Область техники, к которой относится изобретение
Изобретение относится к новым соединениям, композициям, содержащим эти соединения, и их применению в терапии, например при лечении микобактериальных инфекций или при лечении вызванных микобактериями заболеваний, таких как туберкулез (также известный как TB).
Предпосылки создания изобретения
В соответствии с отчетом, опубликованным Всемирной Организации Здравоохранения в 2014 г., каждый год почти десять миллионов людей инфицируются туберкулезом (ТВ), вызывая 1,5 миллиона смертей в год. Несмотря на доступные методы лечения туберкулеза, заболеваемость по-прежнему начинает расти вследствие инфекции Mycobacterium tuberculosis из-за того, что бактериальный агент, вызывающий TB, становятся устойчивыми ко многим препаратам первой линии, таким как изониазид и рифампицин.
Этионамид, структурный аналог изониазида, который является таким же эффективным, как изониазид, часто назначают для лечения резистентного ко многим лекарственным средствам TB (MDR TB). Тем не менее, недостатком, связанным с использованием этионамида, является то, что для получения приемлемой концентрации лекарственного средства в крови, требуется доза до 1 г/день, что связанно с серьезными побочными эффектами, включая нейротоксичность и фатальную гепатотоксичность. Таким образом, существует потребность в снижении клинической дозы и уменьшении воздействия этионамида.
Следовательно, одна цель настоящего изобретения состоит в создании новых соединений, которые могут усиливать активность препаратов, используемых при лечении туберкулеза, в частности, лекарственных средств, которые активируются через сигнальный путь EthA, как и этионамид.
В публикации РСТ WO 2014/0966378 описаны пиперидиновые и пирролидиновые соединения, в которых пиперидиновое или пирролидиновое кольцо замещено различными бензильными, фенильными или гетероциклическими группами, например незамещенной пиридиновой группой. Считается, что такие соединения полезны при лечении TB.
Сущность изобретения
В первом аспекте настоящего изобретения предлагается соединение формулы (I) или его фармацевтически приемлемая соль:
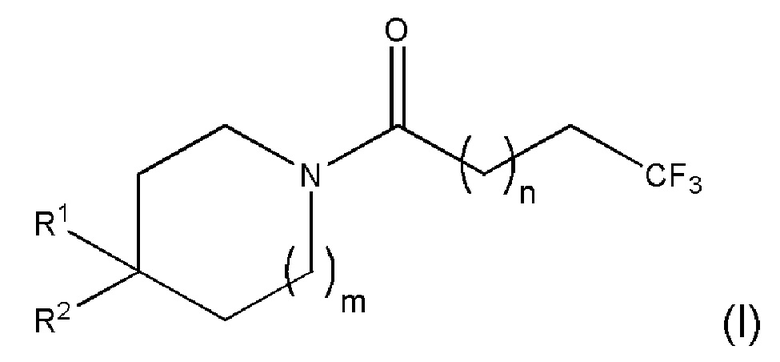
где
n равно 1 или 2;
m равно 0 или 1;
R1 представляет собой Н или F; и
R2 представляет собой пиридил, необязательно замещенный одним или двумя заместителями, независимо выбранными из фтора, хлора, брома, циано, метила, необязательно замещенного одним или несколькими атомами фтора, и метокси, необязательно замещенного одним или несколькими атомами фтора, или
R2 представляет собой пиразинил, необязательно замещенный в мета-положении заместителем, выбранным из фтора, хлора, брома, циано, метила, необязательно замещенного одним или несколькими атомами фтора, и метокси, необязательно замещенного одним или несколькими атомами фтора, или
R2 представляет собой пиразинил, необязательно замещенный в пара-положении фтором или хлором,
при этом, когда R1 представляет собой Н, то R2 является замещенным, и когда m равно 0, то R1 представляет собой H.
Во втором аспекте настоящего изобретения предлагается соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии.
В соответствии с третьим аспектом настоящего изобретения предлагается соединение формулы (I) или его фармацевтически приемлемая соль для применения при лечении микобактериальной инфекции или для применения в лечении заболевания, вызванного микобактериальной инфекцией.
В соответствии с четвертым аспектом настоящего изобретения предлагается соединение формулы (I) или его фармацевтически приемлемая соль для применения при лечении туберкулеза.
В пятом аспекте настоящего изобретения предлагается способ лечения микобактериальной инфекции у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В шестом аспекте настоящего изобретения предлагается способ лечения заболевания, вызванного микобактериальной инфекции, у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В седьмом аспекте настоящего изобретения предлагается применение соединения формулы (I) или его фармацевтически приемлемой соли при изготовлении лекарственного средства, применяемого при лечении микобактериальной инфекции или заболевания, вызванного микобактериальной инфекцией.
В восьмом аспекте настоящего изобретения предлагается фармацевтическая композиция, содержащая (а) соединение формулы (I), описанное выше, или его фармацевтически приемлемую соль; и (b) фармацевтически приемлемый эксципиент.
В девятом аспекте настоящего изобретения, предлагается комбинация (а) соединения формулы (I), описанного выше, или его фармацевтически приемлемой соли; и (b) по меньшей мере одного другого агента против микобактерий.
Подробное описание изобретения
Как укахзано выше, один аспект настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли:
где
n равно 1 или 2;
m равно 0 или 1;
R1 представляет собой Н или F; и
R2 представляет собой пиридил, необязательно замещенный одним или двумя заместителями, независимо выбранными из фтора, хлора, брома, циано, метила, необязательно замещенного одним или несколькими атомами фтора, и метокси, необязательно замещенного одним или несколькими атомами фтора, или
R2 представляет собой пиразинил, необязательно замещенный в мета-положении заместителем, выбранным из фтора, хлора, брома, циано, метила, необязательно замещенного одним или несколькими атомами фтора, и метокси, необязательно замещенного одним или несколькими атомами фтора, или
R2 представляет собой пиразинил, необязательно замещенный в пара-положении фтором или хлором,
при этом, когда R1 представляет собой Н, то R2 является замещенным, и когда m равно 0, то R1 представляет собой H.
В одном варианте осуществления изобретения, соединение по изобретению представляет собой соединение формулы (I), как определено выше.
В одном варианте осуществления n равно 1.
В одном варианте осуществления m равно 1.
В одном варианте осуществления изобретения R1 представляет собой H.
В одном варианте осуществления n равно 1, m равно 1 и R1 представляет собой H.
В одном варианте осуществления изобретения, в частности, когда R1 представляет собой Н, R2 представляет собой замещенный пиридил, который может представлять собой 2-пиридил, 3-пиридил или 4-пиридил, где заместители являются такими, как определено в пункте 1.
В одном варианте осуществления изобретения, когда R2 представляет собой пиридил, этот пиридил может быть замещенным одним или двумя заместителями, независимо выбранными из фтора, хлора, брома, циано, метила, необязательно замещенного одним или несколькими атомами фтора, и метокси, необязательно замещенного одним или несколькими атомами фтора.
В одном варианте осуществления изобретения, когда R2 представляет собой пиридил, этот пиридил может быть замещенным одним или двумя заместителями, независимо выбранными из фтора, хлора, брома, циано, метила, трифторметила и метокси.
В одном варианте осуществления изобретения, когда R2 представляет собой пиридил, этот пиридил может быть замещенным одним или двумя заместителями, независимо выбранными из фтора, хлора, метила, необязательно замещенного одним или несколькими атомов фтора и метокси, необязательно замещенного одним или несколькими атомами фтора.
В одном варианте осуществления изобретения, когда R2 представляет собой пиридил, этот пиридил может быть замещенным одним или двумя заместителями, независимо выбранными из фтора, хлора, метила, трифторметила и метокси.
В одном варианте осуществления изобретения, когда R2 представляет собой 2-пиридил или 4-пиридил, он может быть замещен одним или двумя заместителями, независимо выбранными из хлора, фтора, брома, циано, метила, необязательно замещенного одним или несколькими атомами фтора, и метокси, необязательно замещенного одним или несколькими атомами фтора.
В одном варианте осуществления изобретения, когда R2 представляет собой 2-пиридил, он может быть замещен одним или двумя заместителями, независимо выбранными из хлора, фтора и трифторметила.
В одном варианте осуществления изобретения, когда R2 представляет собой 4-пиридил, он может быть замещен одним или двумя заместителями, независимо выбранными из фтора, хлора, метила и трифторметила.
В одном варианте осуществления изобретения, когда R2 представляет собой 3-пиридил, он может быть замещен одним или двумя заместителями, независимо выбранными из хлора, фтора, брома, циано, метила, необязательно замещенного одним или несколькими атомами фтора, и метокси, необязательно замещенного одним или несколькими атомами фтора, где, когда заместитель представляет собой трифторметил, он присоединен в положении 5 пиридинового кольца, и когда заместитель представляет собой метокси, он присоединен в положении 6 пиридинового кольца.
В одном варианте осуществления изобретения, когда R2 представляет собой 3-пиридил, он может быть замещен одним или двумя заместителями, независимо выбранными из хлора, фтора, метокси и трифторметила, где, когда заместитель представляет собой трифторметил, он присоединен в положении 5 пиридинового кольца, и когда заместитель представляет собой метокси, он присоединен в положении 6 пиридинового кольца. В этом варианте осуществления, этот заместитель также может быть присоединен в положении 5 пиридинового кольца.
В одном варианте осуществления изобретения, когда R2 представляет собой пиразинил, он замещен хлором в мета-положении или в пара-положении, или он замещен трифторметилом в мета-положении.
В одном варианте осуществления изобретения n равно 1, m равно 1 и R2 представляет собой пиридил, замещенный одним или двумя заместителями, где
когда R2 представляет собой 2-пиридил или 4-пиридил, то заместители независимо выбраны из хлора, фтора, брома, циано, метила, необязательно замещенного одним или несколькими атомами фтора, и метокси, необязательно замещенного одним или несколькими атомами фтора; и
когда R2 представляет собой 3-пиридил, то заместители независимо выбраны из хлора, фтора, брома, циано, метила, необязательно замещенного одним или несколькими атомами фтора, и метокси, необязательно замещенного одним или несколькими атомами фтора, и когда заместитель представляет собой трифторметил, он присоединен в положении 5 пиридинового кольца, и когда заместитель представляет собой метокси, он присоединен в положении 6 пиридинового кольца.
В одном варианте осуществления изобретения n равно 1, m равно 1 и R2 представляет собой пиридил, замещенный одним или двумя заместителями, где
когда R2 представляет собой 2-пиридил, то заместители независимо выбраны из хлора, фтора и трифторметила;
когда R2 представляет собой 3-пиридил, то заместители независимо выбраны из хлора, фтора, метокси и трифторметила, и когда заместитель представляет собой трифторметил, он присоединен в положении 5 пиридинового кольца, и когда заместитель представляет собой метокси, он присоединен в положении 6 пиридинового кольца; и
когда R2 представляет собой 4-пиридил, то заместители независимо друг от друга выбирают из фтора, хлора, метила и трифторметила.
В одном варианте осуществления изобретения n равно 1, m равно 1 и R2 представляет собой 3-пиридил, замещенный одним или двумя заместителями, независимо выбранными из хлора, фтора, метокси и трифторметила, где
когда заместитель представляет собой трифторметил, он присоединен в положении 5 пиридинового кольца, и когда заместитель представляет собой метокси, он присоединен в положении 6 пиридинового кольца.
В одном варианте осуществления изобретения R2 представляет собой 3-пиридил, замещенный хлором, фтором или трифторметилом, или R2 представляет собой 4-пиридил, замещенный фтором или трифторметилом.
В одном варианте осуществления изобретения n равно 1, m равно 1 и R2 представляет собой 3-пиридил, замещенный хлором или фтором, или R2 представляет собой 4-пиридил, замещенный фтором или трифторметилом.
В одном варианте осуществления изобретения R2 представляет собой 4-пиридил, замещенный одним заместителем, который представляет собой трифторметил.
В одном варианте осуществления изобретения R2 представляет собой пиридил, необязательно замещенный одним или двумя заместителями, независимо выбранными из фтора, хлора, метила, трифторметила и метокси, и когда заместитель представляет собой трифторметил, он присоединен в мета-положении пиридинового кольца; или R2 представляет собой пиразинил, замещенный в мета-положении одним или двумя заместителями, независимо выбранными из фтора, хлора и трифторметила; или R2 представляет собой пиразинил, замещенный в пара-положении фтором или хлором.
В одном варианте осуществления изобретения n равно 1, m равно 1, R1 представляет собой Н, и R2 представляет собой пиридил, замещенный одним или двумя заместителями, независимо выбранными из фтора, хлора, метила, трифторметила и метокси, и когда заместитель представляет собой трифторметил, он присоединен в мета-положении пиридинового кольца; или R2 представляет собой пиразинил, замещенный в мета-положении одним или двумя заместителями, независимо выбранными из фтора, хлора и трифторметила; или R2 представляет собой пиразинил, замещенный в пара-положении фтором или хлором.
В одном варианте осуществления изобретения R2 представляет собой пиридил, замещенный одним или двумя заместителями, независимо выбранными из фтора, хлора, метила, трифторметила и метокси, и когда заместитель представляет собой трифторметил, он присоединен в мета-положении пиридинового кольца; или R2 представляет собой пиразинил, замещенный в мета-положении одним или двумя заместителями, независимо выбранными из фтора, хлора и трифторметила; или R2 представляет собой пиразинил, замещенный в пара-положении фтором или хлором.
В одном варианте осуществления изобретения n равно 1, m равно 1, R1 представляет собой Н и R2 представляет собой пиридил, замещенный одним заместителем, выбранным из фтора, хлора, метила, трифторметил и метокси, и когда заместитель представляет собой трифторметил, он присоединен в мета-положение пиридинового кольца; или R2 представляет собой пиразинил, замещенный в мета-положении одним заместителем, выбранным из фтора, хлора и трифторметила; или R2 представляет собой пиразинил, замещенный в пара-положении фтором или хлором.
В одном варианте осуществления изобретения n равно 1, m равно 1, R1 представляет собой Н и R2 представляет собой 2-пиридил, замещенный одним или двумя заместителями, независимо выбранными из фтора, хлора и трифторметила; или R2 представляет собой 3-пиридил, замещенный одним или двумя заместителями, независимо выбранными из фтора, хлора, метокси и трифторметила; или R2 представляет собой 4-пиридил, замещенный одним или двумя заместителями, независимо выбранными из фтора, хлора, метила и трифторметила; или R2 представляет собой пиразинил, замещенный в мета-положении одним заместителем, выбранным из фтора, хлора и трифторметила; или R2 представляет собой пиразинил, замещенный в пара-положении фтором или хлором.
В одном варианте осуществления изобретения n равно 1, m равно 1, R1 представляет собой Н и R2 представляет собой 4-пиридил, замещенный одним заместителем, который представляет собой трифторметил.
В другом варианте осуществления изобретения R2 представляет собой 3-пиридил, замещенный одним заместителем, который представляет собой хлор.
В одном варианте осуществления изобретения n равно 1 и m равно 0. Как уже отмечалось выше, когда m равно 0, то R1 представляет собой H. В этом варианте осуществления изобретения R2 предпочтительно представляет собой пиридил, замещенный одним или двумя заместителями, независимо выбранными из фтора, хлора, брома, циано, метила, необязательно замещенного одним или несколькими атомами фтора, и метокси, необязательно замещенного одним или несколькими атомами фтора. Пиридил может представлять собой 2-пиридил, 3-пиридил или 4-пиридил. В этом варианте осуществления изобретения R2 предпочтительно представляет собой 3-пиридил или 4-пиридил, замещенный одним или двумя заместителями.
В одном варианте осуществления изобретения n равно 1, m равно 0 и R2 представляет собой замещенный пиридил, который может представлять собой 2-пиридил, 3-пиридил или 4-пиридил, где заместитель выбран из хлора и трифторметила. В этом варианте осуществления изобретения R2 предпочтительно представляет собой замещенный 3-пиридил или 4-пиридил, в частности, 3-пиридил.
В одном варианте осуществления изобретения n равно 1, m равно 0, R1 представляет собой Н, и R2 представляет собой замещенный пиридил, который может представлять собой 2-пиридил, 3-пиридил или 4-пиридил, где заместители являются такими, как определено в пункте 1. В этом варианте осуществления изобретения R2 предпочтительно представляет собой замещенный 3-пиридил или 4-пиридил. В частности, когда m равно 0, то R2 представляет собой 3-пиридил, замещенный одним заместителем, который представляет собой хлор.
В одном варианте осуществления изобретения R1 представляет собой F. В этом конкретном варианте осуществления изобретения, в котором R1 представляет собой F, R2 представляет собой пиридил, замещенный одним заместителем, выбранным из фтора, хлора и трифторметила. В качестве альтернативы, R2 может представлять собой пиразинил, замещенный хлором. В другом варианте осуществления изобретения R2 представляет собой 3-пиридил или 4-пиридил, замещенный одним заместителем, где заместитель представляет собой хлор, фтор или трифторметил. В альтернативном варианте осуществления изобретения R2 представляет собой незамещенный пиридил, в частности, 2-пиридил.
В одном варианте осуществления изобретения, когда R1 представляет собой F, R2 выбран из незамещенного пиридила или пиридила, замещенного одним заместителем, выбранным из фтора, хлора и трифторметила.
В одном варианте осуществления изобретения, когда R1 представляет собой F, R2 выбран из незамещенного 2-пиридила и 3-пиридила; или 2-пиридила, замещенного трифторметилом; или 3-пиридила, замещенного фтором, хлором или трифторметилом; или 4-пиридила, замещенного фтором, хлором или трифторметилом.
Альтернативно, когда R1 представляет собой F, R2 представляет собой 3-пиридил или 4-пиридил, замещенный одним или двумя заместителями, независимо выбранными из фтора, хлора, брома, циано, метила, необязательно замещенного одним или несколькими атомами фтора, или метокси, необязательно замещенного одним или несколькими атомами фтора. В одном конкретном варианте осуществления заместитель представляет собой фтор или хлор.
В одном варианте осуществления изобретения R2 замещен одним заместителем, выбранным из фтора, хлора, брома, циано, метила, необязательно замещенного одним или несколькими атомами фтора, или метокси, необязательно замещенного одним или несколькими атомами фтора.
Во всех описанных выше вариантах осуществления изобретения предпочтительно, чтобы R2, когда он является замещенным, замещение было в мета-положении.
В одном варианте осуществления изобретения, когда R2 представляет собой пиридил, он замещен одним заместителем в мета-положении, и заместитель выбран из хлора, фтора, метила, метокси и трифторметила.
В одном варианте осуществления изобретения, когда R2 представляет собой пиразинил, он замещен одним заместителем в мета-положении, и заместителем является фтор, хлор или трифторметил.
Во всех описанных выше вариантах осуществления изобретения предпочтительно, чтобы R2, когда он является замещенным, был замещен только одним заместителем.
Конкретные соединения, которые являются полезными в настоящем изобретении, включают следующие соединения:
4,4,4-трифтор-1-[4-фтор-4-(3-пиридил)-1-пиперидил]бутан-1-он;
4,4,4-трифтор-1-[4-(5-фтор-3-пиридил)-1-пиперидил]бутан-1-он;
4,4,4-трифтор-1-[4-(6-фтор-3-пиридил)-1-пиперидил]бутан-1-он;
4,4,4-трифтор-1-[4-[6-(трифторметил)-3-пиридил]-1-пиперидил]бутан-1-он;
4,4,4-трифтор-1-[4-[4-(трифторметил)-2-пиридил]-1-пиперидил]бутан-1-он;
4,4,4-трифтор-1-[4-[5-(трифторметил)-3-пиридил]-1-пиперидил]бутан-1-он;
4,4,4-трифтор-1-[4-[6-(трифторметил)-2-пиридил]-1-пиперидил]бутан-1-он;
4,4,4-трифтор-1-[4-(6-фтор-2-пиридил)-1-пиперидил]бутан-1-он;
4,4,4-трифтор-1-[4-(6-метокси-3-пиридил)-1-пиперидил]бутан-1-он;
4,4,4-трифтор-1-[4-[2-(трифторметил)-3-пиридил]-1-пиперидил]бутан-1-он;
4,4,4-трифтор-1-[4-(5-метокси-3-пиридил)-1-пиперидил]бутан-1-он;
1-[4-(3,5-дифтор-2-пиридил)-1-пиперидил]-4,4,4-трифтор-бутан-1-он;
1-[4-(2,6-дифтор-3-пиридил)-1-пиперидил]-4,4,4-трифтор-бутан-1-он;
4,4,4-трифтор-1-[4-(5-фтор-2-пиридил)-1-пиперидил]бутан-1-он;
4,4,4-трифтор-1-(4-(2-(трифторметил)пиридин-4-ил)пиперидин-1-ил)бутан-1-он;
4,4,4-трифтор-1-(4-(2-фторпиридин-4-ил)пиперидин-1-ил)бутан-1-он;
4,4,4-трифтор-1-(4-(5-(трифторметил)пиразин-2-ил)пиперидин-1-ил)бутан-1-он;
4,4,4-трифтор-1-(4-(2-метилпиридин-4-ил)пиперидин-1-ил)бутан-1-он;
1-(4-(5,6-дифторпиридин-3-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-он;
4,4,4-трифтор-1-(4-(6-(трифторметил)пиразин-2-ил)пиперидин-1-ил)бутан-1-он;
1-(4-(2-хлорпиридин-4-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-он;
4,4,4-трифтор-1-(4-(3-фторпиридин-4-ил)пиперидин-1-ил)бутан-1-он;
1-(4-(6-хлорпиридин-2-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-он;
1-(4-(5-хлорпиридин-3-ил)-4-фторпиперидин-1-ил)-4,4,4-трифторбутан-1-он;
1-(4-(5-хлорпиридин-3-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-он;
1-(4-(5-хлорпиридин-3-ил)пиперидин-1-ил)-5,5,5-трифторпентан-1-он;
1-(4-(6-хлорпиразин-2-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-он;
4,4,4-трифтор-1-(4-фтор-4-(пиридин-2-ил)пиперидин-1-ил)бутан-1-он;
4,4,4-трифтор-1-(4-фтор-4-(5-(трифторметил)пиридин-3-ил)пиперидин-1-ил)бутан-1-он;
4,4,4-трифтор-1-(4-фтор-4-(2-(трифторметил)пиридин-4-ил)пиперидин-1-ил)бутан-1-он;
1-(4-(6-хлорпиразин-2-ил)-4-фторпиперидин-1-ил)-4,4,4-трифторбутан-1-он;
4,4,4-трифтор-1-(4-фтор-4-(2-фторпиридин-4-ил)пиперидин-1-ил)бутан-1-он;
4,4,4-трифтор-1-[4-фтор-4-[4-(трифторметил)-2-пиридил]-1-пиперидил]бутан-1-он;
4,4,4-трифтор-1-[4-фтор-4-[4-(трифторметил)-2-пиридил]-1-пиперидил]бутан-1-он;
1-(3-(5-хлорпиридин-3-ил)пирролидин-1-ил)-4,4,4-трифторбутан-1-он; и
1-[4-(2-хлор-4-пиридил)-4-фтор-1-пиперидил]-4,4,4-трифтор-бутан-1-он.
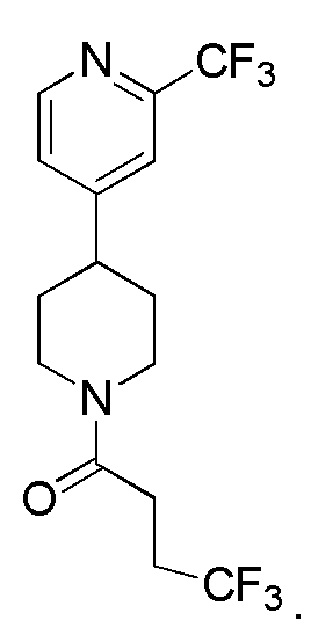
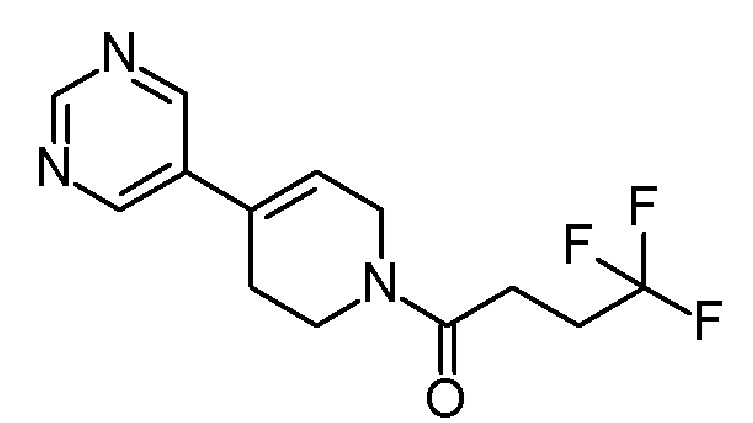
В одном варианте осуществления изобретения, соединение по изобретению представляет собой 4,4,4-трифтор-1-(4-(2-(трифторметил)пиридин-4-ил)пиперидин-1-ил)бутан-1-он, имеющее следующую структуру:
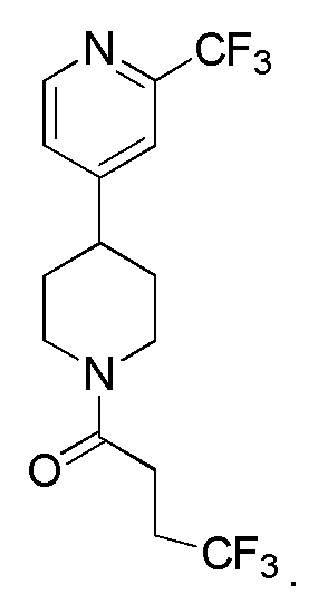
Также раскрыто соединение Формулы (II) или его фармацевтически приемлемая соль:
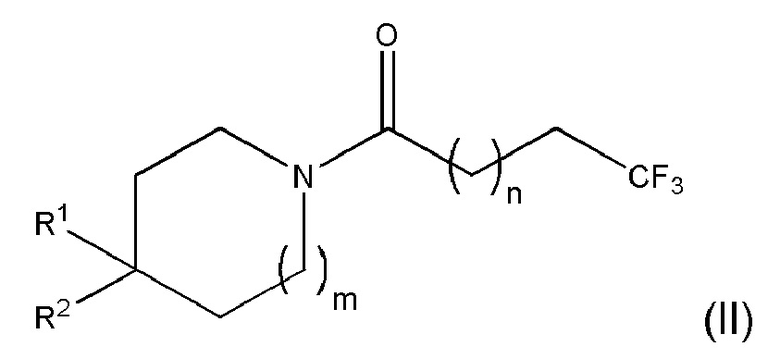
где n равно 1 или 2; m равно 0 или 1; R1 представляет собой Н или F; и
R2 представляет собой пиридил, пиразинил, пиримидинил или пиридазинил, каждый из которых необязательно замещен одним или двумя заместителями, независимо выбранными из фтора, хлора, брома, циано, метила, необязательно замещенного одним или несколькими атомами фтора, и метокси, необязательно замещенного одним или несколькими атомами фтора, и при этом, когда R1 представляет собой Н и R2 представляет собой пиридил, то пиридил является замещенным, и когда m равно 0, то R1 представляет собой Н,
при условии, что соединение является иным, чем
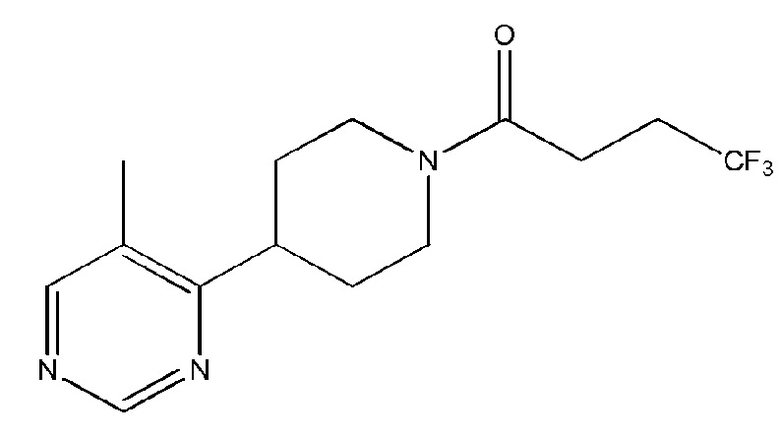
Во избежание ошибочного понимания, соединение, имеющее структуру
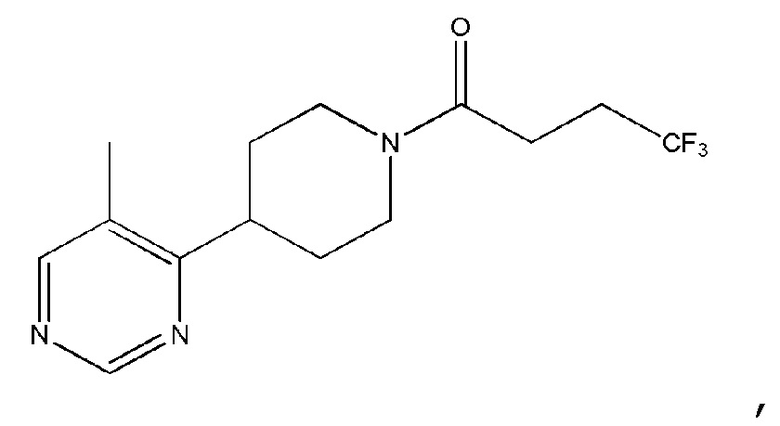
имеет название 4,4,4-трифтор-1-(4-(5-метилпиримидин-4-ил)пиперидин-1-ил)бутан-1-он.
Каждый из описанных выше вариантов осуществления изобретения также применим в отношении соединений формулы (II).
Термины и определения
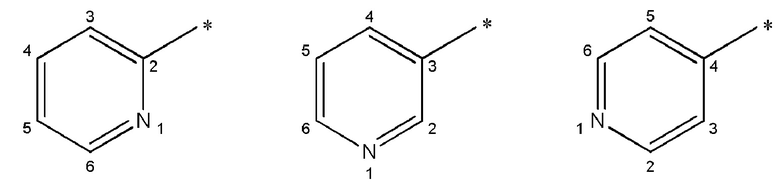
Как используется в данном описании, термин «пиридил» относится к пиридиновой замещающей группе, которая включает 2-пиридил, 3-пиридил и 4-пиридил. Во избежание ошибочного понимания в отношении 2-пиридила, 3-пиридила и 4-пиридила, используется следующая система нумерации IUPAC:
Как показано выше, * обозначает точку присоединения.
Когда в отношении пиридила или пиразинила используется термин «мета» или «пара», то он имеет свое обычное значение в данной области, т.е., мета-замещение или пара-замещение относительно точки присоединения.
Во избежание ошибочного понимания, когда термин «мета» используется в отношении замещения 2-пиридила, он обозначает замещение в положении 4 или 6, как определено выше.
Во избежание ошибочного понимания, когда термин «мета» используется по отношению замещения 3-пиридила, он обозначает замещение в положении 5, как определено выше.
Во избежание ошибочного понимания, когда термин «мета» используется в отношении замещения 4-пиридила, он обозначает замещение в положении 4 или 6, как определено выше.
Как используется в данном описании, термин «пиразинили» относится к пиразиновой замещающей группе. Когда термин «мета» или «пара» используются в отношении замещения пиразинила, то он имеет свое обычное значение в данной области, т.е., мета-замещение или пара-замещение относительно точки присоединения.
Во избежание ошибочного понимания, когда термин «пара» используется в отношении замещения пиразинила, то он означает, что замещение представлено в противоположном положении по отношению точки присоединения, то есть в положении 5, как показано ниже:
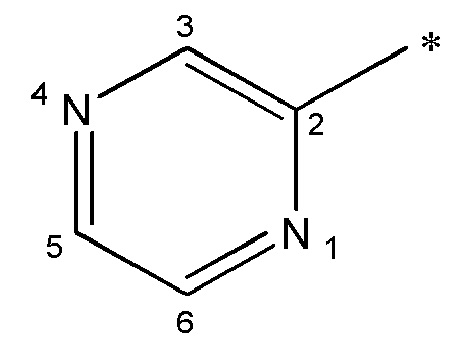
Во избежание ошибочного понимания, когда термин «мета» используется в отношении замещения пиразинила, предполагается, то он означает, что замещение представлено в положении 6, как показано выше.
Как используются в данном описании, термины «пиримидинил» и «пиридазинил» относятся пиримидиновой или пиридазиновой группе-заместителю.
Как используется в данном описании, термин «циано» относится к группе -CN.
Как используется в данном описании, термин «метил, необязательно замещенный одним или несколькими атомами фтора» относится к метильной группе, которая может быть замещена одним, двумя или тремя атомами фтора. Следовательно, термин «метил, необязательно замещенный одним или несколькими атомами фтора» включает метил, монофторметил (-CH2F), дифторметил (-CHF2) и трифторметил (-CF3).
Как используется в данном описании, термин «метокси, необязательно замещенного одним или несколькими атомами фтора» относится к группе метокси, в которой углерод метильной группы может быть замещен одним, двумя или тремя атомами фтора. Таким образом, термин «метокси, необязательно замещенный одним или несколькими атомами фтора» включает метокси, монофторметокси (-OCH2F), дифторметокси (-OCHF2) и трифторметокси (-OCF3).
Термин «соединения по изобретению», используемый в данном описании, означает соединение формулы (I) или его фармацевтически приемлемую соль. Термин «соединение по изобретению» означает любое одно из соединений по изобретению, определенное выше.
Кроме того, следует понимать, что указания, такие как «соединение формулы (I) или его фармацевтически приемлемая соль» или «соединения по изобретению», подразумевают соединение формулы (I) его фармацевтически приемлемую соль или сольват соединения формулы (I) или их любую фармацевтически приемлемую комбинацию. Таким образом, в качестве неограничивающего примера, используемого в данном описании с целью иллюстрации, указание «соединение формулы (I) или его фармацевтически приемлемая соль» охватывает фармацевтически приемлемую соль соединения формулы (I), которая присутствует в виде сольвата, и это указание также охватывает смесь соединения формулы (I) и фармацевтически приемлемой соли соединения формулы (I).
Следует понимать, что в данном описании ссылки на соединение формулы (I) или его фармацевтически приемлемая соль охватывают соединение формулы (I) в виде свободного основания или в виде его фармацевтически приемлемой соли. Таким образом, в одном варианте осуществления настоящее изобретение относится к соединению формулы (I). В другом варианте осуществления настоящее изобретение относится к фармацевтически приемлемой соли соединения формулы (I).
Термин «фармацевтический приемлемый» относится к соединениям (включая соль), материалам, композициям и лекарственным формам, которые в пределах объема знаний в области медицины подходят для использования при контакте с тканями человека и животных без чрезмерной токсичности, раздражения или других проблем или осложнений, соразмерных с разумным соотношением польза/риск.
Фармацевтически приемлемые соли включают, среди прочего, такие, которые описаны в Berge, J. Pharm. Sci, 1977, 66, 1-19, или такие, которые перечислены в справочнике P.H. Stahl & C.G. Wermuth: Handbook of Pharmaceutical Salts; Properties, Selection and Use, Second Edition Stahl/Wermuth: Wiley-VCH/VHCA, 2011 (см. http://www.wiley.com/WileyCDA/WileyTitle/productCd-3906390519.html).
Подходящие фармацевтически приемлемые соли могут включать кислотно-аддитивные соли. Такие соли могут быть образованы путем взаимодействия с соответствующей кислотой, возможно в подходящем растворителе, таком как органический растворитель, с получением соли, которая может быть выделена путем кристаллизации и фильтрации.
Типовые фармацевтически приемлемые кислотно-аддитивные соли включают, но без ограничения, 4-ацетамидобензоат, ацетат, адипат, альгинат, аскорбат, аспартат, бензолсульфонат (безилат), бензоат, бисульфат, битартрат, бутират, кальций-эдетат, камфорат, камфоросульфонат (камсилат), капрат (деканоат), капроат (гексаноат), каприлат (октаноат), циннамат, цитрат, цикламат, диглюконат, 2,5-дигидроксибензоат, дисукцинат, додецилсульфат (эстолат), эдетат (этилендиаминтетраацетат), эстолат (лаурилсульфат), этан-1,2-дисульфонат (эдисилат), этансульфонат (эзилат), формиат, фумарат, галактарат (мукат), гентизат (2,5-дигидроксибензоат), глюкогептонат (глюцептат), глюконат, глюкуронат, глутамат, глутарат, глицерофосфонат, гликолят, гексилрезорцинат, гиппурат, гидрабамин (N, N'- ди(дегидроабиэтил)этилендиамин), гидробромид, гидрохлорид, гидроиодид, гидроксинафтоат, изобутират, лактат, лактобионат, лаурат, малат, малеат, малонат, манделат, метансульфонат (мезилат), метилсульфат, мукат, нафталин-1,5-дисульфонат (нападизилат), нафталин-2-сульфонат (напсилилат), никотинат, нитрат, олеат, пальмитат, п-аминобензолсульфонат, п-аминосалицилат, памоат (эмбонат), пантотенат, пектинат, персульфат, фенилацетат, фенилэтилбарбитурат, фосфат, полигалактуронат, пропионат, п-толуолсульфонат (тозилат), пироглутамат, пируват, салицилат, себацинат, стеарат, субацетат, сукцинат, сульфамат, сульфат, таннат, тартрат, теоклат (8-хлортеофиллинат), тиоцианат, треэтиодид, ундеканоат, ундециленат и валерат.
Используемый в данном описании термин «терапевтически эффективное количество» означает любое количество, которое, при сравнении с соответствующим субъектом, который не получил такое количество, приводит к улучшению лечения, выздоровлению, профилактике или облегчению тяжести заболевания, расстройства или побочного эффекта, или к снижению развития болезни или расстройства.
Соответствующее «терапевтически эффективное количество» будет зависеть от целого ряда факторов, в том числе, например, от возраста и массы субъекта, определенного состояния, требующего лечения и его тяжесть, природы препарата, а также от способа введения, и оно будет, в конечном счете, определяться в соответствии с мнением лечащего врача.
Соединения формулы (I), где m представляет собой 0, может содержать один или несколько асимметрических центров (также называемых хиральными центрами), и, следовательно, они могут существовать в виде отдельных энантиомеров, диастереоизомеров или других стереоизомерных форм или в виде их смесей. Хиральные центры, такие как хиральные атомы углерода, также могут присутствовать в заместителе, таком как алкильная группа. В случае, когда не указана стереохимия хирального центра, присутствующего в соединении формулы (I), или в какой-либо химической структуре, представленной в данном документе, то такая структура охватывает любые стереоизомеры и все их смеси. Таким образом, соединения формулы (I), содержащие один или несколько хиральных центров, могут быть использованы в виде рацемических модификаций, включая рацемические смеси и рацематы, энантиомерно-обогащенные смеси или в виде энантиомерно-чистых индивидуальных стереоизомеров.
Получение соединений
Соединения по изобретению могут быть получены различными методами, в том числе стандартными химическими методами. Любые указанные выше определения групп, радикалов и атомов имеют приведенные выше значения, если не указано иное. Иллюстративные методы общего синтеза показаны ниже на следующих схемах, которые могут быть легко адаптированы для получения других соединений по изобретению. Конкретные соединения по изобретению могут быть получены в соответствии с экспериментальными процедурами, описанными в разделе Примеры.
Общие процедуры, используемые для синтеза соединений формулы (I), показаны ниже на схемах реакций 1-17, и они проиллюстрированы в Примерах.
Получение соединений формулы (I)
Соединения формулы (I), где m=1, n=1 или 2, R1 представляет собой Н и R2 являются такими, как определено выше, могут быть получены по Схеме 1 путем удаления защитной группы BOC у аминогруппы соединений формулы (III) с помощью хлористого водорода с последующим сочетанием соответствующей HCl соли соединения формулы (II) с коммерчески доступной 4,4,4-трифторбутановой кислотой или 5,5,5-трифторпентановой кислотой или с промежуточным соединением формулы (IV). Альтернативно, соединения формулы (I) могут быть получены взаимодействием соответствующих коммерчески доступных аминосоединений, таких как HCl соли соединений формулы (II), с 4,4,4-трифторбутаноилбензотриазолом.
Схема 1
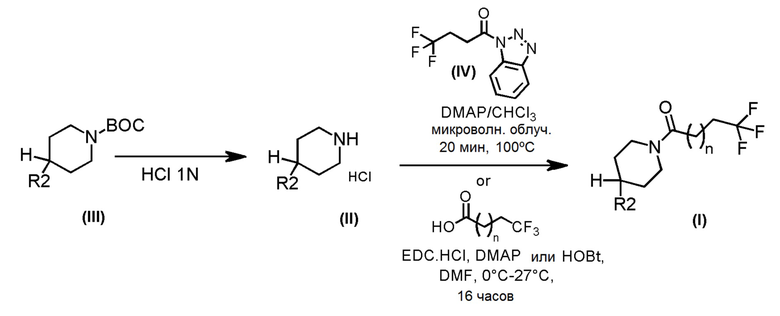
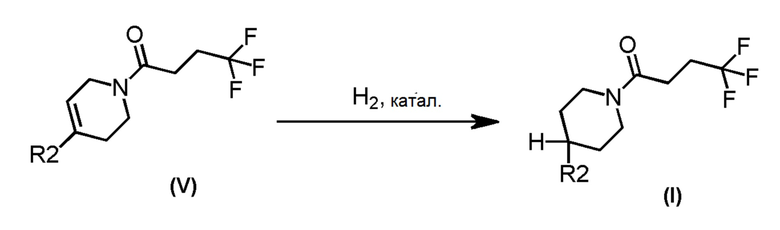
Альтернативно, соединения формулы (I), где m=1, n=1, R1 представляет собой Н и R2 имеет значения, определенные выше, могут быть получены по Схеме 2 путем каталитического гидрирования соединений формулы (V).
Схема 2
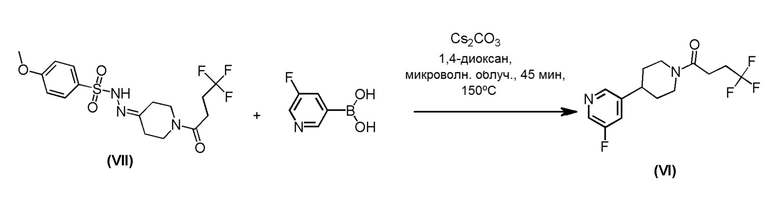
Соединение формулы (VI), которое представляет собой пиридиновое соединение формулы (I), где m=1, n=1, R1 представляет собой Н и R2 представляет собой 5-фтор-3-пиридил, может быть получено по Схеме 3 путем связывания соединения формулы (VII) с (5-фтор-3-пиридил) борной кислоты.
Схема 3
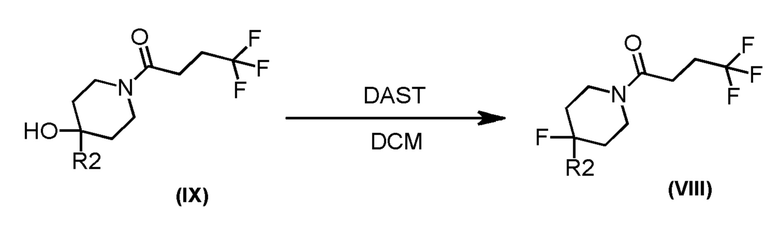
Соединения формулы (VIII), которые являются 4-фторпиперидиновыми соединениями формулы (I), где m=1, n=1, R1 представляет F и R2 имеет значения, определенные выше, могут быть получены в соответствии со Схемой 4 фторированием соответствующих 4-гидроксипиперидинов формулы (IX).
Схема 4
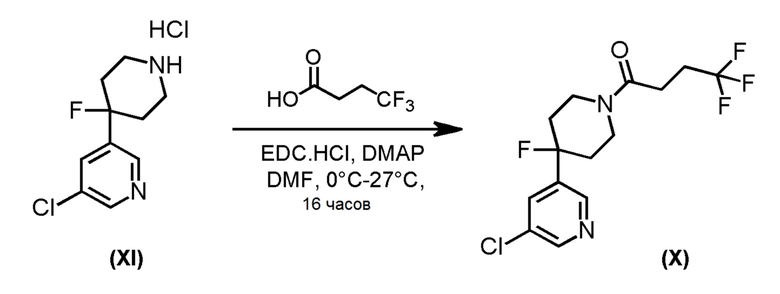
Альтернативно, соединение формулы (X), которое представляет собой 4-фторпиперидиновое соединение формулы (I), где m=1, n=1, R1 представляет F и R2 представляет собой 5-хлорпиридин-3-ил, может быть получено в соответствии со Схемой 5 путем сочетания соответствующего амина HCl соли соединения формулы (XI) с 4,4,4-трифторбутановой кислоты.
Схема 5
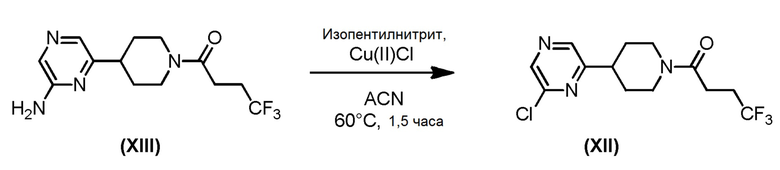
Соединение формулы (XII), которое представляет собой 6- хлорпиразин-2-иловое соединение формулы (I), где m=1, n=1, R1 представляет собой Н и R2 представляет собой 6- хлорпиразин-2-ил, может быть получено по Схеме 6 путем хлорирования соответствующего 6-аминопиразинил-2-илового соединения формулы (XIII).
Схема 6
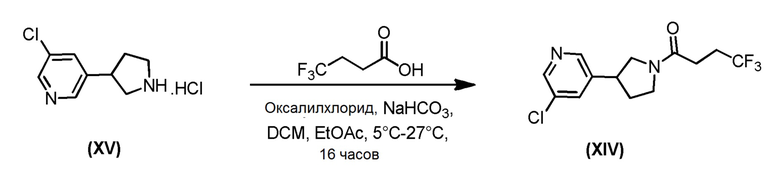
Соединение формулы (XIV), которое представляет собой пирролидиновое соединение формулы (I), где m=0, n=1, R1 представляет собой Н и R2 представляет собой 5-хлорпиридин-2-ил, может быть получено по Схеме 7 путем сочетания соответствующей HCl соли соединения формулы (XV) с 4,4,4-трифторбутановой кислотой.
Схема 7
Получение промежуточных соединений
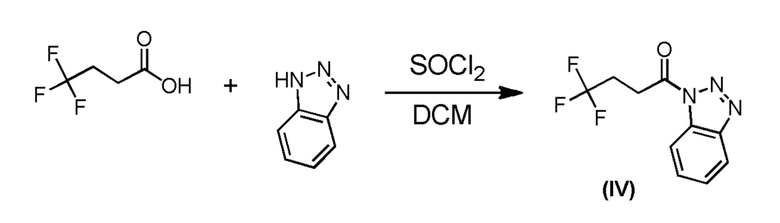
Промежуточное соединение формулы (IV) может быть получено в соответствии со Схемой 8 путем сочетания коммерчески доступной 4,4,4-трифторбутановой кислоты с 1H-бензотриазолом в присутствии тионилхлорида.
Схема 8
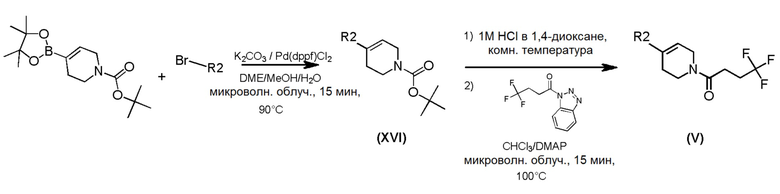
Промежуточные пиперидиновые соединения формулы (V), где m=1, n=1, R1 представляет собой Н и R2 имеет значения, определенные выше, могут быть получены в соответствии со Схемой 9 путем удалением защитной группы BOC у амино-соединений формулы (XVI) с использованием хлористого водорода, с последующим сочетанием соответствующей HCl соли с промежуточным соединением формулы (IV), т.е., 4,4,4-трифторбутаноилбензотриазолом. Промежуточные соединения формулы (XVI) могут быть получены путем сочетания коммерчески доступного трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2Н)карбоксилата с соответствующим бром-производным.
Схема 9
Альтернативно, промежуточные пиперидиновые соединения формулы (V), где m=1, n=1, R1 представляет собой Н и R2 имеет значения, определенные выше, могут быть получены в соответствии со Схемой 10 путем связывания промежуточного соединения формулы (XVII) с соответствующим коммерчески доступным бром-производным. Промежуточное соединение формулы (XVII) может быть получено путем удаления защитной группы ВОС у коммерчески доступного трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2Н)карбоксилата с использованием хлористого водорода и с последующим сочетанием соответствующей HCl соли соединения формулы (XVIII) с 4,4,4-трифторбутановой кислотой.
Схема 10
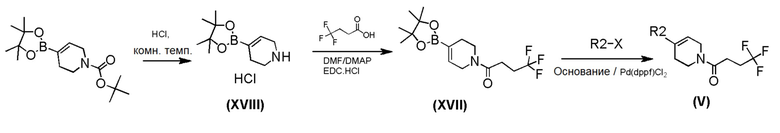
Промежуточное соединение формулы (VII) может быть получено в соответствии со Схемой 11 путем взаимодействием коммерчески доступного 4-метоксибензолсульфонгидразида с защищенным аминокетоном соединением формулы (XIX) в метаноле. Промежуточное соединение формулы (XIX) может быть быстро синтезирован путем реакции сочетания бензотриазола формулы (IV) с коммерчески доступным гидрохлорида пиперидин-4-она в присутствии 4-диметиламинопиридина.
Схема 11
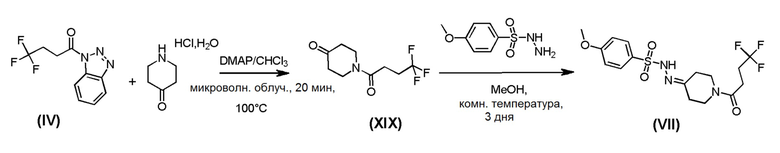
Промежуточное соединение формулы (ХХ) могут быть получено в соответствии со Схемой 12 путем отщепления N-Вос-защитной группы у промежуточного соединения формулы (XXI) в стандартных кислотных условиях. Промежуточное соединение формулы (XXI), может быть получено из алкена формулы (XXII) путем гидрирования в каталитических условиях. Промежуточное соединение формулы (XXII) может быть получено путем сочетания Сузуки коммерчески доступного трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2Н)карбоксилата и 2-бромпиримидина в стандартных условиях.
Схема 12
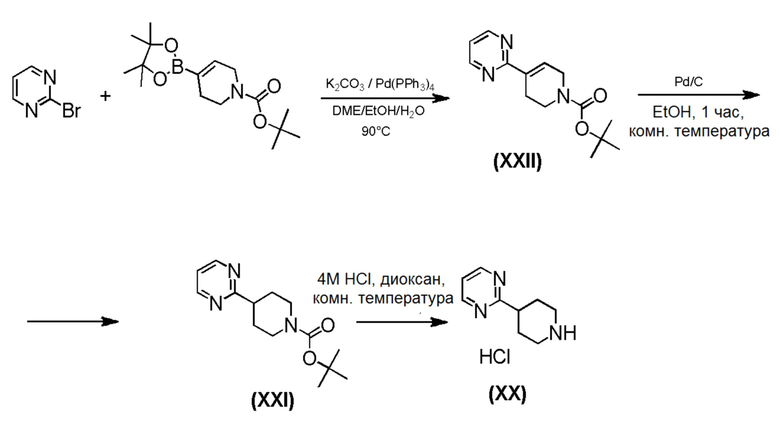
Промежуточные соединения формулы (IX), где m=1, n=0, R1 представляет собой ОН и R2 имеет значения, определенные выше, могут быть получены в соответствии со Схемой 13 путем удаления защитной группы BOC у амино-соединений формулы (XXIV) с использованием хлористого водорода, с последующим сочетанием соответствующей HCl соли соединения формулы (XXIII) с коммерчески доступной 4,4,4-трифторбутановой кислотой. Промежуточные соединения формулы (XXIV) могут быть получены посредством связывания бутиллитием соответствующего бромидного производного с коммерчески доступным кетоном трет-бутил-4-оксопиперидин-1-карбоксилата.
Схема 13
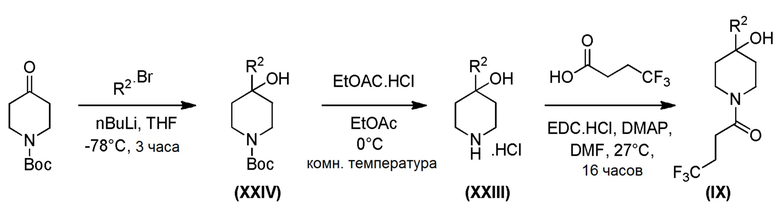
Промежуточное соединение формулы (XI) может быть получено в соответствии со Схемой 14 фторированием 4-гидроксипиперидина формулы (XXVI) с последующим соединением соответствующей HCl соли соединения формулы (XXV) с коммерчески доступной 4,4,4-трифторбутановой кислотой. Промежуточное соединение Формулы (XXVI) может быть получено посредством соединения 3-бром-5-хлорпиридина с трет-бутил-4-оксопиперидин-1-карбоксилатом, опосредованного бутиллитием.
Схема 14
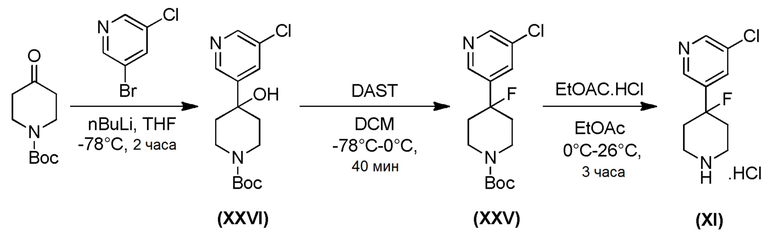
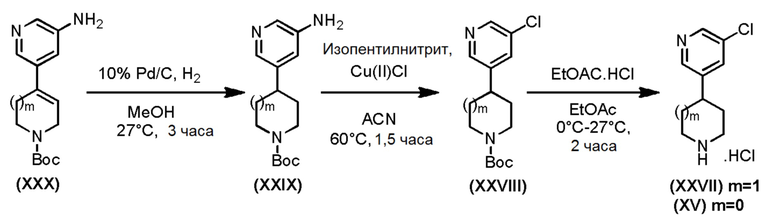
Промежуточное соединение формулы (XXVII), где m=1 и промежуточное соединение формулы (XV), где m=0, могут быть получены в соответствии со Схемой 15 хлорированием соответствующего аминопиридинового соединения формулы (XXIX) с последующим отщеплением N-Вос-защитной группы у промежуточного соединения формулы (XXVIII) в стандартных кислотных условиях. Аминопиридиновое соединение формулы (XXIX), где m=0, 1, может быть получено каталитическим восстановлением двойной связи соединения формулы (XXX).
Схема 15
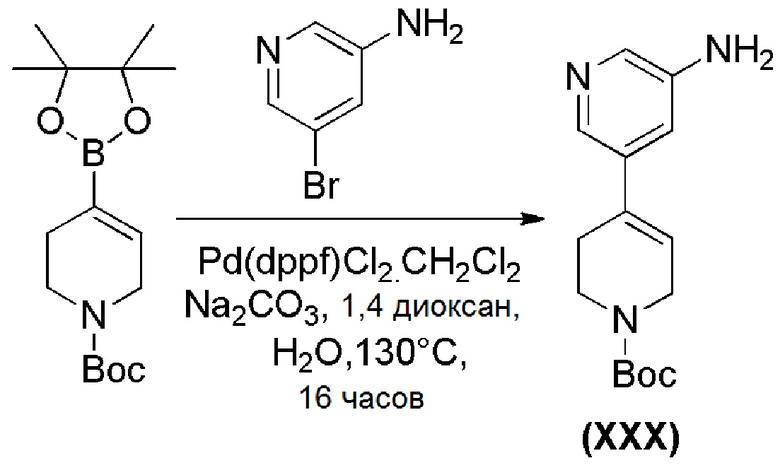
Промежуточное соединение формулы (XXX), где m=1, может быть получено в соответствии со Схемой 16 путем сочетания коммерчески доступного трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)карбоксилата и 5-бромпиразин-3-амина.
Схема 16
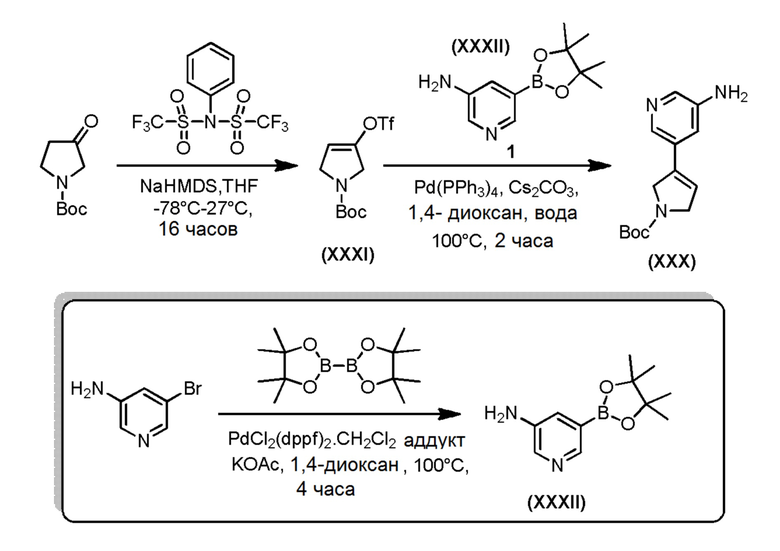
Промежуточное пирролидиновое соединение формулы (XXX), где m=0, может быть получено в соответствии со Схемой 17 посредством сочетания Сузуки аминопиридилборонатного соединения формулы (XXXII) и трифлатного соединения формулы (XXXI) в стандартных условиях. Трифлатное соединение формулы (XXXI) может быть получено путем реакции алкилирования коммерчески доступного трет-бутил-3-оксопирролидин-1-карбоксилата с 1,1,1-трифтор-N-фенил-N-((трифторметил)сульфонил)метансульфонамида в присутствии NaHMDS. Промежуточное соединение формулы (XXXII) может быть легко получено с помощью Pd-опосредованного сочетания коммерчески доступного 5-бромпиридин-3-амина с 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) в стандартных основных условиях.
Схема 17
Способы применения
В одном аспекте настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения его в терапии.
В одном аспекте настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения его при лечении микобактериальной инфекции. Микобактериальная инфекция является инфекцией, которая вызвана микобактериями.
Микобактерия может быть членом одной из следующих групп микобактерий: комплекса Mycobacterium tuberculosis (MTC), комплекса Mycobacterium avium (MAC), клада Mycobacterium gordonae, клада Mycobacterium kansasii, клада Mycobacterium chelonae, клада Mycobacterium fortuitum, клада Mycobacterium parafortuitum или клада Mycobacterium vaccae. Микобактерии могут также относиться к Mycobacterium ulcerans или Mycobacterium leprae.
В одном варианте осуществления микобактерии являются членом комплекса Mycobacterium tuberculosis (MTC).
Члены комплекса Mycobacterium tuberculosis (MTC) включают Mycobacterium tuberculosis, Mycobacterium africanum, Mycobacterium bovis, Mycobacterium bovis BCG, Mycobacterium canetti, Mycobacterium caprae, Mycobacterium microti и Mycobacterium pinnipedii. Эти микобактерии являются возбудителями туберкулеза у человека и животных. Микобактерии туберкулеза являются основной причиной туберкулеза у людей.
В одном варианте осуществления изобретения, инфекция является инфекцией, вызываемой Mycobacterium tuberculosis. Другими словами, микобактериальная инфекция вызвана инфекцией Mycobacterium tuberculosis.
В одном варианте осуществления изобретения, Mycobacterium tuberculosis имеют множественную лекарственную устойчивость. В другом варианте Mycobacterium tuberculosis устойчивы к этионамиду.
Члены комплекса Mycobacterium avium (MAC) включают Mycobacterium avium, Mycobacterium avium paratuberculosis, Mycobacterium avium silaticum, Mycobacterium avium hominissuis, Mycobacterium columbiense и Mycobacterium indicus pranii.
Члены клада Mycobacterium gordonae включают Mycobacterium asiaticum and Mycobacterium gordonae.
Члены клада Mycobacterium kansasii включают Mycobacterium gastri и Mycobacterium kansasii.
Члены клада Mycobacterium chelonae включают Mycobacterium abscessus, Mycobacterium bolletii и Mycobacterium chelonae.
Члены клада Mycobacterium fortuitum включают Mycobacterium boenickei, Mycobacterium brisbanense, Mycobacterium cosmeticum, Mycobacterium fortuitum, Mycobacterium fortuitum subspecies acetamidolyticum, Mycobacterium houstonense, Mycobacterium mageritense, Mycobacterium neworleansense, Mycobacterium peregrinum, Mycobacterium porcinum, Mycobacterium senegalense и Mycobacterium septicum.
Члены клада Mycobacterium parafortuitum включают Mycobacterium austroafricanum, Mycobacterium diernhoferi, Mycobacterium frederiksbergense, Mycobacterium hodleri, Mycobacterium neoaurum и Mycobacterium parafortuitum.
Таким образом, микобактериальные инфекции могут быть вызваны микобактериями, выбранными из следующих: Mycobacterium tuberculosis, Mycobacterium africanum, Mycobacterium bovis, Mycobacterium bovis BCG, Mycobacterium canetti, Mycobacterium caprae, Mycobacterium microti, Mycobacterium pinnipedii, Mycobacterium avium, Mycobacterium avium paratuberculosis, Mycobacterium avium silaticum, Mycobacterium avium hominissuis, Mycobacterium columbiense, Mycobacterium indicus pranii, Mycobacterium asiaticum, Mycobacterium gordonae, Mycobacterium gastri, Mycobacterium kansasii, Mycobacterium abscessus, Mycobacterium bolletii, Mycobacterium chelonae, include Mycobacterium boenickei, Mycobacterium brisbanense, Mycobacterium cosmeticum, Mycobacterium fortuitum, Mycobacterium fortuitum subspecies acetamidolyticum, Mycobacterium houstonense, Mycobacterium mageritense, Mycobacterium neworleansense, Mycobacterium peregrinum, Mycobacterium porcinum, Mycobacterium senegalense, Mycobacterium septicum, Mycobacterium austroafricanum, Mycobacterium diernhoferi, Mycobacterium frederiksbergense, Mycobacterium hodleri, Mycobacterium neoaurum, Mycobacterium parafortuitum, Mycobacterium ulcerans и Mycobacterium leprae.
В другом аспекте настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения при лечении заболевания, вызванного микобактериальной инфекцией, где микобактерия выбрана из микобактерий, которые описаны выше. Заболевания, вызванные микобактериальной инфекцией, включают, но без ограничения, туберкулез (например, вызванный Mycobacterium tuberculosis), лепру (например, вызванную Mycobacterium leprae), болезнь Джона (например, вызванную Mycobacterium avium subspecies paratuberculosis), язву Бурули или Бернсдейла (например, вызванную Mycobacterium ulceran), болезнь Крона (например, вызванную Mycobacterium avium subspecies paratuberculosis), заболевание легких или инфекцию легких, пневмонию, инфекции бурсы, синовия, сухожильных влагалищ, локализованный абсцесс, лимфаденит, инфекции кожи и мягких тканей, синдром леди Уиндермир (например, вызванный комплексом Mycobacterium avium (MAC), заболевание легких, вызванное комплексом Mycobacterium avium (MAC), диссеминированная инфекция, вызванная комплексом Mycobacterium avium intracellulare (DMAIC), легочная болезнь джакузи (например, вызванную комплексом Mycobacterium avium), мастит, вызванный MAC, пиомиозит, вызванный MAC, или гранулему.
В одном варианте осуществления изобретения, заболевание представляет собой туберкулез. Таким образом, один аспект настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения при лечении туберкулеза.
В другом варианте осуществления настоящее изобретение относится к способу лечения микобактериальной инфекции у млекопитающего, нуждающегося в этом, где указанное лечение включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Как описано здесь, микобактериальные инфекции является инфекциями, вызванными микобактериями. Микобактерии являются такими, как описано выше.
В одном варианте осуществления, настоящее изобретение относится к способу лечения инфекции, вызванной Mycobacterium tuberculosis.
В другом варианте осуществления, настоящее изобретение относится к способу лечения заболевания, вызванного микобактериальной инфекцией у млекопитающего, нуждающегося в этом, где указанное лечение включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В одном варианте осуществления изобретения, заболевание представляет собой туберкулез. Таким образом, изобретение предоставляет способ лечения туберкулеза у млекопитающего, нуждающегося в этом, где указанное лечение включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В одном варианте осуществления настоящего изобретения, млекопитающим является человек.
Специалистам в данной области техники понятно, что указания в отношении лечение относятся к лечению установленных патологических состояний. Тем не менее, соединения по изобретению могут, в зависимости от состояния, также быть полезны при профилактике некоторых заболеваний. Таким образом, в одном варианте осуществления, настоящее изобретение обеспечивает лечение или предотвращение заболевания, такого как туберкулез (TB). В другом варианте осуществления, настоящее изобретение обеспечивает лечение заболевания, такого как туберкулез. В другом варианте осуществления, настоящее изобретение обеспечивает предотвращение заболевания, такого как туберкулез.
В другом варианте осуществления, настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли при изготовлении лекарственного средства, применяемого при лечении микобактериальной инфекции или при лечении заболевания, вызванного микобактериальной инфекцией.
Кроме того, здесь также описано применение соединения формулы (I) или его фармацевтически приемлемой соли при изготовлении лекарственного средства, применяемого при лечении туберкулеза.
В одном варианте осуществления изобретения, соединение формулы (I) или его фармацевтически приемлемую соль, применяемые при лечении туберкулеза, вводят совместно с тиоамидом. В другом варианте осуществления изобретения, тиоамид представляет собой этионамид. В альтернативном варианте осуществления изобретения, тиоамид представляет собой протионамид.
Таким образом, в одном варианте осуществления изобретения предлагается фармацевтическая композиция для применения при лечении туберкулеза, где указанная композиция содержит (а) соединение формулы (I); (b) тиоамид, например, этионамид или протионамид; и, необязательно, (c) фармацевтически приемлемый эксципиент.
В другом варианте осуществления, настоящее изобретение относится к способу лечения микобактериальной инфекции у млекопитающего, нуждающегося в этом, где указанное лечение включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли в комбинация с тиоамидом, где указанный тиоамид может представлять собой этионамид. В альтернативном варианте осуществления изобретения, тиоамид представляет собой протионамид. Как описано здесь, микобактериальные инфекции является инфекциями, вызванными микобактериями. Микобактерии являются такими, как описано выше.
В одном варианте осуществления изобретения, микобактериальной инфекцией является инфекция, вызванная Mycobacterium tuberculosis.
В другом варианте осуществления, настоящее изобретение относится к способу лечения заболевания, вызванного микобактериальной инфекцией, у млекопитающего, нуждающегося в этом, где указанное лечение включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтический приемлемой соли в сочетании с тиоамидом, где указанный тиоамид может представлять собой этионамид. В альтернативном варианте осуществления изобретения, тиоамид представляет собой протионамид.
В одном варианте осуществления изобретения, заболевание представляет собой туберкулез. Таким образом, здесь также предоставляется способ лечения туберкулеза у млекопитающего, нуждающегося в этом, где указанное лечение включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли в сочетании с тиоамидом, где указанный тиоамид может представлять собой этионамид. В альтернативном варианте осуществления изобретения, тиоамид представляет собой протионамид.
В другом варианте осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли в сочетании с тиоамидом (например, этионамидом) при изготовлении лекарственного средства, применяемого при лечении микобактериальной инфекции или при лечении заболевания, вызванного микобактериальной инфекцией. В альтернативном варианте осуществления изобретения, тиоамид представляет собой протионамид.
Кроме того, здесь описано применение соединения формулы (I) или его фармацевтически приемлемой соли в сочетании с тиоамидом (например, этионамидом) при изготовлении лекарственного средства, применяемого при лечении туберкулеза. В альтернативном варианте осуществления изобретения, тиоамид представляет собой протионамид.
В одном варианте осуществления изобретения, соединение формулы (I), используемое в описанных выше способах и при лечении, представляет собой 4,4,4-трифтор-1-(4-(2-(трифторметил)пиридин-4-ил)пиперидин-1-ил)бутан-1-он, имеющий следующую структуру:
Фармацевтические композиции
Соединения формулы (I) и их фармацевтически приемлемые соли, перед их введением пациенту, как правило, но не обязательно, могут быть представлены в виде фармацевтических композиций. Соответственно, в другом аспекте настоящего изобретения предлагается фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент.
Фармацевтические композиции могут быть введены любым подходящим способом, например пероральным (в том числе буккальным или сублингвальным), ректальным, ингаляционным, интраназальным, местным (включая трансбуккальным, сублингвальным или трансдермальным) или парентеральным (включая подкожным, внутримышечным, внутривенным или внутрикожным) путем. В частности, фармацевтические композиции по изобретению могут быть введены перорально или внутривенно.
Подходящие фармацевтически приемлемые эксципиенты включают следующие виды эксципиентов: носители, разбавители, наполнители, связующие, дезинтегранты, лубриканты, глиданты, гранулирующие агенты, покрывающие агенты, смачивающие агенты, растворители, сорастворители, суспендирующие агенты, эмульгаторы, подсластители, флаворанты, вещества, маскирующие вкус, красители, вещества, предотвращающие слипание, увлажнители, хелатирующие агенты, пластификаторы, агенты, повышающие вязкость, антиоксиданты, консерванты, стабилизаторы, поверхностно-активные вещества и буферирующие агенты.
Подходящие способы получения препаратов и композиций из соединений по изобретению известны специалистам в данной области техники, и они описаны в справочнике Remington: The Science and Practice of Pharmacy, 21st Edition, 2006.
Фармацевтические композиции могут быть представлены в виде единичных дозированных форм, содержащих заранее определенное количество активного ингредиента в единичной дозированной форме. Предпочтительные единичные дозированные формы композиций содержат суточную дозу активного ингредиента или его суб-дозу, или подходящую часть дозы. Следовательно, такие единичные дозированные формы можно вводить более одного раза в день. Предпочтительными единичными дозированными формами композиции являются такими, которые содержат суточную дозу активного ингредиента или его суб-дозу (для введения чаще, чем один раз в день), как описано здесь выше, или подходящую часть дозы.
Когда соединения по изобретению или их фармацевтически приемлемые соли используют при лечении туберкулеза, то они могут применяться отдельно или в сочетании с дополнительным терапевтическим агентом, таким как дополнительным агентом против микобактерий, например, агентом для борьбы с туберкулезом и/или противовирусным агентом, включая антиретровирусные агенты.
Например, настоящее изобретение относится к соединениям формулы (I) или их фармацевтически приемлемым солям в комбинации с дополнительным агентом против туберкулеза. В одном варианте осуществления изобретения, такая комбинация содержит два, три, четыре, пять, шесть или семь дополнительных противотуберкулезных агентов. Например, при лечении туберкулеза с множественной лекарственной устойчивостью, пациентам обычно вводят комбинацию из четырех или более лекарственных препаратов. Например, при лечении туберкулеза, чувствительного к лекарственным средствам, пациентам обычно вводят комбинацию из трех или четырех лекарственных препаратов.
Дополнительный противотуберкулезный агент представляет собой средство или агент, находящийся на стадии разработке, или средство, одобренное или рекомендованное для лечения туберкулеза, и оно может быть выбрано из изониазида, рифампицина, пиразинамида, этамбутола, моксифлоксацина, рифапентина, клофазимина, этионамида, протионамида, изоксила, триацетазона, рифабутина, диарилхинолина, такого как бедаквилин (TMC207) или TBAJ-587, нитроимидазо-оксазина PA-824, деламанида (OPC-67683), оксазолидинона, такого как линезолид, тедизолид, радезолид, сутезолид (PNU-100480), позизолид (AZD-5847) или TBI-223, аналога EMB SQ109, OPC-167832, GSK3036656 (также известного как GSK070), GSK2556286, GSK3211830, бензотиазинона, такого как BTZ043 или PBTZ169, азаиндола, такого как TBA-7371, динитробензамида или бета-лактама, такого как меропенем, фаропенем, эртапенем, тебипенем, или бета-лактамные комбинации, такие как аугментин (амоксициллин-клавуланат).
В одном варианте осуществления изобретения, противотуберкулезный агент/средство может быть выбрано из изониазида, рифампина, пиразинамида, этамбутола, моксифлоксацина, рифапентина, клофазимина, этионамида, протионамида, изоксазолила, тиазетазозона, бедаквилина (TMC207), нитроимидазо-оксазина РА-824, деламанида (OPC-67683), оксазолидинона, такого как линезолид, тедизолид, радезолид, сутезолид (PNU-100480) или позизолид (AZD-5847), аналога EMB SQ109, OPC-167832, GSK3036656 (также известный как GSK070), GSK2556286, GSK3211830 и бензотиазинона или динитробензамида.
В соответствии с настоящим изобретением комбинация может дополнительно содержать противовирусный агент, включая антиретровирусный агент.
Такие антиретровирусные агенты/средства могут быть выбраны из зидовудина, диданозина, ламивудина, залцитабина, абакавира, ставудина, адефовира, дипивоксил адефовира, фозивудина, тодоксила, эмтрицитабина, аловудина, амдоксовира, элвуцитабина, невирапина, делавирдина, эфавиренца, ловирида, иммунокала, олтипраза, каправирина, лерсивирина, GSK2248761, ТМС-278, ТМС-125, этравирина, саквинавира, ритонавира, индинавира, нелфинавира, ампренавира, фозампренавира, бреканавира, дарунавира, атазанавира, типранавира, палинавира, лазинавира, энфувиртида, Т-20, Т-1249, PRO-542, PRO-140, TNX-355, BMS-806, BMS-663068 и BMS-626529, 5-геликса, ралтегравира, элвитегравира, GSK1349572, GSK1265744, викривирока (Sch-C), Sch-D, TAK779, маравирока, TAK449, диданозина, тенофовира, лопинавира или дарунавира.
Соединение по изобретению (т.е. соединение формулы (I) или его фармацевтически приемлемая соль) может использоваться в комбинации с противотуберкулезным агентом, который активируется через сигнальный путь EthA. Специалист в данной области техники может определить, является ли конкретное соединение активируемым через сигнальный путь EthA, используя для этого, например, метод, описанный в публикации: "Activation of the prodrug ethionamide is regulated by mycobacteria" A. R. Baulard et al., Journal of Biological Chemistry, 2000, pp. 28326-28331.
Более конкретно, противотуберкулезный агент/средство может быть выбрано из семейства тиоамидов, таких как этионамид, протионамид, изоксил и тиазетазон.
В одном варианте осуществления изобретения, соединение по изобретению (т.е. соединение формулы (I) или его фармацевтический приемлемая соль) используется в комбинации с этионамидом. В этом варианте осуществления изобретения, соединения по изобретению (т.е. соединение формулы (I) или его фармацевтически приемлемая соль) показали способность потенцировать активность этионамида.
Комбинации, для удобства их использования, могут быть представлены в форме фармацевтической композиции или препарата. Таким образом, здесь также представляется фармацевтическая композиция, содержащая (а) соединение по изобретению (т.е. соединение формулы (I) или его фармацевтически приемлемая соль), как описано здесь, вместе с (b) одним или несколькими фармацевтически приемлемыми носителями, как описано здесь, и (c) по меньшей мере одним другим противотуберкулезным лекарственным средством, и (d) необязательно, противовирусным агентом/средством, включая антиретровирусные агенты.
Соединение по изобретению (т.е. соединение формулы (I) или его фармацевтически приемлемая соль) и дополнительный терапевтический агент можно вводить вместе или по-отдельности, и при введении по-отдельности введение можно выполнять раздельно или последовательно в любом порядке (этим же или другим путем введения). Количество соединения по изобретению (т.е. соединение формулы (I) или его фармацевтически приемлемая соль) и дополнительный терапевтически активный агент (или агенты) и соответствующие временные показатели введения выбирают таким образом, чтобы достичь желаемого комбинированного терапевтического эффекта.
Примеры
Далее изобретение будет проиллюстрировано с помощью следующих неограничивающих примеров. Несмотря на то, что ниже описаны конкретные варианты осуществления изобретения, специалисту в данной области техники понятно, что могут быть сделаны различные изменения и модификации. Указания в отношении способов получения препаратов, полученных способом, аналогичным или подобным способом, или общим способом получения других препаратов, могут включать изменения обычных параметров, таких как время, температура, условия обработки, незначительные изменения количеств реагентов и т.п.
Сокращения
Ниже приведены определения некоторых сокращений и символов, используемые здесь. Следует иметь в виду, что этот список не является исчерпывающим, и смысл используемых сокращений и символов, не вошедших в перечень определений, приведенный ниже, является очевидным для специалистов в данной области техники. В описании изобретения химические элементы идентифицированы в соответствии с периодической таблицей элементов.
ACN/MeCN ацетонитрил
anh., безводн. безводный
aq., водн. водный
CDCl3 дейтерированный хлороформ
CD2Cl2 дейтерированный дихлорметан
CyHex циклогексан
DAST диэтиламиносульфотрифторид
DCM дихлорметан
DIPEA диизопропилэтиламин
DMAP 4-диметиламинопиридин
DME диметоксиэтан
DMF диметилформамид
DMSO-d6 дейтерированный диметилсульфоксид
Dppf 1,1'-бис(дифенилфосфино)ферроцен
EDC.HCl гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида
EtOAc этилацетат
EtOH этанол
HBTU гексафторфосфат N, N,N′,N′-тетраметил-O-(1H-бензотриазол-1-ил)урония
HOBt гидрат 1-гидроксибензотриазола
HPLC, ВЭЖХ высокоэффективная жидкостная хроматографии
Int. промежуточное соединение
M молярность
MeOH метанол
MS масс-спектроскопия
min, мин минуты
N нормальность
NaH гидрид натрия
NaHMDS бис(триметилсилил)амид натрия
NMR, ЯМР спектр ядерного магнитного резонанса
pet. смесь жидких углеводородов
Ref. Ex. ссылочный (референсный) пример
Rt комнатная температура
TFA трифторуксусная кислота
TEA триэтиламин
THF тетрагидрофуран
При записи результатов протонного ядерного магнитного резонанса (1Н-ЯМР) химические сдвиги (δ) представлены в миллионных долях по отношению к тетраметилсилану (TMS), используемому в качестве внутреннего стандарта. Для данных ЯМР использованы следующие сокращения: с=синглет, д=дублет, т=триплет, кв=квартет, м=мультиплет, набл. = наблюдаемый, ш=широкий, уш. - уширенный. Масс-спектры были получены с использованием методов ионизации с электрораспылением (ES). Все температуры приведены в градусах Цельсия.
В некоторых из нижеприведенных примеров получения промежуточных соединений и соединений по изобретению, исходные материалы идентифицированы путем ссылки на другие номера примеров промежуточных соединений или примеров соединений по изобретению. Это не означает, что материал какого-либо конкретного промежуточного соединения или соединения по примеру обязательно используется на последующей стадии описываемого примера, поскольку такое указание используется для обозначения названия соответствующего соединения.
Промежуточные соединения
Промежуточное соединение 1: 1-(бензотриазол-1-ил)-4,4,4-трифтор-бутан-1-он
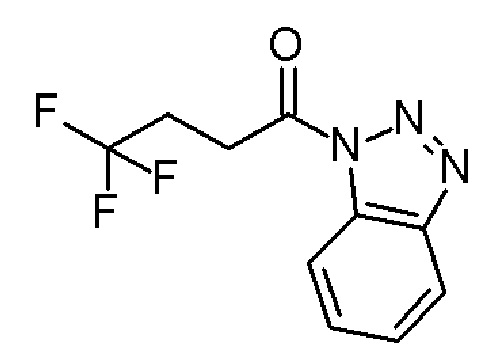
Тионилхлорид (SIGMA-ALDRICH, 6,74 мл, 93 ммоль) и 1Н-бензотриазол (ALFA-AESAR, 31,2 г, 262 ммоль) в DCM (150 мл) добавляли по каплям к раствору 4,4,4-трифторбутановой кислоты (Fluorochem, 12 г, 85 ммоль) в DCM (150 мл). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Осадок отфильтровывали, и фильтрат сушили в вакууме, с получением указанного в заголовке соединения (19,6 г, 94%) в виде не совсем белого твердого вещества.
1H ЯМР (300 МГц, CD2Cl2) δ м.д.: 8,28 (д, J=8,3 Гц, 1H), 8,15 (д, J=8,4 Гц, 1H), 7,75-7,69 (м, 1H), 7,60-7,54 (м, 1H), 3,77 (т, J=7,8 Гц, 2H), 2,91-2,73 (м, 2H); [ES+MS] m/z 244 (MH+).
Промежуточное соединение 2: 1-(4,4,4-трифторбутаноил)пиперидин-4-он
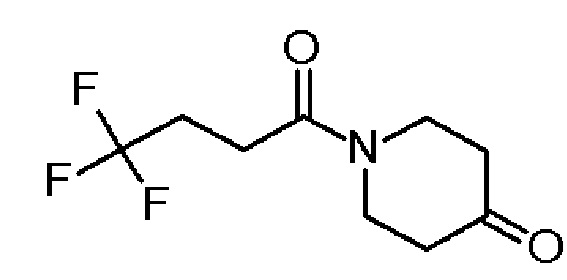
К раствору гидрата гидрохлорида пиперидин-4-она (SIGMA-ALDRICH, 7,05 г, 45,9 ммоль) и 4-DMAP (SIGMA-ALDRICH, 5,6 г, 45,84 ммоль) в хлороформе (16 мл) добавляли промежуточное соединение 1 (10,0 г, 41,12 ммоль). Раствор подвергали воздействию микроволнового излучения в течение 15 мин при 100°C. Реакционную смесь промывали насыщенным раствором Na2CO3 (три раза) и водным 1М раствором HCl. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 (безводн.) и выпаривали, с получением указанного в заголовке соединения 1-(4,4,4-трифторбутаноил)пиперидин-4-она (4,63 г, 49,2%) в виде оранжевого масла.
1H ЯМР (300 МГц, CD2Cl2) δ м.д.: 3,86 (т, J=6,4 Гц, 2H), 3,73 (т, J=6,3 Гц, 2H), 2,69-2,61 (м, 2H), 2,58-2,42 (м, 6H); [ES+MS] m/z 224 (MH+).
Промежуточное соединение 3: 4-метокси-N-[[1-(4,4,4-трифторбутаноил)-4-пиперидилиден]амино]бензол-сульфонамид
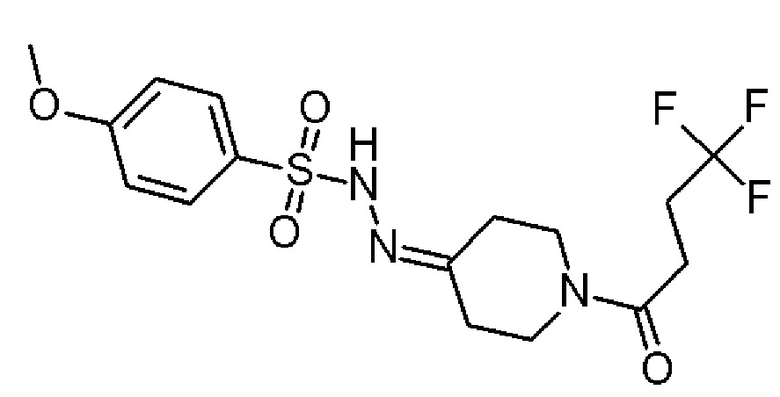
К раствору 4-метоксибензолсульфонгидразида (полученного в соответствии со способом, описанным в J.Org.Chem. 2014, pp. 328-338) (4,2 г, 20,77 ммоль) в MeOH (90 мл) добавляли промежуточное соединение 2 (4,63 г, 20,74 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3-х дней. Растворители удаляли в вакууме, с получением указанного в заголовке соединения (8,39 г, 96,1%) в виде твердого вещества белого цвета.
1H ЯМР (300 МГц, CD2Cl2) δ м.д.: 7,87-7,81 (м, 2H), 7,25-7,20 (м, 1H), 7,03-6,98 (м, 2H), 3,87 (с, 3H), 3,73-3,62 (м, 2H), 3,58-3,53 (м, 2H), 2,60-2,31 (м, 8H); [ES+MS] m/z 408 (MH+).
Промежуточное соединение 4: трет-бутил-4-(6-фтор-3-пиридил)-3,6-дигидро-2Н-пиридин-1-карбоксилат
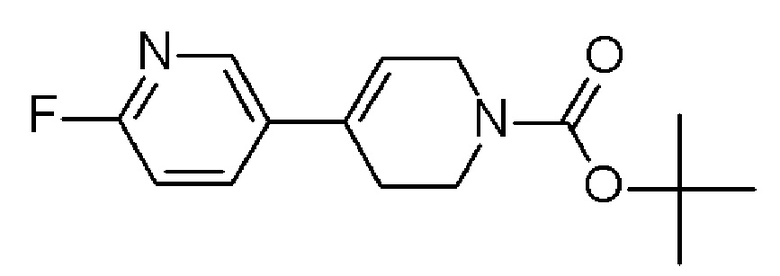
Трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидро-2Н-пиридин-1-карбоксилат (ENAMINE, 1,01 г, 3,27 ммоль), 5-бром-2-фтор-пиридин (ALFA-AESAR, 0,33 мл, 3,23 ммоль) и K2CO3 (SIGMA-ALDRICH, 903,5 мг, 6,54 ммоль) суспендировали в смеси DME/МеОН/Н2O (2/1/2, 5 мл) в инертной атмосфере. Затем добавляли Pd(dppf)Cl2 (ACROS, 264,2 мг, 0,32 ммоль) в атмосфере аргона, и закрытую пробирку подвергали воздействию микроволнового излучения при 90°C в течение 15 минут. К реакционной смеси добавляли воду и экстрагировали с использованием EtOAc (x2). Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 (безводн.) и выпаривали, с получением 1,45 г неочищенного вещества в виде пурпурного масла. Остаток очищали на силикагеле, используя в качестве элюента линейный градиент DCM/MeOH, с получением непрозрачного коричневого масла. Остаток затем очищали с помощью препаративной HPLC (колонка OmniSpher С18, 10 мкм, 41 х 250 мм), градиент от 10% до 100% ACN/Н2О (0,1% муравьиная кислота), 30 минут, с получением указанного в заголовке соединения (587,1 мг, 53,7%) в виде оранжевого масла.
[ES+MS] m/z 279 (MH+).
Промежуточные соединения 5-15 были получены с помощью методов, аналогичных тем, которые описаны для промежуточного соединения 4, но с заменой 5-бром-2-фтор-пиридина на соединение, указанное в Таблице 1. Также указаны изменения, использованные на стадии очистки.
Таблица 1
см. примечание a)
FLUOROCHEM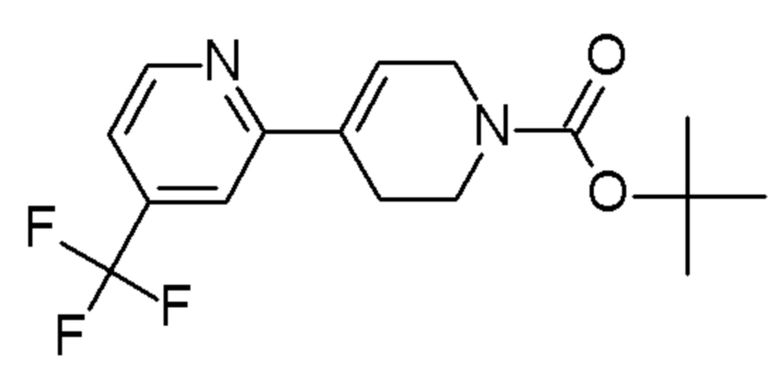
см. примечание a)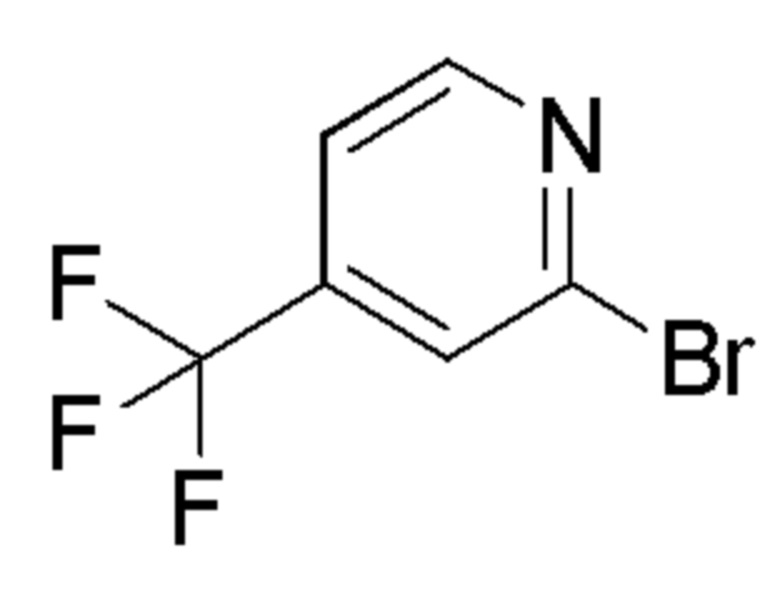
FLUOROCHEM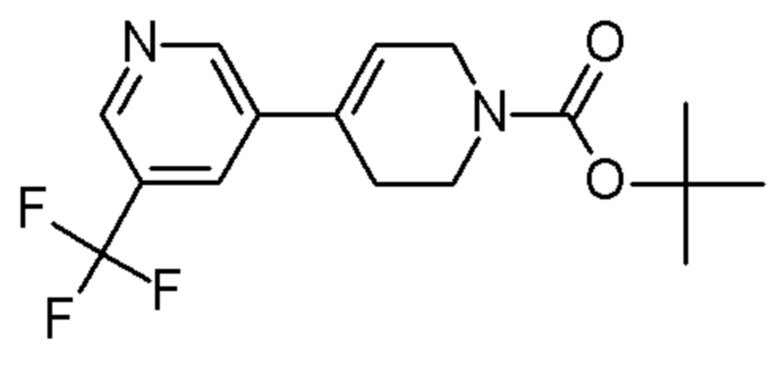
см. примечание a)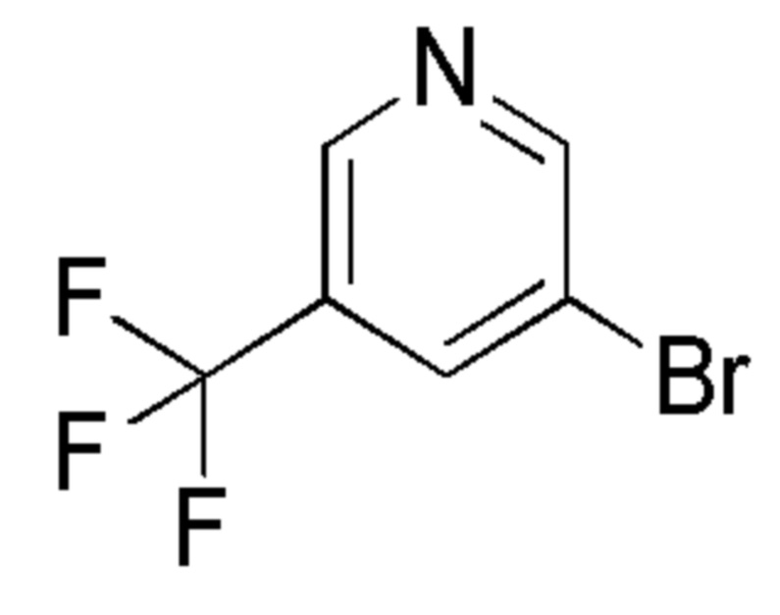
FLUOROCHEM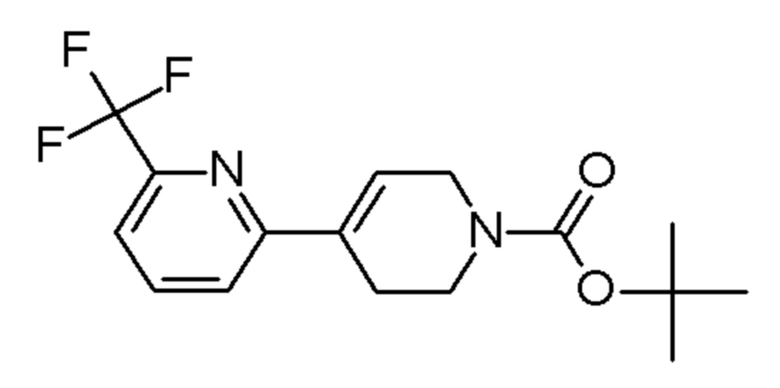
см. примечание a)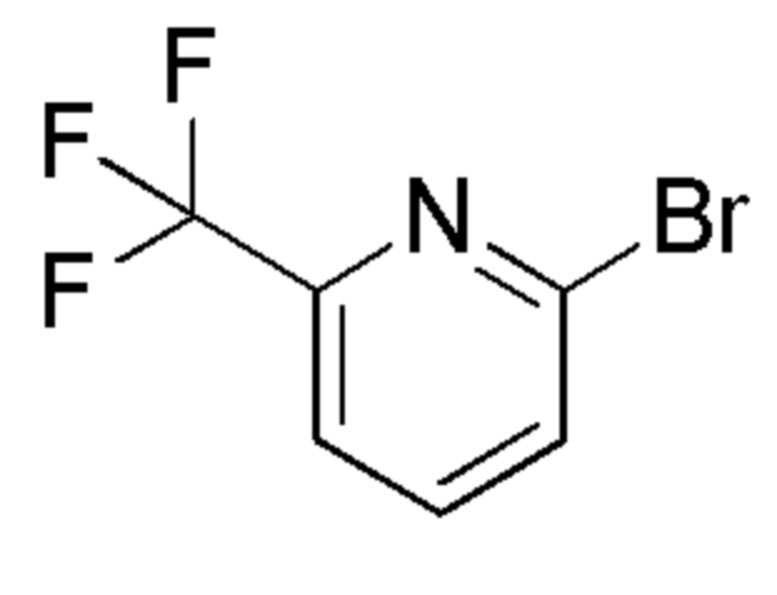
FLUOROCHEM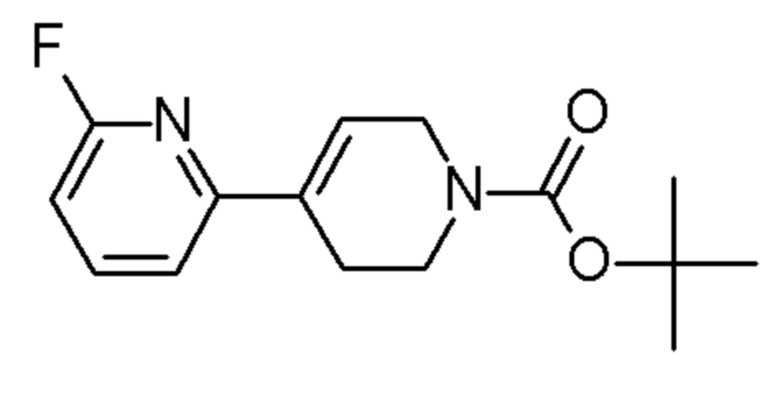
см. примечание a)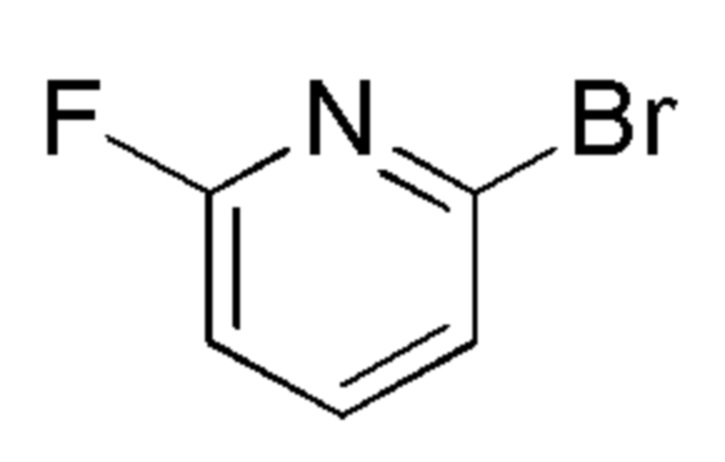
ENAMINE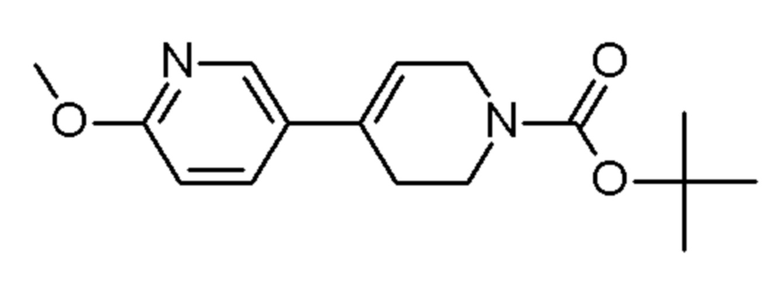
см. примечание a)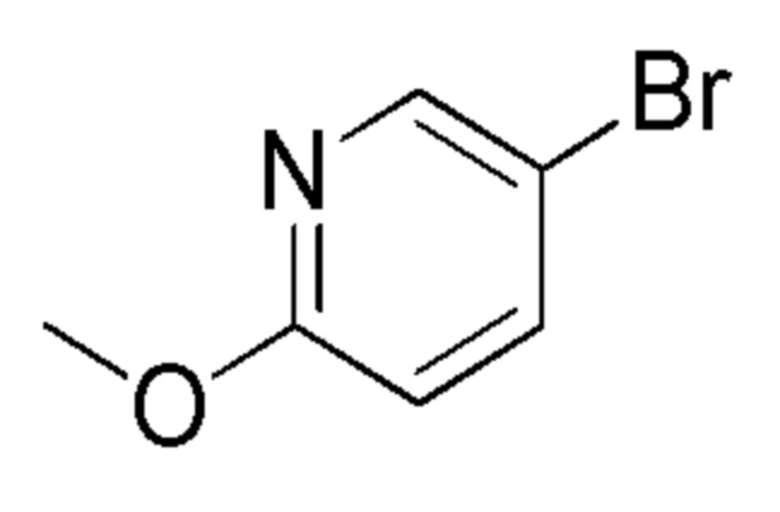
ALFA-AESAR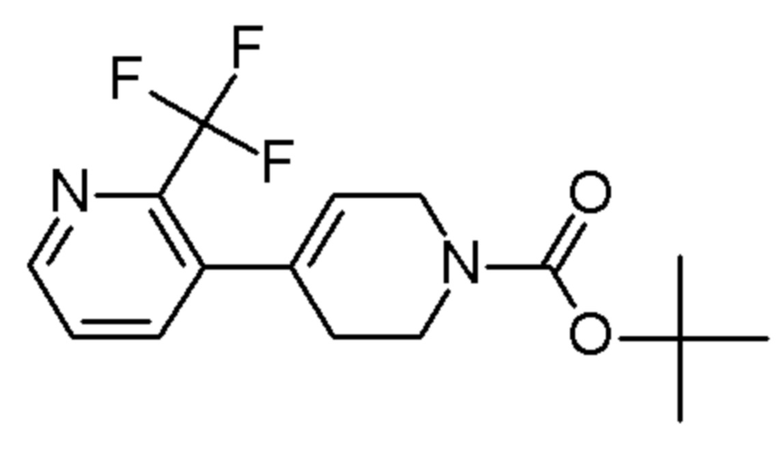
см. примечание a)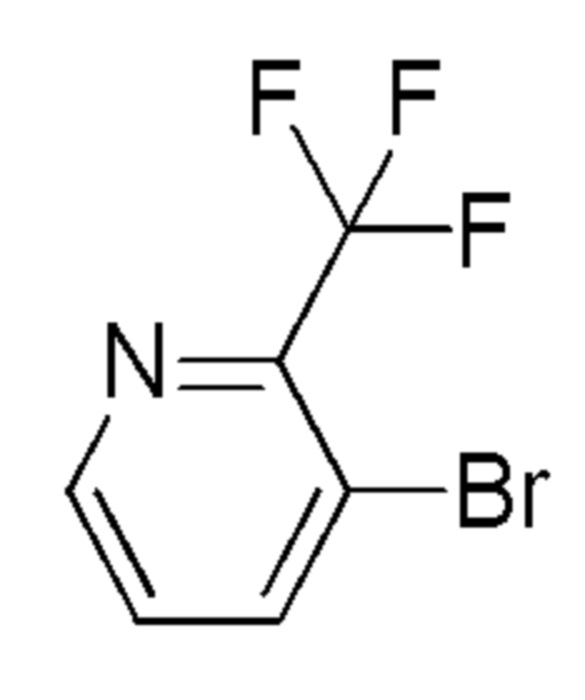
MATRIX SCIENTIFIC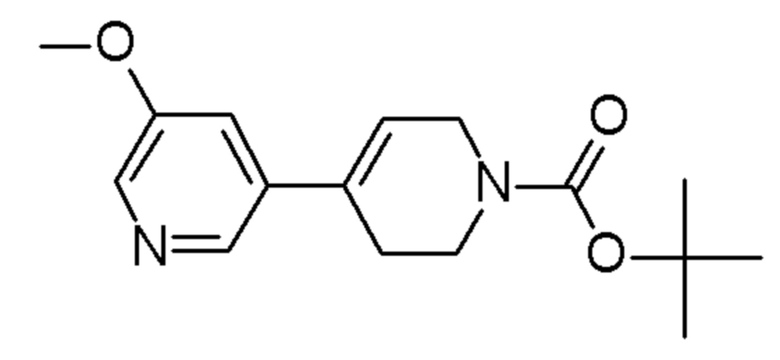
см. примечание a)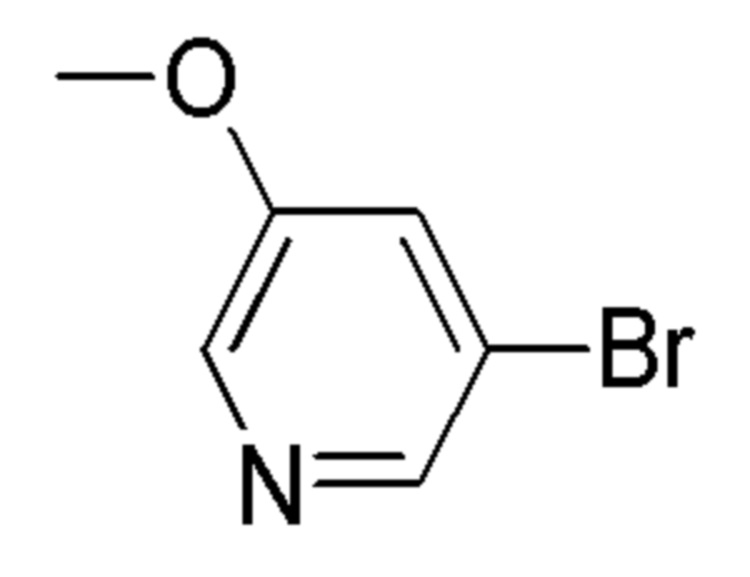
FLUOROCHEM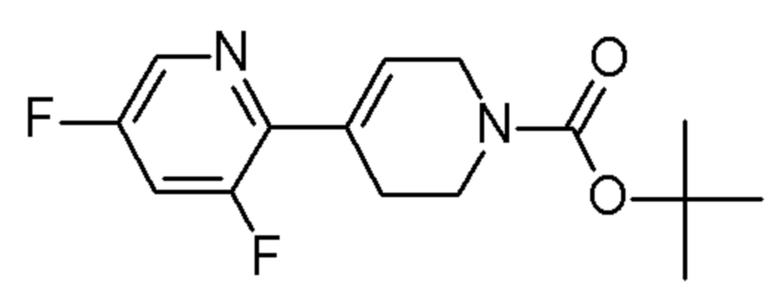
см. примечание a)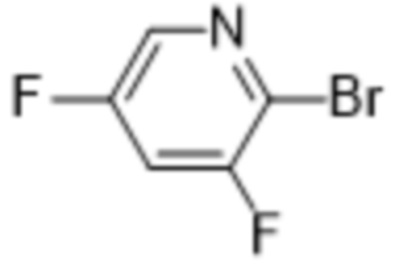
FLUOROCHEM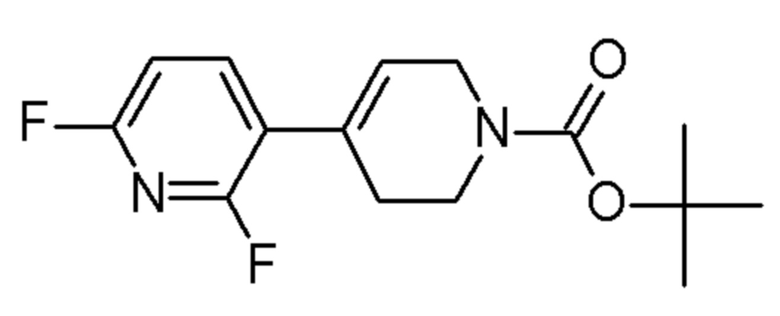
см. примечание a)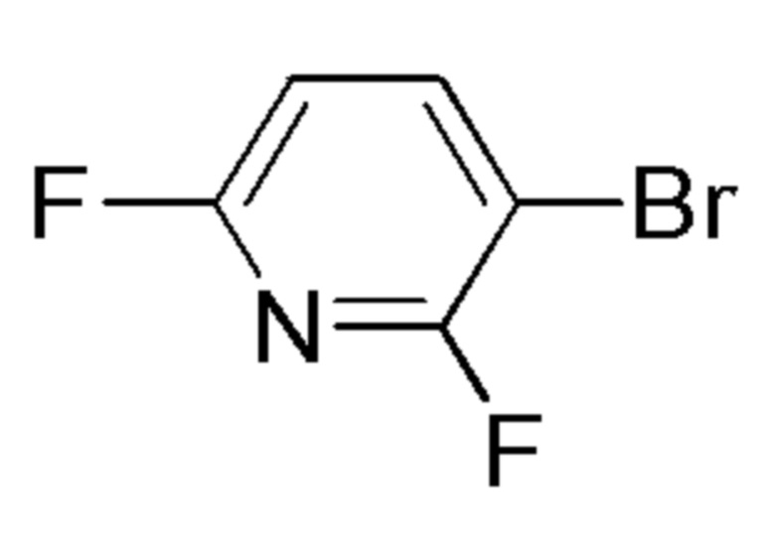
FLUOROCHEM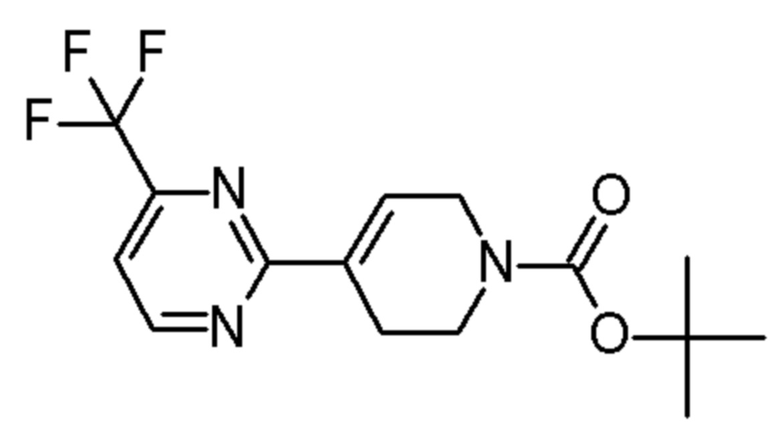
см. примечание a)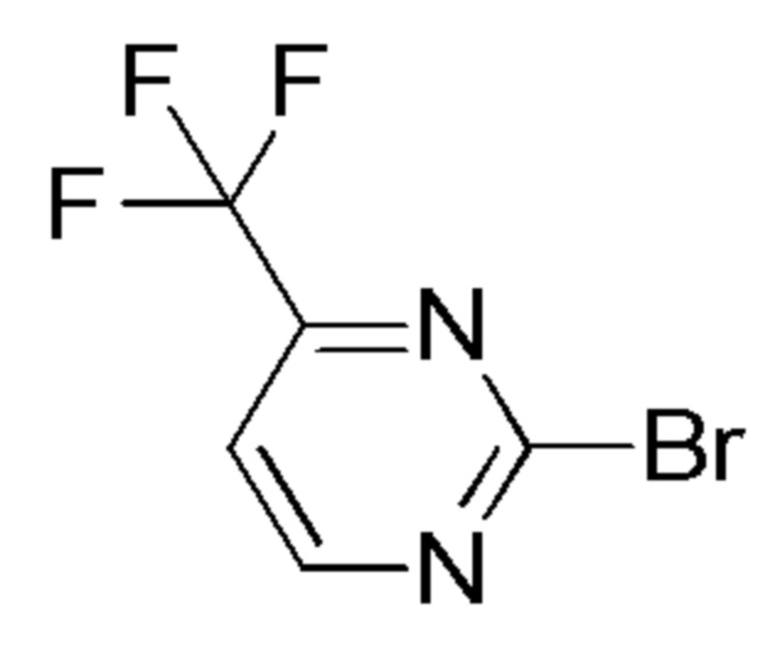
FLUOROCHEM
Примечание:
a) очистка на силикагеле с использованием линейного градиента DCM/MeOH.
Промежуточное соединение 16: 4,4,4-трифтор-1-[4-(6-фтор-3-пиридил)-3,6-дигидро-2Н-пиридин-1-ил]бутан-1-он
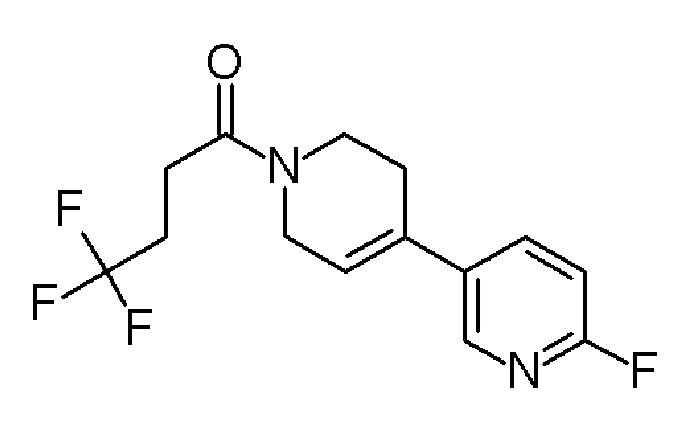
К раствору промежуточного соединения 4 (587,1 мг, 1,76 ммоля) в DCM (9 мл) добавляли по каплям 4M раствор HCl в диоксане (SIGMA-ALDRICH, 5,27 мл, 12 экв.) при комнатной температуре. Затем реакционную смесь перемешивали в течение ночи, и растворитель выпаривали, с получением 519,4 мг гидрохлорида 2-фтор-5-(1,2,3,6-тетрагидропиридин-4-ил)пиридина в виде оранжевого твердого вещества, которое сушили в вакууме. Хлороформ (4,6 мл), DMAP (SIGMA-ALDRICH, 305,8 мг, 2,5 ммоль) и промежуточное соединение 1 (547,6 мг, 2,25 ммоль) добавляли к оранжевому остатку, и раствор подвергали воздействию микроволнового излучения в течение 15 минут при 100°C. Реакционную смесь промывали насыщенным раствором Na2CO3 (три раза) и водным 1М раствором HCl. Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 (безводн.) и упаривали. Остаток очищали с помощью препаративной HPLC (колонка OmniSpher С18, 10 мкм, 41 х 250 мм), градиент от 10% до 100% ACN/Н2О (0,1% муравьиная кислота), 30 минут, с получением указанного в заголовке соединения 4,4,4-трифтор-1-[4-(6-фтор-3-пиридил)-3,6-дигидро-2Н-пиридин-1-ил]бутан-1-она (121,0 мг, 17,8%) в виде твердого вещества белого цвета.
1H ЯМР (300 МГц, CD2Cl2) δ м.д.: 8,22-8,20 (м, 1H), 7,82-7,76 (м, 1H), 6,94-6,90 (м, 1H), 6,13-6,03 (м, 1H), 4,24-4,21 (м, 1H), 4,14-4,11 (м, 1H), 3,82 (т, J=5,8 Гц, 1H), 3,67 (т, J=5,8 Гц, 1H), 2,68-2,49 (м, 6H), 1,53 (с, 9H); [ES+MS] m/z 303 (MH+).
Промежуточное соединение 16 также получали методом, описанным в Таблице 3.
Промежуточные соединения 17-27 получали способами, аналогичными способу, описанному для промежуточного соединения 16, но с заменой промежуточного соединения 4 на соединения, указанные в Таблице 2. Также указаны изменения, использованные на стадии очистки.
Таблица 2
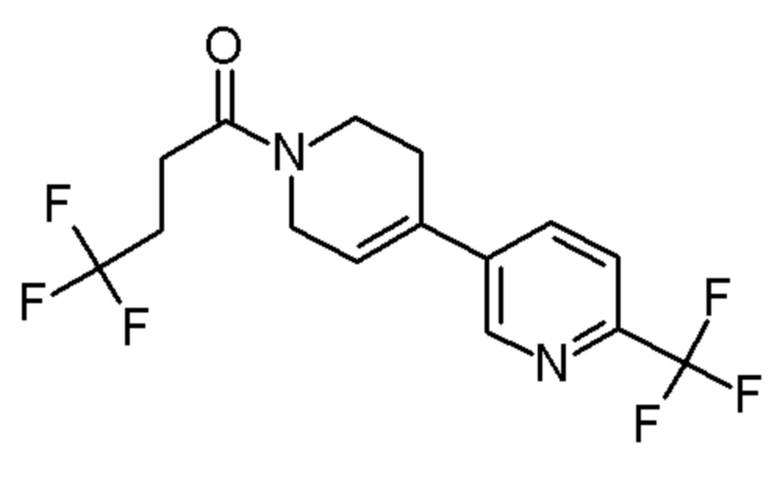
см. примечание a)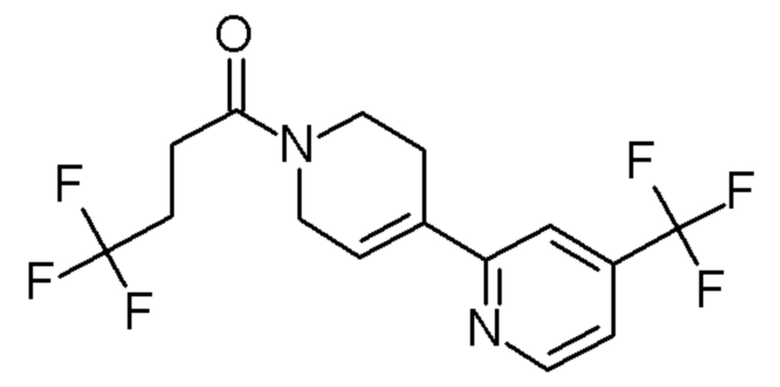
см. примечание a)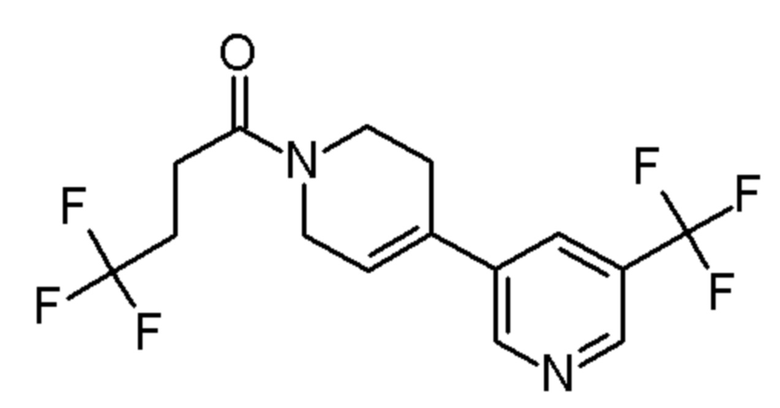
см. примечание a)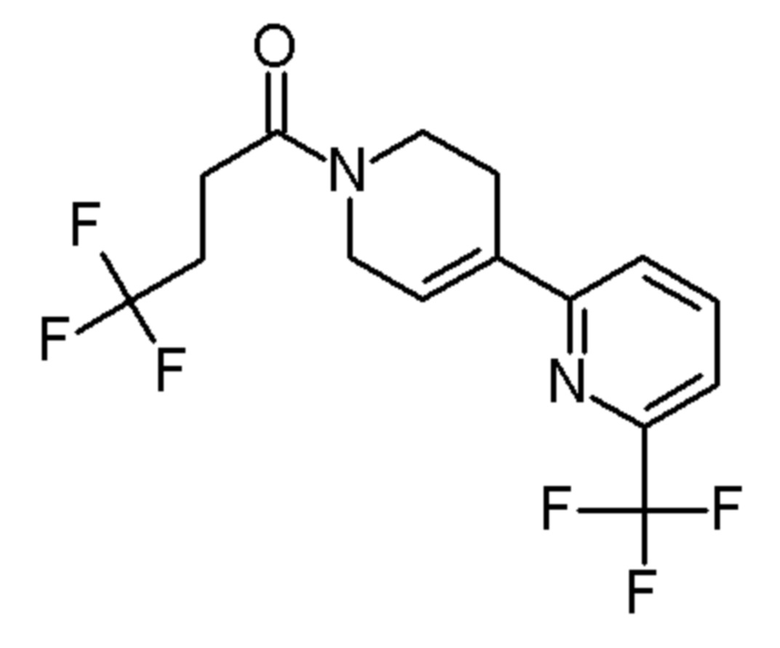
см. примечание a)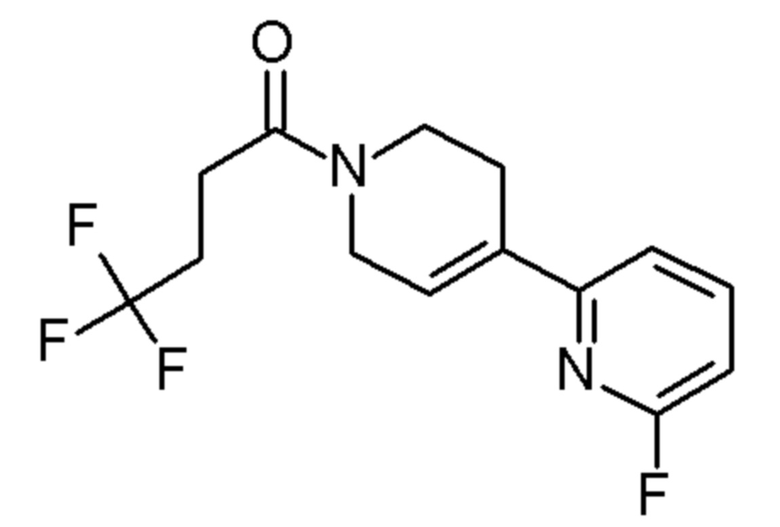
см. примечание a)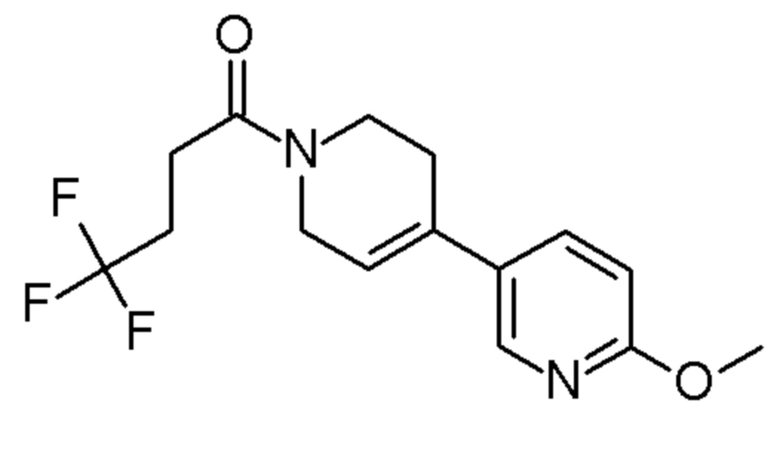
см. примечание a)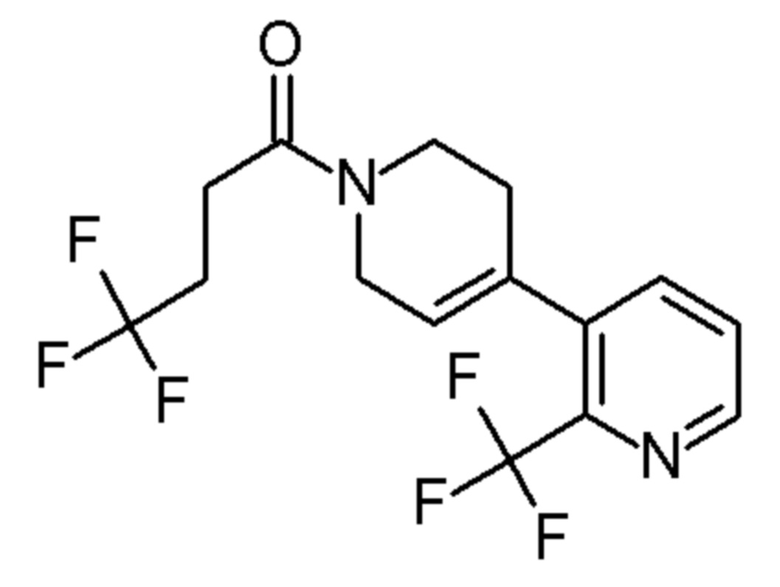
см. примечание a)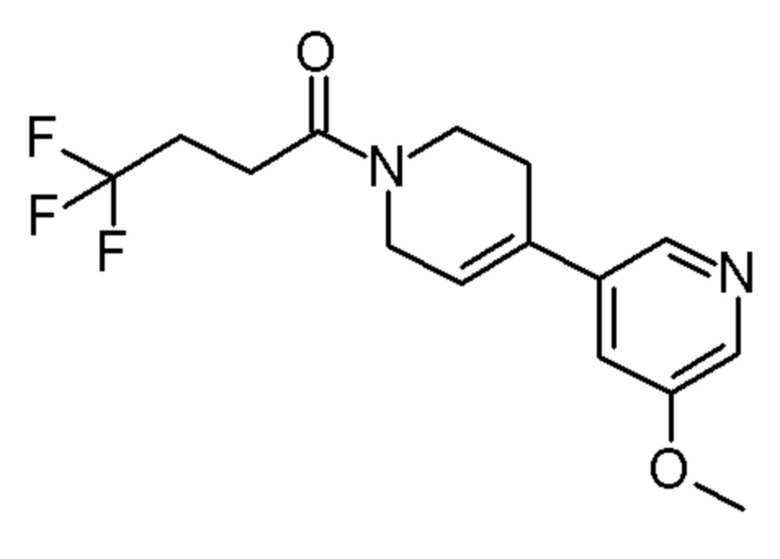
см. примечание b)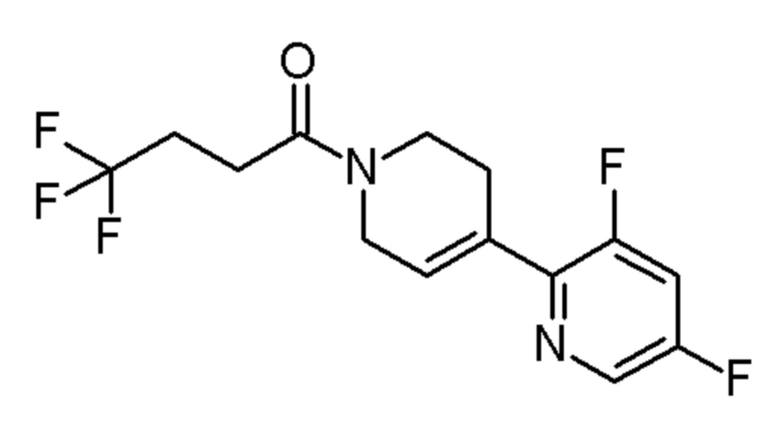
см. примечание b)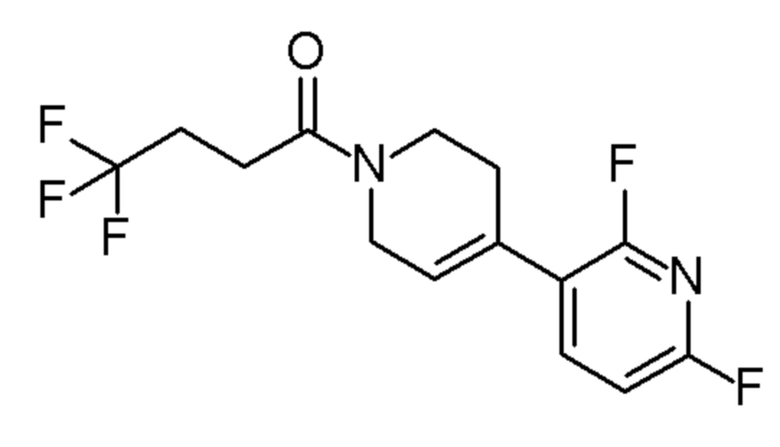
см. примечание b)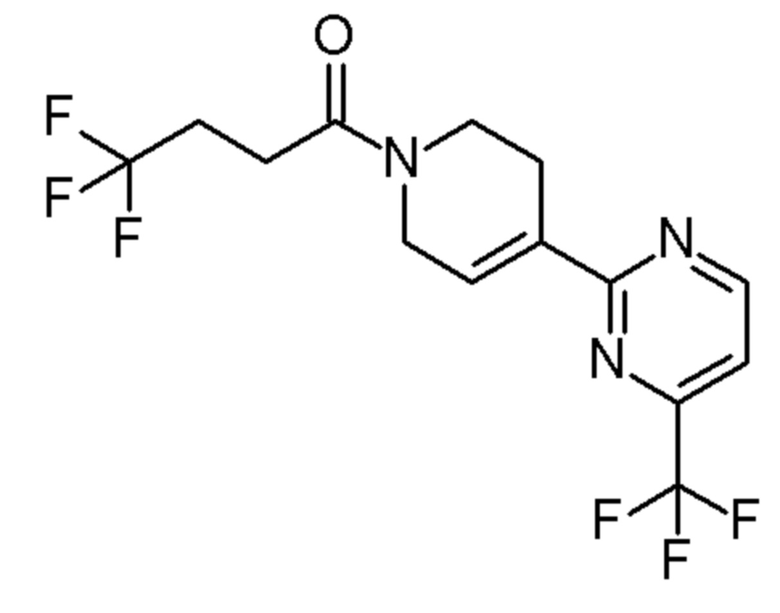
см. примечание b)
Примечания:
a) Препаративная HPLC (колонка OmniSpher C18, 10 мкм, 41×250 мм), градиент 10% - 100% ACN/H2O (0,1% муравьиная кислота), 30 мин
b) Препаративная HPLC (колонка OmniSpher C18, 10 мкм, 41×250 мм) градиент 10% - 100% ACN/H2O (0,1% муравьиная кислота), 35 мин
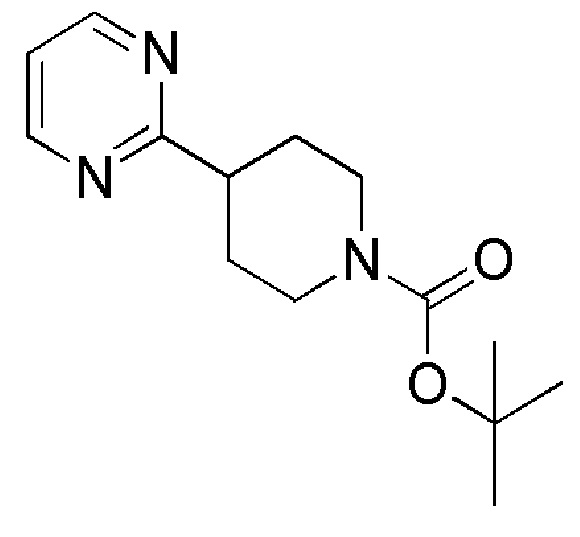
Промежуточное соединение 28: трет-бутил-4-(пиримидин-2-ил)-1,2,3,6-тетрагидропиридин-1-карбоксилат
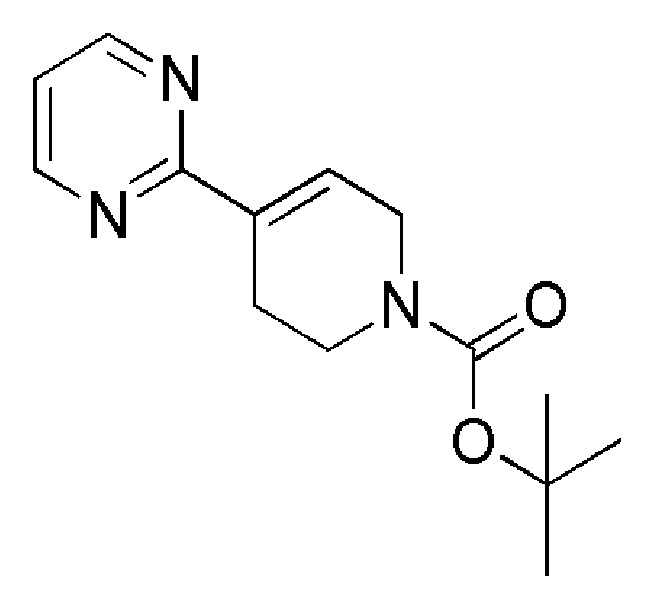
Тетракис(трифенилфосфин)палладий (0) (ALFA-AESAR, 148 мг, 0,128 ммоль) добавляли к суспензии трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (ALFA-AESAR, 381 мг, 1,23 ммоль), 2-бромпиримидина (ALFA-AESAR, 195 мг, 1,23 ммоль) и карбоната калия (ALFA-AESAR, 351 мг, 2,54 ммоль) в DME/EtOH/Н2O 2:1:2 (5 мл). Смесь нагревали в течение ночи при 90°C. UPLC-МС показал полное превращение исходных материалов. Реакционную смесь разбавляли водой и экстрагировали с помощью EtOAc. Объединенные органические слои сушили над Na2SO4 (безводн.), фильтровали и концентрировали при пониженном давлении, с получением сырой реакционной смеси. Остаток очищали с помощью флэш-хроматографии на силикагеле, используя в качестве элюента линейный градиент циклогексан/EtOAc, с получением указанного в заголовке соединения.
1H ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,80 (д, J=4,8 Гц, 2H), 7,36 (т, J=4,8 Гц, 1 H), 7,19 (уш. с, 1H), 4,16-4,05 (м, 2H), 3,55 (т, J=5,6 Гц, 2H), 2,66-2,57 (м, 2H), 1,45 (с, 9H), ES+MS] m/z 262 (MH+).
Промежуточное соединение 29: трет-бутил-4-(пиримидин-2-ил)пиперидин-1-карбоксилат
Промежуточное соединение 28 (400 мг, 1 ммоль 0,53) растворяли в этаноле (30 мл), затем добавляли 10% Pd/C (ALFA-AESAR, 80 мг) и смесь перемешивали в атмосфере H2 при давлении окружающей среды в течение 1 часа. Контроль методом UPLC показал полное расходование исходного материала. Катализатор удаляли фильтрованием, растворитель выпаривали и получали неочищенное соединение, которое очищали с помощью флэш-хроматографии на силикагеле, используя качестве элюента линейный градиент циклогексан/AcOEt, с получением указанного в заголовке соединения (336,8 мг, 84%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,76 (д, J=4,8 Гц, 2H), 7,35 (т, J=4,8 Гц, 1H), 4,02 (уш. д, J=12,3 Гц, 2H), 3,06-2,96 (м, 1 H), 2,88 (уш. с, 2H), 1,97-1,86 (м, 2H), 1,68-1,59 (м, 2H), 1,45 (с, 9H).
Промежуточное соединение 30: гидрохлорид 2-(пиперидин-4-ил)пиримидина
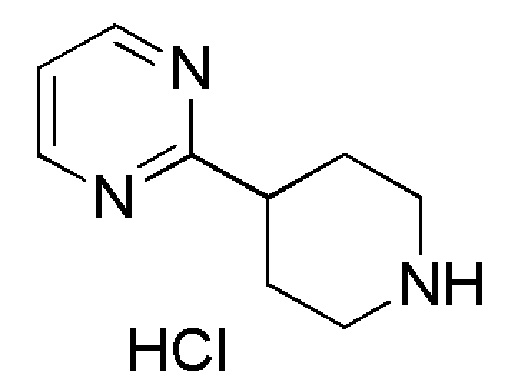
К раствору промежуточного соединения 29 (337 мг, 1,28 ммоль) в 1,4-диоксане (3,2 мл), при 0°C добавляли 4M раствор HCl в 1,4-диоксане (ALFA-AESAR, 3,2 мл, 12,8 ммоль), и полученную смесь перемешивали при комнатной температуре в течение ночи. Контроль с помощью UPLC и TLC показал, что реакция была завершена. Растворитель удаляли в вакууме, с получением указанного в заголовке соединения (295 мг, количественный выход), которое использовали на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, DMSO-d6) δ м.д.: 9,12 (уш. с, 5H), 8,88 (уш. с, 4H), 8,79 (д, J=5,0 Гц, 7H), 7,40 (т, J=4,9 Гц, 4H), 3,38-3,28 (м, 2H), 3,21-3,11 (м, 1H), 3,09-2,97 (м, 2H), 2,17-2,08 (м, 2H), 2,05-1,92 (м, 2H); [ES+MS] m/z 164 (MH+).
Промежуточное соединение 31: гидрохлорид 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,6-тетрагидропиридино (коммерчески доступен)
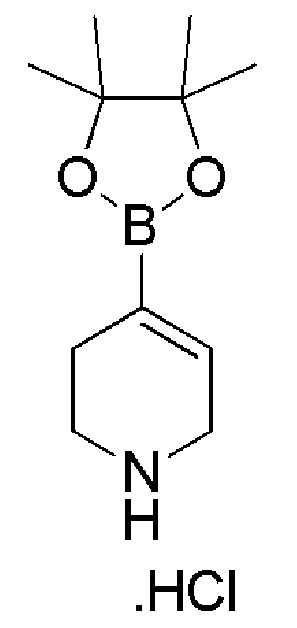
Трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)карбоксилат (АРК PHARMA, 25 г, 80,851 ммоль) растворяли в 250 мл EtOAc и 4Н HCl в EtOAc (Symax Fine Chemicals, 250 мл) при 0°C. Смесь оставляли при 26°C и перемешивали в течение 3 часов. Реакционную смесь выпаривали при пониженном давлении. Неочищенный продукт промывали диэтиловым эфиром и фильтровали, с получением указанного в заголовке соединения (20 г, количественный выход).
1H ЯМР (400 МГц, DMSO-d6) δ м.д.: 9,30 (уш. с, 2H), 6,40-6,30 (м, 1H), 3,64-3,52 (м, 2H), 3,15-3,00 (м, 2H), 2,34-2,22 (м, 2H), 1,21 (с, 12H).
Промежуточное соединение 32: 4,4,4-трифтор-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-ил)бутан-1-он
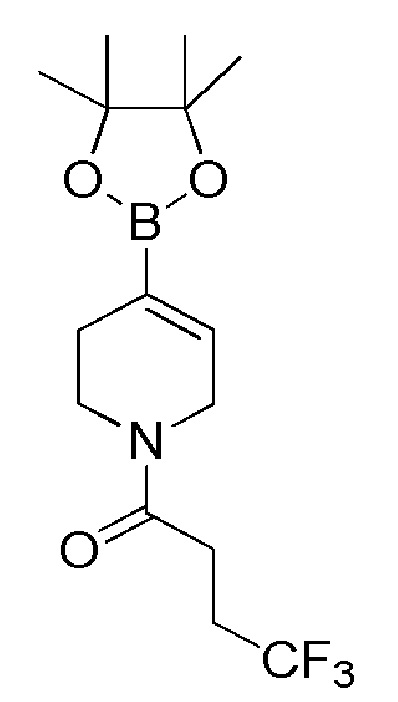
К раствору промежуточного соединения 31 (750 мг, 3,0 ммоль), 4,4,4-трифторбутановой кислоты (COMBIBLOCKS, 477 мг, 3,36 ммоль) в DMF (10 мл) добавляли DMAP (AVRA, 1117 мг, 9,162 ммоль) и EDC.HCl (SILVERY CHEMICALS, 1458 мг, 7,63 ммоль) при 0°C. Реакционную смесь оставляли при 27°C и перемешивали в течение 16 часов. Реакционную смесь гасили холодной водой со льдом (100 мл) и экстрагировали EtOAc (3 х 30 мл). Органический слой промывали насыщенным солевым раствором (30 мл), сушили над Na2SO4 (безводн.), фильтровали и фильтрат выпаривали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на колонке с силикагелем, используя в качестве элюента линейный градиент петролейный эфир/EtOAc, с получением указанного в заголовке соединения (410 мг, 41%) в виде бесцветной жидкости.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 6,55-6,40 (м, 1H), 4,15-3,96 (м, 2H), 3,69-3,43 (м, 2H), 2,63-2,42 (м, 4H), 2,35-2,22 (м, 2H), 1,31-1,22 (м, 12H); [ES+MS] m/z 332 (MH-).
Промежуточное соединение 33: 4,4,4-трифтор-1-(4-пиримидин-5-ил-3,6-дигидро-2Н-пиридин-1-ил)бутан-1-он
Промежуточное соединение 32 (400 мг, 1,2 ммоль), 5-бромпиримидин (ALFA-AESAR, 191 мг, 1 ммоль 0,2) и K2CO3 (SIGMA-ALDRICH, 332 мг, 2,4 ммоль) суспендировали в смеси DME/MeOH/H2O (1/0,5/1, 2,5 мл) в инертной атмосфере. В атмосфере аргона добавляли Pd(dppf)Cl2 (ACROS, 88 мг, 0,12 ммоль) и закрытую пробирку подвергали воздействию микроволнового излучения при 90°C в течение 15 минут. К реакционной смеси добавляли воду и экстрагировали с использованием EtOAc (x2). Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 (безводн.) и упаривали. Остаток очищали с помощью препаративной HPLC (колонка OmniSpher С18, 10 мкм, 41 х 250 мм), градиент от 10% до 100% ACN/Н2O (0,1% муравьиная кислота), 30 минут, с получением указанного в заголовке соединения (160 мг, 46,7%) в виде бледно-желтого масла.
1H ЯМР (300 МГц, CD2Cl2) δ м.д.: 9,05 (с, 1H), 8,74 (с, 2H), 6,25-6,19 (м, 1H), 4,26 (д, J=3,6 Гц, 1H), 4,18 (д, J=3,6 Гц, 1H), 3,84 (т, J=5,8 Гц, 1H), 3,71 (т, J=5,8 Гц, 1H), 2,69-2,55 (м, 6H); [ES+MS] m/z 286 (MH+).
Промежуточные соединения 34-35 были получены методами, аналогичными методу, описанному для промежуточного соединения 33, но с заменой 5-бромпиримидина на соединения, которые указаны в Таблице 3. Также указаны изменения, использованные на стадии очистки.
Таблица 3
ALFA-AESAR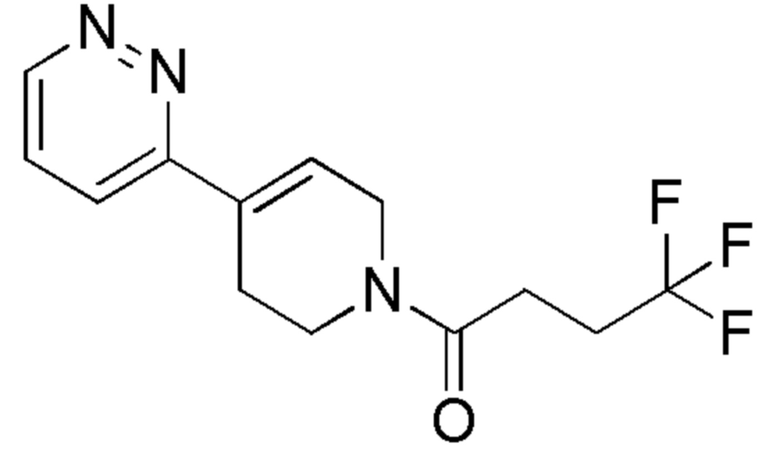
см. примечание a)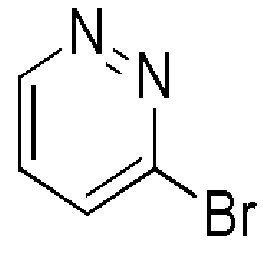
SIGMA-ALDRICH
Примечание:
а) в DME/EtOH/H2O (1/0.5/1, 3 мл), микроволновое облучение при 80°C в течение 90 минут. Очистка флэш-хроматографией на силикагеле с использованием в качестве элюента линейный градиент EtOAc/MeOH.
Промежуточное соединение 36: (4,4,4-трифтор-1-(2'-(трифторметил)-5,6-дигидро-[4,4'-бипиридин]-1(2H)-ил)бутан-1-он)
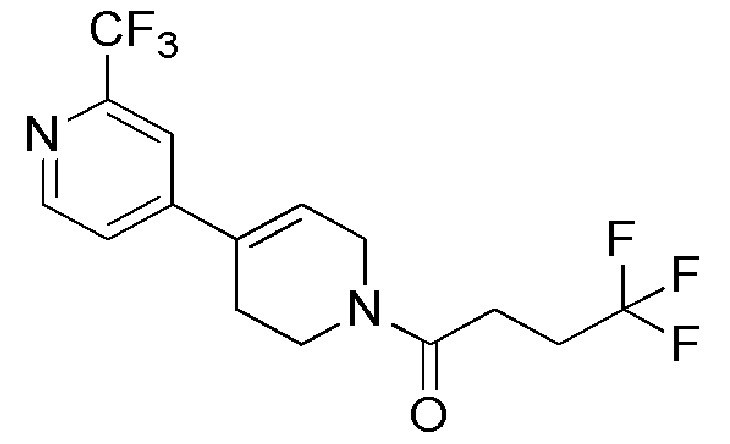
Раствор промежуточного соединения 32 (110 г, 330,33 ммоль), 4-хлор-2-(трифторметил)пиридина (FRAPP´S CHEMICALS, 80,95 г, 445,94 ммоль) в 1,4-диоксане (1100 мл) продували азотом в течение 30 минут. Затем добавляли раствор карбоната натрия (CHEMLABS, 70 г, 660,66 ммоль) и комплекс Pd(dppf)2Cl2.CH2Cl2 (JOHNSON MATTHEY CATALYSTS, 26,9 г, 33,033 ммоль) при 27°С. Реакционную смесь нагревали до 120°C и перемешивали в течение 4 часов при этой же температуре. Ход реакции контролировали с помощью TLC. По завершении реакции реакционную смесь фильтровали через целит и промывали EtOAc (4 х 500 мл). Фильтрат выпаривали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии с использованием в качестве элюента линейный градиент петролейный эфир/EtOAc. Чистые фракции собирали и концентрировали при пониженном давлении, с получением указанного в заголовке соединения (65 г, 53%) в виде густой жидкости коричневого цвета.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,74-8,67 (м, 1H), 7,69-7,61 (м, 1H), 7,47-7,40 (м, 1H), 6,47-6,30 (м, 1Н), 4,38-4,30 (м, 1H), 4,24-4,18 (м, 1H), 3,93-3,85 (м, 1H), 3,75-3,68 (м, 1H), 2,76-2,46 (м, 6H); [ES+ MS] m/z 353 (MH+).
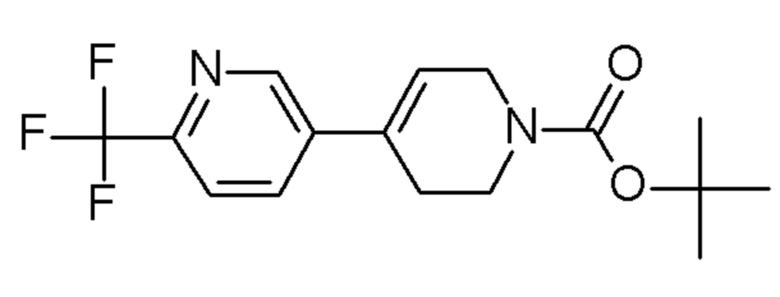
Промежуточные соединения 37-44 были получены способами, аналогичными способу, описанному для промежуточного соединения 36, но с заменой 4-хлор-2-(трифторметил)пиридина на соединения, указанные в Таблице 4.
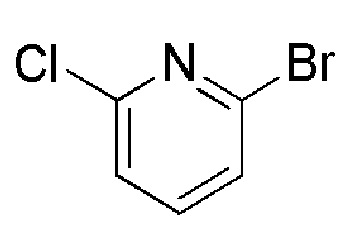
Таблица 4
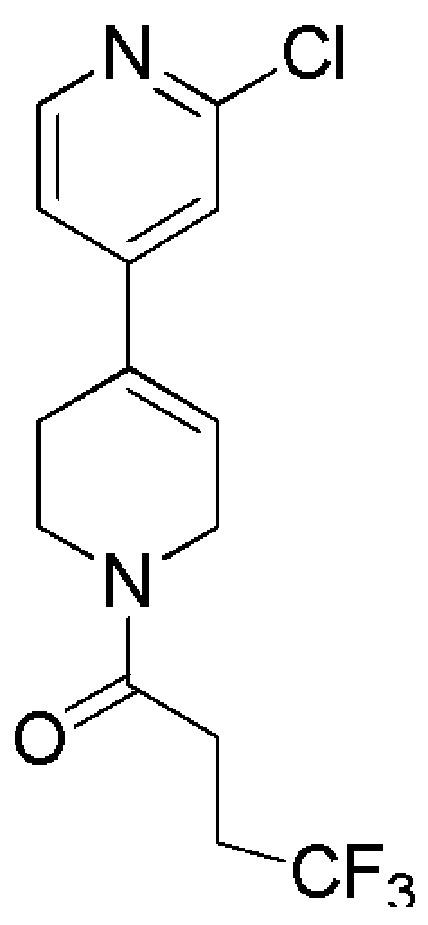
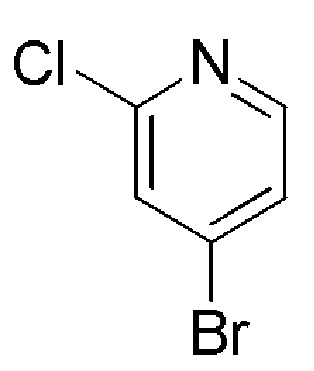
COMBI BLOCKS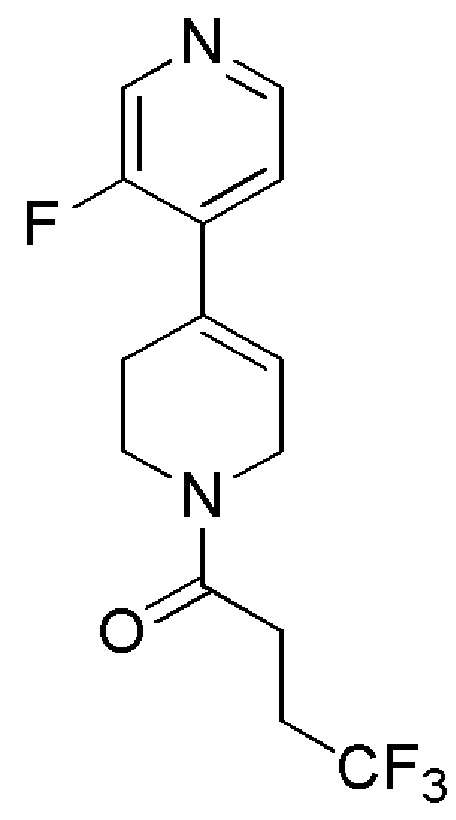
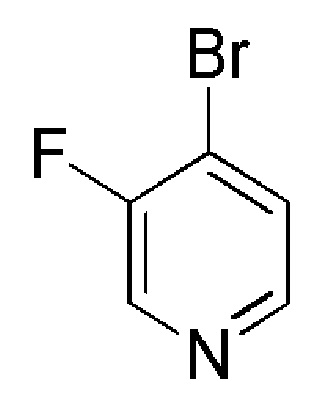
COMBI BLOCKS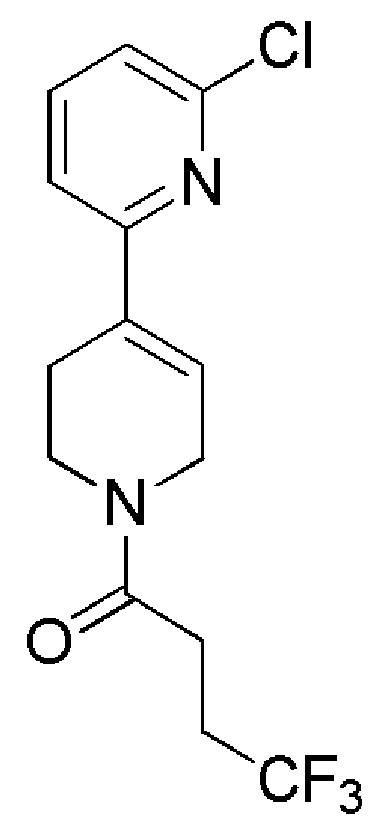
COMBI BLOCKS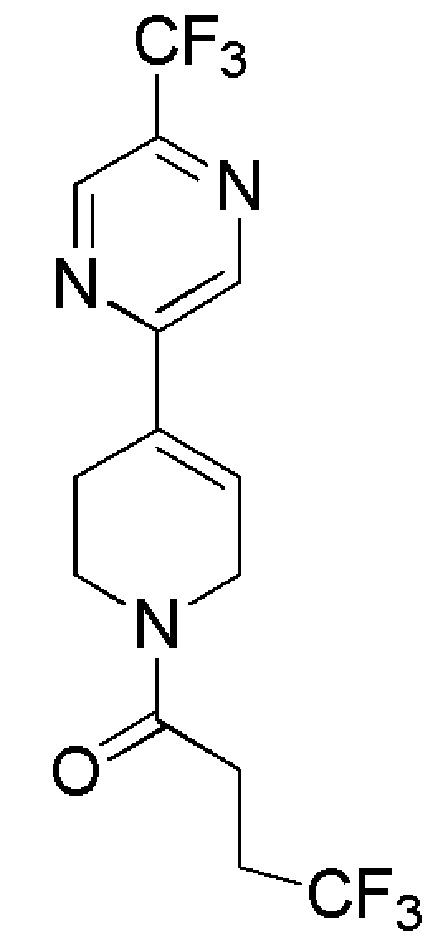
см. примечание a)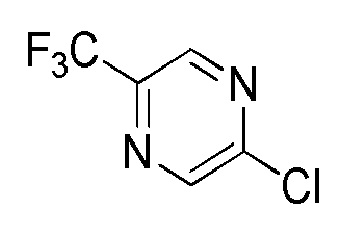
OAKWOOD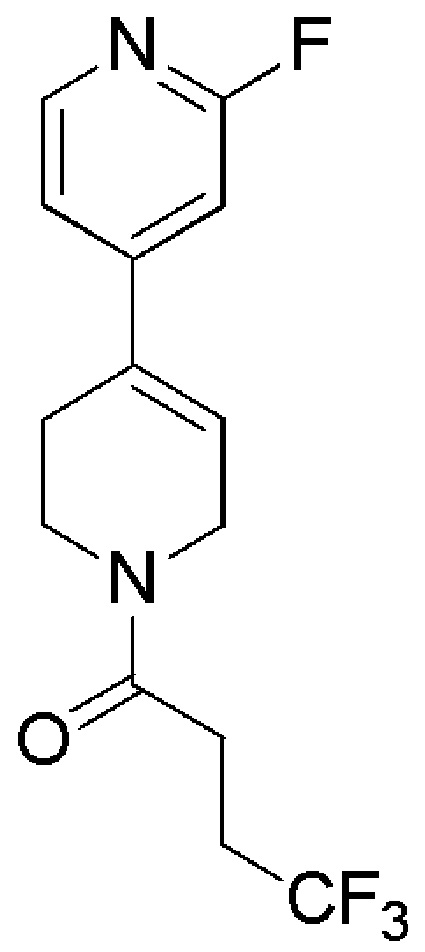
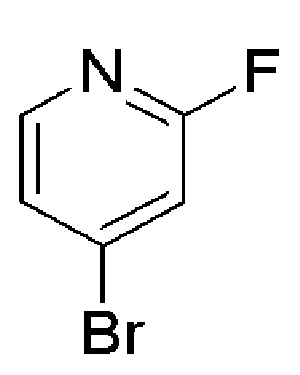
COMBI BLOCKS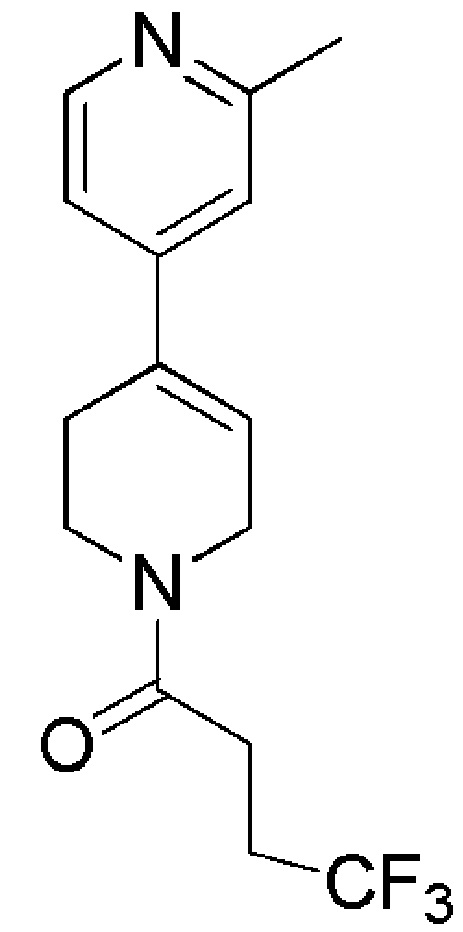
см. примечание a)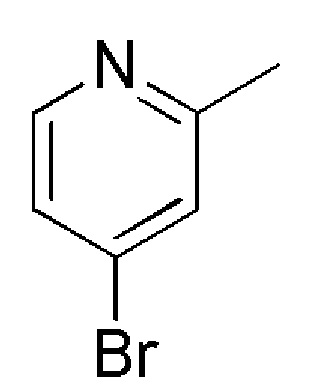
COMBI BLOCKS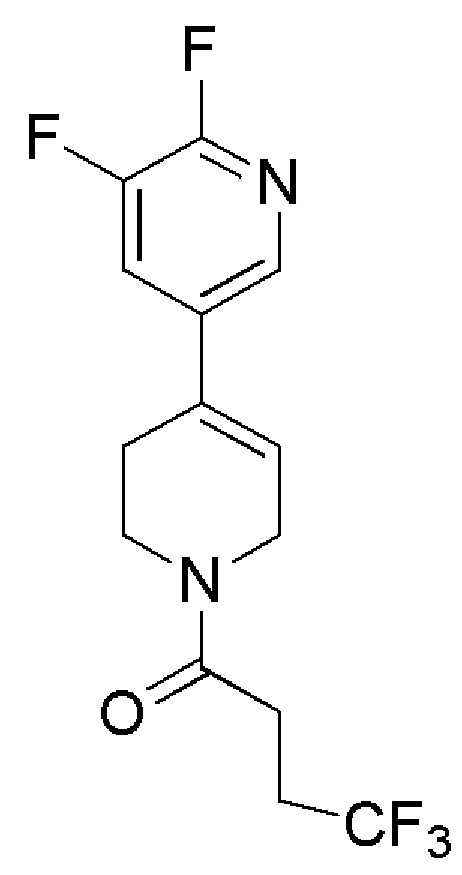
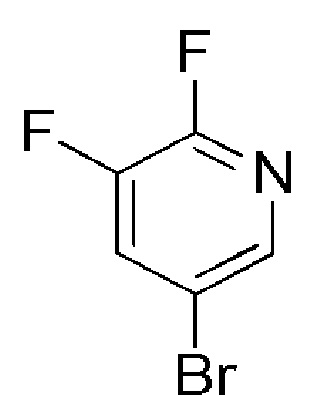
COMBI BLOCKS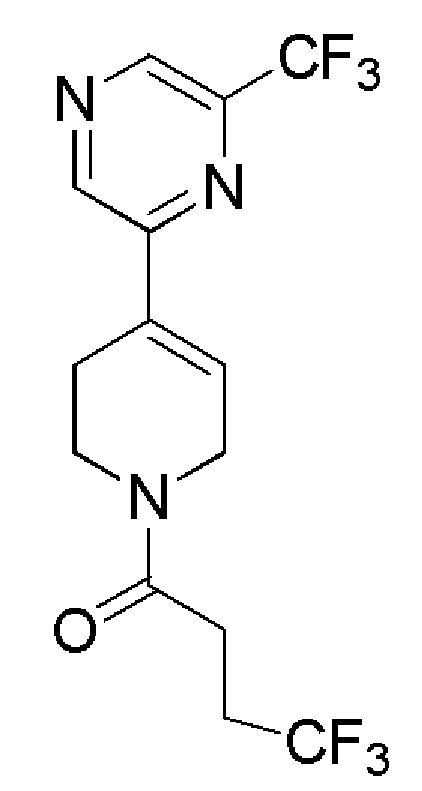
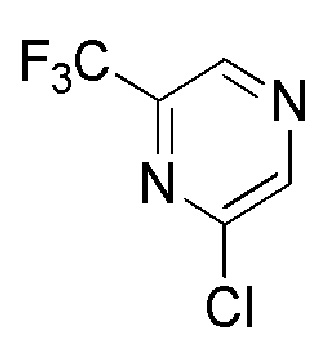
OAKWOOD
Примечание:
a) Очистка флэш-хроматографией на силикагеле с использованием в качестве элюента линейный градиент DCM/MeOH.
Промежуточное соединение 45: 1-(4-(6-аминопиразин-2-ил)-5,6-дигидропиримидин-(2H)-ил)4,4,4-трифторбутан-1-он
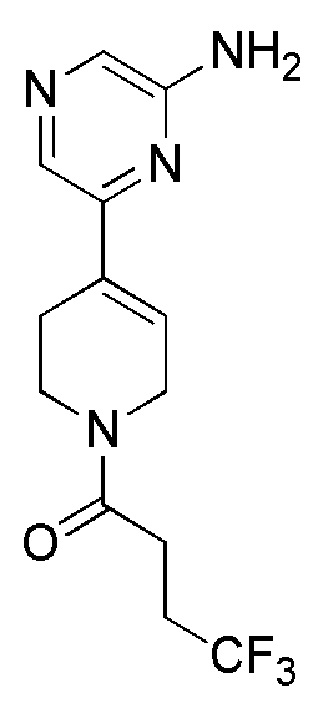
К перемешиваемому раствору промежуточного соединения 32 (600 мг, 1,8018 ммоль) в 1,4-диоксане (10 мл) добавляли 6-бромпиразин-2-амин (COMBI BLOCKS, 314 мг, 1,802 ммоль), карбонат натрия (CHELABS, 477 мг, 4,504 ммоль) и воду (1 мл), а затем реакционную смесь дегазировали аргоном в течение 10 минут при 26°C. После этого при 26°C добавляли Pd(dppf)2Cl2.CH2Cl2 (ALFA-AESAR, 147 мг, 0,1802 ммоль), и реакционную смесь нагревали при 130°С в микроволновой печи в течение 1 часа. Реакционную смесь разбавляли смесью MeOH/DCM (50 мл) и фильтровали через слой целита. Фильтрат концентрировали при пониженном давлении, и неочищенный продукт очищали с помощью колоночной хроматографии на нейтральном глиноземе с использованием в качестве элюента линейный градиент DCM/MeOH, с получением указанного в заголовке соединения (600 мг, 53%) в виде черной смолистой жидкости.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,07-7,95 (м, 1H), 7,87 (д, J=6,1 Гц, 1H), 6,71-6,58 (м, 1H), 4,52 (уш. с, 2H), 4,35-4,25 (м, 1H), 4,23-4,10 (м, 1H), 3,84 (т, J=5,8 Гц, 1H), 3,72-3,62 (м, 1H), 2,90-2,10 (м, 4H); [ES+MS] m/z 301 (MH+).
Промежуточное соединение 46: трет-бутил-5-амино-5',6'-дигидро-[3,4'-бипиридин]-1'(2'H)-карбоксилат
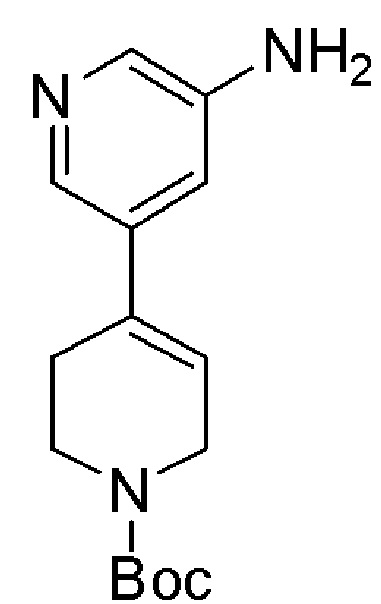
Промежуточное соединение 46 получали, используя метод, аналогичный тому, который описан для промежуточного соединения 45, но с заменой промежуточного соединение 32 на трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилат, а также с заменой 6-бромпиразин-2-амина на 5-бромпиридин-3-амин (COMBI BLOCKS, 560 мг, 3,2362 ммоль).
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,06 (д, J=1,5 Гц, 1H), 7,98 (д, J=2,6 Гц, 1H), 6,92 (т, J=2,2 Гц, 1H), 6,04 (уш. с, 1H), 4,07 (уш. д, J=2,8 Гц, 2H), 3,78-3,66 (м, 2H), 3,63 (т, J=5,7 Гц, 2H), 2,55-2,41 (м, 2H), 1,49 (с, 9H); [ES+MS] m/z 276 (MH+).
Промежуточное соединение 47: трет-бутил-4-(5-аминопиридин-3-ил)пиперидин-1-карбоксилат
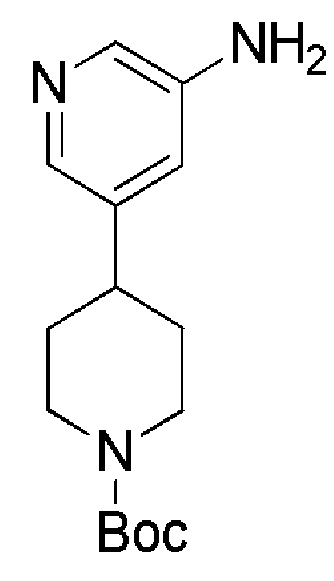
К раствору промежуточного соединения 46 (800 мг, 2,909 ммоль) в MeOH (20 мл) при 27°С добавляли 10% Pd/C (HINDUSTAN, 400 мг). Реакционную смесь перемешивали в течение 3 часов в атмосфере водорода (давление баллона) при той же температуре. Реакционную смесь фильтровали через слой целита в атмосфере азота, и фильтрат концентрировали при пониженном давлении, с получением указанного в заголовке соединения (600 мг, 67%) в виде черной смолистой жидкости.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,04-7,83 (м, 2H), 6,80 (с, 1H), 4,32-4,15 (м, 2H), 3,98-3,35 (м, 2H), 2,89-2,70 (м, 2H), 2,64-2,57 (м, 1H), 1,80 (уш. д, J=12,9 Гц, 2H), 1,69-1,56 (м, 2H), 1,48 (с, 9H); [ES+MS] m/z 278 (MH+).
Промежуточное соединение 48: трет-бутил-4-(5-хлорпиридин-3-ил)пиперидин-1-карбоксилат
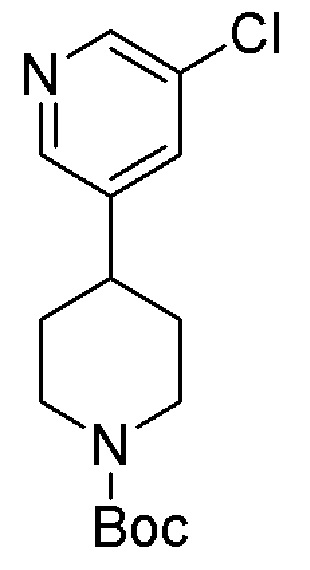
К раствору промежуточного соединения 47 (600 мг, 2,166 ммоль) в ACN (10 мл) добавляли при 27°C по каплям изопентилнитрит (РНР, 381 мг, 3,249 ммоль) и перемешивали в течение 10 минут с последующим добавлением хлорида меди (II) (ALFA-AESAR, 437 мг, 3,249 ммоль) при той же температуре. Реакционную смесь нагревали до 60°С и перемешивали в течение 1,5 часа при той же температуре. Реакционная смесь концентрировали при пониженном давлении, остаток растворяли в EtOAc (50 мл) и фильтровали через слой целита. К фильтрату добавляли воду (60 мл) и экстрагировали EtOAc (3 х 50 мл). Объединенный органический слой сушили над Na2SO4 (безводн.), фильтровали, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента градиент DCM/MeOH, с получением указанного в заголовке соединения (350 мг) в виде коричневой жидкости.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,57-8,26 (м, 2H), 7,51 (с, 1H), 2,95-2,73 (м, 2H), 2,73-2,59 (м, 1H), 1,92-1,78 (м, 2H), 1,70-1,56 (м, 4H), 1,48 (с, 9H); [ES+MS] m/z 297 (MH+).
Промежуточное соединение 49: гидрохлорид 3-хлор-5-(пиперидин-4-ил)пиридина
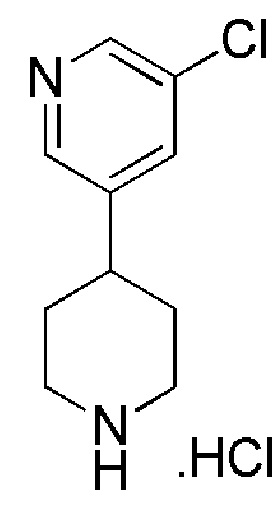
К раствору промежуточного соединения 48 (350 мг, 1,182 ммоль) при 0°C добавляли 4М HCl в EtOAc (HYCHEM, 3 мл). Реакционную смесь оставляли нагреваться до 27°C и перемешивали в течение 2 часов при этой же температуре. Реакционную смесь концентрировали при пониженном давлении, полученный остаток промывали диэтиловым эфиром и сушили в вакууме с получением указанного в заголовке соединения (300 мг) в виде не совсем белого вязкого твердого вещества.
1H ЯМР (400 МГц, DMSO-d6) δ м.д.: 9,05-8,67 (м, 2H), 8,59-8,38 (м, 2H), 7,80 (с, 1H), 3,45-3,25 (м, 2H), 2,98 (кв, J=11,5 Гц, 3H), 2,06-1,80 (м, 4H); [ES+MS] m/z 197 (MH+).
Промежуточное соединение 50: 1-(4-(6-аминопиразин-2-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-он
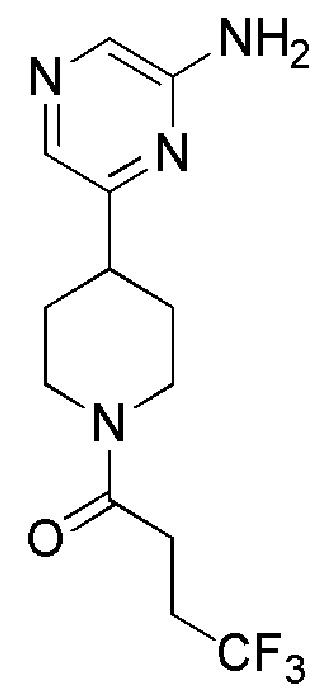
К раствору промежуточного соединения 45 (600 мг, 2,0 ммоль) в MeOH (10 мл) при 27°С добавляли 10% Pd/C (HINDUSTAN, 300 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода (давления баллона) при той же температуре. Реакционную смесь фильтровали через слой целита в атмосфере азота, и фильтрат концентрировали при пониженном давлении, с получением указанного в заголовке соединения (400 мг, 42%) в виде черной смолистой жидкости.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 7,84 (с, 1H), 7,79 (с, 1H), 4,51 (уш. с, 2H), 4,02-3,90 (м, 2H), 3,24-3,14 (м, 1H), 2,60-2,44 (м, 4H), 1,91 (уш. д, J=15,3 Гц, 2H), 1,76-1,70 (м, 2H); [ES+MS] m/z 303 (MH+).
Промежуточное соединение 51: трет-бутил-4-гидрокси-4-(пиридин-2-ил)пиперидин-1-карбоксилат
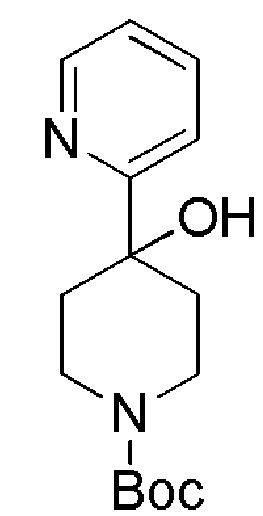
К суспензии трет-бутилового эфира 4-оксопиперидин-1-карбоксилат (ASHVARSHA, 1 г, 5,018 ммоль) и 2-бромпиридина (Мерк, 1.03 г, 6,524 ммоль) в DCM (10 мл) при -78°С добавляли н-бутиллитий (2,5 М в гексане) (HYCHEM, 2 мл, 5,018 ммоль). Реакционную смесь перемешивали в течение 3 часов при -78°С. Реакционную смесь гасили насыщенным раствором хлорида аммония (50 мл) и экстрагировали DCM (3 х 70 мл). Органический слой промывали солевым раствором (50 мл), сушили над Na2SO4 (безводн.), фильтровали, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на колонке с силикагелем, используя в качестве элюента линейный градиент петролейный эфир/EtOAc, с получением указанного в заголовке соединения (750 мг, 50,7%) в виде бледно-желтой жидкости.
1H ЯМР (400МГц, CDCl3) δ м.д.: 8,54 (д, J=4,6 Гц, 1H), 7,77-7,69 (м, 1H), 7,33 (д, J=7,9 Гц, 1H), 7,25-7,19 (м, 1H), 4,16-4,01 (м, 2H), 3,29 (уш. т, J=12,3 Гц, 2H), 2,00-1,87 (м, 2H), 1,60 (уш. д, J=12,1 Гц, 2H), 1,49 (с, 9H); [ES+MS] m/z 279 (MH+).
Промежуточные соединения 52-55 были получены способом, аналогичным способу, описанного для промежуточного соединения 51, но с заменой бромпроизводных на соединения, указанные в Таблице 5.
Таблица 5
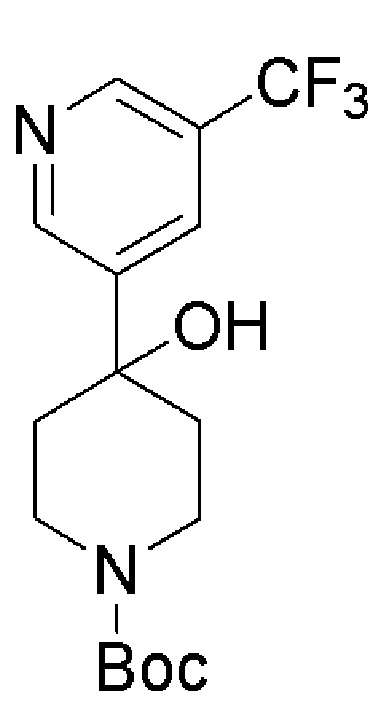
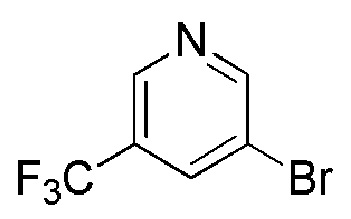
COMBI BLOCKS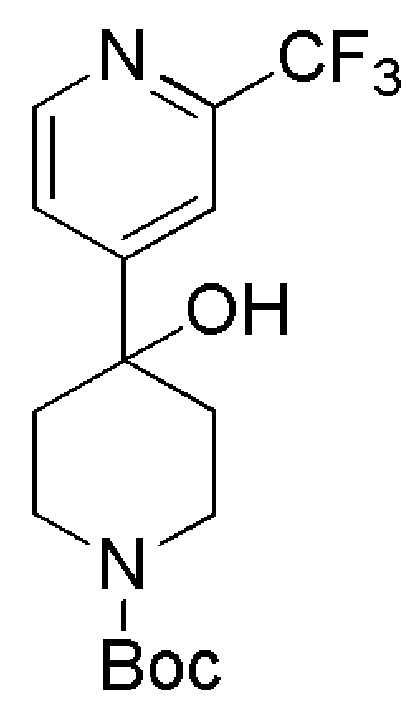
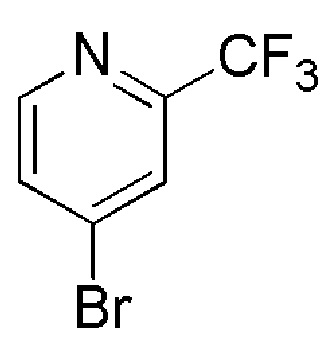
ALFA-AESAR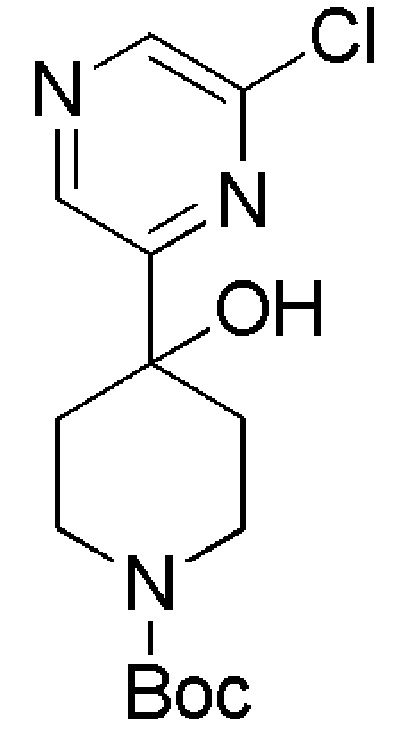
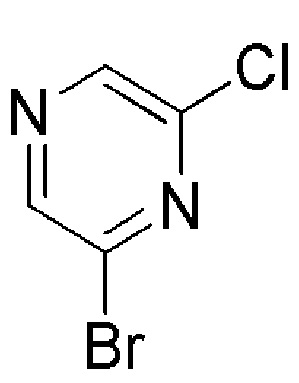
COMBI BLOCKS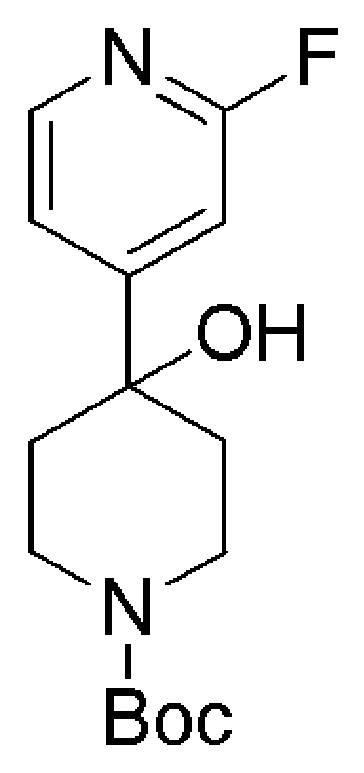
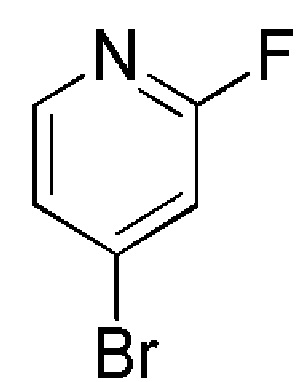
COMBI BLOCKS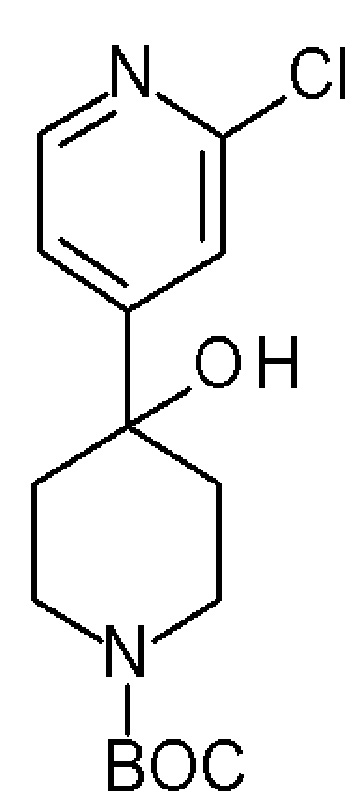
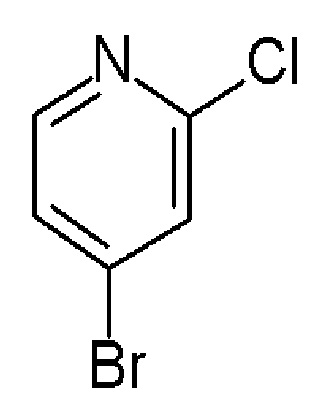
ENAMINE
Промежуточное соединение 56: трет-бутил-4-(5-хлорпиридин-3-ил)-4-гидроксипиперидин-1-карбоксилат
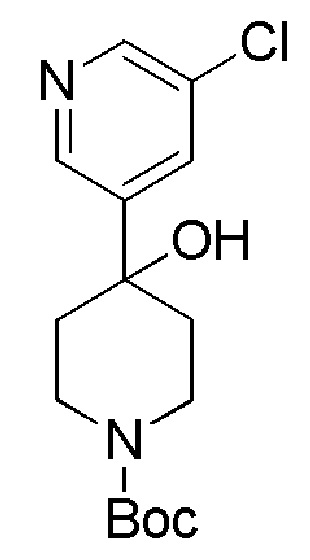
К перемешиваемому раствору трет-бутил-4-оксопиперидин-1-карбоксилата (VASUDHA CHEMICALS, 3,0 г, 15,056 ммоль) и 3-бром-5-хлорпиридина (COMBI BLOCKS, 2,89 г, 15,056 ммоль) в THF (30 мл) при -78°C добавляли по каплям н-бутиллитий (1,6 М в гексане) (HYCHEM, 9,4 мл, 15,056 ммоль). Реакционную смесь перемешивали в течение 2 часов при -78°С. Реакционную смесь гасили насыщенным раствором хлорида аммония (50 мл) и экстрагировали EtOAc (2 х 50 мл). Объединенные органические слои промывали насыщенным солевым раствором (100 мл), сушили над Na2SO4 (безводн.) и фильтровали. Фильтрат концентрировали при пониженном давлении, и неочищенный продукт очищали с помощью хроматографии на колонке с силикагелем, используя в качестве элюента линейный градиент петролейный эфир/EtOAc, с получением указанного в заголовке соединения (2,0 г, 40%) в виде коричневого густой жидкости.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,60 (д, J=2,1 Гц, 1H), 8,49 (д, J=2,3 Гц, 1H), 7,81 (т, J=2,2 Гц, 1H), 4,17-4,02 (м, 2H), 3,22 (т, J=13,1 Гц, 2H), 2,03-1,92 (м, 2H), 1,78-1,70 (м, 2H), 1,49 (с, 9H); [ES+MS] m/z 313 (MH+).
Промежуточное соединение 57: гидрохлорид 4-(пиридин-2-ил)пиперидин-4-ола
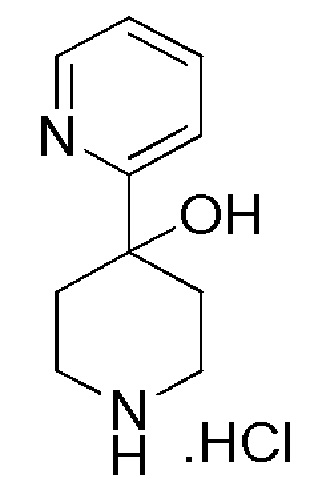
К раствору промежуточного соединени 51 (750 мг, 3,493 ммоль) в EtOAc (7 мл) при 0°C добавляли 4М HCl в EtOAc (HYCHEM, 7 мл). Реакционную смесь оставляли нагреваться до 26°C и перемешивали в течение 3 часов при той же температуре. Реакционную смесь концентрировали при пониженном давлении с получением указанного в заголовке соединения (550 мг, 39%) в виде твердого вещества белого цвета.
1H ЯМР (400МГц, DMSO-d6) δ м.д.: 9,29-8,93 (м, 2H), 8,64 (уш. д, J=3,9 Гц, 1H), 8,13 (уш. с, 1H), 7,82 (уш. д, J=7,9 Гц, 1H), 7,56 (уш. с, 1H), 3,33-3,09 (м, 4H), 2,46-2,34 (м, 2H), 1,82 (уш. д, J=14,3 Гц, 2H); [ES+MS] m/z 179 (MH+).
Промежуточные соединения 58-61 были получены способами, аналогичными способу, описанному для промежуточного соединения 57, но с заменой промежуточного соединения 51 на промежуточные соединения, указанные в Таблице 6.
Таблица 6
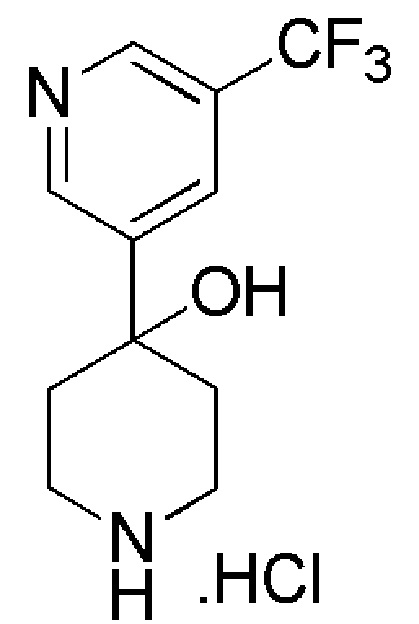
см. примечание a)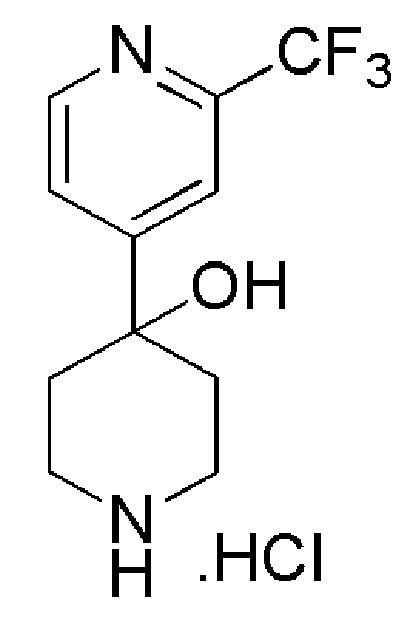
см. примечание b)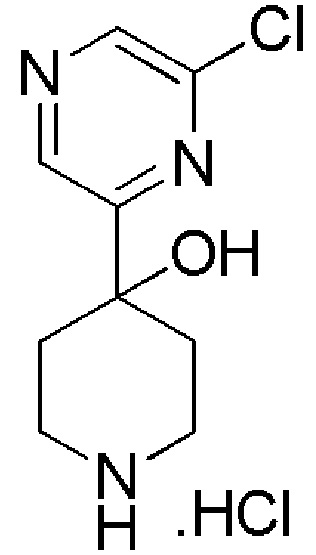
см. примечание a)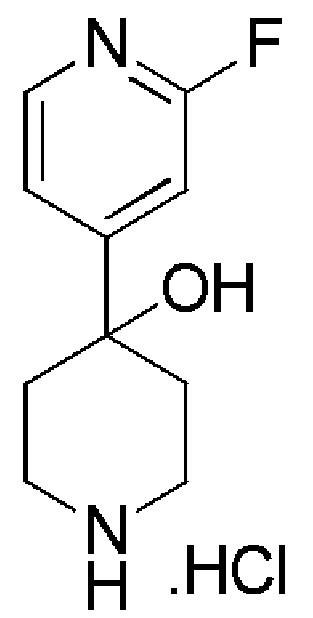
см. примечание a)
Примечания:
Очистка промывкой диэтиловым эфиром
Очистка промывкой н-пентаном.
Промежуточное соединение 62: 4,4,4-трифтор-1-(4-гидрокси-4-(пиридин-2-ил)пиперидин-1-ил)бутан-1-он
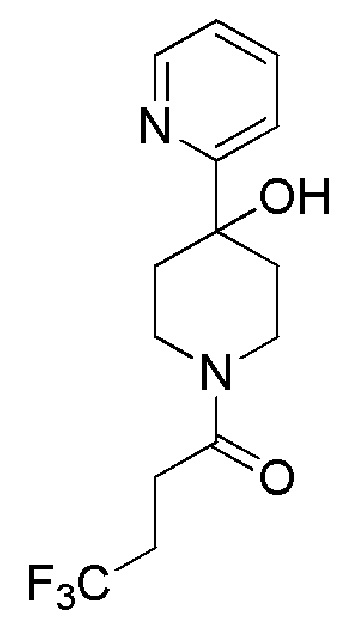
К раствору промежуточного соединения 57 (550 мг, 2,570 ммоль), EDC.HCl (SILVERY, 1,22 г, 6,425 ммоль) и 4,4,4-трифторбутановой кислоты (MATRIX, 437 мг, 3,084 ммоль) в DMF при 27°C добавляли DMAP (AVRA, 940 мг, 7,710 ммоль). Полученную реакционную смесь перемешивали в течение 16 часов при температуре 26°C. Реакционную смесь разбавляли водой (50 мл) и экстрагировали EtOAc (3 х 100 мл). Объединенные органические слои промывали солевым раствором (100 мл), сушили над Na2SO4 (безводн.), фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на колонке с силикагелем, используя в качестве элюента линейный градиент петролейный эфир/EtOAc, с получением указанного в заголовке соединения (450 мг, 53,69%) в виде бледно-желтой жидкости.
1H ЯМР (400МГц, CDCl3) δ м.д.: 8,56 (д, J=5,0 Гц, 1H), 7,78-7,71 (м, 1H), 7,32 (д, J=8,1 Гц, 1H), 7,25 (уш. д, J=5,0 Гц, 1H), 5,32 (с, 1H), 4,72-4,61 (м, 1H), 3,88-3,74 (м, 1H), 3,74-3,62 (м, 1H), 3,26-3,07 (м, 1H), 2,67-2,48 (м, 4H), 2,01-1,86 (м, 2H), 1,79-1,60 (м, 2H); [ES+MS] m/z 303 (MH+).
Промежуточные соединения 63-66 получали способами, аналогичными способу, описанному для промежуточного соединения 62, но с заменой промежуточного соединения 57 на промежуточные соединения, указанные в Таблице 7.
Таблица 7
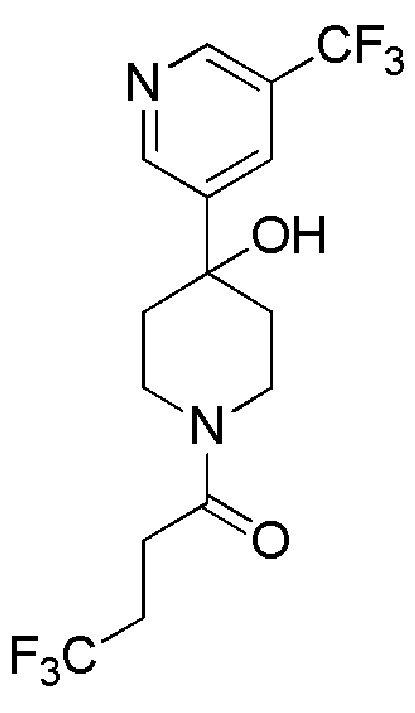
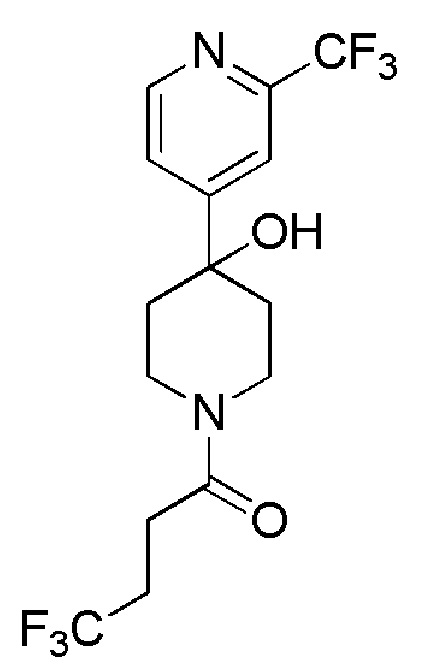
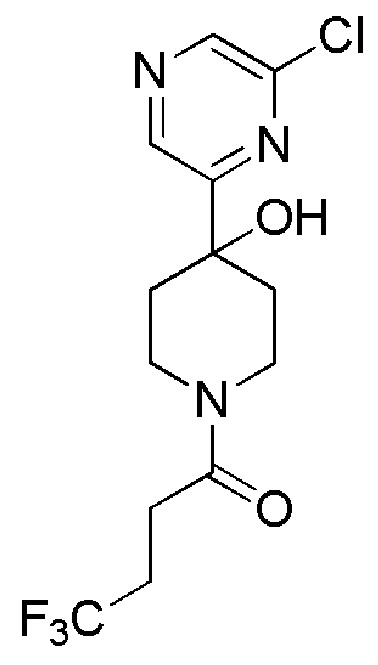
см. примечание a)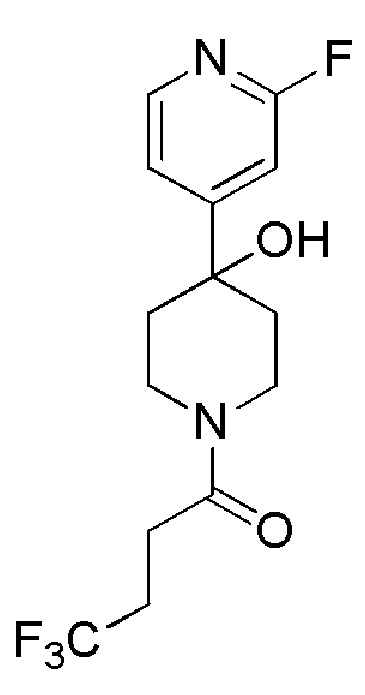
Примечание:
использовали на следующей стадии без очистки
Промежуточное соединение 66а: трет-бутил-4-гидрокси-4-[4-(трифторметил)-2-пиридил] пиперидин-1-карбоксилат
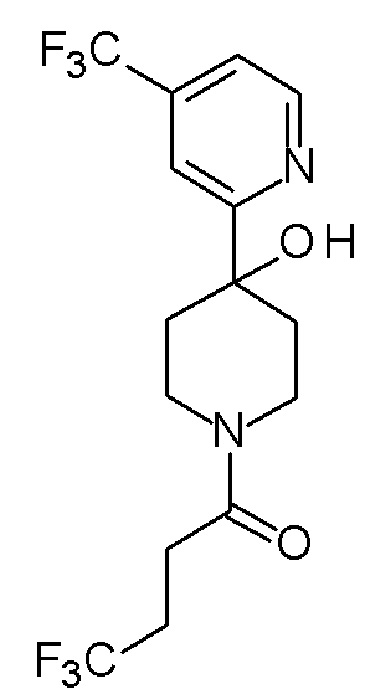
Раствор 2-бром-4-(трифторметил)пиридина (FLUOROCHEM, 139 мкл, 1,12 ммоль) в безводном DCM (3 мл) охлаждали до -78°С в атмосфере аргона. Затем по каплям добавляли 2,5 М раствор н-BuLi в гексане (0,450 мл, 1,12 ммоль), и полученный оранжевый раствор перемешивали при -78°C в течение 30 минут. При -78°С медленно добавляли промежуточное соединение 2 (250 мг, 1,12 ммоль) в безводном DCM (1,5 мл+1,5 мл для ополаскивания), и реакционную смесь перемешивали в течение 2 часов. (контроль LCMS). Реакцию гасили насыщенным раствором NH4Cl (15 мл), экстрагировали DCM и органическую фазу промывали насыщенным солевым раствором (25 мл). Органическую фазу сушили над MgSO4 и концентрировали в вакууме. Сырую смесь очищали препаративной HPLC (ACN/Н2O/НСООН, от 10/90 до 100/0, 32 мин), с получением указанного в заголовке соединения (111 мг, 27%) в виде бесцветного масла.
1H ЯМР (300 МГц, CDCl3) δ м.д.: 8,75 (д, J=5,3 Гц, 1H), 7,58 (с, 1H), 7,49 (д, J=5,3 Гц; 1,3 Гц, 1H), 4,67 (д, J=13,1 Гц, 1H), 3,82 (д, J=13,1 Гц, 1H), 3,71-3,61 (м, J=12,8 Гц, 2,5 Гц, 1H), 3,21-3,11 (м, J=12,8 Гц, 2,8 Гц, 1H), 2,69-2,60 (м, 2H), 2,60-2,46 (м, 2H), 2,06-1,93 (м, 2H), 1,72 (уш. т, J=12,1 Гц, 2H); [ES+MS] m/z 371 (MH+).
Промежуточное соединение 66b: трет-бутил-4-гидрокси-4-[4-(трифторметил)-2-пиридил]пиперидин-1-карбоксилат
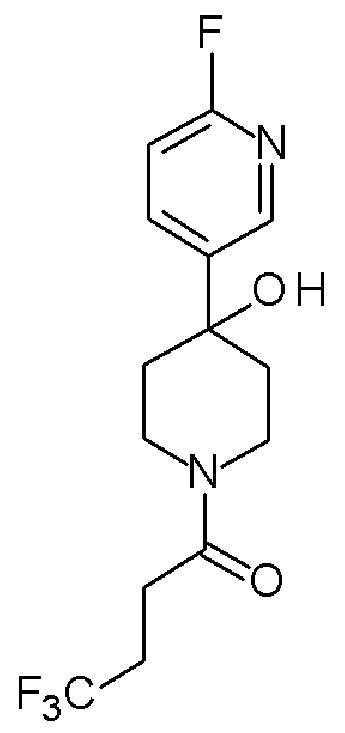
Промежуточное соединение 66b получали способом, аналогичным способу, описанному для промежуточного соединения 66а, но с заменой 2-бром-4-(трифторметил)пиридина на 5-бром-2-фтор-пиридин (FLUOROCHEM).
1H ЯМР (300 МГц, CD2Cl2) δ м.д.: 8,28-8,27 (м, 1H), 7,96-7,89 (м, 1 H), 6,95-6,90 (м, 1 H), 4,51-4,45 (м, 1 H), 3,77-3,71 (м, 1 H), 3,63-3,53 (м, 1 H), 3,32 (с, 1H), 3,14-3,05 (м, 1H), 2,64-2,39 (м, 4 H), 1,97-1,5 (м, 4H); [ES+MS] m/z 321 (MH+).
Промежуточное соединение 67: трет-бутил-4-(5-хлорпиридин-3-ил)-4-фторпиперидин-1-карбоксилат
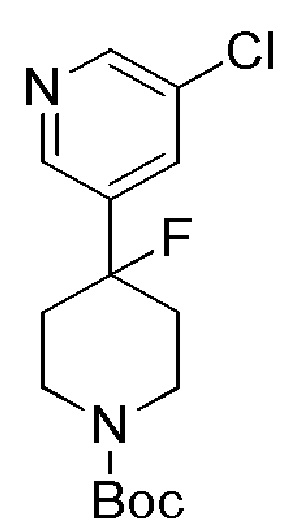
К раствору промежуточного соединения 56 (1,5 г, 4,796 ммоль) в DCM (27 мл) при -78°C добавляли по каплям раствор DAST (ALFA-AESAR, 0,628 мл, 4,796 ммоль) в DCM (3 мл). Реакционную смесь оставляли нагреваться до 0°C и перемешивали в течение 40 минут при той же температуре. Реакционную смесь гасили насыщенным раствором бикарбоната натрия (200 мл) и экстрагировали с помощью DCM (2 х 50 мл). Объединенные органические слои сушили над Na2SO4 (безводн.), фильтровали и концентрировали при пониженном давлении. Полученный продукт очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента линейный градиент петролейный эфир/EtOAc, с получением указанного в заголовке соединения (800 мг, 63%) в виде бесцветной густой жидкости.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,54 (д, J=2,3 Гц, 1H), 8,49 (д, J=1,9 Гц, 1H), 7,71 (т, J=2,2 Гц, 1H), 4,15 (с, 2H), 3,17 (с, 2H), 2,08-1,85 (м, 4H), 1,49 (с, 9H); [ES+MS] m/z 315 (MH+).
Промежуточное соединение 67а: трет-бутил-4-(2-хлор-4-пиридил)-4-фтор-пиперидин-1-карбоксилат
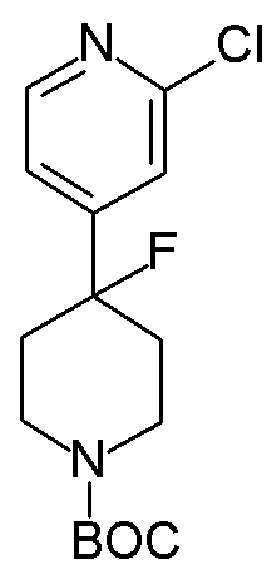
Промежуточное соединение 67а получали методом, аналогичным методу, описанному для промежуточного соединения 67, с заменой промежуточного соединения 56 на промежуточное соединение 55a.
1H ЯМР (300 МГц, CD2Cl2) δ м.д.: 8,37 (д, J=5,2 Гц, 1H), 7,34 (д, J=1,4 Гц, 1H), 7,25-7,15 (м,1H), 4,13 (д, J=12,4 Гц, 2H), 3,11 (д, J=11,6 Гц, 2H), 2,06-1,81 (м, 4H), 1,46 (с, 9H); [ES+MS] m/z 315 (MH+).
Промежуточное соединение 67b: соединение 2-хлор-4-(4-фтор-4-пиперидил)пиридина с 2,2,2-трифторуксусной кислотой
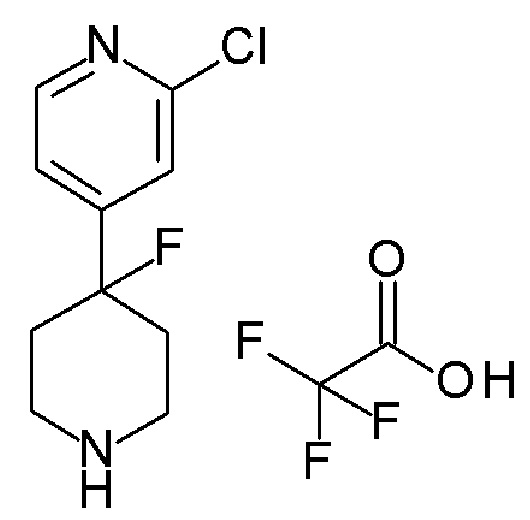
Промежуточное соединение 67а (22,0 мг, 0,07 ммоль) растворяли в DCM (1,0 мл). Раствор 50:50 DCM/TFA (0,2 мл) медленно добавляли при комнатной температуре. Раствор перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли при пониженном давлении с получением указанного в заголовке соединения (23 мг, 100%) в виде не совсем белого твердого вещества. Полученное соединение было оценено как достаточно чистое для использования его на следующей стадии.
ES+MS] m/z 215 (MH+).
Промежуточное соединение 68: гидрохлорид 3-хлор-5-(4-фторпиперидин-4-ил)пиридина
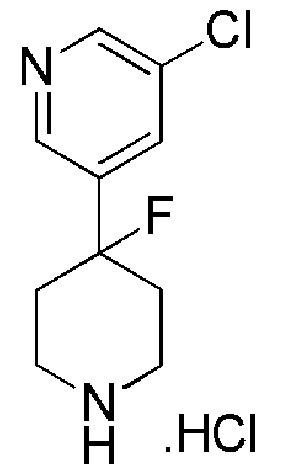
К раствору промежуточного соединения 67 (800 мг, 2,541 ммоль) в EtOAc (8 мл) добавляли 4М раствор HCl в EtOAc (SYMAX FINE CHEMICALS, 8 мл) при 0°С. Реакционную смесь оставляли нагреваться до 26°C и перемешивали в течение 3 часов при той же температуре. Реакционную смесь концентрировали при пониженном давлении, остаток промывали диэтиловым эфиром (3 х 10 мл) и сушили в вакууме, с получением указанного в заголовке соединения (650 мг) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, DMSO-d6) δ м.д.: 9,64 (с, 1H), 9,38 (с, 1H), 8,74-8,60 (м, 2H), 7,97 (т, J=2,1 Гц, 1H), 3,42-3,31 (м, 2H), 3,21-3,04 (м, 2H), 2,69-2,55 (м, 2H), 2,29-2,16 (м, 2H); [ES+MS] m/z 215 (MH+-HCl).
Промежуточное соединение 69: 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-амин
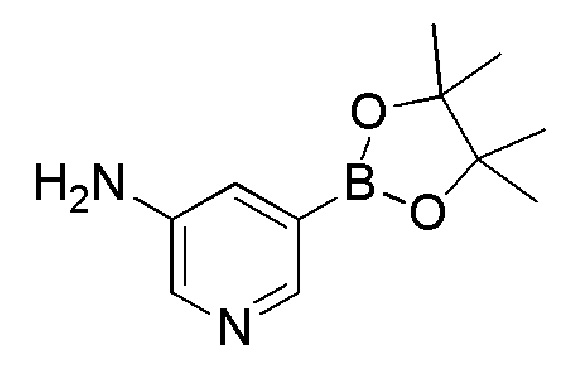
К перемешиваемому раствору 5-бромпиридин-3-амина (MANCHESTER ORGANICS, 1 г, 5,78 ммоль) в 1,4-диоксане (15 мл) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (ALFA-AESAR, 1,46 г, 5,78 ммоль), ацетат калия (AVRA, 1,1 г, 11,56 ммоль), и реакционную смесь дегазировали аргоном в течение 10 минут. Затем добавляли Pd(dppf)Cl2.CH2Cl2 (ALFA-AESAR, 200 мг, 0,289 ммоль). Реакционную смесь нагревали до 100°C и перемешивали в течение 4 часов. Реакционную смесь охлаждали до 27°C и использовали на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,36 (с, 1H), 8,22-8,13 (м, 1H), 8,10-7,95 (м, 1H), 7,36 (уш. с, 2H), 1,27 (с, 12H).
Промежуточное соединение 70: трет-бутил-3-(((трифторметил)сульфонил)окси)-2,5-дигидро-1Н-пиррол-1-карбоксилат
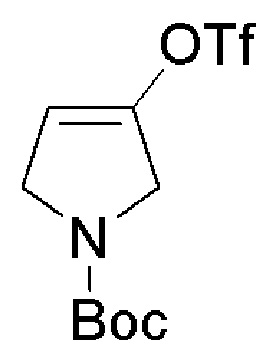
К раствору трет-бутил-3-оксопирролидин-1-карбоксилата (COM ACCERA CHEMBIO, 500 мг, 2,699 ммоль) и N-фенил-бис(трифторметансульфонамид) (COMBI BLOCKS, 1,9 г, 5,398 ммоль) в THF (10 мл) при -78°C добавляли по каплям NaHMDS (1 М в THF) (HYCHEM, 5,39 мл, 5,398 ммоль). Реакционную смесь оставляли нагреваться до 27°C и перемешивали в течение 16 часов при той же температуре. Реакционную смесь гасили насыщенным раствором хлорида аммония (20 мл) и экстрагировали с помощью EtOAc (2 х 20 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 (безводн.), фильтровали и упаривали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на колонке с силикагелем с использованием линейного градиента петролейный эфир/этилацетат, с получением указанного в заголовке соединения (800 мг) в виде бледно-желтой жидкости.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 7,01-6,94 (м, 1H), 4,28-4,17 (м, 4H), 1,48 (с, 9H).
Промежуточное соединение 71: трет-бутил-3-(5-аминопиридин-3-ил)-2,5-дигидро-1Н-пиррол-1-карбоксилат
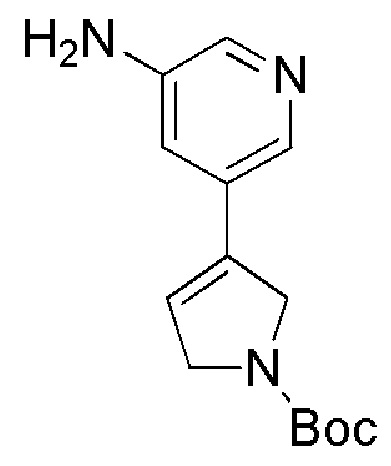
К перемешиваемому раствору промежуточного соединения 69 (реакционная смесь) в 1,4-диоксане добавляли промежуточное соединение 70 (800 мг, 2,521 ммоль), карбонат цезия (ALFA-AESAR, 7,4 г, 22,692 ммоль) и воду (3,0 мл). Реакционную смесь дегазировали аргоном в течение 10 минут. Затем при 27°C добавляли Pd(PPh3)4 (ALFA-AESAR, 200 мг, 0,176 ммоль). Реакционную смесь нагревали до 100°C и перемешивали в течение 2 часов при той же температуре. Реакционную смесь фильтровали через слой целита, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на колонке с силикагелем, используя в качестве элюента линейный градиент петролейный эфир/EtOAc, с получением указанного в заголовке соединения (300 мг) в виде коричневого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,13-8,04 (м, 1H), 8,03-7,97 (м, 1H), 7,02-6,90 (м, 1H), 6,24-6,12 (м, 1H), 4,53-4,39 (м, 2H), 4,37-4,23 (м, 2H), 1,51 (с, 9H); [ES+MS] m/z 262 (MH+).
Промежуточное соединение 72: трет-бутил-3-(5-аминопиридин-3-ил)пирролидин-1-карбоксилат
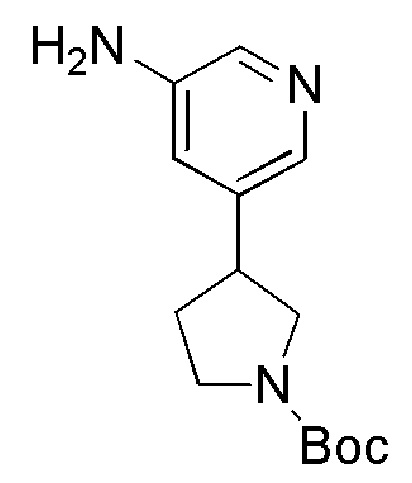
К раствору промежуточного соединения 71 (300 мг, 1 ммоль 0,148) в MeOH (10 мл) при 27°С добавляли 10% Pd/C (HINDUSTAN, 150 мг). Реакционную смесь перемешивали в течение 4 часов в атмосфере водорода (давления баллона) при той же температуре. Реакционную смесь фильтровали через слой целита в атмосфере азота, и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (300 мг) в виде бледно-желтой густой жидкости.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 7,97 (д, J=2,6 Гц, 1H), 7,92 (д, J=1,9 Гц, 1H), 6,83 (т, J=2,2 Гц, 1H), 3,90-3,50 (м, 2H), 3,48-3,24 (м, 2H), 2,30-2,20 (м, 1H), 2,02-1,87 (м, 2H), 1,48 (с, 9H); [ES+MS] m/z 264 (MH+).
Промежуточное соединение 73: трет-бутил-3-(5-хлорпиридин-3-ил) пирролидин-1-карбоксилат
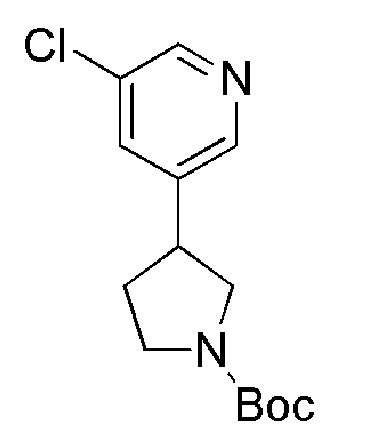
К раствору промежуточного соединения 72 (300 мг, 1 ммоль 0,139) в ACN (5 мл) при 27°C добавляли порциями хлорид меди (II) (ALFA-AESAR, 230 мг, 1,708 ммоль) и перемешивали в течение 20 минут при той же температуре. Затем при 27°C добавляли по каплям изоамилнитрит (RNR, 0,25 мл, 1,708 ммоль). Реакционную смесь нагревали до 65°С и перемешивали в течение 1,5 часа. Реакционную смесь гасили холодной водой со льдом (20 мл) и экстрагировали с помощью EtOAc (2 х 20 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 (безводн.), фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на колонке с силикагелем, используя в качестве элюента линейный градиент петролейный эфир/EtOAc, с получением указанного в заголовке соединения (160 мг, 49%) в виде бледно-желтой густой жидкости.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,47 (д, J=2,3 Гц, 1H), 8,39 (д, J=1,9 Гц, 1H), 7,54 (с, 1H), 3,93-3,52 (м, 2H), 3,51-3,24 (м, 2H), 2,37-2,26 (м, 1H), 1,73-1,53 (м, 2H), 1,48 (с, 9H); [ES+MS] m/z 283 (MH+).
Промежуточное соединение 74: гидрохлорид 3-хлор-5-(пирролидин-3-ил)пиридина
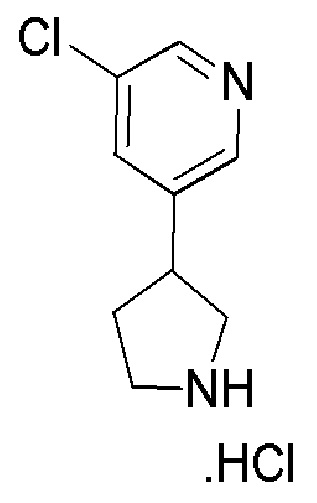
К раствору промежуточного соединения 73 (160 мг, 0,566 ммоль) в EtOAc (2 мл) при 0°С добавляли 4М раствор HCl в EtOAc (SYMAX FINE CHEMICALS, 2 мл). Реакционную смесь оставляли нагреваться до 27°C и перемешивали в течение 3 часов при той же температуре. Реакционная смесь концентрировали при пониженном давлении с получением указанного в заголовке соединения (120 мг) в виде густой жидкости коричневого цвета.
1H ЯМР (400 МГц, DMSO-d6) δ м.д.: 9,66 (уш. с, 2H), 8,64-8,55 (м, 2H), 8,13 (с, 1H), 3,68-3,50 (м, 2H), 3,47-3,35 (м, 1H), 3,28-3,09 (м, 2H), 2,46-2,35 (м, 1H), 2,09-1,94 (м, 1H); [ES+MS] m/z 183 (MH+).
Примеры
Ссылочный пример 1: 4,4,4-трифтор-1-(4-пиразин-2-ил-1-пиперидил)бутан-1-он
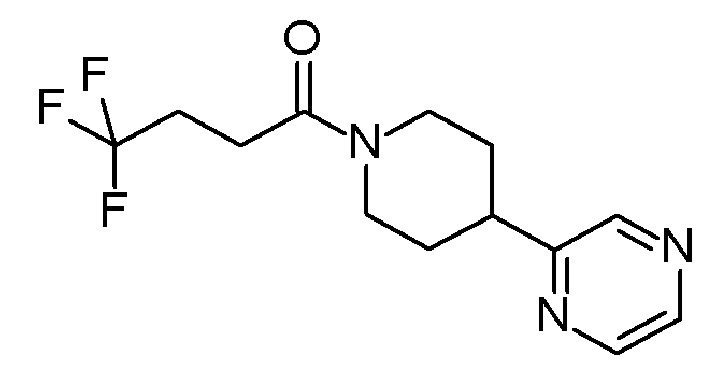
К раствору дигидрохлорида 2-(4-пиперидил)пиразина (ENAMINE, 397,5 мг, 1,68 ммоль) и DMAP (SIGMA-ALDRICH, 422,7 мг, 3,46 ммоль) в хлороформе (3,4 мл) добавляли промежуточное соединение 1 (399,3 мг, 1,64 ммоль). Раствор подвергали воздействию микроволнового излучения в течение 15 минут при 100°C. Реакционную смесь промывали насыщенным раствором Na2CO3 (х 2). Органический слой промывали насыщенным солевым раствором, сушили над MgSO4 (безводн.) и упаривали. Остаток очищали с помощью препаративной HPLC (колонка колонка OmniSpher С18, 10 мкм, 41 х 250 мм), градиент от 10% до 100% ACN/Н2O (0,1% муравьиная кислота), 15 мин, с получением указанного в заголовке соединения (353,3 мг, 74,9%) в виде белого твердого вещества.
1H ЯМР (300 МГц, CD2Cl2) δ м.д.: 8,48-8,47 (м, 2H), 8,41-8,39 (м, 1H), 4,74-4,67 (м, 1H), 3,99-3,92 (м, 1H), 3,23-3,14 (м, 1H), 3,05-2,94 (м, 1H), 2,77-2,67 (м, 1H), 2,64-2,57 (м, 2H), 2,55-2,42 (м, 2H), 2,00-1,92 (м, 2H), 1,87-1,65 (м, 2H); [ES+MS] m/z 288 (MH+).
Пример 2: 4,4,4-трифтор-1-[4-фтор-4-(3-пиридил)-1-пиперидил]бутан-1-он
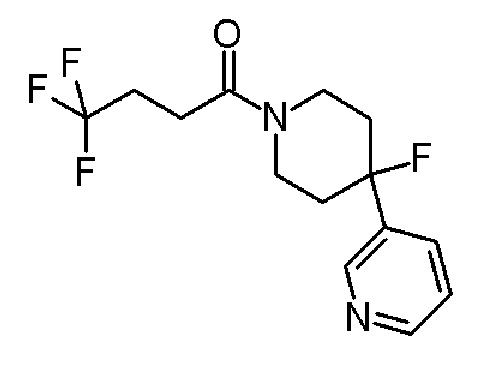
Соединение по Примеру 2 получали способом, аналогичным способу, описанному в Примере 1, но с заменой дигидрохлорида 2-(4-пиперидил)пиразина на дигидрохлорид 3-(4-фтор-4-пиперидил)пиридина (ENAMINE, 0,38 ммоль).
1H ЯМР (300 МГц, CD2Cl2) δ м.д.: 8,63-8,62 (м, 1H), 8,57-8,54 (м, 1H), 7,71-7,67 (м, 1H), 7,35-7,31 (м, 1H), 4,68-4,62 (м, 1H), 3,87-3,80 (м, 1H), 3,57-3,47 (м, 1H), 3,07-2,98 (м, 1H), 2,67-2,45 (м, 4H), 2,14-1,86 (м, 4H); [ES+MS] m/z 305 (MH+).
Пример 3: 4,4,4-трифтор-1-[4-(5-фтор-3-пиридил)-1-пиперидил]бутан-1-он
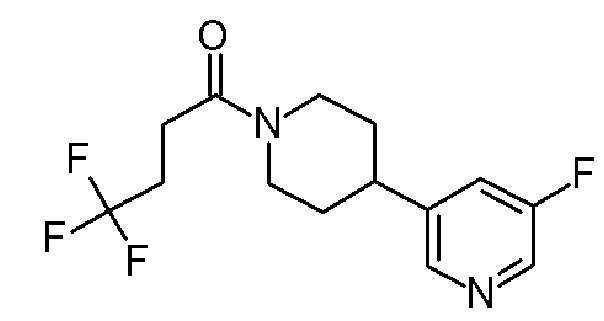
Пробирку укупоривали, заполняли и дегазировали аргоном, а затем добавляли 1,4-диоксан (4 мл). Затем добавляли промежуточное соединение 3 (1,1 г, 2,7 ммоль), (5-фтор-3-пиридил)борную кислоту (ENAMINE, 575,0 мг, 4,08 ммоль) и Cs2СO3 (SIGMA-ALDRICH, 1,35 г, 4,15 ммоль). Раствор подвергали воздействию микроволнового излучения в течение 45 минут при 150°C. Реакционную смесь гасили насыщенным раствором Na2CO3 (2 мл) и экстрагировали DCM (3 раза). Органический слой сушили над безводным MgSO4 и упаривали. Остаток очищали на силикагеле, используя линейный градиент DCM/MeOH, с получением 121,9 мг белого твердого вещества. Остаток очищали с помощью препаративной HPLC (колонка OmniSpher С18, 10 мкм, 41 х 250 мм), градиент от 10% до 100% ACN/Н2О (0,1% муравьиная кислота), 30 мин, с получением указанного в заголовке соединения (95,0 мг, 11,5%) в виде твердого вещества белого цвета.
1H ЯМР (300 МГц, CD2Cl2) δ м.д.: 8,32-8,30 (м, 2H), 7,28-7,23 (м, 1H), 4,78-4,71 (м, 1H), 3,99-3,92 (м, 1H), 3,20-3,11 (м, 1H), 2,90-2,80 (м, 1H), 2,71-2,43 (м, 5H), 1,96-1,87 (м, 2H), 1,68-1,51 (м, 2H); [ES+MS] m/z 305 (MH+).
Пример 4: 4,4,4-трифтор-1-[4-(6-фтор-3-пиридил)-1-пиперидил]бутан-1-он
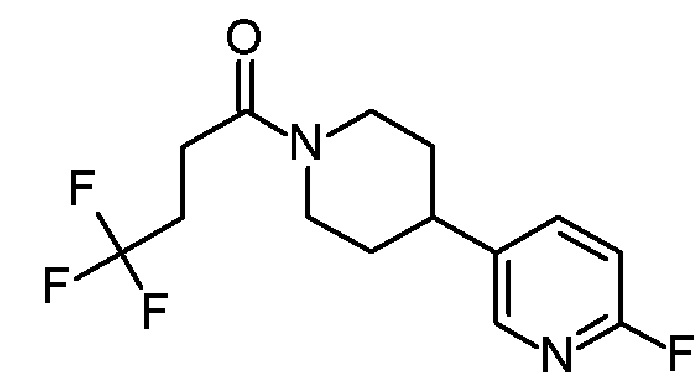
Промежуточное соединение 16 (121 мг 0,0, 0,4 ммоль) растворяли в MeOH (2 мл). Затем медленно добавляли формиат аммония (SIGMA-ALDRICH, 86,9 мг, 1,38 ммоль) и 10% Pd/C (ALFA-AESAR, 44,0 мг, 0,04 ммоль). Реакционную смесь нагревали при 65°С в течение 3 часов, охлаждали до комнатной температуры, фильтровали через целит и фильтрат выпаривали. Остаток очищали с помощью препаративной HPLC (колонка OmniSpher С18, 10 мкм, 41 х 250 мм), градиент от 10% до 100% ACN/Н2О (0,1% муравьиная кислота), 30 мин, с получением указанного в заголовке соединения (49,4 мг, 40,1%) в виде бесцветного масла.
1H ЯМР (300 МГц, CD2Cl2) δ м.д.: 8,06-8,04 (м, 1H), 7,67-7,57 (м, 1H), 6,91-6,87 (м, 1H), 4,78-4,70 (м, 1H), 3,99-3,91 (м, 1H), 3,20-3,10 (м, 1H), 2,85-2,77 (м, 1H), 2,70-2,43 (м, 5H), 1,95-1,83 (м, 1H), 1,67-1,50 (м, 1H); [ES+MS] m/z 305 (MH+).
Соединения по Примерам 5-16 получали способом, аналогичным способу, описанному в Примере 4, но с заменой промежуточного соединение 16 на соединения, которые указаны в Таблице 8. Также указаны изменения, внесенные в протокол получения, и используемые стадии очистки.
Таблица 8
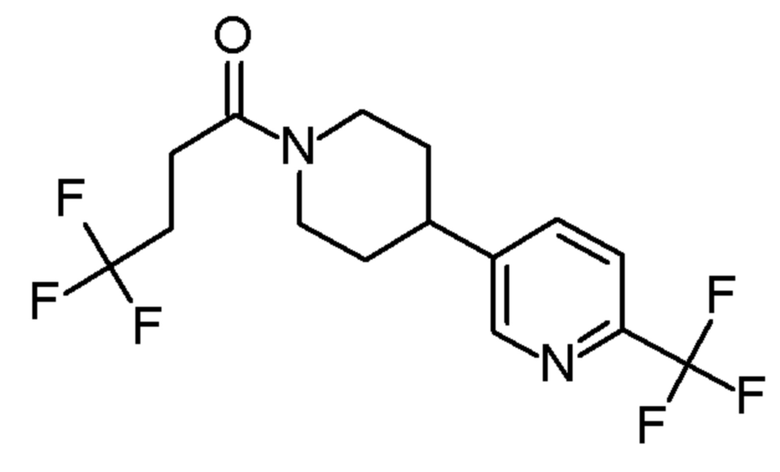
4,4,4-трифтор-1-[4-[6-(трифторметил)-3-пиридил]-1-пиперидил]бутан-1-он
см. примечания a) и d)
1,97 ммоль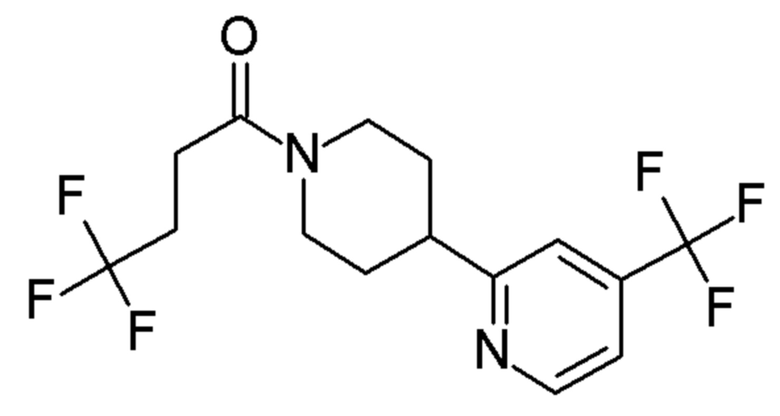
4,4,4-трифтор-1-[4-[4-(трифторметил)-2-пиридил]-1-пиперидил]бутан-1-он
см. примечания a) и d)
1,96 ммоль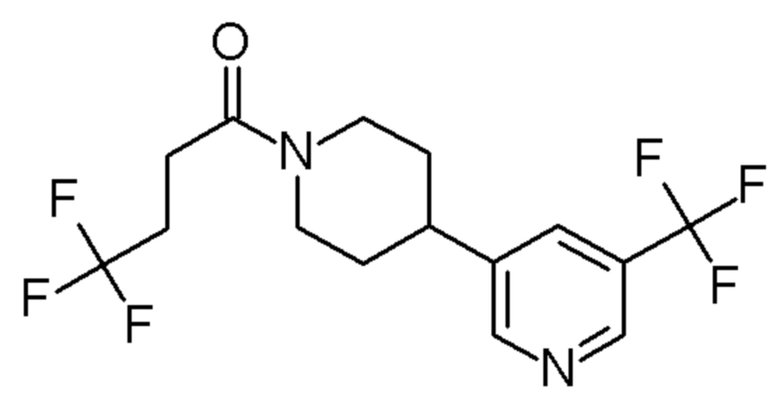
4,4,4-трифтор-1-[4-[5-(трифторметил)-3-пиридил]-1-пиперидил]бутан-1-он
см. примечания a) и d)
1,74 ммоль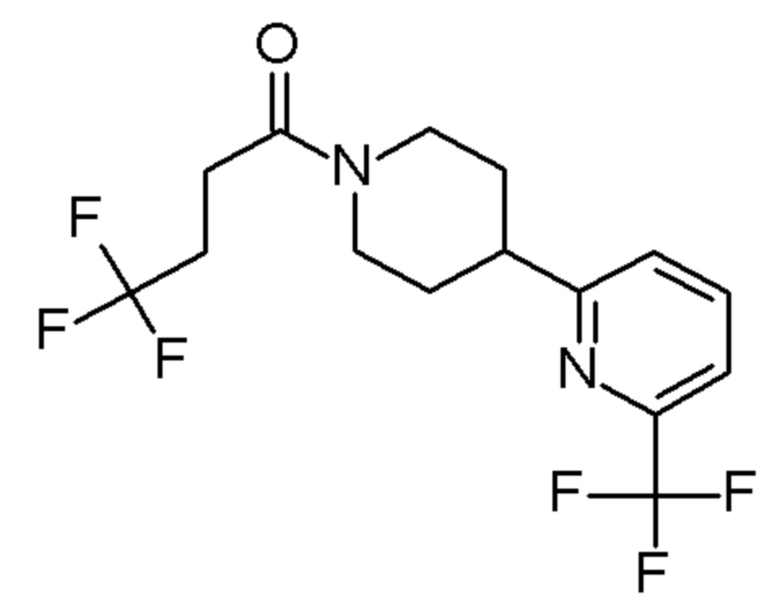
4,4,4-трифтор-1-[4-[6-(трифторметил)-2-пиридил]-1-пиперидил]бутан-1-он
см. примечания a) и d)
1,57 ммоль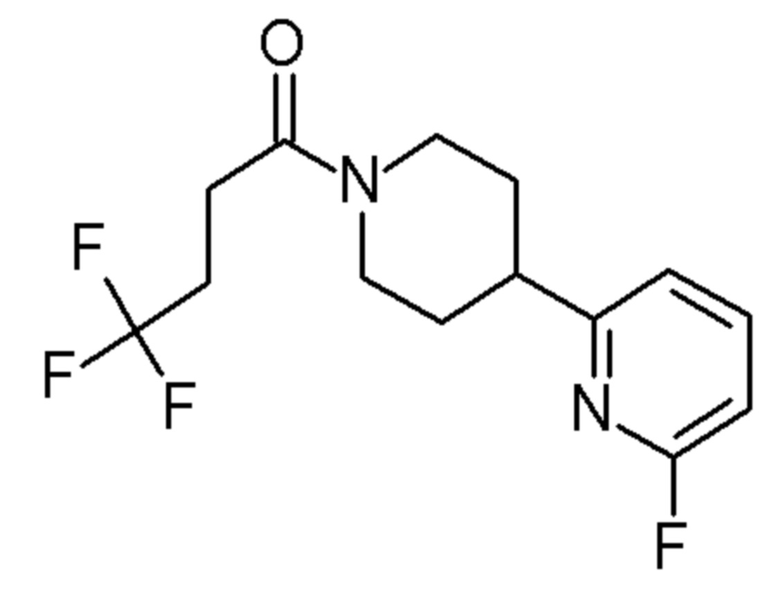
4,4,4-трифтор-1-[4-(6-фтор-2-пиридил)-1-пиперидил]бутан-1-он
см. примечания a) и d)
1,82 ммоль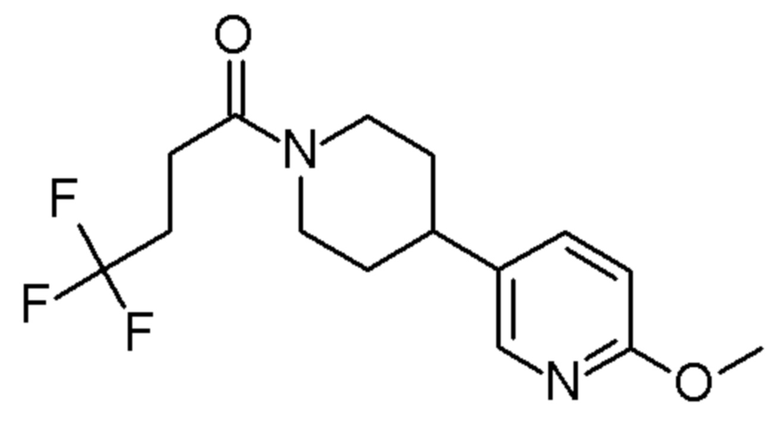
4,4,4-трифтор-1-[4-(6-метокси-3-пиридил)-1-пиперидил]бутан-1-он
см. примечания a) и d)
2,07 ммоль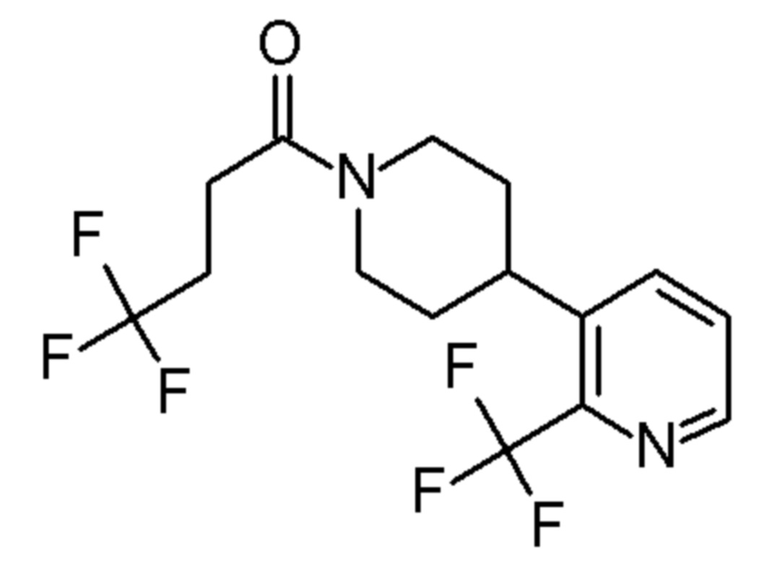
4,4,4-трифтор-1-[4-[2-(трифторметил)-3-пиридил]-1-пиперидил]бутан-1-он
см. примечания b) и d)
1,54 ммоль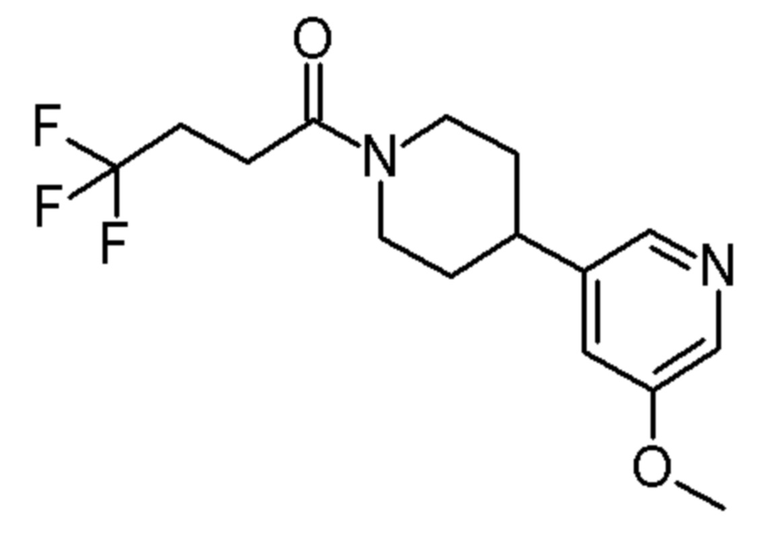
4,4,4-трифтор-1-[4-(5-метокси-3-пиридил)-1-пиперидил]бутан-1-он
см. примечания a) и e)
1,86 ммоль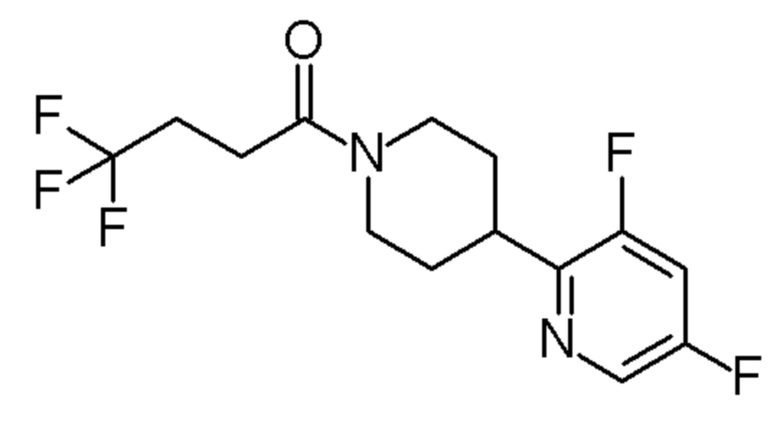
1-[4-(3,5-дифтор-2-пиридил)-1-пиперидил]-4,4,4-трифтор-бутан-1-он
см. примечания c) и e)
1,32 ммоль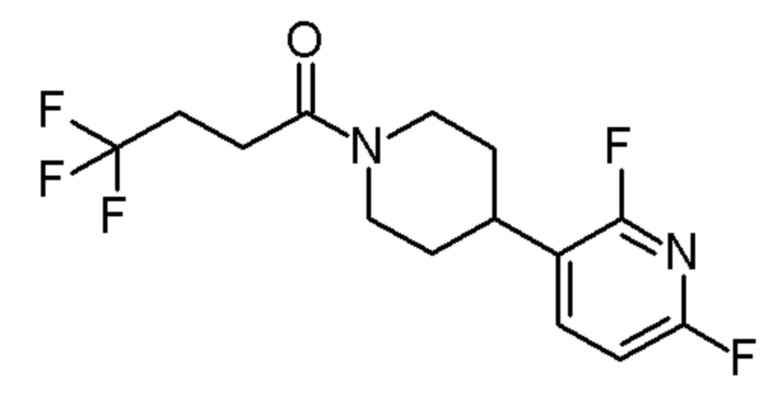
1-[4-(2,6-дифтор-3-пиридил)-1-пиперидил]-4,4,4-трифтор-бутан-1-он
см. примечания c) и e)
1,95 ммоль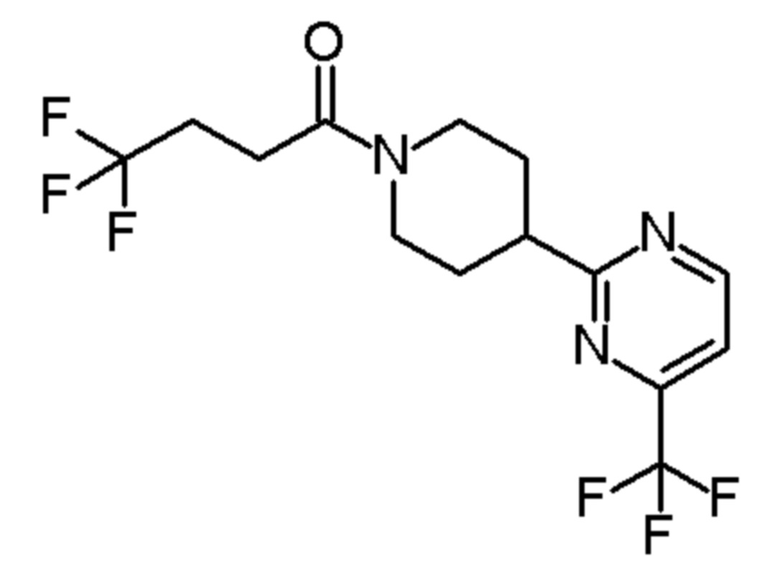
4,4,4-трифтор-1-[4-[4-(трифторметил)
пиримидин-2-ил]-1-пиперидил]бутан-1-он
см. примечания a) и d)
2,30 ммоль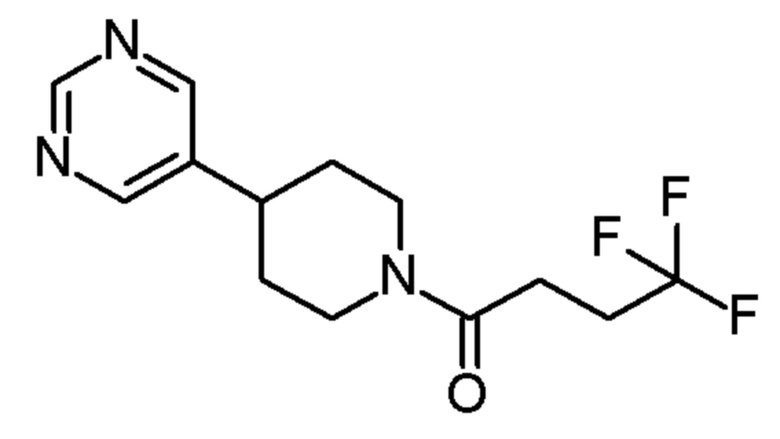
4,4,4-трифтор-1-(4-пиримидин-5-ил-1-пиперидил)бутан-1-он
см. примечание f)
0,53 ммоль
Примечания:
Реакционную смесь нагревали при 65°C в течение 1 часа
Реакционную смесь нагревали при 65°C в течение 1 часа 30 минут
Реакционную смесь нагревали при 65°C в течение 30 минут
Осадок очищали препаративной HPLC (колонка OmniSpher C18, 10 мкм, 41×250 мм) градиент 10%-100% ACN/H2O (0,1% муравьиная кислота), 30 мин
Осадок очищали препаративной HPLC (колонка OmniSpher C18, 10 мкм, 41×250 мм) градиент 10%-100% ACN /H2O (0,1% муравьиная кислота), 35 мин
Реакционную смесь нагревали при кипячении с обратным холодильником в течение 6 часов
Пример 17: 4,4,4-трифтор-1-[4-(5-фтор-2-пиридил)-1-пиперидил]бутан-1-он
Промежуточное соединение 34 (150 мг, 0,49 ммоль) растворяли в MeOH (1 мл), затем медленно добавляли формиат аммония (SIGMA-ALDRICH, 94 мг, 1,49 ммоль) и 10% Pd/C (ALFA-AESAR, 53 мг, 0,05 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 6 часов, и охлаждали до комнатной температуры. Поскольку оставался исходный материал, то к смеси добавляли формиат аммония (SIGMA-ALDRICH, 94 мг, 1,49 ммоль) и 10% Pd/C (ALFA-AESAR, 53 мг, 0,05 ммоль), и реакционную смесь снова нагревали с обратным холодильником в течение 6 часов, затем охлаждали до комнатной температуры и фильтровали через слой целита. Фильтрат упаривали с получением коричневого масла, которое очищали с помощью препаративной HPLC (колонка OmniSpher С18, 10 мкм, 41 х 250 мм), градиент от 10% до 100% ACN/Н2O (0,1% муравьиная кислота), 19 мин, с получением указанного в заголовке соединения (95 мг, 62%) в виде бесцветного масла.
1H ЯМР (300 МГц, CD2Cl2) δ м.д.: 8,07 (д, J=2,5 Гц, 1H), 7,69-7,62 (м, 1H), 6,93-6,89 (м, 1H), 4,79-4,73 (м, 1H), 4,01-3,95 (м, 1H), 3,22-3,18 (м, 1H), 2,89-2,78 (м, 1H), 2,72-2,59 (м, 5H), 2,58-2,45 (м, 2H), 1,96-1,87 (м, 2H); [ES+MS] m/z 305 (MH+).
Пример 18: 4,4,4-трифтор-1-(4-(2-(трифторметил)пиридин-4-ил)пиперидин-1-ил)бутан-1-он
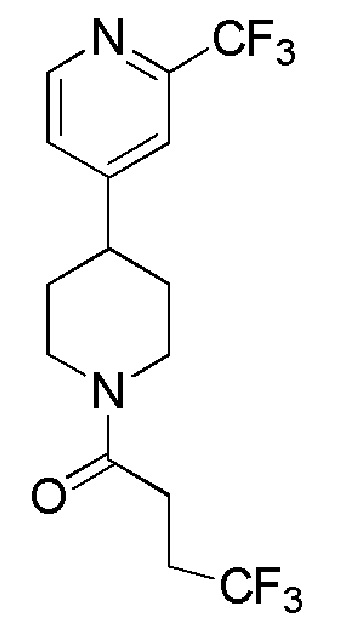
К раствору промежуточного соединения 36 (75 г, 212,904 ммоль) в MeOH (750 мл) при 27°C добавляли 10% Pd/C (HINDUSTAN, 22,5 г). Реакционную смесь перемешивали при той же температуре в атмосфере водорода (30 фунтов на квадратный дюйм (2,068 бар)) в течение 1 часа в смесителе Парра. Ход реакции контролировали с помощью TLC. По завершении реакции реакционную смесь фильтровали через целит и промывали MeOH (4 х 50 мл). Фильтрат концентрировали при пониженном давлении. Полученное соединение очищали с помощью колоночной хроматографии (силикагель 100-200 меш), используя в качестве элюента линейным градиентом петролейный эфир/EtOAc. Чистые фракции собирали и концентрировали при пониженном давлении, с получением указанного в заголовке соединения (55 г, 73%) в виде бледно-желтой густой жидкости. Примечание: Реакцию проводили в нескольких партиях: 3 раза по 20 г и 1 раз по 15 г.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,69-8,64 (м, 1H), 7,52 (с, 1H), 7,35-7,29 (м, 1H), 4,87-4,79 (м, 1H), 4,05-3,96 (м, 1H), 3,25-3,15 (м, 1H), 2,92-2,82 (м, 1H), 2,75-2,47 (м, 5H), 2,03-1,92 (м, 2H), 1,73-1,58 (м, 2H); [ES+MS] m/z 355 (MH+).
Ссылочный пример 19: 4,4,4-трифтор-1-[4-(пиридазин-3-ил)пиперидин-1-ил]бутан-1-он
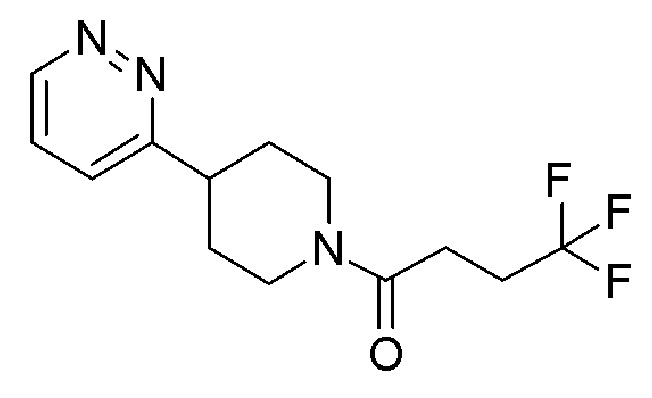
Промежуточное соединение 35 (44,5 мг, 0,16 ммоль) растворяли в этаноле (5 мл), затем добавляли Pd/C 10% (10 мг) и смесь перемешивали в атмосфере водорода при давлении окружающей среды в течение 5 часов. Контроль с помощью UPLC показал образование целевого продукта. Катализатор удаляли фильтрацией, растворитель выпаривали, и полученный неочищенный продукт очищали с помощью препаративной LCMS, с получением указанного в заголовке соединения (8,5 мг, 19%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, DMSO-d6) δ м.д.: 9,04-9,14 (м, 1H), 7,57-7,69 (м, 2H), 4,45-4,63 (м, 1H), 3,96-4,07 (м, 1H), 3,10-3,25 (м, 2H), 2,43-2,79 (м, 5H), 1,85-1,98 (м, 2H), 1,85-1,67 (м, 1H), 1,65-1,52 (м, 1H); [ES+MS] m/z 288 (MH+).
Пример 20: 4,4,4-трифтор-1-(4-(2-фторпиридин-4-ил)пиперидин-1-ил)бутан-1-он
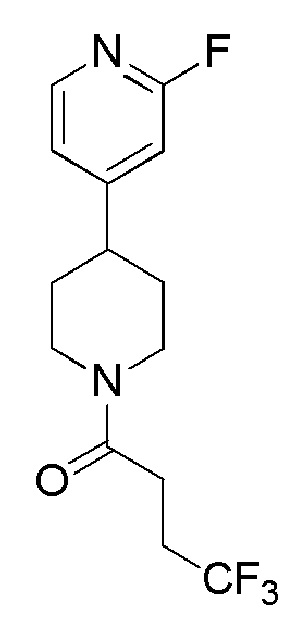
К раствору промежуточного соединения 41 (350 мг, 1,158 ммоль) в MeOH (5 мл) при 26°С добавляли 10% Pd/C (HINDUSTAN, 350 мг). Реакционную смесь перемешивали в течение 3 часов в атмосфере водорода при температуре 26°C. Реакционную смесь фильтровали через целит и промывали MeOH (20 мл). Фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью препаративной HPLC (колонка Kinetex С18, 5 мкм, 30 х 150 мм), градиент от 10% до 100% ACN/бикарбонат аммония (10 мМ водный раствор), 13 мин, получая указанное в заголовке соединение (173 мг, 52%).
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,15 (д, J=5,2 Гц, 1H), 7,05-6,98 (м, 1H), 6,76 (с, 1H), 4,85-4,76 (м, 1H), 4,04-3,94 (м, 1H), 3,22-3,16 (м, 1H), 2,88-2,76 (м, 1H), 2,75-2,41 (м, 5H), 2,03-1,88 (м, 2H), 1,70-1,58 (м, 2H); [ES+MS] m/z 305 (MH+).
Соединения по Примерам 21-24 получены способами, аналогичными способу, описанному в Примере 20, но вместо промежуточного соединения 41 использовали соединения, указанные в Таблице 9. Также указаны изменения, использованные на стадиях очистки.
Таблица 9
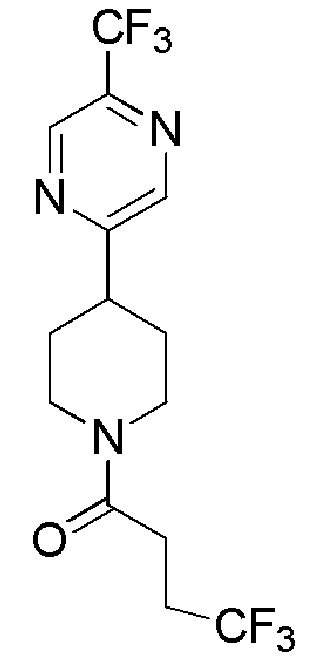
4,4,4-трифтор-1-(4-(5-(трифторметил)
пиразин-2-ил)пиперидин-1-ил)бутан-1-он
см. примечание a)
0,849 ммоль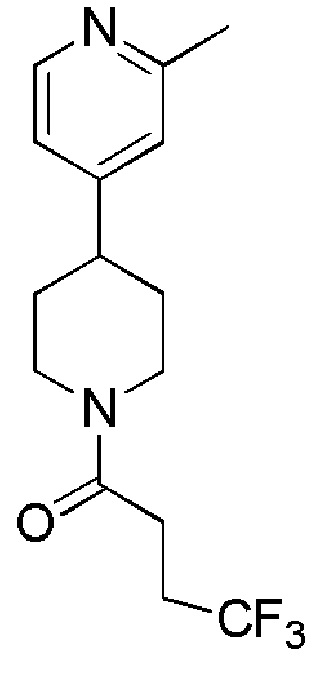
ТВ110
4,4,4-трифтор-1-(4-(2-метилпиридин-4-ил)пиперидин-1-ил)бутан-1-он
см. примечание b)
1,34 ммоль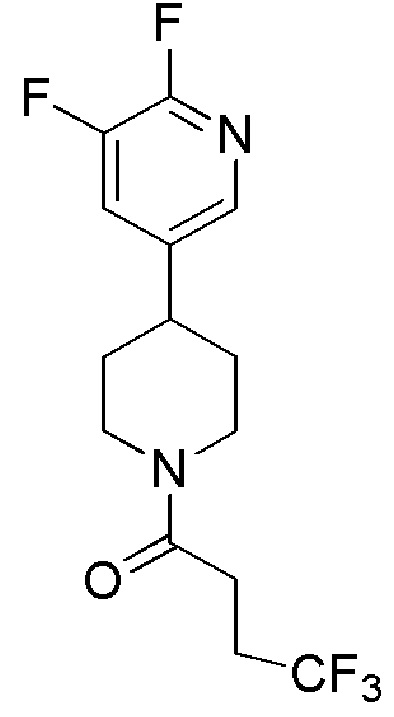
1-(4-(5,6-дифторпиридин-3-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-он
см. примечание c)
0,999 ммоль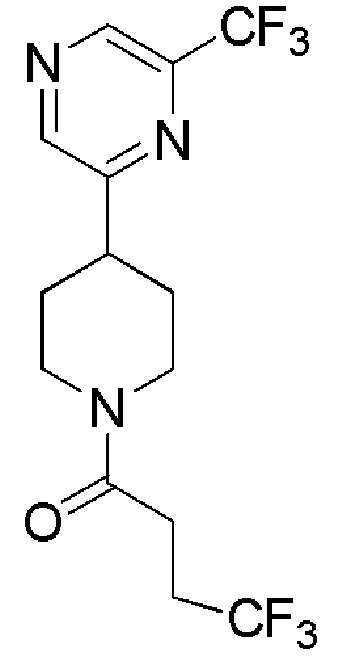
4,4,4-трифтор-1-(4-(6-(трифторметил)
пиразин-2-ил)пиперидин-1-ил)бутан-1-он
см. примечание d)
1,416 ммоль
Примечания:
Очистка препаративной HPLC (колонка Kromasil, 10 мкм, 21.2×250 мм) градиент 20% - 100% ACN/бикарбонат аммония (10 мкМ водный раствор), 16 мин
Очистка препаративной HPLC (колонка Kinetex C-8, 5 мкм, 30×150 мм) градиент 10% - 100% ACN/бикарбонат аммония (10 мкМ водный раствор), 14 мин
Очистка хроматографией на силикагеле с использованием в качестве элюента линейного градиента петролейный эфир/EtOAc, с последующей препаративной HPLC (колонка Sunfire C18, 5 мкм, 19×150 мм) градиент 10% - 100% ACN/бикарбонат аммония (10 мкМ водный раствор), 15 мин:
Очистка препаративной HPLC (колонка Kromasil, C18, 5 мкм, 25×250 мм) градиент 40% - 100% ACN/бикарбонат аммония (10 мкМ водный раствор), 15 мин
Пример 25: 1-(4-(2-хлорпиридин-4-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-он
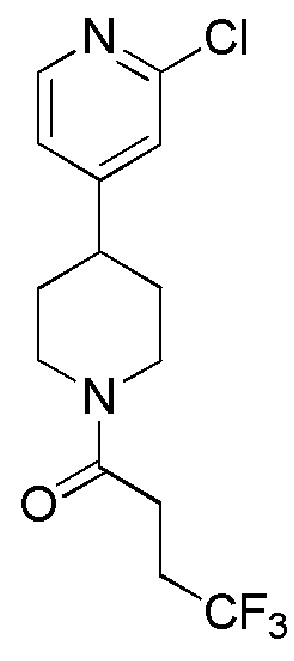
К раствору промежуточного соединения 37 (370 мг, 1,16 ммоль) в MeOH (10 мл) при 26°С добавляли оксид платины (HINDUSTAN, 35 мг). Реакционную смесь перемешивали в течение 4 часов в атмосфере водорода при температуре 26°C. Реакционную смесь фильтровали через целит и промывали MeOH (20 мл). Фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента линейный градиент петролейный эфир/EtOAc, а затем с помощью препаративной HPLC (колонка Kinetex С18, 5 мкм, 30 х 150 мм), градиент от 10% до 100% ACN/ бикарбонат аммония (10 мМ водный раствор), 13 мин, получая указанное в заголовке соединение (15 мг 1, 30%).
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,32 (д, J=5,2 Гц, 1H), 7,19-7,15 (м, 1H), 7,07-7,02 (м, 1H), 4,85-4,76 (м, 1H), 4,03-3,93 (м, 1H), 3,25-3,10 (м, 1H), 2,83-2,72 (м, 1H), 2,71-2,46 (м, 5H), 1,99-1,88 (м, 2H), 1,69-1,57 (м, 2H); [ES+ MS] m/z 321 (MH+).
Соединения по Примерам 26-27 получены способами, аналогичными способу, описанному в Примере 25, но с заменой промежуточного соединения 37 на соединения, указанные в Таблице 10. Также указаны изменения на стадии очистки.
Таблица 10
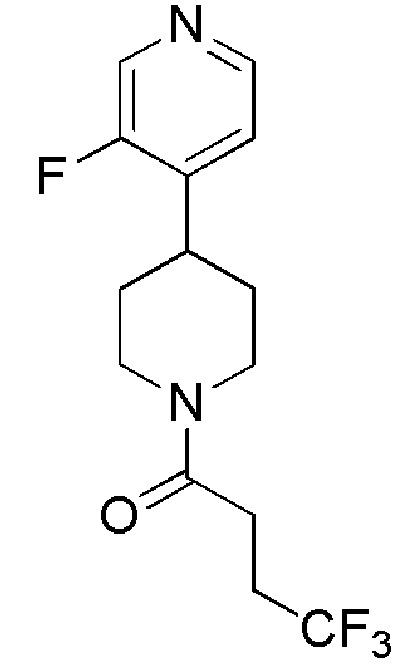
4,4,4-трифтор-1-(4-(3-фторпиридин-4-ил)пиперидин-1-ил)бутан-1-он
см. примечание a)
0,86 ммоль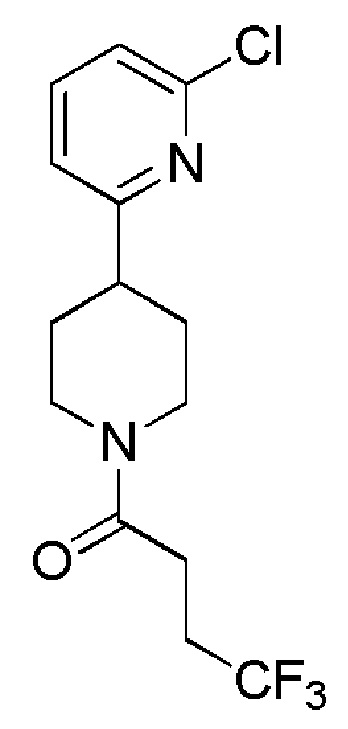
1-(4-(6-хлорпиридин-2-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-он
см. примечание b)
0,878 ммоль
Примечания:
a) Очистка с помощью хроматографии на колонке с силикагелем с использованием в качестве элюента линейный градиент петролейныйо эфир/EtOAc
b) Очистка с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента линейный градиент петролейный эфир/EtOAc, а затем с помощью препаративной HPLC (колонка Atlantis Т3, 5 мкм, 19 х 250 мм), градиент от 10% до 100% ACN/H2O (0,1% муравьиная кислота), 15 мин
Ссылочный пример 28: 4,4,4-трифтор-1-[4-(пиримидин-2-ил)пиперидин-1-ил]бутан-1-он
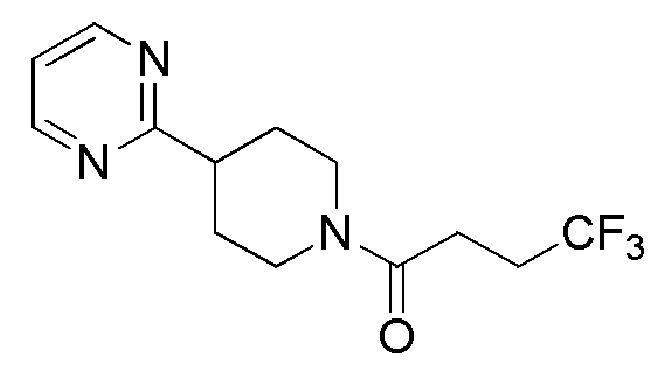
Раствор EDC.HCl (ALFA-AESAR, 340 мг, 1,77 ммоль), TEA (ALFA-AESAR, 0,25 мл, 1 ммоль) 0,77, HOBt (ALDRICH, 240 мг, 1 ммоль) 0,77, промежуточного соединения 30 (295,3 мг, 1,48 ммоль) и 4,4,4-трифтормасляной кислоты (ALFA-AESAR, 252 мг, 1,77 ммоль) в DMF (15 мл) перемешивали при комнатной температуре в течение ночи. Смесь затем промывали насыщенным раствором Na2CO3, добавляли EtOAc, разделяли на две фазы, и водную фазу дополнительно экстрагировали EtOAc. Собранный органический слой сушили над Na2SO4 (безводн.), фильтровали и упаривали. Неочищенный продукт, полученный таким образом, очищали с помощью флэш-хроматографии (Si SNAP 50, CyHex/EtOAc от 1/1 до 0/10, а затем DCM/MeOH 8/2), с получением указанного в заголовке соединения (141 мг, 33%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, DMSO-d6) δ м.д.: 8,75 (д, J=4,9 Гц, 2H), 7,35 (т, J=4,9 Гц, 1H), 4,44 (д, J=13,2 Гц, 1H), 3,94 (д, J=13,7 Гц, 1H), 3,22-3,13 (м, 1H), 3,13-3,05 (м, 1H), 2,82-2,72 (м, 1H), 2,70-2,57 (м, 2H), 2,57-2,45 (м, 2H), 2,02-1,90 (м, 1H), 1,79-1,67 (м, 1H),1,65-1,51 (м, 1H); [ES+MS] m/z 288 (MH+).
Пример 29: 1-(4-(5-хлорпиридин-3-ил)-4-фторпиперидин-1-ил)-4,4,4-трифторбутан-1-он
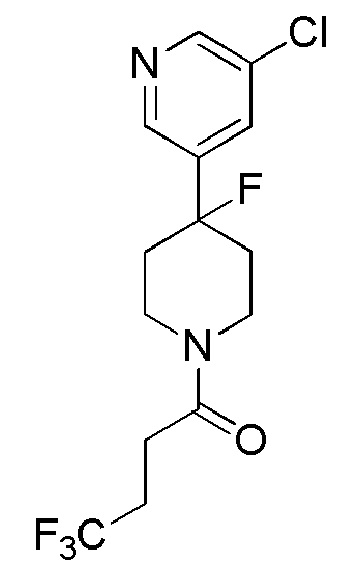
К раствору промежуточного соединения 68 (650 мг, 2,588 ммоль), 4,4,4-трифторбутановой кислоты (MATRIX, 550 мг, 3,882 ммоль) в DMF (20 мл) при 0°C добавляли DMAP (AVRA, 940 мг, 7,765 ммоль) и EDC.HCl (ASHVARSHA, 1,23 г, 6,471 ммоль). Полученную реакционную смесь оставляли перемешиваться при температуре 26°C в течение 16 часов. Реакционную смесь гасили холодной водой со льдом (50 мл), и экстрагировали EtOAc (2 х 50 мл). Объединенные органические слои промывали 1N раствором HCl (50 мл) и холодным насыщенным солевым раствором (100 мл). Органический слой сушили над Na2SO4 (безводн.), фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на колонке с силикагелем, используя в качестве элюента линейный градиент петролейный эфир/EtOAc, с получением указанного в заголовке соединения (400 мг, 45%) в виде бесцветной жидкости.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,56 (д, J=2,3 Гц, 1H), 8,51-8,45 (м, 1H), 7,71 (т, J=2,2 Гц, 1H), 4,82-4,66 (м, 1H), 3,92-3,82 (м, 1H), 3,65-3,48 (м, 1H), 3,12-2,94 (м, 1H), 2,71-2,47 (м, 4H), 2,18-1,86 (м, 4H); [ES+MS] m/z 339 (MH+).
Пример 30: 1-(4-(5-хлорпиридин-3-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-он
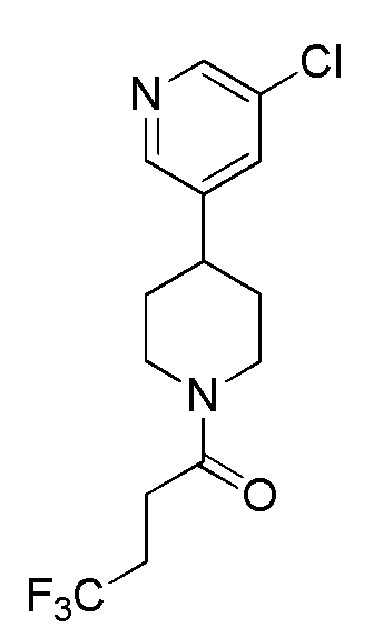
Соединение по Примеру 30 получали способом, аналогичным способу, описанному в Примере 29, но вместо промежуточного соединения 68 использовали промежуточное соединение 49 (343,34 ммоль). Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента линейный градиент петролейный эфир/EtOAc, с получением указанного в заголовке соединения (59,47 г, 54%).
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,46 (д, J=2,3 Гц, 1H), 8,42-8,31 (м, 1H), 7,50 (т, J=2,1 Гц, 1H), 4,89-4,73 (м, 1H), 3,99 (д, J=13,5 Гц, 1H), 3,24-3,13 (м, 1H), 2,87-2,76 (м, 1H), 2,74-2,44 (м, 5H), 1,95 (т, J=14,3 Гц, 2H), 1,70-1,61 (м, 2H); [ES+MS] m/z 321 (MH+).
Пример 31: 1-(4-(5-хлорпиридин-3-ил)пиперидин-1-ил)-5,5,5- трифторпентан-1-он
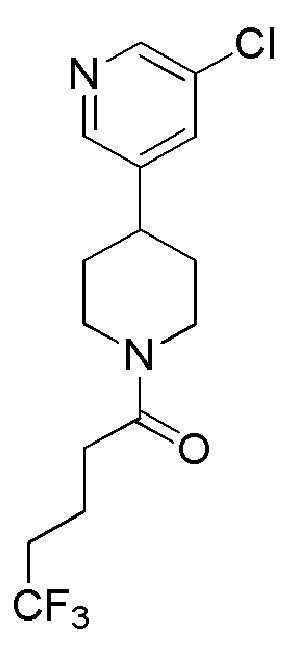
Соединение по Примеру 31 получали способом, аналогичным способу, описанному в Примере 30, но вместо 4,4,4-трифторбутановой кислоты использовали 5,5,5-трифторпентановую кислоту (OAKWOOD, 10,7082 ммоль). Неочищенный продукт очищали с помощью препаративной HPLC (колонка Luna C18, 5 мкм, 21,2 х 250 мм), градиент от 10% до 100% ACN/бикарбонат аммония (10 мМ водный раствор), 12,7 мин, с получением указанного в заголовке соединения (85 мг, 37%) в виде коричневой смолы.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,45 (д, J=2,2 Гц, 1H), 8,36 (д, J=2,0 Гц, 1H), 7,49 (т, J=2,1 Гц, 1H), 4,82 (уш. д, J=13,6 Гц, 1H), 3,98 (уш. д, J=13,6 Гц, 1H), 3,25-3,06 (м, 1H), 2,85-2,75 (м, 1H), 2,65 (уш. с, 1H), 2,45 (т, J=7,2 Гц, 2H), 2,31-2,13 (м, 2H), 2,03-1,83 (м, 4H), 1,68-1,57 (м, 2H); [ES+MS] m/z 335 (MH+).
Пример 32: 1-(4-(6-хлорпиразин-2-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-он
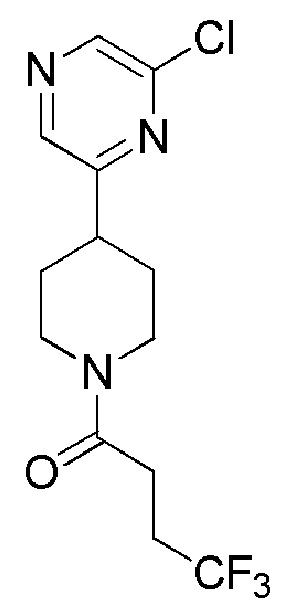
К раствору промежуточного соединения 50 (400 мг, 1,324 ммоль) в ACN (8 мл) при 27°С добавляли по каплям изопентилнитрит (РНР, 233 мг, 1,987 ммоль), с последующим добавлением хлорида меди (II) (ALFA-AESAR, 267 мг, 1,987 ммоль). Реакционную смесь нагревали до 60°С и перемешивали в течение 1,5 часа при той же температуре. Реакционная смесь концентрировали при пониженном давлении, остаток растворяли в EtOAc (50 мл) и фильтровал через слой целита. К фильтрату добавляли воду (50 мл) и экстрагировали с помощью EtOAc (2 х 40 мл). Объединенный органический слой сушили над Na2SO4 (безводн.), фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью препаративной высокоэффективной жидкостной хроматографии (колонка Kromsil С18, 10 мкм, 25 х 150 мм), градиент от 40% до 100% ACN/бикарбонат аммония (10 мМ водный раствор), 18 мин, получая указанное в заголовке соединение (17 мг, 4%) в виде не совсем белого твердого вещества.
1H ЯМР (400 МГц, DMSO-d6) δ м.д.: 8,65 (д, J=6,4 Гц, 2H), 4,52 (уш. д, J=12,7 Гц, 1H), 3,99 (уш. д, J=13,6 Гц, 1H), 3,21-3,03 (м, 2H), 2,76-2,60 (м, 3H), 2,59-2,51 (м, 2H), 1,88 (уш. с, 2H), 1,75-1,63 (м, 1H), 1,59-1,46 (м, 1H); [ES+MS] m/z 322 (MH+).
Пример 33: 4,4,4-трифтор-1-(4-фтор-4-(пиридин-2-ил)пиперидин-1-ил)бутан-1-он
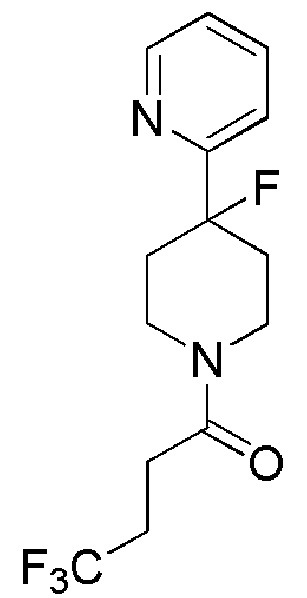
К промежуточному соединению 62 (400 мг, 1,323 ммоль) в DCM (5 мл) при -78°C добавляли по каплям раствор DAST (ALFA-AESAR, 213 мг, 1,587 ммоль) в DCM (5 мл). Реакционную смесь оставляли нагреваться до 27°С и перемешивали в течение 2 часов при той же температуре. Реакционную смесь разбавляли насыщенным раствором бикарбоната натрия (30 мл) и экстрагировали DCM (3 х 100 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 (безводн.), фильтровали и упаривали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии на колонке с силикагелем, используя в качестве элюента линейный градиент петролейный эфир/EtOAc, с получением указанного в заголовке соединения (160 мг, 31,68%) в виде бесцветной жидкости.
1H ЯМР (400МГц, CDCl3) δ м.д.: 8,60-8,51 (м, 1H), 7,80-7,70 (м, 1H), 7,62-7,50 (м, 1H), 7,25-7,20 (м, 1H), 4,79-4,58 (м, 1H), 3,97-3,77 (м, 1H), 3,60-3,47 (м, 1H), 3,10-3,00 (м, 1H), 2,70-2,10 (м, 6H), 2,05-1,84 (м, 2H); [ES+MS] m/z 305 (MH+).
Соединения по Примерам 34-37, 39 и 40 получали способами, аналогичным способу, описанному в Примере 33, но вместо промежуточного соединения 62 использовали промежуточные соединения, указанные в Таблице 11.
Таблица 11
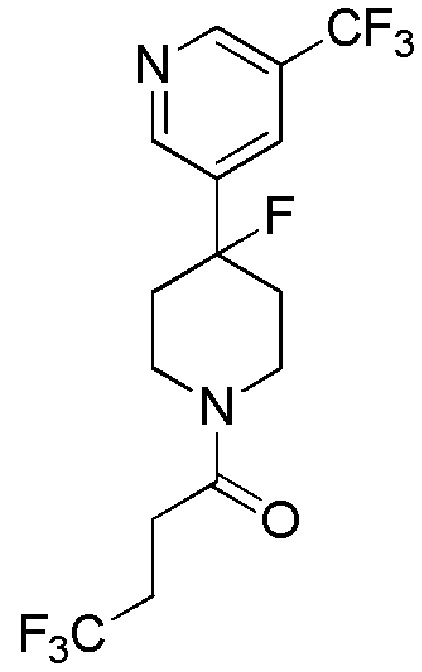
4,4,4-трифтор-1-(4-фтор-4-(5-(трифторметил)
пиридин-3-ил)пиперидин-1-ил)бутан-1-он
18,90 ммоль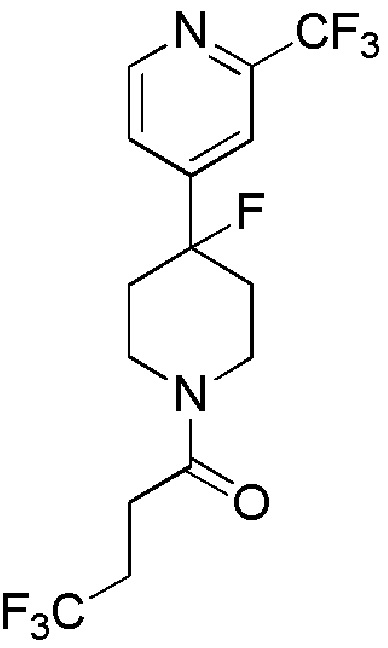
4,4,4-трифтор-1-(4-фтор-4-(2-(трифторметил)
пиридин-4-ил)пиперидин-1-ил)бутан-1-он
0,81 ммоль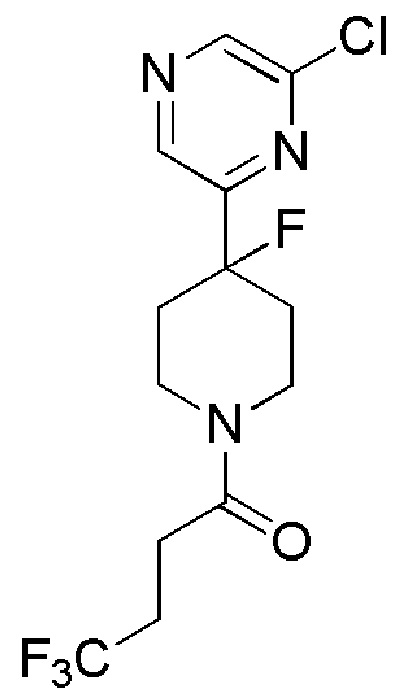
1-(4-(6-хлорпиразин-2-ил)-4-фторпиперидин-1-ил)-4,4,4-трифторбутан-1-он
1,184 ммоль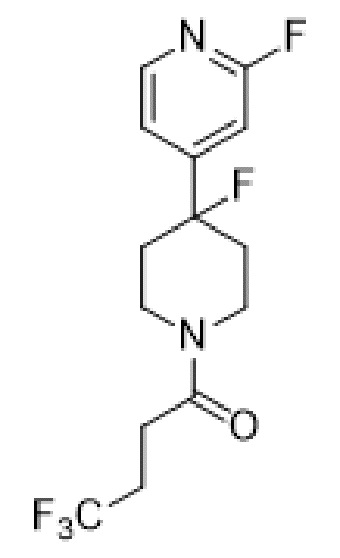
4,4,4-трифтор-1-(4-фтор-4-(2-фторпиридин-4-ил)пиперидин-1-ил)бутан-1-он
1,875 ммоль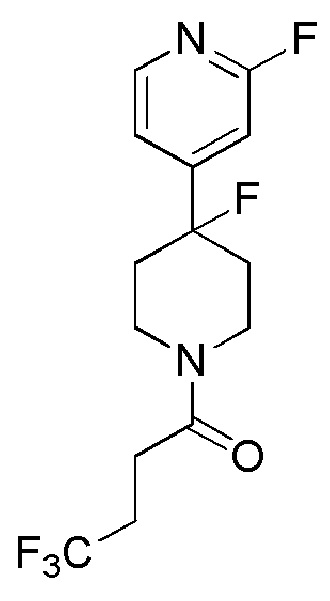
4,4,4-трифтор-1-[4-фтор-4-[4-(трифторметил)-2-пиридил]-1-пиперидил]бутан-1-он
0,24 ммоль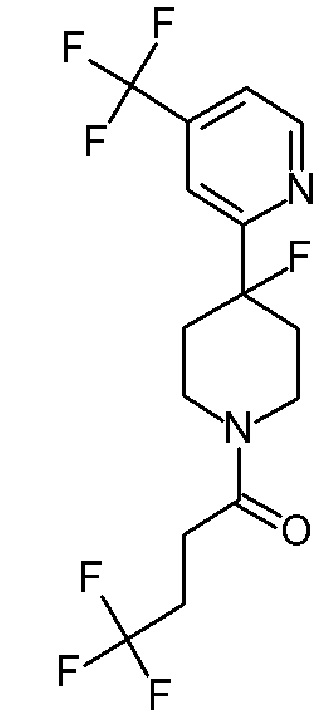
4,4,4-трифтор-1-[4-фтор-4-[4-(трифторметил)-2-пиридил]-1-пиперидил]бутан-1-он
0,15 ммоль
Пример 38: 1-(3-(5-хлорпиридин-3-ил)пирролидин-1-ил)-4,4,4-трифторбутан-1-он
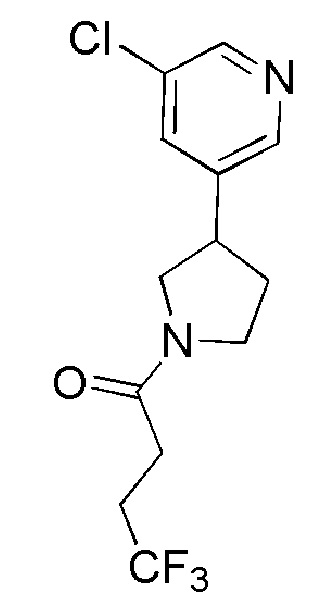
К раствору 4,4,4-трифторбутановой кислоты (OAKWOOD CHEMICALS, 116 мг, 0,821 ммоль) в DCM (2 мл) при температуре 27°C добавляли по каплям каталитическое количество DMF с последующим добавлением оксалилхлорида (AVRA, 0,056 мл, 0,657 ммоль), и перемешивали в течение 2 часов при той же температуре. Указанную выше реакционную смесь добавляли к раствору промежуточного соединения 74 (120 мг, 0,548 ммоль) в смеси насыщенного раствора бикарбоната натрия (2 мл) и EtOAc (2 мл) при 5°С. Реакционную смесь оставляли нагреваться до 27°C и перемешивали в течение 16 часов при той же температуре. Реакционную смесь гасили насыщенным раствором бикарбоната натрия (5 мл) и экстрагировали с помощью DCM (2 х 10 мл). Органический слой сушили над Na2SO4 (безводн.), фильтровали и упаривали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, с использованием в качестве элюента линейного градиента петролейный эфир/EtOAc, с получением указанного в заголовке соединения (75 мг), который снова очищали препаративной HPLC (колонка XBridge С18, 5 мкм, 4,6 х 250 мм), градиентн от 10% до 98% ACN/бикарбонат аммония (10 мМ водный раствор), 18 мин, с получением указанного в заголовке соединения (35 мг, 21%) в виде бесцветной жидкости.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,52-8,47 (м, 1H), 8,42-8,39 (м, 1H), 7,57-7,52 (м, 1H), 4,14-3,65 (м, 2H), 3,64-3,32 (м, 3H), 2,65-2,31 (м, 5H), 2,21-1,95 (м, 1H); [ES+MS] m/z 307 (MH-).
Пример 41: 1-[4-(2-хлор-4-пиридил)-4-фтор-1-пиперидил]-4,4,4-трифтор-бутан-1-он
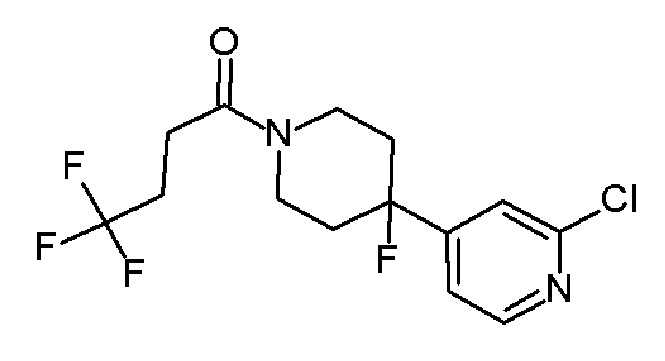
Соединение по Примеру 41 получали способом, аналогичным способу, описанному в Примере 2, но с заменой дигидрохлорида 2-(4-пиперидил)пиразина на промежуточное соединение 67b.
1H ЯМР (300 МГц, CD2Cl2) δ м.д.: 8,40 (д, J=5,2 Гц, 1H), 7,37 (д, J=1,6 Гц, 1H), 7,23-7,19 (м, 1H), 4,74-4,62 (м, 1H), 3,92-3,81 (м, 1H), 3,57-3,44 (м, 1H), 3,07-2,95 (m , 1H), 2,70-2,60 (м, 2H), 2,60-2,46 (2H), 2,08-1,99 (2H), 1,99-1,81 (м, 2H); [ES+MS] m/z 339 (MH+).
Биологическая активность
Анализ 1
Сравнительные данные, полученные на грызунах
Для определения пероральной биодоступности (F%) у крыс, значение которых приведено в Таблице 12 ниже, использовали следующий протокол испытаний.
Для исследования PK внутривенных и пероральных доз испытуемых соединений использовали самцов крыс SD (n=3/соединение/путь введения, для одной дозы). Соединения вводили внутривенно (инфузия в течение 30 минут) в виде раствора (состав: 5% DMSO: 20% энкапсина в физиологическом солевом растворе; целевая доза 1 мг/кг) и перорально в виде суспензии (состав: 1% метилцеллюлозы; целевая доза 5 мг/кг). После дозирования собирали образцы крови (25 мл). Время отбора проб были следующими:
0,25, 0,5 (перед концом инфузии), 0,58, 0,75, 1, 1,5, 2, 3, 5, 7 и 24 часа; n=3 крысы (внутривенное введение инфузией);
0,25, 0,5, 1, 2, 4, 6, 8 и 24 часа; n=3 крыс (пероральное введение через желудочный зонд).
Все образцы крови были разбавлены 25 мкл воды, и их хранили при -80°C до проведения анализа. Образцы крови крыс анализировали для каждого соединения с использованием способа осаждения белка с последующим анализом LC-MS/MS. Все фармакокинетические параметры были получены путем безкомпартаментного анализа различных профилей зависимости "концентрация в крови от времени", используя программное обеспечение WinNonlin Phoenix, версия 6.3.
При определении безопасности в условиях in vivo на мышах (результаты приведены в Таблице 12 ниже) использовали следующий протокол испытаний.
В этом исследовании одну дозу каждого соединения вводили 6 самцам мышей Swiss (Crl:CD-1(ICR)) в возрасте по меньшей мере 6 недель. Дозы вводили перорально, один раз в день в течение 7 дней, в виде суспензии в 1% растворе метилцеллюлозы при объеме введения 10 мл/кг. Уровень дозы в мг/кг для каждого соединения выбирали на основе показателей ADME (всасывание, распределение, метаболизм, выделение), полученных в рамках ранее выполненных исследований по переносимости и эффективности, и предсказанных в PBPK (фармакокинетическая модель, основанная на физиологии), для достижения эквивалентных воздействий на кровь.
Токсикокинетическую (обобщенную) оценку проводили на основании результатов, полученных с использованием образцов крови, полученных от животных, получивших дозу соединения, через 0,5, 1, 3, 7 и 24 часа после введения дозы в дни 1 и 7. Концентрации соединений в цельной крови определяли с помощью метода HPLC-MS/MS с использованием безкомпартаментного метода анализа для получения оценок параметров токсикокинетики (TK): максимальные наблюдаемой концентрации соединения в крови (Cmax), время доcтижения Cmax (Tmax), и площадь под кривой зависимости "концентрация соединения в крови от времени" (AUC). Клинические наблюдения и массу тела регистрировали один раз в день в течение всего периода лечения и в день аутопсии, через 24 часа после последнего введения. Животных умерщвляли путем воздействия сверхдоз СО2 и обескровливанием путем внутрисердечной пункции. При аутопсии взвешивали печень и хранили ее в фиксаторе до проведения анализа методом оптической и электронной микроскопии, а часть печени замораживали до проведения анализа экспрессии генов. 3 из 6 животных использовали для исследования с анализом экспрессии генов. Анализ HepatoTaq© состоял из 16 субпанелей, направленных на различные токсические проявления или состояния печени, при этом каждая субпанель основана на измерении от 4 до 18 различных уровней мРНК гена с использованием микрообъемных технологий Taqman.
Таблица 12
(in vivo на мышах)
Анализ 2
Измерение ингибирования роста штаммов M. tuberculosis GFP при использовании комбинации этионамида (ETH) и соединений по Примерам 1-41
1. Конструирование микобактерий рекомбинантных штаммов.
Штамм M. tuberculosis H37Rv-GFP. Рекомбинантный штамм M. tuberculosis H37Rv, экспрессирующий зеленый флуоресцентный белок (GFP-H37Rv), получали путем трансформации интегративной плазмиды pNIP48 (Abadie et al., 2005; Cremer et al., 2002). В этой плазмиде, полученной из микобактериофага MS6, ген GFP был клонирован под контролем сильного микобактериального промотора pBlaF, и она конститутивно экспрессирует GFP. Эта плазмида также содержит ген устойчивости к гигромицину.
Штамм M. tuberculosis W4-E1-GFP (мутанатный). Штамм M. tuberculosis Е1 получен из штамма Beijing W4, который подвергали селекции на чашках с агаром, содержащим этионамид (20 мкг/мл). Этот штамм имеет мутацию Gly343Ala в EthA. Штамм W4-Е1 трансформировали с использованием pNIP48, как описано выше, с получением флуоресцентного штамма W4-E1-GFP.
2. Рост и подготовка флуоресцентных микобактерий
Бактериальные культуры хранили при -80ºC, и их инокулировали в 5 мл среды Middlebrook 7H9, дополненной коктейлем олеиновая кислота-альбумин-декстроза-каталаза (OADC, Difco, Sparks MD, США) и 50 мкг/мл гигромицина (Invitrogen, Carlsbad, CA, США) в колбы для культуры тканей объемом 25 см2. Колбы инкубировали при 37°С без встряхивания в течение 7 дней. Затем культуры разбавляли свежей культуральной средой до достижения оптической плотности OD600, равной 0,1. Эти разбавленные культуры в объеме 50 мл переносили в культуральные флаконы (75 см2), и их инкубировали в течение 7 дней при 37°C без встряхивания.
3. Подготовка микропланшетов
Этионамид (Sigma, E6005) разводили в DMSO до концентраций 0,1 мг/мл и 0,8 мг/мл; аликвоты хранили в замороженном состоянии при -20°С. Испытуемые соединения ресуспендировали в DMSO до конечной концентрации 10 мкМ. Этионамид и испытуемые соединения переносили в полипропиленовый 384-луночный планшет малого объема (Corning, кат. No. 3672), и их использовали для подготовки аналитических планшетов. Десять 3-кратны серийных разведений соединений (как правило, в диапазоне от 30 до 4,5 e-3 мкМ) приготавливали в черных 384-луночных полистироловых планшетах Greiner с прозрачным дном (Greiner, кат. No. 781091), используя ручные жидкостные дозаторы Echo 550 (Labcyte). Компенсировали объем DMSO, чтобы концентрация во всех лунках составляла 0,3%.
Затем в 384-луночные планшеты переносили этионамид, используя дозаторы Echo. Конечная концентрация ETH для анализов, включающих H37Rv-GFP, составляла 0,1 мкг/мл, и 0,8 мкг/мл для анализов, включающих W4-E1-GFP. Конечное количество DMSO в планшете для анализа составляло <1% об/об для каждой лунки.
Контрольные образцы в планшетах для анализа включали DMSO в концентрации 0,3% (отрицательный контроль) и INH в количестве 1 мкг/мл (положительный контроль). Референсный планшет включал рифампицин, INH (изониазид) и ETH (этионамид) в диапазоне концентраций от 30 до 1,8 e-3 мкг/мл (15 точек, разведения 2x).
Культуры H37Rv-GFP или W4-E1-GFP, предназначенные для добавления в планшеты для анализа, промывали два раза в PBS (Gibco, 14190), ресуспендировали в свежей культуральной среде (без гигромицина), и выращивали в течение 5 дней при температуре 37°C.
Наконец, культуры разводили до оптической плотности при 600 нм (OD600), равной 0,02 с использованием свежей культуральной среды без добавления гигромицина, и в каждый планшет для анализа переносили 50 мкл полученной культуры. Планшеты инкубировали при 37°С в течение 5 дней. Флуоресцентный сигнал регистрировали на планшет-ридере Victor 3 MultiLabel (Perkin Elmer), при длине волны возбуждения 485 нм и длине волны излучения 535 нм.
Все соединения по Примерам испытывали в основном в соответствии с процедурой, описанной выше для Анализа 2, и полученные значения активности представлены ниже.
Показатель EC50_H37Rv отражает способность соединений по изобретению усиливать активность этионамида в отношении штаммов H37Rv, а показатель EC50_мутант отражает способность соединений по изобретению потенцировать активность этионамида против штаммов микобактерий туберкулеза, устойчивых к этионамиду.
(Ссыл. пример)
(Ссыл. прим.)
(Ссыл. прим.)
(Ссыл. прим.)
(Ссыл. прим.)
(Ссыл. прим.)
<50 нМ = +++++
≥50 нМ - <250 нМ = ++++
≥250 нМ - <500 нМ = +++
≥500 нМ - <1,0 мкM (≥500 нМ - <1000 нМ) = ++
≥1,0 мкм,M - ≤10 мкM (≥1000 нМ - ≤10000 нМ) = +
>10 = †
В частности, установлено, что соединения по Примерам 2, 3, 4, 6-10, 13, 14, 17, 18, 20, 22, 23, 24, 25-27, 29-35, 37, 38, 39, 40 и 41 показали среднее значение EC50_H37Rv <150 нМ, а среднее значение EC50_мутант - <1,3 мкМ.
Конкретные результаты для иллюстративных соединения более подробно указаны ниже.
Пример 18: определено среднее значение EC50_H37Rv, равное 11 нМ, и среднее значение EC50_мутант - 37 нМ.
Пример 20: определено среднее значение EC50_H37Rv, равное 18 нМ, и среднее значение EC50_мутант - 119 нМ.
Пример 29: определено среднее значение EC50_H37Rv, равное 8 нМ, и среднее значение EC50_мутант - 17 нМ.
Пример 30: определено среднее значение EC50_H37Rv, равное 30 нМ, и среднее значение EC50_мутант - 93 нМ.
Пример 32: определено среднее значение EC50_H37Rv, равное 33 нМ, и среднее значение EC50_мутант - 211 нМ.
Пример 35: определено среднее значение EC50_H37Rv, равное 6 нМ, и среднее значение EC50_мутант - 24 нМ.
Пример 37: определено среднее значение EC50_H37Rv, равное 7 нМ, и среднее значение EC50_мутант - 29 нМ.
Пример 38: определено среднее значение EC50_H37Rv, равное 15 нМ. и среднее значение EC50_мутант - 20 нМ.
В этом же анализе, как описано выше, испытывали для сравнения соединение по Примеру BDM_70542 из WO 2014/096378, и определили, что значение EC50_мутант для этого соединения равно 1,6 мкМ, т.е. 1600 нМ.
Анализ 3
Анализ ингибирования Mycobacterium tuberculosis H37Rv в человеческих макрофагах ТНР-1 (внутриклеточный анализ) в условиях in vitro
Внутриклеточный скрининг является ценным инструментом для идентификации новых соединений против туберкулеза, которые активны в макрофагах человека. Это ex vivo анализ может отражать физиологические условия, имитирующие заболевание, и позволять оценивать благоприятный вклад для клеток-хозяев (Sorrentino, F. et al. (2016) Antimicrob. Agents Chemother. 60 (1), 640-645).
Процедура осуществляли в соответствии с описанием, представленным в публикации Sorrentino, F. et al. (2016) Antimicrob. Agents Chemother. 60 (1), 640-645, за исключением того, что перед высеванием инфицированных клеток ТНР-1 в 384-луночные планшеты, инфицированные макрофаги на последней стадии промывки отфильтровывали с использованием фильтра с размером пор 40 мкм для удаления клеточных комков и получения суспензии из одиночных клеток.
Соединения по Примерам испытывали в целом в соответствии с протоколом указанного выше анализа (но при отсутствии этионамида). Полученные данные представлены в Таблице, приведенной ниже.
50 нМ = +++++
≥50 нМ - <250 нМ = ++++
≥250 нМ - <500 нМ = +++
≥500 нМ - <1,0 мкM (≥500 нМ - <1000 нМ) = ++
≥1,0 мкM - ≤10 мкM (≥1000 нМ - ≤10,000 нМ) = +
Конкретные результаты для иллюстративных соединений более подробно указаны ниже.
Пример 18: определено, что IC50 равно 20 нМ.
Пример 20: определено, что IC50 равно IC50 30 нМ.
Пример 29: определено, что IC50 равно 2 нМ.
Пример 30: определено, что IC50 равно 13 нМ.
Пример 32: определено, что IC50 равно 40 нМ.
Пример 35: определено, что IC50 равно 4 нМ.
Пример 37: определено, что IC50 равно 5 нМ.
Пример 38: определено, что IC50 равно 1,26 мкМ.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ | 2018 |
|
RU2783078C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АМИНОВ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ЕР4 | 2011 |
|
RU2565596C2 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
| ИНГИБИТОРЫ УРИДИН-5-ДИФОСФАТ (UDP) ГЛИКОЗИЛТРАНСФЕРАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2810928C2 |
| СПИРОЦИКЛИЧЕСКИЕ ПИРРОЛОПИРАЗИН(ПИПЕРИДИН)АМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2012 |
|
RU2634900C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2005 |
|
RU2409582C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 2021 |
|
RU2826628C1 |
| ПОЛИЦИКЛИЧЕСКИЕ АНТАГОНИСТЫ TLR7/8 И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ИММУННЫХ РАССТРОЙСТВ | 2016 |
|
RU2783748C2 |
| СОЕДИНЕНИЯ 2,3-ДИГИДРОХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ NaV1.8 | 2020 |
|
RU2833870C2 |
Изобретение относится к области органических соединений. Предложено соединение или его фармацевтически приемлемая соль, выбранное из следующих соединений: 4,4,4-трифтор-1-[4-фтор-4-(3-пиридил)-1-пиперидил]бутан-1-она; 4,4,4-трифтор-1-[4-(5-фтор-3-пиридил)-1-пиперидил]бутан-1-она; 4,4,4-трифтор-1-[4-(6-фтор-3-пиридил)-1-пиперидил]бутан-1-она; 4,4,4-трифтор-1-[4-[6-(трифторметил)-3-пиридил]-1-пиперидил]бутан-1-она; 4,4,4-трифтор-1-[4-[4-(трифторметил)-2-пиридил]-1-пиперидил]бутан-1-она; 4,4,4-трифтор-1-[4-[5-(трифторметил)-3-пиридил]-1-пиперидил]бутан-1-она; 4,4,4-трифтор-1-[4-[6-(трифторметил)-2-пиридил]-1-пиперидил]бутан-1-она; 4,4,4-трифтор-1-[4-(6-фтор-2-пиридил)-1-пиперидил]бутан-1-она; 4,4,4-трифтор-1-[4-(6-метокси-3-пиридил)-1-пиперидил]бутан-1-она; 4,4,4-трифтор-1-[4-[2-(трифторметил)-3-пиридил]-1-пиперидил]бутан-1-она; 4,4,4-трифтор-1-[4-(5-метокси-3-пиридил)-1-пиперидил]бутан-1-она; 1-[4-(3,5-дифтор-2-пиридил)-1-пиперидил]-4,4,4-трифтор-бутан-1-она; 1-[4-(2,6-дифтор-3-пиридил)-1-пиперидил]-4,4,4-трифтор-бутан-1-она; 4,4,4-трифтор-1-[4-(5-фтор-2-пиридил)-1-пиперидил]бутан-1-она; 4,4,4-трифтор-1-(4-(2-(трифторметил)пиридин-4-ил)пиперидин-1-ил)бутан-1-она; 4,4,4-трифтор-1-(4-(2-фторпиридин-4-ил)пиперидин-1-ил)бутан-1-она; 4,4,4-трифтор-1-(4-(5-(трифторметил)пиразин-2-ил)пиперидин-1-ил)бутан-1-она; 4,4,4-трифтор-1-(4-(2-метилпиридин-4-ил)пиперидин-1-ил)бутан-1-она; 1-(4-(5,6-дифторпиридин-3-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-она; 4,4,4-трифтор-1-(4-(6-(трифторметил)пиразин-2-ил)пиперидин-1-ил)бутан-1-она; 1-(4-(2-хлорпиридин-4-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-она; 4,4,4-трифтор-1-(4-(3-фторпиридин-4-ил)пиперидин-1-ил)бутан-1-она; 1-(4-(6-хлорпиридин-2-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-она; 1-(4-(5-хлорпиридин-3-ил)-4-фторпиперидин-1-ил)-4,4,4-трифторбутан-1-она; 1-(4-(5-хлорпиридин-3-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-она; 1-(4-(5-хлорпиридин-3-ил)пиперидин-1-ил)-5,5,5-трифторпентан-1-она; 1-(4-(6-хлорпиразин-2-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-она; 4,4,4-трифтор-1-(4-фтор-4-(пиридин-2-ил)пиперидин-1-ил)бутан-1-она; 4,4,4-трифтор-1-(4-фтор-4-(5-(трифторметил)пиридин-3-ил)пиперидин-1-ил)бутан-1-она; 4,4,4-трифтор-1-(4-фтор-4-(2-(трифторметил)пиридин-4-ил)пиперидин-1-ил)бутан-1-она; 1-(4-(6-хлорпиразин-2-ил)-4-фторпиперидин-1-ил)-4,4,4-трифторбутан-1-она; 4,4,4-трифтор-1-(4-фтор-4-(2-фторпиридин-4-ил)пиперидин-1-ил)бутан-1-она; 4,4,4-трифтор-1-[4-фтор-4-[4-(трифторметил)-2-пиридил]-1-пиперидил]бутан-1-она; 1-(3-(5-хлорпиридин-3-ил)пирролидин-1-ил)-4,4,4-трифторбутан-1-она; и 1-[4-(2-хлор-4-пиридил)-4-фтор-1-пиперидил]-4,4,4-трифтор-бутан-1-она. Также предложены фармацевтическая композиция и комбинация, содержащие указанное соединение, применение и способы лечения микобактериальной инфекции и заболевания, вызванного инфекцией Mycobacterium tuberculosis. Технический результат заключается в разработке новых соединений, пригодных при лечении микобактериальных инфекций и заболеваний, вызванных микобактериями. 6 н. и 8 з.п. ф-лы, 14 табл., 41 пр.
1. Соединение или его фармацевтически приемлемая соль, выбранное из следующих соединений:
4,4,4-трифтор-1-[4-фтор-4-(3-пиридил)-1-пиперидил]бутан-1-она;
4,4,4-трифтор-1-[4-(5-фтор-3-пиридил)-1-пиперидил]бутан-1-она;
4,4,4-трифтор-1-[4-(6-фтор-3-пиридил)-1-пиперидил]бутан-1-она;
4,4,4-трифтор-1-[4-[6-(трифторметил)-3-пиридил]-1-пиперидил]бутан-1-она;
4,4,4-трифтор-1-[4-[4-(трифторметил)-2-пиридил]-1-пиперидил]бутан-1-она;
4,4,4-трифтор-1-[4-[5-(трифторметил)-3-пиридил]-1-пиперидил]бутан-1-она;
4,4,4-трифтор-1-[4-[6-(трифторметил)-2-пиридил]-1-пиперидил]бутан-1-она;
4,4,4-трифтор-1-[4-(6-фтор-2-пиридил)-1-пиперидил]бутан-1-она;
4,4,4-трифтор-1-[4-(6-метокси-3-пиридил)-1-пиперидил]бутан-1-она;
4,4,4-трифтор-1-[4-[2-(трифторметил)-3-пиридил]-1-пиперидил]бутан-1-она;
4,4,4-трифтор-1-[4-(5-метокси-3-пиридил)-1-пиперидил]бутан-1-она;
1-[4-(3,5-дифтор-2-пиридил)-1-пиперидил]-4,4,4-трифтор-бутан-1-она;
1-[4-(2,6-дифтор-3-пиридил)-1-пиперидил]-4,4,4-трифтор-бутан-1-она;
4,4,4-трифтор-1-[4-(5-фтор-2-пиридил)-1-пиперидил]бутан-1-она;
4,4,4-трифтор-1-(4-(2-(трифторметил)пиридин-4-ил)пиперидин-1-ил)бутан-1-она;
4,4,4-трифтор-1-(4-(2-фторпиридин-4-ил)пиперидин-1-ил)бутан-1-она;
4,4,4-трифтор-1-(4-(5-(трифторметил)пиразин-2-ил)пиперидин-1-ил)бутан-1-она;
4,4,4-трифтор-1-(4-(2-метилпиридин-4-ил)пиперидин-1-ил)бутан-1-она;
1-(4-(5,6-дифторпиридин-3-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-она;
4,4,4-трифтор-1-(4-(6-(трифторметил)пиразин-2-ил)пиперидин-1-ил)бутан-1-она;
1-(4-(2-хлорпиридин-4-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-она;
4,4,4-трифтор-1-(4-(3-фторпиридин-4-ил)пиперидин-1-ил)бутан-1-она;
1-(4-(6-хлорпиридин-2-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-она;
1-(4-(5-хлорпиридин-3-ил)-4-фторпиперидин-1-ил)-4,4,4-трифторбутан-1-она;
1-(4-(5-хлорпиридин-3-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-она;
1-(4-(5-хлорпиридин-3-ил)пиперидин-1-ил)-5,5,5-трифторпентан-1-она;
1-(4-(6-хлорпиразин-2-ил)пиперидин-1-ил)-4,4,4-трифторбутан-1-она;
4,4,4-трифтор-1-(4-фтор-4-(пиридин-2-ил)пиперидин-1-ил)бутан-1-она;
4,4,4-трифтор-1-(4-фтор-4-(5-(трифторметил)пиридин-3-ил)пиперидин-1-ил)бутан-1-она;
4,4,4-трифтор-1-(4-фтор-4-(2-(трифторметил)пиридин-4-ил)пиперидин-1-ил)бутан-1-она;
1-(4-(6-хлорпиразин-2-ил)-4-фторпиперидин-1-ил)-4,4,4-трифторбутан-1-она;
4,4,4-трифтор-1-(4-фтор-4-(2-фторпиридин-4-ил)пиперидин-1-ил)бутан-1-она;
4,4,4-трифтор-1-[4-фтор-4-[4-(трифторметил)-2-пиридил]-1-пиперидил]бутан-1-она;
1-(3-(5-хлорпиридин-3-ил)пирролидин-1-ил)-4,4,4-трифторбутан-1-она; и
1-[4-(2-хлор-4-пиридил)-4-фтор-1-пиперидил]-4,4,4-трифтор-бутан-1-она.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где соединение представляет собой 4,4,4-трифтор-1-(4-(2-(трифторметил)пиридин-4-ил)пиперидин-1-ил)бутан-1-он, имеющее следующую структуру:
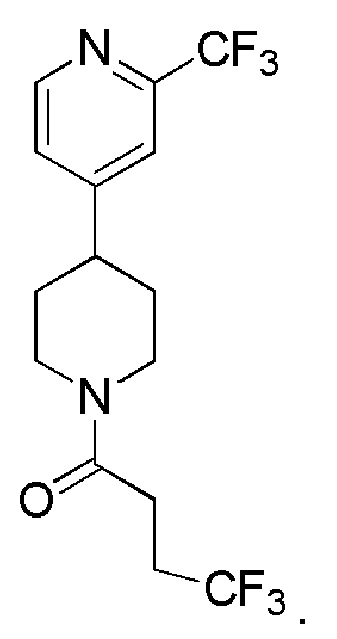
3. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, пригодное в терапии.
4. Соединение или его фармацевтически приемлемая соль по пп. 1-3 для лечения микобактериальной инфекции, вызванной Mycobacterium tuberculosis, или для лечения заболевания, вызванного инфекцией Mycobacterium tuberculosis.
5. Соединение или его фармацевтически приемлемая соль по п. 1 или 2 для лечения туберкулеза.
6. Способ лечения микобактериальной инфекции, вызванной Mycobacterium tuberculosis, у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по п. 1 или 2.
7. Способ лечения заболевания, вызванного инфекцией Mycobacterium tuberculosis, у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по п. 1 или 2.
8. Применение соединения или его фармацевтически приемлемой соли по п. 1 или 2 для изготовления лекарственного средства, пригодного для лечения микобактериальной инфекции, вызванной Mycobacterium tuberculosis, или заболевания, вызванного инфекцией Mycobacterium tuberculosis.
9. Фармацевтическая композиция для лечения микобактериальной инфекции, вызванной заболеванием Mycobacterium tuberculosis, или заболевания, вызванного инфекцией Mycobacterium tuberculosis, содержащая: (а) терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по п. 1 или 2; и (b) фармацевтически приемлемый эксципиент.
10. Комбинация для лечения микобактериальной инфекции, вызванной заболеванием Mycobacterium tuberculosis, или заболевания, вызванного инфекцией Mycobacterium tuberculosis, содержащая терапевтически эффективное количество (а) соединения или его фармацевтически приемлемой соли п. 1 или 2; и (b) по меньшей мере одного другого агента против микобактерий, который активируется через сигнальный путь EthA.
11. Комбинация по п. 10, где по меньшей мере один другой агент против микобактерий представляет собой агент против микобактерий, вызывающих туберкулез.
12. Комбинация по п. 11, где агент против микобактерий, вызывающих туберкулез, выбран из изониазида, рифампицина, пиразинамида, этамбутола, моксифлоксацина, рифапентина, клофазимина, этионамида, протионамида, изоксила, триацетазона, рифабутина, диарилхинолина, такого как бедаквилин (TMC207) или TBAJ-587, нитроимидазо-оксазина PA-824, деламанида (OPC-67683), оксазолидинона, такого как линезолид, тедизолид, радезолид, сутезолид (PNU-100480), позизолид (AZD-5847) или TBI-223, аналога EMB SQ109, OPC-167832, GSK3036656 (также известного как GSK070), GSK2556286, GSK3211830, бензотиазинона, такого как BTZ043 или PBTZ169, азаиндола, такого как TBA-7371, динитробензамида или бета-лактама, такого как меропенем, фаропенем, эртапенем, тебипенем, или бета-лактамные комбинации, такие как аугментин (амоксициллин-клавуланат).
13. Комбинация по любому из пп. 10-12, дополнительно содержащая противовирусный агент, включая антиретровирусный агент.
14. Комбинация по п. 13, где антиретровирусный агент выбран из зидовудина, диданозина, ламивудина, залцитабина, абакавира, ставудина, адефовира, дипивоксил адефовира, фозивудина, тодоксила, эмтрицитабина, аловудина, амдоксовира, элвуцитабина, невирапина, делавирдина, эфавиренца, ловирида, иммунокала, олтипраза, каправирина, лерсивирина, GSK2248761, ТМС-278, ТМС-125, этравирина, саквинавира, ритонавира, индинавира, нелфинавира, ампренавира, фозампренавира, бреканавира, дарунавира, атазанавира, типранавира, палинавира, лазинавира, энфувиртида, Т-20, Т-1249, PRO-542, PRO-140, TNX-355, BMS-806, BMS-663068 и BMS-626529, 5-геликса, ралтегравира, элвитегравира, GSK1349572, GSK1265744, викривирока (Sch-C), Sch-D, TAK779, маравирока, TAK449, диданозина, тенофовира, лопинавира или дарунавира.
| WO 2014096378 A1, 26.06.2014 | |||
| WO 2013060744 A2, 02.05.2013 | |||
| US 20110136823 A1, 09.06.2011 | |||
| КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КАЛЬПАИНОВ | 2007 |
|
RU2485114C2 |
| НОВЫЕ 4-(АЗАЦИКЛОАЛКИЛ)БЕНЗОЛ-1,3-ДИОЛОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНАЗ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ЧЕЛОВЕКА И В КОСМЕТИЧЕСКИХ СРЕДСТВАХ | 2009 |
|
RU2499794C2 |
| Устройство для массового гальванизирования мелких предметов | 1927 |
|
SU9219A1 |

