ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к производным пиридинкарбоксамида, способу их получения и к фармацевтической композиции, содержащей их, а также к применению их в качестве терапевтических средств, особенно в качестве ингибиторов внешнего медуллярного калиевого канала почки (Renal Outer Medullary Potassium, ROMK) и их использования в приготовлении лекарственных препаратов для лечения и/или профилактики расстройств, возникающих в результате чрезмерного удержания соли и воды, включая гипертонию и сердечную недостаточность.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Увеличение почечной реабсорбции соли может вызвать риск гипертонии. Напротив, ингибирование функции почечной реабсорбции может способствовать выведению мочи, что приводит к мочегонному и гипотензивному эффекту. Обычными диуретиками являются тиазидные диуретики, которые являются антигипертензивными препаратами первой линии в США, они в основном действуют на Na+-Cl- транспортер. Петлевые диуретики являются более эффективными для пациентов с недостаточностью почечной функции, которые действуют через Na+-K+-2Cl- трансферный белок. Однако оба типа могут вызывать гипокалиемию (симптомы: слабость, утомляемость, мышечные судороги, запор и проблемы с сердечным ритмом, такие как аритмия), которая повышают риск заболеваемости и смертности от сердечно-сосудистых заболеваний.
Внешний медуллярный калиевый канал почки (ROMK) также известен как калиевый канал внутреннего выпрямления 1.1 (Kir1.1). Калиевый канал ROMK, взаимодействующий с Na+-K+-2Cl- белком ко-транспортером NKCC2 (ответственный за транспорт NaCl) посредством апикальной мембранной проводимости почечного толстого восходящего колена петли Генле (thick ascending limb, TAL) может регулировать реабсорбцию калия. Было обнаружено, что ROMK непосредственно связан с почечным секреторным каналом. Нокаут по гену ROMK приводит к потери мышиных TAL и CCD 35-pS ионных каналов и потери других K+ каналов. Синдром Барттера - это аутосомно-рецессивное заболевание, характеризующееся массивной потерей соли в почках, гипокалиемией и низким кровяным давлением. Синдром Барттера главным образом вызван мутациями в белке ROMK или Na+-K+-2Cl-ко-транспортере. Разница заключается в том, что гипокалиемия при синдроме Барттера, вызванная мутацией ROMK значительно слабее по сравнению с мутацией белка Na+-K+-2Cl-ко-транспортера. Таким образом, ингибирование функции ROMK может эффективно ингибировать функцию реабсорбции соли белком Na+-K+-2Cl-ко-транспортером, способствовать выделению мочи и, таким образом, приводить к мочегонному и гипотензивному эффекту, не вызывая гипокалиемии.
Хотя в настоящее время описан ряд ингибиторов ROMK, таких как WO 2010129379, WO 2012058134, WO 2012058116, WO 2012058134, WO 2013066714, WO 2013028474, WO 2014085210, WO 2014018764, WO 2014015495, WO 2014085210, WO 2013039802, WO 2013062892 и WO 2012058116, необходимо разработать большее количество соединений с лучшей селективностью по hERG. Настоящее изобретение предлагает серию новых соединений, представленных общей формулой (I), где добавлена полярная группа, которая может уменьшать ClogP, усиливать селективность по hERG и является намного более безопасной при сохранении ингибирующей активности ROMK.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению формулы (I),
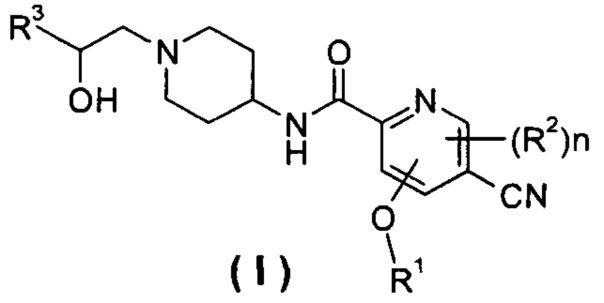
или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси или их фармацевтически приемлемым солям,
где
R1 представляет собой алкил, где указанный алкил необязательно дополнительно замещен одной или более группами, выбранными из группы, состоящей из галогена, гидроксила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и сложного эфира карбоновой кислоты;
R2 выбирают из группы, состоящей из водорода, алкила, галогена, циано, нитро, алкокси, циклоалкила и гетероциклила, где указанный алкил, алкокси, циклоалкил или гетероциклил необязательно дополнительно замещены одной или более группами, выбранными из группы, состоящей из алкила, галогена, гидроксила, гидроксиалкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и эфира карбоновой кислоты;
R3 выбирают из следующих групп:
 и
и  ;
;
R4 и R5, каждый независимо, выбирают из группы, состоящей из водорода, алкила, галогена, циано, нитро, алкокси, циклоалкила, гетероциклила, арила и гетероарила;
R6 выбирают из водорода, алкила и галогена;
n равно 0, 1 или 2.
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или их фармацевтически приемлемые соли, где R1 представляет собой алкил, где указанный алкил необязательно дополнительно замещен одной или более группами, выбранными из группы, состоящей из галогена, гидроксила и алкокси; R1 предпочтительно представляет собой С1-6 алкил, более предпочтительно выбранный из группы, состоящей из метила, этила и пропила.
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (I), или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смеси, или их фармацевтически приемлемые соли, где R4 представляет собой алкил и R5 представляет собой водород.
В другом предпочтительном варианте осуществления настоящего изобретения соединение формулы (I), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или их фармацевтически приемлемые соли, который представляет собой соединение формулы (II),
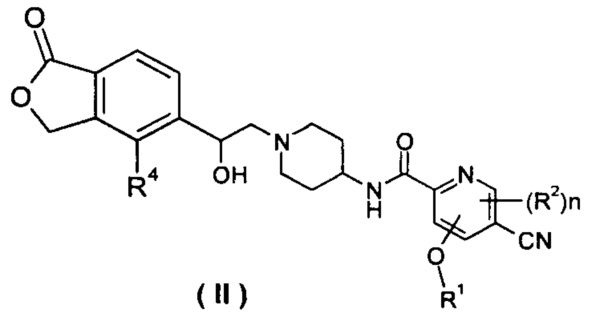
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или их фармацевтически приемлемые соли, где R1, R2, R4 и n такие, как определено в формуле (I).
В другом предпочтительном варианте осуществления настоящего изобретения соединение формулы (I), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или их фармацевтически приемлемые соли, который представляет собой соединение формулы (III),
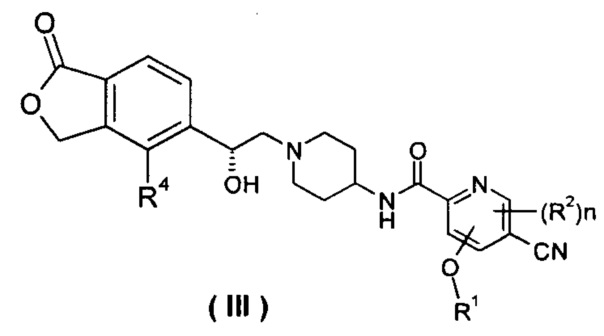
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или их фармацевтически приемлемые соли, где R1, R2, R4 и n такие, как определено в формуле (I).
В другом предпочтительном варианте осуществления настоящего изобретения соединение формулы (I), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или их фармацевтически приемлемые соли, который представляет собой соединение формулы (IV),
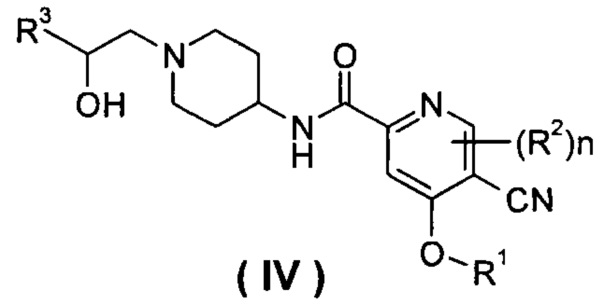
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или их фармацевтически приемлемые соли, где R1, R2, R3 и n такие, как определено в формуле (I).
В другом предпочтительном варианте осуществления настоящего изобретения соединение формулы (I), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или их фармацевтически приемлемые соли, который представляет собой соединение формулы (V),
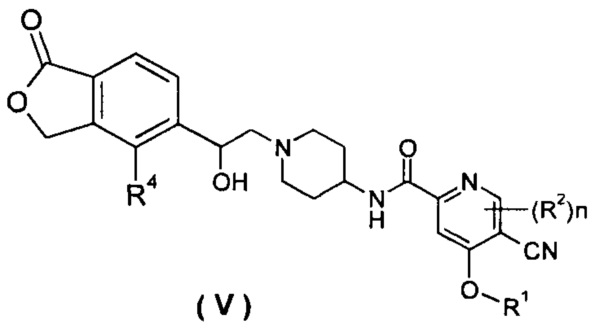
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смеси или их фармацевтически приемлемые соли, где R1, R2, R4 и n такие, как определено в формуле (I).
В другом предпочтительном варианте осуществления настоящего изобретения соединение формулы (I), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или их фармацевтически приемлемые соли, которые представляет собой соединение формулы (VI),
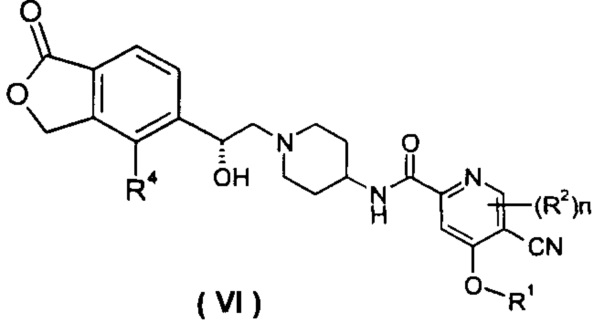
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или их фармацевтически приемлемые соли, где R1, R2, R4 и n такие, как определено в формуле (I).
Типичные соединения согласно настоящему изобретению включают, но не ограничиваются следующими.
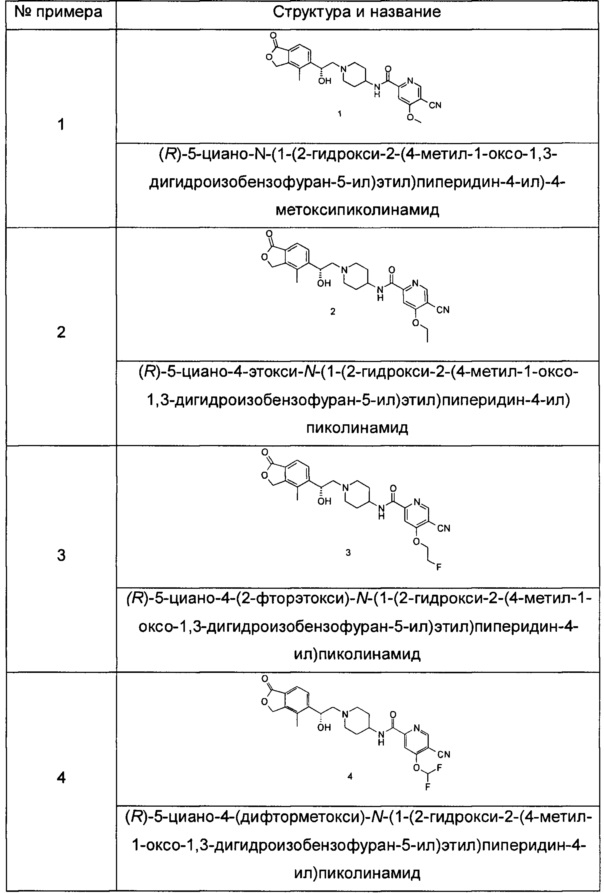
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или их фармацевтически приемлемые соли.
Настоящее изобретение также относится к соединению формулы (IA), или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смесям или их фармацевтически приемлемым солям в качестве промежуточного соединения для получения соединения формулы (I),
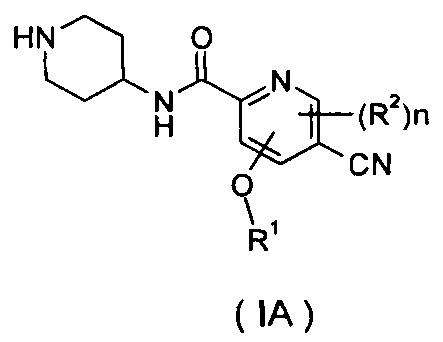
где
R1 представляет собой алкил, где указанный алкил необязательно дополнительно замещен одной или более группами, выбранными из группы, состоящей из галогена, гидроксила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и сложного эфира карбоновой кислоты;
R2 выбран из группы, состоящей из водорода, алкила, галогена, циано, нитро, алкокси, циклоалкила и гетероциклила, где указанный алкил, алкокси, циклоалкил или гетероциклил необязательно дополнительно замещен одной или более группами, выбранными из групп, состоящих из алкила, галогена, гидроксила, гидроксиалкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и сложного эфира карбоновой кислоты;
который может быть использован в качестве промежуточного соединения при получении соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или их фармацевтически приемлемых солей;
n равно 0, 1 или 2.
В другом предпочтительном варианте осуществления настоящего изобретения соединение формулы (IA) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или их фармацевтически приемлемые соли, которые представляют собой соединение формулы (IVA),
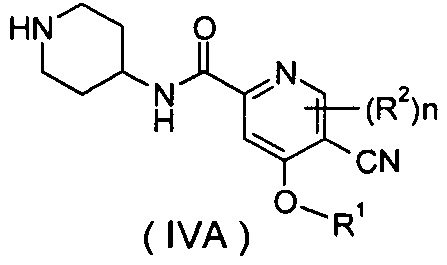
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смеси, или их фармацевтически приемлемые соли; которое может быть использовано в качестве промежуточного соединения при получении соединения формулы (IV) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или их фармацевтически приемлемых солей; где R1, R2 и n такие, как определено в формуле (IA).
Типичные соединения формулы (IA) включают, но не ограничиваются следующими:
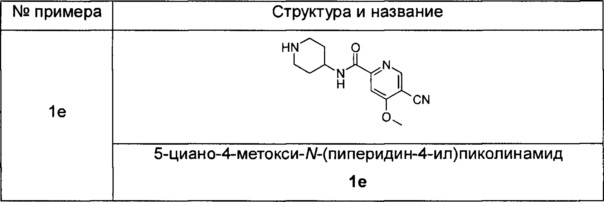
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смеси или их фармацевтически приемлемые соли.
В другом аспекте настоящее изобретение относится к способу получения соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смесей или их фармацевтически приемлемых солей, включающему стадию:
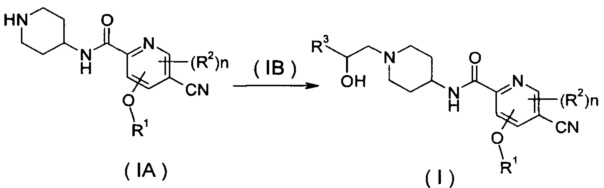
нагревания соединения формулы (IA) с замещенным бензофурановым производным формулы (IB), предпочтительно с (R)-4-метил-5-(оксиран-2-ил)-изобензофуран-1(3H)-она с получением соединения формулы (I);
где R1-R3 и n такие, как определено в общей формуле (I).
Другой аспект настоящего изобретения относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или их фармацевтически приемлемых солей и фармацевтически приемлемые носители, разбавители или эксципиенты.
Другой аспект настоящего изобретения относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или их фармацевтически приемлемых солей или фармацевтической композиции, содержащей их, в приготовлении ингибитора ROMK.
Другой аспект настоящего изобретения относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или их фармацевтически приемлемых солей, или фармацевтической композиции, содержащей их, в приготовлении лекарственного средства для лечения или профилактики гипертонии и/или сердечной недостаточности.
Другой аспект настоящего изобретения относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или их фармацевтически приемлемых солей, или фармацевтической композиции, содержащей их, в приготовлении лекарственного средства для лечения или профилактики опосредованных ROMK заболеваний, причем указанные заболевания предпочтительно выбирают из группы, состоящей из цирроза печени, острой и хронической почечной недостаточности, нефротического синдрома, легочной гипертензии, сердечно-сосудистых болезней, инфаркта миокарда, инсульта, сердечной недостаточности, легочной гипертонии, атеросклероза и мочекаменной болезни.
Другой аспект настоящего изобретения относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смесям, или их фармацевтически приемлемым солям, или к фармацевтической композиции, содержащей их, для использования в качестве ингибитора ROMK.
Другой аспект настоящего изобретения относится к соединению формулы (I), или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смесям, или их фармацевтически приемлемым солям, или к фармацевтической композиции, содержащей их, для использования в лечении или профилактике гипертонии и/или сердечной недостаточности.
Другой аспект настоящего изобретения относится к соединению формулы (I), или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смесям, или их фармацевтически приемлемым солям, или к фармацевтической композиции, содержащей их, для использования в лечении или профилактике заболеваний, опосредованных ROMK, причем указанные заболевания предпочтительно выбирают из группы, состоящей из цирроза печени, острой и хронической почечной недостаточности, нефротического синдрома, легочной гипертензии, сердечно-сосудистых болезней, инфаркта миокарда, инсульта, сердечной недостаточности, легочной гипертонии, атеросклероза и мочекаменной болезни.
Другой аспект настоящего изобретения относится к способу ингибирования ROMK, включающему введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или их фармацевтически приемлемых солей, или фармацевтической композиции, содержащей их.
Другой аспект настоящего изобретения относится к способу лечения или профилактики гипертонии и/или сердечной недостаточности, включающему введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемые соли, или фармацевтической композиции, содержащей их.
Другой аспект настоящего изобретения относится к способу лечения или профилактики, опосредованных ROMK заболеваний или расстройств, включающему введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смесей или их фармацевтически приемлемых солей или фармацевтической композиции, содержащей их, где указанные заболевания или расстройства предпочтительно выбирают из группы, состоящей из цирроза печени, острой и хронической почечной недостаточности, нефротического синдрома, легочной гипертензии, сердечно-сосудистых болезней, инфаркта миокарда, инсульта, сердечной недостаточности, легочной гипертонии, атеросклероза и мочекаменной болезни.
Фармацевтические композиции, содержащие активный ингредиент, могут быть в форме, пригодной для перорального введения, такой как таблетка, пастилка, леденец, водная или масляная суспензия, диспергируемый порошок или гранулят, эмульсия, твердая или мягкая капсула или сироп или эликсир. Пероральные композиции могут быть получены в соответствии с любым известным способом получения фармацевтических композиций в данной области. Такие композиции могут содержать одну или более добавок, выбранных из группы, состоящей из подсластителя, ароматизаторов, красителей и консервантов, чтобы обеспечить приятный и приемлемый фармацевтический состав. Таблетка содержит активный ингредиент и нетоксичный фармацевтически приемлемый эксципиент, пригодный для изготовления таблетки. Эти наполнители могут быть инертными эксципиентами, такими как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующими и дезинтегрирующими агентами, такими как микрокристаллическая целлюлоза, сшитая карбоксиметилцеллюлоза натрия, кукурузный крахмал или альгиновая кислота; связующим, таким как крахмал, желатин, поливинилпирролидон или аравийская камедь; и смазывающим веществом, таким как стеарат магния, стеариновая кислота или тальк. Таблетка может быть непокрытой или покрытой с помощью известной технологии, которая может маскировать вкус лекарства или замедлять распад и абсорбцию активного ингредиента в желудочно-кишечном тракте, обеспечивая тем самым пролонгированное высвобождение в течение длительного периода. Например, можно использовать водорастворимый материал для маскирования вкуса, такой как гидроксипропилметилцеллюлоза или гидроксипропил целлюлоза, или можно использовать материал с пролонгированным периодом, такой как этилцеллюлоза, ацетатбутират целлюлозы.
Пероральная композиция может также быть представлена в виде твердой желатиновой капсулы, где активный ингредиент смешивается с инертным твердым разбавителем, таким как карбонат кальция, фосфат кальция или каолин, или в виде мягкой желатиновой капсулы, где активный ингредиент смешивается с водорастворимым носителем, таким как полиэтиленгликоль или масляная среда, например, арахисовое масло, жидкий парафин или оливковое масло.
Водная суспензия содержит активный ингредиент в смеси с эксципиентами, подходящими для производства водной суспензии. Такой эксципиент является суспендирующим агентом, таким как натрийкарбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон и аравийская камедь; диспергатором или увлажнителем, который может представлять собой встречающийся в природе фосфатид, такой как лецитин, или продукт конденсации алкиленоксида с жирной кислотой, такой как полиоксиэтиленстеарат, или продукт конденсации этиленоксида с длинноцепочечным алифатическим спиртом, таким как гептадекаэтиленоксицетанол или продукт конденсации этиленоксида с частичными сложными эфирами, полученными из жирных кислот и гекситов, таких как моноолеат полиоксиэтиленсорбита, или продукты конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и ангидридов гексита, таких как полиоксиэтиленсорбитанмоноолеат. Водная суспензия может также содержать один или более консервантов, таких как этилпарабен или н-пропилпарабен, один или более красителей, один или более ароматизаторов и один или более подсластителей, таких как сахароза, сахарин или аспартам.
Масляную суспензию можно приготовить суспендированием активного ингредиента в растительном масле, таком как арахисовое масло, оливковое масло, кунжутное масло или кокосовое масло, или в минеральном масле, таком как жидкий парафин. Масляная суспензия может содержать загуститель, такой как пчелиный воск, твердый парафин или цетиловый спирт. Вышеупомянутый подсластитель и ароматизатор могут быть добавлены для обеспечения приемлемого приготовления. Эти композиции могут быть сохранены путем добавления антиоксиданта, такого как бутилированный гидроксианизол или α-токоферол.
Активный ингредиент и диспергирующий агент или увлажняющий агент, суспендирующий агент или один или более консервантов могут быть получены добавлением воды для получения диспергируемого порошка и гранулы, пригодных для приготовления водной суспензии. Примерами подходящих диспергирующих или увлажняющих агентов и суспендирующих агентов являются те, которые были упомянуты выше. Могут также добавляться дополнительные эксципиенты, такие как подсластитель, ароматизатор и краситель. Эти композиции могут быть сохранены путем добавления антиоксиданта, такого как аскорбиновая кислота.
Настоящая фармацевтическая композиция также может быть в форме эмульсии типа «масло в воде». Масляная фаза может представлять собой растительное масло, такое как оливковое масло или арахисовое масло, или минеральное масло, такое как жидкий парафин или их смесь. Подходящим эмульгирующим агентом могут быть природные фосфатиды, такие как лецитин соевых бобов, и сложные эфиры или неполные сложные эфиры, полученные из жирных кислот и ангидридов гексита, такие как моноолеат сорбита, и продукты конденсации указанных неполных эфиров с этиленоксидом, такие как моноолеат полиоксиэтиленсорбита. Эмульсия также может содержать подсластитель, ароматизатор, консервант и антиоксидант. Сироп и эликсир можно приготовить с подсластителем, таким как глицерин, пропиленгликоль, сорбит или сахароза. Такие составы могут также содержать смягчающее средство, консервант, краситель и антиоксидант.
Фармацевтическая композиция может быть в форме стерильного водного раствора для инъекций. Приемлемыми носителями и растворителями, которые могут быть использованы, являются вода, раствор Рингера и изотонический раствор хлорида натрия. Стерильный препарат для инъекций также может быть стерильной микроэмульсией для инъекций типа «масло в воде», где активный ингредиент растворяется в масляной фазе. Например, активный ингредиент может быть сначала растворен в смеси соевого масла и лецитина, затем масляный раствор вводится в смесь воды и глицерина и перерабатывается с образованием микроэмульсии. Инъецируемый раствор или микроэмульсию можно вводить в кровоток пациента путем местной болюсной инъекции. Альтернативно, может быть выгодным введение раствора или микроэмульсии таким образом, чтобы поддерживать постоянную циркулирующую концентрацию данного соединения. Для поддержания такой постоянной концентрации может быть использовано устройство для непрерывного внутривенного введения. Примером такого устройства является насос для внутривенных инъекций Deltec CADD-PLUS. ТМ. 5400.
Фармацевтическая композиция может быть в форме стерильной инъекционной водной или масляной суспензии для внутримышечного и подкожного введения. Такая суспензия может быть приготовлена с использованием подходящего диспергатора или увлажняющих агентов и суспендирующего агента, как описано выше, в соответствии с известными способами. Стерильный инъекционный препарат также может быть стерильным инъекционным раствором или суспензией, приготовленным в нетоксичном парентерально приемлемом разбавителе или растворителе, например, раствор, приготовленный в 1,3-бутандиоле. Кроме того, стерильные нелетучие масла могут быть легко использованы в качестве растворителя или суспендирующей среды. Для этой цели могут быть использованы любые косметические нелетучие масла, в том числе синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, могут быть использованы для приготовления инъекции.
Настоящее соединение может быть введено в форме суппозитория для ректального введения. Эти фармацевтические композиции могут быть получены путем смешивания лекарственного средства с подходящим нераздражающим эксципиентом, который является твердым при обычных температурах, но жидким в прямой кишке, то есть плавящимся в прямой кишке с выделением лекарственного средства. Такие материалы включают какао-масло, глицериновый желатин, гидрированные растительные масла, смесь полиэтиленгликолей и эфиров жирных кислот и полиэтиленгликолей с различными молекулярными массами.
Специалистам в данной области техники хорошо известно, что дозировка лекарственного средства зависит от множества факторов, включая, но не ограничиваясь следующими факторами: активность конкретного соединения, возраст, вес, общее состояние здоровья, поведение и диета пациента, время введения, путь введения, скорость экскреции, комбинация лекарств и тому подобное. Кроме того, наилучшее лечение, такое как режим лечения, суточная дозировка соединения формулы (I) или тип его фармацевтически приемлемой соли могут быть проверены традиционным терапевтическим режимом.
ОПРЕДЕЛЕНИЯ
Если не указано иное, используемые здесь термины имеют следующие значения.
«Алкил» относится к линейной или разветвленной насыщенной алифатической углеводородной группе, содержащей от 1 до 20 атомов углерода, предпочтительно C1-С10-алкил, более предпочтительно C1-С6-алкил. Неограниченные примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-ди метил бутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их разветвленные изомеры. Более предпочтительно алкильная группа представляет собой низший алкил, содержащий от 1 до 6 атомов углерода, и неограниченные примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметил пропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.п. Алкильная группа может быть замещенной или незамещенной. При замещении заместительная группа (группы) может быть замещена в любой доступной точке соединения. Заместительная группа (группы) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклилтио, оксо, амино, галогеналкила, гидроксиалкила, карбоксила, эфира карбоновой кислоты.
«Циклоалкил» относится к насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, содержащей от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 10 атомов углерода и наиболее предпочтительно от 3 до 6 атомов углерода. Неограниченные примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.п., предпочтительно циклопропил и циклогексенил. Полициклический циклоалкил включает циклоалкил, имеющий спироциклическое кольцо, конденсированное кольцо или кольцо с мостиками.
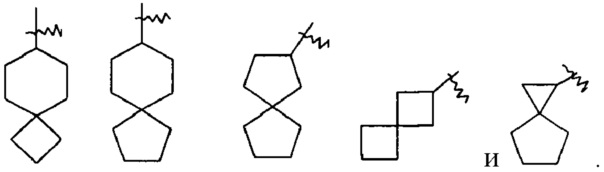
«Спироциклоалкил» относится к 5-20-членной полициклической группе с кольцами, связанными через один общий атом углерода (называемый спиро-атомом), где одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, предпочтителен 6-14-членный спироциклоалкил и более предпочтителен 7-10-членный спироциклоалкил. В соответствии с числом общих спиро-атомов спироциклоалкил может быть подразделен на моноспироциклоалкил, ди-спироциклоалкил или полиспироциклоалкил, и предпочтительны моноспироциклоалкил или ди-спироциклоалкил, более предпочтительны 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноспироциклоалкил. Неограниченные примеры спироциклоалкилов включают, но не ограничиваются ими:
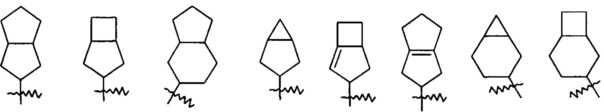
Термин «конденсированный циклоалкил» относится к 5-20-членной полноуглеродной полициклической группе, где каждое кольцо в системе разделяет соседнюю пару атомов углерода с другим кольцом, где одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженную пи-электронную систему, предпочтителен 6-14-членный конденсированный циклоалкил, более предпочтителен 7-10-членный конденсированный циклоалкил. В соответствии с количеством колец в составе конденсированный циклоалкил может быть подразделен на бициклический, трициклический, тетрациклический или полициклический конденсированный циклоалкил, предпочтителен бициклический или трициклический конденсированный циклоалкил и более предпочтителен 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный циклоалкил. Неограниченные примеры конденсированного циклоалкила включают, но не ограничиваются следующими:
 и
и 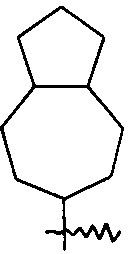 .
.
«Циклоалкил с мостиками» относится к 5-20-членной полноуглеродной полициклической группе, где каждые два кольца в системе имеют два разъединенных атома, причем кольца могут иметь одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, предпочтителен 6-14-членный циклоалкил с мостиками и более предпочтителен 7-10-членный циклоалкил с мостиками. По числу колец в составе циклоалкил с мостиками может быть разделен на бициклический, трициклический, тетрациклический или полициклический циклоалкил с мостиками и предпочтительно бициклический, трициклический или тетрациклический циклоалкил с мостиками и более предпочтителен бициклический или трициклический циклоалкил с мостиками. Неограниченные примеры циклоалкилов с мостиками включают, но не ограничиваются следующими:
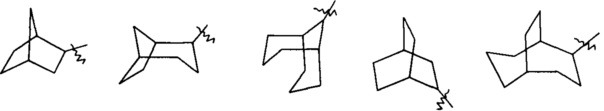
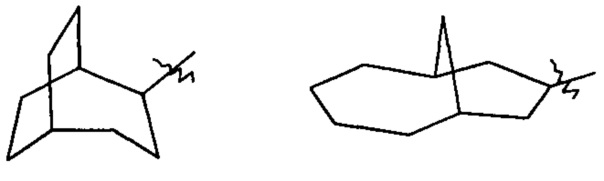 и
и  .
.
Указанный циклоалкил может быть конденсирован с арилом, гетероарилом или гетероциклилом, где кольцо, связанное с исходной структурой, представляет собой циклоалкил. Неограниченные примеры включают инданил, тетрагидронафтил, бензоциклогептил и тому подобное. Циклоалкил может быть необязательно замещенным или незамещенным. В случае замещения замещающая группа (группы) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклилтио, оксо, амино, галогеналкила, гидроксиалкила, карбоксила, эфира карбоновой кислоты.
«Гетероциклил» относится к 3-20-членной насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число, выбранное из диапазона от 0 до 2) в качестве атомов в кольце, но без -O-O-, -O-S- или -S-S- в кольце, а остальные атомы в кольце представляют собой атомы углерода. Предпочтительно гетероциклил имеет от 3 до 12 атомов с 1-4 гетероатомами, более предпочтительно от 3 до 10 атомов с 1-3 гетероатомами и наиболее предпочтительно от 5 до 6 атомов с 1-2 гетероатомами. Неограниченные примеры моноциклического гетероциклила включают, но не ограничиваются, пирролидинил, пиперидил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил, пиранил, тетрагидрофуранил и тому подобное. Полициклический гетероциклил включает гетероциклил, имеющий спиро кольцо, конденсированное кольцо или кольцо с мостиками.
«Спиро гетероциклил» относится к 5-20-членным полициклическим гетероциклилам с кольцами, связанными через один общий атом (называемый спироатомом), где указанные кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число, выбранное из диапазона от 0 до 2) в качестве атомов в кольце, а остальные атомы в кольце представляют собой атомы углерода, причем одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной пи-электронной системы; предпочтителен 6-14-членный спиро-гетероциклил и более предпочтителен 7-10-членный спиро-гетероциклил. В соответствии с числом общих спиро-атомов спиро-гетероциклил может быть разделен на моноспиро гетероциклил, ди-спиро гетероциклил или полиспиро гетероциклил, предпочтителен моноспиро гетероциклил или ди-спиро гетероциклил и более предпочтителен 4-членный/4-членнный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноспиро гетероциклил. Неограниченные примеры спиро-гетероциклилов включают, но не ограничиваются следующими:
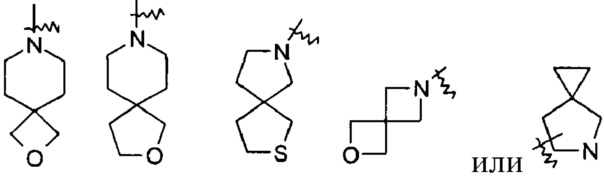
«Конденсированный гетероциклил» относится к 5-20-членной полициклической гетероциклической группе, где каждое кольцо в системе разделяет соседнюю пару атомов с другим кольцом, причем одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, причем указанные кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число, выбранное из диапазона от 0 до 2) в качестве кольцевых атомов, и остальные кольцевые атомы представляют собой атомы углерода; предпочтительно 6-14-членный конденсированный гетероциклил и более предпочтительно 7-10-членный конденсированный гетероциклил. В соответствии с количеством многочленных колец конденсированный гетероциклил может быть разделен на бициклический, трициклический, тетрациклический или полициклический конденсированный гетероциклил, предпочтительно бициклический или трициклический конденсированный гетероциклил и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный гетероциклил. Неограниченные примеры конденсированного гетероциклила включают, но не ограничиваются следующими:
«Гетероциклил с мостиками» относится к 5-14-членной полициклической гетероциклической группе, где каждые два кольца в системе имеют два разъединенных атома, причем кольца могут иметь одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной пи-электронной системы. Кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число, выбранное из диапазона от 0 до 2) в качестве кольцевых атомов, а остальные атомы кольца представляют собой атомы углерода; предпочтителен 6-14-членный гетероциклил с мостиками и более предпочтителен 7-10-членный гетероциклил с мостиками. В связи с количеством многочленных колец гетероциклил с мостиками может быть разделен на бициклический, трициклический, тетрациклический или полициклический гетероциклил с мостиками и предпочтителен бициклический, трициклический или тетрациклический гетероциклил с мостиками, и более предпочтителен бициклический или трициклический гетероциклил с мостиками. Неограниченные примеры гетероциклилов с мостиками включают, но не ограничиваются следующими:
 и
и 
Указанный гетероциклил может быть конденсирован с арилом, гетероарилом или циклоалкилом, где кольцо, связанное с исходной структурой, является гетероциклилом. Неограниченные примеры включают, но не ограничиваются:
 и
и , и т.д.
, и т.д.
Гетероциклил может быть необязательно замещенным или незамещенным. В случае замещения замещающая группа (группы) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклилтио, оксо, амино, галогеналкила, гидроксиалкила, карбоксила, эфира карбоновой кислоты.
«Арил» относится к 6-14-членному полноуглеродному моноциклическому кольцу или полициклическому конденсированному кольцу (то есть каждое кольцо в системе разделяет соседнюю пару атомов углерода с другим кольцом в системе), имеющему полностью сопряженную пи-электронную систему; предпочтителен 6-10-членный арил, более предпочтителен фенил и нафтил и наиболее предпочтителен фенил. Арил может быть конденсирован с гетероарилом, гетероциклилом или циклоалкилом, где кольцо, связанное с исходной структурой, является арилом. Неограниченные примеры включают, но не ограничиваются:
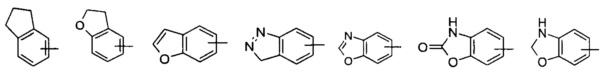
 и
и  .
.
Арил может быть необязательно замещенным или незамещенным. При замещении замещающая группа (группы) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклилтио, амино, галогеналкила, гидроксиалкила, карбоксила, сложного эфира карбоновой кислоты.
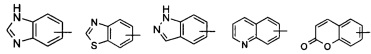
«Гетероарил» относится к 5-14-членным арилам, имеющим от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, S и N в качестве кольцевых атомов, а остальные атомы в кольце представляют собой атомы углерода; предпочтителен 5-10-членный гетероарил, более предпочтителен 5- или 6-членный гетероарил, такой как фурил, тиенил, пиридил, пирролил, N-алкилпирролил, пиримидинил, пиразинил, имидазолил, тетразолил и тому подобное. Гетероарил может быть конденсирован с арилом, гетероциклилом или циклоалкилом, где кольцо, связанное с исходной структурой, является гетероарилом. Неограниченные примеры включают, но не ограничиваются:
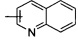 и
и 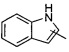 .
.
Гетероарил может быть необязательно замещенным или незамещенным. При замещении замещающая группа (группы) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклилтио, амино, галогеналкила, гидроксиалкила, карбоксила, сложного эфира карбоновой кислоты.
«Алкокси» относится к -О-(алкильной) или -O-(незамещенной циклоалкильной) группе, где алкил является таким, как определено выше. Неограниченные примеры включают, но не ограничиваются, метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси и тому подобное. Алкокси может быть необязательно замещенным или незамещенным. При замещении заместителем предпочтительно является одна или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклилтио, амино, галогеналкила, гидроксиалкила, карбоксила, сложного эфира карбоновой кислоты.
«Галогеналкил» относится к алкилу, замещенному одним или более галогенами, где алкил является таким, как определено выше.
«Гидрокси» относится к группе -ОН.
«Гидроксиалкил» относится к алкилу, замещенному гидроксигруппой, где алкил является таким, как определено выше.
«Галоген» относится к фтору, хлору, брому или йоду.
«Циано» означает группу -CN.
«Карбоксил» относится к-С(O)ОН-группе.
«Сложный эфир карбоновой кислоты» относится к -С(O)O(алкильной) или (циклоалкильной) группе, где алкил и циклоалкил являются такими, как определено выше.
«Необязательно» или «факультативно» означает, что событие или обстоятельство, описанное впоследствии, может, но не обязательно, иметь место, и такое описание включает ситуацию, где событие или обстоятельство может произойти или не произойти. Например, «гетероциклическая группа, необязательно замещенная алкилом», означает, что алкильная группа может быть, но необязательно должна присутствовать, и такое описание включает ситуацию, когда гетероциклическая группа замещена алкилом и гетероциклическая группа не является замещенной алкилом.
«Замещенный» относится к одному или более атомам водорода в группе, предпочтительно до 5, более предпочтительно от 1 до 3 атомов водорода, независимо замещенных соответствующим количеством заместителей. Само собой разумеется, что заместители существуют только в их химически возможном положении. Специалист в данной области способен определить, возможна или невозможна замена путем экспериментов или теории без чрезмерных усилий. Например, когда амино или гидрокси группы, имеющие свободный водород, связаны с атомами углерода, имеющими ненасыщенные связи (такие как олефины), могут быть нестабильными.
«Фармацевтическая композиция» относится к смеси одного или более соединений согласно настоящему изобретению или физиологически/ фармацевтически приемлемых солей или предшественников лекарств и других химических компонентов, таких как физиологически/фармацевтически приемлемые носители и эксципиенты. Целью фармацевтической композиции является облегчение введения соединения в организм и абсорбция активного ингредиента и, таким образом, проявление биологической активности.
СПОСОБ СИНТЕЗА ПО НАСТОЯЩЕМУ ИЗОБРЕТЕНИЮ
Для достижения цели настоящего изобретения настоящее изобретение применяет следующие технические решения синтеза.
Схема 1
Способ получения соединения формулы (I) согласно настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смесей или их фармацевтически приемлемых солей, включающий следующие стадии:
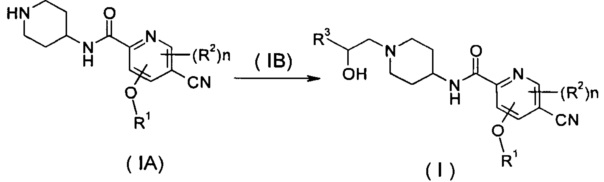
нагревание соединения формулы (IA) с соединением замещенных производных бензофурана (IB), предпочтительно (R)-4-метил-5-(оксиран-2-ил)изобензофуран-1(3Н)-она в органическом растворителе с получением соединения формулы (I), где R1-R3 и n такие, как определено в общей формуле (I).
Схема 2
Способ получения соединения формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смесей или их фармацевтически приемлемых солей, включающий следующие стадии:
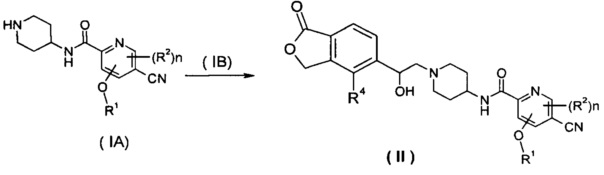
нагревание соединения формулы (IA) с соединением замещенных производных бензофурана (IB) предпочтительно (R)-4-метил-5-(оксиран-2-ил)изобензофуран-1(3Н)-она в органическом растворителе с получением соединения формулы (II), где R1, R2, R4 и n такие, как определено в общей формуле (II).
Схема 3
Способ получения соединения формулы (III) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или их фармацевтически приемлемых солей, включающий следующие стадии:
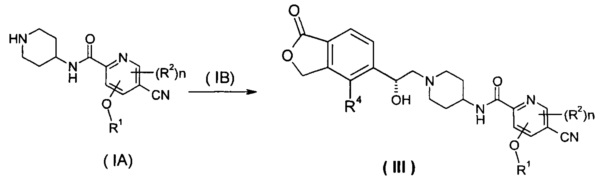
нагревание соединения формулы (IA) с соединением замещенных производных бензофурана (IB), предпочтительно (R)-4-метил-5-(оксиран-2-ил)изобензофуран-1(3H)-она в органическом растворителе с получением соединения формулы (III), где R1, R2, R4 и n такие, как определено в общей формуле (III).
Схема 4
Способ получения соединения формулы (IV) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или их фармацевтически приемлемых солей, включающий следующие стадии:
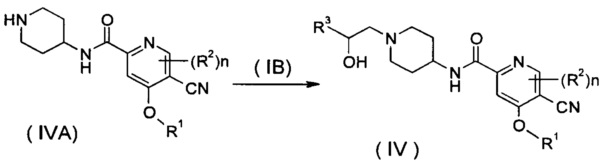
нагревание соединения формулы (IVA) с соединением замещенных производных бензофурана (IB), предпочтительно (R)-4-метил-5-(оксиран-2-ил)изобензофуран-1(3H)-она в органическом растворителе с получением соединения формулы (IV), где R1-R3 и n такие, как определено в общей формуле (I).
Схема 5
Способ получения соединения формулы (V) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смесей или их фармацевтически приемлемых солей, включающий следующие стадии:
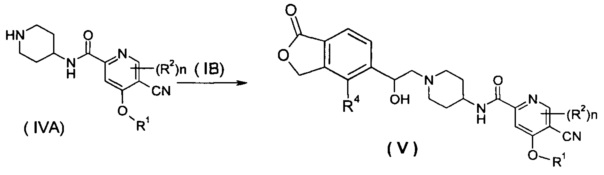
нагревание соединения формулы (IVA) с соединением замещенных производных бензофурана (IB), предпочтительно (R)-4-метил-5-(оксиран-2-ил)изобензофуран-1(3H)-она в органическом растворителе с получением соединения формулы (V), где R1, R2, R4 и n такие, как определено в общей формуле (I).
Схема 6
Способ получения соединения формулы (VI) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смесей, или их фармацевтически приемлемых солей, включающий следующие стадии:
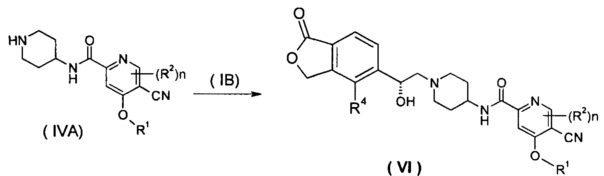
нагревание соединения формулы (IVA) с соединением замещенных производных бензофурана (IB), предпочтительно (R)-4-метил-5-(оксиран-2-ил)изобензофуран-1(3Н)-она в органическом растворителе с получением соединения формулы (VI), где R1, R2, R4 и n такие, как определено в общей формуле (I).
Растворитель включает, но не ограничивается ими, уксусную кислоту, метанол, этанол, ацетонитрил, тетрагидрофуран, дихлорметан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид или N,N-диметилацетамид, предпочтительно неполярный растворитель, более предпочтительно ацетонитрил.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фиг. 1 показывает влияние ингибитора ROMK на объем мочи у крысы SD (Спрег-Доули).
Фиг. 2 показывает влияние ингибитора ROMK на экскрецию натрия с мочой у крысы SD.
Фиг. 3 показывает влияние ингибитора ROMK на экскрецию калия с мочой у крысы SD.
Фиг. 4 показывает влияние ингибитора ROMK на сывороточный натрий у крысы SD.
Фиг. 5 показывает влияние ингибитора ROMK на сывороточный калий у крысы SD.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Далее настоящее изобретение будет описано со следующими примерами, но примеры не должны рассматриваться как ограничивающие объем изобретения.
Условиями, которые не указаны в примерах, будут обычные условия в данной области или рекомендованные производителем продукта условия для сырья. Для реагентов, для которых это не указано, источником будут коммерчески доступные обычные реагенты.
Примеры
Структуру соединений идентифицируют с помощью ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). ЯМР определяют Bruker AVANCE-400. Растворителями являются дейтерированный-диметилсульфоксид (DMSO-d6), дейтерированный-хлороформ (CDCl3) и дейтерированный метанол (CD3OD) с тетраметилсиланом (ТМС) в качестве внутреннего стандарта. Химические сдвиги ЯМР (δ) приведены в 10-6 (ppm).
МС определяют с помощью масс-спектрометра FINNIGAN LCQAd (ESI) (производитель: Thermo, тип: Finnigan LCQ advantage MAX).
Пластину силикагеля Yantai Huanghai HSGF254 или Qingdao GF254 используют для тонкослойной хроматографии на силикагеле (ТСХ). Размер пластины силикагеля, используемой в ТСХ, составляет от 0,15 до 0,2 мм, а размер пластины силикагеля, используемой при очистке продукта, составляет от 0,4 мм до 0,5 мм.
Силикагель Yantai Huanghai 200-300 меш используется в качестве носителя для колоночной хроматографии.
Известные исходные материалы настоящего изобретения могут быть получены известными в данной области техники способами синтеза или могут быть приобретены у ABCR GmbH & Co. KG, Acros Organnics, Aldrich Chemical Company, Accela ChemBio Inc., или Dari chemical Company и т.д.
Если не указано иное, реакции проводят в атмосфере азота или атмосфере аргона.
Термин «атмосфера азота» или «атмосфера аргона» означает, что реакционная колба оснащена 1 л баллоном с азотом или аргоном.
Термин «атмосфера водорода» означает, что реакционная колба оснащена 1 л баллоном водорода.
Микроволновый реактор типа СЕМ Discover-S 908860 используют в микроволновой реакции.
Если не указано иное, «раствор, используемый в реакциях» относится к водному раствору.
Если не указано иное, «реакционная температура» в реакциях относится к комнатной температуре.
Комнатная температура является оптимальной реакционной температурой, которая находится в диапазоне от 20°C до 30°C.
Процесс реакции контролируют тонкослойной хроматографией (ТСХ), и система элюирования включает: А: дихлорметан и метанол, Б: н-гексан и этилацетат, В: петролейный эфир и этилацетат, Г: ацетон. Соотношение объема растворителя можно регулировать в соответствии с полярностью соединений.
Система элюирования для очистки соединений методом колоночной хроматографии и тонкослойной хроматографии включает: А: дихлорметан и метанол, Б: н-гексан и этилацетат, В: н-гексан и ацетон, Г: н-гексан, Д: этилацетат. Соотношение объема растворителя можно регулировать в соответствии с полярностью соединений, и иногда может быть добавлено немного щелочного реагента, такого как триэтиламин, или кислого реагента.
Пример 1
(R)-5-циано-N-(1-(2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)пиперидин-4-ил)-4-метоксипиколинамид
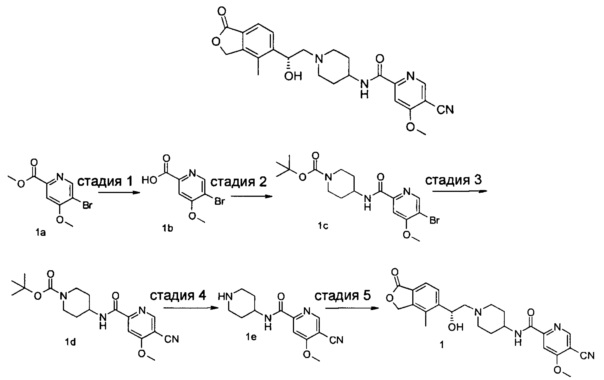
Стадия 1
5-бром-4-метоксипиколиновая кислота
Метил 5-бром-4-метоксипиколинат 1а (250 мг, 1,01 ммоль) растворяли в 10 мл смеси метанола, тетрагидрофурана и воды (V:V:V=3:3:1) и затем добавляли гидроксид натрия 100 мг, (2,5 ммоль) и перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении и к остаткам добавляли 10 мл воды. Полученную смесь доводили до рН 2 с помощью 2 М соляной кислоты и экстрагировали этил ацетатом (20 мл × 3). Органическую фазу промывали насыщенным раствором NaCl (15 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-бром-4-метоксипиколиновой кислоты 1b (200 мг) в виде белого твердого вещества, которое использовали на следующей стадии без дальнейшей очистки.
MS m/z(ESI): 229,9 [М-1].
Стадия 2
Трет-бутил 4-(5-бром-4-метоксипиколинамидо)пиперидин-1-карбоксилат
5-Бром-4-метоксипиколиновую кислоту 1b (150 мг, 0,65 ммоль), 4-амино-1-трет-бутоксикарбонилпиперидин (130 мг, 0,65 ммоль), 1-этил-(3-диметиламинопропил)карбодиимид (190 мг, 1 ммоль), 1-гидроксибензотриазол (20 мг, 0,13 ммоль) и триэтиламин (0,15 мл, 1 ммоль) растворяют в 20 мл N,N-диметилформамида. Реакционную смесь нагревали до 50°С и перемешивали в течение 6 ч при 50°C. Реакционный раствор концентрируют при пониженном давлении. Остатки очищают тонкослойной хроматографией (ТСХ) с помощью элюирующей системы Б с получением указанного в заголовке соединения трет-бутил 4-(5-бром-4-метоксипиколинамидо)пиперидин-1-карбоксилата 1 с (60 мг, 22,4%) в виде светло-желтого масла.
MS m/z(ESI): 414,1 [М+1].
Стадия 3
Трет-бутил 4-(5-циано-4-метоксипиколинамидо)пиперидин-1-карбоксилат
Трет-бутил 4-(5-бром-4-метоксипиколинамидо)пиперидин-1-карбоксилат 1 с (60 мг, 0,15 ммоль), цианид цинка (26 мг, 0,22 ммоль) и тетра(трифенилфосфин)палладий (18 мг, 0,015 ммоль) растворяли в 1,5 мл N,N-диметилацетамида. Смесь перемешивали при микроволновом воздействии в течение 40 мин при 135°C. Реакционный раствор концентрировали при пониженном давлении. Остатки очищали тонкослойной хроматографией (ТСХ) с помощью элюирующей системы Б с получением указанного в заголовке соединения трет-бутил 4-(5-циано-4-метоксипиколинамидо)пиперидин-1-карбоксилата 1d (32 мг, 61,5%) в виде бесцветного масла.
MS m/z(ESI): 361,2 [М+1].
Стадия 4
5-циано-4-метокси-М-(пиперидин-4-ил)пиколинамид
Трет-бутил 4-(5-циано-4-метоксипиколинамидо)пиперидин-1-карбоксилат 1d (32 мг, 0,09 ммоль) растворяли в 5 мл дихлорметана и добавляли 1 мл трифторуксусной кислоты. Реакционную смесь перемешивали в течение 1,5 часов. Реакционную смесь концентрировали при пониженном давлении. К остаткам добавляли 15 мл метанола и доводили до рН 8 насыщенным раствором бикарбоната натрия. Смесь концентрировали при пониженном давлении. Остатки очищали тонкослойной хроматографией (ТСХ) с помощью элюирующей системы А с получением указанного в заголовке соединения 5-циано-4-метокси-N-(пиперидин-4-ил)пиколинамида 1е (23 мг, 100%) в виде белой пасты.
MS m/z(ESI): 261,1 [М+1].
Стадия 5
(R)-5-циано-N-(1-(2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)пиперидин-4-ил)-4-метоксипиколинамид
(R)-4-метил-5-(оксиран-2-ил)изобензофуран-1(3Н)-он (25 мг, 0,09 ммоль, полученный согласно способу, описанному в патентной заявке WO 2010129379) и 5-циано-4-метокси-N-(пиперидин-4-ил)пиколинамид-1е (23 мг, 0,09 ммоль) растворяли в 5 мл ацетонитрила. Реакционную смесь перемешивали при кипячении с обратным холодильником в течение 15 часов. Реакционную смесь концентрировали при пониженном давлении. Остатки очищали тонкослойной хроматографией (ТСХ) с помощью элюирующей системы А с получением указанного в заголовке соединения (R)-5-циано-N-(1-(2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)пиперидин-4-ил)-4-метоксипиколинамида 1 (4,5 мг, 11,3%) в виде светло-желтого твердого вещества.
MS m/z (ESI): 450,2 [М+1].
1Н NMR (400 MHz, DMSO-d6): δ 8,88 (s, 1H), 8,75 (d, 1H), 7,77 (s, 1H), 7,71-7,69 (m, 2H), 5,43-5,40 (m, 2H), 5,35 (s, 1H), 5,08 (s, 1H), 4,09 (s, 3H), 3,78 (s, 1H), 2,95 (s, 3H), 2,38 (s, 1H), 2,27 (s, 3H), 2,25 (s, 2H), 1,72 (s, 4H).
Пример 2
(R)-5-циано-4-этокси-N-(1-(2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)пиперидин-4-ил)пиколинамид
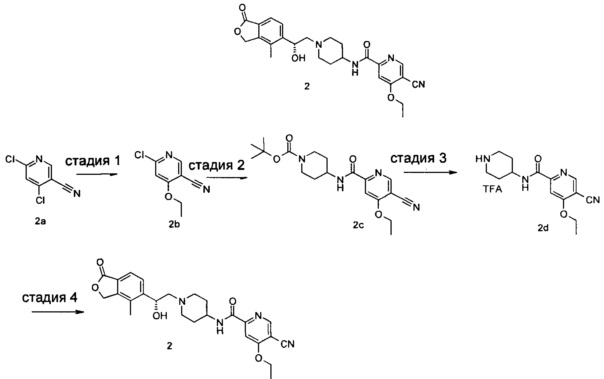
Стадия 1
6-хлор-4-этоксиникотинонитрил
4,6-Дихлорникотинонитрил 2а (500 мг, 2,89 ммоль) растворяли в 20 мл тетрагидрофурана и по каплям добавляли 10 мл раствора этоксида натрия (197 мг, 2,89 ммоль) в этаноле при 0°C. Реакционную смесь нагревали до комнатной температуры и дополнительно перемешивали в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении. Остатки очищали тонкослойной хроматографией (ТСХ) с помощью элюирующей системы Б с получением указанного в заголовке соединения 6-хлор-4-этоксиникотинонитрила 2b (375 мг, 71%) в виде белого твердого вещества. MS m/z(ESI): 183,1 [М+1].
Стадия 2
Трет-бутил 4-(5-циано-4-этоксипиколинамидо)пиперидин-1-карбоксилат
6-Хлор-4-этоксиникотинонитрил 2b (375 мг, 2,05 ммоль), 4-амино-1-трет-бутоксикарбонилпиперидин (422 мг, 2,05 ммоль), ацетат палладия (23 мг, 0,1 ммоль), 1,3-бис(дифенилфосфино)пропан (42 мг, 0,1 ммоль), триэтиламин (0,57 мл, 4,1 ммоль) и 20 мл ацетонитрила загружали в автоклав. Образовавшуюся смесь подвергали реакции в течение 16 часов при 80°C в атмосфере монооксида углерода в 10 бар. Реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении. Остатки очищали тонкослойной хроматографией (ТСХ) с помощью элюирующей системы А с получением указанного в заголовке соединения mpem-бутил 4-(5-циано-4-этоксипиколинамидо)пиперидин-1-карбоксилата 2 с (645 мг, 84%) в виде белого твердого вещества. MS m/z(ESI): 373,2 [М-1].
Стадия 3
5-циано-4-этокси-N-(пиперидин-4-ил)пиколинамид 2,2,2-трифторацетат
Трет-бутил 4-(5-циано-4-этоксипиколинамидо)пиперидин-1-карбоксилат 2 с (100 мг, 0,27 ммоль) растворяли в 5 мл дихлорметана и добавляли 1 мл трифторуксусной кислоты. Реакционную смесь перемешивали в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-циано-4-этокси-N-(пиперидин-4-ил)пиколинамид 2,2,2-трифторацетат 2d (110 мг) в виде желтого масла, используемого на следующей стадии без дополнительной очистки.
Стадия 4
(R)-5-циано-4-этокси-N-(1-(2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)пиперидин-4-ил)пиколинамид
(R)-4-метил-5-(оксиран-2-ил)изобензофуран-1(3H)-он (50,7 мг, 0,27 ммоль) и неочищенный 5-циано-4-этокси-N-(пиперидин-4-ил)пиколинамид 2,2,2-трифторацетат 2d (110 мг, 0,27 ммоль) растворяли в 15 мл ацетонитрила и добавляли карбонат натрия (56,6 мг, 0,53 ммоль). Реакционную смесь нагревали до 80°C и перемешивали в течение 48 часов. Реакционную смесь фильтровали и концентрировали при пониженном давлении. Остатки очищали тонкослойной хроматографией (ТСХ) с помощью элюирующей системы А с получением указанного в заголовке соединения (R)-5-циано-4-этокси-N-(1-(2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)пиперидин-4-ил)пиколинамида 2 (50 мг, 40%) в виде светло-желтого твердого вещества. MS m/z (ESI): 465,2 [М+1].
1Н NMR (400 MHz, CD3OD): δ 8,89 (s, 1Н), 8,74 (d, 1Н), 7,73 (s, 1Н), 7,65 (s, 2Н), 5,41 (d, 2Н), 5,09 (br, 1Н), 4,41 (d, 2Н), 3,71-3,85 (m, 2Н), 2,95 (br, 2H), 2,41-2,55 (m, 2H), 2,31 (s, 3Н), 2,12-2,27 (m, 2H), 1,57-1,81 (m, 4H), 1,40 (t, 3Н).
Пример 3
(R)-5-циано-4-(2-фторэтокси)-N-(1-(2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)пиперидин-4-ил)пиколинамид
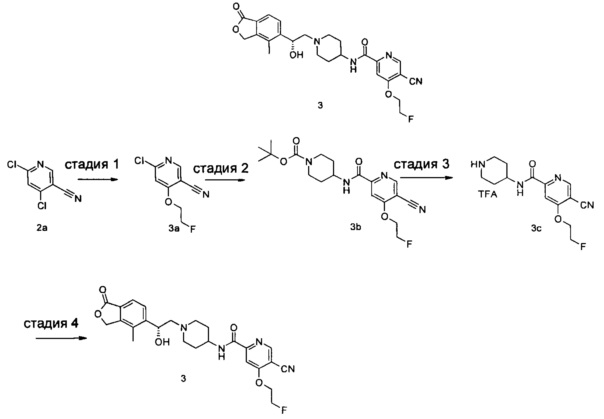
Стадия 1
6-хлор-4-(2-фторэтокси)никотинонитрил
2-Фторэтанол (150 мг, 2,34 ммоль) растворяли в 10 мл тетрагидрофурана, добавляли гидрид натрия (281 мг, 7,02 ммоль), полученную смесь перемешивали в течение 1 часа. 4,6-Дихлорникотинонитрил 2а (405 мг, 2,34 ммоль) растворяли в 25 мл тетрагидрофурана и добавляли по каплям в реакционную смесь при 0°C. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Реакционную смесь гасили 1 мл воды и концентрировали при пониженном давлении. Остатки очищали тонкослойной хроматографией (ТСХ) с помощью элюирующей системы Б с получением указанного в заголовке соединения 6-хлор-4-(2-фторэтокси)никотинонитрила 3а (210 мг, 45%) в виде белого твердого вещества.
МС m/z (ESI): 201,1 [М+1].
Стадия 2
Трет-бутил 4-(5-циано-4-(2-фторэтокси)пиколинамидо)пиперидин-1-карбоксилат 6-Хлор-4-(2-фторэтокси)никотинонитрил 3а (210 мг, 1,05 ммоль), 4-амино-1-трет-бутоксикарбонилпиперидин (216 мг, 1,05 ммоль), ацетат палладия (12 мг, 0,05 ммоль) 1,3-бис(дифенилфосфино)пропан (22 мг, 0,05 ммоль), триэтиламин (0,29 мл, 2,1 ммоль) и 20 мл ацетонитрила загружали в автоклав. Образовавшуюся смесь подвергали реакции в течение 16 часов при 80°C в атмосфере монооксида углерода в 10 бар. Реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении. Остатки очищали тонкослойной хроматографией (ТСХ) с помощью элюирующей системы Б с получением указанного в заголовке соединения трет-бутил 4-(5-циано-4-(2-фторэтокси)пиколинамидо)пиперидин-1-карбоксилата 3b (140 мг, 34%) в виде белого твердого вещества.
МС m/z (ESI): 391,1 [М-1].
Стадия 3
5-циано-4-(2-фторэтокси)-N-(пиперидин-4-ил)пиколинамид 2,2,2-трифторацетат
Трет-бутил 4-(5-циано-4-(2-фторэтокси)пиколинамидо)пиперидин-1-карбоксилат 3b (70 мг, 0,18 ммоль) растворяли в 5 мл дихлорметана и добавляли 1 мл трифторуксусной кислоты. Реакционную смесь перемешивали в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного целевого соединения 5-циано-4-(2-фторэтокси)-N-(пиперидин-4-ил)пиколинамид 2,2,2-трифторацетата 3с (80 мг) в виде желтого масла, которое использовали на следующей стадии без дальнейшей очистки.
Стадия 4
(R)-5-циано-4-(2-фторэтокси)-N-(1-(2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)пиперидин-4-ил)пиколинамид
(R)-4-Метил-5-(оксиран-2-ил)-изобензофуран-1(3Н)-он (34 мг, 0,18 ммоль) и неочищенный 5-циано-4-(2-фторэтокси)-N-(пиперидин-4-ил)пиколинамид 2,2,2-трифторацетат 3с (80 мг, 0,18 ммоль) растворяли в 20 мл ацетонитрила и добавляли карбонат натрия (38 мг, 0,36 ммоль). Реакционную смесь нагревали до 80°C и перемешивали в течение 48 часов. Реакционную смесь концентрировали при пониженном давлении. Остатки очищали тонкослойной хроматографией (ТСХ) с помощью элюирующей системы А с получением указанного в заголовке соединения (R)-5-циано-4-(2-фторэтокси)-N-(1-(2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)пиперидин-4-ил)пиколинамида 3 (10 мг, 12%) в виде белого твердого вещества.
MS m/z (ESI): 481,2 [М-1]
1Н NMR (400 MHz, CD3OD): δ 8,93 (s, 1Н), 8,79 (d, 1Н), 7,82 (s, 1Н), 7,71 (d, 2Н), 5,41 (d, 2Н), 5,14 (br, 1Н), 4,89 (t, 1Н), 4,77 (t, 1Н), 4,72 (t, 1Н), 4,65 (t, 1Н), 3,71-3,82 (m, 2Н), 2,85-3,15 (m, 2Н), 2,40-2,54 (m, 2Н), 2,31 (s, 3Н), 2,12-2,26 (m, 2Н), 1,61-1,90 (m, 4Н).
Пример 4
(R)-5-циано-4-(дифторметокси)-N-(1-(2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)пиперидин-4-ил)пиколинамид
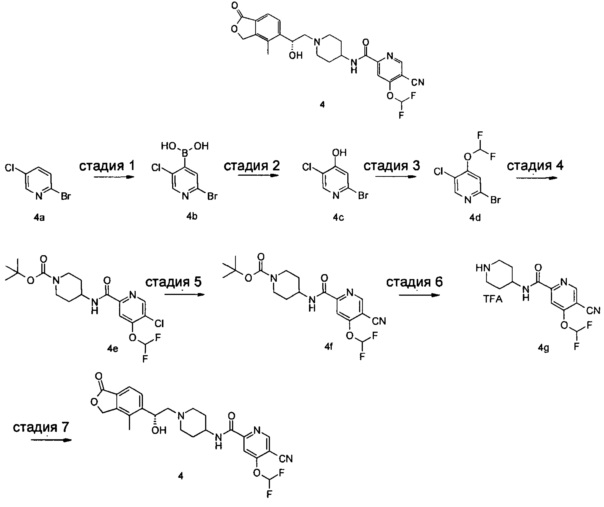
Стадия 1
(2-бром-5-хлорпиридин-4-ил)бороновой кислоты
2-Бром-5-хлорпиридин 4а (2 г, 10,4 ммоль) растворяли в 40 мл тетрагидрофурана и затем добавляли по каплям 7,8 мл 2 М диизопропиламида лития при -78°C. Полученную смесь перемешивали в течение 1 часа. Добавляли триизопропилборат (2,94 мг, 15,6 ммоль) и реакционную смесь перемешивали в течение 30 минут при -78°C. Реакционную смесь затем нагревали до комнатной температуры и дополнительно перемешивали в течение 16 часов. Добавляли 50 мл 4%-ного раствора гидроксида натрия. Смесь перемешивали в течение 30 мин. Водную фазу отделяли и доводили до рН 3-4 6 М раствором гидроксида натрия на ледяной бане. Затем водную фазу экстрагировали этилацетатом (50 мл × 2). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения (2-бром-5-хлорпиридин-4-ил) бороновой кислоты 4b (1,3 г, 53%) в виде белого твердого вещества.
Стадия 2
2-бром-5-хлорпиридин-4-ол
(2-Бром-5-хлорпиридин-4-ил)бороновую кислоту 4b (1,3 г, 5,51 ммоль) растворяли в 40 мл дихлорметана и добавляли перекись водорода (1,87 мл, 16,5 ммоль). Полученную смесь перемешивали в течение 16 часов. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 2-бром-5-хлорпиридин-4-ола 4 с (1 г, 88%) в виде белого твердого вещества.
MS m/z (ESI): 205,9/207,9 [М+1].
Стадия 3
2-бром-5-хлор-4-(дифторметокси)пиридин
Неочищенный 2-бром-5-хлорпиридин-4-ол 4 с (320 мг, 1,54 ммоль), 2-хлор-2,2-дифторацетат натрия (470 мг, 3,08 ммоль) и карбонат калия (470 мг, 3,39 ммоль) растворяли в 5 мл N,N-диметилацетамида. Реакционную смесь нагревали до 120°C и перемешивали в течение 1 часа под микроволновым воздействием. Реакционную смесь концентрировали при пониженном давлении. Остатки очищали тонкослойной хроматографией (ТСХ) с помощью элюирующей системы Б с получением указанного в заголовке соединения 2-бром-5-хлор-4-(дифторметокси)пиридина 4d (950 мг, 60%) в виде бесцветного масла.
Стадия 4
Трет-бутил 4-(5-хлор-4-(дифторметокси)пиколинамидо)пиперидин-1-карбоксилат
2-Бром-5-хлор-4-(дифторметокси)пиридин 4d (1,03 г, 3,99 ммоль), 4-амино-1-трет-бутоксикарбонилпиперидин (800 мг, 3,99 ммоль), ацетат палладия (45 мг, 0,2 ммоль) 1,3-бис(дифенилфосфино)пропан (82 мг, 0,2 ммоль), триэтиламин (1,1 мл, 7,98 ммоль) и 30 мл ацетонитрила загружали в автоклав. Полученная смесь была реакцией в течение 16 часов при 80°C в атмосфере монооксида углерода в 10 бар. Реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении. Остатки очищали тонкослойной хроматографией (ТСХ) с помощью элюирующей системы Б с получением указанного в заголовке соединения трет-бутил 4-(5-хлор-4-(дифторметокси)пиколинамидо)пиперидин-1-карбоксилата 4е (809 мг, 50%) в виде белого твердого вещества.
MS m/z (ESI): 404,1 [М-1].
Стадия 5
Трет-бутил 4-(5-циано-4-(дифторметокси)пиколинамидо)пиперидин-1-карбоксилат
Трет-бутил 4-(5-хлор-4-(дифторметокси)пиколинамидо)пиперидин-1-карбоксилат 4е (100 мг, 0,25 ммоль), цианид цинка (57,6 мг, 0,49 ммоль) и тетра(трифенилфосфин)палладий (88 мг, 0,07 ммоль) растворяли в 5 мл N,N-диметилацетамида. Смесь перемешивали под микроволновым воздействием в течение 30 мин при 170°C. Реакционный раствор концентрировали при пониженном давлении. Остатки очищали тонкослойной хроматографией (ТСХ) с помощью элюирующей системы Б с получением указанного в заголовке соединения трет-бутил 4-(5-циано-4-(дифторметокси)пиколинамидо)пиперидин-1-карбоксилата 4f (83 мг, 85%) в виде белого твердого вещества.
МС m/z(ESI): 395,0 [М-1].
Стадия 6
5-циано-4-(дифторметокси)-N-(пиперидин-4-ил)пиколинамид 2,2,2-трифторацетат
Трет-бутил 4-(5-циано-4-(дифторметокси)пиколинамидо)пиперидин-1-карбоксилат 4f (250 мг, 0,63 ммоль) растворяли в 5 мл дихлорметана и добавляли 2 мл трифторуксусной кислоты. Реакционную смесь перемешивали в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 5-циано-4-(дифторметокси)-N-(пиперидин-4-ил)пиколинамид 2,2,2-трифторацетата 4 г (540 мг), который использовали на следующей стадии без дальнейшей очистки.
MS m/z (ESI): 297,2 [М+1].
Стадия 7
(R)-5-циано-4-(дифторметокси)-N-(1-(2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)пиперидин-4-ил)пиколинамид
(R)-4-Метил-5-(оксиран-2-ил)-изобензофуран-1(3Н)-он (57,7 мг, 0,3 ммоль), неочищенный 5-циано-4-(дифторметокси)-N-(пиперидин-4-ил)пиколинамид 2,2,2-трифторацетат 4 г (260 мг, 0,3 ммоль) и N,N-диизопропилэтиламин (78,4 мг, 0,61 ммоль) растворяли в 3 мл этанола. Реакционную смесь нагревали до 135°C и перемешивали в течение 1 часа под микроволновым воздействием. Реакционную смесь концентрировали при пониженном давлении. Остатки очищали тонкослойной хроматографией (ТСХ) с помощью элюирующей системы А с получением указанного в заголовке соединения (R)-5-циано-4-(дифторметокси)-N-(1-(2-гидрокси-2-(4-метил-1-оксо-1,3-дигидроизобензофуран-5-ил)этил)пиперидин-4-ил)пиколинамида 4 (30 мг, 20%) в виде белого твердого вещества.
MS m/z (ESI): 487.2 [М+1]
1Н NMR (400 MHz, CD3OD): δ 9,13 (s, 1Н), 8,87 (d, 1Н), 7,98 (t, 1Н), 7,83 (s, 1Н), 7,63-7,78 (m, 2Н), 5,40 (d, 2Н), 5,08 (br, 1Н), 3,70-3,81 (m, 2Н), 2,96 (br, 2Н), 2,40-2,54 (m, 2Н), 2,28 (s, 3Н), 2,11-2,26 (m, 2Н), 1,61-1,75 (m, 4Н).
ПРИМЕРЫ ИСПЫТАНИЙ БИОЛОГИЧЕСКИЙ АНАЛИЗ
Тестовый пример 1. Ингибирующая активность настоящих соединений в каналах ROMK человека и каналах ROMK крысы
Способ, описанный ниже, используют для определения ингибирующей активности настоящих соединений в каналах ROMK у человека и каналах ROMK у крысы.
1. Материалы и инструменты
(1) Набор для анализа калиевого ионного канала FluxOR™ (F10016, Invitrogen)
(2) Ouabain (O3125-1G, sigma)
(3) Считывающее устройство для микропланшетов FlexStation3 (молекулярное устройство)
(4) Клетки ROMK/HEK293 человека: клеточная линия HEK293, стабильно экспрессирующая канал ROMK, трансфецированная кДНК ROMK человека (NCBI SEQ ID NO. NM-000220.4)
(5) Клетки ROMK/HEK293 крысы: клеточная линия HEK293, трансфицированная кДНК ROMK крысы (NCBI SEQ ID NO. NM-017023.1), стабильно экспрессирующую канал РОМК
(6) Клеточная линия HEK293: клеточный банк Китайской академии наук, GNHu43
2. Экспериментальная процедура
За исключением ddH2O и Ouabain, все реагенты для экспериментов взяты из калибровочного набора для анализа калиевого ионного канала FluxOR™, а способы образования также относятся к инструкциям набора.
(1) Клетки человека ROMK/HEK293 засевали на планшеты с покрытием PDL (поли-D-лизин) в количестве 20000 клеток на лунку в предыдущий день;
(2) После культивирования в течение ночи среду с планшета убирали; затем в соответствии с инструкциями набора для анализа калиевого ионного канала FluxOR™, добавляли краситель в количестве 100 мкл на лунку и затем инкубировали в течение 90 минут при комнатной температуре;
(3) Затем краситель декантировали и в каждую лунку добавляли 100 мкл буфера для анализа, содержащего Ouabain (300 мкМ) и пробенецида;
(4) 1 мкл соединения или DMSO добавляли в соответствующие лунки, встряхивали в течение 30 секунд, инкубировали в течение 30 мин при комнатной температуре;
(5) Планшеты помещали в считывающее устройство для микропланшетов FlexStation3 и затем добавляли буфер для стимуляции (K2SO4 : Tl2SO4 : 1XFluxOR буфер, не содержащий хлориды: ddH2O=3:12:40:125) в количестве 25 мкл в лунку, затем значение считывали непрерывно 5 мин при ЕХ/ЕМ 490/525 нм сразу;
(6) IC50 настоящих соединений на канале ROMK человека был получен с помощью программного обеспечения для обработки данных Graphpad.
Вышеописанные процедуры повторялись, за исключением замены клеток человека ROMK/HEK293 клетками крысы ROMK/HEK 293, чтобы определить ингибирование IC50 присутствующих соединений в канале ROMK крысы.
Ингибирующую активность настоящих соединений в канале ROMK человека или канале ROMK крысы исследовали с помощью анализа, описанного выше. Значения IC50 показаны в таблице 1 ниже.

Заключение. Соединения согласно настоящему изобретению обладают значительной ингибирующей активностью в отношении каналов ROMK человека или каналов ROMK крысы.
Тестовый пример 2. Ингибирующая активность настоящих соединений на hERG
Способ, описанный ниже, используют для определения ингибирующей активности настоящих соединений на hERG.
1. Материалы и инструменты
(1) Набор для анализа калиевого ионного канала FluxOR™ (F10016, Invitrogen)
(2) Считывающее устройство для микропланшетов FlexStation3 (молекулярное устройство)
(3) клетки hERG/HEK293: клеточная линия HEK293, стабильно экспрессирующая hERG-канал, трансфецированная кДНК hERG (NCBI SEQ ID NO. NM-000238 (RC215928, origene)).
2. Экспериментальная процедура
За исключением ddH2O, все экспериментальные реагенты взяты из калибровочного набора для анализа калиевого ионного канала FluxOR™, а способы образования также относятся к инструкциям набора.
(1) Клетки hERG/HEK293 человека высевали на планшеты с покрытием PDL (поли-D-лизин) в количестве 25000 клеток на лунку в предыдущий день;
(2) После культивирования в течение ночи среду с планшета убирали; затем в соответствии с инструкциями набора для анализа калиевого ионного канала FluxOR™, добавляли краситель в количестве 100 мкл на лунку и затем инкубировали в течение 90 минут при комнатной температуре;
(3) Затем краситель декантировали и в каждую лунку добавляли 100 мкл буфера для анализа, содержащего 100 мкл пробенецида;
(4) 1 мкл соединения или DMSO добавляли в соответствующие лунки, встряхивали в течение 30 секунд, инкубировали в течение 30 мин при комнатной температуре;
(5) Планшеты помещали в считывающее устройство для микропланшетов FlexStation3 и затем добавляли буфер для стимуляции ((K2SO4 : Tl2SO4 : 1XFluxOR хлоридный буфер, не содержащий хлоридов: ddH2O=2:1:2:5) в количестве 25 мкл на лунку, затем значение считывали непрерывно 5 минут при ЕХ/ЕМ 490/525 нм сразу;
(6) IC50 настоящих соединений на канале ионов hERG человека был получен с помощью программного обеспечения для обработки данных Graphpad.
Ингибирующую активность настоящих соединений на hERG тестировали с помощью анализа, описанного выше. Значения IC50 показаны в таблице 2 ниже.
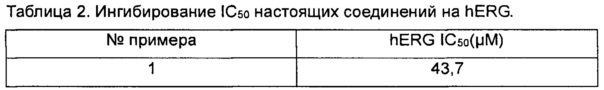
Заключение. Соединения согласно настоящему изобретению оказывают слабое ингибирующее действие на hERG, которое указывают на то, что соединения согласно настоящему изобретению обладают низкой кардиотоксичностью.
Тестовый пример 3. Влияние электрофизиологического ручного метода локальной фиксации потенциала на калиевый канал ROMK
1. Протокол
Эксперимент был разработан для проверки влияния соединений на калиевый канал ROMK в НЕK293 in vitro. Калиевый канал ROMK стабильно экспрессируется на клетках HEK293 настоящей заявки. После того, как поток ионов калия стабилизировался, эффект настоящего соединения на калиевый канал был получен сравнением потока калия, полученного до и после использования настоящего соединения в различных концентрациях.
2. Материалы и инструменты
(1) Клеточная линия НЕК293: клеточный банк китайской академии наук, GNHu43;
(2) клетки человека ROMK/HEK293: клеточная линия HEK293, стабильно экспрессирующая канал ROMK, трансфецированная кДНК ROMK человека (NCBI SEQ ID NO. NM-000220.4);
(3) внеклеточная жидкость (мМ): NaCl, 137; KCl, 4; CaCl2, 1,8; MgCl2, 1; HEPES, 10; глюкоза, 10; pH 7,4 (титрование NaOH);
(4) внутриклеточная жидкость (мМ): аспартат K, 130; MgCl2, 5; EGTA 5; HEPES, 10; Трис-АТФ, 4; рН 7,2 (титрование КОН);
соединения были приобретены в фирме Sigma (Сент-Луис, Миссури) в дополнение к NaOH и KOH для кислотно-основного титрования.
Среда для культивирования клеток: среда F12 Ham (Invitrogen), 10% (по объему) инактивированная фетальная бычья сыворотка, 100 мкг/мл гигромицина В, 100 мкг/мл Geneticin;
ручная система метода локальной фиксации потенциала: усилитель сигнала HEKA ЕРС-10 и цифровая система преобразования, приобретенная у Germany HEKA Electronics;
приборы микроконтроля: МР-225;
инструмент для рисования электродов: РС-10 (Narishige, Япония).
3. Экспериментальная процедура
Испытуемые соединения растворяли в диметилсульфоксиде (DMSO) и затем хранили при комнатной температуре. В день эксперимента тестируемые соединения разбавляли до следующей конечной концентрации (3, 10, 30, 100, 300 нМ) с использованием внеклеточной жидкости. Конечная концентрация тестируемых соединений в DMSO составляла 0,3%.
Клетки человека ROMK/HEK293 выращивали в культуральной чашке, содержащей указанную выше среду для культивирования клеток, и культивировали в инкубаторе, содержащем 5% СО2 при 37°C. Клетки человека ROMK/HEK293 переносили на круглую стеклянную пластинку, помещенную в чашку для культивирования за 24-48 часов до эксперимента, и выращивали при той же культуральной среде и условиях, как указано выше. Плотность клеток человека ROMK/HEK293 на каждой круглой стеклянной пластине требовалась для достижения того, чтобы подавляющее большинство клеток были независимыми и индивидуальными.
В этом эксперименте для текущей регистрации целых клеток использовали ручную систему метода локальной фиксации потенциала. Круглую стеклянную пластинку с человеческими клетками ROMK/HEK293, выращенными на поверхности, помещали в электрофизиологическую регистрирующую емкость под инвертированным микроскопом. В регистрирующей емкости поддерживали непрерывную перфузию с внеклеточной жидкостью (приблизительно 1 мл в минуту). В эксперименте применяли методику текущей регистрации целых клеток метода локальной фиксации потенциала. Если не указано иное, испытания проводили при комнатной температуре (приблизительно 25°C). Клетки фиксировали при минус 80 мВ. Напряжение клеточного зажима деполяризовали до плюс 20 мВ в течение 5 секунд для активации калиевого канала ROMK, а затем фиксировали до минус 50 мВ для устранения инактивации и генерации следового тока. Пиковое значение следового потока использовали в качестве значения потока РОМК. После того, как калиевый поток ROMK, записанный на вышеуказанных стадиях, стабилизировался при непрерывной перфузии внеклеточной жидкостью в регистрирующей емкости, тестируемое лекарственное средство можно перфузировать до тех пор, пока ингибирование лекарственным средством потока ROMK не достигнет устойчивого состояния. Как правило, повторное включение трех последовательных линий записи потока использовали в качестве критериев для определения стабильного состояния. После стабилизации клетки перфузировали внеклеточной жидкостью до тех пор, пока поток ROMK не возвращался к значению до добавления лекарственного средства. Одна клетка может быть протестирована на один или более препаратов или на более концентраций одного и того же препарата, но ее необходимо промыть внеклеточной жидкостью между различными лекарственными средствами.
4. Анализ данных
Данные анализировали с помощью программного обеспечения анализа данных HEKA Patchmaster, XLFit и Graphpad Prism. Значения IC50 показаны в таблице 3 ниже.

Заключение. Соединения согласно настоящему изобретению оказывают сильное ингибирующее действие на калиевый канал ROMK.
Тестовый пример 4. Влияние на калиевый канал hERG, определяемое электрофизиологическим ручным методом локальной фиксации потенциала
1. Цель
Целью этого эксперимента является испытание влияния соединений на hERG калиевый канал клетки СНО in vitro. В настоящем изобретении калиевый канал hERG стабильно экспрессируется на клетках СНО. После стабилизации потока ионов калия влияние соединения на калиевый канал было получено путем сравнения величины потока калия до и после применения различных концентраций соединений.
1. Материалы и инструменты
(1) Клеточная линия СНО: компания Sophion Bioscience, Дания;
(2) клетки hERG/CHO: клеточная линия СНО, стабильно экспрессирующая hERG-канал, трансфицированная кДНК РОМК человека (NCBI SEQ ID NO. NM-000238 (RC215928, origene));
(3) внеклеточная жидкость (мМ): EC 0.0.0 раствор NaCl-Рингера, NaCl, 145; KCl, 4; CaCl2, 2; MgCl2, 1; HEPES, 10; глюкоза, 10; PH 7,4 (титрование NaOH), осмотическое давление приблизительно 305 мОсм;
(4) внутриклеточная жидкость (мМ): 1С 0.0.0 раствор KCl-Рингера, KCl, 120; CaCl2, 5,374; MgCl2, 1,75; EGTA5; HEPES, 10; Na-АТФ 4; РН 7,25 (титрование KOH), осмотическое давление приблизительно 305 мОсм;
соединения были приобретены у Sigma (Сент-Луис, штат Миссури) в дополнение к NaOH и KOH для кислотно-щелочного титрования.
Среда для культивирования клеток: среда F12 Ham (Invitrogen), 10% (по объему) инактивированная фетальная бычьея сыворотка, 100 мкг/мл гигромицина В, 100 мкг/мл генетицин;
ручная система метода локальной фиксации потенциала: усилитель сигнала HEKA ЕРС-10 и цифровая система преобразования, приобретенная у HEKA Electronics, Германия;
приборы микроконтроля: МР-225;
инструмент для рисования электродов: РС-10 (Narishige, Япония).
2. Экспериментальная процедура
Испытуемые соединения градиентно разбавляли диметилсульфоксидом (DMSO) до 30, 10, 3, 1, 0,3 и 0,1 мМ и затем предварительно выдерживали при комнатной температуре. Затем исходный раствор разбавляли до следующей конечной концентрации (30, 10, 3, 1, 0,3 и 0,1 мкМ) с использованием внеклеточной жидкости. Конечная концентрация испытуемого соединения в DMSO составляла 0,1%. Все исходные растворы и тестируемые растворы подвергали воздействию ультразвуком в течение 5-10 минут для обеспечения полного растворения соединений.
Клетки СНО hERG выращивали в культуральной чашке, содержащей указанную выше культуральную среду для клеток, и культивировали в инкубаторе, содержащем 5% СО2 при 37°C. Клетки СНО hERG переносили на круглую стеклянную пластинку, помещенную в чашку для культивирования за 24-48 часов до эксперимента, и выращивали в той же культуральной среде и в условиях, которые указаны выше. Плотность клеток СНО hERG на каждой круглой стеклянной пластинке требовалась для того, чтобы большинство клеток были независимыми и индивидуальными.
В этом эксперименте для текущей регистрации целых клеток использовали ручную систему метода локальной фиксации потенциала. Круглую стеклянную пластинку с клетками СНО hERG, выращенными на поверхности, помещали в электрофизиологическую регистрирующую емкость под инвертированным микроскопом. В регистрирующей емкости поддерживали непрерывную перфузию внеклеточной жидкостью (приблизительно 1 мл в минуту). В эксперименте применяли методику текущей регистрации целых клеток метода локальной фиксации потенциала. Если не указано иное, испытания проводили при комнатной температуре (приблизительно 25°C). Клетки фиксировали при минус 80 мВ. Напряжение клеточного зажима деполяризовали до плюс 20 мВ в течение 5 секунд для активации калиевого канала hERG, а затем клеточный зажим фиксировали до минус 50 мВ для устранения инактивации и генерации следового тока. Пиковое значение следового тока использовали в качестве значения тока hERG. После того, как калиевый поток hERG, записанный на вышеуказанных стадиях, стабилизировался при непрерывной перфузии внеклеточной жидкостью в регистрирующей емкости, испытуемое лекарственное средство можно перфузировать до тех пор, пока ингибирование лекарственным средством потока hERG не достигнет устойчивого состояния. Как правило, повторное включение трех последовательных линий записи потока использовалось в качестве критериев для определения стабильного состояния. После стабилизации клетки перфузировали внеклеточной жидкостью до тех пор, пока ток hERG не возвращался к значению до добавления лекарственного средства. Одну клетку можно протестировать на один или более препаратов или на более концентраций одного и того же препарата, но ее необходимо промыть внеклеточной жидкостью между различными лекарственными средствами.
4. Анализ данных
Данные анализировали с помощью программного обеспечения анализа данных HEKA Patchmaster, XLFit и Graphpad Prism. Значения IC50 показаны в таблице 4 ниже.
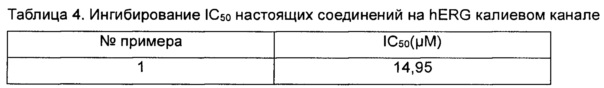
Заключение. Соединения согласно настоящему изобретению оказывают слабое ингибирующее действие на калиевый канал hERG, что указывает, что соединения согласно настоящему изобретению обладают низкой кардиотоксичностью.
Тестовый пример 5. Фармакокинетический анализ соединений по настоящему изобретению
1. Аннотация
Крыс использовали в качестве подопытных животных. Концентрацию лекарственного средства в плазме в разные моменты времени определяли LC/MS/MS после введения соединений крысам. Фармакокинетическое поведение соединений по настоящему изобретению изучали и оценивали на крысах.
2. Протокол
2.1 Образцы
Соединения примера 1
2.2 Испытательные животные
4 здоровых взрослых крысы Спрег-Доули (SD), самцы и самки, половина на половину, которые были приобретены у SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., СО, с сертификатом №: SCXK (Shanghai) 2008-0016.
2.3. Получение тестируемых соединений
Соответствующее количество тестируемых соединений взвешивали и добавляли 0,5% CMC-Na до конечного объема для приготовления суспензии 0,5 мг/мл путем ультразвуковой обработки.
2.4 Введение
После голодания в течение ночи 4 крысам SD, самкам и самцам, половина на половину, вводили внутрижелудочно с дозировкой 5,0 мг/кг и с объемом введения 10 мл/кг.
3. Процесс
Кровь (0,1 мл) отбирали из орбитального синуса перед введением и через 0,5 ч, 1,0 ч, 2,0 ч, 4,0 ч, 6,0 ч, 8,0 ч, 11,0 ч и 24 ч после введения. Образцы хранили в антикоагуляционных ЭДТА пробирках и центрифугировали в течение 10 минут при 3500 оборотах в минуту для отделения плазмы крови. Образцы плазмы хранили при минус 20°C. Крыс кормили через 2 часа после введения.
Концентрацию в плазме испытуемых соединений на крысах после внутрижелудочного введения определяли LC/MS/MS. Образцы плазмы анализировали после предварительной обработки осаждением белка.
4. Результаты фармакокинетических параметров
Фармакокинетические параметры соединений по настоящему изобретению представлены в таблице 5 ниже.
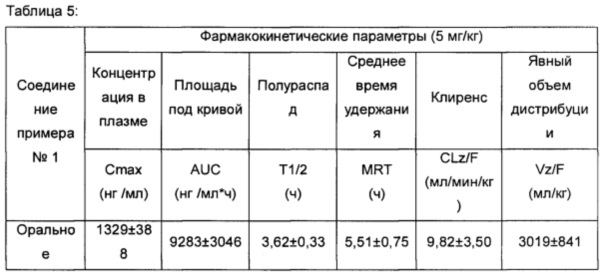
Тестовый пример 6. Диуретическая эффективность ингибиторов ROMK у крыс SD
1. Цель
Оценивали диуретическую эффективность соединения 1 и положительного контрольного препарата ингибитора ROMK на крысах SD.
2. Способ и материалы
2.1. Подопытные животные и условия питания
Самцов крыс SD закупали у SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., СО (Шанхай, Китай, сертификат №2008001647752, лицензия SCXK (Shanghai) 2013-0016). Крысы имели вес 120-130 г и их кормили в условиях 5 крыс на клетку, регулирование света/темноты 12/12 часов при постоянной температуре 23±1°C, влажности 50-60% и свободным доступом к воде и пище. Самцов крыс SD акклиматизировали до этого состояния в течение 7 дней до их использования в экспериментах с диурезом.
2.2 Испытуемый препарат
Соединение 1;
Структура препарата для положительного контроля следующая:
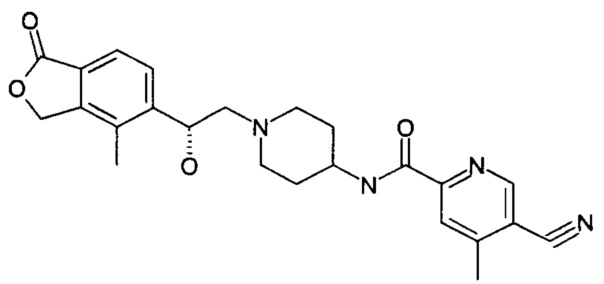
0,9% раствор NaCl (500 мл: 4,5 г).
CMC.Na: Партия №20131022, Sinopharm Group Chemical Reagent Co., Ltd.
Набор для обнаружения натрия: партия №20150203 от Nanjing Jiancheng Biotechnology Company.
Набор для обнаружения калия: партия №20141112 от Nanjing Jiancheng Biotechnology Company.
Дозировку препарата рассчитывали в соответствии со свободным основанием.
2.3. Экспериментальная конструкция и способ
2.3.1 Группирование животных
После адаптивного кормления животных группировали следующим образом:
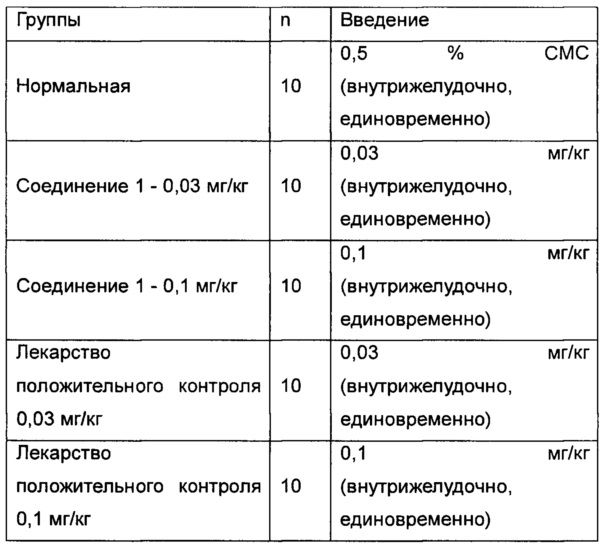
2.3.2 Способ эксперимента
Эксперимент проводили в соответствии со способом, описанным в патентной заявке (WO 2010129379 А1). После адаптивного кормления крыс помещали в камеры для исследования метаболизма и оставляли голодать в течение ночи. Крыс взвешивали и случайным образом разделяли на следующие группы: холостую контрольную группу, группу тестирования соединения 1 с дозировкой 1 0,03 мг/кг, группы тестирования соединения 1 с дозировкой 0,1 мг/кг, группу положительного контроля с дозировкой 0,03 мг/кг, группу положительного контроля с дозировкой 0,1 мг/кг, с 10 крысами для каждой группы. Каждой крысе вводили внутрижелудочно каждое соединение (внутрижелудочно, 1 мл/кг). Крысам в пустой контрольной группе вводили соответствующий растворитель. После внутрижелудочного введения крыс помещали в нормальную клетку. Через 30 мин вводили 25 мл/кг нормального солевого раствора. Крыс помещали в камеры для исследования метаболизма, немедленно оставляли без еды и воды. Общий объем мочи через 4 часа собирали и измеряли. Измеряли также экскрецию калия с мочой в течение 4 часов. И орбитальную сыворотку собирали после сбора мочи для проверки концентраций натрия и калия в сыворотке крови.
2.4. Экспериментальная установка
Центрифуга для комнатной температуры: модель 5417С, поставленная Eppendorf.
2.5 Представление данных и статистическая обработка
Экспериментальные данные были выражены как среднее ± стандартное отклонение (S.D.). Данные были статистически сопоставлены с использованием t-теста Excel. Данные между группой лекарства и контрольной группой анализировали и сравнивали, чтобы определить, имело ли место статистически значимое различие. *Р менее 0,05 указывает на то, что существует значимое различие между группой лекарств и контрольной группой, а **Р менее 0,01 указывает на то, что существует большое значимое различие между группой лекарств и контрольной группой.
3. Результат
Результаты показывают, что по сравнению с холостой контрольной группой, объем мочи для групп положительного контрольного препарата с дозировкой 0,03 мг/кг и 0,1 мг/кг значительно увеличивается (Р менее 0,05), при этом выход мочи увеличивается в 1,41 раза и в 1,46 раза соответственно; объем мочи для тестируемого соединения 1 для групп с дозировкой 0,03 мг/кг и 0,1 мг/кг значительно увеличивается (Р менее 0,01), при этом выход мочи увеличивается в 2,76 раза и в 3,22 раза (см. Фиг. 1). Группа лекарства положительного контроля и группа соединения 1 значительно увеличивали экскрецию натрия с мочой (Р менее 0,01), при которой экскреция натрия с мочой увеличивалась в 1,57 раза, в 1,65 раза, в 3,12 раза и в 3,31 раза (см. Фиг. 2). По сравнению с нормальной контрольной группой, калий в моче при лекарстве положительного контроля и тестируемом препарате был слегка повышен, но статистически не значимо (см. Фиг. 3). Одновременно с этим сыворотчный натрий и калий для лекарства положительного препарата и каждой из тестируемых групп немного изменялся (Р более 0,05) (см. Фиг. 4 и 5).
4. Объяснение
Согласно функциональному характеру, K+-каналы могут быть разделены на следующие четыре типа: медленные (с задержкой) K+ каналы (K-каналы), быстрые (ранние) K+ каналы (А-каналы), активированные Са2+ K+ каналы [K(Са)] и каналы K+ внутреннего выпрямления. K+ каналы внутреннего выпрямления (Kir) можно дополнительно разделить на семь типов: Kir1-Kir7, с различным кодированием гена KCNJ. Внешний медуллярный калиевый канал почки (ROMK) относится к типу Kir1. Есть по крайней мере три подтипа ROMK в почке крысы: ROMK1, ROMK2 и ROMK3. ROMK2 в основном распределяется в толстом сегменте восходящей ветви медуллярной петли. ROMK1 и ROMK3 в основном экспрессируются в собирающих канальцах.
ROMK, экспрессируемый в толстом сегменте восходящей ветви медуллярной петли, регулирует секрецию и реабсорбцию калия вместе с транспортерами Na/K/Cl. ROMK, экспрессируемый в корковых собирательных канальцах, регулирует секрецию калия вместе с Na/K-транспортером. Блокирование места ROMK может способствовать секреции NaCl в просвет без чрезмерной гипокалиемии, приводящей к гипокалиемии. Это хорошее направление исследований диуретиков для пациентов с гипертонической болезнью. Этот эксперимент должен изучить мочегонный эффект ингибитора ROMK.
В этом эксперименте растворимость тестируемого соединения 1 является очень хорошей. Явления расслоения нет. Но при взвешивании положительного контрольного лекарства возникает статическое электричество, которое нелегко взвесить. При начальном измельчении наблюдают образование сгустков и плохую растворимость. После полного измельчения его растворимость становится лучше. Результаты также показывают, что однократное пероральное введение соединения 1 и лекарства положительного контроля крысам имеет значимый диуретический эффект и влияние на экскрецию натрия в сравнении с нормальной группой. Кроме того, они отображают зависимость от дозировки между каждой дозировкой тестируемого соединения 1 и лекарства положительного контроля.
5. Вывод
Соединение 1 и лекарство положительного контроля оба обладают значительным диуретическим действием и влиянием на экскрецию натрия, но не оказывают влияния на сывороточный калий. Однако диуретическое действие соединения 1 лучше, чем диуретическое действие лекарства положительного контроля. Эффективность препарата для каждой группы зависит от дозировки.
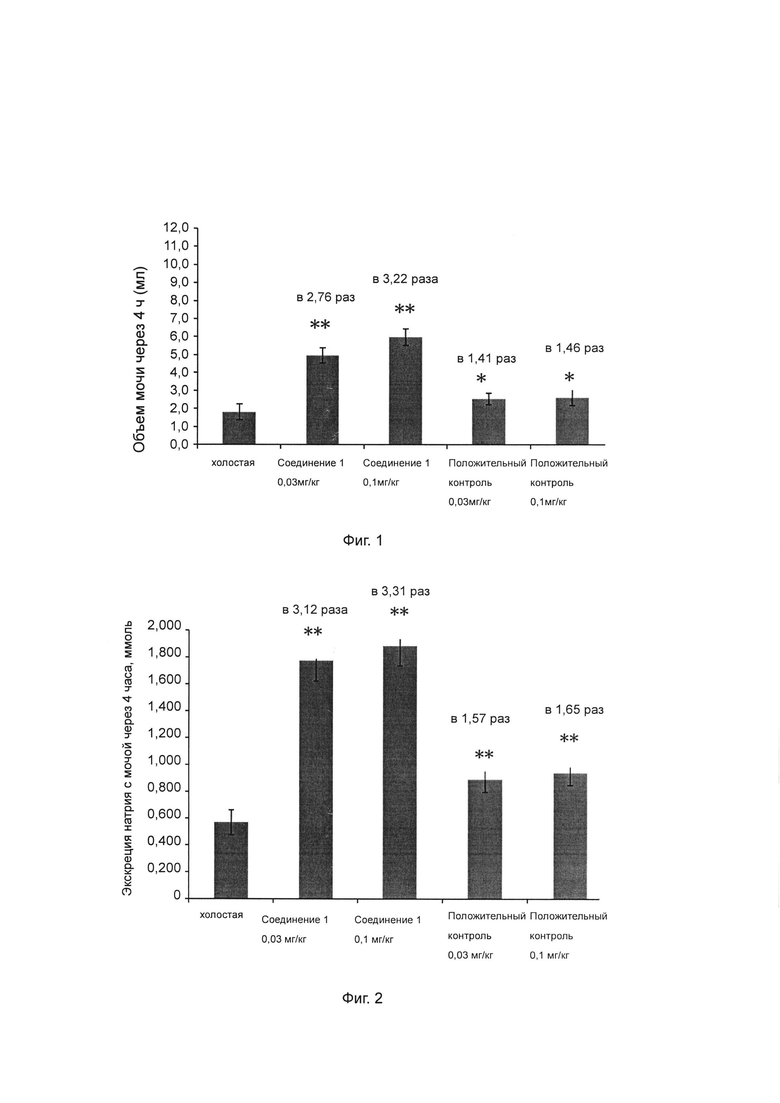
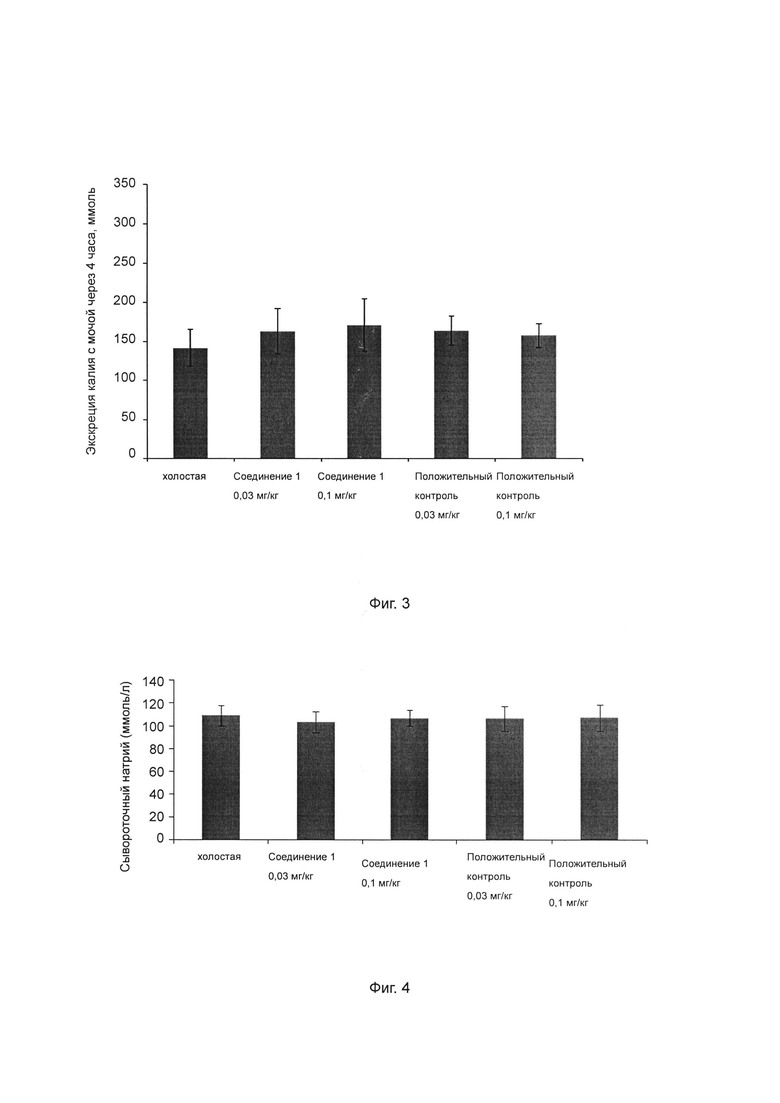
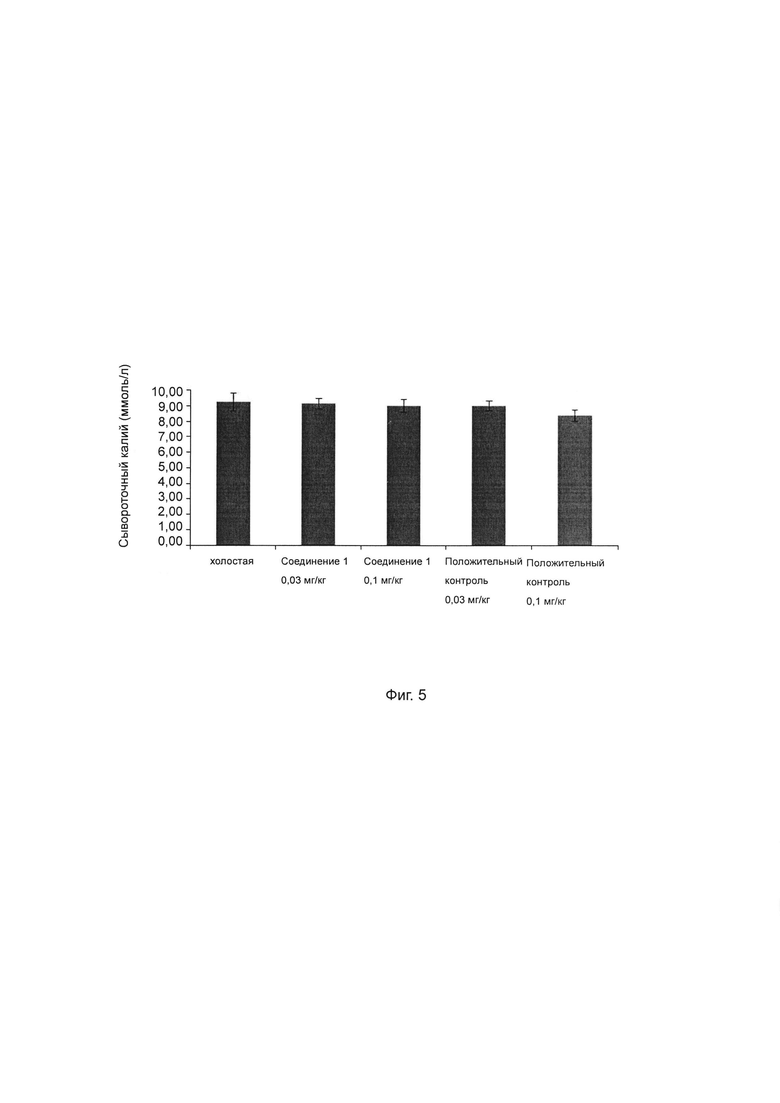
Изобретение относится к производным пиридинкарбоксамида, представленным общей формулой (I), к их фармацевтически приемлемым солям, где R1 представляет собой C1-C6 алкил, где указанный C1-C6 алкил необязательно дополнительно замещен одним или более галогеном; R2 представляет собой водород; R3 представляет собой
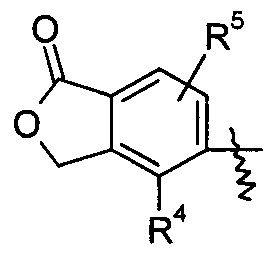 ; R4 и R5 каждый независимо выбирают из группы, состоящей из водорода, C1-C6 алкила; n равно 0, 1 или 2. Изобретение также относится к способу получения указанных соединений, к фармацевтической композиции, ингибирующей внешний медуллярный калиевый канал почки (ROMK) на основе указанных соединений. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для лечения и/или профилактики заболеваний почек, гипертонии и сердечной недостаточности. 10 н. и 8 з.п. ф-лы, 5 ил., 5 табл., 4 пр.
; R4 и R5 каждый независимо выбирают из группы, состоящей из водорода, C1-C6 алкила; n равно 0, 1 или 2. Изобретение также относится к способу получения указанных соединений, к фармацевтической композиции, ингибирующей внешний медуллярный калиевый канал почки (ROMK) на основе указанных соединений. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для лечения и/или профилактики заболеваний почек, гипертонии и сердечной недостаточности. 10 н. и 8 з.п. ф-лы, 5 ил., 5 табл., 4 пр.
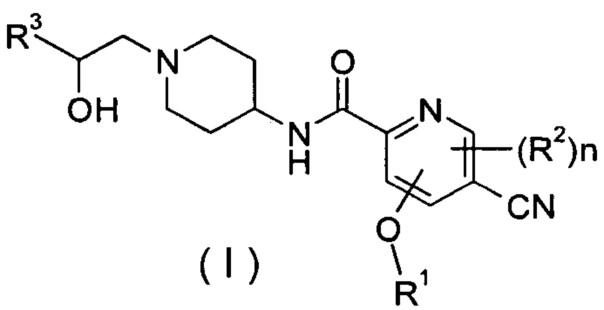
1. Соединение формулы (I)
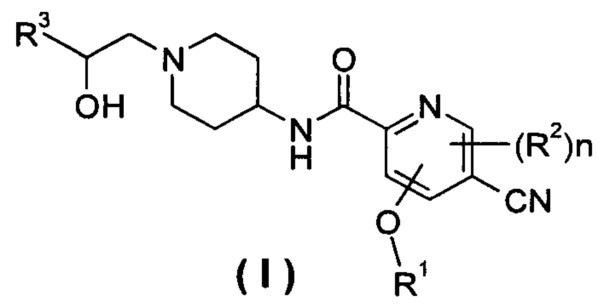
или его фармацевтически приемлемые соли,
где
R1 представляет собой C1-C6 алкил, где указанный C1-C6 алкил необязательно дополнительно замещен одним или более галогеном;
R2 представляет собой водород;
R3 представляет собой
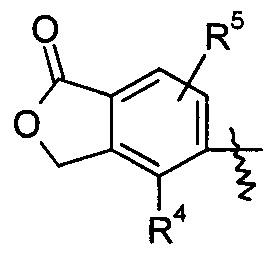 ;
;
R4 и R5 каждый независимо выбирают из группы, состоящей из водорода, C1-C6 алкила;
n равно 0, 1 или 2.
2. Соединение формулы (I) согласно п. 1, где R1 выбирают из группы, состоящей из метила, этила и пропила.
3. Соединение формулы (I) согласно п. 1, где R4 представляет собой C1-C6 алкил и R5 представляет собой водород.
4. Соединение формулы (I) согласно п. 1, которое представляет собой соединение формулы (II)
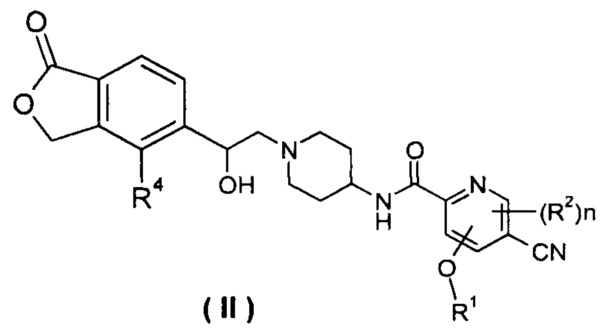 ,
,
или его фармацевтически приемлемые соли,
где R1, R2, R4 и n являются такими, как определено в п. 1.
5. Соединение формулы (I) согласно п. 1, которое представляет собой соединение формулы (III)
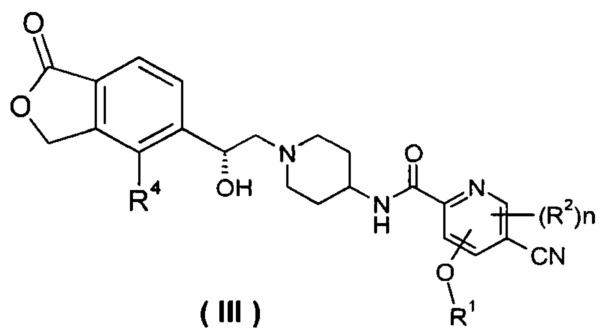 ,
,
или его фармацевтически приемлемые соли,
где R1, R2, R4 и n являются такими, как определено в п. 1.
6. Соединение формулы (I) согласно любому из пп. 1-5, где указанные соединения выбраны из:
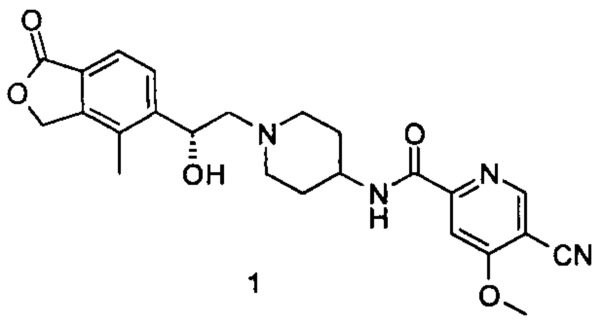
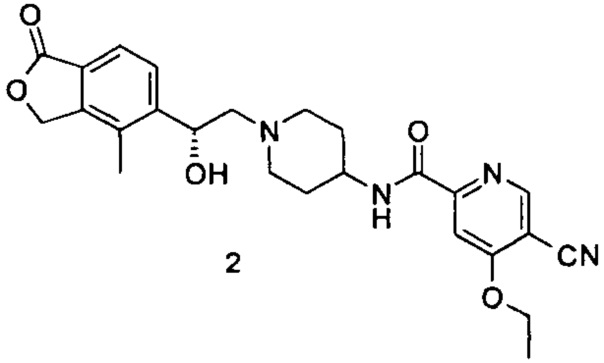
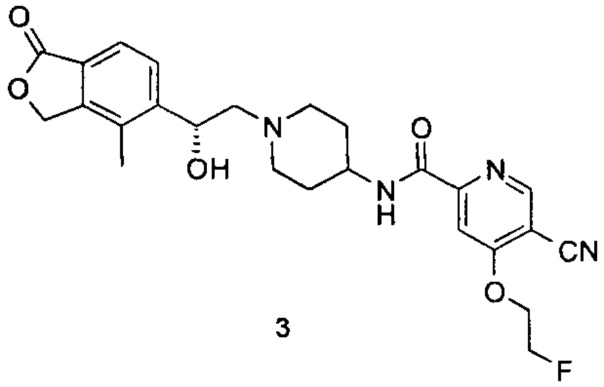 и
и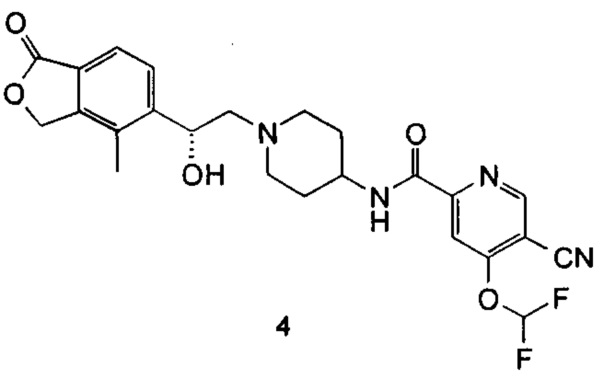 .
.
7. Соединение формулы (IA)
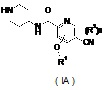
или его фармацевтически приемлемые соли,
где
R1 представляет собой C1-C6 алкил, где указанный C1-C6 алкил необязательно дополнительно замещен одним или более галогеном;
R2 представляет собой водород;
n равно 0, 1 или 2.
8. Соединение формулы (IA) согласно п. 7, где указанное соединение представляет собой:
 .
.
9. Способ получения соединения формулы (I) согласно п. 1, включающий стадию:
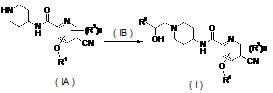
нагревания соединения формулы (IA) с замещенным бензофурановым производным формулы (IB) (R)-4-метил-5-(оксиран-2-ил)изобензофуран-1(3H)-он в органическом растворителе с получением соединения формулы (I);
где растворитель выбран из группы, состоящей из уксусной кислоты, метанола, этанола, ацетонитрила, тетрагидрофурана, дихлорметана, диметилсульфоксида, 1,4-диоксана, воды, N,N-диметилформамида или N,N-диметилацетамида;
R1 - R5 и n являются такими, как определено в п. 1.
10. Способ по п. 9, где органический растворитель представляет собой неполярный растворитель.
11. Способ по п. 10, где органический растворитель представляет собой ацетонитрил.
12. Фармацевтическая композиция, ингибирующая ROMK (внешний медуллярный калиевый канал почки), содержащая терапевтически эффективное количество соединения формулы (I) согласно любому из пп. 1-6 и фармацевтически приемлемый носитель, разбавители или эксципиент.
13. Применение соединения формулы (I) согласно любому из пп. 1-6, или фармацевтической композиции согласно п. 12 для приготовления ингибитора ROMK.
14. Применение соединения формулы (I) согласно любому из пп. 1-6 или фармацевтической композиции согласно п. 12 для приготовления лекарственного средства для лечения или профилактики гипертонии и/или сердечной недостаточности, опосредованной ROMK.
15. Применение соединения формулы (I) согласно любому из пп. 1-6 или фармацевтической композиции согласно п. 12 для приготовления лекарственного средства для лечения или профилактики опосредованных ROMK заболеваний, где указанные заболевания предпочтительно выбраны из группы, состоящей из острой и хронической почечной недостаточности, нефротического синдрома, легочной гипертензии, сердечно-сосудистого заболевания, инфаркта миокарда, инсульта, сердечной недостаточности, легочной гипертонии, атеросклероза и мочекаменной болезни.
16. Способ ингибирования ROMK, включающий введение терапевтически эффективного количества соединения формулы (I) согласно любому из пп. 1-6 или фармацевтической композиции согласно п. 12 пациенту, нуждающемуся в этом.
17. Способ лечения или профилактики гипертонии и/или сердечной недостаточности, опосредованной ROMK, включающий введение терапевтически эффективного количества соединения формулы (I) согласно любому из пп. 1-6, или фармацевтической композиции согласно п. 12 пациенту, нуждающемуся в этом.
18. Способ лечения или профилактики опосредованных ROMK заболеваний, включающий введение терапевтически эффективного количества соединения формулы (I) согласно любому из пп. 1-6, или фармацевтической композиции согласно п. 12 пациенту, нуждающемуся в этом, где указанные заболевания предпочтительно выбирают из группы, состоящей из острой и хронической почечной недостаточности, нефротического синдрома, легочной гипертензии, сердечно-сосудистого заболевания, инфаркта миокарда, инсульта, сердечной недостаточности, легочной гипертонии, атеросклероза и мочекаменной болезни.
| WO 2014085210 A1, 05.06.2014 | |||
| WO 2010129379 A1, 11.11.2010 | |||
| WO 2014018764 A1, 30.01.2014 | |||
| Устройство выдвижного театрального портала | 1929 |
|
SU19509A1 |

