Настоящее изобретение относится к способам получения глицерофосфатидилхолина (ХАС). Глицерофосфатидилхолин является активным переносчиком холина и холиноподобных соединений в мозг человека и животных для лечения различных патологических состояний, в том числе сопровождаемых ишемией (инсульт).
Глицерофосфатидилхолин представляет собой соединений со структурой:
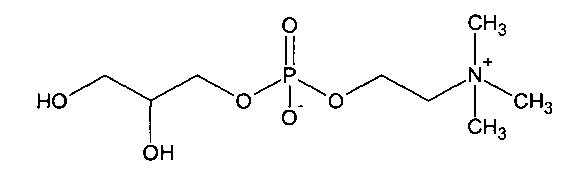
Из уровня техники, соответствующего настоящему изобретению, известен патент США US 5250719, в котором раскрыт способ получения глицерилфосфорилхолина (глицерофосфатидилхолина) путем деацилирования в метаноле деолеотированного (обезжиренного) соевого лецитина.
В данном документе суспензия деолеотированного соевого лецитина в безводном метаноле, содержащем CH3ONa, перемешивается при комнатной температуре около 3 часов. Смесь фильтруется и остаток промывается метанолом. Фильтрат нейтрализуется (рН≈6) уксусной кислотой, концентрируется и отделяются жирные кислоты.
Метанольный раствор обесцвечивается с помощью активированного угля и загружается в смолу Amberlyst.RTM. 15, уравновешенную в метаноле. Смола промывается в метаноле до удаления примесей (контроль по ТСХ), затем смола промывается водой (около 1,2 л). Водный раствор дважды обрабатывается активированным углем (2 г). Водный раствор (содержащий около 44 г глицерофосфатидилхолина и глицерофосфатидилэтаноламина) выделяется сначала на смоле IR 93 (120 мл), в ОН форме уравновешенной в воде и затем на смоле IR 401 (120 мл в ОН форме) уравновешенной в воде и, наконец, на смоле IRC50 (40 мл, в кислой форме) уравновешенной в воде. Полученный раствор концентрируется и содержит 26 г чистого GPC (глицерилфосфорилхолина) (содержание воды 10.1% по результатам неводного титрования с помощью НСlO4 в уксусной кислоте 99.8%).
Путем кристаллизации в этаноле был получен кристаллический глицерилфосфорилхолин со следующими характеристиками:
Угол вращения [α]D=-2,92 (с=10% в воде).
Содержание фосфора Р%=12,18% (расчетное 12.15%).
Содержание азота N%=5,49% (расчетное 5.58%).
Недостатком существующего способа является тот факт, что способ работает только с сырьем с высоким содержанием лецитина (соевый лецитин), так как он содержит большее количество фосфатидилхолина и меньшее количество других производных (фосфатидилэтаноламина, фосфатидилсерина, фосфатидилинозитола).
Также недостатком является то, что в известном способе реакционную массу в метаноле пропускают сначала через колонку с сильнокислым катионитом, в котором происходит сорбция глицерофосфатидилхолина, затем колонку промывают метанолом от примесей и затем продукт (глицерофосфатидилхолин) эллюируют с колонки водой. В результате этой операции образуется большое количество сложно утилизируемых стоков, содержащих водно-метанольною смесь.
Достигаемыми техническими результатами настоящего способа являются расширение сырьевой базы для производства глицерофосфатидилхолина, снижение количества опасных и наносящих вред окружающей среде отходов производства, а также расширение арсенала способов получения кристаллического глицерофосфатидилхолина.
Заявленные технические результаты достигаются с помощью способа получения кристаллического глицерофосфатидилхолина, содержащего следующие этапы: этап синтеза глицерофосфатидилхолина из фосфатидилхолин содержащего сырья и получения водного раствора глицерофосфатидилхолина; этап очистки водного раствора глицерофосфатидилхолина, с использованием сильноосновного сорбента и с извлечением эллюата; и этап кристаллизации глицерофосфатидилхолина с отгонкой азеотропной смеси, на котором эллюат предварительно разбавляют вторым растворителем, в котором глицерофосфатидилхолин является нерастворимым или малорастворимым и который образует с водой азеотропную смесь, температура кипения такой азеотропной смеси ниже температуры кипения самого растворителя и образованная азеотропная смесь является двухфазной, количество растворителя выбирается так, что его количество в смеси с водой больше, чем его содержание в азеотропной точке полученной смеси, и далее осуществляют отгонку азеотропной смеси с последующим выпадением кристаллического глицерофосфатидилхолина.
Получение водного раствора глицерофосфатидилхолина и этап очистки водного раствора глицерофосфатидилхолина, с использованием сильноосновного сорбента с извлечением эллюата вместе обеспечивают повышение выхода глицерофосфатидилхолина и обеспечивают возможность использование сырья с низким содержанием фосфатидилхолина.
Так в способе по US 5250719, первой отчисткой в колонне является катионная очистка. Особенностью данной очистки тот факт, что целевое вещество, глицерофосфатидилхолин, осаждается на сорбенте и удерживается на нем, затем целевое вещество смывается с сорбента. Однако процесс осаждения на сорбенте чувствителен к исходной концентрации целевого вещества, т.е. снижение содержания фосфатидилхолина в исходном сырье и, соответственно, снижение содержания глицерофосфатидилхолина, существенно ухудшает параметры процесса катионной очистки. Кроме того, в данном случае, катионная очистка приводит к потерям целевого вещества, т.к. не все целевое вещество осаждается на сорбенте и часть проходит через колонну в составе исходной очищаемой смеси, а также, поскольку не весь объем целевого вещества эффективно удаляется с сорбента.
В способе согласно настоящему изобретению используется очистка водного раствора фосфатидилхолина на сильноосновном сорбенте. В данном случае фосфатидилхолин не взаимодействует с сорбентом, что устраняет известные недостатки. Также отпадает необходимость утилизации опасных и наносящих вред окружающей среде стоков, содержащих метанол.
Отсутствие многократной очистки смеси и использованием сорбентов, в соответствии с настоящим изобретением, компенсируется эффективным этапом кристаллизации глицерофосфатидилхолина с отгонкой смеси.
В известном способе проводят кристаллизацию глицерофосфатидилхолина в этиловом спирте. Глицерофосфатидилхолин-содержащая смесь растворяется в этиловом спирте, далее понижается температура раствора и начинается выпадение кристаллов глицерофосфатидилхолина. Общий состав данной системы является неизменным.
Очевидно, что в данном случае требуется достаточно высокая чистота глицерофосфатидилхолин-содержащей смеси, которая достигается многократной очисткой на различных сорбентах, которая ведет к описанным выше потерям целевого вещества, чувствительна к содержанию целевого вещества в исходной смеси и производит к образованию большого количества опасных и наносящих вред окружающей среде стоков.
Этап кристаллизации глицерофосфатидилхолина, согласно настоящему изобретению, проводится с отгонкой смеси. Сам глицерофосфатидилхолин является нелетучим и остается в кубовом остатке.
Для водного раствора глицерофосфатидилхолина выбирается растворитель, который образует с водой двухфазную азеотропную смесь, у которой температура кипения в точке азеотропа ниже температуры кипения растворителя. То есть формируют положительный гетероазеотроп. В результате формируется две фазы, в одной преимущественно содержание воды, в другой - второго растворителя. Глицерофосфатидилхолин распределен между фазами. Полярные примеси содержатся преимущественно в водной фазе, неполярные - в фазе второго растворителя.
Отличительной особенностью таких смесей является то, что при перегонке двухфазной жидкой смеси любого состава конденсат (пар) будет иметь постоянную концентрацию до тех пор, пока в кубе сохраняются обе жидкие фазы.
Таким образом, благодаря тому, что количество второго растворителя выбирается больше, чем в азеотропной смеси, и тому, что температура кипения азеотропа ниже температуры кипения второго растворителя, удается отогнать всю водную фазу смеси. Также вместе с водой и вторым растворителем удаляются примеси.
После удаления водной фазы фактически получается смесь с очень низким содержанием воды. Поскольку глицерофосфатидилхолин нерастворим или малорастворим во втором растворителе, происходит его кристаллизация в безводной форме.
Показатели чистоты и содержания воды полученного в результате экспериментов глицерофосфатидилхолина были намного выше показателей, требуемых в фармацевтике.
Применение двухфазной азеотропной смеси также положительно сказывается на следующем.
В любом производстве, для целей снижения ресурсоемкости и выбросов (стоков) требуется регенерация второго растворителя.
Для удаления водной фазы дистиллята, полученного на этапе кристаллизации с отгонкой, достаточно осуществить простую операцию расслаивания смеси с удалением одной из жидких фаз.
Таким образом, объем смеси для дальнейшей очистки второго растворителя (например, перегонки) будет меньше, что положительно сказывается на энергоемкости, количестве используемых реагентов и формируемых при очистке стоков.
Кроме того, применение двухфазной азеотропной смеси обеспечивает возможность использование растворителей, у которых азеотропная точка находится далеко от индивидуального растворителя.
Так, в случае истинной азеотропной смеси, для эффективного удаления воды требуется смесь с азеотропной точкой, соответствующей максимально низкому содержанию воды и максимально высокому содержанию растворителя.
Однако это означает, что требуется использовать растворитель в количестве, во много раз превышающем количество воды, содержащейся в растворе для кристаллизации.
Поскольку, согласно изобретению, имеется возможность отогнать водную фазу в любом случае, расположение азеотропной точки максимально близко к индивидуальному второму растворителю, не является критическим.
Это позволяет использовать такие растворители, как 1-бутанол (вода 44,50 : 1-бутанол 55,50), бутилацетат (27,10 : 72,90), 2-этилбутилацетат (52,40 : 47,60), толуол (20.20 : 79.80) и другие.
Таким образом, предоставляется возможность снизить потребление второго растворителя и, соответственно, снизить негативные последствия его применения для природы и человека.
Предпочтительно, этап кристаллизации с отгонкой азеотропной смеси проводят в, по меньшей мере, две стадии, при этом на каждой из стадий осуществляют разбавление вторым растворителем.
При этом на первой стадии осуществляют неполную отгонку азеотропной смеси с получением глицерофосфатидилхолина в гидратированной форме, а на второй стадии проводят отгонку азеотропной смеси до получения глицерофосфатидилхолина в кристаллической форме.
Предпочтительно, в качестве второго растворителя используют 1-бутанол, так как он показал хорошие результаты в экспериментах, наименее опасен для природы и человека и имеет хорошее значение содержания в азеотропной смеси.
В результате осуществления способа был получен кристаллический глицерофосфатидилхолин.
На его основе изготавливалась фармацевтическая композиция для лечения и\или профилактики нарушений, связанных ишемическими состояниями, в том числе с инсультом, содержащая в качестве активных компонентов глицерофосфатидилхолин и фармацевтически приемлемый носитель.
Указанная фармацевтическая композиция также выполнялась в пероральной форме.
В результате осуществления способа был получен кристаллический глицерофосфатидилхолин, содержащий следы или остатки вещества, в котором глицерофосфатидилхолин нерастворим или малорастворим, которое образовывает с водой азеотропную смесь, температура кипения такой азеотропной смеси ниже, чем температура кипения этого вещества, и образованная азеотропная смесь является двухфазной.
На его основе изготавливалась фармацевтическая композиция для лечения и\или профилактики нарушений, связанных ишемическими состояниями, в том числе с инсультом, содержащая в качестве активных компонентов глицерофосфатидилхолин и фармацевтически приемлемый носитель.
Указанная фармацевтическая композиция также выполнялась пероральной форме.
Далее приводится подробное описание конкретных вариантов осуществления изобретения.
Согласно настоящему изобретению осуществляют следующие этапы.
Этап 1 обезжиривания.
Фосфатидилхолин содержащее сырье, такое как соевый, подсолнечниковый и рапсовый лецитины, обезжиривают.
Для обезжиривания применяют многократную обработку ацетоном. Далее обезжиренный концентрат сушат.
Этап 2 синтеза.
Синтез глицерофосфатидилхолина осуществляют путем проведения реакции переэтерификации в любом алифатическом спирте под действием любых алкоголятов щелочных и/или щелочноземельных металлов.
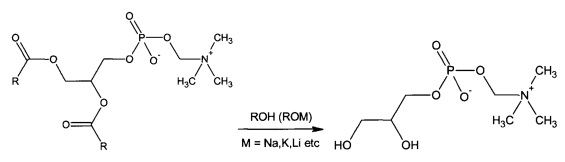
Полученную реакционную массу охлаждают и отделяют выделившийся осадок.
Фильтрат нейтрализуют, например, с помощью ортофосфорной кислоты.
Выпавший осадок фильтруют. Раствор, содержащий глицерофосфатидилхолин, упаривают под вакуумом при температуре не выше 37°С.
Этап 3 очистки в колонне.
К раствору добавляют воду, тем самым получая водный раствор глицерофосфатидилхолина.
В отличие от таких примесей, как глицерофосфатидилэтаноламин, глицерофосфатидилсерин, глицерофосфатидилинозитол, в водном растворе имеют отрицательный заряд, получаемый из фофатидилхолина глицерофосфатидилхолин имеет нейтральный заряд.
Водный раствор глицерофосфатидилхолина пропускают через колонну, заполненную сильноосновным сорбентом. Например, сильнощелочным анионитом в гидроксильной форме.
В результате все отрицательно заряженные примеси задерживаются сорбентом, а глицерофосфатидилхолин свободно проходит через сорбент и остается в водном растворе.
Затем колонну промывают водой для вымывания остатков глицерофосфатидилхолина.
Как видно из указанного выше, в настоящем способе отсутствуют какие-либо водно-метанольные стоки, что является бесспорным преимуществом.
Этап 4 кристаллизации с отгонкой.
К полученному на предыдущем этапе эллюату добавляют растворитель, который отвечает следующим требованиям:
- глицерофосфатидилхолин должен быть нерастворим или малорастворим в растворителе;
- растворитель должен образовывать с водой азеотропную смесь;
- температура кипения азеотропной смеси должна быть ниже, чем температура кипения растворителя;
- образованная азеотропная смесь должна быть двухфазной.
Количество растворителя выбирается так, что его количество в смеси с водой больше, чем его содержание в азеотропной точке полученной смеси.
Очевидно, что растворитель не должен вступать в химическую реакцию с глицерофосфатидилхолином.
К таким растворителям, в частности, относятся 1-бутанол, толуол, ксилол, гексанол, бутилацетат, амилацетат, хлорбензол, анизол и т.д.
После добавления растворителя образуется двухфазная жидкая система, в одном слое которой выше содержание добавленного растворителя, а во втором выше содержание воды, глицерофосфатидилхолин распределен между слоями. При этом содержание глицерофосфатидилхолина выше во втором слое.
Далее начинают отгонку азеотропа.
Как известно, состав пара таких систем всегда равен составу азеотропа и при перегонке такой двухфазной жидкой смеси любого состава конденсат (пар) будет иметь постоянную концентрацию до тех пор, пока в кубе сохраняются обе жидкие фазы. Таким образом, поскольку содержание растворителя в смеси больше, чем в азеотропной точке, при отгонке азеотропа удаляется вся вода и глицерофосфатидилхолин остается в растворителе и кристаллизуется в безводной форме.
Полученный глицерофосфатидилхолин промывают, например, ацетоном для удаления остатков растворителя.
Благодаря использованию сильноосновного сорбента и нового способа кристаллизации становится возможным использовать сырье с низким содержанием фосфатидилхолина, поскольку не требуется осаждение на сорбенте, что ведет к потерям вещества и применяется более эффективный способ кристаллизации.
Для получения еще более чистого глицерофосфатидилхолина, этап кристаллизации проводят в две стадии.
Сначала удаляется азеотроп до содержания воды в смеси 1-1,5% и получают глицерофосфатидилхолин в гидратированной форме (паста).
Глицерофосфатидилхолин в гидратированной форме промывают, например, ацетоном для удаления остатков растворителя.
Далее в полученный глицерофосфатидилхолин снова добавляют растворитель и повторяют процесс отгонки и кристаллизации.
Предпочтительно, второй раз отгонку и кристаллизацию проводить под вакуумом. Это обеспечивает получение менее окрашенного продукта (более чистого продукта).
Глицерофосфатидилхолин в кристаллической безводной форме промывают, например, ацетоном для удаления остатков растворителя и сушат под вакуумом.
В данном случае получают более чистый глицерофосфатидилхолин с более низким содержанием воды.
Как указывается выше, для обезжиривания сырья и для промывки глицерофосфатидилхолина применяют ацетон. Однако очевидно, что для этих целей может использоваться любое вещество, обладающее следующими характеристиками: растворять жиры и спирты, не растворять глицерофосфатидилхолин и фосфатидилхолин. Также очевидно, такое вещество не должно вступать в реакцию с компонентами очищаемой смеси. К таким веществам можно отнести, в частности, диэтиловый эфир, метиэтилкетон и метилтретбутиловый эфир. В данном случае, ацетон выбран из практических соображений: его доступности, степени опасности, экологичности и т.д.
Предварительное обезжиривание сырья обеспечивает снижение примесей, образующихся на этапе синтеза глицерофосфатидилхолина, что положительно сказывается дальнейших этапах очистки.
Промежуточная (после первой стадии кристаллизации с отгонкой азеотропной смеси) и финальная промывка (после второй стадии кристаллизации азеотропной смеси) обеспечивают более полное удаления оставшейся воды и загрязненного растворителя из глицерофосфатидилхолина.
Пример 1.
Этап 1 обезжиривания.
100 кг соевого лецитина с содержанием фосфатидилхолина 13% обрабатывают 3 раза по 100 л ацетона для обезжиривания. Обезжиренный концентрат сушат и получают 62 кг обезжиренного концентрата с содержанием фосфатидилхолина 19%.
Этап 2 синтеза.
62 кг обезжиренного концентрата с содержанием фосфатидилхолина 19% растворяют в 40 л безводного метанола, добавляют 2 кг метилата натрия и перемешивают при 30°С 6 часов.
Полученную реакционную массу охлаждают до 0°С, отделяют выделившийся осадок. Фильтрат нейтрализуют, добавляя 1,21 кг ортофосфорной кислоты. Выпавший осадок фильтруют и упаривают под вакуумом при температуре не выше 37°С.
Этап 3 очистки в колонне.
К кубовому остатку добавляют 14 литров воды и полученный раствор пропускают через колонну высотой 80 см и диаметром 10 см, заполненную сильнощелочным анионитом в гидроксильной форме, затем колонну промывают 1 литром воды.
Этап 4 кристаллизации с отгонкой.
К эллюату добавляют 50 л 1-бутанола и отгоняют 30 литров азеотропа бутанола с водой до остаточного содержания воды в растворе 1-1,5%. Выпавший глицерофосфатидилхолин в гидратированной форме фильтруют, промывают два раза по 30 л ацетона.
Полученную пасту растворяют в 30 л 1-бутанола, и отгоняют под вакуумом 10 литров азеотропа бутанола с водой. Кубовый остаток охлаждают до 10°С, перемешивают 2 часа, фильтруют выпавший безводный глицерофосфатидилхолин, промывают его 2 раза по 10 литров ацетона, сушат под вакуумом.
В результате был получен чистый кристаллический глицерофосфатидилхолин с содержанием воды 0,18% и содержанием основного вещества 98,6% (в пересчете на сухое, свободное от органических растворителей вещество).
Пример 2.
Этап 1 обезжиривания.
150 кг подсолнечникового лецитина с содержанием фосфатидилхолина 9% обрабатывают 3 раза по 100 л ацетона для обезжиривания. Обезжиренный концентрат сушат и получают 91 кг обезжиренного концентрата с содержанием фосфатидилхолина 13%.
Этап 2 синтеза.
91 кг обезжиренного концентрата с содержанием фосфатидилхолина 13% растворяют в 60 л безводного метанола, добавляют 2.1 кг метилата натрия и перемешивают при 30°С 6 часов.
Полученную реакционную массу охлаждают до 0°С, отделяют выделившийся осадок. Фильтрат нейтрализуют, добавляя 1,21 кг ортофосфорной кислоты. Выпавший осадок фильтруют и упаривают под вакуумом при температуре не выше 37°С.
Этап 3 очистки в колонне.
К кубовому остатку добавляют 16 литров воды и полученный раствор пропускают через колонну высотой 100 см и диаметром 12 см, заполненную сильнощелочным анионитом в гидроксильной форме, затем колонну промывают 1,5 литром воды.
Этап 4 кристаллизации с отгонкой.
К эллюату добавляют 64 л 1-бутанола и отгоняют 34 литров азеотропа бутанола с водой до остаточного содержания воды в растворе 1-1,5%. Выпавший глицерофосфатидилхолин в гидратированной форме фильтруют, промывают два раза по 35 л ацетона.
Полученную пасту растворяют в 36 л 1-бутанола, и отгоняют под вакуумом 12 литров азеотропа бутанола с водой. Кубовый остаток охлаждают до 10°С, перемешивают 2 часа, фильтруют выпавший безводный глицерофосфатидилхолин, промывают его 2 раза по 12 литров ацетона, сушат под вакуумом.
В результате был получен чистый кристаллический глицерофосфатидилхолин с содержанием воды 0,15% и содержанием основного вещества 98,3% (в пересчете на сухое, свободное от органических растворителей вещество).
Пример 3.
Этап 1 обезжиривания
230 кг подсолнечникового лецитина с содержанием фосфатидилхолина 8% обрабатывают 3 раза по 150 л ацетона для обезжиривания. Обезжиренный концентрат сушат и получают 91 кг обезжиренного концентрата с содержанием фосфатидилхолина 12%.
Этап 2 синтез
140 кг обезжиренного концентрата с содержанием фосфатидилхолина 12% растворяют в 90 л безводного метанола, добавляют 3.6 кг метилата натрия и перемешивают при 35°С 5 часов.
Полученную реакционную массу охлаждают до 0°С, отделяют выделившийся осадок. Фильтрат нейтрализуют, добавляя 1,85 кг ортофосфорной кислоты. Выпавший осадок фильтруют и упаривают под вакуумом при температуре не выше 37°С.
Этап 3 очистка в колонке
К кубовому остатку добавляют 25 литров воды и полученный раствор пропускают через колонну высотой 150 см и диаметром 14 см, заполненную сильнощелочным анионитом в гидроксильной форме, затем колонну промывают 2.5 литром воды.
Этап 4 кристаллизации с отгонкой
К эллюату добавляют 100 л бутанола и отгоняют 51 литр азеотропа бутанола с водой до остаточного содержания воды в растворе 1-1,5%. Выпавший глицерофосфатидилхолин в гидратированной форме фильтруют, промывают два раза по 50 л ацетона.
В результате был получен чистый кристаллический глицерофосфатидилхолин с содержанием воды 4,2% и содержанием основного вещества 97,6% (в пересчете на сухое, свободное от органических растворителей вещество).
Также были проведены эксперименты по получению глицерофосфатидилхолина из других источников сырья, например, фосфатидный концентрат, полученный из рапсового или подсолнечного масла, с содержанием лецитина до 4%, а также с использованием других растворителей, например, бутилацетат или толуол. Во всех случаях был получен чистый кристаллический глицерофосфатидилхолин с содержанием воды не более 0,5% и содержанием основного вещества не менее 97,0%.
При этом во всех пробах кристаллического глицерофосфатидилхолина, полученного способом, соответствующем настоящему изобретению обнаруживались следы или остатки растворителя, а именно, вещества в котором глицерофосфатидилхолин нерастворим, которое образовывает с водой азеотропную смесь, температура кипения такой азеотропной смеси ниже, чем температура кипения этого вещества, и образованная азеотропная смесь является двухфазной.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ БЕТУЛИНА | 2013 |
|
RU2524778C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5(6)-АМИНО-2(4'-АМИНОФЕНИЛ)-БЕНЗИМИДАЗОЛА | 2005 |
|
RU2283307C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАТРИЕВОЙ СОЛИ 6-[3-(2-ХЛОРФЕНИЛ)-5-МЕТИЛИЗОКСАЗОЛ-4-КАРБАМИНО]-ПЕНИЦИЛЛАНОВОЙ КИСЛОТЫ | 2001 |
|
RU2201419C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2',4,4'-ТРИНИТРОБЕНЗАНИЛИДА | 2006 |
|
RU2348612C2 |
| СПОСОБ ВЫДЕЛЕНИЯ ТРИЭТИЛЕНДИАМИНА | 1994 |
|
RU2087474C1 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРЗАМЕЩЕННЫХ 4,4-ДИАМИНОБЕНЗАНИЛИДОВ | 2007 |
|
RU2385861C2 |
| СПОСОБ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОГО ДИМЕТИЛДИТИОКАРБАМАТА НАТРИЯ | 2003 |
|
RU2248968C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАТРИЕВОЙ СОЛИ 6-(3-ФЕНИЛ-5-МЕТИЛИЗОКСАЗОЛ-4-КАРБАМИНО)-ПЕНИЦИЛЛАНОВОЙ КИСЛОТЫ | 2002 |
|
RU2221801C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2', 4, 4'-ТРИНИТРОБЕНЗАНИЛИДА | 2007 |
|
RU2394810C2 |
| СПОСОБ ПОЛУЧЕНИЯ НАТРИЯ 10-МЕТИЛЕНКАРБОКСИЛАТ-9-АКРИДОНА ИЛИ 10-МЕТИЛЕНКАРБОКСИ-9-АКРИДОНА ИЗ АКРИДОНА | 1998 |
|
RU2162843C2 |
Изобретение относится к области органической химии и раскрывает способ получения кристаллического глицерофосфатидилхолина. Способ характеризуется тем, что содержит следующие этапы: получение водного раствора глицерофосфатидилхолина из фосфатидилхолинсодержащего сырья с последующей его очисткой с использованием сильноосновного сорбента и с извлечением эллюата, этап кристаллизации глицерофосфатидилхолина с отгонкой азеотропной смеси, на котором эллюат предварительно разбавляют вторым растворителем, в котором глицерофосфатидилхолин является нерастворимым или малорастворимым и который образует с водой азеотропную смесь, температура кипения такой азеотропной смеси ниже температуры кипения самого растворителя и образованная азеотропная смесь является двухфазной, количество растворителя выбирается так, что его количество в смеси с водой больше, чем его содержание в азеотропной точке полученной смеси, далее осуществляют отгонку азеотропной смеси с последующим выпадением кристаллического глицерофосфатидилхолина. Изобретение обеспечивает расширение арсенала способов получения кристаллического глицерофосфатидилхолина. расширение сырьевой базы, снижение количества опасных и наносящих вред окружающей среде отходов при получении глицерофосфатидилхолина. 3 з.п. ф-лы, 3 пр.
1. Способ получения кристаллического глицерофосфатидилхолина, содержащий следующие этапы:
этап синтеза глицерофосфатидилхолина из фосфатидилхолинсодержащего сырья и получения водного раствора глицерофосфатидилхолина;
этап очистки водного раствора глицерофосфатидилхолина с использованием сильноосновного сорбента и с извлечением эллюата; и
этап кристаллизации глицерофосфатидилхолина с отгонкой азеотропной смеси, на котором эллюат предварительно разбавляют вторым растворителем, в котором глицерофосфатидилхолин является нерастворимым или малорастворимым и который образует с водой азеотропную смесь, температура кипения такой азеотропной смеси ниже температуры кипения самого растворителя и образованная азеотропная смесь является двухфазной, количество растворителя выбирается так, что его количество в смеси с водой больше, чем его содержание в азеотропной точке полученной смеси, и далее осуществляют отгонку азеотропной смеси с последующим выпадением кристаллического глицерофосфатидилхолина.
2. Способ по п. 1, отличающийся тем, что этап кристаллизации с отгонкой азеотропной смеси проводят в по меньшей мере две стадии, при этом на каждой из стадий осуществляют разбавление вторым растворителем.
3. Способ по п. 2, отличающийся тем, что на первой стадии осуществляют неполную отгонку азеотропной смеси с получением глицерофосфатидилхолина в гидратированной форме, а на второй стадии проводят отгонку азеотропной смеси до получения глицерофосфатидилхолина в кристаллической форме.
4. Способ по любому из пп. 1-3, отличающийся тем, что в качестве второго растворителя используют 1-бутанол.
| US 5250719 A, 05.10.1993 | |||
| Устройство для наблюдения влажных образцов при помощи растрового электронного микроскопа | 1976 |
|
SU575717A1 |
| RU 2008134143 А (KOZHOKA TG), 27.02.2010 | |||
| US 8658401, 25.02.2014 | |||
| ИПАТОВА О.М | |||
| Фосфоглив: механизм действия и применение в клинике | |||
| Издательство Москва | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Устройство для обжатия канатов | 1974 |
|
SU486100A1 |
| Регистр Лекарственных Средств России (РЛС) | |||
| Энциклопедия лекарственных средств | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |

