Предлагаемое изобретение относится к химии акридона, в частности, к усовершенствованию способа синтеза натрия 10-метиленкарбоксилат-9-акридона и 10-метиленкарбокси-9-акридона, общей формулы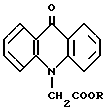
где R = Na, (I), H (Ia),
являющихся полупродуктами для синтеза фармацевтических препаратов (I, Ia) или субстанциями (I) для приготовления лекарственных форм, соль (I) применяется в медицинской и ветеринарной практике в качестве противовирусного средства: индуктор интерферона обладает противовирусным действием в отношении как ДНК-, так и РНК-содержащих вирусов, что обусловлено его способностью резко стимулировать образование интерферона. Предложен в качестве противочумного средства для животных (Патент 2031650, РФ, 1995, Szule B. И at al.//Arch. Immun. Therapie Exper. 1985. Vol. 33. P. 287-297).
Все известные методы получения продуктов (I, Ia) основаны на щелочном гидролизе эфиров 10-метиленкарбокси-9-акридона (II) при их обработке едким натром или алкоголятом натрия в спиртах и последующем выделении соли (I) как целевого продукта или подкислении ее раствора с выделением кислоты (Ia). Поскольку последняя стадия - выделение кислоты (Ia) достаточно проста, то определяющими этапами общей цепи являются получение эфира (II) и его гидролиз с образованием соли (I).
Эфир (II), обычно, синтезируют при алкилировании 10-метоксиакридина [Lyakhov S. A. at al.//Pharmazie. 1994. Vol. 45. N 5. P. 367-3683] либо щелочной соли акридона (III) эфирами хлор- или бромуксусной кислот, используя в случае акридона общую схему [Пат. 3681360. США, 1972, Postescu J. et al. // J. Prakt. Chem. 1977. Bd. 319. N 2. S. 347-352; Патент 2033413. РФ, 1995, Inglot F.D. et al. //Arch. Immun. Therapie Exper. 1985. Vol. 33. P. 275-285] . Кислоту (Ia) получают при обработке раствора соли (I) минеральной кислотой [Postescu J. // J. Prakt. Chem. 1977. Bd. 319. N 2. S. 347-352. Патент 2029764. РФ. 1995].
Способ получения продукта (I) через 9-метоксиакридин состоит в нагревании (120oC) последнего с метилбромацетатом в нитробензоле. В этом варианте вместо планировавшегося замещения по положению 10 с образованием O-метилированного аналога эфира (II) имело место N9-алкилирование по етероатому цикла с образованием промежуточной четвертичной соли, которая при щелочном гидролизе метилатом натрия в метаноле дала целевую соль (I) /60%/ [Lyakhov S.A. и др. // Pharmazie. 1994. Vol. 45. N 5. P. 367-3683], этот способ не технологичен ввиду применения малодоступного 9-этоксиакридина и использования нитробензола, метилата натрия и метанола.
Более перспективны в плане технологии методы, основанные на алкилировании акридона (III) этилгалогенацетатом при нагревании в растворителе в присутствии оснований.
Способ получения целевой соли (I) из эфира (II) описан в [Postescu J. et al. // J. Prakt. Chem. 1977. Bd. 319. N 2. S. 347-352; Inglot F.D. и др. // Arch. Immun. Therapie Exper. 1985. Vol. 33. P. 275-285. Патент 2029764. РФ. 1995] . В двух последних работах предыдущая стадия - алкилирование, не рассматривается вовсе, хотя, как показывает опыт, именно качество продукта этой стадии предопределяет способ его преобразования в целевую соль или кислоту и чистоту последних. Кислоту (Ia) выделяют при подкислении раствора чистой соли (I) минеральной кислотой с выходом >90%.
Методики синтеза соли (I) на базе эфира (II) по [// J. Prakt. Chem. 1977. Bd. 319. N 2. S. 347-352 7] близки: горячий раствор эфира (II) в абсолютном спирте обрабатывают избытком концентрированного водного раствора едкого натра, выход технического вещества 85%, перекристаллизованного из смеси ДМФА/метанол - 70%.
Недостатки этого способа:
1) использование абсолютированного спирта в большом количестве /на 1 г соли (I) до 35 мл/.
2) необходимость использовать очищенный эфир, свободный от акридона, так как последний в принятых условиях щелочного гидролиза перейдет в целевые продукты (I, Ia) практически нацело;
3) невысокий выход очищенного вещества.
Способ [Патент 2029764. РФ. 1995] применяет щелочной гидролиз эфира (II) при кипячении последнего в водной щелочи, что проще и экономичнее, но при этом авторы используют катализаторы - пористые алюмосиликаты (цеолиты, молекулярные сита с размером пор 3-9 A); целевую соль (I) после упаривания профильтрованного раствора высаживает ацетоном, выход >90%. 10-Метиленкарбокси-9-акридон (Ia) в случае необходимости выделяют при подкислении неупаренного раствора соли (I) разбавленной соляной кислотой.
Недостатки этого варианта:
1) применение дополнительно катализаторов - молекулярных сит с определенным размером пор;
2) необходимость при масштабном синтезе регенерации или утилизации катализатора;
3) синтез эфира (II) в патенте не рассматривается, но предполагается применение продукта только высокого качества (т.пл. 176-178oC); превращение технического продукта, содержание акридона в котором существенно выше 2% по этому варианту не предлагается; отметим, что даже в случае положительного результата возврат непрореагировавшего акридона в цикл окажется проблематичным и, как минимум, потребуется разработка методики отделения его от катализатора. Стадия, предшествующая целевой - синтез эфира (II), рассмотрена теми же авторами отдельно в патенте [Патент 2033413. РФ. 1995].
Здесь сложные эфиры 10-метиленкарбокси-9-акридона получали при кипячении акридона с эфирами монохлоруксусной кислоты в среде диметилацетамида (ДМАА) в присутствии катализатора межфазного переноса (четвертичные аммониевые соли) и при применении лишь 0.5-4 экв безводного карбоната щелочного металла. Предварительно смесь акридона, карбоната и катализатора кипятили (165oC) с последующей отгонкой части растворителя, после чего проводили дозировку этилхлорацетата и давали выдержку при кипячении; реакционную массу, охлажденную до 60-70oC, выливали в воду со льдом, выпавший целевой продукт промывали на фильтре этанолом и гексаном и сушили при 120oC; примесь акридона в техническом эфире, по данным патента, была не выше 2%, он не требовал, по мнению авторов, дополнительной очистки, а его выход составлял 82-88%.
К недостаткам этого способа относятся:
1) введение предварительного кипячения основания и катализатора в растворителе и отгонка 1/3 ДМАА; это дополнительная операция, что нетехнологично, при этом сохранность катализатора уже на этом этапе сомнительна;
2) использование очень жестких условий реакции, в частности высокой температуры синтеза 165oC, что требует лишних энергозатрат, но главное - способствует разложению малостабильных катализатора и этилхлорацетата и ведет к частичному осмолению массы. Нужно отметить, что в других случаях алкилирование акридона иными алкилирующими агентами при использовании карбоната калия проводилось в относительно мягких условиях за счет введения четвертичных аммониевых солей; в данном же случае при введении катализатора применены неоправданно жесткие условия алкилирования - ДМФА сменен на более высоко кипящий ДМАА, и реакция проводится при кипении последнего (165oC);
3) применение очень высокой для технологических целей концентрации реагентов (1.43 г реагентов на 1 мл растворителя на старте, более 0.7 г целевого эфира на 1 мл ДМАА плюс не растворимые в ДММФА неорганические соли в конце синтеза); при этом: а/ не достигается полное растворение акридона (или его соли) в малом количестве растворителя на старте, а это важно для более быстрого и целевого расхода, малостабильного при столь высокой температуре этилхлорацетата; б/ реакционная масса трудно перемешивается и очень быстро густеет при охлаждении, что затрудняет ее слив даже при 70oC; это ведет к значительным механическим потерям в конце синтеза;
4) невысокое качество эфира (II); а/ продукт очень темный, что обусловлено высокой концентрацией реагентов и чрезвычайно высокой (165oC, кипение ДМАА) температурой реакции при том, что применяются термически недостаточно стабильные катализатор и этилхлорацетат; полученный продукт требует неоднократной чистки с применением активированного угля (а.у.), что ведет к дополнительным и большим потерям; промывка продукта на фильтре этанолом, как это предлагается в патенте, не дает существенного повышения качества, но ведет к заметному падению выхода; б/ эфир включает заметное количество непрореагировавшего акридона - при выходе технического продукта 85% он содержит более 10% акридона (1. ТСХ; 2. гидролиз щелочью в воде - 58% соли (I). УФ спектроскопия); выход от акридона в пересчете на целевое вещество при этом не достигает и 50%;
5) количество соды, введенное в синтез эфира (II) в конкретном примере, недостаточно даже для связывания галогена;
6) синтез целевых продуктов (I, Ia) в патенте не приводится.
Прототипом предлагаемого решения является синтез натриевой соли 10-метилленкарбокси-9-акридона (I) и 10-метиленкарбокси-9-акридона (Ia) из акридона (III) через эфир (II) [. // J. Prakt. Chem. 1977. Bd. 319. N 2. S. 347-352].
В этом варианте синтез соли (I) проводился при алкилировании акридона (III) этилхлорацетатом при кипячении реагентов в ДМФА и применении KOH в качестве основания с последующим переводом полученного эфира (II) в целевой продукт (I) при обработке его теплого раствора в абсолютном спирте конц. раствором щелочи; соль (I) выделялась, растворялась в воде, а ее раствор при подкислении давал кислоту (Ia); при этом при синтезе эфира (II):
1) предварительно готовилась K-соль акридона в ДМФА (10-минутное кипячение в ДМФА акридона с 2.5-кратным количеством KOH с последующей отгонкой воды в виде ее азеотропа с ДМФА);
2) к охлажденной суспензии соли дозировался этилхлорацетат, реакционная масса нагревалась до кипения, кипятилась 20 мин, выливалась в воду со льдом, продукт отфильтровывался;
3) после сушки осадок дважды экстрагировали хлороформом, после испарения которого получали технический эфир с выходом 79% (качество продукта не оценено); скорее всего именно введение этой трудоемкой операции позволяло отделить целевой препарат от большей части непрореагировавшего акридона и получать эфир (II) после кристаллизации из спирта с очень высокой т.пл. (182.5-183oC); на стадии синтеза целевого продукта (I): полученный эфир (II) растворялся в абсолютном спирте и обрабатывался концентрированным раствором щелочи; для получения продукта (Ia) соль (I) растворяли в воде и подкисляли 5%-ным HCl.
К недостаткам способа следует отнести:
1. использование KOH на стадии алкилирования, что ведет к образованию воды при синтезе K-соли акридона, удаление которой необходимо и выполняется путем отгонки в виде ее азеотропа с ДМФА; контролируемая отгонка части растворителя - прием, существенно усложняющий технологию; в синтезе применяется 2.5 кратное количество щелочи - ее большой избыток, оставаясь в реакционной массе, неизбежно вызывает побочные реакции: превращение этилхлорацетата и эфира (II) в соответствующие соли (RCOOOEt + KOJ ---> RCOOK + EtOH), что загрязняет продукт этой стадии /эфир (II)/ и снижает его выход; при апробации варианта отмечено вспенивание реакционной массы с угрозой выброса, развитие побочных процессов, а примеси, обусловленные применением KOH, усложняли выделение и очистку эфира (II), что резко снижало его выход (около 50%);
2. заметную примесь акридона в техническом эфире (II) - до 30%;
3. введение дополнительной операции - экстракции технического продукта хлороформом с последующим его удалением и кристаллизацией остатка;
4. необходимость применения только очищенного эфира (II) при переводе его в целевые соль (I) и далее - кислоту (Ia);
5. использование абсолютного спирта при высоком модуле на операции гидролиза эфира (II) в целевую соль (I).
Все это делает технологию достаточно сложной, трудоемкой и малоэффективной.
Таким образом,
1. полученный по прототипу при алкилировании акридона технический эфир (II) мало пригоден для перевода его в целевые продукты (I, Ia) по методу, предложенному в прототипе; поэтому требуется его обработка хлороформом, а затем неоднократная кристаллизация полученного после отгонки хлороформа продукта с соответствующими потерями в выходе, большие затраты растворителей и их регенерация;
2. предложенный вариант щелочного гидролиза в спирте требует больших затрат абсолютного спирта, а следовательно, его регенерации, но, главное, он не решает проблемы применения технического продукта (II) для получения целевых веществ (I, Ia).
Как следует из вышеизложенного, определяющей стадией многоэтапного синтеза соединений (I, Ia) из акридона (III) является получение эфира (II), качество которого предопределяет выход и чистоту фармакопейных препаратов (I, Ia). При кажущейся простоте схемы применения общепринятого для NH-кислот варианта - алкилирование акридона галогенацетатом в присутствии основания в апротонной среде и последующем щелочном гидролизе полученного эфира - все приведенные методики имеют недостатки: они не всегда технологичны и их результаты часто трудно воспроизводимы (в первую очередь, это касается качества промежуточного продукта (II)).
Задачей, на решение которой направлено предлагаемое изобретение, является устранение указанных недостатков и разработка технологически приемлемого способа получения натриевой соли 10-метиленкарбокси-9-акридона (I) или 10-метиленкарбокси-9-акридона (Ia) из акридона (III), позволяющего синтезировать целевые препараты (I, Ia) фармакопейной чистоты с высоким выходом.
Задача может быть решена при:
1) доработке стадии алкилирования акридона для обеспечения высокого выхода (>90%) технического эфира (II) с небольшим содержанием акридона (не >10%);
2) разработке способа перевода технического эфира (II) в натриевую соль 10-метиленкарбокси-9-акридона, обеспечивающего удаление остаточного акридона из нее и синтез целевых препаратов (I, Ia) высокого качества.
Сущность решения состоит в том, что
на стадии алкилирования акридона этилхлорацетатом в ДМФА предлагается применять карбонаты калия или натрия, проводя процесс при соотношении акридон-ДМФА и температуре дозировки этилхлорацетата, обеспечивающих полную растворимость акридона (акридон-ДМФА в соотношении 1:10-12 при дозировке этилхлорацетата при температуре 100-130oC), и используя реагенты в соотношении - акридон:карбонат:этилхлорацетат = 1:2-3:2-3 (моль) с последующей выдержкой при температуре ниже температуры кипения ДМФА (130-140oC) и выделением эфира 10-метиленкарбокси-9-акридона обычным способом;
на стадии щелочного гидролиза полученного эфира 10-метиленкарбокси-9-акридона (II) предлагается обрабатывать технический эфир водной щелочью и отделять образовавшийся раствор целевой натриевой соли от непрореагировавшего акридона, например, отфильтровывать последний; фильтрат - раствор целевого продукта, дополнительно обработать сорбентом, например, активированным углем, с последующей фильтрацией от него; фильтрат предлагается использовать для приготовления водных растворов целевого продукта требуемой концентрации (а именно в таком виде он используется как субстанция в лекарственном препарате [1]), а на его основе:
а/ получать натрий 10-метиленкарбоксилат-9-акридона в кристаллическом виде при добавлении спиртов (этилового, изопропилового) к его концентрированному водному раствору;
б/ выделять из него 10-метиленкарбоксилат-9-акридон при подкислении минеральной кислотой.
В результате предлагаемого решения при алкилировании акридона этилхлорацетатом при нагревании в системе ДМФА-карбонат щелочного металла (K2CO3):
а/ карбонат, в частности карбонат калия, в отличие от KOH, при образовании соли акридона и ее алкилировании превращается в MeHCO3 и MeCl (KHCO3, KCl), т.е. не продуцирует воду, и стадия азеотропной отгонки воды с растворителем снимается вовсе;
б/ карбонат калия - хорошее осушающее средство, и применение его избытка (III: K2CO3 = 1:2-3) обеспечивает удаление воды, внесение которой возможно с исходными продуктами или растворителем.
В то же время применение даже большого избытка карбоната калия в отсутствие воды не вызывает в отличие от KOH интенсивного перевода имеющихся в реакционной смеси целевого и алкилирующего эфиров в соли даже при длительном пребывании в условиях высокой температуры (соли на завершающем этапе при применении KOH конечно уйдут с отработанным водным растворителем, но это существенно скажется на выходе продукта стадии алкилирования).
Т.о. применение карбоната калия в избытке обеспечивает синтез K-соли акридона, удаление влаги, необходимое связывание HCl и обеспечивает пониженную щелочность среды и, следовательно, степень прохождения побочных реакций, что в совокупности ведет к повышению выхода и чистоты продукты алкилирования.
При алкилировании с использованием модуля по растворителю - акридон (III): ДМФА = 1:10-12, дозировке этилхлорацетата при повышенной температуре (до 37oC) и применении его избытка (III: ClCH2COOEt = 1:2-3) достигается высокая концентрация соли акридона в растворе именно к моменту дозировки, что обеспечивает быстрый расход малостабильного при повышенной температуре этилхлорацетата в основном по целевому направлению и практически за время выдержки, при этом нет необходимости в длительном кипячении реакционной массы, что обычно ведет к ее частичному осмолению; введение же катализатора межфазного переноса не приводит к ускорению процесса или повышению выхода или качества эфира.
Именно эти приемы обеспечивают быструю и высокую конверсию акридона в эфир (II), а следовательно, его высокий выход и достаточную чистоту технического продукта (выход эфира (II) >90%, содержание акридона (III) в нем - <10%).
И все же поскольку, как правило, не удается достичь полной конверсии, то эфир реально может содержать 5-10% акридона, и при разбавлении эти продукты будут выпадать совместно. Отделять акридон кристаллизацией сложно и неэкономично. Даже при примеси акридона <5% в техническом эфире, применять последний для синтеза соли (I) без дополнительной очистки опасно, так как всегда есть возможность перехода акридона в целевой продукт (содержание примесей в выходном препарате не должно быть >1.5%).
В масштабной практике колебания в содержании акридона в техническом эфире могут быть значительны (от следовых до 20-30% при сбоях при выполнении технологии), и необходим способ, пригодный для переработки и таких "бракованных" партий.
Отсюда необходимо - отработать условия гидролиза технического эфира, обеспечивающие отделение акридона и образование чистой натриевой соли.
При спиртово-щелочном гидролизе технического эфира (II) акридон (III) переходит в целевую соль фактически нацело. А этот этап должен обеспечивать фармакопейную чистоту целевых препаратов. Поэтому спиртовый вариант гидролиза по прототипу не приемлем для технического эфира вовсе.
Акридон, как оказалось, заметно растворим в спиртовой и практически не растворим в водной щелочи.
В настоящем решении отделение акридона предлагается перенести на этап гидролиза эфира, проводя последний в водно-щелочной среде: обрабатывать эфир водной щелочью или содой при нагревании и отфильтровывать непрореагировавший акридон, а полученный фильтрат либо подкислять с последующей фильтрацией выпавшей целевой кислоты (Ia), либо доводить до нужной концентрации соли в воде или выделять целевой продукт (I) в кристаллическом виде.
Именно предложенные условия проведения стадий алкилирования и гидролиза в комплексе позволяют использовать технический эфир, исключив операцию его очистки, отделить и вернуть на предшествующую стадию непрореагировавший и оставшийся в техническом эфире акридон, дочистить водный раствор натриевой соли сорбентом, исключить применение абсолютного спирта, сократить расход растворителя на операциях очистки и выделения и получать чистые целевые препараты с высоким выходом.
Для лучшего понимания сущности предлагаемого изобретения приводится пример его конкретной реализации.
Пример 1. Синтез этилового эфира 10-метиленкарбокси-9-акридона (II). К 150-180 мл ДМФА присыпают 15 г (0.077 моль) акридона (III) и 21-32 г (0.154-0.231 моль) карбоната калия (или 0.154-0.231 моль соды), смесь нагревают при перемешивании до кипения, кипятят 15-20 мин, охлаждают и добавляют 16-24 мл (0.15-0.23 моль) этилхлорацетата так, чтобы температура в реакторе поддерживалась на уровне 100-130oC. По окончании дозировки реакционную массу выдерживают при 130-140oC в течение 0.5-1 ч и контролируют содержание в ней исходного акридона /ТСХ, система хлороформ:метанол = 95:5). В случае необходимости дают дополнительную выдержку. Реакционную массу охлаждают, выливают при перемешивании в воду, выпавший продукт отфильтровывают и промывают на фильтре водой. На стадию щелочного гидролиза продукт поступает без очистки.
Синтез натриевой соли 10-метиленкарбокси-9-акридона (I).
К раствору 3.7 г (0.092 моль) едкого натра в 70 мл дистиллированной воды добавляют эфир (II), полученный на предыдущем этапе, выдерживают при 95-100oC 0.5-1 ч, охлаждают, отфильтровывают непрореагировавший акридон и промывают его дистиллированной водой. После сушки акридон возвращают на стадию алкилирования. Полученный фильтрат обрабатывают активированным углем (а.у.) при нагревании, охлаждают, отфильтровывают от а.у. и промывают на фильтре дистиллированной водой; полученный фильтрат A-раствор целевого продукта (I) в воде; выход 90-94% (УФ-спектроскопия фильтрата); в случае необходимости операцию очистки а.у. повторяют; раствор упаривают под вакуумом, отгоняя основную часть воды, к остатку добавляют спирт, образовавшуюся суспензию доводят до кипения, выдерживают при кипении 0.5 ч, профильтровывают и оставляют фильтрат для кристаллизации. Выпавший продукт отфильтровывают, промывают на фильтре спиртом и сушат; выход продукта (I) 90-94% (с учетом остаточного продукта в маточнике). Водный маточный растворитель может быть частично возвращен в цикл на стадию выделения соли, остаток подлежит регенерации. Выделение 10-метиленкарбокси-9-акридона (Ia). Охлажденный до 5-10oC фильтрат A - раствор соли (I) подкисляют 5%-ной серной (соляной) кислотой до pH 1, выдерживают при перемешивании при этой температуре 0.5 ч, выпавший желтый осадок отфильтровывают, промывают на фильтре водой и сушат, выход 80-87% на акридон, т. пл. 277-279oC; при необходимости продукт кристаллизуют (ДМФА - вода = 1/3); характеристики препарата идентичны описанным в [5].
Строение и индивидуальность полученной натриевой соли 10-метиленкарбокси-9-акридона подтверждена методами ТСХ, элементного анализа, ИК- и УФ-спектроскопии.
УФ-спектр (0.02 р-р в растворе 0.01 моль/л едкого натра), л, нм, (Ig E): 257 (4.68), 388 (3.88), 407 (3.91);
IR, cm-1: 760, 940, 1180, 1270, 1290, 1395 (COO-), 1460, 1490, 1595 (COar, COO-), 3020;
Спектр ПМР, б, м.д.: 4.83 с (CH2, 2H), 7.26-7.68 мм (ArH, 4H), 7.70-7.80 м (2H, 4H и 5H), 8.30 д (2H, J = 8 Гц, 1H и 8H), D2O; ЯМР-C13, б, м.д.: 50.9 (1C), 116.2, 124.4, 122.9, 127.2, 135.9 (12C - ароматика), 175.8, 180.1 (2C - карбонильные), D2O + 0.01% диоксана.
ТСХ (Silufol-UV-254): Rf 0.34-0.36 (ИПС: этилацетат: NH4OH = 59:25:16).
Анализ
Найдено, %: C 65.12, 65.32; H 4.01, 3.88; N 5.23, 5.11
C15H10NO3Na
Вычислено, %: C 65.46; H 3.66; N 5.09
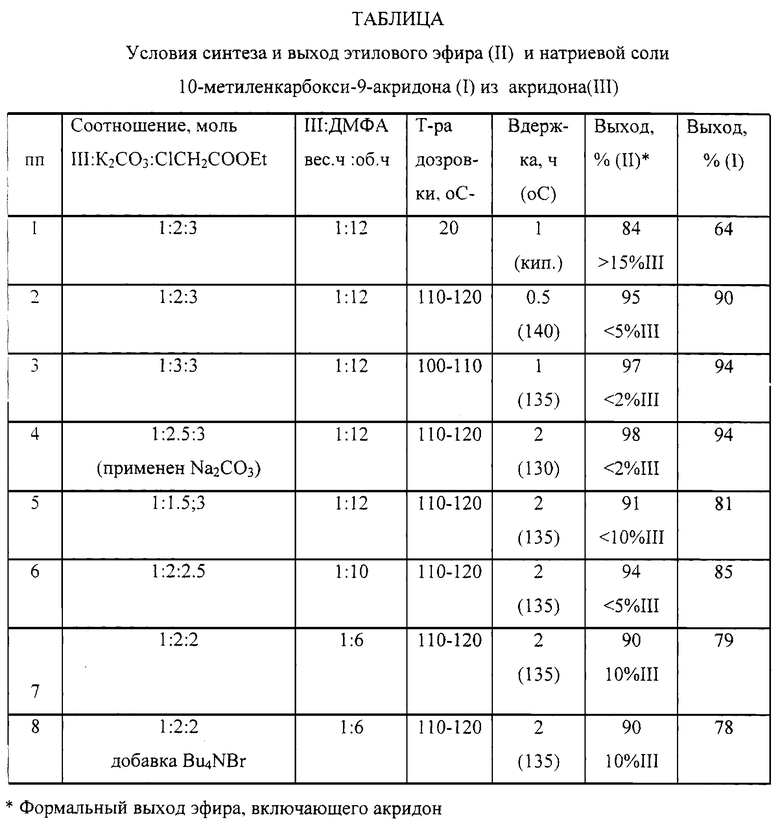
Аналогично осуществляли алкилирование акридона при варьировании параметров процесса с последующим переводом технического продукта в целевую соль по приведенной выше методике; результаты приведены в таблице.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ЭФИРОВ N-МЕТИЛЕНКАРБОКСИ-9-АКРИДОНА | 1993 |
|
RU2033413C1 |
| ТВЕРДОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2001 |
|
RU2198659C1 |
| СПОСОБ ПОЛУЧЕНИЯ N-МЕТИЛЕНКАРБОКСИ-9-АКРИДОНА ИЛИ ЕГО НАТРИЕВОЙ СОЛИ | 1992 |
|
RU2029764C1 |
| ЛЕКАРСТВЕННОЕ СРЕДСТВО, ОБЛАДАЮЩЕЕ ПРОТИВОЯЗВЕННЫМ, КАРДИОПРОТЕКТИВНЫМ И ПРОТИВОШОКОВЫМ ДЕЙСТВИЕМ | 2001 |
|
RU2211213C2 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ЭФИРОВ L-КАРНОЗИНА И ИХ СОЛЕЙ | 2001 |
|
RU2188204C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОЛИ 9-МЕЗИТИЛ-10-МЕТИЛАКРИДИНИЯ | 2015 |
|
RU2582126C1 |
| СПОСОБ ПОЛУЧЕНИЯ НИТРИЛОВ АЛКОКСИФЕНИЛУКСУСНЫХ КИСЛОТ (ВАРИАНТЫ) | 1997 |
|
RU2133736C1 |
| СПОСОБ ПОЛУЧЕНИЯ 17-ЦИКЛОПРОПИЛМЕТИЛ-6,14-ЭНДО-ЭТАНО-7-(2-ГИДРОКСИ-3,3-ДИМЕТИЛБУТ-2-ИЛ)ТЕТР АГИДРО-17-НОРОРИПАВИНА | 1999 |
|
RU2138502C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕКСАГИДРАТА ТРИНАТРИЕВОЙ СОЛИ ФОСФОНМУРАВЬИНОЙ КИСЛОТЫ | 1996 |
|
RU2110521C1 |
| СРЕДСТВО, ОБЛАДАЮЩЕЕ АНТИИШЕМИЧЕСКОЙ, ГИПОТЕНЗИВНОЙ И АНТИГИПОКСИЧЕСКОЙ АКТИВНОСТЬЮ | 2001 |
|
RU2192858C1 |
Изобретение касается усовершенствованного способа получения 10-метиленкарбоксилат-9-акридона или 10-метиленкарбокси-9-акридона, предназначенного для синтеза лекарственных субстанций или приготовления лекарственных форм (натрий 10-метиленкарбоксилат-9-акридон), применяемых в медицинской и ветеринарной практике в качестве противовирусного средства. Предложен технологически приемлемый способ получения натрия 10-метиленкарбоксилат-9-акридона или 10-метиленкарбокси-9-акридона путем алкилирования акридона этилхлорацетатом в присутствии карбонатов натрия или калия в условиях, позволяющих получать технический эфир 10-метиленкарбокси-9-акридона с высоким выходом и высоким содержанием основного вещества и переводить его путем щелочного гидролиза и отделения исходного продукта в целевые продукты высокой чистоты. Технический результат состоит в повышении выхода и чистоты целевых продуктов. 9 з.п. ф-лы, 1 табл.
| POSTESCU J.D., GSAVASSY G.H., J | |||
| PRAKT | |||
| Chem., 1977, т.319, n2, с.347-352 | |||
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ЭФИРОВ N-МЕТИЛЕНКАРБОКСИ-9-АКРИДОНА | 1993 |
|
RU2033413C1 |
| СПОСОБ ПОЛУЧЕНИЯ N-МЕТИЛЕНКАРБОКСИ-9-АКРИДОНА ИЛИ ЕГО НАТРИЕВОЙ СОЛИ | 1992 |
|
RU2029764C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОТИВОВИРУСНОГО СРЕДСТВА | 1992 |
|
RU2020941C1 |
| ПРОТИВОВИРУСНОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2031650C1 |
| US 3681360 A, 01.08.1972 | |||
| Экономайзер | 0 |
|
SU94A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| JP 63310826 A, 19.12.1988 | |||
| Устройство для измерения поверхностной скорости течения в потоках | 1982 |
|
SU1040426A1 |

