ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет временной заявки на патент США 62/627435, поданной 7 февраля 2018 года, которая полностью включена в настоящее описание в качестве ссылки.
УРОВЕНЬ ТЕХНИКИ
Коагуляция крови представляет собой первую линию защиты от потери крови после травмы. В «каскаде» коагуляции крови задействован ряд циркулирующих зимогенов сериновых протеаз, регуляторных кофакторов и ингибиторов. Каждый фермент, однажды полученный из его зимогена, специфически расщепляет следующий зимоген в каскаде для продуцирования активной протеазы. Этот процесс повторяется до тех пор, пока, наконец, тромбин не расщепляет фибринопептиды из фибриногена для продуцирования фибрина, который полимеризуется с образованием сгустка крови. Хотя эффективная коагуляция ограничивает потерю крови в месте травмы, она также создает риск системной коагуляции, приводящей к массивному тромбозу. В нормальных условиях гемостаз поддерживает баланс между образованием сгустка (коагуляцией) и растворением сгустка (фибринолизом). Однако при некоторых патологических состояниях, таких как острый инфаркт миокарда и нестабильная стенокардия, разрыв сформировавшейся атеросклеротической бляшки приводит к аномальному тромбообразованию в сосудистой сети коронарных артерий.
Заболевания, вызванные коагуляцией крови, такие как инфаркт миокарда, нестабильная стенокардия, мерцательная аритмия, инсульт, эмболия легочной артерии и тромбоз глубоких вен, являются основными причинами смерти в развитых странах. Современнные методы лечения антигоагулянтными терапевтическими средствами, такими как нефракционированный гепарин для инъекций, низкомолекулярный (LMW) гепарин и варфарин для перорального введения (кумадин), могут быть опасными вследствие риска эпизодов кровотечения и оказывают разное воздействие на пациентов, что приводит к необходимости тщательного мониторинга и подбора терапевтических доз. Следовательно, на сегодняшний день в медицине имеет место потребность в новых антикоагулянтных лекарственных средствах, не проявляющих некоторых или всех побочных эффектов, которыми обладают доступные в настоящее время лекарственные средства.
Фактор XIa является привлекательной терапевтической мишенью, вовлеченной в метаболизм, связанный с этими заболеваниями. Повышенные уровни фактора XIa или повышенная активность фактора XIa наблюдались при некоторых тромбоэмболических заболеваниях, включая венозный тромбоз (Meijers et al., N. Engl. J. Med. 342:696, 2000), острый инфаркт миокарда (Minnema et al., Arterioscler Thromb Vasc Biol 20:2489, 2000), острый коронарный синдром (Butenas et al., Thromb. Haemost. 99:142, 2008), коронарную болезнь сердца (Butenas et al., Thromb. Haemost. 99:142, 2008), хроническую обструктивную болезнь легких (Jankowski et al., Thromb. Res. 127:242, 2011), аортальный стеноз (Blood Coagul Fibrinolysis, 22:473, 2011), острую цереброваскулярную недостаточность (Undas et al., Eur. J. Clin. Invest., 42:123, 2012) и систолическую сердечную недостаточность вследствие имешической кардиомиопатии (Zabcyk et al., Pol. Arch. Med. Wewn. 120:334, 2010). Пациенты, у которых отсутствует фактор XI вследствие генетического дефицита фактора XI, практически не страдают ишемическими инсультами (Salomon et al., Blood, 111:4113, 2008). В то же время потеря активности фактора XIa, который оставляет интактным один из путей метаболизма, инициирующим коагуляцию, не нарушает гемостаза. У людей дефицит фактора XI может привести к легкому или умеренному нарушению свертываемости, в частности в тканях с высоким уровнем местной фибринолитической активности, таких как мочевыводящие пути, нос, ротовая полость и миндалины. Кроме того, гемостаз почти в норме у мышей с дефициром фактора XI (Gailani, Blood Coagul Fibrinolysis, 8:134, 1997). Далее, было обнаружено, что ингибирование фактора XI также облегчает артериальную гипертензию и другие заболевания и дисфункции, включая воспаление сосудов (Kossmann et al. Sci. Transl. Med. 9, eaah4923 (2017)).
Следовательно, соединения, ингибирующие фктор XIa, обладают потенциалом превотвращения или лечения широкого спектра расстройств без побочных эффектов и терапевтических проблем, которые являются недостатком лекарственных средств, ингибирующих другие компоненты каскада коагуляции. Кроме того, вследствие ограниченной эффективности и неблагоприятных побочных эффектов некоторых современных терапевтических средств для ингибирования нежелательного тромбоза (например, тромбоза глубоких вен, тромбоза печеночных вен и инсульта), необходимы более эффективные соединения и способы (например, соединения и способы, связанные с фактором XIa) для предотвращения или лечения нежелательного тромбоза.
Другой терапевтической мишенью является венорасширяющий фермент калликреин. Калликреин плазмы человека представляет собой сериновую протеазу, которая может быть ответственной за активацию нескольких нижестоящих факторов (например, брадикинина и плазмина), имеющих решающее значение для коагуляции и контроля, например, артериального давления, воспаления и боли. Калликреины экспрессируются, например, в предстательной железе, эпидермисе и центральной нервной системе (ЦНС) и могут участвовать, например, в регуляции разжижения спермы, расщепления белков клеточной адгезии и пластичности нейронов в ЦНС. Кроме того, калликреины могут быть вовлечены в онкогенез и развитие рака и ангионевротического отека, например наследственного ангионевротического отека. Избыточная активация калллинкреин-кининового пути метаболизма может приводить к ряду расстройств, включая ангионевротический отек, например наследственный ангионевротический отек (НАО) (Schneider et al., J. Allergy Clin. Immunol. 120:2, 416, 2007). На сегодняшний день существуют ограниченные варианты лечения НАО (см., например, WO2003/076458). Таким образом, существует потребность в терапевтических средствах для предотвращения или лечения этих заболеваний.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, которые ингибируют фктор XIa или калликреин, и способам предотвращения или лечения нежелательного тромбоза или ангионевротического отека (например, наследственного ангионевротического отека) введением одного или нескольких соединений отдельно или в комбинации с другими действующими веществами. Изобретение также относится к способам разработки и отбора дополнительных ингибиторов фактора XIa или калликреина с использованием этих структур. Желательно, чтобы эти соединения содержали определенные структуры с физическими и пространственными характеристиками, которые позволяют соединениям взаимодействовать со специфическими остатками активного сайта фактора XIa или калликреина.
В одном аспекте настоящее изобретение относится к соединению формулы (II):

или его фармацевтически приемлемой соли, где R1 представляет собой водород или -NR8R9; Ra представляет собой C1-6 алкил, C1-6 галогеналкил, галоген, циано или -OR6; Rb представляет собой водород или C1-6 алкил; R2 представляет собой необязательно замещенный 5-членный гетероарил или необязательно замещенный 5-членный гетероциклил; R3 представляет собой водород, C1-6 алкил или C1-6 галогеналкил; R4 представляет собой C1-6 алкил, C1-6 галогеналкил, циклоалкил, гетероциклил, арил или гетероарил, где циклоалкил, гетероциклил, арил или гетероарил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена, C1-6 алкила, C1-6 галогеналкила, циано или -OR6; R5 представляет собой водород, C1-6 алкил, C1-6 галогеналкил, циклоалкил или арил, где циклоалкил или арил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена, C1-6 алкила, C1-6 галогеналкила, циано или -OR6, или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют кольцо; R6 представляет собой водород, C1-6 алкил, или C1-6 галогеналкил; каждый R8 и R9 независимо представляет собой водород, C1-6 алкил, -C(O)R10 или -C(O)OR10; R10 представляет собой C1-6 алкил или C1-6 галогеналкил; Rx представляет собой -O или отсутствует, где когда Rx представляет собой -O, атом азота пиридильного кольца является положительно заряженным и Rx является отрицательно заряженным, образуя таким образом пиридил-N-оксид; m равно 0, 1, 2 или 3; и n равно 0 или 1, где если n равно 0, тогда R5 представляет собой водород и R4 отсутствует.
В одном аспекте соединение, описанное в настоящем изобретении, представляет собой соединение формулы (II):

или его фармацевтически приемлемую соль, где R1 представляет собой водород или -NR8R9; Ra представляет собой C1-6 алкил, C1-6 галогеналкил, галоген, циано или -OR6; Rb представляет собой водород или C1-6 алкил; R2 представляет собой необязательно замещенный 5-членный гетероарил; R3 представляет собой водород, C1-6 алкил или C1-6 галогеналкил; R4 представляет собой C1-6 алкил, C1-6 галогеналкил, циклоалкил, гетероциклил, арил или гетероарил, где циклоалкил, гетероциклил, арил или гетероарил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена, C1-6 алкила, C1-6 галогеналкила, циано или -OR6; R5 представляет собой водород, C1-6 алкил, C1-6 галогеналкил, циклоалкил или арил, где циклоалкил или арил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена, C1-6 алкила, C1-6 галогеналкила, циано или -OR6, или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют кольцо; R6 представляет собой водород, C1-6 алкил или C1-6 галогеналкил; каждый R8 и R9 независимо представляет собой водород, C1-6 алкил, -C(O)R10 или -C(O)OR10; R10 представляет собой C1-6 алкил или C1-6 галогеналкил; m равно 0, 1, 2 или 3; и n равно 0 или 1, где если n равно 0, тогда R5 представляет собой водород и R4 отсутствует.
В некоторых вариантах осуществления соединение представляет собой соединение формулы (I):

В некоторых вариантах осуществления R6 представляет собой C1-6 алкил или C1-6 галогеналкил. В некоторых вариантах осуществления R6 представляет собой -CH3 или -CF3.
В некоторых вариантах осуществления R1 представляет собой водород, -NH2 или -NHCH3.
В некоторых вариантах осуществления R2 представляет собой 5-членный гетероарил, необязательно замещенный одним или двумя независимыми заместителями, выбранными из C1-6 алкила. В некоторых вариантах осуществления R2 представляет собой пиразолил, имидазолил, тиенил, изотиазолил или тиазолил, где имидазолил, тиенил, изотиазолил или тиазолил является необязательно замещенным одним или двумя независимыми заместителями, выбранными из C1-6 алкила. В некоторых вариантах осуществления R2 представляет собой пиразолил, имидазолил, тиенил, изотиазолил или тиазолил, где имидазолил, тиенил, изотиазолил или тиазолил является необязательно замещенным одним или двумя заместителями, выбранными из -CH3.
В некоторых вариантах осуществления R2 представляет собой

В некоторых вариантах осуществления R3 представляет собой -CH3.
В некоторых вариантах осуществления R4 представляет собой C1-6 алкил, циклоалкил, арил или гетероарил, где циклоалкил, арил или гетероарил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена или C1-6 алкила. В некоторых вариантах осуществления R4 представляет собой циклогексил, циклопропил, фенил, пиридил, пиразолил, тиенил или C1-6 алкил, где циклогексил, циклопропил, фенил, пиридил, пиразолил или тиенил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена или C1-6 алкила. В некоторых вариантах осуществления R4 представляет собой циклогексил, фенил, пиридил, пиразолил или тиенил, где циклогексил, фенил, пиридил, пиразолил или тиенил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена или C1-6 алкила. В некоторых вариантах осуществления R4 представляет собой фенил, необязательно замещенный одним, двумя или тремя заместителями, выбранными из -F, -Cl или C1-6 алкила.
В некоторых вариантах осуществления R5 представляет собой водород, -CH3, -CH2CH3, -CH2CH2CH3, -CH(CH3)2, -CH2CH2CH2CH3, -CF3 или незамещенный циклопропил. В некоторых вариантах осуществления R5 представляет собой -CH2CH3, -CF3 или незамещенный циклопропил.
В некоторых вариантах осуществления R4 представляет собой C1-6 алкил, C1-6 галогеналкил, циклоалкил, гетероциклил, арил или гетероарил, где циклоалкил, гетероциклил, арил или гетероарил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена, C1-6 алкила, C1-6 галогеналкила, циано или -OR6, и R5 представляет собой водород, C1-6 алкил, C1-6 галогеналкил, циклоалкил или арил, где циклоалкил или арил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена, C1-6 алкила, C1-6 галогеналкила или -OR6; или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют кольцо.
В некоторых вариантах осуществления соединение представляет собой соединение формулы (I-a):

В некоторых вариантах осуществления соединение представляет собой соединение формулы (I-b):
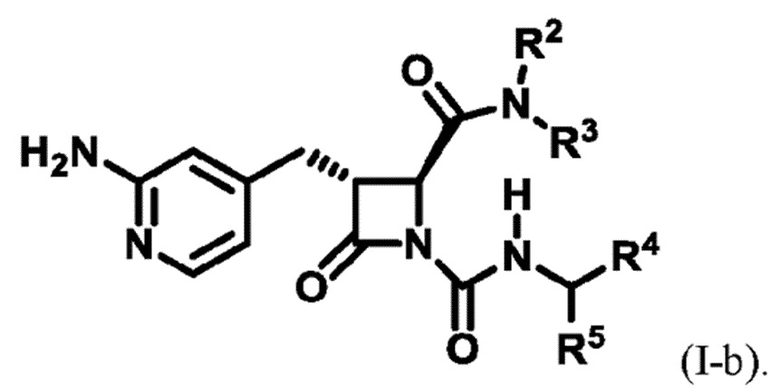
В некоторых вариантах осуществления соединение представляет собой соединение формулы (I-c), формулы (I-d), формулы (I-e), формулы (I-f), формулы (I-g), формулы (I-i) или формулы (I-j):
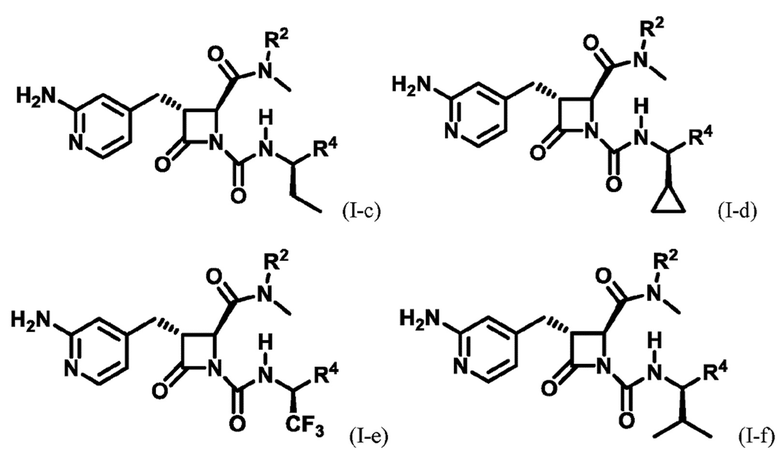

В некоторых вариантах осуществления соединение представляет собой соединение формулы (I-c):

В некоторых вариантах осуществления соединение представляет собой соединение формулы (I-e), формулы (I-k), формулы (I-l), формулы (I-m), формулы (I-o) или формулы (I-p):
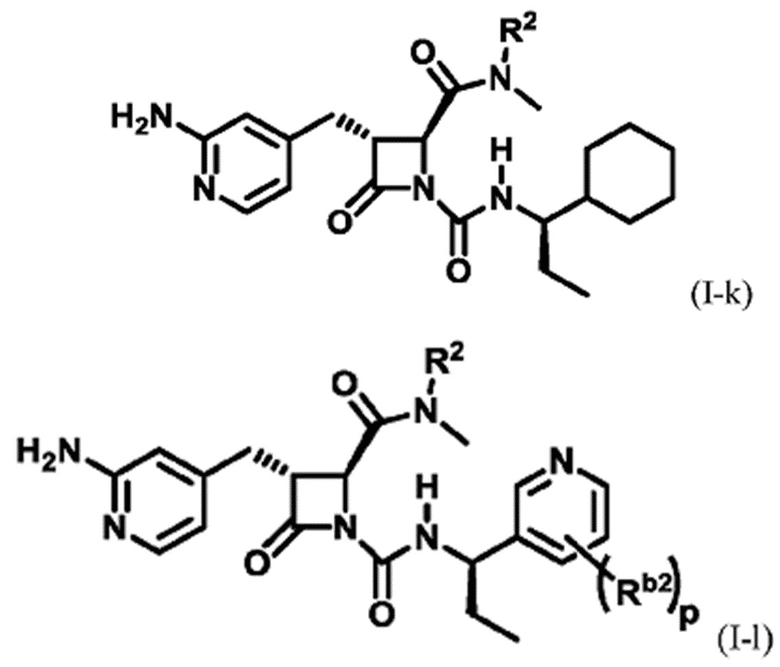

где каждый из Rb2 независимо представляет собой галоген, C1-6 алкил, C1-6 галогеналкил, циано или -OR6; и p равно 0, 1, 2 или 3. В некоторых вариантах осуществления Ra представляет собой -C1-6 алкил.
В некоторых вариантах осуществления Ra представляет собой -CH3. В некоторых вариантах осуществления Ra представляет собой -CH3, и m равно 1. В некоторых вариантах осуществления m равно 1. В некоторых вариантах осуществления Rb представляет собой C1-6 алкил. В некоторых вариантах осуществления Rb представляет собой -CH3.
В некоторых вариантах осуществления соединение представляет собой соединение, выбранное из соединений, представленных в таблице 1.
В некоторых вариантах осуществления соединение представляет собой фармацевтически приемлемую соль (например, гидрохлоридную (HCl), гидробромидную (HBr), тартратную, олеатную или цитратную соль). В предпочтительном варианте осуществления фармацевтически приемлемая соль представляет собой гидрохлоридную (HCl) соль.
В одном аспекте настоящее изобретение отосится к фармацевтической композиции, включающей соединение формулы (II) или его фармацевтически приемлемую соль и один или несколько фармацевтически приемлемых эксципиентов. В некоторых вариантах осуществления композиция представлена в форме жидкого препарата (например, раствора). В некоторых вариантах осуществления композиция представлена в форме твердого препарата (например, капсулы, пилюли, таблетки или порошка).
В одном аспекте настоящее изобретение относится к способу снижения риска инсульта (например, ишемии, в частности транзиторной ишемической атаки) у пациента, который перенес ишемическую атаку (например, транзиторную ишемическую атаку), включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления введение снижает риск инсульта у пациента по сравнению с пациентом, которому не вводят соединение. В некоторых вариантах осуществления введение снижает риск мерцательной аритмии у пациента по сравнению с пациентом, которому не вводят соединение.
В одном аспекте настоящее изобретение относится к способу снижения риска системной эмболии за пределами ЦНС (например, ишемии, в частности транзиторной ишемической атаки) у пациента, который перенес ишемическую атаку (например, транзиторную ишемическую атаку), включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления введение снижает риск системной эмболии за пределами ЦНС у пациента по сравнению с пациентом, которому не вводят соединение.
В одном аспекте настоящее изобретение относится к способу лечения тромбоза глубоких вен, включающему введение пациенту, который перенес ишемическую атаку (например, транзиторную ишемическую атаку), эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В одном аспекте настоящее изобретение относится к способу лечения острого коронарного синдрома у пациента, включающему введение пациенту, нуждающемуся в этом, соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу профилактики тромбоза глубоких вен, включающему введение пациенту, который перенес тромбоз глубоких вен (например, пациенту, который ранее лечил тромбоз глубоких вен), эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В одном аспекте настоящее изобретение относится к способу снижения риска рецидива тромбоза глубоких вен, включающему введение пациенту, который перенес тромбоз глубоких вен (например, пациенту, который ранее лечил тромбоз глубоких вен), эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления введение снижает риск рецидива тромбоза глубоких вен у пациента по сравнению с пациентом, которому не вводят соединение.
В одном аспекте настоящее изобретение относится к способу профилактики венозной тромбоэмболии, например тромбоза глубоких вен или эмболии легочной артерии у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления пациент подвергается хирургической операции. В некоторых вариантах осуществления пациенту вводится соединение, его фармацевтически приемлемая соль или композиция до, во время или после хирургической операции. В некоторых вариантах осуществления пациент подвергается операции эндопротезирования коленного или тазобедренного сустава. В некоторых вариантах осуществления пациент подвергается ортопедической операции. В некоторых вариантах осуществления пациент подвергается операции на легких. В некоторых вариантах осуществления пациент лечится от рака, например с помощью хирургической операции. В некоторых вариантах осуществления пациент страдает хроническим заболеванием. В некоторых вариантах осуществления венозная тромбоэмболия связана с раком. В некоторых вариантах осуществления соединение, его фармацевтически приемлемая соль или композиция по настоящему изобретению представляет собой первичное лекарственное средство в профилактике тромбоза глубоких вен или венозной тромбоэмболии. В некоторых вариантах осуществления соединение, его фармацевтически приемлемая соль или композиция по настоящему изобретению используется в качестве расширенного терапевтического лечения. В одном аспекте настоящее изобретение относится к способу снижения у пациента риска венозной тромбоэмболии, например тромбоза глубоких вен или эмболии легочной артерии, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления пациент подвергается хирургической операции. В некоторых вариантах осуществления пациенту вводится соединение, его фармацевтически приемлемая соль или композиция после хирургической операции. В некоторых вариантах осуществления пациент подвергается операции эндопротезирования коленного или тазобедренного сустава. В некоторых вариантах осуществления пациент подвергается ортопедической операции. В некоторых вариантах осуществления пациент подвергается операции на легких. В некоторых вариантах осуществления пациент лечится от рака, например с помощью хирургической операции. В некоторых вариантах осуществления пациент страдает хроническим заболеванием. В некоторых вариантах осуществления тромбоэмболическое расстройство связано с раком. В некоторых вариантах осуществления соединение, его фармацевтически приемлемая соль или композиция по настоящему изобретению представляет собой первичное лекарственное средство в снижении риска тромбоэмболического расстройства. В некоторых вариантах осуществления соединение, его фармацевтически приемлемая соль или композиция по настоящему изобретению используется в качестве расширенного терапевтического лечения.
В одном аспекте настоящее изобретение относится к способу снижения риска инсульта или системной эмболии у пациента, нуждающегося в этом, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления пациент страдает мерцательной аритмией (например, неклапанной фибрилляцией предсердий). В некоторых вариантах осуществления пациент страдает нарушением функции почек (например, терминальной почечной недостаточностью).
В одном аспекте настоящее изобретение относится к способу профилактики инсульта или системной эмболии у пациента, нуждающегося в этом, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления пациент страдает мерцательной аритмией (например, неклапанной фибрилляцией предсердий). В некоторых вариантах осуществления пациент страдает нарушением функции почек (например, терминальной почечной недостаточностью).
В одном аспекте настоящее изобретение относится к способу снижения риска рецидива эмболии легочной артерии (например, симптоматической эмболии легочной артерии), включающему введение пациенту, который перенес эмболию легочной артерии (например, пациенту, который ранее лечил эмболию легочной артерии), эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления введение снижает риск рецидива эмболии легочной артерии у пациента по сравнению с пациентом, которому не вводят соединение.
В одном аспекте настоящее изобретение относится к способу профилактики эмболии легочной артерии у пациента, который перенес эмболию легочной артерии (например, пациенту, который ранее лечил эмболию легочной артерии), включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В одном аспекте настоящее изобретение относится к способу снижения риска рецидива эмболии легочной артерии (например, симптоматической эмболии легочной артерии), включающему введение пациенту, который перенес тромбоз глубоких вен (например, пациенту, который ранее лечил тромбоз глубоких вен), эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления введение снижает риск рецидива эмболии легочной артерии у пациента по сравнению с пациентом, которому не вводят соединение.
В одном аспекте настоящее изобретение относится к способу профилактики эмболии легочной артерии у пациента, который перенес тромбоз глубоких вен (например, у пациента, который ранее лечил тромбоз глубоких вен), включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В одном аспекте настоящее изобретение относится к способу лечения эмболии легочной артерии у пациента, который перенес тромбоз глубоких вен, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу лечения тромбоза глубоких вен у пациента, которому ранее вводился антикоагулянт, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления антикоагулянт вводился парентерально в течение 5-10 дней.
В одном аспекте настоящее изобретение относится к способу лечения эмболии легочной артерии у пациента, которому ранее вводился антикоагулянт, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления антикоагулянт вводился парентерально в течение 5-10 дней.
В одном аспекте настоящее изобретение относится к способу лечения пациента, который перенес ишемическую атаку (например, транзиторную ишемическую атаку), включающему введение соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)) пациенту. В некоторых вариантах осуществления соединение вводится пациенту в пределах 24 часов или менее, например 12, 10, 9, 8, 7, 6 часов или менее, после начала ишемической атаки у пациента.
В одном аспекте настоящее изобретение относится к способу лечения пациента, который перенес ишемическую атаку (например, транзиторную ишемию), включающему введение соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)) пациенту. В некоторых вариантах осуществления соединение вводится пациенту в интервале от более 2 часов до 12 часов, например от более 2 часов до 10 часов или менее, от более 2 часов до 8 часов или менее, после начала ишемической атаки у пациента.
В одном аспекте настоящее изобретение относится к способу лечения гипертензии, например артериальной гипертензии, у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления гипертензия, например артериальная гипертензия, приводит к атеросклерозу. В некоторых вариантах осуществления гипертензия представляет собой легочную артериальную гипертензию.
В одном аспекте настоящее изобретение относится к способу снижения риска гипертензии, например артериальной гипертензии, у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления гипертензия, например артериальная гипертензия, приводит к атеросклерозу. В некоторых вариантах осуществления гипертензия представляет собой легочную артериальную гипертензию.
В одном аспекте настоящее изобретение относится к способу профилактики гипертензии, например артериальной гипертензии, у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления гипертензия, например артериальная гипертензия, приводит к атеросклерозу. В некоторых вариантах осуществления гипертензия представляет собой легочную артериальную гипертензию.
В одном аспекте настоящее изобретение относится к способу снижения воспаления у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления воспаление представляет собой воспаление сосудов. В некоторых вариантах осуществления воспаление сосудов сопровождается атеросклерозом. В некоторых вариантах осуществления воспаление сосудов у пациента сопровождается тромбоэмболическим заболеванием. В некоторых вариантах осуществления воспаление сосудов представляет собой воспаление сосудов, индуцированное ангиотензином II.
В одном аспекте настоящее изобретение относится к способу предотвращения васкулярной инфильтрации лейкоцитов у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В одном аспекте настоящее изобретение относится к способу предотвращения у пациента эндотелиальной дисфункции, индуцированной ангиотензином II, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В одном аспекте настоящее изобретение относится к способу предотвращения избыточного продуцирования тромбина у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления избыточное продуцирование тромбина происходит на тромбоцитах.
В одном аспекте настоящее изобретение относится к способу лечения у пациента ренальной дисфункции, связанной с гипертензией, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В одном аспекте настоящее изобретение относится к способу профилактики ренальной дисфункции, связанной с гипертензией, у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В одном аспекте настоящее изобретение относится к способу снижения риска ренальной дисфункции, связанной с гипертензией, у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В одном аспекте настоящее изобретение относится к способу лечения фиброза почек у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В одном аспекте настоящее изобретение относится к способу профилактики фиброза почек у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В одном аспекте настоящее изобретение относится к способу снижения риска фиброза почек у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В одном аспекте настоящее изобретение относится к способу лечения почечной недостаточности у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В одном аспекте настоящее изобретение относится к способу профилактики почечной недостаточности у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В одном аспекте настоящее изобретение относится к способу снижения риска почечной недостаточности у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В одном аспекте настоящее изобретение относится к способу ингибирования фактора XIa у пациента, включающему введение пациенту, который перене ишемию, эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления ишемия представляет собой коронарную ишемию. В некоторых вариантах осуществления пациентом является млекопитающее (например, человек). В некоторых вариантах осуществления пациент подвергается хирургической операции (например, эндопротезированию коленного сустава ил эндопротезированию тазобедренного сустава). В некоторых вариантах осуществления ишемия представляет собой коронарную ишемию. В некоторых вариантах осуществления пациентом является пациент с неклапанной фибрилляцией предсердий. В некоторых вариантах осуществления у пациента имеется один или несколько из следующих факторов риска инсульта: инсульт в анамнезе (например, ишемический, неизвестный, геморрагический), транзиторная ишемическая атака или системная эмболия за пределами ЦНС. В некоторых вариантах осуществления у пациента имеется один или несколько из следующих факторов риска: возраст 75 лет или более, гипертензия, сердечная недостаточность или фракция выброса левого желудочка (например, менее или равная 35%) или сахарный диабет.
В некоторых вариантах осуществления соединение вводится пероральным или парентеральным (например, внутривенным) способом введения. В некоторых вариантах осуществления соединение вводится пероральным способом введения.
В некоторых вариантах осуществления соединение вводится перед ишемической атакой (например, пациенту с риском ишемической атаки).
В некоторых вариантах осуществления соединение вводится после ишемической атаки (в частности транзиторной ишемической атаки). В некоторых вариантах осуществления соединение вводится примерно через 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14 дней или более после ишемической атаки (в частности транзиторной ишемической атаки). В некоторых вариантах осуществления соединение вводится примерно через 1, 2, 3, 4, 5, 6, 7 или 8 недель или более после ишемической атаки (в частности транзиторной ишемической атаки).
В некоторых вариантах осуществления соединение вводится в комбинации с дополнительным терапевтическим средством. В некоторых вариантах осуществления дополнительное терапевтическое средство вводится после введения соединения. В некоторых вариантах осуществления дополнительное терапевтическое средство вводится перорально. В некоторых вариантах осуществления дополнительное терапевтическое средство вводится по меньшей мере через 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 14, 16, 18, 20 или 24 часов или более после введения соединения. В некоторых вариантах осуществления дополнительное терапевтическое средство вводится по меньшей мере через 1, 2, 3, 4, 5, 6, 7, 14, 21 или 28 дней или более после введения соединения. В некоторых вариантах осуществления дополнительное терапевтическое средство вводится примерно через 1 день, примерно через 2 дня, примерно через 3 дня, примерно через 4 дня, примерно через 5 дней, примерно через 6 дней, примерно через 7 дней или более после введения соединения.
В некоторых вариантах осуществления дополнительное терапевтическое средство вводится постоянно (например, в течение примерно 1 дня, примерно 2 дней, примерно 3 дней, примерно 4 дней, примерно 5 дней, примерно 6 дней, примерно 7 дней, примерно 8 дней, примерно 9 дней, примерно 10 дней, примерно 11 дней, примерно 12 дней, примерно 13 дней или примерно 14 дней или более) после введения соединения.
В некоторых вариантах осуществления дополнительное терапевтическое средство лечит побочный эффект (например, активное патологическое кровотечение или тяжелые реакции гиперчувствительности (например, анафилактические реакции), спинальную и или эпидуральную гематому, желудочно-кишечное расстройство (например, боль в верхней части живота, диспепсию, зубную боль), общие расстройства и состояния места введения (например, усталость), инфекции и инвазии (например, синусит, инфекции мочевыводящих путей), патологии опорно-двигательного аппарата и соединительных тканей (например, боль в спине, остеоартрит), нарушения со стороны дыхательной системы, органов грудной клетки и средостению (например, боль в ротоглотке), травму, заражение и осложнение после процедуры (например, выделение из раны), нарушения со стороны скелетной мускулатуры и соединительной ткани (например, боли в конечности, мышечный спазм), расстройства нервной системы (например, синкопального состояния), нарушения кожной и подкожной ткани (например, рурита, волдырей), нарушения со стороны системы кроветворения и лимфатической системы (например, агранулоцитоза), желудочно-кишечные расстройства (например, забрюшинное кровоизлияние), гепатобилиарные расстройства (например, желтуху, холестаз, цитолитический гепатит), расстройства иммунной системы (например, гиперчувствительность, анафилатическую реакцию, анафилактический шок, ангионевротический отек), расстройства нервной системы (например, кровоизлияние в мозг, субдуральную гематому, эпидуральную гематому, гемипарез), заболевания кожи и подкожной клетчатки (например, синдрома Стивенса-Джонсона)).
В некоторых вариантах осуществления дополнительное терапевтическое средство представляет собой нестероидное противовоспалительное лекарственное средство (НПВЛС) (например, аспирин или напроксен), ингибитор агрегации тромбоцитов (например, клопидогрел) или антикоагулянт (например, варфарин или эноксапарин).
В некоторых вариантах осуществления дополнительное терапевтическое средство приводит к аддитивному терапевтическому эффекту. В некоторых вариантах осуществления дополнительное терапевтическое средство приводит к синергическому терапевтическому эффекту.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, включающей соединение по настоящему изобретению (например, соединение формулы (II)) и фармацевтически приемлемый эксципиент.
В другом аспекте настоящее изобретение относится к способу модуляции (например, ингибирования) фактора XIa в организме пациента. Способ включает стадию введения эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)) пациенту, нуждающемуся в этом, и модулирование таким образом (например, ингибируя) фактор XIa.
В другом аспекте настоящее изобретение относится к способу лечения у пациента, нуждающегося в этом, тромбоэмболического осложнения. Способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). Тромбоэмболическое осложнение может представлять собой артериальные кардиоваскулярные тромбоэмболические осложнения, артериальный тромбоз, венозные кардиоваскулярные тромбоэмболические осложнения и тромбоэмболические осложнения в камерах сердца, в том числе нестабильную стенокардию, острый коронарный синдром, первичный инфаркт миокарда, рецидив инфаркта миокарда, ишемию (например, коронарную ишемию, внезапную ишемическую смерть или транзиторную ишемическую атаку), инсульт, атеросклероз, окклюзионное заболевание периферических артерий, венозную тромбоэмболию, венозный тромбоз, тромбоз глубоких вен, тромбофлебит, артериальную эмболию, тромбоз коронарных артерий, тромбоз церебральных артерий, церебральную эмболию, почечную эмболию, эмболию легочной артерии и тромбоз, являющийся результатом введения (a) искусственных клапанов или других имплантов, (b) полостных катетеров, (c) стентов, (d) сердечно-легочного шунтирования, (e) гемодиализа или (f) других процедур, при которых кровь подвергается воздействию искусственной поверхности, что способствует тромбозу.
В другом аспекте настоящее изобретение относится к способу профилактики тромбоэмболического осложнения у пациента. Способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). Тромбоэмболическое осложнение может представлять собой артериальные кардиоваскулярные тромбоэмболические осложнения, артериальный тромбоз, венозные кардиоваскулярные тромбоэмболические осложнения и тромбоэмболические осложнения в камерах сердца, включая нестабильную стенокардию, острый коронарный синдром, первичный инфаркт миокарда, рецидив инфаркта миокарда, ишемию (например, коронарную ишемию, внезапную ишемическую смерть или транзиторную ишемическую атаку), инсульт, атеросклероз, окклюзионное заболевание периферических артерий, венозную тромбоэмболию, венозный тромбоз, тромбоз глубоких вен, тромбофлебит, артериальную эмболию, тромбоз коронарных артерий, тромбоз церебральных артерий, церебральную эмболию, почечную эмболию, эмболию легочной артерии и тромбоз, являющийся результатом введения (a) искусственных клапанов или других имплантов, (b) полостных катетеров, (c) стентов, (d) сердечно-легочного шунтирования, (e) гемодиализа или (f) других процедур, при которых кровь подвергается воздействию искусственной поверхности, что способствует тромбозу.
В другом аспекте настоящее изобретение относится к способу снижения риска тромбоэмболического осложнения у пациента. Способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). Тромбоэмболическое осложнение может представлять собой артериальные кардиоваскулярные тромбоэмболические осложнения, артериальный тромбоз, венозные кардиоваскулярные тромбоэмболические осложнения и тромбоэмболические осложнения в камерах сердца, включая нестабильную стенокардию, острый коронарный синдром, первичный инфаркт миокарда, рецидив инфаркта миокарда, ишемию (например, коронарную ишемию, внезапную ишемическую смерть или транзиторную ишемическую атаку), инсульт, атеросклероз, окклюзионное заболевание периферических артерий, венозную тромбоэмболию, венозный тромбоз, тромбоз глубоких вен, тромбофлебит, артериальную эмболию, тромбоз коронарных артерий, тромбоз церебральных артерий, церебральную эмболию, почечную эмболию, эмболию легочной артерии и тромбоз, являющийся результатом введения (a) искусственных клапанов или других имплантов, (b) полостных катетеров, (c) стентов, (d) сердечно-легочного шунтирования, (e) гемодиализа или (f) других процедур, при которых кровь подвергается воздействию искусственной поверхности, что способствует тромбозу.
В одном аспекте настоящее изобретение относится к способу лечения терминальной почечной недостаточностью у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В одном аспекте настоящее изобретение относится к способу профилактики терминальной почечной недостаточностью у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В одном аспекте настоящее изобретение относится к способу снижения риска терминальной почечной недостаточности у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В другом аспекте настоящее изобретение относится к способу лечения тромбоэмболического осложнения у пациента, нуждающегося в этом, причем способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)), где пациент подвергается воздействию искусственной поверхности. В некоторых вариантах осуществления искусственная поверхность контактирует с кровью пациента. В некоторых вариантах осуществления искусственная поверхность представляет собой экстракорпоральную поверхность. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность имплантируемого устройства, например механического клапана. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность диализного катетера. В некоторых вариантах осуществления искусственная поверхность представляет собой контур искусственного кровообращения. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность искусственного сердечного клапана. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность устройства для поддержания работы желудочков. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность трансплантата малого размера. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность центрального венозного катетера. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность аппарата экстракорпоральной мембранной оксигенации (ЭКМО). В некоторых вариантах осуществления искусственная поверхность вызывает тромбоэмболические осложнения или связана с ними. В некоторых вариантах осуществления тромбоэмболическое осложнение представляет собой венозную тромбоэмболию. В некоторых вариантах осуществления тромбоэмболическое осложнение представляет собой тромбоз глубоких вен. В некоторых вариантах осуществления тромбоэмболическое осложнение представляет собой эмболию легочной артерии.
В другом аспекте настоящее изобретение относится к способу снижения риска тромбоэмболического осложнения у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)), где пациент подвергается воздействию искусственной поверхности. В некоторых вариантах осуществления искусственная поверхность контактирует с кровью пациента. В некоторых вариантах осуществления искусственная поверхность представляет собой экстракорпоральную поверхность. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность имплантируемого устройства, например механического клапана. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность диализного катетера. В некоторых вариантах осуществления искусственная поверхность представляет собой контур искусственного кровообращения. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность искусственного сердечного клапана. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность устройства для поддержания работы желудочков. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность трансплантата малого размера. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность центрального венозного катетера. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность аппарата экстракорпоральной мембранной оксигенации (ЭКМО). В некоторых вариантах осуществления искусственная поверхность вызывает тромбоэмболические осложнения или связана с ними. В некоторых вариантах осуществления тромбоэмболическое осложнение представляет собой венозную тромбоэмболию. В некоторых вариантах осуществления тромбоэмболическое осложнение представляет собой тромбоз глубоких вен. В некоторых вариантах осуществления тромбоэмболическое осложнение представляет собой эмболию легочной артерии.
В другом аспекте настоящее изобретение относится к способу профилактики тромбоэмболического осложнения у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)), где пациент подвергается воздействию искусственной поверхности. В некоторых вариантах осуществления искусственная поверхность контактирует с кровью пациента. В некоторых вариантах осуществления искусственная поверхность представляет собой экстракорпоральную поверхность. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность имплантируемого устройства, например механического клапана. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность диализного катетера. В некоторых вариантах осуществления искусственная поверхность представляет собой контур искусственного кровообращения. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность искусственного сердечного клапана. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность устройства для поддержания работы желудочков. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность трансплантата малого размера. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность центрального венозного катетера. В некоторых вариантах осуществления искусственная поверхность представляет собой поверхность аппарата экстракорпоральной мембранной оксигенации (ЭКМО). В некоторых вариантах осуществления искусственная поверхность вызывает тромбоэмболические осложнения или связана с ними. В некоторых вариантах осуществления тромбоэмболическое осложнение представляет собой венозную тромбоэмболию. В некоторых вариантах осуществления тромбоэмболическое осложнение представляет собой тромбоз глубоких вен. В некоторых вариантах осуществления тромбоэмболическое осложнение представляет собой эмболию легочной артерии.
В другом аспекте настоящее изобретение относится к способу лечения мерцательной аритмии у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления пациент также нуждается в диализе, например почечном диализе. В некоторых вариантах осуществления соединение по настоящему изобретению вводится пациенту в во время диализа. В некоторых вариантах осуществления соединение или его фармацевтически приемлемая соль или композиция вводится пациенту перед диализом или после него. В некоторых вариантах осуществления пациент страдает терминальной почечной недостаточностью. В некоторых вариантах осуществления пациент не нуждается в диализе, например почечном диализе. В некоторых вариантах осуществления пациент подвержен высокому риску кровотечения. В некоторых вариантах осуществления мерцательная аритмия связана с другим тромбоэмболическим осложнением, например кровяным сгустком (тромбом).
В другом аспекте настоящее изобретение относится к способу снижения риска мерцательной аритмии у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления у пациента высокий риск развития мерцательной аритмии. В некоторых вариантах осуществления пациент также нуждается в диализе, например почечном диализе. В некоторых вариантах осуществления соединение по настоящему изобретению вводится пациенту во время диализа. В некоторых вариантах осуществления соединение или его фармацевтически приемлемая соль или композиция вводится пациенту перед диализом или после него. В некоторых вариантах осуществления пациент страдает терминальной почечной недостаточностью. В некоторых вариантах осуществления пациент не нуждается в диализе, например почечном диализе. В некоторых вариантах осуществления пациент подвержен высокому риску кровотечения. В некоторых вариантах осуществления мерцательная аритмия связана с другим тромбоэмболическим осложнением, например кровяным сгустком.
В другом аспекте настоящее изобретение относится к способу профилактики мерцательной аритмии у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления у пациента высокий риск развития мерцательной аритмии. В некоторых вариантах осуществления пациент также нуждается в диализе, например почечном диализе. В некоторых вариантах осуществления соединение по настоящему изобретению вводится пациенту во время диализа. В некоторых вариантах осуществления соединение или его фармацевтически приемлемая соль или композиция вводится пациенту перед диализом или после него. В некоторых вариантах осуществления пациент страдает терминальной почечной недостаточностью. В некоторых вариантах осуществления пациент не нуждается в диализе, например почечном диализе. В некоторых вариантах осуществления пациент подвержен высокому риску кровотечения. В некоторых вариантах осуществления мерцательная аритмия связана с другим тромбоэмболическим осложнением, например кровяным сгустком.
В другом аспекте настоящее изобретение относится к способу лечения гепарин-индуцированной тромбоцитопении у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В другом аспекте настоящее изобретение относится к способу снижения риска гепарин-индуцированной тромбоцитопении у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В другом аспекте настоящее изобретение относится к способу профилактики гепарин-индуцированной тромбоцитопении у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В другом аспекте настоящее изобретение относится к способу лечения гепарин-индуцированной тромбоцитопении и тромбоза у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В другом аспекте настоящее изобретение относится к способу снижения риска гепарин-индуцированной тромбоцитопении и тромбоза у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В другом аспекте настоящее изобретение относится к способу профилактики гепарин-индуцированной тромбоцитопении и тромбоза у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В другом аспекте настоящее изобретение относится к способу профилактики томбоэмболического осложнения у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)), где пациент болен раком или проходит курс химиотерапии. В некоторых вариантах осуществления пациент одновременно получает химиотерапию. В некоторых вариантах осуществления у пациента имеется повышенный уровень лактатдегидрогеназы. В некоторых вариантах осуществления тромбоэмболическое осложнение представляет собой венозную тромбоэмболию. В некоторых вариантах осуществления тромбоэмболическое осложнение представляет собой тромбоз глубоких вен. В некоторых вариантах осуществления тромбоэмболическое осложнение представляет собой эмболию легочной артерии.
В другом аспекте настоящее изобретение относится к способу лечения тромботической микроангиопатии у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления тромботическая микроангиопатия представляет собой гемолитический уремический синдром (HUS). В некоторых вариантах осуществления тромботическая микроангиопатия представляет собой тромботическую тромбоцитопеническую пурпуру (TTP).
В другом аспекте настоящее изобретение относится к способу снижения риска тромботической микроангиопатии у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления тромботическая микроангиопатия представляет собой гемолитический уремический синдром (HUS). В некоторых вариантах осуществления тромботическая микроангиопатия представляет собой тромботическую тромбоцитопеническую пурпуру (TTP).
В другом аспекте настоящее изобретение относится к способу профилактики тромботической микроангиопатии у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления тромботическая микроангиопатия представляет собой гемолитический уремический синдром (HUS). В некоторых вариантах осуществления тромботическая микроангиопатия представляет собой тромботическую тромбоцитопеническую пурпуру (TTP).
В другом аспекте настоящее изобретение относится к способу профилактики рецидива ишемии у пациента, нуждающегося в этом, причем указанный способ включает введение пациенту эффективного количества соединения по настоящему изобретению (например, соединения формулы (II)) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)), где у пациента имеется острый коронарный синдром. В некоторых вариантах осуществления пациент страдает мерцательной аритмией. В некоторых вариантах осуществления пациент не страдал мерцательной аритмией. В другом аспекте настоящее изобретение относится к способу лечения пациента, идентифицированного как находящегося в группе риска, например высокого риска, инсульта или тромбоза, который снижает вероятность инсульта или тромбоза у пациента. В некоторых вариантах осуществления пациент также идентифицирован как находящийся в группе риска кровотечения (например, обильного кровотечения) или сепсиса. В некоторых вариантах осуществления лечение эффективно без склоности к кровотечению. В некоторых вариантах осуществления лечение является эффективным для поддержания проходимости инфузионных портов и линий. Кроме того, соединения по настоящему изобретению (например, соединения формулы (II)) могут применяться в лечении и предотвращении других заболеваний, в которых образование тромбина играет физиологическую роль. Например, тромбин содействует заболеваемости и смертности от хронических и дегенеративных заболеваний, таких как рак, артрит, атеросклероз, сосудистая деменция и болезнь Альцгеймера, благодаря его способности регулировать фунцию клеток различных типов посредством специфического расщепления и активации рецептора клеточной поверхности тромбина, митогенных эффектов, различных клеточных функций, таких как клеточная пролиферация, например аномальная пролиферация клеток сосудов, приводящая к рестенозу или ангиогенезу, высвобождение PDGF и синтез ДНК. Ингибирование фактора XIa эффективно блокирует генерацию тромбина и, следовательно, нейтрализует любые физиологические эффекты тромбина в отношении клеток различных типов. Типичные показания, рассмотренные выше, включают некоторые, но не все, потенциальные клинические ситуации, поддающиеся лечению ингибитором фактора XIa.
В другом аспекте настоящее изобретение относится к способу лечения пациента, у которого наблюдается отек (например, ангионевротический отек, в частности наследственный ангионевротический отек), включающему введение соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)) пациенту.
В другом аспекте настоящее изобретение относится к способу профилактики отека (например, ангионевротического аллергического отека, в частности наследственного ангионевротического отека) у пациента, включающему введение соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)) пациенту.
В другом аспекте настоящее изобретение относится к способу снижения риска отека (например, ангионевротического отека, в частности наследственного ангионевротического отека) у пациента, включающему введение соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)) пациенту.
В другом аспекте настоящее изобретение относится к способу ингибирования калликреина у пациента, включающему введение пациенту с отеком (например, ангионевротическим отеком, в частности наследственным ангионевротическим отеком) эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В другом аспекте настоящее изобретение относится к способу лечения тромбоэмболического последствия или осложнения у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления тромбоэмболическое последствие или осложнение связано с вмешательством в периферические кровеносные сосуды (например, конечностей), гемодиализом, катетерной абляцией, цереброваскулярным вмешательством, трансплантацией органа (например, печени), хирургической операцией (например, ортопедической операцией, операцией на легких, операцией на органах брюшной полости или операцией на сердце (например, операцией на открытом сердце)), транс-катетерной имплантацией аортального клапана, вмешательством большого диаметра, используемым для лечения аневризмы, чрескожным коронарным вмешательством или терапией гемофилии. В некоторых вариантах осуществления хирургическая операция представляет собой ортопедическую операцию, операцию на легких, операцию на органах брюшной полости или операцию на сердце.
В другом аспекте настоящее изобретение относится к способу профилактики тромбоэмболического последствия или осложнения у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления тромбоэмболическое последствие или осложнение связано с вмешательством в периферические кровеносные сосуды (например, конечностей), гемодиализом, катетерной абляцией, цереброваскулярным вмешательством, трансплантацией органа (например, печени), хирургической операцией (например, ортопедической операцией, операцией на легких, операцией на органах брюшной полости или операцией на сердце (например, операцией на открытом сердце)), транс-катетерной имплантацией аортального клапана, вмешательством большого диаметра, используемым для лечения аневризмы, чрескожным коронарным вмешательством или терапией гемофилии. В некоторых вариантах осуществления хирургическая операция представляет собой ортопедическую операцию, операцию на легких, операцию на органах брюшной полости или операцию на сердце.
В другом аспекте настоящее изобретение относится к способу снижения риска тромбоэмболического последствия или осложнения у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления тромбоэмболическое последствие или осложнение связано с вмешательством в периферические кровеносные сосуды (например, конечностей), гемодиализом, катетерной абляцией, цереброваскулярным вмешательством, трансплантацией органа (например, печени), хирургической операцией (например, ортопедической операцией, операцией на легкие, операцией на органах брюшной полости или операцией на сердце (например, операцией на открытом сердце)), транс-катетерной имплантацией аортального клапана, вмешательством большого диаметра, используемым для лечения аневризмы, чрескожным коронарным вмешательством или терапией гемофилии. В некоторых вариантах осуществления хирургическая операция представляет собой ортопедическую операцию, операцию на легких, операцию на органах брюшной полости или операцию на сердце.
В другом аспекте изобретение относится к способу лечения рестеноза после повреждения артерии у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления повреждение артерии происходит после стентирования черепной артерии.
В другом аспекте настоящее изобретение относится к способу профилактики рестеноза после повреждения артерии у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления повреждение артерии происходит после стентирования черепной артерии.
В другом аспекте настоящее изобретение относится к способу снижения риска рестеноза после повреждения артерии у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления повреждение артерии происходит после стентирования черепной артерии.
В другом аспекте настоящее изобретение относится к способу лечения тромбоза сосудов печени у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В другом аспекте настоящее изобретение относится к способу профилактики тромбоза сосудов печени у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В другом аспекте настоящее изобретение относится к способу снижения риска тромбоза сосудов печени у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В другом аспекте настоящее изобретение относится к способу лечения инфаркта миокарда без повышения ST-сегмента или инфаркта миокарда с повышением ST-сегмента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В другом аспекте настоящее изобретение относится к способу профилактики инфаркта миокарда без повышения ST-сегмента или инфаркта миокарда с повышением ST-сегмента у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В другом аспекте настоящее изобретение относится к способу снижения риска инфаркта миокарда без повышения ST-сегмента или инфаркта миокарда с повышением ST-сегмента у пациента, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)).
В другом аспекте настоящее изобретение относится к способу поддержания проходимости кровеносных сосудов, включающему введение пациенту эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли или композиции по настоящему изобретению (например, композиции, включающей соединение формулы (II)). В некоторых вариантах осуществления пациент страдает острой почечной недостаточностью. В некоторых вариантах осуществления пациент дополнительно получает непрерывную заместительную почечную терапию.
В некоторых из представленных выше вариантов осуществления соединение по настоящему изобретению или его композиция вводится перорально или парентерально. В некоторых вариантах осуществления соединение или его композиция вводится перорально. В некоторых вариантах осуществления соединение или его композиция вводится после прекращения использования пациентом перорального антикоагулянта прямого действия. В некоторых вариантах осуществления пациент использовал пероральный коагулянт примерно в течение 2,5 лет. В некоторых вариантах осуществления пациентом является млекопитающее, например человек.
ПОДРОБНОЕ ОПИСАНИЕ
Определения
Термин «алкил», сам по себе или как часть другого заместителя означает, если не заявлено иное, прямую или разветвленную цепь или циклический углеводородный радикал или их комбинацию, которые могут быть полностью насыщенными, моно- или полиненасыщенными и могут включать ди- и мультивалентные радикалы, содержащие указанное количество атомов углерода (т.е., C1-C10 означает от одного до десяти атомов углерода). Примеры насыщенных углеводородный радикалов включают, но без ограничения, такие группы, как метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, циклогексил, (циклогексил)метил, циклопропилметил, гомологи и изомеры, например, н-пентила, н-гексила, н-гептила, н-октила и т.п. Ненасыщенная алкильная группа представляет собой группу, содержащую одну или несколько двойных связей или тройных связей. Примеры ненасыщенных алкильных групп включают, но без ограничения, винил, 2-пропенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и их высшие гомологи и изомеры. Термин «алкил», если не указано иное, также включает производные алкила, определенные более подробно ниже, такие как «гетероалкил». Алкильные группы, которые ограничены углеводородными группами, называются «гомоалкил». Если не указано иное, каждый пример алкильной группы является необязательно замещенным, т.е. незамещенным («незамещенный алкил») или замещенным («замещенный алкил») одним или несколькими заместителями; например 1-5 заместителями, 1-3 заместителями или 1 заместителем. В некоторых вариантах осуществления алкильная группа представляет собой незамещенный C1-10 алкил (например, -CH3). В некоторых вариантах осуществления алкильная группа представляет собой замещенный C1-10 алкил. Обычные аббревиатуры включают Me (-CH3), Et (-CH2CH3), iPr (-CH(CH3)2), nPr (-CH2CH2CH3), n-Bu (-CH2CH2CH2CH3) или i-Bu (-CH2CH(CH3)2).
Термин «алкилен» сам по себе или как часть другого заместителя означает двухвалентный радикал, полученный из алкана, например, но без ограничения, -CH2CH2CH2CH2-, и дополнительно включает группы, описанные ниже как «гетероалкилен». Обычно алкильная (или алкиленовая)группа будет содержать от 1 до 24 атомов углерода, причем в настоящем изобретении группы, содержащие 10 или меньше атомов углерода являются предпочтительными. «Низший алкил» или «низший алкилен» представляет собой алкильную или алкиленовую группу с более короткой цепью, обычно содержащую восемь или меньшее количество или атомов углерода.
Термин «алкенил» относится к прямой или разветвленной углеводородной цепи, содержащей 2-12 атомов углерода (если не указано другое) и включающей одну или несколько двойных связей. Примеры алкенильных групп включают, но без ограничения, аллильную, пропенильную, 2-бутенильную, 3-гексенильную и 3-октенильную группы. Один из атомов углерода двойной связи может необязательно быть точкой присоединения алкенильного заместителя.
Термин «алкенилен» относится к двухвалентному алкенилу, например -CH=CH-, -CH2-CH=CH- и -CH=CH-CH2-.
Термин «алкинил» относится к прямой или разветвленной углеводородной смеси, содержащей 2-12 атомов углерода (если не указано другое) и отличающейся наличиет одной или нескольких тройных связей. Примеры алкинильных групп включают, но без ограничения, этинил, пропаргил и 3-гексинил. Один из атомов углерода тройной связи необязательно может точкой присоединения алкинильного заместителя.
Термин «алкинилeн» относится к двухвалентному алкинилу, например -CH≡CH-, -CH2-CH≡CH- и -CH≡CH-CH2-.
Термины «алкокси», «алкиламино» и «алкилтио» (или «тиоалкокси») используются в их обычном значении и относятся к алкильным группам, присоединенным к остальной части молекулы через атом кислорода, аминогруппу или атом серы, соответственно.
Термины «циано» и «нитрил» относятся к радикалу -CN.
Термины «циклоалкил», «гетероциклоалкил» или «гетероциклил», сами по себе или в сочетании с другими терминами, означают, если не указано иное, циклические варианты «алкила» и «гетероалкила», соответственно. Кроме того, в гетероциклоалкиле или гетероциклиле гетероатом может занимать положение, в котором гетероцикл присоединен к остальной части молекулы. Примеры циклоалкила включают, но без ограничения, циклопропил, циклобутил, циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил, циклооктанил и т.п. Примеры гетероциклоалкила и гетероциклила включают, но без ограничения, 1-1,2,5,6-тетрагидропиридил, 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, тетрагидротиен-2-ил, тетрагидротиен-3-ил, 1-пиперазинил, 2-пиперазинил, 2-тетрагидропиранил, 3-тетрагидропиранил, 4-тетрагидропиранил, 1-метил-1,5-дигидро-2-имидазол-4-онил и т.п.
Термин «гетероалкил», когда используется в настоящем описании, относится к алкильной группе, которая определена в настоящем описании, дополнительно вклчающей 1 или несколько (например, 1, 2, 3 или 4) гетероатомов (например, кислорода, серы, азота, бора, кремния, фосфора) в основной цепи, причем или несколько гетероатомов вставлены между соседними атомами углерода в основной углеродной цепи и/или один или несколько гетероатомов вставлены между атомом углерода и основной молекулой, т.е. между точкой присоединения. В некоторых вариантах осуществления гетероалкильная группа относится к насыщенной группе, содержащей от 1 до 10 атомов углерода и 1, 2, 3 или 4 гетероатома («гетеро-C1-10 алкил»). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, содержащую от 1 до 9 атомов углерода и 1, 2, 3 или 4 гетероатома («гетеро-C1-9 алкил»). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, содержащую от 1 до 8 атомов углерода и 1, 2, 3 или 4 гетероатома («гетеро-C1-8 алкил»). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, содержащую от 1 до 7 атомов углерода и 1, 2, 3 или 4 гетероатома («гетеро-C1-7 алкил»). В некоторых вариантах осуществления гетероалкильная группа представляет собой группу, содержащую от 1 до 6 атомов углерода и 1, 2 или 3 гетероатома («гетеро-C1-6 алкил»). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группы, содержащую от 1 до 5 атомов углерода и 1 или 2 гетероатома («гетеро-C1-5 алкил»). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, содержащую 1 дo 4 атомов углерода и 1 или 2 гетероатома («гетеро-C1-4 алкил»). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, содержащую 1 до 3 атомов углерода и 1 гетероатом («гетеро-C1-3 алкил»). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, содержащую 1 или 2 атома углерода и 1 гетероатом («гетеро-C1-2 алкил»). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, содержащую 1 атом углерода и 1 гетероатом («гетеро-C1 алкил»). В некоторых вариантах осуществления гетероалкильная группа представляет собой насыщенную группу, содержащую от 2 до 6 атомов углерода и 1 или 2 гетероатома («гетеро-C2-6 алкил»). Если не указано иное, каждый пример гетероалкильной группы является независимо незамещенным («незамещенный гетероалкил») или замещенным («замещенный гетероалкил») одним или несколькими заместителями. В некоторых вариантах осуществления гетероалкильная группа представляет собой незамещенный гетеро-C1-10 алкил. В некоторых вариантах осуществления гетероалкильная группа представляет собой замещенный гетеро-C1-10 алкил.
Термин «гетероциклил», когда используется в сочетании с другими терминами (например, гетероциклилалкил), включает гетероциклильные кольца, которые определены выше. Таким образом, термин «гетероциклилалкил» включает радикалы, в которых гетероциклильная группа присоединена к алкильной группе, включая алкильные группы, в которых атом углерода (например, метиленовую группу) замещен атомом кислорода.
Термины «галоген», сам по себе или как часть другого заместителя, означает, если не указано иное, атом фтора, хлора, брома или йода.
Термин «галогеналкил», когда используется в настоящем описании, относится к алкильной группе, которая определена в настоящем описании, дополнительно включающей 1 или несколько (например, 1, 2, 3 или 4) атомов галогенов (например, фтора, хлора, брома или йода), где алкильная группа является замещенной одним или несколькими атомами галогенов. В некоторых вариантах осуществления галогеналкильная группа относится к насыщенной группе, содержащей от 1 до 10 атомов углерода и 1, 2, 3 или 4 атомов галогенов («галоген-C1-10 алкил»). Кроме того, термин «галогеналкил» включает моногалогеналкил и полигалогеналкил. Например, термин «галогеналкил» включает, но без ограничения, трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т.п.
Термины «галогеналкокси», когда используется в настоящем описании, относится к алкоксигруппе, которая определена в описании, дополнительно включающей 1 или несколько (например, 1, 2, 3 или 4) атомов галогенов (например, фтора, хлора, брома или йода), где алкоксигруппа является замещенной одним или несколькими атомами галогенов.
Термин «гидроксил» относится к радикалу -OH.
Термин «арил» означает, если не заявлено иное, полиненасыщенный ароматический углеводородный заместитель, который может представлять собой единственное кольцо или включать множество колец (предпочтительно от 1 до 3 колец), которые являются конденсированными или соединены ковалентно. Термин «гетероарил» относится к арильным группам (или кольцам), которые содержат от одного до четырех гетероатомов, выбранных из атомов N, O и S, где атомы азота и серы являются необязательно окисленными, и атом(ы) азота является(ются) необязательно кватернизированным(и). Гетероарильная группа может присоединяться к остальной части молекулы через гетероатом. Неограничивающие примеры арильной и гетероарильной групп включают фенил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 2-имидазолил, 4-имидазолил, 5-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 1,3,4-оксадиазолил, 1,2,3-триазолил, 1,2,4-триазолил, 3-изотиазолил, 4-изотиазолил, 5-изотиазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 1,3,4-тиадиазолил, 3-пиразолил, 4-пиразолил, 5-пиразолил, 2-тиенил, 3-тиенил, 2-фурил, 3-фурил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидил, 4-пиримидил, 5-бензотиазолил, пуринил, 2-бензимидазолил, 5-индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалинил, 5-хиноксалинил, 3-хинолил, 6-хинолил и тетразолил. Каждая из указанных выше арильных или гетероарильных кольцевых систем может быть замещенной, и заместители выбраны из группы приемлемых заместителей, описанной ниже. Примеры замещенного пиразолила включают, но без ограничения, 1-метил-3-пиразолил, 1-метил-4-пиразолил, 1-метил-5-пиразолил; примеры замещенного имидазолила включают, но без ограничения, 1-метил-2-имидазолил, 1-метил-4-имидазолил и 1-метил-5-имидазолил. Для краткости, термин «арил», когда используется в сочетании с другими терминами (например, арилокси, арилтио, арилалкил, аралкил, гетероаралкил), включает арильные и гетероарильные кольца, которые описаны выше. Таким образом, термины «арилалкил», «аралкил» и «гетероаралкил» включают радикалы, в которых арильная или гетероарильная группа присоединена к алкильной группе (например, бензил, фенэтил, пиридилметил и т.п.), включая алкильные группы, в которых атом углерода (например, метиленоую группу) замещен, например, атомом кислорода (например, феноксиметил, 2-пиридилоксиметил, 3-(1-нафтилокси)пропил, и т.п.).
Термин «нитро» относится к радикалу -NO2.
Термин «защитная группа», когда используется в настоящем описании, относится к части соединения, которая по существу стабильна в конкретных условиях реакции, но которая удаляется из соединения в других условиях реакции. Защитная группа также может выбираться таким образом, что она участвует в прямом окислении ароматического кольцевого компонента соединений по настоящему изобретению. Примеры защитных групп, применимых в соответствии с настоящим изобретением, представлены в монографии Greene et al., Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
Алкильная, алкенильная, алкинильная, циклоалкильная, гетероциклильная, арильная и гетероарильная группы, которые определены в настоящем описании, являются необязательно замещенными (например, «замещенная» или «незамещенная» алкильная, «замещенная» или «незамещенная» алкенильная, «замещенная» или «незамещенная» алкинильная, «замещенная» или «незамещенная» циклоалкильная, «замещенная» или «незамещенная» гетероциклильная, «замещенная» или «незамещенная» арильная или «замещенная» или «незамещенная» гетероарильная группа). Обычно термин «замещенный», независимо от того, предшествует ли ему термин «необязательно» или нет, означает, что по меньшей мере один атом водорода, присутствующий на группе (например, атоме углерода или азота), замещен допустимым заместителем, например заместителем, который приводит к полученю стабильного соединения, например соединения, которое не подвергается самопроизвольной трансформации, такой как перегруппировка, циклизация, отыщепление или другая реакция. Если не указано иное, в «замещенной» группе заместитель находится в одном или нескольких замещаемых положениях, и когда в любой данной группе более одного замещаемого положения, заместители в каждом положении одинаковые или разные. Подразумевается, что термин «замещенный» включает замещение всеми допустимыми заместителями органических соединений по настоящему изобретению, которое приводит к образованию стабильного соединения. Для целей настоящего изобретения гетероатомы, такие как атом азота, могут иметь водородные заместители и/или любой подходящий заместитель, как описано в настоящем документе, который удовлетворяет валентностям гетероатомов и приводит к образованию стабильного фтагмента.
Примеры заместителей атома углерода вкючают, но без ограничения, галоген, -CN, -NO2, -N3, -SO2H, -SO3H, -OH, -ORaa, -ON(Rbb)2, -N(Rbb)2, -N(Rbb)3+X-, -N(ORcc)Rbb, -SH, -SRaa, -SSRcc, -C(=O)Raa, -CO2H, -CHO, -C(ORcc)2, -CO2Raa, -OC(=O)Raa, -OCO2Raa, -C(=O)N(Rbb)2, -OC(=O)N(Rbb)2, -NRbbC(=O)Raa, -NRbbCO2Raa, -NRbbC(=O)N(Rbb)2, -C(=NRbb)Raa, -C(=NRbb)ORaa, -OC(=NRbb)Raa, -OC(=NRbb)ORaa, -C(=NRbb)N(Rbb)2, -OC(=NRbb)N(Rbb)2, -NRbbC(=NRbb)N(Rbb)2, -C(=O)NRbbSO2Raa, -NRbbSO2Raa, -SO2N(Rbb)2, -SO2Raa, -SO2ORaa, -OSO2Raa, -S(O)Raa-S(=O)Raa, -OS(=O)Raa, -Si(Raa)3, -Osi(Raa)3 -C(=S)N(Rbb)2, -C(=O)SRaa, -C(=S)SRaa, -SC(=S)SRaa, -SC(=O)SRaa, -OC(=O)SRaa, -SC(=O)ORaa, -SC(=O)Raa, -P(=O)2Raa, -OP(=O)2Raa, -P(=O)(Raa)2, -OP(=O)(Raa)2, -OP(=O)(ORcc)2, -P(=O)2N(Rbb)2, -OP(=O)2N(Rbb)2, -P(=O)(NRbb)2, -OP(=O)(NRbb)2, -NRbbP(=O)(ORcc)2, -NRbbP(=O)(NRbb)2, -P(Rcc)2, -P(Rcc)3, -OP(Rcc)2, -OP(Rcc)3, -B(Raa)2, -B(ORcc)2, -BRaa(ORcc), C1-10 алкил, C1-10 пергалогеналкил, C2-10 алкенил, C2-10 алкинил, C3-10циклоалкил, 3-14 членный гетероциклил, C6-14 арил и 5-14 членный гетероарил, где каждый алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил и гетероарил является независимо замещенным 0, 1, 2, 3, 4 или 5 группами Rdd;
каждый пример Raa независимо выбран из C1-10 алкила, C1-10 пергалогеналкила, C2-10 алкенила, C2-10 алкинила, C3-10циклоалкила, 3-14 членного гетероциклила, C6-14 арила и 5-14 членного гетероарила, или две группы Raa соединены с образованием 3-14 членного гетероциклильного или 5-14 членного гетероарилльного кольца, где каждый алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил и гетероарил является независимо замещенным 0, 1, 2, 3, 4 или 5 группами Rdd;
каждый пример Rbb независимо выбран из водорода, -OH, -ORaa, -N(Rcc)2, -CN, -C(=O)Raa, -C(=O)N(Rcc)2, -CO2Raa, -SO2Raa, -C(=NRcc)ORaa, -C(=NRcc)N(Rcc)2, -SO2N(Rcc)2, -SO2Rcc, -SO2ORcc, -SORaa, -C(=S)N(Rcc)2, -C(=O)SRcc, -C(=S)SRcc, -P(=O)2Raa, -P(=O)(Raa)2, -P(=O)2N(Rcc)2, -P(=O)(NRcc)2, C1-10 алкила, C1-10 пергалогеналкила, C2-10 алкенила, C2-10 алкинила, C3-10 циклоалкила, 3-14 членного гетероциклила, C6-14 арила и 5-14 членного гетероарила, или две группы Rbb соединены с образованием 3-14 членного гетероциклильного или 5-14 членного гетероарильного кольца, где каждый алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил и гетероарил является независимо замещенным 0, 1, 2, 3, 4 или 5 группами Rdd;
каждый пример Rcc независимо выбран из водорода, C1-10 алкила, C1-10 пергалогеналкила, C2-10 алкенила, C2-10 алкинила, C3-10 циклоалкила, 3-14 членного гетероциклила, C6-14 арила и 5-14 членного гетероарила, или две группы Rcc соединены с образованием 3-14 членного гетероциклильного или 5-14 членного гетероарильного кольца, где каждый алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил и гетероарил является независимо замещенным 0, 1, 2, 3, 4 или 5 группами Rdd;
каждый пример Rdd независимо выбран из галогена, -CN, -NO2, -N3, -SO2H, -SO3H, -OH, -ORee, -ON(Rff)2, -N(Rff)2, -N(Rff)3+X-, -N(ORee)Rff, -SH, -SRee, -SSRee, -C(=O)Ree, -CO2H, -CO2Ree, -OC(=O)Ree, -OCO2Ree, -C(=O)N(Rff)2, -OC(=O)N(Rff)2, -NRffC(=O)Ree, -NRffCO2Ree, -NRffC(=O)N(Rff)2, -C(=NRff)ORee, -OC(=NRff)Ree, -OC(=NRff)ORee, -C(=NRff)N(Rff)2, -OC(=NRff)N(Rff)2, -NRffC(=NRff)N(Rff)2,-NRffSO2Ree, -SO2N(Rff)2, -SO2Ree, -SO2ORee, -OSO2Ree, -S(=O)Ree, -Si(Ree)3, -Osi(Ree)3, -C(=S)N(Rff)2, -C(=O)SRee, -C(=S)SRee, -SC(=S)SRee, -P(=O)2Ree, -P(=O)(Ree)2, -OP(=O)(Ree)2, -OP(=O)(ORee)2, C1-6 алкила, C1-6 пергалогеналкила, C2-6 алкенила, C2-6 алкинила, C3-10циклоалкила, 3-10 членного гетероциклила, C6-10 арила, 5-10 членный гетероарила, где каждый алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил и гетероарил является независимо замещенным 0, 1, 2, 3, 4 или 5 группами Rgg, или два концевых заместителя Rdd могут соединяться с образованием =O или =S;
каждый пример Ree независимо выбран из C1-6 алкила, C1-6 пергалогеналкила, C2-6 алкенила, C2-6 алкинила, C3-10 циклоалкила, C6-10 арила, 3-10 членного гетероциклила и 3-10 членного гетероарила, где каждый алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил и гетероарил является независимо замещенным 0, 1, 2, 3, 4, или 5 группами Rgg;
каждый пример Rff независимо выбран из водорода, C1-6 алкила, C1-6 пергалогеналкила, C2-6 алкенила, C2-6 алкинила, C3-10 циклоалкила, 3-10 членного гетероциклила, C6-10 арила и 5-10 членного гетероарила, или две группы Rff соединены с образованием 3-14 членного гетероциклильного или 5-14 членного гетероарильного кольца, где каждый алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил и гетероарил является независимо замещенным 0, 1, 2, 3, 4 или 5 группами Rgg; и
каждый пример Rgg независимо представляет собой галоген, -CN, -NO2, -N3, -SO2H, -SO3H, -OH, -OC1-6 алкил, -ON(C1-6 алкил)2, -N(C1-6 алкил)2, -N(C1-6 алкил)3+X-, -NH(C1-6 алкил)2+X-, -NH2(C1-6 алкил) +X-, -NH3+X-, -N(OC1-6 алкил)(C1-6 алкил), -N(OH)(C1-6 алкил), -NH(OH), -SH, -SC1-6 алкил, -SS(C1-6 алкил), -C(=O)(C1-6 алкил), -CO2H, -CO2(C1-6 алкил), -OC(=O)(C1-6 алкил), -OCO2(C1-6 алкил), -C(=O)NH2, -C(=O)N(C1-6 алкил)2, -OC(=O)NH(C1-6 алкил), -NHC(=O)(C1-6 алкил), -N(C1-6 алкил)C(=O)(C1-6 алкил), -NHCO2(C1-6 алкил), -NHC(=O)N(C1-6 алкил)2, -NHC(=O)NH(C1-6 алкил), -NHC(=O)NH2, -C(=NH)O(C1-6 алкил),-OC(=NH)(C1-6 алкил), -OC(=NH)OC1-6 алкил, -C(=NH)N(C1-6 алкил)2, -C(=NH)NH(C1-6 алкил), -C(=NH)NH2, -OC(=NH)N(C1-6 алкил)2, -OC(NH)NH(C1-6 алкил), -OC(NH)NH2, -NHC(NH)N(C1-6 алкил)2, -NHC(=NH)NH2, -NHSO2(C1-6 алкил), -SO2N(C1-6 алкил)2, -SO2NH(C1-6 алкил), -SO2NH2,-SO2C1-6 алкил, -SO2OC1-6 алкил, -OSO2C1-6 алкил, -SOC1-6 алкил, -Si(C1-6 алкил)3, -OSi(C1-6 алкил)3 -C(=S)N(C1-6 алкил)2, C(=S)NH(C1-6 алкил), C(=S)NH2, -C(=O)S(C1-6 алкил), -C(=S)SC1-6 алкил, -SC(=S)SC1-6 алкил, -P(=O)2(C1-6 алкил), -P(=O)(C1-6 алкил)2, -OP(=O)(C1-6 алкил)2, -OP(=O)(OC1-6 алкил)2, C1-6 алкил, C1-6 пергалогеналкил, C2-6 алкенил, C2-6 алкинил, C3-10 циклоалкил, C6-10 арил, 3-10 членный гетероциклил, 5-10 членный гетероарил; или два концевых заместителя Rgg могут соединяться с образованием =O или =S; где X- представляет собой противоион.
Эти и другие типичные примеры заместителей более подробно описаны в Подробном описании изобретения, Примерах и Формуле изобретения.
Соединения
Соединения по настоящему изобретению представляют собой соединения, которые ингибируют фактор XIa или калликреин.
В одном аспекте соединение по настоящему изобретению представляет собой соединение формулы (II):
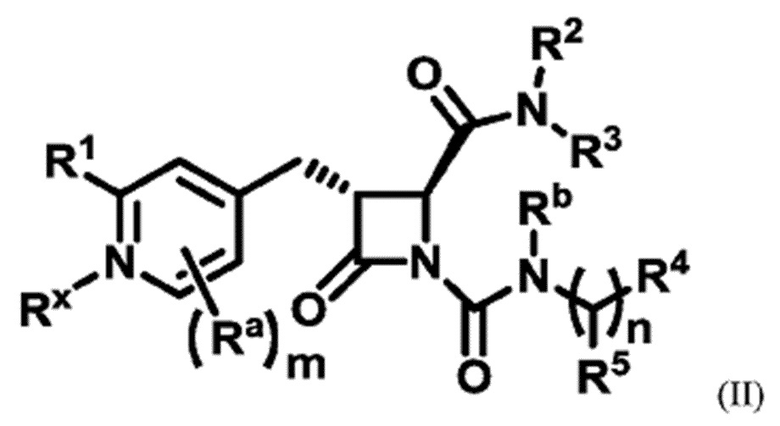
или его фармацевтически приемлемую соль, где R1 представляет собой водород или -NR8R9; Ra представляет собой C1-6 алкил, C1-6 галогеналкил, галоген, циано или -OR6; Rb представляет собой водород или C1-6 алкил; R2 представляет собой необязательно замещенный 5-членный гетероарил или необязательно замещенный 5-членный гетероциклил; R3 представляет собой водород, C1-6 алкил, или C1-6 галогеналкил; R4 представляет собой C1-6 алкил, C1-6 галогеналкил, циклоалкил, гетероциклил, арил или гетероарил, где циклоалкил, гетероциклил, арил или гетероарил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена, C1-6 алкила, C1-6 галогеналкила, циано или -OR6; R5 представляет собой водород, C1-6 алкил, C1-6 галогеналкил, циклоалкил или арил, где циклоалкил или арил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена, C1-6 алкила, C1-6 галогеналкила, циано или -OR6, или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют кольцо; R6 представляет собой водород, C1-6 алкил, или C1-6 галогеналкил; каждый R8 и R9 независимо представляет собой водород, C1-6 алкил, -C(O)R10, или -C(O)OR10; R10 представляет собой C1-6 алкил или C1-6 галогеналкил; Rx представляет собой -O или отсутствует, где когда Rx представляет собой -O, атом азота пиридильного кольца является положительно заряженным и Rx является отрицательно заряженным, образуя таким образом пиридин-N-оксид; m равно 0, 1, 2 или 3; и n равно 0 или 1, где если n равно 0, тогда R5 представляет собой водород и R4 отсутствует.
В одном аспекте настоящее изобретение относится к соединению формулы (II):

или его фармацевтически приемлемой соли, где R1 представляет собой водород или -NR8R9; Ra представляет собой C1-6 алкил, C1-6 галогеналкил, галоген, циано или -OR6; Rb представляет собой водород или C1-6 алкил; R2 представляет собой необязательно замещенный 5-членный гетероарил; R3 представляет собой водород, C1-6 алкил или C1-6 галогеналкил; R4 представляет собой C1-6 алкил, C1-6 галогеналкил, циклоалкил, гетероциклил, арил или гетероарил, где циклоалкил, гетероциклил, арил или гетероарил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена, C1-6 алкила, C1-6 галогеналкила, циано или -OR6; R5 представляет собой водород, C1-6 алкил, C1-6 галогеналкил, циклоалкил или арил, где циклоалкил или арил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена, C1-6 алкила, C1-6 галогеналкила, циано или -OR6, или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют кольцо; R6 представляет собой водород, C1-6 алкил или C1-6 галогеналкил; каждый R8 и R9 независимо представляет собой водород, C1-6 алкил, -C(O)R10 или -C(O)OR10; R10 представляет собой C1-6 алкил или C1-6 галогеналкил; m равно 0, 1, 2 или 3; и n равно 0 или 1, где если n равно 0, тогда R5 представляет собой водород и R4 отсутствует.
В некоторых вариантах осуществления Rx отсутствует.
В некоторых вариантах осуществления соединение представляет собой соединение формулы (I):
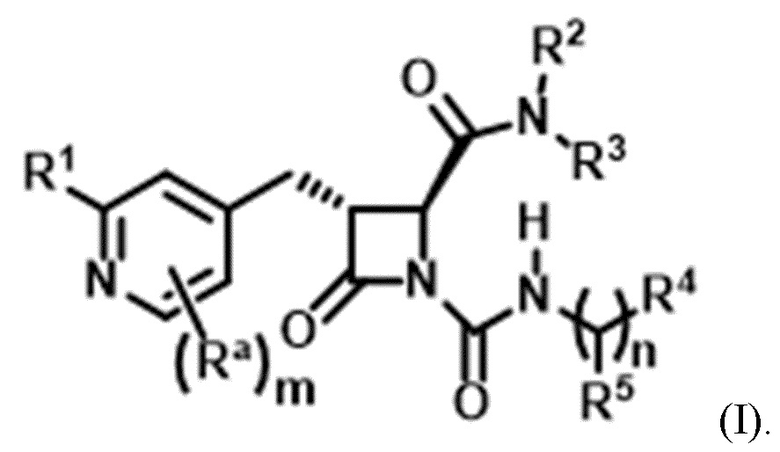
В некоторых вариантах осуществления R6 представляет собой C1-6 алкил или C1-6 галогеналкил. В некоторых вариантах осуществления R6 представляет собой -CH3 или -CF3.
В некоторых вариантах осуществления R1 представляет собой водород, -NH2 или -NHCH3. В некоторых вариантах осуществления R1 представляет собой водород. В некоторых вариантах осуществления R1 представляет собой -NH2. В некоторых вариантах осуществления R1 представляет собой -NHCH3.
В некоторых вариантах осуществления 5-членный гетероциклил представляет собой азот-содержащий 5-членный гетероциклил. В некоторых вариантах осуществления азот-содержащий 5-членный гетероциклил является насыщенным или ненасыщенным.
В некоторых вариантах осуществления R2 представляет собой 5-членный гетероциклил, необязательно замещенный одним, двумя, тремя или четырьмя независимыми заместителями, выбранными из C1-6 алкила, C2-6алкенила, C2-C6 алкинила, C1-6 алкоксила, C1-6 галогеналкила, C1-6 галогеналкоксила, галогена, циано, C3-8 карбоциклила, C3-8 гетероциклила, арила или гетероарила. В некоторых вариантах осуществления азот-содержащий 5-членный гетероциклил является необязательно замещенным оксо-группой (=O). В некоторых вариантах осуществления R2 представляет собой 5-членный гетероциклил, необязательно замещенный одним или двумя независимыми заместителями, выбранными из C1-6 алкила.
В некоторых вариантах осуществления R2 представляет собой 5-членный гетероарил, необязательно замещенный одним, двумя, тремя или четырьмя независимыми заместителями, выбранными из C1-6 алкила, C2-6 алкенила, C2-C6 алкинила, C1-6 алкоксила, C1-6 галогеналкила, C1-6 галогеналкоксила, галогена, циано, C3-8 карбоциклила, C3-8гетероциклила, арила или гетероарила.
В некоторых вариантах осуществления R2 представляет собой 5-членный гетероарил, необязательно замещенный одним или двумя независимыми заместителями, выбранными из C1-6 алкила. В некоторых вариантах осуществления R2 представляет собой пиразолил, имидазолил, тиенил, изотиазолил или тиазолил, где имидазолил, тиенил, изотиазолил или тиазолил является необязательно замещенным одним или двумя независимыми заместителями, выбранными из C1-6 алкила. В некоторых вариантах осуществления R2 представляет собой пиразолил, имидазолил, тиенил, изотиазолил, тиазолил, оксадиазолил или тиадиазолил, где имидазолил, тиенил, изотиазолил, тиазолил, оксадиазолил или тиадиазолил является необязательно замещенным одним или двумя независимыми заместителями, выбранными из C1-6 алкила. В некоторых вариантах осуществления R2 представляет собой пиразолил, имидазолил, тиенил, изотиазолил или тиазолил, где имидазолил, тиенил, изотиазолил или тиазолил является необязательно замещенным одним или двумя заместителями, выбранными из -CH3. В некоторых вариантах осуществления R2 представляет собой пиразолил, имидазолил, тиенил, изотиазолил, тиазолил, оксадиазолил или тиадиазолил, где имидазолил, тиенил, изотиазолил, тиазолил, оксадиазолил или тиадиазолил является необязательно замещенным одним или двумя заместителями, выбранными из -CH3.
В некоторых вариантах осуществления R2 представляет собой
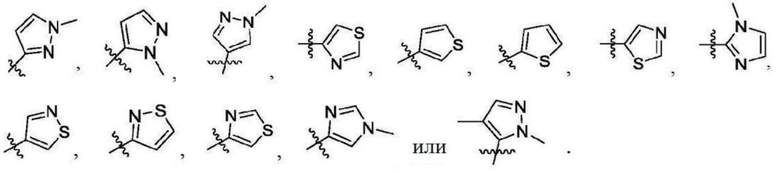
В некоторых вариантах осуществления R2 представляет собой
 .
.
В некоторых вариантах осуществления R2 представляет собой
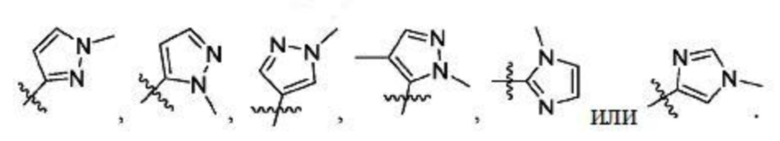
В некоторых вариантах осуществления R2 представляет собой
 В некоторых вариантах осуществления R2 представляет собой
В некоторых вариантах осуществления R2 представляет собой 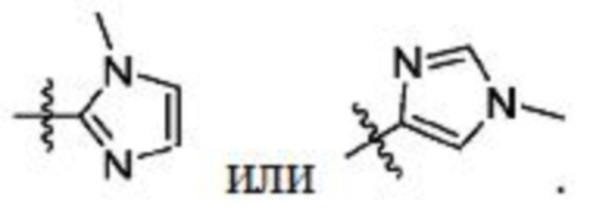
В некоторых вариантах осуществления R3 представляет собой C1-6 алкил. В некоторых вариантах осуществления R3 представляет собой -CH3.
В некоторых вариантах осуществления R4 представляет собой C1-6 алкил, циклоалкил, арил или гетероарил, где циклоалкил, арил или гетероарил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена или C1-6 алкил. В некоторых вариантах осуществления R4 представляет собой циклогексил, циклопропил, фенил, пиридил, пиразолил, тиенил или C1-6 алкил, где циклогексил, циклопропил, фенил, пиридил, пиразолил или тиенил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена или C1-6 алкила. В некоторых вариантах осуществления R4 представляет собой циклогексил, фенил, пиридил, пиразолил или тиенил, где циклогексил, фенил, пиридил, пиразолил или тиенил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена или C1-6 алкила. В некоторых вариантах осуществления R4 представляет собой фенил, необязательно замещенный одним, двумя или тремя заместителями, выбранными из -F, -Cl или C1-6 алкила.
В некоторых вариантах осуществления R4 представляет собой C1-6 алкил, C1-6 галогеналкил, циклоалкил, гетероциклил, арил или гетероарил, где циклоалкил, гетероциклил, арил или гетероарил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена, C1-6 алкила, C1-6 галогеналкила, циано или -OR6, и R5 представляет собой водород, C1-6 алкил, C1-6 галогеналкил, циклоалкил или арил, где циклоалкил или арил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена, C1-6 алкила, C1-6 галогеналкила или -OR6, или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют кольцо.
В некоторых вариантах осуществления R4 представляет собой C1-6 алкил, циклоалкил, гетероциклил, арил или гетероарил, где циклоалкил, арил или гетероарил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена или C1-6 алкила. В некоторых вариантах осуществления R4 представляет собой циклогексил, циклопропил, фенил, тетрагидропиранил, пиридил, пиразолил, тиенил или C1-6 алкил, где циклогексил, циклопропил, фенил, тетрагидропиранил, пиридил, пиразолил или тиенил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена или C1-6 алкила. В некоторых вариантах осуществления R4 представляет собой циклогексил, фенил, пиридил, пиразолил или тиенил, где циклогексил, фенил, пиридил, пиразолил или тиенил является необязательно замещенным одним, двумя или тремя независимыми заместителями, выбранными из галогена или C1-6 алкила. В некоторых вариантах осуществления R4 представляет собой фенил, необязательно замещенный одним, двумя или тремя заместителями, выбранными из -F, -Cl или C1-6 алкила.
В некоторых вариантах осуществления R5 представляет собой водород, -CH3, -CH2CH3, -CH2CH2CH3,-CH(CH3)2, -CH2CH2CH2CH3, -CF3 или незамещенный циклопропил. В некоторых вариантах осуществления R5 представляет собой -CH2CH3, -CF3 или незамещенный циклопропил.
В некоторых вариантах осуществления соединение представляет собой соединение формулы (I-a):
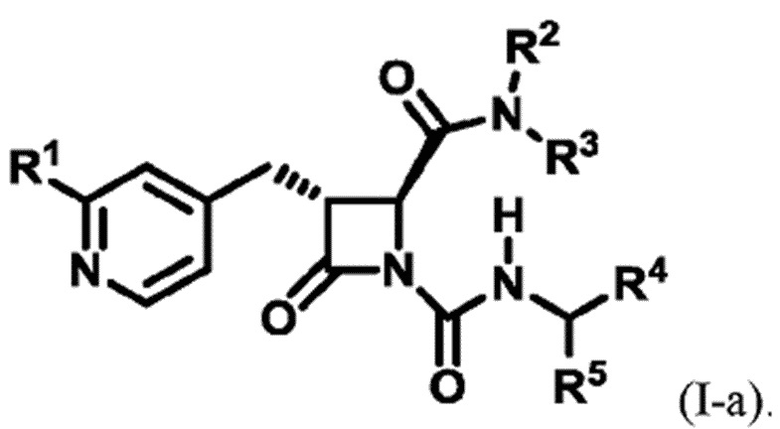
В некоторых вариантах осуществления соединение представляет собой соединение формулы (I-b):

В некоторых вариантах осуществления соединение представляет собой соединение формулы (I-c), формулы (I-d), формулы (I-e), формулы (I-f), формулы (I-g), формулы (I-i) или формулы (I-j):

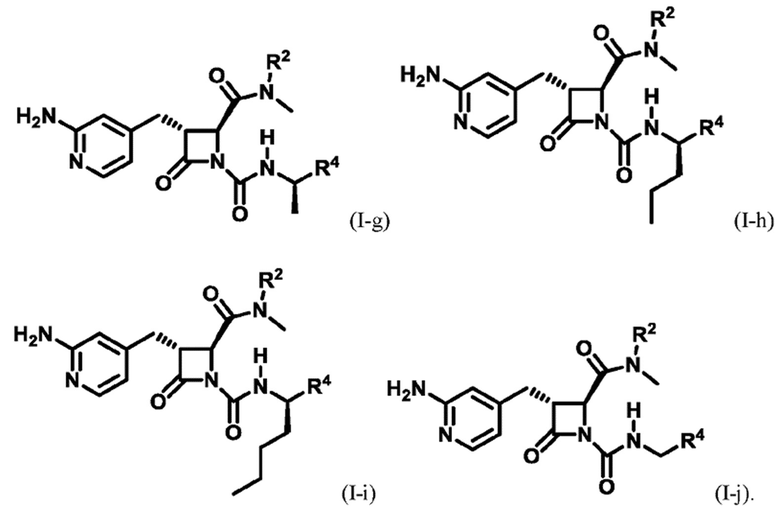
В некоторых вариантах осуществления соединение представляет собой соединение формулы (I-c):
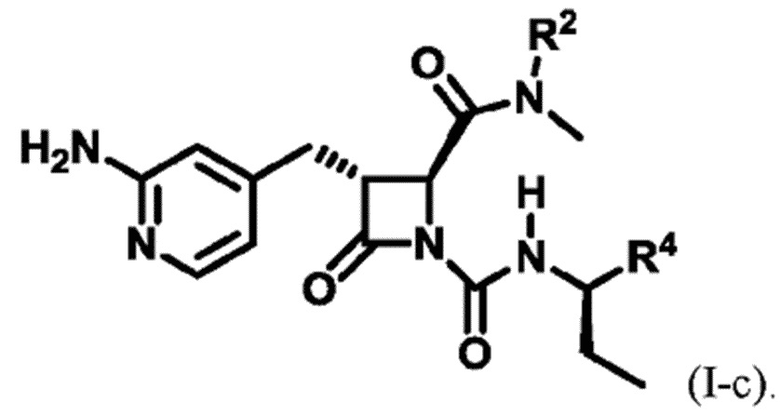
В некоторых вариантах осуществления соединение представляет собой соединение формулы (I-e), формулы (I-k), формулы (I-l), формулы (I-m), формулы (I-o), формулы (I-p), формулы (I-t), формулы (I-u) или формулы (I-v):

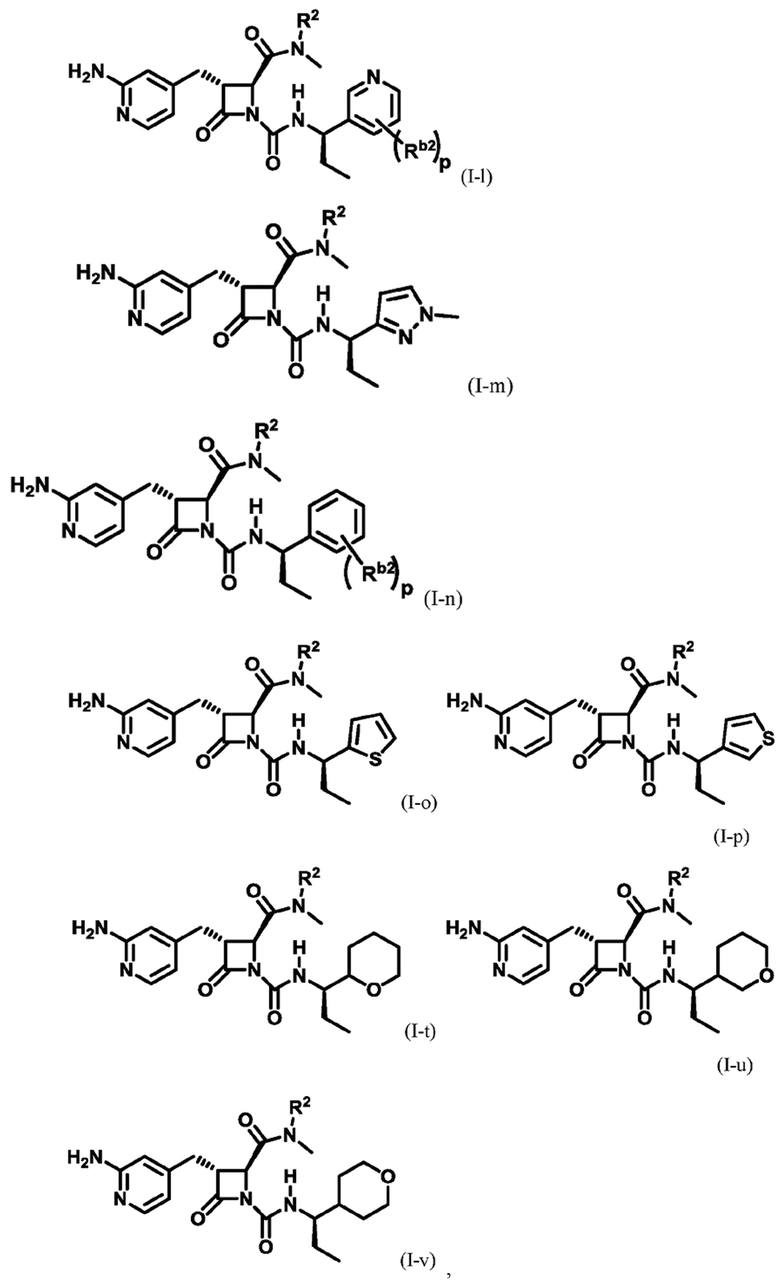
где каждый из Rb2 независимо представляет собой галоген, C1-6 алкил, C1-6 галогеналкил, циано или -OR6; и p равно 0, 1, 2 или 3.
В некоторых вариантах осуществления Ra представляет собой -C1-6 алкил. В некоторых вариантах осуществления Ra представляет собой -CH3. В некоторых вариантах осуществления Ra представляет собой -CH3 и m равно 1. В некоторых вариантах осуществления m равно 1. В некоторых вариантах осуществления Ra представляет собой галоген или -C1-6 алкил. В некоторых вариантах осуществления Ra представляет собой F или -CH3. В некоторых вариантах осуществления Rb представляет собой C1-6 алкил. В некоторых вариантах осуществления Rb представляет собой -CH3.
В некоторых вариантах осуществления соединение представляет собой соединение формулы (I-q):
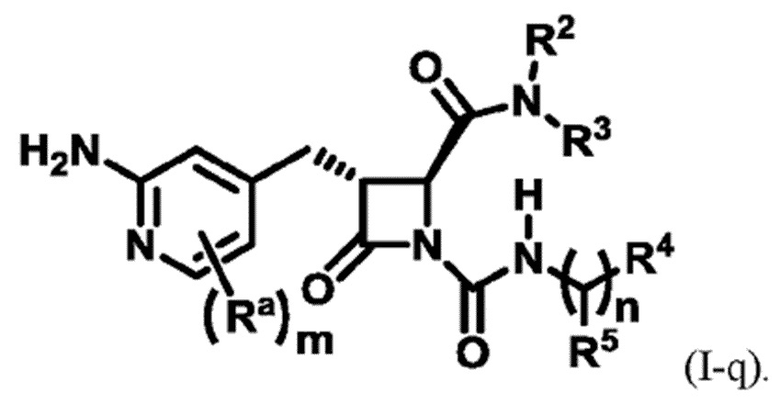
В некоторых вариантах осуществления соединение представляет собой соединение формулы (I-r):

В некоторых вариантах осуществления соединение представляет собой соединение формулы (I-s):
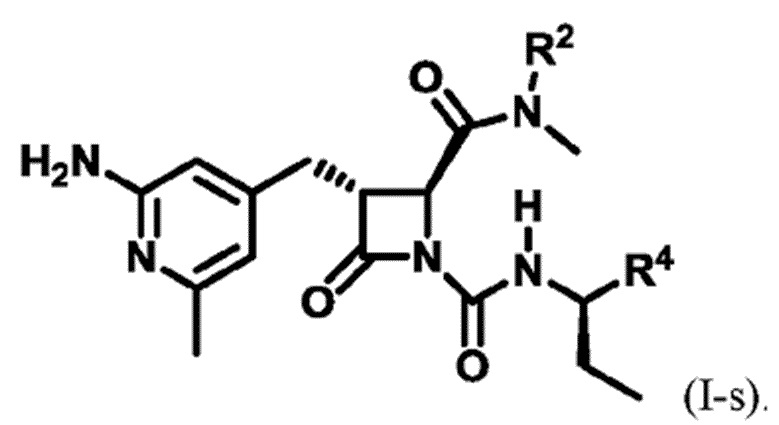
В некоторых вариантах осуществления соединение представляет собой соединение формулы (I-s1) или (I-s2):


где каждый из Rb2 независимо представляет собой галоген, C1-6 алкил, C1-6 галогеналкил, циано или -OR6; и p равно 0, 1, 2 или 3.
В некоторых вариантах осуществления Rb представляет собой C1-6 алкил. В некоторых вариантах осуществления Rb представляет собой -CH3.
Типичные примеры соединений включают, но без ограничения, соединения, представленные в таблице 1 ниже.
Таблица 1. Типичные примеры соединений


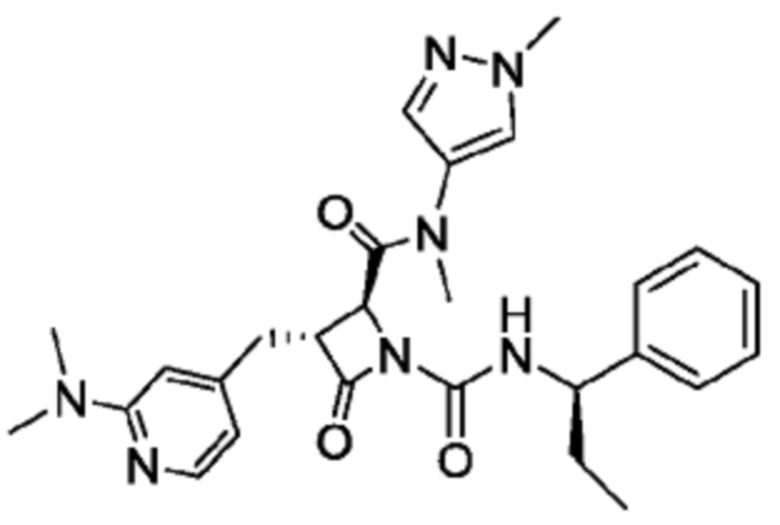




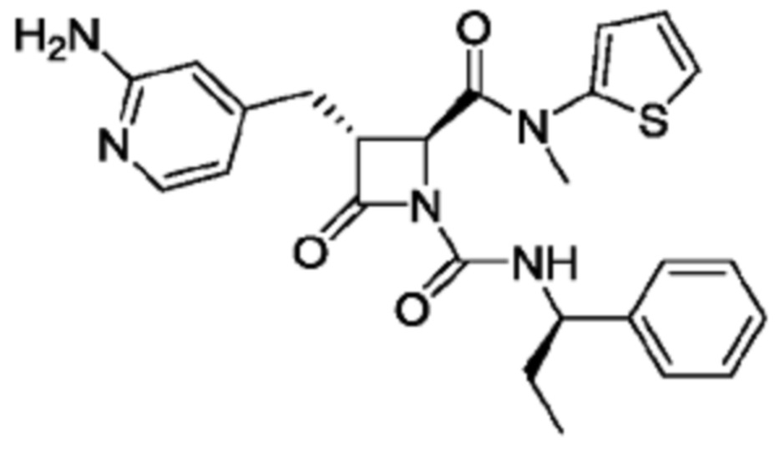
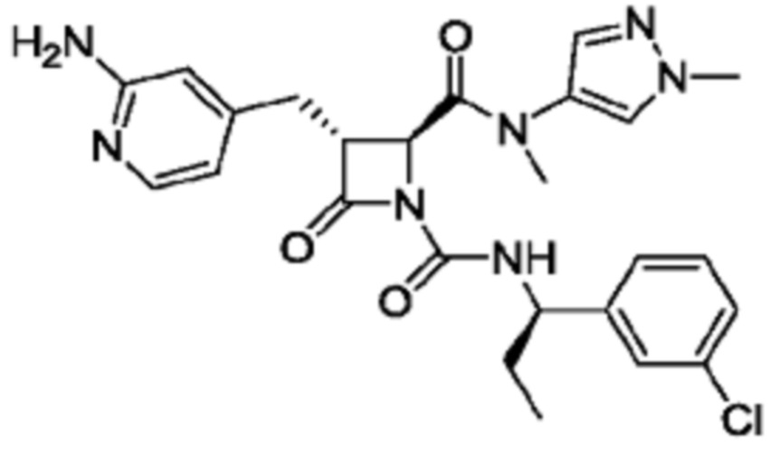
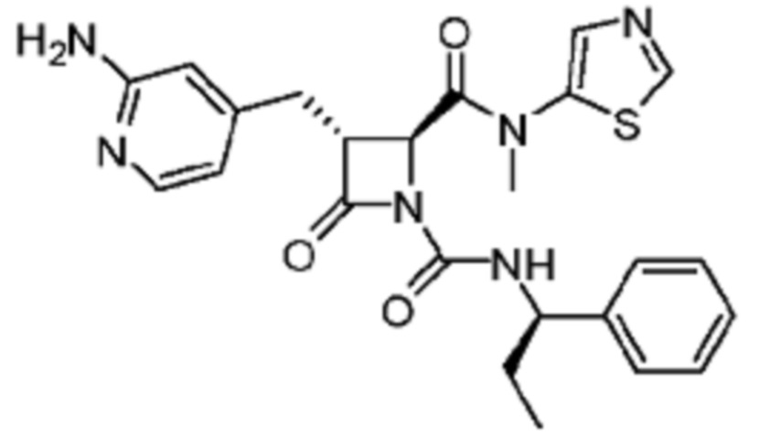
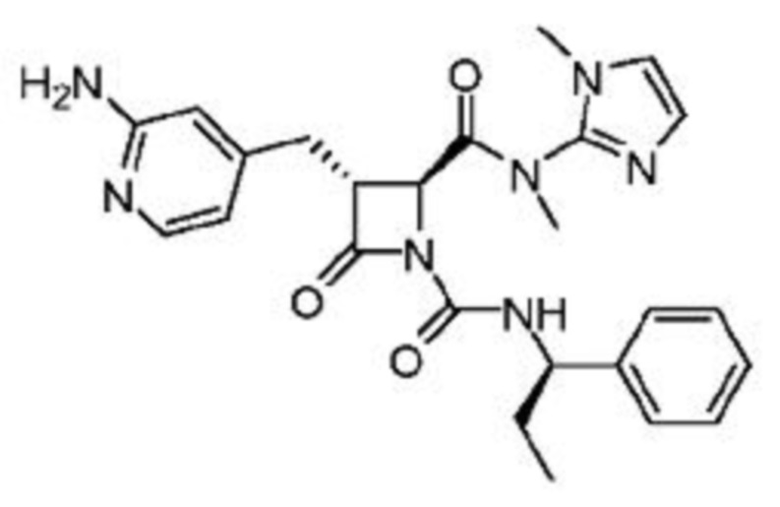

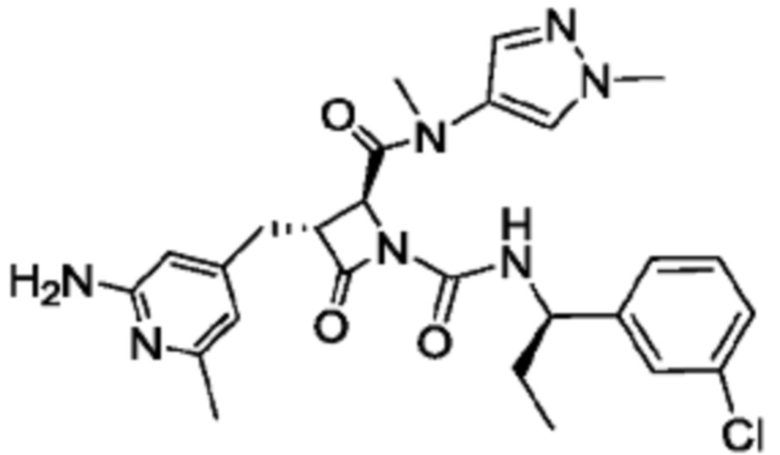

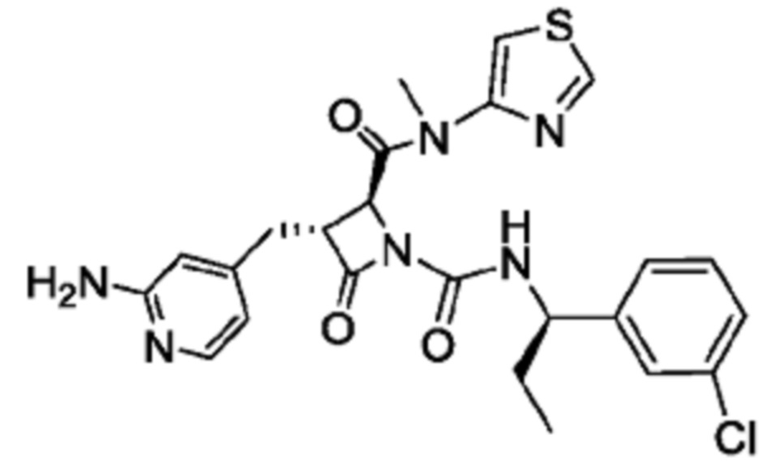


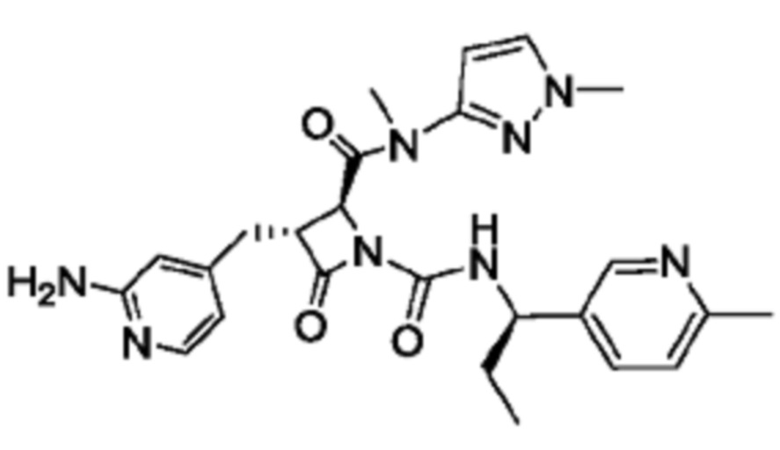
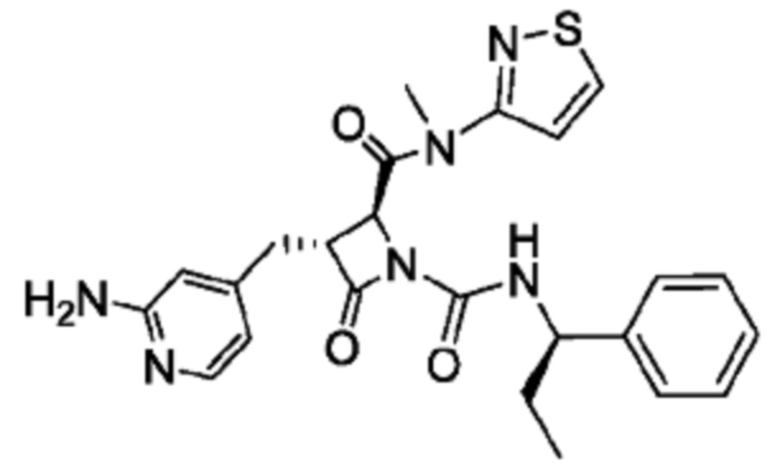


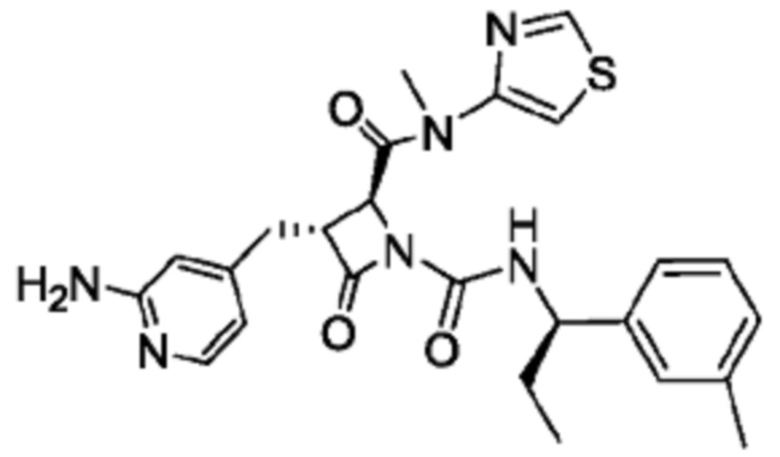



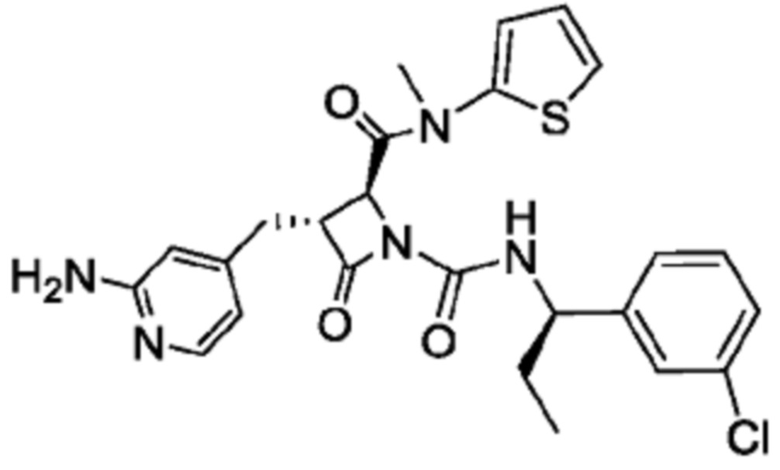






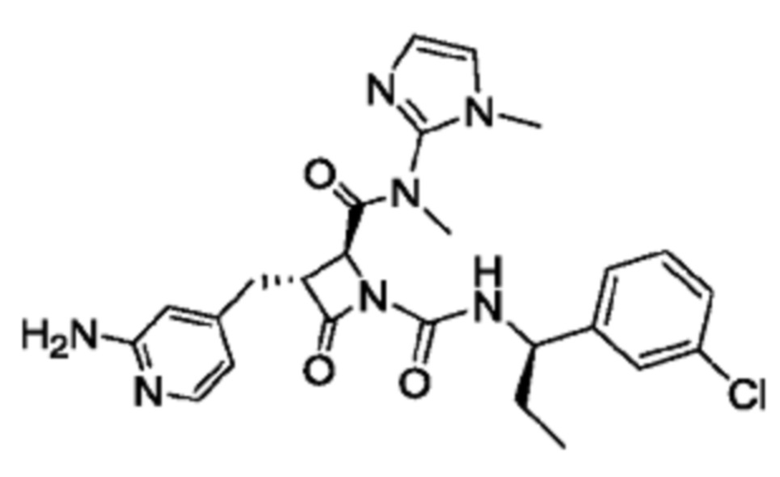


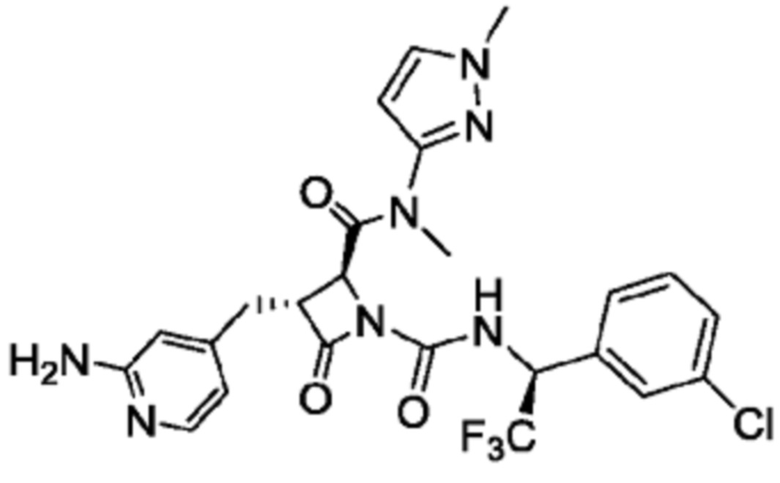

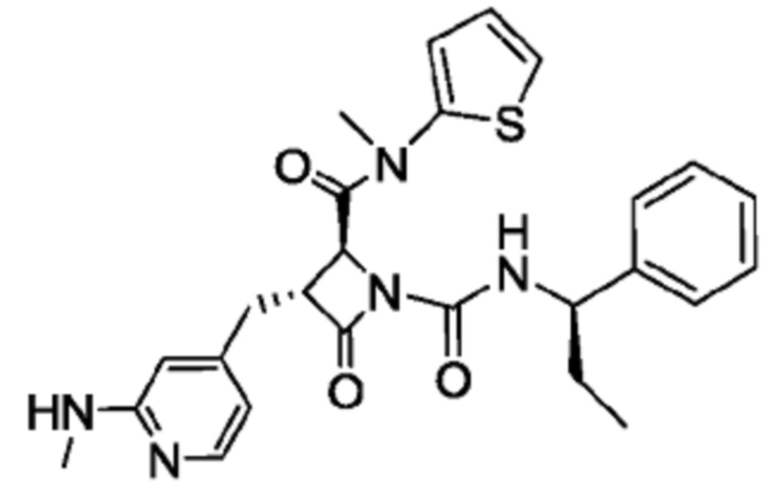

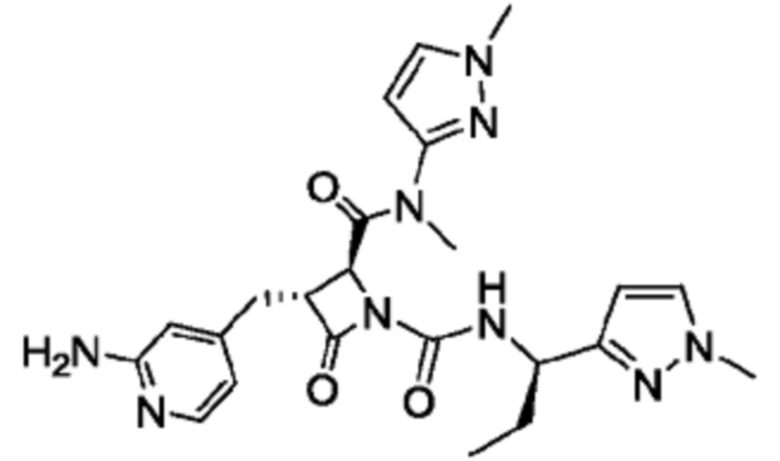



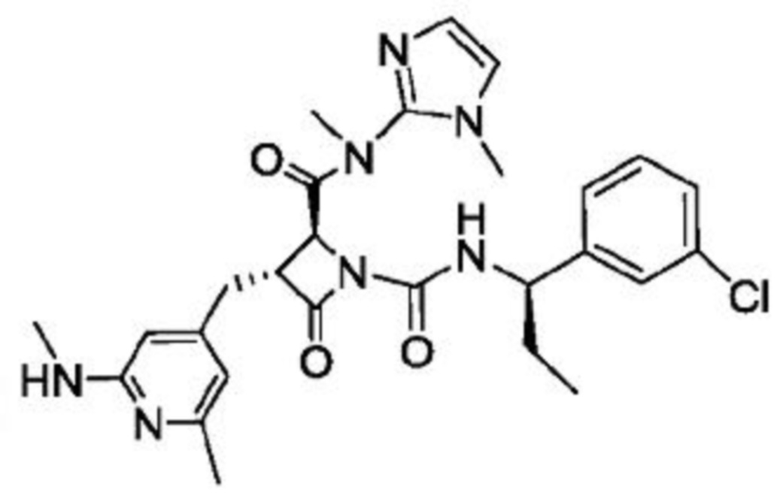
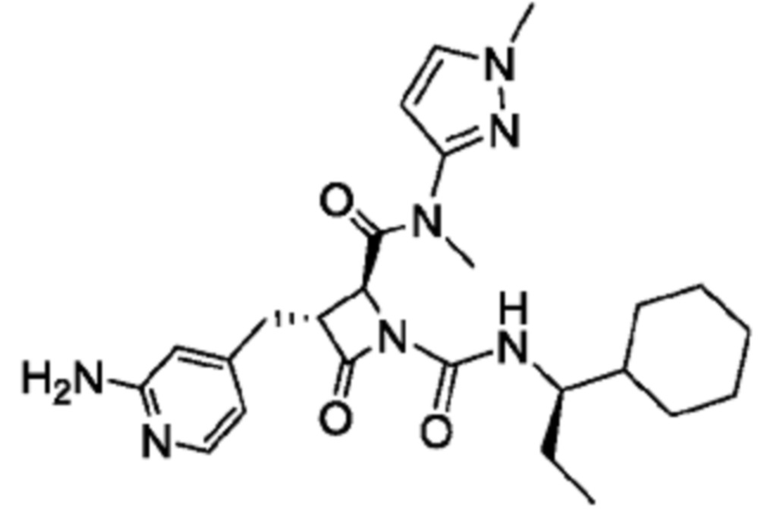

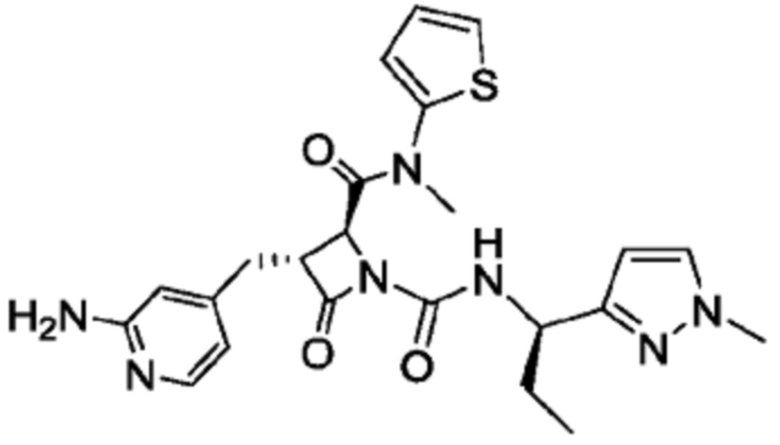
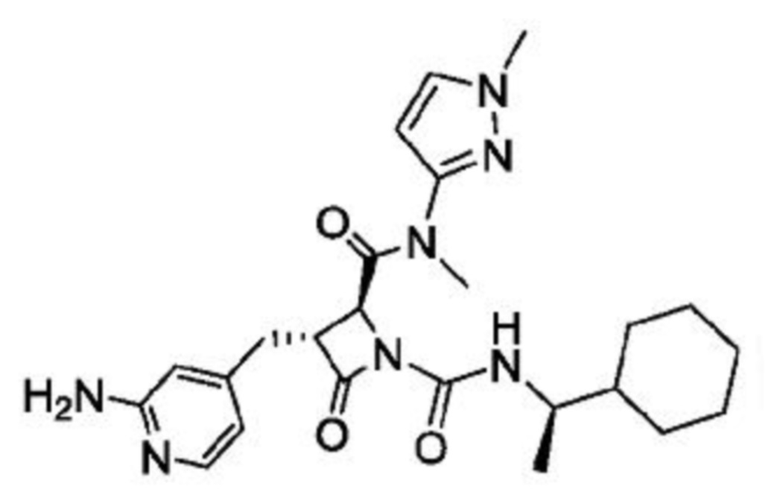

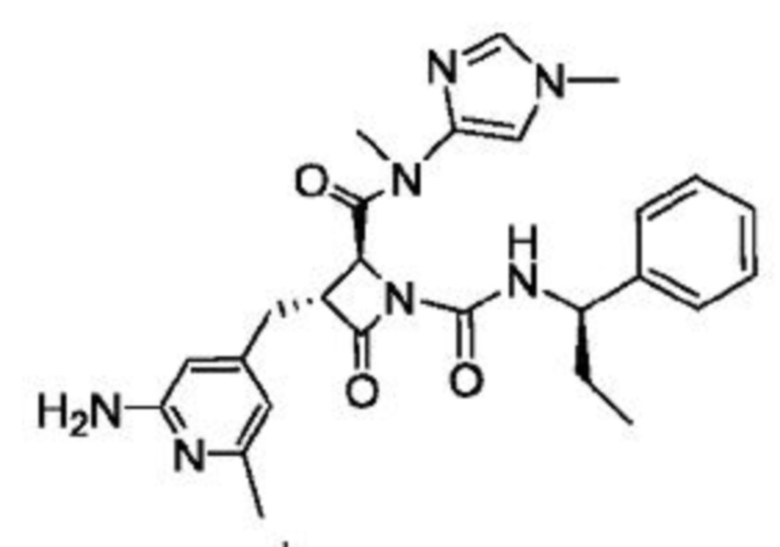

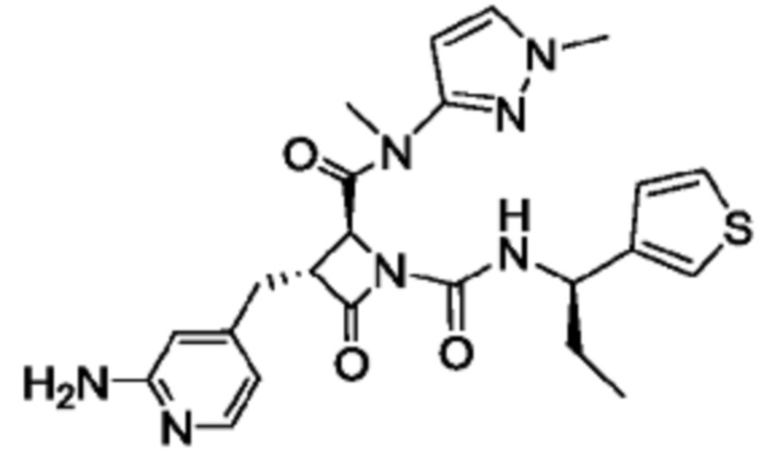
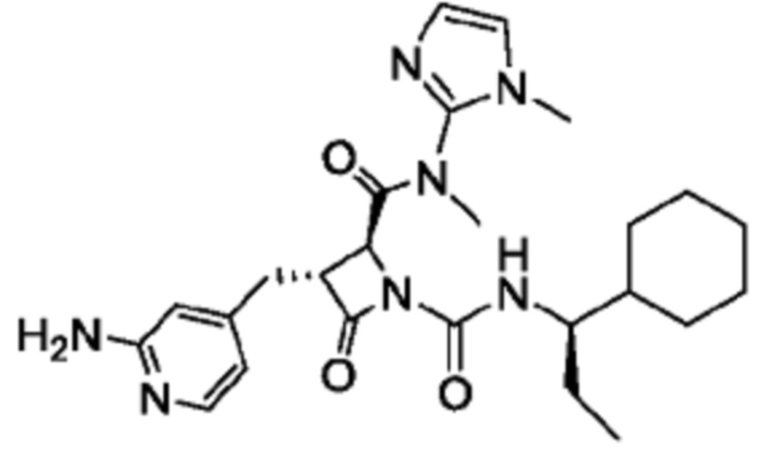

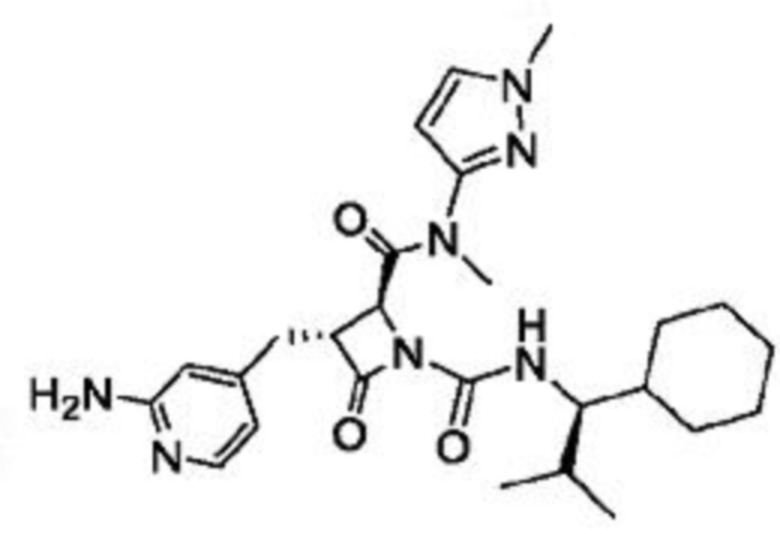
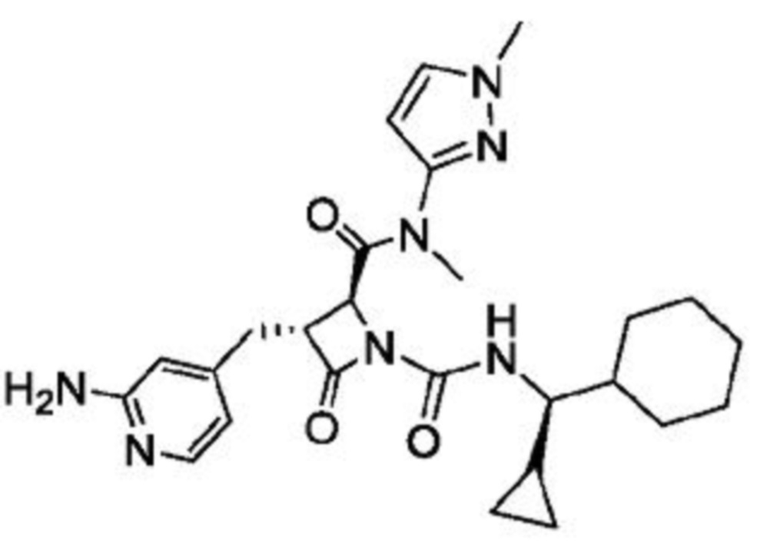



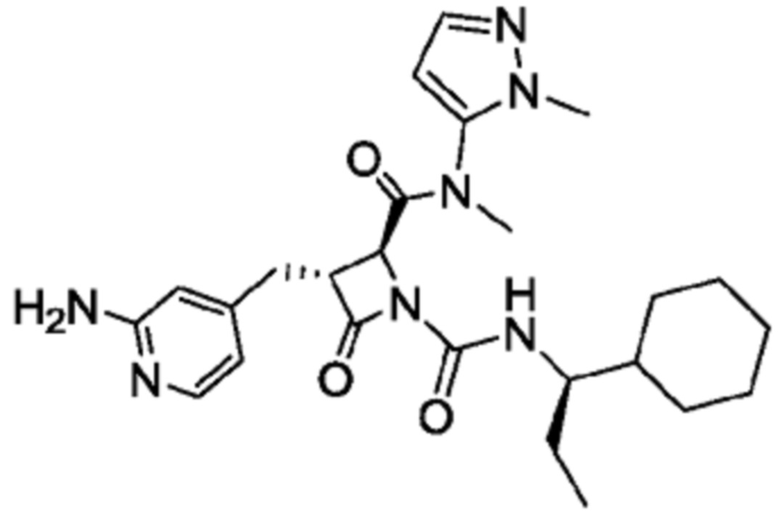






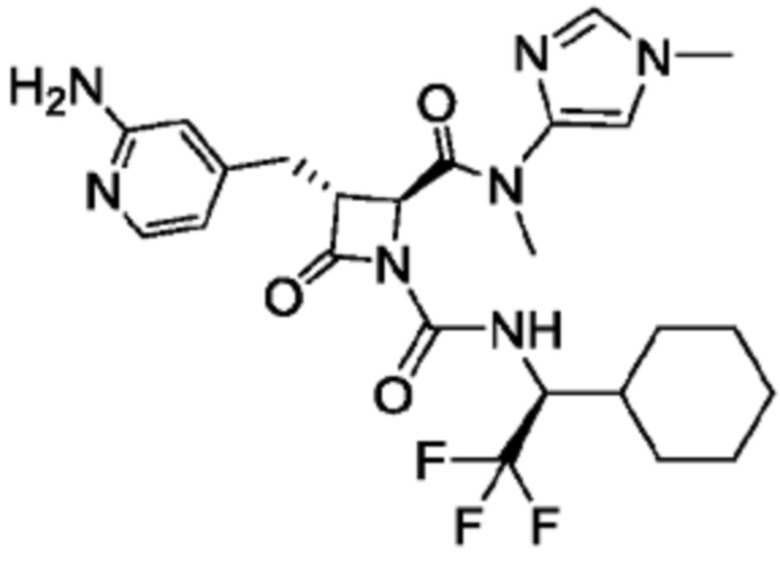
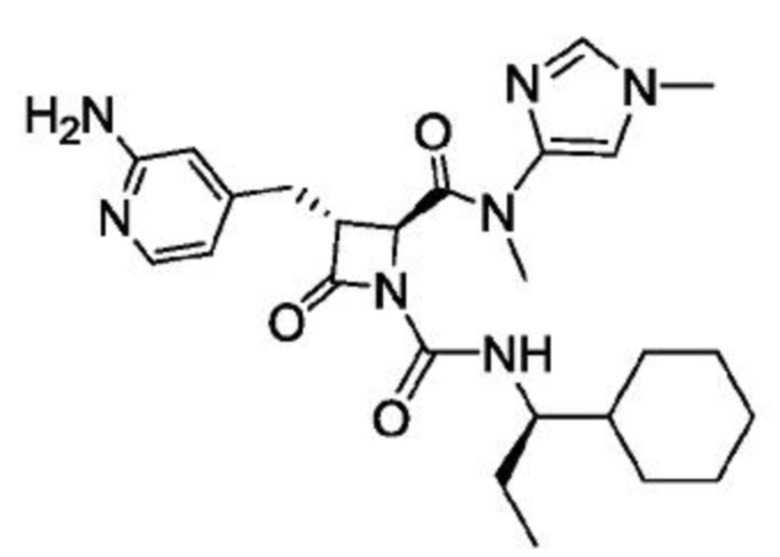

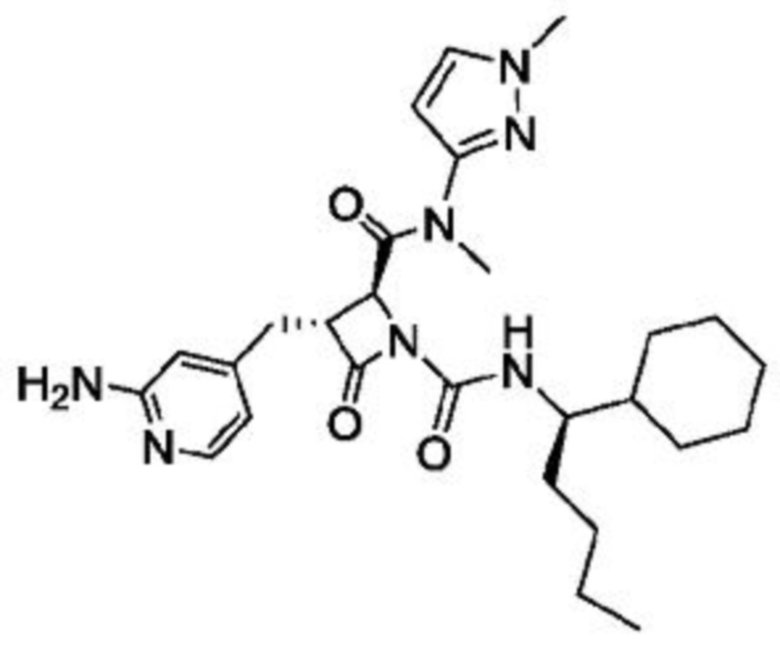




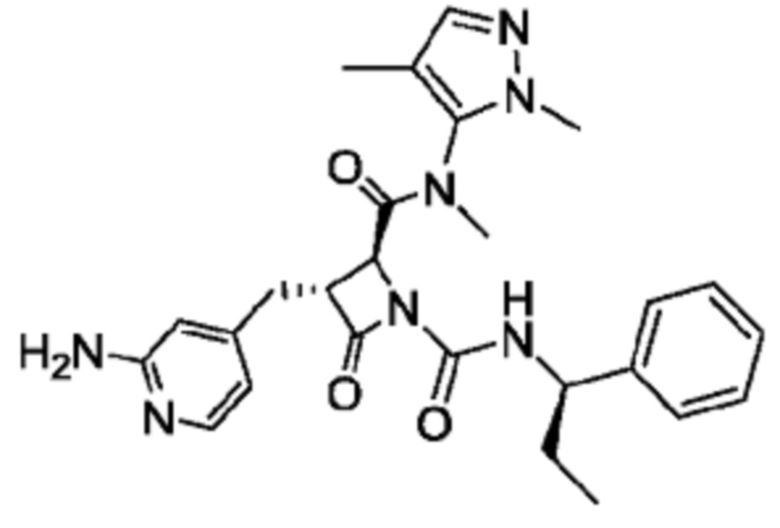






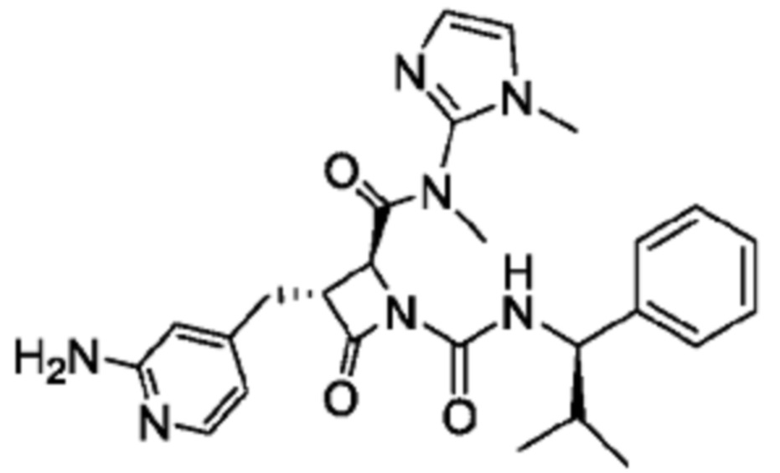
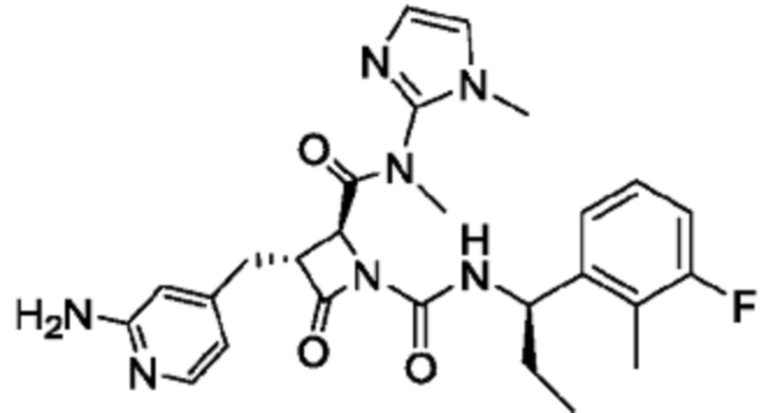

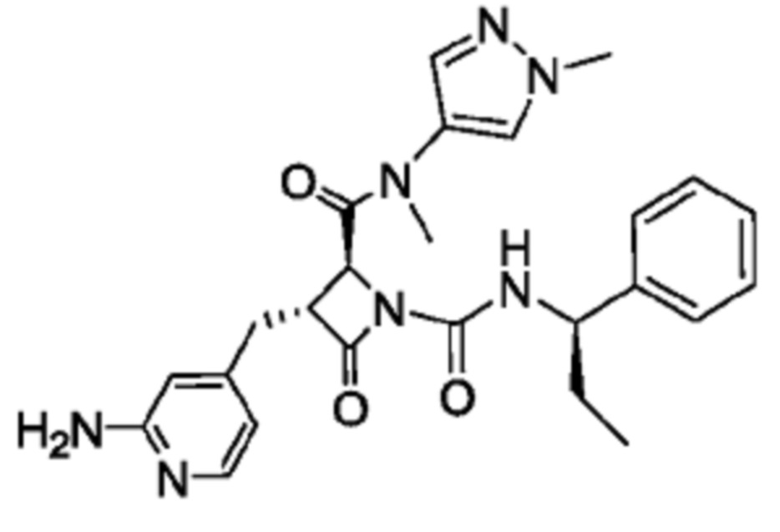

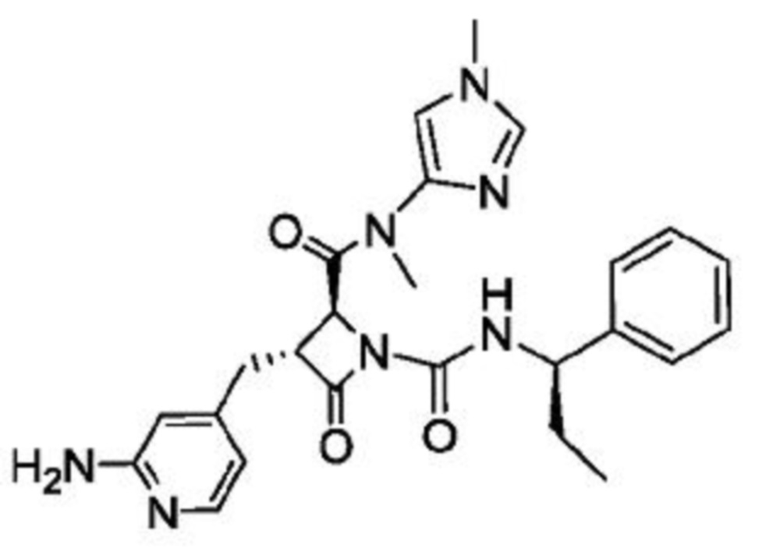

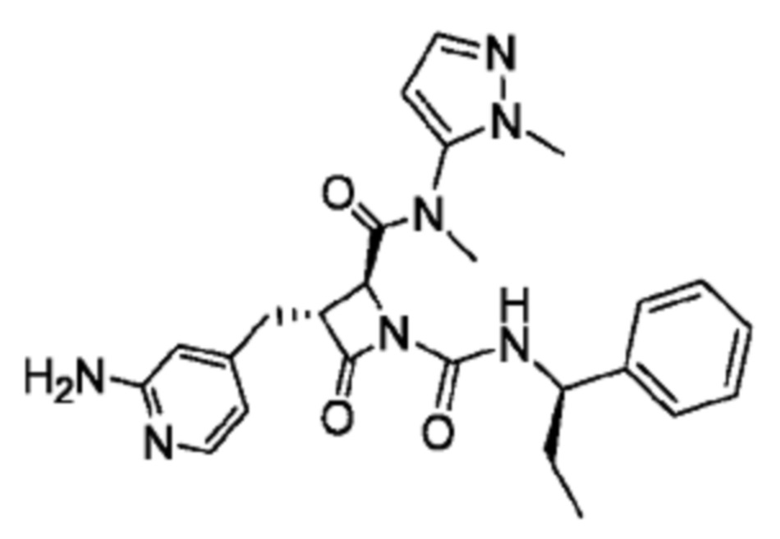
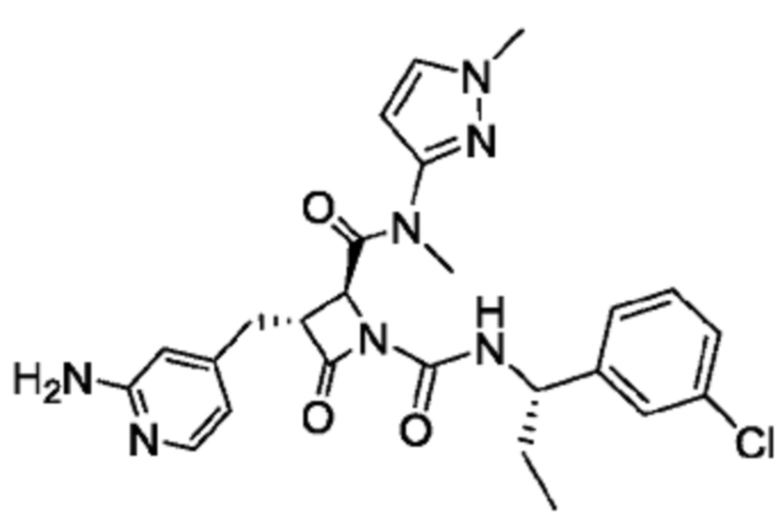



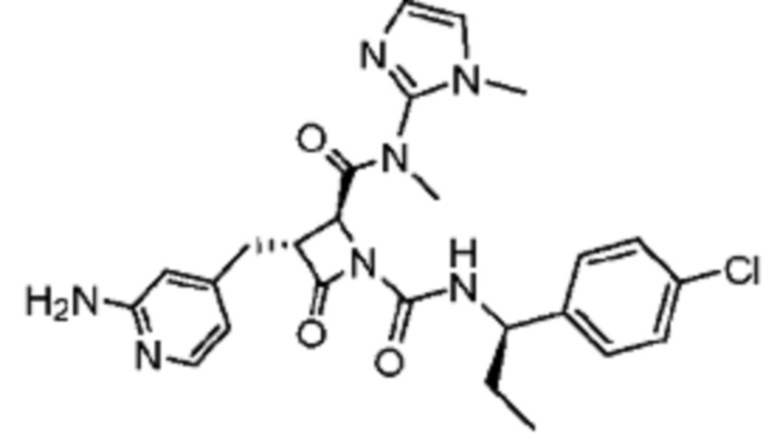





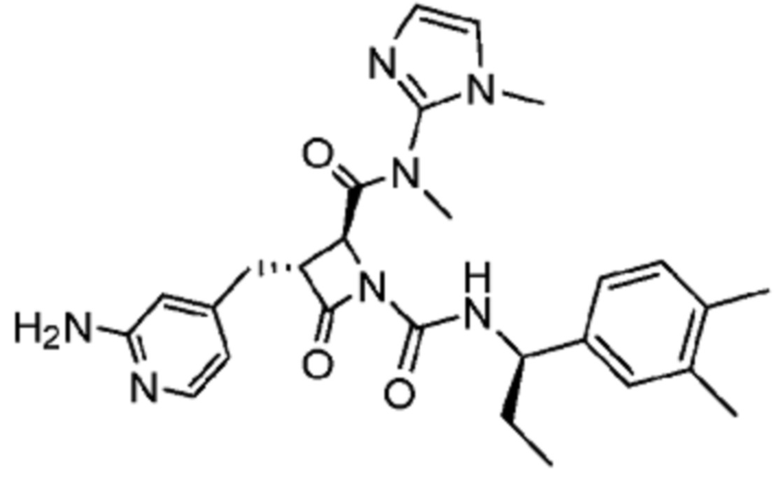





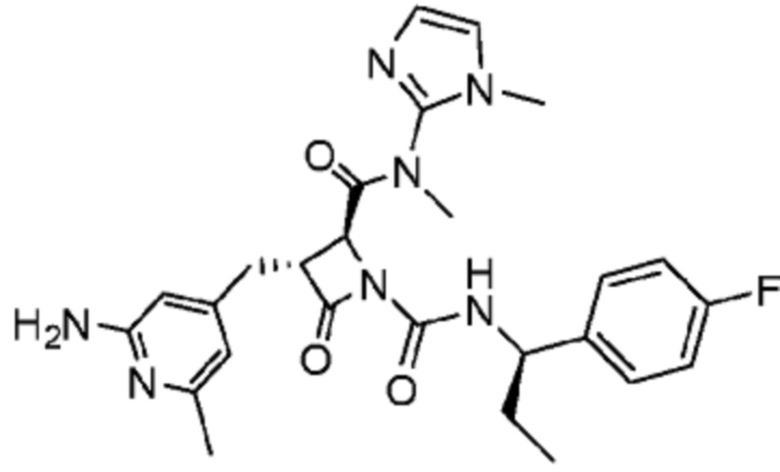
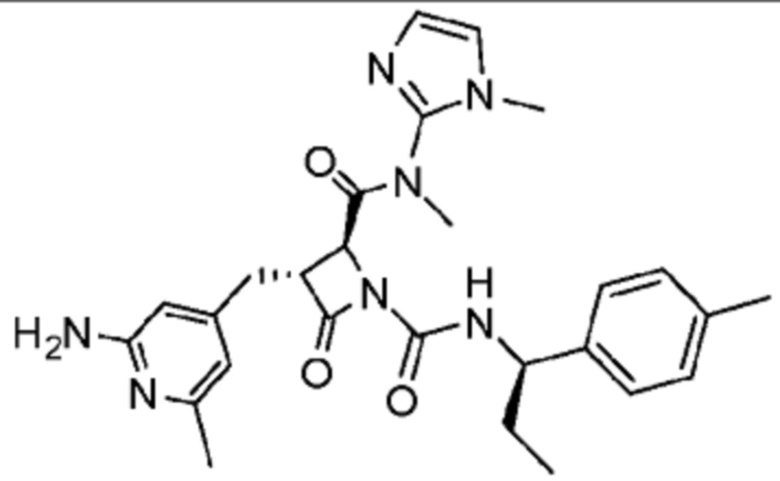
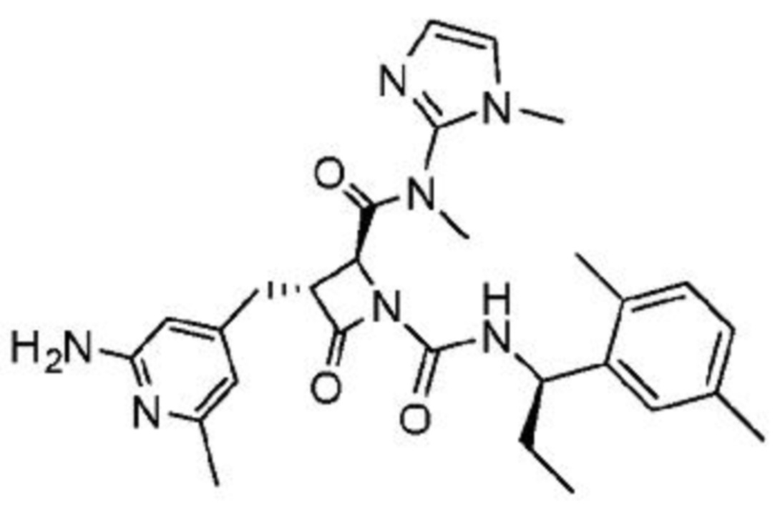






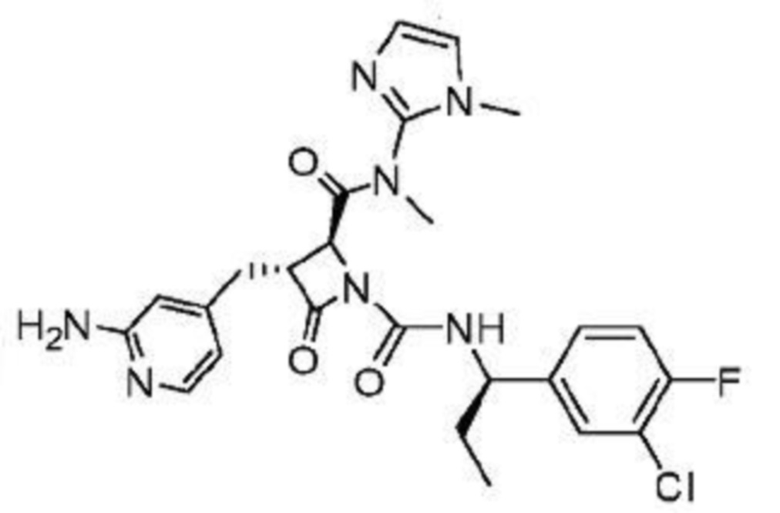

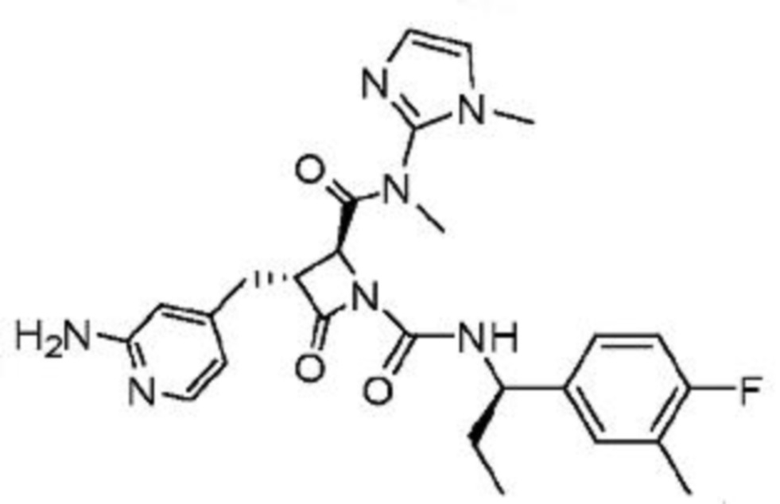



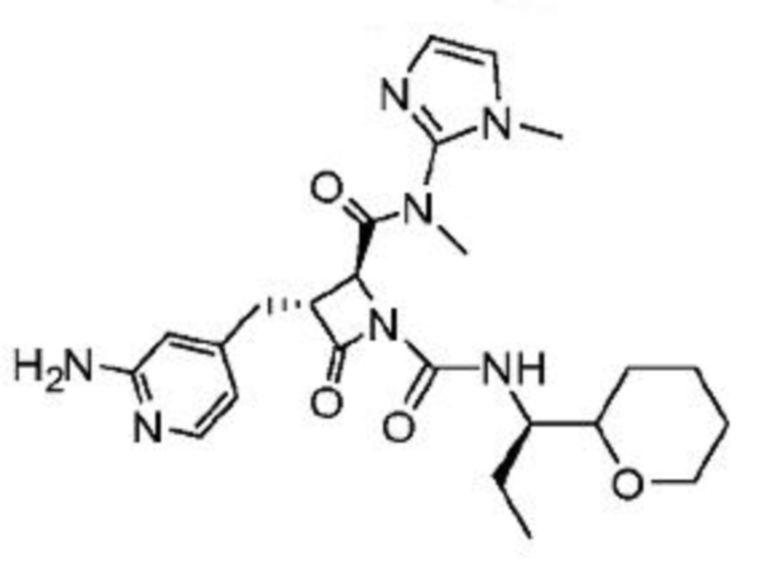
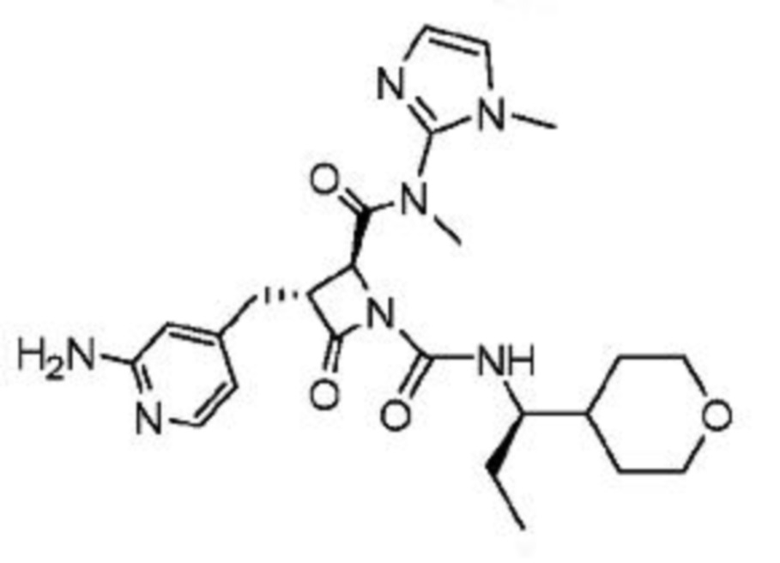


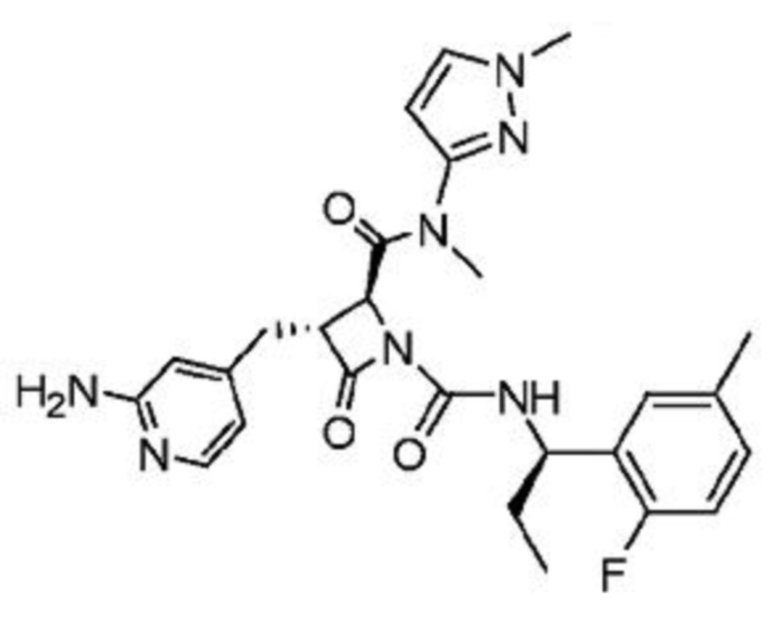



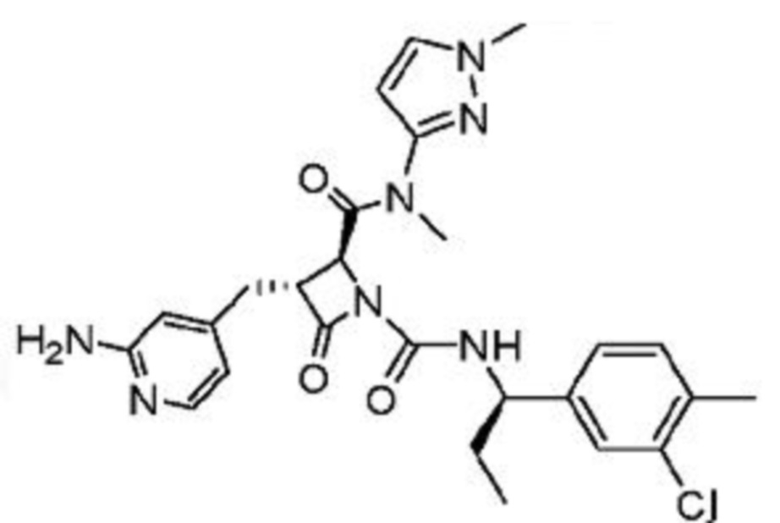
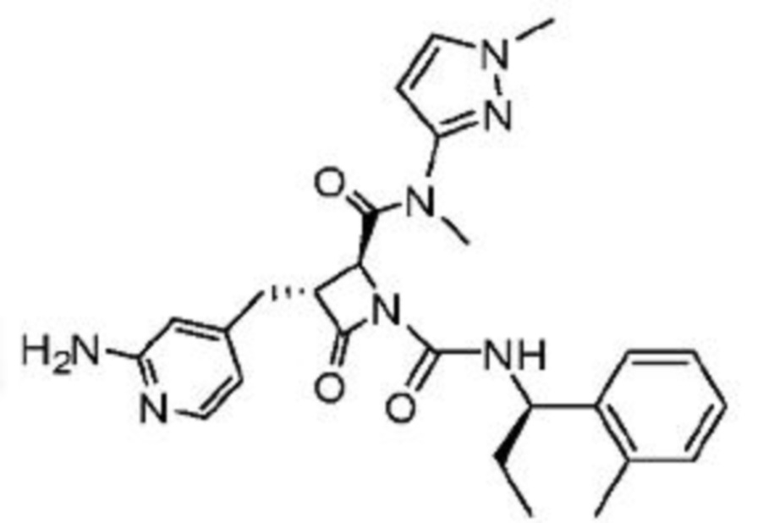
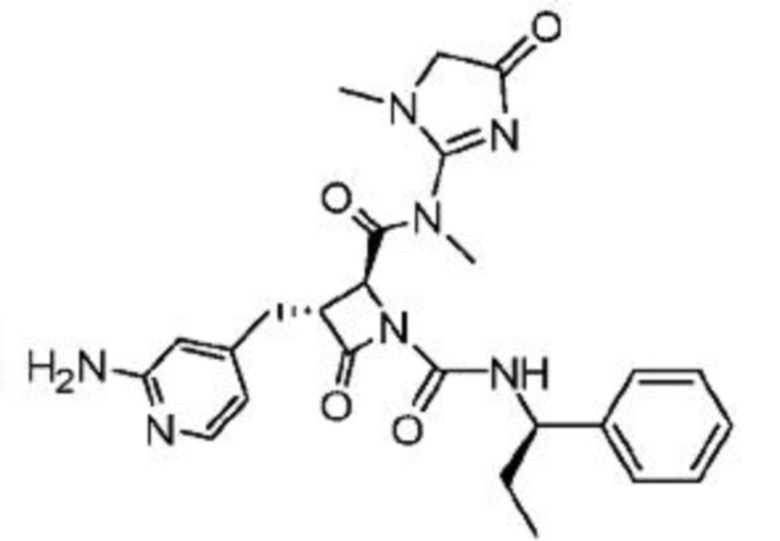

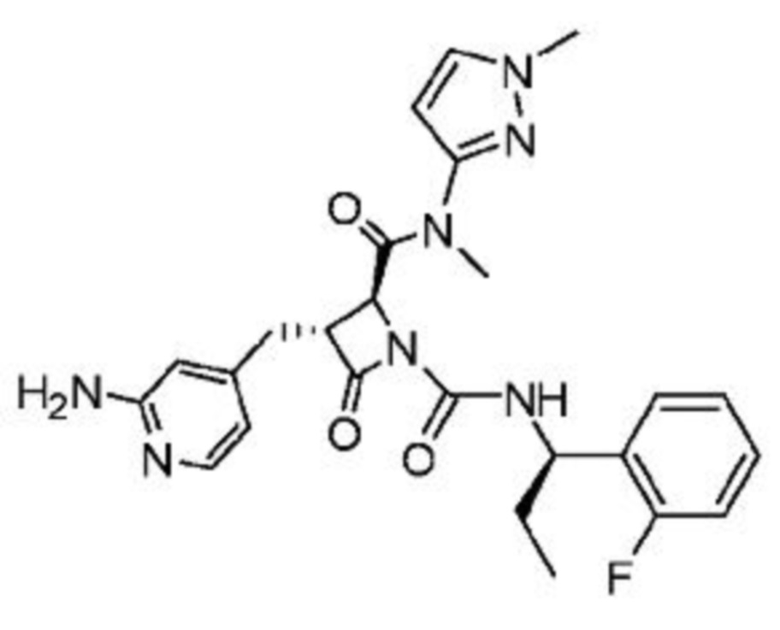
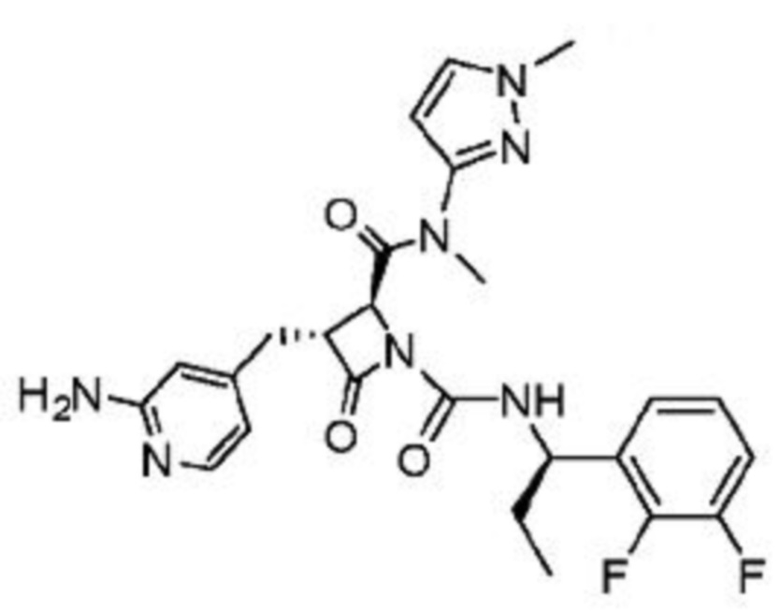
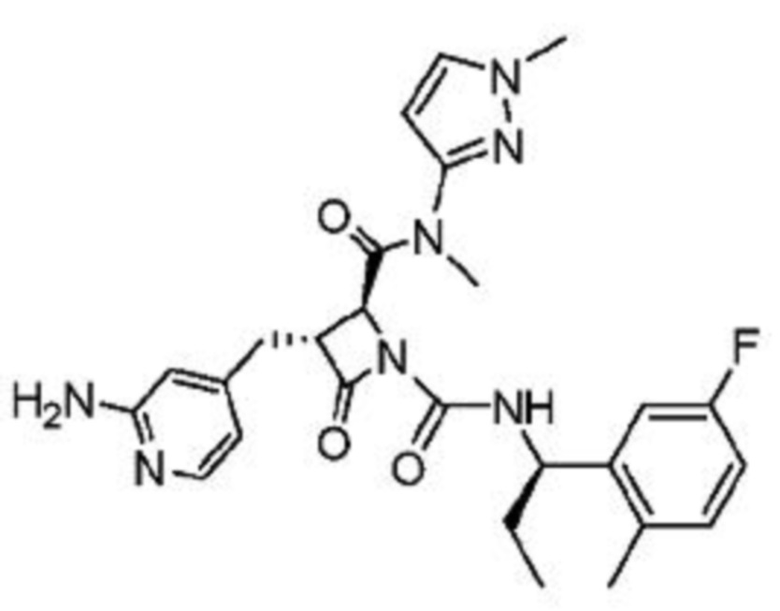
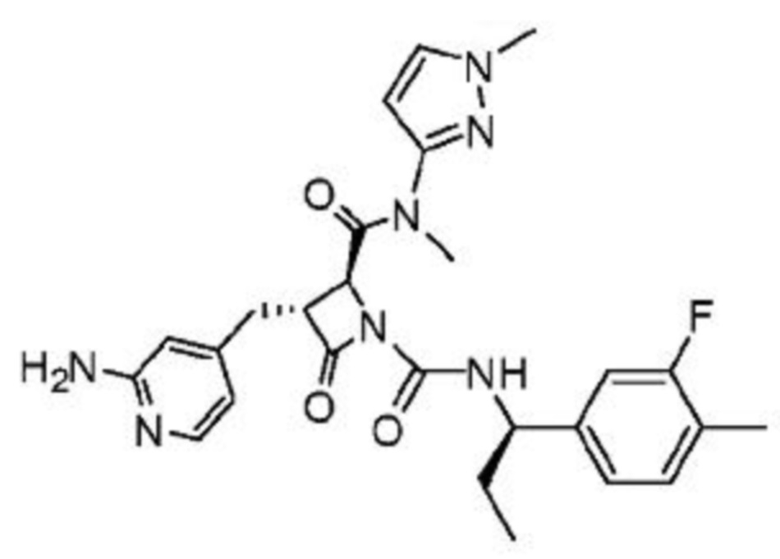





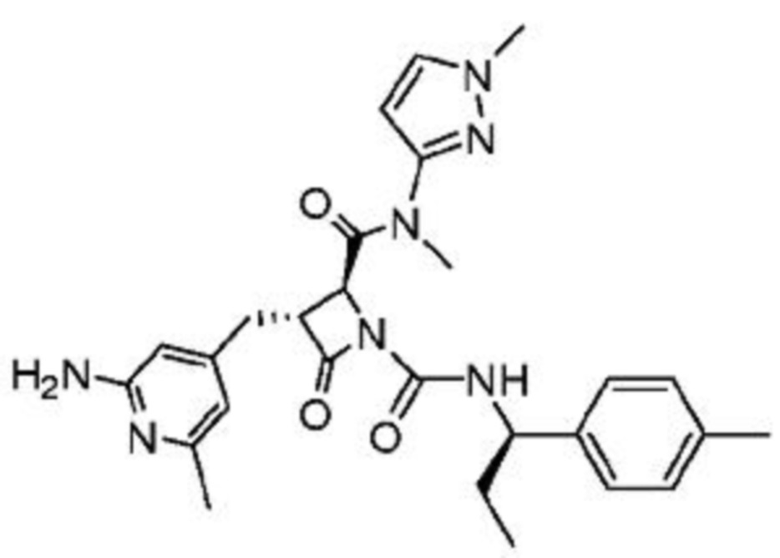
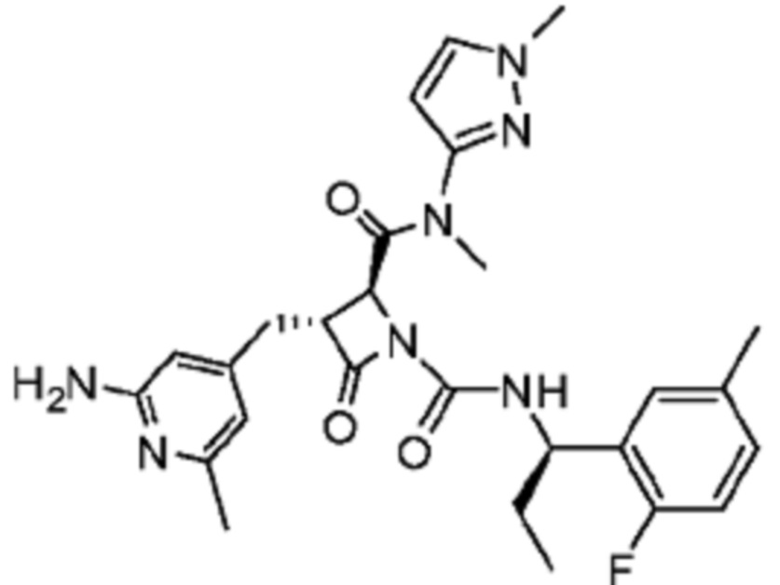




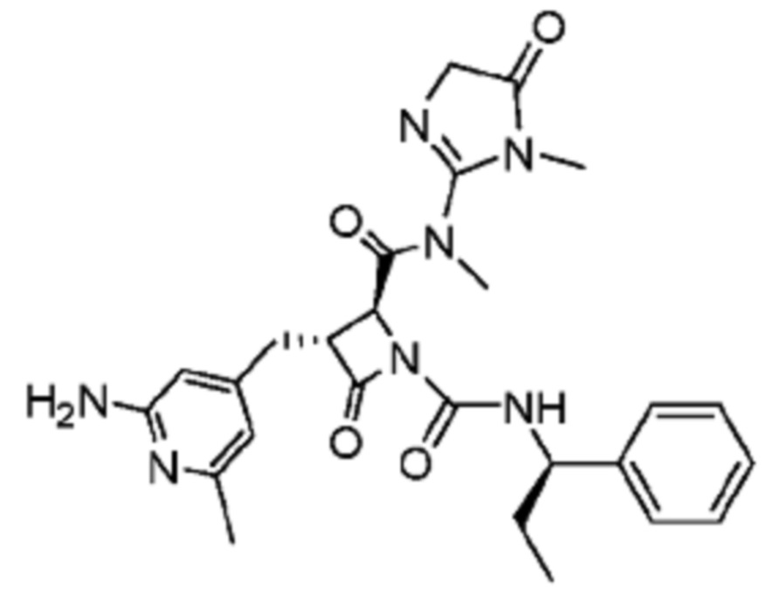





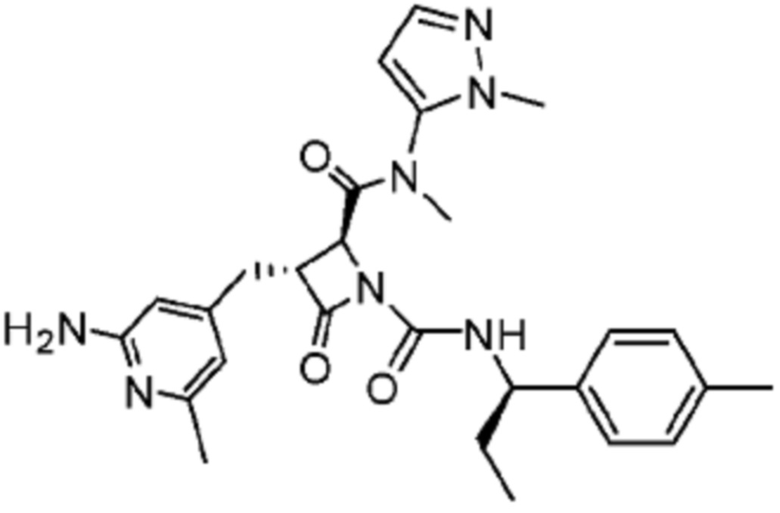

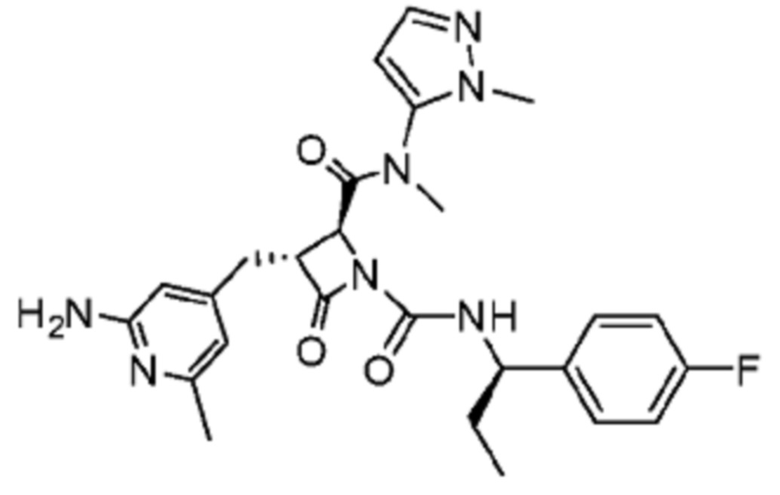
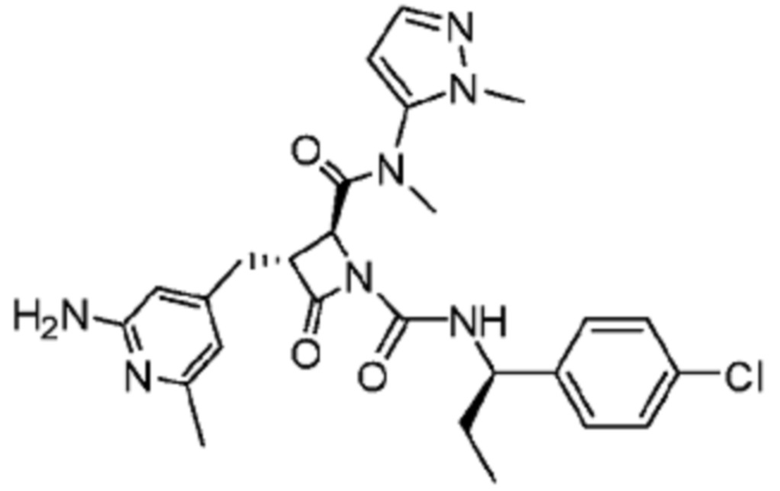





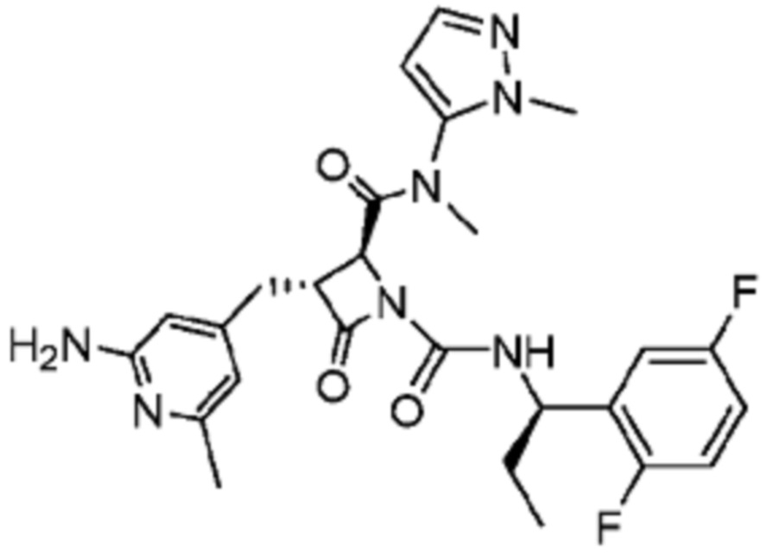
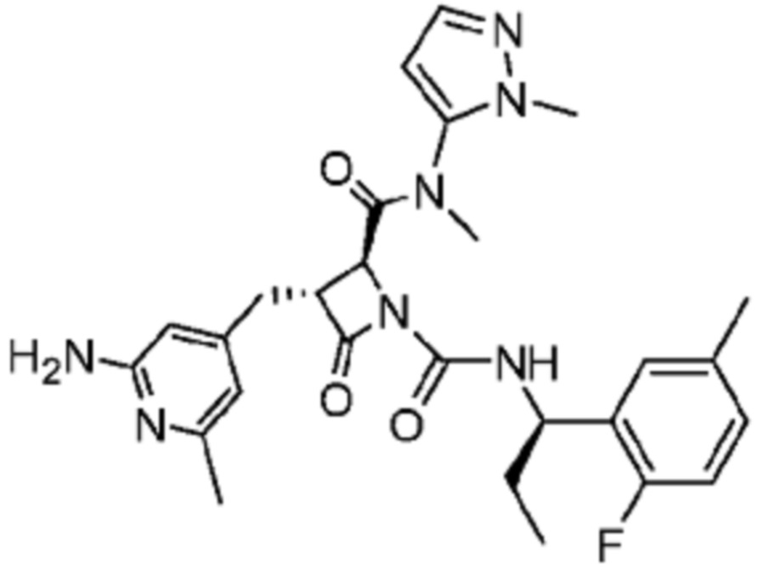
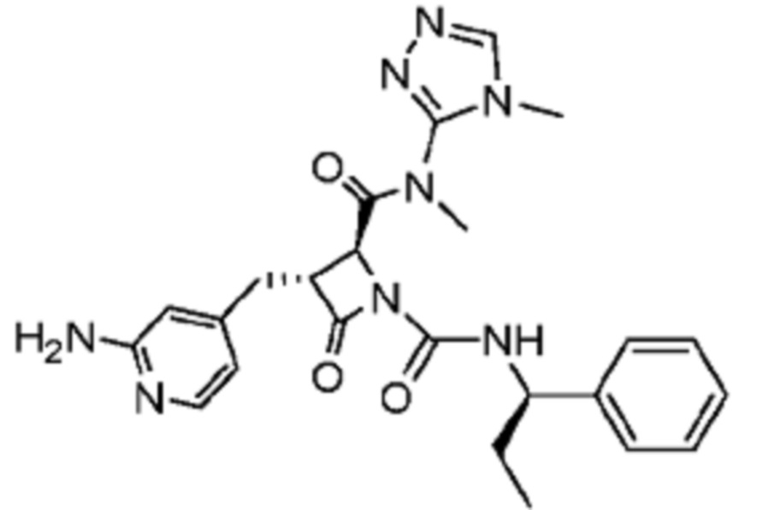
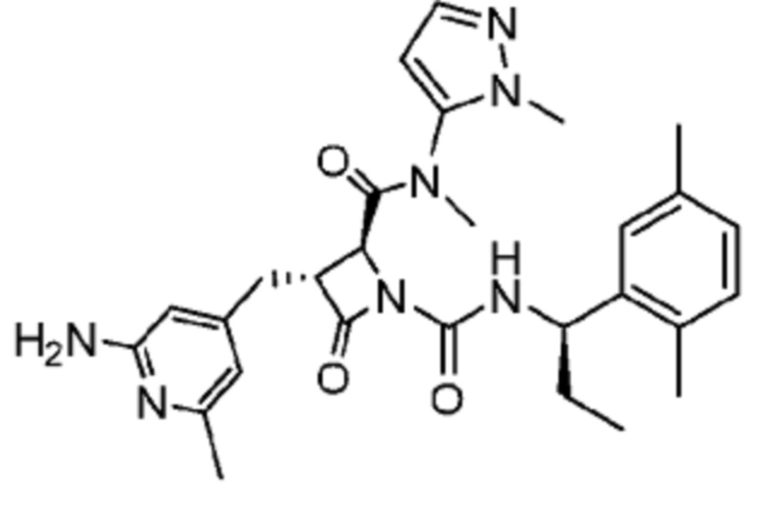

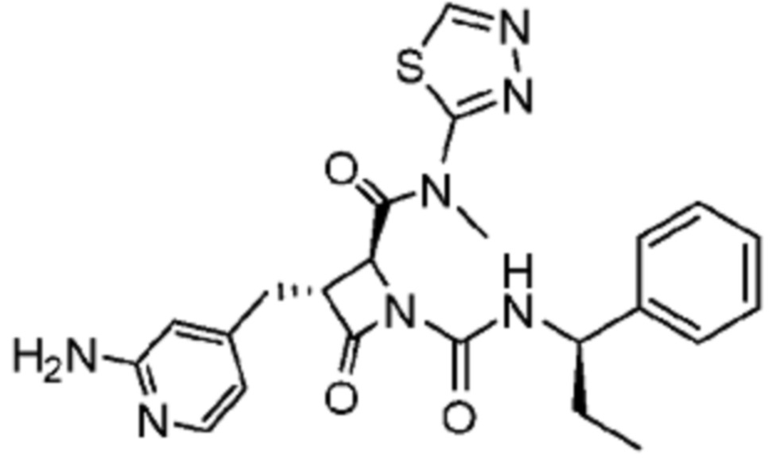
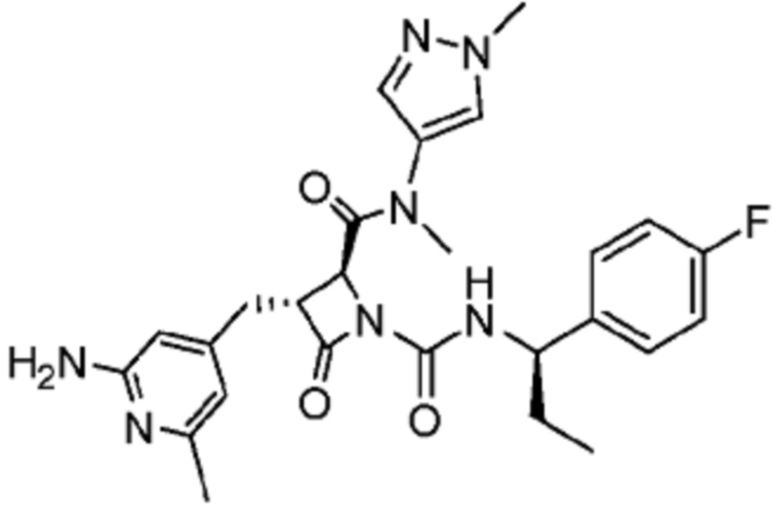
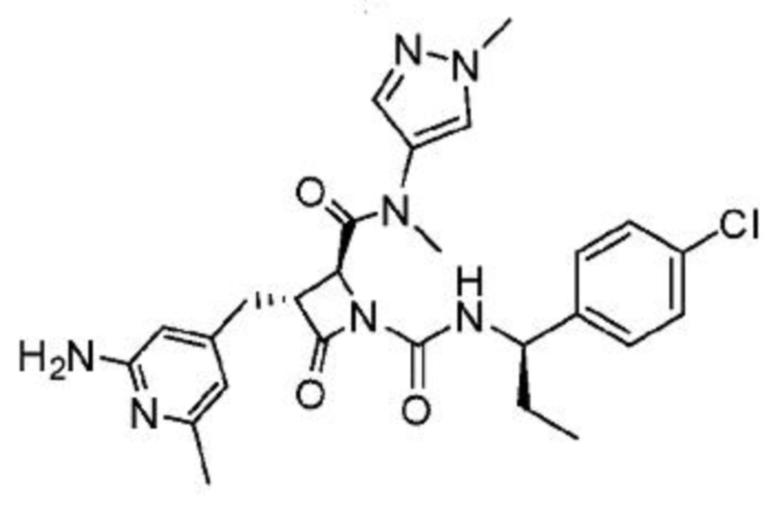




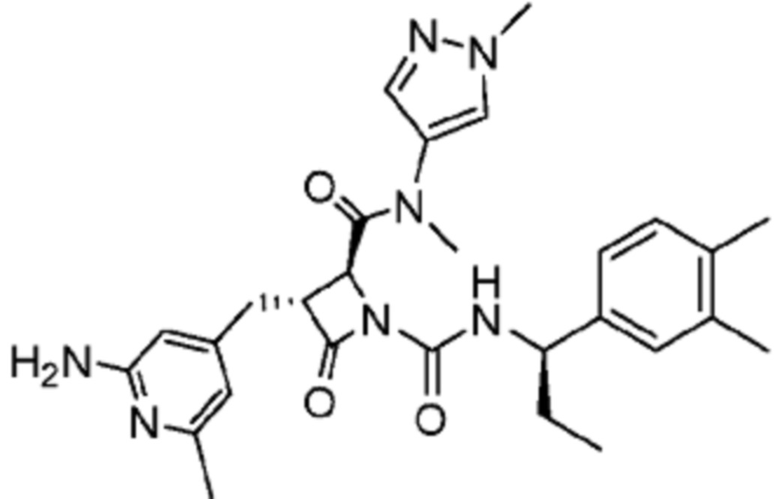
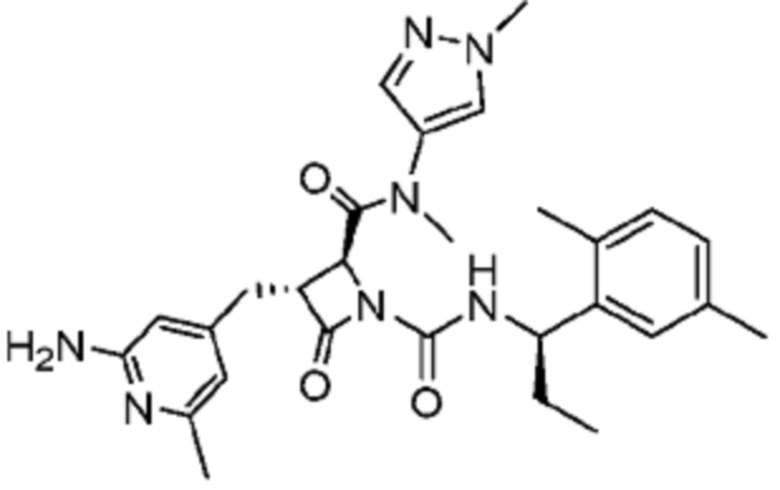

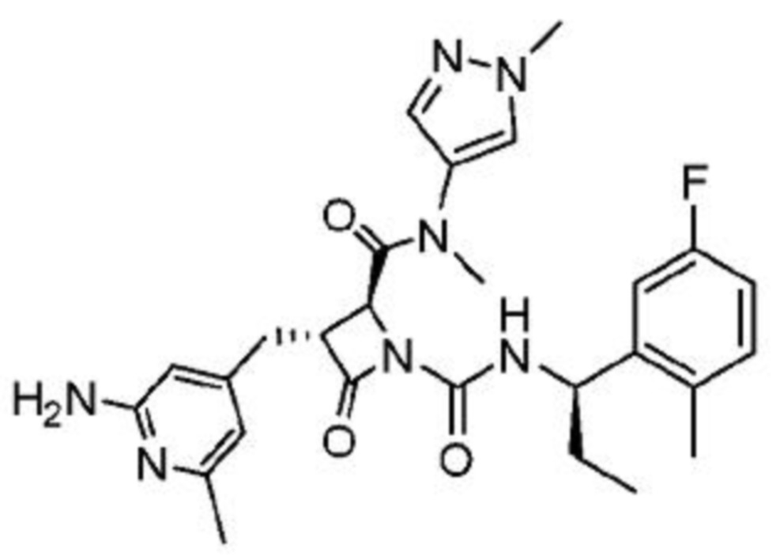




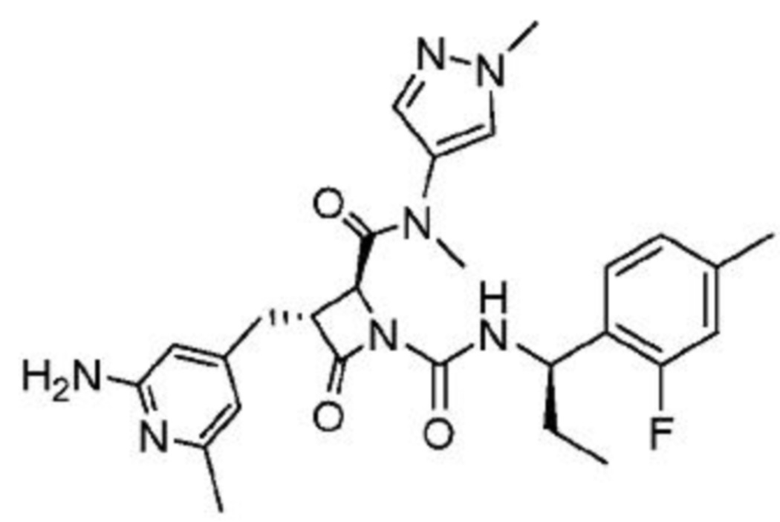

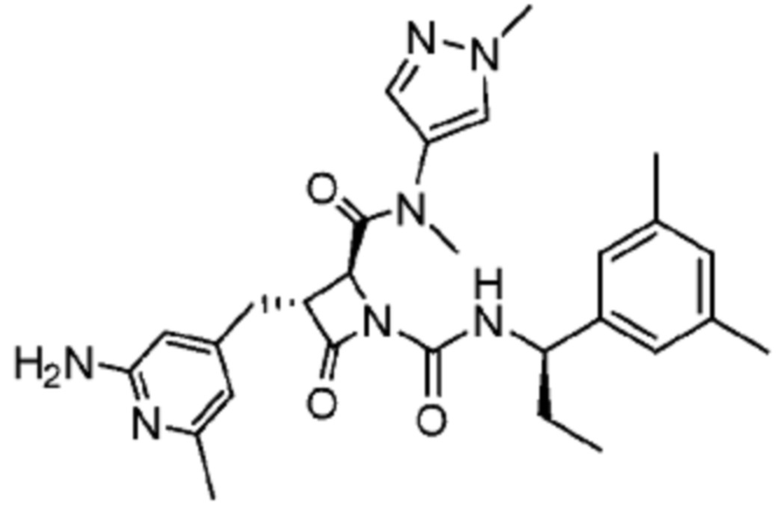
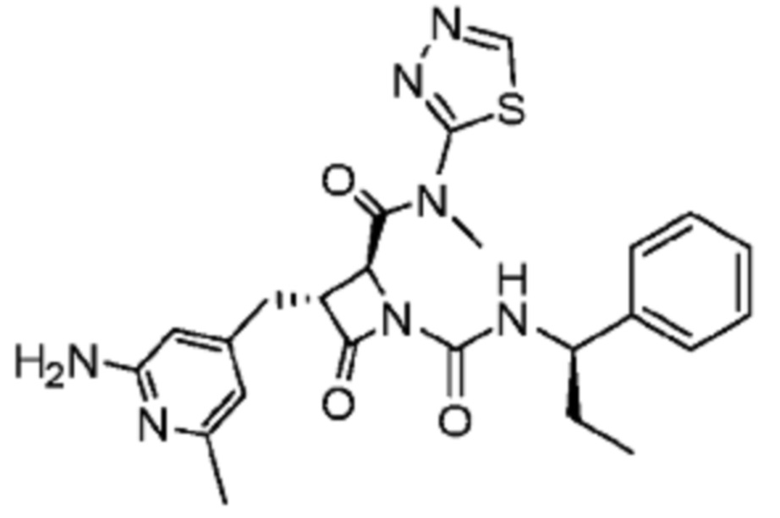
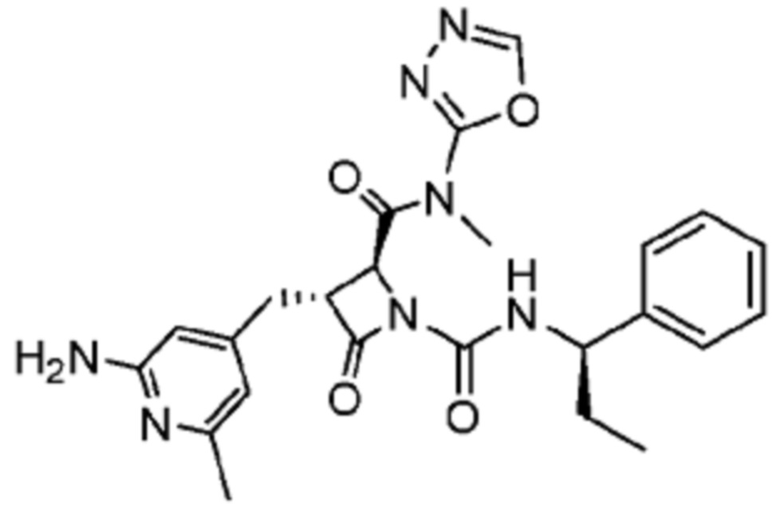
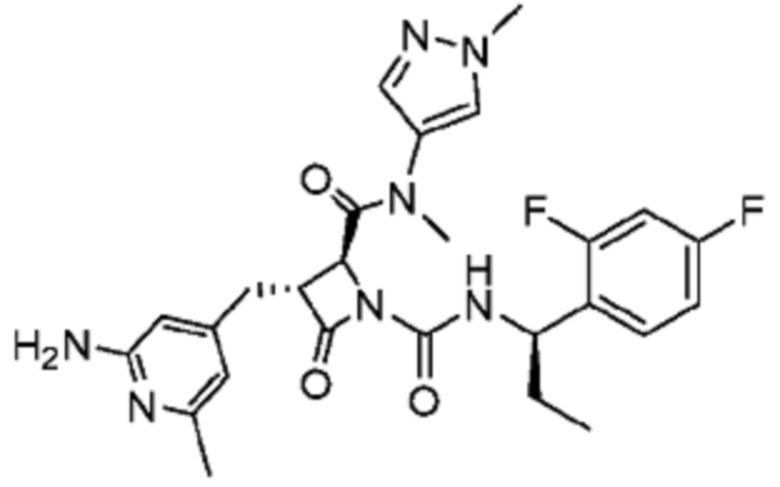
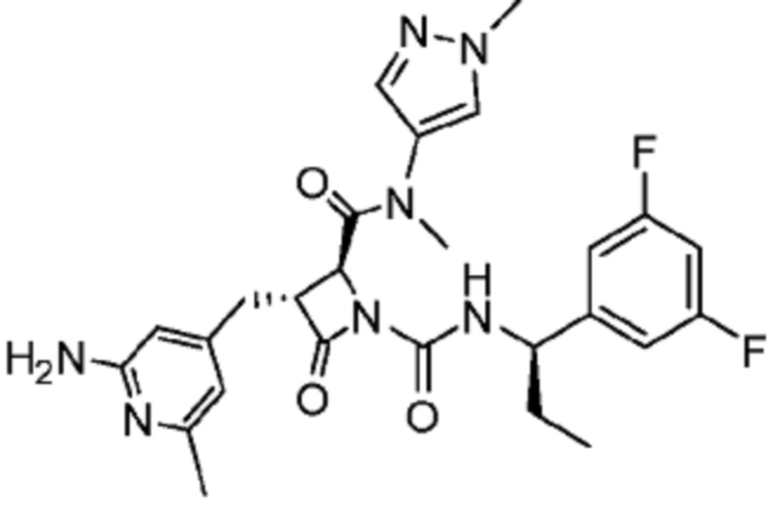
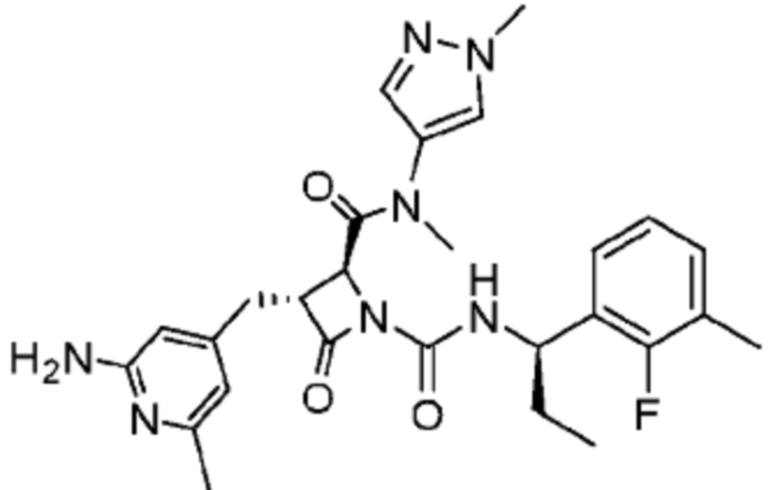
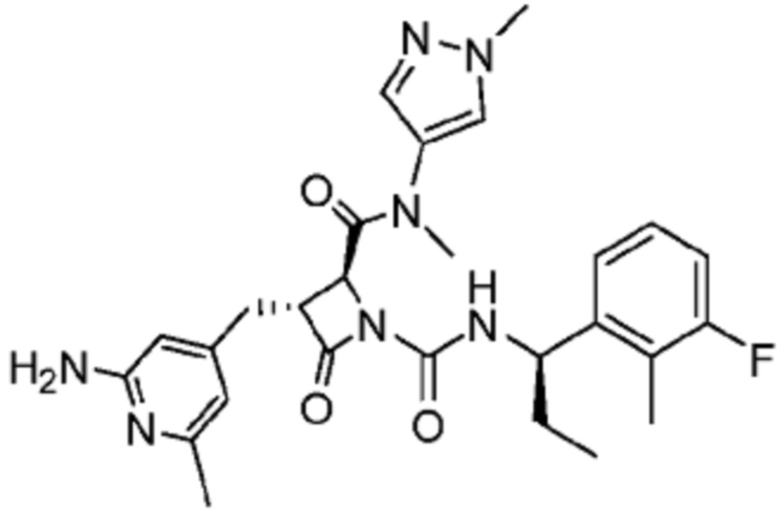


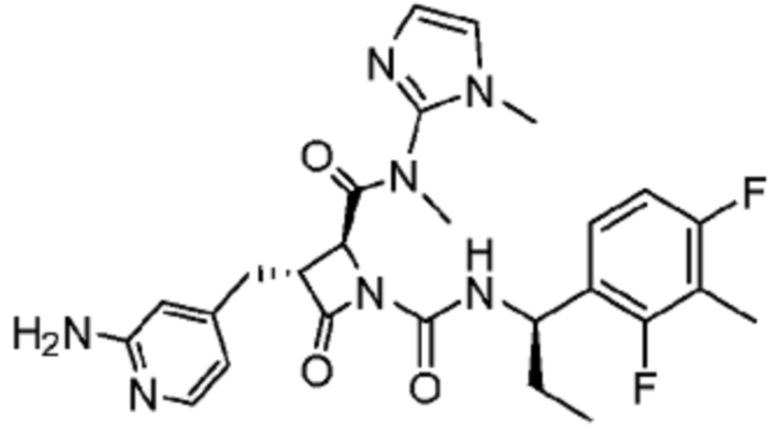

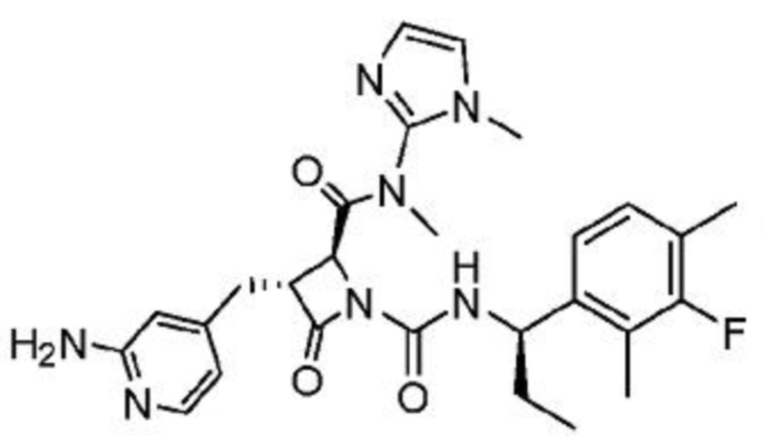



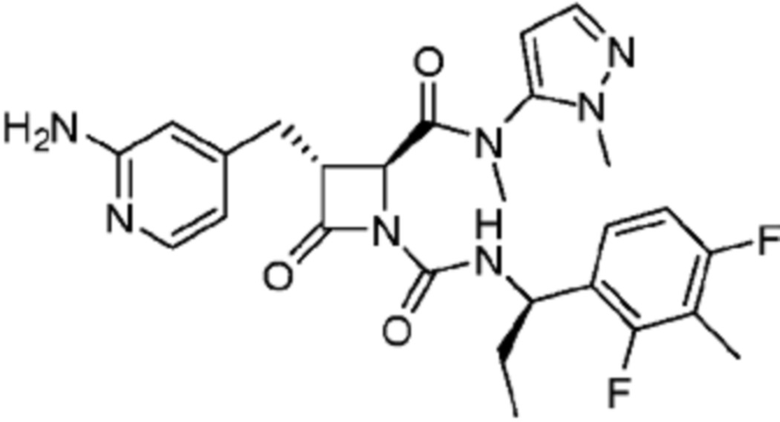
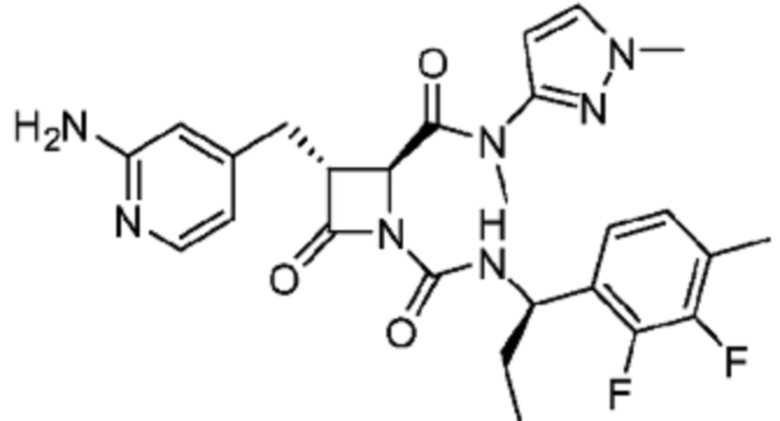

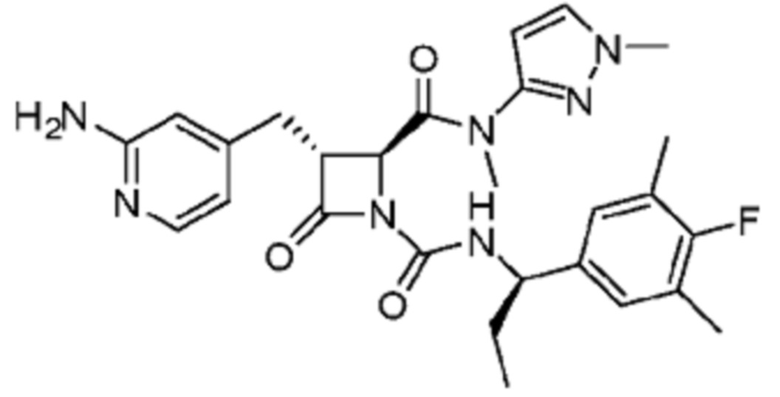

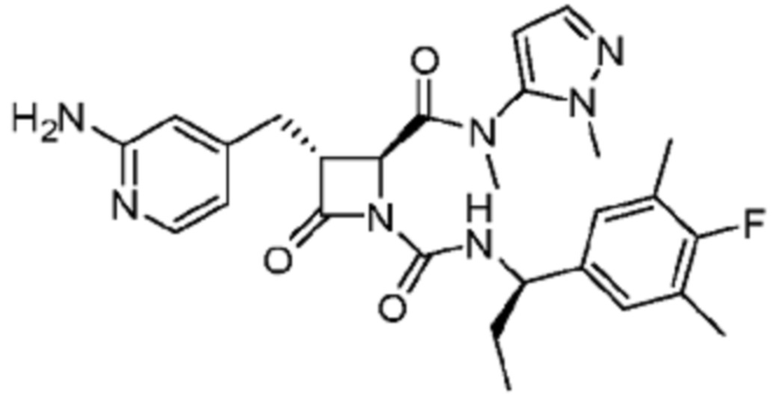


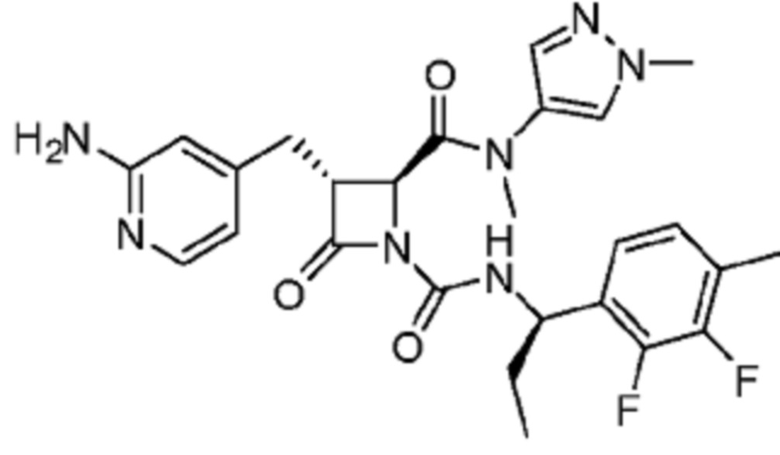
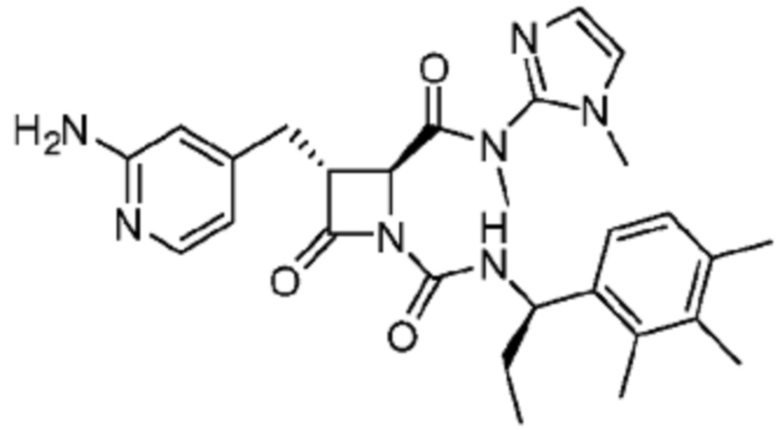

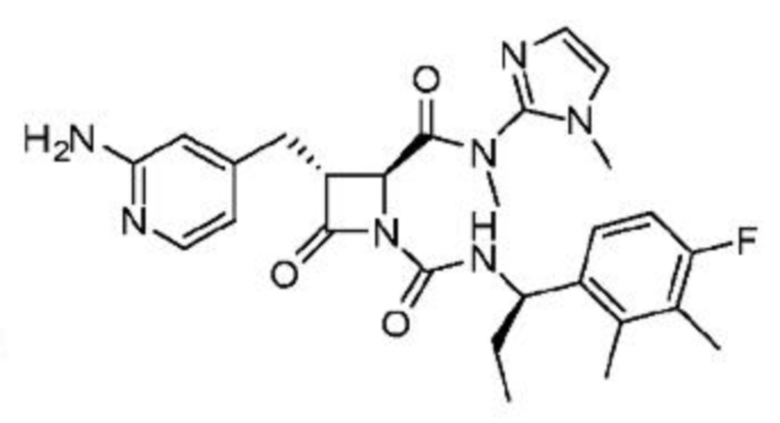
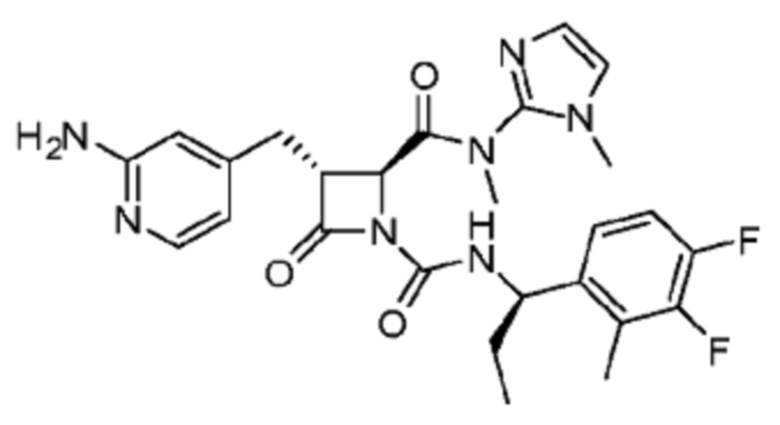



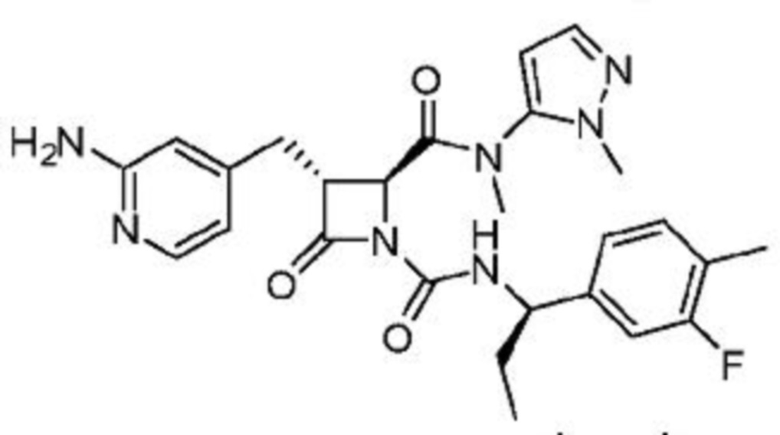





В некоторых вариантах осуществления соединение по настоящему изобретению представлено в форме соли. Соединение по настоящему изобретению может вводиться в форме свободной кислоты, цветтериона или соли. Соль также может образовывать катион и отрицательно заряженный заместитель на соединении по настоящему изобретению. Подходящие катионные противоионы включают ионы натрия, ионы калия, ионы магния, ионы кальция и ионы аммония (например, татраалкиламмониевый катион, такой как ион тетраметиламмония). В соединениях, включающих положительно заряженный заместитель или основной заместитель, соль может образовывать анион и положительно заряженный заместитель (например, аминогруппа) или основной заместитель (например, пиридил) на соединении по настоящему изобретению. Подходящие анионы включают хлор, бром, йод, сульфат, нитрат, фосфат, цитрат, метансульфонат, трифторацетат и ацетат.
Фарацевтически приемлемые соли соединений по настоящему изобретению (например, соединения формулы (II), формулы (I), формулы (I-a), формулы (I-b), формулы (I-c), формулы (I-d), формулы (I-e), формулы (I-f), формулы (I-g), формулы (I-h), формулы (I-i), формулы (I-j), формулы (I-k), формулы (I-l), формулы (I-m), формулы (I-n), формулы (I-o), формулы (I-p), формулы (I-r), формулы (I-с), формулы (I-s1), формулы (I-s2), формулы (I-t), формулы (I-u), формулы (I-v) или соединения, представленного в таблице 1) также включают соли, полученные из фармацевтически приемлемых неорганических и органических кислот и оснований. Примеры подходящих кислотно-аддитивных солей включают ацетат, 4-ацетамидобензoaт, адипат, альгинат, 4-аминосалицилат, аспартат, аскорбат, бензoат, бензолсулльфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, карбонат, циннамат, цикламат, деканоат, декандиоат, 2,2-дихлорацетат, диглюконат, додецилсульфат, этансульфонат, этан-1,2-дисульфонат, формат, фумарат, галактарат, глюкогептаноат, глюконат, глюкогептонат, глюкоронат, глютамат, глютарат, глицерофосфат, гликолят, гемисульфат, гептаноат, гексаноат, гиппурат, гидрохлорид, гидробромид, гидройодид, 1-гидрокси-2-нафтоат, 2-гидроксиэтансульфонат, изобутират, лактат, лактобионат, лаурат, малат, малеат, малонат, манделат, метансульфонат, нафталин-1,5-дисульфонат, 2-нафталинсультонат, никотинат, нитрат, октаноат, олеат, оксалат, 2-оксоглютарат, пальмитат, пальмоат, пектинат, персульфат, 3-фенилпропионат, фосфат, фосфонат, пикрат, пивалат, пропионат, пироглютамат, салицилат, себакат, сукцинат, стеарат, сульфат, тартрат, тиоцианат, толуолсульфонат, тозилат, трифторацетат и ундеканоат.
Соли, полученные из подходящих оснований, включают соли щелочных металлов (например, натрия), щелочно-земельных металлов (например, магния), аммония и соли N-(алкила)4+. Настоящее изобретение также предусматривает кватернизацию любых основных азотсодержащих групп соединений, раскрытых в настоящем документе. При такой кватернизации могут быть получены водорастворимые или диспергируемые в масле соединения.
В контексте настоящего описания соединения по настоящему изобретению, включая соединения формулы (II), определены как включающие их фармацевтически приемлемые производные или пролекарства. Термин «фармацевтически приемлемое производное или пролекарство» означает любую фармацевтически премлемую соль, сложный эфир, соль сложного эфира или другое производное соединения по настоящему изобретению, которое после введения реципиенту способно образовывать (непосредственно или опосредовано) соединение по настоящему изобретению. Особенно предпочтительными производными и пролекарствами являются производные и пролекарства, которые повышают биодоступность соединений по настоящему изобретению, когда такие соединения вводятся млекопитающему (например, позволяя перорально введенному соединению легче всасываться в кровь), или которые повышают доставку исходного соединения в биологический компартмент (например, мозг или лимфатическую систему) относительно исходных видов. Предпочтительные пролекарства включают производные, в которых к структуре формул, описанных в настоящем изобретении, добавлена группа, которая повышает их растворимость в воде или активный транспорт через кишечную мембрану.
Любая формула или соединение по настоящему изобретению также включают как немеченые, так и изотопно меченые формы соединений, где структуры изотопно меченых соединений представлены приведенными формулами, но с тем отличием, что один или несколько атомов замещены атомом(ами) с выбранной атомной массой или выбранным атомным числом. Примеры изотопов, которые могут вводиться в соединения по настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2H, 3H, 11C, 13C, 14C, 15N, 18F 51P, 32P, 35S, 36Cl, 125I, соответственно. Изобретение включает различные изотопно меченые соединения, которые определены в настоящем изобретении, например соединения, в которых присутствуют радиоактивные изотомы, такие как 3H, 13C, и 14C. Такие изотопно меченые соединения применимы в метаболических исследованиях (с 14C), исследования кинетики реакций (например 'H или 3H), методах обнаружения или визуализации, таких как позитронно-эмиссионная томография (PET) или однофотонная эмиссионная компьютерная томография (SPECT), включая методи анализа лекарственного средства или тканевого субстрата или в методах радиоактивного лечения пациентов. В частности, 18F или меченое соединение может быть особенно желательным для PET-или SPECT-исследований, причем изотопно меченые соединения по настоящему изобретения и их пролекарства могут быть получены осуществлением методик, раскрытых в схемах и в примерах и препаратах, описанных ниже, замещением легкодоступным изотопно меченым реагентом изотопно немеченого реагента.
Кроме того, замещение более тяжелыми изотопами, в частности дейтерием (т.е. 2H или D), может обеспечить определенные терапевтические преимущества, обусловленные большей метаболической стабильностью, например увеличением периода полувыведения в условиях in vivo, снижением необходимой дозы и улучшением терапевтического индекса. Понятно, что дейтерий в данном контексте рассматривается как заместитель соединения формулы, описанной в настоящем изобретении. Концентрация такого более тяжелого изотопа, в частности дейтерия, может быть определена коэффициентом изотомного обогащения. Термин «коэффициент изотопного обогащения», когда используется в настоящем описании, означает соотношение содержания указанного изотопа и природного содержания указанного конкретного изотопа, Если заместитель в соединении по настоящему изобретению обозначен как дейтерий, коэффициент изотопного обогащения такого соединения по каждому обозначенному атому дейтерия составляет по меньшей мере 3500 (52,5% дейтeрия введено на каждый обозначенный атом дейтерия), по меньшей мере 4000 (введено 60% дейтерия), по меньшей мере 4500 (введено 67,5% дейтерия), по меньшей мере 5000 (введено 75% дейтерия), по меньшей мере 5500 (введено 82,5% дейтерия), по меньшей мере 6000 дейтерия), по меньшей мере 6333,3 (введено 95% дейтерия), по меньшей мере 6466,7 (введено 97% дейтерия), по меньшей мере 6600 (введено 99% дейтерия) или по меньшей мере 8633,3 (введено 99,5% дейтерия).
Изотопно меченые соединения по настоящему изобретению обычно могут быть получены стандартными методами, известными специалисту в данной области, или способами, аналогичными описанным в прилагаемых примерах и примерах получения, с использованием подходящих изотопно меченых реагентов вместо ранее использованных изотопно немеченых. Фармацевтически приемлемые сольваты в соответствии с настоящим изобретением включают сольваты, в которых растворитель кристаллизации может быть изотопно замещенным, например D2O, D2-ацетон, D2-ДМСО.
Любой асимметричный атом (например, атом углерода или т.п.) соединения(й) по настоящему изобретению может присутствовать в рацемической или энантиомерно обогащенной смеси, например (R)-, (S)- или (RS)-конфигурации. В некоторых вариантах осуществления каждый асимметрический атом имеет по меньшей мере 50% энантиомерный избыток, по меньшей мере 60% энантиомерный избыток, по меньшей мере 70% энантиомерный избыток, по меньшей мере 80% энантиомерный избыток, по меньшей мере 90% энантиомерный избыток, по меньшей мере 95% энантиомерный избыток, или по меньшей мере 99% энантиомерный избыток в (R)- или (S)-конфигурации. Заместители при атомах с незамещенными связями могут, если это возможно, присутствовать в цис-(Z)- или транс-(E)-форме. Соответственно, термин «соединение по настоящему изобретению», когда используется в настоящем описании, означает соединение по настоящему изобретению, которое может быть в форме одного из возможных изомеров, ротамеров, атропизомеров, таутомеров и их смесей, например по-существу чистых геометрических (цис или транс) изомеров, диастереомеров, оптических изомеров (антиподов), рацематов и их смесей. Любые полученные смеси изомеров могут быть разделены, исходя из различий физико-химических свойств составляющих их соединений, на чистые или по-существу чистые геометрические или оптические изомеры, диастереомеры, рацематы, например с помощью хроматографии или фракционной кристаллизации.
Методы синтеза соединений по настоящему изобретению (например, соединения формулы (II)) могут независимо приводить к получению композиций, включающих смеси двух или нескольких диастереомеров указанного соединения. Термин «диастереомерный избыток» («d.e.») или «% дистереомерного избытка» («% d.e.») композиции, когда используется в настоящем описании, относится к избытку одного диастереомера относительно другого или нескольких различных диастереомеров, присутствующих в композиции.
В некоторых вариантах осуществления термин «дистереомерный избыток (d.e.) композиции», относящийся к диастереомеру по настоящему изобретению относительно другого, означает диастереомерный избыток более 90%. В некоторых вариантах осуществления термин «d.e. композиции», относящийся к диастереомеру по настоящему изобретению, означает диастереомерный избыток более 95%. В некоторых вариантах осуществления термин «d.e. композиции», относящийся к диастереомеру по настоящему изобретению, означает диастереомерный избыток более 97%. В некоторых вариантах осуществления термин «d.e. композиции», относящийся к диастереомеру по настоящему изобретению, означает диастереомерный избыток более 99%. В некоторых вариантах осуществления термин «d.e. композиции», относящийся к диастереомеру по настоящему изобретению, означает диастереомерный избыток более 99,9%.
Любые полученные рацематы конечных продуктов или промежуточных соединений могут быть разделены на оптические антиподы известными методами, например разделением из диастереомерных солей, полученных с оптически активной кислорой или оптически активным основанием, и выделением оптически активного кислотного или основного соединения. В частвности, a основной фрагмент молекулы может таким образом применяться для разделения соединений по настоящему изобретению на их оптиеские антиподы, например фракционной кристаллизацией соли, полученной с оптически активной кислотой, например винной кислотой, дибензoилвинной кислотой, диацетилвинной кислотой, (+)-O, O′-ди-п-толуоил-D-винной кислотой, миндальной кислотой, яблочной кислотой или камфор-10-сульфоновой кислотой. Рацемические продукта также могут быть разделены хиральной хроматографией, например жидкостной хроматографией высокого давления (ЖХВД) с использованием хирального адсорбента.
Соединения по настоящему изобретению (например, соединение формулы (II)) также могут быть представлены множеством таутомерных форм, например как соединение формул (II), (I), (I-a), (I-b), (I-c), (I-d), (I-e), (I-f), (I-g), (I-h), (I-i), (I-j), (I-k), (I-l), (I-m), (I-n), (I-o), (I-p), (I-q), (I-r), (I-с), (I-s1), (I-s2), (I-t), (I-u) или (I-v). В таких случаях настоящее изобретение включает все таутомерные формы соединений по настоящему изобретению. Все кристаллические формы соединений по настоящему изобретению включены в настоящее изобретение.
Соединение по настоящему изобретению (например, соединение формулы (II)) может быть испытано на его способность модулировать (например, ингибировать) фактор XIa или калликреин.
Способы синтеза соединений
Соединения по настоящему изобретению могут быть синтезироны стандартными способами с использованием коммерчески доступных исходных веществ и реагентов. Например, соединения могут быть синтезированы с использованием способов, раскрытых в Патенте США № 7501404, или как описано в способах, представленных в настоящем документе.
Способы лечения, профилактики и снижения риска
Соединения по настоящему изобретению (например, соединения формулы (II)) могут ингибировать фактор XIa или калликреин. В некоторых вариантах осуществления соединение по настоящему изобретению могут ингибировать фактор XIa и калликреин. Как результат, указанные соединения могут применяться для лечения, профилактики или снижения риска нарушений, описанных в настоящем документе. Примеры нарушений включают тромботические процессы, связанные с заболеваниями коронарной артерии и сердечно-сосудистыми заболеваниями, венозный или артериальный тромбоз, синдромы коагуляции, ишемию (например, ишемию коронарной артерии) и стенокардию (стабильныю и нестабильную), тромбоз глубоких вен (DVT), тромбоз печеночных вен, диссеминированную внутрисосудистую коагулопатию, синдром Казабаха-Мерритт, эмболию легочной артерии, инфаркт миокарда (например, инфаркт миокарда с повышением ST-сегмента или инфаркт миокарда без повышения ST-сегмента (например, инфаркт миокарда без повышения ST-сегмента перед катетеризацией), церебральный инфаркт, тромбоз сосудов головного мозга, транзиторные ишемические атаки, мерцательную аритмию (например, неклапанную фибрилляцию предсердий), церебральную эмболию, тромбоэмболические осложнения хирургического вмешательства (например, эндопротезирования тазобедренного или коленного сустава, ортопедической операции, операции на сердце, операции на легких, операции на органах брюшной полости или эндартерэктомии) и непроходимость периферических артерий, и могут также применяться для лечения или предотвращения инфаркта миокарда, инсульта, стенокардии и других последствий отрыва атероклеротической бляшки. Соединения по настоящему изобретению, обладающие активностью ингибирования фактора XIa или калликреина, также могут применяться для предотвращения тромбоэмболических осложнений, например венозной тромбоэмболии, у онкологических пациентов, включая пациентов, получающих химиотерапию, и/или пациентов с повышенными уровнями лактатдегидрогеназы (LDH), и для предотвращения тромбоэмболических осложнений во время или после восстановления с помощью тканевого активатора плазминогена или механического восстановления проходимости кровеносных сосудов. Соединения по настоящему изобретению, обладающие ингибирующей активности в отношении фактора XIa или калликреина, также могут применяться в качестве ингибиторов коагуляции крови, например при получении, хранении и фракционировании цельной крови. Кроме того, соединения по настоящему изобретению могут применяться в условиях срочной госпитализации или перипроцедурно, когда пациент подвержен риску тромбоэмболического расстройства или осложнения, а также у пациентов, которые находятся в состоянии повышенной коагуляции, например у онкологических пациентов.
Ингибирование фактора XIa в соответствии с настоящим изобретением может быть более эффективным и безопасным методом ингибирования тромбоза в сравнении с ингибированием других сериновых протеаз свертывания, таких как тромбин или фактор Ха. Введение низкомолекулярного ингибирора фактора XIa должно показывать эффект ингибирования образования тромбина и образования сгустков без какого-либо влияния или по-существу без эффекта на время кровотечения и с незначительным эффектом или по существу не влияя на время кровотечения и незначительно нарушая или не нарушая гомеостаза. Эти результаты существенно отличаются от результатов, которые показывают другие ингибиторы протеазы «прямого действия» (например, ингибиторы активного сайта тромбина и фактора Xa), которые показывают увеличение продолжительности кровотечения и меньшее разделение между антитромботической эффективностью и увеличение продолжительности кровотечения. Предпочтительный способ по настоящему изобретению включает введение млекопитающему фармацевтической композиции, включающей по меньшей мере одно соединение по настоящему изобретению
Соединения по настоящему изобретению (например, соединение формулы (II)), в частности соединения формул (II), (I), (I-a), (I-b), (I-c), (I-d), (I-e), (I-f), (I-g), (I-h), (I-i), (I-j), (I-k), (I-l), (I-m), (I-n), (I-o), (I-p), (I-q), (I-r), (I-с), (I-s1), (I-s2), (I-t), (I-u) или (I-v), могут ингибировать калликреин. Такие образом, указанные соединения могут применяться для лечения, профилактики или снижения риска заболеваний, связанных с воспалением, таких как отек (например, отек головного мозга, макулярный отек и ангионевротический отек (например, наследственный ангионевротический отек)). В некоторых вариантах осуществления соединения по настоящему изобретению могут применяться для лечения или предотвращения наследственного ангионевротического отека. Соединения по настоящему изобретению (например, соединения формулы (II)) также могут применяться для лечения, профилактики или снижения риска, например, инсульта, ишемии (например, ишемической болезни сердца) и периоперационной потери крови, например соединение формулы (II), такое как соединение формул (I), (I-a), (I-b), (I-c), (I-d), (I-e), (I-f), (I-g), (I-h), (I-i), (I-j), (I-k), (I-l), (I-m), (I-n), (I-o), (I-p), (I-q), (I-r), (I-с), (I-s1), (I-s2), (I-t), (I-u) или (I-v). Способы по настоящему изобретения могут применяться для лечения или предотврадения состояний, связанных с действием фактора XIa или калликреина. Соответственно, способы по настоящему изобретению могут применяться для лечения последствий разрыва атеросклеротической блашки, включая серчено-сосудистые заболевания, связанные с активацией каскада коагуляции при тромботических или тромбофильных состояниях.
Точнее, способы по настоящему изобретению могут применяться для лечения, профилактики или снижения риска острых коронарных синдромов, таких как коронарная болезнь сердца, инфаркт миокарда, нестабильная стенокардия (включая нарастающую стенокардию), ишемия (например, ишемия в результате окклюзии сосудов) и церебральный инфаркт. Способы по настоящему изобретению могут дополительно применяться для лечения, профилактики или снижения риска инсульта и связанных с ним заболеваний сосудов головного мозга (включая нарушение мозгового кровообращения, сосудистую деменцию и транзиторную ишемическую атаку); венозного тромбоза и тромбоэмболии, такой как тромбоз глубоких вен (DVT) и эмболия легочной артерии; тромбоза, связанного с мерцательной аритмией, гипертрофии желудочков, распространенная кардиомиопатии или сердечной недостаточности; заболевания заболевания периферических артерий и перемежающейся хромоты; образования атеросклеротических бляшек и атеросклероза трансплантата; рестеноз после повреждения артерий, индуцированный эндогенно (разрывом аторосклеротической бляшки) или экзогенно (инвазивными кардиологическими процедурами, такими как посреждение стенки сосуда в результате ангиопластики или посткраниального стентирования артерии); диссеминированной внутрисосудистой коагулопатии, синдрома Касабаха-Меррита, тромбоза сосудов головного мозга и церебральной эмболии.
Кроме того, способы по настоящему изобретению могут применяться для лечения, профилактики (например, для предотвращения) или снижения риска тромбоэмболических последствий или осложнений, связанных к раком, хирургическим вмешательством (например, эндопротезированием тазобедренного сустава, ортопедической операцией), эндартерэктомией, введением искусственных клапанов сердца, оперативными вмешательствами в периферические сосуды (например, конечностей), оперативными сердечно-сосудистыми вмешательствами, оперативными вмешательствами большого диаметра при лечении аневризмов, сосудистыми транслантатами, механическими органами и имплантацией (например, транскатетерной имплантацией аортального клапана) или трансплантацией органов (например, трансплантацией печени), ткани или клеток); чрескожными коронарными вмешательствами; катетерной абляцией; терапией гемофилии; гемодиализом; лекарственными средствами (такими как тканевой активатор плазминогена или аналогичные лекарственные средства и лекарственными средствами при оперативном восстановлении проходимости кровеносных сосудов) у пациентов с инфарктом миокарда, инсультом, эмболией легочной артерии и т.п. состояниями; лекарственными средствами (такими как контрацептивы перорального введения, гормон-заместительная терапия и гепатин, например для лечения гепарин-индуцированной тромбоцитопеии); сепсисом (таким как сепсис, связанный с синдромом диссеминированной внутрисосудистой коагуляцией); беременностью или родами; и другим хроническим медицинским состоянием. Способы по настоящему изобретению могут применяться для лечения тромбоза вследствие ограничения движения (например, иммобилизации, госпитализации, постельного режима или иммобилизации конечностей, например с помощью иммобилизирующих повязок и т.д.).
Кроме того, соединения по настоящему изобретению (например, соединение формулы (II)) или их фармацевтически приемлемые соли или композиции могут применяться для лечения, профилактики и снижения риска тромбоэмболического осложнения, например венозной тромбоэмболии, тромбоза глубоких вен или эмболии легочной артерии, или связанного с ней осложнения у пациента, где пациент подвергается воздействию искусственной поверхности. Искусственная поверхность может контактировать с кровью пациента, например в качестве экстракорпоральной поверхности или поверхности имплантируемого устройства. Такие искусственные поверхности включают, но без ограничения, поверхности диализных катетеров, контуров искусственного кровообращения, искусственных клапанов сердца, например механических клапанов сердца (MHV), вспомогательных устройств для желудочков, трансплантатов малого размера, катетеров центральных вен, аппаратов экстракорпоральной мембранной оксигенации (ЭКМО). Кроме того, тромбоэмболическое нарушение или тромбоэмболическое осложнение может быть вызвано искусственной поверхностью или связано с искусственной поверхностью. Например, инородные поверхности и различные компоненты механических клапанов сердца (MHV) являются протромботическими и способствуют образованию тромбина через внутренний метаболический путь свертывания крови. Кроме того, ингибиторы тромбина и FXa противопоказаны при тромбоэмболических расстройствах или связанных с ними осложнениях, вызванных искусственными поверхностями, такими как поверхности модфицированного гемостатического клапана (MHV), поскольку эти ингибиторы неэффективны при блокировании внутреннего пути метаболизма на уровнях плазмы, которые не будут вызывать сильного кровотечения. Таким образом, соединения по настоящему изобретению, которые могут применяться, например, в качестве ингибиторов фактора XIa, aрассматриваются в качестве альтернативных терапевтических средств для этих целей.
Соединения по настоящему изобретению (например, соединение формулы (II)) или их фармацевтически приемлемые соли или композиции также могут применяться для лечения, профилактики или снижения риска мерцательной аритмии у пациента, нуждающегося в этом. Например, у пациента может быть высокий риск развития мерцательной аритмии. Пациент также может нуждаться в диализе, таком как почечный диализ. Соединения по настоящему изобретению (например, соединение формулы (II)) или их фармацевтически приемлемые соли или композиции могут вводиться перед, во время или после диализа. Пероральные коагулянты прямого действия (DOAC), доступные на сегодняшний день, такие как некоторые ингибиторы FXa или тромбина, противопоказаны при мерцательной аритмии в таком состоянии. Поэтому соединения по настоящему изобретению, которые могут применяться, например, в качестве ингибиторов фактора XIa, рассматриваются в качестве альтернативных терапевтических средств для этих целей. Кроме того, у пациентп может быть высокий риск кровотечения. В некоторых вариантах осуществления пациент может страдать терминальной почечной недостаточностью. В других случаях пациент не нуждается в диализе, таком как почечный диализ. Кроме того, мерцательная аритмия может быть связана с другим тромбоэмболическим осложнением, таким как тромб.
Далее, соединения по настоящему изобретению (например, соединение формулы (II)) или их фармацевтически приемлемые соли или композиции могут применяться для лечения, профилактики или снижения риска гипертензии, например артериальной гипертензии, у пациента. В некоторых вариантах осуществления гипертензия, например артериальная гипертензия, может приводить к атеросклерозу. В некоторых вариантах осуществления гипертензия может представлять собой легочную артериальную гипертензию.
Более того, соединения по настоящему изобретению (например, соединение формулы (II)) или их фармацевтически приемлемые соли или композиции могут применяться для лечения, профилактики или снижения риска расстройств, таких как гепарин-индуцированная тромбоцитопения, гепарин-индуцированная тромбоцитопения и тромбоз или тромботическая микроангиопатия, например гемолитического уремического синдрома (HUS) или тромботической тромбоцитопенической пурпуры (TTP).
Соединения по настоящему изобретению (например, соединение формулы (II)) или их фармацевтически приемлемые соли или композиции могут применяться для снижения воспаления у пациента. В некоторых вариантах осуществления воспаление может представлять собой воспаление сосудов. В некоторых вариантах осуществления воспаление сосудов может сопровождаться атеросклерозом. В некоторых вариантах осуществления воспаление сосудов может сопровождаться тромботическим заболеванием у пациента. В некоторых вариантах осуществления воспаление сосудов может представлять собой воспаление сосудов, индуцированное ангиотензином II.
Соединения по настоящему изобретению (например, соединение формулы (II)) или их фармацевтически приемлемые соли или композиции могут применяться для лечения, профилактики или снижения риска нарушений функции почек или почечных дисфункций, включая терминальную почечную недостаточность, ренальную дисфункцию, связанную с гипертензией у пациента, фиброз почек и почечную недостаточность.
Способы по настоящему изобретению могут также применяться для поддержания проходимости кровеносных сосудов, например у пациентов, перенесших транслюминальную коронарную ангиопластику, или в связи с хирургической операцией на сосуды, такой как шунтирование, реконструкция артерий, атерэктомия, введение трансплантатов сосудов, восстановление проходимости стента, а также имплантация и трансплантация органов, тканей или клеток. Способы по настоящему изобретению могут применяться для ингибирования коагуляции крови при получении, хранении и фракционировании, а также применении цельной крови. Например, способы по настоящему изобретению могут применяться для сохранения цельной и фракционированной крови в жидкой фазе, как необходимо для аналитического и биологического тестирования, например для исследования ex vivo функции тромбоцитов и других клеток, биоаналитических методик и количественного анализа компонентов крови или для поддержания экстракорпоральных контуров кровообращения, как в заместительном почечном растворе (например, гемодиализа) или при хирургической операции (например, операции на открытом сердце, например аорто-коронарном шунтировании). В некоторых вариантах осуществления заместительный почечный раствор может применяться для лечения пациентов с острой почечной недостаточностью. В некоторых вариантах осуществления заместительный почечный раствор может представлять собой непрерывную заместительную почечную терапию.
Кроме того, способы по настоящему изобретению могут применяться для лечения и предотвращения протромботический осложнений рака. Способы могут применяться для лечения роста опухоли в качестве дополнения к химиотерапии, для предотвращения ангиогенеза и для лечения рака, в частности рака легких, рака предстательной железы, рака толстой кишки, рака молочной железы, рака яичников и рака костей.
В одном аспекте настоящее изобретение относится к способу снижения риска инсульта у пациента, который перенес ишемическую атаку, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления введение снижает риск инсульта у пациента по сравнению с пациентом, которому не вводят соединение.
В одном аспекте настоящее изобретение относится к способу снижения риска инсульта или системной эмболии у пациента, страдающего мерцательной аритмией, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В другом аспекте настоящее изобретение относится к способу снижения риска системной эмболии за пределами ЦНС у пациента, который перенес ишемическую атаку, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления введение снижает риск системной эмболии за пределами ЦНС у пациента по сравнению с пациентом, которому не вводят соединение.
В одном аспекте настоящее изобретение относится к способу лечения тромбоза глубоких вен, включающему введение пациенту, который перенес ишемическую атаку, эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу снижения риска рецидива тромбоза глубоких вен, включающему введение пациенту, у которого наблюдался тромбоз глубоких вен, эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления введение снижает риск рецидива тромбоза глубоких вен у пациента по сравнению с пациентом, которому не вводят соединение.
В одном аспекте настоящее изобретение относится к способу профилактики тромбоза глубоких вен у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления пациент подвергается ортопедической операции или операции эндопротезирования коленного или тазобедренного сустава.
В одном аспекте настоящее изобретение относится к способу снижения риска рецидива эмболии легочной артерии, включающему введение пациенту, который страдает легочной эмболией, эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу снижения риска рецидива эмболии легочной артерии, включающему введение пациенту, который страдал тромбозом глубоких вен, эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления введение снижает риск рецидива эмболии легочной артерии у пациента по сравнению с пациентом, которому не вводят соединение.
В одном аспекте настоящее изобретение относится к способу лечения эмболии легочной артерии у пациента, который страдал тромбозом глубоких вен, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу профилактики эмболии легочной артерии у пациента, который страдал легочной эмболией, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу профилактики эмболии легочной артерии у пациента, который страдал легочной эмболией, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу профилактики эмболии легочной артерии у пациента, который страдал тромбозом глубоких вен, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу лечения пациента, который перенес ишемическую атаку, включающему введение соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II), пациенту в пределах 12 часов или менее, 10 часов или менее, 9 часов или менее, 8 часов или менее, 7 часов или менее, 6 часов или менее или 1 часа или менее, после начала ишемической атаки у пациента.
В одном аспекте настоящее изобретение относится к способу лечения тромбоза глубоких вен у пациента, которому ранее вводился антикоагулянт, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу лечения эмболии легочной артерии у пациента, которому ранее вводился антикоагулянт, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления антикоагулянт вводился парентерально в течение 5-10 дней.
В одном аспекте настоящее изобретение относится к способу лечения пациента, который перенес ишемическую атаку, включающему: введение соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II), пациенту в интервале от более 2 часов до 12 часов, например от более 2 часов до 10 часов или менее, от более 2 часов до 8 часов или менее, после начала ишемической атаки у пациента.
В одном аспекте настоящее изобретение относится к способу лечения острого коронарного синдрома у пациента, включающему введение пациенту, нуждающемуся в этом, соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу ингибирования фактора XIa у пациента, включающему введение пациенту, который перенес ишемию, эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления ишемия представляет собой коронарную ишемию.
В одном аспекте настоящее изобретение относится к способу лечения пациента с отеком, включающему введение соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу ингибирования калликреина у пациента, включающему введение пациенту с отеком эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу лечения последствия тромбоэмболического последствия или тромбоэмболического осложнения у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления тромбоэмболическое последствие или тромбоэмболическое осложнение связано с вмешательством в периферические кровеносные сосуды (например, конечностей), гемодиализом, катетерной абляцией, цереброваскулярным вмешательством, трансплантацией органа (например, печени), хирургической операцией (например, ортопедической операцией, операцией на легких, хирургической операцией на обрганы брюшной полости или операцией на сердце (например, операцией на открытом сердце)), транс-катетерной имплантацией аортального клапана, вмешательством большого диаметра, используемым для лечения аневризмы, чрескожным коронарным вмешательством или терапией гемофилии. В некоторых вариантах осуществления хирургическая операция представляет собой ортопедическую хирургическую операцию, операцию на легких, операцию на органах брюшной полости или операцию на сердце.
В одном аспекте настоящее изобретение относится к способу лечения рестеноза после повреждения артерии у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления повреждение артерии происходит после стентирования черепной артерии.
В одном аспекте настоящее изобретение относится к способу лечения тромбоза сосудов печени у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу лечения инфаркта миокарда без повышения ST-сегмента или инфаркта миокарда с повышением ST-сегмента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу поддержания проходимости кровеносных сосудов, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления пациент страдает острой почечной недостаточностью. В некоторых вариантах осуществления пациент дополнительно получает непрерывную заместительную почечную терапию.
В одном аспекте настоящее изобретение относится к способу лечения гипертензии у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления гипертензия представляет собой артериальную гипертензию.
В одном аспекте настоящее изобретение относится к способу снижения риска гипертензии у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления гипертензия представляет собой артериальную гипертензию.
В одном аспекте настоящее изобретение относится к способу профилактики гипертензии у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления гипертензия представляет собой артериальную гипертензию.
В одном аспекте настоящее изобретение относится к способу снижения воспаления у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II). В некоторых вариантах осуществления воспаление представляет собой воспаление сосудов. В некоторых вариантах осуществления воспаление сосудов представляет собой воспаление сосудов, индуцированное ангиотензином II.
В одном аспекте настоящее изобретение относится к способу предотвращения васкулярной инфильтрации лейкоцитов у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу предотвращения у пациента эндотелиальной дисфункции, индуцированной ангиотензином II, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу предотвращения избыточного продуцирования тромбина у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу лечения у пациента ренальной дисфункции, связанной с гипертензией, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу профилактики у пациента ренальной дисфункции, связанной с гипертензией, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу снижения у пациента риска ренальной дисфункции, связанной с гипертензией, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу лечения у пациента ренальной дисфункции, связанной с гипертензией, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу профилактики у пациента ренальной дисфункции, связанной с гипертензией, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу снижения у пациента риска ренальной дисфункции, связанной с гипертензией, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу лечения фиброза почек у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу профилактики фиброза почек у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу снижения риска фиброза почек у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу лечения почечной недостаточности у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу профилактики почечной недостаточности у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу снижения риска почечной недостаточности у пациента, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу лечения тромбоэмболического осложнения у пациента, нуждающегося в этом, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II), где пациент подвергается воздействию искусственной поверхности.
В одном аспекте настоящее изобретение относится к способу снижения риска тромбоэмболического осложнения у пациента, нуждающегося в этом, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II), где пациент подвергается воздействию искусственной поверхности.
В одном аспекте настоящее изобретение относится к способу профилактики тромбоэмболического осложнения у пациента, нуждающегося в этом, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II), где пациент подвергается воздействию искусственной поверхности.
В одном аспекте настоящее изобретение относится к способу лечения гепарин-индуцированной тромбоцитопении у пациента, нуждающегося в этом, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу снижения риска гепарин-индуцированной тромбоцитопении у пациента, нуждающегося в этом, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу профилактики гепарин-индуцированной тромбоцитопении у пациента, нуждающегося в этом, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу лечения гепарин-индуцированной тромбоцитопении и тромбоза у пациента, нуждающегося в этом, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу снижения риска гепарин-индуцированной тромбоцитопении и тромбоза у пациента, нуждающегося в этом, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу профилактики гепарин-индуцированной тромбоцитопении и тромбоза у пациента, нуждающегося в этом, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу профилактики тромбоэмболического осложнения у пациента, нуждающегося в этом, включающему введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II), где пациент страдает раком.
В одном аспекте настоящее изобретение относится к способу лечения тромботической микроангиопатии у пациента, нуждающегося в этом, причем способ включает введение пациенту эффективного количества соединения формулы (II) по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В одном аспекте настоящее изобретение относится к способу снижения риска тромботической микроангиопатии у пациента, нуждающегося в этом, причем способ включает введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В другом аспекте настоящее изобретение относится к способу профилактики тромботической микроангиопатии у пациента, нуждающегося в этом, причем способ включает введение пациенту эффективного количества соединения по настоящему изобретению, например соединения формулы (II) или его фармацевтически приемлемой соли, или композиции по настоящему изобретению, например композиции, включающей соединение формулы (II).
В некоторых вариантах осуществления пациентом является пациент с неклапанной фибрилляцией предсердий. В некоторых вариантах осуществления у пациента имеется один или несколько из следующих факторов риска: инсульт в анамнезе, транзиторная ишемическая атака или системная эмболия за пределами ЦНС. В некоторых вариантах осуществления у пациента имеется один или несколько из следующих факторов риска: возраст 75 лет или более, гипертензия, сердечная недостаточность или фракция выброса левого желудочка или сахарный диабет.
Экстракорпоральная мембранная оксигенация (ЭКМO)
Соединения по настоящему изобретению могут применяться для лечения, профилактики или снижения риска тромбоэмболического осложнения у пациента, нуждающегося в этом, где пациент подвергается воздействию искусственной поверхности, такой как аппарат экстракорпоральной мембранной оксигенации (ЭКМО) (vide supra), который может применяться в качестве терапии спасения при сердечной или легочной недостаточности. Поверхность ЭКМО аппарата, которая непосредственно контактирует с пациентом, может представлять собой протромботическую поверхность, которая может вызывать тромбоэмболическое осложнение, такое как венозная тромбоэмболия, например тромбоз глубоких вен или эмболию легочной артерии, приводя к трудностям в лечении пациента, нуждающегося в ЭКМО. Сгустки крови в контуре являются наиболее распространенным механическим осложнением (19%). Крупные сгустки крови могут вызывать сбой в работе оксигенатора и приводить к легочной или системной эмболии.
ЭКМО зачастую вводят с непрерывной инфузией гепарина в качестве антикоагулянта для предотвращения образования тромбов. Однако, установка канюли может вызвать повреждение внутренней яремной вены, что вызывает сильное внутреннее кровотечение. Кровотечение имеет место у 30-40% пациентов, получающих ЭКМО, и может быть опасным для жизни. Это сильное кровотечение связано как с необходимостью непрерывной инфузии гепарина, так и с дисфункцией тромбоцитов. Примерно 50% зарегистрированных смертей обусловлено тяжелыми осложнениями кровотечения. Aubron с соавторами (Critical Care, 2013, 17:R73) проанализировал факторы, связанные с результатами применения ЭКМО.
Соединения по настоящему изобретению, которые могут применяться в качестве, например, ингибиторов фактора XIa, рассматриваются в качестве альтернативной замены гепатиру в ЭКМО терапии. Соединения по настоящему изобретению рассматриваются в качестве эффективных лекарственных средств для блокирования внутреннено пути метаболизма на уровнях плазмы, которые не вызывают сильного кровотечения.
Ишемия
Термин «ишемия» или «ишемическая атака» означает заболевание сосудов, обычно включающее окклюзию сосудов или ограничение кровоснабжения ткане. Ишемия может вызвать нехватку кислорода и глюкозы, необходимых для клеточного метаболизма. Ишемия обычно обусловлена проблемными кровеносными сосудами, что приводит к повреждению или дисфункции ткани. Ишемия также может быть связана с локальной потерей крови или кислорода в данной части тела в результате застоя (например, вазоконстрикции или эмболии). Причины включают эмболию, тромбоз атеросклерозной артерии, травму, венозные проблемы, аневризму, сердечные заболевания (например, инфаркт миокарда, заболевание митрального клапана, хроническую мерцательную аритмию, кардиомиопатию и протезирование), травму или травматическое повреждение (например, повреждение конечности, вызывающее частичную или полную окклюзию сосуда), синдром грудного выхода, атеросклероз, гипогликемию, тахикардию, гипотензию, внешнюю компрессию кровеносного сосуда (например, опухолью), серповидно-клеточную анемию, локализованное сильное охлаждение (например, обморожение), наложение жгута, стимуляцию глутаматных рецепторов, артериовенозные мальформации, разрыв важных кровеносных сосудов, снабжающих ткан или орган, и анемию.
Транзиторная ишемическая атака обычно относится к a преходящему (например, кратковременному) эпизоду неврологической дисфункции, вызванному потерей кровотока (например, в головном мозге, спинном мозге или сетчатке) без острого инфаркта (например, гибели ткани). В некоторых вариантах осуществления транзиторная ишемическая атака длится в течение менее 72 часов, 48 часов, 24 часов, 12 часов, 10 часов, 8 часов, 4 часов, 2 часов, 1 часа, 45 минут, 30 минут, 20 минут, 15 минут, 10 минут, 5 минут, 4 минут, 3 минут, 2 минут или 1 минуты.
Ангионевротический отек
Ангионевротический отек представляет собой быстрое разбухание дермы, подкожной ткани, слизистой оболочки и подслизистых тканей. Ангионевротический отек обычно классифицируется как наследственный или как приобретенный.
«Приобретенный ангионевротический отек» может быть иммунологическим, неиммунологическим или идиопатическим; который вызывается, например, аллергией как побочным эффектом лекарственных средств, например ингибиторов АПФ.
Термин «наследственный ангионевротический отек» или «HAО» относится к генерическому расстройству, которое периодически приводит к острому отеку (например, разбуханию), который может возникать почти во всех частях тела, включая лицо, конечности, шею, горло, гортань, кисти и стопы, желудочно-кишечный тракт и гениталии. Приступы НАО зачастую могут быть опасными для жизни, их тяжесть зависит области поражения, например приступы в брюшной области могут привести к интенстинальной непроходимости, в то время как отек гортани и верхних дыхательных путей может привести к асфиксии. Патогенез наследственного ангионевротического отека может быть связан с безальтернативной активацией контактного пути метаболизма посредством начальной выработки калликреина или факторов свертывания (например, фактора XII).
Объективные признаки заболевания и симптомы включают разбухание (отек), например, лица, слизистой оболочки ротовой полости или горла и языка. Зуд, боль, снижение чувствительности на пораженных участках, уртрикария (т.е. крапивница) или стридор дыхательных путей также могут быть объективными признаками ангионевротического отека. Однако не может быть никакого сопутствующего зуда или крапивницы, например, при наследственном ангионевротическом отеке. Пациенты с НАО могут испытывать боль в животе (например, боль в животе, длящуюся в течение от одного до пяти дней, приступы боли в животе с повышением количества лейкоцитов в крови пациента), рвоту, слабость, водянистую диарею или сыпь.
Брадикинин играет важную роль в ангионевротическом отеке, особенно в наследственном ангионевротическом отеке. Брадикинин высвобождается клетками различных типов в ответ на ряд различных стимулов и является медиатором боли. Вмешательство в продуцирование брадикинина или его разложение может приводить к ангионевротическому отеку. При наследственном ангионевротическом отеке непрерывная выработка фермента калликреина может способствовать образованию брадикинина. Ингибирование калликреина может мешать выработке брадикинина и лечить или предотвращать ангионевротический отек.
Фармацевтические композиции
Композиции по настоящему изобретению включают соединение по настоящему изобретению (например, соединение формулы (II), например соединение формул (I), (I-a), (I-b), (I-c), (I-d), (I-e), (I-f), (I-g), (I-h), (I-i), (I-j), (I-k), (I-l), (I-m), (I-n), (I-o), (I-p), (I-q), (I-r), (I-с), (I-s1), (I-s2), (I-t), (I-u) или (I-v), а также дополнительные терапевтические средства, если таковые присутствуют, в количествах, эффективных для обеспечения лечения заболевания или симптомов заболевания (например, такого как заболевания, связанное с фактором Ia или калликреином).
Фармацевтически приемлемые носители, адъюванты и наполнители, которые могут использоваться в фармацевтической композиции по настоящему изобретению, включают, но без ограничения, ионно-обменные смолы, оксид алюминия, стеарат алюминия, лецитин, самоэмульгирующиеся системы лекарственной доставки (self-emulsifying drug delivery systems - SEDDS), такие как d-α-токоферолполиэтиленгликоль 1000 сукцинат, поверхностно-активные вещества, используемые в фармацевтических лекарственных формах, такие как Tweens или другие аналогичные полимерные матрицы доставки, белки сыворотки, такие как альбумин сыворотки крови человека, буферные системы, такие как фосфаты, глицин, сорбиновая кислота, сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как сульфат протамина, динатриевый гидрофосфат, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натриевую соль карбоксиметилцеллюлозы, полиакрилаты, воски, блок сополимеры полиэтилена и полиоксипропилена, полиэтиленгликоль и ланолин. Циклодекстрины, такие как α-, β-, и γ-циклодекстрин или их химически модифицированные производные, такие как гидроксиалкилциклодекстрины, включая 2- и 3-гидроксипропил-β-циклодекстрины или другие растворимые производные, также могут успешно применяться для улучшения доставки соединений формул по настоящему изобретению.
Способы введения
Фармацевтические композиции по настоящему изобретению могут вводиться перорально, ректально или парентерально (например, внутривенным вливанием, внутривенной болюсной инъекцией, ингаляцией, имплантацией). Термин «парентерально», когда используется в настоящем описании, включает подкожную, внутрикожную, внутривенную (например, внутривенную инфузию, внутривенную болюсную инъекцию), назальную, ингаляционую, легочную, чрескожную, внутримышечную, внутрисуставную, внутриартериальную, интрасиновинальную, внутригрудинную, внутриоболочечную, внутриочаговую и внутричерепную инъекцию или другие методы вливания. Фармацевтические композиции по настоящему изобретению могут содержать любые стандартные нетоксичные фармацевтически приемлемые носители, адъюванты или наполнители. В некоторых случаях pH препарата может регулироваться с помощью фармацевтически приемлемых кислот, оснований или буферов для повышения стабильности соединения в препарате или его формы доставки.
Фармацевтические композиции могут быть представлены в форме стерильного препарата для инъекции, например в форме стерильного водного или масляного раствора или стерильной водной или масляной суспензии для инъекций. Эта суспензия может быть получена в соответствии с методами, известными в данной области техники, с использованием подходящих дисергирующих или смачивающих агентов (таких как, например, Tween 80) и суспендирующий агентов. Стерильный препарат для инъекций также может представлять собой стерильный раствор или стерильную суспензию для инъекций в нетоксичном парентерально приемлемом разбавителе или растворителе, например раствор в 1,3-бутандиоле. Приемлемыми разбавителями и растворителями, которые могут применяться, являются маннит, вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла успешно применяются в качестве растворители или суспендирующей среды. Для этой цели может применяться любое легкое нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, могут применяться для получения препаратов для инъекций, как и природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно их полиоксиэтилированные формы. Эти масляные растворы или суспензии также могут содержать разбавитель или дисперсант на основе длинноцепочечного спирта или карбоксиметилцеллюлозу или подобные добавки, которые обычно используются в препаратах фармацевтически приемлемлемых лекарственных форм, таких как эмульсии и/или суспензии. Другие, традиционно используемые поверхностно-активные вещества, такие как Tweens или Spans или другие аналогичные эмульгаторы, или добавки повышающие биодоступность, которые традиционно применяются при производнстве фармацевтически приемлемых твердых, жидких или других лекарственных форм, могут также применяться для получения препарата.
Фармацевтические композиции по настоящему изобретению могут вводиться перорально в любой перорально приемлемой лекарственной форме, включая, но без ограничения, капсулы, таблетки, эмульсии и водные суспензии, дисперсии и растворы. В случае таблеток для перорального применения, носители, которые обычно используются, включают лактозу и кукурузный крахмал. Смазывающие вещества, такие как стеарат магния, также обычно добавляются. Для перорального введения в форме капсул применимые разбавители включают лактозу и высушенный кукурузный крахма. Когда водные суспензии или эмульсии вводятся перорально, активный ингредиент может суспендироваться или растворяться в масляной фазе в сочетании с эмульгаторами или суспендирующими агентами. При желании могут добавляться определенные подсластители, вкусовые добавки, красители или добавки, маскирующие вкус.
Соединения по настоящему изобретению могут вводиться, например, инъекцией внутривенно (например, внутривенной инфузией, внутривенной болюсной инъекцией), внутриартериально, подкожно, внутрибрюшинно, внутримышечно или подкожно; или перорально, буккально, назально, через слизистую оболочку, местно в дозе в интервале от примерно 0,5 до примерно 100 мг/кг массы тела, альтернативно в дозах в интервале от 1 мг до 1000 мг/доза, каждые 4-120 часов или в соответствии с требованиями конкретного лекарственного средства. Способы, описанные в настоящем изобретении, подразумевают введение эффективного количества соединения или композиции соединения для обеспечения желательного или заявленного эффекта. Обычно фармацевтические композиции по настоящему изобретению будут вводиться от примерно 1 до примерно 6 раз в день (например, внутривенной болюсной инъекцией) или, альтернативно, непрерывной инфузией. Такое введение может применяться в качестве длительной или краткосрочной терапии. Количество активного ингредиента, которое может объединяться с носителем для получения лекарственной формы стандартной дозы, будет изменяться в зависимости от организма-хозяина, подлежащего лечению, и конкретного способа введения. Типичный препарат будет содержать от примерно 5% до примерно 95% активного соединения (масс./масс). Альтернативно, такие препараты содержат от примерно 20% до примерно 80% активного соединения.
В некоторых вариантах осуществления фармацевтические композиции, приготовленные для перорального введения или внутривенного введения, вводятся пациенту от 1 раза в день до 6 раз в день (например, 2 раза в день или 4 раза в день). В некоторых вариантах осуществления фармацевтическая композиция, приготовленная для перорального введения, вводится пациенту от 1 раза в день до 6 раз в день (например, 2 раза в день или 4 раза в день) в течение примерно от 3 дo 9 месяцев. В некоторых вариантах осуществления фармацевтическая композиция, приготовленная для перорального введения, вводится пациенту от 1 раза в день до 6 раз в день (например, 2 раза в день или 4 раза в день) до конца его или ее жизни.
В некоторых вариантах осуществления соединение вводится перорально или парентерально. В некоторых вариантах осуществления соединение вводится перорально. В некоторых вариантах осуществления соединение вводится парентерально. В некоторых вариантах осуществления соединение или его фармацевтически приемлемая соль или фармацевтическая композиция вводится после прекращения использования пациентом перорального антикоагулянта прямого действия. В некоторых вариантах осуществления пациент использовал пероральный антикоагулянт. В некоторых вариантах осуществления пациентом является млекопитающее. В некоторых вариантах осуществления млекопитающим является человек.
Комбинации
При осуществлении способов по настоящему изобретению может быть желательно вводить соединения по настоящему изобретению (например, ингибиторы фактора XIa или калликреина) в комбинации с каждым другим и одним или несколькими другими лекарственными средствами для достижения лечебного действия, такимио как антитромботическими или антикоагулянтными средствами, гипотензивными средствами, противоишемическими средствми, противоарритрическими средствми, ингибиторами тромбоцитарной функции и т.д. Например, способы по настоящему изобретению осуществляться введением низкомолекулярных ингибиторов фактора XIa или калликреина в комбинации с низкомолекулярным ингибитором фактора XIa или калликреина. Точнее, способы по настоящему изобретению могут осуществляться введением низкомолекулярных ингибиторов фактора XIa или калликреина в комбинации с аспирином, клопидогрелем, тиклопидином или CS-747, варфарином, низкомолекулярными гепаринами (такими как LOVENOX), блокаторами GPIIb/GPIIIa, ингибиторами PAI-1, такими как XR-330 и T-686, антагонистами рецепторов P2Y1 и P2Y12; антагонистами рецепторов тромбоксана (такими как ифетробан), миметиками простациклина, ингибиторами тромбоксан A синтетазы (такими как пикотамид), антагонистами серотонин-2-рецептора (такими как кетансерин); соединениями, которые ингибируют другие факторы коагуляции, такие как FVII, FVIII, FIX, FX, протромбин, TAFI и фибриноген, или другими соединениями, которые ингибируют FXI или калликреин; фибринолитиками, такими как TPA, стрептокиназа, ингибиторы PAI-1, и ингибиторами α-2-антиплазмина, такими как антитела против α-2-антиплазмина, антагонистами рецептора фибриногена, ингибиторами α-1-антитрипсина, гиполипидемическими средствами, такими как ингибиторы HMG-CoA редуктазы (например, правастатин, симвастатин, аторвастатин, флувастатин, церивастатин, AZ4522 и итавастатин), и ингибиторами микросомального триглицерид-переносящего белка (такими как раскрытые в Патентах США №№ 5739135, 5712279 и 5760246); гипотензивными средствами, такими как ингибиторы ангиотензинпревращающего фермента (например, каптоприл, лизиноприл или фозиноприл); антагонистами рецепторов ангиотензина II (например, ирбесартаном, лозартаном или валсартаном); ингибиторами ACE/NEP (например, омапатрилатом и гемопатрилатом); или β-блокаторами (такими как пропранолол, надолол и карведилол). Способы по настоящему изобретению могут осуществляться введением низкомолекулярных ингибиторов фактора XIа или каликреина в комбинации с противоаритмическим средствами, такими как средства для лечения мерцательной аритмии, например амиодарон или дофетилид. Способы по настоящему изобретению также могут осуществляться в комбинации с непрерывной заместительной почечной терапией для лечения, например, острой почечной недостаточности.
При осуществлении способов по настоящему изобретению может быть желательно вводить соединения по настоящему изобретению (ингибиторы фактора XIa или калликреина) в комбинации с лекарственными средствами, которые повышают уровни cAMP или cGMP в клетках для получения терапевтического полезного действия. Например, соединения по настоящему изобретению могут обладать полезным действие при применении в комбинации с ингибиторами фосфодиэстеразы, включая ингибиторы PDE1 (такие, как описанные в Journal of Medicinal Chemistry, Vol. 40, pp. 2196-2210 [1997]), ингибиторы PDE2, ингибиторы PDE3 (такие как ревизинон, пимобендан или олпринон), ингибиторы PDE4 (такие как ролипрам, циломиласт или пикламиласт), ингибиторы PDE7 или другие ингибиторы PDE, такие как дипиридамол, цилостазол, силденафил, денбутилин, теофиллин (1,2-диметилксантин), ARIFLOT.TM. (т.е., цис-4-циано-4-[3-(циклопентилокси)-4-метоксифенил]циклогексан-1-карбоновая кислота), арофилин, рофлумиласт, C-11294A, CDC-801, BAY-19-8004, ципамфиллин, SCH351591, YM-976, PD-189659, мезиопрам (mesiopram), пумафентрин, CDC-998, IC-485 и KW-4490.
Способы по настоящему изобретению могу осуществляться введением соединений по настоящему изобретению в комбинации с протромбиновыми лекарственными средствами, такими как тканевый активатор плазминогена (природный или рекомбинантный), стрептокиназа, ретеплаза, активаза, ланотеплаза, урокиназа, проурокиназа, ASPAC (anisolated streptokinase plasminogen activator complex), активаторы плазминогена слюнных желез животных и т.п. Способы по настоящему изобретению могут осуществляться введением соединений по настоящему изобретению в комбинации с β-адренергическими агонистами, такими как албутерол, тербуталин, формотерол, салметерол, битолтерол, пилбутерол или фенотерол; aантихолинергетиками, такими как бромид ипратропия; противовоспалительными кортикостероидами, такими как беклометазон, триамцинолон, будесонид, флутиказон, флунизолид или дексаметазон; и противовоспалительными средствами, такими как кромолин, недокромил, теофиллин, зилеурон, зафирлукаст, монтелеукаст и пранлеукаст. Низкомолекулярные ингибиторы фактора XIa или калликреина могут действоватьт синергически с одним или несколькими вышеуказанными средствами. Таким образом, могут использоваться уменьшенные дозы тромболитического(их) средства(средств), получая полезное действие введения указанных соединений при минимизации возможных геморрагических и других побочных эффектов.
Курс лечения
Композиции по настоящему изобретению включают эффективное количество соединения по настоящему изобретению (например, ингибитора фактора XIa или калликреина) в комбинации с одним или несколькими другими лекарственными средствами (например, дополнительным терапевтическим средством), такими как противотромботические или противокоагулянтные средства, гипотензивные средства, противоишемические средства, противоаритмические средства, ингибиторы тромбоцитарной функции и т.д., для достижения терапевтического полезного действия.
В некоторых вариантах осуществления дополнительное терапевтическое средство вводится после введения соединения по настоящему изобретению (например, ингибитора фактора XIa или калликреина). В некоторых вариантах осуществления дополнительное терапевтическое средство вводится через 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 10 часов, 12 часов, 14 часов, 18 часов, 24 часов, 48 часов, 72 часа или через более длительный промежуток времени после введения соединения по настоящему изобретению (например, ингибитора фактора XIa или калликреина). В некоторых вариантах осуществления дополнительное терапевтическое средство вводится (например, перорально) после выписки из медицинского учреждения (например, больницы).
В некоторых вариантах осуществления соединение по настоящему изобретению (например, ингибитор фактора XIa или калликреина) и дополнительное терапевтическое средство вводятся в одну композицию или лекарственную форму. В некоторых вариантах осуществления соединение по настоящему изобретению (например, ингибитор фактора XIa или калликреина) и дополнительное терапевтическое средство вводятся отдельно. В некоторых вариантах осуществления соединение по настоящему изобретению (например, ингибитор фактора XIa или калликреина) и дополнительное терапевтическое средство вводятся последовательно. В некоторых вариантах осуществления соединение по настоящему изобретению (например, ингибитор фактора XIa или калликреина) и дополнительное терапевтическое средство вводятся раздельно и последовательно. Обычно по меньшей мере одно соединение по настоящему изобретению (например, ингибитор фактора XIa или калликреина) и дополнительное терапевтическое средство вводятся парентерально (например, назально, внутримышечно-буккально, ингаляцией, имплантацией, чрескожно, внутривенно (например, внутривенной инфузией, внутривенной болюсной инъекцией), подкожно, внутрикожно, интраназально, пульмонально, чрескожно, внутрисуставным способом, внутриартериально, интрасиновинально, внутригрудинно, интратекально, внутриочаговым способом и внутричерепной инъекцией или другими методами инфузии); перорально; или ректально, например, внутримышечной инъекцией или внутривенно (например, вутривенной инфузией, внутривенной болюсной инъекцией)). В некоторых вариантах осуществления соединение по настоящему изобретению вводится парентерально (например, назально, буккально, внутривенно (например, внутривенной инфузией, внутривенной болюсной инъекцией) или внутримышечно). В некоторых вариантах осуществления дополнительное терапевтическое средство вводится перорально. В некоторых вариантах осуществления соединение по настоящему изобретению (например, ингибитор фактора XIa или калликреина) вводится парентерально (например, назально, буккально, внутривенно (например, внутривенной инфузией, внутривенной болюсной инъекцией или внутримышечно), а дополнительное терапевтическое средство вводится перорально.
В некоторых вариантах осуществления соединение по настоящему изобретению (например, ингибитор фактора XIa или калликреина) может вводиться один раз или несколько раз в день. Продолжительность лечения может составлять, например, при введении один раз в день период примерно 1, 2, 3, 4, 5, 6, 7 дней или более. В некоторых вариантах осуществления лечение является постоянным (например, в течение всей жизни). В некоторых вариантах осуществления вводят либо однократную дозу в виде отдельной лекарственной формы, лебо несколько лекарственных форм меньшей дозы или многократным введением разделенных доз через определенные промежутки времени. Например, лекарственная форма может вводиться через от прмерно 0 часов до примерно 1час, от примерно 1 час до примерно 24 часа, от примерно 1 до примерно 72 часов, от примерно 1 до примерно 120 часов или от примерно 24 часов до по меньшей мере примерно 120 часов после травмы. Альтернативно, лекарственная форма может вводиться через примерно 0,5, 1, 1,5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 30, 40, 48, 72, 96, 120 часов боле после травмы. Последующие лекарственные формы могут вводиться в любое время после первоначального введения таким образом, что достигается терапевтическое действие. В некоторых вариантах осуществления исходная доза вводится перорально. В некоторых вариантах осуществления последующие дозы после начальной дозы вводятся парентерально (например, назально, внутримышечно-буккально, ингаляцией, имплантацией, чрескожно, внутривенно (например, внутривенной инфузией, внутривенной болюсной инъекции), подкожной, внутрикожной, назальной, пульмональной, чрескожной, внутрисуставной, внутриартериальной, интрасиновинальной, внутригрудинной, внутриоболочечной, внутриочаговой и внутричерепной инъекцией или другими методами инфузии); перорально; или ректально.
В некоторых вариантах осуществления соединения по настоящему изобретению (например, ингибитор фактора XIa или калликреина) вводятся перорально, например в виде жидкой или твердой лекарственной формы для проглатывания в течение от примерно 5 минут до примерно 1 недели; от примерно 30 минут до примерно 24 часов, от примерно 1 часа до примерно 12 часов, от примерно 2 часов до примерно 12 часов, от примерно 4 часов до примерно 12 часов, от примерно 6 часов до примерно 12 часов, от примерно 6 часов до примерно 10 часов; от примерно 5 минут до примерно 1 часа, от примерно 5 минут до примерно 30 минут; от примерно 12 часов до примерно 1 недели, от примерно 24 часов до примерно 1 недели, от примерно 2 дней до примерно 5 дней или от примерно 3 дней до примерно 5 дней. В одном варианте осуществления соединение по настоящему изобретению (например, ингибитор фактора XIa или калликреина) вводится перорально в виде жидкой лекарственной формы. В другой варианте осуществления соединение по настоящему изобретению (например, ингибитор фактора XIa или калликреина) вводится перорально в виде твердой лекарственной формы.
Если у пациента, проходящего терапию, наблюдается неполный ответ или рецидив после завершения первого цикла терапевтического лечения, могут потребоваться последующие курсы терапии для достижения ограниченного или полного терапевтического ответа (например, постоянное лечение, например, в течение всей жизни).
В некоторых вариантах осуществления соединение по настоящему изобретению (например, ингибитор фактора XIa или калликреина) вводится внутривенно, например в виде внутривенной инфузии или внутривенной болюсной инъекции, в течение от примерно 5 минут до примерно 1 недели; от примерно 30 минут до примерно 24 часов, от примерно 1 часа до примерно 12 часов, от примерно 2 часов до примерно 12 часов, от примерно 4 часов до примерно 12 часов, от примерно 6 часов до примерно 12 часов, от примерно 6 часов до примерно 10 часов; от примерно 5 минут до примерно 1 часа, от мерно 5 минут до примерно 30 минут; от примерно 12 часов до примерно 1 недели, от примерно 24 часов до примерно 1 недели, от примерно 2 дней до примерно 5 дней или от примерно 3 дней до примерно 5 дней. В одном варианте осуществления соединение по настоящему изобретению (например, ингибитор фактора XIa или калликреина) вводится в виде внутривенной инфузии в течение примерно 5, 10, 15, 30, 45 или 60 минут или дольше; примерно 1, 2, 4, 6, 8, 10, 12, 16 или 24 часов или дольше; примерно 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 дней или дольше.
В некоторых вариантах осуществления соединение вводится после ишемической атаки. В некоторых вариантах осуществления соединение вводится примерно через 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14 дней или более после ишемической атаки. В некоторых вариантах осуществления соединение вводится примерно через 1, 2, 3, 4, 5, 6, 7 или 8 недель или более после ишемической атаки. В некоторых вариантах осуществления соединение вводится примерно через 1, 2, 3, 4, 5 или 6 месяцев или более после ишемической атаки.
В некоторых вариантах осуществления соединение вводится в комбинации с дополнительным терапевтическим средством. В некоторых вариантах осуществления дополнительное терапевтическое средство вводится после введения соединения. В некоторых вариантах осуществления дополнительное терапевтическое средство вводится перорально. В некоторых вариантах осуществления дополнительное терапевтическое средство вводится по меньшей мере через 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 14, 16, 18, 20 или 24 часа или более после введения соединения. В некоторых вариантах осуществления дополнительное терапевтическое средство вводится по меньшей мере через 1, 2, 3, 4, 5, 6, 7, 14, 21 или 28 дней или более после введения соединения. В некоторых вариантах осуществления дополнительное терапевтическое средство вводится примерно через 1 день, примерно через 2 дня, примерно через 3 дня, примерно через 4 дня, примерно через 5 дней, примерно через 6 дней, примерно через 7 дней или более после введения соединения. В некоторых вариантах осуществления дополнительное терапевтическое средство вводится постоянно после введения соединения. В некоторых вариантах осуществления дополнительное терапевтическое средство представляет собой НПВЛС, ингибитор агрегации тромбоцитов или антикоагулянт. В некоторых вариантах осуществления дополнительное терапевтическое средство приводит к аддитивному терапевтическому эффекту. В некоторых вариантах осуществления дополнительное терапевтическое средство приводит к синергическому терапевтическому эффекту.
Дозы и схемы лечения
Эффективное количество низкомолекулярного ингибитора фактора XIa или калликреина, вводимого в соответствии с настоящим изобретением, может быть определено специалистом в данной области техники. Конкретная доза и частота введения для любого конкретного пациента может изменяться и будет зависеть от различных факторов, включая активность конкретного используемого соединения, метаболическую стабильность и продолжительность действия этого соединения, вид, возраст, массу тела, общее состояние здоровья, пол и пищевой рацион пациента, способ и время введения, скорость выведения, комбинированные лекарственные средства и тяжесть конкретного состояния.
При улучшении состояния пациента при необходимости может быть введена поддерживающая доза соединения, композиции или комбинации, предусмотренных здесь. Впоследствии дозировка или частота введения или и то, и другое могут быть уменьшены в зависимости от симптомов до уровня, при котором улучшенное состояние сохраняется, когда симптомы ослаблены до желаемого уровня. Однако пациентам может потребоваться периодическое лечение на долгосрочной основе при любом рецидиве симптомов заболевания.
ПРИМЕРЫ
Исходные материалы и различные промежуточные соединения, описанные в представленных далее примерах, могут быть получены из коммерческих источников, из коммерчески доступных органических соединений или с использованием известных методов синтеза. Типичные примеры способов, подходящих для получения промежуточных соединений по изобретению, также приведены ниже.
Общие методики
Все неводные реакции проводили в атмосфере азота для сохранения безводной атмосферы и получения максимальных выходов. Все реакционные смеси перемешивали с использованием перемешивающего устройства с верхним приводом или магнистой мешалки с тефлоновым покрытием. Термин «сушат над» относится к сушке раствора продукта реакции над конкретным осушителем, например сульфатом магния или сульфатом натрия, и последующей фильтрации раствора через подходящую фильтровальную бумагу или через шоттовскую воронку. Термины «концентрируют», «концентрируют при пониженном давлении» или «упаривают» относятся к удалению растворителей при пониженном давлении с использованием роторного испарителя. Термины «хроматография» или «хроматографирование» относится к применению колоночной флэш-хроматографии на силикагеле, если не указано иное. Флэш-хроматография может выполняться с применением давления газа (например, азота) или с применением механического насоса для подачи растворителя под давлением, такого как коммерческая система от Biotage или других поставщиков. Термин «препаративная ТСХ» относится к тонкослойной хроматографии на силикагельных пластинах. Термин «флэш-хроматграфия» относится к колоночной хроматографии под давлением и может проводиться с использованием коммерческих установок, такой как установка от Biotage. Термин «время удерживания» относится времени элюирования продукта с помощью ВЭЖХ в специфических условиях. Если не указано иное, все ЯМР спектры представляют собой are спектры протонного (1H) ЯМР в указанном растворителе.
В приведенных далее примерах высокоэффективную жидкостную хроматографию/масс-спектрометрию (ВЭЖХ/МС) для характеристики соединений выполняют в следующих условиях:
1. Аппаратура ВЭЖХ/МС: Анализ проводят с использованием модуля бинарного градиента Waters 2545 (Waters Corporation, Milford, MA), Waters SFO System Fluidics Organizer, детектора на диодной матрице Waters 2996 и автоматического пробоотборника Waters 2767, масс-детектора 3100. Данные получают с использованием программного обеспечения MassLynxTM 4.0 с обработкой с помощью OpenLynxTM и AutoLynxTM.
2. Условия аналитической ВЭЖХ: колонка 4,6 × 50 мм; УФ 10 спектров/сек., 220-340 нм; объемная скорость потока 2,0 мл/мин.; объем впрыска 5 мкл;
условия градиента A: подвижная фаза A - вода с 0,1% муравьиной кислоты; подвижная фаза B - ацетонитрил с 0,1% муравьиной кислоты; градиент: 1,50 мин. от 99,0% A до 95,0% B; 0,5 мин. выдерживание; затем возврат до 99,0% A в течение 0,5 мин.;
условия градиента B: подвижная фаза A - вода с 0,1% муравьиной кислоты; подвижная фаза B - ацетонитрил с 0,1% муравьиной кислоты, градиент: 3,00 мин. от 95,0% A до 95,0% B; 2,0 мин. выдерживание; затем возврат до 95,0% A в течение 0,5 мин.
Значения используемых в экспериментальных примерах аббревиатур представлены в таблице ниже.
Таблица аббревиатур
Схема 1
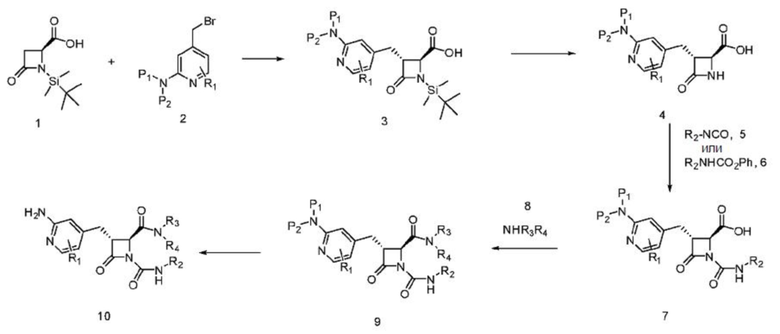
Схема 1 иллюстрирует общий способ получения (2S,3R)-транс-дизамещенных-4-оксоазетидинов общей структуры 10, содержащих амидную группу в 2-положении. Алкилирование дианиона транс 1 с помощью коммерчески доступного или легко синтезируемого соединения 2 (как описано в получении промежуточных соединений 7 и 8) приводит к получению желаемого соединения 3 в форме преимущественно транс-продукта при сохранении стереохимии в положении 2 (Baldwin, J.E., et al., Tetrahedron, 1990, 46, 4733). Удаление TBS защитной группы при N-1 приводит к получению соединения 4, которое затем подвергается взаимодествию с изоцианатом 5 или фенилкарбаматом 6 с получением соединения 7. Связывание карбоновой кислоты 7 с коммерчески доступным или легко синтезируемым амином 8 (как описано в получении промежуточного соединения 6) в присутствии основания и связывающего реагента приводит к получению соединения 9. Последующее удаление защитных групп P1 и P2 приводит к получению соединения 10.
Схема 2

Схема 2 иллюстрирует альтернативный способ получения соединения 10. Связывание карбоновой кислоты 3 с коммерчески доступным или легко синтезируемым амином 8 (как описано в получении промежуточного соединения 6) в присутствии основания и реагента связывания приводит к получению соединения 11. Удаление TBS защитной группы при N-1 приводит к получению соединения 12, которое затем подвергают взаимодействию с изоцианатом 5 или фенилкарбаматом 6 приводит к получению соединения 9. Последующее удаление защитных групп P1 и P2 обеспечивает получение примера общего соединения 10.
Схема 3

Схема 3 иллюстрирует способ получения соединения 18. Алкилирование дианиона соединения 1 с коммерчески доступным или легко синтезируемым соединение 13 (полученным как описано для получния промежуточного соединения 9) приводит к получению желаемого соединения 14. Связывания карбоновой кислоты 14 с коммерчески доступным или легко синтезируемым амином 8 (как описано в получении промежуточного соединения 6) в присутствии основания и реагента связывания приводит к получению соединения 15. Удаление TBS защитной группы при N-1 приводит к получению соединения 16, которое затем подвергается взаимодействию с изоцианатом 5 или фенилкарбаматом 6 с получением соединения 17. Последующее удаление защитной группы P2 приводит к получению соединения 18.
Схема 4

Схема 4 иллюстрирует способ получения соединения 19. Алкилирование соединения 18 (R5=водород или алкил) с использованием алкилгалогенида в присутствии основания или the взаимодействие соединения 18 с алкилальдегидом в присутствии восстановителя (такого как NaBH3CN и т.д.) приводит к получению соединения 19.
Схема 5

Схема 5 иллюстрирует способ получения соединения 21. Алкилирование соединение 9 с использованием алкилгалогенида в присутствии основания приводит к получению соединения 20. Последующее удаление защитных групп P1 и P2 приводит к получению соединения 21.
Пример 1. Получение промежуточных соединений.
Промежуточное соединение 1: Фенил-N-[(1S)-1-циклогексил-2,2,2-трифторэтил]карбамат
К перемешиваемому раствору фенилхлорформиата (714 мкл, 5,69 ммоль) в сухом ТГФ (30 мл) при 0°C по каплям в течение 45 минут добавляют раствор (1S)-1-циклогексил-2,2,2-трифторэтанамина(1,00 г, 5,52 ммоль) и пиридина (536 мкл, 6,63 ммоль) в ТГФ (18 мл) После добавления в течение 45 минут при 0°C смесь разбавляют этилацетатом (EA) (50 мл), промывают водным раствором NaHCO3 (50 мл), водой (40 мл) и насыщенным водным раствором соли (30 мл), сушат над MgSO4 и концентрируют. Полученный твердый остаток растирают с гексанами (35 мл), собирают фильтрацией, промывают небольшими количествами гексанов и сушат с получением 1,51 г (91%) указанного в заголовке соединения в виде твердого белого вещества, которое используют далее без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 7,40 (м, 2H), 7,24 (м, 1H), 7,17 (м, 2H), 5,20 (уш. д, 1H), 4,25 (м, 1H), 1,92-1,68 (м, 6 H), 1,40-1,05 (м, 5H); МС (ESI+) m/z 302,1 (M+H)+, время удерживания: 2,03 мин. (метод A).
Промежуточное соединение 2: Фенил N-[(1R)-1-циклогексилпропил]карбамат

Схема 6
Стадия 1. Получение [N(E),S(S)]-N-(циклогексилметилен)-2-метил-2-пропансульфонамида
К раствору циклогексанкарбальдегида (25 г, 0,223 моль) и (S)-(-)-2-метил-2-пропансульфонамида (28,4 г, 0,234 моль) в ТГФ (300 мл) добавляют этоксид титана (IV) (101,7 г, 0,446 моль). Полученную реакционную смесь нагревают и выдерживают при 75°C в течение 2 часов. Смесь охлаждают до КТ. Добавляют насыщенный раствор соли (250 мл) и смесь энергично перемешивают, затем фильтруют через Celite® и промывают EA (300 мл). Водный слой экстрагируют EA (2 × 200 мл). Объединенную органическую фазу сушат над MgSO4, фильтруют и концентрируют. Сырой продукт очищают хроматографией (элюирование: 4:1 гексан:EA) с получением продукта в виде бесцветного масла (45,6 г, 95%). 1H ЯМР (400 МГц, CDCl3) δ 7,97 (1H, д, J=4,6 Гц), 2.47 (1H, м), 1,92-1,68 (7H, м), 1,67-1,35 (3H, м), 1,20 (9H, с).
Стадия 2. Получение [S(S)]-N-((1R)-1-циклогексилпропил)-2-метил-2-пропансульфонамида

[N(E),S(S)]-N-(циклогексилметилен)-2-метил-2-пропансульфонамид (32,3 г, 0,15 моль) растворяют в TBME (500 мл). Реакционную смесь охлаждают до -40°C, к смеси по каплям добавляют бромид этилмагния (3M в эфире, 100 мл, 0,3 моль) и перемешивают реакционную смесь при -25°C в течение 4 часов. Добавляют водный раствор хлорида аммония и затем EA. Слои разделяют и водный слой экстрагируют EA два раза. Объединенную органическую фазу промывают насыщенным водным раствором соли, сушат над сульфатом натрия, концентрируют и очищают хроматографией (элюирование: 2:1 гексан:EA) с получением [S(S)]-N-((1R)-1-циклогексилпропил)-2-метил-2-пропансульфонамида в виде бесцветного масла (22 г, 60%). 1H ЯМР (400 МГц, CDCl3) δ 0,90-1,00 (м, 4H), 1,12 (м, 1H), 1,20 (с, 9 H), 1,44 (м, 1H), 1,52-1,80 (м, 9H), 2,88-3,00 (м, 2H).
Стадия 3. Получение гидрохлорида (R)-1-циклогексилпропан-1-амина
[S(S)]-N-((1R)-1-циклогексилпропил)-2-метил-2-пропансульфонамид (31,86 г, 0,129 моль) растворяют в метаноле (200 мл) и медленно добавляют 4M HCl в 1,4-диоксане (100 мл). Реакционный раствор перемешивают в течение 60 мин. Реакционную смесь концентрируют с получением твердого остатка. К твердому остатку добавляют гексан (200 мл) и перемешивают полученную суспензию при КТ в течение 60 мин. Твердое вещество собирают фильтрацией, промывают гексанами и сушат с получением гидрохлорида (R)-1-циклогексилпропан-1-амина в виде твердого белого вещества (12,8 г, 56%).
Стадия 4. Получение фенил-N-[(1R)-1-циклогексилпропил]карбамата

К перемешиваемой суспензии гидрохлорида (R)-1-циклогексилпропан-1-амина (5 г, 28,1 ммоль) в ДХМ (100 мл) при 0°C добавляют TEA (6,82 г, 67,4 ммоль) и затем медленно добавляют фенилхлорформиат (4,84 г, 31 ммоль). После перемешивания в течение дополнительных 45 мин. при КТ смесь разбавляют ДХМ (50 мл), промывают водным раствором NaHCO3 (50 мл), водой (40 мл) и насыщенным водным раствором соли (30 мл), сушат над MgSO4 и концентрируют. Полученный твердый остаток растирают с гексаном (35 мл). Твердый продукт собирают фильтрацией, промывают небольшими количествами гексана и сушат с получением 6,3 г (85%) указанного в заголовке соединения в виде твердого белого вещества, которое используют далее без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 7,40 (м, 2H), 7,24 (м, 1H), 7,17 (м, 2H), 4,75 (уш. д, 1H), 3,50 (м, 1H), 1,92-1,68 (м, 6 H), 1,50-1,05 (м, 10H). МС (ESI+) 262,2 (M+H)+, время удерживания: 2,09 мин. (метод A).
Промежуточное соединение 3. Дигидрохлорид (R)- 1-(3-пиридинил)-1-пропанамина
Схема 7
Стадия 1. Получение [S(S)]-N-[1-(пиридин-2-ил)пропилиден]-2-метилпропан-2-сульфинамида

Раствор 3-пропионилпиридина (5 г, 37 ммоль) , (S)-(-)-2-метилпропан-2-сульфинамида (4,7 г, 38,8 ммоль) и этоксида титана (IV) (17,8 г, 78 ммоль) в ТГФ (100 мл) нагревают и выдерживают при 80°C в течение 3 дней. После охлаждения добавляют насыщенный водный раствор соли (100 мл) и EA (100 мл). Смесь перемешивают при КТ в течение 1 часа. Смесь фильтруют через Celite® и промывают фильтр EA. Органическую фазу отделяют, концентрируют и остаток очищают хроматографией (элюирование: 2:1 гексан:EA) с получением продукта в виде бесцветного масла (8,15 г, 92%).
Стадия 2. Получение [S(S),R]-N-[1-(пиридин-2-ил)пропил]-2-метилпропан-2-сульфинамида

К раствору [(S(S)]-N-[1-(пиридин-2-ил)пропилиден]-2-метилпропан-2-сульфинамида (8,15 г, 34,2 ммоль) в ТГФ (150 мл) при -78°C добавляют раствор три-втор-бутилборогидрида (L-selectride, 1M раствор в ТГФ, 68 мл, 68 ммоль). Реакционную смесь перемешивают в течение 3 часов при -78°C. Реакционную смесь гасят водным раствором хлорида аммония (150 мл). Смесь фильтруют через Celite® и промывают фильтр EA. Органическую фазу отделяют, сушат над Na2SO4 и концентрируют. Остаток очищают хроматографией (элюирование: 1:1 гексан:EA) с получением продукта в виде бесцветного масла (4,3 г, 53%).
Стадия 3. Дигидрохлорид (R)-1-(3-пиридинил)-1-пропанамина

К раствору [S(S),R]-N-[1-(пиридин-2-ил)пропил]-2-метилпропан-2-сульфинамида (4,3 г, 17,9 ммоль) в метаноле (50 мл) медленно добавляют 4M HCl в 1,4-диоксане (22 мл). Реакционную смесь перемешивают в течение 1 часа. Реакционную смесь концентрируют с получением твердого остатка. Гексан (50 мл) добавляют к твердому остатку и перемешивают полученную суспензию при КТ в течение 1 часа. Твердое вещество собирают фильтрацией, промывают гексанами и сушат с получением дигидрохлорида (R)-1-(3-пиридинил)-1-пропанамина в виде твердого белого вещества (3,3 г, 90%). 1H ЯМР (400 Гц, ДМСО-d6) δ 0,80 (т, 2H), 1,90 (м, 1H), 2,10 (м,1H), 4,20 (м, 1H), 8,04 (д, 1H), 8,80 (д, 1H), 8,90 (м, 2H), 9,20 (уш., 4 H), МС (ESI+) m/z 137,1 (M+H)+. Время удерживания: 0,35 мин. (метод A).
Промежуточное соединение 4. Гидрохлорид (S)-1-циклопропил-2,2,2-трифторэтиламина
Схема 8
Стадия 1. Получение [N(E),R(S)]-N-(циклопропилметилен)-2-метил-2-пропансульфонамида
К раствору циклопропанкарбоксальдегида (10 г, 0,14 моль) в ТГФ (100 мл) добавляют (R)-2-метил-2-пропансульфонамид (18,2 г, 0,15 моль), и этоксид титана (IV) (65,1 г, 0,285 моль). Полученную смесь нагревают и выдерживают при 75°C в течение 2 часов. Смесь охлаждают до КТ. Добавляют насыщенный раствор соли (150 мл) и энергично перемешивают полученную смесь; затем смесь фильтруют через Celite® и слой фильтра промывают EA (300 мл). Водный слой экстрагируют EA (2 × 200 мл). Объединенные органические экстракты сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 4:1 гексан:EA) с получением продукта в виде бесцветного масла (22,2 г, 90%).
Стадия 2. Получение [R(S)]-N-((1S)-1-циклопропил-2,2,2-трифторэтил)-2-метил-2-пропансульфонамида

К раствору [N(E),R(S)]-N-(циклопропилметилен)-2-метил-2-пропансульфонамида (7,8 г, 45 ммоль) в ТГФ (150 мл) добавляют TMAF (5 г, 53,7 ммоль). Раствор дегазируют азотом и затем охлаждают до -55°C. По каплям добавляют раствор трифторметилтриметилсилана (9,6 г, 67,5 ммоль) в ТГФ (150 мл) и перемешивают реакционную смесь при -55°C в течение 2 часов. Реакционной смеси дают возможность медленно нагреться до -10°C и затем реакцию гасят водным раствором хлорида аммония. Водный слой экстрагируют EA, объединенный органический слой сушат и концентрируют с получением продукта в виде масла желтого цвета, который используют далее без дополнительной очистки (10,7 г, 90%).
Стадия 3. Получение гидрохлорида (S)-1-циклопропил-2,2,2-трифторэтиламина
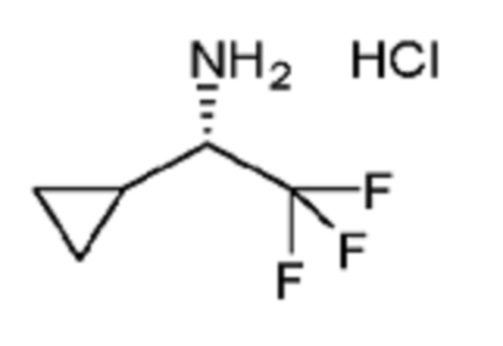
[R(S)]-N-((1S)-1-циклопропил-2,2,2-трифторэтил)-2-метил-2-пропансульфонамид (10,7 г, 44 ммоль) растворяют в метаноле (100 мл) и медленно добавляют 4M HCl в 1,4-диоксане (44 мл). Раствор перемешивают в течение 1 часа. Растворитель выпаривают с получением твердого остатка. К твердому остатку добавляют гексан (100 мл) с получением суспензии, которую перемешивают при КТ в течение 1 часа. Твердое вещество собирают фильтрацией, промывают гексанами и сушат с получением гидрохлорида (S)-1-циклопропил-2,2,2-трифторэтиламина в виде твердого белого вещества (5,4 г, 70%). 1H ЯМР (400 Гц, ДМСО-d6) δ 0,50-0,70 (м,4H), 1,04 (м, 1H), 3,60 (м, 1H) 9,20 (уш., 3H).
Промежуточное соединение 5. Синтез (R)-(+)-1-фенилпропилизоцианата

К раствору R-(+)-1-фенилпропиламина (5 г, 37 ммоль) в ДХМ (80 мл) добавляют 1N водный раствор NaHCO3 (86 мл). Смесь охлаждают до 0°C и добавляют трифосген (3,73 г, 12,6 ммоль). Реакционную смесь перемешивают в течение 15 мин. при 0°C. Реакционную смесь дважды экстрагируют ДХМ. Объединенную органическую фазу сушат над Na2SO4 и концентрируют с получением (R)-(+)-1-фенилпропилизоцианата в виде бесцветного масла (5,4 г, 91%), который используют в следующей стадии без дополнительной очистки. 1H ЯМР (400 Гц, CDCl3) δ 0,97 (т, 3H), 1,85 (м, 2H), 4,52 (т, 1H), 7,29 (м, 3H), 7,34 (м, 2H).
Промежуточное соединение 6. Гидрохлорид N,1-диметил-1H-пиразол-5-амина

Схема 9
Стадия 1. Получение трет-бутил-N-(1-метил-1H-пиразол-5-ил)карбамата
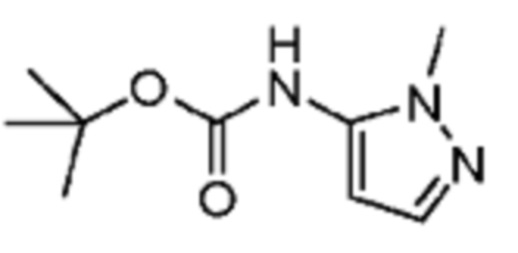
К раствору 1-метил-1H-пиразол-5-иламина (5 г, 51 ммоль) в ДХМ (30 мл) при КТ добавляют ди-трет-бутилдикарбонат (12,4 г, 56,8 ммоль) и затем TEA (10,4 г, 103 ммоль). Полученную смесь перемешивают при КТ в течение ночи. Смесь промывают водой. Органический слой сушат и упаривают с получением продукта в виде твердого белого вещества (10 г, 98%), который используют в следующей стадии без дополнительной очистки. 1H ЯМР (400 Гц, CDCl3) δ 1,40 (с, 9H), 1,74 (с, 1H), 3,16 (с, 3H), 6,02 (д, 1H), 7,40 (д, 1H).
Стадия 2. Получение трет-бутил-N-метил-N-(1-метил-1H-пиразол-5-ил)карбамата

К раствору трет-бутил-N-(1-метил-1H-пиразол-5-ил)карбамата (10 г, 51 ммоль) в ТГФ (100 мл) при 0°C одной порцией добавляют гидрид натрия (2,5 г, 60% в минеральном масле). Смеси дают возможность нагреться до КТ и перемешивают смесь в течение 30 мин. К реакционной смеси медленно добавляют йодметан (10,8 г, 76 ммоль). Полученную смесь перемешивают при КТ в течение ночи. Реакционную смесь гасят водным раствором хлорида аммония (30 мл). Смесь экстрагируют EA (50 мл × 3). Объединенные органические фазы промывают насыщенным водным раствором соли, сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 4:1 гексан:EA) с получением продукта в виде бесцветного масла (9,5 г, 89%). 1H ЯМР (400 Гц, CDCl3) δ 1,40 (с, 9H), 3,16 (с, 3H), 3,64 (с, 3H), 6,10 (д, 1H), 7,42 (д, 1H).
Стадия 2. Получение гидрохлорида N,1-диметил-1H-пиразод-5-амина
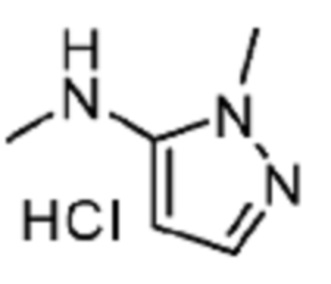
К раствору трет-бутил-N-метил-N-(1-метил-1H-пиразол-5-ил)карбамата (9,5 г, 45 ммоль) в ДХМ (50 мл) добавляют 4N HCl в диоксане (45 мл). Полученную смесь перемешивают при КТ в течение ночи. Растворители удаляют в вакууме с получением продукта в виде твердого белого вещества (6 г, 90%). 1H ЯМР (400 Гц, ДМСО-d6) δ 2,80 (с, 3H), 3,64 (с, 3H), 4,20 (уш., 2H), 5,84 (д, 1H), 7,94 (д, 1H).
Промежуточное соединение 7: трет-Бутил-[4-(бромметил)-6-метилпиридин-2-ил](4-метоксибензил)карбамат

Схема 10
Стадия 1. Получение метил-2-((трет-бутоксикарбонил)амино)-6-метилпиридин-4-карбоксилата

Смесь метил-2-хлор-6-метилпиридин-4-карбоксилата (20 г, 107,8 ммоль), трет-бутилкарбамата (15,2 г, 129,7 ммоль), карбоната цезия (70,2 г, 215,.5 ммоль), X-phos (5,14 г, 10,8 ммоль) и ацетата палладия (1,2 г, 5,3 ммоль) в 1,4-диоксане (300 мл) дегазируют азотом и затем перемешивают при 90°C в атмосфере азота в течение двух часов. Смесь охлаждают до КТ, разбавляют водой (300 мл) и затем экстрагируют EA (150 мл × 2). Органические экстракты объединяют и промывают насыщенным водным раствором соли, сушат (MgSO4) и концентрируют. Остаток очищают хроматографией (элюирование: 0-30% EA в гексане) с получением метил-2-((трет-бутоксикарбонил)амино)-6-метилпиридин-4-карбоксилата в виде твердого белого вещества (25,8 г, 90%). 1H ЯМР (400 Гц, CDCl3) δ 1,52 (с, 9H), 2,48 (с, 3H), 3,92 (с, 3H), 7,36 (с, 1H), 7,40 (с, 1H), 8,24 (с, 1H).
Стадия 2. Получение метил-2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-карбоксилата
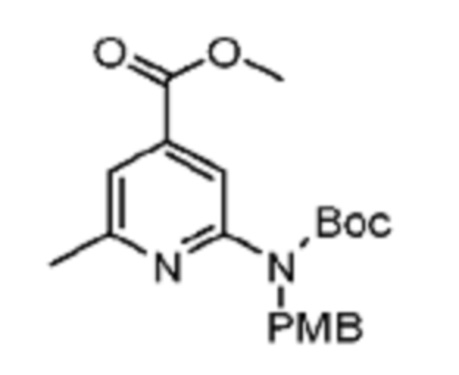
К раствору метил-2-((трет-бутоксикарбонил)амино)-6-метилпиридин-4-карбоксилата (12,3 г, 46,2 ммоль) в ДМФА (100 мл) добавляют одной порцией трет-бутоксид калия (7,3 г, 60,2 ммоль). Смесь перемешивают при КТ в течение 5 мин. и затем добавляют п-метоксибензилхлорид (9,8 г, 59,9 ммоль). Полученную смесь перемешивают при КТ в течение 12 часов. Добавляют воду (150 мл) и экстрагируют полученную смесь EA (100 мл × 2). Объединенную органическую фазу промывают насыщенным водным раствором соли, сушат (MgSO4) и концентрируют. Остаток очищают хроматографией (элюирование: 0-20% EA в гексане) с получением метил-2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-карбоксилата в виде твердого белого вещества (12,8 г, 72%). МС (ESI+) m/z 387,2 (M+H) +, время удерживания: 2,15 мин. (метод A) . 1H ЯМР (400 Гц, CDCl3) δ 1,45 (с, 9H), 2,52 (с, 3H), 3,76 (с, 3H), 3,91 (с, 3H), 5,14 (с, 2H), 6,78 (д, 2H), 7,22 (д, 2H), 7,38 (с, 1H), 7,97 (с, 1H).
Стадия 3. Получение трет-бутил-(4-(гидроксиметил)-6-метилпиридин-2-ил)(4-метоксибензил)карбамата
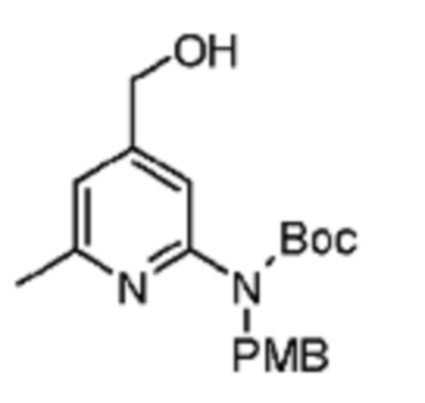
К раствору метил 2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-карбоксилата (12,8 г, 33,1 ммоль) в ТГФ (100 мл) при КТ добавляют борогидрид лития (1,5 г, 68,9 ммоль). Смесь нагревают и выдерживают при 40°C в течение 4 часов. Реакционную смесь охлаждают до 0°C и осторожно гасят медленным добавлением по каплям водного раствора NaHCO3 (30 мл). Смесь разбавляют водой (100 мл) и экстрагируют EA (50 мл × 2). Объединенную органическую фазу промывают насыщенным водным раствором соли (50 мл), сушат (MgSO4) и концентрируют. Остаток очищают хроматографией (элюирование: 40% EA в гексане) с получением трет-бутил-(4-(гидроксиметил)-6-метилпиридин-2-ил)(4-метоксибензил)карбамата в виде бесцветного масла (11,3 г, 95%). МС (ESI+) m/z 359,1 (M+H) +, время удерживания: 1,76 мин. (метод A). 1H ЯМР (400 Гц, CDCl3) δ 1,40 (с, 9H), 2,48 (с, 3H), 3,76 (с, 3H), 4,62 (м, 2H), 5,14 (с, 2H), 6,80 (д, 2H), 6,84 (с, 1H), 7,20 (д, 2H), 7,40 (с, 1H).
Стадия 4. Получение трет-бутил-(4-(бромметил)-6-метилпиридин-2-ил)(4-метоксибензил)карбамата
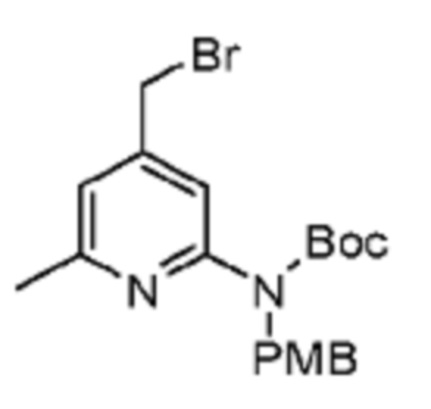
К раствору трет-бутил-(4-(гидроксиметил)-6-метилпиридин-2-ил)(4-метоксибензил)карбамата (11,3 г, 31,5 ммоль) и тетрабромида углерода (12,3 г, 37,1 ммоль) в ДХМ (150 мл) при 0°C одной порцией добавляют трифенилфосфин (9,7 г, 37,1 ммоль). Реакционную смесь перемешивают в течение 20 мин., смеси дают возможность нагреться до КТ и затем перемешивают смесь в течение одного часа. Смесь разбавляют ДХМ (100 мл) и экстрагируют водным раствором NaHCO3 (50 мл × 1). Органический слой сушат (MgSO4) и концентрируют. Остаток очищают хроматографией (элюирование: 0- 20% EA в гексане) с получением трет-бутил-(4-(бромметил)-6-метилпиридин-2-ил)(4-метоксибензил)карбамата в виде твердого белого вещества (10,6 г, 80%). МС (ESI+) m/z 422,1 (M+H)+. Время удерживания: 2,18 мин. (метод A). 1H ЯМР (400 Гц, CDCl3) δ 1,40 (с, 9H), 2,46 (с, 3H), 3,78 (с, 3H), 4,38 (с, 2H), 5,14 (с, 2H), 6,80 (д, 2H), 6,84 (с, 1H), 7,22 (д, 2H), 7,46 (с, 1H).
Промежуточное соединение 8: 4-(бромметил)-N, N-бис(4- метоксибензил)пиридин-2-амин

Схема 11
Стадия 1. Получение метилового эфира 2-[бис-(4-метоксибензил)амино]изоникотиновой кислоты

Смесь метилового эфира 2-аминопиридин-4-карбоновой кислоты (5,5 г) и 4-метоксибензилхлорида (14,64 г) в 33 мл ацетонитрила кипятят с обратным холодильником в течение 3 часов; затем в кипящую смесь медленно добавляют TEA (7,3 г). Реакционной смеси дают возможность охладиться до комнатной температуры и перемешивают смесь в течение ночи. Реакционную смесь концентрируют, удаляя растворители, и к остатку добавляют воду с образованием твердого осадка. Твердые продукты собирают фильтрацией и перекристаллизовывают из изопропанола с получением метилового эфира 2-[бис-(4-метоксибензил)амино]изоникотиновой кислоты (3 г, 21%) в виде твердого белого вещества.
Стадия 2. Получение {2-[бис-(4-метоксибензил)амино]пиридин-4-ил}метанола

Алюмогидрид лития (388 мг) порциями добавляют к ТГФ (16 мл), предварительно охлажденному до 0°C. Затем к смеси по каплям добавляют 2-[бис(4-метоксибензил)амино]изоникотиновой кислоты (4 г) в ТГФ (16 мл), поддерживая температуру смеси в интервале от 0 до -5°C. После этого к реакционной смеси медленно последовательно добавляют EA (900 мг), воду (388 мг) и 15% водный раствор NaOH (388 мг). Смесь перемешивают в течение 10 мин. и затем добавляют безводный Na2SO4 (1,3 г). Смесь перемешивают в течение 30 мин. Твердый осадок собирают фильтрацией и промывают ТГФ (12 мл). Фильтрат концентрируют с получением {2-[бис(4-метоксибензил)амино]пиридин-4-ил}метанола, который используют в следующей стадии без дополнительной очистки
Стадия 3. Получение 4-(бромметилпиридин-2-ил)-бис(4-метоксибензил)амина
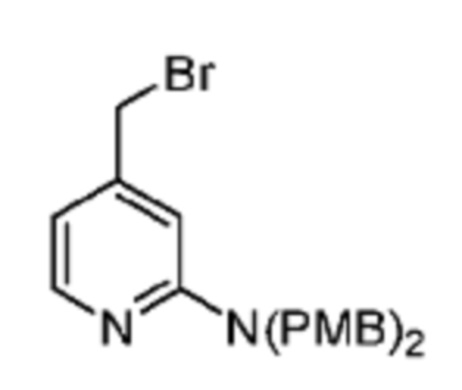
Раствор {2-[бис(4-метоксибензил)амино]пиридин-4-ил}метанола (5 г) и CBr4 (5 г) в ДХМ (25 мл) охлаждают до 0-10°C. По каплям добавляют раствор PPh3 (4,32 г) в ДХМ (10 мл). Реакционную смесь анализируют ТСХ. Если присутстувует исходный спирт, добавляют дополнительную порцию PPh3 до полного завершения реакции. Реакционную смесь концентрируют, маслянистый остаток обрабатывают 50% водным этанолом (16 мл) и перемешивают в течение 1 часа при КТ. Твердый продукт фильтруют и промывают небольшим количеством 50% водного этанола. Полученный осадок на фильтре суспендируют с перемешиванием в 50% водном этаноле (8 мл) в течение 1 часа при КТ. Твердый продукт снова фильтруют и сушат с получением указанного в заголовке соединения в виде твердого не совсем белого кристаллического вещества (5,2 г). 1H ЯМР (CDCl3): 8,17 (д, 1H), 7,14 (д, 4H), 6,84 (д, 4H), 6,60 (д, 1H), 6,45 (с, 1H), 4,69 (с, 4H), 4,23 (с, 2H), 3,79 (с, 6H). МС (ESI+) m/z 427,2 (M+H)+; время удерживания: 1,2 мин. (метод A).
Промежуточное соединение 9. трет-Бутил-(4-(бромметил)пиридин-2-ил)метилкарбамат
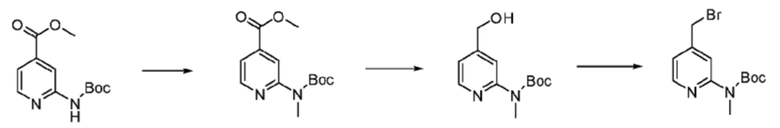
Схема 12
Стадия 1. Получение метил-2-(трет-бутоксикарбонил)(метил)амино)изоникотината
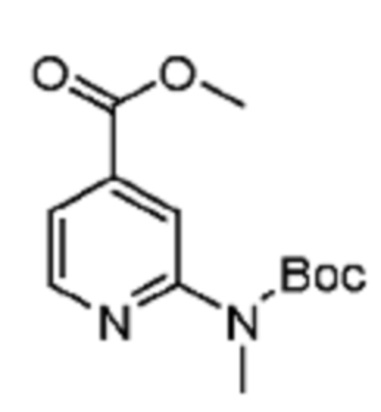
Суспензию метил-2-((трет-бутоксикарбонил)амино)изоникотинат (50,4 г, 200 ммоль) в ДМФА (500 мл) охлаждают до 0°C и порциями добавляют гидрид натрия (10,4 г, 60% в минеральном масле, 260 ммоль). Смеси дают возможность нагреться до КТ и перемешивают смесь в течение 30 мин. К смеси медленно добавляют йодметан (37,2 г, 262 ммоль). Реакционную смесь перемешивают при КТ в течение ночи. Реакционную смесь гасят добавлением водного раствора хлорида аммония (100 мл) и разбавляют водой (400 мл). Смесь экстрагируют EA (250 мл × 2). Объединенные органические фазы промывают водой (100 мл) и насыщенным водным раствором соли (100 мл), сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 5:1 гексан:EA) с получением продукта в виде бесцветного масла (48,9 г, 92%). 1H ЯМР (400 Гц, CDCl3) δ 1,54 (с, 9H), 3,42 (с, 3H), 3,94 (с, 3H), 7,52 (д, 1H), 8,27 (с, 1H), 8,48 (д, 1H).
Стадия 2. Получение трет-бутил-(4-(гидроксиметил)пиридин-2-ил)метилкарбамата
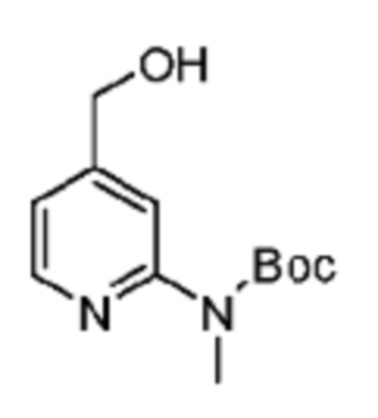
К раствору метил-2-(трет-бутоксикарбонил)(метил)амино)изоникотинат (48,48 г, 182,1 ммоль) в ТГФ (500 мл) при КТ добавляют борогидрид лития (5,15 г, 236,5 ммоль). Смесь нагревают и выдерживают при 40°C в течение 4 часов. Реакционную смесь охлаждают до 0°C и осторожно гасят медленным добавлением по каплям водного раствора NaHCO3 (50 мл). Смесь разбавляют водой (500 мл) и экстрагируют EA (250 мл × 2). Объединенную органическую фазу промывают насыщенным водным раствором соли (150 мл), сушат (MgSO4) и концентрируют. Остаток очищают хроматографией (элюирование: 40% EA в гексане) с получением трет-бутил-(4-(гидроксиметил)пиридин-2-ил)метилкарбамата в виде бесцветного масла (41,2 г, 95%). МС (ESI+) m/z 239,1 (M+H)+, время удерживания: 1,36 мин. (метод A). 1H ЯМР (400 Гц, CDCl3) δ 1,54 (с, 9H), 3,40 (с, 3H), 4,68 (с, 2H), 7,02 (д, 1H), 7,64 (с, 1H), 8,34 (д, 1H).
Стадия 3. Получение трет-бутил-(4-(бромметил)пиридин-2-ил)метилкарбамата

К раствору трет-бутил-(4-(гидроксиметил)пиридин-2-ил)метилкарбамата (20,52 г, 86,1 ммоль) и тетрабромида углерода (33,41 г, 100,7 ммоль) в ДХМ (400 мл) при 0°C одной порцией добавляют трифенилфосфин (26,43 г, 100,7 ммоль). Реакционную смесь перемешивают в течение 20 мин., смеси дают возможность нагреться до КТ и затем перемешивают в течение одного часа. Смесь разбавляют ДХМ (100 мл) и промывают водным раствором NaHCO3 (150 мл) и насыщенным водным раствором соли (150 мл). Органический слой сушат (MgSO4) и концентрируют. Остаток очищают хроматографией (элюирование: 0-20% EA в гексане) с получением трет-бутил-(4-(бромметил)пиридин-2-ил)метилкарбамата в виде бесцветного масла (20,7 г, 80%). 1H ЯМР (400 МГц, CDCl3) δ 1,54 (с, 9H), 3,41 (с, 3H), 4,39 (с, 2H), 6,92-7,12 (м, 1H), 7,66-7,84 (м, 1H), 8,24-8,43 9 (м, 1H).
Промежуточное соединение 10. трет-Бутил-(4-(бромметил)-6-метилпиридин-2-ил)метилкарбамат
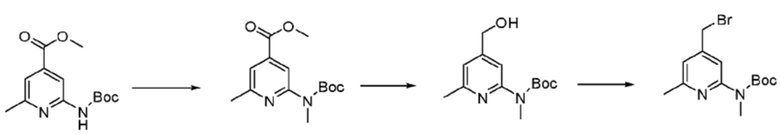
Схема 13
Стадия 1. Получение метил-2-(трет-бутоксикарбонил)(метил)амино)-6-метилпиридин-4-карбоксилата

Суспензию метил-2-((трет-бутоксикарбонил)амино)-6-метилпиридин-4-карбоксилата (20 г, 75,2 ммоль) в ДМФА (200 мл) охлаждают до 0°C и небольшими порциями добавляют гидрид натрия (3,9 г, 60% в минеральном масле, 97,5 ммоль). Смеси дают возможность нагреться до КТ и перемешивают смесь в течение 30 мин. К реакционной смеси медленно добавляют йодметан (12,8 г, 90,1 ммоль). Реакционную смесь перемешивают при КТ в течение ночи. Реакцию гасят добавлением водного раствора хлорида аммония (50 мл) и разбавляют водой (200 мл). Смесь экстрагируют EA (250 мл × 2). Объединенные органические фазы промывают водой (100 мл) и насыщенным водным раствором соли (100 мл), сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 5:1 гексан:EA) с получением продукта в виде бесцветного масла (18,9 г, 90%). МС (ESI+) m/z 281,1 (M+H)+. Время удерживания: 2,03 мин. (метод A).
Стадия 2. Получение трет-бутил-(4-(гидроксиметил)-6-метилпиридин-2-ил)метилкарбамата

К раствору метил-2-(трет-бутоксикарбонил)(метил)амино)-6-метилпиридин-4-карбоксилата (27 г, 96,4 ммоль) в ТГФ (300 мл) при КТ добавляют борогидрид лития (2,1 г, 96,4 ммоль). Смесь нагревают и выдерживают при 40°C в течение 4 часов. Реакционную смесь охлаждают до 0°C и осторожно гасят медленным добавлением по каплям водного раствора NaHCO3 (50 мл). Смесь разбавляют водой (200 мл) и экстрагируют EA (250 мл × 2). Объединенную органическую фазу промывают насыщенным водным раствором соли (150 мл), сушат (MgSO4) и концентрируют. Остаток очищают хроматографией (элюирование: 40% EA в гексане) с получением трет-бутил-(4-(гидроксиметил)-6-метилпиридин-2-ил)метилкарбамата в виде бесцветного масла (20 г, 82%). МС (ESI+) m/z 253,1 (M+H)+, время удерживания: 1,31 мин. (метод A).
Стадия 3. Получение трет-бутил-(4-(бромметил)-6-метилпиридин-2-ил)метилкарбамата

К раствору трет-бутил-4-(гидроксиметил)-6-метилпиридин-2-ил)метилкарбамата (20 г, 79,4 ммоль) и тетрабромида углерода (30,8 г, 92,8 ммоль) в ДХМ (400 мл) при 0°C одной порцией добавляют трифенилфосфин (224,4 г, 93,0 ммоль). Реакционную смесь перемешивают в течение 20 мин., смеси дают возможность нагреться до КТ и затем перемешивают в течение одного часа. Смесь разбавляют ДХМ (100 мл) и экстрагируют водным раствором NaHCO3 (150 мл) и насыщенным водным раствором соли (150 мл). Органический слой сушат (MgSO4) и концентрируют. Остаток очищают хроматографией (элюирование: 0- 20% EA в гексане) с получением трет-бутил-(4-(бромметил)-6-метилпиридин-2-ил)метилкарбамата в виде бесцветного масла (20,7 г, 80%). МС (ESI+) m/z 315,2 (M+H)+, время удерживания: 2,02 мин. (метод A).
Промежуточное соединение 11: трет-Бутил-[4-(бромметил)-5-фторпиридин-2-ил](4-метоксибензил)карбамат
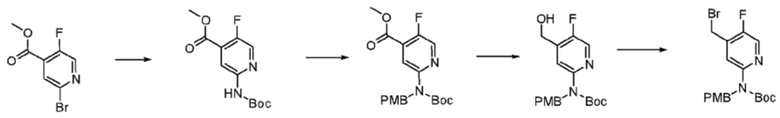
Схема 14
Стадия 1. Получение метил-2-((трет-бутоксикарбонил)амино)-5-фторпиридин-4-карбоксилата
Смесь метил-2-бром-5-фторпиридин-4-карбоксилата (6,64 г, 28,4 ммоль), трет-бутилкарбамата (4,0 г, 34,1 ммоль), карбоната цезия (13,0 г, 39,8 ммоль), X-phos (657 мг, 1,1 ммоль) и Pd(dba)2 (520 мг, 0,9 ммоль) в 1,4-диоксане (100 мл) продувают азотом и затем перемешивают при 100°C в атмосфере азота в течение 6 часов. Смесь охлаждают до КТ, разбавляют водой (100 мл) и экстрагируют EA (150 мл × 2). Объединенную органическую фазу промывают насыщенным водным раствором соли, сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 0-30% EA в гексане) с получением метил-2-((трет-бутоксикарбонил)амино)-5-фторпиридин-4-карбоксилата в виде твердого белого вещества (4,5 г, 56%). МС (ESI+) m/z 271,1 (M+H)+, время удерживания: 1,81 мин. (метод A). 1H ЯМР (400 МГц, CDCl3) δ 1,52 (с, 9H), 3,96 (с, 3H), 8,24 (с, 1H), 8,40 (д, 1H), 9,00 (с, 1H).
Стадия 2. Получение метил-2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-5-фторпиридин-4-карбоксилата

К раствору метил-2-((трет-бутоксикарбонил)амино)-5-фторпиридин-4-карбоксилата (4,5 г, 16,6 ммоль) в ДМФА (50 мл) порциями добавляют трет-бутоксид калия (2,33 г, 19,2 ммоль). Смесь перемешивают при КТ в течение 5 мин.; затем добавляют п-метоксибензилхлорид (3,2 г, 20,4 ммоль). Полученную смесь перемешивают при КТ в течение 12 часов. Добавляют воду (50 мл) и экстрагируют смесь EA (50 мл × 2). Объединенную органическую фазу промывают насыщенным водным раствором соли, сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 0-20% EA в гексане) с получением метил-2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-5-фторпиридин-4-карбоксилата в виде твердого белого вещества (4,8 г, 75%). МС (ESI+) m/z 391,0 (M+H) +, время удерживания: 2,09 мин. (метод A).
Стадия 3. Получение трет-бутил-(4-(гидроксиметил)-5-фторпиридин-2-ил)(4-метоксибензил)карбамата

К раствору метил-2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-5-фторпиридин-4-карбоксилата (7,7 г, 19,7 ммоль) в ТГФ (100 мл) при КТ добавляют борогидрид лития (0,5 г, 23,0 ммоль). Смесь нагревают и выдерживают при 40°C в течение 4 часов. Реакционную смесь охлаждают до 0°C и осторожно гасят медленным добавлением по каплям водного раствора NaHCO3 (30 мл). Смесь разбавляют водой (100 мл) и экстрагируют EA (50 мл × 2). Объединенную органическую фазу промывают насыщенным водным раствором соли (50 мл), сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 40% EA в гексане) с получением трет-бутил-(4-(гидроксиметил)-5-фторпиридин-2-ил)(4-метоксибензил)карбамата в виде бесцветного масла (5,5 г, 77%). МС (ESI+) m/z 363,2 (M+H)+, время удерживания: 1,86 мин. (метод A). 1H ЯМР (400 МГц, CDCl3) δ 1,40 (с, 9H), 2,48 (с, 3H), 3,76 (с, 3H), 4,62 (м, 2H), 5,14 (с, 2H), 6,80 (д, 2H), 6,84 (с, 1H), 7,20 (д, 2H), 7,40 (с, 1H).
Стадия 4. Получение трет-бутил-[4-(бромметил)-5-фторпиридин-2-ил] (4-метоксибензил)карбамата
К раствору трет-бутил-(4-(гидроксиметил)-5-фторпиридин-2-ил)(4-метоксибензил)карбамата (5,5 г, 15,2 ммоль) и тетрабромидауглерода (5,9 г, 17,8 ммоль) в ДХМ (60 мл) при 0°C добавляют трифенилфосфин (4,7 г, 17,9 ммоль) одной порцией. Реакционную смесь перемешивают в течение 20 мин., смеси дают возможность нагреться до КТ и перемешивают смесь в течение 1 часа. Смесь разбавляют ДХМ (100 мл), экстрагируют водным раствором NaHCO3 (50 мл × 1), сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 0-20% EA в гексане) с получением трет-бутил-[4-(бромметил)-5-фторпиридин-2-ил](4-метоксибензил)карбамата в виде твердого белого вещества (10,6 г, 80%). МС (ESI+) m/z 421,1 (M+H)+. Время удерживания: 2,18 мин. (метод A). 1H ЯМР (400 МГц, CDCl3) δ 1,40 (с, 9H), 2,46 (с, 3H), 3,78 (с, 3H), 4,38 (с, 2H), 5,14 (с, 2H), 6,80 (д, 2H), 6,84 (с, 1H), 7,22 (д, 2H), 7,46 (с, 1H).
Промежуточное соединение 12: трет-Бутил[4-(бромметил)-5-метилпиридин-2-ил] (4-метоксибензил)карбамат

Схема 15
Стадия 1. Получение метил-2-((трет-бутоксикарбонил)амино)-5-метилпиридин-4-карбоксилата

К раствору метил-2-амино-5-метилпиридин-4-карбоксилата (6 г, 36,1 ммоль) в ацетоне (20 мл) и трет-бутиловом спирте (60 мл) добавляют 4-диметиламинопиридин (220 мг, 1,8 мл) и ди-трет-бутилдикарбонат (9,5 г, 43,5 ммоль). Смесь перемешивают при КТ в течение ночи. Реакционную смесь концентрируют и остаток очищают хроматографией (элюирование: 0-30% EA в гексане) с получением метил-2-((трет-бутоксикарбонил)амино)-5-метилпиридин-4-карбоксилата в виде твердого белого вещества (7,7 г, 85%). 1H ЯМР (400 МГц, CDCl3) δ 1,52 (с, 9H), 2,44 (с, 3H), 3,92 (с, 3H), 8,04 (с, 1H), 8,20 (с, 1H), 8,32 (с, 1H).
Стадия 2. Получение метил-2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-5-метилпиридин-4-карбоксилата

К раствору метил-2-((трет-бутоксикарбонил)амино)-5-метилпиридин-4-карбоксилата (5 г, 18,8 ммоль) в ДМФА (50 мл) добавляют трет-бутоксид калия (3 г, 24,8 ммоль). Смесь перемешивают при КТ в течение 5 мин. и затем добавляют п-метоксибензилхлорид (3,82 г, 24,4 ммоль). Полученную смесь перемешивают при КТ в течение 12часов. К смеси добавляют воду (50 мл) и экстрагируют смесь EA (50 мл × 2). Объединенную органическую фазу промывают насыщенным водным раствором соли, сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 0-20% EA в гексане) с получением метил-2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-5-метилпиридин-4-карбоксилата в виде твердого белого вещества (6,1 г, 84%). МС (ESI+) m/z 387,2 (M+H) +, время удерживания: 2,14 мин. (метод А).
Стадия 3. Получение трет-бутил-(4-(гидроксиметил)-5-метилпиридин-2-ил)(4-метоксибензил)карбамата

К раствору метил-2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-5-метилпиридин-4-карбоксилата (6 г, 15,5 ммоль) в ТГФ (50 мл) при КТ добавляют борогидрид лития (0,75 г, 34,4 ммоль). Смесь нагревают и выдерживают при 40°C в течение 4 часов. Реакционную смесь охлаждают до 0°C и осторожно гасят медленным добавлением по каплям водного раствора NaHCO3 (20 мл). Смесь разбавляют водой (50 мл) и экстрагируют EA (50 мл × 2). Объединенную органическую фазу промывают насыщенным водным раствором соли (50 мл), сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 40% EA в гексане) с получением трет-бутил-(4-(гидроксиметил)-5-метилпиридин-2-ил)(4-метоксибензил)карбамата в виде бесцветного масла (4,78 г, 89%). МС (ESI+) m/z 359,2 (M+H) +, время удерживания: 1,74 мин. (метод A).
Стадия 4. Получение трет-бутил-4-(бромметил)-5-метилпиридин-2-ил)(4-метоксибензил)карбамата

К раствору трет-бутил-(4-(гидроксиметил)-5-метилпиридин-2-ил)(4-метоксибензил)карбамата (4,78 г, 13,3 ммоль) и тетрабромида углерода (5,17 г, 15,6 ммоль) в ДХМ (50 мл) при 0°C одной порцией добавляют трифенилфосфин (4,1 г, 15,6 ммоль). Реакционную смесь перемешивают в течение 20 мин., смеси дают возможность нагреться до КТ и перемешивают смесь в течение 1 часа. Смесь разбавляют ДХМ (100 мл), промывают водным раствором NaHCO3 (50 мл × 1), сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 0- 20% EA в гексане) с получением трет-бутил(4-(бромметил)-5-метилпиридин-2-ил)(4-метоксибензил)карбамата в виде твердого белого вещества (4,2 г, 75%). МС (ESI+) m/z 422,1 (M+H)+. Время удерживания: 2,18 мин. (метод A). 1H ЯМР (400 МГц, CDCl3) δ 1,40 (с, 9H), 2,34 (с, 3H), 3,76 (с, 3H), 4,38 (с, 2H), 5,14 (с, 2H), 6,80 (д, 2H), 7,20 (д, 2H), 7,60 (с, 1H), 8,20 (с, 1H).
Промежуточное соединение 13. Получение (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)пиридин-4-ил)метил)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновой кислоты

К раствору 4-метоксибензил-(2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)пиридин-4-ил)метил)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоксилата (1,4 г, 2,1 ммоль, получен как описано в WO 2015/120062) в метаноле (20 мл) добавляют 10% Pd/C (0,2 г). Смесь гидрируют при 30 фунтах на кв.дюйм (206,8 кПа) в течение 2 часов. Смесь фильтруют через Celite® и осадок на фильтре промывают метанолом. Растворитель удаляют с получением продукта в виде твердого белого вещества (0,97 г, 95%). МС (ESI+) 556,2 (M+H)+. Время удерживания: 3,64 мин. (метод B).
Промежуточное соединение 14. Получение (2S,3R)-1-[(1,1-диметилэтил)диметилсилил]-4-оксo-3-(пиридин-4-илметил)-2-азетидинкарбоновой кислоты

К раствору гидрохлорида 4-пиколилхлорида (10,0 г, 61 ммоль) в 100 мл воды добавляют NaHCO3 (7,7 г, 91,4 ммоль) при перемешивании. Смесь экстрагируют TBME (3 × 100 мл). Экстракты объединяют, промывают насыщенным водным раствором соли, сушат над MgSO4 и концентрируют с получением 4-пиколилхлорида в виде бесцветного масла (7,5 г, 97%), который используют далее без дополнительной очистки.
К раствору (2S)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновой кислоты (получена как описана в публикации Baldwin et al., Tetrahedron, 1990, 46, 4733-4788) (5 г, 21,8 ммоль) в ТГФ (80 мл) при -25°C по каплям добавляют LDA (2M в ТГФ, 24 мл). Полученную смесь перемешивают при -15°C в течение 30 мин. К смеси по каплям добавляют раствор 4-пиколилхлорида (4 г, 31,3 ммоль) в ТГФ (25 мл). Смесь перемешивают при -15°C в течение 2 часов, затем реакцию гасят водным раствором хлорида аммония (20 мл) и добавляют EA. Органический слой отделяют и экстрагируют водный слой EA. Объединенную органическую фазу промывают насыщенным водным раствором соли, сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: EA) с получением продукта в виде твердого белого вещества (1,5 г, 15%). МС (ESI+) m/z 321,1 (M+H)+, время удерживания: 1,24 мин. (метод A).
Промежуточное соединение 15. Получение (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-4-оксоазетидин-2-карбоновой кислоты
(S)-1-(трет-Бутилдиметилсилил)-4-оксоазетидин-2-карбоновую кислоту (10,0 г, 43 ммоль) растворяют в ТГФ (160 мл) и охлаждают до -20°C. Реакционную смесь обрабатывают LDA (2M в ТГФ, 47 мл, 94 ммоль) при температуре в интервале примерно от -10 до -20°C. Затем добавляют 4-(бромметил)-N, N-бис(4-метоксибензилпиридин-2-амин (21,2 г, 49 ммоль) в ТГФ (80 мл), поддерживая температуру смеси ниже -10°C. Реакционную смесь перемешивают в течение нескольких часов при -15°C, смеси дают возможность нагреться до КТ и перемешивают смесь в течение нескольких часов. Реакционную смесь гасят водой (200 мл) и затем кипятят с обратным холодильником в течение 3 часов. Реакционную смесь охлаждают до КT и обрабатывают 5% водным раствором трикалийфосфата (250 мл). Фазы разделяют и водный слой экстрагируют EA (150 мл × 3) для удаления примесей. Водную фазу подкисляют до pH 3,1 с помощью 6 N HCl и затем экстрагируют EA (300 мл × 3). Органическую фазу сушат и концентрируют. Остаточный EA обрабатывают гептаном(250 мл) с получением суспензии, которую охлаждают и фильтруют. Осадок на фильтре переносят в 40 объемов изопропилового спирта (400 мл) и кипятят с обратным холодильником в течение примерно 1 часа. Смесь охлаждают до КТ и нерастворимые твердые примеси удаляют фильтрацией. В фильтрате изопропилового спирта растворитель заменяют на гептан (250 мл), вызывая осаждение продукта. Гептановую суспензию охлаждают до 5-10°C и фильтруют. Осадок на фильтре сушат с получением (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-4-оксоазетидин-2-карбоновой кислоты (12 г, 59%). МС (ESI+) m/z 462,2 (M+H)+, время удерживания: 1,23 мин. (метод A).
Промежуточное соединение 16. Получение (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-1-(трет-бутил(диметил)силил)-4-оксоазетидин-2-карбоновой кислоты
(S)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновую кислоту (10 г, 43 ммоль) растворяют в ТГФ (160 мл) и охлаждают до -20°C. Ракционную смесь обрабатывают LDA (2M в ТГФ, 47 мл, 94 ммоль) при температуре в интервале примерно от -10 to -20°C и затем 4-(бромметил)-N, N-бис(4-метоксибензилпиридин-2-амином (21,2 г, 49 ммоль) в ТГФ (80 мл), поддерживая температуру смеси ниже -10°C. Реакционную смесь перемешивают в течение нескольких часов при -15°C, смеси дают возможность нагреться до КТ и перемешивают смесь в течение нескольких часов. Реакционную смесь гасят водой (200 мл) и затем подкисляют 10% водной лимонной кислотодй до pH 4. Смесь экстрагируют EA (100 мл × 3). Объединенные экстракты сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 20%-100% EA в гексане) с получением продукта в виде твердого белого вещества (14,5 г, 59%). МС (ESI+) m/z 576,3 (M+H)+, время удерживания: 1,56 мин. (метод A).
Промежуточное соединение 17. Получение (2S,3R)-3-((2-((трет-бутоксикарбонил)(метил)амино)пиридин-4-ил)метил)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновой кислоты

(S)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновую кислоту (12,2 г, 53,2 ммоль) растворяют в ТГФ (195 мл) и охлаждают до -20°C. Реакционную смесь обрабатывают LDA (2M в ТГФ, 60 мл, 120 ммоль) при температуре в интервале примерно от -10 до -20°C и затем трет-бутил(4-(бромметил)пиридин-2-ил)метилкарбаматом (16 г, 53,1 ммоль) в ТГФ (80 мл), поддерживая температуру смеси ниже -10 oC. Реакционную смесь перемешивают в течение нескольких часов при -15°C, смеси дают возможность нагреться до КТ и перемешивают смесь в течение нескольких часов. Реакционную смесь гасят водой (200 мл) и затем подкисляют 10% водной лимонной кислотой до pH 4. Смесь экстрагируют EA (100 мл × 3). Объединенную органическую фазу сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 20%-100% EA в гексане) с получением продукта в виде твердого белого вещества (14,4 г, 60%). МС (ESI+) m/z 450,2 (M+H)+, время удерживания: 1,85 мин. (метод A).
Промежуточное соединение 18. Получение (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-5-метилпиридин-4-ил)метил)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновой кислоты

(S)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновой кислоты (2,42 г, 10,6 ммоль) растворяют в ТГФ (36 мл) и охлаждают до -20°C. Реакционную смесь обрабатывают LDA (2M в ТГФ, 12 мл, 24 ммоль) при температуре в интервале примерно от -10 to -20°C и затем трет-бутил-[4-(бромметил)-5-метилпиридин-2-ил] (4-метоксибензил)карбаматом (4,44 г, 10,5 ммоль) в ТГФ (10 мл), поддерживая температуру смеси ниже -10°C. Реакционную смесь перемешивают в течение нескольких часов при -15°C, затем смеси дают возможность нагреться до КТ и перемешивают смесь в течение нескольких часов. Реакционную смесь гасят водой (20 мл) и затем подкисляют 10% водной лимонной кислотой до pH 4. Смесь экстрагируют EA (30 мл × 3). Объединенную органическую фазу сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 20%-100% EA в гексане) с получением продукта в виде твердого белого вещества (3,12 г, 53%). МС (ESI+) m/z 570,3 (M+H)+, время удерживания: 2,09 мин. (метод A).
Промежуточное соединение 19. Получение (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновой кислоты
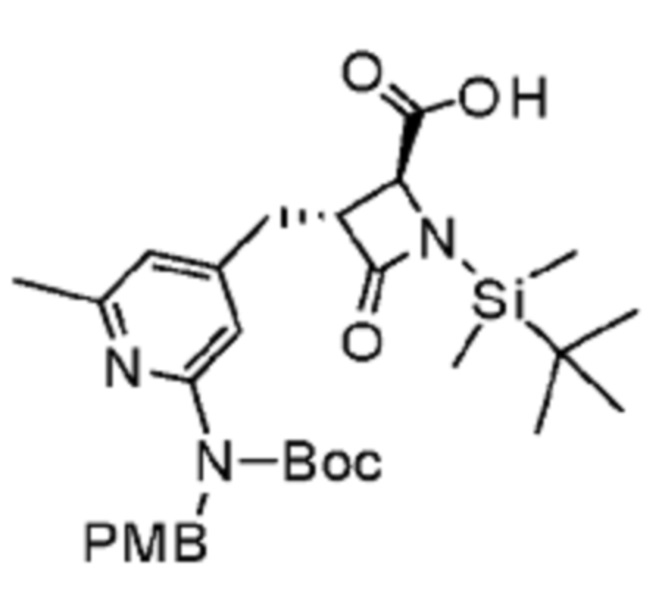
(S)-1-(трет-Бутилдиметилсилил)-4-оксоазетидин-2-карбоновую кислоту (7,1 г, 30,9 ммоль) растворяют в ТГФ (120 мл) и охлаждают до -20°C. Реакционную смесь обрабатывают LDA (2M в ТГФ, 34 мл, 68 ммоль) при температуре в интервале примерно от -10 до -20°C с последующей обработкой трет-бутил-[4-(бромметил)-6-метилпиридин-2-ил](4-метоксибензил)карбаматом (13 г, 30,8 ммоль) в ТГФ (20 мл), поддерживая температуру смеси ниже -10°C. Реакционную смесь перемешивают в течение нескольких часов при -15°C, затем смеси дают возможность нагреться до КТ и перемешивают смесь в течение нескольких часов. Реакционную смесь гасят водой (100 мл) и затем подкисляют 10% водной лимонной кислотой до pH 4. Смесь экстрагируют EA (100 мл × 3). Объединенный органический слой сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 20%-100% EA в гексане) с получением продукта в виде твердого белого вещества (10,5 г, 60%). МС (ESI+) m/z 570,3 (M+H)+, время удерживания: 2,10 мин. (метод A).
Промежуточное соединение 20. Получение (2S,3R)-3-((2-((трет-бутоксикарбонил)(метил)амино)-6-метилпиридин-4-ил)метил)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновой кислоты

(S)-1-(трет-Бутилдиметилсилил)-4-оксоазетидин-2-карбоновую кислоту (4 г, 17,44 ммоль) растворяют в ТГФ (64 мл) и охлаждают до -20°C. Реакционную смесь обрабатывают LDA (2M в ТГФ, 20 мл, 40 ммоль) при температуре в интервале примерно от -10 до -20°C с последующей обработкой трет-бутил-(4-(бромметил)-6-метилпиридин-2-ил)метилкарбаматом (5,5 г, 17,4 ммоль) в ТГФ (15 мл), поддерживая температуру смеси ниже -10°C. Реакционную смесь перемешивают в течение нескольких часов при -15°C затем смеси дают возможность нагреться до КТ и перемешивают смесь в течение нескольких часов. Реакционную смесь гасят водой (40 мл) и затем подкисляют 10% водной лимонной кислотой до pH 4. Смесь экстрагируют EA (60 мл × 3). Объединенную органическую фазу сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 20%-100% EA в гексане) с получением продукта в виде твердого белого вещества (4,44 г, 55%). МС (ESI+) m/z 464,4 (M+H)+, время удерживания: 3,33 мин. (метод B).
Промежуточное соединение 21. Получение (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-5-фторпиридин-4-ил)метил)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновой кислоты
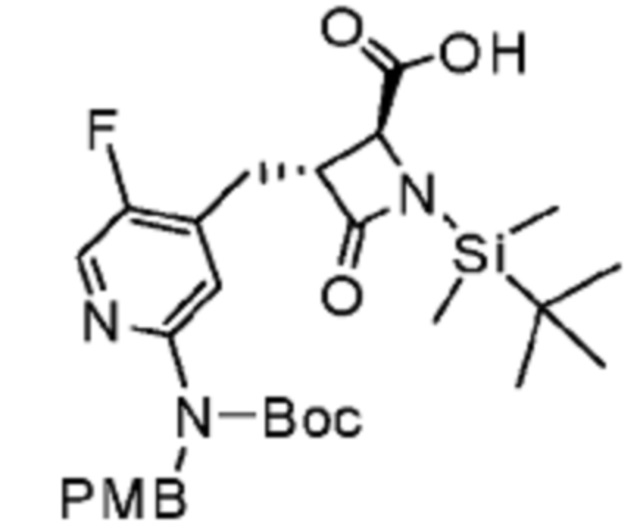
(S)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновую кислоту (2,2 г, 9,6 ммоль) растворяют в ТГФ (36 мл) и охлаждают до -20°C. Реакционную смесь обрабатывают LDA (2M в ТГФ, 11 мл, 22 ммоль) при температуре в интервале примерно от -10 до -20 °C с последующей обработкой трет-бутил-[4-(бромметил)-5-фторпиридин-2-ил] (4-метоксибензил)карбаматом (4,1 г, 9,6 ммоль) в ТГФ (10 мл), поддерживая температуру смеси ниже -10°C. Реакционную смесь перемешивают в течение нескольких часов при -15°C, затем смеси дают возможность нагреться до КТ и перемешивают смесь в течение нескольких часов. Реакционную смесь гасят водой (20 мл) и затем подкисляют 10% водной лимонной кислотой до pH 4. Смесь экстрагируют EA (30 мл × 3). Объединенную органическую фазу сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 20%-100% EA в гексане) с получением продукта в виде твердого белого вещества (2,49 г, 45%). МС (ESI+) m/z 574,4 (M+H)+, время удерживания: 3,79 мин. (метод B).
Промежуточное соединение 22. Синтез (R)-1-(2,5-диметилфенил)пропилизоцианата
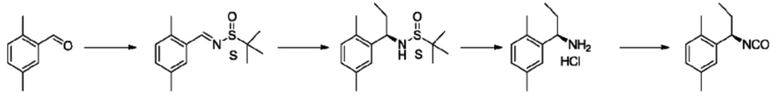
Схема 16
Стадия 1. Получение [N(E),S(S)]-N-[(2,5-диметилфенил)метилен]-2-метил-2-пропансульфонамида

К раствору 2,5-диметилбензальдегида (5 г, 37,3 ммоль) и (S)-(-)-2-метил-2-пропансульфонамида (5 г, 41,2 ммоль) в ТГФ (50 мл) добавляют этоксид титана (IV) (17,8 г, 78,0 ммоль). Полученную реакционную смесь нагревают и выдерживают при 75°C в течение 2 часов. Смесь охлаждают до КТ. К смеси добавляют насыщенный раствор соли (50 мл), смесь энергично перемешивают и затем фильтруют через Celite®. Фильтр промывают EA (200 мл). Фазы разделяют и водный слой экстрагируют EA (2 × 50 мл). Объединенную органическую фазу сушат и концентрируют. Остаток очищают хроматографией (элюирование: 4:1 гексан:EA) с получением [N(E),S(S)]-N-[(2,5-диметилфенил)метилен]-2-метил-2-пропансульфонамида в виде бесцветного масла (8,7 г, 98%). МС (ESI+) 238,1 (M+H)+, время удерживания: 2,05 мин. (метод А).
Стадия 2. Получение [S(S)]-N-((1R)-1-(2,5-диметилфенил)пропил)-2-метил-2-пропансульфонамида

[N(E),S(S)]-N-[(2,5-диметилфенил)метилен]-2-метил-2-пропансульфонамид (8,7 г г, 36,6 моль) растворяют в TBME (50 мл). Реакционную смесь охлаждают до -40°C, по каплям добавляют бромид этилмагния (3M в эфире, 24 мл, 74,3 ммоль) и реакционную смесь перемешивают при -25°C в течение 4 часов. Добавляют насыщенный водный раствор хлорида аммония и затем EA. Слои разделяют и водный слой экстрагируют EA (50 мл). Объединенную органическую фазу промывают насыщенным водным раствором соли, сушат и концентрируют. Остаток очищают хроматографией (элюирование: 2:1 гексан:EA) с получением [S(S)]-N-((1R)-1-(2,5-диметилфенил)пропил)-2-метил-2-пропансульфонамида в виде бесцветного масла (5,2 г, 53%). МС (ESI+) 268,3 (M+H)+, время удерживания: 1,87 мин. (метод А).
Стадия 3. Получение гидрохлорида (R)-1-(2,5-диметилфенил)пропиламина

[S(S)]-N-((1R)-1-(2,5-диметилфенил)пропил)-2-метил-2-пропансульфонамид (5,2 г, 19,4 ммоль) растворяют в метаноле (20 мл) и медленно добавляют 4M HCl в 1,4-диоксане (20 мл). Реакционный раствор перемешивают в течение 60 мин. Реакционную смесь концентрируют с получением твердого остатка. К твердому остатку добавляют гексан (20 мл) и перемешивают полученную суспензию при КТ в течение 60 мин. Твердое вещество собирают фильтрацией, промывают гексанами и сушат с получением гидрохлорида (R)-1-(2,5-диметилфенил)пропиламина (2 г, 54%). МС (ESI+) 164,1 (M+H)+. Время удерживания: 1,14 мин. (метод А).
Стадия 4. Получение (R)-1-(2,5-диметилфенил)пропилизоцианата

К суспензии гидрохлорида (R)-1-(2,5-диметилфенил)пропиламина (2 г, 10,0 ммоль) в ДХМ (30 мл) добавляют 1N водный раствор NaHCO3 (34 мл). Смесь охлаждают до 0°C и добавляют трифосген (1 г, 3,4 ммоль). Реакционную смесь перемешивают в течение 15 мин. при 0°C и в течение 1 часа при КТ. Реакционную смесь два раза экстрагируют ДХМ. Объединенную органическую фазу сушат и концентрируют с получением (R)-1-(2,5-диметилфенил)пропилизоцианата в виде бесцветного масла (1,7 г, 90%), который используют в следующей стадии без дополнительной очистки. 1H ЯМР (400 Гц, CDCl3) δ 7,21 (с, 1H), 7,05 (д, 1H), 7,01 (д, 1H), 4,74 (т, 1H), 2,36 (с, 3H), 2,31 (с, 3H), 1,82 (м, 2H), 1,05 (т, 3H).
Промежуточное соединение 23. Синтез (R)-1-(4-фтор-3-метилфенил)пропилизоцианата

Схема 17
Стадия 1. Получение [S(S)]-N-[1-(4-фтор-3-метилфенил)пропилиден]-2-метилпропан-2-сульфинамида

Раствор 4’-фтор-3’-метилпропиофенона (5 г, 30 ммоль) , (S)-(-)-2-метилпропан-2-сульфинамида (4 г, 33 ммоль) и этоксида титана (IV) (17,4 г, 63 ммоль) в ТГФ (100 мл) нагревают и выдерживают при 80°C в течение ночи. После охлаждения добавляют насыщенный водный раствор соли (100 мл) и EA (100 мл). Смесь перемешивают при КТ в течение 1 часа. Смесь фильтруют через Celite® и фильтр промывают EA. Органическую фазу отделяют, сушат и концентрируют. Остаток очищают хроматографией (элюирование: 2:1 гексан:EA) с получением [S(S)]-N-[1-(4-фтор-3-метилфенил)пропилиден]-2-метилпропан-2-сульфинамида в виде бесцветного масла (7,27 г, 90%). МС (ESI+) 270,1 (M+H)+, время удерживания: 1,97 мин. (метод А).
Стадия 2. Получение [S(S),R]-N-[1-(4-фтор-3-метилфенил)пропил]-2-метилпропан-2-сульфинамида

К раствору [S(S)]-N-[1-(4-фтор-3-метилфенил)пропилиден]-2-метилпропан-2-сульфинамида (7,27 г, 27 ммоль) в ТГФ (80 мл) при 0°C добавляют раствор три-втор-бутилборогидрида (L-selectride, 1M раствор в ТГФ, 81 мл, 81 ммоль). Реакционную смесь перемешивают в течение 3 часов при 0°C. Реакцию гасят насыщенным водным раствором хлорида аммония (80 мл). Смесь фильтруют через Celite® и фильтр промывают EA. Органическую фазу отделяют, сушат и концентрируют. Остаток очищают хроматографией (элюирование: 1:1 гексан:EA) с получением [S(S),R]-N-[1-(4-фтор-3-метилфенил)пропил]-2-метилпропан-2-сульфинамида в виде бесцветного масла (6,1 г, 84%). МС (ESI+) 272,2 (M+H)+, время удерживания: 1,83 мин. (метод А).
Стадия 3. Получение гидрохлорида (R)-1-(4-фтор-3-метилфенил)-1-пропанамина

К раствору [S(S),R]-N-[1-(4-фтор-3-метилфенил)пропил]-2-метилпропан-2-сульфинамид (6,1 г, 22,5 ммоль) в метаноле (50 мл) медленно добавляют 4M HCl в 1,4-диоксане (23 мл). Реакционную смесь перемешивают в течение 1 часа. Реакционную смесь концентрируют с получением твердого остатка. К твердому остатку добавляют гексан (50 мл) добавляют и перемешивают полученную суспензию при КТ в течение 1 часа. Твердое вещество собирают фильтрацией, промывают гексанами и сушат с получением гидрохлорида (R)-1-(4-фтор-3-метилфенил)-1-пропанамина (4,1 г, 90%). МС (ESI+) m/z 168,1 (M+H)+. Время удерживания: 1,13 мин. (метод А).
Стадия 4. Получение (R)-1-(4-фтор-3-метилфенил)пропилизоцианата

К суспензии гидрохлорида (R)-1-(4-фтор-3-метилфенил)-1-пропанамина (1 г, 4,9 ммоль) в ДХМ (30 мл) добавляют 1N водный раствор NaHCO3 (17 мл). Смесь охлаждают до 0°C и добавляют трифосген (0,5 г, 1,7 ммоль). Реакционную смесь перемешивают в течение 15 мин. при 0°C и в течение 1 часа при КТ. Реакционную смесь два раза экстрагируют ДХМ. Объединенную органическую фазу сушат и концентрируют с получением (R)-1-(4-фтор-3-метилфенил)пропилизоцианата в виде бесцветного масла (0,6 г, 63%), который используют в следующей стадии без дополнительной очистки. 1H ЯМР (400 Гц, CDCl3) δ 7,10 (м, 2H), 6,98 (м, 1H), 4,48 (т, 1H), 2,29 (с, 3H), 1,85 (м, 2H), 0,98 (т, 3H).
Промежуточное соединение 24. Синтез гидрохлорида 1,5-дигидро-1-метил-2-(метиламино)-4H-имидазол-4-она

Схема 18
Стадия 1. Получение трет-бутил-N-(1-метил-4-оксо-4,5-дигидро-1H-имидазол-2-ил)карбамата
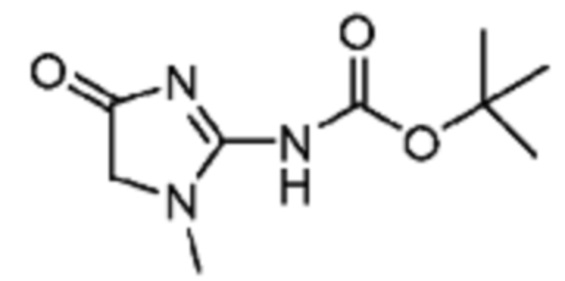
К раствору 2-имино-1-метил-4-имидазолидинона (8,2 г, 72,5 ммоль) в ДМФА (30 мл) при КТ добавляют ди-трет-бутилдикарбонат (17,4 г, 79,7 ммоль). Полученную смесь перемешивают при 60°C в течение ночи. Смесь охлаждают до КТ и гасят водой (30 мл). Смесь экстрагируют EA (30 мл × 2). Органический слой сушат и концентрируют. Остаток очищают хроматографией (элюирование: 1:1 гексан:EA) с получением трет-бутил-N-(1-метил-4-оксо-4,5-дигидро-1H-имидазол-2-ил)карбамата в виде твердого белого вещества (14 г, 90%). МС (ESI+) 214,2 (M+H)+, время удерживания: 1,25 мин. (метод А).
Стадия 2. Получение трет-бутил-N-метил-N-(1-метил-4-оксо-4,5-дигидро-1H-имидазол-2-ил)карбамата

К раствору трет-бутил-N-(1-метил-4-оксо-4,5-дигидро-1H-имидазол-2-ил)карбамата (14 г, 65,7 ммоль) в ТГФ (100 мл) при 0°C порциями добавляют гидрид натрия (2,9 г, 60% в минеральном масле) . Смеси дают возможность нагреться до КТ и перемешивают смесь в течение 30 мин. К реакционной смеси медленно добавляют йодметан (14 г, 95,2 ммоль). Полученную смесь перемешивают при КТ в течение ночи. Реакцию гасят добавлением насыщенного водного раствора хлорида аммония (30 мл). Смесь экстрагируют EA (50 мл × 3). Объединенные органические фазы промывают насыщенным водным раствором соли, сушат и концентрируют. Остаток очищают хроматографией (элюирование: 4:1 гексан:EA) с получением трет-бутил-N-метил-N-(1-метил-4-оксо-4,5-дигидро-1H-имидазол-2-ил)карбамата в виде бесцветного масла (3,5 г, 23%). МС (ESI+) 228,2 (M+H)+, время удерживания: 1,35 мин. (метод А).
Стадия 3. Получение гидрохлорида 1,5-дигидро-1-метил-2-(метиламино)-4H-имидазол-4-она

К раствору трет-бутил-N-метил-N-(1-метил-4-оксо-4,5-дигидро-1H-имидазол-2-ил)карбамата (3,5 г, 15,4 ммоль) в ДХМ (25 мл) добавляют 4N HCl в диоксане (15 мл). Полученную смесь перемешивают при КТ в течение ночи. Растворители удаляют в вакууме с получением гидрохлорида 1,5-дигидро-1-метил-2-(метиламино)-4H-имидазол-4-она в виде твердого белого вещества (1,7 г, 90%). МС (ESI+) 128,0 (M+H)+, время удерживания: 0,33 мин, (метод А).
Промежуточное соединение 25. Синтез (S)-1-(3-хлорфенил)пропилизоцианата
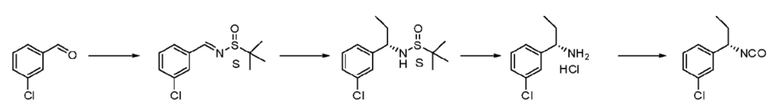
Схема 19
Стадия 1. Получение [N(E),S(S)]-N-[(3-хлорфенил)метилен]-2-метил-2-пропансульфонамида

К раствору 3-хлорбензальдегида (58,5 г, 416 ммоль) и (S)-(-)-2-метил-2-пропансульфонамида (53 г, 437 ммоль) в ТГФ (500 мл) добавляют этоксид титана (IV) (190 г, 832 ммоль). Полученную реакционную смесь нагревают и выдерживают при 75°C в течение 4 часов. Смесь охлаждают до КТ. К смеси добавляют насыщенный раствор соли (500 мл), смесь энергично перемешивают и затем фильтруют через Celite®. Фильтр промывают EA (600 мл). Фазы разделяют и водный слой экстрагируют EA (2 × 150 мл). Объединенную органическую фазу сушат и концентрируют. Остаток очищают хроматографией (элюирование: 4:1 гексан:EA) с получением [N(E),S(S)]-N-[(3-хлорфенил)метилен]-2-метил-2-пропансульфонамида в виде бесцветного масла (100 г, 98%). МС (ESI+) 244,1 (M+H)+, время удерживания: 2,01 мин. (метод А).
Стадия 2. Получение [S(S)]-N-((1S)-1-(3-хлорфенил)пропил)-2-метил-2-пропансульфонамида

[N(E),S(S)]-N-[(3-хлорфенил)метилен]-2-метил-2-пропансульфонамид (100 г, 410 моль) растворяют в TBME (500 мл). Реакционную смесь охлаждают до -20°C, к смеси по каплям добавляют бромид этилмагния (3M в эфире, 274 мл, 820 ммоль) и перемешивают реакционную смесь при -20°C в течение 4 часов. К смеси добавляют насыщенный водный раствор хлорида аммония и затем EA. Слои разделяют и водный слой экстрагируют EA (200 мл). Объединенную органическую фазу промывают насыщенным водным раствором соли, сушат и концентрируют. Остаток очищают хроматографией, элюируя сначала смесью 4:1 гексан:EA с получением [S(S)]-N-((1S)-1-(3-хлорфенил)пропил)-2-метил-2-пропансульфонамида в виде бесцветного масла (65 г, 58%). МС (ESI+) 274,1 (M+H)+, время удерживания: 1,85 мин. (метод А). Колонку дополнительно элюируют этилацетатом с получением [S(S)]-N-((1S)-1-(3-хлорфенил)пропил)-2-метил-2-пропансульфонамида в виде бесцветного масла (33 г, 29%). МС (ESI+) 274,1 (M+H)+, время удерживания: 1,82 мин. (метод А).
Стадия 3. Получение гидрохлорида (S)-1-(3-хлорфенил)пропиламина

[S(S)]-N-((1S)-1-(3-хлорфенил)пропил)-2-метил-2-пропансульфонамид (65 г, 238 ммоль) растворяют в метаноле (200 мл) и к полученному раствору медленно добавляют 4M HCl в 1,4-диоксане (240 мл). Реакционный раствор перемешивают в течение 60 мин. Реакционную смесь концентрируют с получением твердого остатка. К твердому остатку добавляют гексан (200 мл) и перемешивают полученную суспензию при КТ в течение 60 мин. Твердое вещество собирают фильтрацией, промывают гексанами и сушат с получением гидрохлорида (S)-1-(3-хлорфенил)пропиламина (35 г, 71%). МС (ESI+) 170,1 (M+H)+, время удерживания: 1,12 мин. (метод А).
Стадия 4. Получение (S)-1-(3-хлорфенил)пропилизоцианата

К суспензии гидрохлорида (S)-1-(3-хлорфенил)пропиламина (9,7 г, 47 ммоль) в ДХМ (150 мл) добавляют 1N водный раствор NaHCO3 (155 мл). Смесь охлаждают до 0°C и добавляют трифосген (4,8 г, 16 ммоль). Реакционную смесь перемешивают в течение 15 минут при 0°C и в течение 1 часа при КТ. Реакционную смесь два раза экстрагируют ДХМ. Объединенную органическую фазу сушат и концентрируют с получением (S)-1-(3-хлорфенил)пропилизоцианата в виде бесцветного масла (7,3 г, 80%), который используют в следующей стадии без дополнительной очистки. 1H ЯМР (400 Гц, CDCl3) δ 7,31 (м, 3H), 7,16 (с, 1H), 4,54 (т, 1H), 1,86 (м, 2H), 0,99 (т, 3H).
Промежуточное соединение 26. Синтез (R)-1-(3-хлорфенил)пропилизоцианат
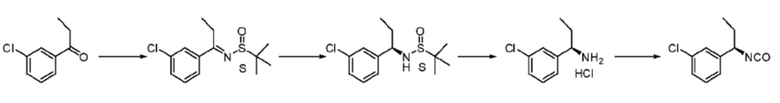
Схема 20
Стадия 1. Получение [S(S)]-N-[1-(3-хлорфенил)пропилиден]-2-метилпропан-2-сульфинамид

Раствор 3’-хлорпропиофенона (75 г, 445 ммоль) , (S)-(-)-2-метилпропан-2-сульфинамида (57 г, 470 ммоль) и этоксида титана (IV) (213 г, 934 ммоль) в ТГФ (700 мл) нагревают и выдерживают при 80°C в течение ночи. После охлаждения добавляют насыщенный водный раствор соли (700 мл) и EA (700 мл). Смесь перемешивают при КТ в течение 1 часа. Смесь фильтруют через Celite® и фильтр промывают EA. Органическую фазу отделяют, сушат и концентрируют. Остаток очищают хроматографией (элюирование: 2:1 гексан:EA) с получением [S(S)]-N-[1-(3-хлорфенил)пропилиден]-2-метилпропан-2-сульфинамида в виде бесцветного масла (100 г, 83%). МС (ESI+) 272,1 (M+H)+, время удерживания: 2,00 мин. (метод А).
Стадия 2. Получение [S(S),R]-N-[1-(3-хлорфенил)пропил]-2-метилпропан-2-сульфинамида
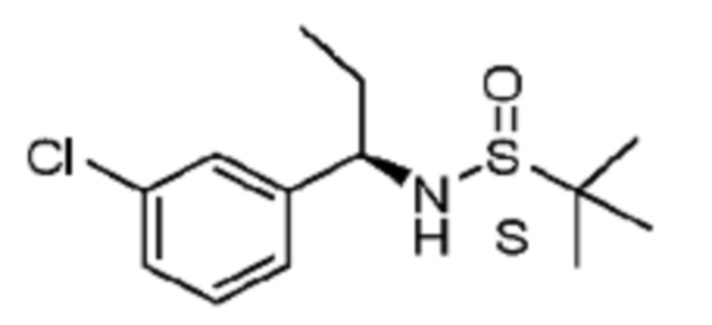
К раствору [S(S)]-N-[1-(3-хлорфенил)пропилиден]-2-метилпропан-2-сульфинамида (25 г, 92 ммоль) в ТГФ (250 мл) при 0°C добавляют раствор три-втор-бутилборогидрида (L-selectride, 1M раствор в ТГФ, 185 мл, 185 ммоль). Реакционную смесь перемешивают в течение 3 часов при 0°C. Реакционную смесь гасят насыщенным водным раствором хлорида аммония (250 мл). Смесь фильтруют через Celite® и фильтр промывают EA. Органическую фазу отделяют, сушат и концентрируют. Остаток очищают хроматографией (элюирование: 1:1 гексан:EA) с получением [S(S),R]-N-[1-(3-хлорфенил)пропил]-2-метилпропан-2-сульфинамида в виде бесцветного масла (20,7 г, 82%). МС (ESI+) 274,1 (M+H)+, время удерживания: 1,83 мин. (метод А).
Стадия 3. Получение гидрохлорида (R)-1-(3-хлорфенил)-1-пропанамина
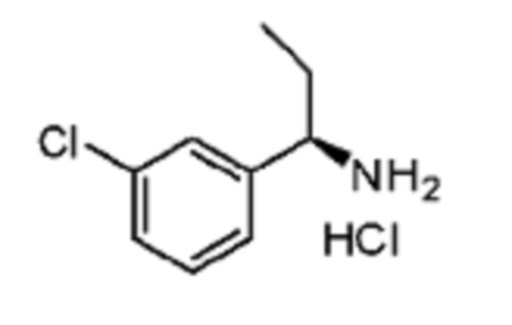
К раствору [S(S),R]-N-[1-(3-хлорфенил)пропил]-2-метилпропан-2-сульфинамида (20,7 г, 75,8 ммоль) в метаноле (150 мл) медленно добавляют 4M HCl в 1,4-диоксане (76 мл). Реакционную смесь перемешивают в течение 1 часа. Реакционную смесь концентрируют с получением твердого остатка. К твердому остатку добавляют гексан (150 мл) и перемешивают полученную суспензию при КТ в течение 1 часа. Твердое вещество собирают фильтрацией, промывают гексанами и сушат с получением гидрохлорида (R)-1-(3-хлорфенил)-1-пропанамина (14 г, 90%). МС (ESI+) m/z 170,1 (M+H)+, время удерживания: 1,12 мин. (метод А).
Стадия 4. Получение (R)-1-(3-хлорфенил)пропилизоцианата

К суспензии гидрохлорида (R)-1-(3-хлорфенил)-1-пропанамина (38,7 г, 188 ммоль) в ДХМ (300 мл) добавляют 1N водный раствор NaHCO3 (646 мл). Смесь охлаждают до 0°C и добавляют трифосген (18,9 г, 63,8 ммоль). Реакционную смесь перемешивают в течение 15 мин. при 0°C и в течение 1 часа при КТ. Реакционную смесь дважды экстрагируют ДХМ. Объединенную органическую фазу сушат и концентрируют с получением (R)-1-(3-хлорфенил)пропилизоцианата в виде бесцветного масла (29,4 г, 80%), который используют в следующей стадии без дополнительной очистки. 1H ЯМР (400 Гц, CDCl3) δ 7,32 (м, 3H), 7,20 (м, 1H), 4,54 (т, 1H), 1,82 (м, 2H), 0,98 (т, 3H).
Пример 2. Получение дополнительных промежуточных соединений
Промежуточные соединения, представленные в таблицах 2, 3 и 4 ниже, синтезируют как показано на схемах 6-20 и в экспериментальных методиках примера 1. В некоторых случаях применяют дополнительные трансформации функциональных групп. Обычно такие трансформации будут понятны специалисту в области органического синтеза. Кроме того, специалисту в области органического синтеза будет понятно, что исходные материалы и условия реакций могут изменяться для получения желательных конечных продуктов.
Таблица 2. Дополнительные промежуточные соединения



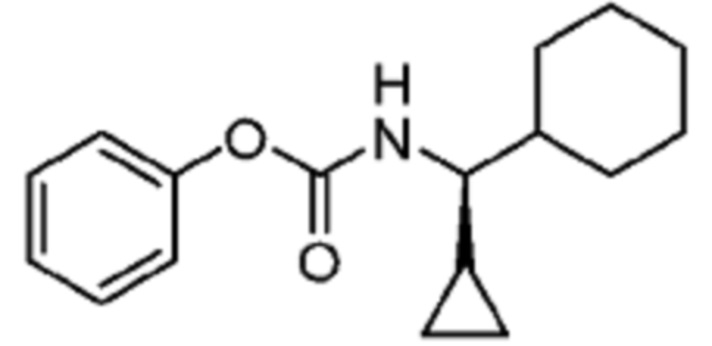

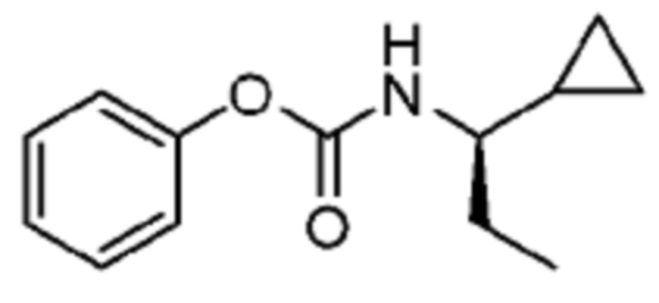
карбамат

Таблица 3. Дополнительные промежуточные соединения



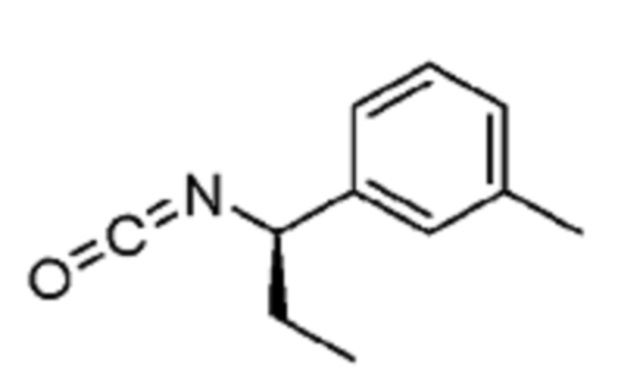




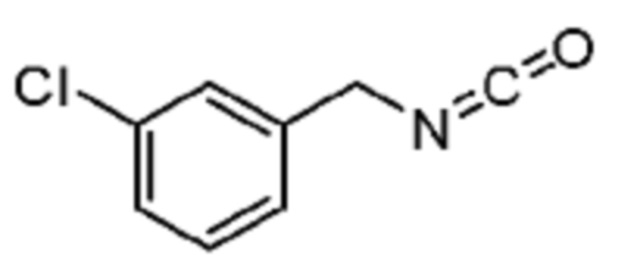


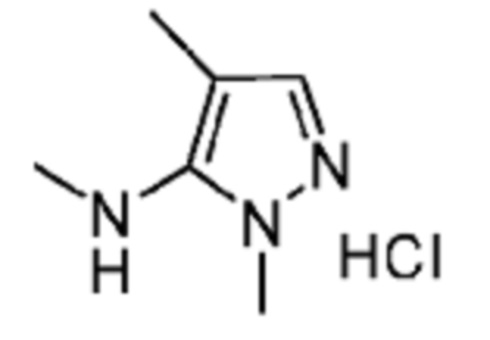
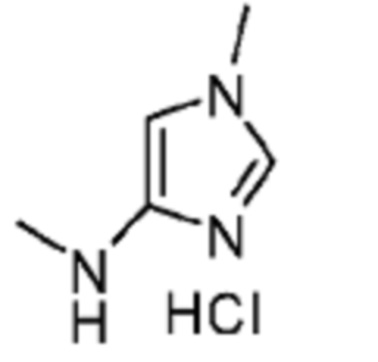
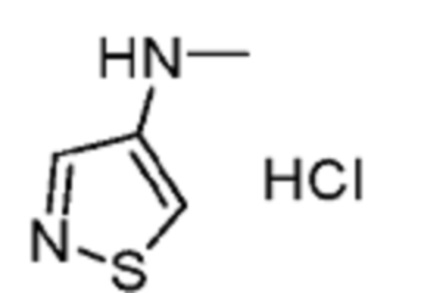
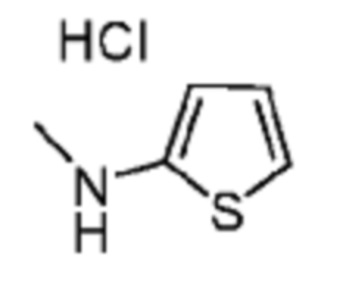

Таблица 4. Дополнительные промежуточные соединения

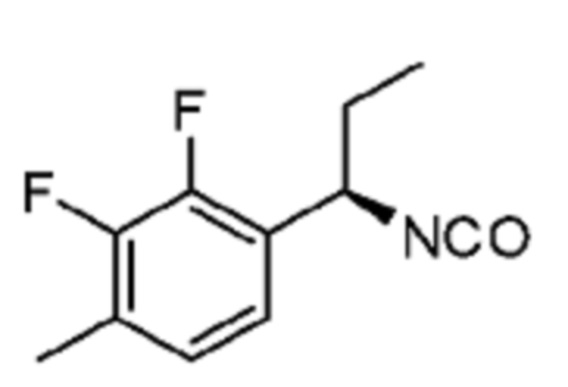

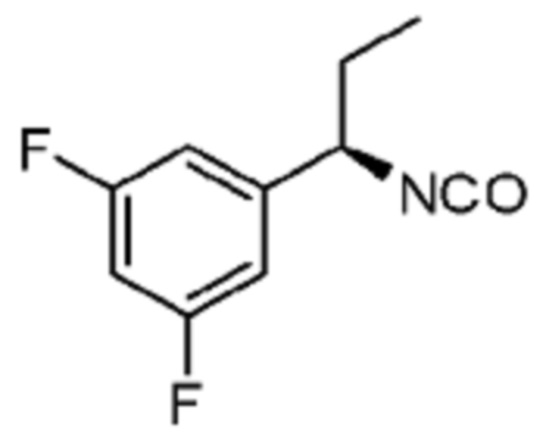




пропилизоцианат
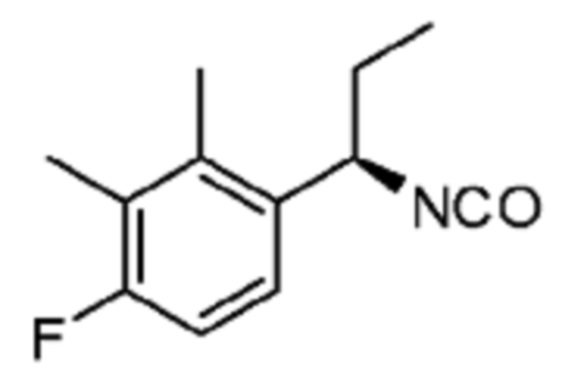
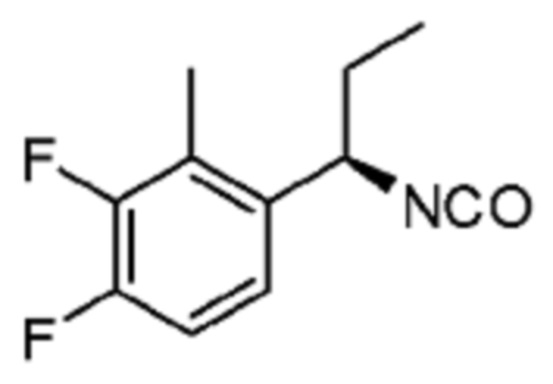



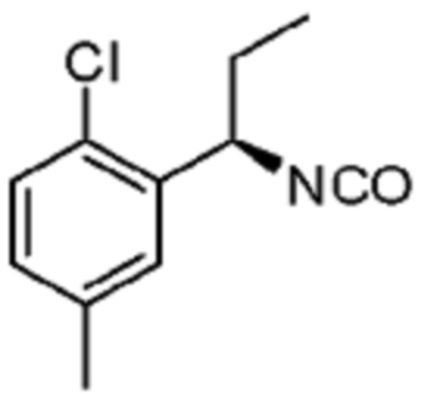
5-метилфенил)
пропилизоцианат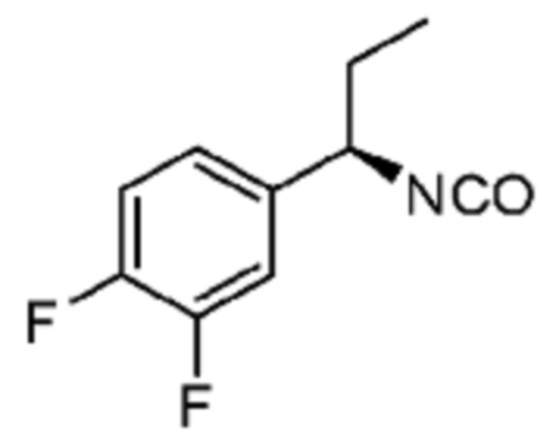
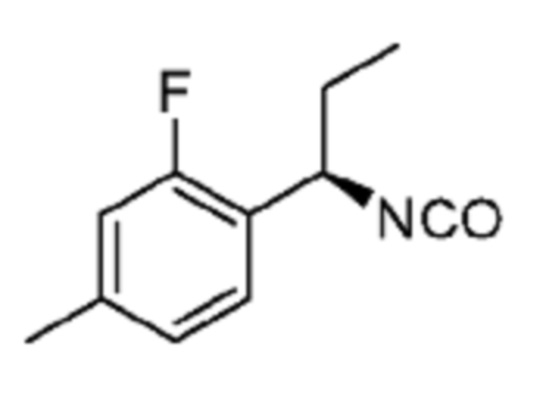
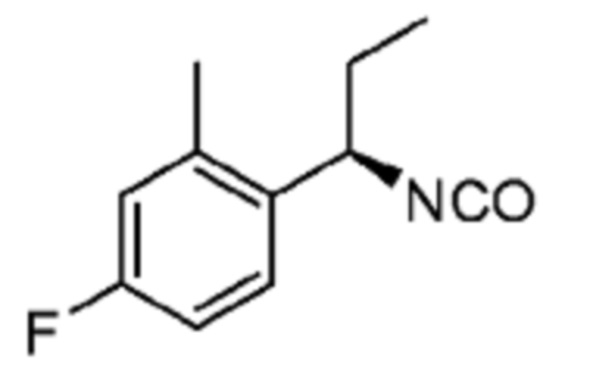
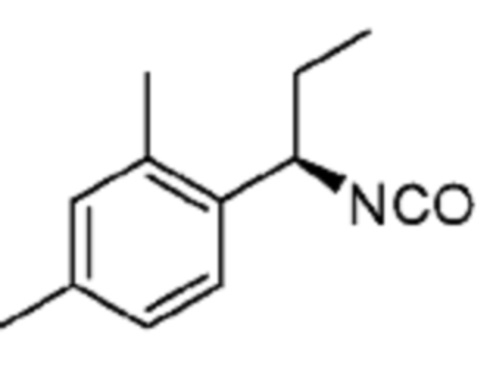
пропилизоцианат


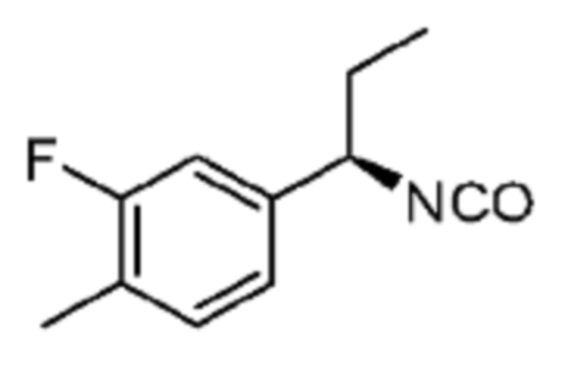

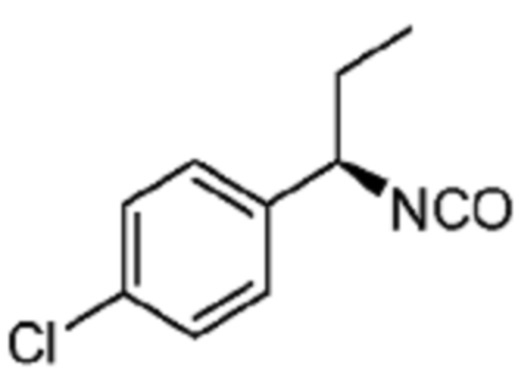

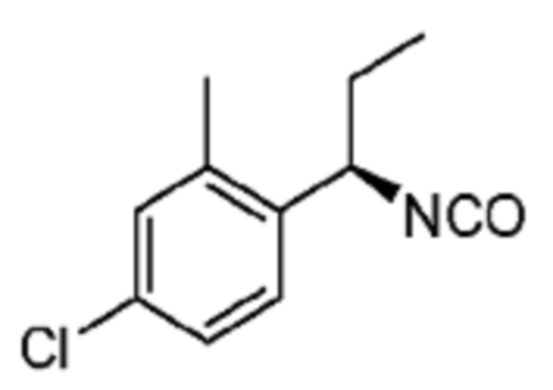


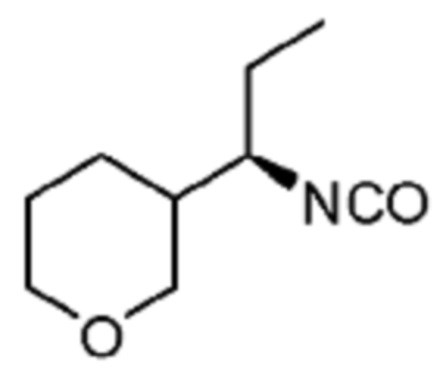


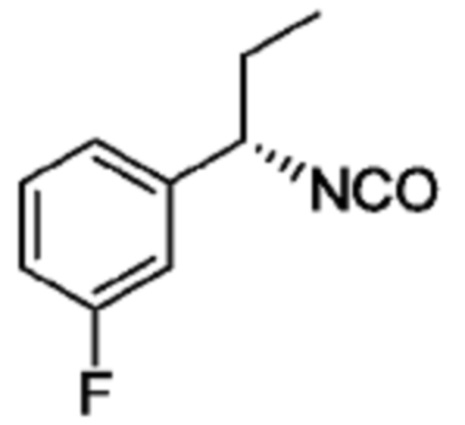

Пример 3. (2S,3R)-3-((2-аминопиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-3-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамид (соединение 1-3)
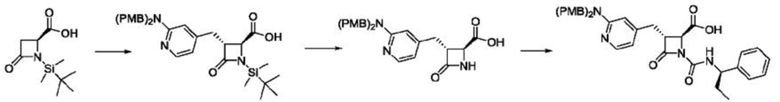
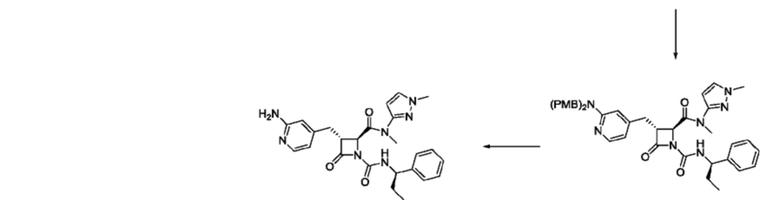
Схема 21
Стадия 1. Получение (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-4-оксоазетидин-2- карбоновой кислоты

(S)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновую кислоту (10,0 г, 43 ммоль) растворяют в ТГФ (160 мл) и охлаждают до -20 °C. Реакционную смесь обрабатывают LDA (92M в ТГФ, 47 мл, 94 ммоль) при температуре в интервале примерно от -10 до -20°C и добавляют 4-(бромметил)-N, N-бис(4-метоксибензилпиридин-2-амин (21,2 г, 49 ммоль) в ТГФ (80 мл), поддерживая температуру смеси ниже -10°C. Реакционную смесь перемешивают в течение нескольких часов при -15°C, затем смеси дают возможность нагреться до КТ и перемешивают смесь в течение нескольких часов. Реакционную смесь гасят водой (200 мл) и затем кипятят с обратным холодильником в течение 3 часов. Реакционную смесь охлаждают до КТ и обрабатывают 5% водным раствором трифосфата калия (250 мл). Фазы разделяют и водный слой экстрагируют EA (150 мл × 3) для удаления примесей. Водную фазу подкисляют до pH 3,1 с помощью 6 N HCl и экстрагируют EA (300 мл × 3). Органичесую фазу сушат над MgSO4 и концентрируют. Остаточный EA заменяют на гептан (250 мл) для получения суспензии, которую охлаждают и фильтруют. Осадок на фильтре переносят в 40 объемов изопропилового спирта (400 мл) и кипятят с обратным холодильником в течение примерно 1 часа. Смесь охлаждают до КТ и нерастворимые твердые примеси удаляют фильтрацией. В фильтрате изопропилового спирта растворитель заменяют на гептан (250 мл), вызывая осаждение продукта. Суспензию охлаждают до 5-10°C и фильтруют. Осадок на фильтре сушат с получением (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-4-оксоазетидин-2-карбоновой кислоты (12 г, 59%). МС (ESI+) m/z 462,2 (M+H)+, время удерживания: 1,23 мин. (метод A).
Стадия 2. Получение (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-1-(((R)-1-фенилэтил)карбамоил)-4-оксоазетидин- 2-карбоновой кислоты

(2S,3R)-3-((2-(Бис(4-метоксибензил)амино)пиридин-4-ил)метил)-4-оксоазетидин-2-карбоновую кислоту (15 г, 32,5 ммоль) растворяют в ДХМ (150 мл) и обрабатывают DBU (17,3 г, 113,6 ммоль) и затем (R)-(+)-1-фенилпропилизоцианат (13,1 г, 81,3 ммоль) при КТ. Реакционную смесь перемешивают при КТ в течение ночи. Смесь разбавляют ДХМ (150 мл) и промывают насколькими порциями 10% водной лимонной кислоты до тех пор, пока ВЭЖХ не будет обнаруживать DBU в органической фазе. Органическую фазу сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 20%-100% EA в гексане) с получением продукта в виде твердого белого вещества (19.2 г, 95%). МС (ESI+) m/z 623,3 (M+H)+, время удерживания: 1,55 мин. (метод A).
Стадия 3. Получение (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-3-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамид

Раствор (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-1-(((R)-1-фенилэтил)карбамоил)-4-оксоазетидин-2-карбоновой кислоты (124 мг, 0,2 ммоль) в ДХМ (3 мл) обрабатывают гидрохлоридом N,1-диметил-1H-пиразол-3-амина (41 мг, 0,3 ммоль) и хлоридом 2-хлор-1,3-диметилимидазолиния (50 мг, 0,3 ммоль). Смесь перемешивают при КТ и добавляют TEA (61 мг, 0,6 ммоль). Реакционную смесь перемешивают в течение ночи; затем смесь разбавляют ДХМ (20 мл). Смесь промывают водным раствором NaHCO3, водой, сушат над MgSO4 и концентрируют. The маслянистый остаток очищают препаративной ТСХ (2:1 гексан/EA) с получением продукта в виде твердого белого вещества (92 мг, 65%). МС (ESI+) m/z 716,4 (M+H)+, время удерживания: 2,95 мин. (метод B).
Стадия 4. Получение (2S,3R)-3-((2-аминопиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-3-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида

Раствор (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-3-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамид (63 мг, 0,09 ммоль) в ДХМ (3 мл) обрабатывают триэтилсиланом (0,1 мл) и охлаждают до 0°C. К смеси по каплям добавляют трифторуксусную кислоты (1 мл) добавляют и медленно нагревают реакционную смесь до КТ. Спустя 21 час ВЭЖХ анализ (метод A) показывает, что исходное вещество израсходовано. Реакционную смесь концентрируют и остаток переносят в 5 мл ДХМ. Смесь промывают водным раствором NaHCO3, водой, сушат над MgSO4 и концентрируют. Остаток очищают препаративной ТСХ (элюирование: EA) с получением указанного в заголовке соединения в виде твердого белого вещества (21 мг, 50%). МС (ESI+) m/z 476,3 (M+H)+, время удерживания: 1,23 мин. (метод A). 1H ЯМР (CDCl3) δ 0,84 (т, 3H), 1,80 (кв, 2H), 2,80 (м, 1H), 2,84 (м, 1H), 3,24 (с, 3H), 3,56 (м, 1H), 3,84 (с, 3H), 4,40 (с, 1H), 4,60 (м, 2H), 4,76 (м, 1H), 6,20-6,32 (м, 3H), 6,74 (д, 1H), 7,20-7,40 (м, 6), 7,94 (д, 1H).
Пример 4. (2S,3R)-3-((2-аминопиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-5-ил)-N1-((R)-1-(3-хлорфенил)пропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамид (соединение 1-4)


Схема 22
Стадия 1. Получение (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-1-(трет-бутил(диметил)силил)-4-оксоазетидин-2-карбоновой кислоты
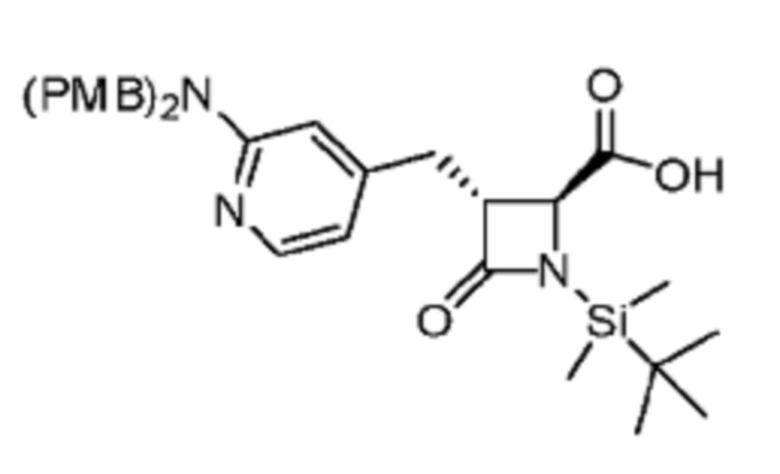
(S)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновой кислоты (10 г, 43 ммоль) растворяют в ТГФ (160 мл) и охлаждают до -20°C. Реакционную смесь обрабатывают LDA (2M в ТГФ, 47 мл, 94 ммоль) при температуре в интервале примерно от -10 до -20°C с последующей обработкой 4-(бромметил)-N, N-бис(4- метоксибензилпиридин-2-амином (21,2 г, 49 ммоль) в ТГФ (80 мл), поддерживая температуру смеси ниже -10°C. Реакционную смесь перемешивают в течение нескольких часов при -15°C; затем дают возможность смеси нагреться до КТ и перемешивают смесь в течение нескольких часов. Реакционную смесь гасят водой (200 мл) и подкисляют 10% лимонной кислотой до pH 4. Смесь экстрагируют EA (100 мл × 3), органичесую фазу сушат над MgSO4 и концентрируют. Остаток очищают хроматографией (элюирование: 20%-100% EA в гексане) с получением продукта в виде твердого белого вещества (14,5 г, 59%). МС (ESI+) m/z 576,3 (M+H)+, время удерживания: 1,56 мин. (метод A).
Стадия 2. Получение (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-1-(трет-бутил(диметил)силил)-N2-(1-метил-1H-пиразол-5-ил)-N2-метил-4-оксоазетидин-2-карбоксамида
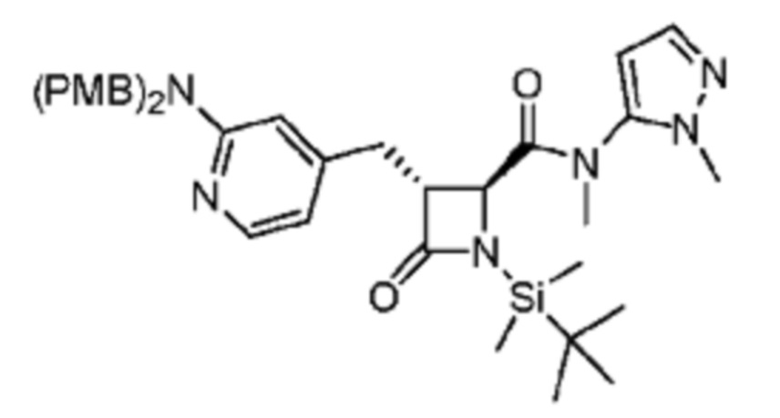
Раствор (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-1-(трет-бутил(диметил)силил)-4-оксоазетидин-2-карбоновой кислоты (4 г, 6,9 ммоль) в ДХМ (30 мл) обрабатывают гидрохлоридом N,1-диметил-1H-пиразол-5-амина (1,6 г, 10,8 ммоль) и хлоридом 2-хлор-1,3-диметилимидазолиния (1,8 г, 10,6 ммоль). Реакционную смесь перемешивают при КТ и к смеси добавляют TEA (2,5 г, 24,7 ммоль). Реакционную смесь перемешивают в течение ночи, затем разбавляют ДХМ (20 мл). Смесь промывают водным раствором NaHCO3 и водой, сушат над MgSO4 и концентрируют. Маслянистый остаток очищают хроматографией (элюирование: 2:1 гексан/EA) с получением продукта в виде твердого белого вещества (3,2 г, 70%). МС (ESI+) m/z 669,6 (M+H)+, время удерживания: 1,77 мин. (метод A).
Стадия 3. Получение (2S,3R)-3-((2-((бис(4-метоксибензил)амино)пиридин-4-ил)метил)- N2-(1-метил-1H-пиразол-5-ил)-N2-метил-4-оксоазетидин-2-карбоксамида

К раствору (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-1-(трет-бутил(диметил)силил)-N2-(1-метил-1H-пиразол-5-ил)-N2-метил-4-оксоазетидин-2-карбоксамида (3,2 г, 4,8 ммоль) в метаноле (30 мл) добавляют 0,5 M NH4F в метаноле (15 мл) и уксусную кислоту (1,5 мл). Реакционную смесь перемешивают при КТ в течение 2 часов. Реакционную смесь концентрируют и остаток растворяют в EA (30 мл). Реакционную смесь экстрагируют водным раствором NaHCO3 (10 мл) и насыщенным водным раствором соли (10 мл), сушат над MgSO4 и концентрируют с получением продукта в виде твердого белого вещества (2,4 г, 90%). МС (ESI+) 555,3 (M+H)+, время удерживания: 1,28 мин. (метод A).
Стадия 4. Получение (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-5-ил)-N1-((R)-1-(3-хлорфенил)пропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида
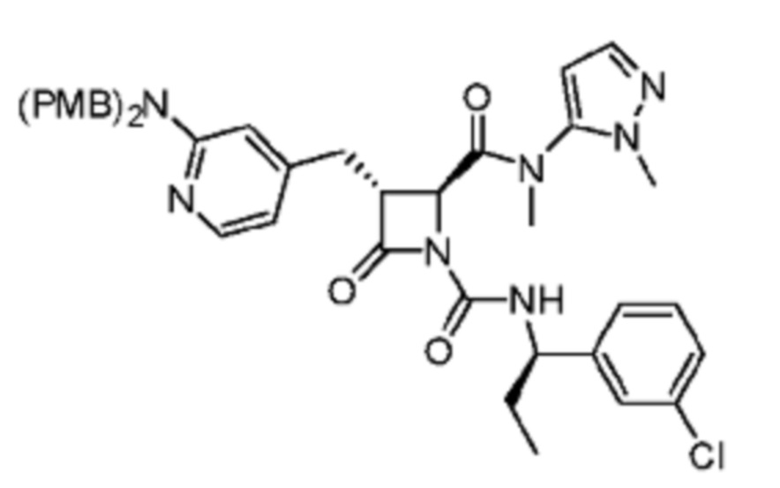
Раствор (2S,3R)-3-((2-((бис(4-метоксибензил)амино)пиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-5-ил)-N2-метил-4-оксоазетидин-2-карбоксамида (1,2 г, 2,2 ммоль) в ДХМ (10 мл) обрабатывают TEA (1,1 г, 10,8 ммоль), к смеси добавляют (R)-1-(3-хлорфенил)пропилизоцианат (1,1 г, 5,6 ммоль) и перемешивают реакционную смесь при 50°C до тех пор, пока анализ ВЭЖХ (метод B) не покажет, что реакция завершена. Реакционную смесь выливают в 40 мл воды и водную фазу экстрагируют ДХМ (3 × 15 мл). Объединенную органическую фазу сушат над MgSO4 и концентрируют. Бесцветный вязкий маслянистый остаток очищают хроматографией (элюирование: 2:1 гексан/EA) с получением указанного в заголовке соединения в виде белой пены (0,9 г, 60%). МС (ESI+) m/z 750,3 (M+H)+, время удерживания: 3,17 мин. (метод B).
Стадия 5. Получение (2S,3R)-3-((2-аминопиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-5-ил)-N1-((R)-1-(3-хлорфенил)пропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида
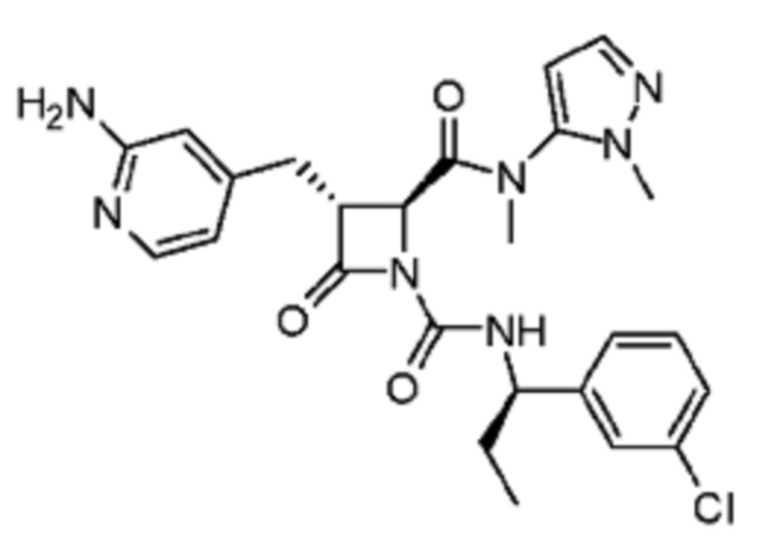
Раствор (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-5-ил)-N1-((R)-1-(3-хлорфенил)пропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида (0,9 г, 1,2 ммоль) в ДХМ (10 мл) обрабатывают трииэтилсиланом (1 мл) и охлаждают до 0°C. К смеси по каплям добавляют трифторуксусную кислоту (2 мл) и медленно нагревают реакционную смесь до КТ. Спустя 21 час ВЭЖХ анализ показывает, что исходное вещество израсходовано. Реакционную смесь концентрируют и остаток переносят в 5 мл ДХМ. Смесь промывают водным раствором NaHCO3 и водой, сушат над MgSO4 и концентрируют. Остаток очищают препаративной ТСХ (10% метанол в ДХМ) с получением указанного в заголовке соединения в виде твердого белого вещества (340 мг, 55%). МС (ESI+) m/z 510,2 (M+H)+, время удерживания: 1,25 мин. (метод A).
Пример 5. (2S,3R)-3-((2-диметиламинопиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-4-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамид (соединение 1-5)
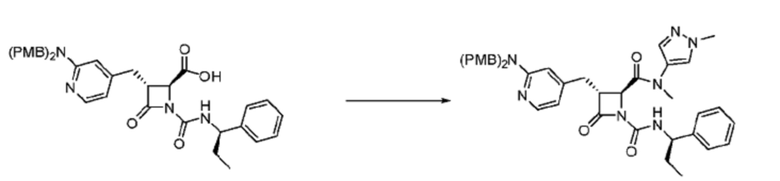
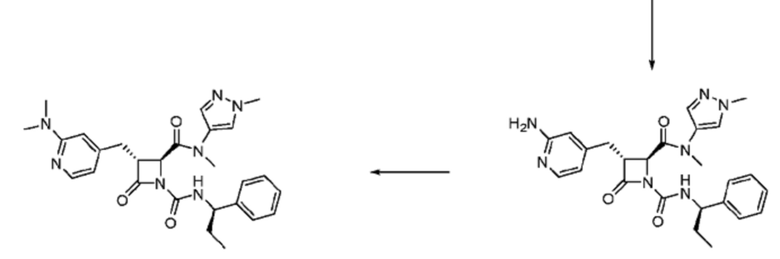
Схема 23
Стадия 1. Получение (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-4-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида

Раствор (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-1-(((R)-1-фенилэтил)карбамоил)-4-оксоазетидин-2-карбоновой кислоты (продукт примера 3, стадии 2, 100 мг, 0,16 ммоль) в ДХМ (5 мл) обрабатывают дигидрохлоридом N,1-диметил-1H-пиразол-4-амина (50 мг, 0,27 ммоль) и хлоридом 2-хлор-1,3-диметилимидазолиния (50 мг, 0.3 ммоль). Смесь перемешивают при КТ и к смеси добавляют TEA (100 мг, 0,98 ммоль). Реакционную смесь перемешивают в течение ночи и затем разбавляют ДХМ (20 мл). Смесь промывают водным раствором NaHCO3, водой, сушат над MgSO4 и концентрируют. Маслянистый остаток очищают препаративной ТСХ (2:1 гексан/EA) с получением продукта в виде твердого белого вещества (69 мг, 60%). МС (ESI+) m/z 716,4 (M+H)+, время удерживания: 2,95 мин. (метод B).
Стадия 2. Получение (2S,3R)-3-((2-аминопиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-4-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида

Раствор (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-4-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамид (69 мг, 0,09 ммоль) в ДХМ (3 мл) обрабатывают трииэтилсилан (0,1 мл) и охлаждают до 0°C. По каплям добавляют трифторуксусную кислоту (1 мл) и реакционную смесь медленно нагревают до КТ. Спустя 21 час ВЭЖХ анализ показывает, что исходное вещество израсходовано. Реакционную смесь концентрируют и остаток переносят в 5 мл ДХМ. Смесь промывают водным раствором NaHCO3, водой, сушат над MgSO4 и концентрируют. Остаток очищают препаративной ТСХ (элюирование: EA) с получением указанного в заголовке соединения в виде твердого белого вещества (38 мг, 83%). МС (ESI+) m/z 476,3 (M+H)+, время удерживания: 1,23 мин. (метод A ), 1H ЯМР (CDCl3) δ 0,86 (т, 3H),1,80 (кв, 2H), 2,10 (м, 2H), 2,62 (м, 1H), 2,84 (м,1H), 3,16 (с, 3H), 3,60 (м, 1H), 3,88 (с, 3H), 4,12 (с, 1H), 4,60 (м, 2H), 4,74 (м, 1H), 6,12 (с, 1H), 6,30 (д, 1H), 6,70 (д, 1H), 7,20-7,40 (м, 5H), 7,96 (д, 1H).
Стадия 3. Получение(2S,3R)-3-((2-диметиламинопиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-4-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамид
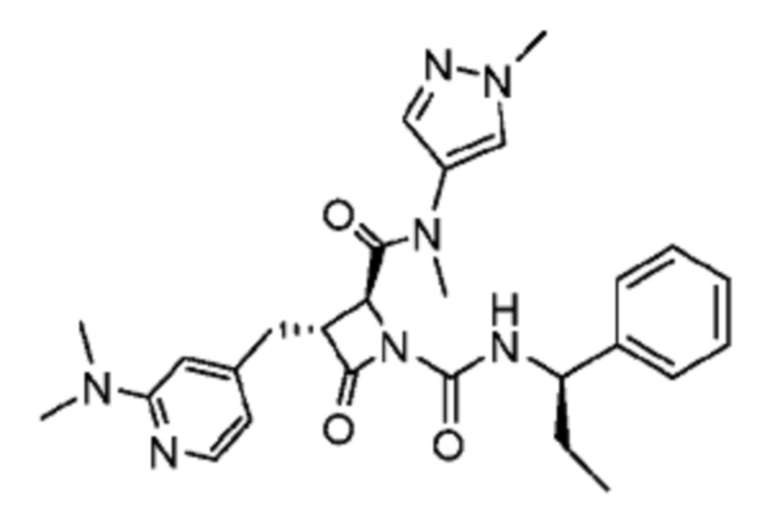
Уксусную кислоты (40 мг, 0,7 ммоль) добавляют к раствору (2S,3R)-3-((2-аминопиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-4-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида (36 мг, 0,08 ммоль), формальдегида (160 мг, 37% водный раствор, 2,0 ммоль) и NaBH3CN (42 мг, 0,7 ммоль) в ACN (5 мл) и воды (1 мл) при КТ. Реакционную смесь перемешивают в течение 2 дней. Реакционную смесь разбавляют водой (10 мл) и подщелачивают до pH 4 1M водным раствором NaOH и затем экстрагируют EA (10 мл × 2). Органический слой промывают насыщенным водным раствором соли (5 мл), сушат над MgSO4 и концентрируют. Остаток очищают препаративной ТСХ (10% метанол в ДХМ) с получением (2S,3R)-3-((2-диметиламинопиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-4-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида в виде твердого белого вещества (25 мг, 66%). МС (ESI+) m/z 504,2 (M+H)+, время удерживания: 1,22 мин. (метод A).
Пример 6. (2S,3R)-3-((2-аминопиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-5-ил)-N1-((R)-1-циклогексил-2,2,2-трифторэтил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамид (соединение 1-6)

Раствор (2S,3R)-3-((2-((бис(4-метоксибензил)амино)пиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-5-ил)-N2-метил-4-оксоазетидин-2-карбоксамид (продукт примера 4, стадия 3, 100 мг, 0,18 ммоль) в ДХМ (10 мл) обрабатывают TEA (100 мг, 0,98 ммоль). Фенил-[(1S)-1-циклогексил-2,2,2-трифторэтил]карбамат (100 мг, 0,48 ммоль) добавляют и перемешивают реакционную смесь при 50°C до тех пор, пока ВЭЖХ анализ (метод B) не покажет, что ракция завершена. Реакционную смесь выливают в 40 мл воду и водную фазу экстрагируют ДХМ (3 × 15 мл). Объединенную органическую фазу сушат над MgSO4 и концентрируют. Бесцветный вязкий маслянистый остаток очищают хроматографией (элюирование: 2:1 гексан/EA) с получением (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-5-ил)-N1-((R)-1-циклогексил-2,2,2-трифторэтил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида в виде белой пены (105 мг, 70%). МС (ESI+) m/z 762,6 (M+H)+, время удерживания: 1,90 мин. (метод A).
Раствор (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-5-ил)-N1-((R)-1-циклогексил-2,2,2-трифторэтил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида (105 мг, 0,14 ммоль) в ДХМ (3 мл) обрабатывают трииэтилсиланом (0,1 мл) и охлаждают до 0°C. К смеси по каплям добавляют трифторуксусную кислоту (1 мл) и реакционную смесь медленно нагревают до КТ. Спустя 21 час ВЭЖХ анализ показывает, что исходное вещество израсходовано. Реакционную смесь концентрируют и остаток переносят в 5 мл ДХМ. Смесь промывают водным раствором NaHCO3 и водой, сушат над MgSO4 и концентрируют. Остаток очищают препаративной ТСХ (элюирование: EA) с получением указанного в заголовке соединения в виде твердого белого вещества (36 мг, 50%). МС (ESI+) m/z 522,4 (M+H)+, время удерживания: 1,28 мин. (метод A).
Пример 7. (2S,3R)-3-((2-аминопиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-3-ил)-N1-((R)-1-(3-хлорфенил)пропил)-N1-метил-N2-метил-4-оксоазетидин-1,2-дикарбоксамид (соединение 1-7)
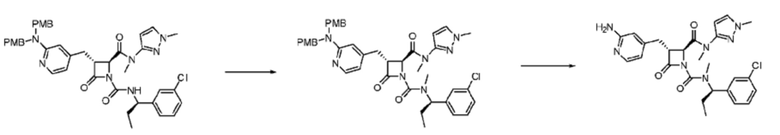
Схема 24
Стадия 1. Получение (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-5-ил)-N1-((R)-1-(3-хлорфенил)пропил)-N1-метил-N2-метил-4-оксоазетидин-1,2-дикарбоксамида
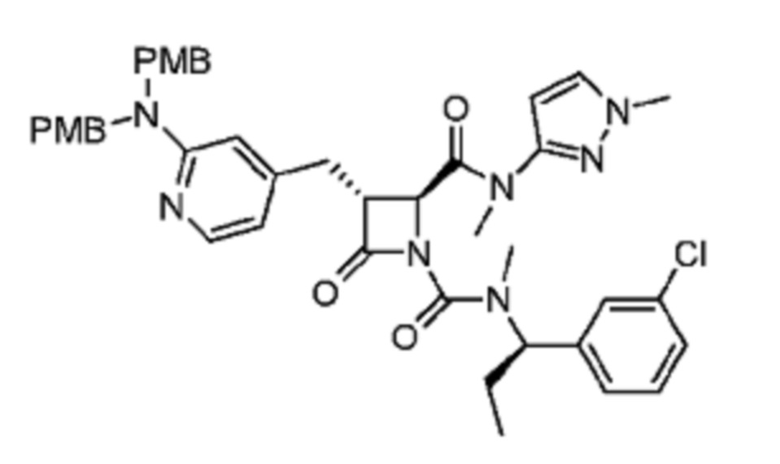
К раствору (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-5-ил)-N1-((R)-1-(3-хлорфенил)пропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида (получен в соответствии с методикой примера 4, стадия 4, 300 мг, 0,4 ммоль) в ДМФА (4 мл) добавляют гидрид натрия (19 мг, 60% в минеральном масле, 0,48 ммоль). Смесь перемешивают в течение 5 мин. К смеси добавляют метилйодид (68 мг, 0,48 ммоль). Смесь перемешивают при КТ в течение 30 мин. Реакционную смесь гасят несколькими каплями воды. Реакционную смесь разбавляют EA (10 мл) и водой (10 мл). Органический слой промывают насыщенным водным раствором соли, сушат над MgSO4 и концентрируют. Сырой продукт очищают препаративной ТСХ (элюирование: EA) с получением указанного в заголовке соединения в виде твердого белого вещества (91 мг, 30%). МС (ESI+) m/z 764,6 (M+H)+, время удерживания: 1,78 мин. (метод А).
Стадия 2. Получение (2S,3R)-3-((2-аминопиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-3-ил)-N1-((R)-1-(3-хлорфенил)пропил)-N1-метил-N2-метил-4-оксоазетидин-1,2-дикарбоксамида
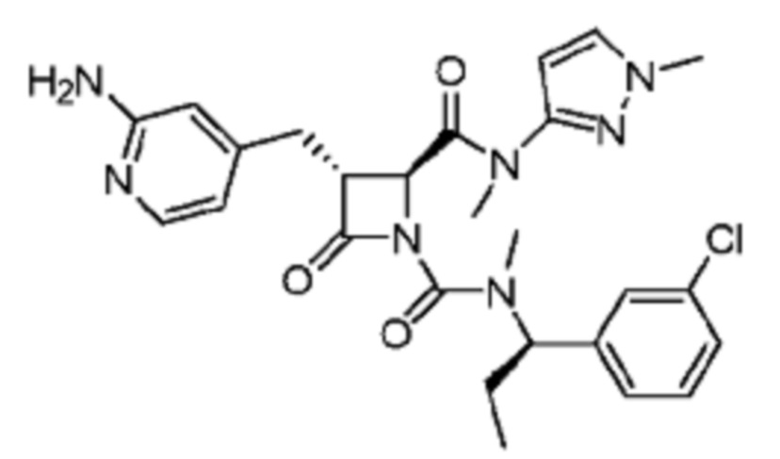
Раствор (2S,3R)-3-((2-(бис(4-метоксибензил)амино)пиридин-4-ил)метил)-N2-(1-метил-1H-пиразол-5-ил)-N1-((R)-1-(3-хлорфенил)пропил)-N1-метил-N2-метил-4-оксоазетидин-1,2-дикарбоксамид (91 мг, 0,12 ммоль) в ДХМ (2 мл) обрабатывают трииэтилсиланом (0,1 мл) и охлаждают до 0°C. Реакционную смесь обрабатывают трифторуксусной кислотой (0,5 мл), добавляя ее по каплям с перемешиванием, и медленно нагревают смесь до КТ. Спустя 21 час ВЭЖХ анализ (метод А) показывает, что исходное вещество израсходовано. Реакционную смесь концентрируют и остаток переносят в ДХМ (15 мл). Смесь промывают насыщенным водным раствором NaHCO3 и водой, сушат над MgSO4 и концентрируют. Сырой продукт очищают препаративной ТСХ (элюирование: EA) с получением указанного в заголовке соединения в виде твердого белого вещества (31 мг, 50%). МС (ESI+) m/z 524,4 (M+H)+, время удерживания: 1,25 мин. (метод А).
Пример 8. Синтез (2S,3R)-3-((2-амино-6-метилпиридин-4-ил)метил)-N2-(1-метил-1H-имидазол-2-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамид

Схема 25
Стадия 1. Получение (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновой кислоты

(S)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновую кислоту (125 г, 545 ммоль) растворяют в ТГФ (2 л) и охлаждают до -20 oC. Реакционную смесь обрабатывают LDA (2M в ТГФ, 600 мл, 1Ю2 моль), поддерживая температуру в интервале от -10 до -20°C. Полученную гелеподобную смесь перемешивают при -15°C в течение 30 мин. Затем добавляют трет-бутил[4-(бромметил)-6-метилпиридин-2-ил] (4-метоксибензил)карбамат (230 г, 546 ммоль) в ТГФ (500 мл), поддерживая температуру смеси ниже -10°C. Реакционную смесь перемешивают при -10°C в течение 2 часов, затем смесь охлаждают до -20°C, гасят водой (200 мл) и затем подкисляют до pH 4 с помощью 10% водной лимонной кислоты, поддерживая температуру смеси ниже 10°C. Смесь экстрагируют EA (500 мл × 3). Объединенный органический слой сушат и концентрируют. Остаток очищают хроматографией (элюирование с градиентом: 20%-50% EA/гексан) с получением (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновой кислоты в виде твердого белого вещества (155 г, 50%). МС (ESI+) m/z 570,3 (M+H)+, время удерживания: 2,10 мин. (метод A).
Стадия 2. Получение (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-1-(трет-бутилдиметилсилил)-N2-(1-метил-1H-имидазол-2-ил)-N2-метил-4-оксоазетидин-2-карбоксамида

К раствору (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновой кислоты (146 г, 256 ммоль) в ДМФА (500 мл) добавляют HATU (146 г, 384 ммоль). Смесь перемешивают в течение 10 мин. при комнатной температуре. К смеси добавляют N,1-диметил-1H-имидазол-2-амин (37 г, 333 ммоль), полученный раствор перемешивают в течение 10 мин. и затем добавляют диизопропилэтиламин (99,5 г, 770 ммоль). Смесь перемешивают при КТ в течение 2 часов и затем добавляют насыщенный водный раствор соли (600 мл). Смесь экстрагируют EA (500 мл × 3). Объединенную органическую фазу промывают водой (300 мл) и концентрируют. Остаток очищают хроматографией (элюирование с градиентом: 20%-50% EA/гексан) с получением (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-1-(трет-бутилдиметилсилил)-N2-(1-метил-1H-имидазол-2-ил)-N2-метил-4-оксоазетидин-2-карбоксамид в виде полутвердого вещества (85 г, 50%). МС (ESI+) m/z 663,7 (M+H)+, время удерживания: 2,19 мин. (метод A).
Стадия 3. Получение (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-N2-(1-метил-1H-имидазол-2-ил)-N2-метил-4-оксоазетидин-2-карбоксамида

К раствору (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-1-(трет-бутилдиметилсилил)-N2-(1-метил-1H-имидазол-2-ил)-N2-метил-4-оксоазетидин-2-карбоксамида (118 г, 178 ммоль) в метаноле (300 мл) добавляют NH4F (12 г, 324 ммоль) и уксусную кислоту (21 г, 333 ммоль). Реакционную смесь перемешивают при КТ в течение 2 часов. Реакционную смесь концентрируют и остаток растворяют в EA (500 мл). Смесь промывают насыщенным водным раствором NaHCO3 (200 мл × 2) и насыщенным водным раствором соли (200 мл), сушат и концентрируют с получением (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-N2-(1-метил-1H-имидазол-2-ил)-N2-метил-4-оксоазетидин-2-карбоксамида в виде твердого белого вещества (85 г, 87%). МС (ESI+) 549,4 (M+H)+, время удерживания: 1,56 мин. (метод A).
Стадия 4. Получение (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-N2-(1-метил-1H-имидазол-2-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида

Раствор (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-N2-(1-метил-1H-имидазол-2-ил)-N2-метил-4-оксоазетидин-2-карбоксамида (119 г, 217 ммоль) в ДХМ (500 мл) обрабатывают TEA (110 г, 1,09 моль). К смеси добавляют (R)-(+)-1-фенилпропилизоцианат (87,4 г, 542 ммоль) и перемешивают реакционную смесь при 50°C в течение 16 часов. Реакционную смесь выливают в воду (400 мл) и фазы разделяют. Водную фазу экстрагируют ДХМ (3 × 150 мл). Объединенную органическую фазу сушат и концентрируют. Бесцветный вязкий маслянистый остаток очищают хроматографией (элюирование: 2:1 гексан/EA) с получением (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-N2-(1-метил-1H-имидазол-2-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида в виде твердого белого вещества (123 г, 80%). МС (ESI+) 710,6 (M+H)+, время удерживания: 2,11 мин. (метод A).
Стадия 5. Получение (2S,3R)-3-((2-амино-6-метилпиридин-4-ил)метил)-N2-(1-метил-1H-имидазол-2-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида

К раствору (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-N2-(1-метил-1H-имидазол-2-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида (140 г, 197 ммоль) в трифторуксусной кислоте (800 мл) медленно добавляют трииэтилсилан (137,6 г, 1,18 моль). Полученную смесь перемешивают при КТ в течение ночи. Растворители удаляют в вакууме. Остаток обрабатывают насыщенным водным раствором NaHCO3 (500 мл). Смесь экстрагируют ДХМ (300 мл × 2). Органическую фазу сушат и концентрируют. Остаток очищают хроматографией (элюирование: 5% метанол/ДХМ) с получением маслянистого вещества, которое растирают со смесью (8:1) гексан/EA с получением (2S,3R)-3-((2-амино-6-метилпиридин-4-ил)метил)-N2-(1-метил-1H-имидазол-2-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида в виде твердого белого вещества (82 г, 85%). МС (ESI+) 490,4 (M+H)+, время удерживания: 2,11 мин. (метод B). 1H ЯМР (400 Гц, CDCl3) δ 0,82 (т, 3H), 1,80 (м, 2H), 2,32 (с, 3H), 2,76 (м, 1H), 2,96 (м, 1H), 3,20 (с, 3H), 3,60 (с, 3H), 3,72 (м, 2H), 4,68 (м, 1H), 5,30 (м, 2H), 6,20 (с, 1H), 6,24 (с, 1H), 6,68 (д, 1H), 6,94 (с, 1H), 7,04 (с, 1H), 7,28 (м, 3H), 7,34 (м, 2H).
Пример 9. Синтез (2S,3R)-3-((1-оксид-2-амино-6-метилпиридин-4-ил)метил)-N2-(1-метил-1H-имидазол-2-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида
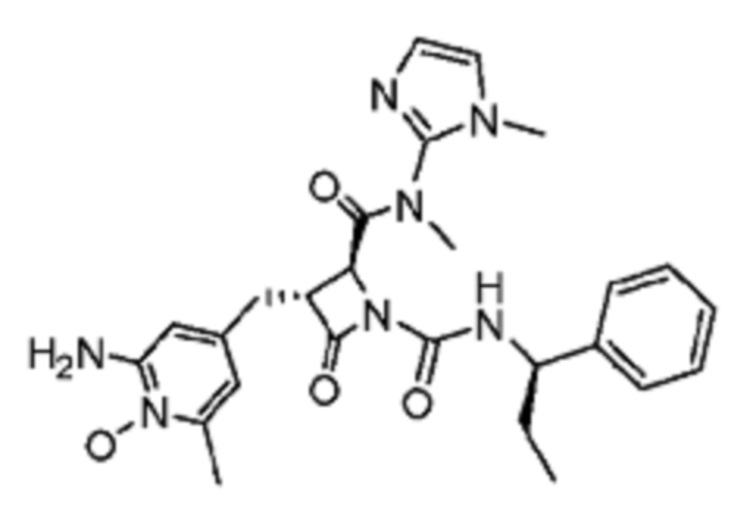
К раствору (2S,3R)-3-((2-амино-6-метилпиридин-4-ил)метил)-N2-(1-метил-1H-имидазол-2-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида (25 мг, 0,05 ммоль, продукт примера 1) в ДХМ (5 мл) добавляют мета-хлорпероксибензойную кислоту (18 мг, 0,08 ммоль). Смесь перемешивают в течение 1 часа при КТ. Смесь разбавляют ДХМ (10 мл). Смесь промывают насыщенным водным раствором NaHCO3 и водой, сушат и концентрируют. Остаток очищают препаративной ТСХ (10% метанол/ДХМ) с получением (2S,3R)-3-((1-оксид-2-амино-6-метилпиридин-4-ил)метил)-N2-(1-метил-1H-имидазол-2-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида в виде твердого белого вещества (15 мг, 58%). МС (ESI+) m/z 510,2 (M+H)+, время удерживания: 1,25 мин. (метод A).
Пример 10. Синтез (2S,3R)-3-((2-амино-6-метилпиридин-4-ил)метил)-N2-(5-оксо-1-метил-4,5-дигидро-1H-имидазол-2-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида

Схема 26
Стадия 1. Получение (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-4-оксоазетидин-2-карбоновой кислоты

К раствору (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-1-(трет-бутилдиметилсилил)-4-оксоазетидин-2-карбоновой кислоты (10 г, 17,6 ммоль) в метаноле (50 мл) добавляют NH4F (1,3 г, 35 ммоль) и уксусную кислоту (2,1 г , 33,3 ммоль). Реакционную смесь перемешивают при КТ в течение 2 часов, концентрируют и остаток растворяют в ДХМ (100 мл). Смесь промывают водой (20 мл) и насыщенным водным раствором соли (20 мл), сушат и концентрируют с получением (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-4-оксоазетидин-2-карбоновой кислоты в виде твердого белого вещества (7 г, 88%). МС (ESI+) m/z 456,2 (M+H)+, время удерживания: 1.55 мин. (метод A).
Стадия 2. Получение (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-1-(((R)-1-фенилэтил)карбамоил)-4-оксоазетидин-2-карбоновой кислоты
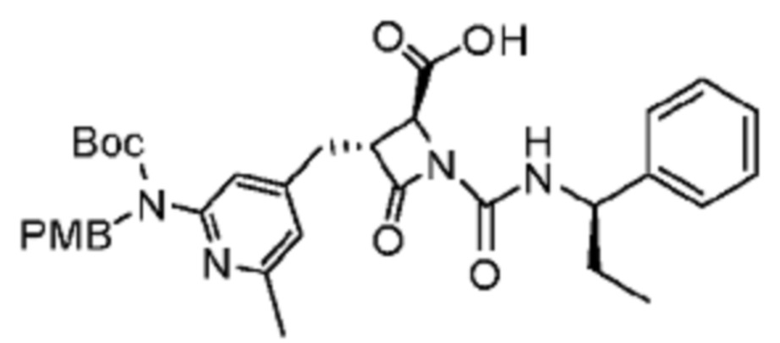
(2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-4-оксоазетидин-2-карбоновую кислоту (7,7 г, 16,9 ммоль) растворяют в ДХМ (100 мл) и обрабатывают DBU (9 г, 59,1 ммоль), а затем (R)-(+)-1-фенилпропилизоцианатом (6,9 г, 42,6 ммоль) при КТ. Реакционную смесь перемешивают при КТ в течение ночи. Смесь разбавляют ДХМ (50 мл) и смесь промывают несколькими порциями 10% водной лимонной кислоты до тех пор, пока ВЭЖХ анализ (метод А) не покажет отсутствие DBU. Органическую фазу сушат и концентрируют. Остаток очищают хроматографией (элюирование с градиентом: от 20% EA/гексан до 100% EA) с получением (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-1-(((R)-1-фенилэтил)карбамоил)-4-оксоазетидин-2-карбоновой кислоты в виде твердого белого вещества (9,3 г, 90%), МС (ESI+) m/z 617,3 (M+H)+, время удерживания: 3,66 мин. (метод B).
Стадия 3. Получение (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-N2-(5-оксо-1-метил-4,5-дигидро-1H-имидазол-2-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида

К раствору (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-1-(((R)-1-фенилэтил)карбамоил)-4-оксоазетидин-2-карбоновой кислоты (100 мг, 0,16 ммоль) в ДМФА (2 мл) добавляют HATU (123 мг, 0,32 ммоль). Смесь перемешивают в течение 10 мин. при КТ. Добавляют гидрохлорид 1-метил-2-метиламино-4,5-дигидроимидазол-5-он (83 мг, 0,5 ммоль, получен как описано в J. Org. Chem., 1968, 33, 552) и полученный раствор перемешивают в течение 10 мин. Добавляют диизопропилэтиламин (120 мг, 0,9 ммоль). Смесь перемешивают при КТ в течение 2 часов и затем добавляют насыщенный водный раствор соли (6 мл). Смесь экстрагируют EA (5 мл × 3). Объединенную органическую фазу промывают водой (3 мл) и концентрируют. Остаток очищают хроматографией (элюирование с градиентом: 20%-50% EA/гексан) с получением (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-N2-(5-оксо-1-метил-4,5-дигидро-1H-имидазол-2-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида в виде полутвердого вещества (58 мг, 50%). МС (ESI+) m/z 726,7 (M+H)+, время удерживания: 2,05 мин. (метод A).
Стадия 4. Получение (2S,3R)-3-((2-амино-6-метилпиридин-4-ил)метил)-N2-(5-оксо-1-метил-4,5-дигидро-1H-имидазол-2-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида

Раствор (2S,3R)-3-((2-((трет-бутоксикарбонил)(4-метоксибензил)амино)-6-метилпиридин-4-ил)метил)-N2-(5-оксо-1-метил-4,5-дигидро-1H-имидазол-2-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида (58 мг, 0,08 ммоль) в ДХМ (3 мл) обрабатывают трииэтилсиланом (0,1 мл) и охлаждают до 0°C. К смеси по каплям добавляют трифторуксусную кислоту (1 мл) и реакционную смесь медленно нагревают до КТ. Спустя 21 час ВЭЖХ анализ (метод А) показывает, что исходное вещество израсходовано. Реакционную смесь концентрируют и остаток переносят в 5 мл ДХМ. Смесь промывают насыщенным водным раствором NaHCO3 и водой, сушат и концентрируют. Остаток очищают препаративной ТСХ (элюирование: EA) с получением (2S,3R)-3-((2-амино-6-метилпиридин-4-ил)метил)-N2-(5-оксо-1-метил-4,5-дигидро-1H-имидазол-2-ил)-N1-((R)-1-фенилпропил)-N2-метил-4-оксоазетидин-1,2-дикарбоксамида в виде твердого белого вещества (20 мг, 50%). МС (ESI+) m/z 506,5 (M+H)+, время удерживания: 1,19 мин. (метод A).
Пример 11. Дополнительные примеры соединений
Соединения, представленные в таблицах 5, 6 и 7 ниже, синтезируют в соответствии со способами, показанными на схемах 1-5 и 21-26, и методиками примеров 3-10.
Таблица 5. Дополнительные примеры полученных соединений
(M+H)+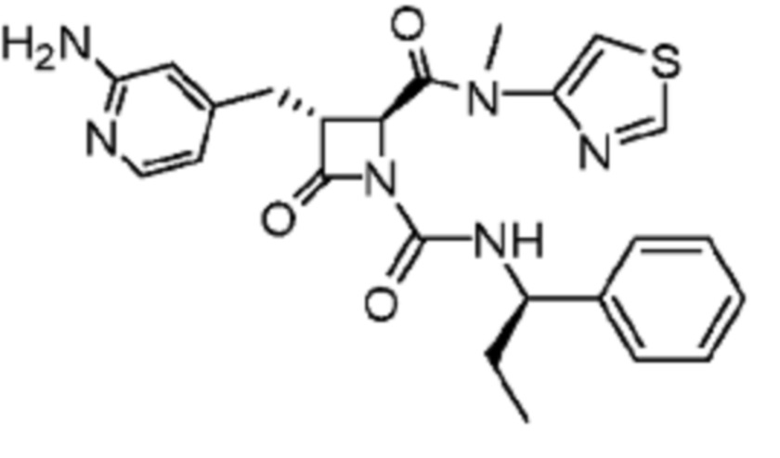
1-8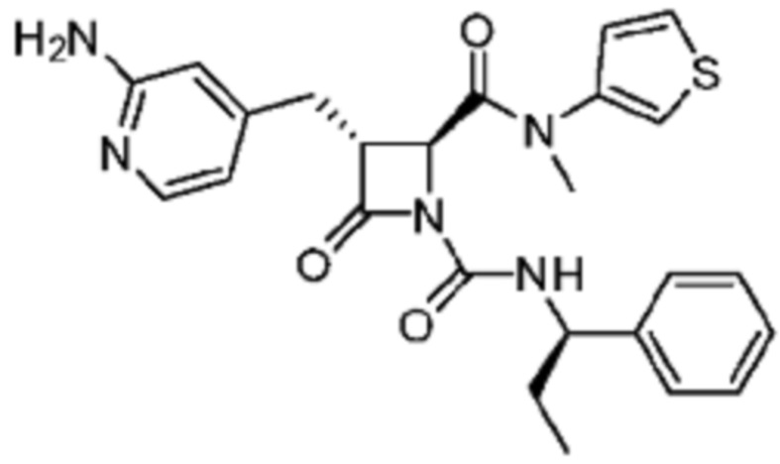
1-9
1-10
1-11
1-12
1-13
1-14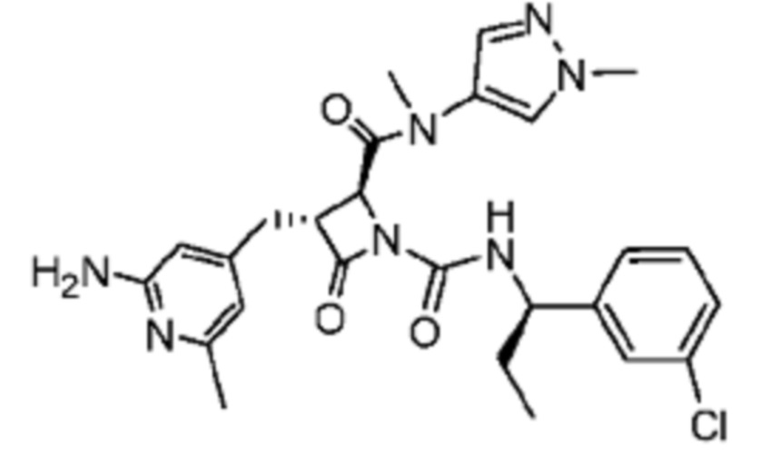
1-15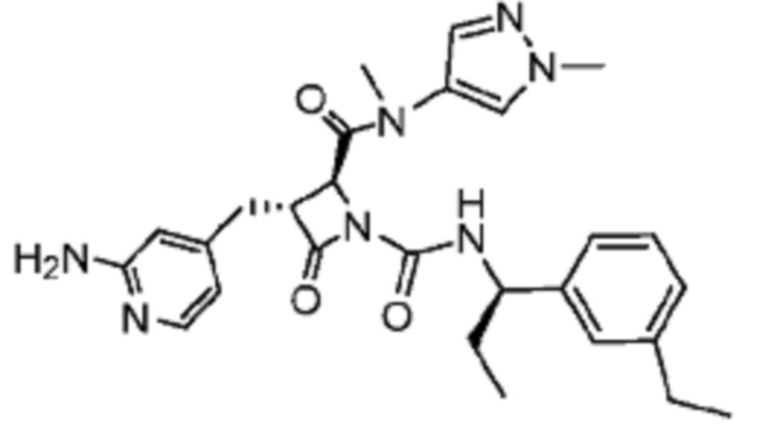
1-16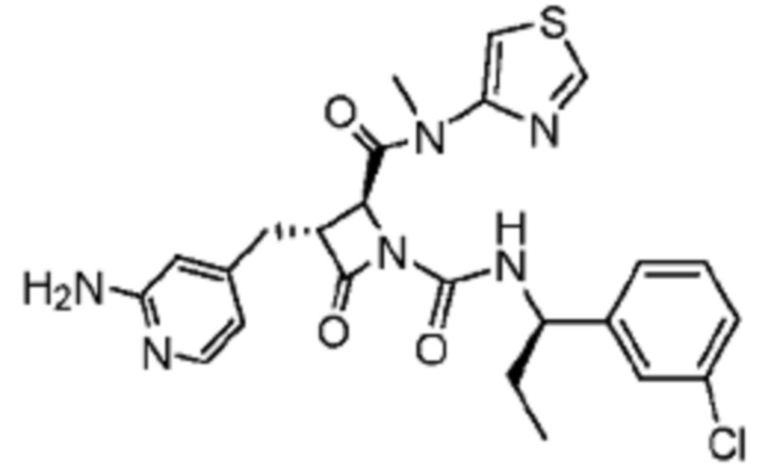 1-17
1-17
1-18
1-19
1-20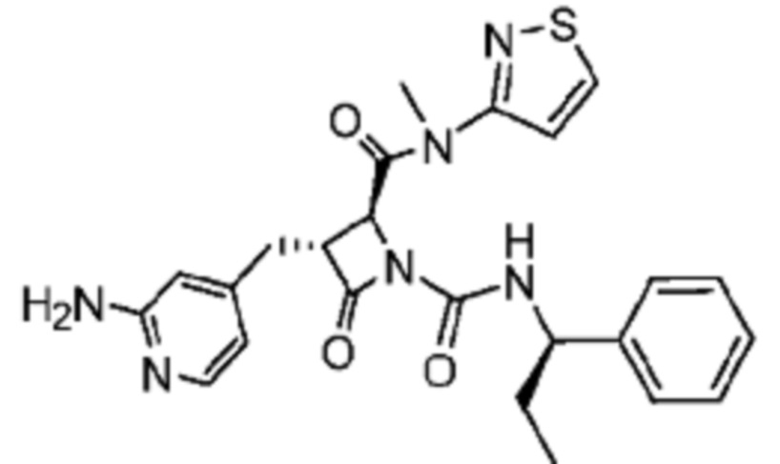
1-21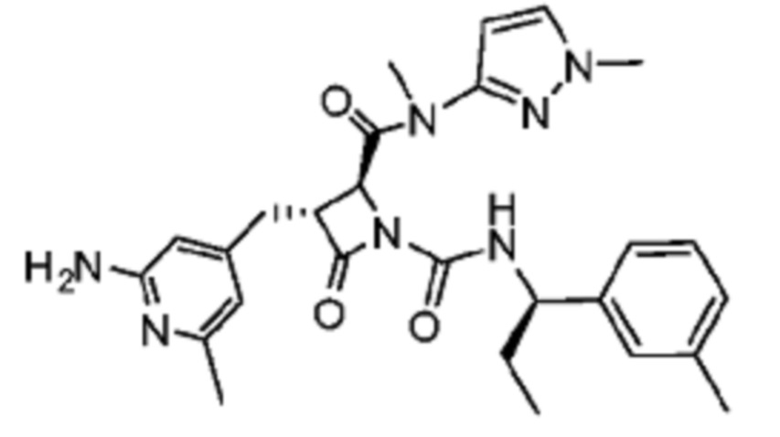
1-22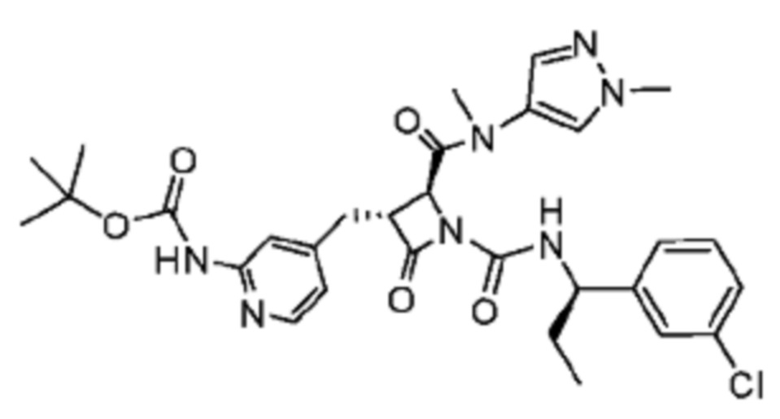
1-23
1-24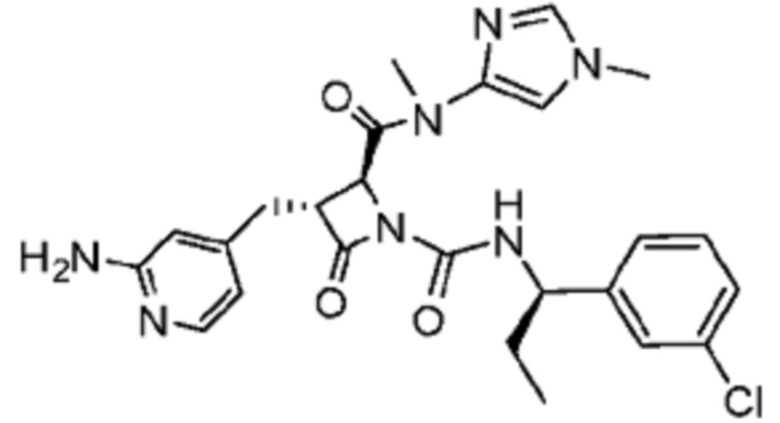
1-25
1-26
1-27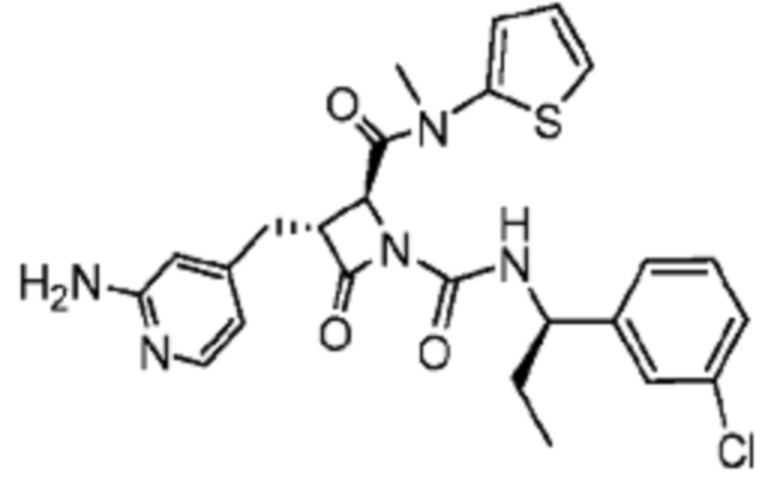
1-28
1-29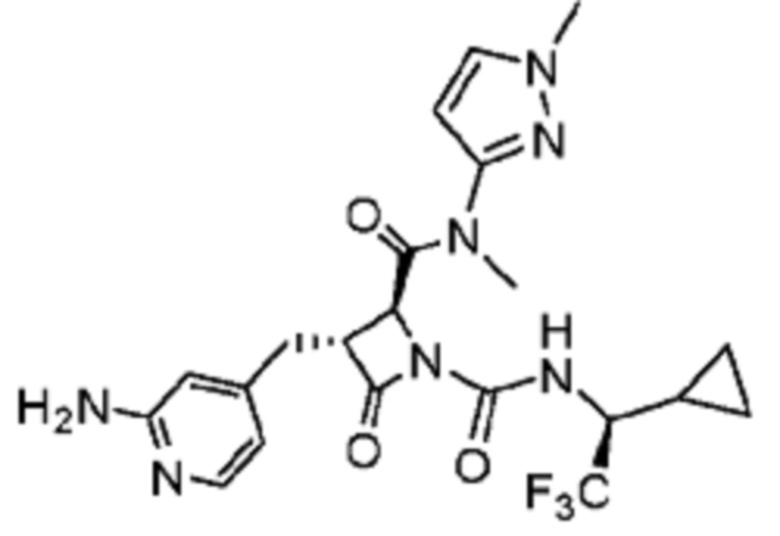
1-30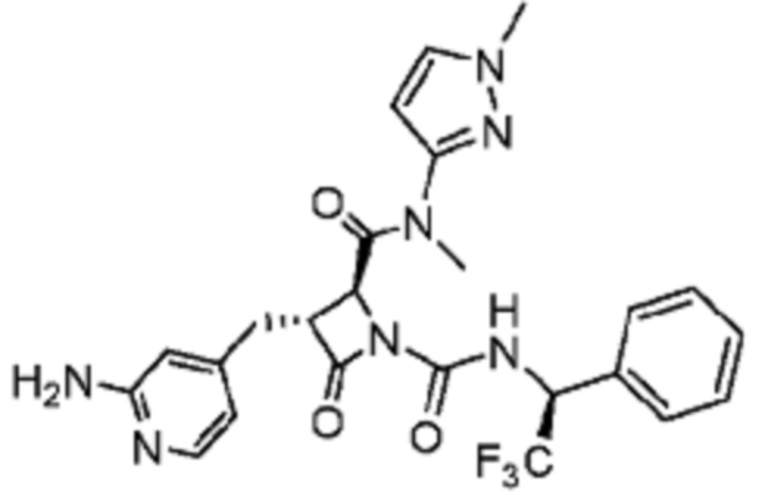
1-31
1-32
1-33
1-34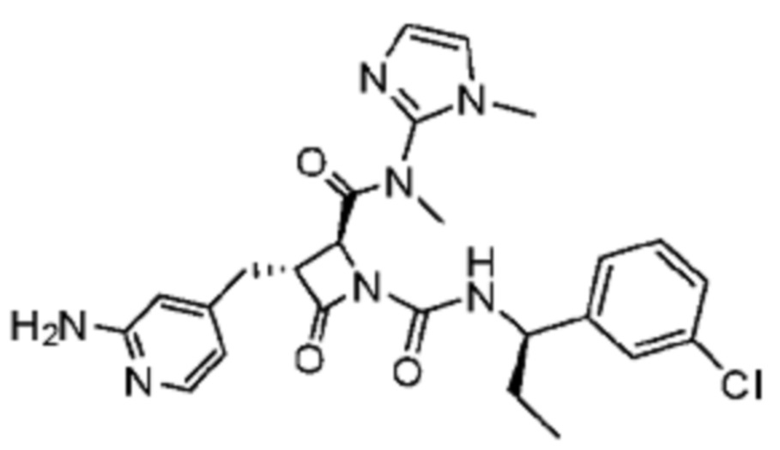
1-35
1-36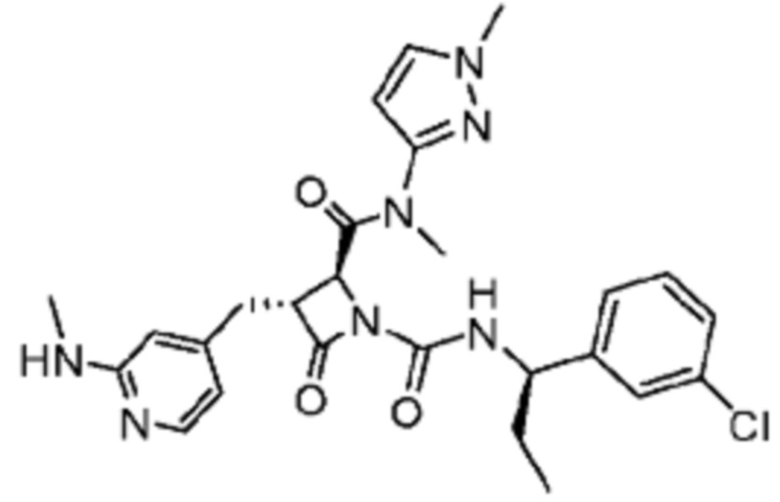
1-37
1-38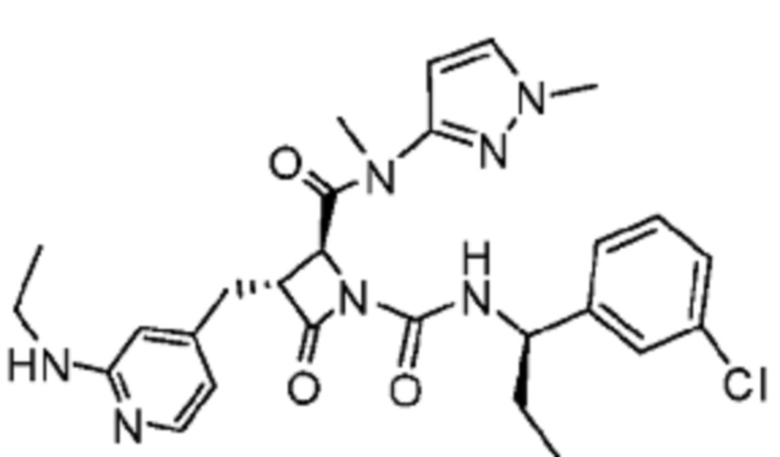
1-39
1-40
1-41
1-42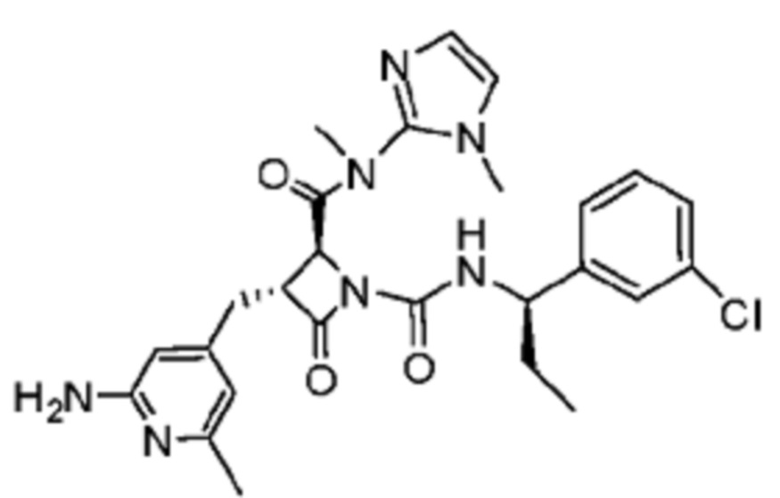
1-43
1-44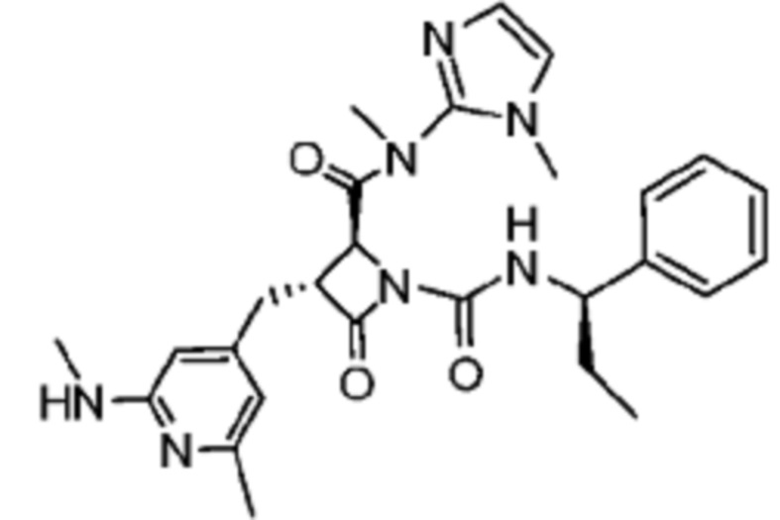
1-45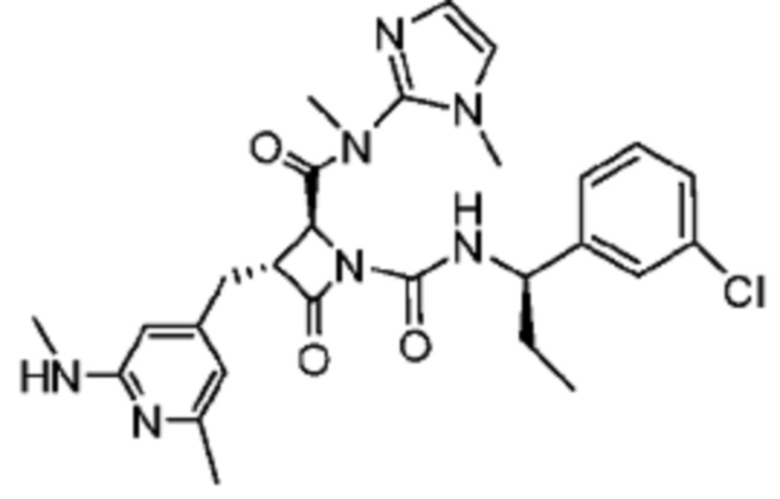
1-46
1-47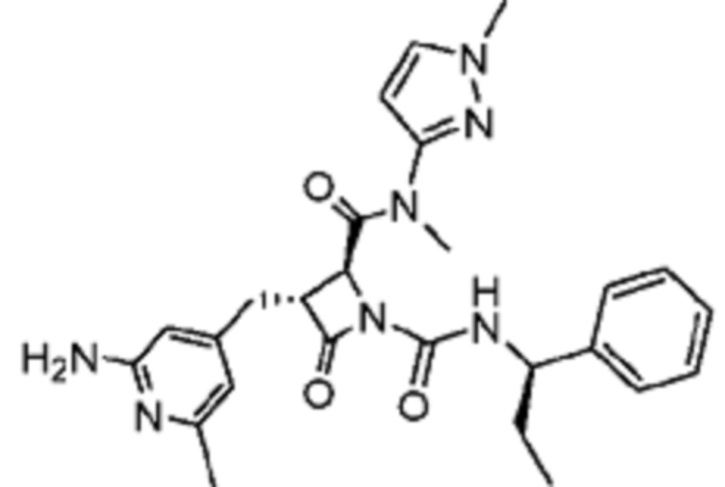
1-48
1-49
1-50
1-51
1-52
1-53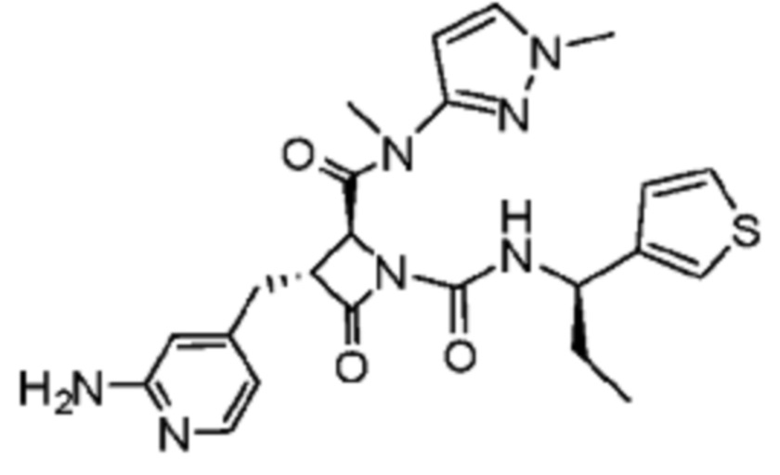
1-54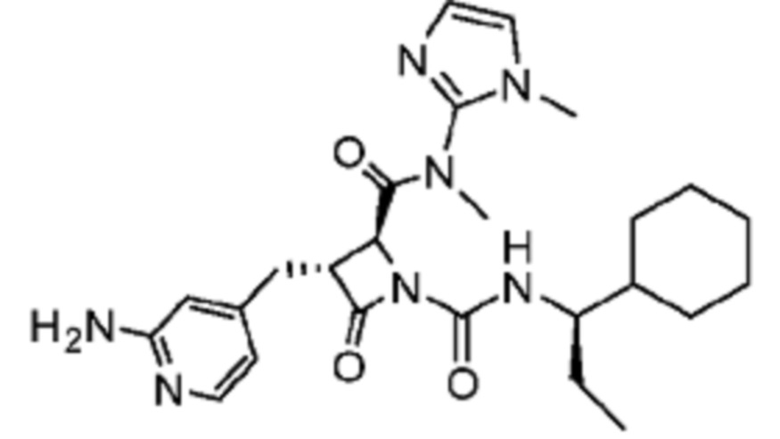
1-55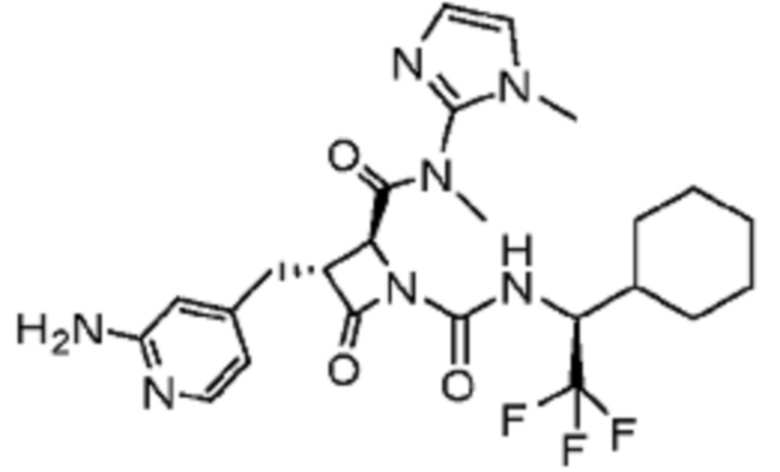
1-56
1-57
1-58
1-60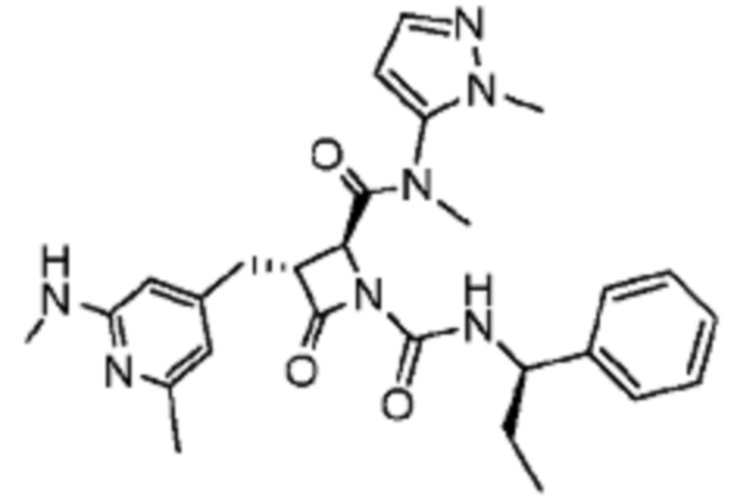
1-61
1-62
1-63
1-64
1-65
1-66
1-67
1-68
1-69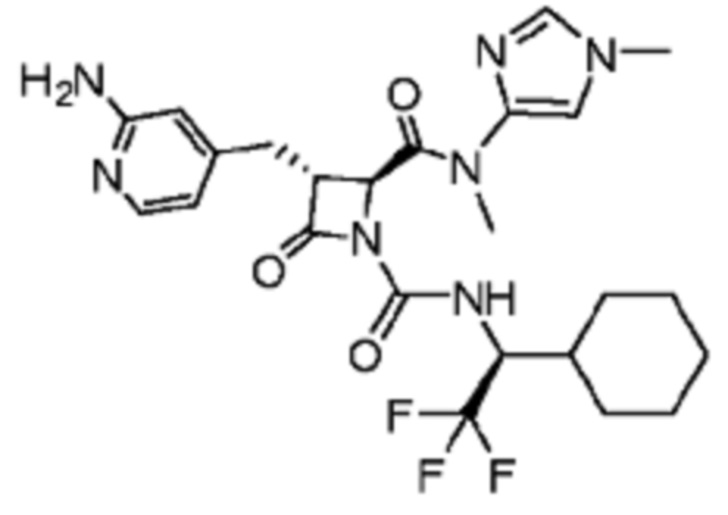
1-70
1-71
1-72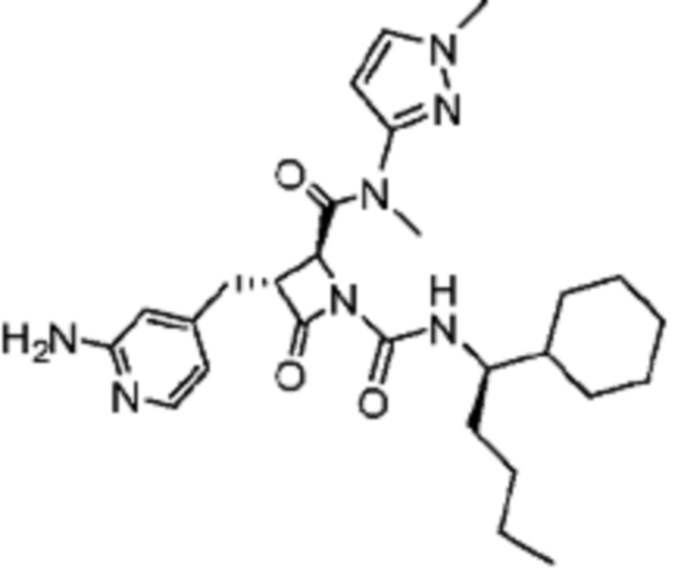
1-73
1-74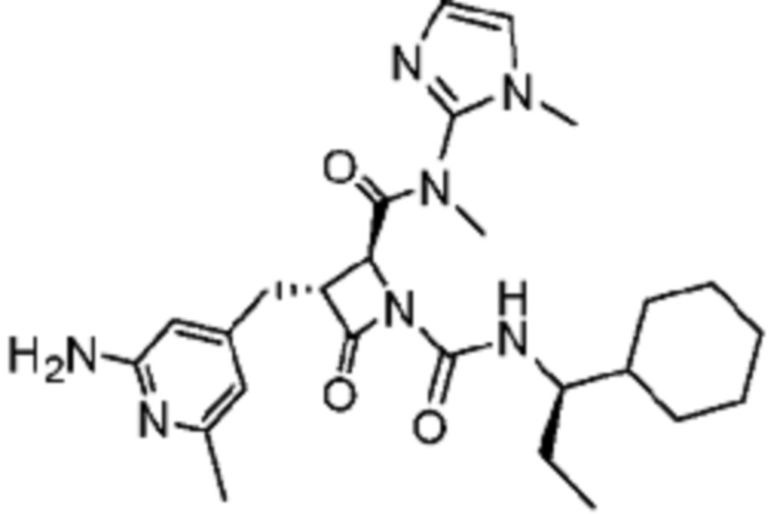
1-75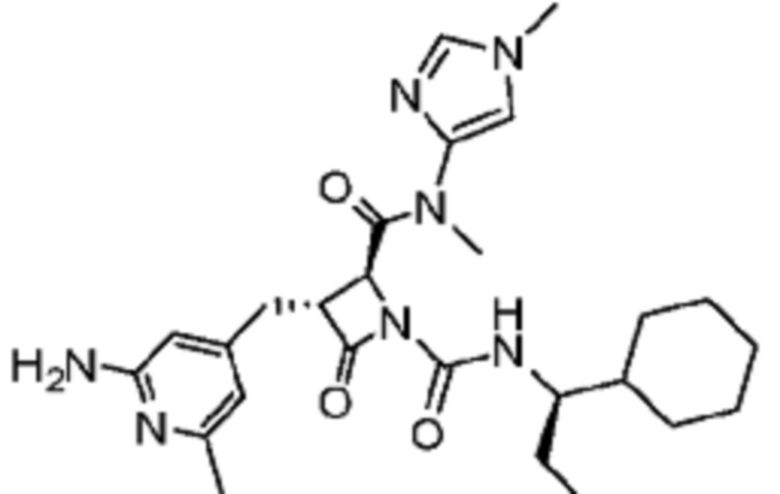
1-76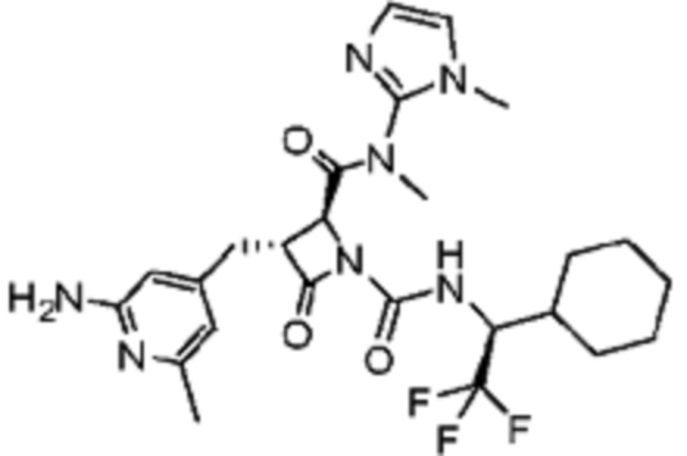
1-77
1-78
1-79
1-80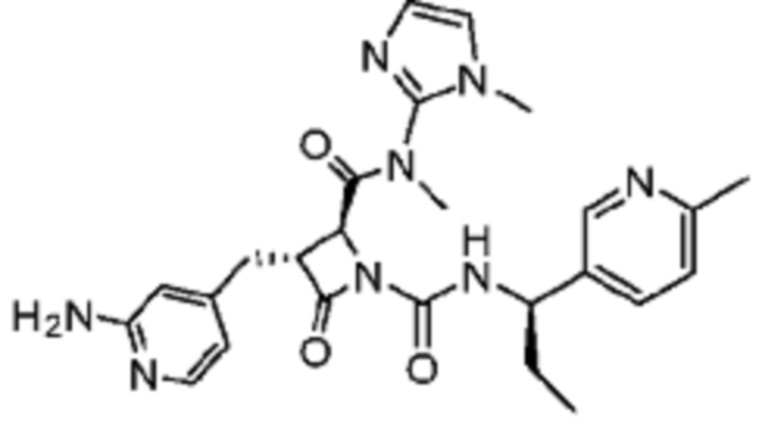
1-81
1-82
1-83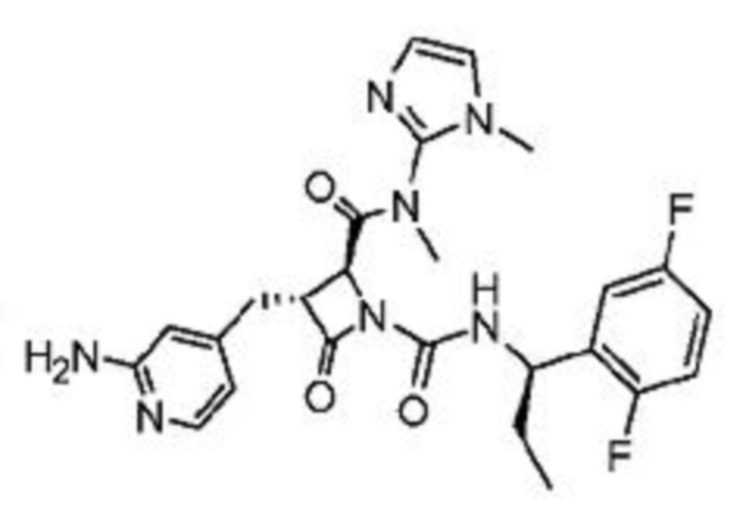
1-84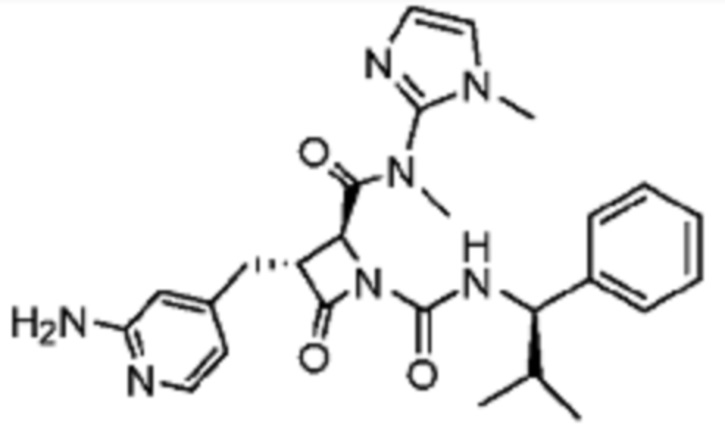
1-85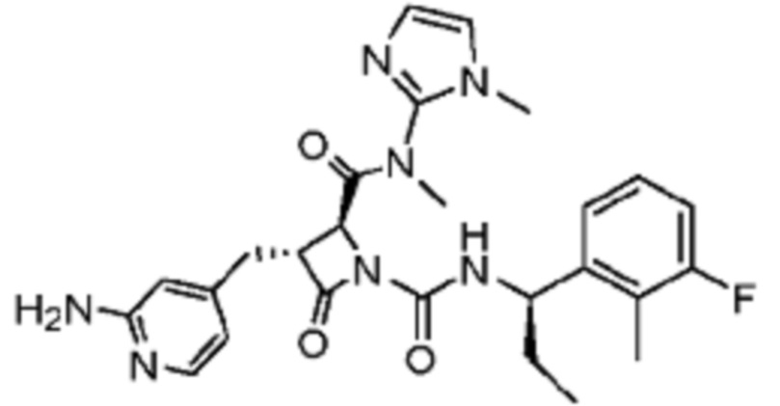
1-86
1-87
Таблица 6. Дополнительные примеры полученных соединений
ЖХ-МС(M+H)+
ЖХ-МС время удерж.(мин.)
1-88
(M+H)+: 476,3
Врем. удерж.: 1,23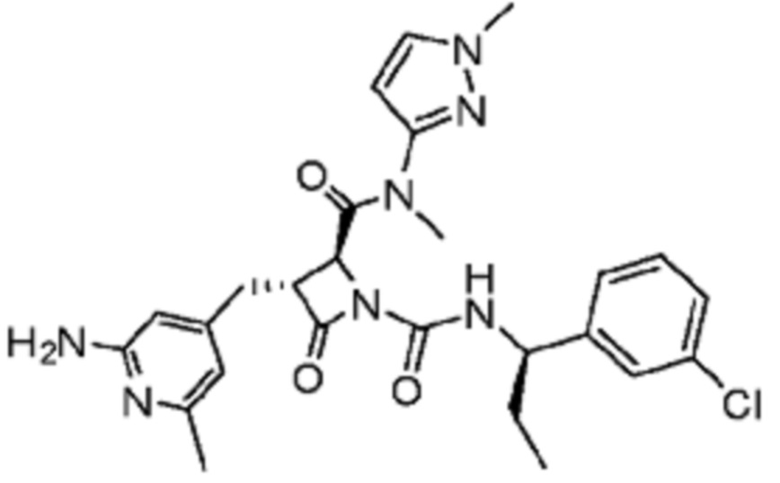
1-89
(M+H)+: 524,2
Врем. удерж.: 1,26 мин, 
1-90
(M+H)+: 476,3
Врем. удерж.: 1,19 мин,
1-91
(M+H)+: 510,2
Врем. удерж.: 1,26 мин,
1-92
(M+H)+: 477,2
Врем. удерж.: 1,22 мин.  1-93
1-93
Врем. удерж.: 1,23 мин,
Таблица 7. Дополнительные примеры полученных соединений
(метод A)
(M+H)+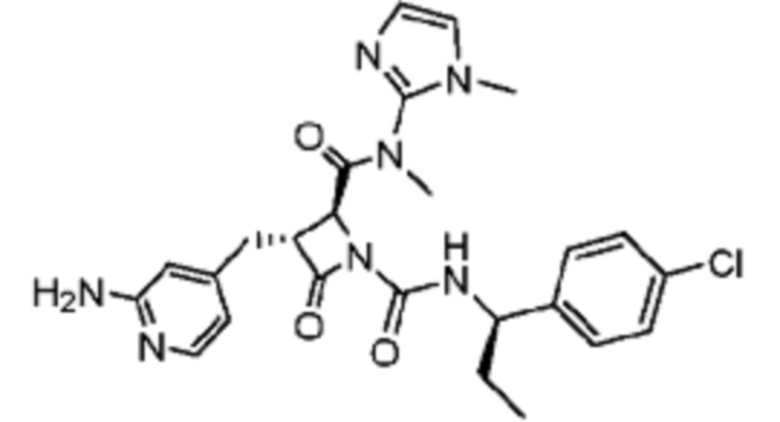
1-94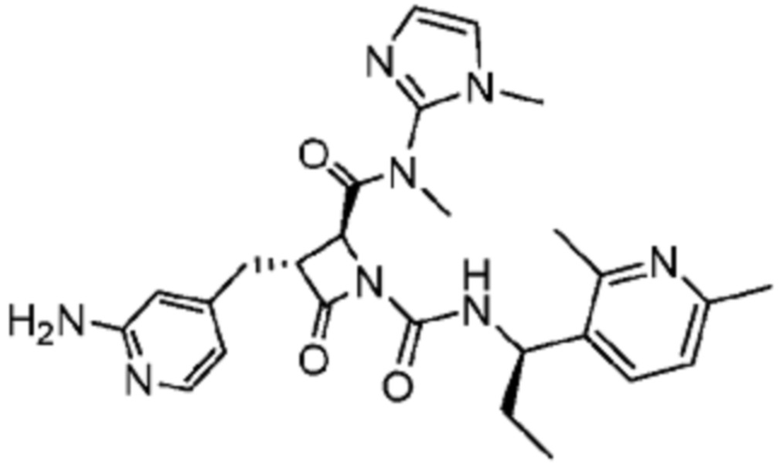
1-95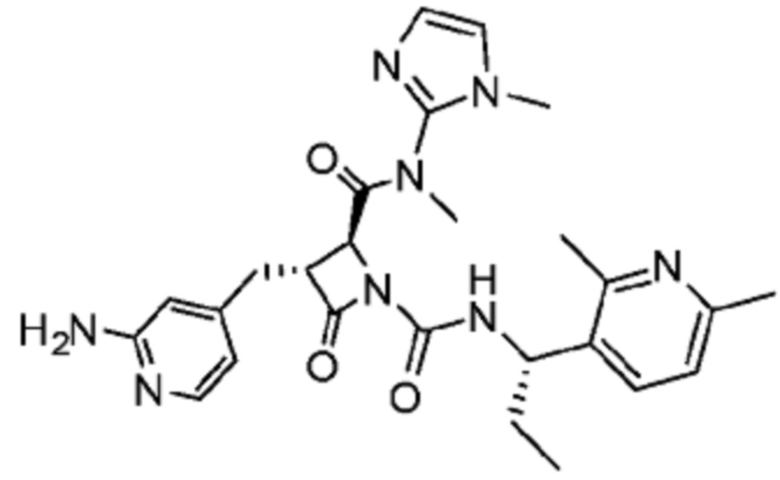
1-96
1-97
1-98
1-99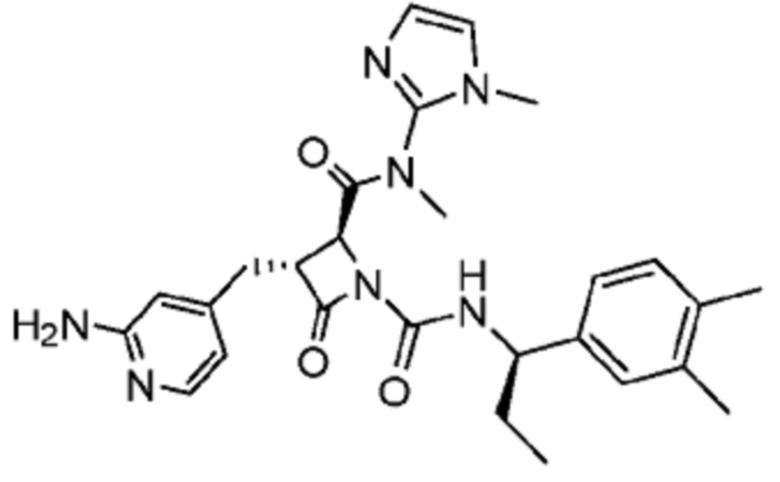
1-100 1-101
1-101
1-102
1-103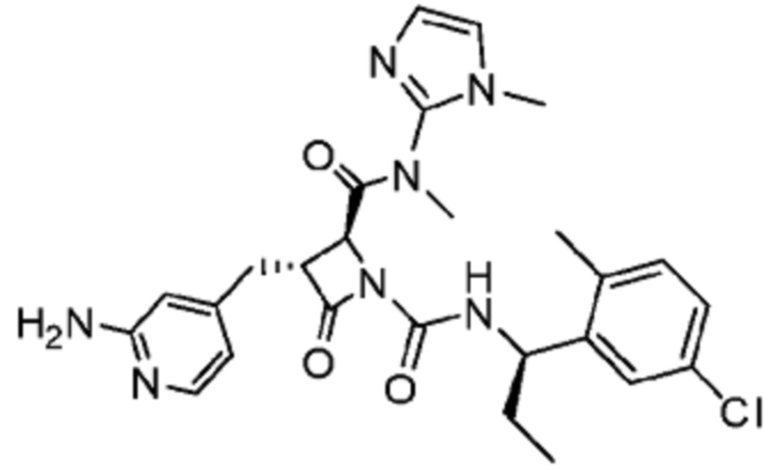
1-104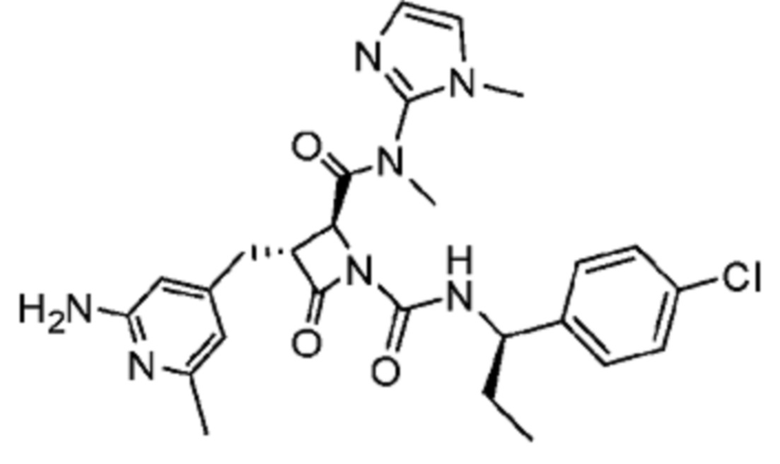
1-105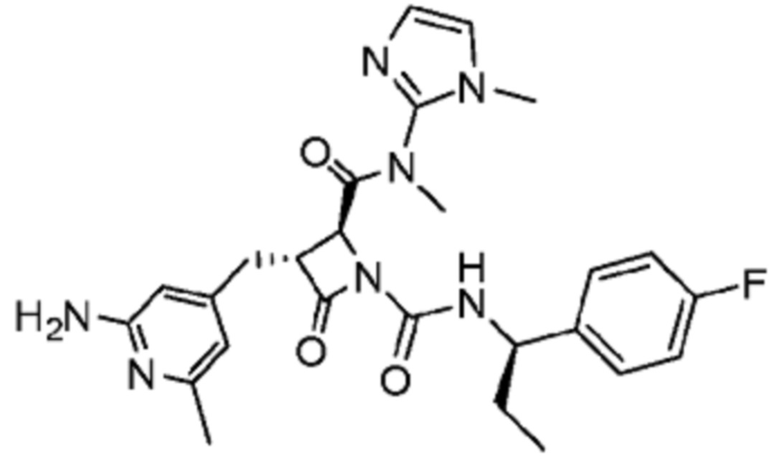
1-106
1-107
1-108
1-109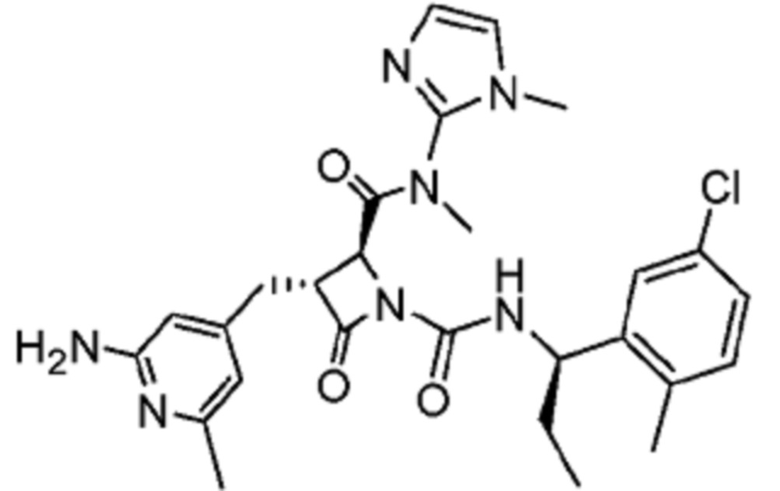
1-110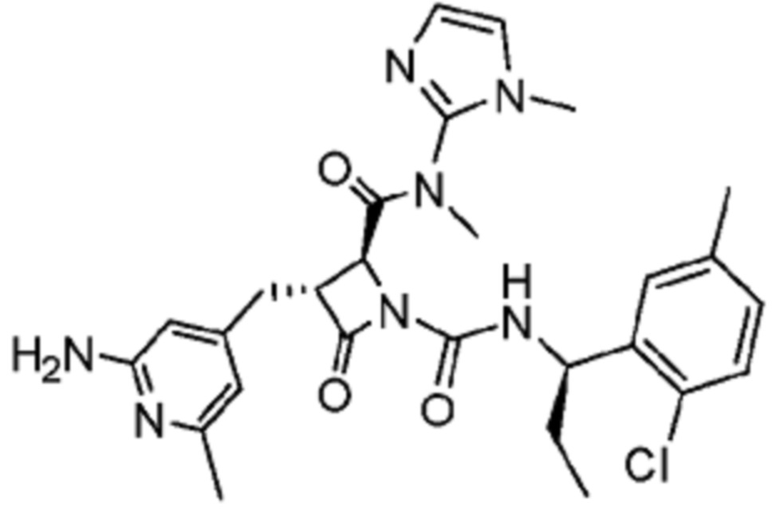
1-111
1-112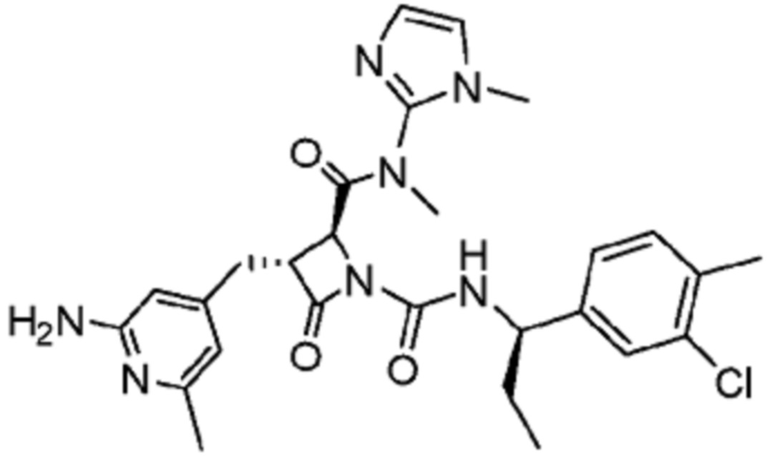
1-113
1-114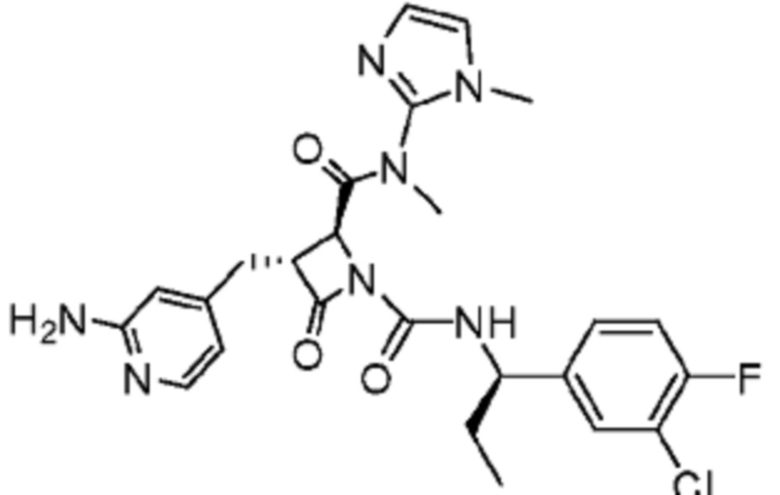
1-115
1-116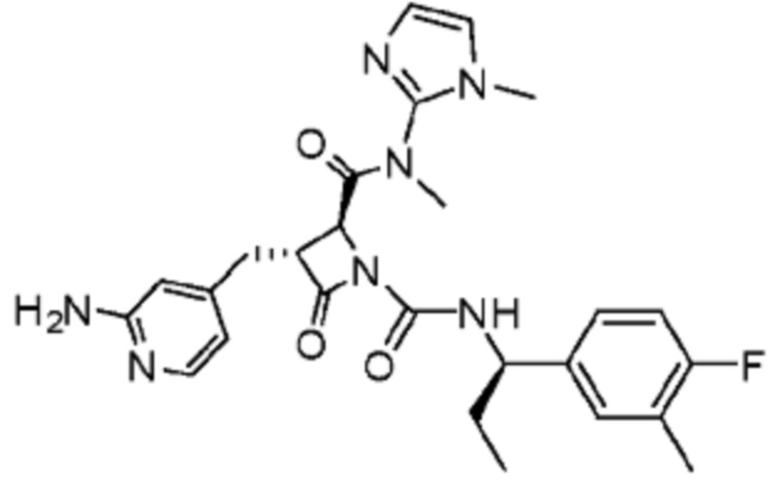
1-117
1-118
1-119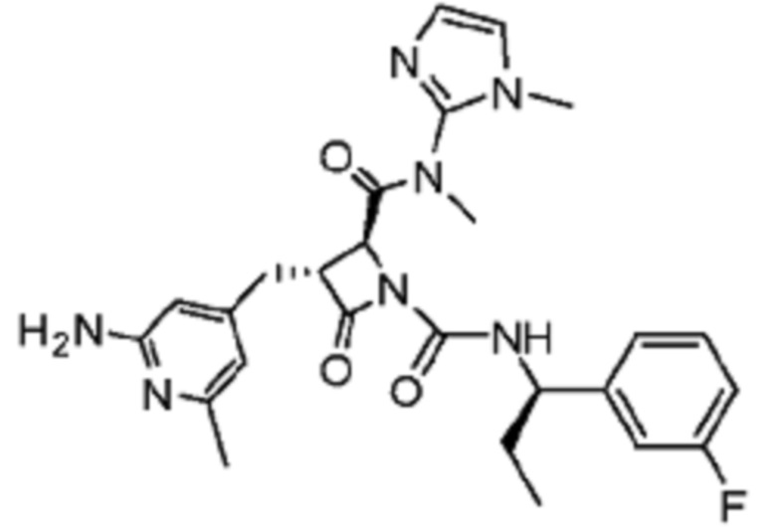
1-120
1-121
1-122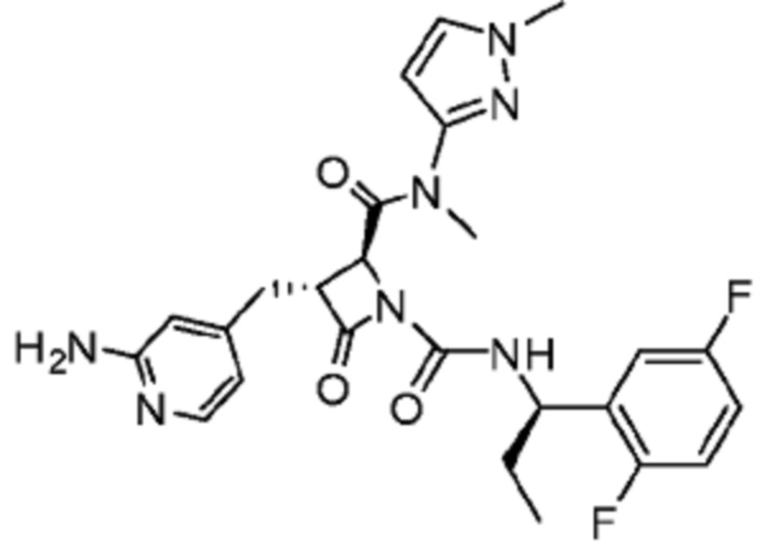
1-123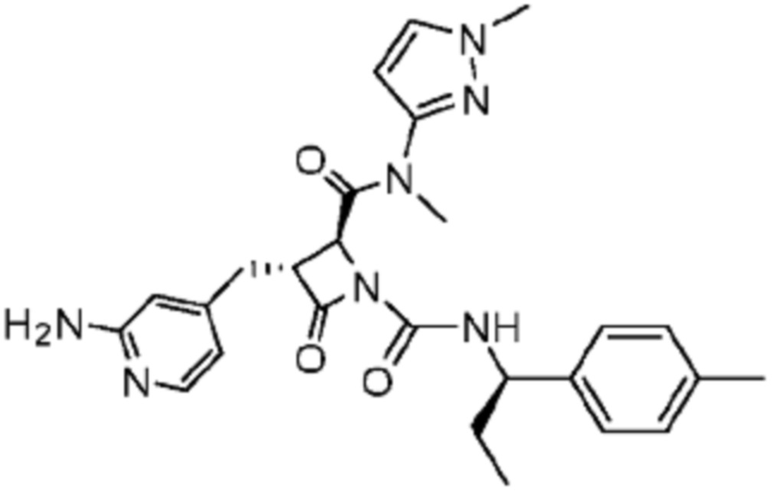
1-124
1-125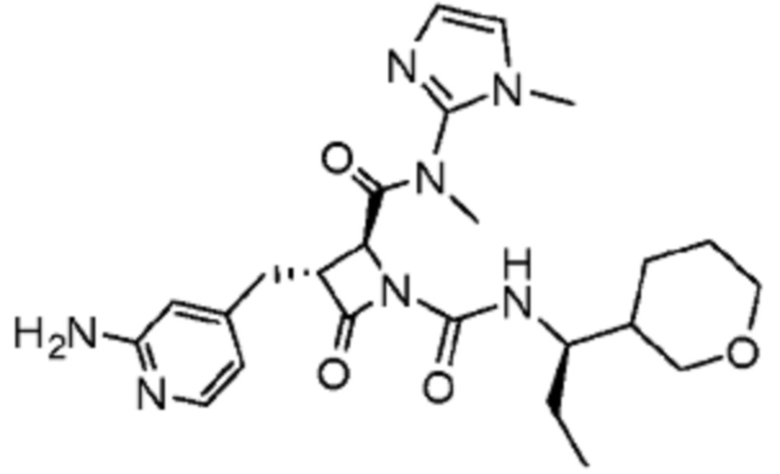
1-126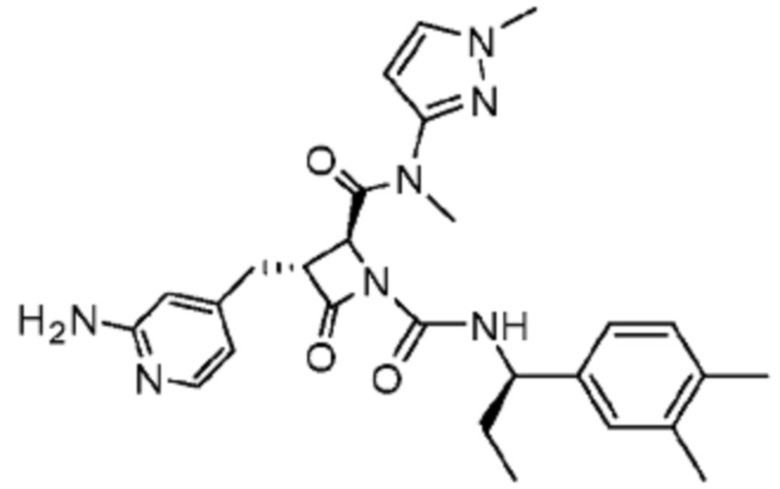
1-127
1-128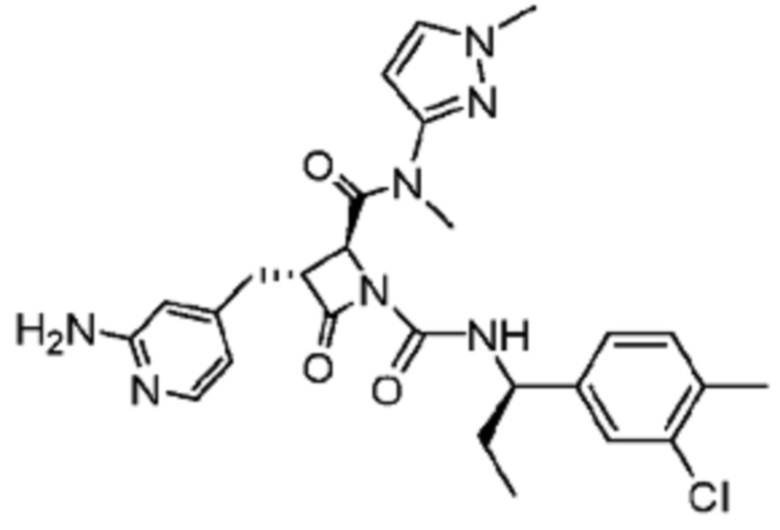
1-129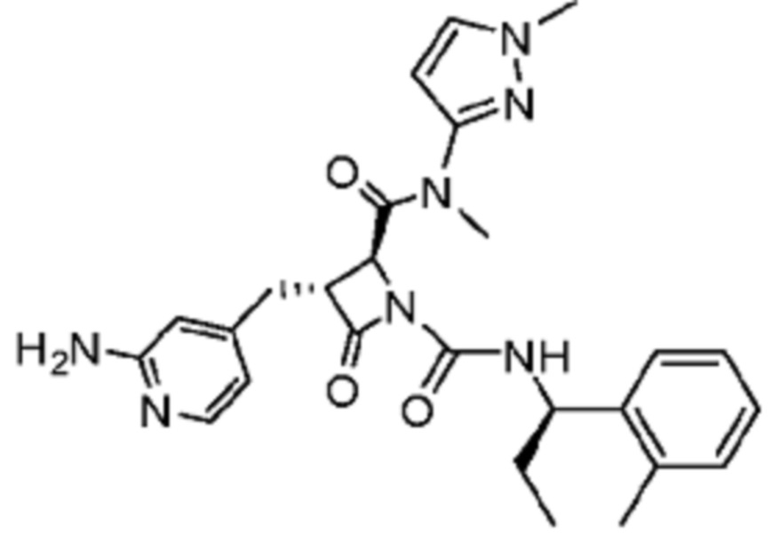
1-130
1-131
1-132
1-133
1-134
1-135
1-136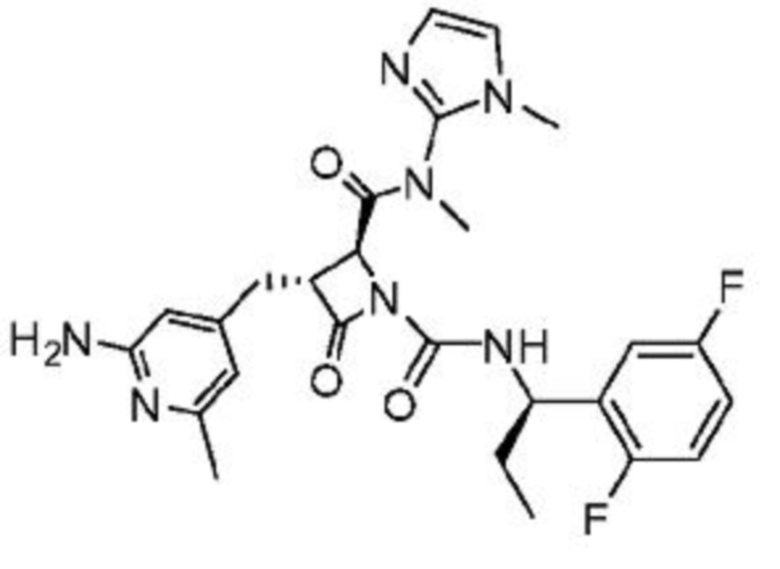
1-137.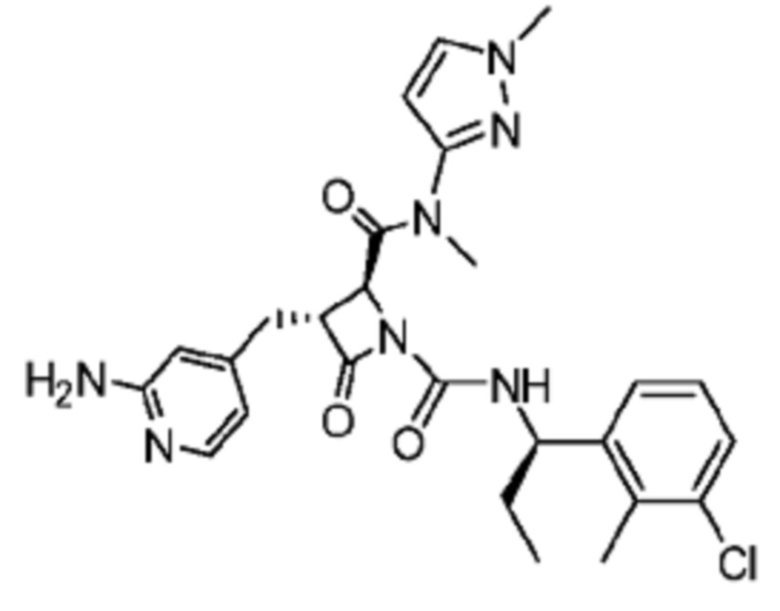
1-138
1-139
1-140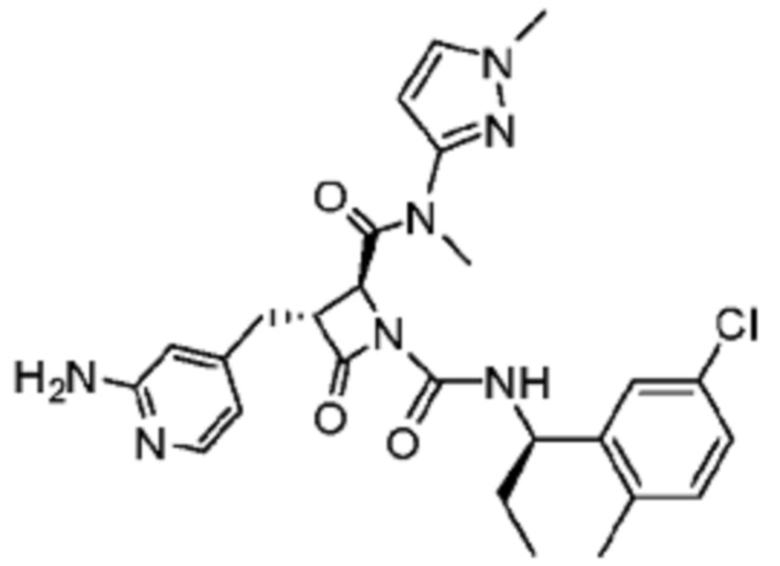
1-141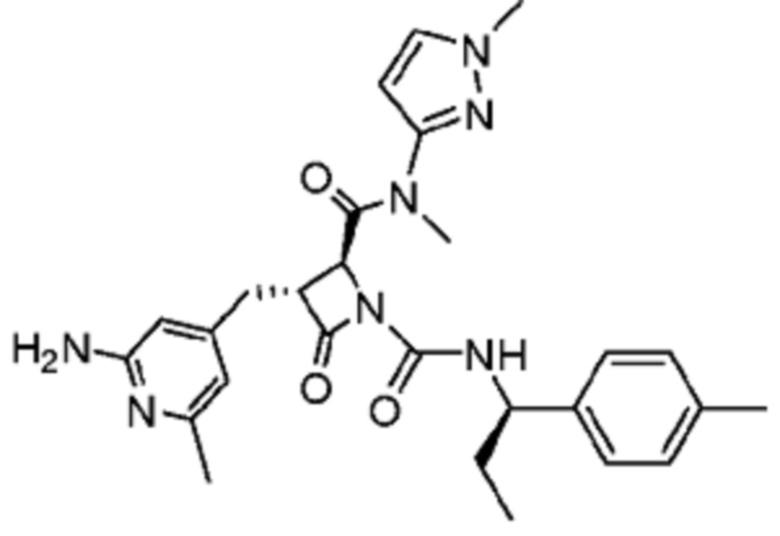
1-142
1-143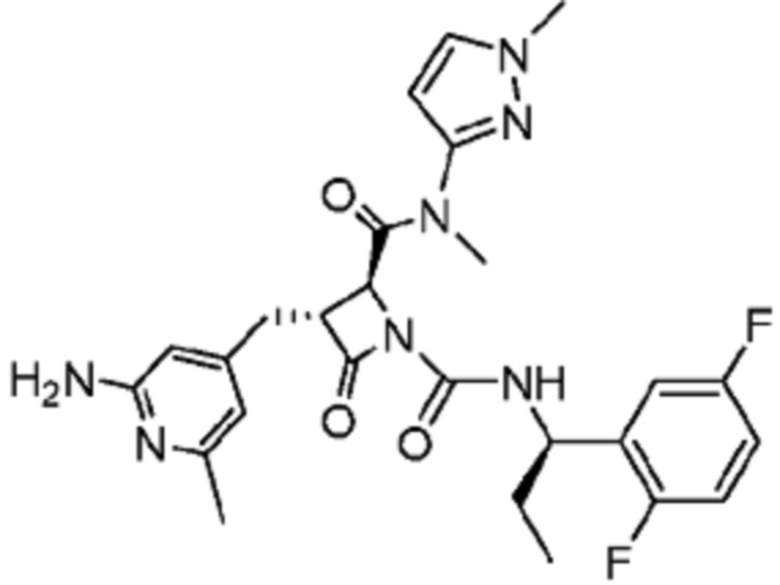
1-144
1-145
1-146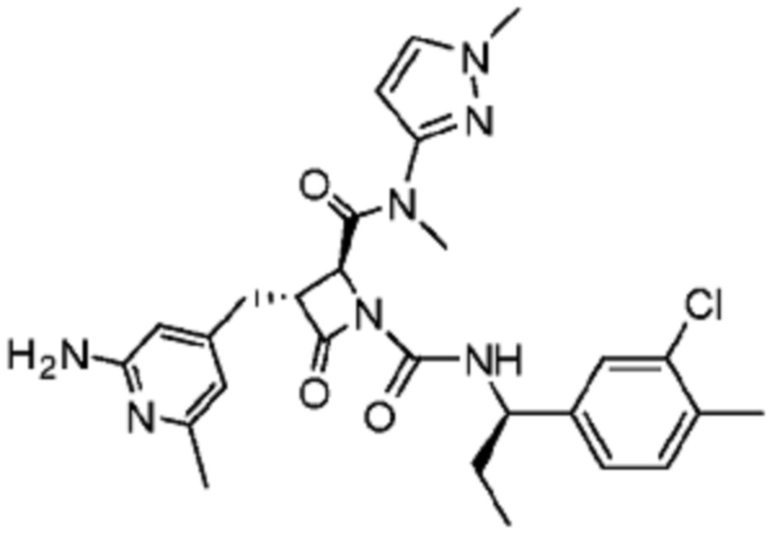
1-147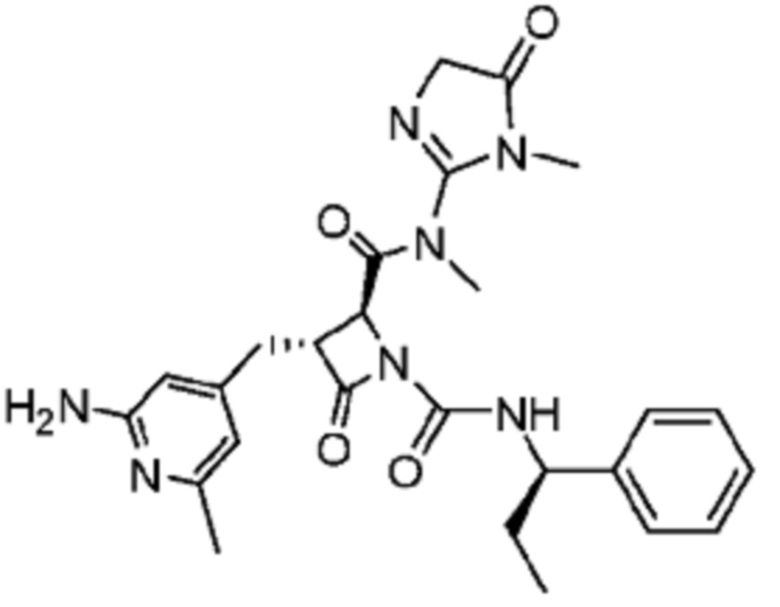
1-148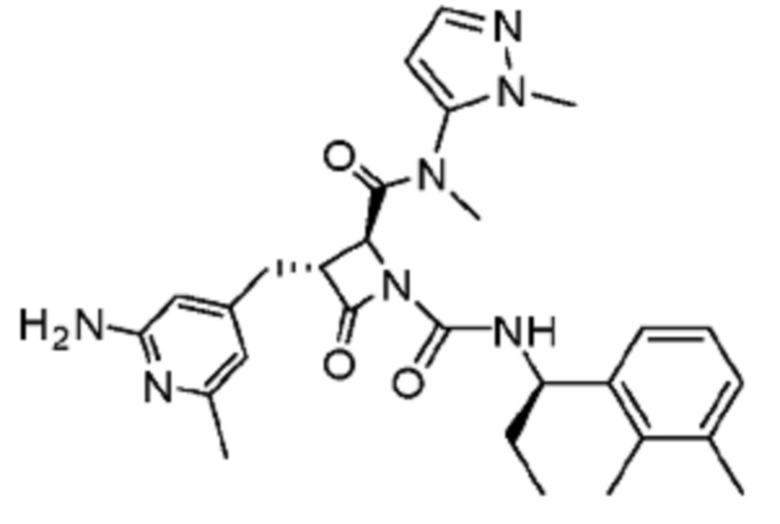
1-149
1-150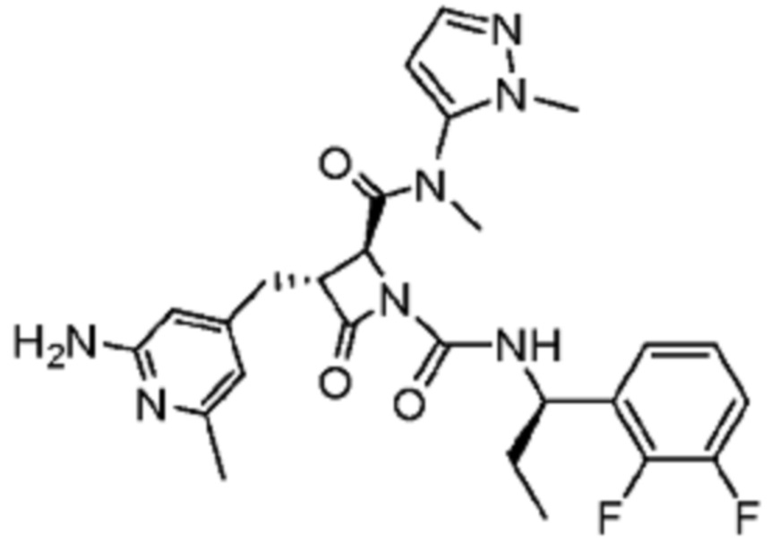
1-151
1-152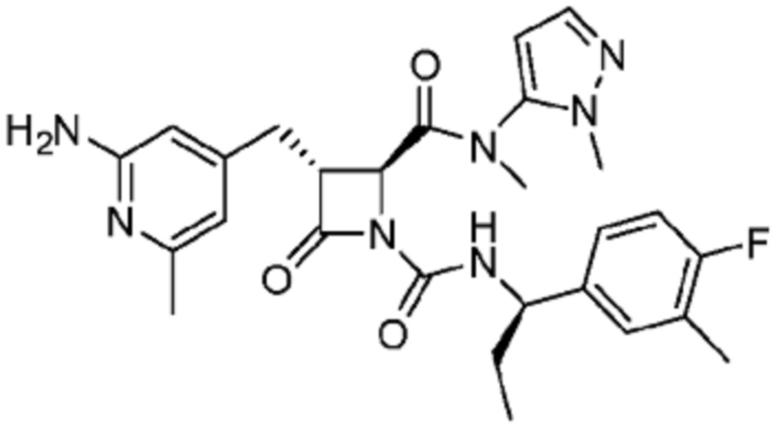
1-153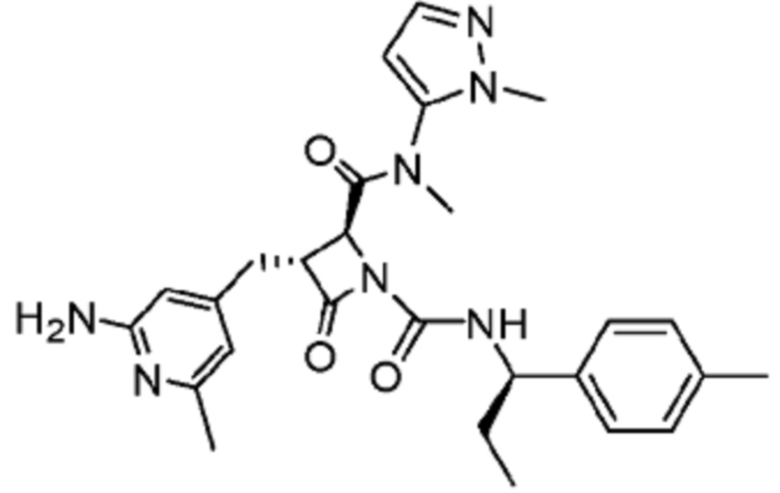
1-154
1-155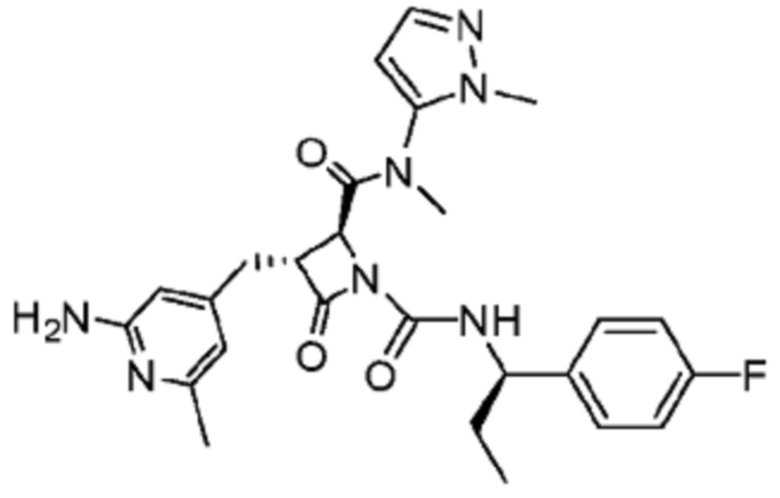
1-156
1-157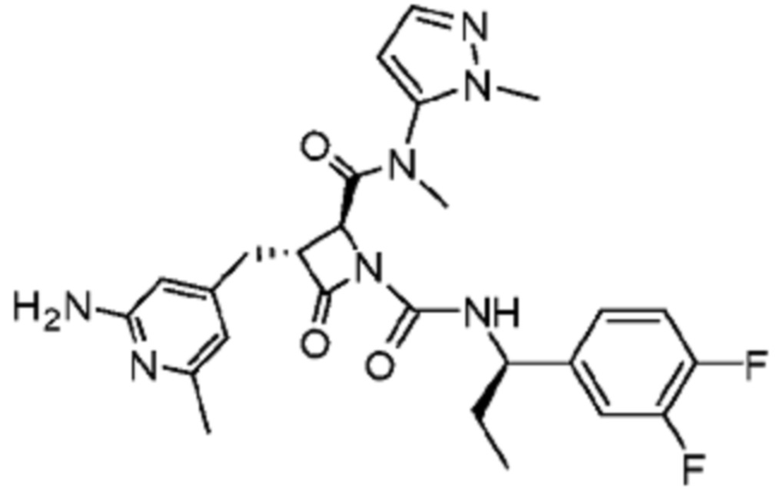
1-158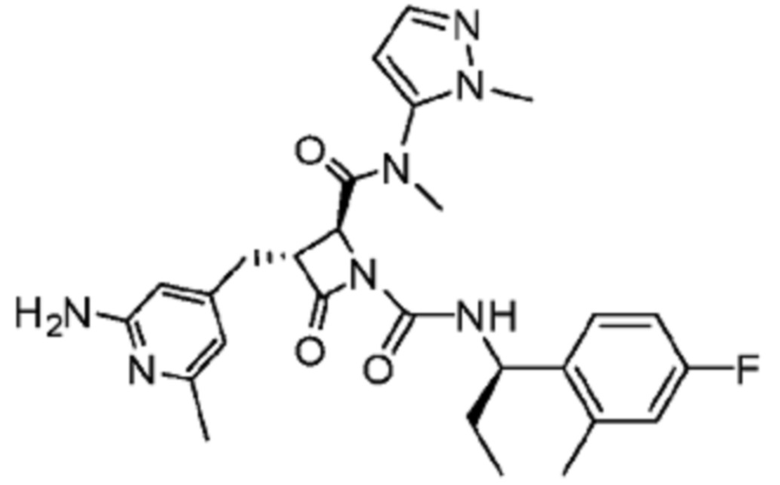
1-159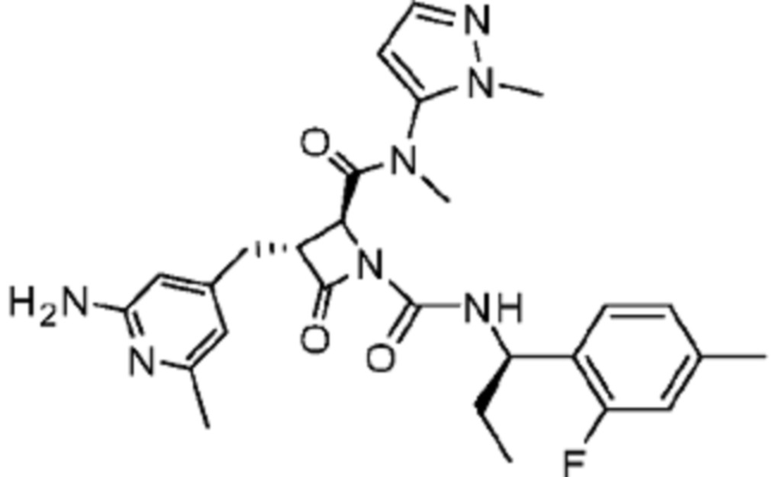
1-160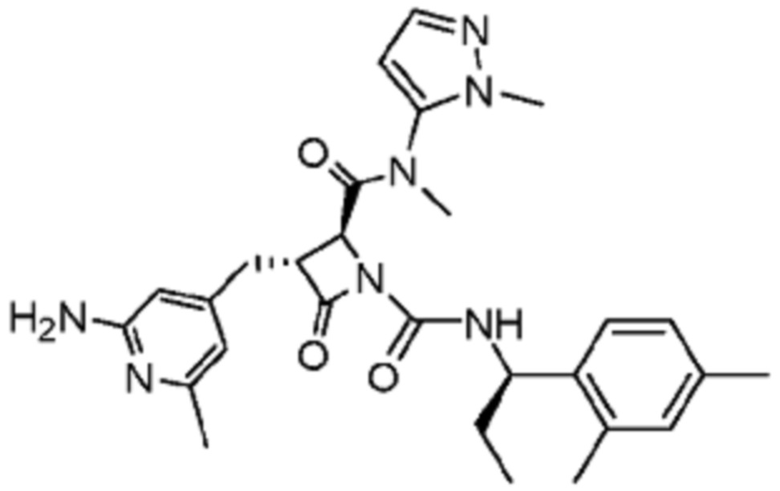
1-161
1-162
1-163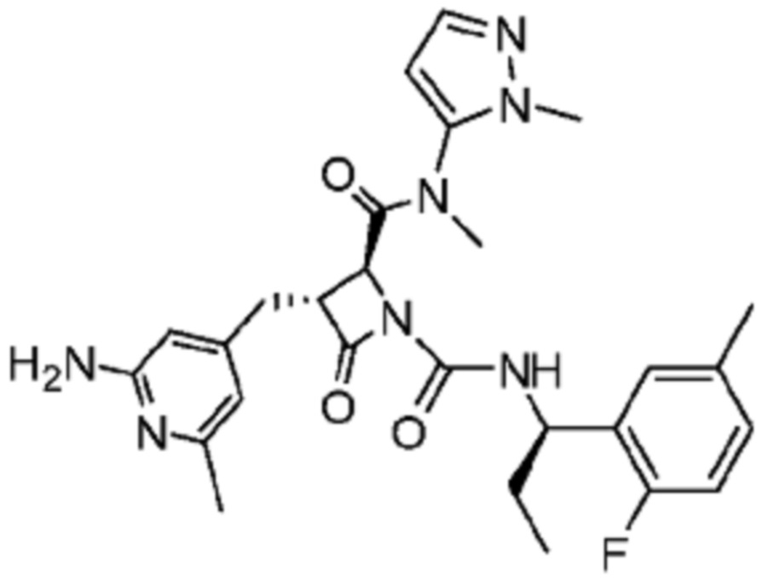
1-164.
1-165
1-166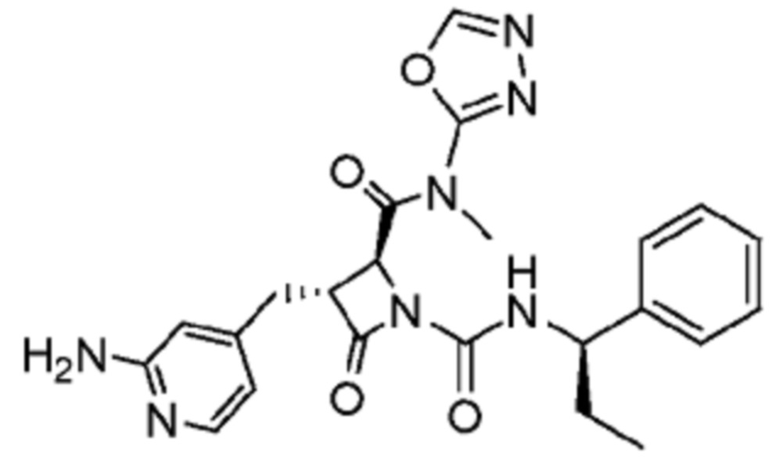
1-167
1-168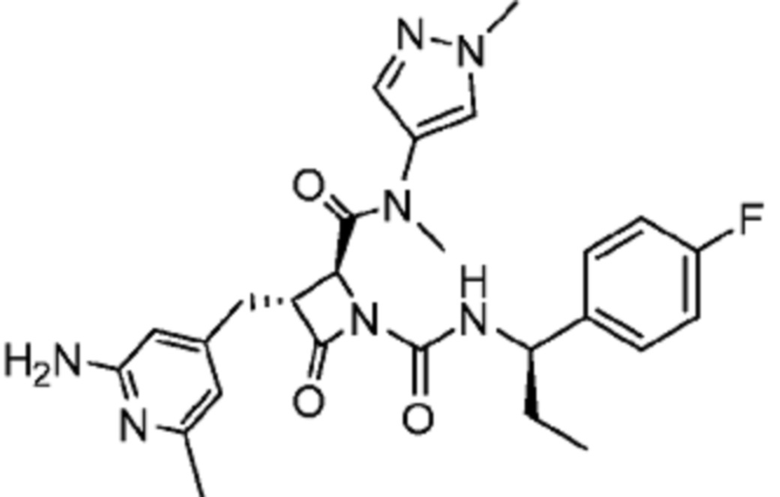
1-169
1-170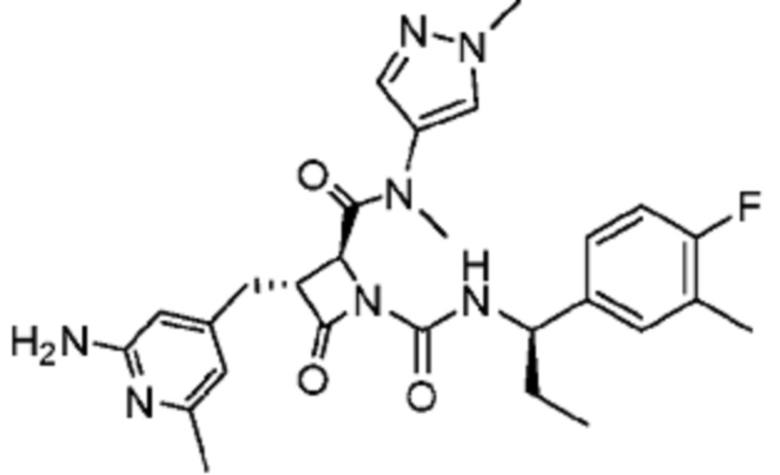
1-171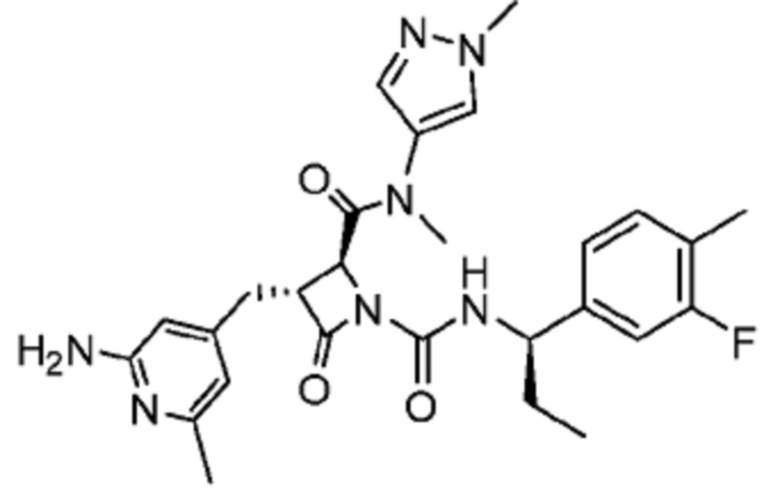
1-172
1-173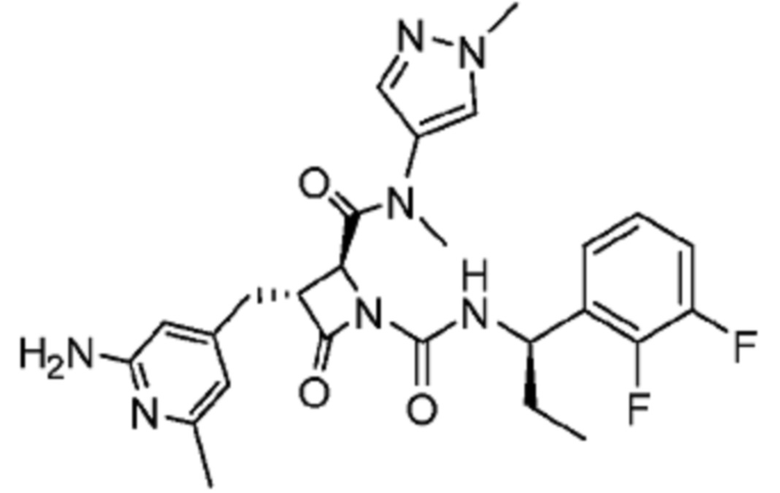
1-174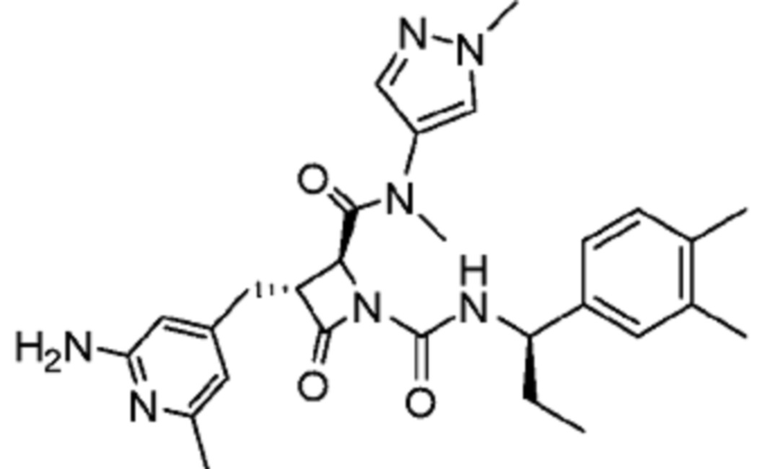
1-175
1-176
1-177
1-178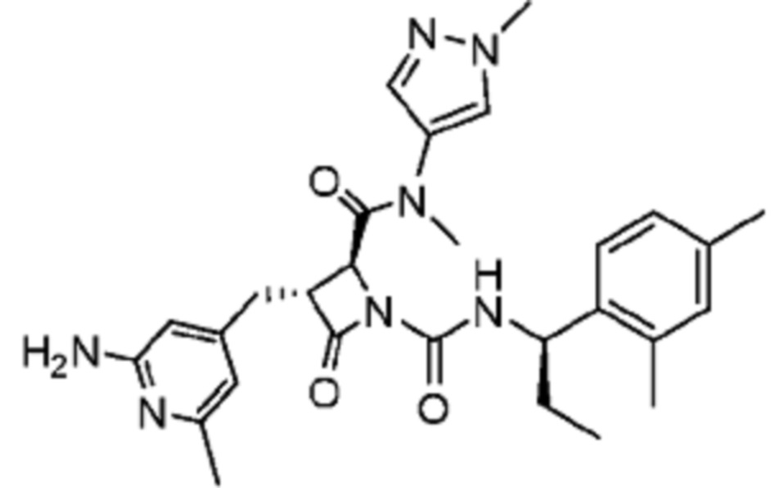
1-179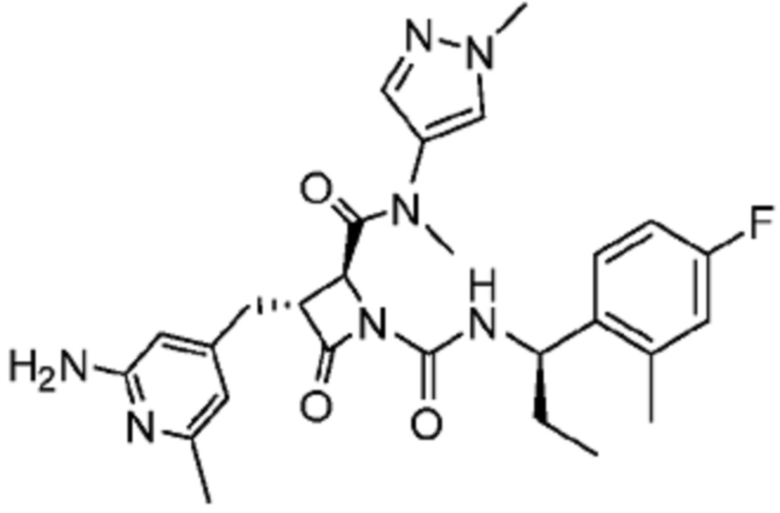
1-180
1-181
1-182
1-183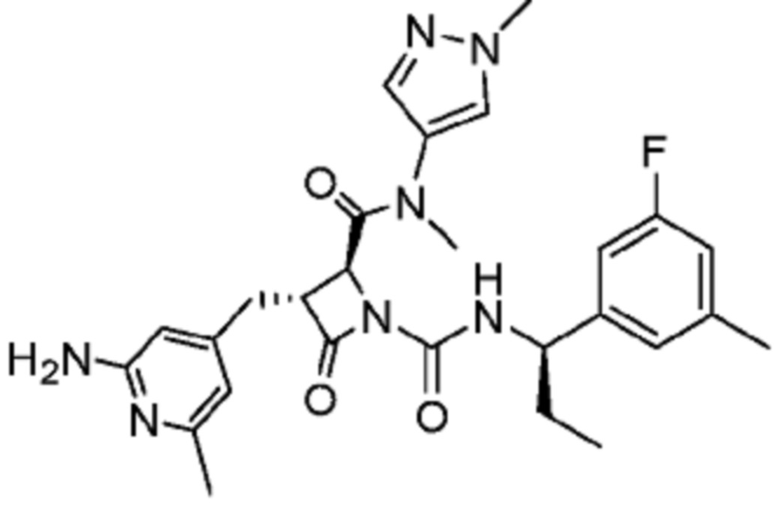
1-184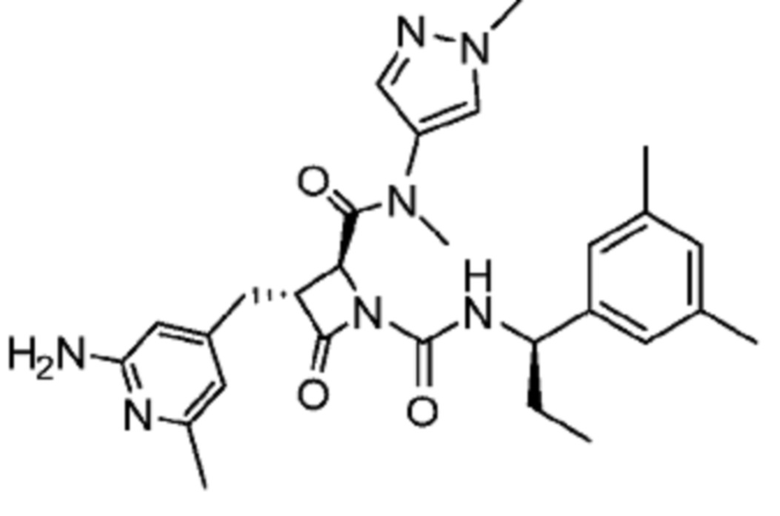
1-185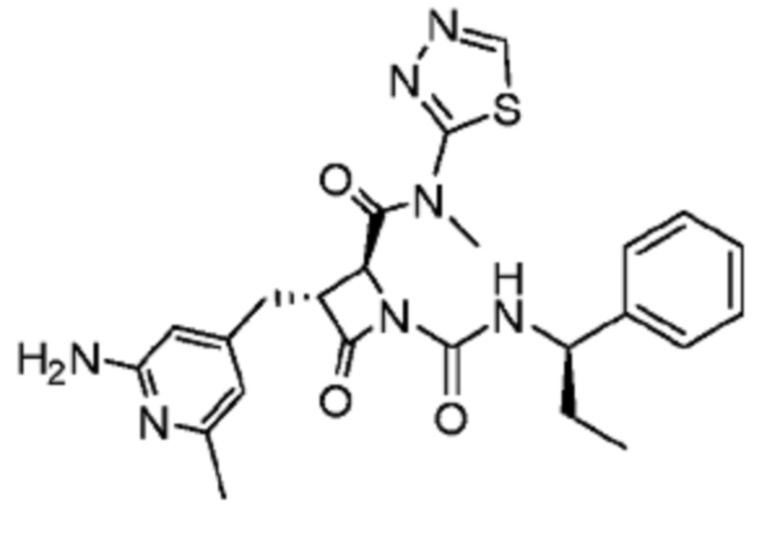
1-186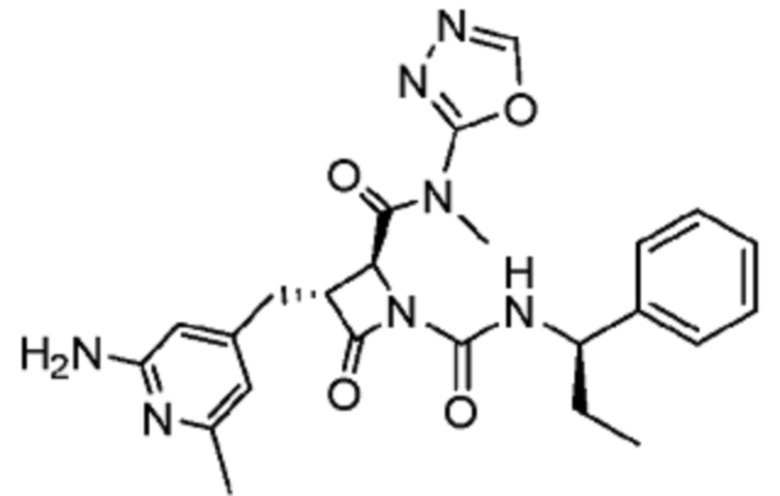
1-187
1-188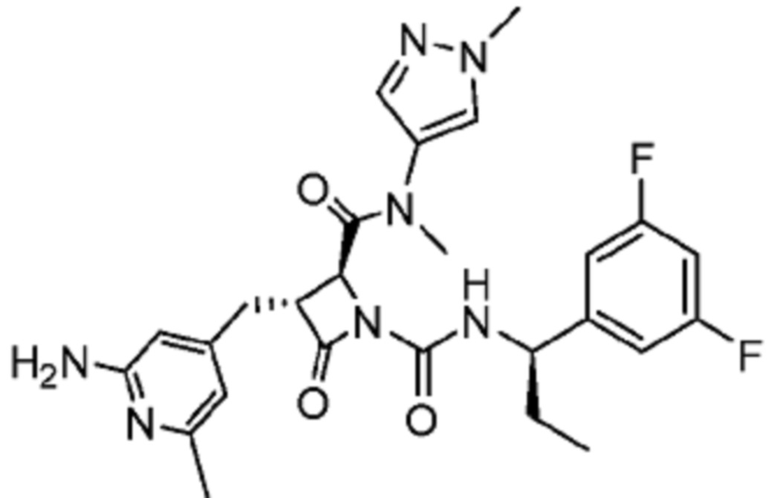
1-189
1-190
1-191
1-192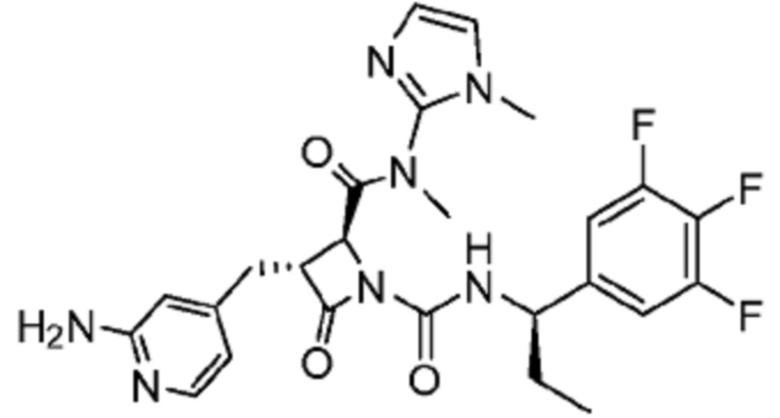
1-193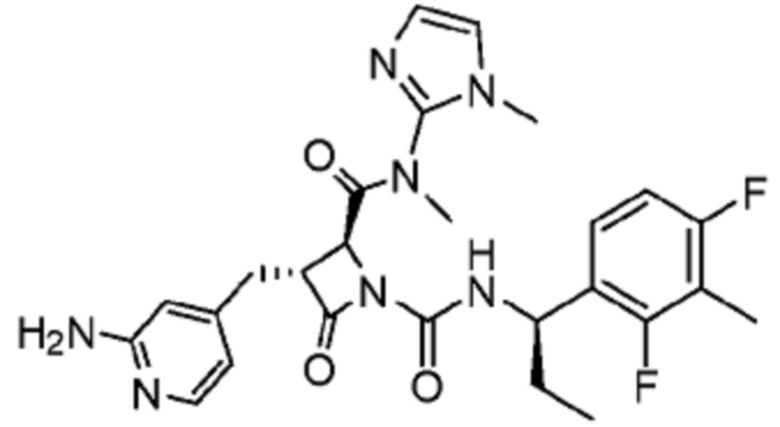
1-194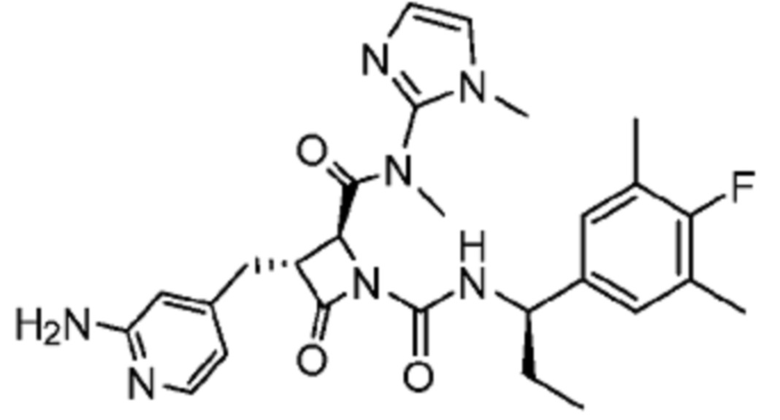
1-195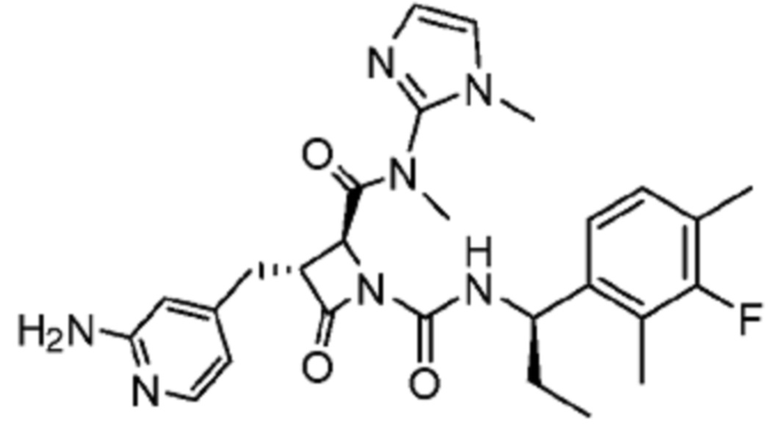
1-196
1-197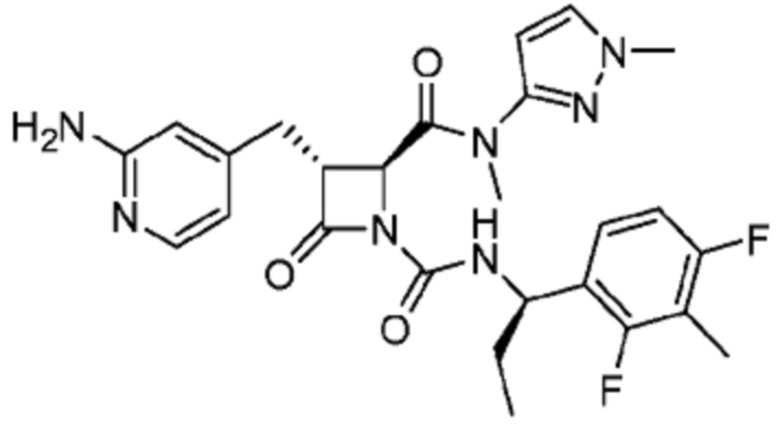
1-198
1-199
1-200
1-201
1-202
1-203
1-204
1-205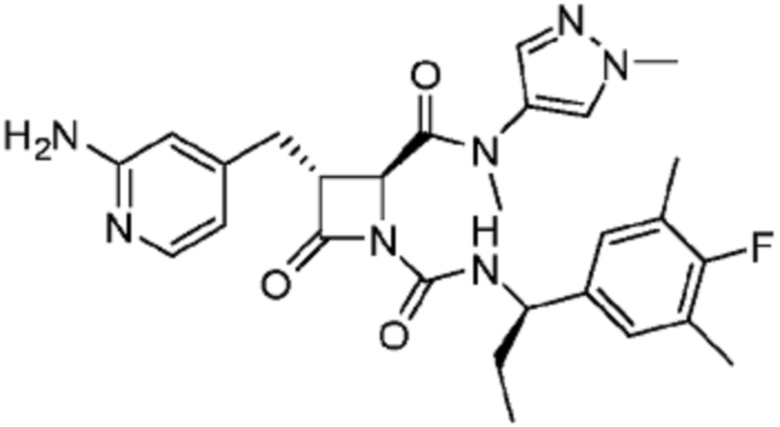
1-206
1-207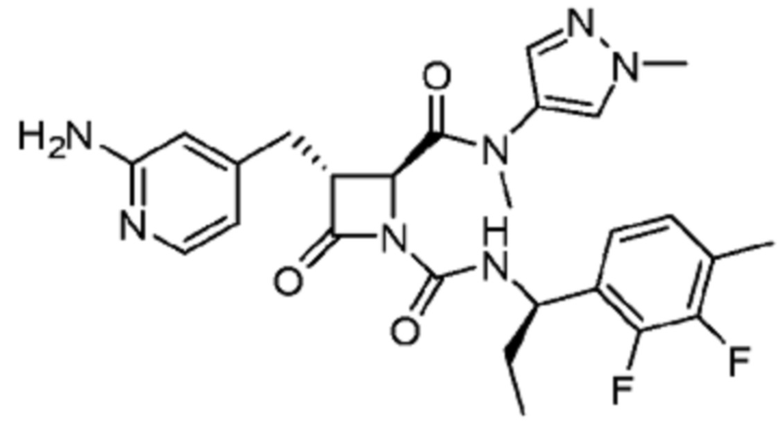
1-208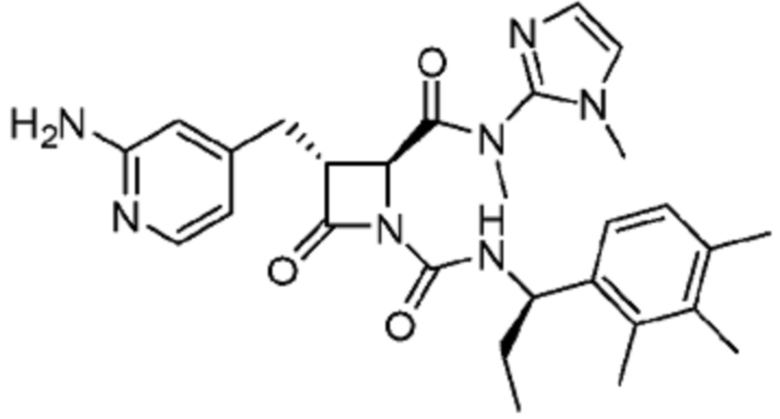
1-209
1-210 1-211
1-211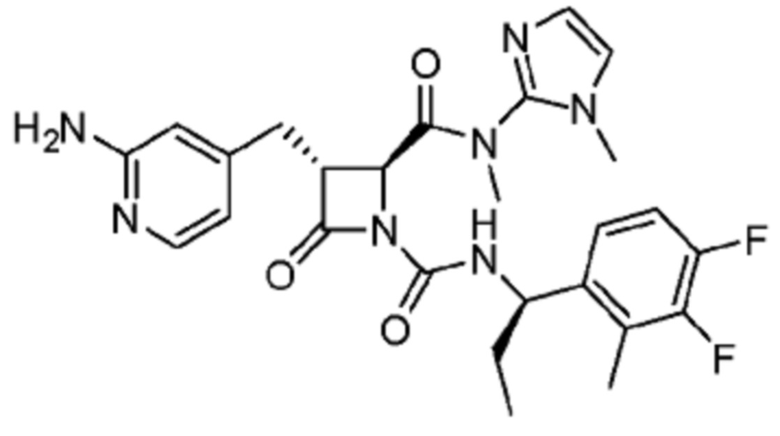
1-212
1-213
1-214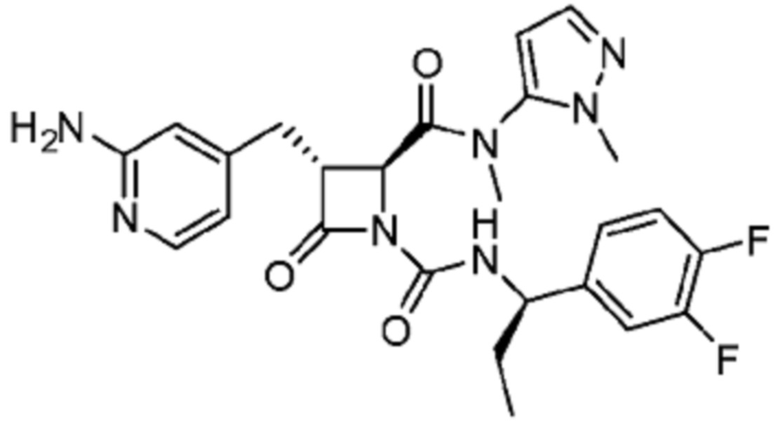
1-215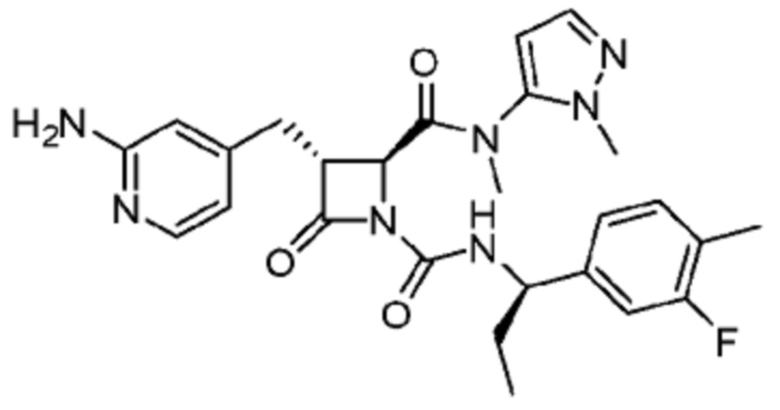
1-216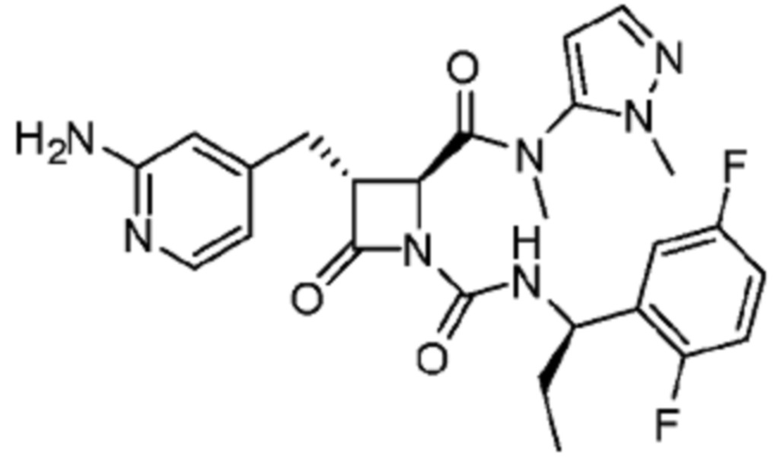
1-217
1-218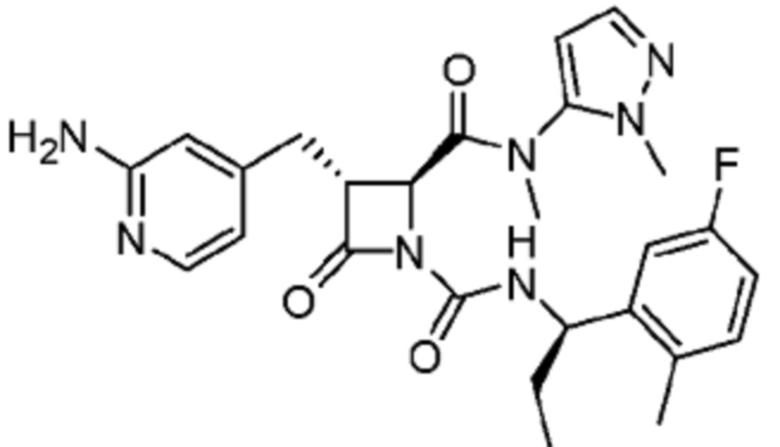
1-219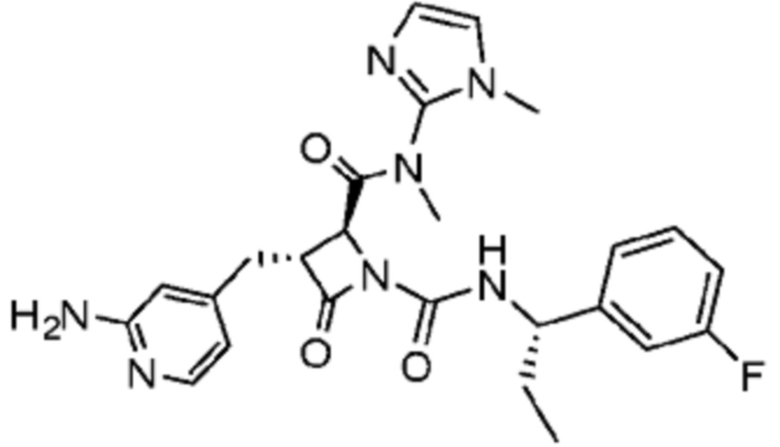
1-220
1-221
Пример 12. Определение зависимости «доза-ответ» для ингибиторов протеазы
Материалы:
Аналитический буфер: 20 мМ Hepes, pH 7,4; 150 мM NaCl; 0,02% Tween 20
Соединения: исходные растворы 10 мM в ДМСО
Субстрат: 20 мМ Glp-Pro-Arg-AMC, 25 мг/2,3 мл H2O (хранят при +4°C) [фактор XIa, тромбин и трипсин]
20 мМ Pro-Phe-Arg-AMC (Bachem I-1295), 25 мг/2,2 мл H2O
или Boc-Leu-Gly-Arg-AMC (Bachem I-1105), 25 мг/2,07 мл H2O
(хранят при при +4°C) [фактор Xa]
Фермент: Фактор Xia, 0,25 мМ в 50% глицерине (20 мкг/мл)
Трипсин: 0,2 мМ в 50% глицерине (4,8 мкг/мл)
Тромбин: 0,2 мМ в 50% глицерине (7,34 μг/мл)
Фактор Xa: 0,2 мМ в 50% глицерине (9,2 мкг/мл)
Исходные растворы разделяют на аликвоты (~100 мкл/аликвота) и хранят при -20° C
Методы:
1. Субстрат разбавляют до 100 мМ в буфере для анализа 30 мкл/6 мл). Фермент разбавляют до 0,5 нM непосредственно перед применением (12 мкл/6 мл для фактора XIa; 15 мкл/6 мл для всех остальных ферментов).
2. С помощью пипетки переносят 50 мкл субстрата в каждую лунку 96-луночного (12 × 8) микротитровального планшета. (Колонку 1 используют в качестве контроля 100% активности, и в нее соединения не добавляют, и колонка 12 является контрольной, в нее не добавляют фермент.) В колонку 12 добавляют дополнительные 46 мкл.
3. С помощью пипетки в соответствующую лунку колонки 2 планшета добавляют 4 мкл каждого соединения (соединения, активность которых не известна, анализируют трижды, стандарт анализируют дважды). Конечная концентрация соединения будет составлять 1/50 исходной.
4. Последовательно двукратно разбавляют раствор соединения смешиванием образца в колонке 2, отбирая 50 мкл и перенося в следующую лунку (колонку 3), смешивая, отбирая и перенося в колонку 4 и т.д. до колонки 11. После смешивания в колонке 11 отбирают 50 мкл и утилизируют.
5. Пипеткой в колонку 12 вносят 50 мкл буфера. Реакцию инициируют добавление 50 мкл раствора фермента в каждую лунку колонок 1-11 как можно быстрее.
6. Планшет считывают в спектрофотометре (SpectraMax) при 30°C, где считывание каждой лунки выполняют каждые 60 секунд в течение 30 мин. Для анализов с фактором Xa считывание каждой лунки проводят каждую 1 мин. в течение 60 мин.
7. Для анализов фактора XIa соединенения испытывают дважды при трех различных исходных концентрациях: 20, 2 и 0,2 мкM; 1:10, 1:100 и 1:1000 разведение исходного раствора 10 мМ или при 2, 0,2 и 0,02 мкM; 1:100, 1:1000 и 1:10,000 разведение исходного раствора 10 мМ. Данные для каждой концентрации объединяют для построения графиков и аппроксимации данных.
8. Полученные данные используют для определения IC50 и вычисления Kon.
Пример 13. Метаболическая стабильность гепатоцитов собак
Описанную далее методику используют для определения стабильности тестируемых соединений в гепатоцитах собак породы бигль в формате 96-луночного планшета. Тестируемые соединения, включая контроль, имипрамин и 7-этоксикумарин, растворяют при 10 мМ в ДМСО с последующим дополнительным 5-кратным разбавлением ДМСО до 2 мМ. Объединенные криоконсервированные гепатоциты собак породы бигль размораживают и переносят в полную среду. Клетки осторожно центрифугируют (700 об./мин. в течение 5 мин.) и пеллет промывают буфером Кребса-Хенселейта (KHB). Клетки снова суспендируют, подсчитывают с использованием трипанового синего и доводят концентрацию до 1 миллиона клеток/мл в KHB буфере. Образцы 2 мМ исходных растворов 10-кратно разбавляют ацетонитрилом с последующим 50-кратным разбавлением KHB буфером. 200 мкл полученных растворов соединений переносят в 96-луночные полипропиленовые планшеты для анализа, добавляют равный объем раствора гепатоцитов и перемешивают для инициирования реакции (конечные концентрации анализа составляют 2 мкM соединения и 500000 клеток/мл). Планшеты для анализа инкубируют при 37°C в 5% CO2 с осторожным перемешиванием. Аликваты 50 мкл отбирают в некоторые временные точки в интервале до 180 минут и гасят 6 объемами охлажденного ацетонитрила (содержащего 250 нг/мл карбутамида и хризина в качестве внутренних стандартов) для прекращения реакции осаждения белка. Облажденные образцы выдерживают на льду. Образцы гашеной смеси центрифугируют при 2000 × g (3100 об./мин.) в течение 10 минут при 4°C. Аликвоты 50 мкл супернатантов отбирают и разбавляют 100 мкл Milli-Q воды для снижения % содержания органических веществ до биоанализа с помощью ЖХ-МС/МС. Определяют площади пиков, соответствущих теституемому соединению. Остальное соединение рассчитывают сравнением площади пика вкажный момент времени с нулевым моментом вемени. Данные анализируют, результаты получают с использованием Microsoft Excel 2010.
Пример 14. Анализы ингибирования фактора XIa
Способность соединений по настоящему изобретению ингибировать фактор XIa оценивают определением концентрации ингибитора, которая приводит к 50% снижению активности фермента (IC50) с использованием очищенного фермента. Потенциальные ингибиторы фактора XIa оценивают с использованием следующего анализа.
PyroGlu-Pro-Arg-7-methylaminocourin (AMC), доступный от CPC Scientific, Inc., наносят на подложку pyro-Glu-Pro-Arg-pNA (S-2366), доступную от Diapharma Group, Inc. (Columbus, Ohio), в котором п-нитроаналиновая группа замещена 7-метиламинокумарином (AMC).
Конечная концентрация субстрата в анализе составляет 50 мкM, конечная концентрация фермента составляета 0,25 нМ. Ингибиторы испытывают последовательным разбавлением в соответствующем диапазоне концентраций для получения кривой «доза-ответ» и определения значения IC50 ингибиторов. Анализируемую смесь считывают каждую минуту в течение 30 минут для получения кривых прогресса. Планшеты считывают в планшет-ридере Spectramax m5 Multimode Plate Reader (Molecular Devices LLC, Sunnyvale, CA). Кривые «доза-ответ» соответствуют приведенному ниже уравнению 1, где А - максимальное ингибирование, В - минимальное ингибирование, С представляет собой IC50, и D - коэффициент Хилла.
Y = [(A-B) / (1 + (X/C)D)] + B (Уравнение 1)
Таблица 8. Эффективность, селективность и стабильность соединений примеров, представленных в таблице 1
(t1/2: мин.)
К таблице 8: FXIa и hFXIa означают фактор XIa и фактор XIa человека, соответственно.
Эффективность: “A” означает < 10 нM, “B” означает 10-100 нM, “C” означает 100-1000 nM, “D” означает > 1000 нM, и “E” означает, что данные недоступны или не были определены.
Селективность: “F” означает < 1, “G” означает 1-100, “H” означает 101-1000; “I” означает > 500, и “J” означает, что данные недоступны или не были определены.
Стабильность гепатоцитов собаки: “K” означает 0-60 мин.; “L” означает 61-120 мин.; “M” означает 121-240 мин.; “N” означает > 240 мин.; “O” означает, что данные не доступны или не были определены.
В таблице 8 активности ингибирования фермента фактор XIa соединения 1-79 и соединения 1-84 были ошибочно отнесены к соединению 1-220 и соединению 1-221, соответственно, в USSN 62/627435, к которой настоящая заявка испрашивает приоритет. Значения ингибирования фермента фактор XIa соединения 1-79, соединения 1-84, соединения 1-220 и соединения 1-221 в настоящем изобретении является правильным.
Пример 15. Анализ проницаемости эпителиальных раковых клеток толстой кишки (Caco-2)
Для определения способности тестируемого соединения к поперечной сшивке Caco-2 клеток (эпителиальных раковых клеток толстой кишки, полученных из колоректальной аденокарциномы). Клетки высевают при плотности 1 × 105 клеток/см2 в 96-луночные микропланшеты MultiscreenTM (Millipore). Анализ протицаемости проводят с клетками через 21-25 дней после высева. Клетки обычно используют для 15 последовательных пассажей в культуре.
Данный анализ может проводиться в направлении от апикального к безолатеральному (A-B) или в направлении от базолатерального к апикальному (B-A). Приготавливают раствор тестируемого соединения в концентрации 10 мкM в HBSS-MES (pH 6,5) или HBSS-HEPES (pH 7,4) с конечной концентрацией ДМСО 1%. Затем рабочий раствор центрифугируют и супернатант добавляют на сторону донора. Планшеты для анализа инкубируют при 37°C осторожным встряхиванием в течение 60 мин. или 40 мин. для A-B или B-A анализа, соответственно. Отбирают аликвоты образцов со стороны донора в нулевой момент времени и в конечной точке, и со стороны акцептора в конечной точке.
В каждый анализ включают эталонные соединения, пропранолол (высокопроницаемый), лабеталол (умеренно проницаемый), ранитидин (плохо проницаемый) и колхицин (P-гликопротеиновый субстрат).
Образцы анализируют с помощью ВЭЖХ-МС/МС с использованием выбранного мониторинга реакции. ВЭЖХ система состоит из бинарного ЖХ насоса с автоматическим пробоотборником, C-18 колонкой и градиентом.
Коэффициент кажущейся проницаемости (Papp) тестируемого соединения (уравнение 2) и его восстановление (уравнение 3) вычисляют следующим образом:
 (Уравнение 2)
(Уравнение 2)
 (Уравнение 3)
(Уравнение 3)
где A - площадь поверхность монослоя клеток (0,11 см2), C - концентрация тестируемого соединения, представленная как площадь пика, D - донор, и R - акцептор. “0”, “mid” и “end” означают время в нулевой, средней и конечной точке инкубации, соответственно. Δt - время инкубации. V - объем донора и акцептора.
Низкое восстановление показывает, что тестируемое соединение теряется во время анализа. Это вероятнее всего связано с неспецифическим связыванием или разложением.
Пример 16. Определение растворимости
Для определения водной растворимости тестируюемого соединения в фосфатно-буферном солевой растворе (PBS - NaCl 137 мМ, KCl 2,7 мМ, Na2HPO4 8,1 мМ, KH2PO4 1,5 мМ, pH 7,4) в формате 96-луночного планшет ипользуют методику ВЭЖХ-УФ/VIS анализа. Из 10 мМ исходного раствора в ДМСО приготавливают 200 мкМ раствор тестируемого соединения в PBS. Конечная ДМСО концентрация составляет 2%. Образец PBS буфера тщательно смешивают с последующей инкубацией при комнатной температуре в течение 24 часов. В конце инкубирования образцы PBS буфера центрифугируют и анализируют супернатанты с помощью ВЭЖХ. Водную растворимость (мкM) тестируемого соединения в PBS определяют сравнением площади основного пика в калибровочном стандарте (200 мкM) с площадью соответствующего пика каждом образцов PBS. Диапазон концентраций анализа составляет приблизительно от 0,5 мкM до 200 мкM. Эталонными соединениями, используемыми в каждом анализе, являются метопролол, рифампицин, кетоконазол, фенитоин, галоперидол, симвастатин, диэтилстилбестрол и тамоксифен, ранжированные от полностью растворимого (200 мкM) до плохо растворимого (< 1 мкM).
Пример 17. Анализы связывания белка плазмы
Представленную далее методику используют для определения связывания белка плазмы тестируемого соединения в объединенной плазме человека (смешанный пол) посредством равновестого диализа в формате 96-луночного планшета. В ячейку диализата загружают фосфатно-буферный раствор воли (pH 7,4), а в боковую часть образца загружают плазму с тестируемым соединением в концентрации 10 мкM. После загрузки образцы закрывают и инкубируют в течение 4 часов при 37°C. После инкубирования из каждой ячейки отбирают образец, разбавляют ацетонитрилом/буфером и центрифугируют. Супернатанты анализируют с помощью ВЭЖХ-МС/МС. Площадь пиков анализа, полученные в помощью ВЭЖХ-МС/МС, используют для вычисления связывния белка в соответствии со следующим уравнением (уравнение 4):
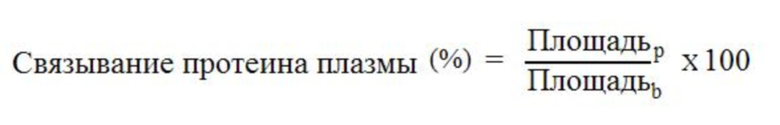 (Уравнение 4)
(Уравнение 4)
где
Площадьp - площадь пика аналита в матрице белка
Площадьb - площадь пика аналита в буфере
Количество, определенное в плазменной ячейке, включает как свободное, так и связанное лекарственное средство, в то время как на стороне буфера представлено только свободое лекарственное средство; разницы изпользуют для расчета процентного содержания связанного белка плазмы. В каждом анализе тестируют три эталонных соединения: ацебутолол, хинидин и варфарин.
Пример 18. Определение биодоступности в организме собаки
Для оценки биодоступности в организме собаки используют следующую методику. Тестируемые соединения вводят в препарат с носителями, совместимыми с внутривенным и пероральным дозированием. Испытивыемые соединения вводят самцам собак породы бигль вформ внутривенных болюсов в дозе 1 мг/мг (1 мг/мл в объеме 1 мл/кг) или через пероральный зонд в дозе 5 мг/кг (1 мг/мл в объеме 5 мл/кг). Образцы крови отбирают через контралатеральную (для внутривенного болюса) яремную вену или через другую доступную вену в различные моменты времени после введения и в интервале до 24 часов после введения дозы. Полученные образцы плазмы (EDTA) центрифугируют (2200 × g в течение 10 минут при 5°C) и хранят замороженными (70°C) до анализа на исходное соединение с помощью ЖХ/МС/МС. Фармаконинетические параметры оценивают исходя из данных концентрации по времени стандартными некомпартментными методами с использованием стандартного программного обеспечения для анализа. Сравнением площади под кривой (AUC) для IV и PO введения получают оценки пероралькой биодоступности.
Таблица 9. Проницаемость клеток Caco-2 (A-B и B-A), водная растворимость, связывание белка плазмы человека (PPB) и биодоступность в организме собаки соединений примеров, представленных в теблице 1.
Для таблицы 9: Caco-2 проницаемость (Papp): “P” означает ≤ 10 × 10-6 см/сек.; “Q” означает 11-30 × 10-6 см/сек.; “R” означает 31-50 × 10-6 см/сек.; “S” означает > 50 × 10-6 см/сек.; “T” означает, что данные не доступны или не были определены.
Водная растворимость: “U” означает ≤ 10 мМ; “V” означает 10-100 мМ; “W” означает > 100 мМ; “Х” означает, что данные недоступны или не были определены.
PPB: “Y” означает > 50%; «Z» означает 50-90%; “AA” означает 91-95%; “BB” означает > 95%; “CC” означает, что данные недоступны или не были определены.
Биодоступность в организме собаки: “DD” означает ≤ 10%; “EE” означает 11-30%; “FF” > 30%; “GG” означает, что данные недоступны или не были определены.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ | 2015 |
|
RU2733405C2 |
| БИАРИЛЬНЫЕ МОНОБАКТАМНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2016 |
|
RU2746129C2 |
| ИНГИБИТОРЫ КАТЕПСИН-ЦИСТЕИНПРОТЕАЗЫ | 2003 |
|
RU2312861C2 |
| СОЕДИНЕНИЯ ПИРИДАЗИНАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ СЕЛЕЗЕНКИ (SYK) | 2013 |
|
RU2627661C2 |
| БЕНЗОТИАЗОЛЬНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ РАССТРОЙСТВ | 2018 |
|
RU2778370C2 |
| ПРОИЗВОДНЫЕ ГИДРОКСАМОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ ИЛИ ВЕТЕРИНАРНАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2126791C1 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| ТЕРАПЕВТИЧЕСКИЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ | 2019 |
|
RU2813780C2 |
| ХИНОЛИНКАРБОКСАМИДНЫЕ И ХИНОЛИНКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ mGLuR2-НЕГАТИВНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ, КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610262C2 |
| ИМИДАЗОПИРИДИНИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ РАССТРОЙСТВ | 2020 |
|
RU2833052C2 |
Изобретение относится к соединениям формулы (II), где R1-R5, Rx, Ra, Rb, m, n определены в формуле изобретения, которые ингибирут фактор XIa или калликреин, и их фармацевтически приемлемым солям и фармацевтическим композициям. Также предложены способы лечения или снижения риска тромбоэмболического осложнения, инсульта, острого коронарного синдрома, тромбоэмболического расстройства или мерцательной аритмии путем ингибирования фактора XIa у пациента. 18 н. и 10 з.п. ф-лы, 9 табл., 221 пр.
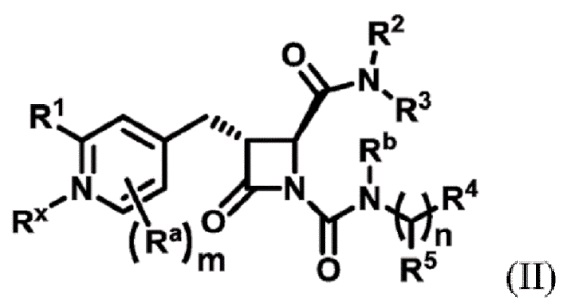
1.Соединение формулы (II)

или его фармацевтически приемлемая соль,
где R1 представляет собой NH2;
Ra представляет собой C1-6 алкил;
Rb представляет собой водород или C1-6 алкил;
R2 представляет собой пиразолил или имидазолил, где пиразолил или имидазолил необязательно замещены одним или двумя независимыми друг от друга С1-6 алкилами;
R3 представляет собой C1-6 алкил;
R4 представляет собой фенил необязательно замещенный одним, двумя или тремя независимыми заместителями, выбранными из галогена или C1-6 алкила;
R5 представляет собой водород, C1-6 алкил, C1-6 галогеналкил, C3-6 циклоалкил
Rx отсутствует;
m равно 0; и
n равно 1.
2. Соединение по п.1, где R2 представляет собой пиразолил илиимидазолил, где пиразолил или имидазолил, является замещенным одним метилом.
3. Соединение по п.1 или 2, где R3 представляет собой метил.
4. Соединение по любому из пп.1-3, где R4 представляет собой фенил, необязательно замещенный одним, двумя или тремя независимыми заместителями, выбранными из метила.
5. Соединение по любому из пп.1-4, где R5 представляет собой водород, -CH3, -CH2CH3, -CH2CH2CH3, -CH(CH3)2, -CH2CH2CH2CH3, -CF3 или незамещенный циклопропил.
6. Соединение по п.1, где соединение представляет собой соединение формулы (I-a)
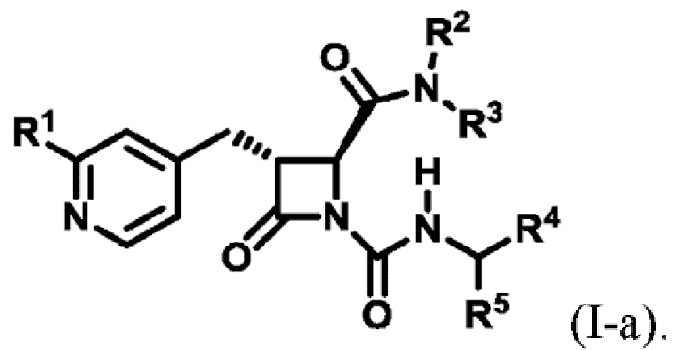
7. Соединение по п.1, где соединение представляет собой соединение формулы (I-c), формулы (I-d), формулы (I-e), формулы (I-f), формулы (I-g), формулы (I-i) или формулы (I-j):

8. Соединение формулы:





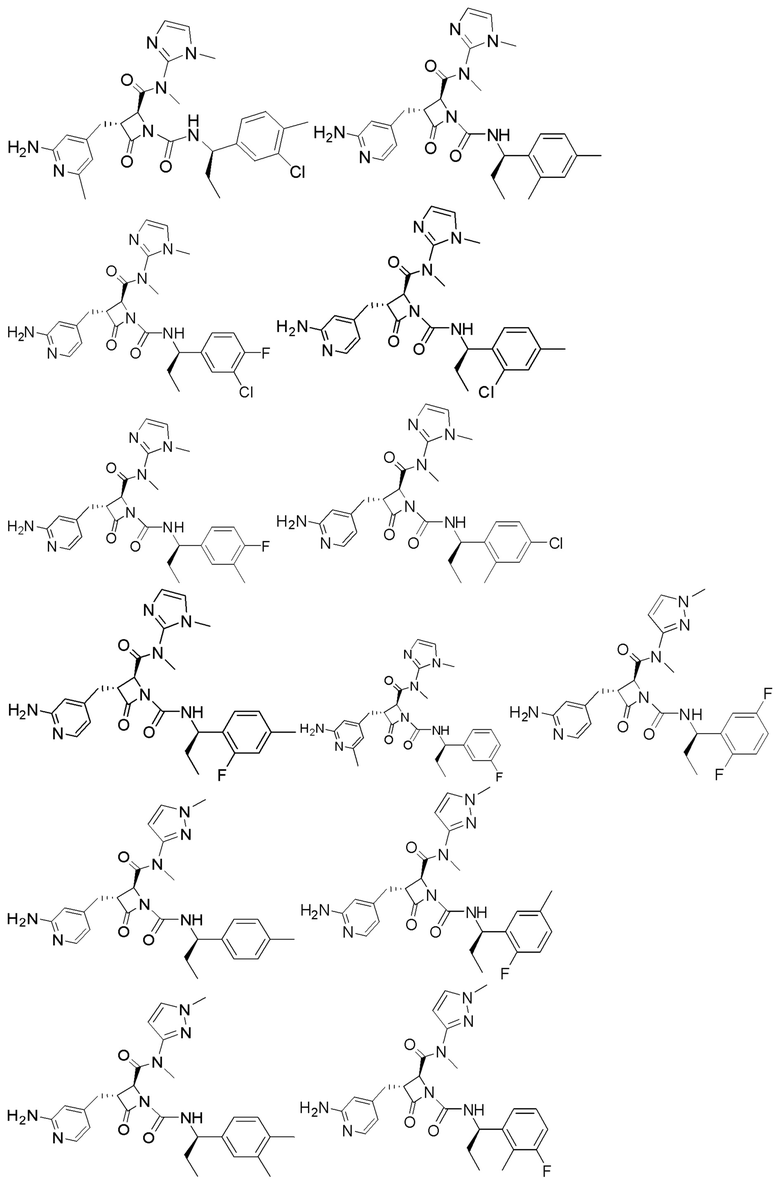
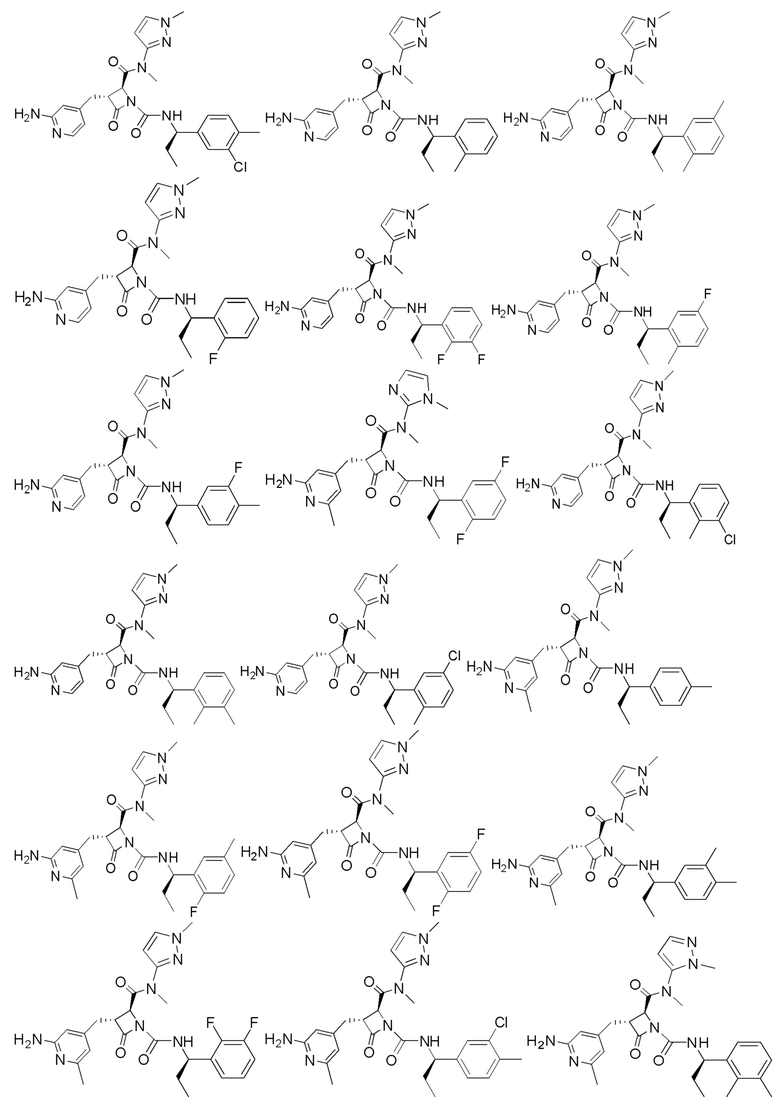
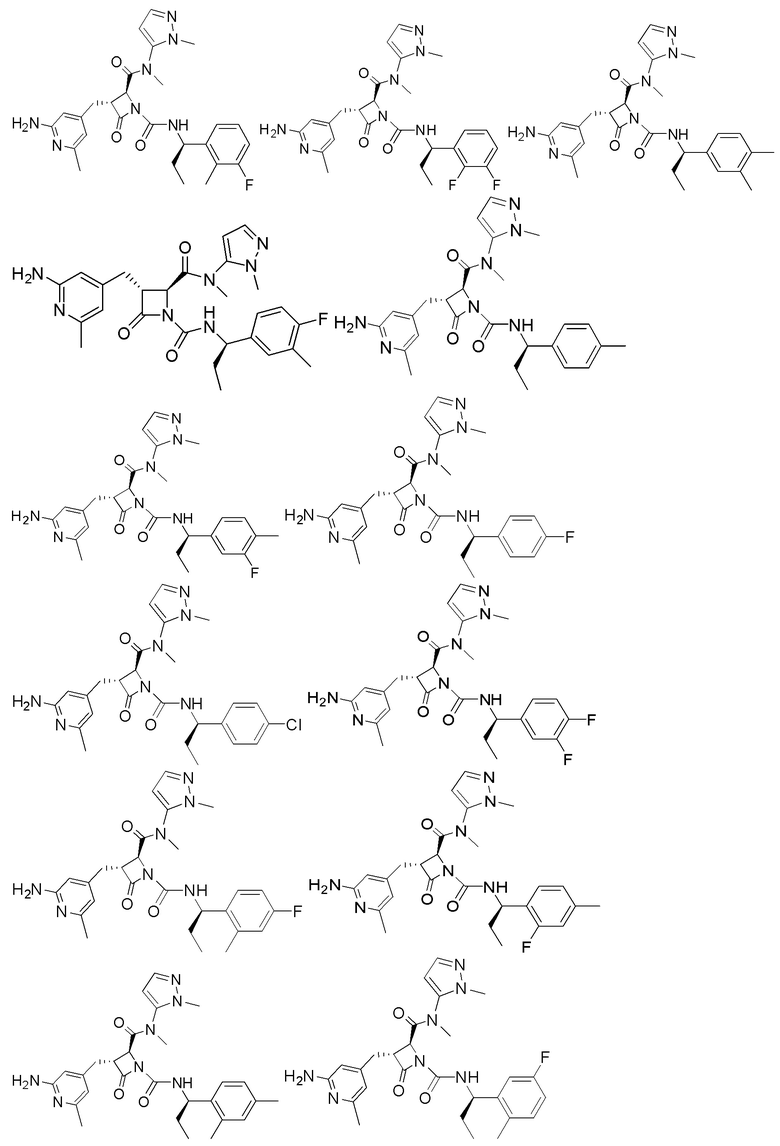

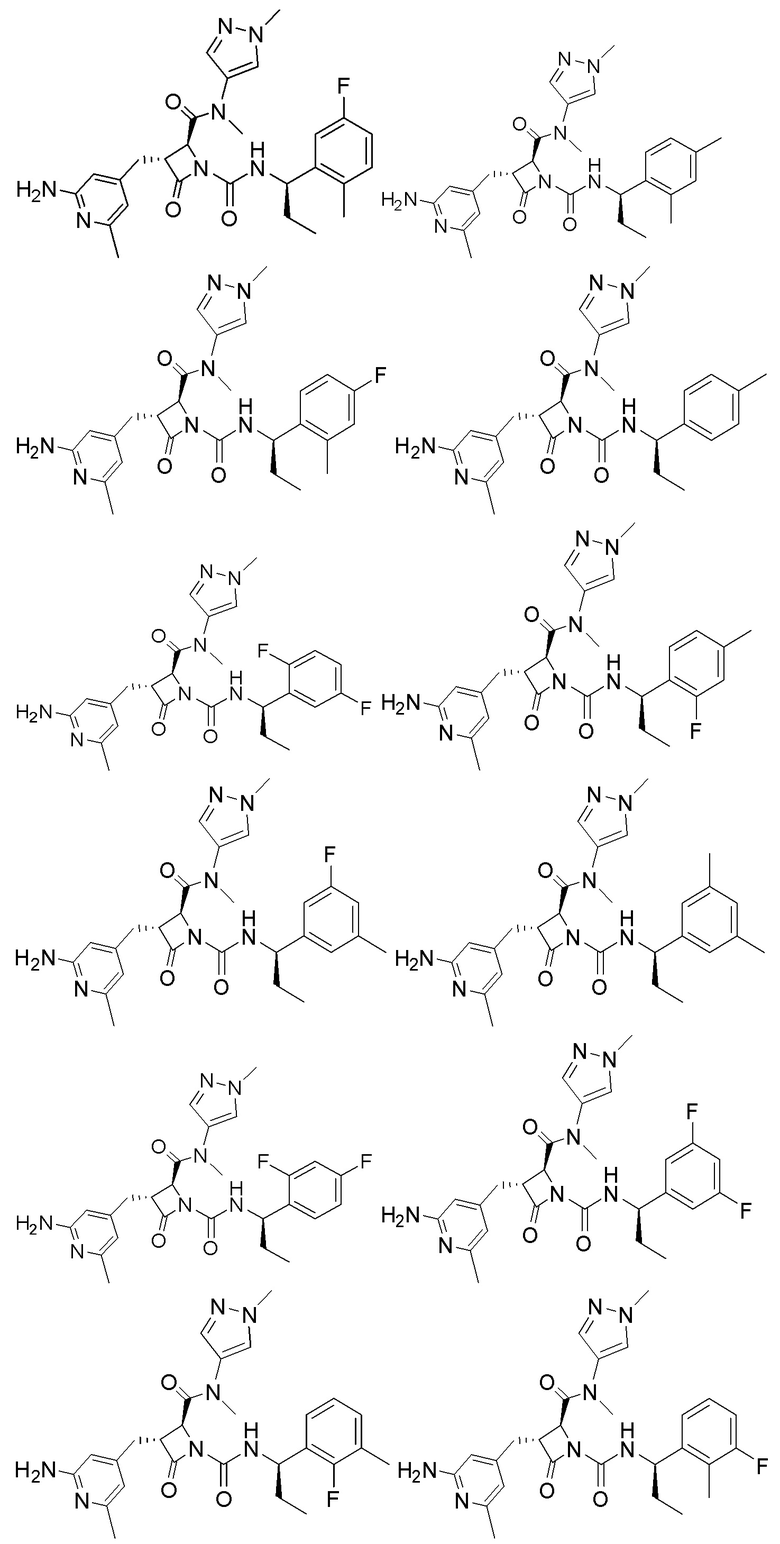



9. Фармацевтическая композиция, имеющая свойство ингибирования фактора XIa или калликреина, включающая эффективное количество соединения по любому из пп.1-8 или его фармацевтически приемлемую соль и один или несколько фармацевтически приемлемых эксципиентов.
10. Фармацевтическая композиция по п.9, которая представляет собой жидкий препарат или твердый препарат.
11. Способ лечения тромбоза глубоких вен путем ингибирования фактора XIa, включающий введение пациенту, который перенес ишемическую атаку, эффективного количества соединения по любому из пп.1-8 или его фармацевтически приемлемой соли или композиции по п.9 или 10.
12. Способ профилактики тромбоза глубоких вен путем ингибирования фактора XIa у нуждающегося в этом пациента, включающий введение пациенту эффективного количества соединения по любому из пп.1-8 или его фармацевтически приемлемой соли или композиции по п.9 или 10.
13. Способ ингибирования фактора XIa у пациента, включающий введение пациенту, который страдал ишемией, эффективного количества соединения по любому из пп.1-8 или его фармацевтически приемлемой соли или композиции по п.9 или 10.
14. Способ лечения тромбоэмболического последствия или осложнения путем ингибирования фактора XIa у нуждающегося в этом пациента, включающий введение пациенту эффективного количества соединения по любому из пп.1-8 или его фармацевтически приемлемой соли или композиции по п.9 или 10.
15. Способ лечения тромбоэмболического осложнения путем ингибирования фактора XIa у пациента, нуждающегося в этом, включающий введение пациенту эффективного количества соединения по любому из пп.1-8 или его фармацевтически приемлемой соли или композиции по п.9 или 10, в котором пациент подвергается воздействию искусственной поверхности.
16. Способ снижения риска тромбоэмболического осложнения путем ингибирования фактора XIa у пациента, нуждающегося в этом, включающий введение пациенту эффективного количества соединения по любому из пп.1-8 или его фармацевтически приемлемой соли или композиции по п.9 или 10, в котором пациент подвергается воздействию искусственной поверхности.
17. Способ снижения риска тромбоэмболического последствия или осложнения путем ингибирования фактора XIa у пациента, нуждающегося в этом, включающий введение пациенту эффективного количества соединения по любому из пп.1-8 или его фармацевтически приемлемой соли или композиции по п.9 или 10.
18. Способ по п.15 или 17, где тромбоэмболическое последствие или осложнение ассоциировано с вмешательством в периферические кровеносные сосуды, гемодиализом, катетерной абляцией, цереброваскулярным вмешательством, трансплантацией органа, хирургической операцией, транс-катетерной имплантацией аортального клапана, вмешательством большого диаметра, используемым для лечения аневризмы, чрескожным коронарным вмешательством или терапией гемофилии.
19. Способ лечения инсульта у пациента путем ингибирования фактора XIa, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения по любому из пп.1-8 или его фармацевтически приемлемой соли или композиции по любому из пп.9, 10.
20. Способ снижения риска инсульта у пациента, который перенес ишемическую атаку, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения по любому из пп.1-8 или его фармацевтически приемлемой соли или композиции по любому из пп.9, 10.
21. Способ лечения острого коронарного синдрома путем ингибирования фактора XIa у пациента, включающий введение пациенту, нуждающемуся в этом, соединения по любому из пп.1-8 или его фармацевтически приемлемой соли или композиции по любому из пп.9, 10.
22. Способ снижения риска острого коронарного синдрома путем ингибирования фактора XIa у пациента, включающий введение пациенту, нуждающемуся в этом, соединения по любому из пп.1-8 или его фармацевтически приемлемой соли или композиции по любому из пп.9, 10.
23. Способ лечения тромбоэмболического расстройства путем ингибирования фактора XIa у пациента, включающий введение пациенту, нуждающемуся в этом, соединения по любому из пп.1-8 или его фармацевтически приемлемой соли или композиции по любому из пп.9, 10.
24. Способ снижения риска тромбоэмболического расстройства путем ингибирования фактора XIa у пациента, включающий введение пациенту, нуждающемуся в этом, соединения по любому из пп.1-8 или его фармацевтически приемлемой соли или композиции по любому из пп.9, 10.
25. Способ по п.23 или 24, где тромбоэмболическое расстройство выбрано из группы, состоящей из артериального кардиоваскулярного тромбоэмболического осложнения, артериального тромбоза, венозного кардиоваскулярного тромбоэмболического осложнения, нестабильной стенокардии, острого коронарного синдрома, первичного инфаркта миокарда, рецидива инфаркта миокарда, ишемии, инсульта, атеросклероза, окклюзионного заболевания периферических артерий, венозной тромбоэмболии, венозого тромбоза, тромбоза глубоких вен, тромбофлебита, артериальной эмболии, тромбоза коронарных артерий, тромбоза церебральных артерий, церебральной эмболии, почечной эмболии и эмболии легочной артерии.
26. Способ лечения мерцательной аритмии путем ингибирования фактора XIa у пациента, включающий введение пациенту, нуждающемуся в этом, соединения по любому из пп.1-8 или его фармацевтически приемлемой соли или композиции по любому из пп.9, 10.
27. Способ снижения риска мерцательной аритмии путем ингибирования фактора XIa у пациента, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения по любому из пп.1-8 или его фармацевтически приемлемой соли или композиции по любому из пп.9, 10.
28. Способ по любому из пп.11-27, в котором соединение вводится перорально или парентерально.
| WO 2015120062 A2, 13.08.2015 | |||
| WO 2006108039 A2, 12.10.2006 | |||
| EP 1099690 A1, 16.05.2001. |

