Настоящее изобретение относится к производным 4-аминохиназолин-7-карбоновой кислоты и касается непосредственно новых производных 4-аминохиназолин-7-карбоновой кислоты, имеющих в составе гидроксамовую кислоту, которые могут использоваться в медицине для лечения ряда заболеваний посредством ингибирования гистоновых деацетилаз, например, при лечении онкологических и нейродегенеративных заболеваний.
Как известно, гистондеацетилазы (HDAC) - это ферменты, катализирующие удаление ацетильной группы ε-N-ацетил-лизина гистонов. HDAC играют важную роль в регуляции экспрессии генов, так как они модифицируют гистоны и изменяют конформацию хроматина. Поэтому HDAC является важной эпигенетической мишенью при терапии рака, а ингибиторы HDAC демонстрируют успешную картину как цитотоксические агенты. Большинство ингибиторов HDAC имеют трехкомпонентную структуру, состоящую из цинк-связывающего участка, линкера, способного занимать канал фермента, и фрагмента, взаимодействующего с аминокислотными остатками у входа в активный центр HDAC. Ингибиторы классических деацетилаз функционируют путем связывания иона цинка в активном центре фермента и, таким образом, инактивируют систему смены зарядов. [Eckschlager, Т.; Plch, J.; Stiborova, М.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414.]
Известно, что эффективными ингибиторами гистондеацетилаз (HDAC) являются производные гидроксамовой кислоты, например вориностат (SAHA), панобиностат (LBH589) и белиностат. ни одобрены FDA USA для лечения Т-клеточной лимфомы кожи (CTCL) и множественной миеломы [Mottamal М, et al. Molecules. 2015; 20(3): 898-941].
Одной из перспективных стратегий в создании новых фармацевтических препаратов в настоящее время является проектирование и синтез гибридных соединений, состоящих из двух или более различных биоактивных фрагментов, и действующих через активацию/блокирование нескольких мишеней. Совмещение двух активных групп в одной молекуле может приводить к более выраженному терапевтическому эффекту, по сравнению с индивидуальными компонентами при комбинированном применении.
Известно, что хиназолин является важным биоактивным фармакологическим фрагментом. Хиназолиновый цикл присутствует как в различных природных соединениях, так и в молекулах многих лекарственных препаратов. Объединение в одной молекуле хиназолиновой и гидроксамовой фармакофорных групп позволяет создавать новые перспективные биоактивные соединения, что подтверждается многочисленными примерами [Osipov V.N., Khachatryan D.S., Balaev A.N. Biologically active quinazoline-based hydroxamic acids. Med Chem Res (2020)].
Патентные публикации [WO 009063054, A61K 1/517, 2009; WO 008033749, A61K 1/517, 2008; WO 018005799, A61K 1/517, 2018; US 008221132, A61K 1/517, 2008; KR 01964810, A61K 1/517, 2019; US 2018098990, A61K 1/517, 2018] каждая, раскрывают ряд соединений, содержащих хиназолиновый цикл и гидроксамовую кислоту, применяемых в качестве бифункциональных ингибиторов тирозинкиназ и гистондеацетилаз.
Одним из возможных направлений присоединения гидроксаматной группы к прозводному хиназолина является 7-е положение хиназолинового цикла.
Известны примеры присоединения гидроксамовой кислоты с помощью линкера через кислород по 7-му положению хиназолинового цикла. Описанные в патентах [US 008221132, A61K 1/40, 2008; US 015284340, C07D 39/94] соединения, заявленны как ингибиторы гистондеацетилаз:
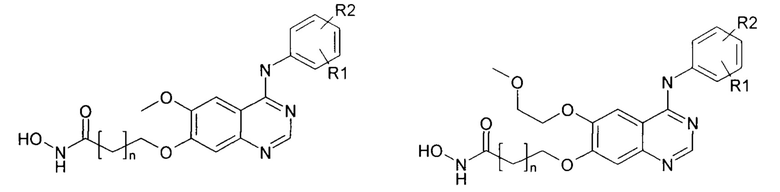
где R1=H, R2=3-этинил; R1=3-С1, R2=4-F; n=3-5
Известные примеры подобных соединений, заявленных как ингибиторы гистондеацетилаз, представлены в патенте [US 2019322643, А61Р 35/00]:
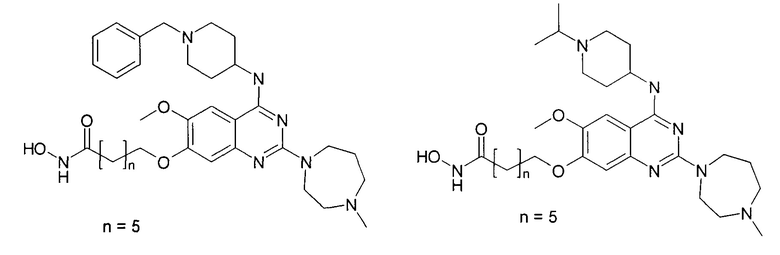
Известны примеры присоединения углеводородного линкера по 7-му положению хиназолинового цикла, описанные в патентах [US 2013267542, A61K 31/517; US 2015196563; RU 2629947, C07D 239/90]:
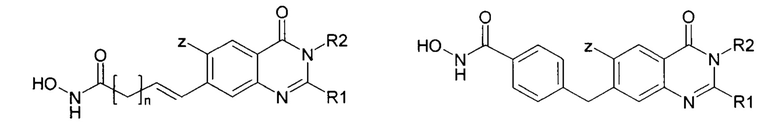
Известен пример производных амидов производных 4-аминохиназолин-7-карбоновой кислоты, заявленных как антитромботические средства в патенте [US 2009181958, A61K 31/47]:
Однако, гидроксамовые кислоты, производные 4-аминохиназолин-7-карбоновой кислоты неизвестны, как и неизвестны способы присоединения гидроксаматной функции через амидную группу в 7-м положении хиназолинового цикла.
Целью предлагаемого изобретения является расширение ассортимента эффективных препаратов, которые могут использоваться в качестве ингибиторов гистондеацетилаз различных изоформ и применяться для лечения онкологических, нейродегенеративных и других заболеваний, а также описание эффективного и промышленно-осуществимого способа получения таких препаратов.
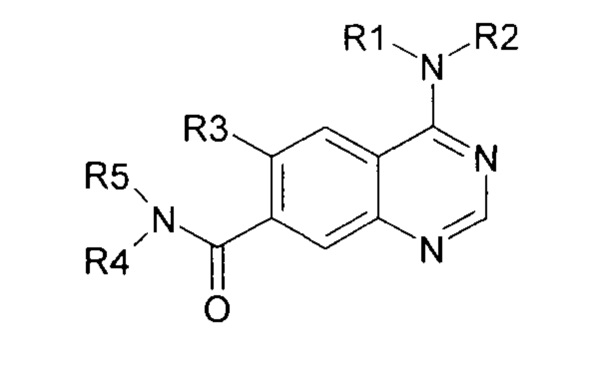
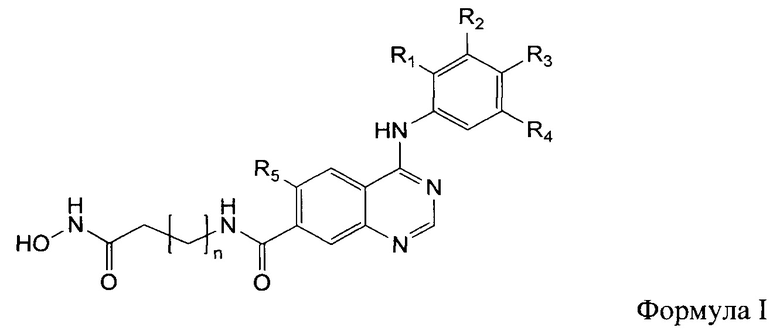
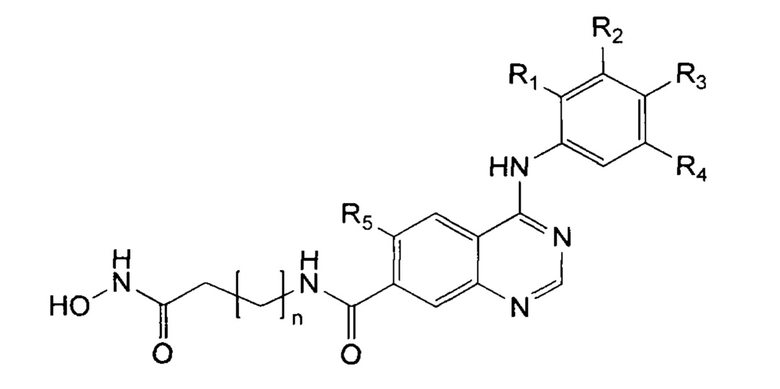
С этой целью предлагаются новые гидроксамовые кислоты, производные 4-аминохиназолин-7-карбоновой кислоты в качестве ингибиторов гистондеацетилаз, имеющие следующую общую формулу:

Формула I
где R1, R2, R3 R4 независимо друг от друга представляют собой водород, галоген, метил, ОМе, этин, а также R2, R3=-ОСН2СН2О-; R5 представляет собой водород или галоген; п=4, 5.
Также заявляется способ получения новых гидроксамовых кислот, производных 4-аминохиназолин-7-карбоновой кислоты, общей формулы I, осуществляемый по следующей схеме: производные метил 4-гидроксихиназолин-7-карбоксилата, получаемые циклизацией производных диметил-2-аминотерефталата в формамиде, взаимодействием с фосфорилхлоридом в присутствии диэтиланилина переводятся в производные метил 4-хлорхиназолин-7-карбоксилата. которые при взаимодействии с замещенными анилинами в диметилфорамиде при комнатной температуре превращаются в производные метил 4-((фенил)амино)хиназолин-7-карбоксилатов, последние подвергаются щелочному гидролизу превращаясь в соответствующие производные 4-[(арил)амино]хиназолин-7-карбоновых кислот, далее к последним присоединяются метиловые эфиры 6-аминогексановой или 7-аминогептановой кислот с использованием 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний тетрафторбората (TBTU) в качестве конденсирующего агента и триметиламина в качестве основания, полученные соединения затем подвергают аминолизу 10-ти кратным мольным избытком гидроксиламина в безводном метаноле в присутствии 3-х эквивалентов 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU), осуществляемому при комнатной температуре в течение 20-26 часов до образования целевого продукта, выделяемого упариванием реакционной массы в вакууме, нейтрализацией ледяной уксусной кислотой до рН 5, фильтрацией с последующей промывкой водой.
Предлагаемый способ иллюстрируется следующей схемой:
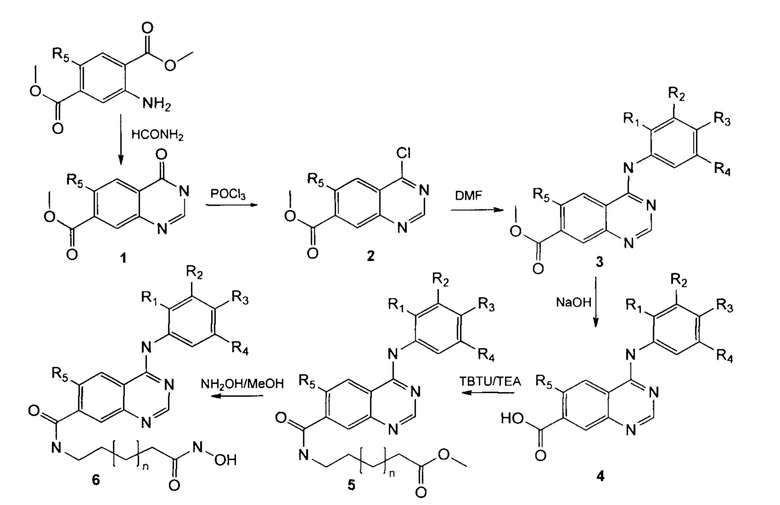
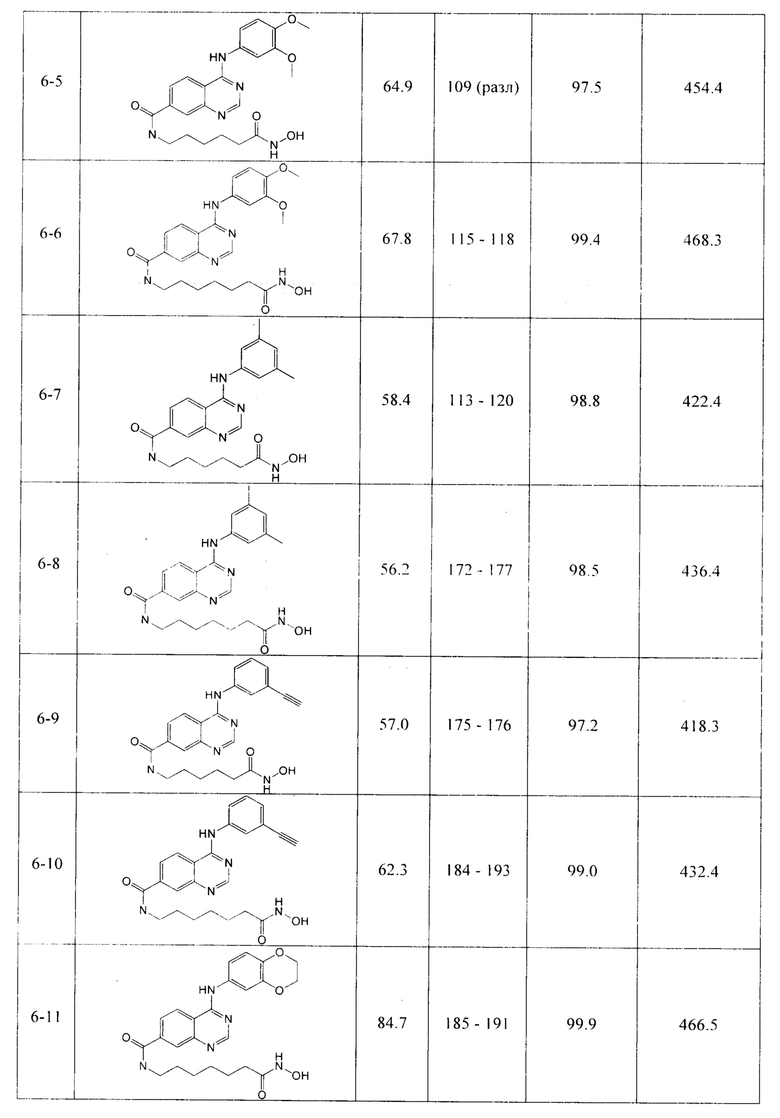
Предлагаемым способом синтезированы, в частности, следующие новые соединения:
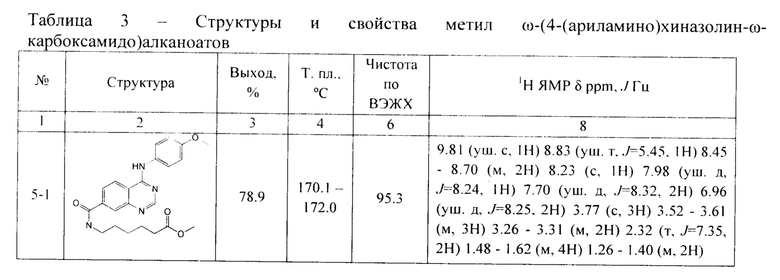
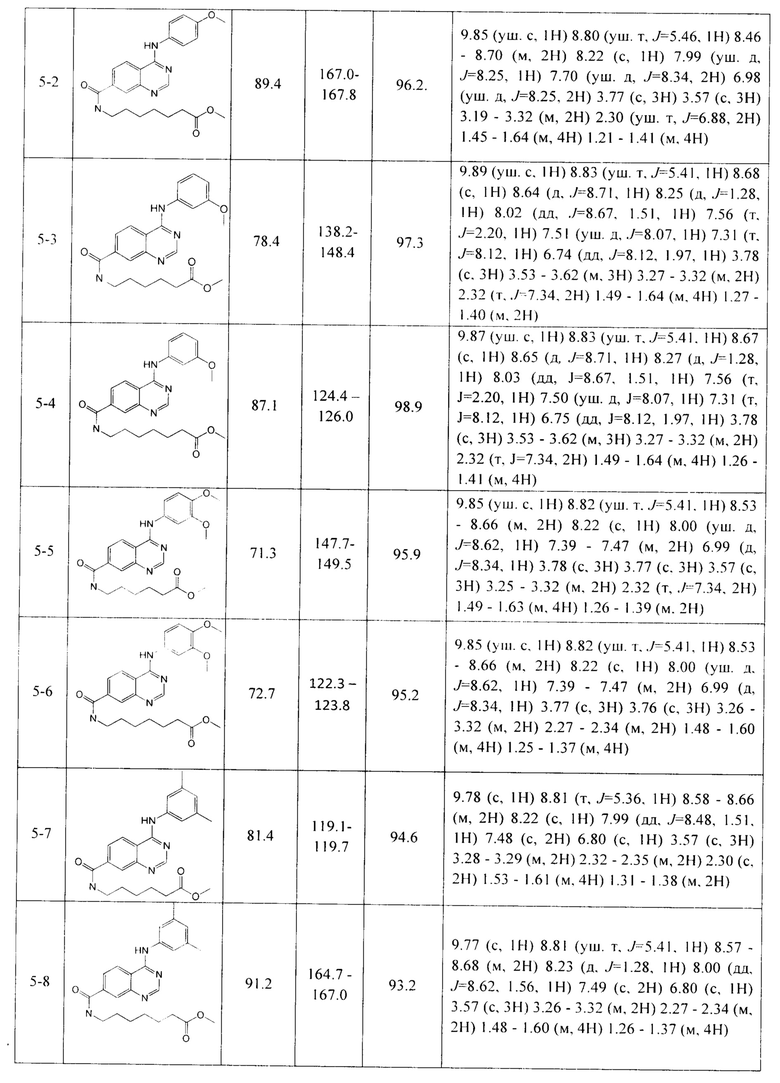
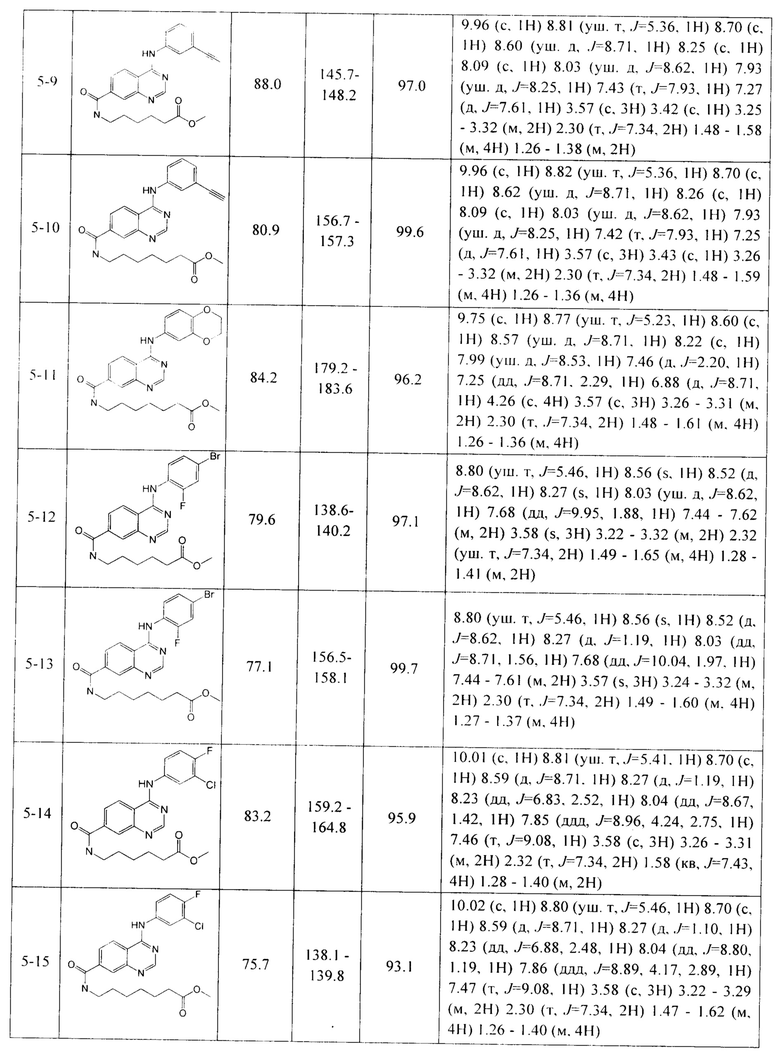
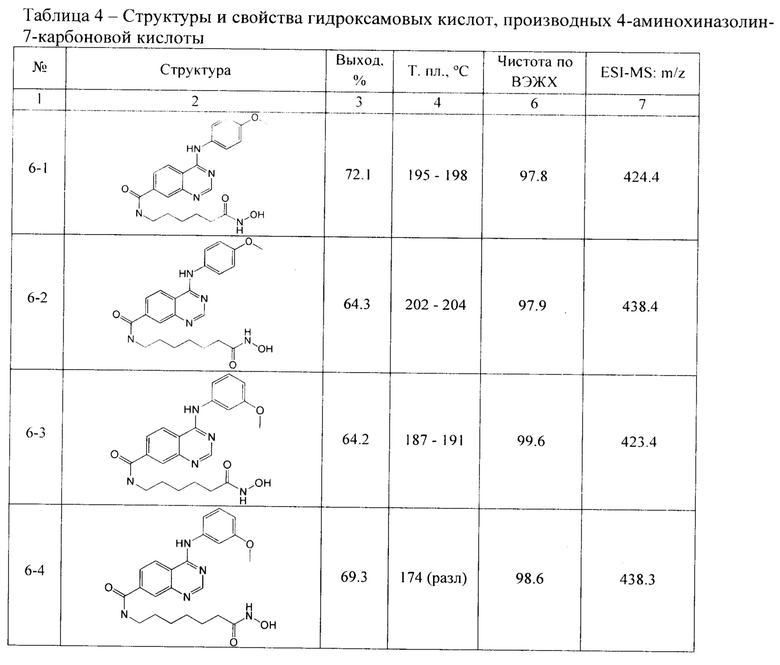
N-(6-(гидроксиамино)-6-оксогексил)-4-((4-метоксифенил)амино)хиназолин-7-карбоксамид (соединение 6-1).
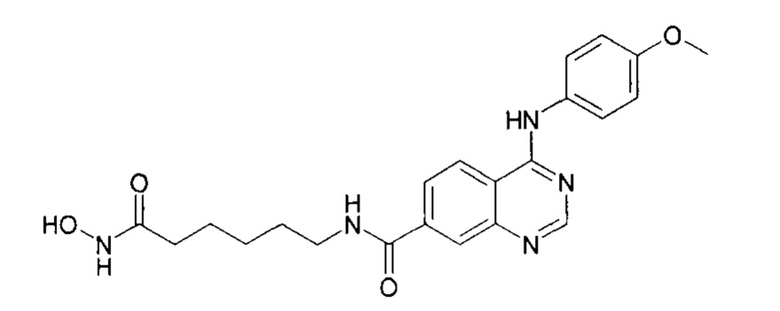
N-(7-(гидроксиамино)-7-оксогептил)-4-((4-метоксифенил)амино)хиназолин-7-карбоксамид (соединение 6-2).
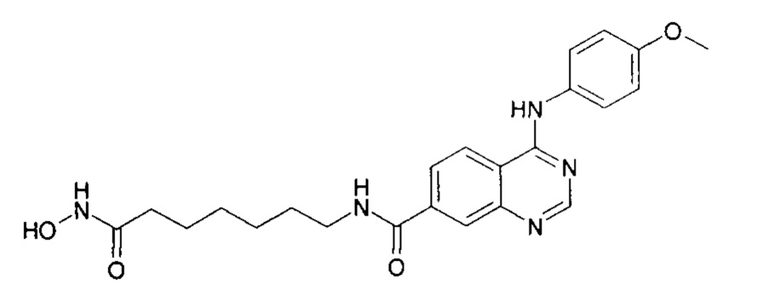
N-(6-(гидроксиамино)-6-оксогексил)-4-((3-метоксифенил)амино)хиназолин-7-карбоксамид (соединение 6-3).
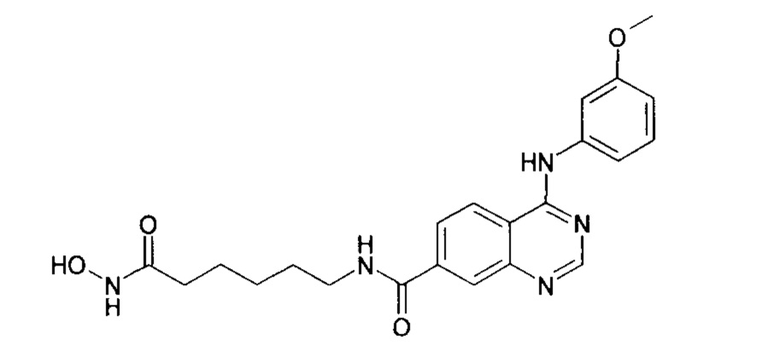
N-(7-(гидроксиамино)-7-оксогептил)-4-((3-метоксифенил)амино)хиназолин-7-карбоксамид (соединение 6-4).
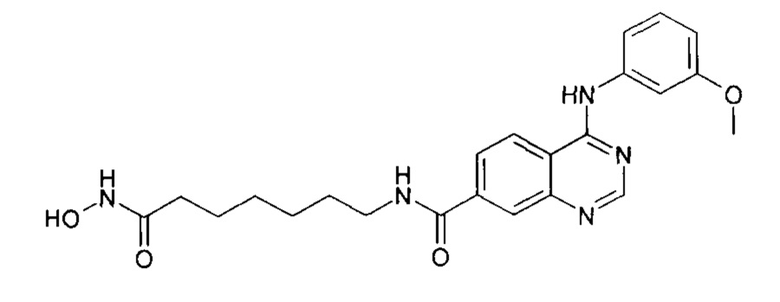
N-(6-(гидроксиамино)-6-оксогексил)-4-((3,4-диметоксифенил)амино)хиназолин-7-карбоксамид (соединение 6-5).
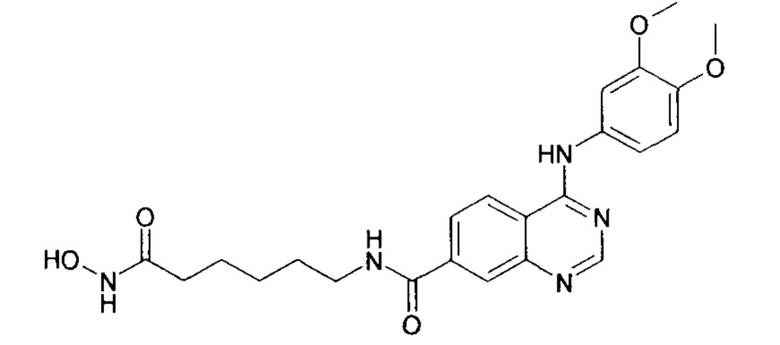
N-(7-(гидроксиамино)-7-оксогептил)-4-((3,4-диметоксифенил)амино)хиназолин-7-карбоксамид (соединение 6-6).
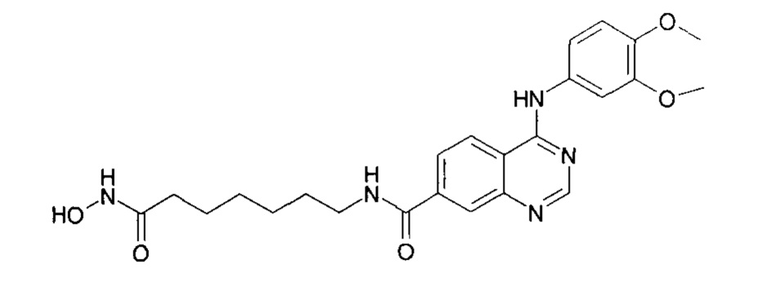
N-(6-(гидроксиамино)-6-оксогексил)-4-((3,5-диметилфенил)амино)хиназолин-7-карбоксамид (соединение 6-7).
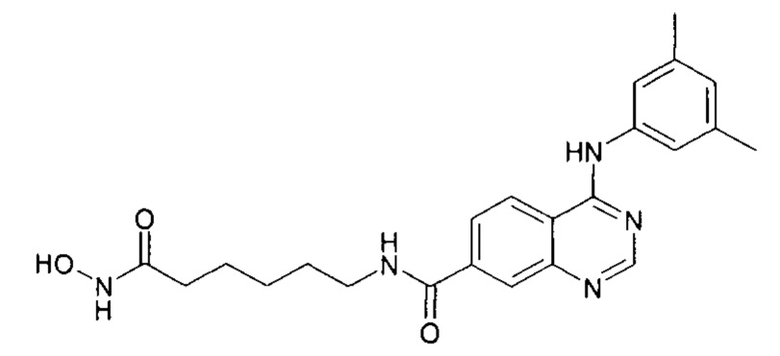
N-(7-(гидроксиамино)-7-оксогептил)-4-((3,5-диметилфенил)амино)хиназолин-7-карбоксамид (соединение 6-8).
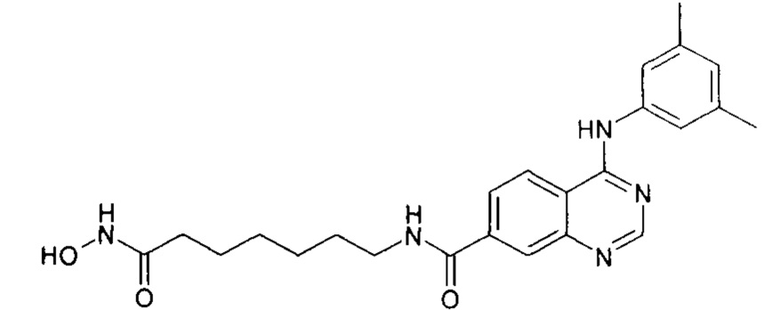
N-(6-(гидроксиамино)-6-оксогексил)-4-((3-этинилфенил)амино)хиназолин-7-карбоксамид (соединение 6-9).

N-(7-(гидроксиамино)-7-оксогептил)-4-((3-этинилфенил)амино)хиназолин-7-карбоксамид (соединение 6-10).
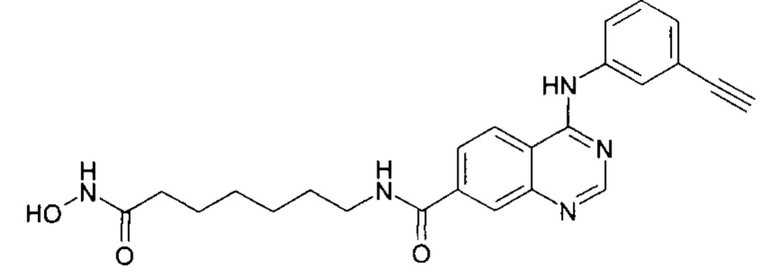
N-(7-(гидроксиамино)-7-оксогептил)-4-((2,3-Дигидробензо[b][1,4]диоксин-6-ил)амино)хиназолин-7-карбоксамид (соединение 6-11).
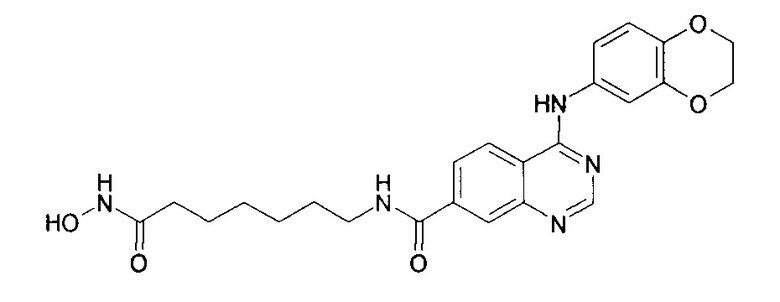
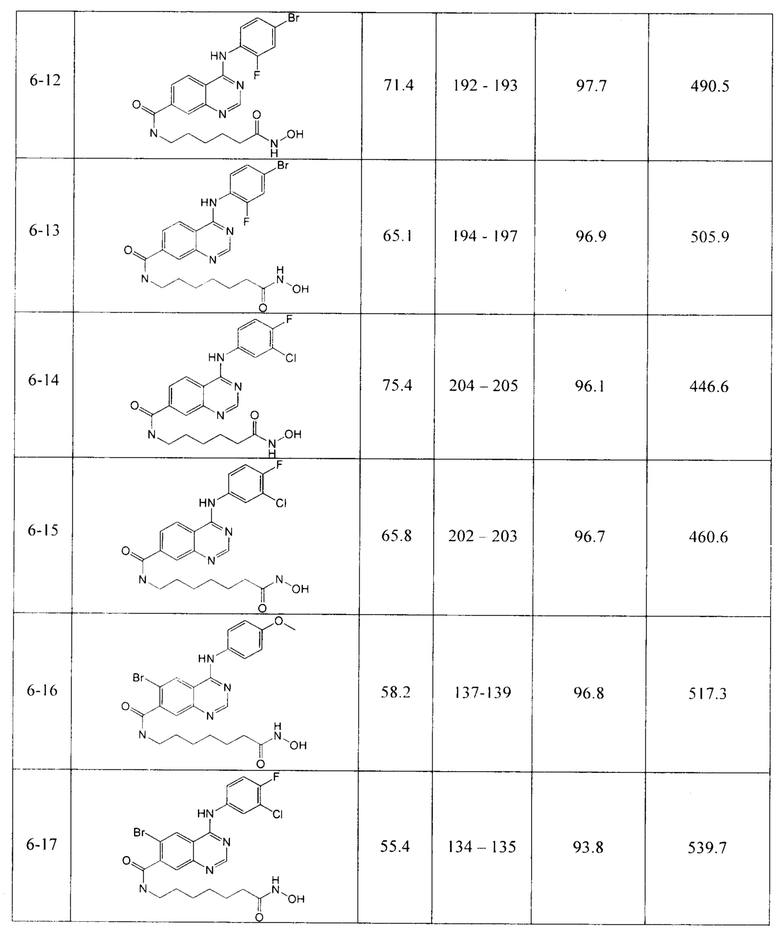
4-((4-бром-2-фторфенил)амино)-N-(6-(гидроксиамино)-6-оксогексил)хиназолин-7-карбоксамид (соединение 6-12).
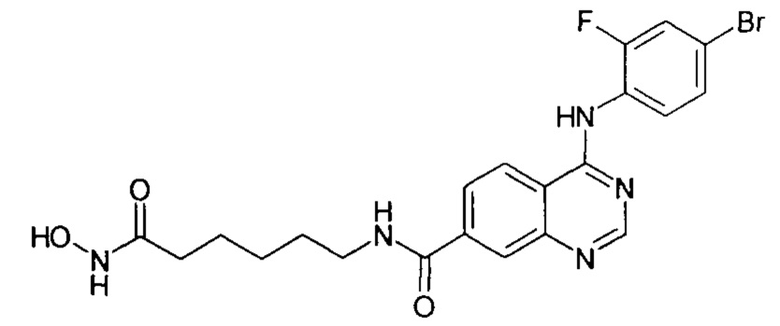
4-((4-бром-2-фторфенил)амино)-N-(7-(гидроксиамино)-7-оксогептил)хиназолин-7-карбоксамид (соединение 6-13).
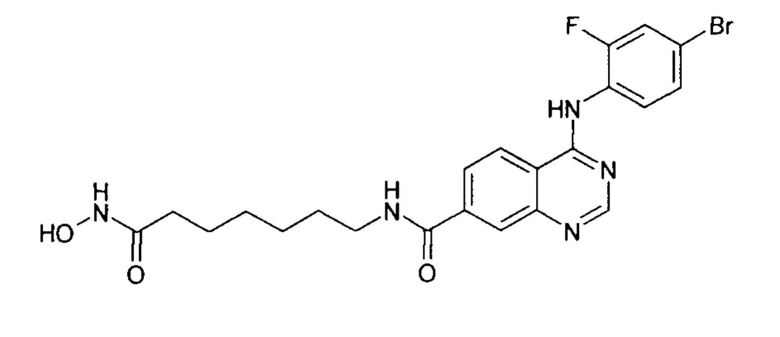
N-(6-(гидроксиамино)-6-оксогексил)-4-((3-хлор-4-фторфенил)амино)хиназолин-7-карбоксамид (соединение 6-14).
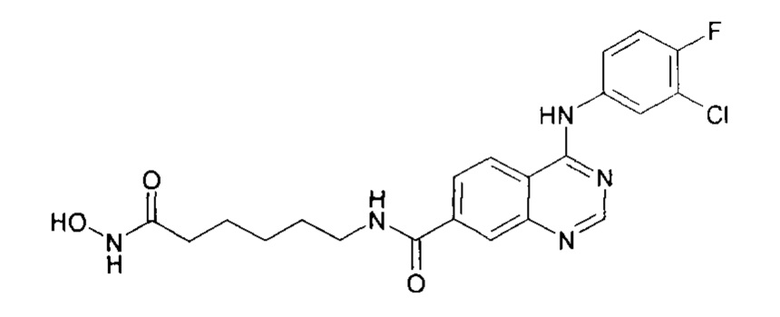
N-(7-(гидроксиамино)-7-оксогептил)-4-((3-хлор-4-фторфенил)амино)хиназолин-7-карбоксамид (соединение 6-15).
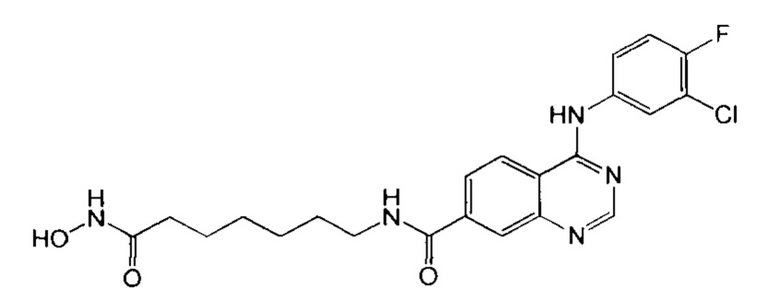
6-Бром-N-[-(6-(гидроксиамино)-6-оксогексил)-4-((3-метоксифенил)амино)хиназолин-7-карбоксамид (соединение 6-16).

N-(7-(гидроксиамино)-7-оксогептил)-4-((3-хлор-4-фторфенил)амино)хиназолин-7-карбоксамид (соединение 6-17).
Заявляемый способ получения новых гидроксамовых кислот, производных 4-аминохиназолин-7-карбоновой кислоты, осуществляется в простых условиях, имеет низкую трудоемкостью процесса, доступность реагентов. Заявляемый способ получения новых гидроксамовых кислот, производных 4-аминохиназолин-7-карбоновой кислоты, также заявляемый способ обеспечивает получение целевых соединений с чистотой, удовлетворяющей требованиям фармацевтической промышленности.
Применяемые аналитические методы и оборудование.
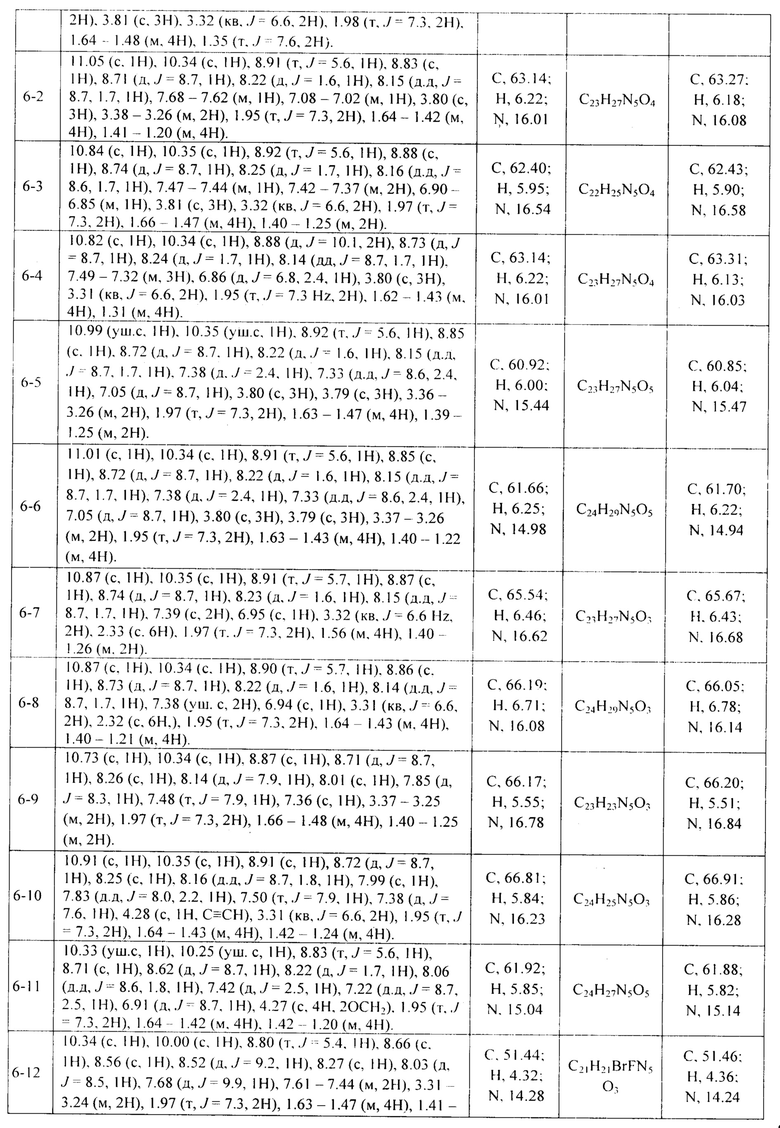
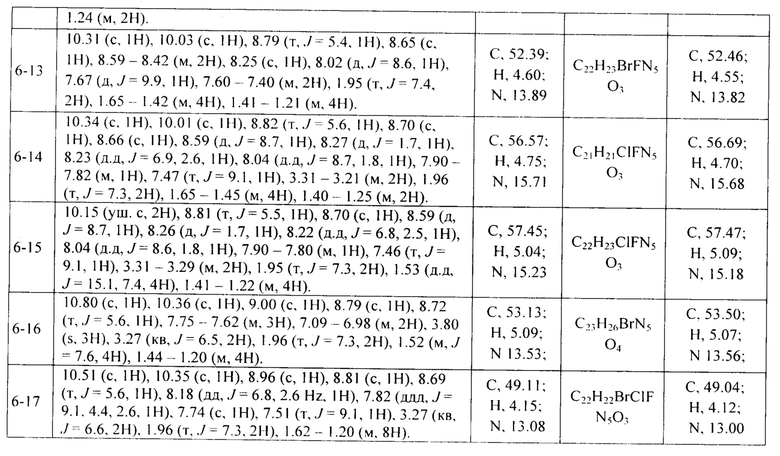
Аналитическую ВЭЖХ проводили на хроматографе фирмы Shimadzu. Колонка: Grom-Sil 12J ODS-4HE, 5 мкм, 250x4.6 мм. Условия: линейный градиент АВ: 5% В (0 мин) 100% В (20 мин). А - 0.01% ТФУ в воде, В - 0.01% ТФУ в ацетонитриле. Спектры ESI-MS регистрировали на приборе "Agilent LC/MS 1200" при ионизации пробы электрораспылением в режиме регистрации положительных ионов. Пробы готовили в системе ацетонитрил/вода 1/1, концентрация 2 мг/мл. Условия анализа: поток 1 мл/мин, давление на нибулайзере 20 psi, температура 360°, скорость потока осушающего газа 9 л/мин, напряжение 3500 В, целевая масса от 100 до 2000. Температуру плавления определяли на приборе марки "Melting Point М-565" (BUCHI). Спектры ЯМР 1Н получены на Фурье ЯМР-спектрометре Bruker AVANCE III NanoBay 300 МГц (для 1Н-ЯМР). Спектры регистрировали в режиме стабилизации по дейтерию, термостабилизация 25°С, внутренний стандарт - тетраметилсилан) в ДМСО-d6. Химические сдвиги приведены в миллионных долях (δ), КССВ - в герцах. ТСХ проводили на пластинах Merck TLC Silica gel 60 F254, проявление в УФ и нингидрином.
Ниже приводятся примеры осуществления изобретения.
Пример 1
Синтез метил 4-гидроксихиназолин-7-карбоксилата (соединение 1-1).
В конической колбе объемом 250 мл, снабженной обратным холодильником, растворяют 20.9 г (0.10 моля) диметилового эфира 2-аминотерефталевой кислоты в 100 мл формамида и перемешивают 5 часов при 150°С. Реакционную смесь выливают в 300 мл воды со льдом, 10%-ным раствором гидрокарбоната натрия доводят рН раствора до 8. Отфильтровывают осадок, промывают 50 мл воды, сушат на воздухе. Получают 20.0 г (98.1%) белого порошка. Т. пл. 270-272°С. 1Н ЯМР (ДМСО-d6, δ м. д., J, Гц) 12.42 (с, 1Н), 8.18 (д, J=8.3, 1Н), 8.15 (с, 1Н), 8.08 (д, J=1.6, 1H), 7.94 (дд, J=8.3, 1.7, 1Н), 3.90 (с, 3Н). Содержание основного продукта по данным ВЭЖХ 99.8%, ESI-MS, m/z 205.2 [М+Н]+. Найдено, %: С, 58.72; Н, 3.91; N, 13.44. C10H8N2O3. Вычислено, %: С, 58.82; Н, 3.92; N, 13.72.
Пример 2
Синтез метил 6-бром-4-гидроксихиназолин-7-карбоксилата (соединение 1-2).
Получают аналогично Примеру 1 из 28.8 г (0.10 моля) диметилового эфира 2-амино-5-бромтерефталевой кислоты. Выход 27.3 г (96.5%), светло-желтый порошок. Т. пл. 228-230°С. 1Н ЯМР (ДМСО-d6, δ м. д., J, Гц) 12.58 (с, 2Н), 8.30 (с, 1H), 8.19 (с, 1Н), 7.96 (с, 1Н), 3.91 (с, 3Н). Содержание основного продукта по данным ВЭЖХ 98.2%, ESI-MS, m/z 282.2 [М+Н]+. Найдено, %: С, 42.12; Н, 2.21; N, 9.49. C10H7BrN2O3. Вычислено, %: С, 42.43; Н, 2.49; N,9.90.
Пример 3
Синтез метил 4-хлорхиназолин-7-карбоксилата (соединение 2-1).
К суспензии метил-4-гидроксихиназолин-7-карбоксилата 1 (4.3 г, 21 ммоль) в 80 мл толуола добавляют диэтиланилин (6.8 мл, 53 ммоль) и затем приливают фосфорилхлорид (4.9 мл, 53 ммоль). Суспензию кипятят 3.5 часа, охлаждают, промывают 50 мл воды, 3×50 мл 8%-ной соляной кислотой, 50 мл воды. Органический слой упаривают в вакууме, кристаллический остаток переносят на фильтр и промывают 2×25 мл гексана, сушат на воздухе. Выход 3.9 г (83.5%), светло-бежевые кристаллы, содержание основного продукта по ВЭЖХ 94.6%. ESI-MS, m/z 223.1 [М+Н]+
Пример 4
Синтез метил 5-бром-4-хлорхиназолин-7-карбоксилата (соединение 2-2).
К суспензии метил 6-бром-гидроксихиназолин-7-карбоксилата 1 (5.7 г, 20 ммоль) в 80 мл толуола добавляют диэтиланилин (6.8 мл, 53 ммоль) и затем приливают фосфорилхлорид (4.9 мл, 53 ммоль). Суспензию кипятят 3.5 часа, охлаждают, промывают 50 мл воды, 3×50 мл 8%-ной соляной кислотой, 50 мл воды. Органический слой упаривают в вакууме, кристаллический остаток переносят на фильтр и промывают 2×25 мл гексана, сушат на воздухе. Выход 4.94 г (82.2%), бежевые кристаллы, содержание основного продукта по ВЭЖХ 95.2%. ESI-MS, m/z 300.7 [М+Н]+
Пример 5
Синтез метил 4-((3-этинилфенил)амино)хиназолин-7-карбоксилата хлоргидрата (соединение 3-4)
К суспензии метил-4-хлорхиназолин-7-карбоксилата (445 мг, 2 ммоль) в 5.5 мл ДМФА добавляют 3-этиниланилина (258 мг, 2.2 моль) и перемешивают 3.5 часа при комнатной температуре. Реакционную смесь разбавляют 10 мл воды, выпавшие кристаллы отфильтровывают, промывают водой и сушат на воздухе. Выход 530 мг (87.5%), Т. пл. 209.5-210.5°С. 1Н ЯМР (ДМСО-d6, δ м. д., J, Гц) 11.97 (уш.с.1 Н) 9.10 (д, J=8.71, 1Н) 9.03 (с, 1Н) 8.50 (д, J=1.47, 1Н) 8.26 (дд, J=8.71, 1.56, 1H) 7.93 (m, J=1.56, 1Н) 7.77-7.86 (м, 1H) 7.52 (m, J=7.79, 1H) 7.44 (дт, J=7.70, 1.17, 1Н) 4.32 (с, 1Н) 3.97 (с, 3Н). Содержание основного по ВЭЖХ 98.8%. ESI-MS: m/z 304.2 [М+Н]+.
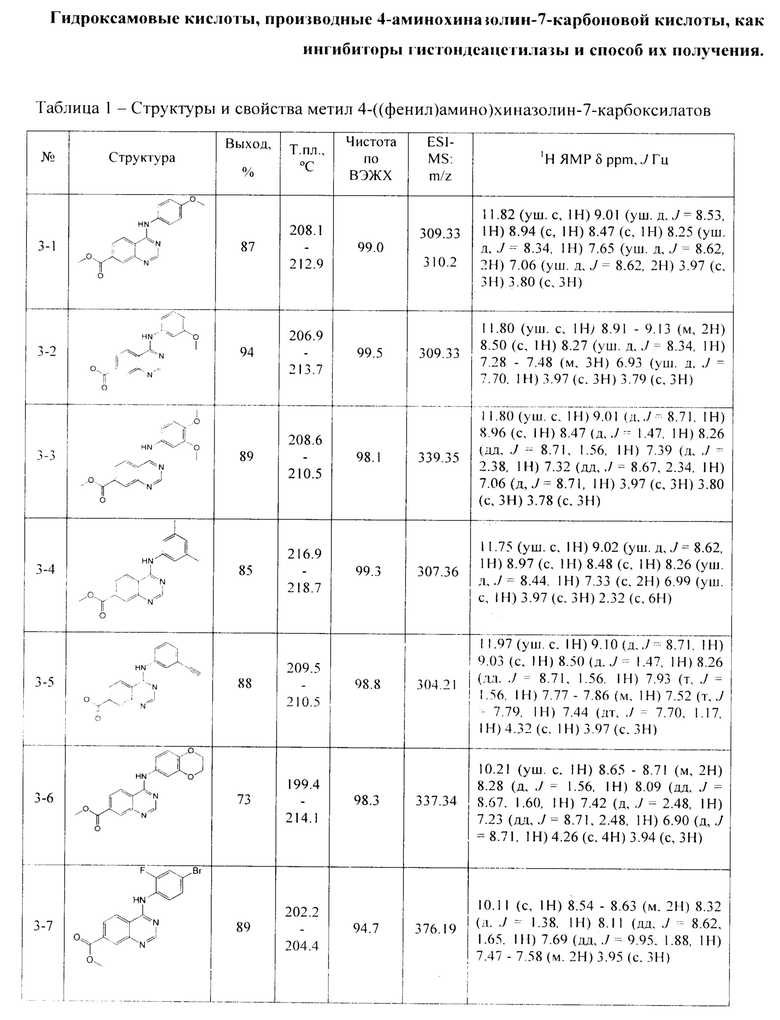
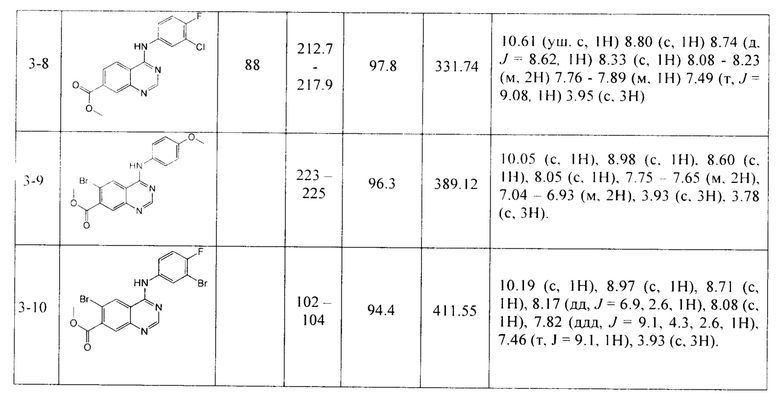
По аналогичной методике получают другие производные метил 4-((фенил)амино)хиназолин-7-карбоксилатов из соответствующих замещенных анилинов (соединения 3-1-3-10) (Таблица 1)
Пример 6
Синтез 4-((3-этинилфенил)амино)хиназолин-7-карбоновой кислоты (соединение 4-5)
К раствору метил 4-((3-этинилфенил)амино)хиназолин-7-карбоксилата (530 мг 1.75 ммоль) в 10 мл метанола добавляют раствор гидроксида натрия (0.21 г, 5.25 ммоль) в 5 мл воды и перемешивают 2.5 часа при 100°С. Реакционную смесь охлаждают, нейтрализуют ледяной уксусной кислотой (0.42 мл, 7 ммоль), добавляют 10 мл воды, выпавшие кристаллы отфильтровывают, промывают 2×5 мл воды и сушат на воздухе. Выход 435 мг (77%), белый порошок. Чистота по ВЭЖХ 98.7% ESI-MS: m/z 290.2 [М+Н]+
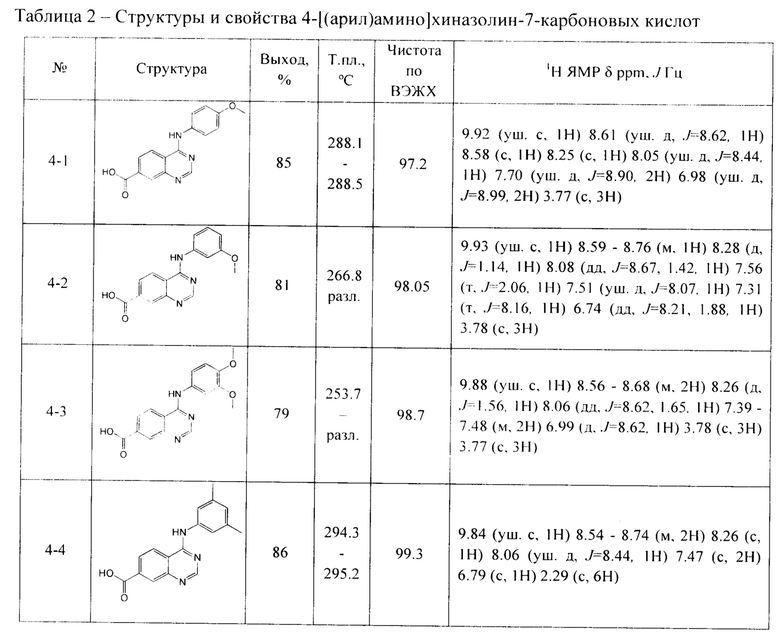
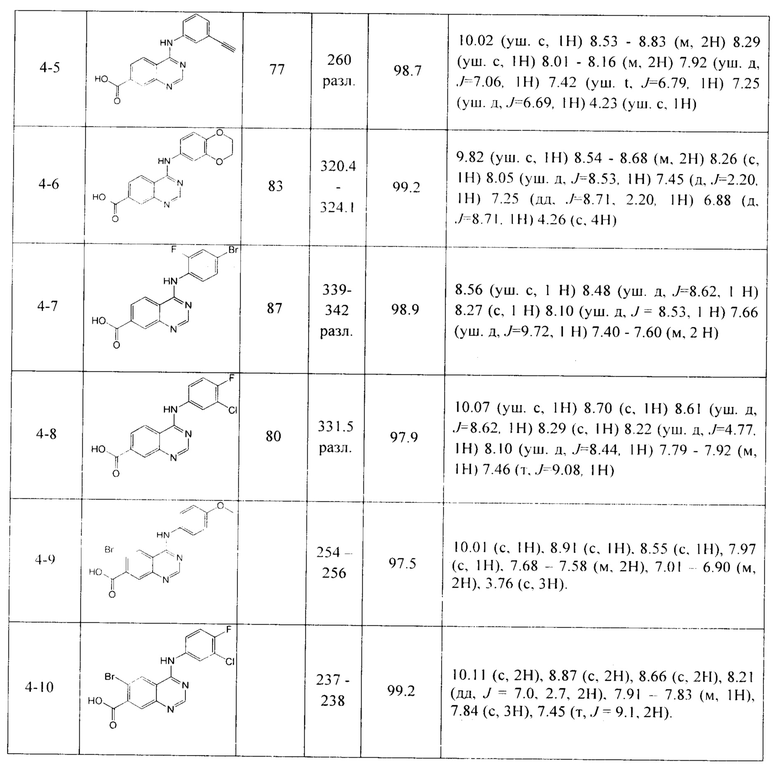
По аналогичной методике получают другие производные 4-[(арил)амино]хиназолин-7-карбоновых кислот (соединения 4-1 - 4-10) (Таблица 2)
Пример 7
Синтез метил 6-(4-((3-этинилфенил)амино)хиназолин-7-карбоксамидо)гексаноата
К суспензии соответствующей 4-((3-этинилфенил)амино)хиназолин-7-карбоновой кислоты 530 мг (0.75 ммоль) в 5 мл ДМФА добавляют TBTU 321 мг (1 ммоль), гидрохлорида метилового эфира 6-аминогексановой кислоты 150 мг (0.83 ммоль), затем добавляют триэтиламин 230 мг (4 ммоль) и перемешивают 3 часа при комнатной температуре. К реакционной смеси добавляют 10 мл воды, выпавшие кристаллы отфильтровывают, промывают 2×5 мл воды и сушат на воздухе. Выход 275 мг (88%), содержание основного продукта по ВЭЖХ 96.9%. ESI-MS: m/z 417.4 [М+Н]+.
По аналогичной методике получают другие производные метил ω-(4-(ариламино)хиназолин-ω-карбоксамидо)алканоатов (соединения 5-1 - 5-16) (Таблица 3).
Пример 8
Синтез 4-((3-этинилфенил)амино)-N-(6-(гидроксиамино)-6-оксогексил)хиназолин-7-карбоксамида (соединение 6-9)
К раствору метил 6-(4-((3-этинилфенил)амино)хиназолин-7-карбоксамидо)гексаноата 417 мг (1 ммоль) в 10 мл метанола добавляют гидрохлорида гидроксиламина 695 мг (10 ммоль) приливают раствор метилата натрия, приготовленный из 230 мг (10 ммоль) натрия и 5 мл метанола и перемешивают 20 минут. Затем к этому раствору добавляют 456 мг (3 ммоль) 1,8-диазабицикло[5.4.0]ундец-7-ена и перемешивают при комнатной температуре. Окончание реакции (исчезновение исходного эфира, примерно 24 часа) определяют с помощью тонкослойной хроматографии (элюент хлороформ-метанол 10:1). Реакционную смесь упаривают в вакууме, добавляют 10 мл воды, нейтрализуют ледяной уксусной кислотой до рН 5, выпавшие кристаллы отфильтровывают, промывают 2×10 мл воды и сушат на воздухе. Остаток кристаллизуют из ДМФА/вода. Выход 156 мг (57%), Содержание основного продукта по ВЭЖХ 97.2%. ESI-MS: m/z 418.3 [М+Н]+
По аналогичной методике получают другие целевые соединения, гидроксамовые кислоты, производные 4-аминохиназолин-7-карбоновой кислоты (соединения 6-1 - 6-17) Структуры и физико-химические целевых соединений представлены в таблицах 4 и 5.
Пример 9
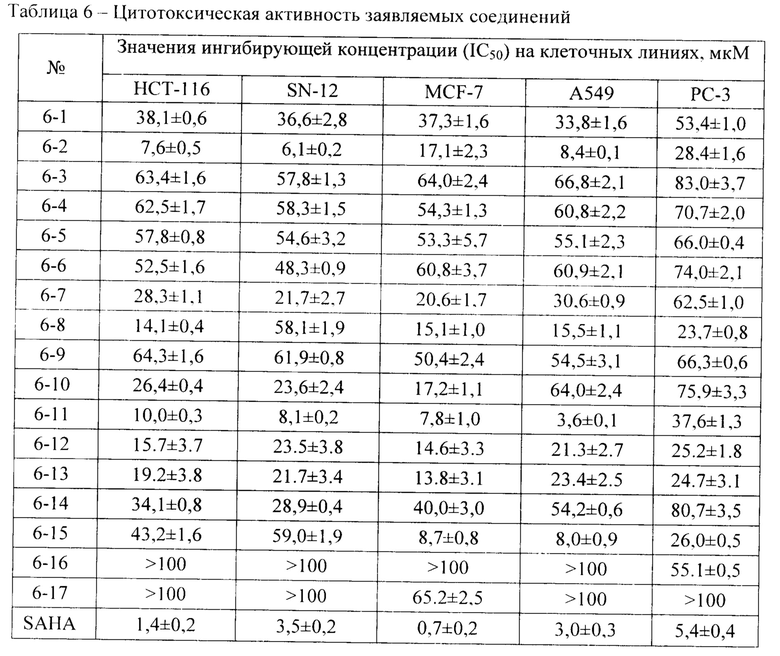
Определение цитотоксической активности
Цитотоксическую активность заявляемых соединений определяли с помощью МТТ-теста. Концентрацию полумаксимального ингибирования (IC50) для синтезированных веществ определяли на клетках рака толстой кишки НТО 116, карциномы легкого А549, рака предстательной железы РС-3, рака молочной железы MCF-7, рака почки SN-12
Для проведения исследования клетки опухолевых линий человека были нанесены в 96-луночный культуральный планшет (104 клеток в каждой лунке) и после 24 часов инкубации к клеткам были добавлены методом раститровки различные концентрации 14 исследуемых соединений в диапазоне 100±0.1 мкМ. В качестве контроля сравнения использовали препарат Вориностат (SAHA), являющийся гидроксамовой кислотой. Планшет с внесенными веществами помещали в СО2-инкубатор на 72 часа. После инкубации вносили в каждую лунку по 20 мкл рабочего раствора МТТ. Инкубировали планшет 3 часа в СО2-инкубаторе. После инкубации заменить в каждой лунке среду на 200 мкл ДМСО.
После растворения кристаллов формазана определяли оптическую плотность каждой лунки при 570 нм, вычитали измеренное фоновое поглощение при 620 нм с помощью планшетного фотометра. Значение концентрации, вызывающее 50% ингибирование роста популяции клеток (IC50), определяли на основе дозозависимых кривых и/или с помощью программного обеспечения (например, GraphPad Pricm 5.0 или OriginPro 9.0). Полученные результаты представлены в таблице 6.
Пример 10
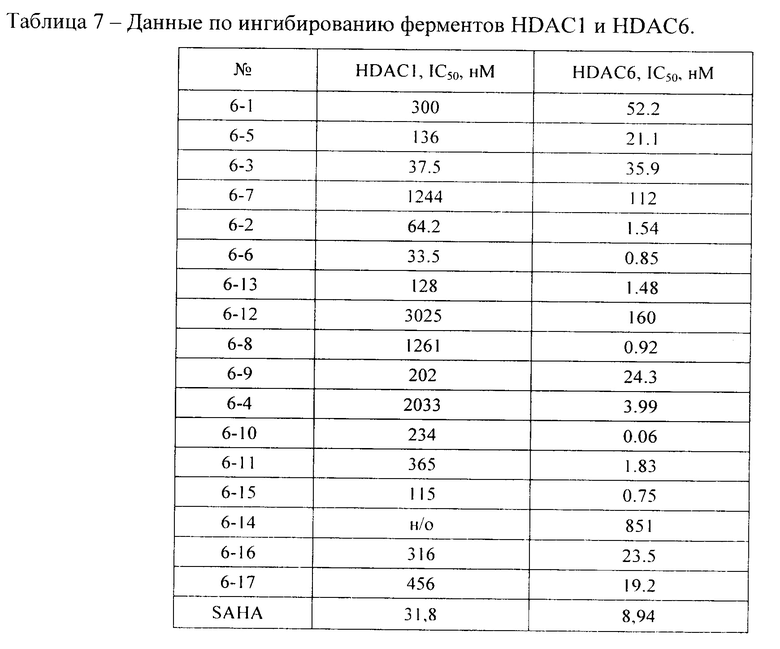
Исследование влияния производных гидроксамовых кислот на активность ферментов HDAC1 и HDAC6.
Делают навески исследуемых веществ и приготовляют растворы с исходной концентрацией 5×10-3 М в подходящем растворителе (вода, спирт, ДМСО). Приготовляют стоковые растворы исследуемых веществ в растворителе для получения концентрационной зависимости с учетом того, что конечная концентрация вещества в инкубационной среде меньше в 4 раза (50 мкл раствора вещества в 200 мкл конечного объема пробы). В качестве эталонных веществ использовался известный ингибитор гистондеацетилазы препарат Вориностат (SAHA).
Эксперимент выполнялся в 96-луночном планшете. Добавляют 50 мкл раствора исследуемых соединений в лунки планшета, согласно схеме соответствующего эксперимента.
Фермент HDAC разбавляют с использованием буфера HDAC-Glo™ до желаемой концентрации. Вносят 50 мкл фермента HDAC в каждую лунку 96-луночного планшета, где уже находятся растворы исследуемых соединений, полученные на предыдущей стадии. Оставляют не менее 3-х лунок без HDAC, для формирования буферного фона, не менее 3-х лунок с HDAC и растворителем, для получения максимального сигнала активности фермента.
Содержимое планшета перемешивают при комнатной температуре в течение 30-60 секунд, используя орбитальный шейкер при 500-700 об/мин, чтобы обеспечить однородность.
Смесь фермент/исследуемое вещество инкубируют при комнатной температуре в течение по меньшей мере 30 минут (но не более 2 часов).
Готовят реагент HDAC-Glo™: 10 мл буфера HDAC-Glo™ добавляют к субстрату HDAC-Glo и осторожно перемешивают. Затем в эту смесь добавляют 10 мкл реагента-проявителя и также осторожно перемешивают.
Равный объем реагента HDAC-Glo™ добавляют в каждую лунку планшета (100 мкл для 96-луночного).
Смешивают содержимое ячеек при комнатной температуре в течение 30-60 секунд, используя орбитальный шейкер при 500-700 об/мин, чтобы обеспечить однородность. Затем содержимое инкубируют при комнатной температуре в течение 15-45 минут.
По истечению необходимого времени инкубации измеряют люминесценцию на многофункциональном планшетном анализаторе EnVicion.
Обработка полученных первичных данных, определение IC50
Так как амплитуды кривых могут различаться от повторения к повторению, полученные абсолютные значения хемилюминесценции нормировали для каждого отдельного повторения. За нулевое значение брали минимальное измеренное значение внутри одной серии разведений, за 100% - максимальное достигнутое значение. Нормированные три повторения вместе аппроксимировали кривой у=у(х), задающейся формулой:
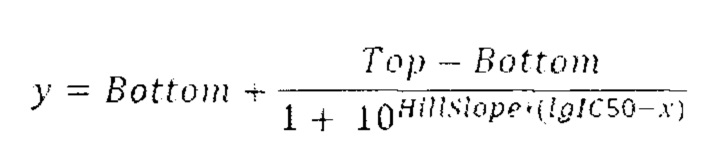
где, у - нормированная интенсивность люминесценции, х - десятичный логарифм концентрации исследуемого вещества.
Параметры Bottom, Top, HillSlope и IgIC50 подбираются так, чтобы сумма квадратов отклонений экспериментальных точек от аппроксимирующей кривой была минимальна.
Обработка данных выполнена с использованием пакета GraphPad Prism 6. Полученные значения IC50 приведены в Таблице 7.
Ряд целевых заявляемых соединений обладает цитотоксической активностью в микромолярном диапазоне на различных линиях опухолевых клеток. Активность ряда соединений по отношению ферменту HDAC6 превышает или сравнима с активностью известного ингибитора гистондеацетилазы - Вориностата (SAHA), при этом необходимо отметить высокую селективность всех соединений к ферменту HDAC6 по сравнению с HDAC1.


| название | год | авторы | номер документа |
|---|---|---|---|
| Производные 4-((арил)(метил)амино)хиназолин-7-карбоновой кислоты с противоопухолевым действием и способ их получения | 2024 |
|
RU2834688C1 |
| Гидроксамовые кислоты, производные 4-аминохиназолина, обладающие противоопухолевой активностью | 2022 |
|
RU2802463C1 |
| Способ получения N-((гидроксиамино)-оксоалкил)-2-(хиназолин-4-иламино)-бензамидов | 2019 |
|
RU2722694C1 |
| Способ получения гидроксамовых кислот, производных 2-арил-2,3-дигидрохиназолин-4(1Н)-онов | 2020 |
|
RU2744750C1 |
| Производные 3-гидроксихиназолин-4(3Н)-она в качестве ингибиторов гистондеацетилазы и способ их получения | 2020 |
|
RU2740503C1 |
| ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ АЛКИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ГИСТОНДЕАЦЕТИЛАЗЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2016 |
|
RU2683022C1 |
| ХИНОЛИЛ-СОДЕРЖАЩЕЕ СОЕДИНЕНИЕ ГИДРОКСАМОВОЙ КИСЛОТЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, А ТАКЖЕ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ АНОМАЛЬНОЙ АКТИВНОСТЬЮ ПРОТЕИНКИНАЗЫ И/ИЛИ ГИСТОНДЕАЦЕТИЛАЗЫ | 2011 |
|
RU2573633C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРРОЛА С ИНГИБИРУЮЩЕЙ ГИСТОНДЕАЦЕТИЛАЗУ АКТИВНОСТЬЮ | 2005 |
|
RU2416600C2 |
| 2,3,5-ТРИЗАМЕЩЕННЫЕ ПИРРОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ (ВАРИАНТЫ) | 2010 |
|
RU2549885C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, ИНДУЦИРУЮЩИЕ ФЕРРОПТОЗ В МЕТАСТАТИЧЕСКИХ КЛЕТКАХ МЕЛАНОМЫ И РАКА ТОЛСТОЙ КИШКИ | 2019 |
|
RU2722308C1 |
Изобретение относится к производным 4-аминохиназолин-7-карбоновой кислоты, и касается непосредственно новых производных 4-аминохиназолин-7-карбоновой кислоты, имеющих в составе гидроксамовую кислоту, которые могут использоваться в медицине для лечения ряда заболеваний посредством ингибирования гистоновых деацетилаз, например при лечении онкологических и нейродегенеративных заболеваний. Предлагаются новые гидроксамовые кислоты, производные 4-аминохиназолин-7-карбоновой кислоты в качестве ингибиторов гистондеацетилаз, имеющие следующую общую формулу I, в которой R1 R2, R3 R4 независимо друг от друга представляют собой водород, галоген, метил, ОМе, этин, а также R2, R3=-ОСН2СН2О-; R5 представляет собой водород или галоген; n=4, 5. Также предлагается способ получения новых гидроксамовых кислот, производных 4-аминохиназолин-7-карбоновой кислоты, общей формулы I, осуществляемый по следующей схеме: производные метил 4-гидроксихиназолин-7-карбоксилата, получаемые циклизацией производных диметил-2-аминотерефталата в формамиде взаимодействием с фосфорилхлоридом в присутствии диэтиланилина, переводятся в производные метил 4-хлорхиназолин-7-карбоксилата, которые при взаимодействии с замещенными анилинами в диметилфорамиде при комнатной температуре превращаются в производные метил 4-((фенил)амино)хиназолин-7-карбоксилатов, последние подвергаются щелочному гидролизу, превращаясь в соответствующие производные 4-[(арил)амино]хиназолин-7-карбоновых кислот, далее к последним присоединяются метиловые эфиры 6-аминогексановой или 7-аминогептановой кислот с использованием 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний тетрафторбората (TBTU) в качестве конденсирующего агента и триметиламина в качестве основания, полученные соединения затем подвергают аминолизу 10-кратным мольным избытком гидроксиламина в безводном метаноле в присутствии 3-х эквивалентов 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU), осуществляемому при комнатной температуре в течение 20-26 часов до образования целевого продукта, выделяемого упариванием реакционной массы в вакууме, нейтрализацией ледяной уксусной кислотой до рН 5, фильтрацией с последующей промывкой водой. Способ получения новых гидроксамовых кислот, производных 4-аминохиназолин-7-карбоновой кислоты, характеризуется низкой трудоемкостью процесса, доступностью реагентов и получением целевых соединений с чистотой, удовлетворяющей требованиям фармацевтической промышленности, и благодаря простоте проводимых операций и применяемого оборудования легко масштабируем до промышленного производства.
2 н. и 1 з.п. ф-лы, 7 табл., 10 пр.
1. Гидроксамовые кислоты, производные 4-аминохиназолин-7-карбоновой кислоты общей формулы:
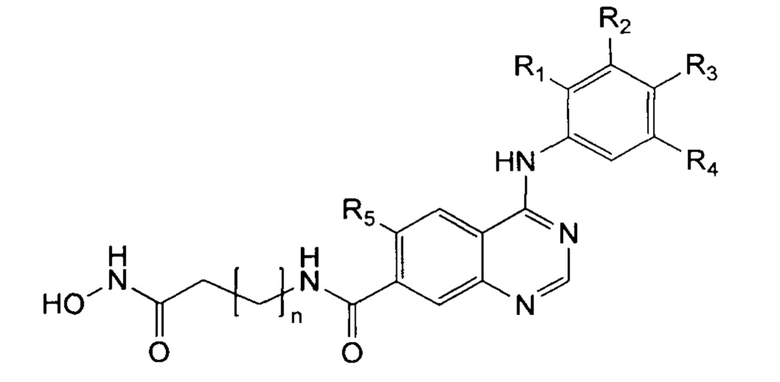 ,
,
где R1, R2, R3 R4 независимо друг от друга представляют собой водород, галоген, метил, ОМе, этин, а также R2, R3=-ОСН2СН2О-;
R5 представляет собой водород или галоген;
n=4, 5,
которые могут использоваться в качестве ингибиторов гистондеацетилаз различных изоформ и применяться для лечения, в частности, онкологических и нейродегенеративных заболеваний
2. Способ получения гидроксамовых кислот, производных 4-аминохиназолин-7-карбоновой кислоты общей формулы:
 ,
,
где R1, R2, R3 R4 независимо друг от друга представляют собой водород, галоген, метил, ОМе, этин, а также R2, R3=-ОСН2СН2О-;
R5 представляет собой водород или галоген;
n=4, 5,
осуществляемый по следующей схеме: производные метил 4-гидроксихиназолин-7-карбоксилата, получаемые циклизацией производных диметил-2-аминотерефталата в формамиде, взаимодействием с фосфорилхлоридом в присутствии диэтиланилина переводятся в производные метил 4-хлорхиназолин-7-карбоксилата, которые при взаимодействии с замещенными анилинами в диметилфорамиде при комнатной температуре превращаются в производные метил 4-((фенил)амино)хиназолин-7-карбоксилатов, последние подвергаются щелочному гидролизу, превращаясь в соответствующие производные 4-[(арил)амино]хиназолин-7-карбоновых кислот, далее к последним присоединяются метиловые эфиры 6-аминогексановой или 7-аминогептановой кислот с использованием 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний тетрафторбората (TBTU) в качестве конденсирующего агента и триметиламина в качестве основания, полученные соединения затем подвергают аминолизу 10-кратным мольным избытком гидроксиламина в безводном метаноле, осуществляемому при комнатной температуре в течение 20-26 часов до образования целевого продукта, выделяемого упариванием реакционной массы в вакууме, нейтрализацией ледяной уксусной кислотой до рН 5, фильтрацией и с последующей промывкой водой
3. Способ получения по п. 2, осуществляемый с использованием 3-х эквивалентов 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU) на стадии аминолиза гидроксиламином.
| US 2013267542 A, 10.10.2013 | |||
| Hieu D.T | |||
| et al "Quinazoline-Based Hydroxamic Acids: Design, Synthesis, and Evaluation of Histone Deacetylase Inhibitory Effects and Cytotoxicity" Chem | |||
| Biodivers | |||
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| Способ получения N-((гидроксиамино)-оксоалкил)-2-(хиназолин-4-иламино)-бензамидов | 2019 |
|
RU2722694C1 |
| US 2009181971 A1, 16.07.2009 | |||
| KR 20190134180 A, 04.12.2019 | |||
| ГИЗАТУЛЛИН А.Р | |||
| и др | |||
| "СИНТЕЗ | |||

