ОБЛАСТЬ ИЗОБРЕТЕНИЯ
[0001] Настоящее изобретение относится к области фармацевтических препаратов, в частности, способу получения промежуточного соединения конъюгата антитело–лекарственное средство.
УРОВЕНЬ ТЕХНИКИ
[0002] Конъюгат антитело–лекарственное средство (сокращенно обозначаемый как ADC) представляет собой класс противоопухолевых лекарственных средств, состоящих из трех компонентов: антитела, линкера и лекарственного средства. Количество лекарственных средств, присоединенных к антителу (соотношение лекарственного средства и антитела, DAR) определяет гомогенность конъюгата антитело–лекарственное средство. Избыточное количество присоединенных лекарственных средств будет приводить к нестабильной фармакодинамике, повышенному метаболизму лекарственного средства, сниженному времени полужизни и повышенной системной токсичности.
[0003] Значение DAR, главным образом, определяется линкером. Для классических ADC есть два способа сочетания: один из них является реакцией сочетания по аминогруппе, а другой – реакцией сочетания по тиоловой группе, являющийся в настоящее время наиболее общеупотребительным способом сочетания. Сочетание по аминогруппе является сочетанием лекарственного средства с остатком лизина (Lys) в антителе через линкер. Т.к. на одном антителе присутствует 80 лизинов, 30 из которых можно соединять, ADC, образованный посредством сочетания с лизином, имеет значение DAR от 0 до 30, и гомогенность продукта является очень плохой. Сочетание по тиоловой группе предназначено для раскрытия межцепочечных дисульфидных связей в антителе для получения свободных остатков цистеина (Cys) перед сочетанием с комплексом линкер–лекарственное средство, способных соединяться с остатками цистеина. Т.к. одно антитело содержит только 4 пары межцепочечных дисульфидных связей, после раскрытия всех межцепочечных дисульфидных связей можно получить только 8 свободных остатков цистеина. Таким образом, ADC, образованные посредством сочетания через остаток цистеина, имеют значения DAR от 0 до 8. Хотя посредством сочетания по тиоловой группе можно лучше контролировать количество связей на антитело, расщепление межцепочечной дисульфидной связи значительно снижает стабильность антитела.
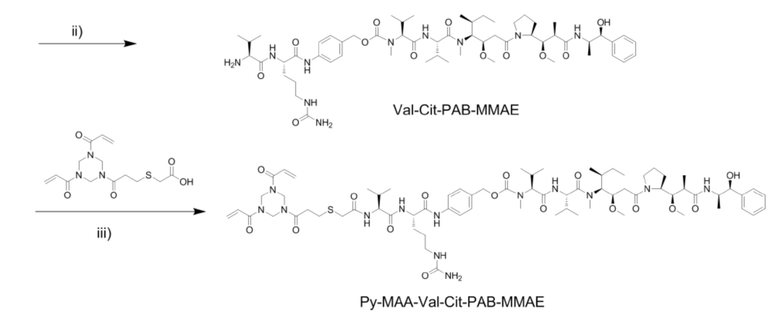
[0004] В настоящее время, в дополнение к классическому сочетанию по аминогруппе и сочетанию по тиоловой группе, есть новый способ сочетания: мостиковое сочетание. Способ мостикового сочетания является новым способом сочетания, разработанным на основе реакции сочетания по тиоловой группе, в котором линкер одновременно сочетают с двумя свободными тиоловыми группами на антителе, и значение DAR полученного таким образом конъюгата антитело–лекарственное средство составляет от 0 до 4. Множество линкеров, которые можно ковалентно связывать с антителом посредством мостикового сочетания, описаны в китайской патентной публикации № CN107921030A, в которой описывают промежуточное соединение конъюгата антитело–лекарственное средство (Py–MAA–Val–Cit–PAB–MMAE) (где Py представляет собой триакрилоилгексагидротриазин, его CAS является 959–52–4, и оно доступно в J&K Scientific и Nanjing Chemlin Chemical Industry Co., Ltd.), как указано ниже на стр. 42 описания.
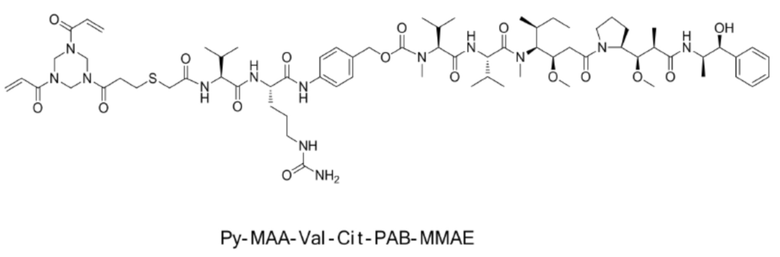
[0005] Кроме того, способ получения промежуточного соединения конъюгата антитело–лекарственное средство (Py–MAA–Val–Cit–PAB–MMAD), в котором лекарственное средство представляет собой MMAD (деметилдоластатин), также описан на стр. 32 патентной заявки:

Способ включает сначала сочетание Val–Cit–PAB с лекарственным средством (MMAD) с образованием конъюгата Val–Cit–PAB–MMAD, а затем реакцию с Py–MAA с образованием промежуточного соединения конъюгата антитело–лекарственное средство Py–MAA–Val–Cit–PAB–MMAD. В результате высокой стоимости лекарственного средства (такого как MMAD/MMAE или MMAF и т.д.), используемого в конъюгате антитело–лекарственное средство, и больших потерь при производстве лекарственного средства с использованием указанного выше способа, производственные затраты являются высокими, что делает этот способ неподходящим для массового промышленного производства.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0006] Настоящее изобретение относится к способу получения промежуточного соединения конъюгата антитело–лекарственное средство, в частности, к способу получения промежуточного соединения конъюгата антитело–лекарственное средство, содержащего линкер и функциональный фрагмент лекарственного средства. Промежуточным соединением конъюгата антитело–лекарственное средство является Py–MAA–Val–Cit–PAB–D, в котором Py–MAA–Val–Cit–PAB является линкером, и D представляет собой присоединенный функциональный фрагмент лекарственного средства. Способ отличается тем, что он включает следующие реакции:
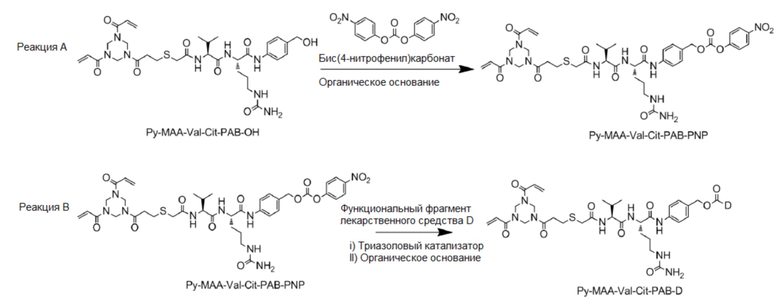
[0007] Кроме того, способ получения включает следующие реакции:
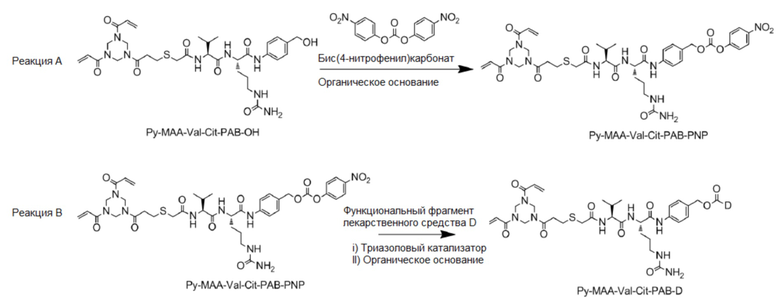
Реакцию A: растворение Py–MAA–Val–Cit–PAB–OH в растворителе, добавление бис(4–нитрофенил)карбоната, перемешивание до растворения или слабой мутности раствора, а затем добавление органического основания;
Реакцию B: после завершения реакции A, добавление триазольного катализатора, функционального фрагмента лекарственного средства D и органического основания и, после завершения реакции, осуществление очистки для получения Py–MAA–Val–Cit–PAB–D. Предпочтительно, для очистки используют препаративную колонку для ВЭЖХ.
[0008] Кроме того, функциональный фрагмент лекарственного средства D является цитотоксическим средством класса ауристатинов, цитотоксическим средством класса антрамицинов, цитотоксическим средством класса антрациклинов или цитотоксическим средством класса пуромицинов.
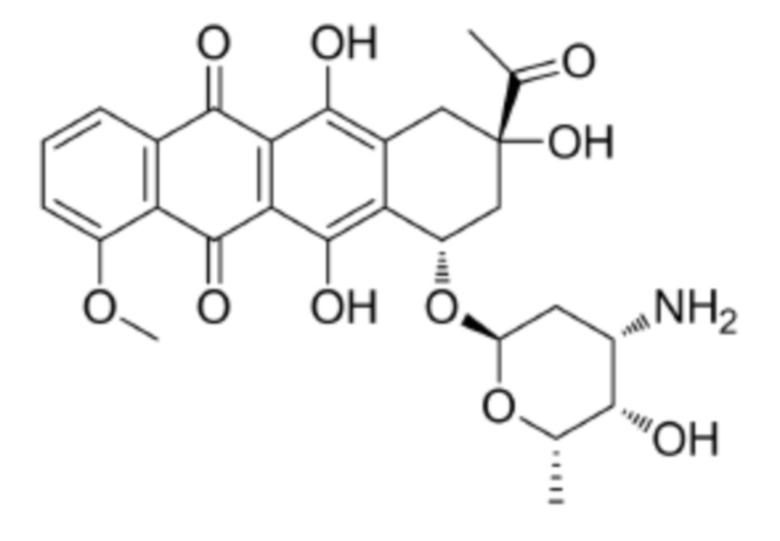
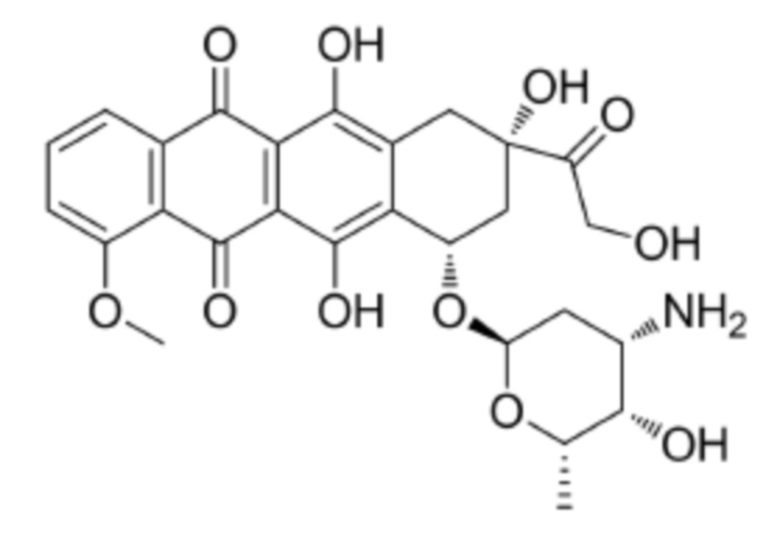
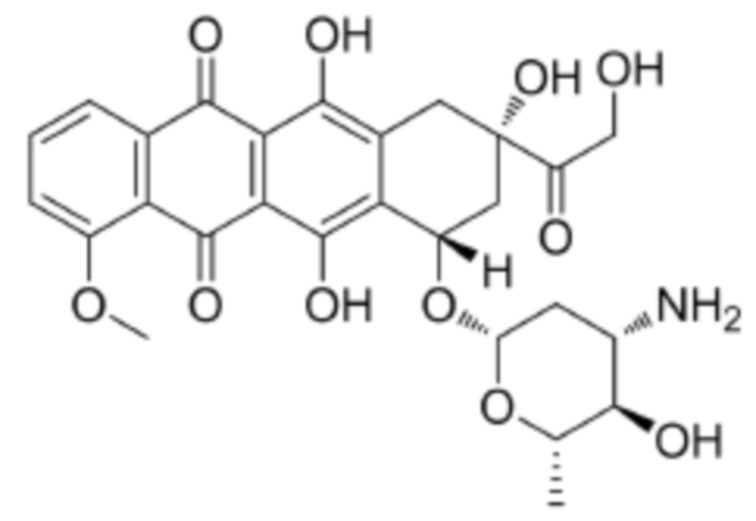
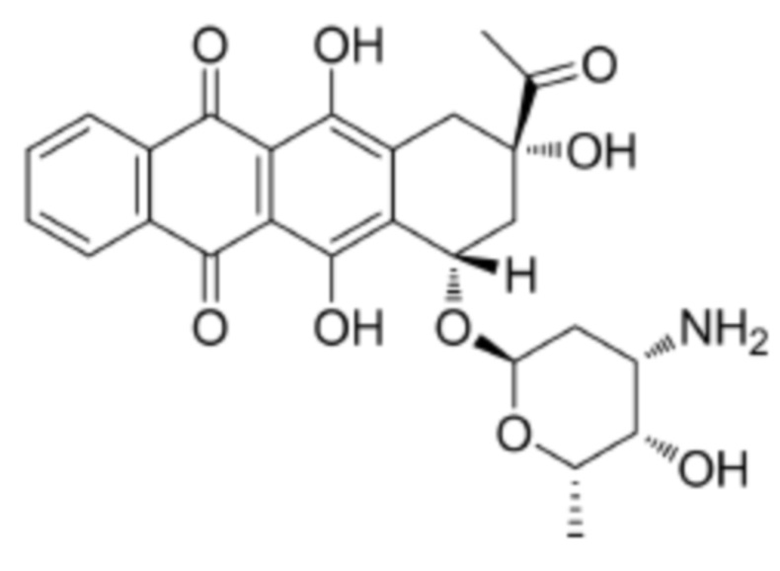
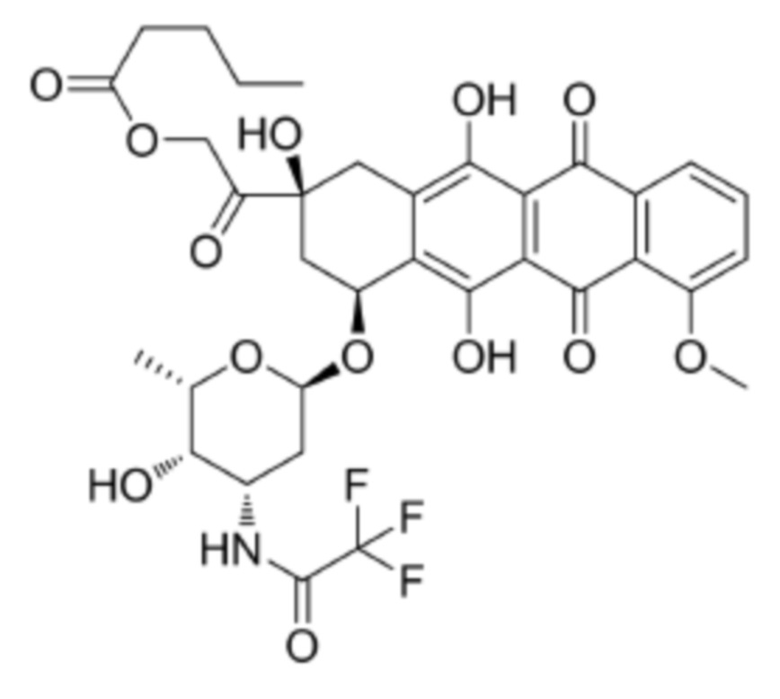
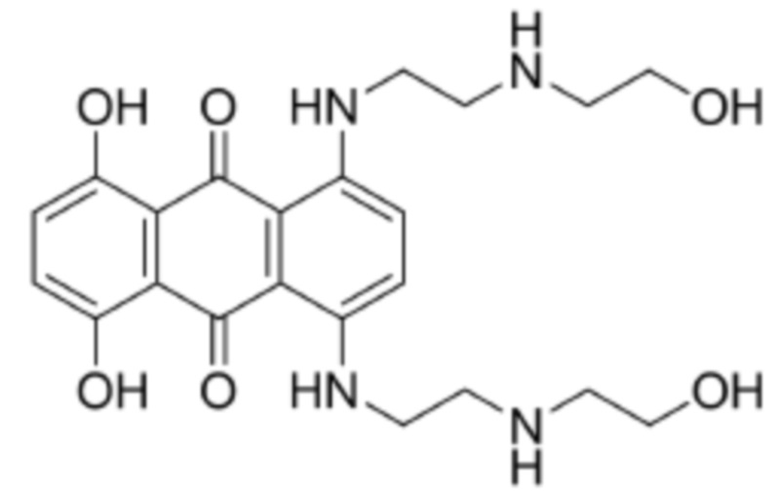
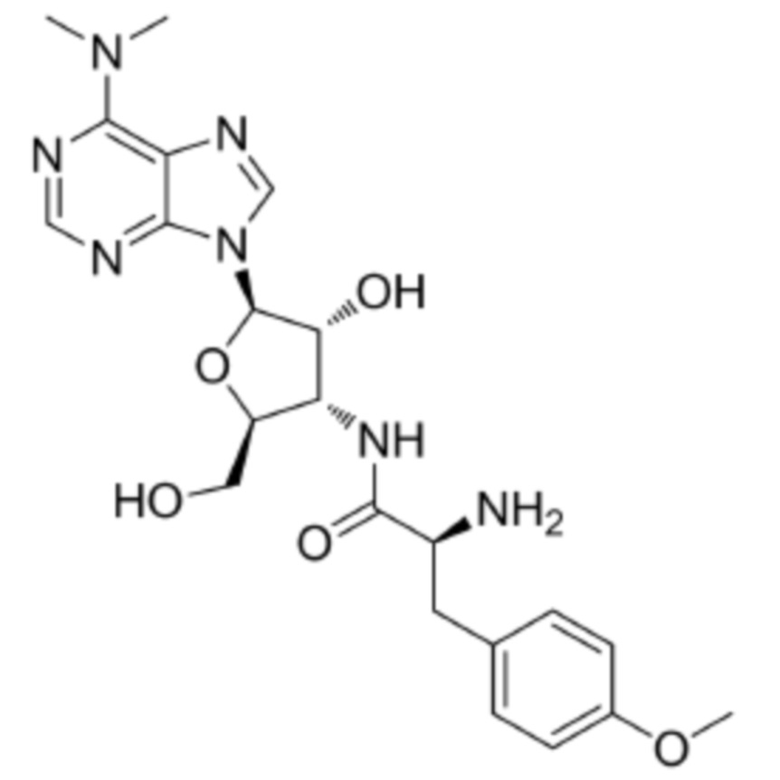
[0009] Предпочтительно, цитотоксическое средство класса ауристатинов выбрано из группы, состоящей из MMAE, MMAF, MMAD и их производных; цитотоксическое средство класса антрамицинов является антрамицином; цитотоксическое средство класса антрациклинов выбрано из группы, состоящей из даунорубицина, доксорубицина, эпирубицина, идарубицина, валрубицина и митоксантрона; и цитотоксическое средство класса пуромицинов является пуромицином.
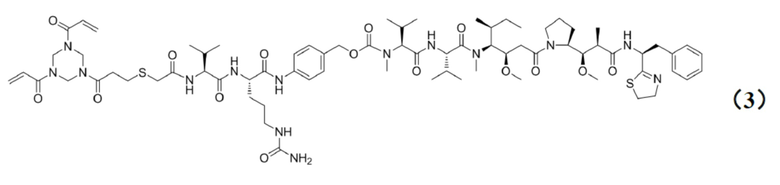
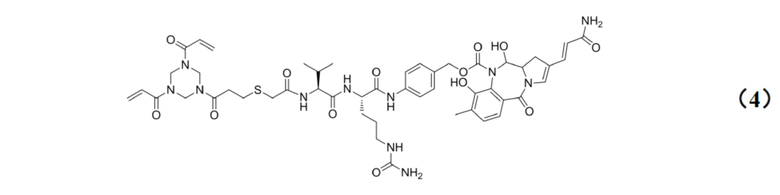
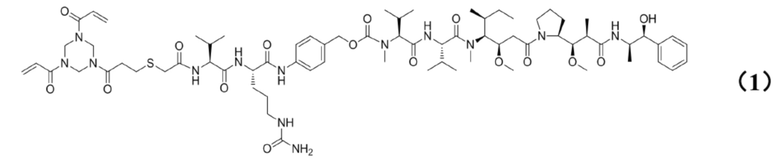
[0010] Предпочтительно, Py–MAA–Val–Cit–PAB–D имеет структуры, представленные формулами (1–11):



 .
.
[0011] Кроме того, растворитель является полярным растворителем и/или неполярным растворителем. Полярный растворитель является одним или более растворителями, выбранными из группы, состоящей из DMF, DMA и NMP; неполярный растворитель является одним или более растворителями, выбранными из группы, состоящей из дихлорметана и тетрахлорида углерода.
[0012] Кроме того, органическое основание является одним или более основаниями, выбранными из группы, состоящей из N,N–диизопропилэтиламина, триэтиламина и пиридина, предпочтительно, одним или двумя выбранными из группы, состоящей из N,N–диизопропилэтиламина и пиридина.
[0013] Кроме того, триазольный катализатор является одним или более катализаторами, выбранными из группы, состоящей из 1–гидроксибензотриазола, 1–гидрокси–7–азабензотриазола, этил–1–гидрокси–1H–1,2,3–триазол–4–карбоксилата, предпочтительно 1–гидроксибензотриазола.
[0014] Кроме того, температура реакции A составляет от 5 до 35°C, и продолжительность реакции составляет от 2 до 10 часов.
[0015] Предпочтительно, в реакции A контрольная температура реакционной системы должна находиться в диапазоне от 5 до 15°C перед добавлением бис(4–нитрофенил)карбоната, и более предпочтительно 10°C.
[0016] Предпочтительно, в реакции A контрольная температура реакционной системы должна находиться в диапазоне от 20 до 35°C после добавления органического основания, предпочтительно 30°C.
[0017] Кроме того, температура реакции B находится в диапазоне от 5 до 30°C, и продолжительность реакции находится в диапазоне от 15 до 48 часов.
[0018] Предпочтительно, в реакции B контрольная температура реакционной системы должна находиться в диапазоне от 5 до 15°C перед добавлением триазольного катализатора, предпочтительно 10°C.
[0019] Предпочтительно, в реакции B контрольная температура реакционной системы должна находиться в диапазоне от 15 до 30°C после добавления органического основания, предпочтительно 20°C. Кроме того, молярное соотношение Py–MAA–Val–Cit–PAB–OH, бис(4–нитрофенил)карбоната и органического основания в реакции A составляет 1:1–5:1–3.
[0020] Кроме того, молярное соотношение триазольного катализатора, MMAE, органического основания и Py–MAA–Val–Cit–PAB–OH в реакции B составляет 1–3:1–3:1,5–30:1.
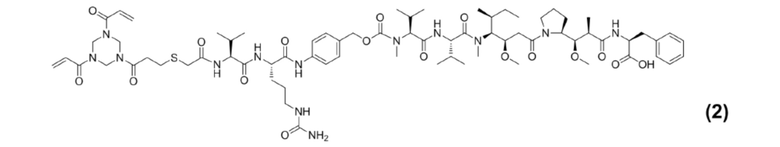
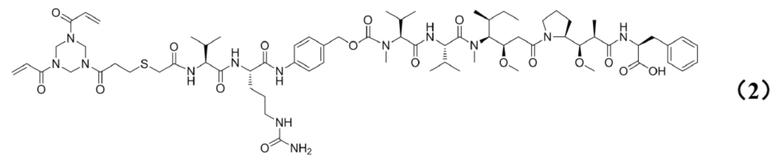
[0021] Кроме того, настоящее изобретение также относится к применению любого из указанных выше способов получения для получения промежуточного соединения конъюгата антитело–лекарственное средство, представляющего собой Py–MAA–Val–Cit–PAB–MMAF, и его структура представлена формулой (2):
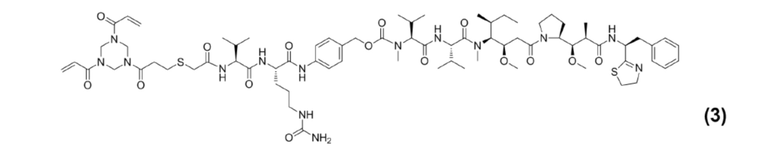
[0022] Кроме того, настоящее изобретение также относится к применению любого из указанных выше способов получения для получения промежуточного соединения конъюгата антитело–лекарственное средство, представляющего собой Py–MAA–Val–Cit–PAB–MMAD, и его структура представлена формулой (3):
[0023] Кроме того, настоящее изобретение также относится к применению любого из указанных выше способов получения для получения промежуточного соединения конъюгата антитело–лекарственное средство, имеющего структуру, представленную формулой (4):
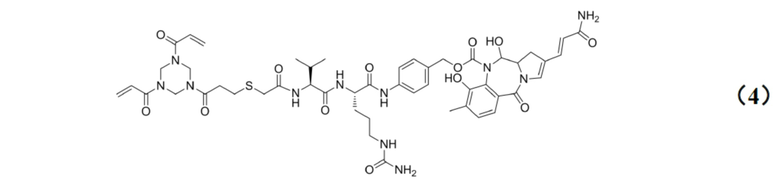
[0024] Кроме того, настоящее изобретение также относится к применению любого из указанных выше способов получения для получения промежуточного соединения конъюгата антитело–лекарственное средство, имеющего структуру, представленную формулой (5):
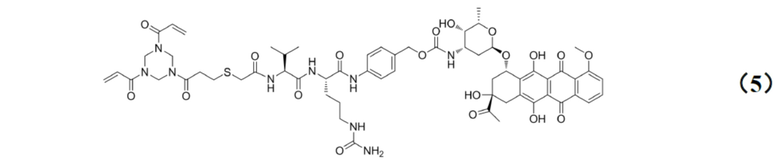
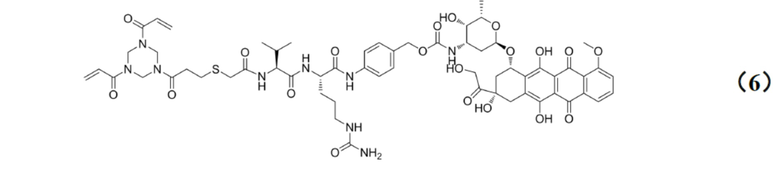
[0025] Кроме того, настоящее изобретение также относится к применению любого из указанных выше способов получения для получения промежуточного соединения конъюгата антитело–лекарственное средство, имеющего структуру, представленную формулой (6):
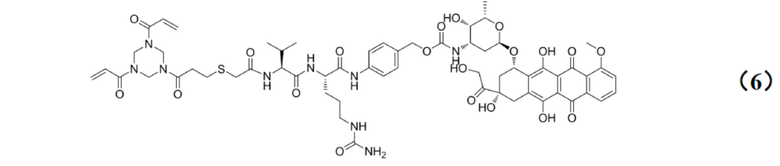
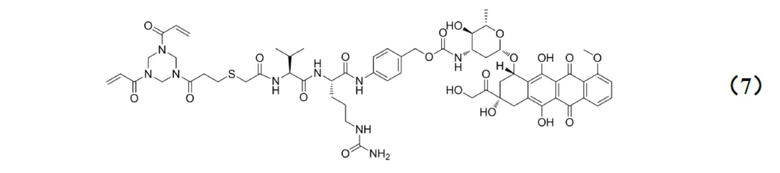
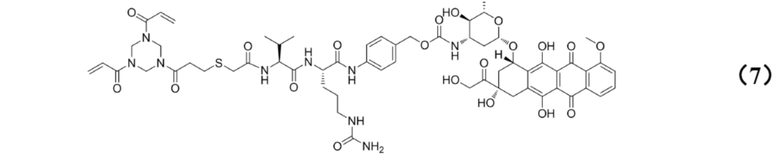
[0026] Кроме того, настоящее изобретение также относится к применению любого из указанных выше способов получения для получения промежуточного соединения конъюгата антитело–лекарственное средство, имеющего структуру, представленную формулой (7):

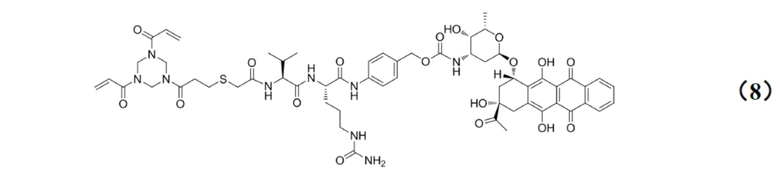
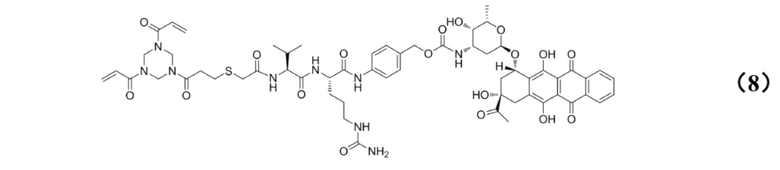
[0027] Кроме того, настоящее изобретение также относится к применению любого из указанных выше способов получения для получения промежуточного соединения конъюгата антитело–лекарственное средство, имеющего структуру, представленную формулой (8):
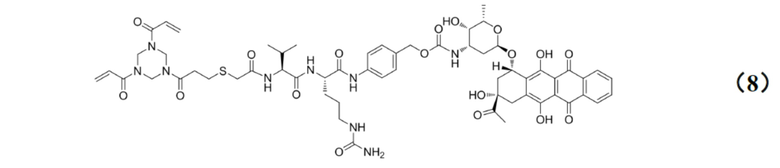
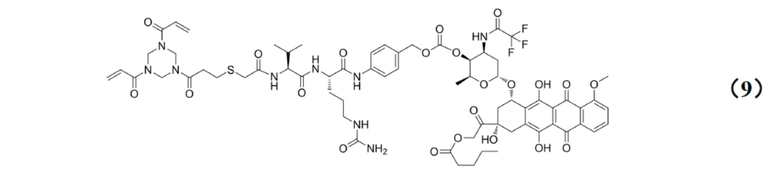
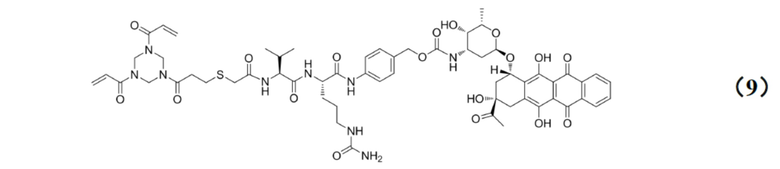
[0028] Кроме того, настоящее изобретение также относится к применению любого из указанных выше способов получения для получения промежуточного соединения конъюгата антитело–лекарственное средство, имеющего структуру, представленную формулой (9):
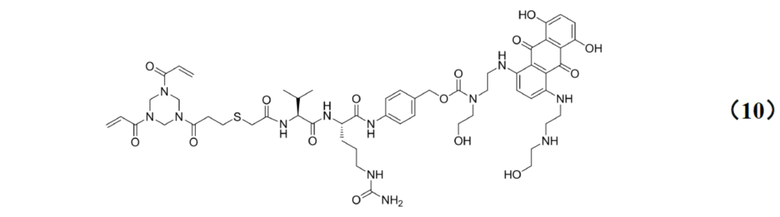
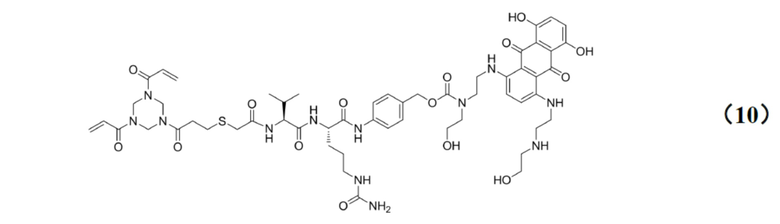
[0029] Кроме того, настоящее изобретение также относится к применению любого из указанных выше способов получения для получения промежуточного соединения конъюгата антитело–лекарственное средство, имеющего структуру, представленную формулой (10):
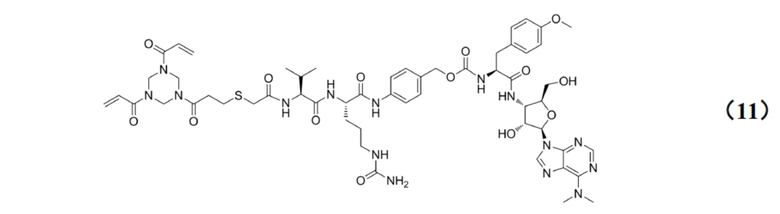
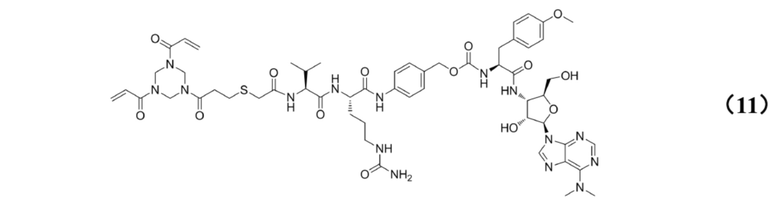
[0030] Кроме того, настоящее изобретение также относится к применению любого из указанных выше способов получения для получения промежуточного соединения конъюгата антитело–лекарственное средство, имеющего структуру, представленную формулой (11):

[0031] В некоторых конкретных вариантах осуществления способ включает следующие стадии:
Реакция A: растворение Py–MAA–Val–Cit–PAB в DMF, добавление бис(p–нитрофенил)карбоната, перемешивание до растворения или небольшой мутности перед добавлением N,N–диизопропилэтиламина по каплям, разделение и очистка для получения Py–MAA–Val–Cit–PAB–PNP;
Реакция B: растворение Py–MAA–Val–Cit–PAB–PNP в DMF, добавление 1–гидроксибензотриазола, MMAE, N,N–диизопропилэтиламина и пиридина, осуществление очистки с помощью препаративной колонки для ВЭЖХ после завершения реакции с получением соединения Py–MAA–Val–Cit–PAB–MMAE, представленного формулой (1), где структура Py–MAA–Val–Cit–PAB–PNP представлена следующим образом:
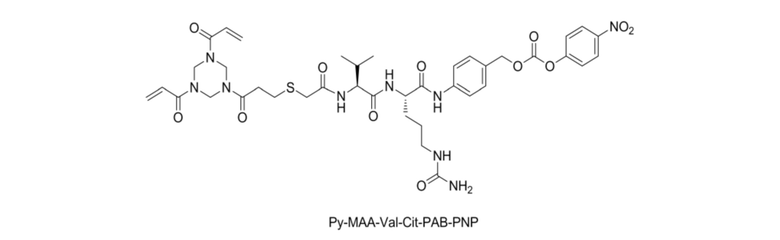
[0032] Кроме того, разделение и очистку в указанном выше способе реакции можно осуществлять следующим образом:
(1) центрифугирование реакционного раствора для реакции A с получением супернатанта;
(2) добавление растворителя 1 к супернатанту и тщательное перемешивание;
(3) добавление растворителя 2 по каплям при температуре от 0 до 10°C и тщательное перемешивание;
(4) фильтрация под вакуумом с получением фильтровального осадка;
(5) повторное растворение фильтровального осадка в смешанном растворе растворителя 3 и растворителя 4;
(6) добавление полученной смеси по каплям к растворителю 5 при температуре от 0 до 10°C и перемешивание;
(7) фильтрация под вакуумом с получением грязно–белого порошка, являющегося очищенным Py–MAA–Val–Cit–PAB–PNP.
При этом растворитель 1 является умеренно полярным растворителем, предпочтительно, выбранным из группы, состоящей из этилацетата, дихлорметана, метанола, метил–трет–бутилового эфира и подобного, наиболее предпочтительно этилацетата;
растворитель 2 является слабо полярным растворителем, предпочтительно, выбранным из группы, состоящей из петролейного эфира, n–гексана, n–пентана, циклогексана и подобного, наиболее предпочтительно петролейного эфира;
растворители 3 и 4 являются растворителями, в которых легко растворяется Py–MAA–Val–Cit–PAB–PNP, предпочтительно, выбранными из группы, состоящей из уксусной кислоты, трифторуксусной кислоты, муравьиной кислоты, метанола, этанола и подобного, наиболее предпочтительно уксусной кислоты и метанола;
растворитель 5 является растворителем, в котором Py–MAA–Val–Cit–PAB–PNP плохо растворяется, предпочтительно, выбранным из группы, состоящей из ацетонитрила, этилацетата, дихлорметана, воды и подобного, наиболее предпочтительно воды.
Объемное/массовое соотношение растворителя 1, растворителя 2, растворителя 3 и растворителя 4 и Py–MAA–Val–Cit–PAB составляет 40–100:80–200:5–15:1–3:15–30:1.
[0033] В некоторых конкретных вариантах осуществления, указанные выше разделение и очистку можно осуществлять следующим образом:
(1) центрифугирование промежуточного продукта A с получением супернатанта;
(2) добавление этилацетата в супернатант и тщательное перемешивание;
(3) добавление петролейного эфира по каплям при температуре от 0 до 10°C и тщательное перемешивание;
(4) фильтрация под вакуумом и получение фильтровального осадка;
(5) перерастворение фильтровального осадка в смешанном растворе уксусной кислоты и метанола;
(6) добавление очищенной воды по каплям при температуре от 0 до 10°C и перемешивание;
(7) фильтрация под вакуумом с получением грязно–белого порошка, являющегося очищенным Py–MAA–Val–Cit–PAB–PNP.
[0034] По сравнению со способом получения промежуточного соединения конъюгата антитело–лекарственное средство (Py–MAA–Val–Cit–PAB–MMAD), описанным в китайской патентной публикации № CN107921030A на стр. 43 описания, с помощью способа получения промежуточного соединения конъюгата антитело–лекарственное средство по настоящему изобретению значительно снижают потери функционального фрагмента лекарственного средства (такого как MMAD/MMAE или MMAF и т.д.), т.к. функциональный фрагмент лекарственного средства используют на последней стадии реакции, таким образом, эффективно снижая производственные затраты, а также повышая эффективность производства. Кроме того, способ по настоящему изобретению является простым, экологически чистым и подходящим для крупномасштабного промышленного производства.
ПОДРОБНОЕ ОПИСАНИЕ
[0035] Сокращения
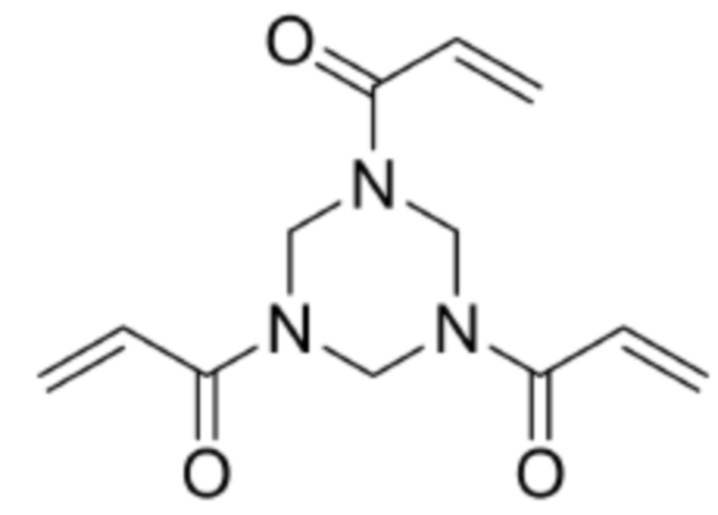
Триакрилоилгексагидротриазин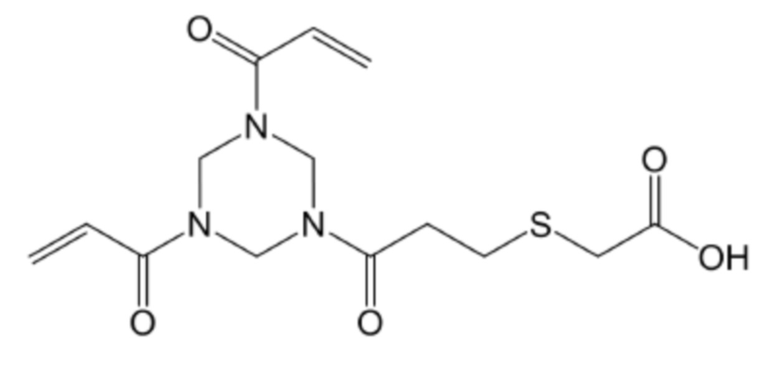
Триакрилоилгексагидротриазин–меркаптоуксусная кислота
Триакрилоилгексагидротриазин–меркаптоуксусная кислота–валин–цитруллин–p–аминобензиловый спирт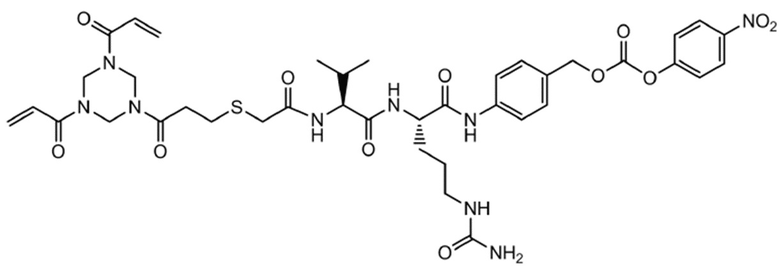
Триакрилоилгексагидротриазин–меркаптоуксусная кислота–валин–цитруллин–p–аминобензилоксикарбонил–p–нитрофениловый сложный эфир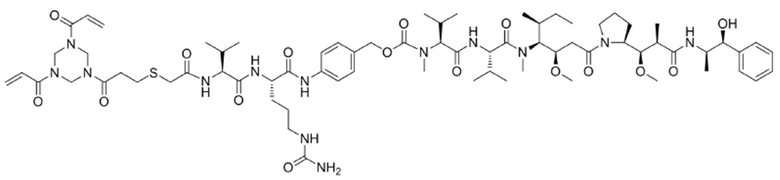
Триакрилоилгексагидротриазин–меркаптоуксусная кислота–валин–цитруллин–p–аминобензилоксикарбонилмонометил–ауристатин E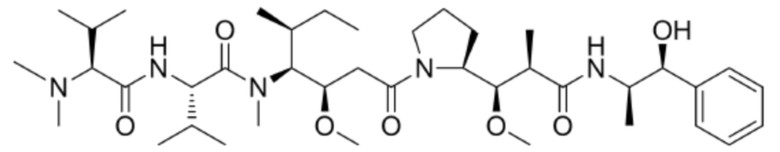
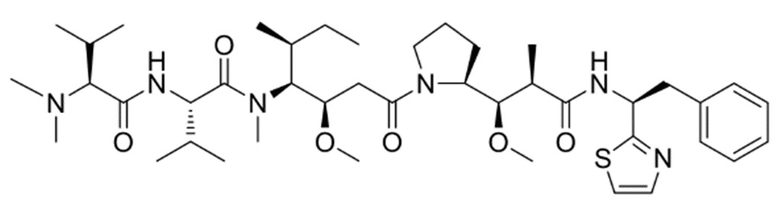
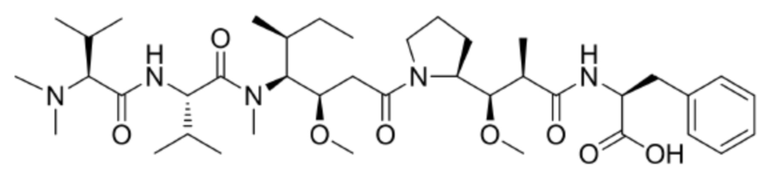
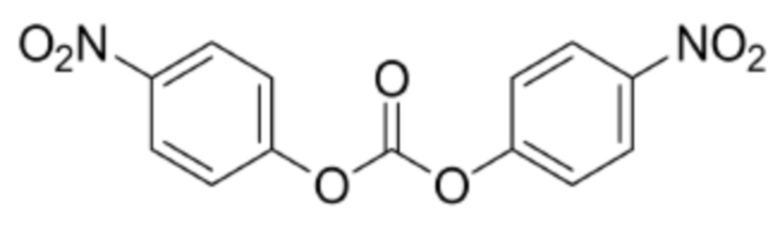
Бис(4–нитрофенил)карбонат
[0036] Определения
[0037] Если не указано иначе, все технические и научные термины, используемые в настоящем описании, обладают значением, общепринято понятным специалисту в этой области.
[0038] Хотя числовые диапазоны и приблизительные значения параметров представлены в настоящем описании в широком объеме, числовые значения, представленные в конкретных вариантах осуществления, описаны настолько точно, насколько это возможно. Однако любое числовое значение по определению содержит некоторые ошибки по причине стандартного отклонения при соответствующем измерении. Кроме того, все диапазоны, представленные в настоящем описании, следует интерпретировать как включающие любые и все поддиапазоны. Например, указанный диапазон "от 1 до 10" следует считать включающим любые и все поддиапазоны от минимума 1 до максимума 10 (включая конечные точки); т.е. все поддиапазоны, начинающиеся с минимума 1 или более, такого как от 1 до 6,1, и поддиапазоны, заканчивающиеся максимумом 10 или менее, таким от как 5,5 до 10. Кроме того, любую ссылку, обозначаемую как "включенная в настоящем описании", интерпретируют как включенную в полном объеме.
[0039] Кроме того, следует отметить, что в рамках изобретения формы в единственном числе включают формы во множественном числе, если четко не указано иное. Термин "или" можно использовать взаимозаменяемо с термином "и/или", если контекст четко не указывает на иное.
[0040] В рамках изобретения термин "конъюгат антитело–лекарственное средство" относится к соединению, в котором антитело/функциональный фрагмент антитела, линкер и функциональный фрагмент лекарственного средства химически соединяют друг с другом, и его структура, как правило, состоит из трех частей: антитела или антительного лиганда, функционального фрагмента лекарственного средства и линкера, с помощью которого соединяют антитело или антительный лиганд и лекарственное средство. В настоящее время получение конъюгатов антитело–лекарственное средство, как правило, разделяют на две стадии: первой стадией является химическая реакция линкера с функциональным фрагментом лекарственного средства с образованием конъюгата "линкер–лекарственное средство", и второй стадией является ковалентное соединение линкерной части конъюгата "линкер–лекарственное средство" с антителом/функциональным фрагментом антитела с помощью тиоловой группы или аминогруппы. В рамках изобретения термин "промежуточное соединение конъюгата антитело–лекарственное средство" относится к конъюгату "линкер–лекарственное средство", как описано выше, и, кроме того, термин "промежуточное соединение конъюгата антитело–лекарственное средство" по изобретению, как правило, относится к конъюгату "линкер–лекарственное средство", в котором линкер и лекарственное средство соединяют с помощью связи "–CO–NH–" посредством аминной переэтерификации.
[0041] В рамках изобретения термины "линкер" и "линкерная часть" относятся к части конъюгата антитело–лекарственное средство, с помощью которой соединяют антитело с лекарственным средством, которая может являться расщепляемой или нерасщепляемой. Расщепляемый линкер (т.е. линкер, являющийся расщепляемым или биодеградируемым линкером) может расщепляться внутри или на клетке–мишени с высвобождением лекарственного средства. В некоторых вариантах осуществления линкер по настоящему изобретению выбран из расщепляемых линкеров, таких как линкер на основе дисульфида (селективно расщепляющийся в опухолевых клетках, имеющих более высокую концентрацию сульфгидрильных групп), пептидный линкер (расщепляемый ферментом в опухолевых клетках) или гидразоновый линкер. В других вариантах осуществления линкер по настоящему изобретению выбран из нерасщепляемых линкеров (т.е. линкеров, не являющихся расщепляемыми), таких как тиоэфирный линкер. В других вариантах осуществления линкер по настоящему изобретению представляет собой комбинацию расщепляемого линкера и нерасщепляемого линкера.
[0042] В рамках изобретения термины "лекарственное средство" и "функциональный фрагмент лекарственного средства", как правило, относятся к любому соединению, имеющему желаемую биологическую активность и реакционноспособную функциональную группу для получения конъюгатов по настоящему изобретению. Желаемая биологическая активность включает диагностику, излечение, облегчение, лечение, профилактику заболеваний у людей или других животных. Т.к. непрерывно открывают и разрабатывают новые лекарственные средства, эти новые лекарственные средства также следует включать в лекарственные средства по настоящему изобретению. В частности, лекарственное средство включает, в качестве неограничивающих примеров, цитотоксические лекарственные средства, факторы дифференцировки клеток, трофические факторы стволовых клеток, стероидные лекарственные средства, лекарственные средства для лечения аутоиммунных заболеваний, противовоспалительные лекарственные средства или лекарственные средства против инфекционных заболеваний. Более конкретно, лекарственное средство включает, в качестве неограничивающих примеров, ингибиторы тубулина или средства, повреждающие ДНК или РНК.
[0043] Примеры
[0044] Далее представлены примеры способов и композиций по настоящему изобретению. Следует понимать, что в свете представленных выше определений и общего описания можно осуществлять различные другие варианты осуществления.
[0045] Пример 1. Получение Py–MAA
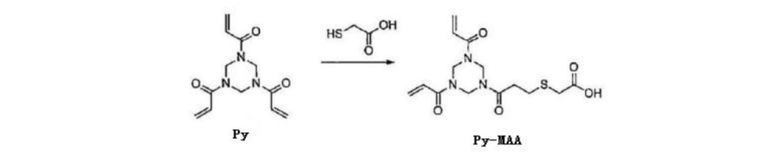
[0046] Соединение Py (1,87 г, 7,51 ммоль) (CAS: 959–52–4) и Et3N (104 мкл, 0,75 ммоль) растворяли в безводном CH2Cl2 (40 мл), по каплям добавляли раствор тиогликолевой кислоты (103,9 мкл, 1,50 ммоль) в CH2Cl2 (40 мл). После завершения добавления температуру системы доводили до комнатной температуры и перемешивали систему в течение ночи. После завершения реакции растворитель удаляли под вакуумом и очищали неочищенный продукт посредством хроматографии на колонках для получения белого твердого Py–MAA (1,87 г, 40,1%).
[0047] Пример 2. Сравнительный пример: Получение Py–MAA–Val–Cit–PAB–MMAE (представляющий собой способ, описанный на стр. 32 описания китайской патентной публикации № CN107921030A).
[0048] Способ представлен ниже:
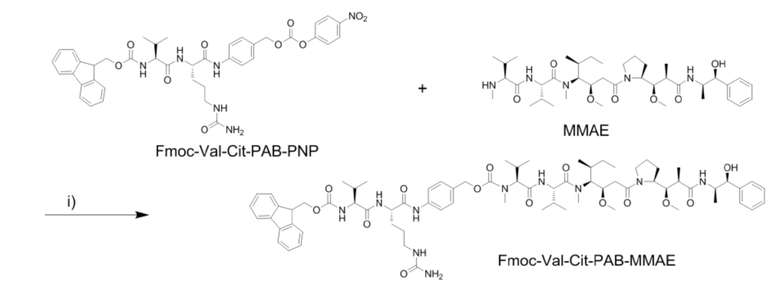
[0049] (1) Получение Fmoc–Val–Cit–PAB–MMAE
[0050] Fmoc–Val–Cit–PAB–PNP (388,4 мг, 0,507 ммоль) получали и растворяли в DMF (40 м) в защитной атмосфере азота при 0°C и добавляли HOBt (68,4 мг, 0,507 ммоль) и соединение MMAE (400,0 мг, 0,558 ммоль). Через 15 минут добавляли пиридин (8 мл) и DIPEA (106,2 мкл, 0,608 ммоль) и проводили реакцию при 0°C в течение 30 минут. Температуру повышали до комнатной температуры и перемешивали смесь в течение 3 часов. Реакционную смесь дополняли DIPEA (106,2 мкл, 0,608 ммоль) и реакцию продолжали при комнатной температуре в течение 24 часов. После завершения реакции смесь концентрировали под вакуумом. Неочищенный продукт очищали посредством хроматографии на колонках с получением белого твердого соединения Fmoc–Val–Cit–PAB–MMAE (325 мг, выход 47,7%).
[0051] (2) Получение Val–Cit–PAB–MMAE
[0052] Соединение Fmoc–Val–Cit–PAB–MMAE (325 мг, 0,289 ммоль) растворяли в DMF (6 мл) перед добавлением диэтиламина (3 мл) и реакцию продолжали с перемешиванием в течение 2,5 часов. После завершения реакции растворитель удаляли при пониженном давлении с получением неочищенного продукта Val–Cit–PAB–MMAE (220 мг, выход: 67,8%), который можно использовать для следующей реакции напрямую без очистки.
[0053] 3) Синтез Py–MAA–Val–Cit–PAB–MMAE
[0054] Соединение Py–MAA (80,2 мг, 0,235 ммоль), HOBt (31,8 мг, 0,235 ммоль) и DIC (29,6 мг, 0,235 ммоль) получали и растворяли в DMF (10 мл) и охлаждали до 0°C на ледяной бане. В реакционный раствор добавляли соединение Val–Cit–PAB–MMAE (220,0 мг, 0,196 ммоль) и DIPEA (25,3 мг, 0,196 ммоль), температуру доводили до комнатной температуры и проводили реакцию в течение 24 часов. После завершения реакции неочищенный продукт очищали посредством хроматографии на колонках (метанол:дихлорметан=60:1–30:1) с получением белого твердого соединения Py–MAA–Val–Cit–PAB–MMAE (147,4 мг, выход 52%). LCMS m/z(ES+), 1445,78 (M+H)+.
[0055] Py–MAA–Val–Cit–PAB–MMAE получали способом получения Py–MAA–Val–Cit–PAB–MMAD, как описано на стр. 32 описания китайской патентной публикации № CN107921030A. Исходное количество материала MMAE составляло 400 мг, а количество полученного Py–MAA–Val–Cit–PAB–MMAE – 147,4 мг.
[0056] Пример 3. Улучшенный способ получения Py–MAA–Val–Cit–PAB–MMAE
[0057] (1) Получение Py–MAA–Val–Cit–PAB
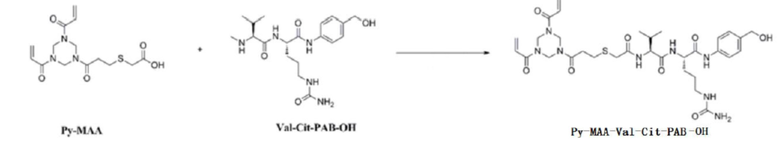
[0058] Соединение Py–MAA (10,00 г, 29,33 ммоль) помещали в тетрагидрофуран (200 мл) перед добавлением N,N'–карбонилдиимидазола (7,13 г, 44,00 ммоль) и Val–Cit–PAB–OH (13,34 г, 35,20 ммоль) и перемешивали при комнатной температуре в течение 24 часов. Добавляли петролейный эфир (200 мл), перемешивали в течение 0,5 часов и фильтровали с получением белого твердого вещества. Белое твердое вещество очищали посредством препаративной высокоэффективной жидкостной хроматографии, полученное вещество подвергали ротационному выпариванию с получением Py–MAA–Val–Cit–PAB–OH (6,67 г, 32,4%, белый твердый порошок).
[0059] (2) Получение Py–MAA–Val–Cit–PAB–PNP
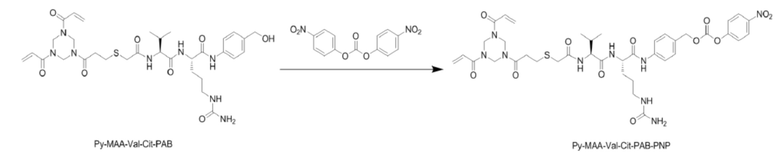
[0060] Py–MAA–Val–Cit–PAB–OH (10,00 г, 1,0 экв.) и DMF (VDMF/WPy–MAA–Val–Cit–PAB=15–25 мл/г) добавляли в трехгорлую колбу в защитной атмосфере азота, перемешивали в защитной атмосфере азота до растворения твердого вещества или получения немного мутного раствора. Температуру в трехгорлой колбе снижали до 10°C, добавляли твердый NPC (1,5–2,5 экв., 6,50–10,83 г) в одну часть и перемешивали до растворения или получения немного мутного раствора. По каплям добавляли DIPEA (1,0–1,5 экв., 1,84–2,76 г) в защитной атмосфере азота. Температуру доводили до 30°C и осуществляли мониторинг реакции посредством UPLC до достижения чистоты Py–MAA–Val–Cit–PAB менее 5,0%, затем реакцию прекращали.
[0061] Реакционный раствор центрифугировали и супернатант наливали в лабораторный стакан подходящего размера, в который добавляли EA (VEA/WPy–MAA–Val–Cit–PAB=60±2) и перемешивали с помощью электрического миксера; по каплям добавляли PE (VPE/WPy–MAA–Val–Cit–PAB=120±2) при температуре от 0 до 6°C и после добавления по каплям перемешивали в течение от 20 до 30 минут. После фильтрации под вакуумом получали фильтровальный осадок и растворяли его уксусной кислотой (VAcOH/WPy–MAA–Val–Cit–PAB=7) и метанолом (VMeOH/WPy–MAA–Val–Cit–PAB) с получением раствора.
[0062] В лабораторный стакан подходящего размера наливали очищенную воду (Vвода/WPy–MAA–Val–Cit–PAB =21), механически перемешивали и охлаждали до приблизительно 3°C. Указанный выше раствор добавляли по каплям в очищенную воду и после добавления по каплям перемешивали в течение 15 минут. После фильтрации под вакуумом получали фильтровальный осадок и лиофилизировали с получением Py–MAA–Val–Cit–PAB–PNP (белого или грязно–белого порошка, 10,69 г, выход 86,54%).
[0063] (3) Получение Py–MAA–Val–Cit–PAB–MMAE
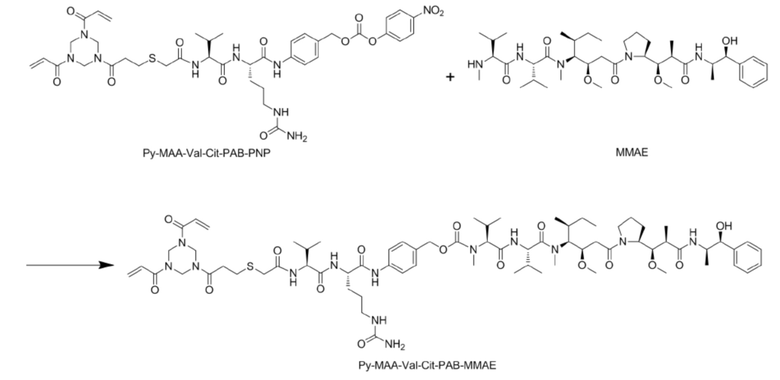
[0064] В реакционный раствор добавляли Py–MAA–Val–Cit–PAB–PNP (9,00 г, 1,0 экв.) и DMF (VDMF/WPy–MAA–Val–Cit–PAB–PNP=15,0–25,0 мл/г), перемешивали в защитной атмосфере азота до получения прозрачного или немного мутного раствора. Поддерживали внутреннюю температуру 10°C, одновременно добавляя HOBt (2,10 г, 1,5 экв.) и перемешивая в течение 20 минут. Добавляли MMAE (8,93 г, 1,2 экв.) и продолжали перемешивание в течение 20 минут. По каплям добавляли смешанный раствор DIPEA (1,34 г, 1,0 экв.) и пиридина (Vпиридин/VDIPEA=5) и доводили температуру для поддержания внутренней температуры 20°C. Через 36 часов реакции осуществляли мониторинг посредством UPLC раз в час и, когда чистота MMAE в реакционном растворе составляла <2,00%, реакцию останавливали.
[0065] Брали лабораторный стакан подходящего размера, в который добавляли насыщенный водный раствор хлорида натрия (обозначаемый как рассол, Vрассол/Vреакционный раствор=10), и снижали температуру до достижения внутренней температуры 5°C. В насыщенный раствор хлорида натрия по каплям добавляли реакционный раствор. Неочищенный продукт получали посредством центрифугирования и очищали посредством препаративной высокоэффективной жидкостной хроматографии. После лиофилизации получали Py–MAA–Val–Cit–PAB–MMAE (белый порошок, 9,79 г, выход 65,27%).
[0066] В этом примере исходное количество MMAE составляло 8,93 г, а полученное количество Py–MAA–Val–Cit–PAB–MMAE составляло 9,79 г.
[0067] При сравнении способов, представленных в примере 2 и примере 3 в терминах количества исходного MMAE и массы конечного продукта (результаты сравнения представлены в таблице 1), из таблицы 1 видно, что полученное количество конечного продукта Py–MAA–Val–Cit–PAB–MMAE, полученного способом получения из примера 2, составляло 147,4 мг, в то время как исходное количество MMAE составляло 400 мг; полученное количество конечного продукта Py–MAA–Val–Cit–PAB–MMAE, полученного способом получения из примера 3, составляло 9,79 г, в то время как исходное количество MMAE составляло 8,93 г.
Таблица 1. Сравнение количества исходного MMAE и полученного количества конечного продукта
[0068] Технологический коэффициент MMAE представляет собой то, как много единиц MMAE необходимо затратить для получения каждой единицы массы Py–MAA–Val–Cit–PAB–MMAE. Как видно из таблицы 1, при использовании способа из примера 2 необходимо 2,71 единицы массы MMAE для получения 1 единицы массы Py–MAA–Val–Cit–PAB–MMAE; в то время как при адаптации способа из примера 3 необходимо лишь 0,91 единиц массы MMAE. Исходное количество снижалось на 66,4%, что значительно снижает производственные затраты.
[0069] Выше описан только предпочтительный вариант осуществления, являющийся исключительно иллюстративным, а не ограничивающим комбинации признаков, необходимых для практического осуществления настоящего изобретения. Представленное название не предназначено для ограничения различных вариантов осуществления изобретения. Термины, такие как "содержащий" и "включающий", не предназначены для ограничения. Кроме того, если не указано иначе, в случае отсутствия числовой модификации включены формы во множественном числе, и термин "или" означает "и/или". Если в настоящем описании не указано иначе, все технические и научные термины, используемые в настоящем описании, обладают значением, общепринято понятным специалисту в этой области.
[0070] Все публикации и патенты, упомянутые в настоящей заявке, включены в нее, таким образом, в качестве ссылки. Специалисту в этой области очевидны многочисленные модификации и варианты описываемых способов и композиций по настоящему изобретению. Хотя настоящее изобретение описано с помощью конкретных предпочтительных вариантов осуществления, следует понимать, что настоящее изобретение, как указано в формуле изобретения, не должно быть чрезмерно ограничено этими конкретными вариантами осуществления. Фактически, эти многочисленные модификации и варианты, используемые для практического осуществления принципов настоящего изобретения, очевидных специалисту в этой области, предназначены для включения в объем формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОДНОРЕАКТОРНЫЙ СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО ПРОДУКТА КОНЪЮГАТА АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2020 |
|
RU2745738C1 |
| АНТИ-МЕЗОТЕЛИН АНТИТЕЛО И ЕГО КОНЪЮГАТ С ЛЕКАРСТВЕННЫМИ СРЕДСТВАМИ | 2019 |
|
RU2747995C1 |
| ЛИНКЕР ДЛЯ КОНЪЮГАТОВ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2792201C2 |
| КОНЪЮГАТ АНТИТЕЛО ПРОТИВ C-MET-ЛЕКАРСТВЕННОЕ СРЕДСТВО И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2822497C1 |
| КОНЪЮГАТ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО, НАГРУЖЕННЫЙ БИНАРНЫМИ ТОКСИНАМИ, И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2795148C2 |
| АНТИТЕЛО ПРОТИВ КЛАУДИНА 18.2 И ЕГО КОНЪЮГАТ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2022 |
|
RU2814164C2 |
| МАТЕРИАЛЫ И СПОСОБЫ, СВЯЗАННЫЕ С ЛИНКЕРАМИ ДЛЯ ПРИМЕНЕНИЯ В КОНЪЮГАТАХ ЛЕКАРСТВЕННОГО СРЕДСТВА И БЕЛКА | 2015 |
|
RU2737553C2 |
| Конъюгаты "антитело - лекарственное средство" с высокой лекарственной нагрузкой | 2015 |
|
RU2674979C2 |
| КОНЪЮГАТ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО, ИМЕЮЩИЙ КИСЛОТНЫЙ САМОСТАБИЛИЗИРУЮЩИЙСЯ УЧАСТОК СОЕДИНЕНИЯ | 2018 |
|
RU2793125C2 |
| АНТИТЕЛА ПРОТИВ TENB2, СКОНСТРУИРОВАННЫЕ С ЦИСТЕИНОМ, И КОНЪЮГАТЫ АНТИТЕЛО - ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2008 |
|
RU2505544C2 |
Изобретение относится к области медицины. Предложен способ получения промежуточного соединения конъюгата антитело–лекарственное средство, содержащего линкер и функциональный фрагмент лекарственного средства, в котором промежуточное соединение конъюгата антитело–лекарственное средство является Py–MAA–Val–Cit–PAB–D, Py–MAA–Val–Cit–PAB является линкером, D представляет собой присоединяемый функциональный фрагмент лекарственного средства и присоединяемый функциональный фрагмент лекарственного средства содержит свободную аминогруппу. Изобретение обеспечивает снижение потери функционального фрагмента лекарственного средства, т.к. функциональный фрагмент лекарственного средства используют на последней стадии реакции. 14 з.п. ф-лы, 2 табл., 3 пр.
1. Способ получения промежуточного соединения конъюгата антитело–лекарственное средство, содержащего линкер и функциональный фрагмент лекарственного средства, в котором промежуточное соединение конъюгата антитело–лекарственное средство является Py–MAA–Val–Cit–PAB–D, Py–MAA–Val–Cit–PAB в промежуточном соединении является линкером, и D в промежуточном соединении представляет собой присоединяемый функциональный фрагмент лекарственного средства, и присоединяемый функциональный фрагмент лекарственного средства содержит свободную аминогруппу, где способ включает следующие реакции:
 .
.
2. Способ по п.1, где
Реакция A: растворение Py–MAA–Val–Cit–PAB–OH в растворителе, добавление бис(4–нитрофенил)карбоната, перемешивание до растворения или получения немного мутного раствора, а затем добавление органического основания;
Реакция B: после завершения реакции A, добавление триазольного катализатора, функционального фрагмента лекарственного средства D и органического основания и, после завершения реакции, осуществление очистки с получением Py–MAA–Val–Cit–PAB–D.
3. Способ по п.1 или 2, где функциональный фрагмент лекарственного средства D является цитотоксическим средством класса ауристатинов, цитотоксическим средством класса антрамицинов, цитотоксическим средством класса антрациклинов или цитотоксическим средством класса пуромицинов.
4. Способ по п.3, где цитотоксическое средство класса ауристатинов выбрано из группы, состоящей из MMAE, MMAF, MMAD и их производного; цитотоксическое средство класса антрамицинов является антрамицином или его производным; цитотоксическое средство класса антрациклинов выбрано из группы, состоящей из даунорубицина, доксорубицина, эпирубицина, идарубицина, валрубицина, митоксантрона и их производных; и цитотоксическое средство класса пуромицинов является пуромицином или его производным.
5. Способ по п.4, где структура Py–MAA–Val–Cit–PAB–D представлена формулами (1–11):
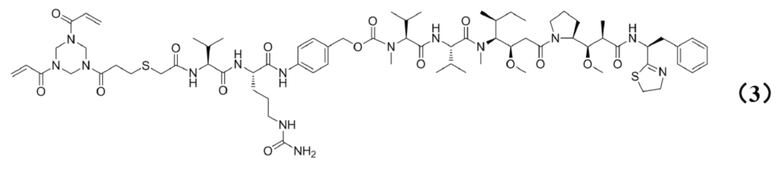
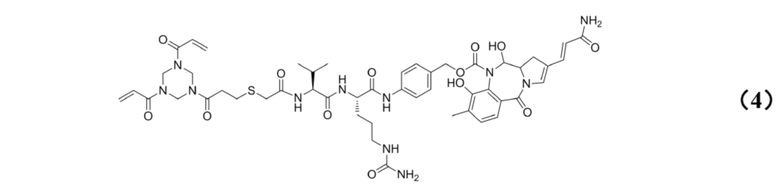
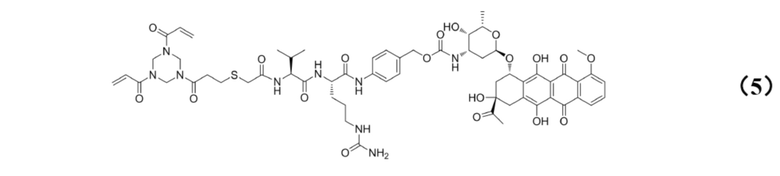
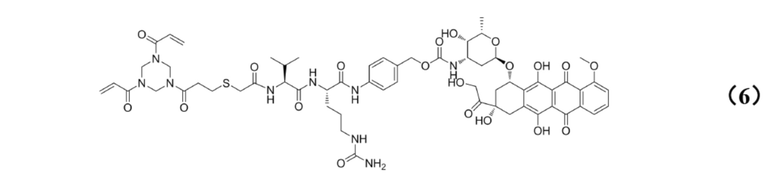


 .
.
6. Способ по любому из предшествующих пунктов, где растворитель является полярным растворителем и/или неполярным растворителем, полярный растворитель является одним или более выбранными из группы, состоящей из DMF, DMA и NMP; неполярный растворитель является одним или более выбранными из группы, состоящей из дихлорметана и тетрахлорида углерода.
7. Способ по любому из предшествующих пунктов, где органическое основание является одним или более выбранными из группы, состоящей из N,N–диизопропилэтиламина, триэтиламина и пиридина, предпочтительно, одним или двумя выбранными из группы, состоящей из N,N–диизопропилэтиламина и пиридина.
8. Способ по любому из предшествующих пунктов, где триазольный катализатор является одним или более выбранными из группы, состоящей из 1–гидроксибензотриазола, 1–гидрокси–7–азабензотриазола и этил–1–гидрокси–1H–1,2,3–триазол–4–карбоксилата, предпочтительно 1–гидроксибензотриазола.
9. Способ по любому из предшествующих пунктов, включающий следующее: в реакции A контрольная температура реакционной системы должна находиться в диапазоне от 5 до 15°C перед добавлением бис(4–нитрофенил)карбоната, предпочтительно 10°C; и более предпочтительно в реакции A контрольная температура реакционной системы должна находиться в диапазоне от 20 до 35°C после добавления органического основания, предпочтительно 30°C.
10. Способ по любому из предшествующих пунктов, включающий следующее: в реакции B контрольная температура реакционной системы должна находиться в диапазоне от 5 до 15°C перед добавлением триазольного катализатора, предпочтительно 10°C; более предпочтительно, в реакции B контрольная температура реакционной системы должна находиться в диапазоне от 15 до 30°C после добавления органического основания, предпочтительно 20°C.
11. Способ получения по любому из предшествующих пунктов, где в реакции A молярное соотношение Py–MAA–Val–Cit–PAB–OH, бис(4–нитрофенил)карбоната и органического основания составляет 1:1–5:1–3.
12. Способ по любому из предшествующих пунктов, где в реакции B молярное соотношение триазольного катализатора, MMAE, органического основания и Py–MAA–Val–Cit–PAB–OH составляет 1–3:1–3:1,5–30:1.
13. Способ по любому из предшествующих пунктов, включающий после завершения реакции B осуществление очистки с помощью препаративной колонки для ВЭЖХ с получением Py–MAA–Val–Cit–PAB–D.
14. Способ по любому из предшествующих пунктов, где очистку осуществляют в реакции A следующим способом с получением Py–MAA–Val–Cit–PAB–PNP:
(1) центрифугирование реакционного раствора реакции A с получением супернатанта;
(2) добавление растворителя 1 к супернатанту и тщательное перемешивание;
(3) добавление растворителя 2 по каплям при температуре от 0 до 10°C и тщательное перемешивание;
(4) фильтрация под вакуумом с получением фильтровального осадка;
(5) повторное растворение фильтровального осадка в смешанном растворе растворителя 3 и растворителя 4;
(6) добавление полученного раствора по каплям к растворителю 5 при температуре от 0 до 10°C и перемешивание;
(7) фильтрация под вакуумом с получением грязно–белого порошка, являющегося очищенным Py–MAA–Val–Cit–PAB–PNP;
где растворитель 1 является умеренно полярным растворителем, предпочтительно, выбранным из группы, состоящей из этилацетата, дихлорметана, метанола, метил–трет–бутилового эфира и подобного, наиболее предпочтительно этилацетата; растворитель 2 является слабо полярным растворителем, предпочтительно, выбранным из группы, состоящей из петролейного эфира, n–гексана, n–пентана, циклогексана и подобного, наиболее предпочтительно петролейного эфира; растворитель 3 и растворитель 4 являются растворителями, в которых легко растворяется Py–MAA–Val–Cit–PAB–PNP, предпочтительно, выбранными из группы, состоящей из уксусной кислоты, трифторуксусной кислоты, муравьиной кислоты, метанола, этанола и подобного, наиболее предпочтительно уксусной кислоты и метанола; растворитель 5, предпочтительно, выбран из группы, состоящей из ацетонитрила, этилацетата, дихлорметана, воды и подобного, наиболее предпочтительно воды.
15. Способ по любому из предшествующих пунктов, где реакции осуществляют в защитной атмосфере азота.
| CN 107921030 A, 17.04.2018 | |||
| АНТИТЕЛА И ИММУНОКОНЪЮГАТЫ И ИХ ПРИМЕНЕНИЯ | 2007 |
|
RU2436796C9 |
| 2005 |
|
RU2404810C2 | |
| КОНЪЮГАТЫ "ПРОИЗВОДНОЕ КАЛИХЕАМИЦИНА-НОСИТЕЛЬ" | 2003 |
|
RU2422157C2 |

