Область изобретения
Настоящее изобретение принадлежит к области медицины и относится к производному пиридопиримидина формулы (I), способу его получения и содержащей его фармацевтической композиции, а также его применению в качестве терапевтического агента, в частности, в качестве агониста TLR8.
Предшествующий уровень техники
Толл-подобные рецепторы (TLR, от англ. toll-like receptors) представляют собой класс ключевых рецепторов, связанных с врожденным иммунитетом. TLR являются одиночными трансмембранными некаталитическими рецепторами, обычно экспрессируемыми на сторожевых клетках, таких как макрофаги и дендритные клетки, и способными распознавать структурно консервативные молекулы, продуцируемые микроорганизмами. Как только эти микроорганизмы преодолевают физические барьеры, такие как кожа или слизистая оболочка кишечного тракта, они будут распознаваться TLR, активируя тем самым иммунные клеточные ответы (Mahla, R.S. et al., Front Immunol., 4: 248 (2013)). Способность иммунной системы распознавать широкий ряд патогенных микроорганизмов частично обусловлена повсеместным присутствием толл-подобных иммунорецепторов.
У млекопитающих имеется по меньшей мере десять типов различных TLR. Для некоторых из этих рецепторов идентифицированы лиганды и соответствующие каскады передачи сигнала. TLR8 является членом подгруппы TLR (TLR3, 7, 8 и 9), расположенных в эндосомальном компартменте клеток, которые специализируются на обнаружении чужеродных нуклеиновых кислот. У человека TLR8 экспрессируется главным образом в моноцитах, натуральных киллерах (NK, от англ. Natural killer) и миелоидных дендритных клетках (mDC). Агонисты TLR8 могут вызывать высвобождение различных провоспалительных цитокинов, таких как интерлейкин (IL)-6, IL-12, фактор некроза опухоли альфа (ФНО-α) и интерферон-гамма (IFN-γ).
В организме TLR8 играет важную роль во врожденном и приобретенном иммунитете. Агонисты TLR8 можно использовать в качестве иммуномодуляторов в лечении различных, связанных с иммунной системой заболеваний, таких как рак яичников, меланома, немелкоклеточный рак легкого, гепатоклеточная карцинома, базальноклеточная карцинома, почечноклеточный рак, миелома, аллергический ринит, астма, хроническая обструктивная болезнь легких (COPD), язвенный колит, фиброз печени, инфекции, вызываемые вирусом гепатита В (HBV), флавивирусом, вирусом гепатита С (HCV), папилломавирусом человека (HPV), респираторно-синтициальным вирусом (RSV), тяжелый острый респираторный синдром (SARS), инфекции, вызываемые вирусом иммунодефицита человека (ВИЧ) или вирусом гриппа и тому подобные заболевания.
Поскольку TLR8 и TLR7 являются высоко гомологичными, агонисты TLR8 в большинстве случаев также являются агонистами TLR7. В связи с этим, во многих патентных заявках, таких как WO 2009111337, WO 2011017611, WO 2011068233, WO 2011139348, WO 2012066336, WO 2013033345 и WO 2017046112, сообщалось об агонистах TLR8 и TLR7 двойного действия. Сообщений о селективных агонистах TLR8 относительно мало, и они в основном включают сообщения о VTX-2337, разработанном VentiRX (WO 2007024612), и GS-9688, разработанном Gilead (WO 2016141092).
Краткое описание сущности изобретения
После интенсивных исследований авторы изобретения разработали и синтезировали ряд пиридопиримидиновых соединений. Эти соединения обладают хорошим активирующим действием в отношении TLR8, но при этом не обладают никаким активирующим действием в отношении TLR7. Следовательно, эти соединения могут быть разработанны в качестве селективных агонистов TLR8 для лечения и/или предупреждения различных заболеваний, связанных с активностью TLR8.
Таким образом, целью настоящего изобретения является получение соединения формулы (I):
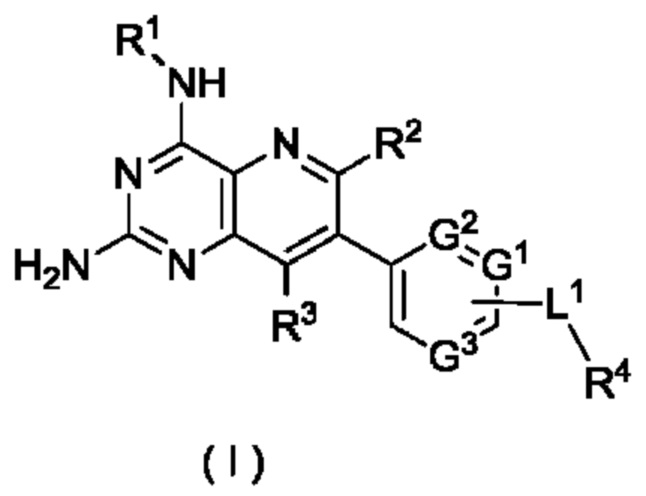
или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли,
где:
G1, G2 и G3 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из СН, CR5 и N;
L1 выбран из группы, состоящей из алкилена и ковалентной связи, при этом указанный алкилен возможно замещен одним или более чем одним заместителем, выбранным(ыми) из группы, состоящей из галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила и гетероциклила;
R1 выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила, при этом алкил, циклоалкил, гетероциклил, арил и гетероарил каждый возможно независимо замещен одним заместителем или несколькими заместителями, выбранным(ыми) из группы, состоящей из галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила;
R2 и R3 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила;
R4 выбран из группы, состоящей из алкила, галогеналкила, циклоалкила, гетероциклила, арила и гетероарила, при этом алкил, циклоалкил, гетероциклил, арил и гетероарил каждый возможно независимо замещен одним заместителем или несколькими заместителями, выбранным(ыми) из группы, состоящей из алкила, алкокси, галогена, амино, циано, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила; и
R5 выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила.
В предпочтительном воплощении настоящего изобретения соединение формулы (I) по настоящему изобретению представляет собой соединение формулы (Ia):
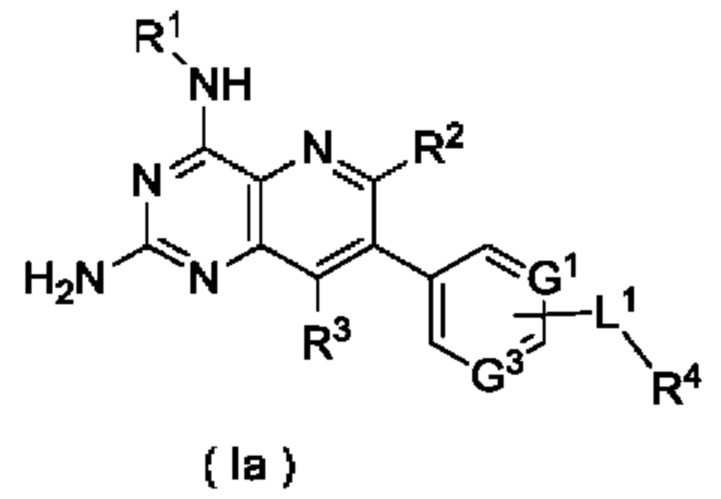
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемую соль,
где:
G1, G3, L1 и R1-R4 являются такими, как определено в формуле (I).
В другом предпочтительном воплощении настоящего изобретения соединение формулы (I) по настоящему изобретению представляет собой соединение формулы (Ib):
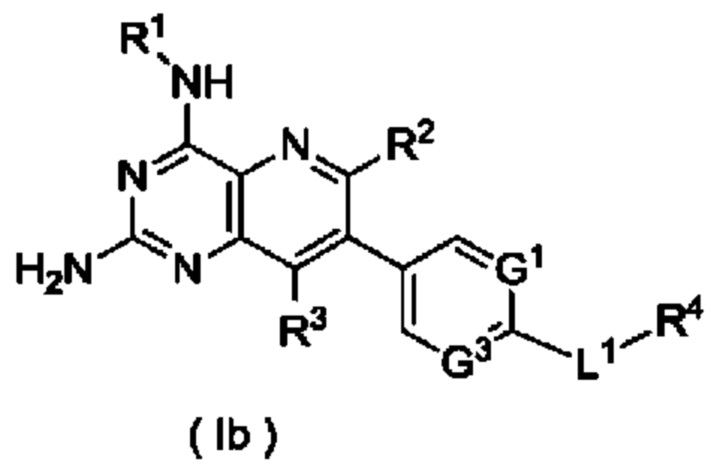
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемую соль,
где:
G1, G3, L1 и R1-R4 являются такими, как определено в формуле (I).
В другом предпочтительном воплощении настоящего изобретения соединение формулы (I) по настоящему изобретению представляет собой соединение формулы (II):
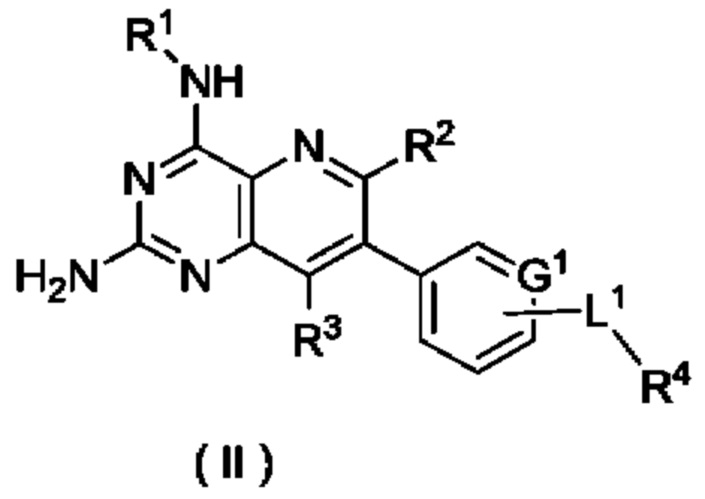
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемую соль,
где:
G1, L1 и R1-R4 являются такими, как определено в формуле (I).
В другом предпочтительном воплощении настоящего изобретения соединение формулы (I) по настоящему изобретению представляет собой соединение формулы (III):
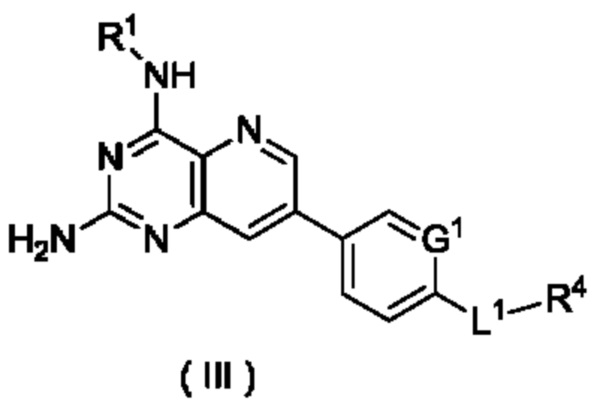
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь либо их фармацевтически приемлемую соль,
где:
G1, L1 и R1 и R4 являются такими, как определено в формуле (I).
В другом предпочтительном воплощении настоящего изобретения предложено соединение формулы (I), формулы (Ia), формулы (Ib), формулы (II) или формулы (III) по настоящему изобретению, где R4 представляет собой гетероциклил, который возможно замещен одним алкилом или несколькими алкилами; R4 предпочтительно представляет собой 4-6-членный гетероциклил, содержащий один гетероатом или два одинаковых или разных гетероатома, выбранный(ых) из группы, состоящей из N, О и S, и данный 4-6-членный гетероциклил возможно замещен одним алкилом или несколькими алкилами; и R4 более предпочтительно представляет собой пирролил, пиперазинил, пиперидинил или морфолинил.
В другом предпочтительном воплощении настоящего изобретения соединение формулы (I), формулы (Ia), формулы (Ib), формулы (II) или формулы (III) по настоящему изобретению представляет собой соединение формулы (IVa):
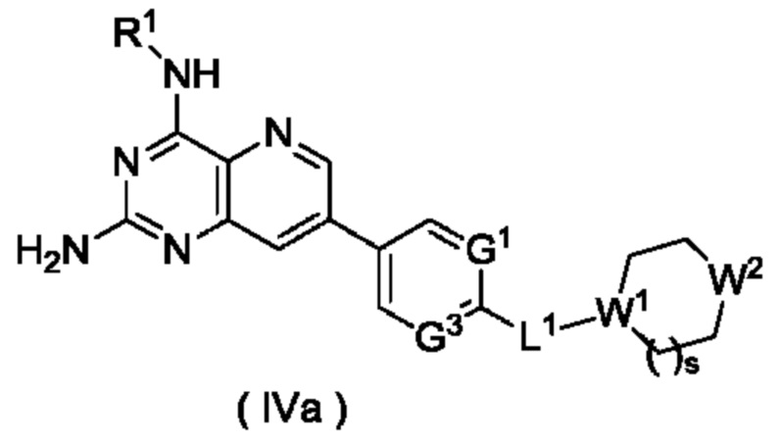
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемую соль,
где:
W1 представляет собой СН, и W2 представляет собой NR6; или
W1 представляет собой N, и W2 представляет собой СН2 или NR6;
R6 выбран из группы, состоящей из атома водорода и алкила, и предпочтительно алкила;
s равно 0 или 1; и
G1, G3, L1 и R1 являются такими, как определено в формуле (I).
В другом предпочтительном воплощении настоящего изобретения соединение формулы (I), формулы (Ia), формулы (Ib), формулы (II) или формулы (III) по настоящему изобретению представляет собой соединение формулы (IV):
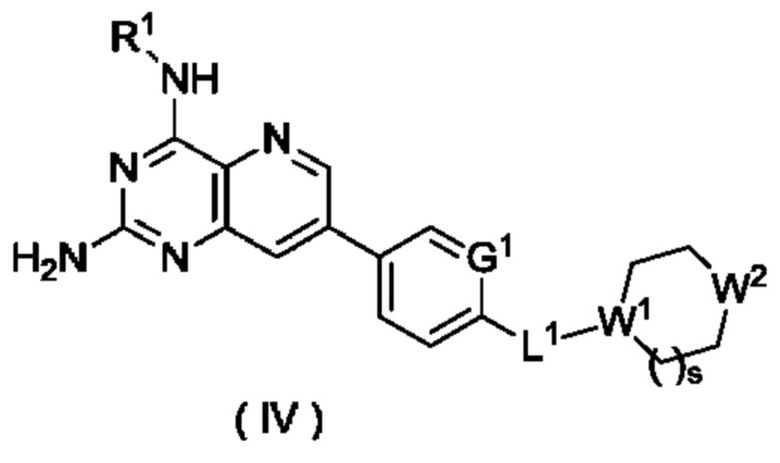
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь либо их фармацевтически приемлемую соль,
где:
W1 представляет собой СН, и W2 представляет собой NR6; или
W1 представляет собой N, и W2 представляет собой СН2 или NR6;
R6 выбран из группы, состоящей из атома водорода и алкила, и предпочтительно алкила;
s равно 0 или 1; и
G1, L1 и R1 являются такими, как определено в формуле (I).
В другом предпочтительном воплощении настоящего изобретения предложено соединение формулы (I), формулы (Ia), формулы (Ib), формулы (II), формулы (III), формулы (IVa) или формулы (IV) по настоящему изобретению, где R1 представляет собой алкил, который возможно замещен одним или несколькими гидрокси; R1 предпочтительно представляет собой С1-12алкил, который возможно замещен одним или несколькими гидрокси.
В другом предпочтительном воплощении настоящего изобретения соединение формулы (I), формулы (Ia), формулы (Ib), формулы (II), формулы (III), формулы (IVa) или формулы (IV) по настоящему изобретению представляет собой соединение формулы (Va):
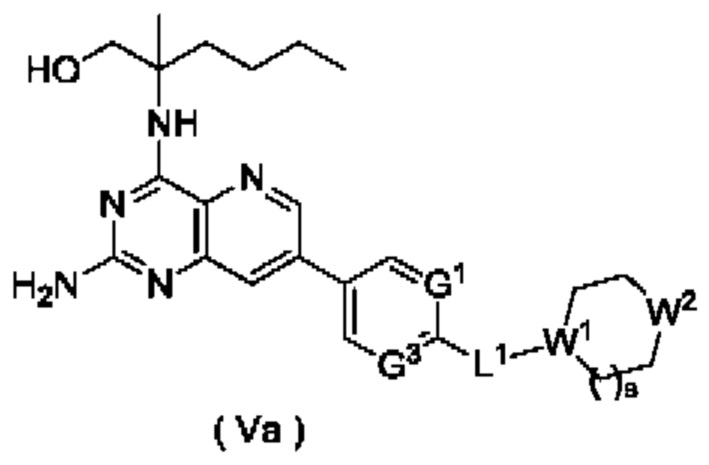
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемую соль,
где:
W1 представляет собой СН, и W2 представляет собой NR6; или
W1 представляет собой N, и W2 представляет собой СН2 или NR6;
R6 выбран из группы, состоящей из атома водорода и алкила, и предпочтительно алкила;
s равно 0 или 1; и
G1, G3 и L1 являются такими, как определено в формуле (I).
В другом предпочтительном воплощении настоящего изобретения соединение формулы (I), формулы (Ia), формулы (Ib), формулы (II), формулы (III), формулы (IVa) или формулы (IV) по настоящему изобретению представляет собой соединение формулы (V):
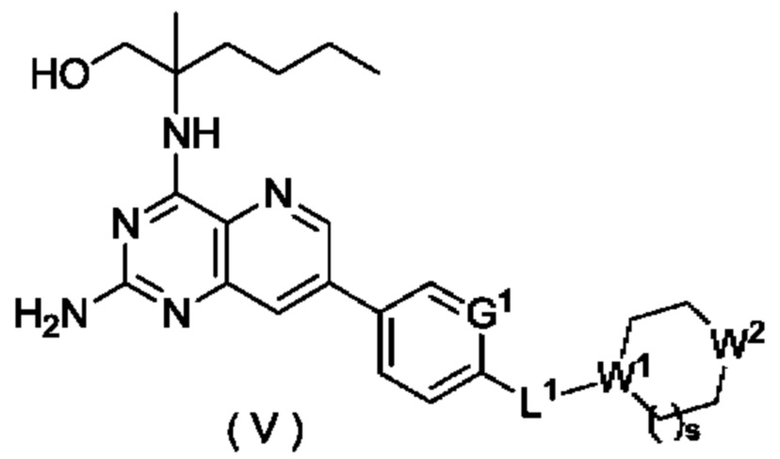
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемую соль,
где:
W1 представляет собой СН, и W2 представляет собой NR6; или
W1 представляет собой N, и W2 представляет собой СН2 или NR6;
R6 выбран из группы, состоящей из атома водорода и алкила, и предпочтительно алкила;
s равно 0 или 1; и
G1 и L1 являются такими, как определено в формуле (I).
В другом предпочтительном воплощении настоящего изобретения предложено соединение формулы (I), формулы (Ia), формулы (Ib), формулы (II), формулы (III), формулы (IVa), формулы (IV), формулы (Va) или формулы (V) по настоящему изобретению, где L1 представляет собой группу -(СН2)n- или ковалентную связь, где n представляет собой целое число от 1 до 6; и L1 предпочтительно представляет собой группу -(СН2)n- или ковалентную связь.
Соединения формулы (I) по настоящему изобретению обычно включают, но не ограничиваются этим, приведенные ниже.
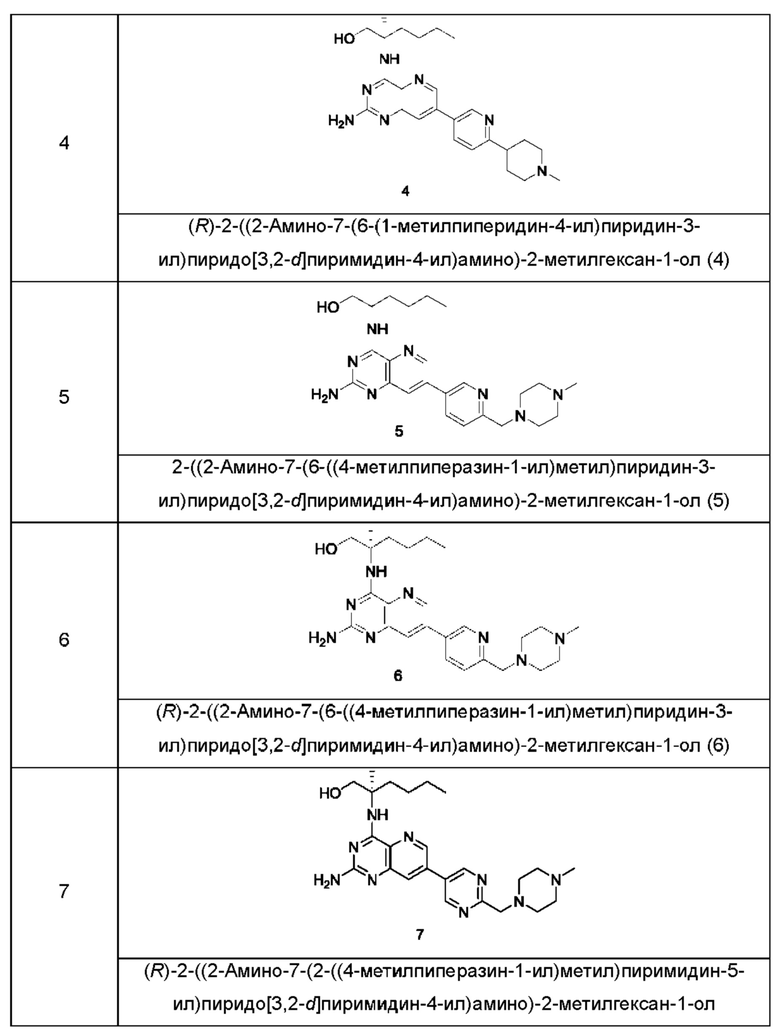
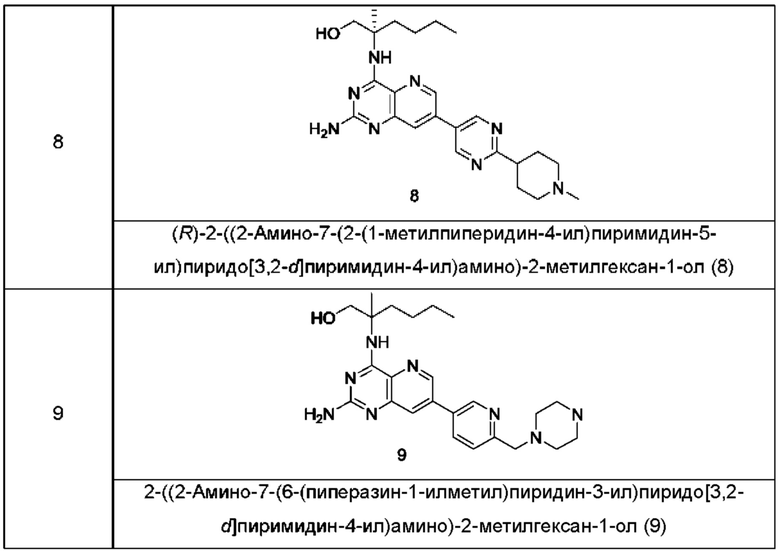
или их таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемую соль.
Согласно другому аспекту настоящее изобретение относится к соединению формулы (IB):
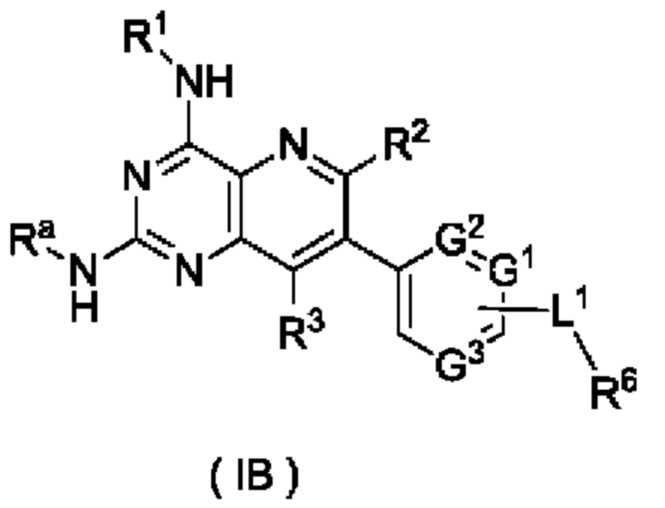
или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смеси, либо их фармацевтически приемлемой соли, которое представляет собой промежуточное соединение для получения соединения формулы (I),
где:
Ra представляет собой амино-защитную группу и предпочтительно 2,4-диметоксибензил;
G1, G2 и G3 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из СН, CR5 и N;
L1 выбран из группы, состоящей из алкилена и ковалентной связи, при этом указанный алкилен возможно замещен одним заместителем или несколькими заместителями, выбранным(ыми) из группы, состоящей из галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила и гетероциклила;
R1 выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила, при этом указанный алкил, циклоалкил, гетероциклил, арил и гетероарил каждый возможно независимо замещен одним заместителем или несколькими заместителями, выбранным(ыми) из группы, состоящей из галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила;
R2 и R3 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила;
R6 выбран из группы, состоящей из алкила, галогеналкила, циклоалкила, гетероциклила, арила и гетероарила, при этом указанный алкил, циклоалкил, гетероциклил, арил и гетероарил каждый возможно независимо замещен одним заместителем или несколькими заместителями, выбранным(ыми) из группы, состоящей из алкила, алкокси, галогена, амино, циано, нитро, гидрокси, гидроксиалкила, трет-бутоксикарбонила (ВОС), циклоалкила, гетероциклила, арила и гетероарила; и
R5 выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила.
В предпочтительном воплощении соединение формулы (IB) по настоящему изобретению представляет собой соединение формулы (IA):
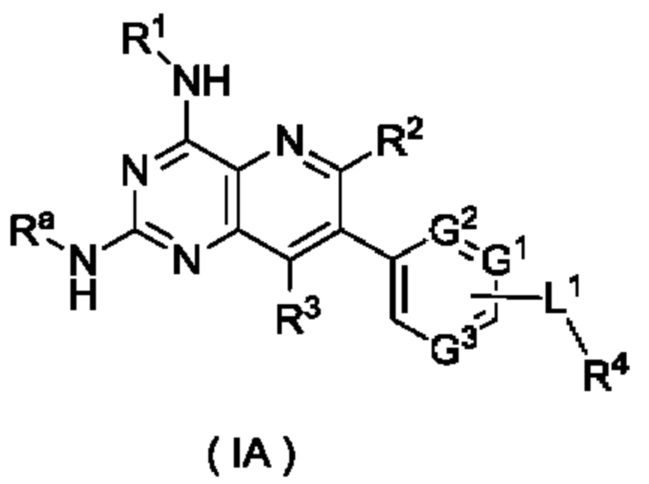
или его таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемую соль, которое представляет собой промежуточное соединение для получения соединения формулы (I),
где:
Ra представляет собой амино-защитную группу и предпочтительно 2,4-диметоксибензил;
G1, G2 и G3 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из СН, CR5 и N;
L1 выбран из группы, состоящей из алкилена и ковалентной связи, при этом указанный алкилен возможно замещен одним заместителем или несколькими заместителями, выбранным(ыми) из группы, состоящей из галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила и гетероциклила;
R1 выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила, при этом указанный алкил, циклоалкил, гетероциклил, арил и гетероарил каждый возможно независимо замещен одним заместителем или несколькими заместителями, выбранным(ыми) из группы, состоящей из галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила;
R2 и R3 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила;
R4 выбран из группы, состоящей из алкила, галогеналкила, циклоалкила, гетероциклила, арила и гетероарила, при этом указанный алкил, циклоалкил, гетероциклил, арил и гетероарил каждый возможно независимо замещен одним заместителем или несколькими заместителями, выбранным(ыми) из группы, состоящей из алкила, алкокси, галогена, амино, циано, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила; и
R5 выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила.
Соединения формулы (IB) по настоящему изобретению обычно включают, но не ограничиваются этим, приведенные ниже.
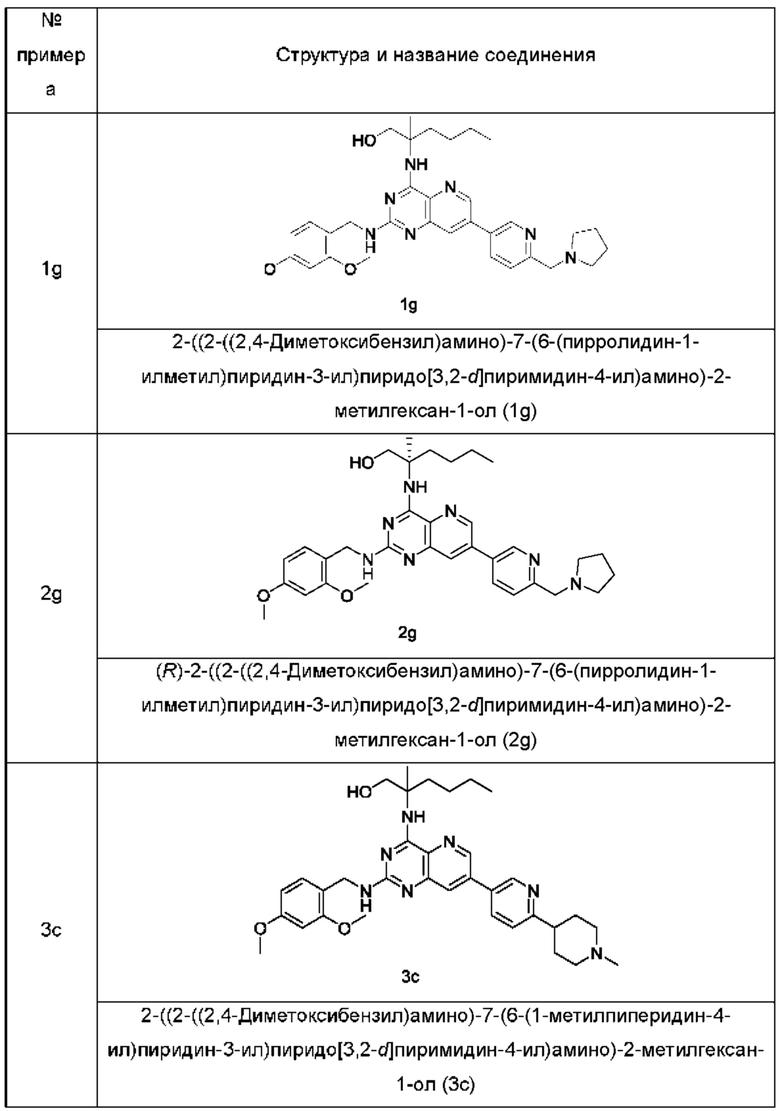
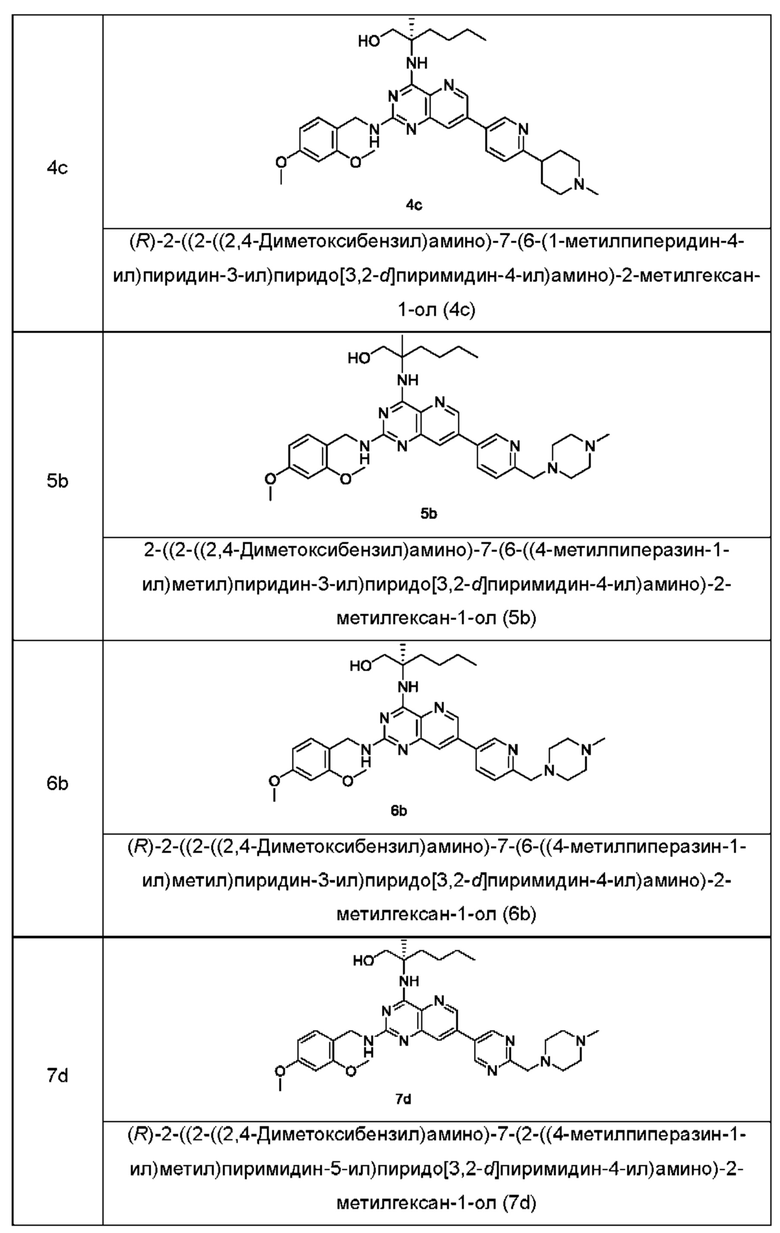
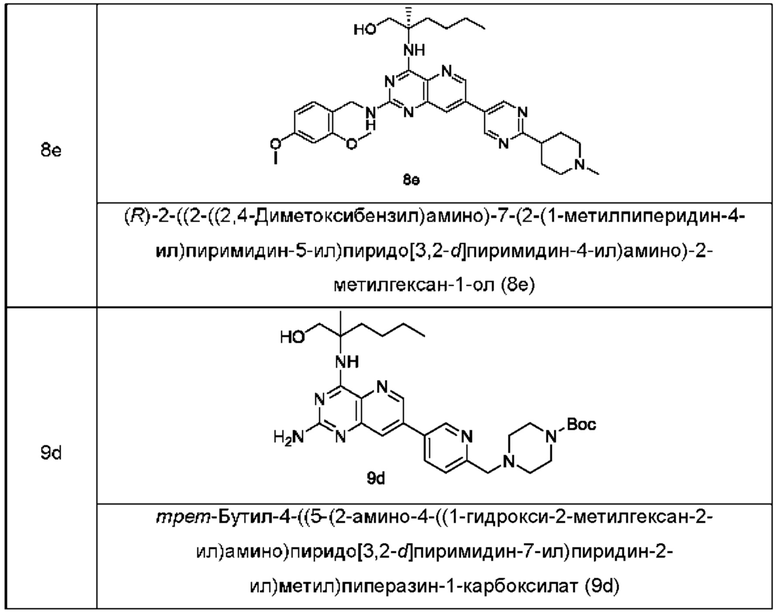
или их таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемую соль.
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (I) по настоящему изобретению, включающему стадию:
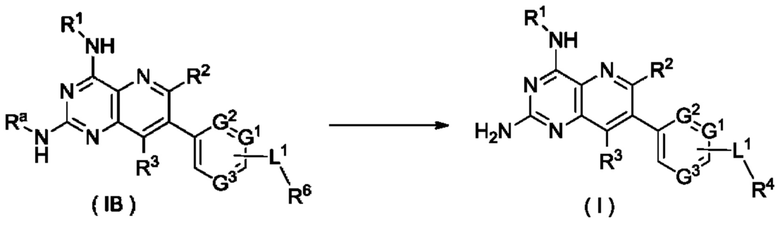
подвергания соединения формулы (IB) реакции удаления защиты с получением соединения формулы (I);
где:
Ra представляет собой амино-защитную группу, и предпочтительно 2,4-диметоксибензил; и
G1, G2 и G3 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из СН, CR5 и N;
L1 выбран из группы, состоящей из алкилена и ковалентной связи, при этом указанный алкилен возможно замещен одним заместителем или несколькими заместителями, выбранным(ыми) из группы, состоящей из галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила и гетероциклила;
R1 выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила, при этом указанный алкил, циклоалкил, гетероциклил, арил и гетероарил каждый возможно независимо замещен одним заместителем или несколькими заместителями, выбранным(ыми) из группы, состоящей из галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила;
R2 и R3 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила;
R4 выбран из группы, состоящей из алкила, галогеналкила, циклоалкила, гетероциклила, арила и гетероарила, при этом указанный алкил, циклоалкил, гетероциклил, арил и гетероарил каждый возможно независимо замещен одним заместителем или несколькими заместителями, выбранным(ыми) из группы, состоящей из алкила, алкокси, галогена, амино, циано, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила; и
R6 выбран из группы, состоящей из алкила, галогеналкила, циклоалкила, гетероциклила, арила и гетероарила, при этом указанный алкил, циклоалкил, гетероциклил, арил и гетероарил каждый возможно независимо замещен одним заместителем или несколькими заместителями, выбранным(ыми) из группы, состоящей из алкила, алкокси, галогена, амино, циано, нитро, гидрокси, гидроксиалкила, трет-бутоксикарбонила, циклоалкила, гетероциклила, арила и гетероарила; и
R5 выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила.
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (I) по настоящему изобретению, включающему стадию:
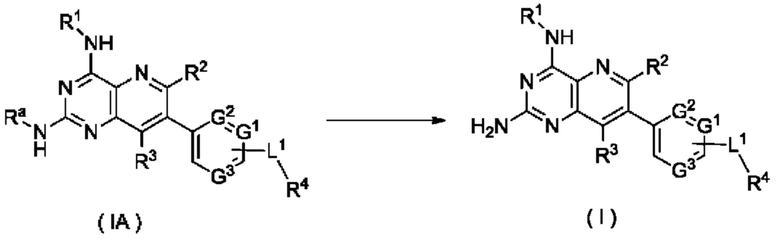
подвергания соединения формулы (IA) реакции удаления защиты с получением соединения формулы (I);
где:
Ra представляет собой амино-защитную группу, и предпочтительно 2,4-диметоксибензил; и
G1-G3, L1 и R1-R4 являются такими, как определено в формуле (I).
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (Ia) по настоящему изобретению, включающему стадию:
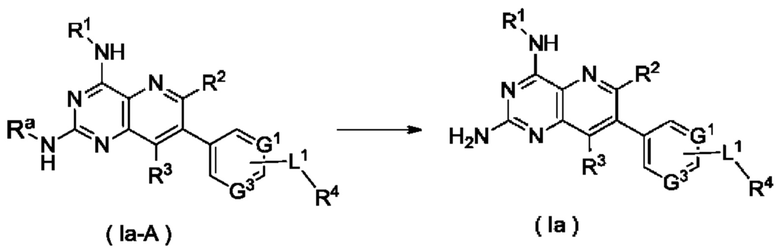
подвергания соединения формулы (Ia-A) реакции удаления защиты с получением соединения формулы (Ia);
где:
Ra представляет собой амино-защитную группу, и предпочтительно 2,4-диметоксибензил; и
G1, G3, L1 и R1-R4 являются такими, как определено в формуле (Ia).
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (Ib) по настоящему изобретению, включающему стадию:
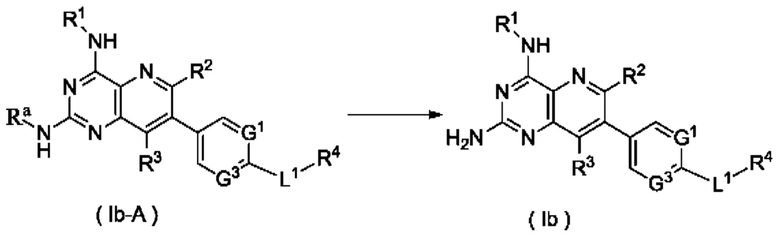
подвергания соединения формулы (Ib-A) реакции удаления защиты с получением соединения формулы (Ib);
где:
Ra представляет собой амино-защитную группу, и предпочтительно 2,4-диметоксибензил; и
G1, G3, L1 и R1-R4 являются такими, как определено в формуле (Ib).
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (II) по настоящему изобретению, включающему стадию:
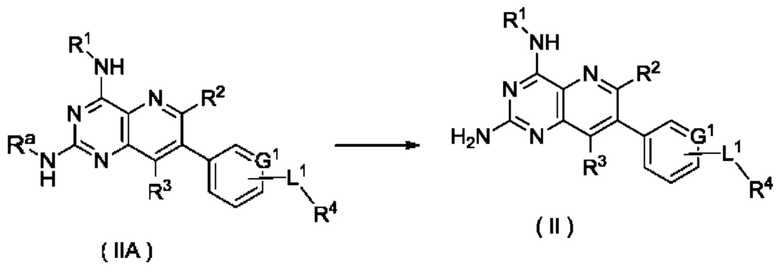
подвергания соединения формулы (IIA) реакции удаления защиты с получением соединения формулы (II);
где:
Ra представляет собой амино-защитную группу, и предпочтительно 2,4-диметоксибензил; и
G1, L1 и R1-R4 являются такими, как определено в формуле (II).
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (III) по настоящему изобретению, включающему стадию:
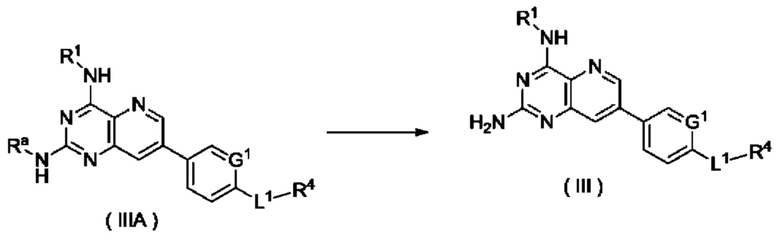
подвергания соединения формулы (IIIA) реакции удаления защиты с получением соединения формулы (III);
где:
Ra представляет собой амино-защитную группу, и предпочтительно 2,4-диметоксибензил; и
G1, L1 и R1-R4 являются такими, как определено в формуле (III).
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (IV) по настоящему изобретению, включающему стадию:
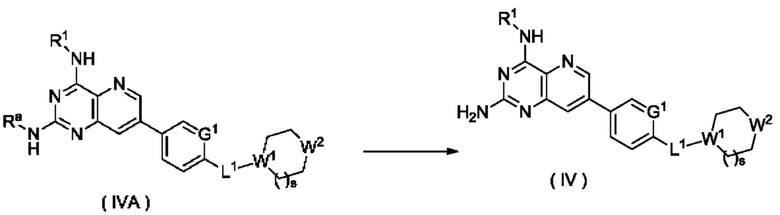
подвергания соединения формулы (IVA) реакции удаления защиты с получением соединения формулы (IV);
где:
Ra представляет собой амино-защитную группу, и предпочтительно 2,4-диметоксибензил; и
G1, L1, R1, W1, W2 и s являются такими, как определено в формуле (IV).
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (Va) по настоящему изобретению, включающему стадию:
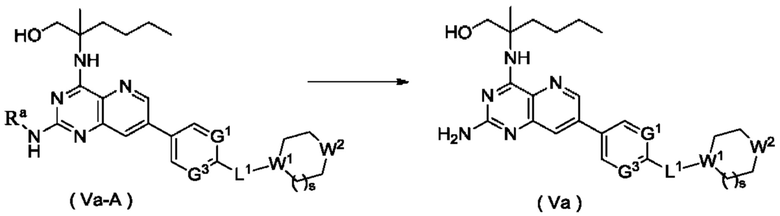
подвергания соединения формулы (Va-A) реакции удаления защиты с получением соединения формулы (Va);
где:
Ra представляет собой амино-защитную группу, и предпочтительно 2,4-диметоксибензил; и
G1, L1, R1, W1, W2 и s являются такими, как определено в формуле (Va).
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (V) по настоящему изобретению, включающему стадию:
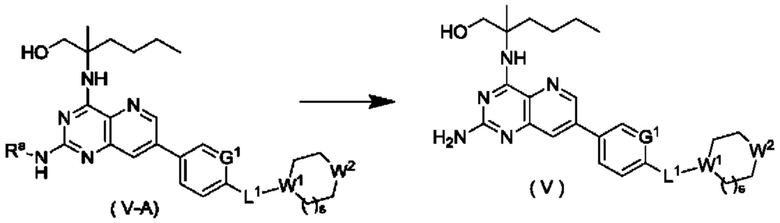
подвергания соединения формулы (V-A) реакции удаления защиты с получением соединения формулы (V);
где:
Ra представляет собой амино-защитную группу, и предпочтительно 2,4-диметоксибензил; и
G1, L1, R1, W1, W2 и s являются такими, как определено в формуле (V).
Согласно настоящему изобретению также предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент.
Настоящее изобретение также относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению или содержащей их фармацевтической композиции для приготовления лекарственного средства для активации TLR8.
Настоящее изобретение также относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению или содержащей их фармацевтической композиции для приготовления лекарственного средства для лечения инфекции, вызываемой вирусом, при этом вирус предпочтительно представляет собой вирус гепатита В, вирус гепатита С, вирус гриппа, вирус герпеса и вирус, вызывающий синдром приобретенного иммунодефицита (СПИД).
Настоящее изобретение также относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению или содержащей их фармацевтической композиции для приготовления лекарственного средства для регулирования иммунной системы.
Настоящее изобретение также относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению или содержащей их фармацевтической композиции для приготовления лекарственного средства для лечения или предупреждения возникновения опухоли.
Настоящее изобретение также относится к способу активации TLR8, включающему стадию приведения в контакт соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению или содержащей их фармацевтической композиции с TLR8.
Настоящее изобретение также относится к способу лечения инфекции, вызываемой вирусом, включающему стадию введения терапевтически эффективной дозы соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению или содержащей их фармацевтической композиции пациенту, нуждающемуся в этом, при этом вирус предпочтительно представляет собой вирус гепатита В, вирус гепатита С, вирус гриппа, вирус герпеса и вирус, вызывающий СПИД.
Настоящее изобретение также относится к способу лечения или предупреждения возникновения опухоли, включающему стадию введения терапевтически или превентивно эффективной дозы соединения формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению или содержащей их фармацевтической композиции пациенту, нуждающемуся в этом.
Настоящее изобретение также относится к соединению формулы (I), или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению или содержащей их фармацевтической композиции для применения в качестве лекарственного средства.
Настоящее изобретение также относится к соединению формулы (I), или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению или содержащей их фармацевтической композиции по настоящему изобретению для применения в качестве агониста TLR8.
Настоящее изобретение также относится к соединению формулы (I), или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению или содержащей их фармацевтической композиции для применения в качестве лекарственного средства для лечения инфекции, вызываемой вирусом, при этом вирус предпочтительно представляет собой вирус гепатита В, вирус гепатита С, вирус гриппа, вирус герпеса и вирус, вызывающий СПИД.
Настоящее изобретение также относится к соединению формулы (I). или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению или содержащей их фармацевтической композиции для применения в качестве лекарственного средства регулирования иммунной системы.
Настоящее изобретение также относится к соединению формулы (I), или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению или содержащей их фармацевтической композиции для применения в качестве лекарственного средства для лечения или предупреждения возникновения опухоли.
Опухоль, согласно настоящему изобретению, предпочтительно представляет собой рак и более предпочтительно рак, выбранный из группы, состоящей из меланомы, рака легкого, рака печени, базальноклеточной карциномы, рака почки, миеломы, рака желчевыводящих путей, рака головного мозга, рака молочной железы, рака шейки матки, хориокарциномы, рака толстой кишки, рака прямой кишки, рака головы и шеи, перитонеальной опухоли, рака фаллопиевых труб, рака эндометрия, рака пищевода, рака желудка, лейкоза, лимфомы, саркомы, нейробластомы, рака полости рта, рака яичников, рака поджелудочной железы, рака предстательной железы, рака яичка, рака кожи и рака щитовидной железы.
Доза соединения или композиции, используемая в способе лечения по настоящему изобретению, обычно будет варьировать в зависимости от тяжести заболевания, массы пациента и относительной эффективности соединения. Однако, как правило, подходящая стандартная доза может составлять от 0,1 до 1000 мг.
Помимо активного соединения фармацевтическая композиция по настоящему изобретению также может содержать одно или более чем одно вспомогательное вещество, включая наполнитель, связующее вещество, увлажняющий агент, разрыхлитель, эксципиент и тому подобное. В зависимости от способа введения композиция может содержать активное соединение в количестве от 0,1 до 99 масс. %.
Фармацевтическая композиция, содержащая активный ингредиент, может быть в форме, подходящей для перорального введения, например, в форме таблетки, лепешки, пастилки, водной или масляной суспензии, диспергируемого порошка или гранулы, эмульсии, твердой или мягкой капсулы, сиропа или эликсира. Композиция для перорального введения может быть приготовлена в соответствии с любым способом, известным в данной области техники для приготовления фармацевтической композиции. Такая композиция может содержать один или более ингредиентов, выбранный(ых) из группы, состоящей из подсластителей, корригентов, красителей и консервантов, для придания фармацевтической композиции привлекательного и приятного вкуса. Таблетка содержит активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, подходящими для изготовления таблеток. Этими эксципиентами могут быть инертные эксципиенты, гранулирующие агенты, разрыхлители, связующие вещества и смазывающие вещества. Таблетка может не иметь покрытия или быть покрыта с использованием метода, известного для маскировки вкуса лекарственного средства или задержки распадаемости и всасывания активного ингредиента в желудочно-кишечном тракте, что будет обеспечивать замедленное высвобождение в течение длительного периода времени.
Композиция для перорального применения также может быть представлена в виде мягких желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем либо активный ингредиент смешан с водорастворимым носителем или масляной средой.
Водная суспензия содержит активный ингредиент в смеси с эксципиентами, подходящими для изготовления водной суспензии. Такие эксципиенты представляют собой суспендирующие агенты, диспергирующие вещества или увлажняющие агенты. Водная суспензия также может содержать один или более консервантов, один или более красителей, один или более корригентов и один или более подсластителей.
Масляная суспензия может быть приготовлена путем суспендирования активного ингредиента в растительном масле или минеральном масле. Масляная суспензия может содержать загуститель. Для придания композиции приятного вкуса могут быть добавлены вышеупомянутые подсластители и корригенты. Сохранение этих композиций может быть обеспечено добавлением антиоксиданта.
Фармацевтическая композиция по настоящему изобретению также может быть в форме эмульсии типа масло-в-воде. Масляная фаза может представлять собой растительное масло или минеральное масло либо их смесь. Подходящими эмульгирующими агентами могут быть природные фосфолипиды. Эмульсия также может содержать подсластитель, корригент, консервант и антиоксидант. Такая композиция также может содержать смягчающее вещество, консервант, краситель и антиоксидант.
Фармацевтическая композиция по настоящему изобретению может быть в форме стерильного инъекционного водного раствора. Приемлемыми наполнителями или растворителями, которые можно использовать, являются вода, раствор Рингера или изотонический раствор хлорида натрия. Стерильная композиция для инъекций может представлять собой стерильную инъекционную микроэмульсию типа масло-в-воде, в которой активный ингредиент растворен в масляной фазе. Раствор или микроэмульсия для инъекций могут быть введены в кровоток пациента посредством местной болюсной инъекции. Альтернативно, раствор и микроэмульсию предпочтительно вводят способом, при котором в циркулирующей крови поддерживается постоянная концентрация соединения по настоящему изобретению. Для поддержания этой постоянной концентрации можно использовать устройство для непрерывной внутривенной доставки. Примером такого устройства является насос для внутривенных инъекций Deltec CADD-PLUS™ 5400.
Фармацевтическая композиция может быть в форме стерильной инъекционной водной или масляной суспензии для внутримышечного и подкожного введения. Такая суспензия может быть приготовлена вместе с подходящими диспергирующими веществами или увлажняющими агентами и суспендирующими агентами, которые описаны выше, согласно известным методам. Стерильная композиция для инъекций также может представлять собой стерильный раствор или суспензию для инъекций, приготовленные в нетоксичном парентерально приемлемом разбавителе или растворителе. Кроме того, в качестве растворителя или суспендирующей среды вполне могут быть использованы стерильные нелетучие масла.
Соединение по настоящему изобретению может быть введено в форме суппозитория для ректального введения. Такие фармацевтические композиции могут быть приготовлены путем смешивания лекарственного средства с подходящим не вызывающим раздражения эксципиентом, который является твердым при обычных температурах, но жидким в прямой кишке, в результате чего претерпевает плавление в прямой кишке с высвобождением лекарственного средства. Такие вещества включают масло какао, глицерин, желатин, гидрогенизированное растительное масло, смесь полиэтиленгликолей различных молекулярных масс и их сложные эфиры с жирными кислотами.
Специалистам в данной области хорошо известно, что дозировка лекарственного средства зависит от ряда факторов, включая, но не ограничиваясь этим, следующие факторы: активность конкретного соединения, возраст пациента, вес пациента, общее состояние здоровья пациента, поведение пациента, рацион пациента, время введения, путь введения, скорость экскреции, комбинация лекарственных средств и тому подобное. Помимо этого, оптимальное лечение, например, способ лечения, суточную дозу соединения формулы (I) или тип его фармацевтически приемлемой соли, можно контролировать с учетом традиционных схем лечения.
Подробное описание изобретения
Если не указано иное, термины, использованные в данном описании и формуле изобретения, имеют описанные ниже значения.
Термин "алкил" относится к насыщенной алифатической углеводородной группе, которая представляет собой группу с линейной или разветвленной цепью, содержащую 1-20 атомов углерода, предпочтительно алкил, имеющий 1-12 атомов углерода и более предпочтительно алкил, имеющий 1-6 атомов углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их различные разветвленные изомеры. Более предпочтительно, алкильная группа представляет собой низший алкил, имеющий 1-6 атомов углерода, и неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и тому подобное. Алкильная группа может быть замещенной или незамещенной. При наличии замещения замещающая группа может быть замещена по любому доступному месту присоединения. Замещающей(ими) группой(ами) предпочтительно являет(ют)ся одна или более групп, независимо выбранная(ых) из группы, состоящей из галогена, алкила, галогеналкила, алкокси, галогеналкокси, алкилтио, алкиламино, алкенила, алкинила, тиола, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио и оксо.
Термин "алкокси" означает группу -О-(алкил) или -O-(незамещенный циклоалкил), где алкил и циклоалкил являются такими, как определено выше. Неограничивающие примеры включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси. Группа алкокси возможно может быть замещенной или незамещенной. При наличии замещения замещаемая группа может быть замещена по любому доступному месту присоединения. Замещающая группа представляет собой одну группу или несколько групп, независимо выбранную(ых) из группы, состоящей из галогена, алкила, галогеналкила, алкокси, галогеналкокси, алкилтио, алкиламино, алкенила, алкинила, тиола, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио и оксо.
Термин "циклоалкил" относится к насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной замещаемой группе, имеющей 3-20 атомов углерода, предпочтительно 3-12 атомов углерода, более предпочтительно 3-6 атомов углерода (например, 3, 4, 5 или 6 атомов углерода) и наиболее предпочтительно 5-6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и тому подобное. Полициклический циклоалкил включает циклоалкил, имеющий спиро-кольцо, конденсированное кольцо или мостиковое кольцо.
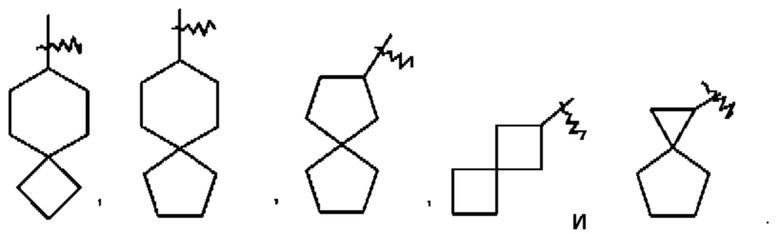
Термин "спиро-циклоалкил" относится к 5-20-членной полициклической группе, состоящей из отдельных колец, соединенных через один общий атом углерода (называемый спиро-атом), при этом данные кольца могут содержать одну двойную связь или несколько двойных связей, но ни одно из этих колец не имеет полностью сопряженной π-электронной системы. Спиро-циклоалкилом предпочтительно является 6-14-членный спиро-циклоалкил и более предпочтительно 7-10-членный спиро-циклоалкил (например, 7-, 8-, 9- или 10-членный спиро-циклоалкил). В зависимости от количества спиро-атомов, общих для этих колец, спиро-циклоалкилы можно подразделять на моно-спиро-циклоалкил, ди-спиро-циклоалкил или поли-спиро-циклоалкил, и спиро-циклоалкилом предпочтительно является моно-спиро-циклоалкил или ди-спиро-циклоалкил, более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моно-спиро-циклоалкил. Неограничивающие примеры спиро-циклоалкилов включают:
Термин "конденсированный циклоалкил" относится к 5-20-членной состоящей полностью из атомов углерода полициклической группе, при этом каждое кольцо в такой системе имеет общую с другим кольцом соседнюю пару атомов углерода, причем одно кольцо или несколько колец могут содержать одну или несколько двойных связей, но ни одно из этих колец не имеет полностью сопряженной π-электронной системы. Конденсированный циклоалкил предпочтительно представляет собой 6-14-членный конденсированный циклоалкил и более предпочтительно 7-10-членный конденсированный циклоалкил (например, 7-, 8-, 9- или 10-членный конденсированный циклоалкил). В зависимости от количества указанных колец конденсированные циклоалкилы можно подразделять на бициклический, трициклический, тетрациклический или полициклический конденсированный циклоалкил, и конденсированный циклоалкил предпочтительно представляет собой бициклический или трициклический конденсированный циклоалкил и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный циклоалкил. Неограничивающие примеры конденсированного циклоалкила включают:
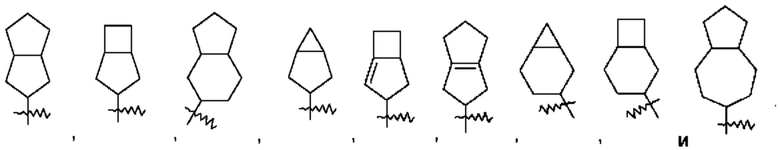
Термин "мостиковый циклоалкил" относится к 5-20-членной состоящей полностью из атомов углерода полициклической группе, при этом каждые два кольца в этой системе имеют два общих несопряженных атома углерода, причем данные кольца могут иметь одну двойную связь или несколько двойных связей, но ни одно из этих колец не имеет полностью сопряженной π-электронной системы. Мостиковый циклоалкил предпочтительно представляет собой 6-14-членный мостиковый циклоалкил и более предпочтительно 7-10-членный мостиковый циклоалкил (например, 7-, 8-, 9- или 10-членный мостиковый циклоалкил). В зависимости от количества указанных колец мостиковые циклоалкилы можно подразделять на бициклический, трициклический, тетрациклический или полициклический мостиковый циклоалкил, и мостиковый циклоалкил предпочтительно представляет собой бициклический, трициклический или тетрациклический мостиковый циклоалкил и более предпочтительно бициклический или трициклический мостиковый циклоалкил. Неограничивающие примеры мостикового циклоалкила включают:
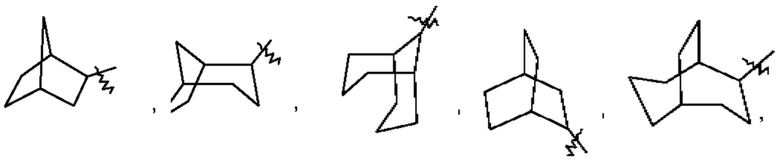
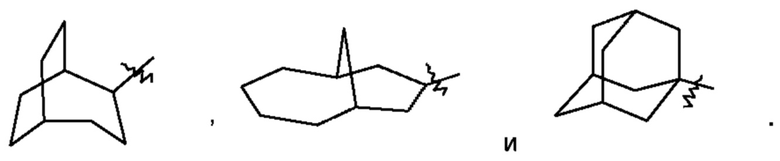
Такое циклоалкильное кольцо может быть конденсировано с кольцом арила, гетероарила или гетероциклила, при этом кольцом, связанным с исходной структурой, является циклоалкил. Неограничивающие примеры включают инданил, тетрагидронафтил, бензоциклогептил и тому подобное и предпочтительно бензоциклогептил, тетрагидронафтил. Циклоалкил возможно может быть замещенным или незамещенным. При наличии замещения замещаемая группа может быть замещена по любому доступному месту присоединения. Замещающей(ими) группой(ами) являет(ют)ся одна группа или несколько групп, независимо выбранная(ых) из группы, состоящей из группы, состоящей из галогена, алкила, галогеналкила, алкокси, галогеналкокси, алкилтио, алкиламино, алкенила, алкинила, тиола, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио и оксо.
Термин "гетероциклил" относится к 3-20-членной насыщенной или частично ненасыщенной моноциклической либо полициклической углеводородной группе, при этом один атом или несколько атомов в кольце представляют собой гетероатомы, выбранные из группы, состоящей из N, О и S(O)m (где m представляет собой целое число, выбранное из 0-2), но за исключением -О-О-, -O-S- или -S-S- в кольце, и при этом остальные атомы в кольце представляют собой атомы углерода. Предпочтительно, гетероциклил имеет 3-12 атомов в кольце, при этом 1-4 атома представляют собой гетероатомы; более предпочтительно 3-8 атомов в кольце, при этом 1-3 атома представляют собой гетероатомы; и наиболее предпочтительно 5-6 атомов в кольце, при этом 1-2 или 1-3 атома представляют собой гетероатомы. Неограничивающие примеры моноциклического гетероциклила включают пирролидинил, имидазолидинил, тетрагидрофуранил, тетрагидропиранил, тетрагидротиенил, дигидроимидазолил, дигидрофуранил, дигидропиразолил, дигидропирролил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и тому подобное и предпочтительно тетрагидропиранил, пиперидинил, пирролинил. Пол и циклический гетероциклил включает гетероциклил, имеющий спиро-кольцо, конденсированное кольцо или мостиковое кольцо.
Термин "спиро-гетероциклил" относится к 5-20-членной полициклической гетеро цикл ильной группе, содержащей отдельные кольца, соединенные через один общий атом (называемый спиро-атомом), при этом один атом или несколько атомов в кольце представляют собой гетероатомы, выбранные из группы, состоящей из N, О и S(O)m (где m представляет собой целое число, выбранное из 0-2) и при этом остальные атомы в кольце представляют собой атомы углерода. Такой спиро-гетероциклил может содержать одну двойную связь или несколько двойных связей, но ни одно из его колец не имеет полностью сопряженной π-электронной системы. Спиро-гетероциклил предпочтительно представляет собой 6-14-членный спиро-гетероциклил и более предпочтительно 7-10-членный спиро-гетероциклил. В зависимости от количества спиро-атомов, общих для этих колец, спиро-гетероциклилы можно подразделять на моно-спиро-гетероциклил, ди-спиро-гетероциклил или поли-спиро-гетероциклил и спиро-гетероциклил предпочтительно представляет собой моно-спиро-гетероциклил или ди-спиро-гетероциклил и более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моно-спиро-гетероциклил. Неограничивающие примеры спиро-гетероциклила включают:
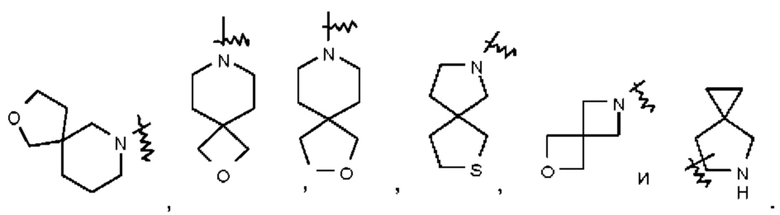
Термин "конденсированный гетероциклил" относится к 5-20-членной полициклической гетеро цикл ильной группе, при этом каждое кольцо в такой системе имеет общую с другим кольцом соседнюю пару атомов, причем одно кольцо или несколько колец могут содержать одну двойную связь или несколько двойных связей, но ни одно из этих колец не имеет полностью сопряженной π-электронной системы и один атом или несколько атомов в кольце представляют собой гетероатомы, выбранные из группы, состоящей из N, О и S(O)m (где m представляет собой целое число, выбранное из 0-2), при этом остальные атомы в кольце представляют собой атомы углерода. Конденсированный гетероциклил предпочтительно представляет собой 6-14-членный конденсированный гетероциклил и более предпочтительно 7-10-членный конденсированный гетероциклил (например, 7-, 8-, 9- или 10-членный конденсированный гетероциклил). В зависимости от количества указанных колец конденсированные гетероциклилы можно подразделять на бициклический, трициклический, тетрациклический или пол и циклический конденсированный гетероциклил и конденсированный гетероциклил предпочтительно представляет собой бициклический или трициклический конденсированный гетероциклил и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный гетероциклил. Неограничивающие примеры конденсированного гетероциклила включают:
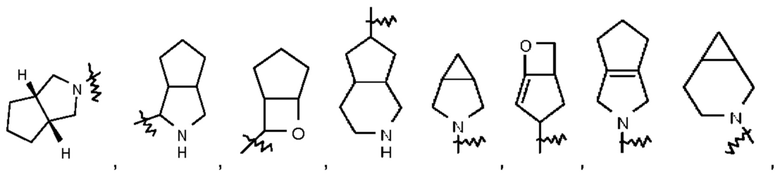
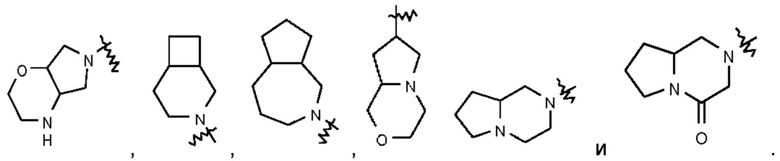
Термин "мостиковый гетероциклил" относится к 5-14-членной полициклической гетеро цикл ильной группе, при этом каждые два кольца в этой системе имеют два общих несопряженных атома, причем данные кольца могут иметь одну двойную связь или несколько двойных связей, но ни одно из этих колец не имеет полностью сопряженной π-электронной системы и один атом или несколько атомов в кольце представляют собой гетероатомы, выбранные из группы, состоящей из N, О и S(O)m (где m представляет собой целое число, выбранное из 0-2), при этом остальные атомы в кольце представляют собой атомы углерода. Мостиковый гетероциклил предпочтительно представляет собой 6-14-членный мостиковый гетероциклил и более предпочтительно 7-10-членный мостиковый гетероциклил (например, 7-, 8-, 9- или 10-членный мостиковый гетероциклил). В зависимости от количества указанных колец мостиковые гетероциклилы можно подразделять на бициклический, трициклический, тетрациклический или полициклический мостиковый гетероциклил и мостиковый гетероциклил предпочтительно представляет собой бициклический, трициклический или тетрациклический мостиковый гетероциклил и более предпочтительно бициклический или трициклический мостиковый гетероциклил. Неограничивающие примеры мостикового гетероциклила включают:
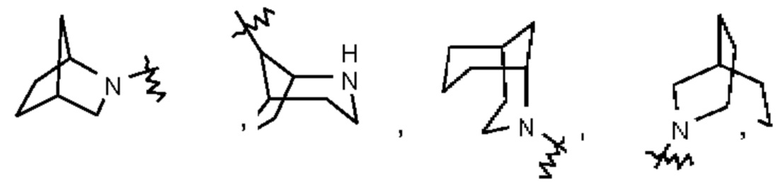
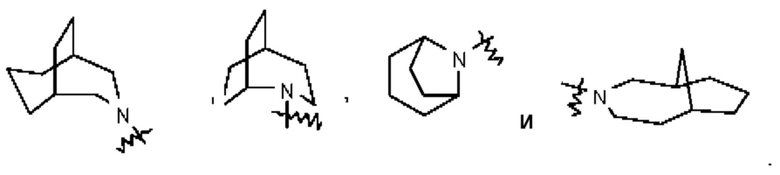
Такое гетероциклильное кольцо может быть конденсировано с арилом, гетероарилом или циклоалкилом, при этом кольцом, связанным с исходной структурой, является гетероциклил. Их неограничивающие примеры включают:
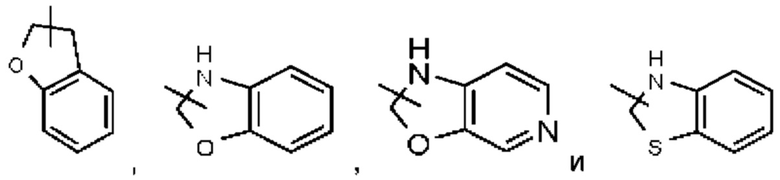 и тому подобное.
и тому подобное.
Гетероциклил возможно может быть замещенным или незамещенным. При наличии замещения замещаемая группа может быть замещена по любому доступному месту присоединения. Замещающей(ими) группой(ами) являет(ют)ся одна группа или несколько групп, возможно независимо выбранная(ых) из группы, состоящей из галогена, алкила, галогеналкила, алкокси, галогеналкокси, алкилтио, алкиламино, алкенила, алкинила, тиола, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио и оксо.
Термин "арил" относится к 6-14-членному состоящему полностью из атомов углерода моноциклическому кольцу или полициклическому конденсированному кольцу (т.е. каждое кольцо в такой системе имеет общую с другим кольцом в данной системе соседнюю пару атомов углерода), имеющему сопряженную π-электронную систему, предпочтительно относится к 6-10-членному арилу и более предпочтительно к 5-6-членному арилу, например, фенилу и нафтилу. Арильное кольцо может быть конденсировано с кольцом гетероарила, гетероциклила или циклоалкила, при этом кольцо, связанное с исходной структурой, представляет собой арильное кольцо. Их неограничивающие примеры включают:
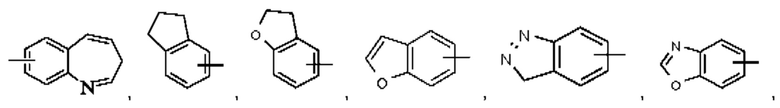

Арил возможно может быть замещенным или незамещенным. При наличии замещения замещаемая группа может быть замещена по любому доступному месту присоединения. Замещающей(ими) группой(ами) являет(ют)ся одна группа или несколько групп, возможно независимо выбранная(ых) из группы, состоящей из галогена, алкила, галогеналкила, алкокси, галогеналкокси, алкилтио, алкиламино, алкенила, алкинила, тиола, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио и оксо.
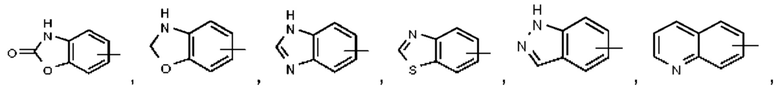
Термин "гетероарил" относится к 5-14-членной гетероароматической системе, имеющей 1-4 гетероатома, выбранных из группы, состоящей из О, S и N. Гетероарил предпочтительно представляет собой 5-10-членный гетероарил, имеющий 1-3 гетероатома, более предпочтительно 5- или 6-членный гетероарил, имеющий 1-2 гетероатома; предпочтительно представляет собой, например, имидазолил, фурил, тиенил, тиазолил, пиразолил, оксазолил, пирролил, 1Н-1,2,3-триазолил, 4Н-1,2,4-триазолил, 4Н-1,2,3-триазолил, 1Н-тетразолил, 2Н-тетразолил, 5Н-тетразолил, пиридил, пиримидинил, тиадиазол, пиразинил и тому подобное, предпочтительно имидазолил, пиразолил, пиримидинил, тиазолил и более предпочтительно пиразолил или имидазолил. Гетероарильное кольцо может быть конденсировано с кольцом арила, гетероциклила или циклоалкила, при этом кольцо, связанное с исходной структурой, представляет собой гетероарильное кольцо. Их неограничивающие примеры включают:
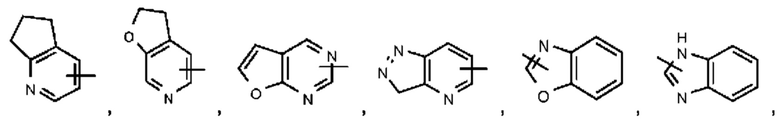
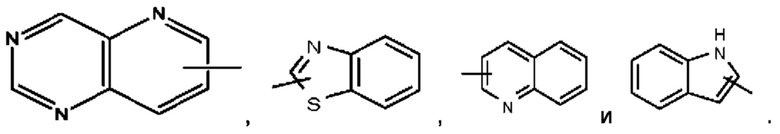
Гетероарил возможно может быть замещенным или незамещенным. При наличии замещения замещаемая группа может быть замещена по любому доступному месту присоединения. Замещающей(ими) группой(ами) являет(ют)ся одна группа или несколько групп, возможно независимо выбранная(ых) из группы, состоящей из галогена, алкила, галогеналкила, алкокси, галогеналкокси, алкилтио, алкиламино, алкенила, алкинила, тиола, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио и оксо.
Термин "амино-защитная группа" относится к группе, которая предохраняет аминогруппу от взаимодействия, когда взаимодействию подвергаются другие части молекулы, и которая может быть легко удалена. Неограничивающие примеры включают трет-бутоксикарбонил, ацетил, бензил, аллил, 2,4-диметоксибензил, пара-метоксибензил и тому подобное. Эти группы возможно могут быть замещены одним-тремя заместителями, выбранными из группы, состоящей из галогена, алкокси и нитро. Амино-защитная группа предпочтительно представляет собой 2,4-диметоксибензил.
Термин "галогеналкил" относится к алкильной группе, замещенной одним атомом или несколькими атомами галогена, при этом алкил является таким, как определено выше.
Термин "галогеналкокси" относится к группе алкокси, замещенной одним атомом или несколькими атомами галогена, при этом группа алкокси является такой, как определено выше.
Термин "гидрокси" относится к группе -ОН.
Термин "гидроксиалкил" относится к алкильной группе, замещенной группой(ами) гидрокси, при этом алкил является таким, как определено выше.
Термин "галоген" относится к фтору, хлору, брому или йоду.
Термин "амино" относится к группе -NH2.
Термин "циано" относится к группе -CN.
Термин "нитро" относится к группе -NO2.
Термин "оксо" относится к группе =O.
"Возможный" или "возможно" означает, что событие или обстоятельство, описываемое впоследствии, может произойти, но происходит не обязательно, и такое описание включает ситуацию, в которой данное событие или обстоятельство может или не может произойти. Например, "гетероциклил, возможно замещенный алкилом", означает, что алкильная группа может присутствовать, но присутствует не обязательно, и такое описание включают ситуацию замещения гетероциклила алкилом и ситуацию, когда гетероциклил не замещен алкилом.
Термин "замещенный" относится к ситуации, когда один или более атомов водорода в группе, предпочтительно до 5 включительно и более предпочтительно от 1 до 3 атомов водорода, независимо замещены соответствующим количеством заместителей. Само собой разумеется, что заместители находятся только в своем возможном с точки зрения химии положении. Специалист в данной области техники способен определить, экспериментально или теоретически, возможно или не возможно такое замещение, без приложения чрезмерных усилий. Например, структура, в которой группа амино или гидрокси со свободным атомом водорода, связана с атомами углерода, имеющими ненасыщенные связи (такие как олефиновые), может быть нестабильной.
"Фармацевтическая композиция" относится к смеси одного или более чем одного из соединений, описанных в данной заявке или их физиологически/фармацевтически приемлемых солей или пролекарств с другими химическими компонентами и другими компонентами, такими как физиологически/фармацевтически приемлемые носители и эксципиенты. Задачей фармацевтической композиции является облегчение введения соединения в организм, что способствует всасыванию активного ингредиента с целью проявления биологической активности.
Термин "фармацевтически приемлемая соль" относится к соли соединения по настоящему изобретению, которая является безопасной и эффективной для млекопитающих и обладает желаемой биологической активностью.
Агонисты TLR8, описанные в предшествующем уровне техники, обладают слабой селективностью в отношении цитохрома Р450 (CYP, от англ. cytochromes Р450) и гена субъединицы калиевого канала сердца человека (hERG, от англ. human Ether a-go-go-Related Gene). Таким образом, по-прежнему существует потребность в продолжении разработки безопасных и терапевтически более эффективных агонистов TLR8.
Принимая во внимание проблемы предшествующего уровня техники, согласно настоящему изобретению предложено фармацевтическое соединение с улучшенной селективностью в отношении CYP и hERG, улучшенной селективностью в отношении TLR8 и более очевидным активирующим действием, которое представляет собой безопасный и более эффективный агонист TLR8.
Способ синтеза соединения по настоящему изобретению
Чтобы выполнить задачу настоящего изобретения, в настоящем изобретении для синтеза применены упомянутые далее технические решения.
Схема I
Способ получения соединения формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению, включающий следующие стадии:
стадию 1:
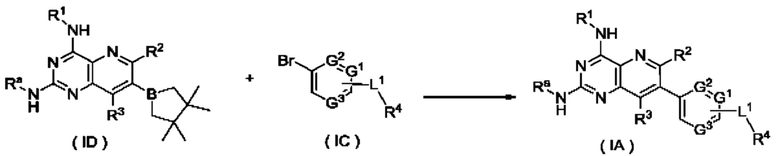
соединение формулы (ID) подвергают реакции сочетания с соединением формулы (IC) в щелочных условиях в присутствии катализатора с получением соединения формулы (I-А);
стадию 2:
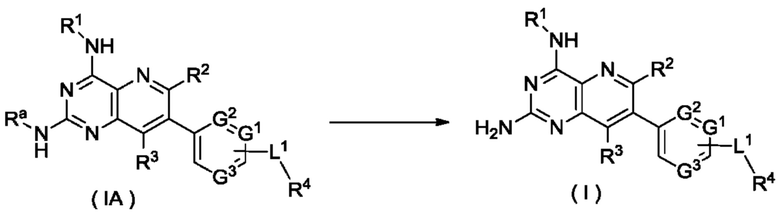
соединение формулы (IA) подвергают реакции удаления защиты в кислых условиях с получением соединения формулы (I);
где:
Ra представляет собой амино-защитную группу и предпочтительно 2,4-диметоксибензил; и
G1-G3, L1 и R1-R4 являются такими, как определено в формуле (I).
Схема II
Способ получения соединения формулы (Ia), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению, включающий следующую стадию:
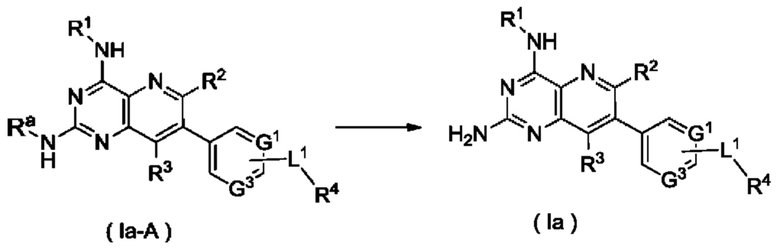
соединение формулы (Ia-А) подвергают реакции удаления защиты в кислых условиях с получением соединения формулы (Ia);
где:
Ra представляет собой амино-защитную группу и предпочтительно 2,4-диметоксибензил; и
G1, G3, L1 и R1-R4 являются такими, как определено в формуле (Ia).
Схема III
Способ получения соединения формулы (Ib), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению, включающий следующую стадию:
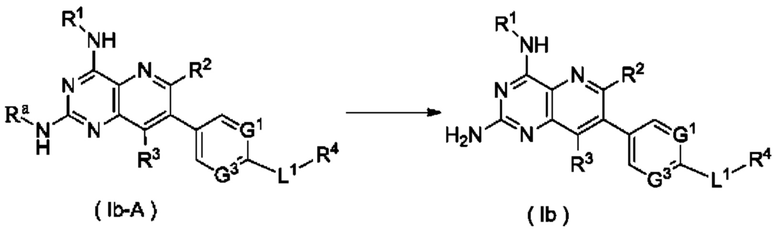
соединение формулы (Ib-A) подвергают реакции удаления защиты в кислых условиях с получением соединения формулы (Ib);
где:
Ra представляет собой амино-защитную группу и предпочтительно 2,4-диметоксибензил; и
G1, G3, L1 и R1-R4 являются такими, как определено в формуле (Ia).
Схема IV
Способ получения соединения формулы (II), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению, включающий следующие стадии:
стадию 1:
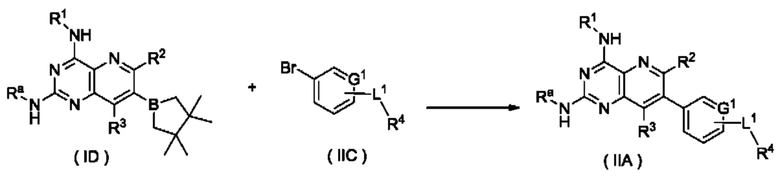
соединение формулы (ID) подвергают реакции сочетания с соединением формулы (IIC) в щелочных условиях в присутствии катализатора с получением соединения формулы (IIA);
стадию 2:
соединение формулы (IIA) подвергают реакции удаления защиты в кислых условиях с получением соединения формулы (II);
где:
Ra представляет собой амино-защитную группу и предпочтительно 2,4-диметоксибензил; и
G1, L1 и R1-R4 являются такими, как определено в формуле (II).
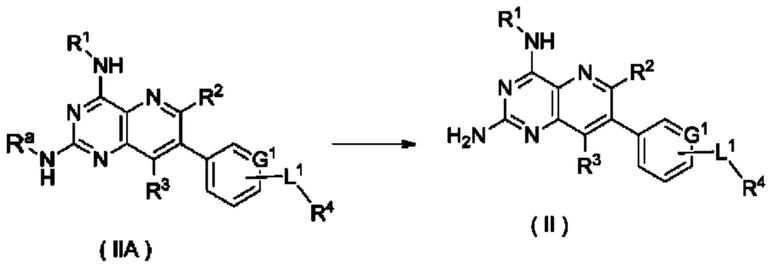
Схема V
Способ получения соединения формулы (III), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению, включающий следующую стадию:
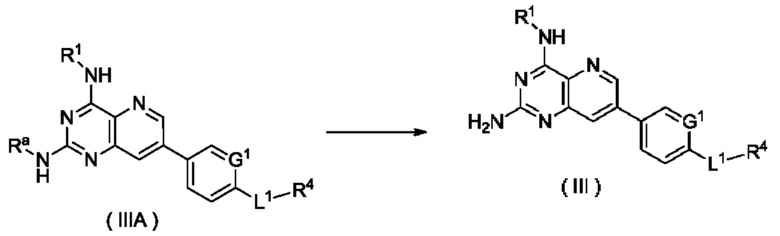
соединение формулы (IIIA) подвергают реакции удаления защиты в кислых условиях с получением соединения формулы (III);
где:
Ra представляет собой амино-защитную группу и предпочтительно 2,4-диметоксибензил; и
G1, L1 и R1-R4 являются такими, как определено в формуле (III).
Схема VI
Способ получения соединения формулы (IVa), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению, включающий следующие стадии:
стадию 1:
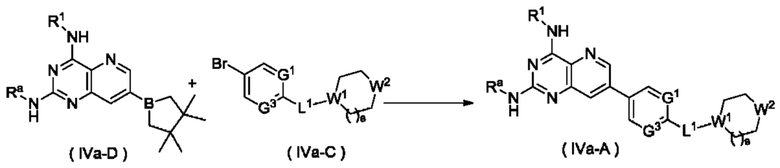
соединение формулы (IVa-D) подвергают реакции сочетания с соединением формулы (IVa-C) в щелочных условиях в присутствии катализатора с получением соединения формулы (IVa-A);
стадию 2:
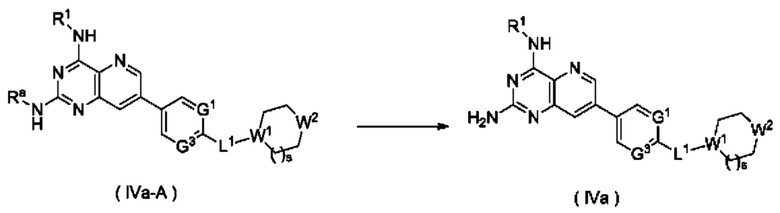
соединение формулы (IVA) подвергают реакции удаления защиты в кислых условиях с получением соединения формулы (IV);
где:
Ra представляет собой амино-защитную группу и предпочтительно 2,4-диметоксибензил; и
G1, G3, L1, R1, W1, W2 и s являются такими, как определено в формуле (IV).
Схема VII
Способ получения соединения формулы (IV), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению, включающий следующие стадии:
стадию 1:
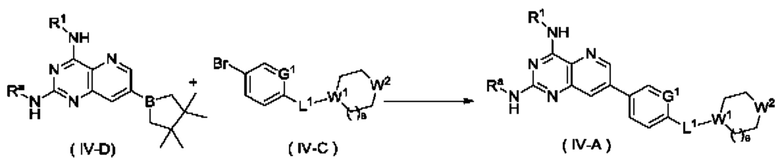
соединение формулы (IV-D) подвергают реакции сочетания с соединением формулы (IV-C) в щелочных условиях в присутствии катализатора с получением соединения формулы (IV-A);
стадию 2:
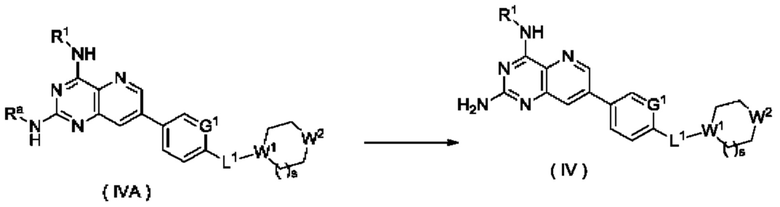
соединение формулы (IVA) подвергают реакции удаления защиты в кислых условиях с получением соединения формулы (IV);
где:
Ra представляет собой амино-защитную группу и предпочтительно 2,4-диметоксибензил; и
G1, L1, R1, W1, W2 и s являются такими, как определено в формуле (IV).
Схема VIII
Способ получения соединения формулы (Va), или его таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, либо их фармацевтически приемлемой соли по настоящему изобретению, включающий следующую стадию:
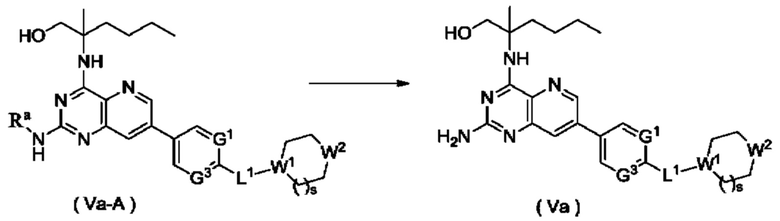
соединение формулы (Va-A) подвергают реакции удаления защиты в кислых условиях с получением соединения формулы (Va);
где:
Ra представляет собой амино-защитную группу и предпочтительно 2,4-диметоксибензил; и
G1, G3, L1, W1, W2 и s являются такими, как определено в формуле (Va).
Реагент, обеспечивающий кислые условия, включает, но не ограничивается этим, хлористый водород, раствор хлористого водорода в 1,4-диоксане, трифторуксусную кислоту, муравьиную кислоту, уксусную кислоту, соляную кислоту, серную кислоту и метансульфоновую кислоту, азотную кислоту, фосфорную кислоту, пара-толуолсульфоновую кислоту, Me3SiCl и триметилсилилтрифторметансульфонат (TMSOTf), и предпочтительно трифторуксусную кислоту.
Вышеупомянутую реакцию предпочтительно осуществляют в растворителе. Использованный растворитель включает, но не ограничивается этим, уксусную кислоту, трифторуксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид и их смеси.
На приведенных выше схемах реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, но не ограничиваются этим, триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, бистриметилсилиламид лития, ацетат калия, трет-бутилат натрия, трет-бутилат калия и н-бутилат натрия. Неорганические основания включают, но не ограничиваются этим, бикарбонат натрия, бикарбонат калия, гидрид натрия, фосфат калия, карбонат натрия, карбонат калия, ацетат калия, карбонат цезия, гидроксид натрия и гидроксид лития, и предпочтительно карбонат калия.
Катализатор включает, но не ограничивается этим, Pd/C, тетракис(трифенилфосфин)палладий, дихлорид палладия, ацетат палладия, бис(дибензилиденацетон)палладий, хлор-(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий, дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия, 1,1'-бис(дибензилфосфор)дихлорферроцен-палладий или трис(дибензилиденацетон)дипалладий и предпочтительно дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия.
Примеры
Структуры соединений устанавливали посредством ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (MS). Сдвиги спектров ЯМР (δ) приведены в 10-6 (млн-1). Спектры ЯМР определяли, используя прибор AVANCE-400 от Bruker. Для определения в качестве растворителей используют дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD), а в качестве внутреннего стандарта используют тетраметилсилан (TMS).
Масс-спектр (MS) определяют на масс-спектрометре FINNIGAN LCQAd (электрораспылительная ионизация (ESI)) (производитель: Thermo, тип: Finnigan LCQ Advantage MAX).
Анализ методом высокоэффективной жидкостной хроматографии (HPLC) проводят на хроматографах для жидкостной хроматографии высокого давления Agilent HPLC 1200DAD, Agilent HPLC 1200VWD и Waters HPLC e2695-2489.
Анализ методом хиральной HPLC проводят на хроматографе для высокоэффективной жидкостной хроматографии Agilent 1260 DAD.
Препаративную высокоэффективную жидкостную хроматографию осуществляют на препаративных хроматографах Waters 2767, Waters 2767-SQ Detecor 2, Shimadzu LC-20AP и Gilson-281.
Препаративную хиральную HPLC осуществляют на препаративном хроматографе LC-20AP от Shimadzu.
В качестве прибора для быстрой препаративной комби-флеш-хроматографии используют систему CombiFlash Rf200 (TELEDYNE ISCO).
В качестве пластинки для тонкослойной хроматографии (TLC) на силикагеле используют пластинку с силикагелем HSGF254 от Yantai Huanghai или GF254 от Qingdao. Размер слоя силикагеля на пластинке, использованной для TLC, составляет от 0,15 мм до 0,2 мм, а размер слоя силикагеля на пластинке, использованной для очистки продукта, составляет от 0,4 мм до 0,5 мм.
В качестве носителя для колоночной хроматографии на силикагеле обычно используют силикагель от Yantai Huanghai с размером частиц 200-300 меш.
Средние значения скоростей ингибирования киназ и значения вызывающей 50%-ное ингибирование концентрации (IC50) определяют с использованием прибора NovoStar для иммуноферментного твердофазного анализа (ELISA) (BMG Co., Германия).
Известные исходные вещества для настоящего изобретения могут быть получены известными в данной области техники методами или могут быть приобретены у ABCR GmbH & Co. KG, Acros Organics, химической компании Aldrich, Accela ChemBio Inc., компании Chembee и тому подобных.
Если не указано иное, реакции проводили в атмосфере аргона или атмосфере азота.
"Атмосфера аргона" или "атмосфера азота" означает, что реакционная колба оснащена баллоном с аргоном или азотом (примерно 1 л).
"Атмосфера водорода" означает, что реакционная колба оснащена баллоном с водородом (примерно 1 л).
Реакции гидрирования под давлением проводят на установке Парра для гидрирования 3916EKX и с использованием генератора водорода Qinglan QL-500 или на установке для гидрирования HC2-SS.
Для проведения реакций гидрирования реакционную систему обычно вакуумируют и заполняют водородом, и указанную выше операцию повторяют три раза.
Для проведения реакций в условиях облучения микроволнами используют микроволновой реактор СЕМ Discover-S 908860.
Если не указано иное, раствор означает водный раствор.
Если не указано иное, температура реакции означает комнатную температуру от 20°С до 30°С.
Мониторинг протекания реакций, приведенных в разделе Примеры, проводили по тонкослойной хроматографии (TLC). Система растворителей, используемая при проведении реакций, система элюентов для колоночной хроматографии и система растворителей для тонкослойной хроматографии, применяемой с целью очистки соединений, включали: А: систему дихлорметан/метанол, В: систему н-гексан/этилацетат и С: систему петролейный эфир/этилацетат. Соотношение объемов растворителей корректируют в соответствии с полярностью соединений, и для этого также может быть добавлено небольшое количество щелочного реагента, такого как триэтиламин, или кислотного реагента, такого как уксусная кислота.
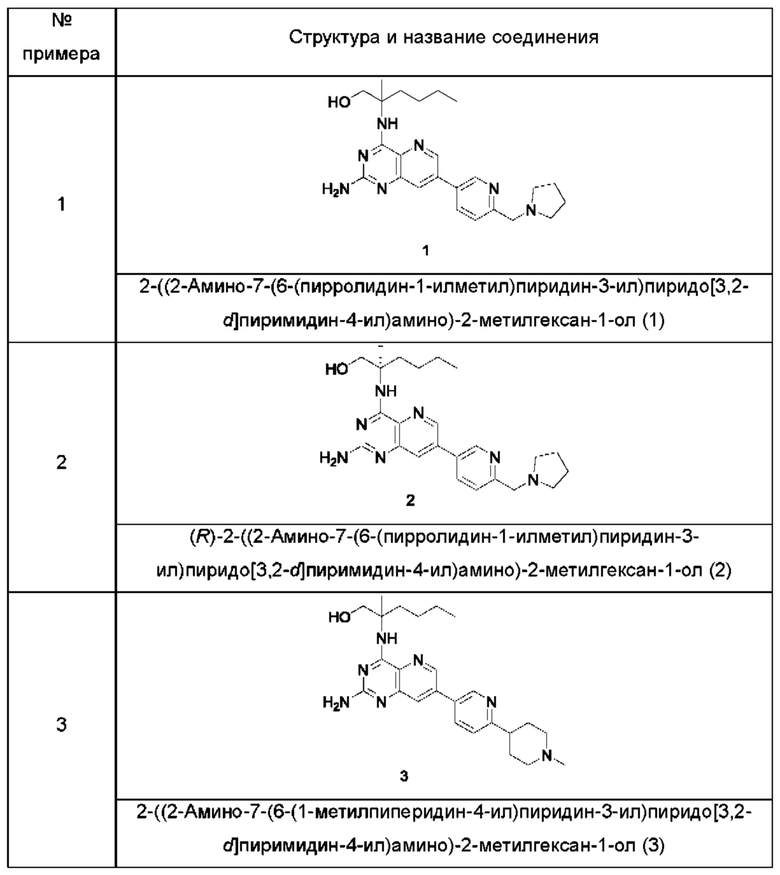
Пример 1
2-((2-Амино-7-(6-(пирролидин-1-илметил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (1)
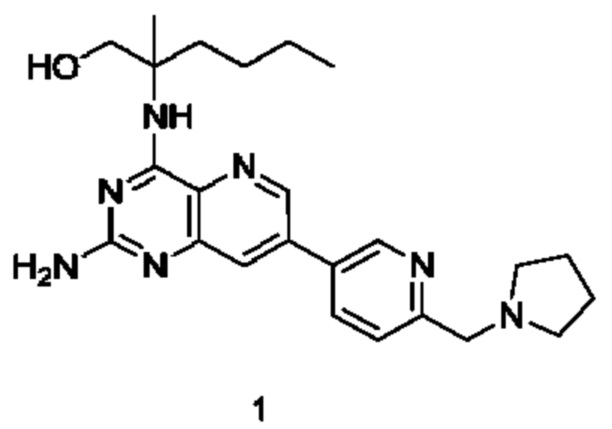
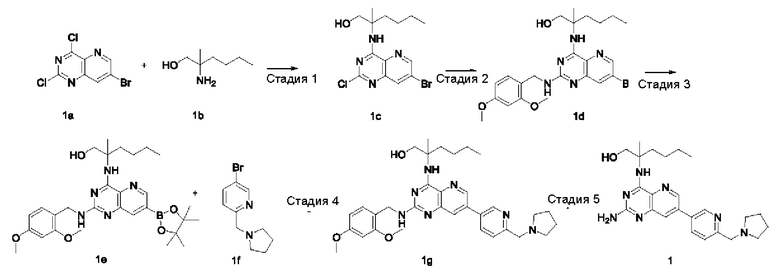
Стадия 1
2-((7-Бром-2-хлорпиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (1с)
7-Бром-2,4-дихлорпиридо[3,2-d]пиримидин 1а (5,4 г; 19,36 ммоль, полученный согласно способу, описанному в патентной заявке WO 2014022728) добавляли к 120 мл ацетонитрила, после чего добавляли 2-амино-2-метилгексан-1-ол 1b (3,8 г; 28,96 ммоль, полученный согласно способу, описанному в патентной заявке WO 2009129097) и карбонат калия (8,027 г; 58,08 ммоль). Реакционный раствор перемешивали при 45°С в течение 16 часов. По завершении реакции нерастворившееся вещество удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой А, получая указанный в заголовке продукт 1с (4,0 г; выход: 55,3%).
MS m/z (ESI): 373,1 [М+1].
Стадия 2
2-((7-Бром-2-((2,4-диметоксибензил)амино)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (1d)
Соединение 1с (4,0 г; 10,71 ммоль) добавляли к 25 мл тетрагидрофурана, после чего добавляли 2,4-диметоксибензиламин (6,0 г; 35,861 ммоль) и N,N-диизопропилэтиламин (4,15 г; 32,11 ммоль). Реакционный раствор герметично закрывали в пробирке и перемешивали при 100°С в течение 16 часов. В реакционный раствор добавляли 20 мл воды, затем проводили экстрагирование дихлорметаном (20 мл×3). Органические фазы объединяли, промывали водой (50 мл) и насыщенным раствором хлорида натрия (50 мл) в указанном порядке, сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая указанный в заголовке продукт 1d (3,5 г; выход: 64,8%).
MS m/z (ESI): 504,1 [M+1].
Стадия 3
2-((2-((2,4-Диметоксибензил)амино)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (1 е)
Соединение 1d (130 мг; 0,237 ммоль) добавляли к 5 мл диметилового эфира этиленгликоля, после чего добавляли бис(пинаколато)дибор (91 мг; 0,358 ммоль), дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия (35 мг; 0,048 ммоль) и ацетат калия (70 мг; 0,713 ммоль). Реакционный раствор три раза продували аргоном, нагревали до 80°С и перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. К полученной системе добавляли 20 мл воды, затем экстрагировали дихлорметаном (10 мл ×3). Органические фазы объединяли, промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл) в указанном порядке, сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении, получая неочищенный указанный в заголовке продукт 1е (130 мг; выход: 99,2%).
Стадия 4
2-((2-((2,4-Диметоксибензил)амино)-7-(6-(пирролидин-1-илметил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (1g)
Неочищенное соединение 1е (130 мг; 0,235 ммоль) добавляли к 10 мл 1,4-диоксана и 2 мл воды, после чего добавляли 5-бром-2-(пирролидин-1-илметил)пиридин 1f (68 мг; 0,282 ммоль, полученный согласно способу, описанному в патентной заявке WO2007084451), карбонат калия (49 мг; 0,355 ммоль) и дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия (18 мг; 0,025 ммоль). Реакционный раствор три раза продували аргоном, нагревали до 80°С и проводили взаимодействие в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. В реакционный раствор добавляли 20 мл воды, затем экстрагировали дихлорметаном (20 мл). Органические фазы объединяли, промывали водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая продукт 1g (60 мг; выход: 43,5%).
MS m/z (ESI): 586,0 [M+1].
Стадия 5
2-((2-Амино-7-(6-(пирролидин-1-илметил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (1)
Соединение 1g (60 мг; 0,102 ммоль) добавляли к 10 мл трифторуксусной кислоты и проводили взаимодействие при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. В реакционный раствор добавляли 20 мл насыщенного раствора бикарбоната натрия, затем экстрагировали дихлорметаном (20 мл ×3). Органические фазы объединяли, промывали водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая продукт 1 (10 мг; выход: 22,4%).
MS m/z (ESI): 436,0 [M+1].
1H ЯМР (400 МГц, ДМСО-d6) δ 8.91 (s, 1H), 8.64 (s, 1H), 8.18-8.20 (m, 1H), 7.83 (s, 1H), 7.56-7.58 (m, 1H), 7.24 (s, 1H), 6.40 (br, 2H), 5.16-5.20 (m, 1H), 3.79 (s, 2H), 3.70-3.73 (m, 1H), 3.51-3.54 (m, 1H), 2.54 (s, 4H), 1.91-1.95 (m, 2H), 1.71-1.75 (m, 4H), 1.43 (s, 3H), 1.23-1.27 (m, 4H), 0.84-0.87 (m, 3H).
Пример 2
(R)-2-((2-Амино-7-(6-(пирролидин-1-илметил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (2)
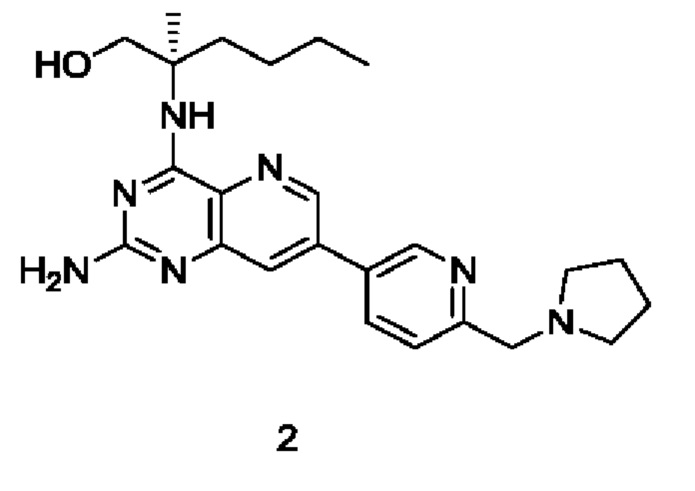
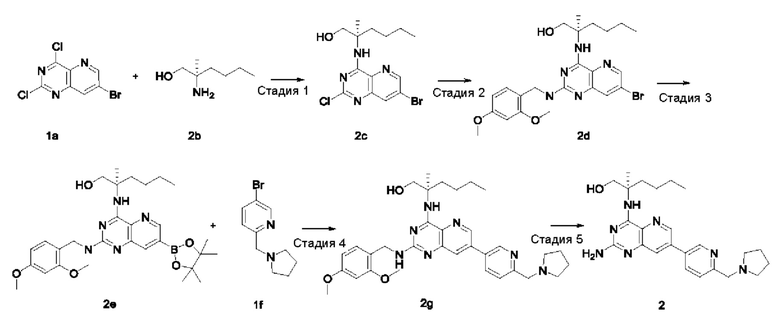
Стадия 1
(R)-2-((7-Бром-2-хлорпиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (2 с)
Соединение 1а (400 мг; 1,434 ммоль) добавляли к 10 мл тетрагидрофурана, после чего добавляли (R)-2-амино-2-метилгексан-1-ола 2b (полученный согласно способу, изложенному в примере 59 на странице 207 описания патентной заявки WO2016141092) (377 мг; 2,873 ммоль) и N,N-диизопропилэтиламин (556 мг; 4,302 ммоль). Реакционный раствор герметично закрывали в пробирке и перемешивали при 100°С в течение 16 часов. По завершении реакции реакционный раствор охлаждали до комнатной температуры и нерастворившееся вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и полученные остатки очищали колоночной хроматографией на силикагеле с элюированием системой А, получая указанный в заголовке продукт 2 с (4,0 г; выход: 55,3%).
MS m/z (ESI): 373,1 [M+1].
Стадия 2
(R)-2-((7-Бром-2-((2,4-диметоксибензил)амино)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (2d)
Соединение 2 с (250 мг; 0,669 ммоль) добавляли к 10 мл тетрагидрофурана, после чего добавляли 2,4-диметоксибензиламин (560 мг; 3,349 ммоль) и N,N-диизопропилэтиламин (259 мг; 2,004 ммоль). Реакционный раствор герметично закрывали в пробирке и перемешивали при 100°С в течение 16 часов. В реакционный раствор добавляли 20 мл воды, затем экстрагировали дихлорметаном (20 мл ×3). Органические фазы объединяли, промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл) в указанном порядке, сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая указанный в заголовке продукт 2d (295 мг; выход: 87,5%).
MS m/z (ESI): 504,1 [М+1].
Стадия 3
(R)-2-((2-((2,4-Диметоксибензил)амино)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (2е)
Соединение 2d (295 мг; 0,54 ммоль) добавляли к 5 мл диметилового эфира этиленгликоля, после чего добавляли бис(пинаколато)дибор (223 мг; 878,169 мкмоль), дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия (43 мг; 0,059 ммоль) и ацетат калия (173 мг; 1,76 ммоль). Реакционный раствор три раза продували аргоном, нагревали до 80°С и перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. К реакционной системе добавляли 20 мл воды, затем экстрагировали дихлорметаном (10 мл ×3). Органические фазы объединяли, промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл) в указанном порядке, сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении, получая неочищенный указанный в заголовке продукт 2е (322 мг; выход: 100%).
Стадия 4
(R)-2-((2-((2,4-Диметоксибензил)амино)-7-(6-(пирролидин-1-илметил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (2g)
Неочищенное соединение 2е (322 мг; 0,584 ммоль) добавляли к 10 мл 1,4-диоксана и 2 мл воды, после чего добавляли соединение 1f (141 мг; 0,584 ммоль), карбонат калия (242 мг; 1,75 ммоль) и дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия (43 мг; 0,059 ммоль). Реакционный раствор три раза продували аргоном, нагревали до 80°С и проводили взаимодействие в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. В реакционный раствор добавляли 20 мл воды, затем экстрагировали дихлорметаном (20 мл). Органические фазы объединяли, промывали водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая продукт 2 g (100 мг; выход: 29,2%).
MS m/z (ESI): 586,0 [М+1].
Стадия 5
(R)-2-((2-Амино-7-(6-(пирролидин-1-илметил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (2)
Соединение 2g (100 мг; 0,170 ммоль) добавляли к 10 мл трифторуксусной кислоты и проводили взаимодействие при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. В реакционный раствор добавляли 20 мл насыщенного раствора бикарбоната натрия, затем экстрагировали дихлорметаном (20 мл ×3). Органические фазы объединяли, промывали водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Полученные остатки очищали колоночной хроматографией на силикагеле с эпюированием системой В, получая продукт 2 (45 мг; выход: 60,5%).
MS m/z (ESI): 436,0 [M+1].
1H ЯМР (400 МГц, ДМСО-d6) δ 8.91 (s, 1H), 8.64 (s, 1H), 8.18-8.20 (m, 1H), 7.83 (s, 1H), 7.56-7.58 (m, 1H), 7.24 (s, 1H), 6.40 (br, 2H), 5.16-5.20 (m, 1H), 3.79 (s, 2H), 3.70-3.73 (m, 1H), 3.51-3.54 (m, 1H), 2.54 (s, 4H), 1.91-1.95 (m, 2H), 1.71-1.75 (m, 4H), 1.43 (s, 3H), 1.23-1.27 (m, 4H), 0.84-0.87 (m, 3H).
Пример 3
2-((2-Амино-7-(6-(1-метилпиперидин-4-ил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (3)
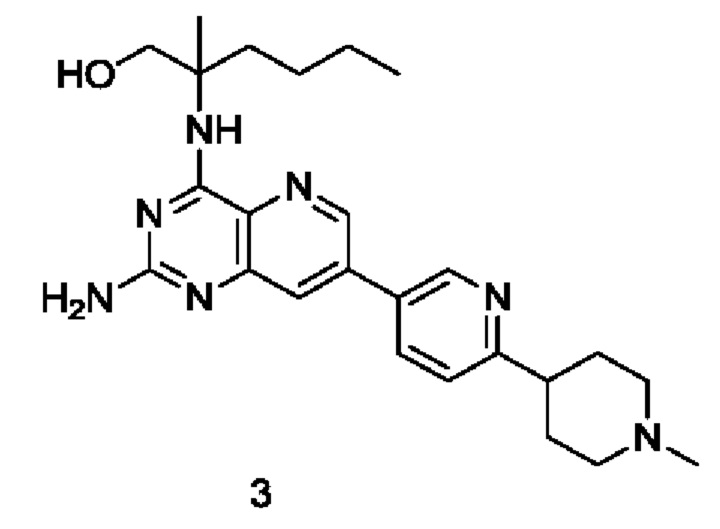
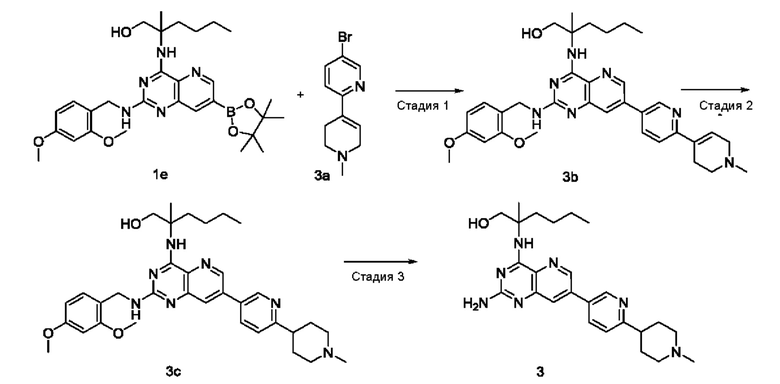
Стадия 1
2-((2-((2,4-Диметоксибензил)амино)-7-(1'-метил-1',2',3',6'-тетрагидро-[2,4'-бипиридин]-5-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (3b)
Соединение 1е (218 мг; 0,395 ммоль) добавляли к 10 мл 1,4-диоксана и 2 мл воды, после чего добавляли 5-бром-1'-метил-1',2',3',6'-тетрагидро-2,4'-бипиридин 3а (100 мг; 0,395 ммоль, полученный согласно способу, изложенному в патентной заявке WO2010054279), дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия (29 мг; 0,040 ммоль) и карбонат калия (164 мг; 1,187 ммоль). Реакционный раствор три раза продували аргоном, нагревали до 80°С и перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. К полученной системе добавляли 20 мл воды, затем экстрагировали дихлорметаном (10 мл ×3). Органические фазы объединяли, промывали водой (20 мл) и насыщенным раствором хлорида натрия (30 мл) в указанном порядке, сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая указанный в заголовке продукт 3b (100 мг; выход: 43,2%).
MS m/z (ESI): 598,0 [M+1].
Стадия 2
2-((2-((2,4-Диметоксибензил)амино)-7-(6-(1-метилпиперидин-4-ил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (3с)
Соединение 3b (100 мг; 0,163 ммоль) добавляли к 10 мл метанола, после чего добавляли Pd/C (20 мг), карбонат калия (49 мг; 0,355 ммоль) и дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия (18 мг; 0,025 ммоль). Реакционный раствор пять раз продували водородом и проводили взаимодействие при комнатной температуре в течение 20 часов. Pd/C удаляли фильтрованием и фильтрат концентрировали при пониженном давлении, получая неочищенный указанный в заголовке продукт 3с (68 мг; выход: 67,8%).
MS m/z (ESI): 600,0 [M+1].
Стадия 3
2-((2-Амино-7-(6-(1-метилпиперидин-4-ил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (3)
Неочищенное соединение 3с (60 мг; 0,100 ммоль) добавляли к 5 мл трифторуксусной кислоты и проводили взаимодействие при комнатной температуре в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении. В реакционный раствор добавляли 20 мл насыщенного раствора бикарбоната натрия, затем экстрагировали дихлорметаном (20 мл ×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая продукт 3 (15 мг; выход: 33,4%).
MS m/z (ESI): 450,0 [M+1].
1H ЯМР (400МГц, ДМСО-d6) δ 8.91 (s, 1H),8.62 (s, 1H), 8.11-8.13 (d, 1H),7.80 (s, 1H), 7.42-7.44 (m, 1H), 7.23 (s, 1H), 6.38 (br, 2H), 3.70-3.72 (m, 1H), 3.50-3.53 (m, 1H), 2.87-2.90 (m, 2H), 2.68-3.72 (m, 1H), 2.00 (s, 3Н), 1.83-1.93 (m, 9H), 1.42 (s, 3H), 1.23-1.27 (m, 4H), 0.83-0.86 (m, 3H).
Пример 4
(R)-2-((2-Амино-7-(6-(1-метилпиперидин-4-ил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (4)
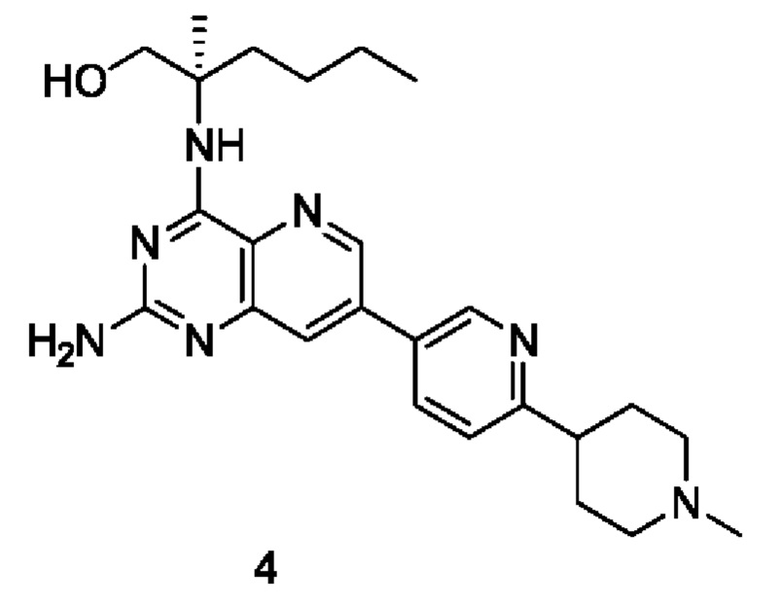
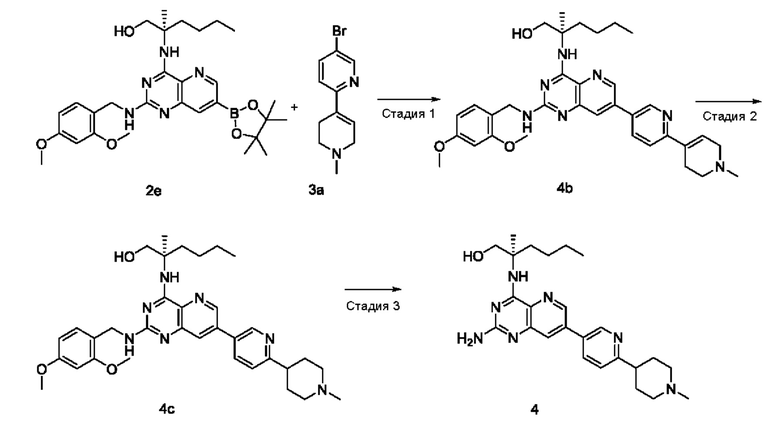
Стадия 1
(R)-2-((2-((2,4-Диметоксибензил)амино)-7-(1'-метил-1',2',3',6'-тетрагидро-[2,4'-бипиридин]-5-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (4b)
Соединение 2е (284 мг; 0,515 ммоль) добавляли к 10 мл 1,4-диоксана и 2 мл воды, после чего добавляли сединение За (130 мг; 0,515 ммоль), дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия (38 мг; 0,052 ммоль) и карбонат калия (214 мг; 1,551 ммоль). Реакционный раствор три раза продували аргоном, нагревали до 80°С и перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. К полученной системе добавляли 20 мл воды, затем экстрагировали дихлорметаном (10 мл ×3). Органические фазы объединяли, промывали водой (20 мл) и насыщенным раствором хлорида натрия (30 мл) в указанном порядке, сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая указанный в заголовке продукт 4b (102 мг; выход: 33,1%).
MS m/z (ESI): 598,0 [M+1].
Стадия 2
(R)-2-((2-((2,4-Диметоксибензил)амино)-7-(6-(1-метилпиперидин-4-ил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (4 с) Соединение 4b (100 мг; 0,163 ммоль) добавляли к 10 мл метанола, после чего добавляли Pd/C (20 мг), карбонат калия (49 мг; 0,355 ммоль) и дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия (18 мг; 0,025 ммоль). Реакционный раствор пять раз продували водородом и проводили взаимодействие при комнатной температуре в течение 20 часов. Pd/C удаляли фильтрованием и фильтрат концентрировали при пониженном давлении, получая неочищенный указанный в заголовке продукт 4 с (85 мг; выход: 84,7%).
MS m/z (ESI): 600,0 [M+1].
Стадия 3
(R)-2-((2-Амино-7-(6-(1-метилпиперидин-4-ил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (4)
Неочищенное соединение 4 с (80 мг; 0,133 ммоль) добавляли к 5 мл трифторуксусной кислоты и проводили взаимодействие при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. В реакционный раствор добавляли 20 мл насыщенного раствора бикарбоната натрия, затем экстрагировали дихлорметаном (20 мл ×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали тонкослойной хроматографией с эпюированием системой В, получая продукт 4 (26 мг; выход: 43,3%).
MS m/z (ESI): 450,0 [M+1].
1H ЯМР (400МГц, ДМСО-d6) δ 8.91 (s, 1H),8.62 (s, 1H), 8.11-8.13 (d, 1H),7.80 (s, 1H), 7.42-7.44 (m, 1H), 7.23 (s, 1H), 6.38 (br, 2H), 3.70-3.72 (m, 1H), 3.50-3.53 (m, 1H), 2.87-2.90 (m, 2H), 2.68-3.72 (m, 1H), 2.00 (s, 3Н), 1.83-1.93 (m, 9H), 1.42 (s, 3H), 1.23-1.27 (m, 4H), 0.83-0.86 (m, 3H).
Пример 5
2-((2-Амино-7-(6-((4-метилпиперазин-1-ил)метил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (5)
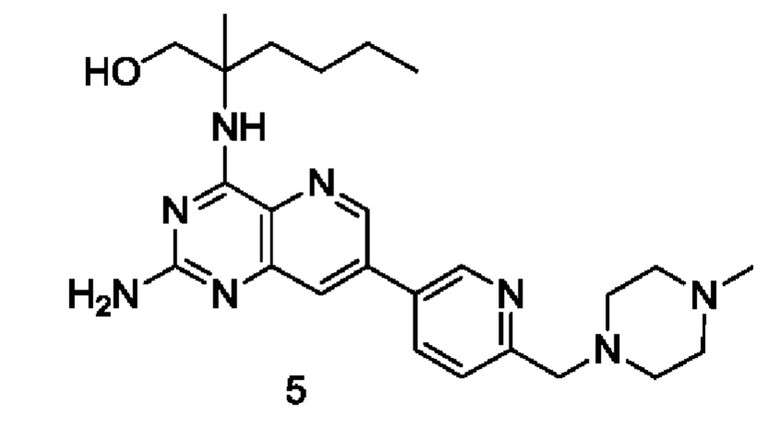
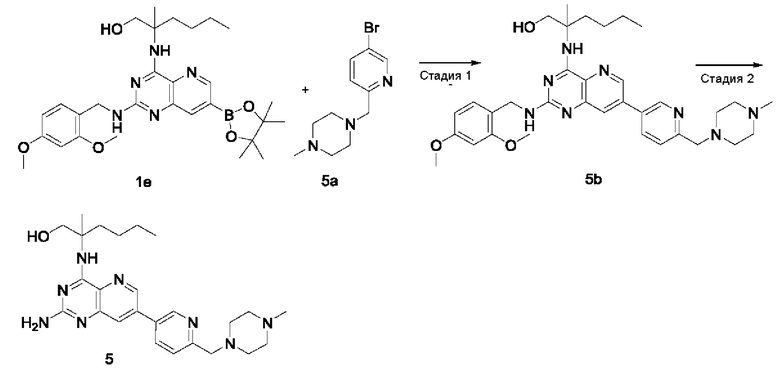
Стадия 1
2-((2-((2,4-Диметоксибензил)амино)-7-(6-((4-метилпиперазин-1-ил)метил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (5b)
Соединение 1е (200 мг; 0,363 ммоль) добавляли к 10 мл 1,4-диоксана и 2 мл воды, после чего добавляли 1-((5-бромпиридин-2-ил)метил)-4-метилпиперазин 5а (108 мг; 0,401 ммоль, полученный согласно способу, описанному в патентной заявке WO20020026052), дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия (27 мг; 0,037 ммоль) и карбонат калия (150 мг; 1,085 ммоль). Реакционный раствор три раза продували аргоном, нагревали до 80°С и проводили взаимодействие в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. К реакционной системе добавляли 20 мл воды, затем экстрагировали дихлорметаном (10 мл ×3). Органические фазы объединяли, промывали водой (20 мл) и насыщенным раствором хлорида натрия (30 мл) в указанном порядке, сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая указанный в заголовке продукт 5b (121 мг; выход: 54,2%).
MS m/z (ESI): 615,1 [M+1].
Стадия 2
2-((2-Амино-7-(6-((4-метилпиперазин-1-ил)метил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (5)
Соединение 5b (85 мг; 0,138 ммоль) добавляли к 5 мл трифторуксусной кислоты и проводили взаимодействие при комнатной температуре в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении. В реакционный раствор добавляли 20 мл насыщенного раствора бикарбоната натрия, затем экстрагировали дихлорметаном (20 мл ×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая продукт 5 (23 мг; выход: 49,5%).
MS m/z (ESI): 465,1 [M+1].
1H ЯМР (400 МГц, ДМСО-d6) δ 8.87-8.88 (d, 1H), 8.60-8.61 (m, 1H), 8.14-8.16 (d, 1H), 7.79-7.80(s, 1H), 7.51-7.53 (d, 1H),7.20(s, 1 H), 6.36 (br, 2H), 5.12-5.15 (t, 1H), 3.66 (s, 2H), 3.68-3.70 (m, 1H), 3.48-2.53 (m, 1H), 2.32-3.42 (m, 8H), 2.12 (s, 3H), 1.90-1.92 (m, 2H), 1.40 (s, 3H), 1.22-1.23 (m, 4H), 0.80-0.84 (m, 3H).
Пример 6
(R)-2-((2-Амино-7-(6-((4-метилпиперазин-1-ил)метил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (6)
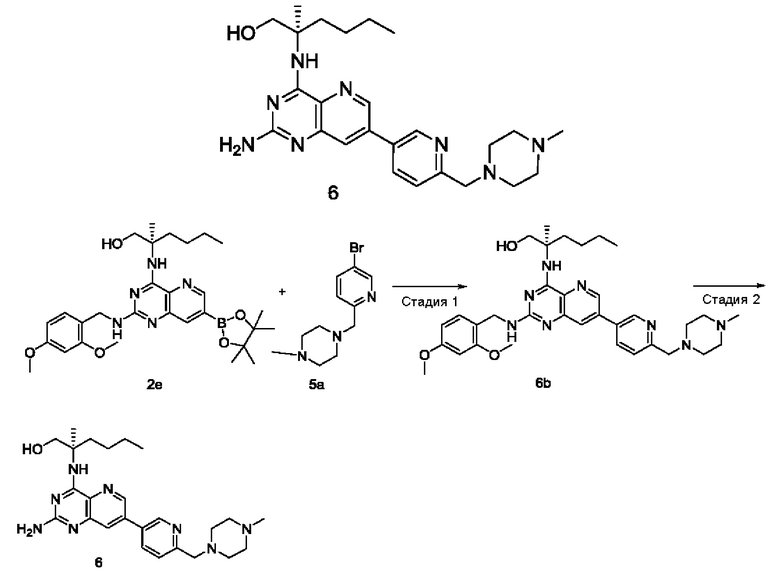
Стадия 1
(R)-2-((2-((2,4-Диметоксибензил)амино)-7-(6-((4-метилпиперазин-1-ил)метил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (6b)
Соединение 2е (650 мг; 1,178 ммоль) добавляли к 20 мл 1,4-диоксана и 4 мл воды, после чего добавляли 1-((5-бромпиридин-2-ил)метил)-4-метилпиперазин 5а (318 мг; 1,170 ммоль), дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия (86 мг; 0,117 ммоль) и карбонат калия (489 мг; 3,538 ммоль). Реакционный раствор три раза продували аргоном, нагревали до 80°С и перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. К реакционной системе добавляли 30 мл воды, затем экстрагировали дихлорметаном (30 мл ×3). Органические фазы объединяли, промывали водой (30 мл) и насыщенным раствором хлорида натрия (30 мл) в указанном порядке, сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая указанный в заголовке продукт 6b (650 мг; выход: 89,70%).
MS m/z (ESI): 615,1 [M+1].
Стадия 2
(R)-2-((2-Амино-7-(6-((4-метилпиперазин-1-ил)метил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (6)
Соединение 6b (650 мг; 0,138 ммоль) добавляли к 5 мл трифторуксусной кислоты и проводили взаимодействие при комнатной температуре в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении. В реакционный раствор добавляли 20 мл насыщенного раствора бикарбоната натрия, затем экстрагировали дихлорметаном (20 мл ×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая продукт 6 (320 мг; выход: 65,14%).
MS m/z (ESI): 465,1 [M+1].
1H ЯМР (400 МГц, ДМСО-d6) δ 8.87-8.88 (d, 1H), 8.60-8.61 (m, 1H), 8.14-8.16 (d, 1H), 7.79-7.80(s, 1H), 7.51-7.53 (d, 1H),7.20(s, 1 H), 6.36 (br, 2H), 5.12-5.15 (t, 1H), 3.66 (s, 2H), 3.68-3.70 (m, 1H), 3.48-2.53 (m, 1H), 2.32-3.42 (m, 8H), 2.12 (s, 3H), 1.90-1.92 (m, 2H), 1.40 (s, 3H), 1.22-1.23 (m, 4H), 0.80-0.84 (m, 3H).
Пример 7
(R)-2-((2-Амино-7-(2-((4-метилпиперазин-1-ил)метил)пиримидин-5-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол
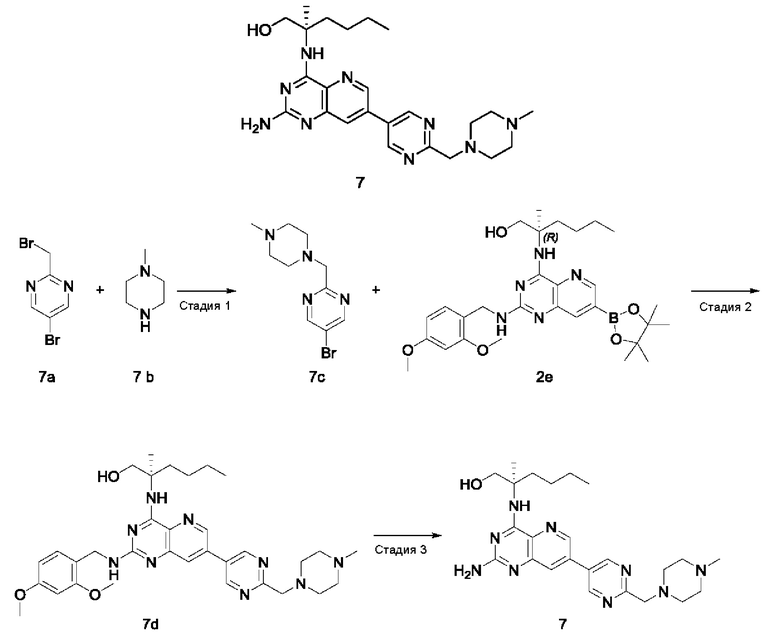
Стадия 1
5-Бром-2-((4-метилпиперазин-1 -ил) метил) пиримидин (7с) Соединение 5-бром-2-(бромметил)пиримидин 7а (200 мг; 0,794 ммоль) добавляли к 5 мл ацетонитрила, после чего добавляли карбонат калия (220 мг; 1,592 ммоль). Добавляли 1-метилпиперазин 7b (120 мг; 1,198 ммоль) при 0°С и реакционный раствор нагревали до комнатной температуры и перемешивали в течение 2 часов. По завершении реакции нерастворившееся вещество удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с эпюированием системой В, получая указанный в заголовке продукт 7 с (200 мг; выход: 92,9%).
MS m/z (ESI): 273,1 [M+1].
Стадия 2
(R)-2-((2-((2,4-Диметоксибензил)амино)-7-(2-((4-метилпиперазин-1-ил)метил)пиримидин-5-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (7d)
Соединение 2е (163 мг; 0,296 ммоль) добавляли к 5 мл 1,4-диоксана и 1 мл воды, после чего добавляли соединение 7 с (81 мг; 0,299 ммоль), тетракис(трифенилфосфоний)палладий (35 мг; 0,030 ммоль) и карбонат калия (82 мг; 0,593 ммоль). Реакционный раствор три раза продували аргоном, нагревали до 100°С и перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. К реакционной системе добавляли 20 мл воды, затем экстрагировали дихлорметаномом (10 мл ×3). Органические фазы объединяли, промывали водой (20 мл) и насыщенным раствором хлорида натрия (30 мл) в указанном порядке, сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая указанный в заголовке продукт 7d (127 мг; выход: 69,8%).
MS m/z (ESI): 616,3 [M+1].
Стадия 3
(R)-2-((2-Амино-7-(2-((4-метилпиперазин-1-ил)метил)пиримидин-5-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (7)
Соединение 7d (127 мг; 0,206 ммоль) добавляли к 3 мл трифторуксусной кислоты и проводили взаимодействие при комнатной температуре в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении. В реакционный раствор добавляли 20 мл насыщенного раствора бикарбоната натрия, затем экстрагировали дихлорметаном (20 мл ×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали высокоэффективной жидкостной хроматографией (Waters-2767; система элюирования: H2O (NH4OAc, 10 ммоль), ACN (ацетонитрил)), получая продукт 7 (34 мг; выход: 35,4%).
MS m/z (ESI): 466,3 [M+1].
1H ЯМР (400 МГц, ДМСО-d6) δ 9.19 (s, 2H), 8.66 (s, 1H), 7.92 (s, 1H), 7.23 (s, 1H), 6.40 (br, 2H), 5.15 (br, 1H), 3.73 (s, 2H), 3.70 (d, 2H), 3.50 (d, 2H), 2.51 (br, 3H), 2.29 (br, 3H), 2.11 (s, 3H), 1.90-1.88 (m, 2H), 1.41 (s, 3H), 1.28-1.20 (m, 4H), 0.83 (t, 3H).
Пример 8
(R)-2-((2-Амино-7-(2-(1-метилпиперидин-4-ил)пиримидин-5-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (8)
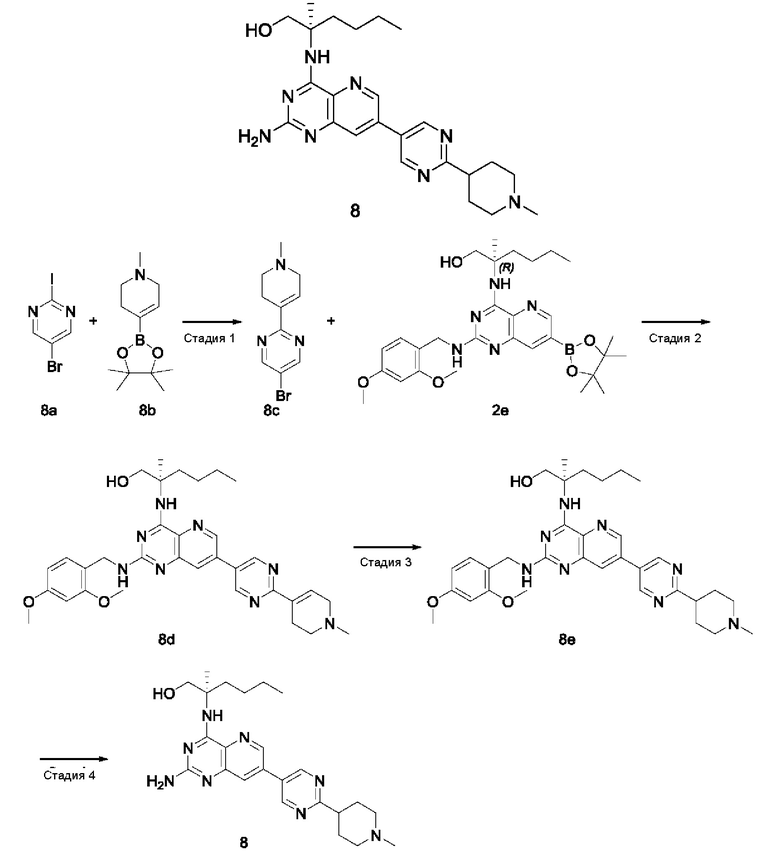
Стадия 1
5-Бром-2-(1 -метил-1,2,3,6-тетрагидропиридин-4-ил)пиримидин (8с) Соединение 5-бром-2-иодпиримидин 8а (4 г; 14,041 ммоль) добавляли к 200 мл 1,4-диоксана и 40 мл воды, после чего добавляли 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,6-тетрагидропиридин 8b (3,45 г; 15,463 ммоль), дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия (1,05 г; 1,435 ммоль) и карбонат калия (3,89 г; 28,146 ммоль). Реакционный раствор три раза продували аргоном, нагревали до 45°С и перемешивали в течение ночи.
Реакционный раствор концентрировали при пониженном давлении. К реакционной системе добавляли 30 мл воды, затем экстрагировали дихлорметаном (60 мл ×3). Органические фазы объединяли, промывали водой (30 мл) и насыщенным раствором хлорида натрия (30 мл) в указанном порядке, сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая указанный в заголовке продукт 8 с (2,5 г; выход: 70,1%).
MS m/z (ESI): 255,9 [M+1].
Стадия 2
(R)-2-((2-((2,4-Диметоксибензил)амино)-7-(2-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)пиримидин-5-ил)пиридо[3,2-с?]пиримидин-4-ил)амино)-2-метилгексан-1-ол (8d)
Соединение 2е (4,15 г; 7,5252 ммоль) добавляли к 80 мл 1,4-диоксана и 16 мл воды, после чего добавляли соединение 8 с (1,53 г; 6,021 ммоль), дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия (551 мг; 0,753 ммоль) и карбонат калия (2,1 г; 15,195 ммоль). Реакционный раствор три раза продували аргоном, нагревали до 95°С и перемешивали в течение 45 минут. Реакционный раствор концентрировали при пониженном давлении. К реакционной системе добавляли 40 мл воды, затем экстрагировали дихлорметаном (40 мл ×3). Органические фазы объединяли, промывали водой (40 мл) и насыщенным раствором хлорида натрия (40 мл) в указанном порядке, сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая указанный в заголовке продукт 8d (3,5 г; выход: 77,7%).
MS m/z (ESI): 599,4 [M+1].
Стадия 3
(R)-2-((2-((2,4-Диметоксибензил)амино)-7-(2-(1-метилпиперидин-4-ил)пиримидин-5-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (8е)
Соединение 8d (3,5 г; 5,846 ммоль) добавляли к 50 мл метанола, после чего добавляли Pd/C (1 г). Реакционный раствор пять раз продували водородом и проводили взаимодействие при комнатной температуре в течение 48 часов. Pd/C удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая указанный в заголовке продукт 8е (1,7 г; выход: 48,4%).
MS m/z (ESI):601,4[M+1].
Стадия 4
(R)-2-((2-Амино-7-(2-(1-метилпиперидин-4-ил)пиримидин-5-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (8)
Неочищенное соединение 8е (1,7 г; 2,830 ммоль) добавляли к 20 мл трифторуксусной кислоты и проводили взаимодействие при комнатной температуре в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении. В реакционный раствор добавляли 50 мл насыщенного раствора карбоната натрия, затем экстрагировали дихлорметаном (50 мл ×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл), сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали высокоэффективной жидкостной хроматографией (Waters-2767; система эпюирования: H2O (NH4OAc, 10 ммоль), ACN), получая продукт 8 (600 мг; выход: 47,1%).
MS m/z (ESI):451,3[M+1].
1H ЯМР (400 МГц, ДМСО-d6) δ 9.16 (s, 2Н), 8.64 (s, 1H), 7.90 (s, 1H), 7.22 (s, 1 H), 6.37 (br,2H), 5.14-5.12 (m, 1H), 3.71-3.67 (m, 1 H), 3.51-3.47 (m, 1H), 2.89-2.72 (m, 3Н), 2.17 (s,3H), 2.05-1.72 (m, 8H), 1.40 (s, 3H), 1.28-1.19 (m, 4H), 0.83 (t, 3H).
Пример 9
2-((2-Амино-7-(6-(пиперазин-1-илметил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (9)
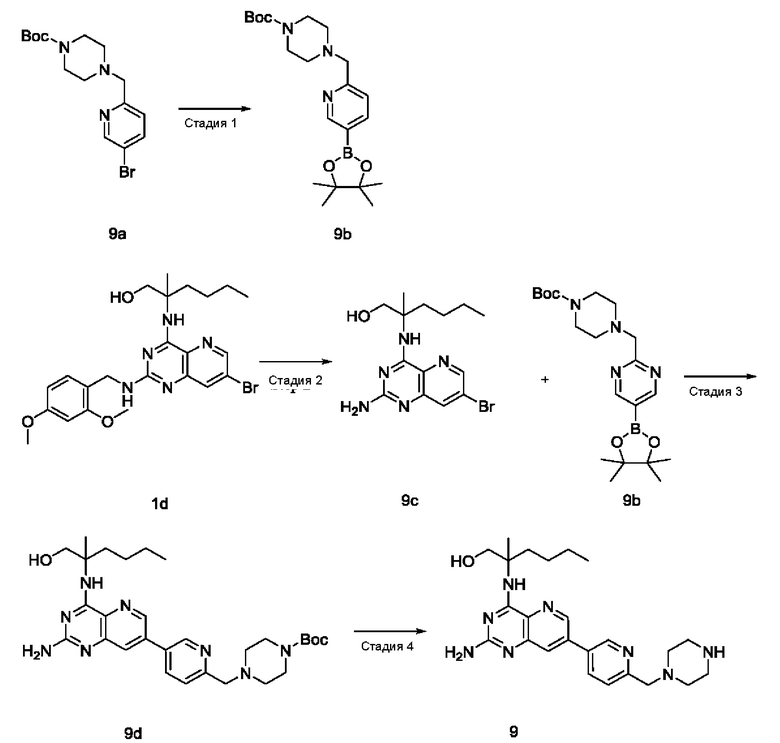
Стадия 1
трет-Бутил-4-((5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)метил)пиперазин-1 -карбоксилат (9b) Соединение 9а (полученное согласно способу, изложенному в примере 121 на странице 220 описания патентной заявки WO2013103973) (180 мг; 0,51 ммоль) добавляли к 5 мл диметилового эфира этиленгликоля, после чего добавляли бис(пинаколато)дибор (193 мг; 0,76 ммоль), дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия (37 мг; 0,050 ммоль) и ацетат калия (149 мг; 1,52 ммоль). Реакционный раствор три раза продували аргоном, нагревали до 80°С и перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. К реакционной системе добавляли 20 мл воды, затем экстрагировали дихлорметаном (10 мл ×3). Органические фазы объединяли, промывали водой (20 мл) и насыщенным раствором хлорида натрия (20 мл) в указанном порядке, сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении, получая неочищенный указанный в заголовке продукт 9b (203 мг; выход: 100%), который использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 404,2 [М+1].
Стадия 2
2-((2-Амино-7-бромпиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (9 с)
Соединение 1d (1,0 г; 1,98 ммоль) добавляли к 15 мл трифторуксусной кислоты и проводили взаимодействие при комнатной температуре в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении. В реакционный раствор добавляли 20 мл насыщенного раствора бикарбоната натрия, затем экстрагировали дихлорметаном (20 мл ×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали высокоэффективной жидкостной хроматографией (Waters-2767; система элюирования: H2O (NH4OAc, 10 ммоль), ACN), получая продукт 9 с (402 мг; выход: 58,0%).
Стадия 3
трет-Бутил-4-((5-(2-амино-4-((1-гидрокси-2-метилгексан-2-ил)амино)пиридо[3,2-d]пиримидин-7-ил)пиридин-2-ил)метил)пиперазин-1-карбоксилат (9d)
Соединение 9 с (193 мг; 0,546 ммоль) добавляли к 10 мл 1,4-диоксана и 2 мл воды, после чего добавляли неочищенного соединение 9b (200 мг; 0,50 ммоль), дихлорид 1,1'-бисдифенилфосфиноферроцен-палладия (37 мг; 0,050 ммоль) и карбонат калия (138 мг; 1,00 ммоль). Реакционный раствор три раза продували аргоном, нагревали до 80°С и перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. К реакционной системе добавляли 20 мл воды, затем экстрагировали дихлорметаном (10 мл ×3). Органические фазы объединяли. Органическую фазу промывали водой (20 мл) и насыщенным раствором хлорида натрия (30 мл) в указанном порядке, сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали колоночной хроматографией на силикагеле с элюированием системой В, получая указанный в заголовке продукт 9d (125 мг; выход: 45,9%).
MS m/z (ESI): 551,3 [М+1].
Стадия 4
2-((2-Амино-7-(6-(пиперазин-1-илметил)пиридин-3-ил)пиридо[3,2-d]пиримидин-4-ил)амино)-2-метилгексан-1-ол (9)
Соединение 9d (100 мг; 0,181 ммоль) добавляли к 5 мл трифторуксусной кислоты и проводили взаимодействие при комнатной температуре в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении. В реакционный раствор добавляли 50 мл насыщенного раствора карбоната натрия, затем экстрагировали дихлорметаном (50 мл ×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл), сушили над безводным сульфатом магния и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остатки очищали высокоэффективной жидкостной хроматографией (Waters-2767; система элюирования: H2O (NH4OAc, 10 ммоль), ACN), получая продукт 9 (30 мг; выход: 36,7%).
MS m/z (ESI):451,2[M+1].
1H ЯМР (400 МГц, ДМСО-d6) δ 8.88 (s, 1Н), 8.60 (s, 1H), 8.14-8.17 (m, 1H), 7.79 (s, 1H), 7.21 (s, 1H), 6.37 (br, 3Н), 3.67-3.71 (m, 1H), 3.71-3.67 (m, 1H), 3.60 (s, 2H), 3.48-3.50 (m, 1H), 2.71-2.74 (m, 3Н), 2.30-2.35 (m, 4H), 1.90-1.92 (m, 2H), 1.40 (s, 3Н), 1.21-1.24 (m, 6H), 0.81-0.84 (t, 3Н).
Примеры тестирования
Биологический анализ
Пример тестирования 1. Определение агонистической активности соединений по настоящему изобретению в отношении TLR8 и TLR7 человека
Агонистическое действие соединений по настоящему изобретению в отношении hTLR8 (TLR8 человека), экспрессированного клетками HEK-Blue™, стабильно трансфицированными hTLR8, определяли с использованием приведенного далее метода эксперимента.
I. Материалы и приборы для эксперимента
1. DMEM (модифицированная Дульбекко среда Игла; Gibco, 10564-029).
2. Фетальная телячья сыворотка (GIBCO, 10099).
3. Раствор трипанового синего (Sigma, T8154-100ML).
4. Многофункциональный микропланшетный ридер Flexstation 3 (Molecular Devices).
5. hTLR8-несущая клеточная линия HEK-Blue™ (InvivoGen, hkb-hTLR8) или hTLR7-несущая клеточная линия HEK-Blue™ (InvivoGen, hkb-hTLR7).
6. Детектирующий реагент HEK-Blue (InvivoGen, hb-det3) и
7. Фосфатный буферный раствор (PBS), рН 7,4 (Shanghai Basalmedia
Technologies Co., Ltd., B320).
II. Экспериментальные методики
а. Определение агонистической активности в отношении TLR8 человека
Сухой порошок из пакета с детектирующим реагентом HEK-Blue растворяли в 50 мл воды, не содержащей эндотоксина, и далее раствор помещали в инкубатор при 37°С на 10 минут, после чего проводили стерилизующую фильтрацию с целью получения детектирующей среды HEK-Blue. Сначала готовили 20 мМ концентрированный раствор соединения, затем разводили неразбавленным ДМСО до максимальной концентрации 6×106 нМ и путем 3-кратного последовательного разведения получали в общей сложности 10 концентраций. Приготовленные как указано выше растворы соединения сначала разбавляли в 20 раз в этой среде, затем в каждую лунку добавляли по 20 мкп разбавленного раствора соединения.
Из суспензии hTLRS-несущих клеток HEK-Blue™ удаляли супернатант, затем добавляли по 2-5 мл предварительно нагретого PBS. Клетки помещали в инкубатор на 1-2 минуты, осторожно пипетировали и подсчитывали с использованием окрашивания трипановым синим. Клетки ресуспендировали в детектирующей среде HEK-Blue с подведением содержания до 2,2×105 клеток/мл. В лунки вышеупомянутого 96-луночного планшета, содержащие по 20 мкл растворов соединения, добавляли по 180 мкл суспензии клеток и инкубировали при 37°С в течение 16 часов.
С использованием микропланшетного ридера на длине волны 620 нм получали соответствующие значения оптической плотности (ОП) и значения полумаксимальной эффективной концентрации (EC50) для соединений рассчитывали, применяя программное обеспечение Graphpad Prism.
b. Определение агонистической активности в отношении TLR7 человека
Сухой порошок из пакета с детектирующим реагентом HEK-Blue растворяли в 50 мл воды, не содержащей эндотоксина, и далее раствор помещали в инкубатор при 37°С на 10 минут, после чего проводили стерилизующую фильтрацию с целью получения детектирующей среды HEK-Blue. Сначала готовили 20 мМ концентрированный раствор соединения, затем разводили неразбавленным ДМСО до максимальной концентрации 6×106 нМ и путем 3-кратного последовательного разведения получали в общей сложности 10 концентраций. Приготовленные как указано выше растворы соединения сначала разбавляли в 20 раз в этой среде, затем в каждую лунку добавляли по 20 мкл разбавленного раствора соединения.
Из суспензии hTLR7-несущих клеток HEK-Blue™ удаляли супернатант, затем добавляли по 2-5 мл предварительно нагретого PBS. Клетки помещали в инкубатор на 1-2 минуты, осторожно пипетировали и подсчитывали с использованием окрашивания трипановым синим. Клетки ресуспендировали в детектирующей среде НЕК-Blue с подведением содержания до 2,2×105 клеток/мл. В лунки вышеупомянутого 96-луночного планшета, содержащие по 20 мкп растворов соединения, добавляли по 180 мкп суспензии клеток и инкубировали при 37°С в течение 16 часов.
С использованием микропланшетного ридера на длине волны 620 нм получали соответствующие значения ОП и значения EC50 для соединений рассчитывали, применяя Graphpad Prism.
Агонистическое действие соединений по настоящему изобретению в отношении TLR8 и TLR7 человека может быть определено с использованием указанного выше метода тестирования, и полученные значения EC50 показаны в Таблице 1.
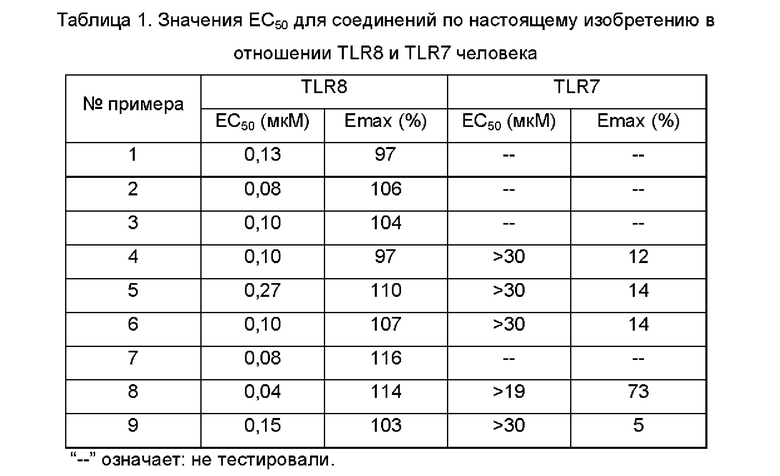
Вывод: соединения по настоящему изобретению оказывают сильное активирующее действие в отношении TLR8 человека, не проявляя при этом никакого активирующего действия в отношении TLR7 человека, что указывает на селективность соединений по настоящему изобретению в отношении TLR8.
Пример тестирования 2. Ингибирующее действие соединений по настоящему изобретению в отношении ферментативной активности сайта метаболизма мидазолама цитохрома Р450 3А4 (CYP3A4) в микросомах печени человека
Действие соединений по настоящему изобретению в отношении ферментативной активности сайта метаболизма мидазолама CYP3A4 в микросомах печени человека, определяли приведенным далее экспериментальным методом.
I. Материалы и приборы для эксперимента
1. Фосфатный буферный раствор (PBS), рН 7,4 (Shanghai Basalmedia Technologies Co., Ltd., B320, далее аналогично).
2. Никотинамидадениндинуклеотидфосфат, восстановленная форма (NADPH) (Sigma, N-1630).
3. Микросомы печени человека (Corning Gentest).
4. Жидкостной хроматограф/масс-спектрометр ABI QTrap 4000 (AB Sciex).
5. Колонка Inertsil C8-3; 4,6×50 мм, 5 мкм (Dikma Technologies Inc., USA) и
6. Маркерный субстрат для CYP (15 мкМ раствор мидазолама, SIGMA, UC429) и ингибитор в качестве положительного контроля (кетоконазол, SIGMA, K1003).
II. Экспериментальные методики
Готовили 100 мМ буфер на основе PBS, который затем использовали для приготовления раствора микросом печени человека в концентрации 2,5 мг/мл и 5 мМ раствора NADPH. Рабочий раствор соединений в 5-кратной концентрации последовательно разбавляли PBS (150; 50; 15; 5; 1,5; 0,15; 0,015; 0 мкМ). Рабочий раствор кетоконазола в 5-кратной концентрации последовательно разбавляли PBS (150; 50; 15; 5; 1,5; 0,15; 0,015; 0 мкМ). Рабочий раствор мидазолама разбавляли PBS до концентрации 15 мкМ.
По 20 мкл раствора микросом (2,5 мг/мл), 20 мкл 15 мкМ рабочего раствора мидазолама, 20 мкл раствора MgCl2 и 20 мкл рабочего раствора соединений (150;
50; 15; 5; 1,5; 0,15; 0,015; 0 мкМ; для каждой концентрации использовали разные реакционные системы) хорошо перемешивали. В случае группы положительного контроля соединение заменяли на кетоконазол в той же концентрации. Эту смесь и 5 мМ раствор NADPH предварительно инкубировали при 37°С в течение 5 минут. Через 5 минут в каждую лунку добавляли по 20 мкл раствора NADPH, начиналась реакция, и планшет инкубировали в течение 30 минут. Все инкубируемые образцы были представлены в двух повторах. Через 30 минут ко всем образцам добавляли по 250 мкл ацетонитрила, содержащего внутренний стандарт (камптотецин, 100 нг/мл), хорошо перемешивали, встряхивали при 800 об./мин в течение 10 минут и затем центрифугировали при 3700 об./мин в течение 10 минут. Отбирали по 80 мкл супернатанта и анализировали посредством жидкостной хроматографии в сочетании стандемной масс-спектрометрией (LC-MS/MS).
Данные рассчитывали с применением Graphpad Prism, получая значения IC50 для соединений в отношении сайта метаболизма мидазолама CYP3A4, которые показаны в Таблице 2.
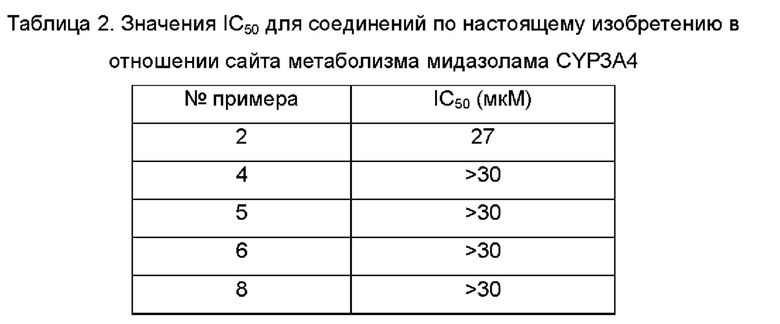
Вывод: соединения по настоящему изобретению не оказывают никакого ингибирующего действия на сайт метаболизма мидазолама CYP3A4 в микросомах печени человека, и демонстрируют хорошую безопасность, показывая, что метаболического взаимодействия лекарственных средств, обусловленного сайтом метаболизма мидазолама CYP3A4, происходить не будет.
Пример тестирования 3. Ингибирующее действие соединений по настоящему изобретению в отношении ферментативной активности цитохрома Р450 2D6 (CYP2D6) в микросомах печени человека
Действие соединений по настоящему изобретению в отношении ферментативной активности CYP2D6 в микросомах печени человека определяли приведенным далее экспериментальным методом.
I. Материалы и приборы для эксперимента
1. Фосфатный буферный раствор (PBS).
2. NADPH (Sigma, N-1630).
3. Микросомы печени человека (Corning Gentest).
4. Жидкостной хроматограф/масс-спектрометр ABI QTrap 4000 (AB Sciex).
5. Колонка Inertsil C8-3; 4,6×50 мм, 5 мкм (Dikma Technologies Inc., USA) и
6. Маркерный субстрат для CYP (20 мкМ раствор декстрометорфана, SIGMA, Q0750) и ингибитор в качестве положительного контроля (хинидин, SIGMA, D9684).
II. Экспериментальные методики
Готовили 100 мМ буфер на основе PBS, который затем использовали для приготовления раствора микросом печени человека в концентрации 2,5 мг/мл и 5 мМ раствора NADPH. Рабочий раствор соединений в 5-кратной концентрации последовательно разбавляли PBS (150; 50; 15; 5; 1,5; 0,15; 0,015; 0 мкМ). Рабочий раствор хинидина в 5-кратной концентрации последовательно разбавляли PBS (150; 50; 15; 5; 1,5; 0,15; 0,015; 0 мкМ). Рабочий раствор декстрометорфана разбавляли PBS до концентрации 20 мкМ.
По 20 мкл раствора микросом (2,5 мг/мл), 20 мкл 20 мкМ рабочего раствора декстрометорфана, 20 мкл раствора MgCl2 и 20 мкл рабочего раствора соединений (150; 50; 15; 5; 1,5; 0,15; 0,015; 0 мкМ; для каждой концентрации использовали разные реакционные системы) хорошо перемешивали. В случае группы положительного контроля соединение заменяли на хинидин в той же концентрации. Эту смесь и 5 мМ раствор NADPH предварительно инкубировали при 37°С в течение 5 минут. Через 5 минут в каждую лунку добавляли по 20 мкл раствора NADPH, начиналась реакция, и планшет инкубировали в течение 30 минут. Все инкубируемые образцы были представлены в двух повторах. Через 30 минут ко всем образцам добавляли по 250 мкл ацетонитрила, содержащего внутренний стандарт (камптотецин, 100 нг/мл), хорошо перемешивали, встряхивали при 800 об./мин в течение 10 минут и затем центрифугировали при 3700 об./мин в течение 10 минут. Отбирали по 80 мкл супернатанта и анализировали посредством LC-MS/MS.
Данные рассчитывали с применением Graphpad Prism, получая значения IC50 для ингибирующего действия соединений по настоящему изобретению в отношении фермента CYP2D6, которые показаны в Таблице 3.
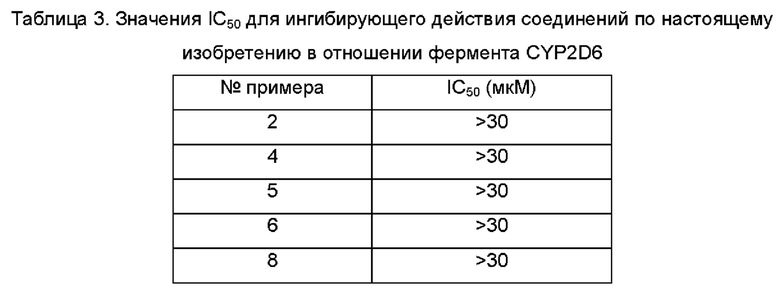
Вывод: соединения по настоящему изобретению оказывают незначительное ингибирующее действие в отношении активности фермента CYP2D6 в микросомах печени человека и демонстрируют хорошую безопасность, показывая, что метаболического взаимодействия лекарственных средств, обусловленного CYP2D6, происходить не будет.
Пример тестирования 4. Ингибирующее действие соединений по настоящему изобретению в отношении ферментативной активности сайта метаболизма тестостерона CYP3A4 в микросомах печени человека
Действие соединений по настоящему изобретению в отношении ферментативной активности сайта метаболизма тестостерона CYP3A4 в микросомах печени человека, определяли приведенным далее экспериментальным методом.
I. Материалы и приборы для эксперимента
1. Фосфатный буферный раствор (PBS).
2. NADPH (Sigma, N-1630).
3. Микросомы печени человека (Corning Gentest).
4. Жидкостной хроматограф/масс-спектрометр ABI QTrap 4000 (AB Sciex).
5. Колонка Inertsil C8-3; 4,6×50 мм, 5 мкм (Dikma Technologies Inc., USA) и
6. Маркерный субстрат для CYP (тестостерон/100 мкМ раствор, SIGMA, К1003) и ингибитор в качестве положительного контроля (кетоконазол, Dr. EhrenstorferGmbH, C17322500).
II. Экспериментальные методики
Готовили 100 мМ буфер на основе PBS, который затем использовали для приготовления раствора микросом печени человека в концентрации 2,5 мг/мл и 5 мМ раствора NADPH. Рабочий раствор соединений в 5-кратной концентрации последовательно разбавляли PBS (150; 50; 15; 5; 1,5; 0,15; 0,015; 0 мкМ). Рабочий раствор кетоконазола в 5-кратной концентрации последовательно разбавляли PBS (150; 50; 15; 5; 1,5; 0,15; 0,015; 0 мкМ). Рабочий раствор декстрометорфана разбавляли PBS до концентрации 50 мкМ.
По 20 мкп раствора микросом (2,5 мг/мл), 20 мкл 50 мкМ рабочего раствора тестостерона, 20 мкп раствора MgCl2 и 20 мкп рабочего раствора соединений (150; 50; 15; 5; 1,5; 0,15; 0,015; 0 мкМ; для каждой концентрации использовали разные реакционные системы) хорошо перемешивали. В случае группы положительного контроля соединение заменяли на кетоконазол в той же концентрации. Эту смесь и 5 мМ раствор NADPH предварительно инкубировали при 37°С в течение 5 минут. Через 5 минут в каждую лунку добавляли по 20 мкл раствора NADPH, начиналась реакция, и планшет инкубировали в течение 30 минут. Все инкубируемые образцы были представлены в двух повторах. Через 30 минут ко всем образцам добавляли по 250 мкл ацетонитрила, содержащего внутренний стандарт (камптотецин, 100 нг/мл), хорошо перемешивали, встряхивали при 800 об./мин в течение 10 минут и затем центрифугировали при 3700 об./мин в течение 10 минут. Отбирали по 80 мкл супернатанта и анализировали посредством LC-MS/MS.
Данные рассчитывали с применением Graphpad Prism, получая значения IC50 для соединений в отношении сайта метаболизма тестостерона CYP3A4, которые показаны в Таблице 4.
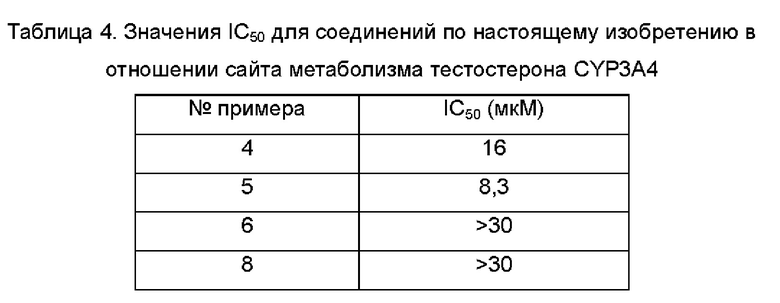
Вывод: соединения по настоящему изобретению не оказывают никакого ингибирующего действия на сайт метаболизма тестостерона CYP3A4 в микросомах печени человека, и демонстрируют хорошую безопасность, показывая, что метаболического взаимодействия лекарственных средств, обусловленного сайтом метаболизма тестостерона CYP3A4, происходить не будет.
Пример тестирования 5. Определение стимулирующего действия соединений по настоящему изобретению в отношении способности мононуклеарных клеток периферической крови (РВМС, от англ. peripheral blood mononuclear cells) секретировать IL12 и IFNγ
Стимулирующее действие соединений по настоящему изобретению в отношении способности РВМС секретировать IL12 и IFNγ определяли приведенным далее экспериментальным методом.
I. Материалы и приборы для эксперимента
1. Среда 1640 от Мемориального института Розуэлла Парка (RPMI 1640) (Invitrogen, 11875).
2. Фетальная телячья сыворотка (FBS) (Gibco, 10099-141).
3. Фиколл-пак PREMIUM (GE, 17-5442-02).
4. Раствор трипанового синего (Sigma, T8154-100ML).
5. SepMate™-50 (Stemcell, 15460).
6. Счетчик форменных элементов крови Bright-Line™ (Sigma, Z359629-1EA).
7. Планшет 96-луночный для культивирования клеток (Corning, 3599).
8. Планшет 96-луночный с лунками с V-образным дном (Corning, 3894).
9. ELISA-набор для определения IL-12 человека (Neobioscience Technologies Co., EHC152.96).
10. Набор для определения IFNγ человека (CISBIO, 62HIFNGPEG) и
11. Многофункциональный микропланшетный ридер PHERAStar (BMG, PHERAStar).
II. Экспериментальные методики
Соединение разводили неразбавленным ДМСО до максимальной концентрации 5 мМ и путем 4-кратного последовательного разведения получали в общей сложности 9 концентраций. Затем по 4 мкл растворов соединения добавляли к 196 мкл среды RPMI 1640, содержащей 10% FBS, и хорошо перемешивали. Отбирали по 50 мкл этой смеси и добавляли в лунки 96-луночного планшета для культивирования клеток.
Все реагенты выдерживали до достижения комнатной температуры. В колбу для культивирования емкостью 250 мл добавляли 60 мл крови здорового человека и такой же объем PBS (содержащего 2% FBS), осторожно пипетировали, хорошо перемешивали и разбавляли. В центрифужную пробирку SepMate™-50 емкостью 50 мл для работы с РВМС добавляли 15 мл раствора фиколл-пака PREMIUM для разделения лимфоцитов, затем добавляли 30 мл указанной выше разбавленной крови. Смесь центрифугировали при 1200×g в течение 10 минут при комнатной температуре. Отбирали супернатант и затем центрифугировали при 300 ×g в течение 8 минут. Клетки ресуспендировали в среде RMPI 1640, содержащей 10% FBS, подсчитывали и количество РВМС подводили до 3,33×106 кпеток/мл. В лунки планшета, содержащие соединения, добавляли по 150 мкл суспензии клеток и выдерживали в инкубаторе при 37°С, 5,0% CO2 в течение 24 часов. Планшет для культивирования клеток помещали в центрифугу и центрифугировали при 1200 об./мин в течение 10 минут при комнатной температуре. Из каждой лунки отбирали по 150 мкл супернатанта.
Реагенты из набора для ELISA IL-12 человека выдерживали до достижения комнатной температуры. Согласно приложенной к данному набору инструкции самая высокая концентрация стандарта составляет 2000 пг/мл, и путем 2-кратного последовательного разведения готовили в общей сложности 8 концентраций. Подлежащий тестированию образец разбавляли в 20 раз. Затем этот раствор добавляли в предварительно покрытый планшет в количестве 100 мкл/лунка. Планшет инкубировали при 37°С в течение 90 минут и промывали. Добавляли конъюгированное с антибиотиком антитело в количестве 100 мкл/лунка, планшет инкубировали при 37°С в течение 60 минут и промывали. Добавляли конъюгированный с пероксидазой хрена (HRP) фермент в количестве 100 мкл/лунка, планшет инкубировали при 37°С в течение 30 минут и промывали. Добавляли тетраметилбензидин (ТМВ) и планшет инкубировали при комнатной температуре в течение 5 минут. В конце добавляли стоп-раствор для остановки реакции и, используя микропланшетный ридер, измеряли поглощение при 450 нм.
Реагенты из набора для тестирования IFNγ человека выдерживали до достижения комнатной температуры. Стандартные и детектирующие антитела готовили согласно приложенной к данному набору инструкции в темноте. В каждую лунку добавляли по 16 мкл супернатанта, полученного путем центрифугирования, и в каждую лунку добавляли по 4 мкл свежеприготовленного путем перемешивания раствора детектирующего антитела. Этот раствор хорошо перемешивали путем встряхивания и инкубировали в течение ночи в темноте при комнатной температуре. Планшет прочитывали, используя многофункциональный микропланшетный ридер PHERAStar.
Концентрацию соединения, которая может стимулировать РВМС, с получением стандартного отклонения (3D, от англ. standard deviation), в 3 раза превышающим среднюю величину для группы без соединения (3D для группы без соединения), принимали за значение минимальной эффективной концентрации (МЕС, от англ. minimum effective concentration) соединения.
Стимулирующее действие соединений по настоящему изобретению в отношении способности РВМС секретировать IL12 и IFNγ определяли согласно приведенному выше методу тестирования, и полученные значения МЕС показаны в Таблице 5.
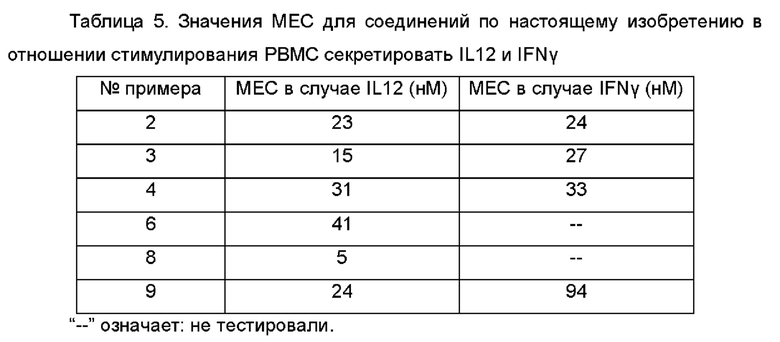
Вывод: из данных по активности соединений по настоящему изобретению в отношении стимулирования РВМС секретировать IL12 и IFNγ следует, что соединения по настоящему изобретению имеют преимущество, заключающееся в более низкой эффективной концентрации.
Пример тестирования 6. Определение ингибирующего действия соединений в отношении калиевых каналов hERG с применением Patchliner
1. Цель эксперимента
Блокирующее действие соединений по настоящей заявке в отношении тока калиевых каналов hERG определяли, используя автоматизированный метод пэтч-кламп на стабильной клеточной линии, трансфицированной геном калиевых каналов hERG.
2. Метод эксперимента
2.1. Материалы и приборы для эксперимента
2.1.1. Материалы эксперимента

2.2. Экспериментальные методики для автоматизированного пэтч-клампа Клеточную линию НЕК293 трансфицировали, используя плазмиду pCDNA3.1(+), в конструкции которой содержался ген hERG. Моноклональную стабильную клеточную линию HEK293-hERG отбирали посредством добавления генетицина (G418). Эту стабильную клеточную линию HEK293-hERG пересевали при плотности 1:4 в среду MEM/EBSS (сбалансированный солевой раствор Эрла) (10% FBS, G418 (400 мкг/мл), 1%-ный раствор MEM с заменимыми аминокислотами (100×), 1%-ный раствор пирувата натрия) и культивировали в течение 48-72 часов для проведения эксперимента с автоматизированным пэтч-клампом. В день эксперимента клетки отщепляли 0,25%-ным раствором трипсина (Life Technologies, 12563-029), собирали центрифугированием и ресуспендировали с использованием внеклеточной жидкости (140 мМ NaCl, 4 мМ KCl, 1 мМ MgCl2, 2 мМ CaCl2, 5 мМ MD глюкозы моногидрат, 10 мМ HEPES (2-гидроксиэтил-пиперазин-2-этансульфоновая кислота), рН 7,4; 298 мОсмоль), получая клеточную суспензию. Эту клеточную суспензию помещали в банк клеток прибора Patchliner; при этом в приборе Patchliner для внесения клеток в чип использовался регулятор вакуума (NPC-16), и индивидуальные клетки прикреплялись к лункам чипа под действием разрежения. После того, как была сформирована конфигурация "целая клетка", прибор регистрировал ток hERG в соответствии с установленной программой "ток-напряжение для hERG" и затем прибор автоматически осуществлял перфузию соединения с изменением концентрации от низкой до высокой. Значения тока для каждой концентрации соединения и холостого контрольного тока анализировали, используя усилитель для пэтч-клампа НЕАК ЕРС 10 (Nanion), программное обеспечение Pathliner и программное обеспечение для анализа данных, предоставленное Pathcontrol HTsoftware.
2.3. Результаты эксперимента
Блокирующее действие соединений по настоящему изобретению в отношении тока калиевых каналов hERG определяли согласно приведенному выше методу тестирования, и полученные значения IC50 показаны в Таблице 6.
Вывод: соединения по настоящей заявке оказывают слабое ингибирующее действие в отношении hERG и могут ослаблять побочные эффекты, обусловленные связанным с hERG путем.
Изобретение относится к производному пиридопиримидина, представленному общей формулой (I), где значения R1-R4, G1-G3, L1 определены в формуле изобретения, способу его получения, фармацевтической композиции, содержащей указанное производное, и его применению в качестве агониста толл-подобного рецептора 8 (TLR8). 9 н. и 6 з.п. ф-лы, 6 табл., 15 пр.
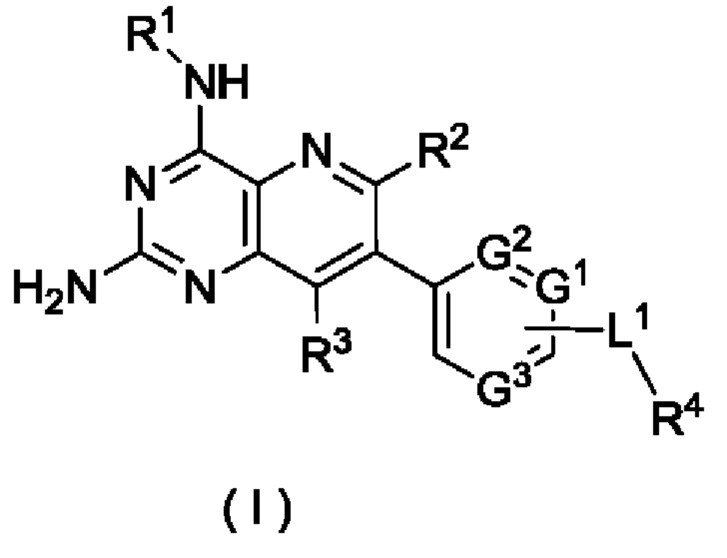
1. Соединение формулы (I):
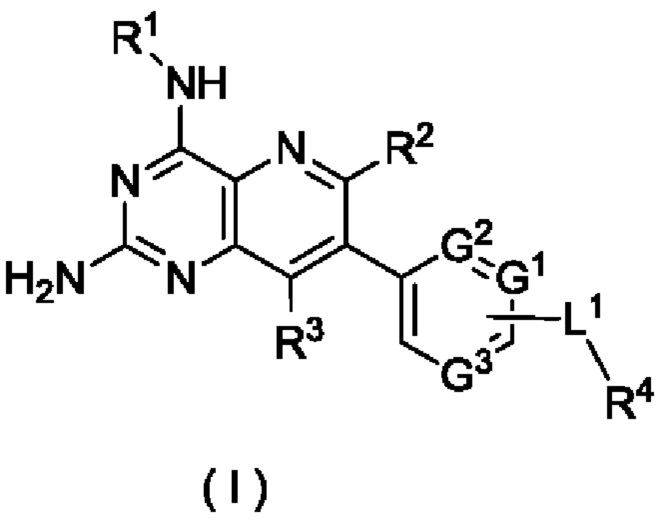
или его рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемая соль, где:
G2 представляет собой СН;
G1 и G3 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из СН и N, при условии, что по меньшей мере один из G1 и G3 представляет собой N;
L1 выбран из группы, состоящей из метилена и ковалентной связи;
R1 представляет собой C1-12 алкил, при этом C1-12 алкил необязательно замещен одной или несколькими гидроксигруппами;
R2 и R3 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена и С1-6 алкила; и
R4 представляет собой 5-6-членный моноциклический гетероциклил, содержащий один или два атома N, при этом 5-6-членный моноциклический гетероциклил необязательно замещен одним или несколькими C1-6 алкилами.
2. Соединение формулы (I) по п. 1, которое представляет собой соединение формулы (II):
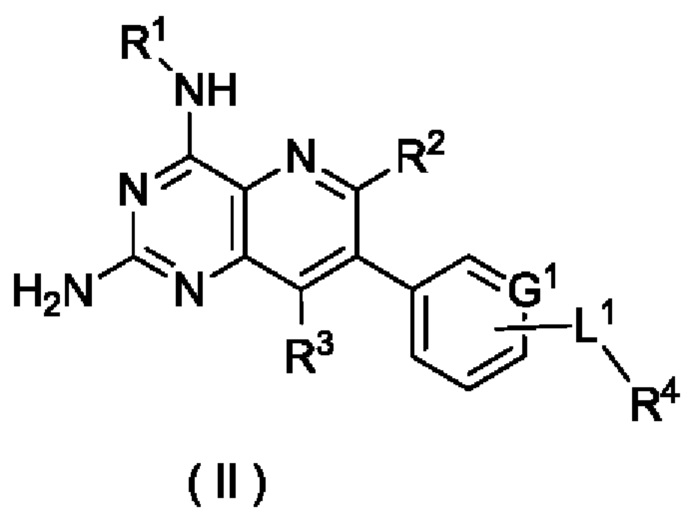
или его рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемую соль, где:
G1, L1 и R1-R4 являются такими, как определено в п. 1.
3. Соединение формулы (I) по п. 1 или 2, которое представляет собой соединение формулы (III):
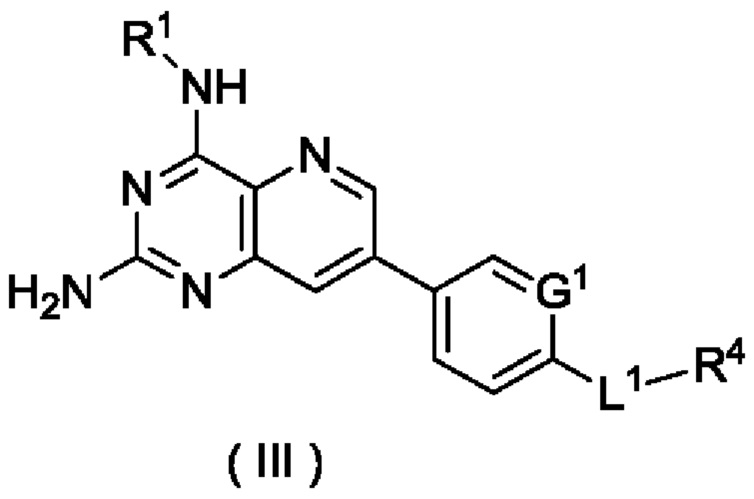
или его рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемую соль, где:
G1, L1, R1 и R4 являются такими, как определено в п. 1.
4. Соединение формулы (I) по любому из пп. 1-3, которое представляет собой соединение формулы (IV):
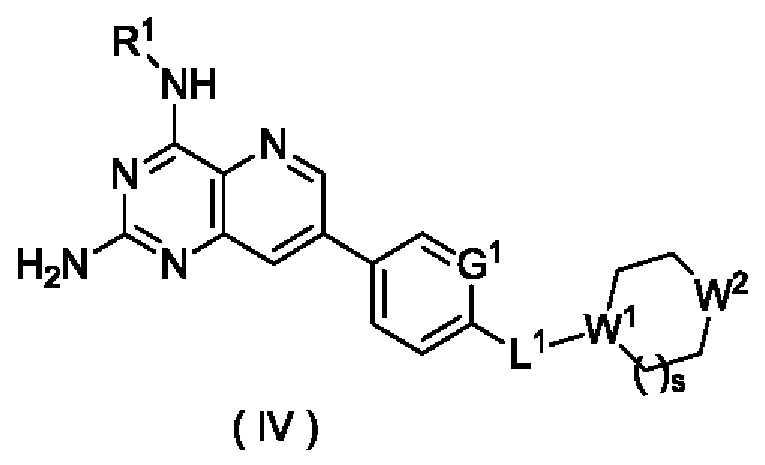
или его рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемую соль, где:
W1 представляет собой СН, и W2 представляет собой NR6; или
W1 представляет собой N, и W2 представляет собой СН2 или NR6;
R6 выбран из группы, состоящей из атома водорода и C1-6 алкила, и предпочтительно C1-6 алкила;
s равно 0 или 1; и
G1, L1 и R1 являются такими, как определено в п. 1.
5. Соединение формулы (I) по любому из пп. 1-4, которое представляет собой соединение формулы (V):
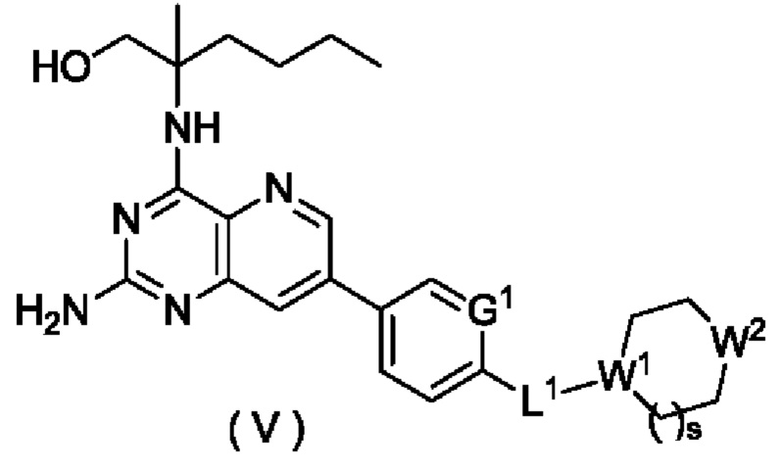
или его рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемую соль, где:
W1 представляет собой СН, и W2 представляет собой NR6; или
W1 представляет собой N, и W2 представляет собой СН2 или NR6;
R6 выбран из группы, состоящей из атома водорода и C1-6 алкила, и предпочтительно C1-6 алкила;
s равно 0 или 1; и
G1 и L1 являются такими, как определено в п. 1.
6. Соединение формулы (I) по любому из пп. 1-5, которое выбрано из группы, состоящей из:
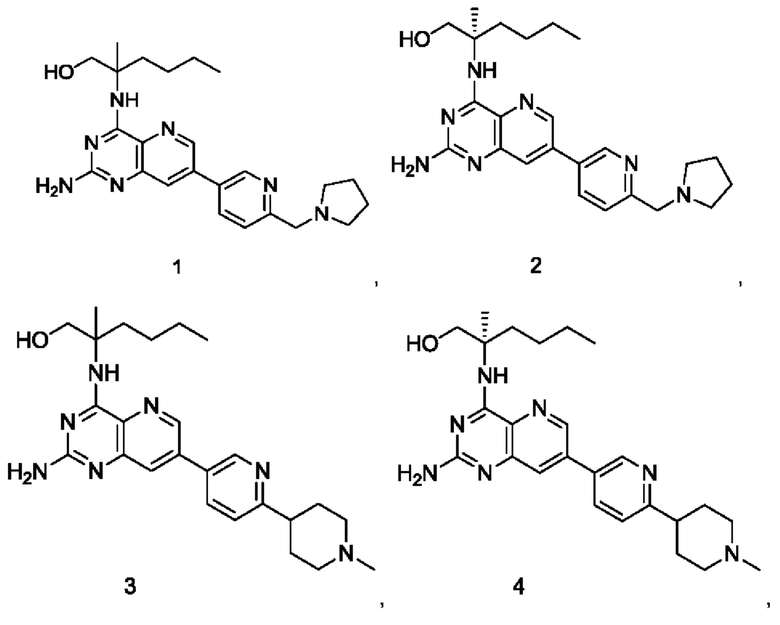
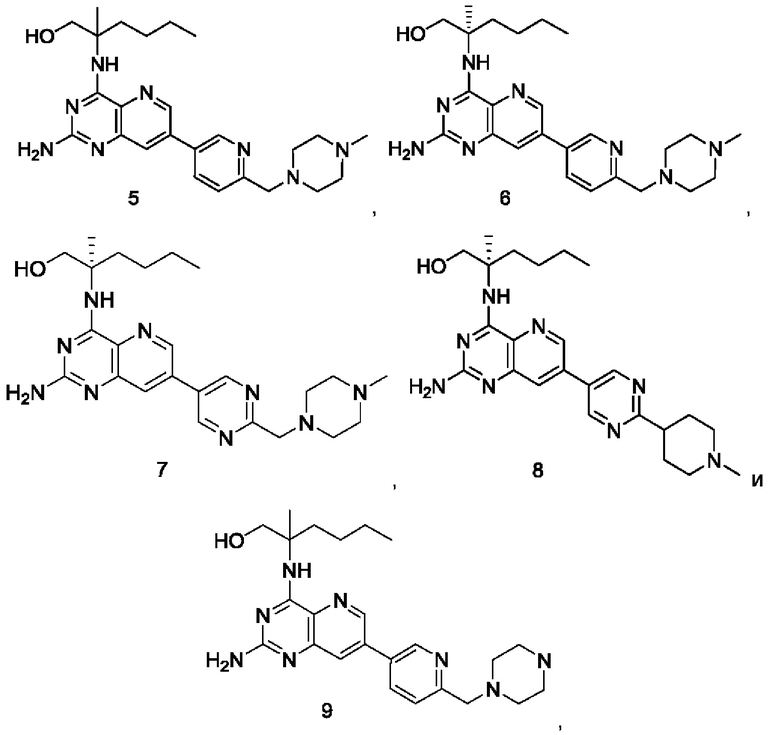
или его рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемая соль.
7. Соединение формулы (IA):
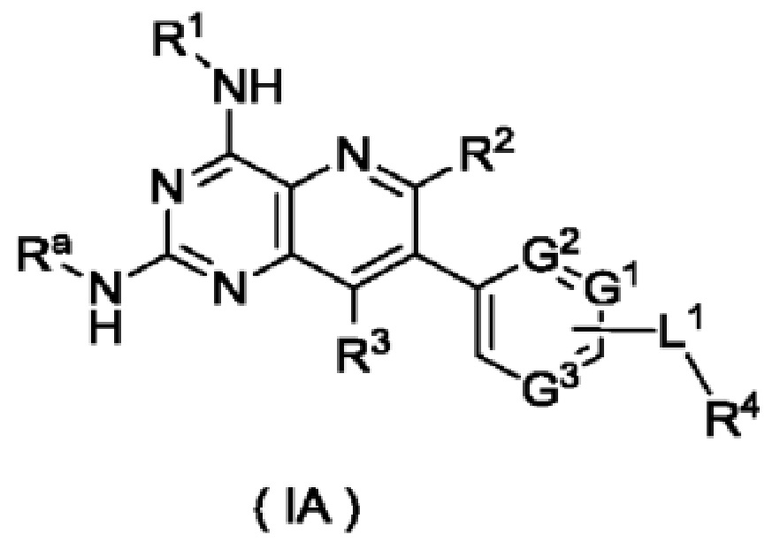
или его рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемая соль, где:
Ra представляет собой 2,4-диметоксибензил;
G2 представляет собой СН;
G1 и G3 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из СН и N, при условии, что по меньшей мере один из G1 и G3 представляет собой N;
L1 выбран из группы, состоящей из метилена и ковалентной связи;
R1 представляет собой С1-12 алкил, при этом С1-12 алкил необязательно замещен одной или несколькими гидроксигруппами;
R2 и R3 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена и C1-6 алкила; и
R4 представляет собой 5-6-членный моноциклический гетероциклил, содержащий один или два атома N, при этом 5-6-членный моноциклический гетероциклил необязательно замещен одним или несколькими С1-6алкилами.
8. Соединение, которое выбрано из группы, состоящей из:
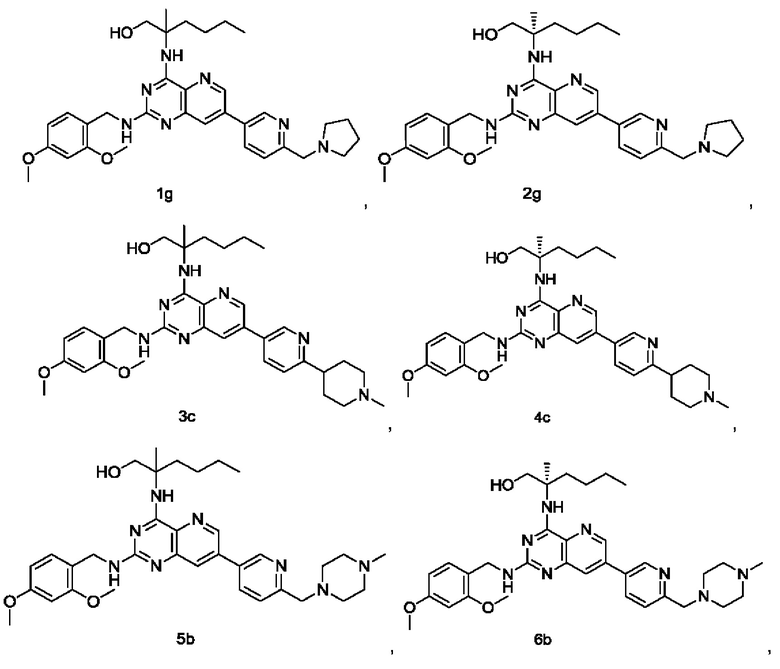
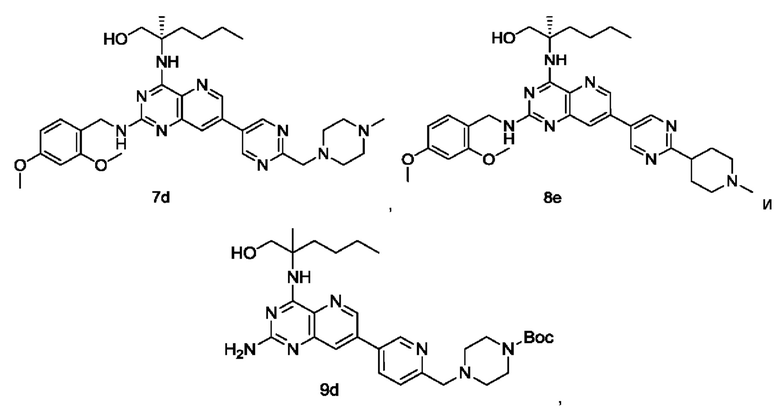
или его рацемат, энантиомер, диастереомер, или их смесь, либо их фармацевтически приемлемая соль.
9. Способ получения соединения формулы (I) по п. 1, включающий стадию:
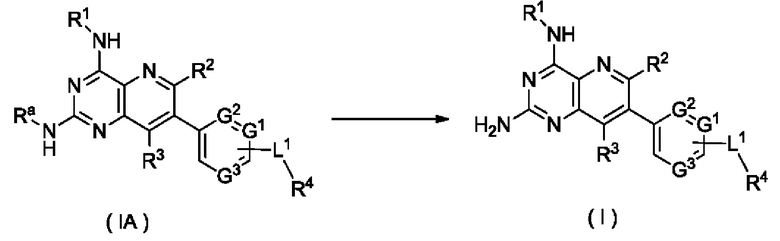
подвергания соединения формулы (IA) реакции удаления защиты с получением соединения формулы (I); где:
Ra представляет собой 2,4-диметоксибензил; и
G1-G3, L1 и R1-R4 являются такими, как определено в п. 1.
10. Фармацевтическая композиция для активации толл-подобного рецептора 8 (TLR8), содержащая терапевтически эффективное количество соединения формулы (I) по любому из пп. 1-6 и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент.
11. Применение соединения формулы (I) по любому из пп. 1-6 или фармацевтической композиции по п. 10 для приготовления лекарственного средства для активации толл-подобного рецептора 8 (TLR8).
12. Применение соединения формулы (I) по любому из пп. 1-6 или фармацевтической композиции по п. 10 для приготовления лекарственного средства для лечения инфекции, вызываемой вирусом, при этом вирус предпочтительно представляет собой вирус гепатита В, вирус гепатита С, вирус гриппа, вирус герпеса и вирус, вызывающий синдром приобретенного иммунодефицита (СПИД).
13. Применение соединения формулы (I) по любому из пп. 1-6 или фармацевтической композиции по п. 10 для приготовления лекарственного средства для регулирования иммунной системы.
14. Применение соединения формулы (I) по любому из пп. 1-6 или фармацевтической композиции по п. 10 для приготовления лекарственного средства для лечения или предупреждения возникновения опухоли.
15. Применение по п. 14, где опухоль представляет собой рак и предпочтительно выбранный из группы, состоящей из меланомы, рака легкого, рака печени, базальноклеточной карциномы, рака почки, миеломы, рака желчевыводящих путей, рака головного мозга, рака молочной железы, рака шейки матки, хориокарциномы, рака толстой кишки, рака прямой кишки, рака головы и шеи, перитонеальной опухоли, рака фаллопиевых труб, рака эндометрия, рака пищевода, рака желудка, лейкоза, лимфомы, саркомы, нейробластомы, рака полости рта, рака яичников, рака поджелудочной железы, рака предстательной железы, рака яичка, рака кожи и рака щитовидной железы.
| US 2013109693 A1, 02.05.2013 | |||
| WO 2006091395 A2, 31.08.2006 | |||
| WO 2006135993 A1, 28.12.2006 | |||
| EA 201791782 A1, 31.05.2018 | |||
| EA 201791305 A1, 29.12.2017. |

