Область техники
Настоящее изобретение относится к области получения промежуточного соединения лекарственного средства, и, в частности, оно относится к новому способу синтеза хирального промежуточного соединения ледипасвира.
Уровень техники
Ледипасвир (Ledipasvir), ранее известный как GS-5885, представляет собой ингибитор протеазы NS5A, разработанный компанией Gilead Sciences, Inc. После завершения III фазы клинических исследований лекарство в таблетках на основе ледипасвира, содержащее фиксированные дозы ледипасвира/софосбувира, применяемое для лечения гепатита С генотипа 1, 10 февраля 2014 г. было включено в Американскую фармакопею. 10 октября 2014 г. Управлением по контролю за продуктами и лекарствами США был утвержден комбинированный препарат на основе ледипасвира/софосбувира под торговым названием Harvoni. Ледипасвир представляет собой первое в мире лекарственное средство, одобренное в качестве применяемого для полностью перорального лечения гепатита С, и оно может исключить необходимость в традиционном введении интерферона (IFN) в качестве лекарственного средства. Механизм действия ледипасвира заключается в ингибирующем действии в отношении белка NS5A, в результате чего прерывается репликация вирусной РНК и достигается эффект лечения гепатита С.
Ледипасвир (Ledipasvir), химическое название - GS-5885; химическое название-
метил-N-[(2S)-1-[(6S)-6-[5-[9,9-дифтор-7-[2-[(1S,2S,4R)-3-[(2S)-2-(метоксикарбониламино)-3-метилбутаноил]-3-азабицикло[2.2.1]гептан-2-ил]-3Н-бензимидазол-5--ил]флуорен-2-ил]-1Н-имидазол-2-ил]-5-азаспиро[2.4]гептан-5-ил]-3-метил-1-оксобутан-2-ил]карбамат (на английском - methyl
N-[(2S)-l-[(6S)-6-[5-[9,9-Difluoro-7-[2-[(1S,2S,4R)-34(2S)-2-(methoxycarbonylamino)-3-methylbutanoyl]-3-azabicyclo[2.2.1]heptan-2-yl]-3Н-benzimidazol-5-yl]fluoren-2-yl]-1H-imidazol-2-yl]-5-azaspiro[2.4]heptan-5-yl]-3-methyl-l-oxobutan-2-yl]carbamate); CAS-номер - 1256388-51-8; химическая формула C49H54F2N8O6; молекулярная масса составляет 889,00; и его торговое название - Harvoni (в комбинации с софосбувиром). Химическая структура указана ниже.
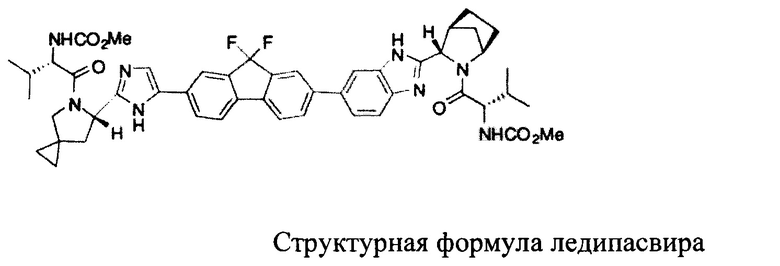
Ниже представлено два известных из литературы способа синтеза, касающиеся промежуточного соединения ледипасвира.
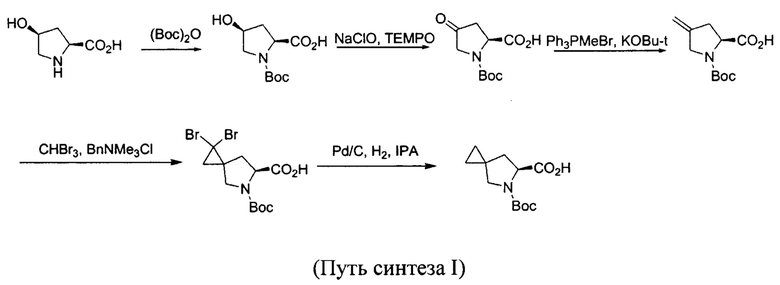
Способ 1. В патенте США №20130324740 описан способ синтеза промежуточного соединения ледипасвира исходя из L-гидроксипролина, защищенного Вос-группой, где его окисление осуществляют с помощью гипохлорида натрия-ТЕМРО с получением из пролина кетона с защитной группой Вос; затем кетон подвергают реакции Виттига с получением производного алкенов; затем проводят реакцию с трибромметаном с получением дибромциклопропильного соединения; и, наконец, гидрогенизируют с помощью палладия на угле для удаления брома с получением целевого промежуточного соединения. Преимущества этого пути заключаются в синтезировании из хирального исходного вещества и в отсутствии необходимости в последующем осуществлении разделения. Недостатки заключаются в сравнительно высокой стоимости исходных веществ, необходимости в безводных и анаэробных условиях реакции, гидрогенизации под высоким давлением и других серьезных операциях; в то же время реакция Виттига характеризуется дорогостоящими реактивами, сложностью очистки путем разделения, сложностью операций и прочими недостатками; гидрогенизация с помощью палладия на угле под высоким давлением во время удаления брома обладает определенным риском, что ограничивает ее применение в масштабе. Путь реакции, например, путь синтеза I, является следующим:
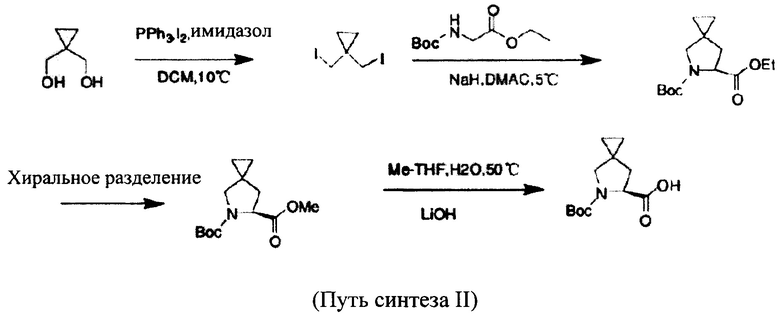
Способ 2. Сначала с применением циклопропандиметанола в качестве исходного вещества проводят реакцию замещения, при этом гидроксильная группа замещается йодом с получением дизамещенного йодида, продукт растворяют при 5°С в DMAC, затем добавляют сложный этиловый эфир N-Boc-глицина и получают азаспиросоединение. Подвергают хиральному разделению, с помощью LiOH гидролизуют и получают производное, представляющее собой (S)-5-азаспиро[2.4]гептан-6-карбоновую кислоту. Посредством хирального разделения с колоночной хроматографией соответственно получают хиральное промежуточное соединение, при этом результаты разделения хорошие и степень чистоты сравнительно высокая, но применение этого способа сравнительно трудное и сложное, выход продукта небольшой, себестоимость относительно более высокая и нет соответствия требованиям промышленного производства. Путь реакции, например, путь синтеза II, является следующим:
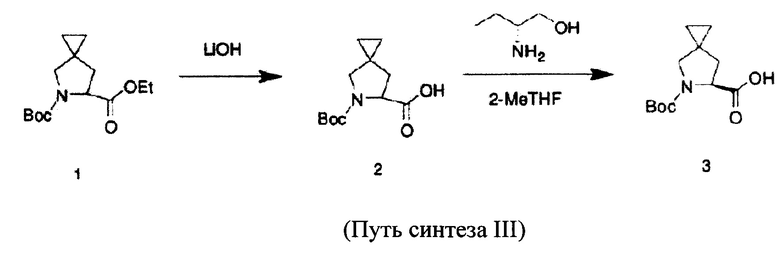
Другой способ разделения заключается в разделении посредством способа химического разделения рацемата азаспиросоединения с помощью хирального аминоспирта, при этом получают продукт (выход 32%), способ представляет собой, например, путь синтеза III, и этот способ представляет собой традиционный способ разделения, при этом применяемые исходные вещества представляют собой традиционные реактивы, которые легко приобрести, их стоимость сравнительно низкая, условия реакции сравнительно мягкие, он прост в применении и относительно соответствует требованиям промышленного производства. Однако его недостатки заключаются в возможности использования лишь половины энантиомеров, что приводит к неразумной трате ресурсов.
Описание
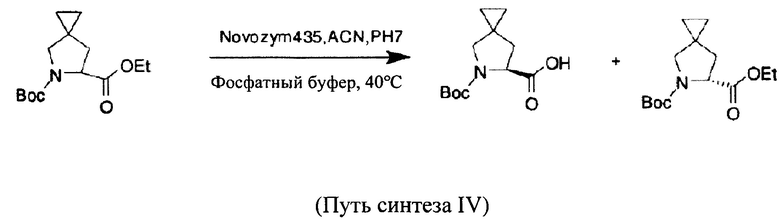
В то же время в патенте США №US 20130324740 описан способ, в котором растворимую смесь рацемата разделяют с применением липазы от компании Novozymes, при этом продукт получают после разделения и окончательной обработки (выход 75,9%, э. и. более 99%). Этот путь разделения представляет собой, например, путь синтеза IV, при этом способ характеризуется высокой производительностью, высоким значением э. и., обеспечением хирального разделения с помощью применяемого фермента, небольшим количеством побочных продуктов, возможностью повторного использования фермента и безвредностью по отношению к окружающей среде.
Однако также имеет место ситуация лишнего расхода энантиомеров.
Ледипасвир представляет собой первое новое, исключительно эффективное лекарство, применяемое для полностью перорального лечения вируса гепатита С, при этом структура лекарственного средства сравнительно сложная, путь синтеза относительно простой, возможно применение в промышленном производстве, общий выход относительно низкий, что делает себестоимость синтеза значительно более высокой. Поэтому разработка полного, безопасного по отношению к окружающей среде пути синтеза с высокой эффективностью становится одной из ключевых задач исследований в химии органического синтеза.
Краткое описание изобретения
Основная задача настоящего изобретения заключается в преодолении недостатков, известных из предшествующего уровня техники, и в предоставлении нового способа синтеза хирального промежуточного соединения ледипасвира, при этом способ характеризуется повышением атомной эффективности, снижением себестоимости продукта, простотой синтеза, удобством получения и выгодностью в отношении широкомасштабного промышленного производства.
Основная задача настоящего изобретения решается посредством представленного ниже технического решения, а именно посредством нового способа синтеза хирального промежуточного соединения ледипасвира, при этом указанное хиральное промежуточное соединение ледипасвира представляет собой S-5-азаспиро[2,4]гептан-6-карбоновую кислоту, при этом путь синтеза является следующим:
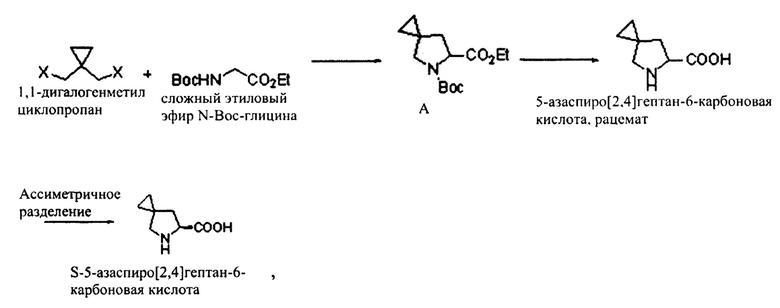
где X представляет собой галоген;
Конкретный способ синтеза включает следующие этапы:
S1: проводят реакцию замыкания цикла между
1,1-дигалогенметилциклопропаном и сложным этиловым эфиром N-Boc-глицина в щелочной среде с получением соединения А;
S2: подвергают омылению соединение А, удаляют защитную группу ВОС и получают 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата;
S3: подвергают асимметричному разделению 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата с получением S-5-азаспиро[2,4]гептан-6-карбоновой кислоты.
Кроме того, путь синтеза указанного 1,1-дигалогенметилциклопропана является следующим:
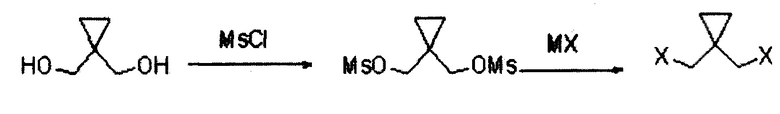 9
9
где в MX М представляет собой металл м, а X представляет собой галоген;
Конкретный способ синтеза является следующим:
(1) посредством реакции циклопропандиметанола с метансульфонилхлоридом получают циклопропилдиметансульфонат;
(2) посредством реакции циклопропилдиметансульфоната с галогенидом металла получают дигалогенциклопропан.
Кроме того, этап S1 осуществляют в реакционном растворителе в присутствии щелочи, при этом указанный реакционный растворитель представляет собой амидный или эфирный растворитель; указанная щелочь представляет собой любую из алкоголята калия, алкоголята натрия, гидрида натрия или гидрида калия; температура реакции составляет -10-100°С.
Кроме того, указанное омыление на этапе S2 осуществляют в растворителе в присутствии щелочи, при этом указанный растворитель представляет собой по меньшей мере один из спиртов или простых эфиров; указанная щелочь представляет собой неорганическую сильную щелочь; кислота, применяемая в указанном удалении защитной группы ВОС, представляет собой любую из хлористоводородной кислоты, серной кислоты, фосфорной кислоты или азотной кислоты; температура реакции составляет 0-100°С
Кроме того, конкретные операции указанного асимметричного разделения на этапе S3 являются следующими: после добавления разделяющего средства нагревают 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата в присутствии органической кислоты и органического альдегида при температуре 40-120°С 4-10 ч.; охлаждают до комнатной температуры и добавляют инертный растворитель с осуществлением кристаллизации; S-5-азаспиро[2,4]гептан-6-карбоновую кислоту и разделяющее средство осаждают в виде твердой фазы; выделенную твердую фазу в виде соли растворяют и после перекристаллизации получают S-5-азаспиро[2,4]гептан-6-карбоновую кислоту.
Предпочтительно, указанное разделяющее средство представляет собой L-виннокаменную кислоту или S-камфорсульфоновую кислоту, предпочтительно представляет собой L-виннокаменную кислоту; указанная органическая кислота представляет собой любую из ледяной уксусной кислоты, орто-пропионовой кислоты или этилуксусной кислоты, предпочтительно представляет собой этилуксусную кислоту; указанный органический альдегид представляет собой любой из н-пропионового альдегида, н-масляного альдегида или салицилового альдегида, предпочтительно представляет собой н-масляный альдегид.
Кроме того, реакцию на этапе (1) проводят в реакционном растворе в присутствии щелочи, при этом указанная щелочь представляет собой пиридин или амин; указанный реакционный растворитель представляет собой один из кетонового растворителя, сложноэфирного растворителя, алканового растворителя, галогенированного углеводорода, ароматического углеводорода, нитрила, амида или простого эфира или многокомпонентной смеси; температура реакции составляет -20-50°C.
Кроме того, указанный на этапе (2) галогенид металла представляет собой бромид натрия, бромид калия, йодид натрия или йодид калия.
Кроме того, на этапе (2) реакцию проводят в растворителе при 20-120°С при этом указанный растворитель представляет собой один из кетонового растворителя, сложноэфирного растворителя, алканового растворителя, галогенированного углеводорода, ароматического углеводорода, нитрила, амида или простого эфира или многокомпонентной смеси.
Настоящее изобретение обладает следующими преимуществами:
(1) в качестве исходного вещества согласно настоящему изобретению можно применять циклопропилдиметилкарбинол, который также является исходным веществом для монтелукаста натрия, при этом отечественная химическая промышленность уже осуществляет его масштабное производство, в то же время другие виды исходных веществ, реактивов и т.п.представляют собой традиционные реактивы, при этом такие исходные вещества легко получить и их себестоимость низкая;
(2) способ синтеза рацемата согласно настоящему изобретению характеризуется одноэтапной реакцией и без побочных реакций; выход высокий; очистка путем разделения не включает сложных операций, а также является простой и легкой в осуществлении;
(3) операции в способе синтеза согласно настоящему изобретению не сложные; требования к технологическому оборудованию низкие; отсутствует коррозионная активность; нет необходимости в безводных, анаэробных, жестких условиях получения, предусматривающих гидрогенизацию под высоким давлением и т.п.; его просто применять в масштабном промышленном производстве;
(4) способ синтеза хирального промежуточного соединения ледипасвира согласно настоящему изобретению характеризуется простотой в осуществлении, коротким производственным циклом, сравнительно высокой эффективностью производства и возможностью применения в масштабном химическом промышленном производстве.
Подробное описание изобретение
Ниже настоящее изобретение дополнительно описано с помощью примеров, при этом объем защиты настоящего изобретения приведенным ниже не ограничивается.
Новый способ синтеза хирального промежуточного соединения ледипасвира, при этом указанное хиральное промежуточное соединение ледипасвира представляет собой S-5-азаспиро[2,4]гептан-6-карбоновую кислоту и путь синтеза является следующим:
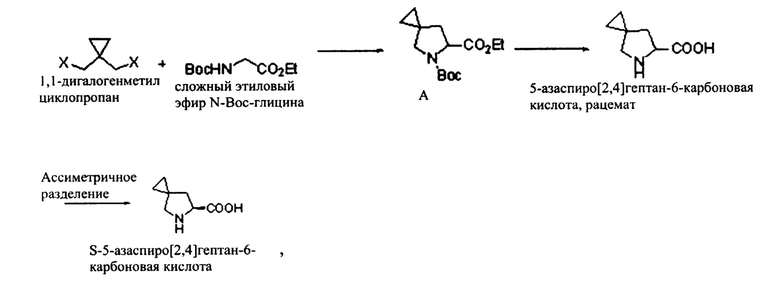
где X представляет собой галоген.
Путь синтеза указанного 1,1-дигалогенметилциклопропана является следующим.
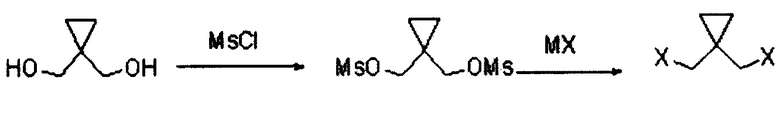
где в MX М представляет собой металл, а X представляет собой галоген; М предпочтительно представляет собой калий или натрий, а X предпочтительно представляет собой бром или йод.
Конкретный способ синтеза является следующим:
(1) посредством реакции циклопропандиметанола с метансульфонилхлоридом получают циклопропилдиметансульфонат;
(2) посредством реакции циклопропилдиметансульфоната с галогенидом металла получают дигалогенциклопропан.
Пример 1. Новый способ синтеза хирального промежуточного соединения ледипасвира, представляющего собой S-5-азаспиро[2,4]гептан-6-карбоновую кислоту.
Конкретный способ синтеза включает следующие этапы:
S1: проводят реакцию замыкания цикла между 1,1-дигалогенметилциклопропаном и сложным этиловым эфиром N-Boc-глицина в реакционном растворителе в присутствии щелочи с получением соединения А; при этом указанный реакционный растворитель представляет собой амидный растворитель; указанная щелочь представляет собой алкоголят калия; температура реакции составляет -10°С;
S2: подвергают омылению соединение А, удаляют защитную группу ВОС и получают 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата; при этом указанное омыление осуществляют в растворителе в присутствии щелочи; указанный растворитель представляет собой спирт; указанная щелочь представляет собой гидроксид натрия; кислота, применяемая в указанном удалении защитной группы ВОС, представляет собой хлористоводородную кислоту; температура реакции составляет 0°С;
S3: подвергают асимметричному разделению
5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата с получением S-5-азаспиро[2,4]гептан-6-карбоновой кислоты, при этом конкретные операции указанного асимметричного разделения являются следующими: после добавления L-виннокаменной кислоты нагревают 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата в присутствии ледяной уксусной кислоты и н-пропионового альдегида при температуре 40°С 4 ч.; охлаждают до комнатной температуры и добавляют инертный растворитель для осуществления кристаллизации; осаждают
S-5-азаспиро[2,4]гептан-6-карбоновую кислоту и L-виннокаменную кислоту в виде твердой фазы; выделенную твердую фазу в виде соли растворяют и после перекристаллизации получают S-5-азаспиро[2,4]гептан-6-карбоновую кислоту.
Пример 2. Новый способ синтеза хирального промежуточного соединения ледипасвира, представляющего собой S-5-азаспиро[2,4]гептан-6-карбоновую кислоту.
Конкретный способ синтеза включает следующие этапы:
S1: проводят реакцию замыкания цикла между 1,1-дигалогенметилциклопропаном и сложным этиловым эфиром N-Boc-глицина в реакционном растворителе в присутствии щелочи с получением соединения А; при этом указанный реакционный растворитель представляет собой эфирный растворитель; указанная щелочь представляет собой алкоголят натрия; температура реакции составляет 100°С;
S2: подвергают омылению соединение А, удаляют защитную группу ВОС и получают 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата; при этом указанное омыление осуществляют в растворителе в присутствии щелочи; указанный растворитель представляет собой простой эфир; указанная щелочь представляет собой гидроксид калия; кислота, применяемая в указанном удалении защитной группы ВОС, представляет собой серную кислоту; температура реакции составляет 100°С;
S3: подвергают асимметричному разделению 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата с получением S-5-азаспиро[2,4]гептан-6-карбоновой кислоты, при этом конкретные операции указанного асимметричного разделения являются следующими: после добавления S-камфорсульфоновой кислоты нагревают 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата в присутствии орто-пропионовой кислоты и н-масляного альдегида при температуре 120°С 10 ч.; охлаждают до комнатной температуры и добавляют инертный растворитель для кристаллизации; осаждают S-5-азаспиро[2,4]гептан-6-карбоновую кислоту и L-виннокаменную кислоту в виде твердой фазы; выделенную твердую фазу в виде соли растворяют и после перекристаллизации получают S-5-азаспиро[2,4]гептан-6-карбоновую кислоту.
Пример 3. Новый способ синтеза хирального промежуточного соединения ледипасвира, представляющего собой S-5-азаспиро[2,4]гептан-6-карбоновую кислоту.
Конкретный способ синтеза включает следующие этапы:
S1: проводят реакцию замыкания цикла между 1,1-дигалогенметилциклопропаном и сложным этиловым эфиром N-Boc-глицина в реакционном растворителе в присутствии щелочи с получением соединения А, при этом указанный реакционный растворитель представляет собой амидный растворитель, указанная щелочь представляет собой гидрид натрия; температура реакции составляет 45°С;
S2: подвергают омылению соединение А, удаляют защитную группу ВОС и получают 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата; при этом указанное омыление осуществляют в растворителе в присутствии щелочи; указанный растворитель представляет собой спирт, указанная щелочь представляет собой оксид натрия, взятый с избытком, кислота, применяемая в указанном удалении защитной группы ВОС, представляет собой фосфорную кислоту, температура реакции составляет 38°C;
S3: подвергают асимметричному разделению 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата с получением S-5-азаспиро[2,4]гептан-6-карбоновой кислоты; конкретные операции указанного асимметричного разделения являются следующими: после добавления L-виннокаменной кислоты нагревают 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата в присутствии этилуксусной кислоты и салицилового альдегида при температуре 65°С 6,5 ч.; охлаждают до комнатной температуры и добавляют инертный растворитель для осуществления кристаллизации; осаждают S-5-азаспиро[2,4]гептан-6-карбоновую кислоту и L-виннокаменную кислоту в виде твердой фазы.
Пример 4. Новый способ синтеза хирального промежуточного соединения ледипасвира, представляющего собой S-5-азаспиро[2,4]гептан-6-карбоновую кислоту.
Конкретный способ синтеза включает следующие этапы:
S1: проводят реакцию замыкания цикла между 1,1-дигалогенметилциклопропаном и сложным этиловым эфиром N-Boc-глицина в реакционном растворителе в присутствии щелочи с получением соединения А; при этом указанный реакционный растворитель представляет собой амидный растворитель или эфирный растворитель; указанная щелочь представляет собой любую из алкоголята калия, алкоголята натрия, гидрида натрия или гидрида калия; температура реакции составляет 86°С;
S2: подвергают омылению соединение А, удаляют защитную группу ВОС и получают 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата; при этом указанное омыление осуществляют в растворителе в присутствии щелочи; указанный растворитель представляет собой простой эфир; указанная щелочь представляет собой гидроксид натрия; кислота, применяемая в указанном удалении защитной группы ВОС, представляет собой азотную кислоту; температура реакции составляет 85°С;
S3: подвергают асимметричному разделению 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата с получением S-5-азаспиро[2,4]гептан-6-карбоновой кислоты, при этом конкретные операции указанного асимметричного разделения являются следующими: после добавления L-виннокаменной кислоты нагревают 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата в присутствии орто-пропионовой кислоты и н-пропионового альдегида при температуре 100°С 9 ч.; охлаждают до комнатной температуры и добавляют инертный растворитель для кристаллизации; осаждают S-5-азаспиро[2,4]гептан-6-карбоновую кислоту и L-виннокаменную кислоту в виде твердой фазы; выделенную твердую фазу в виде соли растворяют и после перекристаллизации получают S-5-азаспиро[2,4]гептан-6-карбоновую кислоту.
Пример 5. Синтез 1,1-дигалогенметилциклопропана, при этом конкретный способ синтеза является следующим:
(1) проводят реакцию циклопропандиметанола с метансульфонилхлоридом в реакционном растворе в присутствии щелочи с получением циклопропилдиметансульфоната; указанная щелочь представляет собой пиридин; указанный реакционный растворитель представляет собой кетоновый растворитель; температура реакции составляет -20°С;
(2) проводят реакцию циклопропилдиметансульфоната с галогенидом металла в растворителе при 20°C с получением дигалогенциклопропана, при этом указанный галогенид металла представляет собой бромид натрия, а указанный растворитель представляет собой кетоновый растворитель.
Пример 6. Синтез 1,1-дигалогенметилциклопропана, при этом конкретный способ синтеза является следующим:
(1) проводят реакцию циклопропандиметанола с метансульфонилхлоридом в реакционном растворе в присутствии щелочи с получением циклопропилдиметансульфоната; указанная щелочь представляет собой амин; указанный реакционный растворитель представляет собой смесь сложноэфирного растворителя и алканового растворителя; температура реакции составляет 50°С;
(2) проводят реакцию циклопропилдиметансульфоната с галогенидом металла в растворителе при 120°С с получением дигалогенциклопропана, при этом указанный галогенид металла представляет собой бромид калия, а указанный растворитель представляет собой смесь сложноэфирного растворителя и алканового растворителя.
Пример 7. Синтез 1,1-дигалогенметилциклопропана, при этом конкретный способ синтеза является следующим:
(1) проводят реакцию циклопропандиметанола с метансульфонилхлоридом в реакционном растворе в присутствии щелочи с получением циклопропилдиметансульфоната; указанная щелочь представляет собой пиридин; указанный реакционный растворитель представляет собой смесь галогенированного углеводорода, ароматического углеводорода и нитрила; температура реакции составляет - 10°С;
(2) проводят реакцию циклопропилдиметансульфоната с галогенидом металла в растворителе при 32°С с получением дигалогенциклопропана, при этом указанный галогенид металла представляет собой йодид натрия, а указанный растворитель представляет собой смесь галогенированного кетона, ароматического углеводорода и нитрила.
Пример 8. Синтез 1,1-дигалогенметилциклопропана, при этом конкретный способ синтеза является следующим:
(1) проводят реакцию циклопропандиметанола с метансульфонилхлоридом в реакционном растворе в присутствии щелочи с получением циклопропилдиметансульфоната; указанная щелочь представляет собой амин; указанный реакционный растворитель представляет собой смесь ароматического углеводорода, нитрила, амида и простого эфира; температура реакции составляет 0°C;
(2) проводят реакцию циклопропилдиметансульфоната с галогенидом металла в растворителе при 45°C с получением дигалогенциклопропана, при этом указанный галогенид металла представляет собой йодид калия, а указанный реакционный растворитель представляет собой смесь ароматического углеводорода, нитрила, амида и простого эфира.
Пример 9. Синтез 1,1-дигалогенметилциклопропана, при этом конкретный способ синтеза является следующим:
(1) проводят реакцию циклопропандиметанола с метансульфонилхлоридом в реакционном растворе в присутствии щелочи с получением циклопропилдиметансульфоната; указанная щелочь представляет собой пиридин или амин; указанный реакционный растворитель представляет собой смесь кетонового растворителя, сложноэфирного растворителя, алканового растворителя, галогенированного углеводорода и ароматического углеводорода; температура реакции составляет 15°С;
(2) проводят реакцию циклопропилдиметансульфоната с галогенидом металла в растворителе при 60°С с получением дигалогенциклопропана, при этом указанный галогенид металла представляет собой бромид натрия, а указанный реакционный растворитель представляет собой смесь кетонового растворителя, сложноэфирного растворителя на основе сложных эфиров, алканового растворителя, галогенированного углеводорода и ароматического углеводорода.
Пример 10. Синтез 1,1-дигалогенметилциклопропана, при этом конкретный способ синтеза является следующим:
(1) проводят реакцию циклопропандиметанола с метансульфонилхлоридом в реакционном растворе в присутствии щелочи с получением циклопропилдиметансульфоната; указанная щелочь представляет собой амин; указанный реакционный растворитель представляет собой смесь кетонового растворителя, сложноэфирного растворителя, алканового растворителя, галогенированного углеводорода, ароматического углеводорода и нитрила; температура реакции составляет 25°С;
(2) проводят реакцию циклопропилдиметансульфоната с галогенидом металла в растворителе при 85°С с получением дигалогенциклопропана, при этом указанный галогенид металла представляет собой бромид калия, а указанный реакционный растворитель представляет собой смесь кетонового растворителя, сложноэфирного растворителя, алканового растворителя, галогенированного углеводорода, ароматического углеводорода и нитрила.
Пример 11. Синтез 1,1-дигалогенметилциклопропана, при этом конкретный способ синтеза является следующим:
(1) проводят реакцию циклопропандиметанола с метансульфонилхлоридом в реакционном растворе в присутствии щелочи с получением циклопропилдиметансульфоната; указанная щелочь представляет собой пиридин; указанный реакционный растворитель представляет собой смесь кетонового растворителя, сложноэфирного растворителя, алканового растворителя, галогенированного углеводорода, ароматического углеводорода, нитрила и амида; температура реакции составляет - 38°С;
(2) проводят реакцию циклопропилдиметансульфоната с галогенидом металла в растворителе при 100°С с получением дигалогенциклопропана, при этом указанный галогенид металла представляет собой йодид натрия, а указанный растворитель представляет собой смесь кетонового растворителя, сложноэфирного растворителя, алканового растворителя, галогенированного углеводорода, ароматического углеводорода, нитрила и амида.
Пример 12. Синтез 1,1-дигалогенметилциклопропана, при этом конкретный способ синтеза является следующим:
(1) проводят реакцию циклопропандиметанола с метансульфонилхлоридом в реакционном растворе в присутствии щелочи с получением циклопропилдиметансульфоната; указанная щелочь представляет собой пиридин; указанный реакционный растворитель представляет собой смесь кетонового растворителя, сложноэфирного растворителя, алканового растворителя, галогенированного углеводорода, ароматического углеводорода, нитрила, амида и простого эфира; температура реакции составляет 47°С;
(2) проводят реакцию циклопропилдиметансульфоната с галогенидом металла в растворителе при 110°С с получением дигалогенциклопропана, при этом указанный галогенид металла представляет собой йодид калия, а указанный растворитель представляет собой смесь кетонового растворителя, сложноэфирного растворителя, алканового растворителя, галогенированного углеводорода, ароматического углеводорода, нитрила, амида и простого эфира.
Положительные эффекты настоящего изобретение объясняются ниже посредством проведенных испытаний.
1. Синтез гидрохлорида сложного этилового эфира глицина
В трехгорлую лабораторную колбу добавляли 4500 мл этанола; при 25°С и ниже по каплям добавляли 1428 г тионилхлорида (12 моль, 871 мл); после добавления по каплям перемешивали полчаса при комнатной температуре; медленно добавляли 751 г глицина (10 моль); температура не превышала 50°С, обеспечивали прохождение реакции затем при 60-65°С в течение 2 ч. Посредством нанесения пятен на пластины для тонкослойной хроматографии (хлороформ:метанол:уксусная кислота = 3:1:3d, трикетогидриндена гидрат) отслеживали протекание реакции с аминокислотой; подвергали центрифугированию; затем применяли этанол еще раз; получали гидрохлорид сложного этилового эфира глицина с весом во влажном состоянии 1415 г, при этом выход составлял 100%, и непосредственно переходили к следующему этапу.
2. Синтез сложного этилового эфира N-BOC-глицина
В одногорлую лабораторную колбу добавляли 5000 мл этанола; перемешивая, добавляли гидрохлорид сложного этилового эфира глицина; с помощью сухого гидрокарбоната натрия, 840 г (10 моль), доводили значение рН до 7-8 (бумага для определения рН не меняет цвет, при смачивании рН 7-8); затем порциями медленно добавляли Вос-ангидрид в количестве 2226 г (10,2 моль) с предотвращением разбавления исходных веществ. Поддерживали внутреннюю температуру не выше 25°С; после добавления ВОС порциями добавляли сухой гидрокарбонат натрия, всего 930 г, и обеспечивали прохождение реакции при 25°С в течение 5 ч. Посредством нанесения пятен образцов на пластины для тонкослойной хроматографии (этил ацетат: петролейный эфир = 1:5, трикетогидриндена гидрат для проявления цвета) отслеживали протекание реакции с исходным веществами, фильтровали, осадок на фильтре промывали этанолом, объединяли фильтрат и подвергали фильтрат центрифугированию. Растворяли в 6000 мл дихлорметана, промывали в 1000 мл воды, осуществляли обратную экстракцию водного слоя с помощью 500 мл дихлорметана, высушивали с помощью центрифугирования, получали продукт в виде светло-желтой маслянистой жидкости в количестве 1992 г, выход 98%.
3. Синтез циклопропилдиметансульфоната
В 50 л реакционную камеру добавляли 1000 г циклопропандиметанола (938 мл, 9,79 моль, 1 эквивалент), 10 л ацетона, 2971 г триэтиламина (4085 мл, 29,37 моль, 3 эквивалента). Понижали температуру, по каплям добавляли 2803 г метилсульфонилхлорида (1893 мл, 24,475 моль, 2,5 эквивалента); температура реакции не превышала 10°С. После добавления по каплям обеспечивали прохождение реакции при 0°С в течение 1 ч., затем повышали температуру до 20°С и обеспечивали прохождение реакции в течение 2 ч.; посредством нанесения пятен на пластины для тонкослойной хроматографии (хлороформ:метанол = 3:1) отслеживали протекание реакции с исходными веществами, добавляли большое количество воды, выделяли продукт, центрифугировали, неочищенный продукт промывали водой. Высушивали при 45°С с получением циклопропилдиметансульфоната в количестве 2282 г, выход 90%.
4. Синтез дийодметилциклопропана
В 50 л реакционную камеру добавляли циклопропилдиметансульфонат в количестве 1290 г (5 моль, 1 эквивалент), 19 л ацетона, 2250 г йодида натрия (15 моль, 3 эквивалента); обеспечивали прохождение реакции при 50°С до полного превращения исходных веществ. Фильтровали, осадок на фильтре промывали ацетоном, концентрировали, полученное маслянистое вещество растворяли в 10 л дихлорметана, промывали водой, промывали 5% тиосульфатом натрия. Высушивали, концентрировали с получением дийодметилциклопропана в количестве 1510 г, выход 93,7%.
5. Синтез спиросоединения А
Гидрид натрия (120 г, 4,3 эквивалента) и DMF (1200 мл) добавляли в колбу и температуру реакции понижали до 0-10°С. Дийодметилциклопропан (383 г, 1 эквивалент) при вышеуказанной температуре добавляли в раствор гидрида натрия в DMF. Посредством нанесения пятен на пластины для тонкослойной хроматографии N-BOC (этилацетат: петролейный эфир = 1:5, трикетогидриндена гидрат для проявления цвета) отслеживают протекание реакции. Поддерживали внутреннюю температуру 4-9°С, медленно по каплям добавляли уксусную кислоту (100 миллилитров, 4,5 эквивалента) для остановки реакции. После добавления по каплям перемешивали при 0-10°С в течение ночи. Обеспечивали внутреннюю температуру 0-10°С и добавляли воду (1400 миллилитров). С помощью 2000 мл петролейного эфира раствор экстрагировали три раза (с помощью нанесения пятен на пластины для тонкослойной хроматографии проверяли, экстрагировали ли продукт). Органический слой сливали, промывали два раза насыщенным солевым раствором (2000 миллилитров), после высушивания концентрировали, получали 320 г светло-желтого маслянистого вещества; выход неочищенного продукта 100%.
6. Синтез 5-азаспиро[2,4]гептан-6-карбоновой кислоты в виде рацемата
К 32 г вышеуказанного светло-желтого маслянистого вещества добавляли 200 мл 20% гидроксида натрия и 15 мл этанола; нагревали при 55°С и перемешивали 2 ч.; после проверки на протекание реакции с исходными веществами посредством нанесения пятен на пластины для тонкослойной хроматографии (этилацетат:петролейный эфир = 1:5, трикетогидриндена гидрат для проявления цвета) останавливали реакцию. Охлаждали до комнатной температуры, с помощью этилацетата собирают побочные продукты, к водной фазе добавляли 60 мл концентрированной хлористоводородной кислоты и 20 мл этанола; при комнатной температуре перемешивали 3 ч. Посредством нанесения пятен на пластины для тонкослойной хроматографии (значение рН довели до 6-7, хлороформ:метанол:уксусная кислота = 3:1/3d, трикетогидриндена гидрат для проявления цвета) отслеживали протекание реакции. После завершения реакции с помощью дихлорметана собирали побочный продукты, с помощью 20% гидроксида натрия доводили значение рН до 6-7, при 50°С концентрировали до сухого состояния, растворяли в теплом этаноле с фильтрацией и деминерализацией (для деминерализации необходим фильтр с диатомовой землей); раствор в этаноле концентрировали до минимального объема, добавляли ацетон в соответствующем количестве и выделяли светло-желтое твердое вещество в количестве 13 г, выход 75%.
7. Синтез S-5-азаспиро[2,4]гептан-6-карбоновой кислоты В 250 мл трехгорлую лабораторную колбу добавляли 6,6 г рацемата (0,0468 моль, 1 эквивалент), 100 мл этилуксусной кислоты, 0,675 г н-масляного альдегида (0,00936 моль, 0,2 эквивалента), L-виннокаменную кислоту в количестве 7,02 г (0,0468 моль, 1 эквивалент); обеспечивали прохождение реакции при 90°С в течение 6 ч.; понижали температуру до комнатной температуры, добавляли соответствующее количество петролейного эфира; и выделяли твердое вещество в большом количестве. Фильтровали, осадок на фильтре промывали соответствующим количеством этанола, затем с помощью смеси этанола и воды осуществляли перекристаллизацию; высушивали, получали соль S-промежуточного соединения и L-TA; соль добавляли к 30 кратному объему раствора метанола, потом добавляли 2 эквивалента триэтиламина; обеспечивали прохождение реакции при 60°С в течение 2 часов и понижали температуру до комнатной температуры. Фильтровали, фильтрат концентрировали до сухого состояния; с помощью растворителя (3:1) в виде смеси этанол-ацетон осуществляли перекристаллизацию и получали S-5-азаспиро[2,4]гептан-6-карбоновую кислоту в количестве 5,3 г; выход 80%, удельное вращение плоскости поляризации -60,0 - -62,0° (4%, H2O).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДСТВО СОЕДИНЕНИЙ И КОМПОЗИЦИЙ ДЛЯ ПОДАВЛЕНИЯ АКТИВНОСТИ SHP2 | 2019 |
|
RU2797951C2 |
| СОЕДИНЕНИЯ АЗЕТИДИНА, КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ РАСТВОРИМОЙ ЭПОКСИДГИДРОЛАЗЫ | 2012 |
|
RU2615995C2 |
| СПОСОБЫ СИНТЕЗА ПРОИЗВОДНЫХ ДИГИДРОПИРИДОФТАЛАЗИНОНА | 2011 |
|
RU2561732C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 8-ХЛОРХИНОЛОНА | 1991 |
|
RU2049778C1 |
| СПИРОСОЕДИНЕНИЕ ИЛИ ЕГО СОЛИ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ, ОБЛАДАЮЩАЯ ПРОТИВОМИКРОБНОЙ АКТИВНОСТЬЮ | 1989 |
|
RU2094432C1 |
| СПОСОБ ПОЛУЧЕНИЯ АМФИФИЛЬНЫХ СОЕДИНЕНИЙ ИМИДАЗОЛИНИЯ | 2016 |
|
RU2753541C2 |
| ПРОИЗВОДНОЕ ПИРИМИДИНА И ПЯТИЧЛЕННОГО АЗОТСОДЕРЖАЩЕГО ГЕТЕРОЦИКЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКИЕ ПРИМЕНЕНИЯ | 2019 |
|
RU2775229C1 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2809257C2 |
| ПРОИЗВОДНЫЕ ХИНОЛОНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1998 |
|
RU2193558C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЗЕТИДИНОНОВЫХ СОЕДИНЕНИЙ И ПРОИЗВОДНЫХ АЗЕТИДИНОНОВЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2650687C1 |
Изобретение относится к способу синтеза S-5-азаспиро[2,4]гептан-6-карбоновой кислоты, которая является хиральным промежуточным соединением ледипасвира. Способ включает следующие этапы: S1: проводят реакцию замыкания цикла между 1,1-дигалогенметилциклопропаном и сложным этиловым эфиром N-Boc-глицина в щелочной среде с получением соединения А; S2: подвергают омылению соединение А, удаляют защитную группу ВОС и получают 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата; S3: подвергают асимметричному разделению 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата с получением S-5-азаспиро[2,4]гептан-6-карбоновой кислоты; при этом конкретные операции указанного асимметричного разделения на этапе S3 являются следующими: после добавления разделяющего средства нагревают 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата в присутствии органической кислоты и органического альдегида при температуре 40-120°С в течение 4-10 ч; охлаждают до комнатной температуры и добавляют инертный растворитель с осуществлением кристаллизации; осаждают S-5-азаспиро[2,4]гептан-6-карбоновую кислоту и разделяющее средство в виде твердой фазы; выделенную твердую фазу в виде соли растворяют и после перекристаллизации получают S-5-азаспиро[2,4]гептан-6-карбоновую кислоту; при этом указанное разделяющее средство представляет собой L-виннокаменную кислоту; при этом указанная органическая кислота представляет собой любую из ледяной уксусной кислоты, орто-пропионовой кислоты или этилуксусной кислоты; указанный органический альдегид представляет собой любой из н-пропионового альдегида, н-масляного альдегида или салицилового альдегида. Способ характеризуется повышением эффективности, простотой синтеза, снижением себестоимости продукта, удобством получения и применим для широкомасштабного промышленного производства. 6 з.п. ф-лы, 12 пр.
1. Способ получения хирального промежуточного соединения ледипасвира, причем указанное хиральное промежуточное соединение ледипасвира представляет собой S-5-азаспиро[2,4]гептан-6-карбоновую кислоту, отличающийся тем, что путь синтеза является следующим:
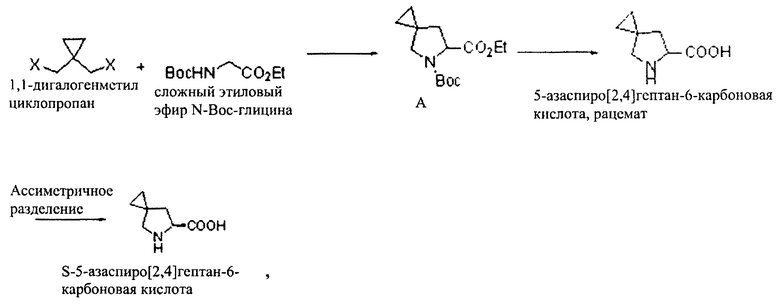
где X представляет собой галоген;
при этом способ синтеза включает следующие этапы, где:
S1: проводят реакцию замыкания цикла между
1,1-дигалогенметилциклопропаном и сложным этиловым эфиром N-Boc-глицина в щелочной среде с получением соединения А;
S2: подвергают омылению соединение А, удаляют защитную группу ВОС и получают 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата;
S3: подвергают асимметричному разделению
5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата с получением S-5-азаспиро[2,4]гептан-6-карбоновой кислоты;
при этом конкретные операции указанного асимметричного разделения на этапе S3 являются следующими: после добавления разделяющего средства нагревают 5-азаспиро[2,4]гептан-6-карбоновую кислоту в виде рацемата в присутствии органической кислоты и органического альдегида при температуре 40-120°С в течение 4-10 ч; охлаждают до комнатной температуры и добавляют инертный растворитель с осуществлением кристаллизации; осаждают S-5-азаспиро[2,4]гептан-6-карбоновую кислоту и разделяющее средство в виде твердой фазы; выделенную твердую фазу в виде соли растворяют и после перекристаллизации получают S-5-азаспиро[2,4]гептан-6-карбоновую кислоту;
при этом указанное разделяющее средство представляет собой L-виннокаменную кислоту; при этом указанная органическая кислота представляет собой любую из ледяной уксусной кислоты, орто-пропионовой кислоты или этилуксусной кислоты; указанный органический альдегид представляет собой любой из н-пропионового альдегида, н-масляного альдегида или салицилового альдегида.
2. Способ по п. 1, отличающийся тем, что путь синтеза указанного 1,1-дигалогенметилциклопропана является следующим:
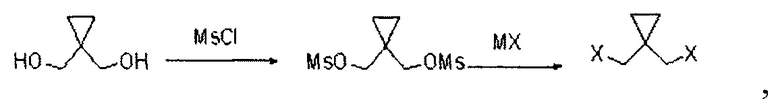
где в MX М представляет собой металл, а X представляет собой галоген;
при этом способ синтеза является следующим:
(1) проводят реакцию циклопропандиметанола с метансульфонилхлоридом с получением циклопропилдиметансульфоната;
(2) проводят реакцию циклопропилдиметансульфоната с галогенидом металла с получением дигалогенциклопропана.
3. Способ по п. 1, отличающийся тем, что этап S1 проводят в реакционном растворителе, представляющем собой амидный или эфирный растворитель, в присутствии любого из алкоголята калия, алкоголята натрия, гидрида натрия или гидрида калия, температура реакции составляет -10-100°С.
4. Способ по п. 1, отличающийся тем, что указанное омыление на этапе S2 проводят в реакционном растворителе в присутствии щелочи, при этом указанный растворитель представляет собой по меньше мере один из спиртов или простых эфиров, указанная щелочь представляет собой неорганическую сильную щелочь, кислота, применяемая в указанном удалении защитной группы ВОС, представляет собой любую из хлористоводородной кислоты, серной кислоты, фосфорной кислоты или азотной кислоты, температура реакции составляет 0-100°С.
5. Способ по п. 2, отличающийся тем, что на этапе (1) реакцию проводят в реакционном растворе в присутствии пиридина или амина; указанный реакционный растворитель представляет собой один из кетонового растворителя, сложноэфирного растворителя, алканового растворителя, галогенированного углеводорода, ароматического углеводорода, нитрила, амида или простого эфира или многокомпонентной смеси; температура реакции составляет -20-50°С.
6. Способ по п. 2, отличающийся тем, что указанный галогенид металла на этапе (2) представляет собой бромид натрия, бромид калия, йодид натрия или йодид калия.
7. Способ по п. 2, отличающийся тем, что реакцию на этапе (2) проводят в растворителе при 20-120°С, при этом указанный растворитель представляет собой один из кетонового растворителя, сложноэфирного растворителя, алканового растворителя, галогенированного углеводорода, ароматического углеводорода, нитрила, амида или простого эфира или многокомпонентной смеси.
| WO 2012158861 A2, 22.11.2012 | |||
| WO 2009102325 A1, 20.08.2009 | |||
| RU 2014150435 A, 27.07.2016 | |||
| WO 2009091561 A1, 23.07.2009 | |||
| CN 103288699 A, 11.09.2013 | |||
| CN 104478877 A, 01.04.2015 | |||
| WO 2013059281 A2, 25.04.2013. |

