Область техники, к которой относится изобретение
Настоящее изобретение относится к молекулярным ситам, в частности к ультрамакропористому молекулярному ситу. Настоящее изобретение также относится к способу получения молекулярного сита и его применению в качестве адсорбента или катализатора и подобного.
Уровень техники
Молекулярные сита могут широко использоваться в различных способах применения, и различные способы применения часто накладывают различные требования на структуру пор в каркасе молекулярного сита. В каркасе молекулярных сит существует четыре типа структур пор: микропористый, мезопористый, макропористый и ультрамакропористый: микропористые молекулярные сита имеют диаметр пор в диапазоне от 3Å до 5Å, такие как CHA, LEV, SOD, LTA, ERI, KFI; мезопористые молекулярные сита имеют диаметр пор в диапазоне от 5Å до 7Å, такие как MFI, MEL, EUO, MWW, TON, MTT, MFS, AEL, AFO, HEU, FER; макропористые молекулярные сита имеют диаметр пор 7Å, такие как FAU, BEA, MOR, LTL, VFI, MAZ и ультрамакропористые молекулярные сита имеют диаметр пор более 7Å. В данных молекулярных ситах с различными структурами пор в каркасе ультрамакропористые молекулярные сита нарушали границы пор молекулярных сит и показали ряд преимуществ в улучшении реакционной способности макромолекул, продлении срока службы молекулярных сит и повышении селективности продукта, и, как ожидается, будут целесообразно применяться при переработке тяжелых масел и производстве органического химического сырья.
Среди существующих на данный момент 232 видов молекулярных сит со структурой пор в каркасе имеется только 10 видов ультрамакропористых молекулярных сит, в основном включая три типа: алюминий-фосфор/галлиевые молекулярные сита, такие как AlPO-8 (AET, 14-кольцевое, 7,9×8,7Å), VPI-5 (VFI, 18-кольцевое, 12,1Å), Кловерит (-CLO, 20-кольцевое, 13,2Å), JDF-20 (20-кольцевое) и ND-1 (24-кольцевое, 10,5Å); кремний-германий/галлиевые молекулярные сита, такие как OSB-1 (OSO, 14-кольцевое, Si/Be=2, 7,3×5,4Å), ECR-34 (ETR, 18-кольцевое, 10,5Å, Si/Ga=3), ITQ-37 (30-кольцевое), ITQ-43 (28-кольцевое), ITQ-33 (18-кольцевое), ITQ-44 (18-кольцевое), ITQ-40 (16-кольцевое) SSZ-53 (14-кольцевое) и SSZ-59 (14-кольцевое); и алюмосиликатные молекулярные сита, такие как UTD-1 (DON, 14-кольцевое, Si/Al2=∞, 10×7,5Å) и CIT-5 (CFI, 14-кольцевое, 7,5×7,2 Å, Si/Al2=190).
Ввиду их высокой производительности и перспектив применения в данной области техники все еще существует потребность в разработке новых типов ультрамакропористых молекулярных сит.
Сущность изобретения
На основе известного уровня техники изобретатели разработали новое ультрамакропористое молекулярное сито и новый способ получения молекулярных сит посредством тщательных исследований, тем самым отвечая вышеупомянутой потребности в данной области техники.
В частности, настоящее изобретение направлено на следующие аспекты:
Первый вариант осуществления
1. Молекулярное сито, имеющее схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид» или формулой «первый оксид ⋅ второй оксид ⋅ органический темплат ⋅ вода», в которой молярное соотношение первого оксида и второго оксида находится в диапазоне от 5 до ∞, предпочтительно от 25 до 95, более предпочтительно от 30 до 70; первый оксид представляет собой, по меньшей мере, один, выбранный из группы, состоящей из диоксида кремния, диоксида германия, диоксида олова, диоксида титана и диоксида циркония, предпочтительно диоксида кремния или комбинации диоксида кремния и диоксида германия; второй оксид представляет собой, по меньшей мере, один, выбранный из группы, состоящей из оксида алюминия, оксида бора, оксида железа, оксида галлия, оксида редкоземельного элемента, оксида индия и оксида ванадия, предпочтительно оксида алюминия; молярное соотношение воды и указанного первого оксида находится в диапазоне от 5 до 50, предпочтительно от 5 до 15; молярное соотношение указанного органического темплата и указанного первого оксида находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,15 до 0,5 или от 0,3 до 0,5, и указанное молекулярное сито имеет рентгеновскую дифрактограмму по существу, как показано в следующей таблице,
2. Молекулярное сито по любому одному из предшествующих аспектов, в котором рентгеновская дифрактограмма дополнительно содержит рентгенодифракционный пик по существу, как показано в следующей таблице,
3. Молекулярное сито по любому одному из предшествующих аспектов, в котором рентгеновская дифрактограмма дополнительно содержит рентгенодифракционный пик по существу, как показано в следующей таблице,
4. Молекулярное сито по любому одному из предшествующих аспектов, имеющее морфологию столбчатой (предпочтительно призматической, более предпочтительно гексагональной) кристаллической частицы.
5. Молекулярное сито по любому одному из предшествующих аспектов, в котором морфология кристаллической частицы имеет размер, определяемый эффективным диаметром в диапазоне от 100 нм до 1000 нм, предпочтительно от 300 нм до 700 нм, высотой в диапазоне от 100 нм до 1000 нм, предпочтительно от 150 нм до 700 нм и соотношением сторон от 1/3 до 8, предпочтительно от 1,5 до 5 или от 2 до 5.
6. Молекулярное сито по любому одному из предшествующих аспектов, в котором молекулярное сито имеет общую удельную площадь поверхности в диапазоне от 400 м2⋅г-1 до 600 м2⋅г-1, предпочтительно от 450 м2⋅г-1 до 580 м2⋅г-1 и объем пор в диапазоне от 0,3 мл/г до 0,5 мл/г, предпочтительно от 0,30 мл/г до 0,40 мл/г.
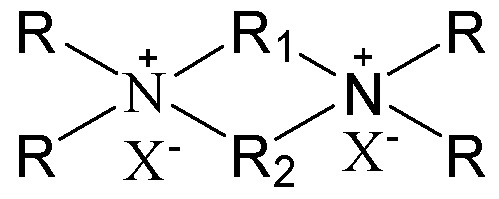
7. Способ получения молекулярного сита, включающий стадию приведения в контакт источника первого оксида, источника второго оксида, необязательного щелочного источника, органического темплата и воды в условиях кристаллизации с получением молекулярного сита и необязательно стадию обжига полученного молекулярного сита, в котором органический темплат содержит соединение, представленное следующей формулой (I),
 (I)
(I)
в которой группы R1 и R2 являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп и С3-12линейных или разветвленных оксаалкиленовых групп, предпочтительно каждая независимо выбрана из группы, состоящей из С3-12линейных алкиленовых групп и С3-12линейных оксаалкиленовых групп или предпочтительно одна из них выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп и С3-12линейных или разветвленных оксаалкиленовых групп, более предпочтительно одна из них выбрана из группы, состоящей из С3-12линейных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных алкиленовых групп и С3-12линейных оксаалкиленовых групп, в частности предпочтительно одна из них выбрана из группы, состоящей из С3-12линейных алкиленовых групп, и другая выбрана из группы, состоящей из C4-6линейных алкиленовых групп и C4-6линейных оксаалкиленовых групп (предпочтительно C4-6линейных монооксаалкиленовых групп, более предпочтительно -(CH2)m-O-(CH2)m-, в которой каждое значение m, являющееся одинаковым или отличным друг от друга, независимо представляет собой 2 или 3); множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, предпочтительно каждая независимо выбрана из группы, состоящей из метила и этила, более предпочтительно всего метила; и X представляет собой ОН.
8. Способ по любому одному из предшествующих аспектов, в котором источник первого оксида представляет собой, по меньшей мере, один, выбранный из группы, состоящей из источника диоксида кремния, источника диоксида германия, источника диоксида олова, источника диоксида титана и источника диоксида циркония, предпочтительно источника диоксида кремния или комбинации источника диоксида кремния и источника диоксида германия, и источник второго оксида представляет собой, по меньшей мере, один, выбранный из группы, состоящей из источника оксида алюминия, источника оксида бора, источника оксида железа, источника оксида галлия, источника оксида редкоземельного элемента, источника оксида индия и источника оксида ванадия, предпочтительно источника оксида алюминия.
9. Способ по любому одному из предшествующих аспектов, в котором условия кристаллизации включают: температуру кристаллизации в диапазоне от 80°С до 120°С, предпочтительно от 120°С до 170°С или от 120°С до 200°С, период кристаллизации, по меньшей мере, 1 день, предпочтительно, по меньшей мере, 2 дня, предпочтительно от 3 дней до 8 дней, от 5 дней до 8 дней или от 4 дней до 6 дней и условия обжига включают: температуру обжига в диапазоне от 300°C до 750°C, предпочтительно от 400°C до 600°C, период обжига в диапазоне от 1 часа до 10 часов, предпочтительно от 3 часов до 6 часов.
10. Способ по любому одному из предшествующих аспектов, в котором молярное соотношение источника первого оксида (рассчитанное на основе первого оксида) и источника второго оксида (рассчитанное на основе второго оксида) находится в диапазоне от 5 до ∞, предпочтительно от 25 до 95, более предпочтительно от 30 до 70; молярное соотношение воды и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 5 до 50, предпочтительно от 5 до 15; молярное соотношение органического темплата и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,15 до 0,5 или от 0,3 до 0,5; молярное соотношение щелочного источника (рассчитанное на основе OH-) и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 0 до 1, предпочтительно от 0,04 до 1, от 0,1 до 1, от 0,2 до 1, от 0,3 до 0,7 или от 0,45 до 0,7.
Второй вариант осуществления
1. Молекулярное сито, имеющее (нативную) губчатую структуру и имеющее рентгеновскую дифрактограмму по существу, как показано в следующей таблице,
2. Молекулярное сито по любому одному из предшествующих аспектов, в котором рентгеновская дифрактограмма дополнительно содержит рентгенодифракционный пик по существу, как показано в следующей таблице,
3. Молекулярное сито по любому одному из предшествующих аспектов, в котором рентгеновская дифрактограмма дополнительно содержит рентгенодифракционный пик по существу, как показано в следующей таблице,
4. Молекулярное сито по любому одному из предшествующих аспектов, в котором губчатая структура содержит крупные поры и/или мезопоры, предпочтительно крупные поры и/или мезопоры открываются на торцевой поверхности и/или боковой поверхности губчатой структуры.
5. Молекулярное сито по любому одному из предшествующих аспектов, в котором крупные поры имеют диаметр в диапазоне от 80 нм до 2 мкм, предпочтительно от 80 нм до 1,5 мкм и мезопоры имеют диаметр в диапазоне от 2 нм до 30 нм, предпочтительно от 2 нм до 4 нм и/или от 7 нм до 15 нм (предпочтительно от 8 нм до 9 нм).
6. Молекулярное сито по любому одному из предшествующих аспектов, в котором мезопоры имеют общую удельную площадь поверхности в диапазоне от 50 м2⋅г-1 до 250 м2⋅г-1, предпочтительно от 100 м2⋅г-1 до 150 м2⋅г-1 и объем пор в диапазоне от 0,05 мл/г до 0,40 мл/г, предпочтительно от 0,15 мл/г до 0,30 мл/г и крупные поры имеют общую удельную площадь поверхности в диапазоне от 10 м2⋅г-1 до 100 м2⋅г-1, предпочтительно от 50 м2⋅г-1 до 100 м2⋅г-1 и объем пор в диапазоне от 0,5 мл/г до 3,0 мл/г, предпочтительно от 1,0 мл/г до 2,0 мл /г.
7. Молекулярное сито по любому одному из предшествующих аспектов, в котором губчатая структура содержит микропоры, в котором микропоры имеют диаметр в диапазоне от 0,5 нм до менее 2 нм, предпочтительно от 0,5 нм до 0,8 нм и/или от 1,1 нм до 1,8 нм, общую удельную площадь поверхности в диапазоне от 100 м2⋅г-1 до 300 м2⋅г-1, предпочтительно от 150 м2⋅г-1 до 250 м2⋅г-1 и объем пор в диапазоне от 0,03 мл/г до 0,20 мл/г, предпочтительно от 0,05 мл/г до 0,15 мл/г.
8. Молекулярное сито по любому одному из предшествующих аспектов, имеющее морфологию столбчатой (предпочтительно призматической, более предпочтительно гексагональной) кристаллической частицы, предпочтительно имеющее морфологию полой столбчатой кристаллической частицы.
9. Молекулярное сито по любому одному из предшествующих аспектов, в котором морфология кристаллической частицы имеет размер, определяемый эффективным диаметром в диапазоне от 100 нм до 5000 нм, предпочтительно от 1000 нм до 3000 нм, высотой в диапазоне от 500 нм до 3000 нм, предпочтительно от 1000 нм до 3000 нм, соотношением сторон в диапазоне от 1/3 до 5, предпочтительно от 1/3 до 3.
10. Молекулярное сито по любому одному из предшествующих аспектов, имеющее схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид» или формулой «первый оксид ⋅ второй оксид ⋅ органический темплат ⋅ вода», в которой молярное соотношение первого оксида и второго оксида находится в диапазоне от 30 до 100, предпочтительно от 55 до 100; первый оксид представляет собой, по меньшей мере, один, выбранный из группы, состоящей из диоксида кремния, диоксида германия, диоксида олова, диоксида титана и диоксида циркония, предпочтительно диоксида кремния или комбинации диоксида кремния и диоксида германия; второй оксид представляет собой, по меньшей мере, один, выбранный из группы, состоящей из оксида алюминия, оксида бора, оксида железа, оксида галлия, оксида редкоземельного элемента, оксида индия и оксида ванадия, предпочтительно оксида алюминия; молярное соотношение воды и первого оксида находится в диапазоне от 5 до 50, предпочтительно от 5 до 15; молярное соотношение органического темплата и первого оксида находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,15 до 0,5 или от 0,3 до 0,5.
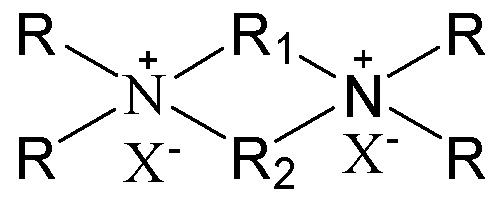
11. Способ получения молекулярного сита, включающий стадию приведения в контакт источника первого оксида, источника второго оксида, необязательного щелочного источника, органического темплата и воды в условиях кристаллизации с получением молекулярного сита и необязательно стадию обжига полученного молекулярного сита, в котором органический темплат содержит соединение, представленное следующей формулой (I)
 (I)
(I)
в которой группы R1 и R2 являются отличными друг от друга, одна из которых выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных или разветвленных оксаалкиленовых групп, предпочтительно одна из которых выбрана из группы, состоящей из С3-12линейных алкиленовых групп, другая выбрана из группы, состоящей из С3-12линейных оксаалкиленовых групп (предпочтительно С4-6линейных оксаалкиленовых групп, более предпочтительно С4-6линейных монооксаалкиленовых групп, более предпочтительно -(CH2)m-O-(CH2)m-, в которой каждое значение m, являющееся одинаковым или отличным друг от друга, независимо представляет собой 2 или 3); множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, предпочтительно каждая независимо выбрана из группы, состоящей из метила и этила, более предпочтительно всего метила; и X представляет собой ОН.
12. Способ по любому одному из предшествующих аспектов, в котором источник первого оксида представляет собой, по меньшей мере, один, выбранный из группы, состоящей из источника диоксида кремния, источника диоксида германия, источника диоксида олова, источника диоксида титана и источника диоксида циркония, предпочтительно источника диоксида кремния или комбинации источника диоксида кремния и источника диоксида германия, и источник второго оксида представляет собой, по меньшей мере, один, выбранный из группы, состоящей из источника оксида алюминия, источника оксида бора, источника оксида железа, источника оксида галлия, источника оксида редкоземельного элемента, источника оксида индия и источника оксида ванадия, предпочтительно источника оксида алюминия.
13. Способ по любому одному из предшествующих аспектов, в котором условия кристаллизации включают: температуру кристаллизации в диапазоне от 80°С до 120°С, предпочтительно от 120°С до 170°С или от 120°С до 200°С, период кристаллизации, по меньшей мере, 1 день, предпочтительно, по меньшей мере, 2 дня, предпочтительно от 3 дней до 8 дней, от 5 дней до 8 дней или от 4 дней до 6 дней и условия обжига включают: температуру обжига в диапазоне от 300°C до 750°C, предпочтительно от 400°C до 600°C и период обжига в диапазоне от 1 часа до 10 часов, предпочтительно от 3 часов до 6 часов.
14. Способ по любому одному из предшествующих аспектов, в котором молярное соотношение источника первого оксида (рассчитанное на основе первого оксида) и источника второго оксида (рассчитанное на основе второго оксида) находится в диапазоне от 30 до 100, предпочтительно от 55 до 100; молярное соотношение воды и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 5 до 50, предпочтительно от 5 до 15; молярное соотношение органического темплата и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,15 до 0,5 или от 0,3 до 0,5, молярное соотношение щелочного источника (рассчитанное на основе OH-) и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 0 до 1, предпочтительно от 0,04 до 1, от 0,1 до 1, от 0,2 до 1, от 0,3 до 0,7 или от 0,45 до 0,7.
Третий вариант осуществления
1. Молекулярное сито, имеющее морфологию (нативной) кристаллической частицы от плоской призматической формы до плоской цилиндрической формы, предпочтительно один или два контура его торцевой поверхности в продольном сечении имеют выпуклую форму, и имеющее рентгеновскую дифрактограмму по существу, как показано в следующей таблице,
2. Молекулярное сито по любому одному из предшествующих аспектов, в котором рентгеновская дифрактограмма дополнительно содержит рентгенодифракционный пик по существу, как показано в следующей таблице,
3. Молекулярное сито по любому одному из предшествующих аспектов, в котором рентгеновская дифрактограмма дополнительно содержит рентгенодифракционный пик по существу, как показано в следующей таблице,
4. Молекулярное сито по любому одному из предшествующих аспектов, в котором морфология кристаллической частицы имеет размер, определяемый эффективным диаметром в диапазоне от 100 нм до 1000 нм, предпочтительно от 100 нм до 500 нм, высотой в диапазоне от 100 нм до 1000 нм, предпочтительно от 150 нм до 300 нм, соотношением сторон в диапазоне от 0,1 до 0,9, предпочтительно от 0,4 до 0,7.
5. Молекулярное сито по любому одному из предшествующих аспектов, в котором молекулярное сито имеет общую удельную площадь поверхности в диапазоне от 400 м2⋅г-1 до 600 м2⋅г-1, предпочтительно от 450 м2⋅г-1 до 580 м2⋅г-1 и объем пор в диапазоне от 0,3 мл/г до 0,5 мл/г, предпочтительно от 0,30 мл/г до 0,40 мл/г.
6. Молекулярное сито по любому одному из предшествующих аспектов, имеющее схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид» или формулой «первый оксид ⋅ второй оксид ⋅ органический темплат ⋅ вода», в которой молярное соотношение первого оксида и второго оксида находится в диапазоне от 40 до 200, предпочтительно от 40 до 150; первый оксид представляет собой, по меньшей мере, один, выбранный из группы, состоящей из диоксида кремния, диоксида германия, диоксида олова, диоксида титана и диоксида циркония, предпочтительно диоксида кремния или комбинации диоксида кремния и диоксида германия; второй оксид представляет собой, по меньшей мере, один, выбранный из группы, состоящей из оксида алюминия, оксида бора, оксида железа, оксида галлия, оксида редкоземельного элемента, оксида индия и оксида ванадия, предпочтительно оксида алюминия; молярное соотношение воды и первого оксида находится в диапазоне от 5 до 50, предпочтительно от 5 до 15; молярное соотношение органического темплата и первого оксида находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,08 до 0,5 или от 0,3 до 0,5.
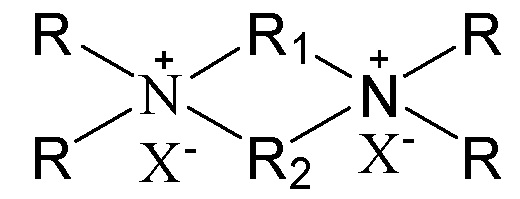
7. Способ получения молекулярного сита, включающий стадию приведения в контакт источника первого оксида, источника второго оксида, необязательного щелочного источника, органического темплата и воды в условиях кристаллизации с получением молекулярного сита и необязательно стадию обжига полученного молекулярного сита, в котором органический темплат содержит соединение, представленное следующей формулой (I),
 (I)
(I)
в которой группы R1 и R2 являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп, предпочтительно каждая независимо выбрана из группы, состоящей из С3-12линейных алкиленовых групп, в частности предпочтительно одна из них выбрана из группы, состоящей из С3-12линейных алкиленовых групп, и другая выбрана из группы, состоящей из С4-6линейных алкиленовых групп; множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, предпочтительно каждая независимо выбрана из группы, состоящей из метила и этила, более предпочтительно всего метила; и X представляет собой ОН.
8. Способ по любому одному из предшествующих аспектов, в котором источник первого оксида представляет собой, по меньшей мере, один, выбранный из группы, состоящей из источника диоксида кремния, источника диоксида германия, источника диоксида олова, источника диоксида титана и источника диоксида циркония, предпочтительно источника диоксида кремния или комбинации источника диоксида кремния и источника диоксида германия, и источник второго оксида представляет собой, по меньшей мере, один, выбранный из группы, состоящей из источника оксида алюминия, источника оксида бора, источника оксида железа, источника оксида галлия, источника оксида редкоземельного элемента, источника оксида индия и источника оксида ванадия, предпочтительно источника оксида алюминия.
9. Способ по любому одному из предшествующих аспектов, в котором условия кристаллизации включают: температуру кристаллизации в диапазоне от 80°С до 120°С, предпочтительно от 120°С до 170°С или от 120°С до 200°С, период кристаллизации, по меньшей мере, 1 день, предпочтительно, по меньшей мере, 2 дня, предпочтительно от 3 дней до 8 дней, от 5 дней до 8 дней или от 4 дней до 6 дней и условия обжига включают: температуру обжига в диапазоне от 300°C до 750°C, предпочтительно от 400°C до 600°C и период обжига в диапазоне от 1 часа до 10 часов, предпочтительно от 3 часов до 6 часов.
10. Способ по любому одному из предшествующих аспектов, в котором молярное соотношение источника первого оксида (рассчитанное на основе первого оксида) и источника второго оксида (рассчитанное на основе второго оксида) находится в диапазоне от 40 до 200, предпочтительно от 40 до 150; молярное соотношение воды и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 5 до 50, предпочтительно от 5 до 15; молярное соотношение органического темплата и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,08 до 0,5 или от 0,3 до 0,5, молярное соотношение щелочного источника (рассчитанное на основе OH-) и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 0 до 1, предпочтительно от 0,04 до 1, от 0,1 до 1, от 0,2 до 1, от 0,3 до 0,7 или от 0,45 до 0,7.
Четвертый вариант осуществления и пятый вариант осуществления
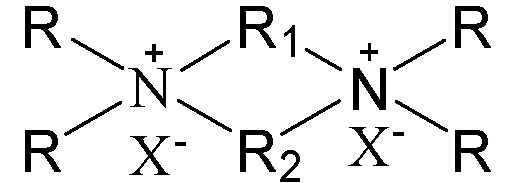
1. Способ получения молекулярного сита, включающий стадию приведения в контакт источника первого оксида, источника второго оксида, необязательного щелочного источника, органического темплата и воды в условиях кристаллизации с получением молекулярного сита и необязательно стадию обжига полученного молекулярного сита, в котором органический темплат содержит соединение, представленное следующей формулой (I),
 (I)
(I)
в которой группы R1 и R2 являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп и С3-12линейных или разветвленных оксаалкиленовых групп, предпочтительно каждая независимо выбрана из группы, состоящей из С3-12линейных алкиленовых групп и С3-12линейных оксаалкиленовых групп или предпочтительно одна из них выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп и С3-12линейных или разветвленных оксаалкиленовых групп, более предпочтительно одна из них выбрана из группы, состоящей из С3-12линейных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных алкиленовых групп и С3-12линейных оксаалкиленовых групп, в частности предпочтительно одна из них выбрана из группы, состоящей из С3-12линейных алкиленовых групп, и другая выбрана из группы, состоящей из C4-6линейных алкиленовых групп и C4-6линейных оксаалкиленовых групп (предпочтительно C4-6линейных монооксаалкиленовых групп, более предпочтительно -(CH2)m-O-(CH2)m-, в которой каждое значение m, являющееся одинаковым или отличным друг от друга, независимо представляет собой 2 или 3); множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, предпочтительно каждая независимо выбрана из группы, состоящей из метила и этила, более предпочтительно всего метила; и X представляет собой ОН.
2. Способ по любому одному из предшествующих аспектов, в котором группы R1 и R2 являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп, предпочтительно каждая независимо выбрана из группы, состоящей из С3-12линейных алкиленовых групп, в частности предпочтительно одна из них выбрана из группы, состоящей из С3-12линейных алкиленовых групп, и другая выбрана из группы, состоящей из С4-6линейных алкиленовых групп; множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, предпочтительно каждая независимо выбрана из группы, состоящей из метила и этила, более предпочтительно всего метила; и X представляет собой ОН.
3. Способ по любому одному из предшествующих аспектов, в котором группы R1 и R2 являются отличными друг от друга, одна из которых выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных или разветвленных оксаалкиленовых групп, предпочтительно одна выбрана из группы, состоящей из С3-12линейных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных оксаалкиленовых групп (предпочтительно С4-6линейных оксаалкиленовых групп, более предпочтительно С4-6линейных монооксаалкиленовых групп, более предпочтительно -(CH2)m-O-(CH2)m-, в которой каждое значение m, являющееся одинаковым или отличным друг от друга, независимо представляет собой 2 или 3); множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, предпочтительно каждая независимо выбрана из группы, состоящей из метила и этила, более предпочтительно всего метила; и X представляет собой ОН.
4. Способ по любому одному из предшествующих аспектов, в котором источник первого оксида представляет собой, по меньшей мере, один, выбранный из группы, состоящей из источника диоксида кремния, источника диоксида германия, источника диоксида олова, источника диоксида титана и источника диоксида циркония, предпочтительно источника диоксида кремния или комбинации источника диоксида кремния и источника диоксида германия, и источник второго оксида представляет собой, по меньшей мере, один, выбранный из группы, состоящей из источника оксида алюминия, источника оксида бора, источника оксида железа, источника оксида галлия, источника оксида редкоземельного элемента, источника оксида индия и источника оксида ванадия, предпочтительно источника оксида алюминия.
5. Способ по любому одному из предшествующих аспектов, в котором условия кристаллизации включают: температуру кристаллизации в диапазоне от 80°С до 120°С, предпочтительно от 120°С до 170°С или от 120°С до 200°С, период кристаллизации, по меньшей мере, 1 день, предпочтительно, по меньшей мере, 2 дня, предпочтительно от 3 дней до 8 дней, от 5 дней до 8 дней или от 4 дней до 6 дней и условия обжига включают: температуру обжига в диапазоне от 300°C до 750°C, предпочтительно от 400°C до 600°C и период обжига в диапазоне от 1 часа до 10 часов, предпочтительно от 3 часов до 6 часов.
6. Способ по любому одному из предшествующих аспектов, в котором молярное соотношение источника первого оксида (рассчитанное на основе первого оксида) и источника второго оксида (рассчитанное на основе второго оксида) находится в диапазоне от 5 до ∞, в частности от 5 до менее 40 (например, от 20 до менее 40), от 40 до 200 (например, от 40 до 150), от более 200 до ∞ (например, от более 200 до 700); молярное соотношение воды и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 5 до 50, предпочтительно от 5 до 15; молярное соотношение органического темплата и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,08 до 0,5 или от 0,3 до 0,5; молярное соотношение щелочного источника (рассчитанное на основе OH-) и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 0 до 1, предпочтительно от 0,04 до 1, от 0,1 до 1, от 0,2 до 1, от 0,3 до 0,7 или от 0,45 до 0,7.
7. Способ по любому одному из предшествующих аспектов, в котором молярное соотношение источника первого оксида (рассчитанное на основе первого оксида) и источника второго оксида (рассчитанное на основе второго оксида) находится в диапазоне от 5 до ∞, в частности от 5 до менее 30 (например, от 10 до менее 30), от 30 до 100 (например, от 55 до 100) и от более 100 до ∞ (например, от 200 до ∞ или от 200 до 700); молярное соотношение воды и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 5 до 50, предпочтительно от 5 до 15; молярное соотношение органического темплата и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,15 до 0,5 или от 0,3 до 0,5; молярное соотношение щелочного источника (рассчитанное на основе OH-) и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 0 до 1, предпочтительно от 0,04 до 1, от 0,1 до 1, от 0,2 до 1, от 0,3 до 0,7 или от 0,45 до 0,7.
8. Молекулярное сито, полученное с помощью способа по любому одному из предшествующих аспектов.
9. Молекулярное сито по любому одному из предшествующих аспектов, имеющее рентгеновскую дифрактограмму по существу, как показано в следующей таблице,
Шестой вариант осуществления
1. Композиция молекулярного сита, содержащая молекулярное сито по любому одному из предшествующих аспектов или молекулярное сито, полученное с помощью способа по любому одному из предшествующих аспектов, и связующее вещество.
2. Способ конверсии углеводорода, включающий стадию подвергания углеводорода реакции конверсии в присутствии катализатора, в котором катализатор содержит или его получают из молекулярного сита по любому одному из предшествующих аспектов, молекулярного сита, полученного с помощью способа по любому одному из предшествующих аспектов, или композиции молекулярного сита по любому одному из предшествующих аспектов.
3. Способ по любому одному из предшествующих аспектов, в котором реакция конверсии выбрана из группы, состоящей из реакций каталитического крекинга, гидрокрекинга, диспропорционирования, алкилирования, олигомеризации и изомеризации.
Положительные эффекты
Молекулярное сито в соответствии с настоящим изобретением имеет ультрамакропористую структуру пор в каркасе, которая может отражаться, по меньшей мере, более высоким значением ее объема пор.
Молекулярное сито в соответствии с настоящим изобретением обладает хорошей термической/гидротермической стабильностью и большим объемом пор. Таким образом, молекулярное сито в соответствии с настоящим изобретением способно адсорбировать больше/более крупные молекулы, тем самым демонстрируя превосходные адсорбционные/каталитические свойства.
Молекулярное сито в соответствии с настоящим изобретением в одном варианте осуществления имеет уникальную рентгеновскую дифрактограмму (XRD) и уникальное соотношение Si/Al2. Данное молекулярное сито не было получено в предшествующем уровне техники.
Молекулярное сито в соответствии с настоящим изобретением в одном варианте осуществления имеет уникальную рентгеновскую дифрактограмму (XRD) и уникальную морфологию нативных кристаллических частиц, такую как морфология кристаллической частицы от плоской призматической формы до плоской цилиндрической формы. Данное молекулярное сито не было получено в предшествующем уровне техники.
Молекулярное сито в соответствии с настоящим изобретением в одном варианте осуществления имеет уникальную рентгеновскую дифрактограмму (XRD) и уникальную морфологию нативных кристаллических частиц, такую как морфология кристаллической частицы губчатой структуры. Данное молекулярное сито не было получено в предшествующем уровне техники. В результате в дополнение к характеристикам микропористых материалов (т. е. присущим характеристикам стандартных молекулярных сит) молекулярное сито в соответствии с настоящим изобретением дополнительно демонстрирует характеристики мезопористых материалов и/или макропористых материалов и способно адсорбировать больше/более крупные молекулы, тем самым демонстрируя превосходные адсорбционные/каталитические свойства.
Молекулярное сито в соответствии с настоящим изобретением в одном варианте осуществления имеет относительно сильную кислотность, в частности относительно большее количество L-кислотных центров. Данное молекулярное сито не было получено в предшествующем уровне техники. В результате молекулярное сито в соответствии с настоящим изобретением демонстрирует лучшую производительность, в частности в катализируемой кислотой реакции.
В соответствии со способом получения молекулярного сита настоящего изобретения используют органический темплат, имеющий определенную химическую структуру. Таким образом, способ обладает характеристиками, что условия способа являются простыми и продукт молекулярного сита может быть легко синтезирован.
Подробное описание изобретения
Определенные варианты осуществления настоящего изобретения будут подробно описаны ниже, но следует отметить, что объем настоящего изобретения не ограничивается данными определенными вариантами осуществления, но определяется прилагаемой формулой изобретения.
Все публикации, заявки на патент, патенты и другие справочные документы, упомянутые в настоящем описании, включены в настоящее описание посредством ссылки. Если не указано иначе, все технические и научные термины, используемые в описании, имеют значение, обычно понимаемое специалистом в данной области техники. В случае конфликта определение настоящего описания будет иметь преимущественную силу.
В настоящем описании, когда материал, вещество, способ, стадия, устройство или компонент и т.д. модифицируется в соответствии с формулировкой «известный специалисту в данной области техники», «предшествующий уровень техники» или подобное, он должен интерпретироваться как охватывающий не только который обычно используется в области техники настоящей заявки на момент подачи заявки, но также который обычно не используется в настоящее время, но будет признан в данной области техники полезным для подобных целей.
В контексте настоящего описания символ «/» обычно понимается как «и/или», например, выражение «более/больше» означает «более и/или больше», если такое понимание не противоречит общим знаниям специалиста в данной области техники.
В контексте настоящего описания термин «органический темплат» иногда упоминается в данной области техники как структурообразующий агент или органический направляющий агент.
В контексте настоящего описания в качестве примеров C1-4линейных или разветвленных алкильных групп могут быть упомянуты в том числе метильная группа, этильная группа или пропильная группа.
В контексте настоящего изобретения термин «линейная или разветвленная оксаалкиленовая группа» относится к двухвалентной группе, в которой структура углеродной цепи линейной или разветвленной алкиленовой группы прерывается одной или более (например, от 1 до 3, от 1 до 2 или 1) гетерогруппой -O-. С точки зрения структурной стабильности предпочтительно, когда имеется множество гетерогрупп, любые две группы не связаны непосредственно. Очевидно, что термин «прерывание» означает, что гетерогруппа отсутствует на любом конце линейной или разветвленной алкиленовой группы или линейной или разветвленной оксаалкиленовой группы. В качестве примера C4-линейная алкиленовая группа (-CH2-CH2-CH2-CH2-) может быть прервана одной гетерогруппой -O- для получения C4-линейной монооксаалкиленовой группы, такой как -CH2-O-CH2-CH2-CH2- или -CH2-CH2-O-CH2-CH2- и подобная, может быть прервана двумя гетерогруппами -O- для получения C4-линейной диоксаалкиленовой группы, такой как -CH2-O-CH2-O-CH2-CH2- или --CH2-O-CH2-O-CH2-CH2- и подобная и может быть прервана тремя гетерогруппами -O- для получения C4-линейной триоксаалкиленовой группы, такой как -CH2-O-CH2-O-CH2-O-CH2- и подобная. Альтернативно, в качестве другого примера С4-разветвленная алкиленовая группа (-CH2(CH3)-CH2-CH2-) может быть прервана одной гетерогруппой -O- для получения С4-разветвленной монооксаалкиленовой группы, такой как -CH2(CH3)-O-CH2-CH2-, -CH2(CH3)-CH2-O-CH2- или -CH2(-O-CH3)-CH2-CH2- и подобная, может быть прервана двумя гетерогруппами -O- для получения С4-разветвленной диоксаалкиленовой группы, такой как -CH2(CH3)-O-CH2-O-CH2-, -CH2(-O-CH3)-O-CH2-CH2- или -CH2(-O-CH3)-CH2-O-CH2- и подобная и может быть прервана тремя гетерогруппами -O- для получения С4-разветвленной триоксаалкиленовой группы, такой как -CH2(-O-CH3)-O-CH2-O-CH2- и подобная.
В контексте настоящего описания термин «общая удельная площадь поверхности» относится к общей площади на единицу массы молекулярного сита, включая площадь внутренней поверхности и площадь внешней поверхности. Непористые материалы, такие как портландцемент, некоторые глинистые минеральные частицы и т.д. имеют только площадь внешней поверхности, тогда как пористые материалы, такие как асбестовые волокна, диатомовая земля и молекулярные сита имеют как площадь внешней поверхности, так и площадь внутренней поверхности.
В контексте настоящего описания термин «объем пор», также известный как объем пористости относится к объему пор на единицу массы молекулярного сита. Кроме того, термин «объем микропор» относится к объему всех микропор (то есть пор, имеющих диаметр пор менее 2 нм) на единицу массы молекулярного сита.
В контексте настоящего описания, как хорошо известно специалисту в данной области техники, w, m, s, vs используются в данных рентгеновской дифракции молекулярного сита для описания интенсивности дифракционного пика, в которых w означает слабую, m означает среднюю, s означает сильную и vs означает очень сильную. В общем случае w означает менее 20; m означает 20-40; s означает 40-70; vs означает более 70.
Все проценты, части, соотношения и т.д., упомянутые в настоящем описании, являются массовыми, если не указано иначе, или являются нецелесообразными на основе массы в соответствии с общепринятыми знаниями специалиста в данной области техники.
В контексте настоящего описания любые два или более аспектов настоящего изобретения могут быть произвольно объединены, и техническое решение, полученное таким образом, является частью первоначального раскрытия настоящего описания и также подпадает под объем настоящего изобретения.
В соответствии с настоящим изобретением предоставляют, по меньшей мере, следующие с первого по шестой варианты осуществления.
Первый вариант осуществления
В соответствии с одним аспектом настоящего изобретения предоставляют молекулярное сито, в котором молекулярное сито имеет схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид». Известно, что молекулярные сита иногда могут содержать определенное количество влаги (особенно сразу после их синтеза), но в настоящей заявке не считается необходимым указывать количество влаги, поскольку наличие или отсутствие влаги не оказывает существенного влияния на спектр рентгеновской дифракции молекулярного сита. Ввиду этого схематическая химическая композиция фактически представляет собой не содержащую воду химическую композицию молекулярного сита. Кроме того, очевидно, что схематическая химическая композиция представляет собой химическую композицию каркаса молекулярного сита.
В соответствии с одним аспектом настоящего изобретения молекулярное сито может обычно дополнительно содержать в своей композиции органический темплат и воду и т.д., такие как те, которые заполнены в его порах сразу после его синтеза. Таким образом, молекулярное сито может иногда иметь схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид ⋅ органический темплат ⋅ вода». В настоящем описании молекулярное сито, имеющее схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид ⋅ органический темплат ⋅ вода», может быть подвергнуто обжигу для удаления любого органического темплата и воды, присутствующей в его порах, так что может быть получено молекулярное сито, имеющее схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид». Кроме того, обжиг может быть осуществлен любым общепринятым способом, известным в данной области техники, например, при температуре обжига, обычно находящейся в диапазоне от 300°С до 750°С, предпочтительно от 400°С до 600°С, и в течение периода обжига, обычно находящегося в диапазоне от 1 часа до 10 часов, предпочтительно от 3 часов до 6 часов. Кроме того, обжиг обычно проводят в кислородсодержащей атмосфере, такой как атмосфера воздуха или кислорода.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции первый оксид обычно представляет собой четырехвалентный оксид и может представлять собой, например, по меньшей мере, один, выбранный из группы, состоящей из диоксида кремния, диоксида германия, диоксида олова, диоксида титана и диоксида циркония, предпочтительно диоксида кремния (SiO2) или комбинации диоксида кремния и диоксида германия. Указанные первые оксиды могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из первых оксидов может представлять собой, например, от 20:200 до 35:100. В качестве примера комбинации диоксид кремния и диоксид германия можно использовать в комбинации, в данном случае молярное соотношение между диоксидом кремния и диоксидом германия может представлять собой, например, от 20:200 до 35:100.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции второй оксид обычно представляет собой трехвалентный оксид и может представлять собой, например, по меньшей мере, один, выбранный из группы, состоящей из оксида алюминия, оксида бора, оксида железа, оксида галлия, оксида редкоземельного элемента, оксида индия и оксида ванадия, предпочтительно оксида алюминия (Al2O3). Указанные вторые оксиды могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из вторых оксидов может представлять собой, например, от 30:200 до 60:150.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции в качестве примера органического темплата может быть упомянут любой органический темплат, пригодный для получения молекулярных сит, и особенно может быть упомянут органический темплат, используемый при получении молекулярного сита в соответствии с настоящим вариантом осуществления (см. подробное описание ниже). Указанные органические темплаты могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. В частности, примеры органического темплата включают соединения, представленные следующей формулой (I).
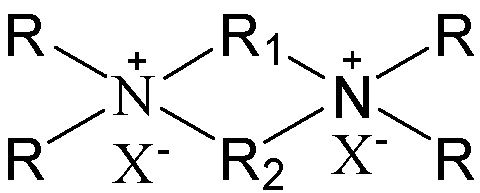 (I)
(I)
В соответствии с одним аспектом настоящего изобретения в формуле (I) группы R1 и R2 являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп и С3-12линейных или разветвленных оксаалкиленовых групп, множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, и Х представляет собой ОН.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции молярное соотношение первого оксида и второго оксида (например, молярное соотношение SiO2 и Al2O3) обычно находится в диапазоне от 5 до ∞, предпочтительно от 25 до 95, более предпочтительно от 30 до 70. В настоящем описании, когда молярное соотношение представляет собой ∞, это означает, что второй оксид отсутствует или содержание второго оксида в схематической химической композиции является незначительным. Авторы настоящего изобретения обнаружили при тщательном исследовании, что молекулярное сито имеет молярное соотношение (такое как молярное соотношение SiO2 и Al2O3), в частности от 25 до 95 (более определенно от 30 до 70) не было получено в предшествующем уровне техники.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции молярное соотношение воды и первого оксида обычно находится в диапазоне от 5 до 50, предпочтительно от 5 до 15.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции молярное соотношение органического темплата и первого оксида обычно находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,15 до 0,5 или от 0,3 до 0,5.
В соответствии с одним аспектом настоящего изобретения в зависимости от исходных материалов, используемых в способе получения, молекулярное сито может иногда дополнительно содержать катион металла, такой как катион щелочного металла и/или щелочноземельного металла в качестве компонента в его композиции (как правило, заполненный в его поры). В данном случае содержание катиона металла, например, массовое соотношение катиона металла и первого оксида обычно находится в диапазоне от 0 до 0,02, предпочтительно от 0,0002 до 0,006, но не ограничивается ими.
В соответствии с одним аспектом настоящего изобретения молекулярное сито имеет рентгеновскую дифрактограмму по существу, как показано в следующей таблице.
В соответствии с одним аспектом настоящего изобретения предпочтительно, что рентгеновская дифрактограмма молекулярного сита дополнительно включает рентгенодифракционный пик по существу, как показано в следующей таблице.
В соответствии с одним аспектом настоящего изобретения предпочтительно, что рентгеновская дифрактограмма молекулярного сита дополнительно включает рентгенодифракционный пик по существу, как показано в следующей таблице.
В соответствии с одним аспектом настоящего изобретения молекулярное сито обычно имеет морфологию столбчатой кристаллической частицы при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «морфология кристаллической частицы» относится к (общей) внешней форме, проявляемой монокристаллической частицей молекулярного сита в поле наблюдения сканирующего электронного микроскопа. Кроме того, в качестве столбчатой формы предпочтительной является призматическая форма, в частности гексагональная призматическая форма. В настоящем описании призма относится к выпуклой призме и обычно относится к прямоугольной призме и правильной многоугольной призме (такой как правильная гексагональная призма). Следует особо отметить, что поскольку процесс роста кристалла молекулярного сита может быть нарушен различными факторами, может быть определенная степень отклонения, когда его реальная морфология кристаллической частицы сравнивается с (истинно) прямоугольной призмой или (истинно) правильной многоугольной призмой в геометрическом смысле, такое как отклонение 30%, 20% или 5%, что приводит к получению скошенной призмы или неправильной многоугольной (или даже искривленной многоугольной) призмы, но настоящее изобретение не предназначено для определения степени отклонения. Более того, любые большие или меньшие отклонения не отступают от объема настоящего изобретения.
В соответствии с одним аспектом настоящего изобретения молекулярное сито (монокристаллическая частица) имеет эффективный диаметр, обычно находящийся в диапазоне от 100 нм до 1000 нм, предпочтительно от 300 до 700 нм, при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «эффективный диаметр» означает, что две точки произвольно выбираются в поперечном сечении молекулярного сита (монокристаллической частицы) вдоль контура (края) поперечного сечения и измеряется линейное расстояние между двумя точками, где наибольшее линейное расстояние принимается за эффективный диаметр. Если профиль поперечного сечения молекулярного сита представлен как многоугольник, такой как шестиугольник, эффективный диаметр обычно относится к линейному расстоянию между двумя вершинами многоугольника, расположенными дальше друг от друга, т.е. диагональному расстоянию. Вкратце, эффективный диаметр по существу соответствует диаметру описанной окружности многоугольника, представляющего контур поперечного сечения.
В соответствии с одним аспектом настоящего изобретения высота молекулярного сита (монокристаллическая частица) обычно находится в диапазоне от 100 нм до 1000 нм, предпочтительно от 150 нм до 700 нм при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «высота» относится к линейному расстоянию между центрами двух торцевых поверхностей монокристаллической частицы (столбчатой кристаллической частицы) молекулярного сита. В обычном случае две торцевые поверхности столбца молекулярного сита по существу параллельны друг другу, и линейное расстояние представляет собой вертикальное расстояние между двумя торцевыми поверхностями, но настоящее изобретение не ограничивается этим.
В соответствии с одним аспектом настоящего изобретения соотношение сторон молекулярного сита (монокристаллическая частица) обычно находится в диапазоне от 1/3 до 8, предпочтительно от 1,5 до 5 или от 2 до 5 при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «соотношение сторон» относится к соотношению высоты и эффективного диаметра.
В соответствии с одним аспектом настоящего изобретения молекулярное сито имеет общую удельную площадь поверхности, обычно находящуюся в диапазоне от 400 м2⋅г-1 до 600 м2⋅г-1, предпочтительно от 450 м2⋅г-1 до 580 м2⋅г-1. В настоящем описании общую удельную площадь поверхности получают с использованием метода адсорбции жидкого азота, рассчитанного по модели БЭТ.
В соответствии с одним аспектом настоящего изобретения молекулярное сито имеет объем пор (объем микропор), обычно находящийся в диапазоне от 0,3 мл/г до 0,5 мл/г, предпочтительно от 0,30 мл/г до 0,40 мл/г. Молекулярное сито в соответствии с настоящим изобретением имеет очень большой объем микропор, что указывает на то, что оно относится к ультрамакропористому молекулярному ситу. В настоящем описании объем пор получают с использованием метода адсорбции жидкого азота, рассчитанного по модели Хорвата-Кавадзоэ.
В соответствии с одним аспектом настоящего изобретения молекулярное сито может быть получено с помощью следующего способа. В настоящем описании способ получения включает стадию приведения в контакт источника первого оксида, источника второго оксида, необязательного щелочного источника, органического темплата и воды в условиях кристаллизации с получением молекулярного сита (в дальнейшем именуемую как стадия взаимодействия).
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита стадия взаимодействия может быть проведена с помощью любого стандартного способа, известного в данной области техники, например, с использованием способа, включающего смешивание источника первого оксида, источника второго оксида, необязательного щелочного источника, органического темплата и воды и подвергание смеси кристаллизации в условиях кристаллизации.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия органический темплат содержит, по меньшей мере, соединение, представленное следующей формулой (I). В настоящем описании соединение, представленное формулой (I), может быть использовано отдельно или в комбинации двух или более из них в любом соотношении.
 (I)
(I)
В соответствии с одним аспектом настоящего изобретения в формуле (I) группы R1 и R2 являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп и С3-12линейных или разветвленных оксаалкиленовых групп.
В соответствии с другим вариантом осуществления настоящего изобретения в формуле (I) группы R1 и R2 являются одинаковыми или отличными друг от друга, одна из которых выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп и С3-12линейных или разветвленных оксаалкиленовых групп.
В соответствии с другим вариантом осуществления настоящего изобретения в формуле (I) группы R1 и R2 являются одинаковыми или отличными друг от друга, одна из которых выбрана из группы, состоящей из С3-12линейных алкиленовых групп, и другая выбрана из группы, состоящей из С4-6линейных алкиленовых групп и С4-6линейных оксаалкиленовых групп.
В соответствии с другим вариантом осуществления настоящего изобретения в формуле (I) группы R1 и R2 являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп.
В соответствии с другим вариантом осуществления настоящего изобретения в формуле (I) группы R1 и R2 являются одинаковыми или отличными друг от друга, одна из которых выбрана из группы, состоящей из С3-12линейных алкиленовых групп, и другая выбрана из группы, состоящей из С4-6линейных алкиленовых групп.
В соответствии с другим вариантом осуществления настоящего изобретения в формуле (I) группы R1 и R2 являются отличными друг от друга, одна из которых выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных или разветвленных оксаалкиленовых групп.
В соответствии с одним аспектом настоящего изобретения в качестве примера С3-12линейных или разветвленных алкиленовых групп может быть упомянута С3-12линейная алкиленовая группа, и ее определенные примеры включают н-пропилен, изопропилиден, н-бутилен, изобутилен, трет-бутилен, н-пентилен, изопентилен, неопентилен, н-гексилен, изогексилен, н-октилен, изооктилен, неооктилен, нонилен (или его изомер), децилен (или его изомер), ундецилен (или его изомер) или додецилен (или его изомер), предпочтительно н-пропилен, н-бутилен, н-пентилен, н-гексилен, н-гептилен, н-октилен, н-нонилен, н-децилен, н-ундецилен или н-додецилен. Кроме того, в качестве примера С3-12линейных алкиленовых групп может быть упомянута С4-6линейная алкиленовая группа, и в частности могут быть упомянуты н-бутилен, н-пентилен или н-гексилен.
В соответствии с одним аспектом настоящего изобретения в качестве С3-12линейных или разветвленных оксаалкиленовых групп, например, может быть упомянута С3-12линейная оксаалкиленовая группа, и ее конкретные примеры включают -(CH2)2-O-(CH2)-, -(CH2)2-O-(CH2)2-, -(CH2)-O-(CH2)3-, -(CH2)2-O-(CH2)3-, -(CH2)-O-пропилен-, -(CH2)-O-(CH2)4-, -(CH2)-O-(CH2)2-O-(CH2)-, -(CH2)-O-(CH2)2-O-(CH2)2-, -(CH2)-O-трет-бутилен-, -(CH2)2-O-(CH2)4-, -(CH2)3-O-(CH2)3-, -(CH2)-O-неопентилен-, -(CH2)2-O-(CH2)6-, -(CH2)2-O-(CH2)7-, -(CH2)-O-(CH2)8-, -(CH2)-O-изооктилен-, -(CH2)-O-(CH2)10-, -(CH2)2-O-децилен или его изомер, -(CH2)-O-(CH2)6-, -(CH2)-O-(CH2)7-, -(CH2)-O-(CH2)8-, -(CH2)-O-(CH2)11-, -(CH2)-O-(CH2)2-O-(CH2)-, -(CH2)2-O-(CH2)2-O-(CH2)2-, -(CH2)2-O-(CH2)4-O-(CH2)2-, -(CH2)2-O-(CH2)6-O-(CH2)2- или -(CH2)2-O-(CH2)8-O-(CH2)2-. Кроме того, в качестве С3-12линейных оксаалкиленовых групп, например, может быть упомянута С4-6линейная оксаалкиленовая группа, в частности может быть упомянута С4-6линейная монооксаалкиленовая группа, и особенно может быть упомянута монооксаалкиленовая группа, представленная формулой -(CH2)m-O-(CH2)m- (где каждое значение m, являющееся одинаковым или отличным друг от друга, независимо представляет собой 2 или 3, например, 2). Более определенно могут быть упомянуты -(CH2)2-O-(CH2)2-, -(CH2)2-O-(CH2)3-, -(CH2)3-O-(CH2)3- или -(CH2)2-O-(CH2)4-.
В соответствии с одним аспектом настоящего изобретения в формуле (I) множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, предпочтительно каждая независимо выбрана из группы, состоящей из метила и этила, более предпочтительно всего метила.
В соответствии с одним аспектом настоящего изобретения в формуле (I) X представляет собой ОН.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение органического темплата и источника первого оксида (рассчитанное на основе первого оксида) обычно находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,15 до 0,5 или от 0,3 до 0,5.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в дополнение к соединению, представленному формулой (I), другие органические темплаты, обычно используемые в данной области техники для получения молекулярных сит, могут быть дополнительно использованы в качестве органического темплата. Предпочтительно на стадии взаимодействия только соединение, представленное формулой (I), используют в качестве органического темплата. В настоящем описании соединение, представленное формулой (I), может быть использовано отдельно или в комбинации двух или более из них в любом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия источник первого оксида обычно представляет собой источник четырехвалентного оксида и может представлять собой, например, по меньшей мере, один, выбранный из группы, состоящей из источника диоксида кремния, источника диоксида германия, источника диоксида олова, источника диоксида титана и источника диоксида циркония, предпочтительно источника диоксида кремния (SiO2) или комбинации источника диоксида кремния и источника диоксида германия. Указанные источники первого оксида могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из источников первого оксида может представлять собой, например, от 20:200 до 35:100. В качестве примера комбинации может быть упомянута комбинация источника диоксида кремния и источника диоксида германия, в данном случае молярное соотношение между источником диоксида кремния и источником диоксида германия может представлять собой, например, от 20:200 до 35:100.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве источника первого оксида может быть использован любой соответствующий источник оксида, обычно используемый для этой цели в данной области техники, включая, но не ограничиваясь ими, в том числе оксиды, гидроксиды, алкоксиды, соли оксикислот металлов, ацетаты, оксалаты, соли аммония, сульфаты, галогенированные соли и нитраты соответствующего металла в первом оксиде. Например, когда первый оксид представляет собой диоксид кремния, примеры источника первого оксида включают золь кремниевой кислоты, неочищенный силикагель, тетраэтилортосиликат, жидкое стекло, белую сажу, кремниевую кислоту, силикагель, силикат калия или подобное. Когда первый оксид представляет собой диоксид германия, примеры источника первого оксида включают тетраалкоксид германия, оксид германия, нитрат германия или подобное. Когда первый оксид представляет собой диоксид олова, примеры источника первого оксида включают хлорид олова, сульфат олова, нитрат олова и подобное. Когда первый оксид представляет собой диоксид титана, примеры источника первого оксида включают тетраалкоксид титана, диоксид титана, нитрат титана и подобное. Когда первый оксид представляет собой диоксид циркония, примеры источника первого оксида включают сульфат циркония, хлорид циркония, нитрат циркония и подобное. Указанные источники первого оксида могут быть использованы отдельно или в комбинации двух или более из них в желаемом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия источник второго оксида обычно представляет собой источник трехвалентного оксида, такой как, по меньшей мере, один, выбранный из группы, состоящей из источника оксида алюминия, источника оксида бора, источника оксида железа, источника оксида галлия, источника редкоземельного оксида, источника оксида индия и источника оксида ванадия, предпочтительно источника оксида алюминия (Al2O3). Указанные источники второго оксида могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из источников второго оксида может представлять собой, например, от 30:200 до 60:150.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве источника второго оксида может быть использован любой соответствующий источник оксида, обычно используемый для этой цели в данной области техники, включая, но не ограничиваясь ими, в том числе оксиды, гидроксиды, алкоксиды, соли оксикислот металлов, ацетаты, оксалаты, соли аммония, сульфаты, галогенированные соли и нитраты соответствующего металла во втором оксиде. Например, когда второй оксид представляет собой оксид алюминия, примеры источника второго оксида включают хлорид алюминия, сульфат алюминия, гидрат алюминия, метаалюминат натрия, золь алюминия, гидроксид алюминия или подобное. Когда второй оксид представляет собой оксид бора, примеры источника второго оксида включают борную кислоту, борат, буру, триоксид бора и подобное. Когда второй оксид представляет собой оксид железа, примеры источника второго оксида включают нитрат железа, хлорид железа, оксид железа и подобное. Когда второй оксид представляет собой оксид галлия, примеры источника второго оксида включают нитрат галлия, сульфат галлия, оксид галлия и подобное. Когда второй оксид представляет собой редкоземельный оксид, примеры источника второго оксида включают оксид лантана, оксид неодима, оксид иттрия, оксид церия, нитрат лантана, нитрат неодима, нитрат иттрия, сульфат церия-аммония и подобное. Когда второй оксид представляет собой оксид индия, примеры источника второго оксида включают хлорид индия, нитрат индия, оксид индия и подобное. Когда второй оксид представляет собой оксид ванадия, примеры источника второго оксида включают хлорид ванадия, метаванадат аммония, ванадат натрия, диоксид ванадия, ванадилсульфат и подобное. Указанные источники второго оксида могут быть использованы отдельно или в комбинации двух или более из них в желаемом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение источника первого оксида (рассчитанное на основе первого оксида, такого как SiO2) и источника второго оксида (рассчитанное на основе второго оксида, такого как Al2O3) обычно находится в диапазоне от 5 до ∞, предпочтительно от 25 до 95, более предпочтительно от 30 до 70. В настоящем описании, когда молярное соотношение представляет собой ∞, это означает, что источник второго оксида не используется или источник второго оксида не преднамеренно вводится на стадии взаимодействия.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение воды и источника первого оксида (рассчитанное на основе первого оксида) обычно находится в диапазоне от 5 до 50, предпочтительно от 5 до 15.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия щелочной источник может использоваться или не использоваться. Когда щелочной источник не преднамеренно используется, группа X, содержащаяся в соединении, представленном формулой (I), может быть использована для обеспечения ОН-, требуемого в настоящем описании. В настоящем описании в качестве щелочного источника может быть использован любой щелочной источник, обычно применяемый для этой цели в данной области техники, включая, но не ограничиваясь ими, неорганические основания, содержащие щелочной металл или щелочноземельный металл в качестве катиона, в частности гидроксид натрия и гидроксид калия. Указанные щелочные источники могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение щелочного источника (рассчитанное на основе OH-) и источника первого оксида (рассчитанное на основе первого оксида) обычно находится в диапазоне от 0 до 1, предпочтительно от 0,04 до 1, от 0,1 до 1, от 0,2 до 1, от 0,3 до 0,7 или от 0,45 до 0,7.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве условия кристаллизации температура кристаллизации обычно находится в диапазоне от 80°С до 120°С, предпочтительно от 120°С до 170°С или от 120°С до 200°С.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве условия кристаллизации период кристаллизации обычно составляет, по меньшей мере, 1 день, предпочтительно, по меньшей мере, 2 дня, предпочтительно от 3 дней до 8 дней, от 5 дней до 8 дней или от 4 дней до 6 дней.
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита после завершения стадии взаимодействия молекулярное сито может быть отделено в виде продукта из полученной реакционной смеси с помощью любого общеизвестного способа разделения. В настоящем описании продукт молекулярного сита содержит молекулярное сито в соответствии с настоящим изобретением. Кроме того, в качестве способа разделения, например, может быть упомянут способ, включающий фильтрование, промывание и сушку полученной реакционной смеси.
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита фильтрование, промывание и сушка могут быть проведены с помощью любого стандартного способа, известного в данной области техники. В качестве примера фильтрования, например, полученная реакционная смесь может быть просто подвергнута фильтрованию с отсасыванием. В качестве примера промывания, например, промывание деионизированной водой может проводиться до тех пор, пока рН фильтрата не достигнет 7-9, предпочтительно 8-9. Температура сушки составляет, например, от 40 до 250°С, предпочтительно от 60 до 150°С, и время сушки составляет, например, от 8 до 30 часов, предпочтительно от 10 до 20 часов. Сушка может быть проведена при нормальном давлении или при пониженном давлении.
В соответствии с одним аспектом настоящего изобретения способ получения молекулярного сита может дополнительно включать стадию обжига полученного молекулярного сита (в дальнейшем именуемую как стадия обжига) по мере необходимости для удаления органического темплата и возможной влаги и подобного, чтобы можно было получить кальцинированное молекулярное сито. В контексте настоящего описания молекулярные сита до и после обжига совместно называются как молекулярное сито настоящего изобретения или молекулярное сито в соответствии с настоящим изобретением.
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита обжиг может быть проведен с помощью любого стандартного способа, известного в данной области техники, например, при температуре обжига, обычно находящейся в диапазоне от 300°С до 750°С, предпочтительно от 400°С до 600°С, в течение периода обжига, обычно находящегося в диапазоне от 1 часа до 10 часов, предпочтительно от 3 часов до 6 часов. Кроме того, обжиг обычно проводят в кислородсодержащей атмосфере, такой как атмосфера воздуха или кислорода.
В соответствии с одним аспектом настоящего изобретения, если желательно, молекулярное сито в соответствии с настоящим изобретением или любое молекулярное сито, полученное с помощью способа получения молекулярного сита в соответствии с настоящим изобретением (в контексте настоящего описания оба в совокупности называются как молекулярное сито настоящего изобретения или молекулярное сито в соответствии с настоящим изобретением), могут дополнительно быть подвергнуты ионному обмену с помощью любых стандартных средств, известных в данной области техники, например, с использованием процесса ионного обмена или пропитки раствором (см., например, патенты США 3140249 и 3140253 и т.д.), так что катион металла (такой как ион Na или ион K в зависимости от способа получения), содержащийся в нем, может быть полностью или частично заменен другим катионом(ами). Примеры указанного другого катиона включают ион водорода, другие ионы щелочных металлов (включая ион K, ион Rb и т.д.), ионы аммония (включая ион NH4, ионы четвертичного аммония, такие как ион тетраметиламмония и ион тетраэтиламмония и т.д.), ионы щелочноземельных металлов (включая ион Mg, ион Ca), ион Mn, ион Zn, ион Cd, ионы благородных металлов (включая ион Pt, ион Pd, ион Rh и т.д.), ион Ni, ион Co, ион Ti, ион Sn, ион Fe и/или ионы редкоземельных металлов и подобные.
Молекулярное сито в соответствии с настоящим изобретением может быть дополнительно обработано разбавленным раствором кислоты или подобным по мере необходимости для увеличения соотношения диоксид кремния - оксид алюминия или обработано водяным паром для улучшения устойчивости кристаллов молекулярного сита к воздействию кислот.
Молекулярное сито в соответствии с настоящим изобретением обладает хорошей термической/гидротермической стабильностью и имеет больший объем пор. Таким образом, молекулярное сито в соответствии с настоящим изобретением способно адсорбировать больше/более крупные молекулы, тем самым демонстрируя превосходные адсорбционные/каталитические свойства.
Молекулярное сито в соответствии с настоящим изобретением имеет относительно сильную кислотность, в частности большое количество L-кислотных центров. Данное молекулярное сито не было получено в предшествующем уровне техники. В результате молекулярное сито в соответствии с настоящим изобретением демонстрирует лучшую производительность, в частности в катализируемой кислотой реакции.
Молекулярное сито в соответствии с настоящим изобретением может находиться в любой физической форме, такой как порошки, гранулы или формованные изделия (например, полоски, клеверы и т.д.). Данные физические формы могут быть получены любым стандартным способом, известным в данной области техники, и без конкретного ограничения.
Второй вариант осуществления
В соответствии с одним аспектом настоящего изобретения предоставляют молекулярное сито, имеющее рентгеновскую дифрактограмму по существу, как показано в следующей таблице.
В соответствии с одним аспектом настоящего изобретения предпочтительно, что рентгеновская дифрактограмма молекулярного сита дополнительно включает рентгенодифракционный пик по существу, как показано в следующей таблице.
В соответствии с одним аспектом настоящего изобретения предпочтительно, что рентгеновская дифрактограмма молекулярного сита дополнительно включает рентгенодифракционный пик по существу, как показано в следующей таблице.
В соответствии с одним аспектом настоящего изобретения молекулярное сито (в отношении его монокристаллической частицы) имеет морфологию кристаллической частицы губчатой структуры, в частности морфологию нативной кристаллической частицы губчатой структуры при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «морфология кристаллической частицы» относится к (общей) внешней форме, проявляемой монокристаллической частицей молекулярного сита в области наблюдения сканирующего электронного микроскопа. Кроме того, термин «нативный» относится к структуре молекулярного сита, объективно и непосредственно представленной после получения, а не к структуре молекулярного сита, представленной после искусственной обработки после получения.
Авторы настоящего изобретения обнаружили посредством тщательного исследования, что молекулярное сито, имеющее как вышеупомянутую определенную рентгеновскую дифрактограмму, так и вышеупомянутую морфологию определенных (нативных) кристаллических частиц, не было получено в предшествующем уровне техники.
В соответствии с одним аспектом настоящего изобретения губчатая структура обычно содержит микропоры (поры в каркасе). Это является неотъемлемым свойством молекулярных сит в качестве микропористых материалов.
В соответствии с одним аспектом настоящего изобретения диаметр (средний диаметр) микропор обычно находится в диапазоне от 0,5 нм до менее 2 нм. В предпочтительном случае микропоры имеют диаметр в диапазоне от 0,5 нм до 0,8 нм или от 1,1 нм до 1,8 нм. В более предпочтительном случае диаметр микропор демонстрирует бимодальное распределение, включая оба диаметра в диапазоне от 0,5 нм до 0,8 нм и от 1,1 нм до 1,8 нм. В настоящем описании диаметр получен с использованием метода адсорбции жидкого азота, рассчитанного по модели теории функционала плотности ТФП. Ввиду большого диаметра микропор, молекулярное сито в соответствии с настоящим изобретением считается ультрамакропористым молекулярным ситом.
В соответствии с одним аспектом настоящего изобретения общая удельная площадь поверхности микропор обычно находится в диапазоне от 100 м2⋅г-1 до 300 м2⋅г-1, предпочтительно от 150 м2⋅г-1 до 250 м2⋅г-1. В настоящем описании общую удельную площадь поверхности получают с использованием метода адсорбции жидкого азота, рассчитанного по модели БЭТ.
В соответствии с одним аспектом настоящего изобретения объем пор микропор обычно находится в диапазоне от 0,03 мл/г до 0,20 мл/г, предпочтительно от 0,05 мл/г до 0,15 мл/г. В настоящем описании объем пор измеряют с помощью метода Хорвата-Кавадзоэ. Кроме того, не будучи привязанными к какой-либо теории, авторы настоящего изобретения полагают, что объем пор микропор имеет такое низкое значение, поскольку исходное положение микропор было занято крупными порами и/или мезопорами, как описано ниже. Следовательно, если данные крупные поры и мезопоры заменяют микропорами, объем пор микропор будет иметь очень высокое значение.
В соответствии с другим вариантом осуществления настоящего изобретения губчатая структура может также содержать крупные поры при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). Это можно понять, например, со ссылкой на Фигуры V-11 (a) и V-11 (b). В настоящем описании Фигуры V-11 (a) и V-11 (b) предназначены только для иллюстрации настоящего изобретения и не предназначены для ограничения настоящего изобретения. В губчатой структуре молекулярного сита (монокристаллической частицы) настоящего изобретения крупные поры и микропоры сообщаются друг с другом и пересекаются друг с другом с образованием пористой структуры со сложной сетью. Данное крупнопористое ультрамакропористое молекулярное сито не было получено в предшествующем уровне техники. В результате молекулярное сито в соответствии с настоящим изобретением проявляет характеристики макропористых материалов в дополнение к характеристикам микропористых материалов.
В соответствии с другим вариантом осуществления настоящего изобретения губчатая структура может также содержать мезопоры при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В губчатой структуре молекулярного сита (монокристаллической частицы) настоящего изобретения мезопоры и микропоры сообщаются друг с другом и пересекаются друг с другом с образованием пористой структуры со сложной сетью. Данное мезопористое ультрамакропористое молекулярное сито не было получено в предшествующем уровне техники. В результате молекулярное сито в соответствии с настоящим изобретением проявляет характеристики мезопористых материалов в дополнение к характеристикам микропористых материалов.
В соответствии с другим вариантом осуществления настоящего изобретения губчатая структура может также содержать как крупные поры, так и мезопоры при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). Данное многопористое ультрамакропористое молекулярное сито не было получено в предшествующем уровне техники. В результате молекулярное сито в соответствии с настоящим изобретением проявляет характеристики микропористых материалов в сочетании с характеристиками макропористых материалов и мезопористых материалов.
В соответствии с другим вариантом осуществления настоящего изобретения крупные поры открываются на одной торцевой поверхности или на обеих торцевых поверхностях губчатой структуры (в данном случае крупная пора становится полным сквозным отверстием или полусквозным отверстием) при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В данном случае губчатая структура может проявлять, например, морфологию кристаллической частицы, подобную сотообразному углю. Кроме того, губчатая структура имеет открытую или полуоткрытую пористую губчатую структуру. Кроме того, крупные поры также могут быть открыты на одной или более боковых поверхностях губчатой структуры, что приводит к выемчатой боковой поверхности(ям), при этом дополнительно увеличивая проницаемость губчатой структуры.
В соответствии с другим вариантом осуществления настоящего изобретения при наблюдении с использованием сканирующего электронного микроскопа (СЭМ) мезопоры открываются на одной или обеих торцевых поверхностях губчатой структуры (в данном случае мезопора становится полным сквозным отверстием или полусквозным отверстием). В данном случае губчатая структура может проявлять, например, морфологию кристаллической частицы, подобную сотообразному углю. Кроме того, открытая пористая губчатая структура имеет открытую пористую или полуоткрытую пористую губчатую структуру. Кроме того, мезопоры также могут быть открыты на одной или более боковых поверхностях губчатой структуры, что приводит к выемчатой боковой поверхности(ям), при этом дополнительно увеличивая проницаемость губчатой структуры.
В соответствии с одним аспектом настоящего изобретения диаметр (средний диаметр) крупных пор обычно находится в диапазоне от 80 нм до 2 мкм, предпочтительно от 80 нм до 1,5 мкм. В настоящем описании диаметр измеряют с помощью метода интрузии ртути.
В соответствии с одним аспектом настоящего изобретения общая удельная площадь поверхности крупных пор обычно находится в диапазоне от 10 м2⋅г-1 до 100 м2⋅г-1, предпочтительно от 50 м2⋅г-1 до 100 м2⋅г-1. В настоящем описании общую удельную площадь поверхности измеряют с помощью метода интрузии ртути.
В соответствии с одним аспектом настоящего изобретения объем пор крупных пор обычно находится в диапазоне от 0,5 мл/г до 3,0 мл/г, предпочтительно от 1,0 мл/г до 2,0 мл/г. В настоящем описании объем пор измеряют с помощью метода интрузии ртути.
В соответствии с одним аспектом настоящего изобретения диаметр (средний диаметр) мезопор обычно находится в диапазоне от 2 нм до 30 нм. В предпочтительном случае мезопоры имеют диаметр в диапазоне от 2 нм до 4 нм или от 7 нм до 15 нм, и последний является более предпочтительным от 8 нм до 9 нм. В более предпочтительном случае диаметр мезопор имеет бимодальное распределение, включая оба диаметра в диапазоне от 2 нм до 4 нм и от 7 нм до 15 нм. В настоящем описании диаметр получают с использованием метода адсорбции жидкого азота, рассчитанного по модели БЭТ.
В соответствии с одним аспектом настоящего изобретения общая удельная площадь поверхности мезопор обычно находится в диапазоне от 50 м2⋅г-1 до 250 м2⋅г-1, предпочтительно от 100 м2⋅г-1 до 150 м2⋅г-1. В настоящем описании общую удельную площадь поверхности получают с использованием метода адсорбции жидкого азота, рассчитанного по модели БЭТ.
В соответствии с одним аспектом настоящего изобретения объем пор мезопор обычно находится в диапазоне от 0,05 мл/г до 0,40 мл/г, предпочтительно от 0,15 мл/г до 0,30 мл/г. В настоящем описании объем пор получают с использованием метода адсорбции жидкого азота, рассчитанного по модели БЭТ.
В соответствии с другим вариантом осуществления настоящего изобретения губчатая структура содержит крупные поры, мезопоры и микропоры, как в то же время описано ранее. В губчатой структуре молекулярного сита (монокристаллической частицы) настоящего изобретения крупные поры, мезопоры и микропоры сообщаются друг с другом и пересекаются друг с другом с образованием пористой структуры со сложной сетью. Данное многопористое ультрамакропористое молекулярное сито не было получено в предшествующем уровне техники. В результате молекулярное сито в соответствии с настоящим изобретением проявляет характеристики микропористых материалов в сочетании с характеристиками мезопористых материалов и/или макропористых материалов, так что оно способно адсорбировать больше/более крупные молекулы, тем самым демонстрируя превосходную адсорбционную/каталитическую производительность.
В соответствии с одним аспектом настоящего изобретения молекулярное сито обычно также имеет морфологию столбчатой кристаллической частицы при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании в качестве столбчатой формы предпочтительной является призматическая форма, в частности гексагональная призматическая форма. Кроме того, призма относится к выпуклой призме и обычно относится к прямоугольной призме и правильной многоугольной призме (такой как правильная гексагональная призма). Следует особо отметить, что поскольку процесс роста кристалла молекулярного сита может быть нарушен различными факторами, может быть определенная степень отклонения, когда его реальная морфология кристаллической частицы сравнивается с (истинно) прямоугольной призмой или (истинно) правильной многоугольной призмой в геометрическом смысле, такое как отклонение 30%, 20% или 5%, что приводит к получению скошенной призмы или неправильной многоугольной (или даже искривленной многоугольной) призмы, но настоящее изобретение не предназначено для определения степени отклонения. Более того, любые большие или меньшие отклонения не отступают от объема настоящего изобретения.
В соответствии с одним аспектом настоящего изобретения молекулярное сито (монокристаллическая частица) имеет эффективный диаметр, обычно находящийся в диапазоне от 100 нм до 5000 нм, предпочтительно от 1000 нм до 3000 нм, при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «эффективный диаметр» означает, что две точки произвольно выбираются в поперечном сечении молекулярного сита (монокристаллической частицы) вдоль контура (края) поперечного сечения и измеряется линейное расстояние между двумя точками, где наибольшее линейное расстояние принимается за эффективный диаметр. Если профиль поперечного сечения молекулярного сита представлен как многоугольник, такой как шестиугольник, эффективный диаметр обычно относится к линейному расстоянию между двумя вершинами многоугольника, расположенными дальше друг от друга, т.е. диагональному расстоянию. Вкратце, эффективный диаметр по существу соответствует диаметру описанной окружности многоугольника, представляющего контур поперечного сечения.
В соответствии с одним аспектом настоящего изобретения молекулярное сито может иметь морфологию полых столбчатых кристаллических частиц, где диаметр крупных пор является достаточно большим (например, близко к эффективному диаметру молекулярного сита). Это можно понять, например, со ссылкой на Фигуры V-12(a) и V-12(b). В настоящем описании Фигуры V-12(a) и V-12(b) предназначены только для иллюстрации настоящего изобретения и не предназначены для ограничения настоящего изобретения. В настоящем описании термин «полый столбчатый» означает трубчатую структуру. В настоящем описании толщина стенки трубчатой структуры может составлять, например, от 50 нм до 400 нм, но настоящее изобретение не ограничивается ими и не предназначено для указания толщины стенки.
В соответствии с одним аспектом настоящего изобретения молекулярное сито (монокристаллическая частица) имеет высоту, обычно находящуюся в диапазоне от 500 нм до 3000 нм, предпочтительно от 1000 нм до 3000 нм, при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «высота» относится к линейному расстоянию между центрами двух торцевых поверхностей монокристаллической частицы (столбчатой кристаллической частицы) молекулярного сита. В обычном случае две торцевые поверхности столбца молекулярного сита по существу параллельны друг другу, и линейное расстояние представляет собой вертикальное расстояние между двумя торцевыми поверхностями, но настоящее изобретение не ограничивается этим.
В соответствии с одним аспектом настоящего изобретения молекулярное сито (монокристаллическая частица) имеет соотношение сторон, обычно находящееся в диапазоне от 1/3 до 5, предпочтительно от 1/3 до 3 при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «соотношение сторон» относится к соотношению высоты и эффективного диаметра.
В соответствии с одним аспектом настоящего изобретения молекулярное сито обычно имеет схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид». Известно, что молекулярные сита иногда могут содержать определенное количество влаги (особенно сразу после их синтеза), но в настоящей заявке не считается необходимым указывать количество влаги, поскольку наличие или отсутствие влаги не оказывает существенного влияния на спектр рентгеновской дифракции молекулярного сита. Ввиду этого схематическая химическая композиция фактически представляет собой не содержащую воду химическую композицию молекулярного сита. Кроме того, очевидно, что схематическая химическая композиция представляет собой химическую композицию каркаса молекулярного сита.
В соответствии с одним аспектом настоящего изобретения молекулярное сито может обычно дополнительно содержать в своей композиции органический темплат и воду и т.д., такие как те, которые заполнены в его порах сразу после его синтеза. Таким образом, молекулярное сито может иногда иметь схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид ⋅ органический темплат ⋅ вода». В настоящем описании молекулярное сито, имеющее схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид ⋅ органический темплат ⋅ вода», может быть подвергнуто обжигу для удаления любого органического темплата и воды, присутствующей в его порах, так что может быть получено молекулярное сито, имеющее схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид». Кроме того, обжиг может быть осуществлен любым общепринятым способом, известным в данной области техники, например, при температуре обжига, обычно находящейся в диапазоне от 300°С до 750°С, предпочтительно от 400°С до 600°С, в течение периода обжига, обычно находящегося в диапазоне от 1 часа до 10 часов, предпочтительно от 3 часов до 6 часов. Кроме того, обжиг обычно проводят в кислородсодержащей атмосфере, такой как атмосфера воздуха или кислорода.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции первый оксид обычно представляет собой четырехвалентный оксид и может представлять собой, например, по меньшей мере, один, выбранный из группы, состоящей из диоксида кремния, диоксида германия, диоксида олова, диоксида титана и диоксида циркония, предпочтительно диоксида кремния (SiO2) или комбинации диоксида кремния и диоксида германия. Указанные первые оксиды могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из первых оксидов может представлять собой, например, от 20:200 до 35:100. В качестве примера комбинации диоксид кремния и диоксид германия можно использовать в комбинации, в данном случае молярное соотношение между диоксидом кремния и диоксидом германия может представлять собой, например, от 20:200 до 35:100.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции второй оксид обычно представляет собой трехвалентный оксид и может представлять собой, например, по меньшей мере, один, выбранный из группы, состоящей из оксида алюминия, оксида бора, оксида железа, оксида галлия, оксида редкоземельного элемента, оксида индия и оксида ванадия, предпочтительно оксида алюминия (Al2O3). Указанные вторые оксиды могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из вторых оксидов может представлять собой, например, от 30:200 до 60:150.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции в качестве примера органического темплата может быть упомянут любой органический темплат, пригодный для получения молекулярных сит, и особенно может быть упомянут органический темплат, используемый при получении молекулярного сита в соответствии с настоящим вариантом осуществления (см. подробное описание ниже). Указанные органические темплаты могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. В частности, примеры органического темплата включают соединения, представленные следующей формулой (I).
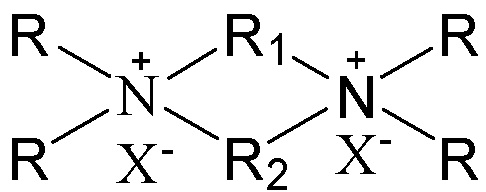 (I)
(I)
В соответствии с одним аспектом настоящего изобретения в формуле (I) группы R1 и R2 являются отличными друг от друга, одна из которых выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных или разветвленных оксаалкиленовых групп, множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, и Х представляет собой ОН.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции молярное соотношение первого оксида и второго оксида (например, молярное соотношение SiO2 и Al2O3) обычно находится в диапазоне от 30 до 100, предпочтительно от 55 до 100.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции молярное соотношение воды и первого оксида обычно находится в диапазоне от 5 до 50, предпочтительно от 5 до 15.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции молярное соотношение органического темплата и первого оксида обычно находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,15 до 0,5 или от 0,3 до 0,5.
В соответствии с одним аспектом настоящего изобретения в зависимости от исходных материалов, используемых в способе получения, молекулярное сито может иногда дополнительно содержать катион металла, такой как катион щелочного металла и/или щелочноземельного металла в качестве компонента в его композиции (как правило, заполненный в его поры). В данном случае содержание катиона металла, например, массовое соотношение катиона металла и первого оксида обычно находится в диапазоне от 0 до 0,02, предпочтительно от 0,0002 до 0,006, но не ограничивается ими.
В соответствии с одним аспектом настоящего изобретения молекулярное сито может быть получено с помощью следующего способа. В настоящем описании способ получения включает стадию взаимодействия источника первого оксида, источника второго оксида, необязательного щелочного источника, органического темплата и воды в условиях кристаллизации с получением молекулярного сита (в дальнейшем именуемую как стадия взаимодействия).
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита стадия взаимодействия может быть проведена с помощью любого стандартного способа, известного в данной области техники, например, с использованием способа, включающего смешивание источника первого оксида, источника второго оксида, необязательного щелочного источника, органического темплата и воды и подвергание смеси кристаллизации в условиях кристаллизации.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия органический темплат содержит, по меньшей мере, соединение, представленное следующей формулой (I). В настоящем описании соединение, представленное формулой (I), может быть использовано отдельно или в комбинации двух или более из них в любом соотношении.
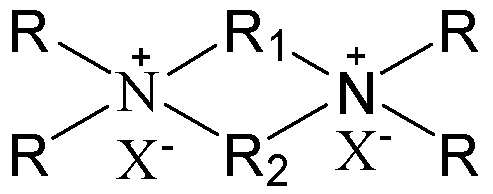 (I)
(I)
В соответствии с одним аспектом настоящего изобретения в формуле (I) группы R1 и R2 являются отличными друг от друга, одна из которых выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных или разветвленных оксаалкиленовых групп.
В соответствии с одним аспектом настоящего изобретения в качестве примера С3-12линейных или разветвленных алкиленовых групп может быть упомянута С3-12линейная алкиленовая группа, и ее определенные примеры включают н-пропилен, изопропилиден, н-бутилен, изобутилен, трет-бутилен, н-пентилен, изопентилен, неопентилен, н-гексилен, изогексилен, н-октилен, изооктилен, неооктилен, нонилен (или его изомер), децилен (или его изомер), ундецилен (или его изомер) или додецилен (или его изомер), предпочтительно н-пропилен, н-бутилен, н-пентилен, н-гексилен, н-гептилен, н-октилен, н-нонилен, н-децилен, н-ундецилен или н-додецилен.
В соответствии с одним аспектом настоящего изобретения в качестве С3-12линейных или разветвленных оксаалкиленовых групп, например, может быть упомянута С3-12линейная оксаалкиленовая группа, и ее конкретные примеры включают -(CH2)2-O-(CH2)-, -(CH2)2-O-(CH2)2-, -(CH2)-O-(CH2)3-, -(CH2)2-O-(CH2)3-, -(CH2)-O-пропилен-, -(CH2)-O-(CH2)4-, -(CH2)-O-(CH2)2-O-(CH2)-, -(CH2)-O-(CH2)2-O-(CH2)2-, -(CH2)-O-трет-бутилен-, -(CH2)2-O-(CH2)4-, -(CH2)3-O-(CH2)3-, -(CH2)-O-неопентилен-, -(CH2)2-O-(CH2)6-, -(CH2)2-O-(CH2)7-, -(CH2)-O-(CH2)8-, -(CH2)-O-изооктилен-, -(CH2)-O-(CH2)10-, -(CH2)2-O-децилен или его изомер, -(CH2)-O-(CH2)6-, -(CH2)-O-(CH2)7-, -(CH2)-O-(CH2)8-, -(CH2)-O-(CH2)11-, -(CH2)-O-(CH2)2-O-(CH2)-, -(CH2)2-O-(CH2)2-O-(CH2)2-, -(CH2)2-O-(CH2)4-O-(CH2)2-, -(CH2)2-O-(CH2)6-O-(CH2)2- или -(CH2)2-O-(CH2)8-O-(CH2)2-. Кроме того, в качестве С3-12линейных оксаалкиленовых групп может быть упомянута С4-6линейная оксаалкиленовая группа, и в частности может быть упомянута С4-6линейная монооксаалкиленовая группа, и особенно может быть упомянута монооксаалкиленовая группа, представленная формулой -(CH2)m-O-(CH2)m- (где каждое значение m, являющееся одинаковым или отличным друг от друга, независимо представляет собой 2 или 3, например, 2). Более определенно могут быть упомянуты -(CH2)2-O-(CH2)2-, -(CH2)2-O-(CH2)3-, -(CH2)3-O-(CH2)3- или -(CH2)2-O-(CH2)4-.
В соответствии с одним аспектом настоящего изобретения в формуле (I) множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, предпочтительно каждая независимо выбрана из группы, состоящей из метила и этила, более предпочтительно всего метила.
В соответствии с одним аспектом настоящего изобретения в формуле (I) X представляет собой ОН.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение органического темплата и источника первого оксида (рассчитанное на основе первого оксида) обычно находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,15 до 0,5 или от 0,3 до 0,5.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в дополнение к соединению, представленному формулой (I), другие органические темплаты, обычно используемые в данной области техники для получения молекулярных сит, могут быть дополнительно использованы в качестве органического темплата. Предпочтительно на стадии взаимодействия только соединение, представленное формулой (I), используют в качестве органического темплата. В настоящем описании соединение, представленное формулой (I), может быть использовано отдельно или в комбинации двух или более из них в любом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия источник первого оксида обычно представляет собой источник четырехвалентного оксида и может представлять собой, например, по меньшей мере, один, выбранный из группы, состоящей из источника диоксида кремния, источника диоксида германия, источника диоксида олова, источника диоксида титана и источника диоксида циркония, предпочтительно источника диоксида кремния (SiO2) или комбинации источника диоксида кремния и источника диоксида германия. Указанные источники первого оксида могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из источников первого оксида может представлять собой, например, от 20:200 до 35:100. В качестве примера комбинации может быть упомянута комбинация источника диоксида кремния и источника диоксида германия, в данном случае молярное соотношение между источником диоксида кремния и источником диоксида германия может представлять собой, например, от 20:200 до 35:100.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве источника первого оксида может быть использован любой соответствующий источник оксида, обычно используемый для этой цели в данной области техники, включая, но не ограничиваясь ими, в том числе оксиды, гидроксиды, алкоксиды, соли оксикислот металлов, ацетаты, оксалаты, соли аммония, сульфаты, галогенированные соли и нитраты соответствующего металла в первом оксиде. Например, когда первый оксид представляет собой диоксид кремния, примеры источника первого оксида включают золь кремниевой кислоты, неочищенный силикагель, тетраэтилортосиликат, жидкое стекло, белую сажу, кремниевую кислоту, силикагель, силикат калия или подобное. Когда первый оксид представляет собой диоксид германия, примеры источника первого оксида включают тетраалкоксид германия, оксид германия, нитрат германия или подобное. Когда первый оксид представляет собой диоксид олова, примеры источника первого оксида включают хлорид олова, сульфат олова, нитрат олова и подобное. Когда первый оксид представляет собой диоксид титана, примеры источника первого оксида включают тетраалкоксид титана, диоксид титана, нитрат титана и подобное. Когда первый оксид представляет собой диоксид циркония, примеры источника первого оксида включают сульфат циркония, хлорид циркония, нитрат циркония и подобное. Указанные источники первого оксида могут быть использованы отдельно или в комбинации двух или более из них в желаемом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия источник второго оксида обычно представляет собой источник трехвалентного оксида, такой как, по меньшей мере, один, выбранный из группы, состоящей из источника оксида алюминия, источника оксида бора, источника оксида железа, источника оксида галлия, источника редкоземельного оксида, источника оксида индия и источника оксида ванадия, предпочтительно источника оксида алюминия (Al2O3). Указанные источники второго оксида могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из источников второго оксида может представлять собой, например, от 30:200 до 60:150.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве источника второго оксида может быть использован любой соответствующий источник оксида, обычно используемый для этой цели в данной области техники, включая, но не ограничиваясь ими, в том числе оксиды, гидроксиды, алкоксиды, соли оксикислот металлов, ацетаты, оксалаты, соли аммония, сульфаты, галогенированные соли и нитраты соответствующего металла во втором оксиде. Например, когда второй оксид представляет собой оксид алюминия, примеры источника второго оксида включают хлорид алюминия, сульфат алюминия, гидрат алюминия, метаалюминат натрия, золь алюминия, гидроксид алюминия или подобное. Когда второй оксид представляет собой оксид бора, примеры источника второго оксида включают борную кислоту, борат, буру, триоксид бора и подобное. Когда второй оксид представляет собой оксид железа, примеры источника второго оксида включают нитрат железа, хлорид железа, оксид железа и подобное. Когда второй оксид представляет собой оксид галлия, примеры источника второго оксида включают нитрат галлия, сульфат галлия, оксид галлия и подобное. Когда второй оксид представляет собой редкоземельный оксид, примеры источника второго оксида включают оксид лантана, оксид неодима, оксид иттрия, оксид церия, нитрат лантана, нитрат неодима, нитрат иттрия, сульфат церия-аммония и подобное. Когда второй оксид представляет собой оксид индия, примеры источника второго оксида включают хлорид индия, нитрат индия, оксид индия и подобное. Когда второй оксид представляет собой оксид ванадия, примеры источника второго оксида включают хлорид ванадия, метаванадат аммония, ванадат натрия, диоксид ванадия, ванадилсульфат и подобное. Указанные источники второго оксида могут быть использованы отдельно или в комбинации двух или более из них в желаемом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение источника первого оксида (рассчитанное на основе первого оксида, такого как SiO2) и источника второго оксида (рассчитанное на основе второго оксида, такого как Al2O3) обычно находится в диапазоне от 30 до 100, предпочтительно от 55 до 100.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение воды и источника первого оксида (рассчитанное на основе первого оксида) обычно находится в диапазоне от 5 до 50, предпочтительно от 5 до 15.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия щелочной источник может использоваться или не использоваться. Когда щелочной источник не преднамеренно используется, группа X, содержащаяся в соединении, представленном формулой (I), может быть использована для обеспечения ОН-, требуемого в настоящем описании. В настоящем описании в качестве щелочного источника может быть использован любой щелочной источник, обычно применяемый для этой цели в данной области техники, включая, но не ограничиваясь ими, неорганические основания, содержащие щелочной металл или щелочноземельный металл в качестве катиона, в частности гидроксид натрия и гидроксид калия. Указанные щелочные источники могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение щелочного источника (рассчитанное на основе OH-) и источника первого оксида (рассчитанное на основе первого оксида) обычно находится в диапазоне от 0 до 1, предпочтительно от 0,04 до 1, от 0,1 до 1, от 0,2 до 1, от 0,3 до 0,7 или от 0,45 до 0,7.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве условия кристаллизации температура кристаллизации обычно находится в диапазоне от 80°С до 120°С, предпочтительно от 120°С до 170°С или от 120°С до 200°С.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве условия кристаллизации период кристаллизации обычно составляет, по меньшей мере, 1 день, предпочтительно, по меньшей мере, 2 дня, предпочтительно от 3 дней до 8 дней, от 5 дней до 8 дней или от 4 дней до 6 дней.
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита после завершения стадии взаимодействия молекулярное сито может быть отделено в виде продукта из полученной реакционной смеси с помощью любого общеизвестного способа разделения. В настоящем описании продукт молекулярного сита содержит молекулярное сито в соответствии с настоящим изобретением. Кроме того, в качестве способа разделения, например, может быть упомянут способ, включающий фильтрование, промывание и сушку полученной реакционной смеси.
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита фильтрование, промывание и сушка могут быть проведены с помощью любого стандартного способа, известного в данной области техники. В качестве примера фильтрования, например, полученная реакционная смесь может быть просто подвергнута фильтрованию с отсасыванием. В качестве примера промывания, например, промывание деионизированной водой может проводиться до тех пор, пока рН фильтрата не достигнет 7-9, предпочтительно 8-9. Температура сушки составляет, например, от 40 до 250°С, предпочтительно от 60 до 150°С, и время сушки составляет, например, от 8 до 30 часов, предпочтительно от 10 до 20 часов. Сушка может быть проведена при нормальном давлении или при пониженном давлении.
В соответствии с одним аспектом настоящего изобретения способ получения молекулярного сита может дополнительно включать стадию обжига полученного молекулярного сита (в дальнейшем именуемую как стадия обжига) по мере необходимости для удаления органического темплата и возможной влаги и подобного, чтобы можно было получить кальцинированное молекулярное сито. В контексте настоящего описания молекулярные сита до и после обжига совместно называются как молекулярное сито настоящего изобретения или молекулярное сито в соответствии с настоящим изобретением.
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита обжиг может быть проведен с помощью любого стандартного способа, известного в данной области техники, например, при температуре обжига, обычно находящейся в диапазоне от 300°С до 750°С, предпочтительно от 400°С до 600°С, в течение периода обжига, обычно находящегося в диапазоне от 1 часа до 10 часов, предпочтительно от 3 часов до 6 часов. Кроме того, обжиг обычно проводят в кислородсодержащей атмосфере, такой как атмосфера воздуха или кислорода.
В соответствии с одним аспектом настоящего изобретения, если желательно, молекулярное сито в соответствии с настоящим изобретением или любое молекулярное сито, полученное с помощью способа получения молекулярного сита в соответствии с настоящим изобретением (в контексте настоящего описания оба в совокупности называются как молекулярное сито настоящего изобретения или молекулярное сито в соответствии с настоящим изобретением), могут дополнительно быть подвергнуты ионному обмену с помощью любых стандартных средств, известных в данной области техники, например, с использованием процесса ионного обмена или пропитки раствором (см., например, патенты США 3140249 и 3140253 и т.д.), так что катион металла (такой как ион Na или ион K в зависимости от способа получения), содержащийся в нем, может быть полностью или частично заменен другими катионами. Примеры указанного другого катиона включают ион водорода, другие ионы щелочных металлов (включая ион K, ион Rb и т.д.), ионы аммония (включая ион NH4, ионы четвертичного аммония, такие как ион тетраметиламмония и ион тетраэтиламмония и т.д.), ионы щелочноземельных металлов (включая ион Mg, ион Ca), ион Mn, ион Zn, ион Cd, ионы благородных металлов (включая ион Pt, ион Pd, ион Rh и т.д.), ион Ni, ион Co, ион Ti, ион Sn, ион Fe и/или ионы редкоземельных металлов и подобные.
Молекулярное сито в соответствии с настоящим изобретением может быть дополнительно обработано разбавленным раствором кислоты или подобным по мере необходимости для увеличения соотношения диоксид кремния-оксид алюминия или обработано водяным паром для улучшения устойчивости кристаллов молекулярного сита к воздействию кислот.
Молекулярное сито в соответствии с настоящим изобретением обладает хорошей термической/гидротермической стабильностью и имеет больший объем пор. Таким образом, молекулярное сито в соответствии с настоящим изобретением способно адсорбировать больше/более крупные молекулы, тем самым демонстрируя превосходные адсорбционные/каталитические свойства.
Молекулярное сито в соответствии с настоящим изобретением имеет относительно сильную кислотность, в частности большое количество L-кислотных центров. Данное молекулярное сито не было получено в предшествующем уровне техники. В результате молекулярное сито в соответствии с настоящим изобретением демонстрирует лучшую производительность, в частности в катализируемой кислотой реакции.
Молекулярное сито в соответствии с настоящим изобретением может находиться в любой физической форме, такой как порошки, гранулы или формованные изделия (например, полоски, клеверы и т.д.). Данные физические формы могут быть получены любым стандартным способом, известным в данной области техники, и без конкретного ограничения.
Третий вариант осуществления
В соответствии с одним аспектом настоящего изобретения предоставляют молекулярное сито, имеющее рентгеновскую дифрактограмму по существу, как показано в следующей таблице.
В соответствии с одним аспектом настоящего изобретения предпочтительно, что рентгеновская дифрактограмма молекулярного сита дополнительно включает рентгенодифракционный пик по существу, как показано в следующей таблице.
В соответствии с одним аспектом настоящего изобретения предпочтительно, что рентгеновская дифрактограмма молекулярного сита дополнительно включает рентгенодифракционный пик по существу, как показано в следующей таблице.
В соответствии с одним аспектом настоящего изобретения при наблюдении с использованием сканирующего электронного микроскопа (СЭМ) молекулярное сито (в отношении его монокристаллической частицы) имеет морфологию кристаллической частицы от плоской призматической формы до плоской цилиндрической формы, в частности морфологию нативной кристаллической частицы от плоской призматической формы до плоской цилиндрической формы. В настоящем описании термин «морфология кристаллической частицы» относится к (общей) внешней форме, проявляемой монокристаллической частицей молекулярного сита в области наблюдения сканирующего электронного микроскопа. Термин «нативный» относится к морфологии молекулярного сита, объективно и непосредственно представленной после получения, а не к морфологии молекулярного сита, представленной после искусственной обработки после получения. Термин «призма» относится к выпуклой призме и обычно относится к прямоугольной призме и правильной многоугольной призме (такой как правильная гексагональная призма). Следует особо отметить, что поскольку процесс роста кристалла молекулярного сита может быть нарушен различными факторами, может быть определенная степень отклонения, когда его реальная морфология кристаллической частицы сравнивается с (истинно) прямоугольной призмой или (истинно) правильной многоугольной призмой в геометрическом смысле, такое как отклонение 30%, 20% или 5%, что приводит к получению скошенной призмы или неправильной многоугольной (или даже искривленной многоугольной) призмы, но настоящее изобретение не предназначено для определения степени отклонения. Более того, любые большие или меньшие отклонения не отступают от объема настоящего изобретения. Термин «плоский» означает, что соотношение высоты и ширины (или диаметра) (например, соотношение сторон, как описано ниже) представляет собой менее единицы. Выражение «от плоской призматической формы до плоской цилиндрической формы» означает, что морфология кристаллической частицы молекулярного сита может представлять собой плоскую призматическую форму, плоскую цилиндрическую форму или любую промежуточную форму между плоской призматической формой и плоской цилиндрической формой. Примеры промежуточной формы включают форму, полученную с помощью закругления одного или более краев плоской призмы. Очевидно, что с помощью закругления всех краев плоской призмы можно получить плоский цилиндр.
В соответствии с другим вариантом осуществления настоящего изобретения, как описано выше, молекулярное сито имеет морфологию столбчатой кристаллической частицы при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). Продольный разрез столбца может быть получен, если он продольно разрезан вдоль его осевой линии. Продольный разрез имеет контуры торцевых поверхностей на верхней и нижней сторонах (диапазон вне области, окруженной двумя ломаными линиями) и боковые контуры на левой и правой сторонах (диапазон в пределах области, окруженной двумя ломаными линиями). Молекулярное сито настоящего другого варианта осуществления уникально тем, что один или оба из двух контуров торцевой поверхности имеют выпуклую форму, то есть радиус его кривизны имеет положительное значение. Настоящее изобретение не предназначено для указания диапазона значений радиуса кривизны в случае, если оно имеет положительное значение. В качестве альтернативы можно сказать, что молекулярное сито другого варианта осуществления обычно имеет внешнюю форму, полученную с помощью закругления или скашивания кромки одной торцевой поверхности или обеих торцевых поверхностей столбца. Это можно понять, например, со ссылкой на Фигуры VI-14(a) и VI-14(b). В настоящем описании Фигуры VI-14(a) и VI-II(b) предоставлены только для иллюстрации настоящего изобретения и не предназначены для ограничения настоящего изобретения. Кроме того, на Фигуре VI-14(c) представлен пример, когда контур торцевой поверхности не имеет выпуклой формы, а имеет плоскую форму.
Авторы настоящего изобретения обнаружили посредством тщательного исследования, что молекулярное сито, имеющее как вышеупомянутую определенную рентгеновскую дифрактограмму, так и вышеупомянутую морфологию определенных (нативных) кристаллических частиц, не было получено в предшествующем уровне техники.
В соответствии с одним аспектом настоящего изобретения молекулярное сито (монокристаллическая частица) имеет эффективный диаметр, обычно находящийся в диапазоне от 100 нм до 1000 нм, предпочтительно от 100 нм до 500 нм, при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «эффективный диаметр» означает, что две точки произвольно выбираются в поперечном сечении молекулярного сита (монокристаллической частицы) вдоль контура (края) поперечного сечения и измеряется линейное расстояние между двумя точками, где наибольшее линейное расстояние принимается за эффективный диаметр. Если профиль поперечного сечения молекулярного сита представлен как многоугольник, такой как шестиугольник, эффективный диаметр обычно относится к линейному расстоянию между двумя вершинами многоугольника, расположенными дальше друг от друга, т.е. диагональному расстоянию. Вкратце, эффективный диаметр по существу соответствует диаметру описанной окружности многоугольника, представляющего контур поперечного сечения. Альтернативно, если профиль поперечного сечения молекулярного сита представлен в виде круга, эффективный диаметр относится к диаметру круга.
В соответствии с одним аспектом настоящего изобретения молекулярное сито (монокристаллическая частица) имеет высоту, обычно находящуюся в диапазоне от 100 нм до 1000 нм, предпочтительно от 150 нм до 300 нм, при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «высота» относится к линейному расстоянию между центрами двух торцевых поверхностей монокристаллической частицы (столбчатой кристаллической частицы) молекулярного сита.
В соответствии с одним аспектом настоящего изобретения молекулярное сито (монокристаллическая частица) имеет соотношение сторон, обычно находящееся в диапазоне от 0,1 до 0,9, предпочтительно от 0,4 до 0,7, при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «соотношение сторон» относится к соотношению высоты и эффективного диаметра. Молекулярное сито, имеющее как вышеупомянутую определенную рентгеновскую дифрактограмму, так и вышеупомянутое определенное соотношение сторон, не было получено в предшествующем уровне техники. Например, морфология кристаллической частицы молекулярного сита в данном случае аналогична таблетке для перорального применения.
В соответствии с одним аспектом настоящего изобретения молекулярное сито имеет общую удельную площадь поверхности, обычно находящуюся в диапазоне от 400 м2⋅г-1 до 600 м2⋅г-1, предпочтительно от 450 м2⋅г-1 до 580 м2⋅г-1. В настоящем описании общую удельную площадь поверхности получают с использованием метода адсорбции жидкого азота, рассчитанного по модели БЭТ.
В соответствии с одним аспектом настоящего изобретения молекулярное сито имеет объем пор, обычно находящийся в диапазоне от 0,3 мл/г до 0,5 мл/г, предпочтительно от 0,30 мл/г до 0,40 мл/г. Молекулярное сито в соответствии с настоящим изобретением имеет очень большой объем пор, что указывает на то, что оно относится к ультрамакропористому молекулярному ситу. В настоящем описании объем пор получают с использованием низкотемпературной адсорбции азота, рассчитанной по модели БЭТ.
В соответствии с одним аспектом настоящего изобретения молекулярное сито может иметь схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид». Известно, что молекулярные сита иногда могут содержать определенное количество влаги (особенно сразу после их синтеза), но в настоящей заявке не считается необходимым указывать количество влаги, поскольку наличие или отсутствие влаги не оказывает существенного влияния на спектр рентгеновской дифракции молекулярного сита. Ввиду этого схематическая химическая композиция фактически представляет собой не содержащую воду химическую композицию молекулярного сита. Кроме того, очевидно, что схематическая химическая композиция представляет собой химическую композицию каркаса молекулярного сита.
В соответствии с одним аспектом настоящего изобретения молекулярное сито может обычно дополнительно содержать в своей композиции органический темплат и воду и т.д., такие как те, которые заполнены в его порах сразу после его синтеза. Таким образом, молекулярное сито может иногда иметь схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид ⋅ органический темплат ⋅ вода». В настоящем описании молекулярное сито, имеющее схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид ⋅ органический темплат ⋅ вода», может быть подвергнуто обжигу для удаления любого органического темплата и воды, присутствующей в его порах, так что может быть получено молекулярное сито, имеющее схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид». Кроме того, обжиг может быть осуществлен любым общепринятым способом, известным в данной области техники, например, при температуре обжига, обычно находящейся в диапазоне от 300°С до 750°С, предпочтительно от 400°С до 600°С, в течение периода обжига, обычно находящегося в диапазоне от 1 часа до 10 часов, предпочтительно от 3 часов до 6 часов. Кроме того, обжиг обычно проводят в кислородсодержащей атмосфере, такой как атмосфера воздуха или кислорода.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции первый оксид обычно представляет собой четырехвалентный оксид и может представлять собой, например, по меньшей мере, один, выбранный из группы, состоящей из диоксида кремния, диоксида германия, диоксида олова, диоксида титана и диоксида циркония, предпочтительно диоксида кремния (SiO2) или комбинации диоксида кремния и диоксида германия. Указанные первые оксиды могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из первых оксидов может представлять собой, например, от 20:200 до 35:100. В качестве примера комбинации диоксид кремния и диоксид германия можно использовать в комбинации, в данном случае молярное соотношение между диоксидом кремния и диоксидом германия может представлять собой, например, от 20:200 до 35:100.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции второй оксид обычно представляет собой трехвалентный оксид и может представлять собой, например, по меньшей мере, один, выбранный из группы, состоящей из оксида алюминия, оксида бора, оксида железа, оксида галлия, оксида редкоземельного элемента, оксида индия и оксида ванадия, предпочтительно оксида алюминия (Al2O3). Указанные вторые оксиды могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из вторых оксидов может представлять собой, например, от 30:200 до 60:150.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции в качестве примера органического темплата может быть упомянут любой органический темплат, пригодный для получения молекулярных сит, и особенно может быть упомянут органический темплат, используемый при получении молекулярного сита в соответствии с настоящим вариантом осуществления (см. подробное описание ниже). Указанные органические темплаты могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. В частности, примеры органического темплата включают соединения, представленные следующей формулой (I).
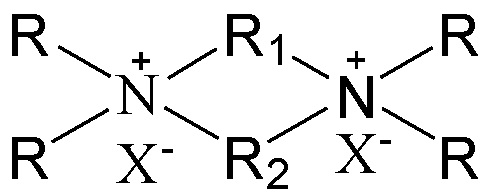 (I)
(I)
В соответствии с одним аспектом настоящего изобретения в формуле (I) группы R1 и R2 являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп, и множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, и X представляет собой ОН.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции молярное соотношение первого оксида и второго оксида (например, молярное соотношение SiO2 и Al2O3) обычно находится в диапазоне от 40 до 200, предпочтительно от 40 до 150.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции молярное соотношение воды и первого оксида обычно находится в диапазоне от 5 до 50, предпочтительно от 5 до 15.
В соответствии с одним аспектом настоящего изобретения в вышеупомянутой схематической химической композиции молярное соотношение органического темплата и первого оксида обычно находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,15 до 0,5 или от 0,3 до 0,5.
В соответствии с одним аспектом настоящего изобретения в зависимости от исходных материалов, используемых в способе получения, молекулярное сито может иногда дополнительно содержать катион металла, такой как катион щелочного металла и/или щелочноземельного металла в качестве компонента в его композиции (как правило, заполненный в его поры). В данном случае содержание катиона металла, например, массовое соотношение катиона металла и первого оксида обычно находится в диапазоне от 0 до 0,02, предпочтительно от 0,0002 до 0,006, но не ограничивается ими.
В соответствии с одним аспектом настоящего изобретения молекулярное сито может быть получено с помощью следующего способа. В настоящем описании способ получения включает стадию взаимодействия источника первого оксида, источника второго оксида, необязательного щелочного источника, органического темплата и воды в условиях кристаллизации с получением молекулярного сита (в дальнейшем именуемую как стадия взаимодействия).
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита стадия взаимодействия может быть проведена с помощью любого стандартного способа, известного в данной области техники, например, с использованием способа, включающего смешивание источника первого оксида, источника второго оксида, необязательного щелочного источника, органического темплата и воды и подвергание смеси кристаллизации в условиях кристаллизации.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия органический темплат содержит, по меньшей мере, соединение, представленное следующей формулой (I). В настоящем описании соединение, представленное формулой (I), может быть использовано отдельно или в комбинации двух или более из них в любом соотношении.
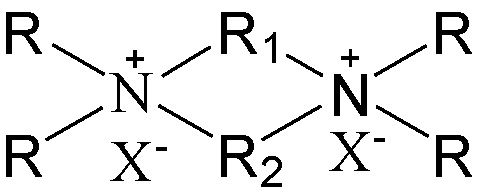 (I)
(I)
В соответствии с одним аспектом настоящего изобретения в формуле (I) группы R1 и R2 являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп.
В соответствии с другим вариантом осуществления настоящего изобретения в формуле (I) группы R1 и R2 являются одинаковыми или отличными друг от друга, одна из которых выбрана из группы, состоящей из С3-12линейных алкиленовых групп, и другая выбрана из группы, состоящей из С4-6линейных алкиленовых групп.
В соответствии с одним аспектом настоящего изобретения в качестве примера С3-12линейных или разветвленных алкиленовых групп может быть упомянута С3-12линейная алкиленовая группа, и ее определенные примеры включают н-пропилен, изопропилиден, н-бутилен, изобутилен, трет-бутилен, н-пентилен, изопентилен, неопентилен, н-гексилен, изогексилен, н-октилен, изооктилен, неооктилен, нонилен (или его изомер), децилен (или его изомер), ундецилен (или его изомер) или додецилен (или его изомер), предпочтительно н-пропилен, н-бутилен, н-пентилен, н-гексилен, н-гептилен, н-октилен, н-нонилен, н-децилен, н-ундецилен или н-додецилен.
В соответствии с одним аспектом настоящего изобретения в качестве С4-6линейных алкиленовых групп могут быть в частности упомянуты н-бутилен, н-пентилен или н-гексилен.
В соответствии с одним аспектом настоящего изобретения в формуле (I) множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, предпочтительно каждая независимо выбрана из группы, состоящей из метила и этила, более предпочтительно всего метила.
В соответствии с одним аспектом настоящего изобретения в формуле (I) X представляет собой ОН.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение органического темплата и источника первого оксида (рассчитанное на основе первого оксида) обычно находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,15 до 0,5 или от 0,3 до 0,5.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в дополнение к соединению, представленному формулой (I), другие органические темплаты, обычно используемые в данной области техники для получения молекулярных сит, могут быть дополнительно использованы в качестве органического темплата. Предпочтительно на стадии взаимодействия только соединение, представленное формулой (I), используют в качестве органического темплата. В настоящем описании соединение, представленное формулой (I), может быть использовано отдельно или в комбинации двух или более из них в любом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия источник первого оксида обычно представляет собой источник четырехвалентного оксида и может представлять собой, например, по меньшей мере, один, выбранный из группы, состоящей из источника диоксида кремния, источника диоксида германия, источника диоксида олова, источника диоксида титана и источника диоксида циркония, предпочтительно источника диоксида кремния (SiO2) или комбинации источника диоксида кремния и источника диоксида германия. Указанные источники первого оксида могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из источников первого оксида может представлять собой, например, от 30:200 до 60:150. В качестве примера комбинации может быть упомянута комбинация источника диоксида кремния и источника диоксида германия, в данном случае молярное соотношение между источником диоксида кремния и источником диоксида германия может представлять собой, например, от 20:200 до 35:100.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве источника первого оксида может быть использован любой соответствующий источник оксида, обычно используемый для этой цели в данной области техники, включая, но не ограничиваясь ими, в том числе оксиды, гидроксиды, алкоксиды, соли оксикислот металлов, ацетаты, оксалаты, соли аммония, сульфаты, галогенированные соли и нитраты соответствующего металла в первом оксиде. Например, когда первый оксид представляет собой диоксид кремния, примеры источника первого оксида включают золь кремниевой кислоты, неочищенный силикагель, тетраэтилортосиликат, жидкое стекло, белую сажу, кремниевую кислоту, силикагель, силикат калия или подобное. Когда первый оксид представляет собой диоксид германия, примеры источника первого оксида включают тетраалкоксид германия, оксид германия, нитрат германия или подобное. Когда первый оксид представляет собой диоксид олова, примеры источника первого оксида включают хлорид олова, сульфат олова, нитрат олова и подобное. Когда первый оксид представляет собой диоксид титана, примеры источника первого оксида включают тетраалкоксид титана, диоксид титана, нитрат титана и подобное. Когда первый оксид представляет собой диоксид циркония, примеры источника первого оксида включают хлорид циркония, сульфат циркония, нитрат циркония и подобное. Указанные источники первого оксида могут быть использованы отдельно или в комбинации двух или более из них в желаемом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия источник второго оксида обычно представляет собой источник трехвалентного оксида, такой как, по меньшей мере, один, выбранный из группы, состоящей из источника оксида алюминия, источника оксида бора, источника оксида железа, источника оксида галлия, источника редкоземельного оксида, источника оксида индия и источника оксида ванадия, предпочтительно источника оксида алюминия (Al2O3). Указанные источники второго оксида могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из источников второго оксида может представлять собой, например, от 20:200 до 35:100.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве источника второго оксида может быть использован любой соответствующий источник оксида, обычно используемый для этой цели в данной области техники, включая, но не ограничиваясь ими, в том числе оксиды, гидроксиды, алкоксиды, соли оксикислот металлов, ацетаты, оксалаты, соли аммония, сульфаты, галогенированные соли и нитраты соответствующего металла во втором оксиде. Например, когда второй оксид представляет собой оксид алюминия, примеры источника второго оксида включают хлорид алюминия, сульфат алюминия, гидрат алюминия, метаалюминат натрия, золь алюминия, гидроксид алюминия или подобное. Когда второй оксид представляет собой оксид бора, примеры источника второго оксида включают борную кислоту, борат, буру, триоксид бора и подобное. Когда второй оксид представляет собой оксид железа, примеры источника второго оксида включают нитрат железа, хлорид железа, оксид железа и подобное. Когда второй оксид представляет собой оксид галлия, примеры источника второго оксида включают нитрат галлия, сульфат галлия, оксид галлия и подобное. Когда второй оксид представляет собой редкоземельный оксид, примеры источника второго оксида включают оксид лантана, оксид неодима, оксид иттрия, оксид церия, нитрат лантана, нитрат неодима, нитрат иттрия, сульфат церия-аммония и подобное. Когда второй оксид представляет собой оксид индия, примеры источника второго оксида включают хлорид индия, нитрат индия, оксид индия и подобное. Когда второй оксид представляет собой оксид ванадия, примеры источника второго оксида включают хлорид ванадия, метаванадат аммония, ванадат натрия, диоксид ванадия, ванадилсульфат и подобное. Указанные источники второго оксида могут быть использованы отдельно или в комбинации двух или более из них в желаемом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение источника первого оксида (рассчитанное на основе первого оксида, такого как SiO2) и источника второго оксида (рассчитанное на основе второго оксида, такого как Al2O3) обычно находится в диапазоне от 40 до 200, предпочтительно от 40 до 150.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение воды и источника первого оксида (рассчитанное на основе первого оксида) обычно находится в диапазоне от 5 до 50, предпочтительно от 5 до 15.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия щелочной источник может использоваться или не использоваться. Когда щелочной источник не преднамеренно используется, группа X, содержащаяся в соединении, представленном формулой (I), может быть использована для обеспечения ОН-, требуемого в настоящем описании. В настоящем описании в качестве щелочного источника может быть использован любой щелочной источник, обычно применяемый для этой цели в данной области техники, включая, но не ограничиваясь ими, неорганические основания, содержащие щелочной металл или щелочноземельный металл в качестве катиона, в частности гидроксид натрия и гидроксид калия. Указанные щелочные источники могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение щелочного источника (рассчитанное на основе OH-) и источника первого оксида (рассчитанное на основе первого оксида) обычно находится в диапазоне от 0 до 1, предпочтительно от 0,04 до 1, от 0,1 до 1, от 0,2 до 1, от 0,3 до 0,7 или от 0,45 до 0,7.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве условия кристаллизации температура кристаллизации обычно находится в диапазоне от 80°С до 120°С, предпочтительно от 120°С до 170°С или от 120°С до 200°С.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве условия кристаллизации период кристаллизации обычно составляет, по меньшей мере, 1 день, предпочтительно, по меньшей мере, 2 дня, предпочтительно от 3 дней до 8 дней, от 5 дней до 8 дней или от 4 дней до 6 дней.
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита после завершения стадии взаимодействия молекулярное сито может быть отделено в виде продукта из полученной реакционной смеси с помощью любого общеизвестного способа разделения. В настоящем описании продукт молекулярного сита содержит молекулярное сито в соответствии с настоящим изобретением. Кроме того, в качестве способа разделения, например, может быть упомянут способ, включающий фильтрование, промывание и сушку полученной реакционной смеси.
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита фильтрование, промывание и сушка могут быть проведены с помощью любого стандартного способа, известного в данной области техники. В качестве примера фильтрования, например, полученная реакционная смесь может быть просто подвергнута фильтрованию с отсасыванием. В качестве примера промывания, например, промывание деионизированной водой может проводиться до тех пор, пока рН фильтрата не достигнет 7-9, предпочтительно 8-9. Температура сушки составляет, например, от 40 до 250°С, предпочтительно от 60 до 150°С, и время сушки составляет, например, от 8 до 30 часов, предпочтительно от 10 до 20 часов. Сушка может быть проведена при нормальном давлении или при пониженном давлении.
В соответствии с одним аспектом настоящего изобретения способ получения молекулярного сита может дополнительно включать стадию обжига полученного молекулярного сита (в дальнейшем именуемую как стадия обжига) по мере необходимости для удаления органического темплата и возможной влаги и подобного, чтобы можно было получить кальцинированное молекулярное сито. В контексте настоящего описания молекулярные сита до и после обжига совместно называются как молекулярное сито настоящего изобретения или молекулярное сито в соответствии с настоящим изобретением.
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита обжиг может быть проведен с помощью любого стандартного способа, известного в данной области техники, например, при температуре обжига, обычно находящейся в диапазоне от 300°С до 750°С, предпочтительно от 400°С до 600°С, в течение периода обжига, обычно находящегося в диапазоне от 1 часа до 10 часов, предпочтительно от 3 часов до 6 часов. Кроме того, обжиг обычно проводят в кислородсодержащей атмосфере, такой как атмосфера воздуха или кислорода.
В соответствии с одним аспектом настоящего изобретения, если желательно, молекулярное сито в соответствии с настоящим изобретением или любое молекулярное сито, полученное с помощью способа получения молекулярного сита в соответствии с настоящим изобретением (в контексте настоящего описания оба в совокупности называются как молекулярное сито настоящего изобретения или молекулярное сито в соответствии с настоящим изобретением), могут дополнительно быть подвергнуты ионному обмену с помощью любых стандартных средств, известных в данной области техники, например, с использованием процесса ионного обмена или пропитки раствором (см., например, патенты США 3140249 и 3140253 и т.д.), так что катион металла (такой как ион Na или ион K в зависимости от способа получения), содержащийся в нем, может быть полностью или частично заменен другими катионами. Примеры указанного другого катиона включают ион водорода, другие ионы щелочных металлов (включая ион K, ион Rb и т.д.), ионы аммония (включая ион NH4, ионы четвертичного аммония, такие как ион тетраметиламмония и ион тетраэтиламмония и т.д.), ионы щелочноземельных металлов (включая ион Mg, ион Ca), ион Mn, ион Zn, ион Cd, ионы благородных металлов (включая ион Pt, ион Pd, ион Rh и т.д.), ион Ni, ион Co, ион Ti, ион Sn, ион Fe и/или ионы редкоземельных металлов и подобные.
Молекулярное сито в соответствии с настоящим изобретением может быть дополнительно обработано разбавленным раствором кислоты или подобным по мере необходимости для увеличения соотношения диоксид кремния - оксид алюминия или обработано водяным паром для улучшения устойчивости кристаллов молекулярного сита к воздействию кислот.
Молекулярное сито в соответствии с настоящим изобретением обладает хорошей термической/гидротермической стабильностью и имеет больший объем пор. Таким образом, молекулярное сито в соответствии с настоящим изобретением способно адсорбировать больше/более крупные молекулы, тем самым демонстрируя превосходные адсорбционные/каталитические свойства.
Молекулярное сито в соответствии с настоящим изобретением имеет относительно сильную кислотность, в частности большое количество L-кислотных центров. Данное молекулярное сито не было получено в предшествующем уровне техники. В результате молекулярное сито в соответствии с настоящим изобретением демонстрирует лучшую производительность, в частности в катализируемой кислотой реакции.
Молекулярное сито в соответствии с настоящим изобретением может находиться в любой физической форме, такой как порошки, гранулы или формованные изделия (например, полоски, клеверы и т.д.). Данные физические формы могут быть получены любым стандартным способом, известным в данной области техники, и без конкретного ограничения.
Четвертый вариант осуществления
В соответствии с одним аспектом настоящего изобретения предоставляют способ получения молекулярного сита. В настоящем описании способ получения включает стадию взаимодействия источника первого оксида, источника второго оксида, необязательного щелочного источника, органического темплата и воды в условиях кристаллизации с получением молекулярного сита (в дальнейшем именуемую как стадия взаимодействия).
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита стадия взаимодействия может быть проведена с помощью любого стандартного способа, известного в данной области техники, например, с использованием способа, включающего смешивание источника первого оксида, источника второго оксида, необязательного щелочного источника, органического темплата и воды и подвергание смеси кристаллизации в условиях кристаллизации.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия органический темплат содержит, по меньшей мере, соединение, представленное следующей формулой (I). В настоящем описании соединение, представленное формулой (I), может быть использовано отдельно или в комбинации двух или более из них в любом соотношении.
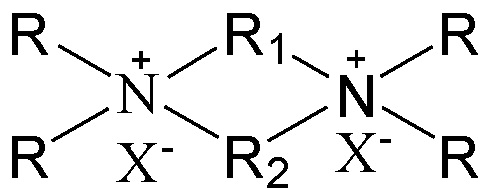 (I)
(I)
В соответствии с одним аспектом настоящего изобретения в формуле (I) группы R1 и R2 являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп.
В соответствии с другим вариантом осуществления настоящего изобретения в формуле (I) группы R1 и R2 являются одинаковыми или отличными друг от друга, одна из которых выбрана из группы, состоящей из С3-12линейных алкиленовых групп, и другая выбрана из группы, состоящей из С4-6линейных алкиленовых групп.
В соответствии с одним аспектом настоящего изобретения в качестве примера С3-12линейных или разветвленных алкиленовых групп может быть упомянута С3-12линейная алкиленовая группа, и ее определенные примеры включают н-пропилен, изопропилиден, н-бутилен, изобутилен, трет-бутилен, н-пентилен, изопентилен, неопентилен, н-гексилен, изогексилен, н-октилен, изооктилен, неооктилен, нонилен (или его изомер), децилен (или его изомер), ундецилен (или его изомер) или додецилен (или его изомер), предпочтительно н-пропилен, н-бутилен, н-пентилен, н-гексилен, н-гептилен, н-октилен, н-нонилен, н-децилен, н-ундецилен или н-додецилен.
В соответствии с одним аспектом настоящего изобретения в качестве С4-6линейных алкиленовых групп могут быть в частности упомянуты н-бутилен, н-пентилен или н-гексилен.
В соответствии с одним аспектом настоящего изобретения в формуле (I) множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, предпочтительно каждая независимо выбрана из группы, состоящей из метила и этила, более предпочтительно всего метила.
В соответствии с одним аспектом настоящего изобретения в формуле (I) X представляет собой ОН.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение органического темплата и источника первого оксида (рассчитанное на основе первого оксида) обычно находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,15 до 0,5 или от 0,3 до 0,5.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в дополнение к соединению, представленному формулой (I), другие органические темплаты, обычно используемые в данной области техники для получения молекулярных сит, могут быть дополнительно использованы в качестве органического темплата. Предпочтительно на стадии взаимодействия только соединение, представленное формулой (I), используют в качестве органического темплата. В настоящем описании соединение, представленное формулой (I), может быть использовано отдельно или в комбинации двух или более из них в любом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия источник первого оксида обычно представляет собой источник четырехвалентного оксида и может представлять собой, например, по меньшей мере, один, выбранный из группы, состоящей из источника диоксида кремния, источника диоксида германия, источника диоксида олова, источника диоксида титана и источника диоксида циркония, предпочтительно источника диоксида кремния (SiO2) или комбинации источника диоксида кремния и источника диоксида германия. Указанные источники первого оксида могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из источников первого оксида может представлять собой, например, от 20:200 до 35:100. В качестве примера комбинации может быть упомянута комбинация источника диоксида кремния и источника диоксида германия, в данном случае молярное соотношение между источником диоксида кремния и источником диоксида германия может представлять собой, например, от 20:200 до 35:100.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве источника первого оксида может быть использован любой соответствующий источник оксида, обычно используемый для этой цели в данной области техники, включая, но не ограничиваясь ими, в том числе оксиды, гидроксиды, алкоксиды, соли оксикислот металлов, ацетаты, оксалаты, соли аммония, сульфаты, галогенированные соли и нитраты соответствующего металла в первом оксиде. Например, когда первый оксид представляет собой диоксид кремния, примеры источника первого оксида включают золь кремниевой кислоты, неочищенный силикагель, тетраэтилортосиликат, жидкое стекло, белую сажу, кремниевую кислоту, силикагель, силикат калия или подобное. Когда первый оксид представляет собой диоксид германия, примеры источника первого оксида включают тетраалкоксид германия, оксид германия, нитрат германия или подобное. Когда первый оксид представляет собой диоксид олова, примеры источника первого оксида включают хлорид олова, сульфат олова, нитрат олова и подобное. Когда первый оксид представляет собой диоксид титана, примеры источника первого оксида включают тетраалкоксид титана, диоксид титана, нитрат титана и подобное. Когда первый оксид представляет собой диоксид циркония, примеры источника первого оксида включают хлорид циркония, сульфат циркония, нитрат циркония и подобное. Указанные источники первого оксида могут быть использованы отдельно или в комбинации двух или более из них в желаемом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия источник второго оксида обычно представляет собой источник трехвалентного оксида, такой как, по меньшей мере, один, выбранный из группы, состоящей из источника оксида алюминия, источника оксида бора, источника оксида железа, источника оксида галлия, источника редкоземельного оксида, источника оксида индия и источника оксида ванадия, предпочтительно источника оксида алюминия (Al2O3). Указанные источники второго оксида могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из источников второго оксида может представлять собой, например, от 30:200 до 60:150.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве источника второго оксида может быть использован любой соответствующий источник оксида, обычно используемый для этой цели в данной области техники, включая, но не ограничиваясь ими, в том числе оксиды, гидроксиды, алкоксиды, соли оксикислот металлов, ацетаты, оксалаты, соли аммония, сульфаты, галогенированные соли и нитраты соответствующего металла во втором оксиде. Например, когда второй оксид представляет собой оксид алюминия, примеры источника второго оксида включают хлорид алюминия, сульфат алюминия, гидрат алюминия, метаалюминат натрия, золь алюминия, гидроксид алюминия или подобное. Когда второй оксид представляет собой оксид бора, примеры источника второго оксида включают борную кислоту, борат, буру, триоксид бора и подобное. Когда второй оксид представляет собой оксид железа, примеры источника второго оксида включают нитрат железа, хлорид железа, оксид железа и подобное. Когда второй оксид представляет собой оксид галлия, примеры источника второго оксида включают нитрат галлия, сульфат галлия, оксид галлия и подобное. Когда второй оксид представляет собой редкоземельный оксид, примеры источника второго оксида включают оксид лантана, оксид неодима, оксид иттрия, оксид церия, нитрат лантана, нитрат неодима, нитрат иттрия, сульфат церия-аммония и подобное. Когда второй оксид представляет собой оксид индия, примеры источника второго оксида включают хлорид индия, нитрат индия, оксид индия и подобное. Когда второй оксид представляет собой оксид ванадия, примеры источника второго оксида включают хлорид ванадия, метаванадат аммония, ванадат натрия, диоксид ванадия, ванадилсульфат и подобное. Указанные источники второго оксида могут быть использованы отдельно или в комбинации двух или более из них в желаемом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение источника первого оксида (рассчитанное на основе первого оксида, такого как SiO2) и источника второго оксида (рассчитанное на основе второго оксида, такого как Al2O3) обычно находится в диапазоне от 5 до ∞, в частности от 5 до менее 40 (например, от 20 до менее 40), от 40 до 200 (например, от 40 до 150) и от более 200 до ∞ (например, от более 200 до 700). В настоящем описании, когда молярное соотношение представляет собой ∞, это означает, что источник второго оксида не используется или источник второго оксида не преднамеренно вводится на стадии взаимодействия.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение воды и источника первого оксида (рассчитанное на основе первого оксида) обычно находится в диапазоне от 5 до 50, предпочтительно от 5 до 15.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия щелочной источник может использоваться или не использоваться. Когда щелочной источник не преднамеренно используется, группа X, содержащаяся в соединении, представленном формулой (I), может быть использована для обеспечения ОН-, требуемого в настоящем описании. В настоящем описании в качестве щелочного источника может быть использован любой щелочной источник, обычно применяемый для этой цели в данной области техники, включая, но не ограничиваясь ими, неорганические основания, содержащие щелочной металл или щелочноземельный металл в качестве катиона, в частности гидроксид натрия и гидроксид калия. Указанные щелочные источники могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение щелочного источника (рассчитанное на основе OH-) и источника первого оксида (рассчитанное на основе первого оксида) обычно находится в диапазоне от 0 до 1, предпочтительно от 0,04 до 1, от 0,1 до 1, от 0,2 до 1, от 0,3 до 0,7 или от 0,45 до 0,7.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве условия кристаллизации температура кристаллизации обычно находится в диапазоне от 80°С до 120°С, предпочтительно от 120°С до 170°С или от 120°С до 200°С.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве условия кристаллизации период кристаллизации обычно составляет, по меньшей мере, 1 день, предпочтительно, по меньшей мере, 2 дня, предпочтительно от 3 дней до 8 дней, от 5 дней до 8 дней или от 4 дней до 6 дней.
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита после завершения стадии взаимодействия молекулярное сито может быть отделено в виде продукта из полученной реакционной смеси с помощью любого общеизвестного способа разделения. В настоящем описании продукт молекулярного сита содержит молекулярное сито в соответствии с настоящим изобретением. Кроме того, в качестве способа разделения, например, может быть упомянут способ, включающий фильтрование, промывание и сушку полученной реакционной смеси.
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита фильтрование, промывание и сушка могут быть проведены с помощью любого стандартного способа, известного в данной области техники. В качестве примера фильтрования, например, полученная реакционная смесь может быть просто подвергнута фильтрованию с отсасыванием. В качестве примера промывания, например, промывание деионизированной водой может проводиться до тех пор, пока рН фильтрата не достигнет 7-9, предпочтительно 8-9. Температура сушки составляет, например, от 40 до 250°С, предпочтительно от 60 до 150°С, и время сушки составляет, например, от 8 до 30 часов, предпочтительно от 10 до 20 часов. Сушка может быть проведена при нормальном давлении или при пониженном давлении.
В соответствии с одним аспектом настоящего изобретения способ получения молекулярного сита может дополнительно включать стадию обжига полученного молекулярного сита (в дальнейшем именуемую как стадия обжига) по мере необходимости для удаления органического темплата и возможной влаги и подобного, чтобы можно было получить кальцинированное молекулярное сито. В контексте настоящего описания молекулярные сита до и после обжига совместно называются как молекулярное сито настоящего изобретения или молекулярное сито в соответствии с настоящим изобретением.
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита обжиг может быть проведен с помощью любого стандартного способа, известного в данной области техники, например, при температуре обжига, обычно находящейся в диапазоне от 300°С до 750°С, предпочтительно от 400°С до 600°С, в течение периода обжига, обычно находящегося в диапазоне от 1 часа до 10 часов, предпочтительно от 3 часов до 6 часов. Кроме того, обжиг обычно проводят в кислородсодержащей атмосфере, такой как атмосфера воздуха или кислорода.
В соответствии с одним аспектом настоящего изобретения, если желательно, любое молекулярное сито, полученное, как описано ранее (называемое как молекулярное сито настоящего изобретения или молекулярное сито в соответствии с настоящим изобретением), может дополнительно быть подвергнуто ионному обмену с помощью любых стандартных средств, известных в данной области техники, например, с использованием процесса ионного обмена или пропитки раствором (см., например, патенты США 3140249 и 3140253 и т.д.), так что катион металла (такой как ион Na или ион K в зависимости от способа получения), содержащийся в нем, может быть полностью или частично заменен другим катионом(ами). Примеры указанного другого катиона включают ион водорода, другие ионы щелочных металлов (включая ион K, ион Rb и т.д.), ионы аммония (включая ион NH4, ионы четвертичного аммония, такие как ион тетраметиламмония и ион тетраэтиламмония и т.д.), ионы щелочноземельных металлов (включая ион Mg, ион Ca), ион Mn, ион Zn, ион Cd, ионы благородных металлов (включая ион Pt, ион Pd, ион Rh и т.д.), ион Ni, ион Co, ион Ti, ион Sn, ион Fe и/или ионы редкоземельных металлов и подобные.
Молекулярное сито в соответствии с настоящим изобретением может быть дополнительно обработано разбавленным раствором кислоты или подобным по мере необходимости для увеличения соотношения диоксид кремния-оксид алюминия или обработано водяным паром для улучшения устойчивости кристаллов молекулярного сита к воздействию кислот.
Молекулярное сито в соответствии с настоящим изобретением обычно имеет рентгеновскую дифрактограмму по существу, как показано в следующей таблице.
В соответствии с одним аспектом настоящего изобретения предпочтительно, что рентгеновская дифрактограмма молекулярного сита дополнительно включает рентгенодифракционный пик по существу, как показано в следующей таблице.
В соответствии с одним аспектом настоящего изобретения предпочтительно, что рентгеновская дифрактограмма молекулярного сита дополнительно включает рентгенодифракционный пик по существу, как показано в следующей таблице.
В соответствии с одним аспектом настоящего изобретения молекулярное сито обычно имеет морфологию столбчатой кристаллической частицы при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «морфология кристаллической частицы» относится к (общей) внешней форме, проявляемой монокристаллической частицей молекулярного сита в поле наблюдения сканирующего электронного микроскопа. Кроме того, в качестве столбчатой формы предпочтительной является призматическая форма, в частности гексагональная призматическая форма. В настоящем описании призма относится к выпуклой призме и обычно относится к прямоугольной призме и правильной многоугольной призме (такой как правильная гексагональная призма). Следует особо отметить, что поскольку процесс роста кристалла молекулярного сита может быть нарушен различными факторами, может быть определенная степень отклонения, когда его реальная морфология кристаллической частицы сравнивается с (истинно) прямоугольной призмой или (истинно) правильной многоугольной призмой в геометрическом смысле, такое как отклонение 30%, 20% или 5%, что приводит к получению скошенной призмы или неправильной многоугольной (или даже искривленной многоугольной) призмы, но настоящее изобретение не предназначено для определения степени отклонения. Более того, любые большие или меньшие отклонения не отступают от объема настоящего изобретения.
В соответствии с одним аспектом настоящего изобретения молекулярное сито (монокристаллическая частица) имеет эффективный диаметр, обычно находящийся в диапазоне от 100 нм до 5000 нм при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «эффективный диаметр» означает, что две точки произвольно выбираются в поперечном сечении молекулярного сита (монокристаллической частицы) вдоль контура (края) поперечного сечения и измеряется линейное расстояние между двумя точками, где наибольшее линейное расстояние принимается за эффективный диаметр. Если профиль поперечного сечения молекулярного сита представлен как многоугольник, такой как шестиугольник, эффективный диаметр обычно относится к линейному расстоянию между двумя вершинами многоугольника, расположенными дальше друг от друга, т.е. диагональному расстоянию. Вкратце, эффективный диаметр по существу соответствует диаметру описанной окружности многоугольника, представляющего контур поперечного сечения.
В соответствии с одним аспектом настоящего изобретения молекулярное сито (монокристаллическая частица) имеет высоту, обычно находящуюся в диапазоне от 100 нм до 3000 нм, при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «высота» относится к линейному расстоянию между центрами двух торцевых поверхностей монокристаллической частицы (столбчатой кристаллической частицы) молекулярного сита. В обычном случае две торцевые поверхности столбца молекулярного сита по существу параллельны друг другу, и линейное расстояние представляет собой вертикальное расстояние между двумя торцевыми поверхностями, но настоящее изобретение не ограничивается этим.
В соответствии с одним аспектом настоящего изобретения молекулярное сито (монокристаллическая частица) имеет соотношение сторон, обычно находящееся в диапазоне от 0,1 до 8, при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «соотношение сторон» относится к соотношению высоты и эффективного диаметра.
В соответствии с одним аспектом настоящего изобретения молекулярное сито имеет общую удельную площадь поверхности, обычно находящуюся в диапазоне от 400 м2⋅г-1 до 600 м2⋅г-1, предпочтительно от 450 м2⋅г-1 до 580 м2⋅г-1. В настоящем описании общую удельную площадь поверхности получают с использованием низкотемпературной адсорбции азота, рассчитанной по модели БЭТ.
В соответствии с одним аспектом настоящего изобретения молекулярное сито имеет объем пор, обычно находящийся в диапазоне от 0,3 мл/г до 0,5 мл/г, предпочтительно от 0,30 мл/г до 0,40 мл/г. Молекулярное сито в соответствии с настоящим изобретением имеет очень большой объем пор, что указывает на то, что оно относится к ультрамакропористому молекулярному ситу. В настоящем описании объем пор получают с использованием низкотемпературной адсорбции азота, рассчитанной по модели БЭТ.
Молекулярное сито в соответствии с настоящим изобретением обладает хорошей термической/гидротермической стабильностью и имеет больший объем пор. Таким образом, молекулярное сито в соответствии с настоящим изобретением способно адсорбировать больше/более крупные молекулы, тем самым демонстрируя превосходные адсорбционные/каталитические свойства.
Молекулярное сито в соответствии с настоящим изобретением имеет относительно сильную кислотность, в частности большое количество L-кислотных центров. Данное молекулярное сито не было получено в предшествующем уровне техники. В результате молекулярное сито в соответствии с настоящим изобретением демонстрирует лучшую производительность, в частности в катализируемой кислотой реакции.
Молекулярное сито в соответствии с настоящим изобретением может находиться в любой физической форме, такой как порошки, гранулы или формованные изделия (например, полоски, клеверы и т.д.). Данные физические формы могут быть получены любым стандартным способом, известным в данной области техники, и без конкретного ограничения.
Пятый вариант осуществления
В соответствии с одним аспектом настоящего изобретения предоставляют способ получения молекулярного сита. В настоящем описании способ получения включает стадию взаимодействия источника первого оксида, источника второго оксида, необязательного щелочного источника, органического темплата и воды в условиях кристаллизации с получением молекулярного сита (в дальнейшем именуемую как стадия взаимодействия).
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита стадия взаимодействия может быть проведена с помощью любого стандартного способа, известного в данной области техники, например, с использованием способа, включающего смешивание источника первого оксида, источника второго оксида, необязательного щелочного источника, органического темплата и воды и подвергание смеси кристаллизации в условиях кристаллизации.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия органический темплат содержит, по меньшей мере, соединение, представленное следующей формулой (I). В настоящем описании соединение, представленное формулой (I), может быть использовано отдельно или в комбинации двух или более из них в любом соотношении.
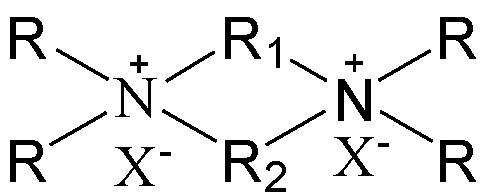 (I)
(I)
В соответствии с одним аспектом настоящего изобретения в формуле (I) группы R1 и R2 являются отличными друг от друга, одна из которых выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных или разветвленных оксаалкиленовых групп.
В соответствии с одним аспектом настоящего изобретения в качестве примера С3-12линейных или разветвленных алкиленовых групп может быть упомянута С3-12линейная алкиленовая группа, и ее определенные примеры включают н-пропилен, изопропилиден, н-бутилен, изобутилен, трет-бутилен, н-пентилен, изопентилен, неопентилен, н-гексилен, изогексилен, н-октилен, изооктилен, неооктилен, нонилен (или его изомер), децилен (или его изомер), ундецилен (или его изомер) или додецилен (или его изомер), предпочтительно н-пропилен, н-бутилен, н-пентилен, н-гексилен, н-гептилен, н-октилен, н-нонилен, н-децилен, н-ундецилен или н-додецилен.
В соответствии с одним аспектом настоящего изобретения в качестве С3-12линейных или разветвленных оксаалкиленовых групп, например, может быть упомянута С3-12линейная оксаалкиленовая группа, и ее конкретные примеры включают -(CH2)2-O-(CH2)-, -(CH2)2-O-(CH2)2-, -(CH2)-O-(CH2)3-, -(CH2)2-O-(CH2)3-, -(CH2)-O-пропилен-, -(CH2)-O-(CH2)4-, -(CH2)-O-(CH2)2-O-(CH2)-, -(CH2)-O-(CH2)2-O-(CH2)2-, -(CH2)-O-трет-бутилен-, -(CH2)2-O-(CH2)4-, -(CH2)3-O-(CH2)3-, -(CH2)-O-неопентилен-, -(CH2)2-O-(CH2)6-, -(CH2)2-O-(CH2)7-, -(CH2)-O-(CH2)8-, -(CH2)-O-изооктилен-, -(CH2)-O-(CH2)10-, -(CH2)2-O-децилен или его изомер, -(CH2)-O-(CH2)6-, -(CH2)-O-(CH2)7-, -(CH2)-O-(CH2)8-, -(CH2)-O-(CH2)11-, -(CH2)-O-(CH2)2-O-(CH2)-, -(CH2)2-O-(CH2)2-O-(CH2)2-, -(CH2)2-O-(CH2)4-O-(CH2)2-, -(CH2)2-O-(CH2)6-O-(CH2)2- или -(CH2)2-O-(CH2)8-O-(CH2)2-. Кроме того, в качестве С3-12линейных оксаалкиленовых групп может быть упомянута С4-6линейная оксаалкиленовая группа, и в частности может быть упомянута С4-6линейная монооксаалкиленовая группа, и особенно может быть упомянута монооксаалкиленовая группа, представленная формулой -(CH2)m-O-(CH2)m- (где каждое значение m, являющееся одинаковым или отличным друг от друга, независимо представляет собой 2 или 3, например, 2). Более определенно могут быть упомянуты -(CH2)2-O-(CH2)2-, -(CH2)2-O-(CH2)3-, -(CH2)3-O-(CH2)3- или -(CH2)2-O-(CH2)4-.
В соответствии с одним аспектом настоящего изобретения в формуле (I) множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, предпочтительно каждая независимо выбрана из группы, состоящей из метила и этила, более предпочтительно всего метила.
В соответствии с одним аспектом настоящего изобретения в формуле (I) X представляет собой ОН.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение органического темплата и источника первого оксида (рассчитанное на основе первого оксида) обычно находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,15 до 0,5 или от 0,3 до 0,5.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в дополнение к соединению, представленному формулой (I), другие органические темплаты, обычно используемые в данной области техники для получения молекулярных сит, могут быть дополнительно использованы в качестве органического темплата. Предпочтительно на стадии взаимодействия только соединение, представленное формулой (I), используют в качестве органического темплата. В настоящем описании соединение, представленное формулой (I), может быть использовано отдельно или в комбинации двух или более из них в любом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия источник первого оксида обычно представляет собой источник четырехвалентного оксида и может представлять собой, например, по меньшей мере, один, выбранный из группы, состоящей из источника диоксида кремния, источника диоксида германия, источника диоксида олова, источника диоксида титана и источника диоксида циркония, предпочтительно источника диоксида кремния (SiO2) или комбинации источника диоксида кремния и источника диоксида германия. Указанные источники первого оксида могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из источников первого оксида может представлять собой, например, от 20:200 до 35:100. В качестве примера комбинации может быть упомянута комбинация источника диоксида кремния и источника диоксида германия, в данном случае молярное соотношение между источником диоксида кремния и источником диоксида германия может представлять собой, например, от 20:200 до 35:100.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве источника первого оксида может быть использован любой соответствующий источник оксида, обычно используемый для этой цели в данной области техники, включая, но не ограничиваясь ими, в том числе оксиды, гидроксиды, алкоксиды, соли оксикислот металлов, ацетаты, оксалаты, соли аммония, сульфаты, галогенированные соли и нитраты соответствующего металла в первом оксиде. Например, когда первый оксид представляет собой диоксид кремния, примеры источника первого оксида включают золь кремниевой кислоты, неочищенный силикагель, тетраэтилортосиликат, жидкое стекло, белую сажу, кремниевую кислоту, силикагель, силикат калия или подобное. Когда первый оксид представляет собой диоксид германия, примеры источника первого оксида включают тетраалкоксид германия, оксид германия, нитрат германия или подобное. Когда первый оксид представляет собой диоксид олова, примеры источника первого оксида включают хлорид олова, сульфат олова, нитрат олова и подобное. Когда первый оксид представляет собой диоксид титана, примеры источника первого оксида включают тетраалкоксид титана, диоксид титана, нитрат титана и подобное. Когда первый оксид представляет собой диоксид циркония, примеры источника первого оксида включают хлорид циркония, сульфат циркония, нитрат циркония и подобное. Указанные источники первого оксида могут быть использованы отдельно или в комбинации двух или более из них в желаемом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия источник второго оксида обычно представляет собой источник трехвалентного оксида, такой как, по меньшей мере, один, выбранный из группы, состоящей из источника оксида алюминия, источника оксида бора, источника оксида железа, источника оксида галлия, источника редкоземельного оксида, источника оксида индия и источника оксида ванадия, предпочтительно источника оксида алюминия (Al2O3). Указанные источники второго оксида могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. Когда два или более используют в комбинации, молярное соотношение между любыми двумя из источников второго оксида может представлять собой, например, от 30:200 до 60:150.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве источника второго оксида может быть использован любой соответствующий источник оксида, обычно используемый для этой цели в данной области техники, включая, но не ограничиваясь ими, в том числе оксиды, гидроксиды, алкоксиды, соли оксикислот металлов, ацетаты, оксалаты, соли аммония, сульфаты, галогенированные соли и нитраты соответствующего металла во втором оксиде. Например, когда второй оксид представляет собой оксид алюминия, примеры источника второго оксида включают хлорид алюминия, сульфат алюминия, гидрат алюминия, метаалюминат натрия, золь алюминия, гидроксид алюминия или подобное. Когда второй оксид представляет собой оксид бора, примеры источника второго оксида включают борную кислоту, борат, буру, триоксид бора и подобное. Когда второй оксид представляет собой оксид железа, примеры источника второго оксида включают нитрат железа, хлорид железа, оксид железа и подобное. Когда второй оксид представляет собой оксид галлия, примеры источника второго оксида включают нитрат галлия, сульфат галлия, оксид галлия и подобное. Когда второй оксид представляет собой редкоземельный оксид, примеры источника второго оксида включают оксид лантана, оксид неодима, оксид иттрия, оксид церия, нитрат лантана, нитрат неодима, нитрат иттрия, сульфат церия-аммония и подобное. Когда второй оксид представляет собой оксид индия, примеры источника второго оксида включают хлорид индия, нитрат индия, оксид индия и подобное. Когда второй оксид представляет собой оксид ванадия, примеры источника второго оксида включают хлорид ванадия, метаванадат аммония, ванадат натрия, диоксид ванадия, ванадилсульфат и подобное. Указанные источники второго оксида могут быть использованы отдельно или в комбинации двух или более из них в желаемом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение источника первого оксида (рассчитанное на основе первого оксида, такого как SiO2) и источника второго оксида (рассчитанное на основе второго оксида, такого как Al2O3) обычно находится в диапазоне от 5 до ∞, в частности от 5 до менее 30 (например, от 10 до менее 30), от 30 до 100 (например, от 55 до 100) и от более 100 до ∞. (например, от 200 до ∞ или от 200 до 700). В настоящем описании, когда молярное соотношение представляет собой ∞, это означает, что источник второго оксида не используется или источник второго оксида не преднамеренно вводится на стадии взаимодействия.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение воды и источника первого оксида (рассчитанное на основе первого оксида) обычно находится в диапазоне от 5 до 50, предпочтительно от 5 до 15.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия щелочной источник может использоваться или не использоваться. Когда щелочной источник не преднамеренно используется, группа X, содержащаяся в соединении, представленном формулой (I), может быть использована для обеспечения ОН-, требуемого в настоящем описании. В настоящем описании в качестве щелочного источника может быть использован любой щелочной источник, обычно применяемый для этой цели в данной области техники, включая, но не ограничиваясь ими, неорганические основания, содержащие щелочной металл или щелочноземельный металл в качестве катиона, в частности гидроксид натрия и гидроксид калия. Указанные щелочные источники могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия молярное соотношение щелочного источника (рассчитанное на основе OH-) и источника первого оксида (рассчитанное на основе первого оксида) обычно находится в диапазоне от 0 до 1, предпочтительно от 0,04 до 1, от 0,1 до 1, от 0,2 до 1, от 0,3 до 0,7 или от 0,45 до 0,7.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве условия кристаллизации температура кристаллизации обычно находится в диапазоне от 80°С до 120°С, предпочтительно от 120°С до 170°С или от 120°С до 200°С.
В соответствии с одним аспектом настоящего изобретения на стадии взаимодействия в качестве условия кристаллизации период кристаллизации обычно составляет, по меньшей мере, 1 день, предпочтительно, по меньшей мере, 2 дня, предпочтительно от 3 дней до 8 дней, от 5 дней до 8 дней или от 4 дней до 6 дней.
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита после завершения стадии взаимодействия молекулярное сито может быть отделено в виде продукта из полученной реакционной смеси с помощью любого общеизвестного способа разделения. В настоящем описании продукт молекулярного сита содержит молекулярное сито в соответствии с настоящим изобретением. Кроме того, в качестве способа разделения, например, может быть упомянут способ, включающий фильтрование, промывание и сушку полученной реакционной смеси.
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита фильтрование, промывание и сушка могут быть проведены с помощью любого стандартного способа, известного в данной области техники. В качестве примера фильтрования, например, полученная реакционная смесь может быть просто подвергнута фильтрованию с отсасыванием. В качестве примера промывания, например, промывание деионизированной водой может проводиться до тех пор, пока рН фильтрата не достигнет 7-9, предпочтительно 8-9. Температура сушки составляет, например, от 40 до 250°С, предпочтительно от 60 до 150°С, и время сушки составляет, например, от 8 до 30 часов, предпочтительно от 10 до 20 часов. Сушка может быть проведена при нормальном давлении или при пониженном давлении.
В соответствии с одним аспектом настоящего изобретения способ получения молекулярного сита может дополнительно включать стадию обжига полученного молекулярного сита (в дальнейшем именуемую как стадия обжига) по мере необходимости для удаления органического темплата и возможной влаги и подобного, чтобы можно было получить кальцинированное молекулярное сито. В контексте настоящего описания молекулярные сита до и после обжига совместно называются как молекулярное сито настоящего изобретения или молекулярное сито в соответствии с настоящим изобретением.
В соответствии с одним аспектом настоящего изобретения в способе получения молекулярного сита обжиг может быть проведен с помощью любого стандартного способа, известного в данной области техники, например, при температуре обжига, обычно находящейся в диапазоне от 300°С до 750°С, предпочтительно от 400°С до 600°С, в течение периода обжига, обычно находящегося в диапазоне от 1 часа до 10 часов, предпочтительно от 3 часов до 6 часов. Кроме того, обжиг обычно проводят в кислородсодержащей атмосфере, такой как атмосфера воздуха или кислорода.
В соответствии с одним аспектом настоящего изобретения, если желательно, любое молекулярное сито, полученное, как описано ранее (называемое как молекулярное сито настоящего изобретения или молекулярное сито в соответствии с настоящим изобретением), может дополнительно быть подвергнуто ионному обмену с помощью любых стандартных средств, известных в данной области техники, например, с использованием процесса ионного обмена или пропитки раствором (см., например, патенты США 3140249 и 3140253 и т.д.), так что катион металла (такой как ион Na или ион K в зависимости от способа получения), содержащийся в нем, может быть полностью или частично заменен другим катионом(ами). Примеры указанного другого катиона включают ион водорода, другие ионы щелочных металлов (включая ион K, ион Rb и т.д.), ионы аммония (включая ион NH4, ионы четвертичного аммония, такие как ион тетраметиламмония и ион тетраэтиламмония и т.д.), ионы щелочноземельных металлов (включая ион Mg, ион Ca), ион Mn, ион Zn, ион Cd, ионы благородных металлов (включая ион Pt, ион Pd, ион Rh и т.д.), ион Ni, ион Co, ион Ti, ион Sn, ион Fe и/или ионы редкоземельных металлов и подобные.
Молекулярное сито в соответствии с настоящим изобретением может быть дополнительно обработано разбавленным раствором кислоты или подобным по мере необходимости для увеличения соотношения диоксид кремния-оксид алюминия или обработано водяным паром для улучшения устойчивости кристаллов молекулярного сита к воздействию кислот.
Молекулярное сито в соответствии с настоящим изобретением обычно имеет рентгеновскую дифрактограмму по существу, как показано в следующей таблице.
В соответствии с одним аспектом настоящего изобретения предпочтительно, что рентгеновская дифрактограмма молекулярного сита дополнительно включает рентгенодифракционный пик по существу, как показано в следующей таблице.
В соответствии с одним аспектом настоящего изобретения предпочтительно, что рентгеновская дифрактограмма молекулярного сита дополнительно включает рентгенодифракционный пик по существу, как показано в следующей таблице.
В соответствии с одним аспектом настоящего изобретения молекулярное сито обычно имеет морфологию столбчатой кристаллической частицы при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «морфология кристаллической частицы» относится к (общей) внешней форме, проявляемой монокристаллической частицей молекулярного сита в поле наблюдения сканирующего электронного микроскопа. Кроме того, в качестве столбчатой формы предпочтительной является призматическая форма, в частности гексагональная призматическая форма. В настоящем описании призма относится к выпуклой призме и обычно относится к прямоугольной призме и правильной многоугольной призме (такой как правильная гексагональная призма). Следует особо отметить, что поскольку процесс роста кристалла молекулярного сита может быть нарушен различными факторами, может быть определенная степень отклонения, когда его реальная морфология кристаллической частицы сравнивается с (истинно) прямоугольной призмой или (истинно) правильной многоугольной призмой в геометрическом смысле, такое как отклонение 30%, 20% или 5%, что приводит к получению скошенной призмы или неправильной многоугольной (или даже искривленной многоугольной) призмы, но настоящее изобретение не предназначено для определения степени отклонения. Более того, любые большие или меньшие отклонения не отступают от объема настоящего изобретения.
В соответствии с одним аспектом настоящего изобретения молекулярное сито (монокристаллическая частица) имеет эффективный диаметр, обычно находящийся в диапазоне от 100 нм до 5000 нм при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «эффективный диаметр» означает, что две точки произвольно выбираются в поперечном сечении молекулярного сита (монокристаллической частицы) вдоль контура (края) поперечного сечения и измеряется линейное расстояние между двумя точками, где наибольшее линейное расстояние принимается за эффективный диаметр. Если профиль поперечного сечения молекулярного сита представлен как многоугольник, такой как шестиугольник, эффективный диаметр обычно относится к линейному расстоянию между двумя вершинами многоугольника, расположенными дальше друг от друга, т.е. диагональному расстоянию. Вкратце, эффективный диаметр по существу соответствует диаметру описанной окружности многоугольника, представляющего контур поперечного сечения.
В соответствии с одним аспектом настоящего изобретения молекулярное сито (монокристаллическая частица) имеет высоту, обычно находящуюся в диапазоне от 100 нм до 3000 нм, при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «высота» относится к линейному расстоянию между центрами двух торцевых поверхностей монокристаллической частицы (столбчатой кристаллической частицы) молекулярного сита. В обычном случае две торцевые поверхности столбца молекулярного сита по существу параллельны друг другу, и линейное расстояние представляет собой вертикальное расстояние между двумя торцевыми поверхностями, но настоящее изобретение не ограничивается этим.
В соответствии с одним аспектом настоящего изобретения молекулярное сито (монокристаллическая частица) имеет соотношение сторон, обычно находящееся в диапазоне от 0,1 до 8, при наблюдении с использованием сканирующего электронного микроскопа (СЭМ). В настоящем описании термин «соотношение сторон» относится к соотношению высоты и эффективного диаметра.
В соответствии с одним аспектом настоящего изобретения молекулярное сито имеет общую удельную площадь поверхности, обычно находящуюся в диапазоне от 400 м2⋅г-1 до 600 м2⋅г-1, предпочтительно от 450 м2⋅г-1 до 580 м2⋅г-1. В настоящем описании общую удельную площадь поверхности получают с использованием метода адсорбции жидкого азота, рассчитанного по модели БЭТ.
В соответствии с одним аспектом настоящего изобретения молекулярное сито имеет объем пор, обычно находящийся в диапазоне от 0,3 мл/г до 0,5 мл/г, предпочтительно от 0,30 мл/г до 0,40 мл/г. Молекулярное сито в соответствии с настоящим изобретением имеет очень большой объем микропор, что указывает на то, что оно относится к ультрамакропористому молекулярному ситу. В настоящем описании объем пор получают с использованием метода адсорбции жидкого азота, рассчитанного по модели БЭТ.
Молекулярное сито в соответствии с настоящим изобретением обладает хорошей термической/гидротермической стабильностью и имеет больший объем пор. Таким образом, молекулярное сито в соответствии с настоящим изобретением способно адсорбировать больше/более крупные молекулы, тем самым демонстрируя превосходные адсорбционные/каталитические свойства.
Молекулярное сито в соответствии с настоящим изобретением имеет относительно сильную кислотность, в частности большое количество L-кислотных центров. Данное молекулярное сито не было получено в предшествующем уровне техники. В результате молекулярное сито в соответствии с настоящим изобретением демонстрирует лучшую производительность, в частности в катализируемой кислотой реакции.
Молекулярное сито в соответствии с настоящим изобретением может находиться в любой физической форме, такой как порошки, гранулы или формованные изделия (например, полоски, клеверы и т.д.). Данные физические формы могут быть получены любым стандартным способом, известным в данной области техники, и без конкретного ограничения.
Шестой вариант осуществления
Молекулярное сито в соответствии с настоящим изобретением может быть использовано в комбинации с другими материалами, тем самым получая композицию молекулярного сита. В качестве таких других материалов могут быть упомянуты, например, активные материалы и неактивные материалы. Примеры активных материалов включают в том числе синтетический цеолит и природный цеолит. Примеры неактивных материалов (обычно называемых как связующее вещество) включают в том числе глину, карклазит, силикагель и оксид алюминия. Указанные другие материалы могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. В отношении количества указанных других материалов не существует особых ограничений, и любое количество, обычно используемое в данной области техники, может быть непосредственно принято.
Молекулярное сито или композиция молекулярного сита в соответствии с настоящим изобретением в частности подходит для применения в качестве адсорбента, например, для отделения, по меньшей мере, одного компонента от смеси компонентов в газовой фазе или в жидкой фазе.
Молекулярное сито или композиция молекулярного сита в соответствии с настоящим изобретением в частности подходит для применения в качестве катализатора в реакции конверсии углеводородов. Примеры реакции конверсии углеводородов включают в том числе реакции каталитического крекинга, гидрокрекинга, диспропорционирования, алкилирования, олигомеризации и изомеризации.
Молекулярное сито или композиция молекулярного сита в соответствии с настоящим изобретением в частности подходит для применения в качестве носителя или компонента носителя катализаторов, и активный компонент(ы) может быть нанесен на него с помощью любого стандартного способа, известного в данной области техники, такого как пропитка раствором. Указанный активный компонент включает, но не ограничивается ими, активные металлические компоненты (включая Ni, Co, Mo, W или Cu и т.д.), реакционноспособные неорганические вспомогательные вещества (такие как F, P и т.д.) и органические соединения (такие как органические кислоты, органические амины и т.д.). Указанные активные компоненты могут быть использованы отдельно или в комбинации двух или более из них в любом соотношении. В отношении количества активного компонента, не существует особых ограничений, и любое количество, обычно используемое в данной области техники, может быть непосредственно принято.
Примеры
Настоящее изобретение дополнительно иллюстрируется со ссылкой на следующие примеры, но настоящее изобретение не ограничивается этим.
В контексте настоящего описания, включая следующие примеры и сравнительные примеры, был использован температурно-программируемый десорбер Autochem II2920, Microtek, Соединенные Штаты. Условия испытаний были следующими: 0,2 г молекулярного сита 20-40 меш взвешивали в пробирке для образцов, помещали в нагревательную печь с использованием газа Не (25 мл/мин) в качестве газа-носителя, повышали до температуры 600°С при 20°С/мин и продували в течение 60 мин для удаления примесей, адсорбированных на поверхности молекулярного сита; затем температуру понижали до 100°С, температуру поддерживали в течение 10 мин и газ переключали на смесь NH3-He (10% NH3+90% He) для проведения адсорбции в течение 30 мин; и затем полученный продукт продували газом Не в течение 90 мин до тех пор, пока исходный уровень не был стабильным для десорбции физически адсорбированного NH3; температуру повышали до 600°С со скоростью нагревания 10°С/мин в течение десорбции, и десорбцию проводили в течение 30 мин, и затем завершали; детектор TCD использовали для обнаружения изменений в композиции газа и интеграцию автоматически проводили с помощью прибора для обеспечения распределения количества кислоты.
В контексте настоящего описания, включая следующие примеры и сравнительные примеры, XRD-исследование было проведено с использованием оборудования Netherland, PANalytical Corporation. Условия испытаний: мишень Cu, излучение Kα, фильтр Ni, напряжение на трубке 40 кВ, ток трубки 40 мА и диапазон сканирования 2-50°.
В контексте настоящего описания, включая следующие примеры и сравнительные примеры, использовали сканирующий электронный микроскоп TECNAIG2F20 (200 кВ), FEI Corporation, Соединенные Штаты. Условия испытаний были следующими: образец получали, используя метод суспензии, и в частности 0,01 г образца молекулярного сита помещали в 2 мл стеклянный флакон, диспергировали с абсолютным этанолом и равномерно перемешивали; одну каплю полученного раствора брали с помощью пипетки, капали на сетку образца диаметром 3 мм, высушивали, помещали в инжектор и затем вводили в электронный микроскоп для наблюдения. Наблюдение может быть проведено с использованием увеличения в 10000 раз или увеличения в 50000 раз. Кроме того, молекулярное сито наблюдали при увеличении в 50000 раз, поле наблюдения выбирали случайным образом, и вычисляли среднее значение суммы эффективных диаметров и среднее значение суммы высот всех кристаллов молекулярного сита в поле наблюдения. Операцию повторяли в общей сложности 10 раз. Среднее значение суммы средних значений, полученных за 10 раз проведения операции, принимали как эффективный диаметр и высоту соответственно.
В контексте настоящего описания, включая следующие примеры и сравнительные примеры, использовали спектрометр ядерного магнитного резонанса Varian UNITY INOVA 500 МГц, Varian, Соединенные Штаты. Условия испытаний: твердотельный двухрезонансный зонд, Φ4 мм ZrO2 ротор. Экспериментальные параметры: температура исследования является комнатной температурой, время сканирования nt=5000, ширина импульса pw=3,9 мкс, ширина спектра sw=31300 Гц, резонансная частота ядерного наблюдения Sfrq=125,64 МГц, время между измерениями=0,5 с, калибрование химического замещения δTMS=0, время задержки d1=4,0 с, режим расщепления dm=nny (обратная стробированная развязка), поле блокирования дейтерированного хлороформа.
В контексте настоящего описания, включая следующие примеры и сравнительные примеры, использовали рентгеновский флуоресцентный спектрометр Модель 3013, Japanese Science and Technology Co., Ltd. Условия испытаний: вольфрамовая мишень, напряжение возбуждения 40 кВ, ток возбуждения 50 мА. Экспериментальная методика заключалась в следующем: образец таблетировали, затем устанавливали на рентгеновский флуоресцентный спектрометр и возбуждали при облучении рентгеновских лучей с целью испускания флуоресценции; соотношение между длиной волны флуоресценции λ и атомным номером Z элемента было следующим: λ=K(Z-S)-2, причем K представляет собой константу, так что в каждом случае, когда измеряли длину волны λ флуоресценции, может быть определен элемент. Интенсивность характеристических линий каждого элемента измеряли с помощью сцинтилляционного счетчика и пропорционального счетчика и проводили элементный или полуколичественный анализ.
В контексте настоящего описания, включая следующие примеры и сравнительные примеры, использовали ИК-Фурье спектрометр типа FTS3O00, BIO-RAD Company, Соединенные Штаты. Условия испытаний были следующими: создавали вакуум до 10-3 Па при 350°С, и диапазон волновых чисел составлял 1300-3900 см-1. Образец таблетировали, затем помещали in-situ в ячейку инфракрасного спектрометра и герметизировали, создавали вакуум до 10-3 Па при 350°С в течение 1 ч для десорбции молекул газа на поверхности образца и охлаждали до комнатной температуры. Пиридин/2,4,6-триметилпиридин при давлением 2,67 Па вводили в ячейку in-situ и после равновесной адсорбции в течение 30 мин температуру повышали до 200°С и снова создавали вакуум до 10-3 Па в течение 30 мин, и затем охлаждали до комнатной температуры. Инфракрасный спектр поглощения адсорбции пиридина/2,4,6-триметилпиридина при 200°С регистрировали путем сканирования в диапазоне волновых чисел 1300-3900 см-1. Образец в инфракрасной абсорбционной ячейке перемещали в зону термической обработки, нагревали до 350°С, создавали вакуум до 10-3 Па, выдерживали в течение 30 мин, охлаждали до комнатной температуры и регистрировали инфракрасный спектр адсорбции пиридина при 350°С.
В контексте настоящего описания, включая следующие примеры и сравнительные примеры, все фармацевтические препараты и сырьевые материалы являются или коммерчески доступными, или могут быть получены на основе имеющихся знаний.
Первый вариант осуществления, четвертый вариант осуществления и шестой вариант осуществления
В контексте настоящего варианта осуществления, включая следующие примеры и сравнительные примеры, общую удельную площадь поверхности, объем пор и диаметр пор молекулярного сита измеряли с помощью следующих аналитических методов.
Оборудование: Статический адсорбер азота Micromeritics ASAP2010.
Условия измерения: образец помещали в систему для обработки образца, создавали вакуум до 1,35×10-2 Па при 350°С и выдерживали при данной температуре и давлении в течение 15 ч для очистки образца; при температуре жидкого азота -196°С измеряли количество адсорбции и количество десорбции азота очищенного образца в условиях различного удельного давления P/P0, и получали изотерму адсорбции-десорбции; и затем общую удельную площадь поверхности рассчитывали с помощью двухпараметрического уравнения БЭТ, и адсорбционную емкость при удельном давлении P/P0≈0,98 принимали за объем пор образца, и распределение пор по диаметру рассчитывали по модели BJH.
Пример I серии
Пример I-1
Получение Темплата A
В 500 мл трехгорлую колбу добавляли 15 г (0,087 моль) тетраметилгексаметилендиамина, добавляли 250 мл изопропанола и добавляли по каплям 18,8 г (0,087 моль) 1,4-дибромбутана при комнатной температуре. Добавление завершали через 15 минут и температуру повышали до температуры нагревания с обратным холодильником. Раствор постепенно менялся от бесцветного и прозрачного до белого и мутного. Реакция сопровождалась высокоэффективной жидкостной хроматографией (ВЭЖХ). После завершения реакции к реакционной смеси добавляли 200 мл этилацетата и смесь нагревали с обратным холодильником в течение 1 часа, охлаждали и фильтровали с отсасыванием. Полученное твердое вещество промывали этилацетатом и затем диэтиловым эфиром с получением 30 г белого твердого продукта в виде 1,1,6,6-тетраметил-1,6-диаза-12-членного кольца-1,6-дибромида (соединение, в котором n равно 4, m равно 6, R представляет собой метил, X представляет собой Br), имеющего относительную молекулярную массу 388,2, температуру плавления 273,7°C. Спектр 1H-ЯМР показывает химический сдвиг (300 МГц, CDCl3) δ 1,50 (т, 4H), 1,90 (т, 8H), 3,14 (с, 12H), 3,40 (т, 8H).
Получение Темплата B: Br в Темплате A заменяли OH- с помощью ионного обмена; ионообменная смола представляла собой сильную основную анионообменную смолу стирольного типа, рабочий раствор представлял собой 15 масс.% водного раствора Темплата А, рабочая температура составляла 25°С, массовое соотношение рабочей жидкости и ионообменной смолы составляло 1:3; скорость потока составляла 3 капли/секунда; обменный раствор дегидратировали с помощью роторного испарителя с получением продукта, который представляет собой соединение формулы (I), в которой n равно 4, m равно 6, R представляет собой метил и X представляет собой ОН, имеющее относительную молекулярную массу 262,2 и чистоту 99,21%. Содержание брома в нем составляло 0,79 масс.%.
Пример I-2
Получение Темплата C
В 500 мл трехгорлую колбу добавляли 10 г (0,058 моль) тетраметилгексаметилендиамина, добавляли 250 мл изопропанола и добавляли по каплям 16,6 г (0,058 моль) 1,9-дибромдекана при комнатной температуре. Добавление завершали через 15 минут и температуру повышали до температуры нагревания с обратным холодильником. Раствор постепенно менялся от бесцветного и прозрачного до белого и мутного. Реакция сопровождалась высокоэффективной жидкостной хроматографией (ВЭЖХ). После завершения реакции к реакционной смеси добавляли 200 мл этилацетата и смесь нагревали с обратным холодильником в течение 1 часа, охлаждали и фильтровали с отсасыванием. Полученное твердое вещество промывали этилацетатом и затем диэтиловым эфиром с получением 25 г белого твердого продукта в виде 1,1,8,8-тетраметил-1,8-диаза-17-членного кольца-1,8-дибромида (соединение, в котором n равно 9, m равно 6, R представляет собой метил, X представляет собой Br), имеющего относительную молекулярную массу 458,4. Спектр 1H-ЯМР показывает химический сдвиг (300 МГц, CDCl3) δ 1,51 (т, 14H), 1,92 (т, 8H), 3,16 (с, 12H), 3,40 (т, 8H).
Получение Темплата D: Br в Темплате C заменяли OH- с помощью ионного обмена; ионообменная смола представляла собой сильную основную анионообменную смолу стирольного типа, рабочий раствор представлял собой 15 масс.% водного раствора Темплата C, рабочая температура составляла 25°С, массовое соотношение рабочей жидкости и ионообменной смолы составляло 1:3; скорость потока составляла 3 капли/секунда; обменный раствор дегидратировали с помощью роторного испарителя с получением продукта, который представляет собой соединение формулы (I), в которой n равно 9, m равно 6, R представляет собой метил, X представляет собой ОН, имеющее относительную молекулярную массу 332,4 и чистоту 99,8%. Содержание брома в нем составляло 0,2 масс.%.
Пример I-3
В 45 мл тефлоновый контейнер добавляли 5,35 г Темплата D, добавляли 0,157 г гидроксида натрия, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98%, содержание Al2O3 0,253%) и 0,033 г твердого NaAlO2, и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: H2O/SiO2=6, Темплат D/SiO2=0,10, NaOH/SiO2=0,08.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем повышали до 180°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре I-1. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 600 нм, высотой 800 нм и соотношением сторон 1,33. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 560 м2⋅г-1 и объем пор 0,360 мл/г. Дифрактограмма продукта была показана на Фигуре I-2. РФА показал, что молекулярное сито имеет Si/Al2=210.
Пример I-4
В 45 мл тефлоновый контейнер добавляли 8,024 г Темплата D, добавляли 0,157 г гидроксида натрия, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г белой сажи (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: H2O/SiO2=10, Темплат D/SiO2=0,15, NaOH/SiO2=0,08.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 110°С в течение 1 дня, и затем нагревали до 160°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре I-3. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 1800 нм, высотой 2400 нм и соотношением сторон 1,33. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 560 м2⋅г-1 и объем пор 0,496 мл/г. Дифрактограмма продукта была показана на Фигуре I-4. РФА показал, что молекулярное сито имеет Si/Al2=∞.
Пример I-5
В 45 мл тефлоновый контейнер добавляли 7,157 г Темплата В, добавляли 0,157 г гидроксида натрия, перемешивали в течение 30 минут до образования гомогенности и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98%, содержание Al2O3 0,253%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: H2O/SiO2=7,3, Темплат D/SiO2=0,15, NaOH/SiO2=0,08. РФА показал, что молекулярное сито имеет Si/Al2=625.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 130°С в течение 2 дней, и затем нагревали до 160°С для взаимодействия в течение 4 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре I-5. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 700 нм, высотой 950 нм и соотношением сторон 1,36. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 558 м2⋅г-1 и объем пор 0,443 мл/г. Дифрактограмма продукта была показана на Фигуре I-6.
Пример I-6
В 45 мл тефлоновый контейнер добавляли 8,024 г Темплата D, добавляли 0,157 г гидроксида натрия, перемешивали в течение 30 минут до образования гомогенности и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98%, содержание Al2O3 0,253%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: H2O/SiO2=7,3, Темплат D/SiO2=0,15, NaOH/SiO2=0,08.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре I-7. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 1200 нм, высотой 1400 нм и соотношением сторон 1,17. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 533 м2⋅г-1 и объем пор 0,295 мл/г. Дифрактограмма продукта была показана на Фигуре I-8. Результаты NH3-TPD показывают (Фигуры I-12), что молекулярные сита имели значительную кислотность. Результаты инфракрасного спектра показывают (Фигура I-13), что молекулярное сито имеет низкое содержание В-кислоты и высокое содержание L-кислоты.
Пример I-7
В 45 мл тефлоновый контейнер добавляли 7,157 г Темплата B, добавляли 0,314 г гидроксида натрия, и перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98%, содержание Al2O3 0,253%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: H2O/SiO2=7,3, Темплат B/SiO2=0,15, NaOH/SiO2=0,16.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали, и автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин и подвергали взаимодействию при 160°С в течение 6 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре I-9. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 400 нм, высотой 600 нм и соотношением сторон 1,50. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 568 м2⋅г-1 и объем пор 0,309 мл/г. РФА показал Si/Al2=521. Кривая адсорбции изотермы БЭТ и кривая распределения пор по диаметру показаны на Фигуре I-10 и Фигуре I-11 соответственно.
Краткое описание чертежей
На Фигуре I-1 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере I-3.
На Фигуре I-2 показана дифрактограмма молекулярного сита, полученного в Примере I-3.
На Фигуре I-3 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере I-4.
На Фигуре I-4 показана дифрактограмма молекулярного сита, полученного в Примере I-4.
На Фигуре I-5 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере I-5.
На Фигуре I-6 показана дифрактограмма молекулярного сита, полученного в Примере I-5.
На Фигуре I-7 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере I-6.
На Фигуре I-8 показана дифрактограмма молекулярного сита, полученного в Примере I-6.
На Фигуре I-9 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере I-7.
На Фигуре I-10 показана изотерма адсорбции-десорбции молекулярного сита, полученного в Примере I-7.
На Фигуре I-11 показана кривая распределения пор по диаметру молекулярного сита, полученного в Примере I-7.
На Фигуре I-12 показана модель NH3-TPD молекулярного сита, полученного в Примере I-6.
На Фигуре I-13 показан ИК-спектр молекулярного сита, полученного в Примере I-6.
Пример II серии
Пример II-1
Получение Темплата A
В 500 мл трехгорлую колбу добавляли 15 г (0,087 моль) тетраметилгексаметилендиамина, добавляли 250 мл изопропанола и добавляли по каплям 18,8 г (0,087 моль) 1,4-дибромбутана при комнатной температуре. Добавление завершали через 15 минут и температуру повышали до температуры нагревания с обратным холодильником. Раствор постепенно менялся от бесцветного и прозрачного до белого и мутного. Реакция сопровождалась высокоэффективной жидкостной хроматографией (ВЭЖХ). После завершения реакции к реакционной смеси добавляли 200 мл этилацетата и смесь нагревали с обратным холодильником в течение 1 часа, охлаждали и фильтровали с отсасыванием. Полученное твердое вещество промывали этилацетатом и затем диэтиловым эфиром с получением 30 г белого твердого продукта в виде 1,1,6,6-тетраметил-1,6-диаза-12-членного кольца-1,6-дибромида (соединение, в котором n равно 4, m равно 6, R представляет собой метил, X представляет собой Br), имеющего относительную молекулярную массу 388,2, температуру плавления 273,7°C. Спектр 1H-ЯМР показывает химический сдвиг (300 МГц, CDCl3) δ 1,50 (т, 4H), 1,90 (т, 8H), 3,14 (с, 12H), 3,40 (т, 8H).
Получение Темплата B: Br в Темплате A заменяли OH- с помощью ионного обмена; ионообменная смола представляла собой сильную основную анионообменную смолу стирольного типа, рабочий раствор представлял собой 15 масс.% водного раствора Темплата А, рабочая температура составляла 25°С, массовое соотношение рабочей жидкости и ионообменной смолы составляло 1:3; скорость потока составляла 3 капли/секунда; обменный раствор дегидратировали с помощью роторного испарителя с получением продукта, который представляет собой соединение формулы (I), в которой n равно 4, m равно 6, R представляет собой метил и X представляет собой ОН, имеющее относительную молекулярную массу 262,2 и чистоту 99,21%. Содержание брома в нем составляло 0,79 масс.%.
Пример II-2
Получение Темплата C
В 500 мл трехгорлую колбу добавляли 10 г (0,058 моль) тетраметилгексаметилендиамина, добавляли 250 мл изопропанола и добавляли по каплям 16,6 г (0,058 моль) 1,9-дибромдекана при комнатной температуре. Добавление завершали через 15 минут и температуру повышали до температуры нагревания с обратным холодильником. Раствор постепенно менялся от бесцветного и прозрачного до белого и мутного. Реакция сопровождалась высокоэффективной жидкостной хроматографией (ВЭЖХ). После завершения реакции к реакционной смеси добавляли 200 мл этилацетата и смесь нагревали с обратным холодильником в течение 1 часа, охлаждали и фильтровали с отсасыванием. Полученное твердое вещество промывали этилацетатом и затем диэтиловым эфиром с получением 25 г белого твердого продукта в виде 1,1,8,8-тетраметил-1,8-диаза-17-членного кольца-1,8-дибромида (соединение, в котором n равно 9, m равно 6, R представляет собой метил, X представляет собой Br), имеющего относительную молекулярную массу 458,4. Спектр 1H-ЯМР показывает химический сдвиг (300 МГц, CDCl3) δ 1,51 (т, 14H), 1,92 (т, 8H), 3,16 (с, 12H), 3,40 (т, 8H).
Получение Темплата D: Br в Темплате C заменяли OH- с помощью ионного обмена; ионообменная смола представляла собой сильную основную анионообменную смолу стирольного типа, рабочий раствор представлял собой 15 масс.% водного раствора Темплата C, рабочая температура составляла 25°С, массовое соотношение рабочей жидкости и ионообменной смолы составляло 1:3; скорость потока составляла 3 капли/секунда; обменный раствор дегидратировали с помощью роторного испарителя с получением продукта, который представляет собой соединение формулы (I), в которой n равно 9, m равно 6, R представляет собой метил, X представляет собой ОН, имеющее относительную молекулярную массу 332,4 и чистоту 99,8%. Содержание брома в нем составляло 0,2 масс.%.
Пример II-3
В 45 мл тефлоновый контейнер добавляли 1,467 г метаалюмината натрия, добавляли 1,925 г Темплата В и затем добавляли 3 г силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=35, H2O/SiO2=6,5, Темплат B/SiO2=0,15, NaOH/SiO2=0,08.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре II-1. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 650 нм, высотой 600 нм и соотношением сторон 1. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 553 м2⋅г-1 и объем пор 0,295 мл/г. Дифрактограмма продукта была показана на Фигуре II-2. Продукт имеет соотношение диоксид кремния-оксид алюминия 35,20.
Пример II-4
В 45 мл тефлоновый контейнер добавляли 1,23 г метаалюмината натрия, добавляли 1,925 г Темплата В и затем добавляли 9 г золя диоксида кремния (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 30%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=35, H2O/SiO2=7,1, Темплат B/SiO2=0,15, NaOH/SiO2=0,12. Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре II-3. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 1200 нм, высотой 1000 нм и соотношением сторон 0,833. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 558 м2⋅г-1 и объем пор 0,51 мл/г. Дифрактограмма продукта была показана на Фигуре II-4. Продукт имеет соотношение диоксид кремния-оксид алюминия 36,38.
Пример II-5
В 45 мл тефлоновый контейнер добавляли 1,957 г метаалюмината натрия, добавляли 2,44 г Темплата D, 0,157 г гидроксида натрия, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 9 г золя диоксида кремния (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 30%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=30, H2O/SiO2=7,1, Темплат D/SiO2=0,15, NaOH/SiO2=0,2.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре II-5. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 900 нм, высотой 1000 нм и соотношением сторон 1,11. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 543 м2⋅г-1 и объем пор 0,304 мл/г. Дифрактограмма продукта была показана на Фигуре II-6. Продукт имеет соотношение диоксид кремния-оксид алюминия 33,68.
Пример II-6
В 45 мл тефлоновый контейнер добавляли 1,23 г метаалюмината натрия, добавляли 2,44 г Темплата D, 0,353 г гидроксида натрия и затем 3 г белой сажи (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=35, H2O/SiO2=6,5, Темплат D/SiO2=0,15, NaOH/SiO2=0,30. Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 4 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре II-7. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 1200 нм, высотой 1300 нм и соотношением сторон 1,08. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 534 м2⋅г-1 и объем пор 0,304 мл/г. Дифрактограмма продукта была показана на Фигуре II-8. Продукт имеет соотношение диоксид кремния-оксид алюминия 30,21.
Пример II-7
В 45 мл тефлоновый контейнер добавляли 0,75 г порошка SB (импортированного из Германии, содержание Al2O3 76,5%), добавляли 2,85 г Темплата А и затем добавляли 3 г силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=30, H2O/SiO2=6,5, Темплат B/SiO2=0,15, NaOH/SiO2=0,08. Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре II-9. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 1500 нм, высотой 2000 нм и соотношением сторон 1,33. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 560 м2⋅г-1 и объем пор 0,342 мл/г. Дифрактограмма продукта была показана на Фигуре II-10. Продукт имеет соотношение диоксид кремния-оксид алюминия 35,29.
Пример II-8
В 45 мл тефлоновый контейнер добавляли 1,957 г метаалюмината натрия, добавляли 3,37 г Темплата С, 0,274 г гидроксида натрия и затем 3 г силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=30, H2O/SiO2=6,5, Темплат B/SiO2=0,15, NaOH/SiO2=0,30. Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре II-11. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 1000 нм, высотой 1400 нм и соотношением сторон 1,4. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 498 м2⋅г-1 и объем пор 0,403 мл/г. Дифрактограмма продукта была показана на Фигуре II-12. Продукт имеет соотношение диоксид кремния-оксид алюминия 34,20.
Пример II-9
В 45 мл тефлоновый контейнер добавляли 1,957 г метаалюмината натрия, добавляли 3,365 г Темплата C, 0,0784 г гидроксида натрия и затем 9 г золя диоксида кремния (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 30%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=30, H2O/SiO2=6,5, Темплат C/SiO2=0,15, NaOH/SiO2=0,20. Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре II-13. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной столбчатой кристаллической частицы с эффективным диаметром 800 нм, высотой 900 нм и соотношением сторон 1,125. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 564 м2⋅г-1 и объем пор 0,350 мл/г. Дифрактограмма продукта была показана на Фигуре II-14. Продукт имеет соотношение диоксид кремния-оксид алюминия 35,28.
Пример II-10
В 45 мл тефлоновый контейнер добавляли 0,187 г порошка SB (импортированного из Германии, содержание Al2O3 76,5%), добавляли 1,925 г Темплата B и затем добавляли 3 г силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=20, H2O/SiO2=6,5, Темплат B/SiO2=0,15, NaOH/SiO2=0,08. Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 110°С в течение 1 дня и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре II-15. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 300 нм, высотой 900 нм и соотношением сторон 3,0. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 473 м2⋅г-1 и объем пор 0,356 мл/г. Дифрактограмма продукта была показана на Фигуре II-16. Продукт имеет соотношение диоксид кремния - оксид алюминия 35,38.
Пример II-11
В 45 мл тефлоновый контейнер добавляли 5,87 г метаалюмината натрия, добавляли 1,925 г Темплата В и затем добавляли 3 г силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=10, H2O/SiO2=6,5, Темплат B/SiO2=0,15, NaOH/SiO2=0,487.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Дифрактограмма данного продукта была показана на Фигуре II-17, который представляет собой молекулярное сито ANA.
Пример II-12
Данный пример был приведен для иллюстрации термической стабильности (XRD) молекулярных сит, полученных в Примерах от II-3 до II-8. На Фигуре II-18 показано, что соотношение диоксид кремния - оксид алюминия составляло 30 или 40 и молекулярное сито с соотношением «натрий-кремний» 0,08-0,30 обладает хорошей термической стабильностью после обжига при 550°C, 650°C или 750°C в течение 6 ч.
Пример II-13
Данный пример был приведен для иллюстрации кислотности молекулярных сит, полученных в Примерах II-6 и II-8. В Таблице II-1 показано, что молекулярные сита с соотношением диоксид кремния-оксид алюминия 30 или 40 имеют более высокое соотношение B/L, и ожидается, что они будут использованы в кислотных каталитических реакциях.
Таблица II-1
Краткое описание чертежей
На Фигуре II-1 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере II-3.
На Фигуре II-2 показана дифрактограмма молекулярного сита, полученного в Примере II-3.
На Фигуре II-3 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере II-4.
На Фигуре II-4 показана дифрактограмма молекулярного сита, полученного в Примере II-4.
На Фигуре II-5 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере II-5.
На Фигуре II-6 показана дифрактограмма молекулярного сита, полученного в Примере II-5.
На Фигуре II-7 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере II-6.
На Фигуре II-8 показана дифрактограмма молекулярного сита, полученного в Примере II-6.
На Фигуре II-9 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере II-7.
На Фигуре II-10 показана дифрактограмма молекулярного сита, полученного в Примере II-7.
На Фигуре II-11 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере II-8.
На Фигуре II-12 показана дифрактограмма молекулярного сита, полученного в Примере II-8.
На Фигуре II-13 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере II-9.
На Фигуре II-14 показана дифрактограмма молекулярного сита, полученного в Примере II-9.
На Фигуре II-15 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере II-10.
На Фигуре II-16 показана дифрактограмма молекулярного сита, полученного в Примере II-10.
На Фигуре II-17 показана дифрактограмма молекулярного сита, полученного в Примере II-11.
На Фигуре II-18 показана дифрактограмма молекулярных сит, полученных в Примерах от II-3 до II-8 после обжига.
Первый вариант осуществления, пятый вариант осуществления и шестой вариант осуществления
В контексте настоящего варианта осуществления, включая следующие примеры и сравнительные примеры, общую удельную площадь поверхности, объем пор и диаметр пор молекулярного сита измеряли с помощью следующих аналитических методов.
Оборудование: Статический адсорбер азота Micromeritics ASAP2010.
Условия измерения: образец помещали в систему для обработки образца, создавали вакуум до 1,35×10-2 Па при 350°С и выдерживали при данной температуре и давлении в течение 15 ч для очистки образца. При температуре жидкого азота -196°С измеряли количество адсорбции и количество десорбции азота очищенного образца в условиях различного удельного давления P/P0 и получали изотерму адсорбции-десорбции. Затем общую удельную площадь поверхности рассчитывали с помощью двухпараметрического уравнения БЭТ, и адсорбционную емкость при удельном давлении P/P0≈0,98 принимали за объем пор образца, и диаметр пор рассчитывали в соответствии с моделью BJH.
Пример III серии
Пример III-1
Получение Темплата А: в двухгорлую колбу добавляли 15 г (0,094 моль) бис[2-(N,N-диметиламиноэтил)]простого эфира, добавляли 100 мл изопропанола и добавляли по каплям 9,5 г (0,047 моль) 1,3-дибромпропана при перемешивании при 25°С. После завершения добавления температуру повышали до температуры нагревания с обратным холодильником и полученную смесь нагревали с обратным холодильником в течение 30 мин. Раствор менялся от бесцветного до белого и мутного, и затем его подвергали взаимодействию при температуре нагревания с обратным холодильником в течение 12 ч, охлаждали до 25°С и добавляли 50 мл этилацетата. Полученную смесь перемешивали в течение 15 мин с образованием белой мутной жидкости, и отфильтровывали, и полученное твердое вещество промывали этилацетатом с получением 13,2 г продукта в виде соединения формулы (I), в которой n равно 1, m равно 2, R представляет собой метил и X представляет собой Br, имеющего температуру плавления 250,3°С, чистоту 99,9 масс.%, относительную молекулярную массу 362,2. Спектр 1H-ЯМР показывает химический сдвиг (300 МГц, внутренний стандарт ТМС, растворитель CDCl2) δ (м.д.): 1,49 (2H, м), 2,27 (4H, м), 2,36 (4H, т), 2,53 (4H, т) 3,47 (4H, т).
Получение Темплата B: Br в Темплате A заменяли OH- с помощью ионного обмена; ионообменная смола представляла собой сильную основную анионообменную смолу стирольного типа, рабочий раствор представлял собой 15 масс.% водного раствора Темплата А, рабочая температура составляла 25°С, массовое соотношение рабочей жидкости и ионообменной смолы составляло 1:3; скорость потока составляла 3 капли/секунда; обменный раствор дегидратировали с помощью роторного испарителя с получением продукта, который представляет собой соединение формулы (I), в которой n равно 1, m равно 2, R представляет собой метил и X представляет собой ОН, имеющее относительную молекулярную массу 236,2 и чистоту 98,2%. Содержание брома в нем составляло 0,79 масс.%.
Пример III-2
Получение Темплата C: соединение формулы (I), в которой n равно 6, m равно 2, R представляет собой метил и X представляет собой Br, получали в соответствии со способом, описанным в Примере III-1 для Темплата A, за исключением того, что 12,78 г (0,047 моль) 1,8-дибромоктана использовали вместо 1,3-дибромпропана. Испытание предоставило 17,6 г продукта, имеющего температуру плавления 288,2°С, относительную молекулярную массу 432,2, чистоту 99,9 масс.%. Спектр 1H-ЯМР показывает химический сдвиг (300 МГц, внутренний стандарт ТМС, растворитель CDCl2) δ (м.д.): 1,29 (2H, с), 1,39 (2H, м), 1,43 (2H, с), 2,27 (2H, м), 2,36 (2H, м), 2,55 (2H, м), 3,63 (4H, м).
Получение Темплата D: Br в Темплате C заменяли OH- с помощью ионного обмена; ионообменная смола представляла собой сильную основную анионообменную смолу стирольного типа, рабочий раствор представлял собой 15 масс.% водного раствора Темплата C, рабочая температура составляла 25°С, массовое соотношение рабочей жидкости и ионообменной смолы составляло 1:3; скорость потока составляла 3 капли/секунда; обменный раствор дегидратировали с помощью роторного испарителя с получением продукта, который представляет собой соединение формулы (I), в которой n равно 6, m равно 2, R представляет собой метил, X представляет собой ОН, имеющее относительную молекулярную массу 306,2 и чистоту 99,5 масс.%. Содержание брома в нем составляло 0,2 масс.%.
Пример III-3
В 45 мл тефлоновый контейнер добавляли 0,134 г метаалюмината натрия, добавляли 1,81 г Темплата B, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%) и 6,3 г деионизированной воды, и тщательно перемешивали в течение 5 минут. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=25, H2O/SiO2=7, Темплат B/SiO2=0,16, OH-/SiO2=0,31.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали, и автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин и подвергали взаимодействию при 150°С в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре III-3. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 1000 нм, высотой 1200 нм и соотношением сторон 1,2. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 523 м2⋅г-1 и объем пор 0,356 мл/г. РФА показал Si/Al2=23. Дифрактограмма данного продукта была показана на Фигуре III-4. На Фигуре III-1 показана кривая адсорбции 2,2-диэтилбутана на продукте после обжига при 550°С в течение 3 ч. Из кривой можно увидеть, что адсорбционная емкость продукта для 2,2-диэтилбутана составляет ~55 мг/г.
Пример III-4
В 45 мл тефлоновый контейнер добавляли 0,134 г метаалюмината натрия, добавляли 3,0 г Темплата D и 9,31 г деионизированной воды, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 4 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%) и тщательно перемешивали в течение 5 минут. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=21, H2O/SiO2=8, Темплат D/SiO2=0,15, OH-/SiO2=0,30. Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали, и автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин и подвергали взаимодействию при 150°С в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре III-6. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной столбчатой кристаллической частицы с эффективным диаметром 2200 нм, высотой 3000 нм и соотношением сторон 1,36. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 573 м2⋅г-1 и объем пор 0,387 мл/г. РФА показал Si/Al2=25.
Пример III-5
В 45 мл тефлоновый контейнер добавляли 0,134 г метаалюмината натрия, добавляли 1,78 г Темплата А, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), 6,98 г деионизированной воды и 0,4 г NaOH, и тщательно перемешивали в течение 5 минут. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=23, H2O/SiO2=8, Темплат A/SiO2=0,10, OH-/SiO2=0,20. Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали, и автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин и подвергали взаимодействию при 150°С в течение 4 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре III-7. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 900 нм, высотой 1200 нм и соотношением сторон 1,33. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 520 м2⋅г-1 и объем пор 0,367 мл/г. РФА показал Si/Al2=24.
Пример III-6
В 45 мл тефлоновый контейнер добавляли 0,134 г метаалюмината натрия, добавляли 3,70 г Темплата В, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%) и 6,11 г деионизированной воды, тщательно перемешивали в течение 5 минут. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=26, H2O/SiO2=7, Темплат B/SiO2=0,32, OH-/SiO2=0,64.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали, и автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин и подвергали взаимодействию при 150°С в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре III-8. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной столбчатой кристаллической частицы с эффективным диаметром 1000 нм, высотой 1500 нм и соотношением сторон 1,5. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 537 м2⋅г-1 и объем пор 0,389 мл/г. РФА показал Si/Al2=24.
Продукт подвергали обжигу отдельно при 550°C, 650°C и 750°C в течение 3 ч, и дифрактограмма продукта после обжига была показана на Фигуре III-5, причем существовал каждый характерный пик. На Фигуре III-2 показана кривая адсорбции 3-пропил-4-бутилоктана на продукте после обжига при 550°С в течение 3 ч. Из кривой можно увидеть, что адсорбционная емкость продукта для 3-пропил-4-бутилоктила представляет собой до ~102 мг/г.
Пример III-7
В 45 мл тефлоновый контейнер добавляли 0,134 г метаалюмината натрия, и добавляли 7,0 г Темплата D и 9,31 г деионизированной воды, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 4 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), и тщательно перемешивали в течение 5 минут. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=22, H2O/SiO2=8, Темплат D/SiO2=0,35, OH-/SiO2=0,70. Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали, и автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин и подвергали взаимодействию при 150°С в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре III-9. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 1200 нм, высотой 1400 нм и соотношением сторон 1,17. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 538 м2⋅г-1 и объем пор 0,408 мл/г. РФА показал Si/Al2=23. Результаты NH3-TPD показывают (Фигура III-11), что молекулярное сито имеет значительную кислотность. Результаты инфракрасного спектра показывают (Фигура III-12), что молекулярное сито имеет низкое содержание В-кислоты и высокое содержание L-кислоты.
Пример III-8
В 45 мл тефлоновый контейнер добавляли 0,134 г метаалюмината натрия, добавляли 7,41 г Темплата С, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), 6,98 г деионизированной воды и 0,4 г NaOH, и тщательно перемешивали в течение 5 минут. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=21, H2O/SiO2=8, Темплат C/SiO2=0,35, OH-/SiO2=0,20. Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали, и автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин и подвергали взаимодействию при 150°С в течение 4 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре III-10. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 1200 нм, высотой 1600 нм и соотношением сторон 1,33. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 546 м2⋅г-1 и объем пор 0,397 мл/г. РФА показал Si/Al2=28.
Краткое описание чертежей
На Фигуре III-1 показана кривая адсорбции 2,2-диэтилбутана на молекулярном сите, полученном в Примере III-3, после обжига.
На Фигуре III-2 показана кривая адсорбции 3-пропил-4-бутилоктана на молекулярном сите, полученном в Примере III-6, после обжига.
На Фигуре III-3 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере III-3.
На Фигуре III-4 показана дифрактограмма молекулярного сита, полученного в Примере III-3.
На Фигуре III-5 показана дифрактограмма молекулярного сита, полученного в Примере III-6, после обжига.
На Фигуре III-6 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере III-4.
На Фигуре III-7 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере III-5.
На Фигуре III-8 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере III-6.
На Фигуре III-9 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере III-7.
На Фигуре III-10 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере III-8.
На Фигуре III-11 показана модель NH3-TPD молекулярного сита, полученного в Примере III-7.
На Фигуре III-12 показаны ИК-спектры молекулярного сита, полученного в Примере III-7.
Пример IV серии
Пример IV-1
Получение Темплата А: в двухгорлую колбу добавляли 15 г (0,094 моль) бис[2-(N,N-диметиламиноэтил)]простого эфира, добавляли 100 мл изопропанола и добавляли по каплям 9,5 г (0,047 моль) 1,3-дибромпропана при перемешивании при 25°С. После завершения добавления температуру повышали до температуры нагревания с обратным холодильником и полученную смесь нагревали с обратным холодильником в течение 30 мин. Раствор менялся от бесцветного до белого и мутного, и затем его подвергали взаимодействию при температуре нагревания с обратным холодильником в течение 12 ч, охлаждали до 25°С и добавляли 50 мл этилацетата. Полученную смесь перемешивали в течение 15 мин с образованием белой мутной жидкости, и отфильтровывали, и полученное твердое вещество промывали этилацетатом с получением 13,2 г продукта в виде соединения формулы (I), в которой n равно 1, m равно 2, R представляет собой метил и X представляет собой Br, имеющего температуру плавления 250,3°С, чистоту 99,9 масс.%, относительную молекулярную массу 362,2. Спектр 1H-ЯМР показывает химический сдвиг (300 МГц, внутренний стандарт ТМС, растворитель CDCl2) δ (м.д.): 1,49 (2H, м), 2,27 (4H, м), 2,36 (4H, т), 2,53 (4H, т) 3,47 (4H, т).
Получение Темплата B: Br в Темплате A заменяли OH- с помощью ионного обмена; ионообменная смола представляла собой сильную основную анионообменную смолу стирольного типа, рабочий раствор представлял собой 15 масс.% водного раствора Темплата А, рабочая температура составляла 25°С, массовое соотношение рабочей жидкости и ионообменной смолы составляло 1:3; скорость потока составляла 3 капли/секунда; обменный раствор дегидратировали с помощью роторного испарителя с получением продукта, который представляет собой соединение формулы (I), в которой n равно 1, m равно 2, R представляет собой метил и X представляет собой ОН, имеющее относительную молекулярную массу 236,2 и чистоту 99,21%. Содержание брома в нем составляло 0,79 масс.%.
Пример IV-2
Получение Темплата C: соединение формулы (I), в которой n равно 6, m равно 2, R представляет собой метил и X представляет собой Br, получали в соответствии со способом, описанным в Примере IV-1 для Темплата A, за исключением того, что 12,78 г (0,047 моль) 1,8-дибромоктана использовали вместо 1,3-дибромпропана. Испытание предоставило 17,6 г продукта, имеющего температуру плавления 288,2°С, относительную молекулярную массу 432,2, чистоту 99,9 масс.%. Спектр 1H-ЯМР показывает химический сдвиг (300 МГц, внутренний стандарт ТМС, растворитель CDCl2) δ (м.д.): 1,29 (2H, с), 1,39 (2H, м), 1,43 (2H, с), 2,27 (2H, м), 2,36 (2H, м), 2,55 (2H, м), 3,63 (4H, м).
Получение Темплата D: Br в Темплате C заменяли OH- с помощью ионного обмена; ионообменная смола представляла собой сильную основную анионообменную смолу стирольного типа, рабочий раствор представлял собой 15 масс.% водного раствора Темплата C, рабочая температура составляла 25°С, массовое соотношение рабочей жидкости и ионообменной смолы составляло 1:3; скорость потока составляла 3 капли/секунда; обменный раствор дегидратировали с помощью роторного испарителя с получением продукта, который представляет собой соединение формулы (I), в которой n равно 6, m равно 2, R представляет собой метил, X представляет собой ОН, имеющее относительную молекулярную массу 306,2 и чистоту 99,8%. Содержание брома в нем составляло 0,2 масс.%.
Пример IV-3
В 45 мл тефлоновый контейнер добавляли 6,975 г Темплата D, добавляли 0,259 г метаалюмината натрия, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98%, содержание Al2O3 0,253%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=201, H2O/SiO2=5,8, Темплат D/SiO2=0,15, NaOH/SiO2=0,05.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 160°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре IV-1. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 800 нм, высотой 1000 нм и соотношением сторон 1,25. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 564 м2⋅г-1 и объем пор 0,394 мл/г. Дифрактограмма продукта была показана на Фигуре IV-2. РФА показал, что молекулярное сито имеет Si/Al2=203.
Пример IV-4
В 45 мл тефлоновый контейнер добавляли 3,71 г Темплата В, добавляли 0,246 г метаалюмината натрия, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г белой сажи (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98%) и 6,02 г деионизированной воды, и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=240, H2O/SiO2=7,1, Темплат B/SiO2=0,15, NaOH/SiO2=0,04.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 110°С в течение 1 дня, и затем нагревали до 170°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре IV-3. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 800 нм, высотой 1000 нм и соотношением сторон 1,25. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 483 м2⋅г-1 и объем пор 0,285 мл/г. Дифрактограмма продукта была показана на Фигуре IV-4. РФА показал, что молекулярное сито имеет Si/Al2=226.
Пример IV-5
В 45 мл тефлоновый контейнер добавляли 4,65 г Темплата D, добавляли метаалюминат натрия до 0,245 г, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98%, содержание Al2O3 составляло 0,253%) и 6 г деионизированной воды, и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=242, H2O/SiO2=9,5, Темплат D/SiO2=0,10, NaOH/SiO2=0,03.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 160°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре IV-5. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 700 нм, высотой 900 нм и соотношением сторон 1,285. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 464 м2⋅г-1 и объем пор 0,384 мл/г. Дифрактограмма продукта была показана на Фигуре IV-6. РФА показал, что молекулярное сито имеет Si/Al2=253.
Пример IV-6
В 45 мл тефлоновый контейнер добавляли 5 г Темплата B, добавляли 0,159 г метаалюмината натрия, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98%, содержание Al2O3 0,253%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=300, H2O/SiO2=7,3, Темплат B/SiO2=0,15, NaOH/SiO2=0,03.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре IV-7. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию гексагональной призматической кристаллической частицы с эффективным диаметром 1000 нм, высотой 1000 нм и соотношением сторон 1,0. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 538 м2⋅г-1 и объем пор 0,376 мл/г. Дифрактограмма продукта была показана на Фигуре IV-8. РФА показал, что молекулярное сито имеет Si/Al2=304.
Пример IV-7
В 45 мл тефлоновый контейнер добавляли 6,975 г Темплата D, добавляли 0,295 г метаалюмината натрия, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98%, содержание Al2O3 0,253%) и 3,5 г деионизированной воды, и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=201, H2O/SiO2=9,8, Темплат D/SiO2=0,15, NaOH/SiO2=0,025.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 2 дней, и затем нагревали до 150°С для взаимодействия в течение 3 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта. РФА показал Si/Al2=207.
Пример IV-8
В 45 мл тефлоновый контейнер добавляли 3,70 г Темплата В, добавляли 0,288 г метаалюмината натрия, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98%, содержание Al2O3 0,253%) и 3,2 г деионизированной воды, и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=205, H2O/SiO2=8,2, Темплат В/SiO2=0,15, NaOH/SiO2=0,025.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 6 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта. РФА показал Si/Al2=211. Результаты NH3-TPD показывают (Фигура IV-9), что молекулярное сито имеет значительную кислотность. Результаты инфракрасного спектра показывают (Фигура IV-10), что молекулярное сито имеет низкое содержание В-кислоты и высокое содержание L-кислоты.
Пример IV-9
В 45 мл тефлоновый контейнер добавляли 4,65 г Темплата D, добавляли 0,297 г метаалюмината натрия, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г белой сажи (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98%) и 4 г деионизированной воды, и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=204, H2O/SiO2=8,4, Темплат D/SiO2=0,10, NaOH/SiO2=0,05.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 160°С для взаимодействия в течение 4 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта. РФА показал Si/Al2=207.
Краткое описание чертежей
На Фигуре IV-1 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере IV-3.
На Фигуре IV-2 показана дифрактограмма молекулярного сита, полученного в Примере IV-3.
На Фигуре IV-3 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере IV-4.
На Фигуре IV-4 показана дифрактограмма молекулярного сита, полученного в Примере IV-4.
На Фигуре IV-5 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере IV-5.
На Фигуре IV-6 показана дифрактограмма молекулярного сита, полученного в Примере IV-5.
На Фигуре IV-7 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере IV-6.
На Фигуре IV-8 показана дифрактограмма молекулярного сита, полученного в Примере IV-6.
На Фигуре IV-9 показана модель NH3-TPD молекулярного сита, полученного в Примере IV-8.
На Фигуре IV-10 показаны ИК-спектры молекулярного сита, полученного в Примере IV-8.
Второй вариант осуществления и шестой вариант осуществления
В контексте настоящего варианта осуществления, включая следующие примеры и сравнительные примеры, общую удельную площадь поверхности, объем пор и диаметр пор молекулярного сита измеряли с помощью следующих аналитических методов.
Оборудование: Статический адсорбер азота Micromeritics ASAP2010
Условия измерения: образец помещали в систему для обработки образца, и создавали вакуум до 1,33×10-2 Па при 300°С, и выдерживали при данной температуре и давлении в течение 8 ч для очистки образца. При температуре жидкого азота -196°С измеряли количество адсорбции и количество десорбции азота очищенного образца в условиях различного удельного давления P/P0 и получали изотерму адсорбции-десорбции. Затем удельную площадь поверхности рассчитывали с помощью двухпараметрической формулы Хорвата-Кавадзоэ. Адсорбционную емкость при удельном давлении P/P0≈0,983 принимали за объем пор образца и диаметр пор рассчитывали в соответствии с моделью теории функционала плотности ТФП.
В данных вариантах осуществления, включая следующие примеры и сравнительные примеры, общую удельную площадь поверхности, объем пор и диаметр пор мезопор молекулярного сита измеряли с помощью следующих аналитических методов.
Оборудование: Статический адсорбер азота Micromeritics ASAP2010
Условия измерения: образец помещали в систему для обработки образца, создавали вакуум до 1,35×10-2 Па при 350°С и выдерживали при данной температуре и давлении в течение 15 ч для очистки образца. При температуре жидкого азота -196°С измеряли количество адсорбции и количество десорбции азота очищенного образца в условиях различного удельного давления P/P0 и получали изотерму адсорбции-десорбции. Затем удельную площадь поверхности рассчитывали с помощью двухпараметрического уравнения БЭТ, и адсорбционную емкость при удельном давлении P/P0≈0,98 принимали за объем пор образца, и диаметр пор рассчитывали в соответствии с моделью Хорвата-Кавадзоэ.
В данных вариантах осуществления, включая следующие примеры и сравнительные примеры, общую удельную площадь поверхности, объем пор и диаметр пор крупных пор молекулярного сита измеряли с помощью следующих аналитических методов.
Оборудование: Ртутный порозиметр AutoPore IV 9510 Micromeritics
Условия измерения: соответствующее количество сухого образца помещали в пробирку для образцов, помещали в прибор, создавали вакуум до 50 мкг для работы при низком давлении, и взвешивание завершали при низком давлении. Пробирку с образцом, заполненную ртутью, помещали в камеру высокого давления и дополнительно создавали давление до 60000 pisa, чтобы заставить ртуть войти в поры. Основываясь на созданном давлении P, можно рассчитать соответствующий диаметр пор r (нм). Объем пор, имеющих соответствующий размер, может быть рассчитан на основе количества введенной ртути, и может быть получена кривая объема пор в зависимости от диаметра пор, тем самым обеспечивая кривую распределения пор по диаметру. Длина поры рассчитывалась из объема пор и диаметра пор при условии, что пора представляла собой столбчатое сквозное отверстие, и площадь поверхности была получена путем умножения окружности поры на ее длину.
Пример V-1
Получение Темплата А: в двухгорлую колбу добавляли 15 г (0,094 моль) бис[2-(N,N-диметиламиноэтил)]простого эфира, добавляли 100 мл изопропанола и добавляли по каплям 9,5 г (0,047 моль) 1,3-дибромпропана при перемешивании при 25°С. После завершения добавления температуру повышали до температуры нагревания с обратным холодильником и полученную смесь нагревали с обратным холодильником в течение 30 мин. Раствор менялся от бесцветного до белого и мутного, и затем его подвергали взаимодействию при температуре нагревания с обратным холодильником в течение 12 ч, охлаждали до 25°С и добавляли 50 мл этилацетата. Полученную смесь перемешивали в течение 15 мин с образованием белой мутной жидкости, и отфильтровывали, и полученное твердое вещество промывали этилацетатом с получением 13,2 г продукта в виде соединения формулы (I), в которой n равно 1, m равно 2, R представляет собой метил и X представляет собой Br, имеющего температуру плавления 250,3°С, чистоту 99,9 масс.%, относительную молекулярную массу 362,2. Спектр 1H-ЯМР показывает химический сдвиг (300 МГц, внутренний стандарт ТМС, растворитель CDCl2) δ (м.д.): 1,49 (2H, м), 2,27 (4H, м), 2,36 (4H, т), 2,53 (4H, т) 3,47 (4H, т).
Получение Темплата B: Br в Темплате A заменяли OH- с помощью ионного обмена; ионообменная смола представляла собой сильную основную анионообменную смолу стирольного типа, рабочий раствор представлял собой 15 масс.% водного раствора Темплата А, рабочая температура составляла 25°С, массовое соотношение рабочей жидкости и ионообменной смолы составляло 1:3; скорость потока составляла 3 капли/секунда; обменный раствор дегидратировали с помощью роторного испарителя с получением продукта, который представляет собой соединение формулы (I), в которой n равно 1, m равно 2, R представляет собой метил и X представляет собой ОН, имеющее относительную молекулярную массу 236,2 и чистоту 98,2%. Содержание брома в нем составляло 0,79 масс.%.
Пример V-2
Получение Темплата C: соединение формулы (I), в которой n равно 6, m равно 2, R представляет собой метил и X представляет собой Br, получали в соответствии со способом, описанным в Примере V-1 для Темплата A, за исключением того, что 12,78 г (0,047 моль) 1,8-дибромоктана использовали вместо 1,3-дибромпропана. Испытание предоставило 17,6 г продукта, имеющего температуру плавления 288,2°С, относительную молекулярную массу 432,2, чистоту 99,9 масс.%. Спектр 1H-ЯМР показывает химический сдвиг (300 МГц, внутренний стандарт ТМС, растворитель CDCl2) δ (м.д.): 1,29 (2H, с), 1,39 (2H, м), 1,43 (2H, с), 2,27 (2H, м), 2,36 (2H, м), 2,55 (2H, м), 3,63 (4H, м).
Получение Темплата D: Br в Темплате C заменяли OH- с помощью ионного обмена; ионообменная смола представляла собой сильную основную анионообменную смолу стирольного типа, рабочий раствор представлял собой 15 масс.% водного раствора Темплата C, рабочая температура составляла 25°С, массовое соотношение рабочей жидкости и ионообменной смолы составляло 1:3; скорость потока составляла 3 капли/секунда; обменный раствор дегидратировали с помощью роторного испарителя с получением продукта, который представляет собой соединение формулы (I), в которой n равно 6, m равно 2, R представляет собой метил, X представляет собой ОН, имеющее относительную молекулярную массу 306,2 и чистоту 99,5 масс.%. Содержание брома в нем составляло 0,2 масс.%.
Пример V-3
В 45 мл тефлоновый контейнер добавляли 0,134 г метаалюмината натрия, добавляли 1,81 г Темплата B, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%) и 6,3 г деионизированной воды, и тщательно перемешивали в течение 5 минут. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=61, H2O/SiO2=7, Темплат B/SiO2=0,16, OH-/SiO2=0,31.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали, и автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин и подвергали взаимодействию при 160°С в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре V-3. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию кристаллической частицы гексагональной призматической формы и губчатой структуры и имеет эффективный диаметр 2500 нм, высоту 1000 нм и соотношение сторон 0,4. В соответствии с измерением молекулярное сито содержит крупные поры, мезопоры и микропоры, в котором крупные поры имеют диаметр 150 нм, общую удельную площадь поверхности 89 м2⋅г-1 и объем пор 1,36 мл/г; мезопоры имеют диаметр 4 нм, общую удельную площадь поверхности 126 м2⋅г-1 и объем пор 0,29 мл/г; и микропоры имеют диаметр 0,5 нм и 1,2 нм, общую удельную площадь поверхности 163 м2⋅г-1 и объем пор 0,07 мл/г. РФА показал Si/Al2=48.
Дифрактограмма данного продукта была показана на Фигуре V-4. На Фигуре V-1 показана кривая адсорбции 2,2-диэтилбутана на продукте после обжига при 550°С в течение 3 ч. Из кривой можно увидеть, что адсорбционная емкость продукта для 2,2-диэтилбутана составляет ~55 мг/г.
Пример V-4
В 45 мл тефлоновый контейнер добавляли 0,134 г метаалюмината натрия, добавляли 3,0 г Темплата D, 9,31 г деионизированной воды, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 4 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), и тщательно перемешивали в течение 5 минут. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=81, H2O/SiO2=8, Темплат D/SiO2=0,15, OH-/SiO2=0,30.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали, и автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин и подвергали взаимодействию при 160°С в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре V-6. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию кристаллической частицы гексагональной призматической формы и губчатой структуры с эффективным диаметром 2500 нм, высотой 850 нм и соотношением сторон 0,34. В соответствии с измерением молекулярное сито содержит крупные поры, мезопоры и микропоры, в котором крупные поры имеют диаметр 400 нм, общую удельную площадь поверхности 65 м2⋅г-1 и объем пор 0,387 мл/г; мезопоры имеют диаметр 5 нм, общую удельную площадь поверхности 116 м2⋅г-1 и объем пор 0,28 мл/г; и микропоры имеют диаметр 0,5 нм и 1,2 нм, общую удельную площадь поверхности 149 м2⋅г-1 и объем пор 0,107 мл/г. РФА показал Si/Al2=75.
Пример V-5
В 45 мл тефлоновый контейнер добавляли 0,134 г метаалюмината натрия, добавляли 1,78 г Темплата А, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), 6,98 г деионизированной воды и 0,4 г NaOH, и тщательно перемешивали в течение 5 минут. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=61, H2O/SiO2=8, Темплат A/SiO2=0,10, OH-/SiO2=0,20.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали, и автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин и подвергали взаимодействию при 160°С в течение 4 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре V-7. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию кристаллической частицы гексагональной призматической формы и губчатой структуры с эффективным диаметром 2200 нм, высотой 3500 нм и соотношением сторон 1,59. В соответствии с измерением молекулярное сито содержит крупные поры, мезопоры и микропоры, в котором крупные поры имеют диаметр 100 нм, общую удельную площадь поверхности 365 м2⋅г-1 и объем пор 0,365 мл/г; мезопоры имеют диаметр 8 нм, общую удельную площадь поверхности 115 м2⋅г-1 и объем пор 0,22 мл/г; и микропоры имеют диаметр 4 нм и 1,2 нм, общую удельную поверхность 280 м2⋅г-1 и объем пор 0,145 мл/г. РФА показал Si/Al2=56.
Пример V-6
В 45 мл тефлоновый контейнер добавляли 0,134 г метаалюмината натрия, добавляли 3,70 г Темплата В, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%) и 6,11 г деионизированной воды, тщательно перемешивали в течение 5 минут. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=61, H2O/SiO2=7, Темплат B/SiO2=0,32, OH-/SiO2=0,64.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали, и автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин и подвергали взаимодействию при 160°С в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре V-8. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию кристаллической частицы гексагональной призматической формы и губчатой структуры с эффективным диаметром 1750 нм, высотой 4000 нм и соотношением сторон 2,29. В соответствии с измерением молекулярное сито содержит крупные поры, мезопоры и микропоры, в котором крупные поры имеют диаметр 200 нм, общую удельную площадь поверхности 65 м2⋅г-1 и объем пор 0,390 мл/г; мезопоры имеют диаметр 9 нм, общую удельную площадь поверхности 145 м2⋅г-1 и объем пор 0,16 мл/г; и микропоры имеют диаметр 4 нм и 1,2 нм, общую удельную площадь поверхности 220 м2⋅г-1 и объем пор 0,130 мл/г РФА показал Si/Al2=54.
Продукт подвергали обжигу отдельно при 550°C, 650°C и 750°C в течение 3 ч, и дифрактограмма продукта после обжига показана на Фигуре V-5, причем существовал каждый характерный пик. На Фигуре V-2 показана кривая адсорбции 3-пропил-4-бутилоктана на продукте после обжига при 550°С в течение 3 ч. Из кривой можно увидеть, что адсорбционная емкость продукта для 3-пропил-4-бутилоктила представляет собой до ~102 мг/г.
Пример V-7
В 45 мл тефлоновый контейнер добавляли 0,134 г метаалюмината натрия, и добавляли 7,0 г Темплата D и 9,31 г деионизированной воды, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 4 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), и тщательно перемешивали в течение 5 минут. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=82, H2O/SiO2=8, Темплат D/SiO2=0,35, OH-/SiO2=0,70.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали, и автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин и подвергали взаимодействию при 160°С в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре V-9. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию кристаллической частицы гексагональной призматической формы и губчатой структуры с эффективным диаметром 1200 нм, высотой 1500 нм и соотношением сторон 1,25. В соответствии с измерением молекулярное сито содержит крупные поры, мезопоры и микропоры, в котором крупные поры имеют диаметр 200 нм, общую удельную площадь поверхности 67 м2⋅г-1 и объем пор 0,354 мл/г; мезопоры имеют диаметр 8 нм, общую удельную площадь поверхности 116 м2⋅г-1 и объем пор 0,18 мл/г; и микропоры имеют диаметр 4,2 нм и 1,2 нм, общую удельную площадь поверхности 151 м2⋅г-1 и объем пор 0,074 мл/г. РФА показал Si/Al2=74. Результаты NH3-TPD показывают (Фигура V-13), что молекулярное сито имеет значительную кислотность. Результаты инфракрасного спектра показывают (Фигура V-14), что молекулярное сито имеет низкое содержание В-кислоты и высокое содержание L-кислоты.
Пример V-8
В 45 мл тефлоновый контейнер добавляли 0,134 г метаалюмината натрия, добавляли 7,41 г Темплата С, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), 6,98 г деионизированной воды и 0,4 г NaOH, и тщательно перемешивали в течение 5 минут. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=61, H2O/SiO2=8, Темплат C/SiO2=0,35, OH-/SiO2=0,20.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали, и автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин и подвергали взаимодействию при 160°С в течение 4 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре V-10. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию кристаллической частицы гексагональной призматической формы и губчатой структуры с эффективным диаметром 1200 нм, высотой 1700 нм и соотношением сторон 1,42. В соответствии с измерением молекулярное сито содержит крупные поры, мезопоры и микропоры, в котором крупные поры имеют диаметр 1000 нм, общую удельную площадь поверхности 26 м2⋅г-1 и объем пор 0,253 мл/г; мезопоры имеют диаметр 8 нм, общую удельную поверхность 142 м2⋅г-1 и объем пор 0,216 мл/г; и микропоры имеют диаметр 4 нм и 1,2 нм, общую удельную площадь поверхности 194 м2⋅г-1 и объем пор 0,037 мл/г. РФА показал Si/Al2=54.
Краткое описание чертежей
На Фигуре V-1 показана кривая адсорбции 2,2-диэтилбутана на молекулярном сите, полученном в Примере V-3 после обжига.
На Фигуре V-2 показана кривая адсорбции 3-пропил-4-бутилоктана на молекулярном сите, полученном в Примере V-6 после обжига.
На Фигуре V-3 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере V-1.
На Фигуре V-4 показана дифрактограмма молекулярного сита, полученного в Примере V-3.
На Фигуре V-5 показана дифрактограмма молекулярного сита, полученного в Примере V-6 после обжига.
На Фигуре V-6 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере V-4.
На Фигуре V-7 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере V-5.
На Фигуре V-8 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере V-6.
На Фигуре V-9 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере V-7.
На Фигуре V-10 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере V-8.
На Фигуре V-11(а) показан схематический вид губчатой структуры, содержащей крупные поры и/или мезопоры, и на Фигуре V-11(b) показана сканирующая электронная микрофотография губчатой структуры, содержащей крупные поры и/или мезопоры.
На Фигуре V-12(а) показан схематический вид молекулярного сита, имеющего морфологию полых столбчатых кристаллических частиц, и на Фигуре V-12(b) показана сканирующая электронная микрофотография молекулярного сита, имеющего морфологию полых столбчатых кристаллических частиц.
На Фигуре V-13 показана модель NH3-TPD молекулярного сита, полученного в Примере V-7.
На Фигуре V-14 показаны ИК-спектры молекулярного сита, полученного в Примере V-7.
Третий вариант осуществления и шестой вариант осуществления
В контексте настоящего варианта осуществления, включая следующие примеры и сравнительные примеры, общую удельную площадь поверхности, объем пор и диаметр пор молекулярного сита измеряли с помощью следующих аналитических методов.
Оборудование: Статический адсорбер азота Micromeritics ASAP2010
Условия измерения: образец помещали в систему для обработки образца, создавали вакуум до 1,35×10-2 Па при 350°С и выдерживали при данной температуре и давлении в течение 15 ч для очистки образца. При температуре жидкого азота -196°С измеряли количество адсорбции и количество десорбции азота очищенного образца в условиях различного удельного давления P/P0 и получали изотерму адсорбции-десорбции. Затем общую удельную площадь поверхности рассчитывали с помощью двухпараметрического уравнения БЭТ. Адсорбционную емкость при удельном давлении P/P0≈0,98 принимали за объем пор образца и распределение пор по диаметру рассчитывали в соответствии с моделью BJH.
Пример VI-1
Получение Темплата A:
В 500 мл трехгорлую колбу добавляли 15 г (0,087 моль) тетраметилгексаметилендиамина, добавляли 250 мл изопропанола и добавляли по каплям 18,8 г (0,087 моль) 1,4-дибромбутана при комнатной температуре. Добавление завершали через 15 минут и температуру повышали до температуры нагревания с обратным холодильником. Раствор постепенно менялся от бесцветного и прозрачного до белого и мутного. Реакция сопровождалась высокоэффективной жидкостной хроматографией (ВЭЖХ). После завершения реакции к реакционной смеси добавляли 200 мл этилацетата и смесь нагревали с обратным холодильником в течение 1 часа, охлаждали и фильтровали с отсасыванием. Полученное твердое вещество промывали этилацетатом и затем диэтиловым эфиром с получением 30 г белого твердого продукта в виде 1,1,6,6-тетраметил-1,6-диаза-12-членного кольца-1,6-дибромида (соединение, в котором n равно 4, m равно 6, R представляет собой метил, X представляет собой Br), имеющего относительную молекулярную массу 388,2, температуру плавления 273,7°C. Спектр 1H-ЯМР показывает химический сдвиг (300 МГц, CDCl3) δ 1,50 (т, 4H), 1,90 (т, 8H), 3,14 (с, 12H), 3,40 (т, 8H).
Получение Темплата B: Br в Темплате A заменяли OH- с помощью ионного обмена; ионообменная смола представляла собой сильную основную анионообменную смолу стирольного типа, рабочий раствор представлял собой 15 масс.% водного раствора Темплата А, рабочая температура составляла 25°С, массовое соотношение рабочей жидкости и ионообменной смолы составляло 1:3; скорость потока составляла 3 капли/секунда; обменный раствор дегидратировали с помощью роторного испарителя с получением продукта, который представляет собой соединение формулы (I), в которой n равно 4, m равно 6, R представляет собой метил и X представляет собой ОН, имеющее относительную молекулярную массу 262,2 и чистоту 99,21%. Содержание брома в нем составляло 0,79 масс.%.
Пример VI-2
Получение Темплата C
В 500 мл трехгорлую колбу добавляли 10 г (0,058 моль) тетраметилгексаметилендиамина, добавляли 250 мл изопропанола и добавляли по каплям 16,6 г (0,058 моль) 1,9-дибромдекана при комнатной температуре. Добавление завершали через 15 минут и температуру повышали до температуры нагревания с обратным холодильником. Раствор постепенно менялся от бесцветного и прозрачного до белого и мутного. Реакция сопровождалась высокоэффективной жидкостной хроматографией (ВЭЖХ). После завершения реакции к реакционной смеси добавляли 200 мл этилацетата и смесь нагревали с обратным холодильником в течение 1 часа, охлаждали и фильтровали с отсасыванием. Полученное твердое вещество промывали этилацетатом и затем диэтиловым эфиром с получением 25 г белого твердого продукта в виде 1,1,8,8-тетраметил-1,8-диаза-17-членного кольца-1,8-дибромида (соединение, в котором n равно 9, m равно 6, R представляет собой метил, X представляет собой Br), имеющего относительную молекулярную массу 458,4. Спектр 1H-ЯМР показывает химический сдвиг (300 МГц, CDCl3) δ 1,51 (т, 14H), 1,92 (т, 8H), 3,16 (с, 12H), 3,40 (т, 8H).
Получение Темплата D: Br в Темплате C заменяли OH- с помощью ионного обмена; ионообменная смола представляла собой сильную основную анионообменную смолу стирольного типа, рабочий раствор представлял собой 15 масс.% водного раствора Темплата C, рабочая температура составляла 25°С, массовое соотношение рабочей жидкости и ионообменной смолы составляло 1:3; скорость потока составляла 3 капли/секунда; обменный раствор дегидратировали с помощью роторного испарителя с получением продукта, который представляет собой соединение формулы (I), в которой n равно 9, m равно 6, R представляет собой метил, X представляет собой ОН, имеющее относительную молекулярную массу 332,4 и чистоту 99,8%. Содержание брома в нем составляло 0,2 масс.%.
Пример VI-3
В 45 мл тефлоновый контейнер добавляли 0,132 г метаалюмината натрия, добавляли 8,024 г Темплата В, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=60, H2O/SiO2=7,8 и Темплат B/SiO2=0,15.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре VI-1. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию плоской призматической или плоской цилиндрической кристаллической частицы с эффективным диаметром 600 нм, высотой 300 нм и соотношением сторон 0,5. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 518 м2⋅г-1 и объем пор 0,351 мл/г. РФА показал Si/Al2=63. Дифрактограмма продукта была показана на Фигуре VI-2. Результаты NH3-TPD показывают (Фигура VI-3), что молекулярное сито имеет значительную кислотность. Результаты инфракрасного спектра показывают (Фигура VI-4), что молекулярное сито имеет низкое содержание В-кислоты и высокое содержание L-кислоты.
Пример VI-4
В 45 мл тефлоновый контейнер добавляли 0,735 г метаалюмината натрия, добавляли 8,024 г Темплата В, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=80, H2O/SiO2=7,5 и Темплат B/SiO2=0,15. Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре VI-5. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию плоской призматической или плоской цилиндрической кристаллической частицы с эффективным диаметром 300 нм, высотой 200 нм и соотношением сторон 0,67. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 482 м2⋅г-1 и объем пор 0,346 мл/г. РФА показал Si/Al2=84. Дифрактограмма продукта была показана на Фигуре VI-6.
Пример VI-5
В 45 мл тефлоновый контейнер добавляли 0,132 г метаалюмината натрия, добавляли 8,731 г Темплата D, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=60, H2O/SiO2=8 и Темплат D/SiO2=0,15. Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 4 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре VI-7. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию плоской призматической или плоской цилиндрической кристаллической частицы с эффективным диаметром 300 нм, высотой 200 нм и соотношением сторон 0,67. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 452 м2⋅г-1 и объем пор 0,385 мл/г. РФА показал Si/Al2=62.
Пример VI-6
В 45 мл тефлоновый контейнер добавляли 0,132 г метаалюмината натрия, добавляли 4,1 г Темплата D, перемешивали в течение 30 минут до образования гомогенности, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=60, H2O/SiO2=7 и Темплат D/SiO2=0,32.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре VI-8. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию плоской призматической или плоской цилиндрической кристаллической частицы с эффективным диаметром 600 нм, высотой 400 нм и соотношением сторон 0,67. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 487 м2⋅г-1 и объем пор 0,387 мл/г. РФА показал Si/Al2=63.
Пример VI-7
В 45 мл тефлоновый контейнер добавляли 0,132 г метаалюмината натрия, добавляли 6,0 г Темплата С, и затем добавляли 4 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=80, H2O/SiO2=5 и Темплат C/SiO2=0,2. Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 110°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре VI-9. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию плоской призматической или плоской цилиндрической кристаллической частицы с эффективным диаметром 400 нм, высотой 200 нм и соотношением сторон 0,5. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 412 м2⋅г-1 и объем пор 0,372 мл/г. РФА показал Si/Al2=83.
Пример VI-8
В 45 мл тефлоновый контейнер добавляли 0,132 г метаалюмината натрия, добавляли 4 г Темплата А, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=60, H2O/SiO2=5 и Темплат C/SiO2=0,2. Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Сканирующая электронная микрофотография продукта была показана на Фигуре VI-10. На фигуре можно легко увидеть, что молекулярное сито имеет морфологию плоской призматической или плоской цилиндрической кристаллической частицы с эффективным диаметром 400 нм, высотой 250 нм и соотношением сторон 0,625. В соответствии с измерением молекулярное сито имело общую удельную площадь поверхности 427 м2⋅г-1 и объем пор 0,418 мл/г. РФА показал Si/Al2=58.
Пример VI-9
В 45 мл тефлоновый контейнер добавляли 0,588 г метаалюмината натрия, добавляли 8,024 г Темплата В, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 98,05%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=100, H2O/SiO2=7,6 и Темплат B/SiO2=0,15.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта.
Дифрактограмма данного продукта была показана на Фигуре VI-11.
Пример VI-10
В 45 мл тефлоновый контейнер добавляли 0,49 г метаалюмината натрия, добавляли 8,024 г Темплата В, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 составляло 98,05%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=120, H2O/SiO2=7,5 и Темплат B/SiO2=0,15.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта. Дифрактограмма данного продукта была показана на Фигуре VI-12.
Пример VI-11
В 45 мл тефлоновый контейнер добавляли 0,392 г метаалюмината натрия, добавляли 8,024 г Темплата В, и затем добавляли 3 г неочищенного силикагеля (Qingdao Haiyang Chemical Co., Ltd., промышленный продукт, содержание SiO2 составляло 98,05%), и полученную смесь оставляли и выдерживали в течение 1 часа для достижения полного смешения. Молярные соотношения компонентов представляли собой следующие: SiO2/Al2O3=150, H2O/SiO2=7,3, Темплат B/SiO2=0,15.
Вышеуказанную смесь помещали в 45 мл стальной автоклав с тефлоновым вкладышем, закрывали и герметизировали. Автоклав помещали во вращающуюся конвекционную печь со скоростью вращения 20 об/мин, и подвергали взаимодействию при 120°С в течение 1 дня, и затем нагревали до 150°С для взаимодействия в течение 5 дней. Автоклав вынимали, и быстро охлаждали до комнатной температуры, и смесь отделяли на высокоскоростной центрифуге при 5000 об/мин, и твердое вещество собирали, тщательно промывали деионизированной водой, и высушивали при 100°С в течение 5 часов с получением продукта. Дифрактограмма данного продукта была показана на Фигуре VI-13.
Краткое описание чертежей
На Фигуре VI-1 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере VI-3.
На Фигуре VI-2 показана дифрактограмма молекулярного сита, полученного в Примере VI-3.
На Фигуре VI-3 показана диаграмма NH3-TPD молекулярного сита, полученного в Примере VI-3.
На Фигуре VI-4 показан инфракрасный спектр молекулярного сита, полученного в Примере VI-3.
На Фигуре VI-5 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере VI-4.
На Фигуре VI-6 показана дифрактограмма молекулярного сита, полученного в Примере VI-4.
На Фигуре VI-7 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере VI-5.
На Фигуре VI-8 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере VI-6.
На Фигуре VI-9 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере VI-7.
На Фигуре VI-10 показана сканирующая электронная микрофотография молекулярного сита, полученного в Примере VI-8.
На Фигуре VI-11 показана дифрактограмма молекулярного сита, полученного в Примере VI-9.
На Фигуре VI-12 показана дифрактограмма молекулярного сита, полученного в Примере VI-10.
На Фигуре VI-13 показана дифрактограмма молекулярного сита, полученного в Примере VI-11.
На Фигуре VI-14(а) показан схематический вид, представляющий контур торцевой поверхности молекулярного сита в соответствии с настоящим изобретением, имеющего выпуклую форму; на Фигуре VI-14(b) показан схематический вид, представляющий контур торцевой поверхности молекулярного сита в соответствии с настоящим изобретением, имеющего другую выпуклую форму, и на Фигуре VI-14(с) показан схематический вид, представляющий контур торцевой поверхности молекулярного сита в соответствии с настоящим изобретением, имеющего плоскую форму вместо выпуклой формы.
Многочисленные подробности представлены в описании настоящей заявки, но следует понимать, что раскрытые в настоящем описании варианты осуществления могут быть осуществлены на практике без данных подробностей. В некоторых случаях хорошо известные способы, структуры и методики не показаны подробно, чтобы не затруднять понимание описания.
Подобным образом, при описании примерных вариантов осуществления настоящего раскрытия различные отличительные признаки иногда группируются вместе в отдельный вариант осуществления, отдельную фигуру или их описание, с тем чтобы сделать раскрытие точным и помочь понять сущность или больше аспектов, как раскрыто в настоящем описании. Однако раскрытие не следует интерпретировать как отражение намерения, что заявленное изобретение требует больше отличительных признаков, чем те, которые явно изложены в каждом пункте формулы изобретения. Скорее, как отражено в формуле изобретения, заявленное техническое решение может содержать меньше отличительных признаков по сравнению с отдельным вариантом осуществления, представленным в описании. Следовательно, формула изобретения после определенных вариантов осуществления таким образом явно включена в определенные варианты осуществления, и каждый из пунктов формулы изобретения может рассматриваться как отдельный вариант осуществления настоящего изобретения.
Следует также отметить, что в данном контексте родственные термины, такие как первый, и второй, и т.д., используются просто для того, чтобы отличать один объект или действие от другого объекта или действия, не обязательно требуя или подразумевая, что на самом деле существует такая взаимосвязь или последовательность выполнения между указанными объектами или действиями. Кроме того, термин «содержать», «содержащий» или любые другие его варианты предназначены для покрытия неэксклюзивного включения, так что процесс, способ, изделие или устройство, содержащее множество элементов, может содержать не только данные элементы, но и также другие элементы или элементы, которые присущи такому процессу, способу, изделию или устройству. При отсутствии дополнительного ограничения элемент, определяемый фразой «содержащий один...», не исключает наличия дополнительного количества того же элемента в процессе, способе, изделии или устройстве.
Вышеуказанные примеры приведены только для иллюстрации технических решений вариантов осуществления настоящего раскрытия и не предназначены для ограничения; хотя настоящее изобретение было подробно описано со ссылкой на вышеупомянутые варианты осуществления, специалист в данной области техники поймет, что технические решения, описанные в вариантах осуществления, могут быть модифицированы или некоторые технические характеристики, содержащиеся в них, могут быть заменены их эквивалентами; и такая модификация или замещение не будет отклонять соответствующее техническое решение от сущности или объема вариантов осуществления настоящего раскрытия.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИЛИКОАЛЮМОФОСФАТНОЕ МОЛЕКУЛЯРНОЕ СИТО, ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2019 |
|
RU2811839C2 |
| МОЛЕКУЛЯРНОЕ СИТО NaY С ОБОГАЩЕННОЙ АЛЮМИНИЕМ ПОВЕРХНОСТЬЮ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2019 |
|
RU2792150C2 |
| Катализатор гидрирования и способ его получения и его применения, и способ проведения реакции гидрирования нефтепродуктов | 2022 |
|
RU2838958C2 |
| ФОСФОРСОДЕРЖАЩЕЕ МОЛЕКУЛЯРНОЕ СИТО С ВЫСОКИМ СОДЕРЖАНИЕМ ДИОКСИДА КРЕМНИЯ, ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2019 |
|
RU2796542C2 |
| МОДИФИЦИРОВАННОЕ МАГНИЕМ МОЛЕКУЛЯРНОЕ СИТО ТИПА Y, ЕГО ПОЛУЧЕНИЕ И СОДЕРЖАЩИЙ ЕГО КАТАЛИЗАТОР | 2018 |
|
RU2770421C2 |
| КАТАЛИЗАТОР КРЕКИНГА И СПОСОБ ДЛЯ ЕГО ПРИГОТОВЛЕНИЯ | 2021 |
|
RU2832734C1 |
| МОЛЕКУЛЯРНОЕ СИТО SSZ-91, СПОСОБЫ ПОЛУЧЕНИЯ SSZ-91 И ПРИМЕНЕНИЕ SSZ-91 | 2016 |
|
RU2785394C2 |
| КАТАЛИЗАТОР КАТАЛИТИЧЕСКОГО КРЕКИНГА И ЕГО ПОЛУЧЕНИЕ | 2018 |
|
RU2755891C2 |
| МОЛЕКУЛЯРНОЕ СИТО SCM-34, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2838325C1 |
| МОДИФИЦИРОВАННОЕ МОЛЕКУЛЯРНОЕ СИТО ТИПА Y, СОДЕРЖАЩИЙ ЕГО КАТАЛИЗАТОР КАТАЛИТИЧЕСКОГО КРЕКИНГА, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2019 |
|
RU2798995C2 |
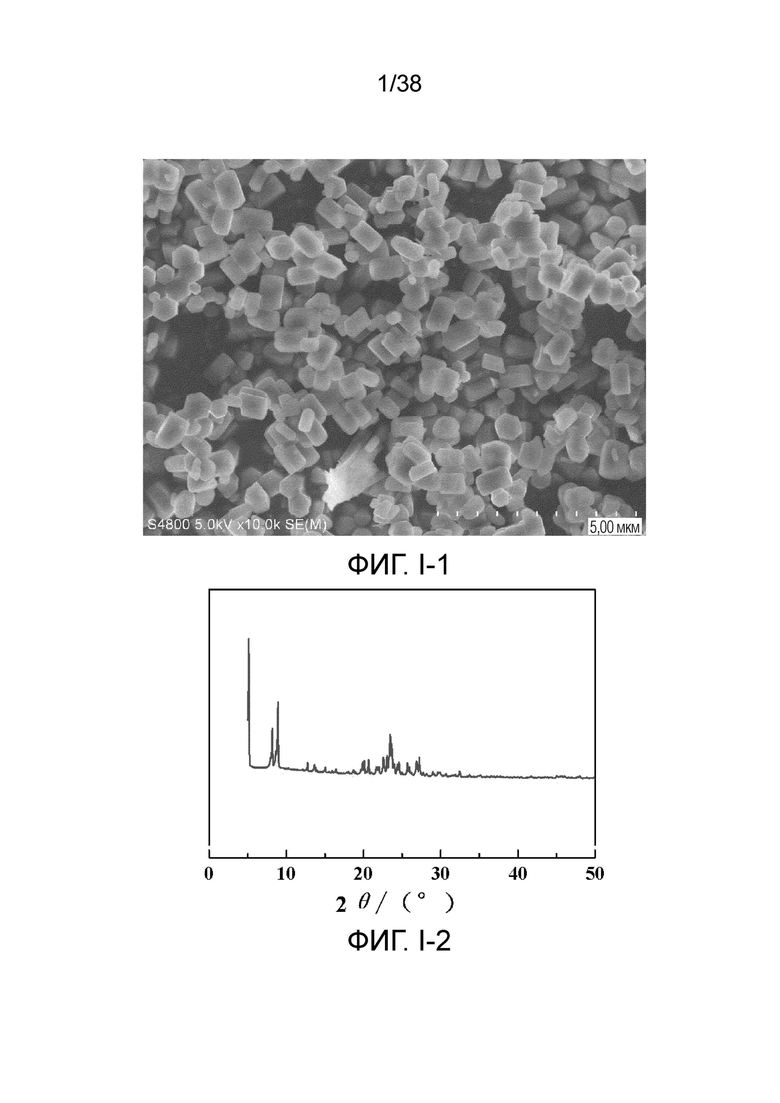
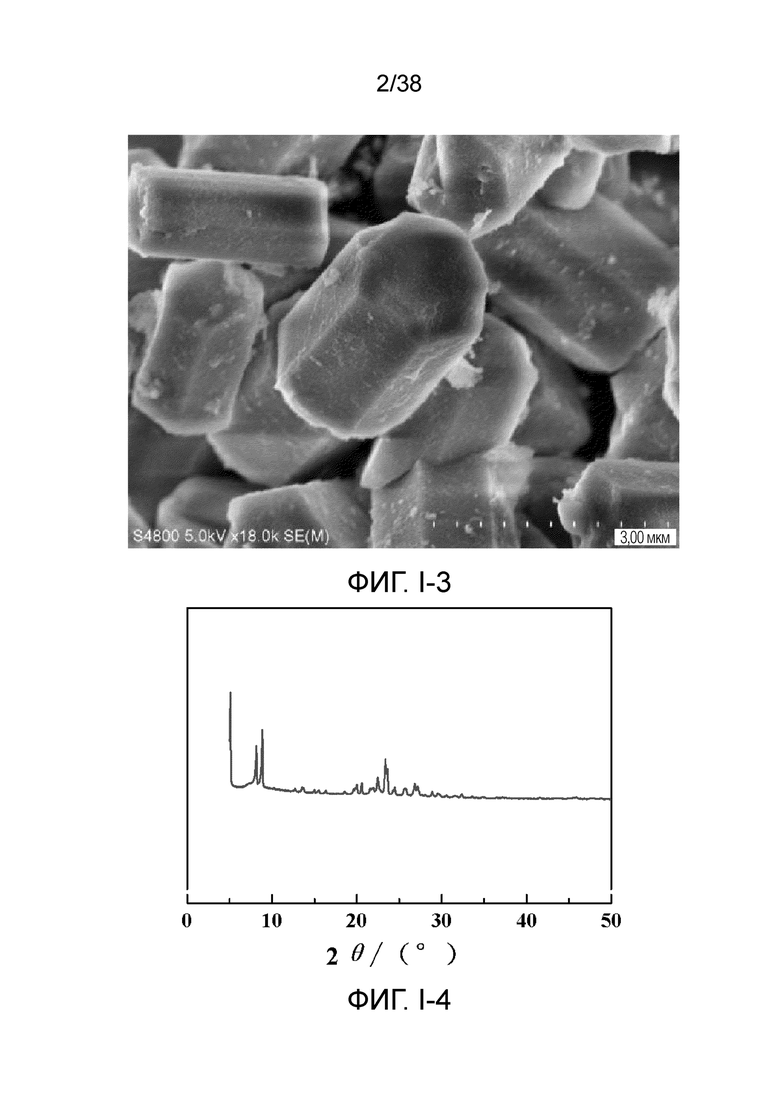
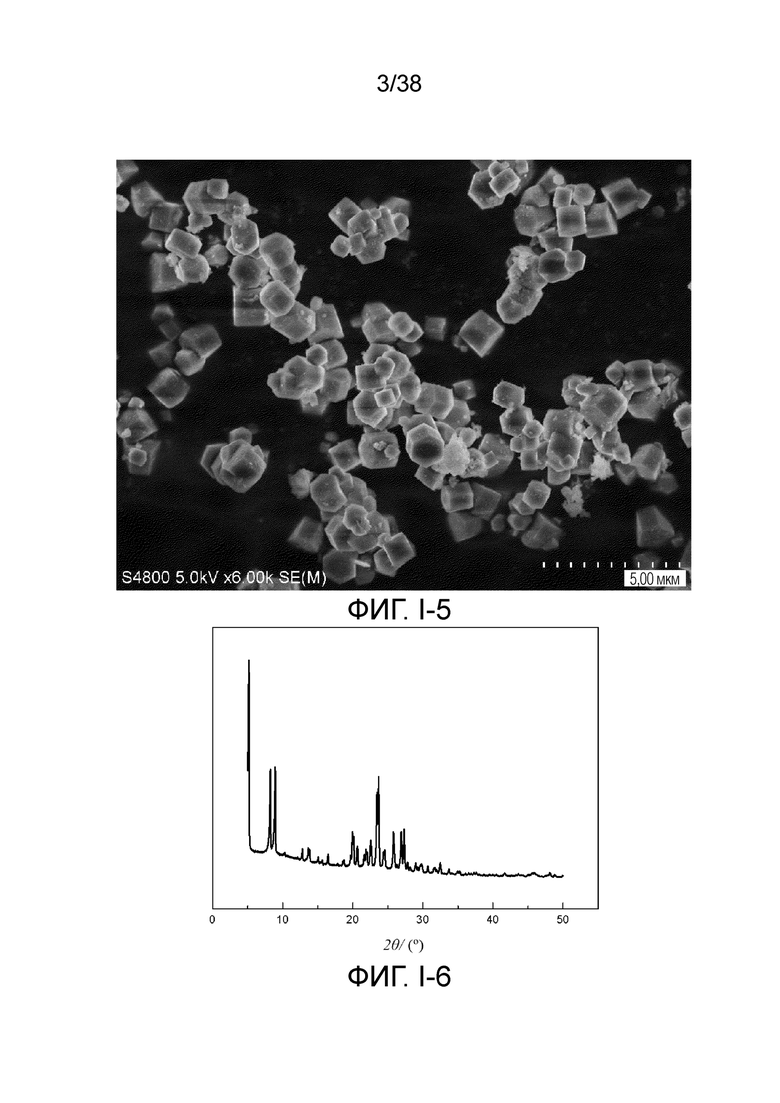

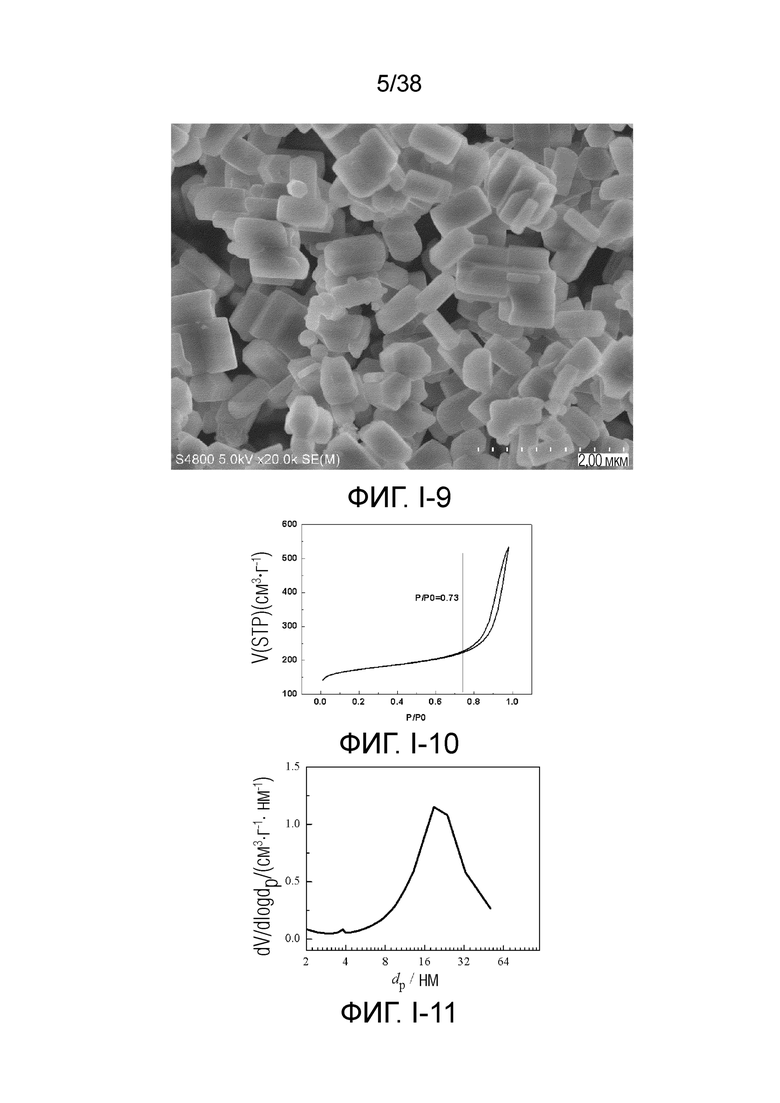
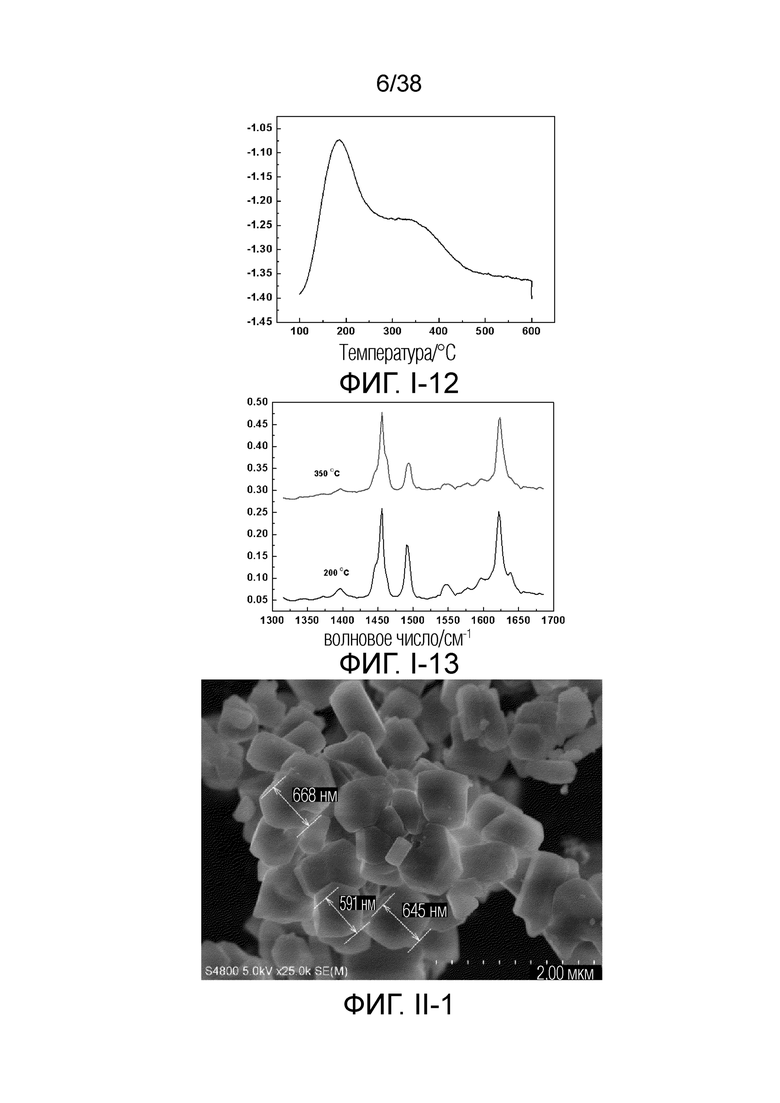

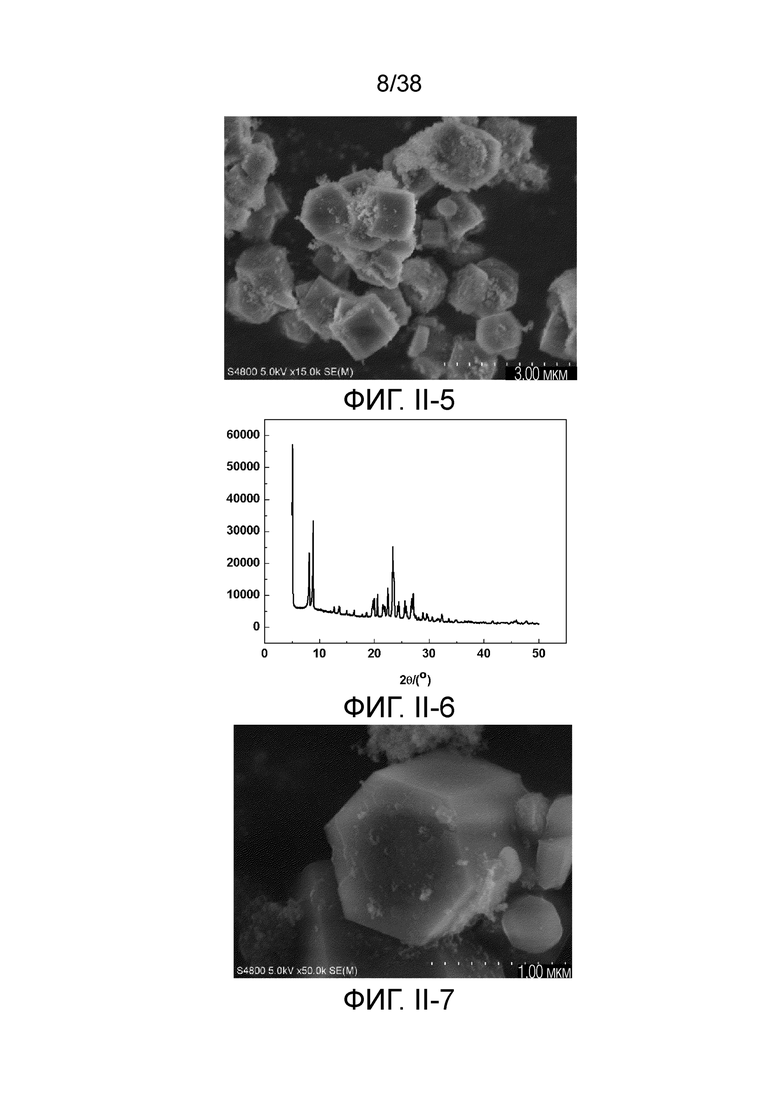
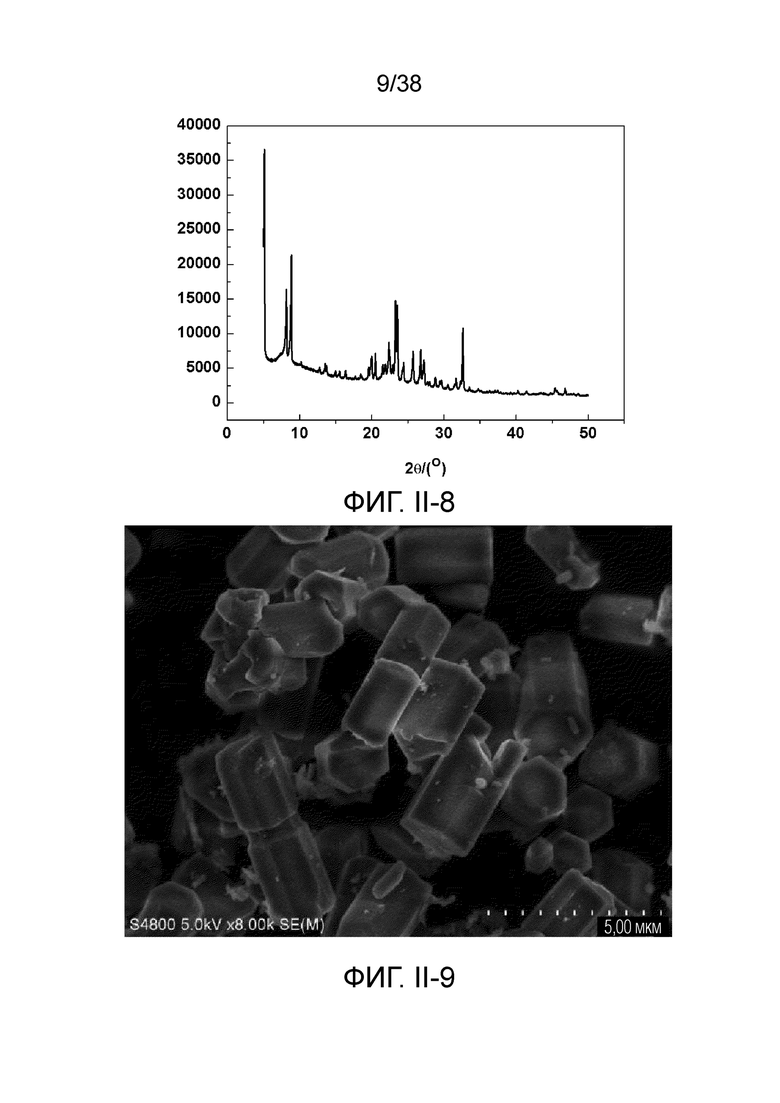
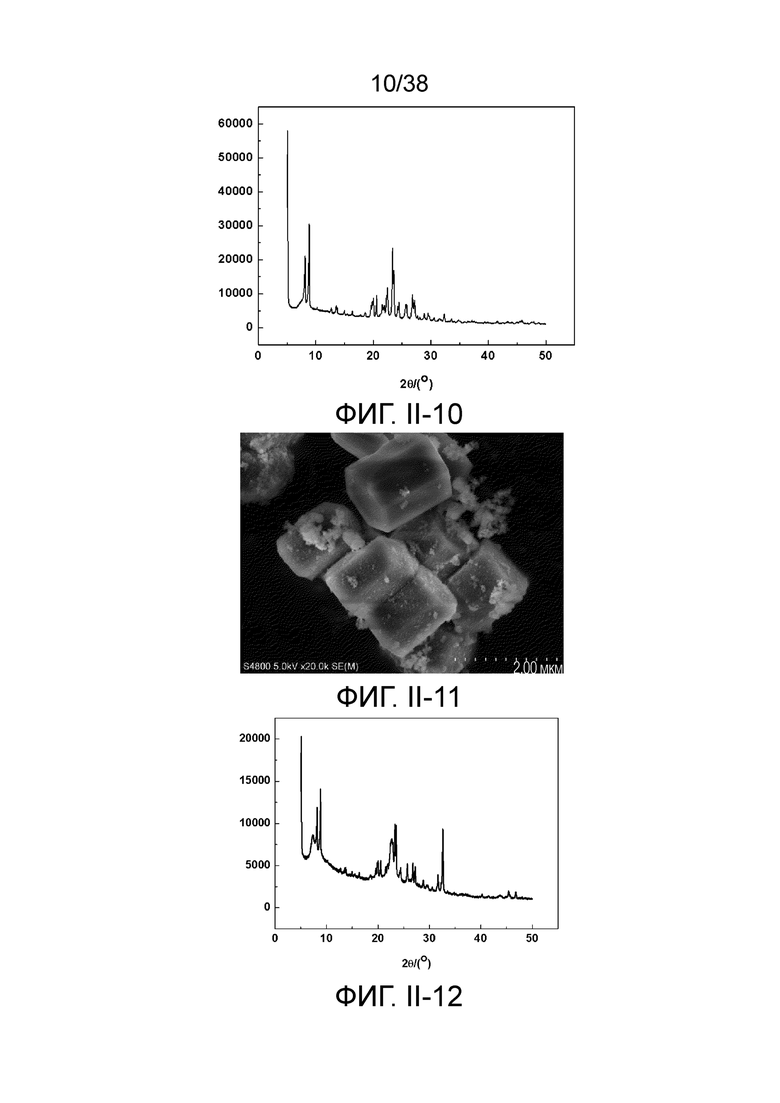
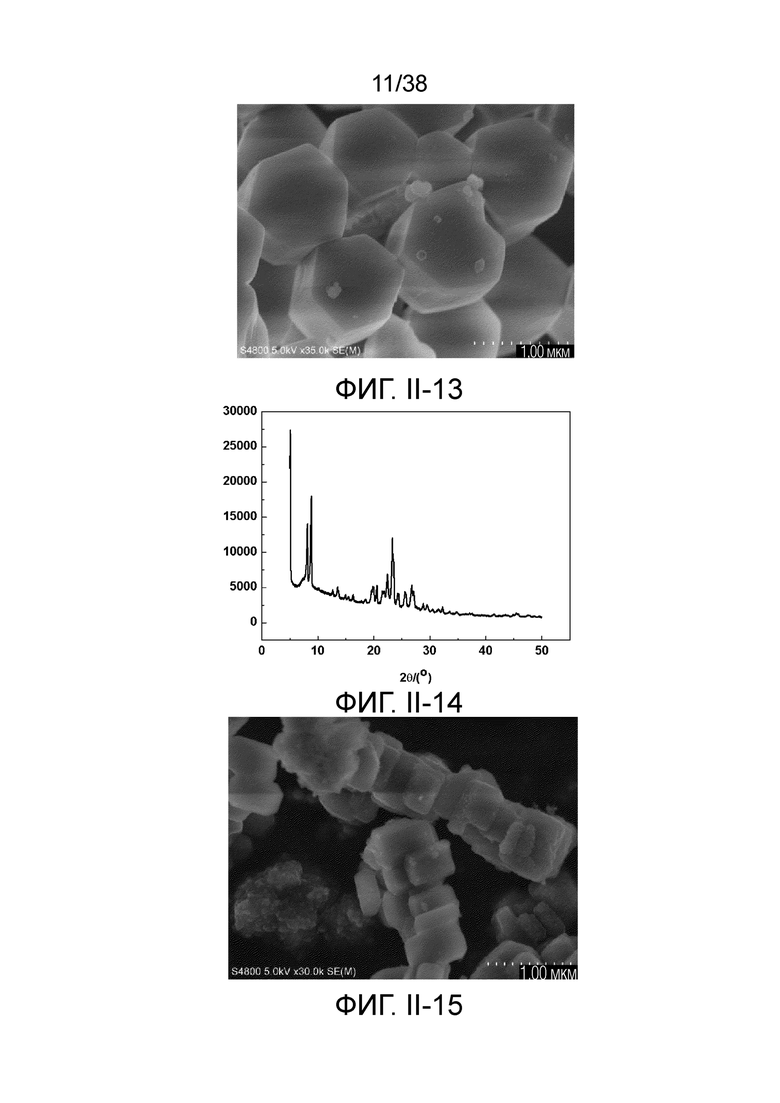
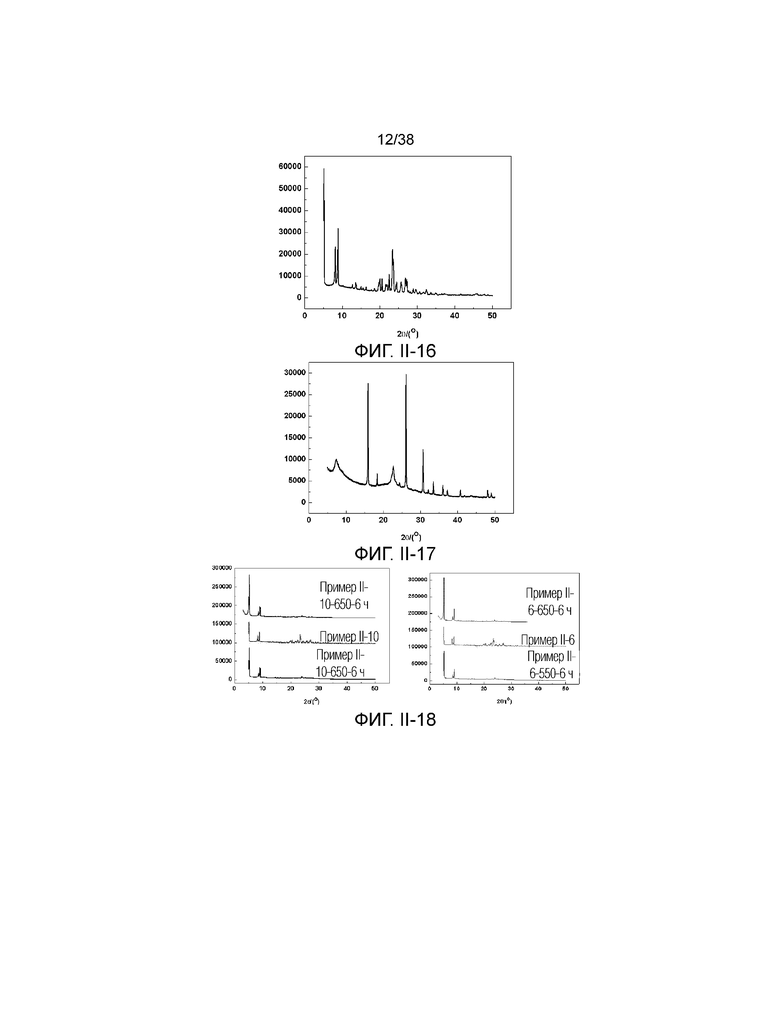

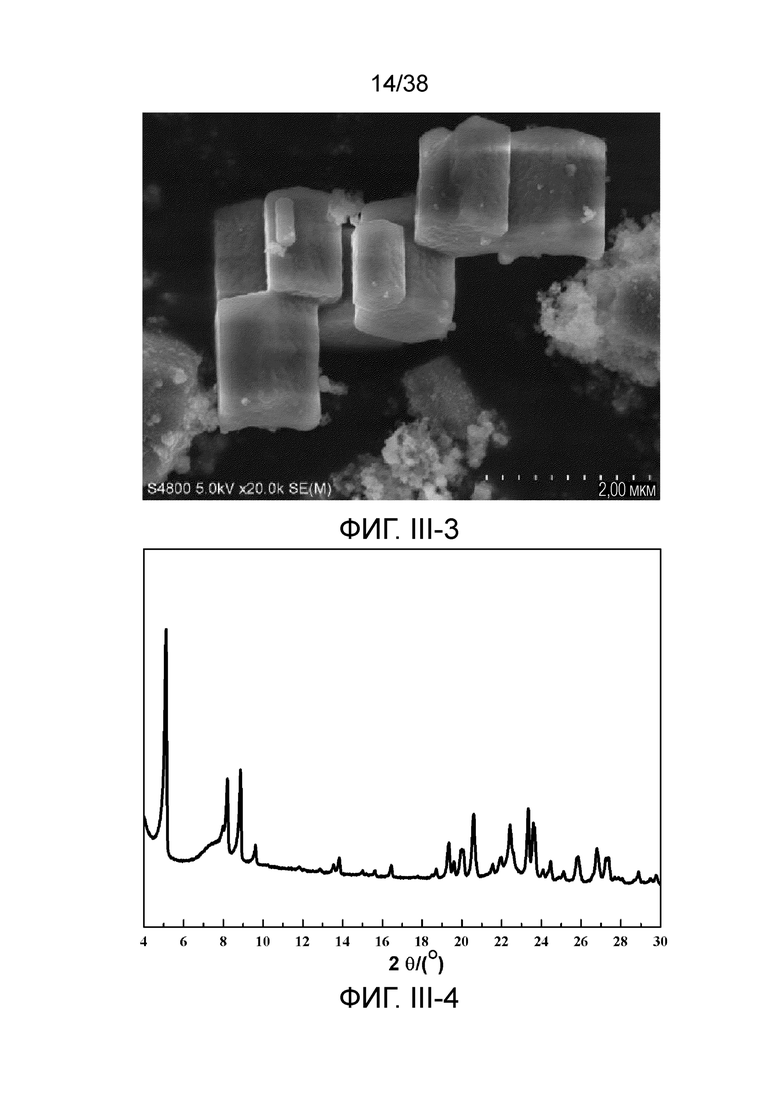
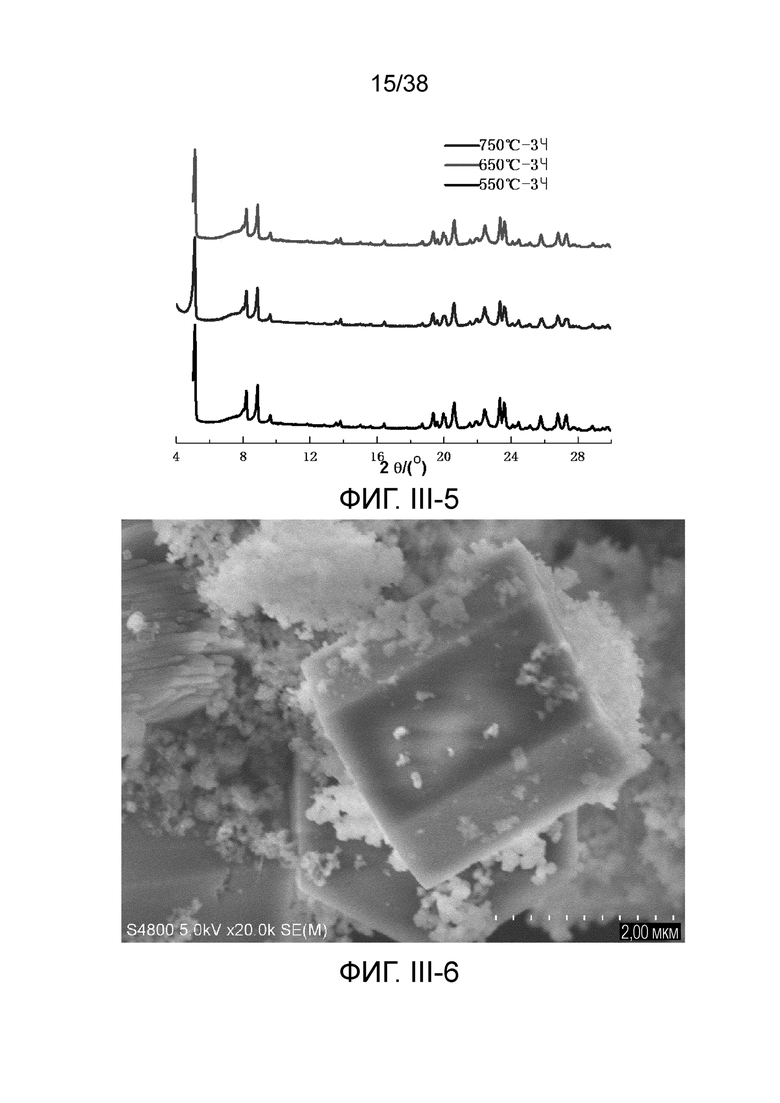


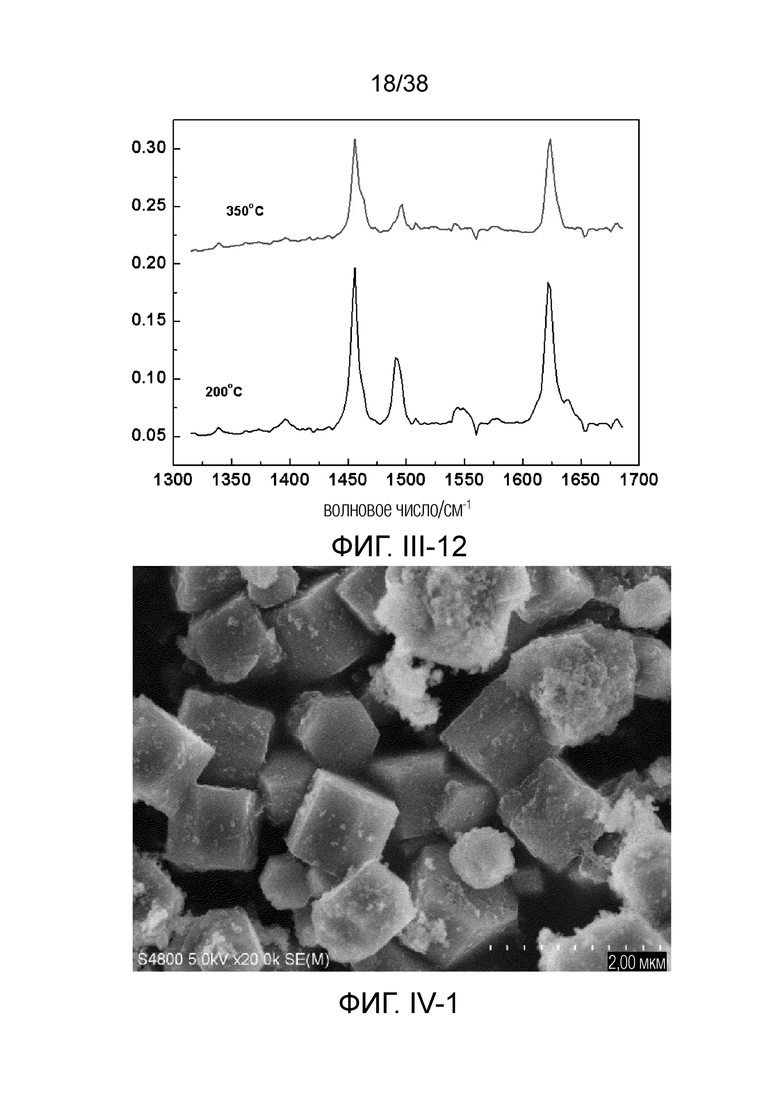
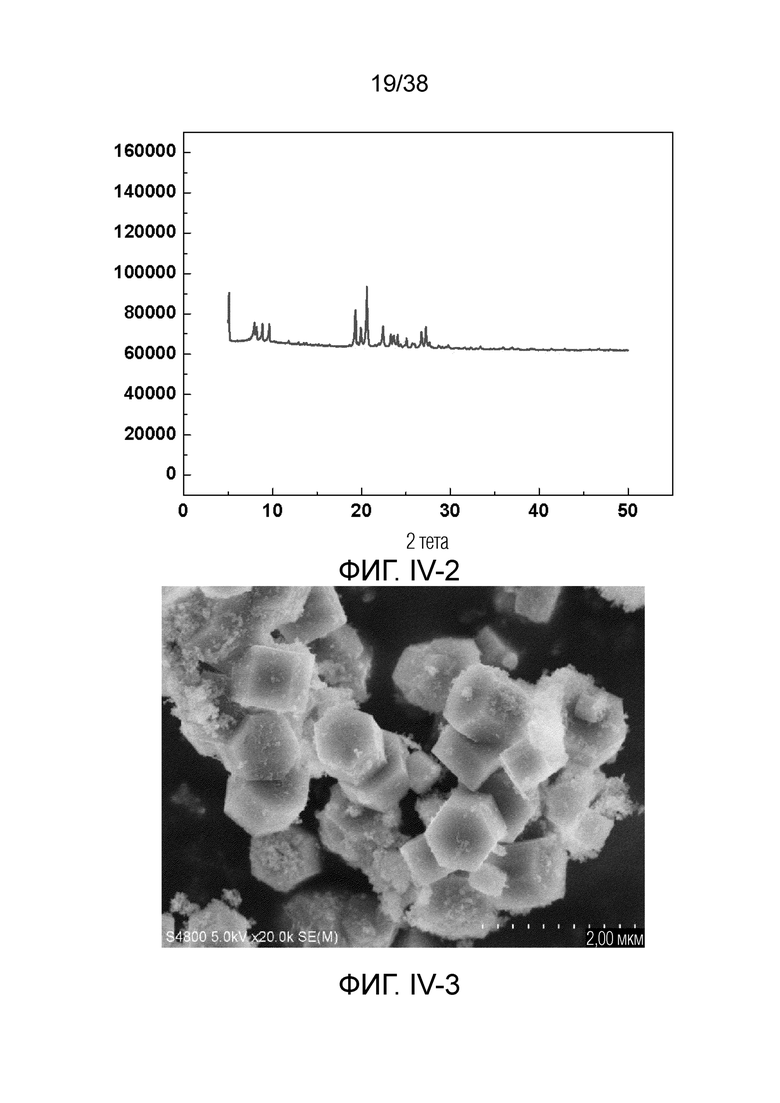
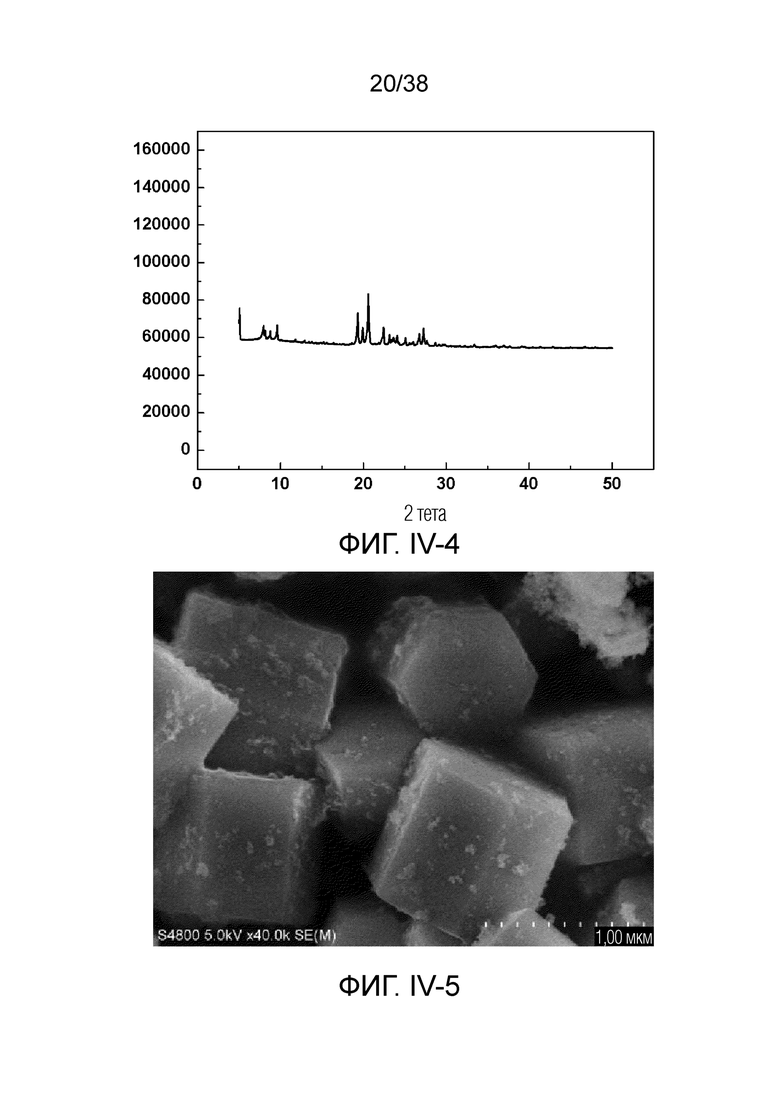

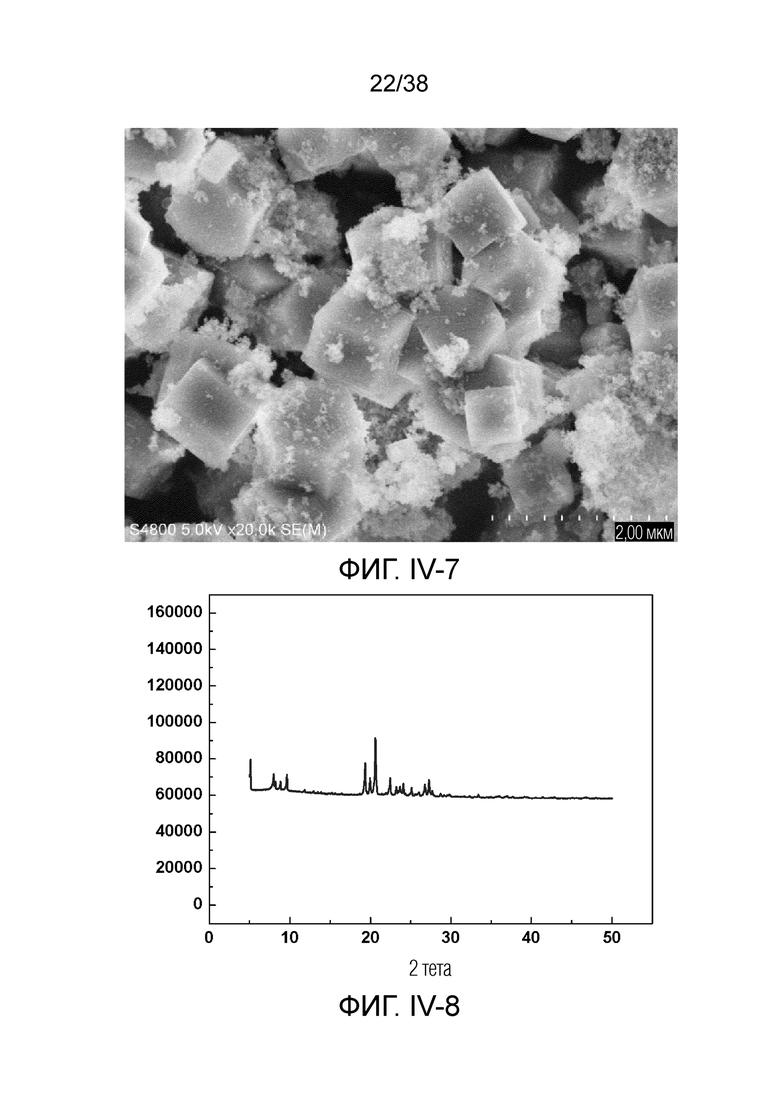
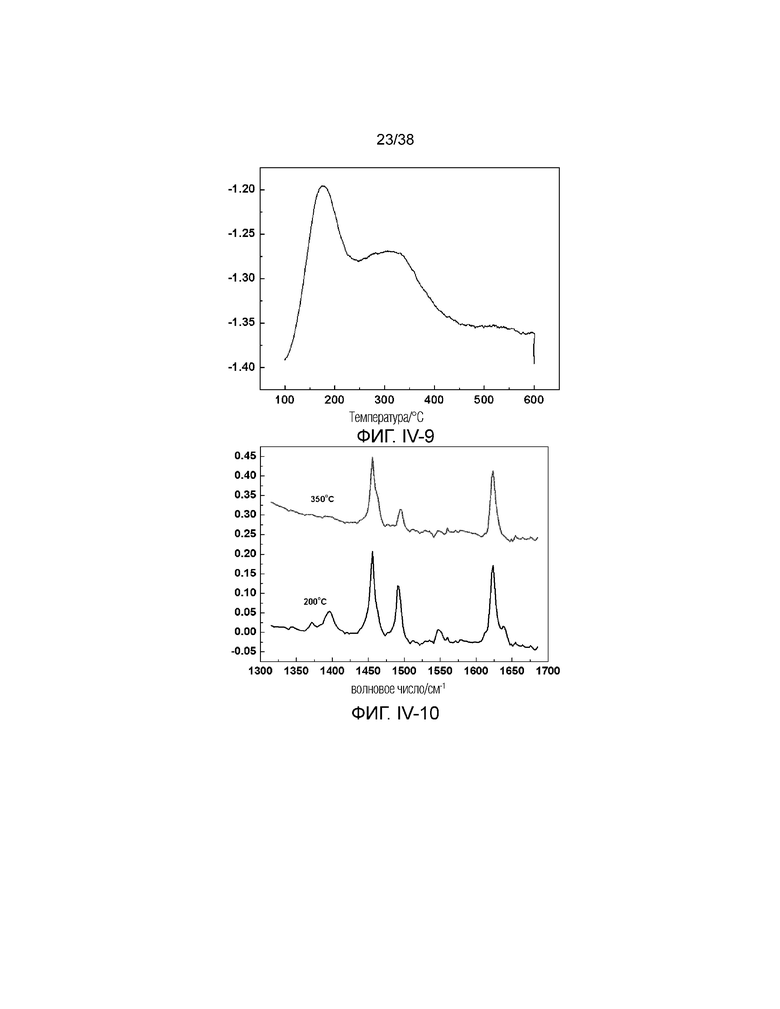

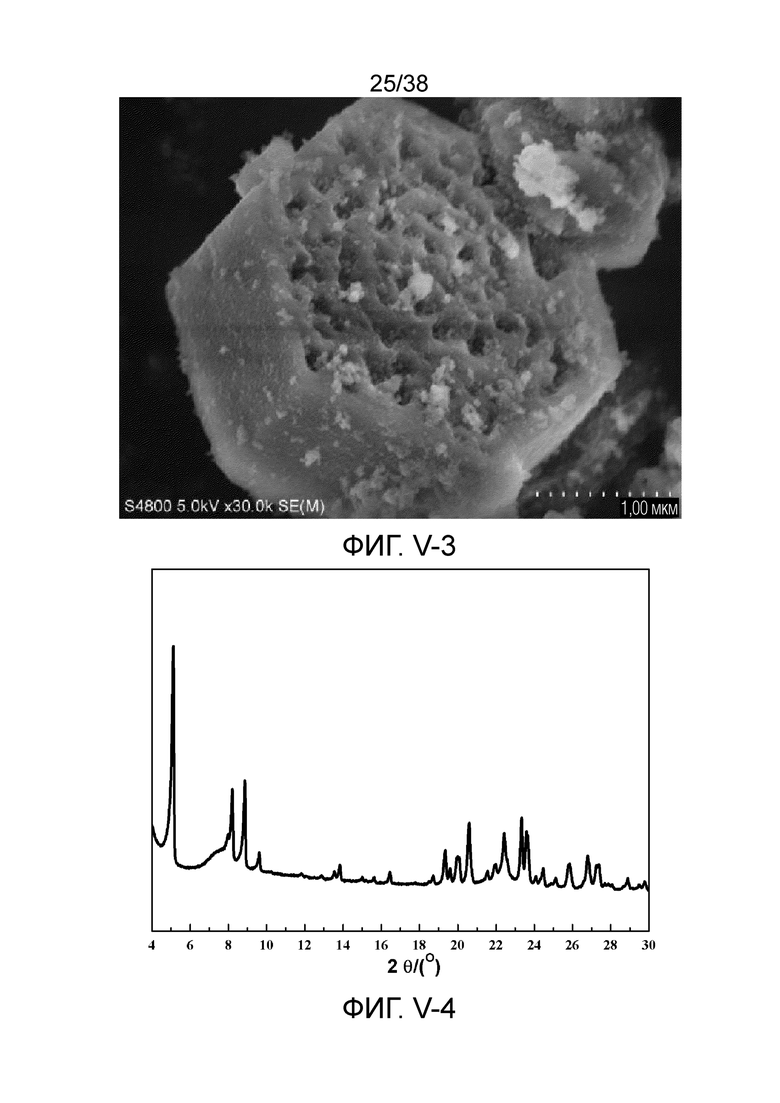
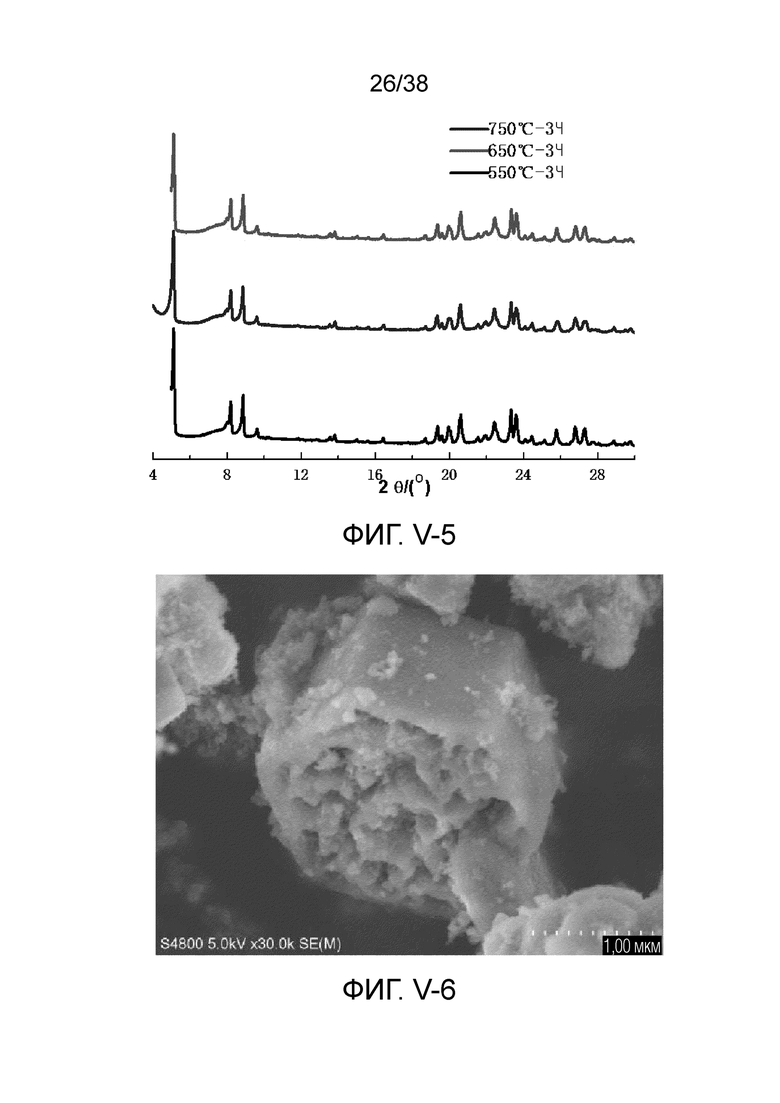




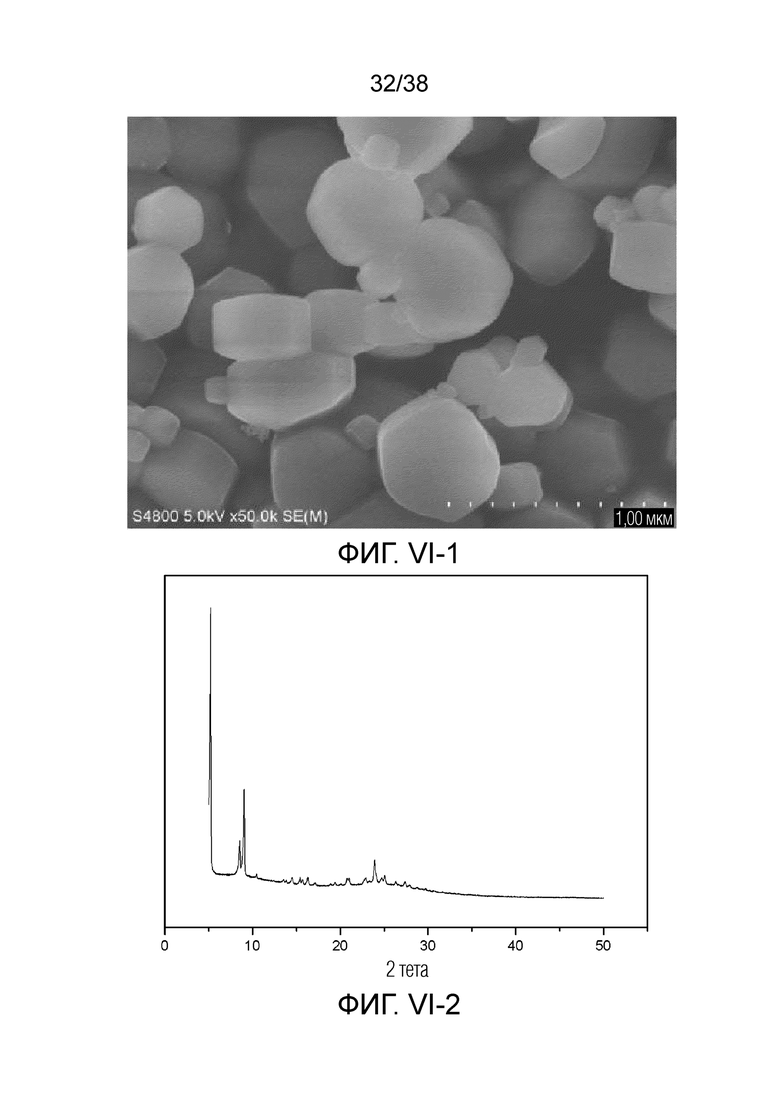



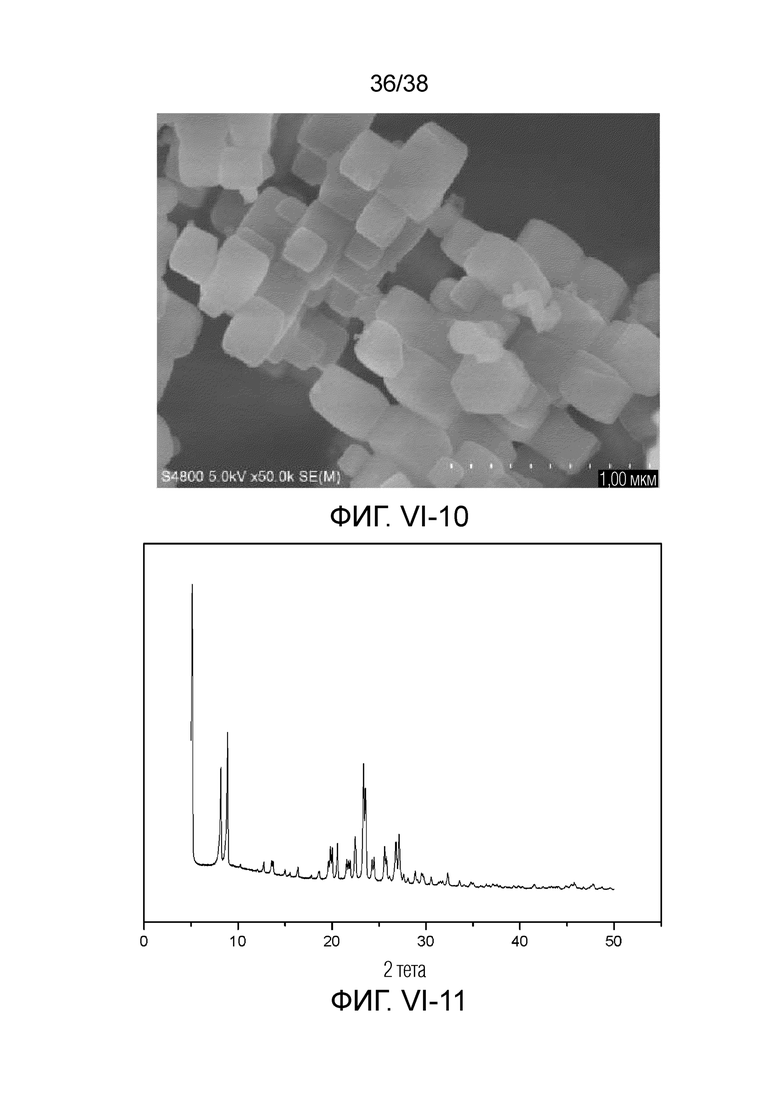

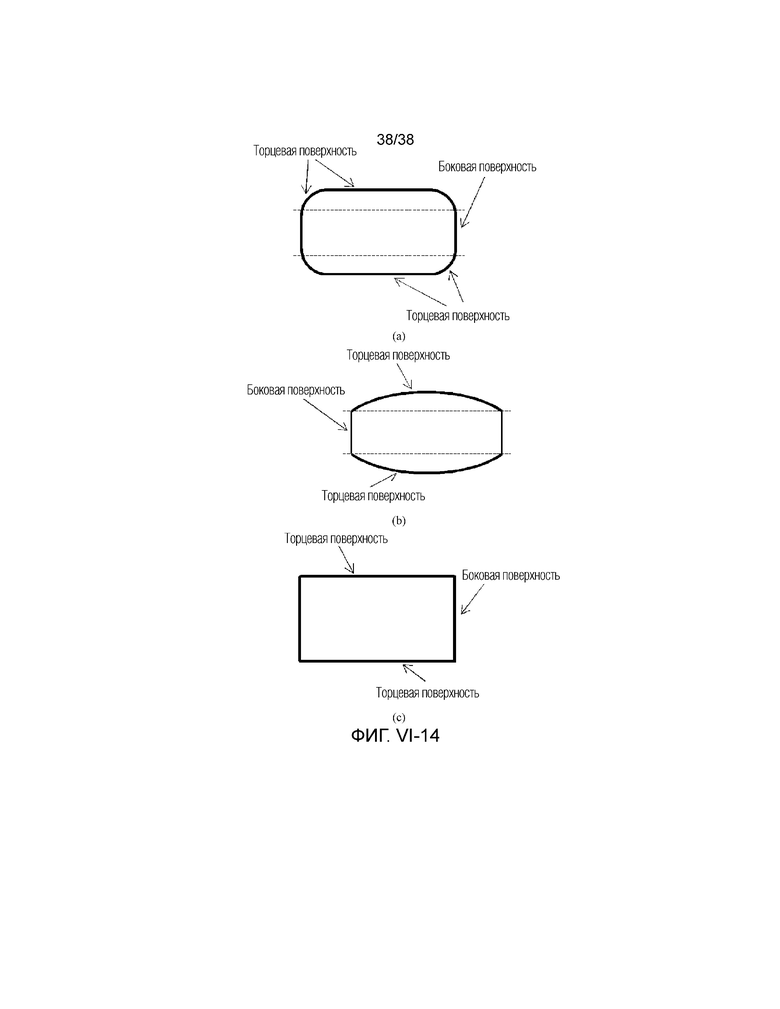
Группа изобретений относится к молекулярному ситу, в частности к ультрамакропористому молекулярному ситу, а также к способу получения молекулярного сита и его применению в качестве катализатора. Молекулярное сито имеет схематическую химическую композицию представленную формулой «первый оксид ⋅ второй оксид» или формулой «первый оксид ⋅ второй оксид ⋅ органический темплат ⋅ вода», в которой молярное соотношение первого оксида и второго оксида находится в диапазоне от 5 до ∞, первый оксид выбран из группы, состоящей из диоксида кремния, диоксида германия, диоксида олова, диоксида титана и диоксида циркония, предпочтительно диоксида кремния или комбинации диоксида кремния и диоксида германия, второй оксид выбран из группы, состоящей из оксида алюминия, оксида бора, оксида железа, оксида галлия, оксида редкоземельного элемента, оксида индия и оксида ванадия, предпочтительно оксида алюминия, молярное соотношение воды и первого оксида находится в диапазоне от 5 до 50, молярное соотношение темплата и первого оксида находится в диапазоне от 0,02 до 0,5, и молекулярное сито имеет определенную рентгеновскую дифрактограмму. Молекулярное сито обладает превосходными адсорбционными и каталитическими свойствами. 4 н. и 16 з.п. ф-лы, 26 табл., 56 пр., 81 ил.
1. Молекулярное сито, имеющее схематическую химическую композицию, представленную формулой «первый оксид ⋅ второй оксид» или формулой «первый оксид ⋅ второй оксид ⋅ органический темплат ⋅ вода»,
в которой молярное соотношение первого оксида и второго оксида находится в диапазоне от 5 до ∞, предпочтительно от 25 до 95, более предпочтительно от 30 до 70;
первый оксид представляет собой, по меньшей мере, один, выбранный из группы, состоящей из диоксида кремния, диоксида германия, диоксида олова, диоксида титана и диоксида циркония, предпочтительно диоксида кремния или комбинации диоксида кремния и диоксида германия;
второй оксид представляет собой, по меньшей мере, один, выбранный из группы, состоящей из оксида алюминия, оксида бора, оксида железа, оксида галлия, оксида редкоземельного элемента, оксида индия и оксида ванадия, предпочтительно оксида алюминия;
молярное соотношение воды и указанного первого оксида находится в диапазоне от 5 до 50, предпочтительно от 5 до 15;
молярное соотношение указанного органического темплата и указанного первого оксида находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,15 до 0,5 или от 0,3 до 0,5, и
указанное молекулярное сито имеет рентгеновскую дифрактограмму по существу, как показано в следующей таблице.
2. Молекулярное сито по п. 1, имеющее (нативную) губчатую структуру.
3. Молекулярное сито по п. 2, в котором губчатая структура содержит крупные поры и/или мезопоры, предпочтительно крупные поры и/или мезопоры открываются на торцевой поверхности и/или боковой поверхности губчатой структуры.
4. Молекулярное сито по п. 3, в котором крупные поры имеют диаметр в диапазоне от 80 нм до 2 мкм, предпочтительно от 80 нм до 1,5 мкм, и мезопоры имеют диаметр в диапазоне от 2 нм до 30 нм, предпочтительно от 2 нм до 4 нм и/или от 7 нм до 15 нм (предпочтительно от 8 нм до 9 нм).
5. Молекулярное сито по п. 3, в котором мезопоры имеют общую удельную площадь поверхности в диапазоне от 50 м2⋅г-1 до 250 м2⋅г-1, предпочтительно от 100 м2⋅г-1 до 150 м2⋅г-1, и объем пор в диапазоне от 0,05 мл/г до 0,40 мл/г, предпочтительно от 0,15 мл/г до 0,30 мл/г, и крупные поры имеют общую удельную площадь поверхности в диапазоне от 10 м2⋅г-1 до 100 м2⋅г-1, предпочтительно от 50 м2⋅г-1 до 100 м2⋅г-1, и объем пор в диапазоне от 0,5 мл/г до 3,0 мл/г, предпочтительно от 1,0 мл/г до 2,0 мл /г.
6. Молекулярное сито по п. 2, в котором губчатая структура содержит микропоры, при этом микропоры имеют диаметр в диапазоне от 0,5 нм до менее 2 нм, предпочтительно от 0,5 нм до 0,8 нм и/или от 1,1 нм до 1,8 нм, общую удельную площадь поверхности в диапазоне от 100 м2⋅г-1 до 300 м2⋅г-1, предпочтительно от 150 м2⋅г-1 до 250 м2⋅г-1, и объем пор в диапазоне от 0,03 мл/г до 0,20 мл/г, предпочтительно от 0,05 мл/г до 0,15 мл/г.
7. Молекулярное сито по п. 2, имеющее морфологию столбчатой (предпочтительно призматической, более предпочтительно гексагональной) кристаллической частицы, предпочтительно имеющее морфологию полой столбчатой кристаллической частицы.
8. Молекулярное сито по п. 7, в котором морфология кристаллической частицы имеет размер, определяемый эффективным диаметром в диапазоне от 100 нм до 5000 нм, предпочтительно от 1000 нм до 3000 нм, высотой в диапазоне от 500 нм до 3000 нм, предпочтительно от 1000 нм до 3000 нм, и соотношением сторон в диапазоне от 1/3 до 5, предпочтительно от 1/3 до 3.
9. Молекулярное сито по п. 2, в котором молярное соотношение первого оксида и второго оксида находится в диапазоне от 30 до 100, предпочтительно от 55 до 100.
10. Молекулярное сито по п. 1, имеющее морфологию (нативной) кристаллической частицы от плоской призматической формы до плоской цилиндрической формы, предпочтительно один или два контура его торцевой поверхности в продольном сечении имеют выпуклую форму.
11. Молекулярное сито по п. 1, в котором рентгеновская дифрактограмма дополнительно содержит рентгенодифракционный пик по существу, как показано в следующей таблице.
12. Молекулярное сито по п. 1, в котором рентгеновская дифрактограмма дополнительно содержит рентгенодифракционный пик по существу, как показано в следующей таблице.
13. Способ получения молекулярного сита по п. 1, включающий стадию приведения в контакт источника первого оксида, источника второго оксида, необязательного щелочного источника, органического темплата и воды в условиях кристаллизации с получением молекулярного сита и необязательно стадию обжига полученного молекулярного сита, в котором органический темплат содержит соединение, представленное следующей формулой (I),
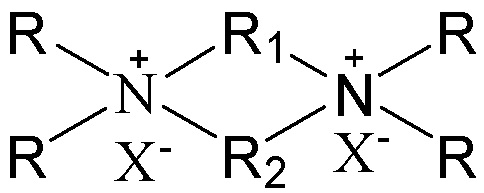 (I)
(I)
в которой группы R1 и R2 являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп и С3-12линейных или разветвленных оксаалкиленовых групп, предпочтительно каждая независимо выбрана из группы, состоящей из С3-12линейных алкиленовых групп и С3-12линейных оксаалкиленовых групп или предпочтительно одна из них выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп и С3-12линейных или разветвленных оксаалкиленовых групп, более предпочтительно одна из них выбрана из группы, состоящей из С3-12линейных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных алкиленовых групп и С3-12линейных оксаалкиленовых групп, в частности предпочтительно одна из них выбрана из группы, состоящей из С3-12линейных алкиленовых групп, и другая выбрана из группы, состоящей из C4-6линейных алкиленовых групп и C4-6линейных оксаалкиленовых групп (предпочтительно C4-6линейных монооксаалкиленовых групп, более предпочтительно -(CH2)m-O-(CH2)m-, в которой каждое значение m, являющееся одинаковым или отличным друг от друга, независимо представляет собой 2 или 3);
множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, предпочтительно каждая независимо выбрана из группы, состоящей из метила и этила, более предпочтительно всего метила; и X представляет собой ОН,
при этом источник первого оксида представляет собой, по меньшей мере, один, выбранный из группы, состоящей из источника диоксида кремния, источника диоксида германия, источника диоксида олова, источника диоксида титана и источника диоксида циркония, предпочтительно источника диоксида кремния или комбинации источника диоксида кремния и источника диоксида германия, и
источник второго оксида представляет собой, по меньшей мере, один, выбранный из группы, состоящей из источника оксида алюминия, источника оксида бора, источника оксида железа, источника оксида галлия, источника оксида редкоземельного элемента, источника оксида индия и источника оксида ванадия, предпочтительно источника оксида алюминия.
14. Способ по п. 13, в котором группы R1 и R2 являются отличными друг от друга, одна из которых выбрана из группы, состоящей из С3-12линейных или разветвленных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных или разветвленных оксаалкиленовых групп, предпочтительно одна выбрана из группы, состоящей из С3-12линейных алкиленовых групп, и другая выбрана из группы, состоящей из С3-12линейных оксаалкиленовых групп (предпочтительно С4-6линейных оксаалкиленовых групп, более предпочтительно С4-6линейных монооксаалкиленовых групп, более предпочтительно -(CH2)m-O-(CH2)m-, в которой каждое значение m, являющееся одинаковым или отличным друг от друга, независимо представляет собой 2 или 3);
множественные группы R являются одинаковыми или отличными друг от друга, каждая независимо выбрана из группы, состоящей из C1-4линейных или разветвленных алкильных групп, предпочтительно каждая независимо выбрана из группы, состоящей из метила и этила, более предпочтительно всего метила; и X представляет собой ОН.
15. Способ по п. 13, в котором щелочной источник представляет собой неорганическое основание, содержащее щелочной металл или щелочноземельный металл в качестве катиона, предпочтительно гидроксид натрия и гидроксид калия.
16. Способ по п. 13, в котором условия кристаллизации включают: температуру кристаллизации в диапазоне от 80°С до 120°С, предпочтительно от 120°С до 170°С или от 120°С до 200°С, и период кристаллизации, по меньшей мере, 1 день, предпочтительно, по меньшей мере, 2 дня, предпочтительно от 3 дней до 8 дней, от 5 дней до 8 дней или от 4 дней до 6 дней; и
условия обжига включают: температуру обжига в диапазоне от 300°C до 750°C, предпочтительно от 400°C до 600°C, и период обжига в диапазоне от 1 часа до 10 часов, предпочтительно от 3 часов до 6 часов.
17. Способ по п. 13, в котором молярное соотношение источника первого оксида (рассчитанное на основе первого оксида) и источника второго оксида (рассчитанное на основе второго оксида) находится в диапазоне от 5 до ∞, предпочтительно от 25 до 95, более предпочтительно от 30 до 70;
молярное соотношение воды и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 5 до 50, предпочтительно от 5 до 15;
молярное соотношение органического темплата и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 0,02 до 0,5, предпочтительно от 0,05 до 0,5, от 0,15 до 0,5 или от 0,3 до 0,5;
молярное соотношение щелочного источника (рассчитанное на основе OH-) и источника первого оксида (рассчитанное на основе первого оксида) находится в диапазоне от 0 до 1, предпочтительно от 0,04 до 1, от 0,1 до 1, от 0,2 до 1, от 0,3 до 0,7 или от 0,45 до 0,7.
18. Композиция молекулярного сита, содержащая молекулярное сито по любому одному из пп. 1-12 или молекулярное сито, полученное с помощью способа по любому одному из пп. 13-17, и связующее вещество.
19. Способ конверсии углеводорода, включающий стадию подвергания углеводорода реакции конверсии в присутствии катализатора, в котором катализатор содержит или его получают из молекулярного сита по любому одному из пп. 1-12, молекулярного сита, полученного с помощью способа по любому одному из пп. 13-17, или композиции молекулярного сита по п. 18.
20. Способ по п. 19, в котором реакция конверсии выбрана из группы, состоящей из реакций каталитического крекинга, гидрокрекинга, диспропорционирования, алкилирования, олигомеризации и изомеризации.
| CN 105129816 A, 09.12.2015 | |||
| CN 104370296 A, 25.02.2015 | |||
| US 5538711 A, 23.07.1996 | |||
| US 5593655 A, 14.01.1997 | |||
| US 5800800 A, 01.09.1998 | |||
| BAERLOCHER CH., et al Atlas of Zeolite Framework Types, 6-th edition, 2007 | |||
| БРЕК Д | |||
| Цеолитовые молекулярные сита, М., 1976. |

