Область техники
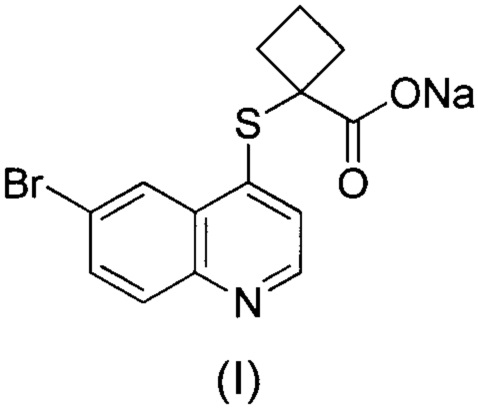
Настоящее изобретение относится к 1-((6-бромхинолин-4-ил)тио)цикло-бутан-1-карбоксилату натрия в кристаллической форме I, способу его получения и применения. Соединение формулы (I), полученное способом по настоящему изобретению полезно для лечения подагры.
Предшествующий уровень техники
В последнее время распространенность подагры возрастает с каждым годом, и наблюдается тенденция к изменению возраста манифестации в сторону более раннего, ввиду улучшения жизненных стандартов. Подагре подвержены мужчины, а также женщины после менопаузы, и возраст, на который приходится пик частоты возникновения заболевания, составляет 40-50 лет. Клиническими признаками подагры являются гиперурикемия, рецидив подагрического острого артрита, отложение подагрического узла, характерный хронический артрит и суставная деформация; как правило, поражаются почки, что вызывает хронический интерстициальный нефрит и уратический нефролитиаз. Предпосылкой подагры является гиперурикемия: насыщенная концентрация мочевой кислоты в сыворотке при 37°С составляет примерно 420 мкмоль/л (70 мг/л), и пациент страдает гиперурикемией, если концентрация мочевой кислоты у него выше указанной величины. Тем не менее, лишь у части пациентов с гиперурикемией развивается подагра, и ее механизм не ясен. Считается, что только гиперурикемические пациенты с отложениями кристаллов уратов, артритом и/или заболеванием почек, камнями в почках и т.п. страдают подагрой. Таким образом, гиперурикемия представляет собой важный биохимический базовый показатель подагры, и он тесно связан с проявлением подагры. Гиперурикемия тесно связана с проявлением гипертонии, гиперлипидемии, атеросклероза, тучности и инсулиновой резистентности, и она стала серьезным метаболическим заболеванием, которое угрожает здоровью человека.
Мочевая кислота представляет собой конечный продукт метаболизма пурина у человека. У человека отсутствует уриказа из-за мутации гена уриказы в ходе эволюции, и поэтому мочевая кислота не может быть метаболизирована до растворимого аллантоина для выведения из организма. Поэтому у пациентов с гиперурикемией имеется избыток сывороточной концентрации мочевой кислоты. Причинами проявления гиперурикемии могут быть: (1) увеличение продукции мочевой кислоты, что составляет от 15% до 20% случаев проявления подагры (например, при употреблении в избытке пищи, обогащенной пурином; или же большее количество мочевой кислоты синтезируется из аминокислоты и нуклеотида in vivo, и избыточная мочевая кислота продуцируется в ходе катаболизма нуклеиновой кислоты); (2) снижение экскреции мочевой кислоты и повышение реабсорбции мочевой кислоты, что представляют собой основной путь патогенеза гиперурикемии и подагры и составляет примерно от 80% до 85% случаев. Около 95% реабсорбции мочевой кислоты осуществляется транспортером мочевой кислоты 1 (URAT1, от англ. Ureic Acid Transporter 1), который локализуется в клетках эпителия проксимальных почечных канальцев. URAT1 представляет собой полностью мембранный белок, локализующийся в почках, который принадлежит к семейству 22 переносчиков растворенных веществ (SLC22, от англ. Solute Carrier). Он осуществляет урат-анионный обмен и отвечает за регуляцию уровня мочевой кислоты в крови. Таким образом, ингибитор URAT1 может повышать экскрецию мочевой кислоты за счет ингибирования такой реабсорбции.
На данный момент имеется лишь небольшое количество лекарственных средств против подагры на фармацевтическом рынке Китая, при этом не было разработано новых и улучшенных противоподагрических лекарственных препаратов. Основными лекарственными средствами до сих пор являются аллопуринол и бензбромарон. Фебуксостат, одобренный управлением по контролю за продуктами и лекарствами США в 2009, относится к ингибитору ксантиноксидазы (ХО). Он лечит подагру за счет снижения продукции мочевой кислоты. RDEA-594 (Лезинурад), разработанный Ardea Biosciences Inc., повышает экскрецию мочевой кислоты за счет ингибирования транспортера мочевой кислоты 1 (URAT1), достигая таким образом целевого снижения концентрации мочевой кислоты в сыворотке. На его эффективность не влияют функция почек и доза аллопуринола. Он не влияет на транспортный эффект транспортера органических анионов 1/3 (ОАТ1/ОАТ3) в пределах клинических дозировок. Кроме того, он более специфичен к целевым молекулам, по сравнению с другими урикозурическими препаратами, и меньше взаимодействует с другими препаратами.
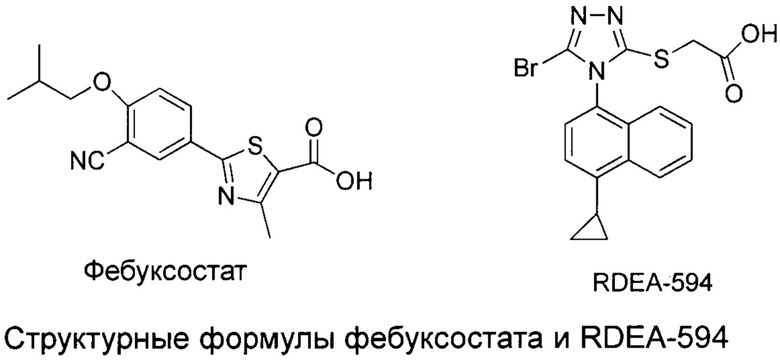
Тем не менее, в ходе клинических испытаний препаратов для лечения ВИЧ-инфекции было обнаружено, что активность RDEA-594 против транспортера мочевой кислоты URAT1 не высока (IC50 составляет около 7 мкМ). Более того, его дозировка при применении в клинической практике сравнительно высока. Кроме этого, до сих пор остается обширное поле для исследования в области нацеливания на транспортер мочевой кислоты URAT1.
В WO 2014183555 раскрыт ряд соединений с более высокой ингибиторной активностью по отношению к транспортеру мочевой кислоты URAT1. Эти соединения могут эффективно ингибировать реабсорбцию мочевой кислоты и выводить мочевую кислоту из организма, длительно снижая при этом содержание мочевой кислоты в крови, достигая при этом целевого лечения подагры. В том числе, включено соединение, показанное ниже.
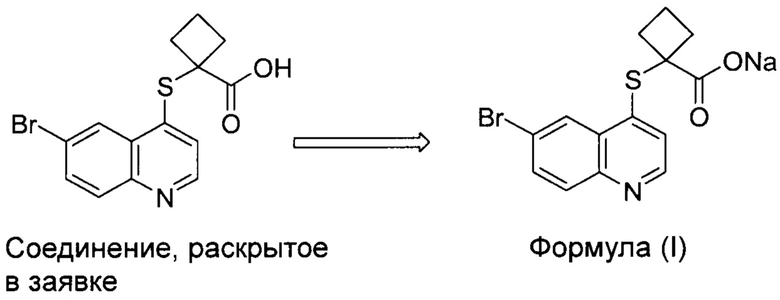
Для того чтобы улучшить растворимость в воде данного соединения, заявители разработали его натриевую соль (формула I). Растворимость в воде возросла от почти нерастворимого до 0,14 мг/мл. С одной стороны, кристаллическая структура фармацевтически активного ингредиента часто влияет на химическую стабильность лекарственного вещества. Различные условия кристаллизации и условия хранения могут приводить к изменениям в кристаллической структуре соединения и иногда - к побочному образованию других кристаллических форм. В целом, аморфное лекарственное вещество не имеет упорядоченной кристаллической структуры и часто обладает другими нежелательными свойствами, такими как низкая стабильность продукта, меньший размер частиц, сложность фильтрации, подверженность агломерации и слабая текучесть. В связи с этим необходимо улучшать различные свойства указанного выше продукта. Основываясь на открытии его новой разрабатываемой формы, необходимо найти новую кристаллическую форму с высокой степенью чистоты и хорошей химической стабильностью.
Сущность изобретения
Цель настоящего изобретения - предложить соединение формулы (I), т.е. 1-((6-бромхинолин-4-ил)тио)циклобутан-1-карбоксилат натрия. Оно в известной мере улучшает желаемые свойства соединения, раскрытого в WO 2014183555, которое применяется в качестве фармацевтически активного ингредиента.
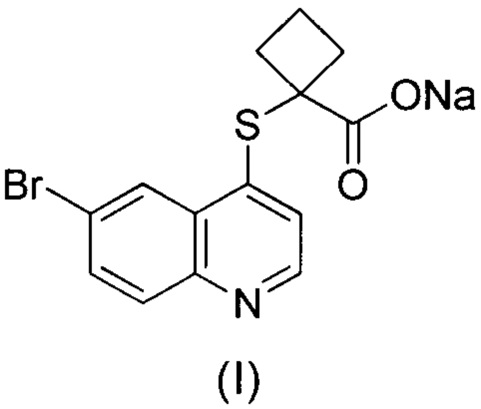
Соединение формулы (I) можно получить вступлением в реакцию 1-((6-бромхинолин-4-ил)тио)циклобутан-1-карбоновой кислоты с гидроксидом натрия.
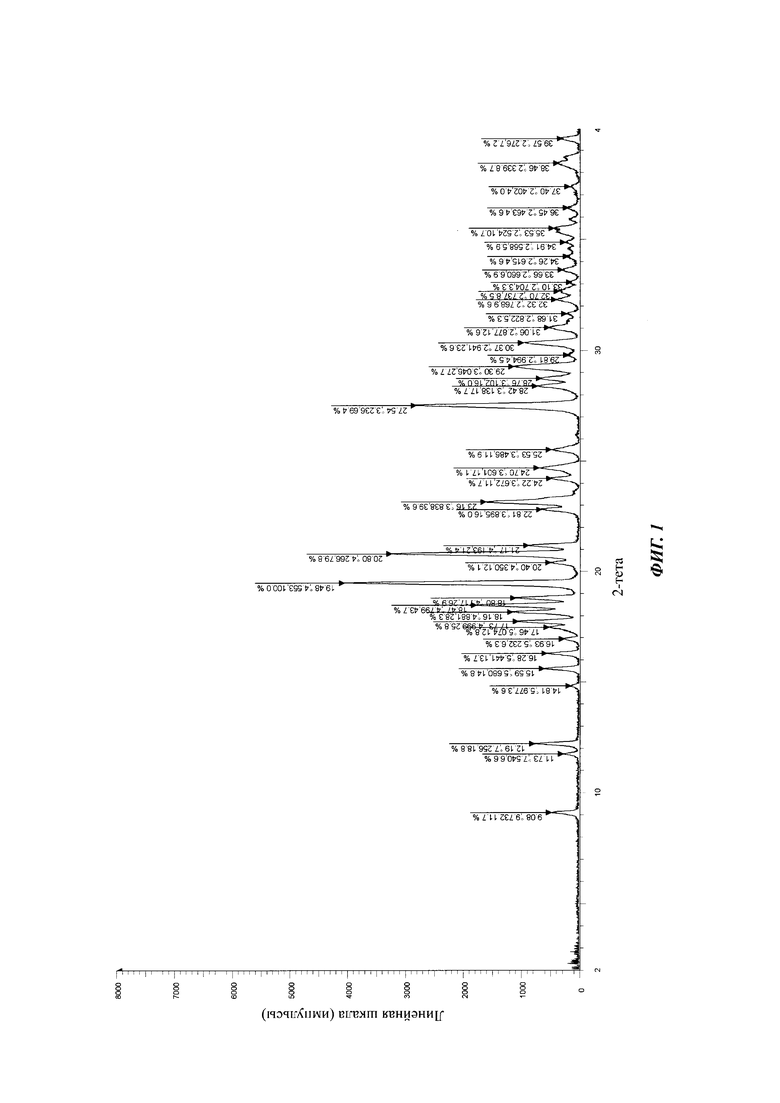
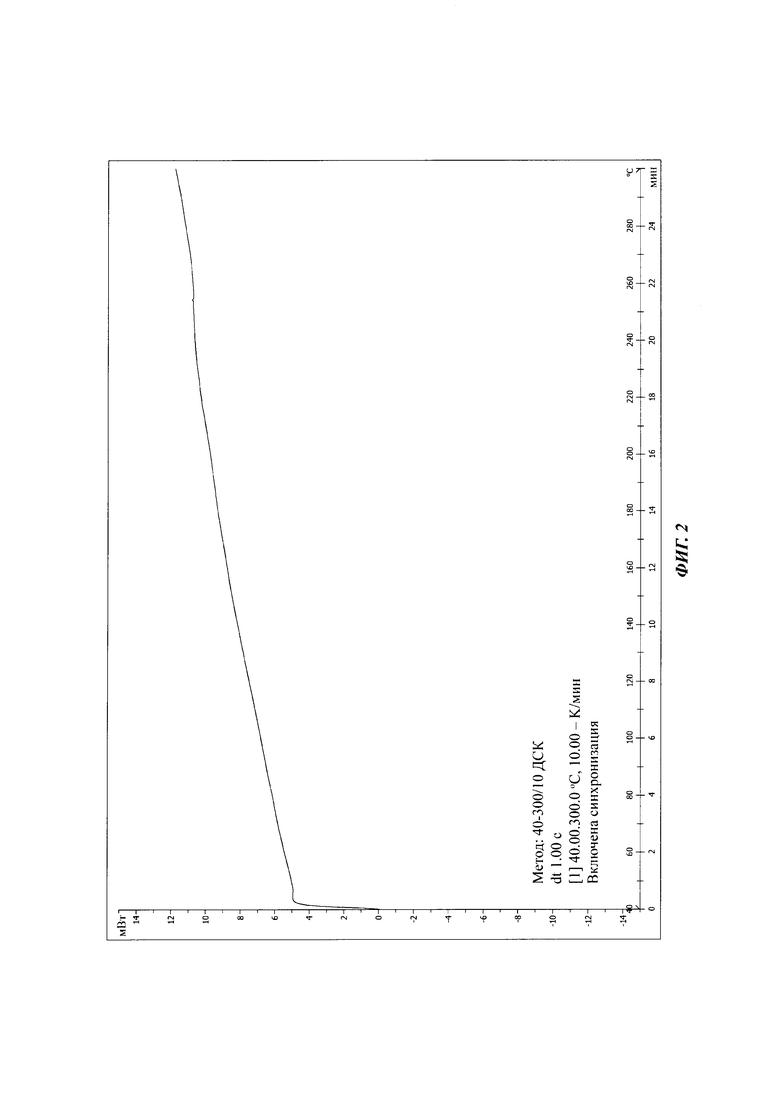
Заявитель исследовал ряд кристаллических продуктов соединения формулы (I), полученных в различных условиях кристаллизации, и были проведены исследования полученных кристаллических продуктов с помощью рентгеновской дифракции и дифференциальной сканирующей калориметрии (ДСК). Было обнаружено, что стабильную кристаллическую форму, которая обозначается как кристаллическая форма I, можно получить в особых условиях кристаллизации. На спектре ДСК кристаллической формы I по настоящему изобретению отсутствует абсорбция в пределах 300°С, и это говорит о том, что точка плавления находится выше 300°С. Спектр порошковой рентгеновской дифракции, который получен с использованием излучения Cu-Kа и представлен как угол 2θ и межплоскостное расстояние (величина d), показан на Фиг. 1, где характеристические пики расположены при значениях 9,08 (9,73), 11,73 (7,54), 12,19 (7,26), 15,59 (5,68), 16,28 (5,44), 17,73 (5,00), 18,16 (4,88), 18,80 (4,72), 19,48 (4,55), 20,80 (4,27), 23,16 (3,84), 27,54 (3,24) и 30,37 (2,94).
Настоящим изобретением предложен также способ получения кристаллической формы I соединения формулы (I). В частности, указанный способ включает следующие стадии:
(1) растворение твердого 1-((6-бромхинолин-4-ил)тио)циклобутан-1-карбоксилата натрия в любой кристаллической форме или аморфной форме в подходящем количестве растворителя при нагревании, затем охлаждение раствора для осаждения кристалла;
(2) фильтрация кристалла, затем его промывка и сушка.
Растворитель на стадии (1) представляет собой смесь растворителей: воды и любого спирта и кетона, которые содержат 3 или менее атомов углерода; более предпочтительна смесь вода/изопропиловый спирт, вода/ацетон, ацетон/вода/ацетон, ацетон/вода/изопропиловый спирт.
В одном воплощении настоящего изобретения предпочтительный смешанный растворитель представляет собой смешанный растворитель ацетон/вода/ацетон, при этом соотношение особым образом не ограничено. В предпочтительном воплощении настоящего изобретения объемное соотношение этих трех компонентов составляет 1:1:5. Если смешанный растворитель представляет собой смесь ацетон/вода/ацетон, это означает, что 1-((6-бром-хинолин-4-ил)тио)циклобутан-1-карбоксилат натрия растворяют в смешанном растворителе ацетон/вода до тех пор, пока раствор не станет прозрачным, затем добавляют другую часть ацетона для осаждения кристалла. Смесь ацетон/вода/изопропиловый спирт употребляется в аналогичном смысле.
Способ перекристаллизации конкретным образом не ограничен, и его можно проводить обычным способом перекристаллизации. Например, вещество, т.е. соединение формулы (I), можно растворять в органическом растворителе при нагревании, и затем раствор медленно охлаждать при перемешивании для осаждения кристалла. По завершении кристаллизации, желаемый кристалл можно получить путем фильтрации и сушки. В частности, кристалл, полученный фильтрацией, обычно сушат в вакууме при пониженном давлении при температуре нагревания примерно 30~100°С, предпочтительно 40~60°С, для удаления растворителя после перекристаллизации.
Полученную кристаллическую форму определяют с помощью дифференциальной сканирующей калориметрии (ДСК) и спектра рентгеновской дифракции. При этом также определяют остаточный растворитель в полученном кристалле.
Кристаллическая форма соединения формулы (I), полученная способом по настоящему изобретению, не содержит или содержит лишь сравнительно небольшое количество остаточного растворителя, что отвечает требованиям Национальной фармакопеи в отношении ограничения остаточного растворителя в лекарственных продуктах. Таким образом, кристалл по настоящему изобретению подходит для применения в качестве фармацевтически активного ингредиента.
Результаты исследования показывают, что кристаллическая форма I соединения формулы (I), полученная по настоящему изобретению, стабильна в условиях освещения, высокой температуры и высокой влажности, кристаллическая форма I также стабильна в условиях помола, давления и нагревания, что соответствует технологическим требованиям, требованиям транспортировки и хранения лекарственных продуктов. Способ его получения стабилен, воспроизводим и его можно контролировать, что делает его подходящим для промышленного производства.
Описание графических материалов
На Фиг. 1 показан спектр порошковой рентгеновской дифракции кристаллической формы I соединения формулы (I).
На Фиг. 2 показан спектр ДСК кристаллической формы I соединения формулы (I).
Подробное описание изобретения
Настоящее изобретение детально представлено в следующих примерах. Примеры по настоящему изобретению предназначены лишь для описания технического решения настоящего изобретения, и их не следует рассматривать как ограничение объема настоящего изобретения.
Приборы для проведения исследования, использованные в экспериментах:
1. Спектр ДСК
Тип прибора: Mettler Toledo DSC 1 Staree System
Газ для продувки: азот
Скорость нагревания: 10,0°С/мин
Диапазон температур: 40-300°С
2. Спектр рентгеновской дифракции
Тип прибора: порошковый рентгеновский дифрактометр Bruker D8 Focus
Излучение: монохроматическое излучение Cu-Kа (λ равно 1,5406)
Способ сканирования: θ/2θ, угол сканирования: 2-40°
напряжение: 40 кВ, электрический ток: 40 мА.
Пример 1
1-((6-Бромхинолин-4-ил)тио)циклобутан-1-карбоновую кислоту (полученную способом, раскрытым в WO 2014/183555) (1,0 г; 2,96 ммоль) вносили в трехгорловую реакционную колбу на 50 мл при 25°С, затем добавляли 4,0 г безводного этанола. По каплям, при перемешивании вносили 0,5 мл водного раствора гидроксида натрия (118 мг; 2,96 ммоль), затем реакционную смесь перемешивали. Реакционную смесь фильтровали, осадок на фильтре промывали безводным этанолом и высушивали в вакууме при 40°С. Получали 850 мг порошка цвета от белого до бледно-желтого с выходом 84,0%. Спектр порошковой рентгеновской дифракции кристаллического образца показан на Фиг. 1, где характеристические пики расположены примерно при значениях 9,08 (9,73), 11,73 (7,54), 12,19 (7,26), 15,59 (5,68), 16,28 (5,44), 17,73 (5,00), 18,16 (4,88), 18,80 (4,72), 19,48 (4,55), 20,80 (4,27), 23,16 (3,84), 27,54 (3,24) и 30,37 (2,94). Спектр ДСК, представленный на Фиг. 2, показывает отсутствие абсорбции в районе 300°С, т.е. точка плавления находится выше 300°С. Кристаллическая форма определена как кристаллическая форма I.
Пример 2
Соединение формулы (I) (полученное по Примеру 1) (1,0 г; 2,78 ммоль) вносили в одногорловую колбу на 250 мл, затем вносили 30 мл воды. Эту смесь нагревали до температуры флегмы до тех пор, пока раствор на становился прозрачным, затем концентрировали примерно до 3 мл при пониженном давлении. Медленно, при перемешивании добавляли 150 мл изопропилового спирта для осаждения кристалла. На следующий день смесь фильтровали и высушивали с получением 689 мг белого твердого вещества с выходом 68,9%. Кристаллический образец после изучения и сравнения спектров рентгеновской дифракции и ДСК был идентифицирован как кристаллическая форма I.
Пример 3
Соединение формулы (I) (полученное по Примеру 1) (1,0 г; 2,78 ммоль) вносили в одногорловую колбу на 150 мл, затем вносили 30 мл воды. Смесь нагревали до температуры флегмы, пока раствор не становился прозрачным, затем концентрировали досуха при пониженном давлении. Для осаждения кристалла непосредственно в смесь вносили при перемешивании 30 мл изопропилового спирта. На следующий день смесь фильтровали и высушивали с получением 812 мг белого твердого вещества с выходом 81,2%. После изучения и сравнения спектров рентгеновской дифракции и ДСК кристаллический образец был идентифицирован как кристаллическая форма I.
Пример 4
Соединение формулы (I) (полученное по Примеру 1) (1,0 г; 2,78 ммоль) вносили в 150 мл одногорловую колбу, затем вносили 30 мл воды. Смесь нагревали до температуры флегмы до тех пор, пока раствор не становился прозрачным, затем концентрировали примерно до 3 мл при пониженном давлении. Медленно, при перемешивании вносили 30 мл ацетона для осаждения кристалла. На следующий день смесь фильтровали и высушивали с получением 918 мг белого твердого вещества с выходом 91,8%. После изучения и сравнения спектров рентгеновской дифракции и ДСК кристаллический образец был идентифицирован как кристаллическая форма I.
Пример 5
Соединение формулы (I) (полученное по Примеру 1) (1,0 г; 2,78 ммоль) вносили в одногорловую колбу на 150 мл, затем добавляли 24 мл смеси ацетон/вода (объемное соотношение составляет 1:1). Эту смесь нагревали до температуры флегмы до тех пор, пока раствор не становился прозрачным, затем медленно вносили 60 мл ацетона. Эту смесь непрерывно нагревали с обратным холодильником в течение 10 мин, после чего нагревание прекращали. Затем смесь перемешивали для осаждения кристалла. На следующий день смесь фильтровали и высушивали с получением 688 мг белого твердого вещества с выходом 68,8%. После изучения и сравнения спектров рентгеновской дифракции и ДСК кристаллический образец идентифицировали как кристаллическая форма I.
Пример 6
Соединение формулы (I) (полученное по Примеру 1) (1,0 г; 2,78 ммоль) вносили одногорловую колбу на 150 мл, затем вносили 24 мл смеси ацетона/вода (объемное соотношение составляет 1:1). Эту смесь нагревали до температуры флегмы до тех пор, пока раствор не становился прозрачным, затем медленно вносили 60 мл изопропилового спирта. Эту смесь непрерывно нагревали с обратным холодильником в течение 10 мин, после чего нагревание прекращали. Затем эту смесь перемешивали для осаждения кристалла. На следующий день смесь фильтровали и высушивали с получением 752 мг белого твердого вещества с выходом 75,2%. После изучения и сравнения спектров рентгеновской дифракции и ДСК кристаллический образец был идентифицирован как кристаллическая форма I.
Пример 7
Соединение формулы (I) (полученное по Примеру 1) (1,0 г; 2,78 ммоль) вносили в одногорловую колбу на 500 мл, затем вносили 30 мл воды. Эту смесь нагревали до температуры флегмы до тех пор, пока раствор не становился прозрачным, затем медленно, при перемешивании добавляли 300 мл ацетона для осаждения кристалла. На следующий день смесь фильтровали и высушивали с получением 728 мг белого твердого вещества с выходом 72,8%. После изучения и сравнения спектров рентгеновской дифракции и ДСК кристаллический образец был идентифицирован как кристаллическая форма I.
Пример 8
Образец кристаллической формы I, полученный в Примере 1 раскладывали слоем на воздухе для изучения его стабильности в условиях освещенности (4500 Люкс), нагревания (40°С, 60°С) и высокой влажности (отн. влажность 75%, отн. влажность 90%). Забор образцов проводили на 5 и 10 сутки. Степень чистоты, определенная с помощью ВЭЖХ, показана в табл. 1.
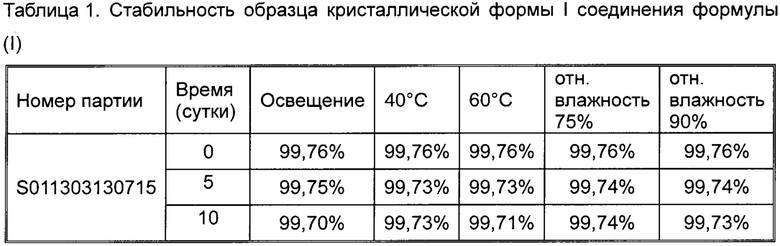
Результаты исследования стабильности показали, что образец кристаллической формы I имеет хорошую стабильность, если его разложить слоем на воздухе в условиях освещенности, высокой температуры и высокой влажности.
Пример 9
Кристаллическую форму I соединения формулы (I), полученную способом по Примеру 1, перемалывали, нагревали и прессовали. Полученные результаты показывают, что такая кристаллическая форма стабильна. Более подробно данные эксперимента показаны ниже в табл. 2.
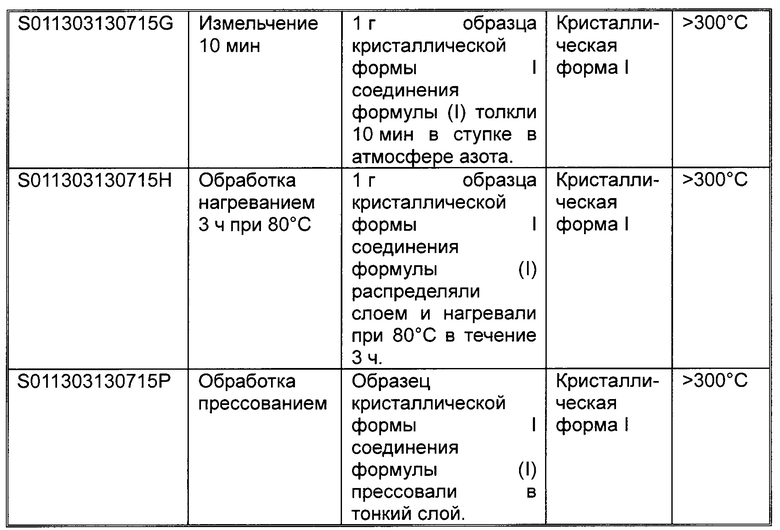
Пример 10
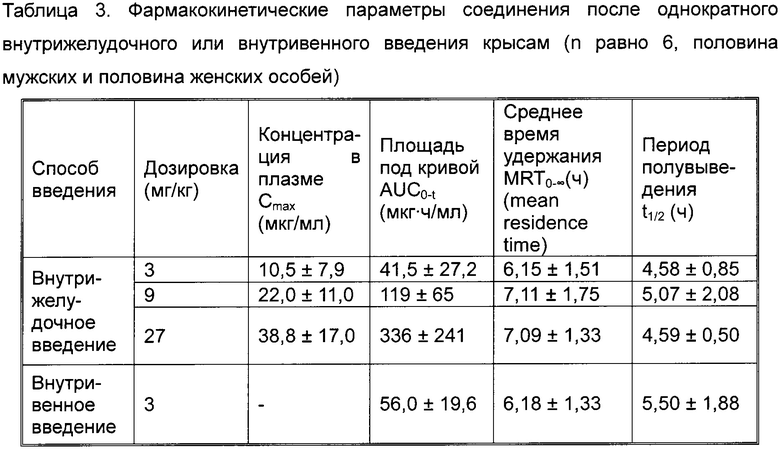
В фармакокинетическом исследовании соединения по Примеру 1 по настоящему изобретению в качестве тестовых животных использовали крыс линии Спраг Доули (СД). Соединения по Примеру 1 вводили крысам внутрижелудочно и внутривенно, затем определяли концентрацию в плазме крови лекарственного вещества в различные моменты времени с помощью жидкостной хроматографии с тандемной масс-спектрометрией (LC/MS/MS) для исследования фармакокинетического профиля и оценки фармакокинетических параметров соединения по настоящему изобретению у крыс. Фармакокинетические параметры соединения по настоящему изобретению представлены в табл. 3. Эти результаты показали, что соединение по настоящему изобретению хорошо абсорбируется и обладает значительным эффектом пероральной абсорбции. Согласно средней величине площади под кривой (AUC0-t), абсолютная биодоступность соединения после однократного внутрижелудочного введения дозой 3 мг/кг крысам по расчетам составила 74,1%.
Пример 11
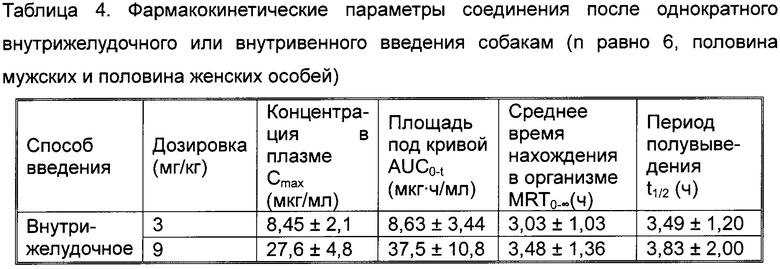
В фармакокинетическом исследовании соединения по Примеру 1 по настоящему изобретению в качестве тестовых животных использовали собак породы бигль. Соединение по Примеру 1 вводили собакам внутрижелудочно и внутривенно, затем определяли концентрацию лекарственного вещества в плазме крови в различные моменты времени с помощью LC/MS/MS для исследования фармакокинетического профиля и оценки фармакокинетических параметров соединения по настоящему изобретению у собак. Фармакокинетические параметры соединения по настоящему изобретению показаны в табл. 4. Результаты показали, что соединение по настоящему изобретению хорошо абсорбируется и обладает значительным эффектом пероральной абсорбции. Согласно среднему значению AUC0-t, абсолютная биодоступность соединения после однократного внутрижелудочного введения в дозе 3 мг/кг собакам по расчетам составила 59,5%.

Изобретение относится к области органической химии, а именно к 1-((6-Бромхинолин-4-ил)тио)циклобутан-1-карбоксилат натрия формулы (I). Также изобретение относится к кристаллической форме соединения формулы (I), способу получения соединения формулы (I) и его кристаллической формы, фармацевтической композиции на основе соединения формулы (I) или его кристаллической формы, а также применению соединения формулы (I) или его кристаллической формы. Технический результат: получено новое соединение, в том числе и в кристаллической форме, полезное при лечении заболевания, связанного с транспортером мочевой кислоты (URAT1). 6 н. и 1 з.п. ф-лы, 2 ил., 4 табл., 11 пр.
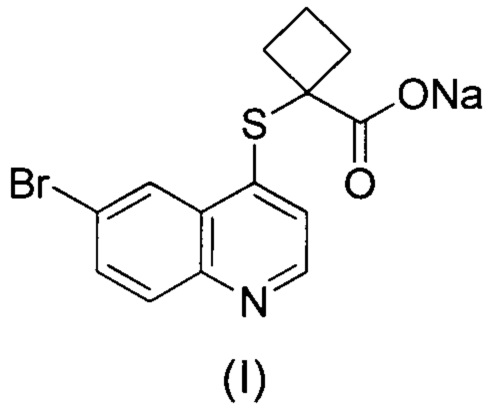
1. 1-((6-Бромхинолин-4-ил)тио)циклобутан-1-карбоксилат натрия формулы (I),
2. Кристаллическая форма I 1-((6-бромхинолин-4-ил)тио)цикло-бутан-1-карбоксилата натрия формулы (I) по п. 1, отличающаяся тем, что такой кристалл имеет спектр порошковой рентгеновской дифракции, который получен с помощью излучения Cu-Ka и представлен углом 2θ и межплоскостным расстоянием, как показано на Фиг. 1, и в котором характеристические пики расположены примерно при значениях 9,08 (9,73), 11,73 (7,54), 12,19 (7,26), 15,59 (5,68), 16,28 (5,44), 17,73 (5,00), 18,16 (4,88), 18,80 (4,72), 19,48 (4,55), 20,80 (4,27), 23,16 (3,84), 27,54 (3,24) и 30,37 (2,94).
3. Способ получения 1-((6-бромхинолин-4-ил)тио)циклобутан-1-карбоксилата натрия формулы (I) по п. 1, включающий стадию вступления в реакцию 1-((6-бромхинолин-4-ил)тио)циклобутан-1-карбоновой кислоты с гидроксидом натрия.
4. Способ получения кристаллической формы I по п. 2, включающий следующие стадии:
1) растворение твердого 1-((6-бромхинолин-4-ил)тио)циклобутан-1-карбоксилата натрия в растворителе при нагревании до температуры флегмы, затем охлаждение раствора для осаждения кристалла, при этом растворитель представляет собой смесь вода/изопропиловый спирт, вода/ацетон, ацетон/вода/ацетон, ацетон/вода/изопропиловый спирт;
2) фильтрация кристалла, затем его промывка и сушка.
5. Фармацевтическая композиция для лечения заболевания, связанного с транспортером мочевой кислоты (URAT1), включающая 1-((6-бромхинолин-4-ил)тио)циклобутан-1-карбоксилат натрия формулы (I) по п.1 или кристаллическую форму I по п.2, и фармацевтически приемлемый носитель.
6. Применение 1-((6-бромхинолин-4-ил)тио)циклобутан-1-карбоксилата натрия формулы (I) по п.1, кристаллической формы I по п.2 или фармацевтической композиции по п.5 в изготовлении лекарственного средства для лечения заболевания, связанного с транспортером мочевой кислоты (URAT1).
7. Применение по п.6, отличающееся тем, что указанное заболевание представляет собой подагру.
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Sherry L.Morissette et al.: "High-throughput crystallization: polymorphs, salts, co-crystals and solvates of pharmaceutical solids", ADVANCED DRUG DELIVERY REVIEWS, 2004, v.56, pp.275-300 (section 1; 3.1) (DOI:10.1016/J.ADDR.2003.10.020) | |||
| STEPHEN M.BERGE et al., Pharmaceuticals Salts, JOURNAL of PHARMACEUTICAL | |||

