Область техники
Настоящее изобретение имеет отношение к области медицины и относится к производному фенилпропанамида, к способу его получения и к его применению в медицине. В частности, настоящее изобретение относится к производному фенилпропанамида формулы (I), к способу его получения и к содержащей его фармацевтический композиции, к его применению в качестве агониста κ-опиоидного рецептора (KOR) и его применению для получения лекарственного средства для лечения и/или профилактики боли и связанных с болью заболеваний.
Предшествующий уровень техники
Опиоидные рецепторы являются важным классом рецепторов, связанных с G-белком, и являются целью комбинации эндогенных опиоидных пептидов и опиоидов. Активированные опиоидные рецепторы играют регуляторную роль в иммунитете нервной системы и в эндокринной системе. В настоящее время опиоиды являются самым сильным и наиболее часто используемым центральным анальгетиком. Эндогенные опиоидные пептиды являются природными опиоидными активными веществами у млекопитающих. Известные в настоящее время эндогенные опиоидные пептиды грубо подразделяют на энкефалины, эндорфины, динорфины и неоморфины (Pharmacol Rev 2007; 59: 88-123). В центральной нервной системе имеются соответствующие опиоидные рецепторы, а именно μ, δ, κ-рецепторы и тому подобное.
K-опиоидный рецептор (KOR) состоит из 380 аминокислот, а динорфин является его эндогенным лигандом. Он экспрессируется в сенсорных нейронах, ганглиозных клетках дорсального корешка и первичных афферентных нейронах и включен в важные физиологические процессы, такие как боль, нейроэндокринная система, эмоциональное поведение и познание. В настоящее время известно, что человеческий KOR кодируется геном OPRK1 и находится в хромосоме 8q11-12 (Simonin F, Gaveriaux Ruff С, Kieffer BL с соавт. Proc Natl Acad Sci USA 1995, 92(15): 7006-10). Активация KOR связана с G-белком Gi/G0, который повышает активность фосфодиэстеразы, ингибирует активность аденилатциклазы и снижает уровни внутриклеточного цАМФ, тем самым вызывая ингибирование нейронов. Агонисты KOR многократно воздействуют на рецепторы, вызывая десенсибилизацию и уменьшая ингибирование активности аденилатциклазы (Raynor K, Kong Н, Hines J, et al. J Pharmacol Exp Ther, 1994, 270: 1381-6). KOR также связан с калиевыми каналами внутреннего выпрямления и кальциевыми ионными каналами N-типа (Henry DJ, Grny DK, Lester HA, Davidson N, Chavkin С (Mar 1995) Molecular Pharmacology 47 (3): 551-7). Агонисты KOR способны ингибировать (кальций-зависимо) высвобождение про-болевого и провоспалительного вещества Р из периферических чувствительных нервных окончаний, что может быть причиной их антиноцицептивного и противовоспалительного действия. В дополнение к динорфинам различные природные алкалоиды и синтетические лиганды могут также связываться с KOR. KOR обеспечивает естественный механизм контроля зависимости, поэтому лекарство, являющееся агонистом рецептора, обладает потенциалом для лечения наркомании.
Эти наблюдения, например, эффект агониста KOR асимадолина при диабетической невропатии грызунов (Jolivalt et al. Diabetologia 2006, 49 (11): 2775-85; Epub 19 августа) и эффект действия агониста KOR U-50488 на модели хронического компрессионного повреждения (CCI) у крыс с невропатической болью и блокадой опиоидного антагониста налоксона в отношении его действия (Bileviciute-Ljungar с соавт. Eur. J. Pharm 2004. 494: 139-46), поддерживают использование агонистов KOR в лечении невропатической боли, вызванной диабетом, вирусами и химиотерапией. Применение агонистов KOR для лечения или профилактики висцеральной боли, включая гинекологические состояния, такие как дисменорея и эндометриоз, также оценивалось (Riviere, Br. J. Pharmacol 2004. 141: 1331-4).
К-опиоидные агонисты повышают почечную экскрецию воды и снижают экскрецию натрия с мочой (то есть вызывают селективный водный диурез, также известный как водо-стимулирующий). Многие исследователи считают, что этот эффект обусловлен ингибированием секреции вазопрессина гипофизом. Исследование, сравнивающее центрально действующие и предполагаемые периферические селективные κ-опиоиды, пришло к выводу, что KOR в пределах гематоэнцефалического барьера ответственен за опосредование этого эффекта. Некоторые исследователи предложили лечить гипонатриемию пептидом ноцицептина или заряженным пептидным конъюгатом, который действует на рецептор ноцицептина на периферии, а рецептор ноцицептина относится к KOR, но отличается (DR Kapusta, Life Sci., 60:15-21, 1997).
Патентные заявки, в настоящее время раскрывающие агонисты KOR, включают WO 20071398, WO 2008060552, WO 09932510, WO 2013184794, WO 2014089019, WO 2014184356 и WO 2015065867.
Агонисты к-опиоидного рецептора (KOR-рецептора) имеют хорошие перспективы применения в фармацевтической промышленности. Для достижения лучшего терапевтического эффекта и лучшего удовлетворения рыночного спроса авторы изобретения надеются разработать новое поколение агонистов рецептора KOR с высоким эффектом и низкой токсичностью. Настоящее изобретение предлагает новое соединение агониста κ-опиоидного рецептора (KOR-рецептора) (с дальнейшей модификацией аминогруппы глицина в структуре ядра), которое неожиданно демонстрирует превосходные эффекты и функции. В частности, когда заместитель в аминогруппе глицина представляет собой замещенную или незамещенную этиленовую группу, это соединение имеет неожиданный эффект.
Сущность изобретения
Настоящее изобретение относится к соединению формулы (I):
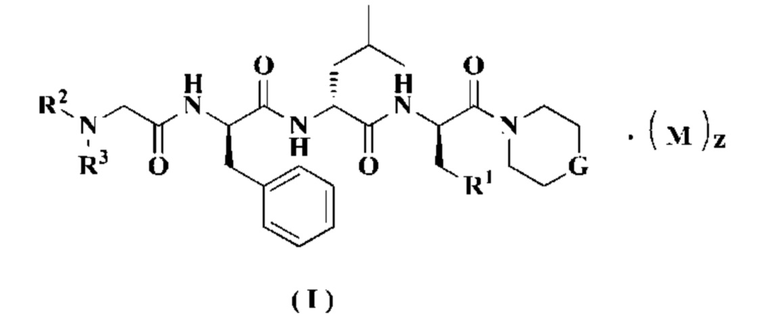
или к его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли, где:
М представляет собой неорганическую кислоту или органическую кислоту, предпочтительно - органическую кислоту и более предпочтительно трифторуксусную кислоту;
G выбран из группы, состоящей из О, -NR4 и -CR5R6;
R1 выбран из группы, состоящей из водорода, алкила, алкокси, галоалкила, галогена, амино, нитро, гидрокси, циано, циклоалкила, гетероциклила, арила, гетероарила, -OR7, -C(O)R7, -C(O)OR7, -S(O)mR7 и -NR8R9, где каждый алкил, галоалкил, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, галоалкила, галогена, амино, нитро, циано, гидрокси, алкокси, галоалкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R2 выбран из группы, состоящей из водорода, алкила, алкокси, галоалкила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, арилалкила, гетероарила, гетероарилалкила, -OR7, -C(O)R7 и -C(O)OR7, где каждый алкил, галоалкил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, арилалкил, гетероарил и гетероарилалкил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, галоалкила, галогена, амино, нитро, циано, гидрокси, алкокси, галоалкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R3 выбран из группы, состоящей из водорода, алкила, алкокси, галоалкила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, арила, арилалкила, гетероарила, гетероарилалкила, -OR7, -C(O)R7 и -C(O)OR7, где каждый алкил, галоалкил, циклоалкил, циклоалкилалкил, гетероциклил, гетероциклилалкил, арил, арилалкил, гетероарил и гетероарилалкил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, галоалкила, галогена, амино, нитро, циано, гидрокси, алкокси, галоалкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R4 выбран из группы, состоящей из водорода, алкила, галоалкила, циклоалкила, алкокси, гидроксиалкила, гидрокси, амино, алкоксикарбонила, гетероциклила, арила, гетероарила, -OR7, -C(O)R7, -C(O)OR7, -S(O)mR7, -NR8R9 и -NHC(O)NR8R9, где каждый алкил, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, галогена гидрокси, амино, алкоксикарбонила, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R5 и R6 каждый независимо выбран из группы, состоящей из водорода, алкила, алкокси, гидроксиалкила, гидрокси, амино, алкоксикарбонила, циклоалкила, гетероциклила, арила, гетероарила, -OR7, -C(O)R7, -C(O)OR7, -S(O)mR7, -NR8R9 и -NHC(O)NR8R9, где каждый алкил, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, галогена, гидрокси, амино, алкоксикарбонила, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R7 выбран из группы, состоящей из водорода, алкила, амино, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый алкил, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, галогена, гидрокси, амино, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R8 и R9 каждый независимо выбран из группы, состоящей из водорода, алкила, алкокси, гидроксиалкила, гидрокси, амино, алкоксикарбонила, циклоалкила, гетероциклила, арила и гетероарила, где каждый алкил, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, галогена, гидрокси, амино, алкоксикарбонила, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
z равно 0, 1, 2, 3 или 4; и
m равно 0, 1 или 2.
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (I) дополнительно представляет собой соединение формулы (II):
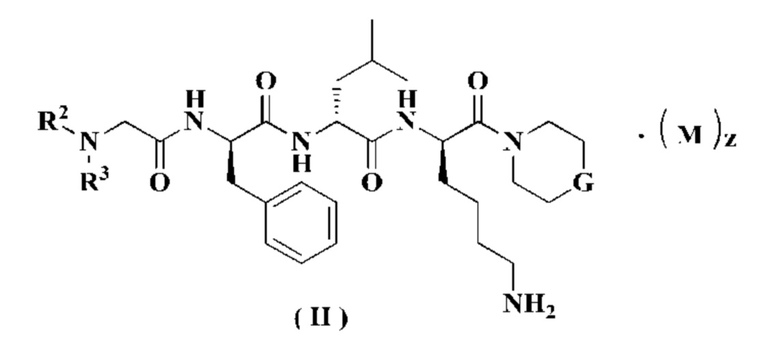
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или его фармацевтически приемлемую соль,
где:
М, G, R2, R3 и z являются такими, как определено в формуле (I).
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (I) или (II) дополнительно представляет собой соединение формулы (III):
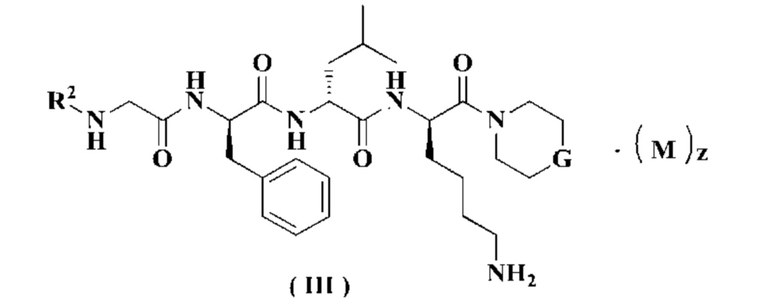
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или его фармацевтически приемлемую соль,
где:
М, G, R2, R3 и z являются такими, как определено в формуле (I).
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (I), (II) или (III) дополнительно представляет собой соединение формулы (IV):
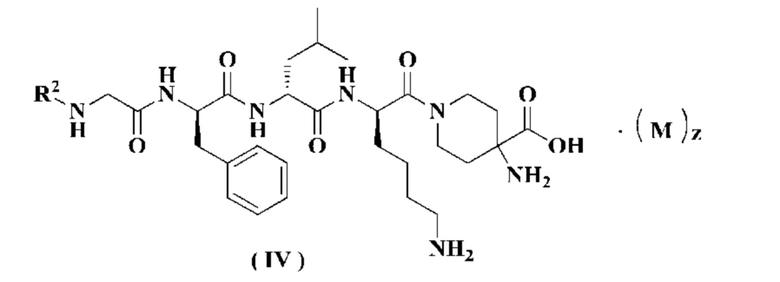
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или его фармацевтически приемлемую соль,
где:
М, R2 и z являются такими, как определено в формуле (I).
В предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I), (II), (III) или (IV) R2 выбран из группы, состоящей из арилалкила, циклоалкилалкила и циклоалкила, где каждый арилалкил, циклоалкилалкил и циклоалкил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, циклоалкила и арила.
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (I), (II) или (III) дополнительно представляет собой соединение формулы (III-A):
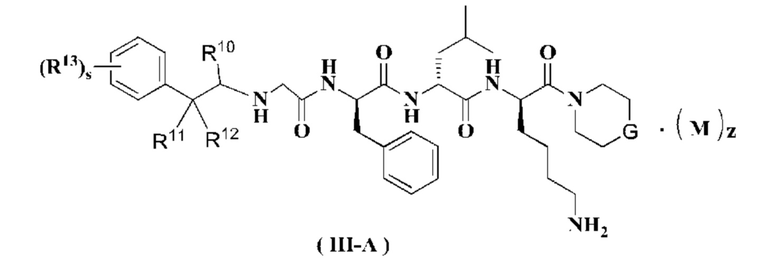
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или его фармацевтически приемлемую соль,
где:
G представляет собой О или CR5R6; предпочтительно CR5R6;
R10 выбран из группы, состоящей из водорода, алкила, галоалкила, галогена, амино, нитро, циано, гидрокси, алкокси, галоалкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R11 и R12 являются одинаковыми или различными, и каждый независимо выбран из группы, состоящей из водорода, алкила, галоалкила, галогена, амино, нитро, циано, гидрокси, алкокси, галоалкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
или R10 и R11, взятые вместе, образуют циклоалкил;
или R11 и R12, взятые вместе, образуют циклоалкил;
R13 выбран из группы, состоящей из водорода, алкила, галоалкила, галогена, амино, нитро, циано, гидрокси, алкокси, галоалкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
s равно 0, 1 или 2; и
R5 - R6, М и z являются такими, как определено в формуле (I).
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (I), (II), (III), (IV) или (III-A) дополнительно представляет собой соединение формулы (IV-A):
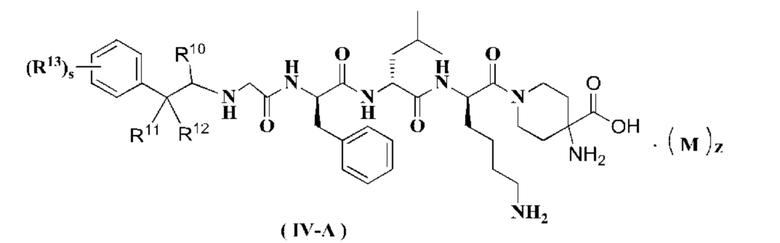
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или его фармацевтически приемлемую соль,
где:
R10 - R13, М, z и s являются такими, как определено в формуле (III-A).
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (I), (II), (III), (IV), (III-A) или (IV-A) дополнительно представляет собой соединение формулы (IV-B):
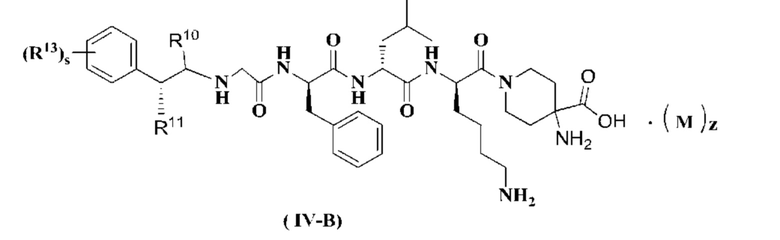
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или его фармацевтически приемлемую соль,
где:
R10 - R11, R13, М, z и s являются такими, как определено в формуле (III-A).
В предпочтительном варианте осуществления настоящего изобретения в соединении формулы (I), (II), (III), (IV), (III-A), (IV-A) или (IV-B) z равно 0 или 1.
Типичные соединения формулы (I) включают, но не ограничиваются этим:
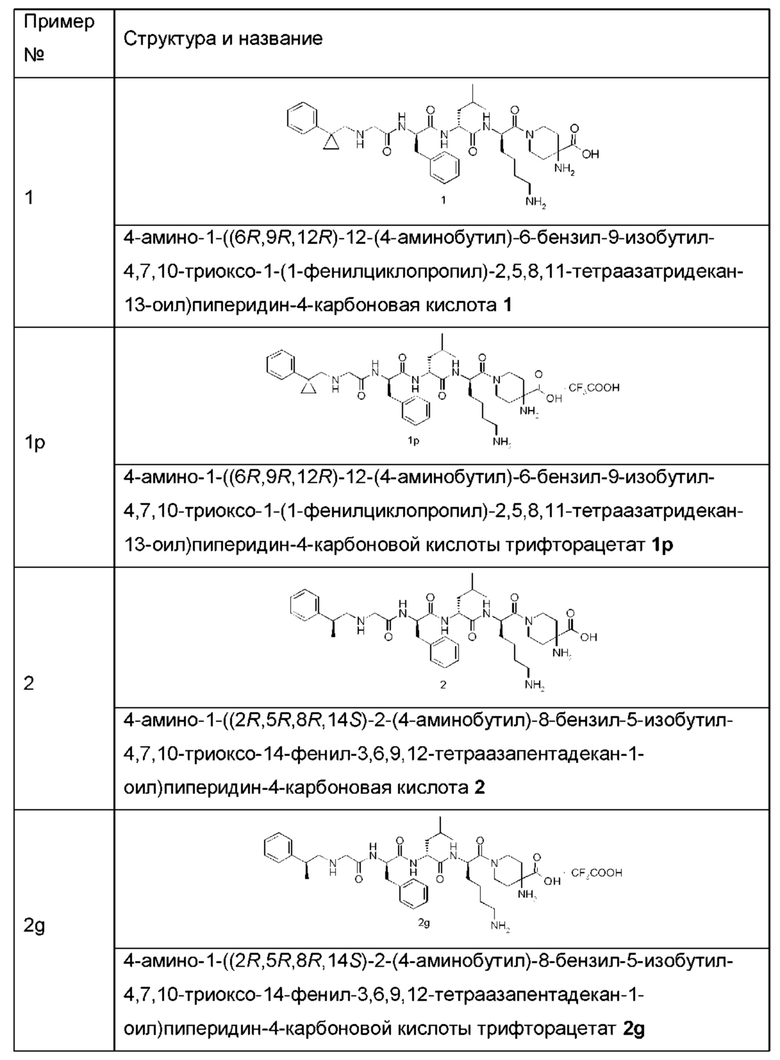
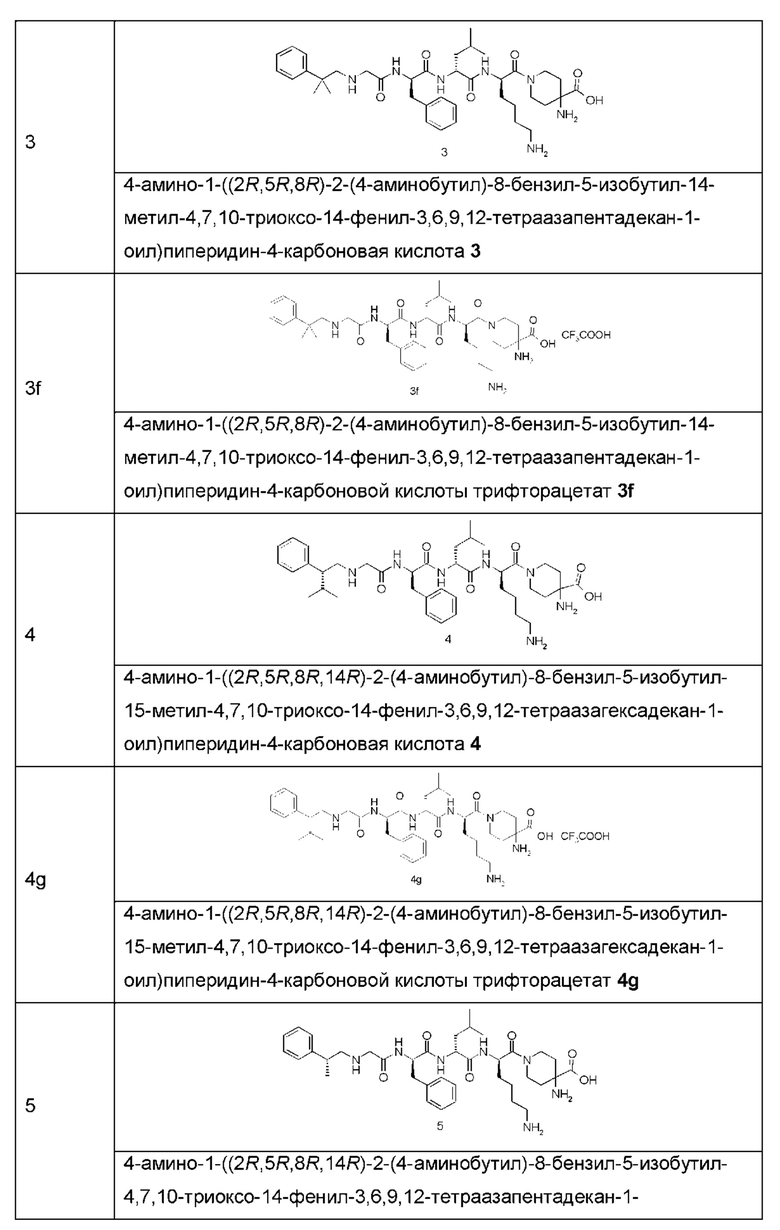
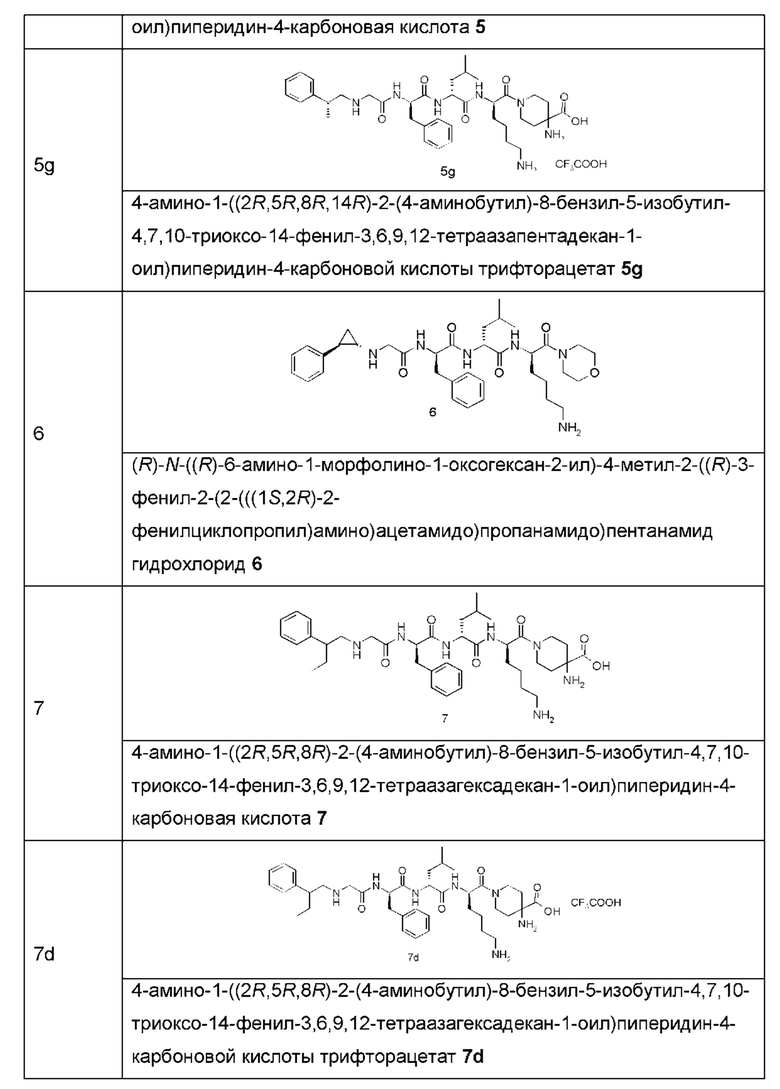
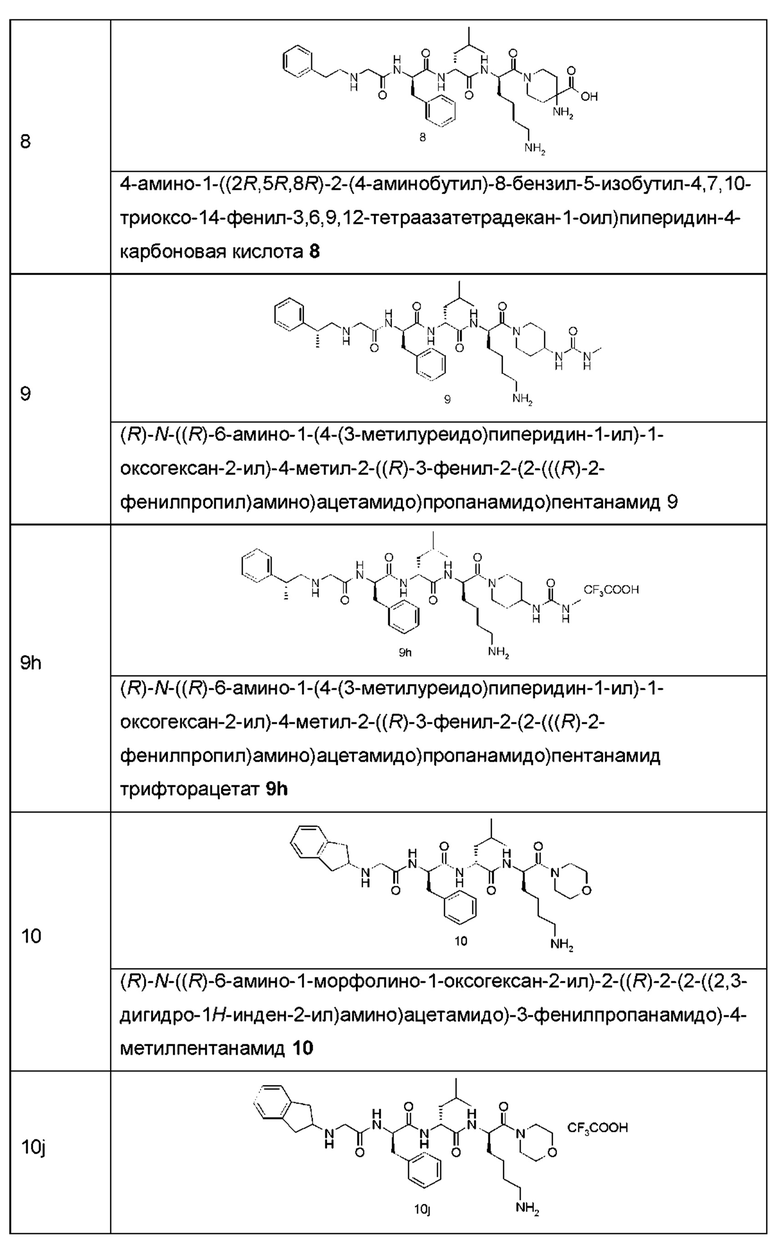
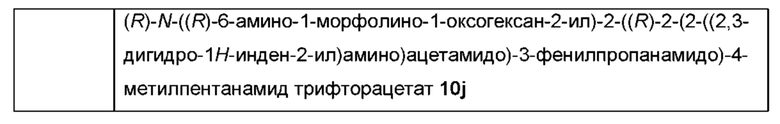
или их таутомеры, мезомеры, рацематы, энантиомеры, диастереомеры или их смеси, или их фармацевтически приемлемые соли.
В другом аспекте настоящее изобретение также относится к соединению формулы (V), которое является промежуточным соединением для получения соединения формулы (II):
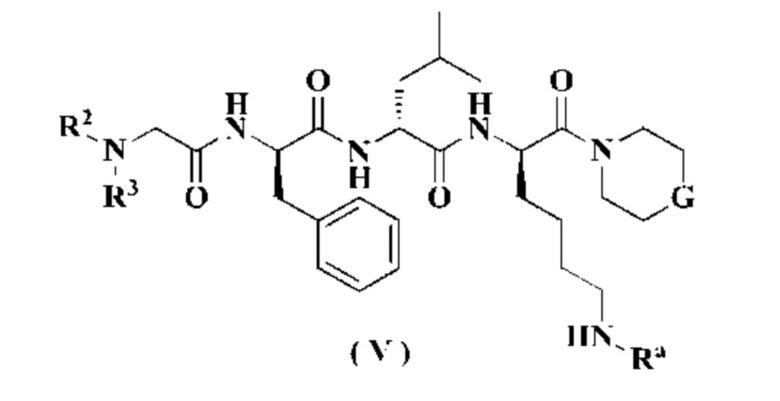
или к его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли,
где:
Ra является аминозащитной группой, предпочтительно представляет собой трет-бутоксикарбонил, 9-флуоренилметоксикарбонил, аллилоксикарбонил, трихлорэтоксикарбонил, триметилсилилэтоксикарбонил, бензилоксикарбонил, пара-метилбензолсульфонил, пара-нитробензолсульфонил или трет-бутил-(т.е., Boc, Fmoc, Alloc, Теос, CBz, Тозил, Нозил и t-Bu); и
G, R2 и R3 являются такими, как определено в формуле (II).
В другом аспекте настоящее изобретение также относится к способу получения соединения формулы (II), включающему стадию:
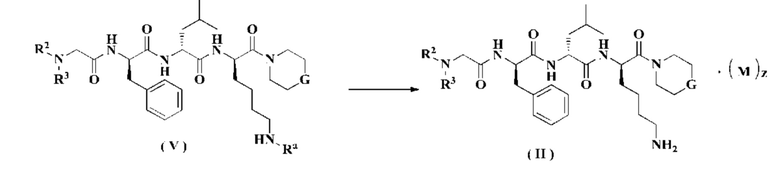
удаления защитной группы Ra из соединения формулы (V) в кислых условиях с получением соединения формулы (II); где:
М, G, z, R2 и R3 являются такими, как определено в формуле (II), и Ra является таким, как определено в формуле (V).
В другом аспекте настоящее изобретение также относится к соединению формулы (VI), которое является промежуточным соединением для получения соединения формулы (III):
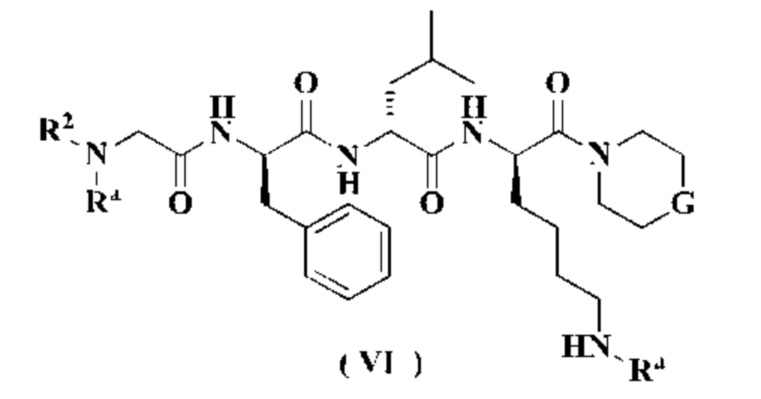
или к его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли,
где:
Ra является аминозащитной группой, предпочтительно представляет собой трет-бутоксикарбонил, 9-флуоренилметоксикарбонил, аллилоксикарбонил, трихлорэтоксикарбонил, триметилсилилэтоксикарбонил, бензилоксикарбонил, пара-метилбензолсульфонил, пара-нитробензолсульфонил или трет-бутил; и
G и R2 являются такими, как определено в формуле (III).
В другом аспекте настоящее изобретение также относится к способу получения соединения формулы (III), включающему стадию:
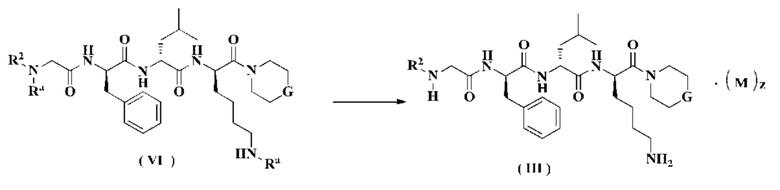
удаления защитной группы Ra из соединения формулы (VI) в кислых условиях с получением соединения формулы (II);
где:
М, G, z и R2 являются такими, как определено в формуле (III), и Ra является таким, как определено в формуле (VI).
Кислотным реагентом, обеспечивающим кислые условия, является предпочтительно раствор трифторуксусной кислоты или хлористого водорода в 1,4-диоксане.
Кроме того, когда z не равен нулю в соединениях формул (I), (II), (III), (IV), (III-А), (IV-A) или (IV-B) можно добавлять слабое основание для проведения свободной реакции с получением продукта в свободном состоянии, т.е. соединения формулы (I), (II), (III), (IV), (III-A), (IV-A) или (IV-B).
В другом аспекте настоящее изобретение также относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения вышеуказанной формулы (I), (II), (III), (IV), (III-A), (IV-A) или (IV-B), или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
Настоящее изобретение также относится к способу получения указанной композиции, включающему стадию смешивания соединения формулы (I), (II), (III), (IV), (III-A), (IV-A) или (IV-B), его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, с одним или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
В одном варианте осуществления фармацевтическая композиция согласно настоящему изобретению дополнительно содержит одно или более из следующих соединений: опиоиды, каннабиноиды, антидепрессанты, противосудорожные препараты, транквилизаторы, кортикостероиды, блокаторы ионных каналов или нестероидные противовоспалительные лекарственные средства (НПВЛС).
Изобретение дополнительно относится к применению соединения формулы (I), (II), (III), (IV), (III-A), (IV-A) или (IV-B), или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для получения лекарственного средства для агонизации или антагонизации κ-опиоидного рецептора (KOR-рецептора).
Изобретение дополнительно относится к применению соединения формулы (I), (II), (III), (IV), (III-A), (IV-A) или (IV-B), или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для получения лекарственного средства для профилактики и/или лечения заболевания, опосредованного агонистом κ-опиоидного рецептора и связанного с агонистом κ-опиоидного рецептора (KOR-рецептора), где заболевание, опосредованное агонистом κ-опиоидного рецептора и связанное с агонистом κ-опиоидного рецептора (KOR-рецептора), предпочтительно выбрано из группы, состоящей из боли, воспаления, зуда, отека, гипонатриемии, гипокалиемии, кишечной непроходимости, кашля и глаукомы, и более предпочтительно - боли.
Изобретение дополнительно относится к применению соединения формулы (I), (II), (III), (IV), (III-A), (IV-A) или (IV-B), или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для получения лекарственного средства для профилактики и/или лечения боли и связанных с болью заболеваний у млекопитающих (например, людей), где боль может представлять собой послеоперационную боль, боль, вызванную раком, невропатическую боль, травматическую боль и боль, вызванную воспалением, и тому подобное.
Настоящее изобретение также относится к способу агонизации или антагонизации κ-опиоидного рецептора (KOR-рецептора), включающему стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I), (II), (III), (IV), (III-A), (IV-A) или (IV-B) согласно настоящему изобретению, или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли.
Настоящее изобретение также относится к способу профилактики и/или лечения заболевания, опосредованного агонистом KOR-рецептора и связанного с агонистом KOR-рецептора, включающему стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I), (II), (III), (IV), (III-A), (IV-A) или (IV-B) согласно настоящему изобретению, или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли. Этот способ демонстрирует заметную эффективность и меньшее количество побочных эффектов. Где заболевание, опосредованное агонистом κ-опиоидного рецептора и связанное с агонистом κ-опиоидного рецептора (KOR-рецептора), выбрано из группы, состоящей из боли, воспаления, зуда, отека, гипонатриемии, гипокалиемии, кишечной непроходимости, кашля и глаукомы, предпочтительно - боли.
Настоящее изобретение дополнительно относится к соединению формулы (I), (II), (III), (IV), (III-A), (IV-A) или (IV-B), в частности формулы (I), или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или их фармацевтически приемлемой соли для применения в качестве лекарственного средства.
Настоящее изобретение дополнительно относится к соединению формулы (I), (II), (III), (IV), (III-A), (IV-A) или (IV-B), в частности формулы (I), или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или их фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для применения в агонизации или антагонизации κ-опиоидного рецептора (KOR-рецептора).
Настоящее изобретение дополнительно относится к соединению формулы (I), (II), (III), (IV), (III-A), (IV-A) или (IV-B), или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или их фармацевтически приемлемой соли для применения в профилактике и/или лечении заболевания, опосредованного агонистом KOR-рецептора и связанного с агонистом KOR-рецептора.
Настоящее изобретение дополнительно относится к соединению формулы (I), (II), (III), (IV), (IIIA), (IV-A) или (IV-B), или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или их фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для применения в профилактике и/или лечении боли и связанных с болью заболеваний у млекопитающих (например, людей). Заболевание, расстройство или состояние опосредованное агонистом κ-опиоидного рецептора (KOR-рецептора) и связанное с агонистом κ-опиоидного рецептора (KOR-рецептора), может представлять собой любое состояние, опосредованное агонистом κ-опиоидного рецептора (KOR-рецептора) и связанное с агонистом κ-опиоидного рецептора (KOR-рецептора), включая, но не ограничиваясь этим, острую или хроническую боль, воспаление, зуд, гипонатриемию, отек, кишечную непроходимость, кашель и глаукому. Например, боль, связанная с κ-опиоидным рецептором (KOR-рецептором), может представлять собой невропатическую боль, соматическую боль, висцеральную боль или кожную боль. Некоторые заболевания, расстройства или состояния связаны с более чем одним типом боли. Например, послеоперационная боль может представлять собой любой тип или все типы нейропатической боли, соматической боли, висцеральной боли или кожной боли, в зависимости от типа и степени используемой хирургии.
Воспаление, связанное с κ-опиоидным рецептором (KOR-рецептором), включенное в настоящее изобретение, может представлять собой любое воспалительное заболевание или состояние, включая, но не ограничиваясь этим, синусит, ревматоидный артрит, теносиновит, бурсит, тендинит, плечевой эпикондилит, адгезивный капсулит, остеомиелит, остеоартрит. воспалительное заболевание кишечника (IBD), синдром раздраженного кишечника (IBS), воспаление глаз, воспаление уха или аутоиммунное воспаление.
Зуд, связанный с κ-опиоидным рецептором (KOR-рецептором), включенный в настоящее изобретение, может представлять собой любое зудящее заболевание и состояние, например, глазной зуд, такой как конъюнктивный глазной зуд, зуд, и зуд, связанный с терминальной стадией почечной недостаточности (при которой многие пациенты подвергаются почечному диализу), и другие формы холестаза, включая первичный билиарный цирроз печени, внутрипеченочный холестаз беременности, хроническое холестериновое заболевание печени, уремию, злокачественный холестаз, желтуху, и кожные заболевания, такие как экзема (дерматит), включая атопический дерматит или контактный дерматит, пятна на коже, полицитемию, красный плоский лишай, хронический простой мох, педикулез, тиреотоксикоз, эпидермофитию стопы, крапивницу, чесотку, вагинит, связанный с акне анальный зуд, зуд укусов насекомых и зуд, вызванный лекарственными средствами, такой как зуд, вызванный μ-опиоидами.
Отек, связанный с κ-опиоидным рецептором (KOR-рецептором), включенный в настоящее изобретение, может представлять собой любое отечное заболевание или состояние, такое как отек, вызванный застойной болезнью сердца, или отек, вызванный синдромом неадекватной секреции антидиуретического гормона (ADH).
Кишечная непроходимость, связанная с κ-опиоидным рецептором (KOR-рецептором), включенная в настоящее изобретение, может представлять собой любое кишечное обструктивное заболевание или состояние, включая, но не ограничиваясь этим, послеоперационную кишечную непроходимость или кишечную дисфункцию, вызванную опиоидом.
Невропатическая боль, связанная с κ-опиоидным рецептором (KOR-рецептором), включенная в настоящее изобретение, может представлять собой любую невропатическую боль, например, невралгию тройничного нерва, диабетическую боль, вызванную вирусом боль, такую как боль, вызванная герпесом зостер, боль, вызванная химиотерапией, боль, вызванная инвазией нерва метастазами рака, невропатическая боль, связанная с травмой и хирургическим вмешательством, а также различные варианты головной боли с невропатологическими факторами, такие как мигрень.
Боли, связанные с κ-опиоидным рецептором (KOR-рецептором), включенные в настоящее изобретение, включают глазную боль, например, боль при рефракционной кератэктомии (PRK), разрыве глаза, переломе дна глазницы, химическом ожоге, абразии эпителия роговицы, или боль в глазах после раздражения, или глазную боль, связанную с конъюнктивитом, язвой роговицы, склеритом, воспалением склеры, склеральным кератитом, глазным герпесом зостер, интерстициальным кератитом, острым притом, сухим кератоконъюнктивитом, флегмоной орбиты, псевдопухолью орбиты, пузырчаткой, трахомой или увеитом.
Боли, связанные с κ-опиоидным рецептором (KOR-рецептором), включенные в настоящее изобретение, также включают боль в горле, особенно боль в горле, связанную с воспалительными состояниями, такими как аллергический ринит, острый бронхит, ОРВИ, контактные язвы, повреждение вирусом простого герпеса, инфекционный мононуклеоз, грипп, рак гортани, острый ларингит, острый язвенно-некротический гингивит, абсцесс миндалин, жжение в глотке, фарингит, рефлюкс-фарингит, острый синусит и тонзиллит.
Боль, связанная с κ-опиоидным рецептором (KOR-рецептором), может быть болью при артрите, камнях в почках, камнях мочевого пузыря и камнях желчных протоков, при гистероспазме, дисменорее, эндометриозе, мастите, расстройстве желудка, послеоперационной болью (например, болью при аппендэктомии, открытой колоректальной хирургии, пластике грыжи, простатэктомии, колонэктомии, гастрэктомии, спленэктомии, колэктомии, колостомии, тазовой лапароскопии, при перевязке маточных труб, гистерэктомии, вазэктомии или послеоперационной болью, вызванной холецистэктомией), болью после медицинского вмешательства (например, болью после колоноскопии, цистоскопии, гистероскопии или биопсии шейки матки или эндометрия), болью при отите, болью при скоротечном раке и болью, связанной с нарушениями желудочно-кишечного тракта, такими как воспалительное заболевание кишечника (IBD) или синдром раздраженного кишечника (IBS), или с другими воспалительными состояниями, особенно болью, связанной с висцеральным воспалением (например, с гастроэзофагеальной рефлюксной болезнью, панкреатитом, острым пиелонефритом, язвенным колитом, холециститом, циррозом печени, кистой печени, гепатитом, язвой двенадцатиперстной кишки или язвой желудка, эзофагитом, гастритом, гастроэнтеритом, колитом, дивертикулитом, кишечной непроходимостью, кистой яичника, воспалением тазовых органов, перфорацией язвы, перитонитом, простатитом, интерстициальным циститом), или болью, вызванной контактом с ядом (например, токсинами насекомых или лекарствами, такими как салицилаты (соли) или НПВЛС).
Связанная с опиоидным рецептором (KOR-рецептором) гипонатриемия может представлять собой любое заболевание или состояние, при котором присутствует гипонатриемия (состояние с низким содержанием натрия), например, у людей, когда концентрация натрия в плазме ниже 135 ммоль/л, могут встречаются отдельные аномалии, либо более часто она наблюдается как осложнение других заболеваний, или в результате использования лекарственного средства, которое вызывает дефицит натрия, где заболевания, связанные с гипонатриемией, включают, но не ограничиваются этим: факторы опухоли, которые вызывают избыточную секрецию АДГ включая рак легких, двенадцатиперстной кишки, поджелудочной железы, яичника, мочевого пузыря и мочеточника, тимому, мезотелиому, бронхиальную аденому, карциноидную опухоль, ганглионеврому и саркому Юинга; инфекцию, например, пневмонию (бактериальную или вирусную), абсцесс (легкое или мозг), вакуолизацию (аспергиллез), туберкулез (легкого или мозга), менингит (бактериальный или вирусный), энцефалит и СПИД; сосудистые факторы, например: цереброваскулярный инфаркт или кровоизлияние и эмболию кавернозного синуса; неврологические факторы, например синдром Гийена-Барре, рассеянный склероз, белую горячку, мышечный коллатеральный склероз, гидроцефалию, психоз, периферическую невропатию, травму головы (закрытую и проникающую), опухоль или инфекцию ЦНС, повреждение ЦНС, влияющее на осморецепторы гипоталамуса; врожденные пороки развития, в том числе: агенез мозолистого тела, расщелины губы и неба и другие срединные дефекты; метаболические факторы, например, острую перемежающуюся порфирию, астму, пневмоторакс и дыхание с положительным давлением; лекарственные средства, например, тиазидные диуретики, парацетамол, барбитураты, холин, эстроген, оральные гипогликемические агенты, вазопрессин или десмопрессин, высокую дозу окситоцина хлорпропамида, винкристины, карбамазепин, никотин, фенотиазин, циклофосфамид, трициклические антидепрессанты, ингибиторы моноаминоксидазы и ингибиторы обратного захвата серотонина; например, введение избытка гипотонической жидкости во время госпитализации, во время операции или во время или после физической активности (т.е. гипонатриемия, связанная с физической нагрузкой), и применение пищевых добавок с низким содержанием натрия у пожилых людей, другие состояния, связанные с гипонатриемией, включая почечную недостаточность, нефротический синдром (модельную нефропатию и минимальное поражение), злокачественную природу, недоедание, рабдомиолиз, хирургическое лечение, селективную катетеризацию сердца, кровопотерю, гиперкальциемию, гипокалиемию и гипергликемию гликозурию, которые могут вызвать осмотический диурез.
Настоящее изобретение также относится к способу профилактики и/или лечения заболевания, расстройства или состояния, опосредованного κ-опиоидным рецептором и связанного с агонистом κ-опиоидного рецептора (KOR-рецептора), включающему стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения любой из формул, в частности соединения формулы (I), или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли. Этот способ демонстрирует заметную эффективность и меньшее количество побочных эффектов. Где заболевание, опосредованное опиоидным рецептором и связанное с агонистом κ-опиоидного рецептора (KOR-рецептора), включает, но не ограничивается этим, острую или хроническую боль, воспаление, зуд, гипонатриемию, отек, непроходимость кишечника, кашель и глаукому.
Настоящее изобретение также относится к способу профилактики и/или лечения боли и связанных с болью заболеваний у млекопитающих, включающему стадию введения млекопитающим, нуждающимся в этом, терапевтически эффективного количества соединения формулы (I), (II), (III)) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли. Этот способ демонстрирует заметную эффективность и меньшее количество побочных эффектов. Где боль может быть послеоперационной болью, болью, вызванной раком, невропатической болью, травматической болью, соматической болью, висцеральной болью, кожной болью и болью, вызванной воспалением, например, послеоперационная боль может быть любым одним или всеми факторами невропатической боли, соматической боли, висцеральной боли или кожной боли, в зависимости от типа и степени используемой операции; рак может быть выбран из группы, состоящей из рака молочной железы, рака эндометрия, рака шейки матки, рака кожи, рака простаты, рака яичника, опухоли маточной трубы, опухоли яичника, гемофилии и лейкемии.
Фармацевтические композиции, содержащие активный ингредиент, могут быть в форме, подходящей для перорального введения, например, в форме таблетки, троше, пастилки, водной или масляной суспензии, диспергируемого порошка или гранулы, эмульсии, твердой или мягкой капсулы, или сиропа, или эликсира. Пероральные композиции могут быть получены в соответствии с любым способом, известным в данной области техники для приготовления фармацевтических композиций. Такие композиции могут содержать один или более агентов, выбранных из группы, состоящей из подсластителей, ароматизаторов, красителей и консервантов, чтобы обеспечить приятную и удобоваримую на вкус фармацевтическую композицию. Таблетка содержит активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, подходящими для изготовления таблетки. Этими эксципиентами могут быть инертные эксципиенты, гранулирующие агенты, разрыхлители и лубриканты. Таблетка может быть непокрытой или покрытой оболочкой с помощью известного метода для маскировки вкуса лекарственного средства или замедления распада и абсорбции активного ингредиента в желудочно-кишечном тракте, тем самым обеспечивая замедленное высвобождение в течение длительного периода.
Пероральные композиции могут быть представлены в виде мягких желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, или активный ингредиент смешан с водорастворимым носителем, или масляной средой, или оливковым маслом.
Водная суспензия содержит активный ингредиент в смеси с эксципиентами, пригодными для изготовления водной суспензии. Такими эксципиентами являются суспендирующие агенты, диспергаторы или увлажнители. Водная суспензия также может содержать один или более консервантов, таких как этилпарабен или н-пропилпарабен, один или более красителей, один или более ароматизаторов и один или более подсластителей.
Масляная суспензия может быть приготовлена путем суспендирования активного ингредиента в растительном масле. Масляная суспензия может содержать загуститель. Вышеупомянутые подсластители и ароматизаторы могут быть добавлены, чтобы обеспечить приятный на вкус препарат. Эти композиции могут быть сохранены путем добавления антиоксиданта.
Активный ингредиент в смеси с диспергирующими или смачивающими агентами, суспендирующим агентом или одним или несколькими консервантами может быть приготовлен в виде диспергируемого порошка или гранул, подходящих для приготовления водной суспензии путем добавления воды. Подходящие диспергаторы или смачивающие агенты и суспендирующие агенты иллюстрируются теми, которые уже упоминались выше. Также могут быть добавлены дополнительные эксципиенты, такие как подсластители, ароматизаторы и красители.
Фармацевтическая композиция согласно настоящему изобретению также может быть в форме эмульсии масло-в-воде. Масляная фаза может быть растительным маслом или минеральным маслом, таким как жидкий парафин, или их смесью. Подходящими эмульгирующими агентами могут быть встречающиеся в природе фосфолипиды или неполные эфиры. Эмульсии также могут содержать подсластители, ароматизаторы, консерванты и антиоксиданты.
Фармацевтическая композиция согласно настоящему изобретению может быть в форме стерильного водного раствора. Приемлемыми носителями или растворителями, которые можно использовать, являются вода, раствор Рингера и изотонический раствор хлорида натрия. Стерильный инъецируемый препарат также может быть стерильной инъецируемой микроэмульсией типа «масло в воде», в которой активный ингредиент растворен в масляной фазе. Раствор для инъекций или микроэмульсия могут быть введены в кровоток человека путем местной болюсной инъекции.
Фармацевтическая композиция согласно настоящему изобретению может быть в форме стерильной инъецируемой водной или масляной суспензии для внутримышечного и подкожного введения. Такая суспензия может быть приготовлена с подходящими диспергаторами или смачивающими агентами и суспендирующими агентами, как описано выше, в соответствии с известными методиками. Стерильный инъекционный препарат также может представлять собой стерильный инъекционный раствор или суспензию, приготовленную в нетоксичном парентерально приемлемом разбавителе или растворителе. Кроме того, стерильные жирные масла могут легко использоваться в качестве растворителя или суспендирующей среды.
Настоящее соединение можно вводить в форме суппозитория для ректального введения. Эти фармацевтические композиции могут быть получены путем смешивания лекарственного средства с подходящим нераздражающим наполнителем, который является твердым при обычных температурах, но жидким в прямой кишке, тем самым плавясь в прямой кишке для высвобождения лекарственного средства.
Специалистам в данной области хорошо известно, что дозировка лекарственного средства зависит от множества факторов, включая, но не ограничиваясь этим, следующие факторы: активность конкретного соединения, возраст пациента, вес пациента, общее состояние здоровья пациента, поведение пациента, диета пациента, время введения, путь введения, скорость выведения, комбинацию лекарств и тому подобное. Кроме того, наилучшее лечение, такое как режим лечения, суточная доза соединения формулы (I) или тип его фармацевтически приемлемой соли, может быть проверено традиционными терапевтическими схемами лечения.
Подробное описание изобретения
Если не указано иное, термины, используемые в описании и формуле изобретения, имеют значения, описанные ниже.
«Алкил» относится к насыщенной алифатической углеводородной группе, включающей группы с прямой и разветвленной цепью C1-С20, предпочтительно - алкил с 1-12 атомами углерода и более предпочтительно - алкил с 1-6 атомами углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил, и их разветвленные изомеры. Более предпочтительно, алкильная группа представляет собой низший алкил, имеющий от 1 до 6 атомов углерода, и неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.п. Алкильная группа может быть замещенной или незамещенной. Будучи замещенной, заместительная группа может быть замещена в любой доступной точке соединения. Заместительная группа (группы) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилкилтио, оксо, карбокси и алкоксикарбонила.
«Циклоалкил» относится к насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 6 атомов углерода и наиболее предпочтительно от 5 до 6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.п. Полициклический циклоалкил включает циклоалкил, имеющий спирокольцо, конденсированное кольцо или кольцо с внутренним мостиком.
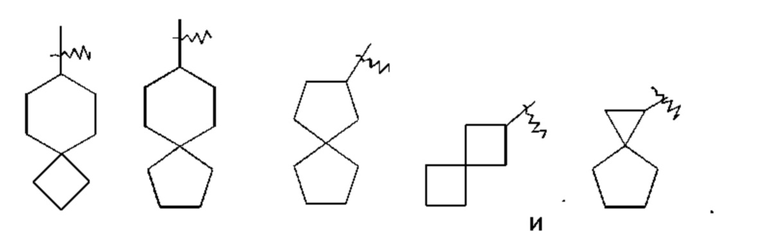
«Спироциклоалкил» относится к 5-20-членной полициклической группе с кольцами, связанными через общий атом углерода (называемый спироатомом), где одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, предпочтительно - к 6-14-членному спироциклоалкилу и более предпочтительно - к 7-10-членному спироциклоалкилу. В соответствии с количеством спироатомов, поделенных между кольцами, спироциклоалкилы можно подразделить на моноспироциклоалкил, диспироциклоалкил или полиспироциклоалкил, и предпочтительно - моноспироциклоалкил или диспироциклоалкил, и более предпочтительно - 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноспироалкил. Неограничивающие примеры спироциклоалкилов включают:
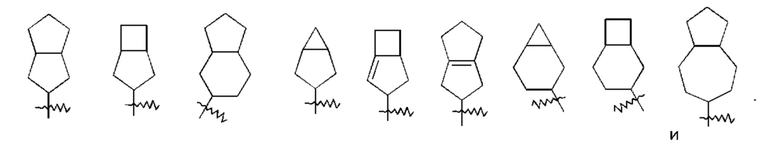
«Конденсированный циклоалкил» относится к 5-20-членной полностью углеродной полициклической группе, где каждое кольцо в системе имеет общую соседнюю пару атомов углерода с другим кольцом, где одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, предпочтительно - к 6-14-членному конденсированному циклоалкилу, и еще более предпочтительно - к 7-10-членному конденсированному циклоалкилу. По количеству членных колец конденсированные циклоалкилы можно подразделить на бициклические, трициклические, тетрациклические или полициклические конденсированные циклоалкилы, предпочтительно - бициклические или трициклические конденсированные циклоалкилы и более предпочтительно - 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный циклоалкил. Неограничивающие примеры конденсированных циклоалкилов включают:
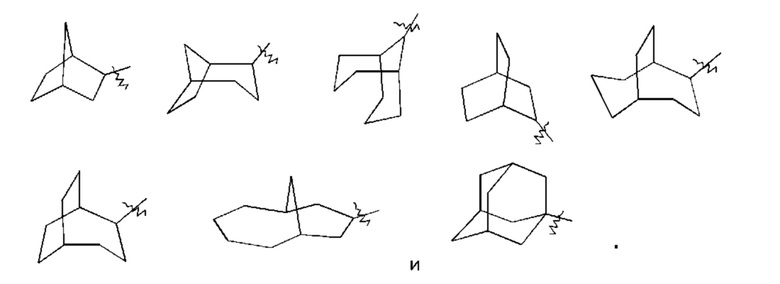
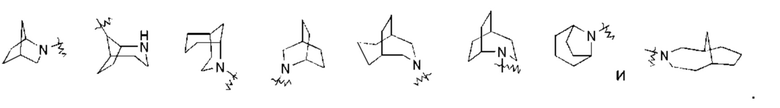
«Мостиковый циклоалкил» относится к 5-20-членной полностью углеродной полициклической группе, где каждые два кольца в системе имеют два общих несвязанных друг с другом атома углерода, где кольца могут иметь одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, предпочтительно - к 6-14-членному мостиковому циклоалкилу и более предпочтительно - к 7-10-членному мостиковому циклоалкилу. Мостиковые циклоалкилы можно подразделить на бициклический, трициклический, тетрациклический или полициклический мостиковый циклоалкил, и предпочтительно - бициклический, трициклический или тетрациклический мостиковый циклоалкил, и более предпочтительно - бициклический или трициклический мостиковый циклоалкил. Неограничивающие примеры мостиковых циклоалкилов включают:
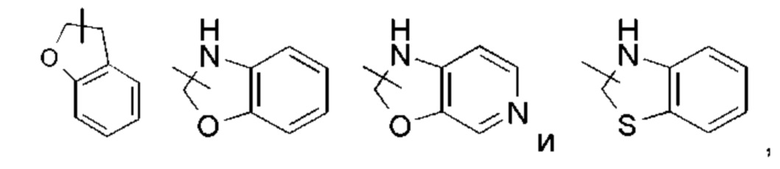
Кольцо циклоалкила может быть сконденсировано с кольцом арила, гетероарила или гетероциклила, где кольцо, присоединенное к исходной структуре, представляет собой циклоалкил. Неограничивающие примеры включают инданил, тетрагидронафтил, бензоциклогептил и тому подобное, предпочтительно - бензоциклопентил, тетрагидронафтил. Циклоалкил может быть необязательно замещенным или незамещенным. При замещении группа (группы) заместителя предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, оксо, карбокси и алкоксикарбонила.
«Гетероциклил» относится к 3-20-членной насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2), в качестве атомов кольца, но исключая -OO-, -OS- или -SS- в кольце, причем остальные атомы кольца являются атомами углерода. Предпочтительно гетероциклил имеет от 3 до 12 атомов, где от 1 до 4 атомов представляют собой гетероатомы, более предпочтительно - от 3 до 8 атомов, где от 1 до 3 атомов представляют собой гетероатомы, и наиболее предпочтительно - от 5 до 6 атомов, где от 1 до 2 или от 1 до 3 атомов представляют собой гетероатомы. Неограничивающие примеры моноциклических гетероциклилов включают пирролидинил, имидазолидинил, тетрагидрофуранил, тетрагидропиранил, тетрагидротиенил, дигидроимидазолил, дигидрофуранил, дигидропиразолил, дигидропирролил, пиперидил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и тому подобное, предпочтительно - тетрагидропиранил, пиперидил или пирролидинил. Полициклический гетероциклил включает гетероциклил, имеющий спиро-кольцо, конденсированное кольцо или кольцо с внутренним мостиком.
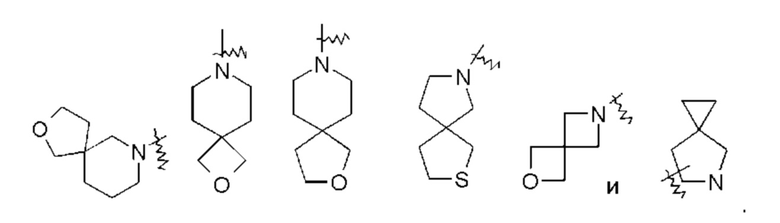
«Спирогетероциклил» относится к 5-20-членному полициклическому гетероциклилу с кольцами, связанными через общий атом углерода (называемый спироатомом), где кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2), в качестве атомов кольца, причем остальные атомы кольца являются атомами углерода, где одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, предпочтительно - к 6-14-членному спирогетероциклилу и более предпочтительно - к 7-10-членному спирогетероциклилу. В соответствии с количеством спироатомов, поделенных между кольцами, спирогетероциклилы можно подразделить на моноспирогетероциклил, диспироспирогетероциклил или полиспирогетероциклил, предпочтительно - моноспирогетероциклил или диспирогетероциклил, и более предпочтительно - 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноспирогетероциклил. Неограничивающие примеры спирогетероциклилов включают:
«Конденсированный гетероциклил» относится к 5-20-членной полициклической гетероциклильной группе, где каждое кольцо в системе имеет общую пару соседних атомов с другим кольцом, где одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, и где кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2), в качестве атомов кольца, причем оставшиеся атомы кольца являются атомами углерода; предпочтительно - к 6-14-членному конденсированному гетероциклилу, и более предпочтительно - к 7-10-членному конденсированному гетероциклилу. По количеству членных колец конденсированные гетероциклилы можно подразделить на бициклический, трициклический, тетрациклический или полициклический конденсированный гетероциклил, предпочтительно - бициклический или трициклический конденсированный гетероциклил и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный гетероциклил. Неограничивающие примеры конденсированных гетероциклилов включают:
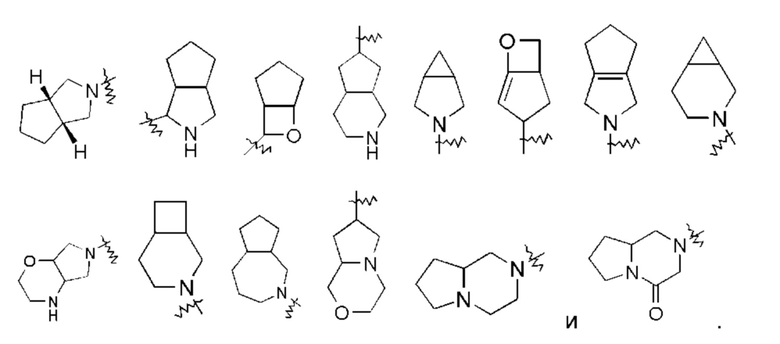
«Мостиковый гетероциклил» относится к 5-14-членной полициклической гетероциклильной группе, где каждые два кольца в системе имеют два общих несвязанных друг с другом атома углерода, где кольца могут иметь одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, и кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2), в качестве атомов кольца, а остальные атомы кольца представляют собой атомы углерода, предпочтительно - к 6-14-членному мостиковому гетероциклилу, более предпочтительно - к 7-10-членному мостиковому гетероциклилу. В зависимости от количества членных колец, мостиковые гетероциклилы можно подразделить на бициклический, трициклический, тетрациклический или полициклический мостиковый гетероциклил, и предпочтительно - бициклический, трициклический или тетрациклический мостиковый гетероциклил и более предпочтительно бициклический или трициклический мостиковый гетероциклил. Неограничивающие примеры мостиковых гетероциклилов включают:
Гетероциклильное кольцо может быть сконденсировано с кольцом арила, гетероарила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой гетероциклил. Неограничивающие примеры включают:
 и т.п.
и т.п.
Гетероциклил может быть необязательно замещенным или незамещенным. При замещении группа заместителя (заместители) представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, оксо, карбокси и алкоксикарбонила.
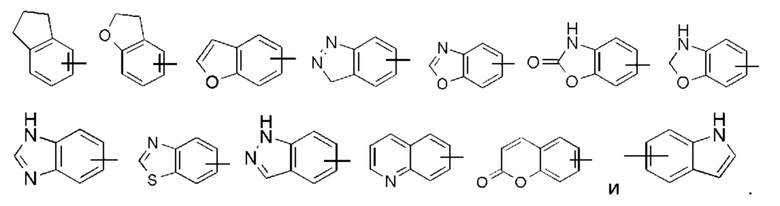
«Арил» относится к 6-14-членному полностью углеродному моноциклическому кольцу или полициклическому конденсированному кольцу (т.е. каждое кольцо в системе имеет общую пару соседних атомов углерода с другим кольцом в системе), имеющему полностью сопряженную пи-электронную систему, предпочтительно - к 6-10-членному арилу, более предпочтительно - к 5-6-членному арилу, например, к фенилу и нафтилу. Арил может быть сконденсирован с кольцом гетероарила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, является арилом. Неограничивающие примеры включают:
Арил может быть необязательно замещенным или незамещенным. При замещении группа заместителя (заместители) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, карбокси и алкоксикарбонила.
«Гетероарил» относится к 5-14-членной гетероароматической системе, имеющей от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, S и N, в качестве кольцевых атомов, предпочтительно - к 5-10-членному гетероарилу, имеющему от 1 до 3 гетероатомов, и более предпочтительно - к 5-ти или 6-членному гетероарилу, имеющему от 1 до 2 гетероатомов, например, к имидазолилу, фурилу, тиенилу, тиазолилу, пиразолилу, оксазолилу, пирролилу, тетразолилу, пиридилу, пиримидинилу, тиадиазолилу, пиразинилу и тому подобному, предпочтительно - к имидазолилу, пиразолилу, пиримидинилу или тиазолилу, более предпочтительно - к пиразолилу. Гетероарильное кольцо может быть сконденсировано с кольцом арила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой гетероарил. Неограничивающие примеры включают в себя:
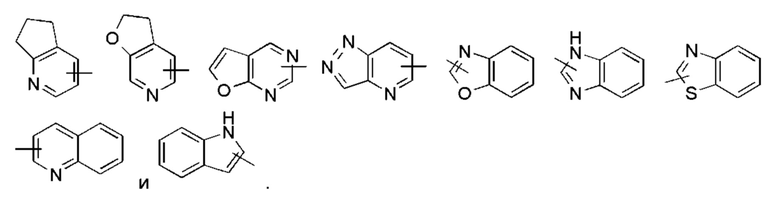
Гетероарил может быть необязательно замещенным или незамещенным. При замещении группа заместителя (заместители) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, карбокси и алкоксикарбонила.
«Алкокси» относится к группе -О-(алкил) или -O-(незамещенный циклоалкил), где алкил такой, как определено выше. Неограничивающие примеры включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси и т.п. Алкокси может быть необязательно замещенным или незамещенным. При замещении заместитель предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, карбокси и алкоксикарбонила.
«Гидроксиалкил» относится к алкилу, замещенному гидрокси (гидроксилами), где алкил такой, как определено выше.
«Галоалкил» относится к алкилу, замещенному одним или более галогенами, где алкил такой, как определено выше.
«Циклоалкилалкил» относится к алкилу, замещенному одним или более циклоалкилами, где циклоалкил и алкил имеют значения, определенные выше.
«Гетероциклилалкил» относится к алкилу, замещенному одним или более гетероциклилами, где гетероциклил и алкил имеют значения, определенные выше.
«Арилалкил» относится к алкилу, замещенному одним или более арилами, где арил и алкил имеют значения, определенные выше.
"Гидрокси" относится к -ОН группе.
"Галоген" относится к фтору, хлору, брому и иоду.
"Амино" относится к группе -NH2.
"Циано" относится к группе -CN.
"Нитро" относится к группе -NO2.
"Карбокси" относится к группе -С(O)ОН.
"Алкоксикарбонил" относится к группе -С(O)O(алкил) или -С(O)O(циклоалкил), где алкил и циклоалкил имеют значения, определенные выше.
«Ацилгалид» относится к соединению, содержащему группу -С(O)-галоген.
Все выражения «X выбран из группы, состоящей из А, В, или С», «X выбран из группы, состоящей из А, В и С», «X представляет собой А, В или С», «X представляет собой А, В и С "и тому подобное, имеют одинаковое значение. Это означает, что X может быть любым или более из А, В и С.
«Необязательный» или «необязательно» означает, что описанное впоследствии событие или обстоятельство может происходить, но не обязательно, и это описание включает ситуацию, в которой событие или обстоятельство возникает или не возникает. Например, «гетероциклическая группа, необязательно замещенная алкилом» означает, что алкильная группа может присутствовать, но не обязательно, и это описание включает ситуацию, когда гетероциклическая группа замещена алкилом и гетероциклическая группа не замещена алкилом.
«Замещенный» относится к одному или более атомам водорода в группе, предпочтительно до 5, более предпочтительно - к 1-3 атомам водорода, независимо замещенным соответствующим числом заместителей. Само собой разумеется, что заместители существуют только в их возможных химических положениях. Специалист в данной области способен определить, необязательно или не необязательно замещение, с помощью экспериментов или теории, не прилагая чрезмерных усилий. Например, комбинация амино или гидрокси, имеющих свободные атомы водорода и атомы углерода, имеющие ненасыщенные связи (такие как олефиновая), может быть нестабильной.
«Фармацевтическая композиция» относится к смеси одного или более соединений согласно настоящему изобретению или их физиологически/фармацевтически приемлемых солей или пролекарств с другими химическими ингредиентами и другими компонентами, такими как физиологически/фармацевтически приемлемые носители и эксципиенты. Назначение фармацевтической композиции состоит в том, чтобы облегчить введение соединения в организм, что способствует абсорбции активного ингредиента и, таким образом, проявлению биологической активности.
«Фармацевтически приемлемая соль» относится к соли соединения согласно настоящему изобретению, которая безопасна и эффективна для млекопитающих и обладает желаемой биологической активностью.
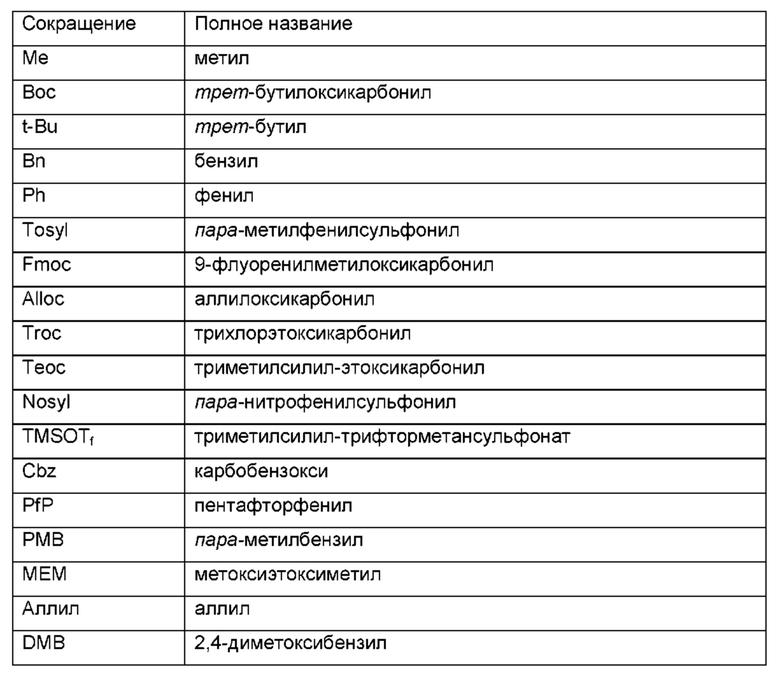
Таблица сокращений:
Способ синтеза соединения согласно настоящему изобретению
Для решения задачи настоящего изобретения в настоящем изобретении применяются следующие технические решения.
Способ получения соединения формулы (II) согласно настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси или его фармацевтически приемлемой соли включает следующие стадии:
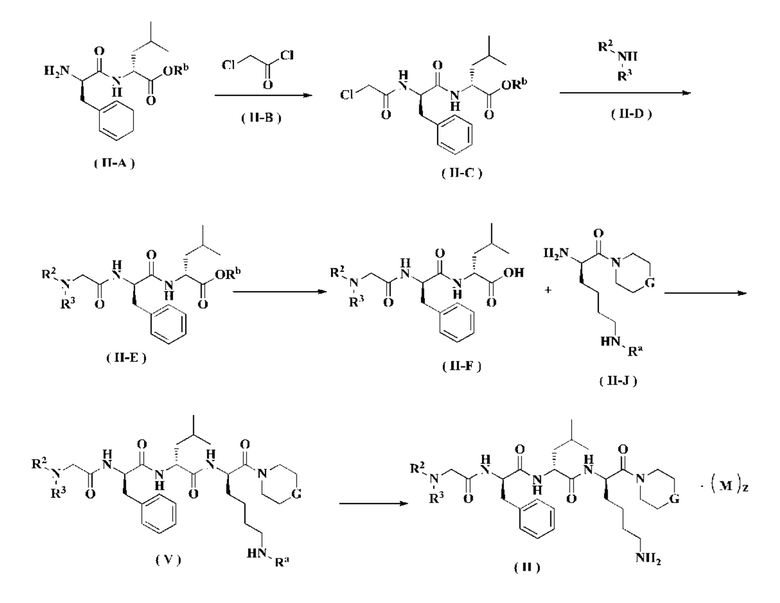
Соединение формулы (II-A) реагирует с соединением формулы (II-В) в щелочных условиях с получением соединения формулы (II-С), где щелочным реагентом в этих условиях является предпочтительно триэтиламин. Полученное соединение формулы (II-С) реагирует с соединением формулы (II-D) в присутствии йодида калия в щелочных условиях и при нагревании с получением соединения формулы (II-Е), где щелочной реагент в этих условиях представляет собой предпочтительно карбонат калия. Полученное соединение формулы (II-Е) подвергают снятию защиты с получением соединения формулы (II-F). Полученное соединение формулы (II-F) реагирует с соединением формулы (II-J) в присутствии конденсирующего агента с получением соединения формулы (V), где конденсирующим реагентом в этих условиях является предпочтительно 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат. Полученное соединение формулы (V) подвергают удалению защитной группы на аминогруппе в кислых условиях с получением соединения формулы (II), где кислотным реагентом в этих условиях является предпочтительно трифторуксусная кислота или раствор соляной кислоты в 1,4-диоксане.
Кроме того, когда z не равен нулю в соединении формулы (II), необязательно добавляют слабое основание для проведения свободной реакции с получением продукта в свободном состоянии, то есть соединения формулы (II).
Реагент, который обеспечивает щелочные условия, включает органическое основание и неорганическое основание, где указанное органическое основание включает, но не ограничивается этим, пиридин, пиперидин, триэтиламин, N,N-дизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия, где неорганическое основание включает, но не ограничивается этим, гидрид натрия, фосфат калия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия и гидроксид лития.
Реагент, который обеспечивает кислые условия, включает, но не ограничивается этим, хлористый водород, трифторуксусную кислоту, муравьиную кислоту, уксусную кислоту, соляную кислоту, серную кислоту и метансульфоновую кислоту.
Конденсирующий агент выбирают из группы, состоящей из 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида, N,N'-дициклогексилкарбодиимида, N,N'-диизопропилкарбодиимида, О-бензотриазол-N,N,N',N'-тетраметилурония тетрафторбората, 1-гидроксибензотриазола, 1-гидрокси-7-азабензотриазола, O-бензотриазол-N,N,N',N'-тетраметилурония гексафторфосфата, 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилмочевины гексафторфосфата, бензотриазол-1-илокситрис(диметиламино)фосфония гексафторфосфата и бензотриазол-1-ил-окситрипирролидино-фосфония гексафторфосфата, предпочтительно - 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата.
Используемый растворитель включает, но не ограничивается этим, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, диметилсульфоксид, 1,4-диоксан, воду и N,N-диметилформамид.
Где:
Ra представляет собой амино-защитную группу, предпочтительно трет-бутоксикарбонил, 9-флуоренилметоксикарбонил, аллилоксикарбонил, трихлорэтоксикарбонил, триметилсилилоксикарбонил, бензилоксикарбонил, пара-метилбензолсульфонил, пара-нитробензолсульфонил и трет-бутил;
Rb представляет собой карбокси-защитную группу, предпочтительно DM В, Bn, аллил, Pfp, Me, РМВ, MEM и t-Bu; и
М, G, z, R2 и R3 являются такими, как определено в формуле (II).
Способ получения соединения формулы (III) согласно настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси или его фармацевтически приемлемой соли включает следующие стадии:
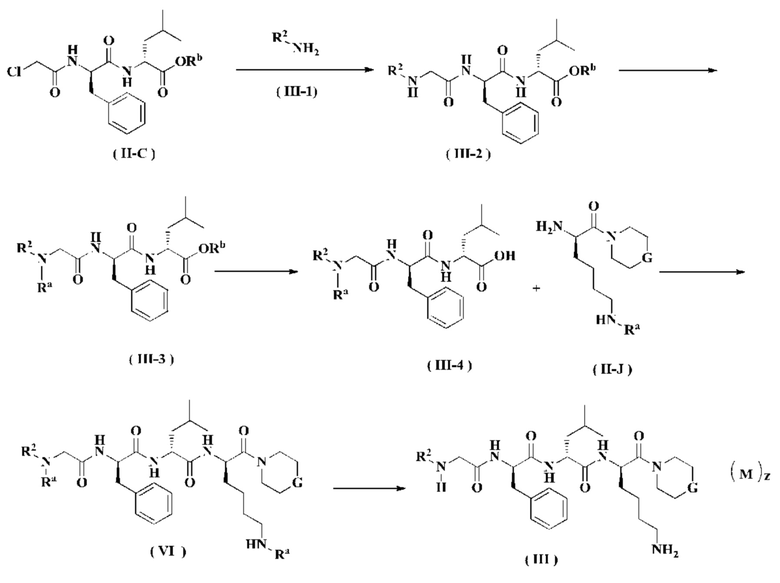
Соединение формулы (II-С) реагирует с соединением формулы (III-1) в присутствии йодида калия в щелочных условиях и при нагревании с получением соединения формулы (III-2), где щелочным реагентом в этих условиях является предпочтительно карбонат калия. В полученное соединение формулы (III-2) добавляют амино-защитную группу с получением соединения формулы (III-3). Полученное соединение формулы (III-3) реагирует с соединением формулы (II-J) в присутствии конденсирующего агента с получением соединения формулы (VI), где конденсирующим реагентом в этих условиях является предпочтительно 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат. Полученное соединение формулы (VI) подвергают удалению защитной группы на аминогруппе в кислых условиях с получением соединения формулы (III), где кислотным реагентом в этих условиях является предпочтительно трифторуксусная кислота или раствор соляной кислоты в 1,4-диоксане.
Кроме того, когда z не равен нулю в соединении формулы (III), необязательно добавляют слабое основание для проведения свободной реакции с получением продукта в свободном состоянии, то есть соединения формулы (III).
Реагент, который обеспечивает щелочные условия, включает органическое основание и неорганическое основание, где указанное органическое основание включает, но не ограничивается этим, пиридин, пиперидин, триэтиламин, N,N-дизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия, где неорганическое основание включает, но не ограничивается этим, гидрид натрия, фосфат калия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия и гидроксид лития.
Реагент, который обеспечивает кислые условия, включает, но не ограничивается этим, хлористый водород, трифторуксусную кислоту, муравьиную кислоту, уксусную кислоту, соляную кислоту, серную кислоту и метансульфоновую кислоту.
Конденсирующий агент выбирают из группы, состоящей из 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида, N,N'-дициклогексилкарбодиимида, N,N'-диизопропилкарбодиимида, О-бензотриазол-N,N,N',N'-тетраметилурония тетрафторбората, 1-гидроксибензотриазола, 1-гидрокси-7-азабензотриазола, O-бензотриазол-N,N,N',N'-тетраметилурония гексафторфосфата, 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилмочевины гексафторфосфата, бензотриазол-1-илокситрис(диметиламино)фосфония гексафторфосфата и бензотриазол-1-ил-окситрипирролидино-фосфония гексафторфосфата, предпочтительно - 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата.
Используемый растворитель включает, но не ограничивается этим, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, диметилсульфоксид, 1,4-диоксан, воду и N,N-диметилформамид.
Где:
Ra представляет собой амино-защитную группу, предпочтительно трет-бутоксикарбонил, 9-флуоренилметоксикарбонил, аллилоксикарбонил, трихлорэтоксикарбонил, триметилсилилоксикарбонил, бензилоксикарбонил, пара-метилбензолсульфонил, пара-нитробензолсульфонил и трет-бутил;
Rb представляет собой карбокси-защитную группу, предпочтительно DM В, Bn, аллил, Pfp, Me, РМВ, MEM и t-Bu; и
М, G, z и R2 являются такими, как определено в формуле (III).
Способ получения соединения формулы (III-А) согласно настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси или его фармацевтически приемлемой соли включает следующие стадии:
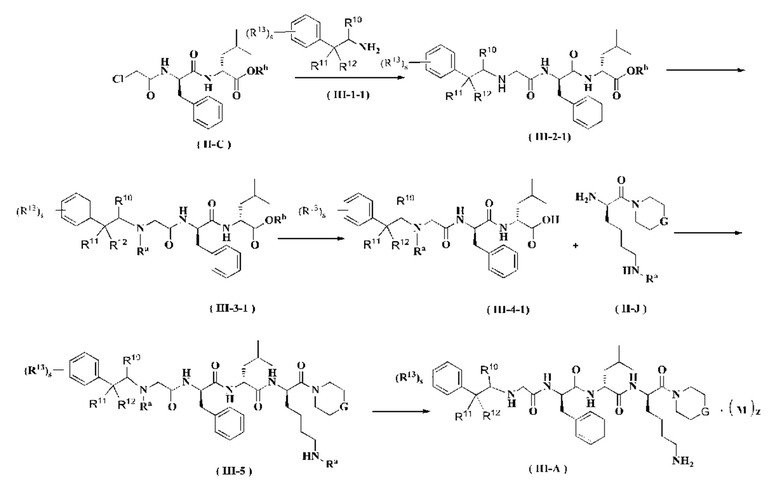
Соединение формулы (II-C) реагирует с соединением формулы (III-1-1) в присутствии йодида калия в щелочных условиях и при нагревании с получением соединения формулы (III-2-1), где щелочным реагентом в этих условиях является предпочтительно карбонат калия. В полученное соединение формулы (III-2-1) добавляют амино-защитную группу с получением соединения формулы (III-3-1). Полученное соединение формулы (III-3-1) реагирует с соединением формулы (II-J) в присутствии конденсирующего агента с получением соединения формулы (III-5), где конденсирующим реагентом в этих условиях является предпочтительно 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат. Полученное соединение формулы (III-5) подвергают удалению защитной группы на аминогруппе в кислых условиях с получением соединения формулы (III-А), где кислотным реагентом в этих условиях является предпочтительно трифторуксусная кислота или раствор соляной кислоты в 1,4-диоксане.
Кроме того, когда z не равен нулю в соединении формулы (III-А), необязательно добавляют слабое основание для проведения свободной реакции с получением продукта в свободном состоянии, то есть соединения формулы (III-А).
Реагент, который обеспечивает щелочные условия, включает органическое основание и неорганическое основание, где указанное органическое основание включает, но не ограничивается этим, пиридин, пиперидин, триэтиламин, N,N-дизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия, где неорганическое основание включает, но не ограничивается этим, гидрид натрия, фосфат калия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия и гидроксид лития.
Реагент, который обеспечивает кислые условия, включает, но не ограничивается этим, хлористый водород, трифторуксусную кислоту, муравьиную кислоту, уксусную кислоту, соляную кислоту, серную кислоту и метансульфоновую кислоту.
Конденсирующий агент выбирают из группы, состоящей из 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида, N,N'-дициклогексилкарбодиимида, N,N'-диизопропилкарбодиимида, О-бензотриазол-N,N,N',N'-тетраметилурония тетрафторбората, 1-гидроксибензотриазола, 1-гидрокси-7-азабензотриазола, O-бензотриазол-N,N,N',N'-тетраметилурония гексафторфосфата, 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилмочевины гексафторфосфата, бензотриазол-1-илокситрис(диметиламино)фосфония гексафторфосфата и бензотриазол-1-ил-окситрипирролидино-фосфония гексафторфосфата, предпочтительно - 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата.
Используемый растворитель включает, но не ограничивается этим, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, диметилсульфоксид, 1,4-диоксан, воду и N,N-диметилформамид.
Где:
Ra представляет собой амино-защитную группу, предпочтительно трет-бутоксикарбонил, 9-флуоренилметоксикарбонил, аллилоксикарбонил, трихлорэтоксикарбонил, триметилсилилоксикарбонил, бензилоксикарбонил, пара-метилбензолсульфонил, пара-нитробензолсульфонил и трет-бутил;
Rb представляет собой карбокси-защитную группу, предпочтительно DM В, Bn, аллил, Pfp, Me, РМВ, MEM и t-Bu; и
R10 - R13, М, G и z являются такими, как определено в формуле (III-A).
Способ получения соединения формулы (IV-A) согласно настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси или его фармацевтически приемлемой соли включает следующие стадии:
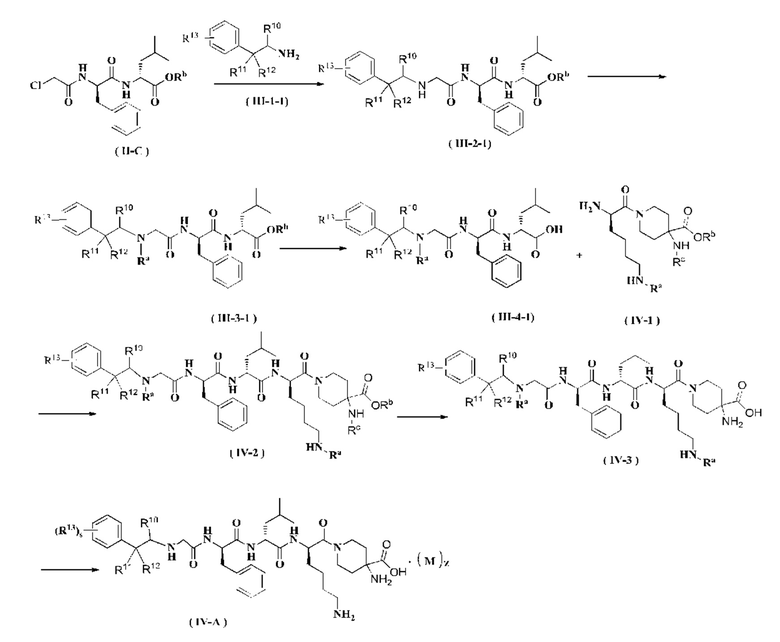
Соединение формулы (II-С) реагирует с соединением формулы (III-1-1) в присутствии йодида калия в щелочных условиях и при нагревании с получением соединения формулы (III-2-1), где щелочным реагентом в этих условиях является предпочтительно карбонат калия. В полученное соединение формулы (III-2-1) добавляют амино-защитную группу с получением соединения формулы (III-3-1). Полученное соединение формулы (III-3-1) реагирует с соединением формулы (IV-1) в присутствии конденсирующего агента с получением соединения формулы (IV-2), где конденсирующим реагентом в этих условиях является предпочтительно 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат. Полученное соединение формулы (IV-2) подвергают удалению защитных групп Rc и Rb в кислых условиях с получением соединения формулы (IV-3). Полученное соединение формулы (IV-3) дополнительно подвергают удалению защитной группы Ra с получением соединения формулы (IV-A), где кислотным реагентом в этих условиях является предпочтительно трифторуксусная кислота или раствор соляной кислоты в 1,4-диоксане.
Кроме того, когда z не равен нулю в соединении формулы (IV-A), необязательно добавляют слабое основание для проведения свободной реакции с получением продукта в свободном состоянии, то есть соединения формулы (IV-A).
Реагент, который обеспечивает щелочные условия, включает органическое основание и неорганическое основание, где указанное органическое основание включает, но не ограничивается этим, пиридин, пиперидин, триэтиламин, N,N-дизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия, где неорганическое основание включает, но не ограничивается этим, гидрид натрия, фосфат калия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия и гидроксид лития.
Реагент, который обеспечивает кислые условия, включает, но не ограничивается этим, хлористый водород, трифторуксусную кислоту, муравьиную кислоту, уксусную кислоту, соляную кислоту, серную кислоту и метансульфоновую кислоту.
Конденсирующий агент выбирают из группы, состоящей из 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида, N,N'-дициклогексилкарбодиимида, N,N'-диизопропилкарбодиимида, О-бензотриазол-N,N,N',N'-тетраметилурония тетрафторбората, 1-гидроксибензотриазола, 1-гидрокси-7-азабензотриазола, O-бензотриазол-N,N,N',N'-тетраметилурония гексафторфосфата, 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилмочевины гексафторфосфата, бензотриазол-1-илокситрис(диметиламино)фосфония гексафторфосфата и бензотриазол-1-ил-окситрипирролидино-фосфония гексафторфосфата, предпочтительно - 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата.
Используемый растворитель включает, но не ограничивается этим, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, диметилсульфоксид, 1,4-диоксан, воду и N,N-диметилформамид.
Где:
Ra представляет собой амино-защитную группу, предпочтительно трет-бутоксикарбонил, 9-флуоренилметоксикарбонил, аллилоксикарбонил, трихлорэтоксикарбонил, триметилсилилоксикарбонил, бензилоксикарбонил, пара-метилбензолсульфонил, пара-нитробензолсульфонил и трет-бутил;
Rb представляет собой карбокси-защитную группу, предпочтительно DM В, Bn, аллил, Pfp, Me, РМВ, MEM и t-Bu; и
Rc представляет собой амино-защитную группу, предпочтительно бензилоксикарбонил, трет-бутоксикарбонил, 9-флуоренилметоксикарбонил, аллилоксикарбонил, трихлорэтоксикарбонил, триметилсилилоксикарбонил, пара-метилбензолсульфонил, пара-нитробензолсульфонил или трет-бутил; и
R10 - R13, М и z являются такими, как определено в формуле (IV-A).
Краткое описание графических материалов
На фигуре 1 показано влияние соединений согласно настоящей заявке на воспалительную боль, вызванную каррагинаном у крыс.
Предпочтительные варианты осуществления изобретения
Настоящее изобретение будет дополнительно описано со ссылкой на следующие примеры, но примеры не следует рассматривать как ограничивающие объем изобретения.
Примеры
Структуры соединений определяют с помощью ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). Химические сдвиги ЯМР (δ) приведены в 10-6 м.д. (ppm). ЯМР определяют на приборе Bruker AVANCE-400. Растворителями для определения являются дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD), а внутренним стандартом является тетраметилсилан (TMS).
МС определяют масс-спектрометром FINNIGAN LCQAd (ESI) (производитель: Thermo, тип: Finnigan LCQ advantage MAX).
Анализ хиральной высокоэффективной жидкостной хроматографии (ВЭЖХ) определяют с помощью LC-10A vp (Shimadzu) или SFC-analytical (Berger Instruments Inc.).
Пластина силикагеля Yantai Huanghai HSGF254 или Qingdao GF254 используется для тонкослойной хроматографии на силикагеле (ТСХ). Размер пластины силикагеля, используемой в ТСХ, составляет от 0,15 мм до 0,2 мм, а размер пластины силикагеля, используемой при очистке продукта, составляет от 0,4 мм до 0,5 мм.
В качестве носителя для колоночной хроматографии используют силикагель Yantai Huanghai размером от 200 до 300 меш.
Prep Star SD-1 (Varian Instruments Inc.) или SFC-multigram (Berger Instruments Inc.) используют для хиральной препаративной колоночной хроматографии.
Средние уровни ингибирования киназы и значения IC50 определяют с помощью NovoStar ELISA (BMG Co., Германия).
Известные исходные материалы согласно настоящему изобретению могут быть получены обычными способами синтеза, известными в данной области техники, или могут быть приобретены у ABCR GmbH & Co. KG, Acros Organnics, Aldrich Chemical Company, Accela ChemBio Inc. или Dari chemical Company и т.д.
Если не указано иное, реакции проводят в атмосфере азота или атмосфере аргона.
Термин «атмосфера азота» или «атмосфера аргона» означает, что реакционная колба оснащена баллоном азота или аргона объемом 1 л.
Термин «атмосфера водорода» означает, что реакционная колба оснащена баллоном водорода объемом 1 л.
Реакции гидрирования под давлением проводят с помощью устройства для гидрирования Parr 3916EKX и генератора водорода QL-500 или устройства для гидрирования HC2-SS.
В реакциях гидрирования реакционную систему обычно вакуумируют и заполняют водородом, и вышеуказанную операцию повторяют три раза.
Микроволновый реактор типа СЕМ Discover-S 908860 используют в микроволновых реакциях.
Если не указано иное, понятие «раствор» относится к водному раствору.
Если не указано иное, температура реакции в реакциях относится к комнатной температуре в диапазоне от 20 до 30°С.
Процесс реакции контролируют с помощью тонкослойной хроматографии (ТСХ), и система проявляющего растворителя включает в себя: А: систему дихлорметана и метанола, В: систему н-гексана и этилацетата, С: систему петролейного эфира и этилацетата, D: ацетон. Соотношение объема растворителя можно регулировать в соответствии с полярностью соединений.
Система элюирования для очистки соединений с помощью колоночной хроматографии и тонкослойной хроматографии включает: А: систему дихлорметан и метанол, В: систему н-гексан и этилацетат, С: систему дихлорметан и ацетон. Соотношение объема растворителя можно регулировать в соответствии с полярностью соединений, и иногда можно добавлять немного щелочного реагента, такого как триэтиламин, или кислотного реагента, такого как уксусная кислота.
Жидкостный хроматограф высокого давления, используемый в высокоэффективной жидкостной хроматографии в примерах, представляет собой Gilson-281, хроматографическая колонка - Shim-pack PREПAPA-ODS от Shimadzu, в качестве подвижной фазы используется буферная система с трифторуксусной кислотой, т.е. водой, содержащей 0,05% трифторацетат, и ацетонитрилом.
Каждое из соединений в форме трифторацетатной соли в примерах может быть получено в свободном состоянии следующим общим способом: трифторацетатную соль растворяют в подходящем растворителе (например, метаноле, этаноле, тетрагидрофуране, ацетоне и т.д.) и добавляют слабое основание (такое как бикарбонат натрия, карбонат натрия, карбонат калия и т.д.), чтобы довести pH до нейтральности, смесь концентрируют при пониженном давлении, и остаток очищают, чтобы получить свободное состояние.
Пример 1
4-амино-1-((6R,9R,12R)-12-(4-аминобутил)-6-бензил-9-изобутил-4,7,10-триоксо-1-(1-фенилциклопропил)-2,5,8,11-тетраазатридекан-13-оил)пиперидин-4-карбоновая кислота 1
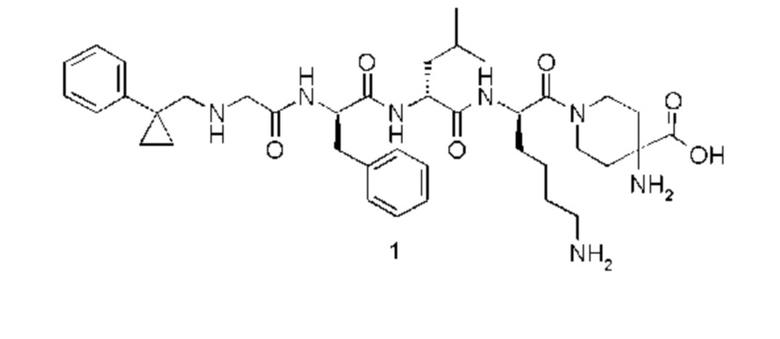
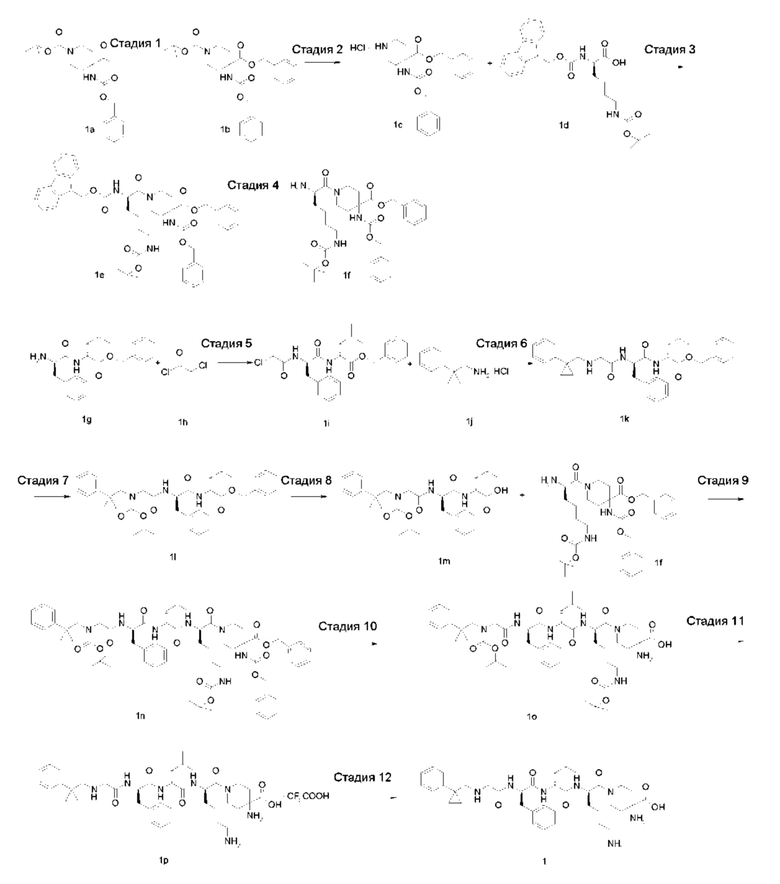
Стадия 1
4-бензил-1-трет-бутил-4-(((бензилокси)карбонил)амино)пиперидин-1,4-дикарбоксилат 1b
4-(((бензилокси)карбонил)амино)-1-(трет-бутоксикарбонил)пиперидин-4-карбоновую кислоту 1а (1,2 г, 0,0032 моль, полученную известным способом, раскрытым в «Bioorganic Medicinal Chemistry Letters, 2007, 7 (9), 2448-2451»), бензилбромид (0,65 г, 0,0038 моль) и карбонат цезия (2,1 г, 0,0064 моль) растворяли в 20 мл N,N-диметилформамида и перемешивали в течение 12 часов при комнатной температуре. Реакционный раствор выливали в воду и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали тонкослойной хроматографией с элюирующей системой В с получением указанного в заголовке соединения 1b (800 мг, выход: 53%).
Стадия 2
Бензил-4-(((бензилокси)карбонил)амино)пиперидин-4-карбоксилата гидрохлорид 1с
1b (800 мг, 1,71 ммоль) растворяли в 2 мл дихлорметана и добавляли 2 мл раствора 4М соляной кислоты в 1,4-диоксане. После перемешивания в течение 4 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении, чтобы получить указанное в заголовке неочищенное соединение 1 с (800 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 3
(R)-бензил-1-(2-((((9H-флуорен-9-ил)метокси)карбонил)амино)-6-((трет-бутоксикарбонил)амино)гексаноил)-4-(((бензилокси)карбонил)амино)пиперидин-4-карбоксилат 1е
Неочищенное соединение 1с (800 мг, 1,97 ммоль) и (R)-2-(((((9Н-флуорен-9-ил)метокси)карбонил)амино)-6-((трет-бутоксикарбонил)амино)гексановую кислоту 1d (926 мг, 1,97 ммоль, полученную известным способом, раскрытым в «ChemMedChem, 2015, 10 (7), 1232-1239»), растворяли в 20 мл N,N-диметилформамида. Добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (1,12 г, 3,0 ммоль) и N,N-диизопропилэтиламин (0,7 мл, 3,94 ммоль). После перемешивания в течение 12 часов при комнатной температуре реакционный раствор выливали в 2 н. раствор лимонной кислоты и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором бикарбоната натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения 1е (1,6 г), указанного в заголовке, которое использовали непосредственно на следующей стадии без очистки.
Стадия 4
(R)-бензил-1-(2-амино-6-((трет-бутоксикарбонил)амино)гексаноил)-4-(((бензилокси) карбонил)амино)пиперидин-4-карбоксилат 1f
Неочищенное соединение 1е (1,6 г, 0,002 моль) растворяли в 10 мл дихлорметана, затем добавляли 10 мл пиперидина. После перемешивания в течение 2 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с элюирующей системой А с получением указанного в заголовке соединения 1f (900 мг, выход: 77%).
Стадия 5
(R)-бензил-2-((R)-2-(2-хлорацетамидо)-3-фенилпропионамидо)-4-метилпентаноат 1i
(R)-бензил-2-((R)-2-амино-3-фенилпропанамидо)-4-метилпентаноат 1g (500 мг, 1,36 ммоль, полученный способом, раскрытым в заявке на патент US 20110212882 А1) и триэтиламин (275 мг, 2,72 ммоль) растворяли в 10 мл дихлорметана, затем добавляли хлорацетилхлорид (230 мг, 2 ммоль) по каплям. После перемешивания в течение 12 часов при комнатной температуре реакционный раствор выливали в воду и промывали насыщенным раствором хлорида аммония. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, чтобы получить неочищенный продукт 1i, указанный в заголовке (500 мг), который использовали непосредственно на следующей стадии без очистки.
Стадия 6
(R)-бензил-4-метил-2-((R)-3-фенил-2-(2-(((1-фенилциклопропил)метил)амино)ацетамидо)пропанамидо)пентаноат 1k
Неочищенное соединение 1i (150 мг, 0,33 ммоль) и (1-фенилциклопропил)метанамина гидрохлорид 1j (74 мг, 0,4 ммоль, полученный известным способом, раскрытым в «Journal of American Chemical Society», 2015, 137 (5), 2042-2046»), растворяли в 10 мл N,N-диметилформамида и затем добавляли йодид калия (110 мг, 0,67 ммоль) и карбонат калия (139 мг, 1 ммоль). Реакционный раствор нагревали до 60°С и перемешивали в течение 5 часов, затем концентрировали при пониженном давлении. Полученный остаток добавляли в воду и экстрагировали дихлорметаном (30 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, чтобы получить неочищенное указанное в заголовке соединение 1k (187 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 7
(9R,12R)-бензил-9-бензил-12-изобутил-2,2-диметил-4,7,10-триоксо-5-((1-фенилциклопропил)метил)-3-окса-5,8,11-триазатридекан-13-оат 1I
Неочищенное соединение 1k (187 мг, 0,337 ммоль) растворяли в дихлорметане, затем добавляли ди-трет-бутилдикарбонат (147 мг, 0,67 ммоль) и N,N-диизопропилэтиламин (130 мг, 1,01 ммоль). После перемешивания в течение 12 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с элюирующей системой А с получением указанного в заголовке соединения 11 (100 мг, выход 45,5%).
Стадия 8
(9R,12R)-9-бензил-12-изобутил-2,2-диметил-4,7,10-триоксо-5-((1-фенилциклопропил)метил)-3-окса-5,8,11-триазатридекан-13-овая кислота 1m
1I (100 мг, 0,152 ммоль) растворяли в 10 мл этанола, затем добавляли палладий на угле (100 мг, каталитическое количество). После завершения добавления реакционную систему продували водородом три раза и перемешивали в течение 5 часов при комнатной температуре. Реакционный раствор фильтровали через целит, и фильтрат концентрировали при пониженном давлении, чтобы получить неочищенное указанное в заголовке соединение 1m (86 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 9
Бензил-1-((9R,12R,15R)-9-бензил-15-(4-((трет-бутоксикарбонил)амино)бутил)-12-изобутил-2,2-диметил-4,7,10,13-тетраоксо-5-((1-фенилциклопропил)метил)-3-окса-5,8,11,14-тетраазагексадекан-16-оил)-4-(((бензилокси)карбонил)амино)пиперидин-4-карбоксилат 1n
Неочищенное соединение 1m (86 мг, 0,152 ммоль), 1f (91 мг, 0,152 ммоль) и 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (115 мг, 0,3 ммоль) растворяли в 10 мл N,N-диметилформамида. После перемешивания в течение 5 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с элюирующей системой А с получением указанного в заголовке соединения 1n (100 мг, выход: 57,5%).
Стадия 10
4-амино-1-((9R,12R,15R)-9-бензил-15-(4-((трет-бутоксикарбонил)амино)бутил)-12-изобутил-2,2-диметил-4,7,10,13-тетраоксо-5-((1-фенилциклопропил)метил)-3-окса-5,8,11,14-тетраазагексадекан-16-оил)пиперидин-4-карбоновая кислота 1o 1n (100 мг, 0,087 ммоль) растворяли в 10 мл этанола, затем добавляли палладий на угле (100 мг, каталитическое количество). После завершения добавления реакционную систему продували водородом три раза и перемешивали в течение 12 часов при комнатной температуре. Реакционный раствор фильтровали через целит, и фильтрат концентрировали при пониженном давлении, чтобы получить неочищенное указанное в заголовке соединение 1о (80 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 11
4-амино-1-((6R,9R,12R)-12-(4-аминобутил)-6-бензил-9-изобутил-4,7,10-триоксо-1-(1-фенилциклопропил)-2,5,8,11-тетраазатридекан-13-оил)пиперидин-4-карбоновой кислоты трифторацетат 1р
Неочищенное соединение 1о (80 мг, 0,087 ммоль) растворяли в 10 мл дихлорметана, затем добавляли 2 мл трифторуксусной кислоты. После перемешивания в течение 5 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали высокоэффективной жидкостной хроматографией с получением указанного в заголовке соединения 1р (10 мг, выход: 15,9%).
МС m/z (ESI): 720.4 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8.47-8.40 (m, 2Н), 7.42-7.27 (m, 11Н), 4.84-4.81 (m, 1Н), 4.70-4.67 (m, 1Н), 4.40-4,38 (m, 1Н), 4.25-4.10 (m, 1Н), 3.95-3.85 (m, 2Н), 3.78-3.70 (m, 2Н), 3.61-3,52 (m, 1Н), 3,5-3.41 (m, 1Н), 3.25-3.10 (m, 3Н), 3.10-2.95 (m, 2Н), 2.95-2.89 (m, 2Н), 2.89-2.75 (m, 2Н), 2,31-2.24 (m, 2Н), 1.95-1.45 (m, 13Н), 1.1-0.9 (m, 6Н), 0.9-0.86 (m, 4Н).
Стадия 12
4-амино-1-((6R,9R,12R)-12-(4-аминобутил)-6-бензил-9-изобутил-4,7,10-триоксо-1-(1-фенилциклопропил)-2,5,8,11-тетраазатридекан-13-оил)пиперидин-4-карбоновая кислота 1
1р (10 мг, 0,012 ммоль) растворяли в 1 мл смешанного раствора дихлорметана и метанола (об/об = 10:1), затем по каплям добавляли насыщенный водный раствор карбоната натрия для доведения pH до примерно 7. Реакционный раствор перемешивали в течение 30 минут при комнатной температуре и оставляли для отстаивания и разделения. Органическую фазу собирали, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 1 (8,6 мг, выход: 100%).
МС m/z (ESI): 720.4 [М+1]
Пример 2
4-амино-1-((2R,5R,8R,14S)-2-(4-аминобутил)-8-бензил-5-изобутил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазапентадекан-1-оил)пиперидин-4-карбоновая кислота 2
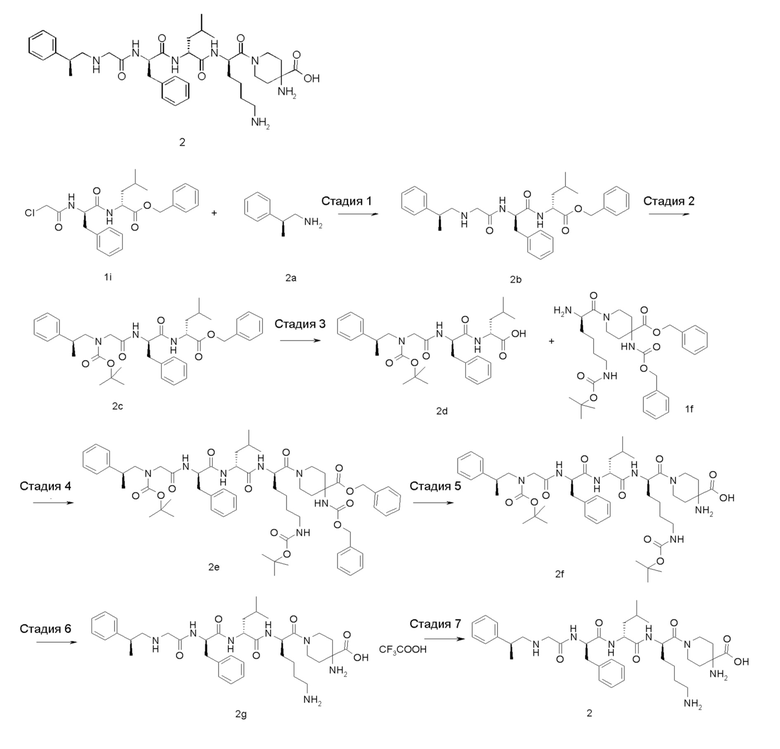
Стадия 1
(R)-бензил-4-метил-2-((R)-3-фенил-2-(2-(((S)-2-фенилпропил)амино)-ацетамидо)пропанамидо)пентаноат 2b
1i (500 мг, 1,12 ммоль) и (S)-2-фенилпропан-1-амин 2а (228 мг, 1,68 ммоль), полученный известным способом, раскрытым в «Advanced Synthesis & Catalysis, 2015, 357 (18), 3875-3879») растворяли в 10 мл N,N-диметилформамида, затем добавляли йодид калия (372 мг, 2,24 ммоль) и карбонат калия (309 мг, 2,24 ммоль). Реакционный раствор нагревали до 60°C и перемешивали в течение 12 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли воду и экстрагировали дихлорметаном (30 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, чтобы получить неочищенное указанное в заголовке соединение 2b (600 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 2
(9R,12R)-бензил-9-бензил-12-изобутил-2,2-диметил-4,7,10-триоксо-5-((S)-2-фенилпропил)-3-окса-5,8,11-триазатридекан-13-оат 2с
Неочищенное соединение 2b (600 мг, 1,1 ммоль) растворяли в 10 мл дихлорметана, затем добавляли ди-трет-бутилдикарбонат (360 мг, 1,65 ммоль) и триэтиламин (222 мг, 2,2 ммоль). После перемешивания в течение 12 часов реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением указанного в заголовке соединения 2с (380 мг, выход: 54%).
Стадия 3
(9R,12R)-9-бензил-12-изобутил-2,2-диметил-4,7,10-триоксо-5-((S)-2-фенилпропил)-3-окса-5,8,11-триазатридекан-13-овая кислота 2d
2с (380 мг, 0,59 ммоль) растворяли в 15 мл метанола, затем добавляли палладий на угле (40 мг, каталитическое количество). После завершения добавления реакционную систему продували водородом три раза и перемешивали при комнатной температуре в течение 12 часов. Реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении, чтобы получить неочищенное указанное в заголовке соединение 2d (300 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 4
Бензил-1-((9R,12R,15R)-9-бензил-15-(4-((трет-бутоксикарбонил)амино)бутил)-12-изобутил-2,2-диметил-4,7,10,13-тетраоксо-5-((S)-2-фенилпропил)-3-окса-5,8,11,14-тетраазагексадекан-16-оил)-4-(((бензилокси)карбонил)амино)пиперидин-4-карбоксилат 2е
Неочищенное соединение 2d (300 мг, 0,54 ммоль), 1f (356 мг, 0,6 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (308 мг, 0,81 ммоль) и N,N-диизопропилэтиламин (104 мг, 0,81 ммоль) растворяли в 10 мл N,N-диметилформамида. После перемешивания в течение 1,5 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с элюирующей системой А с получением указанного в заголовке соединения 2е (500 мг, выход: 81,8%).
Стадия 5
4-амино-1-((9R,12R,15R)-9-бензил-15-(4-((трет-бутоксикарбонил)амино)бутил)-12-изобутил-2,2-диметил-4,7,10,13-тетраоксо-5-((S)-2-фенилпропил)-3-окса-5,8,11,14-тетраазагексадекан-16-оил)пиперидин-4-карбоновая кислота 2f
2е (500 мг, 0,44 ммоль) растворяли в 10 мл метанола, затем добавляли палладий на угле (50 мг, каталитическое количество). После завершения добавления реакционную систему продували водородом три раза и перемешивали в течение 12 часов при комнатной температуре. Реакционный раствор фильтровали через целит, и фильтрат концентрировали при пониженном давлении, чтобы получить неочищенное указанное в заголовке соединение 2f (400 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 6
4-амино-1-((2R,5R,8R,14S)-2-(4-аминобутил)-8-бензил-5-изобутил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазапентадекан-1-оил)пиперидин-4-карбоновой кислоты трифторацетат 2g
Неочищенное соединение 2f (400 мг (0,44 ммоль) растворяли в 5 мл дихлорметана, затем добавляли 2 мл раствора 4М соляной кислоты в 1,4-диоксане. После перемешивания в течение 2 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали высокоэффективной жидкостной хроматографией с получением указанного в заголовке соединения 2g (150 мг, выход: 48%).
МС m/z (ESI): 708.6 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.40-7.17 (m, 11Н), 4.89-4.82 (m, 1Н), 4.79-4.74 (m, 1Н), 4.40-4,39 (m, 1Н), 4.22-4.15 (m, 1Н), 4.01-3.95 (m, 1Н), 3.85-3.60 (m, 5Н), 3.49-3,36 (m, 1Н), 3.21-3.09 (m, 5Н), 2.96-2.92 (m, 4Н), 2.27-2.25 (m, 3Н), 1.83-1.45 (m, 14Н), 1,34-1,33 (m, 3Н), 1.01-0.92 (m, 6Н)
Стадия 7
4-амино-1-((2R,5R,8R,14S)-2-(4-аминобутил)-8-бензил-5-изобутил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазапентадекан-1-оил)пиперидин-4-карбоновая кислота 2
2g (150 мг, 0,182 ммоль) растворяли в 1 мл смешанного раствора дихлорметана и метанола (об/об = 10:1), затем по каплям добавляли насыщенный водный раствор карбоната натрия для доведения pH до примерно 7. Реакционный раствор перемешивали в течение 30 минут при комнатной температуре и оставляли для отстаивания и разделения. Органическую фазу собирали, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 2 (129 мг, выход: 100%).
МС m/z (ESI): 708.6 [М+1]
Пример 3
4-амино-1-((2R,5R,8R)-2-(4-аминобутил)-8-бензил-5-изобутил-14-метил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазапентадекан-1-оил)пиперидин-4-карбоновая кислота 3
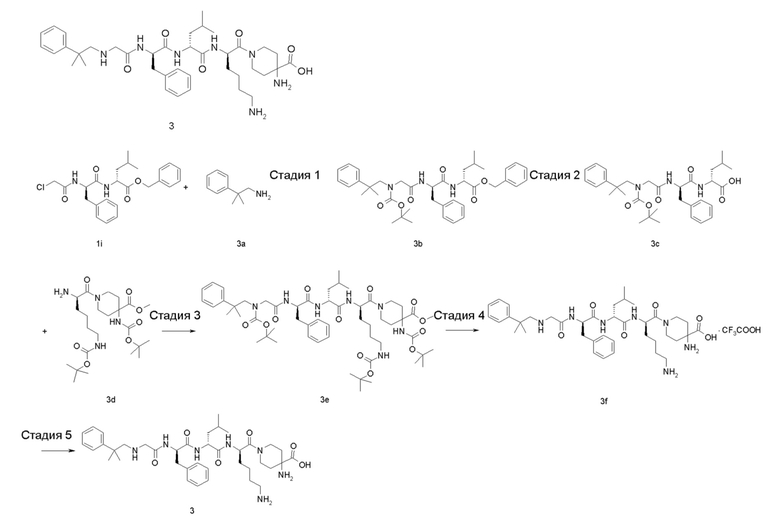
Стадия 1
(9R,12R)-бензил-9-бензил-12-изобутил-2,2-диметил-5-(2-метил-2-фенилпропил)-4,7,10-триоксо-3-окса-5,8,11-триазатридекан-13-оат 3b
Неочищенное соединение 1i (130 мг, 0,293 ммоль) и 2-метил-2-фенилпропан-1-амин 3а (130 мг, 0,878 ммоль, полученный способом, раскрытым в заявке на патент WO 2007030582), растворяли в 2 мл N,N-диметилформамида затем добавляли йодид калия (73 мг, 0,44 ммоль) и карбонат калия (121 мг, 0,878 ммоль). Реакционный раствор нагревали до 80°C и перемешивали в течение 12 часов. Реакционный раствор охлаждали до комнатной температуры, затем добавляли 1 мл тетрагидрофурана и 1 мл воды. После равномерного перемешивания добавляли ди-трет-бутилдикарбонат (96 мг, 0,439 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 2 часов, затем концентрировали при пониженном давлении. К остатку добавляли воду и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с элюирующей системой В с получением указанного в заголовке соединения 3b (110 мг, выход: 57%).
Стадия 2
(9R,12R)-9-бензил-12-изобутил-2,2-диметил-5-(2-метил-2-фенилпропил)-4,7,10-триоксо-3-окса-5,8,11-триазатридекан-13-овая кислота 3с
3b (110 мг, 0,162 ммоль) растворяли в метаноле, затем добавляли палладий на угле (20 мг, каталитическое количество). После завершения добавления реакционную систему продували водородом три раза, нагревали до 30°C и перемешивали в течение 12 часов. Реакционный раствор фильтровали через целит, и фильтрат концентрировали при пониженном давлении, чтобы получить неочищенное указанное в заголовке соединение 3с (74 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 3
Метил-1-((9R,12R,15R)-9-бензил-15-(4-((трет-бутоксикарбонил)амино)бутил)-12-изобутил-2,2-диметил-5-(2-метил-2-фенилпропил)-4,7,10,13-тетраоксо-3-окса-5,8,11,14-тетраазагексадекан-16-оил)-4-((трет-бутоксикарбонил)амино)пиперидин-4-карбоксилат 3е
Неочищенное соединение 3с (74 мг, 0,13 ммоль), (R)-метил-1-(2-амино-6-((трет-бутоксикарбонил)амино)гексаноил)-4-((трет-бутоксикарбонил)амино)-пиперидин-4-карбоксилат 3d (70 мг, 0,143 ммоль, полученный способом, раскрытым в заявке на патент JP 5807140 B1), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (74 мг, 0,195 ммоль) и N,N-диизопропилэтиламин (50 мг, 0,39 ммоль) растворяли в 2 мл N,N-диметилформамида и перемешивали при 0°C в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. Полученный остаток добавляли в воду и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, чтобы получить неочищенное заглавное соединение 3е (134 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 4
4-амино-1-((2R,5R,8R)-2-(4-аминобутил)-8-бензил-5-изобутил-14-метил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазапентадекан-1-оил)пиперидин-4-карбоновой кислоты трифторацетат 3f
Неочищенное соединение 3е (134 мг, 0,13 ммоль) растворяли в 2 мл смешанного раствора тетрагидрофурана и метанола (об/об = 3:1), затем добавляли 0,65 мл 1 М гидроксида лития. После перемешивания в течение 2 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали высокоэффективной жидкостной хроматографией с получением указанного в заголовке соединения 3f (40 мг, выход: 30%).
МС m/z (ESI): 722.6 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.90-7.759 (m, 2Н), 7.45-7.15 (m, 10Н), 4.80-4.71 (m, 1Н), 4.45-4,37 (m, 1Н), 4.17-4.10 (m, 1Н), 4.02-3.85 (m, 2Н), 3.80-3.72 (m, 3Н), 3.65-3,50 m, 1Н), 3.48-3.40 (m, 1Н), 3.25-3.15 (m, 3Н), 3.05-2.80 (m, 5Н), 2,38-2.20 (m, 3Н), 2.02-1.40 (m, 20Н), 1.05-0.92 (m, 6Н).
Стадия 5
4-амино-1-((2R,5R,8R)-2-(4-аминобутил)-8-бензил-5-изобутил-14-метил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазапентадекан-1-оил)пиперидин-4-карбоновая кислота 3
3f (40 мг, 0,048 ммоль) растворяли в 1 мл смешанного раствора дихлорметана и метанола (об/об = 10:1). Насыщенный водный раствор карбоната натрия добавляли по каплям, чтобы довести pH до примерно 7. Реакционный раствор перемешивали в течение 30 минут при комнатной температуре и оставляли для отстаивания и разделения. Органическую фазу собирали, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 3 (34,6 мг, выход: 100%).
МС m/z (ESI): 722.6 [М+1]
Пример 4
4-амино-1-((2R,5R,8R,14R)-2-(4-аминобутил)-8-бензил-5-изобутил-15-метил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазагексадекан-1-оил)пиперидин-4-карбоновая кислота 4
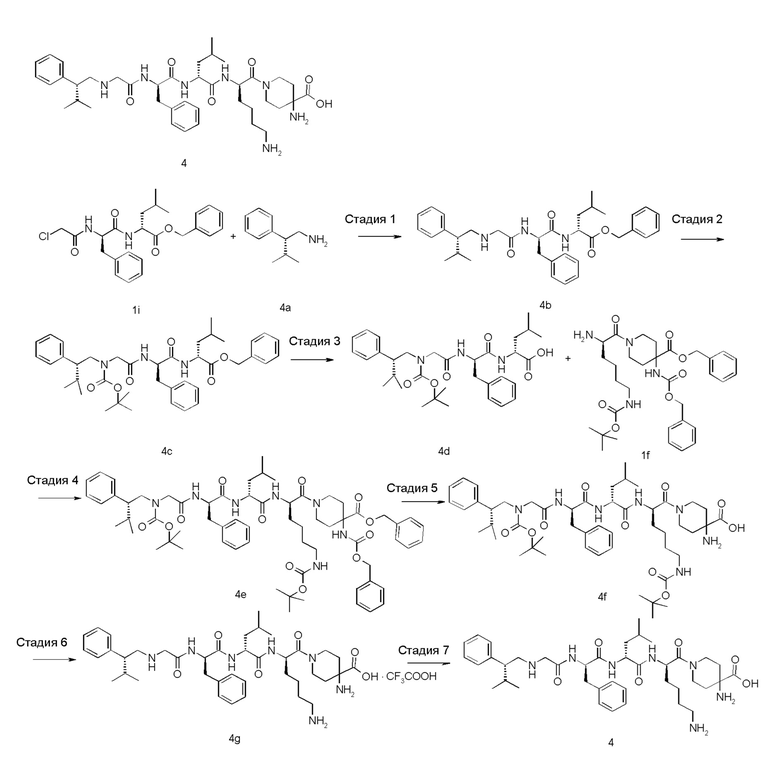
Стадия 1
(R)-бензил-4-метил-2-((R)-2-(2-(((R)-3-метил-2-фенилбутил)амино)ацетамидо)-3-фенилпропанамидо)пентаноат 4b
Неочищенное соединение 1i (1 г, 2,45 ммоль) и (R)-3-метил-2-фенилбутан-1-амин 4а (500 мг, 3 ммоль, полученный известным способом, раскрытым в "Tempahedron:Asymmetry, 2003, 14(16), 2401-2406") растворяли в 10 мл N,N-диметилформамида, затем добавляли йодид калия (1 г, 6 ммоль) и карбонат калия (1,3 г, 9,2 ммоль). Реакционный раствор нагревали до 60°C и перемешивали в течение 12 часов, затем концентрировали при пониженном давлении. Полученный остаток добавляли в воду и экстрагировали дихлорметаном (30 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, чтобы получить неочищенное указанное в заголовке соединение 4b (500 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 2
(9R,12R)-бензил-9-бензил-12-изобутил-2,2-диметил-5-((R)-3-метил-2-фенилбутил)-4,7,10-триоксо-3-окса-5,8,11-триазатридекан-13-оат 4с
Неочищенное соединение 4b (500 мг, 0,875 ммоль) растворяли в дихлорметане, затем добавляли ди-трет-бутилдикарбонат (380 мг, 1,75 ммоль) и N,N-диизопропилэтиламин (340 мг, 2,62 ммоль). После перемешивания в течение 12 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с элюирующей системой А с получением указанного в заголовке соединения 4с (300 мг, выход: 51,1%).
Стадия 3
(9R,12R)-9-бензил-12-изобутил-2,2-диметил-5-((R)-3-метил-2-фенилбутил)-4,7,10-триоксо-3-окса-5,8,11-триазатридекан-13-овая кислота 4d
4с (300 мг, 0,447 ммоль) растворяли в 10 мл этанола, затем добавляли палладий на угле (100 мг, каталитическое количество). После завершения добавления реакционную систему продували водородом три раза и перемешивали в течение 12 часов при комнатной температуре. Реакционный раствор фильтровали через целит, фильтрат концентрировали при пониженном давлении, чтобы получить неочищенное указанное в заголовке соединение 4d (260 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 4
Бензил-1-((9R,12R,15R)-9-бензил-15-(4-((трет-бутоксикарбонил)амино)-бутил)-12-изобутил-2,2-диметил-5-((R)-3-метил-2-фенилбутил)-4,7,10,13-тетраоксо-3-окса-5,8,11,14-тетраазагексадекан-16-оил)-4-(((бензилокси)карбонил)амино)пиперидин-4-карбоксилат 4е
Неочищенное соединение 4d (260 мг, 0,447 ммоль), 1f (270 мг, 0,447 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (500 мг, 1,34 ммоль) и 3 мл триэтиламина растворяли в 10 мл N,N-диметилформида. После перемешивания в течение 2 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с элюирующей системой А с получением указанного в заголовке соединения 4е (100 мг, выход: 19,2%).
Стадия 5
4-амино-1-((9R,12R,15R)-9-бензил-15-(4-((трет-бутоксикарбонил)амино)бутил)-12-изобутил-2,2-диметил-5-((R)-3-метил-2-фенилбутил)-4,7,10,13-тетраоксо-3-окса-5,8,11,14-тетраазагексадекан-16-оил)пиперидин-4-карбоновая кислота 4f
4е (100 мг, 0,086 ммоль) растворяли в 20 мл этанола, добавляли палладий на угле (100 мг, каталитическое количество). После завершения добавления реакционную систему продували водородом три раза и перемешивали в течение 12 часов при комнатной температуре. Реакционный раствор фильтровали через целит, и фильтрат концентрировали при пониженном давлении, чтобы получить неочищенное указанное в заголовке соединение 4f (81 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 6
4-амино-1-((2R,5R,8R,14R)-2-(4-аминобутил)-8-бензил-5-изобутил-15-метил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазагексадекан-1-оил)пиперидин-4-карбоновой кислоты трифторацетат 4g
Неочищенное соединение 4f (81 мг, 0,086 ммоль) растворяли в 10 мл дихлорметана, затем добавляли 3 мл трифторуксусной кислоты. После перемешивания в течение 2 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали высокоэффективной жидкостной хроматографией с получением указанного в заголовке соединения 4g (8 мг, выход: 12,7%).
МС m/z (ESI): 736.4 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.45-7.15 (m, 10Н), 4.9-4.8 (m, 1Н), 4.8-4.62 (m, 1Н), 4.41-4.28 (m, 1Н), 4.1-4.0 (m, 1Н), 3.98-3.85 (m, 1Н), 3.85-3.65 (m, 3Н), 3.65-3,55 (m, 1Н), 3,55-3.45 (m, 1Н) 3,38-3.28 (m, 2Н), 3.25-3.08 (m, 2Н), 3.25-3.05 (m, 2Н), 2.95-2.87 (m, 2Н), 2.85-2.75 (m, 1Н), 2.75-2.65 (m, 2Н), 2.4-2.15 (m, 3Н), 2-1,35 (m, 14Н), 1.2-0.83 (m, 9Н), 0.71-0.62 (d, 3Н).
Стадия 7
4-амино-1-((2R,5R,8R,14R)-2-(4-аминобутил)-8-бензил-5-изобутил-15-метил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазагексадекан-1-оил)пиперидин-4-карбоновая кислота 4
4g (8 мг, 0,009 ммоль) растворяли в 1 мл смешанного растворителя дихлорметана и метана (об./об. = 10:1). Насыщенный водный карбонат натрия добавляли по каплям для доведения pH до примерно 7. Реакционный раствор перемешивали в течение 30 минут при комнатной температуре и оставляли для отстаивания и разделения. Органическую фазу собирали, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 4 (6,9 мг, выход: 100%).
MC m/z (ESI): 736.4 [M+1]
Пример 5
4-амино-1-((2R,5R,8R,14R)-2-(4-аминобутил)-8-бензил-5-изобутил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазапентадекан-1-оил)пиперидин-4-карбоновая кислота 5
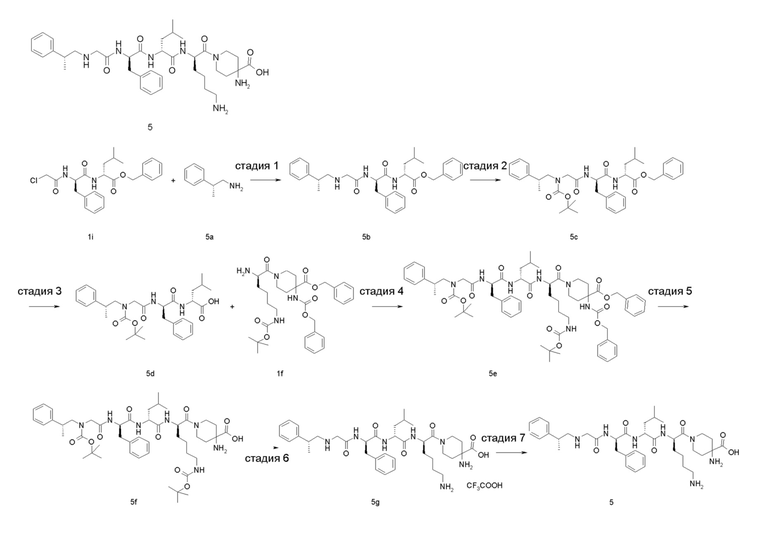
Стадия 1
(R)-бензил-4-метил-2-((R)-3-фенил-2-(2-(((R)-2-фенилпропил)амино)-ацетамидо)пропанамидо)пентаноат 5b
1i (500 мг, 1,12 ммоль) и (R)-2-фенилпропан-1-амин 5а (228 мг, 1,68 ммоль), полученный известным способом, раскрытым в «Angewete Chemie, International Edition, 2003, 42 (39), 4793-4795») растворяли в 10 мл N,N-диметилформамида, затем добавляли йодид калия (372 мг, 2,24 ммоль) и карбонат калия (309 мг, 2,24 ммоль). Реакционный раствор нагревали до 60°C и перемешивали в течение 12 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли воду и экстрагировали дихлорметаном (30 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, чтобы получить указанное в заголовке неочищенное соединение 5b (600 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 2
(9R,12R)-бензил-9-бензил-12-изобутил-2,2-диметил-4,7,10-триоксо-5-((R)-2-фенилпропил)-3-окса-5,8,11-триазатридекан-13-оат 5с
Неочищенное соединение 5b (600 мг, 1,1 ммоль) растворяли в 20 мл дихлорметана, затем добавляли ди-трет-бутилдикарбонат (361 мг, 1,66 ммоль) и триэтиламин (222 мг, 2,2 ммоль). После перемешивания в течение 12 часов при комнатной температуре реакционный раствор концентрировали под давлением. Полученный остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением указанного в заголовке соединения 5с (580 мг, выход: 82%).
Стадия 3
(9R,12R)-9-бензил-12-изобутил-2,2-диметил-4,7,10-триоксо-5-((R)-2-фенилпропил)-3-окса-5,8,11-триазатридекан-13-овая кислота 5d
5с (580 мг, 0,9 ммоль) растворяли в 10 мл метанола, затем добавляли палладий на угле (60 мг, каталитическое количество). После завершения добавления реакционную систему продували водородом три раза и перемешивали в течение 12 часов при комнатной температуре. Реакционный раствор фильтровали через целит, и фильтрат концентрировали при пониженном давлении, чтобы получить указанное в заголовке неочищенное соединение 5d (500 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 4
Бензил-1-((9R,12R,15R)-9-бензил-15-(4-((трет-бутоксикарбонил)амино)-бутил)-12-изобутил-2,2-диметил-4,7,10,13-тетраоксо-5-((R)-2-фенилпропил)-3-окса-5,8,11,14-тетраазагексадекан-16-оил)-4-(((бензилокси)карбонил)амино)-пиперидин-4-карбоксилат 5е
Неочищенное соединение 5d (365 мг, 0,66 ммоль), 1f (393 мг, 0,66 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (376 мг, 0,99 ммоль) и N,N-диизопропилэтиламин (0,16 мл, 0,99 ммоль) растворяли в 10 мл N,N-диметилформамида. После перемешивания в течение 2 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением указанного в заголовке соединения 5е (170 мг, выход: 23%).
Стадия 5
4-амино-1-((9R,12R,15R)-9-бензил-15-(4-((трет-бутоксикарбонил)амино)-бутил)-12-изобутил-2,2-диметил-4,7,10,13-тетраоксо-5-((R)-2-фенилпропил)-3-окса-5,8,11,14-тетраазагексадекан-16-оил)пиперидин-4-карбоновая кислота 5f
5е (80 мг, 0,0706 ммоль) растворяли в 10 мл метанола, затем добавляли палладий на угле (10 мг, каталитическое количество). После завершения добавления реакционную систему продували водородом три раза и перемешивали в течение 12 часов при комнатной температуре. Реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке неочищенного соединения 5f (60 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 6
4-амино-1-((2R,5R,8R,14R)-2-(4-аминобутил)-8-бензил-5-изобутил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазапентадекан-1-оил)пиперидин-4-карбоновой кислоты трифторацетат 5g
Неочищенный продукт 5f (60 мг (0,066 ммоль) растворяли в 2 мл дихлорметана, затем добавляли 1 мл раствора 4М соляной кислоты в 1,4-диоксане. После перемешивания в течение 2 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали высокоэффективной жидкостной хроматографией с получением указанного в заголовке соединения 5g (30 мг, выход: 55,6%).
МС m/z (ESI): 708.6 [М+1]
Стадия 7
4-амино-1-((2R,5R,8R,14R)-2-(4-аминобутил)-8-бензил-5-изобутил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазапентадекан-1-оил)пиперидин-4-карбоновая кислота 5
5g (30 мг, 0,028 ммоль) растворяли в 5 мл смешанного растворителя метанол/вода, затем добавляли твердый бикарбонат натрия (10 мг) для доведения pH до 7. Реакционный раствор перемешивали в течение 30 минут, затем концентрировали при пониженном давлении. К полученному остатку добавляли 10 мл дихлорметана, перемешивали в течение 30 минут и фильтровали. Осадок на фильтре промывали 10 мл дихлорметана и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 5 (17 мг, выход: 85,9%).
МС m/z (ESI): 708.6 [М+1]
1Н ЯМР (400 МГц, CD3OD): δ 7,33-7.19 (m, 10Н), 4.90-4.84 (m, 2Н), 4.64-4.61 (m, 2Н), 4.42-4,39 (m, 1Н), 3.86-3.74 (m, 5Н), 3.20-3.12 (m, 4Н), 2.94-2.84 (m, 4Н), 2.61-2,54 (m, 2H), 2.20-2.15 (m, 3Н), 1.79-1.70 (m, 2H), 1.68-1.60 (m, 8H), 1.45-1.40 (m, 3Н), 1,30-1.20 (m, 5H), 0.99-0.76 (m, 6H).
Пример 6
(R)-N-((R)-6-амино-1-морфолино-1-оксогексан-2-ил)-4-метил-2-((R)-3-фенил-2-(2-(((1S,2R)-2-фенилциклопропил)амино)ацетамидо)пропанамидо)-пентанамид 6
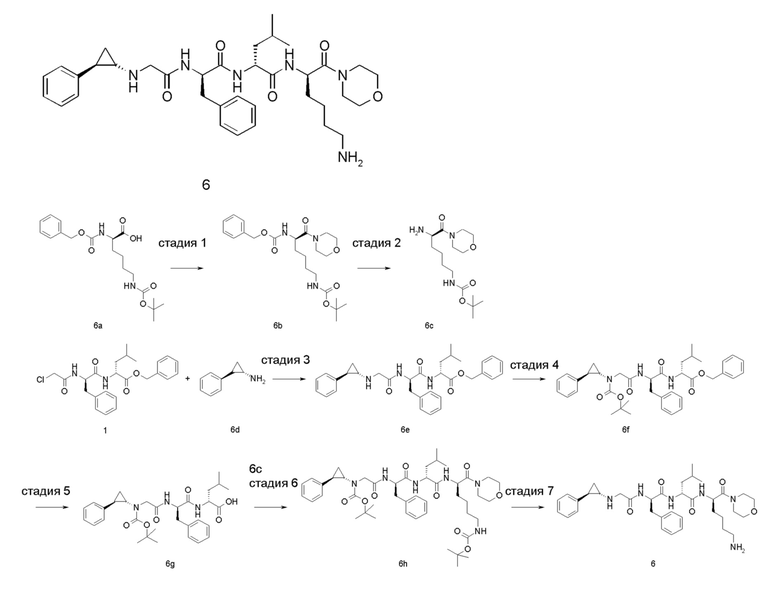
Стадия 1
(R)-бензил-трет-бутил-(6-морфолино-6-оксогексан-1,5-диил)дикарбамат 6b
(R)-2-(((бензилокси)карбонил)амино)-6-((трет-бутоксикарбонил)амино)-гексановую кислоту 6а (1,14 г, 3 ммоль, полученную известным способом, раскрытым в African Journal of pure и Applied Chemistry, 2009, 3 (6), 108-115), морфолин (0,31 мл, 3,6 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (1,73 г, 4,5 ммоль) и N,N-диизопропилэтиламин (0,8 мл, 4,5 ммоль) растворяли в 10 мл N,N-диметилформамида и перемешивали в течение 2 часов при комнатной температуре. К реакционному раствору добавляли 50 мл этилацетата, последовательно промывали насыщенным раствором хлорида аммония, насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 6b (1,3 г), которое использовали непосредственно на следующей стадии без очистки.
Стадия 2
(R)-трет-бутил-(5-амино-6-морфолино-6-оксогексил)карбамат 6с
Неочищенное соединение 6b (1,3 г, 2,9 ммоль) растворяли в 15 мл метанола, затем добавляли палладий на угле (350 мг, каталитическое количество). После завершения добавления реакционную систему продували водородом три раза и перемешивали в течение 12 часов при комнатной температуре. Реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением сырого указанного в заголовке соединения 6с (914 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 3
(R)-бензил-4-метил-2-((R)-3-фенил-2-(2-(((1S,2R)-2-фенилциклопропил)-амино)ацетамидо)пропанамидо)пентаноат 6е
Неочищенное соединение 1i (300 мг, 0,675 ммоль) и (1S,2S)-2-фенилциклопропанамин 6d (120 мг, 0,68 ммоль, полученный способом, раскрытым в заявке на патент US 20060116370 A1), растворяли в 10 мл N,N-диметилформамида, затем добавляли йодид калия (560 мг, 3,375 ммоль) и карбонат калия (465 мг, 3,375 ммоль). Реакционный раствор нагревали до 60°C и перемешивали в течение 5 часов, затем концентрировали при пониженном давлении. Полученный остаток добавляли в воду и экстрагировали дихлорметаном (30 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 6е (200 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 4
(9R,12R)-бензил-9-бензил-12-изобутил-2,2-диметил-4,7,10-триоксо-5-((1S,2R)-2-фенилциклопропил)-3-окса-5,8,11-триазатридекан-13-оат 6f
Неочищенное соединение 6е (200 мг, 0,35 ммоль) растворяли в дихлорметане, затем добавляли ди-трет-бутилдикарбонат (100 мг, 0,525 ммоль) и триэтиламин (110 мг, 1,05 ммоль). После перемешивания в течение 12 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с элюирующей системой А с получением указанного в заголовке соединения 6f (140 мг, выход: 62,5%)
Стадия 5
(9R,12R)-9-бензил-12-изобутил-2,2-диметил-4,7,10-триоксо-5-((1S,2R)-2-фенилциклопропил)-3-окса-5,8,11-триазатридекан-13-овая кислота 6g
6f (140 мг, 0,218 ммоль) растворяли в 4,5 мл смешанного растворителя тетрагидрофурана, метанола и воды (об/об/об = 4:4:1), затем добавляли моногидрат гидроксида лития (55 мг, 1,31 ммоль). После перемешивания в течение 12 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении, чтобы удалить растворители метанол и тетрагидрофуран. Добавляли воду и по каплям добавляли 1 М соляную кислоту для доведения pH до 6. Реакционный раствор экстрагировали дихлорметаном (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, чтобы получить неочищенное указанное в заголовке соединение 6g (130 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 6
трет-бутил-((10R,13R,16R)-16-фенил-13-изобутил-2,2-диметил-10-(морфолин-4-карбонил)-4,12,15,18-тетраоксо-3-окса-5,11,14,17-тетраазанонадекан-19-ил)((1S,2R)-2-фенилциклопропил)карбамат 6h
Неочищенное соединение 6g (130 мг, 0,218 ммоль), неочищенное соединение 6с (85 мг, 0,26 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (125 мг, 0,327 ммоль) и N,N-диизопропилэтиламин (85 мг, 0,654 ммоль) растворяли в 5 мл N,N-диметилформамида и перемешивали в течение 12 часов при комнатной температуре. В реакционный раствор добавляли 30 мл ацетилацетата, последовательно промывали насыщенным раствором хлорида аммония, насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением указанного в заголовке соединения 6h (130 мг, выход: 70%).
Стадия 7
(R)-N-((R)-6-амино-1-морфолино-1-оксогексан-2-ил)-4-метил-2-((R)-3-фенил-2-(2-(((1S,2R)-2-фенилциклопропил)амино)ацетамидо)пропанамидо)-пентанамид 6
6h (60 мг, 0,071 ммоль) растворяли в 3 мл дихлорметана, затем добавляли 0,8 мл раствора 4М соляной кислоты в 1,4-диоксане. После перемешивания в течение 2 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток растворяли в смешанном растворителе метанола и воды (об:об = 20:1). Карбонат натрия добавляли для доведения pH до более 8. Раствор концентрировали при пониженном давлении. К полученному остатку добавляли 10 мл дихлорметана, перемешивали в течение 10 минут и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 6 (19 мг, выход: 43,5%).
МС m/z (ESI): 649,3 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7,26-7,23 (m, 10Н), 4.95-4.93 (m, 1Н), 4.80-4.78 (m, 1Н), 4.70-4.68 (m, 1Н), 3.66-3.60 (m, 8Н), 3,32-3,30 (m, 6Н), 3,28-3,26 (m, 1Н), 3.18-3.16 (m, 1Н), 2.94-2.91 (m, 1Н), 2.65-2.63 (m, 1Н), 2,26-2,23 (m, 1Н), 1.71-1.68 (m, 5Н), 1.48-1.42 (m, 6Н), 0.95-0.93 (dd, 6Н).
Пример 7
4-амино-1-((2R,5R,8R)-2-(4-аминобутил)-8-бензил-5-изобутил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазагексадекан-1-оил)пиперидин-4-карбоновая кислота 7
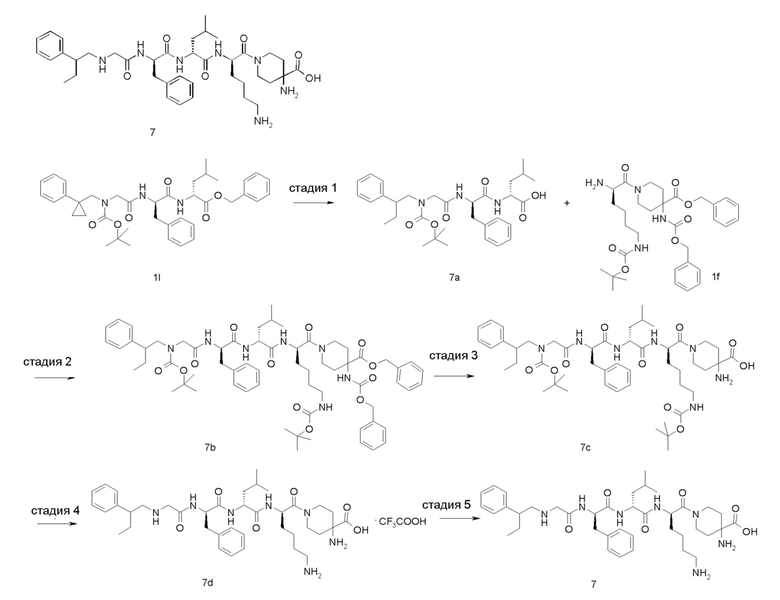
Стадия 1
(9R,12R)-9-бензил-12-изобутил-2,2-диметил-4,7,10-триоксо-5-(2-фенилбутил)-3-окса-5,8,11-триазатридекан-13-овая кислота 7а
1I (300 мг, 0,458 ммоль) растворяли в 10 мл метанола, затем добавляли каталитическое количество палладия на угле. После завершения добавления реакционную систему продували водородом три раза и перемешивали в течение 12 часов при комнатной температуре. Реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением сырого указанного в заголовке соединения 7а (189 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 2
Бензил-1-((9R,12R,15R)-9-бензил-15-(4-((трет-бутоксикарбонил)амино)-бутил)-12-изобутил-2,2-диметил-4,7,10,13-тетраоксо-5-(2-фенилбутил)-3-окса-5,8,11,14-тетраазагексадекан-16-оил)-4-(((бензилокси)карбонил)амино)-пиперидин-4-карбоксилат 7b
Неочищенное соединение 7а (189 мг, 0,34 ммоль), 1f (200 мг, 0,34 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (194 мг, 0,51 ммоль) и триэтиламин (67 мг, 0,68 ммоль) растворяли в 10 мл N,N-диметилформамида. После перемешивания в течение 12 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с элюирующей системой А с получением указанного в заголовке соединения 7b (80 мг, выход: 20%).
Стадия 3
4-амино-1-((9R,12R,15R)-9-бензил-15-(4-((трет-бутоксикарбонил)амино)-бутил)-12-изобутил-2,2-диметил-4,7,10,13-тетраоксо-5-(2-фенилбутил)-3-окса-5,8,11,14-тетраазагексадекан-16-оил)пиперидин-4-карбоновая кислота 7с
7b (80 мг, 0,07 ммоль) растворяли в 10 мл метанола, затем добавляли палладий на угле (10 мг, каталитическое количество). После завершения добавления реакционную систему продували водородом три раза и перемешивали в течение 12 часов при комнатной температуре. Реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением сырого указанного в заголовке соединения 7с (50 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 4
4-амино-1-((2R,5R,8R)-2-(4-аминобутил)-8-бензил-5-изобутил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазагексадекан-1-оил)пиперидин-4-карбоновой кислоты трифторацетат 7d
Неочищенное соединение 7с (50 мг, 0,054 ммоль) растворяли в 10 мл дихлорметана, затем добавляли 2 мл раствора 4М соляной кислоты в 1,4-диоксауне. После перемешивания в течение 1 часа при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали высокоэффективной жидкостной колоночной хроматографией с получением указанного в заголовке соединения 7d (10 мг, выход: 25,6%).
МС m/z (ESI): 722.6 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.42-7,25 (m, 10Н), 4.84-4.69 (m, 3Н), 4,39-4,38 (m, 2Н), 3.90-3.73 (m, 8Н), 3,22-3.19 (m, 4Н), 2.94-2.67 (m, 5Н), 2,24-2.19 (m, 3Н), 1.79-1,58 (m, 15Н), 0.99-0.93 (m, 6Н), 0.78 (t, 3Н)
Стадия 5
4-амино-1-((2R,5R,8R)-2-(4-аминобутил)-8-бензил-5-изобутил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазагексадекан-1-оил)пиперидин-4-карбоновая кислота 7
7d (10 мг, 0,012 ммоль) растворяли в 1 мл смешанного растворителя из дихлорметана и метанола (об/об = 10:1), затем добавляли насыщенный водный раствор карбоната натрия для доведения pH до примерно 7. Реакционный раствор перемешивали в течение 30 минут при комнатной температуре и оставляли для отстаивания и разделения. Органическую фазу собирали, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 7 (8,6 мг, выход: 100%).
МС m/z (ESI): 722.6 [М+1]
Пример 8
4-амино-1-((2R,5R,8R)-2-(4-аминобутил)-8-бензил-5-изобутил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазатетрадекан-1-оил)пиперидин-4-карбоновая кислота 8
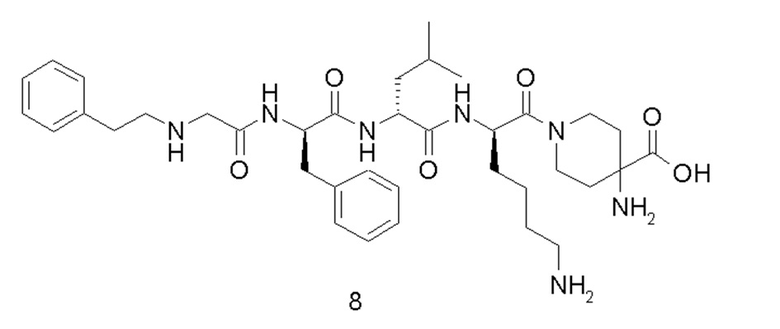
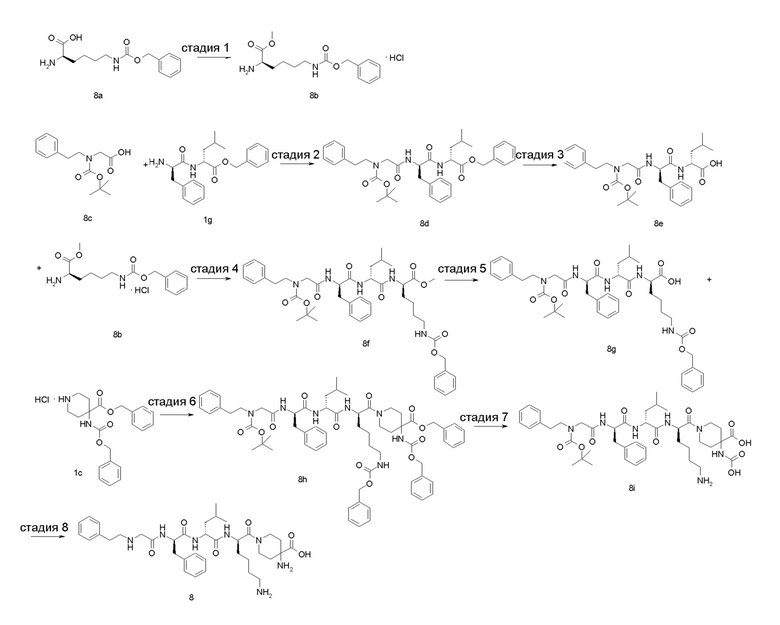
Стадия 1
(R)-метил 2-амино-6-(((бензилокси)карбонил)амино)гексаноат гидрохлорид 8b
1,3 мл дихлорсульфоксида растворяли в 20 мл метанола и охлаждали до 0°C на ледяной бане. Добавляли (R)-2-амино-6-(((бензилокси)карбонил)амино)-гексановую кислоту 8а (2 г, 7,1 ммоль). После перемешивания в течение 12 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении с получением указанного в заголовке неочищенного соединения 8b (2,09 г), которое использовали непосредственно на следующей стадии без очистки.
Стадия 2
(9R,12R)-бензил-9-бензил-12-изобутил-2,2-диметил-4,7,10-триоксо-5-фенилэтил-3-окса-5,8,11-триазатридекан-13-оат 8d
2-((трет-бутоксикарбонил)(фенилэтил)амино)уксусную кислоту 8с (332 мг, 1,19 ммоль, полученную способом, раскрытым в заявке на патент US 6245746 B1) и 1g (439 мг, 1,19 ммоль), растворяли в 6,6 мл N,N-диметилформамида, затем добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (679 мг, 1,785 ммоль) и N,N-диизопропилэтиламин (615 мг, 4,76 ммоль). Раствор перемешивали при 0°C в течение 2 часов, затем концентрировали при пониженном давлении. Полученный остаток добавляли в воду и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с элюирующей системой В с получением указанного в заголовке соединения 8d (410 мг, выход: 55%).
Стадия 3
(9R,12R)-9-бензил-12-изобутил-2,2-диметил-4,7,10-триоксо-5-фенилэтил-3-окса-5,8,11-триазатридекан-13-овая кислота 8е
8d (410 мг, 0,65 ммоль) растворяли в 20 мл метанола, затем добавляли палладий на угле (60 мг, каталитическое количество). После завершения добавления реакционную систему продували водородом три раза и перемешивали в течение 12 часов при комнатной температуре. Реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 8е (313 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 4
(9R,12R,15R)-метил-9-бензил-15-(4-(((бензилокси)карбонил)амино)бутил)-12-изобутил-2,2-диметил-4,7,10,13-тетраоксо-5-фенилэтил-3-окса-5,8,11,14-тетраазагексадекан-16-оат 8f
Неочищенное соединение 8е (100 мг, 0.125 ммоль) и неочищенное соединение 8b (85 мг, 0,231 ммоль) растворяли в 5 мл N,N-диметилформамида, затем добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (105 мг, 0,277 ммоль) и N,N-диизопропилэтиламин (72 мг, 0,555 ммоль). После перемешивания в течение 2 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток добавляли в воду и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, последовательно промывали насыщенным раствором хлорида аммония (30 мл × 3), насыщенным раствором бикарбоната натрия (30 мл × 3) и насыщенным раствором хлорида натрия (30 мл × 3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения, которое использовали непосредственно на следующей стадии без очистки.
Стадия 5
(9R,12R,15R)-9-бензил-15-(4-(((бензилокси)карбонил)амино)бутил)-12-изобутил-2,2-диметил-4,7,10,13-тетраоксо-5-фенилэтил-3-окса-5,8,11,14-тетраазагексадекан-16-овая кислота 8g
Неочищенное соединение 8f (140 мг, 0,17 ммоль) растворяли в 7 мл смешанного растворителя тетрагидрофурана, метанола и воды (об/об/об = 3:3:1), затем добавляли моногидрат гидроксида лития (40 мг, 0,85 ммоль). После перемешивания в течение 0,5 часа при комнатной температуре реакционный раствор концентрировали при пониженном давлении для удаления растворителей метанола и тетрагидрофурана. Добавляли воду, затем по каплям добавляли 1 М соляную кислоту для доведения pH до 6. Реакционный раствор экстрагировали дихлорметаном (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 8g (220 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 6
Бензил-1-((9R,12R,15R)-9-бензил-15-(4-(((бензилокси)карбонил)амино)-бутил)-12-изобутил-2,2-диметил-4,7,10,13-тетраоксо-5-фенилэтил-3-окса-5,8,11,14-тетраазагексадекан-16-оил)-4-(((бензилокси)карбонил)амино)-пиперидин-4-карбоксилат 8h
Неочищенное соединение 8g (220 мг, 0,17 ммоль) и неочищенное соединение 1с (130 мг, 0,26 ммоль) растворяли в 5 мл N,N-диметилформамида, затем добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (100 мг, 0,26 ммоль) и N,N-диизопропилэтиламин (66 мг, 0,51 ммоль). После перемешивания в течение 3 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток добавляли в воду и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, последовательно промывали насыщенным раствором хлорида аммония (30 мл × 3), насыщенным раствором бикарбоната натрия (30 мл × 3) и насыщенным раствором хлорида натрия (30 мл × 3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением указанного в заголовке соединения 8h (170 мг, выход: 87%).
Стадия 7
1-((9R,12R,15R)-15-(4-аминобутил)-9-бензил-12-изобутил-2,2-диметил-4,7,10,13-тетраоксо-5-фенилэтил-3-окса-5,8,11,14-тетраазагексадекан-16-оил)-4-(карбоксиамино)пиперидин-4-карбоновая кислота 8i
8h (170 мг, 0,147 ммоль) растворяли в 5 мл метанола, затем добавляли палладий на угле (50 мг, каталитическое количество). После завершения добавления реакционную систему продували водородом три раза и перемешивали в течение 12 часов при комнатной температуре. Реакционный раствор фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 8i (140 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 8
4-амино-1-((2R,5R,8R)-2-(4-аминобутил)-8-бензил-5-изобутил-4,7,10-триоксо-14-фенил-3,6,9,12-тетраазатетрадекан-1-оил)пиперидин-4-карбоновая кислота 8
Неочищенное соединение 8i (140 мг, 0,167 ммоль) растворяли в 3 мл дихлорметана, затем добавляли 0,5 мл раствора 4М соляной кислоты в 1,4-диоксане. После перемешивания в течение 1 часа при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток растворяли в смешанном растворителе метанола и воды (об/об = 20:1), затем добавляли карбонат натрия, чтобы довести pH до более чем 8. Реакционный раствор концентрировали при пониженном давлении. К полученному остатку добавляли 10 мл дихлорметана, перемешивали в течение 10 минут и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 8 (19 мг, выход: 17,4%).
МС m/z (ESI): 694.4 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7,32-7,01 (m, 10Н), 4.68-4.66 (m, 1Н), 4.44-4.42 (m, 1Н), 3.75-3.65 (m, 6Н), 3,35 (s, 2Н), 3,32-3,28 (m, 6Н), 3,20-3,18 (m, 2Н), 3,00 (s, 2Н), 2.70-2,58 (m, 5Н), 2,10-1.98 (m, 3Н), 1,55-1,50 (m, 6Н), 0.95-0.93 (dd, 6Н).
Пример 9
(R)-N-((R)-6-амино-1-(4-(3-метилуреидо)пиперидин-1-ил)-1-оксогексан-2-ил)-4-метил-2-((R)-3-фенил-2-(2-(((R)-2-фенилпропил)амино)ацетамидо)-пропанамидо)пентанамид 9
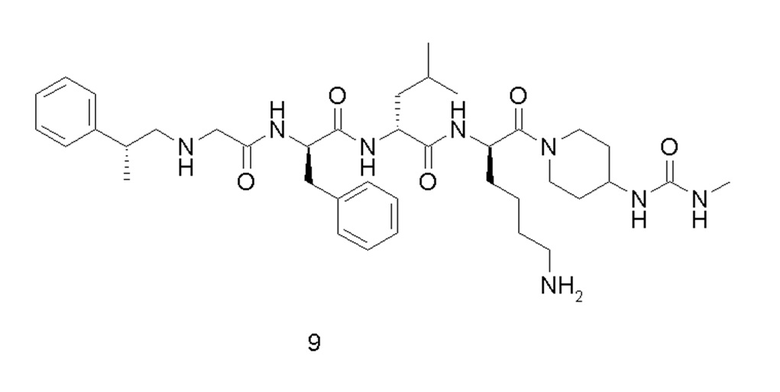
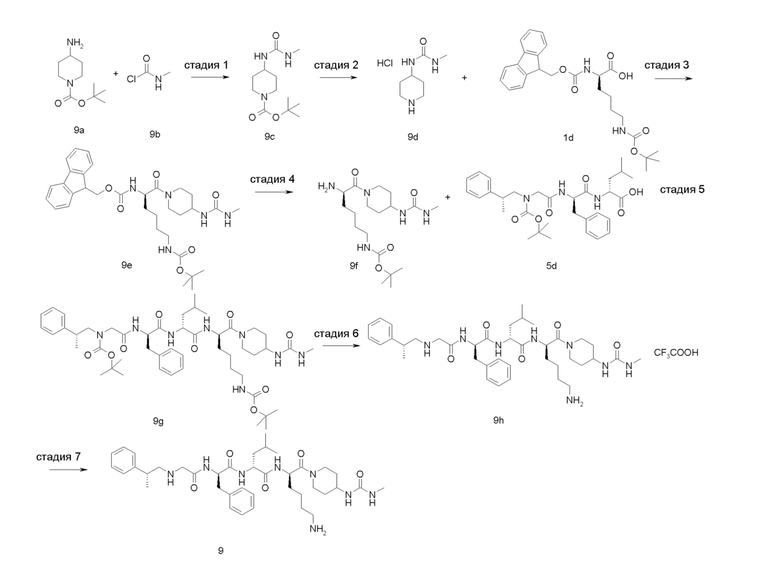
Стадия 1
трет-Бутил-4-(3-метилуреидо)пиперидин-1-карбоксилат 9с
9а (8,11 г, 40 ммоль, полученное способом, раскрытым в заявке на патент WO 2006115353), растворяли в 130 мл дихлорметана, затем добавляли N,N-диизопропилэтиламин (15,51 г, 120 ммоль). После охлаждения до 0°C к реакционному раствору добавляли 9b (3,74 г, 40 ммоль) и перемешивали в течение 2 часов при комнатной температуре. В реакционный раствор добавляли 200 мл насыщенного раствора бикарбоната натрия и экстрагировали дихлорметаном (200 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (200 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, чтобы получить указанное в заголовке неочищенное соединение 9с (9,3 г), которое использовали непосредственно на следующей стадии без очистки.
Стадия 2
1-метил-3-(пиперидин-4-ил)мочевины гидрохлорид 9d
Неочищенное соединение 9с (1 г, 4 ммоль) растворяли в 10 мл дихлорметана, затем добавляли 2 мл раствора 4М соляной кислоты в 1,4-диоксане. После перемешивания в течение 2 часов реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 9d (1 г, белое твердое вещество), которое использовали непосредственно на следующей стадии без очистки.
Стадия 3
(R)-(9H-флуорен-9-ил)метил трет-бутил-(6-(4-(3-метилуреидо)пиперидин-1-ил)-6-оксогексан-1,5-диил)дикарбамат 9е
Неочищенное соединение 9d (1 г, 5,16 ммоль) и 1d (2,42 г, 5,16 ммоль) растворяли в 20 мл N,N-диметилформамида, затем добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (2,94 г, 7,74 ммоль) и триэтиламин (1,03 г, 10,32 ммоль). После перемешивания в течение 4 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением указанного в заголовке соединения 9е (1 г, выход: 32%).
Стадия 4
(R)-трет-бутил-(5-амино-6-(4-(3-метилуреидо)пиперидин-1-ил)-6-оксогексил)карбамат 9f
9е (304 мг, 0,5 ммоль) растворяли в 2 мл N,N-диметилформамида, затем добавляли 6 мл триэтиламина. Реакционный раствор перемешивали в течение 12 часов, затем использовали непосредственно на следующей стадии без очистки.
Стадия 5
трет-Бутил-((10R,13R,16R)-16-бензил-13-изобутил-2,2-диметил-10-(4-(3-метилуреидо)пиперидин-1-карбонил)-4,12,15,18-тетраоксо-3-окса-5,11,14,17-тетраазанонадекан-19-ил)((R)-2-фенилпропил)карбамат 9g
Неочищенное соединение 9f (193 мг, 0,5 ммоль), неочищенное соединение 5d (277 мг, 0,5 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (380 мг, 1 ммоль) и N,N-диизопропилэтиламин (129 мг, 1 ммоль) растворяли в 30 мл N,N-диметилформамида и перемешивали в течение 12 часов при комнатной температуре. К реакционному раствору добавляли 10 мл насыщенного раствора лимонной кислоты и 20 мл воды и экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, последовательно промывали насыщенным раствором бикарбоната натрия (20 мл) и насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, чтобы получить неочищенное указанное в заголовке соединение 9g (500 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 6
(R)-N-((R)-6-амино-1-(4-(3-метилуреидо)пиперидин-1-ил)-1-оксогексан-2-ил)-4-метил-2-((R)-3-фенил-2-(2-(((R)-2-фенилпропил)амино)ацетамидо)-пропанамидо)пентанамида трифторацетат 9
Неочищенное соединение 9g (184 мг, 0,2 ммоль) растворяли в 10 мл дихлорметана, затем добавляли 2 мл раствора 4М соляной кислоты в 1,4-диоксане. После перемешивания в течение 12 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали высокоэффективной жидкостной хроматографией с получением указанного в заголовке соединения 9 (20 мг, выход: 14%).
МС m/z (ESI): 721.6 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8,37-8,25 (m, 1Н), 7,73-7.65 (m, 1Н), 7.41-7,37 (m, 2Н), 7,39-7,29 (m, 7Н), 7,18-7,16 (m, 2Н), 4,93-4,90 (m, 1Н), 4.83-4.82 (m, 1Н), 4,54-4,50 (m, 2Н), 4,00-3,92 (m, 1Н), 3.88-3.60 (m, 6Н), 3,15-3,08 (m, 5Н), 3,05-2,98 (m, 4Н), 2,70 (s, 3Н), 2,15-1.88 (m, 3Н), 1,79-1.61 (m, 15Н), 1,01-0,96 (m, 6Н).
Стадия 7
(R)-N-((R)-6-амино-1-(4-(3-метилуреидо)пиперидин-1-ил)-1-оксогексан-2-ил)-4-метил-2-((R)-3-фенил-2-(2-(((R)-2-фенилпропил)амино)ацетамидо)-пропанамидо)пентанамид 9
9h (20 мг, 0,024 ммоль) растворяли в 1 мл смешанного растворителя дихлорметан и метанол (об/об = 10:1), затем по каплям добавляли насыщенный водный раствор карбоната натрия для доведения pH до примерно 7. Реакционный раствор перемешивали в течение 30 минут при комнатной температуре и оставляли для отстаивания и разделения. Органическую фазу собирали, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 9 (17 мг, выход: 100%).
МС m/z (ESI): 721.6 [М+1]
Пример 10
(R)-N-((R)-6-амино-1-морфолино-1-оксогексан-2-ил)-2-((R)-2-(2-((2,3-дигидро-1H-инден-2-ил)амино)ацетамидо)-3-фенил пропанамидо)-4-метилпентанамид 10
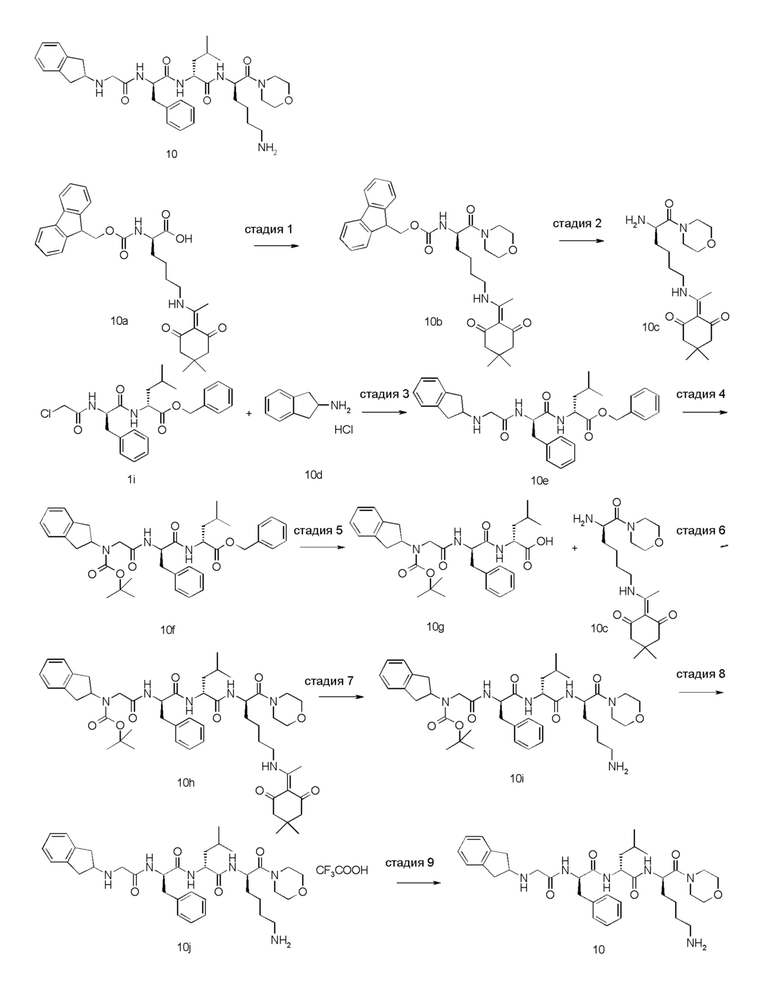
Стадия 1
(R)-(9H-флуорен-9-ил)метил(6-((1-(4,4-диметил-2,6-диоксо-циклогексилиден)этил)амино)-1-морфолино-1-оксогексан-2-ил)карбамат 10b
(R)-2-((((9H-флуорен-9-ил)метокси)карбонил)амино)-6-((1-(4,4-диметил-2,6-диоксоциклогексилиден)этил)амино)гексановую кислоту 10а (1,06 г, 2 ммоль, приобретенную у Accela ChemBio Inc.) и морфолин (200 мг, 2,4 ммоль) растворяли в 10 мл N,N-диметилформамида, затем добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (1,51 г, 4 ммоль) и триэтиламин (400 мг, 4 ммоль). После перемешивания в течение 3 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. К полученному остатку добавляли 5 мл насыщенного раствора лимонной кислоты и 50 мл воды и экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли, промывали водой (100 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого указанного в заголовке соединения 10b (1,3 г), которое использовали непосредственно на следующей стадии без очистки.
Стадия 2
(R)-2-(1-((5-амино-6-морфолино-6-оксогексил)амино)этилиден)-5,5-диметилциклогексан-1,3-дион 10с
Неочищенное соединение 10b (1,3 г, 2 ммоль) растворяли в 5 мл дихлорметана, затем добавляли 2 мл пиперидина. После перемешивания в течение 12 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с элюирующей системой А с получением указанного в заголовке соединения 10с (500 мг, выход: 75%).
Стадия 3
(R)-бензил-2-((R)-2-(2-((2,3-дигидро-1H-инден-2-ил)амино)ацетамидо)-3-фенилпропанамидо)-4-метил пентаноат 10е
Неочищенное соединение 1i (222 мг, 0,5 ммоль) и 2,3-дигидро-1H-инден-2-амин гидрохлорид 10d (127 мг, 0,4 ммоль, полученный известным способом, описанным в Tempahedron, 2005, 61 (28), 6801-6807), растворяли в 5 мл N,N-диметилформамида, затем добавляли йодид калия (415 мг, 2,5 ммоль) и карбонат калия (345, 2,5 ммоль). Реакционный раствор нагревали до 60°C и перемешивали в течение 12 часов, затем добавляли, концентрировали при пониженном давлении. Полученный остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением указанного в заголовке соединения 10е (300 мг, выход: 100%).
Стадия 4
(9R,12R)-бензил-9-бензил-5-(2,3-дигидро-1H-инден-2-ил)-12-изобутил-2,2-диметил-4,7,10-триоксо-3-окса-5,8,11-триазатридекан-13-оат 10f
10е (300 мг, 0,55 ммоль) растворяли в 10 мл дихлорметана, затем добавляли ди-трет-бутилдикарбонат (181 мг, 083 ммоль) и N,N-диизопропилэтиламин (0,3 мл, 1,65 ммоль). После перемешивания в течение 12 часов при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с элюирующей системой А с получением указанного в заголовке соединения 10f (170 мг, выход: 48%).
Стадия 5
(9R,12R)-9-бензил-5-(2,3-дигидро-1H-инден-2-ил)-12-изобутил-2,2-диметил-4,7,10-триоксо-3-окса-5,8,11-триазатридекан-13-овая кислота 10g
10f (170 мг, 0,26 ммоль) растворяли в 10 мл метанола, затем добавляли палладий на угле (50 мг, 10%). Реакционную систему продували водородом три раза и перемешивали в течение 12 часов при комнатной температуре. Реакционный раствор фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 10g (112 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 6
трет-Бутил-((4R,7R,10R)-4-бензил-16-(4,4-диметил-2,6-диоксоциклогексилиден)-7-изобутил-10-(морфолин-4-карбонил)-2,5,8-триоксо-3,6,9,15-тетраазагептадецил)(2,3-дигидро-1H-инден-2-ил)карбамат 10h
Неочищенное соединение 10g (112 мг, 0,2 ммоль), 10с (78 мг, 0,2 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (114 мг, 0,3 ммоль) и N,N-диизопропилэтиламин (0,1 мл, 0,6 ммоль) растворяли в 15 мл N,N-диметилформамида и перемешивали в течение 2 часов при комнатной температуре. Реакционный раствор концентрировали при пониженном давлении. К полученному остатку добавляли 100 мл этилацетата, последовательно промывали водой (50 мл × 3) и насыщенным раствором хлорида аммония (50 мл × 3), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, чтобы получить неочищенное указанное в заголовке соединение 10h (183 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 7
трет-Бутил-(2-(((R)-1-(((R)-1-(((R)-6-амино-1-морфолино-1-оксогексан-2-ил)амино)-4-метил-1-оксопентан-2-ил)амино)-1-оксо-3-фенилпропан-2-ил)амино)-2-оксоэтил)(2,3-дигидро-1H-инден-2-ил)карбамат 10i
Неочищенное соединение 10h (183 мг, 0,2 ммоль) растворяли в 10 мл метанола, затем добавляли 0,5 мл гидразингидрата. После перемешивания при комнатной температуре в течение 1 часа реакционный раствор концентрировали при пониженном давлении, чтобы получить неочищенное указанное в заголовке соединение 10i (150 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 8
(R)-N-((R)-6-амино-1-морфолино-1-оксогексан-2-ил)-2-((R)-2-(2-((2,3-дигидро-1H-инден-2-ил)амино)ацетамидо)-3-фенилпропанамидо)-4-метилпентанамид трифторацетат 10j
Неочищенное соединение 10i (150 мг, 1,31 ммоль) растворяли в 5 мл дихлорметана, затем добавляли 1 мл трифторуксусной кислоты. После перемешивания в течение 1 часа при комнатной температуре реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали высокоэффективной жидкостной хроматографией с получением указанного в заголовке соединения 10j (5 мг, выход: 4%).
МС m/z (ESI): 649.4 [М+1]
1Н ЯМР (400 МГц, ЦМСО-d6) δ 8,36 (d, 1Н), 7.88 (d, 2Н), 7,21-7,33 (m, 9Н), 4.61-4.64 (m, 1Н), 4,36-4.47 (m, 1Н), 4,02-4,15 (m, 2Н), 3.60-3,70 (m, 3Н), 3.45-3,59 (m, 5Н), 3,35-3.44 (m, 3Н), 2,90-3,25 (m, 9Н), 1,55-1.80 (m, 6Н), 1,30-1.45 (m, 4Н), 0,98 (d, 3Н), 0,92 (d, 3Н).
Стадия 9
(R)-N-((R)-6-амино-1-морфолино-1-оксогексан-2-ил)-2-((R)-2-(2-((2,3-дигидро-1H-инден-2-ил)амино)ацетамидо)-3-фенил пропанамидо)-4-метил пентанамид 10
10j (5 мг, 0,0057 ммоль) растворяли в 2 мл смешанного растворителя дихлорметана и метанола (об/об = 10:1), затем добавляли насыщенный карбонат натрия по каплям для доведения pH до примерно 7. Реакционный раствор перемешивали в течение 30 минут при комнатной температуре и оставляли отстаиваться и разделяться. Органическую фазу собирали, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 10 (4 мг, выход: 100%).
МС m/z (ESI): 649.4 [М+1]
БИОЛОГИЧЕСКИЕ ИСПЫТАНИЯ
Настоящее изобретение будет дополнительно описано со ссылкой на следующие примеры испытаний, но эти примеры не должны рассматриваться как ограничивающие объем изобретения.
Тестовый пример 1
1. Цель эксперимента
Целью этого эксперимента является определение агонистического действия соединений согласно настоящему изобретению на рецепторы KOR (h-KOR) человека и оценка активности соединений in vitro в соответствии со значениями ЕС50.
2. Тест на активность h-KOR
2.1. Цель эксперимента
Соединения согласно настоящему изобретению могут активировать рецептор h-KOR, тем самым снижая внутриклеточные уровни цАМФ. Второй мессенджер цАМФ входит в ядро и связывается с CRE ДНК, тем самым инициируя экспрессию нисходящей люциферазы. Люцифераза реагирует с субстратом, испуская флуоресценцию, и измеренные сигналы флуоресценции отражают агонистическую активность соединений.
2.2. Метод эксперимента
Активность соединений тестового примера на агонизацию h-KOR и влияние на снижение уровней цАМФ тестировали следующим способом.
2.1.1. Материалы и приборы эксперимента
2.2.2. Методика эксперимента
1) Получение моноклональных клеточных линий HEK293/KOR/CRE
KOR/pcDNA3,1 (+) и CRE/pGL4,29 трансфецировали в клетки HEK293. G418, гигромицин добавляли в культуральную среду и моноклональные клеточные линии HEK293/KOR/CRE подвергали скринингу в 96-луночном планшете для культивирования клеток.
2) Агонистический эффект иллюстративных соединений на h-KOR
Моноклональные клетки HEK293/h-KOR/CRE культивировали в среде DMEM с высоким содержанием глюкозы (10% FBS, 1 мг/мл G418, 200 мкг/мл гигромицина, равномерно смешанные) и пересеивали каждые 3 дня. В день эксперимента готовили клеточную суспензию со свежей клеточной средой, добавляли в 96-луночный планшет (BD, №356692) 20000 клеток/лунку и инкубировали в 5% CO2 при 37°C. На второй день соединение растворяли в чистом ДМСО в концентрации 20 мМ, затем смешивали с ДМСО до первой концентрации 200 нМ и разбавляли в 3-кратном градиенте концентрации до 8 концентраций. В холостые и контрольные лунки добавляли 90 мкл ДМСО. Раствор соединения разбавляли в 20 раз средой DMEM с высоким содержанием глюкозы (SH30243,01B, Hyclone), содержащей 10 мкМ форсколина. Планшеты для культивирования клеток, инокулированные в первый день, извлекали и в каждую лунку добавляли 10 мкл разбавленного лекарства или контроля (0,5% ДМСО). Планшет осторожно встряхивали и помещали при 37°C на 4 часа. В 96-луночный планшет для культивирования клеток в каждую лунку добавляли 100 мкл раствора для анализа люциферазы (Promega, # Е6110). Планшет помещали на 5 минут при комнатной температуре. Значение хемилюминесценции измеряли с использованием Victor 3.0. Значения ЕС50 для соединений рассчитывали с использованием программного обеспечения Graphpad Prism на основе каждой концентрации соединения и соответствующего значения сигнала.
2.3. Результаты испытаний
Активность соединений согласно настоящему изобретению на агонизацию h-KOR и влияние на снижение уровней цАМФ определяли с помощью вышеописанного теста, значения ЕС50 показаны в таблице 1.1.
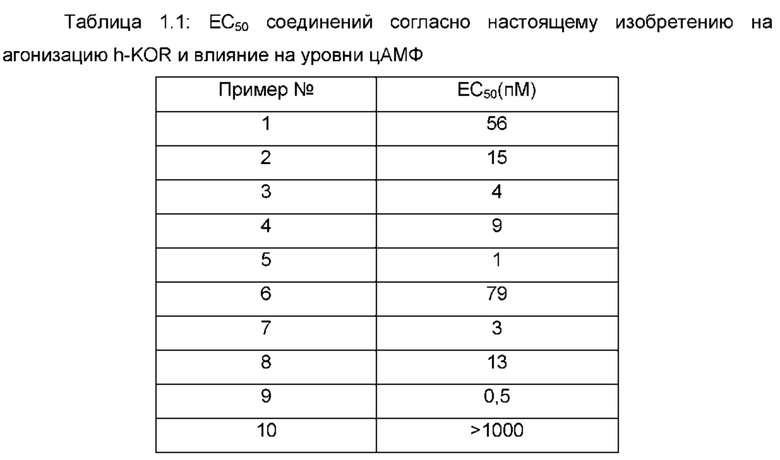
Вывод. Соединения согласно настоящему изобретению оказывают значительное агонистическое действие на рецептор h-KOR. В частности, когда заместитель в аминогруппе глицина представляет собой замещенную или незамещенную этиленовую группу, соединение оказывает неожиданный эффект.
Оценка фармакокинетики
Тестовый пример 2. Фармакокинетический анализ соединений примеров 2, 5 и 8 согласно настоящему изобретению на крысах
1. Краткое описание
В качестве подопытных животных использовали крыс. Концентрацию лекарства в плазме в разные моменты времени определяли методом ЖХ/МС/МС после внутривенного введения крысам соединений примеров 2, 5 и 8. Фармакокинетическое поведение соединений согласно настоящему изобретению изучали и оценивали на SD крысах.
2. Протокол
2.1. Тестовые соединения
Соединения примеров 2, 5 и 8
2.2. Тестовые животные
12 крыс Sprague-Dawley (SD), половина самцов и половина самок, были приобретены у SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., СО, с лицензией №: SCXK (Шанхай) 2008-0016.
2.3. Приготовление тестовых соединений
Соответствующее количество тестовых соединений взвешивали и последовательно добавляли 5% 5%ДМСО + 5% ПЭГ400 + 90% нормального физиологического раствора.
2.4. Введение
После ночного голодания 12 крыс SD, половина самцов и половина самок, разделяли на 3 группы поровну, и им вводили дозу внутривенно 5 мл/кг.
3. Процесс
В венозной группе кровь (0,2 мл) брали из орбитального синуса до введения и через 0,25, 0,5, 1,0, 2,0, 4,0, 8,0, 11,0 и 24,0 часа после введения. Образцы хранили в гепаринизированных пробирках и центрифугировали в течение 10 минут при 3500 об/мин для отделения плазмы крови. Образцы плазмы хранили при -20°C.
Концентрацию тестовых соединений в плазме крыс SD после внутривенного введения определяли методом ЖХ-МС/МС.
4. Результаты фармакокинетических показателей у SD крыс
Фармакокинетические параметры соединений примеров 2, 5 и 8 согласно настоящему изобретению приведены ниже.
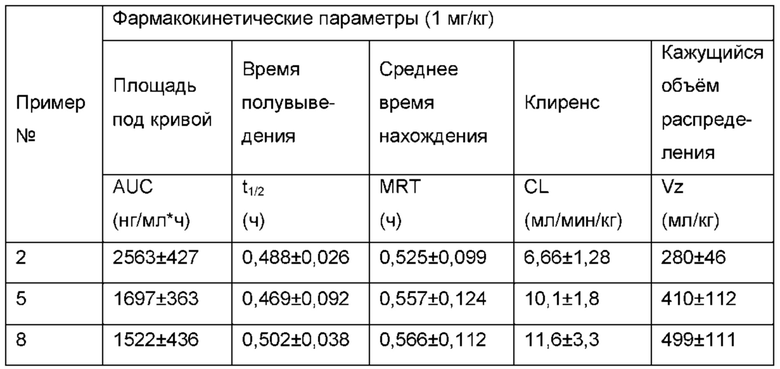
Вывод: соединения согласно настоящему изобретению обладают хорошими фармакокинетическими свойствами у крыс.
Тестовый пример 3. Фармакокинетический анализ соединения примера 5 согласно настоящему изобретению на собаках
1. Краткое описание
В качестве подопытных животных использовали собак породы бигль. Концентрацию лекарства в плазме в разные моменты времени определяли методом ЖХ/МС/МС после внутривенного введения собакам породы бигль соединения примера 5. Фармакокинетическое поведение соединений согласно настоящему изобретению изучали и оценивали на собаках породы бигль.
2. Протокол
2.1. Тестовое соединение
Соединение примера 5
2.2. Тестовые животные
3 собаки породы бигль в одной группе, самцы, были приобретены у компании Medicilon Pharmaceutical Technology (Shanghai) Co., Ltd.
2.3. Приготовление тестового соединения
Соответствующее количество тестового соединения взвешивали и добавляли со 100% физиологическим раствором.
2.4. Введение
После ночного голодания 3 собакам породы бигль в одной группе, самцам, вводили внутривенно дозу 2 мл/кг.
3. Процесс
В венозной группе кровь (1 мл) брали из яремной вены до введения и через 5 минут, 15 минут, 0,5, 1,0, 2,0, 4,0, 8,0, 12,0 и 24,0 часа после введения. Образцы хранили в гепаринизированных пробирках и центрифугировали в течение 10 минут при 3500 об/мин для отделения плазмы крови. Образцы плазмы хранили при -80°C.
Концентрацию тестовых соединений в плазме собак породы бигль после внутривенного введения определяли методом ЖХ-МС/МС.
4. Результаты фармакокинетических показателей у собак породы бигль
Фармакокинетические параметры соединения примера 5 согласно настоящему изобретению приведены ниже.
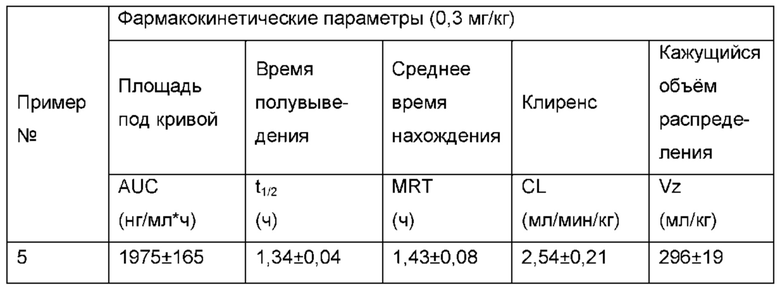
Вывод: соединение согласно настоящему изобретению обладает хорошими фармакокинетическими свойствами у собак породы бигль.
Тестовый пример 4. Экспериментальный отчет по агонисту KOR при лечении каррагинан-индуцированной воспалительной боли у крыс
1. Цель эксперимента
Модель каррагинан-индуцированной воспалительной боли у крыс была разработана для оценки терапевтического эффекта агонистов KOR на воспалительную боль у крыс.
2. Метод и материалы эксперимента
2.1. Подопытные животные и условия кормления
Самцов крыс Wistar приобретали у Shanghai Slac Laboratory Animal Co., Ltd. (Шанхай, Китай, сертификат №2015000513408, лицензия № SCXK (Shanghai) 2012-0002). Крысы весили 150-180 г, их содержали по 5 на клетку в условиях цикла 12/12 часов свет/темнота при постоянной температуре 23±1°C, влажности 50~60% и при свободном доступе к пище и воде. После приобретения животных адаптировали к этому состоянию в течение 7 дней до начала эксперимента.
2.2. Тестовое соединение
Соединение Примера 5;
λ-Каррагинан, партия № BCBP8978V, приобретенный у Sigma.
0,9% раствор хлорида натрия (500 мл, 4,5 г)
1% λ-каррагинана помещали в физиологический раствор, выдерживали в течение ночи для образования желеобразной суспензии.
Дозу соединения рассчитывали по базовым условиям.
2.3. Процедура и метод эксперимента
2.3.1. Группировка животных
После адаптивного кормления крыс группировали следующим образом:
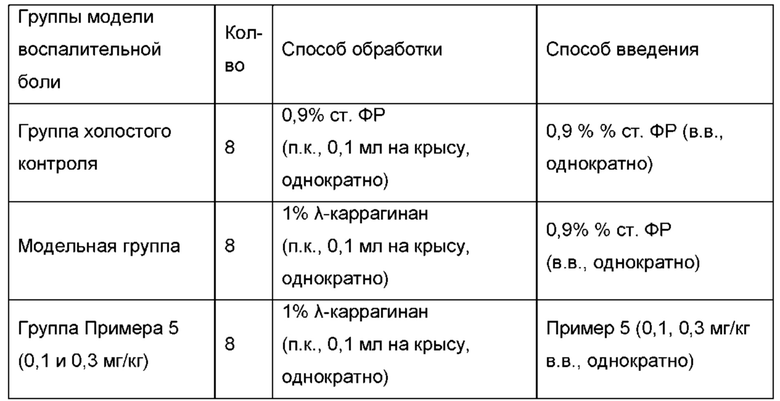
Примечание: ст. ФР: стандартный физиологический раствор, используемый для приготовления раствора каррагинана; в.в.: внутривенное введение; п. к.: подкожное введение.
2.3.2. Метод эксперимента[1] [2]:
Метод эксперимента модифицировали в соответствии с методом Документа 1 (Kazunari Nakao et al.). Перед экспериментом по воспалительной боли крысы были в основном разделены на следующие группы в зависимости от массы тела: группа холостого контроля, модельная группа, группа Примера 5-0,1 мг/кг и группа Примера 5-0,3 мг/кг. В каждой группе было по 8 крыс. Модель воспалительной боли была сделана на лапках крыс Wistar, которым подкожно вводили 1%-ный каррагенан (100 мкл). Через 4 часа крыс подвергали тесту подошвенной чувствительности для оценки порога механической боли. Однократное введение препарата в хвостовую вену (1 мл/кг) проводили за 30 минут до детектирования, контрольной группе и модельной группе вводили соответствующие растворители.
Примечание: Документы 1, CJ-023,423, a Novel, Potent and Selective Prostaglandin KOR Receptor Antagonist with Anti-hyperalgesic Properties[J]. The Journal of Pharmacology and Experimental Therapeutics, 2007, 322(2):686-694.
2.4. Прибор, использованный в эксперименте
Электронный прибор измерения тактильных ощущений: UGO BASILE, тип 38450.
2.5. Представление данных и статистическая обработка
Экспериментальные данные выражали как среднее ± стандартное отклонение (S.D.). Статистические сравнения выполняли с использованием программного теста Excel. Данные между модельной группой и контрольной группой анализировали и сравнивали для определения существенной статистической значимости. *Р<0,05 указывает, что существует значительная разница между модельной группой и контрольной группой, **Р<0,01 указывает, что существует высокая значительная разница между модельной группой и контрольной группой, #Р<0,05 указывает, что существует значительная разница между модельной группой и контрольной группой, ##Р<0,01 указывает, что существует высокая значительная разница между модельной группой и группой введения.
3. Результаты. Влияние соединения согласно настоящему изобретению на каррагинан-индуцированную воспалительную боль у крыс.
Результаты экспериментов на крысах показали, что порог чувствительности в контрольной группе составлял около 20 г, а порог чувствительности в модельной группе составлял 7,6 г. По сравнению с группой холостого контроля, порог чувствительности в модельной группе был значительно снижен (Р<0,01). По сравнению с модельной группой все препараты могли значительно увеличить порог чувствительности у крыс с воспалением (Р<0,01). Порог чувствительности в группе Примера 5 (0,1 мг/кг) и Примера 5 (0,3 мг/кг) составил 13,7 г и 23,2 г соответственно. Увеличение составило 79,5% и 204,5% соответственно, со значительной зависимостью от дозы (см. фиг. 1).
4. Обсуждение
λ-Каррагинан представляет собой коллоидное вещество, извлекаемое из водных растений и обладающее аллергическим стимулирующим действием. Один только каррагинан может вызвать воспаление и боль. В этом эксперименте модель каррагинановой воспалительной боли была разработана для наблюдения за изменениями порога чувствительности после введения агониста KOR крысам и для оценки анальгетического эффекта препарата на подострую воспалительную боль и ее интенсивность. В эксперименте использовали электронный измерительный прибор для измерения реакции крысы на чувствительность. Электронный прибор измерения тактильных ощущений (e-VF) был разработан с использованием оригинальной конструкции Ugo Basile для оценки аллергии и аллодинии у крыс и мышей. Этот прибор автоматически регистрирует время стимуляции и интенсивность стимуляции животных. Уникальная конструкция призмы позволяет легко наблюдать за подошвой испытуемых животных во время эксперимента. Во время детектирования прибор может почувствовать, как тестируемое животное отводит тестовый коготь, или это можно определить по педальному контроллеру. Более сфокусированное позиционирование больше подходит для измерения локальной боли и нейропатической боли.
5. Вывод
Тестируемое соединение может уменьшать воспалительную боль у крыс дозозависимым образом.
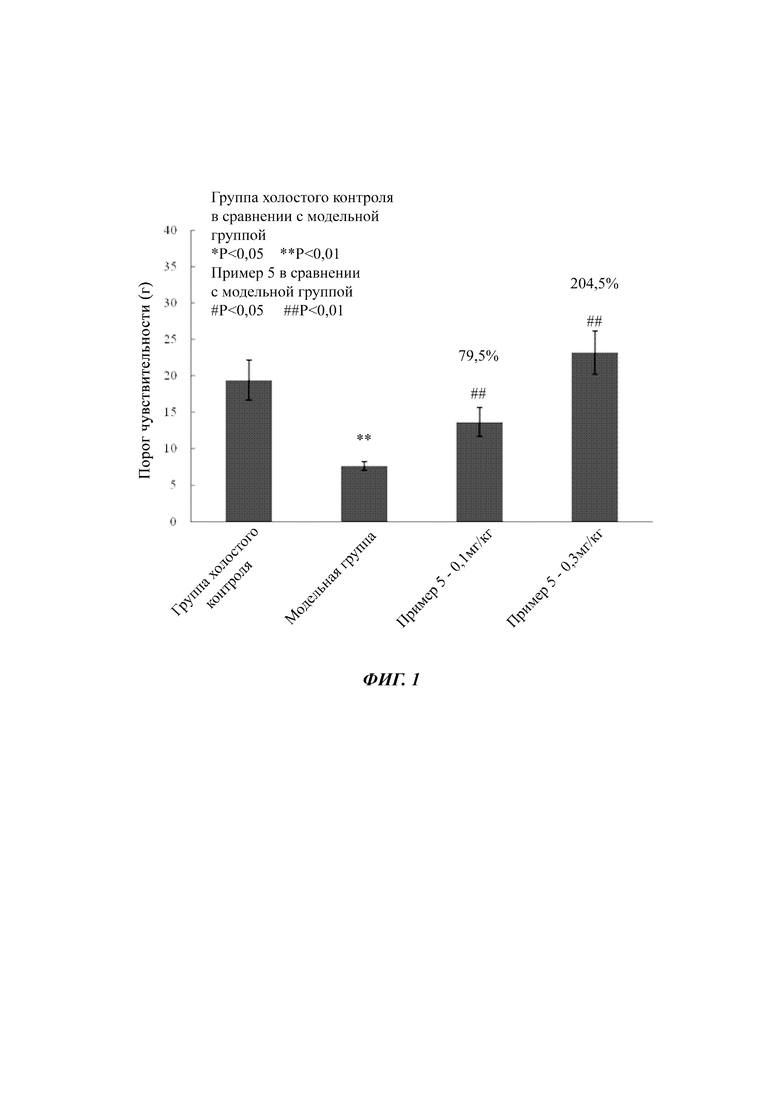
Изобретение относится к производному фенилпропанамида формулы (I), к способу его получения и к его применению в качестве агониста κ-опиоидного рецептора (KOR), и к его применению для получения лекарственного средства для лечения и/или профилактики боли и связанных с болью заболеваний. 8 н. и 6 з.п. ф-лы, 1 табл., 10 пр., 1 ил.
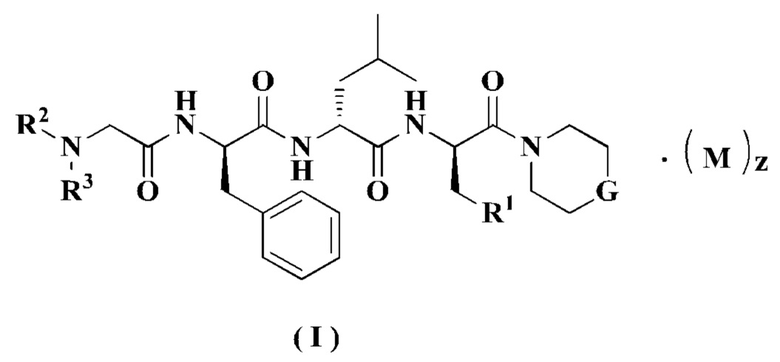
1. Соединение формулы (III-А):
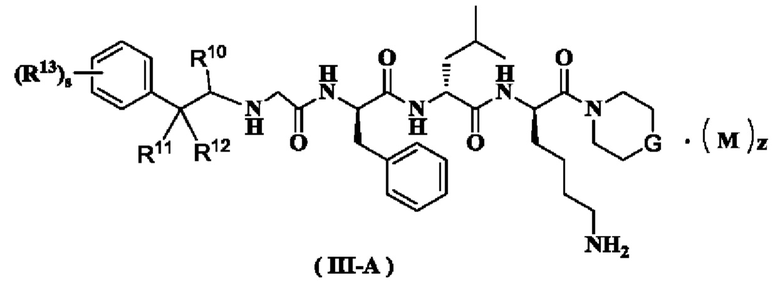
или энантиомер, диастереомер или их смесь, или его фармацевтически приемлемая соль,
где:
G представляет собой О или CR5R6;
R10 представляет собой водород;
R11 и R12 являются одинаковыми или различными, и каждый независимо выбран из группы, состоящей из водорода и С1-6алкила;
или R11 и R12, взятые вместе, образуют С3-6циклоалкил;
R13 представляет собой водород;
s равно 0;
каждый из R5 и R6 независимо выбран из группы, состоящей из водорода, амино, -C(O)OR7 и -NHC(O)NR8R9;
R7 представляет собой водород;
каждый из R8 и R9 независимо выбран из группы, состоящей из водорода и С1-6алкила;
М представляет собой трифторуксусную кислоту; и
z равен 0 или 1.
2. Соединение формулы (III-А) по п. 1, где G представляет собой CR5R6, где R5 и R6 являются такими, как определено в п. 1.
3. Соединение формулы (III-А) по любому из пп. 1, 2, представляющее собой соединение формулы (IV-A):
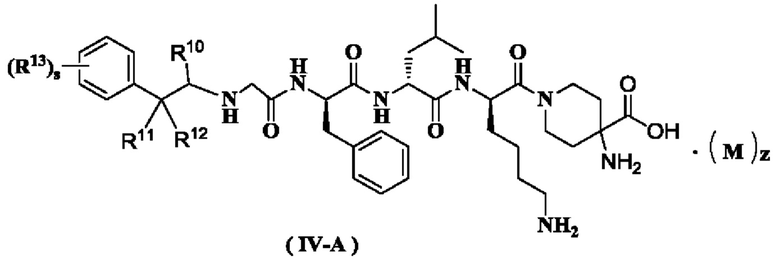
или энантиомер, диастереомер или их смесь, или его фармацевтически приемлемую соль,
где
R10-R13, М, z и s являются такими, как определено в п. 1.
4. Соединение формулы (III-А) по любому из пп. 1, 2, представляющее собой соединение формулы (IV-B):
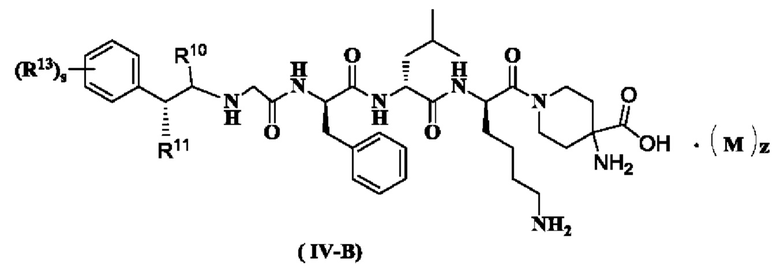
или энантиомер, диастереомер или их смесь, или его фармацевтически приемлемую соль,
где
R10-R11, R13, М, z и s являются такими, как определено в п. 1.
5. Соединение, выбранное из группы, состоящей из:
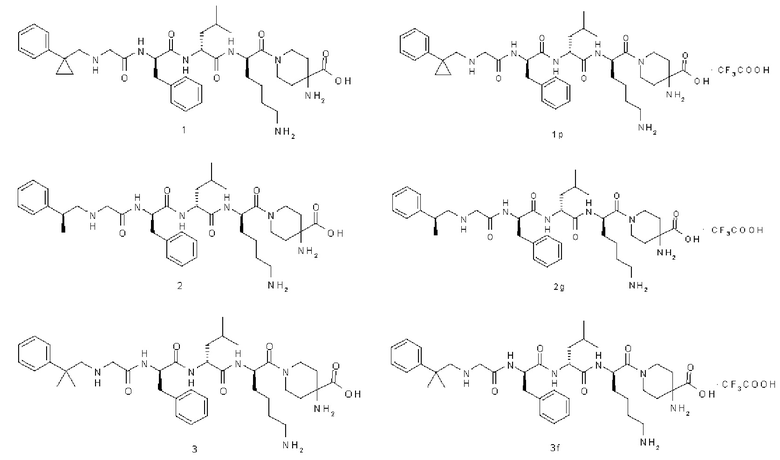
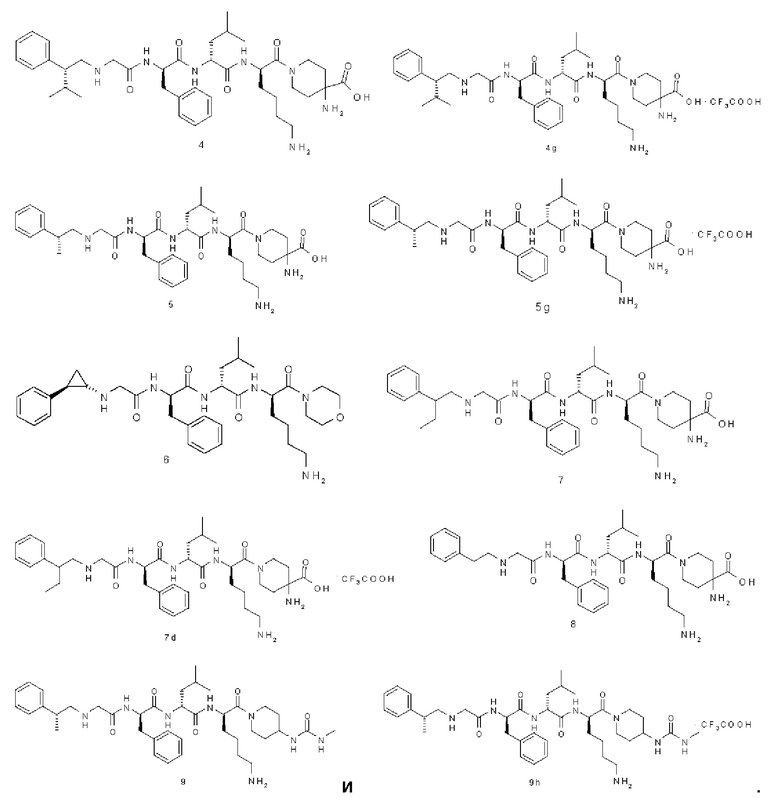
6. Соединение формулы (III-5):
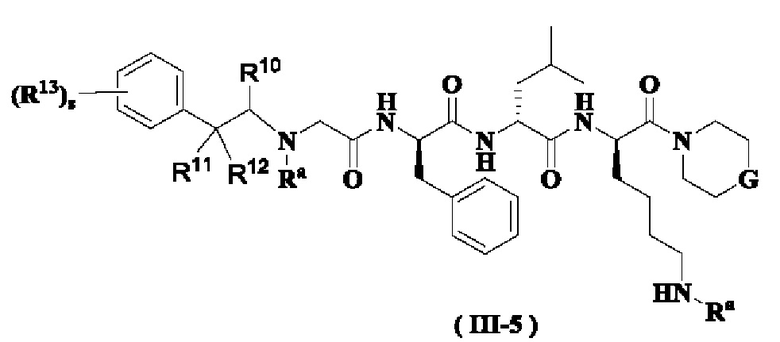
или энантиомер, диастереомер или его фармацевтически приемлемая соль,
где:
Ra представляет собой трет-бутоксикарбонил или бензилоксикарбонил; и
R10-R13, G и s являются такими, как определено в п. 1.
7. Способ получения соединения формулы (III-A) по п. 1, включающий стадию:
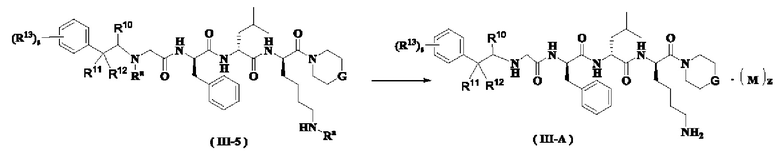
удаления защитной группы Ra из соединения формулы (III-5) в кислых условиях с получением соединения формулы (III-А);
где:
R10-R13, М, G, z и s являются такими, как определено в п. 1, и Ra является таким, как определено в п. 6.
8. Фармацевтическая композиция, обладающая агонистической активностью в отношении κ-опиоидного рецептора, содержащая терапевтически эффективное количество соединения формулы (III-A) по любому из пп. 1-5 и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
9. Применение соединения формулы (III-A) по любому из пп. 1-5, или фармацевтической композиции по п. 8 для получения лекарственного средства для профилактики и/или лечения заболевания, опосредованного агонистом κ-опиоидного рецептора и связанного с агонистом κ-опиоидного рецептора.
10. Применение по п. 9, где заболевание, опосредованное агонистом κ-опиоидного рецептора и связанное с агонистом κ-опиоидного рецептора, выбрано из группы, состоящей из боли, воспаления, зуда, отека, гипонатриемии, гипокалиемии, кишечной непроходимости, кашля и глаукомы.
11. Применение по п. 9, где заболевание, опосредованное агонистом κ-опиоидного рецептора и связанное с агонистом κ-опиоидного рецептора, представляет собой боль.
12. Применение соединения формулы (III-A) по любому из пп. 1-5 или фармацевтической композиции по п. 8 для получения лекарственного средства для профилактики и/или лечения боли и связанных с болью заболеваний.
13. Применение по п. 10 или 12, где боль выбрана из группы, состоящей из невропатической боли, боли в туловище, висцеральной боли, кожной боли, боли в суставах, боли при мочекаменной болезни, спазма матки, дисменореи, эндометриоза, диспепсии, боли после хирургической операции, боли после медикаментозного лечения, боли в глазах, боли при отите, боли при скоротечном раке и боли, связанной с нарушениями желудочно-кишечного тракта.
14. Применение соединения формулы (III-A) по любому из пп. 1-5 или фармацевтической композиции по п. 8 для получения лекарственного средства для агонизации κ-опиоидного рецептора.
| СИНТЕТИЧЕСКИЕ ПЕПТИДНЫЕ АМИДЫ | 2007 |
|
RU2500685C2 |
| WO 2013184794 A2, 12.12.2013 | |||
| WO 2010105636 A1, 23.09.2010. | |||

