Область техники
Настоящая заявка относится к области фармацевтической химии и, в частности, относится к классу ингибиторов PI3K, их фармацевтическим композициям, способу их получения и их применению.
Уровень техники
Фосфоинозитид-3-киназы (PI3K) представляют собой убиквитарные липидкиназы, функционирующие не только в качестве трансдуктора сигнала ниже рецептора на поверхности клетки, но также в качестве трансдуктора сигнала в составной внутриклеточной мембране и путях направленной миграции белков (Vanhaesebroeck, В. et al., Nature Rev. Mol. Cell Biol., 2010, 11, 329-341). Семейство PI3K липидкиназ может быть подразделено, исходя из их физиологической субстратной специфичности, на три класса: Класс I, Класс II и Класс III. Среди этих трех классов широко был исследован Класс I. В Класс I PI3K входят гетеродимеры, составленные из каталитической субъединицы р110 и регуляторной субъединицы. Класс I PI3K дополнительно подразеделены на киназы Класса Ia и киназы Класса Ib. Киназы Класса Ia составлены из трех различных каталитических субъединиц (р110α, р110β и р110δ), которые димеризуются с пятью различными регуляторными субъединицами (р85α, р55α, р50α, р85β, и р55γ), причем все каталитические субъединицы могут взаимодействовать со всеми регуляторными субъединицами для образования различных гетеродимеров, которые называются PI3Kα, PI3Kβ и PI3Kδ. р110α и р110β экспрессируются по существу в клетках всех типов, тогда как р110δ экспрессируется преимущественно в лейкоцитах. Однотипные киназы Класса Ib составлены из каталитических субъединиц р110γ, взаимодействующих с регуляторными субъединицами р101, и они называются PI3Kγ. Как и р110δ, киназы Класса Ib экспрессируются преимущественно в лейкоцитах.
PI3K играют роль в опухолеобразовании во многих типах рака ввиду дисрегуляции или сверхактивации пути PI3K/AKT (Vivanco and Sawyers, Nature Rev. Cancer, 2002, 2, 489-501). Среди четырех изоформ, PI3Kδ играет роль в контроле за выживаемостью В-клеток в некоторых типах В-клеточного рака. Например, PI3Kδ важен для выживаемости неходжкинской лимфомы (НХЛ) и хронического лимфоцитарного лейкоза (ХЛЛ). Клинически было доказано, что ингибитор PI3Kδ, иделалисиб, способен лечить ХЛЛ (Furman, R.R., et al., The New Englang Jounal of Medicine, 2014, 370, 997-1007; O'Brien, S., et al., Blood, 2015, 126, 2686-2694), и, таким образом, он был одобрен со стороны US FDA для лечения этих заболеваний. Это адекватным образом показывает, что В-клеточная лимфома и лейкоз, в том числе НХЛ и ХЛЛ, могут быть вылечены путем ингибирования активности PI3Kδ.
В дополнение, ингибирование PI3Kδ может разрушить опосредованную регуляторными Т-клетками иммунную толерантность к опухолям и увеличить иммунные ответы, тем самым обеспечивая регрессию опухоли в модели животного (Ali, K. et al., Nature, 2014, 510, 407-411). Эти результаты показывают, что ингибирование PI3Kδ является значимым для лечения опухолей, особенно таковых с недостаточным иммунным ответом, таких как рак молочной железы, рак легких (в том числе мелко клеточный рак легких, немелкоклеточный рак легких и бронхиолоальвеолярная карцинома), рак простаты, холангиокарцинома, рак костей, рак мочевого пузыря, рак головы и шеи, рак почки, рак печени, рак желудочно кишечной ткани, рак пищевода, рак яичников, рак поджелудочной железы, рак кожи, рак яичка, рак щитовидной железы, рак матки, рак шейки матки, рак влагалища, лейкоз, множественная миелома, лимфома и т.д.
В дополнение к опухолям, доказательными материалами также показано, что PI3Kδ также играет важную роль в воспалениях и аутоиммунных заболеваниях (Puri, K.D. et al., J. Immunol, 2009, 182 (Suppl. 50), 14; Maxwell, M. J. et al., J. Autoimmunity, 2012, 38, 381-391; Suarez-Fueyo, A. et al., J. Immunol., 2011, 187, 2376-2385). Таким образом, ингибирование PI3Kδ может быть использовано для лечения воспалительных заболеваний и аутоиммунных заболеваний, в том числе, но без ограничения, дерматита, ревматоидного артрита, аллергического ринита, астмы, болезни Крона, хронической обструктивной болезни легких (ХОБЛ), системной красной волчанки, псориаза, множественного склероза, синдрома активированной PI3Kδ, синдрома Шегрена и т.д.
Настоящая заявка относится к новому поколению ингибитора PI3K, который может быть использован при лечении рака, воспалительных заболеваний и аутоиммунных заболеваний.
РАСКРЫТИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящая заявка, в одном аспекте, представляет соединение Формулы (I):
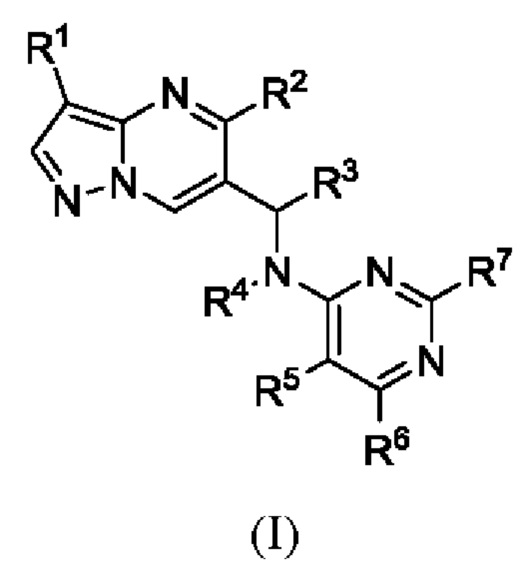
или его фармацевтическую соль, причем:
R1 является галогеном;
R2 является фенилом, причем R2 необязательно может быть замещен R2a от одного до пяти;
R2a выбран из Н или галогена;
R3 является С1-С4 алкилом;
R4 является Н;
R5 является циано;
R6 выбран из амино или алкила, a R7 выбран из Н или амино;
Предпочтительно, настоящая заявка представляет соединение Формулы (II):
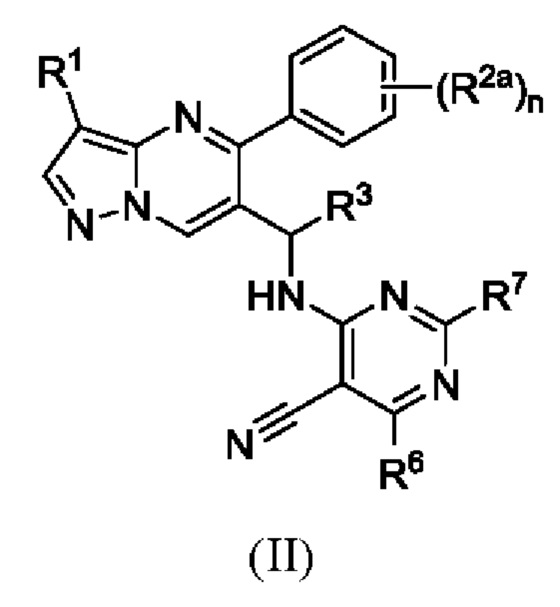
или его фармацевтическую соль, причем:
R1 является хлором;
R2a выбран из Н или галогена;
R3 является метилом;
R6 выбран из амино или метила, a R7 выбран из Н или амино; и
n равняется 1, 2 или 3.
В предпочтительном варианте реализации, R1 выбран из группы, состоящей из галогена, циано, алкила, галогеналкила, циклоалкила, гетероциклоалкила, арила и гетероарила.
В более предпочтительном варианте реализации, R1 выбран из галогена.
В предпочтительном варианте реализации, R2a выбран из Н и галогена.
В более предпочтительном варианте реализации, R2a выбран из Н и фтора.
В предпочтительном варианте реализации, R3 выбран из группы, состоящей из алкила, галогеналкила и циклоалкила.
В более предпочтительном варианте реализации, R3 выбран из алкила.
В предпочтительном варианте реализации, R4 выбран из Н и алкила.
В более предпочтительном варианте реализации, R4 выбран из Н.
В предпочтительном варианте реализации, R5 выбран из группы, состоящей из галогена, алкинила, циклоалкилалкинила, арилалкинила, гетероарилалкинила, арила и гетероарила
В более предпочтительном варианте реализации, R5 выбран из циано.
В предпочтительном варианте реализации, R6 выбран из группы, состоящей из Н, амино и алкила.
В предпочтительном варианте реализации, R7 выбран из Н и амино.
Термин «гало» или «галоген» включает фтор, хлор, бром и йод.
Термин «алкил» относится к прямоцепочечной или разветвленной насыщенной углеводородной группе. Примеры алкила включают метил (Me), этил (Et), пропил (такой как н-пропил и изопропил), бутил (такой как н-бутил, изобутил, трет-бутил), пентил (такой как н-пентил, изопентил, неопентил), гексил (такой как н-гексил, 2-гексил, 3-гексил, 2-метилпентил, 3-метилпентил, 2,2-диметилбутил, 3-этилпентил-1 и т.д.), гептил (такой как н-гептил, 2-гептил, 3-гептил, 4-гептил, 2-метилгексил, 3-метилгексил, 2,2-диметилпентил, 3,3-диметилпентил, 3-этилпентил-1 и т.д.), октил (такой как 1-октил, 2-октил, 2-этилгексил и т.д.), нонил (такой как 1-нонил), децил (такой как н-децил) и подобные группы. Термин «алкил» относится, в частности, к прямоцепочечному или разветвленному алкилу, имеющему от одного до двадцати атомов углерода, и более конкретно, к прямоцепочечному или разветвленному алкилу, имеющему от одного до десяти атомов углерода, и более предпочтительно, к прямоцепочечному или разветвленному алкилу, имеющему от одного до шести атомов углерода. Если не указано иное, определения всех групп в настоящей заявке определены в тексте настоящего документа.
Термин «гидроксиалкил» относится к алкильной группе, замещенной гидроксилом, причем алкильная группа является такой, как определено выше.
Термин «галогеналкил» относится к алкильной группе, имеющей один или более заместителей галогена, причем алкильная группа и гало или галоген являются такими, как определено выше. Примеры галогеналкила включают CH2F, CHF2, CF3, C2F5, CCl3 и подобные группы.
Термин «цианоалкил» относится к алкильной группе, замещенной цианогруппой (-CN).
Термин «алкенил» относится к углеводородной группе, имеющей один или более двойных связей С=С. Примеры алкенила включают винил, пропенил, аллил, 1-бутенил, 2-бутенил, 1,3-бутадиенил, 1-пентенил, 2-пентенил, 1,3-пентадиенил, 1-гексенил, 2-гексенил и т.д., и подобные группы.
Термин «алкинил» относится к углеводородной группе, имеющей один или более тройных связей С=С. Примеры алкинила включают этинил, пропинил, пропаргил, 1-бутинил, 2-бутинил, 1-пентинил, 2-пентинил, 1-гексинил, 2-гексинил и т.д., и подобные группы.
Термин «циклоалкил» относится к неароматическому карбоциклическому кольцу, включая циклизированную алкильную группу, циклизированную алкенильную группу и циклизированную алкинильную группу. Циклоалкильная группа может представлять собой моноциклическую или полициклическую (например, имеющую два, три или четыре сочлененных кольца) систему, включая спирокольца. В некоторых вариантах реализации, циклоалкильная группа может иметь от трех до двадцати атомов углерода. Циклоалкильная группа дополнительно может иметь ноль, одну, две или три двойных связи С=С и/или ноль, одну или две тройных связи С=С. Также в определение циклоалкила включены фрагменты, имеющие одно или более ароматических колец, сочлененных с циклоалкильным кольцом (например, имеющих общую связь), такие как бензопроизводные пентала, пентена, гексана, гексена и подобные соединения. Циклоалкильные группы, имеющие одно или более сочлененных ароматических колец, могут быть связаны ароматической частью или неароматической частью. Примеры циклоалкильных групп включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклопентенил, циклогексенил, циклогексадиенил, циклогептенил, циклогептадиенил, адамантил и подобные группы.
Термин «гетероциклоалкил» относится к неароматическому гетероциклическому кольцу, в котором один или более атомов, образующие кольцо, являются гетероатомами, такими как О, N, S или фосфора. Циклоциклическая группа может включать моноциклическую или полициклическую (например, имеющую два, три или четыре сочлененных кольца) систему и спирокольца. Предпочтительно, примеры «гетероциклоалкила» включают, но без ограничения: азиридинил (азациклопропил), азетидинил (азациклобутил), тетрагидрофуранил, тетрагидротиенил, пирролидинил, оксазолидинил, тиазолидинил, имидазолидинил, изоксазолидинил, изотиазолидинил, пиразолидинил, морфолинил, тиоморфолинил, пиперазинил, пиперидинил и подобные группы. Также в определение гетероциклоалкила включены фрагменты, имеющие одно или более ароматических колец, сочлененных с неароматическим гетероциклоалкильным кольцом (например, имеющих общую связь), такие как 2,3-дигидробензофуранил, 1,3-бензодиоксолил, бензо-1,4-диоксанил, фталимидо, нафталимидо и подобные группы. Гетероциклоалкильные группы, имеющие одно или более сочлененных ароматических колец, могут быть связаны ароматической частью или неароматической частью. Атомы азота и серы в гетероциклоалкилке могут присутствовать в окисленной форме.
Термин «арил» относится к монокольцевому или многокольцевому (например, имеющему два, три или четыре сочлененных кольца) ароматическому углеводороду, такому как фенил, нафтил, антрил, фенантрил, инденил и подобные группы.
Термин «гетероарил» относится к ароматическому гетероциклическому кольцу, имеющему по меньшей мере один кольцевой член в виде гетероатома (такой как О, N или S). Гетероарильная группа включает моноциклическую или полициклическую (например, имеющую два, три или четыре сочлененных кольца) кольцевую систему. Любые атомы N, образующие кольцо в гетероциклической группе, также могут быть окислены для образования N-оксида. Предпочтительно, примеры «гетероарила» включают, но без ограничения: пиридил, пиримидинил, пиразинил, пиридазинил, триазинил, фуранил, тиенил, имидазолил, триазолил, тетразолил, тиазолил, изотиазолил, 1,2,4-тиадиазолил, пирролил, оксазолил, изоксазолил, оксадиазолил, бензофуранил, бензотиенил, бензотиазолил, индолил, индазолил, хинолил, изохинолил, пуринил, карбазолил, бензоимидазолил, пирропиридил, пирропиримидинил, пиразолопиридил, пиразолопиримидинил и подобные группы.
Предполагается, что термин «соединение», используемый в настоящем документе, включает все стереоизомеры, геометрические изомеры, таутомеры и изотопы.
Соединение по настоящей заявке может быть ассиметрическим, например, имеющим один или более стереоцентров. Если не указаное иное, все стереоизомеры представляют собой, например, энантиомеры и диастереоизомеры. Соединение по настоящей заявке, имеющее асимметрически замещенные атомы углерода, могут быть выделены в оптически активные или рацемические формы. Оптически активные формы могут быть приготовлены путем разложения рацемических смесей или путем использования хирального синтона или хирального реагента.
Соединение по настоящей заявке также может включать таутометрические формы. Новые таутометрические формы являются результатом обмена одинарной связи на смежную двойную связь вместе с миграцией протона.
Соединение по настоящей заявке также может включать все изотопические формы атомов, присутствующих в промежуточном или конечном соединении. Изотопы включают те атомы, которые имеют такое же атомное число, но другие массовые числа. Например, изотопы водорода включают дейтерий и тритий.
Настоящая заявка также включает фармацевтическую соль соединения Формулы (I). Фармацевтическая соль относится к производному соединения, в котором исходное соединение модифицировано путем преобразования фрагмента основания в форму его соли, или производному соединения, в котором исходное соединение модифицировано путем преобразования фрагмента кислоты в форму его соли. Примеры фармацевтической соли включают, но без ограничения, соли неорганических или органических кислот основных групп (таких как амины) или соли неорганического или органического основания кислотных групп (таких как карбоновые кислоты). Фармацевтическая соль по настоящей заявке может быть синтезирована путем приведения свободного основания исходного соединения Формулы (I) в реакцию с 1-4 эквивалентами подходящей кислоты в системе растворителя. Подходящие соли перечислены в Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, Pa., 1985, p.1418 and Journal of Pharmaceutical Science, 66, 2 (1977).
Соединение по настоящей заявке и его фармацевтическая соль дополнительно включают сольватированные формы или гидратированные формы. В целом, сольватированные формы или гидратированные формы и несольватированные формы или негидратированные формы эквивалентны и обе включены в объем настоящей заявки. Некоторые соединения по настоящей заявке могут присутствовать в различных кристаллических формах или аморфных формах. В целом, все физические формы соединений включены в объем в настоящей заявки.
Настоящая заявка также включает пролекарство соединения Формулы (I). Пролекарство представляет собой фармакологическое вещество (а именно, лекарственное средство), которое получено из исходного лекарственного средства. После введения, пролекарство метаболизируется в исходное лекарственное средство in vivo. Пролекарство может быть приготовлены путем замещения одной или более функциональных групп в соединении, причем заместитель в пролекарстве удаляется in vivo таким образом, что пролекарство преобразуется в исходное соединение. С приготовлением и использованием пролекарства можно ознакомиться в Т. Higuchi and V. Stella, "Pro-drugs as Novel Delivery Systems," Vol. 14 of the A.C.S. Symposium Series и Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987.
В настоящей заявке также представлена композиция, включающая соединение Формулы (I) и фармацевтический носитель или вспомогательное вещество. Композиция по настоящей заявке может быть введена перорально, парентерально (путем инъекции), путем вдыхания аэрозоля, местного применения, ректального введения, назального введения, вагинального введения, интраперитонеального введения или через имплантированный резервуар.
В другом аспекте настоящей заявки представлен способ регулирования активности киназы с помощью соединения Формулы (I). Как используется в настоящем документе, термин «регулирование активности киназы» относится к пониженной активности киназы при контакте с соединением Формулы (I) по настоящей заявке по сравнению с активностью киназы в отсутствии соединения Формулы (I). Таким образом, в настоящей заявке представлен способ регулирования активности киназы путем приведения киназы в контакт с соединением Формулы (I) по настоящей заявке.
В некоторых вариантах реализации киназа представляет собой липидкиназу, такую как PI3Kα, PI3Kβ, PI3Kγ или PI3Kδ.
В некоторых вариантах реализации киназа представляет собой PI3Kδ.
Еще в одном другом аспекте настоящей заявки представлен способ лечения PI3K-опосредованных заболеваний с помощью соединения по настоящей заявке. PI3K-связанные заболевания включают рак, воспалительные заболевания и аутоиммунные заболевания.
В некоторых вариантах реализации, рак, описанный в настоящей заявке, представляет собой солидную опухоль, лейкоз, лимфому и множественную миелому.
В некоторых вариантах реализации, лейкоз, описанный в настоящей заявке, выбран из группы, состоящей из острого лимфоцитарного лейкоза (ОЛЛ), острого миелоидного лейкоза (ОМЛ), хронического лимфоцитарного лейкоза (ХЛЛ) и хронического миелоидного лейкоза (ХМЛ). Лимфома выбрана из группы, состоящей из лейкоза Ходжкина, неходжкинского лейкоза (НХЛ), мантийноклеточного лейкоза (МКЛ), фолликулярного лейкоза, В-клеточной лимфомы, Т-клеточной лимфомы и диффузной В-крупноклеточной лимфомы.
В некоторых вариантах реализации, аутоиммунные заболевания и воспалительные заболевания выбраны из группы, состоящей из дерматита, ревматоидного артрита, аллергического ринита, астмы, болезни Крона, хронической обструктивной болезни легких (ХОБЛ), системной красной волчанки, псориаза, множественного склероза, синдрома активированной PI3Kδ и синдрома Шегрена.
В другом аспекте настоящей заявки представлено применение соединения или его фармацевтической соли, или фармацевтической композиции по настоящей заявке, скомбинированной с одним или более лекарственных средств, для лечения рака, воспалительных заболеваний и аутоиммунных заболеваний.
В некоторых вариантах реализации, лекарственные средства, используемые в комбинации, включают соединение, имеющее малую молекулу, и лекарственное средство-антитело, имеющее макромолекулу. Соединение, имеющее малую молекулу, выбрано из, но без ограничения, различных ингибиторов киназы, таких как ингибиторы ВТК и другие некиназные ингибиторы, имеющие малую молекулу, и так далее. Лекарственное средство-антитело, имеющее макромолекулу, включает анти-CD20, анти-CTLA4, анти-PD-1 и анти-PD-L1, и так далее.
Еще в одном другом аспекте настоящей заявки представлен способ получения соединения Формулы (I). Соединение по настоящей заявке может быть получено посредством схемы реакции и способов, которые описаны ниже.
Схема реакции 1
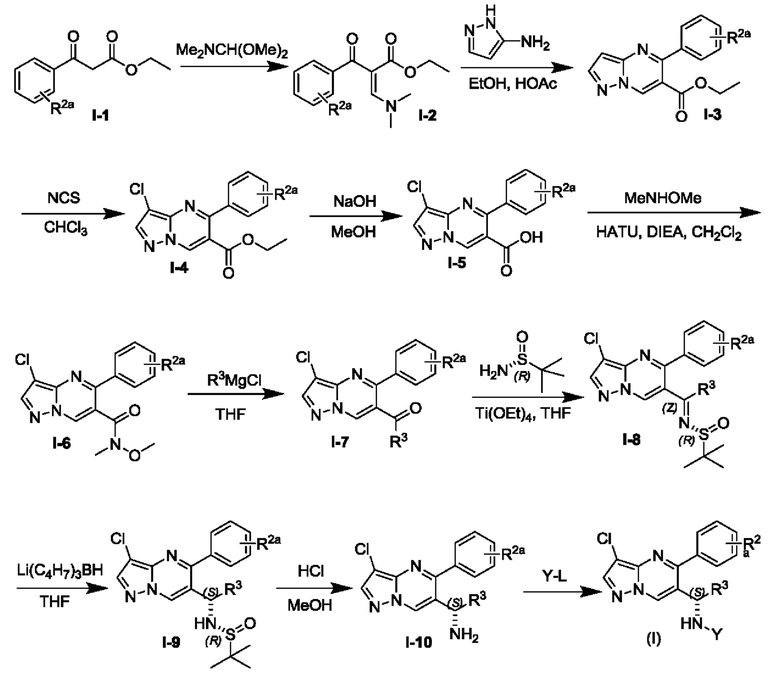
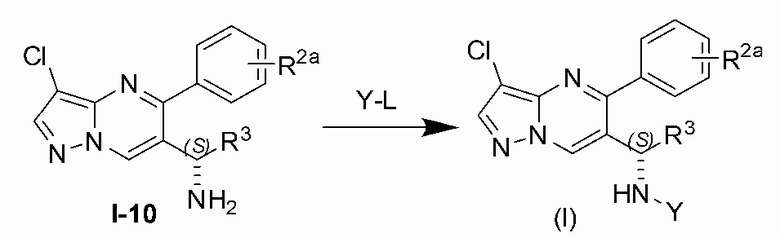
Соединение Формулы (I) может быть получено, согласно пути синтезирования, перечисленного в схеме реакции 1. Исходные материалы I-1 и N,N-диметилформамида диметилацеталь нагревают с обратным холодильником для получения соединения I-2. Соединение I-2 реагирует с 3-аминопиразолом в уксусной кислоте для получения продукта циклизации пиразолопиримидина I-3. Хлорирование пиразолопиримидин а I-3 с NCS дает хлорированный продукт I-4. Сложноэфирную группу хлорированного продукта I-4 подвергают гидролизу для образования карбоновой кислоты I-5, и карбоновую кислоту I-5 и N,O-диметилгидроксиламин подвергают реакции конденсации для получения амидного соединения I-6. Реактив Гриньяра R3MgCl и амидную связь в амидном соединении I-6 подвергают реакции присоединения для образования кетона I-7. Кетон I-7 и (R)-сульфенамид подвергают реакции конденсации для получения соединения I-8. Иминогруппу в соединении I-8 понижают с помощью три-втор-бутил борогидрида лития для получения соединения I-9 сульфонамида S-конфигурации. Сульфинил в соединении I-9 удаляют с помощью хлористоводородной кислоты для получения свободного амина I-10. Свободный амин I-10 вступает в реакцию с Y-L (Y представляет собой пиримидин, замещенный на R5, R6 и R7, a L представляет собой отходящую группу) для получения конечного продукта (I).
Подробное описание
Следующие варианты реализации используются для иллюстрации предпочтительных вариантов реализации настоящей заявки. Специалистам в данной области техники будет ясно, что технологии, раскрытые в следующих вариантах реализации, представляют собой технологии, разработанные автором изобретения по настоящей заявке и играющие важнейшую роль в вариантах реализации настоящей заявки, и, следовательно, их следует рассматривать как составляющие предпочтительные варианты реализации настоящей заявки. Однако исходя из содержания, раскрытого в настоящей заявке, специалистам в данной области техники будет ясно, что в раскрытых конкретных вариантах реализации могут быть сделаны различные вариации для достижения подобных или таких же результатов без выхода за рамки сущности и объема настоящей заявки.
Пример 1
(S)-4-амино-6-(1-(3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-ил)этиламино)пиримидин-5-формонитрил
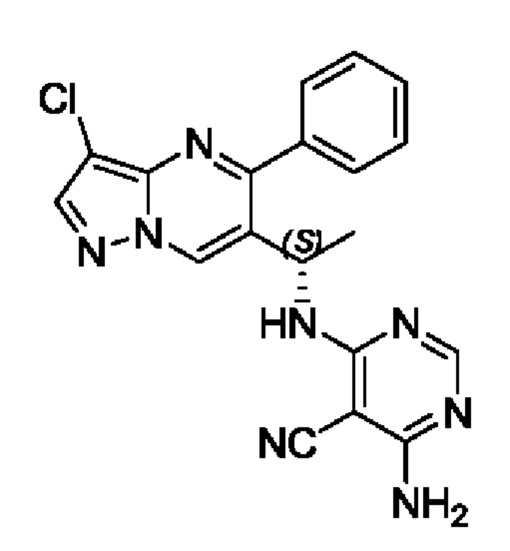
Этап 1. этил5-фенилпиразоло[1,5-а]пиримидин-6-карбоксилат
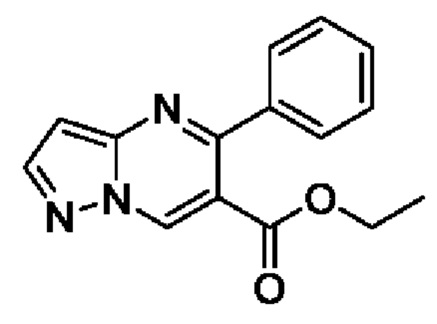
В 250 мл круглодонную колбу добавляли этил бензоилформат (10 г, 56,2 ммоль), N,N-диметилформамида диметилацеталь (6,7 г, 56,2 ммоль) и 120 мл толуола для реакции рециркуляции на протяжении 2 часов. После завершения реакции полученную смесь подвергали дистилляции с пониженным давлением для удаления толуола. Полученный остаток растворяли в 200 мл этанола для получения раствора. В этот раствор добавляли 3-аминопиразол (4,7 г, 56,6 ммоль) и 40 мл ледяной уксусной кислоты при перемешивании с последующей реакцией при комнатной температуре на протяжении 8 часов. После завершения реакции полученную смесь подвергали дистилляции с пониженным давлением для удаления этанола для получения остатка. Остаток разбавляли путем добавления H2O и затем экстрагировали с помощью CH2Cl2. Полученную органическую фазу промывали последовательно с помощью насыщенного раствора Na2CO3 и насыщенного раствора NaCl, сушили с помощью безводного Na2SO4 и затем фильтровали и концентрировали для получения 14,2 г желтого твердого вещества для прямого использования на следующем этапе реакции. Выход составил 94%. ЖХМС (ИЭ): масса/заряд=268 (М+Н)+.
Этап 2. этил 3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-карбоксилат
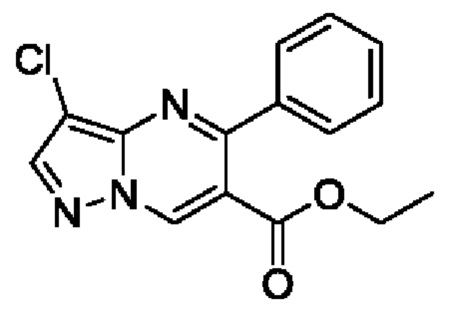
Этил 5-фенилпиразоло[1,5-а]пиримидин-6-карбоксилат (14,2 г, 53,1 ммоль) растворяли в 150 мл CHCl3 с последующим добавлением NCS (7,1 г, 53,2 ммоль). Полученную смесь нагревали до 60°С для реакции на протяжении 6 часов. После завершения реакции полученную смесь разбавляли с помощью CH2Cl2 и затем промывали последовательно с помощью водного раствора 0,1 N HCl и насыщенного раствора NaCl. Полученную органическую фазу сушили с помощью безводного Na2SO4, а также фильтровали и концентрировали для получения 15,5 г желтого твердого вещества для прямого использования на следующем этапе реакции. Выход составил 97%. ЖХМС (ИЭ): масса/заряд=302 (М+Н)+.
Этап 3. этил 3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-карбоксилат
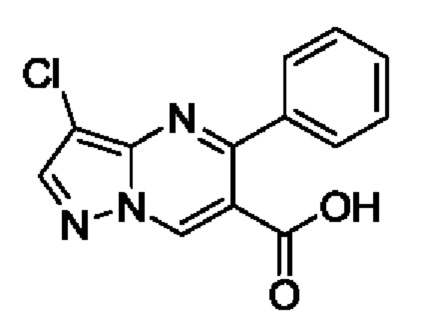
В 250 мл круглодонную колбу добавляли этил 3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-карбоксилат (15,5 г, 51,5 ммоль), 50 мл метанола и 100 мл водного раствора 2 N NaOH для реакции при комнатной температуре на протяжении 5 часов. После завершения реакции полученную смесь подвергали дистилляции с пониженным давлением для удаления метанола, а затем фильтровали для получения остатка на фильтре и фильтрата. Остаток на фильтре промывали с помощью H2O. Фильтрат регулировали с использованием водного раствора 4 N HCl до получения значения рН 3 для осаждения твердого вещества и затем фильтровали для получения 8,2 г твердого вещества. Выход составил 58%. ЖХМС (ИЭ): масса/заряд=274 (М+Н)+.
Этап 4. 3-хлор-N-метокси-N-метил-5-фенилпиразоло[1,5-а]пиримидин-6-амид
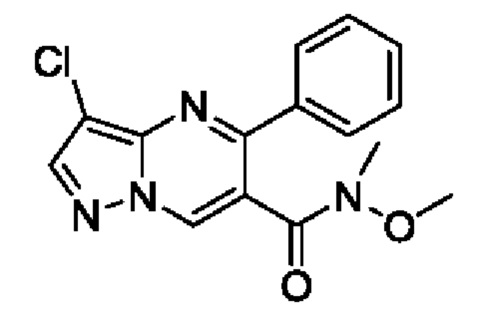
3-Хлор-5-фенилпиразоло[1,5-а]пиримидин-6-карбоновую кислоту (8,2 г, 30 ммоль) и DIEA (11,6 г, 90 ммоль) растворяли в 100 мл CH2Cl2 с последующим добавлением N,O-диметилгидроксиламин гидрохлорида (3,2 г, 33 ммоль) и HATU (12,5 г, 33 ммоль) для реакции при комнатной температуре на протяжении 2 часов. После завершения реакции полученную смесь разбавляли с помощью CH2Cl2. Полученную органическую фазу промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и концентрировали для получения остатка. Остаток очищали колоночной хроматографией (РЕ:EtOAc (об/об)=2:1) для получения 8,5 г твердого вещества.
Выход составил 90%. ЖХМС (ИЭ): масса/заряд=317 (М+Н)+.
Этап 5. 1-(3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-ил)этанон
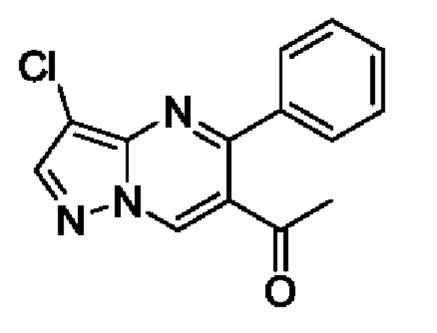
В атмосфере азота, 3-хлор-N-метокси-N-метил-5-фенилпиразоло[1,5-а]пиримидин-6-амид (8,5 г, 26,9 ммоль) растворяли в 80 мл THF, а затем помещали и охлаждали в ледяной бане. В верхний раствор медленно по каплям добавляли хлорид метилмагния в THF (3М, 14 мл) и добавление завершали за 20 минут. После завершения добавления по каплям полученную смесь поддерживали при температуре для реакции на протяжении 1 часа. После завершения реакции в полученную смесь аккуратно добавляли насыщенный раствор NH4Cl для гашения реакции, и полученную смесь подвергали дистилляции с пониженным давлением для удаления THF. Полученную водную фазу экстрагировали с помощью EtOAc. Органические фазы комбинировали и затем промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и затем концентрировали для получения 6,5 г твердого вещества. Выход составил 89%. ЖХМС (ИЭ): масса/заряд=272 (М+Н)+.
Этап 6. (R,E)-N-(1-(3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-ил)этилдиен)-2-метилпропан-2-сульфонамид
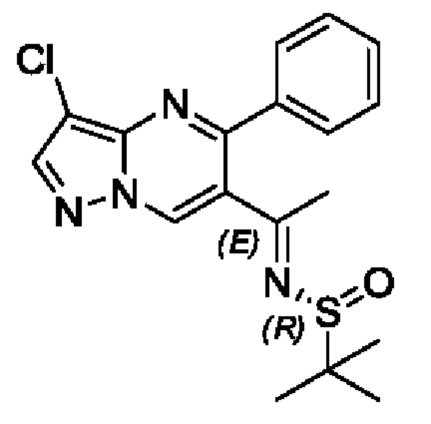
(R)-трет-бутилсульфенамид (3 г, 24,8 ммоль) растворяли в 30 мл THF с последующим добавлением по каплям раствора THF (50 мл), содержащего 1-(3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-ил)этанон (6,5 г, 24 ммоль) и тетраэтил титаната (11 г, 48,2 ммоль). Полученную смесь нагревали для реакции рециркуляции на протяжении 4 часов. После завершения реакции полученную смесь охлаждали до комнатной температуры с последующим добавлением насыщенного раствора NH4Cl. Полученную смесь подвергали дистилляции с пониженным давлением для удаления THF. Полученную водную фазу экстрагировали с помощью EtOAc. Органические фазы комбинировали и затем промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и затем концентрировали для получения остатка. Остаток очищали колоночной хроматографией (PE:EtOAc (об/об)=1:1) для получения 4,2 г твердого вещества. Выход составил 45%. ЖХМС (ИЭ): масса/заряд=375 (М+Н)+.
Этап 7. (R)-N-((5)-1-(3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-ил)этил)-2-метилпропан-2-сульфенамид
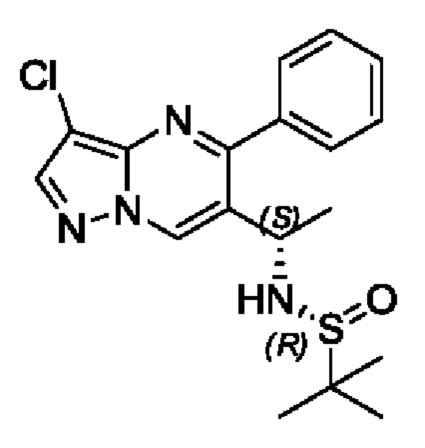
В атмосфере азота, (R,E)-N-(1-(3-хлор-5-фенилпиразоло[1,5-a]пиримидин-6-ил)этилдиен)-2-метилпропан-2-сульфенамид (4,2 г, 11,2 ммоль) растворяли в 40 мл THF, охлаждали до -40°С и поддерживали при -40°С с последующим добавлением по каплям раствора THF (1 М, 22 мл), содержащего три-втор-бутилборогидрид лития. После добавления по каплям, полученную смесь поддерживали при температуре для реакции на протяжении 1 часа. После завершения реакции добавляли насыщенный раствор NH4Cl и перемешивали на протяжении 5 минут. Полученную смесь нагревали до комнатной температуры, подвергали дистилляции с пониженным давлением для удаления THF. Полученную водную фазу экстрагировали с помощью EtOAc. Органические фазы комбинировали и затем промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и затем концентрировали для получения остатка. Остаток очищали колоночной хроматографией (РЕ:EtOAc (об/об)=1:1) для получения 2 г твердого вещества. Выход составил 47%. ЖХМС (ИЭ): масса/заряд=377 (М+Н)+.
Этап 8. (S)-1-(3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-ил)этиламин
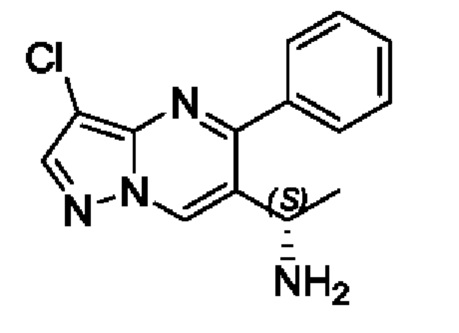
(R)-N-(S)-1-(3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-ил)этил)-2-метилпропан-2-сульфенамид (2 г, 5,3 ммоль) растворяли в 10 мл МеОН с последующим добавлением концентрированной хлористоводородной кислоты (4 мл) для реакции на протяжении 1 часа при перемешивании при комнатной температуре. После завершения реакции полученную смесь помещали и охлаждали в ледяной бане, регулировали путем использования насыщенного раствора Na2CO3 для получения значения рН 9 и подвергали дистилляции с пониженным давлением для удаления метанола. Полученную водную фазу экстрагировали с помощью ЕtOАс. Органические фазы комбинировали и затем промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и затем концентрировали для получения 1,2 г твердого вещества. Выход составил 83%. ЖХМС (ИЭ): масса/заряд=273 (М+Н)+.
Этап 9. (S)-4-амино-6-(1-(3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-ил)этиламино)пиримидин-5-формонитрил
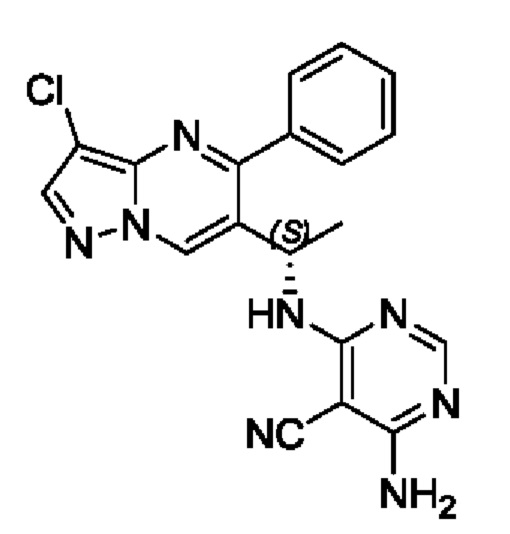
В 25 мл круглодонную колбу добавляли (S)-1-(3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-ил)этиламин (150 мг, 0,55 ммоль), 4-амино-5-нитрил-6-хлорпиримидин (85 мг, 0,55 ммоль), KF (64 мг, 1,1 ммоль), DIEA (355 мг, 2,75 ммоль) и DMSO (5 мл), и полученную смесь нагревали до 90°С для реакции на протяжении 2 часов. После завершения реакции полученную смесь разбавляли с помощью H2O и экстрагировали с помощью EtOAc. Органические фазы комбинировали и затем промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и затем концентрировали для получения остатка. Остаток очищали колоночной хроматографией (РЕ:EtOAc (об/об)=1:1) для получения 178 мг твердого вещества. Выход составил 83%. ЖХМС (ИЭ): масса/заряд=391 (М+Н)+ 1Н ЯМР (400 МГц, CDCl3) δ 8,69 (s, 1H), 8,03 (s, 1H), 8,01 (s, 1H), 7,84-7,69 (m, 1H), 7,66-7,55 (m, 3Н), 7,52-7,32 (m, 1H), 5,59 (d, J=6,2 Гц, 1H), 5,51 (s, 2Н), 5,26-5,14 (m, 1H), 1,57 (d, J=7,1 Гц, 3Н).
1Н ЯМР (400 МГц, CDCl3) δ 8,69 (s, 1H), 8,03 (s, 1H), 8,01 (s, 1H), 7,84-7,69 (m, 1H), 7,66-7,55 (m, 3Н), 7,52-7,32 (m, 1H), 5,59 (d, J=6,2 Гц, 1H), 5,51 (s, 2Н), 5,26-5,14 (m, 1H), 1,57 (d, J=7,1 Гц, 3Н).
Пример 2
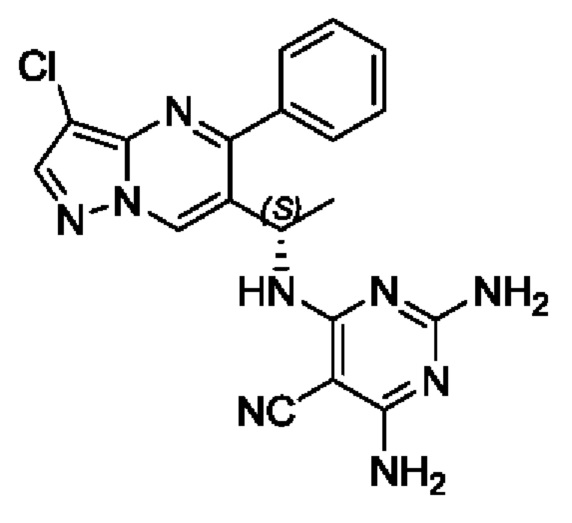
(S)-2,4-диамино-6-(1-(3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-ил)этиламино)пиримидин-5-формонитрил
Этап 1. 2,4,6-трихлорпиримидин-5-карбоксальдегид
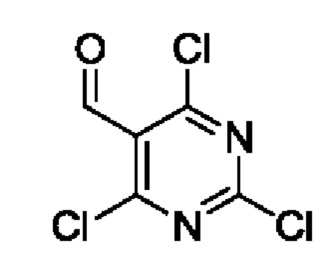
POCl3 (76,5 г, 0,5 ммоль) добавляли в 150 мл круглодонную колбу, а затем помещали и охлаждали в ледяной бане с последующим добавлением DMF (5,7 г, 78 ммоль) по каплям. Полученную смесь поддерживали при температуре для реакции на протяжении 20 минут. Барбитуровую кислоту (10 г, 78 ммоль) добавляли в верхний раствор и ледяную баню удаляли. Полученную смесь нагревали для реакции рециркуляции на протяжении 12 часов. После завершения реакции полученную смесь подвергали дистилляции с пониженным давлением для удаления оставшегося POCl3. Остаток добавляли в дробленый лед и экстрагировали с помощью ЕtOАс. Органические фазы комбинировали, промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4 и затем фильтровали и концентрировали для получения 10 г масляного вещества для прямого использования на следующем этапе реакции. Выход составил 61%.
Этап 2. 2,4,6-трихлорпиримидин-5-формонитрил
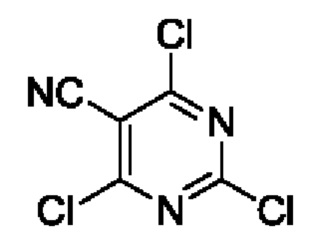
2,4,6-трихлорпиримидин-5-карбоксальдегид (10 г, 47,4 ммоль), гидроксиламина гидрохлорид (3,3 г, 47,5 ммоль), ледяную уксусную кислоту (100 мл) и Н2О (5 мл) добавляли в 150 мл круглодонную колбу и нагревали до 60°С для реагирования на протяжении 1 часа. После завершения реакции реакционный раствор выливали в дробленый лед и экстрагировали с помощью CH2Cl2. Полученные органические фазы комбинировали, промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, и затем фильтровали и концентрировали. Полученное вещество растворяли в SOCl2 (60 мл), вводили в реакцию при комнатной температуре на протяжении 10 минут, а затем нагревали для реакции рециркуляции на протяжении 2 часов. После завершения реакции полученную смесь подвергали дистилляции с пониженным давлением для удаления SOCl2 для получения остатка, который затем растворяли в ЕtOАс и промывали с помощью Н2О. Полученную органическую фазу сушили с помощью безводного Na2SO4, фильтровали и концентрировали для получения 9 г масляного вещества. Выход составил 92%.
Этап 3. 2,4-диамино-6-хлорпиримидин-5-формонитрил
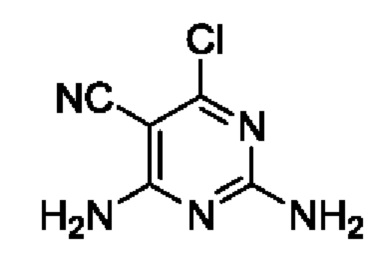
2,4,6-трихлорпиримидин-5-формонитрил (9 г, 43,5 ммоль), диоксан (40 мл) и гидроксид аммония (36%, 40 мл) добавляли в 150 мл круглодонную колбу и нагревали до 50°С для реагирования на протяжении 1 часа. После завершения реакции полученную смесь охлаждали до комнатной температуры с последующим добавлением 50 мл ледяной воды, и затем помещали и охлаждали в ледяной бане и перемешивали на протяжении 1,5 часов. Полученную смесь фильтровали для получения остатка на фильтре. Остаток на фильтре промывали с помощью H2O для получения 3,8 г легкого желтого твердого вещества. Выход составил 51%. ЖХМС (ИЭ): масса/заряд=170 (М+Н)+.
Этап 4.
(S)-2,4-диамино-6-(1-(3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-ил)этиламино)пиримидин-5-формонитрил
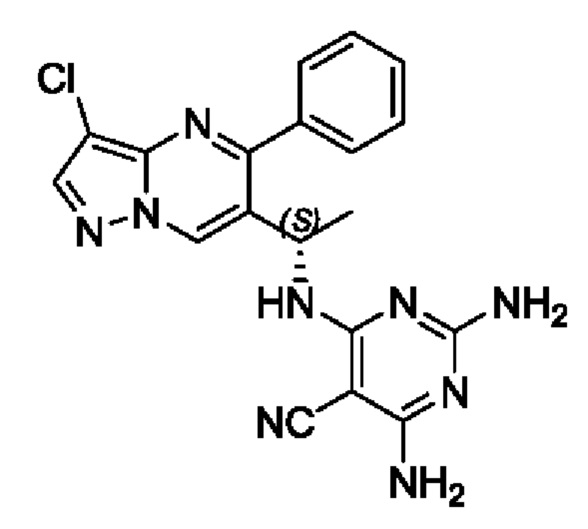
(S)-1-(3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-ил)этиламин (150 мг, 0,55 ммоль), 2,4-амино-6-хлорпиримидин-5-формонитрил (93 мг, 0,55 ммоль), KF (64 мг, 1,1 ммоль), DIEA (355 мг, 2,75 ммоль) и DMSO (5 мл) добавляли в 25 мл круглодонную колбу и нагревали до 90°С для реакции на протяжении 2 часов. После завершения реакции полученную смесь разбавляли с помощью H2O и экстрагировали с помощью ЕtOАс. Органические фазы комбинировали, промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и затем концентрировали для получения остатка. Остаток очищали колоночной хроматографией (РЕ:ЕtOАс (об/об)=1:1) для получения 156 мг твердого вещества. Выход составил 70%. ЖХМС (ИЭ): масса/заряд=406 (М+Н)+. 1H ЯМР (400 МГц, CDCl3) δ 8,71 (s, 1Н), 8,02 (s, 1H), 7,79-7,69 (m, 1H), 7,69-7,56 (m, 3H), 7,50-7,38 (m, 1H), 5,47 (d, J=6,7 Гц, 1H), 5,31-5,08 (m, 3H), 4,80 (s, 2H), 1,57 (d, J=7,1 Гц, 3Н).
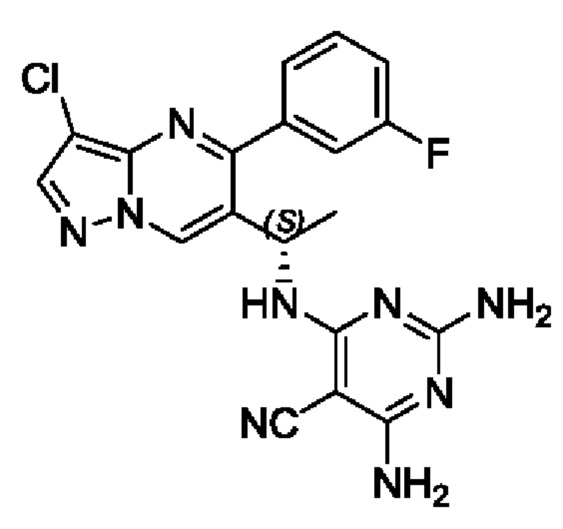
Пример 3
(S)-2,4-диамино-6-(1-(3-хлор-5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-ил)этиламино)пиримидин-5-формонитрил
Этап 1. этил 3-(3-фторфенил)-3-оксопропаноат
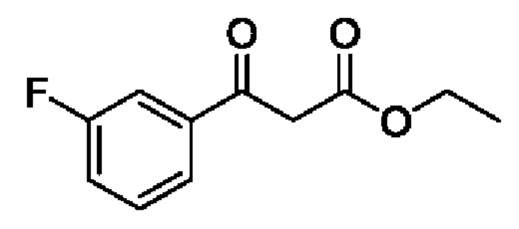
Трет-бутоксид калия (20 г, 178,6 ммоль) растворяли в 200 мл THF и охлаждали до -20°С с последующим добавлением этил 3-фторбензоата (10 г, 59,5 ммоль) по каплям, поддерживали при температуре и вводили в реакцию на протяжении 30 минут. Затем медленно по каплям добавляли этил ацетат (15,7 г, 178,4 ммоль), после чего полученную смесь поддерживали при температуре и вводили в реакцию на протяжении 1 часа. После завершения рекции добавляли водный раствор 3 N HCl (75 мл) и полученную смесь подвергали дистилляции с пониженным давлением для удаления THF. Полученную водную фазу экстрагировали с помощью ЕtOАс. Органические фазы комбинировали, промывали последовательно с помощью насыщенного раствора Na2CO3 и насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и концентрировали для получения 5 г маслянистого вещества для прямого использования на следующем этапе реакции. Выход составил 40%. ЖХМС (ИЭ): масса/заряд=211 (М+Н)+.
Этап 2. этил 5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-карбоксилат
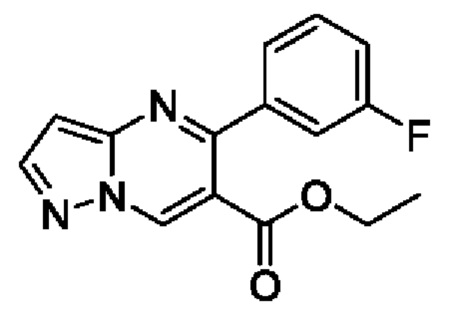
Этил 3-(3-фторфенил)-3-оксопропаноат (5 г, 23,8 ммоль), N,N-диметилформамида диметилацеталь (2,9 г, 24,4 ммоль) и 50 мл толуола добавляли в 250 мл круглодонную колбу для реакции рециркуляции на протяжении 2 часов. После завершения реакции полученную смесь подвергали дистилляции с пониженным давлением для удаления метилбензола. Полученный остаток растворяли в 100 мл этанола с последующим добавлением 3-аминопиразола (2 г, 24 ммоль) и 20 мл ледяной уксусной кислоты при перемешивании, и затем вводили в реакцию на протяжении 8 часов при комнатной температуре. После завершения реакции полученную смесь подвергали дистилляции с пониженным давлением для удаления этанола для получения остатка. Остаток разбавляли путем добавления H2O и затем экстрагировали с помощью CH2Cl2. Полученную органическую фазу промывали последовательно с помощью насыщенного раствора Na2CO3 и насыщенного раствора NaCl, сушили с помощью безводного Na2SO4 и затем фильтровали и концентрировали для получения 4,3 г желтого твердого вещества для прямого использования на следующем этапе реакции. Выход составил 63%. ЖХМС (ИЭ): масса/заряд=286 (М+Н)+.
Этап 3. этил 3-хлор-5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-карбоксилат
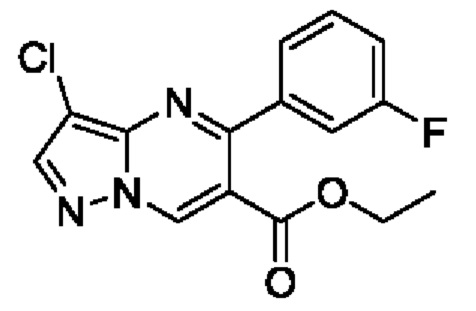
Этил 5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-карбоксилат (4,3 г, 15,1 ммоль) растворяли в 50 мл CHCl3 с последующим добавлением NCS (2,1 г, 15,7 ммоль). Полученную смесь нагревали до 60°С для реакции на протяжении 6 часов. После завершения реакции полученную смесь разбавляли с помощью CH2Cl2 и затем промывали последовательно с помощью водного раствора 0,1 N HCl и насыщенного раствора NaCl. Полученную органическую фазу сушили с помощью безводного Na2SO4, а также фильтровали и концентрировали для получения 4,6 г желтого твердого вещества для прямого использования на следующем этапе реакции. Выход составил 95%. ЖХМС (ИЭ): масса/заряд=320 (М+Н)+.
Этап 4. 3-хлор-5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-карбоновая кислота
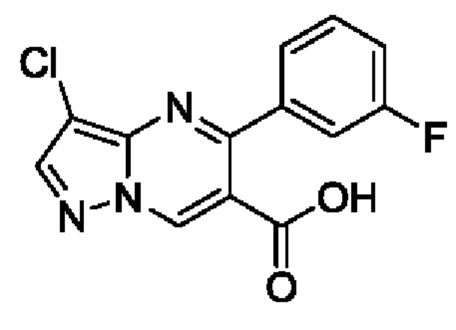
Этил 3-хлор-5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-карбоксилат (4,6 г, 14,4 ммоль), 20 мл метанола и 60 мл водного раствора 2 N NaOH добавляли в 100 мл круглодонную колбу для реакции при комнатной температуре на протяжении 5 часов. После завершения реакции полученную смесь подвергали дистилляции с пониженным давлением для удаления метанола, а затем фильтровали для получения остатка на фильтре и фильтрата. Остаток на фильтре промывали с помощью Н2О. Фильтрат регулировали с использованием раствора 4 N HCl до получения значения рН 3 для осаждения твердого вещества и затем фильтровали для получения 2,3 г твердого вещества. Выход составил 55%. ЖХМС (ИЭ): масса/заряд=292 (М+Н)+.
Этап 5. 3-хлор-5-(3-фторфенил)-N-метокси-N-метилпиразоло[1,5-а]пиримидин-6-амид
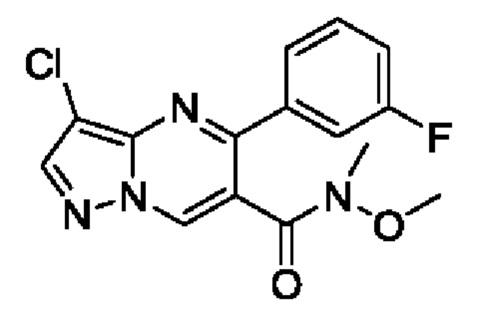
3-хлор-5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-карбоновую кислоту (2,3 г, 7,9 ммоль), и DIEA (3 г, 23,3 ммоль) растворяли в 20 мл CH2Cl2 с последующим добавлением N,O-диметилгидкросиламин гидрохлорида (850 мг, 8,7 ммоль) и HATU (3,3 г, 8,7 ммоль) при перемешивании для реакции при комнатной температуре на протяжении 2 часов. После завершения реакции полученную смесь разбавляли с помощью CH2Cl2. Полученную органическую фазу промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и концентрировали для получения остатка. Остаток очищали колоночной хроматографией (РЕ:EtOAc (об/об)=2:1) для получения 2 г твердого вещества. Выход составил 76%. ЖХМС (ИЭ): масса/заряд=335 (М+Н)+.
Этап 6. 1-(3-хлор-5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-ил)этанон
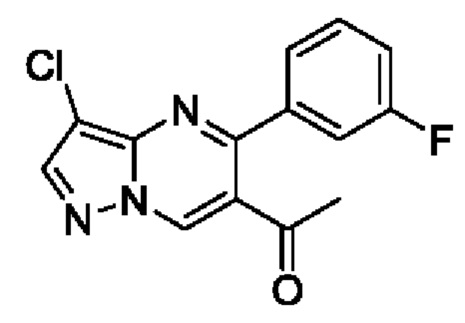
В атмосфере азота, 3-хлор-5-(3-фторфенил)-N-метокси-N-метилпиразоло[1,5-а]пиримидин-6-амид (2 г, 6 ммоль) растворяли в 20 мл THF, а затем помещали и охлаждали в ледяной бане. Раствор метилмагний хлорида в THF (3М, 3 мл) добавляли медленно по каплям в верхний раствор и добавление завершали через 20 минут. После завершения добавления по каплям полученную смесь поддерживали при температуре и вводили в реакцию на протяжении 1 часа. После завершения реакции в полученную смесь аккуратно добавляли насыщенный раствор NH4Cl для гашения реакции, и полученную смесь подвергали дистилляции с пониженным давлением для удаления THF. Полученную водную фазу экстрагировали с помощью EtOAc. Органические фазы комбинировали и затем промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и затем концентрировали для получения 1,5 г твердого вещества. Выход составил 87%. ЖХМС (ИЭ): масса/заряд=290 (М+Н)+.
Этап 7. (R,E)-N-(1-(3-хлор-5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-ил)этилдиен)-2-метилпропан-2-сульфенамид
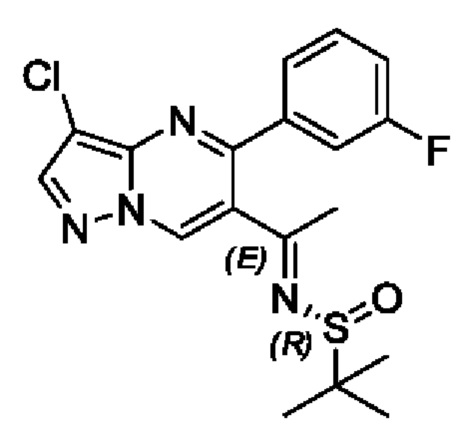
(R)-трет-бутилсульфенамид (692 мг, 5,7 ммоль) растворяли в 10 мл THF с последующим добавлением по каплям 1-(3-хлор-5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-ил)этанона (1,5 г, 5,2 ммоль) и раствора тетраэтил титаната (2,4 г, 10,5 ммоль) в THF (20 мл). Полученную смесь нагревали для реакции рециркуляции на протяжении 4 часов. После завершения реакции полученную смесь охлаждали до комнатной температуры с последующим добавлением насыщенного раствора NH4Cl. Полученную смесь подвергали дистилляции с пониженным давлением для удаления THF. Полученную водную фазу экстрагировали с помощью EtOAc. Органические фазы комбинировали и затем промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и затем концентрировали для получения остатка. Остаток очищали колоночной хроматографией (РЕ:EtOAc (об/об)=1:1) для получения 840 мг твердого вещества. Выход составил 41%. ЖХМС (ИЭ): масса/заряд=393 (М+Н)+.
Этап 8. (R)-N-((S)-1-(3-хлор-5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-ил)этил)-2-метилпропан-2-сульфенамид
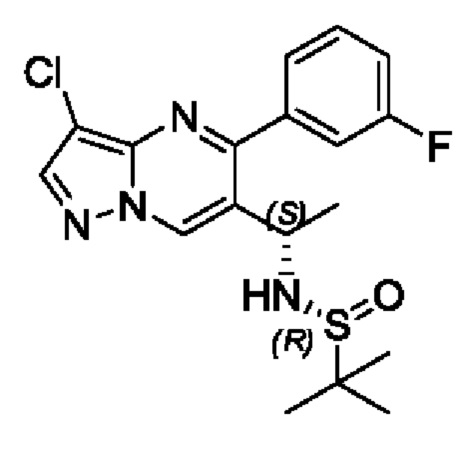
В атмосфере азота, (R,E)-N-(1-(3-хлор-5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-ил)этилдиен)-2 -метилпропан-2-сульфенамид (840 мг, 2,14 ммоль) растворяли в 10 мл THF и охлаждали до -40°С. Полученную смесь поддерживали при температуре с последующим добавлением по каплям раствора три-втор-бутилборогидрида лития в THF (1 М, 4,3 мл). После добавления по каплям, полученную смесь поддерживали при температуре для реакции на протяжении 1 часа. После завершения реакции добавляли насыщенный раствор NH4Cl и перемешивали на протяжении 5 минут. Полученную смесь нагревали до комнатной температуры, подвергали дистилляции с пониженным давлением для удаления THF. Полученную водную фазу экстрагировали с помощью EtOAc. Органические фазы комбинировали и затем промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и затем концентрировали для получения остатка. Остаток очищали колоночной хроматографией (РЕ:EtOAc (об/об)=1:1) для получения 600 мг твердого вещества. Выход составил 71%. ЖХМС (ИЭ): масса/заряд=395 (М+Н)+.
Этап 9. (S)-1-(3-хлор-5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-ил)этиламин
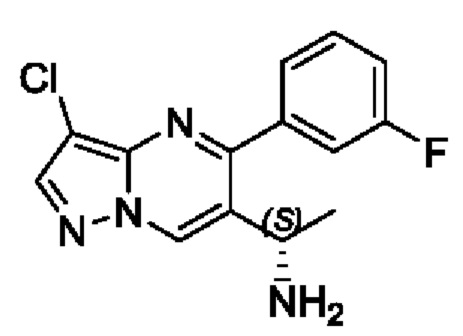
(R)-N-(S)-1-(3-хлор-5-(3-фторфенил)пиразоло[1,5-а]иримидин-6-ил)этил)-2-метилпропан-2-сульфенамид (600 мг, 1,52 ммоль) растворяли в 5 мл МеОН с последующим добавлением концентрированной хлористоводородной кислоты (2 мл) для реакции на протяжении 1 часа при перемешивании при комнатной температуре. После завершения реакции полученную смесь помещали и охлаждали в ледяной бане, регулировали путем использования насыщенного раствора Na2CO3 для получения значения рН 9 и подвергали дистилляции с пониженным давлением для удаления метанола. Полученную водную фазу экстрагировали с помощью EtOAc. Органические фазы комбинировали и затем промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и затем концентрировали для получения 400 мг твердого вещества. Выход составил 90%. ЖХМС (ИЭ): масса/заряд=291 (М+Н)+.
Этап 10.
(S)-2,4-диамино-6-(1-(3-хлор-5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-ил)этиламино)пиримидин-5-формонитрил
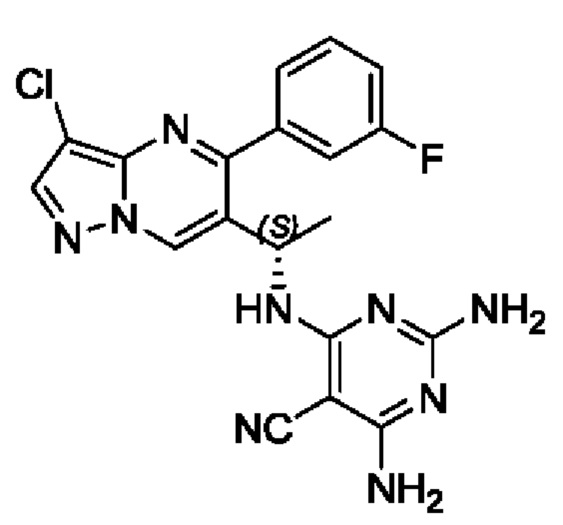
(S)-1-(3-хлор-5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-ил)этиламин (100 мг, 0,34 ммоль), 2,4-диамино-5-циано-6-хлорпиримидин (58 мг, 0,34 ммоль), KF (40 мг, 0,69 ммоль), DIEA (219 мг, 1,7 ммоль) и DMSO (5 мл) добавляли в 25 мл круглодонную колбу и нагревали до 90°С для реакции на протяжении 2 часов. После завершения реакции полученную смесь разбавляли с помощью Н2О и экстрагировали с помощью EtOAc. Органические фазы комбинировали и затем промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и затем концентрировали для получения остатка. Остаток очищали колоночной хроматографией (РЕ:EtOAc (об/об)=1:1) для получения 70 мг твердого вещества. Выход составил 51%. ЖХМС (ИЭ): масса/заряд=406 (М+Н)+ 1H ЯМР (400 МГц, CDCl3) δ 8,70 (s, 1Н), 8,03 (s, 1H), 7,72-7,52 (m, 2H), 7,45-7,28 (m, 2H), 5,44-5,29 (m, 1H), 5,26-5,13 (m, 1H), 5,09 (s, 2H), 4,78 (s, 2H), 1,55 (d, J=7,0 Гц, 3Н).
Пример 4
(S)-4-амино-6-(1-(3-хлор-5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-ил)этиламино)пиримидин-5-формонитрил
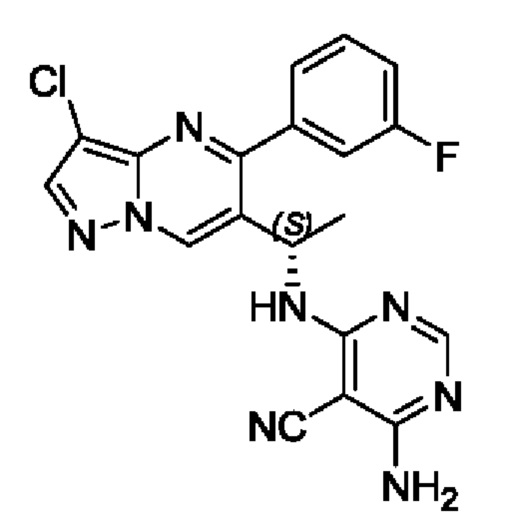
(S)-1-(3-хлор-5-(3-фторфенил)пиразоло[1,5-а]пиримидин-6-ил)этиламин (100 мг, 0,34 ммоль), 4-амино-5-циано-6-хлорпиримидин (53 мг, 0,34 ммоль), KF (40 мг, 0,69 ммоль), DIEA (219 мг, 1,7 ммоль) и DMSO (5 мл) добавляли в 25 мл круглодонную колбу и нагревали до 90°С для реакции на протяжении 2 часов. После завершения реакции полученную смесь разбавляли с помощью H2O и экстрагировали с помощью EtOAc. Органические фазы комбинировали и затем промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и затем концентрировали для получения остатка. Остаток очищали колоночной хроматографией (РЕ:EtOAc (об/об)=1:1) для получения 50 мг твердого вещества. Выход составил 36%. ЖХМС (ИЭ): масса/заряд=409 (М+Н)+ 1Н ЯМР (400 МГц, CDCl3) δ 8,67 (s, 1H), 8,10-7,98 (m, 2Н), 7,68-7,54 (m, 2Н), 7,42-7,28 (m, 2Н), 5,54 (d, J=6,0 Гц, 1Н), 5,41 (s, 2H), 5,22-5,10 (m, 1H), 1,57 (d, J=7,1 Гц, 3Н).
Пример 5
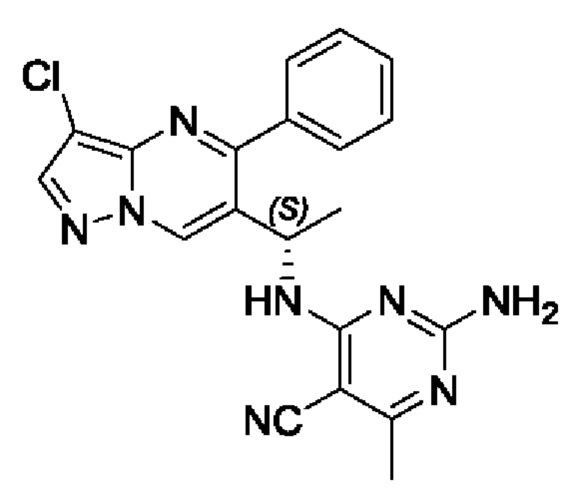
(S)-2-амино-4-((1-(3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-ил)этил)амин)-6-метилпиримидин-5-формонитрил
Этап 1. 4-хлор-5-йод-6-метилпиримидин-2-амин
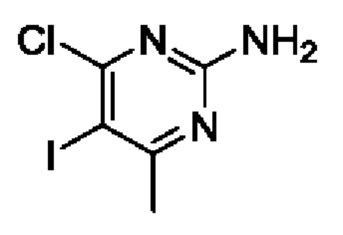
4-хлор-6-метилпиримидин-2-амин (3 г, 21,0 ммоль) растворяли в 40 мл ледяной уксусной кислоты в 100 мл круглодонной колбе, а затем помещали в ледяную баню и охлаждали до ниже 10°С с последующим добавлением NIS (2,4 г, 10,6 ммоль) в верхний раствор для реакции. После течения реакции на протяжении 1 часа, дополнительно добавляли NIS (2,4 г, 10,6 ммоль). После течения реакции на протяжении 1 часа, полученные продукты оставляли нагреваться естественным образом до комнатной температуры и вводили в реакцию на протяжении 8 часов. После завершения реакции раствор реагента выливали в ледяную воду и экстрагировали с помощью EtOAc. Полученную органическую фазу промывали последовательно с помощью 5% раствора Na2SO3, 10% раствора NaHCO3 и насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали, а затем подвергали концентрации с пониженным давлением для удаления растворителя для получения 4,9 г твердого вещества, которое представляло собой обозначенное соединение. Выход составил 87%. ЖХМС (ИЭ): масса/заряд=270 (М+Н)+.
Этап 2. 2-амино-4-хлор-6-метилпиримидин-5-формонитрил
В атмосфере азота, 4-хлор-5-йод-6-метилпиримидин-2-амин (4,9 г, 18,2 ммоль), Zn(CN)2 (4,3 г, 36,6 ммоль), Pd(PPh3)4 (2,1 г, 1,8 ммоль), CuI (1,7 г, 8,9 ммоль) и DMF (60 мл) добавляли в 150 мл круглодонную колбу, нагревали до 80°С для реакции на протяжении 16 часов. После завершения реакции полученную смесь разбавляли ЕtOАс и фильтровали через целит для получения остатка на фильтре и фильтрата. Остаток на фильтре промывали с помощью EtOAc. В фильтрат добавляли H2O и фильтрат экстрагировали с помощью EtOAc. Полученную органическую фазу промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтрвали, а затем концентрировали для получения остатка, который очищали колоночной хроматографией (РЕ:EtOAc (об/об)=5:1) для получения 1,1 г твердого вещества. Выход составил 36%. ЖХМС (ИЭ): масса/заряд=169 (М+Н)+.
Этап 3.
(S)-2-амино-4-((1-(3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-ил)этил)амин)-6-метилпиримидин-5-формонитрил
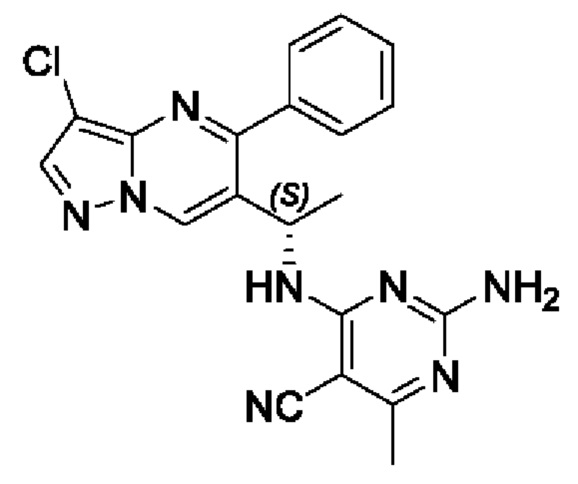
(S)-1-(3-хлор-5-фенилпиразоло[1,5-а]пиримидин-6-ил)этиламин (100 мг, 0,37 ммоль), 2-амино-4-хлор-6-метилпиримидин-5-формонитрил (62 мг, 0,37 ммоль), KF (43 мг, 0,74 ммоль), DIEA (232 мг, 1,8 ммоль) и DMSO (5 мл) добавляли в 25 мл круглодонную колбу и нагревали до 90°С для реакции на протяжении 2 часов. После завершения реакции полученную смесь разбавляли с помощью H2O и экстрагировали с помощью EtOAc. Органические фазы комбинировали и затем промывали с помощью насыщенного раствора NaCl, сушили с помощью безводного Na2SO4, фильтровали и затем концентрировали для получения остатка. Остаток очищали колоночной хроматографией (РЕ:EtOAc (об/об)=1:1) для получения 102 мг твердого вещества. Выход составил 68%. ЖХМС (ИЭ): масса/заряд=405 (М+Н)+ 1H ЯМР (400 МГц, CDCl3) δ 8,70 (s, 1H), 8,03 (s, 1H), 7,79-7,31 (m, 5Н), 5,76 (d, J=6,7 Гц, 1Н), 5,66-4,71 (m, 3Н), 2,39 (s, 3Н), 1,63 (d, J=7,1 Гц, 3Н).
Применение соединений по настоящей заявке для регулирования активности киназы и ингибирования пролиферации клеток протестировано методами, описанными ниже.
Пример А: Анализ активности киназы PI3K
Активность киназы PI3Kδ тестировали методом ADP-Glo. ADP-Glo получали от Promega (#V9101). p110δ/р85α получали от Millipore (#14-604-K). IC50 каждого из соединений оценивали путем тестирования соединения при 10 концентрациях. Исходная концентрация составляла 1 мкМ, а затем выполняли 3-кратное серийное разведение. Буферными растворами для тестирования были 50 мМ HEPES рН 7,5, 3 мМ MgCl2, 1 мМ EGTA, 100 мМ NaCl, 0,03% CHAPS и 2 мМ DTT. Конечная концентрация киназы PI3Kδ составила 17 нМ; конечная концентрация PIP2 составила 50 мкМ; и конечная концентрация АТР составила 25 мкМ.
1. Получение 10 мкл реакционного раствора киназы: 2,5 мкл раствора тестируемого соединения, 2,5 мкл раствора киназы и 5 мкл раствора субстрата добавляли в каждую лунку 384-луночного планшета (Зернение #4512).
2. 384-луночный планшет закрывали и инкубировали при комнатной температуре на протяжении 60 минут.
3. 5 мкл реакционного раствора переносили из каждой лунки в лунки нового 384-луночного планшета.
4. 5 мкл реагента ADP-Glo добавляли в лунки 384-луночного планшета для прекращения реакции.
5. 384-луночный планшет осторожно встряхивали на приборе для встряхивания.
6. 10 мкл реагента для обнаружения киназы добавляли в каждую лунку с последующим встряхиванием на протяжении 1 минуты и позволяли выстояться при комнатной температуре на протяжении 30 минут.
8. Значения люминесцентности образца считывали с помощью Synegy.
9. Данные обрабатывали с помощью Excel и к ним подбирали кривые с помощью программы XLFit для получения значения IC50 каждого из соединений (Таблица 1).
Пример В. Анализ ингибирования активности клеток соединений
Метод тестирования CellTiter-Glo использовали для тестирования ингибирования пролиферации клетки лимфомы SU-DHL-6 (АТСС, Номер по каталогу: CRL-2959) соединениями. Раствором клеточной культуры, использованным при тесте, был RPMI1640 (Invitrogen, Номер по каталогу: 11875-093). 10% фетальной телячьей сыворотки (Invitrogen, Номер по каталогу: 10099-141) использовали при тесте.
100 мкл культурального раствора, содержащего 15000 клеток, распределяли в лунки 96-луночного планшета (Зернение #3903). Планшет помещали в инкубатор с углекислым газом на ночь. 0,5 мкл соединения (которое получали с помощью DMSO для обеспечения восьми непрерывных градиентов концентраций), подлежащего тестированию, добавляли в каждую из лунок на следующий день. Каждую концентрацию устанавливали с помощью двух репликатов, а также лунки без клетки (бланковая проба) и лунки с DMSO (контроль растворителем). После дозирования лунки культивировали при 37°С в атмосфере 5% СO2 на протяжении 72 часов. Наконец, 100 мкл агента CellTiter-Glo (Promega, Номер по каталогу: G7571) добавляли в каждую из лунок. Сигнал люминесценции обнаруживали с помощью Flex Station3 (Molecular Devices), а значение IC50 каждого из соединений, ингибирующего пролиферацию клеток, вычисляли с помощью программы XLfit (Таблица 1).
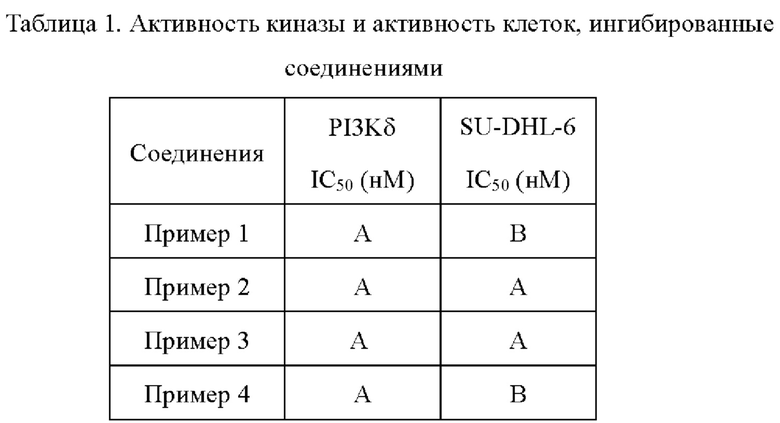
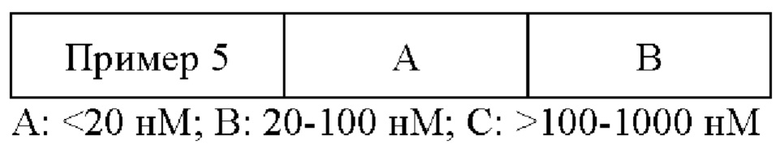
Как можно увидеть из Таблицы 1, соединения по настоящей заявке обладают превосходной способностью к ингибированию активности киназы PI3Kδ. Соединения по настоящей заявке также способны к эффективному ингибированию пролиферации клеток лимфомы.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ АМИНОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2014 |
|
RU2675105C9 |
| ЗАМЕЩЕННЫЕ АМИНОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2014 |
|
RU2683793C2 |
| ПИРАЗОЛПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2016 |
|
RU2735522C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2811612C2 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2829484C2 |
| Ингибитор ДНК-зависимой протеинкиназы | 2020 |
|
RU2835434C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| ГЕТЕРОАРИЛЬНОЕ ПРОИЗВОДНОЕ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С PI3 КИНАЗАМИ, СОДЕРЖАЩАЯ ДАННОЕ ДЕЙСТВУЮЩЕЕ ВЕЩЕСТВО | 2016 |
|
RU2719367C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЕСПЕЧИВАЮЩИЕ РАЗРУШЕНИЕ БЕЛКА IKAROS И БЕЛКА AIOLOS | 2020 |
|
RU2833608C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
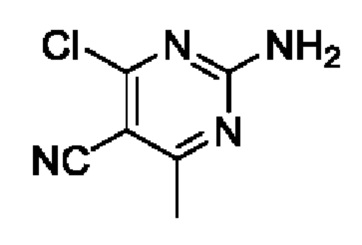
Изобретение относится к области органической химии, а именно к соединению Формулы (I):
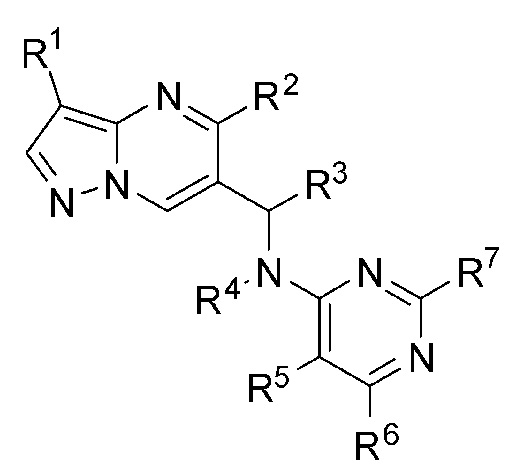 , где R1 является галогеном; R2 является фенилом, причем R2 необязательно замещен R2a от одного до пяти;R2a выбран из H или галогена;R3 является С1-4 алкилом;R4 является Н;R5 является циано;R6 выбран из амино или алкила, а R7 выбран H или амино; причем любой атом водорода в соединении Формулы (I) может представлять собой дейтерий. Также предложена фармацевтическая композиция, применение и способ получения соединения формулы (I). Технический результат: получены новые соединения, которые могут быть использованы для ингибирования активности липидной киназы PI3K. 4 н. и 5 з.п. ф-лы, 1 табл., 7 пр.
, где R1 является галогеном; R2 является фенилом, причем R2 необязательно замещен R2a от одного до пяти;R2a выбран из H или галогена;R3 является С1-4 алкилом;R4 является Н;R5 является циано;R6 выбран из амино или алкила, а R7 выбран H или амино; причем любой атом водорода в соединении Формулы (I) может представлять собой дейтерий. Также предложена фармацевтическая композиция, применение и способ получения соединения формулы (I). Технический результат: получены новые соединения, которые могут быть использованы для ингибирования активности липидной киназы PI3K. 4 н. и 5 з.п. ф-лы, 1 табл., 7 пр.
1. Соединение Формулы (I):
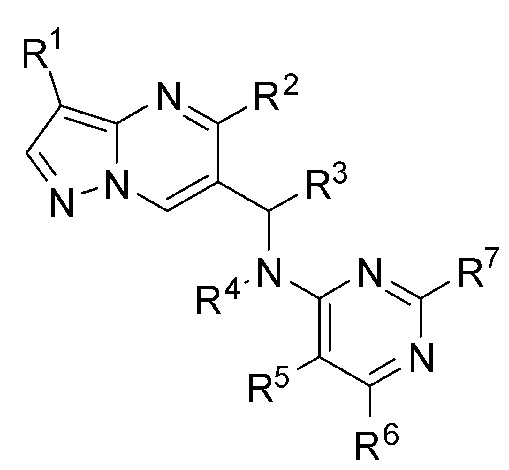
(I)
отличающееся тем, что:
R1 является галогеном;
R2 является фенилом, причем R2 необязательно замещен R2a от одного до пяти;
R2a выбран из H или галогена;
R3 является С1-4 алкилом;
R4 является Н;
R5 является циано;
R6 выбран из амино или алкила, а R7 выбран из H или амино;
причем любой атом водорода в соединении Формулы (I) может представлять собой дейтерий.
2. Соединение по п. 1, отличающееся тем, что соединение имеет структуру Формулы (II):
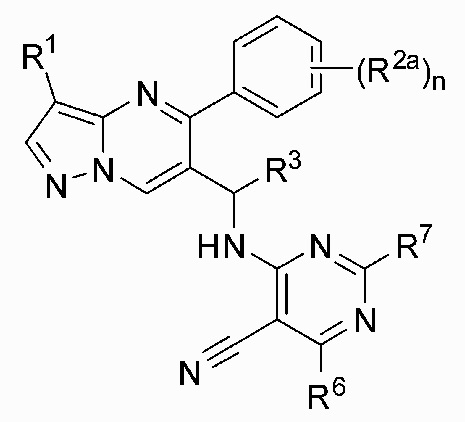
(II)
причем:
R1 является хлором;
R2a выбран из H или галогена;
R3 является метилом;
R6 выбран из амино или метила, а R7 выбран из H или амино;
n равняется 1, 2 или 3.
3. Соединение по любому из пп. 1, 2, отличающееся тем, что соединение выбрано из группы, состоящей из:
(S)-4-амино-6-(1-(3-хлор-5-фенилпиразоло[1,5-a]пиримидин-6-ил)этиламино)пиримидин-5-карбонитрил, (S)-2,4-диамин-6-(1-(3-хлор-5-фенилпиразоло[1,5-a]пиримидин-6-ил)этиламино)пиримидин-5-карбонитрил, (S)-2,4-диамино-6-(1-(3-хлор-5-(3-фторфенил)пиразоло[1,5-a]пиримидин-6-ил)этиламино)пиримидин-5-карбонитрил, (S)-4-амино-6-(1-(3-хлор-5-(3-фторфенил)пиразоло[1,5-a]пиримидин-6-ил)этиламино)пиримидин-5-карбонитрил, (S)-2-амино-4-((1-(3-хлор-5-фенилпиразоло[1,5-a]пиримидин-6-ил)этил)амино)-6-метилпиримидин-5-карбонитрил.
4. Фармацевтическая композиция, обладающая свойствами ингибитора киназы PI3K, содержащая в эффективном количестве соединение по любому из пп. 1-3 и по меньшей мере один фармацевтический носитель или вспомогательное вещество.
5. Применение соединения по любому из пп. 1-3 или фармацевтической композиции по п. 4, при получении эффективного количества лекарственного средства для лечения связанных с PI3K заболеваний.
6. Применение по п. 5, отличающееся тем, что связанные с PI3K заболевания выбраны из группы, состоящей из рака, воспалительных заболеваний и аутоиммунных заболеваний.
7. Применение по п. 6, отличающееся тем, что рак выбран из группы, состоящей из острого лимфоцитарного лейкоза, острого миелоидного лейкоза, хронического лимфоцитарного лейкоза, хронического миелоидного лейкоза, лейкоза Ходжкина, неходжкинского лейкоза, мантийноклеточного лейкоза, фолликулярного лейкоза, B-клеточной лимфомы, T-клеточной лимфомы и диффузной В-крупноклеточной лимфомы.
8. Применение по п. 6, отличающееся тем, что аутоиммунные заболевания и воспалительные заболевания выбраны из группы, состоящей из дерматита, ревматоидного артрита, аллергического ринита, астмы, болезни Крона, хронической обструктивной болезни легких, системной красной волчанки, псориаза, множественного склероза, синдрома активированной PI3Kδ и синдрома Шегрена.
9. Способ получения соединения по любому из пп. 1-3, включающий следующие этапы:
1) подвергают исходный материал I-1 и N,N-диметилформамида диметилацеталя реакции рециркуляции в толуоле для получения соединения I-2;
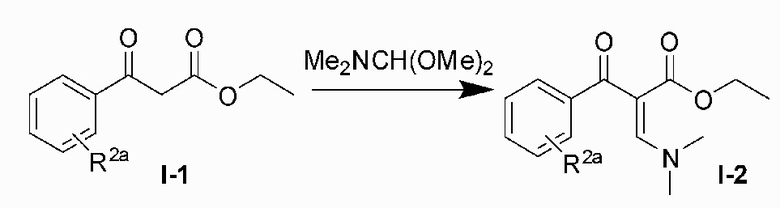
2) подвергают соединение I-2 и 3-аминопиразола реакции в уксусной кислоте для получения продукта циклизации пиразолопиримидина I-3;
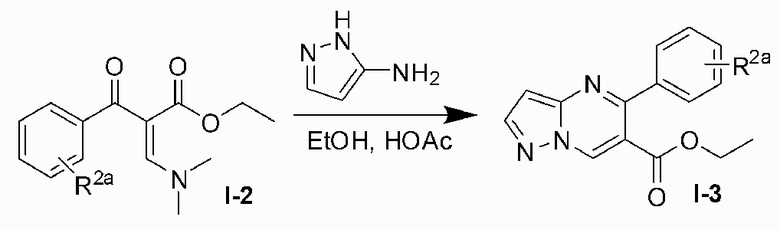
3) хлорируют пиразолопиримидина I-3 с NCS для получения хлорированного продукта I-4;
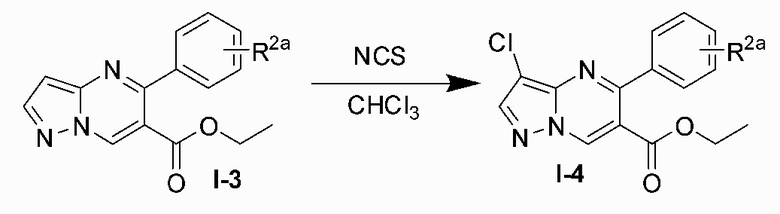
4) гидролизуют сложноэфирной группы хлорированного продукта I-4 с помощью щелочи для образования карбоновой кислоты I-5;
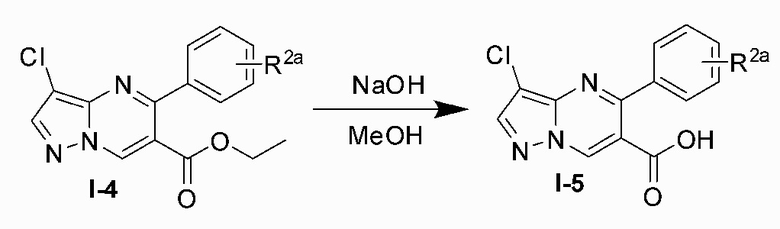
5) подвергают карбоновую кислоту I-5 и N,O-диметилгидроксиламина реакции конденсации для получения амидного соединения I-6;
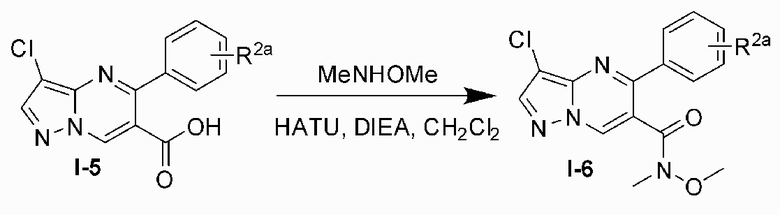
6) подвергают реактив Гриньяра R3MgCl и амидной связи в амидном соединении I-6 реакции присоединения для образования кетона I-7;
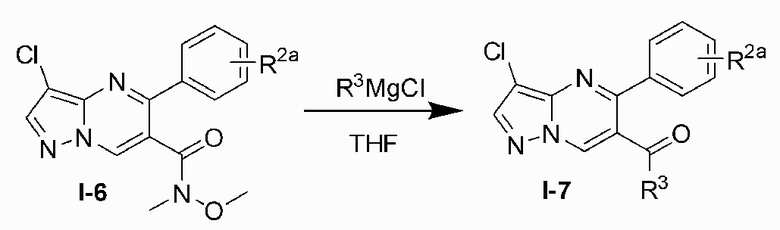
7) подвергают кетон I-7 и (R)-сульфенамида реакции конденсации для получения соединения I-8;
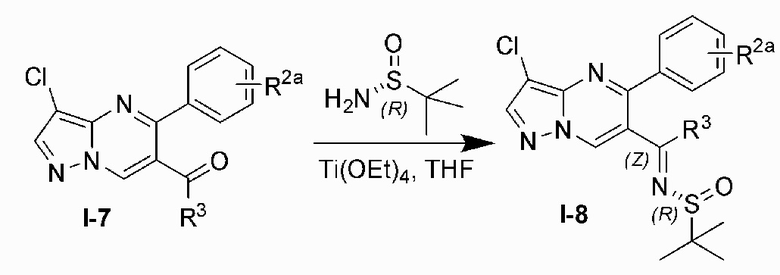
8) восстанавливают иминогруппу в соединении I-8 с помощью три-втор-бутил борогидрида лития для получения соединения I-9 сульфонамида S-конфигурации;
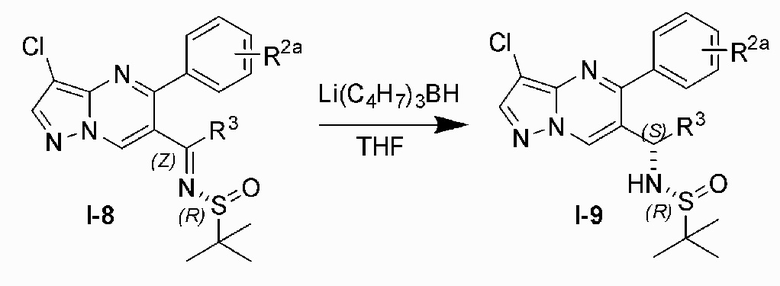
9) удаляют сульфинил в соединении I-9 с помощью хлористоводородной кислоты для получения свободного амина I-10; и
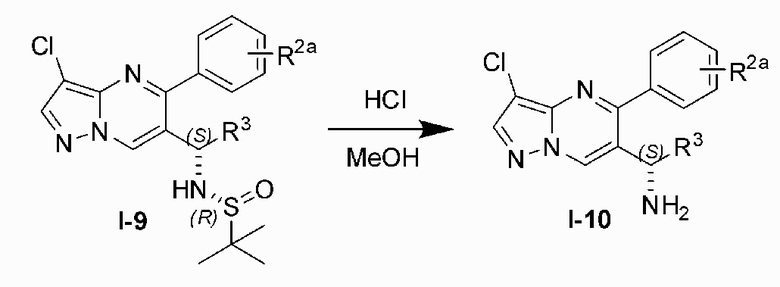
10) подвергают свободный амин I-10 и Y-L (Y представляет собой пиримидин, замещенный на R5, R6 и R7, а L представляет собой отходящую группу) реакции для получения конечного продукта (I).
| WO 2007044401 A2, 19.04.2007 | |||
| WO 2004022561 А1, 10.07.2011 | |||
| WO 200123387 А1, 31.10.2002 | |||
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИМИДИНА, КАК ИНГИБИТОРЫ ЦИКЛИН-ЗАВИСИМОЙ КИНАЗЫ | 2005 |
|
RU2414472C9 |

