Область техники, к которой относится изобретение
Настоящее изобретение относится к улучшенному способу синтеза промежуточных продуктов для синтеза аналогов анти-ВИЧ агента каланолид А.
Уровень техники
Каланолид А в настоящий момент является перспективным лекарственным кандидатом и проходит фазу II клинических испытаний против ВИЧ-инфекции. Однако были синтезированы аналоги Каланолида А, существенно превосходящие родоначальное соединение. Например, соединение 6 характеризуется анти-ВИЧ активностью (EC50 = 2.85 нМ, ТИ > 10526), превосходящей таковую для каланолида А (EC50 = 640 нM, ТИ 47–140) приблизительно в 220 раз, см. D1. Было описано две схемы синтеза соединения 6 (D1), одна из которых включает исчерпывающее тозилирование 1 с получением дитозильного производного 2 и последующее удаление одной из тозильных групп. Далее соединение 3а превращают в 6. Такой способ требует дополнительной стадии снятия защиты с получением 3a и, как результат, увеличение количества стадий и снижение выхода. Кроме того, описанная очистка соединения 3а проводится при помощи хроматографии. С другой стороны, воспроизводя процедуру, описанную в D1, мы обнаружили в реакционной смеси существенное количество непрореагировавшего 2, дигидроксипроизводного 1 и побочного изомера 3b (Схема. 1).
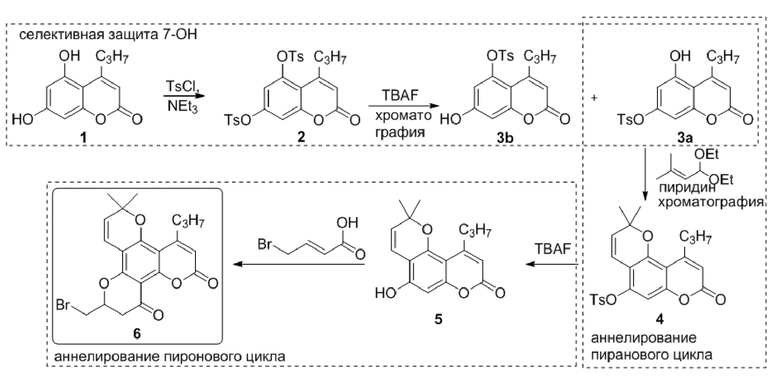
Схема 1. Одна из известных схем синтеза аналога каланолида А
Более того, на стадии аннелирования пиранового цикла также требует очистки с помощью хроматографии.
Таким образом, существует потребность в эффективных методах синтеза полупродуктов для получения аналогов каланолида А.
Цитированные документы уровня техники
D1 Ma, T. et al. J. Med. Chem. 2008, 51, 1432–1446.
Раскрытие изобретения
Настоящее изобретение относится к улучшенному способу синтеза промежуточных продуктов для синтеза аналогов анти-ВИЧ препарата каланолид А. Было обнаружено, что двухстадийную процедуру тозилирования/детозилирования можно заменить одностадийной процедурой никотиноилирования, которое позволяет легко и с высоким выходом сразу получать селективно защищенный полупродукт (I) в виде твердого вещества. Присутствие пиридильного фрагмента никотиновой кислоты позволяет также достичь других преимуществ. В частности, возможно проведение стадии аннелирования пиранового цикла (которое обычно проводят в пиридиновом растворителе) в условиях «без растворителя». Более того, продукт с аннелированным пирановым циклом может быть легко выделен в форме соли. Не желая быть связанным теорией, авторы полагают, что основный пиридиновый атом азота может быть использован для образования твердых кристаллических солей, которые легко выделяются из реакционной смеси и могут быть легко очищены.
Таким образом, в одном из вариантов осуществления настоящий способ включает одну или несколько из следующих стадий:
Стадия 1. Введение защитной никотиноильной группы. Проводят взаимодействие соединения формулы (I)
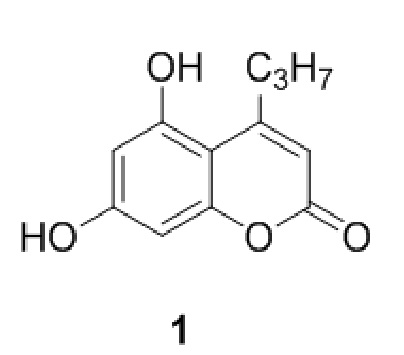 ,
,
с соединением формулы (I)
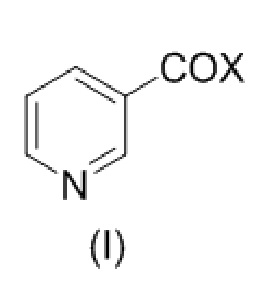 ,
,
где Х представляет собой –ОН, –N3 или бензотриазолил;
с образованием соединения (II)
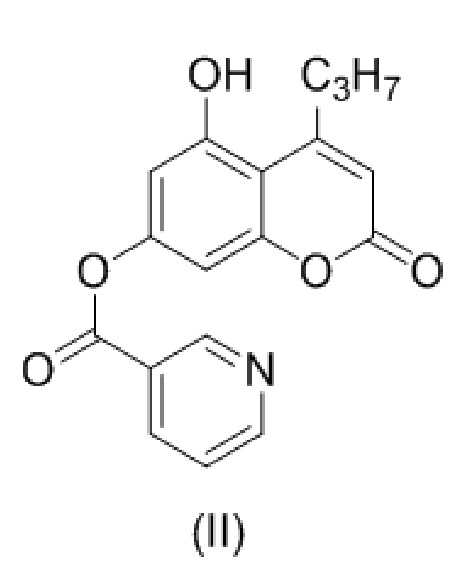 .
.
Стадия 2. Проводят взаимодействие соединения формулы (II) с соединением формулы (III)
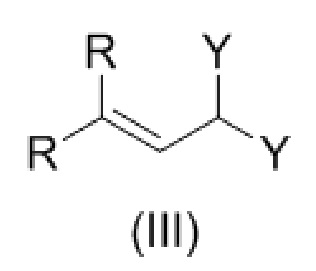 ,
,
где Y представляет собой OR, или два Y образуют вместе группу =O; и
R, в каждом случае независимо, представляет собой С1-6 алкил, в частности, метил или этил;
с образованием соединения (IV)
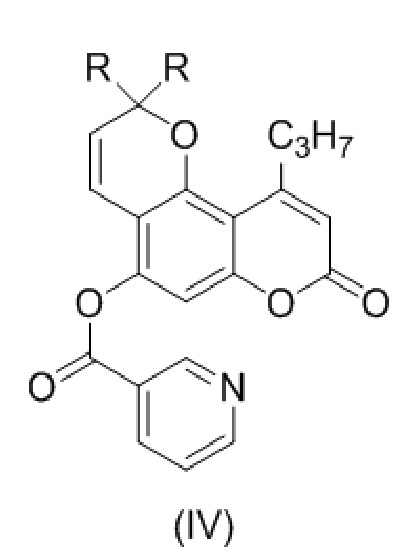 ,
,
где R такой как описан выше.
Стадия 3. Удаление защитной никотиноильной группы из соединения (IV) с кислотой или основанием с получением соединения 5.
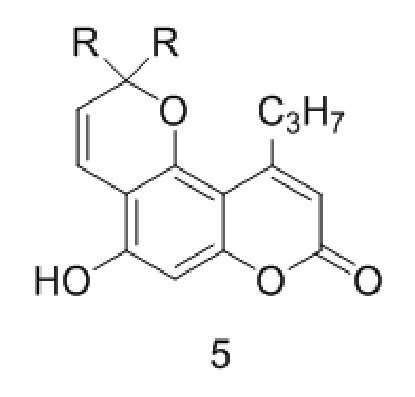 ,
,
где R такой как описан выше.
В другом варианте настоящее изобретение относится к соединению формулы (II)
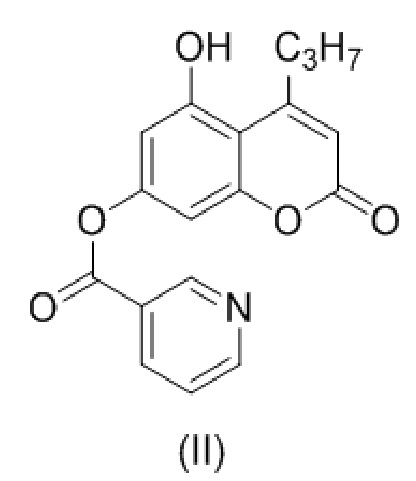 .
.
В еще одном варианте настоящее изобретение относится к соединению формулы (IV)
,
где R, в каждом случае независимо, представляет собой С1-6 алкил, в частности, метил или этил.
Предпочтительные варианты осуществления
Стадия 1
Соотношение соединений 1 и (I) может составлять от 1:5 до 5:1, однако наиболее предпочтительно соотношение составляет примерно 1:1.
Взаимодействие на стадии 1 можно проводить в инертном апротонном растворителе, в частности, в тетрагидрофуране, ацетоне, ацетонитриле, ДМФА, этилацетате или диоксане.
Взаимодействие на стадии 1 можно проводить в присутствии основания, в частности, органического амина; например, триэтиламина, N-метилморфолина, N-этилдиизопропиламина, 1,4-диазабицикло[2.2.2]октана, этилендиамина и т.п. При этом оличество основания может составлять от 0,01 до 10 эквивалентов. Наиболее предпочтительно использовать около 1 эквивалента основания, например, от 1,05 до 1,10 эквивалента основания.
Взаимодействие на стадии 1 можно проводить в присутствии агентов сочетания, таких как 1,3-дициклогексилкарбодиимид (DDC), гексафторфосфат азабензотриазол тетраметил-урония (HATU), 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC), 1,3-диизопропилкарбодиимид и т.п. Количество используемого агента сочетания не ограничено специально, но наиболее предпочтительно использовать 1,0–5,0 эквивалента.
Температура проведения реакции стадии 1 не ограничивается специальным образом, однако, с практической точки зрения, предпочтительная температура составляет 0–60°С, наиболее предпочтительно около 25°С.
Время реакции стадии 1 обычно составляет от 1 часа до 72 часов, наиболее предпочтительно от 6 до 12 часов.
Образующееся на стадии 1 соединение (II) предпочтительно отфильтровывают от раствора, содержащего побочные продукты и/или непрореагировавшие исходные соединения. Соединение 1, отфильтрованное из реакционной смеси предпочтительно является достаточно чистым, поэтому нет необходимости в его дополнительной очистке. Однако такая очистка может быть проведена с использованием стандартных методов, например, кристаллизации из органического растворителя.
Стадия 2
Взаимодействие на стадии 2 можно проводить без использования растворителя или в инертном апротонном высококипящем растворителе, в частности, в пиридине, ДМФА, толуоле, ксилоле или подобном.
Количество используемого соединения (III) специально не ограничено, наиболее предпочтительным количеством является 1-10 эквивалентов, например, 4–5 эквивалентов.
Температура проведения стадии 2 не ограничивается специальным образом, однако, с точки зрения технологии, предпочтительная температура составляет 50–200°С, наиболее предпочтительно 135–150 °С.
Время реакции стадии 2 обычно составляет от 5 минут до 2 часов, наиболее предпочтительно от 15 до 45 минут.
Образующуюся на стадии 2 реакционную смесь, содержащую соединение (IV), после охлаждения можно разбавить инертным органическим разбавителем, таким как ацетон или этилацетат в количестве от 1 до 5 объемов, после чего добавляют кислоту или ее раствор в органическом растворителе, таком как ацетон, этилацетат или С1-С6 алкиловый спирт. Через некоторое время из полученного раствора выпадает кристаллический осадок соли соединения (IV) с кислотой. Могут быть использованы различные органические и неорганические кислоты в количестве от 1 до 20 эквивалентов, однако предпочтительной кислотой является пикриновая кислота в количестве 1,0–2,0 эквивалента, которая образует кристаллический осадок соли соединения (IV). Полученная соль может быть собрана, например, фильтрованием и необязательно перекристаллизована из растворителя, такого как о-ксилол или 1,4-диоксан с получением очищенной соли (IV).
Стадия 3
Взаимодействие на стадии 3 можно проводить в подходящем инертном растворителе, в частности, в С1-6 спирте, воде, ДМФА, толуоле, ксилоле или подобном.
Используемым основанием может быть KOH, NaOH, NH3, метилат натрия, этилат натрия, триэтиламин и подобное.
Используемой кислотой может быть соляная кислота, трифторуксусная кислота, кислый полистиролсульфокислотный катионит и подобное.
Температура проведения стадии 2 не ограничивается специальным образом, однако, с точки зрения технологии, предпочтительная температура составляет 20–100°С, наиболее предпочтительно 25–70°С или 50–60°С.
Время реакции стадии 3 обычно составляет от 1 часа до 48 часов, наиболее предпочтительно от 3 до 12 часов, в частности 6 часов.
Полученное соединение 5 может быть выделено из реакционной смеси фильтрованием, экстракцией и т.п.
Полноту протекания реакции можно установить при помощи тонкослойной хроматографии (ТСХ), спектроскопии ядерного магнитного резонанса ЯМР или других общеизвестных методов.
Примеры
Представленные ниже примеры иллюстрируют некоторые предпочтительные варианты осуществления настоящего изобретения, но не ограничивают его.
Пример 1а
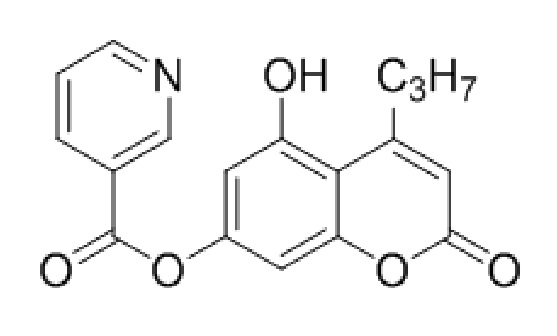
5-Гидрокси-4-пропилкумарин-7-ил никотинат
К раствору 5,7-дигидрокси-4-пропилкумарина (1 ммоль, 220 мг) и триэтиламина (1,1 ммоль, 111 мг) в 10 мл ацетона при перемешивании добавили 1-никотиноилбензотриазол (1,0 ммоль, 224 мг). Через сутки отфильтровали выпавший осадок 5-гидрокси-4-пропилкумарин-7-ил никотината и промыли ацетоном. Выход 78%.
Пример 1б
5-Гидрокси-4-пропилкумарин-7-ил никотинат
К раствору 5,7-дигидрокси-4-пропилкумарина (1 ммоль, 220 мг) и триэтиламина (1,1 ммоль, 111 мг) в 10 мл ацетона при перемешивании добавили 1-никотиноилазид (1,0 ммоль, 148 мг). Через сутки отфильтровали выпавший осадок 5-гидрокси-4-пропилкумарин-7-ил никотината.
Пример 1в
5-Гидрокси-4-пропилкумарин-7-ил никотинат
К раствору 5,7-дигидрокси-4-пропилкумарина (1 ммоль, 220 мг) и триэтиламина (1,1 ммоль, 111 мг) в 10 мл диоксана при перемешивании добавили никотиновую кислоту (1,0 ммоль, 123 мг) и 1,3-дициклогексилкарбодиимид (1,5 ммоль, 309 мг). Через неделю выдерживания реакционной смеси при комнатной температуре отфильтровали выпавший осадок 5-гидрокси-4-пропилкумарин-7-ил никотината и дициклогексилмочевины и перекристаллизовали из этилового спирта с получением чистого 5-гидрокси-4-пропилкумарин-7-ил никотината.
Т.пл. 229–231°С.
1Н-ЯМР: (ДМСО-d6+CCl4) 10,88 (с, 1H, OH-5), 9,25 (д, J=1,5 Гц, 1H, H2’), 8,84 (дд, J=1,5 Гц, J=4,8 Гц, 1H, H6’), 8,44 (дт, Jд=7,6 Гц, Jт=1,9 Гц, 1H, H4’), 7,59 (дд, J=4,9 Гц, J=7,6 Гц, 1H, H5’), 6,73 (с, 2H, H6, H8), 5,98 (с, 1H, H3), 2,96 (м, 2H, CH2), 1,70 (м, 2H, CH2), 1,04 (м, 3H, CH3);
ИК: 1082, 1150, 1290, 1434, 1613, 1739 см-1.
Пример 2
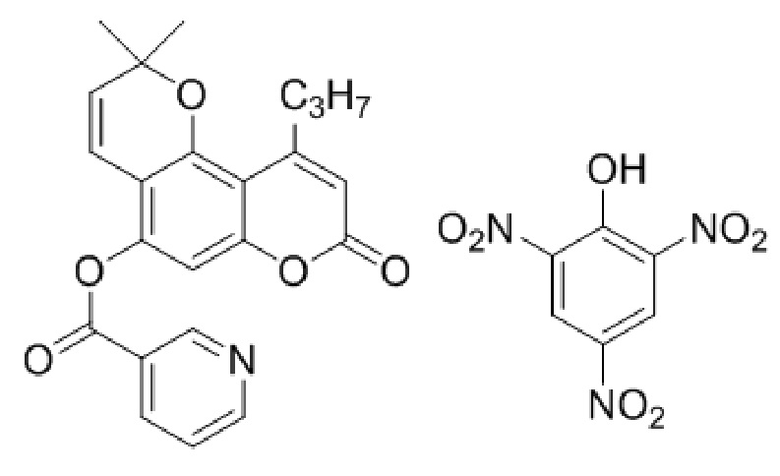
Пикрат 2,2-диметил-8-оксо-10-пропил-2H,8H-пирано[2,3-f]хромен-5-ил никотината
Смесь 5-гидрокси-4-пропилкумарин-7-ил никотината (2 ммоль, 650 мг) и диэтилового ацеталя акролеина (1,266 г, 8 ммоль) нагревали при 145°С с перемешиванием в течение 40 минут. Затем реакционную смесь разбавляли 10 мл этилацетата и добавляли раствор пикриновой кислоты (458 мг, 2 ммоль) в 10 мл этилацетата. Выпавшие светло-желтые кристаллы пикрата были отфильтрованы и перекристаллизованы из о-ксилола или диоксана. Выход 464 мг, 45%.
1H-ЯМР (ДМСО-d6+CCl4): 9,38 (уш с, 1H), 8,98 (уш с, 1H), 8,84 (уш с, 1H), 8,65–8,67 (м, 1H), 8,63 (с, 2H), 7,77–7,80 (м, 1H), 6,90 (с, 1H), 6,72 (с, 2H), 6,46 (д, J=10 Гц, 1H), 6,08 (с, 1H), 5,77 (д, J=10 Гц, 1H), 2,92–2,96 (м, 2H, CH2), 1,68–1,73 (м, 2H), 1,55 (с, 6H), 1,06–1,10 (м, 3H).
Пример 3
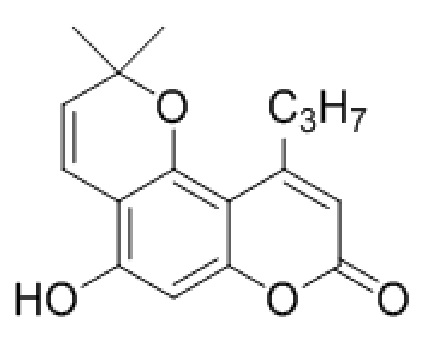
5-Гидрокси-2,2-диметил-10-пропил-2H,8H-пирано[2,3-f]хромен-8-он
Пикрат 2,2-диметил-8-оксо-10-пропил-2H,8H-пирано[2,3-f]хромен-5-ил никотината (1 ммоль, 621 мг) смешивали с 20 мл метанола и полистиролсулфонатным катионитом, нагревали при 55°С с перемешиванием в течение 8 часов. Затем реакционную смесь фильтровали и фильтрат разбавляли 60 мл воды. Выпавший осадок продукта отфильтровывали и сушили на воздухе. Выход 281 мг, 98%.
1H-ЯМР (ДМСО-d6+CCl4): 10,73 (с, 1H), 6,56 (д, J=9,9 Гц, 1H), 6,33 (с, 1H), 5,90 (с,1H), 5,64 (д, J=9,9 Гц, 1H, H-3), 2,83–2,77 (м, 2H), 1,66–1,51 (м, 2H), 1.43 (с, 6H), 1,10–0,91 (м, 3H).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ СЕЛЕКТИВНОГО ВВЕДЕНИЯ ЗАЩИТНЫХ ГРУПП В ПРОИЗВОДНЫЕ РЕЗОРЦИНА | 2019 |
|
RU2738408C1 |
| МЕХАНОХИМИЧЕСКИЙ СПОСОБ СИНТЕЗА КУМАРИНОВ | 2022 |
|
RU2799566C1 |
| СИНТЕЗ ПРЕДШЕСТВЕННИКА ИНГИБИТОРА ПРОТЕАЗЫ | 2006 |
|
RU2421459C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНИЛПЕНТАМИДА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ ВИЧ-ПРОТЕАЗЫ | 1992 |
|
RU2131416C1 |
| СПОСОБЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ МАКРОЦИКЛИЧЕСКИХ ИНГИБИТОРОВ ПРОТЕАЗЫ HCV | 2011 |
|
RU2628081C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИПЕРАЗИНИЛПЕНТАНАМИДА И ПРОИЗВОДНОЕ ПИПЕРАЗИНИЛПЕНТАНАМИДА | 1992 |
|
RU2171254C2 |
| ПРОИЗВОДНЫЕ ГИДРОКСАМОВЫХ КИСЛОТ И ГИДРОКСИМОЧЕВИН И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2108324C1 |
| СПОСОБ СИНТЕЗИРОВАНИЯ НОВОГО ХИРАЛЬНОГО ЛИГАНДА, ХЕЛАТА МЕТАЛЛА, РАЗЛИЧНЫХ НЕПРИРОДНЫХ АМИНОКИСЛОТ, МАРАВИРОКА И ЕГО ОСНОВНЫХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2017 |
|
RU2727723C1 |
| БЕНЗОКСАЗИН-ОКСАЗОЛИДИНОНОВОЕ СОЕДИНЕНИЕ, ЗАМЕЩЕННОЕ АЗОТСОДЕРЖАЩИМ ГЕТЕРОЦИКЛОМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2018 |
|
RU2744784C1 |
| КОМБИНАЦИЯ ИНГИБИТОРА ВИЧ-ПРОТЕАЗЫ С ДРУГИМИ АНТИВИРУСНЫМИ СОЕДИНЕНИЯМИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ | 1994 |
|
RU2139052C1 |
Изобретение относится к области органической химии, а именно к способу получения соединения 5, соединение общей формулы (II) 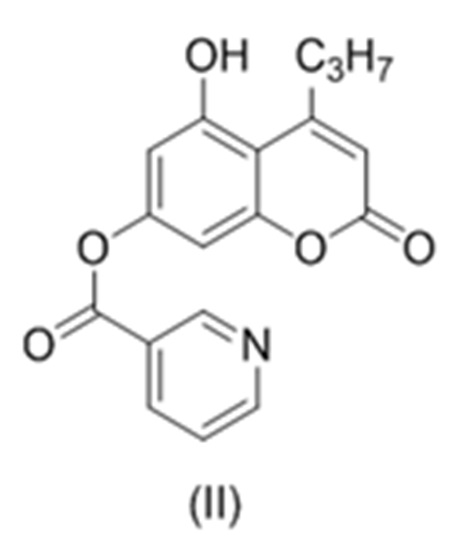 , соединение общей формулы (IV)
, соединение общей формулы (IV) 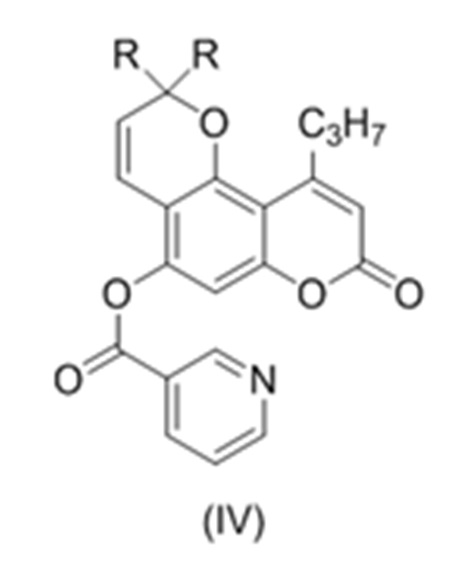 , где R, в каждом случае независимо, представляет собой С1-6 алкил. Технический результат: получен улучшенный способ получения промежуточных продуктов для синтеза аналогов анти-ВИЧ агента каланолид А и его аналогов, а также получены новые гетероциклические соединения. 3 н. и 7 з.п. ф-лы, 1 ил., 3 пр.
, где R, в каждом случае независимо, представляет собой С1-6 алкил. Технический результат: получен улучшенный способ получения промежуточных продуктов для синтеза аналогов анти-ВИЧ агента каланолид А и его аналогов, а также получены новые гетероциклические соединения. 3 н. и 7 з.п. ф-лы, 1 ил., 3 пр.
1. Способ получения соединения 5, включающий следующие стадии:
стадия 1: приводят в контакт в инертном апротонном растворителе в присутствии основания соединение формулы 1 и соединение формулы (I) с образованием соединения формулы (II):
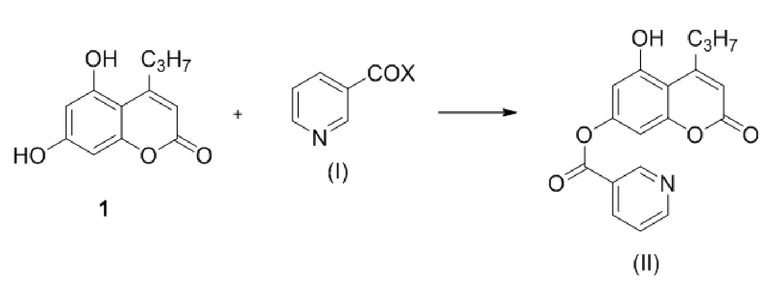 ,
,
где Х представляет собой –ОН, –N3 или бензотриазолил;
стадия 2: приводят в контакт в инертном растворителе или без растворителя соединение формулы (II) и соединение формулы (III) с образованием соединения формулы (IV):
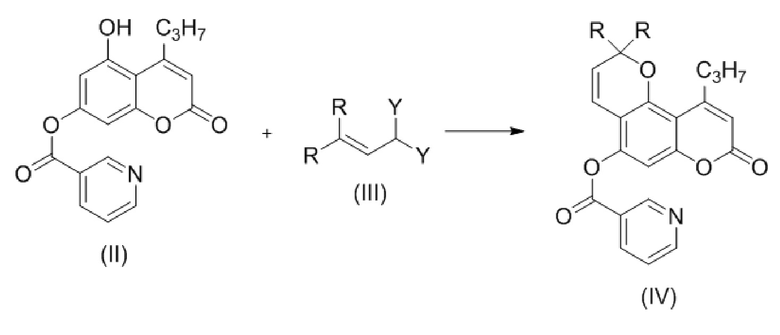 ,
,
где Y представляет собой OR, или два Y образуют вместе группу =O; и
R, в каждом случае независимо, представляет собой С1-6 алкил, в частности метил или этил;
после завершения реакции к реакционной смеси, содержащей соединение (IV) добавляют кислоту или ее раствор в органическом растворителе с получением кристаллического осадка соли соединения (IV) с кислотой;
стадия 3. приводят в контакт в полярном растворителе соединение формулы (IV) с кислотой или основанием с получением соединения 5:
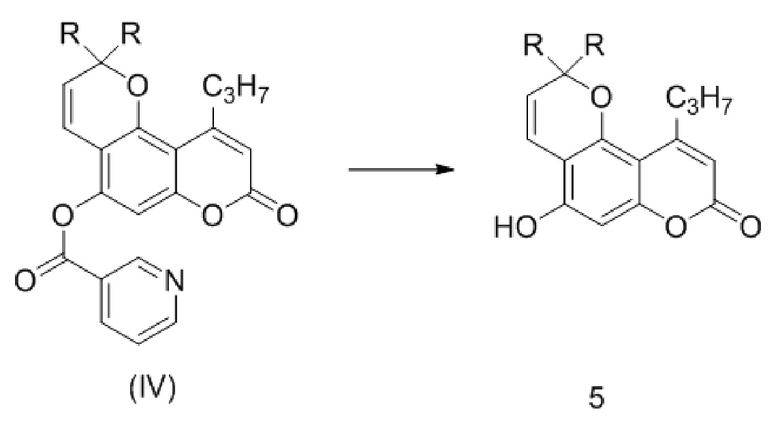
где R, в каждом случае независимо, представляет собой С1-6 алкил.
2. Способ по п.1, где на стадии 1 соотношение соединений 1 и (I) составляет примерно 1:1, растворителем является ацетон или диоксан, основанием является триэтиламин.
3. Способ по п.1 или 2, где стадию 1 проводят в присутствии агента сочетания, такого как 1,3-дициклогексилкарбодиимид (DDC) или 1-этил-3-(3-диметиламинопропил)карбодиимид.
4. Способ по любому из пп.1–3, где взаимодействие на стадии 2 проводят без использования растворителя при температуре 135–150°С.
5. Способ по любому из пп.1–4, где на стадии 2 кислотой является пикриновая кислота.
6. Способ по любому из пп.1–5, где на стадии 3 кислотой является кислый полистиролсулфокислотный катионит.
7. Способ по любому из пп.1–6, где стадию 3 проводят в С1-С6 алкиловом спирте, в частности в метаноле при температуре 50–60°С.
8. Соединение формулы (II)
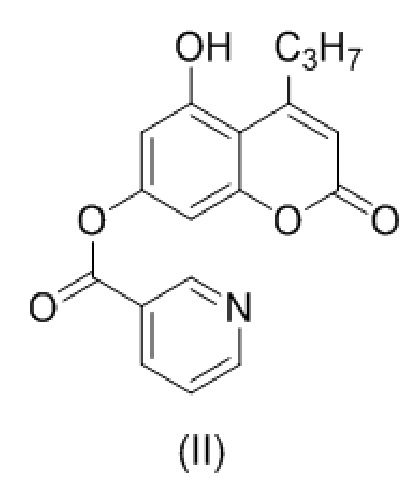 .
.
9. Соединение формулы (IV)
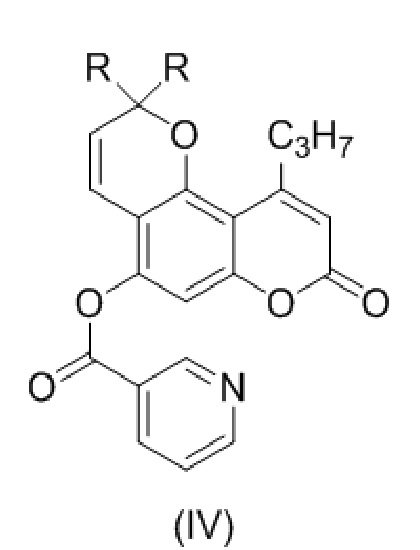 ,
,
или его соль;
где R, в каждом случае независимо, представляет собой С1-6 алкил.
10. Соединение по п.9, где R представляет собой метил.
| US 6313320 B1, 06.11.2001 | |||
| Устройство для обмотки лентой предметов | 1979 |
|
SU956356A1 |
| ПРОИЗВОДНЫЕ КУМАРИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1997 |
|
RU2135490C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3-АРИЛ-1Н-БЕНЗО[f]ХРОМЕНОВ | 2014 |
|
RU2597363C2 |

