Уровень техники
Галихондрин B представляет собой структурно сложное макроциклическое соединение, которое изначально было выделено из морской губки Halichondria okadai, а затем было обнаружено в Axinella sp., Phakellia carteri и Lissodendoryx sp. Эрибулин является синтетическим аналогом галихондрина B. Мезилатная соль эрибулина (мезилат эрибулина, который продается под торговым наименованием HALAVEN®) одобрена для лечения пациентов с раком молочной железы, которые ранее получали по меньшей мере два курса химиотерапии для лечения метастатического заболевания, которые включали в себя антрациклин и таксан или в адъювантном, или в метастатическом режиме.
Сущность изобретения
Настоящее изобретение предлагает эрибулин и его диастереомеры, содержащие углерод-11, а также способы синтеза эрибулина с помощью двухстадийного процесса, включающего в себя нитроальдольную реакцию и восстановление.
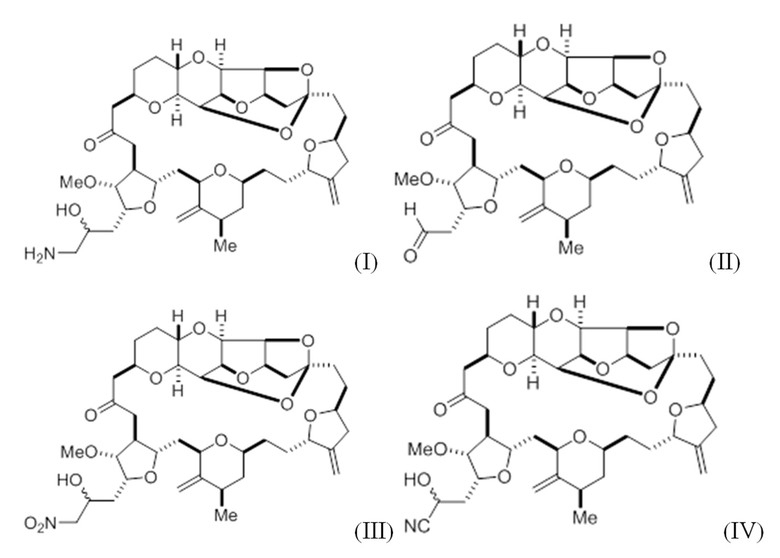
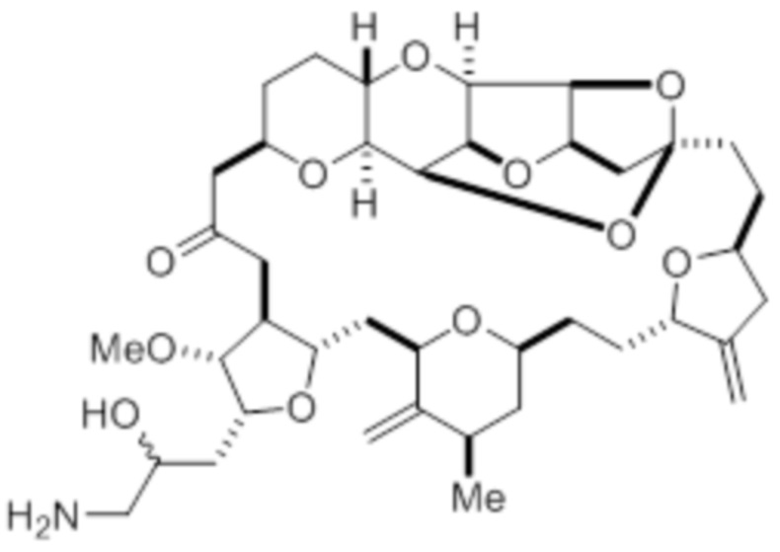
В первом аспекте настоящее изобретение предлагает соединение в соответствии с формулой (I),
 (I)
(I)
или его фармацевтически приемлемую соль, причем данное соединение содержит углерод-11.
В некоторых вариантах осуществления стереохимическая конфигурация в положении 34 соединения представляет собой (S). В некоторых вариантах осуществления стереохимическая конфигурация в положении 34 соединения представляет собой (R). В некоторых вариантах осуществления углерод-11 расположен в положении 35 соединения формулы (I).
В некоторых вариантах осуществления соединение представляет собой мезилатную соль формулы (I).
Соединение формулы (I) может также иметь форму композиции, обогащенной изотопно, т.е. углеродом-11. Термин "изотопно обогащенный", как используется в настоящем документе, относится композиции, включающей в себя изотоп, например 11C, в некотором положении в соединении в большем количестве, чем другие изотопы, например 12C, в том же положении. Как правило, в зависимости от изотопа композиции, обогащенные конкретным изотопом, могут иметь коэффициент изотопного обогащения по меньшей мере 5, по меньшей мере 10, по меньшей мере 50, по меньшей мере 500, по меньшей мере 2000, по меньшей мере 3000, по меньшей мере 6000 или по меньшей мере 6600, например, относительно 12C.
В дополнительном аспекте настоящее изобретение относится к способу синтеза соединения в соответствии с формулой (I) путем приведения в реакцию альдегида в соответствии с формулой (II):
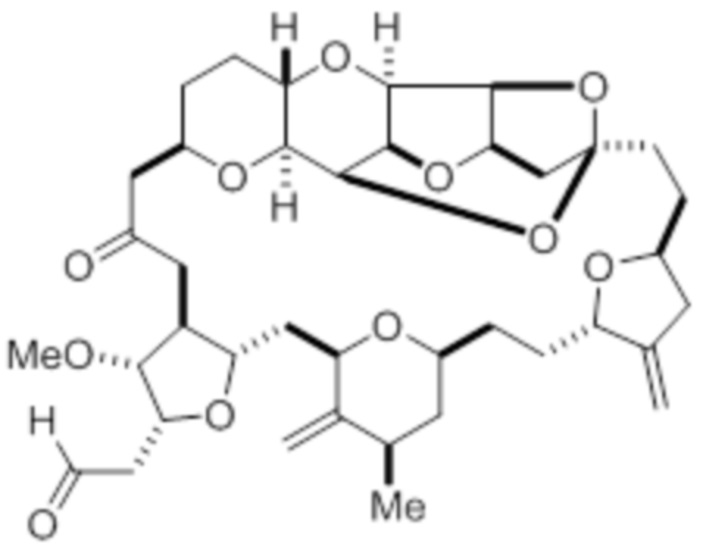 (II)
(II)
с нитрометаном, например содержащим углерод-11, в условиях реакции Генри с образованием соединения в соответствии с формулой (III):
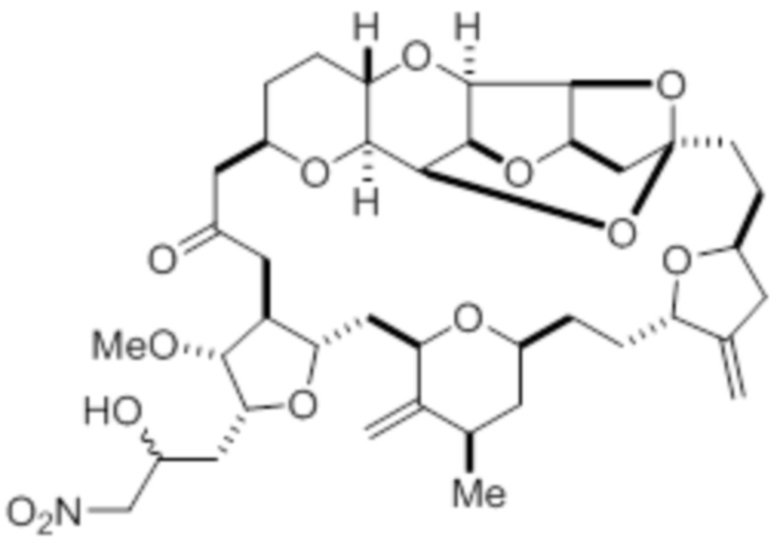 (III)
(III)
и восстановления соединения в соответствии с формулой (III) с помощью восстанавливающего средства с образованием соединения в соответствии с формулой (I).
В некоторых вариантах осуществления данный способ включает в себя растворение альдегида в спирте, например метаноле, с образованием раствора. В некоторых вариантах осуществления раствор имеет концентрацию от 0,01 М до 0,5 М, например приблизительно 0,1 М. В некоторых вариантах осуществления данный способ включает в себя добавление от 1 до 10000 молярных эквивалентов нитрометана к альдегиду, например приблизительно 1000 молярных эквивалентов нитрометана к альдегиду.
В некоторых вариантах осуществления данный способ включает в себя добавление основания, например гидроксида натрия, к смеси, содержащей альдегид и нитрометан.
В некоторых вариантах осуществления соединение в соответствии с формулой (III) восстанавливают с помощью соли лантанида, такой как йодид самария (II). В некоторых вариантах осуществления данный способ включает в себя растворение соединения в соответствии с формулой (III) в спирте, например метаноле, с образованием раствора. В некоторых вариантах осуществления раствор имеет концентрацию соединения в соответствии с формулой (III) от 0,01 мМ до 1 мМ, например приблизительно 0,7 мМ. В некоторых вариантах осуществления восстанавливающее средство присутствует в растворе в концентрации от 0,01 М до 1 М, например приблизительно 0,1 М. В некоторых вариантах осуществления данный способ включает в себя смешивание от 2 молярных эквивалентов до 1000 молярных эквивалентов восстанавливающего средства с соединением в соответствии с формулой (III).
В другом аспекте настоящее изобретение предлагает способ синтеза соединения в соответствии с формулой (I) путем приведения в реакцию альдегида в соответствии с формулой (II):
(II)
с цианидной солью, например цианидом калия, в условиях образования цианогидрина в соответствии с формулой (IV):
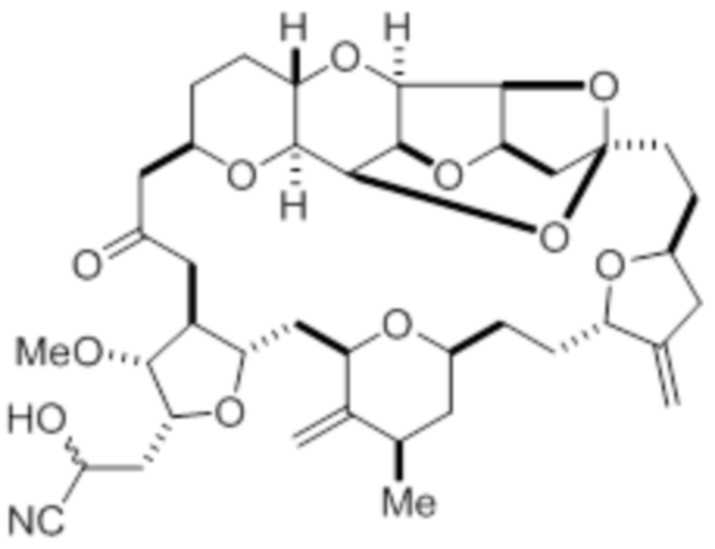 (IV)
(IV)
и восстановления цианогидрина в соответствии с формулой (IV) с помощью восстанавливающего средства с образованием соединения в соответствии с формулой (I). В некоторых вариантах осуществления восстанавливающее средство представляет собой силан, такой как триэтилсилан, используемый, например, в комбинации с кислотой Льюиса, такой как трис(перфторфенил)боран. В некоторых вариантах осуществления цианидная соль содержит углерод-11 (например, K11CN).
В некоторых вариантах осуществления альдегид синтезируют путем приведения в реакцию диола в соответствии с формулой (V):
 (V)
(V)
с окисляющим средством, например перйодатом натрия.
В некоторых вариантах осуществления любой из способов может дополнительно включать в себя образование соли соединения формулы (I) с получением его фармацевтически приемлемой соли, например мезилатной соли. Способы могут также включать в себя разделение диастереомеров соединения формулы (I) для выделения эрибулина или его фармацевтически приемлемой соли, например мезилатной соли.
В другом аспекте настоящее изобретение предлагает соединение в соответствии с формулой (II):
(II).
В другом аспекте настоящее изобретение предлагает соединение в соответствии с формулой (IV):
(IV).
Настоящее изобретение дополнительно предлагает фармацевтическую композицию, содержащую эффективное количество содержащего углерод-11 соединения формулы (I), или изотопно обогащенную композицию соединения формулы (I) и фармацевтически приемлемый носитель.
В дополнительном аспекте настоящее изобретение предлагает способ применения содержащего углерод-11 соединения формулы (I) для визуализации субъекта, например человека. В некоторых вариантах осуществления данный способ включает в себя введение субъекту соединения и детектирование присутствия соединения. В некоторых вариантах осуществления детектирование включает в себя анализ субъекта с помощью позитронной эмиссионной томографии.
Краткое описание фигур
Фигура 1 демонстрирует серию хроматограмм, изображающих превращение соединения (II) в соединение (III), как описано в примере 1. Сверху вниз хроматограммы соответствуют отбору реакционной смеси через приблизительно 5 минут, приблизительно 30 минут, приблизительно 1 час и 50 минут и приблизительно 3 часа от начала добавления нитрометана и гидроксида натрия к соединению (II), как описано в примере 1.
Фигура 2 демонстрирует серию хроматограмм, изображающих превращение соединения (III) в соединение (I), как описано в примере 2. Сверху вверх хроматограммы соответствуют отбору реакционной смеси в начале реакции, приблизительно через 17 минут после добавления SmI2 к нитро-аддукту (III) и после добавления раствора соли Рошель к реакционной смеси.
Фигура 3A представляет собой хроматограмму, полученную из анализа реакционной смеси после добавления нитро-аддукта к восстанавливающему средству в обратном порядке, как описано в примере 3.
Фигура 3B представляет собой хроматограмму, полученную из анализа реакционной смеси после добавления восстанавливающего средства к нитро-аддукту, как описано в примере 2.
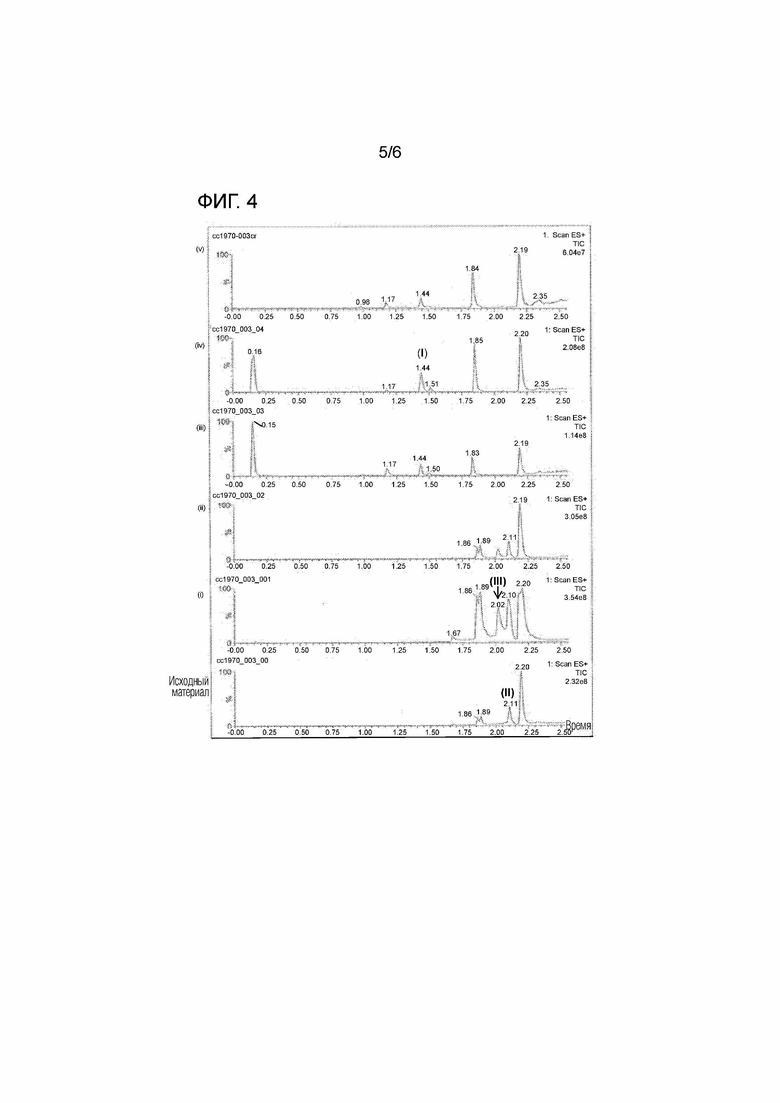
Фигура 4 демонстрирует серию хроматограмм, изображающих превращение соединения (II) в соединение (I), как описано в примере 4. Снизу вверх хроматограммы соответствуют исходному материалу, (i) реакционной смеси после добавления нитрометана и гидроксида натрия, (ii) реакционной смеси после гашения уксусной кислотой, (iii) и (iv) реакционной смеси после добавления SmI2 и (v) реакционной смеси после выделения продукта.
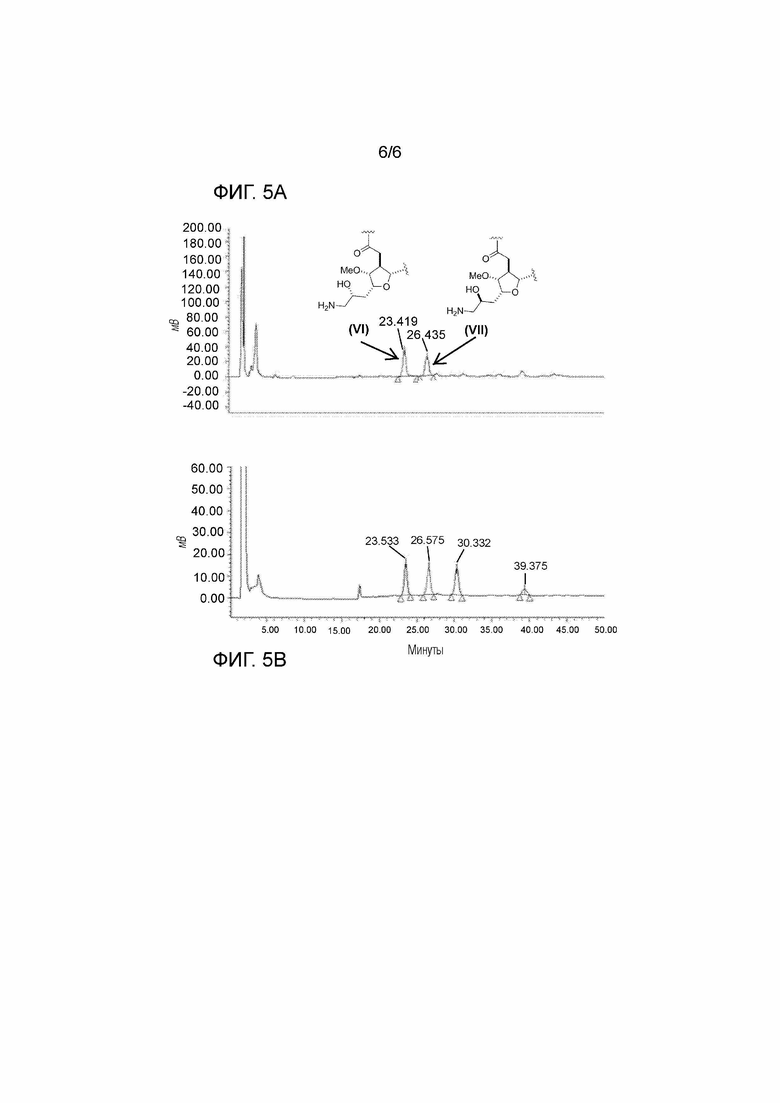
Фигура 5A представляет собой хроматограмму, полученную из ВЭЖХ-анализа продукта, полученного с помощью реакции эквимолярных количеств соединения (II) и нитрометана с использованием тандемного процесса нитроальдольная реакция/восстановление, как описано в примере 4. Эрибулин отделяли с помощью данного процесса от его диастереомера C34.
Фигура 5B представляет собой хроматограмму, полученную из ВЭЖХ-анализа продукта, полученного с помощью реакции соединения (II) с 0,2 молярного эквивалента нитрометана с использованием тандемного процесса нитроальдольная реакция/восстановление, как описано в примере 4. Эрибулин отделяли с помощью данного процесса от его диастереомера C34.
Подробное описание
Настоящее изобретение предлагает эрибулин и его диастереомеры, представленные формулой (I) ниже, которые содержат углерод-11, а также соответствующие способы синтеза.
 (I).
(I).
Настоящее изобретение дополнительно предлагает фармацевтически приемлемую соль соединения формулы (I), например мезилатную соль.
Соединения, получаемые с помощью способов настоящего изобретения, такие как эрибулин или его диастереомер, содержащие углерод-11 в положении 35 молекулы, можно использовать для различных целей. Эрибулин является известным химиотерапевтическим средством, и эрибулин, синтезированный с помощью способов настоящего изобретения, можно вводить пациенту-человеку, страдающему от рака, необязательно в комбинации с дополнительными химиотерапевтическими средствами, для лечения рака. Кроме того, поскольку углерод-11 является хорошо известной радиометкой для позитронной эмиссионной томографии, эрибулин, содержащий углерод-11, можно вводить пациенту для визуализации образца внутри субъекта, такого как конкретный орган или ткань внутри субъекта. Например, эрибулин, содержащий углерод-11, можно вводить пациенту для визуализации одной или нескольких солидных опухолей внутри субъекта, например подвергаемого химиотерапевтическому лечению.
С помощью способов настоящего изобретения эрибулин может быть синтезирован в условиях реакции Генри, например следующим образом:
Схема 1
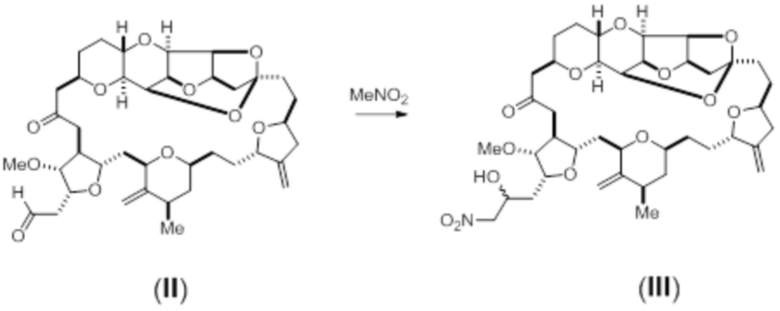
В соответствии со способами настоящего изобретения, соединение (III) может быть восстановлено, например, с использованием соли лантанида, такой как йодид самария (II). В данной области техники известны другие подходящие восстанавливающие средства. Например, соединение (III) может быть восстановлено в соответствии со следующей схемой реакции:
Схема 2
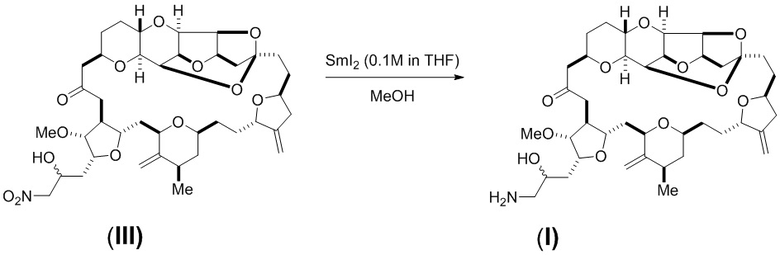
Необязательно, эрибулин может быть синтезирован с использованием одно- или двухреакторной процедуры, например, как показано на схеме 3 ниже. В этом способе соединение (II) превращается в соединение (III) с помощью нитроальдольного процесса с использованием нитрометана. Восстанавливающее средство, такое как SmI2, может затем быть смешано непосредственно с реакционной смесью, содержащей соединение (III). Как показано на схеме 3 ниже, и как описано более подробно в примерах, этот способ может быть осуществлен или путем добавления восстанавливающего средства непосредственно к реакционной смеси после реакции Генри, или путем переноса реакционной смеси, содержащей соединение (III), в сосуд, содержащий восстанавливающее средство. Кислотную обработку также можно проводить с помощью пивалиновой кислоты вместо уксусной кислоты.
Схема 3
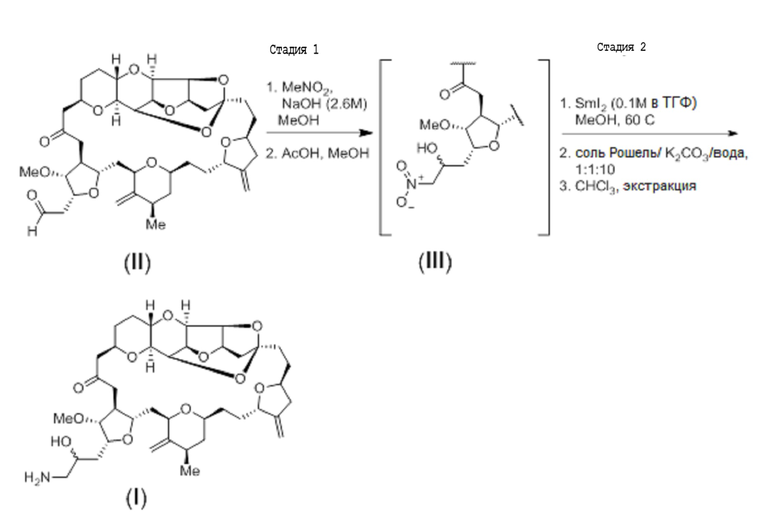
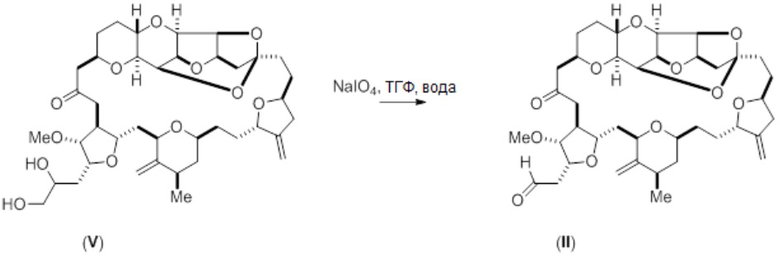
С помощью способов настоящего изобретения соединение (II) может быть синтезировано путем окисления диола в соответствии с формулой (V). Например, соединение (II) может быть получено с помощью опосредованного перйодатом натрия окисления диола (V), как показано на схеме реакции 4 ниже.
Схема 4
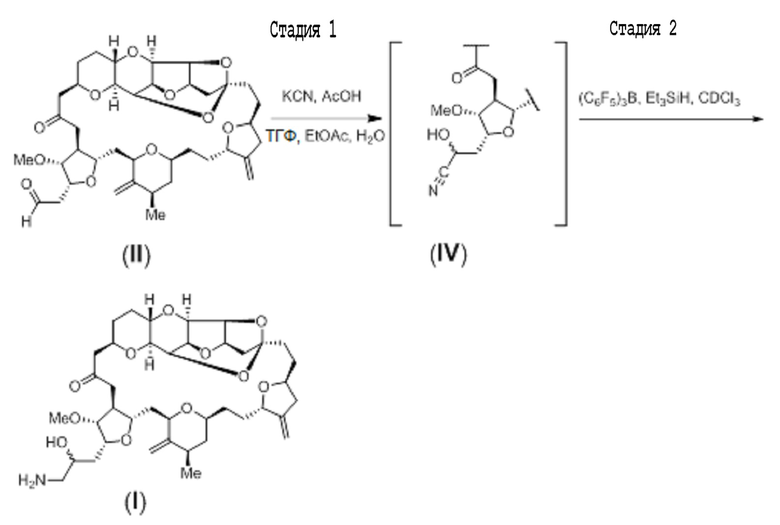
С помощью способов настоящего изобретения эрибулин можно альтернативно синтезировать путем приведения альдегида (II) в реакцию с цианидной солью, такой как цианид калия, для образования цианогидрина, представленного формулой (IV) ниже. Полученный таким образом цианогидрин может затем быть восстановлен для генерации эрибулина. Этот двухстадийный процесс изображен на схеме 5 ниже. Нитрильный заместитель циангидрина (IV) может быть восстановлен до амина, например с помощью реакции цианогидрина с восстанавливающим средством, таким как силан, например диэтилсилан или триэтилсилан, в присутствии кислоты Льюиса, такой как трис(перфторфенил)боран (Chang et al. J. Org. Chem. 2015, 80, 7281-7287).
Схема 5
Эрибулин может быть отделен от своего диастереомера C-34 с использованием стандартных методов, таких как ВЭЖХ.
В данной области техники известны условия реакции солеобразования. Образование соли эрибулина может давать фармацевтически приемлемую соль эрибулина (например, мезилат эрибулина). В частности, реакция солеобразования может включать в себя приведение эрибулина в контакт с кислотой Бренстеда (например, фармацевтически приемлемой кислотой Бренстеда (например, метансульфоновой кислотой)) для получения фармацевтически приемлемой соли эрибулина (например, Handbook of Pharmaceutical Salts: Properties, Selection and Use, ed.: Stahl and Wermuth, Wiley-VCH/VHCA, Weinheim/Zurich, 2002). Фармацевтически приемлемые соли эрибулина, например мезилат эрибулина, могут быть образованы с помощью способов, известных в данной области техники, например in situ во время окончательного выделения и очистки соединения или отдельно, путем приведения свободной основной группы в реакцию с подходящей органической кислотой. В одном примере эрибулин обрабатывают раствором MsOH и NH4OH в воде и ацетонитриле. Смесь концентрируют. Остаток растворяют в ДХМ-пентане, и раствор добавляют к безводному пентану. Образовавшийся осадок отфильтровывают и сушат в высоком вакууме с получением мезилата эрибулина.
Композиции
Соединения или изотопно обогащенные композиции настоящего изобретения могут быть составлены в виде фармацевтически приемлемых солей, которые являются солями с медицинской точки зрения, подходящих для применения в контакте с тканями людей и животных без чрезмерной токсичности, раздражения, аллергической реакции и т.п. и соразмерно с разумным соотношением риск/польза. Фармацевтически приемлемые соли хорошо известны в данной области техники. Например, фармацевтически приемлемые соли описаны в: Berge et al., J. Pharmaceutical Sciences 66:1-19, 1977, и в Pharmaceutical Salts: Properties, Selection, and Use, (Eds. P.H. Stahl and C.G. Wermuth), Wiley-VCH, 2008. Типичные соли присоединения кислоты включают соли ацетат, адипат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, фумарат, глюкогептонат, глицерофосфат, гемисульфат, гептонат, гексаноат, гидробромид, гидрохлорид, гидройодид, 2-гидроксиэтансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, толуолсульфонат, ундеканоат, валерат и т.п. Предпочтительной солью является мезилатная соль.
Соединения или изотопно обогащенные композиции настоящего изобретения могут также быть составлены в виде фармацевтических композиций, например, путем объединения эффективного количества соединения или изотопно обогащенной композиции с фармацевтически приемлемым носителем. Эффективное количество, как правило, представляет собой количество, необходимое для визуализации субъекта с помощью позитронной эмиссионной томографии.
Фармацевтические композиции могут быть получены с использованием стандартных способов, известных в данной области техники, или могут быть получены из коммерческих источников. Соединение формулы (I), например, эрибулин, обычно получают в жидкой форме для внутривенного введения.
Фармацевтические композиции, используемые в настоящем изобретении, могут быть получены, например, путем смешивания или растворения активного ингредиента(ов) с требуемой степенью чистоты в физиологически приемлемом носителе (смотри, например, Remington's Pharmaceutical Sciences (20th edition), ed. A. Gennaro, 2000, Lippincott, Williams & Wilkins, Philadelphia, PA). Приемлемые носители включают воду и солевой раствор, необязательно включающие в себя буферы, такие как фосфатный, цитратный или из других органических кислот; антиоксиданты, включая бутилированный гидрокситолуол (BHT), бутилированный гидроксианизол (BHA), аскорбиновую кислоту; низкомолекулярные (менее приблизительно 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон, аминокислоты, такие как глицин, глутамин, аспарагины, аргинин или лизин; моносахариды, дисахариды или другие углеводы, включая глюкозу, маннозу или декстрины; хелатирующие средства, такие как ЭДТА; сахарные спирты, такие как маннит или сорбит; солеобразующие противоионы, такие как натрий; и/или неионогенные поверхностно-активные вещества, такие как TWEEN™, PLURONICS™ или ПЭГ.
Необязательно, композиции настоящего изобретения содержат фармацевтически приемлемый консервант. В некоторых вариантах осуществления концентрация консерванта составляет от 0,1 до 2,0%, обычно об/об. Подходящие консерванты включают известные в области фармацевтики, такие как бензиловый спирт, фенол, м-крезол, метилпарабен и пропилпарабен. Кроме того, композиции соединения формулы (I), например, эрибулин, могут, необязательно, включать в себя фармацевтически приемлемую соль, такую как хлорид натрия, например, в приблизительно физиологический концентрации. Поэтому в одном примере соединение формулы (I), например, эрибулин (например, мезилат эрибулина), составляют в виде инъекции в 0,9% хлориде натрия (USP).
Композиции, отмеченные выше (и другие) можно использовать для парентерального введения лекарственных средств. Таким образом, лекарственные средства можно вводить разными способами, включая внутривенный, внутриопухолевый, перитуморальный, внутриартериальный, внутрикожный, внутрипузырный, офтальмический, внутримышечный, внутрикожный, внутрибрюшинный, легочный, подкожный и чрескожный способы. Можно также использовать другие способы, включая, например, трансмукозальный, трансдермальный, ингаляционный, внутривагинальный, ректальный и пероральный способы введения.
Дозировка вводимого соединения формулы (I), например эрибулина, может заметно различаться в зависимости от типа целевого заболевания, выбора способа доставки, а также возраста, пола и веса пациента, тяжести симптомов, а также других факторов.
Примеры
Следующие способы иллюстрируют синтез соединений формулы (I) с использованием реагентов с природным изотопным составом по углероду. Эти способы могут быть модифицированы путем использования 11C-меченого нитрометана или 11C-меченого цианида с получением 11C-меченых соединений формулы (I).
Пример 1. Синтез соединения формулы (III) с помощью нитроальдольной реакции
Альдегид формулы (II) (278 мг, 0,398 ммоль) растворяли в метаноле (2,780 мл), и последовательно добавляли в раствор альдегида нитрометан (21,45 мкл, 1 экв., 0,398 ммоль) и гидроксид натрия (2,6 М, 161 мкл, 1,05 экв., 0,418 ммоль). Возникающей реакции давали протекать в течение приблизительно 3 часов, и за реакционной смесью периодически наблюдали с помощью жидкостной хроматографии/масс-спектрометрии (ЖХ/МС). Через 5 минут отношение продукта, соединения формулы (III), к исходному материалу составляло приблизительно 2:1. Через приблизительно 30 минут оставалось только 5-10% альдегида, причем реакционная смесь содержала преимущественно нитросодержащее соединение формулы (III). Через приблизительно 1 час 50 минут не наблюдали никаких изменений состава реакционной смеси по сравнению с реакционной смесью, проанализированной через 30 минут. Кроме того, через приблизительно 3 часа не наблюдали никаких изменений состава реакционной смеси по сравнению с реакционной смесью, проанализированной через 30 минут (фигура 1).
Реакцию затем гасили добавлением уксусной кислоты (23,91 мкл, 0,418 ммоль) с последующим добавлением по каплям воды (7 мл) для получения белой суспензии. Суспензию фильтровали и сушили в потоке N2 с получением 235 мг соединения формулы (III) в виде белого порошка (0,31 ммоль, 78%).
Пример 2. Восстановление соединения формулы (III) путем стандартного процесса добавления
Соединение формулы (III) (6,40 мг, 8,422 мкмоль) растворяли в метаноле (1,28 мл) и дегазировали пропусканием через него N2 в течение 2 минут. Затем раствор постепенно нагревали до 55-60°C. Затем к раствору соединения формулы (III) добавляли раствор SmI2 (0,1 М в ТГФ, 0,84 мл, 10 экв.) в течение 2 минут. Температура реакционной смеси в начале добавления составляла 40°C и при добавлении постоянно повышалась до 50°C. Через 8 минут от начала добавления раствора SmI2 к соединению формулы (III) наблюдали завершение реакции. Через дополнительных 10 минут не наблюдали никаких изменений состава реакции. Затем к реакционной смеси добавляли раствор, содержащий соль Рошель (тартрат калия натрия), K2CO3 и воду (молярное соотношение 1:1:10, 1 мл), и полученную суспензию перемешивали в течение 2 минут. Затем добавляли хлороформ (1 мл), и реакционную смесь перемешивали в течение дополнительных 2 минут (фигура 2). Нижний слой удаляли, и водный слой дважды экстрагировали хлороформом. Анализ экстрактов с помощью тонкослойной хроматографии (ТСХ) показал, что соединение (I) содержалось в первых двух растворах хлороформа. Экстракты объединяли и концентрировали с получением 9 мг неочищенного продукта в виде бледно-желтого твердого вещества.
Пример 3. Восстановление соединения формулы (III) путем процесса добавления в обратном порядке
Соединение формулы (III) (6,40 мг, 8,422 мкмоль) растворяли в метаноле (1,28 мл) и дегазировали пропусканием через него N2 в течение 2 минут. Раствор SmI2 (0,1 М в ТГФ, 0,84 мл, 10 экв.) готовили в отдельной колбе и постепенно нагревали до 55-60°C. Затем раствор соединения формулы (III) добавляли к раствору SmI2 в течение 3 минут. Температура реакционной смеси в начале добавления составляла 64°C, а в конце добавления составляла 60°C. За реакционной смесью через 12 минут от начала добавления раствора SmI2 к соединению формулы (III) наблюдали с помощью ЖХ/МС, и результаты сравнивали с графиком ЖХ/МС, полученным после стандартного процесса добавления, описанного в примере 2. Это сравнение показано на фигурах 3A и 3B.
Через 12 минут от начала добавления раствора SmI2 к соединению формулы (III) смесь охлаждали до комнатной температуры. Затем к реакционной смеси добавляли раствор, содержащий соль Рошель, K2CO3 и воду (молярное соотношение 1:1:10, 1 мл), и полученную суспензию перемешивали в течение 2 минут. Затем добавляли хлороформ (1 мл), и реакцию перемешивали в течение дополнительных 2 минут. Нижний слой удаляли, и водный слой дважды экстрагировали хлороформом. Анализ экстрактов с помощью тонкослойной хроматографии (ТСХ) показал, что соединение формулы (I) содержалось в первых двух растворах хлороформа. Экстракты объединяли и концентрировали с получением 16 мг неочищенного продукта в виде бледно-желтого твердого вещества.
Пример 4. Однореакторная процедура синтеза соединения формулы (I) с помощью тандемного процесса нитроальдольная реакция/восстановление
Стадия 1: Исходный раствор углерод-11-меченого нитрометана (3,5 мкл) добавляли в метанол (1,5 мл), продуваемый N2. Соединение формулы (II) (15,00 мг, 0,02 ммоль) растворяли в растворе нитрометан/метанол (0,15 мл раствора, 0,262 мг нитрометана, 4,293 мкмоль, 0,2 экв.) во флаконе объемом 1,5 мл, через который пропускали N2, снабженный мешалкой и мембраной из силикона-политетрафторэтилена (PTFE). Аликвоту полученной смеси 5-10 мкл удаляли и разбавляли в 0,25 мл метанола для анализа. Затем добавляли к смеси раствор гидроксида натрия (2,6 М, 1,65 мкл, 4,29 мкмоль), и возникающей реакции давали протекать в течение 16 минут. Затем добавляли к реакционной смеси уксусную кислоту (12,28 мкл, 0,215 ммоль, 10 экв.), которую затем разбавляли метанолом, через который пропускали N2 (3 мл).
Стадия 2: Раствор SmI2 (0,1 М в ТГФ, 2,15 мл, 0,215 ммоль) добавляли в продуваемый N2 конический микроволновый флакон объемом 7 мл, снабженный винтовым колпачком из силикона-PTFE и мешалкой. Затем смесь нагревали до 60°C. Затем в течение 2 минут к раствору SmI2 медленно добавляли нитро-аддукт, образовавшийся на стадии 1. Температура получающейся смеси в начале процесса добавления составляла 38°C, а температура смеси в конце процесса добавления составляла 45°C и повышалась. Через 12 минут от начала процесса добавления наблюдали завершение восстановления. Затем реакционную смесь постепенно охлаждали до комнатной температуры, и метанол выпаривали потоком N2. Затем к реакционной смеси добавляли раствор, содержащий соль Рошель, K2CO3 и воду (молярное соотношение 1:1:10, 3 мл). Через 5 минут к смеси добавляли хлороформ (2 мл), и затем смесь энергично перемешивали. Слоям давали разделиться, и удаляли нижний слой (хлороформ). Экстракцию хлороформом повторяли второй раз, и экстракты анализировали с помощью ТСХ. Наблюдали, что продукт (соединение формулы (I)) находился преимущественно в первом экстракте, а во втором экстракте наблюдали следовые количества продукта. Было обнаружено, что водный слой не содержит продукта. Графики ЖХ/МС, записанные на различных этапах стадии 1 и стадии 2, показаны на фигуре 4.
Пример 5. Синтез соединения формулы (II) путем окисления диольного предшественника
Диол формулы (V) (126 мг, 0,172 ммоль) растворяли в ТГФ (1,6 мл) и воде (0,88 мл). Добавляли перйодат натрия (55,3 мг, 0,259 ммоль) с образованием смеси, которую затем перемешивали при комнатной температуре. За ходом возникающей реакции окисления наблюдали с помощью ЖХ/МС-анализа. В течение 10 минут перемешивания образовывался белый осадок. Через 2 часа добавляли воду (15 мл), и смесь экстрагировали этилацетатом (3×15 мл). К смеси добавляли этанол 1 (мл), что вызывало разделение слоев. Затем объединенные экстракты промывали водой, а затем концентрировали с получением альдегида формулы (II) в виде белого твердого вещества (115 мг, 0,9 вес., выход 95%).
Пример 6. Очистка эрибулина с помощью жидкостной хроматографии высокого давления с обращенной фазой (ОФ-ВЭЖХ)
После однореакторной процедуры описанной в примере 4, соединение формулы (I) очищали с помощью ОФ-ВЭЖХ с использованием двухкомпонентной подвижной фазы и профиля градиентного элюирования, как описано в таблицах 1 и 2 ниже. Колонки C18 были получены от ACE® (Абердин, Шотландия). Растворитель A: 760 мл воды и 240 мл ацетонитрила смешивали вместе, добавляли 7,0±0,2 г трифторметансульфоната аммония, добавляли 3,0 мл 1,0 М водного раствора дигидрофосфата тетрабутиламмония, и доводили pH до 6,9-7,1 с помощью или 5,6% раствора гидроксида аммония, или 1 М HCl. Растворитель B: 300 мл воды, 7000 мл ацетонитрила и 20 мл 2-пропанола смешивали вместе, добавляли 7,0±0,2 г трифторметансульфоната аммония, 3,0 мл 1,0 М водного раствора дигидрофосфата тетрабутиламмония, и доводили pH до 6,9-7,1 с помощью или 5,6% раствора гидроксида аммония, или 1 М HCl.
Таблица 1. Сводка параметров, используемых для ВЭЖХ-анализа
(например, ACE® C18, размер частиц 3 мкм)
Таблица 2. Профиль градиента ВЭЖХ
Образец хроматограммы, полученной с помощью очистки на ОФ-ВЭЖХ, показан на фигуре 5A (однореакторнвй процесс, молярное соотношение альдегид:нитрометан 1:1) и фигуре 5B (однореакторный процесс, молярное соотношение альдегид:нитрометан 5:1).
Пример 7. Очистка соединения формулы (I) с помощью хроматографии на силикагеле
Неочищенное соединение формулы (I) (смесь диастереомеров, полученная с использованием однореакторной процедуры, как описано в примере 4) очищали с помощью колоночной хроматографии на силикагеле (230-400 SiO2, 0,5 см d × 5,5 см h). Соединение формулы (I) элюировали с помощью смеси 3:2 гептан:этилацетат и этилацетата по 25 мл, 40 мл смеси 4:1 ацетонитрил:вода, содержащей 0,2% NH4OH. Колонку с силикагелем кондиционировали с помощью смеси 3:2 гептан:этилацетат, и смесь неочищенных продуктов вводили со смесью дихлорметан/гептан. Элюенты гептан:этилацетат и этилацетат собирали по одной фракции каждого (фракции 1 и 2). Элюент ацетонитрил:вода собирали в 4 фракциях по 10 мл (фракции 3-6). Затем каждую фракцию анализировали с помощью ТСХ и ЖХ/МС. Фракцию 4 концентрировали получением смеси 1:1 эрибулина и его диастереомера C-34 (2,5 мг, 3,56 мкмоль, 83% по нитрометану, незначительное содержание примеси амида).
Пример 8. Синтез соединения формулы (I) с помощью цианогидрина
Соединение формулы (II) (78 мг, 0,112 ммоль) растворяли в (EtOAc:ТГФ:AcOH:вода; 2:1:1,6:0,4 (об/об), 25 объемов). Добавляли цианид калия (25 мг, 0,384 ммоль) в воде (0,17 мл), раствор перемешивали при комнатной температура, и наблюдали с помощью ТСХ (4:1 толуол/ацетонитрил) и ЖХ/МС. Когда полагали, что реакция завершена, добавляли воду (20 мл), и реакционную смесь перемешивали. Удаляли органический слой, и водный слой экстрагировали с помощью EtOAc (3×15 мл). Объединенные экстракты промывали 10% водным бикарбонатом натрия (20 мл). Водный слой подвергали обратной экстракции с помощью ДХМ (10 мл). Объединенные органические слои концентрировали и подвергали азеотропной перегонке с толуолом с получением неочищенных диастереомеров продукта формулы (IV). Остаток очищали с помощью флеш-роматографии (толуол/ацетонитрил от 6:1 до 4:1) с получением β-изомера (4 мг) и смеси α- (эпи-34) и β-изомеров (19 мг).
Исходный раствор трис(перфторфенил)борана и триэтилсилана получали растворением трис(перфторфенил)борана (10,6 мг) и триэтилсилана (0,275 мл) в D-хлороформе (0,75 мл). 70 мкл раствора реагентов (10 экв. силана, 0,03 экв. трис(перфторфенил)борана) добавляли к α-изомеру (5,00 мг, 6,888 мкмоль) при комнатной температуре, и следили за реакцией с помощью ЖХ/МС. Через 10 минут добавляли HCl (0,01 н, 0,2 мл), и реакционную смесь перемешивали в течение 5 минут. Реакционную смесь экстрагировали с помощью ДХМ (0,2 мл) для удаления липофильных примесей. Водный слой обрабатывали бикарбонатом натрия (0,5 мл) и затем экстрагировали с помощью ДХМ (2×0,4 мл). Объединенные экстракты концентрировали с получением соединения формулы (I) (эпи-34 эрибулина).
Другие варианты осуществления
Все публикации, патенты и патентные заявки, упомянутые в данном описании изобретения, включены в настоящий документ посредством ссылки в той же степени, как если бы каждая отдельная публикация или патентная заявка были специально и индивидуально указаны для включения посредством ссылки.
Хотя настоящее изобретение было описано в связи с конкретными вариантами его осуществления, следует понимать, что его можно подвергать дальнейшим модификациям, и настоящая заявка предназначена для охвата любых вариантов, применений или адаптаций настоящего изобретения, следующих в общих чертах принципам настоящего изобретения, и включающих в секбя такие отклонения от настоящего изобретения, которые входят в известную или обычную практику в области, к которой относится настоящее изобретение, и могут быть применены к основным признакам, изложенным выше в настоящем документе, и соответствует объему формулы изобретения.
Другие варианты осуществления приведены в формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ, ПРЕДНАЗНАЧЕННЫЕ ДЛЯ СИНТЕЗА АНАЛОГОВ ГАЛИХОНДРИНА B | 2014 |
|
RU2676486C1 |
| СПОСОБ ПОЛУЧЕНИЕ АМИНОЭПОКСИДА ПУТЕМ НЕПРЕРЫВНОГО ПРОЦЕССА СИНТЕЗА IN SITU | 1997 |
|
RU2194045C2 |
| СПОСОБ СИНТЕЗА 2-ТИОГИСТИДИНА И ЕГО АНАЛОГОВ | 2010 |
|
RU2548153C2 |
| ПРОИЗВОДНЫЕ ФУРО[3,2-В]ПИРАНА, ПРИМЕНИМЫЕ В СИНТЕЗЕ АНАЛОГОВ | 2011 |
|
RU2579511C2 |
| СИНТЕЗ АРИЛЬНЫХ СОЕДИНЕНИЙ | 2016 |
|
RU2755580C2 |
| ПРОИЗВОДНЫЕ ГЛИКОПЕПТИДА ИЛИ ИХ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2145609C1 |
| СПОСОБЫ СИНТЕЗА СПИРО-ОКСИНДОЛЬНЫХ СОЕДИНЕНИЙ | 2010 |
|
RU2544852C2 |
| СПОСОБ СИНТЕЗА АМИНОАЛКИЛЕНФОСФОНОВОЙ КИСЛОТЫ | 2013 |
|
RU2694047C2 |
| ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ СИНТЕЗА ИНГИБИТОРОВ ВИЧ-ПРОТЕАЗ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2125561C1 |
| СПОСОБ СИНТЕЗА ГИДРАЗИНА, КОТОРЫЙ МОЖНО ПРИМЕНЯТЬ ПРИ ЛЕЧЕНИИ ВИРУСА ПАПИЛЛОМЫ | 2013 |
|
RU2649006C2 |




Изобретение относится к способам синтеза соединения формулы (I) или его фармацевтически приемлемой соли. В одном варианте способ включает взаимодействие альдегида формулы (II) с нитрометаном в условиях реакции Генри в присутствии основания с образованием соединения формулы (III) и восстановление соединения формулы (III) с помощью соли лантанида с образованием соединения формулы (I). В другом варианте способ включает взаимодействие альдегида формулы (II) с цианидной солью в условиях образования цианогидрина формулы (IV) и восстановление цианогидрина формулы (IV) с помощью силана с образованием соединения формулы (I). Изобретение также относится к промежуточному соединению формулы (IV). Соединение формулы (I) является лекарственным средством эрибулином, которое находит применение в химиотерапевтическом лечении рака. 3 н. и 10 з.п. ф-лы, 2 табл., 8 пр., 5 ил.
1. Способ синтеза соединения в соответствии с формулой (I)
 (I)
(I)
или его фармацевтически приемлемой соли, причем способ включает взаимодействие альдегида в соответствии с формулой (II)
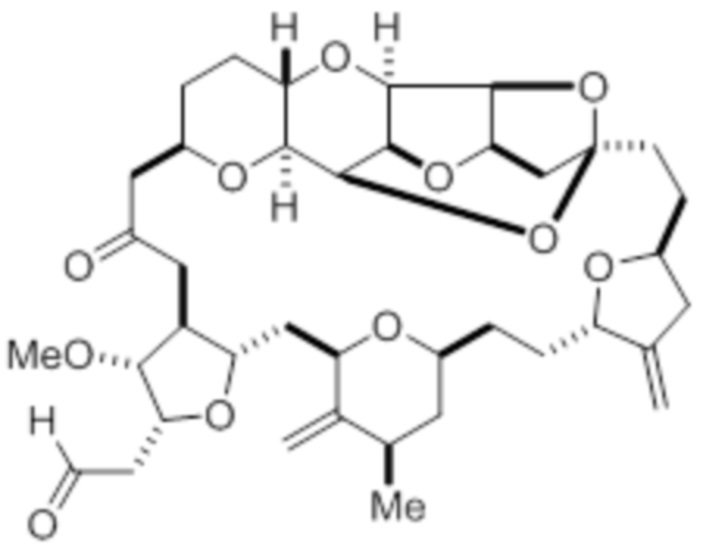 (II)
(II)
с нитрометаном в условиях реакции Генри в присутствии основания с образованием соединения в соответствии с формулой (III)
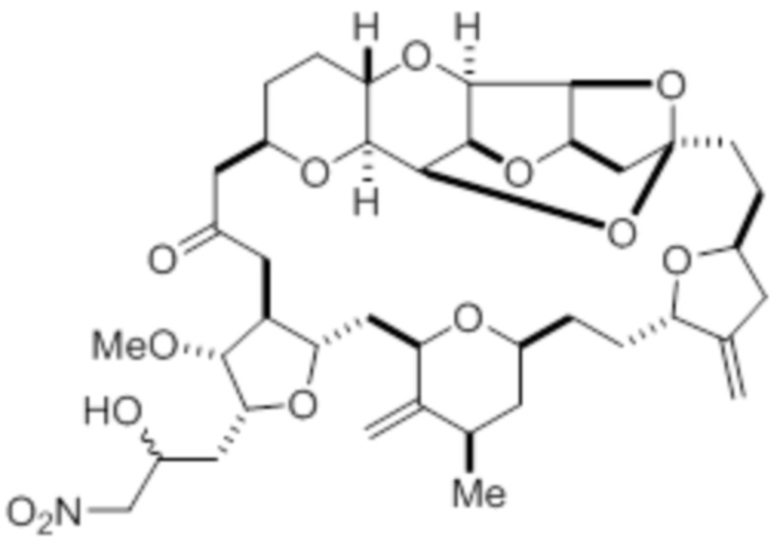 (III)
(III)
и восстановление соединения в соответствии с формулой (III) с помощью соли лантанида с образованием соединения в соответствии с формулой (I).
2. Способ по п. 1, в котором основание представляет собой гидроксид натрия.
3. Способ по п. 1 или 2, в котором соль лантанида представляет собой йодид самария (II).
4. Способ синтеза соединения в соответствии с формулой (I)
(I)
или его фармацевтически приемлемой соли, причем способ включает взаимодействие альдегида в соответствии с формулой (II)
(II)
с цианидной солью в условиях образования цианогидрина в соответствии с формулой (IV)
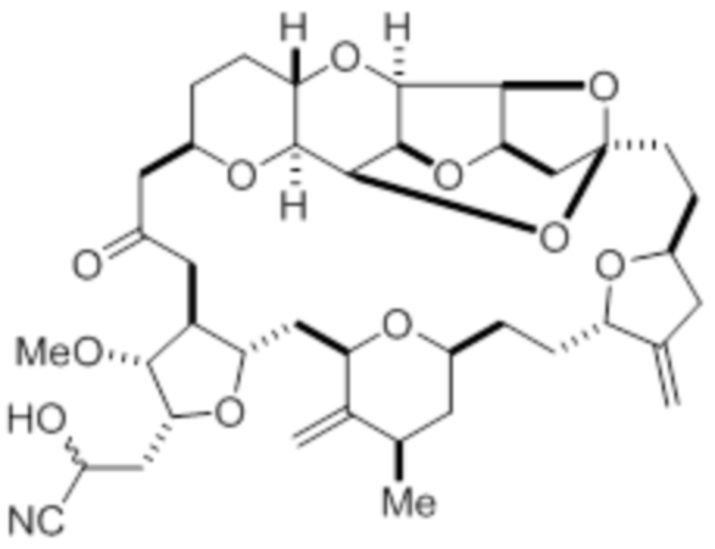 (IV)
(IV)
и восстановление цианогидрина в соответствии с формулой (IV) с помощью силана с образованием соединения в соответствии с формулой (I).
5. Способ по п. 4, в котором цианидная соль представляет собой цианид калия.
6. Способ по п. 4 или 5, в котором силан представляет собой диэтилсилан или триэтилсилан.
7. Способ по любому из пп. 4-6, дополнительно включающий добавление кислоты Льюиса к смеси цианогидрина и силана.
8. Способ по п. 7, в котором кислота Льюиса представляет собой трис(перфторфенил)боран.
9. Способ по любому из пп. 1-8, дополнительно включающий синтез альдегида в соответствии с формулой (II) путем взаимодействия диола в соответствии с формулой (V)
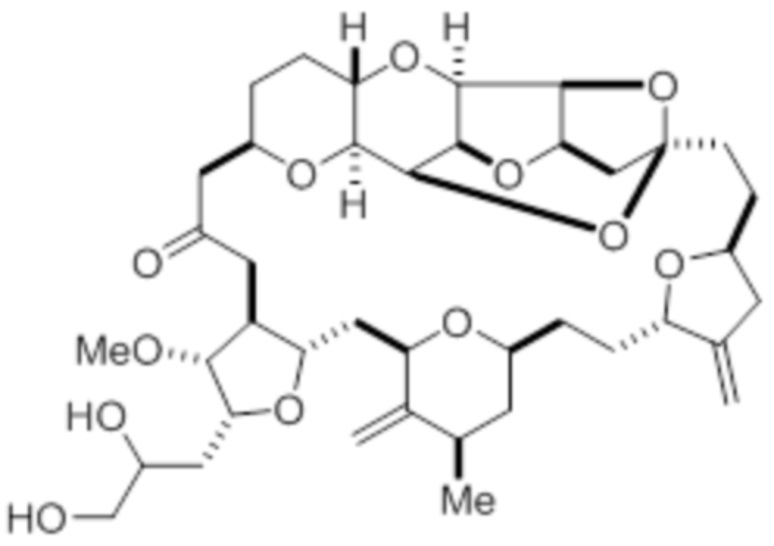 (V)
(V)
с окисляющим средством с образованием альдегида.
10. Способ по п. 9, в котором окисляющее средство представляет собой перйодат натрия.
11. Способ по любому из пп. 1-10, дополнительно включающий образование соли соединения формулы (I) с получением его фармацевтически приемлемой соли.
12. Способ по п. 11, в котором фармацевтически приемлемая соль представляет собой мезилатную соль.
13. Соединение в соответствии с формулой (IV)
(IV).
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ СИНТЕЗА АНАЛОГОВ ГАЛИХОНДРИНА В | 2008 |
|
RU2489437C2 |

