящегося к классу биологически активных веществ - комплексной соли Gd3+ с DTPA-Lys-дигликолат-Ahx-Abu-TGIRIS-Abu-NH2, где DTPA обозначает диэтилентриаминопентауксусную кислоту (хелатирующий агент), Ahx обозначает аминогексановую кислоту, Abu обозначает α-аминомасляную кислоту.
Ранее авторами был получен пептид с последовательностью Abu-Ser-Ser-Val-Lys-Val-Ser-Abu (Abu - α-аминомасляная кислота, далее обозначается как Abu-SSKVKS-Abu), соответствующий консервативному участку VH-домена тяжелой цепи иммуноглобулинов человека, обладающий иммуносупрессорными свойствами, и предназначенный для лечения рассеянного склероза у человека. Пептид Abu-SSKVKS-Abu при внутривенном введении крысам предотвращает развитие у них экспериментального аутоиммунного энцефаломиелита (ЭАЭ) - распространенной животной модели рассеянного склероза, а также подавляет конканавалин А зависимую и миелин (антиген) - зависимую пролиферацию лимфоцитов крысы с в диапазоне от 10-4 до 10-6 М. Снижение скорости пролиферации лимфоцитов в два раза по сравнению с исходным уровнем (IC50) достигается при концентрации пептида Abu-SSVKVS-Abu 11,8±2,1 мкМ. Терапевтическое и иммуносупрессорное действие пептида объясняется тормозным эффектом в отношении популяций лимфоцитов, специфичных в отношении белков миелина и способствующих разрушению миелиновых оболочек центральной нервной системы, что и приводит к возникновению симптомов рассеянного склероза. Таким образом, пептид Abu-SSVKVS-Abu может рассматриваться не только как средство, непосредственно подавляющее активность лимфоцитов, ответственных за аутоиммунный механизм поражения ЦНС при рассеянном склерозе, но и как специфических лиганд-зонд, способный избирательно узнавать такие лимфоциты.
Также был синтезирован пептид с последовательностью Abu-TGIRIS-Abu, (Abu - α-аминомасляная кислота), который при внутривенном введении крысам предотвращает развитие у них экспериментального аутоиммунного энцефаломиелита (ЭАЭ) - распространенной животной модели рассеянного склероза, превосходя по эффективности ранее синтезированный прототип - пептид Abu-SSVKVS-Abu, вводимый в равной концентрации.
Последовательность Abu-TGIRIS-Abu получена путем модификации прототипа таким образом, чтобы увеличить его способность подавлять развитие симптомов ЭАЭ у крыс при системном введении. Раскрыты способы твердофазного и жидкофазного синтеза пептида Abu-TGIRIS-Abu. Показано, что наибольшей эффективностью (выход 30% от теоретического) обладает способ жидкофазного синтеза пептида Abu-TGIRIS-Abu методом конденсации фрагментов.
В рамках настоящего изобретения была поставлена цель создать производное пептида Abu-TGIRIS-Abu, меченное ионами Gd3+, для дальнейшего его использования в качестве средства визуализации лимфоцитов, потенциально принимающих участие в аутоиммунном поражении миелиновых оболочек ЦНС in vivo методом электронного парамагнитного резонанса. Этот же конъюгат может использоваться для избирательного уничтожения таких лимфоцитов методом рентген-индуцированной термоабляции как in vitro, так и in vivo (Dinger et al, 2016). Параллельно ставилась задача получения меченного ионами Gd3+ пептида Р0 с последовательностью Abu-VSS-Abu-KSV для использования его в качестве контрольного препарата для выявления неспецифических эффектов ионов Gd3+ и координирующей его молекулы хелатирующего агента in vitro и in vivo.
Комплексные соединения гадолиния (3+) с DTPA (Gd3+-DTPA) находят широкое применение в качестве диагностических реагентов в медицине и в биологических исследованиях (Caravan et al, 1999; Merbach &  , 2001). Комплекс Gd3+-DTPA, в отличие от свободных ионов Gd3+ не токсичен, обладая при этом высокой активностью в качестве зонда для спектроскопии парамагнитного резонанса (Goischke, 2016). Это позволяет использовать Gd3+-DTPA в качестве контрастирующего агента для проведения диагностики методом ПР-имиджинга (Позднякова и соавт., 2016).
, 2001). Комплекс Gd3+-DTPA, в отличие от свободных ионов Gd3+ не токсичен, обладая при этом высокой активностью в качестве зонда для спектроскопии парамагнитного резонанса (Goischke, 2016). Это позволяет использовать Gd3+-DTPA в качестве контрастирующего агента для проведения диагностики методом ПР-имиджинга (Позднякова и соавт., 2016).
В литературе описан ряд способов функционализации Gd3+-DTPA различными лигандами с целью придания им специфической аффинности в отношении тканей, подвергшихся патогенетической трансформации вследствие развития различных заболеваний (Desreux et al, 2002). При этом многие источники описывают способы синтеза биоконъюгатов с использованием одной из пяти имеющихся карбоксильных групп DTPA для образования ковалентной связи с лигандом (Hnatowich et al 1983; Edwards et al. 1994; Fichna & Janecka, 2003). Этот способ имеет два существенных недостатка. Во-первых, активация карбоксильных групп неселективна, что приводит к образованию смеси изомеров моноамидов и диамидов DTPA (Maisano et al. 1992). Во-вторых, в литературе имеются сведения, что термодинамическая стабильность комплексных соединений при превращении одной из карбоксильных групп в амидную, значительно уменьшается. Для получения стабильных и нетоксичных комплексов гадолиния с DTPA необходимо сохранять все пять карбоксильных групп в свободном состоянии, обеспечивающем их участие в образовании комплекса с ионом Gd3+(Sherry et al, 1988; Anellet al. 1999; Fossheim et al, 1991).
Известен также патент US №4479930, в котором описывается способ присоединения DTPA к N-концу пептидной цепи с применением дициклического диангидрида. Известен патент US №5804157, в котором к свободной N-аминогруппе пептида D-Phe-Cys-Phe-D-Trp-Lys-Thr-Cys-Thr-OH (производное соматостатина), иммобилизованного на твердой фазе в виде смолы SASRIN (Bachem Biosciences), присоединяли активированное триаза-производное DTPA - 1,1,4-Трис(т-бутилоксикарбонилметил)-7,7-бис(карбоксиметил)-1,4,7-триазапентан. Эти способы обеспечивают возможность получения основного продукта с однозначно заданной структурой (без образования смеси изомеров) при хорошем выходе. Однако, они требуют использования дорогостоящих реагентов, не доступных для крупнотоннажного производства. Кроме того, эти способы не обеспечивают образования линкера достаточной длины гибкости между пептидом и DTPA, что может вызвать падение аффиности связывания пептида со специфическим лигандов на поверхности клетки-мишени.
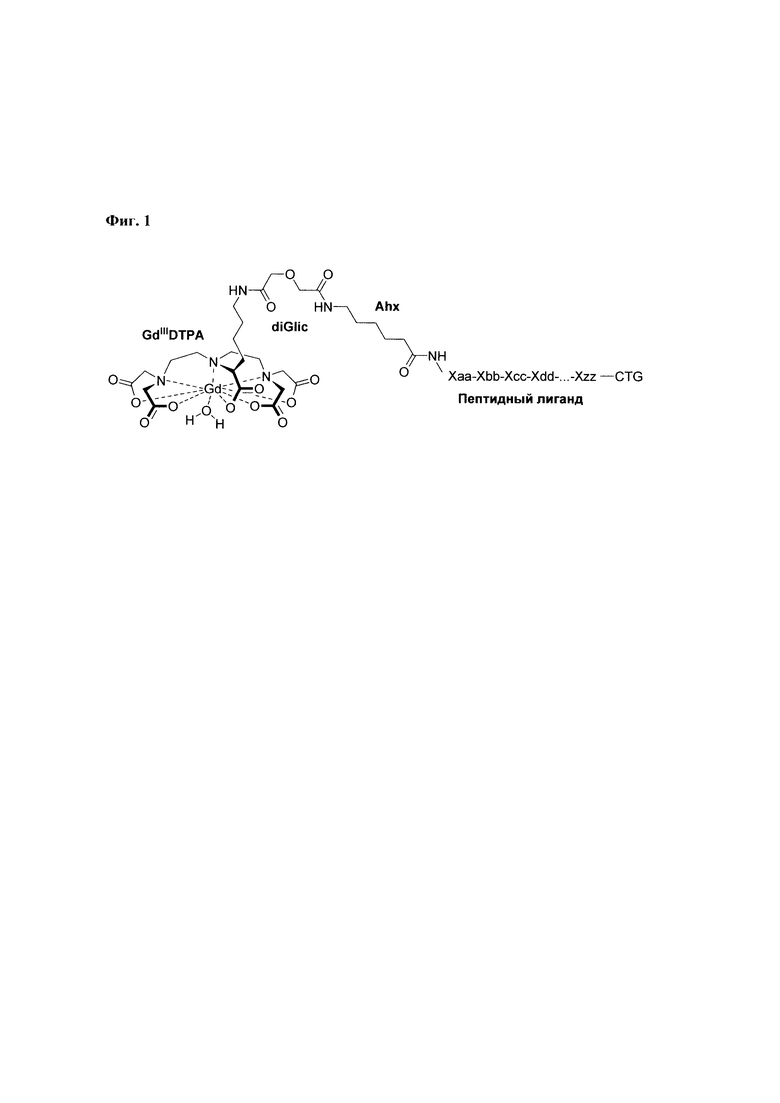
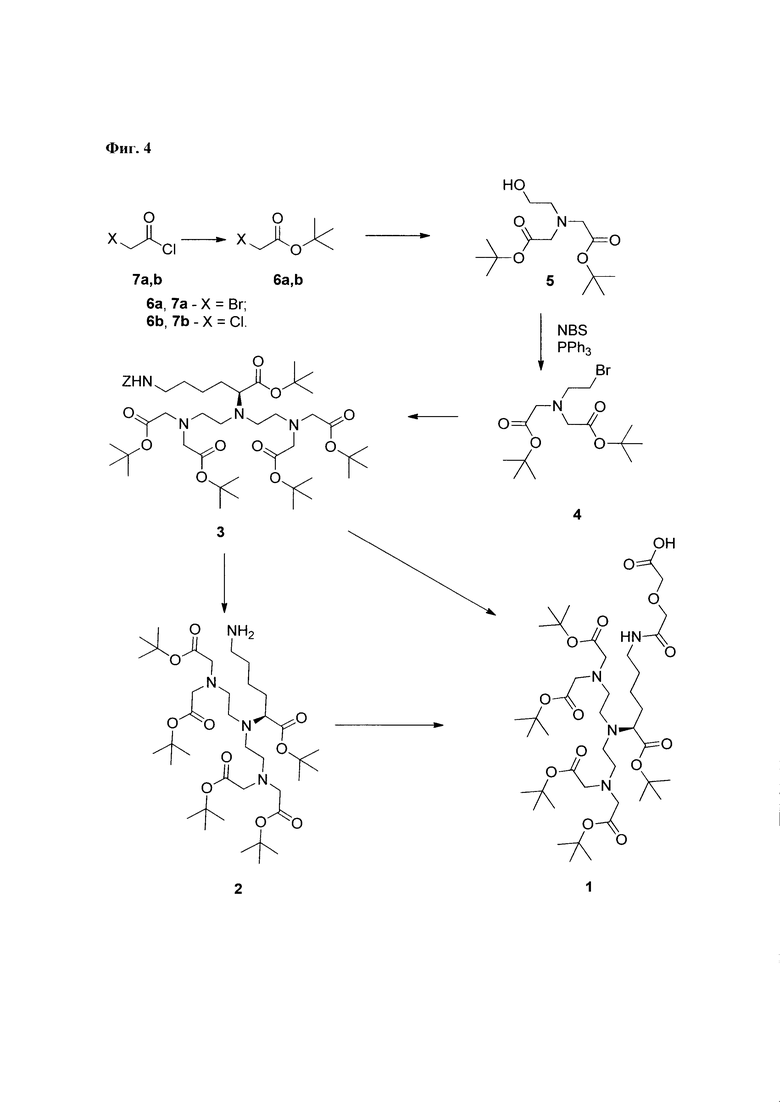
При разработке структуры конъюгата координационного комплекса Gd3+-DTPA с пептидами Abu-TGIRIS-Abu и Р0, в качестве аналога использовался способ (Langereis et al. 2005), предназначенный для получения ковалентных конъюгатов DTPA с аминокислотой Lys. Существенным отличием в структуре заявляемого способа образования связи между комплексом Gd3+ и пептидным лигандом по сравнению с аналогом является использование комбинации дигликолевой кислоты и аминогексановой кислоты для получения четырехчленного соединяющего агента, обладающего центральной симметрией (Фиг. 1).
Дигликолевая кислота используется для образования устойчивой ковалентной химической связи между комплексным соединением Gd3+-DTPA и пептидом, N-конец которого модифицирован аминогексановой кислотой. Использование аминогексановой кислоты увеличивает расстояние между ионом Gd3+ и пептидным лигандом, что снижает риск возникновения стерических затруднений при связывании пептидного лиганда с его рецептором на поверхности клетки.
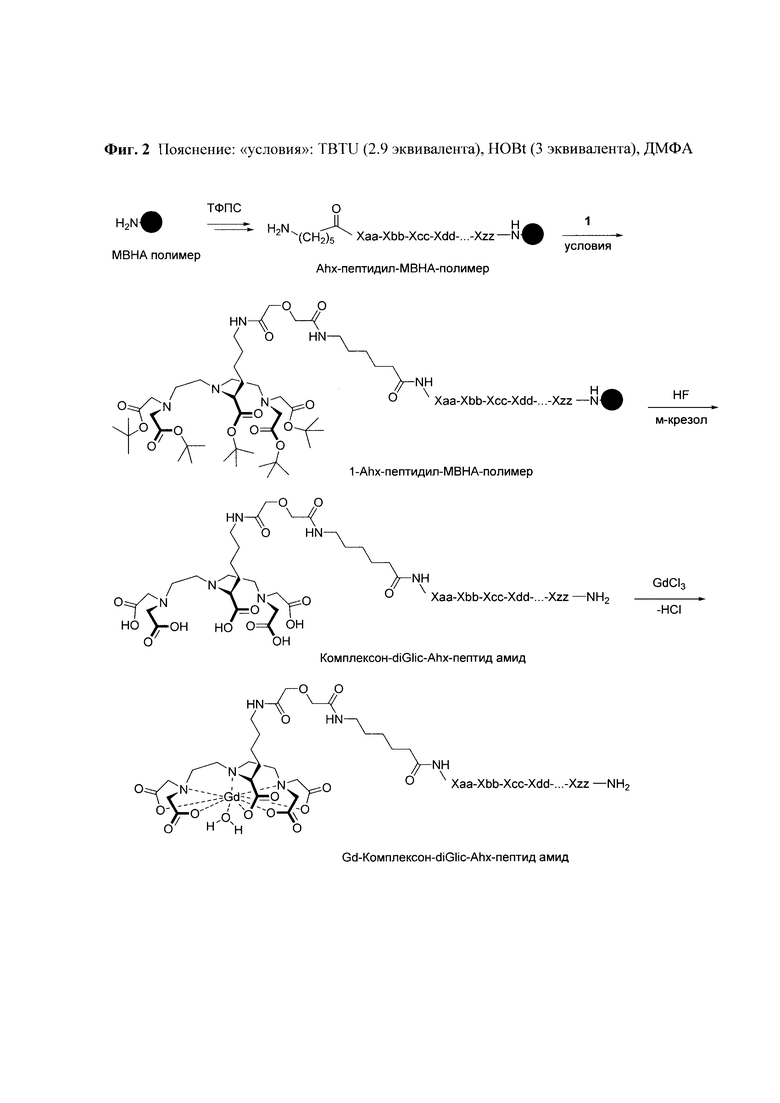
Синтез пептида осуществляют твердофазным методом, что снимает необходимость очистки промежуточных продуктов от избытка реагентов. По окончании очередной стадии синтеза их удаление осуществляется путем фильтрования нерастворимого пептидил-полимера. Присоединение аминогексановой кислоты осуществляется также с использованием метода твердофазного пептидного синтеза. При этом образуется соединение «Ahx-пептидил-МВНА-полимер» (Фиг. 2). Наконец, после деблокирования N-конца, пептидил-полимер вводят во взаимодействие с соединением 1 (Фиг. 3).
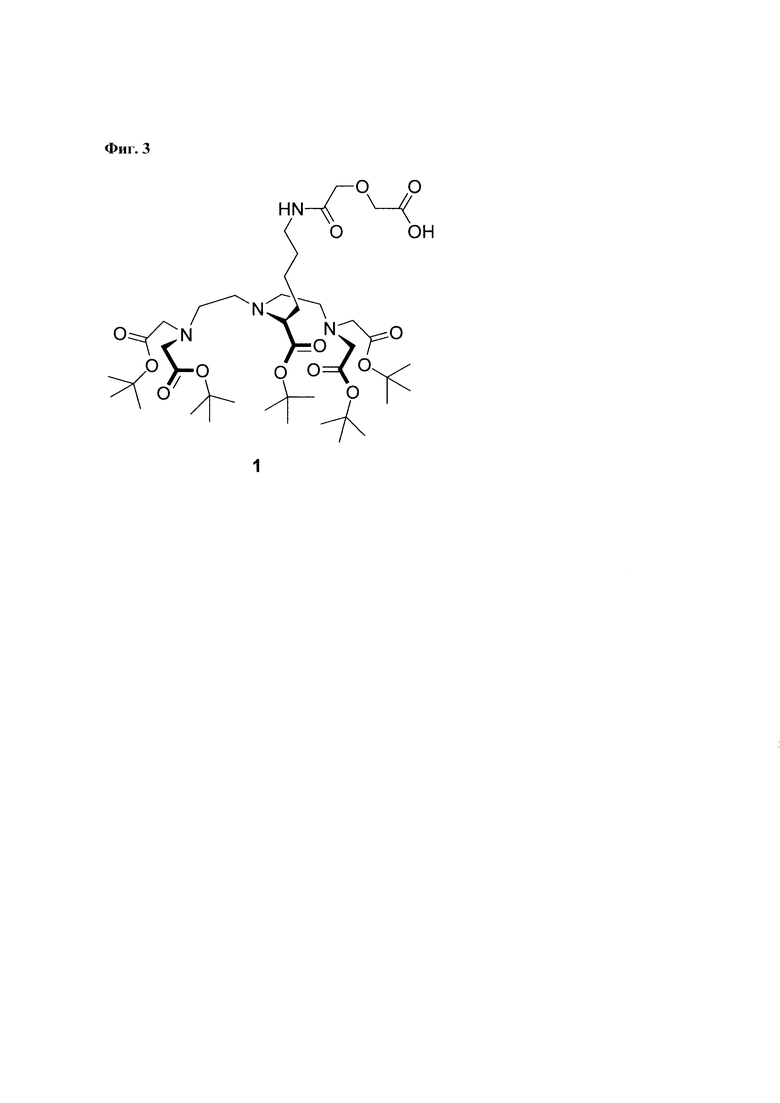
Соединение 1 представляет собой защищенное производное DTPA (Фиг. 3). Остаток аминокислоты лизина ацилирован дигликолевой кислотой. Единственная имеющаяся карбоксильная группа может быть активирована, и в активированном виде использована для введения в реакцию с аминогруппой пептида на полимерной подложке. При этом остальные 5 карбоксильных групп присутствуют в виде сложных эфиров и, вследствие чего в химическое взаимодействие с аминогруппами пептидил-полимера в используемых условиях реакции вступать не могут.
Продуктом реакции на полимерной положке является вещество «1-Ahx-пептидил-МВНА-полимер». Пептид, соединенный с DTPA, снимают с полимерной подложки с использованием раствора, состоящего из безводного фтористого водорода и ловушек электрофильных частиц. При этом третичные бутиловые эфиры DTPA превращаются в свободные карбоксильные группы (Фиг. 2).
Конечным продуктом этой реакции является вещество DTPA-Lys-дигликолат-Ahx-Abu-TGIRIS-Abu-NH2. При необходимости это вещество может быть очищено от побочных продуктов различными хроматографическими методами, включая ОФ ВЭЖХ и ионообменную хроматографию.
Очищенный полупродукт вводится в реакцию образования комплекса с ионами Gd3+. Для этого в водный раствор вещества DTPA-Lys-дигликолат-Ahx-Abu-TGIRIS-Abu-NH2 добавляют соль Gd3+, например хлорид, нейтрализуя выделяющуюся HCl различными основаниями. Продуктом этой реакции является целевое вещество - комплексная соль Gd3+ с DTPA-Lys-дигликолат-Ahx-Abu-TGIRIS-Abu-NH2, структура которого показана на Фиг 1. Строение полученного соединения подтверждают методом масс-спектрометрии при ионизации электрораспылением.
Осуществление изобретения
1. Исходные реагенты
Ацетонитрил, дихлорметан, тетрагидрофуран, трет-бутанол (ХиммидСинтез, РФ). Гидрокарбонат калия, сульфат магния (ХиммедСинтез, РФ), хлорангидрид бромуксусной кислоты (Fluka, Buchs, Switzerland), хлорангидрид хлоруксусной кислоты (Реахим), триэтиламин (Реахим), 2-этаноламин (Fluka, Buchs, Switzerland), трифенилфосфин (Fluka, Buchs, Switzerland), Pd/C (Aldrich, USA), циклогексен (Fluka, Buchs, Switzerland), NBS (Pierce, Rockford, II, USA), HCl⋅H-(L)-Lys(Z)-OBut (Reanal, Budapest, Hungary), дигликолевый ангидрид (Sigma, USA).
ЯМР спектры регистрируют на приборе Bruker Avance III (600 МГц для ядер 1Н, 150 МГц для ядер 13С).
Масс-спектры высокого разрешения регистрируют на приборе Thermo Finnigan Orbitrap Elite, метод ионизации - электрораспыление.
2. Получение трет-бутил бромацетата (соединение 6а) и трет-бутил хлорацетата (соединение 6b)
Трет-бутил бромацетат 6а получают согласно известному протоколу с незначительными изменениями [Wolfe et al, 2003] (Фиг. 4). 71 ммоль трет-бутанола (5,26 г,) и 77 ммоль триэтиламина (10,71 мл,) растворяют в дихлорметане (45 мл). Смесь охлаждают до 0°С, добавляют свежеперегнанный 84,06 ммоль хлорангидрида бромуксусной кислоты 7а (т.к. = 126-127°С при 760 мм.рт.ст.) (7,0 мл). Полученный раствор перемешивают в течение 45 мин. при 0°С и 3 часа при комнатной температуре. К реакционной смеси добавляют воду (60 мл), органический слой отделяют, водную фазу промывают дихлорметаном (двукратно по 15 мл), объединенные органические фазы промывают насыщенным раствором NaHCO3 (50 мл), высушивают над MgSO4, фильтруют, неорганический осадок на фильтре промывают дихлорметаном (20 мл), растворитель удаляют на роторном испарителе, остаток перегоняют. Выход целевого вещества на стадии составляет 3,7 г (26,7% от теоретического). Вещество представляет собой бесцветную жидкость с т.к.=75-76,5°С (при 37 мм.рт.ст.). Выход реакции при масштабировании на 21 грамм хлорангидрида меняется незначительно.
Трет-бутил хлорацетат 6b получают из 7b согласно опубликованному протоколу [Westheimer & Shookhoff, 1940]. Выход - 54%. Бесцветная жидкость, т.к.=78°С (52 мм. рт.ст).
3. Получение ди-трет-бутил 2,2'-((2-гидроксиэтш)азандиил)диацетата (соединение 5)
Метод А. Реакцию проводят согласно опубликованному протоколу [Williams & Rapoport] с незначительными модификациями (Фиг. 4). 88,1 ммоль соединения 6а растворяют в диметилформамиде (50 мл), к раствору добавляют KHCO3 (10 г, мелкоизмельченный), охлаждают до 0°С, добавляют 39,7 ммоль 2-этаноламина (2,4 мл). Перемешивают при 0°С в течение 30 мин, 22 ч при комнатной температуре. К смеси добавляют диэтиловый эфир (120 мл) и NaHCO3 (нас.) (100 мл). После растворения органический слой отделяют, промывают NaHCO3 (нас.) (100 мл), объединенные водные фазы экстрагируют абсолютным диэтиловым эфиром (80 мл), объединенную эфирную фазу промывают 100 мл NaCl (нас). Сушат над MgSO4. Органический раствор фильтруют и упаривают на роторном испарителе до консистенции вязкого масла. Выход на стадии составляет 14,43 г, Rf=0,10 (2:1, Hept/EtOAc). Продукт имеет вид масла. Продукт используют для получения вещества 4 без очистки.
Метод Б. Соединение 5 может быть получено из 6b согласно вышеописанной методике. Время реакции 72 часа.
4. Получение ди-трет-бутил 2,2'-((2-бромоэтил)азандиил)диацетата (соединение 4)
Полученное на предыдущей стадии вещество 5 (14,4 г) растворяют в дихлорметане (120 мл), добавляют 50,9 ммоль трифенилфосфина (13,36 г) (Фиг. 4). Раствор охлаждают до 0°С и прибавляют 50,7 ммоль N-бромосукцинимида (9,03 г) порциями в течении 5 мин. Реакционную смесь перемешивают в течении 1,5 часа при 0°С. Реакционную смесь упаривают на роторном испарителе, к остатку темно-красного цвета добавляют диэтиловый эфир (100 мл). Эфирную фазу декантируют с образовавшегося осадка, который промывают эфиром (50 мл), эфирные фазы объединяют. Раствор в эфире выдерживают при 4°С в течение 16 часов. Образовавшийся осадок отфильтровывают, промывают эфиром (20 мл), эфирные фазы объединяют, к полученной смеси добавляют бензол (20 мл) и гептан до начала помутнения раствора. Раствор выдерживают при -8°С в течение 16 часов. Осадок отфильтровывают, промывают 20 мл смеси толуол/гептан в соотношении 1:1, смесь упаривают. Получают масло в количестве 15,35 г, представляющее собой смесь веществ. По данным ТСХ: (5:1, гептан: EtOAc); Rf=0,12 (5), 0,57 (4), 0,84 (трифенилфосфин). ТСХ (2:1, гептан: EtOAc); Rf=0,10 (5), 0,6 (4).
Для 1Н ЯМР анализа аликвоту синтезированного продукта очищают хроматографически. Очистку методом флэш-хроматографии проводят на силикагеле., используя в качестве элюента смесь этилацетат-гептан 1:10. Определяют спектры 1Н ЯМР и масс-спектры соединения 4, контролируя их совпадение с литературными данными [Williams, & Rapoport, 1993].
5. Получение (S)-трет-бутил 10-(2-(бис(2-(трет-бутокси)-2-оксоэтил)амино)этил)-13-(2-(трет-бутокси)-2-оксоэтил)-9-(трет-бутоксикарбонил)-3-оксо-1-фенил-2-окс-4,10,13-триазапентадекан-15-оата (соединение 3)
Готовят фосфатный буферный раствор, смешивая 0,1 моль KH2PO4 (13,6 г) и 0,1 моль КОН (5,6 г), растворяют смесь в H2O (40 мл). Контролируют рН полученного раствора: должен быть 7,8±0,1. Добавляют КОН (2 М), доводя рН до 8,1, доводят объем до 50 мл (рН=8,04).
0,77 ммоль HCl⋅H-(L)-Lys(Z)-OBut (288 мг) и 1,9 ммоль вещества 4 (0,67 г) растворяют в ацетонитриле (3 мл), добавляют фосфатный буфер (3 мл), смесь перемешивают 2 часа, водный слой удаляют, прибавляют фосфатный буфер (3 мл) (Фиг. 4). Смесь перемешивают в течение 48 часов. Органический слой отделяют, водный слой промывают ацетонитрилом, органические фазы объединяют, растворитель упаривают на роторном испарителе. Маслянистый остаток растворяют в дихлорметане (15 мл), промывают водой (2×5 мл), сушат над сульфатом магния. Неорганические соли отфильтровывают, промывают дихлорметаном, растворитель упаривают на роторном испарителе. Выход на стадии - 615 мг, внешний вид - масло, Rf=0,76 (МеОН). Соединение 3 очищают методом флэш-хроматографии на силикагеле, используя в качестве элюента используют этилацетат-гептан 1:4. 1Н ЯМР и масс-спектры соединения 3 находятся в соответствии с литературными данными [Williams et al., 1993; Anelli et al., 1999].
6. Получение (S) -тетра-трет-бутил 2,2',2'',2'''-((((6-амино-1-(трет-бутокси)-1-оксогексан-2-ил)азандиил)бис(этан-2,1-диил)бис)(азантриил))тетраацетата (соединение 2)
В колбе на 100 мл суспендируют Pd/C (350 мг) в изопропаноле (15 мл), добавляют 20 ммоль циклогексена (2 мл,), полученную смесь перемешивают при кипячении 7 минут, охлаждают до 30°С (Фиг. 4). К реакционной смеси добавляют 0,7 ммоль соединения 3 (615 мг,) в виде раствора в изопропаноле (6 мл) и 10 ммоль циклогексен в (1 мл). Смесь перемешивают 10 мин при кипячении, охлаждают до комнатной температуры, фильтруют, катализатор промывают этанолом (10 мл), объединенные растворы упаривают на роторном испарителе, к маслянистому осадку добавляют толуол (15 мл), смесь упаривают. Выход на стадии - 485 мг, внешний вид - масло, Rf=0,2 (МеОН). 1Н ЯМР и масс-спектры соединения 2 должны соответствовать опубликованным данным [Anelli et al, 1999].
7. Получение (S)-9-(2-(бис(2-(трет-бутокси)-2-оксоэтил)амино)этил)-6-(2-(трет-бутокси)-2-оксоэтил)-10-(трет-бутоксикарбонил)-2,2-диметил-4,16-диоксо-3,18-диокса-6,9,15-триазикозан-20-олевой кислоты (1)
Метод А. Полученное на предыдущей стадии соединение 2 растворяют в 9 мл ацетонитрила. К 5 мл полученного раствора добавляют 0,32 ммоль ангидрида дигликолевой кислоты (37,78 мг). Смесь перемешивают до исчезновения исходного соединения и упаривают на роторном испарителе (Фиг. 4). Полученную смесь разделяют методом флэш-хроматографии на силикагеле с использованием в качестве элюента смеси «хлороформ-метанол-вода» в соотношении 60: 11:1. Выход на данной стадии составляет около 160 мг. Rf=0,37 (хлороформ-метанол 4:1).
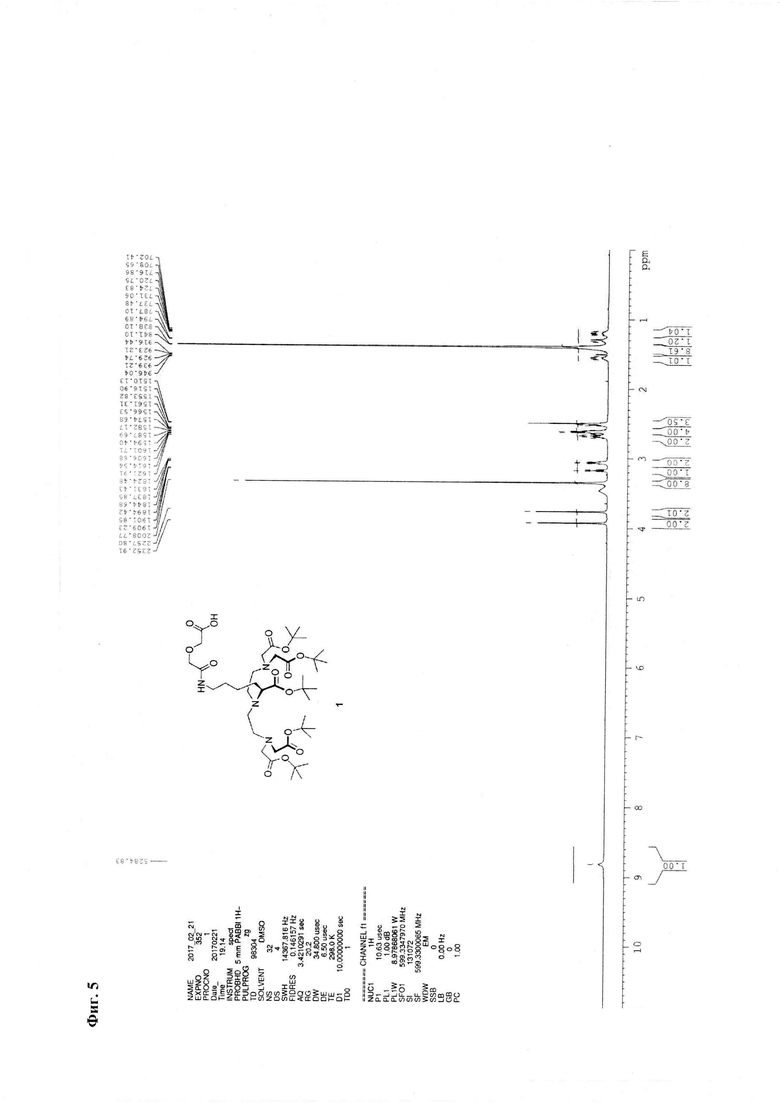
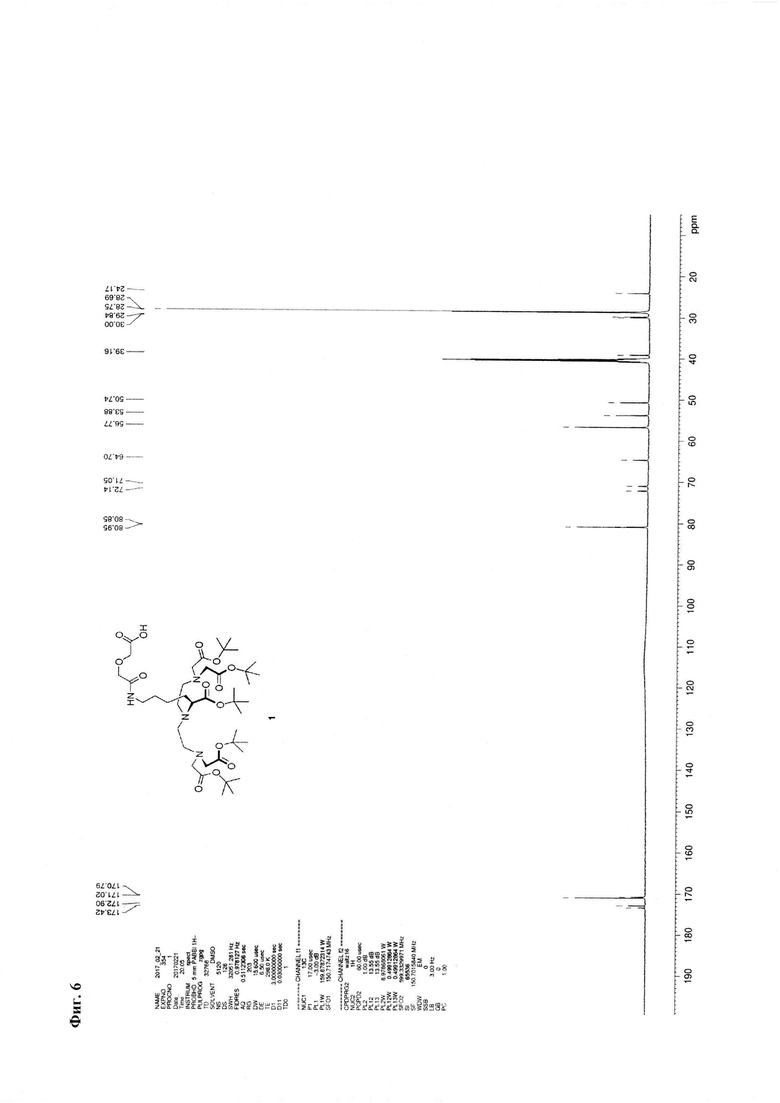
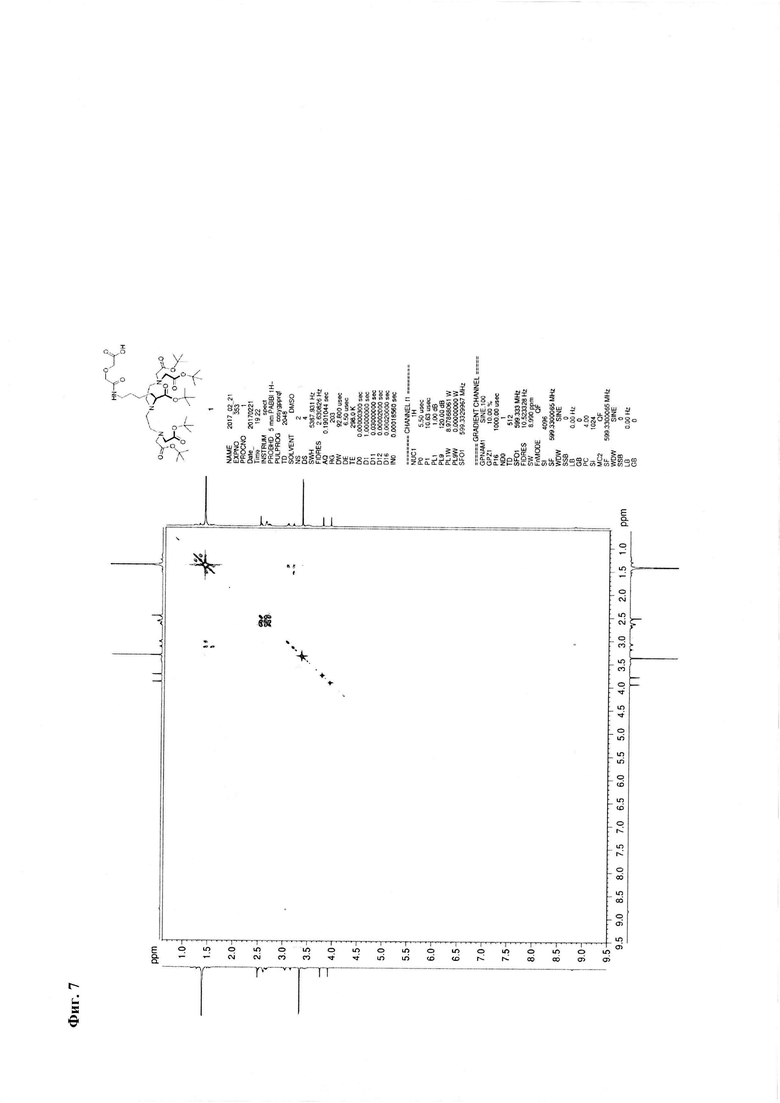
Метод В. Катализатор Pd/C (2,5 г) суспендируют в 20 мл тетрагидрофурана. К суспензии добавляют 30 ммоль циклогексена (3 мл), перемешивали при температуре кипения в течение 15 минут, охлаждают до 30°С. К реакционной смеси добавляют 3,24 ммоль соединения 2 (2,85 г) и 3,24 ммоль ангидрида дигликолевой кислоты (376 мг) в виде раствора в тетрагидрофуране (10 мл). Реакционную смесь перемешивают при кипении в течение 30 мин и далее при комнатной температуре в течение 2,5 часов. Образовавшийся осадок отделяют на фильтре Celite, промывают абсолютным этанолом. Объединенный раствор в смеси органических растворителей упаривают. Соединение 7 очищают флэш-хроматографией на силикагеле с использованием в качестве элюента смеси «хлороформ-метанол-вода» 60:11:1. Выход на данной стадии - 2,0 г. Основное вещество имеет вид масла. Для 1: Rƒ=0,3 (силикагель, 60:11:1 об/об/об CHCl3/МеОН/H2O); 1Н ЯМР (ДМСО-d6, 600 МГц) δ 1,14-1,25 (m, 1Н, CHγLys), 1,25-1,35 (m, 1Н, CHγLys), 1,35-1,48 (m, 48Н, tBuO, CHβLys, CHδLys), 1,52-1,59 (m, 1H, CHβLys), 2,47-2,56 (m, 3,5H, CH-Lys, (CHD2)2SO), 2,56-2,64 (m, 4H, NCH2), 2,64-2,72 (m, 2H, CH-Lys), 3,06 (dd, J=6,65, 13,01 Гц, 2H, CHεLys), 3.17 (ψt, J=7,21 Гц, 1H, CHαLys), 3,35 (s, 8H, CH2CO2tBu), 3,77 (s, 2H, CH2CO2H), 3,93 (s, 2H, NHC(O)CH2), 8,82 (br.s., 1H, NHC(O)); 13C NMR (DMSO-d6, 600 МГц) 173,42, 172,90, 171,02, 170,79, 80.95, 80,85, 72,14, 71,05, 64,70, 56,77, 53,88, 50,74, 39,16, 30,00, 29,84, 28,75, 28,69, 24,17. HRMS (+ESI-orbitrap) m/z для C42H77N4O14 [M+H+] вычислено: 861,5431, экспериментально найдено: 861,5436 (Фиг. 5 - Фиг. 8).
8. Получение соединения DTPA-Lys-дигликолат-Ahx-Abu-TGIRIS-Abu-NH2
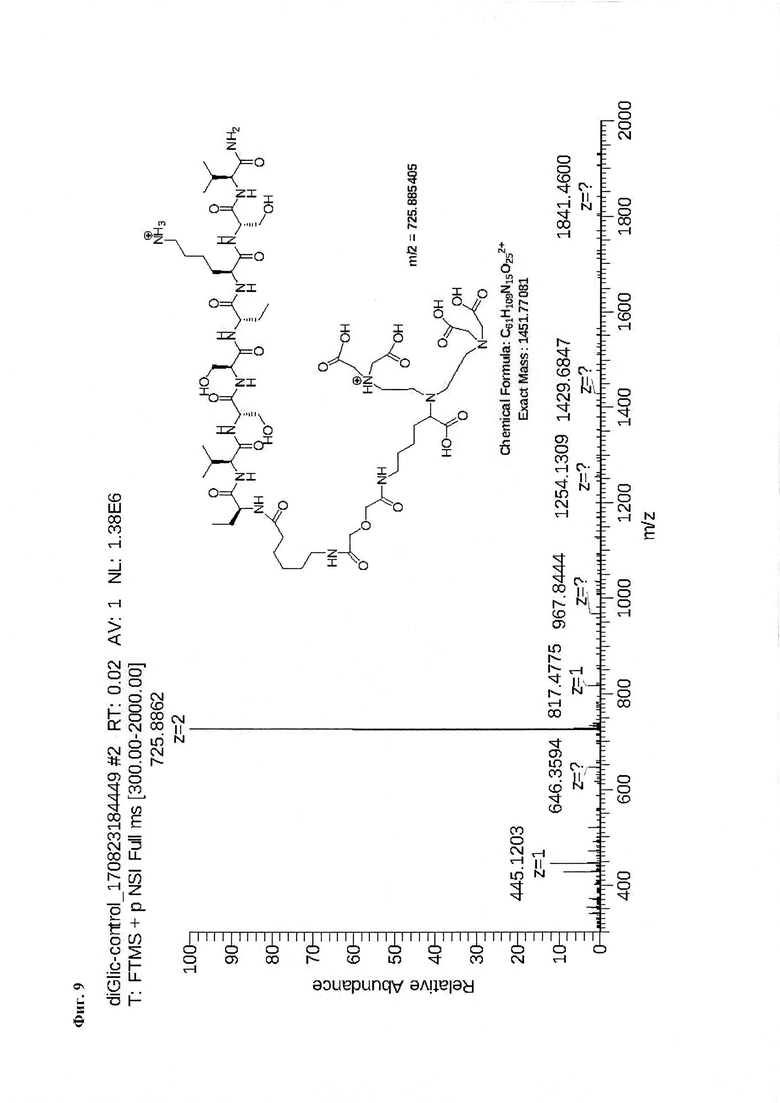
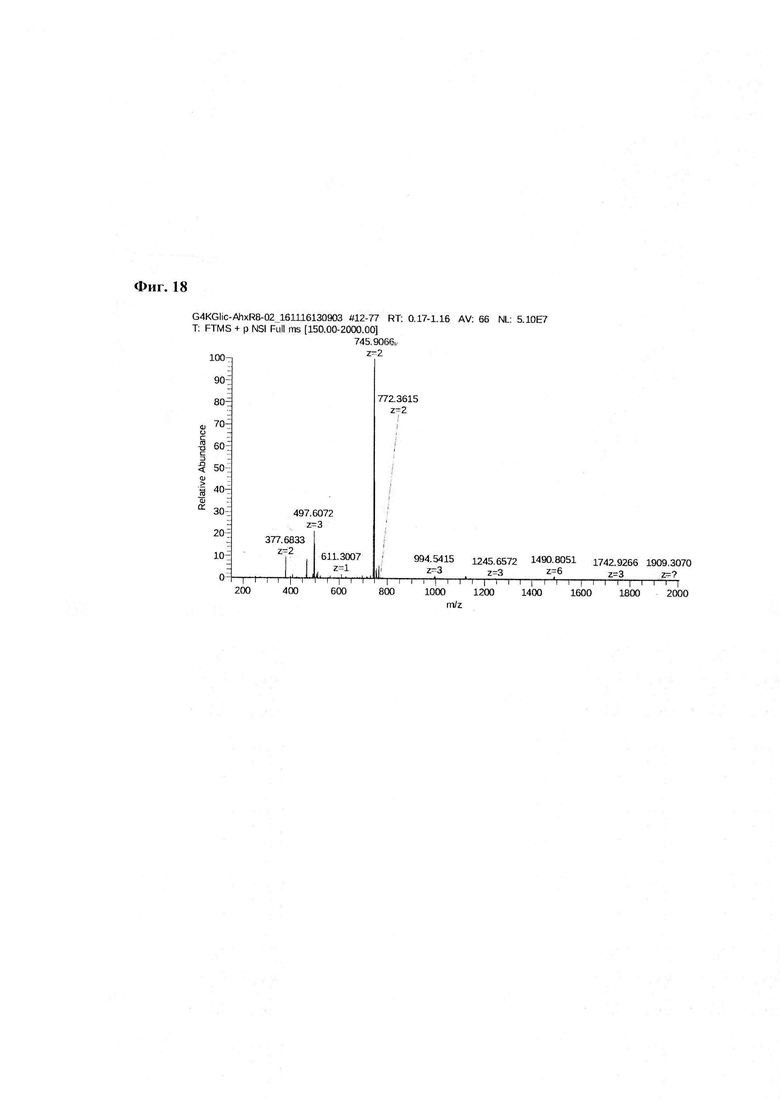
Для проведения синтеза используют пептид P1 - Ahx-Abu-TGIRIS-Abu или пептид Р0 - Ahx-Abu-VSS-Abu-KSV, полученные в результате твердофазного синтеза, и через остаток α-аминомасляной кислоты присоединенный аминогруппу МВНА-модифицированного полистирола (Фиг. 2). К подложке массой 1,53 г, содержащей ~1,4 ммоль пептида Р1 или Р0 добавляют конденсирующую смесь в виде 4,2 ммоль раствора соединения 1 (3 эквивалента) в диметилформамиде, которое предварительно активируют добавлением 12,2 ммоль (2,9 эквивалента) реагента сочетания O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (TBTU - 2,9 эквивалента) в присутствии 12,6 ммоль 1-гидроксибензотриазола (HOBt) (3 эквивалента). Реакцию конденсации проводят в течение 18 часов при перемешивании при комнатной температуре. Степень конверсии в реакции ацилирования аминогруппы контролируют качественно с использованием нингидринового теста и количественно с помощью пикриновой кислоты. Удаление защитных групп и снятие пептида с подложки осуществляют с помощью безводной фтористоводородной кислоты (10 мл на 1 грамм пептидилполимера) с добавкой скэвенджера м-крезол (1 мл на 1 грамм пептидилполимера) в течение 1 часа при 0°С. По окончании реакции фтористый водород упаривают в вакууме, к остатку добавляют диэтиловый эфир (50 мл на 1 грамм пептидилполимера) и полученную суспензию выдерживают при -34°С в течение 14 часов. Эфир отфильтровывают через стеклянный пористый фильтр с размером пор 40 микрон. Осадок на фильтре промывают эфиром (трижды по 50 мл), а затем высушивают. Пептид отделяют от полимера посредством растворения в 1М водной уксусной кислоте. Полученный раствор лиофилизуют. Продукт очищают с помощью обращеннофазной ВЭЖХ в системе ацетонитрил - вода - 0,1% ТФУ (Фиг. 9, Фиг. 18).
9. Получение комплексной соли Gd3+ с DTPA-Lys-дигликолат-Ahx-Abu-TGIRIS-Abu-NH2
Синтез комплексной соли Gd3+ с DTPA-Lys-дигликолат-Ahx-Abu-TGIRIS-Abu-NH2 (Фиг. 2) осуществляют путем растворения субстрата в воде при перемешивании. Значение рН полученного водного раствора доводят до 6,5-7,5 с помощью 5% водного раствора аммиака или другого органического основания со значением константы основности 9-12. К полученному раствору прибавляют порциями водный раствор соли Gd3+ (0,95 эквивалента) в диапазоне концентраций 1-6%, контролируя значение рН реакционной смеси в пределах значения 6,5-7,5, внося при необходимости необходимый объем 5% водного раствора основания. После добавления всего раствора соли Gd3+ реакционную смесь перемешивают дополнительно 3 часа. Контроль за степенью полноты комплексообразования осуществляют масс-спектрометрически (Фиг 10-17; Фиг. 19-24). Полученный раствор лиофилизируют.
Описание графических изображений
Фиг. 1. Общая схема структуры заявляемого конъюгата координационного комплекса Gd3+-DTPA с пептидом. Обозначения: Ahx - аминогексановая кислота, CTG - С-концевая группа пептида.
Фиг 2. Общая схема синтеза конъюгатов состава «Gd3+-DTPA-Lys-дигликолат-Ahx-Abu-TGIRIS-Abu-NH2».
Фиг 3. Структура соединения 1, (S)-9-(2-(бис(2-(трет-бутокси)-2-оксоэтил)амино)этил)-6-(2-(трет-бутокси)-2-оксоэтил)-10-(трет-бутоксикарбонил)-2,2-диметил-4,16-диоксо-3,18-диокса-6,9,15-триазикозан-20-олевая кислота.
Фиг. 4. Общая схема синтеза соединения 1, (S)-9-(2-(бис(2-(трет-бутокси)-2-оксоэтил)амино)этил)-6-(2-(трет-бутокси)-2-оксоэтил)-10-(трет-бутоксикарбонил)-2,2-диметил-4,16-диоксо-3,18-диокса-6,9,15-триазикозан-20-олевая кислота.
Фиг. 5. 1Н ЯМР-спектр соединения 1.
Фиг. 6. 13С ЯМР-спектр соединения 1.
Фиг. 7. 1Н-1Н - корреляционный ЯМР-спектр соединения 1.
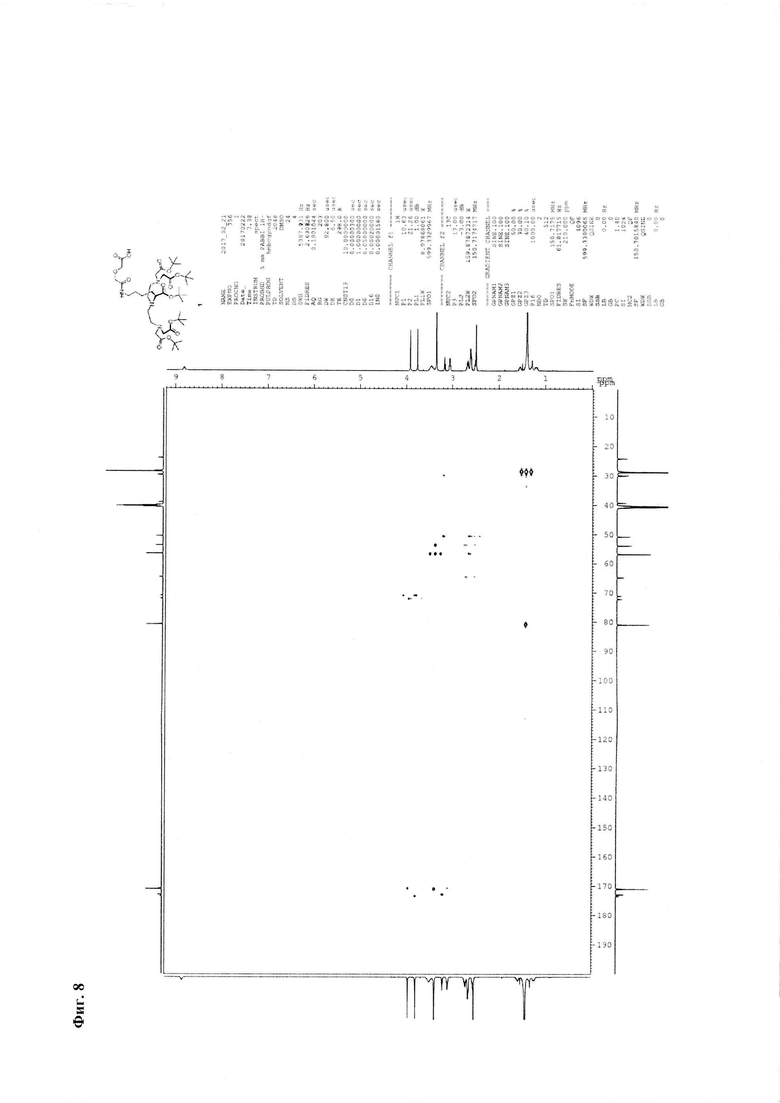
Фиг. 8. 1H-13C корреляционный НМВС ЯМР-спектр соединения 1.
Фиг. 9. Масс-спектр соединения на основе пептида Р0 «DTPA-дигликолат-Ahx-Abu-VSS-Abu-KSV».
Фиг. 10. Масс-спектр соединения на основе пептида Р0 «Gd3+-DTPA-дигликолат-Ahx-Abu-VSS-Abu-KSV» в области 500-2000 а.е.м.
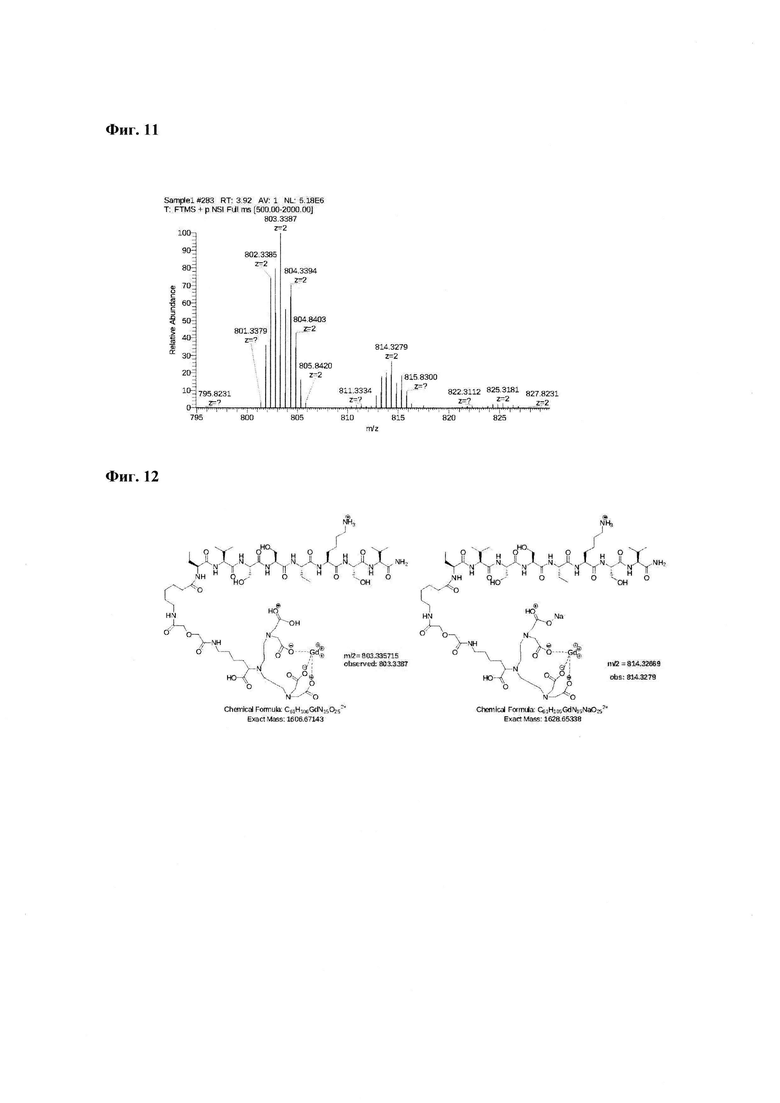
Фиг. 11. Масс-спектр соединения пептида Р0 «Gd3+-DTPA-дигликолат-Ahx-Abu-VSS-Abu-KSV» 795-830 а.е.м. Видно изотопное распределение, характерное для соединений гадолиния.
Фиг. 12. Интерпретация сигналов в масс-спектре соединения пептида Р0 «Gd3+-DTPA-дигликолат-Ahx-Abu-VSS-Abu-KSV»в области m/z=803,3387 и 814,3270.
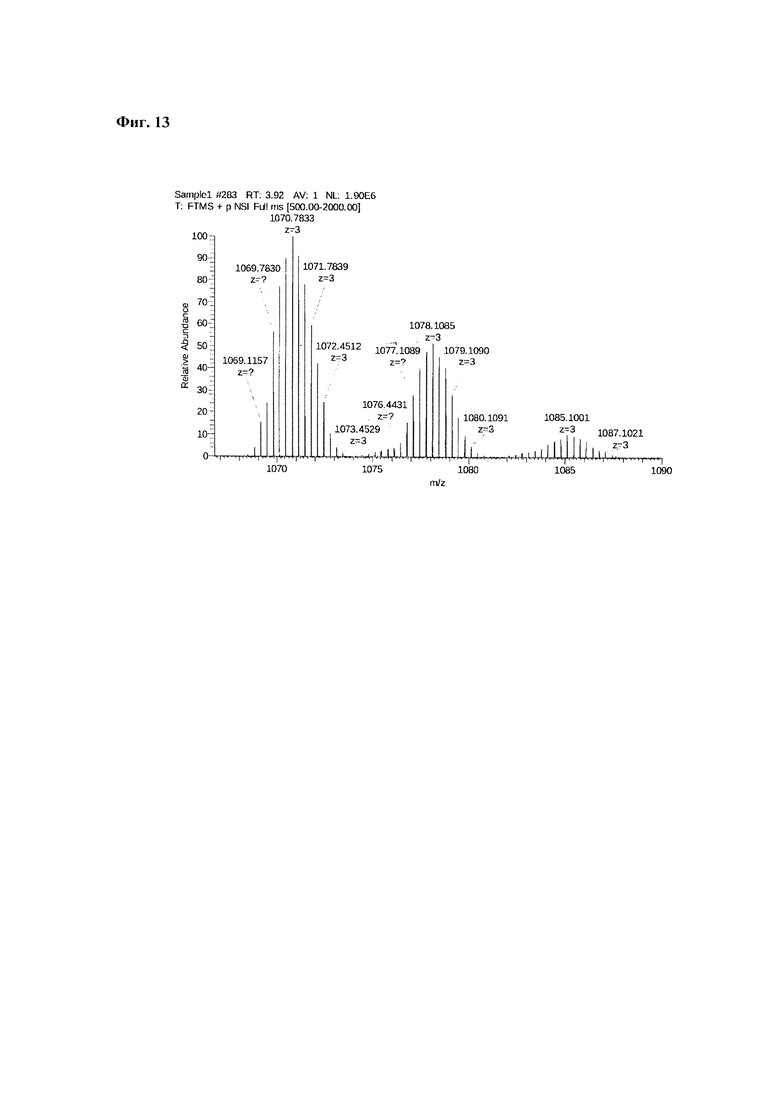
Фиг. 13. Масс-спектр соединения пептида Р0 «Gd3+-DTPA-дигликолат-Ahx-Abu-VSS-Abu-KSV» в области 1067-1090 а.е.м.
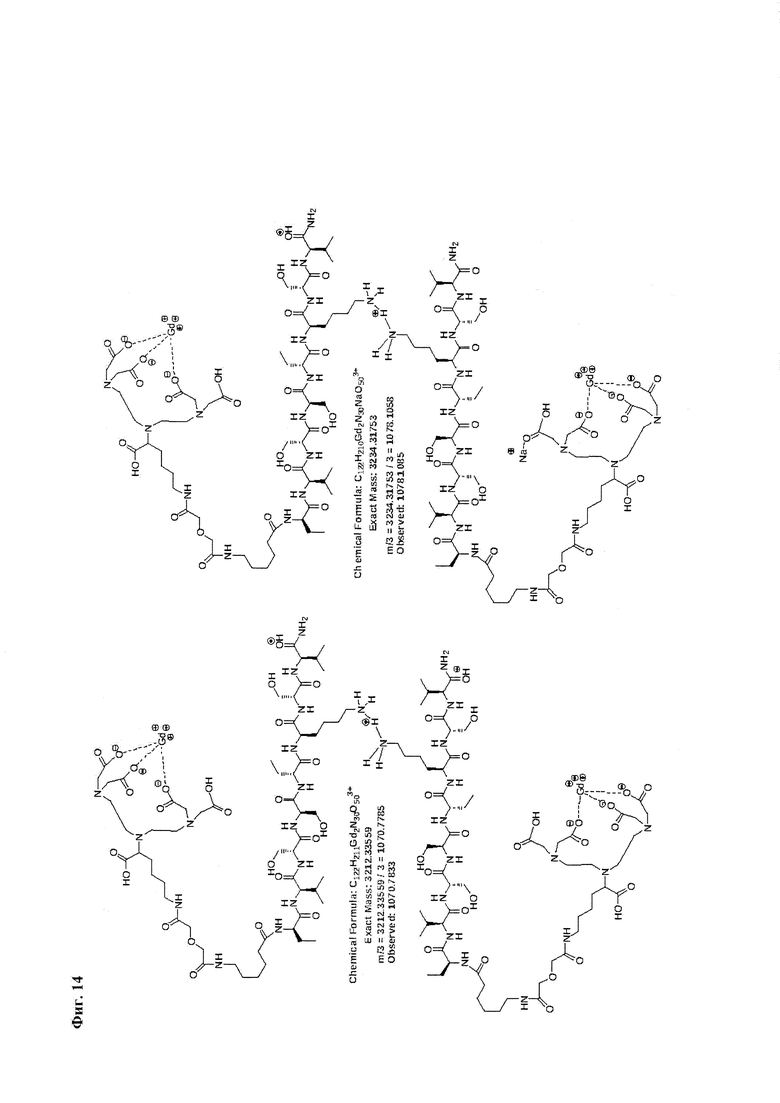
Фиг. 14. Интерпретация сигналов в масс-спектре соединения пептида Р0 «Gd3+-DTPA-дигликолат-Ahx-Abu-VSS-Abu-KSV» при m/z=1070,78 и 1078,11.
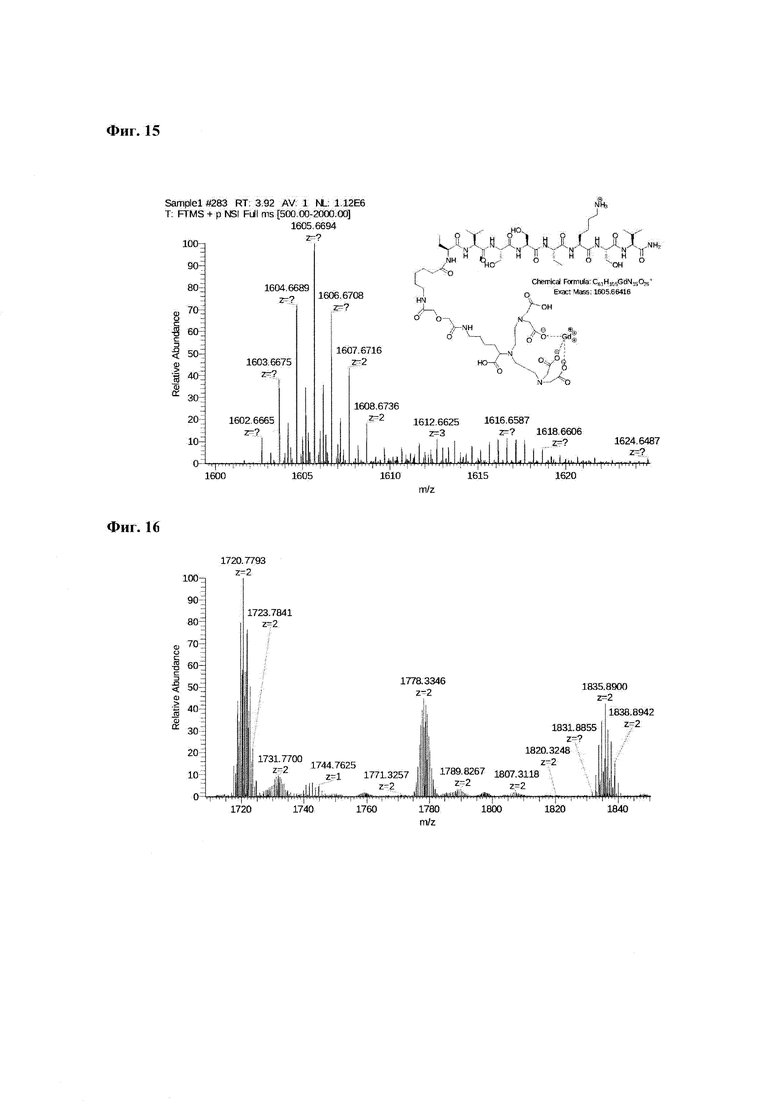
Фиг. 15. Масс-спектр соединения пептида Р0 «Gd3+-DTPA-дигликолат-Ahx-Abu-VSS-Abu-KSV» в области 1600-1625 а.е.м и интерпретация сигнала при m/z=1066,94.
Фиг. 16. Масс-спектр соединения пептида Р0 «Gd3+-DTPA-дигликолат-Ahx-Abu-VSS-Abu-KSV» в области 1718-1842 а.е.м.
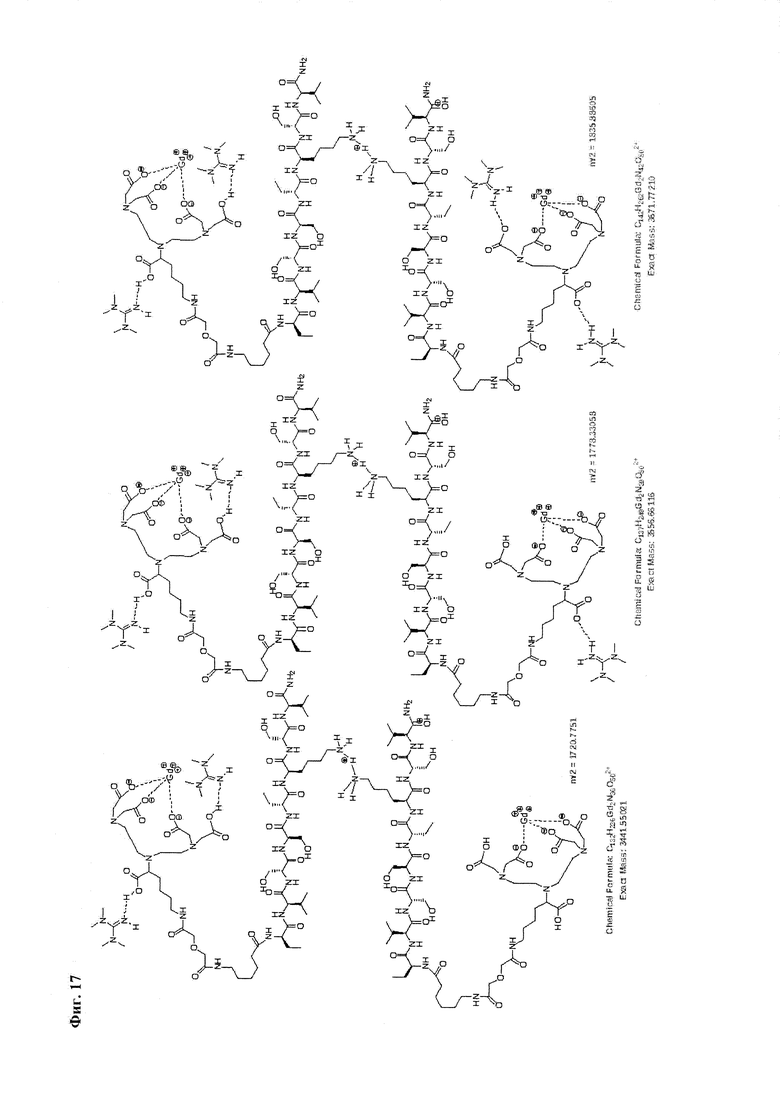
Фиг. 17. Интерпретация сигналов в масс-спектре соединения пептида Р0 «Gd3+-DTPA-дигликолат-Ahx-Abu-VSS-Abu-KSV» при m/z=1720,77, 1778,33 и 1835,89. Наблюдаются аддукты с противоионом внешней координационной сферы.
Фиг. 18. Масс-спектр соединения пептида P1 - DTPA-Abu-TGIRIS-Abu в области 150-2000 а.е.м.
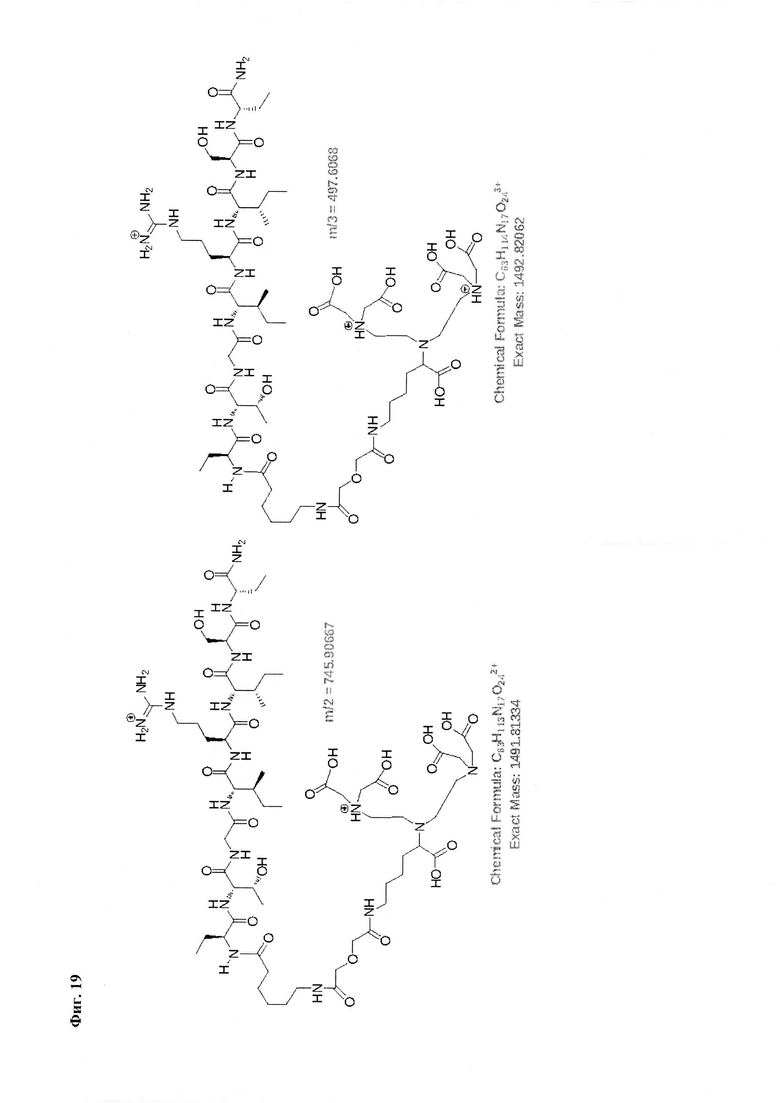
Фиг. 19. Интерпретация сигналов в масс-спектре комплексной соли Gd3+ с DTPA-Lys-дигликолат-Ahx-Abu-TGIRIS-Abu-NH2 при m/z=497,60 и 745,90.
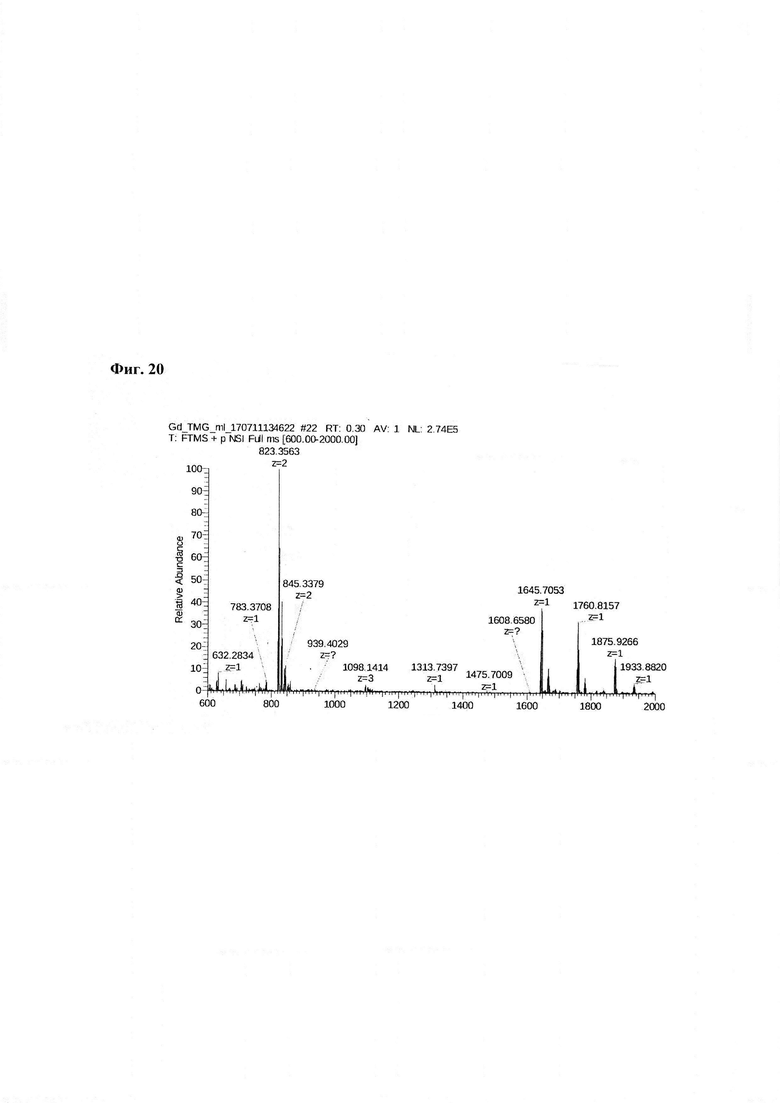
Фиг. 20. Масс-спектр комплексной соли Gd3+ с DTPA-Lys-дигликолат-Ahx-Abu-TGIRIS-Abu-NH2 в области 600-2000 а.е.м.
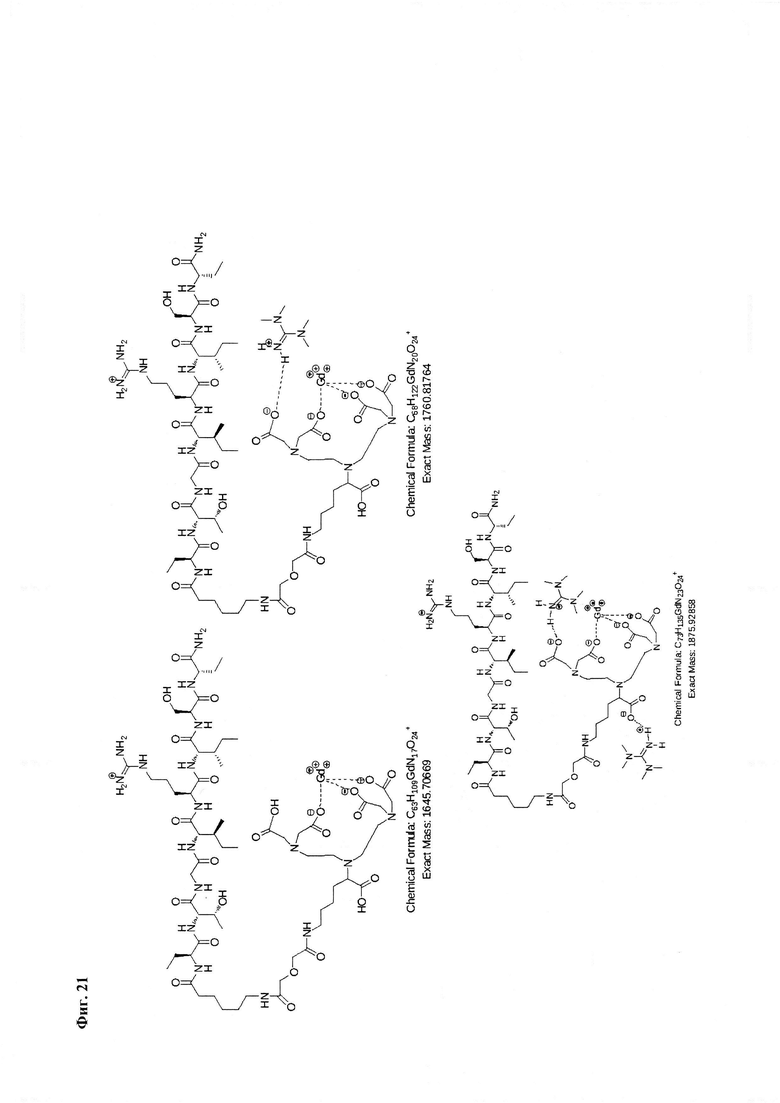
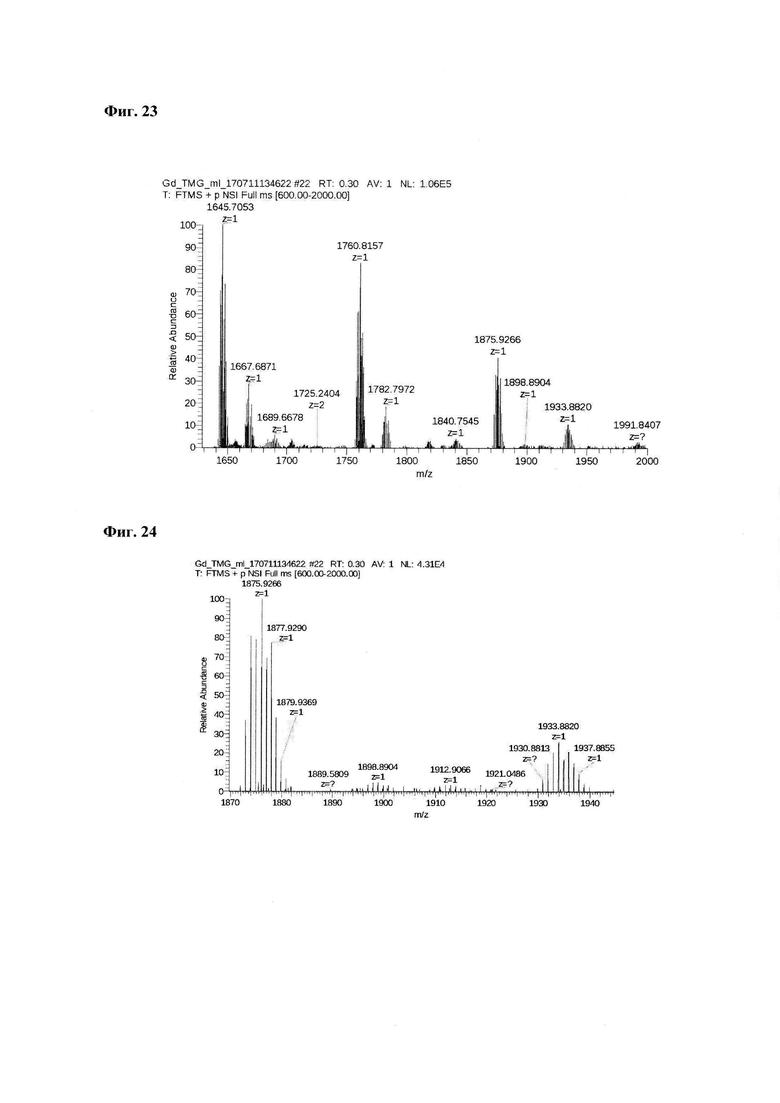
Фиг. 21. Интерпретация сигналов в масс-спектре соединения пептида P1 «Gd3+-DTPA-Abu-TGIRIS-Abu» при m/z=1645,70, 1760,81 и 1876,92. Наблюдаются аддукты с противоионами внешней координационной сферы.
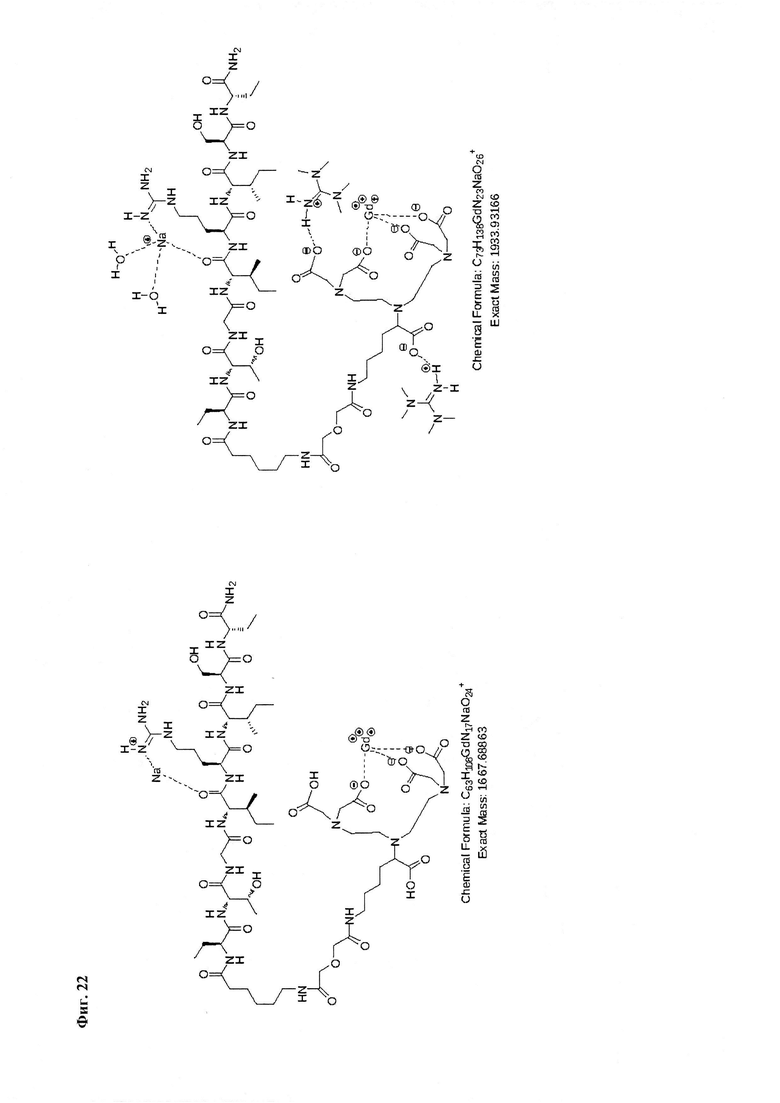
Фиг. 22. Интерпретация сигналов в масс-спектре соединения пептида P1 «Gd3+-DTPA-Abu-TGIRIS-Abu» при m/z=1667,68 и 1933,93. Наблюдаются аддукты с противоионами внешней координационной сферы и катионами натрия из стекла капилляров ионизации.
Фиг. 23. Масс-спектр соединения пептида P1 «Gd3+-DTPA-Abu-TGIRIS-Abu» в области 1630-2000 а.е.м.
Фиг. 24. Масс-спектр комплексной соли Gd3+ с DTPA-Lys-дигликолат-Ahx-Abu-TGIRIS-Abu-NH2 в области 1630-2000 а.е.м. Видно изотопное распределение, характерное для соединений гадолиния.
Использованные источники
1. Позднякова Н.В., Григорьева Е.Ю., Шевелев А.Б. Мицеллярные композиции на основе функционализированных α-фетопротеином амфифильных блок-сополимеров, содержащих гадолиний и куркумин. Вестник Российского государственного медицинского университета. 2016. №3. С. 30-37.
2. Anelli P.L., Fedeli F., Gazzotti О., Lattuada L., G Lux., Rebasti F., Bioconjugate Chem. 1999, 10, 137-140.
3. Anelli P.L., Fedeli F., Gazzotti O., Lattuada L., Lux G. and Rebasti F. 1-Glutamic Acid and 1-Lysine as Useful Building Blocks for the Preparation of Bifunctional DTPA-like Ligands. Bioconjugate Chem., 1999, 10(1), 137-140.
4. Brechbiel M.W., Gansow O.A., Bioconjugate Chem. 1991, 2, 187-194.
5. Caravan P., Ellison J.J., McMurry T.J., Lauffer R.B., Chem. Rev. 1999, 99, 2293-2352.
6. Desreux J.F., Jacques V., Top. Curr. Chem. 2002, 221, 123-164.
7. Dinger SC, Fridjhon P, Rubin DM. Thermal Excitation of Gadolinium-Based Contrast Agents Using Spin Resonance. PLoS One., 2016, 11 (6): e0158194.
8. Edwards W.B., Fields C.G., Anderson C.J., Pajeau T.S., Welch M.J., Fields G.B., J. Med. Chem. 1994, 37, 3749-3757.
9. Fichna J., Janecka A., Bioconjugate Chem. 2003, 14, 3-17.
10. Fossheim R., Dugstad H., Dahl S.G., J. Med. Chem. 1991, 34, 819-826.
11. Goischke H.K. MRI with gadolinium-based contrast agents: practical help to ensure patient safety. J Am CollRadiol., 2016, pii: S1546-1440(16), 30315-5.
12. Hnatowich D.J., Layne W.W., Childs R.L., Lanteigne D., Davis M.A., Griffin T.W., Doherty P.W., Science 1983, 220, 613-615.
13. Langereis S., Dirksen A., De Waal B.F.M., Van Gerden M.H.P.,. De Lussanet Q.G, Hackeng T.M., Meijer E.W. Eur. J. Org. Chem. 2005, 2534-2538.
14. Maisano F., Gozzini L., De Haen C, Chem Bioconjugate. 1992, 3, 212-217.
15. Merbach A.E., E., The Chemistry of Contrast Agents, in: Medical Magnetic Resonance Imaging, John Wiley & Sons: New York, 2001.
16. Sherry A.D., Cacheris W.P., Kuan K.T., Magn. Reson. Med 1988, 8, 180-190.
17. Westheimer F.H., Shookhoff M.W. The Electrostatic Influence of Substituents on Reactions Rates. I. J. Am. Chem. Soc, 1940, 62(2), 269-275.
18. Williams, M.A., Rapoport H. Synthesis of enantiomerically pure diethylenetriaminepentaacetic acid analogs. L-Phenylalanine as the educt for substitution at the central acetic acid. J. Org. Chem. 1993, 58(5), 1151-1158.
19. Wolfe S., Akuche Ch., Ro S., Wilson M.-C, Kim C.-K., Shi Zh. 5-Hydroxy[1,2]oxazinan-3-ones as potential carbapenem and D-ala-D-ala surrogates. Canadian Journal of Chemistry, 2003, 81(8): 915-936.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПЛЕКСЫ ТЕХНЕЦИЯ И РЕНИЯ С БИС(ГЕТЕРОАРИЛАМИ) И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2009 |
|
RU2539584C2 |
| ТЕХНЕЦИЙ- И РЕНИЙ-БИС(ГЕТЕРОАРИЛЬНЫЕ) КОМПЛЕКСЫ И МЕТОДЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ИНГИБИРОВАНИЯ PSMA | 2009 |
|
RU2532912C2 |
| КОНЪЮГАТЫ ЦИТОТОКСИЧЕСКИХ СРЕДСТВ С ПЕПТИДАМИ | 2007 |
|
RU2457218C2 |
| ПЕПТИДНЫЕ ВЕКТОРЫ | 2004 |
|
RU2361876C2 |
| ДВУГЛАВЫЙ ИНГИБИТОР ПРОТЕАЗЫ | 2018 |
|
RU2780491C2 |
| АГОНИСТЫ РЕЦЕПТОРОВ НЕЙРОМЕДИНА В И СОМАТОСТАТИНА | 2000 |
|
RU2263680C2 |
| СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ MASP, И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2840441C2 |
| КОНЪЮГАТ, ВКЛЮЧАЮЩИЙ ЛИГАНД, СПЕЙСЕР, ПЕПТИДНЫЙ ЛИНКЕР И БИОМОЛЕКУЛУ | 2020 |
|
RU2807081C2 |
| ПРОИЗВОДНЫЕ ДЕКАПЕПТИДОВ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2124023C1 |
| СОЕДИНЕНИЯ ДЛЯ НАЦЕЛЕННОЙ ДОСТАВКИ ЛЕКАРСТВЕННОГО СРЕДСТВА И УСИЛЕНИЯ АКТИВНОСТИ siPHK | 2012 |
|
RU2769872C2 |

Изобретение относится к области медицинской химии, иммунологии и нейробиологии и направлено на разработку нового средства для лечения рассеянного склероза, относящегося к классу биологически активного вещества – конъюгата иммуносупрессорного пептида Abu-TGIRIS-Abu с хелатирующим агентом - диэтилентриаминопентауксусной кислотой (DTPA), находящейся в форме координационного комплекса с ионом Gd3+. В рамках настоящего изобретения был предложен способ получения комплексной соли Gd3+ с DTPA-Lys-дигликолат-Ahx-Abu-TGIRIS-Abu-NH2, где DTPA обозначает диэтилентриаминопентауксусную кислоту, Ahx обозначает аминогексановую кислоту, Abu обозначает α-аминомасляную кислоту, включающий следующие стадии: (1) к пептиду Abu-TGIRIS-Abu, синтезируемому твердофазным методом с использованием в качестве носителя МВНА-полистирола, не снимая с полимерной подложки, присоединяют аминогексановую кислоту, с получением пептида Ahx-Abu-TGIRIS-Abu-NH2, присоединенного через остаток α-аминомасляной кислоты к аминогруппе МВНА-модифицированного полистирола, (2) получают (S)-9-(2-(бис(2-(трет-бутокси)-2-оксоэтил)амино)этил)-6-(2-(трет-бутокси)-2-оксоэтил)-10-(трет-бутоксикарбонил)-2,2-диметил-4,16-диоксо-3,18-диокса-6,9,15-триазикозан-20-олевую кислоту, (3) к пептиду Ahx-Abu-TGIRIS-Abu, присоединенному к аминогруппе МВНА-модифицированного полистирола через остаток α-аминомасляной кислоты, добавляют конденсирующую смесь в виде раствора (S)-9-(2-(бис(2-(трет-бутокси)-2-оксоэтил)амино)этил)-6-(2-(трет-бутокси)-2-оксоэтил)-10-(трет-бутоксикарбонил)-2,2-диметил-4,16-диоксо-3,18-диокса-6,9,15-триазикозан-20-олевой кислоты в диметилформамиде, (4) проводят удаление защитных групп и снятие пептида с подложки, (5) получают комплексную соль Gd3+ с DTPA-Lys-дигликолат-Ahx-Abu-TGIRIS-Abu-NH2 путем реакции модифицированного пептида с солью Gd3+. Конъюгат может быть использован в качестве средства визуализации лимфоцитов, потенциально принимающих участие в аутоиммунном поражении миелиновых оболочек ЦНС in vivo методом парамагнитного резонанса или для избирательного уничтожения таких лимфоцитов методом рентген-индуцированной термоабляции in vitro и in vivo. Способ позволяет получать конъюгат пептида Abu-TGIRIS-Abu с DTPA, сочлененный через длинный умеренно гидрофильный гибкий линкер. Структура линкера исключает риск нарушения связывания пептидного лиганда с поверхностным рецептором лимфоцитов за счет стерических затруднений, связанных с большим размером и жесткостью координационного комплекса DTPA c ионами Gd3+. 24 ил., 1 пр.
Способ получения комплексной соли Gd3+ с DTPA-Lys-дигликолат-Ahx-Abu-TGIRIS-Abu-NH2, где DTPA обозначает диэтилентриаминопентауксусную кислоту, Ahx обозначает аминогексановую кислоту, Abu обозначает α-аминомасляную кислоту, включающий следующие стадии: (1) к пептиду Abu-TGIRIS-Abu, синтезируемому твердофазным методом с использованием в качестве носителя МВНА-полистирола, не снимая с полимерной подложки, присоединяют аминогексановую кислоту (которую предварительно активируют добавлением 2,9 эквивалентов реагента сочетания O-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат (TBTU) в присутствии 12,6 ммоль скэвенджера 1-гидроксибензотриазол (HOBt) (3 эквивалента)) с получением пептида Ahx-Abu-TGIRIS-Abu-NH2, присоединенного через остаток α-аминомасляной кислоты к аминогруппе МВНА-модифицированного полистирола; (2) получают (S)-9-(2-(бис(2-(трет-бутокси)-2-оксоэтил)амино)этил)-6-(2-(трет-бутокси)-2-оксоэтил)-10-(трет-бутоксикарбонил)-2,2-диметил-4,16-диоксо-3,18-диокса-6,9,15-триазикозан-20-олевую кислоту по следующей схеме: (2а) трет-бутил бромацетат получают из трет-бутанола и хлорангидрида бромуксусной кислоты путем инкубации при 0°С в присутствии триэтиламина в дихлорметане; (2б) ди-трет-бутил 2,2'-((2-гидроксиэтил)азандиил)диацетат получают из трет-бутил бромацетата путем инкубации при комнатной температуре с KHCO3 и 2-этаноламином с последующей экстракцией диэтиловым эфиром, насыщенным NaHCO3; (2в) ди-трет-бутил 2,2'-((2-бромоэтил)азандиил)диацетат получают из ди-трет-бутил 2,2'-((2-гидроксиэтил)азандиил)диацетата и N-бромосукцинимида в присутствии трифенилфосфина инкубацией при перемешивании в течение 1,5 часа при 0°C с последующей экстракцией продукта диэтиловым эфиром; (2г) (S)-трет-бутил 10-(2-(бис(2-(трет-бутокси)-2-оксоэтил)амино)этил)-13-(2-(трет-бутокси)-2-оксоэтил)-9-(трет-бутоксикарбонил)-3-оксо-1-фенил-2-окс-4,10,13-триазапентадекан-15-оат получают из ди-трет-бутил 2,2'-((2-бромоэтил)азандиил)диацетата путем конденсации с HCl×H-(L)-Lys(Z)-OBut в смеси ацетонитрила с фосфатным буфером при комнатной температуре при перемешивании в течение 48 часов; (2д) S) -тетра-трет-бутил 2,2',2'',2'''-((((6-амино-1-(трет-бутокси)-1-оксогексан-2-ил)азандиил)бис(этан-2,1-диил)бис)(азантриил)) тетраацетат получают из (S)-трет-бутил 10-(2-(бис(2-(трет-бутокси)-2-оксоэтил)амино)этил)-13-(2-(трет-бутокси)-2-оксоэтил)-9-(трет-бутоксикарбонил)-3-оксо-1-фенил-2-окс-4,10,13-триазапентадекан-15-оата путем реакции с циклогексеном в присутствии катализатора Pd/C при кипячении в изопропаноле в течение 7 минут; (2е) заключительную стадию получения (S)-9-(2-(бис(2-(трет-бутокси)-2-оксоэтил)амино)этил)-6-(2-(трет-бутокси)-2-оксоэтил)-10-(трет-бутоксикарбонил)-2,2-диметил-4,16-диоксо-3,18-диокса-6,9,15-триазикозан-20-олевую кислоты получают в реакции (S)-тетра-трет-бутил 2,2',2'',2'''-((((6-амино-1-(трет-бутокси)-1-оксогексан-2-ил) азандиил)бис(этан-2,1-диил)бис)(азантриил))тетраацетата с ангидридом дигликолевой кислоты путем перемешивания при комнатной температуре с последующей очисткой методом флэш-хроматографии на силикагеле с использованием в качестве элюента смеси «хлороформ-метанол-вода» в соотношении 60:11:1; (3) к пептиду Ahx-Abu-TGIRIS-Abu, присоединенному к аминогруппе МВНА-модифицированного полистирола через остаток α-аминомасляной кислоты, добавляют конденсирующую смесь в виде раствора (S)-9-(2-(бис(2-(трет-бутокси)-2-оксоэтил)амино)этил)-6-(2-(трет-бутокси)-2-оксоэтил)-10-(трет-бутоксикарбонил)-2,2-диметил-4,16-диоксо-3,18-диокса-6,9,15-триазикозан-20-олевой кислоты (3 эквивалента) в диметилформамиде, которое предварительно активируют добавлением 2,9 эквивалентов реагента сочетания TBTU (2,9 эквивалента) в присутствии HOBt (3 эквивалента), после чего инкубируют в течение 18 часов при перемешивании при комнатной температуре; (4) проводят удаление защитных групп и снятие пептида с подложки путем инкубации подложки с помощью безводной фтористоводородной кислоты (10 мл на 1 грамм пептидилполимера) с добавкой м-крезола (1 мл на 1 г пептидилполимера) в течение 1 часа при 0°С; (5) получают комплексную соль Gd3+ с DTPA-Lys-дигликолат-Ahx-Abu-TGIRIS-Abu-NH2 путем реакции модифицированного пептида с солью Gd3+, взятой в недостатке (0,95 эквивалента) по отношению к модифицированному пептиду в диапазоне концентраций от 1 до 6% в водной среде, при рН 6,5-7,5.
| СМЕСИТЕЛЬНОЕ УСТРОЙСТВО (ВАРИАНТЫ) | 2011 |
|
RU2454272C1 |
| КОНЪЮГАТЫ АНТАГОНИСТА ПЕПТИДА АНАЛОГА БОМБЕЗИНА | 2009 |
|
RU2523531C2 |
| Устройство для обработки металлов путем штампования, резания и т.п. | 1927 |
|
SU6239A1 |

