Область техники, к которой относится изобретение
Изобретение относится к области химико-фармацевтической промышленности и касается новых способов получения кристаллической формы 8 софосбувира. Предложенные способы получения характеризуется высокими выходами и чистотой полученной формы, а также простотой осуществления для реализации в промышленных масштабах. Полученные по данным способам кристаллическая форма 8 софосбувира может использоваться в качестве противовирусного средства в терапии вирусного гепатита С.
Уровень техники
Гепатит С представляет собой заболевание печени, вызываемое вирусом гепатита С (HCV): вирус может приводить к развитию как острого, так и хронического гепатита с разной степенью тяжести - от легкой болезни, длящейся несколько недель, до серьезной пожизненной болезни. В РФ, по разным оценкам, доля людей, которые встречались с вирусом гепатита С, составляет от 2 до 4 процентов, а хроническим гепатитом С больны около 3,5 миллиона человек. Примерно у 30% инфицированных вирус исчезает самопроизвольно без какого-либо лечения в течение шести месяцев после заражения. У остальных 70% инфицированных развивается хроническая инфекция, среди пациентов с хронической инфекцией HCV риск развития цирроза печени в течение следующих 20 лет составляет от 15% до 30%.
Заболевание вызывает РНК-содержащий вирус из семейства Flaviviridae, обнаруженный учеными в 1989 году. Вирус размножается в основном в клетках печени (гепатоцитах). Проникая в них, он использует внутриклеточный механизм репликации (самокопирования генома): каждая вирусная частица производит в день до 50 реплик, которые впоследствии выходят за пределы клетки-хозяина. Основная цель лечение гепатита С - эррадикация вируса (удаление вируса из организма). При невозможности эррадикации целями лечения могут быть: прекращение или замедление воспалительных процессов в печени, предотвращение перехода заболевания в цирроз или рак. Выбор лечения зависит от многих факторов: пола, возраста, вирусной нагрузки, состояния печени, характера течения заболевания (острый, хронический). Если риск развития цирроза высокий, то лечение должно быть назначено как можно скорее. Основным методом лечения гепатита С в настоящее время является противовирусная терапия (ПВТ) с помощью препаратов прямого действия (ПППД), основными мишенями которых являются вирусные белки, необходимые для его размножения (эффективность более 90-95%). ВОЗ рекомендует использовать пангенотипные комбинации ПППД для лечения лиц с хронической HCV-инфекцией в возрасте 18 лет и старше, в частности комбинации софосбувира с велпатасвиром и даклатасвиром в течение 12 недель, для лечения подростков от 12 до 17 лет рекомендуются комбинации софосбувира с ледипасвиром или рибавирином в течение 12 или 24 недель в зависимости от генотипа вируса. Таким образом, применение различных лекарственных комбинаций софосбувира является основой современного подхода к лечению вирусного гепатита С у взрослых и детей старше 12 лет.
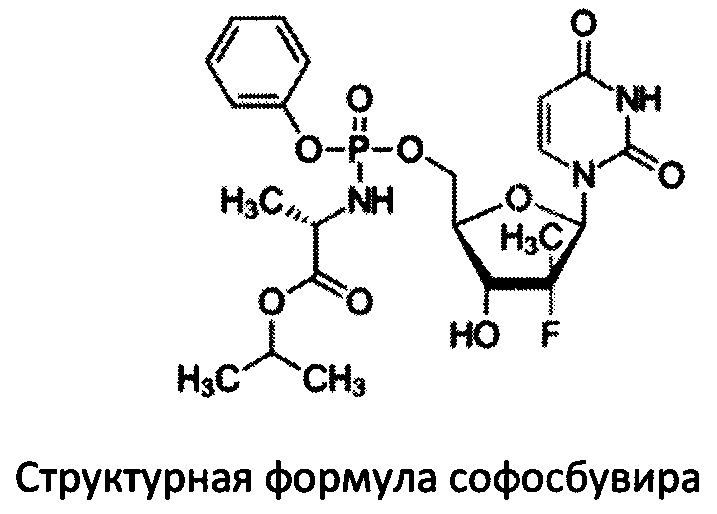
Софосбувир (изопропил-(2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-диоксипиримидин-1-ил)-4-фторо-3-гидрокси-4-метил-тетрагидрофуран-2-ил]-метокси-фенокси-фосфорил]-амино]пропаноат) (см. WO2008/121634, RU2478104) является пангенотипическим ингибитором РНК-зависимой РНК-полимеразы NS5B HCV, необходимой для репликации вируса. Это нуклеотидное пролекарство, которое подвергается внутриклеточному метаболизму, в процессе которого формируется фармакологически активный аналог - 2'-дезокси-2'-α-фтор-β-С-метилуридин-5'-трифосфат (GS-461203). С помощью NS5B полимеразы GS-461203 может встраиваться в строящуюся цепочку РНК HCV и действовать как обрыватель цепи (Fung Α., et. al., July 2014, Antimicrobial Agents and Chemotherapy. 58 (7): 3636 - 45).
Единственный препарат с МНН софосбувир зарегистрирован в России в марте 2016 года под торговым названием «Совальди» (номер регистрационного удостоверения ЛП-003527; дата выдачи 25.03.2016 года). Ввиду своей высокой эффективности в отношении многих генотипов HCV, сопряженной с низкой частотой возникновения побочных эффектов, препарат пользуется высоким спросом среди конечных потребителей, несмотря на высокую стоимость. Таким образом, существует острая необходимость в разработке и выводе в гражданский оборот воспроизведенных препаратов софосбувира, обладающих фармакологическими и технологическими свойствами, не уступающими оригинальному препарату.
Известно (Η.М. Mande, 2017, Acta Crystallographica Section A Foundations and Advances. 73. P. 434), что софосбувир проявляет полиморфизм. Сообщается, что софосбувир имеет более чем 15 полиморфов, включая аморфную форму.
Полиморфные формы 1, б, 7 и 8 являются несольватированными формами, тогда как формы 2, 3, 4 и 5 являются нестабильными сольватами хлористого метилена, хлороформа, ацетонитрила и анизола соответственно. Исходя из представленных выше данных очевидно, что для проведения технологического процесса изготовления твердых лекарственных форм подходят исключительно стабильные несольватированные формы софосбувира. Применение формы 1 из-за ее гигроскопичности имеет ряд недостатков, так при повышенной влажности воздуха форма 1 имеет тенденцию к образованию маслянистого материала, следовательно, препараты, содержащие форму 1 требуют мер предосторожности для защиты от влаги, делая тем самым процессы разработки, упаковки и хранения сложными и дорогостоящими. Кроме того, использование кристаллической формы 1 в составе воспроизведенных препаратов затруднено на территории РФ из-за имеющегося патентного ограничения (ЕА26731) до 20.05.2030 года.
На сегодняшний день наиболее технологически подходящей кристаллической формой софосбувира является форма 6 (WO 2011123645), она входит в композицию оригинального препарата «Совальди» (WO 2013082003; ЕА27296). Однако, использование в воспроизведенных препаратах на территории РФ кристаллической формы 6 затруднено из-за имеющегося патентного ограничения (ЕА26341) до 31.03.2031 года, что обуславливает высокую цену препарата в РФ (зарегистрированная цена составляет 126000 рублей за упаковку из 28 таблеток, минимально необходимый 12 недельный курс лечения обойдется примерно в 380000 рублей), в то время как в странах (например, Индия), в которых налажено производство воспроизведенных препаратов стоимость курса приближается к 100$ США (https://undark.org/2018/02/07/hepatitis-c-sub-sofosbuvir-india/). Таким образом, существует острая необходимость в получении патентно чистых форм софосбувира, отличающихся стабильностью, фармакологическими и техническими свойствами, не уступающими форме 6. С этой точки зрения наибольший интерес представляют формы 7 и 8, не имеющие патентной защиты на территории РФ (заявка ЕА201691029 отозвана без возможности восстановления). Данные формы не уступают форме 6, а даже несколько опережают в плане скорости растворения (см. ЕР3524234).

В WO 2015099989 в качестве способа получения кристаллической формы 8 раскрывается перекристаллизация софосбуфира из изопропилацетата с добавлением затравочных кристаллов. В тексте патентной заявки нет данных о выходе целевого продукта, полученного описанным методом. Попытка авторов настоящего изобретения воспроизвести экспериментально методики, описанной в WO 2015099989, показала, что образование кристаллов на поверхности затравки идет крайне медленно и с образованием загрязненного продукта (см. Сравнительный пример 1).
Мин и др. (CN 109053840) предлагают в качестве способа получения формы 8 переосаждение софосбувира в кристаллической форме 1 или 6 н-гептаном из этилацетата и изопропилацетата с добавлением затравочных кристаллов, данный метод характеризуется невысоким (от 65,5 до 79,0 %) выходом целевого продукта и длительностью более 24 часов (около 6 часов занимает перемешивание раствора софосбувира с н-гептаном при добавлении затравочных кристаллов и 24 часа продолжается осаждение кристаллов формы 8).
Другой способ получения формы 8 в CN 109053840 представляет собой получение расплава софосбувира нагреванием до 135°С на масляной бане с последующим охлаждением до комнатной температуры. Методика характеризуется высоким выходом целевого продукта (99,3 %), однако, при этом является энергоемкой и не подходящей для масштабного промышленного применения. Кроме того, попытка авторов настоящего изобретения воспроизвести экспериментально способ получения формы 8 софосбувира по вышеописанной методике показала, что полученный продукт имеет темно желтый цвет, что свидетельствует об образовании продуктов осмоления. Из этого следует, что получаемый непосредственно при осуществлении методики продукт не соответствует фармакопейному качеству и требует дополнительной стадии очистки, например, хроматографией. Для воплощения хроматографической очистки требуется перевод вещества в растворенное состояние, которое приведет к потере веществом кристаллической формы (см. Сравнительный пример 2).
Таким образом перед авторами настоящего изобретения стояла задача разработки способа получения формы 8 софосбувира, характеризующегося высоким выходом и чистотой получаемой формы, а также возможностью применения для производства софосбувира в промышленных масштабах.
Раскрытие изобретения
Авторами изобретения было неожиданно обнаружено, что кристаллическая форма 8 софосбувира образуется при затирании смеси софосбувира известной формы и органического растворителя, в котором она растворима или умеренно растворима с осаждающим агентом, причем количество в смеси органического растворителя составляет от 5 до 60% от массы смеси.
В одном варианте изобретения смесь известной формы софосбувира и органического растворителя, в котором она растворима или умеренно растворима, получают растворением известной формы софосбувира в достаточном для растворения формы количестве органического растворителя с последующим упариванием органического растворителя до необходимого количества в смеси органического растворителя.
В еще одном варианте изобретения смесь известной формы софосбувира и органического растворителя, в котором она растворима или умеренно растворима, получают смешением известной формы софосбувира и необходимого количества в смеси органического растворителя с последующим нагреванием на водяной бане до однородного гомогенного состояния.
В предпочтительном варианте изобретения в способе получения кристаллической формы 8 софосбувира используют известную форму софосбувира выбранную из группы, включающей кристаллические формы 1, 6, 7, α, Z1 и аморфную форму.
В более предпочтительном варианте изобретения в качестве органического растворителя, в котором известная форма софосбувира растворима или умеренно растворима, используют C1-С6алифатический спирт, С1-С6алкиловый эфир С1-С6алкиловой кислоты или их смеси.
В наиболее предпочтительном варианте изобретения в качестве органического растворителя, в котором известная форма софосбувира растворима или умеренно растворима, используют растворитель, выбранный из изопропанола, этанола, этилацетата.
В еще одном варианте изобретения в качестве осаждающего агента используют органические растворители, в которых софосбувир мало растворим или практически нерастворим, представляющие собой алифатические углеводороды и эфиры, выбранные из группы алифатических углеводородов или эфиров, включающие диэтиловый эфир, диизопропиловый эфир, диметиловый эфир, метилтрет-бутиловый эфир, петролейный эфир, н-гексан, н-гептан, н-октан и их смеси.
Авторами также было неожиданно обнаружено, что кристаллическая форма 8 образуется при обработке ультразвуком известной формы софосбувира суспендированной в органическом растворителе, в котором она очень мало растворима или практически нерастворима. При этом обработку ультразвуком проводят до тех пор, пока известная форма софосбувира не перейдет в аморфное маслообразное состояние, а затем опять в твердое кристаллическое состояние.
В одном варианте изобретения в способе получения кристаллической формы 8 софосбувира используют известную форму софосбувира выбранную из группы, включающей кристаллические формы 1, 6, 7, α, Z1 и аморфную форму.
В предпочтительном варианте изобретения в качестве органического растворителя, в котором известная форма софосбувира мало растворима или практически нерастворима используют неполярные апротонные растворители, выбранные из группы, включающей н-гептан, н-гексан, н-октан, их изомеры или их смеси, петролейный эфир.
Краткое описание фигур и чертежей
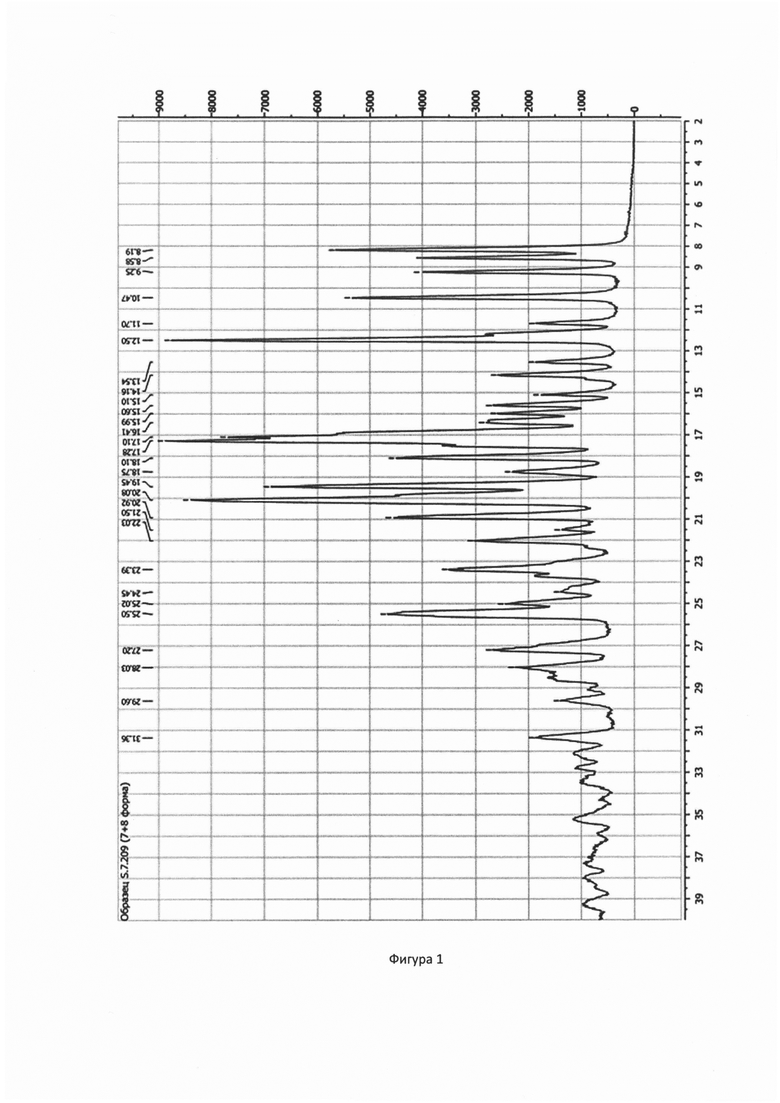
На фигуре 1 представлена порошковая рентгенограмма кристаллической формы софосбувира, полученной согласно WO 2015099989.
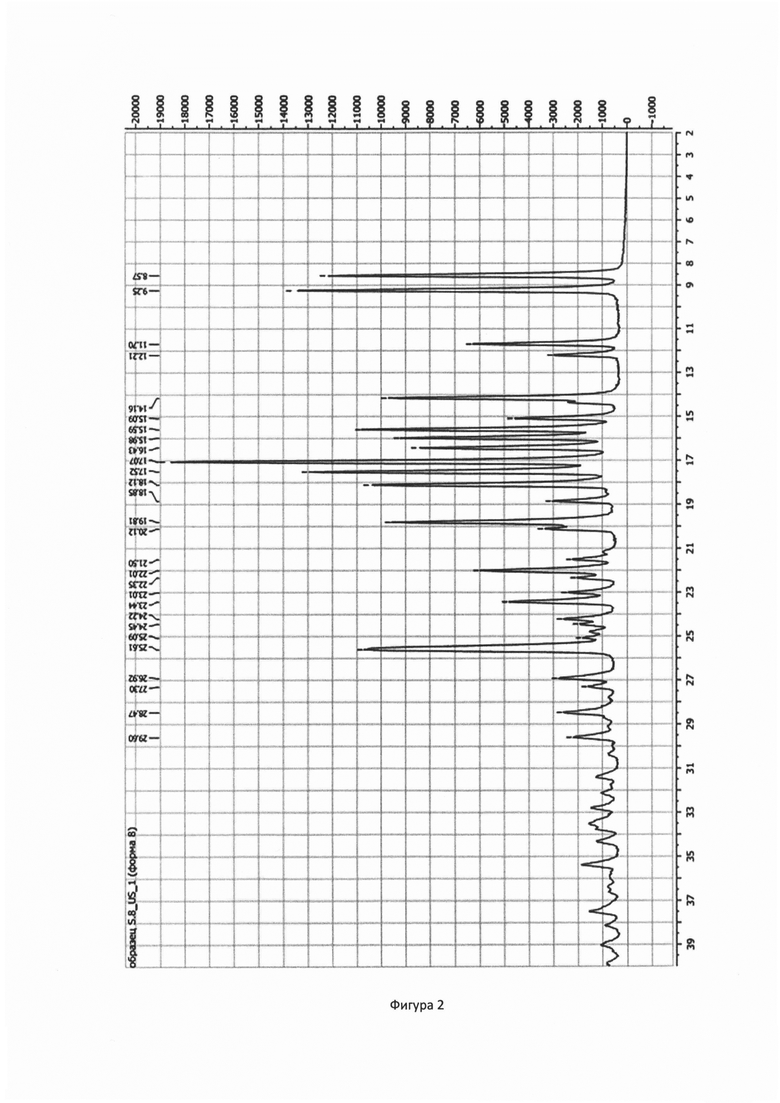
На фигуре 2 представлена порошковая рентгенограмма формы 8 софосбувира, полученного ультразвуковой обработкой суспензии софосбувира аморфной формы в петролейном эфире.
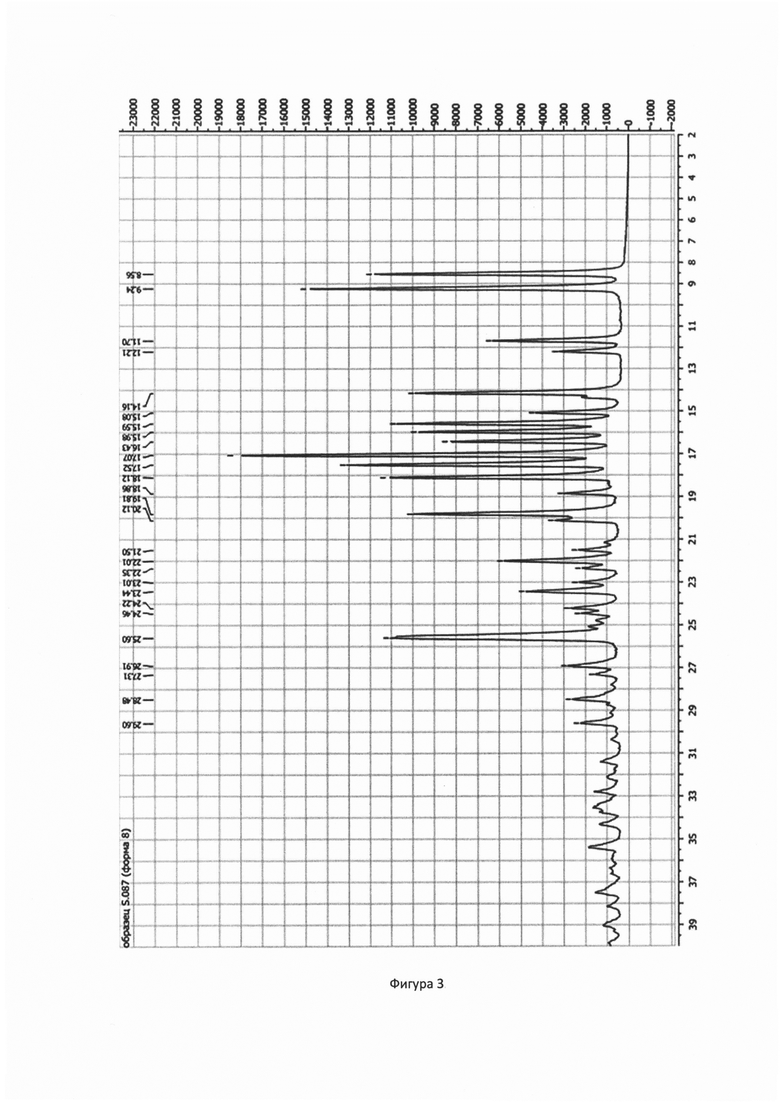
На фигуре 3 представлена порошковая рентгенограмма формы 8 софосбувира, полученного затиранием аморфной формы софосбувира в изопропиловом спирте с МТБЭ.
Осуществление изобретения
Данное изобретение направлено на два способа получения кристаллической формы 8 софосбувира (варианты изобретения).
Первый из способов получения кристаллической формы 8 софосбувира включает затирание смеси софосбувира известной формы и органического растворителя, в котором она растворима или умеренно растворима с осаждающим агентом, причем количество в смеси органического растворителя составляет от 5 до 60% от массы смеси.
Смесь известной формы софосбувира и органического растворителя, в котором она растворима или умеренно растворима, может быть получена любым подходящим способом, известным в уровне технике.
Предпочтительно, смесь известной формы софосбувира и органического растворителя, в котором она растворима или умеренно растворима, может быть получена смешением подходящих количеств растворителя и формы софосбувира, чтобы в смеси было необходимое для осуществления способа количество органического растворителя. Более предпочтительно, после смешения известной формы софосбувира и органического растворителя их можно нагреть, например, на водяной бане. Еще более предпочтительно, нагрев продолжают до тех пор, пока смешанные известная форма софосбувира и органический растворитель не превратятся в однородную смесь.
Также предпочтительно, смесь известной формы софосбувира и органического растворителя, в котором она растворима или умеренно растворима, может быть получена растворением известной формы софосбувира в достаточном для растворения формы количестве органического растворителя с последующим упариванием органического растворителя до необходимого для осуществления способа количества в смеси органического растворителя. Достаточное для растворения формы софосбувира количество органического растворителя подбирают известными методами, исходя из растворимости соответствующей формы софосбувира в соответствующем органическом растворителе.
Для осуществления первого способа получения кристаллической формы 8 софосбувира и достижения заявленных технических результатов (высокие выход и чистота, простота осуществления) необходимо, чтобы количество органического растворителя в смеси органического растворителя и известной формы софосбувира составляло от 5 до 60% от массы смеси, предпочтительно от 9 до 33% от массы смеси, более предпочтительно от 16 до 20% от массы смеси, наиболее предпочтительно примерно 20% от массы смеси.
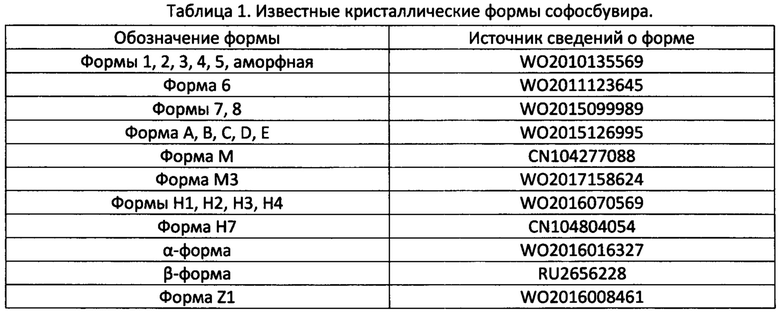
Под известной формой софосбувира, которая может использоваться в первом способе получения кристаллической формы 8 софосбувира по настоящему изобретению, понимаются формы софосбувира, которые на сегодняшний день описаны в литературе: WO 2010135569 (кристаллические формы 1, 2, 3, 4, 5, аморфная), WO 2011123645 (кристаллическая форма 6), WO 2015099989 (кристаллическая форма 7), WO 2015126995 (кристаллические формы А, В, С, D, Е), CN 104277088 (кристаллическая форма М), WO 2017158624 (кристаллическая форма М3), WO 2016070569 (кристаллические формы H1, Н2, Н3, Н4), CN 104804054 (кристаллическая форма Н7), WO 2016016327 (кристаллическая форма α), RU2656228 (кристаллическая форма β), WO 2016008461 (кристаллическая форма Ζ1).
Наиболее предпочтительными формами софосбувира для осуществления первого способа являются кристаллические формы 1, 6, 7, α, Ζ1 и аморфная форма. Аморфная форма может быть получена известными в WO 2010135569 способами, а также при помощи сублимационной сушки растворов софосбувира. Кристаллическая форма 1 может быть получена известными в WO 2010135569 способами, а также при помощи перекристаллизации кристаллической формы 6 из жидкого галогензамещенного алкана, например, хлористого метилена, хлороформа, 1,1,2,2-тетрахлорэтана и других. Кристаллическая форма 6 может быть получена известными в WO 2011123645 способами. Кристаллическая форма 7 может быть получена известными в WO 2015099989, WO 2015189386, WO 2015191945, WO 2016055576, WO 2016097173 и WO 2016156512 способами. Кристаллическая форма α может быть получена известными в WO 2016016327 способами. Кристаллическая форма Ζ1 может быть получена известными в WO 2016008461 способами.
Для осуществления первого способа получения кристаллической формы 8 софосбувира по настоящему изобретению в качестве органического растворителя, в котором известная форма софосбувира растворима или умеренно растворима, используют С1-С6алифатические спирты, С1-С6алкиловые эфиры С1-С6алкиловой кислоты или их смеси. В качестве С1-С6алифатических спиртов могут использоваться метанол, этанол, пропанолы, бутанолы, пентанолы, гексанолы, а также их изомеры (например, 2-метилпропанол-1, 2,2-диметилпропанол-1, 2-метилбутанол-1) и их смеси. В качестве С1-С6алкиловых эфиров С1-С6алкиловой кислоты могут использоваться С1-С6алкиловые эфиры муравьиной кислоты, уксусной кислоты, пропионовой кислоты, масляной кислоты, пентановой кислоты, гексановой кислоты, а также их изомеры и смеси. Наиболее предпочтительными С1-С6алкиловыми эфирам С1-С6алкиловой кислоты являются С1-С6алкиловые эфиры уксусной и пропионовой кислот, например, этилацетат, пропилацетат, изопропилацетат, изобутилацетат, этилпропионат, изопропилпропионат.
Для осуществления первого способа получения кристаллической формы 8 софосбувира по настоящему изобретению в качестве осаждающего агента используют органический растворитель, в котором софосбувир мало растворим или практически нерастворим. В качестве такового органического растворителя могут использоваться углеводороды и эфиры (также известны как простые эфиры). В качестве углеводородов могут использоваться любые жидкие алифатические и ароматические углеводороды, предпочтительно выбранные из пентана, гексана, гептана, октана, петролейного эфира, бензола, толуола, этилбензола, кумол, ксилолы (о-, м-, п-) или их изомеров (например, 2-метилпентан, 2,2-диметилгексан и другие) и смесей. Наиболее предпочтительно использовать алифатические углеводороды, выбранные из пентана, гексана, гептана, октана, петролейного эфира или их изомеров (например, 2-метилпентан, 2,2-диметилгексан и другие) и смесей. В качестве эфиров могут использоваться любые жидкие эфиры, предпочтительно, выбранные из диэтилового эфира, диизопропилового эфира, диметилового эфира, метилтретбутилового эфира.
Кристаллизацию формы 8 софосбувира по первому способу получения проводят при перемешивании, предпочтительно использовать лопастную мешалку или якорную мешалку, которая способствует вращению всех жидкостей, причем диаметр вращения данной мешалки предпочтительно составляет от 2/5 до 4/5 от диаметра используемого реактора. Закристаллизовавшуюся форму 8 софосбувира выделяют общепринятыми методами, например, отделяют от жидкой фазы при помощи фильтрования, при необходимости, сушат, например, в сушильном или вакуумном шкафу, предпочтительно, под вакуумом. Было установлено, что стадия сушки, проводимая в печи или в сушилке с перемешиванием, не изменяет кристаллическую структуру, полученную в результате кристаллизации. Выделенную форму 8 софосбувира при необходимости измельчают и используют для лечения или для получения готовой лекарственной формы (ГЛФ), например, в виде таблетки или капсулы.
Второй из способов получения кристаллической формы 8 софосбувира включает обработку ультразвуком известной формы софосбувира суспендированной в органическом растворителе, в котором она очень мало растворима или практически нерастворима.
Под известной формой софосбувира, которая может использоваться во втором способе получения кристаллической формы 8 софосбувира по настоящему изобретению, понимаются формы софосбувира, которые на сегодняшний день описаны в литературе: WO 2010135569 (кристаллические формы 1, 2, 3, 4, 5, аморфная), WO 2011123645 (кристаллическая форма 6), WO 2015099989 (кристаллическая форма 7), WO 2015126995 (кристаллические формы А, В, С, D, Е), CN 104277088 (кристаллическая форма М), WO 2017158624 (кристаллическая форма М3), WO 2016070569 (кристаллические формы H1, Н2, Н3, Н4), CN 104804054 (кристаллическая форма Н7), WO 2016016327 (кристаллическая форма α), RU2656228 (кристаллическая форма β), WO 2016008461 (кристаллическая форма Ζ1).
Наиболее предпочтительными формами софосбувира для осуществления второго способа являются кристаллические формы 1, 6, 7, α, Ζ1 и аморфная форма. Аморфная форма может быть получена известными в WO 2010135569 способами, а также при помощи сублимационной сушки растворов софосбувира. Кристаллическая форма 1 может быть получена известными в WO 2010135569 способами, а также при помощи перекристаллизации кристаллической формы 6 из жидкого хлорзамещенного алкана, например, хлористого метилена, хлороформа, 1,1,2,2-тетрахлорэтана и других. Кристаллическая форма 6 может быть получена известными в WO 2011123645 способами. Кристаллическая форма 7 может быть получена известными в WO 2015099989, WO 2015189386, WO 2015191945, WO 2016055576, WO 2016097173 и WO 2016156512 способами. Кристаллическая форма α может быть получена известными в WO 2016016327 способами. Кристаллическая форма Ζ1 может быть получена известными в WO 2016008461 способами.
Для осуществления второго способа получения кристаллической формы 8 софосбувира по настоящему изобретению в качестве осаждающего агента используют органический растворитель, в котором софосбувир мало растворим или практически нерастворим. В качестве такового органического растворителя могут использоваться углеводороды и эфиры (также известны как простые эфиры). В качестве углеводородов могут использоваться любые жидкие алифатические и ароматические углеводороды, предпочтительно выбранные из пентана, гексана, гептана, октана, петролейного эфира, бензола, толуола, этилбензола, кумол, ксилолы (о-, м-, п-) или их изомеров (например, 2-метилпентан, 2,2-диметилгексан и другие) и смесей. Наиболее предпочтительно использовать алифатические углеводороды, выбранные из пентана, гексана, гептана, октана, петролейного эфира или их изомеров (например, 2-метилпентан, 2,2-диметилгексан и другие) и смесей. В качестве эфиров могут использоваться любые жидкие эфиры, предпочтительно, выбранные из диэтилового эфира, диизопропилового эфира, диметилового эфира, метилтретбутилового эфира.
Для осуществления второго способа получения кристаллической формы 8 софосбувира по настоящему изобретению при обработке суспензии известной формы софосбувира в органическом растворителе, в котором она очень мало растворима или практически нерастворима, могут быть использованы любые подходящие лабораторные или промышленные ультразвуковые ванны, работающие с частотой от 22 до 60 кГц. В наиболее предпочтительном варианте исполнения используются ультразвуковые ванны, работающие с частотой от 35 до 44 кГц.
Образовавшуюся по второму способу форму 8 софосбувира выделяют общепринятыми методами, например, отделяют от жидкой фазы при помощи фильтрования, при необходимости, сушат, например, в сушильном или вакуумном шкафу, предпочтительно, под вакуумом. Было установлено, что стадия сушки, проводимая в печи или в сушилке с перемешиванием, не изменяет кристаллическую структуру, полученную в результате кристаллизации. Выделенную форму 8 софосбувира при необходимости измельчают и используют для лечения или для получения готовой лекарственной формы (ГЛФ), например, в виде таблетки или капсулы.
При получении ГЛФ в виде таблеток или желатиновых капсул кристаллическую форму 8 софосбувира, полученную по любому из двух способов настоящего изобретения, смешивают с фармацевтическими носителями, такими как желатины, крахмалы, лактоза, микрокристаллическая целлюлоза, производные винилпирролидона, стеариновая кислота и стеараты, тальк, кремния диоксид и другими. Кроме того, ГЛФ могут иметь пролонгированное или замедленное действие и, таким образом, непрерывно высвобождать определенное количество активного ингредиента.
Кристаллическая форма 8 софосбувира, полученная по любому из двух способов настоящего изобретения, а также содержащие ее ГЛФ, может применяться в способах лечения пациентов, инфицированных вирусом гепатита С генотипа 1 (включая 1а и 1b), генотипа 2, генотипа 3, генотипа 4, генотипа 5 и генотипа 6, предпочтительно в комбинации с другими средствами, включающими, но не ограниченными следующими: интерфероны, рибавирин, ингибиторы NS5A, ингибиторы NS5B, ингибиторы протеазы, ингибиторы альфа-глюкозидазы, гепатопротекторы, агонисты TLR-7, ингибиторы циклофиллина, ингибиторы IRES HCV и другие агенты против HCV.
Примеры
Представленные ниже примеры иллюстрируют (без ограничения объема притязаний) наиболее предпочтительные варианты осуществления изобретения, а именно подтверждают возможность получения заявляемой формы 8 софосбувира и достижение заявленных преимуществ способа получения.
Методы
Дифрактограммы (РФА) регистрировали на приборе D2 PHASER фирмы Bruker, излучение Cu-Κα, фильтр - Ni, с графитовым монохроматором 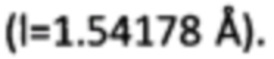 Режим трубки (Cu) 10 мА, 30 кВ. Диапазон значений угла 2Θ - от 2 до 40°, шаг 0.025°, щель 0.6 мм, выдержка в точке - 500 мсек, дискриминатор по энергиям - 0.17-0.23 кэВ.
Режим трубки (Cu) 10 мА, 30 кВ. Диапазон значений угла 2Θ - от 2 до 40°, шаг 0.025°, щель 0.6 мм, выдержка в точке - 500 мсек, дискриминатор по энергиям - 0.17-0.23 кэВ.
Чистоту полученных образцов контролировали методом ВЭЖХ. Хроматограммы регистрировали на приборе Shimadzu LC-20A Prominence.
Хроматографические условия:
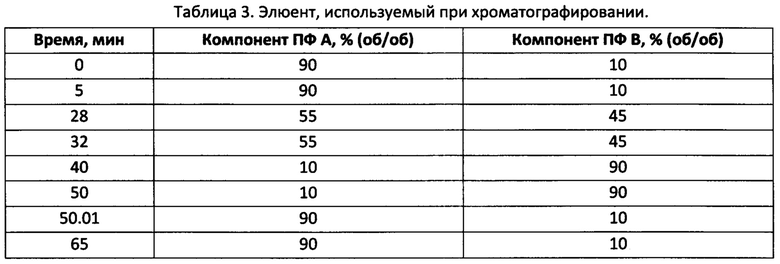
• Колонка: 250x 4,6 мм, заполненная окстадецилсилильным силикагелем для хроматографии с размером частиц 5 мкм (ProjectSil Target);
• Подвижная фаза А: 0,05%-ный раствор Н3РО4;
• Подвижная фаза В: ацетонитрил: метанол (80:20);
• Скорость потока: 1,35 мл/мин;
• Температура колонки: 35°С;
• Детектор: спектрофотометрический, 210 нм;
• Объем вводимой пробы: 10 мкл;
• Время хроматографирования: 65 мин;
• Режим элюирования: градиентный
Ультразвуковая ванна, используемая при получении кристаллической формы 8, имеет рабочую частоту 37 кГц. Модель Elmasonic S30H. При проведении опытов температура ванны поддерживалась в диапазоне 25-30°С.
Сублимационная сушка проводилась на приборе Labconco 4.5 Freezone Benchtop Freeze Dryer при поддержании вакуума 0.2-0.1 мбар.
Сравнительный пример 1. Получение кристаллической формы 8 софосбувира согласно WO 2015099989.
Раствор софосбувира в изопропилацетате (20 об./масс) нагревали до примерно 68°С и перемешивали в течение 1 часа. После осветления фильтрованием фильтрат охлаждали до температуры примерно 40°С и добавляли затравку формы 8. Смесь охлаждали до примерно 20°С без перемешивания. Авторами изобретения было отмечено, что рост кристаллов на поверхности затравки идет крайне медленно. Спустя 2 дня после проведения эксперимента, было обнаружено образование ромбических кристаллов. Данные кристаллы в реакционном сосуде были оставлены в морозильной камере (около -15°С), однако спустя 1 неделю после инициации дальнейшей кристаллизации не было обнаружено. При оставлении реакционной массы в контакте с окружающей средой, часть растворителя испарилась, в результате чего был получен твердый продукт, который, согласно визуальному исследованию был неоднороден: помимо изначальных кристаллов ромбической формы на дне колбы был налет игольчатого кристаллического порошка, который в большей степени можно отнести к смеси нескольких форм из-за большой неоднородности. Согласно полученной дифрактограмме образца (фигура 1) он представляет собой смесь 7 и 8 кристаллических форм софосбувира.
Сравнительный пример 2. Получение кристаллической формы 8 софосбувира согласно CN 109053840.
Навеску софосбувира кристаллической формы 1 (1 грамм) поместили в трехгорлую колбу, образец нагревали на масляной бане до 95°С, после того как происходило частичное расплавление материала температуру непрерывно повышали до 135°С до полного расплавления софосбувира. Далее температуру понижали до 130°С и выдерживали при этой температуре 40 минут. По истечении времени массу охлаждали до комнатной температуры. Полученное соединение имело темно желтый цвет, что свидетельствует о наличии продуктов осмоления. Выход составил 0,99 г (99%), чистота (ВЭЖХ) - 93.3%.
На дифрактограмме образца получены следующие пики: 8.45°, 9.11°, 14.06°, 15.53°, 15.88°, 16.96°, 17.43°, 18.02°, 19.70°, 20.12°, 25.60° 2θ.
Пример 1. Получение кристаллической формы 8 софосбувира затиранием формы 6 в изопропиловом спирте с МТБЭ.
Навеску софосбувира 6 формы (0.500 г, 0,944 ммоль) (Nantong Chanyoo Pharmatech Co., Ltd) помещали в круглодонную колбу объемом 25 мл, снабженную магнитным перемешивающим устройством и обратным холодильником (контроль температуры осуществляется с помощью термопары, помещенной в реакционную массу). К навеске добавляли 4 мл изопропилового спирта (ХЧ), реакционную массу нагревали при перемешивании до 40-45°С, после полного растворения осадка реакционную массу охлаждали до комнатной температуры. Затем часть растворителя упаривали с применением роторно-пленочного испарителя (РПИ) до остаточной массовой доли спирта (0.25, 0.125, 0.045 г изопропанола для 0.5 г софосбувира соответственно) в реакционной массе. К осадку добавляли МТБЭ в количестве согласно выполняемому опыту (5, 10, 15 мл соответственно). Маслянистые жидкости затирались магнитным якорем в течение 3 ч (для 1/10 масс, доли спирта затирка проводится в течение 4-5 ч). Выпавший осадок фильтровали и промывали 5 мл МТБЭ и сушили в вакуумном шкафу (0,07-0,08 МПа) при 25°С. Выход согласно таблице 4.
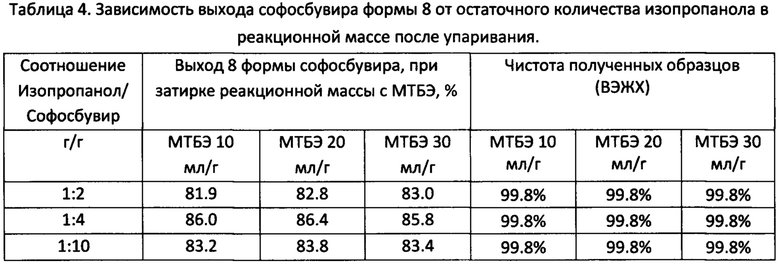
На дифрактограмме образца получены следующие пики: 8.55°, 9.24°, 14.17°, 15.59°, 15.98°, 17.13°, 17.48°, 18.05°, 19.80°, 20.26°, 25.61° 2θ.
Пример 2. Получение кристаллической формы 8 софосбувира из формы 6 ультразвуковой обработкой суспензии в петролейном эфире.
Навеску софосбувира 6 формы (0.500 г, 0,944 ммоль) (Nantong Chanyoo Pharmatech Co., Ltd) помещали в круглодонную колбу объемом 25 мл. К навеске добавляли 5 мл петролейного эфира, реакционную массу подвергали УЗ-облучению, через еще 15 минут вносились дополнительно 5 мл петролейного эфира, через 5 минут в реакционной массе на дне колбы образовывалось масло, которое при продолжении облучения еще в течение 20 минут переходило в состояние твердого вещества. Выпавший осадок фильтровали и промывали 10 мл МТБЭ и сушили в вакуумном шкафу (0,07-0,08 МПа) при 25°С. Выход составил 0.46 г (92%), чистота (ВЭЖХ) - 99.8%.
На дифрактограмме образца получены следующие пики: 8.59°, 9.22°, 14.19°, 15.58°, 16.08°, 17.0°, 17.54°, 18.13°, 19.79°, 20.23°, 25.60° 2θ.
Пример 3. Получение кристаллической формы 8 софосбувира из формы 1 ультразвуковой обработкой суспензии в петролейном эфире.
Форма 1 софосбувира была получена при перекристаллизации формы 6 (Nantong Chanyoo Pharmatech Co., Ltd) из хлористого метилена.
Навеску софосбувира 1 формы (0.250 г, 0,472 ммоль) помещают в круглодонную колбу объемом 10 мл. К навеске добавляют 5 мл петролейного эфира, реакционную массу подвергают УЗ-облучению, за 20 минут осадок не растворяется, переходит в аморфное состояние и потом кристаллизуется в течение следующих 20 минут. Выпавший осадок фильтруют и промывают 10 мл МТБЭ и сушат в вакуумном шкафу (0,07-0,08 МПа) при 25°С. Выход составляет 0.241 г (96.5%), чистота (ВЭЖХ) - 99.9%.
На дифрактограмме образца получены следующие пики: 8.61°, 9.28°, 14.20°, 15.63°, 16.01°, 17.10°, 17.56°, 18.15°, 19.84°, 20.13°, 25.61° 2**Θ.
Пример 4. Получение кристаллической формы 8 софосбувира затиранием формы 1 в изопропиловом спирте с МТБЭ.
Форма 1 софосбувира была получена при перекристаллизации формы 6 (Nantong Chanyoo Pharmatech Co., Ltd) из хлористого метилена.
Навеску софосбувира 1 формы (0.250 г, 0,472 ммоль) помещали в круглодонную колбу объемом 10 мл, снабженную магнитным перемешивающим устройством и обратным холодильником. К навеске добавляли 2 мл изопропилового спирта (ХЧ), реакционную массу нагревали при перемешивании до 40-45°С, после полного растворения осадка реакционную массу охлаждали до комнатной температуры. Затем часть растворителя упаривали с применением РПИ до остаточного количества спирта 40% от массы смеси. К осадку добавляли МТБЭ в количестве 5 мл. Маслянистые жидкости затирались магнитным якорем в течение 3 ч. Выпавший осадок фильтровали и промывали 5 мл МТБЭ и сушили в вакуумном шкафу (0,07-0,08 МПа) при 25°С. Выход составил 0.217 г (86.8%), чистота (ВЭЖХ) - 99.8%.
На дифрактограмме образца получены следующие пики: 8.58°, 9.24°, 14.23°, 15.60°, 16.11°, 17.17°, 17.52°, 18.10°, 19.82°, 20.22°, 25.61° 2θ.
Пример 5. Получение кристаллической формы 8 софосбувира затиранием аморфной формы в изопропиловом спирте с МТБЭ.
Аморфная форма софосбувира была получена сублимационной сушкой раствора софосбувира в смеси ацетонитрила с метанолом и водой, полученного при разделывании реакционной смеси после химического синтеза софосбувира.
Кристаллическую форму 8 получали согласно примеру 4, использовав вместо формы 1 аморфную форму. Выход составил 0.218 г (87.2%), чистота (ВЭЖХ) - 99.8%.
На дифрактограмме образца получены следующие пики: 8.62°, 9.19°, 14.26°, 15.64°, 16.02°, 16.98°, 17.50°, 18.12°, 19.88°, 20.26°, 25.62° 2θ.
Пример 6. Получение кристаллической формы 8 софосбувира из аморфной формы ультразвуковой обработкой суспензии в петролейном эфире.
Аморфная форма софосбувира была получена сублимационной сушкой раствора софосбувира в смеси ацетонитрила с метанолом и водой, полученного при разделывании реакционной смеси после химического синтеза софосбувира.
Форму 8 получали согласно примеру 3, использовав вместо формы 1 аморфную форму. Выход составляет 0.242 г (96.8%), чистота (ВЭЖХ) - 99.8%.
На дифрактограмме образца получены следующие пики: 8.57°, 9.25°, 14.16°, 15.59°, 15.98°, 17.07°, 17.52°, 18.12°, 19.81°, 20.12°, 25.61° 2θ (фигура 2).
Пример 7. Получение кристаллической формы 8 софосбувира из формы 1 ультразвуковой обработкой суспензии в н-гексане.
Форма 1 софосбувира была получена при перекристаллизации формы 6 (Nantong Chanyoo Pharmatech Co., Ltd) из хлористого метилена.
Форму 8 получали согласно примеру 3, использовав вместо петролейного эфира н-гексан. Выход составляет 0.239 г (95.6%), чистота (ВЭЖХ) - 99.9%.
На дифрактограмме образца получены следующие пики: 8.62°, 9.29°, 14.18°, 15.60°, 16.11°, 17,10°, 17.54°, 18.10°, 19.78°, 20.22°, 25.59° 2θ.
Пример 8. Получение кристаллической формы 8 софосбувира затиранием аморфной формы в изопропиловом спирте с МТБЭ.
Аморфная форма софосбувира была получена сублимационной сушкой раствора формы 6 софосбувира (Nantong Chanyoo Pharmatech Co., Ltd) в смеси ацетонитрила с водой.
Навеску софосбувира аморфной формы (0.750 г, 1,416 ммоль) помещали в круглодонную колбу объемом 25 мл, снабженную магнитным перемешивающим устройством и обратным холодильником (контроль температуры осуществляется с помощью термопары, помещенной в реакционную массу). К навеске добавляли 7 мл изопропилового спирта (ХЧ), реакционную массу нагревали при перемешивании до 40-45°С, после полного растворения осадка реакционную массу охлаждали до комнатной температуры. Затем часть растворителя упаривали с применением роторно-пленочного испарителя (РПИ) до остаточной массовой доли спирта (1.125, 0.15, 0.04 г изопропанола для 0.75 г софосбувира соответственно) в реакционной массе. К осадку добавляли МТБЭ в количестве согласно выполняемому опыту (7.5,15, 22.5 мл соответственно). Маслянистые жидкости затирались магнитным якорем в течение 2-3 ч (для 1/19 масс. доли спирта затирка проводится в течение 5 ч). Выпавший осадок фильтровали и промывали 5 мл МТБЭ и сушили в вакуумном шкафу (0,07-0,08 МПа) при 25°С. Выход согласно таблице 5.
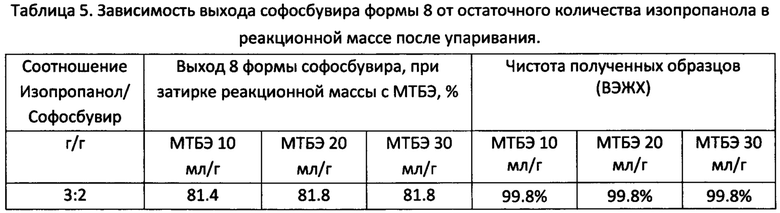

На дифрактограмме образца получены следующие пики: 8.56°, 9.24°, 14.16°, 15.59°, 15.98°, 17.07°, 17.52°, 18.12°, 19.81°, 20.12°, 25.60° 2θ (фигура 3).
Пример 9. Получение кристаллической формы 8 софосбувира затиранием формы α в этиловом спирте с н-гептаном.
Кристаллическая форма α софосбувира была получена перекристаллизацией формы 6 (Nantong Chanyoo Pharmatech Co., Ltd) из метилэтилкетона.
Кристаллическую форму 8 получали согласно примеру 4, использовав вместо формы 1 форму α, этиловый спирт вместо изопропилового и н-гептан вместо МТБЭ. Выход составил 0.221 г (88.4%), чистота (ВЭЖХ) - 99.8%.
На дифрактограмме образца получены следующие пики: 8.59°, 9.17°, 14.22°, 15.64°, 16.12°, 16.98°, 17.52°, 18.12°, 19.75°, 20.23°, 25.60° 2θ.
Пример 10. Получение кристаллической формы 8 софосбувира затиранием формы α в изопропиловом спирте с н-гексаном.
Кристаллическая форма α софосбувира была получена перекристаллизацией формы 6 (Nantong Chanyoo Pharmatech Co., Ltd) из метилэтилкетона.
Кристаллическую форму 8 получали согласно примеру 4, использовав вместо формы 1 форму α и н-гексан вместо МТБЭ. Выход составил 0.219 г (87.6%), чистота (ВЭЖХ) - 99.9%.
На дифрактограмме образца получены следующие пики: 8.58°, 9.24°, 14.23°, 15.60°, 16.12°, 16.98°, 17.52°, 18.12°, 19.79°, 20.23°, 25.60° 2θ.
Пример 11. Получение кристаллической формы 8 софосбувира затиранием аморфной формы в этилацетате с МТБЭ.
Аморфная форма софосбувира была получена сублимационной сушкой раствора софосбувира в смеси ацетонитрила с метанолом и водой, полученного при разделывании реакционной смеси после химического синтеза софосбувира.
Кристаллическую форму 8 получали согласно примеру 4, использовав вместо формы 1 аморфную форму и этилацетат вместо изопропилового спирта. Выход составил 0.216 г (86.4%), чистота (ВЭЖХ) - 99.8%.
На дифрактограмме образца получены следующие пики: 8.61°, 9.17°, 14.21°, 15.69°, 16.10°, 16.94°, 17.59°, 18.14°, 19.80°, 20.31°, 25.66° 2θ.
Пример 12. Получение кристаллической формы 8 софосбувира затиранием аморфной формы в этиловом спирте с МТБЭ.
Аморфная форма софосбувира была получена сублимационной сушкой раствора софосбувира в смеси ацетонитрила с метанолом и водой, полученного при разделывании реакционной смеси после химического синтеза софосбувира.
Навеску софосбувира аморфной формы (1.000 г, 1,888 ммоль) помещали в круглодонную колбу объемом 10 мл, снабженную магнитным перемешивающим устройством и обратным холодильником. К навеске добавляли 1,27 мл (1 г) этилового спирта (ХЧ), реакционную массу нагревали при перемешивании до 50-55°С, после гомогенизации реакционной массы ее охлаждали до комнатной температуры. Затем к смеси добавляли 6 мл МТБЭ и маслянистые жидкости затирались магнитным якорем в течение 3,5 ч. Выпавший осадок фильтровали и промывали 5 мл МТБЭ и сушили в вакуумном шкафу (0,07-0,08 МПа) при 25°С. Выход составил 0.221 г (88.4%), чистота (ВЭЖХ) - 99.8%.
На дифрактограмме образца получены следующие пики: 8.58°, 9.14°, 14.2°, 15.61°, 16.07°, 17.0°, 17.54°, 18.19°, 19.79°, 20.22°, 25.54° 2θ.
Изобретение относится к вариантам способа получения кристаллической формы 8 софосбувира. Один из вариантов способа включает затирание смеси известной формы софосбувира, выбранной из кристаллических форм 1, 6, 7, α и аморфной формы, и органического растворителя, в котором она растворима или умеренно растворима, выбранного из изопропанола, этанола, этилацетата, с осаждающим агентом, выбранным из группы, включающей диэтиловый эфир, диизопропиловый эфир, диметиловый эфир, метилтрет-бутиловый эфир, петролейный эфир, н-гексан, н-гептан, н-октан и их смеси, причем количество в смеси органического растворителя составляет от 5 до 60% от массы смеси известной формы софосбувира и органического растворителя. При этом смесь известной формы софосбувира и органического растворителя, в котором она растворима или умеренно растворима, получают растворением известной формы софосбувира в достаточном для растворения формы количестве органического растворителя с последующим упариванием органического растворителя до указанного количества в смеси органического растворителя. Другой вариант способа включает ультразвуковую обработку известной формы софосбувира, выбранной из кристаллических форм 1, 6, 7, α и аморфной формы, суспендированной в органическом растворителе, в котором она очень мало растворима или практически нерастворима, выбранном из группы, включающей н-гептан, н-гексан, н-октан, их изомеры или их смеси, петролейный эфир. При этом обработку ультразвуком проводят до тех пор, пока известная форма софосбувира не перейдет в аморфное маслообразное состояние, а затем опять в твердое кристаллическое состояние. Предлагаемые варианты способа позволяют получить кристаллическую форму 8 софосбувира с высоким выходом и чистотой. 2 н.п. ф-лы, 3 ил., 5 табл., 12 пр.
1. Способ получения кристаллической формы 8 софосбувира, включающий затирание смеси известной формы софосбувира, выбранной из кристаллических форм 1, 6, 7, α и аморфной формы, и органического растворителя, в котором она растворима или умеренно растворима, выбранного из изопропанола, этанола, этилацетата, с осаждающим агентом, выбранным из группы, включающей диэтиловый эфир, диизопропиловый эфир, диметиловый эфир, метилтрет-бутиловый эфир, петролейный эфир, н-гексан, н-гептан, н-октан и их смеси, причем количество в смеси органического растворителя составляет от 5 до 60% от массы смеси известной формы софосбувира и органического растворителя, отличающийся тем, что смесь известной формы софосбувира и органического растворителя, в котором она растворима или умеренно растворима, получают растворением известной формы софосбувира в достаточном для растворения формы количестве органического растворителя с последующим упариванием органического растворителя до указанного количества в смеси органического растворителя.
2. Способ получения кристаллической формы 8 софосбувира, включающий ультразвуковую обработку известной формы софосбувира, выбранной из кристаллических форм 1, 6, 7, α и аморфной формы, суспендированной в органическом растворителе, в котором она очень мало растворима или практически нерастворима, выбранном из группы, включающей н-гептан, н-гексан, н-октан, их изомеры или их смеси, петролейный эфир, отличающийся тем, что обработку ультразвуком проводят до тех пор, пока известная форма софосбувира не перейдет в аморфное маслообразное состояние, а затем опять в твердое кристаллическое состояние.
| CN 0105732751 A, 06.07.2016 | |||
| CN 109053840 A, 21.12.2018 | |||
| WO 2015099989 A1, 02.07.15 | |||
| Sofia, Michael J | |||
| Et al | |||
| Journal of Medicinal Chemistry, 2010, 53(19), 7202-7218 | |||
| US 8642756 B2, 04.02.2014 | |||
| US 20180346506 A1, 06.12.2018 | |||
| Приемник с стопстартной коррекцией для телеграфных аппаратов типа Бодо | 1931 |
|
SU26731A1 |
| WO 2011123645 A2, 06.10.2011 | |||
| Mino R | |||
| Caira | |||
| Crystalline Polymorphism of Organic Compounds | |||
| Design | |||

