ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области медицинской химии, в частности, к кристаллической форме малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты и способу получения и применения новой кристаллической формы.
УРОВЕНЬ ТЕХНИКИ
Сахарный диабет (СД) является хроническим, системным метаболическим заболеванием, вызванным длительным взаимодействием генетических факторов и факторов окружающей среды. Он характеризуется повышенным уровнем глюкозы в плазме крови и представляет собой заболевание, влияющее на нормальную физиологическую активность, вызванное нарушением обмена сахаров, жиров и белков, главным образом, в связи с недостаточной секрецией или дисфункцией инсулина (резистентность к инсулину) в организме. Осложнения диабета можно разделить на острые осложнения и хронические осложнения, при которых острые осложнения включают в себя диабетический кетоацидоз, диабетическую гиперосмолярную кому, различные острые инфекции и лактоацидоз и др. (гипогликемия, возникающая во время лечения диабета, также является одним из наиболее распространенных острых осложнений); и хронические осложнения включают в себя диабетическое заболевание глаз, диабетическую нефропатию, диабетическую нейропатию, диабетическое сердечно-сосудистое заболевание конечностей, диабетические заболевания кожи стоп и тому подобные. Основными клиническими проявлениями диабета являются полидипсия, полиурия, полифагия и потеря веса и тому подобное.
Диабет подразделяется на инсулинозависимый сахарный диабет (СД-1, т.е. диабет I типа) и инсулиннезависимый сахарный диабет (СД-2, т.е. диабет II типа), причем диабет II типа является наиболее распространенным, на его долю приходится более 90% пациентов с диабетом. Точная этиология и патогенез диабета типа I до сих пор недостаточно выяснены, причем заболевание вызывается сочетанием генетических факторов и факторов окружающей среды, главным образом, из-за разрушения островковых β-клеток в организме, что приводит к неспособности вырабатывать инсулин в организме. Пациентам необходимо ежедневно вводить инсулин для контроля уровня инсулина в крови. Диабет II типа - это разновидность метаболического синдрома, который не может контролировать уровень глюкозы в крови человека. Он в основном характеризуется гипергликемией, инсулинорезистентностью и недостаточной секрецией инсулина. Причиной диабета типа II является, главным образом, резистентность к инсулину, которая делает организм неспособным эффективно использовать инсулин, или вызывает снижение секреции инсулина, так что потребности организма не могут быть удовлетворены. Поскольку у пациентов с диабетом инсулин может секретироваться, инсулиновая терапия, как правило, не требуется, и уровень глюкозы в крови может контролироваться только с помощью диеты или пероральных гипогликемических агентов.
Лекарственные средства, доступные в настоящее время для лечения диабета типа II, в основном включают в себя инсулин и его аналоги, сульфонилмочевины, бигуаниды, ингибиторы α-глюкозидазы, тиазолидиндионы, аналоги глюкагоноподобного пептида-1 (GLP-1), ингибиторы дипептидилпептидазы IV (DPP IV), и подобные. Хотя существующие лекарства могут контролировать уровень сахара в крови и снижать частоту осложнений, большинство из них имеют серьезные побочные эффекты, такие как желудочно-кишечная токсичность, увеличение веса, отеки, гипогликемия и т.д. Поэтому лечение сахарного диабета II типа все еще остается сложной задачей. Поиск и разработка терапевтических лекарств с новым механизмом действия и меньшими побочными эффектами не теряет своей актуальности, привлекая внимание специалистов как из академических, так и из промышленных кругов.
Ингибиторы DPP IV могут в значительной степени снижать уровень глюкозы в крови в организме, повышать переносимость глюкозы, стимулировать секрецию инсулина, снижать уровни глюкагона, задерживать резистентность к инсулину и повышать уровень ответа на инсулин у пациентов с диабетом II типа при повышении уровня глюкозы в крови. По сравнению с существующими пероральными лекарствами от диабета, ингибиторы DPP IV имеют следующие характеристики: (1) Ингибиторы DPP IV не обязательно вводить инъекционным путем и они могут непрерывно снижать уровни гликозилированного гемоглобина при пероральном введении; (2) длительное применение ингибиторов DPP IV имеет хорошую переносимость; (3) улучшается секреция инсулина и высвобождение глюкагона; (4) улучшается чувствительность к инсулину при одновременном повышении функции β-клеток поджелудочной железы; (5) уменьшается частота гипогликемии, при этом не наблюдается увеличения веса, тошноты, рвоты и желудочно-кишечной дисфункции; (6) ингибиторы DPP IV оказывают синергетический эффект при использовании в комбинации с другими лекарственными средствами для лечения диабета II типа.
(R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновая кислота (соединение формулы I) является новым ингибитором DPP IV с сильной гипогликемической активностью in vivo. Однако общие характеристики (R)-метил-2-3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофена[3,2-d]пиримидин-6-карбоновой кислоты в существующей форме свободного основания не являются удовлетворительными.
Следовательно, на известном уровне техники существует острая потребность в разработке полиморфа малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты (соединение формулы I), который является высокоэффективным соединением, обладает низкой токсичностью и длительным действием, способ его получения являлся бы простым, термостабильность высокой, гигроскопичность низкой, и при этом была бы возможность получения такого полиморфа в крупных масштабах для производства фармацевтически активных ингредиентов с лучшими характеристиками.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Задачей настоящего изобретения является получение полиморфа малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофена[3,2-d]пиримидин-6-карбоновой кислоты (соединение формулы I), который обладает высокой эффективностью, низкой токсичностью и длительным действием.
В первом аспекте изобретения предложена кристаллическая форма соединения формулы I, причем кристаллическая форма представляет собой кристалл высокой стабильности и низкой гигроскопичности, и такая кристаллическая форма выбрана из группы, состоящей из кристаллической формы А, кристаллической формы В и кристаллической формы С.
В другом предпочтительном варианте осуществления изобретения «низкая гигроскопичность» означает, что увеличение веса кристаллической формы А, кристаллической формы В или кристаллической формы С составит не более ≤1% после помещения в эксикатор на 24 часа при температуре 80°С и влажности 80% и дальнейшего извлечения.
В другом предпочтительном варианте осуществления изобретения увеличение веса составляет ≤0,5%, более предпочтительно - ≤0,3%.
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы А содержит 3 или более 2θ значений, выбранных из группы, состоящей из: 3,72±0,2°, 7,47±0,2°, 11,44±0,2°, 12,28±0,2°, 21,59±0,2°.
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы А включает в себя 3 или более 2θ значений, выбранных из группы, состоящей из: 3,72±0,2°, 7,47±0,2°, 10,74±0,2°, 11,44±0,2°, 12,28±0,2°, 14,30±0,2°, 15,20±0,2°, 17,11±0,2°, 17,32±0,2°, 18,16±0,2°, 19,22±0,2°, 21,59±0,2°, 23,15±0,2°, 25,76±0,2°, 28,02±0,2°, 32,82±0,2°.
В другом предпочтительном варианте осуществления изобретения характерный пик поглощения, представленный значением 2θ на порошковой рентгеновской дифрактограмме кристаллической формы А, имеет отклонение ± 0,5, предпочтительно - отклонение ± 0,3, более предпочтительно - отклонение ± 0,1.
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы А по существу показана на фиг. 1.
В другом предпочтительном варианте осуществления изобретения спектр дифференциальной сканирующей калориметрии (спектр ДСК) кристаллической формы А имеет характерный пик в диапазоне 118±5°С.
В другом предпочтительном варианте осуществления изобретения спектр дифференциальной сканирующей калориметрии кристаллической формы А имеет характерный пик поглощения при 118,37°С.
В другом предпочтительном варианте осуществления изобретения спектр дифференциальной сканирующей калориметрии кристаллической формы А по существу характеризуется, как на фиг. 3.
В другом предпочтительном варианте осуществления изобретения спектр термогравиметрического анализа кристаллической формы А содержит 3 или более характерных пика поглощения, выбранных из группы, состоящей из: 63±5°С, 184±5°С, 215±5°С, 287±5°С, 325±5°C.
В другом предпочтительном варианте осуществления изобретения спектр термогравиметрического анализа кристаллической формы А содержит 3 или более характерных пика поглощения, выбранных из группы, состоящей из: 62,6°С, 184,3°С, 214,9°С, 286,6°С, 324,6°С.
В другом предпочтительном варианте осуществления изобретения спектр термогравиметрического анализа (спектр ТГА) кристаллической формы А по существу показан на фиг. 2.
В другом предпочтительном варианте осуществления изобретения кристаллическая форма А имеет потерю теплового веса от 47 до 48 мас.% при 400°С.
В другом предпочтительном варианте осуществления изобретения кристаллическая форма А имеет потерю теплового веса 47,97 мас.% при 400°С.
В другом предпочтительном варианте осуществления изобретения начальное значение температуры эндотермического перехода кристаллической формы А составляет 115±2°С, предпочтительно - 115,04°С.
В другом предпочтительном варианте осуществления изобретения гигроскопичность кристаллической формы А составляет ≤3%, предпочтительно - ≤1%, более предпочтительно - ≤0,5%, наиболее предпочтительно - ≤0,3%.
В другом предпочтительном варианте осуществления изобретения профиль динамической сорбции паров кристаллической формы А по существу показан на фиг. 4.
В другом предпочтительном варианте осуществления изобретения спектр комбинационного рассеяния кристаллической формы А включает в себя следующие характерные пики поглощения, выраженные длиной волны λ: 2226±2 см-1, 1716±2 см-1, 1689±2 см-1, 1604±2 см-1, 1566±2 см-1, 1536±2 см-1, 1486±2 см-1, 1393±2 см-1.
В другом предпочтительном варианте осуществления изобретения спектр комбинационного рассеяния кристаллической формы А включает в себя следующие характерные пики поглощения, выраженные длиной волны λ: 2226 см-1, 1716 см-1, 1689 см-1, 1604 см-1, 1566 см-1, 1536 см-1, 1486 см-1, 1393 см-1.
В другом предпочтительном варианте осуществления изобретения спектр комбинационного рассеяния кристаллической формы А по существу показан на фиг. 5.
В другом предпочтительном варианте осуществления изобретения профиль ИК-излучения кристаллической формы А включает в себя следующие характерные пики поглощения, выраженные длиной волны λ: 3435±2 см-1, 2952±2 см-1, 2225±2 см-1, 1718±2 см-1, 1685±2 см-1, 1558±2 см-1, 1531±2 см-1, 1452±2 см-1, 1234±2 см-1, 1064±2 см-1, 862±2 см-1, 756±2 см-1.
В другом предпочтительном варианте осуществления изобретения профиль ИК-излучения кристаллической формы А включает в себя следующие характерные пики поглощения, выраженные длиной волны λ: 3435 см-1, 2952 см-1, 2225 см-1, 1718 см-1, 1685 см-1, 1558 см-1, 1531 см-1, 1452 см-1, 1234 см-1, 1064 см-1,862 см-1, 756 см-1.
В другом предпочтительном варианте осуществления изобретения профиль ИК-излучения кристаллической формы А по существу показан на фиг. 6.
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы В включает 3 или более 2θ значений, выбранных из группы, состоящей из: 5,37±0,2°, 12,01±0,2°, 14,93±0,2°, 16,04±0,2°, 20,09±0,2°.
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы В включает 3 или более 2θ значений, выбранных из группы, состоящей из: 5,37±0,2°, 7,85±0,2°, 11,20±0,2°, 12,01±0,2°, 14,93±0,2°, 16,04±0,2°, 20,09±0,2°, 22,10±0,2°, 22,61±0,2°, 24,19±0,2°, 30,16±0,2°, 32,12±0,2°, 32,39±0,2°.
В другом предпочтительном варианте осуществления изобретения характерный пик поглощения, представленный значением 2θ на порошковой рентгеновской дифрактограмме кристаллической формы В, имеет отклонение ± 0,5, предпочтительно - отклонение ± 0,3, более предпочтительно - отклонение ± 0,1.
В предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы В по существу показана на фиг. 7.
В другом предпочтительном варианте осуществления изобретения спектр дифференциальной сканирующей калориметрии (спектр ДСК) кристаллической формы В имеет характерный пик в диапазоне 137±5°С.
В другом предпочтительном варианте осуществления изобретения спектр дифференциальной сканирующей калориметрии (спектр ДСК) кристаллической формы В имеет характерный пик поглощения при 136,77°С.
В другом предпочтительном варианте осуществления изобретения спектр дифференциальной сканирующей калориметрии кристаллической формы В по существу показан на фиг. 9.
В другом предпочтительном варианте осуществления изобретения начальное значение температуры эндотермического перехода кристаллической формы В составляет 127±2°С.
В другом предпочтительном варианте осуществления изобретения начальное значение температуры эндотермического перехода кристаллической формы В составляет 127,14°С.
В другом предпочтительном варианте осуществления изобретения профиль термогравиметрического анализа кристаллической формы В содержит 3 или более характерных пика поглощения, выбранных из группы, состоящей из: 119±5°С, 202±5°С, 284±5°С, 318±5°С.
В другом предпочтительном варианте осуществления изобретения профиль термогравиметрического анализа кристаллической формы В содержит 3 или более характерных пика поглощения, выбранных из группы, состоящей из: 118,7°С, 202,5°С, 283,7°C; 317,9°С.
В другом предпочтительном варианте осуществления изобретения профиль термогравиметрического анализа кристаллической формы В по существу показан на фиг. 8.
В другом предпочтительном варианте осуществления изобретения кристаллическая форма В имеет потерю теплового веса от 51 до 52 мас.% при 400°С.
В другом предпочтительном варианте осуществления изобретения кристаллическая форма В имеет потерю теплового веса 51,38 мас.% при 400°С.
В другом предпочтительном варианте осуществления изобретения гигроскопичность кристаллической формы В составляет ≤3%, предпочтительно - ≤1%, более предпочтительно - ≤0,5%, наиболее предпочтительно - ≤0,3%.
В другом предпочтительном варианте осуществления изобретения профиль динамической сорбции паров кристаллической формы В по существу показан на фиг. 10.
В другом предпочтительном варианте осуществления изобретения спектр комбинационного рассеяния кристаллической формы В включает в себя следующие характерные пики поглощения, выраженные длиной волны λ: 2234±2 см-1, 1718±2 см-1, 1693±2 см-1, 1607±2 см-1, 1565±2 см-1, 1536±2 см-1, 1476±2 см-1, 1386±2 см-1, 1349±2 см-1, 1216±2 см-1, 1174±2 см-1, 1047±2 см-1, 813±2 см-1, 730±2 см-1, 690±2 см-1, 641±2 см-1.
В другом предпочтительном варианте осуществления изобретения спектр комбинационного рассеяния кристаллической формы В включает в себя следующие характерные пики поглощения, выраженные длиной волны λ: 2234 см-1, 1718 см-1, 1693 см-1, 1607 см-1, 1565 см-1, 1536 см-1, 1476 см-1, 1386, 1349 см-1, 1216 см-1, 1174 см-1, 1047 см-1, 813 см-1, 730 см-1, 690 см-1, 641 см-1.
В другом предпочтительном варианте осуществления изобретения спектр комбинационного рассеяния кристаллической формы В по существу показан на фиг. 11.
В другом предпочтительном варианте осуществления изобретения профиль ИК-излучения кристаллической формы В включает в себя следующие характерные пики поглощения, выраженные длиной волны λ: 3404±2 см-1, 2954±2 см-1, 2222±2 см-1, 1718±2 см-1, 1678±2 см-1, 1558±2 см-1, 1531±2 см-1, 1369±2 см-1, 1290±2 см-1, 1219±2 см-1, 1063±2 см-1, 862±2 см-1, 775±2 см-1.
В другом предпочтительном варианте осуществления изобретения профиль ИК-излучения кристаллической формы В включает в себя следующие характерные пики поглощения, выраженные длиной волны λ: 3404 см-1, 2954 см-1, 2222 см-1, 1718 см-1, 1678 см-1, 1558 см-1, 1531 см-1, 1369 см-1, 1290 см-1, 1219 см-1, 1063 см-1, 862 см-1, 775 см-1.
В другом предпочтительном варианте осуществления изобретения профиль ИК-излучения кристаллической формы В по существу показан на фиг. 12.
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы С включает 3 или более 2θ значений, выбранных из группы, состоящей из: 6,63±0,2°, 11,16±0,2°, 17,06±0,2°, 19,46±0,2°, 20,84±0,2°, 25,74±0,2°.
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы С включает 3 или более 2θ значений, выбранных из группы, состоящей из: 6,63±0,2°, 8,67±0,2°, 11,16±0,2°, 11,64±0,2°, 15,24±0,2°, 16,43±0,2°, 17,06±0,2°, 17,41±0,2°, 18,00±0,2°, 18,61±0,2°, 18,90±0,2°, 19,46±0,2°, 19,96±0,2°, 20,84±0,2°, 21,35±0,2°, 22,81±0,2°, 23,11±0,2°, 23,59±0,2°, 24,7±0,2°, 25,18±0,2°, 25,74±0,2°, 27,62±0,2°, 28,32±0,2°, 31,17±0,2°.
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы С по существу показана на фиг. 13.
В другом предпочтительном варианте осуществления изобретения характерный пик поглощения, представленный значением 2θ на порошковой рентгеновской дифрактограмме кристаллической формы С, имеет отклонение ± 0,5, предпочтительно - отклонение ± 0,3, более предпочтительно - отклонение ± 0,1.
В другом предпочтительном варианте осуществления изобретения спектр дифференциальной сканирующей калориметрии (спектр ДСК) кристаллической формы С имеет характерный пик в диапазоне 161±5°С.
В другом предпочтительном варианте осуществления изобретения профиль дифференциальной сканирующей калориметрии кристаллической формы С имеет характерный пик поглощения при 160,90°С.
В другом предпочтительном варианте осуществления изобретения начальное значение температуры эндотермического перехода кристаллической формы С составляет 147±2°С.
В другом предпочтительном варианте осуществления изобретения начальное значение температуры эндотермического перехода кристаллической формы С составляет 147,02°С.
В другом предпочтительном варианте осуществления изобретения спектр дифференциальной сканирующей калориметрии кристаллической формы С по существу характеризуется, как на фиг. 15.
В другом предпочтительном варианте осуществления изобретения профиль термогравиметрического анализа кристаллической формы С содержит характерный пик поглощения, выбранный из группы, состоящей из: 183±5°С, 212±5°С, 289±5°С.
В другом предпочтительном варианте осуществления изобретения профиль термогравиметрического анализа кристаллической формы С содержит характерный пик поглощения, выбранный из группы, состоящей из: 183,3°С, 212,4°С, 288,8°С.
В другом предпочтительном варианте осуществления изобретения спектр термогравиметрического анализа кристаллической формы С по существу показан на фиг. 14.
В другом предпочтительном варианте осуществления изобретения кристаллическая форма С имеет потерю теплового веса от 46 до 47 мас.% при 400°С.
В другом предпочтительном варианте осуществления изобретения кристаллическая форма С имеет потерю теплового веса 46,30 мас.% при 400°С.
В другом предпочтительном варианте осуществления изобретения гигроскопичность кристаллической формы С составляет ≤3%, предпочтительно - ≤1%, более предпочтительно - ≤0,5%, наиболее предпочтительно - ≤0,3%.
В другом предпочтительном варианте осуществления изобретения профиль динамической сорбции паров кристаллической формы С по существу показан на фиг. 16.
В другом предпочтительном варианте осуществления изобретения спектр комбинационного рассеяния кристаллической формы С включает в себя следующие характерные пики поглощения, выраженные длиной волны λ: 3383±2 см-1, 3378±2 см-1, 3370±2 см-1, 3288±2 см-1, 3163±2 см-1, 2963±2 см-1, 2230±2 см-1, 1719±2 см-1, 1558±2 см-1, 1478±2 см-1, 662±2 см-1.
В другом предпочтительном варианте осуществления изобретения спектр комбинационного рассеяния кристаллической формы С включает в себя следующие характерные пики поглощения, выраженные длиной волны λ: 3383 см-1, 3378 см-1, 3370 см-1, 3288 см-1, 3163 см-1, 2963 см-1, 2230 см-1, 1719 см-1, 1558 см-1, 1478 см-1, 662 см-1.
В другом предпочтительном варианте осуществления изобретения спектр комбинационного рассеяния кристаллической формы С по существу показан на фиг. 17.
В другом предпочтительном варианте осуществления изобретения профиль ИК-излучения кристаллической формы С включает в себя следующие характерные пики поглощения, выраженные длиной волны λ: 3428±2 см-1, 2936±2 см-1, 1640±2 см-1, 1514±2 см-1, 1463±2 см-1, 1418±2 см-1, 1385±2 см-1, 1272±2 см-1, 1136±2 см-1, 1022±2 см-1, 874±2 см-1, 766±2 см-1, 599±2 см-1.
В другом предпочтительном варианте осуществления изобретения профиль ИК-излучения кристаллической формы С включает в себя следующие характерные пики поглощения, выраженные длиной волны λ: 3428 см-1, 2936 см-1, 1640 см-1, 1514 см-1, 1463 см-1, 1418 см-1, 1385 см-1, 1272 см-1, 1136 см-1, 1022 см-1, 874 см-1, 766 см-1, 599 см-1.
В другом предпочтительном варианте осуществления изобретения профиль ИК-излучения кристаллической формы С по существу показан на фиг. 18.
Во втором аспекте изобретения предложен способ получения кристаллической формы А с характеристиками, указанными выше, включающий этапы:
(1) получение первого раствора, содержащего первый растворитель и соединение формулы I, причем отношение массы соединения формулы I к объему первого растворителя составляет от 0,1 г/л до 100 г/л (предпочтительно - от 1 г/л до 90 г/л, более предпочтительно - от 10 до 60 г/л, наиболее предпочтительно - от 15 до 40 г/л);
(2) смешивание второго растворителя с первым раствором и кристаллизирование при 0-50°С с получением кристаллической формы А.
В другом предпочтительном варианте осуществления изобретения концентрация соединения формулы I составляет 0,1 г/л насыщенной концентрации в первом растворе.
В другом предпочтительном варианте осуществления изобретения первый растворитель выбирают из группы, состоящей из С1-С4 спиртов, С2-С8 кетонов (предпочтительно - С3-С5 кетонов), сложных эфиров С1-С10 (предпочтительно - сложных эфиров С1-С7, более предпочтительно - сложных эфиров С1-С5), галогенированного С1-С6-алкана, воды или их сочетания.
В другом предпочтительном варианте осуществления изобретения второй растворитель выбирают из группы, состоящей из простых эфиров С1-С10 (предпочтительно - простых эфиров С1-С8, более предпочтительно - простых эфиров С1-С6), С2-С15-алкана (предпочтительно - С3-С10-алкана, более предпочтительно - С4-С8-алкана), тетрагидрофурана, 1,4-диоксана или их сочетания.
В другом предпочтительном варианте осуществления изобретения С2-С8 кетоны выбирают из группы, состоящей из ацетона, метилэтилкетона, изобутанолбутанона или их сочетания.
В другом предпочтительном варианте осуществления изобретения сложные эфиры С1-С10 выбирают из группы, состоящей из метилформиата, этилацетата, изобутилформиата или их сочетания.
В другом предпочтительном варианте осуществления изобретения галогенированный С1-С6-алкан выбирают из группы, состоящей из дихлорметана, трихлорметана или их сочетания, предпочтительно - дихлорметана
В другом предпочтительном варианте осуществления изобретения сложные эфиры С1-С10 выбирают из группы, состоящей из петролейного эфира, трет-бутилметилового эфира, этилового эфира, изопропилового эфира, диэтилового эфира или их сочетания.
В другом предпочтительном варианте осуществления изобретения С2-С15-алкан выбирают из группы, состоящей из n-пентана, n-гексана, n-гептана или их сочетания.
В другом предпочтительном варианте осуществления изобретения соединение формулы I находится в аморфной форме.
В другом предпочтительном варианте осуществления изобретения после этапа (2), способ дополнительно содержит следующие этапы: (3) фильтрация и/или сушка кристаллической формы А, полученной на этапе (2).
В другом предпочтительном варианте осуществления изобретения на этапе (3) температура сушки составляет от 10 до 70°С, предпочтительно - от 20 до 80°С, более предпочтительно - от 25 до 40°С.
В другом предпочтительном варианте осуществления изобретения на этапе (3) давление сушки составляет от 0 до 20 кПа, предпочтительно - от 0 до 10 кПа, более предпочтительно - от 5 до 10 кПа.
В другом предпочтительном варианте осуществления изобретения на этапе (3) время сушки составляет от 5 до 150 часов, предпочтительно - от 30 до 100 часов, более предпочтительно - от 60 до 80 часов.
В другом предпочтительном варианте осуществления изобретения выход кристаллической формы А составляет от 50 до 99%, предпочтительно - от 75 до 99%, более предпочтительно - от 85 до 99%.
В другом предпочтительном варианте осуществления изобретения концентрация соединения формулы I составляет от 0,1 г/л до 1000 г/л в первом растворе.
В другом предпочтительном варианте осуществления изобретения концентрация соединения формулы I составляет >1000 г/л в первом растворе.
В другом предпочтительном варианте осуществления изобретения на этапе (2) кристаллизацию проводят при температуре от 0 до 50°С, предпочтительно - от 0 до 40°С, более предпочтительно - от 20 до 30°С.
В другом предпочтительном варианте осуществления изобретения на этапе (2) кристаллизацию проводят с перемешиванием.
В третьем аспекте изобретения предложен способ получения кристаллической формы В с характеристиками, указанными выше, включающий этапы:
(1) получение первого раствора, содержащего первый растворитель и соединение формулы I, причем отношение массы соединения формулы I к объему первого растворителя составляет от 0,1 г/л до 100 г/л, предпочтительно - от 1 г/л до 900 г/л, более предпочтительно - от 10 до 600 г/л, наиболее предпочтительно - от 50 до 400 г/л;
(2) смешивание второго растворителя с первым раствором и кристаллизирование при 5-80°С с получением кристаллической формы В.
В другом предпочтительном варианте осуществления изобретения первый растворитель выбирают из группы, состоящей из сложных эфиров С1-С10 (в предпочтительном варианте осуществления изобретения сложных эфиров С1-С7, более предпочтительно - сложных эфиров С1-С5), ароматического углеводорода С5-С10 (предпочтительно - ароматического углеводорода С5-С6), простых эфиров С1-С10 (предпочтительно - простого эфира С1-С8, более предпочтительно - простого эфира С1-С6), галогенированного С1-С6-алкана или их сочетания.
В другом предпочтительном варианте осуществления изобретения сложные эфиры С1-С10 выбирают из группы, состоящей из метилформиата, этилацетата, изобутилформиата или их сочетания.
В другом предпочтительном варианте осуществления изобретения ароматический углеводород С5-С10 выбирают из группы, состоящей из бензола, толуола, фурана, тиофена, нафталина или их сочетания.
В другом предпочтительном варианте осуществления изобретения простые эфиры С1-С10 выбирают из группы, состоящей из петролейного эфира, трет-бутилметилового эфира, этилового эфира, изопропилового эфира, диэтилового эфира или их сочетания.
В другом предпочтительном варианте осуществления изобретения галогенированный С1-С6-алкан представляет собой дихлорметан, трихлорметан или их сочетание, предпочтительно - дихлорметан.
В другом предпочтительном варианте осуществления изобретения соединение формулы I находится в аморфной форме.
В другом предпочтительном варианте осуществления изобретения второй растворитель выбирают из группы, состоящей из С2-С15-алкана (предпочтительно - С3-С10-алкана, более предпочтительно-С4-С8-алкана), тетрагидрофурана, 1,4-диоксана или их сочетания.
В другом предпочтительном варианте осуществления изобретения С2-С15-алкан выбирают из группы, состоящей из n-пентана, n-гексана, n-гептана или их сочетания.
В другом предпочтительном варианте осуществления изобретения после этапа (2), следующий этап дополнительно содержит стадию:
(3) фильтрация и/или сушка кристаллической формы В, полученной на этапе (2).
В другом предпочтительном варианте осуществления изобретения температура сушки составляет от 10 до 70°С, предпочтительно - от 20 до 80°С, более предпочтительно - от 25 до 40°С.
В другом предпочтительном варианте осуществления изобретения давление для сушки составляет от 0 до 20 кПа, предпочтительно - от 0 до 10 кПа, более предпочтительно - от 5 до 10 кПа.
В другом предпочтительном варианте осуществления изобретения время для сушки составляет от 5 до 150 часов, предпочтительно - от 30 до 100 часов, в более предпочтительном варианты осуществления изобретения - от 60 до 80 часов.
В другом предпочтительном варианте осуществления изобретения выход кристаллической формы В составляет от 50 до 99%, предпочтительно - от 75 до 99%, более предпочтительно - от 85 до 99%.
В другом предпочтительном варианте осуществления изобретения концентрация соединения формулы I составляет от 0,1 г/л до 1000 г/л в первом растворе.
В другом предпочтительном варианте осуществления изобретения концентрация соединения формулы I составляет >1000 г/л в первом растворе.
В другом предпочтительном варианте осуществления изобретения кристаллизация проходит при 0-50°С.
В другом предпочтительном варианте осуществления изобретения кристаллизацию проводят при температуре 0-40°С, предпочтительно - 20-30°С, предпочтительно - при комнатной температуре.
В другом предпочтительном варианте осуществления изобретения кристаллизация проходит с перемешиванием.
В четвертом аспекте изобретения предложен способ получения кристаллической формы С с характеристиками, указанными выше, включающий этапы:
(1) получение первого раствора, содержащего первый растворитель и соединение формулы I, причем отношение массы соединения формулы I к объему первого растворителя составляет от 0,1 г/л до 100 г/л, предпочтительно - от 1 г/л до 900 г/л, более предпочтительно - от 10 до 600 г/л, наиболее предпочтительно - от 50 до 400 г/л;
(2) смешивание второго растворителя с первым раствором и кристаллизирование при 5-80°С с получением кристаллической формы С.
В другом предпочтительном варианте осуществления изобретения первый растворитель выбирают из группы, состоящей из нитрилов С1-С10 (предпочтительно - нитрилов С1-С8, более предпочтительно - нитрилов С1-С5), С2-С8 кетонов (предпочтительно - С3-С5 кетонов), сложных эфиров С1-С10 (предпочтительно - сложных эфиров С1-С7, более предпочтительно - сложных эфиров С1-С5), галогенированного С1-С6-алкана или их сочетания.
В другом предпочтительном варианте осуществления изобретения второй растворитель выбирают из группы, состоящей из С2-С15-алканов (предпочтительно - С3-С10-алканов, более предпочтительно - С4-С8-алканов), простых эфиров С1-С10 (предпочтительно - простых эфиров С1-С8, более предпочтительно - простых эфиров С1-С6), тетрагидрофурана, 1,4-диоксана или их сочетания.
В другом предпочтительном варианте осуществления изобретения С2-С15-алкан выбирают из группы, состоящей из n-пентана, n-гексана, n-гептана или их сочетания.
В другом предпочтительном варианте осуществления изобретения нитрилы С1-С10 выбирают из группы, состоящей из формионитрила, ацетонитрила, n-пропионитрила, изопропионитрила или их сочетания.
В другом предпочтительном варианте осуществления изобретения С2-С8 кетоны выбирают из группы, состоящей из ацетона, метилэтилкетона, изобутанолбутанона или их сочетания.
В другом предпочтительном варианте осуществления изобретения сложные эфиры С1-С10 выбирают из группы, состоящей из метилформиата, этилацетата, изобутилформиата или их сочетания.
В другом предпочтительном варианте осуществления изобретения простые эфиры С1-С10 выбирают из группы, состоящей из петролейного эфира, трет-бутилметилового эфира, этилового эфира, изопропилового эфира, диэтилового эфира или их сочетания.
В другом предпочтительном варианте осуществления изобретения галогенированный С1-С6-алкан представляет собой дихлорметан, трихлорметан или их сочетание, предпочтительно - дихлорметан.
В другом предпочтительном варианте осуществления изобретения соединение формулы I находится в аморфной форме.
В другом предпочтительном варианте осуществления изобретения после этапа (2), следующий этап дополнительно содержит:
(3) фильтрация и/или сушка кристаллической формы С, полученной на этапе (2).
В другом предпочтительном варианте осуществления изобретения температура сушки составляет от 10 до 70°С, предпочтительно - от 20 до 80°С, более предпочтительно - от 25 до 40°С.
В другом предпочтительном варианте осуществления изобретения давление для сушки составляет от 0 до 20 кПа, предпочтительно - от 0 до 10 кПа, более предпочтительно - от 5 до 10 кПа.
В другом предпочтительном варианте осуществления изобретения время для сушки составляет от 5 до 150 часов, предпочтительно - от 30 до 100 часов, в более предпочтительном варианты осуществления изобретения - от 60 до 80 часов.
В другом предпочтительном варианте осуществления изобретения выход кристаллической формы С составляет от 50 до 99%, предпочтительно - от 75 до 99%, более предпочтительно - от 85 до 99%.
В другом предпочтительном варианте осуществления изобретения концентрация соединения формулы I составляет от 0,1 г/л до 1000 г/л в первом растворе.
В другом предпочтительном варианте осуществления изобретения концентрация соединения формулы I составляет >1000 г/л в первом растворе.
В другом предпочтительном варианте осуществления изобретения кристаллизация проходит при 0-50°С.
В другом предпочтительном варианте осуществления изобретения кристаллизацию проводят при температуре 0-40°С, предпочтительно - 20-30°С, более предпочтительно - при комнатной температуре.
В другом предпочтительном варианте осуществления изобретения кристаллизация проходит с перемешиванием.
В пятом аспекте настоящего изобретения представлена фармацевтическая композиция, содержащая:
(a) кристаллическую форму А, В или С соединения формулы I с характеристиками, указанными выше, и
(b) фармацевтически приемлемый носитель.
В другом предпочтительном варианте осуществления изобретения фармацевтически приемлемый носитель выбирают из группы, состоящей из наполнителя, разрыхлителя, связующего вещества, смазывающего вещества (лубриканта) или их сочетания.
В другом предпочтительном варианте осуществления изобретения наполнитель выбирают из группы, состоящей из крахмала, лактозы, микрокристаллической целлюлозы, декстрина, маннитола, оксида магния, сульфата кальция или их сочетания.
В другом предпочтительном варианте осуществления изобретения разрыхлитель выбирают из группы, состоящей из карбоксиметилцеллюлозы и ее соли, перекрестно сшитой карбоксиметилцеллюлозы и ее соли, перекрестно сшитого повидона, натрий-карбоксиметилкрахмала, низкозамещенной гидроксипропилцеллюлозы или их сочетания.
В другом предпочтительном варианте осуществления изобретения связующее вещество выбирают из группы, состоящей из повидона, гидроксипропилметилцеллюлозы, крахмальной пульпы или их сочетания.
В другом предпочтительном варианте осуществления изобретения смазывающее вещество выбирают из группы, состоящей из магния стеарата, кальция стеарата или их сочетания.
В шестом аспекте изобретения предлагают применение кристаллической формы в соответствии с первым аспектом изобретения или фармацевтической композиции в соответствии с пятым аспектом изобретения для получения лекарственного средства для профилактики и/или лечения диабета типа II и/или осложнений диабета типа II.
В другом предпочтительном варианте осуществления изобретения осложнения сахарного диабета II типа выбирают из группы, состоящей из заболевания коронарных артерий, инсульта, гипертонии, нефропатии, заболевания периферических сосудов, неврологического заболевания и ретинопатии.
В седьмом аспекте изобретения предлагают способ лечения или профилактики диабета типа II и/или осложнений диабета типа II, содержащий введение пациенту терапевтически эффективного количества кристаллической формы согласно первому аспекту изобретения или фармацевтической композиции согласно пятому аспекту изобретения.
Следует понимать, что в настоящем изобретении любой из технических признаков, изложенных выше и ниже (например, в Примерах), могут быть объединены друг с другом, тем самым образуя новые или предпочтительные технические решения, которые не требуют повторного описания каждого из них в настоящем документе.
ОПИСАНИЕ ФИГУР
Фиг. 1 представляет собой рентгеновскую дифрактограмму кристаллической формы А кристалла из Примера 1 настоящего изобретения.
Фиг. 2 представляет собой профиль термогравиметрического (ТГ) анализа кристаллической формы А кристалла из Примера 1 настоящего изобретения.
Фиг. 3 представляет собой спектр дифференциальной сканирующей калориметрии (ДСК) кристаллической формы А кристалла из Примера 1 настоящего изобретения.
Фиг. 4 представляет собой спектр анализа гигроскопичности (ДСП) кристаллической формы А кристалла из Примера 1 настоящего изобретения.
Фиг. 5 представляет собой диаграмму спектра комбинационного рассеяния кристаллической формы А кристалла из Примера 1 настоящего изобретения.
Фиг. 6 представляет собой профиль инфракрасного спектра (ИК) кристаллической формы А кристалла из Примера 1 настоящего изобретения.
Фиг. 7 представляет собой рентгеновскую дифрактограмму кристаллической формы В кристалла из Примера 4 настоящего изобретения.
Фиг. 8 представляет собой профиль ТГ анализа кристаллической формы В кристалла из Примера 4 настоящего изобретения.
Фиг. 9 представляет собой спектр дифференциальной сканирующей калориметрии (ДСК) кристаллической формы В кристалла из Примера 4 настоящего изобретения.
Фиг. 10 представляет собой спектр анализа гигроскопичности (ДСП) кристаллической формы В кристалла из Примера 4 настоящего изобретения.
Фиг. 11 представляет собой диаграмму спектра комбинационного рассеяния кристаллической формы В кристалла из Примера 4 настоящего изобретения.
Фиг. 12 представляет собой профиль инфракрасного спектра (ИК) кристаллической формы В кристалла из Примера 4 настоящего изобретения.
Фиг. 13 представляет собой рентгеновскую дифрактограмму кристаллической формы С кристалла из Примера 7 настоящего изобретения.
Фиг. 14 представляет собой профиль ТГ анализа кристаллической формы С кристалла из Примера 7 настоящего изобретения.
Фиг. 15 представляет собой спектр дифференциальной сканирующей калориметрии (ДСК) кристаллической формы С кристалла из Примера 7 настоящего изобретения.
Фиг. 16 представляет собой спектр анализа гигроскопичности (ДСП) кристаллической формы С кристалла из Примера 7 настоящего изобретения.
Фиг. 17 представляет собой диаграмму спектра комбинационного рассеяния кристаллической формы С кристалла из Примера 7 настоящего изобретения.
Фиг. 18 представляет собой профиль инфракрасного спектра (ИК) кристаллической формы С кристалла из Примера 7 настоящего изобретения.
ОСУЩЕСТВЛЕНИЕ ИЗОБРЕТЕНИЯ
В результате обширных и интенсивных исследований авторы изобретения неожиданно обнаружили полиморф кристалла малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофена[3,2-d]пиримидин-6-карбоновой кислоты с лучшими фармацевтическими свойствами, т.е. кристаллическими формами А, В и С. Все они имеют хорошую термостойкость и негигроскопичность, а процесс получения прост и эффективен. Была продемонстрирована хорошая воспроизводимость, и может быть реализовано крупное промышленное производство. Исходя из вышеизложенного, было сформулировано настоящее изобретение.
ТЕРМИНЫ
Если не указано иное, все технические и научные термины, используемые в данном документе, имеют те же значения, которые обычно используют специалисты на данном уровне техники, к которому относится данное изобретение.
При использовании в настоящем документе в отношении определенного значения термин «около» означает, что это значение может варьироваться не более чем на 1% от указанного значения. Например, как используется в настоящем документе, выражение «около 100» включает в себя все значения от 99 до 101 (например, 99,1, 99,2, 99,3, 99,4 и т.д.).
Используемые здесь термины «содержит» или «включает (содержит)» могут быть открытыми, полузакрытыми и закрытыми. То есть эти термины также включают в себя «состоящий по существу из» или «состоящий из».
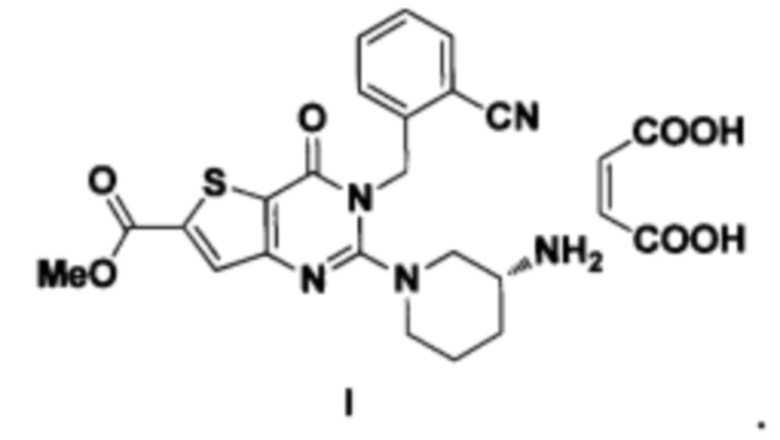
Соединение формулы I
Соединение формулы I согласно изобретению представляет собой (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновая кислота
(R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоноюй кислоты (Формула I) является новым селективным, обратимым конкурентным ингибитором DPP IV с сильной гипогликемической активностью in vivo. Его ингибирующая активность достигает наномолярного уровня, а его ингибирующая активность DPP IV и селективность in vitro превосходят имеющиеся на рынке препараты ситаглиптин и вильдаглиптин.
У животных соединение формулы I эффективно ингибирует активность DPP IV в плазме здоровой мыши и крысы, и его ингибирующая активность DPP IV превосходит продаваемый лекарственный препарат алоглиптин. Соединение формулы I может увеличить толерантность к глюкозе при пероральном применении у здоровых ICR мышей в зависимости от дозы, и его минимальная эффективная доза составляет всего 0,1 мг/кг, что превосходит действие алоглиптина. При постоянном введении соединения мышам линии ob/ob оно может эффективно снижать уровень глюкозы в крови натощак у мышей линии ob/ob, что превосходит лекарственное средство с положительным контролем алоглиптином. При постоянном введении соединения мышам линии db/db с дефицитом генов оно может снизить уровень глюкозы в крови натощак, что сравнимо с положительным контрольным препаратом алоглиптином.
Исследования фармакокинетики и безопасности показали, что соединение формулы I обладает хорошими фармакокинетическими свойствами и безопасностью у крыс и собак, а его период полувыведения и AUC0-t превосходят таковые у имеющегося в продаже алоглиптина у крыс и собак. Соединение является безопасным, и испытание на острую токсичность у мышей ICR показывает, что при введении 300 мг/кг ни одно животное в группе не умирает. Испытание на острую токсичность у собак породы бигль показал, что в группе, получавшей 1 г/кг, гибели животных не было. Испытание подострой токсичности на крысах показывает, что в группе, получавшей перорально 150 мг/кг, явной токсичности не обнаружено.
Обобщая результаты исследований фармакодинамической оценки in vitro, фармакологической оценки in vivo, фармакокинетических исследований и оценки безопасности и т.д., гипогликемический эффект соединения in vivo лучше, чем у клинически используемых ингибиторов DPPIV.
Полиморф
Полиморфизм лекарственного средства относится к двум или более различным кристаллическим состояниям лекарственного средства. Для твердых химических лекарств, из-за их различного молекулярного расположения и законов симметрии, одно лекарство может образовывать множество различных кристаллических состояний твердого вещества.
Аморфная форма (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты была приготовлена способом получения, описанным в CN 201210262331.3. 1Н ЯМР(CDCl3): δ 7.76 (s, 1Н), 7.610 (d, 1Н), 7.493 (t, 1Н), 7.320 (t, 1Н), 7.180 (d, 1Н), 5.500 (quartet, 2Н), 3.895 (s, 3Н), 3.680 (d, 2Н), 3.355 (m, 1Н), 3.010 (m, 2Н), 2.150 (m, 1Н), 1.894 (m, 2Н), 1.644 (m, 1Н); LC-MS m/z 424.1 [M+H]+.
В настоящем документе термины «кристаллическая форма А», «кристаллическая форма А малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофена[3,2-d]пиримидин-6-карбоновой кислоты», «кристаллическая форма А соединения формулы I» являются взаимозаменяемыми. «Кристаллическая форма В», «кристаллическая форма В малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофена[3,2-d]пиримидин-6-карбоновой кислоты», «кристаллическая форма В соединения формулы I» являются взаимозаменяемыми. «Кристаллическая форма С», «кристаллическая форма С малеата (R)-метил-2-(3-аминопипфидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофена[3,2-d]пиримидин-6-карбоновой кислоты», «кристаллическая форма С соединения формулы I» являются взаимозаменяемыми.
Кристаллическая форма А
Порошковая рентгеновская дифрактограмма кристаллической формы А включает 3 или более 2θ значений, выбранных из группы, состоящей из: 3,72±0,2°, 7,47±0,2°, 11,44±0,2°, 12,28±0,2°, 21,59±0,2°.
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы А включает в себя 3 или более 2θ значений, выбранных из группы, состоящей из: 3,72±0,2°, 7,47±0,2°, 10,74±0,2°, 11,44±0,2°, 12,28±0,2°, 14,30±0,2°, 15,20±0,2°, 17,11±0,2°, 17,32±0,2°, 18,16±0,2°, 19,22±0,2°, 21,59±0,2°, 23,15±0,2°, 25,76±0,2°, 28,02±0,2°, 32,82±0,2°.
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы А по существу показана на фиг. 1.
Кристаллическая форма В
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы В включает 3 или более 2θ значений, выбранных из группы, состоящей из: 5,37±0,2°, 12,01±0,2°, 14,93±0,2°, 16,04±0,2°, 20,09±0,2°.
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы В включает 3 или более 2θ значений, выбранных из группы, состоящей из: 5,37±0,2°, 7,85±0,2°, 11,20±0,2°, 12,01±0,2°, 14,93±0,2°, 16,04±0,2°, 20,09±0,2°, 22,10±0,2°, 22,61±0,2°, 24,19±0,2°, 30,16±0,2°, 32,12±0,2°, 32,39±0,2°.
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы В по существу показана на фиг. 7.
Кристаллическая форма С
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы С включает 3 или более 2θ значений, выбранных из группы, состоящей из: 6,63±0,2°, 11,16±0,2°, 17,06±0,2°, 19,46±0,2°, 20,84±0,2°, 25,74±0,2°.
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы С включает 3 или более 2θ значений, выбранных из группы, состоящей из: 6,63±0,2°, 8,67±0,2°, 11,16±0,2°, 11,64±0,2°, 15,24±0,2°, 16,43±0,2°, 17,06±0,2°, 17,41±0,2°, 18,00±0,2°, 18,61±0,2°, 18,90±0,2°, 19,46±0,2°, 19,96±0,2°, 20,84±0,2°, 21,35±0,2°, 22,81±0,2°, 23,11±0,2°, 23,59±0,2°, 24,7±0,2°, 25,18±0,2°, 25,74±0,2°, 27,62±0,2°, 28,32±0,2°, 31,17±0,2°.
В другом предпочтительном варианте осуществления изобретения порошковая рентгеновская дифрактограмма кристаллической формы С по существу показана на фиг. 13.
СПОСОБ ПОЛУЧЕНИЯ
Изобретение также предлагает способ получения кристаллических форм.
Способ получения кристаллической формы А согласно настоящему изобретению включает следующие этапы:
(1) приготовление первого раствора, содержащего первый растворитель и соединение формулы I, причем отношение массы к объему соединения формулы I к первому растворителю составляет от 0,1 г/л до 100 г/л (предпочтительно - от 1 г/л до 90 г/л, более предпочтительно - от 10 до 60 г/л, наиболее предпочтительно - от 15 до 40 г/л);
(2) смешивание второго растворителя с первым раствором и кристаллизирование при 0-50°С с получением кристаллической формы А.
В другом предпочтительном варианте осуществления изобретения концентрация соединения формулы I составляет 0,1 г/л насыщенной концентрации в первом растворе.
В другом предпочтительном варианте осуществления изобретения первый растворитель выбирают из группы, состоящей из С1-С4 спирта, С2-С8 кетонов (предпочтительно - С3-С5 кетонов), сложных эфиров С1-С10 (предпочтительно - сложных эфиров С1-С7, более предпочтительно - сложных эфиров С1-С5), галогенированного C1-С6-алкана, воды или их сочетания.
В другом предпочтительном варианте осуществления изобретения второй растворитель выбирают из группы, состоящей из простых эфиров С1-С10 (предпочтительно - простых эфиров С1-С8, более предпочтительно - простых эфиров С1-С6), С2-С15-алкана (предпочтительно - С3-С10-алкана, более предпочтительно - С4-С8-алкана), тетрагидрофурана, 1,4-диоксана или их сочетания.
В другом предпочтительном варианте осуществления изобретения С2-С8 кетоны выбирают из группы, состоящей из ацетона, метилэтилкетона, изобутанолбутанона или их сочетания.
В другом предпочтительном варианте осуществления изобретения сложные эфиры С1-С10 выбирают из группы, состоящей из метилформиата, этилацетата, изобутилформиата или их сочетания.
В другом предпочтительном варианте осуществления изобретения галогенированный С1-С6-алкан выбирают из группы, состоящей из дихлорметана, трихлорметана или их сочетания, предпочтительно - дихлорметана.
В другом предпочтительном варианте осуществления изобретения сложные эфиры С1-С10 выбирают из группы, состоящей из петролейного эфира, трет-бутилметилового эфира, этилового эфира, изопропилового эфира, диэтилового эфира или их сочетания.
В другом предпочтительном варианте осуществления изобретения С2-С15-алкан выбирают из группы, состоящей из n-пентана, n-гексана, n-гептана или их сочетания.
В другом предпочтительном варианте осуществления изобретения соединение формулы I находится в аморфной форме.
В другом предпочтительном варианте осуществления изобретения после этапа (2), способ дополнительно содержит следующие этапы: (3) фильтрация и/или сушка кристаллической формы А, полученной на этапе (2).
В другом предпочтительном варианте осуществления изобретения на этапе (3) температура сушки составляет от 10 до 70°С, предпочтительно - от 20 до 80°С, более предпочтительно - от 25 до 40°С.
В другом предпочтительном варианте осуществления изобретения на этапе (3) давление сушки составляет от 0 до 20 кПа, предпочтительно - от 0 до 10 кПа, более предпочтительно - от 5 до 10 кПа.
В другом предпочтительном варианте осуществления изобретения на этапе (3) время сушки составляет от 5 до 150 часов, предпочтительно - от 30 до 100 часов, более предпочтительно - от 60 до 80 часов.
В другом предпочтительном варианте осуществления изобретения выход кристаллической формы А составляет от 50 до 99%, предпочтительно - от 75 до 99%, более предпочтительно - от 85 до 99%.
В другом предпочтительном варианте осуществления изобретения концентрация соединения формулы I составляет от 0,1 г/л до 1000 г/л в первом растворе.
В другом предпочтительном варианте осуществления изобретения концентрация соединения формулы I составляет >1000 г/л в первом растворе.
В другом предпочтительном варианте осуществления изобретения на этапе (2) кристаллизацию проводят при температуре от 0 до 50°С, предпочтительно - от 0 до 40°С, более предпочтительно - от 20 до 30°С.
В другом предпочтительном варианте осуществления изобретения на этапе (2), кристаллизацию проводят с перемешиванием.
Способ получения кристаллической формы В согласно настоящему изобретению, способ получения включает следующие этапы:
(1) приготовление первого раствора, содержащего первый растворитель и соединение формулы I, причем отношение массы соединения формулы I к объему к первого растворителя составляет от 0,1 г/л до 100 г/л, предпочтительно - от 1 г/л до 900 г/л, более предпочтительно - от 10 до 600 г/л, наиболее предпочтительно - от 50 до 400 г/л;
(2) смешивание второго растворителя с первым раствором и кристаллизирование при 5-80°С с получением кристаллической формы В.
Способ получения кристаллической формы С по настоящему изобретению, способ получения включает следующие этапы:
(1) приготовление первого раствора, содержащего первый растворитель и соединение формулы I, причем отношение массы соединения формулы I к объему первого растворителя составляет от 0,1 г/л до 100 г/л, предпочтительно - от 1 г/л до 900 г/л, более предпочтительно - от 10 до 600 г/л, наиболее предпочтительно - от 50 до 400 г/л;
(2) смешивание второго растворителя с первым раствором и кристаллизирование при 5-80°С с получением кристаллической формы С.
ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ПРИМЕНЕНИЕ
Настоящее изобретение также представляет фармацевтическую композицию, состоящую из активного компонента в безопасном и эффективном количестве, и фармацевтически приемлемый носитель.
«Активный ингредиент», используемый в настоящем изобретении, означает кристаллическую форму А, В или С настоящего изобретения.
«Активный ингредиент» и фармацевтическая композиция настоящего изобретения используются для приготовления лекарственного средства для профилактики и/или лечения диабета II типа и/или осложнений диабета II типа.
В другом предпочтительном варианте осуществления изобретения осложнения сахарного диабета II типа выбирают из группы, состоящей из заболеваний коронарных артерий, инсульта, гипертонии, нефропатии, заболеваний периферических сосудов, неврологического заболевания и ретинопатии.
Следует понимать, что в настоящем изобретении фармацевтически приемлемый носитель конкретно не ограничен и может быть выбран из обычных материалов на известном уровне техники, или получен общепринятым способом, или получен из имеющихся в продаже.
В частности, фармацевтически приемлемый носитель включает, без ограничений, наполнитель, разрыхлитель, связующее вещество, смазывающее вещество или их сочетание.
Обычно наполнитель включает (без ограничений) крахмал, лактозу, микрокристаллическую целлюлозу, декстрин, маннитол, оксид магния, сульфат кальция или их сочетание.
Обычно разрыхлитель включает (без ограничения) карбоксиметилцеллюлозу и ее соль, перекрестно сшитую карбоксиметилцеллюлозу и ее соль, перекрестно сшитый повидон, натрий-карбоксиметилкрахмал, низкозамещенную гидроксипропилцеллюлозу или их сочетание.
Как правило, связующее вещество включает (без ограничения) повидон, гидроксипропилметилцеллюлозу, крахмальную пульпу или их сочетание.
Обычно смазывающее вещество включает (без ограничения) магния стеарат, кальция стеарат или их сочетание.
«Безопасное и эффективное количество» относится к: количеству активного ингредиента, достаточного для значительного улучшения состояния, но без серьезных побочных эффектов. Как правило, фармацевтическая композиция содержит от 1 до 2000 мг активного ингредиента на дозу, более предпочтительно от 10 до 200 мг активного ингредиента на дозу. В предпочтительном варианте осуществления изобретения «одна доза» представляет собой одну таблетку.
«Фармацевтически приемлемый носитель» означает один или несколько совместимых твердых или жидких наполнителей или желатиновых материалов, которые подходят для использования человеком и должны иметь достаточную чистоту и достаточно низкую токсичность. «Совместимость» означает, что каждый компонент в композиции может быть смешан с соединениями в настоящем изобретении и друг с другом без существенного снижения эффективности соединений. Некоторые примеры фармацевтически приемлемых носителей включают в себя целлюлозу и ее производные (такие как натриевая соль карбоксиметилцеллюлозы, этилцеллюлоза натрия, ацетат целлюлозы и т.д.), желатин, тальк, твердые смазывающие вещества (такие как стеариновая кислота, магния стеарат), сульфат кальция, растительный масла (такие как соевое масло, кунжутное масло, арахисовое масло, оливковое масло и т.д.), полиолы (такие как пропиленгликоль, глицерин, маннит, сорбит и т.д.), эмульгаторы (такие как Tween®), смачивающие вещества (такие как додецилсульфат натрия), красители, ароматизаторы, стабилизаторы, антиоксиданты, консерванты, апирогенная вода и др.
Способ введения соединения или фармацевтической композиции в настоящем изобретении конкретно не ограничен. Способ введения активного ингредиента или фармацевтической композиции конкретно в настоящем изобретении не ограничен, и репрезентативные способы введения включают в себя, без ограничения, пероральный, внутриопухолевый, ректальный, парентеральный (внутривенный, внутримышечный или подкожный) и тому подобное. Соединения по изобретению можно вводить отдельно или в комбинации с другими фармацевтически приемлемыми соединениями. Выбирают те же или аналогичные способы введения обычной (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофена[3,2-d]пиримидин-6-карбоновой кислоты, включающие в себя (без ограничения) пероральный, трансдермальный, внутривенный, внутримышечный, специальный способ применения и тому подобное.
Твердая лекарственная форма для перорального введения включает в себя капсулы, таблетки, пилюли, порошки и гранулы.
В этих твердых лекарственных формах активный ингредиент смешивают по меньшей мере с одним обычным инертным наполнителем (или носителем), таким как цитрат натрия или дикальцийфосфат, или с любым из следующих компонентов: (а) наполнители или компатибилизаторы, например крахмал, лактоза, сахароза, глюкоза, маннит и кремниевая кислота; (б) связующие вещества, например, гидроксиметилцеллюлоза, альгинат, желатин, поливинилпирролидон, сахароза и аравийская камедь; (в) увлажнители, такие как глицерин; (d) разрыхлители, такие как агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, конкретные сложные силикаты и карбонат натрия; (е) замедляющие растворение агенты, такие как парафин; (f) ускорители поглощения, например, четвертичные аммониевые соединения; (ж) смачивающие агенты, такие как цетиловый спирт и глицерилмоностеарат; (h) адсорбенты, например каолин; и (i) смазывающие вещества, такие как тальк, кальция стеарат, магния стеарат, твердый полиэтиленгликоль, лаурилсульфат натрия или их смесь. В капсулах, таблетках и пилюлях лекарственные формы могут также содержать буферные агенты.
Твердые лекарственные формы также готовят с использованием таких покрытий и материалов, как оболочки и другие материалы на известном уровне техники. Они могут содержать непрозрачные агенты, а активный ингредиент в таких композициях может высвобождаться в части пищеварительного тракта с замедлением. Примерами встраиваемых компонентов, которые можно использовать, являются полимерные и воскообразные материалы.
Жидкие лекарственные формы для перорального введения включают в себя фармацевтические приемлемые эмульсии, растворы, взвеси, сиропы или вытяжки. В дополнение к активному ингредиенту жидкая лекарственная форма может содержать любой обычный инертный разбавитель на известном уровне техники, такой, как вода или другие растворители, солюбилизаторы и эмульгаторы, например, этанол, изопропанол, этилкарбонат, этилацетат, пропиленгликоль, 1,3-бутандиол, диметилформамид, а также масло, в частности, хлопковое масло, арахисовое масло, масло кукурузного зародыша, оливковое масло, касторовое масло и кунжутное масло, или их смесь и т.д. В дополнение к этим инертным разбавителям композиции могут содержать такие вспомогательные вещества, как смачивающие агенты, эмульгирующие и суспендирующие агенты, подслащивающие агенты, ароматизаторы и специи.
В дополнение к активному ингредиенту суспензия может содержать суспендирующий агент, например, этоксилированный изооктадеканол, полиоксиэтиленсорбитол и сложный эфир дегидратированного сорбита, микрокристаллическую целлюлозу, метоксид алюминия, агар или их смесь и т.д.
Композиции для парентеральных инъекций могут содержать физиологические приемлемые стерильные водные или безводные растворы, дисперсии, взвеси или эмульсии, а также стерильные порошки, которые можно повторно растворить в стерильных инъекционных растворах или дисперсиях. Подходящие водные и безводные носители, разбавители, растворители или эксципиенты включают в себя воду, этанол, полиолы и их подходящие смеси.
Когда используют фармацевтическую композицию, безопасное и эффективное количество соединений изобретения вводят млекопитающему (например, человеку), нуждающемуся в лечении, где дозировка представляет собой фармацевтически приемлемую эффективную дозировку, которая варьируется в зависимости от возраста, пола этнической принадлежности, состояния больного. Для человека весом 60 кг суточная доза обычно составляет от 1 до 2000 мг, предпочтительно от 20 до 500 мг. Конечно, конкретные дозы также должны учитывать такие факторы, как путь введения, здоровье пациента и т.д., о чем известно специалисту в данной области.
Применение
В настоящем изобретении также предлагают применение кристаллической формы А, кристаллической формы В и кристаллической формы С малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты или фармацевтическая композиция для приготовления лекарственных средств для профилактики и/или лечения диабета II типа и/или осложнений сахарного диабета II типа.
Как правило, к осложнениям сахарного диабета II типа включают в себя (без ограничения) заболевание коронарной артерии, инсульт, гипертонию, нефропатию, заболевание периферических сосудов, неврологическое заболевание и ретинопатию.
По сравнению с аналогами из области техники настоящее изобретение имеет следующие основные преимущества:
(1) кристаллическая форма настоящего изобретения имеет более высокую степень гомогенности;
(2) кристаллическая форма настоящего изобретения обладает превосходной стабильностью, особенно термостабильностью;
(3) кристаллическая форма настоящего изобретения имеет более низкую гигроскопичность, и когда относительная влажность (ОВ) составляет менее 50%, кристаллическая форма имеет гигроскопичность менее 0,3%;
(4) кристаллическая форма настоящего изобретения стабильна и трудно разлагается в обычных условиях;
(5) способ получения кристаллической формы изобретения достаточно прост для осуществления и контроля, имеет хорошую воспроизводимость и подходит для промышленного производства;
(6) кристаллическая форма настоящего изобретения обладает превосходной пероральной гипогликемической активностью;
(7) кристаллическая форма настоящего изобретения может улучшать растворимость соединения, улучшать оральную абсорбционную способность, улучшать биодоступность и является более эффективной в профилактике или лечении диабета II типа.
Настоящее изобретение будет дополнительно проиллюстрировано ниже со ссылкой на конкретные примеры. Следует понимать, что данные примеры предназначены только для иллюстрации изобретения, а не для ограничения объема изобретения. Способы проведения экспериментов без особых условий, описанные в следующих примерах, обычно подразумевают проведение в обычных условиях или в соответствии с инструкциями изготовителя. Если не указано иное, части и процентные доли рассчитываются по весу.
Если не указано иное, вся профессиональная и научная терминология, используемая в тексте, имеет те значения, которые уже известны специалистам в уровне техники. Кроме того, любые способы и материалы, подобные или аналогичные раскрытым в описании заявки, могут применяться к способам изобретения. Описанные в настоящем документе способ предпочтительного варианта осуществления изобретения и вещество предназначены только для демонстрационных целей.
ОБЩИЕ МЕТОДЫ И ПАРАМЕТРЫ ИСПЫТАНИЙ
В настоящем изобретении кристалл подвергается серии общих испытаний следующим образом.
Рентгеновская дифракция (XRD) - это метод структурного анализа пространственного распределения внутренних атомов в веществе с использованием дифракции рентгеновских лучей при помощи кристаллов. Когда рентгеновскими лучами, имеющими определенную длину волны, облучают кристаллическое вещество, рентгеновские лучи рассеиваются из-за присутствия в кристалле регулярно расположенных атомов или ионов, а рассеянное рентгеновское излучение усиливается в некоторых направлениях, чтобы показать уникальную дифракцию, соответствующую данной кристаллической структуре.
В настоящем изобретении параметры испытания XRD являются следующими: модель прибора: Bruker D8 advance; цель: Cu-Kα (40 кВ, 40 мА); расстояние от образца до детектора: 30 см; диапазон сканирования: 3° ~ 40° (значение 2 тега); шаг сканирования: 0,1 с.
Термогравиметрический анализ (ТГА) - это аналитическая методика определения изменения массы вещества с температурой в условиях запрограммированного контроля температуры. Термогравиметрический анализ может быть использован для получения тепла, выделяемого при тепловых изменениях образца. Он подходит для проверки потери кристаллизационного растворителя или молекул кристаллической воды, или процессов сублимации и разложения образца в кристаллическом материале. Он также может эффективно различать, содержит ли материал кристаллизационный растворитель или кристаллическую воду.
В настоящем изобретении параметры испытания ТГА являются следующими: Тип прибора: Netzsch TG 209F3; Тигель: алюминиевый тигель (оксид алюминия); Диапазон температур: От 30 до 400°С; Скорость сканирования: 10 К/мин; продувочный газ: 25 мл/мин; Защитный газ: 15 мл/мин.
Дифференциальный сканирующий калориметр (ДСК) - это метод определения изменения разности температур между образцом и инертным стандартным образцом (обычно используют α-Al2O3) с помощью программы управления нагревом или охлаждением. Анализ ДСК подходит для анализа состояния разложения расплава, состояния смешанного кристаллического вещества, состояния кристаллического превращения образца и т.д.
В настоящем изобретении параметры испытания ДСК являются следующими: Тип прибора: Perkin Elmer ДСК 8500; Тигель: алюминиевый тигель; Сканирование от 50° до 280°С со скоростью нагрева 10°С/мин при продувке азотом.
Спектроскопия комбинационного рассеяния (СКР) - это метод изучения молекулярных колебаний, основанный на эффектах комбинационного рассеяния. В отличие от инфракрасного спектра поглощения, спектроскопия комбинационного рассеяния исследует частоту рассеянного света, генерируемого взаимодействием молекулы и света. Неполярные группы, которые обычно имеют незначительное инфракрасное поглощение, имеют ярко выраженные спектры комбинационного рассеяния.
В настоящем изобретении параметры испытания СКР являются следующими: Тип прибора: Рамановский микроскоп Thermo DXR (Рамановский спектрометр с конфокальным микроскопом); длина волны лазера: 532 нм; время воздействия: 1,0 сек; количество воздействия: 10.
Инфракрасная спектрометрия (ИК) - это первый аналитический метод, используемый для распознавания и идентификации кристаллических веществ. Из-за разного электрического окружения ковалентной связи в разных кристаллических молекулах сила ковалентной связи может изменяться, и изменение прочности ковалентной связи неизбежно приведет к различным ИК-спектрам разных кристаллических форм.
В настоящем изобретении параметры испытания ИК являются следующими: модель прибора: Инфракрасный спектрометр с преобразованием Nicolet 6700 Fourier; одноточечный метод ATR, разрешающая способность 4,0 см-1.
Испытание на динамическую сорбцию паров (ДСП)/испытание на водопоглощение быстро измеряет увеличение и потерю влаги в образце, вызванную потоком газа-носителя с заданной относительной влажностью (ОВ), при этом образец помещается в цифровой микробаланс с высокой чувствительностью и высокой стабильностью в состоянии самоподвески, а затем измеряют адсорбцию/десорбцию водяных паров путем измерения увеличения/уменьшения массы материала, таким образом определяя гигроскопичность образца.
В настоящем изобретении параметры испытаний ДСП являются следующими: Тип прибора: SMS DVS Intrinsic; безводное соединение: От 0 до 95% - 0% относительной влажности; Температура: 25°С; водное соединение: От 40 до 95% - 0% относительной влажности; Температура: 25°С.
Пример 1
Получение кристаллической формы А малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофена[3,2-d]пиримидин-6-карбоновой кислоты (№1)
200 мг малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты растворяли в 10 мл этанола, и кристаллы кристаллизовали при перемешивании при комнатной температуре до тех пор, пока не кристаллизовались твердые вещества, и время кристаллизации составляло около 2 часов. Полученные твердые вещества фильтровали, помещали в вакуумную сушильную печь и сушили в вакууме в течение 70 часов при температуре 25°С, 5 кПа, чтобы получить 110 мг кристалл кристаллической формы А малеата (R)-метил-2-(3-аминопиперидина-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты.
РЕЗУЛЬТАТ
Кристаллическая форма А кристалла, полученного в Примере 1, была подвергнута таким испытаниям, как XRD, ТГА, ДСК, ДСП, СКР и ИК и т.д.
Фигура 1 представляет собой порошковую рентгеновскую дифрактограмму кристаллической формы А из Примера 1, из Фигуры 1 видно, что кристаллическая форма А имеет пики поглощения при температуре 3,72°, 7,47°, 10,74°, 11,44°, 12,28°, 14,30°, 15,20°. 17,11°, 17,32°, 18,16°, 19,22°, 21,59°, 23,15°, 25,76°, 28,02°, 32,82°.
Фигура 2 представляет собой ТГ диаграмму кристаллической формы А из Примера 1, из Фигуры 2 видно, что кристаллическая форма А имеет потерю веса 47,97% при температуре 210-400°С.
Фигура 3 представляет собой спектр анализа дифференциальной сканирующей калориметрии (ДСК) кристаллической формы А из Примера 1, из Фигуры 3 видно, что соответствующий ДСК кристаллической формы А показывает температуру плавления 118,37°С.
Фигура 4 представляет собой диаграмму гигроскопического анализа (ДСП) кристаллической формы А из Примера 1. Из Фигуры 4 видно, что кристаллическая форма А имеет небольшую гигроскопичность в диапазоне обычной влажности при хранении, с малым диапазоном изменения (менее 2,0%).
Фигура 5 представляет собой диаграмму спектра комбинационного рассеяния кристаллической формы А из Примера 1, из Фигуры 5 видно, что кристаллическая форма А имеет характерные пики поглощения при 2226 см-1, 1716 см-1, 1689 см-1, 1604 см-1, 1566 см-1, 1536 см-1, 1486 см-1, 1393 см-1.
Фигура 6 представляет собой диаграмму инфракрасного спектра (ИК) кристаллической формы А из Примера 1, и на фигуре 6 видно, что кристаллическая форма А имеет характерные пики поглощения при 3435 см-1, 2952 см-1, 2225 см-1, 1718 см-1, 1685 см-1, 1558 см-1, 1531 см-1, 1452 см-1, 1234 см-1, 1064 см-1, 862 см-1, 756 см-1.
Пример 2
Получение кристаллической формы А малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофена[3,2-d]пиримидин-6-карбоновой кислоты (№2)
200 мг (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты растворяли в 10 мл воды, и кристаллы кристаллизовали при перемешивании при комнатной температуре до тех пор, пока твердые вещества не кристаллизовались. Полученные твердые вещества фильтруют, помещают в вакуумную сушильную печь и сушат в вакууме в течение 70 часов при температуре 25°С при 5 кПа, получая 100 мг кристаллической формы А малеата (R)-метил-2-(3-аминопиперидин-1-ила)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты.
Порошковая рентгеновская дифрактограмма полученного продукта была практически такой же, как в Примере 1.
Пример 3
Получение кристаллической формы А малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофена[3,2-d]пиримидин-6-карбоновой кислоты (№3)
200 мг (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты растворяли в 10 мл метанола, и кристаллы кристаллизовали при перемешивании при комнатной температуре до завершения процесса кристаллизации. Полученные твердые вещества фильтруют, помещают в вакуумную сушильную печь и сушат в вакууме в течение 70 часов при температуре 25°С при 5 кПа, получая 100 мг кристаллической формы А малеата (R)-метил-2-(3-аминопиперидин-1-ила)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты.
Порошковая рентгеновская дифрактограмма полученного продукта была практически такой же, как в Примере 1.
Пример 4
Получение кристаллической формы В малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофена[3,2-d]пиримидин-6-карбоновой кислоты (№4)
200 мг (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-малеат 6-карбоновой кислоты растворяли в 10 мл этилацетата и кристаллы кристаллизовали при перемешивании при комнатной температуре до тех пор, пока не кристаллизовались твердые вещества, а время кристаллизации составляло около 2 часов. Полученные твердые вещества отфильтровали, поместили в вакуумную сушильную печь и сушили в вакууме в течение 70 часов при температуре 25°С, 5 кПа, чтобы получить 110 мг кристалла В малеата (R)-метил-2-(3-аминопиперидин-1-)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты.
РЕЗУЛЬТАТ
Кристаллическая форма В кристалла, полученного в Примере 4, подвергалась таким испытаниям, как XRD, ТГА, ДСК, ДСП, СКР и ИК и т.д.
Фигура 7 представляет собой порошковую рентгеновскую дифрактограмму кристаллической формы В из Примера 4, из Фигуры 7 видно, что кристаллическая форма В имеет пики поглощения при температуре 5,37°, 7,85°, 11,20°, 12,01°, 14,93°, 16,04°, 20,09°, 22,10°, 22,61°, 24,19°, 30,16°, 32,12°, 32,39°.
Фигура 8 представляет собой диаграмму ТГ кристаллической формы В из Примера 4, и из фигуры 8 видно, что кристаллическая форма В имеет потерю массы 51,38% при температуре 210-400°С.
Фигура 9 представляет собой спектр анализа дифференциальной сканирующей калориметрии (ДСК) кристаллической формы В Примера 4. Из Фигуры 9 можно увидеть, как соответствующий ДСК кристаллической формы В показывает температуру плавления 136,77°С.
Фигура 10 представляет собой диаграмму гигроскопического анализа (ДСП) кристаллической формы В из Примера 4. Из Фигуры 10 можно увидеть, как кристалл из В имеет небольшую гигроскопичность в пределах обычной влажности хранения, с малым диапазоном изменения (менее 2,0%).
Фигура 11 представляет собой спектр комбинационного рассеяния кристаллической формы В из Примера 4, из Фигуры 11 видно, что кристаллическая форма В имеет характерные пики поглощения при 2234 см-1, 1718 см-1, 1693 см-1, 1607 см-1, 1565 см-1, 1536 см-1, 1476 см-1, 1386 см-1, 1349 см-1, 1216 см-1, 1174 см-1, 1047 см-1, 813 см-1, 730 см-1, 690 см-1, 641 см-1.
Фигура 12 представляет собой инфракрасный спектр (ИК) кристаллической формы В из Примера 1, из Фигуры 12 видно, что кристаллическая форма В имеет характерные пики поглощения при 3404 см-1, 2954 см-1, 2222 см-1. 1718 см-1, 1678 см-1, 1558 см-1, 1531 см-1, 1369 см-1, 1290 см-1, 1219 см-1, 1063 см-1, 862 см-1, 775 см-1.
Пример 5
Получение кристаллической формы В малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофена[3,2-d]пиримидин-6-карбоновой кислоты (№5)
200 мг (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты растворяли в 10 мл толуола, и кристаллы кристаллизовали при перемешивании при комнатной температуре до тех пор, пока не кристаллизовались твердые вещества. Полученные твердые вещества отфильтровывают, помещают в вакуумную сушильную печь и сушат в вакууме в течение 70 часов при 25°С при 5 кПа, получая 120 мг кристаллической формы В малеата (R)-метил-2-(3-аминопиперидин-1-ила)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты.
Порошковая рентгеновская дифрактограмма полученного продукта была практически такой же, как в Примере 4.
Пример 6
Получение кристаллической формы В малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофена[3,2-d]пиримидин-6-карбоновой кислоты (№6)
200 мг (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты растворяли в 10 мл метил-трет-бутилового эфира, и кристаллы кристаллизовали при перемешивании при комнатной температуре до тех пор, пока не кристаллизовались твердые вещества. Полученные твердые вещества отфильтровывают, помещают в вакуумную сушильную печь и сушат в вакууме в течение 70 часов при температуре 25°С при 5 кПа, получая 100 мг кристаллической формы В малеата (R)-метил-2-(3-аминопиперидин-1-ила)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты.
Порошковая рентгеновская дифрактограмма полученного продукта была практически такой же, как в Примере 4.
Пример 7
Получение кристаллической формы С малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофена[3,2-d]пиримидин-6-карбоновой кислоты (№7)
200 мг малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты растворяли в 10 мл ацетонитрила, добавляли 12 мл n-гексана, и кристаллы кристаллизовали при перемешивании при комнатной температуре до тех пор, пока не кристаллизовались твердые вещества, и время кристаллизации составляло около 2 часов. Полученные твердые вещества отфильтровывают, помещают в вакуумную сушильную печь и сушат в вакууме в течение 70 часов при температуре 25°С, 5 кПа, чтобы получить 110 мг кристалла С малеата (R)-метил-2-(3-аминопиперидин-1-ила)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен[3,2-d]пиримидин-6-карбоновой кислоты.
РЕЗУЛЬТАТ
Кристаллическая форма С кристалла, полученная в Примере 7, была подвергнута испытаниям, таким как XRD, ТГА, ДСК, ДСП, СКР и ИК и т.д.
Фигура 13 представляет собой порошковую рентгеновскую дифрактограмму кристаллической формы С из Примера 7, на Фигуре 13 видно, что кристаллическая форма С имеет пики поглощения при температуре 6,63°, 8,67°, 11,16°, 11,64°, 15,24°, 16,43°, 17,06°, 17,41°, 18,00°, 18,61°, 18,90°, 19,46°, 19,96°, 20,84°, 21,35±0,2°, 22,81±0,2°, 23,11±0,2°, 23,59±0,2°, 24,7°, 25,18°, 25,74°, 27,62°, 28,32°, 31,17°.
Фигура 14 представляет собой диаграмму ТГ кристаллической формы С из Примера 7, из Фигуры 14 видно, что кристаллическая форма С имеет потерю веса 46,30% при температуре 210-400°С.
Фигура 15 представляет собой спектр анализа дифференциальной сканирующей калориметрии (ДСК) кристаллической формы С из Примера 7, из Фиг. 15 видно, что соответствующий ДСК кристаллической формы А показывает температуру плавления 160,90°С.
Фигура 16 представляет собой диаграмму гигроскопического анализа (ДСП) кристаллической формы С из Примера 7. Из Фигуры 16 видно, что кристалл из С имеет небольшую гигроскопичность в диапазоне обычной влажности при хранении, с малым диапазоном изменения (менее 2,0%).
Фигура 17 представляет собой диаграмму спектра комбинационного рассеяния кристаллической формы С из Примера 7, из Фигуры 17 видно, что кристаллическая форма С имеет характерные пики поглощения при 3383 см-1, 3378 см-1, 3370 см-1, 3288 см-1, 3163 см-1, 2963 см-1, 2230 см-1, 1719 см-1, 1558 см-1, 1478 см-1, 662 см-1.
Фигура 18 представляет собой инфракрасный спектр (ИК) кристаллической формы С из Примера 7, из Фигуры 18 видно, что кристаллическая форма С имеет характерные пики поглощения при 3428 см-1, 2936 см-1, 1640 см-1, 1514 см-1, 1463 см-1, 1418 см-1, 1385 см-1, 1272 см-1, 1136 см-1, 1022 см-1, 874 см-1, 766 см-1, 599 см-1.
Все источники, упомянутые в настоящем изобретении, включены в описание посредством ссылки, как если бы они были включены отдельно посредством ссылки. Кроме того, следует понимать, что после прочтения упомянутого описания специалистами в данной области техники очевидны многие изменения и модификации, и эти эквиваленты также попадают в объем, определенный в прилагаемой формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛЯНАЯ ФОРМА ИНГИБИТОРА ДИПЕПТИДИЛПЕПДИДАЗЫ IV И СПОСОБ ПОЛУЧЕНИЯ СОЛЕВОЙ ФОРМЫ | 2017 |
|
RU2734549C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА А СОЕДИНЕНИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2016 |
|
RU2699030C2 |
| НОВОЕ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЙ ТИЕНО[3,2-D]ПИРИМИДИН-4-ОНА | 2019 |
|
RU2768828C1 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА α МОНОБЕНЗОАТА СОЕДИНЕНИЯ А, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ЕЕ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2015 |
|
RU2677660C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ОРГАНИЧЕСКОГО СОЕДИНЕНИЯ | 2009 |
|
RU2572529C2 |
| Способ получения полиморфа гидрохлорида 2-[4-(метиламинометил)фенил]-5-фтор-бензофуран-7-карбоксамида | 2018 |
|
RU2783418C1 |
| СОЛЬ И ПОЛИМОРФ ФЕНИЛ-ПИРИМИДОНОВОГО СОЕДИНЕНИЯ, ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2018 |
|
RU2761213C2 |
| ФЕНИЛАМИНОПИРИМИДИН ИЛИ ПОЛИМОРФНАЯ ФОРМА СОЛИ ФЕНИЛАМИНОПИРИМИДИНА | 2016 |
|
RU2712226C2 |
| КРИСТАЛЛИЧЕСКАЯ ИЛИ АМОРФНАЯ ФОРМА АГОНИСТОВ FXR, ПРЕДСТАВЛЯЮЩИХ СОБОЙ ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2800751C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ИНГИБИТОРА PARP-1 И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2018 |
|
RU2792620C2 |
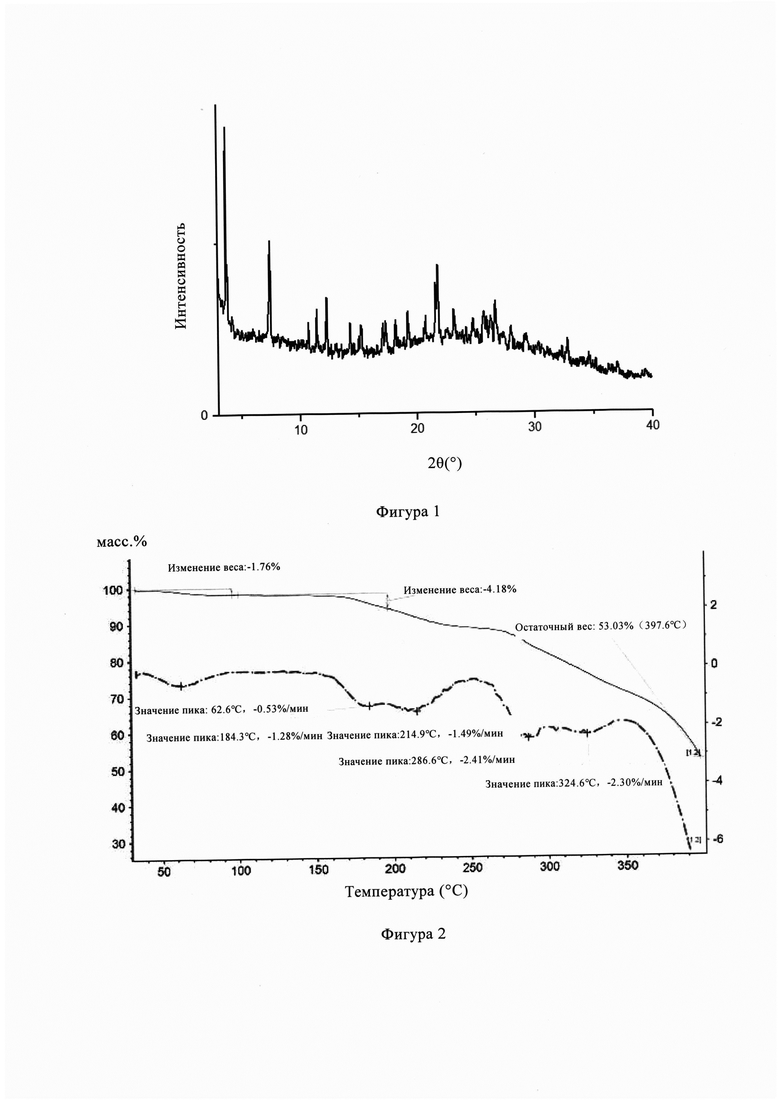
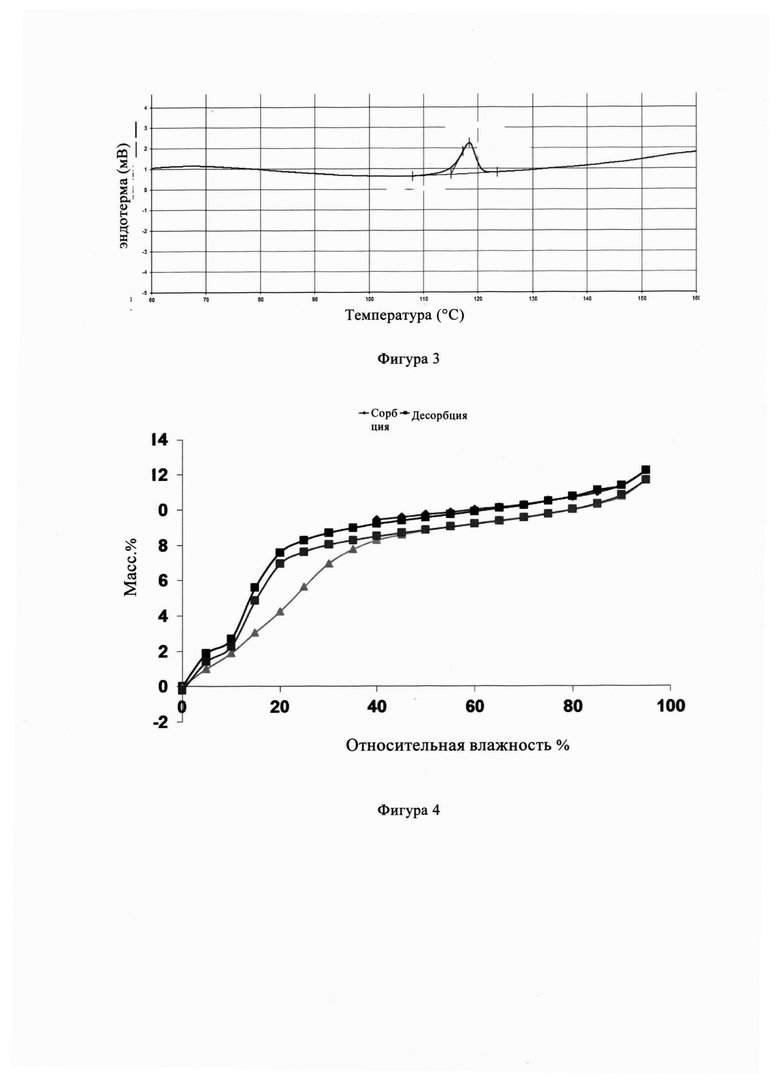

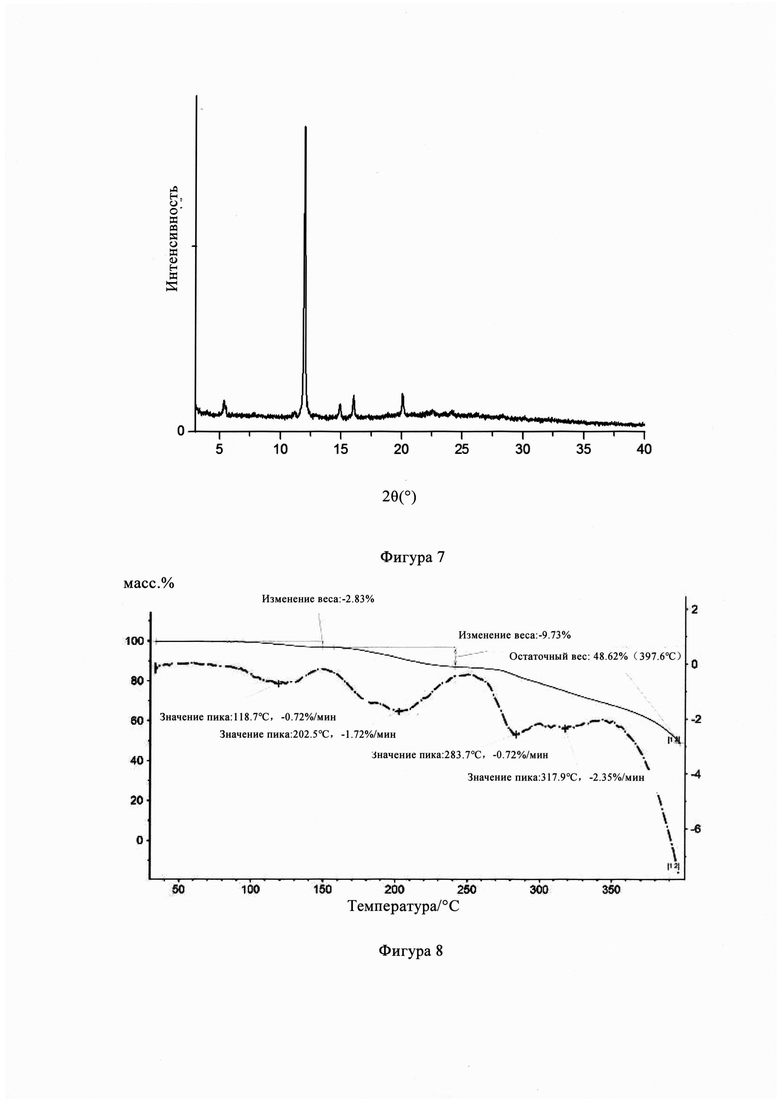
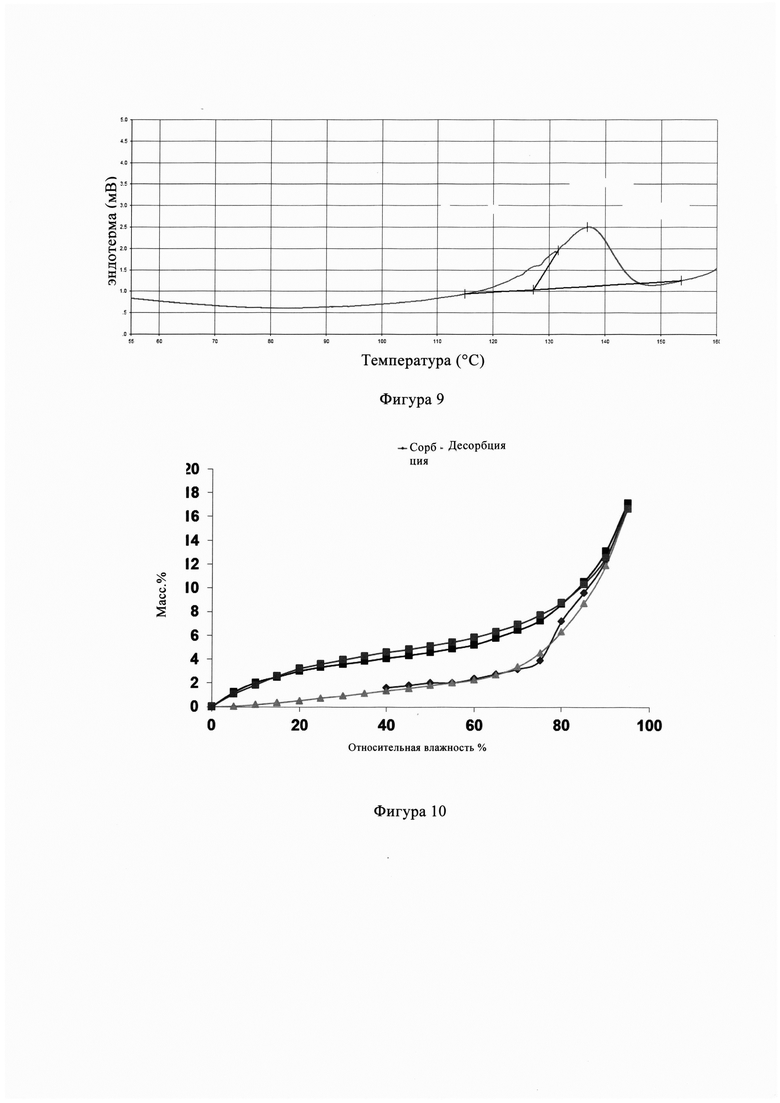
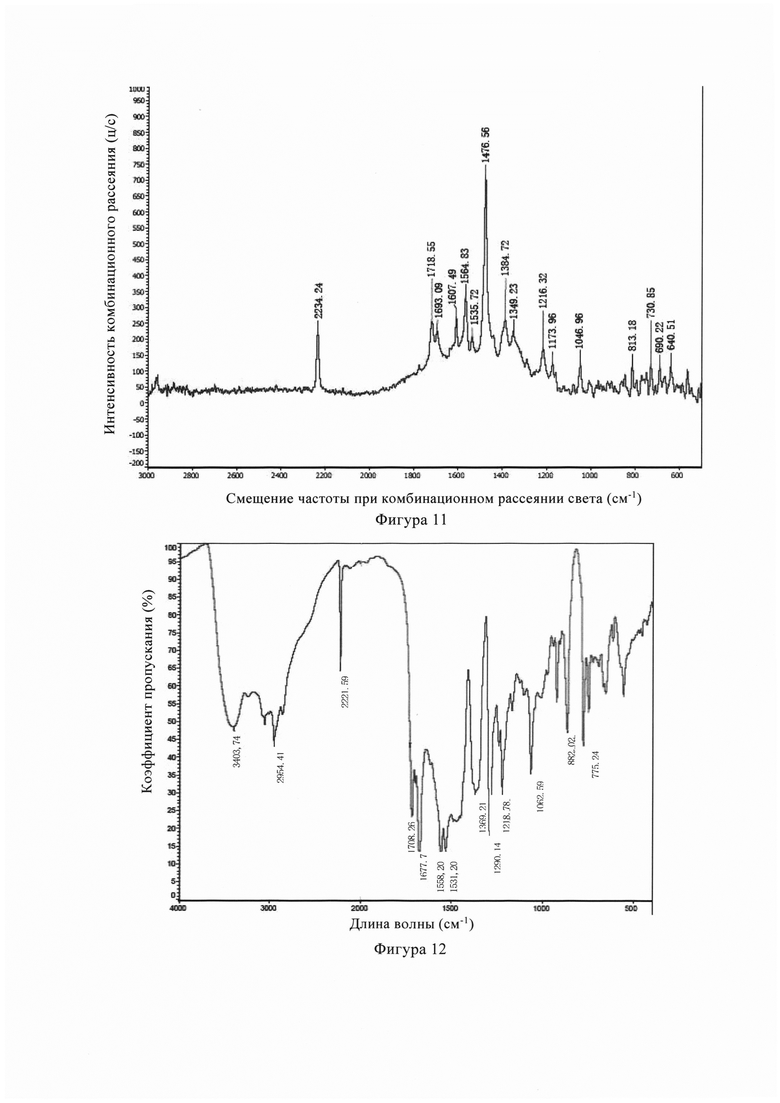
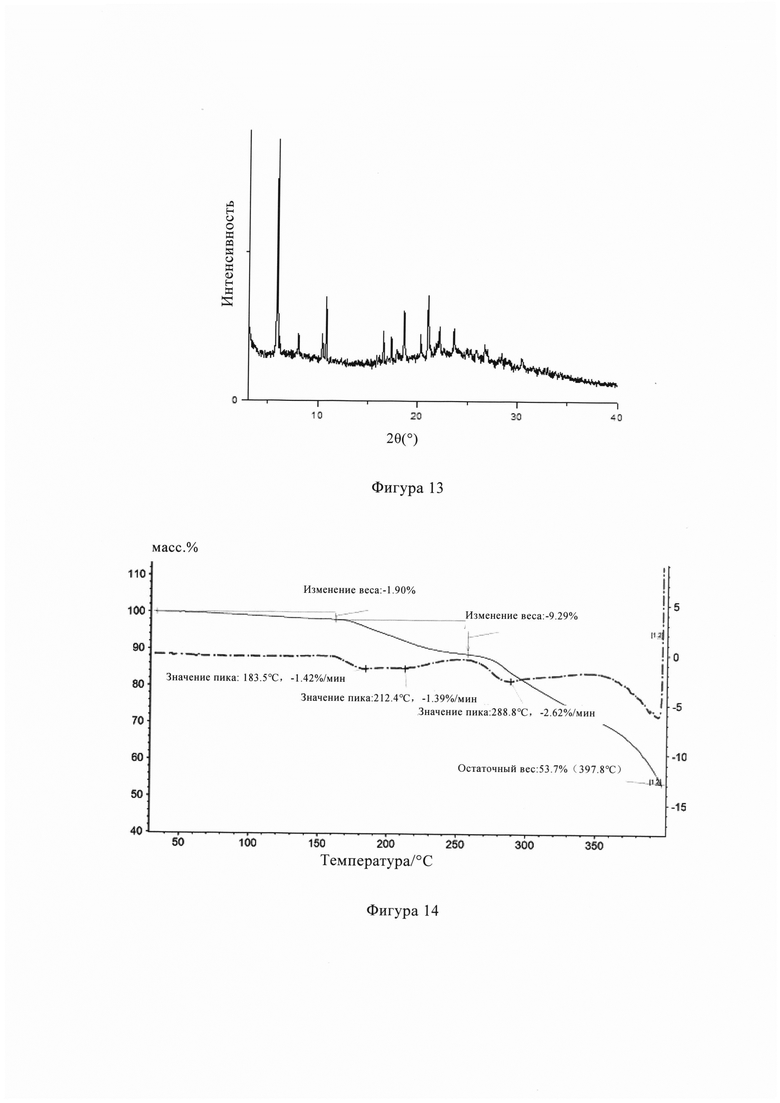
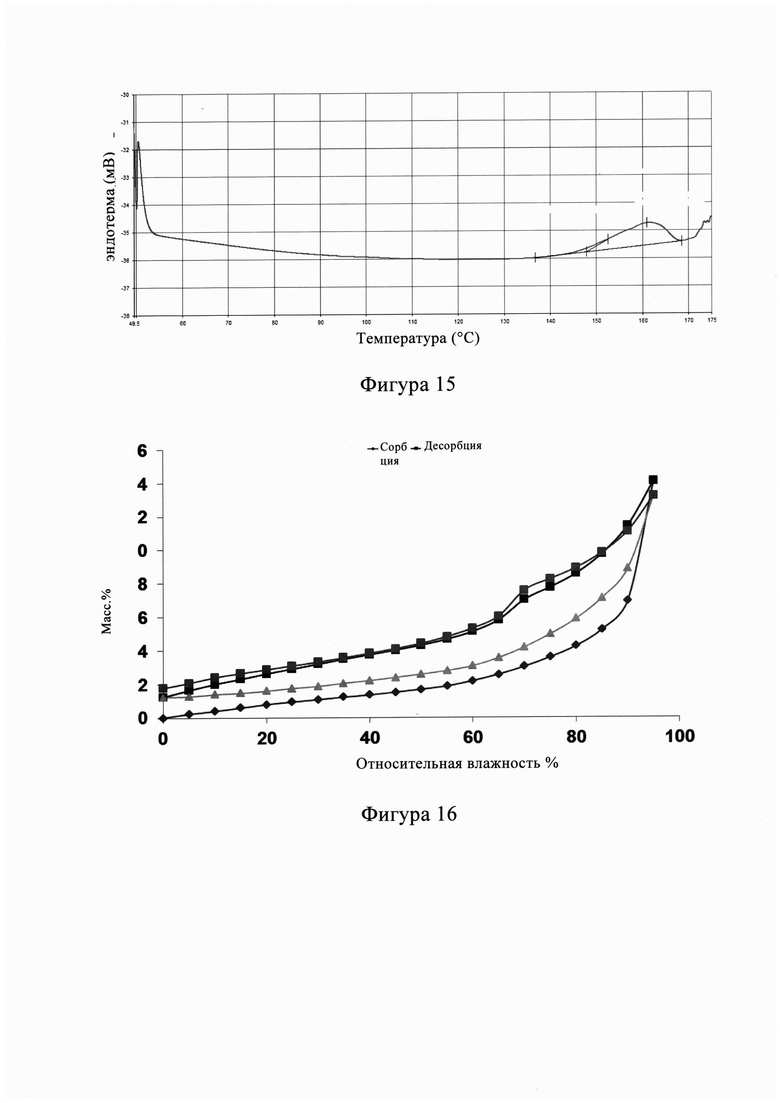
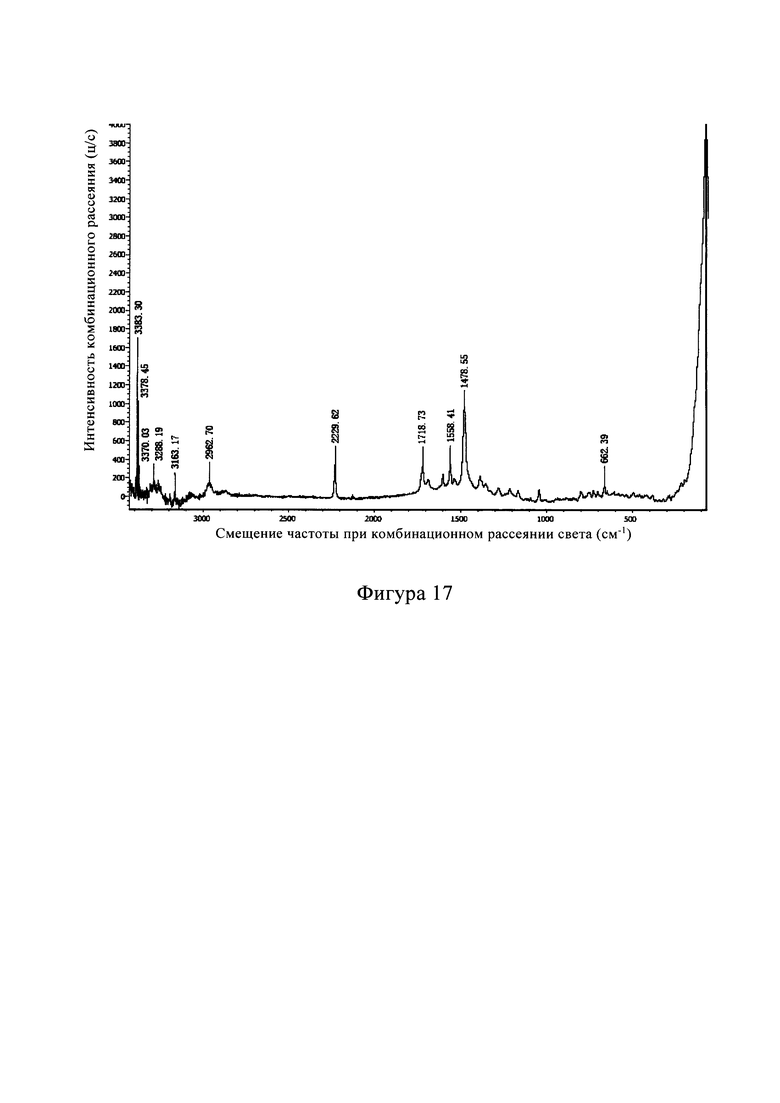
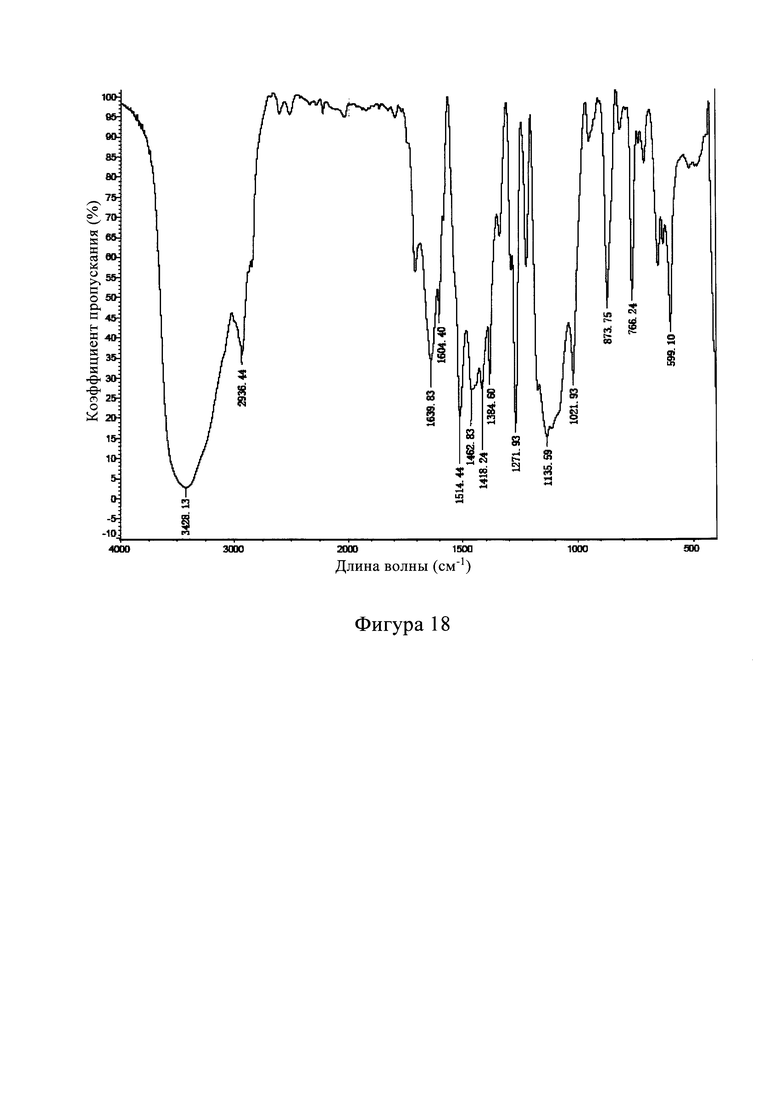
Изобретение относится к полиморфу малеата (R)-метил-2-(3-аминопиперидин-1-ил)-3-(2-цианобензил)-4-карбонил-3,4-дигидротиофен [3,2-d] пиримидин-6-карбоновой кислоты и способам его получения и фармацевтической композиции. Полиморф представляет собой кристалл, имеющий высокую стабильность и низкую гигроскопичность, причем кристаллическую форму выбирают из кристаллической формы А, кристаллической формы В и кристаллической формы С. Кристаллическая форма обладает гипогликемической активностью и может использоваться для получения нового лекарственного средства для лечения или профилактики сахарного диабета типа II и/или осложнений сахарного диабета типа II. 6 н. и 4 з.п. ф-лы, 7 пр., 18 ил.
1. Кристаллическая форма соединения формулы I, в которой кристаллическая форма представляет собой кристалл, имеющий высокую стабильность и низкую гигроскопичность, причем кристаллическую форму выбирают из группы, состоящей из кристаллической формы A, кристаллической формы B и кристаллической формы C 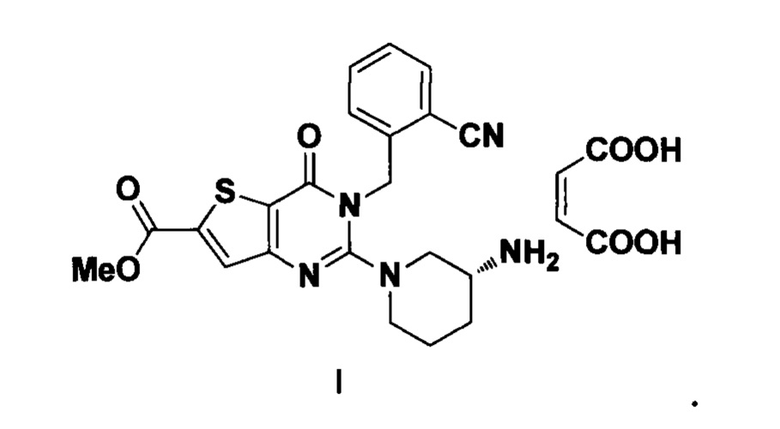
где порошковая рентгеновская дифрактограмма кристаллической формы A включает в себя 3 или более 2θ значений, выбранных из группы, состоящей из 3,72 ± 0,2°, 7,47 ± 0,2°, 11,44 ± 0,2°, 12,28 ± 0,2°, 21,59 ± 0,2°;
порошковая рентгеновская дифрактограмма кристаллической формы B включает 3 или более 2θ значений, выбранных из группы, состоящей из: 5,37±0,2°, 12,01±0,2°, 14,93±0,2°, 16,04±0,2°, 20,09±0,2°;
порошковая рентгеновская дифрактограмма кристаллической формы С включает 3 или более 2θ значений, выбранных из группы, состоящей из: 6,63±0,2°, 11,16±0,2°, 17,06±0,2°, 19,46±0,2°, 20,84±0,2°, 25,74±0,2°.
2. Кристаллическая форма по п. 1, в котором «низкая гигроскопичность» означает, что увеличение веса кристаллической формы A, кристаллической формы B или кристаллической формы C составит не более ≤1% после помещения в эксикатор на 24 часа при температуре 80°С и влажности 80% и дальнейшего извлечения.
3. Кристаллическая форма по п.1, в котором кристаллическая форма А имеет одну или несколько характеристик, выбранных из группы, состоящей из
(1) порошковой рентгеновской дифрактограммы кристаллической формы A, включающей в себя 3 или более 2θ значений, выбранных из группы, состоящей из 3,72 ± 0,2°, 7,47 ± 0,2°, 10.74± 0,2°, 11,44 ± 0,2°, 12,28 ± 0,2°, 14.30± 0,2°, 15.20± 0,2°, 17.11± 0,2°, 17.32± 0,2°, 18.16± 0,2°, 19.22± 0,2°, 21,59 ± 0,2°, 23.15± 0,2°, 25.76± 0,2°, 28.02± 0,2°, 32.82± 0,2°; и/или
(2) спектра дифференциальной сканирующей калориметрии (спектр ДСК) кристаллической формы А, имеющего характерный пик в диапазоне 118 ± 5°С; и/или
(3) профиля термогравиметрического анализа кристаллической формы А, включающего в себя 3 или более характерных пика поглощения, выбранных из группы, состоящей из 63 ± 5°С, 184 ± 5°С, 215 ± 5°С, 287 ± 5°С, 325 ± 5°С; и/или
(4) потери теплового веса кристаллической формы A, составляющей 47-48 мас.% при 400°C; и/или
(5) начального значения температуры эндотермического перехода кристаллической формы A, составляющего 115 ± 2°C; и/или
(6) спектра комбинационного рассеяния кристаллической формы A, включающего в себя следующие характерные пики поглощения, выраженные длиной волны λ: 2226±2 см-1, 1716±2 см-1, 1689±2 см-1, 1604±2 см-1, 1566±2 см-1, 1536±2 см-1, 1486±2 см-1, 1393±2 см-1; и/или
(7) профиля ИК-излучения кристаллической формы A, включающего в себя следующие характерные пики поглощения, выраженные длиной волны λ: 3435±2 см-1, 2952±2 см-1, 2225±2 см-1, 1718±2 см-1, 1685±2 см-1, 1558±2 см-1, 1531±2 см-1, 1452±2 см-1, 1234±2 см-1, 1064±2 см-1, 862±2 см-1, 756±2 см-1.
4. Кристаллическая форма по п.1, в которой кристаллическая форма В имеет одну или несколько характеристик, выбранных из группы, состоящей из
(1) порошковой рентгеновской дифрактограммы кристаллической формы B, включающей 3 или более 2θ значений, выбранных из группы, состоящей из: 5,37±0,2°, 7.85± 0,2°, 11.20± 0,2°, 12,01±0,2°, 14,93±0,2°, 16,04±0,2°, 20,09±0,2°, 22.10± 0,2°, 22.61± 0,2°, 24.19± 0,2°, 30.16± 0,2°, 32.12± 0,2°, 32.39± 0,2°; и/или
(2) спектра дифференциальной сканирующей калориметрии (спектр ДСК) кристаллической формы В, имеющего характерный пик в диапазоне 137 ± 5°С; и/или
(3) начального значения температуры эндотермического перехода кристаллической формы B, составляющего 127 ± 2°C; и/или
(4) профиля термогравиметрического анализа кристаллической формы В, включающего в себя 3 или более характерных пика поглощения, выбранных из группы, состоящей из: 119±5°C, 202±5°C, 284±5°C, 318±5°C; и/или
(5) потери теплового веса кристаллической формы В, составляющей 51-52 мас.% при 400°C; и/или
(6) спектра комбинационного рассеяния кристаллической формы B, включающего в себя следующие характерные пики поглощения, выраженные длиной волны λ: 2234±2 см-1, 1718±2 см-1, 1693±2 см-1, 1607±2 см-1, 1565±2 см-1, 1536±2 см-1, 1476±2 см-1, 1386±2 см-1, 1349±2 см-1, 1216±2 см-1, 1174±2 см-1, 1047±2 см-1, 813±2 см-1, 730±2 см-1, 690±2 см-1, 641±2 см-1; и/или
(7) профиля ИК-излучения кристаллической формы B, включающего в себя следующие характерные пики поглощения, выраженные длиной волны λ: 3404±2 см-1, 2954±2 см-1, 2222±2 см-1, 1718±2 см-1, 1678±2 см-1, 1558±2 см-1, 1531±2 см-1, 1369±2 см-1, 1290±2 см-1, 1219±2 см-1, 1063±2 см-1, 862±2 см-1, 775±2 см-1.
5. Кристаллическая форма по п.1, в которой кристаллическая форма С имеет одну или несколько характеристик, выбранных из группы, состоящей из
(1) порошковой рентгеновской дифрактограммы кристаллической формы С, включающей 3 или более 2θ значений, выбранных из группы, состоящей из: 6,63±0,2°, 8.67 ± 0,2°, 11,16±0,2°, 11.64 ± 0,2°, 15.24± 0,2°, 16.43 ± 0,2°, 17,06±0,2°, 17.41± 0,2°, 18.00± 0,2°, 18.61± 0,2°, 18.90± 0,2°, 19,46±0,2°, 19.96± 0,2°, 20,84±0,2°, 21.35± 0,2°, 22.81± 0,2°, 23.11± 0,2°, 23.59± 0,2°, 24.7± 0,2°, 25.18± 0,2°, 25,74±0,2°, 27.62± 0,2°, 28.32± 0,2°, 31.17± 0,2°; и/или
(2) спектра дифференциальной сканирующей калориметрии (спектр ДСК) кристаллической формы С, имеющего характерный пик в диапазоне 161 ± 5°С; и/или
(3) начального значения температуры эндотермического перехода кристаллической формы С, составляющего 147 ± 2°C; и/или
(4) профиля термогравиметрического анализа кристаллической формы С, включающего характерный пик поглощения, выбранный из группы, состоящей из: 183±5°C, 212±5°C, 289±5°; и/или
(5) потери теплового веса кристаллической формы С, составляющей 46-47 мас.% при 400°C; и/или
(6) спектра комбинационного рассеяния кристаллической формы С, включающего в себя следующие характерные пики поглощения, выраженные длиной волны λ: 3383±2 см-1, 3378±2 см-1, 3370±2 см-1, 3288±2 см-1, 3163±2 см-1, 2963±2 см-1, 2230±2 см-1, 1719±2 см-1, 1558±2 см-1, 1478±2 см-1, 662±2 см-1; и/или
(7) профиля ИК-излучения кристаллической формы В, включающего в себя следующие характерные пики поглощения, выраженные длиной волны λ: 3428±2 см-1, 2936±2 см-1, 1640±2 см-1, 1514±2 см-1, 1463±2 см-1, 1418±2 см-1, 1385±2 см-1, 1272±2 см-1, 1136±2 см-1, 1022±2 см-1, 874±2 см-1, 766±2 см-1, 599±2 см-1.
6. Способ получения кристаллической формы А по п.1, содержащий этапы:
(1) получают первый раствор, содержащий первый растворитель и соединение формулы I, причем отношение массы соединения формулы I к объему первого растворителя составляет от 0,1 г/л до 100 г/л (предпочтительно - от 1 г/л до 90 г/л, более предпочтительно - от 10 до 60 г/л, наиболее предпочтительно - от 15 до 40 г/л);
(2) смешивают второй растворитель с первым раствором и кристаллизуют смесь при 0-50°С с получением кристаллической формы А;
где первый растворитель выбирают из группы, состоящей из C1-C4 спиртов, C2-C8 кетонов, C1-C10 сложных эфиров, галогенированного C1-C6-алкана, воды или их комбинации;
второй растворитель выбирают из группы, состоящей из C1-C10 простых эфиров, C2-C15-алкана, тетрагидрофурана, 1,4-диоксана или их комбинации.
7. Способ получения кристаллической формы В по п.1, содержащий этапы:
(1) получают первый раствор, содержащий первый растворитель и соединение
формулы I, причем отношение массы соединения формулы I к объему первого растворителя составляет от 0,1 г/л до 100 г/л, предпочтительно - от 1 г/л до 900 г/л, более предпочтительно - от 10 до 600 г/л, наиболее предпочтительно - от 50 до 400 г/л;
(2) смешивают второй растворитель с первым раствором и кристаллизуют смесь при 5-80°С с получением кристаллической формы В;
где первый растворитель выбирают из группы, состоящей из C1-C10 сложных эфиров, С5-С10 ароматического углеводорода, C1-C10 простых эфиров, галогенированного C1-C6-алкана или их комбинации;
второй растворитель выбирают из группы, состоящей из C2-C15-алкана, тетрагидрофурана, 1,4-диоксана или их комбинации.
8. Способ получения кристаллической формы С по п.1, содержащий этапы:
(1) получают первый раствор, содержащий первый растворитель и соединение формулы I, причем отношение массы соединения формулы I к объему первого растворителя составляет от 0,1 г/л до 100 г/л, предпочтительно - от 1 г/л до 900 г/л, более предпочтительно - от 10 до 600 г/л, наиболее предпочтительно - от 50 до 400 г/л;
(2) смешивают второй растворитель с первым раствором и кристаллизуют смесь при 5-80°С с получением кристаллической формы C;
где первый растворитель выбирают из группы, состоящей из C1-C10 нитрилов, С2-С8 кетонов, C1-C10 сложных эфиров, галогенированного C1-C6-алкана или их комбинации;
второй растворитель выбирают из группы, состоящей из C2-C15-алканов, C1-C10 простых эфиров, тетрагидрофурана, 1,4-диоксана или их сочетания.
9. Фармацевтическая композиция для профилактики и/или лечения диабета типа II и/или осложнений диабета типа II, содержащая:
(a) эффективное количество кристаллической формы А, В или С по п. 1, и
(b) фармацевтически приемлемый носитель.
10. Применение кристаллической формы по п. 1 или фармацевтической композиции по п. 9 для производства лекарственного средства для профилактики и/или лечения диабета типа II и/или осложнений диабета типа II.
| WO 2013078765 A1 06.06.2013 | |||
| Berge, S | |||
| M., Bighley, L | |||
| D., & Monkhouse, D | |||
| C | |||
| Шеститрубный элемент пароперегревателя в жаровых трубках | 1918 |
|
SU1977A1 |
| Pharmaceutical Salts | |||
| Journal of Pharmaceutical Sciences, 66(1), 1-19 | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Morissette, S | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| High-throughput crystallization: polymorphs, salts, co-crystals and solvates of pharmaceutical solids | |||
| Advanced Drug Delivery Reviews, | |||

