Перекрестная ссылка на родственные заявки
Эта не являющаяся предварительной заявка, поданная в соответствии с §1.53(b) п.37 Свода федеральных правил (CFR), испрашивает, согласно §119(e) главы 35 Свода законов США (USC), приоритет на основании предварительной заявки США с серийным № 62/188029, поданной 2 июля 2015 г., которая включена посредством ссылки во всей своей полноте.
Область изобретения
В целом данное изобретение относится к соединениям бензоксазепиноксазолидинонов с активностью, направленной против гиперпролиферативных расстройств, таких как рак. Изобретение также относится к способам применения этих соединений для диагностики или обработки in vitro, in situ и in vivo клеток млекопитающих или лечения ассоциированных патологических состояний.
Предшествующий уровень техники
Активация опосредованного фосфоинозитид-3-киназой(PI3K)/Akt (протеинкиназа B) сигнального пути является общим признаком большинства раковых заболеваний (Yuan and Cantley (2008) Oncogene, 27: 5497-510). Генетические отклонения, касающиеся данного пути, были обнаружены во многих случаях рака у человека (Osaka et al. (2004) Apoptosis, 9: 667-76), и их влияние преимущественно связано со стимулированием пролиферации, миграции и выживаемости клеток. Активация данного пути происходит вследствие активирующих точечных мутаций или амплификаций гена PIK3CA (от англ. phosphatidylinositol-3-kinase, catalytic subunit alpha - каталитическая альфа-субъединица фосфатидилинозитол-3-киназы), кодирующего p110a (альфа) изоформ PI3K (Hennessy et al. (2005) Nat. Rev. Drug Discov., 4: 988-1004). Генетическая делеция или приводящие к потере функциональности мутации в гене онкосупрессора PTEN (от англ. phosphatase and tensin homolog - гомолог фосфатазы и тензина), фосфатазы с функцией, противоположной PI3K, также усиливает PI3K-опосредованный путь передачи сигнала (Zhang and Yu (2010) Clin. Cancer Res., 16: 4325-30). Эти нарушения приводят к усилению последующих звеньев передачи сигнала с участием таких киназ, как Akt и mTOR (мишень рапамицина у млекопитающих), и высказано предположение, что повышение активности PI3K-пути является признаком резистентности к лечению рака (Opel et al. (2007) Cancer Res., 67: 735-45; Razis et al. (2011) Breast Cancer Res. Treat., 128: 447-56).
Фосфатидилинозитол-3-киназа (PI3K) является основным узлом пути передачи сигнала для ключевых сигналов, связанных с выживаемостью и ростом в случае лимфом, и она обладает активностью, противоположной активности фосфатазы PTEN. Зависимый от фосфоинозитид-3-киназы (PI3K) сигнальный путь является самым разрегулированным путем в случае положительного в отношении рецепторов гормонов рака молочной железы (HR+ BC). PI3K-путь также разрегулирован при агрессивных формах лимфомы (Abubaker (2007) Leukemia, 21: 2368-2370). В восьми процентах случаев рака типа DLBCL (диффузной B-клеточной крупноклеточной лимфомы) имеются миссенс-мутации в гене PIK3CA (каталитической альфа-субъединицы фосфатидилинозитол-3-киназы), а 37% случаев по данным иммуногистохимического тестирования являются PTEN-отрицательными.
Фосфатидилинозит является одним из ряда фосфолипидов, обнаруженных в клеточных мембранах и принимающих участие во внутриклеточной передаче сигнала. Передача сигнала в клетках, опосредованная 3'-фосфорилированными фосфоинозитидами, вовлечена в целый ряд клеточных процессов, например, малигнизацию, передачу сигнала с участием ростовых факторов, воспаление и иммунитет (Rameh et al. (1999) J. Biol. Chem., 274: 8347-8350). Фермент, отвечающий за образование таких фосфорилированных продуктов сигнального пути, фосфатидилинозитол-3-киназу (также обозначаемую как PI 3-киназа или PI3K), изначально идентифицировали по активности, ассоциированной с вирусными онкобелками и тирозинкиназами, являющимися рецепторами ростовых факторов, которые фосфорилируют фосфатидилинозит (PI) и его фосфорилированные производные по 3'-гидроксилу кольца инозита (Panayotou et al. (1992) Trends Cell Biol., 2: 358-60). Фосфоинозитид-3-киназы (PI3K) представляют собой липидные киназы, которые фосфорилируют липиды по 3-гидроксильному остатку кольца инозита (Whitman et al. (1988) Nature, 332: 664). 3http://www.piramed.com/science_fig1.html-Фосфорилированные фосфолипиды (PIP3 (фосфатидилинозит-(3,4,5)-трифосфат)), образованные с помощью PI3-киназ, действуют в качестве вторичных мессенджеров, вовлекающих киназы с липид-связывающими доменами (включая плекстрин-гомологичные (PH) участки), такие как Akt и фосфоинозитид-зависимая киназа-1 (PDK1) (Vivanco et al. (2002) Nature Rev. Cancer, 2: 489; Phillips et al. (1998) Cancer, 83: 41).
Семейство PI3-киназ содержит по меньшей мере 15 разных ферментов, систематизированных по структурной гомологии и разделенных на 3 класса на основе гомологии последовательностей и продукта, образующегося в результате ферментативного катализа. PI3-киназы I класса состоят из 2 субъединиц: каталитической субъединицы с молекулярной массой (ММ) 110 килодальтон (кДа) и регуляторной субъединицы с ММ 85 кДа. Регуляторные субъединицы содержат SH2-домены (src-гомологичные домены 2) и связываются с остатками тирозина, фосфорилированными под действием рецепторов ростовых факторов, обладающих тирозинкиназной активностью, или продуктов онкогенов, индуцируя тем самым PI3K-активность каталитической субъединицы p110, которая фосфорилирует свой липидный субстрат. PI3-киназы I класса вовлечены в важные события передачи сигнала вниз по пути цитокинов, интегринов, ростовых факторов и иммунорецепторов, что предполагает, что регулирование этого пути может приводить к получению важных терапевтических эффектов, таких как модулирование клеточной пролиферации и канцерогенеза. PI3K I класса могут фосфорилировать фосфатидилинозит (PI), фосфатидилинозит-4-фосфат и фосфатидилинозит-4,5-бифосфат (PIP2) с получением фосфатидилинозит-3-фосфата (PIP), фосфатидилинозит-3,4-бифосфата и фосфатидилинозит-3,4,5-трифосфата, соответственно. PI3K II класса фосфорилируют PI и фосфатидилинозит-4-фосфат. PI3K III класса могут фосфорилировать только PI. Ключевой изоформой PI3-киназ при раке является PI3-киназа I класса, содержащая p110α, на что указывают неоднократно повторяющиеся онкогенные мутации в p110α (Samuels et al. (2004) Science, 304: 554; US 5824492; US 5846824; US 6274327). Другие изоформы могут играть важную роль при раке и также вовлечены в сердечно-сосудистые и иммуновоспалительные заболевания (Workman P (2004) Biochem. Soc. Trans., 32: 393-396; Patel et al. (2004) Proc. Am. Assoc. of Cancer Res. (Abstract LB-247) 95th Annual Meeting, March 27-31, Orlando, Florida, USA; Ahmadi K. and Waterfield M.D. (2004) “Phosphoinositide 3-Kinase: Function and Mechanisms” Encyclopedia of Biological Chemistry (Lennarz W.J., Lane M.D. eds) Elsevier/Academic Press). Онкогенные мутации p110α (альфа) со значительной частотой были обнаружены в солидных опухолях толстой кишки, молочной железы, головного мозга, печени, яичников, желудка, легкого и головы и шеи. Примерно 35-40% случаев раковых опухолей молочной железы, положительных в отношении рецепторов гормонов (HR+), содержат мутацию в PIK3CA. Нарушения в PTEN обнаруживаются при глиобластоме, меланоме, раке предстательной железы, эндометрия, яичников, молочной железы, легкого, головы и шеи, гепатоклеточном раке и раке щитовидной железы.
PI3-киназа (PI3K) представляет собой гетеродимер, состоящий из субъединиц p85 и p110 (Otsu et al. (1991) Cell, 65: 91-104; Hiles et al. (1992) Cell, 70: 419-29). Были идентифицированы четыре разных PI3K I класса, обозначенных как PI3K-α (альфа), -β(бета), -δ(дельта) и -γ(гамма), каждая из которых содержит индивидуальную каталитическую субъединицу с ММ 110 кДа и регуляторную субъединицу. Каждая из трех каталитических субъединиц, т.е. p110-альфа, p110-бета и p110-дельта, взаимодействует с одной и той же регуляторной субъединицей, p85; тогда как p110-гамма взаимодействует с другой регуляторной субъединицей, p101. Картины экспрессии для каждой из этих PI3K в клетках и тканях человека различаются. В случае каждого из подтипов PI3K, альфа, бета и дельта, субъединица p85 действует таким образом, чтобы локализовать PI3-киназу в плазматической мембране посредством взаимодействия ее SH2-домена с фосфорилированными остатками тирозина (имеющимися в соответствующем окружении последовательностей) в белках-мишенях (Rameh et al. (1995) Cell, 83: 821-30; Volinia et al. (1992) Oncogene, 7: 789-93).
Путь PI3-киназа/Akt/PTEN представляет собой привлекательную мишень для разработки противораковых лекарственных средств, поскольку ожидается, что такие агенты будут ингибировать клеточную пролиферацию, подавлять сигналы от стромальных клеток, обеспечивающих раковым клеткам выживаемость и химиорезистентность, реверсировать подавление апоптоза и преодолевать внутреннюю резистентность раковых клеток к цитотоксическим агентам. PI3K активируется в результате включения опосредованного рецепторными тирозинкиназами сигнального пути, а также активирующих мутаций в каталитической субъединице p110 PI3K, потери функциональности онкосупрессора PTEN или в результате редко встречающихся активирующих мутаций в AKT.
Таселисиб (GDC-0032, Roche RG7604, регистрационный № в Химической реферативной службе (CAS) 1282512-48-4, Genentech Inc.) с названием 2-(4-(2-(1-изопропил-3-метил-1H-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1H-пиразол-1-ил)-2-метилпропанамид обладает сильной активностью в отношении PI3K (Ndubaku C.O. et al. (2013) J. Med. Chem., 56: 4597-4610; WO 2011/036280; US 8242104; US 8343955), и в настоящее время проходят его исследования на пациентах с локально распространенными или метастатическими солидными опухолями. Таселисиб (GDC-0032) представляет собой умеренный ингибитор каталитической субъединицы бета-изоформы PI3K, в 31 раз более селективный в отношении альфа-субъединицы по сравнению с бета-субъединицей. Таселисиб проявляет более высокую селективность в отношении мутантных изоформ PI3Kα, чем в отношении PI3Kα дикого типа (Olivero A.G. et al., AACR 2013. Abstract DDT02-01). В настоящее время таселисиб разрабатывается с целью лечения пациентов с диагнозом рецептор эстрогенов(ER)-положительного, HER2 (от англ. human epidermal growth factor receptor 2 - рецептор эпидермального фактора роста человека 2 типа)-отрицательного метастатического рака молочной железы (mBC, metastatic breast cancer) и немелкоклеточного рака легкого (NSCLC, non-small cell lung cancer). В исследовании на фазе Ia с использованием единственного агента таселисиба наблюдали частичные ответы (PR, partial responses) у 6/34 включенных в исследование пациентов. Все 6 ответов наблюдали у пациентов, имеющих опухоли с мутацией в PIK3CA (Juric D. et al. AACR, 2013), что указывает на необходимость определения PIK3CA-мутационного статуса для пациентов, подвергаемых лечению таселисибом.
Благодаря недавно полученным клиническим данным с использованием ингибиторов PI3K отмечено участие активности PI3K-дельта в качестве источника желудочно-кишечных токсических эффектов (Akinleye et al. “Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics”, Journal of Hematology & Oncology, 2013, 6: 88-104; C. Saura et al., “Phase Ib Study of the PI3K Inhibitor Taselisib (GDC-0032) in Combination with Letrozole in Patients with Hormone Receptor-Positive Advanced Breast Cancer”, San Antonio Breast Cancer Symposium - December 12, 2014, PD5-2; Lopez et al., “Taselisib, a selective inhibitor of PIK3CA, is highly effective on PIK3CA-mutated and HER2/neu amplified uterine serous carcinoma in vitro and in vivo”, (2014) Gynecologic Oncology).
Иделалисиб (GS-1101, CAL-101, ZYDELIG®, Gilead Sciences Inc., регистрационный № в CAS 870281-82-6, 5-фтор-3-фенил-2-[(1S)-1-(7H-пурин-6-иламино)пропил]-4(3H)-хиназолинон) представляет собой селективный ингибитор PI3Kδ (дельта) и одобрен к применению для лечения хронического лимфоидного лейкоза, CLL (Somoza J.R. et al. (2015) J. Biol. Chem., 290: 8439-8446; US 6800620; US 6949535; US 8138195; US 8492389; US 8637533; US 8865730; US 8980901; RE44599; RE44638). Среди самых общеизвестных неблагоприятных событий после лечения иделалисибом, отмечают диарею и колит (Brown et al. “Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110d, for relapsed/refractory chronic lymphocytic leukemia” (2014) Blood, 123(22): 3390-3397; инструкция по применению препарата Zydelig®, 2014; информационный бюллетень REMS (Risk Evaluation and Mitigation Strategy - Стратегия оценки и снижения рисков) для Zydelig®). Существенные желудочно-кишечные (ЖК) токсические эффекты, обнаруженные после лечения иделалисибом, не противоречат гипотезе о том, что ингибирование PI3Kδ (дельта) является источником желудочно-кишечных токсических эффектов. Дополнительные серьезные побочные эффекты наблюдали при клинических испытаниях иделалисиба (Zydelig®) в комбинации с другими терапиями. Неблагоприятные события, включая случаи смерти, были связаны с такими инфекциями, как пневмония. В марте 2016 года Комитет по оценке фармаконадзорных рисков (PRAC, Pharmacovigilance Risk Assessment Committee) Европейского агентства по лекарственным средствам (EMA) опубликовал предварительную предупреждающую информацию и рекомендацию для пациентов относительно необходимости совмещения лечения антибиотиками с постоянным мониторингом наличия инфекции при приеме зиделига (иделалисиба). В марте 2016 года Управление по контролю качества пищевых продуктов и лекарственных средств США опубликовало оповещение, что “шесть клинических испытаний по исследованию иделалисиба (Zydelig®) в комбинации с другими терапиями, были остановлены по причине появления сообщений об увеличении числа неблагоприятных событий, включая смерть”.
Существует потребность в дополнительных модуляторах PI3Kα, полезных для лечения раковых заболеваний, в частности, в ингибиторе PI3Kα, который избирательно воздействует на опухоли, экспрессирующие мутантную PI3Kα, по сравнению с клетками, экспрессирующими немутантную PI3Kα. Существует особая потребность в таком агенте, который избирательно ингибирует изоформу PI3Kα по сравнению с изоформами PI3Kβ, PI3Kδ и PI3Kγ, что, как можно ожидать, приведет к расширению терапевтического окна.
Сущность изобретения
Данное изобретение в целом относится к соединениям бензоксазепиноксазолидинонов с активностью, избирательной в отношении модулирования мутантных форм изоформы PI3Kα (альфа), и имеющим структуру формулы I:
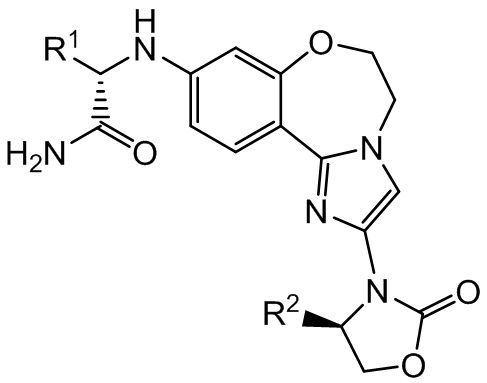 I
I
и стереоизомерам, геометрическим изомерам, таутомерам и их фармацевтически приемлемым солям. Различные заместители являются такими, как определено в данном описании.
Другой аспект изобретения относится к фармацевтической композиции, содержащей соединение из семейства бензоксазепиноксазолидинонов формулы I и фармацевтически приемлемый носитель, вещество, способствующее скольжению, разбавитель или эксципиент.
Другой аспект изобретения относится к способу лечения рака у пациента, имеющего диагноз рак, включающему введение указанному пациенту терапевтически эффективного количества соединения из семейства бензоксазепиноксазолидинонов формулы I.
Другой аспект изобретения относится к набору для терапевтического лечения рака молочной железы, содержащему:
a) соединение из семейства бензоксазепиноксазолидинонов формулы I; и
b) инструкции по применению в терапевтическом лечении рака молочной железы.
Краткое описание графических материалов
На Фиг. 1 показаны полученные с использованием рентгеновского излучения совместные кристаллические структуры PI3Kα с: A) таселисибом (GDC-0032), B) соединением 529 из US 8242104, C) соединением 101 и D) соединением 103.
На Фиг. 2 показаны полученные с использованием рентгеновского излучения совместные кристаллические структуры PI3Kα с: A) таселисибом (GDC-0032) и B) соединением 101.
На Фиг. 3A показаны данные вестерн-блоттинга с изображением уровней p110α (p110a, p110 альфа) через 24 часа после обработки соединением 101, соединением 103 и соединением 436 из US 8242104, клеток HCC-1954 (с мутацией H1047R в PI3Kα).
На Фиг. 3B показаны данные вестерн-блоттинга с изображением уровней p110α (p110a, p110 альфа) через 24 часа после обработки соединением 101, соединением 103 и соединением 436 из US 8242104 клеток HDQ-P1 (PI3Kα дикого типа).
Подробное описание иллюстративных вариантов осуществления
Теперь будет приведена подробная ссылка на некоторые воплощения изобретения, примеры которых иллюстрируются сопровождающими структурами и формулами. Несмотря на то, что изобретение будет описано в сочетании с приведенными воплощениями, очевидно, что изобретение не предполагает ограничения этими воплощениями. Напротив, подразумевается, что данное изобретение охватывает все альтернативные варианты, модификации и эквиваленты, которые могут быть включены в объем настоящего изобретения, определенный формулой изобретения. Специалисту в данной области техники будут известны многие способы и материалы, аналогичные или эквивалентные изложенным в данном описании, которые могут быть использованы при практическом применении настоящего изобретения. Настоящее изобретение никоим образом не ограничено описанными способами и материалами. В том случае, если в одном или более чем одном из включенных литературных источников, патентов и аналогичных материалов имеется отличие от этой заявки или противоречие с этой заявкой, включая, но не ограничиваясь этим, определенные термины, употребление терминов, описанные методы или тому подобное, то эта заявка является контрольным документом. Если не указано иное, то все технические и научные термины, использованные в данном описании, имеют значение, обычно понимаемое средним специалистом в области техники, к которой данное изобретение относится. Несмотря на то, что при практическом применении или тестировании данного изобретения могут быть использованы способы и материалы, аналогичные или эквивалентные таковым, описанным в данной заявке, подходящие способы и материалы описаны ниже. Все публикации, патентные заявки, патенты и другие ссылки, упомянутые в данном описании, включены посредством ссылки во всей своей полноте. Использованная в данной заявке номенклатура основывается на систематической номенклатуре IUPAC (Международный союз по теоретической и прикладной химии), если не указано иное.
Определения
При указании числа заместителей термин “один или более” относится к диапазону от одного заместителя до самого наиболее возможного числа заместителей, т.е. от замены одного атома водорода вплоть до замены всех атомов водорода на заместители. Термин “заместитель” означает атом или группу атомов, заменяющих атом водорода в исходной молекуле. Термин “замещенный” означает, что конкретная группа имеет один или несколько заместителей. Если какая-либо группа может иметь несколько заместителей и предложен ряд возможных заместителей, то такие заместители выбраны независимо и необязательно будут одинаковыми. Термин “незамещенный” означает, что конкретная группа не имеет никаких заместителей. Термин “возможно замещенный” означает, что конкретная группа является незамещенной или замещена одним или несколькими заместителями, независимо выбранными из группы возможных заместителей. При указании числа заместителей термин “один или более” означает замену на заместители в количестве от одного заместителя до самого наиболее возможного числа заместителей, т.е. замену от одного атома водорода вплоть до замены всех атомов водорода на заместители.
Термин “алкил”, использованный в данном описании, относится к насыщенному линейному или разветвленному одновалентному углеводородному радикалу из одного-двенадцати атомов углерода (C1-C12), причем этот алкильный радикал возможно может быть независимо замещен одним или несколькими заместителями, описанными ниже. В другом воплощении алкильный радикал содержит от одного до восьми атомов углерода (C1-C8) или от одного до шести атомов углерода (C1-C6). Примеры алкильных групп включают, но не ограничиваются этим, метил (Me, -CH3), этил (Et, -CH2CH3), 1-пропил (н-Pr, н-пропил, -CH2CH2CH3), 2-пропил (i-Pr, изопропил, -CH(CH3)2), 1-бутил (н-Bu, н-бутил, -CH2CH2CH2CH3), 2-метил-1-пропил (i-Bu, изобутил, -CH2CH(CH3)2), 2-бутил (втор-Bu, втор-бутил, -CH(CH3)CH2CH3), 2-метил-2-пропил (t-Bu, трет-бутил, -C(CH3)3), 1-пентил (н-пентил, -CH2CH2CH2CH2CH3), 2-пентил (-CH(CH3)CH2CH2CH3), 3-пентил (-CH(CH2CH3)2), 2-метил-2-бутил (-C(CH3)2CH2CH3), 3-метил-2-бутил (-CH(CH3)CH(CH3)2), 3-метил-1-бутил (-CH2CH2CH(CH3)2), 2-метил-1-бутил (-CH2CH(CH3)CH2CH3), 1-гексил (-CH2CH2CH2CH2CH2CH3), 2-гексил (-CH(CH3)CH2CH2CH2CH3), 3-гексил (-CH(CH2CH3)(CH2CH2CH3)), 2-метил-2-пентил (-C(CH3)2CH2CH2CH3), 3-метил-2-пентил (-CH(CH3)CH(CH3)CH2CH3), 4-метил-2-пентил (-CH(CH3)CH2CH(CH3)2), 3-метил-3-пентил (-C(CH3)(CH2CH3)2), 2-метил-3-пентил (-CH(CH2CH3)CH(CH3)2), 2,3-диметил-2-бутил (-C(CH3)2CH(CH3)2), 3,3-диметил-2-бутил (-CH(CH3)C(CH3)3, 1-гептил, 1-октил и тому подобное.
Термины “карбоцикл”, “карбоциклил”, “карбоциклическое кольцо” и “циклоалкил” относятся к одновалентному неароматическому, насыщенному или частично ненасыщенному кольцу, имеющему от 3 до 12 атомов углерода (C3-C12) в виде моноциклического кольца или от 7 до 12 атомов углерода в виде бициклического кольца. Бициклические карбоциклы, имеющие от 7 до 12 атомов, могут быть организованы, например, в виде бицикло-[4,5]-, -[5,5]-, -[5,6]- или -[6,6]-систем, а бициклические карбоциклы, имеющие 9 или 10 атомов в кольце, могут быть организованы в виде бицикло-[5,6]- или -[6,6]-систем либо в виде мостиковых систем, таких как бицикло[2.2.1]гептан, бицикло[2.2.2]октан и бицикло[3.2.2]нонан. Спиро-карбоциклильные группировки также включены в объем этого определения. Примеры спиро-карбоциклильных группировок включают [2.2]пентанил, [2.3]гексанил и [2.4]гептанил. Примеры моноциклических карбоциклов включают, но не ограничиваются этим, циклопропил, циклобутил, циклопентил, 1-циклопент-1-енил, 1-циклопент-2-енил, 1-циклопент-3-енил, циклогексил, 1-циклогекс-1-енил, 1-циклогекс-2-енил, 1-циклогекс-3-енил, циклогексадиенил, циклогептил, циклооктил, циклононил, циклодецил, циклоундецил, циклододецил и тому подобное. Карбоциклильные группы возможно независимо замещены одним или несколькими заместителями, описанными в данной заявке.
“Арил” означает одновалентный ароматический углеводородный радикал из 6-20 атомов углерода (C6-C20), образуемый в результате удаления одного атома водорода от единственного атома углерода исходной ароматической кольцевой системы. Некоторые арильные группы представлены в приведенных в качестве примера структурах как “Ar”. Арил включает бициклические радикалы, содержащие ароматическое кольцо, конденсированное с насыщенным, частично ненасыщенным кольцом или ароматическим карбоциклическим кольцом. Типичные арильные группы включают, но не ограничиваются этим, радикалы, происходящие из бензола (фенил), замещенных бензолов, нафталина, антрацена, бифенила, инденила, инданила, 1,2-дигидронафталина, 1,2,3,4-тетрагидронафтила и тому подобное. Арильные группы возможно независимо замещены одним или несколькими заместителями, описанными в данной заявке.
Термины “гетероцикл”, “гетероциклил” и “гетероциклическое кольцо” используются в данном описании взаимозаменяемо и относятся к насыщенному или частично ненасыщенному (т.е. имеющему одну или несколько двойных и/или тройных связей в пределах кольца) карбоциклическому радикалу, содержащему от 3 до примерно 20 атомов в кольце, при этом по меньшей мере один атом в кольце представляет собой гетероатом, выбранный из атомов азота, кислорода, фосфора и серы, а остальные атомы в кольце представляют собой атомы C, причем один или несколько атомов в кольце возможно независимо замещены одним или несколькими заместителями, описанными ниже. Гетероцикл может представлять собой моноцикл, имеющий от 3 до 7 членов в кольце (от 2 до 6 атомов углерода и от 1 до 4 гетероатомов, выбранных из N, O, P и S), или бицикл, имеющий от 7 до 10 членов в кольце (от 4 до 9 атомов углерода и от 1 до 6 гетероатомов, выбранных из N, O, P и S), например: бицикло-[4,5]-, -[5,5]-, -[5,6]- или -[6,6]-систему. Гетероциклы описаны в Paquette Leo A.; “Principles of Modern Heterocyclic Chemistry” (W.A. Benjamin, New York, 1968), особенно в главах 1, 3, 4, 6, 7 и 9; “The Chemistry of Heterocyclic Compounds, A series of Monographs” (John Wiley & Sons, New York, с 1950 года по настоящее время), в частности, в томах 13, 14, 16, 19 и 28; и в J. Am. Chem. Soc. (1960) 82: 5566. Термин “гетероциклил” также включает в себя радикалы, при этом гетероциклические радикалы конденсированы с насыщенным, частично ненасыщенным кольцом либо ароматическим карбоциклическим или гетероциклическим кольцом. Примеры гетероциклических колец включают, но не ограничиваются этим, морфолин-4-ил, пиперидин-1-ил, пиперазинил, пиперазин-4-ил-2-он, пиперазин-4-ил-3-он, пирролидин-1-ил, тиоморфолин-4-ил, S-диоксотиоморфолин-4-ил, азокан-1-ил, азетидин-1-ил, октагидропиридo[1,2-a]пиразин-2-ил, [1,4]диазепан-1-ил, пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидротиенил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, пиперидино, морфолино, тиоморфолино, тиоксанил, пиперазинил, гомопиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 2-пирролинил, 3-пирролинил, индолинил, 2H-пиранил, 4H-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинил, имидазолинил, имидазолидинил, 3-азабицикло[3.1.0]гексанил, 3-азабицикло[4.1.0]гептанил, азабицикло[2.2.2]гексанил, 3H-индолил, хинолизинил и N-пиридилмочевины. Спиро-гетероциклильные группировки также включены в объем этого определения. Примеры спиро-гетероциклильных группировок включают азаспиро[2.5]октанил и азаспиро[2.4]гептанил. Примерами гетероциклической группы, где 2 атома в кольце замещены группировками оксо (=O), являются пиримидинонил и 1,1-диоксо-тиоморфолинил.
Термин “гетероарил” относится к одновалентному ароматическому радикалу, состоящему из 5-, 6- или 7-членных колец, и включает в себя конденсированные кольцевые системы (по меньшей мере одно из колец в которых является ароматическим) из 5-20 атомов, содержащие один или несколько гетероатомов, независимо выбранных из атомов азота, кислорода и серы. Примерами гетероарильных групп являются пиридинил (включая, например, 2-гидроксипиридинил), имидазолил, имидазопиридинил, пиримидинил (включая, например, 4-гидроксипиримидинил), пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксадиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, тетрагидроизохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, триазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил и фуропиридинил.
Термины “лечить” и “лечение” относятся к терапевтическому лечению, при котором у объекта должно быть ослаблено (облегчено) нежелательное физиологическое изменение или расстройство, такое как развитие или распространение артрита или рака. Для целей данного изобретения полезные или желаемые клинические результаты включают, но не ограничиваются этим, облегчение симптомов, уменьшение степени заболевания, стабильное (т.е. без ухудшения) состояние заболевания, задержку или замедление прогрессирования заболевания, уменьшение интенсивности симптомов или временное облегчение болезненного состояния и ремиссию (будь то частичную или полную), как детектируемые, так и недетектируемые. “Лечение” также может означать продление выживаемости по сравнению с ожидаемой выживаемостью в отсутствие получения лечения. Нуждающиеся в таком лечении, включают уже имеющих данное состояние или расстройство.
Фраза “терапевтически эффективное количество” означает количество соединения по настоящему изобретению, которое (1) лечит конкретное заболевание, состояние или расстройство, (2) ослабляет, уменьшает интенсивность или устраняет один или более симптомов конкретного заболевания, состояния или расстройства или (3) предотвращает или задерживает начало развития одного или более симптомов конкретного заболевания, состояния или расстройства, описанного в данной заявке. В случае рака терапевтически эффективное количество лекарственного средства может уменьшать количество раковых клеток; уменьшать размер опухоли; ингибировать (т.е. замедлять до некоторой степени и предпочтительно останавливать) инфильтрацию раковых клеток в периферические органы; ингибировать (т.е. замедлять до некоторой степени и предпочтительно останавливать) опухолевый метастаз; ингибировать до некоторой степени опухолевый рост; и/или ослаблять до некоторой степени один или более симптомов, ассоциированных с раком. В зависимости от возможности лекарственного средства предотвращать рост и/или уничтожать существующие раковые клетки, оно может быть цитостатическим и/или цитотоксическим. Что касается терапии рака, то эффективность может быть измерена, например, путем оценки времени до прогрессирования заболевания (TTP) и/или определения коэффициента ответа (RR).
Термин “рак” относится к физиологическому состоянию или описывает физиологическое состояние у млекопитающих, которое обычно характеризуется нерегулируемым клеточным ростом. Термин “опухоль” подразумевает наличие одного или более типов раковых клеток. Примеры рака включают, но не ограничиваются этим, карциному, лимфому, бластому, саркому и лейкоз или лимфолейкозы. Более конкретные примеры таких видов рака включают плоскоклеточный рак (например, плоскоклеточный рак эпителиальной ткани), рак легкого, в том числе мелкоклеточный рак легкого, немелкоклеточный рак легкого (“NSCLC”), аденокарциному легкого и плоскоклеточный рак легкого, рак брюшины, гепатоклеточный рак, рак желудочно-кишечного тракта или желудка, в том числе гастроинтестинальный рак, рак поджелудочной железы, глиобластому, рак шейки матки, рак яичников, рак печени, рак мочевого пузыря, гепатому, рак молочной железы, рак толстой кишки, рак прямой кишки, колоректальный рак, рак эндометрия или матки, рак слюнных желез, рак почки или почечный рак, рак предстательной железы, рак вульвы, рак щитовидной железы, печеночный рак, анальный рак, рак пениса, а также рак головы и шеи.
“Гематологические злокачественные новообразования” (британское написание “haematological” отличается от принятого в США написания “hematological”) представляют собой типы рака, которые воздействуют на кровь, костный мозг и лимфатические узлы. Поскольку эти три системы тесно связаны через иммунную систему, то заболевание, затрагивающее одну из них, в равной степени часто будет затрагивать и остальные: несмотря на то, что лимфома является заболеванием лимфатических узлов, зачастую она распространяется в костный мозг, воздействуя на кровь. Гематологические злокачественные новообразования представляют собой злокачественные неоплазии (“рак”), и как правило их лечат специалисты в области гематологии и/или онкологии. В некоторых центрах “гематологию/онкологию” относят к одной из узких специализаций терапии внутренних болезней, тогда как в других считают отдельными подразделениями (имеются также специалисты в области хирургической и радиационной онкологии). Не все гематологические расстройства являются злокачественными (“раковыми”); помощь при этих иных заболеваниях крови также может быть оказана гематологом. Гематологические злокачественные новообразования могут возникать из любой из двух основных линий клеток крови: линий миелоидных и лимфоидных клеток. Линия миелоидных клеток обычно продуцирует гранулоциты, эритроциты, тромбоциты, макрофаги и тучные клетки; линия лимфоидных клеток продуцирует B-, T-, NK-клетки (природные киллерные клетки) и плазматические клетки. Лимфомы, лимфоцитарные лейкозы и миелома происходят из лимфоидной линии, в то время как острый и хронический миелогенный лейкоз, миелодиспластические синдромы и миелопролиферативные расстройства имеют миелоидное происхождение. Лейкозы включают острый лимфобластный лейкоз (ALL, acute lymphoblastic leukemia), острый миелогенный лейкоз (AML, acute myelogenous leukemia), хронический лимфоцитарный лейкоз (CLL, chronic lymphocytic leukemia), хронический миелогенный лейкоз (CML, chronic myelogenous leukemia), острый моноцитарный лейкоз (AMOL, acute monocytic leukemia) и мелкоклеточную лимфоцитарную лимфому (SLL, small lymphocytic lymphoma). Лимфомы включают лимфомы Ходжкина (все четыре подтипа) и неходжкинские лимфомы (NHL (Non-Hodgkin's lymphoma), все подтипы).
“Химиотерапевтический агент” представляет собой химическое соединение, полезное в лечении рака, независимо от механизма действия. Классы химиотерапевтических агентов включают, но не ограничиваются этим: алкилирующие агенты, антиметаболиты, алкалоиды растительного происхождения, являющиеся “веретенными ядами”, цитотоксические/противоопухолевые антибиотики, ингибиторы топоизомеразы, антитела, фотосенсибилизирующие вещества и ингибиторы киназ. Химиотерапевтические агенты включают соединения, используемые в “терапии направленного действия” и традиционной химиотерапии. Примеры химиотерапевтических агентов включают: ибрутиниб (IMBRUVICA™, APCI-32765, Pharmacyclics Inc./Janssen Biotech Inc.; регистрационный № в CAS 936563-96-1, US 7514444), иделалисиб (ранее называвшийся CAL-101, GS 1101, GS-1101, Gilead Sciences Inc.; регистрационный № в CAS 1146702-54-6), эрлотиниб (TARCEVA®, Genentech/OSI Pharm.), доцетаксел (TAXOTERE®, Sanofi-Aventis), 5-FU (фторурацил, 5-фторурацил, регистрационный № в CAS 51-21-8), гемцитабин (GEMZAR®, Lilly), PD-0325901 (№ в CAS 391210-10-9, Pfizer), цисплатин (Platinol®, (SP-4-2)-диаминдихлорплатина(II), цис-диамин, дихлорплатина(II), № в CAS 15663-27-1), карбоплатин (№ в CAS 41575-94-4), паклитаксел (TAXOL®, Bristol-Myers Squibb Oncology, Princeton, N.J.), трастузумаб (HERCEPTIN®, Genentech), темозоломид (4-метил-5-оксо-2,3,4,6,8-пентазабицикло[4.3.0]нона-2,7,9-триен-9-карбоксамид, № в CAS 85622-93-1, TEMODAR®, TEMODAL®, Schering Plough), тамоксифен ((Z)-2-[4-(1,2-дифенилбут-1-енил)фенокси]-N,N-диметилэтанамин, NOLVADEX®, ISTUBAL®, VALODEX®) и доксорубицин (АDRIАMYCIN®, № в CAS 23214-92-8, Аkti-1/2 (ингибитор Аkt-1/2), HPPD (оксим 5-(1-(2-гидроксиэтил)-3-(пиридин-4-ил)-1H-пиразол-4-ил)-2,3-дигидро-1H-инден-1-она) и рапамицин.
Химиотерапевтические агенты включают ингибиторы мишеней B-клеточных рецепторов, такие как ингибиторы BTK (тирозинкиназа Брутона), Bcl-2 (B-cell lymphoma 2 - B-клеточная лимфома 2) и JAK (янус-киназа).
Другие примеры химиотерапевтических агентов включают: оксалиплатин (ELOXATIN®, Sanofi), бортезомиб (VELCADE®, Millennium Pharm.), сутент (SUNITINIB®, SU11248, Pfizer), летрозол (FEMARA®, Novartis), иматиниба мезилат (GLEEVEC®, Novartis), XL-518 (ингибитор Mek (Mitogen-activated protein kinase kinase - киназа митоген-активируемой протеинкиназы), Exelixis, WO 2007/044515), ARRY-886 (ингибитор Mek, AZD6244, Array BioPharma, Astra Zeneca), SF-1126 (ингибитор PI3K, Semafore Pharmaceuticals), BEZ-235 (ингибитор PI3K, Novartis), XL-147 (ингибитор PI3K, Exelixis), PTK787/ZK 222584 (Novartis), фулвестрант (FASLODEX®, AstraZeneca), лейковорин (фолиновая кислота), рапамицин (сиролимус, RAPAMUNE®, Wyeth), лапатиниб (TYKERB®, GSK572016, Glaxo Smith Kline), лонафарниб (SARASAR™, SCH 66336, Schering Plough), сорафениб (NEXAVAR®, BAY43-9006, Bayer Labs), гефитиниб (IRESSA®, AstraZeneca), иринотекан (CAMPTOSAR®, CPT-11, Pfizer), типифарниб (ZARNESTRA™, Johnson & Johnson), ABRAXANE™ (без кремофора), препараты наночастиц паклитаксела, сконструированных с использованием альбумина (American Pharmaceutical Partners, Schaumberg, Il), вандетаниб (rINN, ZD6474, ZACTIMA®, AstraZeneca), хлорамбуцил, AG1478, AG1571 (SU 5271; Sugen), темсиролимус (TORISEL®, Wyeth), пазопаниб (GlaxoSmithKline), канфосфамид (TELCYTA®, Telik), тиотепа и циклофосфамид (CYTOXAN®, NEOSAR®); алкилсульфонаты, такие как бусульфан, импросульфан и пипосульфан; азиридины, такие как бензодопа, карбоквон, метуредопа и уредопа; этиленимины и метиламеламины, в том числе алтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметиломеламин; ацетогенины (особенно буллатацин и буллатацинон); камптотецин (в том числе синтетический аналог топотекан); бриостатин; каллистатин; CC-1065 (в том числе его синтетические аналоги адозелезин, карзелезин и бизелезин); криптофицины (в частности, криптофицин 1 и криптофицин 8); доластатин; дуокармицин (в том числе синтетические аналоги, KW-2189 и CB1-TM1); элеутеробин; панкратистатин; саркодиктиин; спонгистатин; азотистые иприты, такие как хлорамбуцил, хлорнафазин, хлорфосфамид, эстрамустин, ифосфамид, мехлоретамин, гидрохлорид мехлоретаминоксида, мелфалан; новембихин, фенестерин, преднимустин, трофосфамид, урациловый иприт; нитрозомочевины, такие как кармустин, хлорозотоцин, фотемустин, ломустин, нимустин и ранимустин; антибиотики, такие как энедииновые антибиотики (например, калихеамицин, калихеамицин гамма 1I, калихеамицин омега I1 (Angew Chem. Intl. Ed. Engl. (1994) 33: 183-186); динемицин, динемицин А; бисфосфонаты, такие как клодронат; эсперамицин; а также неокарциностатиновый хромофор и родственные хромофоры хромопротеиновых энедииновых антибиотиков), аклациномизины, актиномицин, аутрамицин, азасерин, блеомицины, кактиномицин, карабицин, карминомицин, карцинофилин, хромомицины, дактиномицин, даунорубицин, деторубицин, 6-диазо-5-оксо-L-норлейцин, морфолино-доксорубицин, цианоморфолино-доксорубицин, 2-пирролинo-доксорубицин и дезоксидоксорубицин), эпирубицин, эзорубицин, идарубицин, неморубицин, марцелломицин, митомицины, такие как митомицин С, микофеноловую кислоту, ногаламицин, оливомицины, пепломицин, порфиромицин, пуромицин, квеламицин, родорубицин, стрептонигрин, стрептозоцин, туберцидин, убенимекс, зиностатин, зорубицин; антиметаболиты, такие как метотрексат и 5-фторурацил (5-FU); аналоги фолиевой кислоты, такие как деноптерин, метотрексат, птероптерин, триметрексат; аналоги пурина, такие как флударабин, 6-меркаптопурин, тиамиприн, тиогуанин; аналоги пиримидина, такие как анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидезоксиуридин, доксифлуридин, эноцитабин, флоксуридин; андрогены, такие как калустерон, дромостанолона пропионат, эпитиостанол, мепитиостан, тестолактон; антиадренергические средства, такие как аминоглутетимид, митотан, трилостан; средство для восполнения фолиевой кислоты, такое как фролиновая кислота; ацеглатон; альдофосфамидгликозид; аминолевулиновую кислоту; энилурацил; амсакрин; бестрабуцил; бисантрен; эдатраксат; дефофамин; демеколцин; диазиквон; элфорнитин; эллиптиния ацетат; эпотилон; этоглуцид; галлия нитрат; гидроксимочевину; лентинан; лонидаинин; майтансиноиды, такие как майтансин и ансамитоцины; митогуазон; митоксантрон; мопиданмол; нитраэрин; пентостатин; фенамет; пирарубицин; лозоксантрон; подофиллиновую кислоту; 2-этилгидразид; прокарбазин; полисахаридный комплекс PSK® (JHS Natural Products, Eugene, OR); разоксан; ризоксин; сизофиран; спирогерманий; тенуазоновую кислоту; триазиквон; 2,2',2''-трихлортриэтиламин; трихотецены (особенно, токсин Т-2, верракурин А, роридин А и ангуидин); уретан; виндезин; дакарбазин; манномустин; митобронитол; митолактол; пипоброман; гацитозин; арабинозид (“Ara-C”); циклофосфамид; тиотепа; 6-тиогуанин; меркаптопурин; метотрексат; аналоги платины, такие как цисплатин и карбоплатин; винбластин; этопозид (VP-16); ифосфамид; митоксантрон; винкристин; винорелбин (NAVELBINE®); новантрон; тенипозид; эдатрексат; дауномицин; аминоптерин; капецитабин (XELODA®, Roche); ибандронат; СРТ-11; ингибитор топоизомеразы RFS 2000; дифторметилорнитин (DMFO); ретиноиды, такие как ретиноевая кислота; и фармацевтически приемлемые соли, кислоты и производные любого из упомянутого выше.
В определение “химиотерапевтического агента” также включены: (1) антигормональные агенты, действие которых заключается в регуляции или ингибировании воздействия гормонов на опухоли, как например, антиэстрогены и селективные модуляторы рецепторов эстрогенов (SERM, selective estrogen receptor modulator), в том числе, например, тамоксифен (включая NOLVADEX®; тамоксифена цитрат), ралоксифен, дролоксифен, 4-гидрокситамоксифен, триоксифен, кеоксифен, LY117018, онапристон, FARESTON® (торемифина цитрат) и селективные негативные регуляторы рецепторов эстрогенов (SERD, selective estrogen receptor down-regulator), такие как фулвестрант (FASLODEX®, Astra Zeneca); (2) ингибиторы ароматазы, ингибирующие фермент ароматазу, которая регулирует образование эстрогенов в надпочечниках, такие как, например, 4(5)-имидазолы, аминоглутетимид, MEGASE® (мегестрола ацетат), AROMASIN® (экземестан; Pfizer), форместан, фадрозол, RIVISOR® (ворозол), FEMARA® (летрозол; Novartis) и ARIMIDEX® (анастрозол; AstraZeneca); (3) антиандрогены, такие как флутамид, нилутамид, бикалутамид, лейпролид и гозерелин; а также троксацитабин (1,3-диоксолановый аналог нуклеозида цитозина); (4) ингибиторы протеинкиназ, такие как ингибиторы MEK, например, кобиметиниб (WO 2007/044515); (5) ингибиторы липидных киназ, такие как таселисиб (GDC-0032, Genentech Inc.); (6) антисмысловые олигонуклеотиды, в частности такие, которые ингибируют экспрессию генов в сигнальных путях, участвующих в аберрантной пролиферации клеток, например, PKC-альфа (протеинкиназа C), Raf и H-Ras, такие как облимерсен (GENASENSE®, Genta Inc.); (7) рибозимы, такие как ингибиторы экспрессии VEGF (сосудистый эндотелиальный фактор роста) (например, ANGIOZYME®) и ингибиторы экспрессии HER2; (8) вакцины, как например, генотерапевтические вакцины, например, ALLOVECTIN®, LEUVECTIN® и VAXID®; rIL-2 (рекомбинантный интерлейкин-2) PROLEUKIN®; ингибиторы топоизомеразы 1, такие как LURTOTECAN®; rmRH ABARELIX®; (9) антиангиогенные агенты, такие как бевацизумаб (AVASTIN®, Genentech); и фармацевтически приемлемые соли, кислоты и производные любого из упомянутого выше.
В определение “химиотерапевтического агента” также включены терапевтические антитела, такие как алемтузумаб (Кэмпас (англ. - campath)), бевацизумаб (AVASTIN®, Genentech); цетуксимаб (ERBITUX®, Imclone); панитумумаб (VECTIBIX®, Amgen), ритуксимаб (RITUXAN®, Genentech/Biogen Idec), пертузумаб (PERJETA™, 2C4, Genentech), трастузумаб (HERCEPTIN®, Genentech), трастузумаба эмтанзин (KADCYLA®, Genentech Inc.) и тозитумомаб (бексар (bexxar), Corixia).
Термин “метаболит” относится к продукту, образуемому в процессе метаболизма конкретного соединения или его соли в организме. Метаболиты соединения могут быть идентифицированы с использованием общепринятых методик, известных в данной области техники, а их активности определены с использованием таких тестов, которые изложены в данном описании. Такие продукты могут получаться из вводимого соединения, например, в результате его окисления, восстановления, гидролиза, амидирования, дезамидирования, этерификации, деэтерификации, ферментативного расщепления и тому подобного. Соответственно, изобретение включает метаболиты соединений по изобретению, в том числе соединения, получаемые способом, включающим приведение соединения формулы I по данному изобретению в контакт с млекопитающим в течение периода времени, достаточного для получения продукта его метаболизма.
Термин “инструкция по применению” обычно относится к инструкциям, традиционно включаемым в промышленные упаковки терапевтических продуктов, которые содержат информацию о показаниях, применении, дозировке, введении, противопоказаниях и/или предупреждениях касательно применения таких терапевтических продуктов.
Термин “хиральный” относится к молекулам, обладающим свойством не совпадать при наложении на зеркально отображаемого партнера, тогда как термин “ахиральный” относится к молекулам, совпадающим при наложении на своего зеркально отображаемого партнера.
Термин “стереоизомеры” относится к соединениям, которые имеют идентичный химический состав, но различаются расположением атомов или групп в пространстве.
“Диастереомер” относится к стереоизомеру с двумя или более центрами хиральности, и такие молекулы не являются зеркальными отображениями друг друга. Диастереомеры имеют разные физические свойства, например точки плавления, точки кипения, спектральные свойства и реакционные способности. Смеси диастереомеров можно разделить с помощью аналитических методик высокого разрешения, таких как электрофорез и хроматография.
Термин “энантиомеры” относится к двум стереоизомерам соединения, которые не совпадают с зеркальными отображениями друг друга.
В данном описании в основном использованы стереохимические определения и правила, представленные в S.P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984), McGraw-Hill Book Company, New York; и Eliel E. and Wilen S., “Stereochemistry of Organic Compounds”, John Wiley & Sons, Inc., New York, 1994. Соединения по изобретению могут содержать асимметрические или хиральные центры и ввиду этого существовать в разных стереоизомерных формах. Предполагается, что все стереоизомерные формы соединений по изобретению, включая, но не ограничиваясь этим, диастереомеры, энантиомеры и атропизомеры, а также их смеси, такие как рацемические смеси, составляют часть настоящего изобретения. Многие органические соединения существуют в оптически активных формах, т.е. они обладают способностью вращать плоскость плоскополяризованного света. При описании оптически активного соединения для обозначения абсолютной конфигурации молекулы относительно ее хирального центра(ов) используют префиксы D и L или R и S. Для обозначения знака направления вращения плоскополяризованного света соединением применяют префиксы d и l или (+) и (-), при этом (-) или l означает, что соединение является левовращающим. Соединение с префиксом (+) или d является правовращающим. Для данной химической структуры эти стереоизомеры являются идентичными, за исключением того, что они являются зеркальными отображениями друг друга. Конкретный стереоизомер также может быть определен как энантиомер, и смесь таких изомеров часто называют энантиомерной смесью. Смесь с соотношением энантиомеров 50:50 называется рацемической смесью или рацематом, которая может образовываться, если не соблюдалось никакой стереоизбирательности или стереоспецифичности в химической(ом) реакции или процессе. Термины “рацемическая смесь” и “рацемат” относятся к эквимолярной смеси двух энантиомерных разновидностей, не обладающей оптической активностью. Энантиомеры можно выделить по отдельности из рацемической смеси методом хирального разделения, таким как сверхкритическая жидкостная хроматография (SFC). Присвоение конфигурации при хиральных центрах в разделенных энантиомерах может быть ориентировочным и отражено в структурах, приведенных в Таблице 1 в целях иллюстрации, тогда как стереохимическая конфигурация является однозначно устанавленной, например, по данным рентгеноструктурного анализа кристаллов.
Термин “таутомер” или “таутомерная форма” относится к структурным изомерам, обладающим разной энергией, взаимопревращение которых протекает через низкий энергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) характеризуются взаимопревращениями в результате миграции протона, как например при кето-енольной и имин-енаминной изомерии. Валентные таутомеры включают взаимопревращения посредством реогранизации некоторых из связывающих электронов.
Термин “фармацевтически приемлемые соли” означает соли, которые не являются нежелательными ни в биологическом, ни в других отношениях. Фармацевтически приемлемые соли включают соли присоединения как кислоты, так и основания. Фраза “фармацевтически приемлемые” указывает на то, что вещество или композиция должны быть совместимы химически и/или токсикологически с другими ингредиентами, входящими в состав композиции, и/или с млекопитающим, подвергаемым лечению ими.
Термин “фармацевтически приемлемая соль присоединения кислоты” означает такие фармацевтически приемлемые соли, которые образуются с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, угольная кислота, фосфорная кислота, и органическими кислотами, выбранными из классов алифатических, циклоалифатических, ароматических, арил-алифатических, гетероциклических, карбоновых и сульфоновых органических кислот, таких как муравьиная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, глюконовая кислота, молочная кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, аспарагиновая кислота, аскорбиновая кислота, глутаминовая кислота, антраниловая кислота, бензойная кислота, коричная кислота, миндальная кислота, эмбоновая кислота, фенилуксусная кислота, метансульфоновая кислота (“мезилат”), этансульфоновая кислота, пара-толуолсульфоновая кислота и салициловая кислота.
Термин “фармацевтически приемлемая соль присоединения основания” означает такие фармацевтически приемлемые соли, которые образуются с органическим или неорганическим основанием. Примеры приемлемых солей с неорганическими основаниями включают соли натрия, калия, аммония, кальция, магния, железа, цинка, меди, марганца и алюминия. Соли, происходящие из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, в том числе природных замещенных аминов, циклических аминов и основных ионообменных смол, таких как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, 2-диэтиламиноэтанол, триметамин, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилглюкамин, теобромин, пурины, пиперазин, пиперидин, N-этилпиперидин и полиаминные смолы.
“Сольват” относится к ассоциации или комплексу одной или более молекул растворителя и соединения по изобретению. Примеры растворителей, которые образуют сольваты, включают, но не ограничиваются этим, воду, изопропанол, этанол, метанол, ДМСО (диметилсульфоксид), этилацетат, уксусную кислоту и этаноламин.
Термин “EC50” представляет собой “полумаксимальную эффективную концентрацию” и означает концентрацию конкретного соединения в плазме крови, необходимую для получения конкретного эффекта in vivo, равного 50% от максимального.
Термин “Ki” относится к константе ингибирования и означает абсолютную аффинность связывания конкретного ингибитора с рецептором. Ее измеряют, используя анализы конкурентного связывания, и она эквивалентна концентрации, при которой этот конкретный ингибитор будет занимать 50% рецепторов в случае отсутствия какого-либо конкурирующего лиганда (например, радиоактивного лиганда). Величины Ki можно преобразовать логарифмически в величины pKi (-log Ki), при этом более высокие значения указывают на экспоненциально более высокую эффективность.
Термин “IC50” относится к “концентрации, вызывающей половину от максимального ингибирования” и означает концентрацию конкретного соединения, необходимую для получения 50% ингибирования биологического процесса in vitro. Величины IC50 можно преобразовать логарифмически в величины pIC50 (-log IC50), при этом более высокие значения указывают на экспоненциально более высокую эффективность. Величина IC50 не является абсолютной величиной, а зависит от экспериментальных условий, например, от используемых концентраций, и ее можно преобразовать в абсолютную константу ингибирования (Ki), используя уравнение Ченга-Пруссофа (Biochem. Pharmacol. (1973) 22: 3099). Можно рассчитать параметры для другого процента ингибирования, такие как IC70, IC90 и т.д.
Термины “соединение по данному изобретению”, и “соединения по настоящему изобретению”, и “соединения формулы I” включают в себя соединения формул I и стереоизомеры, геометрические изомеры, таутомеры, сольваты, метаболиты и фармацевтически приемлемые соли и их пролекарства.
Подразумевается, что любая формула или структура, приведенная в данном описании, включая соединения формулы I, также представляет гидраты, сольваты и полиморфы таких соединений и их смеси.
Подразумевается, что любая формула или структура, приведенная в данном описании, включая соединения формулы I, также представляет немеченые формы, равно как и меченные изотопом формы этих соединений. Меченные изотопом соединения имеют структуры, изображенные формулами, приведенными в данном описании, но с учетом того, что один или более чем один атом заменен на атом, имеющий выбранную(ое) атомную массу или массовое число. Примеры изотопов, которые могут быть инкорпорированы в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как, но не ограничиваясь этим, 2H (дейтерий, D), 3H (тритий), 11C, 13C, 14C, 15N, 18F, 31P, 32P, 35S, 36Cl и 125I. Включены различные меченные изотопом соединения по настоящему изобретению, например, соединения, в которые инкорпорированы такие радиоактивные изотопы, как 3H, 13C и 14C. Такие меченные изотопом соединения могут быть полезны в исследованиях метаболизма, исследованиях кинетики реакций, методах детекции или визуализации, таких как позитронно-эмиссионная томография (PET) или однофотонная эмиссионная компьютерная томография (SPECT), в том числе в анализах распределения лекарственного средства или субстрата в тканях, или в лечении пациентов с применением радиоактивных средств. Меченные или замещенные дейтерием терапевтические соединения по изобретению могут иметь улучшенные свойства с точки зрения DMPK (метаболизма и фармакокинетики лекарственных средств), связанные с всасыванием, распределением, метаболизмом и экскрецией (ADME, англ. absorbtion, distribution, metabolism and excretion). Замена на более тяжелые изотопы, такие как дейтерий, может давать некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенный in vivo период полувыведения или снижение дозировки. Для исследований с использованием PET или SPECT может быть полезно 18F-меченное соединение. В общем случае, меченные изотопом соединения по данному изобретению и их пролекарства могут быть получены путем осуществления методик, описанных на схемах или в разделах Примеры и Подготовительные примеры, изложенных ниже, в результате замены не меченного изотопом реагента легко доступным меченным изотопом реагентом. Кроме того, замена на более тяжелые изотопы, в частности, дейтерий (т.е. 2Н или D), может давать некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличенный in vivo период полувыведения, или снижение дозировки, или улучшение терапевтического индекса. Очевидно, что в этом случае дейтерий считается заместителем в соединении формулы (I). Концентрация такого более тяжелого изотопа, в частности, дейтерия, может быть определена по фактору изотопного обогащения. Подразумевается, что любой атом в соединениях по данному изобретению, специально не обозначенный как конкретный изотоп, представляет собой любой стабильный изотоп этого атома. Если не указано иное, то когда положение конкретно обозначено как “H” или “ атом водорода”, понимают, что атом водорода в данном положении имеет свой природный изотопный состав. Соответственно подразумевается, что любой атом в соединениях по данному изобретению, конкретно обозначенный как дейтерий (D), представляет собой дейтерий.
Соединения бензоксазепиноксазолидинонов
Согласно настоящему изобретению предложены соединения бензоксазепиноксазолидинонов формулы I и фармацевтические композиции на их основе, которые являются потенциально полезными в лечении рака, имеющие структуру:
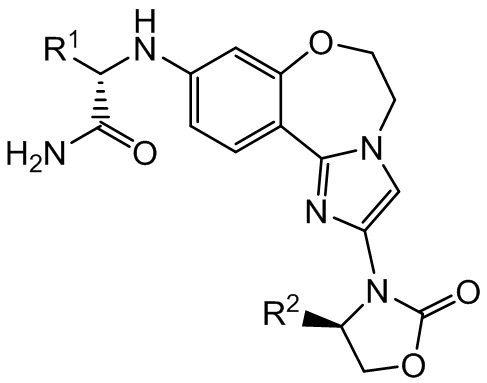 I
I
и стереоизомеры, геометрические изомеры, таутомеры и их фармацевтически приемлемые соли, где:
R1 выбран из -CH3, -CH2CH3, циклопропила и циклобутила;
R2 выбран из -CH3, -CHF2, -CH2F и -CF3.
Иллюстративные варианты осуществления соединений формулы I включают соединения, где R1 представляет собой циклопропил.
Иллюстративные варианты осуществления соединений формулы I включают соединения, где R1 представляет собой CH3 или циклопропил.
Иллюстративные варианты осуществления соединений формулы I включают соединения, где R1 представляет собой CH3.
Иллюстративные варианты осуществления соединений формулы I включают соединения, где R2 представляет собой -CHF2.
Иллюстративные варианты осуществления соединений формулы I включают соединения, где R2 представляет собой -CH2F.
Иллюстративные варианты осуществления соединения формулы I включают соединение, где R1 представляет собой циклопропил, а R2 представляет собой -CHF2.
Иллюстративные варианты осуществления соединения формулы I включают соединение, где R1 представляет собой циклопропил, а R2 представляет собой -CH2F.
Иллюстративные варианты осуществления соединения формулы I включают соединение, где R1 представляет собой CH3, а R2 представляет собой -CHF2
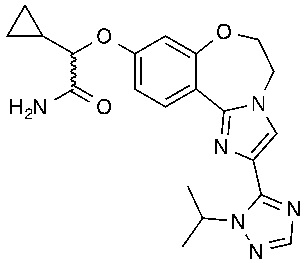
Иллюстративные варианты осуществления соединений формулы I включают соединения из Таблицы 1.
Соединения формулы I по изобретению могут содержать асимметрические или хиральные центры и ввиду этого существовать в разных стереоизомерных формах. Подразумевается, что все стереоизомерные формы соединений по изобретению, включая, но не ограничиваясь этим, диастереомеры, энантиомеры и атропоизомеры, а также их смеси, такие как рацемические смеси, составляют часть настоящего изобретения. В некоторых случаях стереохимическая конфигурация не была определена или была присвоена ориентировочно.
Помимо этого, настоящее изобретение охватывает все диастереомеры, включая цис/транс- (геометрические) и конформационные изомеры. Например, если соединение формулы I содержит двойную связь или конденсированное кольцо, то в объем изобретения включены цис- и транс-формы, а также их смеси.
Если в структурах, приведенных в данном описании, стереохимическая конфигурация какого-либо конкретного хирального атома не конкретизирована, то в качестве соединений по изобретению охвачены и включены все стереоизомеры. Если стереохимическая конфигурация указана сплошной клиновидной или пунктирной линией, изображающей конкретную конфигурацию, то указан и определен именно этот стереоизомер.
Соединения по настоящему изобретению могут существовать в несольватированных, а также сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и тому подобное, и подразумевается, что данное изобретение охватывает как сольватированные, так и несольватированные формы.
Соединения по настоящему изобретению также могут существовать в разных таутомерных формах, и все такие формы включены в объем изобретения. Термин “таутомер” или “таутомерная форма” относится к структурным изомерам, обладающим разной энергией, взаимопревращение которых протекает через низкий энергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) характеризуются взаимопревращениями в результате миграции протона, как например, при кето-енольной и имин-енаминной изомерии. Валентные таутомеры включают взаимопревращения посредством реогранизации некоторых из связывающих электронов.
Биологическая оценка
Величины относительной эффективности соединений формулы I в качестве ингибиторов ферментативной активности (или другой биологической активности) можно определить, установив концентрацию, при которой каждое соединение ингибирует активность до предварительно оговоренной степени и затем сравнив результаты. В типичном случае, предпочтительно определяют концентрацию, при которой активность в биохимическом анализе ингибируется на 50%, т.е. ингибирующую на 50% концентрацию или “IC50”. Определение величин IC50 можно провести, используя традиционные методы, известные в данной области техники. В общем случае IC50 можно определить посредством измерения активности заданного фермента в присутствии изучаемого ингибитора, взятого в диапазоне концентраций. Затем строят график зависимости экспериментально полученных значений ферментативной активности от используемых концентраций ингибитора. Концентрацию ингибитора, при которой наблюдают 50% ферментативной активности (по сравнению с активностью в отсутствие какого-либо ингибитора), принимают за величину IC50. Аналогично, концентрации, вызывающие другой ингибирующий эффект, можно определить посредством соответствующих определений активности. Например, в некоторых случаях постановки задачи может оказаться желательным ввести концентрацию, вызывающую ингибирование на 90%, т.е. IC90, и т.д.
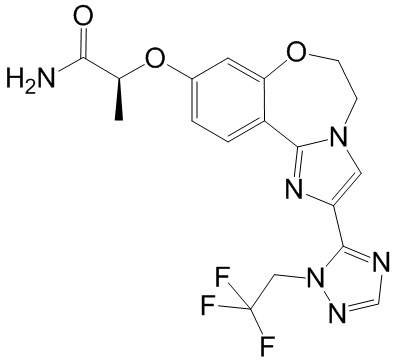
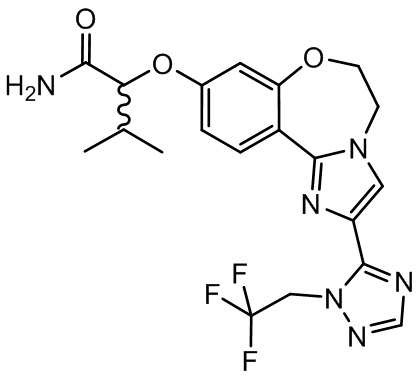
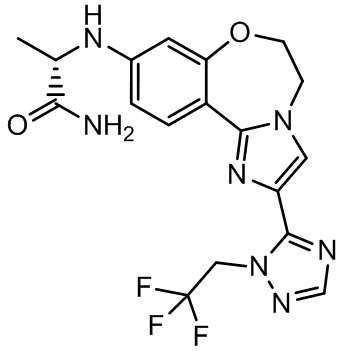
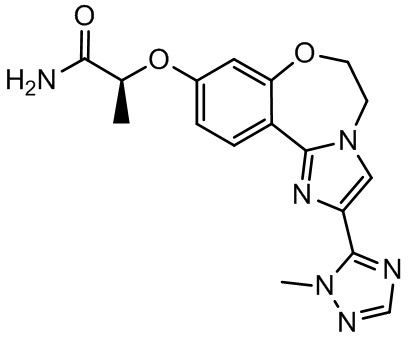
Получали типичные соединения формулы I, приведенные в Таблице 1, определяли их характеристики и тестировали на предмет связывания с различными изоформами и мутантными формами PI3K в соответствии со способами по данному изобретению, и они имеют приведенные далее структуры, соответствующие названия (ChemBioDraw, версия 12.0.02, CambridgeSoft Corp., Cambridge, MA) и биологическую активность. В том случае, когда соединение формулы I или промежуточное соединение соотносится более чем с одним названием, такое соединение будет определяться по химической структуре.
Таблица 1. Соединения формулы I





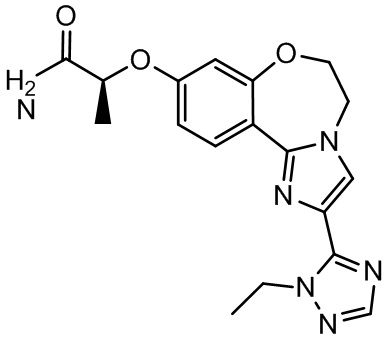
Таселисиб
Соединение, известное как таселисиб, GDC-0032 и Roche RG7604 (регистрационный № в CAS 1282512-48-4, Genentech Inc.), имеет название по IUPAC: 2-(4-(2-(1-изопропил-3-метил-1H-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1H-пиразол-1-ил)-2-метилпропанамид и следующую структуру:
 таселисиб,
таселисиб,
в том числе стереоизомеры, геометрические изомеры, таутомеры и их фармацевтически приемлемые соли.
Таселисиб может быть получен и охарактеризован так, как описано в WO 2011/036280, US 8242104 и US 8343955.
Пиктилисиб
Соединение, известное как пиктилисиб (pictilisib) GDC-0941, Roche, RG-7321, и пиктрелисиб (pictrelisib) (CAS Регистр. № 957054-30-7, Genentech Inc.,) представляет собой сильнодействующий нацеленный на многие мишени ингибитор изоформ PI3K пан-класса I (class I (pan)). В настоящее время GDC-0941 проходит фазу II клинических испытаний на предмет лечения распространенных форм солидных опухолей. GDC-0941 имеет название 4-(2-(1H-индазол-4-ил)-6-((4-(метилсульфонил)пиперазин-1-ил)метил)тиено[3,2-d]пиримидин-4-ил)морфолин (US 7781433; US 7750002; Folkes et al. (2008) Jour. of Med. Chem., 51(18): 5522-5532) и следующую структуру:
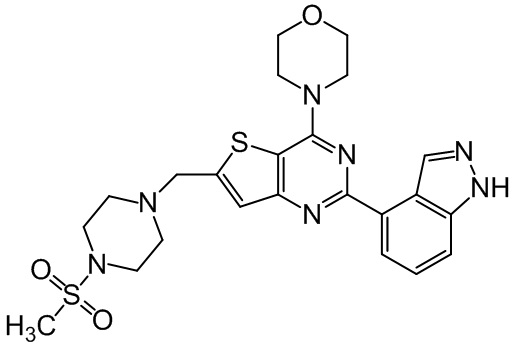 пиктилисиб,
пиктилисиб,
в том числе стереоизомеры, геометрические изомеры, таутомеры и их фармацевтически приемлемые соли.
Алпелисиб
Соединение, известное как алпелисиб (BYL719, Novartis, № в CAS: 1217486-61-7), представляет собой пероральный селективный ингибитор изоформы PI3K-альфа и проходит клинические испытания, касающиеся возможности лечения различного типа опухолей, включая фазу III испытания, в комбинации с фулвестрантом в качестве терапии второй линии в случае положительного к рецепторам гормонов, HER2- распространенного метастатического рака молочной железы (Furet P. et al. (2013) Bioorg. Med. Chem. Lett., 23: 3741-3748; US 8227462; US 8476268; US 8710085). Алпелисиб имеет название (S)-N1-(4-метил-5-(2-(1,1,1-трифтор-2-метилпропан-2-ил)пиридин-4-ил)тиазол-2-ил)пирролидин-1,2-дикарбоксамид) и следующую структуру:
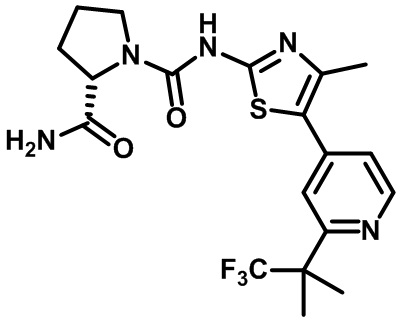 алпелисиб.
алпелисиб.
Биохимические данные, характеризующие ингибирование изоформ pi3k
Способность соединения по изобретению действовать в качестве ингибитора PI3Kα с селективностью, превышающей таковую в отношении PI3Kβ, PI3Kδ и PI3Kγ, определяли, используя методы из примера 901. Значения Ki, показанные в Таблицах 2A и 2B, представляют собой среднее геометрическое значение как минимум из трех независимых экспериментов, если не указано иное.
В Таблице 2A показаны биохимические данные, характеризующие ингибирование четырех изоформ PI3K соединениями формулы I из Таблицы 1. Помимо этого, в качестве соединений сравнения включены два клинически протестированных в отношении PI3K соединения, таселисиб и пиктилисиб. Репрезентативные соединения по изобретению демонстрируют сильную PI3Kα-ингибирующую активность и, по сравнению с таселисибом (GDC-0032) и пиктилисибом (GDC-0941), демонстрируют существенно более высокую селективность в отношении других изоформ PI3Kβ, PI3Kδ и PI3Kγ. В частности, показатели селективности, приведенные во втором справа столбце Таблицы 2A, демонстрируют, что каждое из соединений 101-107 формулы I имеет показатель селективности в отношении PI3K, от альфа до дельта, намного более высокий, чем таселисиб или пиктилисиб. Фактически, и таселисиб, и пиктилисиб проявляют более высокую PI3K-дельта-ингибирующую активность, чем в отношении PI3K-альфа, т.е. их показатели селективности для них меньше 1. Показатели селективности соединений 101-107 формулы I лежат в диапазоне от 301 до 634.
В Таблице 2B показаны биохимические данные, характеризующие ингибирование двух изоформ PI3K, альфа и дельта, и показатели селективности в отношении PI3K, от альфа до дельта, по сравнению с некоторыми соединениями сравнения из US 8242104 и соединением, несущим группу диметилоксазолидин-2-она, из US 8263633 (соединение 356, колонка 149). Эти соединения сравнения, показанные в данном описании в Таблице 2B, представляют собой примеры многочисленных классов соединений, описанных в каждом из US 8242104 и US 8263633. Ни в US 8242104, ни в US 8263633 не описано соединение, входящее в объем соединений формулы I по изобретению. Хотя репрезентативные примеры соединений сравнения из US 8242104, которые описаны в Таблице 2B, демонстрируют показатели селективности в отношении PI3Kα (альфа) относительно PI3Kδ (дельта), превышающие 1, максимальный наблюдаемый показатель селективности составляет 46,9. Таким образом, при применении соединений 101-107 формулы I достигают значительно более высоких показателей селективности, чем в случае соединений из примеров в US 8242104. Ни в US 8242104, ни в US 8263633 нет никаких указаний на то, как осуществить выбор структурных элементов соединений формулы I для приобретения свойства высокой селективности в отношении PI3K-альфа по сравнению с PI3K-дельта. Это неожиданное свойство, заключающееся в приобретении в больше чем в 300 раз более высокой селективности в отношении PI3K-альфа, сохраняется для всего спектра соединений, приведенных в качестве примера в Таблице 1.
Ингибиторы PI3K, проходящие в настоящее время клинические испытания, такие как таселисиб (WO 2011/036280; US 8242104; US 8343955), и другие репрезентативные соединения из примеров в US 8242104 демонстрируют значительную активность против изоформы PI3Kδ (дельта). При этом, отсутствие селективности в отношении PI3Kδ (дельта) согласуется с ЖК токсическим эффектом таселисиба, наблюдаемым в клинических испытаниях. Существует потребность в ингибиторах PI3Kα (альфа), обладающих благоприятными характеристиками, свойственными соединениям из примеров в US 8242104, у которых одновременно с этим отсутствует активность против PI3Kδ (дельта). Согласно данному изобретению предложены соединения, которые соответствуют такой активности и такому профилю селективности.
Это неожиданное свойство селективности в отношении PI3K-альфа является благоприятным в плане устранения желудочно-кишечного токсического эффекта, наблюдаемого в клинических испытаниях кандидатов в ингибиторы PI3K. Благодаря недавно полученным клиническим данным для ингибиторов PI3K отмечено участие активности PI3K-дельта в качестве источника желудочно-кишечных токсических эффектов (Akinleye et al. “Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics”, Journal of Hematology & Oncology, 2013, 6:88-104). См. Таблицу 2 с данными клинических испытаний ингибиторов PI3K - таселисиба и пиктилисиба.
Таким образом, ввиду значительно более высокой селективности ингибирования PI3Kα (альфа) по сравнению с ингибированием PI3Kδ (дельта) можно ожидать, что соединения 101-107 формулы I будут иметь больший интервал между клинической активностью, вызываемой ингибированием PI3Kα (альфа), и токсическими эффектами, вызываемыми ингибированием PI3Kδ (дельта), по сравнению с проходящими клинические испытания таселисибом и пиктилисибом или любым из соединений примеров из US 8242104 или US 8263633. Соответственно, соединения формулы I по изобретению могут быть полезны в качестве терапевтических агентов со сниженным профилем токсичности по сравнению с агентами, которые демонстрируют более сильное ингибирование обычного функционирования PI3Kβ, PI3Kδ или PI3Kγ.
Таблица 2A. Биохимические данные, характеризующие ингибирование изоформ PI3K соединениями формулы I и соединениями сравнения таселисибом и пиктилисибом
Ki (нМ)
Ki (нМ)
Ki (нМ)
GDC-0032
GDC-0941
Таблица 2B. Биохимические данные, характеризующие ингибирование изоформ PI3K соединениями сравнения
(US 8242104)
таселисиб
GDC-0032


(US 8263633)
* Значение Ki представляет собой среднее по двум экспериментам, ** значение Ki представляет собой результат одного эксперимента.
Взаимодействия соединений с pi3k
Рациональная основа селективности соединений формулы I в отношении PI3Kα может заключаться в определенных обуславливающих связывание взаимодействиях.
Способность соединения по изобретению специфически взаимодействовать с PI3Kα оценивали посредством разрешения полученной с использованием рентгеновского излучения совместной кристаллической структуры репрезентативных соединений вместе с PI3Kα (альфа), применяя методы из примера 908. Оптимизированный структурный дизайн ингибиторов PI3K с селективностью в отношении изоформы PI3Kα, превышающей селективность в отношении других изоформ, может включать точную ориентацию и точное расположение атомов и функциональных групп, необходимые для взаимодействия со специфичными для данной изоформы остатками в сайте связывания. В частности, обнаружено, что замещение в положении 9 и в положении 2 5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепиновой кольцевой системы оказывает существенно важное воздействие на специфическую активность соединений против PI3Kα.
На Фиг. 1A-D показаны полученные с использованием рентгеновского излучения совместные кристаллические структуры таселисиба (GDC-0032), соединения сравнения 529 (US 8242104) и двух репрезентативных соединений по изобретению с PI3Kα. Как показано на Фиг. 1A, таселисиб (GDC-0032) содержит функциональную группу первичного амида, которая находится в тесном контакте как с Gln859, так и с Ser854, и по-видимому дает возможность взаимодействий посредством образования водородных связей. Остаток Gln859 специфичен для изоформы PI3Kα, при этом в других изоформах это положение занимает другой остаток (в PI3Kβ это Asp, в PI3Kδ это Asn, в PI3Kγ это Lys). Однако, несмотря на такой тесный контакт с PI3Kα-специфичным остатком, GDC-0032 по результатам измерений в биохимическом анализе имеет одинаковую активность против обеих изоформ PI3Kα и PI3Kδ и лишь слегка сниженную активность против изоформы PI3Kγ (см. Таблицу 2A).
Как показано на Фиг. 1B, соединение сравнения 529 (US 8242104) содержит функциональную группу первичного амида в том же положении, что и таселисиб. Эта функциональная группа находится на соответствующем расстоянии как от Ser854, так и от Gln859, и характеризуется соответствующей геометрией расположения относительно них обоих, чтобы осуществить взаимодействия посредством образования водородных связей. В 46,9 раз более высокий показатель селективности в отношении PI3Kα по сравнению с PI3Kδ (см. Таблицу 2B) может быть логически объяснен с учетом этих взаимодействий и того знания, что PI3Kδ не содержит остатка Gln в положении 859, и поэтому эти взаимодействия должны быть специфичными для PI3Kα.
На Фиг. 1C и 1D показано, что каждый первичный амид (S)-2-аминопропанамидной группы соединения 101 и (S)-2-амино-2-циклопропилацетамидной группы соединения 103 занимает очень схожее по сравнению с первичными амидами GDC-0032 и соединения сравнения 529 (US 8242104) положение в сайте связывания. Эта функциональная группа первичного амида в каждом репрезентативном соединении по изобретению находится на соответствующем расстоянии как от Ser854, так и от Gln859, и характеризуется соответствующей геометрией расположения относительно них обоих, чтобы осуществить взаимодействия посредством образования идеальных водородных связей. Несмотря на явное сходство в размещении и ориентации функциональных групп, репрезентативные соединения из примеров, показанные на Фиг. 1C и 1D, а также другие соединения по данному изобретению со схожими заместителями и функциональными группами демонстрируют улучшенное взаимодействие первичного амида по сравнению как с таселисибом, так и с соединением сравнения 529 (US 8242104), в результате чего соединения по данному изобретению, как установлено, имеют существенно более высокую селективность в отношении PI3Kα по сравнению с PI3Kδ, по результатам измерений в биохимическом анализе. Соединение 101 оказывается в 361 раз более селективным, а соединение 103 оказывается в 634 раза более селективным, что является существенным увеличением по сравнению с соединением сравнения 529 (US 8242104), которое оказывается лишь в 46,9 раза более селективным. С учетом сходства в ориентации функциональной группы первичного амида при сравнении с таселисибом и другими соединениями из US 8242104 (примером которых является соединение сравнения 529) повышенная селективность к PI3Kα по сравнению с PI3Kδ, демонстрируемая соединениями по данному изобретению, является неожиданным свойством. В US 8242104 нет никаких указанийна то, как осуществить выбор структурных элементов соединений формулы I для приобретения свойства высокой (больше чем в 300 раз более высокой) селективности в отношении PI3K-альфа. Соединения формулы I по изобретению демонстрируют улучшенное взаимодействие первичного амида по сравнению с GDC-0032, в результате чего соединения по данному изобретению, как установлено, имеют существенно более высокую селективность в отношении PI3Kα по сравнению с PI3Kδ, по результатам измерений в биохимическом анализе, относительно соединений сравнения (см. Таблицы 2A и 2B).
На Фиг. 2A показана полученная с использованием рентгеновского излучения структура таселисиба, связанного в активном центре PI3Kα (альфа). Атом N2 триазольного кольца не может взаимодействовать непосредственно с боковой цепью Tyr836 (расстояние 4,04 Å) или Ser774 (расстояние 2,74 и 2,82 Å, отсутствие какой-либо взаимодополняющей полярности между лигандом и остатком). На Фиг. 2B показана полученная с использованием рентгеновского излучения структура соединения 101, связанного в активном центре PI3K-альфа, и показано, что оксазолидиноновое кольцо способно осуществлять многочисленные улучшенные взаимодействия с белком по сравнению с триазольным кольцом. Карбонильная функциональная группа располагается близко к боковой цепи Tyr836 (2,67 Å) и способна принимать участие в благоприятном дипольном взаимодействии. Атом фтора заместителя на оксазолидиноновом кольце находится в тесном контакте (2,21 Å) с гидроксильной группой Ser774, что согласуется с возможностью дипольного взаимодействия или образования неклассической водородной связи, благоприятного взаимодействия, обеспечиваемого за счет поляризации связи углерод-фтор (Böhm et al., Fluorine in Medicinal Chemistry, (2004) ChemBioChem, 5: 637-643; Zhou et al., “Fluorine Bonding - How Does it Work In Protein-Ligand Interactions”, (2009) J. Chem. Inf. Model., 49: 2344-2355).
Все соединения по изобретению содержат оксазолидиноновое кольцо и способны принимать участие в улучшенном взаимодействии с Tyr836 в PI3Kα (альфа). Некоторые соединения из примеров по изобретению также содержат фторированный заместитель на оксазолидиноновом кольце и способны принимать участие в улучшенном взаимодействии с Ser774 в PI3Kα. Оба эти обуславливающие связывание взаимодействия могут вносить вклад в повышенную селективность к PI3Kα, наблюдаемую у соединений из примеров по изобретению по сравнению с соединениями из примеров в US 8242104. Как показано в Таблицах 2A и 2B, соединения, которые содержат оксазолидиноновое кольцо, обладают более высокой селективностью в отношении изоформ, чем соединения сравнения, которые содержат триазольное кольцо. Остатки Ser774 и Tyr836 не являются уникальными для изоформы PI3Kα; PI3Kδ содержит эти же остатки в тех же самых положениях, и повышенную селективность для ингибиторов с оксазолидиноновым кольцом в отношении изоформ нельзя предсказать, исходя из этих кристаллических структур. Тонкие различия между разными изоформами в расположении и ориентации одних и тех же остатков могут вытекать из тонких изменений во вторичной и третичной структуре белка. Эти различия трудно предсказать и интерпретировать даже с учетом знания кристаллических структур, полученных с использованием рентгеновского излучения, для обеих изоформ белка. Эти удивительные и неожиданные свойства ингибиторов, содержащих оксазолидиноновое кольцо, принимать участие в улучшенных молекулярных взаимодействиях и проявлять повышенную селективность в отношении изоформ сохраняются для всего спектра соединений, приведенных в качестве примера в Таблице 1.
Оксазолидинон структурно отличается от триазола тем, что оксазолидинон имеет карбонильную группу, является более полярным и не обладает ароматическими свойствами. Триазол не имеет карбонильной группы, является менее полярным и обладает ароматическими свойствами.
Оксазолидиноновое кольцо обеспечивает дополнительное преимущество по сравнению с триазольным кольцом с точки зрения усиления характера гибридизации в сторону образования sp3-орбиталей и меньшего числа ароматических колец. Согласно литературным данным принято считать, что более высокое число ароматических колец коррелирует с повышенным риском случайного связывания. Напротив, увеличение доли атомов углерода в sp3-гибридизации (число атомов углерода в sp3-гибридизации/общее число атомов углерода) коррелирует с улучшенными физико-химическими свойствами и более низкой вероятностью случайного связывания, снижением риска побочного токсикологического действия. Эти сведения приведены в ссылках Lovering et al., “Escape From Flatland”, (2009) J. Med. Chem., 52: 6752-6756 и Ritchie and Macdonald, “Physicochemical Descriptors of Aromatic Character and Their Use in Drug Discovery”, (2014) J. Med. Chem., 57: 7206-7215. Замена триазольного ароматического кольца, примерами которого являются репрезентативные соединения из US 8242104, на насыщенное гетероциклическое кольцо, т.е. оксазолидиноновое кольцо, содержащееся в каждом соединении из примеров по изобретению, представляет собой благоприятное снижение риска побочного токсикологического действия. Совокупность соединений, приведенных в качестве примеров в US 8242104, в подавляющем большинстве случаев представлена соединениями с ароматическими кольцами в этом положении, 4-мя примерами карбоксамидной функциональной группы, замещающей ароматическое кольцо, и при этом нет ни одного примера насыщенных циклических или гетероциклических систем. Вследствие значительно различающихся обуславливающих связывание взаимодействий и стерических требований в случае ароматических и насыщенных гетероциклов, обычно они не являются взаимозаменяемыми. В связи с отсутствием каких-либо примеров насыщенных гетероциклических систем в положении 2 5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепинового кольца, в US 8242104 не приводится никаких указаний относительно способа замены ароматического кольца насыщенным гетероциклом с одновременным сохранением активности против PI3Kα.
Соответственно, соединения по изобретению содержат оптимизированные заместители и функциональные группы как в положении 9, так и в положении 2 5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина. Эти оптимизированные соединения обеспечивают значительное и до настоящего момента неизвестное преимущество, касающееся улучшенных молекулярных взаимодействий и повышенной избирательной активности против PI3Kα, со сниженной активностью против PI3Kδ. Соединения по изобретению могут быть полезны в качестве терапевтических агентов с улучшенным терапевтическим окном по сравнению со схожими агентами, такими как таселисиб (GDC-0032).
Селективное ингибирование мутантной формы pi3kα (альфа)
Способность соединения по изобретению действовать предпочтительно на клетки, содержащие мутантную форму PI3Kα, определяли посредством измерения ингибирования PI3K-пути в изогенных клеточных линиях SW48, содержащих: PI3Kα дикого типа (исходную форму), с мутацией E545K в спиральном домене и с мутацией H1047R в киназном домене, как описано в методах из примера 902.
Статистический анализ: значения EC50 представляют собой среднее геометрическое значение как минимум из 4-х независимых экспериментов, если не указано иное. Все статистические выкладки выполняли с использованием программного обеспечения KaleidaGraph (версия 4.1.3). t-Критерий Стьюдента для сравнения активности в отношении клеток с мутациями и клеток дикого типа определяли с использованием непарных данных с равными дисперсиями. Значение P менее 0,05 считается статистически значимым.
В Таблице 3A показано ингибирование фосфорилированного обогащенного остатками пролина субстрата Akt с ММ 40 кДа (p-PRAS40 - Proline-rich Akt Substrate of 40 kDa) в изогенных клетках SW48 соединениями формулы I из Таблицы 1. Все эти соединения проявляют повышенную активность против клеток с мутантной формой PI3Kα по сравнению с клетками с PI3Kα дикого типа. Соединения по изобретению демонстрируют активность аналогично таселисибу в клетках SW48 с мутантной формой PI3Kα, с селективностью, равной или превышаюшей таковую таселисиба, по сравнению с активностью в клетках с PI3Kα дикого типа (см. Таблицу 3B).
В Таблице 3B показано ингибирование образования p-PRAS40 в изогенных клетках SW48 некоторыми соединениями сравнения из US 8242104, соединением, несущим группу диметилоксазолидин-2-она, из US 8263633 (соединением 356, колонка 149) и пиктилисибом. Соединения сравнения, показанные в данном описании в Таблице 3B, представляют собой примеры многочисленных классов соединений, описанных в каждом из US 8242104 и US 8263633. Ни в US 8242104, ни в US 8263633 не описано соединение, входящее в объем соединений формулы I по данному изобретению. Соединения сравнения включают соединения из примеров, которые не обладают значительно повышенной активностью в отношении клеток с мутантной формой PI3Kα по сравнению с клетками с PI3Kα дикого типа (см. соединения сравнения 436 и 549; p больше 0,05). Эти соединения структурно очень похожи на соединения сравнения, которые демонстрируют значительно более высокую активность в отношении клеток с мутантной формой PI3Kα по сравнению с клетками с PI3Kα дикого типа (см. соединение сравнения 529). У данных соединений сравнения нет никакого общего структурного элемента, который бы давал указания в отношении селективного ингибирования клеток с мутантной формой PI3Kα (альфа). В более широком смысле, ни в US 8242104, ни в US 8263633 нет никаких указаний на то, как осуществить выбор структурных элементов соединений формулы I для приобретения повышенной или эквивалентной активности против клеток с мутантной формой PI3Kα по сравнению с клетками с PI3Kα дикого типа. Это неожиданное свойство сохраняется для всего спектра соединений, приведенных в качестве примера в Таблице 1.
Таблица 3A. Ингибирование Р-PRAS40 в изогенных клетках SW48 соединениями формулы I
Таблица 3B. Ингибирование P-PRAS40 в изогенных клетках SW48 соединениями сравнения
(US 8242104)
таселисиб
GDC-0032
GDC-0941
GDC-0326
*EC50 представляет собой результат одного эксперимента.
Антипролиферативная активность в опухолевых клетках с мутантной формой pi3k
Способность соединения по изобретению действовать на опухолевые клетки, содержащие мутантную форму PI3K, определяли посредством измерения EC50 по жизнеспособности клеток на клетках HCC1954 (PI3Kα с мутацией H1047R) и клетках MCF7 (PI3Kα с мутацией E545K), используя методы из примера 903. В Таблице 4 показано, что репрезентативные соединения формулы I, 101 и 103 по данному изобретению, способны ингибировать PI3K-путь и ингибировать пролиферацию клеток HCC1954 и клеток MCF7 с одинаковым уровнем эффективности, что и соединения сравнения, таселисиб (соединение 196, US 8242104), пиктилисиб и соединение 436 (US 8242104).
Таблица 4. Антипролиферативная активность PI3K-ингибирующих соединений в опухолевых клетках с мутантной формой PI3K-альфа
196 (US 8242104)
GDC-0032
GDC-0941
Эффективность in vivo
В Таблицах 5-8 показаны данные исследований ингибирования роста опухолей (TGI) in vivo PI3K-ингибирующими соединениями. Изменение объема опухолей измеряли в течение 20 суток или больше в группах мышей с ослабленным иммунитетом, несущих ксенотрансплантаты рака молочной железы, которым один раз в сутки перорально (п/о) вводили разбавитель и PI3K-ингибирующие соединения (пример 904).
В Таблице 5 показано, что в максимальных переносимых дозах (MTD) каждое из соединений, GDC-0032 (таселисиб), соединение 103 и соединение 101, оказывается более эффективным, чем алпелисиб (BYL-719) в модели опухоли с мутантной формой PI3K.
В Таблице 6 показано, что в дозах ниже максимальной переносимой дозы (т.е. 25 мг/кг) соединение 101 оказывается более эффективным, чем GDC-0032 в модели опухоли с мутантной формой PI3K. Существует потенциальная возможность для достижения более высокого терапевтического индекса (TI) при использовании соединения 101, поскольку максимальная эффективнная доза достигается раньше максимальной переносимой дозы.
В Таблице 7 показано, что в максимальных переносимых дозах более высокие ответы (PR (Partial Response - частичный ответ) и CR (Complete Response - полный ответ)) наблюдаются при использовании соединения 101 по сравнению с GDC-0032 в модели опухоли с мутантной формой PI3K. Кроме того, в максимальных переносимых дозах GDC-0032 и BYL-719 оказываются одинаково эффективными.
В Таблице 8 показано, что в максимальных переносимых дозах GDC-0032 и соединение 101 оказываются более эффективными, чем BYL-719, и соединение 101 приблизительно столь же эффективно, как и GDC-0032, в модели опухоли с мутантной формой PI3K.
Таблица 5. Сравнение максимальных переносимых доз (MTD) PI3K-ингибирующих соединений в (ER-, PI3KH1047R)-ксенотрансплантатной модели рака молочной железы HCC-1954x1
Таблица 6. Исследование с целью определения оптимальной дозы соединения 101 в (ER-, PI3KH1047R)-ксенотрансплантатной модели рака молочной железы HCC-1954x1
Таблица 7. Исследование с целью определения оптимальной дозы соединения 101 в (ER-, PI3KH1047R)-ксенотрансплантатной модели рака молочной железы KPL-4
Таблица 8. Сравнение GDC-0032, соединения 101 и BYL-719 в (ER+, PI3KH1047R)-ксенотрансплантатной модели на основе выделенных из пациентов (PDX, от англ. patient-derived xenograft) раковых опухолей молочной железы HCI-003
Ингибирование пути в выделенных B-клетках
Способность соединения по изобретению ингибировать PI3K-путь в
B-клетках оценивали по влиянию соединений на уровни CD69 после агонистической обработки a-IgM, используя методы из примера 906. Считается, что экспрессия CD69 в B-клетках, являющаяся результатом обработки a-IgM, управлятся посредством передачи сигнала через PI3Kδ (дельта). В Таблице 9 показано, что репрезентативные соединения формулы I являются более селективными ингибиторами пути передачи сигнала в линии с мутантной формой PI3K (SW48 (H1047R)) относительно B-клеток (колонка 3) по сравнению с таселисибом, пиктилисибом, алпелисибом, соединением 436 (US US8242104) и иделалисибом.
Таблица 9. Ингибирование экспрессии CD69 в B-клетках отдельными соединениями
* IC50 в случае экспрессии CD69, измеренная в цельной крови человека и скорректированная путем умножения на величину fu (доля соединения, не связанная с белками; от англ. fraction of compound unbound to proteins) для человека из эксперимента по связыванию с белками плазмы крови.
Путь ингибирования в опухолевых клетках с мутантной формой pi3k и с pi3k дикого типа
Способность соединения по изобретению ингибировать PI3K-путь передачи сигнала в опухолевых клетках оценивали путем измерения уровней
p-PRAS40 в линиях HCC1954 (PI3Kα с мутацией H1047R) и HDQ-P1 (PI3Kα дикого типа), используя методы из примера 907. В Таблице 10 показано, что репрезентативные соединения 101, 103 и 105 формулы I способны избирательно ингибировать PI3K-путь в опухолевых клетках с мутантной формой PI3Kα (HCC1954, PI3Kα с мутацией H1047R) по сравнению с опухолевыми клетками с PI3Kα дикого типа (HDQ-P1, PI3Kα дикого типа). Соединения 101, 103 и 105 обладают большей селективностью в отношении мутантной формы по сравнению с формой дикого типа, чем соединения сравнения - таселисиб, пиктилисиб, алпелисиб и соединение 436 (US 8242104).
Таблица 10. Ингибирование p-PRAS40 отдельными соединениями в линиях HCC1954 и HDQ-P1
в HDQ-P1 MSD
Деградация p110α в опухолевых клетках с мутантной формой pi3k
Способность соединения по изобретению снижать уровни p110a определяли в экспериментах с линиями HCC1954 (PI3Kα с мутацией H1047R) и HDQ-P1 (PI3Kα дикого типа), используя методы из примера 905. На Фиг. 3A и 3B показаны репрезентативные соединения 101 и 103 формулы I, способные избирательно стимулировать снижение уровней p110a в опухолевых клетках с мутантной формой PI3Kα (HCC1954, с мутацией H1047R) по сравнению с опухолевыми клетками с PI3Kα дикого типа (HDQ-P1, PI3Kα дикого типа) зависимым от концентрации образом. На Фиг. 3A показаны данные вестерн-блоттинга, отображающие уровни p110α (p110a, p110-альфа) в клетках HCC-1954 (PI3Kα с мутацией H1047R) после обработки в течение 24 часов соединением 101, соединением 103 и соединением 436 из US 8242104. На Фиг. 3B показаны данные вестерн-блоттинга, отображающие уровни p110α (p110a, p110-альфа) в HDQ-P1 клетках (PI3Kα дикого типа) после обработки в течение 24 часов соединением 101, соединением 103 и соединением 436 из US 8242104. Соединения 101 и 103 оказывают более сильное влияние на уровни p110α по сравнению с соединением 436 (US 8242104).
Многодневное пероральное введение доз собакам
Способность соединения по изобретению стимулировать желудочно-кишечное и/или системное воспаление либо вызывать истощение лимфоидной ткани оценивали с точки зрения клинической и анатомической патологии после многодневного (7-14 суток) введения доз собакам породы бигль. Соединения 101 и 103 формулы I при более чем 5-ти кратном воздействии для свободного соединения по сравнению с TGI60 (дозой, вызывающей ингибирование роста опухолей на 60%, в исследовании ксенотрансплантатной модели с мутантной формой PI3K) не стимулируют провоспалительной картины по результатам оценки с точки зрения клинической и анатомической патологии (Таблицы 11a, 11b). Аналогично, соединения 101 и 103 при высоких кратностях воздействия вызывают лишь незначительное истощение лимфоидной ткани. Напротив, эксперименты с соединением сравнения таселисибом указывают на значительные провоспалительные эффекты и значительное истощение лимфоидной ткани при менее чем 0,3-кратном воздействии для свободного соединения по сравнению с TGI60 (Таблица 11c). Соединения сравнения алпелисиб (BYL-719) и соединение 436 (US 8242104) также вызывают воспаление и истощение лимфоидной ткани в дозах с более низкими кратностями воздействия по сравнению с соединениями 101 и 103 формулы I (Таблицы 11d, 11e). Величина и степень достоверности полученных результатов согласуются с усилением ингибирования PI3Kδ (дельта) в дозах, соответствующих кратностям воздействия по сравнению с IC50, в отношении экспрессии CD69 для соединений сравнения.
Таблица 11. Многодневное введение доз собаке соединения формулы I и соединений сравнения
(a)
Доза мг/кг/сутки
1х/сут
(общ./свободн., AUC (площадь под кривой))
(общ., Cmin)
14-е сут
(мкМ.ч, общ./свободн.)
14-е сут
(мкМ, общ./свободн.)
14-е сут
(мкМ, общ.)
Fu для собак составляет 0,692; Fu для мышей составляет 0,252 (Fu означает долю несвязанного соединения в образцах плазмы крови).
* Кратности воздействия с использованием значения TGI60 из 21-дневного исследования на модели ксенотрансплантатов KPL4; TGI60 дозе 2 мг/кг; общее воздействие 1,04 мкМ.ч; воздействие для свободного соединения 0,26 мкМ.ч.
** IC50 составляет 67 нМ для a-IgM-стимулированной экспрессии CD69 (в цельной крови).
+ Данные, связанные с воспалением и лимфоидными органами.
(b)
Доза мг/кг/сут
1х/сут
(общ./свободн., AUC)
(общ., Cmin)
14-е сут
(мкМ.ч, общ./ свободн.)
14-е сут
(мкМ, общ./свободн.)
14-е сут
(мкМ, общ.)
0,066
слизистые, 4/4).
Fu для собак составляет 0,507; Fu для мышей составляет 0,03 (Fu означает долю несвязанного соединения в образцах плазмы крови).
* Кратности воздействия с использованием значения TGI60 из 21-дневного исследования TGI на модели с HCC1954; TGI60 в дозе 3 мг/кг; общее воздействие 1,13 мкМ.ч; воздействие для свободного соединения 0,03 мкМ.ч.
** IC50 составляет 142 нМ для a-IgM-стимулированной экспрессии CD69 (в цельной крови).
+ Данные, связанные с воспалением и лимфоидными органами.
(c)
Доза мг/кг/ сутки
1х/сут
(общ./сво-бодн., AUC)
(общ., Cmin)
7-е сут
(мкМ.ч, общ./свободн.)
7-е сут
(мкМ, общ./свободн.)
7-е сут
(мкМ, общ.)
Воспаление ЖК тракта: желудок, нейтрофильное воспаление.
Воспаление ЖК тракта: темно-красные участки в желудке и прямой кишке, соответствующие воспалению в желудке.
нейтрофилы ↑;
моноциты ↑;
глобулины ↑;
соотнош. A:G (альбумин : глобулины) ↓;
фибриноген ↑.
Воспаление ЖК тракта: темно-красные участки в желудке и прямой кишке, соответствующие воспалению в желудке, прямой кишке, слепой кишке.
Системное воспаление: нейтрофильные инфильтраты в лимфатич. узлах, селезенке, тимусе, печени, легком, почке.
Fu для собак составляет 0,28; Fu для мышей составляет 0,10 (Fu означает долю несвязанного соединения в образцах плазмы крови).
* Кратности воздействия с использованием значения TGI60 из 21-дневного исследования на модели ксенотрансплантатов KPL4; TGI60 при общем воздействии 4,3 мкМ.ч или воздействии для свободного соединения 0,44 мкМ.ч.
** IC50 составляет 3,1 нМ для a-IgM-стимулированной экспрессии CD69 (в цельной крови).
+ Данные, связанные с воспалением и лимфоидными органами.
(d)
Доза мг/кг/сут
1х/сут
(общ./свободн., AUC)
(общ., Cmin)
7-е сут
(мкМ.ч, общ./свободн.)
7-е сут
(мкМ, общ./ свободн.)
7-е сут
(мкМ, общ.)
глобулины ↑.
Гипофункция, обильное слюноотделение, усиленная рвота, аномальные фекалии.
нейтрофилы ↑;
фибриноген ↑;
глобулины ↑.
тимус, лимфатич. узлы, селезенка, GALT.
Воспаление ЖК тракта: пищевод, желудок, толстая кишка, слепая кишка.
Системное воспаление: сердце, аорта, оболочки головного мозга.
Fu для собак составляет 0,68; Fu для мышей составляет 0,36 (Fu означает долю несвязанного соединения в образцах плазмы крови).
* Кратности воздействия с использованием значения TGI60 из 21-дневного исследования на модели ксенотрансплантатов KPL4; TGI60 при общем воздействии 2,8 мкМ.ч или воздействии для свободного соединения 1,0 мкМ.ч.
** IC50 составляет 45 нМ для a-IgM-стимулированной экспрессии CD69 (в цельной крови).
+ Данные, связанные с воспалением и лимфоидными органами.
(e)
Доза мг/кг/ сутки
1х/сут
(общ./свободн., AUC)
(общ., Cmin)
7-е сут
(мкМ.ч, общ./свободн.)
7-е сут
(мкМ, общ./свободн.)
7-е сут
(мкМ, общ.)
моноциты ↑;
глобулины ↑;
фибриноген ↑.
Воспаление ЖК тракта: нейтрофильные инфильтраты в толстом и тонком кишечнике; образование язв в прямой кишке.
Fu для собак составляет 0,072; Fu для мышей составляет 0,082 (Fu означает долю несвязанного соединения в образцах плазмы крови).
* Кратности воздействия с использованием значения TGI60 из 21-дневного исследования на модели ксенотрансплантатов KPL4; TGI60 при общем воздействии 72 мкМ.ч или воздействии для свободного соединения 5,9 мкМ.ч.
** IC50 составляет 613 нМ для a-IgM-стимулированной экспрессии CD69 (в цельной крови).
+ Данные, связанные с воспалением и лимфоидными органами.
Введение соединений формулы I
Соединения по изобретению можно вводить любым путем, соответствующим подвергаемому лечению состоянию. Подходящие пути включают пероральный, парентеральный (в том числе подкожный, внутримышечный, внутривенный, интраартериальный, интрадермальный, интратекальный и эпидуральный), трансдермальный, ректальный, назальный, местный (в том числе трансбуккальный и сублингвальный), вагинальный, внутрибрюшинный, внутрилегочный и интраназальный. Что касается местного иммуносупрессивного лечения, то соединения можно вводить в область поражения, включая перфузию трансплантата ингибитором или иное приведение трансплантата в контакт с ингибитором перед трансплантацией. Очевидно, что предпочтительный путь может варьировать в зависимости, например, от состояния реципиента. Если соединение предназначено для перорального введения, то на его основе может быть изготовлена композиция в виде пилюли, капсулы, таблетки и т.д. вместе с фармацевтически приемлемым носителем или эксципиентом. Если соединение предназначено для парентерального введения, то на его основе может быть изготовлена композиция вместе с фармацевтически приемлемым парентеральным разбавителем и в стандартной инъекционной лекарственной форме, как подробно описано ниже.
Доза для лечения пациентов, являющихся людьми, может изменяться от примерно 1 мг до примерно 1000 мг соединения формулы I. Типичная доза может составлять от примерно 10 мг до примерно 300 мг соединения. Дозу можно вводить один раз в сутки (QD, лат. quaque die), два раза в сутки (BID, лат. bis in die) или чаще, в зависимости от фармакокинетических и фармакодинамических свойств, включая всасывание, распределение, метаболизм и экскрецию конкретного соединения. К тому же, на дозировку и схему введения могут влиять факторы, определяющие токсичность. При пероральном введении пилюлю, капсулу или таблетку можно принимать путем проглатывания один раз в сутки или с меньшей частотой в течение определенного периода времени. Эта схема может быть повторена для ряда циклов терапии.
Способы лечения соединениями формулы I
Соединения формулы I по настоящему изобретению полезны для лечения пациента - человека или животного, страдающего от заболевания или расстройства, обусловленного аномальным(ой) клеточным(ой) ростом, функционированием или характером изменений, ассоциированного с PI3K, такого как рак, который таким образом можно лечить способом, включающим введение ему соединения по настоящему изобретению, определенного выше. Пациента - человека или животного, страдающего от рака, также можно лечить способом, включающим введение ему соединения по настоящему изобретению, определенного выше. Тем самым можно нормализовать или улучшить состояние пациента.
Способы по изобретению также включают лечение рака, выбранного из рака молочной железы, яичника, шейки матки, предстательной железы, яичка, мочеполового тракта, пищевода, гортани, глиобластомы, нейробластомы, рака желудка, кожи, кератоакантомы, рака легкого, эпидермоидной карциномы, крупноклеточной карциномы, немелкоклеточного рака легкого (NSCLC), мелкоклеточной карциномы, аденокарциномы легкого, рака кости, толстой кишки, аденомы, рака поджелудочной железы, аденокарциномы, рака щитовидной железы, фолликулярной карциномы, недифференцированной карциномы, папиллярной карциномы, семиномы, меланомы, саркомы, рака мочевого пузыря, рака печени и желчных протоков, рака почки, панкреатического рака, миелоидных расстройств, лимфомы, волосатоклеточного рака, рака щечной полости, носоглотки, глотки, губы, языка, полости рта, тонкого кишечника, толстой кишки-прямой кишки, толстого кишечника, прямой кишки, головного мозга и центральной нервной системы, болезни Ходжкина, лейкоза, рака бронха, щитовидной железы, печени и внутрипеченочных желчных протоков, гепатоклеточного рака, желудочного рака, глиомы/глиобластомы, рака эндометрия, меланомы, рака почки и почечной лоханки, мочевого пузыря, тела матки, шейки матки, множественной миеломы, острого миелогенного лейкоза, хронического миелогенного лейкоза, лимфоцитарного лейкоза, хронического лимфоидного лейкоза (CLL), миелоидного лейкоза, рака ротоглотки, неходжкинской лимфомы, меланомы и ворсинчатой аденомы толстой кишки.
Основываясь на данных анализа экспрессии, иммуногистохимического анализа и на профилирования клеточных линий, можно полагать, что злокачественные новообразования толстой кишки, молочной железы, шейки матки, желудка, легкого и множественная миелома с большой долей вероятности будут поддаваться лечению модуляторами или ингибиторами PI3K.
Изобретение относится к применению соединения, которое описано выше, для лечения рака у пациента.
Изобретение относится к применению соединения, которое описано выше, для изготовления лекарственного средства для лечения рака у пациента.
Изобретение относится к соединению, которое описано выше, для применения для лечения рака у пациента.
Изобретение относится к применению соединения, которое описано выше, для лечения рака у пациента, при этом указанный рак выбран из рака молочной железы и немелкоклеточного рака легкого.
Изобретение относится к применению соединения, которое описано выше, для изготовления лекарственного средства для лечения рака у пациента, при этом указанный рак выбран из рака молочной железы и немелкоклеточного рака легкого.
Изобретение относится к соединению, которое описано выше, для применения для лечения рака у пациента, при этом указанный рак выбран из рака молочной железы и немелкоклеточного рака легкого.
Изобретение, как оно описано выше.
Фармацевтические композиции
Чтобы применить соединение по данному изобретению для терапевтического лечения млекопитающих, в том числе людей, обычно на его основе изготавливают фармацевтическую композицию в соответствии с обычной фармацевтической практикой. Согласно этому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение по данному изобретению вместе с фармацевтически приемлемым разбавителем или носителем.
Типичную композицию изготавливают путем смешивания соединения по настоящему изобретению и носителя, разбавителя или эксципиента. Подходящие носители, разбавители и эксципиенты хорошо известны специалистам в данной области техники и включают такие вещества, как углеводы, воски, растворимые и/или набухаемые в воде полимеры, гидрофильные или гидрофобные вещества, желатин, масла, растворители, вода и тому подобное. Выбор конкретного используемого носителя, разбавителя или эксципиента будет зависеть от средства и цели предполагаемого применения соединения по настоящему изобретению. Как правило, растворители выбирают из растворителей, признанных специалистами в данной области техники безвредными (GRAS) для введения млекопитающему. В общем случае, безвредными растворителями являются нетоксичные водные растворители, такие как вода и другие нетоксичные растворители, которые растворимы в воде или смешиваются с водой. Подходящие водные растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, ПЭГ400, ПЭГ300) и т.д. и их смеси. Композиции также могут включать в себя одно или более чем одно из следующего: буферы, стабилизирующие агенты, поверхностно-активные вещества, увлажняющие агенты, смазывающие вещества, эмульгаторы, суспендирующие агенты, консерванты, антиоксиданты, придающие непрозрачность агенты, вещества, способствующие скольжению, технологические добавки, красители, подсластители, отдушки, корригенты и другие известные вспомогательные вещества для обеспечения наилучшей презентации лекарственного средства (т.е. соединения по настоящему изобретению или фармацевтической композиции на его основе) или для содействия производству фармацевтического продукта (т.е. лекарственного средства).
Композиции могут быть изготовлены с использованием традиционных методик растворения и смешивания. Например, нерасфасованную лекарственную субстанцию (т.е. соединение по настоящему изобретению или стабилизированную форму соединения (например, комплекс с производным циклодекстрина или другим известным агентом комплексообразования)) растворяют в подходящем растворителе в присутствии одного или более эксципиентов, описанных выше. При изготовлении, соединение по настоящему изобретению обычно вводят в состав фармацевтических лекарственных форм, обеспечивающих получение легко контролируемой дозировки лекарственного средства и облегчающих соблюдение больным предписанной схемы лечения.
Фармацевтическая композиция (или препарат) для применения может быть упакована по-разному в зависимости от способа, используемого для введения лекарственного средства. В общем случае, изделие, предназначенное для распространения, содержит контейнер с размещенной в нем фармацевтической композицией в соответствующей форме. Подходящие контейнеры хорошо известны специалистам в данной области техники и включают такие материалы, как бутылки (пластиковые и стеклянные), саше, ампулы, пластиковые пакеты, металлические цилиндрические сосуды и тому подобное. Контейнер также может включать в себя блок, предохраняющий от неумелого обращения, для предупреждения неосторожного доступа к содержимому упаковки. Помимо этого, контейнер снабжен этикеткой с описанием содержимого контейнера. Этикетка также может содержать соответствующие предупредительные надписи.
Фармацевтические композиции на основе соединений по настоящему изобретению могут быть изготовлены для различных путей и типов введения. Например, соединение формулы I желаемой степени чистоты возможно может быть смешано с фармацевтически приемлемыми разбавителями, носителями, эксципиентами или стабилизаторами (Remington's Pharmaceutical Sciences (1980) 16-е издание, ред. Osol A.) в форме лиофилизированной композиции, измельченного порошка или водного раствора. Изготовление композиций может быть осуществлено путем смешивания при температуре окружающей среды при соответствующем pH и желаемой степени чистоты с физиологически приемлемыми носителями, т.е. носителями, являющимися нетоксичными для реципиентов в применяемых дозировках и концентрациях. Величина pH композиции зависит главным образом от конкретного применения и концентрации соединения, но может изменяться в диапазоне от примерно 3 до примерно 8. Подходящим воплощением является композиция, изготовленная в ацетатном буфере, при pH 5.
Как правило, соединение можно хранить в виде твердой композиции, лиофилизированной композиции или в виде водного раствора.
Изготовление, дозирование и введение фармацевтических композиций по изобретению будет осуществлено способом, согласующимся с надлежащей медицинской практикой, т.е. с учетом количеств, концентраций, схем, порядка, наличия разбавителей и путей введения. Факторы, рассматриваемые в этом контексте, включают конкретное подлежащее лечению расстройство, конкретное подлежащее лечению млекопитающее, клиническое состояние отдельного пациента, причину расстройства, место доставки агента, способ введения, схему введения и другие факторы, известные врачам. “Терапевтически эффективное количество” подлежащего введению соединения будет обусловлено такого рода соображениями и составляет минимальное количество, необходимое для уменьшения интенсивности симптомов или лечения гиперпролиферативного расстройства.
Из общих соображений первоначальное фармацевтически эффективное количество ингибитора, вводимого парентерально, из расчета на одну дозу будет находиться в диапазоне примерно 0,01-100 мг/кг, а именно, от примерно 0,1 до 20 мг/кг массы тела пациента в сутки, при этом типичный начальный диапазон для используемого соединения составляет 0,3-15 мг/кг/сутки.
Приемлемые разбавители, носители, эксципиенты и стабилизаторы нетоксичны для реципиентов в применяемых дозировках и концентрациях и включают буферы, например, на основе фосфата, цитрата и других органических кислот; антиоксиданты, в том числе аскорбиновую кислоту и метионин; консерванты (такие как октадецилдиметилбензиламмония хлорид; гексаметонийхлорид; бензалконийхлорид, бензетонийхлорид; фенол, бутиловый или бензиловый спирт; алкилпарабены, такие как метил- или пропил-парабен; катехол; резорцин; циклогексанол; 3-пентанол и м-крезол); низкомолекулярные (менее чем примерно 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатирующие агенты, такие как EDTA (этилендиаминтетрауксусная кислота); сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как ион натрия; комплексы с металлами (например, комплексы Zn-белок); и/или неионные поверхностно-активные вещества, такие как TWEEN™, PLURONICS™ или полиэтиленгликоль (ПЭГ). Кроме того, активные фармацевтические ингредиенты могут быть заключены в микрокапсулы, приготовленные, например, с использованием методов коацервации или путем межфазной полимеризации, например, в микрокапсулы из гидроксиметилцеллюлозы или желатина и микрокапсулы из поли-(метилметакрилата), соответственно, в коллоидные системы лекарственной доставки (например, липосомы, микросферы из альбумина, микроэмульсии, наночастицы и нанокапсулы) или в макроэмульсии. Такие методы описаны в Remington's Pharmaceutical Sciences, 16е издание, ред. Osol A. (1980).
На основе соединений формулы I можно изготовить препараты с длительным высвобождением. Подходящие примеры препаратов с длительным высвобождением включают полупроницаемые матрицы из гидрофобных полимеров в твердой форме, содержащие соединение формулы I, причем эти матрицы существуют в форме штампованных изделий, например, пленок или микрокапсул. Примеры матриц с длительным высвобождением включают сложные полиэфиры, гидрогели (например, поли(2-гидроксиэтил-метакрилат) или поли(виниловый спирт)), полилактиды (US 3773919), coполимеры L-глутаминовой кислоты и гамма-этил-L-глутамата, неразлагаемый этилен-винилацетат, разлагаемые coполимеры молочной кислоты и гликолевой кислоты, такие как LUPRON DEPOT™ (инъецируемые микросферы, состоящие из coполимера молочной кислоты и гликолевой кислоты и из ацетата лейпролида), и поли-D-(-)-3-гидроксимасляную кислоту.
Данные композиции включают композиции, подходящие для путей введения, подробно изложенных в данном описании. Может быть удобным представление композиций в стандартной лекарственной форме, и они могут быть изготовлены любым из способов, хорошо известных в области фармацевтики. Методы и композиции обычно можно найти в Remington's Pharmaceutical Sciences (Mack Publishing Co., Easton, PA). Такие способы включают стадию приведения активного ингредиента в ассоциацию с носителем, который состоит из одного или более дополнительных ингредиентов. В общем случае композиции изготавливают путем непрерывного и равномерного приведения активного ингредиента в ассоциацию с жидкими носителями или тонкоизмельченными твердыми носителями или ими обоими и затем, при необходимости, придания формы продукту.
Композиции на основе соединения формулы I, подходящие для перорального введения, могут быть изготовлены в виде дискретных единиц, таких как пилюли, капсулы, облатки или таблетки, каждая из которых содержит предварительно заданное количество соединения формулы I. Прессованные таблетки могут быть изготовлены в подходящей машине путем прессования активного ингредиента в свободно текучей форме, такой как порошок или гранулы, возможно в смеси со связующим веществом, смазывающим веществом, инертным разбавителем, консервантом, поверхностно-активным или диспергирующим агентом. Формованные таблетки могут быть изготовлены в подходящей машине путем формования смеси порошкообразного активного ингредиента, увлажненного инертным жидким разбавителем. Возможно, что таблетки могут иметь покрытие или риску и возможно могут быть изготовлены таким образом, чтобы обеспечить медленное или регулируемое высвобождение из нее активного ингредиента. Для перорального применения можно изготовить таблетки, лепешки, пастилки, водные или масляные суспензии, диспергируемые порошки или гранулы, эмульсии, твердые или мягкие капсулы, например желатиновые капсулы, сиропы или эликсиры. Композиции на основе соединения формулы I, предназначенные для перорального применения, могут быть изготовлены в соответствии с любым способом, известным в области изготовления фармацевтических композиций, и такие композиции могут содержать один или более агентов, включая подсластители, корригенты, красители и консерванты, с целью придания препарату приятного вкуса. Приемлемыми являются таблетки, содержащие активный ингредиент в смеси с нетоксичным фармацевтически приемлемым эксципиентом, подходящим для изготовления таблеток. Этими эксципиентами могут быть, например, инертные разбавители, такие как карбонат кальция или натрия, лактоза, фосфат кальция или натрия; гранулирующие и разрыхляющие агенты, такие как кукурузный крахмал или альгиновая кислота; связующие вещества, такие как крахмал, желатин или аравийская камедь; и смазывающие вещества, такие как стеарат магния, стеариновая кислота или тальк. Таблетки могут не иметь покрытия или могут быть покрыты оболочкой с использованием известных методов, включая микроинкапсулирование, для задержки распадаемости и всасывания в желудочно-кишечном тракте и тем самым для обеспечения длительного действия в течение большего периода времени. Например, можно использовать такое вещество-замедлитель, как глицерилмоностеарат или глицерилдистеарат, сами по себе или вместе с воском.
Для лечения глазных или других внешних тканей, например, полости рта и кожи, предпочтительно применяют композиции в виде мази или крема для местного применения, содержащие активный(ые) ингредиент(ы) в количестве, например, 0,075-20% масс./масс. При изготовлении композиции в виде мази активные ингредиенты могут быть использованы либо с парафиновой, либо со смешивающейся с водой мазевой основой. Альтернативно, на основе активных ингредиентов могут быть изготовлены композиции в виде крема с использованием основы для крема типа масло-в-воде. При желании водная фаза основы для крема может включать многоатомный спирт, т.е. спирт, имеющий две или более чем две гидроксильные группы, такой как пропиленгликоль, бутан-1,3-диол, маннит, сорбит, глицерин и полиэтиленгликоль (включая ПЭГ400) и их смеси. Композиции для местного применения при желании могут включать в себя соединение, которое усиливает всасывание или проникновение активного ингредиента через кожу или другие зоны воздействия. Примеры таких усилителей проникновения через кожу включают диметилсульфоксид и родственные аналоги. Масляная фаза эмульсий по данному изобретению может быть составлена из известных ингредиентов известным образом. При том, что эта фаза может содержать только эмульгатор, желательно, чтобы она содержала смесь по меньшей мере одного эмульгатора с жиром или маслом либо и с жиром, и с маслом обоими. Предпочтительно, гидрофильный эмульгатор включают вместе с липофильным эмульгатором, который действует как стабилизатор. Также предпочтительно включение в состав как масла, так и жира. Эмульгатор(ы) совместно со стабилизатором(ами) или без него(них) образуют так называемый эмульгирующий воск, и этот воск вместе с маслом и жиром составляет так называемую эмульгирующую мазевую основу, которая образует масляную дисперсионную фазу композиций в форме крема. Эмульгаторы и стабилизаторы эмульсий, подходящие для использования в композиции по изобретению, включают Tween® 60, Span® 80, цетостеариловый спирт, бензиловый спирт, миристиловый спирт, глицерилмоностеарат и лаурилсульфат натрия.
Водные суспензии соединений формулы I содержат активные вещества в смеси с эксципиентами, подходящими для изготовления водных суспензий. Такие эксципиенты включают суспендирующий агент, такой как карбоксиметилцеллюлозы натриевая соль, кроскармелоза, повидон, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь, и диспергирующие или увлажняющие агенты, такие как природный фосфатид (например, лецитин), продукт конденсации алкиленоксида с жирной кислотой (например, полиоксиэтиленстеарат), продукт конденсации этиленоксида с длинноцепочечным алифатическим спиртом (например, гептадекаэтиленокси-цетанол), продукт конденсации этиленоксида с неполным сложным эфиром, являющимся производным жирной кислоты и ангидрида гексита (например, полиоксиэтиленсорбитан-моноолеат). Водная суспензия также может содержать один или более консервантов, таких как этил- или н-пропил-пара-гидроксибензоат, один или более красителей, один или более корригентов и один или более подсластителей, таких как сахароза или сахарин.
Фармацевтические композиции на основе соединений формул I могут быть в форме стерильного препарата для инъекций, такого как стерильная водная или масляная суспензия для инъекций. Эта суспензия может быть изготовлена согласно известным в данном области техники способам с использованием таких подходящих диспергирующих или увлажняющих агентов и суспендирующих агентов, которые упомянуты выше. Стерильный препарат для инъекций также может представлять собой стерильный инъекционный раствор или стерильную инъекционную суспензию в нетоксичном приемлемом для парентерального введения разбавителе или растворителе, как например, раствор в 1,3-бутандиоле, или может быть изготовлен из лиофилизированного порошка. Среди приемлемых разбавителей и растворителей, которые могут быть использованы, находится вода, раствор Рингера и изотонический раствор хлорида натрия. Помимо этого, в качестве растворителя или суспендирующей среды традиционно можно использовать стерильные нелетучие масла. Для этой цели может быть использовано любое маловязкое нелетучее масло, включая синтетические моно- или диглицериды. В дополнение к этому, в изготовлении инъекционных средств таким же образом могут быть использованы жирные кислоты, такие как олеиновая кислота.
Количество активного ингредиента, которое можно комбинировать с веществом-носителем для изготовления разовой лекарственной формы, будет варьировать в зависимости от подвергаемого лечению реципиента и конкретного способа введения. Например, композиция с замедленным высвобождением, предназначенная для перорального введения людям, может содержать приблизительно 1-1000 мг активного вещества в смеси с соответствующим и удобным количеством вещества-носителя, которое может варьировать от примерно 5 до примерно 95% от массы всей композиции (масса:масса). Фармацевтическая композиция может быть изготовлена таким образом, чтобы беспрепятственно обеспечить введение измеряемого количества. Например, водный раствор, предназначенный для внутривенной инфузии, может содержать от примерно 3 до 500 мкг активного ингредиента на один миллилитр раствора с тем, чтобы можно было осуществлять инфузию подходящего объема со скоростью примерно 30 мл/ч.
Композиции, подходящие для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатические средства и растворенные вещества, делающие композицию изотоничной крови предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать в себя суспендирующие агенты и загустители.
Композиции, подходящие для местного введения в глаз, также включают глазные капли, где активный ингредиент растворен или суспендирован в подходящем носителе, в частности, в водном растворителе для активного ингредиента. Активный ингредиент предпочтительно присутствует в таких композициях в концентрации приблизительно 0,5-20% масс./масс., например, приблизительно 0,5-10% масс./масс., например, приблизительно 1,5% масс./масс.
Композиции, подходящие для местного введения в полость рта, включают пастилки, содержащие активный ингредиент в ароматизированной основе, обычно сахарозе и аравийской камеди или трагаканте; пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и аравийская камедь; и жидкости для полоскания рта, содержащие активный ингредиент в подходящем жидком носителе.
Композиции для ректального введения могут быть представлены в виде суппозитория с подходящей основой, содержащей, например масло какао или салицилат.
Композиции, подходящие для внутрилегочного или назального введения, имеют размер частиц, например, в диапазоне 0,1-500 микрон (в том числе размеры частиц в диапазоне от 0,1 до 500 микрон с таким шагом, как 0,5, 1, 30 микрон, 35 микрон и т.д.), которые вводят посредством быстрой ингаляции через носовой проход или посредством ингаляции через рот, чтобы достичь альвеолярных мешочков. Подходящие композиции включают водные или масляные растворы активного ингредиента. Композиции, подходящие для введения аэрозоля или сухого порошка, могут быть изготовлены традиционными способами и могут быть доставлены вместе с другими терапевтическими агентами, такими как соединения, до сих пор используемые в лечении или профилактике расстройств, которые описаны ниже.
Композиции, подходящие для вагинального введения, могут быть представлены в виде композиций в форме пессариев, тампонов, кремов, гелей, паст, пенок или спрея, содержащих помимо активного ингредиента такие носители, целесообразность использования которых в данной области техники известна.
Композиции могут быть упакованы в однодозовые или многодозовые контейнеры, например, герметично закрытые ампулы и флаконы, и их можно хранить в высушенном сублимационной сушкой (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например воды, для инъекций непосредственно перед применением. Экстемпоральные растворы и суспензии для инъекций готовят из стерильных порошков, гранул и таблеток описанного ранее вида. Предпочтительными стандартными дозированными лекарственными формами являются такие, которые содержат суточную дозу или стандартную суточную субдозу активного ингредиента, как приведено в данном описании выше, или соответствующую ее долю.
Согласно изобретению также предложены ветеринарные композиции, содержащие по меньшей мере один активный ингредиент, который определен выше, вместе с приемлемым в ветеринарии соответствующим носителем. Приемлемыми в ветеринарии носителями являются вещества, полезные для введения композиции, и они могут быть твердыми, жидкими или газообразными веществами, которые в остальном инертны или приемлемы в области ветеринарии и совместимы с активным ингредиентом. Эти ветеринарные композиции можно вводить парентерально, перорально или любым другим желаемым путем.
Комбинированная терапия
Соединения формулы I могут быть применены по отдельности или в комбинации с дополнительными терапевтическими агентами для лечения заболевания или расстройства, описанного в данной заявке, такого как воспаление или гиперпролиферативное расстройство (например, рак). В некоторых воплощениях соединение формулы I объединяют в фармацевтической комбинированной композиции, или схеме введения в виде комбинированной терапии, с дополнительным, вторым терапевтическим соединением, которое обладает противовоспалительными или антигиперпролиферативными свойствами, или таким соединением, которое полезно для лечения воспаления, нарушения иммунной системы или гиперпролиферативного расстройства (например, рака). Дополнительным терапевтическим агентом может быть ингибитор Bcl-2, ингибитор JAK, противовоспалительный агент, иммуномодулирующий агент, химиотерапевтический агент, усилитель апоптоза, нейротропный фактор, агент для лечения сердечно-сосудистого заболевания, агент для лечения заболевания печени, противовирусный агент, агент для лечения заболевания крови, агент для лечения диабета и агент для лечения иммунодефицитных состояний. Вторым терапевтическим агентом может быть противовоспалительный агент NSAID (нестероидное противовоспалительное лекарственное средство). Вторым терапевтическим агентом может быть химиотерапевтический агент. Второе соединение в такой фармацевтической комбинированной композиции или схеме введения предпочтительно обладает взаимодополняющими активностями по отношению к соединению формулы I, такими, что они не оказывают неблагоприятного влияния друг на друга. Такие соединения соответственно присутствуют в комбинации в количествах, эффективных для предполагаемой задачи. В одном из воплощений композиция по данному изобретению содержит соединение формулы I либо его стереоизомер, таутомер, сольват, метаболит либо фармацевтически приемлемую соль или их пролекарство в комбинации с таким терапевтическим агентом, как NSAID.
Введение при комбинированной терапии может быть осуществлено по одновременной или последовательной схеме. В случае последовательного введения комбинация может быть введена за два или более чем два раза. Введение при комбинированной терапии включает совместное введение, при котором используются отдельные композиции или единая фармацевтическая композиция, и последовательное введение в любом порядке, при котором, предпочтительно, есть период времени, когда оба активных агента (или все активные агенты) одновременно проявляют свои биологические активности.
Подходящими дозировками для любого из вышеупомянутых вводимых совместно агентов являются дозировки, используемые в настоящее время, и они могут быть снижены вследствие объединенного действия (синергизма) нового идентифицированного агента и других терапевтических агентов или схем лечения.
Комбинированная терапия может обеспечивать “синергизм” и проявлять “синергетический характер”, т.е. эффект, достигаемый при совместном использовании активных ингредиентов, больше суммы эффектов, получаемых в результате использования этих соединений по отдельности. Синергетический эффект может быть получен тогда, когда активные ингредиенты: (1) совместно включают в состав композиции при ее изготовлении и вводят или доставляют одновременно в комбинированной, стандартной дозированной лекарственной форме; (2) доставляют путем чередования или параллельно в виде отдельных композиций; или (3) доставляют с использованием какой-либо другой схемы (введения). При доставке с использованием чередующейся терапии синергетический эффект может быть получен тогда, когда соединения вводят или доставляют последовательно, например, посредством разных инъекций в отдельных шприцах, отдельных пилюлях или капсулах либо отдельных капельницах. В общем случае, при чередующейся терапии эффективную дозировку каждого активного ингредиента вводят последовательно, т.е., сериями, тогда как при комбинированной терапии эффективные дозировки двух или более активных ингредиентов вводят совместно.
В конкретном воплощении, относящемся к терапии, соединение формулы I либо его стереоизомер, таутомер, сольват, метаболит либо фармацевтически приемлемая соль или их пролекарство могут быть скомбинированы с другими терапевтическими агентами, гормональными агентами или агентами на основе антител, как например, агенты, описанные в данной заявке, а также скомбинированы с хирургической терапией и лучевой терапией. Таким образом, комбинированные терапии по настоящему изобретению включают введение по меньшей мере одного соединения формулы I либо его стереоизомера, таутомера, сольвата, метаболита или фармацевтически приемлемой соли или их пролекарства и применение по меньшей мере одного или более чем одного другого способа лечения рака. Количества соединения(ий) формулы I и другого(их) фармацевтически активного(ых) терапевтического(их) агента(ов) и относительные временные интервалы введения будут выбраны с целью достижения желаемого комбинированного терапевтического эффекта.
Дополнительные терапевтические агенты, применяемые в комбинации с соединением формулы I, включают 5-FU, доцетаксел, эрибулин, гемцитабин, кобиметиниб, ипатасертиб, паклитаксел, тамоксифен, фулвестрант, GDC-0810, дексаметазон, палбоциклиб, бевацизумаб, пертузумаб, трастузумаба эмтанзин, трастузумаб и летрозол.
Метаболиты соединений формулы i
Кроме того, в объем данного изобретения включены продукты метаболизма in vivo соединений формулы I, описанных в данной заявке. Такие продукты могут получаться из вводимого соединения, например, в результате его окисления, восстановления, гидролиза, амидирования, дезамидирования, этерификации, деэтерификации, ферментативного расщепления и тому подобного. Соответственно, изобретение включает метаболиты соединений формулы I, в том числе соединений, получаемых способом, включающим приведение соединения по данному изобретению в контакт с млекопитающим в течение периода времени, достаточного для получения продукта его метаболизма.
Продукты метаболизма обычно идентифицируют посредством получения меченного радиоактивным (например, 14C или 3H) изотопом соединения по изобретению, введения его парентеральным способом в детектируемой дозе (например, более чем примерно 0,5 мг/кг) животному, такому как крыса, мышь, морская свинка, обезьяна, или человеку, с обеспечением достаточного для осуществления метаболизма времени (обычно примерно от 30 секунд до 30 часов) и выделения продуктов его превращения из мочи, крови или других биологических образцов. Эти продукты легко выделить, поскольку они являются мечеными (другие выделяют посредством применения антител, способных к связыванию с эпитопами, сохранившимися в метаболите). Структуры метаболитов определяют традиционным образом, например, анализом с использованием масс-спектрометрии (MS), жидкостной хроматографии в сочетании с MS (LC/MS) или ядерного магнитного резонанса (ЯМР). В общем случае, анализ метаболитов осуществляют так же, как и традиционные исследования метаболизма лекарственных средств, хорошо известные специалистам в данной области техники. Продукты метаболизма, при условии, что в других случаях они не обнаруживаются in vivo, полезны в диагностических анализах для определения терапевтической дозы соединений по изобретению.
Изделие производства
В другом воплощении изобретения предложены изделие производства или “набор”, содержащие вещества, полезные для лечения описанных выше заболеваний и расстройств. В одном из воплощений набор включает в себя контейнер, содержащий соединение формулы I или его стереоизомер, таутомер, сольват, метаболит либо фармацевтически приемлемую соль или их пролекарство. Кроме того, набор может содержать этикетку или инструкцию по применению, находящуюся на контейнере или вместе с ним. Термин “инструкция по применению” обычно относится к инструкциям, традиционно включаемым в коммерческие упаковки терапевтических продуктов, которые содержат информацию о показаниях, применении, дозировке, введении, противопоказаниях и/или предупреждениях касательно применения таких терапевтических продуктов. Подходящие контейнеры включают, например, бутылки, флаконы, шприцы, блистерную упаковку и т.д. Контейнер может быть сделан из разнообразных материалов, таких как стекло или пластик. Контейнер может включать в себя соединение формулы I или композицию на его основе, которые эффективны для лечения указанного состояния, и может иметь стерильное входное отверстие (например, контейнер может представлять собой мешочек или флакон для внутривенных растворов, имеющий пробку, поддающуюся прокалыванию иглой для подкожных инъекций). По меньшей мере одним активным агентом в композиции является соединение формулы I. На этикетке или в инструкции по применению указывается, что данную композицию применяют для лечения выбранного состояния, такого как рак. Помимо этого, на этикетке или в инструкции по применению может быть указано, что подлежащим лечению пациентом является пациент с таким расстройством, как гиперпролиферативное расстройство, нейродегенерация, гипертрофия сердца, боль, мигрень или нейротравматическое заболевание или событие. В одном из воплощений на этикетке или в инструкциях по применению указывается, что композицию, содержащую соединение формулы I, можно использовать для лечения расстройства, являющегося результатом аномального клеточного роста. На этикетке или в инструкции по применению также может быть указано, что данную композицию можно использовать для лечения других расстройств. Альтернативно или помимо этого такое изделие производства может дополнительно включать в себя второй контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), забуференный фосфатом физиологический раствор, раствор Рингера и раствор декстрозы. Кроме того, оно может включать в себя другие материалы, подходящие с коммерческой точки зрения и точки зрения потребителя, в том числе другие буферы, разбавители, фильтры, иглы и шприцы.
Набор может дополнительно содержать указания относительно введения соединения формулы I и второй фармацевтической композиции, если она присутствует. Например, если набор включает в себя первую композицию, содержащую соединение формулы I, и вторую фармацевтическую композицию, то такой набор может дополнительно содержать указания относительно одновременного, последовательного или раздельного введения первой и второй фармацевтических композиций нуждающемуся в этом пациенту.
В другом воплощении наборы подходят для доставки твердых пероральных форм на основе соединения формулы I, таких как таблетки или капсулы. Такой набор предпочтительно содержит ряд стандартных дозировок. Такие наборы могут включать в себя карточку с дозировками, расположенными в порядке их предполагаемого применения. Примером такого набора является “блистерная упаковка”. Блистерные упаковки хорошо известны в упаковочной индустрии и широко используются для упаковывания фармацевтических стандартных лекарственных форм. При желании может быть придана памятка, например, в виде чисел, букв или других маркировок либо с календарем-вкладышем, где указаны дни в схеме лечения, в которые можно вводить дозировки.
Согласно одному из воплощений набор может включать в себя (a) первый контейнер с содержащимся в нем соединением формулы I; и возможно (b) второй контейнер с содержащейся в нем второй фармацевтической композицией, при этом вторая фармацевтическая композиция содержит второе соединение, обладающее антигиперпролиферативной активностью. Альтернативно или помимо этого набор может дополнительно включать в себя третий контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекций (BWFI), забуференный фосфатом физиологический раствор, раствор Рингера и раствор декстрозы. Кроме того, он может включать в себя другие материалы, подходящие с коммерческой точки зрения и точки зрения потребителя, в том числе другие буферы, разбавители, фильтры, иглы и шприцы.
В некоторых других воплощениях, где набор включает в себя композицию на основе соединения формулы I и второй терапевтический агент, такой набор может содержать контейнер для помещения в него отдельных композиций, такой как разделенная бутылка или разделенный пакет из фольги, однако, отдельные композиции также могут содержаться в едином неразделенном контейнере. Обычно, набор содержит указания по введению отдельных компонентов. Форма набора имеет особое преимущество в тех случаях, когда отдельные компоненты предпочтительно вводят в разных лекарственных формах (например, пероральной и парентеральной), вводят с различными интервалами дозирования или в тех случаях, когда согласно предписанию врача желательно провести титрование индивидуальных компонентов комбинации.
Получение соединений формулы i
Соединения формулы I могут быть синтезированы с использованием путей синтеза, включающих способы, аналогичные хорошо известным в области химии, в частности, с учетом содержащегося в данной заявке описания, и способы, используемые для синтеза других гетероциклических соединений, описанные в работах: Comprehensive Heterocyclic Chemistry II, редакторы Katritzky и Rees, Elsevier, 1997, например в томе 3; Liebigs Annalen der Chemie, (9): 1910-16, (1985); Helvetica Chimica Acta, 41: 1052-60, (1958); Arzneimittel-Forschung, 40(12): 1328-31, (1990), каждая из которых явным образом включена посредством ссылки. Обычно, исходные вещества приобретают в коммерческих источниках, таких как Aldrich Chemicals (Milwaukee, WI), или легко получают, используя способы, хорошо известные специалистам в данной области техники (например, получают способами, в целом описанными в Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-23, Wiley, N.Y. (изд. 1967-2006 г.г.) или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая приложения (также доступные через онлайн-базу данных Бейльштейна).
Методологии химических преобразований в ходе синтеза и использования защитных групп (введения и удаления защиты), полезные в синтезе соединений формулы I, и необходимые реагенты и промежуточные соединения известны в данной области техники и включают, например, таковые, описанные в R. Larock, Comprehensive Organic Transformations, VCH Publishers (1989); T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd Ed., John Wiley and Sons (1999); и L.Paquette, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995) и в их последующих изданиях.
В разделе Примеры приведены типичные способы получения соединений формулы I. Специалистам в данной области техники будет очевидно, что для синтеза соединений формулы I можно использовать и другие пути синтеза. Хотя на фигурах и в разделе Примеры изображены и описаны конкретные исходные вещества и реагенты, они могут быть легко заменены на другие исходные вещества и реагенты для получения целого ряда производных и/или реакционных условий. Помимо этого, многие типичные соединения, полученные описанными способами, могут быть дополнительно модифицированы с учетом данного описания с использованием общепринятых методов химии, хорошо известных специалистам в данной области техники.
При получении соединений формул I может потребоваться защита функциональной группы (например, первичного или вторичного амина) в промежуточных соединениях. Необходимость в такой защите будет варьировать в зависимости от природы такой функциональной группы и условий способов получения. Подходящие амино-защитные группы включают ацетил, трифторацетил, трет-бутоксикарбонил (BOC), бензилоксикарбонил (CBz) и 9-флуоренилметиленоксикарбонил (Fmoc). Необходимость в такой защите легко определяется специалистом в данной области техники. Общее описание защитных групп и их применение см. в T. W.Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
В способах получения соединений формулы I предпочтительным может быть отделение продуктов реакции друг от друга и/или от исходных веществ. Желаемые продукты с каждой стадии или серии стадий разделяют и/или очищают до желаемой степени гомогенности методами, общеизвестными в данной области техники. Обычно такие методы разделения включают в себя многофазную экстракцию, кристаллизацию из растворителя или смеси растворителей, дистилляцию, сублимацию или хроматографию. Хроматография может включать в себя любое количество методов, в том числе, например: обращенно-фазовую и нормально-фазовую; гель-проникающую; ионообменную хроматографию; методы и устройство для жидкостной хроматографии высокого, среднего и низкого давления; мелкомасштабную аналитическую хроматографию; хроматографию с псевдодвижущимся слоем (SMB) и препаративную тонкослойную и толстослойную хроматографию, а также методы мелкомасштабной тонкослойной и флэш-хроматографии.
Другой класс методов разделения включает обработку смеси реагентом, выбранным для связывания с желаемым продуктом, непрореагировавшим исходным веществом, побочным продуктом реакции или тому подобным либо оказывающим на все вышеперечисленное какое-либо иное действие, способствующее разделению. Такие реагенты включают адсорбенты или абсорбенты, как например, активированный уголь, молекулярные сита, ионообменные среды или тому подобное. Альтернативно, такими реагентами могут быть кислоты в случае основного вещества, основания в случае кислотного вещества, связывающие реагенты, такие как антитела, связывающие белки, селективные хелаторы, такие как краун-эфиры, реагенты для жидкость/жидкостной ионной экстракции (LIX) или тому подобное. Выбор соответствующих методов разделения зависит от природы рассматриваемых веществ, например, от точки кипения и молекулярной массы при дистилляции и сублимации, присутствия или отсутствия полярных функциональных групп при хроматографии, стабильности веществ в кислотных и щелочных средах при многофазной экстракции и тому подобного.
Диастереомерные смеси могут быть разделены на свои индивидуальные диастереомеры на основе различий их физико-химических свойств методами, хорошо известными специалистам в данной области техники, например, посредством хроматографии и/или фракционной кристаллизации. Энантиомеры можно разделить путем превращения энантиомерной смеси в диастереомерную смесь в результате взаимодействия с соответствующим оптически активным соединением (например, хиральным вспомогательным веществом, таким как хиральный спирт или хлорангидрид кислоты Мошера), разделения диастереомеров и превращения (например, посредством гидролиза) индивидуальных диастереоизомеров в соответствующие чистые энантиомеры. Кроме того, некоторые соединения по настоящему изобретению могут быть атропоизомерами (например, замещенные биарилы), и они рассматриваются как часть данного изобретения. Энантиомеры также могут быть разделены с применением колонки для хиральной HPLC (high pressure liquid chromatography).
Индивидуальный стереоизомер, например энантиомер, по существу не содержащий своего стереоизомера, можно получить путем разделения рацемической смеси, используя такой метод, как образование диастереомеров с применением оптически активных разделяющих агентов (Eliel E. and Wilen S., “Stereochemistry of Organic Compounds”, John Wiley & Sons, Inc., New York, 1994; Lochmuller C.H. (1975) J. Chromatogr., 113(3): 283-302). Рацемические смеси хиральных соединений по изобретению могут быть разделены и соединения выделены любым подходящим методом, включая: (1) образование ионных диастереомерных солей с хиральными соединениями и разделение фракционной кристаллизацией или другими методами, (2) образование диастереомерных соединений с использованием хиральных дериватизирующих реагентов, разделение диастереомеров и превращение в чистые стереоизомеры и (3) разделение по существу чистых или обогащенных стереоизомеров непосредственно в хиральных условиях. См.: “Drug Stereochemistry, Analytical Methods and Pharmacology”, Irving W. Wainer, Ed., Marcel Dekker, Inc., New York (1993).
Согласно методу (1) диастереомерные соли могут быть образованы в результате взаимодействия энантиомерно чистых хиральных оснований, таких как бруцин, хинин, эфедрин, стрихнин, α-метил-β-фенилэтиламин (амфетамин) и тому подобное, с ассиметрическими соединениями, несущими кислотную функциональную группу, такую как группа карбоновой кислоты и сульфоновой кислоты. Разделение диастереомерных солей можно осуществить фракционной кристаллизацией или ионообменной хроматографией. Что касается разделения оптических изомеров аминосоединений, то к образованию диастереомерных солей может приводить добавление хиральных карбоновых или сульфоновых кислот, таких как камфорсульфоновая кислота, винная кислота, миндальная кислота или молочная кислота.
Альтернативно, согласно методу (2) подлежащее разделению вещество приводят во взаимодействие с одним из энантиомеров хирального соединения с образованием диастереомерной пары (Eliel E. and Wilen S., “Stereochemistry of Organic Compounds”, John Wiley & Sons, Inc., New York, 1994, p. 322). Диастереомерные соединения могут образовываться в результате взаимодействия ассиметрических соединений с энантиомерно чистыми хиральными дериватизирующими реагентами, такими как ментильные производные, с последующим разделением диастереомеров и гидролизом с получением чистого или обогащенного энантиомера. Метод определения оптической чистоты включает в себя получение хиральных сложных эфиров, таких как сложный ментиловый эфир, например (-)-ментил-хлорформиат, в присутствии основания, или эфир Мошера, α-метокси-α-(трифторметил)фенилацетат (Jacob III, J. Org. Chem. (1982) 47: 4165), в рацемической смеси и анализ 1H ЯМР-спектра на наличие двух атропоизомерных энантиомеров или диастереомеров. Стабильные диастереомеры атропоизомерных соединений можно разделить и выделить нормально-фазовой и обращенно-фазовой хроматографией, следуя способам разделения атропоизомерных нафтил-изохинолинов (WO 96/15111). Согласно методу (3) рацемическую смесь двух энантиомеров можно разделить хроматографией с использованием хиральной неподвижной фазы (“Chiral Liquid Chromatography” (1989), W. J. Lough, Ed., Chapman and Hall, New York; Okamoto, J. Chromatogr. (1990) 513: 375-378). Обогащенные или очищенные энантиомеры можно различить методами, используемыми для различения других хиральных молекул с ассиметрическими атомами углерода, такими как оптическое вращение и круговой дихроизм.
Соединения по изобретению получали так, как проиллюстрировано на общих схемах 1 и 2.
Схема 1

a) MgCl2, триэтиламин, параформальдегид, ацетонитрил, нагревание; b) оксальдегид, гидроксид аммония, нагревание; c) карбонат цезия, 1,2-дибромэтан, DMF (диметилформамид), нагревание; d) N-иодсукцинимид, DMF, нагревание; e) 1. EtMgBr, THF (тетрагидрофуран), -20°C; 2. водный раствор хлорида аммония.
Как показано на схеме 1, 4-бром-2-гидроксибензальдегид 2 может быть получен путем формилирования имеющегося в продаже 3-бромфенола. После нагревания соединения 2 с оксальдегидом и гидроксидом аммония получают соединение 3. Оксазепиновое кольцо может быть образовано в результате нагревания соединения 3 с 1,2-дибромэтаном. Бис-иодинирование может быть индуцировано посредством взаимодействия с N-иодсукцинимидом, а группа 3-иод может быть избирательно удалена путем обработки этилмагнийбромидом при пониженной температуре с получением соединения 6.
Схема 2
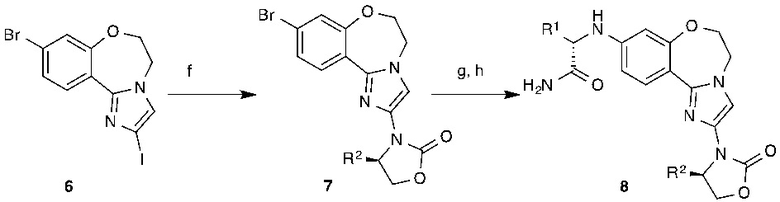
f) 4-замещенный оксазолидин-2-он, Cu(OAc)2, транс-N,N'-диметилциклогексан-1,2-диамин, карбонат калия, диоксан, нагревание; g) HN(R2)CH(R1)CO2H, CuI, K3PO4, ДМСО, нагревание; h) хлорид аммония, триэтиламин, HATU (1-[бис(диметиламино)метилен]-1H-1,2,3-триазолo[4,5-b]пиридиний-3-оксида гексафторфосфат).
Как показано на схеме 2, можно провести реакцию сочетания соединения 6 с соответствующим образом замещенным оксазолидин-2-оном с использованием катализатора на основе меди для получения соединения 7. Можно провести реакцию сочетания бром-содержащего промежуточного соединения 7 с соответствующим образом замещенными аминокислотами с использованием катализатора на основе меди, после чего осуществить HATU-опосредуемую реакцию амидного сочетания с хлоридом аммония для получения соединений 8.
ПРИМЕРЫ
Сокращения
Метод A LCMS: эксперименты проводили на квадрупольном масс-спектрометре Micromass ZQ2000 от Waters, соединенном с системой для сверхэффективной жидкостной хроматографии (UPLC) Acquity от Waters с ультрафиолетовым (УФ) детектором на фотодиодной матрице (PDA). Спектрометр имеет электрораспылительный источник, работающий в режиме положительных и отрицательных ионов. В этой системе используется колонка Acquity BEH C18, 1,7 мкм, 100 x 2,1 мм, поддерживаемая при 40°C, или колонка Acquity BEH Shield RP18, 1,7 мкм, 100 x 2,1 мм, поддерживаемая при 40°C, и скорость потока 0,4 мл/минута. В качестве исходной системы растворителей использовали смесь, состоящую из 95% воды, содержащей 0,1% муравьиной кислоты, (растворителя A) и 5% ацетонитрила, содержащего 0,1% муравьиной кислоты, (растворителя B), в течение первых 0,4 минуты, затем градиент до 5% растворителя A и 95% растворителя B в течение следующих 5,6 минуты. Эту смесь поддерживали в течение 0,8 минуты, после чего снова возвращались к смеси из 95% растворителя A и 5% растворителя B в течение следующих 0,2 минуты. Общее время хроматографирования составляло 8 минут.
Метод B LCMS: эксперименты проводили на HPLC-системе Agilent 1100, соединенной с масс-спектрометром Agilent MSD, используя ESI в качестве источника ионизации. Разделение методом LC проводили, используя колонку XB-C18, 1,7 мкм, 50 × 2,1 мм, от Phenomenex, при скорости потока 0,4 мл/минута. В качестве растворителя A использовали воду с 0,1% муравьиной кислоты, а в качестве растворителя B использовали ацетонитрил с 0,1% муравьиной кислоты. В качестве градиента использовали градиент 2→98% растворителя B в течение 7 минут с поддержанием уровня 97% B в течение 1,5 минуты, после уравновешивания. Температура колонки для LC составляет 40°C. Данные по УФ поглощению собирали при 220 нм и 254 нм и во всех экспериментах применяли масс-спектрометрическое сканирование во всем диапазоне масс.
Пример 101. (S)-2-((2-((S)-4-(Дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид 101
Стадия 1. 4-Бром-2-гидроксибензальдегид
В 4-горлую круглодонную колбу емкостью 20 л, дегазированную и поддерживаемую в атмосфере инертного газа азота, помещали 3-бромфенол (1300 г; 7,51 моль), дихлормагний (1078 г; 11,3 моль), триэтиламин (3034 г; 30,0 моль) и ацетонитрил (7,8 л). Смесь перемешивали в течение 30 минут при 40°C. К смеси добавляли параформальдегид (676 г; 22,6 моль) при 80°C. Полученный раствор перемешивали в течение 6 часов при 76°C. Эту реакцию повторяли 5 раз. Объединенные реакционные смеси гасили путем добавления 12 л водного раствора хлористого водорода (4 н.). Значение pH раствора подводили до 5, используя концентрированный водный раствор хлористого водорода (12 н.). Полученный раствор экстрагировали этилацетатом (1 x 20 л). Органические экстракты упаривали в вакууме. Остаток очищали посредством флэш-хроматографии на силикагеле (элюировали: 15%-ным этилацетатом в петролейном эфире), получая неочищенный продукт, который промывали 2,4 л смеси метил-трет-бутиловый эфир:гексан (1:4). Полученные твердые вещества собирали фильтрованием, получая 7,0 кг (78%) указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 2. 5-Бром-2-(1H-имидазол-2-ил)фенол
В 4-горлую круглодонную колбу емкостью 20 л помещали раствор 4-бром-2-гидроксибензальдегида (700 г; 3,50 моль) в метаноле (7,0 л) и оксальдегид (40%-ный) (2540 г; 17,5 моль), затем по каплям добавляли в течение 4 часов водный раствор аммиака (25-28%-ный, 3500 г) с перемешиванием и поддержанием температуры ниже 40°C. Полученный раствор перемешивали в течение 15 часов при 30-35°C. Эту реакцию повторяли 9 раз. Объединенные 9 реакционных смесей упаривали в вакууме, поддерживая температуру ниже 45°C. Остаток разбавляли 100 л этилацетата с перемешиванием в течение 30 минут. Твердые вещества отфильтровывали и полученный раствор разбавляли водой. Водную фазу экстрагировали 35 л этилацетата. Органические экстракты упаривали под вакуумом и остаток очищали посредством флэш-хроматографии на силикагеле (градиент растворителей: 5→75% этилацетата в петролейном эфире), получая 2,4 кг (29%) указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 3. 9-Бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин
В 4-горлую круглодонную колбу емкостью 20 л помещали раствор 5-бром-2-(1H-имидазол-2-ил)фенола (1,4 кг; 5,86 моль) в N,N-диметилформамиде (14 л) и карбонат цезия (7,2 кг; 22,1 моль). Смесь перемешивали в течение 20 минут. К реакционной смеси добавляли 1,2-дибромэтан (4,1 кг; 21,8 моль). Полученный раствор перемешивали в течение 4-12 часов при 85-90°C, охлаждали до 15°C и фильтровали. Осадок на фильтре промывали 3,0 л этилацетата. Фильтрат разбавляли 14 л этилацетата. Объединенные органические экстракты промывали рассолом (4 x 14 л), сушили над безводным сульфатом натрия, фильтровали и упаривали в вакууме, получая 1,1 кг (71%) указанного в заголовке соединения в виде светло-желтого твердого вещества. LCMS (ESI): [M+H]+ = 265; 1H ЯМР (400 МГц, ДМСО-d6) δ 8.32 (d, J = 8,4 Гц, 1H), 7.35-7.24 (m, 3H), 7.06 (s, 1H), 4.47-4.42 (m, 4H).
Стадия 4. 9-Бром-2,3-дииод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин
В 4-горлую круглодонную колбу емкостью 20 л помещали 9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (2,5 кг; 9,43 моль) и N,N-диметилформамид (12,5 л), затем при перемешивании добавляли N-иодсукцинимид (6,0 кг; 26,7 моль) в виде нескольких порций. Полученный раствор перемешивали в течение 12 часов при 60°C, охлаждали до 15°C в бане со смесью вода/лед, разбавляли, используя 12,5 л смеси вода/лед, и фильтровали. Отфильтрованные твердые вещества перекристаллизовывали из петролейного эфира, получая 4,0 кг (82%) указанного в заголовке соединения в виде желтого твердого вещества.
Стадия 5: 9-Бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин
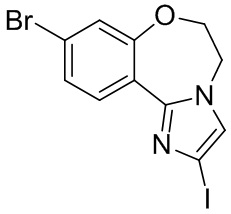
В 4-горлую круглодонную колбу емкостью 20 л, дегазированную и поддерживаемую в атмосфере инертного газа азота, помещали 9-бром-2,3-дииод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (800 г; 1,55 моль) и тетрагидрофуран (2,4 л), затем по каплям добавляли этилмагнийбромид (1 н. раствор в эфире; 1,7 л) с перемешиванием при -20°C в течение 3,5 часа. Реакционную смесь перемешивали в течение 3 часов, поддерживая температуру при -15°C с использованием бани со смесью лед/соль. Полученную смесь гасили путем добавления 3,0 л насыщенного водного раствора хлорида аммония и экстрагировали этилацетатом (2 x 8,0 л). Объединенные органические экстракты промывали рассолом (2 x 10 л), сушили над безводным сульфатом натрия, фильтровали и упаривали в вакууме. Неочищенный остаток растирали с 8,0 л смеси этилацетат:петролейный эфир (1:5), фильтровали и промывали петролейным эфиром, получая 501 г (83%) указанного в заголовке соединения в виде коричневого твердого вещества. LCMS (ESI): [M+H]+ = 391; 1H ЯМР (400 МГц, ДМСО-d6) δ 8.22 (d, J = 8,7 Гц, 1H), 7.55 (s, 1H), 7.30-7.25 (m, 2H), 4.45-4.41 (m, 4H).
Стадия 6. (R)-2,2-Диметил-[1,3]диоксолан-4-карбальдегид
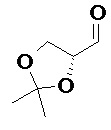
Периодат натрия (57,0 г; 270 ммоль) растворяли в горячей воде (115 мл) и добавляли диоксид кремния (200 г; 60 Å, 220-440 меш, размер частиц 35-75 мкм). Смесь энергично перемешивали до получения свободно текучего порошка. Его добавляли к раствору 1,2:5,6-бис-O-(1-метилэтилиден)-D-маннита (50 г; 190 ммоль) в дихлорметане (1,0 л) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Полученную смесь фильтровали через набивку Na2SO4 и твердые вещества тщательно промывали дихлорметаном. Объединенные органические экстракты упаривали в вакууме, получая 37,2 г (75%) указанного в заголовке соединения в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 9.73 (d, J = 1,9 Гц, 1H), 4.38 (ddd, J = 7,4; 4,7; 1,9 Гц, 1H), 4.18 (dd, J = 8,8; 7,4 Гц, 1H), 4.10 (dd, J = 8,8; 4,7 Гц, 1H), 1.49 (s, 3H), 1.43 (s, 3H).
Стадия 7. (R)-4-Дифторметил-2,2-диметил-[1,3]диоксолан
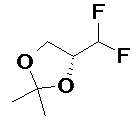
К раствору (R)-2,2-диметил-[1,3]диоксолан-4-карбальдегида (7,08 г; 54 ммоль) в дихлорметане (50 мл), охлажденному в водяной бане, по каплям добавляли трифторид диэтиламиносеры (8,4 мл; 62,6 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Полученную смесь по каплям добавляли к быстро перемешиваемому, охлажденному во льду насыщенному водному раствору бикарбоната натрия. Затем эту смесь экстрагировали дихлорметаном. Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и упаривали в вакууме, получая 6,58 г (79%) неочищенного указанного в заголовке соединения в виде оранжевого масла. 1H ЯМР (400 МГц, CDCl3) δ 5.69 (td, J = 55,8; 4,9 Гц, 1H), 4.27-4.17 (m, 1H), 4.16-4.03 (m, 2H), 1.46 (s, 3H), 1.38 (s, 3H).
Стадия 8. (R)-3-(трет-Бутилдиметилсиланилокси)-1,1-дифторпропан-2-ол

HCl в диоксане (4 н.; 10,8 мл; 43,2 ммоль) добавляли к раствору (R)-4-дифторметил-2,2-диметил[1,3]диоксолана (6,58 г; 43,2 ммоль) в метаноле (40 мл) и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Полученную смесь упаривали в вакууме и азеотропно отгоняли с ацетонитрилом. Остаток растворяли в N,N-диметилформамиде (10 мл) и добавляли трет-бутилдиметилсилилхлорид (6,53 г; 43,2 ммоль), триэтиламин (9,0 мл; 64,9 ммоль) и 4-(диметиламино)пиридин (в каталитических количествах). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Полученную смесь промывали водой и затем экстрагировали дихлорметаном. Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и упаривали в вакууме. Полученный неочищенный остаток очищали посредством флэш-хроматографии на силикагеле (градиент растворителей: 0→30% этилацетата в циклогексане), получая 3,43 г (35%) указанного в заголовке соединения в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ 5.66 (td, J = 56,4; 4,6 Гц, 1H), 3.76-3.60 (m, 2H), 2.46 (d, J = 6,4 Гц, 1H), 0.81 (s, 9H), 0.00 (s, 6H).
Стадия 9. ((S)-2-Азидо-3,3-дифторпропокси)-трет-бутилдиметилсилан

Трифторметансульфоновый ангидрид (2,9 мл; 17,4 ммоль) по каплям добавляли к раствору (R)-3-(трет-бутилдиметилсиланилокси)-1,1-дифторпропан-2-ола (3,43 г; 15,1 ммоль) и пиридина (2,0 мл; 24,2 ммоль) в дихлорметане (50 мл) при -20°C, реакционную смесь перемешивали при -20°C в течение 20 минут и затем при 0°C в течение 1 часа. Полученную смесь разбавляли 0,5 н. водным раствором HCl и экстрагировали дихлорметаном. Объединенные органические экстракты сушили над сульфатом магния и упаривали в вакууме. Неочищенный остаток растворяли в N,N-диметилформамиде (10 мл), добавляли азид натрия (2,96 г; 45,5 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Полученную смесь разбавляли водой и экстрагировали этилацетатом. Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и упаривали в вакууме, получая 4,50 г неочищенного указанного в заголовке соединения. 1H ЯМР (400 МГц, CDCl3) δ 5.74 (td, J = 55,4; 4,4 Гц, 1H), 3.81-3.71 (m, 2H), 3.58-3.47 (m, 1H), 0.81 (s, 9H), 0.00 (s, 6H).
Стадия 10. (S)-1-(трет-Бутилдиметилсиланилоксиметил)-2,2-дифторэтиламин

Гидроксид палладия на угле (200 мг; 20%-ный) добавляли к раствору ((R)-2-азидо-3,3-дифторпропокси)-трет-бутилдиметилсилана (4,50 г; неочищенного, предположительно приблизительно 15,1 ммоль) в этилацетате (20 мл) и метаноле (2,0 мл) и реакционную смесь перемешивали под давлением подаваемого из баллона водорода в течение 16 часов. Реакционную смесь фильтровали, добавляли свежую порцию гидроксида палладия на угле (400 мг; 20%-ного) и реакционную смесь перемешивали под давлением подаваемого из баллона водорода в течение 16 часов. Полученную смесь фильтровали и фильтрат упаривали в вакууме, получая 3,08 г (90%) неочищенного указанного в заголовке продукта в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 5.66 (td, J = 57,0; 4,7 Гц, 1H), 3.71-3.57 (m, 2H), 3.00-2.89 (m, 1H), 1.42 (br s, 2H), 0.82 (s, 9H), 0.00 (s, 6H).
Стадия 11. (S)-4-Дифторметилоксазолидин-2-он

Раствор HCl в диоксане (4 н.; 5,0 мл; 20 ммоль) добавляли к раствору (R)-1-(трет-бутилдиметилсиланилоксиметил)-2,2-дифторэтиламина (Org. Lett., Vol. 9, № 1, 2007, 41-44) (2,30 г; 10,3 ммоль) в метаноле (5,0 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Смесь упаривали в вакууме и полученное масло растирали с диэтиловым эфиром, получая твердое вещество, которое сушили в вакууме. Это твердое вещество растворяли в смеси толуола (20 мл) и KOH (2,50 г; 44,6 ммоль в 20 мл воды) при 0°C. По каплям добавляли фосген (16,3 мл; 20%-ный раствор в толуоле), охлаждающую баню удаляли и реакционную смесь перемешивали в течение 1 часа. Смесь упаривали в вакууме, полученный остаток экстрагировали горячим промышленным денатурированным спиртом и твердое вещество собирали фильтрованием. Фильтрат упаривали в вакууме и полученный остаток очищали посредством флэш-хроматографии на силикагеле (градиент растворителей: 0→100% этилацетата в циклогексане), получая 830 мг (68%) указанного в заголовке соединения в виде беловатого твердого вещества. [α]D = +10,1 (c = 2,37; CHCl3). 1H ЯМР (400 МГц, CDCl3) δ 5.96 (br s, 1H), 5.78 (td, J = 55,3; 4,8 Гц, 1H), 4.54 (t, J = 9,2 Гц, 1H), 4.42 (dd, J = 9,6; 4,4 Гц, 1H), 4.17-4.06 (m, 1H).
Стадия 12. (S)-3-(9-Бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-он

Смесь 9-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (250 мг; 0,64 ммоль), (S)-4-дифторметилоксазолидин-2-она (88 мг; 0,64 ммоль), транс-N,N'-диметил-1,2-циклогександиамина (36 мг; 0,26 ммоль), иодида одновалентной меди (24 мг; 0,13 ммоль) и карбоната калия (177 мг; 1,28 ммоль) в диоксане (3,0 мл) продували аргоном при обработке ультразвуком. Реакционную смесь нагревали при 100°C в течение 5 ч и затем оставляли охлаждаться до комнатной температуры. Полученную смесь разбавляли 15%-ным водным раствором аммиака и экстрагировали этилацетатом. Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и упаривали в вакууме. Полученный остаток растирали с метанолом и затем очищали препаративной HPLC [C18, 60%-ный ацетонитрил (с 0,1% муравьиной кислоты) в воде (с 0,1% муравьиной кислоты), продолжительность хроматографирования 20 минут], получая 20 мг (8%) указанного в заголовке соединения в виде белого твердого вещества. LCMS (ESI): [M+H]+ = 400/402. ¹H ЯМР (400 МГц, CDCl3) δ 8.19 (d, J = 9,2 Гц, 1H), 7.29 (s, 1H), 7.24-7.19 (m, 2H), 6.65 (ddd, J = 57,8; 54,5; 1,0 Гц, 1H), 4.87 (ddd, J = 24,0; 9,2; 4,0 Гц, 1H), 4.73 (dd, J = 9,5; 4,2 Гц, 1H), 4.53 (t, J = 9,2 Гц, 1H), 4.48-4.43 (m, 2H), 4.38-4.33 (m, 2H).
Стадия 13. (S)-2-((2-((S)-4-(Дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид
(S)-3-(9-Бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-он (600 мг; 1,50 ммоль), L-аланин (267 мг; 3,00 ммоль), иодид одновалентной меди (57 мг; 0,30 ммоль) и фосфат калия трехосновный (637 мг; 3,00 ммоль) суспендировали в диметилсульфоксиде (6,0 мл). Реакционную смесь нагревали при 100°C в течение 2 часов. После охлаждения до комнатной температуры добавляли диметилсульфоксид (4,0 мл), хлорид аммония (480 мг; 9,00 ммоль) и триэтиламин (3,1 мл; 22,5 ммоль). К полученной перемешиваемой суспензии порциями в течение 5 минут добавляли 1-[бис(диметиламино)метилен]-1H-1,2,3-триазолo[4,5-b]пиридиний-3-оксида гексафторфосфат (5,10 г; 13,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем фильтровали через Celite®, промывая этилацетатом. Органические экстракты промывали насыщенным водным раствором бикарбоната натрия и водную фазу экстрагировали этилацетатом. Объединенные органические экстракты промывали рассолом, сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Неочищенный остаток очищали посредством флэш-хроматографии на силикагеле (градиент растворителей: 0→5% метанола в дихлорметане) и затем посредством хиральной сверхкритической жидкостной хроматографии, получая 294 мг (46%) соединения 101 в виде беловатого твердого вещества. LCMS (ESI): RT (мин) = 2,89, [M+H]+ = 408, метод A; 1H ЯМР (400 МГц, ДМСО-d6) δ 8.00 (d, J = 8,7 Гц, 1H), 7.38 (br s, 1H), 7.18 (s, 1H), 7.00 (br s, 1H), 6.71 (t, J = 55,9 Гц, 1H), 6.41 (dd, J = 8,8; 2,3 Гц, 1H), 6.16 (d, J = 7,2 Гц, 1H), 6.09 (d, J = 1,9 Гц, 1H), 5.02-4.89 (m, 1H), 4.63-4.52 (m, 2H), 4.39-4.30 (m, 4H), 3.76 (квинтет, J = 7,0 Гц, 1H), 1.30 ( d, J = 7,1 Гц, 3H).
Пример 102. (S)-2-Циклобутил-2-((2-((R)-4-метил-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)ацетамид 102
Стадия 1. (R)-4-Метилоксазолидин-2-он
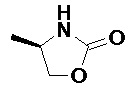
К смеси D-аланинола (8,65 г; 0,12 ммоль) в толуоле и водном растворе KOH (124 мл; 12,5%-ном водн.; 0,28 ммоль) при 0°C добавляли фосген (72,7 мл; 20%-ный раствор в толуоле; 0,14 ммоль) с такой скоростью, чтобы внутренняя температура оставалась ниже 5°C. Реакционную смесь перемешивали при 0°C в течение еще 40 минут, затем упаривали досуха. Неочищенный остаток экстрагировали промышленным денатурированным спиртом, суспензию фильтровали и фильтрат упаривали в вакууме. Полученный остаток очищали посредством флэш-хроматографии на силикагеле (градиент растворителей: 40→100% этилацетата в циклогексане), получая 10,4 г (90%) указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 6.00 (br s, 1H), 4.50 (t, J = 6,5 Гц, 1H), 4.07-3.97 (m, 1H), 3.95 (dd, J = 7,8; 6,2 Гц, 1H), 1.30 (d, J = 6,1 Гц, 3H).
Стадия 2. (R)-3-(9-Бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-метилоксазолидин-2-он и (R)-3-(9-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-метилоксазолидин-2-он
Смесь 9-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (30,0 г; 76,7 ммоль), (R)-4-метилоксазолидин-2-она (7,70 г; 76,7 ммоль), иодида одновалентной меди (1,61 г; 8,40 ммоль), транс-N,N'-диметил-1,2-циклогександиамина (2,7 мл; 16,9 ммоль) и карбоната калия (14,9 г; 107 ммоль) суспендировали в 1,4-диоксане (200 мл) и реакционную смесь продували аргоном при обработке ультразвуком. Полученную смесь нагревали при 100°C в течение 16 ч. Реакционную смесь разбавляли водным раствором аммиака (приблизительно 16%-ным) и экстрагировали этилацетатом. Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и упаривали в вакууме. Полученный остаток очищали посредством флэш-хроматографии на силикагеле (градиент растворителей: 0→100% этилацетата в циклогексане), получая 13,4 г (приблизительно 42%) указанного в заголовке соединения (смесь продуктов 9-Br:9-I в соотношении приблизительно 2:1). 1H ЯМР (400 МГц, CDCl3) δ 8.28 (d, J = 7,6 Гц, 0.33H), 8.11 (d, J = 6,9 Гц, 0.66H), 7.42-7.38 (m, 1H), 7.28-7.24 (m, 1.33H), 7.23-7.18 (m, 0.66H), 4.77-4.68 (m, 1H), 4.58 (t, J = 8,3 Гц, 1H), 4.49-4.39 (m, 2H), 4.37-4.30 (m, 2H), 4.08 (dd, J = 8,4; 4,5 Гц, 1H), 1.57-1.50 (m, 3H).
Стадия 3. (R)-3-(9-Бром-5,6-дигидроимидазо[1,2-d][1,4]бензоксазепин-2-ил)-4-метил-оксазолидин-2-он
80 мг смеси (R)-3-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-метилоксазолидин-2-она и (R)-3-(9-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-метилоксазолидин-2-она разделяли посредством хиральной SFC, получая 27,6 мг указанного в заголовке соединения. LCMS (ESI): [M+H]+ = 364,0/366,0/367,2; 1H ЯМР (400 МГц, ДМСО-d6) δ 8.22 (d, J = 8,7 Гц, 1H), 7.35 (s, 1H), 7.31 (dd, J = 8,7; 2,1 Гц, 1H), 7.25 (d, J = 2,0 Гц, 1H), 4.65-4.54 (m, 2H), 4.49-4.43 (m, 4H), 4.09-4.06 (m, 1H), 1.42 (d, J = 6,0 Гц).
Стадия 4. Метил-(S)-2-циклобутил-2-((2-((R)-4-метил-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)ацетат
Смесь (4R)-3-(9-бром-5,6-дигидроимидазо[1,2-d][1,4]бензоксазепин-2-ил)-4-метил-оксазолидин-2-она (0,2746 ммоль; 100 мг), иодида одновалентной меди (0,084 ммоль; 16 мг), (2S)-2-амино-2-циклобутил-уксусной кислоты (1,10 ммоль; 142 мг) и фосфата калия трехосновного (1,37 ммоль; 297 мг) в диметилсульфоксиде (3 мл) нагревали в условиях облучения микроволнами при 120°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, добавляли иодметан (1,4 ммоль; 0,086 мл) и реакционную смесь экстрагировали дихлорметаном и водой. Объединенные органические экстракты объединяли, промывали рассолом и сушили с использованием сульфата натрия, фильтровали и упаривали в вакууме. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле (24 г диоксида кремния; градиент растворителей: 5-40% смеси 3:1 изопропилацетат:метанол в дихлорметане), получая 100 мг (85%) указанного в заголовке соединения.
Стадия 5. (S)-2-Циклобутил-2-((2-((R)-4-метил-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)ацетамид
К раствору метил-(2S)-2-циклобутил-2-[[2-[(4R)-4-метил-2-оксо-оксазолидин-3-ил]-5,6-дигидроимидазо[1,2-d][1,4]бензоксазепин-9-ил]амино]ацетата (0,234 ммоль; 100 мг) в тетрагидрофуране (5 мл) добавляли воду (0,45 мл) и гидроксида лития моногидрат (0,357 ммоль; 15 мг). Реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь упаривали в вакууме. К раствору полученного остатка в N,N-диметилформамиде (3 мл) добавляли 1-[бис(диметиламино)метилен]-1H-1,2,3-триазолo[4,5-b]пиридиний-3-оксида гексафторфосфат (0,353 ммоль; 137 мг), хлорид аммония (0,71 ммоль; 38 мг) и N,N-диизопропилэтиламин (0,705 ммоль; 0,123 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь упаривали в вакууме и полученный остаток обрабатывали водой, затем экстрагировали дихлорметаном. Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния и упаривали в вакууме. Неочищенный продукт очищали посредством обращенно-фазовой HPLC, затем SFC и лиофилизировали, получая 15,0 мг (15%) соединения 102. LCMS (ESI): RT (мин) = 3,03, [M+H]+ = 412,2, метод D; 1H ЯМР (400 МГц, ДМСО-d6) δ 7.96 (d, J = 8,8 Гц, 1H), 7.39-7.36 (brs, 1H), 7.13 (s, 1H), 7.00-6.97 (brs, 1H), 6.44 (dd, J = 8,9; 2,3 Гц, 1H), 6.14 (d, J = 2,3 Гц, 1H), 5.96 (d, J = 7,7 Гц, 1H), 4.62-4.49 (m, 2H), 4.38-4.28 (m, 4H), 4.06-4.03 (m, 1H), 3.70-3.61 (m, 1H), 2.06-1.75 (m, 6H), 1.42-1.34 (m, 3H).
Пример 103. (S)-2-Циклопропил-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)ацетамид 103
Смесь (S)-3-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-она со стадии 12 примера 101 (400 мг; 1,00 ммоль), L-циклопропилглицина (230 мг; 2,00 ммоль), иодида одновалентной меди (38 мг; 0,20 ммоль) и фосфата калия трехосновного (424 мг; 2,00 ммоль) в диметилсульфоксиде (2,0 мл) продували аргоном при обработке ультразвуком. Смесь нагревали при 100°C в течение 5 часов, затем охлаждали до температуры окружающей среды. Полученную смесь разбавляли диметилсульфоксидом (5,0 мл) и добавляли хлорид аммония (320 мг; 6,00 ммоль) и триэтиламин (1,4 мл; 10,0 ммоль). Затем к перемешиваемой суспензии порциями добавляли 1-[бис(диметиламино)метилен]-1H-1,2,3-триазолo[4,5-b]пиридиний-3-оксида гексафторфосфат (2,28 г; 6,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Полученную смесь разбавляли 15%-ным водным раствором аммиака и экстрагировали этилацетатом. Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и упаривали в вакууме. Неочищенный остаток очищали посредством флэш-хроматографии на силикагеле (градиент растворителей: 0→7% метанола в этилацетате). Остаток растворяли в минимальном количестве ацетонитрила. Затем добавляли воду для осаждения твердого вещества, которое собирали фильтрованием и сушили в вакууме, получая 324 мг (75%) соединения 103 в виде беловатого твердого вещества. LCMS (ESI): RT (мин) = 3,21, [M+H]+ = 434, метод A; 1H ЯМР (400 МГц, ДМСО-d6) δ 7.98 (d, J = 8,6 Гц, 1H), 7.40 (br s, 1H), 7.17 (s, 1H), 7.03 (br s, 1H), 6.71 (t, J = 56,0 Гц, 1H), 6.42 (dd, J = 8,9; 2,4 Гц, 1H), 6.24 (d, J = 7,2 Гц, 1H), 6.09 (d, J = 2,4 Гц, 1H), 5.01-4.89 (m, 1H), 4.63-4.51 (m, 2H), 4.38-4.29 (m, 4H), 3.15 ( t, J = 7,7 Гц, 1H), 1.16-1.05 (m, 1H), 0.56-0.44 (m, 3H), 0.33-0.25 (m, 1H).
Пример 104. (S)-2-Циклопропил-2-((2-((R)-4-метил-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)ацетамид 104
Смесь (R)-3-(9-бром-5,6-дигидроимидазо[1,2-d][1,4]бензоксазепин-2-ил)-4-метил-оксазолидин-2-она (пример 102, стадия 3) (1,098 ммоль; 400 мг), иодида одновалентной меди (0,330 ммоль; 62,8 мг), (2S)-2-амино-2-циклопропил-уксусной кислоты (3,295 ммоль; 379,3 мг) и фосфата калия трехосновного (4,393 ммоль; 951,5 мг) в диметилсульфоксиде (35 ммоль; 2,5 мл) нагревали при 110°C в течение 2 часов в условиях облучения микроволнами. Реакционную смесь охлаждали до комнатной температуры. К реакционной смеси добавляли 1-[бис(диметиламино)метилен]-1H-1,2,3-триазолo[4,5-b]пиридиний-3-оксида гексафторфосфат (12,08 ммоль; 4260 мг), хлорид аммония (12,08 ммоль; 646 мг) и триэтиламин (1,53 мл; 11,0 ммоль). Через 20 минут выдерживания при комнатной температуре реакционную смесь обрабатывали водой, затем экстрагировали дихлорметаном. Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и упаривали в вакууме. Неочищенный продукт очищали посредством обращенно-фазовой HPLC и лиофилизировали, получая 110 мг (25% за 2 стадии) соединения 104. LCMS (ESI): RT (мин) = 2,588, [M+H]+ = 398,2, метод B; 1H ЯМР (400 МГц, ДМСО-d6) δ 7.96 (d, J = 8,8 Гц, 1H), 7.39 (d, J = 2,2 Гц, 1H), 7.13 (s, 1H), 7.02 (d, J = 2,3 Гц, 1H), 6.42 (dd, J = 8,9; 2,4 Гц, 1H), 6.20 (d, J = 7,1 Гц, 1H), 6.09 (d, J = 2,4 Гц, 1H), 4.61-4.49 (m, 2H), 4.40-4.27 (m, 4H), 4.10-3.99 (m, 1H), 3.22-3.09 (m, 1H), 1.42-1.36 (m, 3H), 1.16-1.04 (m, 1H), 0.56-0.42 (m, 3H), 0.32-0.27 (m, 1H).
Пример 105. (S)-2-Циклопропил-2-((2-((S)-4-(фторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)ацетамид 105
Стадия 1. (R)-1-(трет-Бутилдиметилсиланилокси)-3-фторпропан-2-ол
трет-Бутилдиметилсилилхлорид (1,60 г; 10,63 ммоль) добавляли к раствору (R)-3-фторпропан-1,2-диола (1,00 г; 10,6 ммоль), триэтиламина (1,93 мл; 13,8 ммоль) и каталитического количества 4-(диметиламино)пиридина в дихлорметане при 0°C, реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь разбавляли водой и экстрагировали дихлорметаном. Объединенные органические фракции промывали рассолом, сушили над сульфатом магния, фильтровали и упаривали в вакууме. Полученный неочищенный остаток очищали посредством флэш-хроматографии на силикагеле (градиент растворителей: 0→40% этилацетата в циклогексане), получая 1,80 г (81%) указанного в заголовке соединения в виде бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ 4.45-4.36 (m, 1H), 4.34-4.25 (m, 1H), 3.87-3.73 (m, 1H), 3.66-3.56 (m, 2H), 2.30 (d, J = 6,0 Гц, 1H), 0.82 (s, 9H), 0.00 (s, 6H).
Стадия 2. (S)-2-Азидо-3-фторпропокси)-трет-бутилдиметилсилан
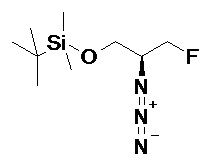
Трифторметансульфоновый ангидрид (1,67 мл; 9,93 ммоль) по каплям добавляли к раствору (R)-1-(трет-бутилдиметилсиланилокси)-3-фторпропан-2-ола (1,80 г; 8,60 ммоль) и пиридина (1,2 мл; 13,8 ммоль) в дихлорметане при -20°C и реакционную смесь перемешивали при -20°C в течение 20 минут, затем при 0°C в течение 30 минут. Реакционную смесь разбавляли 0,5 н. водным раствором HCl и экстрагировали дихлорметаном. Объединенные органические экстракты сушили над сульфатом магния, фильтровали и упаривали в вакууме. Остаток растворяли в N,N-диметилформамиде (5,0 мл) и добавляли азид натрия (1,68 г; 25,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Полученную смесь разбавляли водой и экстрагировали этилацетатом. Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и упаривали в вакууме, получая неочищенное указанное в заголовке соединение, которое переносили на следующую стадию без очистки. 1H ЯМР (400 МГц, CDCl3) δ 4.58-4.26 (m, 2H), 3.75-3.63 (m, 2H), 3.62-3.46 (m, 1H), 0.80 (s, 9H), 0.00 (s, 6H).
Стадия 3. (S)-1-(трет-Бутилдиметилсиланилоксиметил)-2-фторэтиламин
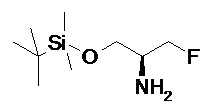
Гидроксид палладия (400 мг; 20%-ный на угле) добавляли к раствору ((S)-2-азидо-3-фторпропокси)-трет-бутилдиметилсилана (неочищенного; предположительно 8,60 ммоль) в этилацетате (15 мл) и метаноле (5,0 мл) и реакционную смесь перемешивали под давлением подаваемого из баллона водорода в течение 16 часов. Полученную смесь фильтровали, добавляли свежую порцию гидроксида палладия (400 мг; 20%-ного на угле) и реакционную смесь перемешивали под давлением подаваемого из баллона водорода в течение еще 16 часов. Полученную смесь фильтровали и фильтрат упаривали в вакууме, получая указанное в заголовке соединение в виде смеси продукт:исходное вещество в соотношении приблизительно 2:1, которую переносили на следующую стадию без очистки.
Стадия 4. (S)-4-Фторметилоксазолидин-2-он
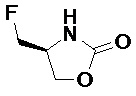
Раствор HCl в диоксане (4 н.; 2,0 мл; 8,00 ммоль) добавляли к раствору (S)-1-(трет-бутилдиметилсиланилоксиметил)-2-фторэтиламина (неочищенного; предположительно 8,60 ммоль) в метаноле (3,0 мл) и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь упаривали в вакууме. Полученный остаток растворяли в смеси толуола (20 мл) и KOH (2,89 г; 51,6 ммоль; 12,5%-ного водн. раствора) при 0°C. К этой смеси по каплям добавляли фосген (13,6 мл; 20%-ный раствор в толуоле), охлаждающую баню удаляли и полученную смесь перемешивали в течение 1 часа. Реакционную смесь упаривали в вакууме и полученный остаток экстрагировали горячим промышленным денатурированным спиртом. Фильтрат упаривали в вакууме и полученный остаток очищали посредством флэш-хроматографии на силикагеле (градиент растворителей: 50→100% этилацетата в циклогексане), получая 450 мг (44%; за 3 стадии) указанного в заголовке соединения в виде беловатого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 5.69 (br s, 1H), 4.59-4.42 (m, 2H), 4.42-4.32 (m, 1H), 4.25-4.08 (m, 2H).
Стадия 5. (S)-3-(9-Бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(фторметил)оксазолидин-2-он и (S)-3-(9-иод-5,6-дигидро-бензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(фторметил)оксазолидин-2-он
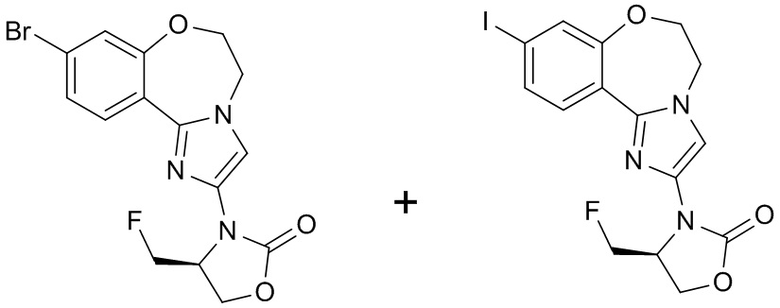
Смесь 9-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (722 мг; 1,85 ммоль), (S)-4-фторметилоксазолидин-2-она (220 мг; 1,85 ммоль), 3,4,7,8-тетраметил-1,10-фенантролина (131 мг; 0,55 ммоль), Cu(OAc)2.H2O (74 мг; 0,37 ммоль), карбоната калия (510 мг; 3,70 ммоль) и диоксана (6,0 мл) герметично закрывали в пробирке и смесь продували аргоном при обработке ультразвуком. Реакционную смесь нагревали при 100°C в течение 72 часов. Полученную реакционную смесь разбавляли 15%-ным водным раствором аммиака и экстрагировали этилацетатом. Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и упаривали в вакууме. Неочищенный остаток очищали посредством флэш-хроматографии на силикагеле (градиент растворителей: 0→100% этилацетата в циклогексане), получая 390 мг (53%) указанных в заголовке соединений (смеси продуктов 9-Br и 9-I в соотношении приблизительно 2:1). LCMS (ESI): [M+H]+ = 382/384/430; 1H ЯМР (400 МГц, CDCl3) δ 8.22 (d, J = 9,3 Гц, 0.7H), 8.05 (d, J = 8,8 Гц, 0.3H), 7.43-7.37 (m, 0.6H), 7.29 (s, 1.2H), 7.23-7.18 (m, 1.2H), 5.03-4.66 (m, 3H), 4.60 (t, J = 8,5 Гц, 1H), 4.54 (dd, J = 8,6; 4,3 Гц, 1H), 4.47-4.43 (m, 2H), 4.37-4.33 (m, 2H).
Стадия 6. (S)-2-Циклопропил-2-((2-((S)-4-(фторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)ацетамид
Смесь (S)-3-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(фторметил)оксазолидин-2-она и (S)-3-(9-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(фторметил)оксазолидин-2-она (195 мг; смесь Br:I в соотношении приблизительно 2:1; приблизительно 0,49 ммоль), L-циклопропилглицина (104 мг; 0,90 ммоль), иодида одновалентной меди (17 мг; 0,09 ммоль) и фосфата калия трехосновного (190 мг; 0,90 ммоль) в диметилсульфоксиде (1,5 мл) продували аргоном при обработке ультразвуком. Реакционную смесь нагревали при 100°C в течение 16 часов, затем охлаждали до комнатной температуры. Полученную смесь разбавляли диметилсульфоксидом (1,0 мл) и добавляли хлорид аммония (144 мг; 2,70 ммоль) и триэтиламин (950 мкл, 6,75 ммоль). Затем к этой смеси порциями добавляли 1-[бис(диметиламино)метилен]-1H-1,2,3-триазолo[4,5-b]пиридиний-3-оксида гексафторфосфат (1,54 г; 4,05 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Полученную смесь разбавляли насыщенным водным раствором бикарбоната натрия и экстрагировали этилацетатом. Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и упаривали в вакууме. Полученный неочищенный остаток очищали посредством флэш-хроматографии на силикагеле (градиент растворителей: 0→5% метанола в дихлорметане) и затем дополнительно очищали флэш-хроматографией на силикагеле (градиент растворителей: 0→100% метилацетата в циклогексане), получая 90 мг (48%) соединения 105 в виде беловатого твердого вещества. LCMS (ESI): RT (мин) = 2,76 [M+H]+ = 416, метод A; 1H ЯМР (400 МГц, ДМСО-d6) δ 7.94 (d, J = 8,8 Гц, 1H), 7.40 (br s, 1H), 7.17 (s, 1H), 7.03 (br s, 1H), 6.41 (dd, J = 8,8; 2,3 Гц, 1H), 6.22 (d, J = 7,1 Гц, 1H), 6.09 (d, J = 2,2 Гц, 1H), 4.99 (ddd, J = 48,3; 9,8; 2,5 Гц, 1H), 4.81-4.56 (m, 3H), 4.40 (dd, J = 8,6; 3,9 Гц, 1H), 4.37-4.29 (m, 4H), 3.15 (t, J = 7,6 Гц, 1H), 1.16-1.05 (m, 1H), 0.54-0.43 (m, 3H), 0.33-0.25 (m, 1H).
Пример 106. (S)-2-((2-((S)-4-(Фторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид 106
Смесь (S)-3-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(фторметил)оксазолидин-2-она и (S)-3-(9-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(фторметил)оксазолидин-2-она (пример 105, стадия 5) (195 мг; смесь 9-Br:9-I в соотношении приблизительно 2:1; приблизительно 0,49 ммоль), L-аланина (87 мг; 0,98 ммоль), иодида одновалентной меди (17 мг; 0,09 ммоль) и фосфата калия трехосновного (208 мг; 0,98 ммоль) в диметилсульфоксиде (3,0 мл) продували аргоном при обработке ультразвуком. Реакционную смесь нагревали при 100°C в течение 4 часов, затем охлаждали до комнатной температуры. Полученную смесь разбавляли диметилсульфоксидом (3,0 мл) и добавляли хлорид аммония (157 мг; 2,94 ммоль) и триэтиламин (683 мкл; 4,8 ммоль). Затем к этой смеси порциями добавляли 1-[бис(диметиламино)метилен]-1H-1,2,3-триазолo[4,5-b]пиридиний-3-оксида гексафторфосфат (1,10 г; 2,94 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Полученную смесь разбавляли насыщенным водным раствором бикарбоната натрия и экстрагировали этилацетатом. Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и упаривали в вакууме. Полученный остаток очищали посредством флэш-хроматографии на силикагеле (градиент растворителей: 0→5% метанола в дихлорметане) и затем дополнительно очищали хиральной сверхкритической жидкостной хроматографией, получая 36 мг (19%) соединения 106 в виде беловатого твердого вещества. LCMS (ESI): RT (мин) = 2,43 [M+H]+ = 390, метод A; 1H ЯМР (400 МГц, ДМСО-d6) δ 7.96 (d, J = 8,8 Гц, 1H), 7.37 (br s, 1H), 7.17 (s, 1H), 7.00 (br s, 1H), 6.39 (dd, J = 8,6; 1,6 Гц, 1H), 6.15 (d, J = 7,0 Гц, 1H), 6.09 (d, J = 1,6 Гц, 1H), 5.08-4.55 (m, 5H), 4.42-4.28 (m, 4H), 3.76 (квинтет, J = 7,2 Гц, 1H), 1.30 (d, J = 7,2 Гц, 3H).
Пример 107. (S)-2-((2-((S)-4-(Дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)бутанамид 107
Смесь (S)-3-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-4-(дифторметил)оксазолидин-2-она (пример 101, стадия 12) (240 мг; 0,60 ммоль), (S)-2-аминомасляной кислоты (124 мг; 1,19 ммоль), иодида одновалентной меди (22,8 мг; 0,119 ммоль), фосфата калия трехосновного (255 мг; 1,19 ммоль) и диметилсульфоксида (6,0 мл) перемешивали в атмосфере аргона при 100°C в течение 6 часов. Полученную смесь оставляли охлаждаться до комнатной температуры и затем добавляли хлорид аммония (188 мг; 3,52 ммоль) и триэтиламин (1,2 мл; 8,80 ммоль). К перемешиваемой суспензии порциями добавляли 1-[бис(диметиламино)метилен]-1H-1,2,3-триазолo[4,5-b]пиридиний-3-оксида гексафторфосфат (2,01 г; 5,28 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Полученную смесь разбавляли этилацетатом, промывали насыщенным раствором хлорида аммония, сушили над сульфатом магния, фильтровали и упаривали в вакууме. Неочищенный продукт очищали посредством флэш-хроматографии на силикагеле (градиент растворителей: 0→10% метанола в этилацетате), дополнительно очищали посредством обращенно-фазовой HPLC, затем хиральной сверхкритической жидкостной хроматографии, получая 73,6 мг (30%) соединения 107 в виде белого твердого вещества. LCMS (ESI): RT (мин) = 3,13, [M+H]+ = 422, метод A; 1H ЯМР (400 МГц, ДМСО-d6) δ 7.99 (d, J = 8,8 Гц, 1H), 7.40 (s, 1H), 7.17 (s, 1H), 7.03 (s, 1H), 6.71 (t, J = 56,0 Гц, 1H), 6.44 (dd, J = 8,8; 2,2 Гц, 1H), 6.13 (d, J = 2,2 Гц, 1H), 6.09 (d, J = 7,6 Гц, 1H), 5.02-4.89 (m, 1H), 4.62-4.53 (m, 2H), 4.41-4.27 (m, 4H), 3.65-3.60 (m, 1H), 1.72-1.59 (m, 2H), 0.94 (t, J = 7,3 Гц, 3H).
Пример 901. Анализ связывания p110α (альфа) PI3K
Анализы связывания с PI3K предназначены для определения биохимической эффективности низкомолекулярных ингибиторов PI3K. Реакцию с участием липидной киназы PI3K проводят в присутствии липидного субстрата PIP2:3PS (смесь 1:3 PIP2 и фосфатидилсерина) (Promega, № V1792) и АТФ (аденозинтрифосфат). После завершения киназной реакции степень превращения АТФ в АДФ (аденозиндифосфат) в результате фосфорилирования липидного субстрата детектируют с использованием набора для анализа ADP-Glo™ от Promega (Promega, № V1792). Реакции проводят, используя для каждой изоформы PI3K следующие далее условия, которые приведены в Таблице 5.
Таблица 5
(мкМ)
(мин)
Когда продолжительность реакции достигает 120 минут, киназную реакцию останавливают. Весь АТФ, оставшийся после прохождения реакции, удаляют, при этом остается только АДФ. Затем добавляют реагент для детекции киназы, чтобы превратить АДФ в АТФ, который используют в сопряженной люциферин/люциферазной реакции. Измеряют выход по люминесценции и соотносят с киназной активностью.
Все реакции проводят при комнатной температуре. Для каждой изоформы PI3K добавляют по 3 мкл смеси раствора фермент/липидный субстрат (1:1) в 384-луночный используемый в анализе белый планшет (Perkin Elmer, № 6007299), содержащий 50 нл раствора тестируемого соединения или ДМСО только для необрабатываемых контролей. Реакцию инициируют путем добавления 2 мкл смеси АТФ/MgCl2. Буфер для киназной реакции содержит 50 мМ HEPES (N-2-гидроксиэтил-пиперазин-N-2-этансульфоновая кислота), 50 мМ NaCl, 3 мМ MgCl2, 0,01% BSA (бычий сывороточный альбумин), 1% ДМСО, а фермент и субстрат используют в концентрациях, указанных в приведенной выше таблице. Реакцию останавливают путем добавления 10 мкл реагента ADP-Glo. Планшеты прочитывают с использованием системы Envision от Perkin Elmer, применяя режим для определения интенсивности люминесценции. Для каждого тестируемого соединения получают кривую зависимости ответов от дозы по 10 точкам. Величины Ki для каждого соединения определяют, используя уравнение Моррисона.
Анализы связывания: начальные эксперименты по поляризации проводили на Analyst НТ 96-384 (Molecular Devices Corp., Sunnyvale, CA). Образцы для измерения аффинности методом поляризации флуоресценции готовили, добавляя p110-альфа Pl3K (Upstate Cell Signaling Solutions, Charlottesville, VA) в серийных разведениях 1:3, начиная с конечной концентрации 20 мкг/мл, в буфере для измерения поляризации (10 мМ Трис, рН 7,5; 50 мМ NaCl; 4 мМ MgCl2; 0,05% CHAPS (3-[(3-холамидопропил)-диметиламмонио]-1-пропан-сульфонат) и 1 мМ DTT (дитиотреит)), к PIP2 (Echelon-Inc., Salt Lake Clty, UT.) в конечной концентрации 10 мМ. После инкубации в течение 30 минут при комнатной температуре реакции останавливали, добавляя GRP-1 (основной рецептор фосфоинозитидов) и зонд PIP3-TAMRA (Echelon-lnc., Salt Lake City, UT) в конечных концентрациях 100 нМ и 5 нМ, соответственно. Измерения проводили, используя стандартные фильтры с ограниченной полосой пропускания для флуорофора родамина (λвозб. = 530 нм; λисп. = 590 нм) в 384-луночных черных планшетах на малые объемы Proxiplates® (PerkinElmer, Wellesley, MA). Строили график зависимости величин поляризации флуоресценции от концентрации белка. Значения ЕС50 получали путем аппроксимации данных к четырехпараметрическому уравнению с использованием программного обеспечения KaleidaGraph® (программное обеспечение для анализа синергетического действия (synergy software), Reading, PA). В этом эксперименте также устанавливают концентрацию белка, подходящую для использования в последующих экспериментах по конкуренции с ингибиторами.
Величины IC50 для ингибиторов определяли, добавляя p110-альфа Pl3K (конечная концентрация 0,04 мг/мл) вместе с PIP2 (конечная концентрация 10 мМ) в лунки, содержащие серийные разведения (1:3) антагонистов в растворе АТФ в конечной концентрации 25 мМ (Cell Signaling Technology, Inc., Danvers, MA) в буфере для измерения поляризации. После инкубации в течение 30 минут при комнатной температуре реакции останавливали, добавляя GRP-1 и зонд PIP3-TAMRA (Echelon-Inc., Salt Lake City, UT) в конечной концентрации 100 нМ и 5 нМ, соответственно. Измерения проводили, используя стандартные фильтры с ограниченной полосой пропускания для флуорофора родамина (λвозб. = 530 нм; λисп. = 590 нм) в 384-луночных черных планшетах на малые объемы Proxiplates® (PerkinElmer, Wellesley, MA). Строили график зависимости величин поляризации флуоресценции от концентрации антагонистов и путем аппроксимации данных к четырехпараметрическому уравнению с использованием программного обеспечения Assay Explorer (MDL, San Ramon, CA) получали значения IС50.
Альтернативно, ингибирование PI3K определяли в радиометрическом анализе с использованием очищенного рекомбинантного фермента и АТФ в концентрации 1 мкМ (микромолярной). Выполняли серийные разведения соединения в 100%-ном ДМСО. Реакционную смесь с киназой инкубировали в течение 1 ч при комнатной температуре и реакцию останавливали, добавляя PBS (забуференный фосфатом физиологический раствор). Затем определяли значения IC50, используя аппроксимацию зависимости доза-ответ к сигмоидальной кривой (с вариабельным наклоном).
Пример 902. Селективное ингибирование мутантных форм PI3Kα (альфа)
Способность соединения по изобретению действовать предпочтительно против клеток, содержащих мутантные формы PI3Kα (альфа), определяли посредством измерения ингибирования PI3K-пути в изогенных клеточных линиях SW48: с PI3Kα дикого типа (родительская линия), с мутацией E545K в спиральном домене и с мутацией H1047R в киназном домене. Приведенные далее анализы предназначены для определения эффективности низкомолекулярных ингибиторов PI3Kα в клетках и их селективности в отношении мутантных форм. В данном анализе используют изогенные клеточные линии, которые экспрессируют PI3Kα дикого типа (WT), PI3Kα с мутацией E545K/+ (Horizon Discovery, 103-001) или PI3Kα с мутацией H1047R/+ (Horizon Discovery, 103-005). Эффективность ингибирования pPRAS40 под действием PI3Kα в каждой клеточной линии измеряют через 24 часа после обработки соединением. Селективность ингибиторов PI3Kα в отношении ее мутантных форм определяют на основании соотношений выраженного в EC50 показателя эффективности для WT по сравнению с E545K-несущими клеточными линиями и WT по сравнению с H1047R-несущими клеточными линиями.
Клеточная культура: клеточные линии поддерживают в инкубаторе для клеточных культур при 37°C и 5% CO2 в среде для культивирования клеток, содержащей RPMI1640 (среду 1640 от Мемориального института Розуэлла Парка; полученную от Genentech), 10% FBS (фетальная телячья сыворотка) (Gibco, 16140-071), 2 мМ L-глутамин (полученный от Genentech) и 10 мМ HEPES, pH 7,2 (полученный от Genentech). Каждые 72 часа клетки отщепляют с последующим разбавлением в соотношении 1:8, используя 0,25%-ный раствор трипсина в EDTA (Gibco, 25200).
Методика анализа: клетки собирают и высевают в используемые в анализе 384-луночные планшеты для работы с тканевыми культурами Greiner, № по каталогу 781091) и инкубируют в течение ночи при 37°C и 5% CO2. Высевание и анализ трех клеточных линий (WT, E545K и H1047R) проводят параллельно. На следующий день выполняют серийные разведения тестируемых соединений в диметилсульфоксиде (ДМСО) и добавляют к клеткам (конечная концентрация ДМСО 0,5%). Затем клетки инкубируют в течение 24 часов при 37°C и 5% CO2. Через 24 часа осуществляют лизис клеток и измеряют уровни pPRAS40, используя специально разработанный набор для анализа pPRAS40 на 384-луночных планшетах от Meso-Scale (Meso Scale Discovery, № по каталогу L21CA-1). Клеточные лизаты добавляют в используемые в анализе планшеты, предварительно покрытые антителами к фосфорилированному PRAS40. Образцы с фосфорилированными PRAS40 оставляют для связывания с захватывающими антителами в течение ночи при 4°C. К лизату после осуществления связывания добавляют детектирующее антитело (антитело к общему PRAS40, меченное электрохемилюминесцентной меткой SULFO-TAG) и инкубируют в течение 1 часа при комнатной температуре. Добавляют буфер для прочтения от MSD, благодаря чему при приложении напряжения к электродам планшета, метки, связавшиеся с поверхностью электродов, излучают свет. Используя прибор Sector от MSD измеряют интенсивность света и выполняют количественные измерения содержания фосфорилированного PRAS40 (фосфо-PRAS40) в образце. Процент ингибирования фосфорилирования PRAS40 под действием тестируемых соединений в различных концентрациях рассчитывают относительно не подвергнутых обработке контролей. Значения EC50 рассчитывают, используя 4-параметрическую логистическую нелинейную регрессионную модель зависимости доза-ответ.
Статистический анализ: значения EC50 представляют собой среднее геометрическое значение как минимум из 4-х независимых экспериментов. Все статистические выкладки выполняли с использованием программного обеспечения KaleidaGraph (версия 4.1.3). t-Критерий Стьюдента для сравнения активности в отношении клеток с мутациями и клеток дикого типа определяли с использованием непарных данных с равными дисперсиями. Значение P < 0,05 считается статистически значимым.
Пример 903. Анализы жизнеспособности клеток in vitro
Клетки высевали (1500 на одну лунку) в 384-луночные планшеты на 16 ч. На 2-е сутки готовили по девять серийных разведений (1:3) соединений в ДМСО в 96-луночном планшете. Дальнейшие разведения соединений выполняли в ростовой среде, используя робот Rapidplate® (Zymark Corp.). Разведенные соединения затем добавляли в четырех повторах в лунки 384-луночных планшетов для клеток и инкубировали при 37°C и 5% CO2. Через 4 суток измеряли относительные количества жизнеспособных клеток на основании люминесценции, используя Cell Titer-Glo® (Promega) в соответствии с инструкциями производителя, и снимали показания на ридере Wallac Multilabel (Perkin-Elmer). Значения EC50 рассчитывали, используя программное обеспечение Prism 6.0 (GraphPad).
Пример 904. Эффективность in vivo в отношении опухолевых ксенотрансплантатов мышей
Мыши: самки мышей с тяжелым комбинированным иммунодефицитом (SCID) (C.B-17 SCID.bg, Charles River Labs, San Diego), NOD.SCID (Charles River Labs, Hollister) или бестимусных NCR.мышей (Taconic) на 0-е сутки исследования находились в возрасте 8-9 недель и имели BW в диапазоне от 18 до 26 граммов. Животные получали воду без ограничения и лабораторный автоклавируемый корм для грызунов 5010 (LabDiet, St. Louis, MO). Мышей содержали в микроизоляторах в условиях 12-часового светового цикла. Genentech точно соответствует рекомендациям Руководства по содержанию и использованию лабораторных животных в отношении ограничения, условий содержания, хирургических процедур, регулирования кормления и приема жидкости и ветеринарного обслуживания. Программа по содержанию и использованию животных в Genentech аккредитована Международной ассоциацией по аттестации и аккредитации (условий) содержания лабораторных животных (AAALAC International), которая обеспечивает соблюдение принятых стандартов по содержанию и использованию лабораторных животных. Мышей в Genentech содержали в стандартных клетках-микроизоляторах для грызунов и давали возможность адаптироваться к условиям исследования в течение по меньшей мере 3 суток перед имплантацией опухолевых клеток. Для исследования использовали только животных, которые оказались здоровыми и у которых не были обнаружены явные нарушения.
Имплантация опухолей: процедуры ксенотрансплантации начинали, используя либо раковые клетки (HCC1954x1 или KPL4), либо пассажем опухолей (HCI-003). Клетки культивировали в среде RPMI 1640, дополненной 10% фетальной телячьей сыворотки, 2 мМ глутамином, пенициллином в концентрации 100 единиц/мл, стрептомицина сульфатом в концентрации 100 мкг/мл (микрограммов в одном мл) и гентамицином в концентрации 25 мкг/мл, собирали в логарифмической фазе роста и ресуспендировали в 50%-ном матригеле (Matrigel, Becton Dickinson Bioscience; San Jose, CA), не содержащем фенолового красного, и сбалансированном солевом растворе Хенкса в количестве 3х106 или 5x106 клеток/мл в зависимости от времени удвоения количества клеток в линии. В случае модели, происходящей от пациента линии HCI-003, выполняли подкожную имплантацию шариков пчелиного воска массой 30 мг, содержащих приблизительно 1 мг 17β(бета)-эстрадиола, за 3 суток до имплантации фрагментов опухоли. Опухолевые клетки или фрагменты имплантировали на глубину 2/3 жировой подушки молочной железы и мониторинг опухолевого роста выполняли до момента попадания среднего размера в целевой диапазон 100-250 мм3. После того, как большинство опухолей достигало целевого диапазона, мышей разделяли на группы из 7-10 мышей с учетом объема опухолей.
Терапевтические агенты: PI3K-ингибирующие соединения поставляли в виде сухого порошка в форме свободного основания и хранили при комнатной температуре с защитой от света. В качестве разбавителя для таселисиба (GDC-0032) и BYL719 использовали смесь 0,5% метилцеллюлозы:0,2% твина 80 (MCT) в деионизованной воде. В качестве контрольного разбавителя для соединения 101 использовали наносуспензию 0,5% метилцеллюлозы/0,2% твина 80 (MCT). Наносуспензию MCT получают, сначала готовя суспензию MCT. После приготовления суспензии используют стеклянные шарики диаметром 1 мм и магнитный стержень из редкоземельного элемента для перемешивания с целью измельчения суспензии MCT в течение примерно 24 часов с получением наносуспензии из мелких частиц. Для проверки конечного размера частиц использовали анализатор размера частиц. Дозы лекарственных средств готовили один раз в неделю и хранили при 4°C.
Обработка: мыши получали п/о с использованием желудочного зонда один раз в сутки в течение 21-28 суток (разбавитель) или заданную дозировку PI3K-ингибирующих соединений в мг/кг (выраженную в виде эквивалента свободного основания) в объеме 100мкл (микролитров) (5 мл/кг).
Ожидаемый результат: объем опухоли измеряли в 2-х направлениях (по длине и ширине), используя циркули Ultra Cal IV (модель 5410111; Fred V. Fowler Company), как приведено ниже: объем опухоли (мм3) = (длина x ширина2) × 0,5, и анализировали с использованием Excel-версии 11.2 (Microsoft Corporation). Чтобы провести анализ повторного измерения объемов опухолей во времени у одних и тех же животных, использовали подход с применением линейной модели со смешанными эффектами (LME) (Pinheiro J., et al., nlme: linear and nonlinear mixed effects models, 2009; пакет программ R, версия 3.2.5). Такой подход предусматривает как повторные измерения, так и незначительные количества выбывших из исследования вследствие любой не связанной с лечением гибели животных до завершения исследования. Для аппроксимации нелинейного профиля зависимости объема опухоли (в log2 координатах) от времени лечения при каждом уровне доз использовали кубическую сплайн-регрессию. Затем эти нелинейные профили соотносили с дозой в рамках модели со смешанными эффектами. Ингибирование роста опухоли в процентном отношении к контролю-разбавителю (% TGI) рассчитывали как процентное отношение площади под кривой, полученной с помощью аппроксимации (area under the fitted curve; AUC), для группы, принимающей соответствующую дозу в сутки, относительно разбавителя, используя следующую формулу: % TGI = 100 × (1 - AUCдоза/AUCразб.). Согласно этой формуле значение TGI, равное 100%, указывает на остановку роста опухоли, значение TGI больше (>) 1%, но меньше (<) 100% указывает на задержку опухолевого роста, а значение TGI больше (>) 100% указывает на регрессию опухоли. Частичный ответ (PR) для животного определяли как регрессию опухоли, составляющую больше (>) 50%, но меньше (<) 100% от начального объема опухоли. Полный ответ (CR) определяли как 100%-ную регрессию опухоли (т.е. как не поддающуюся измерению опухоль) в любой день в ходе исследования.
Токсичность: животных взвешивали один раз в сутки в течение первых пяти суток исследования и после этого два раза в неделю. Массы тела животных измеряли, используя весы Adventurer Pro® AV812 (Ohaus Corporation). Изменение массы в процентах рассчитывали так, как приведено ниже: изменение массы тела (%) = [(массадругие сутки - масса0-е сутки)/масса0-е сутки] × 100. Мышей часто осматривали, отслеживая выраженные признаки любых неблагоприятных, связанных с лечением побочных эффектов, и регистрировали клинические признаки токсичности при их обнаружении. Приемлемую токсичность определяют как токсичность, приводящую к средней потере массы тела (BW) в группе, составляющей менее 20% в ходе исследования, и не более чем к одному случаю смерти в результате лечения (TR death) из десяти подвергнутых лечению животных. Любой режим введения, который приводит к более высокой токсичности, считается превышающим максимальную переносимую дозу (MTD). Смерть классифицируют как смерть, являющуюся результатом лечения (TR), если она связана с побочными эффектами лечения, что подтверждено клиническими признаками и/или результатами вскрытия, или также может быть классифицирована как TR, если она обусловлена неизвестными причинами во время периода введения или в течение 10 суток после введения заключительной дозы. Смерть классифицируют как не являющуюся результатом лечения (NTR), если нет никаких доказательств того, что смерть была связана с побочными эффектами лечения.
Пример 905. Клеточная культура и эксперименты с ингибиторами in vitro
Клеточные линии выращивали в стандартных условиях культивирования тканей в среде RPMI с 10% фетальной телячьей сыворотки, пенициллином (100 ед./мл) и стрептомицином (100 мкг/мл). HCC-1954 и HDQ-P1 представляют собой линии раковых клеток молочной железы (Американская коллекция типовых культур; Manassas, VA). Клетки HCC-1954 и HDQ-P1 помещали в каждую лунку
6-луночного планшета для тканевых культур в количестве 80000 клеток/лунка и инкубировали при 37°C в течение ночи. Клетки инкубировали с каждым из соединений в указанных концентрациях в течение 24 часов. После инкубации клетки промывали один раз холодным забуференным фосфатом физиологическим раствором (PBS) и лизировали в буфере для экстракции клеток Biosource™ (Invitrogen; Carlsbad, CA), дополненном ингибиторами протеаз (F. Hoffman-LaRoche; Mannheim, Germany), 1 мМ фенилметилсульфонил-фторидом и смесями 1 и 2 ингибиторов фосфатаз (Sigma-Aldrich; St. Louis, MO). Концентрации белка определяли, используя набор для анализа белков с применением бицинхониновой кислоты (BCA) Pierce (Thermo Fisher Scientific; Rockford, IL).
Анализы белков
Концентрацию белка определяли, используя используя набор для анализа белков с применением BCA от Pierce (Rockford, IL). Для получения иммуноблотов проводили разделение равных количеств белков электрофорезом в 4-12%-ных бис-трис-градиентных гелях NuPage™ (Invitrogen; Carlsbad, CA); белки переносили на нитроцеллюлозные мембраны, используя систему IBlot и протокол от InVitrogen. Антитела к p110альфа и фосфор-Akt (Ser473) получали от Cell Signaling (Danvers, MA). Антитела к β-актину и GAPDH (глицеральдегид-3-фосфатдегидрогеназа) поступали от Sigma.
Пример 906. Экспрессия CD69 B-клетками, анализ в цельной крови человека в отношении экспресии CD69 в CD19+ CD27- B-клетках
Клеточная культура: цельную кровь человека разливали в 96-луночные планшеты с глубокими лунками в объеме 100 мкл на одну лунку. Соединения разбавляли в ДМСО для получения желаемых исходных концентраций, затем дополнительно разбавляли в PBS до желаемой рабочей концентрации и добавляли в объеме 5,5 мкл на одну лунку. Затем образцы инкубировали в течение 1 часа при 37°C в условиях 5% CO2, после чего добавляли 5 мкг (10 мкл на одну лунку) козьих F(ab')2 против IgM (Southern Biotech, AL) и инкубировали в течение 18 часов при 37°C в условиях 5% CO2. Все варианты обработок тестировали в двух повторах.
Методики выделения и окрашивания клеток: после инкубации уровень экспрессии CD69 в CD19+ CD27--клетках определяли посредством окрашивания образцов цельной крови смесью, содержащей CD27: 10 мкл/лунка (клон L128; BD Biosciences, NJ), CD19: 7,5 мкл/лунка (клон SJ25C1; BD Biosciences, NJ) и CD69: 10 мкл/лунка (клон FN50; BD Biosciences, NJ). Помимо этого цельную кровь человека от каждого донора окрашивали, используя изотипически сходные флуоресцентные контрольные антитела. После добавления смеси соответствующих антител образцы цельной крови окрашивали в течение 30 минут в темноте и затем осуществляли лизис образцов, используя BD Pharm Lysis™ (BD Bioscience, NJ). Далее полученные образцы промывали буфером для FACS (метод сортировки флуоресцентно-активированных клеток) (забуференным фосфатом физиологическим раствором (без Ca/Mg++), содержащим 1 мМ EDTA, 25 мМ HEPES, pH 7,0, 1% фетальной телячьей сыворотки (инактивированной нагреванием)) и фиксировали в буфере для FACS, дополненном 0,1% формальдегида (Polysciences Inc., PA) и 0,1% плуроника F-68 (Sigma, MO). Сбор данных осуществляли, используя установку LSR-II от BD (BD Biosciences) с программным обеспечением FACSDiva от BD.
Экспрессия CD69 в CD19+ CD27- B-клетках. Уровни CD19, CD27 и CD69 в клетках оценивали посредством проточной цитометрии, используя программное обеспечение FACSDiva от BD, и определяли среднюю интенсивность флуоресценции при экспрессии CD69 (CD69 MFI-Mean) в популяции CD19+ CD27- лимфоцитов. Концентрацию соединения, вызывающую уменьшение CD69 MFI-Mean на 50% (IC50), определяли, используя программное обеспечение Genedata (Genedata Screener, MA).
Пример 907. EC50 для pPRAS40 в HCC1954 и HDQP1
Клетки высевают в используемые в анализе 384-луночные планшеты для работы с тканевыми культурами и инкубируют в течение ночи. На следующий день клетки обрабатывают соединениями и инкубируют в течение 24 часов. Через 24 часа осуществляют лизис клеток и измеряют уровни pPRAS40, используя аналитическую платформу т Meso-Scale. Эти клеточные линии являются весьма полезными для характеристики селективности ингибиторов PI3Ka в случае мутантных форм PI3Ka. Клеточная линия HCC1954 экспрессирует мутантную форму PI3Ka (с мутацией E545K) по сравнению с WT в HDQP1.
Принцип анализа: платформа от MSD предоставляет возможность проведения метода измерения уровней фосфорилирования в случае pPRAS40 в одном образце. Клеточные лизаты добавляют в используемые в анализе планшеты, предварительно покрытые антителами к общему PRAS40. После лизиса клеток образцы оставляют для осуществления связывания PRAS40 с захватывающими антителами. Детектирующее антитело (антифосфо-PRAS40), меченное электрохемилюминесцентным соединением SULFO-TAG от MSD, добавляют к лизату, в котором произошло связывание. Добавляют буфер для прочтения от MSD, благодаря чему при приложении напряжения к электродам планшета, метки, связанные с поверхностью электродов, излучают свет. Используя прибор Sector от MSD измеряют интенсивность света и количество фосфо-EGFR (фосфорилированного рецептора эпидермального фактора роста) в образце (принцип анализа Meso Scale).
Материалы
Линия с мутацией E545K: HCC1954 (CL130216)
10% FBS (Gibco 16140-071),
2 мМ L-глутамин (GNE (от англ. Grade not established - марка не установлена), для внутреннего пользования),
1% HEPES (GNE, для внутреннего пользования)
Методика
• Полученные соединения используют в 2 мМ концентрации в ДМСО. Подготовить планшет для титрования раствора соединений в ДМСО в соотношении 1:3 с использованием чистого ДМСО.
• Материнский планшет для разведение в ДМСО содержит по 72 мкл раствора каждого из 13 соединений.
• Контрольное соединение с селективностью в отношении мутаций: добавить по 72 мкл 2 мМ раствора контрольного соединения в лунки B2 каждого используемого в анализе планшета. Это контрольное соединение демонстрирует приблизительно в 20 раз более высокую эффективность в клеточной линии HCC1954 по сравнению с линией HDQP1.
• Используя многоканальную пипетку, перенести по 36 мкл из каждой лунки с соединением в лунку, расположенную непосредственно ниже, (например, из B2 в C2) для построения второй копии кривых зависимости доза-ответ.
• Использовать метод с применением устройства Biomek FX под названием “SLS_serial dilution/1 plate_384_3_13_3x” для выполнения серийных разведений соединений в материнском планшете.
• Герметично закрыть и хранить как материнский, так и дочерний планшеты для разведений в ДМСО, если они непосредственно не используются, в условиях термоизоляции.
1-е сутки: культивирование клеток в планшетах
1. Высеять по 12500 клеток в 45 мкл среды для каждой клеточной линии. Дать возможность клеткам осесть/прикрепиться к планшету в течение 15-20 мин при комнатной температуре.
2. Инкубировать клетки в течение ночи при 37°C в инкубаторе с регулированием влажности и содержания CO2.
2-е сутки: подготовка планшета с соединениями и обработка соединениями
1. Относительно планшета для выполнения промежуточных 10-кратных разведений: добавить по 95 мкл бессывороточной среды в 384-луночный полипропиленовый планшет от Greiner с лунками стандартного профиля.
2. Для выполнения промежуточного разведения соединений в среде и добавления к клеткам использовать протокол для Biomek FX: “SLS Intermed Dil Add 5 ul to Cells July 13 2012”. Согласно этому протоколу Biomek перенести по 5 мкл из дочернего планшета для ДМСО в планшет для промежуточного разведения соединений, содержащий в лунках по 95 мкл среды, и провести перемешивание среды с соединениями. Затем, согласно данному методу, перенести по 5 мкл из планшета для промежуточного разведения соединений в соответствующие лунки планшета для клеток.
3. Инкубировать обработанные клетки при 37 градусах в течение 24 часов в инкубаторе с увлажнением и 5% CO2.
3-и сутки: лизирование клеток и добавление в планшеты от MSD
Блокировать используемый в анализе планшет от MSD, применяя 50 мкл 3%-ного раствора блокирующего средства A в 1X буфере для промывки от MSD, в течение 1-2 ч при комнатной температуре. Этот раствор можно хранить при 4°C в течение до одного месяца включительно. Блокирующий буфер A содержит 1X буфер для промывки от MSD (20 мл 1X трис-буфера для промывки и 600 мг блокирующего средства A).
Приготовление лизирующего буфера
(или смесь с № по каталогу Sigma P2714-1BTL, ресуспендированная в 10 мл PBS)
Удалить отсасыванием среды и выполнить лизирование клеток.
1. Выполнить лизирование клеток в 50 мкл лизирующего буфера. Лизирование выполнять при комнатной температуре в течение 10-20 минут на планшетном шейкере.
2. Пока осуществляется лизирование клеток, промыть блокированные планшеты 1x буфером для промывки от MSD.
3. Перенести по 42 мкл лизатов (21 + 21 мкл) в блокированный используемый в анализе pPRAS40 планшет от MSD.
4. Герметично закрыть планшеты от MSD и инкубировать при 4°C со встряхиванием в течение ночи.
4-е сутки: анализ/детекция согласно протоколу MSD
8. Приготовить раствор, содержащий 1% блокирующего средства A в 1X буфере для промывки от MSD. (20 мл 1X трис-буфера для промывки и 200 мг блокирующего средства A (1%, масс./об.)). Этот раствор можно хранить при 4°C в течение до одного месяца включительно.
9. Промыть планшеты от MSD 1x буфером для промывки от MSD.
10. Добавить в планшеты по 10 мкл раствора разбавленного меченного SULFO-TAG детектирующего антитела. Инкубировать в течение 1 ч со встряхиванием при комнатной температуре.
11. Промыть планшеты 4 раза 1x буфером для промывки от MSD.
12. Добавить по 35 мкл 1x буфера для прочтения, используя реверсивное пипетирование и избегая образования пузырьков.
13. Незамедлительно прочитать планшет на приборе SECTOR от MSD.
Пример 908. Совместная кристаллография с p110α (альфа)
Укороченную по N-концу p110α (альфа) получали согласно Chen и др. и Nacht и др. (Chen P., Y.L. Deng, S. Bergqvist, M.D. Falk, W. Liu, S. Timofeevski and A. Brooun “Engineering of an isolated p110alpha subunit of PI3Kalpha permits crystallization and provides a platform for structure-based drug design”, (2014) Protein Sci., 23(10): 1332-1340; Nacht M. et al. (2013) “Discovery of a potent and isoform-selective targeted covalent inhibitor of the lipid kinase PI3K”, J. Med. Chem. 56(3): 712-721).
Для получения кристаллов в присутствии разрабатотанных соединений применяли стандартные протоколы. Собранные кристаллы до получения дифракционных данных хранили путем погружения в жидкий азот и помещали в канал синхротрона, дающий монохроматическое рентгеновское излучение. Дифракционные данные собирали, обрабатывали и объединяли, используя стандартные протоколы. Элементарные ячейки и пространственная группа кристаллов были изоморфны таковым, приведенным ранее (Nacht, 2013; Chen, 2014). Размещение разработанных соединений в картах электронной плотности и уточнение кристаллической структуры до пределов разрешения в диапазоне от 2,36 до 2,56 Å проводили, используя стандартные протоколы.
В этой заявке единицы мкл, мкмоль и т.д. означают микролитр, микромоль и т.д.
Хотя изложенное выше изобретение для ясности понимания описано довольно подробно посредством иллюстрации и примеров, эти описания и примеры не следует истолковывать как ограничивающие объем данного изобретения. Описания всей патентной и научной литературы, приведенной в данном описании, явным образом включены во всей своей полноте посредством ссылки.
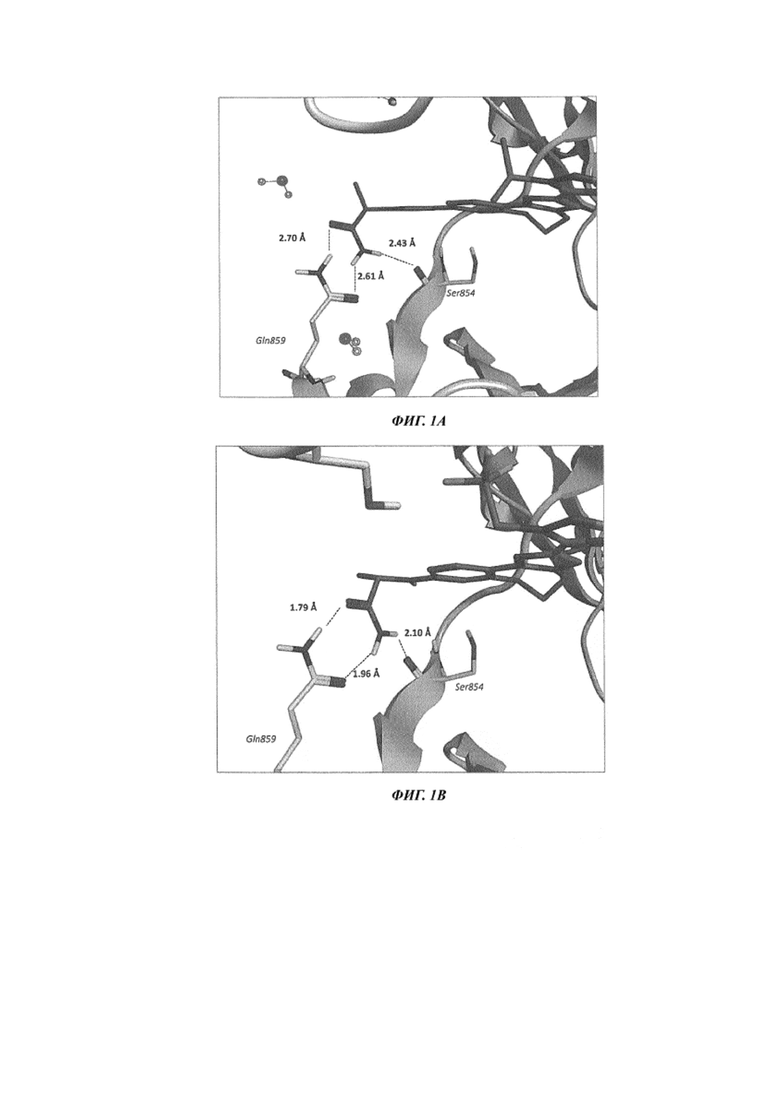
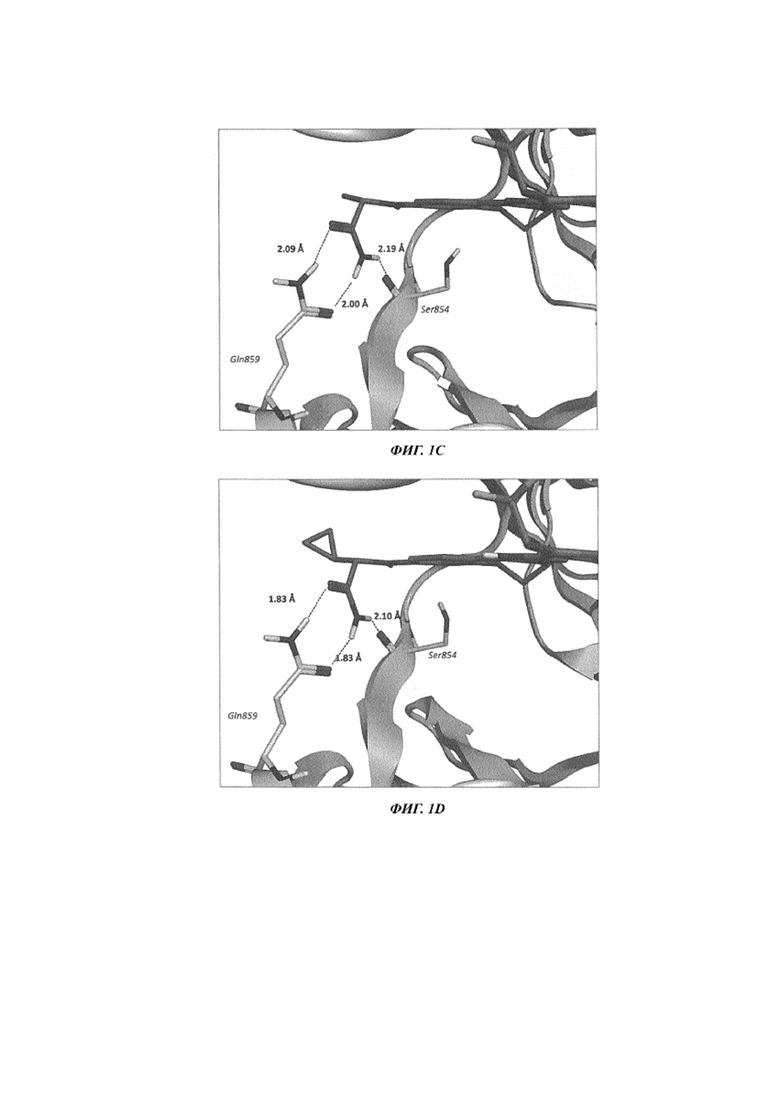
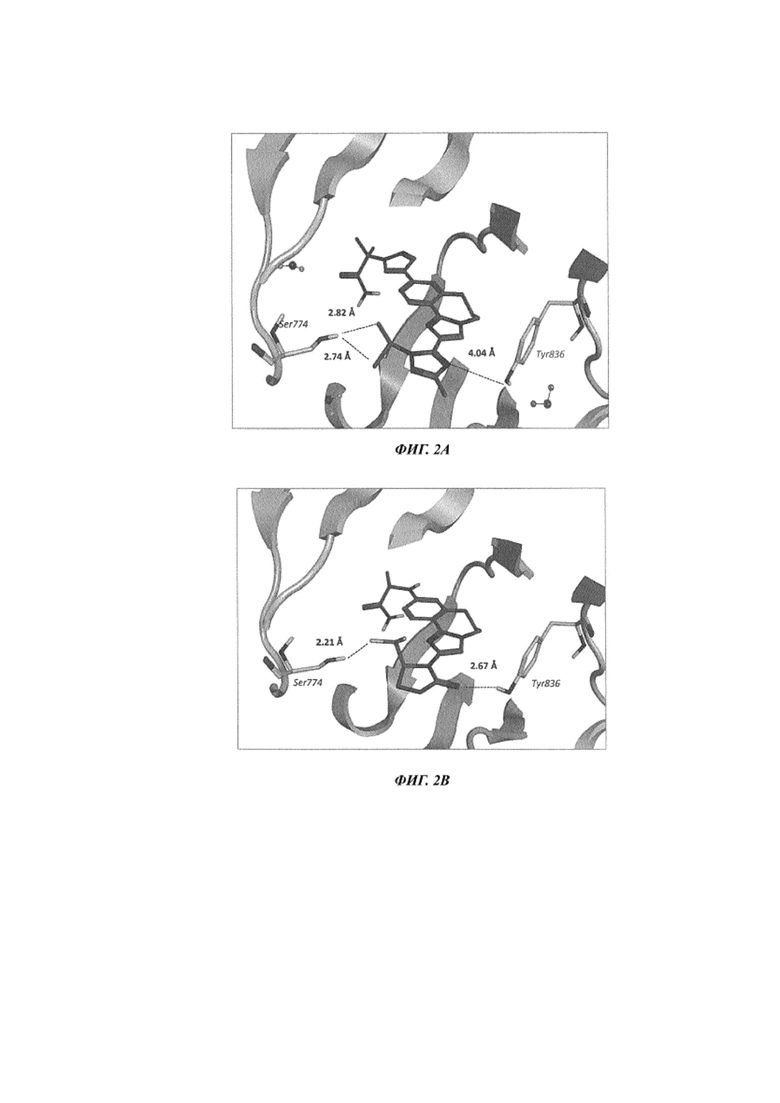
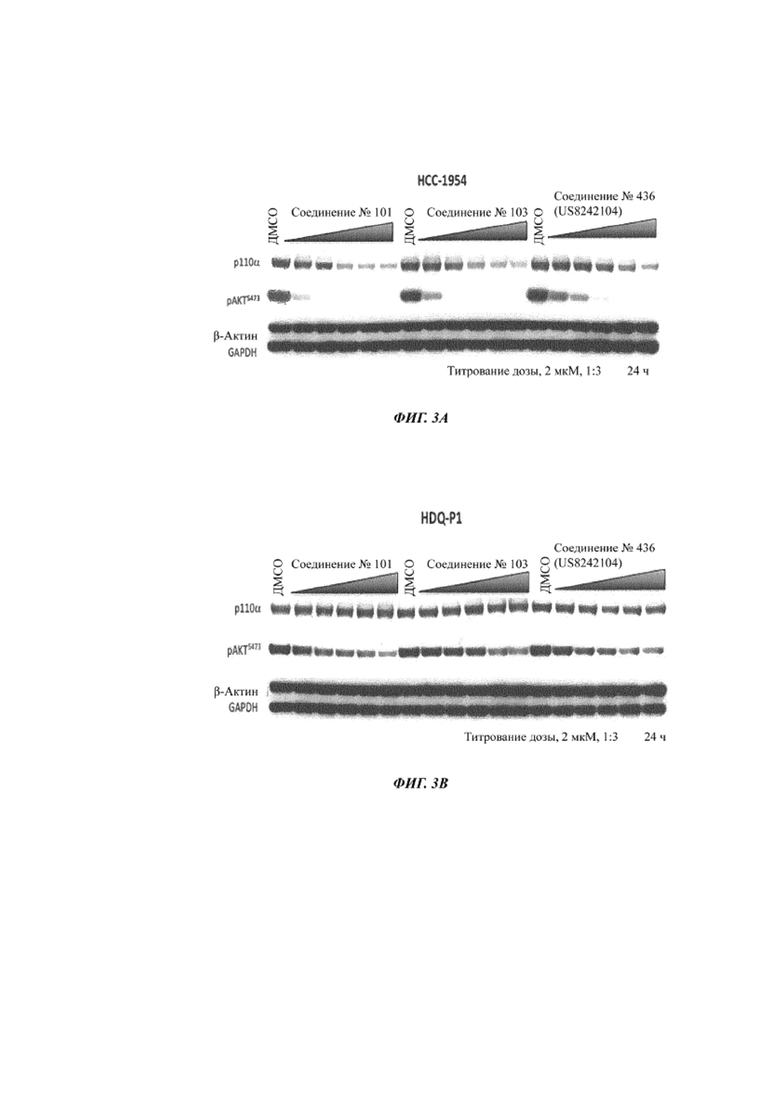
Изобретение относится к бензоксазепиноксазолидинонам формулы I, фармацевтическим композициям на и основе, селективно ингибирующим мутантную форму PI3Kα, способам лечения рака, связанного с экспрессией мутантной формы PI3Kα. Технический результат: получены новые соединения, пригодные для лечения рака, связанного с экспрессией мутантной формы PI3Kα. 11 н. и 12 з.п. ф-лы, 21 табл., 3 ил.
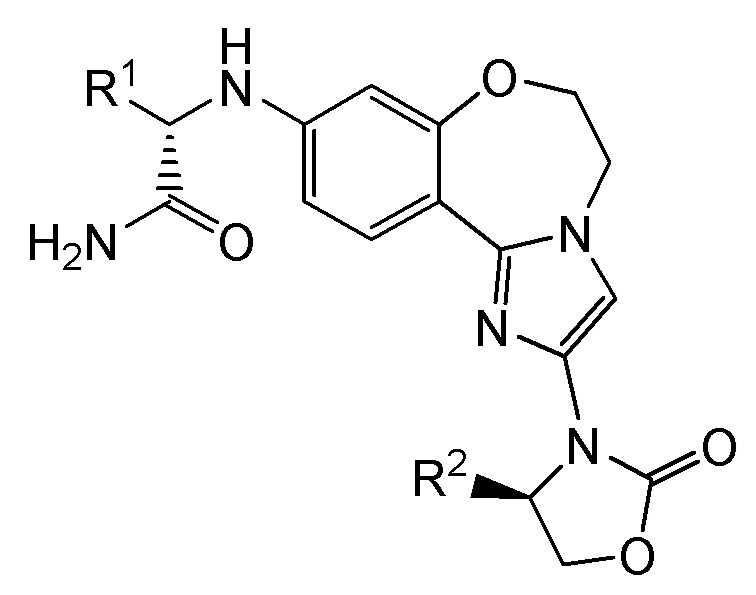 (I)
(I)
1. Соединение, выбранное из соединения формулы I:

и его стереоизомеров, геометрических изомеров и фармацевтически приемлемых солей, где:
R1 выбран из -СН3, -СН2СН3, циклопропила и циклобутила;
R2 выбран из -СН3, -CHF2, -CH2F и -CF3.
2. Соединение по п. 1, где R1 представляет собой СН3 или циклопропил.
3. Соединение по п. 1 или 2, где R2 представляет собой -CHF2.
4. Соединение, выбранное из:
(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида;
(S)-2-циклобутил-2-((2-((R)-4-метил-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)ацетамида;
(S)-2-циклопропил-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)ацетамида;
(S)-2-циклопропил-2-((2-((R)-4-метил-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)ацетамида;
(S)-2-циклопропил-2-((2-((S)-4-(фторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)ацетамида;
(S)-2-((2-((S)-4-(фторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамида;
(S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)бутанамида
и его фармацевтически приемлемых солей.
5. (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид
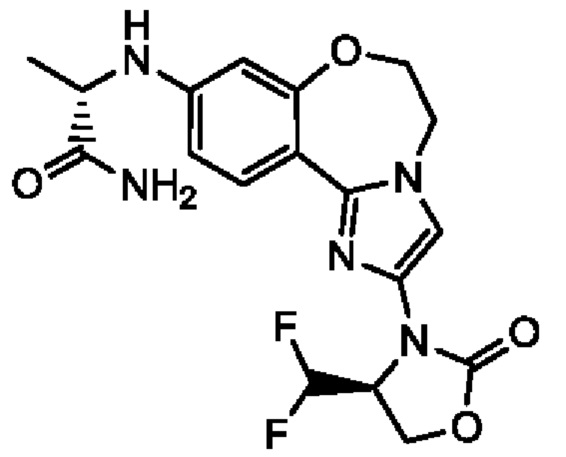
или его фармацевтически приемлемые соли.
6. (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид
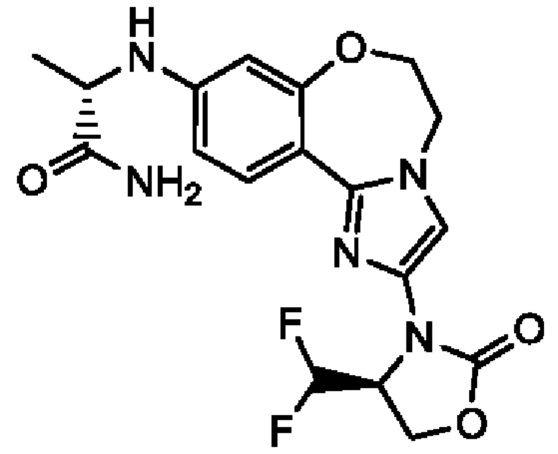
7. Фармацевтическая композиция, селективно ингибирующая мутантную форму PI3Kα, содержащая соединение по любому из пп. 1-6 в эффективном количестве и фармацевтически приемлемый носитель, разбавитель или эксципиент.
8. Фармацевтическая композиция для лечения рака с экспрессией мутантной формы PI3Kα, содержащая (S)-2-((2-((S)-4-(дифторметил)-2-оксооксазолидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)амино)пропанамид
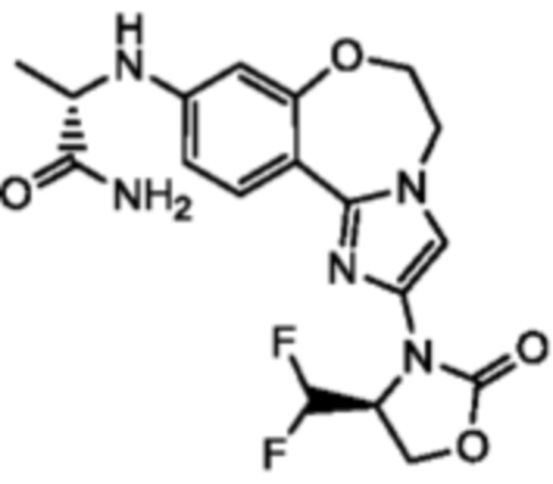
в эффективном количестве и фармацевтически приемлемый носитель, разбавитель или эксципиент.
9. Фармацевтическая композиция по п. 8, где рак представляет собой рак молочной железы.
10. Способ приготовления фармацевтической композиции, включающий комбинирование соединения по любому из пп. 1-6 с фармацевтически приемлемым носителем, разбавителем или эксципиентом.
11. Способ лечения рака у пациента, имеющего рак с экспрессией мутантной формы PI3Kα, включающий введение указанному пациенту терапевтически эффективного количества соединения по любому из пп. 1-6.
12. Способ по п. 11, где рак представляет собой рак молочной железы.
13. Способ по п. 12, где рак молочной железы представляет собой положительный по рецепторам эстрогенов (ER+) рак молочной железы.
14. Способ по п. 12, где раком молочной железы является рак базального или люминального подтипа.
15. Способ по п. 12, где данный вид рака представляет собой HER2-положительный рак.
16. Способ по п. 11, где рак представляет собой немелкоклеточный рак легкого.
17. Способ по п. 11, где при данном виде рака экспрессируется PIK3CA с мутацией, выбранной из E545K и H1047R.
18. Способ по п. 11, где при данном виде рака экспрессируется мутантная форма PTEN.
19. Набор для терапевтического лечения рака молочной железы с экспрессией мутантной формы PI3Kα, содержащий:
a) фармацевтическую композицию по любому из пп. 7-9; и
b) инструкции для применения при терапевтическом лечении рака молочной железы с экспрессией мутантной формы PI3Kα.
20. Применение соединения по любому из пп. 1-6 для лечения рака с экспрессией мутантной формы PI3Kα у пациента.
21. Применение соединения по любому из пп. 1-6 для изготовления лекарственного средства для лечения рака с экспрессией мутантной формы PI3Kα у пациента.
22. Применение по п. 20 или 21, где рак выбран из рака молочной железы и немелкоклеточного рака легкого.
23. Соединение по любому из пп. 1-6 для применения для лечения рака с экспрессией мутантной формы PI3Kα у пациента, причем данный рак выбран из рака молочной железы и немелкоклеточного рака легкого.
| RU 2012117398 A, 10.11.2013 | |||
| 0 |
|
SU181346A1 | |

