Настоящее изобретение относится к производным хиназолина, содержащим гидроксаматный фрагмент, а именно - N-((гидроксиамино)-оксоалкил)-2-(хиназолин-4-иламино)бензамидам, которые могут быть применены в медицине для лечения различных заболеваний, например, злокачественных новообразований, посредством ингибирования гистоновых деацетилаз.
Как известно, гистондеацетилазы (HDAC) - это ферменты, катализирующие удаление ацетильной группы ε-N-ацетил-лизина гистонов. Модифицируя гистоны и изменяя конформацию хроматина, гистондеацетилазы играют важную роль в регуляции экспрессии генов. Таким образом, HDAC является важной эпигенетической мишенью при терапии рака, а ингибиторы HDAC демонстрируют успешную картину как цитотоксические агенты. Большинство ингибиторов HDAC имеют трехкомпонентную структуру, состоящую из цинк-связывающего участка, линкера, способного занимать канал фермента, и фрагмента, взаимодействующего с аминокислотными остатками у входа в активный центр HDAC. Ингибиторы классических деацетилаз функционируют путем связывания иона цинка в активном центре фермента и, таким образом, инактивируют систему смены зарядов.
Известно, что эффективными ингибиторами гистондеацетилаз (HDAC) являются производные гидроксамовой кислоты, например, вориностат (SAHA), панобиностат (LBH589) и белиностат. Они одобрены FDA USA для лечения Т-клеточной лимфомы кожи (CTCL) и множественной миеломы [Mottamal М, et al. Molecules. 2015; 20(3):3898-941].
Одной из перспективных стратегий в создании новых фармацевтических препаратов в настоящее время является проектирование и синтез гибридных соединений, состоящих из двух или более различных биоактивных фрагментов, и действующих через активацию/блокирование нескольких мишеней. Совмещение двух активных групп в одной молекуле может приводить к более выраженному терапевтическому эффекту, по сравнению с индивидуальными компонентами при комбинированном применении
Известно, что хиназолин является важным фармакологическим фрагментом. Хиназолиновый цикл присутствует как в различных природных соединениях, так и в молекулах многих лекарственных препаратов. Соединение в одной молекуле хиназолиновой и гидроксамовой фармакофорных групп может потенциально приводить к новым перспективным соединениям, что подтверждается многочисленными примерами.
Описан ряд соединений, содержащих хиназолиновый цикл и гидроксамовую кислоту, применяемых в качестве бифункциональных ингибиторов тирозинкиназ и гистондеацетилаз (WO2009063054, A61K 31/517, 2009; WO2008033749, A61K 31/517, 2008; WO2018005799, A61K 31/517, 2018; US2008221132, A61K 31/517, 2008; KR101964810, A61K 31/517, 2019; US 2018098990, A61K 31/517, 2018;)
Наибольший интерес представляют известные способы синтеза хиназолингидроксамовых кислот, в которых гидроксамовые кислоты, присоединены к хиназолиновому циклу в 4-м положении.
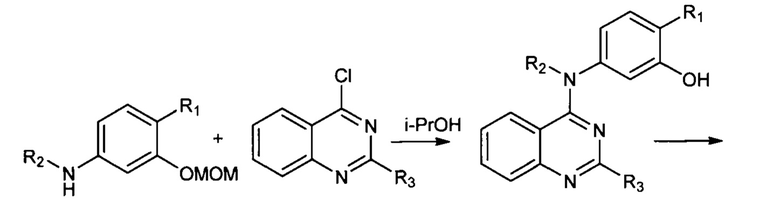
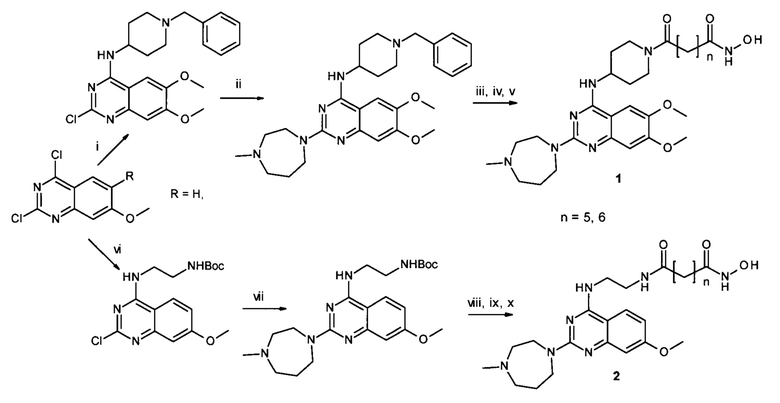
Так, например, описан способ синтеза гидроксамовых кислот, присоединенных к 4-му положению хиназолина, состоящий из трех стадий и протекающий с использованием качестве исходных реагентов производных 4-хлорхиназолина по прилагаемой ниже Схеме 1. (US 20180098990, C07D 239/94, 2018). В процессе осуществления данного способа собственно гидроксамовая кислота получается на последней стадии процесса гидроксиламинолиза, протекающего при взаимодействии этиловых эфиров соответствующих карбоновых кислот с гидроксиламином. Выход конечных продуктов на последней стадии составляет от 50-70%. Для очистки целевых соединений в данном способе предлагается использование высокоэффективной хроматографии.
Процесс протекает по следующей схеме (Схема 1):
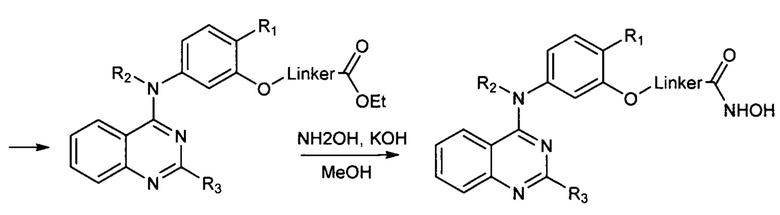
В ряде работ для получения гидроксамовых кислот, присоединенных к 4-му положению хиназолинового фрагмента, предлагается процесс синтеза, осуществляемый в две стадии, исходя из производных хиназолин-4-она (Hieu D.T. et al. Chemistry&Biodiversity. - 2018. Vol. 15, No 6. P.e 1800027; KR101964810, A61K 31/517; C07D 239/88, 2019; KR102000035, C07D 239/94, 2019). Гидроксамовые кислоты в этом случае получаются на последней стадии аминолиза гидроксиламином эфиров соответствующих карбоновых кислот. Выход целевых соединений, полученных данным методом составляет 62-77%. Процесс в этом случае протекает по Схеме 2:
Синтез аналогичных соединений описан в других известных публикациях (Zhang Q., et al. Bioorganic & Medicinal Chemistry Letters. - 2017. - Vol. 27. - P. 4885-4888; WO2013170757, C07D 239/94, 2013). В цитированных работах используется метод, в котором гидроксамовые кислоты получают на последней стадии 3-х стадийного синтеза, протекающего с получением промежуточных производных 4-хлорхиназолина и с выходом основных продуктов 45-69%. Данный процесс протекает по Схеме 3:
Еще одна схема синтеза присоединенных к 4-му положению хиназолинового фрагмента гидроксамовых кислот, являющихся двойными ингибиторами как гистондеацетилазы, так и протеин лизин метилтрансферазы, предлагается в опубликованной заявке США (US2019322643, А61Р 35/00; C07D401/12, 2019). Гидроксаматная функция, согласно данной схеме, появляется на последней стадии многостадийного синтеза в результате реакции гидроксиламинолиза. Выход соединений 1-й группы данным методом составляет 44-45% и для соединений 2-й группы 29-36%. Данный процесс протекает по Схеме 4:
Основными недостатками рассмотренных выше способов являются: трудоемкость их осуществления, одной из причин которой является многостадийность, а также, загрязненность получаемых промежуточных и конечных продуктов и, соответственно, необходимость введения дополнительных стадий очистки. Во всех приведенных примерах фрагмент гидроксамовой кислоты образуется на последней стадии в конечной громоздкой молекуле. Однако известно, что в сложных молекулах реакция прямого гидроксиламинолиза эфиров карбоновых кислот зачастую затруднена и/или сопровождается образованием побочных продуктов. Основным побочным продуктом в данном случае является соответствующая карбоновая кислота, образующаяся в результате гидролиза. Таким образом, стадия гидроксиламинолиза является самой проблемной в рассмотренных выше способах. Эту проблему, в частности, пытаются решать добавкой дополнительных реагентов, таких как, диазабициклоундецен (Beillard, A.etal. Tetrahedron Letters. 2016, Vol. 57(20), P. 2165-2170) или цианистый калий (Но С.Y. et al. J. Org. Chem. 2005, 70, P. 4873-4875), что, в свою очередь, усложняет процесс и не всегда приводит к положительному результату.
Для исключения стадии гидроксиламинолиза сложных эфиров кислот, которая может идти с затруднениями, предлагается осуществлять синтез через образование защищенных гидроксамовых кислот, используя, например, бензильную или тетрагидропиранильную защиту. (Zhang X., Su М., Chen Y. et al. Molecules. - 2013. - Vol. 18. - P. 6491-6503). В этом случае получаются гидроксамовые кислоты, соединенные с производными 4-ариламинохиназолинов. Гидроксаматную функцию получают из соответствующих этиловых эфиров двухстадийными методами: либо через получение тетрагидропиранильного эфира гидроксамовой кислоты с последующим кислотным гидролизом (с выходом 39-91%) [вариант 1], либо через получение бензилового эфира гидроксамовой кислоты с последующим гидрированием (с выходом 16-52%) [вариант 2], согласно Схеме 5:
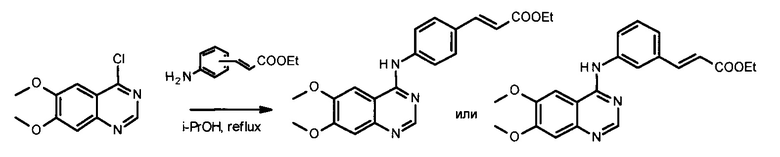
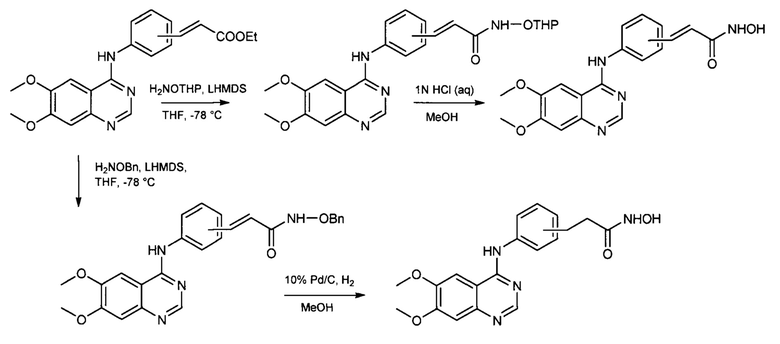
Однако в рассматриваемом выше процессе (Схема 5), несмотря на исключение стадии гидроксиламинолиза, вводятся дополнительные стадии получения промежуточных продуктов - (защищенных гидроксамовых кислот), что усложняет весь процесс. К тому же рассматриваемый процесс является трудоемким и не экономичным, поскольку введение защитной группы проводится при сильном охлаждении (до - 78°С), в безводном растворителе, и в процессе получения используются дорогостоящие реактивы, такие как бис(триметилсилил)амид лития и палладий на угле.
Данным способом получены такие соединения как:
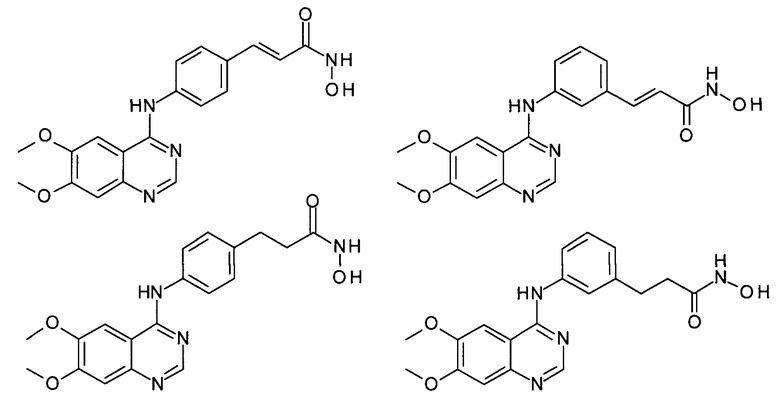
Эти соединения по своей структуре близки к структуре получаемых новых соединений и могут рассматриваться как их ближайшие структурные аналоги. Эти известные соединения проявляют противоопухолевую активность (Zhang X., Su М., Chen Y. et al. Molecules. - 2013. - Vol. 18. - P. 6491-6503)
С целью расширения ассортимента противоопухолевых препаратов и создания наиболее эффективных соединений, получаемых к тому же менее трудоемкими и более экономичными способами, обеспечивающими получение чистых конечных продуктов предлагается способ получения N-((гидроксиамино)-оксоалкил)-2-(хиназолин-4-иламино)бензамидов в качестве потенциальных ингибиторов гистондеацетилаз, имеющих следующую общую структурную формулу:
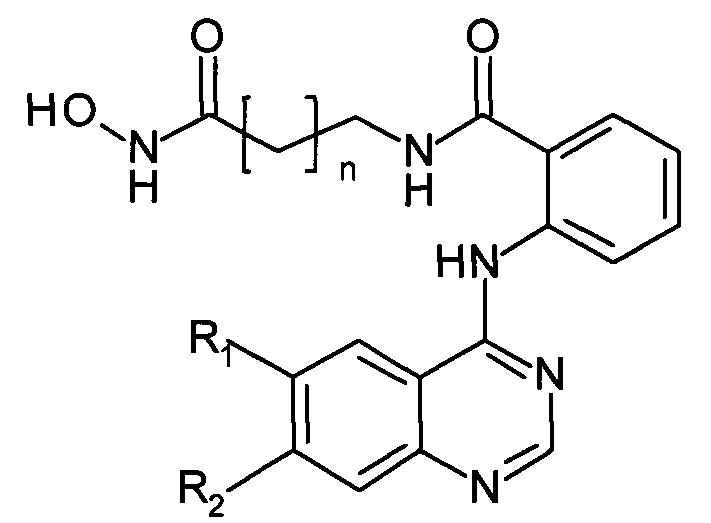
где R1 и R2 независимо друг от друга представляют собой: Н, Cl, Br, ОМе;
n=4, 5
осуществляемый с использованием в качестве исходных соединений производных гидроксамовой кислоты, а именно 2-амино-N-(6-(гидроксиамино)-6-оксогексил)бензамида или 2-амино-N-(7-(гидроксиамино)-7-оксогептил)-бензамида, которые в эквимолярном количестве в виде раствора в органическом растворителе прибавляют к раствору производных 4-хлорхиназолина в идентичном растворителе, выбранных из группы следующих соединений: 4-хлорхиназолин, 4,6-дихлорхиназолин, 6-бром-4-хлорхиназолин, 6,7-диметокси-4-хлорхиназолин, после чего реакционную смесь перемешивают при температуре 20-100°С, охлаждают до комнатной температуры, выпавший осадок отфильтровывают, промывают используемым в реакции растворителем, затем диэтиловым эфиром и сушат.
Способ осуществляют с использованием производных гидроксамовой кислоты, предварительно полученных реакцией изатового ангидрида с аминоэфирами с последующим аминолизом полученных эфиров гидроксиламином.
Оптимально реакция взаимодействия 4-хлорхиназолинов с производными гидроксамовой кислоты проводится в среде диметилформамида при температуре 40-50°С и в течение 20-30 мин.
Предлагаемым способом синтезированы следующие соединения:
N-(6-(гидроксиамино)-6-оксогексил)-2-(хиназолин-4-иламино)бензамид (соединение I-1),
N-(7-(гидроксиамино)-7-оксогептил)-2-(хиназолин-4-иламино)бензамид (соединение I-2),
N-(6-(гидроксиамино)-6-оксогексил)-2-((6-хлорхинозолин-4-ил)амино)бензамид (соединение I-3),
N-(7-(гидроксиамино)-7-оксогептил)-2-((6-хлорхинозолин-4-ил)амино)бензамид (соединение I-4),
2-((6-бромхиназолин-4-ил)амино)-N-(6-(гидроксиамино)-6-оксогексил))бензамид (соединение I-5),
2-((6-бромхинозолин-4-ил)амино)-N-(7-(гидроксиамино)-7-оксогептил)бензамид (соединение I-6),
N-(6-(гидроксиамино)-6-оксогексил)-2-((6,7-диметоксихинозолин-4-ил)амино)бензамид (соединение I-7),
N-(7-(гидроксиамино)-7-оксогептил)-2-((6,7-диметоксихинозолин-4-ил)амино)бензамид (соединение I-8).
Полученные новые соединения общей формулы I могут использоваться в качестве ингибиторов гистондеацетилаз различных изоформ и применяться для лечения, в частности, нейродегенеративных и онкологических заболеваний.
Предлагаемый способ иллюстрируется следующей схемой реакций (Схема 6):
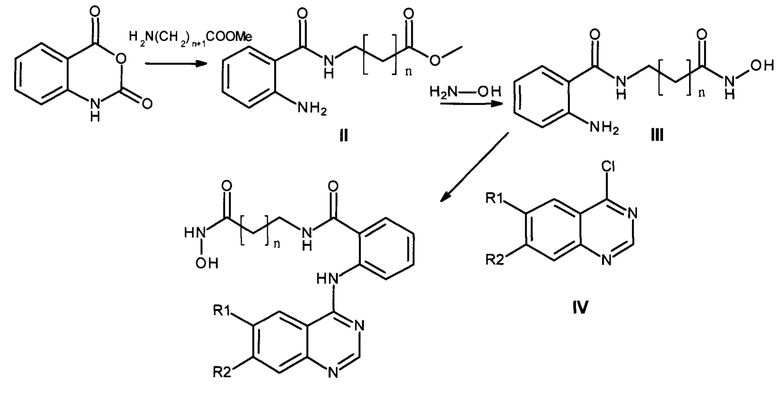
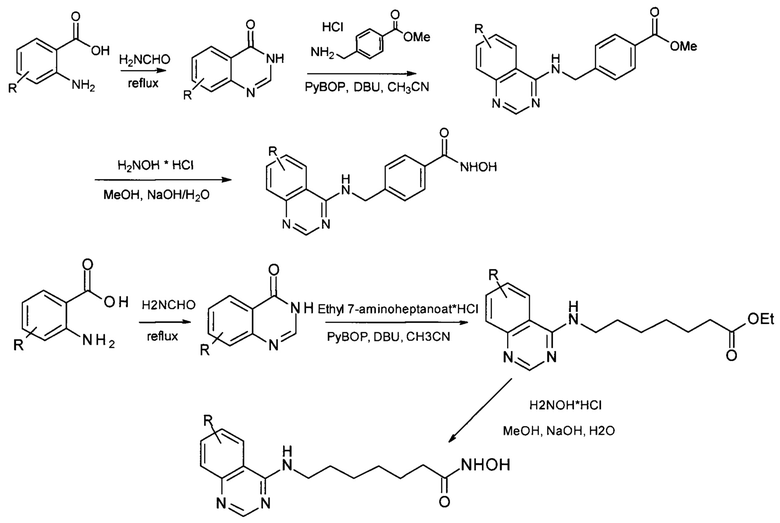
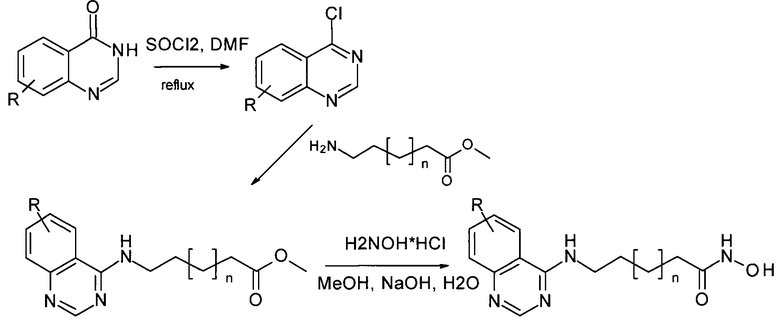
Соединения II, III и IV (Схема 6) могут быть получены любым известным способом, описанным в литературе. Например, соединения II (метил 6-(2-аминобензамидо)гексаноат (II-1) и метил 7-(2-аминобензамидо)гептаноат (II-2)) могут быть получены взаимодействием изатового ангидрида с соответствующим амином в воде, водном спирте, ацетоне или в водном диметилформамиде (Khattab S. et al. Bioorganic and Medicinal Chemistry. - 2015. - vol. 23; No. 13. - P. 3574-3585). Соединения III (2-амино-N-(6-(гидроксиамино)-6-оксогексил)бензамид (III-1) и 2-амино-N-(7-(гидроксиамино)-7-оксогептилил)бензамида (III-2)) могут быть получены любым известным методом, например, обработкой соединения II гидроксиламином в метаноле (Ganeshpurkar А., Kumar D., Singh S. K.. Strategies for the Synthesis of Hydroxamic Acids // Current Organic Synthesis. - 2018. - Vol. 15. - P. 154-165). Соединения IV (производные 4-хлорхиназолина) могут быть получены взаимодействием соответствующих хиназолин-4-онов с хлористым тионилом, хлорокисью фосфора или с другим хлорирующим агентом как, например, описано в статьях (Zhang, Q et al. Bioorganic and Medicinal Chemistry Letters. - 2017. - vol. 27, No 21. - P. 4885 - 4888; Wang Z et al. Bioorganic and Medicinal Chemistry Letters. - 2016. - vol. 26, No 11. - P. 2589-2593; Shi L. et al. Bioorganic and Medicinal Chemistry. - 2014. -vol. 22, No 17. - P. 4735 - 4744; Mphahlele M. et al. Pharmaceuticals. - 2017. - vol. 10, No 4. - Art. No. 87).
Способ получения N-((гидроксиамино)-оксоалкил)-2-(хиназолин-4-иламино)бензамидов формулы:
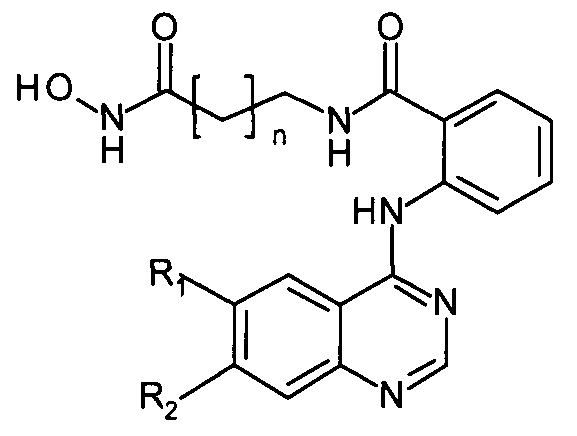
отличается от известных ранее способов синтеза их структурных аналогов тем, что в качестве исходных продуктов в предлагаемом способе используются гидроксамовые кислоты, предварительно полученные реакцией изатового ангидрида с аминоэфирами с дальнейшим гидроксиламинолизом. Полученные гидроксамовые кислоты затем вступают в реакцию с производными 4-хлорхиназолина.
В предлагаемом способе получения N-((гидроксиамино)-оксоалкил)-2-(хиназолин-4-иламино)-бензамидов процесс осуществляется с использованием в качестве исходных соединений производных гидроксамовой кислоты, а именно 2-амино-N-(6-(гидроксиамино)-6-оксогексил)бензамида или 2-амино-N-7-(гидроксиамино)-7-оксогептил)-бензамида в виде их растворов в органическом растворителях, таких как изопропанол, бутанол, диметилформамид, N-метилпирролидон. Производные 4-хлорхиназолина (4-хлорхиназолин, 4,6-дихлорхиназолин, 6-бром-4-хлорхиназолин, 6,7-диметокси-4-хлорхиназолин), вступающие в реакцию с производными гидроксамовой кислоты, вводятся в виде растворов в идентичном органическом растворителе (изопропаноле, бутаноле, диметилформамиде, N-метилпирролидоне). Реакция проводится при температуре от 20°С до 100°С в зависимости от выбранного растворителя. Оптимально осуществление реакции взаимодействия 4-хлорхиназолинов с производными гидроксамовой кислоты в среде диметилформамида при температуре 40-50°С. В ходе реакции целевой продукт выпадает в виде осадка, а окончание реакции контролируется с помощью тонкослойной хроматографии. Время реакции может составлять от 20 минут до 1 часа, что зависит от температуры реакции и используемого растворителя. Целевые продукты выпадают в виде осадка, осадок отфильтровывается, промывается водой, затем диэтиловым эфиром и сушится на воздухе. Выход целевых продуктов близок к количественному (выше 95%). Чистота целевых соединений по данным аналитической высокоэффективной жидкостной хроматорафии 97-99%.
Преимущество предлагаемого способа получения N-((гидроксиамино)-оксоалкил)-2-(хиназолин-4-иламино)-бензамидов перед известными заключается в том, что он позволяет получать большие молекулы с гидроксаматной функцией, и при этом исключается процесс гидроксиламинолиза на последней стадии синтеза, который обычно проходит с затруднениями и приводит к образованию побочных продуктов. В то время как гидроксиламинолиз промежуточных амидоэфиров антраниловой кислоты (соединения II, Схема 6) в предлагаемом способе проходит без затруднений и гидроксамовые кислоты получаются с хорошими выходами и приемлемого качества. Реакция промежуточных гидроксамовых кислот с производными 4-хлорхиназолина протекает быстро, практически количественно, с образованием качественных целевых соединений, легко выделяющихся чистом виде (содержание основного вещества по ВЭЖХ - более 97%) и не требующих дополнительной очистки.
Преимущества заявляемого способа по сравнению со способами, применяемыми при синтезе структурных аналогов, заключаются в эффективности процесса синтеза 4-аминоарильньгх хиназолинов с гидроксамовой кислотой, присоединенной к арильному фрагменту, снижении его трудоемкости и повышении чистоты конечных продуктов
Данный способ позволяет существенно уменьшить количество промежуточных соединений, которые следует выделять или очищать. Получаемые основные промежуточные соединения - 2-амино-N-(ω-(гидроксиамино)-ω-оксоалкил)бензамиды (соединения III-1 и III-2) имеют чистоту выше 95% (по данным ВЭЖХ) и могут использоваться для получения большого числа целевых соединений вышеуказанной структурной формулы.
Аналитическую ВЭЖХ проводили на хроматографе фирмы Shimadzu. Колонка: Grom-Sil 12J ODS-4HE, 5μм, 250*4,6 мм. Условия: линейный градиент АВ: 5% В (0 мин) 100% В (20 мин). А - 0,001% ТФУ в воде, В - 0,001% ТФУ в ацетонитриле. Спектры ESI-MS регистрировали на приборе "Agilent LC/MS 1200" при ионизации пробы электрораспылением в режиме регистрации положительных ионов. Пробы готовили в системе ацетонитрил/вода 1/1, концентрация 2 мг/мл. Условия анализа: поток 1 мл/мин, давление на нибулайзере 20 psi, температура 360°С, скорость потока осушающего газа 9 л/мин, напряжение 3500 В, целевая масса от 100 до 2000. Температуру плавления определяли на приборе марки "Melting Point М-565" (BUCHI). Спектры ЯМР 1Н получены на приборе Bruker АМ-360 (рабочая частота 360,13 Мгц). ТСХ проводили на пластинах Merck TLC Silica gel 60 F254, проявление в УФ и нингидрином.
Ниже приводятся примеры осуществления изобретения.
Пример 1
Синтез метил 6-(2-аминобензамидо)гексаноата (соединение II-1, n=5)
Метиловый эфир 6-аминогексановой кислоты 15 г (83 ммоль) растворяют в 20 мл воды, 10%-ным раствором гидрокарбоната натрия доводят рН раствора до 7-8. Полученный раствор приливают к суспензии 10 г (61 ммоль) изатового ангидрида в 100 мл диметилформамида в плоскодонной колбе объемом 250 мл, полученную смесь перемешивают 2 часа при комнатной температуре. Смесь выливают в 200 мл воды, 10%-ным раствором лимонной кислоты доводят рН раствора до 3-4. Экстрагируют диэтиловым эфиром 2 по 100 мл, органический слой промывают 50 мл воды. Отгоняют растворитель, остаток кристаллизуют из 50 мл гексана. Выход 15.0 г (92%), светло-бежевые кристаллы, т.пл. 53-55°С. 1Н-ЯМР: 8.16 (т, 1Н, J=5.7), 7.46 (дд, 1H, J=7.9, 1.5), 7.15-7.08 (м, 1Н), 6.68 (дд, 1H, J=8.3, 1.2), 6.50 (дд, 1Н, J=8.1, 7.1), 6.36 (с, 2Н), 3.58 (с, 3Н), 3.26-3.13 (м, 2Н), 2.30 (т, 2Н, J=7.4), 1.64-1.42 (м, 4Н), 1.39-1.22 (м, 2Н).
Пример 2
Синтез метил 7-(2-аминобензамидо)гептаноата (соединение II-2, n = 6)
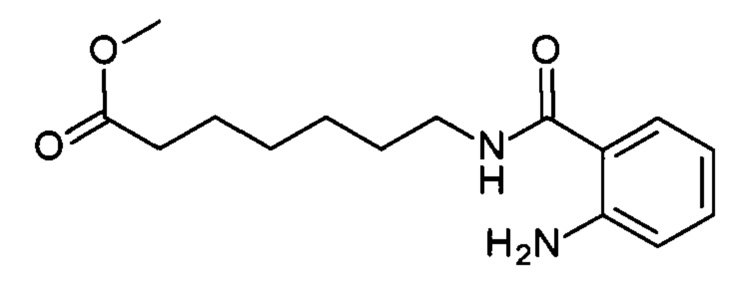
Получают аналогично Примеру 1 из 8.0 г (50 ммоль) метилового эфира 7-аминогептановой кислоты и 6.0 г (37 ммоль) изатового ангидрида. Выход 94%, светло-бежевые кристаллы, т.пл. 48-50°С.
Пример 3
Синтез 2-амино-N-(6-(гидроксиамино)-6-оксогексил)бензамида
(соединение III-1, n=5)
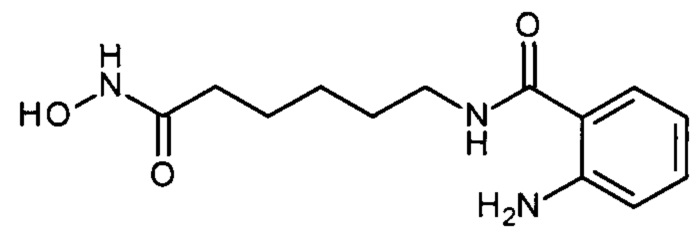
К раствору 2.64 г (0.01 моля) метил 6-((2-аминобензоил)амино)гексаноата в 30 мл метанола прибавляют 30 мл раствора гидроксиламина в метаноле, полученного из 1.15 г (0.05 моля) натрия и 2.10 г (0.03 моля) хлоргидрата гидроксиламина. Перемешивают реакционную смесь 6 часов при комнатной температуре. Отгоняют метанол в вакууме, добавляют 20 мл воды, подкисляют 5%-ной лимонной кислотой до рН 4, отфильтровывают осадок, промывают 10 мл воды. Выход 2.20 г (83.3%), бежевые кристаллы, т.пл. 125-126°С. 1Н ЯМР (ДМСО-d6, δ м.д., J, Гц) 10.28 (с, 1Н, NH-OH), 8.60 (с, 1H,NH-OH) 8.11 (т, 1Н, CH2NH,/=5.6), 7.44 (дд, 1Н, Ar, J=8.0, 1.5), 7.11 (дд, 1H, Ar, J=8.5, 7.1), 6.67 (дд, 1H, Ar, J=8.2, 1.2), 6.49 (дд, 1H, Ar, J=8.0, 7.1), 6.30 (д, 1Н, NH, J=7.4), 3.19 (тд, 2Н, NCH2, J=6.9, 4.7,), 1.95 (т, 2Н, СОСН2, J=7.4), 1.51 (м, 4Н, CH2, 1.28 (п, 2Н, CH2, J=7.9). ESI-MS, m/z 553.1 [2M+Na]+, 266.1 [М]+.
Пример 4
Синтез 2-амино-N-(7-(гидроксиамино)-7-оксогептил)бензамида
(соединение III-2, n=6)
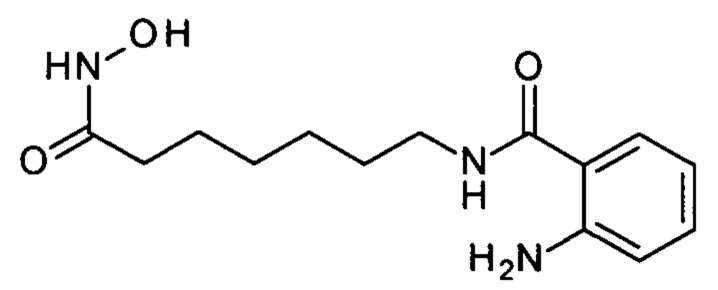
Получают аналогично Примеру 3 из 2.78 г (0.01 моля) метил 7-((2-аминобензоил)амино)гептаноата. Выход 2.41 г (86.3%), бежевые кристаллы, т.пл. 108-110°С. 1Н ЯМР (ДМСО-d6, δ м.д., J, Гц) 10.32 (с, 1H), 8.65 (с, 1Н), 8.15 (т, J=5.6 Hz, 1Н), 7.44 (дд, J=8.0, 1.5 Hz, 1H), 7.17-7.05 (м, 1H), 6.67 (дд, J=8.3, 1.2 Hz, 1H), 6.55-6.44 (м, 1H), 6.31 (с, 2H), 3.18 (т, J=7.2 Hz, 2H), 1.94 (т, J=7.3 Hz, 2H), 1.55-1.39 (м, 4H), 1.35-1.19 (м, 4H). ESI-MS, m/z 280.2 [M]+.
Пример 5
Общая методика синтеза производных 4-хлорхиназолина (соединения IV)
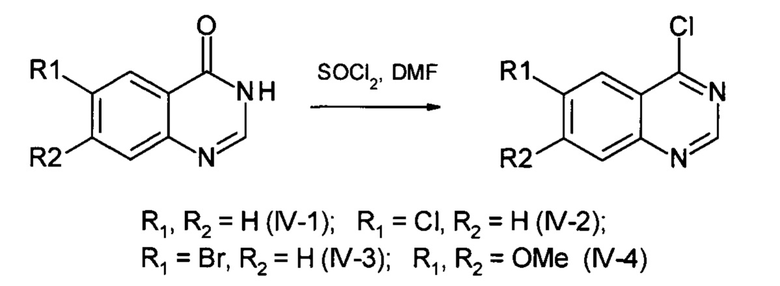
Синтез проводится аналогично описанному в статье (Zhang, Q et al. Bioorganic and Medicinal Chemistry Letters. - 2017. - vol. 27, No 21. - P. 4885-4888). Раствор соответствующего хиназолин-4-она (5 ммоль) кипятят в 10 мл хлористого тионила с 0.05 мл ДМФА в течение 1 часа. Охлаждают до комнатной температуры, добавляют 50 мл диэтилового эфира, декантируют раствор с выпавшего хлоргидрата 4-хлорхиназолина. Добавляют 30 мл хлороформа, промывают 10 мл воды, 2×10 мл 5%-ного раствора бикарбоната натрия. Органический слой сушат над сульфатом натрия, отгоняют досуха. Получают белое кристаллическое вещество. Таким способом получены соединения IV(1-4). Полученные продукты используют далее без дополнительной очистки.
Пример 6
N-(6-(гидроксиамино)-6-оксогексил)-2-(хиназолин-4-иламино)бензамид (соединение I-1)
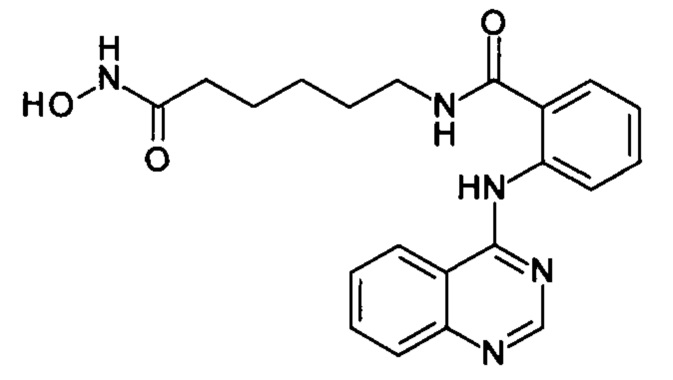
Вариант 1. К раствору 164 мг (1 ммоль) 4-хлорхиназолина (IV-1) в 5 мл диметилформамида прибавляют раствор 265 мг (1 ммоль) 2-амино-N-6-(гидроксиамино)-6-оксогексил)бензамида в 5 мл диметилформамида. Реакционную смесь перемешивают при температуре 40-50°С в течение 20-30 мин. Окончание реакции контролируют с помощью тонкослойной хроматографии (элюент хлороформ-метанол 10:1), Реакционную смесь охлаждают до комнатной температуры, осадок отфильтровывают, промывают 5 мл диметилформамида, 2×10 мл диэтилового эфира. Сушат на воздухе. Выход 384 мг (97.6%), светло-бежевые кристаллы, т.пл. 180.8-187.5°С. 1Н ЯМР (ДМСО-d6, δ м.д., J, Гц) 12.60 (с, 1Н, NH-OH), 10.33 (с, 1H, NH-OH), 8.98 (д, 1Н, NH, J=8.4), 8.90 (т, 1Н, CH2NH, J=5.6), 8.72 (с, 1H, Ar), 8.66 (с, 1Н, Ar), 8.17 (д, 1Н, Ar, J=8.2), 7.95-7.81 (м, 3Н, Ar), 7.75 (т, 1Н, Ar, J=7.5), 7.60 (т, 1Н, Ar, J=7.4), 7.19 (тд, 1Н, Ar, J=7.6, 1.2), 1.94 (т, 2Н, СОСН2, J=7.3), 1.62-1.45 (м, 4Н, CH2), 1.38-1.21 (м, 2Н, CH2).
Вариант 2. К раствору 164 мг (1 ммоль) 4-хлорхиназолина (IV) в 5 мл диметилформамида прибавляют раствор 265 мг (1 ммоль) 2-амино-N-6-(гидроксиамино)-6-оксогексил)бензамида в 7 мл изопропанола. Реакционную смесь кипятят в течение 1 часа. Окончание реакции контролируют с помощью тонкослойной хроматографии (элюент хлороформ-метанол 10:1), Реакционную смесь охлаждают до комнатной температуры, осадок отфильтровывают, промывают 5 мл изопропанола, 2×10 мл диэтилового эфира. Сушат на воздухе. Выход 368 мг (95.4%), светло-бежевые кристаллы, т.пл. 181.3-186.9°С. Спектральные характеристики аналогичны приведенным в Варианте 1.
Пример 7
N-(7-(гидроксиамино)-7-оксогептил)-2-(хиназолин-4-иламино)бензамид
(соединение I-2)
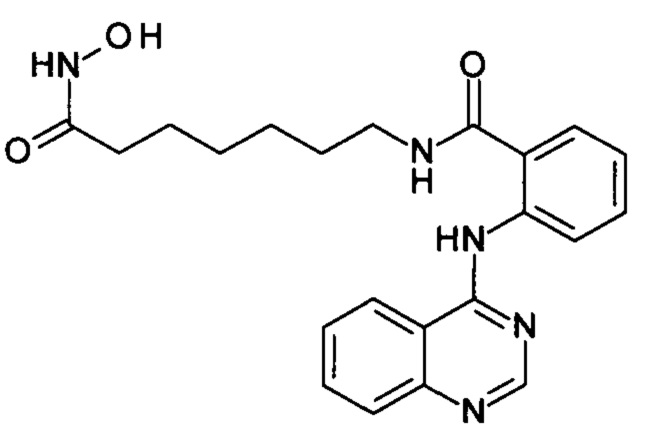
К раствору 164 мг (1 ммоль) 4-хлорхиназолина (IV-1) в 5 мл диметилформамида прибавляют раствор 279 мг (1 ммоль) 2-амино-N-(7-(гидроксиамино)-7-оксогептил)бензамида в 5 мл диметилформамида. Реакционную смесь перемешивают при температуре 20°С в течение 1 часа. Окончание реакции контролируют с помощью тонкослойной хроматографии (элюент хлороформ-метанол 10:1), Выпавший осадок отфильтровывают, промывают 5 мл диметилформамида, 2×10 мл диэтилового эфира. Сушат на воздухе. Выход 97.2%. Т.пл. 193.2°С (разл.). 1Н ЯМР (ДМСО-d6, δ м.д., J, Гц) 10.42 (с, 1Н), 9.31 (с, 1H), 8.88 (д, J=5.5, 1H), 8.76 (д, J=8.0, 1Н), 8.02-7.84 (м, 4Н), 7.78-7.72 (м, 1Н), 7.65-7.60 (м, 1Н), 7.58-7.50 (м, 1Н), 2.72 (м, 2Н), 1.97 (м, 2Н), 1.62-1.39 (м, 4Н), 1.26 (м, 4Н).
Пример 8
N-(6-(гидроксиамино)-6-оксогексил)-2-((6-хлорхинозолин-4-ил)амино)бензамид
(соединение I-3)
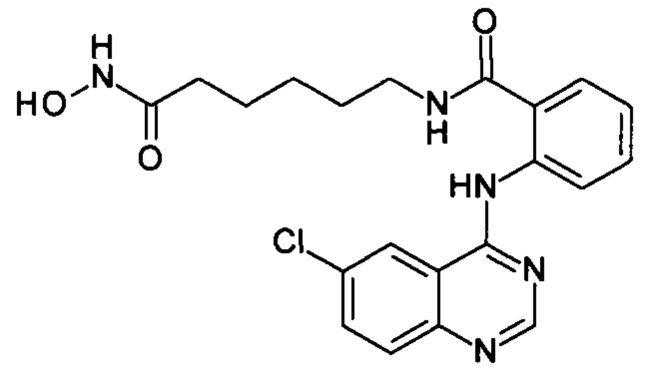
Получают аналогично Примеру 6, Вариант 1. К раствору 205 мг (1 ммоль) 4,6-дихлорхиназолина (IV-2) в 5 мл диметилформамида прибавляют раствор 265 мг (1 ммоль) 2-амино-N-(6-(гидроксиамино)-6-оксогексил)бензамида в 5 мл диметилформамида. Реакционную смесь перемешивают при температуре 40°С в течение 20-30 мин. Окончание реакции контролируют с помощью тонкослойной хроматографии (элюент хлороформ-метанол 10:1), Реакционную смесь охлаждают до комнатной температуры, осадок отфильтровывают, промывают 5 мл диметилформамида, 2×10 мл диэтилового эфира. Сушат на воздухе. Выход 96.8%. Т.пл. 209.9-210.4°С. 1Н ЯМР (ДМСО-d6, δ м.д., J, Гц) 12.59 (s, 1H), 10.78 (s, 1Н), 9.30 (с, 1Н), 8.69 (с, 1Н), 8.65 (д, J=2.4, 1Н), 8.34 (д, J=8.2, 1H), 8.16 (д, J=7.9, 1H), 8.06-7.75 (м, 4Н), 7.71-7.52 (м, Н), 2.73 (м, J=5.8, 2Н), 1.95 (т, J=7.3, 2Н), 1.51 (дп, J=14.7, 7.4, 4Н), 1.26 (м, 2Н).
Пример 9
N-(7-(гидроксиамино)-7-оксогептил)-2-((6-хлорхинозолин-4-ил)амино)бензамид
(соединение I-4)
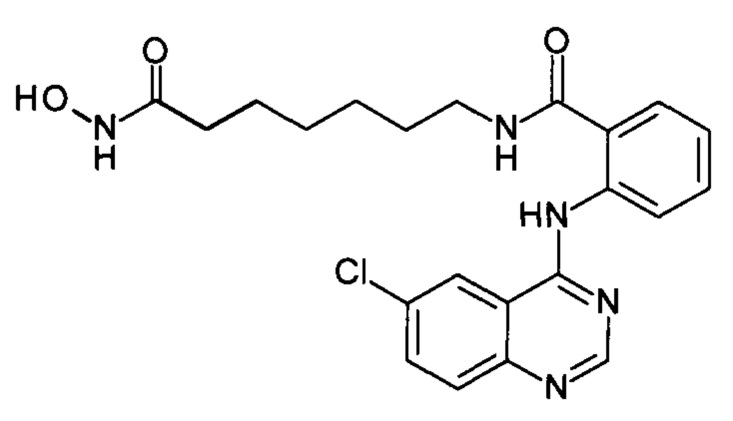
Получают аналогично Примеру 8. и 2. К раствору 205 мг (1 ммоль) 4,6-дихлорхиназолина (IV-2) в 5 мл диметилформамида прибавляют раствор 279 мг (1 ммоль) 2-амино-N-(7-(гидроксиамино)-7-оксогептил)бензамида в 5 мл диметилформамида. Реакционную смесь перемешивают при температуре 40°С в течение 20-30 мин. Окончание реакции контролируют с помощью тонкослойной хроматографии (элюент хлороформ-метанол 10:1), Реакционную смесь охлаждают до комнатной температуры, осадок отфильтровывают, промывают 5 мл диметилформамида, 2×10 мл диэтилового эфира. Сушат на воздухе. Выход 97.4%. Т.пл. 213.4-215.1°С. 1Н ЯМР (ДМСО-d6, δ м.д., J, Гц): 12.59 (s, 1Н), 10.78 (s, 1H), 9.30 (s, 1H), 8.69 (s, 1H), 8.66 (d, J=2.4, 1H), 8.25 (d, J=53.4, 2Н), 8.05-7.75 (m, 4Н), 7.72-7.50 (m, Н), 2.73 (м, 2Н), 1.94 (t, J=7.3, 2Н), 1.50 (р, J=7.3, 4Н), 1.25 (м, 4Н).
Пример 10
2-((6-бромхиназолин-4-ил)амино)-N-(6-(гидроксиамино)-6-оксогексил))бензамид
(соединение I-5)
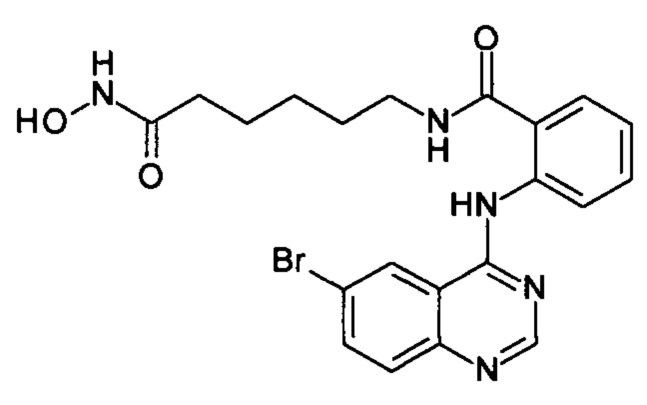
К раствору 243 мг (1 ммоль) 6-бром-4-хлорхиназолина (IV-3) в 8 мл бутанола прибавляют раствор 265 мг (1 ммоль) 2-амино-N-(6-(гидроксиамино)-6-оксогексил)бензамида в 5 мл бутанола. Реакционную смесь перемешивают при температуре 80°С в течение 40 мин. Окончание реакции контролируют с помощью тонкослойной хроматографии (элюент хлороформ-метанол 10:1), Реакционную смесь охлаждают до комнатной температуры, осадок отфильтровывают, промывают 2×5 мл бутанола, 2×10 мл диэтилового эфира. Сушат на воздухе. Выход 95.5%. Т.пл. 224.8-226.1°С. 1Н ЯМР (ДМСО-d6, δ м.д., J, Гц): 12.48 (s, 1Н), 10.36 (s, 1H), 8.88 (s, 1Н), 8.80-8.68 (m, 2Н), 8.28-8.12 (т, 2Н), 7.91-7.76 (т, 2Н), 7.64 (t, J=7.8, 1H), 7.41 (t, J=7.6, 1Н), 3.18 (q, J=6.6, 2H), 1.90 (t ,J=7.4, 2H), 1.44 (p, J=8.2,4H), 1.33-1.11 (m, 2H).
Пример 11
2-((6-бромхинозолин-4-ил)амино)-N-(7-(гидроксиамино)-7-оксогептил)бензамид
(соединение I-6)
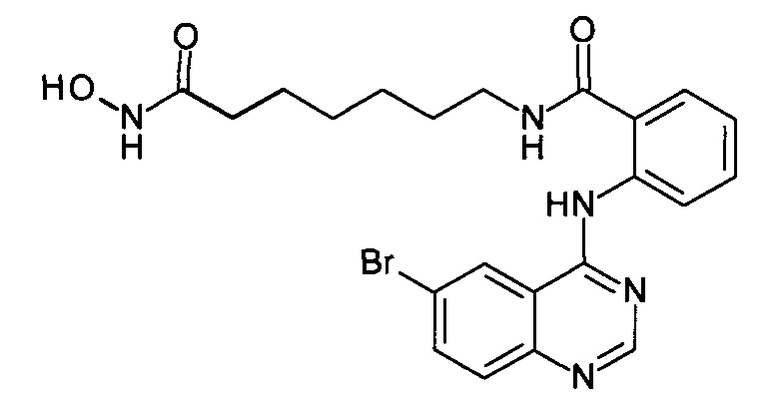
Получают аналогично Примеру 1. К раствору 243 мг (1 ммоль) 6-бром-4-хлорхиназолина (IV-3) в 8 мл диметилформамида прибавляют раствор 279 мг (1 ммоль) 2-амино-N-(7-(гидроксиамино)-7-оксогептил)бензамида в 5 мл диметилформамида. Реакционную смесь перемешивают при температуре 50°С в течение 20-30 мин. Окончание реакции контролируют с помощью тонкослойной хроматографии (элюент хлороформ-метанол 10:1), Реакционную смесь охлаждают до комнатной температуры, осадок отфильтровывают, промывают 5 мл диметилформамида, 2×10 мл диэтилового эфира. Сушат на воздухе. Выход 96.9%. Т.пл. 225.6-227.8°С. 1Н ЯМР (ДМСО-d6, δ м.д., J, Гц): 12.59 (s, 1Н), 10.78 (s, 1H), 10.41 (s, 1Н), 8.81 (d, J=2.3, 1H), 8.69 (s, 1H), 8.40-7.47 (m, 6H), 2.73 (m, 2H), 1.94 (t, J=7.3, 2H), 1.50 (m, 4H), 1.26 (m, 4H).
Пример 12
N-(6-(гидроксиамино)-6-оксогексил)-2-((6,7-диметоксихинозолин-4-ил)амино)бензамид
(соединение I-7)
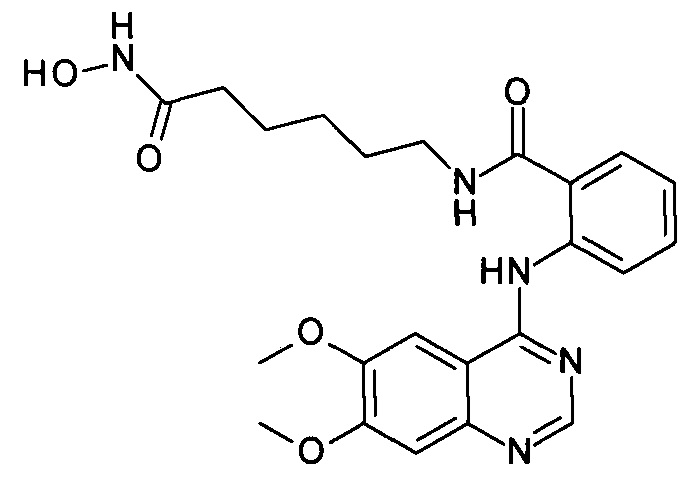
К раствору 164 мг (1 ммоль) 6,7-диметокси-4-хлорхиназолина (IV-4) в 8 мл диметилформамида прибавляют раствор 265 мг (1 ммоль) 2-амино-N-(6-(гидроксиамино)-6-оксогексил)бензамида в 5 мл диметилформамида. Реакционную смесь перемешивают при температуре 50°С в течение 20-30 мин. Окончание реакции контролируют с помощью тонкослойной хроматографии (элюент хлороформ-метанол 10:1), Реакционную смесь охлаждают до комнатной температуры, осадок отфильтровывают, промывают 5 мл диметилформамида, 2×10 мл диэтилового эфира. Сушат на воздухе. Выход 97.9%. Т.пл. 267.0-271.1°С. 1Н ЯМР (ДМСО-d6, δ м.д., J, Гц): 10.39 (s, 1H), 10.34 (s, 1Н), 8.78 (s, 1Н), 8.72 (t, J=5.8 1H), 8.06-7.68 (m, 5H), 7.68-7.53 (t, 1H), 7.39 (d, J=14.7, 1H), 4.07-3.93 (s, 6H), 2.74 (m, 2H), 1.95 (t, J=7.3 2H), 1.50 (m, 4H), 1.34-1.22 (m, 2H).
Пример 13
N-(7-(гидроксиамино)-7-оксогептил)-2-((6,7-диметоксихинозолин-4-ил)амино)бензамид
(соединение I-8)
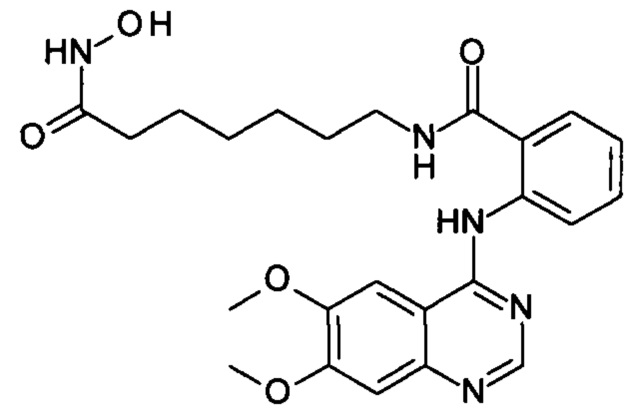
К раствору 164 мг (1 ммоль) 6,7-диметокси-4-хлорхиназолина (IV-4) в 8 мл диметилформамида прибавляют раствор 265 мг (1 ммоль) 2-амино-N-(7-(гидроксиамино)-7-оксогептил)бензамида в 5 мл диметилформамида. Реакционную смесь перемешивают при температуре 50°С в течение 20-30 мин. Окончание реакции контролируют с помощью тонкослойной хроматографии (элюент хлороформ-метанол 10:1), Реакционную смесь охлаждают до комнатной температуры, осадок отфильтровывают, промывают 5 мл диметилформамида, 2×10 мл диэтилового эфира. Сушат на воздухе. Выход 97.1%. Т.пл. 250.0°С (разл.). 1Н ЯМР (ДМСО-d6, δ м.д., J, Гц): 12.22 (s, 1Н), 10.35 (s, 1Н), 8.78 (s, 1Н), 8.68 (t, J=5.7, 1Н), 8.00 (d, J=10.3, 1H), 7.75 (d, J=7.8, 1H), 7.63 (t, J=1.1, 1H), 7.42 (t, J=7.6, 1H), 7.35 (s, 1H), 4.01 (s, 6H), 3.16 (t, J=6.5 Hz, 2H), 1.89 (t, J=7.3, 2H), 1.35 (m, 4H), 1.14 (m, 4H).
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения гидроксамовых кислот, производных 2-арил-2,3-дигидрохиназолин-4(1Н)-онов | 2020 |
|
RU2744750C1 |
| Гидроксамовые кислоты, производные 4-аминохиназолин-7-карбоновой кислоты как ингибиторы гистондеацетилазы и способ их получения | 2021 |
|
RU2779981C1 |
| Производные 4-((арил)(метил)амино)хиназолин-7-карбоновой кислоты с противоопухолевым действием и способ их получения | 2024 |
|
RU2834688C1 |
| Производные 3-гидроксихиназолин-4(3Н)-она в качестве ингибиторов гистондеацетилазы и способ их получения | 2020 |
|
RU2740503C1 |
| Гидроксамовые кислоты, производные 4-аминохиназолина, обладающие противоопухолевой активностью | 2022 |
|
RU2802463C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ 2-(7-ХЛОР-1,8-НАФТИРИДИН-2-ИЛ)-3-(5-МЕТИЛ-2-ОКСОГЕКСИЛ)-1-ИЗОИНДОЛИНОНА | 2003 |
|
RU2318823C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ АЛКИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ГИСТОНДЕАЦЕТИЛАЗЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2016 |
|
RU2683022C1 |
| НОВЫЕ 4-АМИНО-N-ГИДРОКСИБЕНЗАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ HDAC ДЛЯ ЛЕЧЕНИЯ РАКА | 2012 |
|
RU2591190C2 |
| МОДУЛИРУЮЩИЕ JAK КИНАЗУ ХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2529019C2 |
| 2,3,5-ТРИЗАМЕЩЕННЫЕ ПИРРОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ (ВАРИАНТЫ) | 2010 |
|
RU2549885C2 |
Изобретение относится к способу получения производных N-((гидроксиамино)-оксоалкил)-2-(хиназолин-4-иламино)-бензамидов указанной ниже общей формулы, которые могут найти применение в лечении злокачественных новообразований. В общей формуле R1 и R2 независимо друг от друга представляют собой H, Cl, Br, OMe. 2 з.п. ф-лы, 13 пр.
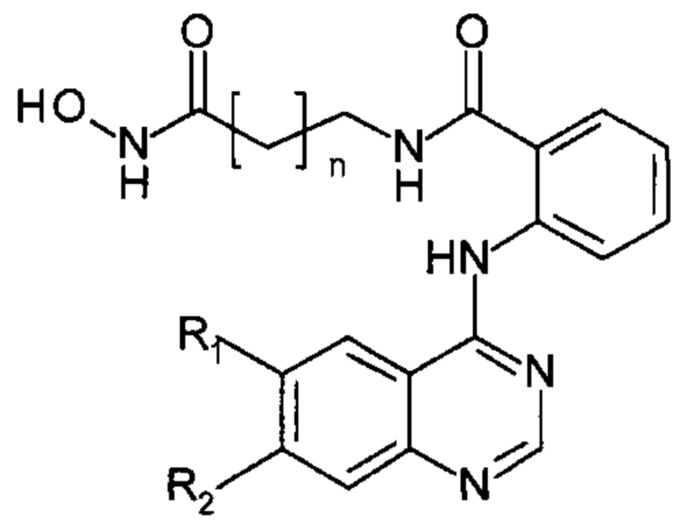
1. Способ получения производных N-((гидроксиамино)-оксоалкил)-2-(хиназолин-4-иламино)-бензамидов общей формулы
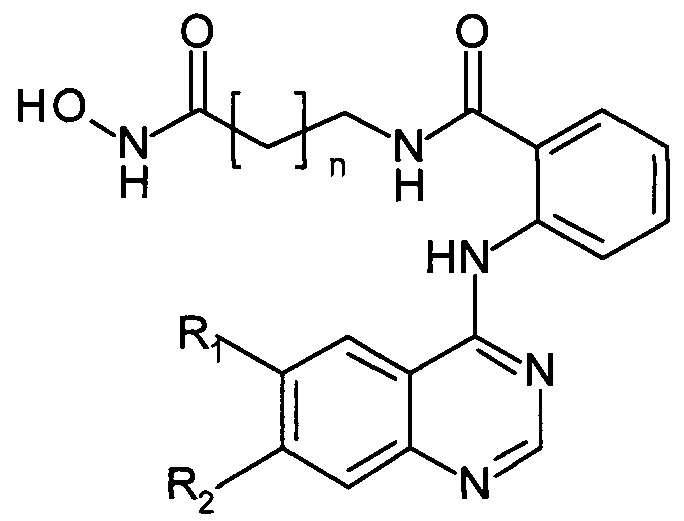 ,
,
где R1 и R2 независимо друг от друга представляют собой Н, Cl, Br, ОМе;
n=4, 5, осуществляемый с использованием в качестве исходных соединений производных гидроксамовой кислоты, а именно 2-амино-N-(6-(гидроксиамино)-6-оксогексил)бензамида или 2-амино-N-(7-(гидроксиамино)-7-оксогептил)бензамида, которые в эквимолярном количестве в виде раствора в органическом растворителе прибавляют к идентичному раствору производных 4-хлорхиназолина, выбранных из группы следующих соединений: 4-хлорхиназолин, 4,6-дихлорхиназолин, 6-бром-4-хлорхиназолин, 6,7-диметокси-4-хлорхиназолин, после чего реакционную смесь перемешивают при температуре 20-100°С, охлаждают до комнатной температуры, выпавший осадок отфильтровывают, промывают используемым в реакции растворителем, затем диэтиловым эфиром и сушат.
2. Способ по п. 1, осуществляемый с использованием производных гидроксамовой кислоты, предварительно полученных реакцией изатового ангидрида с аминоэфирами с последующим аминолизом полученных эфиров гидроксиламином.
3. Способ по п. 1, оптимально осуществляемый реакцией взаимодействия 4-хлорхиназолинов с производными гидроксамовой кислоты, проводимый в среде диметилформамида при температуре 40-50°С.
| A.V.KOLOTAEV et al, Synthesis of hydroxamic acids whith quinazoline moiety, BOOK OF ABSTRACTS "ADVANCES IN SYNTHESIS AND COMPLEXING", ISBN 978-5-209-093947-7, 22-26 April 2019, V.1, p.166 | |||
| J.HAO et al, Research progress in quinazoline derivatives as multi-target tyrosine kinase inhibitors, HETEROCYCL COMMUN, 2018, V.24, p.1-10 | |||
| X.Zhang et al, The |

