Область техники
Настоящее изобретение относится к новым хиназолиноновым производным, ингибирующим PI3K, и способу получения данных производных.
Кроме того, настоящее изобретение относится к фармацевтической композиции для лечения рака крови, заболевания печени или аутоиммунного заболевания, которая содержит хиназолиноновые производные.
Уровень техники
Рак представляет собой вторую основную причину смерти после сердечных заболеваний в Соединенных Штатах (Cancer Facts and Figures 2005, American Cancer Society, Inc.). На ранней стадии развития рака, можно выбрать способ удаления опухолей или уничтожения раковых клеток химиотерапией, радиотерапией и подобными, но в случае пациентов с раком последней стадии, побочные эффекты, обусловленные агрессивными терапиями, являются относительно большими и процент положительного клинического ответа после терапии является низким и, таким образом, можно выбрать терапии для снижения побочных эффектов замедлением развития рака и повышения качества жизни. В данных аспектах, противораковые лекарственные средства предполагаются не только для предотвращения рецидива рака уничтожением раковых клеток, но также увеличения периода выживания ингибированием роста и пролиферации раковых клеток, когда трудно ожидать полного восстановления.
Существующая химиотерапия для метастатического рака не способна обеспечить долгосрочного лечения из-за ее сниженной эффективности. Кроме того, хотя новые химиотерапии введены в медицинскую область, все еще есть необходимость в новых эффективных лекарственных средствах в качестве терапии первой, второй и третьей линии в монотерапии или комбинированной терапии с существующими агентами, для лечения устойчивых опухолей.
Кроме того, даже противораковые соединения с высокой эффективностью не являются применимыми ко всем типам рака и, таким образом, существует острая необходимость в разработке лекарственных средств, улучшающих эффективность лечения.
Направленные терапии представляют интерес, поскольку они являются специфическими к опухолям, эффективными и обладают гораздо меньшими эффектами на нормальные клетки по сравнению с существующими системными противораковыми терапиями. Дисрегуляцию протеинкиназ обычно обнаруживают в раковых клетках и, таким образом, она представляет собой приемлемую мишень для разработки противораковых лекарственных средств.
Среди липидкиназ, структура и функция PI3K (фосфатидилинозитол-3-киназные изомеры) постепенно и четко подтверждены в последние годы. Известно, что PI3K принадлежат к семейству ферментов, которые выполняют жизненно-важную роль в путях передачи внутриклеточных сигналов и вовлечены в большое количество клеточных функций, таких как клеточный рост, пролиферация, дифференциация, подвижность, выживаемость и внутриклеточная миграция.
В течение последних 20 лет, постепенно было обнаружено, что когда PI3K теряют свою регуляторную функцию, проблемы, такие как сверхактивация и подобные, возникают в путях передачи внутриклеточных сигналов, вызывая многие типы заболеваний.
PI3K классифицируют по классам: класс I, класс II и класс III. Класс I снова разделен на подклассы: класс IA и класс IB. PI3K класса I существуют в виде димеров, и димер разделен на каталитическую и регуляторную субъединицы. PI3K класса 1A представляет собой димер, состоящий из p110 каталитической субъединицы и p85 регуляторной субъединицы и, в связи с этим, p110 каталитическая субъединица включает три изоформы, а именно, p110α, p110β и p110δ. Таким образом, изоформы PI3K называют PI3Kα, PI3Kβ и PI3Kδ.
При этом, PI3K класса IB представляет собой димер, состоящий из p110γ каталитической субъединицы и p101 регуляторной субъединицы, и PI3K обычно называют PI3Kγ.
PI3Kδ в основном активируется рецепторными тирозинкиназами (RTK), фосфорилируя PIP2 до PIP3, и PI3Kγ в основном активируется сопряженными с G-белком рецепторами (GPCR), фосфорилируя PIP2 до PIP3. PIP3 активирует протеинкиназу B (Akt/PKB) и постепенно приводит к передачи сигналов в нисходящем направлении, посредством этого, вовлекаясь в регуляцию основный функций клетки, таких как клеточный рост, пролиферация, дифференциация, подвижность, выживаемость и внутриклеточная миграция. Одна из наиболее важных проблем последних лет заключается в том, что возникают различные заболевания в диапазоне от воспаления и аутоиммунных реакций до гематологической злокачественной опухоли и солидного рака, когда PI3Kδ и PI3Kγ неправильно функционируют в регуляции передачи внутриклеточного сигнала, и соответственно, предпринимаются активные усилия по разработке лекарственных средств для лечения воспаления, аутоиммунных реакций, гематологической злокачественной опухоли и солидного рака ингибированием PI3Kδ и PI3Kγ, которые потеряли свои регуляторные функции.
Пример репрезентативных лекарственных средств, разрабатываемых в данной области, представляет собой иделализиб, вещество, которое разработано Gilead Calistoga и селективно ингибирует PI3Kδ. Данное лекарственное средство обладает превосходной эффективностью относительно различных типов гематологических злокачественных образований и, таким образом, привлекло внимание в качестве прорывного лекарственного средства, которое решает проблемы (особенно цитотоксичности относительно нормальных клеток) существующих цитотоксических противораковых лекарственных средств и также компенсирует проблемы эффективности существующих противораковых лекарственных средств. Однако в Европе при клинических испытаниях возникали некоторые случаи серьезной токсичности, в которых пациенты умирали от пневмонии и, таким образом, разработка данного лекарственного средства была приостановлена. Согласно отчету, причина заключается в том, что ингибирующая активность данного лекарственного средства была достаточно селективной и мощной для PI3Kδ, а не для PI3Kα и PI3Kβ, но была недостаточно селективной относительно PI3Kγ. Дувелезиб показал двойную ингибирующую активность относительно PI3Kδ и PI3Kγ и, таким образом, была возможность его разработки в качестве много обещающего лекарственного средства для лечения гематологической злокачественной опухоли, воспаления и аутоиммунного заболевания. Однако в процессе клинических испытаний разработка дувелизиба была остановлена из-за проблем, аналогичных проблемам с иделализибом. Известно, что ингибирующая активность данного вещества является недостаточно селективной относительно PI3Kδ и PI3Kγ, чем относительно PI3Kβ. Следовательно, существует необходимость в разработке лекарственного средства, способного более селективно ингибировать PI3Kδ, чем, по меньшей мере, иделализиб.
При этом, хиназолиноновые производные представляют собой специфические структуры, присутствующие во многих биологически активных соединениях, таких как метквалон, который представляет собой седативно-снотворное средство, хлороквалон, который представляет собой противокашлевое средство, и пириквалон, который представляет собой противосудорожное средство. Хиназолинон и его производные обладают широким диапазоном биологических свойств, таких как гипноз, устранение боли, подавление судорог, подавление кашля и противовоспалительная активность.
В частности, хиназолиноновые производные применяют в лечении пролиферативных заболеваний, включая рак, и они представляют собой одни из терапевтических агентов, которые широко применяют в настоящее время. Например, патенты США регистрационные №5747498 и 5773476 описывают хиназолиноновые производные, применяемые в лечении рака, который вызван сверхактивацией или нарушенной активацией рецепторных тирозинкиназ. Следовательно, хиназолиноновые производные требуется исследовать и разрабатывать с помощью различных подходов для лечения пролиферативных заболеваний.
Описание
Техническая проблема
Цель настоящего изобретения заключается в обеспечении новых хиназолиноновых производных, которые ингибируют PI3K, и способов получения данных производных.
Другая цель настоящего изобретения заключается в обеспечении фармацевтической композиции, содержащей хиназолиноновые производные для предотвращения или лечения гематологической злокачественной опухоли, заболевания печени или аутоиммунного заболевания.
Техническое решение
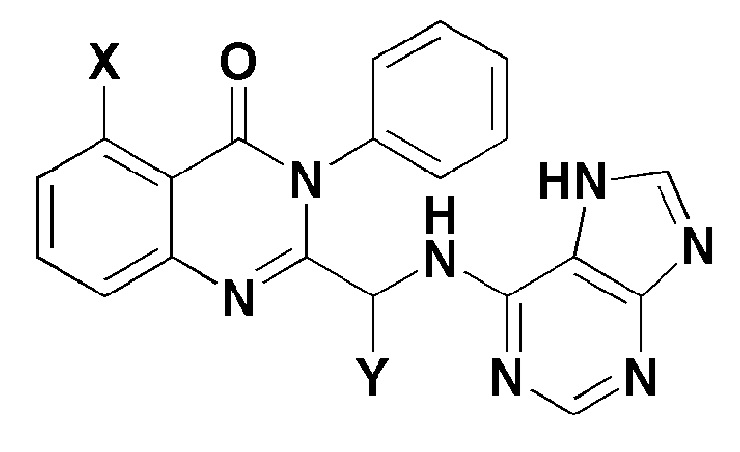
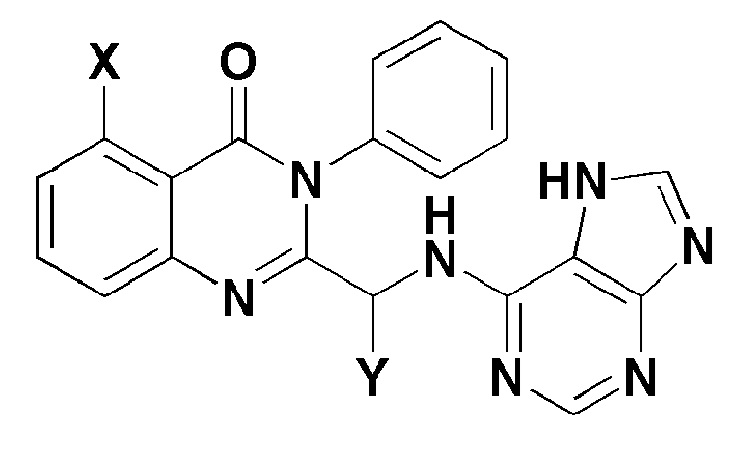
Согласно одному аспекту настоящей заявки, обеспечивают соединение, представленное формулой 1 ниже, или его фармацевтически приемлемую соль:
[Формула 1]
где,
X представляет собой -H, галоген, -CH3, или -NH2; и
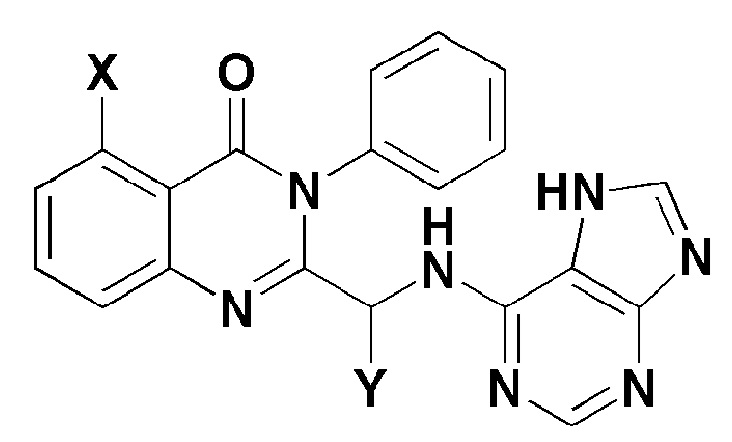
Y представляет собой C1-2 линейный алкил или C3-4 циклоалкил. Согласно другому аспекту настоящего изобретения, обеспечивают фармацевтическую композицию, содержащую соединение, представленное формулой 1 ниже, или его фармацевтически приемлемую соль в качестве активного ингредиента для предотвращения или лечения рака крови, заболевания печени или аутоиммунного заболевания.
[Формула 1]
где,
X представляет собой -H, галоген, -CH3, или -NH2; и
Y представляет собой C1-2 линейный алкил или C3-4 циклоалкил. Рак крови может представлять собой лейкемию или лимфому.
Заболевание печени можно выбрать из группы, состоящей из неалкогольной жировой болезни печени (NAFLD), неалкогольного стеатогепатита (NASH), стеатоза печени, гепатоцирроза, гепатита, аденомы печени, гиперчувствительности к инсулину и рака печени.
Аутоиммунное заболевание можно выбрать из группы, состоящей из аллергического ринита, астмы, хронического обструктивного заболевания легких (COPD) и ревматоидного артрита.
Согласно еще другому аспекту настоящего изобретения, обеспечивают способ получения соединения, представленного формулой 1 ниже, причем способ включает:
реакцию соединения, представленного формулой 2 ниже, с соединением, представленным формулой 3 ниже, получая соединение, представленное формулой 4 ниже;
деблокирование соединения, представленного формулой 4 ниже, получая соединение, представленное формулой 5 ниже; и
реакцию соединения, представленного формулой 5 ниже, с соединением, представленным формулой 6, получая соединение формулы 1.
[Формула 1]

где, в формуле 1,
X представляет собой -H, галоген, -CH3, или -NH2; и
Y представляет собой C1-2 линейный алкил или C3-4 циклоалкил.
[Формула 2]
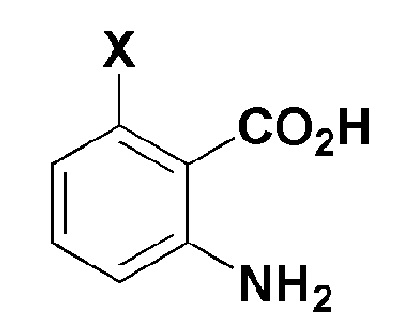
где, в формуле 2,
X представляет собой -H, галоген, -CH3, или -NH2.
[Формула 3]
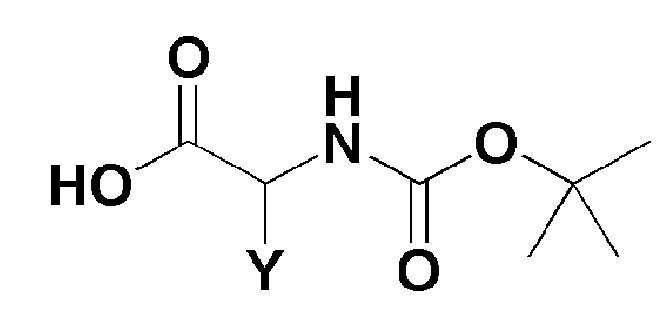
где, в формуле 3,
Y представляет собой C1-2 линейный алкил или C3-4 циклоалкил.
[Формула 4]
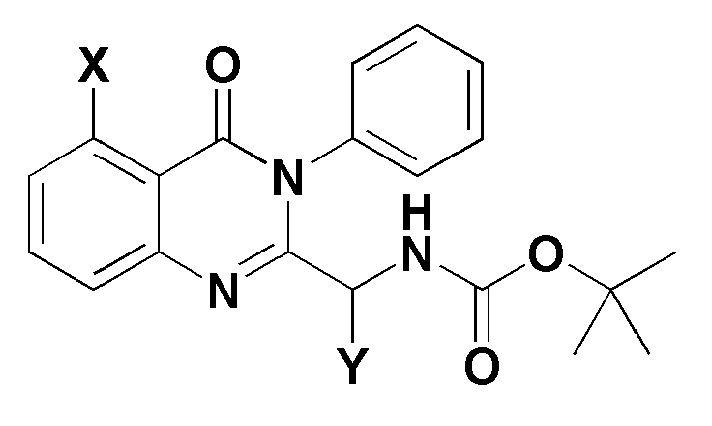
где, в формуле 4,
X представляет собой -H, галоген, -CH3, или -NH2; и
Y представляет собой C1-2 линейный алкил или C3-4 циклоалкил.
[Формула 5]
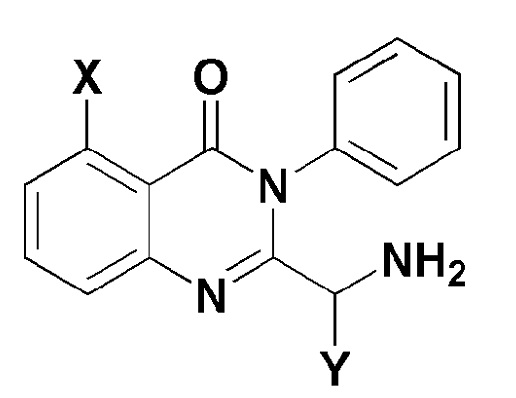
где, в формуле 5,
X представляет собой -H, галоген, -CH3, или -NH2; и
Y представляет собой C1-2 линейный алкил или C3-4 циклоалкил.
[Формула 6]
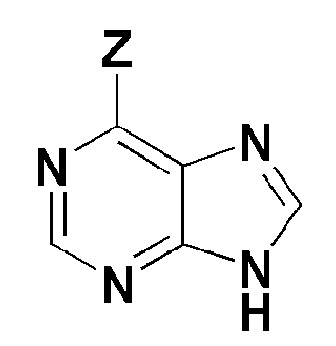
где, в формуле 6,
Z представляет собой галоген.
Полезные эффекты
Новые хиназолиноновые производные согласно настоящему изобретению являются эффективными в лечении рака крови или заболеваний печени.
В частности, по сравнению с существующими ингибиторами PI3Kδ, хиназолиноновые производные настоящего изобретения могут ингибировать PI3Kδ с высокой селективностью, значительно снижая иммунотоксичность, или ингибировать одновременно PI3Kδ и PI3Kγ, таким образом, обеспечивая не только противораковую терапию гематологической злокачественной опухоли и подобных, но также терапию аутоиммунных заболеваний. Данные терапевтические агенты направленного действия могут решить проблемы, такие как побочные эффекты существующих противораковых терапий с тяжелой цитотоксичностью.
Описание чертежей
ФИГУРА 1 иллюстрирует принцип эксперимента Adapta киназного анализа экспериментального примера 1.
ФИГУРА 2 иллюстрирует результаты экспериментального примера 2 для подтверждения эффекта на снижение AKT (Ser473) фосфорилирования в клеточных линиях лейкемии и лимфомы.
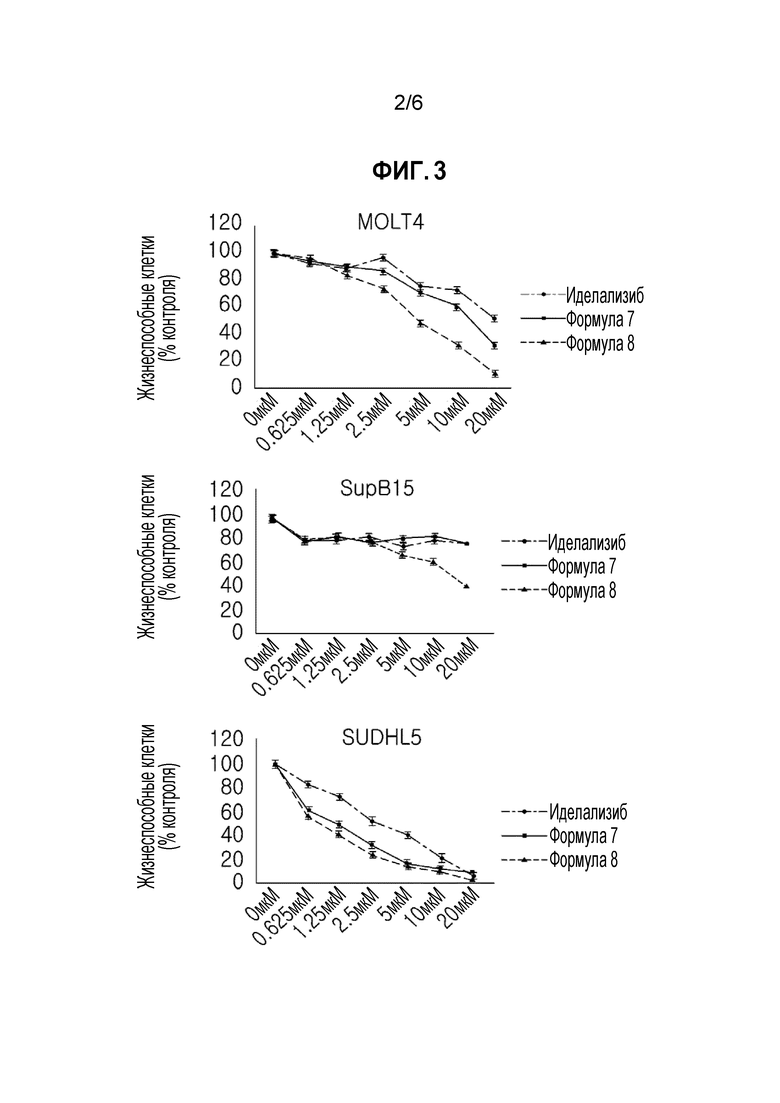
ФИГУРА 3 иллюстрирует результаты экспериментального примера 3 для подтверждения ингибирующего эффекта на рост клеток лейкемии и лимфомы.
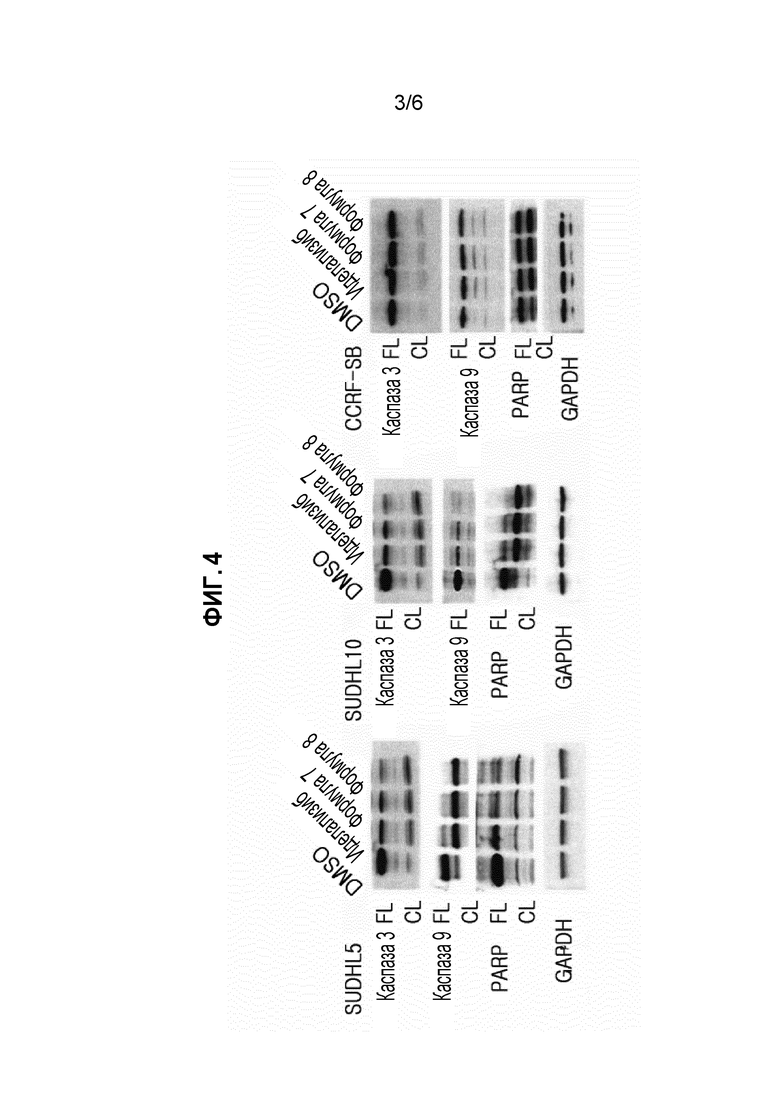
ФИГУРА 4 иллюстрирует результаты SDS-PAGE анализа экспериментального примера 4 для подтверждения эффекта на апоптоз клеток диффузной В-крупноклеточной лимфомы (DLBCL) и клеток острого лимфобластного лейкоза (ALL).
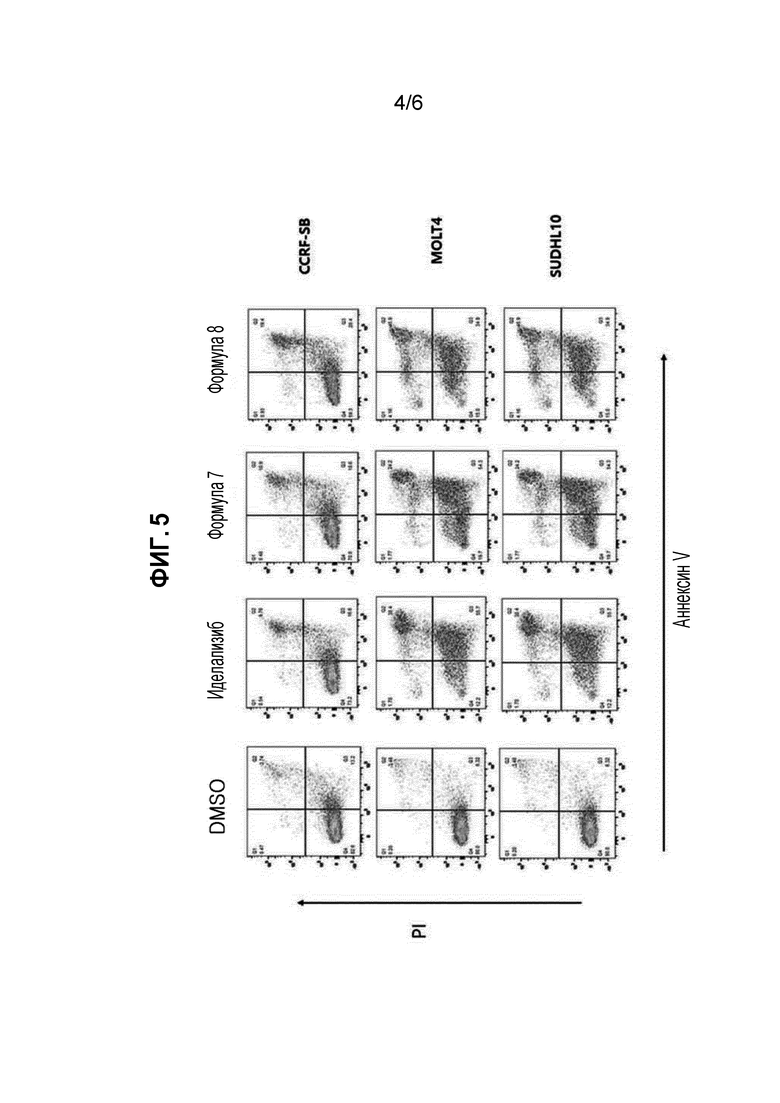
ФИГУРА 5 иллюстрирует результаты проточной цитометрии экспериментального примера 5 для подтверждения эффекта на апоптоз клеток диффузной В-крупноклеточной лимфомы (DLBCL) и клеток острого лимфобластного лейкоза (ALL).
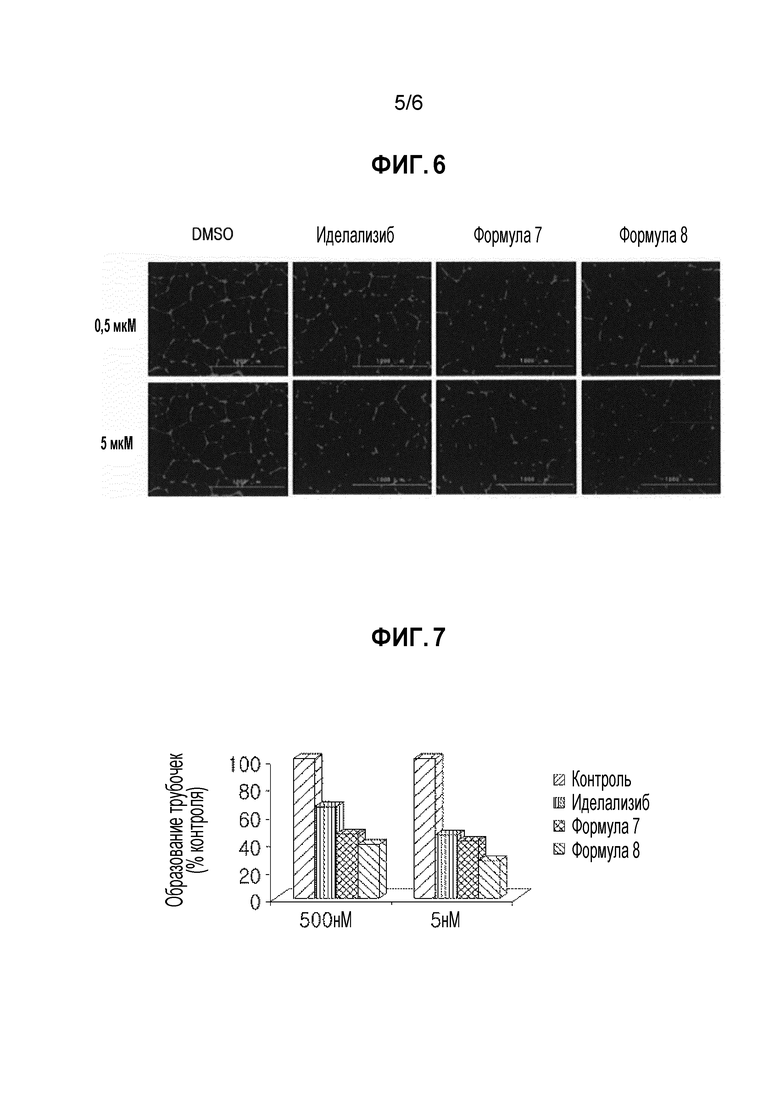
ФИГУРА 6 иллюстрирует изображения Cytation™ 5 флуоресцентной микроскопии (BioTek), показывающие результаты образования экспериментального примера 6 для подтверждения ингибирующего эффекта на ангиогенез.
ФИГУРА 7 представляет собой график, показывающий ингибирующий эффект на ангиогенез в экспериментальном примере 6 для подтверждения ингибирующего эффекта на ангиогенез.
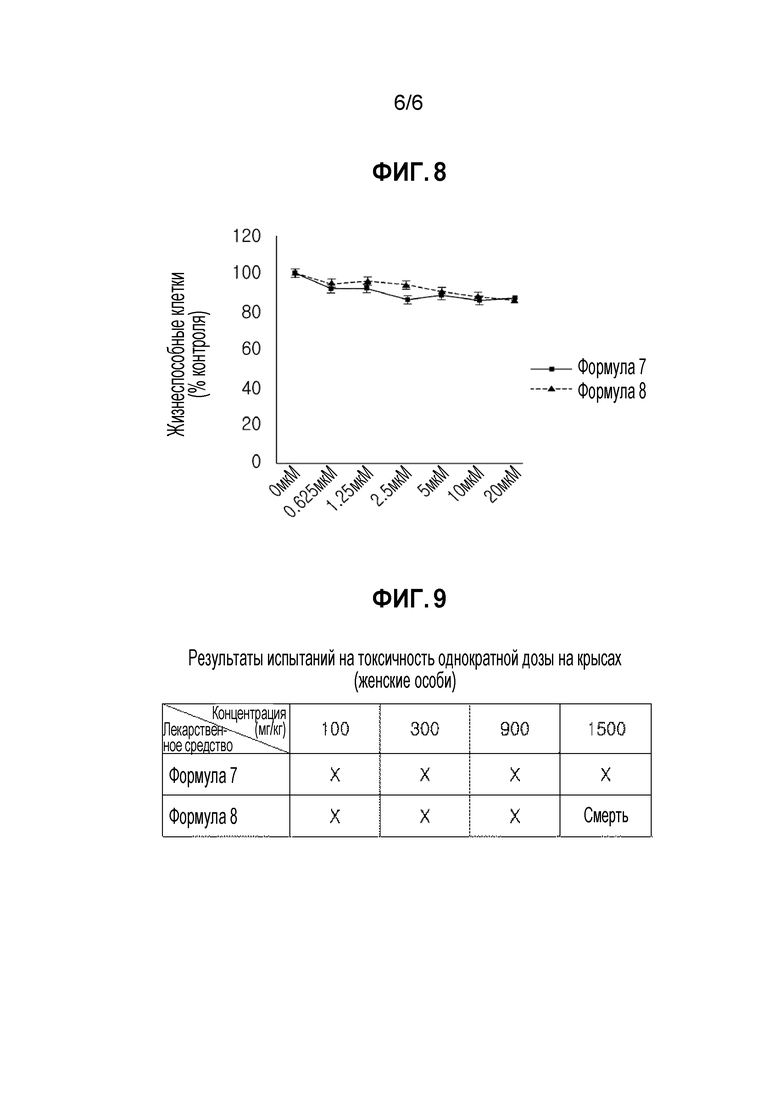
ФИГУРА 8 представляет собой график, показывающий количество жизнеспособных HUVEC, обработанных соединением в экспериментальном примере 6, для подтверждения ингибирующего эффекта на ангиогенез.
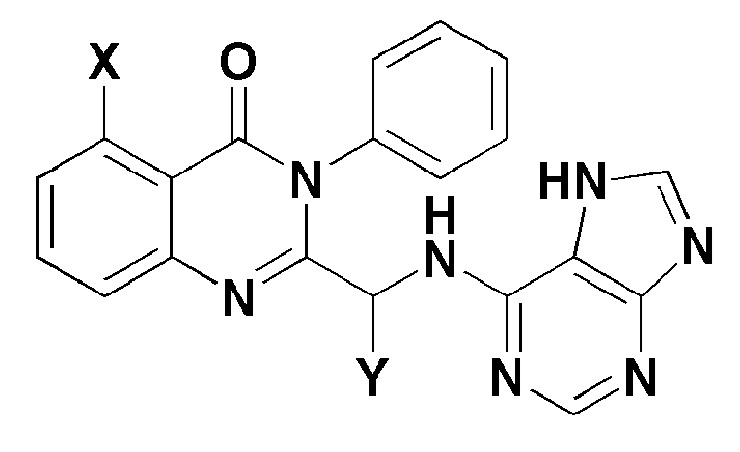
ФИГУРА 9 представляет собой таблицу для подтверждения токсичности в зависимости от концентраций соединения формулы 7 и соединения формулы 8 в экспериментальном примере 7, который представляет собой тест на токсичность при однократной дозе на крысах.
ОСУЩЕСТВЛЕНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение будет описано более подробно ниже. Термины или слова, применяемые в настоящем описании и формуле изобретения, не следует считать ограниченными стандартными или словарными значениями и следует считать значениями и понятиями, согласующимися с сущностью настоящего изобретения, исходя из принципа, что изобретатель может подходящим образом определить понятия терминов для объяснения настоящего изобретения изобретателя наилучшим способом. Таким образом, конфигурации, описанные в вариантах осуществления, изложенные в настоящем изобретении, представляют собой просто примерные варианты осуществления настоящего изобретения и не представляют все технические идеи настоящего изобретения и, таким образом, должно быть ясно, что различные эквиваленты и модификации, которые могут замещать данные варианты осуществления, можно осуществлять в момент регистрации настоящей заявки.
Настоящее изобретение относится к новым соединениям, хиназолиноновым производным, в качестве ингибиторов PI3K. PI3K ингибиторы блокируют PI3K-AKT сигнальный путь стыковкой с АТФ-связывающим сайтом p110δ, и активация PI3K путей опосредована PI3K каталитическими изотипами, т.е., p110α, p110β, p110δ и p110γ.
p110δ играет важную роль при раке крови и развитии B-клеток и преимущественно экспрессируется в гемопоэтических стволовых клетках и экспрессируется при многих типах рака, включая лейкемию, лимфому, колоректальный рак, рак мочевого пузыря, злокачественную глиому и подобные. Она регулирует клеточную пролиферацию за счет стимулирования родственных цитокинов и хемокинов посредством PI3K-AKT сигнального пути.
Кроме того, новые хиназолиноновые производные согласно настоящему изобретению преодолевают существующие проблемы с токсичностью, включая гепатотоксичность. Поскольку PI3K p110δ может экспрессироваться на высоком уровне даже при запущенной гепатоклеточной карциноме, новые хиназолиноновые производные согласно настоящему изобретению являются также эффективными в качестве терапевтического агента для гепатоклеточной карциномы, которая представляет собой солидный рак.
Ввиду проблем существующих хиназолиноновых противораковых лекарственных средств, новые хиназолиноновые производные должны в значительной степени и селективно ингибировать PI3Kδ. Предпочтительно, необходимо, чтобы селективность между PI3K изомерами удовлетворяла критерию, что, применяя величины IC50, каждое из соотношений PI3Kα/PI3Kδ и PI3Kβ/PI3Kδ превышало 150, и соотношение PI3Kγ/PI3Kδ являлось большим, чем соотношение, по меньшей мере, иделализиба.
Кроме того, когда PI3Kδ и PI3Kγ ингибируются одновременно, предпочтительно, чтобы соотношения PI3Kβ/PI3Kδ и PI3Kβ/PI3Kγ были большими, чем соотношения для дувелизиба.
Настоящее изобретение относится к соединению, представленному формулой 1, или его фармацевтически приемлемой соли.
[Формула 1]
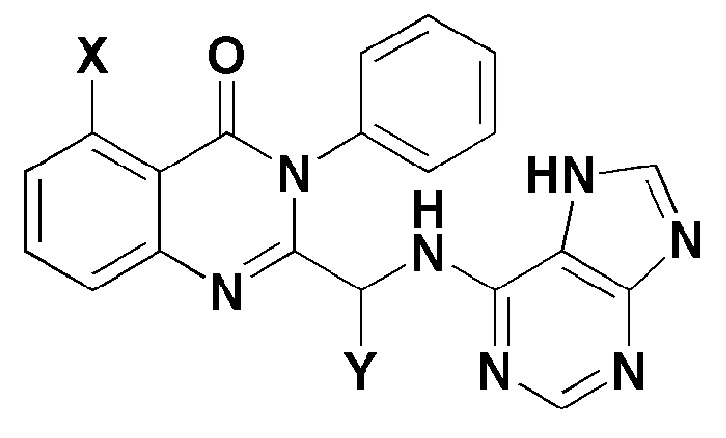
где,
X представляет собой -H, галоген, -CH3 или -NH2; и
Y представляет собой C1-2 линейный алкил или C3-4 циклоалкил.
Термин "галоген", как применяют в настоящем изобретении, относится к фтору (F), брому (Br), хлору (Cl) или йоду (I). В формуле 1, Y может быть соединен в виде (S)-изомера или (R)-изомера, но предпочтительно в виде (S)-изомера.
Конкретные примеры соединения формулы 1 включают соединения, представленные следующими формулами 7-14, но настоящее изобретение не ограничивается ими.
Соединение формулы 7 согласно настоящему изобретению представляет собой соединение формулы 1, в котором X представляет собой -F, и Y представляет собой циклопропил.
[Формула 7]
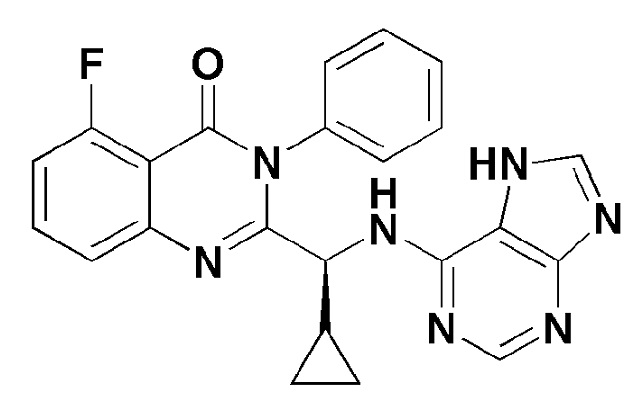
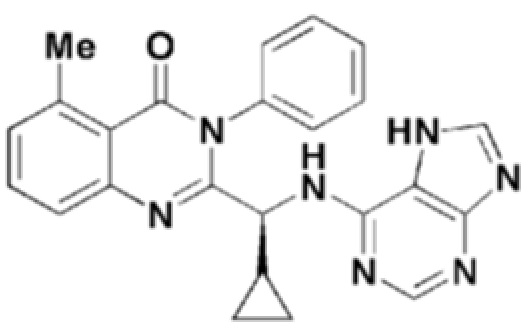
Кроме того, соединение формулы 8 согласно настоящему изобретению представляет собой соединение формулы 1, в котором X представляет собой метил, и Y представляет собой циклопропил.
[Формула 8]
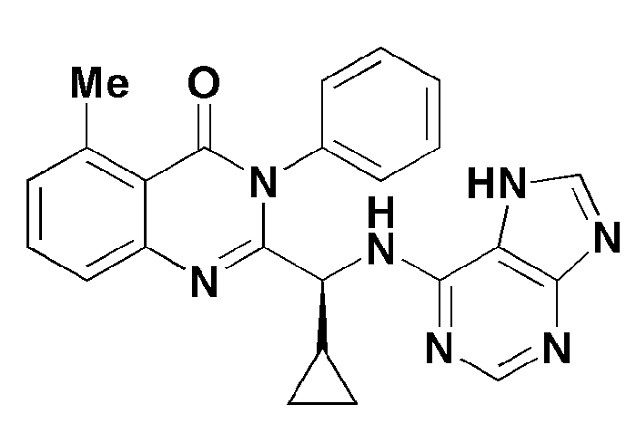
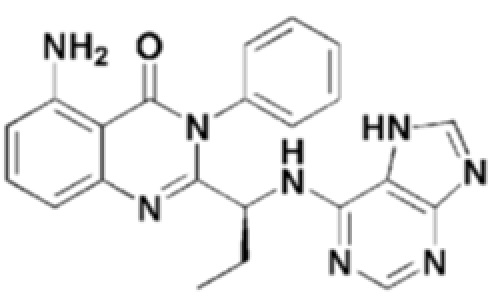
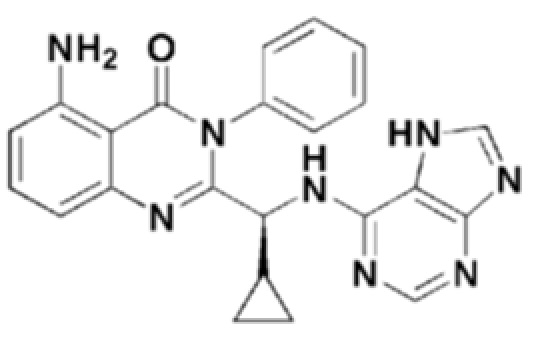
Кроме того, соединение формулы 9 согласно настоящему изобретению представляет собой соединение формулы 1, в котором X представляет собой -NH2, и Y представляет собой циклопропил.
[Формула 9]
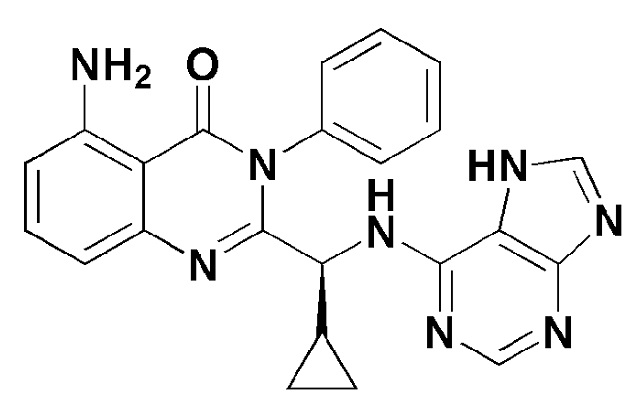
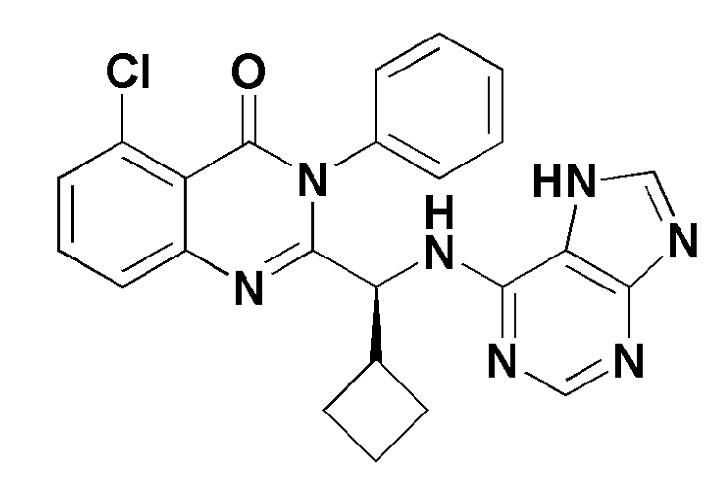
Кроме того, соединение формулы 10 согласно настоящему изобретению представляет собой соединение формулы 1, в котором X представляет собой -NH2, и Y представляет собой метил.
[Формула 10]
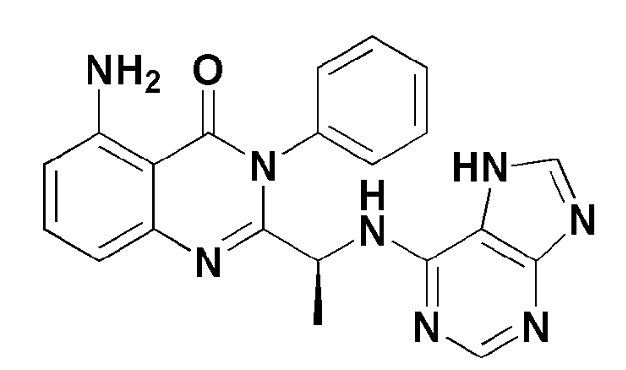
Кроме того, соединение формулы 11 согласно настоящему изобретению представляет собой соединение формулы 1, в котором X представляет собой -NH2, и Y представляет собой этил.
[Формула 11]
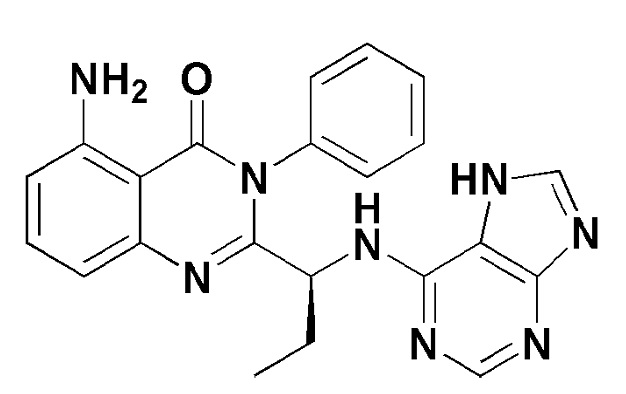
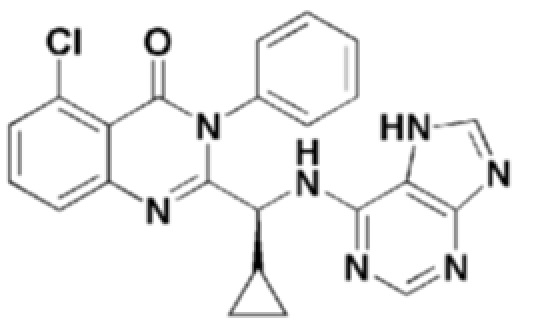
Кроме того, соединение формулы 12 согласно настоящему изобретению представляет собой соединение формулы 1, в котором X представляет собой -Cl, и Y представляет собой циклопропил.
[Формула 12]
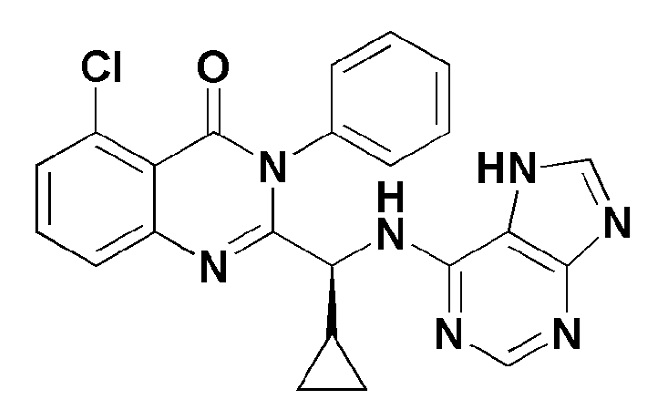
Кроме того, соединение формулы 13 согласно настоящему изобретению представляет собой соединение формулы 1, в котором X представляет собой -F, и Y представляет собой циклобутил.
[Формула 13]
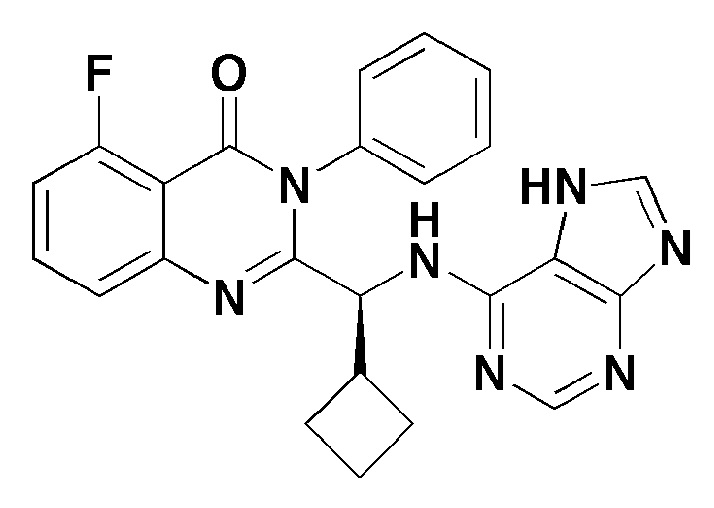
Кроме того, соединение формулы 14 согласно настоящему изобретению представляет собой соединение формулы 1, в котором X представляет собой -Cl, и Y представляет собой циклобутил.
[Формула 14]
Соединение, представленное формулой 1, настоящего изобретения можно применять в виде фармацевтически приемлемой соли, и соль может представлять собой соль присоединения кислоты, образованную фармацевтически приемлемой свободной кислотой. Соль присоединения кислоты получают из: неорганических кислот, таких как хлористоводородная кислота, азотная кислота, фосфорная кислота, серная кислота, бромистоводородная кислота, йодистоводородная кислота, азотистая кислота, фосфористая кислота и подобные; нетоксичных органических кислот, таких как алифатические моно- и дикарбоксилаты, фенил-замещенные алканоаты, гидроксиалканоаты и алкандиоаты, ароматические кислоты, алифатические и ароматические сульфокислоты и подобные; или органических кислот, таких как уксусная кислота, бензойная кислота, лимонная кислота, молочная кислота, малеиновая кислота, глюконовая кислота, 4-толуолсульфокислота, винная кислота, фумаровая кислота и подобные. Примеры данных фармацевтически нетоксичных солей включают сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, нитраты, фосфаты, гидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, йодиды, фториды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капраты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себакаты, фумараты, малеаты, бутен-1,4-диоаты, гексан-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, терефталаты, бензолсульфонаты, толуолсульфонаты, хлорбензолсульфонаты, ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, β-гидроксибутираты, гликоляты, малаты, тартраты, метансульфонаты, пропансульфонаты, нафталин-1-сульфонаты, нафталин-2-сульфонаты, манделаты и подобные.
Соли присоединения кислоты согласно настоящему изобретению можно получить, применяя общепринятый способ. Например, данные соли присоединения кислоты можно получить растворением производного формулы 1 в органическом растворителе, таком как метанол, этанол, ацетон, дихлорметан, ацетонитрил или подобном, добавлением к нему органической кислоты или неорганической кислоты, получая осадок, и фильтрованием и сушкой осадка, или можно получить отгонкой растворителя и избытка кислоты при пониженном давлении, и затем сушкой полученного в результате раствора, с последующей кристаллизацией в присутствии органического растворителя.
Кроме того, фармацевтически приемлемые соли металлов можно получить, применяя основания. Соли щелочных или щелочноземельных металлов получают, например, растворением соединения в избытке раствора гидроксида щелочного металла или гидроксида щелочноземельного металла, фильтрованием нерастворимой соли соединения и упариванием и сушкой фильтрата. В то же время, фармацевтически предпочтительно, чтобы соль натрия, соль калия или соль кальция получали в качестве соли металла. Кроме того, соответствующие соли получают реакцией соли щелочного или щелочноземельного металла с подходящей солью серебра (например, нитратом серебра).
Более того, настоящее изобретение включает не только соединение, представленное формулой 1, и его фармацевтически приемлемые соли, но также сольваты, стереоизомеры, гидраты и подобные, которые можно получить из них.
Настоящее изобретение также относится к фармацевтической композиции для предотвращения или лечения рака крови, заболевания печени и аутоиммунного заболевания, которая содержит соединение, представленное формулой 1 ниже, или его фармацевтически приемлемую соль в качестве активного ингредиента.
[Формула 1]
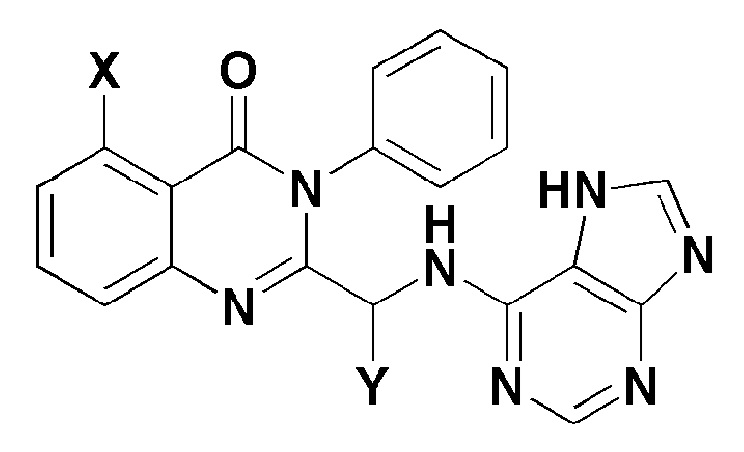
где,
X представляет собой -H, галоген, -CH3, или -NH2; и
Y представляет собой C1-2 линейный алкил или C3-4 циклоалкил.
В формуле 1, Y может быть соединен в виде (S)-изомера или (R)-изомера, но предпочтительно, чтобы Y был соединен в виде (S)-изомера. В фармацевтической композиции согласно настоящему изобретению, соединение, представленное формулой 1, или его фармацевтически приемлемую соль можно вводить перорально или парентерально в виде различных лекарственных форм в процессе клинического введения, и можно формулировать, применяя общепринятые разбавители или вспомогательные вещества, такие как наполнители, разбавители, связующие, смачивающие агенты, разрыхлители, поверхностно-активные вещества и подобные.
Составы для перорального введения могут включать, например, таблетки, пилюли, твердые/мягкие капсулы, жидкости, суспензии, эмульсии, сиропы, гранулы, эликсиры, суспензии, троше и подобные. Данные составы содержат, в добавление к активному ингредиенту, разбавитель (например, лактозу, декстрозу, сахарозу, маннитол, сорбитол, целлюлозу и/или глицин) или смазывающий агент (например, оксид кремния, тальк, стеариновую кислоту и ее магниевую или кальциевую соли, и/или полиэтиленгликоль). Таблетки могут содержать связующее, такое как силикат магния алюминия, крахмальную пасту, желатин, метилцеллюлозу, карбоксиметилцеллюлозу натрия и/или поливинилпирролидон и, в некоторых случаях, могут содержать разрыхлитель, такой как крахмал, агар, альгиновая кислота или ее натриевые соли, или кипящую смесь и/или абсорбент, краситель, ароматизатор и подсластитель.
Фармацевтическую композицию, содержащую соединение, представленное формулой 1, в качестве активного ингредиента можно вводить парентерально, и парентеральное введение осуществляют подкожной инъекцией, внутривенной инъекцией, внутримышечной инъекцией или интраторакальной инъекцией.
В этой связи, для получения составов для парентерального введения, соединение, представленное формулой 1, или его фармацевтически приемлемую соль смешивают со стабилизатором или буфером в воде, получая раствор или суспензию, с последующим получением единичной лекарственной формы, ампулы или флакона. Композицию можно стерилизовать, и/или она может содержать адъювант, такой как консервант, стабилизатор, смачиваемый порошок, соль для осморегуляции, и/или буфер, и другие терапевтически эффективные материалы, и можно формулировать, применяя общепринятый способ, такой как смешение, гранулирование или нанесение покрытия.
Композиция настоящего изобретения может дополнительно содержать, в добавление к хиназолиноновому соединению, один или более эффективных ингредиентов, проявляющих идентичные или аналогичные функции.
Подходящую дозу фармацевтической композиции настоящего изобретения можно подходящим образом выбрать в зависимости от состояния и веса тела пациентов, тяжести симптомов, лекарственной формы, пути введения и периода введения. В композиции настоящего изобретения, предпочтительно, чтобы эффективный ингредиент (ингредиенты) вводили в количестве 0,2 мг/кг-200 мг/кг в день для оптимальной эффективности. Композицию можно вводить один раз в день или в виде нескольких доз в день, но настоящее изобретение не ограничивается этим.
Согласно настоящему изобретению, рак крови может представлять собой лейкемию или лимфому.
Лейкемию можно выбрать из острого лимфоцитарного лейкоза (ALL), острого миелобластного лейкоза (AML), хронического лимфоцитарного лейкоза (CLL) и мелкоклеточной лимфоцитарной лимфомы (SLL), и острый лимфоцитарный лейкоз также известен как острый лимфобластный лейкоз.
Лимфома может представлять собой неоплазию зрелых (периферических) B-клеток, и более конкретно ее можно выбрать из B-клеточного хронического лимфоцитарного лейкоза/мелкоклеточной лимфоцитарной лимфомы; B-клеточного пролимфоцитарного лейкоза; лимфоплазмоцитарной лимфомы; лимфомы маргинальной зоны, например, B-клеточной лимфомы маргинальной зоны селезенки (+/- ворсинчатые лимфоциты), узловой лимфомы маргинальной зоны (+/- моноцитоидные B-клетки), и экстранодальной B-клеточной лимфомы маргинальной зоны типа лимфоидная ткань слизистых оболочек (MALT); волосатоклеточного лейкоза; миеломы клеток плазмы/плазмацитомы; фолликулярной лимфомы; лимфомы из клеток центра фолликула; мантийклеточной лимфомы; диффузной В-крупноклеточной лимфомы (включая медиастинальную B-крупноклеточную лимфому, внутрисосудистую B-крупноклеточную лимфому и первичную выпотную лимфому); и лимфомы Беркитта/клеточной лимфомы Беркитта.
Кроме того, лимфому можно выбрать из множественной миеломы (MM), неходжкинской лимфомы (NHL), мантийклеточной лимфомы (MCL), фолликулярной лимфомы, макроглобулинемии Вальденстрема (WM), B-клеточной лимфомы и диффузной В-крупноклеточной лимфомы (DLBCL).
Заболевание печени настоящего изобретения можно выбрать из группы, состоящей из неалкогольной жировой болезни печени (NAFLD), неалкогольного стеатогепатита (NASH), стеатоза печени, гепатоцирроза, гепатита, аденомы печени, гиперчувствительности к инсулину и рака печени.
Рак печени может, например, представлять собой опухоль печени, гепатоклеточную аденому или гепатоклеточную карциному.
Аутоиммунное заболевание можно выбрать из группы, состоящей из аллергического ринита, астмы, хронического обструктивного заболевания легких (COPD) и ревматоидного артрита.
Настоящее изобретение также относится к способу получения соединения, представленного формулой 1 ниже, где способ включает:
реакцию соединения, представленного формулой 2 ниже, с соединением, представленным формулой 3 ниже, получая соединение, представленное формулой 4 ниже;
деблокирование соединения формулы 4, получая соединение, представленное формулой 5 ниже; и
реакцию соединения, представленного формулой 5 ниже, с соединением, представленным формулой 6, получая соединение, представленное формулой 1.
[Формула 1]
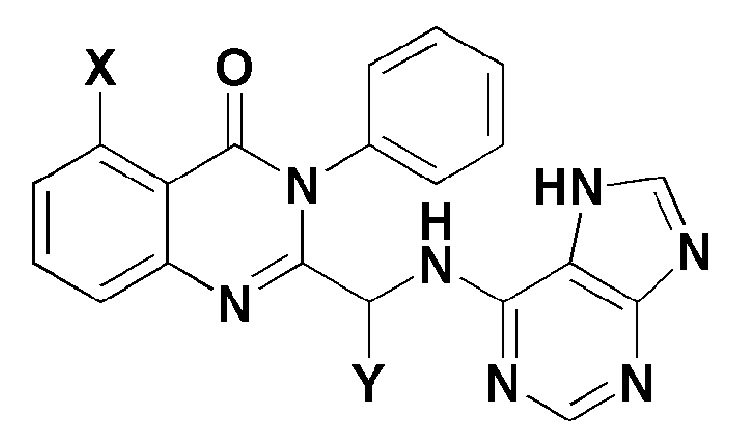
где, в формуле 1,
X представляет собой -H, галоген, -CH3 или -NH2; и
Y представляет собой C1-2 линейный алкил или C3-4 циклоалкил.
[Формула 2]
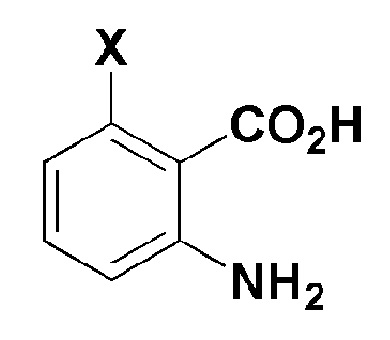
где, в формуле 2,
X представляет собой -H, галоген, -CH3 или -NH2.
[Формула 3]
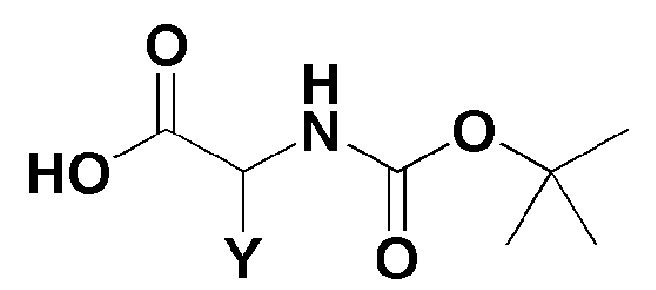
где, в формуле 3,
Y представляет собой C1-2 линейный алкил или C3-4 циклоалкил.
[Формула 4]
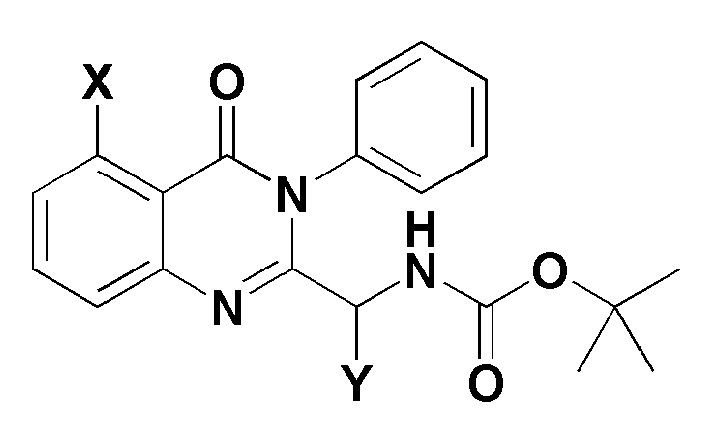
где, в формуле 4,
X представляет собой -H, галоген, -CH3 или -NH2; и
Y представляет собой C1-2 линейный алкил или C3-4 циклоалкил.
[Формула 5]
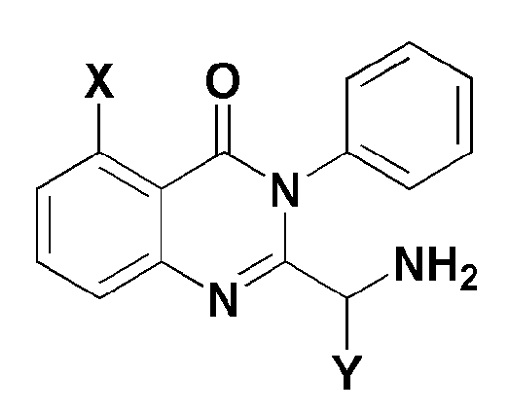
где, в формуле 5,
X представляет собой -H, галоген, -CH3 или -NH2; и
Y представляет собой C1-2 линейный алкил или C3-4 циклоалкил.
[Формула 6]
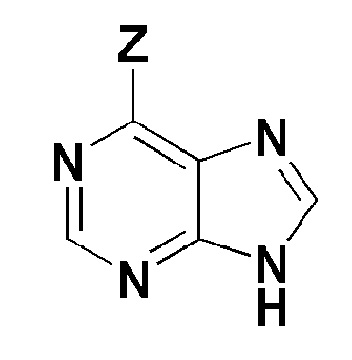
где, в формуле 6,
Z представляет собой галоген.
Стадия 1 представляет собой способ получения соединения, представленного формулой 4, реакцией соединения, представленного формулой 2, с соединением, представленным формулой 3.
Например, трифенилфосфит можно добавлять к раствору, в котором соединение, представленное формулой 2, и соединение, представленное формулой 3, смешивают вместе в присутствии пиридинового растворителя при перемешивании при комнатной температуре.
В это же время, температура конкретно не ограничена, но смесь можно перемешивать при температуре 30°C-100°C, предпочтительно 45°C-80°C и более предпочтительно 55°C-60°C.
Продолжительность перемешивания конкретно не ограничена, но способ перемешивания можно осуществлять в течение 5 часов-20 часов, предпочтительно 8 часов-16 часов и более предпочтительно 10 часов-14 часов.
Затем, можно добавлять анилин, обеспечивая протекание реакции. В это же время, температура конкретно не ограничена, но реакцию можно осуществлять при температуре 50°C-200°C, предпочтительно 90°C-150°C и более предпочтительно 100°C-120°C.
В это же время, продолжительность реакции конкретно не ограничена, но реакцию можно осуществлять в течение 1 часа-20 часов, предпочтительно 3 часов-15 часов и более предпочтительно 5 часов-10 часов.
Стадия 2 представляет собой способ получения соединения, представленного формулой 5, деблокированием соединения, представленного формулой 4.
Например, соединение, представленное формулой 5, можно получить следующим способом: соединение, представленное формулой 4, добавляют к дихлорметановому раствору, в котором растворена трифторуксусная кислота (CF3COOH), и затем оно реагирует при комнатной температуре в течение 0,1 часа-2 часов, предпочтительно 0,2 часов-1,5 часов и более предпочтительно 0,5 часа-1 часа.
Стадия 3 представляет собой способ получения соединения, представленного формулой 1, реакцией соединения, представленного формулой 5, с соединением, представленным формулой 6.
Например, соединение, представленное формулой 5, добавляют к трет-бутанолу, добавляют к нему N,N-диизопропилэтиламин, и затем соединение, представленное формулой 6, добавляют к реакционному раствору, и реакционный раствор можно перемешивать при кипячении с обратным холодильником в течение 10 часов-48 часов, предпочтительно 15 часов-30 часов и более предпочтительно 20 часов-26 часов.
Настоящее изобретение относится к функциональной оздоровительной продукции для предотвращения или облегчения гематологической злокачественной опухоли или заболевания печени, содержащей новое хиназолиноновое соединение или его фармацевтически приемлемую соль в качестве активного ингредиента.
Функциональную оздоровительную продукцию можно получить в виде, но не ограничиваясь, различных типов напитков, жвачки, чая, кондитерских изделий, витаминных комплексов и пищевых добавок.
Ниже, настоящее изобретение будет описано подробно со ссылкой на примеры и экспериментальные примеры.
Однако данные примеры и экспериментальные примеры приводят только для целей иллюстрации, и они не предполагаются ограничивающими объем настоящего изобретения.
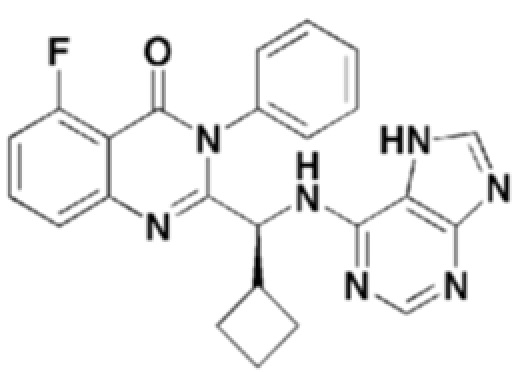
<Пример 1> получение (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-фтор-3-фенилхиназолин-4(3H)-она (формула 7)
[Реакционная схема 1]
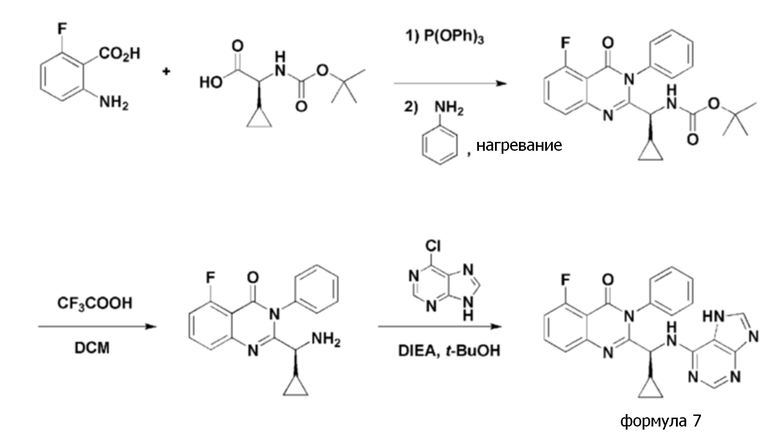
Стадия 1: Получение (S)- трет- бутил циклопропил(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)метилкарбамата
Трифенилфосфит (1,4 экв.) добавляли к раствору, в котором 2-амино-6-фторбензойную кислоту (1,0 экв.) и (S)-2-(трет-бутоксикарбониламино)-2-циклопропилуксусную кислоту (1,0 экв.) смешивали в пиридиновом растворителе, тогда как раствор перемешивали при комнатной температуре. Полученную в результате смесь перемешивали при 55°C-60°C в течение 12 часов. Добавляли к ней анилин (1,4 экв.), и затем она реагировала при приблизительно 110°C в течение 7 часов. Затем, смешанный реакционный раствор охлаждали до комнатной температуры и экстрагировали этилацетатом и водой. Объединенный органический слой обезвоживали безводным сульфатом магния (MgSO4) и концентрировали при пониженном давлении. н-Гептан добавляли к остатку, с последующим перемешиванием в течение 30 минут, приводящем к выпадению осадка, осадок фильтровали и промывали н-гептаном, и затем полученный в результате твердый остаток сушили, получая (S)-трет-бутил циклопропил(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)метилкарбамат с выходом 65%-80%.
1H ЯМР (300 МГц, CDCl3): δ 7,66-7,73 (м, 1H), 7,50-7,61 (м, 4H), 7,32-7,40 (м, 2H), 7,09-7,15 (т, J=18 Гц, 1H), 5,53-5,56 (д, J=9 Гц, 1H), 4,18-4,23 (т, J=15 Гц, 1H), 1,42 (с, 9H), 1,08-1,16 (м, 1H), 0,38-0,42 (м, 2H), 0,24-0,30 (м, 1H), 0,01-0,11 (м, 1H).
Стадия 2: получение (S)-2-(амино(циклопропил)метил)-5-фтор-3-фенилхиназолин-4(3H)-она
Трифторуксусную кислоту (приблизительно 8 кратный вес по отношению к (S)-трет-бутил циклопропил(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)метилкарбамату) добавляли к дихлорметановому раствору (приблизительно 15 кратный вес по отношению к (S)-трет-бутил циклопропил(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)метилкарбамату), в котором растворяли (S)-трет-бутил циклопропил(5-фтор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)метилкарбамат. Реакционный раствор перемешивали при комнатной температуре в течение от приблизительно 0,5 часа до приблизительно 1 часа, и затем pH реакционного раствора регулировали до приблизительно 7, применяя водный раствор карбоната натрия. Дихлорметановый раствор отделяли, обезвоживали, применяя сульфат магния (MgSO4), и фильтровали, и сульфат магния (MgSO4) удаляли, и затем фильтрат концентрировали при пониженном давлении, получая (S)-2-(амино(циклопропил)метил)-5-фтор-3-фенилхиназолин-4(3H)-он с выходом 80%-95%.
1H ЯМР (400 МГц, CDCl3): δ 7,66-7,71 (м, 1H), 7,47-7,57 (м, 4H), 7,27-7,31 (м, 2H), 7,08-7,12 (т, J=16 Гц, 1H), 2,97-2,99 (д, J=8 Гц, 1H), 1,87 (с, 2H), 1,22-1,31 (м, 1H), 0,39-0,53 (м, 2H), 0,01-0,15 (м, 2H).
Стадия 3: получение (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-фтор-3-фенилхиназолин-4(3H)-она
(S)-2-(Амино(циклопропил)метил)-5-фтор-3-фенилхиназолин-4(3H)-он, полученный на стадии 2, добавляли к трет-бутанолу (приблизительно 15 кратный вес по отношению к (S)-2-(амино(циклопропил)метил)-5-фтор-3-фенилхиназолин-4(3H)-ону), добавляли к нему N,N-диизопропиламин (приблизительно 2 эквивалента по отношению к (S)-2-(амино(циклопропил)метил)-5-фтор-3-фенилхиназолин-4(3H)-ону) и 6-бром-9H-пурин, и затем реакционный раствор перемешивали при кипячении с обратным холодильником в течение 24 часов.
Реакционную смесь охлаждали и концентрировали при пониженном давлении, удаляя трет-бутанол. Этилацетат добавляли к концентрату, и затем промывали разбавленным раствором хлористоводородной кислоты и разбавленным раствором карбоната калия. Этилацетатный слой обезвоживали безводным сульфатом магния (MgSO4) и фильтровали, и фильтрат концентрировали при пониженном давлении, получая (S)-2-(((7H-пурине-6-ил)амино)(циклопропил)метил)-5-фтор-3-фенилхиназолине-4(3H)-он (формула 7) в виде твердого остатка с выходом 60%-80%.
1H ЯМР (300 МГц, CDCl3): δ 13,02 (с, 1H), 8,03 (с, 1H), 7,98 (с, 1H), 7,50-7,71 (м, 6H), 7,39-7,42 (дд, J=9 Гц, 1H), 7,08-7,14 (т, J=18 Гц, 1H), 6,76-6,79 (д, J=9 Гц, 1H), 4,93 (уш с, 1H), 1,72 (уш с, 1H), 1,33-1,44 (м, 1H), 0,49-0,53 (м, 2H), 0,37-0,46 (м, 1H), 0,21-0,27 (м, 1H).
ESI-MS m/z 428,45 [M+H]+
<Пример 2> получение (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-метил-3-фенилхиназолин-4(3H)-она (формула 8)
[Реакционная схема 2]
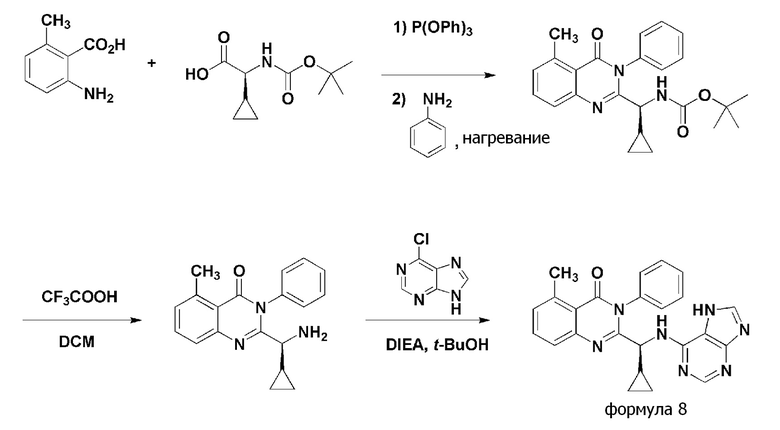
Стадия 1: Получение (S)- трет- бутил циклопропил(5-метил-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)метилкарбамата
(S)-трет-бутил циклопропил(5-метил-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)метилкарбамат получали тем же способом, как в примере 1, за исключением того, что 2-амино-6-метилбензойную кислоту применяли вместо 2-амино-6-фторбензойной кислоты.
1H ЯМР (400 МГц, CDCl3): δ 8,33 (уш с, 3H), 7,50 (д, 4H, J=7,9 Гц), 7,28 (т, 11H, J=7,7 Гц), 7,07 (т, 2H, J=7,3 Гц), 5,35 (уш с, 1H), 3,61 (уш с, 3H), 1,32-1,51 (м, 12H), 1,17-1,30 (м, 1H), 0,51-0,73 (м, 4H), 0,47 (тд, 3H, J=4,7, 9,6 Hz).
Стадия 2: получение (S)-2-(амино(циклопропил)метил)-5-метил-3-фенилхиназолин-4(3H)-она
(S)-2-(амино(циклопропил)метил)-5-метил-3-фенилхиназолин-4(3H)-он получали тем же способом, как в примере 1.
1H ЯМР (400 МГц, DMSO-d6): δ 8,41 (уш с, 2H), 7,79 (т, 1H, J=7,7 Гц), 7,54-7,73 (м, 2H), 7,31-7,46 (м, 1H), 2,74 (с, 3H), 1,23 (уш с, 1H), 1,18 (тт, 1H, J=4,4, 8,7 Гц), 0,51 (с, 1H), 0,32-0,41 (м, 1H, J=4,8, 10, 10 Hz).
Стадия 3: получение (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-метил-3-фенилхиназолин-4(3H)-она
(S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-метил-3-фенилхиназолин-4(3H)-он (формула 8) получали тем же способом, как в примере 1, за исключением того, что (S)-2-(амино(циклопропил)метил)-5-метил-3-фенилхиназолин-4(3H)-он применяли вместо (S)-2-(амино(циклопропил)метил)-5-фтор-3-фенилхиназолин-4(3H)-она.
1H ЯМР (400 МГц, CDCl3): δ 8,29 (с, 1H), 7,96 (уш с, 1H), 7,36-7,71 (м, 7H), 7,19-7,25 (м, 1H), 6,83 (д, 1H, J=6,6 Гц), 4,96 (т, 1H, J=8,1 Гц), 2,82 (с, 3H), 1,24-1,43 (м, 2H), 0,29-0,67 (м, 3H), 0,24 (с, 1H), 0,07 (с, 1H).
ESI-MS m/z 424,48 [M+H]+
<Пример 3> получение (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-амино-3-фенилхиназолин-4(3H)-она (формула 9)
[Реакционная схема 3]
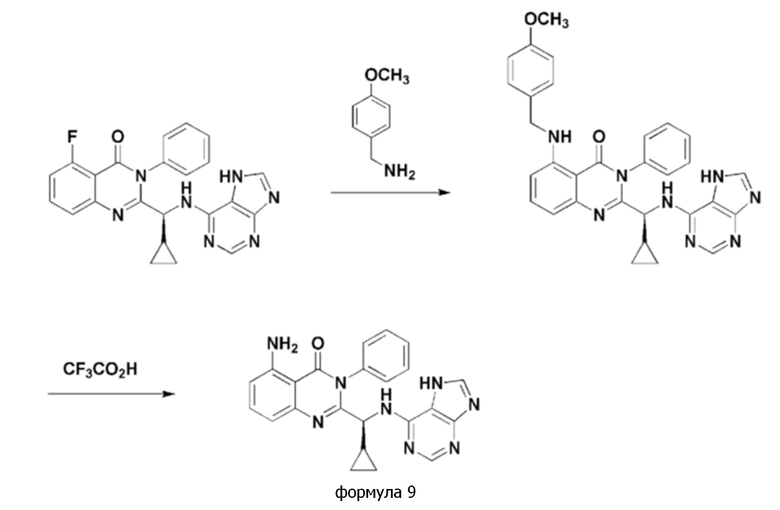
Стадия 1: получение (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-((4-метоксибензил)амино)-3-фенилхиназолин-4(3H)-она
В герметичную пробирку, в которой раствор получали последовательным добавлением (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-фтор-3-фенилхиназолин-4(3H)-она (1,0 экв.), полученного в Примере 1, и триэтиламина (5,0 экв.) к этанолу (15 кратный объем триэтиламина), дополнительно добавляли 4-метоксибензиламин.
Затем, пробирку заполняли азотом и герметично закрывали, и затем реакционную смесь нагревали при 180°C, и она реагировала в течение одного дня. После охлаждения до комнатной температуры, этанольный раствор удаляли при пониженном давлении. Затем, неочищенную смесь подвергали колоночной хроматографии на силикагеле (дихлорметан/метанол 20:1), получая (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-((4-метоксибензил)амино)-3-фенилхиназолин-4(3H)-он в виде желтого твердого остатка (выход: 38%).
1H ЯМР (300 МГц, CDCl3): δ 13,67 (с, 1H), 8,80-8,84 (т, J=12 Гц, 1H), 8,31 (с, 1H), 7,96 (с, 1H), 7,52-7,62 (м, 4H), 7,39-7,48 (м, 2H), 7,23-7,25 (д, J=6 Гц, 2H), 6,84-6,92 (т, J=24 Гц, 2H), 6,80-6,84 (д, J=12 Гц, 2H), 6,46-6,49 (д, J=9 Гц, 1H), 4,92 (с, 1H), 4,31-4,33 (д, J=6 Гц, 2H), 3,76 (с, 3H), 1,37-1,39 (м, 1H), 0,43-0,50 (м, 2H), 0,38-0,40 (м, 1H), 0,20-0,25 (м, 1H).
Стадия 2: получение (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-амино-3-фенилхиназолин-4(3H)-она
К раствору, в котором (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-(4-метоксибензиламино)-3-фенилхиназолин-4(3H)-он (1,0 экв.) растворяли в дихлорметане (6 кратный объем (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-(4-метоксибензиламино)-3-фенилхиназолин-4(3H)-она), добавляли трифторуксусную кислоту (2 кратный объем дихлорметана), и полученный в результате раствор перемешивали при комнатной температуре в течение 0,5 часа-2 часов. Затем, pH неочищенной смеси регулировали до 7 1M NaOH раствором при 0°С. Полученный в результате раствор экстрагировали три раза дихлорметаном, и объединенные органические фазы обезвоживали безводным сульфатом магния (MgSO4) и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле, получая (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-амино-3-фенилхиназолин-4(3H)-он (формула 9) в виде кремового твердого остатка (выход: 21%).
1H ЯМР (300 МГц, CDCl3): δ 13,16 (с, 1H), 8,30 (с, 1H), 7,97 (с, 1H), 7,53-7,64 (м, 4H), 7,40-7,45 (т, J=15 Гц, 2H), 6,92-6,95 (д, J=9 Гц, 1H), 6,84-6,86 (д, J=6 Гц, 1H), 6,54-6,56 (д, J=6 Гц, 1H), 6,15 (с, 2H), 4,93 (с, 1H), 1,32-1,41 (м, 1H), 0,47-0,48 (м, 2H), 0,38-0,43 (м, 1H), 0,22-0,25 (м, 1H).
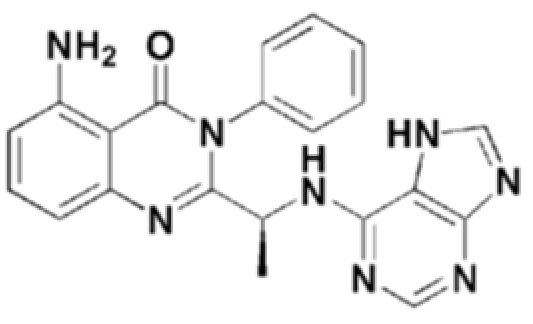
<Пример 4> получение (S)-2-(1-((7H-пурин-6-ил)амино)этил)-5-амино-3-фенилхиназолин-4(3H)-она (формула 10)
[Реакционная схема 4]
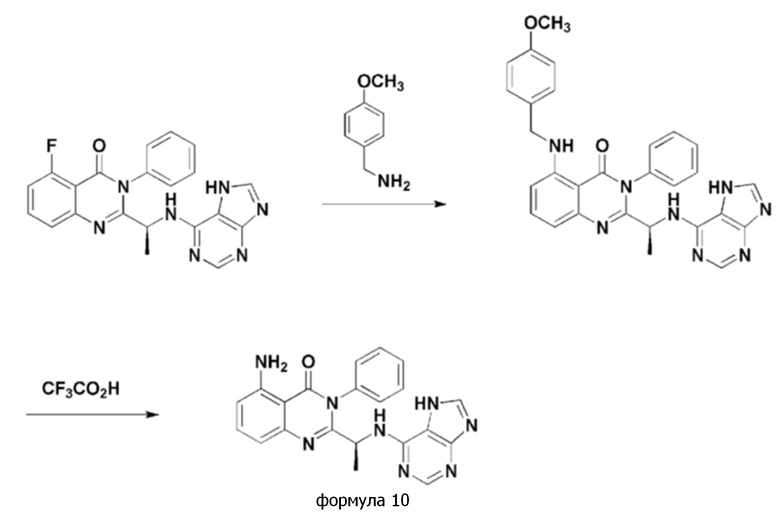
(S)-2-(1-((7H-пурин-6-ил)амино)этил)-5-амино-3-фенилхиназолин-4(3H)-он (формула 10) получали тем же способом, как в примере 3, за исключением того, что (S)-2-(1-((7H-пурин-6-ил)амино)этил)-5-фтор-3-фенилхиназолин-4(3H)-он применяли вместо (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-фтор-3-фенилхиназолин-4(3H)-она.
1H ЯМР (500 МГц, DMSO-d6): δ 8,00-8,22 (м, 1H), 7,32-7,78 (м, 4H), 7,06 (уш с, 1H), 6,55-6,70 (м, 1H), 1,99 (с, 1H), 1,06-1,55 (м, 4H), 0,70-0,93 (м, 2H).
<Пример 5> получение (S)-2-(1-(7H-пурин-6-иламино)пропил)-5-амино-3-фенилхиназолин-4(3H)-она (формула 11)
[Реакционная схема 5]
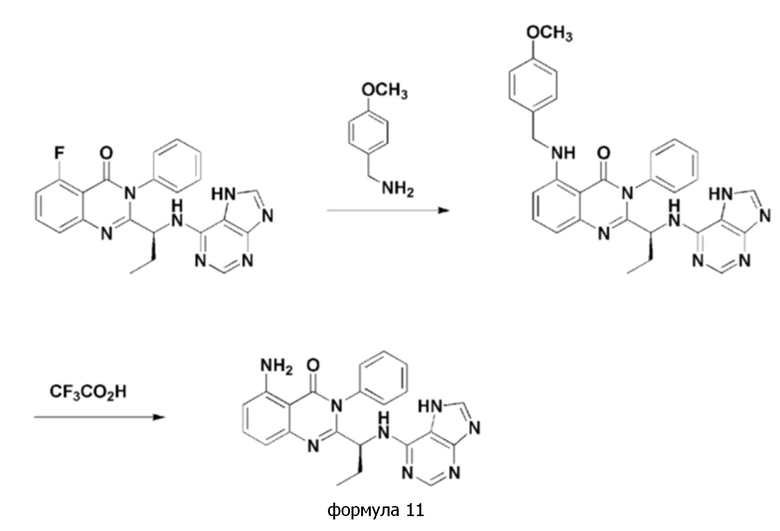
(S)-2-(1-((7H-пурин-6-ил)амино)пропил)-5-амино-3-фенилхиназолин-4(3H)-он (формула 11) получали тем же способом, как в примере 3, за исключением того, что (S)-2-(1-((7H-пурин-6-ил)амино)пропил)-5-фтор-3-фенилхиназолин-4(3H)-он применяли вместо (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-фтор-3-фенилхиназолин-4(3H)-она.
1H ЯМР (300 МГц, CDCl3): δ 8,31 (с, 1H), 7,97 (с, 1H), 7,35-7,68 (м, 6H), 6,91-6,94 (д, J=9 Гц, 1H), 6,82-6,84 (д, J=6 Гц, 1H), 6,53-6,56 (д, J=9 Гц, 1H), 6,15 (с, 2H), 5,16 (с, 1H), 1,91-2,05 (м, 1H), 1,74-1,84 (м, 1H), 0,84-0,89 (т, J=15 Гц, 3H).
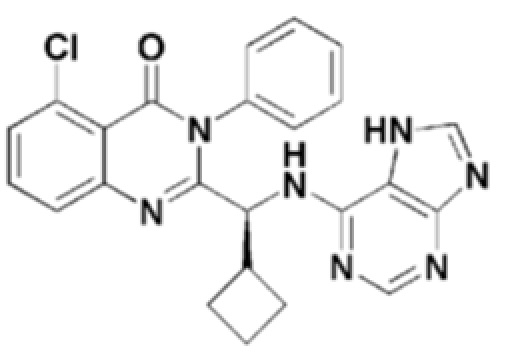
<Пример 6> получение (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-хлор-3-фенилхиназолин-4(3H)-она (формула 12)
[Реакционная схема 6]
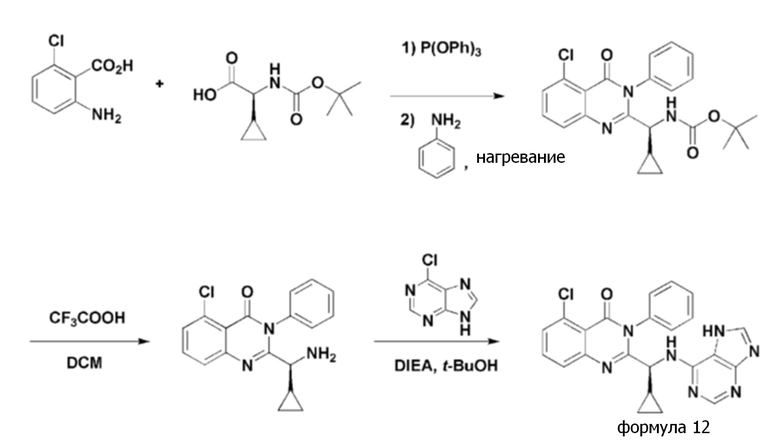
Стадия 1: получение (S)- трет- бутил (5-хлор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)(циклопропил)метилкарбамата
(S)-трет-бутил (5-хлор-4-оксо-3-фенил-3,4-дигидрохиназолин-2-ил)(циклопропил)метилкарбамат получали тем же способом, как в примере 1, за исключением того, что 2-амино-6-хлорбензойную кислоту применяли вместо 2-амино-6-фторбензойной кислоты.
Стадия 2: получение (S)-2-(амино(циклопропил)метил)-5-хлор-3-фенилхиназолин-4(3H)-она
(S)-2-(амино(циклопропил)метил)-5-хлор-3-фенилхиназолин-4(3H)-он получали тем же способом, как в примере 1.
Стадия 3: получение (S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-хлор-3-фенилхиназолин-4(3H)-она
(S)-2-(((7H-пурин-6-ил)амино)(циклопропил)метил)-5-хлор-3-фенилхиназолин-4(3H)-он (формула 12) получали тем же способом, как в примере 1, за исключением того, что (S)-2-(амино(циклопропил)метил)-5-хлор-3-фенилхиназолин-4(3H)-он применяли вместо (S)-2-(амино(циклопропил)метил)-5-фтор-3-фенилхиназолин-4(3H)-она.
1H ЯМР (300 МГц, CDCl3): δ 13,02 (с, 1H), 8,02 (с, 1H), 7,98 (с, 1H), 7,15-7,68 (м, 8H), 6,76-6,79 (д, J=9 Гц, 1H), 4,93 (уш с, 1H), 1,72 (уш с, 1H), 1,33-1,44 (м, 1H), 0,49-0,53 (м, 2H), 0,37-0,46 (м, 1H), 0,21-0,27 (м, 1H).
ESI-MS m/z 444,40 [M+H]+
<Пример 7> получение (S)-2-(((7H-пурин-6-ил)амино)(циклобутил)метил)-5-фтор-3-фенилхиназолин-4(3H)-она (формула 13)
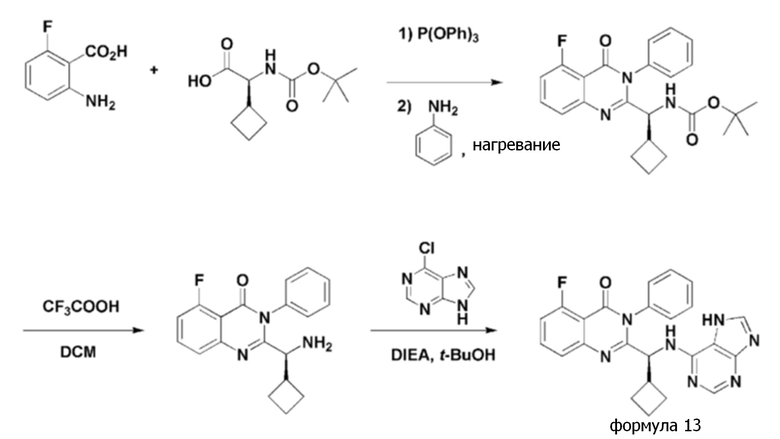
(S)-2-(((7H-пурин-6-ил)амино)(циклобутил)метил)-5-фтор-3-фенилхиназолин-4(3H)-он (формула 13) получали тем же способом, как в примере 1, за исключением того, что (S)-2-(трет-бутоксикарбониламино)-2-циклобутилуксусную кислоту применяли вместо (S)-2-(трет-бутоксикарбониламино)-2-циклопропилуксусной кислоты.
1H ЯМР (300 МГц, DMSO-d6): δ 12,95 (с, 1H), 8,13 (уш с, 1H), 7,85 (уш с, 1H), 7,24-7,60 (м, 8H), 5,18 (уш с, 1H), 3,05 (уш с, 1H), 1,64-2,01 (м, 7H).
<Пример 8> получение (S)-2-(((7H-пурин-6-ил)амино)(циклобутил)метил)-5-хлор-3-фенилхиназолин-4(3H)-она (формула 14)
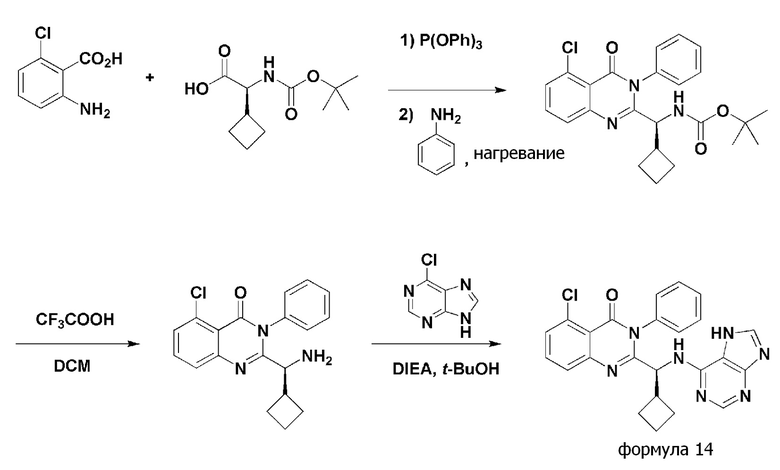
(S)-2-(((7H-пурин-6-ил)амино)(циклобутил)метил)-5-хлор-3-фенилхиназолин-4(3H)-он (формула 14) получали тем же способом, как в примере 1, за исключением того, что 2-амино-6-хлорбензойную кислоту применяли вместо 2-амино-6-фторбензойной кислоты, и (S)-2-(трет-бутоксикарбониламино)-2-циклобутилуксусную кислоту применяли вместо (S)-2-(трет-бутоксикарбониламино)-2-циклопропилуксусной кислоты.
1H ЯМР (300 МГц, DMSO-d6): δ 12,88 (с, 1H), 8,17 (уш с, 1H), 8,00 (с, 1H), 7,12-7,87 (м, 8H), 5,18 (уш с, 1H), 3,06 (уш с, 1H), 1,62-1,99 (м, 7H)
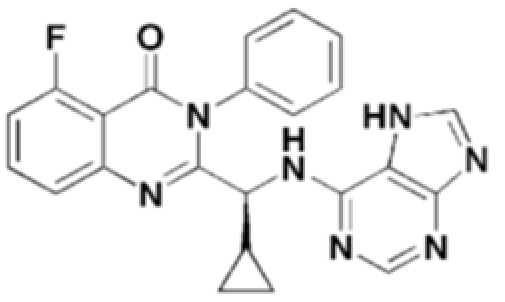
Структуры соединений формул 7-14 показаны в таблице 1 ниже.
[Таблица 1]
<Экспериментальный пример 1> тест на PI3K киназную активность
(1) Экспериментальный способ
Эксперимент проводили, применяя гомогенный флуоресцентный иммуноанализ, который представляет собой Adapta киназный анализ.
Пользовались услугами SelectScreen™, осуществляемыми Thermo Fisher Scientific Inc. Принцип эксперимента показан на фигуре 1.
(2) Экспериментальные результаты
Результаты эксперимента показаны в таблице 2 ниже.
Как показано в таблице 2, соединение формулы 7 и соединение формулы 8 показали большую активность, чем контрольное лекарственное средство, иделализиб. В частности, они показали специфическую ингибирующую активность на p110δ и также показали превосходную активность относительно p110γ.
В частности, соотношения PI3Kα/PI3Kδ и PI3Kβ/PI3Kδ, в качестве величин IC50, были показаны как 412 и 210, соответственно, в случае соединения формулы 7, и 1488 и 1800, соответственно, в случае соединения формулы 8.
Кроме того, подтверждали, что соединение формулы 7 и соединение формулы 8 имеют высокую дельта (δ) селективность. Каждый из них показал соотношение PI3Kγ/PI3Kδ 51 и 95, соответственно, тогда как иделализиб показал соотношение приблизительно 25. Из данных результатов, подтверждали, что соединения формул 7 и 8 могут обладать мощной и достаточной активностью относительно дельта (δ)-зависимых типов рака.
[Таблица 2]
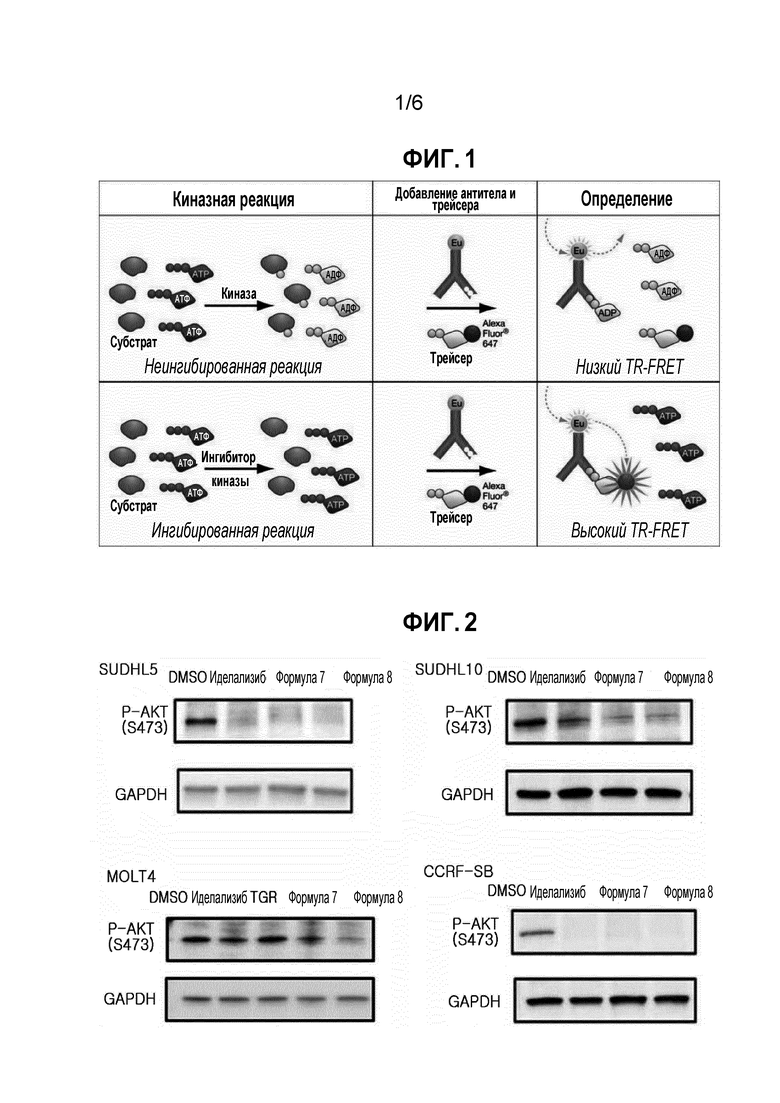
<Экспериментальный пример 2> эксперимент на подтверждение эффекта на снижение AKT(Ser473) фосфорилирования в клеточных линиях лейкемии и лимфомы
(1) Экспериментальный способ
Клеточные линии (SUDHL 5, SUDHL 10, CCRF-SB, и MOLT4) подвергали удалению сыворотки в течение 2 часов, и затем обрабатывали 1 мкМ каждого из соединения формулы 7, соединения формулы 8, иделализиба (сравнительное лекарственное средство 1), TGR1202 (сравнительное лекарственное средство 2), и диметилсульфоксида (DMSO) в течение 1 часа. Затем, клетки лизировали и фракционировали в зависимости от размера, с последующим иммуноблоттингом с антителами к Phospho-Akt (Ser473).
(2) Экспериментальные результаты
Результаты эксперимента показаны на фигуре 2.
Как показано на фигуре 2, подтверждали, что соединение формулы 7 и соединение формулы 8 вызывали снижение AKT фосфорилирования в различных клетках диффузной В-крупноклеточной лимфомы (DLBCL) и острого лимфоцитарного лейкоза (ALL).
<Экспериментальный пример 3> Эксперимент на подтверждение эффекта на ингибирование роста клеток лейкемии и лимфомы
(1) Экспериментальный способ
PI3K p110δ в высокой степени экспрессируются в клеточных линиях лейкемии и лимфомы, и клеточный рост ингибируется подавлением PI3K p110δ.
Таким образом, эксперимент осуществляли для подтверждения эффектов соединений на ингибирование клеточного роста.
Клетки диффузной В-крупноклеточной лимфомы (DLBCL) и клетки острого лимфоцитарного лейкоза (ALL) выращивали с соединением формулы 7, соединением формулы 8 или иделализибом, вместе с контрольной средой в течение 48 часов.
Эффекты по ингибированию клеточного роста на DLBCL-клетках и ALL клетках оценивали измерением поглощения красителя набора для подсчета клеток-8 (CCK-8). В течение последних 3 часов из 48 часов, 10 мкМ CCK-8 красителя добавляли к каждому планшеты, и затем выращивали.
Все данные выражали в виде среднего (± SD) трех независимых экспериментов.
(2) Экспериментальные результаты
Результаты эксперимента показаны на фигуре 3.
Как показано на фигуре 3, рост DLBCL клеток и ALL клеточной линии снижался при концентрации в диапазоне от 0,625 мкМ до 20 мкМ.
В это же время, летальная концентрация 50 (LC50), которая представляет собой концентрацию соединений, являющуюся летальной для 50% клеток, показана в таблице 3, и подтверждали, что соединение формулы 7 и соединение формулы 8 показывают более низкие LC50 величины, чем иделализиб.
[Таблица 3]
<Экспериментальный пример 4> эксперимент на подтверждение эффекта на апоптоз клеток диффузной В-крупноклеточной лимфомы (DLBCL) и острого лимфоцитарного лейкоза (ALL) SDS-PAGE
(1) Экспериментальный способ
2,6×106 клеток выращивали с иделализибом (сравнительное лекарственное средство), соединением формулы 7, или соединением формулы 8 при концентрации 50 мкМ в течение 36 часов, и затем белковый анализ проводили электрофорезом в полиакриламидном геле с додецилсульфатом натрия (SDS-PAGE).
Каспазные 3 и 9 PARP белки, которые представляют собой белки, участвующие в апоптозе, обычно существуют в виде неактивных предшественников (FL), и они активируются расщеплением (CL) при получении сигнала, стимулирующего апоптоз. Иммуноблоттинг анализ проводили, применяя их антитела, и глицероальдегид-3-фосфатдегидрогеназу (GAPDH) применяли в качестве контроля загрузки.
(2) Экспериментальные результаты
Результаты эксперимента показаны на фигуре 4.
Как показано на фигуре 4, соединение формулы 7 и соединение формулы 8 вызывали апоптоз в клетках диффузной В-крупноклеточной лимфомы (DLBCL) и острого лимфоцитарного лейкоза (CLL).
Кроме того, апоптоз возникал более активно с соединением формулы 7 и соединением формулы 8, чем со сравнительным лекарственным средством (иделализибом).
Из данных результатов, подтверждали, что соединение формулы 7 и соединение формулы 8 ингибировали клеточный рост за счет апоптоза.
<Экспериментальный пример 5> эксперимент на подтверждение эффекта на апоптоз клеток диффузной В-крупноклеточной лимфомы (DLBCL) и острого лимфоцитарного лейкоза (ALL) проточной цитометрией
(1) Экспериментальный способ
1×106 клеток диффузной В-крупноклеточной лимфомы (DLBCL) и 1×106 клеток острого лимфоцитарного лейкоза (ALL) выращивали и обрабатывали каждым из сравнительного лекарственного средства (иделализиб), соединения формулы 7 и соединения формулы 8 при концентрации 50 мкМ в течение 24 часов. Клетки промывали PBS, и затем суспендировали в буфере для связывания. Добавляли к ним 5 мкл Annexin V-FITC исходного раствора (Becton Dickinson Science, Inc) и 5 мкл PI (20 мкг/мл), с последующим выдерживанием при комнатной температуре в течение 15 минут при защите от света, и затем целевой материал количественно определяли на FACScan TM (Becton Dickinson) проточной цитометрией.
(2) Экспериментальные результаты
Результаты эксперимента показаны на фигуре 5.
Как показано на фигуре 5, подтверждали, что соединение формулы 7 и соединение формулы 8 более эффективно вызывали апоптоз, чем сравнительное лекарственное средство (иделализиб).
<Экспериментальный пример 6> эксперимент на подтверждение ингибирующего эффекта на ангиогенез
(1) Экспериментальный способ
Для сравнения степеней ингибирования ангиогенеза соединением формулы 7, соединением формулы 8 и сравнительным лекарственным средством (иделализиб), эндотелиальные клетки пупочной вены человека (HUVEC), которые представляют собой эндотелиальные клетки сосудов, и среду для выращивания эндотелиальных клеток получали у Technologies.
Эндотелиальные клетки пупочной вены человека (HUVEC) выращивали вместе с соединением формулы 7, соединением формулы 8 или сравнительным лекарственным средством (иделализиб) на базальной мембранной матрице при 37°C. Через 18 часов, образование трубочек фотографировали, применяя Cytation™ 5 флуоресцентный микроскоп (BioTek), и количество участков, образованных точками ветвления, считали, применяя программное обеспечение (смотри фигуру 6).
(2) Экспериментальные результаты
Результаты эксперимента показаны на фигурах 7 и 8.
Как показано на фигуре 7, соединение формулы 7 и соединение формулы 8 ингибировало ангиогенез в большей степени, чем сравнительное лекарственное средство (иделализиб).
Кроме того, как показано на фигуре 8, соединение формулы 7 и соединение формулы 8 не вызывало значительной цитотоксичности на эндотелиальных клетках пупочной вены человека (HUVEC).
<Экспериментальный пример 7> испытание на токсичность однократной дозы на крысах
(1) Экспериментальный способ
Соединение формулы 7 и соединение формулы 8 вводили перорально восьми шестинедельным самкам крыс, наблюдая пероральную токсичность их однократной дозы и получая приблизительную летальную дозу. Дозировку устанавливали равной 10 мл/кг, и дозу для каждой крысы рассчитывали, исходя из веса тела. Каждое соединение вводили при дозах 100 мг/кг, 300 мг/кг, 900 мг/кг и 1500 мг/кг, и общие симптомы наблюдали один раз в день с 1 по 2 день после введения.
(2) Экспериментальные результаты
Результаты эксперимента показаны на фигуре 9.
Летальная доза соединения формулы 7 была большей, чем 1500 мг/кг, и летальная доза соединения формулы 8 составляла 1500 мг/кг.
При этом, соединение, представленное формулой 1, согласно настоящему изобретению можно формулировать в виде различных форм. Несколько способов формулирования, применяя соединение, представленное формулой 1, согласно настоящему изобретению в качестве активного ингредиента обеспечивают ниже только для целей иллюстрации, но они не предполагаются ограничивающими настоящее изобретение.
<Пример получения 1> состав фармацевтических препаратов
1-1. Получение порошка
Ингредиенты выше смешивали, и заполняли в герметичные упаковки, получая порошок.
1-2. Получение таблеток
Ингредиенты выше смешивали, и затем таблетки получали согласно общему способу получения таблеток.
1-3. Получение капсул
Ингредиенты выше смешивали, и затем желатиновые капсулы заполняли согласно общему способу получения капсул, получая капсулы.
1-4. Получение инъекций
Согласно общему способу получения инъекции, ампулы получали с ингредиентами выше, включенными в одну ампулу (2 мл).
1-5. Получение жидкостей
Каждый ингредиент добавляли к и растворяли в очищенной воде согласно общему способу получения жидкости. Лимонный ароматизатор добавляли в подходящем количестве, и ингредиенты выше смешивали. Добавляли к ним очищенную воду так, чтобы довести суммарное количество полученного в результате раствора до 100 мл. Коричневую бутыль заполняли раствором и стерилизовали, получая жидкости.
Настоящее изобретение относится к новому хиназолиноновому производному формулы 1, обладающему свойствами ингибитора PI3K. Соединение может найти применение для предотвращения или лечения гематологической злокачественной опухоли, заболевания печени или аутоиммунного заболевания. Гематологическая злокачественная опухоль представляет собой лейкемию или лимфому. Заболевание печени выбрано из группы, состоящей из неалкогольной жировой болезни печени (NAFLD), неалкогольного стеатогепатита (NASH), стеатоза печени, гепатоцирроза, гепатита, аденомы печени, гиперчувствительности к инсулину и рака печени. В формуле 1
X представляет собой галоген или -CH3; и Y представляет собой C3-4 циклоалкил. 2 н. и 2 з.п. ф-лы, 9 ил., 3 табл., 6 пр.
1. Соединение, представленное формулой 1
 [Формула 1]
[Формула 1]
где
X представляет собой галоген или -CH3; и
Y представляет собой C3-4 циклоалкил.
2. Фармацевтическая композиция, обладающая свойствами ингибитора PI3K для предотвращения или лечения гематологической злокачественной опухоли, заболевания печени или аутоиммунного заболевания, причем фармацевтическая композиция содержит эффективное количество соединения, представленного формулой 1, в качестве активного ингредиента
[Формула 1]
где
X представляет собой галоген или -CH3; и
Y представляет собой C3-4 циклоалкил.
3. Фармацевтическая композиция по п. 2, где гематологическая злокачественная опухоль представляет собой лейкемию или лимфому.
4. Фармацевтическая композиция по п. 2, где заболевание печени выбрано из группы, состоящей из неалкогольной жировой болезни печени (NAFLD), неалкогольного стеатогепатита (NASH), стеатоза печени, гепатоцирроза, гепатита, аденомы печени, гиперчувствительности к инсулину и рака печени.
| WO 2013082540 A1, 06.06.013 | |||
| WO 2010057048 A1, 20.05.2010 | |||
| WO 2005113556 A1, 01.12.2005 | |||
| US 2010029693 A1, 04.02.2010 | |||
| US 2014121224 A1, 01.05.2014 | |||
| Shah A; Mangaonkar A | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| WO 2005120511 A1, 22.12.2005 | |||
| RU 2016115082 A, 25.10.2017. | |||

