Область техники
Изобретение относится к области органической химии, а именно к способу получения галогенпроизводных ароматических и гетероароматических соединений, где в качестве объектов галогенирования (хлорирования, бромирования, иодирования) могут выступать алкилбензолы, алкоксибензолы, арилоксибензолы, нафталин, замещенные изоксазолы, пиразолы, тиофены.
Галогенирование ароматических и гетероароматических соединений является одним из ключевых процессов органического синтеза. Полученные в результате реализации данного изобретения галогенпроизводные могут применяться в качестве строительных блоков с целью дальнейшей функционализации ароматических или гетероароматических колец и создания более сложных структур с использованием таких основополагающих реакций как нуклеофильное замещение и металлирование, кросс-сочетание (реакции Сузуки, Соногаширы, Стилле, Негиши), реакция Гриньяра.
Уровень техники
Известен способ получения хлор-/бромпроизводных ароматических соединений путем хлорирования/бромирования ароматических углеводородов хлором/бромом в присутствии кислот Льюиса [Бюлер К., Пирсон Д. Органические синтезы, ч. 1, «Мир», 1973, с. 446]. К недостаткам данного способа можно отнести необходимость работы с опасными отравляющими веществами (Cl2, Br2), высокую гигроскопичность кислот Льюиса. Кроме того, применение кислот Льюиса может вызывать побочные процессы: изомеризацию, дезалкилирование, диспропорционирование исходного ароматического соединения, которые усложняют процесс галогенирования, а также сильное осмоление донорных гетероциклов.
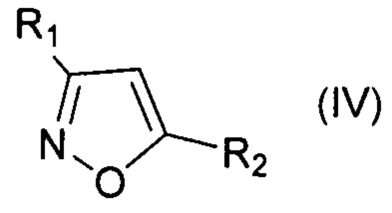
Известен способ синтеза 3,5-диарил-4-галогенизоксазолов галоидированием 3,5-диарилизоксазолов N-галогенсукцинимидами. [Day R.A., Blake J.A., Stepens С.Е., Synthesis, 2003, 10, 1586-1590]. К недостаткам этого способа следует отнести жесткие условия проведения реакции - кипячение в уксусной/трифторуксусной кислоте; необходимость очистки продукта от сукцинимида, образующегося из реагента в ходе реакции; дороговизну N-галогенсункцинимидов; невысокие выходы 4-иодизоксазолов вследствие частичного протодеиодирования.
Известен способ синтеза 4-бром-/4-иодарилов галогенированием ароматических и гетероароматических соединений бромом/иодом при 40-129°С в присутствии азотной кислоты или смеси азотной и серной кислот (в качестве окислителя) в среде ледяной уксусной кислоты и четыреххлористого углерода [В.Т. Слюсарчук, А.И. Новиков. Прямое иодирование фенантренхинона, флуорена и его производных. Журн. Органич. химиии, 1967, 3; 1323; Н.К. Кочетков, С.Д. Соколов, Н.М. Вагуртова. Журн. Общей химиии, 1961, 31; 2326]. К недостаткам данного метода следует отнести жесткие условия проведения реакции, недостаточно высокие выходы целевых продуктов (24-78%).
Наиболее близкими к предлагаемому изобретению по технической сущности и достигаемому результату являются галогенирующие системы Hal2/PhI(OAcf)2, в которых ароматические углеводороды или их простые эфиры галогенируют галоидом (Br2, I2), в присутствии окислителя - фенилиодозотрифторацетата [Меркушев Е.Б., Симахина Н.Д. Способ получения иодароматических углеводородов или их простых эфиров. Авт. свидет. №639846. Бюллет. изобрет. 1978, 8. Меркушев Е.Б., Большакова Т.М. Способ получения бромароматических углеводородов или их простых эфиров. Авт. свидет. №827471. Бюллет. изобрет. 1981, 17.]. Сущность способа - электрофильное галогенирование ароматических соединений. К ограничению данного способа можно отнести невозможность осуществления хлорирования ароматических соединений, относительную дороговизну фенилиодозотрифторацетата, необходимость очистки продукта от образующегося в ходе реакции иодбензола, а также токсичной трифторуксусной кислоты.
Раскрытие изобретения
Технической проблемой, на решение которой направлено заявляемое изобретение, является разработка простого и безопасного в исполнении способа галогенирования ароматических и гетероароматических соединений с высокими выходами целевого продукта.
Техническим результатом заявляемого способа является разработка простого и безопасного в исполнении способа галогенирования ароматических и гетероароматических соединений, который отличается мягкими условиями галогенирования, высокой селективностью, а также универсальностью, позволяя проводить хлорирование, бромирование, а также иодирование ароматических и гетероароматических колец, получением галогенпроизводных ароматических соединений, в том числе конденсированных ароматических соединений, а также соединений ряда бензола, изоксазола, пиразола, тиофена с выходами от 50 до 99% в зависимости от заместителей в ароматическом/гетероароматическом соединении.
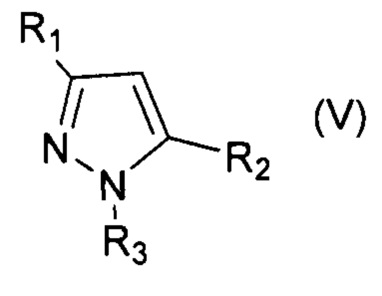
Технический результат достигается заявляемым способом получения галогенпроизводных ароматических и гетероароматических соединений, заключающимся в том, что исходные соединения, выбранные из группы, включающей алкилбензолы, алкоксибензолы, арилоксибензолы, нафталин, замещенные изоксазолы, пиразолы, тиофены галогенируют активной галогенирующей частицей образующейся in situ, посредством смешения галогенидов алкиламмония общей формулы R4N+Hal-, где R=Н, Alk, Hal=Cl, Br, I, с соединением, содержащим нитрозоний катион - NOHSO4 или NOBF4 или NOSO3Cl, в среде органического растворителя, по окончании реакции галогенпроизводные ароматические или гетероароматические соединения выделяют и очищают от примесей (непрореагировавшее исходное соединение, непрореагировавший избыток тетраметиламмоний галогенида) перекристаллизацией соединения или очищают при помощи колоночной хроматографии.
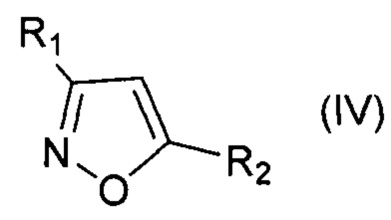
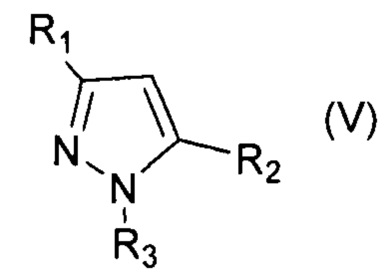
При этом для получения соединения формулы (VII) в качестве исходного соединения используют соединение формулы (IV), для соединения формулы (VIII) - соединение формулы (V), для соединения формулы (IX) - соединение формулы (VI), для соединения формулы (X) - соединение формулы (I), для соединения формулы (XI) - соединение формулы (II), для соединения формулы (XII) - соединение формулы (III):
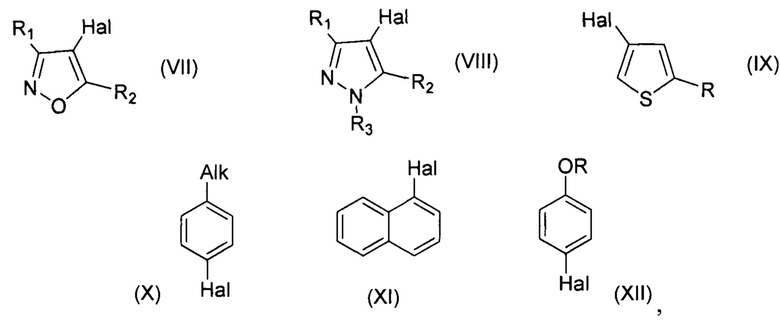
исходные соединения:
 , где R=Ar, Alk
, где R=Ar, Alk
 , где R1=Ar, R2=Ar, Alk
, где R1=Ar, R2=Ar, Alk
 , где R1=Ar, Alk; R2=Ar, Alk; R3=H, Ar;
, где R1=Ar, Alk; R2=Ar, Alk; R3=H, Ar;
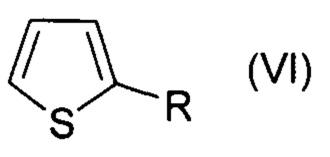 , где R=СОН,
, где R=СОН,
Для проведения реакции используют 1.5-3-хкратный мольный избыток нитрозилсерной кислоты и галогенида тетраалкиламмония по отношению к исходному ароматическому или гетероароматическому соединению, при этом реакцию проводят при температуре 0-60°С в течение времени, необходимого для максимального выхода галогенпроизводного ароматического или гетероароматического соединения. Время максимального выхода галогенпроизводного ароматического или гетероароматического соединения контролируют методом ТСХ. В качестве соединения, содержащего нитрозоний катион используют нитрозилсерную кислоту, тетрафторборат нитрозония, хлорсульфат нитрозония, а в качестве органических растворителей используют нитрометан, бензол, четыреххлористый углерод, хлороформ, ацетонитрил.
Осуществление изобретения
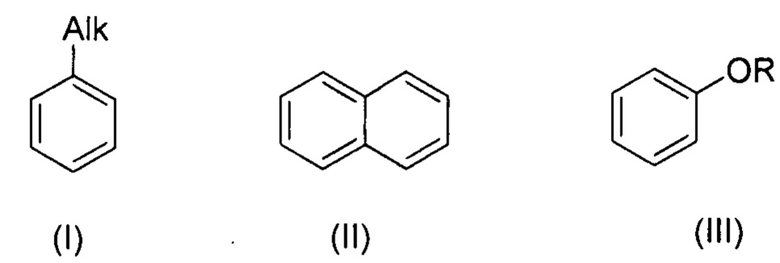
В настоящем изобретении заявляется способ галогенирования ароматических и гетероароматических соединений формул (I-VI):
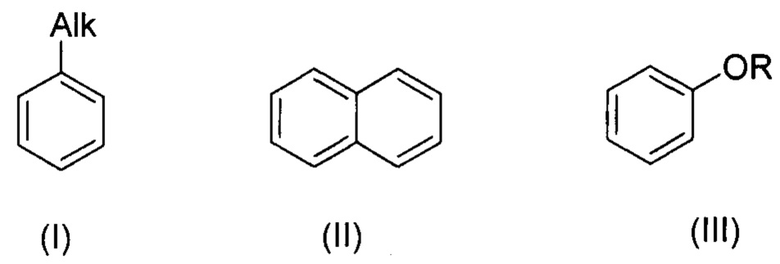 , где R=Ar, Alk
, где R=Ar, Alk
 , где R1=Ar, R2=Ar, Alk
, где R1=Ar, R2=Ar, Alk
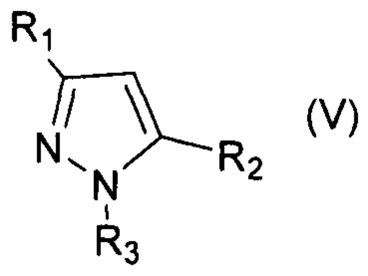 , где R1=Ar, Alk; R2=Ar, Alk; R3=H, Ar;
, где R1=Ar, Alk; R2=Ar, Alk; R3=H, Ar;
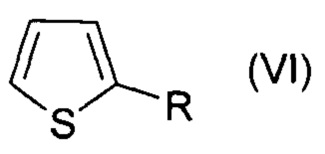 , где R=СОН,
, где R=СОН,
в котором с целью упрощения технологии процесса и его удешевления в качестве источника галогена используются удобные в работе соли алкиламмония.
Полученные соединения строения (VII-IX) могут быть использованы в медицинской химии для синтеза веществ с потенциальной биологической активностью.

В литературе описаны способы получения галоидароматических соединений путем галогенирования соответствующих ароматических производных галоидами (бромом или иодом), в присутствии окислителя - азотной кислоты, или смеси азотной и серной кислот, или фенилиодозотрифторацетата. Однако способы получения галогенароматических соединений, в которых в качестве источника галогена используют галогениды алкиламмония, а в качестве окислителя - соединения, содержащие катион нитрозония, в литературных источниках не описаны. Оптимальными условиями для проведения реакции, обеспечивающими выходы галогенпроизводных конденсированных ароматических соединений, а также соединений ряда бензола, изоксазола, пиразола, тиофена с выходами от 50 до 99% являются:
1. Добавление тетраметиламмоний галогенида, а затем нитрозилсерной кислоты к перемешиваемому раствору ароматического и гетероароматического соединения в подходящем органическом растворителе.
2. Использование в качестве источника галогена хлоридов, бромидов или иодидов алкиламмония: среди них тетраметиламмоний галогениды, диметиламмоний галогениды, н-бутиламмоний галогениды, триэтилбензиламмоний хлорид (ТЭБАХ), триэтилбензиламмоний бромид (ТЭБАБ). При этом предпочтительнее использовать тетраметиламмоний галогениды в силу легкости отделения от продукта реакции и меньшей стоимости.
3. Использование для окисления галогенидов алкиламмония соединений, содержащих нитрозоний катион (NOHSO4, NOBF4, NOSO3Cl). При этом предпочтительнее использовать нитрозилсерную кислоту в силу ее большей доступности (легкости получения как в лабораторных условиях, так и в промышленных масштабах), несколько меньшей гигроскопичности по сравнению с другими перечисленными реагентами, а также значительной дешевизны.
4. Использование трехкратного мольного избытка галогенида тетраметиламмония, а также трехкратного мольного избытка нитрозилсерной кислоты по отношению к исходному ароматическому и гетероароматическому соединению. Использование меньшего количества реагентов приводит к неполной конверсии галогенируемого соединения, либо требует значительного увеличения времени реакции. Исключение составляют соединения, содержащие донорные заместители в ароматических кольцах, для которых количество реагентов может быть снижено до 1.5 эквивалентов.
5. Проведение реакции в течение времени, необходимого для максимального выхода галогенпроизводного ароматического или гетероароматического соединения, как правило реакция проходит в течение часа: за это время реакции достигаются максимальные выходы галогенпроизводных ароматических и гетероароматических соединений (контроль методом ТСХ по исчезновению пятна исходного соединения). При меньшем времени реакции происходит неполная конверсия исходных соединений, что особенно актуально для соединений с дезактивирующими заместителями.
6. Проведение реакции в температурном интервале от 0 до 60°С. Галогенирование ароматических и гетероароматических соединений донорного характера происходит энергично уже при комнатной температуре. В этом случае начинать реакцию следует при 0°С, медленно отогревая реакционную смесь после смешения всех компонентов до комнатной температуры. Галогенирование гетероциклов с акцепторными группами в ароматических заместителях при температуре 60±5°С значительно сокращает время реакции.
7. Использование в качестве растворителя любого, обеспечивающего максимальную гомогенность реакции и способствующего протеканию галогенирования, среди них: бензол, четыреххлористый углерод, хлороформ, ацетонитрил или нитрометан. Последний предпочтительнее, т.к. обеспечивает лучшие выходы и большую чистоту продукта.
По окончании реакции смесь встряхивают с 10 масс. % водным раствором сульфита натрия, взятого в объеме, равном объему реакционной смеси, до полного обесцвечивания, а затем обрабатывают любым подходящим растворителем для извлечения галогенпроизводных ароматических и гетероароматических соединений. Растворитель отгоняют, продукт перекристаллизовывают для удаления органических и неорганических примесей или очищают при помощи колоночной хроматографии для жидких продуктов [Органикум. т. 1, М.: Мир, 1992, 487 с.].
Некоторые синтезированные соединения являются новыми. Строение их подтверждено результатами ЯМР спектроскопии, масс-спектрометрии и данными элементного анализа.
Настоящее изобретение поясняется конкретными примерами выполнения, которые не являются единственно возможным, но наглядно демонстрирует возможность достижения требуемого технического результата.
Пример 1. Получение 4-бром-3,5-дифенилизоксазола (общая методика)
В колбу, снабженную магнитным мешальником, помещали 150 мг (0.67 ммоль) 3,5-дифенилизоксазола в 6.7 мл нитрометана. К раствору при перемешивании последовательно добавляли 310 мг (2.01 ммоль) Me4NBr (или Me4NCl, или Me4NI для некоторых примеров) и 255 мг (2.01 ммоль NOHSO4). Полученную смесь перемешивали 1 ч. По окончании реакции (дополнительный контроль ТСХ) реакционную смесь встряхивали с 10 мл 10% раствора Na2SO3 до полного обесцвечивания. Органический слой отделяли, водный - обрабатывали хлороформом (3×15 мл). Органические фракции объединяли и сушили над сульфатом натрия. Растворитель отгоняли, остаток перекристаллизовывали из этанола (или очищали хроматографированием на силикагеле в системе этилацетат - петролейный эфир 1:10 для некоторых примеров).
Получили 196 мг (98%) 4-бром-3,5-дифенилизоксазола в виде желтоватых кристаллов с Тпл 125°С [Kaewsri, W., Thongsornkleeb, С., Tummatorn, J., Ruchirawat, S., RSC Advances, 2016, 6(54), 48666-48675]
1H ЯМР (400.13 МГц, CDCl3) δ 7.49-7.56 (м, 6H), 7.88-7.92 (м, 2H), 8.08-8.13 (м, 2Н).
13С ЯМР (101 МГц, CDCl3) δ 89.6 (Сизокс-Br), 126.8 (С(1)аром), 127.1 (2СНаром), 127.9 (С(1)аром), 128.6 (2СНаром), 128.7 (2СНаром), 128.9 (2СНаром), 130.2 (СНаром), 130.7 (СНаром), 162.1 (C=N), 165.8 (С-О).
Пример 2. Получение 4-йод-3,5-дифенилизоксазола.
Из 150 мг (0.67 ммоль) 3,5-дифенилизоксазола в условиях, описанных в примере 1, было получено 202 мг (87%) 4-йод-3,5-дифенилизоксазола в виде оранжевых кристаллов с Тпл 160°С [Kaewsri, W., Thongsornkleeb, С., Tummatorn, J., Ruchirawat, S., RSC Advances, 2016, 6(54), 48666-48675]
1H ЯМР (400 МГц, CDCl3) δ 7.50-7.57 (м, 6H), 7.80-7.84 (м, 2H), 8.06-8.12 (м, 2Н).
13С ЯМР (101 МГц, CDCl3) δ 56.2 (Сизокс-I), 127.3 (С(1)аром), 127.8 (2СНаром), 128.6 (2СНаром), 128.7 (С(1)аром), 128.8 (2СНаром), 129.1 (2СНаром), 130.1 (СНаром), 130.8 (СНаром), 164.7 (C=N), 169.0 (С-О).
Пример 3. Получение 4-бром-3,5-бис(4-бромфенил)изоксазола.
Из 100 мг (0.26 ммоль) 3,5-бис(4-бромфенил)изоксазола в условиях, описанных в примере 1, было получено 118 мг (98%) 4-бром-3,5-бис(4-бромфенил)изоксазола в виде светло-желтых кристаллов с Тпл 180°С
1Н ЯМР (400 МГц, CDCl3) δ 7.65-7.72 (м, 4Н), 7.73-7.78 (м, 2Н), 7.97 (д.д, J=8.6, 1.8, 2Н).
13С ЯМР (101 МГц, CDCl3) δ 89.6 (СизоксBr), 124.9, 125.4, 126.5, 128.4 (2СНаром), 130.0 (2СНаром), 132.0 (2СНаром), 132.2 (2СНаром), 161.3 (C=N), 165.1 (С-О). 1 сигнал накладывается на другие.
Вычислено для C15H8Br3NO, %: С 39.34; Н 1.76; N 3.06; Найдено, %: С 39.46; Н 1.84; N 3.15.
Пример 4. Получение 4-бром-3-(2-метилфенил)-5-фенилизоксазола.
Из 100 мг (0.42 ммоль) 3-(2-метилфенил)-5-фенилизоксазола в условиях, описанных в примере 1, было получено 131 мг (98%) 4-бром-3-(2-метилфенил)-5-фенилизоксазола в виде желтоватой прозрачной жидкости.
1Н ЯМР (400 МГц, CDCl3) δ 2.39 (с, 3Н, СН3), 7.32-7.46 (м, 4Н), 7.54-7.58 (м, 3Н), 8.14-8.17 (м, 2Н).
13С ЯМР (101 МГц, CDCl3) δ 20.0 (СН3), 91.3 (СизоксBr), 125.8 (СНаром), 126.8 (С(1)аром), 126.9 (2СНаром), 127.2 (С(1)аром), 128.9 (2СНаром), 130.0 (СНаром), 130.1 (СНаром), 130.5 (СНаром), 130.7 (СНаром), 137.6 (С-СН3), 163.9 (C=N), 164.9 (С-О).
Вычислено для C16H12BrNO, %: С, 61.17; Н, 3.85; N, 4.46; Найдено, %: С, 61.52; Н, 4.09; N, 4.56.
Пример 5. Получение 4-бром-3-(2-метокси-5-хлорфенил)-5-фенилизоксазола.
Из 100 мг 3-(2-метокси-5-хлорфенил)-5-фенилизоксазола (0.35 ммоль) в условиях, описанных в примере 1, было получено 126 мг (74%) 4-бром-3-(2-метокси-5-хлорфенил)-5-фенилизоксазола в виде желтоватой прозрачной жидкости.
1Н ЯМР (400 МГц, CDCl3) δ 3.88 (с, 3Н, СН3), 6.99 (д, 3J=8.8, 1H), 7.44-7.49 (м, 2Н), 7.54-7.55 (м, 3Н), 8.11-8.14 (м, 2Н).
13С ЯМР (101 МГц, CDCl3) δ 55.6 (ОСН3), 91.3 (СизоксBr), 112.1 (СНаром), 117.9 (С(1)аром), 125.1 (С-Cl), 126.3 (С(1)аром), 126.5 (2СНаром), 128.4 (2СНаром), 130.2 (СНаром), 130.6 (СНаром), 131.0 (СНаром), 155.8 (С-ОМе), 160.8 (C=N), 164.6 (СО).
Вычислено для C16H11NO2BrCl, %: С, 52.70; Н, 3.04; N, 3.84. Найдено, %: С, 52.48; Н, 3.35; N, 3.93.
Пример 6. Получение 3-(4-бромфенил)-4-йод-5-(4-метоксифенил)изоксазола.
Из 100 мг (0.30 ммоль) 3-(4-бромфенил)-5-(4-метоксифенил)изоксазола в условиях, описанных в примере 1, было получено 101 мг (74%) 3-(4-бромфенил)-4-йод-5-(4-метоксифенил)изоксазола в виде светло-желтых кристаллов с Тпл 142°С
1Н ЯМР (400 МГц, CDCl3) δ 3.88 (с, 3Н, ОСН3), 7.03 (д, 3J=8.6, 2Н, 2СНаром), 7.65 (уш. с, 4Н, 4СНаром), 8.02 (д, 3J=8.6, 2Н, 2СНаром).
13С ЯМР (101 МГц, CDCl3) δ 54.2 (Сизокс-I), 55.5 (ОСН3), 114.2 (2СНаром), 119.5 (Саром), 124.6 (Саром), 127.8 (Саром), 129.4 (2СНаром), 130.5 (2СНаром), 131.8 (2СНаром), 161.5, 163.7, 169.2 (С-О).
ГХМС (EI, 70 эВ): кластер 455 (16), 457 (14) [М+]; 290 (4); 249 (8) [М-Br-I]+; 135 (100) [CH3O-С6Н4-СО]+; 107 (10) [CH3O-С6Н4]+; 77 (12) [Ph]+.
Вычислено для C16H11BrINO2, %: С, 42.14; Н, 2.43; N, 3.07; Найдено, %: С, 42.08; Н, 2.37; N, 3.05.
Пример 7. Получение 5-(4-бромфенил)-3-фенил-4-хлоризоксазола.
Из 150 мг (0.50 ммоль) 5-(4-бромфенил)-3-фенилизоксазола в условиях, описанных в примере 1, было получено 113 мг (68%) 5-(4-бромфенил)-3-фенил-4-хлоризоксазола в виде бесцветных кристаллов с Тпл 110-111°С [Day, R.A., Blake, J.A., Stephens, С.Е., Synthesis, 2003, 10, 1586-1590]
1Н ЯМР (400 МГц, CDCl3) δ 7.49-7.55 (м, 3Н), 7.65 d (3J=8.6, 2Н), 7.88 (м, 2Н), 7.92 d (3J=8.6, 2Н)
Пример 8. Получение 3-(4-хлорфенил)-4-йод-5-фенилизоксазола.
Из 200 мг (0.78 ммоль) 3-(4-хлорфенил)-5-фенилизоксазола в условиях, описанных в примере 1, было получено 293 мг (98%) 3-(4-хлорфенил)-4-йод-5-фенилизоксазола в виде бесцветных кристаллов с Тпл 165°С [Waldo, J.P., Larock, R.С., J. Org. Chem., 2007, 72(25), 9643-9647.]
1H ЯМР (400 МГц, CDCl3) δ 7.51 (м, 5H), 7.76 (д, 3J=8.1, 2H), 8.09 (м, 2H).
13C ЯМР (101 МГц, CDCl3) δ 55.8 (Сизокс-I), 127.1 (С(1)аром), 127.2 (С(1)аром), 127.8 (2СНаром), 128.8 (2СНаром), 128.9 (2СНаром), 130.3 (2СНаром), 130.9 (СНаром), 136.4 (Саром-Cl), 163.8 (C=N), 169.3 (С-О).
ГХМС (EI, 70 эВ): кластер 381 (4), 383 (1) [М+]; 105 (100) [PhCO]+; 77 (54) [Ph]+.
Пример 9. Получение 4-бром-3-(3-нитрофенил)-5-фенилизоксазола.
Из 100 мг (0.37 ммоль) 3-(3-нитрофенил)-5-фенилизоксазола в условиях, описанных в примере 1, было получено 71 мг (54%) 4-бром-3-(3-нитрофенил)-5-фенилизоксазола в виде бесцветных кристаллов с Тпл 182°С
1Н ЯМР (400 МГц, CDCl3) δ 7.53-7.61 (м, 3Н, CHPh), 7.73 (т, 3J=8.0, 1H, СНаром), 8.09 (д.д, J=6.7, 2.9, 2Н, CHPh), 8.24 (д, 3J=7.7, 1H, СНаром), 8.40 (д, 3J=8.3, 1H, СНаром), 8.81 (уш.с, 1Н, СНаром)
13С ЯМР (101 МГц, CDCl3) δ 89.0 (СизоксBr), 123.6 (СНаром), 124.9 (СНаром), 126.3 (Саром), 127.1 (2СНаром), 129.0 (2СНаром), 129.6 (Саром), 129.9 (СНаром), 131.1 (СНаром), 134.4 (СНаром), 148.4 (C-NO2), 160.2 (C=N), 166.7 (С=O).
ГХМС (EI, 70 эВ): m/z (Iотн (%)): 344 (5), 346 (5) [М+], 105 (100) [Ph-CO]+, 77 (57), 51 (17), 50 (13).
HRMS Вычислено для C15H10BrN2O3 [М+Н]+ 344.9869, 346.9849; Найдено: 344.9857, 346.9863.
Пример 10. Получение 1-йод-4-метоксибензола.
Из 108 мг анизола (1 ммоль) в условиях, описанных в примере 1, было получено 71 мг (54%) 1-йод-4-метоксибензола в виде бесцветных кристаллов с Тпл 50°С [Haszeldine, R.N., Sharpe, A.G., J. Chem. Soc., 1952, 777, 993-1001]
1H ЯМР (400 MHz, CDCl3) δ 3.77 (с, 3Н, ОСН3), 6.69 (д, 3J=8.7, 2Н, 2СНаром), 7.56 (д, 3J=8.7, 2Н, 2СНаром).
Пример 11. Получение 4-йод-3-метил-5-фенилизоксазола.
Из 43 мг (0.27 ммоль) 3-метил-5-фенилизоксазола в условиях, описанных в примере 1, было получено 70 мг (92%) 4-йод-3-метил-5-фенилизоксазола в виде желтоватого масла [Kaewsri, W., Thongsornkleeb, С., Tummatorn, J., Ruchirawat, S., RSC Advances, 2016, 6(54), 48666-48675]
1H ЯМР (400 МГц, CDCl3) δ 2.35 (с, 3H, CH3), 7.48 (м, 3Н, Ph), 8.03 (м, 2H, Ph).
13C ЯМР (101 МГц, CDCl3) δ 12.7 (СН3), 57.9 (CизоксI), 127.2 (C(1)Ph), 127.3 (2CHPh), 128.7 (2CHPh), 130.6 (CHPh), 163.0 (C=N), 167.4 (C-O).
Пример 12. Получение 2-бром-1,3,5-триметилбензола.
Из 146 мг (1.22 ммоль) мезитилена в условиях, описанных в примере 1, было получено 189 мг (78%) 2-бром-1,3,5-триметилбензола в виде бесцветной жидкости [Organic Syntheses, Coll. Vol. 2, p. 95 (1943); Vol. 11, p. 24 (1931)]
1H ЯМР (400 МГц, CDCl3) δ 2.26 (с, 3H, CH3), 2.39 (с, 6H, 2CH3), 6.91 (с, 2H, 2CHаром);
13C ЯМР (101 МГц, CDCl3) δ 20.7 (CH3), 23.7 (2CH3), 124.2 (CBr), 129.1 (2CHаром), 136.3 (C-CH3), 137.9 (С-СН3).
Пример 13. Получение 4-бром-3,5-диметил-1Н-пиразола.
Из 100 мг 3,5-диметил-1Н-пиразола (1.04 ммоль) в условиях, описанных в примере 1, было получено 158 мг 4-бром-3,5-диметил-1Н-пиразола в виде бесцветных кристаллов с Тпл 123°С [Zhao, Z.G., Wang, Z.X., Synthetic communications, 2007, 37(1), 137-147]
1Н ЯМР (400 МГц, CDCl3) δ 2.25 (с, 2СН3);
13С ЯМР (101 МГц, CDCl3) δ 11.2 (2СН3), 94.2 (С-Br), 142.7 (С-СН3).
Пример 14. Получение 1-бензил-3,5-диметил-4-хлор-1Н-пиразола.
Из 100 мг (0.34 ммоль) 1-бензил-3,5-диметил-4-хлор-1Н-пиразола в условиях, описанных в примере 1, было получено 79 мг (70%) 1-бензил-3,5-диметил-4-хлор-1Н-пиразола в виде бесцветных кристаллов с Тпл 104°С
1Н ЯМР (400 МГц, CDCl3) δ 5.33 (с, 2Н, СН2), 7.09 (д, 3J=7.3, 2Н, 2СНаром), 7.24-7.32 (м, 3Н, 3СНаром), 7.34-7.39 (м, 2Н, 2СНаром), 7.41 (д, 3J=7.3, 1Н, СНаром), 7.44-7.53 (м, 5Н, Ph), 8.02 (д, 3J=7.3, 2Н, 2СНаром);
13С ЯМР (101 МГц, CDCl3) δ 54.2 (СН2), 107.2 (CCl), 127.0 (2СНаром), 127.5 (2СНаром), 127.7 (СНаром), 128.2 (СНаром), 128.5 (2СНаром), 128.6 (2СНаром), 128.8 (2СНаром), 129.4 (СНаром), 129.9 (2СНаром), 130.5 (С(1)аром), 131.9 (С(1)аром), 136.9 (С(1)аром), 141.6 (C-N), 146.9 (C=N)
ГХМС (EI, 70 эВ): кластер 344 (70), 346 (23) [М+]; кластер 343 (56), 345 (19) [М-Н]+; кластер 267 (40), 269 (13) [M-Ph]+; 189 (22), 91 (100).
Вычислено для C22H17ClN2, %: С, 76.63; Н, 4.97; N, 8.12. Найдено, %: С, 76.45; Н, 4.80; N, 8.24.
Пример 15. Получение 3,4-дийодтиофенил-2-карбальдегида. Из 100 мг (0.89 ммоль) тиофенил-2-карбальдегида в условиях, описанных в примере 1, было получено 207 мг (64%) 3,4-дийодтиофенил-2-карбальдегида в виде желтоватых кристаллов с Тпл 145°С
1Н ЯМР (400 МГц, CDCl3) δ 7.53 (с, 1H, СНаром), 9.75 (с, СНО).
13С ЯМР (101 МГц, CDCl3) δ 95.0 (C-I), 98.0 (C-I), 143.2 (СНаром), 150.3 (С-СНО), 180.3 (СНО).
ГХМС (EI, 70 эВ): 364 (100) [М+]; 363 (58) [М-Н]+; 208 (17) [М-I-СНО]+; 127 (16) [I+]; 81 (25) [C4HS]+.
Пример 16. Получение 4,4'-дийоддифенилового эфира. Из 50 мг (0.29 ммоль) дифенилового эфира, 198 мг (1.36 ммоль) NOSO3Cl в условиях, описанных в примере 1, было получено 102 мг (82%) 4,4'-дийоддифенилового эфира в виде желтоватых кристаллов. Тпл 140°С.
1Н ЯМР (400 МГц, CDCl3) δ 6.67 (д, 3J=8.8, 4Н, 4СНаром), 7.63 (д, 3J=8.8, 4Н, 4СНаром);
13С ЯМР (101 МГц, CDCl3) δ 86.2 (C-I), 120.7 (4СНаром), 138.4 (4СНаром), 156.3 (С-О).
Пример 17. Получение 2,5-дийод-1,4-диметилбензола. Из 100 мг (0.94 ммоль) пара-ксилола, 360 мг (2.82 ммоль) NOHSO4 в условиях, описанных в примере 1, было получено 170 мг (47%) 1-иоднафталина в виде оранжевых кристаллов. Тпл 102°С.
1Н ЯМР (400 МГц, CDCl3) δ 2.34 (с, 6Н, 2СН3), 7.64 (с, 2Н, 2СНаром);
13С ЯМР (101 МГц, CDCl3) δ 26.6 (2СН3), 100.3 (2C-I), 138.8 (2СНаром), 140.2 (2Саром).
Пример 18. Получение 1-иоднафталина. Из 100 мг (0.78 ммоль) дифенилового эфира, 340 мг (2.44 ммоль) NOSO3Cl в условиях, описанных в примере 1, было получено 96 мг (48%) 1-иоднафталина в виде оранжевого масла.
13С ЯМР (101 МГц, CDCl3) δ 99.3 (C-I), 126.4 (СНаром), 126.5 (СНаром), 127.4 (СНаром), 128.2 (СНаром), 128.6 (СНаром), 131.7 (СНаром), 133.7 (Саром), 133.9 (Саром), 137.0 (СНаром)
Пример 19. Получение 4-бром-3-(3-бром-4-метилфенил)-5-фенилизоксазола. Из 50 мг (0.21 ммоль) дифенилового эфира, 73 мг (0.63 ммоль) NOBF4 в условиях, описанных в примере 1, было получено 82 мг (98%) 4-бром-3-(3-бром-4-метилфенил)-5-фенилизоксазола в виде желтоватых кристаллов. Тпл 110°С.
1Н ЯМР (400 МГц, CDCl3) δ 2.49 (с, 3Н, СН3), 7.38 (д, 3J=7.9, 1H, СНаром), 7.54 (м, 3Н, 3СНаром), 7.74 (д.д., 4J=1.6, 3J=7.9, 1Н, СНаром), 8.08 (м, 3Н, 3СНаром);
13С ЯМР (101 MHz, CDCl3) δ 23.0 (СН3), 89.3 (CisBr), 125.1, 126.6, 127.1 (2СНаром), 127.4 (СНаром), 128.9 (2СНаром), 130.8 (СНаром), 131.0 (СНаром), 132.2 (СНаром), 140.2 (С-СН3), 160.7 (C=N), 166.0 (С-О), 1 сигнал перекрывается с другими.
ГХМС (EI, 70 эВ): кластер 391 (9), 393 (16), 395 (8) [М+]; кластер 284 (5), 286 (5) [М+-Br-С=O]; кластер 272 (4), 274 (4); 233 (21) [М-2Br]+; 105(100) [Ph-C=O]+; 77(41) [С6Н5]+; 51 (5) [С4Н3]+.
Вычислено для C16H11Br2NO, %: С 48.89, Н 2.82; Найдено, %: С 49.04, Н 3.20.
| название | год | авторы | номер документа |
|---|---|---|---|
| 3,5-ДИАРИЛИЗОКСАЗОЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2021 |
|
RU2801861C2 |
| 5-АРИЛ-4-ФТОРИЗОКСАЗОЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2018 |
|
RU2708456C1 |
| Замещенные 4-(азол-1-илметил)-1-фенил-5,5-диалкилспиро-[2.5]октан-4-олы, способ их получения (варианты), фунгицидная и рострегуляторная композиции на их основе | 2016 |
|
RU2648240C1 |
| НОВЫЕ 2',5'-ДИАРИЛСПИРО[ИНДОЛ-3,3'-ПИРРОЛИДИН]-2(1Н)-ОНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2019 |
|
RU2730287C1 |
| ИНДОЛИН-СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ | 2007 |
|
RU2402530C2 |
| Донорно-акцепторные сопряженные молекулы и способ их получения | 2014 |
|
RU2624820C2 |
| Способ получения 8-(алкиламино)-9-ароил-6-(2-гидроксифенил)-1,3-дициклогексил-1,3,6-триазаспиро[4.4]нона-8-ен-2,4,7-трионов | 2021 |
|
RU2758865C1 |
| ЗАМЕЩЁННЫЕ ИЗОКСАЗОЛЫ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ, ОБЛАДАЮЩИЕ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2733945C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 3-АРИЛ-5-ХЛОРИЗОКСАЗОЛОВ | 2015 |
|
RU2637927C2 |
| ПОЛУЧЕНИЕ НОВОГО КЛАССА ЖИДКОКРИСТАЛЛИЧЕСКИХ СОЕДИНЕНИЙ, СОДЕРЖАЩИХ ЧЕТЫРЕХАТОМНЫЙ ФТОРСОДЕРЖАЩИЙ ФРАГМЕНТ МОСТИКОГО ТИПА | 2012 |
|
RU2511009C2 |
Изобретение относится к способу получения галогенпроизводных ароматических и гетероароматических соединений путем галогенирования соответствующих ароматических и гетероароматических соединений галогенидами алкиламмония в присутствии окислителя – соединения, содержащего катион нитрозония. Технический результат: мягкие условия галогенирования, высокая селективность, а также универсальность - хлорирование, бромирование, йодирование ароматических или гетероароматических колец с одинаково хорошими выходами. 7 з.п. ф-лы, 19 пр.
1. Способ получения галогенпроизводных ароматических и гетероароматических соединений путем галогенирования ароматических и гетероароматических соединений, отличающийся тем, что исходные ароматические и гетероароматические соединения, выбранные из группы, включающей алкилбензолы, алкоксибензолы, арилоксибензолы, нафталин, замещенные изоксазолы, пиразолы, тиофены, галогенируют активной галогенирующей частицей, образующейся in situ, посредством смешения галогенидов алкиламмония общей формулы R4N+Hal-, где R=Н, Alk, Hal=Cl, Br, I, с соединением, содержащим нитрозоний катион - NOHSO4 или NOBF4 или NOSO3Cl, в среде органического растворителя, по окончании реакции галогенпроизводные ароматические или гетероароматические соединения выделяют и очищают от примесей.
2. Способ по п. 1, отличающийся тем, что для получения соединения формулы (VII) в качестве исходного соединения используют соединение формулы (IV), для соединения формулы (VIII) - соединение формулы (V), для соединения формулы (IX) - соединение формулы (VI), для соединения формулы (X) - соединение формулы (I), для соединения формулы (XI) - соединение формулы (II), для соединения формулы (XII) - соединение формулы (III):

исходные соединения:
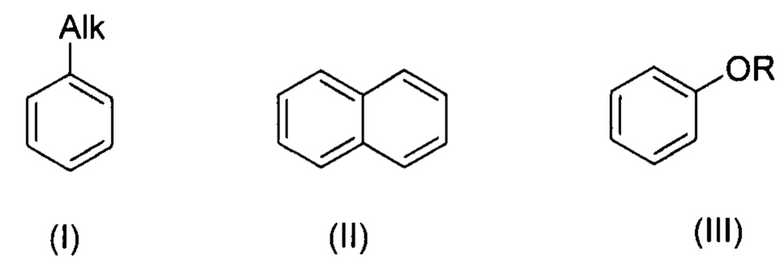 , где R=Ar, Alk
, где R=Ar, Alk
 , где R1=Ar, R2=Ar, Alk
, где R1=Ar, R2=Ar, Alk
 , где R1=Ar, Alk; R2=Ar, Alk; R3=H, Ar;
, где R1=Ar, Alk; R2=Ar, Alk; R3=H, Ar;
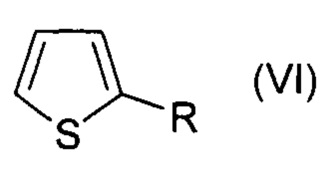 , где R=СОН.
, где R=СОН.
3 Способ по п. l, отличающийся тем, что используют 1.5-3-кратный мольный избыток нитрозилсерной кислоты и галогенида тетраалкиламмония по отношению к исходному ароматическому или гетероароматическому соединению.
4. Способ по п. 1, отличающийся тем, что реакцию проводят в течение времени, необходимого для максимального выхода галогенпроизводного ароматического или гетероароматического соединения.
5. Способ по п. 4, отличающийся тем, что время максимального выхода галогенпроизводного ароматического или гетероароматического соединения контролируют методом ТСХ.
6. Способ по п. 1, отличающийся тем, что реакцию проводят в интервале температур 0-60°С.
7. Способ по п. 1, отличающийся тем, что в качестве соединения, содержащего нитрозоний катион, используют нитрозилсерную кислоту, тетрафторборат нитрозония, хлорсульфат нитрозония.
8. Способ по п. 1, отличающийся тем, что в качестве органических растворителей используют нитрометан, бензол, четыреххлористый углерод, хлороформ, ацетонитрил.
| Способ получения иодароматических углеводородов или их простых эфиров | 1977 |
|
SU639846A1 |
| Способ получения бромсодержащих ароматических или конденсированных N- или О-содержащих гетероциклических соединений | 1988 |
|
SU1817764A3 |
| Способ получения бром- или йод-ароматических углеводородов | 1984 |
|
SU1203082A1 |

