Изобретение относится к фосфорорганической химии, а именно к способу получения флуоресцентных α-гидрокси-бисфосфоновых кислот и их солей для применения в качестве ингибиторов солеотложений, которые могут быть использованы для экспресс-анализа и мониторинга концентрации ингибитора в водооборотных системах без отбора проб и предотвращения отложений солей в водооборотных системах на предприятиях химической, нефтехимической, металлургической промышленности и жилищно-коммунального хозяйства в реальном времени.
Применение ингибиторов солеотложений в теплоэнергетике является в настоящее время наиболее распространенным и динамично развивающимся направлением борьбы с отложением солей. Основными реагентами, используемыми для этих целей, являются фосфонаты и полифосфаты, несмотря на последовательное внедрение полимерных ингибиторов.
Бисфосфонаты и их производные в настоящее время широко и успешно используются в качестве ингибиторов коррозии и солеотложений в теплоэнергетике и в установках обратного осмоса, как в России, так и за рубежом. Дозирование реагентов и контроль содержания ингибиторов в системе осуществляются методом периодического отбора проб и их химического анализа. Такой подход практически исключает возможность организации автоматического мониторинга содержания реагента в режиме реального времени. Однако, именно автоматический непрерывный мониторинг состояния водооборотной системы является залогом ее стабильной работы.
Распространенным подходом для решения проблемы контроля и дозирования при создании новых полимерных ингибиторов является введение флуоресцентного фрагмента в структуру полимера. Эта область является совершенно новым и быстро развивающимся направлением исследований. Однако попытки встраивать флуоресцентные метки в фосфорсодержащий ингибитор коррозии и солеотложений до настоящего времени никем в мире не предпринимались.
Отслеживание флуоресценции активного компонента позволяет эффективно устанавливать концентрацию ингибитора. Этот процесс можно реализовать в режиме реального времени с помощью оборудования, управляемого сигналом датчиков, определяющих интенсивность флуоресценции метки в воде. Также такое оборудование позволяет управлять дозированием реагента.
Наиболее близким к изобретению по технической сущности и достигаемому результату является способ получения ингибитора солеотложений для водооборотных систем, содержащего связанную флуоресцентную метку, заключающийся в том, что проводят радикальную сополимеризацию акриловой кислоты или акриловой кислоты и моноэфира фумаровой кислоты с соединениями формулы 1 или 2 по п. 1 в водной среде при нагревании в присутствии инициатора, сополимеризацию проводят при массовом содержании моноэфира фумаровой кислоты от 20 до 80% от общей массы мономеров и массовом содержании флуорофора от 1 до 10% от общей массы мономеров, а остальное составляет акриловая кислота, причем реакционная масса представляет собой водный раствор с массовой долей мономеров от 15 до 30%. (см. патент RU 2640339, кл. C09K 11/06, С09 В 57/14, С09 В 57/12, C08F 222/22, C08F 220/06, C02F 1/56 опубл. 18.10.2017).

Однако, поскольку данный ингибитор представляет собой полимер, механизм его ингибирующего действия в растворе должен существенно отличаться от механизма действия широко используемых органических бисфосфонатов. Стоит отметить, что процесс перехода от одного класса ингибиторов к другому является достаточно дорогостоящим, ввиду необходимости отработки режима работы оборудования и подбора оптимальных концентраций ингибитора солеотложений. Поэтому разработка флуоресцентно меченого ингибитора на основе бисфосфонатов является наиболее перспективной задачей ввиду более широкого промышленного применения данного класса ингибиторов солеотложений.
Примеры совмещения бисфосфоната с флуорофорным фрагментом представлены в литературе в качестве контрастных препаратов для скрининга микрокальцинозов при раке груди, биологически активных компонентов, позволяющих проводить контроль терапевтического процесса по флуоресцентному отклику. [Robin A. Nadar, Nicola Margiotta, Michele Iafisco, Jeroen J.J.P. van den Beucken, Otto C. Boerman, and Sander C.G. Leeuwenburgh. // Bisphosphonate-Functionalized Imaging Agents, Anti-Tumor Agents and Nanocarriers for Treatment of Bone Cancer. // Adv. Healthcare Mater., 2017, DOI: 10.1002/adhm.201601119.]. Примеры таких бифункциональных соединений: Pam78 [Zaheer A, Lenkinski RE, Mahmood A, Jones AG, Cantley LC, Frangioni JV. In vivo near-infrared fluorescence imaging of osteoblastic activity. // Nat. Biotechnol, 19(12), 1148-1154 (2001)., DOI:10.1038/nbt1201-1148], FAMRIS [Kashemirov BA, Bala JLF, Chen X et al. Fluorescently labeled risedronate and related analogues: 'magic linker' synthesis. // Bioconjugate Chem. 19(12), 2308-2310 (2008)., DOI:10.1359/jbmr.091009], Pam800 [Bhushan KR, Tanaka E, Frangioni JV. // Synthesis of conjugatable bisphosphonates for molecular imaging of large animals. // Angew. Chem. Int. Ed. 46, 7969-7971 (2007)., DOI:10.1002/anie.200701216], Pam-Tc/Re-800 [Bhushan KR, Misra P, Liu F, Mathur S, Lenkinski RE, Frangioni JV. // Detection of breast cancer microcalcifications using a dual-modality SPECT/NIR fluorescent probe. // J. Am. Chem. Soc. 130(52), 17648-17649 (2008)., DOI:10.1021/ja807099s]. Эти соединения предлагаются авторами как эффективные контрастные агенты для флуоресцентной визуализации [
 & Ivan
& Ivan  . // Bone-seeking probes for optical and magnetic resonance imaging. // Future Med. Chem. 2010, 2(3), 521-531]. Тем не менее, представленные в вышеуказанных источниках вещества имеют очень сложное химическое строение, что ограничивает их применение в качестве ингибиторов солеотложения. Низкий квантовый выход флуоресценции и локализация ее максимума в красной области спектра также делают подобные соединения (см. Примеры ниже) малоперспективными для использования в промышленной водоподготовке.
. // Bone-seeking probes for optical and magnetic resonance imaging. // Future Med. Chem. 2010, 2(3), 521-531]. Тем не менее, представленные в вышеуказанных источниках вещества имеют очень сложное химическое строение, что ограничивает их применение в качестве ингибиторов солеотложения. Низкий квантовый выход флуоресценции и локализация ее максимума в красной области спектра также делают подобные соединения (см. Примеры ниже) малоперспективными для использования в промышленной водоподготовке.
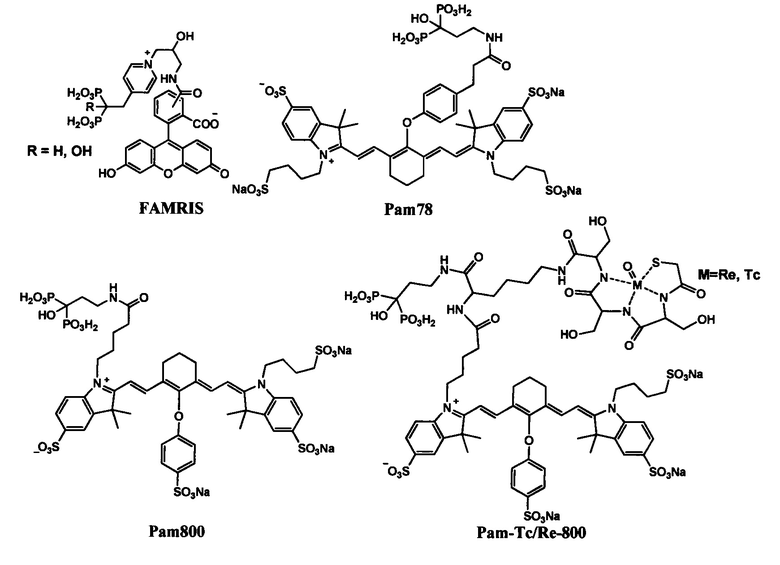
Примеры флуоресцентных бисфосфонатов, предложенных для флуоресцентной визуализации.
Задачей изобретения является устранение указанных недостатков.
Технический результат заключается в том, что достигается возможность получения флуоресцентных производных α-гидрокси-бисфосфонатов, положение максимумов флуоресценции которых локализовано в ультрафиолетовой области спектра в водной среде; также данные фосфонаты должны обладать способностью к ингибированию солеотложений. Таким образом, реализуется процесс предотвращения осадкообразования малорастворимых солей щелочноземельных металлов и, благодаря наличию флуоресцентной метки, может обеспечиваться возможность проведения экспресс-анализа и мониторинга концентрации ингибитора в водооборотных системах в реальном времени, без отбора проб, а окраска рабочего раствора не изменяется.
Поставленная задача решается заявляемыми соединениями общей формулы I (Способ А), представляющими собой производные α-гидрокси-бисфосфонатов 4-метокси-N-алкил-1,8-нафталимида, соединениями формулы 1а-с (Способ Б), представляющими собой производные α-гидрокси-бисфосфонатов 4-бром-N-алкил-1,8-нафталимида, являющихся полупродуктами для соединений формулы I, а технический результат достигается разработкой способов их получения (Способ А; Способ Б).
Способ А.

Способ А, включающий (i) ацилирование первичных алифатических аминокислот с (n=1, 3, 6) 4-бром-1,8-нафталевым ангидридом при кипячении в этаноле в присутствии триэтиламина, метилирование полученых 4-бром-(N-алкилкарбокси)-1,8-нафталимидов карбонатом калия в метаноле (ii), фосфорилирование (iii) хлорангидридов 4-метокси-(N-алкилкарбокси)-1,8-нафталимидов трис(триметилсилил)фосфитом с последующим метанолизом до целевого соединения формулы I, все этапы (iii), включая получение соответствующих хлорангидридов 4-метокси-(N-алкилкарбокси)-1,8-нафталимидов, проводятся in situ. После метанолиза целевые соединения формулы I выделяют известными приемами, заявляемый способ отличается тем, что этап метилирования проводят с более дешевым и технологически более удобным карбонатом калия в метаноле при температуре кипения растворителя. Выход целевых соединений по данному пути не менее 85%.
Способ Б.
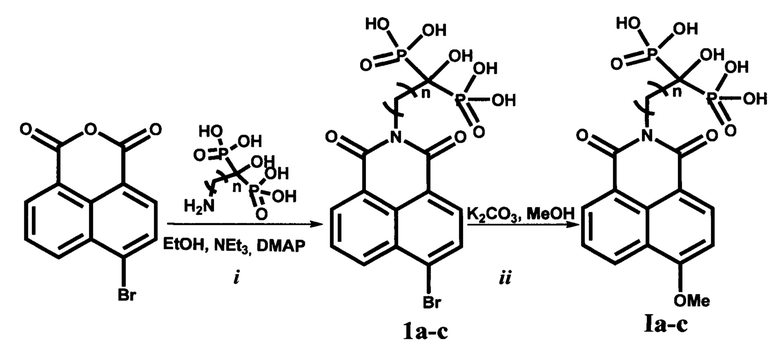
Способ Б, включающий (i) ацилирование α-аминобисфосфоновых кислот с (n=1, 3, 6) 4-бром-1,8-нафталевым ангидридом в присутствии триэтиламина и 4-диметиламинопиридина (ДМАП, DMAP) при кипячении в этаноле, метилирование (ii) α-гидрокси-бисфосфонатов 4-бром-N-алкил-1,8-нафталимида карбонатом калия в метаноле и выделение соединений формулы I известными приемами. Заявляемый способ отличается меньшим количеством стадий, отсутствием необходимости использовать хлористый тионил и трис(триметилсилил)фосфит, которые являются дорогостоящими реагентами, что делает его более технологичным и экономически рентабельным. Выход целевых соединений по данному пути составляет не менее 70%.
Описываемый способ позволяет получить ингибитор, эффективно предотвращающий процесс осадкообразования малорастворимых солей щелочно-земельных металлов. Наличие флуоресцентной группы в структуре ингибитора позволяет проводить экспресс-анализ и мониторинг в реальном времени концентрации ингибитора в водооборотных системах без отбора проб.
Строение всех полученных соединений доказано с помощью методов 1H, 13С, 31Р ЯМР-спектроскопии, масс-спектрометрического анализа высокого разрешения. Необходимые для реализации заявляемых способов исходные соединения, такие как 4-бром-1,8-нафталевый ангидрид, триэтиламин, диметиламинопиридин, карбонат калия, хлористый тионил, трис(триметилсилил)фосфит, аминоуксусная кислота, 4-аминобутановая кислота, 7-аминогептановая кислота; растворители: этанол, метанол, бензол являются коммерчески доступными реагентами (Acros, Sigma Aldrich, Merck, ЭКОС-1). Аминобисфосфоновые кислоты, а именно: 7-амино-1-гидрокси-1,1-бисфосфоногептановую, 4-амино-1-гидрокси-1,1-бисфосфонобутановую получали по известным методикам из соответствующих аминокислот [BY 13720 С1 2010.10.30 «Способ получения алендроновой кислоты»], физико-химические свойства полученных аминобисфосфоновых кислот соответствуют литературным данным.
Изобретение иллюстрируется следующими примерами. Общая методика синтеза 4-бром-(N-алкилкарбокси)-1,8-нафталимидов (Способ A, (i)).
0.072 моль 4-бромнафталевого ангидрида суспендируют в 340 мл этилового спирта, к полученной суспензии добавляют заранее приготовленную смесь следующего состава: 0,08 моль соответствующей аминокислоты, 20-50 мл этилового спирта, 4-10 мл дистиллированной воды и 0,24 моль триэтиламина. После чего кипятят полученную реакционную массу 5-12 часов, контролируя ход реакции с помощью тонкослойной хроматографии. По завершению реакции реакционную массу подкисляют концентрированной соляной кислотой до рН=3-5, фильтруют выпавший осадок, промывают осадок водой до нейтрального значения рН и высушивают при температуре 105°С не менее 6 часов. Получают 4-бром-N-алкилкарбокси-1,8-нафталимиды, которые используют в качестве исходных реагентов на следующей стадии.
Пример 1. По общей методике (Способ A, (i)) получают 17.1 г 4-бром-(N-метилкарбокси)-1,8-нафталимида (Выход 71%). Спектр 1Н ЯМР (300.21 МГц, ДМСО-d6, 26.2°С, δ/м.д., J/Гц): 4.73 (с, 2Н, -СН2-), 8.01 (дд, 1H, Н(6), 3J1=8.5, 3J2=7.3), 8.23 (д, 1H, H(2), 3J=7.9), 8.35 (д, 1H, H(3), 3J=1.9), 8.55-8.61 (мультиплет, 2H, H(5), H(7)), 13.13 (уш. с, 1H, -COOH).
Пример 2. По общей методике (Способ А, (i)) получают 18.8 г 4-бром-(N-пропилкарбокси)-1,8-нафталимида (Выход 72%). Спектр 1Н ЯМР (300.21 МГц, ДМСО-d6, 24.7°С, δ/м.д., J/Гц): 1.89 (квинтет, 2Н, -СН2-, 3J1=7.1, 3J2=6.9), 2.31 (т, 2Н, -СН2-, 3J=7.1), 4.07 (т, 2Н, -СН2-, 3J=6.9), 7.96 (дд, 1Н, Н(6), 3J1=7.4, 3J2=8.5), 8.17 (д, 1Н, Н(3), 3J=7.9), 8.29 (д, 1Н, Н(2), 3J=7.9), 8.47-8.56 (м, 2Н, Н(5), Н(7)), 12.02 (уш. с, 1H, -СООН).
Пример 3. По общей методике (Способ А, (i)) получают 18.6 г 4-бром-(N-гексилкарбокси)-1,8-нафталимида (Выход 64%). Спектр 1Н ЯМР (300.21 МГц, ДМСО-d6, 24.6°С, δ/м.д., J/Гц): 1.27-1.36 (м, 4Н, 2СН2-), 1.50 (т, 2Н, -СН2-, 3J=6.97), 1.60 (м, 2Н, -СН2-), 2.19 (т, 2Н, -СН2-, 3J=7.34), 3.97 (т, 2Н, -СН2-, 3J=7.34), 7.92 (дд, 1Н, Н(6), 3J=7.4, 3J=8.4), 8.13 (д, 1Н, Н(3), 3J=7.7), 8.24 (д, 1H, Н(2), 3J=7.7), 8.44 (д, 1H, Н(7), 3J=8.4), 8.48 (д, 1Н, Н(5), 3J=7.4), 11.97 (уш. с, 1H, -СООН).
Общая методика синтеза 4-метокси-(N-алкилкарбокси)-1,8-нафталимидов (Способ А, (ii)).
Соответствующий 4-бром-(N-алкилкарбокси)-1,8-нафталимид (0.05 моль), полученный на предыдущей стадии суспендируют в 108 мл метилового спирта, добавляют 138 г карбоната калия (1 моль). Полученную реакционную массу кипятят 7-10 часов, контролируя ход реакции с помощью тонкослойной хроматографии. По завершении реакции растворитель удаляют под вакуумом, остаток разбавляют в два раза водой, подкисляют концентрированной соляной кислотой до рН=3-5, полученную суспензию экстрагируют этилацетатом 3 раза по 50 мл, промывают органический экстракт водой, высушивают над безводным сульфатом магния и упаривают. Полученный осадок высушивают при температуре 80°С не менее 5 часов.
Пример 4. По общей методике (Способ А, (ii)) получают 10.7 г 4-метокси-(N-метилкарбокси)-1,8-нафталимида (Выход 75%). Спектр 1Н ЯМР (300.21 МГц, ДМСО-d6, 25.7°С, δ/м.д., J/Гц): 4.15 (с, 2Н, -СН2-), 4.72 (с, 3Н, -ОМе), 7.37 (д, 1Н, Н(3), 3J=8.4), 7.85 (дд, 1Н, Н(6), 3J1=8.3, 3J1=7.4), 8.48-8.62 (м, 3Н, Н(2), Н(5), Н(7)), 13.04 (с, 1Н, -СООН).
Пример 5. По общей методике (Способ A, (ii)) получают 12.7 г 4-метокси-(N-пропилкарбокси)-1,8-нафталимида (Выход 81%). Спектр 1Н ЯМР (300.21 МГц, ДМСО-d6, 26.7°С, δ/м.д., J/Гц): 1.86 (квинтет, 2Н, -СН2-, J=7.3), 2.29 (т, 2Н, -СН2-, J=7.3), 4.03 (т, 2Н, -СН2-, J=7.3), 4.08 (с, 3Н, -ОМе), 7.23 (д, 1Н, Н(3), 3J=8.4), 7.73 (дд, 1Н, Н(6), 3J1=8.3, 3J1=7.4), 8.30-8.46 (м, 3Н, 3Н, Н(2), Н(5), Н(7)), 12.03 (с, 1H, -СООН).
Пример 6. По общей методике (Способ A, (ii)) получают 14 г 4-метокси-(N-гексилкарбокси)-1,8-нафталимида (Выход 79%). Спектр 1Н ЯМР (300.21 МГц, ДМСО-d6, 26.7°С, δ/м.д., J/Гц): 1.32 (м, 4Н, 2СН2-), 1.48 (м, 2Н, -СН2-), 1.58 (м, 2Н, -СН2-), 2.19 (т, 2Н, -СН2-, 3J=7.3), 3.97 (т, 2Н, 3J=7.3, -СН2-), 4.10 (с, 3Н, -ОМе), 7.25 (д, 1H, Н(3), 3J=8.4), 7.74 (дд, 1Н, Н(6), 3J1=8.3, 3J1=7.4), 8.35-8.46 (м, 3Н, Н(2), Н(5), Н(7)), 11.96 (с, 1H, -СООН).
Общая методика синтеза 4-метокси-(N-алкил-1,1-бисфосфоно-α-гидрокси)-1,8-нафталимидов (Ia-с), (Способ А, (iii)).
Соответствующий 4-метокси-(N-алкилкарбокси)-1,8-нафталимид (0.01 моль), полученный на предыдущей стадии суспендируют в 30 мл абсолютного бензола, к полученной суспензии медленно, по каплям добавляют избыток тионилхлорида (0.1 моль), кипятят 7-8 часов, удаляют избыток хлористого тионила и бензола под вакуумом, остаток суспендируют в абсолютном бензоле и медленно по каплям добавляют трис(триметилсилил)фосфит (0.02 моль), перемешивают при комнатной температуре 12 часов, затем удаляют растворитель под вакуумом, к маслообразному остатку добавляют метиловый спирт (30 мл) и перемешивают 3 часа, затем отфильтровывают выпавший осадок, промывают метанолом и высушивают при 80°С.
Пример 7.
По общей методике (Способ A, (iii)) получают 3.75 г 4-метокси-(N-метил-1,1-бисфосфоно-α-гидрокси)-1,8-нафталимида - Ia (Выход 87%). Спектр 1Н ЯМР (300.21 МГц, ДМСО-d6, 25.2°С, δ/м.д., J/Гц): 4.01 (с, 3Н, -ОМе), 4.51 (с, 2Н, СН2-), 6.87 (д, 1Н, Н(3), 3J=8.4), 7.37 (дд, 1H, Н(6), 3J1=8.26, 3J2=7.54) 7.98-8.12 (м, 3Н, Н(2), Н(5), Н(7)). Спектр 13С ЯМР (100.24 МГц, D2O, 22°С, δ/м.д., J/Гц) 40.80, 57.09, 74.35 (т, JC,P=130.11 Гц -СОН), 106.71, 111.7, 119.15, 122.56, 126.40, 126.86, 127.21, 130.02, 132.53, 157.51, 162.20, 163.14. Спектр 31Р ЯМР (121.73 МГц, ДМСО-d6, 30°С, δ/м.д.): 19.7. Масс-спектр (ИЭР), вычислено, m/z: 431.02; найдено: 432.02 ([М+Н]+), 454.02 ([M+Na]+).
Пример 8.
По общей методике (Способ А, (iii)) получают 3.9 г 4-метокси-(N-пропил-1,1-бисфосфоно-α-гидрокси)-1,8-нафталимида - Ib (Выход 85%). Спектр 1Н ЯМР (300.21 МГц, ДМСО-d6, 26.7°С, δ/м.д., J/Гц): 1.48 (м, 4Н, 2СН2-), 3.54 (м, 2Н, СН2-), 3.66 (с, 3Н, ОМе-), 6.84 (д, 1Н, Н(3), 3J=8.4), 7.34 (дд, 1H, Н(6), 3J1=8.25, 3J2=7.52) 7.93-8.08 (м, 3Н, Н(2), Н(5), Н(7)). Спектр 13С ЯМР (100.24 МГц, D2O, 22°С, δ/м.д., J/Гц): 25.40, 27.55, 40.65, 57.07, 74.34 (т, JC,P=130.11 Гц -СОН), 106.42, 111.55, 119.09, 121.76, 126.57, 126.68, 127.31, 130.12, 132.43, 157.71, 162.24, 163.32. Спектр 31Р ЯМР (121.73 МГц, ДМСО-d6, 30°С, δ/м.д.): 20.4. Масс-спектр (ИЭР), вычислено, m/z: 459.05; найдено: 460.05 ([М+Н]+), 482.05 ([M+Na]+).
Пример 9.
По общей методике (Способ А, (iii)) получают 4.3 г 4-метокси-(N-гексил-1,1-бисфосфоно-α-гидрокси)-1,8-нафталимида - Ic (Выход 86%). Спектр 1Н ЯМР (400.13 МГц, ДМСО-d6, 25°С, δ/м.д., J/Гц): 1.17-1.39 (м, 4Н, 2СН2), 1.45-1.70 (м, 4Н, 2СН2), 1.70-1.97 (м, 2Н, СН2), 3.90-4.03 (м, 2Н, СН2), 4.09 (с, 3Н, -ОМе), 7.24 (д, 1Н, Н(3), 3J=8.3,), 7.74 (дд, 1H, 3J1=7.9, 3J2=7.5), 8.47-8.33 (м, 3Н, Н(2), Н(5), Н(7)). Спектр 13С ЯМР (100.61 МГц, D2O, 19.3°C, δ/м. д., J/Гц): 23.64, 26.53, 27.44, 29.65, 33.88, 40.45, 55.96, 74.33 (т, JC,P=129.11 Гц -СОН), 105.29, 111.60, 119.04, 121.06, 125.29, 126.54, 128.35, 130.87, 133.35, 160.30, 163.63, 164.37. Спектр 31Р ЯМР (121.53 МГц, ДМСО-d6, 24.8°С, δ/м.д.): 20.6. Масс-спектр (ИЭР), вычислено, m/z: 501.10; найдено: 502.10 ([МН]+), 524.10 ([M+Na]+).
Общая методика синтеза 4-бром-(N-алкил-1,1-бисфосфоно-α-гидрокси)-1,8-нафталимидов (X), (Способ Б, (i)).
0.048 Моль 4-бромнафталевого ангидрида суспендируют в 370 мл этилового спирта, к полученной суспензии добавляют заранее приготовленную смесь следующего состава: 0,048 моль соответствующей аминобисфосфоновой кислоты, 30-70 мл этилового спирта, 6-13 мл дистиллированной воды, 0,5 моль триэтиламина, 4⋅10-3 моль диметиламинопиридина (ДМАП, DMAP). После чего кипятят полученную реакционную массу 13-18 часов. Затем реакционную массу подкисляют концентрированной соляной кислотой до рН=3-5, разбавляют изопропиловым спиртом в 5-7 раз (по объему), оставляют на 24 часа при +4°С для формирования осадка, который затем фильтруют, промывают смесью изопропанол: вода (80:20 по объему) и высушивают при температуре 110°С не менее 8 часов. Получают 4-бром-N-алкил-1,1-бисфосфоно-α-гидрокси-1,8-нафталимиды (X), которые затем используют для получения целевых соединений (Ia-с).
Пример 10.
По общей методике (Способ Б, (i) получают 13.4 г 4-бром-(N-метил-1,1-бисфосфоно-α-гидрокси)-1,8-нафталимида - 1а (Выход 58%). Спектр 1H ЯМР (300.21 МГц, ДМСО-d6, 25.8°С, δ/м.д., J/Гц): 4.71 (с, 1H, СН2), 7.66 (дд, 1H, Н(6), 3J1=8.44, 3J2=7.52), 7.82 (д, 1Н, Н(3), 3J=8.07), 7.98 (д, 1Н, Н(2), 3J=8.07), 8.21 (д, 1H, Н(5), 3J=8.44), 8.28 (д, Н(7), 1Н, 3J=7.52). Спектр 13С ЯМР (75.50 МГц, D2O, 30°С, δ/м.д., J/Гц): 46.69, 78, 10 (т, JC,P=128.81 Гц -СОН), 123.89, 124.71, 130.57, 131.09, 132.42, 133.36, 134.04, 134.39, 135.29, 136.45, 168.55, 168.64. Спектр 31Р ЯМР (121.52 МГц, D2O, 27°С, δ/м.д.): 18.1. Масс-спектр (ИЭР), вычислено, m/z: 480.92; найдено: 481.92 ([МН]+), 503.92 ([M+Na]+).
Пример 11.
По общей методике (Способ Б, (i) получают 14.8 г 4-бром-(N-пропил-1,1-бисфосфоно-α-гидрокси)-1,8-нафталимида - 1b (Выход 61%).
Спектр 1Н ЯМР (400.13 МГц, D2O, 19.3°С, δ/м.д., J/Гц): 1.73-1.83 (м, 2Н, СН2), 1.86-2.01 (м, 2Н, СН2), 3.58-3.72 (м, 2Н, СН2), 7.18 (дд, 1H, Н(6), 3J1=8.41, 3J2=7.43), 7.25 (д, 1H, Н(3), 3J=7.85), 7.32 (д, 1H, Н(2), 3J=7.85), 7.55 (д, 1H, Н(5), 3J=7.43), 7.67 (д, 1H, Н(7), 3J=8.41). Спектр 13С ЯМР (100.61 МГц, D2O, 20°С, δ/м.д., J/Гц): 22.20, 30.60, 39.93, 73.65 (т, JC,P=134.25 Гц -С(ОН)), 119.15, 120.06, 125.93, 127.90, 128.20, 130.41, 130.65, 130.93, 131.77, 133.15, 163.55, 163.67. Спектр 31Р ЯМР (161.98 МГц, D2O, 19.2°С, δ/м.д.): 18.3. Масс-спектр (ИЭР), вычислено, m/z: 506.95; найдено: 507.95 ([МН]+), 529.95 ([M+Na]+).
Пример 12.
По общей методике (Способ Б, (i) получают 16.3 г 4-бром-(N-гексил-1,1-бисфосфоно-α-гидрокси)-1,8-нафталимида - 1с (Выход 62%). Спектр 1Н ЯМР (300.13 МГц, D2O, 20.2°С, δ/м.д., J/Гц): 1.19-1.40 (м, 6Н, 3СН2), 1.47-1.67 (м, 2Н, СН2), 1.72-1.90 (м, 2Н, СН2), 3.92-4.03 (м, 2Н, СН2), 7.95 (дд, 1Н, Н(6), 3J1=8.45, 3J2=7.54), 8.15 (д, 1Н, Н(3), 3J=7.77), 8.27 (д, 1H, Н(2), 3J=7.77), 8.47 (д, 1H, Н(5), 3J=8.45), 8.51 (д, 1Н, Н(7), 3J=7.54). Спектр 13С ЯМР (100.61 МГц, ДМСО-d6, 22.1°С, δ/м.д., J/Гц): 27.55, 27.82, 28.77, 28.85, 29.46, 40.09, 74.55 (т, JC,P=132.15 Гц -СОН), 118.15, 119.56, 124.98, 128.01, 128.68, 130.41, 130.68, 130.93, 131.77, 133.18, 163.57, 163.77. Спектр 31Р ЯМР (121.49 МГц, ДМСО-d6, 20.1°С, δ/м.д.): 20.1. Масс-спектр (ИЭР), вычислено, m/z: 549.00; найдено: 550.00 ([МН]+), 572.00 ([M+Na]+).
Общая методика синтеза 4-метокси-(N-алкил-1,1-бисфосфоно-α-гидрокси)-1,8-нафталимидов (Ia-с), (Способ Б, (ii)).
Соответствующий 4-бром-N-алкил-1,1-бисфосфоно-α-гидрокси-1,8-нафталимид (0.01 моль), полученный на предыдущей стадии суспендируют в 46 мл метанола, к полученной суспензии добавляют мелко растертый безводный карбонат калия (0.25 моль), кипятят 15-18 часов, затем удаляют избыток метанола под вакуумом, остаток растворяют в воде и подкисляют концентрированной соляной кислотой до рН=3-5, разбавляют изопропиловым спиртом в 10 раз (по объему), оставляют на 48 часов при +4°С для формирования осадка, который затем фильтруют, промывают этанолом и высушивают при температуре 107°С не менее 10 часов.
По общей методике (Способ Б, (ii) получают 3.1 г 4-метокси-(N-метил-1,1-бисфосфоно-α-гидрокси)-1,8-нафталимида - Ia (Выход 71%).
По общей методике (Способ Б, (ii) получают 3.4 г 4-метокси-(N-пропил-1,1-бисфосфоно-α-гидрокси)-1,8-нафталимида - Ib (Выход 73%).
По общей методике (Способ Б, (ii) получают 3.9 г 4-метокси-(N-гексил-1,1-бисфосфоно-α-гидрокси)-1,8-нафталимида - Ic (Выход 77%).
Физико-химические характеристики соединений Ia-с, полученных по Способу Б (ii) идентичны свойствам Ia-с, полученных по Способу A (iii).
Водные растворы полученных соединений Ia-с характеризуются максимумами длин волн поглощения (λemmax=365 нм) и флуоресценции (λflmax=455 нм). Квантовый выход достигает 90% (в воде).
Процедура тестирования ингибирующей способности полученного описанным выше способом ингибитора.
Процесс ингибирования исследовали, используя в качестве базового протокол NACE Standard ТМ0374-2007 protocol. Для получения пересыщенного раствора карбоната кальция готовили два раствора в дистиллированной воде: рассол кальция (12,15 г/дм3 CaCl2⋅2H2O; 3,68 г/дм3 MgCl2⋅6H2O; NaCl 33 г/дм3) и бикарбонатный рассол (7,36 г NaHCO3; 33 г/дм3 NaCl). Состав рассолов для получения пересыщенного раствора сульфата кальция: кальциевый рассол 11,10 г/дм3 CaCl2⋅2H2O, 7,50 г/л NaCl; сульфатный рассол: 10,66 г/дм3 Na2SO4, 7,50 NaCl.
При смешении этих рассолов в объемном соотношении 1:1 получали пересыщенные растворы карбоната или сульфата кальция. Пересыщенные растворы карбоната или сульфата кальция с заранее внесенным количеством ингибитора выдерживали 24 часа при 71°С, охлаждали и определяли остаточное содержание кальция.
Эффективность испытуемых ингибиторов определяли в виде процента ингибирования
I=100⋅([Са]ехр - [Са]fin / ([Ca]init - [Ca]fin])
где
- [Са]exp _ концентрация кальция в фильтрате в присутствии ингибитора по прошествии 24 часов обработки;
- [Ca]fin - концентрация кальция в фильтрате в отсутствии ингибитора по прошествии 24 часов обработки;
- [Ca]init - начальная концентрация кальция.
В таблице 1 приведены результаты тестирования ингибирующей способности флуоресцентных бисфосфонатов Ia-с, в сравнении с наиболее часто используемым в промышленности фосфонатным ингибитором - оксиэтилидендифосфоновой кислотой (ОЭДФ).

| название | год | авторы | номер документа |
|---|---|---|---|
| 4-ЗАМЕЩЕННЫЕ N-АРИЛ-1,8-НАФТАЛИМИДЫ, ПРОЯВЛЯЮЩИЕ СВОЙСТВА ФЛУОРЕСЦЕНТНЫХ СЕНСОРОВ НА КАТИОНЫ МЕТАЛЛОВ, И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2012 |
|
RU2515195C1 |
| АЗАКРАУНСОДЕРЖАЩИЕ N-АРИЛ-1,8-НАФТАЛИМИДЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2017 |
|
RU2656106C1 |
| ЭНАНТИОМЕРНО ЧИСТЫЕ ОСНОВНЫЕ ЭФИРЫ АРИЛ-ЦИКЛОАЛКИЛГИДРОКСИКАРБОНОВЫХ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ | 1997 |
|
RU2238936C2 |
| ТРИФЕНИЛФОСФОНИЕВЫЕ СОЛИ ЛУПАНОВЫХ ТРИТЕРПЕНОИДОВ, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ ВЕЩЕСТВ | 2012 |
|
RU2551647C2 |
| БОРСОДЕРЖАЩИЕ МАЛЫЕ МОЛЕКУЛЫ В КАЧЕСТВЕ ПРОТИВОВОСПАЛИТЕЛЬНЫХ АГЕНТОВ | 2009 |
|
RU2547441C2 |
| Производные кумарина, тиокумарина и хинолинона, обладающие противосудорожной активностью | 2017 |
|
RU2720510C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ С ИХ ИСПОЛЬЗОВАНИЕМ | 2001 |
|
RU2265598C2 |
| (3bR*,7aR*,10bR*,14aR*-cis-14c,14d)-2,9 БИС(ГАЛОГЕНФЕНИЛ)ОКТАДЕКАГИДРО-1Н,8Н-2,3а,7b,9,10a,14b-ГЕКСААЗАДИБЕНЗО[fg,op]ТЕТРАЦЕНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2021 |
|
RU2787455C1 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРЕНКАРБОКСАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ | 2003 |
|
RU2320656C2 |
| ИНГИБИТОРЫ НУКЛЕОЗИДФОСФОРИЛАЗ И НУКЛЕОЗИДАЗ | 2003 |
|
RU2330042C2 |
Изобретение относится к новым флуоресцентным производным α-гидрокси-бисфосфонатов для применения в качестве ингибиторов солеотложений. Описываются соединения (I) в качестве флуоресцентных ингибиторов солеотложений. Способ их получения включает ацилирование первичных алифатических аминокислот с числом СН2- звеньев 1, 3, 6 4-бром-1,8-нафталевым ангидридом при кипячении в этаноле в присутствии триэтиламина, метоксилирование 4-бром-(N-алкилкарбокси)-1,8-нафталимидов карбонатом калия в метаноле, реакцию с хлористым тионилом, фосфорилирование хлорангидридов 4-метокси-(N-алкилкарбокси)-1,8-нафталимидов трис(триметилсилил)фосфитом и метанолиз. Описывается также альтернативный способ их получения, включающий ацилирование α-аминобисфосфоновых кислот с числом СН2- звеньев 1,3,6 4-бром-1,8-нафталевым ангидридом в присутствии триэтиламина и 4-диметиламинопиридина при кипячении в этаноле, метоксилирование α-гидрокси-бисфосфонатов 4-бром-N-алкил-1,8-нафталимида карбонатом калия в метаноле и выделение целевого продукта известными приемами. Изобретение обеспечивает предотвращение осадкообразования малорастворимых солей щелочноземельных металлов и возможность проведения экспресс-анализа и мониторинга концентрации ингибитора в водооборотных системах в реальном времени без отбора проб и изменения окраски рабочего раствора. 4 н. и 2 з.п. ф-лы, 1 табл., 12 пр.
1. Соединения общей формулы I:

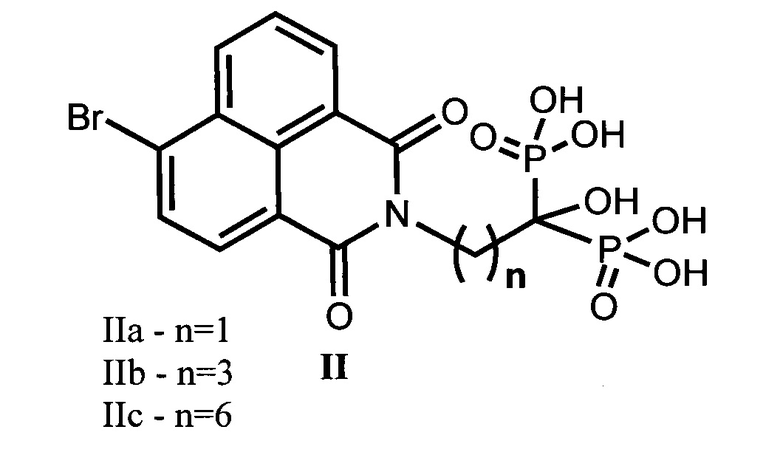
2. Соединения общей формулы II в качестве полупродуктов для получения соединений (I) по п. 1:
3. Соединения по п. 1 (Ia-с) в качестве флуоресцентных ингибиторов солеотложений.
4. Соединения по п. 2 (IIa-с) в качестве полупродуктов для получения соединений по п. 1 (Ia-c).
5. Способ получения флуоресцентных ингибиторов солеотложений формулы (I) по п. 1, заключающийся в том, что проводят (i) ацилирование первичных алифатических аминокислот (n=1, 3, 6, где n - количество звеньев СН2-) 4-бром-1,8-нафталевым ангидридом при кипячении в этаноле в присутствии триэтиламина, метоксилирование полученных 4-бром-(N-алкилкарбокси)-1,8-нафталимидов карбонатом калия в метаноле (ii), образование соответствующих хлорангидридов 4-метокси-(N-алкилкарбокси)-1,8-нафталимидов путем реакции с хлористым тионилом, фосфорилирование полученных (iii) хлорангидридов 4-метокси-(N-алкилкарбокси)-1,8-нафталимидов трис(триметилсилил)фосфитом с последующим добавлением метанола для проведения обменного процесса метанолиза до целевых соединений по п. 1 (Ia-c), все этапы (iii), включая получение соответствующих хлорангидридов 4-метокси-(N-алкилкарбокси)-1,8-нафталимидов, проводятся in situ без выделения, после метанолиза целевые соединения формулы I выделяют известными приемами.
6. Способ получения соединений по п. 1 (I a-с), заключающийся в том, что проводят (i) ацилирование α-аминобисфосфоновых кислот (n=1, 3, 6, где n - количество звеньев СН2-) 4-бром-1,8-нафталевым ангидридом в присутствии триэтиламина и 4-диметиламинопиридина при кипячении в этаноле, метоксилирование (ii) α-гидрокси-бисфосфонатов 4-бром-N-алкил-1,8-нафталимида карбонатом калия в метаноле и выделение соединений по п. 1 (Ia-с) известными приемами.
| DE 3526640 A1, 29.01.1987 | |||
| СПОСОБ ПОЛУЧЕНИЯ (СО)ПОЛИМЕРА ВИНИЛХЛОРИДА | 1985 |
|
RU2012565C1 |
| WO 2014105493 A1, 03.07.2014 | |||
| ФЛУОРОФОР И СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРА СОЛЕОТЛОЖЕНИЙ, СОДЕРЖАЩЕГО ФЛУОРОФОР В КАЧЕСТВЕ ФЛУОРЕСЦЕНТНОЙ МЕТКИ | 2016 |
|
RU2640339C2 |
| ОЩЕПКОВ М.С | |||
| и др | |||
| Ингибиторы солеотложений для водооборотных систем, содержащие флуоресцентную метку | |||
| Сборник докладов VII Научно-Практической конференции "Современные технологии водоподготовки и защиты оборудования от коррозии и накипеобразования", 25-26 октября 2017, Москва, Экспоцентр, с.103-110. | |||

