ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к производному сульфонилбензамида и его конъюгату лиганд-лекарственное средство (ADC). В частности, настоящее изобретение относится к производному сульфонилбензамида с совершенно новой структурой и его конъюгату антитело-лекарственное средство, способу его получения, фармацевтической композиции, содержащей конъюгат, и применению конъюгата или фармацевтической композиции.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Важной особенностью, которая отличает опухолевые клетки от нормальных клеток, является то, что апоптоз в опухолевых клетках ингибируется, что дает им большее преимущество в выживании. Белки семейства BCL-2 являются важными факторами в регуляции ингибирования апоптоза.
Белки семейства BCL-2 в основном присутствуют на митохондриальных мембранах и могут быть классифицированы на две основные группы в соответствии с их функциями: антиапоптотические и проапоптотические белки. Антиапоптотический белок включает BCL-2, BCL-xL, BCL-w, MCL-1 и т.п. Проапоптотический белок включает белки Вах, Bak и белок, содержащий только ВН3. Вах и Bak при активации превращаются из безвредных мономеров в смертоносные олигомеры, которые образуют поры во внешней мембране митохондрий, увеличивают проницаемость митохондриальной мембраны клетки и способствуют высвобождению цитохрома С и тому подобного в цитоплазму, что приводит к гибели клеток. Белок, содержащий только ВН3, содержит только домен ВН3. В выжившем состоянии клетки белок, содержащий только ВН3 (например, Bim) связывается с антиапоптотический белком. Когда клетка подвергается внешнему давлению, баланс связывания нарушается, и белок, содержащий только ВН3, высвобождается для связывания с ВАХ на митохондриях, тем самым способствуя образованию полимеров ВАХ/BAK, способствуя высвобождению цитохрома С и SMAC в цитоплазму и активируя последующий апоптотический путь.
Имеющиеся доклинические данные показывают, что двухцелевой ингибитор BCL2/BCL-xL ABT263 не только эффективен против гематологических опухолей, но также оказывает хорошее ингибирующее действие на солидные опухоли, особенно мелкоклеточный рак легкого. Однако клиническая токсичность АВТ263 для тромбоцитов ограничила его эффективность и в конечном итоге привела к прекращению клинических испытаний.
По-прежнему существует необходимость в исследовании двухцелевого ингибитора BCL2/BCL-xL, чтобы его можно было использовать в качестве монотерапии или в комбинированной терапии или в ADC для удовлетворения клинических требований.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложено соединение общей формулы (D) или его фармацевтически приемлемая соль:
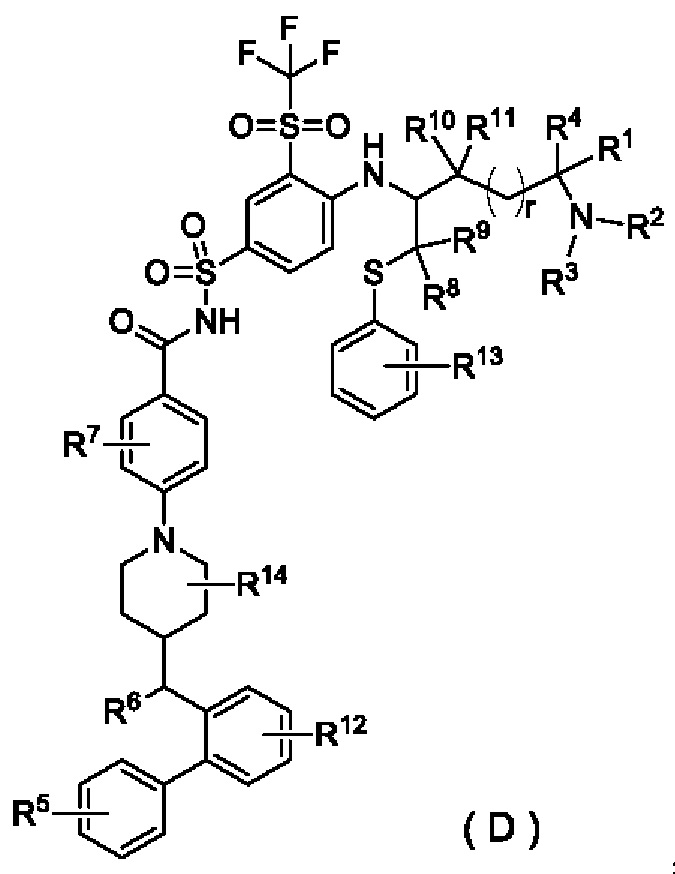
где:
R1 выбран из группы, состоящей из дейтерия, водорода, алкила и дейтерированного алкила;
R2 выбран из группы, состоящей из алкила и гидроксиалкила;
или R1 и R2 вместе с атомом, к которому они присоединены, образуют гетероциклил, необязательно дополнительно замещенный заместителем, выбранным из группы, состоящей из галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила;
R3 выбран из группы, состоящей из водорода, гидроксиалкила, алкила, дейтерированного алкила, циклоалкила и циклоалкилалкила;
R4 выбран из группы, состоящей из водорода, галогена, дейтерированного алкила и алкила;
R5 выбран из группы, состоящей из галогена и галогеналкила;
R6 выбран из группы, состоящей из алкила, амино, гидрокси и алкокси;
R7 выбран из группы, состоящей из водорода, галогена, алкила, дейтерированного алкила, гидрокси и алкокси;
каждый из R8 и R9 независимо выбран из группы, состоящей из водорода, алкила, дейтерированного алкила и циклоалкила; или R8 и R9 вместе с атомом, к которому они присоединены, образуют циклоалкил;
каждый из R10 и R11 независимо выбран из группы, состоящей из водорода, алкила, дейтерированного алкила и циклоалкила; или R10 и R11 вместе с атомом, к которому они присоединены, образуют циклоалкил;
R12 выбран из группы, состоящей из водорода, дейтерия, дейтерированного алкила, галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила;
R13 выбран из группы, состоящей из водорода, дейтерия, дейтерированного алкила, галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила;
R14 выбран из группы, состоящей из водорода, дейтерия, дейтерированного алкила, галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила; и
r выбран из группы, состоящей из 0, 1, 2 и 3;
при условии, что если R1 и R2 вместе с атомом, к которому они присоединены, не образуют гетероциклил, R5 представляет собой галогеналкил.
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, где R1 представляет собой водород, a R2 представляет собой алкил.
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, где R1 и R2 вместе с атомом, к которому они присоединены, образуют гетероциклил; предпочтительно, гетероциклил необязательно дополнительно содержит 1-3 гетероатома, выбранных из группы, состоящей из азота и кислорода; более предпочтительно, R1 и R2 вместе с атомом, к которому они присоединены, образуют 3-6-членный гетероциклил; предпочтительно, 3-6-членный гетероциклил необязательно дополнительно содержит 1-3 гетероатома, выбранных из группы, состоящей из азота и кислорода; и наиболее предпочтительно, R1 и R2 вместе с атомом, к которому они присоединены, образуют пирролидинил.
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, которое представляет собой соединение общей формулы (D-I) или его фармацевтически приемлемую соль:
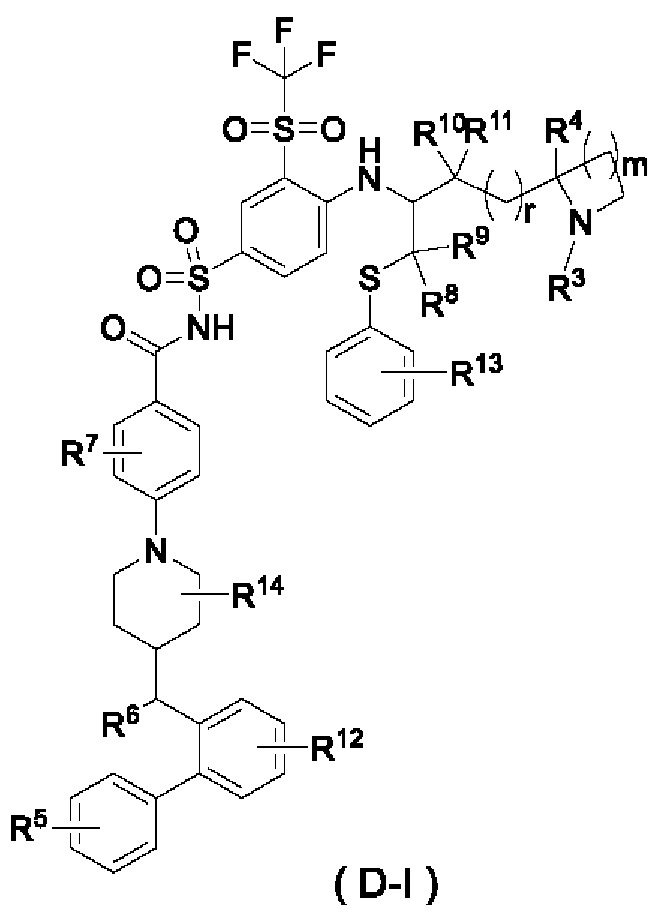
где m выбран из группы, состоящей из 0, 1, 2 и 3, и
r и R3-R14 являются такими, как определено в общей формуле (D).
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, которое представляет собой соединение общей формулы (D-II) или его фармацевтически приемлемую соль:
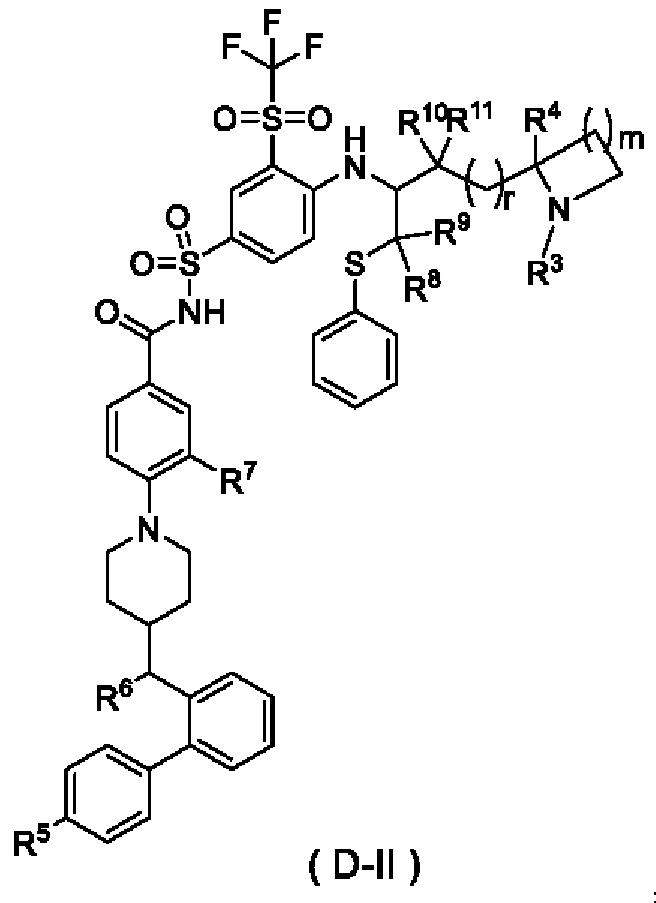
где m выбран из группы, состоящей из 0, 1, 2 и 3, и
r и R3-R11 являются такими, как определено в общей формуле (D).
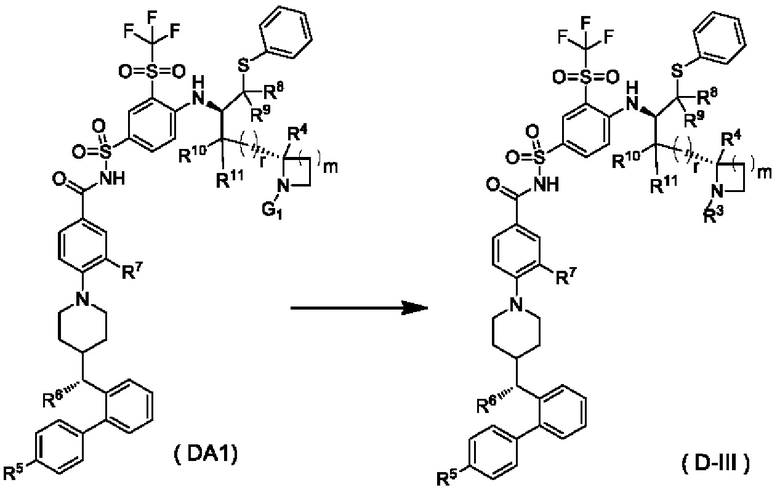
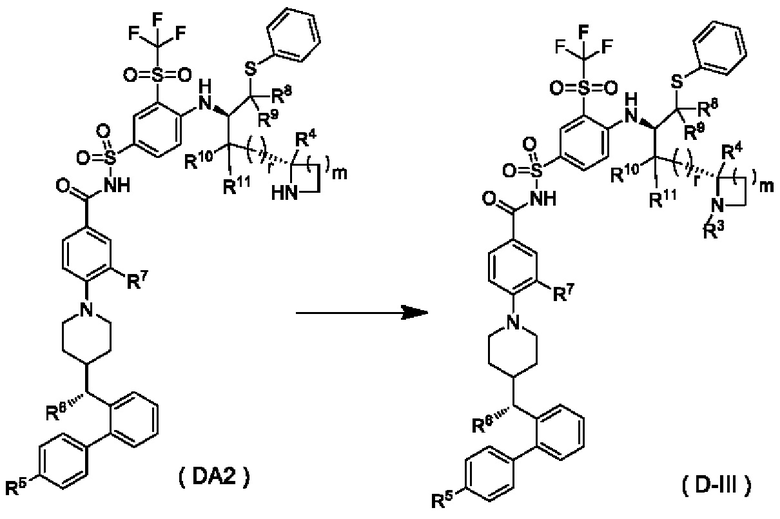
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, которое представляет собой соединение общей формулы (D-III) или его фармацевтически приемлемую соль:
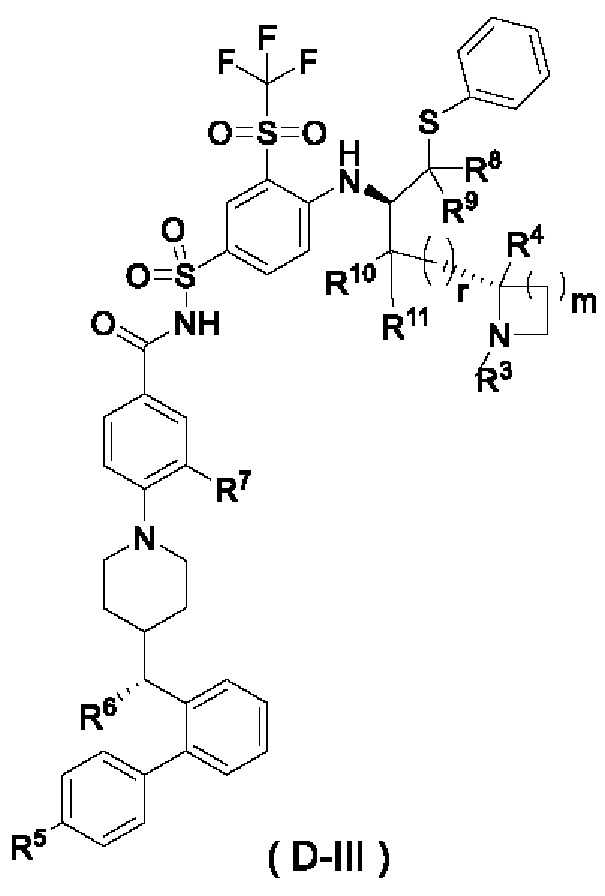
где m выбран из группы, состоящей из 0, 1, 2 и 3, и
r и R3-R11 являются такими, как определено в общей формуле (D). В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, которое представляет собой соединение общей формулы (D-IV) или его фармацевтически приемлемую соль:
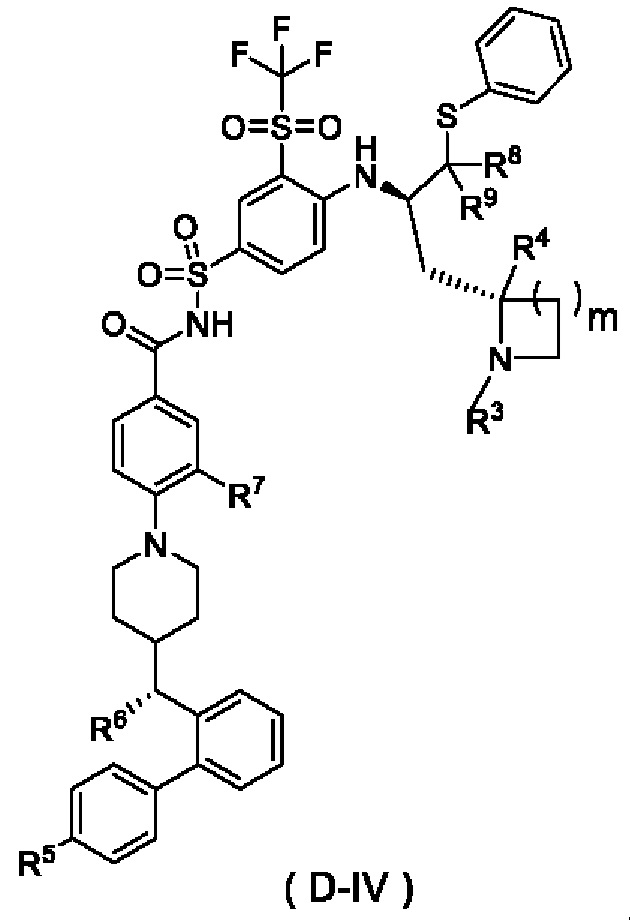
где m выбран из группы, состоящей из 0, 1, 2 и 3, и
R3-R9 являются такими, как определено в общей формуле (D).
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, где m представляет собой 2.
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, где R5 представляет собой хлор.
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, где R5 представляет собой трифторметил.
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, где R3 выбран из группы, состоящей из водорода, гидроксиалкила и алкила; предпочтительно гидроксиалкила; и более предпочтительно C1-6 гидроксиалкила.
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, которое представляет собой соединение общей формулы (D-V) или его фармацевтически приемлемую соль:
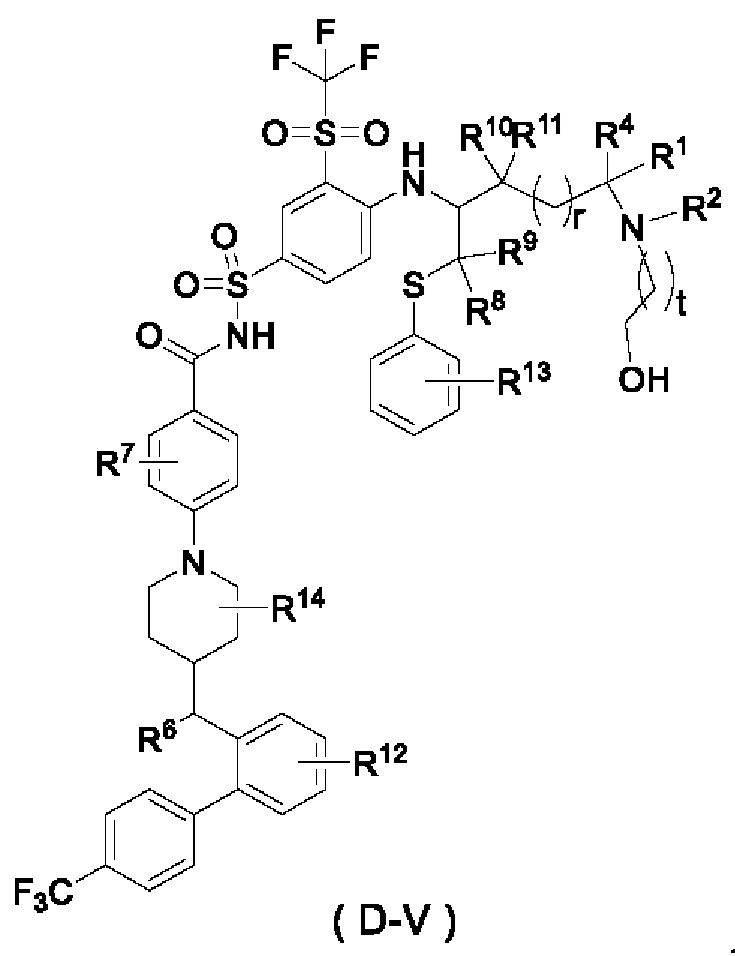
где t выбран из группы, состоящей из 0, 1, 2 и 3, предпочтительно 1, и
r, R1, R2, R4 и R6-R14 являются такими, как определено в общей формуле (D).
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, которое представляет собой соединение общей формулы (D-VI) или его фармацевтически приемлемую соль:
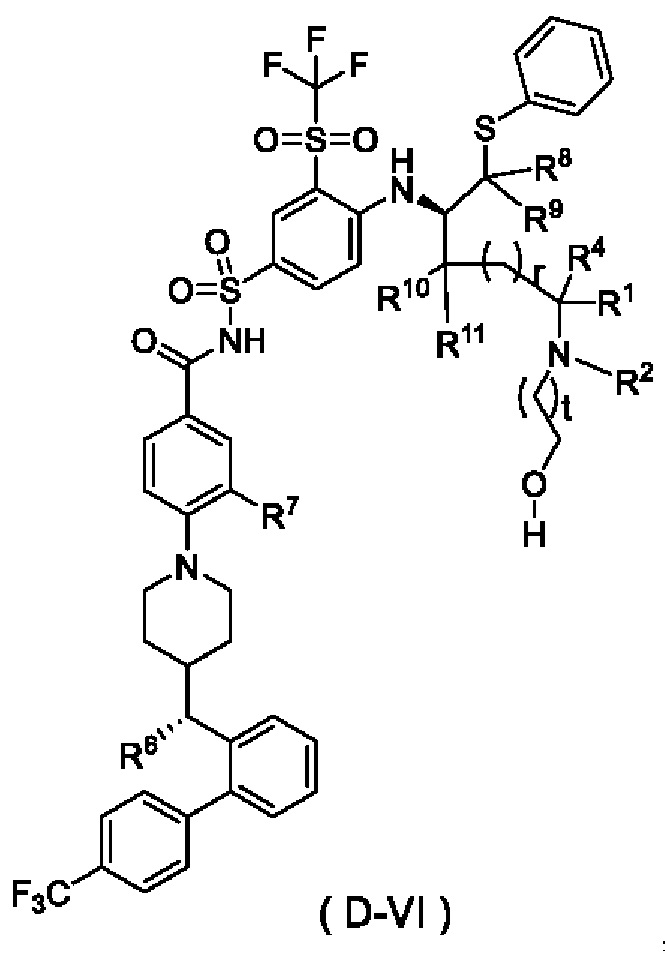
где t выбран из группы, состоящей из 0, 1, 2 и 3, предпочтительно 1, и
r, R1, R2, R4 и R6-R11 являются такими, как определено в общей формуле (D).
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, которое представляет собой соединение общей формулы (D-VII) или его фармацевтически приемлемую соль:
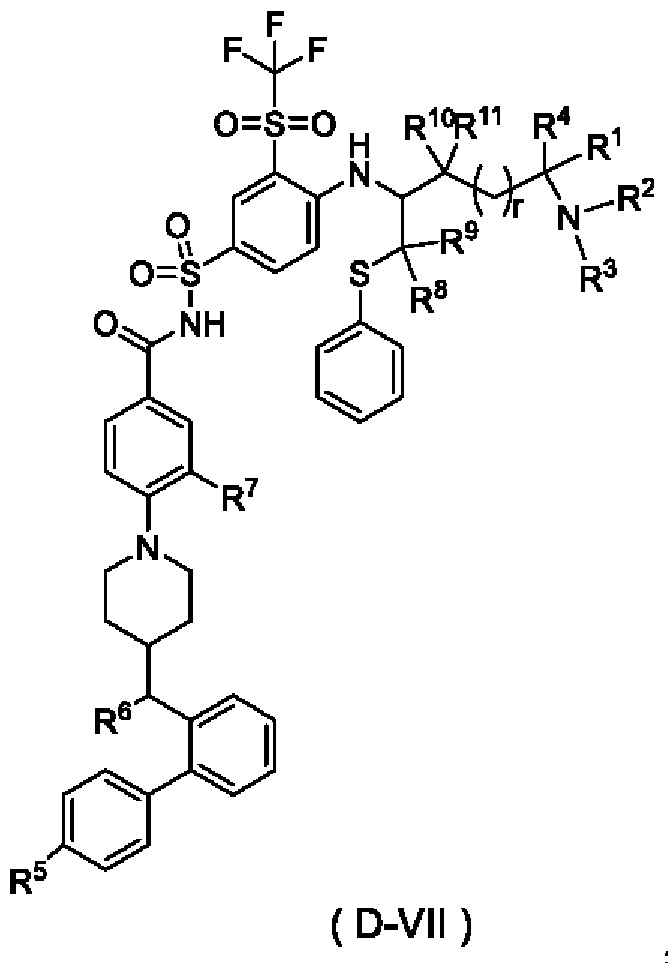
где t выбран из группы, состоящей из 0, 1, 2 и 3, предпочтительно 1, и
r и R1-R11 являются такими, как определено в общей формуле (D).
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, где R4 выбран из группы, состоящей из водорода и алкила; предпочтительно водорода и C1-6 алкила; и более предпочтительно водорода.
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, где R6 выбран из группы, состоящей из гидрокси и алкокси; предпочтительно гидрокси и C1-6 алкокси; и более предпочтительно гидрокси.
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, где R7, R8 и R9 представляют собой водород.
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, где каждый из R10 и R11 независимо выбран из группы, состоящей из водорода и алкила; предпочтительно водорода и С1-6 алкила; и более предпочтительно водорода.
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D) или его фармацевтически приемлемая соль, где r представляет собой 0 или 1, предпочтительно 0.
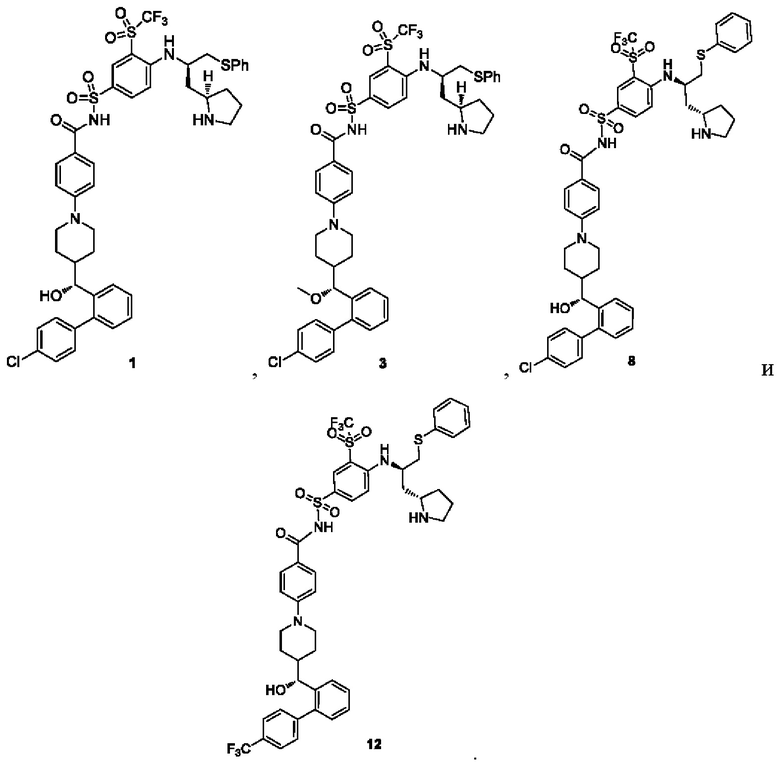
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (D), включая, но не ограничиваясь ими:
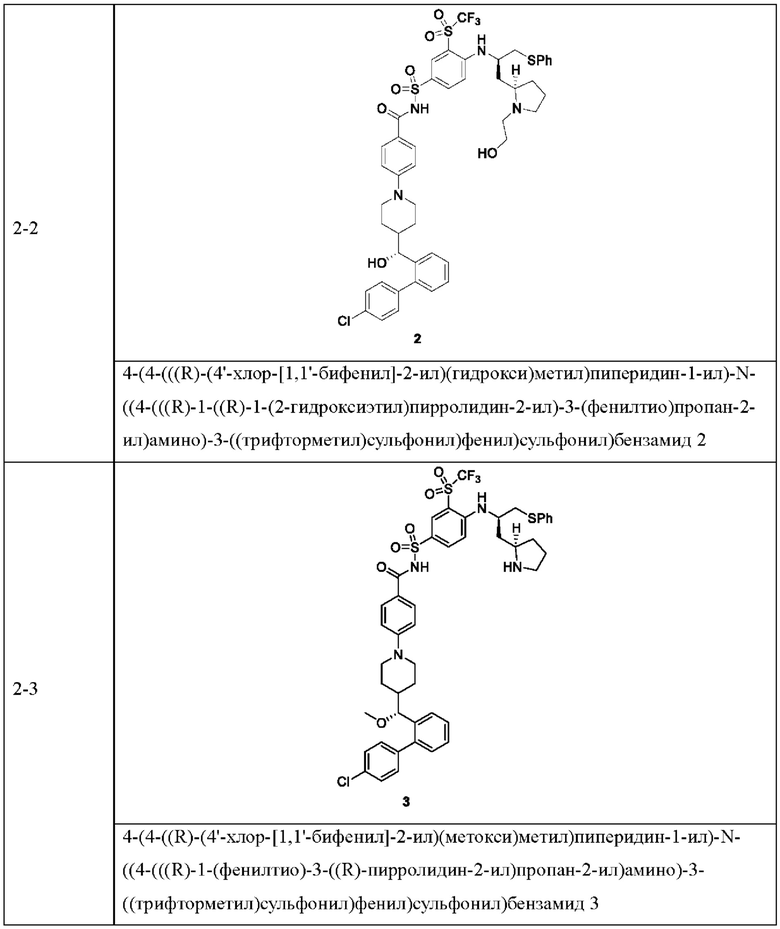
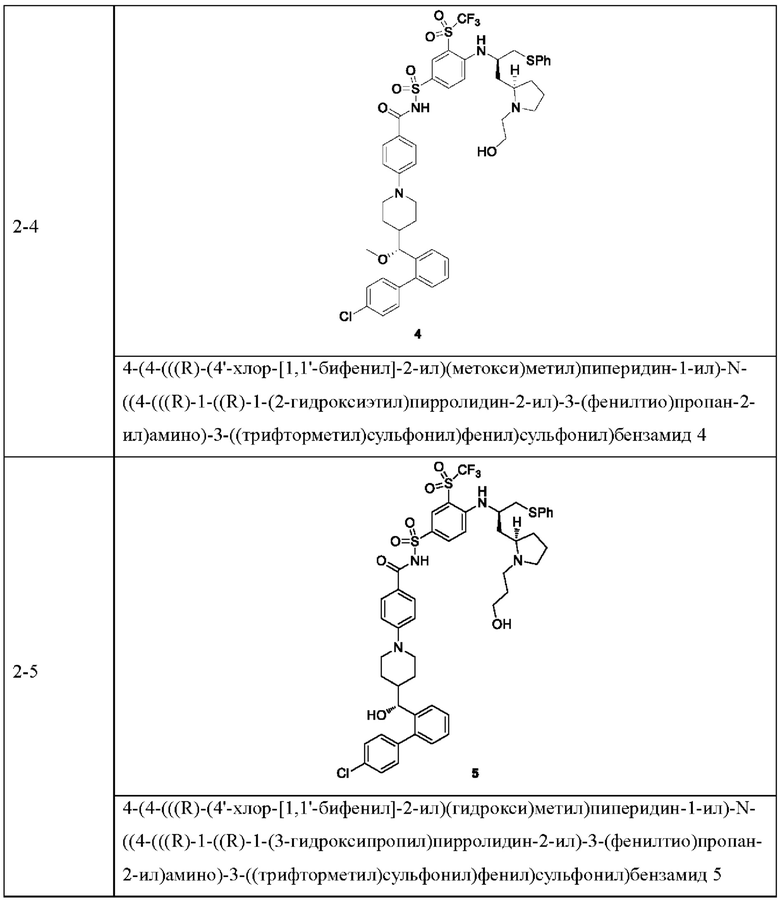

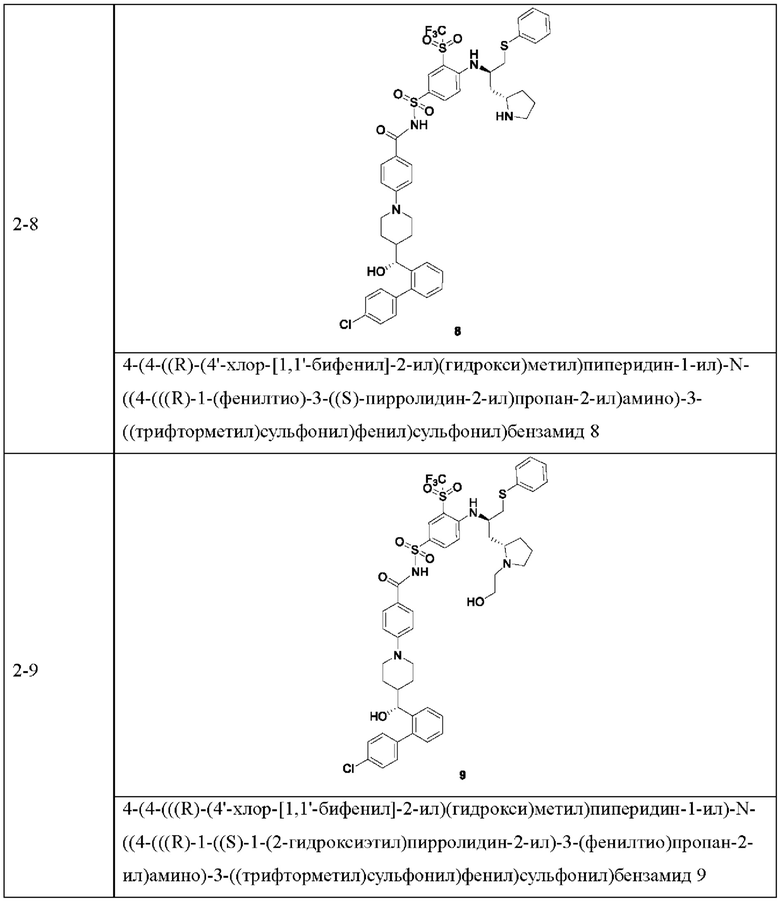
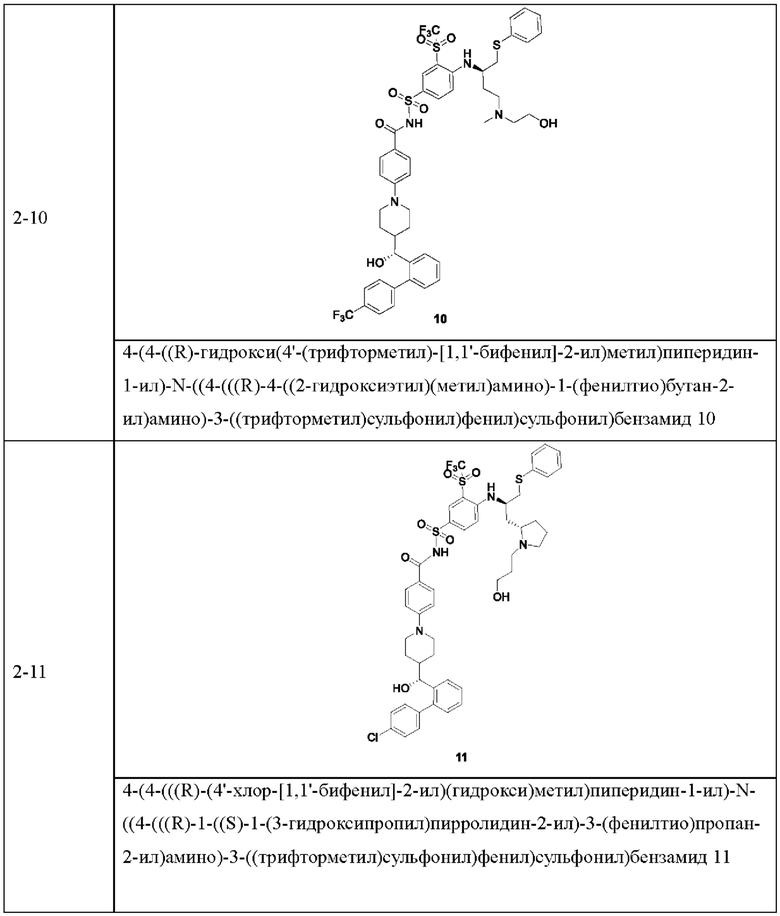
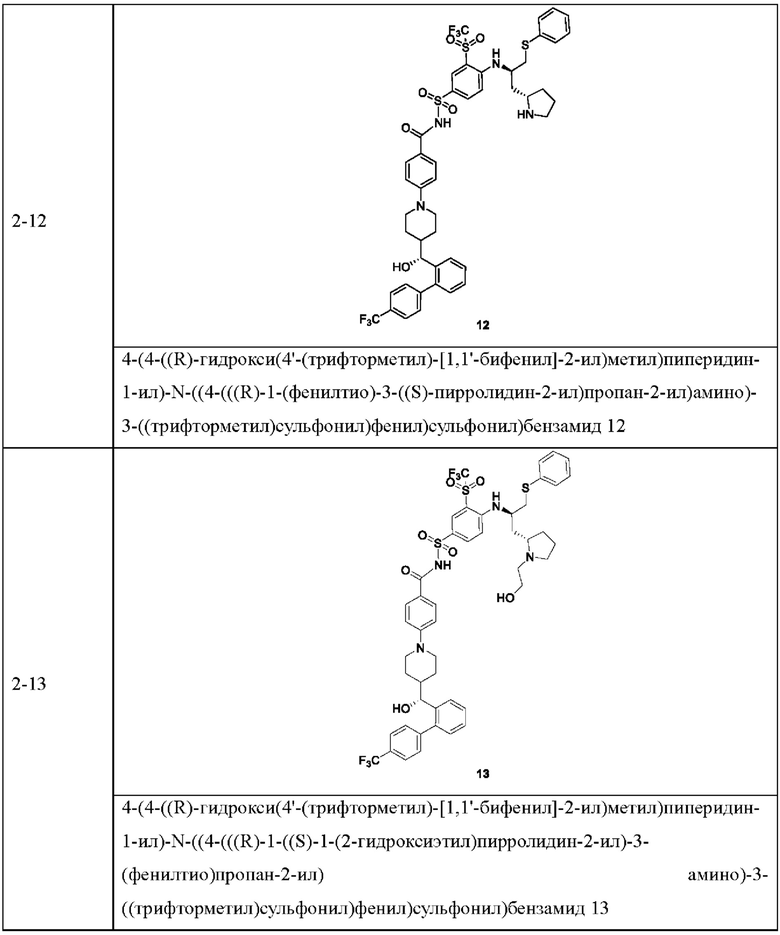
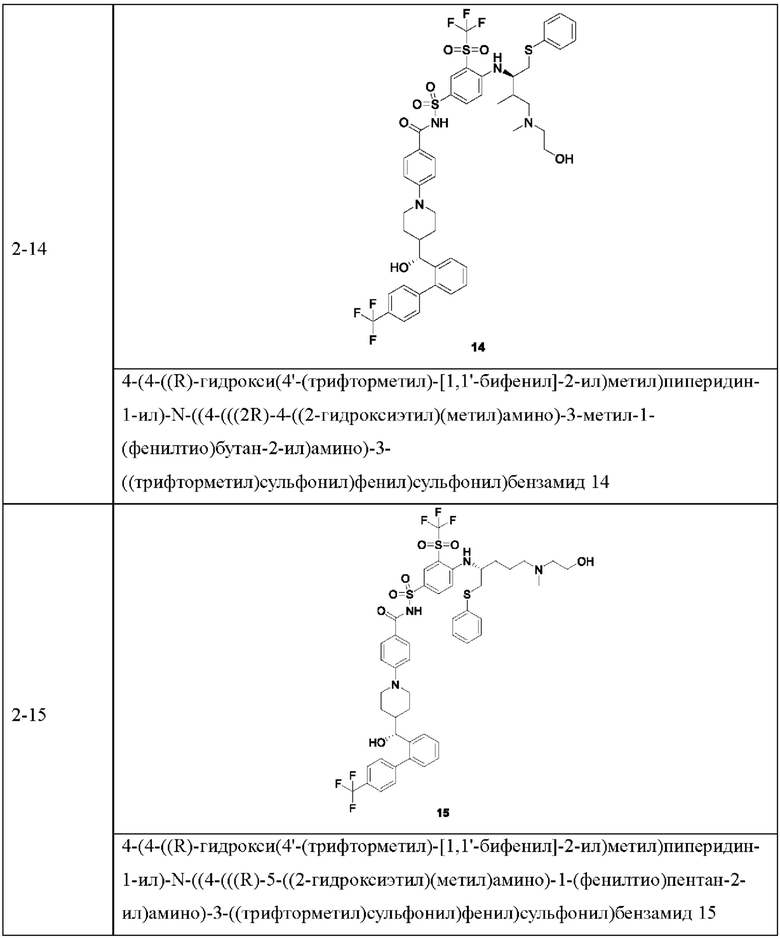
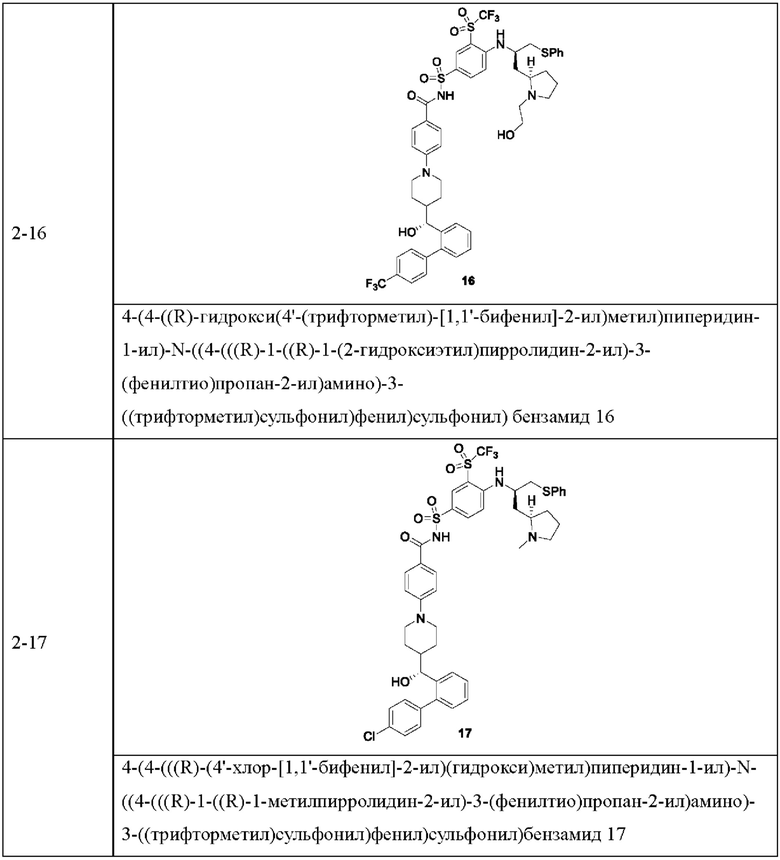
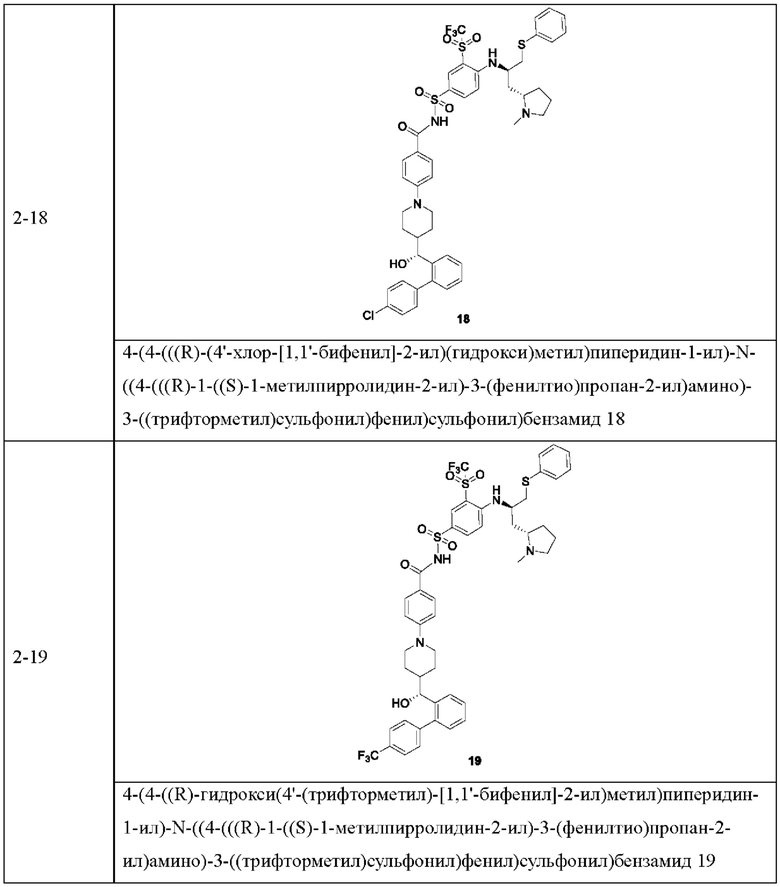
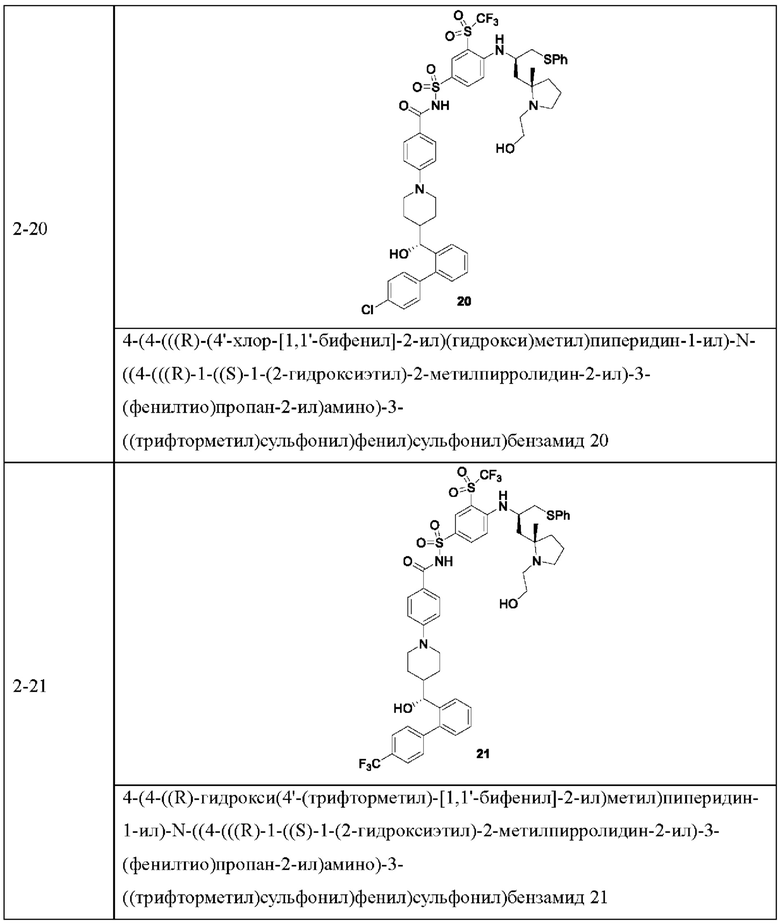
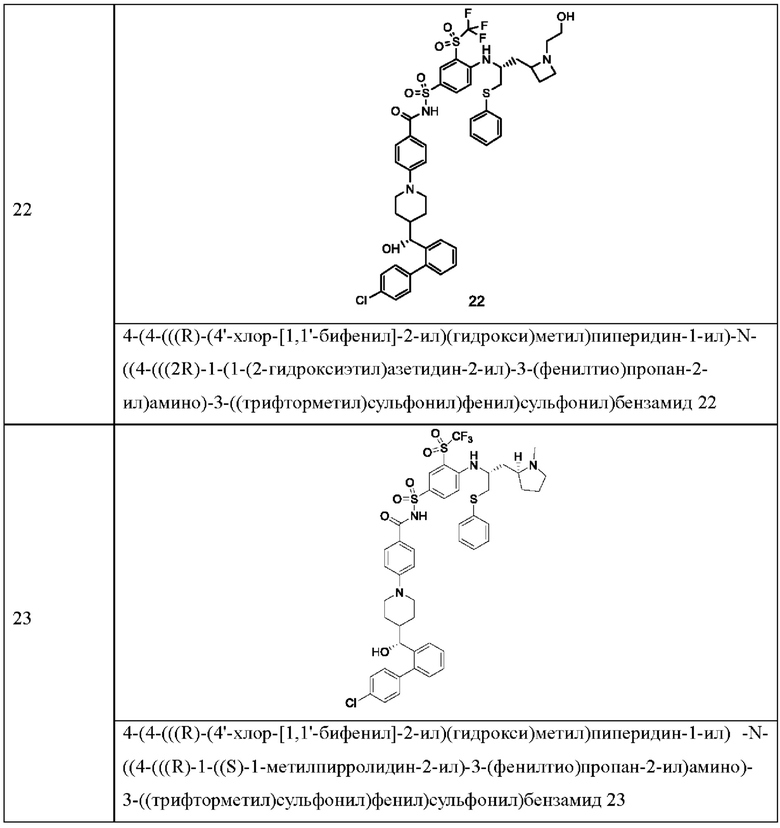
или любой их фармацевтически приемлемой соли.
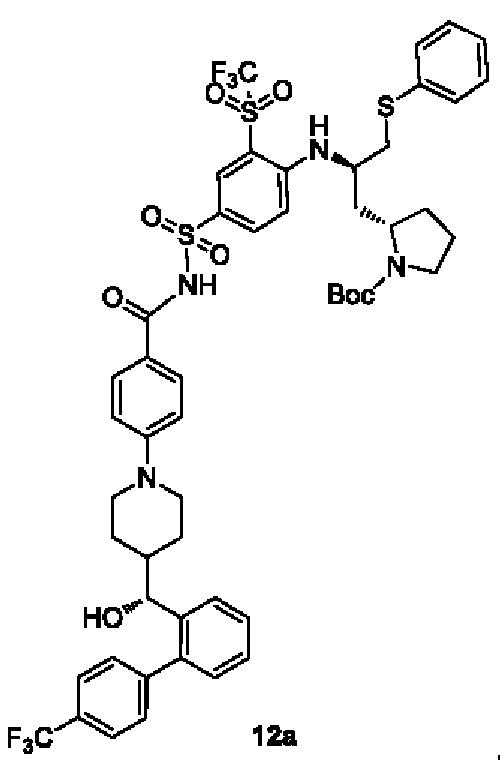
В настоящем изобретении дополнительно предложено соединение общей формулы (DA1) или (DB1) или его фармацевтически приемлемая соль,
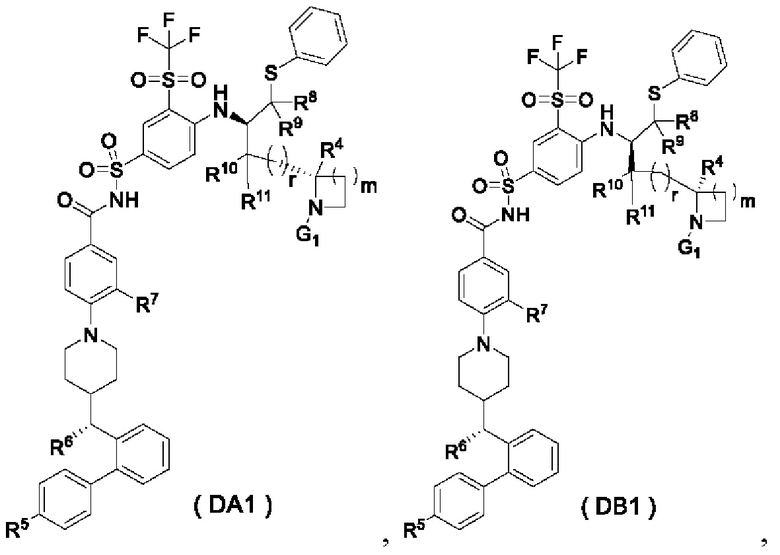
где:
G1 представляет собой аминозащитную группу, предпочтительно Boc (трет-бутоксикарбонильная группа);
m выбран из группы, состоящей из 0, 1, 2 и 3, и
r и R4-R11 являются такими, как определено в общей формуле (D).
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (DA1) или (DB1) или его фармацевтически приемлемая соль, которое выбрано из любого из следующих соединений:
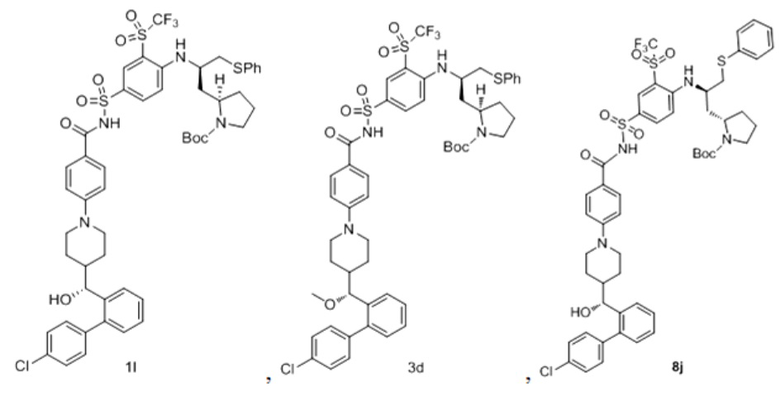 и
и
В настоящем изобретении дополнительно предложен способ получения соединения общей формулы (D-III) или его фармацевтически приемлемой соли, который включает:
подвергание соединения общей формулы (DA1) или его фармацевтически приемлемой соли реакции снятия защиты в кислой среде с получением соединения общей формулы (D-III) или его фармацевтически приемлемой соли; предпочтительно, реагент, используемый в кислой среде, представляет собой HCl;
где:
G1 представляет собой аминозащитную группу, предпочтительно Boc;
m выбран из группы, состоящей из 0, 1, 2 и 3;
R3 представляет собой водород; и
r и R4-R11 являются такими, как определено в общей формуле (D).
В настоящем изобретении дополнительно предложено соединение общей формулы (DA2) или (DB2) или его фармацевтически приемлемая соль,
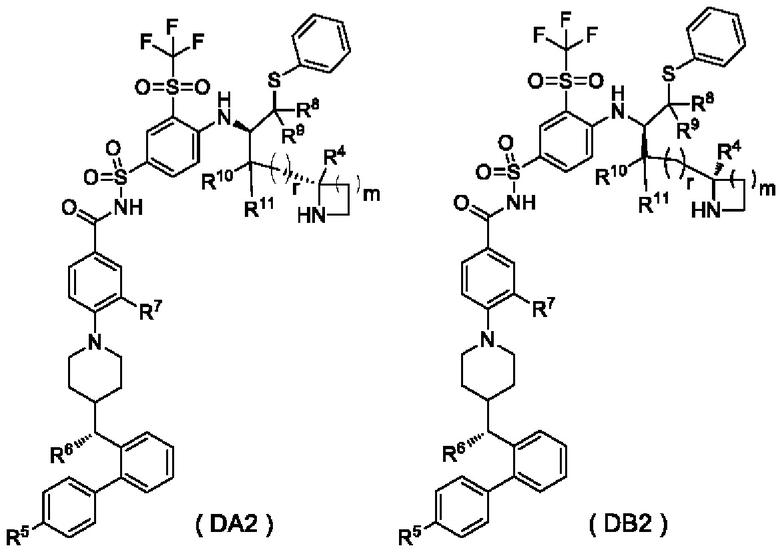
где:
m выбран из группы, состоящей из 0, 1, 2 и 3, и
r и R4-R11 являются такими, как определено в общей формуле (D).
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (DA2) или (DB2) или его фармацевтически приемлемая соль, которое выбрано из любого из следующих соединений:
В настоящем изобретении дополнительно предложен способ получения соединения общей формулы (D-III) или его фармацевтически приемлемой соли, который включает:
подвергание соединения общей формулы (DA2) или его фармацевтически приемлемой соли реакции замещения в щелочной среде с получением соединения общей формулы (D-III) или его фармацевтически приемлемой соли; предпочтительно, реагент, используемый в щелочной среде, представляет собой триэтиламин;
где:
m выбран из группы, состоящей из 0, 1, 2 и 3;
R3 представляет собой гидроксиалкил; и
r и R4-R11 являются такими, как определено в общей формуле (D).
В настоящем изобретении дополнительно предложено соединение общей формулы (DA3), (DB3) или (DC3) или его фармацевтически приемлемая соль,
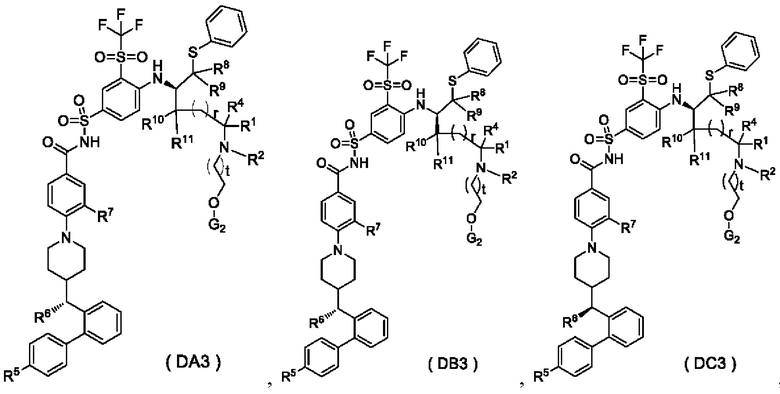
где:
G2 представляет собой гидроксизащитную группу, предпочтительно TBS;
t выбран из группы, состоящей из 0, 1, 2 и 3, и
r, R1, R2, R4-R11 являются такими, как определено в общей формуле (D).
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (DA3), (DB3) или (DC3) или его фармацевтически приемлемая соль, которое выбрано из любого из следующих соединений:
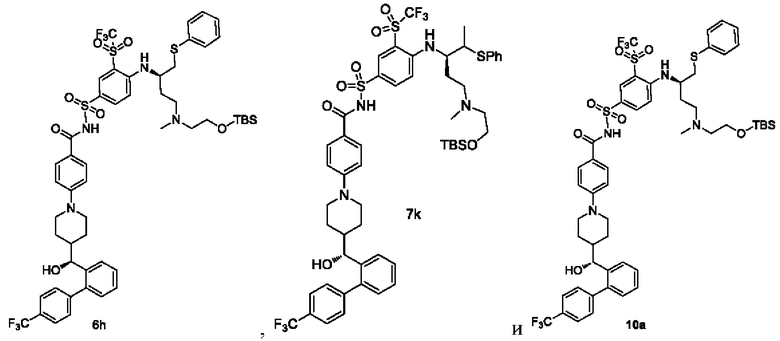
В настоящем изобретением дополнительно предложено способ получения соединения общей формулы (D-VI) или его фармацевтически приемлемой соли, который включает:
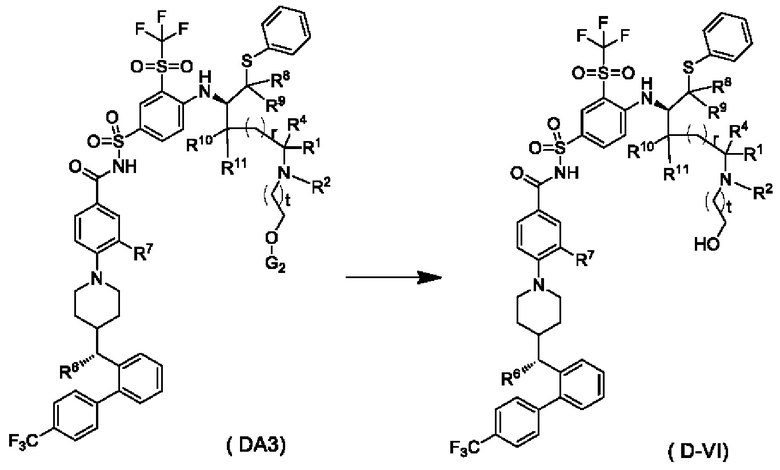
подвергание соединения общей формулы (DA3) или его фармацевтически приемлемой соли реакции снятия защиты в щелочной среде с получением соединения общей формулы (D-VI) или его фармацевтически приемлемой соли; и реагент, используемый в щелочной среде, представляет собой фторид тетрабутиламмония;
где:
G2 представляет собой гидроксизащитную группу, предпочтительно TBS;
t выбран из группы, состоящей из 0, 1, 2 и 3, и
r, R1, R2, R4-R11 являются такими, как определено в общей формуле (D).
В настоящем изобретении дополнительно предложен конъюгат лиганд-лекарственное средство, имеющий структуру формулы (-D), или его фармацевтически приемлемая соль:
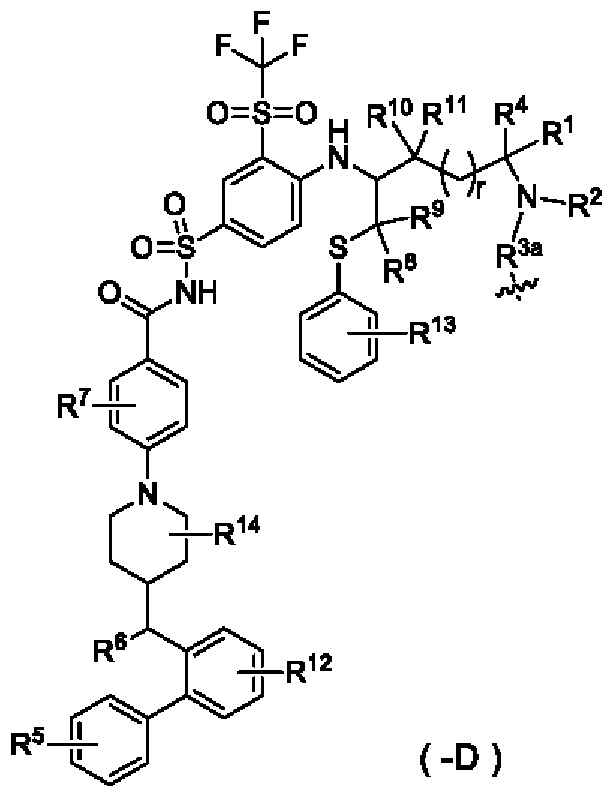
где:
R1 выбран из группы, состоящей из дейтерия, водорода, алкила и дейтерированного алкила;
R2 выбран из группы, состоящей из алкила и гидроксиалкила;
или R1 и R2 вместе с атомом, к которому они присоединены, образуют гетероциклил, необязательно дополнительно замещенный заместителем, выбранным из группы, состоящей из галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила;
R3a выбран из группы, состоящей из связи и -(СН2)t-СН2-О-; алкильный конец связан с атомом N фрагмента лекарственного средства, а -О- конец связан с линкером;
R4 выбран из группы, состоящей из водорода, галогена, дейтерированного алкила и алкила;
R5 выбран из группы, состоящей из галогена и галогеналкила;
R6 выбран из группы, состоящей из алкила, амино, гидрокси и алкокси;
R7 выбран из группы, состоящей из водорода, галогена, алкила, дейтерированного алкила, гидрокси и алкокси;
каждый из R8 и R9 независимо выбран из группы, состоящей из водорода, алкила, дейтерированного алкила и циклоалкила; или R8 и R9 вместе с атомом, к которому они присоединены, образуют циклоалкил;
каждый из R10 и R11 независимо выбран из группы, состоящей из водорода, алкила, дейтерированного алкила и циклоалкила; или R10 и R11 вместе с атомом, к которому они присоединены, образуют циклоалкил;
R12 выбран из группы, состоящей из водорода, дейтерия, дейтерированного алкила, галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила;
R13 выбран из группы, состоящей из водорода, дейтерия, дейтерированного алкила, галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила;
R14 выбран из группы, состоящей из водорода, дейтерия, дейтерированного алкила, галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила;
r выбран из группы, состоящей из 0, 1, 2 и 3;
t выбран из группы, состоящей из 0, 1, 2 и 3, и
волнистая линия указывает на ковалентное связывание с линкерным фрагментом или с лигандом.
В некоторых других вариантах осуществления настоящего изобретения конъюгат лиганд-лекарственное средство, имеющий структуру формулы (-D), или его фармацевтически приемлемая соль, где формула (-D) выбрана из группы, состоящей из структур формул (-di) и (-DII):
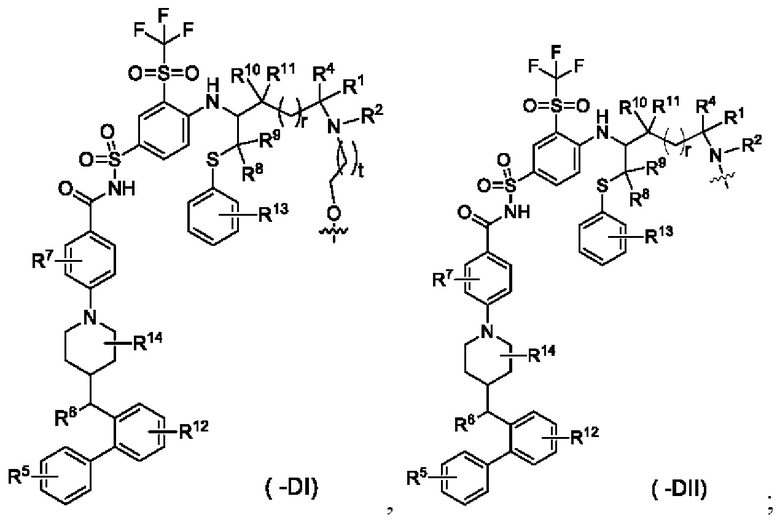
t выбран из группы, состоящей из 0, 1, 2 и 3, и
волнистая линия r, R1, R2 и R4-R14 являются такими, как определено в общей формуле (-D).
В некоторых других вариантах осуществления настоящего изобретения предложен конъюгат лиганд-лекарственное средство, имеющий структуру формулы (-D), или его фармацевтически приемлемая соль, где формула (-D) выбрана из группы, состоящей из структур формул (-DIII), (-DIV) и (-DV):
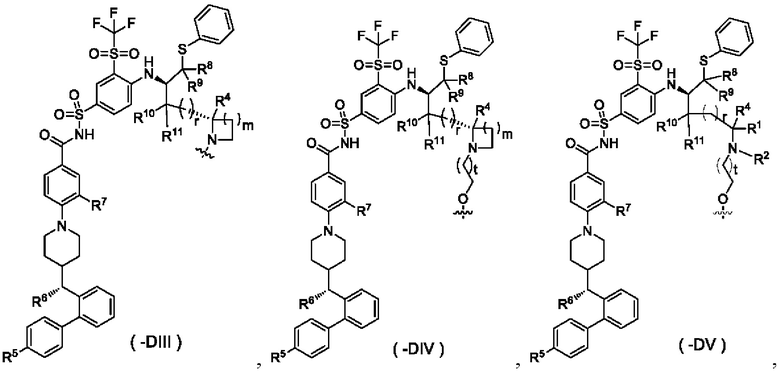
где:
t выбран из группы, состоящей из 0, 1, 2 и 3;
m выбран из группы, состоящей из 0, 1, 2 и 3, и
волнистая линия r, R1, R2 и R4-R11 являются такими, как определено в общей формуле (-D);
предпочтительно в формуле (-DV), R1 и R2 не образуют гетероциклил.
В некоторых других вариантах осуществления настоящего изобретения предложен конъюгат лиганд-лекарственное средство, имеющий структуру формулы (-D), или его фармацевтически приемлемая соль, где формула (-D) выбрана из группы, состоящей из структур формул (-DVI) и (-DVII):
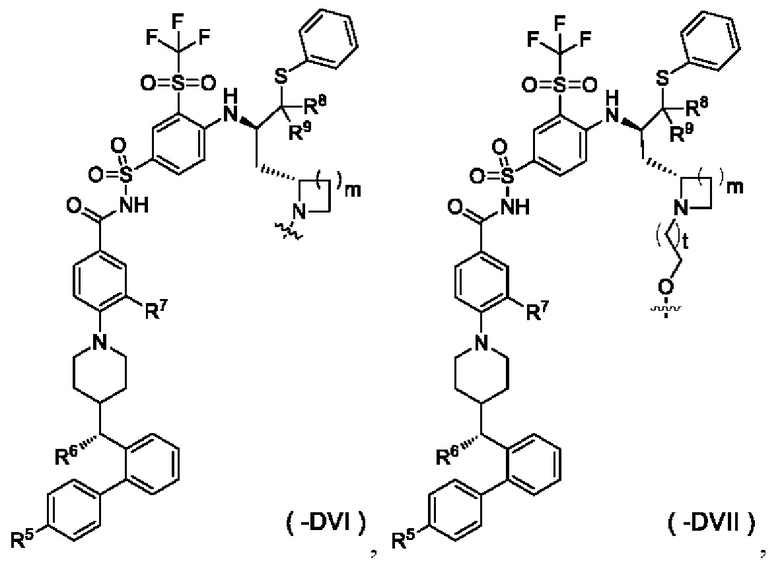
где:
t выбран из группы, состоящей из 0, 1, 2 и 3;
m выбран из группы, состоящей из 0, 1, 2 и 3, и
волнистая линия, r и R5-R9 являются такими, как определено в общей формуле (-D).
В некоторых других вариантах осуществления настоящего изобретения предложен конъюгат лиганд-лекарственное средство, имеющий структуру формулы (-D), или его фармацевтически приемлемая соль, который представляет собой конъюгат лиганд-лекарственное средство общей формулы (Pc-L-D) или его фармацевтически приемлемую соль,
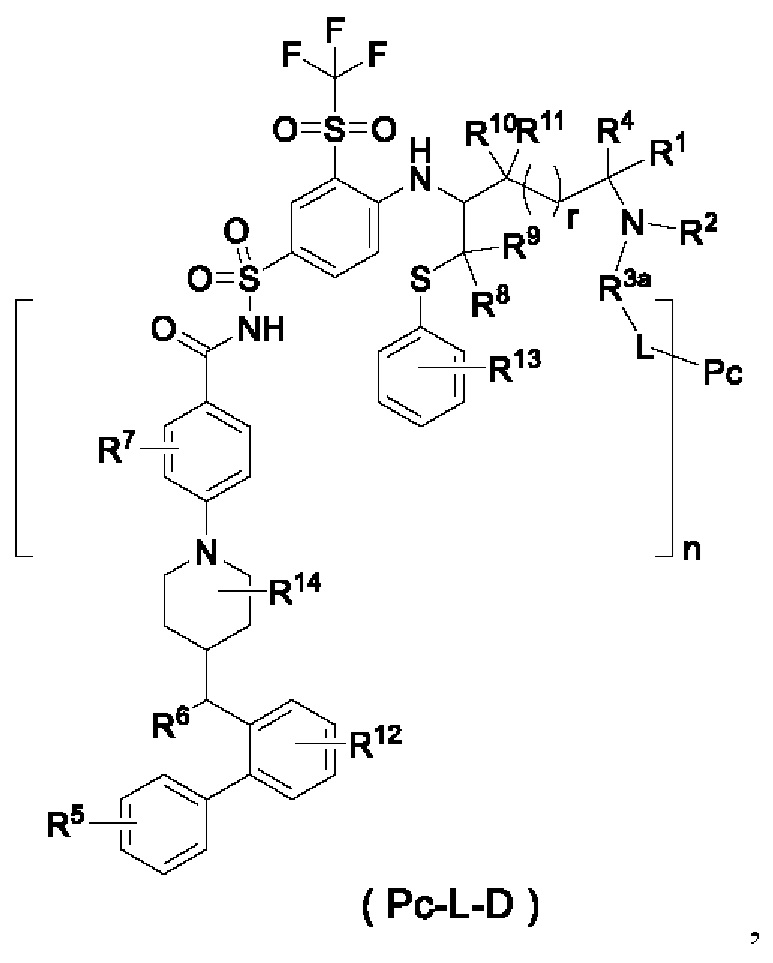
где:
R1 выбран из группы, состоящей из дейтерия, водорода, алкила и дейтерированного алкила;
R2 выбран из группы, состоящей из алкила и гидроксиалкила;
или R1 и R2 вместе с атомом, к которому они присоединены, образуют гетероциклил, необязательно дополнительно замещенный заместителем, выбранным из группы, состоящей из галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила;
R3a выбран из группы, состоящей из связи и -(CH2)t-СН2-О-;
R4 выбран из группы, состоящей из водорода, галогена, дейтерированного алкила и алкила;
R5 выбран из группы, состоящей из галогена и галогеналкила;
R6 выбран из группы, состоящей из алкила, амино, гидрокси и алкокси;
R7 выбран из группы, состоящей из водорода, галогена, алкила, дейтерированного алкила, гидрокси и алкокси;
каждый из R8 и R9 независимо выбран из группы, состоящей из водорода, алкила, дейтерированного алкила и циклоалкила; или R8 и R9 вместе с атомом, к которому они присоединены, образуют циклоалкил;
каждый из R10 и R11 независимо выбран из группы, состоящей из водорода, алкила, дейтерированного алкила и циклоалкила; или R10 и R11 вместе с атомом, к которому они присоединены, образуют циклоалкил;
R12 выбран из группы, состоящей из водорода, дейтерия, дейтерированного алкила, галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила;
R13 выбран из группы, состоящей из водорода, дейтерия, дейтерированного алкила, галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила;
R14 выбран из группы, состоящей из водорода, дейтерия, дейтерированного алкила, галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила;
r выбран из группы, состоящей из 0, 1, 2 и 3;
t выбран из группы, состоящей из 0, 1, 2 и 3;
n представляет собой десятичное число от 1 до 10; и
Рс представляет собой лиганд; и L представляет собой линкерный фрагмент.
В некоторых других вариантах осуществления настоящего изобретения предложен конъюгат лиганд-лекарственное средство, имеющий структуру формулы (-D), или его фармацевтически приемлемая соль, который представляет собой конъюгат лиганд-лекарственное средство общей формулы (Pc-L-DIII), (Pc-L-DIV) или (Pc-L-DV) или его фармацевтически приемлемую соль:
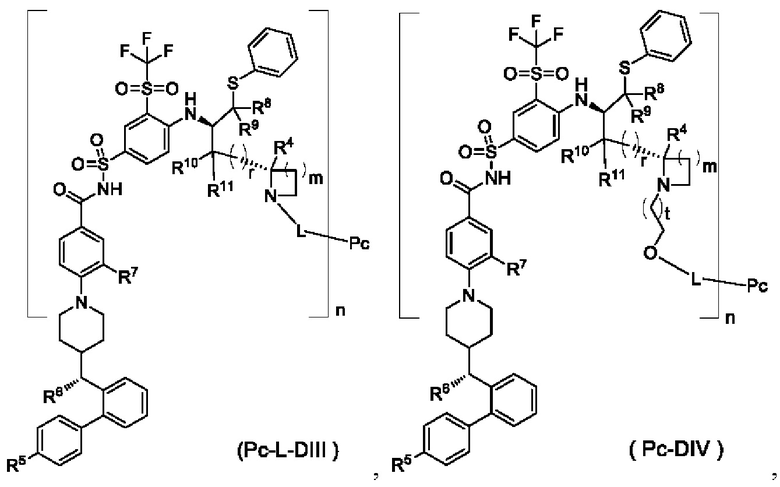
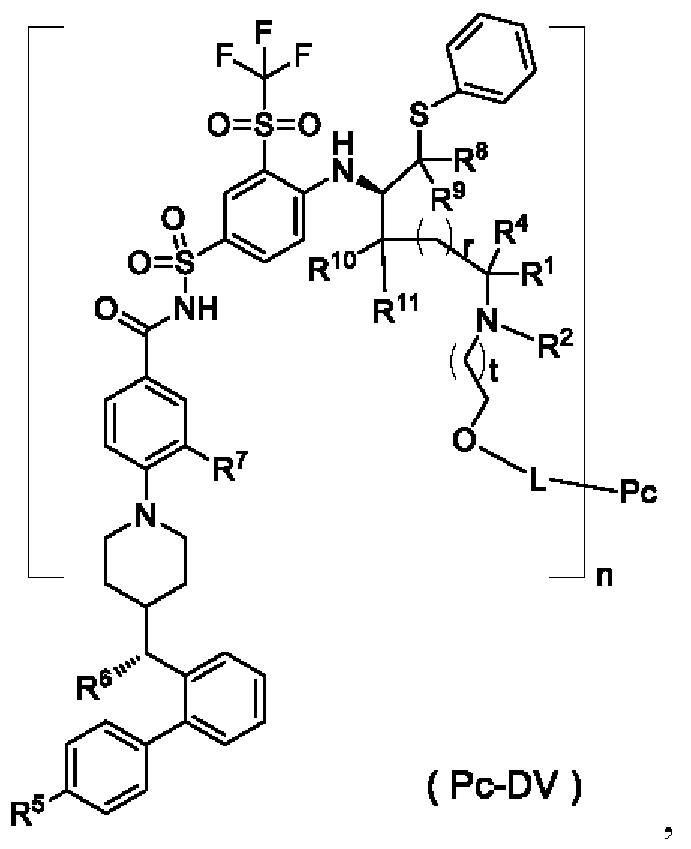
где:
t выбран из группы, состоящей из 0, 1, 2 и 3; m выбран из группы, состоящей из 0, 1, 2 и 3, и
r, R1, R2, R4-R11, Pc, L и n являются такими, как определено в общей формуле (Pc-L-D).
В некоторых других вариантах осуществления настоящего изобретения предложен конъюгат лиганд-лекарственное средство или его фармацевтически приемлемая соль, где п представляет собой целое или десятичное число от 1 до 8; предпочтительно целое или десятичное число от 2 до 8.
В некоторых других вариантах осуществления настоящего изобретения предложен конъюгат лиганд-лекарственное средство или его фармацевтически приемлемая соль, где Рс представляет собой антитело.
В некоторых других вариантах осуществления настоящего изобретения предложен конъюгат лиганд-лекарственное средство или его фармацевтически приемлемая соль, где антитело выбрано из группы, состоящей из антитела к HER2 (ErbB2), антитела к Trop-2, антитела к CD79b, антитела к EGFR, антитела к В7-Н3, антитела к c-Met, антитела к HER3 (ErbB3), антитела к HER4 (ErbB4), антитела к CD20, антитела к CD22, антитела к CD30, антитела к CD33, антитела к CD44, антитела к CD56, антитела к CD70, антитела к CD73, антитела к CD105, антитела к СЕА, антитела к А33, антитела к Cripto, антитела к EphA2, антитела к G250, антитела к MUC1, антитела к Lewis Y, антитела к VEGFR, антитела к GPNMB, антитела к интегрину, антитела к PSMA, антитела к тенасцину-С, антитела к SLC44A4 и антитела к мезотелину; предпочтительно трастузумаба, пертузумаба, нимотузумаба, эноблитузумаба, эмибетузумаба, инотузумаба, пинатузумаба, брентуксимаба, гемтузумаба, биватузумаба, лорвотузумаба, cBR96 и глематумамаба.
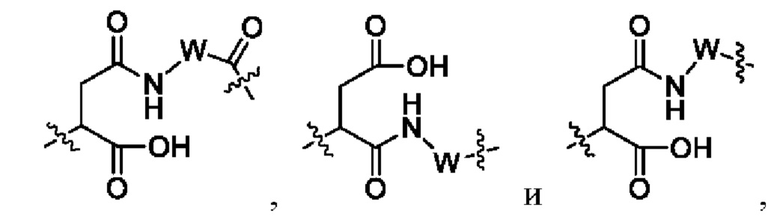
В некоторых других вариантах осуществления настоящего изобретения предложен конъюгат лиганд-лекарственное средство или его фармацевтически приемлемая соль, где линкерный фрагмент -L- представляет собой -La-Lb-Lc-, где
La выбран из группы, состоящей из 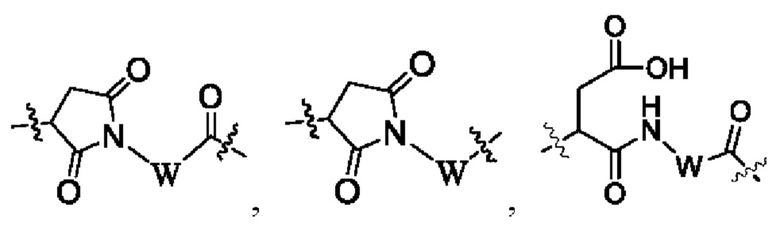
 где W выбран из группы, состоящей из -С1-6 алкила-, -С1-6 алкил-циклоалкила- и линейного -гетероалкила- из 1-6 атомов, где гетероалкил содержит от 1 до 3 гетероатомов, выбранных из группы, состоящей из N, О и S, где -C1-6 алкила-, -С1-6 алкил-циклоалкила- и линейного -гетероалкила- из 1-6 атомов каждый независимо и необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, амино, алкила, хлороалкила, дейтерированного алкила, алкокси и циклоалкила;
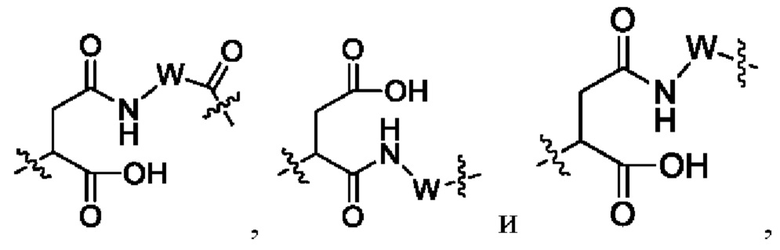
где W выбран из группы, состоящей из -С1-6 алкила-, -С1-6 алкил-циклоалкила- и линейного -гетероалкила- из 1-6 атомов, где гетероалкил содержит от 1 до 3 гетероатомов, выбранных из группы, состоящей из N, О и S, где -C1-6 алкила-, -С1-6 алкил-циклоалкила- и линейного -гетероалкила- из 1-6 атомов каждый независимо и необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, амино, алкила, хлороалкила, дейтерированного алкила, алкокси и циклоалкила;
Lb представляет собой пептидный остаток или химическую связь, состоящий из 2-7 аминокислот, которые являются необязательно замещенными одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, амино, алкила, хлоралкила, дейтерированного алкила, алкокси и циклоалкила; и
Lc выбран из группы, состоящей из -NR17(CR18R19)t-, -NH-C(R18R19)-O-C(R20R21)-С(О)-, -NH-R22-(CH2)q-OC(O)-, -C(O)NR17, -C(O)NR17(CH2)q-и химической связи, где q представляет собой целое число от 1 до 6;
R17 выбран из группы, состоящей из водорода, алкила, галогеналкила, дейтерированного алкила и гидроксиалкила;
R18 и R19 являются одинаковыми или различными и каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, дейтерированного алкила и гидроксиалкила;
R20 выбран из группы, состоящей из алкила, циклоалкилалкила и циклоалкила;
R21 выбран из группы, состоящей из водорода, алкила и галогеналкила; или R20 и R21 вместе с атомом углерода, к которому они присоединены, образуют С3-6 циклоалкил;
R22 выбран из группы, состоящей из арила и гетероарила.
В некоторых других вариантах осуществления настоящего изобретения предложен конъюгат лиганд-лекарственное средство или его фармацевтически приемлемая соль, где W выбран из группы, состоящей из -(СН2)2-и -(СН2)5-.
В некоторых других вариантах осуществления настоящего изобретения предложен конъюгат лиганд-лекарственное средство или его фармацевтически приемлемая соль, где пептидный остаток Lb представляет собой аминокислотный остаток, образованный из одной или более аминокислот, выбранных из группы, состоящей из фенилаланина, глицина, валина, лизина, цитруллина, серина, глутаминовой кислоты и аспарагиновой кислоты; предпочтительно тетрапептидный остаток, дипептидный остаток и химическую связь; и более предпочтительно тетрапептидный остаток глицин-глицин-фенилаланин-глицина или дипептидный остаток валин-цитруллина.
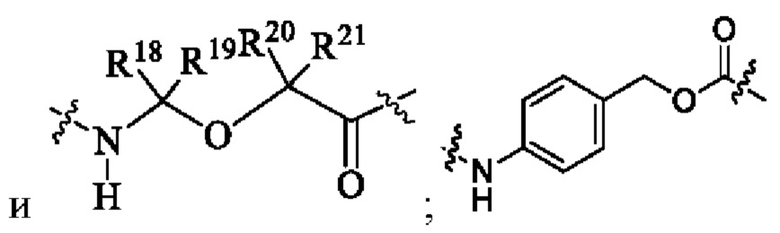
В некоторых других вариантах осуществления настоящего изобретения предложен конъюгат лиганд-лекарственное средство или его фармацевтически приемлемая соль, где линкерная единица Lc выбрана из группы, состоящей из -NH-C(R18R19)-O-C(R20R21)-C(O)-, -NH-R22-(CH2)q-OC(O)- и химической связи, q представляет собой целое число от 1 до 6; и
R22 выбран из группы, состоящей из арила и гетероарила;
предпочтительно Lc выбран из группы, состоящей из следующих структурных формул:
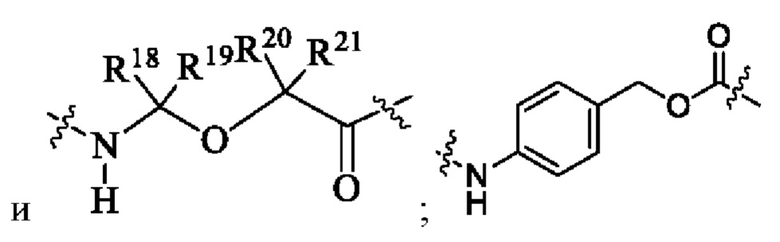
R18 и R19 являются одинаковыми или различными и каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, дейтерированного алкила и гидроксиалкила;
R20 выбран из группы, состоящей из алкила, циклоалкилалкила и циклоалкила;
R21 выбран из группы, состоящей из водорода, алкила и галогеналкила; или R20 и R21 вместе с атомом углерода, к которому они присоединены, образуют С3-6 циклоалкил.
В некоторых других вариантах осуществления настоящего изобретения предложен конъюгат лиганд-лекарственное средство или его фармацевтически приемлемая соль, где линкерный фрагмент -L- представляет собой -La-, где -La- является таким, как определено выше.
В некоторых других вариантах осуществления настоящего изобретения предложен конъюгат лиганд-лекарственное средство или его фармацевтически приемлемая соль, где линкерный фрагмент -L- представляет собой -La-Lb-Lc-, где
La выбраниз группы, состоящей из 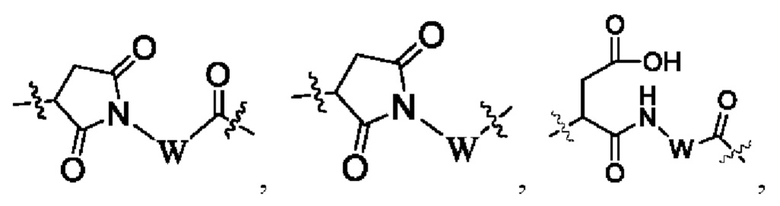
 где W выбран из группы, состоящей из -(СН2)2-и -(СН2)5-;
где W выбран из группы, состоящей из -(СН2)2-и -(СН2)5-;
Lb выбран из группы, состоящей из тетрапептидного остатка и дипептидного остатка; предпочтительно тетрапептидный остаток глицин-глицин-фенилаланин-глицина или дипептидный остаток валин-цитруллина; и
Lc выбран из группы, состоящей из следующих структурных формул:
R18 и R19 являются одинаковыми или различными и каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, дейтерированного алкила и гидроксиалкила;
R20 выбран из группы, состоящей из алкила, циклоалкилалкила и циклоалкила;
R21 выбран из группы, состоящей из водорода, алкила и галогеналкила; или R20 и R21 вместес атомом углерода, к которому они присоединены, образуют С3-6 циклоалкил.
В некоторых других вариантах осуществления настоящего изобретения предложен конъюгат лиганд-лекарственное средство или его фармацевтически приемлемая соль, который содержит связывающий фрагмент -L-, где -L- выбран из группы, состоящей из:
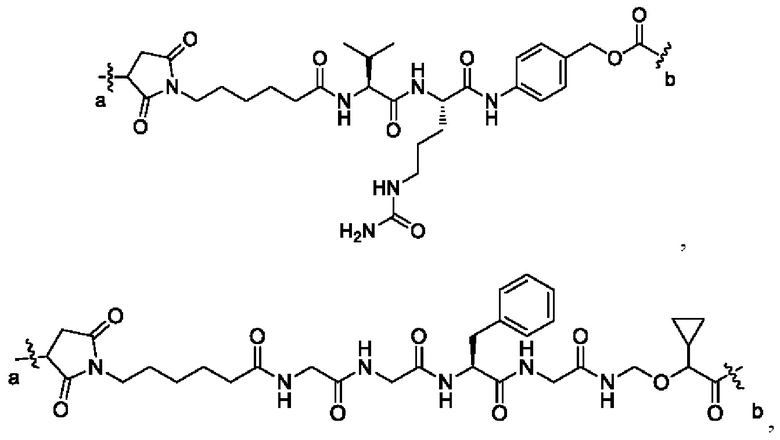
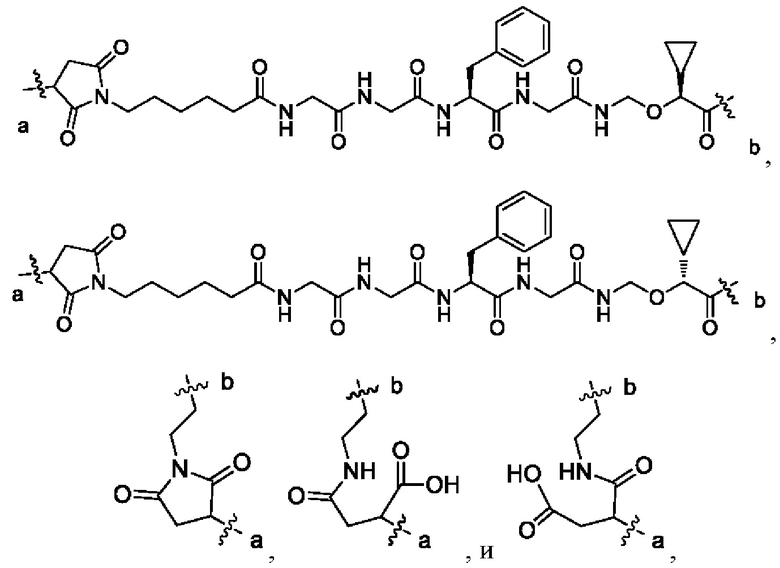
где конец а связан с лигандом Рс, а конец b связан с концом R3a лекарственного средства.
В некоторых других вариантах осуществления настоящего изобретения предложен конъюгат лиганд-лекарственное средство или его фармацевтически приемлемая соль, где линкерный фрагмент -L- представляет собой -La-Lb-Lc-, где
La выбран из группы, состоящей из 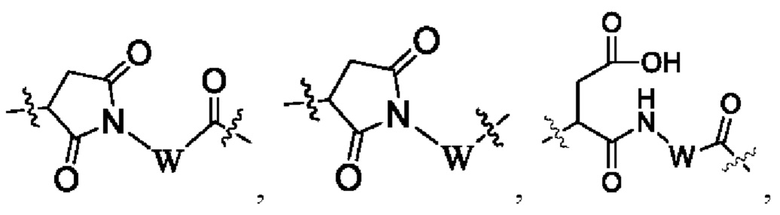
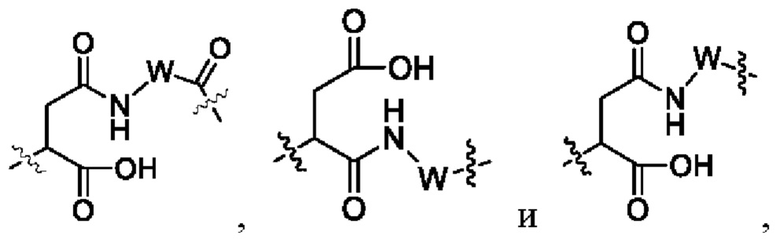 где W выбран из группы, состоящей из -(СН2)2-и -(СН2)5-;
где W выбран из группы, состоящей из -(СН2)2-и -(СН2)5-;
Lb выбран из группы, состоящей из тетрапептидного остатка и дипептидного остатка; предпочтительно тетрапептидный остаток глицин-глицин-фенилаланин-глицина или дипептидный остаток валин-цитруллина; и
Lc имеет следующую структурную формулу:
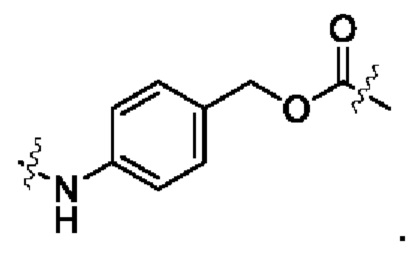
В настоящем изобретении дополнительно предложено соединение общей формулы (Lu-D) или его фармацевтически приемлемая соль:
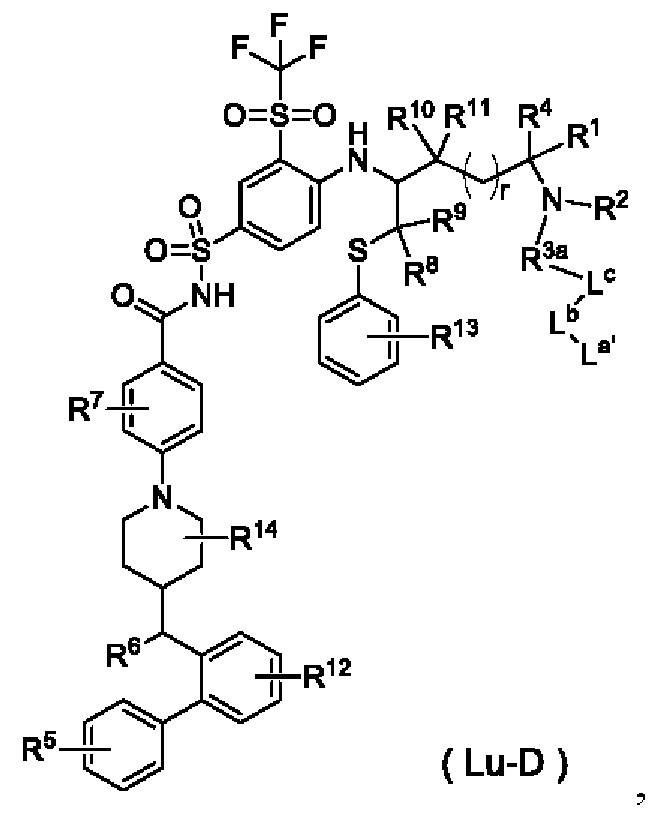
где:
La' выбран из группы, состоящей из, 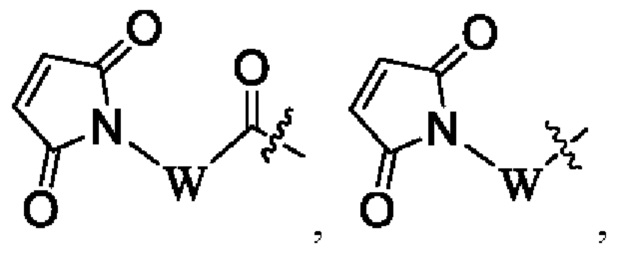 где W выбран из группы, состоящей из -(СН2)2- и -(CH2)5-;
где W выбран из группы, состоящей из -(СН2)2- и -(CH2)5-;
Lb представляет собой пептидный остаток или химическую связь, состоящий из 2-7 аминокислот, которые являются необязательно замещенными одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, амино, алкила, хлоралкила, дейтерированного алкила, алкокси и циклоалкила; и
Lc имеет следующую структурную формулу:
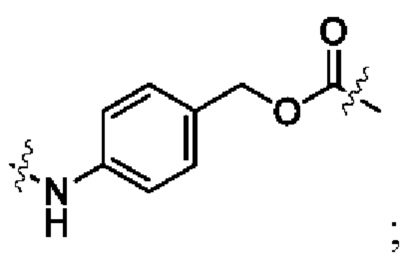
R1 выбран из группы, состоящей из дейтерия, водорода, алкила и дейтерированного алкила;
R2 выбран из группы, состоящей из алкила и гидроксиалкила;
или R1 и R2 вместе с атомом, к которому они присоединены, образуют гетероциклил, необязательно дополнительно замещенный заместителем, выбранным из группы, состоящей из галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила;
R3a выбран из группы, состоящей из связи и -(СН2)t-СН2-О-;
R4 выбран из группы, состоящей из водорода, галогена, дейтерированного алкила и алкила;
R5 выбран из группы, состоящей из галогена и галогеналкила;
R6 выбран из группы, состоящей из алкила, амино, гидрокси и алкокси;
R7 выбран из группы, состоящей из водорода, галогена, алкила, дейтерированного алкила, гидрокси и алкокси;
каждый из R8 и R9 независимо выбран из группы, состоящей из водорода, алкила, дейтерированного алкила и циклоалкила; или R8 и R9 вместе с атомом, к которому они присоединены, образуют циклоалкил;
каждый из R10 и R11 независимо выбран из группы, состоящей из водорода, алкила,
дейтерированного алкила и циклоалкила; или R10 и R11 вместе с атомом, к которому они присоединены, образуют циклоалкил;
R12 выбран из группы, состоящей из водорода, дейтерия, дейтерированного алкила, галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила;
R13 выбран из группы, состоящей из водорода, дейтерия, дейтерированного алкила, галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила;
R14 выбран из группы, состоящей из водорода, дейтерия, дейтерированного алкила, галогена, гидрокси, алкила, гидроксиалкила, алкокси и циклоалкила;
r выбран из группы, состоящей из 0, 1, 2 и 3; и
t выбран из группы, состоящей из 0, 1, 2 и 3.
В некоторых других вариантах осуществления настоящего изобретения предложено соединение общей формулы (Lu-D) или его фармацевтически приемлемая соль, которое представляет собой соединение, как показано ниже:
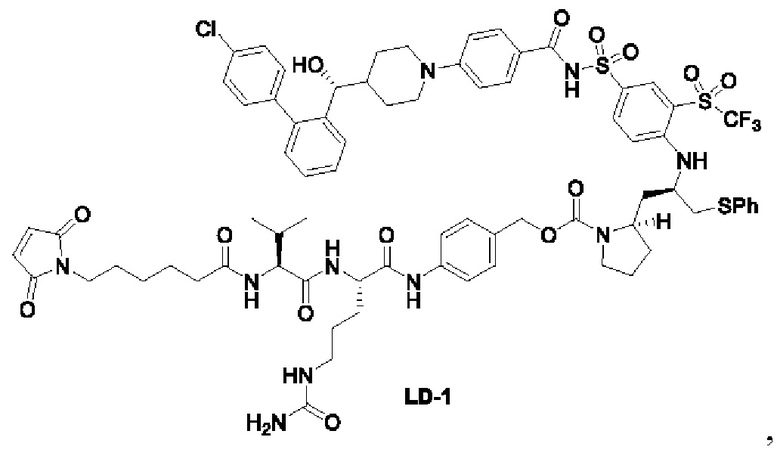
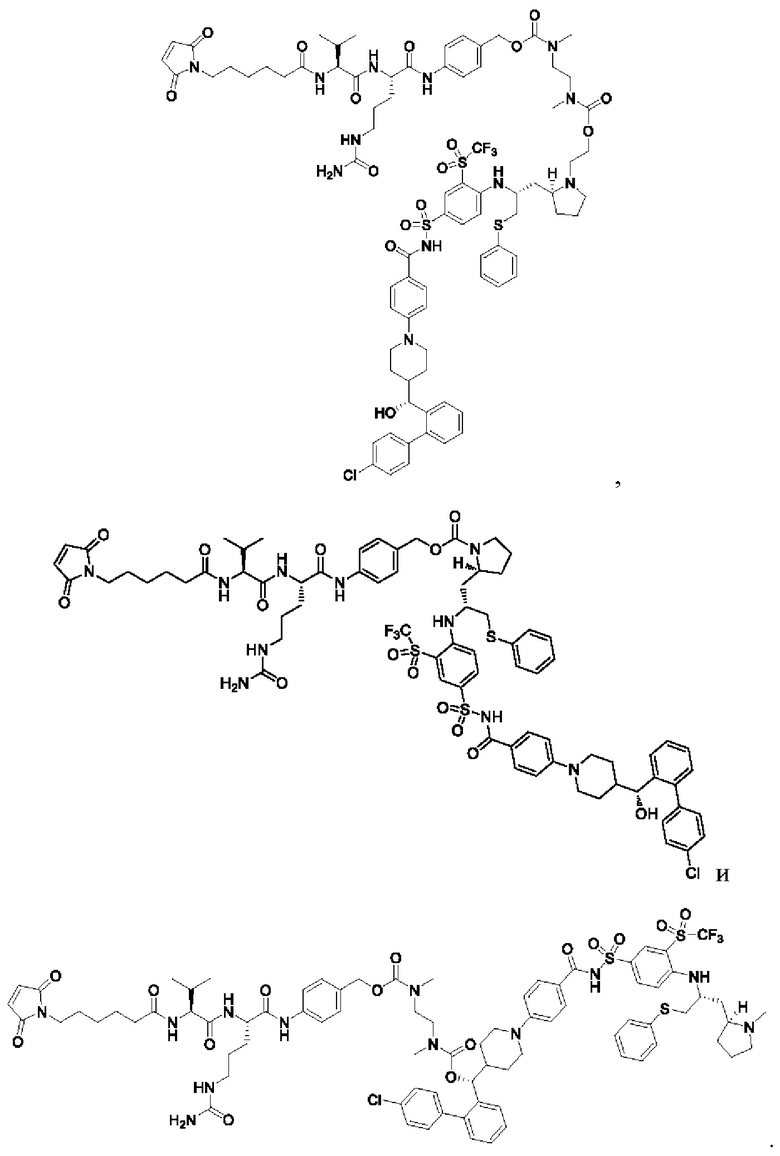
В настоящем изобретении дополнительно предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения общей формулы (D) или его фармацевтически приемлемой соли, или конъюгата лиганд-лекарственное средство, имеющего структуру формулы (-D) или его фармацевтически приемлемой соли, или конъюгата лиганд-лекарственное средство общей формулы (Pc-L-D) или его фармацевтически приемлемой соли, и фармацевтически приемлемого носителя, разбавителя или эксципиента.
В настоящем изобретении дополнительно предложено применение соединения общей формулы (D) или его фармацевтически приемлемой соли, или конъюгата лиганд-лекарственное средство, имеющего структуру формулы (-D), или его фармацевтически приемлемой соли, или конъюгата лиганд-лекарственное средство общей формулы (Pc-L-D), или его фармацевтически приемлемой соли, или фармацевтической композиции, описанной выше, при получении ингибитора Bcl-2 и/или Bcl-XL.
В настоящем изобретении дополнительно предложено применение соединения общей формулы (D) или его фармацевтически приемлемой соли, или конъюгата лиганд-лекарственное средство, имеющего структуру формулы (-D), или его фармацевтически приемлемой соли, или конъюгата лиганд-лекарственное средство общей формулы (Pc-L-D), или его фармацевтически приемлемой соли, или фармацевтической композиции, описанной выше, в получении лекарственного средства для лечения или предотвращения опухоли.
В настоящем изобретении предложено применение соединения общей формулы (D) или его фармацевтически приемлемой соли, или конъюгата лиганд-лекарственное средство, имеющего структуру формулы (-D), или его фармацевтически приемлемой соли, или его фармацевтически приемлемой соли, или конъюгата лиганд-лекарственное средство общей формулы (Pc-L-D) или его фармацевтически приемлемой соли, или фармацевтической композиции, как описано выше, в получении лекарственного средства для лечения и/или предотвращения рака, где рак предпочтительно представляет собой меланому рак печени, рак почки, рак легкого, мелкоклеточный рак легкого, не мелкоклеточный рак легкого, рак носоглотки, колоректальный рак, рак толстой кишки, рак прямой кишки, рак поджелудочной железы, рак шейки матки, рак яичников, рак молочной железы, рак мочевого пузыря, рак предстательной железы, лейкемия, острый миелоидный лейкоз, хронический миелоидный лейкоз, хронический лимфолейкоз, острый лимфоцитарный лейкоз, острый миелогенный лейкоз, мантийно-клеточная лимфома, диффузную крупноклеточную В-клеточную лимфому, фолликулярную центральную лимфому неходжкинскую лимфому, Т-клеточную лимфому, В-клеточную лимфому, плоскоклеточный рак головы и шеи, рак шейки матки, рак щитовидной железы, лимфому, саркому, нейробластому, опухоль головного мозга, миелому, астроцитому и глиому.
Другой аспект настоящего изобретения дополнительно относится к способу лечения или предотвращения рака, который включает введение терапевтически эффективной дозы соединения общей формулы (D) или его фармацевтически приемлемой соли, или конъюгата лиганд-лекарственное средство, имеющего структуру формулы (-D) или его фармацевтически приемлемой соли, или конъюгата лиганд-лекарственное средство общей формулы (Pc-L-D) или его фармацевтически приемлемой соли, или его фармацевтической композиции, описанной выше; где рак предпочтительно представляет собой меланому, рак печени, рак почки, рак легкого, мелкоклеточный рак легкого, не мелкоклеточный рак легкого, рак носоглотки, колоректальный рак, рак толстой кишки, рак прямой кишки, рак поджелудочной железы, рак шейки матки, рак яичников, рак молочной железы рак, рак мочевого пузыря, рак предстательной железы, лейкоз, острый миелоидный лейкоз, хронический миелоидный лейкоз, хронический лимфоцитарный лейкоз, острый лимфоцитарный лейкоз, острый миелогенный лейкоз, мантийно-клеточную лимфому, диффузную В-крупноклеточную лимфому, фолликулярную центральную лимфому, неходжкинскую лимфому, Т-клеточную лимфому, В-клеточную лимфому, плоскоклеточный рак головы и шеи, рак шейки матки, рак щитовидной железы, лимфому, саркому, нейробластому, опухоль головного мозга, миелому, астроцитому и глиому.
Активное соединение (например, конъюгат лиганда-лекарственное средство или его фармацевтически приемлемая соль в соответствии с настоящим изобретением) может быть составлено в форму, подходящую для введения любым подходящим способом, предпочтительно в форме стандартной дозы или в форме однократной дозы, которую субъект может вводить самостоятельно. Стандартная доза активного соединения или композиции по настоящему изобретению может быть в таблетке, капсуле, крахмальной капсуле, флаконе, порошке, грануле, пастилке, суппозитории, порошке для разведения или жидком препарате.
Доза введения активного соединения или композиции, применяемая в способе лечения по настоящему изобретению, обычно варьируется в зависимости от тяжести заболевания, массы субъекта и эффективности активного соединения. Как правило, подходящая однократная доза может составлять от 0,1 до 1000 мг.
Фармацевтическая композиция по настоящему изобретению может содержать, в дополнение к активному соединению, один или более вспомогательных веществ, выбранных из группы, состоящей из наполнителя, разбавителя, связующего вещества, смачивающего агента, разрыхлителя, эксципиента и тому подобного. В зависимости от способа введения композиция может содержать от 0,1 до 99 мас. % активного соединения.
Фармацевтическая композиция, содержащая активный ингредиент, может быть в форме, подходящей для перорального введения, например, в форме таблетки, драже, пастилки, водной или масляной суспензии, диспергируемого порошка или гранулы, эмульсии, твердой или мягкой капсулы или сиропа или настойки. Пероральные композиции могут быть получены в соответствии с любым способом, известным в данной области техники для получения фармацевтических композиций. Такие композиции для перорального применения могут содержать связующее вещество, наполнитель, смазывающее вещество, разрыхлитель или фармацевтически приемлемый смачивающий агент, а также могут содержать один или более ингредиентов, выбранных из группы, состоящей из подсластителя, корригирующего вещества, красителя и консерванта, чтобы обеспечить приятную на вид и вкус фармацевтический состав.
Водная суспензия содержит активное вещество и эксципиент, который используется для смешивания и подходит для получения водной суспензии. Водная суспензия может также содержать один или более консервантов, один или более красителей, один или более корригирующих веществ и один или более подсластителей.
Масляная суспензия может быть составлена путем суспендирования активного ингредиента в растительном масле. Масляная суспензия может содержать загуститель. Подсластители и корригирующие вещества, описанные выше, могут быть добавлены для получения притяного по вкусу состава.
Фармацевтические композиции также могут быть получены следующим образом: диспергируемые порошки или гранулы для получения водных суспензий обеспечивают активный ингредиент, и добавляли воду для смешивания активного ингредиента с одним или более диспергирующими веществами, смачивающими агентами, суспендирующими агентами или консервантами. Также могут быть добавлены другие эксципиенты, такие как подсластители, корригирующие вещества и красители. Для сохранения этих композиций добавляют антиоксиданты, такие как аскорбиновая кислота.
Фармацевтическая композиция по настоящему изобретению также может быть в форме эмульсии масло-в-воде.
Фармацевтическая композиция может быть в форме стерильного водного раствора для инъекций. Доступные и приемлемые носители или растворители включают воду, раствор Рингера и изотонический раствор хлорида натрия. Стерильный препарат для инъекций может представлять собой стерильную микроэмульсию масло-в-воде для инъекций, в которой активный ингредиент растворен в масляной фазе. Например, активный ингредиент растворяют в смеси соевого масла и лецитина. Затем масляный раствор добавляли к смеси воды и глицерина и обрабатывают с образованием микроэмульсии. Инъекция или микроэмульсия может быть введена местно в кровоток субъекта в больших количествах. В качестве альтернативы, может быть целесообразным введение растворов и микроэмульсий таким образом, чтобы поддерживать постоянную циркулирующую концентрацию соединения по настоящему изобретению. Для поддержания такой постоянной концентрации может использоваться устройство для непрерывной внутривенной доставки. Примером такого устройства является насос для внутривенных инъекций Deltec CADD-PLUS.ТМ.5400.
Фармацевтическая композиция может быть в форме стерильной водной или масляной суспензии для инъекции для внутримышечного и подкожного введения.
Суспензия может быть получена в соответствии с предшествующим уровнем техники с использованием тех подходящих диспергирующих веществ или увлажняющих агентов и суспендирующих веществ, как раскрыто выше. Стерильный состав для инъекции также может представлять собой стерильную инъекцию или суспензию, полученную в парентерально приемлемом нетоксичном разбавителе или растворителе. Кроме того, стерильное нелетучее масло может обычно использоваться в качестве растворителя или суспендирующей среды.
Соединение по настоящему изобретению можно вводить в форме суппозитория для ректального введения. Такая фармацевтическая композиция может быть получена путем смешивания лекарственного средства с подходящим не вызывающим раздражения эксципиентом, который представляет собой твердое вещество при температуре окружающей среды, но представляет собой жидкость в прямой кишке и, следовательно, будет растворяться в прямой кишке с высвобождением лекарственного средства. Такие материалы включают масло какао, глицерожелатин, гидрогенизированные растительные масла, полиэтиленгликоли различной молекулярной массы и смеси сложных эфиров жирных кислот полиэтиленгликолей.
Как хорошо известно специалистам в данной области техники, дозировка вводимого лекарственного средства зависит от множества факторов, включая, но не ограничиваясь, активность конкретного используемого соединения, возраст субъекта, масса тела субъекта, состояние здоровья субъекта, поведение субъекта, рацион питания субъекта, время введения, способ введения, скорость выведения, комбинацию лекарственных средств и тому подобное. Кроме того, оптимальная схема лечения, такая как способ введения, суточная доза соединения общей формулы или тип фармацевтически приемлемых солей, может быть проверена в соответствии с обычными схемами лечения.
Настоящее изобретение относится к конъюгатам, содержащим нацеливающий (связывающий) фрагмент и терапевтический фрагмент, который представляет собой лекарственное средство, с улучшенной способностью нацеливаться на различные раковые клетки, инфекционные болезнетворные организмы и/или для применения в лечении аутоиммунных заболеваний. Нацеливающий фрагмент антитела, связан с терапевтическим фрагментом соединения посредством внутриклеточно расщепляемой связи, которая повышает терапевтическую эффективность.
На протяжении многих лет целью ученых в области специфической лекарственной терапии было использование моноклональных антител (MAb) для специфической доставки токсических агентов при раке человека. Были разработаны конъюгаты опухолеассоциированных MAb с соответствующими токсическими агентами, но они имели смешанный успех в лечении рака и мало использовались при других заболеваниях, таких как инфекционные заболевания и аутоиммунные заболевания. Токсичные агенты являются наиболее распространенными химиотерапевтическими препаратами. Существует дальнейшая необходимость в разработке более эффективного конъюгата антитела с внутриклеточно расщепляемым линкером для лечения рака, патогенов и других заболеваний.
Подробное описание изобретения
Если не указано иное, все технические и научные термины, используемые в настоящем изобретении, имеют такое же значение, которое обычно подразумевается специалистом в области техники, к которой относится настоящее изобретение. Хотя любые способы и материалы, аналогичные или эквивалентные описанным в настоящем документе, также могут быть использованы для осуществления или тестирования настоящего изобретения, предпочтительные способы и материалы описаны в настоящем документе. В описании и формуле изобретения по настоящему изобретению используются следующие термины в соответствии с приведенными ниже определениями.
При использовании торгового наименования в настоящем описании предполагается, что оно включает состав коммерческого продукта под указанным торговым наименованием и лекарственное средство и активный компонент лекарственного средства коммерческого продукта под указанным торговым наименованием.
Термин «лиганд» относится к макромолекулярному соединению, способному распознавать и связываться с антигеном или рецептором, связанным с клеткой-мишенью. Роль лиганда заключается в том, чтобы представлять лекарственное средство целевой популяции клеток, с которой связывается лиганд, и лиганды включают, но не ограничиваются ими, белковые гормоны, лектины, факторы роста, антитела или другие молекулы, способные связываться с клетками. В вариантах осуществления настоящего изобретения лиганд обозначен как Рс, и лиганд может образовывать связь со связывающим фрагментом через гетероатом на лиганде и предпочтительно представляет собой антитело или его антигенсвязывающий фрагмент, где антитело выбрано из группы, состоящей из химерного антитела, гуманизированного антитела, полностью человеческого антитела и мышиного антитела; предпочтительно моноклонального антитела.
Термин «лекарственное средство» относится к цитотоксическому лекарственному средству или иммуномодулятору. Цитотоксическое лекарственное средство может иметь химическую молекулу в опухолевой клетке, которая является достаточно сильной, чтобы нарушить ее нормальный рост. Цитотоксическое лекарственное средство может уничтожать опухолевые клетки в целом при достаточно высокой концентрации; однако из-за отсутствия специфичности, цитотоксическое лекарственное средство может вызывать апоптоз нормальных клеток при уничтожении опухолевых клеток, что приводит к серьезным побочным эффектам. Этот термин включает токсины, такие как низкомолекулярные токсины или ферментативно активные токсины бактериального, грибкового, растительного или животного происхождения, радиоизотопы (например, At211, I131, I125, Y90, Re186, Re188, Sm153, Bi212, P32 и радиоактивные изотопы Lu), химиотерапевтические лекарственные средства, антибиотики и нуклеолитические ферменты. В таком варианте осуществления настоящего изобретения лекарственное средство обозначено как D и может представлять собой ингибитор BCL2, ингибитор BCL-XL или ингибитор BCL2/BCL-XL с двумя мишенями.
Термин «линкерная единица», «линкер» или «линкерный фрагмент» относится к химическому структурному фрагменту или связи, который связан с лигандом на одном конце и с лекарственным средством на другом конце, а также может быть связан с другими линкерами, а затем связан с лекарственным средством.
Линкер содержит удлинитель, спейсер и аминокислотный узел и может быть синтезирован способами, известными в данной области техники, такими как описанными в US 2005-0238649 A1. Линкер может представлять собой «расщепляемый линкер», благоприятствующий высвобождению лекарственных средств в клетках. Например, могут быть использованы кислотолабильные линкеры (например, гидразоны), линкеры, чувствительные к протеазе (например, чувствительные к пептидазе), фотолабильные линкеры, диметильные линкеры или дисульфидсодержащие линкеры (Chari et al., Cancer Research 52: 127-131(1992); патент США №5208020).
Термин «конъюгат лиганд-лекарственное средство» означает, что лиганд связан с биологически активным лекарственным средством посредством линкерного фрагмента. В настоящем описании «конъюгат лиганда-лекарственное средство» предпочтительно представляет собой конъюгат антитела и лекарственного средства (ADC), что означает, что моноклональное антитело или фрагмент антитела связаны с биологически активным токсичным лекарственным средством посредством стабильного линкерного фрагмента.
Трехбуквенные и однобуквенные коды для аминокислот, используемые в настоящем описании, раскрыты в J. biol. chem, 243, р3558 (1968).
«Антитело», описанное в настоящем документе, используется в самом широком смысле и включает различные структуры антител, включая, но не ограничиваясь ими, моноклональное антитело, поликлональное антитело, мышиное антитело, химерное антитело, гуманизированное антитело, мультиспецифическое антитело (например, биспецифическое антитело) и антигенсвязывающие фрагменты, при условии, что они проявляют желаемую антигенсвязывающую активность и специфичность.
Способы получения и очистки антител и антигенсвязывающих фрагментов хорошо известны в данной области техники, например, описанные в главах 5-8 и 15 публикации Antibodies: A Laboratory Manual, Cold Spring Harbor Press. Аналогично, антигенсвязывающие фрагменты также могут быть получены с помощью общепринятых способов. Антитело или антигенсвязывающий фрагмент, описанные в данном изобретении, генетически сконструированы таким образом, чтобы содержать одну или более дополнительных FR человека в CDR нечеловеческого происхождения. Последовательности FR зародышевой линии человека можно получить на веб-сайте http://imgt.cines.fr ImMunoGeneTics (IMGT) или в журнале иммуноглобулинов, 2001ISBN012441351, путем сравнения с базой данных генов вариабельной области человеческих антител зародышевой линии с помощью программного обеспечения МОЕ.
Термин «алкил» относится к насыщенной алифатической углеводородной группе, которая представляет собой линейную или разветвленную группу, содержащую от 1 до 20 атомов углерода, предпочтительно к алкилу содержащему от 1 до 12 (например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 и 12) атомов углерода, более предпочтительно к алкилу, содержащему от 1 до 10 атомов углерода, и наиболее предпочтительно к алкилу, содержащему от 1 до 6 атомов углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их различные изомеры с боковыми цепями и т.д. Более предпочтительным является низший алкил, имеющий от 1 до 6 атомов углерода, и неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.д. Алкил может быть замещенным или незамещенным, и при его замещении замещение заместителем может быть выполнено в любом доступном месте соединения, где заместитель предпочтительно представляет собой одну или более следующих групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио и оксо.
Термин «гетероалкил» относится к алкилу, содержащему один или более гетероатомов, выбранных из группы, состоящей из N, О и S, где алкил является таким, как определено выше.
Термин «алкилен» относится к насыщенной линейной или разветвленной алифатической углеводородной группе, имеющей 2 остатка, полученных из исходного алкана путем удаления двух атомов водорода из одного и того же атома углерода или двух различных атомов углерода, которая представляет собой линейную или разветвленную группу, содержащую от 1 до 20 атомов углерода, предпочтительно алкиленовую группу, содержащую от 1 до 12 (например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 и 12) атомов углерода, и более предпочтительно алкиленовую группу, содержащую от 1 до 6 атомов углерода. Неограничивающие примеры алкилена включают, но не ограничиваются, метилен (-СН2-), 1,1-этилиден (-СН(СН3)-), 1,2-этилиден (-СН2СН2)-, 1,1-пропилиден (-СН(СН2СН3)-), 1,2-пропилиден (-СН2СН(СН3)-), 1,3-пропилиден (-СН2СН2СН2-), 1,4-бутилиден (-СН2СН2СН2СН2-), 1,5-бутилиден (-СН2СН2СН2СН2СН2-) и т.п. Алкил может быть замещенным или незамещенным, и при его замещении замещение заместителем может быть выполнено в любом доступном месте соединения, где заместитель предпочтительно независимо и необязательно выбран из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио и оксо.
Термин «алкокси» относится к группе -О-(алкил) и -O-(незамещенный циклоалкил), где алкил или циклоалкил является таким, как определено выше. Неограничивающие примеры алкокси включают: метокси, этокси, пропокси, бутокси, циклопропокси, циклобутокси, циклопентилокси и циклогексилокси. Алкокси может быть необязательно замещенным или незамещенным, и когда он замещен, заместитель предпочтительно представляет собой одну или более из следующих групп, независимо выбранных из группы, состоящей из: алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио и гетероциклоалкилтио.
Термин «циклоалкил» относится к насыщенному или частично ненасыщенному моноциклическому или полициклическому углеводородному заместителю. Циклоалкильное кольцо содержит от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 10 атомов углерода и наиболее предпочтительно от 3 до 8 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и тому подобное. Полициклический циклоалкил включает спироциклоалкил, конденсированный циклоалкил и мостиковый циклоалкил.
Термин «циклоалкилен» относится к циклоалкильной группе, имеющей 2 остатка, полученных путем удаления двух атомов водорода из исходного циклоалкила. Циклоалкильная группа является такой, как определено выше.
Термин «гетероциклил» относится к насыщенному или частично ненасыщенному моноциклическому или полициклическому углеводородному заместителю, содержащему от 3 до 20 кольцевых атомов, где один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода и S(O)m (где m представляет собой целое число от 0 до 2), за исключением циклической части -О-О-, -О-S- или -S-S-, и остальные кольцевые атомы представляют собой атомы углерода. Гетероциклил предпочтительно содержит от 3 до 12 кольцевых атомов, из которых от 1 до 4 (например, 1, 2, 3 и 4) являются гетероатомами; более предпочтительно от 3 до 10 кольцевых атомов; более предпочтительно от 3 до 8 (например, 3, 4, 5, 6, 7 и 8) кольцевых атомов, из которых от 1 до 3 (например, 1, 2 и 3) являются гетероатомами; более предпочтительно от 3 до 6 кольцевых атомов, из которых от 1 до 3 являются гетероатомами; и наиболее предпочтительно от 5 до 6 кольцевых атомов, из которых от 1 до 3 являются гетероатомами. Неограничивающие примеры моноциклического гетероциклила включают пирролидинил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и т.д. Полициклический гетероциклил включает спирогетероциклил, конденсированный гетероциклил и мостиковый гетероциклил.
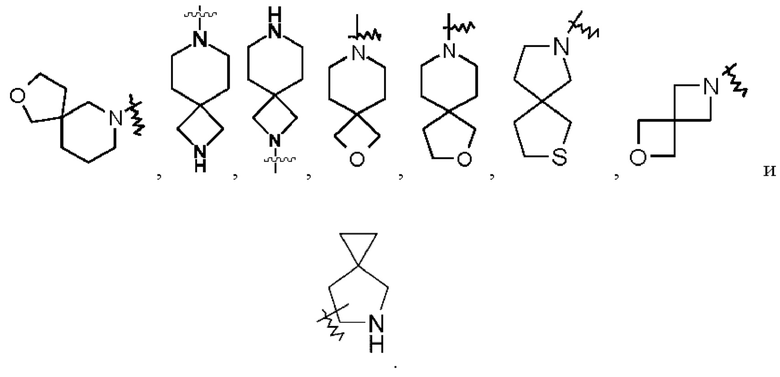
Термин «спирогетероциклил» относится к 5-20-членной полициклической гетероциклильной группе, в которой моноциклические кольца имеют общий один атом (называемый спироатомом), где один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода и S(O)m (где m представляет собой целое число от 0 до 2), а остальные кольцевые атомы представляют собой атомы углерода. Эти кольца могут содержать одну или более двойных связей, но ни одно из них не имеет полностью сопряженной π-электронной системы. Предпочтительно, спирогетероциклил представляет собой 6-14-членный и более предпочтительно 7-10-членный (например, 7-членный, 8-членный, 9-членный или 10-членный). В соответствии с количеством спироатомов, являющихся общими между кольцами, спирогетероциклил может являться моноспирогетероциклилом, биспирогетероциклилом или полиспирогетероциклилом, предпочтительно моноспирогетероциклилом и биспирогетероциклилом и более предпочтительно 4-членным/4-членным, 4-членным/5-членным, 4-членным/6-членным, 5-членным/5-членным или 5-членным/6-членным моноспирогетероциклилом. Неограничивающие примеры спирогетероциклила включают:
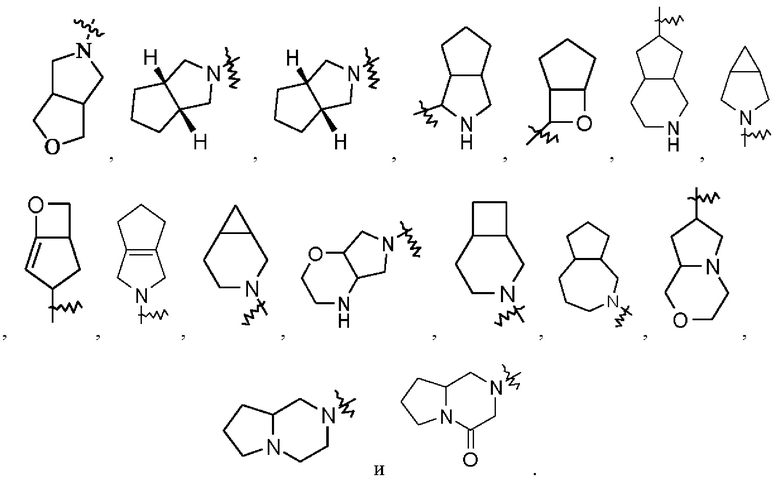
Термин «конденсированный гетероциклил» относится к 5-20-членной полициклической гетероциклльной группе, в которой каждое кольцо имеет общую пару соседних атомов с другими кольцами в системе, где одно или более колец могут содержать одну или более двойных связей, но ни одно из них не имеет полностью сопряженной π-электронной системы, где один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода и S(O)m (где m представляет собой целое число от 0 до 2), а остальные кольцевые атомы представляют собой атомы углерода. Предпочтительно, конденсированный гетероциклил представляет собой 6-14-членный и более предпочтительно 7-10-членный (например, 7-членный, 8-членный, 9-членный или 10-членный). В соответствии с количеством образованных колец, конденсированный гетероциклил может быть бициклическим, трициклическим, тетрациклическим или полициклическим конденсированным гетероциклилом, предпочтительно бициклическим или трициклическим конденсированным гетероциклилом и более предпочтительно 5-членным/5-членным или 5-членным/6-членным бициклическим конденсированным гетероциклилом. Неограничивающие примеры конденсированного гетероциклила включают:
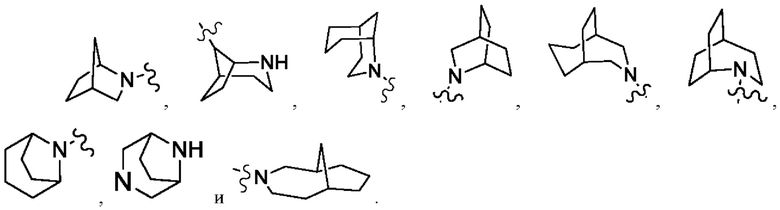
Термин «мостиковый гетероциклил» относится к 5-14-членной полициклической гетероциклильной группе, в которой любые два кольца имеют общих два атома углерода, которые непосредственно не присоединены друг к другу, причем эти кольца могут содержать одну или более двойных связей, но ни одно из них не имеет полностью сопряженной π-электронной системы, причем один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода и S(O)m (где m представляет собой целое от 0 до 2), а остальные кольцевые атомы представляют собой атомы углерода. Предпочтительно, мостиковый гетероциклил представляет собой 6-14-членный и более предпочтительно 7-10-членный (например, 7-членный, 8-членный, 9-членный или 10-членный). В соответствии с количеством образованных колец, мостиковый гетероциклил может быть бициклическим, трициклическим, тетрациклическим или полициклическим, предпочтительно бициклическим, трициклическим или тетрациклическим и более предпочтительно бициклическим или трициклическим. Неограничивающие примеры мостикового гетероциклила включают:
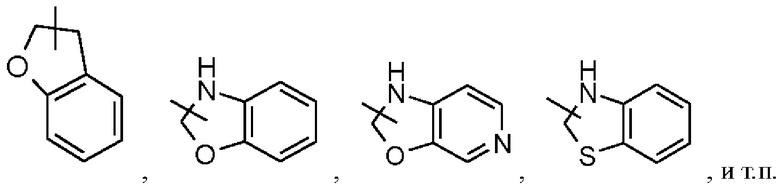
Гетероциклильное кольцо может быть конденсировано с арильным, гетероарильным или циклоалкильным кольцом, причем кольцо, присоединенное к исходной структуре, представляет собой гетероциклил; неограничивающие примеры включают, но не ограничиваются ими:
Гетероциклил может быть необязательно замещенным или незамещенным, и когда он замещен, заместитель предпочтительно представляет собой одну или более из следующих групп, независимо выбранных из группы, состоящей из: алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио и оксо.
Термин «гетероциклилен» относится к гетероциклильной группе, имеющей 2 остатка, полученных путем удаления двух атомов водорода из идентичных или разных кольцевых атомов исходного гетероциклила. Гетероциклил является таким, как определено выше.
Термин «арил» относится к 6-14-членной, предпочтительно 6-10-членной углеродной моноциклической или конденсированной полициклической (т.е., кольца, имеющие общую пару соседних атомов углерода) группе, имеющей сопряженную π-электронную систему, такой как фенил и нафтил, предпочтительно фенил. Арильное кольцо может быть конденсировано с гетероарильным, гетероциклильным или циклоалкильным кольцом, причем кольцо, присоединенное к исходной структуре, представляет собой арильное кольцо; неограничивающие примеры включают, но не ограничиваются ими:
Арил может быть замещен или незамещен, и когда он замещен, заместитель предпочтительно представляет собой одну или более из следующих групп, независимо выбранных из группы, состоящей из: алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио и гетероциклоалкилтио.
Термин «гетероарил» относится к гетероароматической системе, содержащей от 1 до 4 гетероатомов и от 5 до 14 кольцевых атомов, где гетероатомы выбраны из группы, состоящей из кислорода, серы и азота. Гетероарил предпочтительно представляет собой 5-10-членный, более предпочтительно 5- или 6-членный, такой как фуранил, тиенил, пиридинил, пирролил, N-алкилпирролил, пиримидинил, пиразинил, имидазолил и тетразолил. Гетероарильное кольцо может быть конденсировано с арильным, гетероциклильным или циклоалкильным кольцом, причем кольцо, присоединенное к исходной структуре, представляет собой гетероарил; неограничивающие примеры включают, но не ограничиваются ими:
Гетероарил может быть необязательно замещен или незамещен, и когда он замещен, заместитель предпочтительно представляет собой одну или более из следующих групп, независимо выбранных из группы, состоящей из: алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио и гетероциклоалкилтио.
Термин «аминозащитная группа» относится к группе, которая может быть легко удалена и предназначена для защиты аминогруппы от изменения, когда реакция проводится в другом месте молекулы. Неограничивающие примеры аминозащитных групп включают 9-флуоренилметоксикарбонил, трет-бутоксикарбонил, ацетил, бензил, аллил, п-метоксибензили т.д. Эти группы могут быть необязательно замещены 1-3 заместителями, выбранными из группы, состоящей из галогена, алкокси и нитро. Аминозащитная группа предпочтительно представляет собой 9-флуоренилметоксикарбонил.
Термин «алкенил» относится к алкильному соединению, содержащему углерод-углеродные двойные связи в молекуле, где алкил является таким, как определено выше. Алкенил может быть замещенным или незамещенным, и когда он замещен, заместитель предпочтительно представляет собой одну или более групп, независимо выбранных из одного или более заместителей водорода, алкила, алкокси, галогена, галогеналкила, галогеналкокси, циклоалкилокси, гетероциклилокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила.
Термин «алкинил» относится к алкильному соединению, содержащему тройную связь углерод-углерод в молекуле, где алкил является таким, как определено выше. Алкинил может быть замещенным или незамещенным, и когда он замещен, заместитель предпочтительно представляет собой одну или более групп, независимо выбранных из одного или более заместителей водорода, алкила, алкокси, галогена, галогеналкила, галогеналкокси, циклоалкилокси, гетероциклилокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила.
Термин «циклоалкилалкил» относится к алкильной группе, замещенной одной или более циклоалкильными группами, предпочтительно одной циклоалкильной группой, где алкил является таким, как определено выше, и циклоалкил является таким, как определено выше.
Термин «галогеналкил» относится к алкильной группе, замещенной одним или более галогенами, где алкил является таким, как определено выше.
Термин «дейтерированный алкил» относится к алкильной группе, замещенной одним или более атомами дейтерия, где алкил является таким, как определено выше.
Термин «гидрокси» относится к группе -ОН.
Термин «галоген» относится к фтору, хлору, брому или йоду.
Термин «амино» относится к -NH2.
Термин «нитро» относится к -NO2.
Термин «ациламино» относится к -С(O)N(алкил) или -С(O)N(циклоалкил), где алкил и циклоалкил являются такими, как определено выше.
Термин «карбоксилатная группа» относится к -С(O)O(алкилу) или С(O)O(циклоалкилу), где алкил и циклоалкил являются такими, как определено выше.
В химической формуле аббревиатура «Ме» относится к метилу.
В химической формуле аббревиатура «Ph» относится к фенилу.
Настоящее изобретение также содержит различные дейтерированные формы соединения формулы (D). Каждый доступный атом водорода, соединенный с атомом углерода, может быть независимо замещен атомом дейтерия. Специалисты в данной области техники могут синтезировать дейтерированные формы соединения общей формулы (D) согласно соответствующей литературе. Коммерчески доступные дейтерированные исходные материалы могут быть использованы для получения дейтерированных форм соединения формулы (D), или они могут быть синтезированы с использованием обычных методов с дейтерированными реагентами, включая, но не ограничиваясь, дейтерированный боран, тридейтерированный боран в тетрагидрофуране, дейтерированный алюмогидрид лития, дейтерированный йодэтан, дейтерированный йодметан и тому подобное.
Термин «необязательный» или «необязательно» означает, что событие или обстоятельство, описанное далее, может, но не безусловно, иметь место, и что такое описание включает случаи, когда событие или обстоятельство происходит или не происходит.Например, «гетероциклильная группа, необязательно замещенная алкилом» означает, что алкил может присутствовать, но не обязательно, и что описание включает случаи, когда гетероциклильная группа является или не является замещенной алкилом.
Термин «замещен» означает, что один или более, предпочтительно вплоть до 5, и более предпочтительно от 1 до 3 атомов водорода в группе независимо замещены соответствующим количеством заместителей. Само собой разумеется, что заместитель находится только в своем возможном химическом положении, и специалисты в данной области техники смогут определить (экспериментально или теоретически) возможное или невозможное замещение без особых усилий. Например, она может быть нестабильной, когда амино или гидрокси, имеющие свободный водород, связаны с атомом углерода, имеющим ненасыщенную (например, олефиновую) связь.
Термин «фармацевтическая композиция» относится к смеси, содержащей одно или более соединений, описанных в настоящем документе, или их физиологически/фармацевтически приемлемую соль, или пролекарство и другие химические компоненты, например, физиологически/фармацевтически приемлемые носители и эксципиенты. Фармацевтическая композиция предназначена для того, чтобы способствовать введению в организм, что облегчает абсорбцию активного ингредиента, тем самым осуществляя биологическую активность.
Термин «фармацевтически приемлемая соль» относится к соли конъюгата лиганда и лекарственного средства по настоящему изобретению или к соли соединения, описанная в настоящем документе. Такие соли являются безопасными и эффективными при использовании у млекопитающих и обладают требуемой биологической активностью. Конъюгат лиганд-лекарственное средство по настоящему изобретению содержит по меньшей мере одну аминогруппу и, таким образом, может образовывать соль с кислотой. Неограничивающие примеры фармацевтически приемлемых солей включают: гидрохлорид, гидробромид, гидроиодат, сульфат, бисульфат, цитрат, ацетат, сукцинат, аскорбат, оксалат, нитрат, сорбат, гидрофосфат, дигидрофосфат, салицилат, гидроцитрат, тартрат, малеат, фумарат, формиат, бензоат, мезилат, этансульфонат, бензолсульфонат и n-толуолсульфонат.
Термин «содержание лекарственного средства» относится к среднему количеству цитотоксических лекарственных средств, содержащихся в каждом лиганде в молекулах конъюгата, и также может быть обозначен как отношение лекарственного средства к антителу (DAR). Содержание лекарственного средства может составлять от 0 до 12, предпочтительно от 1 до 10, цитотоксических лекарственных средств, прикрепленных к каждому лиганду (Рс). В вариантах осуществления настоящего изобретения содержание лекарственного средства обозначено как n, и иллюстративно, может составлять в среднем 1, 2, 3, 4, 5, 6, 7, 8, 9 и 10, в диапазоне от 0 до 12, предпочтительно от 1 до 10, и более предпочтительно от 1 до 8, или от 2 до 8, или от 2 до 7, или от 3 до 8, или от 3 до 7, или от 3 до 6, или от 4 до 7, или от 4 до 6, или от 4 до 5. Содержание лекарственного средства на молекулу ADC после реакции связывания может быть охарактеризовано обычными методами, такими как спектроскопия в УФ/видимом спектре (УФ - ультрафиолелтовый спектр), масс-спектрометрия, анализы ELISA (иммуноферментный анализ), CE-SDS (капиллярный электрофорез в присутствии додецилсульфата натрия) и HPLC (высокоэффективная жидкостная хроматография).
Содержание конъюгата лиганд-цитотоксическое лекарственное средство может контролироваться следующими неограничивающими способами, включая:
(1) контроль молярного отношения связывающего реагента к лиганду,
(2) контроль времени и температуры реакции, и
(3) выбор различных реагентов реакции.
Для получения обычных фармацевтических композиций см. Китайскую фармакопею.
Термин «носитель» для лекарственного средства по настоящему изобретению относится к системе, которая может изменять способ попадания лекарственного средства в организм человека и распределение лекарственного средства в организме человека, контролировать скорость высвобождения лекарственного средства и доставлять лекарственное средство в орган-мишень. Система высвобождения и нацеливания на основе носителя лекарственного средства может уменьшить деградацию и потерю лекарственного средства, уменьшить побочные эффекты и улучшить биодоступность. Например, полимерные поверхностно-активные вещества, которые могут быть использованы в качестве носителей, могут самостоятельно собираться благодаря своим уникальным амфифильным структурам с образованием различных форм агрегатов, таких как мицеллы, микроэмульсии, гели, жидкие кристаллы и везикулы, в качестве предпочтительных примеров. Агрегаты имеют способность инкапсулировать молекулы лекарственного средства и имеют хорошую проницаемость для мембран, и поэтому могут быть использованы как превосходные носители лекарственного средства.
Термин «эксципиент» представляет собой дополнение, помимо основного лекарственного средства, к фармацевтической композиции. Его также можно назвать вспомогательным веществом. Например, связующие вещества, наполнители, разрыхлители и смазывающие вещества в таблетках; основная часть в полутвердой мази и кремовых препаратах; консерванты, антиоксиданты, корригенты, ароматизаторы, сорастворители, эмульгаторы, солюбилизаторы, агенты, регулирующие тоничность, красители и тому подобное в жидких составах могут быть названы эксципиентами.
Термин «разбавитель», также называемый наполнителем, используется в первую очередь для увеличения массы и объема таблетки. Добавление разбавителя не только обеспечивает определенный объем, но и уменьшает отклонение дозы основных ингредиентов, а также улучшает способность лекарственного средства к прессованию и тому подобное. Когда лекарственное средство в форме таблетки содержит маслянистые компоненты, в обязательном порядке добавляют абсорбент для абсорбции маслянистых компонентов таким образом, чтобы поддерживать «сухое» состояние и, таким образом, облегчать получение таблетки. Примеры включают крахмал, лактозу, неорганические соли кальция, микрокристаллическую целлюлозу и тому подобное.
Реагенты, обеспечивающие щелочную среду, включают органические и неорганические основания, причем органические основания включают, но не ограничиваются, триэтиламин, диэтиламин, N-метилморфолин, пиридин, пиперидин, N, N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксиднатрия фторид тетрабутиламмония или трет-бутоксидкалия, а неорганические основания включают, но не ограничиваются, гидрид натрия, фосфат калия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия и гидроксид лития, предпочтительно N,N-диизопропилэтиламин.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг. 1: ингибирующее действие соединений по настоящему изобретению на RS4;11 человеческого острого лимфобластного лейкоза на мышиной подкожной ксенотрансплантатной опухоли.
Фиг. 2: изменение массы тела, вызванное ингибированием RS4;11 острого лимфобластного лейкоза человека на мышиной подкожной ксенотрансплантатной опухоли.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Следующие примеры дополнительно иллюстрируют настоящее изобретение, но настоящее изобретение не ограничивается этим.
Экспериментальные процедуры без условий, указанные в примерах настоящего изобретения, как правило, проводили в соответствии с общепринятыми условиями или в соответствии с условиями, рекомендованными производителем исходных материалов или коммерческих продуктов. Реагенты без указания конкретного происхождения являются коммерчески доступными общепринятыми реагентами.
I. Синтез соединений
Примеры
Структуру соединения определяли с помощью спектроскопии ядерного магнитного резонанса (NMR) и/или масс-спектрометрии (MS). Сдвиг NMR (δ) приведен в единицах измерения 10-6 (млн-1). Спектры NMR определяли с использованием прибора ядерного магнитного резонанса Bruker AVANCE NEO 500М, с дейтерированным диметилсульфоксидом (DMSO-d6), дейтерированным хлороформом (CDCl3) и дейтерированным метанолом (CD3OD) в качестве растворителей и тетраметилсиланом (TMS) в качестве внутреннего стандарта.
Масс-спектры определяли с использованием системы жидкостной хроматографии-масс-спектрометрии Agilent 1200/1290 DAD-6110/6120 Quadrupole MS (производитель: Agilent; модель MS: 6110/6120 Quadrupole MS), Waters ACQuity UPLC-QD/SQD (производитель: Waters, модель MS: Waters ACQuity Qda Detector/Waters SQ Detector) и THERMO Ultimate 3000-Q Exactive (производитель: THERMO, модель MS: THERMO Q Exactive).
Анализ методом высокоэффективной жидкостной хроматографии (HPLC) проводили с использованием высокоэффективных жидкостных хроматографов Agilent HPLC 1200DAD, Agilent HPLC 1200VWD и Waters HPLC e2695-2489.
Хиральный HPLC -анализ проводят с использованием высокоэффективного жидкостного хроматографа Agilent 1260 DAD.
Высокоэффективную жидкостную препаративную хроматографию проводили с использованием препаративных хроматографов Waters 2545-2767, Waters 2767-SQ Detecor2, Shimadzu LC-20AP и Gilson GX-281.
Хиральную препаративную HPLC проводили с использованием препаративного хроматографа Shimadzu LC-20AP.
Используемый инструмент быстрой препарации CombiFlash (флеш-хроматография) - Combiflash Rf200 (TELEDYNE ISCO).
Силикагелевые пластины Huanghai HSGF254 или Qingdao GF254 со спецификациями от 0,15 мм до 0,2 мм применяли для тонкослойной хроматографии (TLC) и от 0,4 мм до 0,5 мм для разделения и очистки с помощью TLC.
Силикагель Yantai Huanghai 200-300 меш обычно использовали в качестве носителя в колоночной хроматографии.
Среднее ингибирование киназы и значение IC50 определяли с помощью устройства для микропланшетного ридера NovoStar (BMG, Германия).
Известные исходные материалы, описанные в настоящем документе, могут быть синтезированы с использованием или в соответствии со способами, известными в данной области техники, или могут быть приобретены у ABCR GmbH & Co. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc., Chembee Chemicals и других компаний.
В примерах все реакции можно проводить в атмосфере аргона или в атмосфере азота, если не указано иное.
Атмосфера аргона или атмосфера азота означает, что реакционная колба соединена с баллоном, содержащим около 1 л аргона или азота.
Атмосфера водорода означает, что реакционная колба соединена с баллоном, содержащим около 1 л водорода.
Гидрогенизатор Parr 3916EKX, гидрогенизатор Qinglan QL-500 или гидрогенизатор HC2-SS использовали в реакциях гидрирования под давлением.
Реакции гидрирования обычно включают 3 цикла вакуумирования и продувки водородом.
В реакциях, проводимых под воздействием микроволнового излучения, использовали микроволновый реактор СЕМ Discover-S 908860.
В примерах раствор относится к водному раствору, если не указано иное.
В примерах температура реакции относится к комнатной температуре, то есть от 20 до 30°С, если не указано иное.
Мониторинг хода реакции в примерах проводили с помощью тонкослойной хроматографии (TLC). Проявляющий растворитель для реакций, элюирующая система для очистки колоночной хроматографией и система проявляющего растворителя для тонкослойной хроматографии включали: А: дихлорметан/метанольную систему, и В: н-гексан/этилацетатную систему. Объемное соотношение растворителей корректировали в соответствии с полярностью соединения или путем добавления небольшого количества основных или кислотных реагентов, таких как триэтиламин и уксусная кислота.
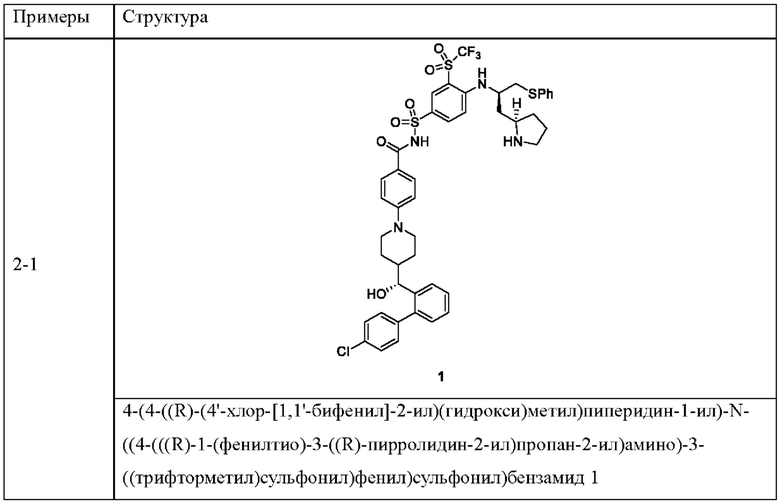
Пример 2-1
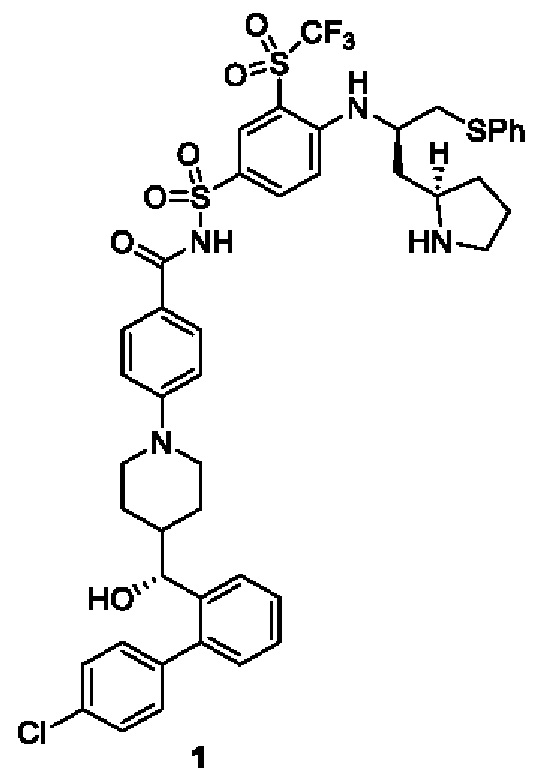
4-(4-((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-(фенилтио)-3-((R)-пирролидин-2-ил)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 1
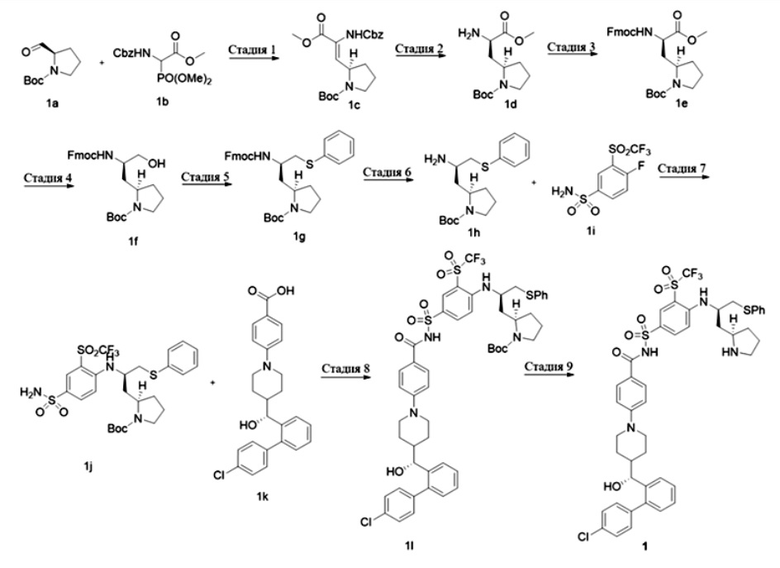
Стадия 1
Трет-бутил(R,Z)-2-(2-(((бензилокси)карбонил)амино)-3-метокси-3-оксопроп-1-ен-1-ил)пирролидин-1-карбоксилат 1с
Метил-2-(((бензилокси)карбонил)амино)-2-(диметоксифосфорил)ацетат 1b (1 г, 3,02 ммоль, Accela ChemBio) растворяли в дихлорметане (10 мл) и охлаждали раствор до 0°С под ледяной баней. 1,8-диазабицикло[5.4.0]ундец-7-ен (356 мг, 2,34 ммоль, Accela ChemBio) добавляли по каплям, и смесь перемешивали при 0°С в течение 20 мин, с последующим добавлением раствора трет-бутил(R)-2-формилпирролидин-1-карбоксилата 1а (500 мг, 2,51 ммоль, PharmaBlock) в дихлорметане (5 мл). После добавления по каплям ледяную баню удаляли и смесь нагревали до комнатной температуры и реагировали в течение 48 ч. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 1с (830 г, выход: 81,8%).
MS m/z (масса/заряд) (ESI (ионизация электроспреем)): 305,0 [М+Н-100].
Стадия 2
Трет-бутил(R)-2-((R)-2-амино-3-метокси-3-оксопропил)пирролидин-1-карбоксилат 1d
1c (520 мг, 1,28 ммоль) растворяли в изопропаноле (10 мл) и добавляли палладий на угле (100 мг, 10% содержания, на сухой основе). Смесь продували водородом 3 раза и перемешивали при комнатной температуре в течение 16 ч. Реакционный раствор фильтровали через целит и фильтрат концентрировали с получением указанного в заголовке продукта 1d (300 мг, выход: 85,7%), который непосредственно использовали на следующей стадии без очистки.
MS m/z(ESI): 273,1 [М+1].
Стадия 3
Трет-бутил(R)-2-((R)-2-((((9Н-флуорен-9-ил)метокси)карбонил)амино)-3-метокси-3-оксопропил)пирролидин-1-карбоксилат 1е
Неочищенный продукт 1d (300 мг, 1,10 ммоль) растворяли в 1,4-диоксане (4 мл) и добавляли воду (1 мл), бикарбонат натрия (278 мг, 3,31 ммоль) и флуоренилметоксикарбонилхлорид (285 мг, 1,10 ммоль, Accela ChemBio). Смесь перемешивали при комнатной температуре в течение 1,5 ч. К реакционному раствору добавляли воду (10 мл) и экстрагировали смесь этилацетатом (10 мл 3 раза). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали для удаления высушивающего вещества, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 1е (396 мг, выход: 72,7%).
MS m/z (ESI): 395,1 [М+Н-100].
Стадия 4
Трет-бутил(R)-2-((R)-2-((((9Н-флуорен-9-ил)метокси)карбонил)амино)-3-гидроксипропил)-(пирролидин-1-карбоксилат 1f
1е (200 мг, 0,40 ммоль) растворяли в тетрагидрофуране (10 мл) и добавляли этанол (10 мл) с последующим последовательным добавлением боргидрида натрия (92 мг, 2,43 ммоль) и хлорида лития (112 мг, 2,64 ммоль). Смесь перемешивали при 28°С в течение 2,5 ч. Реакционный раствор охлаждали на ледяной бане и по каплям добавляли насыщенный раствор хлорида аммония (10 мл) для гашения реакции. Добавляли воду (10 мл) и экстрагировали смесь этилацетатом (10 мл 3 раза). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия и фильтровали для удаления высушивающего вещества, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 1f (144 мг, выход: 76,3%).
MS m/z (ESI): 367,1 [М+Н-100].
Стадия 5
Трет-бутил(R)-2-((R)-2-((((9Н-флуорен-9-ил)метокси)карбонил)амино)-3-(фенилтио)пропил)пирролидин-1-карбоксилат 1g
1f (144 мг, 0,31 ммоль) растворяли в толуоле (5 мл) и добавляли дифенилдисульфид (225 мг, 1,03 ммоль, адамас (adamas)) и трибутилфосфин (208 мг, 1,03 ммоль). Реакционную систему продували азотом 3 раза и нагревали до 80°С и перемешивали в течение 12 ч. Реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 1g (168 мг, 97,4%).
MS m/z (ESI): 459,1 [М+Н-100].
Стадия 6
Трет-бутил(R)-2-((R)-2-амино-3-(фенилтио)пропил)пирролидин-1-карбоксилат 1h
1g (168 мг, 0,30 ммоль) растворяли в дихлорметане (2 мл) и добавляли диэтиламин (2 мл). Смесь перемешивали при комнатной температуре в течение 3 ч. Реакционный раствор концентрировали при пониженном давлении для удаления органического растворителя и концентрировали с толуолом (5 мл) при пониженном давлении для получения неочищенного указанного в заголовке продукта 1h (101 мг), который непосредственно использовали на следующей стадии без очистки.
MS m/z(ESI): 337,1 [М+1].
Стадия 7
Трет-бутил(R)-2-((R)-3-(фенилтио)-2-((4-сульфамоил-2-((трифторметил)сульфонил)фенил)амино)пропил)пирролидин-1-карбоксилат 1j
Неочищенный 1h (101 мг, 0,30 ммоль) растворяли в N,N-диметилформамиде (5 мл) и добавляли 4-фтор-3-((трифторметил)сульфонил)бензолсульфонамид 1i (102 мг, 0,33 ммоль, Bide) и N,N-диизопропилэтиламин (388 мг, 3,00 ммоль). Реакционную систему продували азотом 3 раза и нагревали до 50°С и перемешивали в течение 8 ч. К реакционному раствору добавляли воду (10 мл) и экстрагировали смесь этилацетатом (10 мл 3 раза). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали для удаления высушивающего вещества, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 1j (129 мг, 68,9%).
MS m/z (ESI): 524,0 [М+Н-100].
Стадия 8
Трет-бутил-(R)-2-((R)-2-((4-(N-(4-(4-((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)бензил)сульфамоил)-2-((трифторметил)сульфонил)фенил)амино)-3-(фенилтио)пропил)пирролидин-1-карбоксилат 1l
1j (22 мг, 0,035 ммоль) растворяли в дихлорметане (3 мл) и 1k (16 мг, 0,038 ммоль, синтезировали с помощью способа, представленного для промежуточного соединения 40 на странице 79 описания в патенте «CN 103153954 В»), добавляли 4-диметиламинопиридин (8,6 мг, 0,070 ммоль) и 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (20,3 мг, 0,106 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 4 часов в атмосфере азота, добавляли 1k (11 мг, 0,026 ммоль) и перемешивали при комнатной температуре в течение ночи после добавления. Реакционный раствор последовательно промывали водой (5 мл) и насыщенным раствором хлорида натрия (5 мл), сушили над безводным сульфатом натрия и фильтровали, а фильтрат концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с развивающейся системой растворителей А с получением указанного в заголовке продукта 1 л (28 мг, выход: 77,2%).
MS m/z (ESI): 1027,0 [М+1].
Стадия 9
4-(4-((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-(фенилтио)-3-((R)-пирролидин-2-ил)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 1
1 л (14 мг, 0,014 ммоль) растворяли в дихлорметане (0,2 мл) и добавляли раствор 4 М хлористого водорода в диоксане (0,5 мл). Смесь перемешивали при комнатной температуре в течение 30 мин. Реакционный раствор концентрировали при пониженном давлении для удаления органического растворителя. Остаток суспендировали простым диэтиловым эфиром и фильтровали. Полученное твердое вещество очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: Sharpsil-T Prep С18; подвижная фаза: бикарбонат аммония, вода, ацетонитрил), и соответствующую фракцию собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 1 (5 мг, выход: 39,6%).
MS m/z (ESI): 927,2 [М+1].
1Н NMR (500 МГц, CD3OD) δ 8,25 (s, 1H), 8,06 (d, 1H), 7,82 (d, 2Н), 7,62 (d, 1H), 7,48-7,31 (m, 8Н), 7,26 (t, 2Н), 7,20 (t, 2Н), 6,82 (d, 2Н), 6,74 (d, 1H), 4,44 (d, 1H), 4,00-3,95 (m, 1H), 3,78 (d, 1H), 3,61 (d, 1Н), 3,52-3,45 (m, 2Н), 3,26-3,17 (m, 2Н), 2,68 (t, 2Н), 2,53 (t, 2Н), 2,27-1,92 (m, 6Н), 1,81-1,59 (m, 4Н), 1,27-1,21 (m, 1Н), 1,18-1,12 (m, 1H), 1,02-0,90 (m, 2Н).
Пример 2-2
4-(4-(((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-((R)-1-(2-гидроксиэтил)пирролидин-2-ил)-3-(фенилтио)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 2
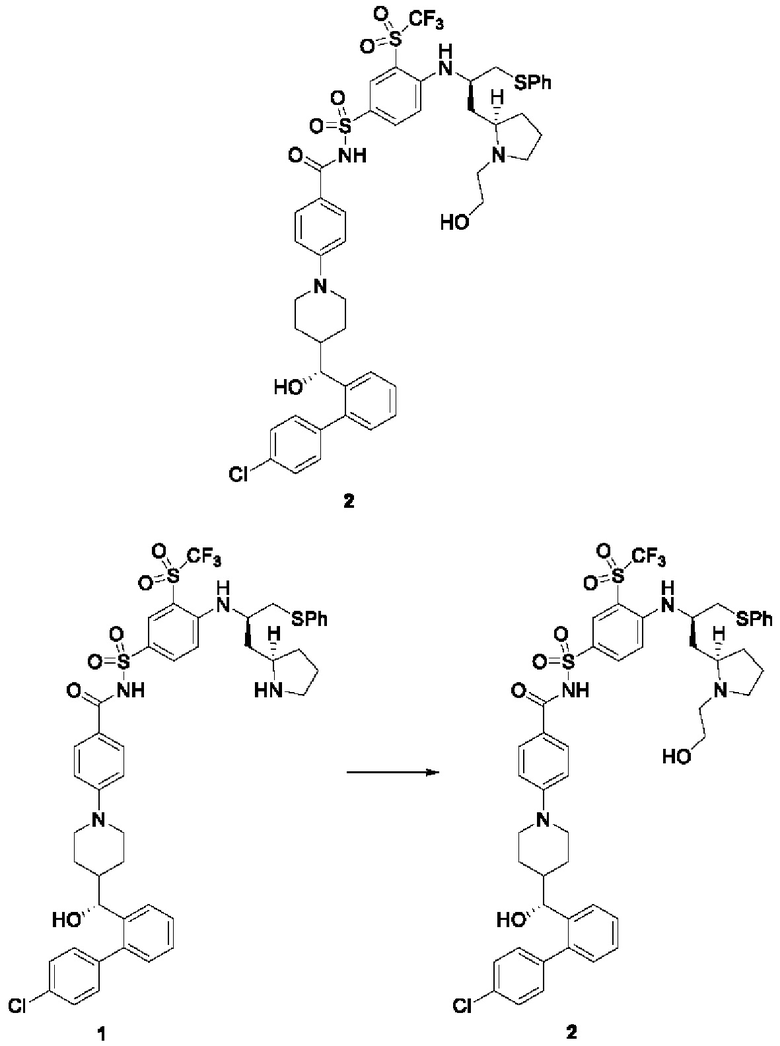
1 (13 мг, 0,014 ммоль) помещали в реакционную колбу и добавляли ацетонитрил (5 мл), затем 2-бромэтанол (26 мг, 0,210 ммоль, адамас) и триэтиламин (11 мг, 0,112 ммоль). Реакционную систему продували азотом 3 раза и нагревали до 70°С и перемешивали в течение 7 ч. Реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: Sharpsil-T Prep С18; подвижная фаза: бикарбонат аммония, вода, ацетонитрил), и соответствующую фракцию собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 2 (3 мг, выход: 22,0%).
MS m/z(ESI): 971,2 [М+1].
1Н NMR (500 МГц, CD3OD) δ 8,26 (s, 1H), 8,03 (d, 1H), 7,80 (d, 2Н), 7,62 (d, 1H), 7,46-7,36 (m, 5H), 7,35-7,27 (m, 5H), 7,24 (d, 1H), 7,18 (d, 1H), 6,81 (d, 2H), 6,63 (d, 1H), 4,42 (d, 1H), 3,91-3,77 (m, 4H), 3,63 (d, 2H), 3,25-3,16 (m, 2H), 2,67 (t, 2H), 2,54 (t, 2H), 2,36-2,29 (m, 2H), 2,23-2,13 (m, 3Н), 2,09-1,97 (m, 4H), 1,81-1,71 (m, 2H), 1,65-1,59 (m, 2H), 1,27-1,21 (m, 1H), 1,17-1,11 (m, 1H), 1,02-0,90 (m, 2H).
Пример 2-3
4-(4-((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(метокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-(фенилтио)-3-((R)-пирролидин-2-ил)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 3
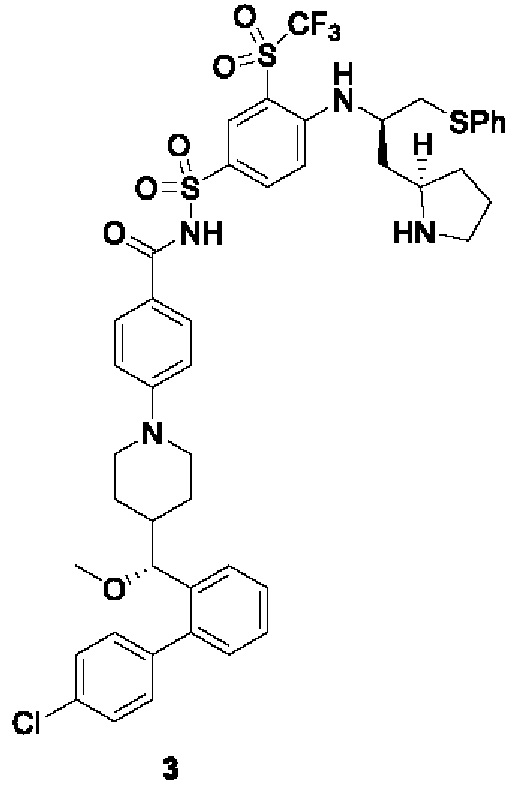
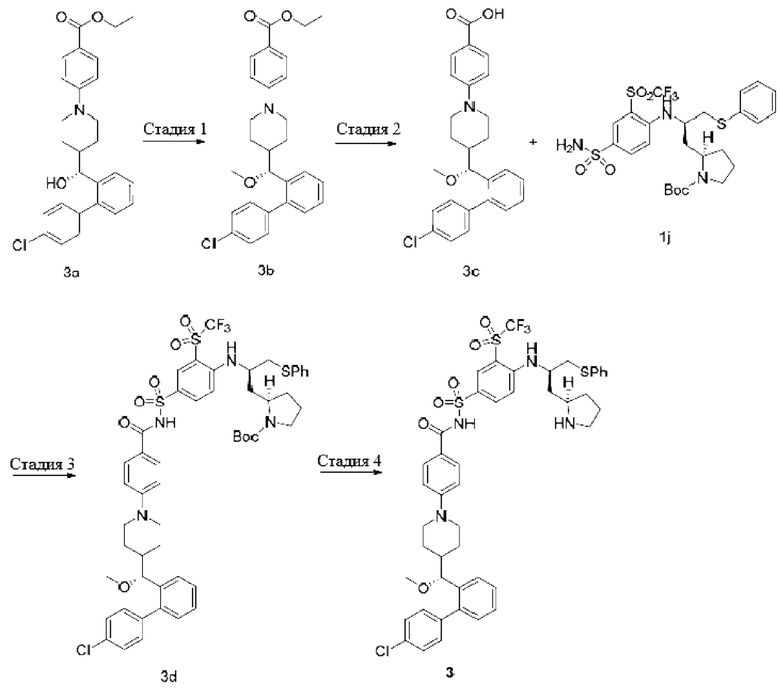
Стадия 1
Этил (R)-4-(4-((4'-хлор-[1,1'-бифенил]-2-ил)(метокси)метил)пиперидин-1-ил)бензоат 3b
Этил (R)-4-(4-((4'-хлор-[1,1'-бнфенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)бензоат 3а (60 мг, 0,133 ммоль, синтезированный со ссыпкой на способ, представленный для промежуточного соединения 11 на стр. 63 описания в патенте «CN 103153954 В»), растворяли в 2 мл смешанного растворителя N,N-диметилформамида и тетрагндрофурана (V:V=1:1). Раствор продували азотом три раза и последовательно добавляли йодметан (190 мг, 1,34 ммоль) н 60% гидрид натрия (11 мг, 0,275 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч. Реакционный раствор охлаждали на ледяной бане и добавляли насыщенный раствор хлорида аммония (5 мл) для гашения реакции. Добавляли воду (5 мл) и экстрагировали смесь этилацетатом (10 мл 3 раза). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия и фильтровали для удаления высушивающего вещества, и фильтрат концентрировали при пониженном давлении.
Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 3b (57 мг, выход: 92,1%).
MS m/z (ESI): 464,1 [М+1].
Стадия 2
(R)-4-(4-((4'-хлор-[1,1'-бифенил]-2-ил)(метокси)метил)пиперидин-1-ил)бензойная кислота 3с
3b (57 мг, 0,122 ммоль) растворяли в 3 мл смешанного растворителя тетрагидрофурана, метанола и воды (V:V:V=4:1:1) и добавляли моногидрат гидроксида лития (26 мг, 0,620 ммоль). Смесь нагревали до 50°С и перемешивали в течение 16 ч. Реакционный раствор концентрировали при пониженном давлении, доводили до рН 2-3 1М соляной кислотой и фильтровали. Фильтрующий осадок растворяли в 10 мл дихлорметана, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке продукта 3 с (53 мг, выход: 98,9%).
MS m/z (ESI): 436,1 [М+1].
Стадия 3
Трет-бутил-(R)-2-((R)-2-((4-(N-(4-(4-((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(метокси)метил)пиперидин-1-ил)бензоил)сульфамоил)-2-((трифторметил)сульфонил)фенил)амино)-3-(фенилтио)пропил)пирролидин-1-карбоксилат 3d
1j (32 мг, 0,051 ммоль) растворяли в дихлорметане (5 мл) и добавляли 3 с (24 мг, 0,055 ммоль), затем 4-диметиламинопиридин (13 мг, 0,106 ммоль) и 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (30 мг, 0,156 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 4 часов в атмосфере азота, добавляли 3с (22 мг, 0,051 ммоль) и перемешивали при комнатной температуре в течение ночи после добавления. Реакционный раствор последовательно промывали водой (10 мл) и насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия и фильтровали, а фильтрат концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с развивающейся системой растворителей А с получением указанного в заголовке продукта 3d (53 мг, выход: 99,6%).
MS m/z (ESI): 1041,1 [М+1].
Стадия 4
4-(4-((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(метокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-(фенилтио)-3-((R)-пирролидин-2-ил)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 3
3d (30 мг, 0,028 ммоль) растворяли в дихлорметане (0,4 мл) и добавляли 4 М раствор хлористого водорода в диоксане (1,2 мл). Смесь перемешивали при комнатной температуре в течение 40 мин. Реакционный раствор концентрировали при пониженном давлении для удаления органического растворителя. Остаток суспендировали простым диэтиловым эфиром и фильтровали. Полученное твердое вещество очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: Sharpsil-T Prep С18; подвижная фаза: бикарбонат аммония, вода, ацетонитрил), и соответствующую фракцию собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 3 (10 мг, выход: 36,9%).
MS m/z (ESI): 941,1 [М+1].
1Н NMR (500 МГц, CD3OD) δ 8,26 (s, 1H), 8,04 (dd, 1H), 7,81 (d, 2Н), 7,50 (d, 1H), 7,47-7,41 (m, 3Н), 7,40-7,32 (m, 3Н), 7,30-7,23 (m, 4Н), 7,22-7,16 (m, 2Н), 6,81 (d, 2Н), 6,72 (d, 1Н), 4,06 (d, 1Н), 3,99-3,93 (m, 1H), 3,77 (d, 1H), 3,66 (d, 1H), 3,55-3,49 (m, 1H), 3,23 (d, 2Н), 3,17 (s, 3Н), 2,61 (t, 2Н), 2,51 (t, 2Н), 2,25-1,93 (m, 6Н), 1,72-1,58 (m, 4Н), 1,27-1,23 (m, 1H), 1,20-1,11 (m, 2Н), 0,91 (t, 1H).
Пример 2-4
4-(4-(((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(метокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-((R)-1-(2-гидроксиэтил)пирролидин-2-ил)-3-(фенилтио)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 4
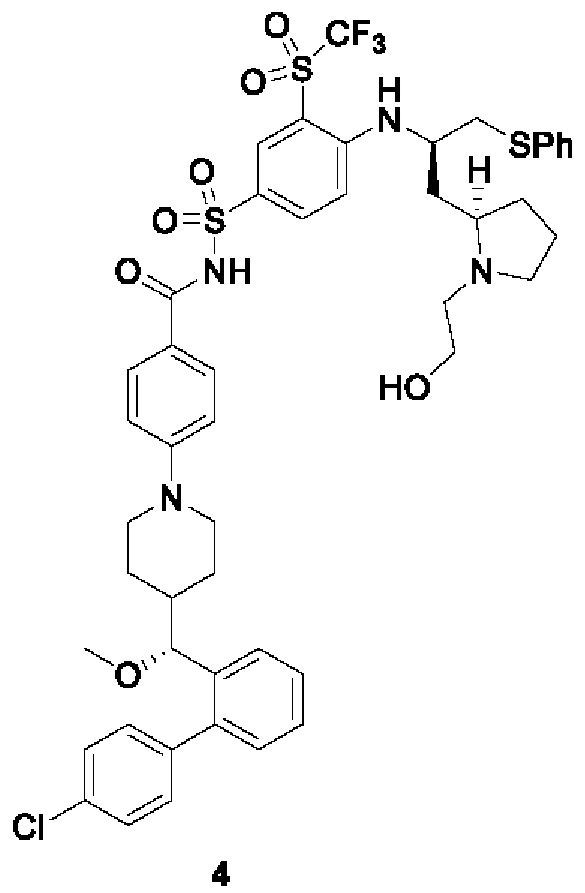
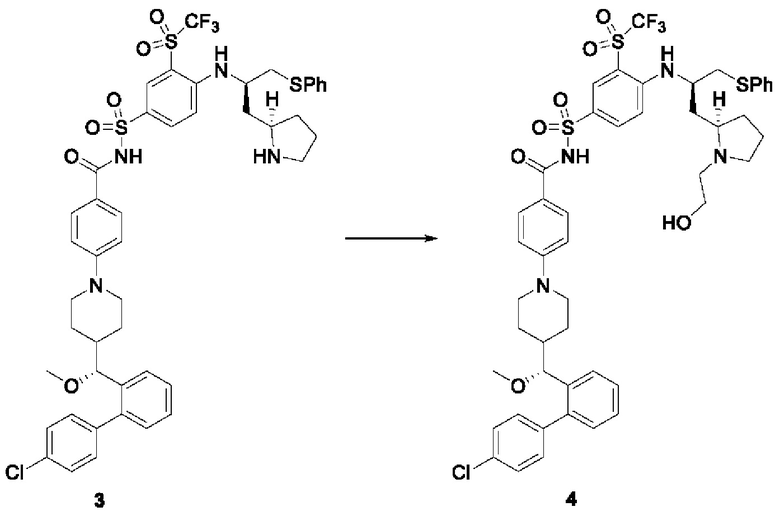
3 (15 мг, 0,015 ммоль) помещали в реакционную колбу и добавляли ацетонитрил (5 мл), затем 2-бромэтанол (29 мг, 0,232 ммоль) и триэтиламин (16, 0,158 ммоль). Реакционную систему продували азотом 3 раза и нагревали до 70°С и перемешивали в течение 16 ч. Реакционный раствор очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: Sharpsil-T Prep С18; подвижная фаза: бикарбонат аммония, вода, ацетонитрил), и соответствующую фракцию собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 4 (2,5 мг, выход: 16,5%).
MS m/z (ESI): 985,1 [М+1].
1H NMR (500 МГц, CD3OD) δ 8,27 (s, 1H), 8,03 (dd, 1H), 7,81 (d, 2H), 7,51 (d, 1H), 7,47-7,39 (m, 5H), 7,35 (t, 1H), 7,32-7,21 (m, 5H), 7,18 (d, 1H), 6,82 (d, 2H), 6,63 (d, 1H), 4,06 (d, 1H), 3,90-3,75 (m, 4H), 3,69-3,63 (m, 1H), 3,27-3,20 (m, 2H), 3,17 (s, 3H), 2,62 (t, 2H), 2,52 (t, 2H), 2,36-2,28 (m, 2H), 2,23-2,13 (m, 3H), 2,09-1,94 (m, 4H), 1,78-1,58 (m, 4H), 1,28-1,24 (m, 1H), 1,21-1,13 (m, 2H), 0,91 (t, 1H).
Пример 2-5
4-(4-(((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-((R)-1-(3-гидроксипропил)пирролидин-2-ил)-3-(фенилтио)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 5
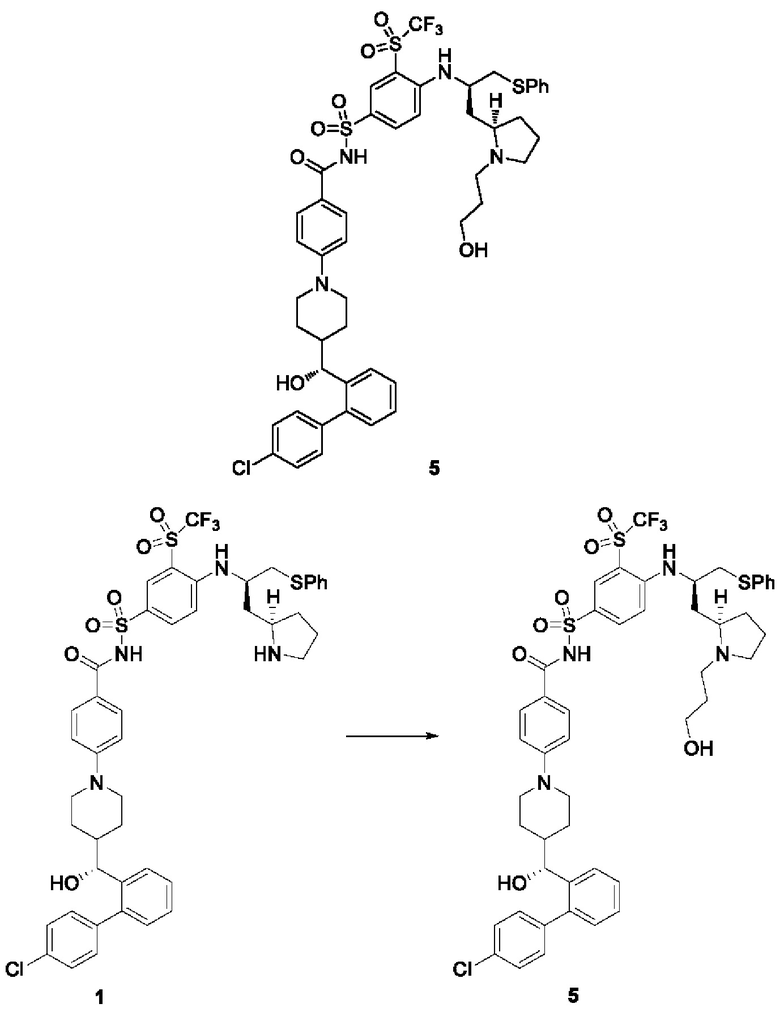
1 (10 мг, 0,011 ммоль) помещали в реакционную колбу и добавляли ацетонитрил (3 мл), затем 3-бромпропанол (15 мг, 0,108 ммоль, адамас) и триэтиламин (11 мг, 0,108 ммоль). Реакционную систему продували азотом 3 раза и нагревали до 70°С и перемешивали в течение 16 ч. Реакционный раствор очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: Sharpsil-T Prep С18; подвижная фаза: бикарбонат аммония, вода, ацетонитрил), и соответствующую фракцию собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 5 (2,5 мг, выход: 23,5%).
MS m/z (ESI): 985,1 [М+1].
1H NMR (500 МГц, CD3OD) δ 8,26 (s, 1H), 8,03 (d, 1H), 7,81 (d, 2H), 7,61 (d, 1H), 7,46-7,39 (m, 5H), 7,35-7,26 (m, 5H), 7,22 (t, 1H), 7,18 (d, 1H), 6,81 (d, 2H), 6,62 (d, 1H), 4,43 (d, 1H), 3,89-3,84 (m, 1H), 3,79 (d, 1H), 3,67 (t, 2H), 3,63 (d, 2H), 3,25-3,15 (m, 2H), 2,67 (t, 2H), 2,53 (t, 2H), 2,32-2,26 (m, 2H), 2,19 (t, 2H), 2,15-2,11 (m, 1H), 2,08-1,95 (m, 4H), 1,92-1,84 (m, 2H), 1,79-1,67 (m, 2H), 1,65-1,58 (m, 2H), 1,26-1,22 (m, 1H), 1,16-1,12 (m, 1H), 1,02-0,96 (m, 1H), 0,91 (t, 1H).
Пример 2-6
4-(4-((S)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)-N-((4-(((R)-4-((2-гидроксиэтил)(метил)амино)-1-(фенилтио)бутан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 6
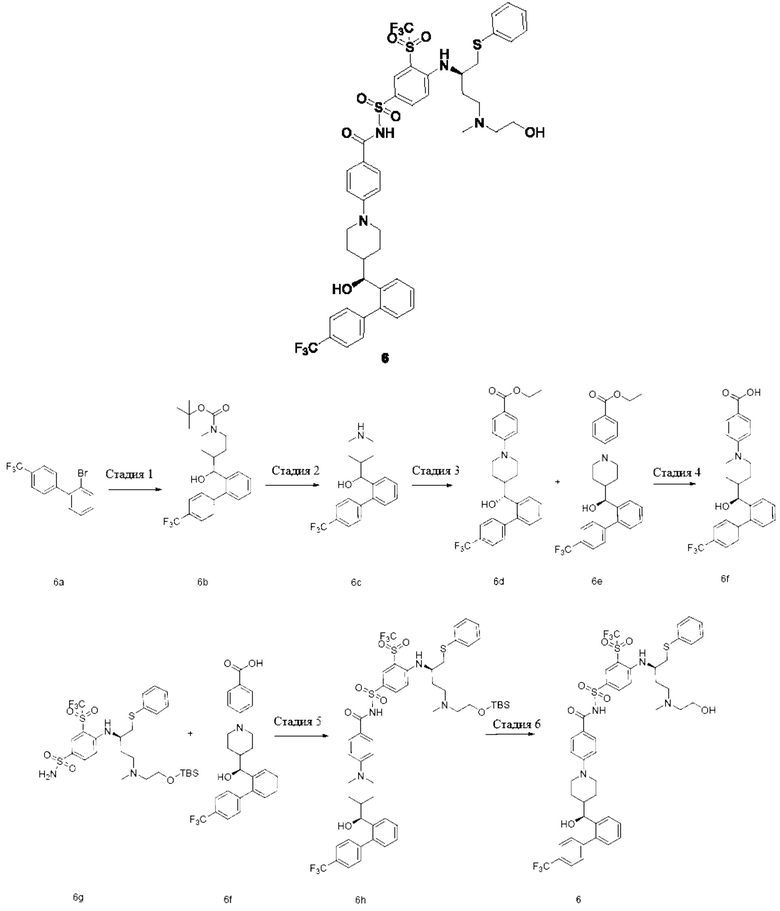
Стадия 1
Трет-бутил-4-(гидрокси(4'-(трифторметил)-[1,1'-дифенил]-2-ил)метил)пиперидин-1-карбоксилат 6b
2-бром-4'-(трифторметил)-1,1'-бифенил 6а (3,3 г, 10,95 ммоль, синтезированный в соответствии со способом, описанным на странице 53 Примера 53 в патенте «US 2004/171833, 2004, А1»), помещенный в колбу, и добавляли тетрагидрофуран (50 мл). После охлаждения смеси до -78°С в атмосфере азота в реакционную колбу по каплям добавляли н-бутиллитий (1,40 г, 21,92 ммоль) при внутренней температуре ниже -65°С, а после добавления смесь перемешивали при -78°С в течение 40 мин с последующим добавлением раствора трет-бутилформилпиперидин-1-карбоксилата (4,67 г, 21,92 ммоль) в тетрагидрофуране (20 мл). После добавления полученную смесь нагревали до 0°С и подвергали реакции в течение трех часов, а затем по каплям добавляли насыщенный водный раствор хлорида аммония для гашения реакции. Реакционный раствор концентрировали при пониженном давлении и экстрагировали этилацетатом (40 мл 3 раза). Органическую фазу объединяли, последовательно промывали водой (40 мл) и насыщенным солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали, а фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 6b (5,5 г, выход: более 100%).
MS m/z (ESI): 434,1 [М-1].
Стадия 2
Пиперидин-4- ил(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метанол 6с
6b (5,5 г, 12,63 ммоль) помещали в реакционную колбу и добавляли раствор 4 М хлористого водорода в диоксане (20 мл) под ледяной баней. После добавления смесь естественным образом нагревали до комнатной температуры и перемешивали в течение 2 ч. Реакционный раствор концентрировали при пониженном давлении и к остатку добавляли толуол (10 мл). Полученную смесь концентрировали при пониженном давлении и сушили масляным насосом с получением неочищенного указанного в заголовке продукта 6с (4,23 г, выход: 100%), который непосредственно использовали на следующей стадии без очистки.
MS m/z (ESI): 336,0 [М+1].
Стадия 3
Этил (R)-4-(4-(гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)бензоат 6d
Этил (S)-4-(4-(гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)бензоат 6е
Неочищенный 6 с (270 мг, 0,805 ммоль) помещали в реакционную колбу и добавляли диметилсульфоксид (3 мл) с последующим последовательным добавлением этил-п-фторбензоата (270 мг, 1,61 ммоль, Accela ChemBio) и N,N-диизопропилэтиламина (1,04 г, 8,05 ммоль). После добавления смесь нагревали до 120°С и реагировали в течение 24 ч в атмосфере азота. К реакционному раствору добавляли 5 мл воды и экстрагировали смесь этилацетатом (4 мл 5 раз). Органические фазы объединяли, последовательно промывали водой (4 мл) и насыщенным солевым раствором (4 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением смеси (360 мг), которую отправляли на хиральную препаративную HPLC (условия препарации: колонка: CHIRALPAK IG, 0,50 см внутр. диам. *25 см L, 0,1 мкм; подвижная фаза: МеОН=100%; длина волны обнаружения: UV 214 нм; температура: 38°С) с получением двух фракций, Пика 1 с меньшим временем удерживания (который составлял 6d (73 мг)) и Пика 2 с большим временем удерживания (который составлял 6е (72 мг)).
MS m/z (ESI): 484,1 [М+1].
Пик 1: анализ хиральной HPLC: время удерживания: 4,008 мин, хиральная чистота: 100% (колонка: CHIRALPAK IG 150*4,6 мм, 5 мкм (с защитной колонкой); подвижная фаза: н-гексан/этанол=70/30 (об./об.)).
Пик 2: анализ хиральной HPLC: время удерживания: 6,289 мин, хиральная чистота: 100% (колонка CHIRALPAK IG 150*4,6 мм, 5 мкм (с защитной колонкой); подвижная фаза: н-гексан/этанол=70/30 (об./об.)).
Стадия 4
(R)-4-(4-(гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)бензойная кислота 6f
6е (70 мг, 0,145 ммоль) помещали в колбу и добавляли тетрагидрофуран (2 мл), воду (0,5 мл) и метанол (0,5 мл), а затем моногидрат гидроксида лития (30,3 мг, 0,723 ммоль). Смесь нагревали до 50°С и подвергали реакции в течение 16 ч в атмосфере азота. Реакционный раствор концентрировали при пониженном давлении и добавляли воду (5 мл) к остатку. Полученную смесь охлаждали на ледяной бане, доводили до рН 4 1н раствором хлористоводородной кислоты и фильтровали. Фильтрующий осадок промывали н-гексаном (5 мл) и сушили масляным насосом с получением указанного в заголовке продукта 6f (63 мг, выход: 95,54%).
MS m/z (ESI): 456,0 [М+1].
Стадия 5
N-((4-(((R)-4-((2-((трет-бутилдиметилсилил)окси)этил)(метил)амино)-1-(фенилтио)бутан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)-4-(4-((S)-гидрокси(4'-(трифторметил)-[1, 1'-бифенил]-2-ил)метил)пиперидин-1-ил)бензамид 6h
6g (30 мг, 0,040 ммоль, синтезированное со ссылкой на способ, представленный для промежуточного соединения 67 на странице 90 описания в патенте «CN 103153954 В»), растворяли в дихлорметане (3 мл) и добавляли 6f (25 мг, 0,055 ммоль), с последующим последовательным добавлением 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (17,5 мг, 0,091 ммоль) и 4-диметиламинопиридина (11,2 мг, 0,091 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 4 часов в атмосфере азота, добавляли 6f (16,6 мг, 0,037 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (10,5 мг, 0,055 ммоль) и 4-диметиламинопиридин (6,7 мг, 0,055 ммоль) и перемешивали при комнатной температуре в течение ночи после добавления. Реакционный раствор последовательно промывали водой (5 мл) и насыщенным рассолом (5 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с системой элюирования А с получением указанного в заголовке продукта 6h (40 мг, выход: 79,9%).
MS m/z (ESI): 1093,4 [М+1].
Стадия 6
4-(4-((S)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)-N-((4-(((R)-4-((2-гидроксиэтил)(метил)амино)-1-(фенилтио)бутан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 6
К тетрагидрофурану (2 мл) добавляли 6h. (40 мг, 0,036 ммоль) и фторид тетрабутиламмония (16,4 мг, 0,073 ммоль). Смесь перемешивали при комнатной температуре в течение 4 ч. Реакционный раствор концентрировали при пониженном давлении и остаток растворяли в дихлорметане (8 мл) и промывали насыщенным раствором хлорида аммония (10 мл 2 раза). Органическую фазу концентрировали при пониженном давлении, а остаток растворяли в этилацетате (8 мл), последовательно промывали насыщенным раствором хлорида аммония (10 мл 6 раз), водой (10 мл) и насыщенным рассолом (10 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученное твердое вещество очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: Sharpsil-T Prep С18; подвижная фаза: бикарбонат аммония, вода, ацетонитрил), и соответствующую фракцию собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 6 (17 мг, выход: 47,5%).
MS m/z (ESI): 979,2 [М+1].
1Н NMR (500 МГц, CD3OD) δ 8,28 (d, 1H), 8,09-8,03 (m, 1H), 7,80 (d, 2Н), 7,74 (d, 2Н), 7,66 (d, 1H), 7,55 (d, 2H), 7,48 (t, 1H), 7,42-7,34 (m, 2H), 7,29-7,16 (m, 3H), 6,81 (dd, 2H), 4,41 (d, 1H), 4,02-3,97 (m, 1H), 3,84-3,75 (m, 2H), 3,64 (d, 1H), 3,37 (s, 2H), 3,30-3,18 (m, 3H), 3,12-3,06 (m, 2H), 2,75 (s, 2H), 2,72-2,63 (m, 1H), 2,59-2,50 (m, 1H), 2,29-2,19 (m, 2H), 2,08-2,02 (m, 2H), 1,82-1,72 (m, 1H), 1,66-1,58 (m, 1H), 1,39-1,31 (m, 4H), 1,27-1,12 (m, 2H), 1,03-0,89 (m, 2H).
Пример 2-7
4-(4-((R)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)-N-((4-(((3R)-1-((2-гидроксиэтил)(метил)амино)-4-(фенилтио)пентан-3-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)сульфонил)бензамид 7
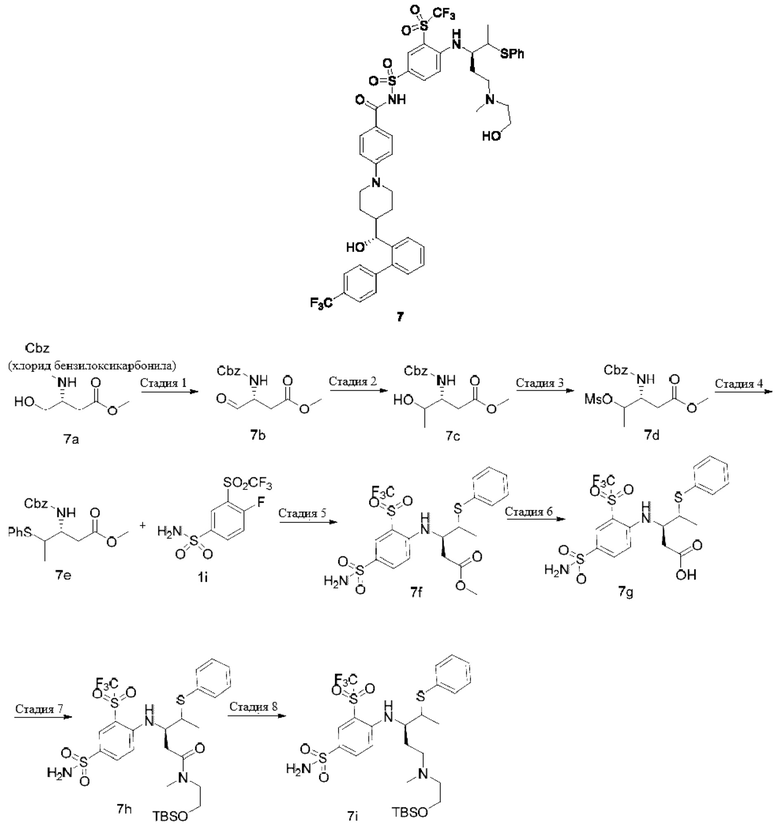
Стадия 1
Метил (R)-3-(((бензилокси)карбонил)амино)-4-оксобутаноат 7b
Оксалилхлорид (772 мг, 6,08 ммоль) растворяли в дихлорметане (5 мл), и раствор трижды продували азотом, охлаждали до -78°С, после чего по каплям добавляли раствор диметилсульфоксида (950 мг, 12,16 ммоль) в дихлорметане (5 мл). После добавления по каплям смесь перемешивали при -78°С в течение 30 минут и по каплям добавляли раствор метил (R)-3-(((бензилокси)карбонил)амино)-4-гидроксибутирата 7а (650 мг, 2,43 ммоль, синтезировали с помощью способа, представленного для промежуточного соединения 4 на странице 59 описания в патенте «CN 103153954 В») в дихлорметане (5 мл). После добавления по каплям смесь перемешивали при -78°С в течение 1 часа и по каплям добавляли N,N-диизопропилэтиламин (3,14 г, 24,29 ммоль). После добавления по каплям полученную смесь нагревали до -60°С и перемешивали в течение 30 мин, а затем нагревали до 0°С и перемешивали в течение 30 мин. 1 М холодную соляную кислоту (20 мл) добавляли при 0°С для гашения реакции, и смесь подвергали разделению жидкостью. Органическую фазу промывали фосфатно-содержащим буферным раствором (30 мл) с рН 7 и насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом натрия и фильтровали, а фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 7b (540 мг, выход: 83,7%).
МС m/z (ESI): 266,2 [М+1].
Стадия 2
Метил (3R)-3-(((бензилокси)карбонил)амино)-4-гидроксипентаноат 7с
7b (490 мг, 1,95 ммоль) растворяли в простом диэтиловом эфире (15 мл), и раствор трижды продували азотом и охлаждали до -78°С, после чего по каплям добавляли раствор 1 М бромида метилмагния в тетрагидрофуране (2 мл, 2,03 ммоль). После добавления по каплям смесь естественным образом нагревали до 0°С и перемешивали в течение 3 ч. При 0°С добавляли насыщенный раствор хлорида аммония (20 мл) для гашения реакции. Добавляли воду (10 мл) и экстрагировали смесь этилацетатом (10 мл 3 раза). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия и фильтровали для удаления высушивающего вещества, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 7с (188 мг, выход: 36,2%).
MS m/z (ESI): 282,1 [М+1].
Стадия 3
Метил (3R-3-(((бензилокси)карбонил)амино)-4-((метилсульфонил)окси)пентаноат 7d
7с (220 мг, 0,782 ммоль) растворяли в дихлорметане (8 мл) и добавляли пиридин (93 мг, 1,176 ммоль) и 4-диметиламинопиридин (9,5 мг, 0,078 ммоль). Смесь трижды продували азотом и охлаждали до 0°С, и медленно добавляли метансульфоангидрид (164 мг, 0,941 ммоль). После добавления смесь нагревали до комнатной температуры и перемешивали в течение 1,5 ч. К реакционному раствору добавляли воду (20 мл) и экстрагировали смесь дихлорметаном (8 мл 3 раза). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия и фильтровали для удаления высушивающего вещества, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 7d (216 мг, выход: 76,8%).
MS m/z (ESI): 360,0 [М+1].
Стадия 4
Метил (3R)-3-(((бензилокси)карбонил)амино)-4-(фенилтио)пентаноат 7е
7d (216 мг, 0,601 ммоль) растворяли в толуоле (8 мл) и добавляли тиофенолат натрия (397 мг, 3,004 ммоль). Смесь трижды продували азотом и нагревали до 105°С и перемешивали в течение 16 ч. После охлаждения реакционного раствора до комнатной температуры добавляли воду (20 мл) и экстрагировали смесь этилацетатом (10 мл 3 раза). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 7е (120 мг, выход: 53.5%).
MS m/z (ESI): 374,1 [М+1].
Стадия 5
Метил (3R)-4-(фенилтио)-3-((4-сульфамоил-2-((трифторметил)сульфонил)фенил)амино)пентаноат 7f
7е (112 мг, 0,300 ммоль) растворяли в тетрагидрофуране (1,5 мл) и добавляли трифторуксусную кислоту (5 мл). Смесь нагревали до 72°С и перемешивали в течение 1,5 ч. Реакционный раствор концентрировали при пониженном давлении дихлорметаном (20 мл) и сушили в вакууме в течение 2 ч. Остаток растворяли в N,N-диметилформамиде (4 мл) и добавляли 1i (93 мг, 0,302 ммоль) и N,N-диизопропилэтиламине (388 мг, 3,00 ммоль). Реакционную систему продували азотом 3 раза и нагревали до 50°С и перемешивали в течение 24 ч. К реакционному раствору добавляли воду (20 мл) и экстрагировали смесь этилацетатом (10 мл 3 раза). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали для удаления высушивающего вещества, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 7f (119 мг, 75.3%).
MS m/z (ESI): 524.9 [М-1].
Стадия 6
(3R)-4-(фенилтио)-3-((4-сульфамоил-2-((трифторметил)сульфонил)фенил)амино)пентановая кислота 7g
7f (119 мг, 0,226 ммоль) растворяли в 5 мл смешанного растворителя тетрагидрофурана, метанола и воды (V:V:V=3:1:1) и добавляли моногидрат гидроксида лития (29 мг, 0,691 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч. Реакционный раствор концентрировали при пониженном давлении, доводили до рН 3-4 1 М соляной кислотой и экстрагировали дихлорметаном (8 мл 3 раза). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали для удаления осушителя, и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке продукта 7 г (115 мг, выход: 99,3%).
MS m/z (ESI): 511,0 [М-1].
Стадия 7
(3R)-N-(2-((трет-бутилдиметилсилил)окси)этил)-N-метил-4-(фенилтио)-3-((4-сульфамоил-2-((трифторметил)сульфонил)фенил)амино)пентанамид 7h
7g (115 мг, 0,224 ммоль) растворяли в тетрагидрофуране (3 мл) и последовательно добавляли 2-((трет-бутилдиметилсилил)окси)-N-метилэтил-1-амин (43 мг, 0,227 ммоль, Accela ChemBio), 3-(диэтоксия-о-ацилокси)-1,2,3-бензотриазин-4-он (134 мг, 0,448 ммоль) и N,N-диизопропилэтиламин (58 мг, 0,448 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 3 часов в атмосфере азота. Реакционный раствор концентрировали, а остаток растворяли в этилацетате (10 мл), последовательно промывали насыщенным водным раствором хлорида аммония (20 мл), насыщенным раствором бикарбоната натрия (20 мл) и насыщенным раствором хлорида натрия (20 мл). Органическую фазу сушили над безводным сульфатом натрия и фильтровали, а фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 7h (107 мг, выход: 69,7%).
MS m/z (ESI): 684,0 [М+1].
Стадия 8
4-(((3R)-1-((2-((трет-бутилдиметилсилил)окси)этил)(метил)амино)-4-(фенилтио)пентан-3-ил)амино)-3-((трифторметил)сульфонил)бензолсульфонамид 7i
7 ч (107 мг, 0,156 ммоль) помещали в реакционную колбу и добавляли 1 М раствор комплекса боран-тетрагидрофуран (5 мл). После добавления смесь нагревали до 60°С и перемешивали в течение 24 ч. Реакционную систему охлаждали до 0°С и по каплям добавляли 4 мл метанола для гашения реакции. Смесь концентрировали при пониженном давлении и к остатку добавляли раствор 7 М аммиака в метаноле (10 мл). Смесь перемешивали при комнатной температуре в течение 48 часов после герметизации и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 7i (10 мг, выход: 9.5%).
MS m/z (ESI): 670,1 [М+1].
Стадия 9
(R)-4-(4-(гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)бензойная кислота 7j
6d (70 мг, 0,145 ммоль) помещали в колбу и добавляли тетрагидрофуран (2 мл), воду (0,5 мл) и метанол (0,5 мл), а затем моногидрат гидроксида лития (30 мг, 0,72 ммоль). Смесь нагревали до 50°С и подвергали реакции в течение 16 ч в атмосфере азота. Реакционный раствор концентрировали при пониженном давлении и добавляли воду (5 мл) к остатку. Полученную смесь охлаждали на ледяной бане, доводили до рН 4 1М раствором хлористоводородной кислоты и фильтровали. Фильтрующий осадок промывали н-гексаном (5 мл) и сушили масляным насосом с получением указанного в заголовке продукта 7j (63 мг, выход: 95,5%).
MS m/z (ESI): 456,0 [М+1].
Стадия 10
N-((4-(((3R)-1-((2-((трет-бутилдиметилсилил)окси)этил)(метил)амино)-4-(фенилтио)пентан-3-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)-4-(4-((R)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)бензамид 7k
7i (16 мг, 0,024 ммоль) растворяли в дихлорметане (4 мл) и добавляли 7j (13 мг, 0,028 ммоль), затем 4-диметиламинопиридин (6 мг, 0,049 ммоль) и 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (14 мг, 0,073 ммоль). После добавления смесь трижды продували азотом, перемешивали при комнатной температуре в течение 4 ч, добавляли 7j (13 мг, 0,028 ммоль) и перемешивали при комнатной температуре в течение 16 ч после добавления. Реакционный раствор последовательно промывали водой (8 мл) и насыщенным раствором хлорида натрия (8 мл), сушили над безводным сульфатом натрия и фильтровали, а фильтрат концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с развивающейся системой растворителей А с получением указанного в заголовке продукта 7k (14 мг, выход: 52,9%).
MS m/z (ESI): 1107,2 [М+1].
Стадия 11
4-(4-((R)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)-N-((4-(((3R)-1-((2-гидроксиэтил)(метил)амино)-4-(фенилтио)пентан-3-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)сульфонил)бензамид 7
7k (14 мг, 0,013 ммоль) растворяли в тетрагидрофуране (3 мл) и добавляли фторид тетрабутиламмония (6 мг, 0,027 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч. Реакционный раствор концентрировали при пониженном давлении и остаток растворяли в дихлорметане (5 мл) и промывали насыщенным раствором хлорида аммония (5 мл 2 раза). Органическую фазу концентрировали при пониженном давлении, а остаток растворяли в этилацетате (8 мл), последовательно промывали насыщенным раствором хлорида аммония (5 мл 6 раз), водой (10 мл) и насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученное твердое вещество очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: Sharpsil-T Prep С18; подвижная фаза: бикарбонат аммония, вода, ацетонитрил), и соответствующую фракцию собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 7 (4,5 мг, выход: 35,8%).
MS m/z (ESI): 993,2 [М+1].
1Н NMR (500 МГц, CD3OD) δ 8,33 (s, 1H), 8,02 (d, 1H), 7,76 (dd, 4Н), 7,65 (d, 1H), 7,56 (dd, 4Н), 7,48 (t, 1H), 7,44-7,33 (m, 4Н), 7,21 (d, 1H), 6,81 (d, 2Н), 6,60 (d, 1H), 4,41 (d, 1H), 3,97-3,92 (m, 1H), 3,84-3,72 (m, 3Н), 3,69-3,56 (m, 3Н), 2,76-2,62 (m, 4Н), 2,55 (t, 2Н), 2,32-2,17 (m, 3Н), 2,10-1,87 (m, 4Н), 1,82-1,72 (m, 2Н), 1,64-1,58 (m, 2Н), 1,27-1,22 (m, 1H), 1,21-1,12 (m, 2Н), 1,03-0,96 (m, 1Н), 0,92 (t, 1H).
Пример 2-8
4-(4-((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-(фенилтио)-3-((S)-пирролидин-2-ил)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 8
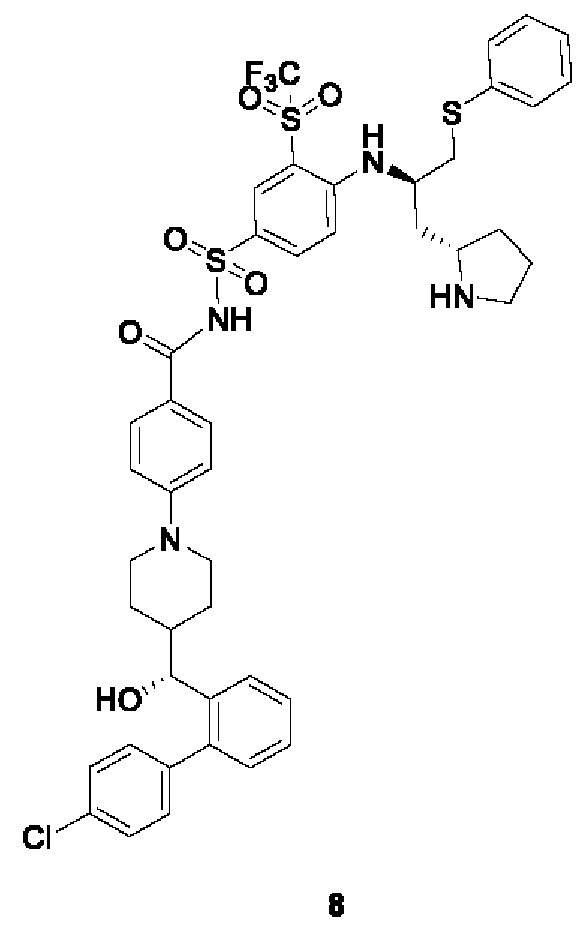
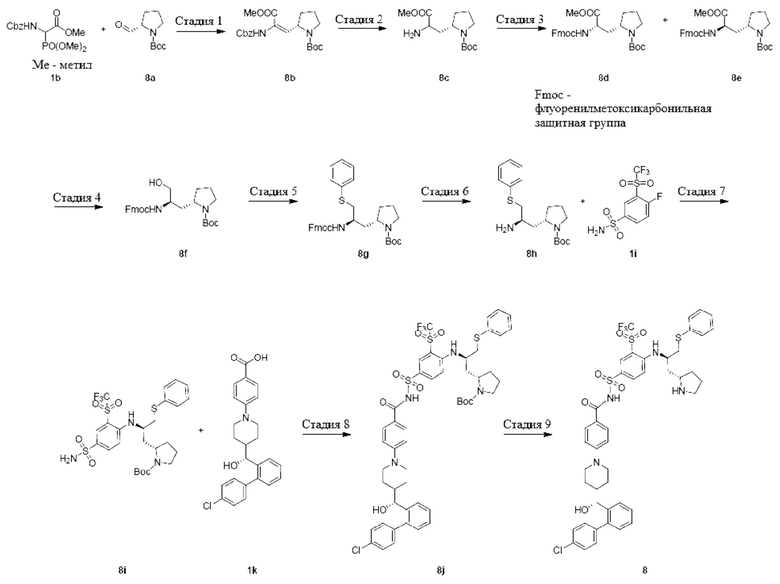
Стадия 1
Трет-бутил(S,)-2-(2-(((бензилокси)карбонил)амино)-3-метокси-3-оксопроп-1-ен-1-ил)пирролидин-1-карбоксилат 8b
1b (2,49 г, 7,53 ммоль, Accela ChemBio) растворяли в тетрагидрофуране (20 мл) и охлаждали раствор до 0°С на ледяной бане с последующим по каплям добавлением N,N-литиядиизопропиламида (661 мг, 6,17 ммоль). Смесь перемешивали при 0°С в течение 1 часа. Реакционную систему охлаждали до -78°С и по каплям добавляли раствор трет-бутил(S)-2-формилпирролидин-1-карбоксилата 8а (1,00 г, 5,02 ммоль, PharmaBlock) в тетрагидрофуране (20 мл). После добавления по каплям, ванну с сухим льдом и ацетоном удаляли, и смесь нагревали до комнатной температуры и реагировали в течение 12 ч. Для гашения реакции добавляли насыщенный раствор хлорида аммония (20 мл), и смесь экстрагировали этилацетатом (20 мл 3 раза). Органическую фазу сушили над безводным сульфатом натрия и фильтровали, а фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 8b (1,90 г, выход: 94,1%).
MS m/z (ESI): 305,0 [М+Н-100].
Стадия 2
Трет-бутил(2S)-2-(2-амино-3-метокси-3-оксопропил)пирролидин-1-карбоксилат 8с
8b (1,90 г, 4,7 ммоль) растворяли в изопропаноле (50 мл) и добавляли палладий на угле (380 мг, 10% содержания, на сухой основе). Смесь продували водородом 3 раза и перемешивали при комнатной температуре в течение 8 ч. Реакционный раствор фильтровали через целит, а осадок на фильтре промывали этилацетатом (20 мл) и метанолом (20 мл). Фильтрат концентрировали с получением неочищенного указанного в заголовке продукта 8с (1,28 г), который непосредственно использовали на следующей стадии без очистки.
Стадия 3
Трет-бутил(S)-2-((S)-2-((((9Н-флуорен-9-ил)метокси)карбонил)амино)-3-метокси-3-оксопропил)пирролидин-1-карбоксилат 8d
Трет-бутил(S)-2-((R)-2-((((9Н-флуорен-9-ил)метокси)карбонил)амино)-3-метокси-3-оксопропил)пирролидин-1-карбоксилат 8е
Неочищенный 8с (1,28 г, 4,7 ммоль) растворяли в 1,4-диоксане (40 мл) и добавляли воду (10 мл), бикарбонат натрия (1,17 г, 14,1 ммоль) и флуоренилметоксикарбонилхлорид (1,21 г, 4,7 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч. Добавляли воду (30 мл) для гашения реакции и смесь экстрагировали этилацетатом (20 мл 3 раза). Органическую фракцию сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов А с получением указанного в заголовке продукта 8d (1,03 г, 44,2%, пик 1) и 8е (513 мг, 22,1%, пик 2).
МС m/z (ESI): 495,2 [М+1].
Пик 1: анализ UPLC: время удерживания: 2,77 мин (колонка: ACQUITY UPLC ВЕНС18 50*2,1 мм, 1,7 мкм; подвижная фаза: ацетонитрил/вода/муравьиная кислота=10/90/0,1 (об./об./об.) до 95/5/0,1 (об./об./об.) градиентное элюирование).
Пик 2: анализ UPLC: время удерживания: 2,69 мин (колонка: ACQUITY UPLC ВЕНС18 50*2,1 мм, 1,7 мкм; подвижная фаза: ацетонитрил/вода/муравьиная кислота=50/50/0,1 (об./об./об.) до 95/5/0,1 (об./об./об.) градиентное элюирование).
Стадия 4
Трет-бутил(S)-2-((R)-2-((((9Н-флуорен-9-ил)метокси)карбонил)амино)-3-гидроксипропил)-(пирролидин-1-карбоксилат 8f
8е (169 мг, 0,34 ммоль) растворяли в тетрагидрофуране (8 мл) и добавляли этанол (8 мл), боргидрид натрия (77 мг, 2,05 ммоль) и хлорид лития (94 мг, 2,22 ммоль). Смесь перемешивали при 28°С в течение 2,5 ч и добавляли насыщенный хлорид аммония (5 мл) для гашения реакции. Растворитель концентрировали досуха, а остаток растворяли в этилацетате (10 мл) и последовательно промывали водой (10 мл) и насыщенным хлоридом натрия (10 мл). Органическую фракцию сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 8f (125 мг, 78.4%).
MS m/z (ESI): 467.2 [М+1].
Стадия 5
Трет-бутил(S)-2-((R)-2-((((9Н-флуорен-9-ил)метокси)карбонил)амино)-3-(фенилтио)пропил)пирролидин-1-карбоксилат 8g
8f (120 мг, 0,26 ммоль) растворяли в толуоле (5 мл) и добавляли дифенилдисульфид (168 мг, 0,77 ммоль) и трибутилфосфин (156 мг, 0,77 ммоль). Смесь нагревали до 80°С и подвергали реакции в течение 12 ч в атмосфере азота. Растворитель концентрировали досуха. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 8g (135 мг, 93,9%).
MS m/z (ESI): 559,2 [М+1].
Стадия 6
Трет-бутил(S)-2-((R)-2-амино-3-(фенилтио)пропил)пирролидин-1-карбоксилат 8h
8g (135 мг, 0,24 ммоль) растворяли в дихлорметане (4 мл) и добавляли диэтиламин (4 мл). Смесь перемешивали при комнатной температуре в течение 4 ч. Реакционный раствор концентрировали при пониженном давлении для удаления органического растворителя с получением указанного в заголовке продукта 8h (81,6 мг), который непосредственно использовали на следующей стадии без очистки.
Стадия 7
Трет-бутил(S)-2-((R)-4-(фенилтио)-3-((4-сульфамоил-2-((трифторметил)сульфонил)фенил)амино)пропил)пирролидин-1-карбоксилат 8i
Неочищенный 8h (81,6 мг, 0,24 ммоль) растворяли в N,N-диметилформамиде (5 мл) и добавляли 1i (82 мг, 0,27 ммоль) и N,N-диизопропилэтиламине (314 мг, 2,4 ммоль). Реакционную систему нагревали до 50°С и перемешивали в течение 12 ч в атмосфере азота. Добавляли воду (10 мл) для гашения реакции, и смесь экстрагировали этилацетатом (5 мл 3 раза). Органическую фазу сушили над безводным сульфатом натрия и фильтровали, а фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с системой элюентов В с получением указанного в заголовке продукта 8i (105 мг, 69.3%).
MS m/z (ESI): 622,1 [М-1].
Стадия 8
Трет-бутил-(S)-2-((R)-2-((4-(N-(4-(4-((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)бензоил)сульфамоил)-2-((трифторметил)сульфонил)фенил)амино)-3-(фенилтио)пропил)пирролидин-1-карбоксилат 8j
8i (30 мг, 0,048 ммоль) растворяли в дихлорметане (3 мл) и добавляли 1k (24 мг, 0,058 ммоль) с последующим последовательным добавлением 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (19 мг, 0,096 ммоль) и 4-диметиламинопиридина (12 мг, 0,096 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 4 часов в атмосфере азота, добавляли 1k (12 мг, 0,038 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (19 мг, 0,096 ммоль) и 4-диметиламинопиридин (12 мг, 0,096 ммоль) и перемешивали при комнатной температуре в течение ночи после добавления. Реакционный раствор последовательно промывали водой (5 мл) и насыщенным рассолом (5 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с развивающейся системой растворителей А с получением указанного в заголовке продукта 8j (30 мг, выход: 60,7%).
MS m/z (ESI): 1027,2 [М+1].
Стадия 9
4-(4-((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-(фенилтио)-3-((S)-пирролидин-2-ил)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 8
8j (30 мг, 0,029 ммоль) растворяли в дихлорметане (2 мл) и добавляли 4 М раствор хлористого водорода в 1,4-диоксане (2 мл). Полученную смесь перемешивали при комнатной температуре в течение 20 мин. Реакционный раствор концентрировали при пониженном давлении для удаления органического растворителя. Остаток растворяли в дихлорметане и концентрировали при пониженном давлении до сухости, что повторяли три раза. Полученное твердое вещество очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: Sharpsil-T Prep С18; подвижная фаза: бикарбонат аммония, вода, ацетонитрил), и соответствующую фракцию собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 8 (25 мг, выход: 92,3%).
MS m/z (ESI): 927,1 [М+1].
1H NMR (500 МГц, CD3OD) δ 8,24 (d, 1H), 8,05-8,01 (m, 1H), 7,82 (d, 2Н), 7,60 (d, 1Н), 7,45-7,39 (m, 3Н), 7,37 (d, 2Н), 7,34-7,28 (m, 3Н), 7,23 (t, 2Н), 7,20-7,14 (m, 2Н), 6,80 (d, 2Н), 6,72 (d, 1Н), 4,42 (d, 1H), 3,97 (br, 1H), 3,84-3,74 (m, 1Н), 3,27-3,21 (m, 2Н), 3,19-3,07 (m, 3Н), 3,06-2,97 (m, 1H), 2,69-2,61 (m, 1H), 2,55-2,48 (m, 1H), 2,16-2,00 (m, 5Н), 1,98-1,89 (m, 2H), 1,88-1,80 (m, 1H), 1,78-1,69 (m, 1H), 1,63-1,57 (m, 2H), 1,54-1,49 (m, 1H), 1,26-1,20 (m, 1H), 1,15-1,04 (m, 1H), 1,01-0,94 (m, 1H).
Пример 2-9
4-(4-(((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-((S)-1-(2-гидроксиэтил)пирролидин-2-ил)-3-(фенилтио)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 9
8 (15 мг, 0,016 ммоль) растворяли в ацетонитриле (2 мл) и последовательно добавляли триэтиламин (16 мг, 0,16 ммоль) и бромэтанол (20 мг, 0,16 ммоль). Реакционную систему нагревали до 70°С и подвергали реакции в течение 12 ч в атмосфере азота. Полученное твердое вещество очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: Sharpsil-T Prep С18; подвижная фаза: бикарбонат аммония, вода, ацетонитрил), и соответствующую фракцию собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 9 (10 мг, выход: 63,4%).
MS m/z (ESI): 971,2 [М+1].
1Н NMR (500 МГц, CD3OD) δ 8,25 (d, 1H), 8,06 (dd, 1H), 7,80 (d, 2Н), 7,61 (dd, 1Н), 7,49-7,35 (m, 5Н), 7,35-7,28 (m, 3Н), 7,24 (dd, 2Н), 7,20-7,14 (m, 2Н), 6,79 (t, 3Н), 4,42 (d, 1Н), 4,02 (t, 1Н), 3,84-3,70 (m, 3Н), 3,61 (d, 2Н), 3,28-3,14 (m, 2Н), 3,06 (br, 1Н), 2,96 (br, 1Н), 2,71-2,59 (m, 1H), 2,56-2,45 (m, 1H), 2,45-2,35 (m, 1Н), 2,32-2,22 (m, 1Н), 2,10-1,97 (m, 4Н), 1,96-1,86 (m, 1H), 1,85-1,69 (m, 2Н), 1,60 (t, 2Н), 1,26-1,18 (m, 1Н), 1,06-1,08 (m, 1H), 1,01-0,92 (m, 1Н), 0,92-1,86 (m, 1H).
Пример 2-10
4-(4-((R)-гидрокси (4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)-N-((4-(((R)-4-((2-гидроксиэтил)(метил)амино)-1-(фенилтио)бутан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 10
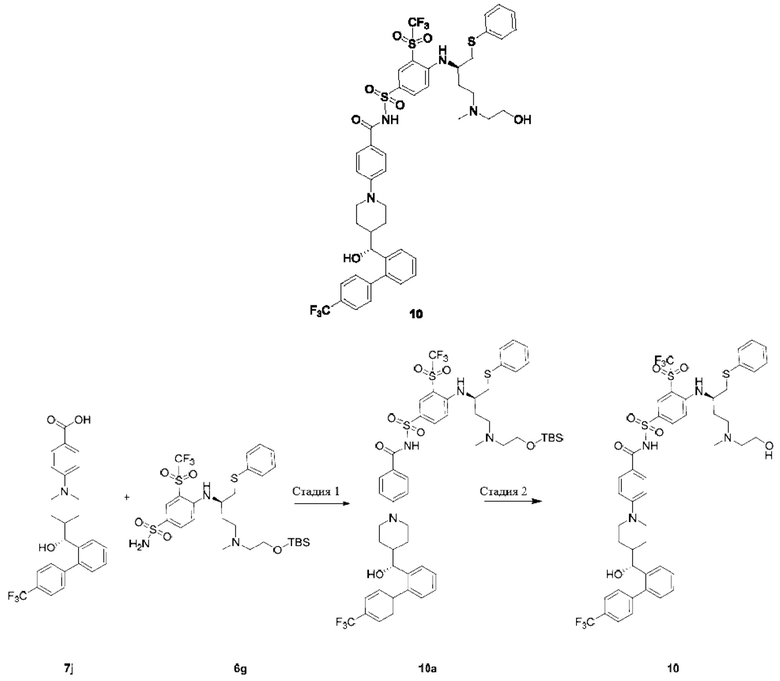
Стадия 1
N-((4-(((R)-4-((2-((трет-бутилдиметилсилил)окси)этил)(метил)амино)-1-(фенилтио)бутан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)-4-(4-((R)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)бензамид 10а
6g (30 мг, 0,040 ммоль) растворяли в дихлорметане (3 мл) и добавляли 7j (25 мг, 0,055 ммоль) с последующим последовательным добавлением 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (17,5 мг, 0,091 ммоль) и 4-диметиламинопиридина (11,2 мг, 0,091 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 4 часов в атмосфере азота, добавляли 7j (16,6 мг, 0,037 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (10,5 мг, 0,055 ммоль) и 4-диметиламинопиридин (6,7 мг, 0,055 ммоль) и перемешивали при комнатной температуре в течение ночи после добавления. Реакционный раствор последовательно промывали водой (5 мл) и насыщенным рассолом (5 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с развивающейся системой растворителей А с получением указанного в заголовке продукта 10а (40 мг, выход: 79,9%).
MS m/z (ESI): 1093,4 [М+1].
Стадия 2
4-(4-((R)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)-N-((4-(((R)-4-((2-гидроксиэтил)(метил)амино)-1-(фенилтио)бутан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 10
К тетрагидрофурану (2 мл) добавляли 10а (40 мг, 0,036 ммоль) и фторид тетрабутиламмония (16 мг, 0,073 ммоль). Смесь перемешивали при комнатной температуре в течение 4 ч. Реакционный раствор концентрировали при пониженном давлении и остаток растворяли в дихлорметане (8 мл) и промывали насыщенным раствором хлорида аммония (10 мл 2 раза). Органическую фазу концентрировали при пониженном давлении, а остаток растворяли в этилацетате (8 мл), последовательно промывали насыщенным раствором хлорида аммония (10 мл 6 раз), водой (10 мл) и насыщенным рассолом (10 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: Sharpsil-T Prep С18; подвижная фаза: бикарбонат аммония, вода, ацетонитрил), и соответствующую фракцию собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 10 (17 мг, выход: 47,5%).
MS m/z (ESI): 979,2 [М+1].
1Н NMR (500 МГц, CD3OD) δ 8,26 (d, 1H), 8,04 (dd, 1Н), 7,79 (d, 2Н), 7,72 (d, 2Н), 7,64 (d, 1H), 7,53 (d, 2Н), 7,46 (t, 1H), 7,40-7,31 (m, 3Н), 7,29-7,12 (m, 4Н), 6,86-6,71 (m, 3Н), 4,39 (d, 1H), 3,98 (br, 1H), 3,84-3,71 (m, 3Н), 3,62 (d, 1H), 3,26-3,16 (m, 3H), 3,14-2,96 (m, 2H), 2,80-2,60 (m, 4H), 2,56-2,49 (m, 1H), 2,19 (t, 1H), 2,11-1,93 (m, 3H), 1,76 (d, 1H), 1,27-1,18 (m, 1H), 1,17-1,10 (m, 1H), 1,03-0,93 (m, 1H), 0,94-0,83 (m, 1H).
Пример 2-11
4-(4-(((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-((S)-1-(3-гидроксипропил)пирролидин-2-ил)-3-(фенилтио)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 11
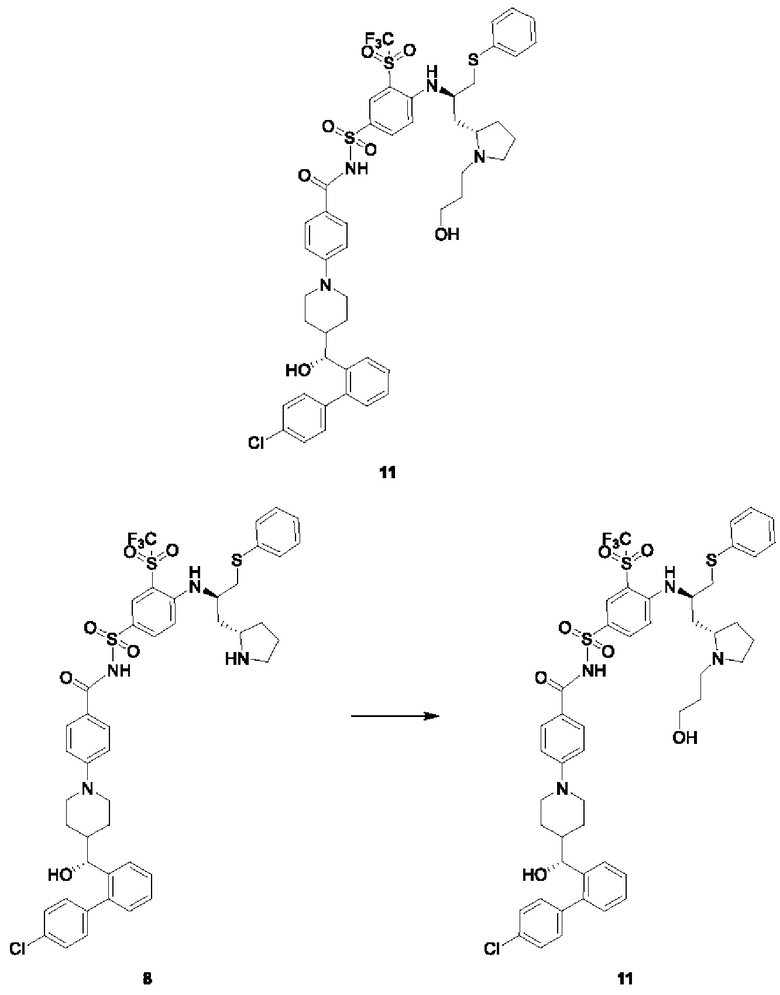
8 (20 мг, 0,021 ммоль) растворяли в ацетонитриле (2 мл) и последовательно добавляли триэтиламин (22 мг, 0,22 ммоль) и бромэтанол (30 мг, 0,22 ммоль). Реакционную систему нагревали до 70°С и подвергали реакции в течение 12 ч в атмосфере азота. Полученное твердое вещество очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: Sharpsil-T Prep С18; подвижная фаза: бикарбонат аммония, вода, ацетонитрил), и соответствующую фракцию собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 11 (13 мг, выход: 61,1%).
MS m/z (ESI): 985,1 [М+1].
1Н NMR (500 МГц, CD3OD) δ 8,28 (d, 1H), 8,09 (dd, 1H), 7,86-7,78 (m, 2Н), 7,63 (dd, 1H), 7,48-7,38 (m, 5Н), 7,37-7,31 (m, 3Н), 7,27 (dd, 2Н), 7,23-7,17 (m, 2Н), 6,81 (t, 3Н), 4,44 (d, 1Н), 4,08-4,00 (m, 1H), 3,81 (d, 1H), 3,72-3,55 (m, 4Н), 3,25 (dd, 2Н), 3,05 (s, 1H), 2,94 (s, 1H), 2,73-2,63 (m, 1H), 2,59-2,50 (m, 1H), 2,48-2,39 (m, 1H), 2,29 (s, 1H), 2,21 (t, 1H), 2,10-2,01 (m, 4Н), 1,97-1,70 (m, 5Н), 1,62 (s, 1H), 1,31-1,20 (m, 3Н), 1,15 (d, 1H), 1,04-0,95 (m, 1H), 0,93 (d, 1H).
Пример 2-12
4-(4-((R)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)-N-((4-(((R)-1-(фенилтио)-3-((S)-пирролидин-2-ил)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 12
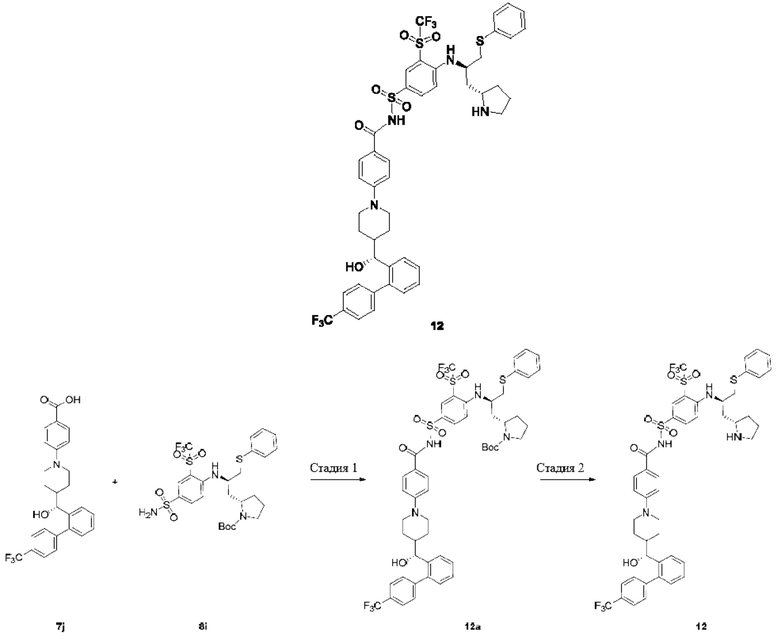
Стадия 1
Трет-бутил(S)-2-((R)-2-((4-(N-(4-(4-((R)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)бензоил)сульфамоил)-2-((трифторметил)сульфонил)фенил)амино)-3-(фенилтио)пропил)пирролидин-1-карбоксилат 12а
8i (80 мг, 0,13 ммоль) растворяли в дихлорметане (3 мл) и добавляли 7j (70 мг, 0,15 ммоль) с последующим последовательным добавлением 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (49 мг, 0,26 ммоль) и 4-диметиламинопиридина (32 мг, 0,26 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 4 часов в атмосфере азота, добавляли 7j (47 мг, 0,10 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (49 мг, 0,26 ммоль) и 4-диметиламинопиридин (32 мг, 0,26 ммоль) и перемешивали при комнатной температуре в течение ночи после добавления. Реакционный раствор последовательно промывали водой (5 мл) и насыщенным рассолом (5 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с развивающейся системой растворителей А с получением указанного в заголовке продукта 12а (80 мг, выход: 58,8%).
MS m/z (ESI): 1059,0 [М-1].
Стадия 2
4-(4-((R)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)-N-((4-(((R)-1-(фенилтио)-3-((S)-пирролидин-2-ил)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 12
12а (50 мг, 0,047 ммоль) растворяли в дихлорметане (2 мл) и добавляли 4 М раствор хлористого водорода в 1,4-диоксане (2 мл). Полученную смесь перемешивали при комнатной температуре в течение 20 мин. Реакционный раствор концентрировали при пониженном давлении для удаления органического растворителя. Остаток растворяли в дихлорметане и концентрировали при пониженном давлении до сухости, что повторяли три раза. Полученный остаток очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: Sharpsil-T Prep С18; подвижная фаза: бикарбонат аммония, вода, ацетонитрил), и соответствующую фракцию собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 12 (6 мг, выход: 66,2%).
MS m/z (ESI): 961,2 [М+1].
1Н NMR (500 МГц, CD3OD) δ 8,23 (s, 1H), 8,03 (d, 1H), 7,80 (d, 2Н), 7,72 (d, 2Н), 7,64 (d, 1H), 7,53 (d, 2Н), 7,47 (t, 1H), 7,35 (t, 3Н), 7,27-7,14 (m, 4Н), 6,79 (d, 2Н), 6,73 (d, 1Н), 4,39 (d, 1H), 3,98 (s, 1H), 3,81-3,73 (m, 1H), 3,63-3,58 (m, 1H), 3,56-3,50 (m, 1H), 3,21-3,13 (m, 2Н), 2,64 (t, 1Н), 2,50 (t, 1H), 2,33-2,25 (m, 1H), 2,25-2,20 (m, 1H), 2,11-1,91 (m, 6Н), 1,80-1,65 (m, 2Н), 1,64-1,54 (m, 2Н), 1,25-1,17 (m, 2Н), 1,16-1,10 (m, 1H), 1,00-0,93 (m, 1H).
Пример 2-13
4-(4-((R)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)-N-((4-(((R)-1-((S)-1-(2-гидроксиэтил)пирролидин-2-ил)-3-(фенилтио)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 13
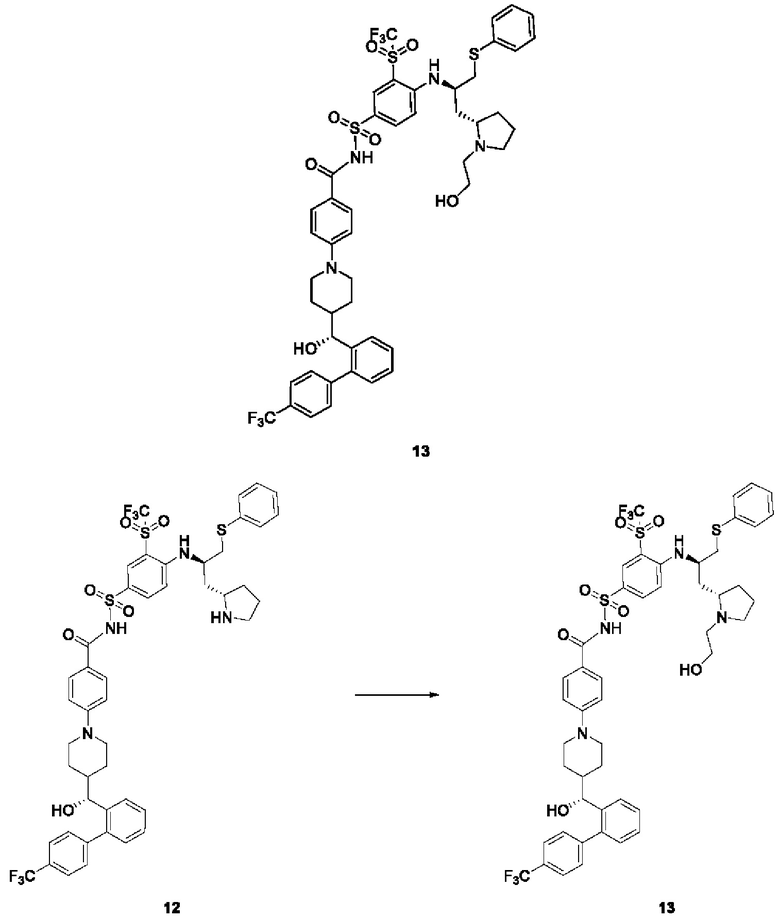
12 (30 мг, 0,031 ммоль) растворяли в ацетонитриле (4 мл) и последовательно добавляли триэтиламин (32 мг, 0,31 ммоль) и бромэтанол (39 мг, 0,31 ммоль). Реакционную систему нагревали до 70°С и подвергали реакции в течение 12 ч в атмосфере азота. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: Sharpsil-T Prep С18; подвижная фаза: бикарбонат аммония, вода, ацетонитрил), и соответствующую фракцию собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 13 (25 мг, выход: 56,9%).
MS m/z (ESI): 1005,1 [М+1].
1H NMR (500 МГц, CD3OD) δ 8,27 (d, 1H), 8,08 (dd, 1H), 7,85-7,79 (m, 2H), 7,74 (d, 2H), 7,68-7,63 (m, 1H), 7,55 (d, 2H), 7,50-7,46 (m, 1H), 7,42-7,35 (m, 3H), 7,29-7,18 (m, 4H), 6,87-6,69 (m, 3H), 4,41 (d, 1H), 4,03 (br, 1H), 3,83-3,73 (m, 3H), 3,67-3,56 (m, 2H), 3,25-3,18 (m, 2H), 3,14-2,86 (m, 2H), 2,72-2,59 (m, 1H), 2,58-2,47 (m, 1H), 2,46-2,36 (m, 1H), 2,33-2,23 (m, 1H), 2,10-1,97 (m, 4H), 1,96-1,87 (m, 1H), 1,84-1,72 (m, 2H), 1,66-1,55 (m, 1H), 1,28-1,19 (m, 1H), 1,16 (d, 1H), 1,03-0,95 (m, 1H), 0,95-0,89 (m, 1H).
Пример 2-14
4-(4-((R)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)-N-((4-(((2R)-4-((2-гидроксиэтил)(метил)амино)-3-метил-1-(фенилтио)бутан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 14
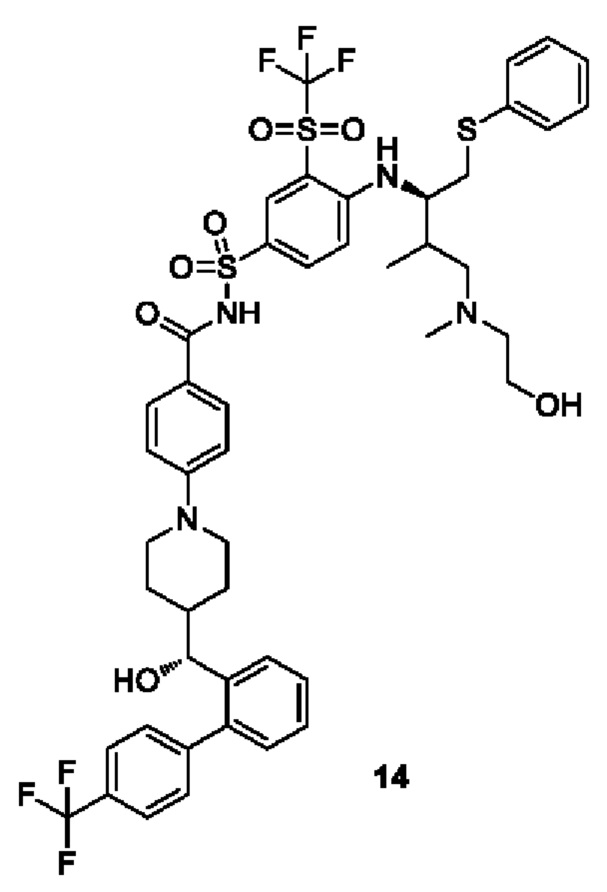
Пример 2-15
4-(4-((R)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)-N-((4-(((R)-5-((2-гидроксиэтил)(метил)амино)-1-(фенилтио)пентан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 15
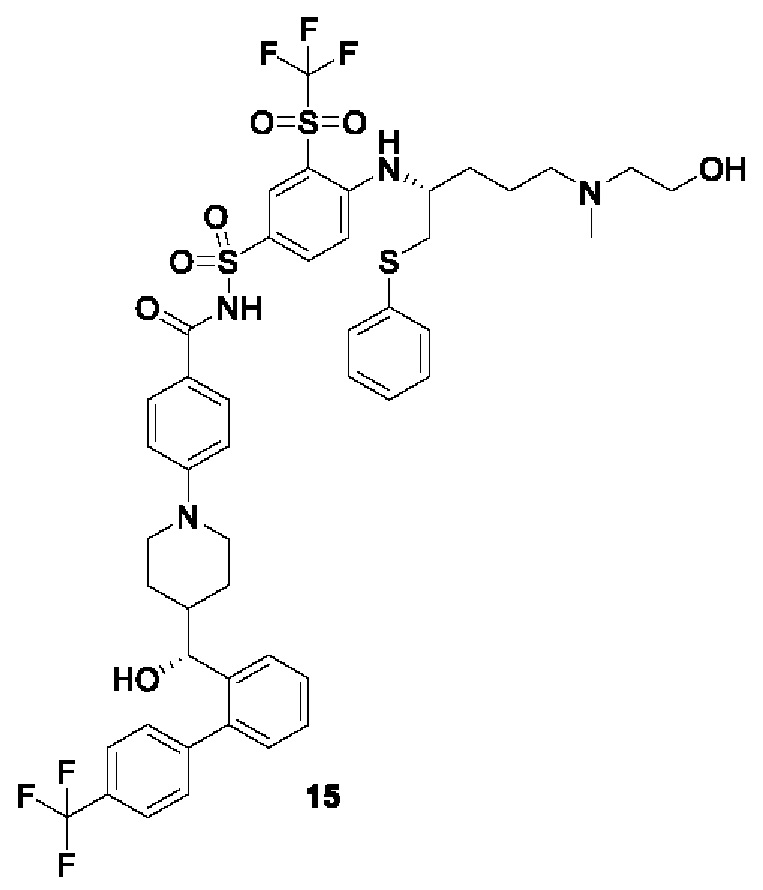
Пример 2-16
4-(4-((R)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)-N-((4-(((R)-1-((R)-1-(2-гидроксиэтил)пирролидин-2-ил)-3-(фенилтио)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 16
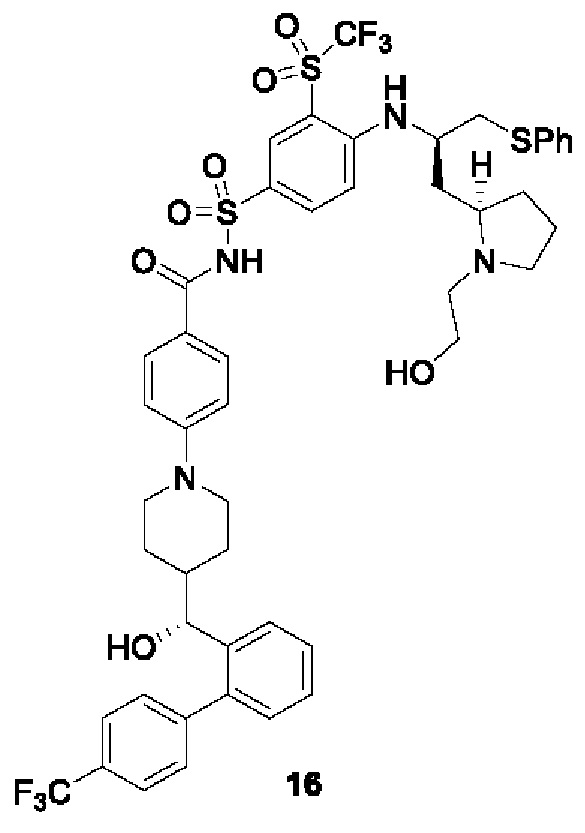
Пример 2-17
4-(4-(((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-((R)-1-метилпирролидин-2-ил)-3-(фенилтио)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 17
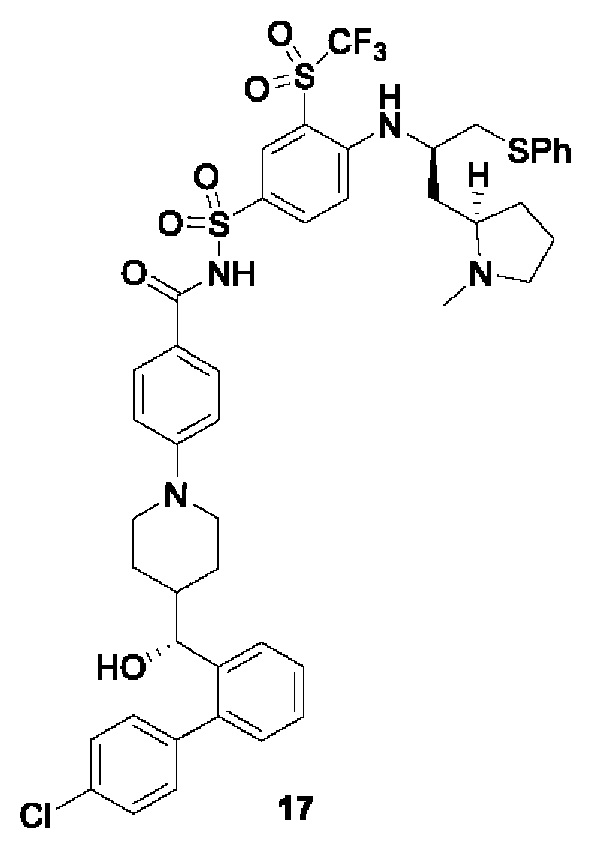
Пример 2-18
4-(4-(((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-((S)-1-метилпирролидин-2-ил)-3-(фенилтио)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 18
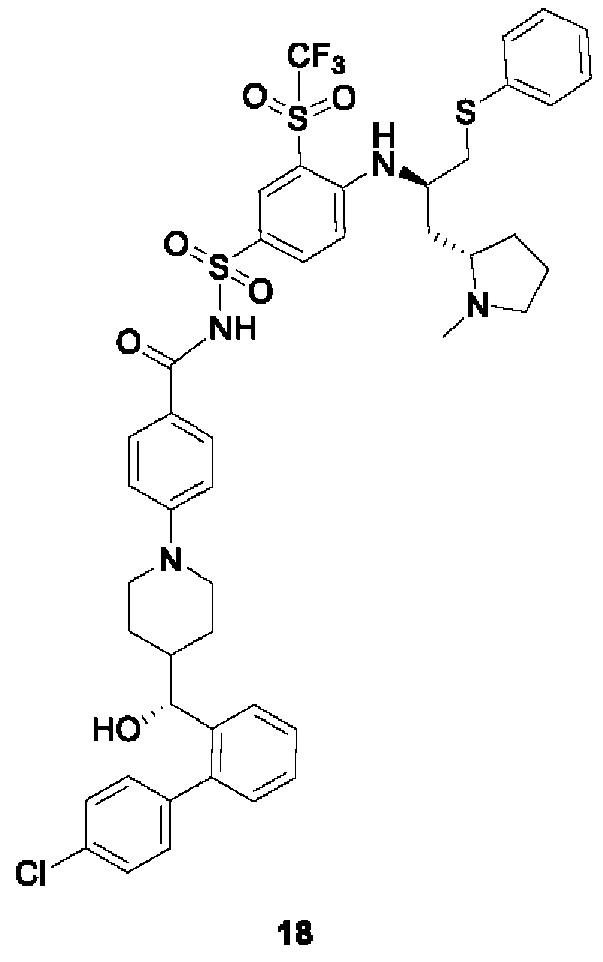
Пример 2-19
4-(4-((R)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)-N-((4-(((R)-1-((S)-1-метилпирролидин-2-ил)-3-(фенилтио)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 19
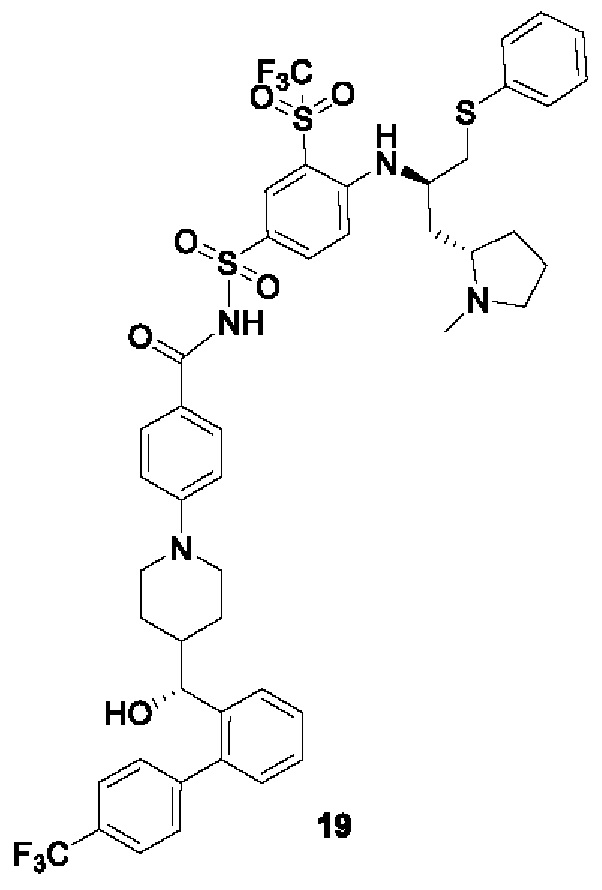
Пример 2-20
4-(4-(((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-((S)-1-(2-гидроксиэтил)-2-метилпирролидин-2-ил)-3-(фенилтио)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 20
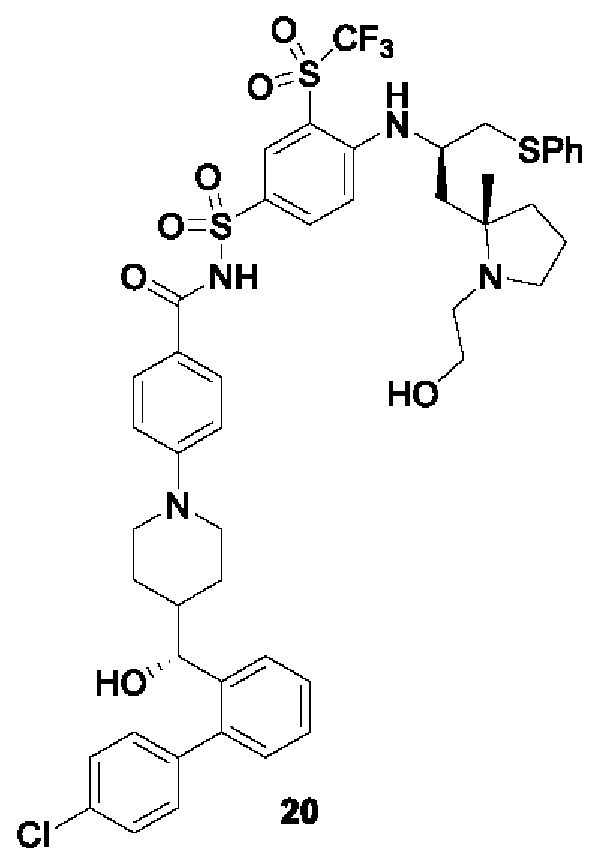
Пример 2-21
4-(4-((R)-гидрокси(4'-(трифторметил)-[1,1'-бифенил]-2-ил)метил)пиперидин-1-ил)-N-((4-(((R)-1-((S)-1-(2-гидроксиэтил)-2-метилпирролидин-2-ил)-3-(фенилтио)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 21
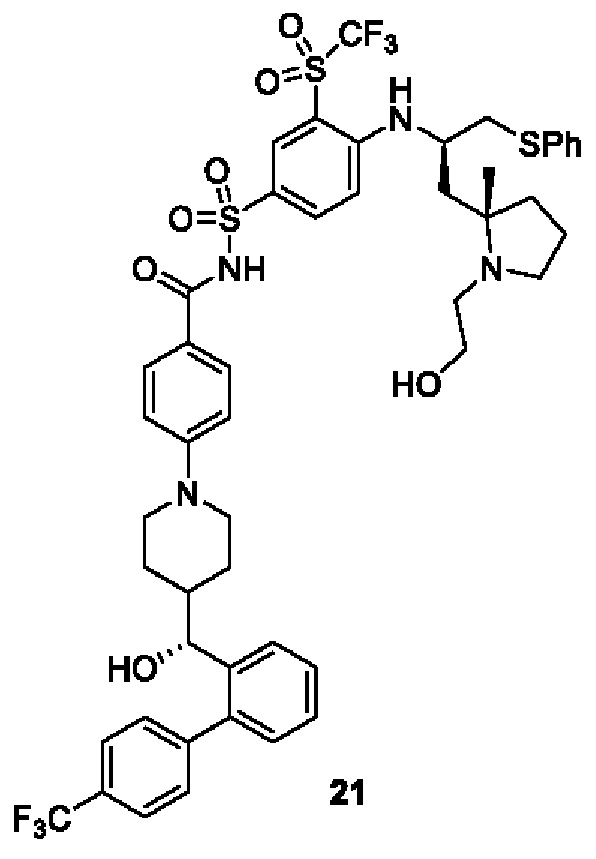
Пример 2-22
4-(4-(((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)-N-((4-(((2R)-1-(1-(2-гидроксиэтил)азетидин-2-ил)-3-(фенилтио)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 22
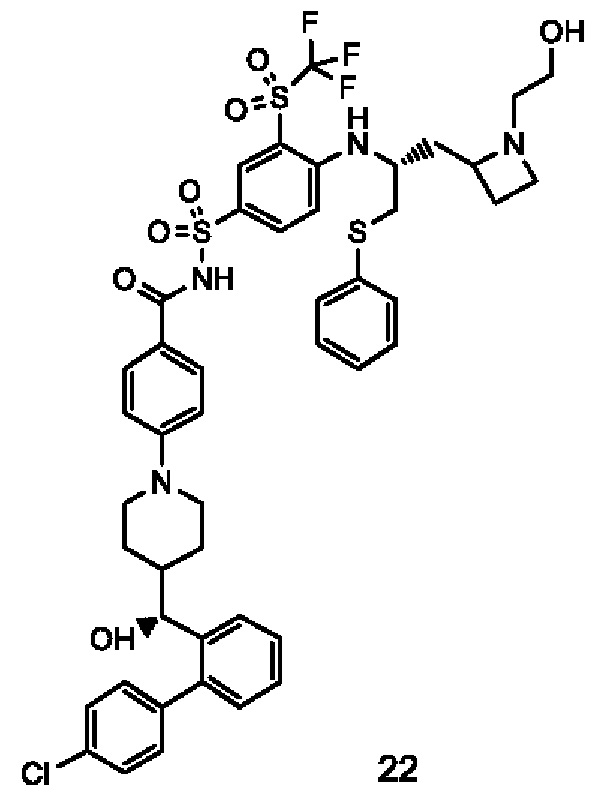
Пример 2-23
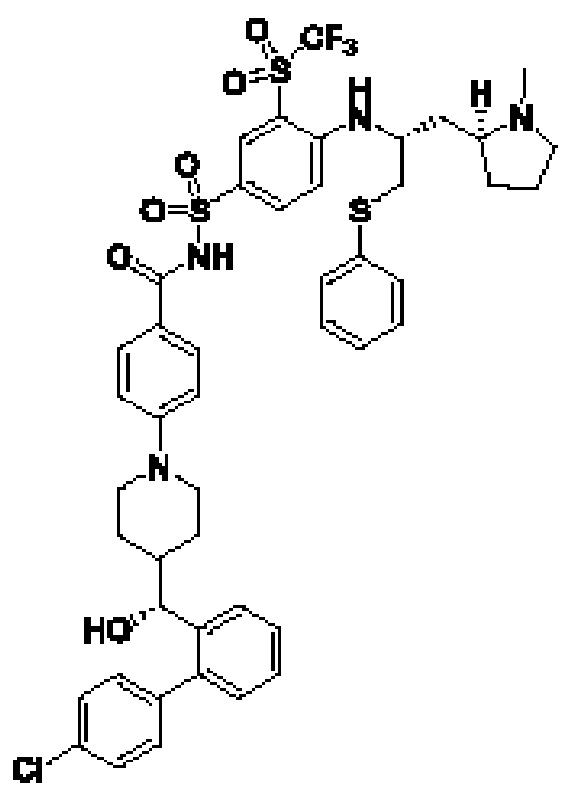
Пример 2-24 AZD4320
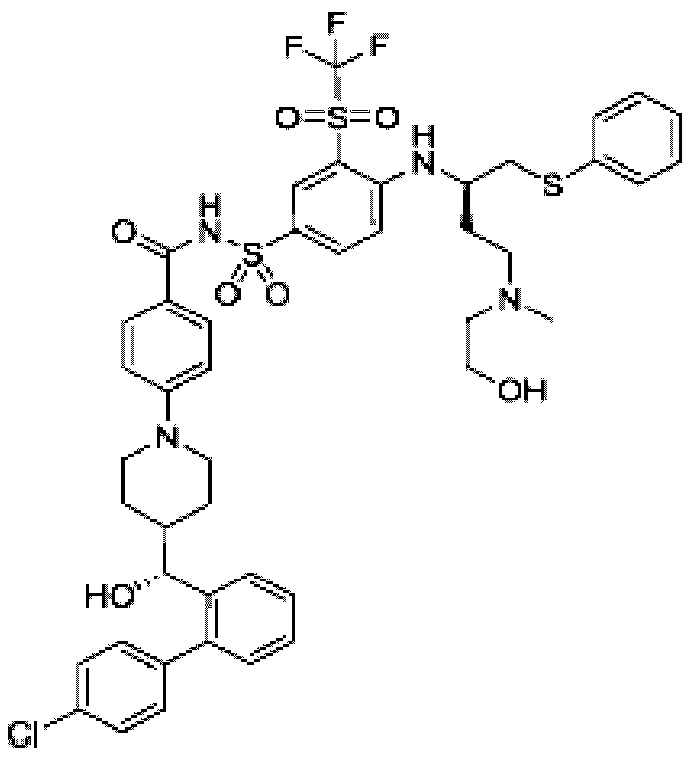
Соединение получали способом, описанным в Примере 3 (соединение положительного контроля AZD4320) на стр. 160-161 патента WO 2017251.
Пример 3-1 LD-1
4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамидо)-3-метил6утанамидо)-5-уреидопентанамидо)6ензил (R)-2-((R)-2-((4-(N-(4-((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)бензоил)сульфамоил)-2-((трифторметил)сульфонил)фенил)амино)-3-(фенилтио)пропил)пирролидин-1-карбоксилат LD-1
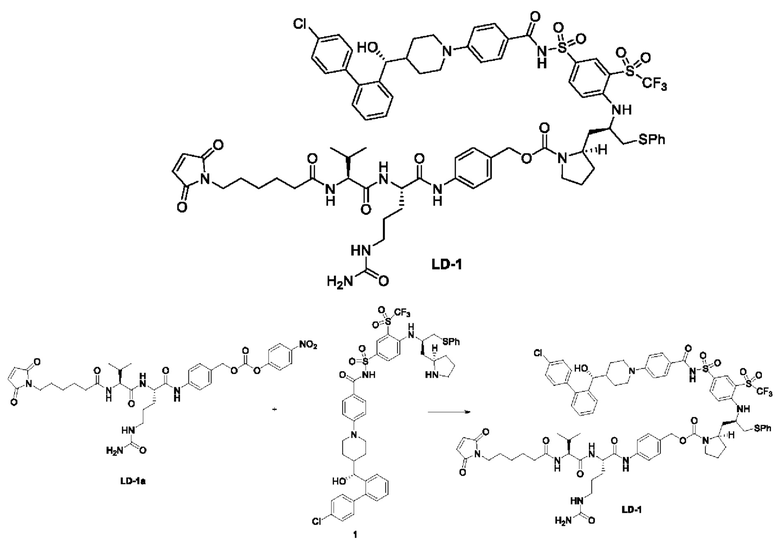
К 0,5 мл N,N-диметилформамида добавляли 1 (3 мг, 0,003 ммоль) и добавляли 4-((S)-2-((S)-2-(6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексанамидо)-3-метилбутанамидо)-5-уреидопентанамидо)бензил (4-нитрофенил) карбонат LD-1a (3,1 мг, 0,004 ммоль, Haoyuan), а затем 0,1 мл пиридина. Смесь продували аргоном три раза и добавляли 1-гидроксибензотриазол (2 мг, 0,014 ммоль) и N,N-диизопропилэтиламин (2 мг, 0,015 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционный раствор очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: XBridge Prep С18; подвижная фаза: ацетат аммония, вода, ацетонитрил), и соответствующие фракции собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта LD-1 (2 мг, выход: 40,5%).
MS m/z (ESI): 1525,0 [М+1].
1Н NMR (500 МГц, CD3OD) δ 8,17 (s, 1H), 7,92-7,80 (m, 3Н), 7,64-7,51 (m, 3Н), 7,46-7,09 (m, 12Н), 6,84-6,74 (m, 3Н), 6,48-6,40 (m, 2Н), 5,35 (t, 4Н), 4,41 (d, 1H), 4,15 (d, 1H), 4,00-3,71 (m, 6Н), 3,66-3,54 (m, 2Н), 3,44 (t, 2Н), 3,22-3,05 (m, 6Н), 2,77 (t, 1H), 2,72-2,62 (m, 2Н), 2,53 (t, 2Н), 2,23 (t, 2Н), 2,18 (t, 4Н), 2,11-2,06 (m, 2Н), 1,96-1,82 (m, 4Н), 1,80-1,71 (m, 2Н), 1,65-1,51 (m, 8Н), 1,17-1,09 (m, 2Н), 1,00-0,89 (m, 9Н).
Биологическая оценка
Тестовый пример 1. Анализ связывающей активности соединений согласно настоящему описанию для BCL-xL
Активность связывания BCL-xLin vitro тестировали следующим способом.
Реагенты, используемые в этом эксперименте: His-меченный белок BCL-xL (R&D, кат. №894-ВХ-050); BIM-белок, меченный биотином (R&D, кат. №3526/1); связывающий буфер (cisbio, кат. №62DLBDDF); буфер для анализа (cisbio, кат. №62DB1FDG); криптат MAb к 6His-Eu (cisbio, кат. №61HI2KLA); и аффинный стрептомицин, связанный с XL665 (cisbio, кат. №611SAXLA).
Анализ связывания in vitro, описанный ниже, может определять активность связывания тестируемых соединений для BCL-xL.
Испытуемые соединения растворяли в диметилсульфоксиде и разбавляли до градиентных концентраций, необходимых для эксперимента, а соединения в различных концентрациях, составленных в диметилсульфоксиде, подвергали 100-кратному разбавлению с помощью связывающего буфера. BCL-xL разбавляли до соответствующей концентрации с помощью связывающего буфера и добавляли в белый 384-луночный планшет при 4 мкл/лунку. Затем разбавленное тестируемое соединение добавляли в белый 384-луночный планшет при 2 мкл/лунку. Планшет инкубировали при 25°С в течение 1 часа после равномерного перемешивания смеси. Меченный биотином BIM-белок разбавляли до соответствующей концентрации с помощью связывающего буфера и добавляли в белый 384-луночный планшет при 4 мкл/лунку. Планшет инкубировали при 25°С в течение 2 ч после равномерного перемешивания смеси. Криптат MAb к 6His-Eu и аффинный стрептомицин, связанный с XL665, каждый разбавляли до соответствующей концентрации буфером для анализа и добавляли в белый 384-луночный планшет при 5 мкл/лунку. Планшет инкубировали при 25°С в течение 2-4 часов после равномерного перемешивания смеси. Значения сигнала считывали с помощью программы HTRF в микропланшетном ридере (BMG Labtech, модель: PHERAstar FS).
Результаты теста приведены в Таблице 1.
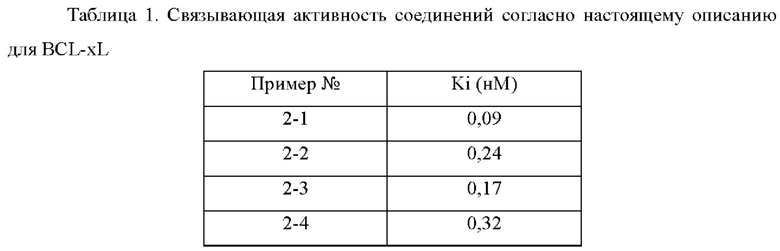
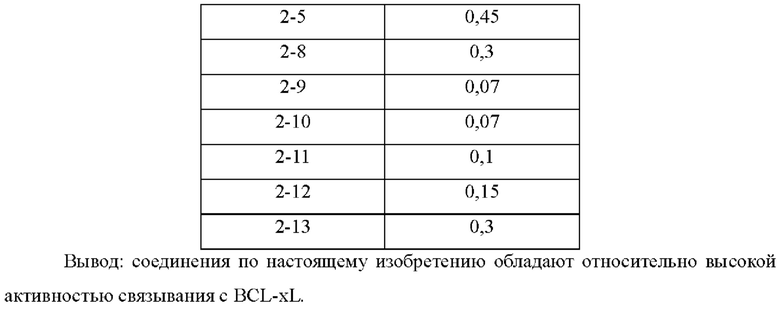
Тестовый пример 2. Анализ связывающей активности соединений согласно настоящему описанию для BCL-2
Активность связывания BCL-2 in vitro тестировали следующим способом. Реагенты, используемые в этом эксперименте: His-меченный белок BCL-2 (R&D systems, кат. №827-ВС-050); BIM-белок, меченный биотином (R&D, кат. №3526/1); связывающий буфер (cisbio, кат. №62DLBDDF); буфер для анализа (cisbio, кат. №62DB1FDG); криптат MAb к 6His-Eu (cisbio, кат. №61HI2KLA); и аффинный стрептомицин, связанный с XL665 (cisbio, кат. №611SAXLA).
Анализ связывания in vitro, описанный ниже, может определять активность связывания тестируемых соединений для BCL-2.
Испытуемые соединения растворяли в диметилсульфоксиде и разбавляли до градиентных концентраций, необходимых для эксперимента, а соединения в различных концентрациях, составленных в диметилсульфоксиде, подвергали 100-кратному разбавлению с помощью связывающего буфера. BCL-2 разбавляли до соответствующей концентрации с помощью связывающего буферного раствора и добавляли в белый 384-луночный планшет при 4 мкл/лунку. Затем разбавленное тестируемое соединение добавляли в белый 384-луночный планшет при 2 мкл/лунку. Планшет инкубировали при 25°С в течение 1 часа после равномерного перемешивания смеси. Меченный биотином BIM-белок разбавляли до соответствующей концентрации с помощью связывающего буфера и добавляли в белый 384-луночный планшет при 4 мкл/лунку. Планшет инкубировали при 25°С в течение 2 ч после равномерного перемешивания смеси. Криптат MAb к 6His-Eu и аффинный стрептомицин, связанный с XL665, каждый разбавляли до соответствующей концентрации буфером для анализа и добавляли в белый 384-луночный планшет при 5 мкл/лунку. Планшет инкубировали при 25°С в течение 2-4 часов после равномерного перемешивания смеси. Значения сигнала считывали с помощью программы HTRF в микропланшетном ридере (BMG Labtech, модель: PHERAstar FS). Результаты теста представлены в таблице 2.
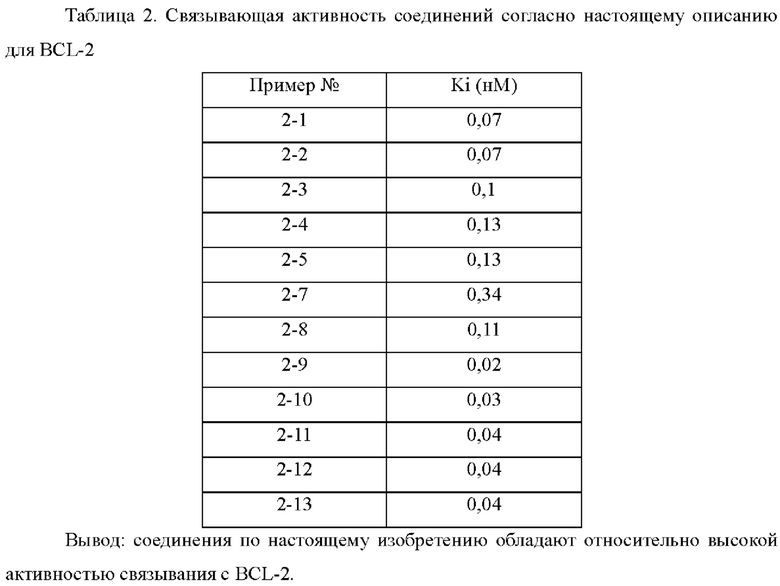
Тестовый пример 3. Определение ингибирующей активности соединений по настоящему изобретению для пролиферации RS4;11 клеток и клеток NCI-H146
Ингибирующую активность in vitro в отношении пролиферации клеток RS4;11 и клеток NCI-H146 испытывали следующим способом.
Реагенты, используемые в этом эксперименте: среда RPMI-1640 (HyCLone, кат. №SH30243018); фетальная бычья сыворотка (Gibco, кат. №10099-141); панкреатин (Gibco, кат. №25200-072); клеточный титр-Glo (Promega, кат. №G7571).
Используемые материалы: клетки RS4;11 (Nanjing Kebai Biotechnology Co., Ltd, кат. №CBP60641); и клетки NCI-H 146 (АТСС, кат. №НТВ-173).
Анализ пролиферации клеток in vitro, описанный ниже, может определять ингибирующую активность тестируемых соединений для пролиферации клеток RS4;11 и клеток NCI-H146.
RS4;11 клеток (или клеток NCI-H146) культивировали в культуральной среде RPMI-1640, содержащей 10% FBS, и пассировали 2-3 раза в неделю в соотношении пассажей 1:3 или 1:5. Во время пассажа клетки переносили в центрифужную пробирку и центрифугировали при 1200 об/мин в течение 3 мин. Супернатант сбрасывали и добавляли свежую питательную среду для ресуспендирования клеток. 90 мкл клеточной суспензии добавляли в 96-луночный планшет для культивирования клеток, где добавляли RS4;11 клеток и клетки NCI-H146 при плотности 2,22×105 клеток/мл. На периферию 96-луночного планшета добавляли только 100 μл полной среды (среда RPMI-1640, содержащая 10% FBS). Планшет инкубировали в инкубаторе в течение 24 часов (37°С, 5% CO2).
Тестовый образец разбавляли до надлежащего градиента концентрации диметилсульфоксидом, как требуется для эксперимента. 5 μл раствора тестирумого соединения, полученного в градиентной концентрации, добавляли к 95 μл свежей культуральной среды и перемешивали равномерно, а затем 10 мкл культуральной среды, содержащей указанное соединение, добавляли в планшет для культивирования клеток. Планшет для культивирования клеток инкубировали в инкубаторе в течение 3 дней (37°С, 5% СО2). 50 мкл реагента CellTiter-Glo добавляли в каждую лунку 96-луночного планшета для культивирования клеток. Планшет оставляли стоять в течение 5-10 минут вдали от света при комнатной температуре. Сигналы хемилюминесценции считывали с помощью микропланшетного ридера (BMG Labtech, модель: PHERAstar FS), а данные обрабатывали с помощью программного обеспечения GraphPad. Результаты приведены в Таблице 3.
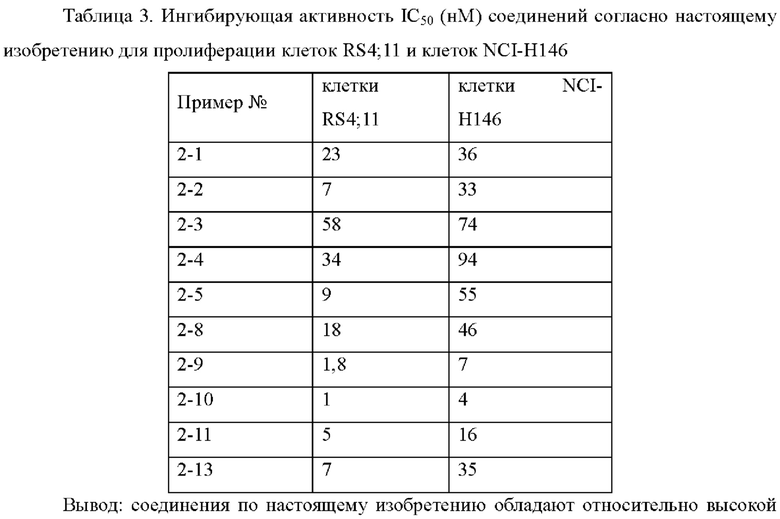

Тестовый пример 4. Испытание на токсичность на крысах SD (Спрег-Доули) путем повторного внутривенного введения в течение 7 дней
1. Цель
Этот эксперимент проводили для оценки токсичности соединения 9 и AZD4320 для крыс SD путем повторного внутривенного введения в течение 7 дней.
2. Тестовые соединения
Условия хранения: хранили в сушилке при комнатной температуре вдали от света. Соединение 9, в 100% чистоте, молекулярная масса: 976,1. Соединение AZD4320, в 100% чистоте, молекулярная масса: 945,5.
3. План эксперимента и процедуры
3.1 Экспериментальные животные и условия содержания
30 крыс SD, весом около 180-200 г и в возрасте 6-7 недель, половина самцов и половина самок, приобретенные у Zhejiang Vital River Laboratory Animal Technology Co., Ltd., класс SPF (не содержащие специфичных патогенов). Номер сертификации животных: 200917Aazz0619000298 и 20200917Aazz0619000104. Номер лицензии на производство: SCXK (Zhejiang) 2019-0001.
Условия содержания: класс SPF, животных размещали в пластиковых и прозрачных крысиных клетках, по 2-3 на клетку. Комнатная температура составляла 20-26°С, а относительная влажность составляла 40-70%. Освещение контролировалось в цикле 12/12 свет/темнота. Гранулированный корм мыши свободно принимали в неограниченном количестве.
3.2 Группировка животных
Крысы были случайным образом разделены на следующие 3 группы: контрольная группа, получающая носитель, группа, получающая соединения 9-1 мг/кг и группа, получающая соединения AZD4320-1 мг/кг, с 6 крысами в каждой группе, половина самцы и половины самки.
3.3 Экспериментальные процедуры
Дозировка испытуемого образца для 7-дневного испытания токсичности на крысах: 1 мг/кг (1 р/сут) в течение 7 дней подряд. Путь введения: внутривенно, объем дозирования: 10 мл/кг, частота введения и период дозирования: ежедневно утром в течение 7 дней.
Носитель: 1,5% DMA+98,5% (10% HS-15), где DMA представляет собой N,N-диметилацетамид (Shanghai Titan Scientific Co., Ltd., кат. №01013630); и HS-15 представляет собой стеарат полиэтиленгликоля 15 (Solutol HS15) (Shanghai Xietai Chemical Co., Ltd., кат. №50253404).
3.4 Оборудование для экспериментов Автоматический биохимический анализатор Hitachi 7100 Автоматический анализатор клеток крови Siemens Advia 2120i Автоматический анализатор коагуляции Sysmex са1500
Сбор токсикокинетических данных: жидкостный хроматограф/масс-спектрометр АВ 4000
3.5 Выражение данных и статистическая обработка
Результаты обнаружения были введены и статистически обработаны в Excel компьютера.
Клинические симптомы, потребление пищи и общие результаты визуального наблюдения при диссекции статистически не рассчитывались, а такие показатели, как масса тела, масса органа и клиническое обследование, подсчитывали с помощью Т-критерия.
4. Результаты
Крысам SD внутривенно вводили 1 мг/кг соединения 9 или 1 мг/кг AZD4320 по 5 мл/кг в течение 7 повторных дней.
Никаких значимых отклонений в клиническом наблюдении за животными в группе введения соединения 9 во время лечения не наблюдалось. По сравнению с контрольной группой, получавшей растворитель, не наблюдалось существенной разницы в массе тела и скорости роста массы тела животных (р>0,05) среди групп дозирования, и не наблюдалось значительных изменений в потреблении пищи.
Тромбоциты крыс в группе введения соединения 9 были слегка снижены, но в пределах фонового нормального диапазона, в то время как тромбоциты крыс AZD 4320 были более значительно снижены (р<0,01). Результаты показаны в таблице 4.
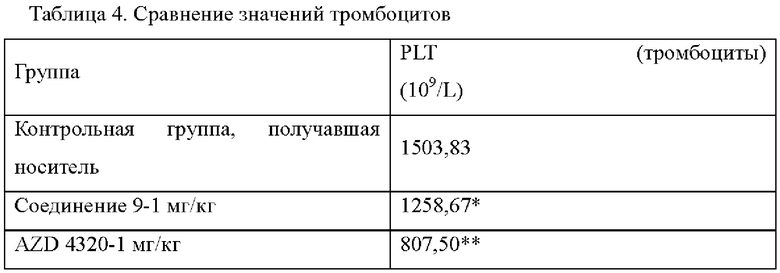
5. Заключение
В условиях этого эксперимента, по сравнению с контрольной группой, получавшей носитель и группой положительного контроля AZD 4320, соединение 9 уменьшает количество тромбоцитов меньше, чем группа положительного контроля, и имеет преимущество более высокой безопасности.
Тестовый пример 5. Фармакокинетическая оценка соединений по настоящему изобретению у мышей
1. Аннотация
Концентрацию лекарственного средства в плазме испытуемых животных (мышей) в разные моменты времени после внутривенного (iv) введения соединения 10 и AZD4320 определяли методом LC/MS/MS. Изучали фармакокинетическое поведение соединений по настоящему изобретению у мышей и оценивали их фармакокинетический профиль.
2. Методология
2.1 Тестовые соединения
Соединение 10
2.2. Тестовые животные
18 самок мышей С57, случайным образом разделенных на 2 группы, приобретенных у Zhejiang Vital River Laboratory Animal Technology Co., Ltd, номер лицензии на производство животных: SCXK (Shanghai) 2017-0005.
2.3. Подготовка лекарственного средства
Определенное количество соединения 10 взвешивали, растворяли путем добавления 5% по объему DMSO и 5% Tween 80 (Shanghai Titan Scientific Co., Ltd.), a затем подготавливали в 0,1 мг/мл прозрачного раствора путем добавления 90% нормального физиологического раствора. Соединение AZD4320 составили так же, как и соединение 10.
2.4. Введение
Внутривенное введение: соединения 10 и AZD4320 вводили мышам раздельно внутривенно в дозе 2,0 мг/кг и объеме 20,0 мл/кг.
3. Процедуры
Мышам внутривенно вводили соединение 10 и собирали 0,1 мл крови перед введением и 5 мин, 0,25 ч, 0,5 ч, 1,0 ч, 2,0 ч, 4,0 ч, 8,0 ч, 11,0 ч и 24,0 ч после введения, помещали в антикоагуляционную пробирку EDTA-K2, центрифугировали при 10000 об/мин в течение 1 мин (4°С). Плазму отделяли в течение 1 часа и хранили при -20°С для тестирования. Процесс от забора крови до центрифугирования проводили на ледяной бане.
Мышам внутривенно вводили соединение AZD4320 и собирали 0,1 мл крови перед введением и 5 мин, 0,25 ч, 0,5 ч, 1,0 ч, 2,0 ч, 4,0 ч, 8,0 ч, 11,0 ч и 24,0 ч после введения, помещали в антикоагуляционную пробирку EDTA-K2, центрифугировали при 10000 об/мин в течение 1 мин (4°С). Плазму отделяли в течение 1 часа и хранили при -20°С для тестирования. Процесс от забора крови до центрифугирования проводили на ледяной бане.
Определяли содержание тестируемых соединений при различных концентрациях в плазме мышей после введения: 10 мкл плазмы мышей в каждый момент времени после введения смешивали с 200 мкл метанола (содержащего раствор внутреннего стандарта камптотецина (100 нг/мл)); смесь перемешивали на вортексе в течение 1 мин и центрифугировали в течение 10 мин (18000 об/мин), и 3 мкл супернатанта брали для анализа LC/MS/MS.
4. Фармакокинетические параметры

Тестовый пример 6. Терапевтическое воздействие на RS4;11 острого лимфобластного лейкоза человека на мышиной подкожной ксенотрансплантатной опухоли
Этот эксперимент проводили для оценки и сравнения терапевтического эффекта соединений по настоящему изобретению на RS4;11 острого лимфобластного лейкоз на мышиной подкожной ксенотрансплантатной опухоли.
1. Экспериментальные животные и условия содержания
Мыши NOD-Scid (диабетические мыши без ожирения с тяжелым комбинированным иммунодефицитом), возраст 5 недель, приобретенные у Beijing Huafukang Biotechnology Co., Ltd. Лицензия на производство №: SCXK (Пекин) 2019-0008, сертификат животных №: 110322211100992176. Среда содержания: класс SPF. 6 мышей в группе носителя и 8 мышей в группе соединения.
2. Тестовые соединения
Группы, получающие соединение 9, соединение 10 и носитель.
5 мг соединения добавляли к 250 мкл DMSO и перемешивали на вортексе с получением раствора 20 мг/мл, который разбавляли до желаемой концентрации 30% гидроксипропил-β-циклодекстрином (HP-β-CD, InnoStar Biotechnology Nantong Co., Ltd, Nantong).
3. План эксперимента и процедуры
Острый лимфобластный лейкоз человека RS4;11 был приобретен в American Туре Culture Collection. RS4;11 клетки культивировали в чашке Петри 10 см с культуральной средой RPMI 1640 (Gibco), содержащей 10% фетальной бычьей сыворотки (Gibco) и пенициллин и стрептомицин, в инкубаторе при 37°С, содержащем 5% СО2. Клетки пассировали два раза в неделю, а затем собирали, подсчитывали и инокулировали, когда они находились в фазе экспоненциального роста.
Каждой мыши подкожно инокулировали 1×107 клеток RS4;11. Когда опухоли выросли до 100-200 мм3, мышей сгруппировали в соответствии с объемом опухоли, внутривенно (в/в) вводили препарат один раз в неделю (1 р/нед) в общей сложности 4 раза, при объеме инъекции 0,1 мл/10 г массы тела и дозе 10 мг/кг.~
4. Результаты
Экспериментальный показатель должен был исследовать влияние препарата на рост опухоли, а удельный показатель составлял Т/С% или ингибирование роста опухоли (TGI%).
Диаметры опухоли измеряли два раза в неделю с помощью штангенциркуля, а объем опухоли (V) рассчитывали по следующей формуле:
V=1/2×а×b2, где а и b обозначают соответственно длину и ширину.
Т/С(%)=(Т-Т0)/(С-С0)×100, где Т и С предсталяли собой объемы опухоли животных в конце эксперимента в группе лечения и контрольной группе, соответственно; Т0 и С0 представляли собой объемы опухоли животных в начале эксперимента в группе лечения и контрольной группе, соответственно.
% ингибирования роста опухоли (TGI%)=100-Т/С (%);
Когда опухоль начала регрессировать, % ингибирования роста опухоли (TGI%)=100-(Т-Т0)/Т0×100;
Если опухоль имела уменьшенный объем по сравнению с исходным объемом, т.е. Т меньше Т0 или С меньше С0, это определяли как частичную регрессию (PR) опухоли; если опухоль полностью исчезала, это определяли как полную регрессию (CR) опухоли.
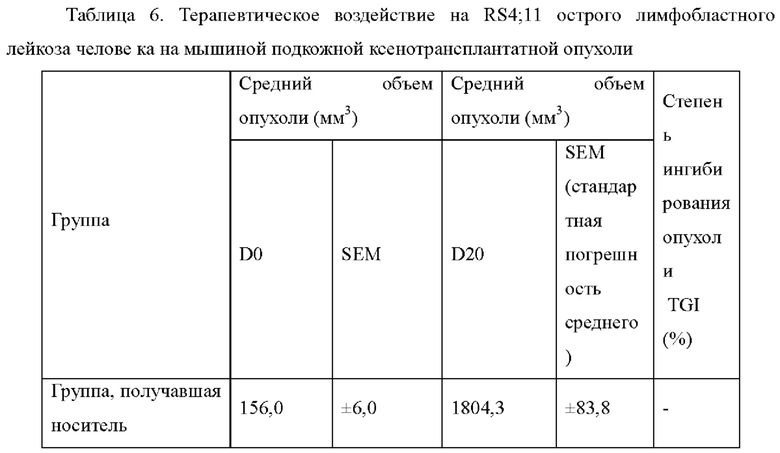

Заключение: соединение 9 (10 мг/кг, в/в, 1 р/нед 3 раза) и соединение 10 (10 мг/кг, в/в, 1 р/нед 3 раза) оказывают значительное ингибирующее действие на рост RS4;11 острого лимфобластного лейкоза человека на мышиной подкожной ксенотрансплантатной опухоль (фиг. 1), при этом частота ингибирования опухоли составляет 72% и 82%, соответственно; все мыши, имеющие опухоль, хорошо переносят вышеуказанные лекарственные средства, и никаких симптомов, таких как значительная потеря веса, не наблюдается (фиг. 2).
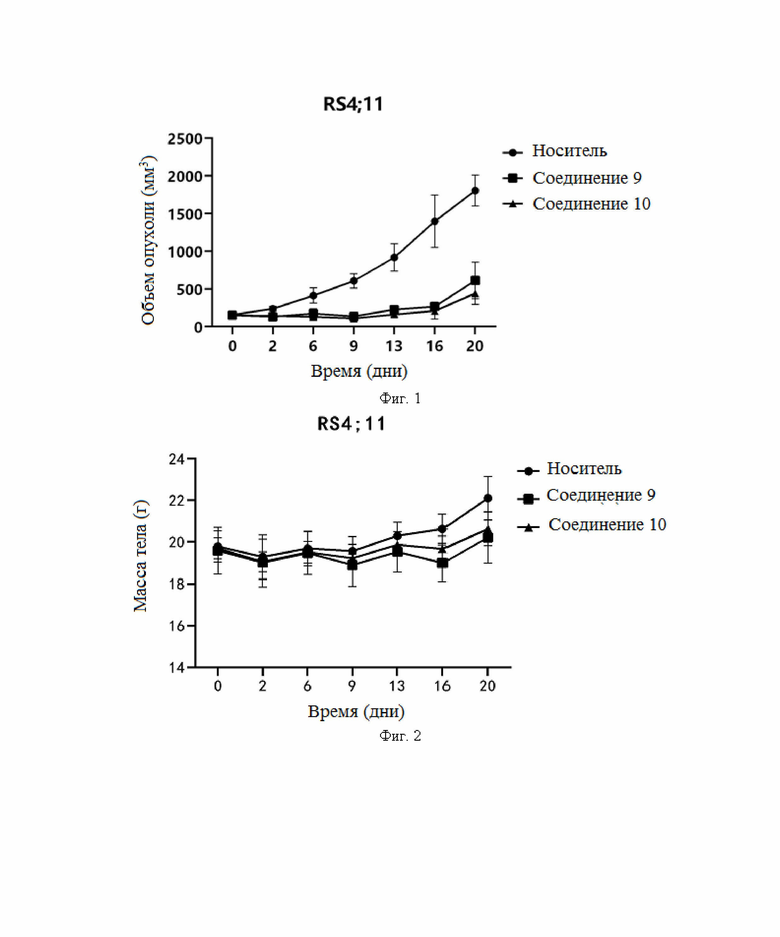
Изобретение относится к соединению формулы (D), где R1-R14 определены в формуле изобретения, или его фармацевтически приемлемой соли, его способу получения, фармацевтической композиции, содержащей указанное соединение, для лечения рака посредством регуляции рецепторов. 3 н. и 21 з.п. ф-лы, 2 ил., 6 табл., 24 пр.
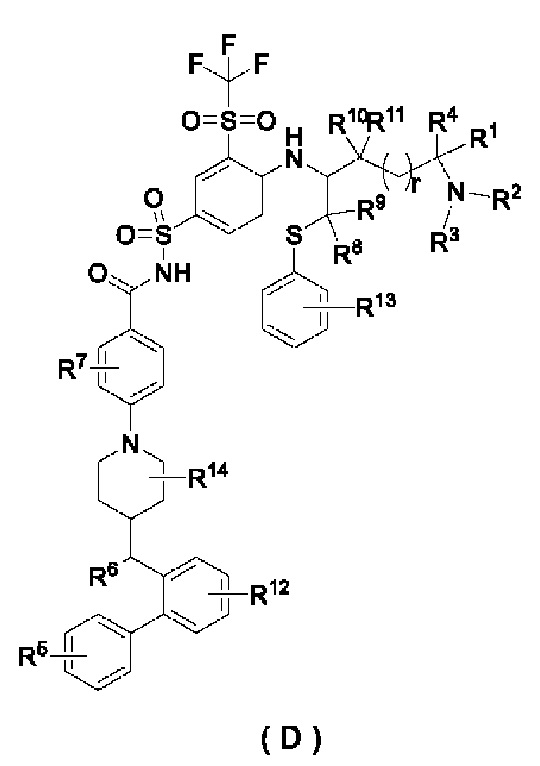
1. Соединение общей формулы (D) или его фармацевтически приемлемая соль:
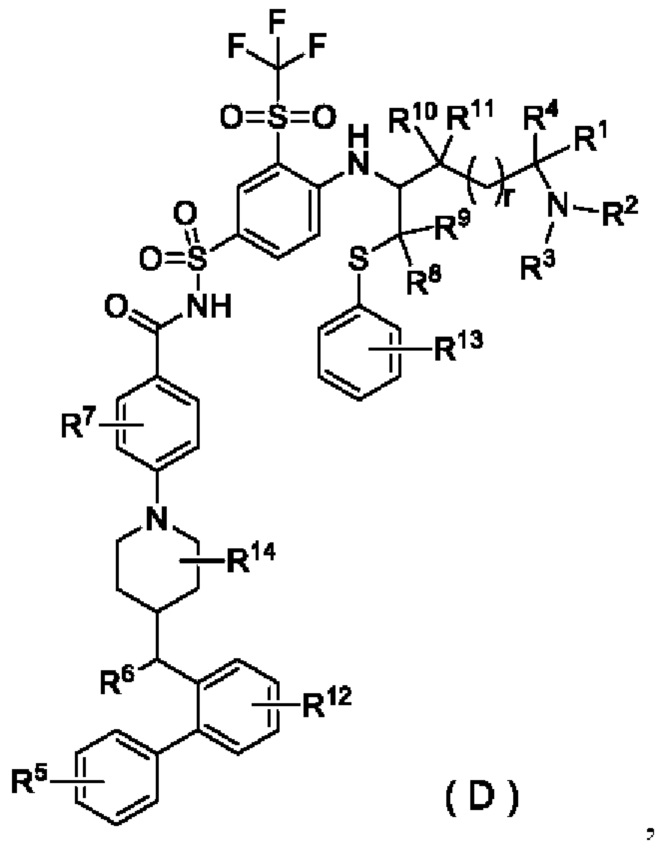
где:
R1 представляет собой водород;
R2 выбран из группы, состоящей из C1-6 алкила и C1-6 гидроксиалкила;
или R1 и R2 вместе с атомом, к которому они присоединены, образуют 3-6-членный гетероциклил;
R3 выбран из группы, состоящей из водорода, C1-6 гидроксиалкила и C1-6 алкила;
R4 выбран из группы, состоящей из водорода и C1-6 алкила;
R5 выбран из группы, состоящей из галогена и C1-6 галогеналкила;
R6 выбран из группы, состоящей из гидрокси и C1-6 алкокси;
R7 представляет собой водород;
каждый из R8 и R9 независимо выбран из группы, состоящей из водорода и C1-6 алкила; каждый из R10 и R11 независимо выбран из группы, состоящей из водорода и C1-6 алкила;
R12 представляет собой водород;
R13 представляет собой водород;
R14 представляет собой водород; и
r выбран из группы, состоящей из 0, 1, 2 и 3;
при условии, что если R1 и R2 вместе с атомом, к которому они присоединены, не образуют 3-6-членный гетероциклил, R5 представляет собой галогеналкил.
2. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, где R1 и R2 вместе с атомом, к которому они присоединены, образуют пирролидинил.
3. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, представляющее собой соединение общей формулы (D-I) или его фармацевтически приемлемую соль:
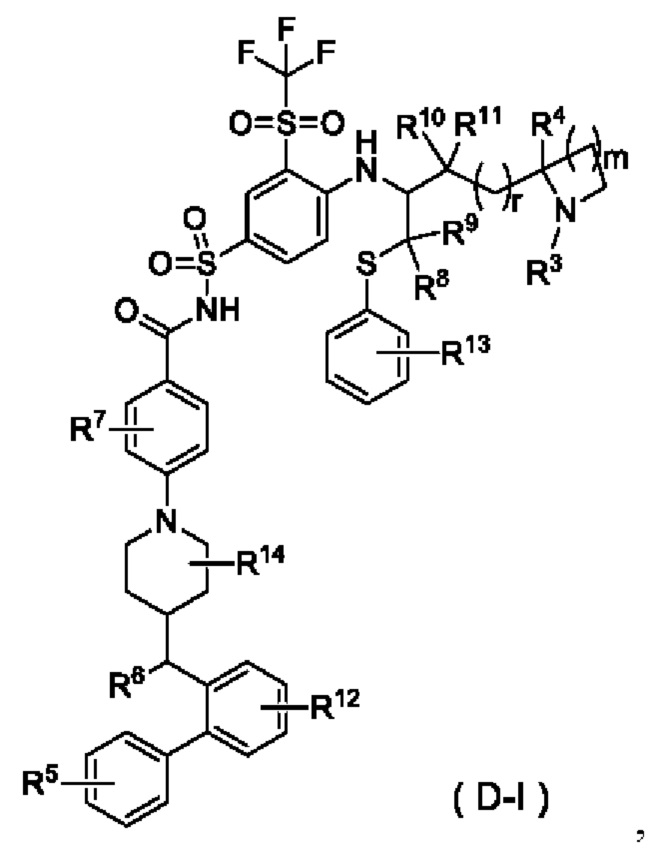
где m выбран из группы, состоящей из 0, 1, 2 и 3, и
r и R3-R14 являются такими, как определено в п. 1.
4. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, представляющее собой соединение общей формулы (D-II) или его фармацевтически приемлемую соль:
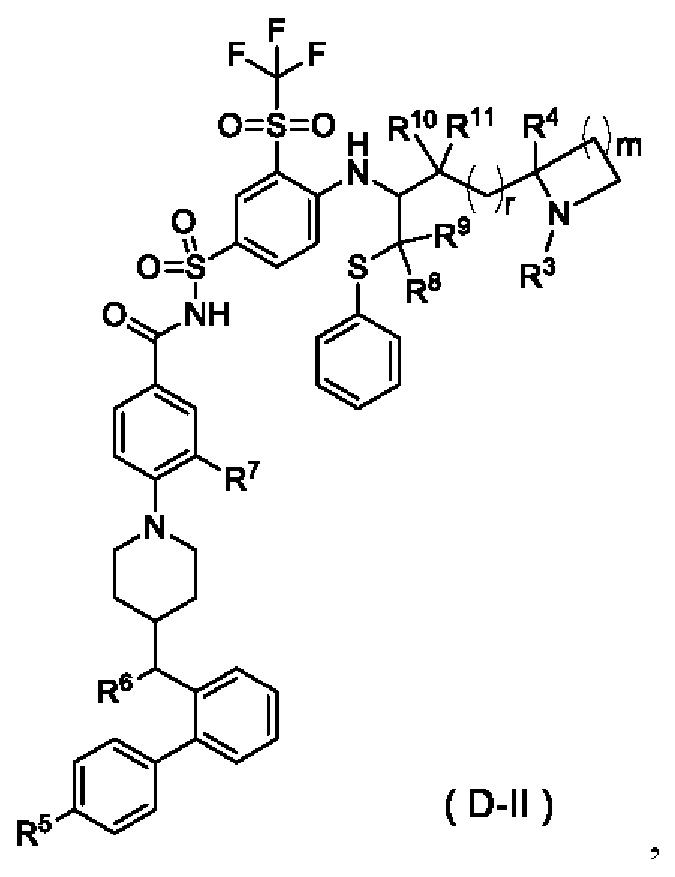
где m выбран из группы, состоящей из 0, 1, 2 и 3, и
r и R3-R11 являются такими, как определено в п. 1.
5. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, представляющее собой соединение общей формулы (D-III) или его фармацевтически приемлемую соль:
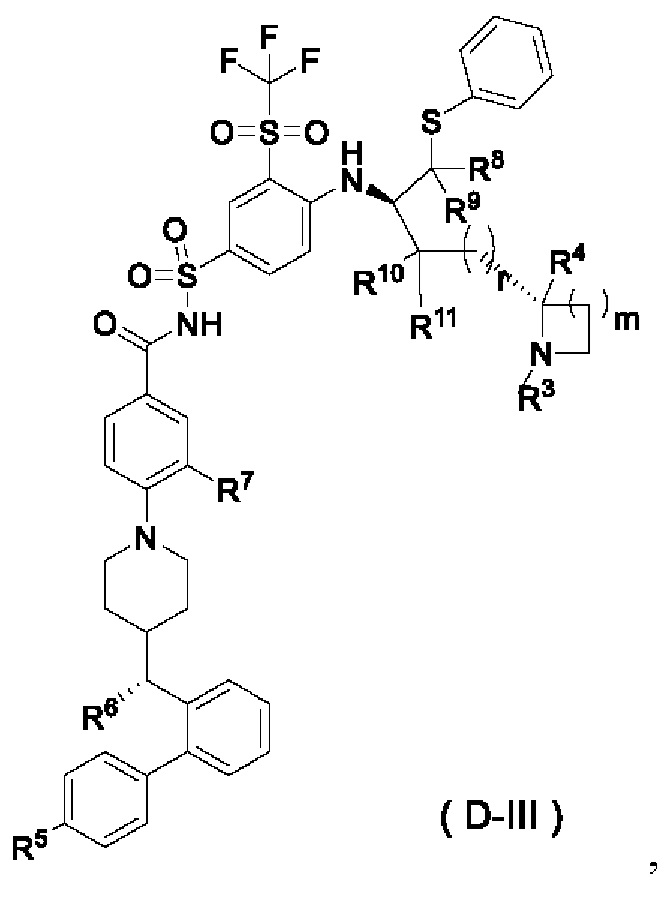
где m выбран из группы, состоящей из 0, 1, 2 и 3, и
r и R3-R11 являются такими, как определено в п. 1.
6. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, представляющее собой соединение общей формулы (D-IV) или его фармацевтически приемлемую соль:
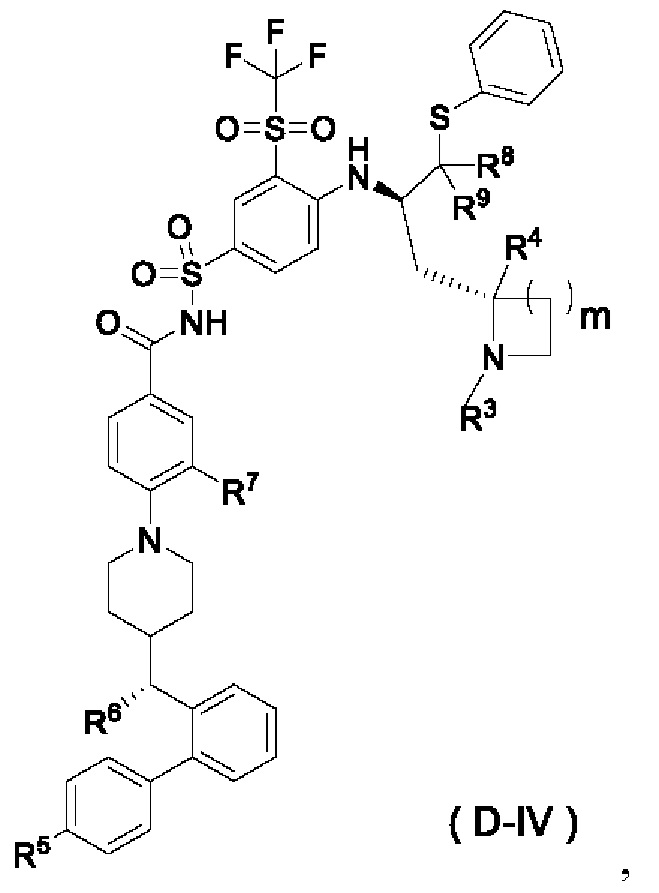
где
m выбран из группы, состоящей из 0, 1, 2 и 3, и
R3-R9 являются такими, как определено в п. 1.
7. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 3, где m равно 2.
8. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, где R5 представляет собой хлор.
9. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, где R5 представляет собой трифторметил.
10. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, где R3 представляет собой C1-6 гидроксиалкил.
11. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, представляющее собой соединение общей формулы (D-V) или его фармацевтически приемлемую соль:
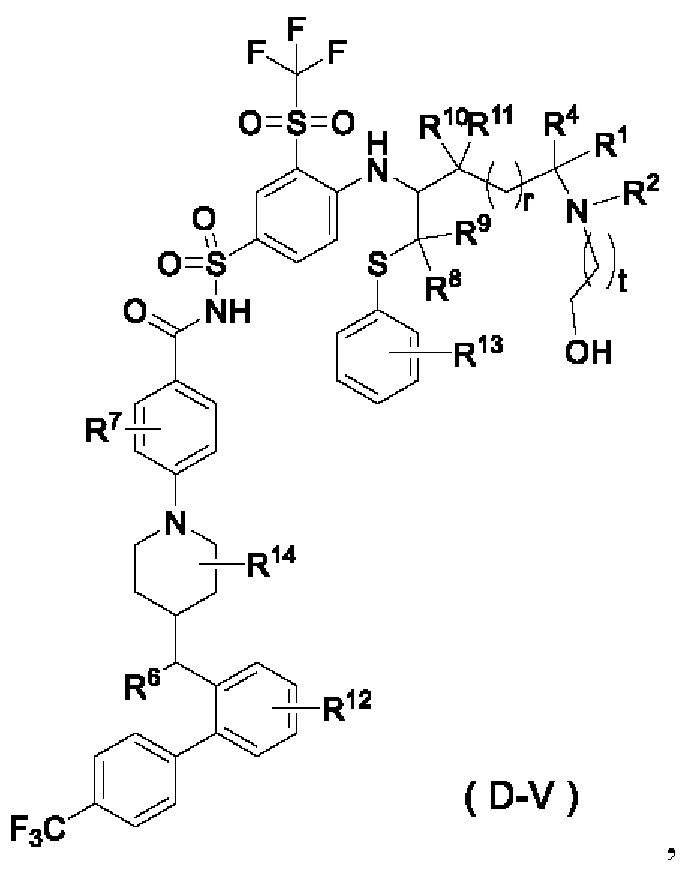
где t выбран из группы, состоящей из 0, 1, 2 и 3, и
r, R1, R2, R4 и R6-R14 являются такими, как определено в п. 1.
12. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, представляющее собой соединение общей формулы (D-VI) или его фармацевтически приемлемую соль:
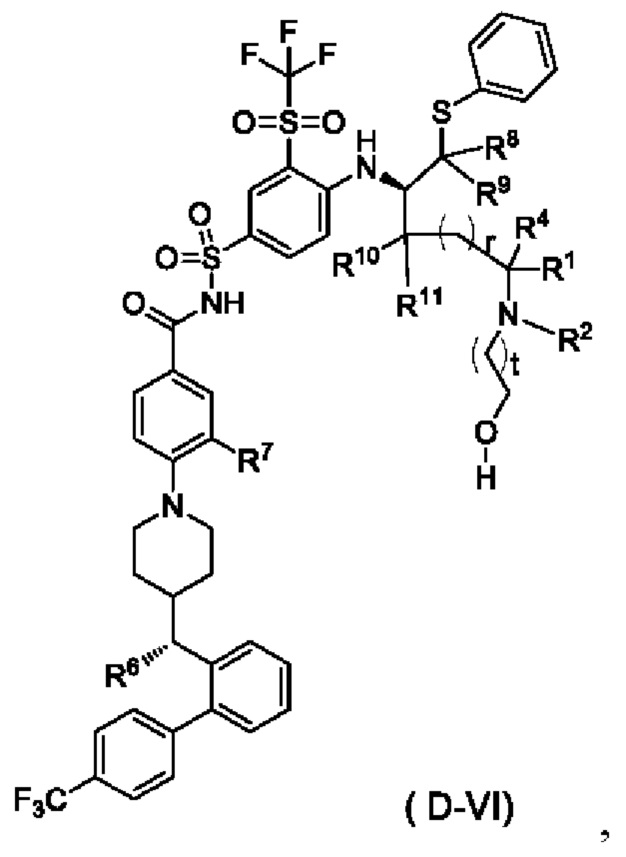
где t выбран из группы, состоящей из 0, 1, 2 и 3, и
r, R1, R2, R4 и R6-R11 являются такими, как определено в п. 1.
13. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 12, представляющее собой соединение общей формулы (D-VI) или его фармацевтически приемлемую соль, где t равно 1.
14. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, представляющее собой соединение общей формулы (D-VII) или его фармацевтически приемлемую соль:
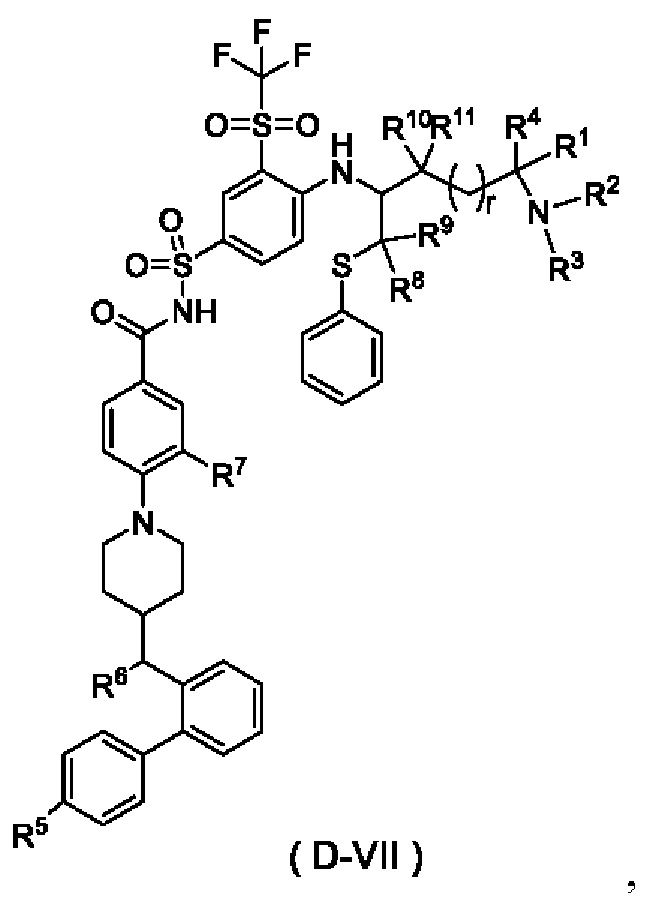
где r и R1-R11 являются такими, как определено в п. 1.
15. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, где R4 представляет собой водород.
16. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, где R6 представляет собой гидрокси.
17. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, где R7, R8, R9, R10 и R11 представляют собой водород.
18. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, где r равно 0 или 1.
19. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, где r равно 0.
20. Соединение общей формулы (D) или его фармацевтически приемлемая соль по п. 1, где соединение выбрано из группы, состоящей из:
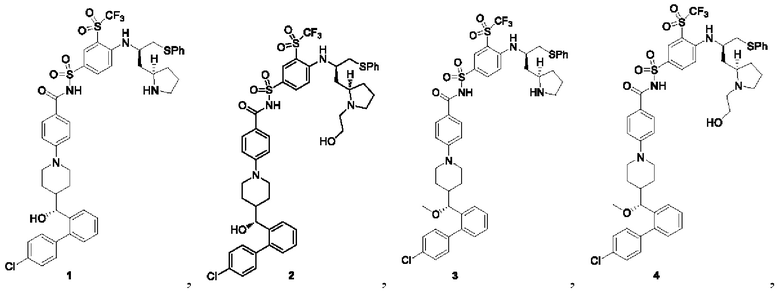
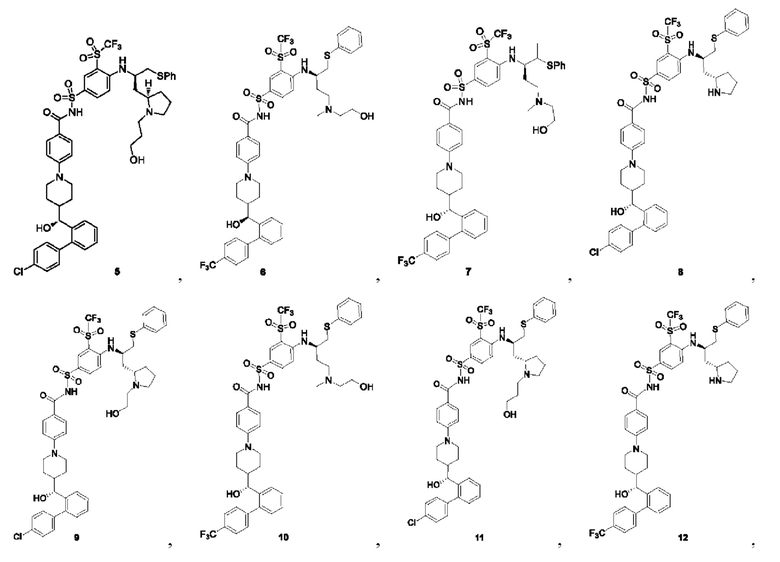
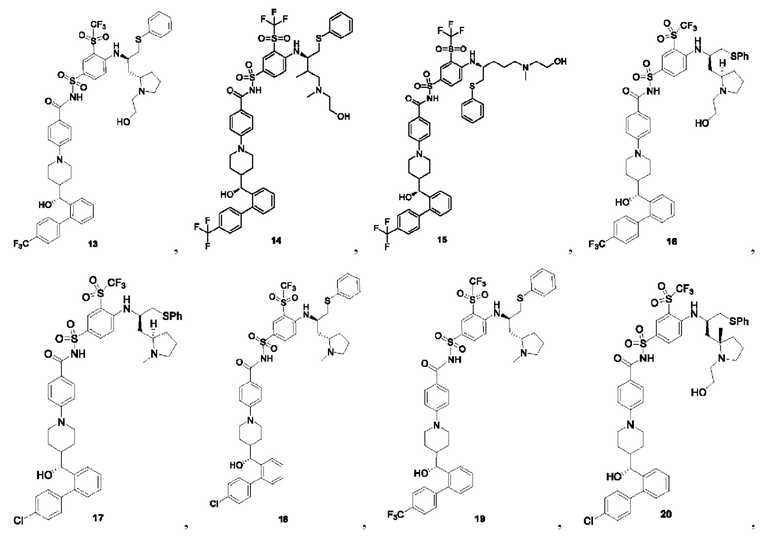
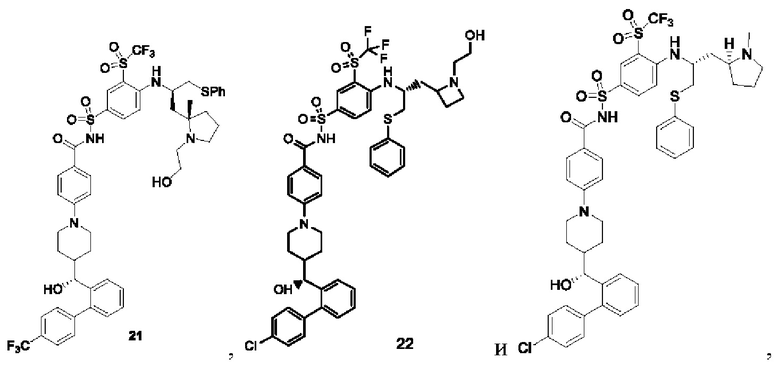
или их фармацевтически приемлемой соли.
21. Способ получения соединения общей формулы (D-III) или его фармацевтически приемлемой соли, включающий:
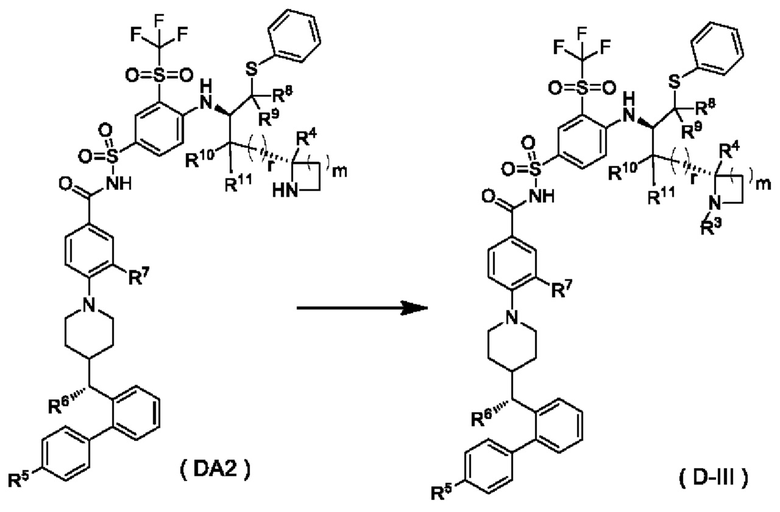
подвергание соединения общей формулы (DA2) или его фармацевтически приемлемой соли реакции замещения в щелочной среде с получением соединения общей формулы (D-III) или его фармацевтически приемлемой соли; причем реагент, используемый в щелочной среде, представляет собой триэтиламин,
где:
m выбран из группы, состоящей из 0, 1, 2 и 3, и
R3 представляет собой C1-6 гидроксиалкил; и
r и R4-R11 являются такими, как определено в п. 1.
22. Фармацевтическая композиция для лечения рака или опухоли, содержащая терапевтически эффективное количество соединения общей формулы (D) или его фармацевтически приемлемой соли по любому из пп. 1-20 и фармацевтически приемлемый носитель, разбавитель или эксципиент.
23. Соединение общей формулы (D) или его фармацевтически приемлемая соль по любому из пп. 1-20 или фармацевтическая композиция по п. 22 для применения в ингибировании Bcl-2 и/или Bcl-XL.
24. Соединение общей формулы (D) или его фармацевтически приемлемая соль по любому из пп. 1-20 или фармацевтическая композиция по п. 22 для применения в лечении рака или опухоли, где рак выбран из группы, состоящей из: меланомы, рака печени, рака почки, рака легкого, мелкоклеточного рака легкого, немелкоклеточного рака легкого, рака носоглотки, колоректального рака, рака толстой кишки, рака прямой кишки, рака поджелудочной железы, рака шейки матки, рака яичников, рака молочной железы, рака мочевого пузыря, рака предстательной железы, лейкоза, острого миелоидного лейкоза, хронического миелоидного лейкоза, хронического лимфоцитарного лейкоза, острого лимфоцитарного лейкоза, острого миелогенного лейкоза, мантийноклеточной лимфомы, диффузной В-крупноклеточной лимфомы, фолликулярной центральной лимфомы, неходжкинской лимфомы, Т-клеточной лимфомы, В-клеточной лимфомы, плоскоклеточного рака головы и шеи, рака шейки матки, рака щитовидной железы, лимфомы, саркомы, нейробластомы, опухоли головного мозга, миеломы, астроцитомы и глиомы.
| WO 2018154004 A9, 27.12.2018 | |||
| WO 2012017251 A1, 09.02.2012 | |||
| WO 2005049593 A2, 02.06.2005 | |||
| BCL-2-СЕЛЕКТИВНЫЕ АПОПТОЗ-ИНДУЦИРУЮЩИЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ РАКА И ИММУННЫХ ЗАБОЛЕВАНИЙ | 2010 |
|
RU2542994C2 |

