Настоящее изобретение относится к новым производным нистатина и к способам их получения. Изобретение, в частности, относится к применению новых производных нистатина в медицине, особенно в качестве противогрибковых агентов.
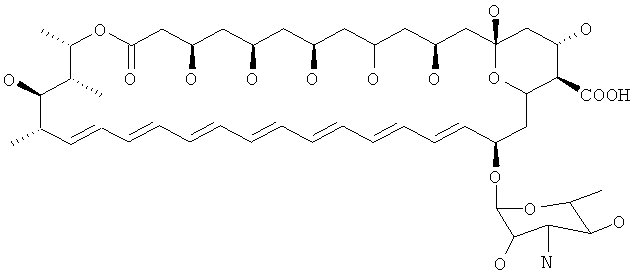
Нистатин представляет собой встречающийся в природе комплекс противогрибковых веществ, продуцируемый некоторыми штаммами грамположительных бактерий Streptomyces noursei. Основной компонент нистатина представляет собой нистатин A1, который представляет собой полиеновый макролид, имеющий структуру, представленную ниже:
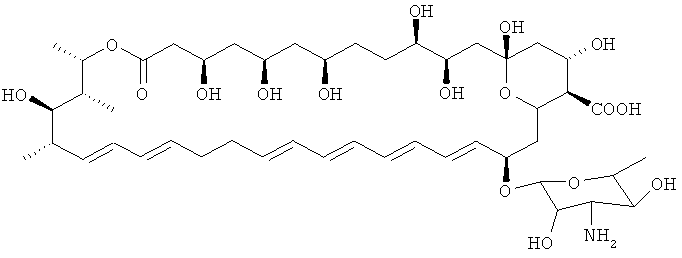
Нистатин представляет собой активный агент, присутствующий во множестве патентованных препаратов (например Infestat, Nyspes, Nystamont, Nystan, Mycostatin и Nilstat) для применения в лечении кандидоза кожи и слизистых оболочек. Его обычно вводят перорально или местно, хотя он плохо всасывается из желудочно-кишечного тракта или через кожу и слизистые оболочки. Причиной этого является его токсичность при парентеральном введении.
Нистатин представляет собой член семейства полиеновых макролидных антибиотиков. Другие члены этого семейства включают пимарицин, римоцидин, кандицидин, ауреофацин, леворин и амфотерицин. Амфотерицин представляет собой наиболее часто используемый противогрибковый агент, поскольку его, как правило, рассматривают как наиболее эффективный. Он обладает широким спектром противогрибковой активности, но, как и нистатин, обладает основным недостатком, заключающемся в токсичности, которая ограничивает пути введения и дозы, которые могут быть использованы.
Таким образом, попытки исследования макролидных поликетидных антибиотиков сфокусированы на амфотерицине и, в частности, на идентификации новых амфотерициновых аналогов, обладающих меньшей токсичностью. В общем, были приняты два отдельных подхода. При первом подходе исследователи используют химические модификации для уменьшения токсичности и/или улучшения противогрибковой активности амфотерицина. Поскольку полный синтез амфотерицина экономически не реализуем, тем не менее, исходное вещество, как правило, представляет собой амфотерицин В, получаемый в культурах Streptomyces nodosus. В результате химические модификации сконцентрированы на дериватизации функциональных групп, присутствующих в кольце амфотерицина В.
Относительно недавно, исследователи приняли другой подход, который направлен на биосинтетический путь амфотерицина, который состоит из сборной цепочки различных ферментов. Углеродную цепь амфотерицина собирают из ацетатных и пропионатных единиц модульными (тип 1) поликетидсинтазами, и образующийся в результате макролид затем модифицируют другими ферментами, которые осуществляют окисление, гликозилирование и гидроксилирование. Гены, кодирующие ферменты, вовлеченные в этот биосинтез, идентифицированы исследователями, как способные вызывать изменения природных биосинтетических путей посредством, например, удаления, добавления или модификации гена, и, таким образом, удаления, добавления или модификации фермента, вовлеченного в синтез. Общий результат представляет собой модифицированные молекулы амфотерицина.
Таким образом, получено и тестировано большое количество модифицированных молекул амфотерицина, особенно химически модифицированных производных. Несмотря на эти попытки, тем не менее, идентифицировано небольшое количество перспективных кандидатов лекарственных средств, имеющих подходящий баланс фармакологических свойств, а именно высокую противогрибковую активность, низкую токсичность и растворимость в воде. Следовательно, сохраняется потребность в новых антибиотиках для лечения инфекционных заболеваний, и, в частности, острых системных грибковых инфекций, частота которых увеличивается. Это требование становится все более насущным, поскольку развивается резистентность к существующим противогрибковым агентам.
По сравнению с амфотерицином проведено очень мало исследований по модификации нистатина, преимущественно из-за того, что он обладает меньшей активностью по сравнению с амфотерицином, при этом имеет те же самые недостатки: токсичность и нерастворимость в воде. В результате его не рассматривают как привлекательную исходную молекулу для такой работы.
В недавнем исследовании (Organic Letters, 2006, 8 (9), 1807-1809) два производных нистатина получали путем восстановительного алкилирования аминогруппы микозамина. Обнаружили, что получающиеся в результате производные, обладают более высокой противогрибковой активностью по сравнению с нистатином, но менее активны по сравнению с производными амфотерицина В, полученными тем же самым путем. Таким образом, модификацию амфотерицина В рассматривали как более перспективную, и токсичность этих протестированных производных, как было продемонстрировано, оказалась значительно меньше по сравнению с родительской молекулой.
В WO 01/59126 описано клонирование и секвенирование генного кластера, кодирующего поликетидсинтазу и другие ферменты, ответственные за синтез нистатина. В WO 01/59126 также предложено множество потенциальных манипуляций, которые теоретически могут быть осуществлены с генным кластером для получения модифицированных структур нистатина. Они включают модификации для удаления или добавления двойных связей из полиенового фрагмента нистатина, модификации для удаления или перемещения С16 карбоксильной группы, модификации для добавления дополнительных гидроксильных групп и модификации для усечения макролактонового кольца.
Из вышеописанного понятно, что множество (приблизительно 1017) различных модифицированных структур нистатина могут быть получены путем осуществления одной или более чем одной из модификаций, предложенных в WO 01/59126. Возможность дополнительной модификации любого из соединений, продуцируемых путем генетической манипуляции с дополнительными химическими модификациями, означает, что теоретически возможны квинтильоны соединений. Поскольку слишком мало работы произведено с нистатином, тем не менее, отсутствуют данные, на основании которых можно сделать предположение о том, какое соединение из этого огромного количества соединений может демонстрировать меньшую токсичность по сравнению с нистатином и/или амфотерицином (то есть теми активными агентами, которые используются в настоящее время).
Тем не менее, в настоящее время неожиданно обнаружено, что некоторые производные нистатина обладают значительно меньшей токсичностью по сравнению с амфотерицином, а также обнаружено, что те же самые производные демонстрируют полезную противогрибковую активность. В отличие от подавляющего большинства полученных модифицированных амфотерициновых соединений, производные нистатина в настоящее время идентифицируют как производные "второго поколения". Другими словами, эти соединения представляют собой производные производного нистатина, и, таким образом, содержат две или более чем две модификации по сравнению со структурой нистатина Ai.
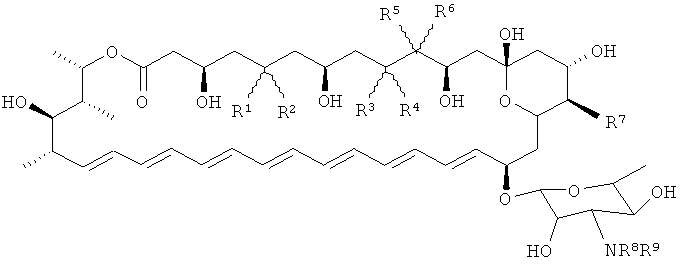
Таким образом, в соответствии с первым аспектом изобретения предложено соединение, представляющее собой производное нистатина, имеющее дополнительную двойную связь, присутствующую между С28 и С29, и дополнительно модифицированное относительно нистатина по одному или более чем одному из положний С5, С9, С10, С16 или по аминогруппе микозамина, или его фармацевтически приемлемая соль.
В соответствии с еще одним аспектом изобретение предложено соединение, представляющее собой производное нистатина, имеющее дополнительную двойную связь, присутствующую между С28 и С29, и дополнительно модифицированное относительно нистатина по одному или более чем одному из положений С5, С7, С9, С10, С11, С16 или по аминогруппе микозамина, или его фармацевтически приемлемая соль.
Предпочтительные соединения по изобретению представляют собой соединения формулы I
 ,
,
где
каждый из R1 и R2 независимо представляет собой атом водорода, гидроксильную группу, алкоксигруппу, группу ацилокси или алкильную группу или вместе образуют карбонильную группу (например каждый из R1 и R2 независимо представляет собой атом водорода или гидроксильную группу или вместе образуют карбонильную группу;
каждый из R3 и R4 независимо представляет собой атом водорода, гидроксильную группу, алкоксигруппу, группу ацилокси или алкильную группу или вместе образуют карбонильную группу (например каждый из R3 и R4 независимо представляет собой атом водорода или гидроксильную группу или вместе образуют карбонильную группу;
каждый из R5 и R6 независимо представляет собой атом водорода, гидроксильную группу, алкоксигруппу, группу ацилокси или алкильную группу (например атом водорода или гидроксильную группу);
R7 представляет собой атом водорода, СООН, алкильную группу, алкоксигруппу, группу сложного эфира карбоновой кислоты или амидную группу (например СООН, алкильную группу, группу сложного эфира карбоновой кислоты или амидную группу);
каждый из R8 и R9 независимо представляет собой атом водорода, группу алкиламино, группу сахара или ацильную группу;
или их фармацевтически приемлемую соль,
при условии, что соединение не представляет собой соединение (II)
 ,
,
или соединение микогептин (изображенное ниже)
 .
.
Еще одно предпочтительное соединение по изобретению, также удовлетворяющее вышеприведенным условиям, имеет формулу (I), но где группа -ОН по атому углерода 7 и/или атому углерода 11 превращена, например в группу оксо. Оно имеет формулу (I′), то есть
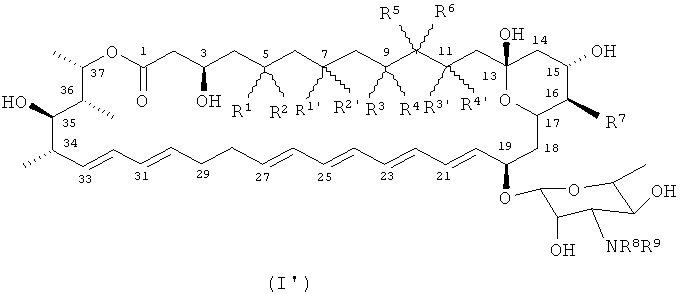 ,
,
где все заместители являются такими, как определено для формулы (I), и каждый из R1, R2, R3 и R4 независимо является таким, как определено для R1-R4 соответственно, и при дополнительном условии, что соединение формулы (I′) не представляет собой кандидин (изображенный ниже)
 .
.
В соответствии с еще одним аспектом изобретения предложен способ получения описанных выше соединений, включающий:
(1) модификацию генного кластера, кодирующего поликетидсинтазную систему, ответственную за синтез нистатина, с получением производного нистатина, имеющего двойную связь между С28 и С29; и
(2) дополнительную модификацию указанного генного кластера с получением производного нистатина, которое дополнительно модифицировано по одному или более чем одному из положений С5, С9, С10, С16 или по аминогруппе микозамина,
или
(3) модификацию получающегося в результате производного по одному или более чем одному из положений С5, С9, С10, С16 или по аминогруппе микозамина путем химического взаимодействия.
В соответствии с еще одним аспектом изобретения предложен способ получения описанных выше соединений, включающий:
(1) модификацию генного кластера, кодирующего поликетидсинтазную систему, ответственную за синтез нистатина, с получением производного нистатина, имеющего двойную связь между С28 и С29; и
(2) дополнительную модификацию указанного генного кластера с получением производного нистатина, которое дополнительно модифицировано по одному или более чем одному из положений С5, С7, С9, С10, С11, С16 или по аминогруппе микозамина,
или
(3) модификацию получающегося в результате производного по одному или более чем одному из положений С5, С7, С9, С10, С11, С16 или по аминогруппе микозамина путем химического взаимодействия.
В соответствии с еще одним аспектом изобретения предложена композиция, содержащая описанное выше соединение и носитель, разбавитель или эксципиент.Предпочтительные композиции представляют собой фармацевтические композиции.
В соответствии с еще одним аспектом изобретения предложено описанное выше соединение для применения в терапии.
Применение описанного выше соединения для изготовления композиции для лечения грибковых инфекций образует еще один аспект изобретения.
Способ лечения грибковой инфекции у животного (например человека), включающий введение указанному животному определенного выше соединения, образует еще один аспект.
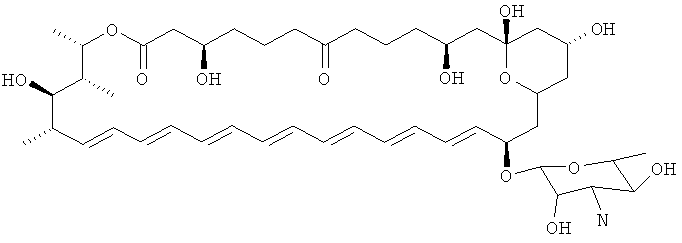
Использованный здесь термин "производное нистатина" охватывает соединения, которые имеют структуру, идентичную структуре нистатина A1 за исключением указанных конкретных модификаций. Таким образом, предпочтительные производные имеют то же самое макролактоновое кольцо, как и нистатин, за исключением добавления двойной связи по С28-С29, и имеют те же самые функциональные группы, как нистатин, за исключением модификации по одному или более чем одному из положений С5, С7, С9, С10, С11, С16 или по аминогруппе микозамина. Таким образом, предпочтительные производные содержат гликозилированный макролактоновый кольцевой скелет, представленный ниже:
 ,
,
или
 ,
,
или
 .
.
Понятно, что в этих формулах могут быть осуществлены модификации по С5, С9, С10, С16 и по N микозамина. Таким образом, то, что представлено, представляют собой только атомы, которые должны присутствовать. Эти формулы демонстрируют химический скелет, который может присутствовать.
Используемый здесь термин "поликетидсинтазная система" обозначает набор ферментов, ответственных за поликетидный синтез и модификации. Они включают поликетидсинтазы, монооксигеназы, гликозилтрансферазу и аминотрансферазу.
Термин "алкильные группа" используют здесь для представления циклической или ациклической, прямоцепочечной или разветвленной насыщенной группы углеводорода. Такие группы могут содержать до 20 атомов углерода, но предпочтительны группы, содержащие от 1 до 12 атомов углерода, более предпочтительно от 1 до 6 атомов углерода.
Алкильные группы могут быть незамещенными или замещенными. Заместители, которые могут присутствовать в замещенных алкильных группах, включают гидрокси, алкокси, ацилокси и амино.
Алкильные группы также могут быть прерваны одной или более чем одной группой, например -арил- (такой как Ph), -О-, -NR12- или -S-, где R12 представляет собой атом водорода или С1-6алкильную группу.
Термин "алкокси" используют здесь для представления группы - OR13, где R13 представляет собой алкильную группу, определенную выше. В предпочтительных алкоксигруппах R13 содержит от 1 до 6 атомов углерода, более предпочтительно от 1 до 4 атомов углерода. Предпочтительная алкоксигруппа представляет собой -OC1-6, например -ОСН3.
Термин "ацилокси" используют здесь для представления группы -OCOR14, где R14 представляет собой алкильную группу, определенную выше. В предпочтительных группах ацилокси R14 содержит от 1 до 6 атомов углерода, более предпочтительно от 1 до 4 атомов углерода. Предпочтительная группа ацилокси представляет собой -OCOC1-6, например -ОСОСН3.
Термин "группа сложного эфира карбоновой кислоты" используют здесь для представления группы -COOR15, где R15 представляет собой алкильную группу, определенную выше. В предпочтительных группах сложного эфира карбоновой кислоты R15 содержит от 1 до 6 атомов углерода, более предпочтительно от 1 до 4 атомов углерода. Предпочтительная группа сложного эфира карбоновой кислоты представляет собой -COOC1-6, например -СООСН3.
Термин "амидная группа" используют здесь для представления группы формулы III:

где каждый из R10 и R11 независимо представляет собой атом водорода или возможно замещенную алкильную группу, определенную выше, или вместе образуют алкильное кольцо, определенное выше.
Термин "группа алкиламино" используют здесь для представления группы -(CH2)xNR16R17, где х равен числу от 1 до 10, предпочтительно от 2 до 6 (например 3) и каждый из R16 и R17 независимо представляет атом водорода или C1-6алкильную группу.
Термин "сахар" используют здесь для представления сахаридов, особенно олиго- и моносахаридов. Сахара, присутствующие в соединениях по изобретению, могут содержать любое количество сахаридных единиц, но предпочтительные соединения содержат от 1 до 10 сахаридных единиц, например 1 или 2 сахаридные единицы.
Термин "ацил" используют здесь для представления группы -COR18, где R18 представляет собой алкильную группу, определенную выше. В предпочтительных ацильных группах R18 представляет собой замещенную алкильную группу, например аминозамещенную алкильную группу. Особенно предпочтительные ацильные группы представляют собой аминоацильные группы.
Описанные выше соединения по настоящему изобретению представляют собой производные нистатина, имеющее дополнительную двойную связь, присутствующую между С28 и С29, и дополнительно модифицированные относительно нистатина по одному или более чем одному из положений С5, С7, С9, С10, С11, С16 или по аминогруппе микозамина.
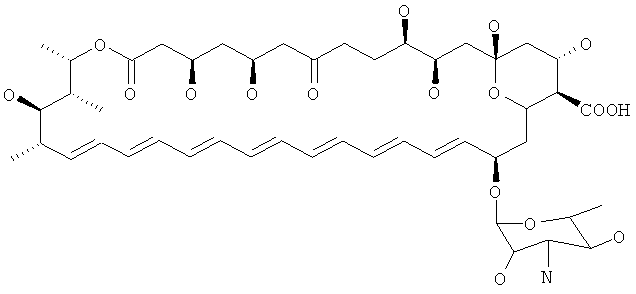
Предпочтительные соединения по настоящему изобретению модифицированы по С5 относительно нистатина. В нистатине С5 замещен гидроксильной группой и атомом водорода и представляет собой стереоцентр R. Таким образом, в предпочтительных соединениях по изобретению С5 замещен группами, отличающимися от гидроксильной группы, и атомом водорода, и/или С5 представляет собой стереоцентр S. В предпочтительных соединениях различные группы присутствуют в С5, например С5 могут быть замещен двумя гидроксильными группами или двумя атомами водорода. В особенно предпочтительном соединении С5 замещен карбонильной группой.
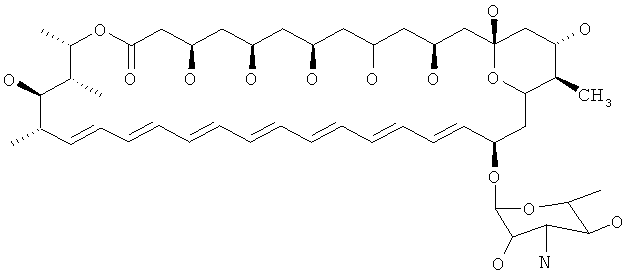
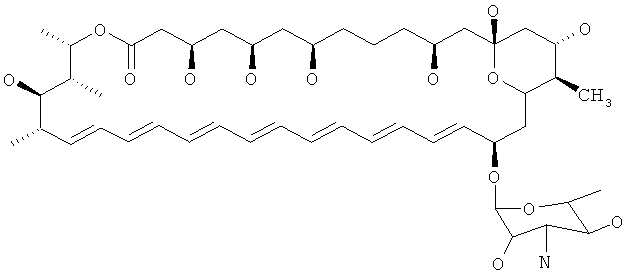
Другие предпочтительные соединения по изобретению модифицированы по С16 относительно нистатина. В нистатине С16 замещен карбоновой кислотой и атомом водорода и представляет собой стереоцентр R. Следовательно, в предпочтительных соединениях по изобретению С16 замещен группами, отличающимися от группы карбоновой кислоты, и атомом водорода, и/или С16 представляет собой стереоцентр S. В предпочтительных соединениях присутствуют различные группы, например алкильная группа (например метил) и атом водорода или амидная группа и атом водорода. В особенно предпочтительных соединениях С16 замещен метильной группой и атомом водорода. В предпочтительных соединениях С16 представляет собой стереоцентр R.
Дополнительные предпочтительные соединения по изобретению модифицированы по С9 относительно нистатина. В нистатине С9 замещен двумя атомами водорода. Следовательно, в предпочтительных соединениях по изобретению С9 замещен группами, отличающимися от двух атомов водорода. В предпочтительных соединениях присутствуют различные группы, например гидроксильная группа и атом водорода, или карбонильная группа.
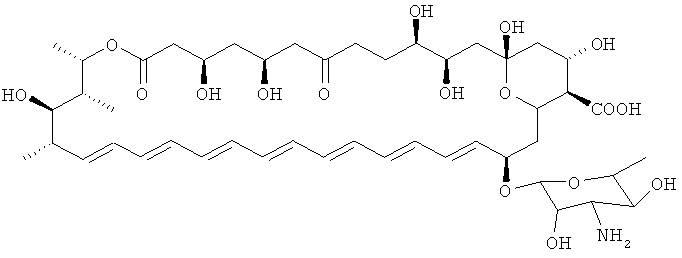
Дополнительные предпочтительные соединения по изобретению могут быть модифицированы по С7 относительно нистатина. В нистатине С7 замещен гидроксилом. Следовательно, в некоторых соединениях по изобретению С7 замещен группами, отличающимися от гидроксила. В предпочтительных соединениях присутствует карбонильная группа. Когда присутствует карбонил, также необходимо, чтобы присутствовала дополнительная модификация соединения по С5, С7, С9, С10, С11, С16 или N микозамина относительно нистатина для того, чтобы исключить изображенное выше соединение кандидин.
Дополнительные предпочтительные соединения по изобретению могут быть модифицированы по С11 относительно нистатина. В нистатине С11 замещен гидроксилом. Следовательно, в некоторых соединениях по изобретению С11 замещен группами, отличающимися от гидроксила. В предпочтительных соединениях присутствует карбонильная группа.
Дополнительные предпочтительные соединения по изобретению модифицированы по С10 относительно нистатина. В нистатине С10 замещен гидроксильной группой и атомом водорода и представляет собой стереоцентр R. Следовательно в предпочтительных соединениях по изобретению С10 замещен группами, отличающимися от гидроксильной группы, и атомом водорода, и/или С10 представляет собой стереоцентр S. В предпочтительных соединениях присутствуют различные группы, например два атома водорода или карбонильная группа. В особенно предпочтительных соединениях С10 замещен двумя атомами водорода.
Другие предпочтительные соединения по изобретению модифицированы по аминогруппе микозамина относительно нистатина. В нистатине аминогруппа микозамина не замещена, то есть она представляет собой -NH2. Следовательно, в предпочтительных соединениях по изобретению аминогруппа микозамина замещена, например одной или более чем одной группой ал килами но или группой сахара.
Когда соединения по изобретению имеют карбонил по С5 или С7, тогда особенно предпочтительно, чтобы также присутствовали дополнительные модификации соединения по С5, С7, С9, С10, С11, С16 или N микозамина относительно нистатина. Например амин микозамина может быть функционализирован таким образом, чтобы нести по меньшей мере один заместитель R8/R9, отличающийся от водорода.
Хотя соединения по изобретению могут содержать только одну замену относительно известных веществ, таких как S44HP, в идеале любое соединение по изобретению содержит две замены по С5, С7, С9, С10, С11, С16 или N микозамина относительно нистатина. Предпочтительные соединения могут содержать три замены по С5, С7, С9, С10, С11, С16 или N микозамина относительно нистатина. Дополнительные предпочтительные соединения содержат по меньшей мере две замены по С5, С7, С9, С10, С11, С16 или N микозамина относительно S44HP. Другие предпочтительные соединения содержат по меньшей мере две замены по С5, С7, С9, С10, С11, С16 или N микозамина относительно микогептина. Дополнительные предпочтительные соединения содержат по меньшей мере две замены по С5, С7, С9, С10, С11, С16 или N микозамина относительно кандидина.
Особенно предпочтительные соединения содержат по меньшей мере две замены по С5, С7, С9, С10, С11, С16 или N микозамина по сравнению с нистатином, S44HP, микогептином и кандидином.
Особенно предпочтительные соединения по изобретению представляют собой соединения формулы (I) или (I′), как изложено выше.
В предпочтительных соединениях формулы (I) R1 представляет собой атом водорода, гидроксильную группу, алкоксигруппу, группу ацилокси или алкильную группу. Еще более предпочтительно, R1 представляет собой атом водорода, гидроксильную группу или алкоксигруппу (например группу -OC1-6), в частности, атом водорода или гидроксильную группу (например гидроксильную группу). В предпочтительных соединениях R2 представляет собой атом водорода. В альтернативных предпочтительных соединениях R1 и R2 вместе образуют карбонильную группу. Когда С5 представляет собой стереоцентр, он предпочтительно представляет собой центр R.
Предпочтительные соединения формулы (I) также представляют собой соединения, где R3 представляет собой атом водорода, гидроксильную группу, алкоксигруппу, группу ацилокси или алкильную группу. Еще более предпочтительно, R3 представляет собой атом водорода, гидроксильную группу или алкоксигруппу (например группу -OC1-6), в особенности, атом водорода или гидроксильную группу (например атом водорода). В предпочтительных соединениях R4 представляет собой атом водорода. В альтернативных предпочтительных соединениях R3 и R4 вместе образуют карбонильную группу. Когда С9 представляет собой стереоцентр, тогда он предпочтительно представляет собой центр R.
Дополнительные предпочтительные соединения формулы (I) представляют собой соединения, где R5 представляет собой атом водорода, гидроксильную группу, алкоксигруппу, группу ацилокси или алкильную группу. Еще более предпочтительно, R5 представляет собой атом водорода, гидроксильную группу или алкоксигруппу (например группу -ОС1-6), в особенности, атом водорода или гидроксильную группу (например гидроксильную группу). В предпочтительных соединениях R6 представляет собой атом водорода. Когда С10 представляет собой стереоцентр, тогда он предпочтительно представляет собой центр R.
Другие дополнительные предпочтительные соединения формулы (I) представляют собой соединения, где R7 представляет собой СООН, алкильную группу, группу сложного эфира карбоновой кислоты или амидную группу. Предпочтительная эфирная группа карбоновой кислоты представляет собой -СООС1-6, более предпочтительно -СООС1-4, например -СООСН3.
Особенно предпочтительные соединения формулы (I) представляют собой соединения, где R7 представляет собой алкильную группу или амидную группу. Предпочтительные алкильные группы представляют собой C1-6алкильные, например метил, этил, пропил или бутил, в особенности, метил.
Предпочтительные амиды представляют собой амиды определенной выше формулы III. Особенно предпочтительные амиды представляют собой амиды, где каждый из R10 и R11 независимо представляет атом водорода или C1-10разветвленную или неразветвленную Смоалкильную группу, возможно замещенную гидроксилом, сложным эфиром карбоновой кислоты или аминогруппами и возможно прерванную атомами кислорода или азота, или R10 и R11 вместе образуют C1-8циклическую алкильную группу, возможно замещенную группами гидрокси, ацилокси или амино и возможно прерванную атомами кислорода или азота.
В некоторых предпочтительных амидах R10 представляет собой водород. В дополнительных предпочтительных амидах R11 представляет собой группу формулы IV:
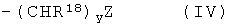 ,
,
где R18 представляет собой водород или алкильную группу, содержащую до 6 атомов углерода (например метил или СН(СН3)2);
y равен числу от 1 до 6, предпочтительно 2-4, например 3; и
Z представляет собой ОН, NR19R20 или COOR21, где каждый из R18, R19 и R21 независимо представляет собой водород или С1-6алкил (например метил).
В особенно предпочтительных амидах R18 представляет собой водород.
В других предпочтительных амидах R10 и R11 вместе образуют циклическую алкильную группу, предпочтительно циклическую алкильную группу формулы V

где каждый из a, b и с независимо равен числу от 1 до 6, предпочтительно 2-4, например 2; и
Z является таким, как определено выше в отношении формулы IV.
Типичные примеры особенно предпочтительных амидных групп формулы III представляют собой представленные ниже:
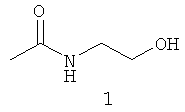
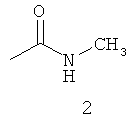
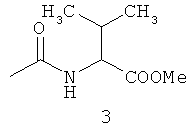
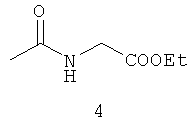

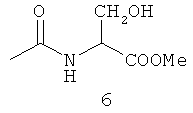
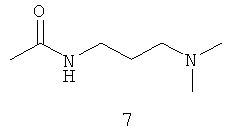
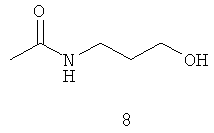
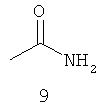
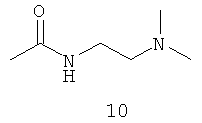
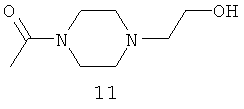
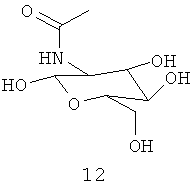 .
.
Амидные группы 4, 6, 7, 8 и 10 являются особенно предпочтительными, в особенности, амидная группа 7 и 10.
Дополнительные предпочтительные соединения формулы (I) представляют собой соединения, где R8 представляет собой атом водорода, группу алкиламино, группу сахара или ацильную группу. В предпочтительных соединениях R9 идентичен R8 или представляет собой атом водорода, например R9 представляет собой атом водорода.
Предпочтительные группы алкиламино представляют собой группы формулы -(CH2)xNH2, где х равен числу от 2 до 6 (например 3), или их алкилированные аналоги -(СН2)xNC1-6алкил2, например -(CH2)xNMe2. Другая предпочтительная группа представляет собой остаток лизина COCH[(CH2)4NH2]NH2.
Предпочтительные группы сахара содержат от 1 до 5 сахаридных единиц, более предпочтительно 2 или 3 сахаридные единицы.
Когда группа сахара представляет собой моносахарид, тогда она может присутствовать в виде линейных, циклических или смеси линейных и циклических конформеров. Когда группа сахара содержит более чем одну сахаридную единицу, тогда каждый моносахарид может являться циклическим, линейным или представлять собой смесь линейных и циклических конформеров. Кроме того, присутствующие сахаридные единицы могут быть одинаковыми или различными.
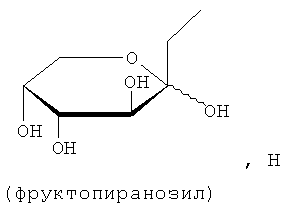
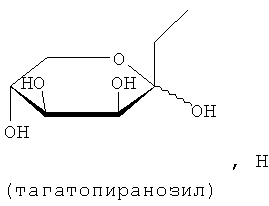
В соединениях формулы (I) предпочтительные моносахариды представляют собой пентозы и гексозы, например глюкозу, галактозу, глюкопиранозу, маннопиранозу, галактопиранозу, фруктопиранозу и таготопиранозу. Предпочтительные ди- и олигосахариды включают лактозу, мелибиозу, сахарозу, мальтозу и целлобиозу. D-глюкоза, D-галактоза и лактоза являются особенно предпочтительными.
Дополнительные предпочтительные ацильные группы (для R8) включают -(CH2)Ph(CH2)xNH2 или -(CH2)Ph(CH2)xNMe2, где х равен 0/1.
Особенно предпочтительные соединения по изобретению представляют собой соединения формулы I, где R1 и R2 вместе образуют карбонильную группу. Более предпочтительно, когда R1 и R2 образуют карбонильную группу, тогда R3, R4 и R6 представляют собой атомы водорода, и R5 представляет собой гидроксильную группу. В таких соединениях предпочтительно по меньшей мере один из R8 и R9 (например R8 и R9) представляет собой атом водорода. Особенно предпочтительно С10 представляет собой стереоцентр R.
Другие особенно предпочтительные соединения по изобретению представляют собой соединения формулы I, где R7 представляет собой C1-6алкил (например метил). Более предпочтительно, когда R7 представляет собой С1-6алкил (например метил), тогда по меньшей мере один из R8 и R9 (например R8 и R9) представляет собой атом водорода. В таких соединениях R5 предпочтительно представляет собой гидроксильную группу, и R6 представляет собой атом водорода. Еще более предпочтительно, R3 и R4 представляют собой атомы водорода. Особенно предпочтительно, С10 представляет собой стереоцентр R.
Другие предпочтительные соединения по изобретению представляют собой соединения формулы I, где R7 представляет собой амидную группу, предпочтительно амидную группу формулы III, как определено выше. Более предпочтительно, когда R7 представляет собой амидную группу, тогда по меньшей мере один из R8 и R9 (например R8 и R9) представляет собой атом водорода. В таких соединениях R5 предпочтительно представляет собой гидроксильную группу, a R6 представляет собой атом водорода. Еще более предпочтительно, R3 и R4 представляют собой атомы водорода. Особенно предпочтительно, С10 представляет собой стереоцентр R.
Другие дополнительные предпочтительные соединения по изобретению представляют собой соединения формулы I, где R8 представляет собой группу сахара или группу алкиламино, определенную выше. Более предпочтительно, когда R8 представляет собой группу сахара или группу алкиламино, тогда R9 представляет собой атом водорода. В таких соединениях R7 предпочтительно представляет собой СООН. Еще более предпочтительно R5 представляет собой гидроксильную группу, a R6 представляет собой атом водорода, и/или R3 и R4 представляют собой атомы водорода. Особенно предпочтительно, С10 представляет собой стереоцентр R.
Для соединений формулы (I′) предпочтительные возможности представляют собой возможности, изложенные выше в связи с формулой (I). Дополнительно, в предпочтительных соединениях формулы (I′) R1 представляет собой гидроксил, a R2 представляет собой атом водорода, или R1′ и R2′ вместе образуют карбонильную группу. Когда С7 представляет собой стереоцентр, тогда он предпочтительно представляет собой центр R.
Предпочтительные соединения формулы (I′) также представляют собой соединения, где R3′ представляет собой гидроксильную группу, a R4′ представляет собой атом водорода, или R3′ и R4′ представляют собой карбонильную группу. Когда С11 представляет собой стереоцентр, тогда он предпочтительно представляет собой центр R.
Предпочтительно, только один из R1/R2 вместе или R3/R4 вместе представляет собой карбонил.
Типичные примеры предпочтительных соединений по изобретению представлены на Фиг.1. Особенно предпочтительные соединения представляют собой соединения под номерами 1, 2, 9, 11, 12, 13, 14, 15, 16, 17, 18, 21 и 22, особенно соединение под номерами 1, 2, 9, 11, 12, 13, 14, 21 и 22, например соединение под номерами 1 и 11.
Дополнительные предпочтительные соединения представляют собой соединения, в которых С16 карбоксильная группа превращена в диметилэтиламид. Соединения, в которых та же самая карбоксильная группа превращена в метильную группу, также являются предпочтительными. Особенно предпочтительные соединения включают соединения с одной из вышеприведенных замен по С16, а также заменами в полиольной области (С5-С11).
Как упомянуто выше, соединения по изобретению могут принимать форму фармацевтически приемлемых солей. Такие соли включают соли присоединения кислоты с физиологически приемлемыми органическими или неорганическими кислотами. Примеры подходящих кислот для образования таких солей включают уксусную, аспарагиновую, бензолсульфоновую, бензойную, биугольную (bicarbonic), бисерную (bisulfuric), бивинную, масляную, эдетат кальция, камсиловую (camsylic), угольную, хлорбензойную, лимонную, этилендиаминтетрауксусную, 1,2-этандисульфоновую (edisylic), лаурилсерную (estolic), этансульфоновую кислоту (esyl), этансульфиновую (esylic), муравьиную, фумаровую, глюкогептоновую (gluceptic), глюконовую, глутаминовую, гликолиларсаниловую (glycollylarsanilic), гексамовую (hexamic), гексилрезорциновую (hexylresorcinoic), гидрабаминовую (hydrabamic), бромоводородную, соляную, йодводородную, гидроксинафтойную (hydroxynaphthoic), изэтионовую, молочную, лактобионовую, малеиновую, яблочную, малоновую, миндальную, метансульфоновую, метилазотную, метилсерную, слизевую, муконовую, 2-нафталинсульфоновую (napsylic), азотную, щавелевую, лара-нитрометансульфоновую, памовую, пантотеновую, фосфорную, двузамещенную соль фосфорной кислоты, однозамещенную соль фосфорной кислоты, фталевую, полигалактуроновую, пропионовую, салициловую, стеариновую, янтарную, сульфаминовую, сульфаниловую, сульфоновую, серную, дубильную, винную, 8-хлортеофиллиновую (teoclic) и толуолсульфоновую. Соли глутаматы являются особенно предпочтительными. Альтернативно, соли могут быть образованы с основаниями. Примеры подходящих оснований для образования таких солей включают первичные, вторичные и третичные амины, замещенные амины, включающие встречающиеся в природе замещенные амины, циклические амины, аргинин, бетаин, кофеин, холин, N,N-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин и трипропиламин. Фармацевтически приемлемые соли могут быть представлены в гидратированной форме. Способы превращения соединений формулы I в такие соли являются традиционными в данной области техники.
Весьма предпочтительные соединения по изобретению могут представлять собой производные следующих скелетов (где группа СООН в положении 16 или аминогруппа функционализирована, как требуется, например где СООН замещена R7). Следует иметь ввиду, что, в общем, атомы водорода не изображены на этих скелетах. Понятно, что они тем не менее присутствуют (то есть образуют гидроксильные группы по атомам -О):
B1
В2 (где группа СООН в положении 16 или группа N функционализирована)
B3
B4
B5

B6
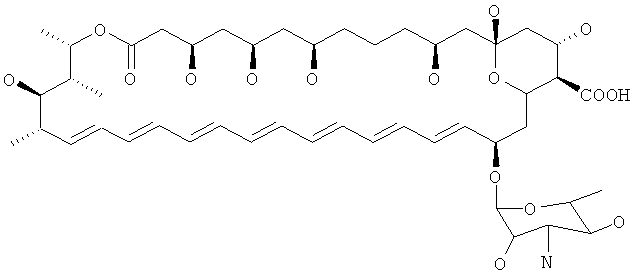
B7
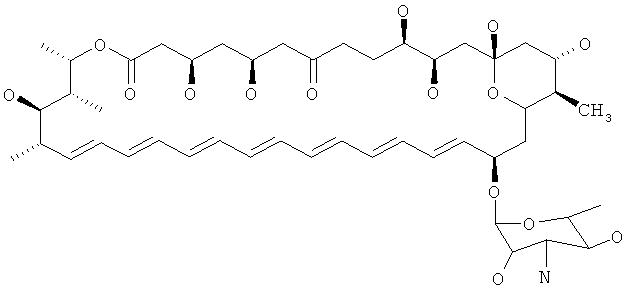
Соединение 1 (из Примеров)
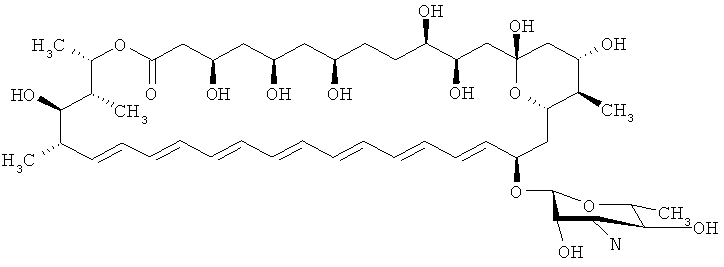
S44HP (где группа СООН в положении 16 или группа N функционализирована)

Понятно, что существует множество переменных в соединениях по изобретению. Для того, чтобы избежать неопределенности, подчеркивается, что каждое описание определения переменной раскрыто в связи со всеми определениями других переменных в заявке. В частности, таким образом, предпочтительные альтернативы для каждой переменной могут быть комбинированы с менее предпочтительными и предпочтительными альтернативами для любой другой переменной.
Соединения по изобретению могут быть получены с использованием способов, известных в уровне техники, включающих обычные способы химии синтеза, генетическую манипуляцию или их комбинацию.
Как упомянуто выше, в аспекте настоящего изобретения предложен способ получения описанных выше соединений, включающий:
(1) модификацию генного кластера, кодирующего поликетидсинтазную систему, ответственную за синтез нистатина, с получением производного нистатина, имеющего двойную связь между С28 и С29; и
(2) дополнительно модификацию указанного генного кластера с получением производного нистатина, которое дополнительно модифицировано по одному или более чем одному из положений С5, С7, С9, С10, С11, С16 или по аминогруппе микозамина,
или
(3) модификацию получающегося в результате производного по одному или более чем одному из положений С5, С7, С9, С10, С11, С16 или по аминогруппе микозамина путем химического взаимодействия.
Предпочтительный способ по изобретению включает стадии (1) и (2), то есть модификацию генного кластера, кодирующего поликетидсинтазную систему, ответственную за синтез нистатина, с получением производного нистатина, имеющего двойную связь между С28 и С29, и дополнительную модификацию относительно нистатина по одному или более чем одному из положений С5, С7, С9, С10, С11, С16 или по аминогруппе микозамина (например по одному или более чем одному из положений С5, С9, С10 или С16).
Второй предпочтительный способ по изобретению включает стадии (1) и (3). Модификация генного кластера, кодирующего поликетидсинтазную систему, ответственную за синтез нистатина, может быть осуществлена в соответствии с обычными способами, например, как описано в WO 01/59126. Генный кластер нистатиновой поликетидсинтазы кодирует множество повторяющихся единиц (модулей), каждый из которых ответственен за один цикл конденсации в синтезе поликетидной цепи. Существует загрузочный модуль (nysA), который определяет природу "стартовой" единицы (карбоновой кислоты) для инициации синтеза поликетидной цепи, и 18 "удлиняющих" модулей, которые включают (конденсируются на цепи) дополнительные "удлиняющие" единицы (следующие карбоновые кислоты, которые добавляются к цепи). Таким образом, каждый такой модуль кодирует несколько ферментативных активностей (например ферментов), которые приводят в результате к синтезу молекулы; модуль содержит домены, которые по отдельности кодируют такие активности. Таким образом, модуль типично может содержать ацилтрансферазный (AT) домен, домен ацилпереносящего белка (АСР) и домен β-кетоацилсинтазы (KS) для синтеза и удлинения (кеторедуктазный (KR), дегидратазный (DH) и еноилредуктазный (ER) домены, которые определяют восстановленное состояние включаемой стартовой/удлиняющей единицы. Генный кластер дополнительно содержит гены (или открытые рамки считывания, ORF), кодирующие другие ферменты (или ферментативные активности), вовлеченные в биосинтез нистатина. Таким образом, тиоэстераза (ТЕ) может обеспечить высвобождение поликетида из PKS (поликетидсинтазы). Кроме того, гены или генные последовательности в генном кластере могут кодировать ферменты или ферментативные активности, которые могут дополнительно модифицировать синтезированную молекулу (поликетидную цепь), например путем гидроксилирования, например монооксигеназы (например nysN или nysL), или путем гликозилирования (например глюкуронозилтрансферазная активность, например nys Dl).
Таким образом, модули и/или домены генного кластера PKS могут быть модифицированы, например путем вставки, делеции или инактивации, или путем замещения домена или модуля, или путем мутации домена или модуля для изменения его активности. Таким образом, количество модулей и/или доменов может быть изменено или они могут быть изменены для изменения их активности. Другие гены или генные последовательности, кодирующие другие ферментативные активности, например монооксигеназную или гликозилтрансферазную, также могут быть модифицированы. Типично, "восстановительные" домены (например ER, DH и/или KR) могут быть модифицированы (например инактивированы) в одном или более чем одном из модулей, или генные последовательности, кодирующие монооксигеназные активности (например NysL, NysN), могут быть модифицированы (например инактивированы).
Например двойная связь между С28 и С29 может быть введена путем инактивации домена ER в модуле 5 поликетидсинтазы, как описано в Примере 2 в WO 01/59126.
Дополнительные модификации относительно нистатина, например, по одному или более чем одному из положений С5, С9, С10 или С16 могут быть введены при помощи других модификаций генного кластера, кодирующего поликетидсинтазную систему. Когда осуществляют дополнительные модификации генного кластера, тогда они предпочтительно приводят к модификации относительно нистатина по одному или более чем одному из положений С5, С9, С10 или С16.
Модификация структуры нистатина по С5 может быть осуществлена путем инактивации кеторедуктазного домена в модуле 17 поликетидсинтазы нистатина, например при помощи способа, описанного в примерах.
Модификация структуры нистатина по С9 может быть осуществлена путем инактивации кеторедуктазного домена в модуле 15 поликетидсинтазы нистатина, например при помощи способа, описанного в примерах.
Модификация структуры нистатина по С10 может быть осуществлена путем инактивации гена NysL монооксигеназы, ответственного за гидроксилирование С10, например при помощи способа, описанного в примерах.
Модификация структуры нистатина по С7 может быть осуществлена путем инактивации доменов KR16.
Модификация структуры нистатина по С11 может быть осуществлена путем инактивации доменов KR14.
Модификация структуры нистатина по С16 может быть осуществлена путем инактивации генов nysN, кодирующих монооксигеназу Р540, например при помощи способа, описанного в примерах.
Дополнительная модификация соединений в результате стадии (1), и, возможно, стадии (2) по одному или более чем одному из положений С5, С7, С9, С10, С11, С16 или по аминогруппе микозамина путем химической реакции может быть осуществлена при помощи любого известного способа. Обычный химизм защитных групп также может быть использован при необходимости. Когда дополнительные модификации вводят путем химической реакции, тогда они предпочтительно приводят к модификации относительно нистатина в положении С16 и/или по аминогруппе микозамина.
Предпочтительное химическое взаимодействие представляет собой образование амида по С16. Превращение группы СООН нистатина в амид может быть осуществлено путем взаимодействия производного нистатина с соответствующим амином, возможно, в присутствии активирующего агента (например гексафторфосфата (бензотриазол-1-илокси)трипирролидинофосфония (PyBOP)).
Еще одно предпочтительное химическое взаимодействие представляет собой восстановительное алкилирование по аминогруппе микозамина. Восстановительное алкилирование может быть осуществлено путем взаимодействия производного нистатина с соответствующим альдегидом с образованием имина и восстановления имина, предпочтительно in situ, путем добавление восстановителя. Предпочтительный восстановитель представляет собой NaBH3CN.
Еще одно предпочтительное химическое взаимодействие представляет собой реакцию Амадори (Amadori) по аминогруппе микозамина. Реакция Амадори может быть осуществлена путем взаимодействия производного нистатина с соответствующим(и) редуцирующим(и) сахаром(ами).
Предпочтительный способ по изобретению позволяет получить описанные выше соединения формулы (I). В таких способах модификации по положению С5 модифицируют R1 и/или R2 относительно нистатина, модификации по С9 модифицируют R3 и/или R4 относительно нистатина, модификации по С10 модифицируют R5 и/или R6 относительно нистатина и модификации по С16 модифицируют R7 относительно нистатина. Модификации по аминогруппе микозамина модифицируют R8 и R9 относительно нистатина. Предпочтительные способы представляют собой способы получения определенных здесь R1-R9, как предпочтительных по сравнению с соединениями формулы I.
Соединения могут быть очищены при помощи любого обычного способа, например путем кристаллизации, хроматографии и так далее. Соединения формулы I по изобретению содержат множество хиральных центров, и, сами по себе, существуют в различных стереоизомерных формах. Особенно предпочтительные соединения представлены на Фиг.1.
Соединения по изобретению могут быть использованы в широком диапазоне применений для ингибирования роста или уничтожения грибов. Например соединения по изобретению могут быть использованы в качестве дезинфицирующих средств или в качестве консервантов (например в продуктах питания и косметических средствах). Применение соединений по изобретению в качестве дезинфицирующего средства или в качестве консерванта образует еще один аспект изобретения.
Для применения в качестве дезинфицирующего средства или в качестве консерванта соединения по изобретению могут быть использованы самостоятельно или в комбинации с другими противогрибковыми и/или антибактериальными агентами. Соединения могут быть использованы самостоятельно или, более предпочтительно, в смеси с носителем, разбавителем или эксципиентом.
Соединения по изобретению, в частности, подходят для лечения или профилактики грибковых инфекций. Соединения и фармацевтические композиции по изобретению особенно подходят для лечения или профилактики грибковых инфекций внутренних и внешних участков организма. Примеры участков организма, которые можно лечить, включают кожу, рот, влагалище и желудочно-кишечный тракт.Соединения и композиции, в частности, подходят для лечения инвазивных (например системных) грибковых инфекций.
Соединения по изобретению идеально обладают широким спектром активности, но особенно подходят для лечения грибковых инфекций, вызванных видами Candida, Cryptococcus, Aspergillus, Colletotrichum, Geotrichum, Hormonema, Lecythophora, Paecilomyces, Penicillium, Rhodotorula, Fusarium, Saccharomyces, Trichoderma, Trichophyton и Scopularilopsis, особенно видами Candida и Cryptococcus (например видами Candida).
Для применения в таком лечении соединения могут быть приготовлены в виде фармацевтических композиций любым обычным образом с одним или более чем одним носителем, эксципиентом и/или разбавителем. Конкретная фармацевтическая композиция зависит от желаемого способа введения и легко может быть определена специалистом в данной области техники. Примеры подходящих носителей, эксципиентов и разбавителей включают воду, этанол, глицерин, полиэтиленгликоль, хлорид натрия, дезоксихолат натрия, сахара (например глюкозу, сахарозу или лактозу), крахмалы (например кукурузный крахмал или маисовый крахмал), микрокристаллическую целлюлозу, камеди (например трагакантовую камедь), сорбит, маннит, ксилит, стеарат магния, поливинилпирролидон, жирные кислоты (например стеариновую кислоту), жиры, воски, карбонат кальция, хлорид кальция и лимонную кислоту.
Такие фармацевтические композиции дополнительно могут содержать один или более чем один увлажнитель, подсластитель (например сахар, аспартам или сахарин), смазывающие агенты, стабилизаторы, эмульгаторы суспендирующие агенты, консерванты, корригенты (например ванилин, масло мяты перечной или фруктовый корригент) и/или агенты, усиливающие всасывание.
Соединения по изобретению при желании могут быть введены вместе с другими (например одним, двумя или тремя) фармацевтически активными веществами. Преимущество, ассоциированное с применением комбинации соединений по изобретению с дополнительным(и) (например одним) активным(и) веществом(ами), заключается в том, что оно может увеличивать спектр заболеваний, для которых композиция подходит для применения в качестве лекарственного средства. Дополнительно или альтернативно, благоприятно может быть достигнуто уменьшение дозы соединения по изобретению и/или дополнительного(ых) активного(ых) вещества(веществ). Последнее, в частности, благоприятно, когда такое дополнительное активное вещество ассоциировано с известными побочными эффектами.
Дополнительные активные вещества, которые могут быть использованы в композициях по изобретению, включают другие противогрибковые агенты и антибиотики. Типичные противогрибковые агенты представляют собой азолы и эхинокандины. Типичные антибиотики включают демеклоциклин, ацетонид триамцинолона, сульфат неомицина, грамицидин, окситетрациклин и эритромицин.
Фармацевтические композиции, используемые по изобретению, могут быть введены перорально, ректально (например с использованием суппозитория), местно или системно. Выбранный путь зависит, например от заболевания и/или субъекта, которого лечат, хотя композиции для перорального и системного введения являются предпочтительными. Композиции для системного введения являются особенно предпочтительными.
Композиции могут быть представлены в любой форме, адаптированной для применения при введении выбранным путем. Формы, подходящие для перорального введения, включают, например обычные таблетки или таблетки с оболочки, таблетки с длительным высвобождением, жевательные таблетки, мягкие капсулы, твердые капсулы, суспензии и сиропы. Предпочтительные формы для применения по изобретению представляют собой таблетки, суспензии и сиропы, в частности таблетки.
Формы, подходящие для системного введения, могут, например представлять собой композиции для интрадермального, интраперитонеального введения или внутривенной инъекции или инфузии. Композиции для внутривенной инъекции являются особенно предпочтительными.
Формы, адаптированные для местного введения, включают композиции для введения в кожу и слизистые оболочки (например гели, кремы, спреи, лосьоны, мази и аэрозоли). Соединения также могут быть приготовлены в виде композиций для ректального или вагинального введения, таких как суппозитории или удерживающие клизмы.
Количество соединения по изобретению, используемое для любого применения, может быть легко определено специалистом в данной области техники. Например, для применения в качестве дезинфицирующего средства или консерванта количество соединения, которое требуется, представляет собой количество, которые ингибирует рост или является летальным для грибов-мишеней. Хотя действительное количество зависит от конкретного гриба-мишени и применения, для применения в качестве дезинфицирующего средства или консерванта соединения по изобретению типично используют в количествах от 0,5 до 5 масс.%, более предпочтительно от 1 до 3 масс.% раствора дезинфицирующего средства или материала, который консервируют.
Для применения для лечения грибковых инфекций количество соединения по изобретению, а также любого(ых) другого(их) возможного(ых) активного(ых) вещества(веществ), присутствующих в смеси, может быть легко определено специалистом в данной области техники и зависит от нескольких факторов, включающих природу любого(ых) возможного(ых) дополнительного(ых) активного(ых) вещества(веществ), способа введения, заболевания, которое лечат, и массы субъекта. Как правило, тем не менее, соединения по изобретению могут быть использованы в количествах, составляющих от 0,01 до 50 масс.% композиции, предпочтительно 1-20 масс.%.
Изобретение описано со ссылкой на следующие неограничивающие объем изобретения примеры и графические материалы, где:
на Фиг.1 представлена структура ряда соединений по изобретению;
на Фиг.2 представлены UV (ультрафиолетовые)-изографики (isoplot) S44HP и экстракта в DMSO (диметилсульфоксиде) мутанта с инактивированным ER5 и NysN, продуцирующего соединение 1;
на Фиг.3 представлен спектр TOF (времяпролетный) основного полиенового пика из экстракта в DMSO мутанта с инактивированными ER5 и NysN. Пики с m/z=980 и 1034 представляют собой внутренние стандарты;
на Фиг.4 представлены UV-изографики экстрактов в DMSO штамма GG5073SP (эталонного штамма), продуцирующего S44HP, и мутанта с инактивированными ER5 и KR17, продуцирующего соединение 2;
на Фиг.5 представлен спектр TOF основного полиенового пика из экстракта в DMSO мутанта с инактивированными ER5 и KR17;
на Фиг.6 представлены UV-изографики экстрактов в DMSO штамма GG5073SP (эталонного штамма), продуцирующего S44HP, и мутанта с инактивированными ER5, KR17 и NysN, продуцирующего соединение 3;
на Фиг.7 представлен спектр TOF основного полиенового пика из экстракта DMSO мутанта с инактивированными ER5, KR17 и NysN. Пики с m/z, равными 980 и 1034, представляют собой внутренние стандарты;
на Фиг.8 представлены UV-изографики экстрактов в DMSO штамма GG5073SP (эталонного штамма), продуцирующего S44HP, и мутанта с инактивированными ER5 и KR15, продуцирующего соединение 4; и
на Фиг.9 представлены результаты тестирования противогрибковой активности in vivo для соединений 1 и 11;
на Фиг.10-16 представлены схемы получения различных соединений по изобретению;
на Фиг.17 представлены результаты тестирования in vivo новых соединений по изобретению в нейтропенической мышиной модели диссеминированного кандидоза.
ПРИМЕРЫ
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ
Получали соединения, перечисленные в таблице 1. Точная стереохимическая конфигурация каждого из этих соединений представлена на Фиг.1.
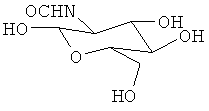
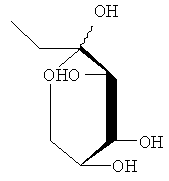

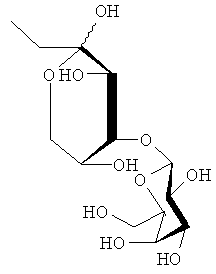

Синтез of S44HP
Это соединение получали, как описано в Borgos S.E. et al., Arch Microbiol. 2006 Apr; 185(3): 165-71.
Синтез Соединений 1-4
Соединения 1-4 получали путем генетической манипуляции с использованием следующих комбинаций сайт-специфических мутаций в генном кластере биосинтеза нистатина (последовательность - в Brautaset, Т., et al., Chem Biol. 2000 Jun; 7(6):395-403:
Соединение 1 мутации по ER5 + NysN;
Соединение 2 мутации по ER5 + KR17;
Соединение 3 мутации по ER5 + KR17 + NysN;
Соединение 4 мутации по ER5 + KR15.
Использовали ER5 инактивирующий вектор, как описано в Borgos S.E. et al., Arch Microbiol. 2006 Apr; 185(3): 165-71.
Авторами изобретения разработаны и получены следующие конструкции:
NysN:
Векторы pSOK201nysN4.1-CL346ST и pSOK201nysN4.1-CL346AS для введения мутаций CL346ST и CL346AS, соответственно, в nysN
Фрагмент длиной 4,1 тысяч пар оснований (kb) Ncol/Xbal рекомбинантного клона лямбда N95 (Brautaset, Т., et al., Chem Biol. 2000 Jun; 7(6):395-403) клонировали в соответствующие сайты плазмиды pLITMUS28. Из получающейся в результате конструкции вырезали полноразмерную вставку 4,1 kb при помощи EcoRI/Hindlll и клонировали в соответствующие сайты pGEM11zf(+). Из получающейся в результате плазмиды, обозначенной pGEM11nysN4.1, область 1,5 kb, включающую активный сайт NysN, подвергали амплификации при помощи PCR (полимеразной цепной реакции) с использованием следующих праймеров:
conA-1F: 5′-ttttgaaTTCTTCAAGCCGATGAGCC-3′ (смысловой), и
conA-1R: 5′-ttttaaqctTGGTCGAACAGGTCCGG-3′ (антисмысловой).
Праймеры вводили сайты EcoRI и Hindlll (подчеркнутые), которые использовали для клонирования получающегося в результате PCR-фрагмента в соответствующие сайты pGEM11zf(+), с получением плазмиды pGEM11nysN1.5. Последнюю плазмиду использовали в качестве матрицы для сайт-направленного мутагенеза (с использованием набора QuickChange, Stratagene), с использованием следующих мутагенных олигонуклеотидов:
Мутация CL346ST:
CL346ST-F: 5′- CTACGGTGTCCACCAGTCGACGGGCCAGAACCTGGTGC-3′
CL346ST-R: 5′-GCACCAGGTTCTGGCCC GTCG ACTGGTGGACACCGTAG-3′
Мутация CL346AS:
CL346AS-F: 5′-TCGGCTACGGTGTCCAC GCTA GCCTGGGCCAGAACCTGG-3′
CL346AS-R: 5′-CCAGGTTCTGGCCCAGGCTAGC GTGGACACCGTAGCCGA-3′
Мутантные нуклеотиды указаны жирным шрифтом, тогда как новый введенный сайт рестрикции (Sail для мутации CL346ST; Nhel для мутации CL346AS) подчеркнут в последовательностях. Мутации идентифицировали при помощи рестрикционного анализа Sail или Nhel и полные вставки правильных мутантных векторов подтверждали при помощи секвенирования ДНК. Мутантные фрагменты 1,3 kb FspAI/Bpu 1102I из получающихся в результате плазмид использовали для замещения соответствующего фрагмента плазмиды pGM11nysN4.1 с получением плазмиды pGM11nysN4.1-CL346ST и pGM11nysN4.1-CL346AS. Из последних конструкций вставки длиной 4,1 kb, содержащие мутантные гены nysN, вырезали при помощи EcoRI/Hindlll, и лигировали в скелет 3,1 kb EcoRI/Hindlll pSOK201, с получением nysN инактивирующих плазмид pSOK201nysN4.1-CL346ST и pSOK201nysN4.1-CL346AS.
Для инактивации nysN в штамме, продуцирующем соединение 1, использовали конструкцию pSOK201nysN4.1-CL346ST. Для инактивации nysN в штамме, продуцирующем соединение 3, использовали конструкцию pSOK201 nysN4.1-CL346AS.
KR17:
Вектор pKR17m для введения мутации YA5145FE в KR17 NysJ
Фрагмент 4,0 kb Pmll/BamHI плазмиды pL98E (Brautaset, Т., et al., Chem Biol. 2000 Jun; 7(6):395-403) вырезали и лигировали в сайты Hincll/BamHI вектора pGEM3zf(+) с получением плазмиды рВВ4.0. Фрагмент ДНК 1,5 kb, включающий область активного сайта KR17, подвергали амплификации при помощи PCR из рВВ4.0 с использованием следующих праймеров:
KR17-F: 5′-ttttctgCAGGCCGCGGTGCGCGC-3′ (смысловой), и
KR17-R: 5′-TCCGGCATGGTCCGTGAAACC-3′ (антисмысловой).
Продукт PCR подвергали расщеплению с конца при помощи Pstl (сайт распознавания подчеркнут в праймере) и Sacl (сайт распознавания в амплифицированном фрагменте ДНК), и фрагмент 1,4 kb лигировали в соответствующие сайты pLITMUS28. Получающуюся в результате плазмиду pLIT1.4 использовали в качестве матрицы для сайт-направленного мутагенеза со следующими мутагенными олигонуклеотидами:
KR17-mut1:
5′-GCCCCGGCCAGGGCAAC TT CGAA GCCGGCAACACGTTCC-3′
KR17-mut2:
5′-GGAACGTGTTGCCGGC TT CGAAGTTGCCCTGGCCGGGGC-3′.
Мутантные нуклеотиды выделены жирным шрифтом, тогда как новый введенный сайт рестрикции BstBI подчеркнут в последовательностях. Правильность мутации подтверждали при помощи расщепления BstBI и полноразмерную вставку мутантной плазмиды подтверждали при помощи секвенирования ДНК. Из получающейся в результате плазмиды pLIT1.4m фрагмент 1072 пар оснований (bp) BcII/AccIII вырезали и использовали для замены соответствующего фрагмента плазмиды рВВ4.0 с получением плазмиды рВВ4.0 т. Полноразмерную вставку 4.0 рВВ4.0 т вырезали при помощи EcoRI + HindIII и лигировали вместе с 3.0 kb EcoRI/HindIII скелетом плазмиды pSOK201, с получением KR17 инактивирующего вектора pKR17m.
KR15:
Вектор pKR15m для введения мутации YA1888FE в KR15 NysJ
Фрагмент 3,6 kb KpnI/PmII pL20X (Brautaset, Т., et al., Chem Biol. 2000 Jun;7(6):395-403) вырезали и лигировали в сайты KpnI/HincII pGEM3zf(+) с получением рКР3.6. Последнюю плазмиду использовали в качестве матрицы для амплификации PCR фрагмента ДНК 1,1 kb с использованием следующих праймеров:
KR15-F1: 5′-ttttqaaTTCCCGACGGCCTCTCCTACC-3′ (смысловой), и
KR15-R1: 5′-ttttaagCTTGCCGAGTCGGTTGCGC-3′ (антисмысловой).
Получающийся в результате продукт PCR подвергали расщеплению по концу при помощи EcoRI/HindIII (сайты рестрикции подчеркнуты в праймерах) и лигировали в соответствующие сайты pLITMUS28 с получением pLIEH1.1. Последняя плазмида служила в качестве матрицы для сайт-направленного мутагенеза с использованием следующих мутагенных олигонуклеотидов:
mutKR15-1F: 5′-CCGGGCCAGGCCAACTTCGAA GCCGGCAACACCTTCCTCG-3′
mutKR15-1R: 5′-CGAGGAAGGTGTTGCCGGC TT CGAAGTTGGCCTGGCCCGG-3′.
Мутантные нуклеотиды показаны жирным шрифтом, и подчеркнут новый введенный сайт BstBI. Правильность мутации подтверждали при помощи расщепления BstBI, и полноразмерную вставку мутантной плазмиды подтверждали при помощи секвенирования ДНК. Из мутантной плазмиды вырезали фрагмент 1045 bp FspA1/AccIII и использовали для замены соответствующей области рКР3.6 с получением pKP3.6mut. Полноразмерную вставку 3,6 kb pKP3.6mut затем вырезали при помощи EcoRI/HindIII и лигировали с фрагментом 3,2 kb EcoRI/HindIII pSOK201 с получением KR15 инактивирующего вектора pKR15m.
Введение векторов замещения в штаммы S.noursei:
Все сконструированные инактивирующие векторы трансформировали в Escherichia coli ET12567 (pUZ8002) и получающиеся в результате рекомбинаниные штаммы использовали для конъюгации инактивирующих векторов в штаммы S.noursei, как описано ранее (Brautaset, Т., et al., Chem Biol. 2000 Jun; 7(6):395-403);
- Проверка 1-го кроссинговера при помощи саузерн-блоттинга + PCR.
- Отбор 2х кандидатов кроссинговера (чувствительных к апрамицину).
- Генетическая характеристика 2 кандидатов кроссинговера при помощи саузерн-блоттинга + PCR.
- Дополнение правильных 2х кроссинговерных мутантов с pNA0 (Brautaset et al., 2000, Chem. Biol., 7: 395-403).
Дополняющие рекомбинантные штаммы подвергали ферментации, и продукцию соответствующих молекул подтверждали анализом посредством высокоэффективной жидкостной хроматографии (HPLC) и времяпролетной масс-спектрометрии (MS-TOF).
Соединение 1
UV (ультрафиолетовые) изографики
S44HP стандарт и экстракт в DMSO (диметилсульфоксид) из мутанта S. noursei с инактивированными ER5 и NysN представлены на изографиках на Фиг.2.
LC-MS (жидкостная хроматография/масс-спектрометрия) изографики:
Колонка: Zorbax SB-C18 2,1×100 мм, 3,5 мкм (Agilent Technologies).
Подвижная фаза А: 10 мМ ацетат аммония (Riedel-de-Haën #25006), рН не доводили, Подвижная фаза Б: 100% ацетонитрил (Rathburn, чистота для HPLC)
Поток: 0,22 мл/мин
Температура колонки: Температура окружающей среды Параметры времяпролетной масс-спектрометрии (TOF-MS): API-ES (ионизация при атмосферном давлении-электрораспыление) ионизация в режиме отрицательных ионов Осушающий газ: 10 л/мин
Давление в распылителе: 40 индикаторных ф./кв.дюйм (279853 Па)
Темп. осушающего газа: 350°С
Напряжение на капилляре: 3000 В
Фрагментор: 200 В
TOF-MS спектр
TOF-MS спектр представлен на Фиг.3, где пики с m/z, равные 966, 980 и 1034, представляют собой внутренние референсные массы. Теоретическое m/z (отрицательный ион) для соединения 1 (C47H75NO15) составляет 892,5063. Это m/z обнаружено с высокой точностью и хорошо коррелирует с гептаеновым UV-пиком. Потеря воды (1 и 2 молекулы) во время ионизации соединения 1 также позволяет получить пики масс с Δm/z=-18 и Δm/z=-36, соответственно.
Очистка при помощи препаративной LC
Очистку осуществляли на препаративной обращенно-фазовой колонке.
Препаративный способ:
Колонка: Agilent Prep-C18 50×250 мм, 10 мкм (Agilent Technologies).
Подвижная фаза: 10 мМ ацетат аммония (Riedel-de-Haën #25006), рН доводили до 4,0 уксусной кислотой (JT Baker #6002), Подвижная фаза Б: 100% метанол (Lab-Scan, чистота для HPLC)
Поток: 85 мл/мин
Температура колонки: Температура окружающей среды
После очистки при помощи препаративной LC соединение 1, как было показано, составляют более 97% полиеновых макролидов в образце, как определили при помощи данных UV и MS.
Способ LC-MS:
Колонка: Zorbax Bonus-RP 2,1×50 мм, 3,5 мкм (Agilent Technologies).
Подвижная фаза А: 10 мМ ацетат аммония (Riedel-de-Haën #25006), рН доводили до 4,0 уксусной кислотой (JT Baker #6002), Подвижная фаза Б: 100% ацетонитрил (Rathbum, чистота для HPLC)
Поток: 0,3 мл/мин
Температура колонки: Температура окружающей среды
Для полиенов без карбоксильной группы (а именно соединение 1), способ также проводили с 20 мМ бикарбонатом аммония (Fluka #09830), рН доводили до 7,0 уксусной кислотой (JT Baker #6002) в качестве подвижной фазы А, поскольку увеличенный рН обеспечивал несколько лучшее разделение между полиенами с карбоксильной ионизируемой группой и без нее.
Параметры MS:
API-ES ионизация в режиме отрицательных ионов
Осушающий газ: 12 л/мин
Давление в распылителе: 35 индикаторных ф./кв.дюйм (244872 Па)
Темп, осушающего газа: 350°С
Напряжение на капилляре: 3000 В
Соединение 2
UV изографики
Экстракты в DMSO штамма GG5073SP (продуцирующего S44HP) и мутанта с инактивированными доменами ER5 и KR17 представлены на изографиках на Фиг.4.
Способ LC-MS и параметры TOF-MS: такие же, как для Соединения 1.
Данные TOF-MS
Спектр TOF-MS представлен на Фиг.5. Теоретическое m/z (отрицательный ион) для соединения 2 (C47H71NO17) составляет 920,4649. Это m/z обнаружено с приемлемой точностью (ошибка менее 3 млн-1) и хорошо коррелирует с гептаеновым UV-пиком.
Очистка при помощи препаративной LC
Очистку осуществляли на препаративной обращенно-фазовой колонке.
Препаративный способ:
Колонка: Agilent Prep-C18 50×250 мм, 10 мкм (Agilent Technologies).
Подвижная фаза: 10 мМ ацетат аммония (Riedel-de-Haën #25006), рН доводили до 4,0 уксусной кислотой (JT Baker #6002), Подвижная фаза Б: 100% метанол (Lab-Scan, чистота для HPLC)
Поток: 100 мл/мин
Температура колонки: Температура окружающей среды
После очистки при помощи препаративной LC соединение 2, как было показано, составляют более 95,2% полиеновых макролидов в образце, как определено при помощи данных UV и MS.
Способ LC-MS: такой же, как для Соединения 1.
Соединение 3
UV изографики
Экстракты в DMSO штамма GG5073SP (продуцирующего S44HP) и мутанта с инактивированными доменами ER5 и KR17 и инактивированным NysN представлены на изографиках на Фиг.6.
Способ LC-MS:
Колонка: Zorbax Bonus-RP 2,1×50 мм, 3,5 мкм (Agilent Technologies).
Подвижная фаза А: 10 мМ ацетат аммония (Riedel-de-Haën #25006), рН доводили до 4,0 уксусной кислотой (JT Baker #6002), Подвижная фаза Б: 100% ацетонитрил (Rathburn, чистота для HPLC)
Поток: 0,3 мл/мин
Температура колонки: Температура окружающей среды
Параметры TOF-MS: такие же, как для Соединения 1
Спектр TOF-MS представлен на Фиг.7, где пики m/z, равные 980 и 1034, представляют собой внутренние референсные массы. Теоретическое m/z (отрицательный ион) для соединения 3 (C47H73NO15) составляет 890,4907. Это m/z обнаружено с высокой точностью (смотри масс-спектр) и хорошо коррелирует с гептаеновым UV-пиком. Потеря воды (1 и 2 молекулы) во время ионизации соединения 3 также позволяет получить массовые пики с Δm/z=-18 и Δm/z=-36, соответственно. Кроме того, карбоксилированный аналог (с m/z=920,4644 в представленном MS-спектре) демонстрирует значительную степень хроматографической соэлюции. Известно, что карбоксилированные соединения обладают значительно более высокой эффективностью ионизации в режиме MS с отрицательными ионами, таким образом, относительное количественное определение не должно быть основано на интенсивностях сигнала в представленном MS-спектре.
Соединение 4
UV изографики
Экстракты в DMSO штамма GG5073SP (продуцирующего S44HP) и мутанта с инактивированными доменами ER5 и KR15 представлены на изографиках на Фиг.8.
Способ LC-MS: такой же, как для Соединения 3.
Синтез соединений 5-23
Общая информация
Реакции контролировали при помощи TLC (тонкослойной хроматографии) на силикагелевых пластинах Merck 60F254 в системе растворителей CHCl3:MeOH:H2O:HCOOH. (13:6:1:0,1). Чистоту получающихся в результате продуктов определяли в соответствии с HPLC, и она составляла 80-90%. Аналитическую обращенно-фазовую HPLC осуществляли на приборе Shimadzu HPLC серии LC 10 на колонке Kromasil 100-C18 (4×250 мм, размер частиц 6 мкм) при инжектируемом объеме 20 мкл и длине волны 408 нм. Система растворителей содержала 0,01 М Н3РО4 при рН 2,6 и ацетонитрил. Доля ацетонитрила варьировала от 30 до 70% в течение 30 мин при скорости потока 1,0 мл/мин.
Синтез Соединений 5-16
К смеси S44HP (18 мг, 0,02 ммоль) и 0,06 ммоль подходящего гидрохлорида амина, растворенного в 0,3 мл DMSO, порциями добавляли EtaN для доведения рН 8-8,5 и после этого в течение 15 минут 0,03 ммоль реагента РуВОР. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Последующее добавление к реакционной смеси диэтилового эфира (приблизительно 3 мл) привело к маслянистому остатку, который непрерывно встряхивали с диэтиловым эфиром (3 мл × 2). После добавления к этому маслу 5 мл ацетона образовывался желтый осадок амида. Осадок фильтровали, промывали ацетоном и затем сушили в вакууме. Все образцы получали с выходами более 90%. Аналитические данные для этих соединений обобщены в таблице 2 ниже.
Синтез Соединений 17-19
Подходящий моносахарид (D-глюкозу или D-галактозу) или дисахарид (лактозу) (0,086 ммоль) добавляли к раствору S44HP (40 мг, 0,043 ммоль) в DMF (диметилформамиде) (2 мл). Реакционную смесь поддерживали при 37°С в течение 20 часов, и затем раствор по каплям добавляли к диэтиловому эфиру (50 мл). Получающийся в результате осадок отфильтровывали, промывали диэтиловым эфиром и сушили. Получающийся желтый осадок очищали путем флэш-хроматографии (CHCl3:MeOH:H2O:HCOOH (13:6:1:0,1)). Фракции, содержащие желаемое соединение, собирали, и раствор концентрировали. Добавление диэтилового эфира затем позволило получить желтый осадок, которые отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме. Все образцы получали с выходами 40-45%. Аналитические данные для этих соединений обобщены в таблице 2 ниже.
Синтез Соединения 20
К раствору 4-N,N-диметиламинобензальдегида (0,065 ммоль) и S44HP (20 мг, 0,022 ммоль) в DMF (2 мл) добавляли NaBH3CN (4,1 мг, 0,065 ммоль). Реакционную смесь поддерживали при 37°С в течение 20 ч. Добавление диэтилового эфира (10 мл) привело к маслянистому остатку, который непрерывно встряхивали с диэтиловым эфиром (10 мл × 2). Желтый осадок образовывался после добавления ацетона (10 мл). Осадок отфильтровывали, промывали диэтиловым эфиром и сушили. Получающееся твердое вещество очищали путем флэш-хроматографии (CHCl3:МеОН:Н2О:НСООН (13:6:1:0,1)) на силикагеле Merck для колоночной хроматографии (0,040-0,063 мм). Фракции, содержащие желаемое соединение, собирали, раствор концентрировали, и добавление диэтилового эфира позволило получить желтый осадок, который отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме.
Синтез Соединений 21 и 22
К раствору N-(9-флуоренилметоксикарбонил)-3-аминопропионового альдегида (160 мг, 0,054 ммоль) и S44HP (100 мг, 0,011 ммоль) в DMF (3 мл) добавляли NaBH3CN (34 мг, 0,054 ммоль). Реакционную смесь поддерживали при 37°С в течение 20 часов, и затем по каплям добавляли к диэтиловому эфиру (200 мл). Желтый осадок смеси соединений отфильтровывали, и индивидуальные соединения разделяли и очищали путем флэш-хроматографии на силикагеле Merck для колоночной хроматографии (0,040-0,063 мм) в системе линейного градиента CHCI3:MeOH:HCOOH (3:1:0,01)→CHCl3:MeOH:H2O:HCOOH (13:6:1:0,1) с получением N-[N-(9-флуоренилметоксикарбонил)-3-аминопропил]-S44НР и N,N-ди-[N-(9-флуоренилметоксикарбонил)-3-аминопропил]-S44НР в виде желтых твердых веществ.
К раствору N-[N-(9-флуоренилметоксикарбонил)-3-аминопропил]-S44НР или N,N-ди-[N-(9-флуоренилметоксикарбонил)-3-аминопропил]-S44НР в DMSO (3 мл) добавляли пиперидин (0,1 мл). Через 2 ч при комнатной температуре добавляли диэтиловый эфир (приблизительно 7 мл) и образовывался маслянистый остаток, которые непрерывно встряхивали с диэтиловым эфиром (7 мл × 2). Добавление ацетона (10 мл) позволило получить желтый осадок соединения 21 или соединения 22, который отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме. Аналитические данные для получающихся в результате соединений 21 и 22 обобщены в таблице 2 ниже.
Синтез Соединения 23
К раствору S44HP (100 мг, 0,11 ммоль) в безводном DMF (2 мл) добавляли N-оксисукцинимидный эфир Nα,Nε-ди-(9-флуоренилметоксикарбонил)-L-лизина (221 мг, 0,33 ммоль) и Et3N (15,3 мкл, 0,11 ммоль). Реакционную смесь поддерживали при 37°С в течение 1 ч, затем добавляли Н2О (5 мл). Смесь экстрагировали с н-BuOH (3×5 мл). Органические фракции объединяли и промывали 0,01 н HCl (1×5 мл) и Н2О (3×5 мл). Раствор концентрировали, и добавление диэтилового эфира позволило получить желтый осадок, который отфильтровывали, промывали диэтиловым эфиром и очищали путем флэш-хроматографии на силикагеле (CHCl3:MeOH:H2O:HCOOH (13:4:0,5:0,01)). Фракции, содержащие желаемое соединение, собирали, раствор концентрировали, и добавление диэтилового эфира позволило получить желтый осадок, который отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме.
К выделенному желтому твердому веществу (30 мг) в DMSO (3 мл) добавляли пиперидин (0,1 мл). Через 2 ч при комнатной температуре добавляли диэтиловый эфир (приблизительно 7 мл), образовывался маслянистый остаток, и его непрерывно встряхивали с диэтиловым эфиром (7 мл × 2). Добавление ацетона (10 мл) позволило получить желтый осадок, который отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме.
Таблица 2. TLC (Rf), HPLC (Rt), масс-спектры MALDI (ионизация лазерной десорбцией с использованием матрицы) и данные по растворимости для соединений 5-23
Растворимость в воде:+водорастворимое,±малораствормое, -нерастворимое
В соответствии с принципами синтеза, изложенными выше или ниже, также синтезировали соединения 24-29. Дополнительно, следующие дополнительные соединения получали на основе протоколов синтеза, изложенных на Фиг.10-16, и следующих схемах и описании.


Соединение 28 представляет собой соль глутамат соединения 48.
Синтез дополнительных производных S44HP (Соединения 30-48)
Общая информация
Реакции контролировали при помощи TLC на силикагелевых пластинах Merck 60F254 в системе растворителей: I - CHCl3:MeOH:H2O:HCOOH (13:6:1:0,1), II - CHCl3:MeOH:H2O:HCOOH (7:1:0,01:0,01), III CHCl3:MeOH:H2O:NH4OH конц. (13:7:1:0,1), IV - AcOEt:n-ProOH:NH4OH конц. (15:10:10).
Аналитическую обращеннофазовую HPLC осуществляли на приборе Shimadzu HPLC серии LC 10 на колонке Kromasil 100-C18 (4×250 мм, размер частиц 6 мкм) при инжектируемом объеме 20 мкл и длине волны 408 нм при скорости потока 1,0 мл/мин. Система растворителей включает: А - 0,2% HCOONH4 pH 4,5 и MeCN, доля MeCN варьирует от 30 до 70% в течение 30 мин.; В - 0,2% HCOONH4 pH 4,5 и MeCN: 25→65%, 40 мин.; С - 0,2% HCOONH4 pH 4,5 и MeCN, 30→90%, 30 мин., 90→90%, 10 мин.; D - изократическая система - 35% MeCN; Е - 0,01 М Н3РО4 при рН 2,6 и MeCN, 30→70%, 30 мин. F - 0,01 М Н3РО4 при рН 2,6 и MeCN, 40→80%, 30 мин.
Димодифицированные производные S44HP
N-Фруктозил S44HP N,N-диметиламинопропиламид (DMAP), - гидроксипропиламид (HP) или -N,N-диметиламиноэтила (DMAE) амид (Соединения 30, 36, 41, соответственно); N-тагатопиранозил S44HP DMAP амид (Соединение 31) или N-тагатопиронозил S44HP DMAE амид (Соединение 42) (Схема 1, левая сторона).
1я Стадия (амидирование) - S44HP DMAP амид (Соединение 11), HP амид (Соединение 12) или DMAE амид (Соединение 48).
К смеси S44HP (36 мг, 0,04 ммоль) и 0,12 ммоль подходящего гидрохлорида амина, растворенного в 0,6 мл DMSO, порциями добавляли Et3N для доведения рН 8-8,5 и после этого в течение 15 минут 0,06 ммоль реагента PyBOP. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Последующее добавление к реакционной смеси диэтилового эфира (приблизительно 5 мл) приводило к маслянистому остатку, который непрерывно встряхивали с диэтиловым эфиром (5 мл × 2). После добавления к этому маслу 8 мл ацетона образовывался желтый осадок амида. Осадок фильтровали, промывали ацетоном и затем сушили в вакууме. Неочищенный амид (чистота 85-90% в соответствии с данными HPLC, и содержащий соли) очищали путем колоночной хроматографии на Sephadex G-25 (Pharmacia Fine Chemicals AB, Uppsala, Sweden). Колоночную хроматографию осуществляли с использованием воды. Чистоту фракции контролировали в соответствии с HPLC, что приводило в результате к целевой чистоте соответствующего амида S44HP более 92% в соответствии с HPLC.
2я Стадия (перегруппировка амидов Амадори): D-Глюкозу или D-галактозу (0,086 ммоль) добавляли к раствору подходящего амида S44HP (Соединение 11, 12, 48) (0,040 ммоль) в DMF (2 мл). Реакционную смесь поддерживали при 37°С в течение 20 часов. Затем реакционную смесь по каплям добавляли к диэтиловому эфиру (50 мл). Получающийся в результате осадок отфильтровывали, промывали диэтиловым эфиром и сушили. Неочищенное соединение (чистота 85-90% в соответствии с данными HPLC, и содержащее соли) очищали путем колоночной хроматографии на Sephadex G-25 (Pharmacia Fine Chemicals AB, Uppsala, Sweden). Колоночную хроматографию осуществляли с использованием воды. Чистоту фракции контролировали при помощи TLC, система IV. Фракции, содержащие желаемое соединение, собирали, и раствор концентрировали. Добавление ацетона позволило получить желтый осадок, которые отфильтровывали, промывали ацетоном и сушили в вакууме. Соединение 41 получали в виде гидрохлорида. К раствору Соединения 41 в метаноле добавляли 0,1 н HCl в метаноле до рН приблизительно 4. Добавление диэтилового эфира позволило получить желтый осадок, который отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме. Чистота N-алкильного производного S44HP амида составляла более 92%. Аналитические данные для этих соединений (30, 36, 41, 31, 42) обобщены в таблице 4 выше.
N-(4-N,N-ди-метиламинобензил) S44HP DMAP амид, -HP амид или -DMAE амид (Соединение 32, 37, 43, соответственно) (Схема 1 (Фиг.10), правая сторона)
1я Стадия (амидирование). S44HP DMAP амид (Соединение 11), HP амид (Соединение 12) или DMAE амид (Соединение 48) получали, как описано выше для Соединения 30 (1я стадия).
2я Стадия (восстановительное N-алкилирование амидов). Раствор 4-N,N-диметиламинобензальдегида (0,065 ммоль) и подходящего S44HP амида (Соединение 11,12 или 48) (0,022 ммоль) в DMF (2 мл) поддерживали при 37°С в течение 2 ч, затем добавляли NaBH3CN (4,1 мг, 0,065 ммоль). Реакционную смесь поддерживали при 37°С в течение 20 ч. Добавление диэтилового эфира (10 мл) приводило к маслянистому остатку, который непрерывно встряхивали с диэтиловым эфиром (10 мл × 2). Желтый осадок образовывался после добавления ацетона (10 мл). Осадок отфильтровывали, промывали диэтиловым эфиром и сушили. Неочищенное соединение, содержащее соли, очищали путем колоночной хроматографии на силикагеле Merck с использованием системы CHCl3:MeOH:H2O:HCOOH (13:6:1:0,1). Чистоту фракций контролировали при помощи TLC, система I. Фракции, содержащие желаемое соединение, собирали, и раствор концентрировали. Добавление ацетона позволило получить желтый осадок, который отфильтровывали, промывали ацетоном и сушили в вакууме. Чистота N-алкильного производного соответствующего S44HP амида составляла более 92%. Аналитические данные для этих соединений обобщены в таблице 4 выше.
N-(N,N-диметилглицил) S44HP DMAE амид, -HP амид или -DMAP амид (Соединение 39, 45, 34, соответственно) (N-аминоацилирование амидов, Схема 2 (Фиг.11, левая сторона)
К смеси N,N-диметилглицина (0,04 ммоль) и подходящего S44HP DMAE амида (Соединение 48), -HP амида (Соединение 12) или -DMAP амида (Соединение 11) (смотри Схему 1) (0,02 ммоль) в 0,5 мл DMSO для доведения рН 8 добавляли Et3N, и после этого 0,03 ммоль реагента PyBOP порциями в течение 15 минут. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Последующее добавление к реакционной смеси диэтилового эфира (приблизительно 3 мл) приводит к маслянистому остатку, который непрерывно встряхивали с диэтиловым эфиром (3 мл × 2). После добавления к этому маслу 5 мл ацетона образовывался желтый осадок амида. Осадок фильтровали, промывали ацетоном и затем сушили в вакууме с получением N-(N,N-диметилглицил) S44HP DMAE амида, -HP амида или -DMAP амида (Соединение 39, 45, 34). Аналитические данные для этих соединений обобщены в таблице 4 выше.
N-(N-L-Лизил) S44HP HP амид и -DMAE амид (Соединение 38 и 44, соответственно) (Схема 2, правая сторона)
1я Стадия: (N-[N-Fmoc-амино]ацилирование) - N-(Nα,Nε-ди-Fmoc-L-лизил) S44HP
К охлажденному приблизительно до +5°С раствору Nα,Nε-ди-(9-флуоренилметоксикарбонил)-L-лизина (180 мг, 0,33 ммоль) добавляли N-гидроксибензотриазол (54 мг, 0,4 ммоль) в безводном DMF (1,2 мл), DCC (62 мг, 0,3 ммоль), и реакционную смесь перемешивали приблизительно при +5°С в течение 1 ч. Остаток DCU отфильтровывали, и получающийся элюат добавляли к S44HP (184 мг, 0,2 ммоль) в DMF (1,0) мл). Затем к реакционной смеси по каплям в течение 10 мин добавляли (i-Pro)2EtN (0,1 мл, 0,81 ммоль) при встряхивании. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч под контролем путем TLC в системе: CHCl3:MeOH:H2O:HCOOH (6:1:0,01:0,02), и затем добавляли 20 мл этилового эфира. Получающийся осадок отфильтровывали, промывали смесью этилового эфира:ацетона (1:1, 10 мл × 3) и сушили в вакууме с получением 202 мг желтого порошка. Неочищенное соединение очищали путем колоночной хроматографии на силикагеле Merck. Колоночную хроматографию осуществляли с использованием системы: CHCl3:MeOH:H2O:HCOOH (9:1:0,01:0,02 → 7:1:0,01:0,02). Чистоту фракции контролировали при помощи TLC в системе II. Фракции, содержащие желаемое соединение, собирали, и раствор концентрировали. Добавление ацетона позволило получить желтый осадок, который отфильтровывали, промывали ацетоном и сушили в вакууме с получением 60 мг чистого N-[Nα,Nε-ди-(9-флуоренилметоксикарбонил)-L-лизил] S44HP (более 96%, HPLC, Rt=22,80, система В). Масс-спектры MALDI. Найдено: 1500,542 [M-H2O+Na]+1. Вычислено для C83H105N3O22 1495,72 (точная масса).
2я Стадия (амидирование N-(Nα,Nε-ди-Fmoc-L-лизил) S44HP - N-(Nα,Мε-ди-Fmoc-L-лизил) S44HP HP амид или -DMAE амид:
К смеси N-(Nα,Nε-ди-Fmoc-L-лизил) S44HP (0,02 ммоль) и 0,06 ммоль 3-гидроксипропиламингидрохлорида или дигидрохлорида 4-(N,N-диметиламино)пропиламина, растворенного в 0,3 мл DMSO, порциями добавляли Et3N для доведения рН 7-7,5 и после этого в течение 15 минут 0,03 ммоль реагента PyBOP. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа под контролем TLC в системе CHCl3:MeOH:H2O:NH4OH конц. (5:1:0,01:0,01). Последующее добавление к реакционной смеси диэтилового эфира (приблизительно 3 мл) приводило к маслянистому остатку, которые непрерывно встряхивали с диэтиловым эфиром (3 мл × 2). После добавления к этому маслу 5 мл ацетона образовывался желтый осадок амида. Осадок фильтровали, промывали ацетоном и затем сушили в вакууме с получением соответствующего N-(Nα,Nε-ди-Fmoc-L-лизил) S44HP HP-амида или (Nα,Nε-ди-Fmoc-L-лизил) S44HP -DMAE амида, имеющего чистоту приблизительно 90%, в соответствии с данными HPLC: Rt=21,43 (С) и 20,39 (В), соответственно. Данные TLC: Rf=0,47 и Rf=0,43, соответственно, в системе CHCl3:MeOH:H2O:NH4OH конц. (5:1:0,01:0,01).
3я Стадия (удаление Fmoc). Подходящий N-(Nα,Nε-ди-Fmoc-L-лизил) S44HP амид (30 мг) растворяли в DMSO (3 мл), добавляли пиперидин (0,1 мл). Через 2 ч при комнатной температуре добавляли диэтиловый эфир (приблизительно 7 мл), образовывался маслянистый остаток, и его непрерывно встряхивали с диэтиловым эфиром (7 мл × 2). Добавление ацетона (10 мл) позволило получить желтый осадок, которые отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме. Соединение 44 получали в виде гидрохлорида. К раствору Соединения 44 в метаноле добавляли 0,1 н HCl в метаноле до рН приблизительно 4. Добавление диэтилового эфира позволило получить желтый осадок, который отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме. Аналитические данные для этих двух соединений обобщены в таблице 4 выше.
N-(4-Аминометилбензоил) S44HP DMAP амид, -HP амид, -DMAE амид (Соединение 35, 40, 46, соответственно) (Схема 3, Фиг.12, левая сторона)
1я Стадия (N-[4-Fmoc-аминометил]бензоилирование) - N-(4-Fmoc-аминометилбензоил) S44HP: Производные получали в условиях, как для Соединения 38 (1я Стадия). К раствору 4-N-Fmoc-аминометилбензойной кислоты (80 мг, 0,022 ммоль) и S44HP (100 мг, 0,11 ммоль) в DMSO (3 мл), добавляли Et3N и PyBOP (90 мг, 0,165 ммоль) для доведения рН до 7. Реакционную смесь поддерживали при комнатной температуре (к.т.) в течение 20 ч, и затем по каплям добавляли к диэтиловому эфиру (200 мл). Желтый осадок отфильтровывали и очищали путем колоночной хроматографии на силикагеле Merck для колоночной хроматографии (0,040-0,063 мм) в системе CHCl3:MeOH:HCOOH (3:1:0,01) с получением N-(4-N-Fmoc-аминометил)бензоил S44HP (Соединение 47), имеющего чистоту приблизительно 95% в соответствии сданными HPLC (Rt=18,18, система В). TLC (Rf=0,61, I)
2я Стадия (Амидирование N-[4-Fmoc-аминометил]бензоил S44HP) -N-(4-Fmoc-аминометилбензоил) S44HP DMAP амид, -HP амид, или -DMAE амид
Производные получали в условиях, как для Соединения 38 (2я Стадия), начиная с N-(4-Fmoc-аминометилбензоил) S44HP и подхоодящего DMAP-, НР- или DMAE амина и с использованием реагента PyBOP. Реакционную смесь перемешивали в течение 4 ч. Чистота составляла приблизительно 85-93% в соответствии с данными HPLC: N-(4-Fmoc-аминометилбензоил) S44HP DMAP амид (Rt=15,88, В; Rf=0,25, I), N-(4-Fmoc-аминометилбензоил) S44HP HP амид (Соединение 12) (Rt=17,14, В; Rf=0,60, I) или N-(4-Fmoc-аминометилбензоил) S44HP DMAE амид (Rt=15,95, В; Rf=0,24, I),
3я Стадия (удаление Fmoc). Производные получали в условиях, как для Соединения 38 (3я Стадия), начиная с N-(4-Fmoc-аминометилбензоил) S44HP DMAP амида, -HP амида или -DMAE амида. Аналитические данные для этих трех соединений обобщены в таблице 2 ниже.
N-L-лизил S44HP DMAP-амид (Соединение 33) (Схема 3, правая сторона)
1я Стадия (Амидирование) - S44HP DMAP-амид (Соединение 11); S44HP DMAP амид получали, как для Соединения 33 {Схема 1, левая сторона). Чистота составляла приблизительно 90% (HPLC).
2я Стадия (N-[N-Fmос-амино]ацилирование DMAP амида) - N-(Nα,Nε-ди-Fmoc-L-лизил) S44HP DMAP-амид; К раствору S44HP DMAP амида (0,02 ммоль) в DMSO (0,5 мл) добавляли Nα,Nε-ди-Fmoc-L-лизин (0,04 ммоль) и Et3N (рН 7-7,5) и после этого в течение 15 минут 0,03 ммоль реагента PyBOP. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Последующее добавление к реакционной смеси диэтилового эфира (приблизительно 3 мл) привело к маслянистому остатку, который непрерывно встряхивали с диэтиловым эфиром (3 мл × 2). После добавления к этому маслу 5 мл ацетона образовывался желтый осадок амида. Осадок фильтровали, промывали ацетоном и затем сушили в вакууме с получением N-(Nα,Nε-ди-Fmoc-L-лизил) S44HP DMAP амида, имеющего чистоту приблизительно 95% в соответствии с данными HPLC (Rt=22,01, система В).
3я Стадия (удаление Fmoc). Производные получали в условиях, как для Соединения 38 (3я Стадия), начиная с N-(Nα,Мε-ди-Fmoc-L-лизил) DMAP амида. Аналитические данные для этих двух соединений обобщены в таблице 4 выше.
Моно-модиФииированные производные S44HP
N-(4-Аминометилбензоил) S44HP (Соединение 47) (смотри Схему 4, Фиг.13, удаление Fmoc) получали в условиях, как для Соединения 38 (3я Стадия), начиная с N-(4-Fmoc-аминометилбензоил) S44HP (полученного в соответствии со Схемой 3, 1я Стадия). Аналитические данные для этих двух соединений обобщены в таблице 4 выше.
N,N-диметиламиноэтил (DMAE) S44HP амид (Соединение 48) (Альтернативный способ, Схема 5, Фиг.14).
1я Стадия (введение Fmoc). N-Fmoc-S44HP. К перемешиваемому раствору 500 мг (0,5 ммоль) неочищенного S44HP в 6 мл DMF:MeOH (5:1) порциями добавляли 0,057 мл (0,65 ммоль) пиридина и 360 мг (1,0 ммоль) FmocOSu. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Последующее добавление к реакционной смеси диэтилового эфира (20 мл) привело к желтому осадку N-Fmoc-S44HP. Осадок фильтровали и промывали диэтиловым эфиром и сушили. Неочищенный N-Fmoc-S44HP очищали путем колоночной хроматографии на силикагеле Merck в системе CHCl3:MeOH:H2O:HCOOH (13:6:1:0,05) с получением приблизительно 30% N-Fmoc-S44HP, имеющего чистоту 94% в соответствии с HPLC (Rt=20,49, система: 0,01 М Н3РО4 при рН 2,6 и MeCN, 40→80%, 30 мин. TLC 0,39 (III).
2я Стадия (Амидирование) N-Fmoc-S44HP DMAE амид. К перемешиваемому раствору 115 мг (0,10 ммоль) N-Fmoc-S44HP в 2 мл DMSO и 37 мг (0,30 ммоль) гидрохлорида N.N-диметиламиноэтиламина и Et3N для доведения рН до 7-7,5 порциями добавляли 78 мг (0,15 ммоль) PyBOP; рН реакционной смеси поддерживали 7-7,5 путем добавления Et3N. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Последующее добавление к реакционной смеси диэтилового эфира (5 мл) привело к маслянистому остатку, который непрерывно встряхивали с диэтиловым эфиром (5 мл). Добавление ацетона (10 мл) привело к желтому осадку, который отфильтровывали, промывали ацетоном и сушили с получением N-Fmoc-S44HP DMAE амида, имеющего чистоту приблизительно 92% в соотвествии с данными HPLC (Rt=14,50, система А).
3я Стадия (удаление Fmoc). Производное - (Соединение 48) получали в условиях, как для Соединения 38 (3я Стадия), начиная с N-Fmoc-S44HP DMAE амида. Аналитические данные для этого соединения обобщены в таблице 4 выше.
Очищенное Соединение 1: очистка Соединения 1. начиная с образца неочищенного Соединения 1 (Схема 6, Фиг.15)
1я Стадия (введение Fmoc и очистка)
N-Fmoc-Соединение 1. К перемешиваемому раствору 2000 мг (2,0 ммоль) неочищенного Соединения 1 (содержащего соединение 1 - 81,5%, S44HP - 8,27% и неизвестные примеси приблизительно 10% в соответствии с данными HPLC, система В) в 40 мл DMSO добавляли Et3N для доведения рН до 7-7,5. Порциями добавляли 1400 мг (4,0 ммоль) FmocOSu; рН реакционной смеси поддерживали 7-7,5 путем добавления Et3N. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Последующее добавление к реакционной смеси диэтилового эфира (200 мл) привело к маслянистому остатку, который непрерывно встряхивали с диэтиловым эфиром (150 мл × 3). После добавления к этому остатку 50 мл ацетона добавляли диэтиловый эфир (50 мл) с получением желтого осадка. Осадок фильтровали и промывали диэтиловым эфиром и сушили в вакууме с получением 2200 мг неочищенного N-Fmoc-соединения 1, которое очищали путем колоночной хроматографии на силикагеле Merck в системе CHCl3:МеОН:Н2О:25% NH4OH (13:6:1:0,05) с получением 650 мг (приблизительно 30%) N-Fmoc-соединения 1, имеющего чистоту 86% в соответствии с HPLC (Rt=18,44, система С).
2я Стадия (удаление Fmoc)
Fmoc-соединение 1 (400 мг) растворяли в DMSO (60 мл), при 15°С добавляли пиперидин (0,2 мл). Через 2 ч при 15°С добавляли диэтиловый эфир (приблизительно 50 мл); образовывался маслянистый остаток, и его непрерывно встряхивали с диэтиловым эфиром (50 мл × 3). Добавление ацетона (10 мл) и диэтилового эфира (20 мл) позволило получить желтый осадок, который отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме с получением 300 мг очищенного соединения 1. Порцию очищенного соединения 1 для HPLC получали в виде гидрохлорида. К раствору соединения 1 (основание) в метаноле добавляли 0,1 н HCl в метаноле до рН приблизительно 4. Добавление диэтилового эфира позволило получить желтый осадок, который отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме, и он имел чистоту 92,1% в соответствии с данными HPLC (гидрохлорид, Rt=11,37, система А).
Моно-модифицированные производные Соединения 1 (Схема 7, Фиг.16)
N-Фруктопиранозил-Соединение 1 (Соединение 49) (перегруппировка Амадори):
D-Глюкозу (0,086 ммоль) добавляли к раствору соединения 1 (0,040 ммоль) в DMF (2 мл). Реакционную смесь поддерживали при 37°С в течение 20 часов. Затем реакционную смесь по каплям добавляли к диэтиловому эфиру (50 мл). Получающийся в результате осадок отфильтровывали, промывали диэтиловым эфиром и сушили. Неочищенное соединение (чистота 85-90% в соответствии с данными HPLC, и содержащее соли) очищали при помощи колоночной хроматографии на Sephadex G-25 (Pharmacia Fine Chemicals AB, Uppsala, Sweden). Колоночную хроматографию осуществляли с использованием воды. Чистоту фракции контролировали при помощи TLC, система IV. Фракции, содержащие желаемое соединение, собирали, и раствор концентрировали. Добавление ацетона позволило получить желтый осадок, который отфильтровывали, промывали ацетоном и сушили в вакууме. Соединение 41 получали в виде гидрохлорида. К раствору Соединения 49 в метаноле добавляли 0,1 н HCl в метаноле до рН приблизительно 4. Добавление диэтилового эфира позволило получить желтый осадок, который отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме. Аналитические данные для N-Фруктопиранозил-Соединения 1 (Соединение 49) обобщены в таблице 2 ниже.
N-(4-N,N-ди-метиламинобензил) Соединение 1 (Соединение 50) (восстановительное N-алкилирование)
Раствор 4-N,N-диметиламинобензальдегида (0,065 ммоль) и Соединения 1 (0,022 ммоль) в DMF (2 мл) поддерживали при 37°С в течение 2 ч, затем добавляли NaBH3CN (4,1 мг, 0,065 ммоль). Реакционную смесь поддерживали при 37°С в течение 20 ч. Добавление диэтилового эфира (10 мл) приводило к маслянистому остатку, который непрерывно встряхивали с диэтиловым эфиром (10 мл × 2). После добавления ацетона (10 мл) образовывался желтый осадок. Осадок отфильтровывали, промывали диэтиловым эфиром и сушили. Неочищенное соединение, содержащее соли, очищали путем колоночной хроматографии на силикагеле Merck с использованием системы CHCl3:MeOH:H2O:HCOOH (13:6:1:0,1). Чистоту фракции контролировали при помощи TLC, система I. Фракции, содержащие желаемое соединение, собирали, и раствор концентрировали. Добавление ацетона позволило получить желтый осадок, который отфильтровывали, промывали ацетоном и сушили в вакууме. Аналитические данные для N-(4-N,N-ди-метиламинобензил) Соединения 1 (Соединение 50) обобщены в таблице 4 выше.
N-L-Лизил-Соединение 1 (Соединение 51)
1я Стадия (N-[N-Fmoc-амино]ацилирование) Раствор Соединения 1 (50 мг, 0,055 ммоль) в безводном DMF (1 мл) охлаждали при 10°С и затем добавляли Et3N (7,5 мкл, 0,055 ммоль) и N-оксисукцинимидный эфир Nα,Nε-ди-Fmoc-L-лизина (57 мг, 0,083 ммоль). Реакционную смесь перемешивали при этой температуре в течение 1 часа. Ход взаимодействия контролировали при помощи TLC в системе растворителей CHCl3-МеОН (6:1). Добавление к реакционной смеси диэтилового эфира (приблизительно 5 мл) привело к желтому осадку, который отфильтровывали, промывали диэтиловым эфиром, сушили в вакууме и очищали путем колоночной хроматографии на силикагеле (система CHCl3-МеОН с градиентом МеОН 0→10%). Фракции, содержащие желаемое соединение, собирали, раствор концентрировали, и добавление диэтилового эфира позволило получить желтый осадок, который отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме. TLC: Rf=0,6 (CHCl3-МеОН 6:1). HPLC: Rt=13,08 (0,2% HCOONH4 pH 4,5 и MeCN: 75→90%, 20 мин, 90→90%, 10 мин).
2я Стадия (удаление Fmoc)
К выделенному желтому твердому веществу (22 мг, 0,015 ммоль) в DMSO/MeOH 10:1 (1 мл) добавляли пиперидин (4,5 мкл, 0,045 ммоль). Через 30 мин перемешивания при комнатной температуре добавляли диэтиловый эфир (приблизительно 3 мл); образовывался маслянистый остаток, и его непрерывно встряхивали с диэтиловым эфиром (3 мл × 2). Добавление ацетона (2 мл) позволило получить желтый осадок, который отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме. Аналитические данные для N-L-лизил-Соединения 1 (Соединение 51) обобщены в таблице 4 выше.
N,N-ди-(3-аминопропил)-соединение 1 (Соединение 52)
1я Стадия (восстановительное N-алкилирование) К раствору N-Fmoc-3-аминопропионового альдегида (160 мг, 0,054 ммоль) и Соединения 1 (100 мг, 0,011 ммоль) в DMF (3 мл) добавляли NaBH3CN (34 мг, 0,054 ммоль). Реакционную смесь поддерживали при 37°С в течение 20 часов и затем по каплям добавляли к диэтиловому эфиру (200 мл). Желтый осадок отфильтровывали и очищали путем флэш-хроматографии на силикагеле Merck для колоночной хроматографии (0,040-0,063 мм) в системе линейного градиента CHCl3:MeOH:HCOOH (3:1:0,01)→CHCl3:MeOH:H2O:HCOOH (13:6:1:0,1) с получением желтого осадка N,N-ди-(N-Fmoc-3-аминопропил) Соединения 1. Данные HPLC: Rt=27,43 (С).
2я Стадия (удаление Fmoc)
К раствору N,N-ди-[3-(N-Fmoc)-аминопропил]-S44НР в DMSO (3 мл), добавляли пиперидин (0,1 мл). Через 2 ч при комнатной температуре добавляли диэтиловый эфир (приблизительно 7 мл), и образовывался маслянистый остаток, который непрерывно встряхивали с диэтиловым эфиром (7 мл × 2). Добавление ацетона (10 мл) позволило получить желтый осадок, который отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме. Аналитические данные для N,N-ди-(3-аминопропил) Соединения 1 (Соединение 52) обобщены в таблице 4 выше.
Соли Соединения 1
L-аспарат (55), Соединение 1 L-глутамат (29), D-глутамат (54)
Соединение 1 (13 мг, 0,015 ммоль) растворяли в 0,2 мл DMSO, и затем добавляли раствор соответствующей аминокислоты (0,015 ммоль) в 0,1 мл воды. Реакционную смесь поддерживали при комнатной температуре в течение 15 минут. Добавление к реакционной смеси диэтилового эфира (приблизительно 2 мл) привело к маслянистому остатку, который непрерывно встряхивали с диэтиловым эфиром (2 мл × 2). После добавления к этому маслу 3 мл ацетона образовывался желтый осадок соли. Осадок фильтровали, промывали ацетоном и затем сушили в вакууме.
Метилсульфонат (56), дихлорацетат (57)
Соответствующую кислоту (0,015 ммоль) растворяли в 0,1 мл DMSO и затем добавляли к раствору Соединения 1 (13 мг, 0,015 ммоль) в 0,1 мл DMSO. Реакционную смесь поддерживали при комнатной температуре в течение 5 минут. Добавление к реакционной смеси диэтилового эфира (приблизительно 2 мл) привело к маслянистому остатку, который непрерывно встряхивали с диэтиловым эфиром (2 мл × 2). После добавления к этому маслу 3 мл ацетона образовывался желтый осадок соли. Осадок фильтровали, промывали ацетоном и затем сушили в вакууме.
Гидрохлорид соединения 1 (53)
К раствору Соединения 1 (13 мг, 0,015 ммоль) в 0,2 мл DMSO добавляли 0,15 мл (0,015 ммоль) водного раствора 0,1 н HCl. Реакционную смесь поддерживали при комнатной температуре в течение 5 минут. Добавление к реакционной смеси диэтилового эфира (приблизительно 2 мл) привело к маслянистому остатку, который непрерывно встряхивали с диэтиловым эфиром (2 мл × 2). После добавления к этому маслу 3 мл ацетона образовывался желтый осадок соли. Осадок фильтровали, промывали ацетоном и затем сушили в вакууме.
Все образцы получали с выходами приблизительно 90%, и они растворимы в воде.
ОПРЕДЕЛЕНИЕ ПРОТИВОГРИБКОВОЙ АКТИВНОСТИ И ТОКСИЧНОСТИ
Исследование чувствительности грибов к противогрибковому действию (1)
Тестирование чувствительности к противогрибковому действию для дрожжей и мицелиальных грибов (плесневые грибы) проводили с использованием способа микроразведения в бульоне, описанного соответственно в документах NCCLS (Национальный комитет по клиническим лабораторным стандартам) М27-А [Reference method for Broth Dilution Antifungal Susceptability Testing of Yeasts, ISBN 1-56238-328-0, Pennsylvania, 1997] и М38-А [Reference method for Broth Dilution Antifungal Susceptability Testing of Filamentous Fungi: Approved Standard, ISBN 1-56238-470-8, Pennsylvania, 2002].
Среда и буфер. В исследовании использовали среду RPMI 1640 с L-глутамином и феноловым красным, без бикарбоната натрия, дополненную 0,2% глюкозой (ICN Biomedicals Inc., Ohio, USA), забуференную 0,165 M морфолинпропансульфоновой кислотой (MOPS; ACROS ORGANICS, New Jersey, USA), pH 7,0.
Противогрибковые агенты. Соединения сначала растворяли в диметилсульфоксиде (DMSO) при исходной концентрации 1600 мкг/мл. Готовили серии разведений (от 1600 до 3,13 мкг/мл) из исходных растворов в том же самом растворителе. Затем эти растворы в DMSO разбавляли в 50 раз (сначала в 10 раз и затем в 5 раз) в контрольной среде. Наконец, их разбавляли в 2 раза при инокуляции, что уменьшало конечную концентрацию растворителя до 1% (конечные концентрации противогрибковых агентов составляли от 16 до 0,13 мкг/мл). Растворы готовили непосредственно перед использованием.
Микроорганизмы. Микроорганизмы, используемые для тестирования чувствительности, представляли собой: Candida albicans ATCC 14053, Cryptococcus humicolus ATCC 9949, Aspergillus niger ATCC 16404 и Fusarium oxysporum VKM F-140 (= CMI, IMI 90473). Микроорганизмы хранили на скошенном агаре из картофельного крахмала/декстрозы и в 50% растворе глицерина при -70°С. Для краткосрочного хранения исходные культуры хранили на скошенном агаре при 4°С. Культуры дрожжей субкультивировали на декстрозном агаре Сабуро (SAB) и инкубировали в течение 24 часов при 35°С (Candida albicans) или в течение 48 часов при 25°С (Cryptococcus humicolus) с получением свежевыращенной чистой культуры. Культуры плесневых грибов субкультивировали на агаре из картофельного крахмала/декстрозы и инкубировали в течение 7 суток при 35°С (Aspergillus niger) или в течение 2 суток при 35°С, затем 5 суток при 25°С (Fusarium oxysporum) для обеспечения максимального спорообразования.
Приготовление инокулята
Для дрожжей исходный инокулят готовили путем суспендирования колоний культур в стерильном 0,85% физиологическом растворе. Получающуюся в результате суспензию подвергали вихреобразному перемешиванию и доводили при помощи спектрофотометра при 530 нм для того, чтобы обеспечить мутность 0,5 по МакФарланду (от 1×106 до 5×106 CFU (колониеобразующих единиц)/мл). Исходную суспензию дрожжей разводили 1:1000 средой с получением разведенного в два раза тестируемого инокулята (от 1×103 до 5×103 CFU/мл).
Для плесневых грибов исходные суспензии готовили путем покрывания зрелых колоний грибов стерильным 0,85% физиологическим раствором и осторожного соскребания. 1% (об./об.) увлажняющего агента Tween 20 добавляли в физиологический раствор для приготовления инокулята Aspergillus. Наконец, суспензии подвергали вихреобразному перемешиванию в течение 15 с для разрушения сгустков клеток и затем фильтровали через четыре слоя стерильной марли. Плотности суспензий конидиальных спор измеряли при 530 нм и доводили до 0,09-0,11 для Aspergillus niger и 0,15-0,17 для Fusarium oxysporum. Эти суспензии разводили 1:50 стандартной средой. Разведения инокулята 1:50 соответствовали 2х плотности, необходимой для достижения приблизительно от 0,4×104 до 5×104 CFU/мл.
Количественное определение инокулята осуществляли при помощи посева методом разведения инокулята на декстрозном агаре Сабуро для определения количества жизнеспособных CFU на миллилитр.
Способ микроразведения
Стерильные 96-луночные плоскодонные микротитровальные планшеты использовали для тестирования. 100 мкл разведенных инокулятов распределяли в каждую лунку для микроразведений, содержащую по 100 мкл двойных разведений соединений. Были включены контроли, не содержащие лекарственные средства и не содержащие дрожжи/плесневые грибы. Планшеты для микроразведения инкубировали при 35°С (Candida albicans и плесневые грибы) или 25°С (Cryptococcus humicolus) без встряхивания и обследовали на присутствие или отсутствие видимого роста. Численную оценку от 0 до 4 присваивали каждой лунке с использованием следующей шкалы 0 = зрительно прозрачные или отсутствие роста, 1 = слабый рост (25% контроль роста), 2 = заметное уменьшение роста (50% контроль роста), 3 = слабое уменьшение роста (75% контроль роста), 4 = отсутствие уменьшения роста. Минимальные ингибирующие концентрации (MIC) считывали через 48 часов.
MIC для каждого тестируемого соединения определяли как наименьшую концентрацию противогрибковых агентов, которая предотвращают какой-либо видимый рост микроорганизма (оценка роста = 0). Для объяснения слабых различий между агентами в некоторых случаях MIC для плесневых грибов дополнительно определяли как наименьшие концентрации, обеспечивающие оценку роста 1 (25% контроль роста). Результаты представлены в таблице 5 ниже.
Результаты демонстрируют, что соединения по изобретению обладают противогрибковой активностью. Конкретнее, эти тесты свидетельствуют о том, что производное нистатина в указанном количестве обладает противогрибковой активностью, сравнимой с активностью амфотерицина В и S44HP (например соединения №1, 9, 11, 12, 13, 14 и 22).
Исследование чувствительности грибов к противогрибковому действию (2)
Противогрибковую активность нескольких производных нистатина исследовали дополнительно. Способ, используемый в исследовании (1), представляет собой принятый во всем мире способ тестирования противогрибковой активности, но не является очень чувствительным и, таким образом, не позволяет легко проводить различия между соединениями, обладающими сравнимой активностью. Таким образом, противогрибковую активность ряда производных нистатина определяли в способе, который разработан таким образом, чтобы быть более чувствительным.
Тестируемый микроорганизм, используемый для дополнительного тестирования биологической активности полиенового макролида, представлял собой Candida albicans ATCC (Американская коллекция типовых культур) 10231, выращенный в 120 мкл стандартной среды М19 без NaCl (с инокулятом 1000 cfu на лунку) и 30 мкл разведенных образцов полиенового макролида. Антибиотики разводили в МеОН последовательно, обеспечивая спектр концентраций, которые обеспечивают результаты, находящиеся в диапазоне от полного ингибирования роста до отсутствия ингибирования роста тестируемого микроорганизма. Культуры тестируемого микроорганизма с разведениями антибиотиков инкубировали в 96-луночных микротитровальных планшетах при 30°С без встряхивания, и рост измеряли как OD490 (оптическую плотность при 490 нм) через 12, 14 и 16 часов с использованием ридера для микротитровальных планшетов SpectraMax Plus. OD наносили на график в зависимости от концентрации антибиотиков, и MIC50 (среднюю минимальную ингибирующую концентрацию) оценивали по кривой регрессии при 50% ингибировании роста.
Результаты представлены в таблице 6 ниже.
Результаты подтверждают, что ряд аналогов нистатина по изобретению действительно обладают улучшенной противогрибковой активностью по сравнению с амфотерицином В и S44HP.
Противогрибковая активность солей Соединения 1 (Таблица 7)
Тестировали следующие растворимые в воде соли
гидрохлорид СОЕДИНЕНИЯ 1 (53),
L-глутамат СОЕДИНЕНИЯ 1 (29),
D-глутамат СОЕДИНЕНИЯ 1 (54)
L-аспартат СОЕДИНЕНИЯ 1 (55),
дихлорацетат СОЕДИНЕНИЯ 1 (57)
L-глутамат соединения 1 (29) представляет собой наиболее активную соль Соединения 1,
Противогрибковая активность производных соединения (таблицы 8 и 9)
Соединение 51 представляет собой наиболее активное производное в этих сериях, обладающее несколько более высокой противогрибковой активностью по сравнению с родительским антибиотиком.
Исследование токсичности in vitro
Токсичность различных производных нистатина тестировали в концентрации 2 и 5 мкг/мл и 25 мкМ.
Соединения растворяли в DMSO в концентрации 5 мг/мл, и 0,1 мл образцов с различными концентрациями готовили в DMSO. Образцы смешивали с 0,9 мл фосфатно-буферного раствора (PBS), содержащего 2,5% лошадиной крови (PBS-HB; на льду) и инкубировали при 37°С в течение 30 мин без встряхивания.
Клетки осаждали путем центрифугирования при 5000 об./мин в течение 5 мин, и гемолиз оценивали путем измерения OD545. 100% гемолиз определяли как величину OD545, получаемую для суспензии 2,5% лошадиной крови в дистиллированной воде.
Результаты представлены в таблицах 10-12 ниже.
Эти результаты демонстрируют, что соединения по изобретению значительно менее токсины по сравнению с S44HP или амфотерицином В. Это весьма благоприятно, поскольку соединения по изобретению, таким образом, можно использовать в больших количествах без отрицательного действия.
IN VIVO ТЕСТИРОВАНИЕ СОЕДИНЕНИЙ 1 И 11 И ДРУГИХ
Животные. Использовали самцов мышей гибридов первого поколения (C57Bl/6xDBA/2)F1 B6D2F1, массой 20-22 г, полученных из центральной фермы "Крюково" Российской Академии Медицинских Наук (РАМН). Животных поддерживали в виварии в пластиковых клетках (с древесной подстилкой в контролируемых условиях окружающей среды: 24±1°С, цикл 12/12 часов свет/темнота) со стандартной диетой с кормом в брикетах при легком доступе к питьевой воде (без ограничения). После периода карантина в течение 2 недель здоровых животных использовали в экспериментальной работе.
Инфекционный агент. Использовали Candida albicans (штамм 14053 АТСС).
Противогрибковые агенты. Растворы соединений 1 и 11 (и другие) готовили "ex temporae" и хранили в темных стеклянных флаконах для того, чтобы избежать воздействия света. Растворы готовили следующим образом: безводные антибиотические вещества (5 мг) смешивали с безводным дезоксихолатом натрия (4,1 мг) в стерильном стеклянном флаконе. 10 мл фосфатного буфера (NaH2PO4 - 1,59 г; Na2HPO4 - 0,96 г; Н2О - до 100 мл) добавляли к смеси, и суспензию немедленно подвергали интенсивному встряхиванию в течение 10 минут до получения гомогенных суспензий. Получающиеся суспензии (2 мл) помещали в новые стерильные стеклянные флаконы, добавляли 6 мл 5% нейтрального стерильного раствора глюкозы, и получающиеся в результате растворы (0,125 мг/мл) использовали для внутривенного введения.
Препараты амфотерицина В и S44HP также готовили тем же самым путем.
Острая токсичность
Определяли острую токсичность соединений 1 и 11 (и других), а также амфотерицина В и S44HP.
Антибиотические препараты однократно инъецировали в хвостовую вену мышей в течение 1-1,5 часов после приготовления растворов. Скорость инъекции не превышала 0,5 мл в минуту. Каждый антибиотик использовали в диапазоне доз, приводящем в результате к от 0% до 100% летальности, и минимально 3 промежуточных доз. Животных случайным образом распределяли по группам, каждая из которых содержала по 6 мышей. Характеризующие токсичность дозы MTD (максимальную переносимую дозу) и LD50 (среднюю летальную дозу) рассчитывали при помощи способа "пробит-анализа" в соответствии с Litchfield - Wilcoxon (J Pharmacol Exp Ther 96:99) с использованием статистической программы анализа "StatPlus-3.5.0. - 2005". Результаты представлены в Таблице 13 ниже.
Противогрибковая активность in vivo
Животных внутривенно инфицировали культурой Candida albicans в дозе 1,0 миллион CFU/мышь (объем 0,1 мл). Через 30 минут после инфицирования мышам внутривенно вводили противогрибковые агенты в различных дозах в объеме 0,2 мл (при скорости 0,2 мл/30 секунд). Каждую дозу вводили ежесуточно в течение четырех суток, включающих сутки инфицирования (0, 1, 2 и 3 сутки).
В качестве контроля использовали группу необработанных мышей (инфицированных Candida albicans тем же самым путем). Кроме того, была группа "плацебо" из неинфицированных животных, которым внутривенно вводили (в том же объеме, как препараты противогрибковых агентов) в 0,2 мл растворителя (фосфатный буфер +5% глюкоза (1:1)).
Отсутствие активности идентифицировали в любой из групп "плацебо". Кроме того, Candida albicans ни разу не обнаружили ни у одного из неинфицированных животных.
На 5-е сутки эксперимента мышей взвешивали и умерщвляли (убивали) при помощи смещения шейных позвонков. Затем в стерильных условиях количество Candida albicans для каждой мыши определяли путем подсчета жизнеспособных гомогенатов почек. Почки извлекали асептически и взвешивали, затем помещали в фарфоровые ступки со стерильным корундом. Готовили разведения получающихся в результате суспензий, и высевали на чашки Петри с агаром Сабуро, инкубировали в течение 24 часов при температуре 35°С. Затем подсчитывали развившиеся колонии Candida albicans, и их количество пересчитывали исходя из 1 г ткани почек.
Статистический анализ осуществляли при помощи Microsoft Office Excel 2003. Значимые различия имели Р≤0,05 при сравнении в соответствии с t-критерием Стьюдента.
Результаты представлены в таблице 14 ниже и на Фиг.9.
Исследование специфической активности соединений 29, 51, 41, 48 в модели кандидозного сепсиса у мышей
Животные. В экспериментах использовали самок мышей гибридов первого поколения (C57BI/6xDBA/2)F1 (B6D2F1), массой 18-20 г, полученных из центральной фермы "Крюково" Российской Академии Медицинских Наук (РАМН). Животных поддерживали в институтском виварии в пластиковых клетках (с древесной подстилкой в контролируемых условиях окружающей среды: 24±1°С, цикл 12/12 часов свет/темнота) со стандартной диетой с кормом в брикетах при легком доступе к питьевой воде (без ограничения). После периода карантина в течение 2 недель здоровых животных использовали в экспериментальной работе.
В качестве инфицирующего агента использовали Candida albicans (штамм 14053 АТСС). Тестируемые препараты представляли собой соединения 29, 51, 41, 48. Препарат для сравнения представлял собой амфотерицин В (чистый). Растворы тестируемых препаратов готовили ex tempore при помощи способа, указанного заказчиком.
Постановка эксперимента
Животных содержали по 3 мыши и внутривенно инфицировали культурой Candida albicans в дозе 1,0 миллион CFU/мышь в объеме 0,1 мл. Необходимо отметить, что доза Candida albicans остается постоянной во всех экспериментах. В течение 30 минут после инфицирования мышам осуществляли первое внутривенное введение тестируемых препаратов в соответствующих дозах в объеме 0,2 мл (со скоростью 0,2 мл/30 секунд).
Эксперименты планировали таким образом, чтобы в каждом эксперименте использовали по одной дозе (для амфотерицина В и для тестируемых препаратов), где каждую дозу вводили раз в сутки в течение четырех суток, начиная с суток инфекции (0, 1, 2 и 3 сутки). В каждом эксперименте существовала необрабатываемая группа животных, инфицированная Candida albicans. Кроме того, была группа "плацебо", состоящая из интактных (неинфицированных) животных, которым внутривенно (в том же самом объеме, как лекарственные препараты) инъецировали 0,2 мл растворителя - фосфатного буфера +5% глюкоза (1:1). Данные по активности "плацебо" не показали какую-либо активность. Candida albicans ни разу не обнаружили ни у одного из неинфицированных животных.
На 5-е сутки эксперимента после заражения мышей взвешивали и умерщвляли (убивали) при помощи смещения шейных позвонков. Затем в стерильных условиях количество Candida albicans определяли путем подсчета жизнеспособных гомогенатов почек. Почки, которые асептически извлекали и взвешивали, помещали в фарфоровые ступки со стерильным корундом.
Готовили разведения получающихся в результате суспензий и высевали на чашки Петри с агаром Сабуро, инкубировали в течение 48 часов при температуре 35°С. Затем подсчитывали развившиеся колонии Candida albicans, и их количество пересчитывали исходя из 1 г ткани почек. Первое разведение составляло 10-1. Нулевой результат при этом выращивании принимали для 5 CFU/г.
В качестве основного критерия оценки исследуемых препаратов принимали максимальный эффект амфотерицина В (эффективная доза, ED).
Статистическую обработку осуществляли при помощи компьютерной программы Microsoft Office Excel 2003. Значимые различия среднего имели Р≤0,05 при сравнении в соответствии с t-критерием Стьюдента.
Полученные результаты представлены в таблице 15. Амфотерицин В продемонстрировал максимальную активность, принятую для критерия активности, в наибольшей из исследуемых доз (1,25 мг/кг/сутки, приблизительно 62% в соответствии с MTD).
Синтез дополнительных соединений на основе генетической манипуляции
Получали различные дополнительные структуры скелетов, с которыми можно проводить манипуляции. С использованием вышеприведенного химизма различные функциональные группы могут быть присоединены к свободным СООН и аминогруппам. Все эти основные структуры образуют дополнительные предпочтительные соединения по изобретению, (атомы Н не изображены)
В1
Инактивация доменов ER5 и DH15 в нистатиновой PKS, отсутствие гидроксилирования по С-10
B2
Инактивация доменов ER5 и KR16 в нистатиновой PKS
B3
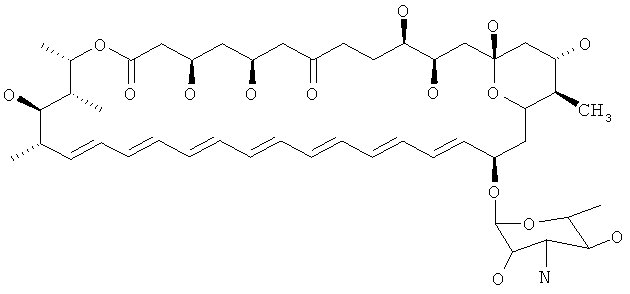
Инактивация доменов ER5, DH15 в нистатиновой PKS и NysN монооксигеназе, отсутствие гидроксилирования по С-10
B4
Инактивация домена ER5 в нистатиновой PKS и NysN и NysL монооксигеназах
B5
Инактивация доменов ER5 и KR14 в нистатиновой PKS и NysN монооксигеназе В6
B6
Инактивация домена ER5 в нистатиновой PKS и NysL монооксигеназе
B7
Инактивация доменов ER5 и KR16 в нистатиновой PKS и NysN монооксигеназе
Конструирование мутанта В2
Конструирование вектора замещения гена для инактивации KR16
Полноразмерную вставку 14 kb рекомбинантного фага N20 (Brautaset et al., 2000) вырезали при помощи Xbal и клонировали в плазмиду pGEM-3zf с получением pL20X. Из этой плазмиды вырезали фрагмент 3,7 kb BamHI и лигировали в pGEM-3zf, приводя в результате к плазмиде pGEMB3.7. Фрагмент ДНК 0,8 kb (включающий остаток Y3404 активного сайта KR16) затем амплифицировали при помощи PCR (полимеразной цепной реакции) из pGEMB3.7 с использованием следующих праймеров:
KR16-F1: 5′-ttttctgCAGCCGACCGGCACCGTCC-3′ (смысловой), и
KR16-1R: 5′-ttttaaGCTTCCTGGACCGCGCGGG-3′ (антисмысловой).
Продукт PCR подвергали расщеплению по концам при помощи PstI и HindIII (новые сайты распознавания подчеркнуты в двух праймерах PCR) и клонировали в соответствующие сайты pLITMUS28 с получением pLITPH0.8. Последняя плазмида служила в качестве матрицы для сайт-направленного мутагенеза активного сайта KR16 с использованием следующих мутагенных олигонуклеотидов:
mutKR16-1F:
5′-CCCCGGCCAGGCCGGCTTCGAA GCCGCCAACGCGGTCC-3′ (смысловой)
mutKR16-1R:
5′-GGACCGCGTTGGCGGC TT CGAA GCCGGCCTGGCCGGGG-3′ (антисмысловой).
Мутантные нуклеотиды показаны жирным шрифтом, а новый введенный сайт распознавания BstBI подчеркнут.С использованием этих двух олигонуклеотидов Туг активного сайта KR16 заменяют на Phe и одновременно следующий в цепи Ala заменяют на Glu, с получением двойной мутации YA3404FE. Плазмиду с правильной мутацией идентифицировали при помощи расщепления BstBI, и полную клонированную вставку секвенировали для обнаружения любых других нежелательных мутаций. Из правильным образом мутированной плазмиды вырезали фрагмент 627 bp Bpu102l/Bpu10l и использовали для замены соответствующего фрагмента плазмиды pGEMB3.7 с получением плазмиды pGEMB3.7m. Из последней плазмиды вырезали полноразмерную вставку 3,7 kb при помощи EcoRI/HindIII и лигировали вместе с 3,1 kb скелетом EcoRI/HindIII pSOK201 с получением вектора замещения гена pKR16m.
Генная замена
Последний вектор вводили в дефектный по nysA мутант S. noursei GG5073SP (Borgos et al., 2006) при помощи двойной гомологичной рекомбинации. Правильность хромосомной мутации KR16 подтверждали с использованием PCR+расщепления BstBI, а также саузерн-блоттинга, а затем продукцию полиенов возобновляли путем введения гена nysA в транс положение, как описано (Brautaset et al., 2003). Получающийся в результате мутант обозначили как В2.
Конструирование мутанта В7
Плазмиду pKOnysN-CL346AS (смотри PNAS-manus) водили в дефектный по nysA B2 путем двойной гомологичной рекомбинации. Правильность хромосомной мутации nysN подтверждали с использованием PCR и расщепления Nhel, а также саузерн-блоттинга, а затем продукцию полнена возобновляли путем введения гена nysA в транс положение, как описано (Brautaset et al., 2003). Получающийся в результате мутант обозначили как В7.
Конструирование мутанта В5
Конструирование вектора замещения гена для инактивации KR14:
Фрагмент 4.0 BcII/EcoRI плазмиды pL20X (смотри стратегию В2, выше) вырезали и лигировали в pGEM-3zf, расщепленный с использованием BamHI/EcoRI с получением рВЕ4.0. Фрагмент 1 kb амплифицировали при помощи PCR из последней плазмиды с использованием следующих праймеров:
KR14-F1: 5′-TTCGGCGGCGTAATCTCC-3′ (смысловой), и
KR14-R1: 5′-ttttaaGCTTCCACGGCGAGGTGC-3′ (антисмысловой).
Фрагмент PCR расщепляли при помощи Sad и Hind III (новый сайт распознавания подчеркнут в праймере) и лигировали в pLITMUS28 с получением pLITHSLO, и последнюю плазмиду использовали в качестве матрицы для сайт-направленного мутагенеза активного сайта KR14 с использованием следующих мутагенных олигонуклеотидов:
KR14-mut1:
5′-CCCGGGCCAGGCCAACTTCGAA GCTGCCAACGCCGTCCTG-3′ (смысловой),
KR14-mut2:
5′-CAGGACGGCGTTGGCAGC TT CGAAGTTGGCCTGGCCCGGG-3′ (антисмысловой).
Мутантные нуклеотиды показаны жирным шрифтом, а новый введенный сайт распознавания BstBI подчеркнут.С использованием этих двух олигонуклеотидов Туг активного сайта KR14 заменяют на Phe и одновременно следующий в цепи Ala заменяют на Glu, с получением двойной мутации YA9197FE. Плазмиду с правильной мутацией идентифицировали при помощи расщепления BstBI, и полную клонированную вставку секвенировали для обнаружения любых других нежелательных мутаций. Из правильным образом мутированной плазмиды вырезали фрагмент 0,7 kb FseI/AscI и использовали для замены соответствующего фрагмента плазмиды рВЕ4.0 с получением плазмиды pBE4.0mut. Из последней плазмиды вырезали полноразмерную вставку 4,0 kb при помощи EcoRI/HindIII и лигировали вместе с 3,1 kb скелетом EcoRI/HindIII pSOK201 с получением вектора замещения гена pKR14m.
Генная замена
Последний вектор вводили в дефектный по nysA мутант S. noursei GG5073SP (Borgos et al., 2006) при помощи двойной гомологичной рекомбинации. Правильность хромосомной мутации KR14 подтверждали с использованием PCR+расщепления BstBI, а также саузерн-блоттинга, а затем продукцию полиенов возобновляли путем введения гена nysA в транс положение, как описано (Brautaset et al., 2003). Получающийся в результате мутант обозначили BSG015.
Затем nysN инактивирующий вектор pKOnysN-CL346AS (смотри PNAS-manus) вводили в дефектный по nysA BSG015 путем двойной гомологичной рекомбинации. Правильность хромосомной мутации nysN подтверждали с использованием PCR и расщепления NheI, а также саузерн-блоттинга, а затем продукцию полнена возобновляли путем введения гена nysA в транс положение, как описано (Brautaset et al., 2003). Получающийся в результате мутант обозначили как В5.
Конструирование мутанта В4
Конструирование вектора замещения гена для инактивации nysL:
Описана заменяющая ген nysL плазмида pNLD1 с внутрирамочной делецией (Volokhan et al., 2006), и клонированная вставка 4,2 kb этой плазмиды включает области nysN-кодирующей области S. noursei. Для введения мутации nysL в скелет мутанта nysN-CL346ST авторы изобретения должны были модифицировать эту плазмиду путем включения также этой мутации nysN. Последнее осуществили в соответствии со следующим: фрамент 1011 bp Agel/Fspal плазмиды pSOK201nysN4.1-CL346ST (смотри завку на патент Biosergen от мая 2007 года) вырезали и использовали для замены на соответствующий фрагмент плазмиды pNLD1 с получением плазмиды замещения гена pNLD2.
Генная замена:
Плазмиду pNLD2 вводили в дефектное по nysA Соединение 1 при помощи двойной гомологичной рекомбинации. Правильность хромосомной мутации nysL подтверждали с использованием PCR и расщепления NheI, a также саузерн-блоттинга, а затем продукцию полнена возобновляли путем введения гена nysA в транс положение, как описано (Brautaset et al., 2003). Получающийся в результате мутант обозначили как В4.
В1 и В3
Инактивация His966 активного сайта DH15 NysJ:
Задача: Ввести сайт-специфическую мутацию в активный сайт nysJ DH15, приводящую к заменяющей мутации H966F в домене DH15
Способ:
- Расщепляют pL20X при помощи BeII+SphI и клонируют 3,33 kb фрагмент в pGEM11-zf, расщепленный при помощи BamHI/SphI=pDH15-A
- Расщепляют pDH15-A при помощи EcoRI+HindIII и клонируют 3,34 kb фрагмент в соответствующие сайты pGEM3-zf=pDH15-B
- Осуществляют сайт-направленный мутагенез с использованием матрицы pDH15-B и при помощи следующих мутагенных олигонуклеотидов:
Mut-DH15-1:
5′-CACCCCTGGCTCGCCGAC TTC GTCGTCGGCGGCATGGTC-3′
Mut-DH15-2:
5′-GACCATGCCGCCGACGAC GAA GTCGGCGAGCCAGGGGTG-3′
- Мутантные нуклеотиды указаны жирным шрифтом. Мутация нарушает природный сайт BtrI в этом положении.
- Подтверждают правильность мутации при помощи расщепления BrtI и секвенирования ДНК=pDH15-Bmut.
- Выделяют 1,6 kb EcoRI/SacI фрагмент-1 из pDH15-B, 1,1 kb HindIII/BgIII фрагмент-2 из pDH15-B и SacI/BgIII фрагмент-3 из pDH15-Bmut, и все три фрагмента лигируют вместе с 3,1 kb pSOK201 EcoRI/HindIII скелетом; pDH15-123
- Осуществляют генную замену в S.noursei.
- Подтверждения правильности мутаций DH15 S.noursei
- 1е кроссинговерные мутанты могут быть подтверждены при помощи саузерн-блоттинга после расщепления NotI тотальной ДНК и с использованием меченой вставки 3,33 pDH15-A в качестве зонда. Дикий тип должен обеспечивать сигналы 5,2+8 kb, тогда как правильный 1й кроссинговерный мутант (оба теоретических варианта) должен обеспечивать сигналы 5,2+6,4+8 kb.
- 2е кроссинговерные мутанты могут быть проанализированы при помощи PCR с использованием следующих праймеров:
DH15-F: 5′-CCTTCCTCGAACTCGGCC-3′
DH15-R: 5′-GCGTAGATCTCGGTGTCG-3′
которые должны обеспечивать продукт 817 bp. Продукт PCR из родительского штамма может быть расщеплен BtrI, обеспечивая фрагменты 392 bp и 425 bp, тогда как продукт PCR из мутанта не может быть расщеплен BtrI. Дополнительно, 2й мутант также может быть подтвержден при помощи саузерн-блоттинга с использованием меченого 817 bp продукта PCR в качестве зонда. ДНК родительского штамма, расщепленая BtrI, должна обеспечить 0,95 kb + 0,45 kb гибридизующий фрагмент, тогда как расщепленная BtrI ДНК из мутанта должна обеспечивать один 1,4 kb гибридизующий фрагмент.
Конструирование мутанта В1
Мутацию DH15 вводили в дефектный по nysA штамм GG5073SP, и затем правильный мутант дополняли геном nysA в транс положении.
Конструирование мутантов В3
Мутацию nysN-CL346ST вводили в дефектную по nysA B1-S1 генетическую структуру, и получающийся в результате правильный мутантный штамм дополняли геном nysA в транс положении с получением мутанта B3-1S.
Эффективность амфотерицина В и новых аналогов S44HP in vivo в нейтропенической модели диссеминированного кандидоза у мышей
У мышей вызывали нейтропению путем интраперитонеальной инъекции моногидрата циклофосфамида (Fluka, BioChemika, Sigma-Aldrich, Switzerland) 2×100 мг/кг массы тела за 1 сутки до инфицирования.
Диссеминированную инфекцию Candida albicans достигали путем инъекции 3×105 CFU/мл инокулята в латеральный отдел хвостовой вены нейтропенических мышей за 2 ч до начала терапии антибиотиками. Эффективность антибиотиков по устранению инфекции С.albicans оценивали путем определения грибковой загрузки почек после 4 суток лечения (1 доза в сутки). Результаты представлены на Фиг.17.
Амфотерицин В вводили в самой высокой дозе 2 мг/кг (чуть ниже MTD).
Эти результаты ясно демонстрируют, что Соединение 28 и соединение 29 значительно лучше, чем амфотерицин В, поскольку:
1) они демонстрируют приблизительно в 5-7 раз меньшую острую токсичность;
2) они демонстрируют в 5-10 раз лучшую эффективность в лечении диссеминированного кандидоза в нейтропенической модели у мышей;
3) они растворимы в 5% глюкозе (более 10 мг/мл), тогда как растворимость амфотерицина В менее 1 мг/мл.
Хотя соединение 25 и соединение 26 менее активны по сравнению с амфотерицином В в нейтропенической модели кандидоза у мышей, они демонстрируют значительно меньшую острую токсичность (приблизительно в 20 раз меньшую по сравнению с амфотерицином В), и, таким образом, рассматриваются как интересные кандидаты.
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИДЫ НАТАМИЦИНА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ГРИБКОВЫХ ИНФЕКЦИЙ | 2022 |
|
RU2803742C1 |
| Способ получения противогрибкового полусинтетического полиенового антибиотика | 2020 |
|
RU2751333C1 |
| Противогрибковый полусинтетический полиеновый антибиотик, его водорастворимые соли и фармацевтические композиции на их основе | 2018 |
|
RU2688658C1 |
| Способ получения метилсульфатов N,N,N-триметиламмониевых производных полиеновых макролидов | 1979 |
|
SU955855A3 |
| Ингибиторы фактора VIIa | 1999 |
|
RU2223967C2 |
| СИНТЕТИЧЕСКИЕ ПЕПТИДЫ, УМЕНЬШАЮЩИЕ ИЛИ УСТРАНЯЮЩИЕ МЕШКИ, ОБРАЗУЮЩИЕСЯ ПОД НИЖНИМ КРАЕМ ГЛАЗ, И ИХ ПРИМЕНЕНИЕ В КОСМЕТИЧЕСКИХ ИЛИ ДЕРМОФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЯХ | 2005 |
|
RU2387666C2 |
| Лиганды PSMA для визуализации и эндорадиотерапии | 2018 |
|
RU2807076C2 |
| ПТГ-СОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ФРАГМЕНТ ДНК И СЛИТЫЙ БЕЛОК | 1993 |
|
RU2130945C1 |
| АНАЛОГИ ОКСИТОЦИНА | 2009 |
|
RU2496788C2 |
| НИЗКОМОЛЕКУЛЯРНЫЕ ПРОИЗВОДНЫЕ ПЕПТИДОВ КАК ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ ЛАМИНИНА/НИДОГЕНА | 2000 |
|
RU2262509C2 |
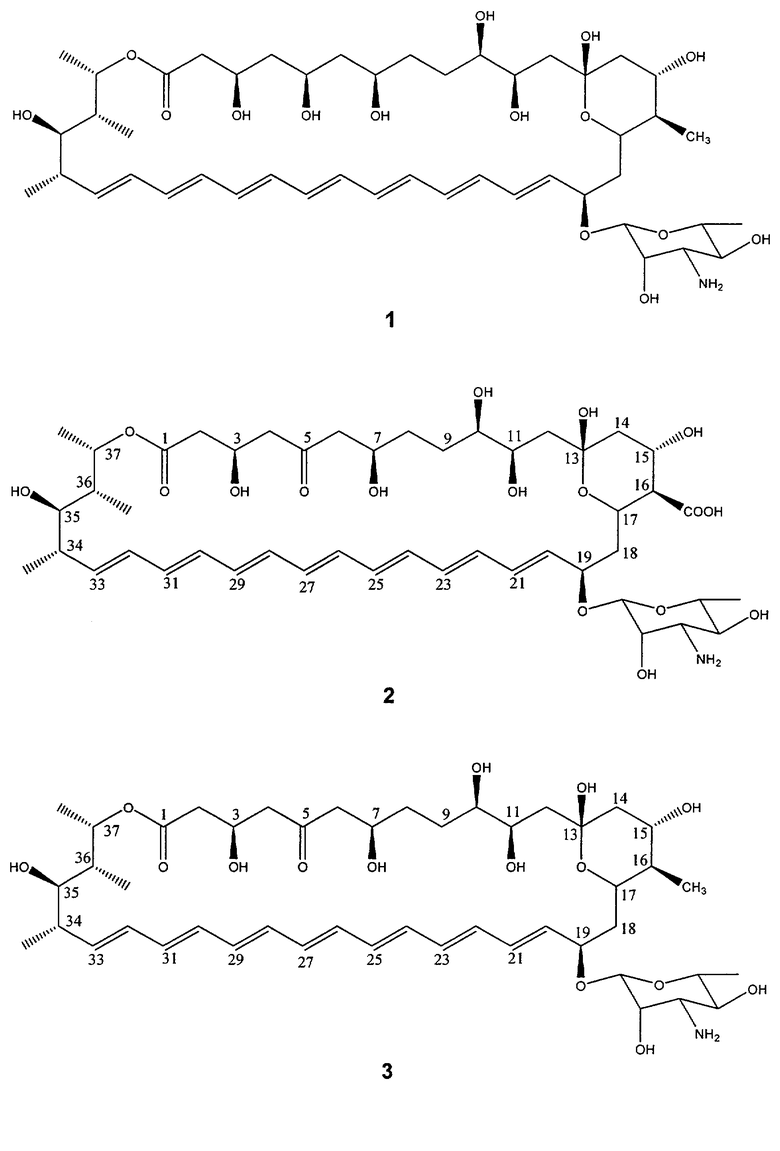

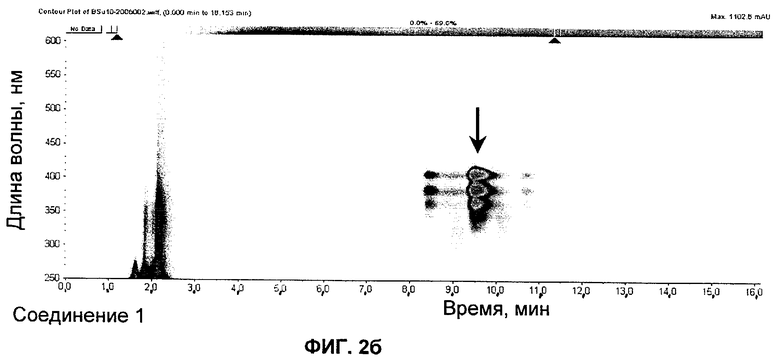

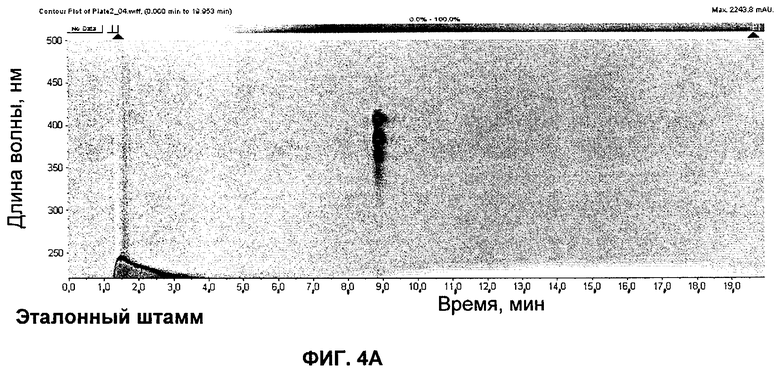


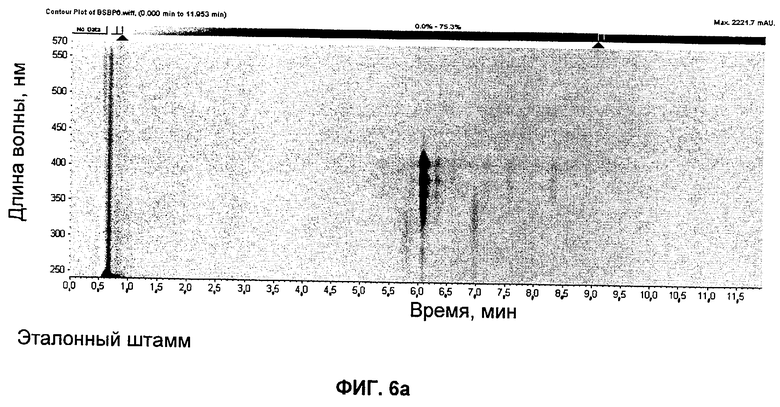

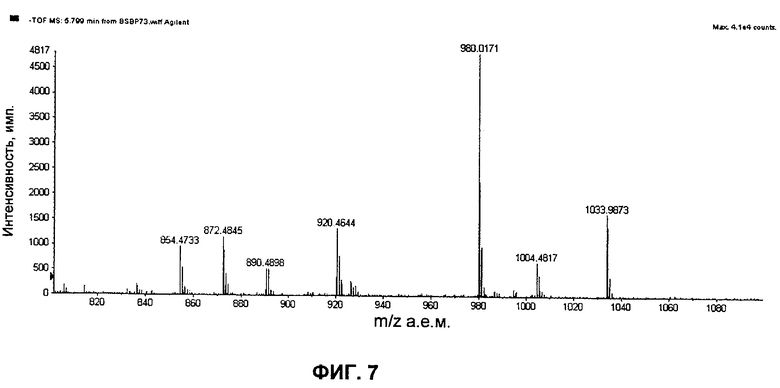
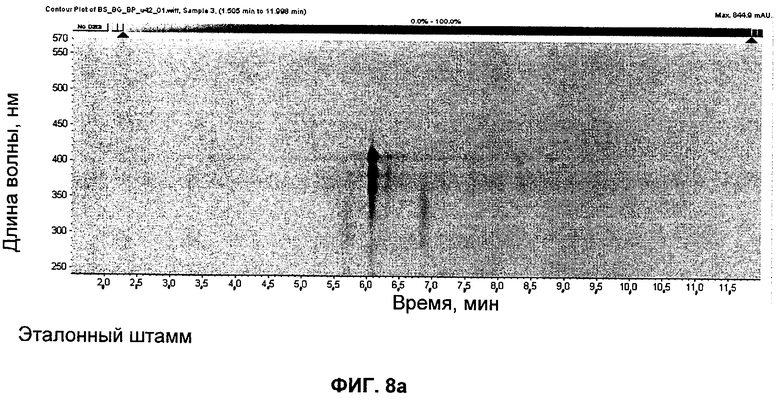
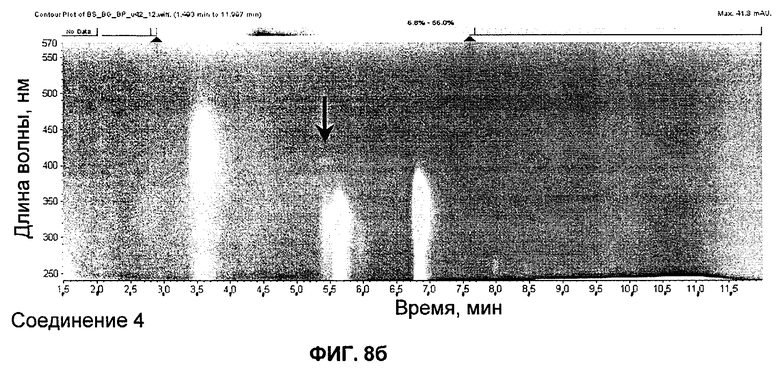
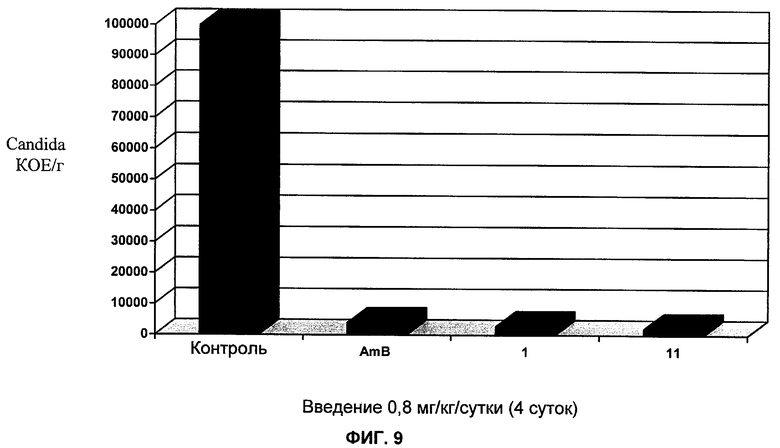
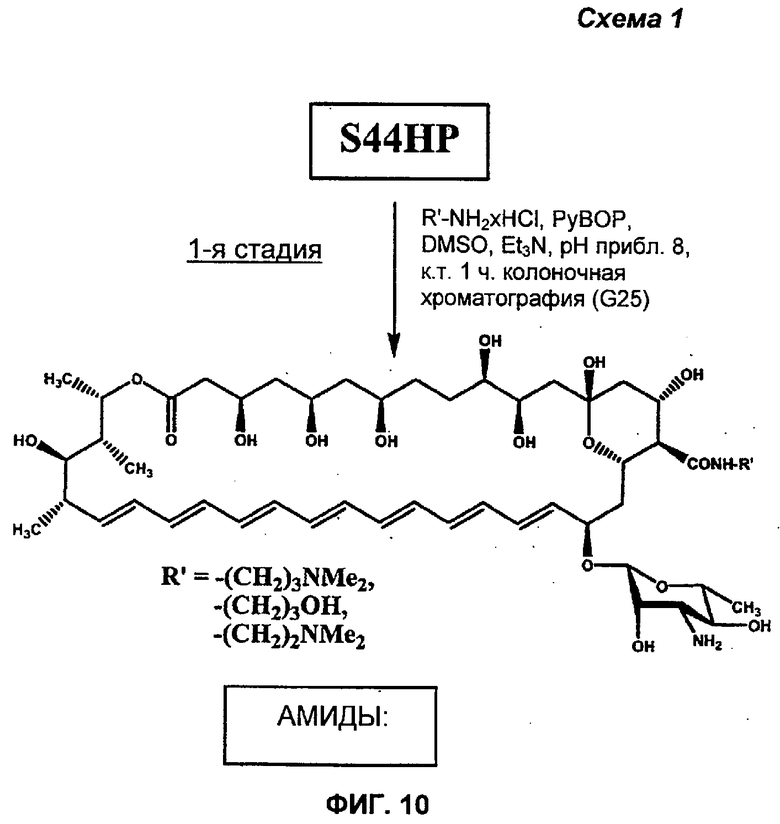

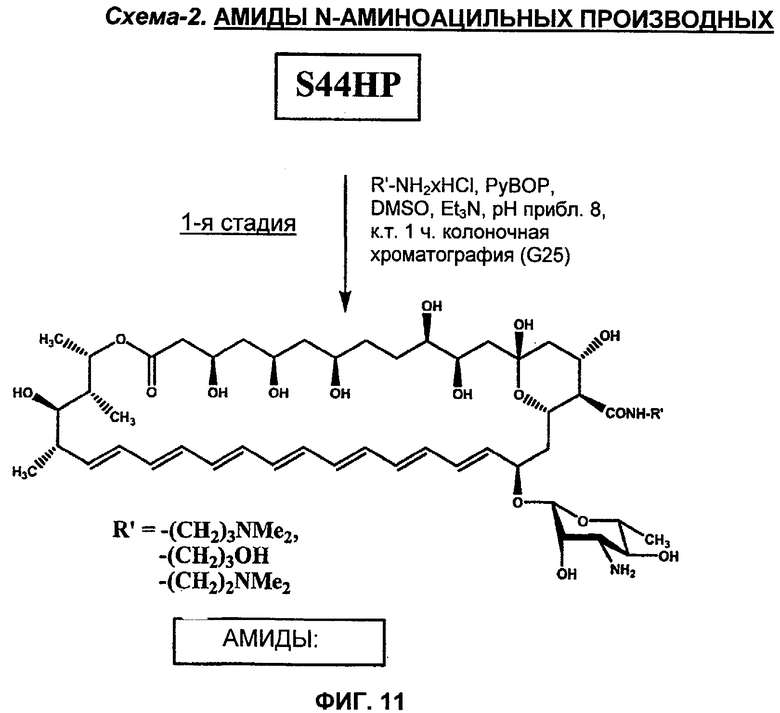
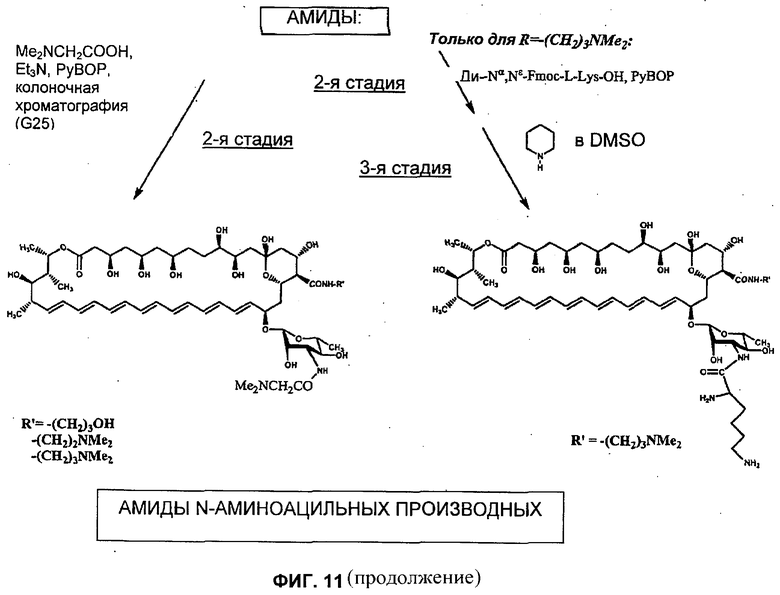

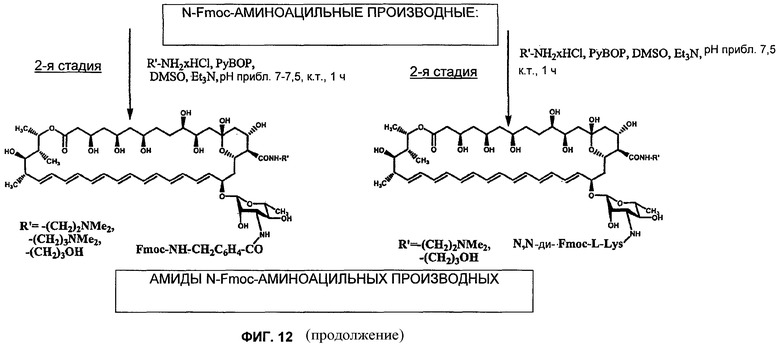
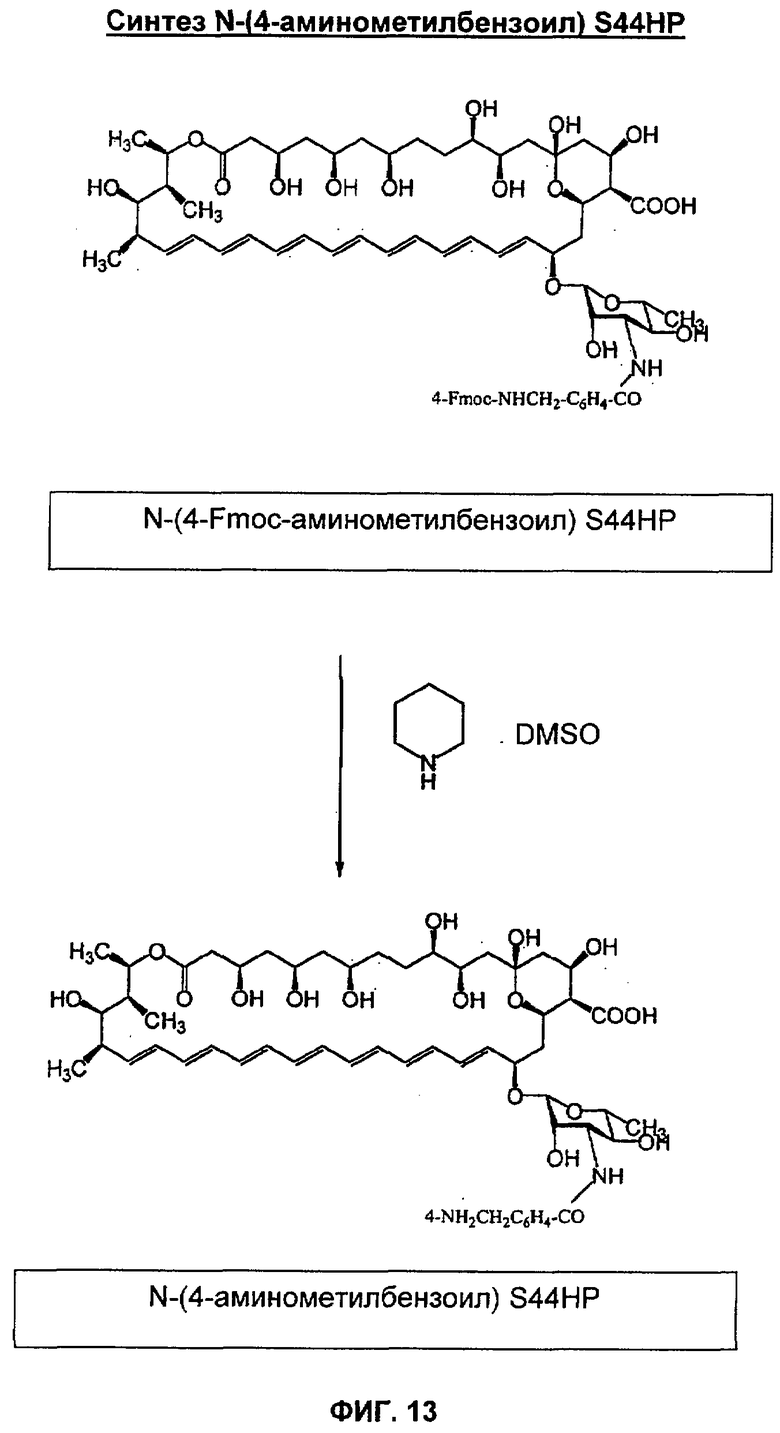
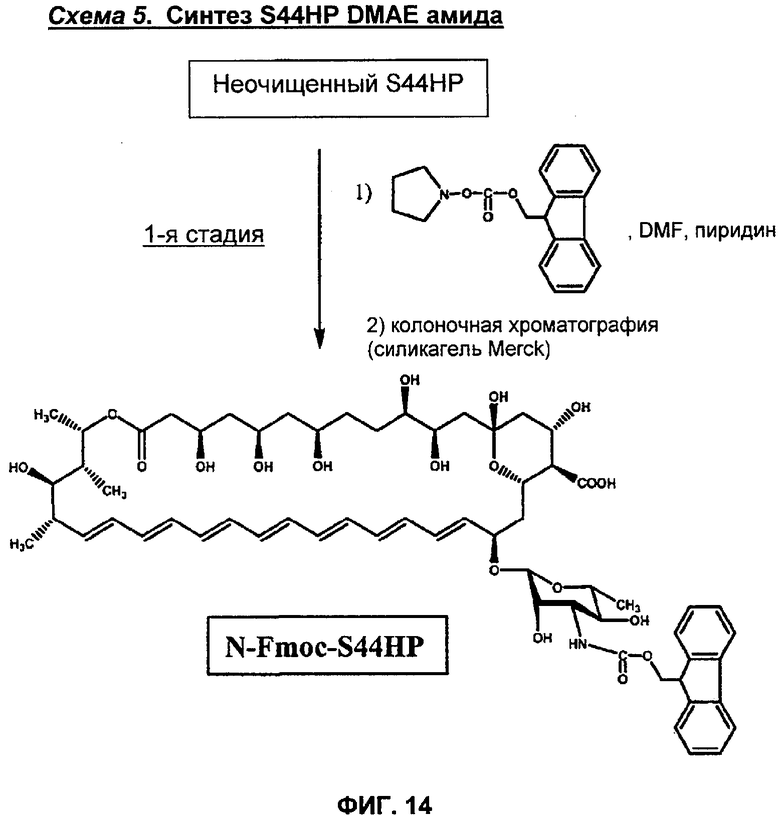
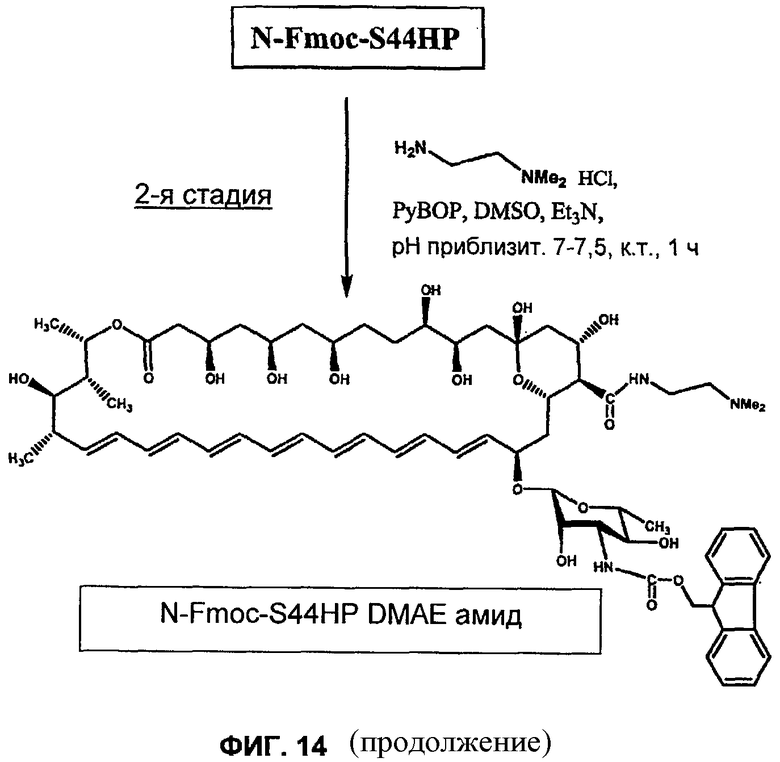
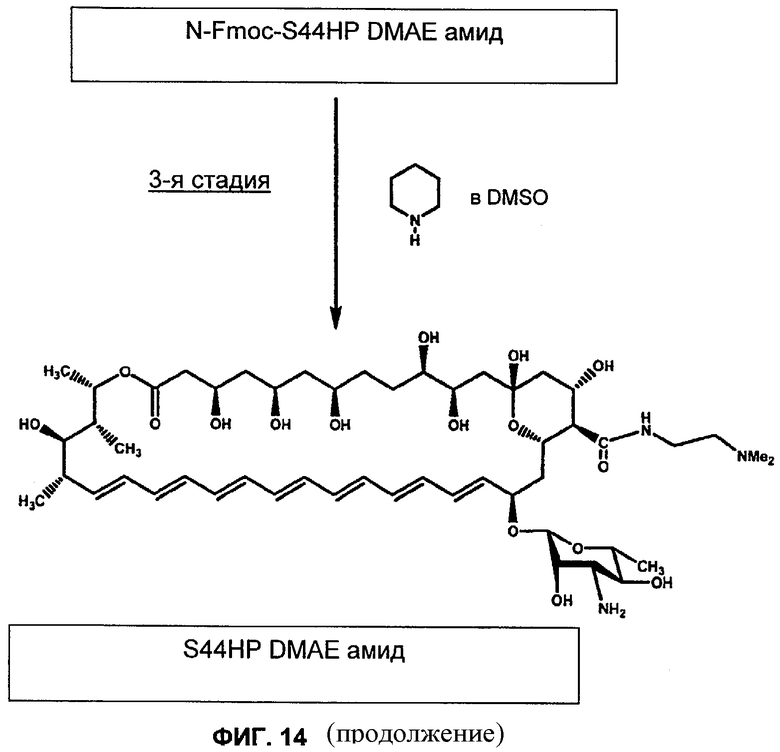
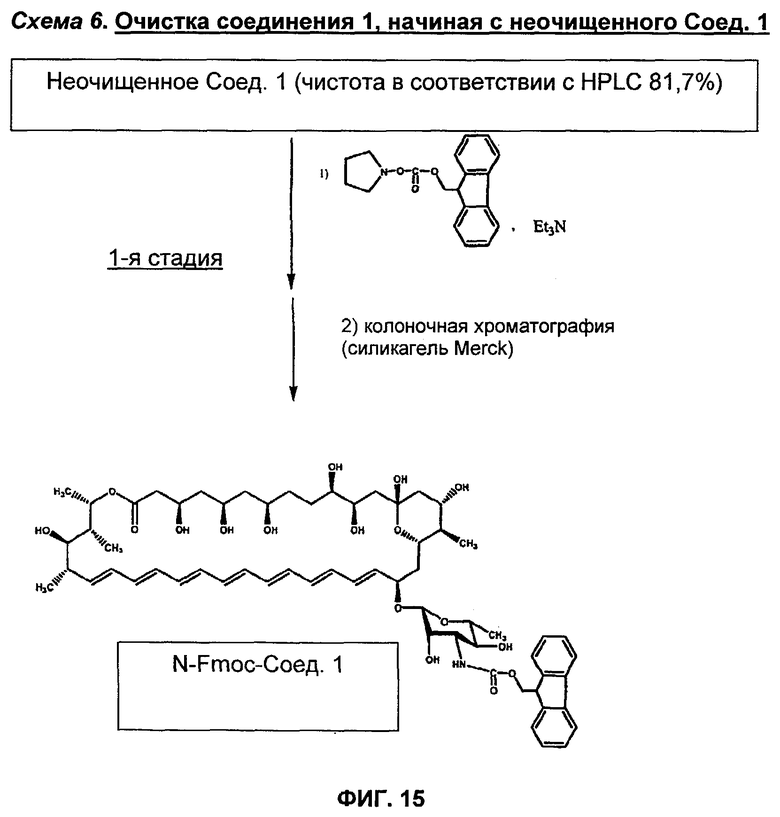
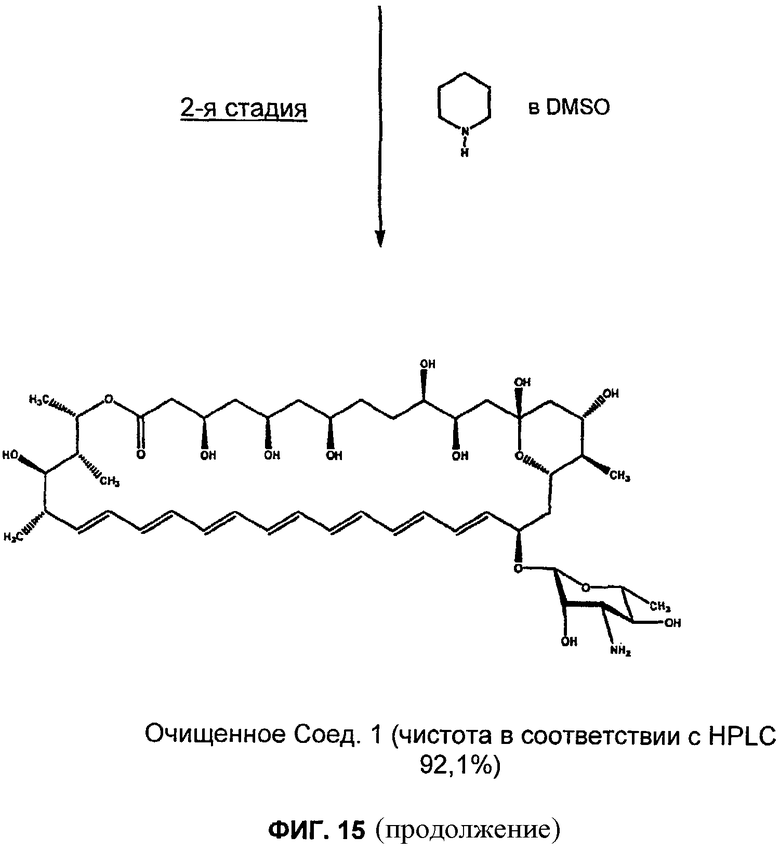
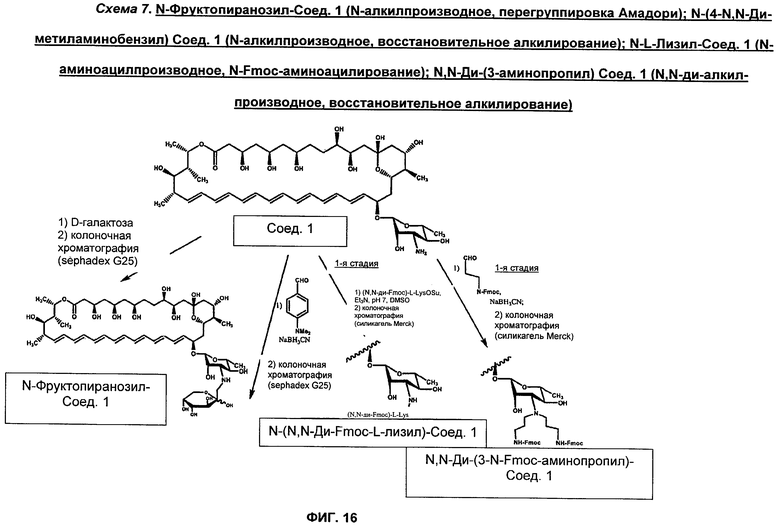

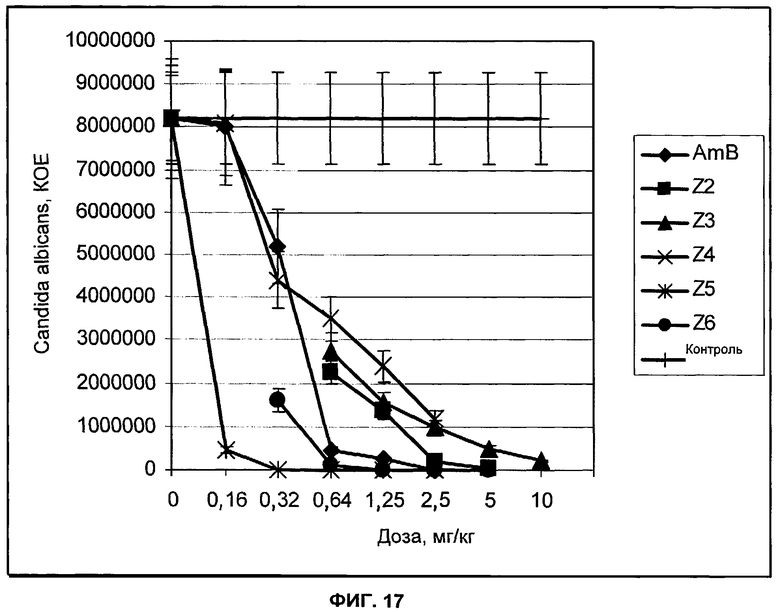
В настоящем изобретении предложено соединение формулы, указанной ниже, или его фармацевтически приемлемая соль, представляющее собой производное нистатина, имеющее дополнительную двойную связь, присутствующую между С28 и С29, и дополнительно модифицированное относительно нистатина по положению С 16. Эти соединения используют в качестве противогрибковых агентов. Также в изобретении заявляется фармацевтическая композиция и ее изготовление для лечения грибковых инфекций и способ лечения грибковой инфекции у животного, включающий введение указанному животному эффективной дозы соединения. 6 н. и 4 з.п. ф-лы, 29 пр., 17 ил., 15 табл.
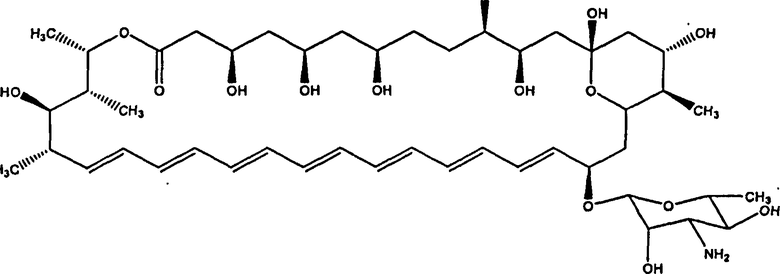
1. Соединение формулы
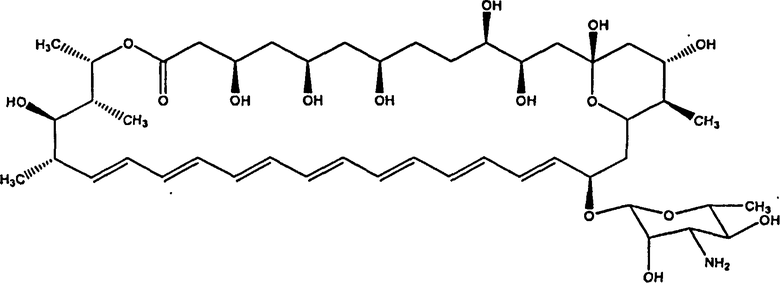
или его фармацевтически приемлемая соль.
2. Соединение по п.1 в форме соли, образованной с физиологически приемлемой органической или неорганической кислотой, выбранной из группы, состоящей из уксусной, аспарагиновой, бензолсульфоновой, бензойной, биугольной, бисерной, бивинной, масляной, эдетата кальция, камсиловой, угольной, хлорбензойной, лимонной, этилендиаминтетрауксусной, 1,2-этандисульфоновой, лаурилсерной, этансульфоновой, этансульфиновой, муравьиной, фумаровой, глюкогептоновой, глюконовой, глутаминовой, гликолиларсаниловой, гексамовой, гексилрезорциновой, гидрабаминовой, бромоводородной, соляной, йодводородной, гидроксинафтойной, изэтионовой, молочной, лактобионовой, малеиновой, яблочной, малоновой, миндальной, метансульфоновой, метилазотной, метилсерной, слизевой, муконовой, 2-нафталинсульфоновой, азотной, щавелевой, пара-нитрометансульфоновой, памовой, пантотеновой, фосфорной, двузамещенной соли фосфорной кислоты, однозамещенной соли фосфорной кислоты, фталевой, полигалактуроновой, пропионовой, салициловой, стеариновой, янтарной, сульфаминовой, сульфаниловой, сульфоновой, серной, дубильной, винной, 8-хлортеофиллиновой и толуолсульфоновой кислоты.
3. Соединение по п.1 в форме соли, образованной с основанием, выбранным из группы, состоящей из первичных, вторичных и третичных аминов, замещенных аминов, циклических аминов, аргинина, бетаина, кофеина, холина, N.N-дибензилэтилендиамина, диэтиламина, 2-диэтиламиноэтанола, 2-диметиламиноэтанола, этаноламина, этилендиамина, N-этилморфолина, N-этилпиперидина, глюкамина, глюкозамина, гистидина, гидрабамина, изопропиламина, лизина, метилглюкамина, морфолина, пиперазина, пиперидина, полиаминовых смол, прокаина, пуринов, теобромина, триэтиламина, триметиламина и трипропиламина.
4. Соединение по п.1 в форме соли глутаматной соли.
5. Соединение по любому из пп.1-4 для применения в противогрибковой терапии.
6. Способ получения соединений по любому из пп.1-4, включающий:
(1) модификацию генного кластера, кодирующего поликетидсинтазную систему, ответственную за синтез нистатина, путем инактивации домена ER в модуле 5 поликетидсинтазы с получением производного нистатина, имеющего двойную связь между С28 и С29; и
(2) дополнительную модификацию указанного генного кластера путем инактивации генов nysN, кодирующих монооксигеназу Р540, с получением производного нистатина, которое модифицировано по С16.
7. Способ получения соединений по любому из пп.1-4, включающий:
(1) модификацию генного кластера, кодирующего поликетидсинтазную систему, ответственную за синтез нистатина, путем инактивации домена ER в модуле 5 поликетидсинтазы с получением производного нистатина, имеющего двойную связь между С28 и С29; и
(2) модификацию получающегося в результате производного по С16 посредством восстановительного алкилирования.
8. Фармацевтическая композиция для лечения грибковых инфекций, содержащая соединение по любому из пп.1-4 и носитель, разбавитель или эксципиент.
9. Применение соединения по любому из пп.1-4 для изготовления фармацевтической композиции для лечения грибковых инфекций.
10. Способ лечения грибковой инфекции у животного, включающий введение указанному животному эффективной дозы соединения по любому из пп.1-4.
| Original Reference No | |||
| Ударно-долбежная врубовая машина | 1921 |
|
SU115A1 |
| Grzybowska, A | |||
| Czerwinski, E | |||
| Borowski, "Process for preparing L-asparaginates of 3-(N,N-dimethylamino)-propylamides of polyene macrolide antibiotics", (Politechnika Gdanska, Pol.) | |||
| Pol | |||
| Установка для светления металла квадратных профилей | 1960 |
|
SU138831A1 |
| (Polish) | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| APPLICATION: | |||
| Гребенчатая передача | 1916 |
|
SU1983A1 |
| V | |||
| Paquet, E | |||
| Carreira, | |||

