Изобретение относится к способам получения перфторалкилгалогенидов, а именно перфторалкилйодидов и бромидов, которые широко используются для получения различных соединений, содержащих перфорированные алифатические заместители, в частности лекарственных препаратов, разнообразных смазок и полимерных продуктов.
Известно большое количество различных способов получения перфторалкилйодидов и бромидов, в частности присоединение галогенов к ненасыщенным перфорированным соединениям, реакция обмена галогенов в перфторалкилгалогенидах, теломеризация тетрафторэтилена с перфторалкилйодидами [Chen Q. et al., Joumal of Fluorine Chemistry, 36 (4), 483-489 (1987); патент CN 103880588 (2014); патентная заявка JP 2013221026 (2013)], декарбоксилирование серебряных солей и других производных перфторкарбоновых кислот [патентная заявка JP 2010018533 (2010); патент CN 102464569 (2012); Szlavik Z. et at., Tetrahedron Letters, 38 (50), 8757-8760 (1997)].
Особый интерес представляет получение перфторалкилйодидов и бромидов из перфторалкилсульфонилфторидов вследствие того, что они доступны электрохимическим фторированием - одним из основных промышленных способов введения фтора в органические молекулы, благодаря чему, в частности, являются важным сырьем для получения разнообразных фторсодержащих органических соединений.
В патенте CN 107556202 (2018) описан двухстадийный способ получения бромидов и йодидов из перфторалкилсульфонилфторидов, включающий взаимодействие сульфонилфторидов с восстановителем (в качестве которого выступает дитионит, боргидрид или сульфит натрия) с образованием натриевых солей соответствующих сульфиновых кислот с выходом около 90% и последующее галогенирование их бромом или йодом с выходом 75-85%. Получать перфторалкилгалогениды можно и не выделяя промежуточные продукты («one pot» синтез).
В патенте US 6391948 (2002) раскрывается способ получения перфтороктилйодида с выходом около 80% (с учетом 90% конверсии), заключающийся в гидролизе перфтороктилсульфонилфторида водой при нагревании в диоксане и последующем взаимодействии образовавшейся сульфокислоты с боратом натрия, калийалюминийсульфатом и йодом при нагревании в воде.
Недостатком всех рассмотренных выше способов является необходимость использования свободных галогенов, представляющих собой токсичные вещества с сильно выраженным удушающим и поражающим ткани действием, которые относятся ко Ч классу опасности (высокоопасные вещества) по ГОСТ 12.1.007-76 (действующему в настоящее время).
В патентной заявке US 185355 (2007) описан способ получения перфторалкилбромидов и йодидов взаимодействием перфторалкил-сульфонилхлоридов и фторидов с соответствующими галогенидами щелочных металов в ДМСО, однако если с сульфонилхлоридами выход реакции близок к количественному, то с сульфонилфторидами он составляет не более 60%.
Наиболее близким к предлагаемому способу является способ, описанный в патенте RU 2359953 (2009). Он включает две стадии, первая из которых - взаимодействие перфтороктилсульфонилфторида с гидразингидратом в апротонном полярном органическом растворителе при температуре, не превышающей 50°С, вторая - взаимодействие полученного продукта с соответствующим свободным галогеном и йодатом калия в том же органическом растворителе при температуре 90-120°С. В качестве растворителя используют N-метилпирролидон, выходы соответствующих галогенидов составляют около 80% в пересчете на чистый продукт.
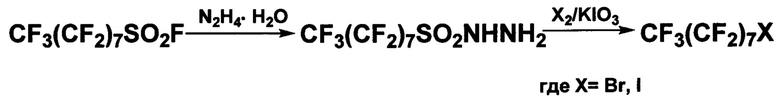
К недостатку данного способа относится использование в качестве галогенирующих агентов свободных йода и брома - дорогих и, как уже отмечалось выше, ядовитых веществ (класс опасности ООН: 8 для брома и 8,3 для йода; класс опасности по ГОСТ: II).
Задачей настоящего изобретения является разработка способа получения перфторалкилбромидов и йодидов, позволяющего исключить использование высокотоксичных галогенирующих агентов, таких, как свободные бром и йод.
Задача решается способом получения перфторалкилйодидов и бромидов, включающим взаимодействие перфторалкилсульфонилфторидов с гидразингидратом в апротонном полярном растворителе и последующее галогенирование промежуточных продуктов, причем продукт взаимодействия перфторалкилсульфонилфторида с гидразингидратом галогенируют соответствующей галогенводородной кислотой в присутствии неорганического окислителя в водной среде.
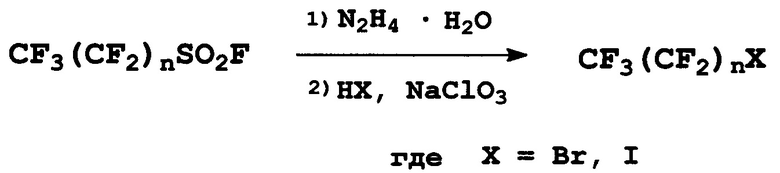
Промежуточный продукт взаимодействия перфторалкилсульфонилфторида с гидразингидратом можно не выделять («one pot»-синтез), а можно выделить в свободном от растворителя и образовавшихся солей виде и затем подвергнуть взаимодействию с галогенводородной кислотой и неорганическим окислителем в воде.
В качестве апротонного полярного растворителя (ацетонитрил, диметилформамид, N-метилпирролидон и др.) предпочтительно использовать ацетонитрил, в качестве неорганического окислителя (хлорат калия или натрия, йодат калия или натрия и др.) - хлорат натрия.
Полученные предложенным способом целевые перфторалкилгалогениды выделяют с выходом около 90%, разбавляя реакционную смесь подкисленной водой и собирая образующийся нижний слой.
Предложенным способом могут быть получены в том числе и перфторалкилбромиды и перфторалкилйодиды, содержащие в алкильной цепочке атомы кислорода, а также алкоксильные заместители.
Способ по изобретению включает две стадии:
1. Взаимодействие перфторалкилсульфонилфторида с гидразингидратом в ацетонитриле.
2. Взаимодействие полученного интермедиата с концентрированной галогенводородной кислотой и водным раствором хлората натрия.
Гидразингидрат растворяют в ацетонитриле и к полученному раствору при перемешивании по каплям прибавляют перфторалкилсульфонилфторид, поддерживая температуру реакционной смеси в интервале 20-25°С. Перемешивание при этой температуре продолжают до окончания выделения газа.
Далее к реакционной смеси приливают концентрированную галогенводородную кислоту и постепенно, при перемешивании, прибавляют водный раствор хлората натрия. Прибавление ведут, постепенно повышая температуру, до прекращения выделения газа. Для получения перфторалкилбромидов достаточно температуры 20-30°С, для получения перфторалкилйодидов необходима более высокая температура (сравни примеры 1 и 2). Температура процесса йодирования различается в зависимости от структуры целевого йодида. Так, при получении CF3(CF2)7I температура увеличивается от 45-60 до 65°С (пример 2), а при получении CH3OCF2CF(CF3)OCF2CF(CF3)OCF2CF2I - от 85-90 до 95-100°С (пример 3).
Промежуточный продукт взаимодействия перфторалкилсульфонилфторида с гидразингидратом можно выделить в свободном от ацетонитрила и образовавшихся солей виде выливанием реакционной смеси в разбавленную соляную кислоту и отделением нижнего слоя, который затем подвергают взаимодействию с галогенводородной кислотой и хлоратом натрия в воде.
Полученный перфторалкилгалогенид выделяют, выливая реакционную смесь в подкисленную соляной кислотой воду и собирая образовавшийся нижний слой.
Несомненным преимуществом предлагаемого способа является использование в качестве галогенирующих агентов доступных йодисто- и бромистоводородной кислот, которые менее опасны, чем высотоксичные йод и бром, применяемые в прототипе: согласно ГОСТ 12.1.007-76, указанные кислоты относятся к умеренно опасным веществам (класс опасности III), тогда как соответствующие галогены - к высокоопасным веществам (класс опасености II).
Техническим результатом изобретения является способ получения перфторалкилйодидов и бромидов из доступного сырья, позволяющий исключить использование высокоопасных галогенирующих агентов и не требующий выделения промежуточных продуктов.
Осуществление изобретение иллюстрируется следующими примерами.
Сигналы 19F и 1H ЯМР-спектров приведены в миллионных долях в шкале δ относительно CFCl3 (в случае ядер 19F) и ТМС (в случае ядер 1Н).
Пример 1. Получение перфтороктилбромида
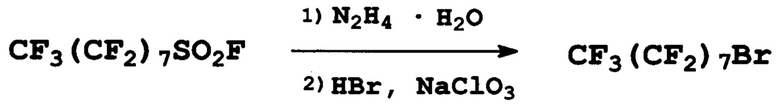
К 32,3 г (0,65 моль) гидразингидрата в 400 мл ацетонитрила, поддерживая температуру 20-25°С, при перемешивании прибавили 150 г (0,30 моль) перфтороктилсульфонилфторида. Смесь перемешивали до прекращения газовыделения, затем прилили 101 г 48%-ной бромистоводородной кислоты (0,60 моль HBr) и при перемешивании прибавили по каплям 63 г (0,60 моль) NaClO3 в 150 мл воды при постепенном повышении температуры от 20 до 30°С. Под конец прибавления смесь пожелтела и разделилась на три слоя. После добавления около 500 мл воды и 20 мл концентрированной соляной кислоты произошло разделение на два слоя. Нижний слой отделили, получили 138 г (92%) перфтороктилбромида. Т. кип. 143°С (лит.: т. кип. 143°С [O.N. Chechina, V.V. Berenblit, S.V. Sokolov, Fluorine Notes, 2 (81), 5-6 (2012)]).
Спектр 19F ЯМР (CDCl3): -65,81 т (2F, CF2Br), -83,87 д (3F, CF3), - 119,56 м (2F, CF2), -123,23 м (2F, CF2), -123,99 м (4F, 2CF2), -124,95 м (2F, CF2), -128,63 м (2F, CF2).
Лит. спектр 19F ЯМР (без растворителя): -64,0; -80,5; -117,5; -121,3; -122,2; -123,2; -126,8 [Wolfgang Robien / University of Vienna (Spectra! Data were obtained from Wiley Subscription Services, inc. (US)].
Пример 2. Получение перфтороктилйодида
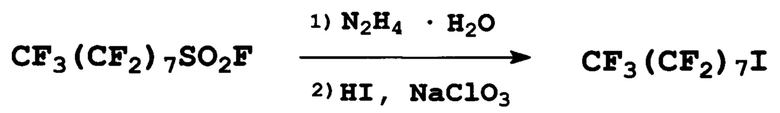
К 40 г (0,80 моль) гидразингидрата в 400 мл ацетонитрила, поддерживая температуру 20-25°С, при перемешивании прибавили по каплям 150 г (0,30 моль) перфтороктилсульфонилфторида. Смесь перемешивали до прекращения газовыделения, после чего прилили 101 г концентрированной йодистоводородной кислоты (0,36 моль Hl) и при перемешивании прибавили по каплям 79 г (0,61 моль) NaClO3 в 120 мл Н2О: первую половину при 45-60°С, остаток при 65°С. Хлорат нужно добавлять медленно, поддерживая равномерное, средней интенсивности газовыделение. После прибавления всего количества хлората натрия смесь перемешивали еще час, за это время наблюдалось поднятие температуры до 75°С и газовыделение. Реакционную смесь охладили до комнатной температуры и прилили около 1 л воды и 20 мл концентрированной соляной кислоты. Нижний слой отделили, промыли водным раствором Na2SO3 до обесцвечивания, получили 142 г (выход 86%) перфтороктилйодида. Т кип. 160-161°С (лит.: т. кип. 160-161°С [Tesevic, V., Green Cnemistry, 7 (12), 833-836 (2005)]).
19F ЯМР-спектр (ацетон-d6):-65,54 т (2F, CF2I), -83,01 д (3F, CF3), -115,15 м (2F, CF2), -122,57 м (2F, CF2), -123,50 м (4F, 2CF2), -124,40 м (2F, CF2), -127,96 м (2F, CF2).
Лит. 19F ЯМР-спектр (CDCl3): -59,2; -80,8; -113,1; -120,9; -121,9; -122,7; -126,2 [Cao, Hai-Ping Chen, Qing-Yun Joumal of Fluorine Chemistry, 128 (10), 1187-1190 (2007)].
Пример 3. Получение 1-йод-8-метоксиперфтор-4,7-диметил-3,6-диоксаоктана
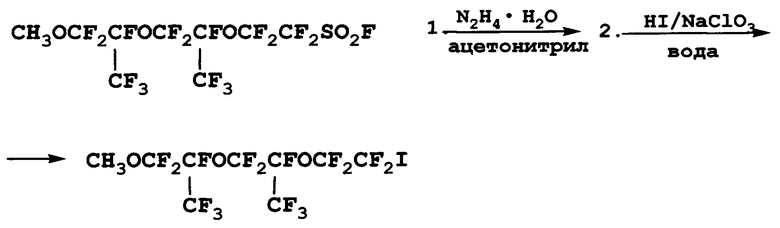
К 620 мл ацетонитрила добавили 65 г (1,30 моль) гидразингидрата. Раствор нагрели на водяной бане до температуры 40°С и из капельной воронки за 3 ч при перемешивании прибавили по каплям 300 г (0,55 моль) 8-метоксиперфтор-4,7-диметил-3,6-диоксаоктилсульфонилфторида. В ходе прибавления реагента наблюдалось выделение газа. Реакционную смесь перемешивали при температуре 40-50°С в течение 3 ч, после чего оставили на ночь. Затем при перемешивании и охлаждении ледяной водой при температуре 20°С реакционную массу вылили в 2,5 л 15%-ной соляной кислоты. Нижний слой массой 365 г отделили и использовали в следующей стадии без очистки.
К 500 мл воды добавили продукт, полученный на предыдущей стадии, и из капельной воронки прилили 339 г 46%-ной йодистоводородной кислоты (1,22 моль Hl). Температуру смеси довели до 85-90°С и при перемешивании по каплям прибавили раствор 127,8 г (1,20 моль) NaClO3 в 195 мл воды. Наблюдалось газовыделение, интенсивность которого контролировали скоростью подачи хлората натрия. По окончании прибавления всего количества раствора NaClO3 реакционную смесь перемешивали при температуре 95-100°С в течение 2 ч до прекращения газовыделения. После охлаждения реакционная масса расслоилась на три слоя. Два нижних слоя отделили, промыли равным объемом 10%-ной соляной кислоты (около 500 мл). Нижний слой массой 290 г отделили и перегнали с водяным паром с насадкой Дина-Старка. Дистиллят массой 210 г чистотой, по данным ГХ, 84% ректифицировали. Получили 183 г 1-йод-8-метоксиперфтор-4,7-диметил-3,6-диоксаоктана чистотой, по данным ГХ и 19F ЯМР-спектроскопии, 96%. Т. кип. 95-96°С/40 Торр. Выход суммарный 56%.
19F ЯМР-спектр (без растворителя): -65,36 д (2F, CF2I), -81,65 м (6F, 2CF3), -80…-82 кв (2F, CFAFBO), -84,95 м (2F, CF2), -89,74 м (2F, CF2), -145,95 м (1F, CF), -146,60 м (1F, CF).
1Н ЯМР-спектр (без растворителя): 3,59 с (3Н, ОСН3).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПЕРФТОРАЛКИЛЙОДИДОВ | 2020 |
|
RU2748928C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕНТАФТОРЙОДЭТАНА | 2017 |
|
RU2642789C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5-(ФТОРСУЛЬФОНИЛ)ПЕРФТОРПЕНТИЛВИНИЛОВОГО ЭФИРА | 2024 |
|
RU2840850C1 |
| СПОСОБ ПОЛУЧЕНИЯ МОНО- И ДИГИДРОКСИПОЛИФТОРБЕНЗОЛОВ | 2013 |
|
RU2536872C1 |
| α-БРОМ-ω-ГАЛОГЕНПЕРФТОРПОЛИЭФИРЫ В КАЧЕСТВЕ ОСНОВЫ ГАЗОТРАНСПОРТНЫХ КОМПОЗИЦИЙ МЕДИКО-БИОЛОГИЧЕСКОГО НАЗНАЧЕНИЯ | 2019 |
|
RU2707081C1 |
| ОГНЕПОДАВЛЯЮЩИЕ СОСТАВЫ, СОДЕРЖАЩИЕ НЕНАСЫЩЕННЫЕ ФТОРУГЛЕРОДЫ, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2434659C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИМЕТИЛ(ТРИФТОРМЕТИЛ)СИЛАНА | 2014 |
|
RU2550139C1 |
| Способ получения фенилперфторалкилсульфидов | 1991 |
|
SU1773909A1 |
| ВИНИЛСУЛЬФОНОВЫЙ АНГИДРИД, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ВИНИЛСУЛЬФОНИЛФТОРИДА | 2019 |
|
RU2762629C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕНТАФТОРПРОПИОНОВОЙ КИСЛОТЫ | 2020 |
|
RU2758675C1 |
Изобретение относится к способу получения перфторалкилйодидов или бромидов из перфторалкилсульфонилфторидов, включающему взаимодействие последних с гидразингидратом в апротонном полярном растворителе - ацетонитриле и последующее галогенирование промежуточных продуктов, причем продукт взаимодействия перфторалкилсульфонилфторида с гидразингидратом без предварительного его выделения из реакционной смеси галогенируют соответствующей галогенводородной кислотой в присутствии неорганического окислителя – хлората натрия в водной среде. 3 пр.
Способ получения перфторалкилйодидов или бромидов из перфторалкилсульфонилфторидов, включающий взаимодействие последних с гидразингидратом в апротонном полярном растворителе - ацетонитриле и последующее галогенирование промежуточных продуктов, отличающийся тем, что продукт взаимодействия перфторалкилсульфонилфторида с гидразингидратом без предварительного его выделения из реакционной смеси галогенируют соответствующей галогенводородной кислотой в присутствии неорганического окислителя – хлората натрия в водной среде.
| СПОСОБ ПОЛУЧЕНИЯ ПЕРФТОР- C-CАЛКИЛГАЛОГЕНИДОВ | 2007 |
|
RU2359953C1 |
| US 6391948 B1, 21.05.2002 | |||
| CN 107556202 A, 09.01.2018 | |||
| Способ получения перфторалкилбромидов | 2015 |
|
RU2606382C2 |

