Область техники, к которой относится изобретение
Настоящее изобретение относится к трициклическим гетероциклическим соединениям, композициям, включающим по меньшей мере одно трициклическое гетероциклическое соединение и способам применения трициклических гетероциклических соединений для лечения или профилактики ВИЧ-инфекции у субъекта.
Предпосылки создания изобретения
Ретровирус, называемый вирусом иммунодефицита человека (ВИЧ), особенно штаммы, известные как вирус ВИЧ типа-1 (ВИЧ-1) и типа-2 (ВИЧ-2), является этиологическим фактором комплексного заболевания, которое включает прогрессирующую деструкцию иммунной системы (синдром приобретенного иммунодефицита; СПИД) и дегенерацию центральной и периферической нервной системы. Типичным признаком репликации ретровирусов является встраивание, осуществляемое посредством вирусно-кодируемой интегразы, +провирусной ДНК в геном клетки-хозяина, необходимая стадия в репликации ВИЧ в человеческих T-лимфоидных и моноцитоидных клетках. Считается, что интеграция опосредуется интегразой в три стадии: сборка стабильного нуклеопротеинового комплекса с вирусными последовательностями ДНК; отщепление двух нуклеотидов от 3'-концов линейной провирусной ДНК и ковалентное соединение сокращенных 3' OH-концов провирусной ДНК на ступенчатом разрыве, сделанном на сайте-мишени хозяина. Четвертая стадия в этом процессе, репарация образовавшегося гэпа, может быть осуществлена посредством клеточных ферментов.
Секвенирование нуклеиновых кислот ВИЧ показывает присутствие pol гена в одной открытой рамке считывания [Ratner, L. et al., Nature, 313, 277(1985)]. Гомология аминокислотных последовательностей подтверждает, что последовательность pol кодирует обратную транскриптазу, интегразу и протеазу ВИЧ [Toh, H. et al., EMBO J. 4, 1267 (1985); Power, M.D. et al., Science, 231, 1567 (1986); Pearl, L.H. et al., Nature, 329, 351 (1987)]. Было показано, что все три фермента являются существенными для репликации ВИЧ.
Известно, что некоторые противовирусные соединения, которые действуют как ингибиторы репликации ВИЧ, являются эффективными средствами для лечения СПИДа и подобных заболеваний, в том числе ингибиторы обратной транскриптазы, такие как азидотимидин (AZT) и эфавиренц, и ингибиторы протеазы, такие как индинавир и нелфинавир. Соединения по настоящему изобретению являются ингибиторами интегразы ВИЧ и ингибиторами репликации ВИЧ.
Следующие ссылочные документы могут представлять интерес в качестве известного уровня техники:
Международные публикации №№ WO 11/045330 и WO 11/121105 раскрывают макроциклические соединения, обладающие ингибиторной активностью в отношении интегразы ВИЧ.
Kinzel et al., Tet. Letters 2007, 48(37): pp. 6552-6555 раскрывает синтез тетрагидропиридопиримидонов в качестве каркаса для ингибиторов интегразы ВИЧ-1.
Ferrara et al., Tet. Letters 2007, 48(37), pp. 8379-8382 раскрывает синтез гексагидропиримидо[1,2-a]азепин-2-карбоксамидного производного, полезного в качестве ингибитора интегразы ВИЧ.
Muraglia et al., J. Med. Chem. 2008, 51: 861-874 раскрывает структуру и синтез бициклических пиримидинонов в качестве сильных и перорально биодоступных ингибиторов интегразы ВИЧ-1.
US2004/229909 раскрывает некоторые соединения, обладающие ингибиторной активностью в отношении интегразы.
US 7232819 и US 2007/0083045 раскрывают некоторые 5,6-дигидроксипиримидин-4-карбоксамиды в качестве ингибиторов интегразы ВИЧ.
US 7169780, US 7217713 и US 2007/0123524 раскрывают некоторые N-замещенные 5-гидрокси-6-оксо-1,6-дигидропиримидин-4-карбоксамиды в качестве ингибиторов интегразы ВИЧ.
US 7279487 раскрывает некоторые гидроксинафтиридинонкарбоксамиды, которые могут быть полезны в качестве ингибиторов интегразы ВИЧ.
US 7135467 и US 7037908 раскрывают некоторые пиримидинкарбоксамиды, которые могут быть полезны в качестве ингибиторов интегразы ВИЧ.
US 7211572 раскрывает некоторые азот-содержащие соединения с конденсированными кольцами, которые являются ингибиторами интегразы ВИЧ.
US 7414045 раскрывает некоторые тетрагидро-4H-пиридо[1,2-a]пиримидинкарбоксамиды, гексагидропиримидо[1,2-a]азепинкарбоксамиды и родственные соединения, которые могут быть полезны в качестве ингибиторов интегразы ВИЧ.
US 8129385 раскрывает некоторые гексагидро-2H-пиридо[1',2':4,5]пиразино[2,1-b][1,3]оксазин-9-карбоксамиды и родственные соединения, которые могут быть полезны в качестве ингибиторов интегразы ВИЧ.
WO 2006/103399 раскрывает некоторые тетрагидро-4H-пиримидооксазепинкарбоксамиды, тетрагидропиразинопиримидинкарбоксамиды, гексагидропиримидодиазепинкарбоксамиды и родственные соединения, которые могут быть полезны в качестве ингибиторов интегразы ВИЧ.
US 2007/0142635 раскрывает способы для получения гексагидропиримидо[1,2-a]азепин-2-карбоксилатов и родственных соединений.
US 2007/0149556 раскрывает некоторые гидроксипиримидиноновые производные, обладающие ингибиторной активностью в отношении интегразы ВИЧ.
Различные пиримидиноновые соединения, полезные в качестве ингибиторов интегразы ВИЧ, также раскрыты в US 7115601, US 7157447, US 7173022, US 7176196, US 7192948, US 7273859 и US 7419969.
US 2007/0111984 раскрывает ряд бициклических пиримидиноновых соединений, полезных в качестве ингибиторов интегразы ВИЧ.
US 2006/0276466, US 2007/0049606, US 2007/0111985, US 2007/0112190, US 2007/0281917, US 2008/0004265 каждый раскрывает ряд бициклических пиримидиноновых соединений, полезных в качестве ингибиторов интегразы ВИЧ.
US7462608 и US7649015 каждый раскрывает фосфат- и фосфонат-замещенные гетероциклы, полезные в качестве nNRTI ингибиторов ВИЧ и ингибиторов протеазы ВИЧ, соответственно.
Сущность изобретения
В одном аспекте настоящее изобретение обеспечивает соединения формулы (I):
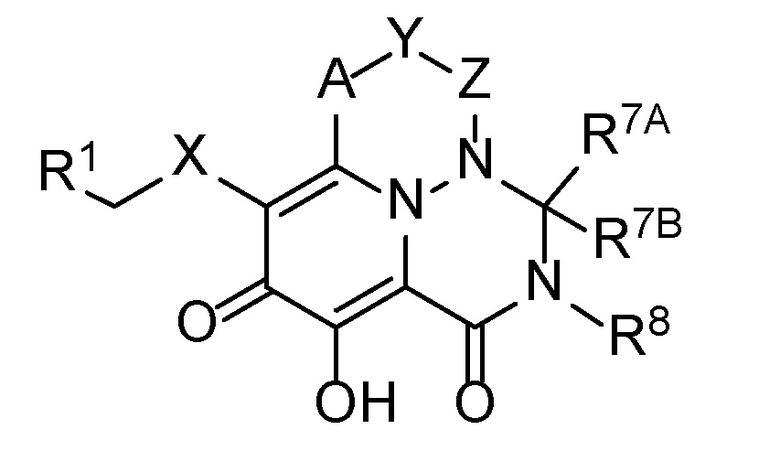
(I)
или их фармацевтически приемлемые соли,
где:
A представляет собой -C(R2)-;
X представляет собой 5- или 6-членный моноциклический гетероарил или -N(R5)C(O)-;
Y выбран из -O-, -N(R5)- или -CH(R3)-, или -A-Y- представляет собой -C(R2)=CH-;
Z представляет собой -C(O)-, -CH(R4)- или связь, таким образом, что: (i) когда Y представляет собой -O- или -N(R5)-, тогда Z представляет собой связь, (b) когда Y представляет собой -CH(R3)-, тогда Z представляет собой связь или -CH(R4), и (iii) когда -A-Y- представляет собой -C(R2)=CH-, тогда Z представляет собой связь;
R1 представляет собой фенильную группу, которая необязательно замещена 1-3 группами, каждая из которых независимо выбрана из C1-C6 алкила, галогена, -O-(C1-C6 алкил), C1-C6 галогеналкила, -O-(C1-C6 галогеналкил), -CN, -NO2, -N(R4)2, -C(O)OR6, -C(O)N(R4)2 и -NHC(O)R6;
R2 выбран из H, C1-C6 алкила, -O-(C1-C6 алкил) и -N(R4)2;
R3 выбран из H, C1-C6 алкила и -O-(C1-C6 алкил);
R4 в каждом случае независимо выбран из H, C1-C6 алкила и -O-(C1-C6 алкил);
R5 в каждом случае независимо представляет собой H или C1-C6 алкил;
R6 в каждом случае независимо выбран из H, C1-C6 алкила и C3-C7 циклоалкила;
R7A представляет собой H;
R7B представляет собой H, или R7A и R7B, вместе с общим для них атомом углерода, к которому они присоединены, объединяются с образованием спироциклической C3-C7 циклоалкильной группы или спироциклической 4-7-членной моноциклической гетероциклоалкильной группы; и
R8 выбран из C1-C6 алкила, -(C1-C6 алкилен)-O-(C1-C6 алкил), C3-C7 циклоалкила и -(C1-C6 алкилен)-C3-C7 циклоалкила.
Соединения формулы (I) (также указанные как ʺТрициклические Гетероциклические Соединенияʺ) и их фармацевтически приемлемые соли или пролекарства могут быть полезны, например, для ингибирования вирусной репликации или активности репликона ВИЧ или для лечения или профилактики ВИЧ-инфекции у субъекта. Не связывая это с какой-либо конкретной теорией, считают, что трициклические гетероциклические соединения ингибируют вирусную репликацию ВИЧ путем ингибирования интегразы ВИЧ.
Соответственно, настоящее изобретение обеспечивает способы лечения или профилактики ВИЧ-инфекции у субъекта, включающие введение субъекту эффективного количества по меньшей мере одного трициклического гетероциклического соединения.
Изобретение подробно изложено в прилагаемом ниже подробном описании.
Хотя любые способы и материалы, подобные описанным в настоящей заявке, можно использовать при практическом применении или испытании настоящего изобретения, далее будут описаны иллюстративные способы и материалы. Другие варианты осуществления, аспекты и признаки настоящего изобретения либо описаны далее, либо будут очевидны из последующего описания, примеров и прилагаемой формулы изобретения.
Подробное описание изобретения
Настоящее изобретение включает трициклические гетероциклические соединения, композиции, включающие по меньшей мере одно трициклическое гетероциклическое соединение, и способы применения трициклических гетероциклических соединений для лечения или профилактики ВИЧ-инфекции у субъекта.
Определения и аббревиатуры
Термины, используемые в настоящей заявке, имеют свое обычное значение, и значение таких терминов является независимым в каждом случае, когда они встречаются в нстоящей заявке. Несмотря на это и за исключением случаев, когда указано иное, следующие определения применимы повсеместно в описании и формуле изобретения. Химические названия, общие названия и химические структуры могут использоваться взаимозаменяемо для описания одной и той же структуры. Эти определения применяются независимо от того, используется ли термин сам по себе или в сочетании с другими терминами, если не указано иное. Следовательно, определение "алкил" относится к "алкилу", а также к "алкильным" частям "гидроксиалкила", "галогеналкила", "-О-алкила" и т.д.
Как они используются в данном разделе и в других разделах настоящего раскрытия, следующие термины, если не указано иное, должны иметь следующие значения:
"Субъект" означает человека или млекопитающее, не являющееся человеком. В одном варианте осуществления субъектом является человек. В другом варианте осуществления субъектом является примат. В другом варианте осуществления субъектом является обезьяна. В другом варианте осуществления субъектом является шимпанзе. В еще одном варианте осуществления субъект представляет собой макака-резуса.
Термин "эффективное количество" в контексте настоящей заявки относится к количеству трициклического гетероциклического соединения и/или дополнительного терапевтического средства, или его композиции, которое эффективно для ингибирования репликации ВИЧ и для достижения желаемого терапевтического, улучшающего, ингибиторного или профилактического эффекта при введении субъекту, страдающему от ВИЧ-инфекции или СПИДа. В комбинированных терапиях по настоящему изобретению эффективное количество может относиться к каждому отдельному средству или к комбинации в целом, где количества всех вводимых средств являются вместе эффективными, но где отдельный компонент комбинации может не присутствовать индивидуально в эффективном количестве.
Термин ʺпрофилактикаʺ в контексте настоящей заявки, в отношении вирусной ВИЧ-инфекции или СПИДа, относится к снижению вероятности заражения или тяжести ВИЧ-инфекции или СПИДа.
Термин "алкилʺ в контексте настоящей заявки относится к алифатической углеводородной группе, в которой один из атомов водорода замещен связью. Алкильная группа может быть линейной или разветвленной и может содержать от около 1 до около 20 атомов углерода. В одном варианте осуществления алкильная группа содержит от около 1 до около 12 атомов углерода. В других вариантах осуществления алкильная группа содержит от 1 до 6 атомов углерода (C1-C6 алкил) или от около 1 до около 4 атомов углерода (C1-C4 алкил). Неограничивающие примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, неопентил, изопентил, н-гексил, изогексил и неогексил. Алкильная группа может быть незамещенной или замещенной одним или несколькими заместителями, которые могут быть одинаковыми или отличными друг от друга, при этом каждый заместитель независимо выбран из группы, состоящей из галогена, алкенила, алкинила, арила, циклоалкила, циано, гидрокси, -O-алкила, -O-арила, -алкилен-O-алкила, алкилтио, -NH2, -NH(алкил), -N(алкил)2, -NH(циклоалкил), -O-C(O)-алкила, -O-C(O)-арила, -O-C(O)-циклоалкила, -C(O)OH и -C(O)O-алкила. В одном варианте осуществления алкильная группа является линейной. В другом варианте осуществления алкильная группа является разветвленной. Если не указано иное, алкильная группа является незамещенной.
Термин "алкенилʺ в контексте настоящей заявки относится к алифатической углеводородной группе, содержащей по меньшей мере одну углерод-углеродную двойную связь, и в которой один из атомов водорода замещен связью. Алкенильная группа может быть линейной или разветвленной и может содержать от около 2 до около 15 атомов углерода. В одном варианте осуществления алкенильная группа содержит от около 2 до около 12 атомов углерода. В другом варианте осуществления алкенильная группа содержит от около 2 до около 6 атомов углерода. Неограничивающие примеры алкенильных групп включают этенил, пропенил, н-бутенил, 3-метилбут-2-енил, н-пентенил, октенил и деценил. Алкенильная группа может быть незамещенной или замещенной одним или несколькими заместителями, которые могут быть одинаковыми или отличными друг от друга, при этом каждый заместитель независимо выбран из группы, состоящей из галогена, алкенила, алкинила, арила, циклоалкила, циано, гидрокси, -O-алкил, -O-арила, -алкилен-O-алкила, алкилтио, -NH2, -NH(алкил), -N(алкил)2, -NH(циклоалкил), -O-C(O)-алкил, -O-C(O)-арила, -O-C(O)-циклоалкила, -C(O)OH и -C(O)O-алкила. Термин ʺC2-C6 алкенилʺ относится к алкенильной группе, содержащей от 2 до 6 атомов углерода. Если не указано иное, алкенильная группа является незамещенной.
Термин "алкинилʺ в контексте настоящей заявки относится к алифатической углеводородной группе, содержащей по меньшей мере одну углерод-углеродную тройную связь, и в которой один из атомов водорода замещен связью. Алкинильная группа может быть линейной или разветвленной и может содержать от около 2 до около 15 атомов углерода. В одном варианте осуществления алкинильная группа содержит от около 2 до около 12 атомов углерода. В другом варианте осуществления алкинильная группа содержит от около 2 до около 6 атомов углерода. Неограничивающие примеры алкинильных групп включают этинил, пропинил, 2-бутинил и 3-метилбутинил. Алкинильная группа может быть незамещенной или замещенной одним или несколькими заместителями, которые могут быть одинаковыми или отличными друг от друга, при этом каждый заместитель независимо выбран из группы, состоящей из галогена, алкенила, алкинила, арила, циклоалкила, циано, гидрокси, -O-алкила, -O-арила, -алкилен-O-алкила, алкилтио, -NH2, -NH(алкил), -N(алкил)2, -NH(циклоалкил), -O-C(O)-алкила, -O-C(O)-арила, -O-C(O)-циклоалкила, -C(O)OH и -C(O)O-алкила. Термин ʺC2-C6 алкинилʺ относится к алкинильной группе, содержащей от 2 до 6 атомов углерода. Если не указано иное, алкинильная группа является незамещенной.
Термин "алкиленʺ в контексте настоящей заявки относится к алкильной группе, определенной выше, где один из атомов водорода алкильной группы замещен связью. Неограничивающие примеры алкиленовых групп включают -CH2-, -CH2CH2-, -CH2CH2CH2-, -CH2CH2CH2CH2-, -CH(CH3)CH2CH2-, -CH(CH3)- и -CH2CH(CH3)CH2-. В одном варианте осуществления алкиленовая группа содержит от 1 до около 6 атомов углерода. В другом варианте осуществления алкиленовая группа содержит от около 3 до около 5 атомов углерода. В другом варианте осуществления алкиленовая группа является разветвленной. В другом варианте осуществления алкиленовая группа является линейной. В одном варианте осуществления алкиленовая группа представляет собой -CH2-. Термин ʺC1-C6 алкиленʺ относится к алкиленовой группе, содержащей от 1 до 6 атомов углерода. Термин ʺC1-C3 алкиленʺ относится к алкиленовой группе, содержащей от 1 до 3 атомов углерода.
Термин "алкениленʺ в контексте настоящей заявки относится к алкенильной группе, определенной выше, где один из атомов водорода алкенильной группы замещен связью. Неограничивающие примеры алкениленовых групп включают -CH=CH-, -CH=CHCH2-, -CH2CH=CH-, -CH2CH=CHCH2-, -CH=CHCH2CH2-, -CH2CH2CH=CH- и -CH(CH3)CH=CH-. В одном варианте осуществления алкениленовая группа содержит от 2 до около 6 атомов углерода. В другом варианте осуществления алкениленовая группа содержит от около 3 до около 5 атомов углерода. В другом варианте осуществления алкениленовая группа является разветвленной. В другом варианте осуществления алкениленовая группа является линейной. Термин ʺC2-C6 алкиленʺ относится к алкениленовой группе, содержащей от 2 до 6 атомов углерода. Термин ʺC3-C5 алкениленʺ относится к алкениленовой группе, содержащей от 3 до 5 атомов углерода.
Термин "арил" в контексте настоящей заявки относится к ароматической моноциклической или мультициклической кольцевой системе, включающей от около 6 до около 14 атомов углерода. В одном варианте осуществления арильная группа содержит от около 6 до около 10 атомов углерода. Арильная группа необязательно замещена одним или несколькими "заместителями кольцевой системы", которые могут быть одинаковыми или отличными друг от друга и которые определены ниже. В одном варианте осуществления арильная группа необязательно может быть конденсирована с циклоалкильной или циклоалканоильной группой. Неограничивающие примеры арильных групп включают фенил и нафтил. В одном варианте осуществления арильная группа представляет собой фенил. Если не указано иное, арильная группа является незамещенной.
Термин "арилен" в контексте настоящей заявки относится к двухвалетной группе, образованной из арильной группы, определенной выше, путем удаления атома водорода у кольцевого углерода арильной группы. Ариленовая группа может происходить из моноциклической или мультициклической кольцевой системы, включающей от около 6 до около 14 атомов углерода. В одном варианте осуществления ариленовая группа содержит от около 6 до около 10 атомов углерода. В другом варианте осуществления ариленовая группа представляет собой нафтиленовую группу. В другом варианте осуществления ариленовая группа представляет собой фениленовую группу. Ариленовая группа необязательно замещена одним или несколькими "заместителями кольцевой системы", которые могут быть одинаковыми или отличными друг от друга и которые определены ниже. Ариленовая группа является двухвалентной, и любая доступная связь на ариленовой группе может соединять с любой группой, фланкирующей ариленовую группу. Например, группа ʺA-арилен-B,ʺ где ариленовая группа представляет собой:
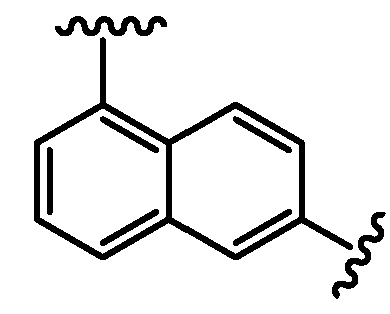 ,
,
как подразумевается, включает обе группы:
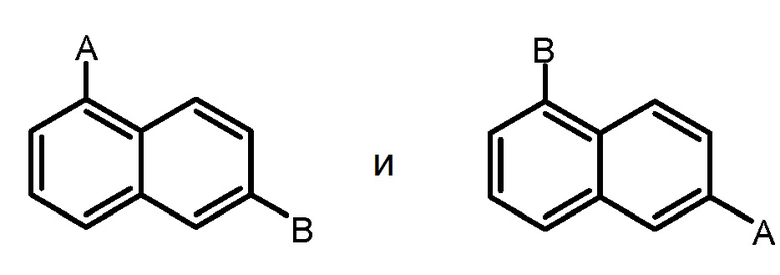 .
.
В одном варианте осуществления ариленовая группа необязательно может быть конденсирована с циклоалкильной или циклоалканоильной группой. Неограничивающие примеры ариленовых групп включают фенилен и нафталин. В одном варианте осуществления ариленовая группа является незамещенной. В другом варианте осуществления ариленовая группа представляет собой:
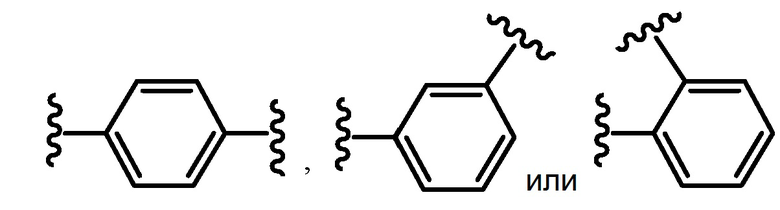 .
.
Если не указано иное, ариленовая группа является незамещенной.
Термин "циклоалкил" в контексте настоящей заявки относится к неароматической моно- или мультициклической насыщенной кольцевой системе, включающей от около 3 до около 10 кольцевых атомов углерода. В одном варианте осуществления циклоалкил содержит от около 5 до около 10 кольцевых атомов углерода. В другом варианте осуществления циклоалкил содержит от около 3 до около 7 кольцевых атомов. В другом варианте осуществления циклоалкил содержит от около 5 до около 6 кольцевых атомов. Термин ʺциклоалкилʺ также охватывает циклоалкильную группу, определенную выше, которая конденсирована с арильным (например, бензольным) или гетероарильным кольцом. Неограничивающие примеры моноциклических циклоалкилов включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. Неограничивающие примеры мультициклических циклоалкилов включают 1-декалинил, норборнил и адамантил. Циклоалкильная группа необязательно замещена одним или несколькими "заместителями кольцевой системы", которые могут быть одинаковыми или отличными друг от друга и которые определены ниже. В одном варианте осуществления циклоалкильная группа является незамещенной. Термин ʺ3-7-членный циклоалкилʺ относится к циклоалкильной группе, содержащей 3-7 кольцевых атомов углерода. Если не указано иное, циклоалкильная группа является незамещенной. Кольцевой атом углерода циклоалкильной группы может быть функционализирован в виде карбонильной группы. Иллюстративный пример такой циклоалкильной группы (также указана как ʺциклоалканоильнаяʺ группа) включает, но не ограничивается этим, циклобутаноил:
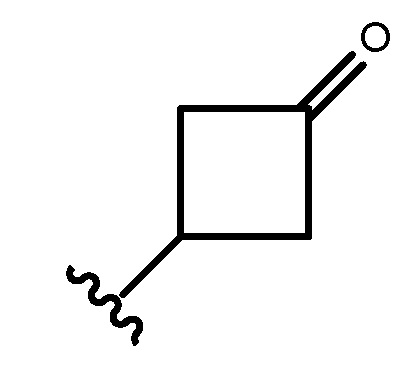 .
.
Термин ʺгалогенʺ в контексте настоящей заявки означает -F, -Cl, -Br или -I.
Термин "галогеналкил" в контексте настоящей заявки относится к алкильной группе, определенной выше, где один или несколько атомов водорода алкильной группы замещены галогеном. В одном варианте осуществления галогеналкильная группа содержит от 1 до 6 атомов углерода. В другом варианте осуществления галогеналкильная группа замещена 1-3 атомами F. Неограничивающие примеры галогеналкильных групп включают -CH2F, -CHF2, -CF3, -CH2Cl и -CCl3. Термин ʺC1-C6 галогеналкилʺ относится к галогеналкильной группе, содержащей от 1 до 6 атомов углерода.
Термин "гидроксиалкил" в контексте настоящей заявки относится к алкильной группе, определенной выше, где один или несколько атомов водорода алкильной группы замещены -OH группой. В одном варианте осуществления гидроксиалкильная группа содержит от 1 до 6 атомов углерода. Неограничивающие примеры гидроксиалкильных групп включают -CH2OH, -CH2CH2OH, -CH2CH2CH2OH и -CH2CH(OH)CH3. Термин ʺC1-C6 гидроксиалкилʺ относится к гидроксиалкильной группе, содержащей от 1 до 6 атомов углерода.
Термин "гетероарилʺ в контексте настоящей заявки относится к ароматической моноциклической или мультициклической кольцевой системе, включающей около 5 до около 14 кольцевых атомов, где от 1 до 4 кольцевых атомов независимо представляют собой O, N или S, а остальные кольцевые атомы являются атомами углерода. В одном варианте осуществления гетероарильная группа содержит 5-10 кольцевых атомов. В другом варианте осуществления гетероарильная группа является моноциклической и содержит 5 или 6 кольцевых атомов. В другом варианте осуществления гетероарильная группа является бициклической. В другом варианте осуществления гетероарильная группа является бициклической и содержит 9 или 10 кольцевых атомов. Гетероарильная группа необязательно замещена одним или несколькими "заместителями кольцевой системы", которые могут быть одинаковыми или отличными друг от друга и которые определены ниже. Гетероарильная группа присоединяется через кольцевой атом углерода, и любой атом азота гетероарила необязательно может быть окислен до соответствующего N-оксида. Термин ʺгетероарилʺ также охватывает гетероарильную группу, определенную выше, которая конденсирована с бензольным кольцом. Неограничивающие примеры гетероарилов включают пиридил, пиразинил, фуранил, тиенил, пиримидинил, пиридон (включая N-замещенные пиридоны), изоксазолил, изотиазолил, оксазолил, оксадиазолил, тиазолил, пиразолил, фуразанил, пирролил, триазолил, 1,2,4-тиадиазолил, пиразинил, пиридазинил, хиноксалинил, фталазинил, оксиндолил, имидазо[1,2-a]пиридинил, имидазо[2,1-b]тиазолил, бензофуразанил, индолил, азаиндолил, бензимидазолил, бензотиенил, хинолинил, имидазолил, бензимидазолил, тиенoпиридил, хиназолинил, тиенoпиримидил, пирролoпиридил, имидазопиридил, изохинолинил, бензоазаиндолил, 1,2,4-триазинил, бензотиазолил и т.п. и все их изомерные формы. Термин ʺгетероарилʺ также относится к частично насыщенным гетероарильным группам, таким как, например, тетрагидроизохинолил, тетрагидрохинолил и т.п. В одном варианте осуществления гетероарильная группа представляет собой 5-членный гетероарил. В другом варианте осуществления гетероарильная группа представляет собой 6-членный моноциклический гетероарил. В другом варианте осуществления гетероарильная группа включает 5-6-членную моноциклическую гетероарильную группу, конденсированную с бензольным кольцом. Если не указано иное, гетероарильная группа является незамещенной.
Термин "гетероциклоалкил" в контексте настоящей заявки относится к неароматической насыщенной моноциклической или мультициклической кольцевой системе, включающей от 3 до около 11 кольцевых атомов, где от 1 до 4 кольцевых атомов независимо представляют собой O, S, N или Si, а остальные кольцевые атомы являются атомами углерода. Гетероциклоалкильная группа может присоединяться через кольцевой углерод, кольцевой атом кремния или кольцевой атом азота. В одном варианте осуществления гетероциклоалкильная группа является моноциклической и содержит от около 3 до около 7 кольцевых атомов. В другом варианте осуществления гетероциклоалкильная группа является моноциклической и содержит от около 5 до около 8 кольцевых атомов. В другом варианте осуществления гетероциклоалкильная группа является бициклической и содержит от около 8 до около 11 кольцевых атомов. Еще в одном варианте осуществления гетероциклоалкильная группа является моноциклической и содержит 5 или 6 кольцевых атомов. В одном варианте осуществления гетероциклоалкильная группа является моноциклической. В другом варианте осуществления гетероциклоалкильная группа является бициклической. Никаких смежных атомов кислорода и/или серы нет в кольцевой системе. Любая -NH группа в гетероциклоалкильном кольце может существовать в защищенной форме, такой как, например, -N(BOC), -N(Cbz), -N(Tos) группа и т.п.; такие защищенные гетероциклоалкильные группы считаются частью настоящего изобретения. Термин ʺгетероциклоалкилʺ также охватывает гетероциклоалкильную группу, определенную выше, которая конденсирована с арильным (например, бензольным) или гетероарильным кольцом. Гетероциклоалкильная группа необязательно замещена одним или несколькими "заместителями кольцевой системы", которые могут быть одинаковыми или отличными друг от друга и которые определены ниже. Атом азота или серы гетероциклоалкила необязательно может быть окислен до соответствующего N-оксида, S-оксида или S,S-диоксида. Неограничивающие примеры моноциклических гетероциклоалкильных колец включают оксетанил, пиперидил, пирролидинил, пиперазинил, морфолинил, тиоморфолинил, тиазолидинил, 1,4-диоксанил, тетрагидрофуранил, тетрагидротиофенил, дельта-лактам, дельта-лактон и т.п. и все их изомеры.
Кольцевой атом углерода гетероциклоалкильной группы может быть функционализирован в виде карбонильной группы. Иллюстративным примером такой гетероциклоалкильной группы является:
 .
.
В одном варианте осуществления гетероциклоалкильная группа представляет собой 5-членный моноциклический гетероциклоалкил. В другом варианте осуществления гетероциклоалкильная группа представляет собой 6-членный моноциклический гетероциклоалкил. Термин ʺ4-7-членный моноциклический гетероциклоалкилʺ относится к моноциклической гетероциклоалкильной группе, содержащей от 4 до 7 кольцевых атомов. Термин ʺ5-8-членный моноциклический гетероциклоалкилʺ относится к моноциклической гетероциклоалкильной группе, содержащей от 5 до 8 кольцевых атомов. Термин ʺ8-11-членный бициклический гетероциклоалкилʺ относится к бициклической гетероциклоалкильной группе, содержащей от 8 до 11 кольцевых атомов. Если не указано иное, гетероциклоалкильная группа является незамещенной.
Термин "гетероциклоалкенил" в контексте настоящей заявки относится к гетероциклоалкильной группе, определенной выше, которая является неароматической и содержит по меньшей мере одну эндоциклическую двойную связь между двумя смежными кольцевыми атомами. Гетероциклоалкенильная группа может присоединяться через кольцевой углерод, кольцевой атом кремния или кольцевой атом азота. В одном варианте осуществления гетероциклоалкенильная группа является моноциклической и содержит от около 3 до около 7 кольцевых атомов. В другом варианте осуществления гетероциклоалкенильная группа является моноциклической и содержит от около 5 до около 8 кольцевых атомов. В другом варианте осуществления гетероциклоалкенильная группа является бициклической и содержит от около 8 до около 11 кольцевых атомов. Еще в одном варианте осуществления гетероциклоалкенильная группа является моноциклической и содержит 5 или 6 кольцевых атомов. В одном варианте осуществления гетероциклоалкенильная группа является моноциклической. В другом варианте осуществления гетероциклоалкенильная группа является бициклической. Никаких смежных атомов кислорода и/или серы нет в кольцевой системе. Любая -NH группа в гетероциклоалкенильном кольце может быть замещена или может существовать в защищенной форме, такой как, например, -N(BOC), -N(Cbz), -N(Tos) группа и т.п.; такие защищенные гетероциклоалкенильные группы считаются частью настоящего изобретения. Термин ʺгетероциклоалкенилʺ также охватывает гетероциклоалкенильную группу, определенную выше, которая конденсирована с арильным (например, бензольным) или гетероарильным кольцом. Гетероциклоалкенильная группа необязательно замещена одним или несколькими "заместителями кольцевой системы", которые могут быть одинаковыми или отличными друг от друга и которые определены ниже. Атом азота или серы гетероциклоалкенила необязательно может быть окислен до соответствующего N-оксида, S-оксида или S,S-диоксида.
Кольцевой атом углерода гетероциклоалкенильной группы может быть функционализирован в виде карбонильной группы. Иллюстративным примером такой гетероциклоалкенильной группы является:
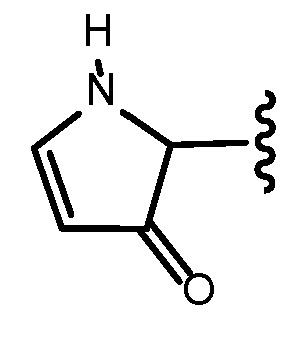 .
.
В одном варианте осуществления гетероциклоалкенильная группа представляет собой 5-членный моноциклический гетероциклоалкенил. В другом варианте осуществления гетероциклоалкенильная группа представляет собой 6-членный моноциклический гетероциклоалкенил. Термин ʺ4-7-членный моноциклический гетероциклоалкенилʺ относится к моноциклической гетероциклоалкенильной группе, содержащей от 4 до 7 кольцевых атомов. Термин ʺ5-8-членный моноциклический гетероциклоалкенилʺ относится к моноциклической гетероциклоалкенильной группе, содержащей от 5 до 8 кольцевых атомов. Термин ʺ8-11-членный бициклический гетероциклоалкенилʺ относится к бициклическй гетероциклоалкенильной группе, содержащей от 8 до 11 кольцевых атомов. Если не указано иное, гетероциклоалкенильная группа является незамещенной.
Термин "заместитель кольцевой системы" в контексте настоящей заявки относится к группе заместителя, присоединенной к ароматической или неароматической кольцевой системе, которая, например, замещает доступный водород на кольцевой системе. Заместители в кольцевой системе могут быть одинаковыми или отличными друг от друга, при этом каждый независимо выбран из группы, состоящей из алкила, алкенила, алкинила, арила, гетероарила, -алкилен-арила, -арилен-алкила, -алкилен-гетероарила, -алкенилен-гетероарила, -алкинилен-гетероарила, -OH, гидроксиалкила, галогеналкила, -O-алкила, -O-галогеналкила, -алкилен-O-алкила, -O-арила, -O-алкилен-арила, ацила, -C(O)-арила, галогена, -NO2, -CN, -SF5, -C(O)OH, -C(O)O-алкила, -C(O)O-арила, -C(O)O-алкилен-арила, -S(O)-алкила, -S(O)2-алкила, -S(O)-арила, -S(O)2-арила, -S(O)-гетероарила, -S(O)2-гетероарила, -S-алкила, -S-арила, -S-гетероарила, -S-алкилен-арила, -S-алкилен-гетероарила, -S(O)2-алкилен-арила, -S(O)2-алкилен-гетероарила, -Si(алкил)2, -Si(арил)2, -Si(гетероарил)2, -Si(алкил)(арил), -Si(алкил)(циклоалкил), - Si(алкил)(гетероарил), циклоалкила, гетероциклоалкила, -O-C(O)-алкила, -O-C(O)-арила, -O-C(O)-циклоалкила, -C(=N-CN)-NH2, -C(=NH)-NH2, -C(=NH)-NH(алкил), -N(Y1)(Y2), -алкилен-N(Y1)(Y2), -C(O)N(Y1)(Y2) и -S(O)2N(Y1)(Y2), где Y1 и Y2 могут быть одинаковыми или отличными друг от друга и независимо выбраны из группы, состоящей из водорода, алкила, арила, циклоалкила и -алкилен-арила. ʺЗаместитель кольцевой системыʺ также может означать одну группу, которая одновременно замещает два доступных водорода на двух смежных атомах углерода (один H на каждом углероде) на кольцевой системе. Примерами такой группы являются метилендиокси, этилендиокси, -C(CH3)2- и т.п., которые образуют группы, такие как, например:
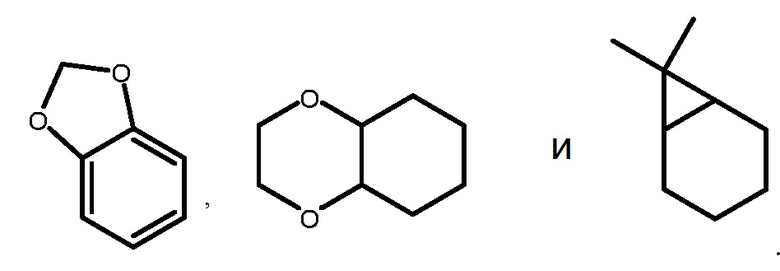 .
.
Термин ʺзамещенныйʺ означает, что один или несколько атомов водорода на указанном атоме замещены выбором из указанной группы, при условии, что нормальная валентность указанного атома при существующих обстоятельствах не превышена и что замещение приводит к стабильному соединению. Комбинации заместителей и/или переменных допустимы, только если такие комбинации приводят к стабильным соединениям. Под "стабильным соединением" или "стабильной структурой" подразумевается соединение, которое является достаточно устойчивым, чтобы выдержать выделение из реакционной смеси до необходимой степени чистоты и формулирование в эффективное терапевтическое средство.
Термин "в по существу очищенной форме" в контексте настоящей заявки относится к физическому состоянию соединения после того, как соединение выделено из процесса синтеза (например, из реакционной смеси), природного источника или их комбинации. Термин "в по существу очищенной форме" также относится к физическому состоянию соединения после того, как соединение получено из процесса очистки или процессов, описанных в настоящей заявке или хорошо известных специалистам в данной области (например, хроматография, перекристаллизация и т.п.), с достаточной чистотой, чтобы его можно было охарактеризовать стандартными аналитическими методами, описанными в настоящей заявке или хорошо известными специалистам в данной области.
Следует также отметить, что любой углерод, а также гетероатом с незанятыми валентностями в тексте, схемах, примерах и таблицах в настоящей заявке, как предполагается, имеют достаточное количество атомов водорода для удовлетворения валентностей.
Когда функциональную группу в соединении называют "защищенной", это означает, что группа находится в модифицированной форме для предотвращения нежелательных побочных реакций на защищенном участке, когда соединение подвергают взаимодействию. Подходящие защитные группы должны быть известны специалистам в данной области, а также их можно найти в стандартных справочниках, таких как, например, T.W. Greene et al., Protective Groups in Organic Synthesis (1991), Wiley, New York.
Когда какой-либо заместитель или переменная (например, R4 и R5) встречаются более одного раза в любом составляющем фрагменте или в формуле (I), их определение в каждом случае не зависит от определения в каждом другом случае, если не указано иное.
Термин "композиция" в контексте настоящей заявки предназначен для охвата продукта, включающего указанные ингредиенты в указанных количествах, а также любого продукта, который получают в результате комбинации указанных ингредиентов в указанных количествах.
Пролекарства и сольваты соединений по изобретению также предусматриваются в настоящем изобретении. Обсуждение пролекарств представлено в T. Higuchi and V. Stella, Pro-drug as Novel Delivery Systems (1987) 14, A.C.S. Symposium Series, и в Bioreversible Carriers in Drug Design, (1987) Edward B. Roche, ed., American pharmaceutical Association and Pergamon Press. Термин "пролекарство" означает соединение (например, предшественник лекарственного средства), которое преобразуется in vivo с обеспечением трициклического гетероциклического соединения или фармацевтически приемлемой соли такого соединения. Преобразование может происходить посредством различных механизмов (например, в результате метаболических или химических процессов), таких как, например, гидролиз в крови. Например, если трициклическое гетероциклическое соединение или фармацевтически приемлемая соль, гидрат или сольват такого соединения содержит карбоновокислотную функциональную группу, пролекарство может включать сложный эфир, образованный путем замещения атома водорода кислотной группы группой, такой как, например, (C1-C8)алкил, (C2-C12)алканоилоксиметил, 1- (алканоилокси)этил, содержащий от 4 до 9 атомов углерода, 1-метил-1-(алканоилокси)этил, содержащий от 5 до 10 атомов углерода, алкоксикарбонилоксиметил, содержащий от 3 до 6 атомов углерода, 1-(алкоксикарбонилокси)этил, содержащий от 4 до 7 атомов углерода, 1-метил-1-(алкоксикарбонилокси)этил, содержащий от 5 до 8 атомов углерода, N-(алкоксикарбонил)аминометил, содержащий от 3 до 9 атомов углерода, 1-(N-(алкоксикарбонил)амино)этил, содержащий от 4 до 10 атомов углерода, 3-фталидил, 4-кротонолактонил, гамма-бутиролактон-4-ил, ди-N,N-(C1-C2)алкиламино(C2-C3)алкил (такой как β-диметиламиноэтил), карбамоил-(C1-C2)алкил, N,N-ди(C1-C2) алкилкарбамоил-(C1-C2)алкил и пиперидино-, пирролидино- или морфолино(С2-С3)алкил и т.п.
Подобным образом, если трициклическое гетероциклическое соединение содержит спиртовую функциональную группу, пролекарство может быть образовано путем замещения одного или нескольких атомов водорода спиртовых групп группой, такой как, например, (C1-C6)алканоилоксиметил, 1-((C1-C6)алканоилокси)этил, 1-метил-1-((C1-C6)алканоилокси)этил, (C1-C6)алкоксикарбонилоксиметил, N-(C1-C6)алкоксикарбониламинометил, сукциноил, (C1-C6)алканоил, α-амино(C1-C4)алкил, α-амино(C1-C4)алкилен-арил, арилацил и α-аминоацил, или α-аминоацил-α-аминоацил, где каждая α-аминоацильная группа независимо выбрана из природных L-аминокислот или гликозила (радикал, образованный в результате удаления гидроксильной группы гемиацетальной формы углевода).
Если трициклическое гетероциклическое соединение включает функциональную амино группу, пролекарство может быть образовано путем замещения атома водорода в амино группе группой, такой как, например, R-карбонил-, RO-карбонил-, NRR'-карбонил-, где R и R' каждый независимо представляет собой (C1-C10)алкил, (C3-C7) циклоалкил, бензил, природный α-аминоацил, -C(OH)C(O)OY1, где Y1 представляет собой H, (C1-C6)алкил или бензил, -C(OY2)Y3, где Y2 представляет собой (C1-C4) алкил и Y3 представляет собой (C1-C6)алкил; карбокси(C1-C6)алкил; амино(C1-C4)алкил или моно-N- или ди-N,N-(C1-C6)алкиламиноалкил; -C(Y4)Y5, где Y4 представляет собой H или метил и Y5 представляет собой моно-N- или ди-N,N-(C1-C6)алкиламино морфолино; пиперидин-1-ил или пирролидин-1-ил и т.п.
Фармацевтически приемлемые сложные эфиры соединений по настоящему изобретению включают следующие группы: (1) эфиры карбоновых кислот, полученные этерификацией гидроксигруппы гидроксильного соединения, в которых некарбонильная часть карбоновокислотной части сложноэфирной группы выбрана из алкила с прямой или с разветвленной цепью (например, метила, этила, н-пропила, изопропила, трет-бутила, втор-бутила или н-бутила), алкоксиалкила (например, метоксиметила), аралкила (например, бензила), арилоксиалкила (например, феноксиметила), арила (например, фенила, необязательно замещенного, например, галогеном, C1-4-алкилом, -O-(C1-4-алкилом) или амино); (2) сульфонатные сложные эфиры, такие как алкил- или аралкилсульфонил (например, метансульфонил); (3) сложные эфиры аминокислот, включая те, которые соответствуют как природным, так и неприродным аминокислотам (например, L-валил или L-изолейцил); (4) фосфонатные сложные эфиры и (5) моно-, ди- или трифосфатные эфиры. Фосфатные эфиры могут быть дополнительно этерифицированы, например, С1-20 спиртом или его реакционноспособным производным или 2,3-ди(С6-24) ацилглицерином.
Одно или несколько соединений по изобретению могут существовать в несольватированных, а также в сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п., и предполагается, что изобретение охватывает как сольватированные, так и несольватированные формы. "Сольват" означает физическую ассоциацию соединения по настоящему изобретению с одной или несколькими молекулами растворителя. Эта физическая ассоциация включает различные степени ионного и ковалентного связывания, включая водородное связывание. В некоторых случаях сольват может быть выделен, например, когда одна или несколько молекул растворителя включены в кристаллическую решетку кристаллического твердого вещества. "Сольват" охватывает как находящиеся в фазе раствора, так и выделяемые сольваты. Неограничивающие примеры сольватов включают этаноляты, метаноляты и т.п. "Гидрат" представляет собой сольват, в котором молекула растворителя представляет собой воду.
Одно или несколько соединений по изобретению необязательно могут быть преобразованы в сольват. Получение сольватов общеизвестно. Так, например, в M. Caira et al., J. Pharmaceutical Sci., 93(3), 601-611 (2004) описано получение сольватов противогрибкового флуконазола в этилацетате, а также из воды. Подобные получения сольватов, гемисольватов, гидратов и т.п. описаны в E.C. van Tonder et al., AAPS PharmSciTech., 5 (1), article 12 (2004 год); и A.L. Bingham et al., Chem. Commun., 603-604 (2001). Типичный неограничивающий способ включает растворение соединения по изобретению в желаемых количествах желаемого растворителя (органического или воды или их смесей) при температуре выше комнатной и охлаждение раствора со скоростью, достаточной для образования кристаллов, которые затем выделяют стандартными методами. Аналитические методы, такие как, например, ИК-спектроскопия, показывают присутствие растворителя (или воды) в кристаллах в виде сольвата (или гидрата).
Трициклические гетероциклические соединения могут образовывать соли, которые также входят в объем настоящего изобретения. Ссылка на трициклическое гетероциклическое соединение в настоящей заявке подразумевает ссылку на его соли, если не указано иное. Термин "соль(соли)" в контексте настоящей заявки обозначает кислотные соли, образованные с неорганическими и/или органическими кислотами, а также оснóвные соли, образованные с неорганическими и/или органическими основаниями. Кроме того, когда трициклическое гетероциклическое соединение содержит как оснóвную часть, такую как, но не ограничиваясь этим, пиридин или имидазол, так и кислотную часть, такую как, но не ограничиваясь этим, карбоновая кислота, могут образовываться цвиттерионы ("внутренние соли"), и они включены в термин "соль(соли)" как он используется в настоящей заявке. В одном варианте осуществления соль представляет собой фармацевтически приемлемую (т.е. нетоксичную, физиологически приемлемую) соль. В другом варианте осуществления соль является отличной от фармацевтически приемлемой соли. Соли соединений формулы (I) могут быть получены, например, путем взаимодействия трициклического гетероциклического соединения с количеством кислоты или основания, такого как эквивалентное количество, в среде, такой как среда, в которой соль осаждается, или в водной среде, с последующей лиофилизацией.
Примеры кислотно-аддитивных солей включают ацетаты, аскорбаты, бензоаты, бензолсульфонаты, бисульфаты, бораты, бутираты, цитраты, камфораты, камфорсульфонаты, фумараты, гидрохлориды, гидробромиды, гидроиодиды, лактаты, малеаты, метансульфонаты, нафталинсульфонаты, нитраты, оксалаты, фосфаты, пропионаты, салицилаты, сукцинаты, сульфаты, тартраты, тиоцианаты, толуолсульфонаты (также известные как тозилаты) и т.п. Кроме того, кислоты, которые обычно считаются подходящими для образования фармацевтически полезных солей из щелочных фармацевтических соединений, обсуждаются, например, в P. Stahl et al., Camille G. (eds.) Handbook of Pharmaceutical Salts. Properties, Selection and Use. (2002) Zurich: Wiley-VCH; S. Berge et al, Journal of Pharmaceutical Sciences (1977) 66(1) 1-19; P. Gould, International J. of Pharmaceuticalics (1986) 33 201-217; Anderson et al, The Practice of Medicinal Chemistry (1996), Academic Press, New York; и в The Orange Book (Food & Drug Administration, Washington, D.C. на их веб-сайте). Эти раскрытия включены в настоящую заявку посредством ссылки.
Примеры солей оснований включают соли аммония, соли щелочных металлов, такие как соли натрия, лития и калия, соли щелочно-земельных металлов, такие как соли кальция и магния, соли с органическими основаниями (например, органическими аминами), такими как дициклогексиламин, трет-бутиламин, холин, и соли с аминокислотами, такими как аргинин, лизин и т.п. Оснóвные азот-содержащие группы могут быть кватернизированы при помощи таких агентов, как низшие алкилгалогениды (например, метил-, этил- и бутил- хлориды, бромиды и йодиды), диалкилсульфаты (например, диметил-, диэтил- и дибутилсульфаты), длинноцепочечные галогениды (например, децил-, лаурил- и стеарил- хлориды, бромиды и йодиды), арилалкилгалогениды (например, бензил- и фенетилбромиды) и др.
Предполагается, что все такие кислотные соли и оснóвные соли являются фармацевтически приемлемыми солями, охватываемыми объемом изобретения, и все кислотные и оснóвные соли считаются эквивалентными свободным формам соответствующих соединений для целей изобретения.
Диастереомерные смеси можно разделить на отдельные диастереомеры на основе их физико-химических различий способами, хорошо известными специалистам в данной области, такими как, например, хроматография и/или фракционная кристаллизация. Энантиомеры можно разделить путем преобразования энантиомерной смеси в диастереомерную смесь путем взаимодействия с соответствующим оптически активным соединением (например, хиральным вспомогательным веществом, таким как хиральный спирт или хлорангидрид кислоты Мошера), разделения диастереомеров и преобразования (например, путем гидролиза) отдельных диастереомеров в соответствующие чистые энантиомеры. Стерохимически чистые соединения также можно получить с использованием хиральных исходных веществ или с использованием методов разделения солей. Также, некоторые из трициклических гетероциклических соединений могут быть атропоизомерами (например, замещенные биарилы) и рассматриваются как часть настоящего изобретения. Энантиомеры также можно непосредственно разделить с использованием методов хиральной хроматографии.
Также возможно существование трициклических гетероциклических соединений в разных таутомерных формах, и все такие формы включены в объем изобретения. Например, все кето-енольные и имин-енаминовые формы соединений включены в изобретение.
Если не указано иное, все стереоизомеры (например, геометрические изомеры, оптические изомеры и т.п.) соединений по настоящему изобретению (в том числе солей, сольватов, гидратов, сложных эфиров и пролекарств соединений, а также солей, сольватов и сложных эфиров пролекарств), например, которые могут существовать благодаря асимметричным атомам углерода на различных заместителях, включая энантиомерные формы (которые могут существовать даже в отсутствие асимметричных атомов углерода), ротамерные формы, атропоизомеры и диастереомерные формы, предусматриваются как охватываемые объемом настоящего изобретения. Если трициклическое гетероциклическое соединение включает двойную связь или конденсированное кольцо, как цис-, так и транс-формы, а также смеси включены в объем изобретения.
Когда заместитель на хиральном атоме углерода представлен в виде рацемата (используя прямую линию, показывающую связь с хиральным центром), следует понимать, что как альфа-, так и бета-конфигурации указанной группы заместителя должны рассматриваться как часть настоящего изобретения. Например, подразумевается, что соединение по настоящему изобретению, которое показано следующим образом:
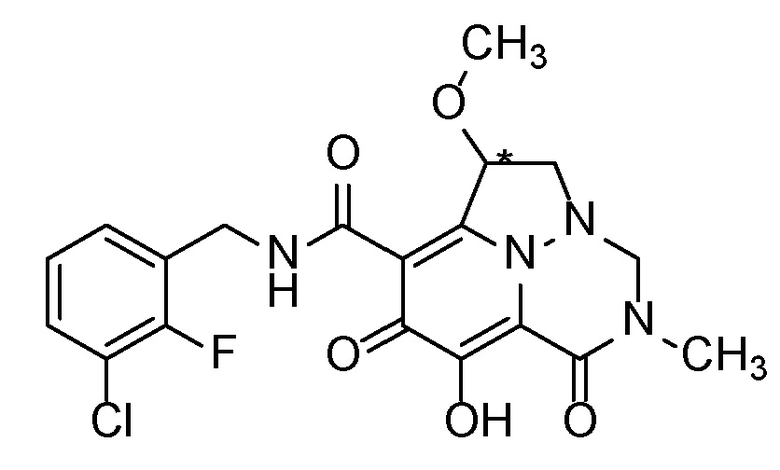 ,
,
охватывает оба стереоизомера при указанном хиральном центре, структуры которых являются следующими:

и
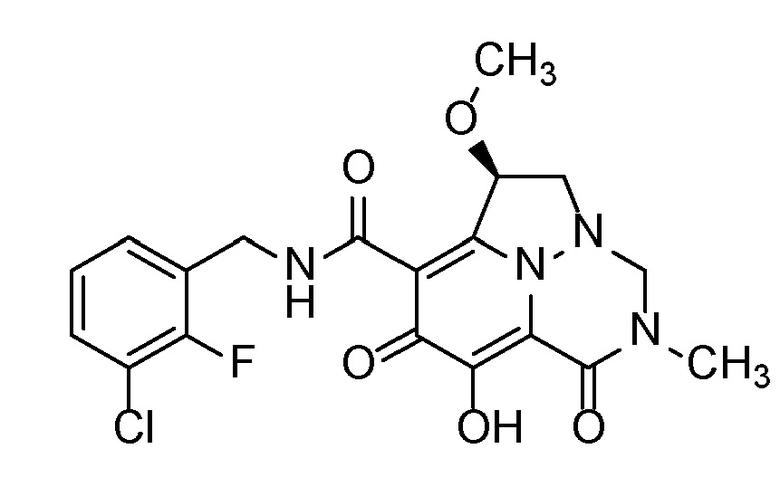 .
.
В разделе Примеры, представленом ниже, соединения по настоящему изобретению, которые были очищены в виде отдельных стереоизомеров, иногда представлены в рацемической форме, но идентифицированы с использованием одного или нескольких из терминов: "диастереомер 1", "диастереомер 2", "энантиомер А" и "энантиомер B". В этом случае абсолютная стереохимия каждого выделенного диастереомера и энантиомерного центра не была определена, и указанные выше термины используются для представления каждого индивидуального очищенного стереохимически чистого соединения.
Индивидуальные стереоизомеры соединений по изобретению могут, например, по существу не содержать других изомеров или могут быть смешаны, например, в виде рацематов или со всеми другими, или другими выбранными, стереоизомерами. Хиральные центры по настоящему изобретению могут иметь S или R конфигурацию, как определено Рекомендациями IUPAC 1974. Используемые термины "соль", "сольват", "сложный эфир", "пролекарство" и т.п. равным образом применимы к соли, сольвату, сложному эфиру и пролекарству энантиомеров, стереоизомеров, ротамеров, таутомеров, рацематов или пролекарств соединений по настоящему изобретению.
В соединениях формулы (I) атомы могут демонстрировать их природный относительный изотопный состав, или один или несколько атомов могут быть искусственно обогащены определенным изотопом, имеющим тот же атомный номер, но атомную массу или массовое число, отличное от атомной массы или массового числа, преимущественно встречающихся в природе. Подразумевается, что настоящее изобретение включает все подходящие изотопные варианты соединений общей формулы I. Например, различные изотопные формы водорода (Н) включают протий (1Н) и дейтерий (2Н). Протий является преобладающим изотопом водорода, встречающимся в природе. Обогащение дейтерием может обеспечить определенные терапевтические преимущества, такие как увеличение периода полувыведения in vivo или снижение требований к дозировке, или может обеспечить соединение, полезное в качестве стандарта для характеризации биологических образцов. Изотопно-обогащенные соединения формулы (I) можно получить без чрезмерного экспериментирования обычными методами, хорошо известными специалистам в данной области техники, или способами, аналогичными описанным на Схемах и в Примерах, представленных в настоящей заявке, с использованием соответствующих изотопно-обогащенных реагентов и/или промежуточных соединений. В одном варианте осуществления соединение формулы (I) имеет один или несколько атомов водорода, замещенных дейтерием.
Трициклические гетероциклические соединения могут быть полезны в медицине и ветеринарии для лечения или профилактики ВИЧ-инфекции у субъекта. В одном варианте осуществления трициклические гетероциклические соединения могут быть ингибиторами вирусной репликации ВИЧ. В конкретном варианте осуществления трициклические гетероциклические соединения являются ингибиторами ВИЧ-1. Соответственно, трициклические гетероциклические соединения могут быть полезны для лечения ВИЧ-инфекций и СПИДа. В соответствии с настоящим изобретением, трициклические гетероциклические соединения можно вводить субъекту, нуждающемуся в лечении или профилактике ВИЧ-инфекции.
Соответственно, в одном варианте осуществления изобретение обеспечивает способы лечения ВИЧ-инфекции у субъекта, включающие введение субъекту эффективного количества по меньшей мере одного трициклического гетероциклического соединения или его фармацевтически приемлемой соли. В конкретном варианте осуществления настоящее изобретение обеспечивает способы лечения СПИДа у субъекта, включающие введение субъекту эффективного количества по меньшей мере одного трициклического гетероциклического соединения или его фармацевтически приемлемой соли.
Перечень аббревиатур
Соединения формулы (I)
Настоящее изобретение обеспечивает трициклические гетероциклические соединения формулы (I):
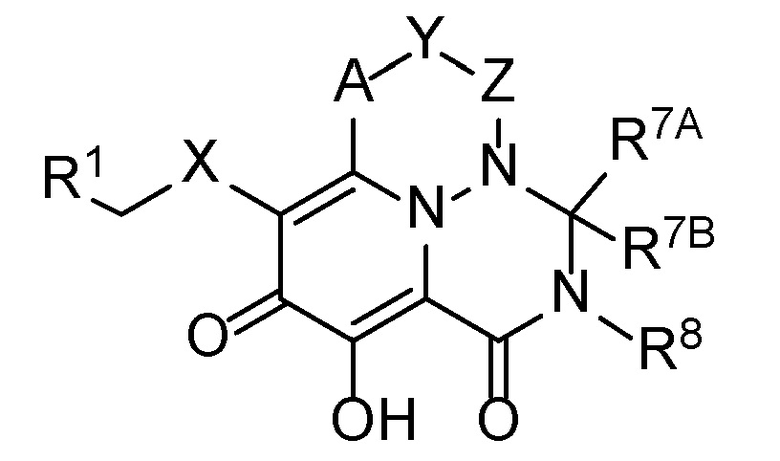
(I)
и их фармацевтически приемлемые соли, где A, X, Y, Z, R1, R7A, R7B и R8 определены выше для соединений формулы (I).
В одном варианте осуществления настоящее изобретение обеспечивает соединения формулы (I):
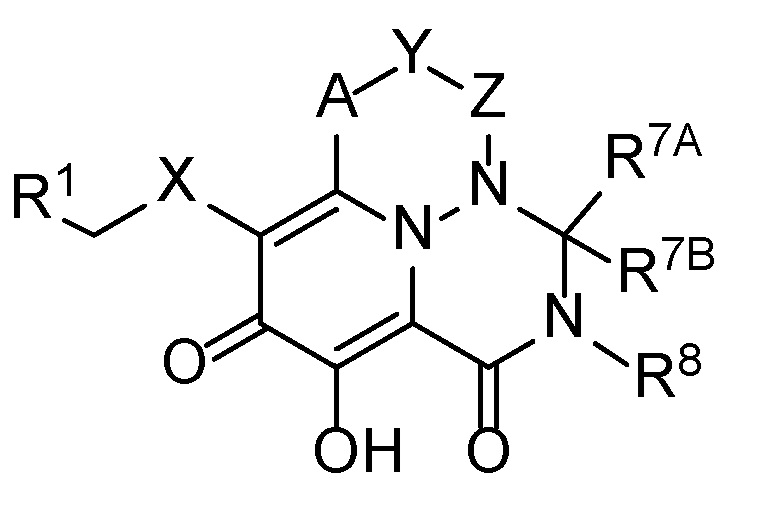
(I)
или их фармацевтически приемлемые соли,
где:
группа -A-Y-Z- выбрана из -CH(R2)-, -CH2-N(R5)-C(O)-CH2-, -CH(R2)-CH(R3)-CH(R4)- и -C(R2)=CH-;
X представляет собой диазолил или -N(R5)C(O)-;
R1 представляет собой фенильную группу, которая необязательно замещена 1-3 группами, каждая из которых независимо выбрана из Cl и F;
R2 представляет собой H или -O-(C1-C6 алкил);
в каждом случае R4 независимо выбран из H и C1-C6 алкила;
в каждом случае R5 независимо представляет собой H или C1-C6 алкил;
R7A представляет собой H;
R7B представляет собой H, или R7A и R7B вместе с общим для них атомом углерода, к которому они присоединены, объединяются с образованием спироциклической 4-7-членной моноциклической гетероциклоалкильной группы; и
R8 выбран из C1-C6 алкила, -(C1-C6 алкилен)-O-(C1-C6 алкил) и -(C1-C6 алкилен)-C3-C7 циклоалкила.
В одном варианте осуществления А представляет собой -CH2-.
В другом варианте осуществления А представляет собой -CH(-O-C1-C6 алкил)-.
В другом варианте осуществления А представляет собой -CH(-OCH3)-.
В одном варианте осуществления Y представляет собой -CH2-.
В другом варианте осуществления Y представляет собой -N(C1-C6 алкил)-.
В другом варианте осуществления Y представляет собой -N(CH3)-.
В одном варианте осуществления Z представляет собой -CH2-.
В другом варианте осуществления Z представляет собой -C(O)-
В другом варианте осуществления Z представляет собой связь.
В одном варианте осуществления -A-Y-Z- группа представляет собой -CH(R2)-CH2-.
В другом варианте осуществления -A-Y-Z- группа представляет собой -CH2-N(R5)-C(O)-CH2-.
В другом варианте осуществления -A-Y-Z- группа представляет собой -CH(R2)-CH(R3)-CH(R4)-.
В другом варианте осуществления -A-Y-Z- группа представляет собой -C(R2)=CH-.
В одном варианте осуществления -A-Y-Z- группа представляет собой -CH(-OCH3)-CH2-.
В другом варианте осуществления -A-Y-Z- группа представляет собой -CH2-N(CH3)-C(O)-.
В другом варианте осуществления -A-Y-Z- группа представляет собой -CH2-CH2-CH2-.
В другом варианте осуществления -A-Y-Z- группа представляет собой -CH=CH-.
В одном варианте осуществления X представляет собой -NHC(O)-.
В другом варианте осуществления X представляет собой 5- или 6-членный гетероарил.
В другом варианте осуществления X представляет собой 5-членный гетероарил.
Еще в одном варианте осуществления X представляет собой диазолил или тиадиазолил.
В другом варианте осуществления X представляет собой диазолил.
В одном варианте осуществления R1 представляет собой фенил, который замещен 1-3 группами, каждая из которых независимо выбрана из C1-C6 алкила, галогена и -O-(C1-C6 алкил);
В одном варианте осуществления R1 представляет собой фенил, который замещен 1-3 группами, каждая из которых независимо выбрана из Cl и F.
В другом варианте осуществления R1 выбран из:
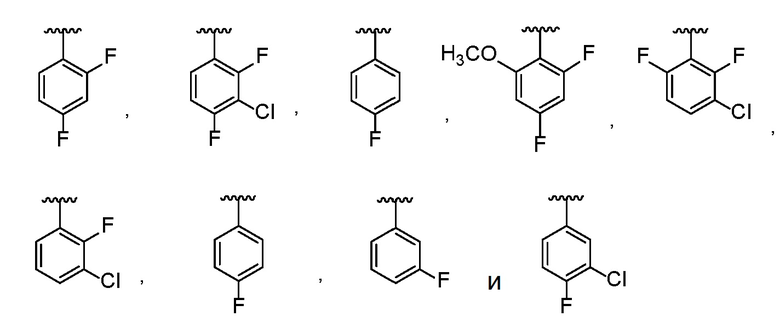 .
.
В одном варианте осуществления R7A и R7B каждый представляет собой H.
В другом варианте осуществления R7A и R7B, вместе с общим для них атомом углерода, к которому они присоединены, объединяются с образованием спироциклической 4-7-членной гетероциклоалкильной группы.
В другом варианте осуществления R7A и R7B вместе с общим для них атомом углерода, к которому они присоединены, объединяются с образованием спироциклической тетрагидрофуранильной группы.
В одном варианте осуществления R8 представляет собой C1-C6 алкил.
В другом варианте осуществления R8 представляет собой -(C1-C6 алкилен)-O-(C1-C6 алкил).
В другом варианте осуществления R8 представляет собой -(C1-C6 алкилен)-C3-C7 циклоалкил.
Еще в одном варианте осуществления R8 выбран из метила, этила, изопропила, -CH2CH2OCH3 и -CH2-циклопропила.
В одном варианте осуществления переменные A, X, Y, Z, R1, R7A, R7B и R8 для соединений формулы (I) выбраны независимо друг от друга.
В другом варианте осуществления соединения формулы (I) находятся в по существу очищенной форме.
Должно быть понятно, что любой из перечисленных выше вариантов осуществления можно комбинировать с одним или несколькими отдельными вариантами осуществления.
Другие варианты осуществления настоящего изобретения включают следующие:
(a) Фармацевтическая композиция, включающая эффективное количество соединения формулы (I) и фармацевтически приемлемый носитель.
(b) Фармацевтическая композиция по пункту (a), дополнительно включающая второе терапевтическое средство, выбранное из группы, состоящей из противовирусных средств против ВИЧ, иммуномодуляторов и противоинфекционных средств.
(c) Фармацевтическая композиция по пункту (b), где противовирусное средство против ВИЧ представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ и NNRTI ингибиторов ВИЧ.
(d) Фармацевтическая комбинация, которая представляет собой (i) соединение формулы (I) и (ii) второе терапевтическое средство, выбранное из группы, состоящей из противовирусных средств против ВИЧ, иммуномодуляторов и противоинфекционных средств; где соединение формулы (I) и второе терапевтическое средство каждое используют в количестве, которое делает комбинацию эффективной для ингибирования репликации ВИЧ или для лечения ВИЧ-инфекции и/или уменьшения вероятности или тяжести симптомов ВИЧ-инфекции.
(e) Комбинация по пункту (d), где противовирусное средство против ВИЧ представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ и NNRTI ингибиторов ВИЧ.
(f) Способ ингибирования репликации ВИЧ у субъекта, нуждающегося в этом, который включает введение субъекту эффективного количества соединения формулы (I).
(g) Способ лечения ВИЧ-инфекции и/или уменьшения вероятности или тяжести симптомов ВИЧ-инфекции у субъекта, нуждающегося в этом, который включает введение субъекту эффективного количества соединения формулы (I).
(h) Способ по пункту (g), где соединение формулы (I) вводят в комбинации с эффективным количеством по меньшей мере одного второго терапевтического средства, выбранного из группы, состоящей из противовирусных средств против ВИЧ, иммуномодуляторов и противоинфекционных средств.
(i) Способ по пункту (h), где противовирусное средство против ВИЧ представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ и NNRTI ингибиторов ВИЧ.
(j) Способ ингибирования репликации ВИЧ у субъекта, нуждающегося в этом, который включает введение субъекту фармацевтической композиции по пункту (a), (b) или (c) или комбинации по пункту (d) или (e).
(k) Способ лечения ВИЧ-инфекции и/или уменьшения вероятности или тяжести симптомов ВИЧ-инфекции у субъекта, нуждающегося в этом, который включает введение субъекту фармацевтической композиции по пункту (a), (b) или (c) или комбинации по пункту (d) или (e).
Дополнительные варианты осуществления настоящего изобретения включают следующие:
(l) Фармацевтическая композиция, включающая эффективное количество фармацевтически приемлемой соли соединения формулы (I) и фармацевтически приемлемый носитель.
(m) Фармацевтическая композиция по пункту (l), дополнительно включающая второе терапевтическое средство, выбранное из группы, состоящей из противовирусных средств против ВИЧ, иммуномодуляторов и противоинфекционных средств.
(n) Фармацевтическая композиция по пункту (m), где противовирусное средство против ВИЧ представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ и NNRTI ингибиторов ВИЧ.
(o) Фармацевтическая комбинация, которая представляет собой (i) фармацевтически приемлемую соль соединения формулы (I) и (ii) второе терапевтическое средство, выбранное из группы, состоящей из противовирусных средств против ВИЧ, иммуномодуляторов и противоинфекционных средств; где фармацевтически приемлемая соль соединения формулы (I) и второе терапевтическое средство каждое используют в количестве, которое делает комбинацию эффективной для ингибирования репликации ВИЧ или для лечения ВИЧ-инфекции и/или уменьшения вероятности или тяжести симптомов ВИЧ-инфекции.
(p) Комбинация по пункту (o), где противовирусное средство против ВИЧ представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ и NNRTI ингибиторов ВИЧ.
(q) Способ ингибирования репликации ВИЧ у субъекта, нуждающегося в этом, который включает введение субъекту эффективного количества фармацевтически приемлемой соли соединения формулы (I).
(r) Способ лечения ВИЧ-инфекции и/или уменьшения вероятности или тяжести симптомов ВИЧ-инфекции у субъекта, нуждающегося в этом, который включает введение субъекту эффективного количества фармацевтически приемлемой соли соединения формулы (I).
(s) Способ по пункту (r), где фармацевтически приемлемую соль соединения формулы (I) вводят в комбинации с эффективным количеством по меньшей мере одного второго терапевтического средства, выбранного из группы, состоящей из противовирусных средств против ВИЧ, иммуномодуляторов и противоинфекционных средств.
(t) Способ по пункту (s), где противовирусное средство против ВИЧ представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ и ингибиторов полимеразы NS5B ВИЧ.
(u) Способ ингибирования репликации ВИЧ у субъекта, нуждающегося в этом, который включает введение субъекту фармацевтической композиции по пункту (l), (m) или (n) или комбинации по пункту (o) или (p).
(v) Способ лечения ВИЧ-инфекции и/или уменьшения вероятности или тяжести симптомов ВИЧ-инфекции у субъекта, нуждающегося в этом, который включает введение субъекту фармацевтической композиции по пункту (l), (m) или (n) или комбинации по пункту (o) или (p).
Другие варианты осуществления настоящего изобретения включают следующие:
(w) Фармацевтическая композиция, включающая эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
(x) Фармацевтическая композиция по пункту (w), дополнительно включающая второе терапевтическое средство, выбранное из группы, состоящей из противовирусных средств против ВИЧ, иммуномодуляторов и противоинфекционных средств.
(y) Фармацевтическая композиция по пункту (x), где противовирусное средство против ВИЧ представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ и NNRTI ингибиторов ВИЧ.
(z) Фармацевтическая комбинация, которая представляет собой (i) соединение формулы (I) и (ii) или его фармацевтически приемлемую соль, второе терапевтическое средство, выбранное из группы, состоящей из противовирусных средств против ВИЧ, иммуномодуляторов и противоинфекционных средств; где соединение формулы (I) и второе терапевтическое средство каждое используют в количестве, которое делает комбинацию эффективной для ингибирования репликации ВИЧ или для лечения ВИЧ-инфекции и/или уменьшения вероятности или тяжести симптомов ВИЧ-инфекции.
(aa) Комбинация по пункту (z), где противовирусное средство против ВИЧ представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ и NNRTI ингибиторов ВИЧ.
(bb) Способ ингибирования репликации ВИЧ у субъекта, нуждающегося в этом, который включает введение субъекту эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
(cc) Способ лечения ВИЧ-инфекции и/или уменьшения вероятности или тяжести симптомов ВИЧ-инфекции у субъекта, нуждающегося в этом, который включает введение субъекту эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
(dd) Способ по пункту (cc), где соединение формулы (I) или его фармацевтически приемлемую соль вводят в комбинации с эффективным количеством по меньшей мере одного второго терапевтического средства, выбранного из группы, состоящей из противовирусных средств против ВИЧ, иммуномодуляторов и противоинфекционных средств.
(ee) Способ по пункту (dd), где противовирусное средство против ВИЧ представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ и NNRTI ингибиторов ВИЧ.
(ff) Способ ингибирования репликации ВИЧ у субъекта, нуждающегося в этом, который включает введение субъекту фармацевтической композиции по пункту (w) (x) или (y) или комбинация по пункту (z) или (aa).
(gg) Способ лечения ВИЧ-инфекции и/или уменьшения вероятности или тяжести симптомов ВИЧ-инфекции у субъекта, нуждающегося в этом, который включает введение субъекту фармацевтической композиции по пункту (w) (x) или (y) или комбинации по пункту (z) или (aa).
Настоящее изобретение также включает соединение по настоящему изобретению для применения (i) для, (ii) в качестве лекарственного средства для, или (iii) для получения лекарственного средства для: (a) терапии; (b) ингибирования репликации ВИЧ или (c) лечения ВИЧ-инфекции и/или уменьшения вероятности или тяжести симптомов ВИЧ-инфекции. В этих применениях соединения по настоящему изобретению необязательно можно использовать в комбинации с одним или несколькими вторыми терапевтическими средствами, выбранными из противовирусных средств против ВИЧ, противоинфекционных средств и иммуномодуляторов.
Дополнительные варианты осуществления изобретения включают фармацевтические композиции, комбинации и способы, изложенные в (а) - (gg) выше, и применения, изложенные в предыдущем абзаце, где используемое в них соединение по настоящему изобретению представляет собой соединение в соответствии с одним из вариантов осуществления, аспектов, классов, подклассов или признаков соединений, описанных выше. Во всех этих вариантах осуществления соединение необязательно можно использовать в форме фармацевтически приемлемой соли или гидрата, в зависимости от ситуации.
Кроме того, должно быть понятно, что варианты осуществления композиций и способов, представленных как (а) - (gg) выше, подразумевают включение всех вариантов осуществления соединений, включая такие варианты осуществления, которые получают при комбинировании вариантов осуществления.
Неограничивающие примеры соединений формулы (I) включают соединения 2-40, описанные в приведенных ниже примерах, и их фармацевтически приемлемые соли.
Способы получения соединений формулы (I)
Соединения формулы (I) можно получить из исходных веществ, которые являются известными или могут быть легко получены, в соответствии со способами, известными специалистам в области органического синтеза. Способы, полезные для получения соединений формулы (I), изложены в приведенных ниже примерах и в общем виде показаны на схемах, приведенных ниже. Альтернативные пути синтеза и аналогичные структуры будут очевидны для специалистов в области органического синтеза.
Схема 1 описывает способы, полезные для получения соединений формулы (I), где -A-Y-Z- группа представляет собой -CH(-O-C1-C6 алкил)-CH2-.
Схема 1
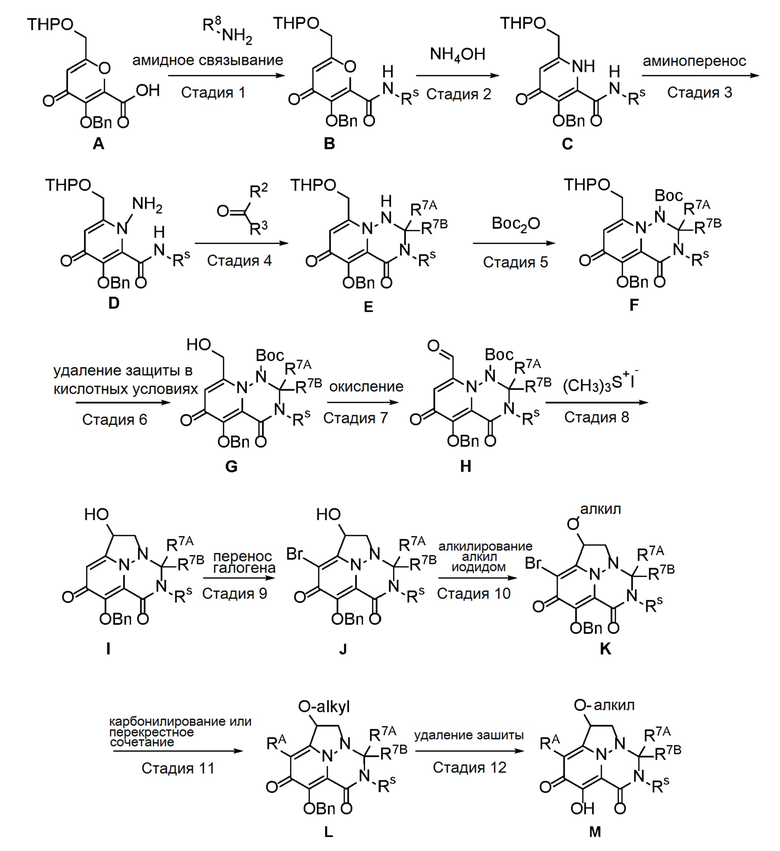
где RA представляет собой -X-CH2-R1.
Пироновое соединение формулы A подвергают взаимодействию с подходяще функционализированым амином с получением амида B, который преобразуют в пиридон C в присутствии гидроксида аммония. Соединение C затем преобразуют в соединение D с использованием подходящего реагента для переноса аминогруппы. Соединение D затем обрабатывают альдегидом или кетоном в присутстви кислоты с получением аминаля E, который защищают по атому азота в виде трет-бутилкарбамата F. Удаление защиты в слабокислотных условиях обеспечивает соединение G, которое окисляют с получением соединения H. Перенос метилена в щелочных условиях обеспечивает соединение I, которое подвергают галогенированию с получением соединения J. O-алкилирование дает соединение K, которое подвергают опосредованному переходным металлом карбонилированию или перекрестному сочетанию с получением соединения L. В завершение, удаление защиты дает соединение M.
Схема 2 описывает способы, полезные для получения соединений формулы (I), где -A-Y-Z- группа представляет собой -CH2-O-C(O)-.
Схема 2
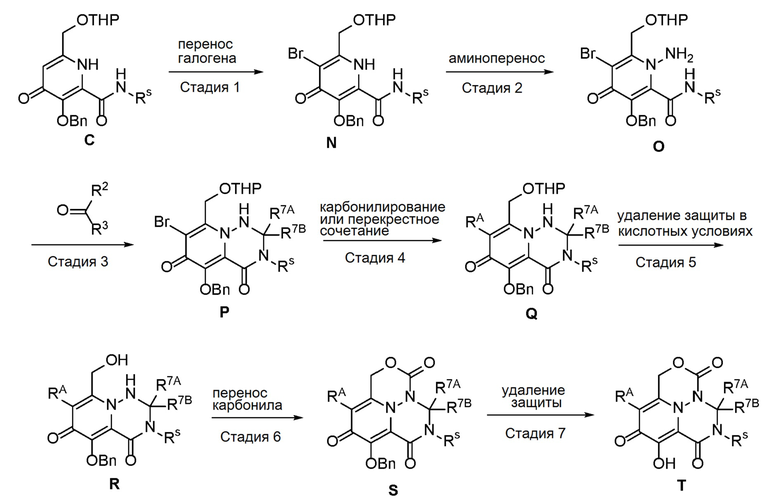
где RA представляет собой -X-CH2-R1.
Соединение C (из Схемы 1) подвергают взаимодействию с подходящим реагентом для переноса галогена с получением соединения N. Соединение N затем преобразуют в соединение O с использованием подходящего реагента для переноса аминогруппы. Соединение O затем обрабатывают альдегидом или кетоном в присутстви кислоты с получением аминаля P, который подвергают опосредованному переходным металлом карбонилированию или перекрестному сочетанию с получением соединения Q. Удаление защиты в слабокислотных условиях обеспечивает соединение R, которое подвергают циклизации с использованием реагента переноса карбонила, с получением соединения S. В завершение, удаление защиты дает соединение T.
Схема 3 описывает способы, полезные для получения соединений формулы (I), где -A-Y-Z- группа представляет собой -CH2-N(R5)-C(O)-.
Схема 3
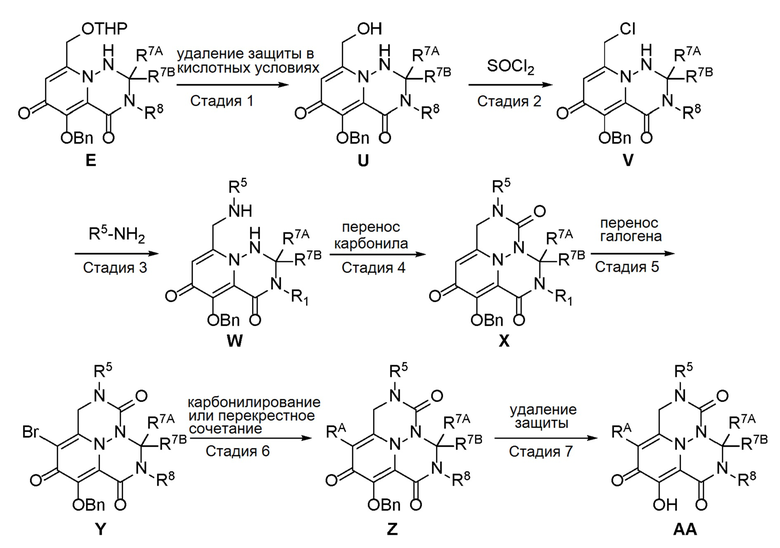
где RA представляет собой -X-CH2-R1.
Удаление защиты соединения E (из Схемы 1) в слабокислотных условиях обеспечивает соединение U, которое преобразуют в соединение V с использованием реагента, такого как тионилхлорид. Соединение V затем обрабатывают подходяще функционализированым амином с получением соединения W, которое подвергают циклизации с использованием реагента переноса карбонила, с получением соединения X. Перенос галогена затем обеспечивает соединение Y, которое подвергают опосредованному переходным металлом карбонилированию или перекрестному сочетанию с получением соединения Z. В завершение, удаление защиты дает соединение AA.
Схема 4 описывает способы, полезные для получения соединений формулы (I), где -A-Y-Z- группа представляет собой -CH2-CH2-.
Схема 4
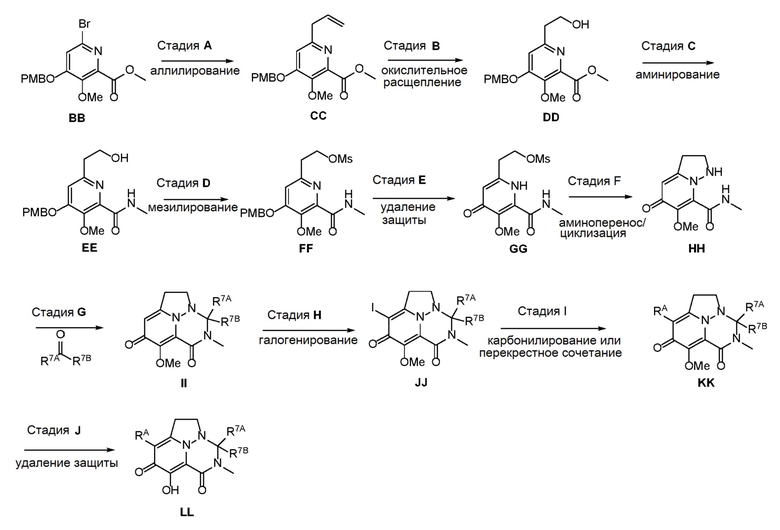
Аллилирование ВВ дает соединение СС, которое затем преобразуют в DD путем окислительного расщепления. DD подвергают аминированию при помощи реагента, такого как метанамин в THF, с получением EE. Мезилирование EE дает соединение FF, которое затем подвергают процедуре удаления защиты, с получением GG. Аминоперенос и циклизация GG дает соединение HH, которое затем обрабатывают альдегидом или кетоном в присутствии кислоты с получением II. Соединение II подвергают галогенированию с использованием реагента, такого как NIS, с получением JJ. Соединение JJ подвергалют либо карбонилированию, либо перекрестному сочетанию с получением соединения KK, которое затем подвергают процедуре удаления защиты, с получением соединения LL.
ПРИМЕРЫ
Общие способы
Соединения, описанные в настоящей заявке, можно получить в соответствии с процедурами следующих схем и примеров, используя соответствующие материалы, и далее они иллюстрируются следующими конкретными примерами. Соединения, проиллюстрированные в примерах, не должны, однако, рассматриваться как образующие единственный род, который рассматривается как изобретение. Примеры дополнительно иллюстрируют подробности получения соединений по настоящему изобретению. Специалисты в данной области техники легко поймут, что можно использовать известные вариации условий и осуществления следующих процедур получения для получения этих соединений. Концентрирование относится к удалению летучих компонентов при пониженном давлении (например, роторное испарение), если не указано иное. Все температуры указаны в градусах Цельсия, если не указано иное. Масс-спектры (МС) измеряли методом масс-спектроскопии с электрораспылительной ионизацией (ESI) в режиме детекции положительных ионов, и m/z относится к [M+H]+ иону, если не указано иное. Спектры 1Н ЯМР регистрировали при 400-500 МГц при температуре окружающей среды, если не указано иное. ОФ-ВЭЖХ относится к обращенно-фазовой ВЭЖХ на С18-функционализированных препаративных или полупрепаративных колонках с градиентным элюированием с использованием ацетонитрила и воды, модифицированной трифторуксусной кислотой, в качестве элюентов, и фракции лиофилизировали или концентрировали в вакууме путем роторного испарения, если не указано иное. Очистку методом колоночной хроматографии на силикагеле осуществляли с использованием системы флэш-хроматографии (например, ISCO или Biotage) и коммерческих предварительно заполненных силикагелем колонках с использованием для элюирования указанных систем растворителей. Описанные в настоящей заявке соединения были синтезированы в виде рацематов, если не указано иное в экспериментальных процедурах и таблицах соединений. Что касается стереоизомеров, энантиомер A относится к более быстро элюирующему энантиомеру, а энантиомер B относится к более медленно элюирующему энантиомеру в точке разделения, и эта номенклатура сохраняется для остальной последовательности синтеза для данного энантиомерного ряда, независимо от возможности того, что последующие промежуточные соединения и конечные соединения могут иметь такой же или противоположный порядок элюирования. Диастереомер 1 относится к более быстро элюирующему диастереомеру, а диастереомер 2 относится к более медленно элюирующему диастереомеру, и эта номенклатура сохраняется для остальной последовательности синтеза для данного диастереомерного ряда, независимо от возможности того, что последующие промежуточные соединения и конечные соединения могут иметь такой же или противоположный порядок элюирования.
Пример 1
Получение промежуточного соединения 1

Стадия A - Синтез промежуточного соединения Int-1a
В 100-л реактор, который продували и поддерживали в инертной атмосфере азота, загружали раствор 5-гидрокси-2-(гидроксиметил)-4H-пиран-4-она (5 кг, 35,18 моль, 1,00 экв.) в дихлорметане (50 л) и 3,4-дигидро-2H-пирана (3,54 кг, 42,08 моль, 1,20 экв.). После этого добавляли моногидрат п-толуолсульфоновой кислоты (60 г, 315 ммоль, 0,01 экв.) несколькими партиями при 10°C за 20 мин. Полученный раствор перемешивали в течение 3 часов при комнатной температуре. Раствор доводили до pH 7 гидроксидом натрия (5 моль/л). Органическую фазу промывали 1×10 л насыщенного солевого раствора и концентрировали в вакууме с получением Int-1a, которое использовали без дополнительной очистки.
Стадия B - Синтез промежуточного соединения Int-1b
В 50-л 4-горлую круглодонную колбу, которую продували и поддерживали в инертной атмосфере азота, загружали раствор Int-1a (5,5 кг, 24,31 моль, 1,00 экв.) в воде (27,5 л), гидроксид натрия (973,5 г, 24,34 моль, 1,00 экв.), формальдегид (2,15 кг, 26,49 моль, 1,09 экв., 37% водный раствор). Полученный раствор перемешивали в течение ночи при комнатной температуре, затем доводили до pH 5 с использованием уксусной кислоты. Полученный раствор экстрагировали при помощи 5×20 л этилацетата и органические слои объединяли. Полученную смесь промывали 5 л насыщенного солевого раствора, затем сушили над безводным сульфатом натрия и концентрировали в вакууме с получением Int-1b, которое использовали без дополнительной очистки.
Стадия C - Синтез промежуточного соединения Int-1c
В 50-л 4-горлую круглодонную колбу, которую продували и поддерживали в инертной атмосфере азота, загружали раствор Int-1b (5,6 кг, 21,85 моль, 1,00 экв.) в N,N-диметилформамиде (20 л), карбонат калия (6,04 кг, 43,70 моль, 2,00 экв.) и бензилбромид (3,93 кг, 22,98 моль, 1,05 экв.). Полученный раствор перемешивали в течение ночи при комнатной температуре. Реакционную смесь затем гасили, выливая в 100 л воды. Полученный раствор экстрагировали при помощи 3×20 л этилацетата и органические слои объединяли и концентрировали в вакууме с получением Int-1c, которое использовали без дополнительной очистки.
Стадия C - Синтез промежуточного соединения 1
В 50-л 4-горлую круглодонную колбу загружали раствор Int-1c (5 кг, 14,44 моль, 1,00 экв.) в дихлорметане (25 л), раствор KBr (343,6 г, 2,89 моль, 0,20 экв.) в воде (5 л), раствор KHCO3 (5,058 кг, 50,58 моль, 3,50 экв.) в воде (20 л) и 2,2,6,6-тетраметилпиперидин 1-оксил (TEMPO) (40,75 г, 0,02 экв.). После этого добавляли по каплям NaClO (30 кг, 32%) при перемешивании при 5°C в течение 4 часов. Полученный раствор перемешивали в течение ночи при комнатной температуре. Полученный раствор экстрагировали при помощи 2×10 л дихлорметана и водные слои объединяли. Значение pH раствора доводили до 3 водным раствором хлористого водорода (6 моль/л). Полученный раствор экстрагировали при помощи 3×20 л этилацетата и органические слои объединяли и сушили над безводным сульфатом натрия и концентрировали в вакууме с получением промежуточного соединения 1. 1H ЯМР (400МГц, CDCl3) δ 7,50 (5H, м), 6,66 (1H, с), 5,65 (2H, с), 4,76 (1H, с), 4,64 (1H, м), 4,45 (1H, м), 3,82 (1H, м), 3,58 (1H, м), 1,69-1,90 (6H, м). Масса, рассчитанная для C19H20O7: 360,1, найдено 361,1 (M+H)+.
Пример 2
Получение соединения 2
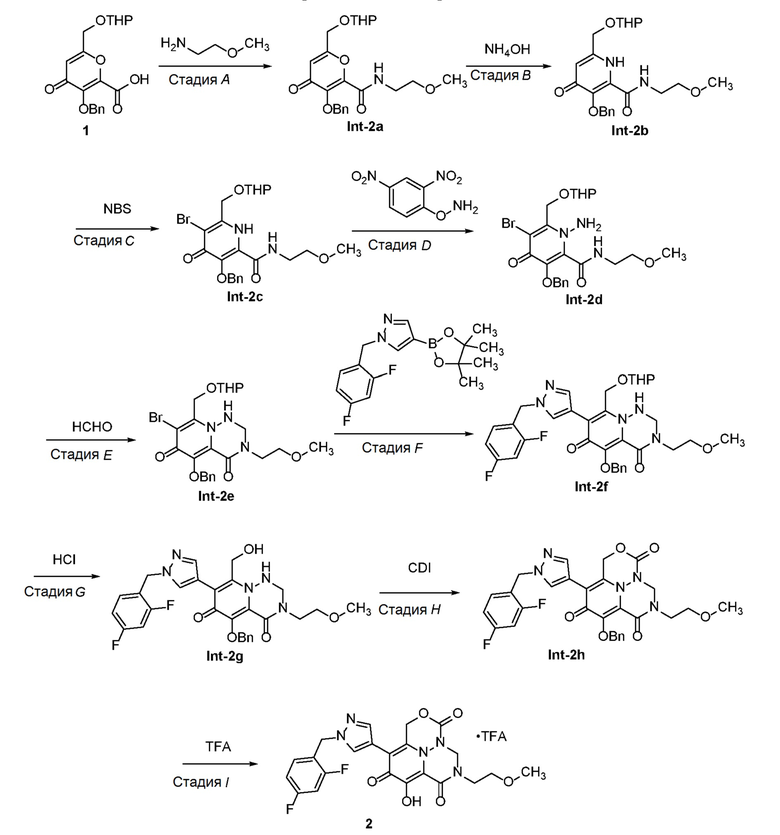
Стадия A - Синтез промежуточного соединения Int-2a
К раствору промежуточного соединения 1 (3,0 г, 8,3 ммоль) в безводном N,N-диметилформамиде (30 мл) добавляли 2-метоксиэтанамин (750 мг, 10,0 ммоль), 1-[бис(диметиламино) метилен]-1H-1,2,3-триазолo[4,5-b]пиридиний 3-оксид гексафторфосфат (1,7 г, 10,0 ммоль) и N,N-диизопропилэтиламин (2,6 г, 20 ммоль). Смесь оставляли для перемешивания при комнатной температуре в течение 16 часов, разбавляли водой и экстрагировали этилацетатом. Объединенные органические части промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Неочищенный продукт очищали с использованием колоночной хроматографии (петролейный эфир:этилацетат=5:1) с получением Int-2a. Масса, рассчитанная для C22H27NO7: 417,2, найдено 418,2 (M+H)+.
Стадия B - Синтез промежуточного соединения Int-2b
К раствору соединения Int-2a (2,5 г, 5,9 ммоль) в этаноле (30 мл) добавляли гидроксид аммония (28% водный раствор, 3 мл) и смесь оставляли для перемешивания при комнатной температуре в течение 2 дней. Смесь концентрировали с получением неочищенного соединения Int-2b, которое использовали без дополнительной очистки. Масса, рассчитанная для C22H28N2O6: 416,2, найдено 417,2 (M+H)+.
Стадия C - Синтез промежуточного соединения Int-2c
К раствору Int-2b (2,0 г, 4,8 ммоль) в дихлорметане (15 мл) добавляли N-бромсукцинимид (885 мг, 5 ммоль) при 0°C. Смесь оставляли для перемешивания при 20°C в течение 16 часов, гасили насыщенным водным раствором NaHCO3, экстрагировали дихлорметаном. Объединенные органические части концентрировали в вакууме с получением Int-2c, которое использовали без дополнительной очистки. Масса, рассчитанная для C22H27BrN2O6: 494,1, 496,1 найдено 495,1, 497,1 (M+H)+.
Стадия D - Синтез промежуточного соединения Int-2d
К раствору Int-2c (1,9 г, 3,85 ммоль) и K2CO3 (690 мг, 5 ммоль) в N,N-диметилформамиде (20 мл) добавляли O-(2,4-динитрофенил)гидроксиламин (895 мг, 4,5 ммоль). Смесь оставляли для перемешивания при 20°C в течение 3 дней. После фильтрования и концентрирования полученный остаток очищали с использованием препаративной ОФ-ВЭЖХ с получением Int-2d. Масса, рассчитанная для C22H28BrN3O6: 509,1, 511,1 найдено 510,1, 512,1 (M+H)+.
Стадия E - Синтез промежуточного соединения Int-2e
К раствору Int-2d (700 мг, 1,38 ммоль) и уксусной кислоты (3 мл) в тетрагидрофуране (20 мл) добавляли параформальдегид (41 мг, 1,38 ммоль). Смесь оставляли для перемешивания при 70°C в течение 12 часов. После концентрирования полученный остаток очищали с использованием препаративной ТСХ на силикагеле (100% этилацетат) с получением Int-2e. 1H ЯМР (400МГц, CD3CN) δ 7,55-7,57(м, 2H), 7,31-7,40(м, 3H), 5,89-5,93 (т, J=8,0 Гц, 1H), 5,17(с, 2H), 5,06-5,09(м, 1H), 4,84-4,87 (м, 1H), 4,75-4,77 (т, J=4,0 Гц, 1H), 4,52-4,54 (м, 2H), 3,80-3,85 (м, 1H), 3,62-3,65 (м, 2H), 3,47-3,54 (м, 3H), 3,32 (с, 3H), 1,58-1,78 (м, 2H), 1,53-1,57 (м, 4H). Масса, рассчитанная для C23H28BrN3O6: 521,1, 523,1, найдено 522,1, 524,1, (M+H)+.
Стадия F - Синтез промежуточного соединения Int-2f
К раствору Int-2e (100 мг, 0,19 ммоль) в диоксане (10 мл) добавляли 1-(2,4-дифторбензил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (96 мг, 0,3 ммоль), Cs2CO3(78 мг, 0,24 ммоль) и Pd(PPh3)4 (21 мг, 0,019 ммоль). Смесь оставляли для перемешивания при 80°C в течение 16 часов, охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом. Объединенные органические части сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ на силикагеле (100% этилацетат) с получением Int-2f, которое использовали без дополнительной очистки. Масса, рассчитанная для C33H35F2N5O6: 635,3, найдено 636,1 (M+H)+.
Стадия G - Синтез промежуточного соединения Int-2g
К раствору Int-2f (80 мг, 0,13 ммоль) в этилацетате (15 мл) добавляли раствор HCl в этилацетате (4 M, 1 мл, 4,0 ммоль) при 0°C. Смесь оставляли для перемешивания при 20°C в течение 1 часа и концентрировали в вакууме с получением Int-2g, которое использовали без дополнительной очистки. Масса, рассчитанная для C28H27F2N5O5: 551,2, найдено 552,1 (M+H)+.
Стадия H - Синтез промежуточного соединения Int-2h
К раствору Int-2g (60 мг, 0,11 ммоль) в дихлорметане (15 мл) добавляли 1,1'-карбонилдиимидазол (24 мг, 0,15 ммоль) при 0°C. Смесь оставляли для перемешивания при 20°C в течение 3 часов и затем концентрировали в вакууме с получением Int-2h, которое использовали без дополнительной очистки. Масса, рассчитанная для C29H25F2N5O6: 577,2, найдено 578,1 (M+H)+.
Стадия I - Синтез соединения 2
К раствору Int-2h (43 мг, 0,07 ммоль) в дихлорметане (5 мл) добавляли трифторуксусную кислоту (1 мл). Смесь оставляли для перемешивания при 20°C в течение 3 часов. Смесь концентрировали в вакууме и полученный остаток очищали с использованием препаративной ОФ-ВЭЖХ с получением соединения 2. 1H ЯМР (400 МГц, CD3CN) 7,98 (с, 1H), 7,56 (с, 1H), 7,28-7,34 (м, 1H), 6,96-7,03 (м, 2H), 5,38 (с, 2H), 5,37 (с, 2H), 5,31 (с, 2H), 3,74-3,76 (т, J=4,0 Гц, 2H), 3,60-3,62 (т, J=4,0 Гц, 2H), 3,34 (с, 3H). Масса, рассчитанная для C22H19F2N5O6: 487,1, найдено 488,1 (M+H)+.
Пример 3
Получение соединения 3
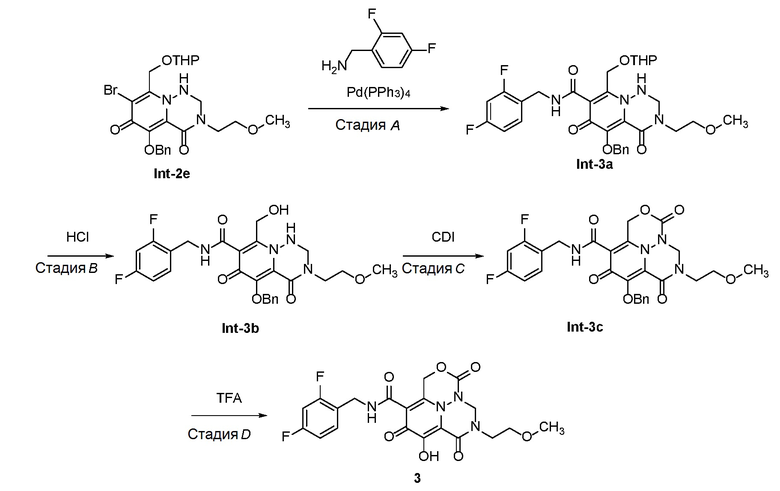
Стадия A - Синтез промежуточного соединения Int-3a
К раствору Int-2e (40 мг, 0,08 ммоль) в диметилсульфоксиде (1 мл) и метаноле (4 мл) добавляли (2,4-дифторфенил)метанамин (55 мг, 0,38 ммоль). Смесь оставляли для перемешивания при 80°C в атмосфере оксида углерода (1 атм) в течение 16 часов. Смесь распределяли между этилацетатом и водой. Органический слой отделяли и концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ на силикагеле (дихлорметан:этилацетат=1:1) с получением соединения Int-3a. 1H ЯМР (400 МГц, CDCl3) δ 10,07 (с, 1H), 7,44-7,63 (м, 2H), 7,30-7,36 (м, 4H), 6,80-7,26 (м, 1H), 5,31-5,54 (м, 1H), 5,23-5,29 (м, 3H), 4,67-4,80 (м, 1H), 4,64 (д, J=5,6 Гц, 2H), 4,51 (д, J=8,0 Гц, 2H), 3,79-3,81 (м, 1H ), 3,67-3,70 (м, 5H), 3,50 (с, 3H), 1,72-1,76 (м, 2H), 1,60 (с, 3H), 1,25 (м, 2H). Масса, рассчитанная для C31H34F2N4O7: 612,2, найдено 613,2 (M+H)+.
Стадия B - Синтез промежуточного соединения Int-3b
К раствору Int-3a (30 мг, 0,05 ммоль) в этилацетате (1 мл) добавляли раствор HCl в этилацетате (4 M, 0,3 мл) при 0°C. Смесь оставляли для перемешивания при комнатной температуре в течение 5 минут и затем концентрировали в вакууме с получением неочищенного Int-3b, которое использовали без дополнительной очистки. Масса, рассчитанная для C26H26F2N4O6: 528,2, найдено 529,1 (M+H)+.
Стадия C - Синтез промежуточного соединения Int-3c
К раствору Int-3b (20 мг, 0,04 ммоль) и 4-диметиламинопиридина (40 мг, 0,32 ммоль) в дихлорметане (2 мл) добавляли 1,1'-карбонилдиимидазол (40 мг, 0,24 ммоль) при 20°C. Смесь оставляли для перемешивания при 20°C в течение 16 часов и затем экстрагировали из воды этилацетатом. Объединенные органические части промывали 10% водным раствором HCl, концентрировали в вакууме и полученный остаток очищали с использованием препаративной ТСХ на силикагеле (дихлорметан:этилацетат=1:1) с получением Int-3c. Масса, рассчитанная для C27H24F2N4O7: 554,2, найдено 555,1 (M+H)+.
Стадия D - Синтез соединения 3
Раствор Int-3c (16 мг, 0,032 ммоль) в трифторуксусной кислоте (2 мл) и дихлорметане (1 мл) оставляли для перемешивания при 20°C в течение 1 часа. Смесь концентрировали в вакууме и полученный остаток очищали с использованием препаративной ОФ-ВЭЖХ с получением соединения 3. 1H ЯМР (400 МГц, CD3OD) δ 7,44-7,48 (м, 1H), 6,93-6,98 (м, 2H), 5,95 (с, 2H), 5,49 (с, 2H), 4,60 (с, 2H), 3,77-3,83 (м, 2H), 3,65-3,67 (м, 2H), 3,38 (с, 3H). Масса, рассчитанная для C20H18F2N4O7: 464,1, найдено 465,1 (M+H)+.
Пример 4
Получение соединения 4
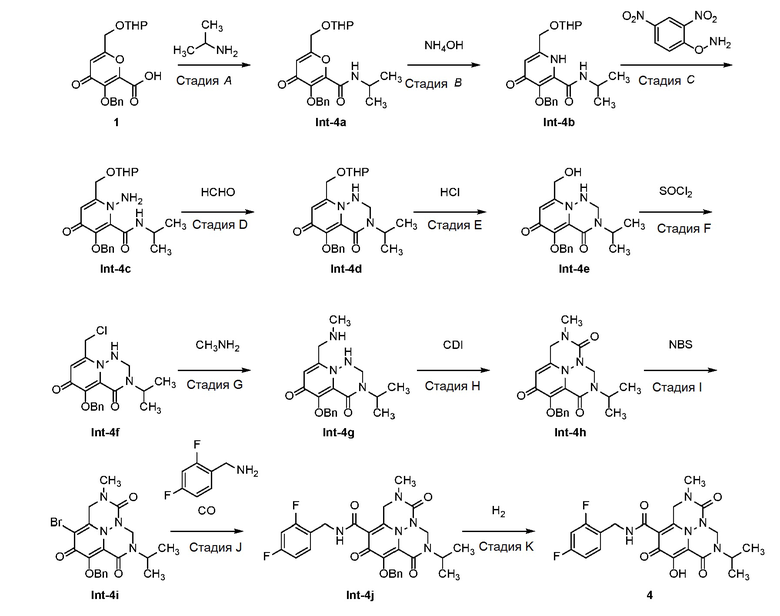
Стадия A - Синтез промежуточного соединения Int-4a
К раствору промежуточного соединения 1 (2,1 г, 6,0 ммоль) в N,N-диметилформамиде (20 мл) добавляли HOAT (1,6 г, 12,0 ммоль), HATU (4,5 г, 12,0 ммоль) и пропан-2-амин (1 мл) при 25°C. Смесь перемешивали в течение 36 часов при 25°C, гасили водой и экстрагировали этилацетатом. Объединенные органические слои промывали водой и насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали. Полученный остаток очищали с использованием колоночной хроматографии на силикагеле (петролейный эфир/этилацетат 5:1 до 1:1) с получением Int-4a. 1H ЯМР (400 МГц, CDCl3) δ 7,56 (д, J=7,2 Гц, 1H), 7,35-7,39 (м, 5H), 6,57 (с, 1H), 5,37 (с, 2H), 4,72 (с, 1H), 4,61-4,57 (м, 1H), 4,39-4,44 (м, 1H), 3,97-4,06 (м, 1H), 3,76-3,82 (м, 1H), 3,49-3,54 (м, 1H), 1,84-1,50 (м, 6H), 0,94 (д, J=6,8 Гц, 6H). Масса, рассчитанная для C22H27NO6: 401,2, найдено 402,2 (M+H)+.
Стадия B - Синтез промежуточного соединения Int-4b
Раствор Int-4a (1,4 г, 3,0 ммоль) в этаноле (10 мл) обрабатывали гидроксидом аммония (28% водный раствор, 30 мл) при 25°C. Смесь перемешивали в течение 20 часов при 25°C и затем концентрировали с получением Int-4b, которое использовали без дополнительной очистки. Масса, рассчитанная для C22H28N2O5: 400,2, найдено 401,2 (M+H)+.
Стадия C - Синтез промежуточного соединения Int-4c
Раствор Int-4b (1,4 г, 3,0 ммоль) в N,N-диметилформамиде (15 мл) обрабатывали K2CO3 (828 мг, 6,0 ммоль) и O-(2,4-динитрофенил)гидроксиламином (716 мг, 3,6 ммоль). Смесь перемешивали в течение 20 часов при 25°C и затем фильтровали. Неочищенный продукт очищали с использованием препаративной ОФ-ВЭЖХ с получением Int-4c. 1H ЯМР (400 МГц, CDCl3) δ 7,62 (д, J=5,6 Гц, 1H), 7,36-7,39 (м, 2H), 7,28-7,30 (м, 3H), 6,39 (с, 1H), 5,33 (с, 2H), 5,07-5,11 (м, 2H), 4,68-4,73 (м, 2H), 4,49 (д, J=14,4 Гц, 1H), 4,03-4,12 (м, 1H), 3,79-3,85 (м, 1H), 3,51-3,56 (м, 1H), 1,54-1,86 (м, 6H), 1,08 (д, J=6,4 Гц, 6H). Масса, рассчитанная для C22H29N3O5: 415,2, найдено 416,2 (M+H)+.
Стадия D - Синтез промежуточного соединения Int-4d
К раствору соединения Int-4c (100 мг, 0,24 ммоль) в тетрагидрофуране (20 мл) добавляли параформальдегид (18 мг, 0,6 ммоль) и уксусную кислоту (0,8 мл). Смесь нагревали до 80°C в течение 3 часов и затем концентрировали в вакууме с получением неочищенного продукта Int-4d, который использовали без дополнительной очистки. 1H ЯМР (400 МГц, CD3OD) δ 7,45-7,46 (м, 2H), 7,29-7,30 (м, 3H), 6,68 (с, 1H), 5,19 (с, 2H), 4,76-4,78 (м, 2H), 4,61-4,64 (с, 3H), 4,07-4,08 (м, 1H), 3,82-3,84 (м, 1H), 3,52-3,55 (м, 1H), 1,57-2,00 (м, 6H), 1,22 (д, J=6,8 Гц, 6H); Масса, рассчитанная для C23H29N3O5: 427,2, найдено 428,2 (M+H)+.
Стадия E - Синтез промежуточного соединения Int-4e
К раствору соединения Int-4d (52 мг, 0,121 ммоль) в этилацетате (5 мл) добавляли раствор HCl в этилацетате (4 M, 1,5 мл) при 0°C. Смесь перемешивали в течение 20 минут при 0°C и затем концентрировали в вакууме с получением неочищенного Int-4e, которое использовали на следующей стадии без дополнительной очистки. Масса, рассчитанная для C18H21N3O4: 343,2, найдено 344,2 (M+H)+.
Стадия F - Синтез промежуточного соединения Int-4f
К раствору соединения Int-4e (40 мг, 0,116 ммоль) в дихлорметане (12 мл) добавляли SOCl2 (0,1 мг, 0,166 ммоль) при 0°C. Смесь перемешивали в течение 4 часов при 25°C и затем концентрировали в вакууме с получением неочищенного Int-4f, которое использовали без дополнительной очистки. Масса, рассчитанная для C18H20ClN3O3: 361,1, найдено 362,2 (M+H)+.
Стадия G - Синтез промежуточного соединения Int-4g
Соединени Int-4f (40 мг, 0,086 ммоль) растворяли с MeNH2 в метаноле (5 мл), смесь оставляли для перемешивания при комнатной температуре в течение 20 мин. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта Int-4g (37 мг, выход: 94,8%), который использовали на следующей стадии без дополнительной очистки. Масса, рассчитанная для C19H24N4O3: 356,2, найдено 357,2 (M+H)+.
Стадия H - Синтез промежуточного соединения Int-4h
К раствору неочищенного Int-4g (37 мг, 0,103 ммоль) в диметилсульфоксиде/дихлорметане (1 мл/10 мл) добавляли 4-диметиламинопиридин (101 мг, 0,82 ммоль) и CDI (101 мг, 0,62 ммоль). Смесь оставляли для перемешивания при комнатной температуре в течение 3 дней и затем разбавляли дихлорметаном (20 мл), промывали 0,5% водным раствором HCl и водой (15 мл), сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного Int-4h, которое использовали без дополнительной очистки. Масса, рассчитанная для C20H22N4O4: 382,2, найдено 383,2 (M+H)+.
Стадия I - Синтез промежуточного соединения Int-4i
К раствору соединения Int-4h (30 мг, 0,078 ммоль) в ацетонитриле (20 мл) добавляли N-бромсукцинимид (20 мг, 0,12 ммоль). Смесь оставляли для перемешивания при комнатной температуре в течение 1 часа и затем концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ (100% этилацетат) с получением соединения Int-4i. 1H ЯМР (400 МГц, CD3OD) δ 7,53-7,55 (м, 2H), 7,34-7,37 (м, 3H), 5,26 (с, 2H), 5,17 (с, 2H), 4,64-4,65 (с, 2H), 4,64 (м, 1H), 3,06 (с, 3H), 1,30 (д, J=8 Гц, 6H). Масса, рассчитанная для C20H21BrN4O4: 460,1, найдено 461,1 (M+H)+.
Стадия J - Синтез промежуточного соединения Int-4j
Раствор Int-4i (17 мг, 0,037 ммоль) в диметилсульфоксиде (2 мл) и метаноле (6 мл) обрабатывали (2,4-дифторфенил)метанамином (26 мг, 0,184 ммоль), N,N-диизопропилэтиламином (24 мг, 0,18 ммоль) и Pd(PPh3)4 (1,53 мг, 0,0013 ммоль). Смесь перемешивали в атмосфере оксида углерода (1 атм) при 80°C в течение 15 часов, охлаждали до комнатной температуры и гасили водой (4 мл). Смесь экстрагировали этилацетатом и органические части промывали водой и концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ на силикагеле (дихлорметан:MeOH=20:1) с получением Int-4j. Масса, рассчитанная для C28H27F2N5O5: 551,2, найдено 552,2 (M+H)+.
Стадия K - Синтез соединения 4
Раствор Int-4j (17 мг, 0,031 ммоль) в дихлорметане (1 мл) обрабатывали трифторуксусной кислотой (2 мл) при 25°C. Смесь оставляли для перемешивания при 25°C в течение 1 часа и затем концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ОФ-ВЭЖХ с получением соединения 4. 1H ЯМР (400 МГц, CD3OD) δ 7,45-7,50 (м, 1H), 6,94-7,00 (м, 2H), 5,37 (с, 2H), 5,13 (с, 2H), 4,75-4,80 (м, 1H), 4,62 (с, 2H), 3,02 (с, 3H), 1,36 (т, J=6,8 Гц, 6H). Масса, рассчитанная для C21H21F2N5O5: 461,2, найдено 462,2 (M+H)+.
Пример 5
Получение соединения 5
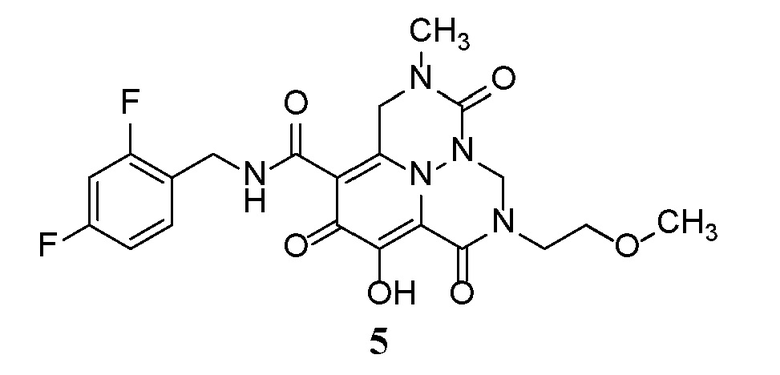
Соединение 5 получали с использованием способов, описанных в Примере 4. 1H ЯМР (400 МГц, CD3OD) δ 7,45-7,51 (м, 1H), 6,94-7,00 (м, 2H), 5,43 (с, 2H), 5,13 (с, 2H), 4,62 (с, 2H), 3,82 (т, J=9,6 Гц, 2H), 3,68 (т, J=9,6 Гц, 2H), 3,40 (м, 3H), 3,02 (с, 3H). Масса, рассчитанная для C21H21F2N5O6: 477,1, найдено 478,2 (M+H)+.
Пример 6
Получение соединения 6
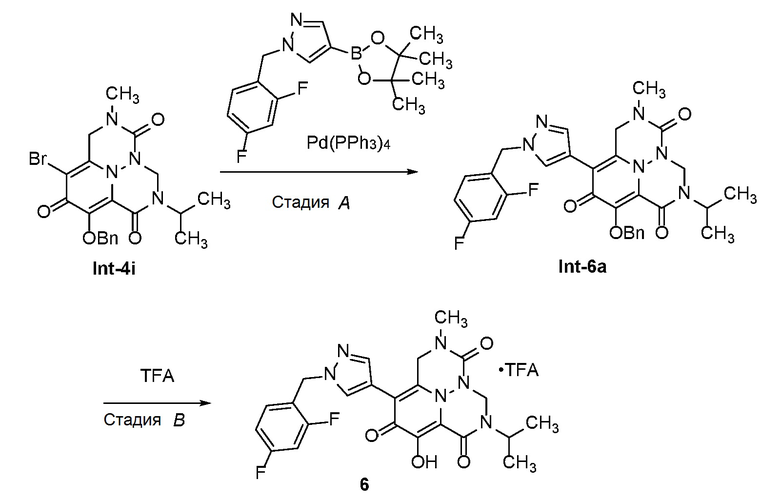
Стадия A - Синтез промежуточного соединения Int-6a
К раствору соединения Int-4i (25 мг, 0,053 ммоль) в диоксане (5 мл) добавляли Cs2CO3 (35 мг, 0,108 ммоль), Pd(PPh3)4 (20 мг, 0,02 ммоль) и 1-(2,4-дифторбензил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (34 мг, 0,108 ммоль) при комнатной температуре. Смесь нагревали в условиях микроволнового облучения при 130°C в течение 1 часа. Смесь фильтровали и фильтрат непосредственно очищали с использованием препаративной ОФ-ВЭЖХ с получением соединения Int-6a. Масса, рассчитанная для C30H28F2N6O4: 574,2, найдено 575,2 (M+H)+.
Стадия B - Синтез соединения 6
К раствору соединения Int-6a (18 мг, 0,031 ммоль) в дихлорметане (1 мл) добавляли трифторуксусную кислоту (2 мл) при 25°C. Смесь оставляли для перемешивания при 25°C в течение 1 часа и затем концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ОФ-ВЭЖХ с получением соединения 6. 1H ЯМР (400 МГц, CD3OD) δ 8,13 (с, 1H), 7,74 (с, 1H), 7,38-7,44 (м, 1H), 6,98-7,07 (м, 2H), 5,45-5,48 (м, 4H), 4,77-4,84 (м, 1H), 4,60 (с, 2H), 2,79 (с, 3H), 1,37 (т, J=6,8 Гц, 6H). Масса, рассчитанная для C23H22F2N6O4: 484,2, найдено 485,2 (M+H)+.
Пример 7
Получение соединения 7

Стадия A - Синтез промежуточного соединения Int-7a
К раствору Int-4d (900 мг, 2,105 ммоль) в дихлорметане (50 мл) добавляли 4-диметиламинопиридин (25,7 мг, 0,211 ммоль) и ди-трет-бутилдикарбонат (689 мг, 3,16 ммоль). Смесь оставляли для перемешивания при комнатной температуре в течение 16 часов и затем концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ на силикагеле (петролейный эфир:этилацетат=1:1) с получением Int-7a. 1H ЯМР (400 МГц, CD3OD) δ 7,47-7,48 (м, 2H), 7,30-7,31 (м, 3H), 6,66 (д, J=16,0 Гц, 1H), 5,25 (с, 2H), 4,71-4,73 (м, 5H), 4,42-4,57 (м, 1H), 3,78-3,83 (м, 1H), 3,52-3,54 (м, 1H), 1,57-1,87 (м, 6H), 1,45 (с, 9H), 1,20 (дд, J=6,8, 6,8 Гц, 6H).
Стадия B - Синтез промежуточного соединения Int-7b
Раствор Int-7a (450 мг, 0,855 ммоль) в этилацетате (20 мл) обрабатывали раствором HCl в этилацетате (4 M, 6 мл). Смесь оставляли для перемешивания при 0°C в течение 2 часов и затем концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ (100% этилацетат) с получением Int-7b. 1H ЯМР (400 МГц, CD3OD) δ 7,46-7,48 (м, 2H), 7,30-7,31 (м, 3H), 6,67 (с, 1H), 5,16-5,34 (м, 2H), 4,60-4,68 (м, 3H), 1,45 (с, 9H), 1,20 (дд, J=6,8, 6,8 Гц, 6H).
Стадия C - Синтез промежуточного соединения Int-7c
К раствору Int-7b (250 мг, 0,564 ммоль) в дихлорметане (15 мл) добавляли N,N-диизопропилэтиламин (947 мг, 7,33 ммоль), диметилсульфоксид (881 мг, 11,27 ммоль) и пиридин-SO3 (219 мг,1,378ммоль). Смесь оставляли для перемешивания при комнатной температуре в течение 16 часов, промывали водным раствором HCl (0,5 м), сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением неочищенного Int-7c, которое использовали без дополнительной очистки. 1H ЯМР (400 МГц, CD3OD) δ 7,46-7,48 (м, 2H), 7,30-7,31 (м, 3H), 6,78 (с, 1H), 5,16-5,19 (м, 2H), 4,56-4,61 (м, 2H), 1,43 (с, 9H), 1,24 (м, 6H).
Стадия D - Синтез промежуточного соединения Int-7d
К раствору триметилсульфоний иодида (285 мг, 1,812 ммоль) в диметилсульфоксиде (6 мл) добавляли гидрид натрия (54,4 мг, 2,265 ммоль) и смесь оставляли для перемешивания при комнатной температуре в течение 40 мин. К смеси добавляли раствор Int-7c (200 мг, 0,453 ммоль) в диметилсульфоксиде (2 мл) и перемешивали при комнатной температуре в течение 30 мин. Смесь разбавляли водой (4 мл) при 0°C и фильтровали. Фильтрат очищали с использованием препаративной ВЭЖХ с получением Int-7d. 1H ЯМР (400 МГц, CD3OD) δ 7,49-7,50 (м, 2H), 7,33-7,34 (м, 3H), 7,15 (с, 1H), 5,54 (т, J=13,2 Гц, 1H), 5,29 (с, 2H), 4,80-4,82 (м, 1H), 4,50-4,59 (м, 2H), 3,82-3,86 (м, 1H), 3,38-3,42 (м, 1H), 1,24 (д, J=6,4 Гц, 1H); Масса, рассчитанная для C19H21N3O4: 355,2, найдено 356,2 (M+H)+.
Стадия E - Синтез промежуточного соединения Int-7e
К раствору Int-7d (30 мг, 0,084 ммоль) в ацетонитриле (8 мл) добавляли N-бромсукцинимид (22,54 мг, 0,127 ммоль). Смесь оставляли для перемешивания при комнатной температуре в течение 30 мин, концентрировали в вакууме и полученный остаток очищали с использованием препаративной ТСХ на силикагеле (100% этилацетат) с получением Int-7e. 1H ЯМР (400 МГц, CD3OD) δ 7,50-7,51 (м, 2H), 7,30-7,32 (м, 3H), 5,51 (т, J=13,2 Гц, 1H), 5,26 (дд, J=6,8, 6,8 Гц, 2H), 4,67-4,70 (м, 3H), 4,18-4,19 (м, 1H), 3,61-3,64 (м, 1H), 1,18-1,26 (м, 6H); Масса, рассчитанная для C19H20BrN3O4: 433,1, найдено 434,2 (M+H)+.
Стадия F - Синтез промежуточного соединения Int-7f
К раствору Int-7e (20 мг, 0,046 ммоль) в N,N-диметилформамиде (3 мл) добавляли гидрид натрия (1,105 мг, 0,046 ммоль) при 0°C. Смесь оставляли для перемешивания при 0°C в течение 30 мин, обрабатывали иодметаном (6,54 мг, 0,046 ммоль), перемешивали при 0°C в течение 2 часов, гасили водой и экстрагировали этилацетатом. Объединенные органические части сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ на силикагеле (100% этилацетат) с получением Int-7f. 1H ЯМР (400 МГц, CD3OD) δ 7,48-7,50 (м, 2H), 7,28-7,30 (м, 3H), 5,51 (т, J=13,2 Гц, 1H), 5,17-5,25 (м, 2H), 4,04-4,12 (м, 3H), 3,83-3,86 (м, 1H), 3,52 (с, 3H), 1,16 (д, J=7,2 Гц, 6H); Масса, рассчитанная для C20H22BrN3O4: 447,1, найдено 448,2 (M+H)+.
Стадия G - Синтез промежуточного соединения Int-7g
К раствору Int-7f (20 мг, 0,045 ммоль) в метаноле (3 мл) и диметилсульфоксиде (1 мл) добавляли Pd(PPh3)4 (11,60 мг, 0,010 ммоль), (2,4-дифторфенил)метанамин (12,8 мг, 0,09 ммоль) и N,N-диизопропилэтиламин (5,6 мг, 0,05 ммоль). Смесь оставляли для перемешивания при 90°C в течение 2 часов в атмосфере оксида углерода (1 атм), фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ (этилацетат) с получением Int-7g. 1H ЯМР (400 МГц, CD3OD) 7,43-7,60 (м, 6H), 6,91-6,95 (м, 2H), 5,95 (д, J=4,2 Гц, 1H), 5,26 (дд, J=6,8, 6,8 Гц, 2H), 4,77-4,79 (м, 2H), 4,58-4,63 (м, 3H), 4,03-4,06 (м, 1H), 3,86-3,89 (м, 1H), 3,45 (с, 1H), 1,16-1,24 (м, 6H); Масса, рассчитанная для C28H28F2N4O5: 538,2, найдено 539,3 (M+H)+.
Стадия H - Синтез соединения 7
Раствор Int-7g (15 мг, 0,028 ммоль) в N,N-диметилформамиде (3 мл) обрабатывали хлоридом лития (11,8 мг, 0,28 ммоль) при комнатной температуре. Смесь оставляли для перемешивания при 110°C в течение 30 мин, охлаждали до комнатной температуры и непосредственно очищали с использованием препаративной ОФ-ВЭЖХ с получением соединения 7. 1H ЯМР (400 МГц, CD3OD) δ 7,45-7,46 (м, 1H), 6,95-6,98 (м, 2H), 5,98-6,00 (д, 1H), 5,10-5,20 (м, 1H), 4,63-4,66 (м, 2H), 4,29-4,32 (м, 3H), 3,94-3,97 (м, 1H), 3,49 (с, 3H), 3,30-3,40 (м, 2H), 1,24-1,35 (м, 6H); Масса, рассчитанная для C21H22F2N4O5: 448,2, найдено 449,2 (M+H)+.
Пример 8
Получение соединения 8
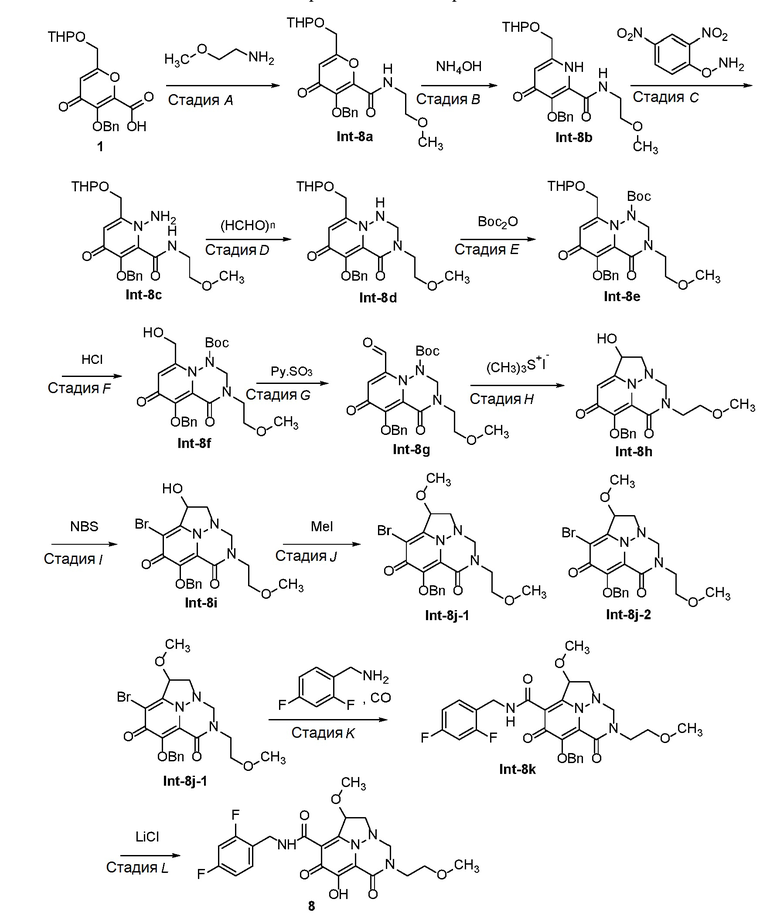
Стадия A - Синтез промежуточного соединения Int-8a
К раствору соединения 1 (72 г, 199,7 ммоль), 2-метоксиэтанамина (30 г, 398 ммоль), HOAT (35,4 г, 259,7 ммоль), HATU (98,78 г, 261,1 ммоль) в N,N-диметилформамиде (500 мл) добавляли N,N-диизопропилэтиламин (71,02 г, 600 ммоль) при 0°C. Смесь оставляли для перемешивания при 20°C в течение 16 часов, разбавляли водой и экстрагировали этилацетатом. Объединенные органические части промывали насыщенным солевым раствором (150 мл), сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием колоночной хроматографии на силикагеле (петролейный эфир:этилацетат=1,5:1) с получением Int-8a. 1H ЯМР (400 МГц, CD3OD) δ 8,09 (шир.с, 1H), 7,28-7,51 (м, 5H), 6,59 (с, 1H), 5,41 (с, 2H), 4,69-4,78 (м, 1H), 4,61 (д, J=15,3 Гц, 1H), 4,42 (д, J=15,3 Гц, 1H), 3,77-3,86 (м, 1H), 3,45-3,58 (м, 3H), 3,36-3,43 (м, 2H), 3,28 (с, 3H), 1,51-1,78 (м, 6H). Масса, рассчитанная для C22H27NO7: 417,2, найдено 418,1 (M+H)+.
Стадия B - Синтез промежуточного соединения Int-8b
Раствор Int-8a (24 г, 57,5 ммоль) и гидроксида аммония (28% водный раствор, 77 мл) в этаноле (50 мл) оставляли для перемешивания при 25°C в течение 20 часов. Смесь концентрировали в вакууме с получением Int-8b, которое использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 8,60 (с, 1H), 7,23-7,52 (м, 5H), 6,29-6,43 (м, 1H), 5,49 (с, 2H), 4,35-4,77 (м, 4H), 3,93 (м, 1H), 3,39-3,69 (м, 4H), 3,18-3,36 (м, 3H), 1,54-1,92 (м, 6H). Масса, рассчитанная для C22H28N2O6: 416,2, найдено 417,2 (M+H)+.
Стадия C - Синтез промежуточного соединения Int-8c
К раствору Int-8b (23 г, 45,2 ммоль) и K2CO3 (12,50 г, 90 ммоль) в N,N-диметилформамиде (200 мл) добавляли O-(2,4-динитрофенил)гидроксиламин (13,51 г, 67,8 ммоль) по порциям при перемешивании при 25°C. Смесь оставляли для перемешивания при 25°C в течение 26 часов. Развитие реакции отслеживали методом ТСХ (этилацетат). Смесь фильтровали и фильтрат очищали с использованием препаративной ОФ-ВЭЖХ (вода с 0,05% NH4OH/ацетонитрил) с получением Int-8c. 1H ЯМР (400 МГц, CDCl3) δ 10,15-10,35 (м, 1H), 8,59 (с, 1H), 7,20-7,48 (м, 5H), 6,36 (с, 1H), 5,47 (с, 2H), 4,45-4,69 (м, 3H), 3,90-3,99 (м, 1H), 3,36-3,64 (м, 4H), 3,21-3,34 (м, 3H), 1,47-1,86 (м, 6H). Масса, рассчитанная для C22H29N3O6: 431,2, найдено 432,3 (M+H)+.
Стадия D - Синтез промежуточного соединения Int-8d
К раствору Int-8c (12,5 г, 29,0 ммоль) в уксусной кислоте (10 мл) и тетрагидрофуране (100 мл) добавляли параформальдегид (0,869 г, 29,0 ммоль). Смесь оставляли для перемешивания при 80°C в течение 18 часов. Смесь концентрировали в вакууме и полученный остаток растворяли в дихлорметане, промывали насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием колоночной хроматографии на силикагеле (этилацетат до этилацетат:метанол=9:1) с получением Int-8d. 1H ЯМР (400 МГц, CD3OD) δ 7,48 (д, J=5,5 Гц, 2H), 7,29 (д, J=5,9 Гц, 3H), 6,65 (с, 1H), 5,37 (т, J=6,3 Гц, 1H), 5,23 (с, 2H), 4,48 (с, 2H), 3,73 (с, 2H), 3,49-3,62 (м, 3H), 3,34 (с, 3H), 3,18-3,24 (м, 1H), 2,88 (с, 2H), 1,47-1,86 (м, 6H). Масса, рассчитанная для C23H29N3O6: 443,2, найдено 444,2 (M+H)+.
Стадия E - Синтез промежуточного соединения Int-8e
Раствор Int-8d (9 г, 20,3 ммоль ), триэтиламина (8,49 мл, 60,9 ммоль) и ди-трет-бутилдикарбоната (9,42 мл, 40,6 ммоль) в дихлорметане (100 мл) обрабатывали 4-диметиламинопиридином (0,248 г, 2,029 ммоль). Смесь оставляли для перемешивания при 25°C в течение 16 часов, концентрировали в вакууме и полученный остаток очищали с использованием колоночной хроматографии на силикагеле (петролейный эфир:этилацетат=1:1 до 1:2) с получением Int-8e. 1H ЯМР (400 МГц, CD3OD) δ 7,20-7,57 (м, 5H), 6,63 (д, J=12,9 Гц, 1H), 5,10-5,36 (м, 3H), 4,84 (д, J=3,5 Гц, 1H), 4,67-4,73 (м, 1H), 4,53 (с, 1H), 3,99 (д, J=13,7 Гц, 1H), 3,51 (д, J=5,1 Гц, 2H), 3,42 (дд, J=6,26, 12,5 Гц, 1H), 3,29 (шир.с, 6H), 1,54-1,98 (м, 6H), 1,06-1,50 (м, 9H). Масса, рассчитанная для C28H37N3O8: 543,3, найдено 544,2 (M+H)+.
Стадия F - Синтез промежуточного соединения Int-8f
К раствору Int-8e (9 г, 16,56 ммоль) в этилацетате (10 мл) добавляли раствор HCl в этилацетате (4 M, 4,14 мл) при 0°C. Смесь оставляли для перемешивания при 25°C в течение 10 минут и затем концентрировали в вакууме. Полученный остаток очищали с использованием колоночной хроматографии на силикагеле (этилацетат:метанол=100:2) с получением Int-8f. 1H ЯМР (400 МГц, CDCl3) δ 7,60 (д, J=6,4 Гц, 2H), 7,30-7,40 (м, 3H), 7,17 (с, 1H), 5,33 (д, J=9,2 Гц, 1H), 5,19 (д, J=9,2 Гц, 1H), 5,12 (д, J=11,2 Гц, 2H), 4,97 (с, 1H), 4,77 (д, J=14,8 Гц, 1H), 4,39 (д, J=14,8 Гц, 1H), 3,94 (д, J=12,8 Гц, 2H), 3,47 (с, 2H), 3,26-3,34 (м, 3H), 1,42 (с, 9H). Масса, рассчитанная для C23H29N3O7: 459,2, найдено 460,2 (M+H)+.
Стадия G - Синтез промежуточного соединения Int-8g
Раствор Int-8f (9 г, 10,88 ммоль), диметилсульфоксида (15,44 мл, 217 ммоль) и N,N-диизопропилэтиламина (24,7 мл, 141,6 ммоль) в дихлорметане (150 мл) обрабатывали комплексом триоксид серы-пиридин (37,4 г, 235 ммоль). Смесь оставляли для перемешивания при 25°C в течение 16 часов, разбавляли дихлорметаном, промывали водным раствором HCl (1 N) и насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением Int-8g, которое использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 9,81 (с, 1H), 7,52 (д, J=6,4 Гц, 2H), 7,21-7,32 (м, 3H), 6,81-6,88 (м, 1H), 5,50 (д, J=10,6 Гц, 1H), 5,24-5,36 (м, 2H), 4,79 (д, J=13,6 Гц, 1H), 4,19 (д, J=14,4 Гц, 1H), 3,43-3,53 (м, 3H), 3,26-3,31 (м, 3H), 1,35 (с, 9H). Масса, рассчитанная для C23H27N3O7: 457,2, найдено 458,2 (M+H)+.
Стадия H - Синтез промежуточного соединения Int-8h
К раствору триметилсульфоний иодида (2140 мг, 10,48 ммоль) в N,N-диметилформамиде (8 мл) добавляли NaH (840 мг, 21 ммоль) и смесь перемешивали в атмосфере азота при 25°C в течение 2 часов. Смесь обрабатывали по каплям раствором Int-8g (1,2 г, 2,62 ммоль) в N,N-диметилформамиде (15 мл) при 0°C и смесь оставляли для перемешивания при 0°C в течение 10 минут в атмосфере азота. Смесь разбавляли водой (4 мл) при 0°C и фильтровали. Фильтрат непосредственно очищали с использованием препаративной ОФ-ВЭЖХ с получением Int-8h. 1H ЯМР (400 МГц, CD3OD) δ 7,48 (д, J=5,1 Гц, 2H), 7,29 (д, J=5,6 Гц, 3H), 6,65 (с, 1H), 5,37 (т, J=6,4 Гц, 1H), 5,23 (с, 2H), 4,48 (с, 2H), 3,73 (с, 2H), 3,62 (с, 1H), 3,55 (т, J=4,8 Гц, 2H), 3,34 (с, 3H), 3,23 (с, 1H). Масса, рассчитанная для C19H21N3O5: 371,1, найдено 372,2 (M+H)+.
Стадия I - Синтез промежуточного соединения Int-8i
N-бромсукцинимид (144 мг, 0,808 ммоль) добавляли к раствору Int-8h (200мг, 0,539 ммоль) в ацетонитриле (10 мл). Смесь оставляли для перемешивания при 25°C в течение 3 часов и затем концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ на силикагеле (этилацетат:метанол=8:1) с получением Int-8i. 1H ЯМР: (400 МГц, CD3OD) δ 7,49-7,51 (м, 2H), 7,29-7,30 (м, 3H), 5,47-5,49 (м, 1H), 5,28 (д, J=10,4 Гц 1H), 5,20 (д, J=10,4 Гц 1H), 4,63-4,66 (м, 1H), 4,37-4,40 (м, 1H), 3,64-3,84 (м, 1H), 3,56-3,63 (м, 4H), 3,32-3,34 (м, 4H). Масса, рассчитанная для C19H20BrN3O5: 449,1, 451,1, найдено 450,1, 452,1 (M+H)+.
Стадия J - Синтез промежуточного соединения Int-8j-1 (энантиомер A) и Int-8j-2 (энантиомер B)
Раствор Int-8i (100 мг, 0,222 ммоль) в N,N-диметилформамиде (5 мл) обрабатывали гидридом натрия (6,92 мг, 0,289 ммоль) при 0°C. После перемешивания при 0°C в течение 30 минут добавляли иодметан (47,3 мг, 0,332 ммоль). Реакционную смесь оставляли для перемешивания при 0°C в течение 10 мин, разбавляли водой и экстрагировали этилацетатом. Объединенные органические части сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ на силикагеле (100% этилацетат) с получением Int-8j в виде рацемата. 1H ЯМР(400 МГц, CD3OD) δ 7,52 (д, J=6,4 Гц, 2H), 7,25-7,36 (м, 3H), 5,25-5,35 (м, 2H), 5,21 (д, J=10,4 Гц, 1H), 4,62-4,73 (м, 2H), 4,39 (д, J=10,4 Гц, 1H), 3,79-3,96 (м, 3H), 3,50-3,64 (м, 5H), 3,36 (с, 3H). Масса, рассчитанная для C20H22BrN3O5: 463,1, 465,1, найдено 464,0, 466,0 (M+H)+.
Разделение энантиомеров осуществляли методом СФХ (AD, 250 мм × 30 мм, 10 мкм, SC-CO2/метанол=55/45 при 80 мл/мин) с получением Int-8j-1(энантиомер A) и Int-8j-1 (энантиомер B).
Стадия K - Синтез промежуточного соединения Int-8k
Раствор Int-8j-1(энантиомер A) (20 мг, 0,043 ммоль), (2,4-дифторфенил)метанамина (18,50 мг, 0,129 ммоль) и N,N-диизопропилэтиламина (0,038 мл, 0,215 ммоль) в диметилсульфоксиде (2 мл) и метаноле (2 мл) обрабатывали при помощи Pd(Ph3P)4 (24,89 мг, 0,022 ммоль). Смесь оставляли для перемешивания при 89°C в атмосфере оксида углерода (1 атм) в течение 3 часов. Смесь охлаждали до комнатной температуры, разбавляли водным раствором HCl (1N, 5 мл) и экстрагировали этилацетатом. Объединенные органические части промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ на силикагеле (100% этилацетат) с получением Int-8k. 1H ЯМР: (400 МГц, CD3OD) δ 7,52-7,59 (м, 2H), 7,45-7,52 (м, 1H), 7,30-7,38 (м, 3H), 6,94-7,04 (м, 2H), 5,95 (д, J=5,6 Гц, 1H), 5,20-5,35 (м, 2H), 4,74 (д, J=11,2 Гц, 1H), 4,65 (д, J=7,2 Гц, 2H), 4,35-4,39 (м, 1H), 3,84-3,94 (м, 2H), 3,61-3,69 (м, 1H), 3,57-3,61 (м, 2H), 3,48-3,52 (м, 3H), 3,38 (с, 3H), 3,12 (дд, J=5,2, 11,0 Гц, 1H). Масса, рассчитанная для C28H28F2N4O6: 554,2, найдено 555,2 (M+H)+.
Стадия L - Синтез соединения 8
Раствор Int-8k (5 мг, 0,009 ммоль) и хлорид лития (3,82 мг, 0,09 ммоль) в N,N-диметилформамиде (2 мл) оставляли для перемешивания при 80°C в течение 5 часов. Смесь охлаждали до комнатной температуры, фильтровали и фильтрат непосредственно очищали с использованием препаративной ОФ-ВЭЖХ с получением соединения 8. 1H ЯМР 0361628-0112-1: (400 МГц, CD3OD) δ 7,36-7,48 (м, 1H), 6,83-7,01 (м, 2H), 5,91 (д, J=4,4 Гц, 1H), 4,44-4,70 (м, 3H), 3,88 (д, J=10,4 Гц, 2H), 3,52-3,75 (м, 3H), 3,30-3,51 (м, 6H), 3,09-3,27 (м, 2H). Масса, рассчитанная для C21H22F2N4O6: 464,2, найдено 465,2 (M+H)+.
Пример 9
Получение соединения 9
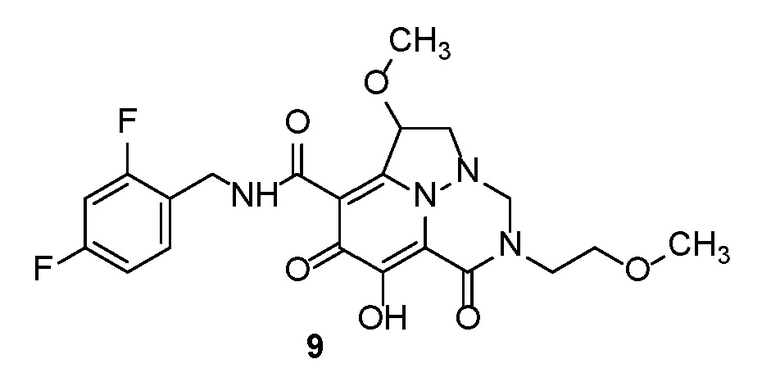
Соединение 9 получали из Int-8j-2 (энантиомер B) с использованием способов, описанных в Примере 8.
1H ЯМР (400 МГц, CD3OD) δ 7,36-7,49 (м, 1H), 6,82-7,02 (м, 2H), 5,90 (д, J=4,4 Гц, 1H), 4,83 (с, 1H), 4,45-4,71 (м, 3H), 3,88 (д, J=10,4 Гц, 2H), 3,50-3,73 (м, 3H), 3,29-3,49 (м, 6H), 3,09-3,19 (м, 1H). Масса, рассчитанная для C21H22F2N4O6: 464,2, найдено 465,2 (M+H)+.
Следующие соединения по настоящему изобретению получали с использованием способов, описанных в Примерах 8 и 9, с соответствующими заменами взаимодействующих веществ и/или реагентов.
[M+H]+
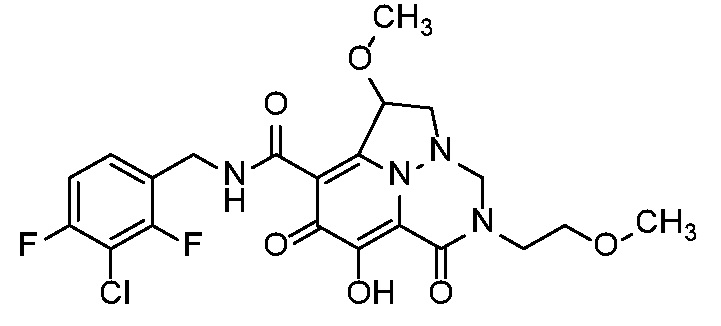
найдено 499,1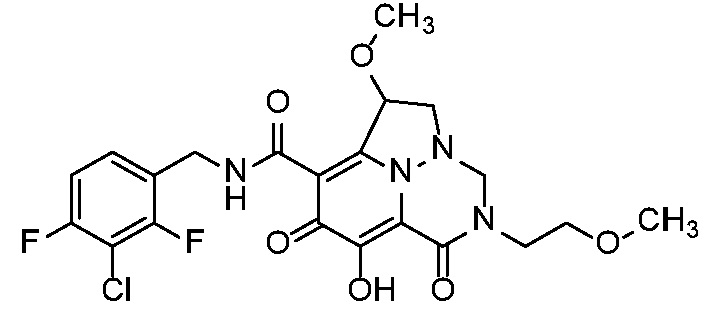
найдено 499,1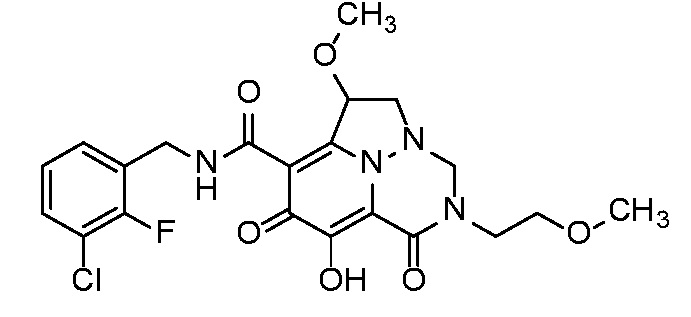
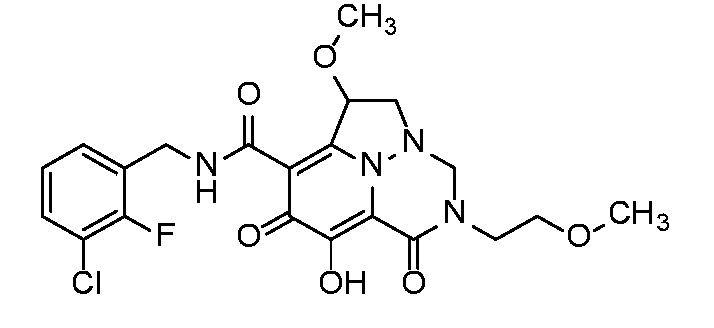
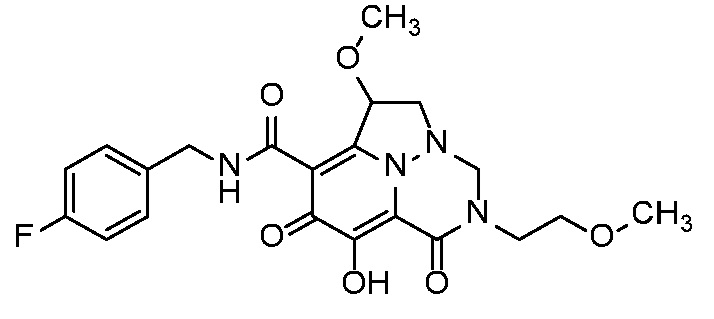
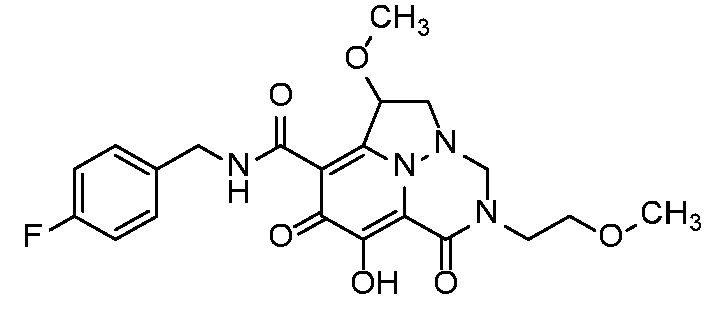
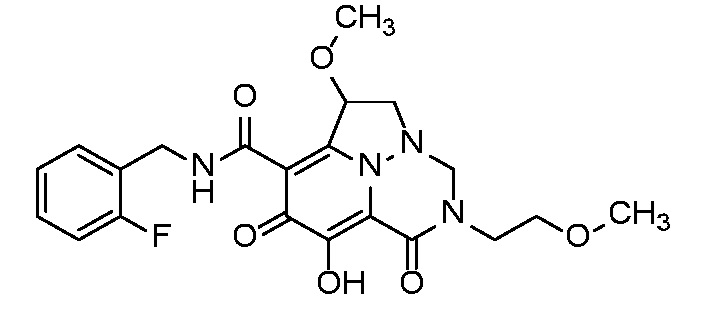
Пример 10
Получение соединения 17
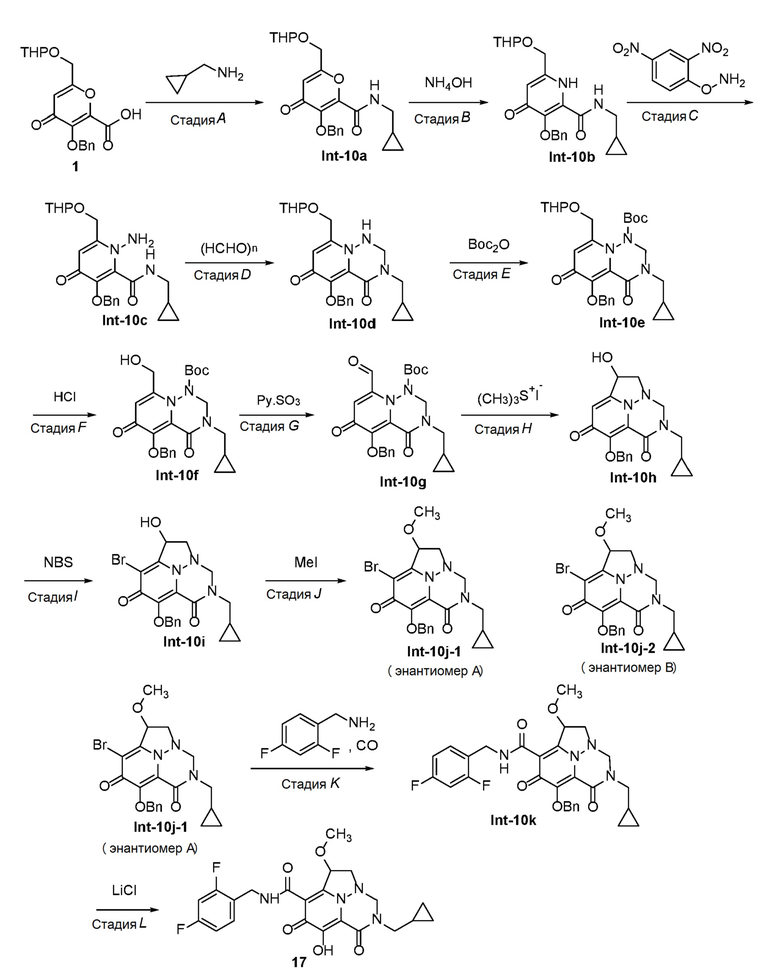
Стадия A - Синтез промежуточного соединения Int-10a
К раствору соединения 1 (15 г, 41,6 ммоль) в N,N-диметилформамиде (200 мл) добавляли 6-хлор-бензотриазол-1-ил-окси-трис-пирролидинoфосфоний гексафторфосфат (34,6 г, 62,4 ммоль), N,N-диизопропилэтиламин (10,76 г, 83 ммоль) и циклопропилметанамин (5,92 г, 83 ммоль), смесь оставляли для перемешивания при 20°C в течение 16 часов. Добавляли воду (100 мл) и смесь экстрагировали этилацетатом. Объединенные органические части промывали насыщенным солевым раствором (100 мл), сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием колоночной хроматографии на силикагеле (петролейный эфир/этилацетат=5:1) с получением соединения Int-10a. 1H ЯМР (400 МГц, CD3OD) δ 7,45-7,46 (м, 2H), 7,36-7,37 (м, 2H), 6,58 (с, 1H), 5,36 (с, 2H), 4,75-4,77 (м, 1H), 4,60 (д, J=16 Гц, 1H), 4,47 (д, J=16 Гц, 1H), 3,83-3,89 (м, 1H), 3,53-3,56 (м, 1H), 3,33-3,37 (м, 2H), 1,57-1,76 (м, 6H), 1,12-1,14 (м, 1H), 0,59-0,60 (м, 2H), 0,36-0,37 (м, 2H). Масса, рассчитанная для C23H27NO6: 413,2, найдено 414,2 (M+H)+.
Стадия B - Синтез промежуточного соединения Int-10b
Раствор соединения Int-10a (8 г, 19,35 ммоль) и гидроксида аммония (28% водный раствор, 60 мл) в этаноле (30 мл) оставляли для перемешивания при 22°C в течение 20 часов. Смесь концентрировали в вакууме с получением неочищенного соединения Int-10b, которое использовали без дополнительной очистки. 1H ЯМР (400 МГц, CD3OD) δ 7,31-7,32 (м, 2H), 7,21-7,22 (м, 3H), 6,44 (с, 1H), 5,28 (с, 2H), 4,42-4,59 (м, 3H), 3,79-3,81 (м, 1H), 3,41-3,44 (м, 1H), 2,97-2,99 (м, 2H), 1,45-1,65 (м, 6H), 0,65-0,75 (м, 1H), 0,28-0,37 (м, 2H), 0,09-0,10 (м, 2H). Масса, рассчитанная для C23H28N2O5: 412,2, найдено 413,2 (M+H)+.
Стадия C - Синтез промежуточного соединения Int-10c
К раствору Int-10b (3 г, 7,3 ммоль) и K2CO3 (3,0 г, 21,8 ммоль) в N,N-диметилформамиде (50 мл) добавляли O-(2,4-динитрофенил)гидроксиламин (2,2 г, 10,9 ммоль) по порциям при перемешивании при 25°C. Смесь оставляли для перемешивания при 25°C в течение 16 часов. Смесь фильтровали и фильтрат очищали с использованием препаративной ОФ-ВЭЖХ (вода с 0,05% NH4OH/ацетонитрил) с получением Int-10c. 1H ЯМР (400 МГц, CD3OD) δ 7,11-7,14 (м, 2H), 7,22-7,24 (м, 3H), 6,45 (с, 1H), 4,94 (с, 2H), 4,43-4,57 (м, 3H), 3,64-3,68 (м, 1H), 3,35-3,38 (м, 1H), 2,96-2,97 (м, 2H), 1,37-1,68 (м, 6H), 0,71-0,75 (м, 1H), 0,22-0,24 (м, 2H), 0,09-0,10 (м, 2H). Масса, рассчитанная для C23H29N3O5: 427,2, найдено 428,2 (M+H)+.
Стадия D - Синтез промежуточного соединения Int-10d
К раствору Int-10c (4 г, 3,96 ммоль) в уксусной кислоте (2 мл) и тетрагидрофуране (40 мл) добавляли параформальдегид (0,309 г, 10,3 ммоль). Смесь оставляли для перемешивания при 80°C в течение 18 часов. Смесь концентрировали в вакууме и полученный остаток растворяли в дихлорметане (50 мл), промывали NaHCO3, насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением Int-10d, которое использовали без дополнительной очистки. Масса, рассчитанная для C24H29N3O5: 439,2, найдено 440,2 (M+H)+.
Стадия E - Синтез промежуточного соединения Int-10e
К раствору Int-10d (3,5 мг, 7,9 ммоль), триэтиламина (2,22 мл, 15,9 ммоль) и ди-трет-бутилдикарбоната (3,70 мл, 15,93 ммоль) в дихлорметане (100 мл) добавляли 4-диметиламинопиридин (0,097 г, 0,796 ммоль). Смесь оставляли для перемешивания при 20°C в течение 16 часов. Смесь концентрировали в вакууме и полученный остаток очищали с использованием колоночной хроматографии на силикагеле (петролейный эфир:этилацетат=1:1 до 1:2) с получением Int-10e. 1H ЯМР (400 МГц, CD3OD) δ 7,47-7,48 (м, 2H), 7,29-7,30 (м, 3H), 6,64 (д, J=14,4 Гц, 1H), 5,18-5,42 (м, 3H), 4,42-4,73 (м, 5H), 3,78-3,80 (м, 1H), 3,50-3,55 (м, 2H), 3,19-2,21 (м, 1H), 1,57-1,77 (м, 6H), 1,45 (с, 1H), 1,05-1,09 (м, 1H), 0,54-0,61 (м, 2H), 0,34-0,35 (м, 2H). Масса, рассчитанная для C29H37N3O7: 539,3, найдено 540,1 (M+H)+.
Стадия F - Синтез промежуточного соединения Int-10f
К раствору Int-10e (3,2 г, 5,93 ммоль) в этилацетате (30 мл) добавляли раствор HCl в этилацетате (4 M, 4 мл) при 0°C. Смесь оставляли для перемешивания при 20°C в течение 10 мин. Смесь концентрировали в вакууме и полученный остаток очищали с использованием колоночной хроматографии на силикагеле (этилацетат:метанол=100:2) с получением Int-10f. 1H ЯМР (400 МГц, CD3OD) δ 7,12-7,13 (м, 2H), 6,95-6,96 (м, 3H), 6,52 (с, 1H), 5,08 (д, J=14 Гц, 1H), 4,92 (д, J=10 Гц, 1H), 4,86 (д, J=10 Гц, 2H), 4,30 (д, J=16 Гц, 1H), 4,10 (д, J=16 Гц, 1H), 3,18-3,23 (м, 1H), 2,98-3,10 (м, 2H), 2,83-2,88 (м, 1H), 1,11 (с, 9H). 0,65-0,75 (м, 1H), 0,20-0,27 (м, 2H), 0,05-0,10 (м, 2H). Масса, рассчитанная для C24H29N3O6: 455,2, найдено 456,2 (M+H)+.
Стадия G - Синтез промежуточного соединения Int-10g
К раствору Int-10f (2,2 г, 4,83 ммоль), диметилсульфоксида (6,86 мл, 97 ммоль) и N,N-диизопропилэтиламина (10,97 мл, 62,8 ммоль) в дихлорметане (50 мл) добавляли комплекс триоксид серы-пиридин (9,22 г, 58 ммоль). Смесь оставляли для перемешивания при 20°C в течение 16 часов. Смесь разбавляли дихлорметаном (150 мл). Органические фазы промывали водным раствором HCl (1 N, 3 × 25 мл) и насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением Int-10g, которое использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 9,54 (с, 1H), 7,23-7,25 (м, 2H), 6,96-7,00 (м, 3H), 6,58 (с, 1H), 5,10-5,23 (м, 2H), 4,36-4,49 (м, 2H), 3,29-3,36 (м, 1H), 2,86-2,92 (м, 1H), 1,07 (с, 9H), 0,65-0,75 (м, 1H), 0,26-0,30 (м, 2H), 0,10-0,16 (м, 2H). Масса, рассчитанная для C24H27N3O6: 453,2, найдено 454,2 (M+H)+.
Стадия H - Синтез промежуточного соединения Int-10h
Раствор триметилсульфоний иодида (360 мг, 1,76 ммоль) в N,N-диметилформамиде (8 мл) обрабатывали при помощи NaH (152 мг, 3,79 ммоль) и смесь перемешивали в атмосфере азота при 20°C в течение 1,5 часов. К смеси добавляли по каплям раствор Int-10g (200 мг, 0,441 ммоль) в N,N-диметилформамиде (12 мл) при 20°C и смесь оставляли для перемешивания при 20°C в течение 30 минут в атмосфере азота. Смесь разбавляли водой (2 мл) при 0°C и фильтровали. Фильтрат непосредственно очищали с использованием препаративной ОФ-ВЭЖХ с получением Int-10h. 1H ЯМР (400 МГц, CD3OD) δ 7,16-7,17 (м, 2H), 6,96-7,00 (м, 3H), 6,84 (с, 1H), 5,20 (т, J=12,4 Гц, 1H), 4,94 (с, 2H), 4,34 (с, 2H), 3,48-3,50 (м, 1H), 3,00-3,16 (м, 3H), 0,70-0,78 (м, 1H), 0,22-0,27 (м, 2H), 0,14-0,26 (м, 2H). Масса, рассчитанная для C20H21N3O4: 367,2, найдено 368,2 (M+H)+.
Стадия I - Синтез промежуточного соединения Int-10i
N-бромсукцинимид (145 мг, 0,817 ммоль) добавляли к раствору Int-10h (200 мг, 0,554 ммоль) в ацетонитриле (5 мл). Смесь оставляли для перемешивания при 25°C в течение 20 минут. Смесь концентрировали в вакууме и полученный остаток очищали с использованием препаративной ТСХ на силикагеле (этилацетат:метанол=14:1) с получением Int-10i. 1H ЯМР (400 МГц, CD3OD) δ 7,54-7,56 (м, 2H), 7,31-7,35 (м, 3H), 5,51-5,54 (м, 1H), 5,31 (д, J=10 Гц, 1H), 5,24 (д, J=10,4 Гц, 1H), 4,76-4,76 (м, 1H), 4,44-4,46 (м, 1H), 3,55-3,62 (м, 2H), 3,36-3,40 (м, 2H), 1,12-1,14 (м, 1H), 0,57-0,61 (м, 2H), 0,35-0,38 (м, 2H). Масса, рассчитанная для C20H20BrN3O4: 445,1, найдено 446,1 (M+H)+.
Стадия J - Синтез промежуточного соединения Int-10j-1 (энантиомер A) и Int-10j-2 (энантиомер B)
К раствору Int-10i (142 мг, 0,318 ммоль) в N,N-диметилформамиде (5 мл) добавляли гидрид натрия (25,5 мг, 0,636 ммоль) при 25°C. После перемешивания при 25°C в течение 2 минут добавляли иодметан (90 мг, 0,636 ммоль). Реакционную смесь оставляли для перемешивания при 25°C в течение 5 минут и затем разбавляли водой (5 мл) и экстрагировали этилацетатом. Объединенные органические части сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ на силикагеле (этилацетат:метанол=12:1) с получением Int-10j в виде рацемата. 1H ЯМР (400 МГц, CD3OD) δ 7,54 (д, J=6 Гц, 2H), 7,32-7,34 (м, 3H), 5,22-5,35 (м, 3H), 4,78 (д, J=10,4 Гц, 1H), 4,43 (д, J=10,4 Гц, 1H), 3,85-3,88 (м, 1H), 3,55-3,62 (м, 4H), 3,36-3,38 (м, 2H), 1,12-1,14 (м, 1H), 0,57-0,62 (м, 2H), 0,35-0,37 (м, 2H). Масса, рассчитанная для C21H22BrN3O4: 459,1, найдено 460,0 (M+H)+.
Разделение осуществляли методом СФХ (AD, 250 мм × 30 мм, 10 мкм, SC-CO2/метанол 60/40 при 80 мл/мин) с получением Int-10j-1 (энантиомер A) и Int-10j-2 (энантиомер B).
Стадия K - Синтез промежуточного соединения Int-10k
К раствору соединения Int-10j-1 (14 мг, 0,030 ммоль), (2,4-дифторфенил)метанамина (8,71 мг, 0,061 ммоль) и N,N-диизопропилэтиламина (0,010 мл, 0,061 ммоль) в диметилсульфоксиде (2 мл) и метаноле (2 мл) добавляли Pd(Ph3P)4 (3,51 мг, 3,04 ммоль). Смесь оставляли для перемешивания при 90°C в атмосфере оксида углерода (1 атм) в течение 3 часов. Смесь охлаждали до комнатной температуры, разбавляли водным раствором HCl (1 N, 4 мл) и экстрагировали этилацетатом. Объединенные органические части промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Неочищенное вещество очищали с использованием препаративной ТСХ на силикагеле (100% этилацетат) с получением Int-10k. 1H ЯМР (400 МГц, CD3OD) δ 7,29-7,52 (м, 6H), 6,95-6,97 (м, 2H), 5,95 (д, J=4,8 Гц, 1H), 5,19-5,32 (м, 2H), 4,60-4,64 (м, 3H), 4,33-4,36 (м, 1H), 3,62-3,89 (м, 2H), 3,57-3,62 (м, 2H), 3,47 (с, 3H), 0,73-0,75 (м, 1H), 0,56-0,58 (м, 2H), 0,35-0,37 (м, 2H). Масса, рассчитанная для C29H28F2N4O5: 550,2, найдено 551,2 (M+H)+.
Стадия L - Синтез соединения 17
Раствор Int-10k (16 мг, 0,029 ммоль) и хлорида лития (4,93 мг, 0,116 ммоль) в N,N-диметилформамиде (5 мл) оставляли для перемешивания при 80°C в течение 3 часов. Смесь фильтровали и фильтрат очищали с использованием препаративной ОФ-ВЭЖХ с получением соединения 17. 1H ЯМР (400 МГц, CD3OD) δ 7,41-7,47 (м, 1H), 7,91-6,99 (м, 2H), 5,95 (д, J=5,60 Гц, 1H), 4,64-4,68 (м, 2H), 4,53-4,61 (м, 2H), 3,92 (д, J=10,06 Гц, 1H), 3,63-3,65 (м, 2H), 3,47 (с, 3H), 3,18-3,21 (м, 1H), 1,12-1,14 (м, 1H), 0,59-0,60 (м, 2H), 0,36-0,37 (м, 2H). Масса, рассчитанная для C22H22F2N4O5: 460,2, найдено 461,2 (M+H)+.
Пример 11
Получение соединения 18
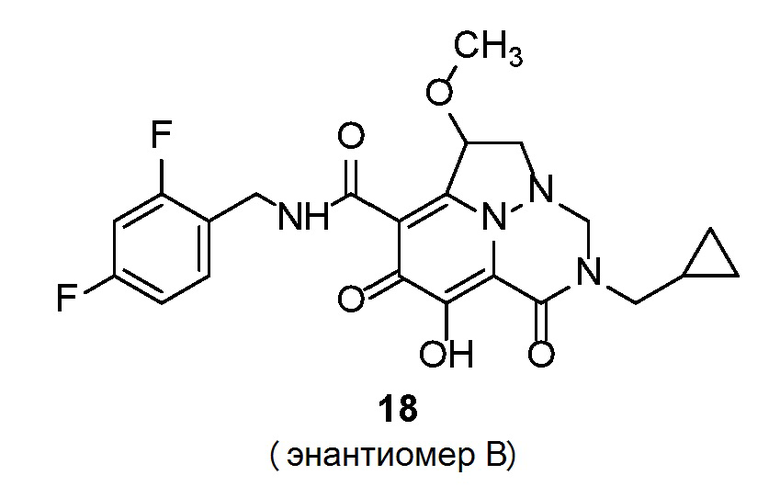
Соединение 18 получали из Int-10j-2(энантиомер B) с использованием способа, описанного в Примере 10. 1H ЯМР (400 МГц, CD3OD) δ 7,41-7,47 (м, 1H), 7,91-6,99 (м, 2H), 5,95 (д, J=5,60 Гц, 1H), 4,64-4,68 (м, 2H), 4,54-4,61 (м, 2H), 3,93 (д, J=10,10 Гц, 1H), 3,63-3,65 (м, 2H), 3,47 (с, 3H), 3,18-3,21 (м, 1H), 1,12-1,14 (м, 1H), 0,59-0,60 (м, 2H), 0,36-0,37 (м, 2H). Масса, рассчитанная для C22H22F2N4O5: 460,2, найдено 461,2 (M+H)+.
Следующие соединения по настоящему изобретению получали с использованием способов, описанных в Примерах 10 и 11, с соответствующими заменами взаимодействующих веществ и/или реагентов.
[M+H]+
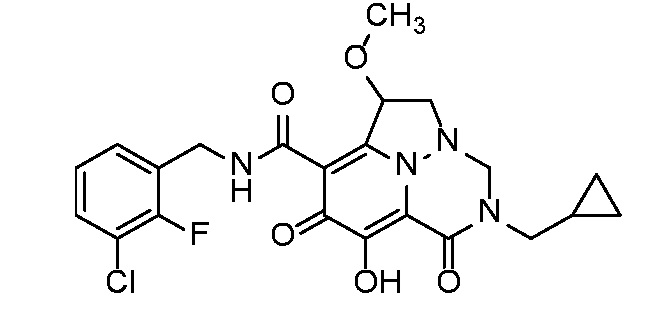
найдено 477,2
найдено 477,2
Пример 12
Получение соединения 22
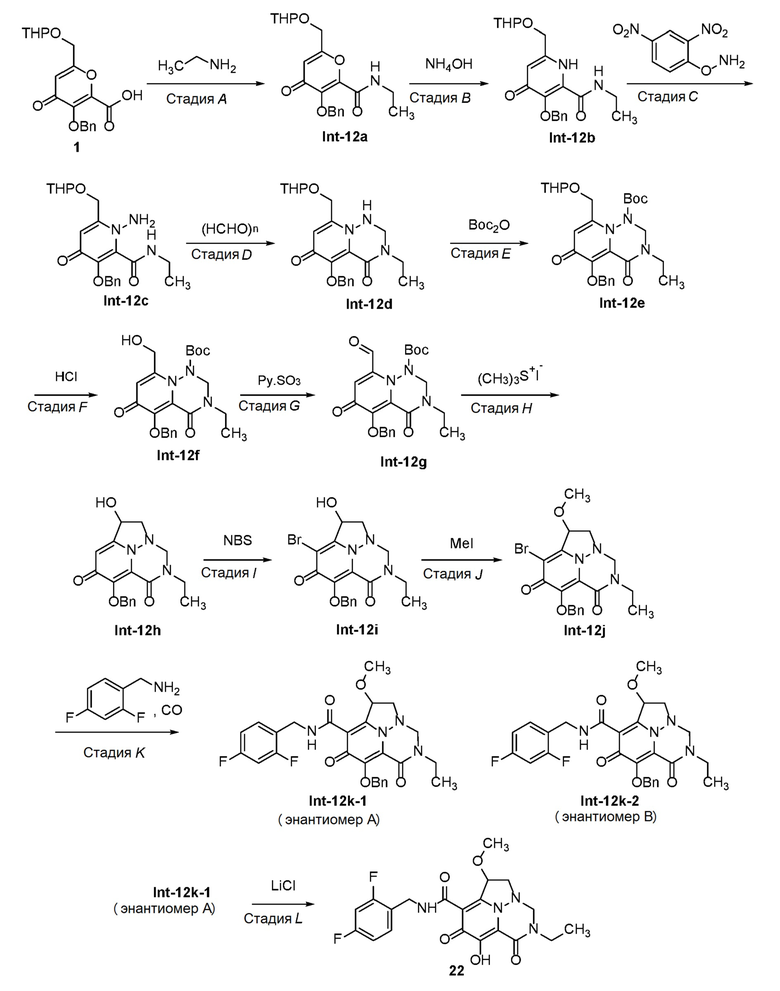
Стадия A - Синтез промежуточного соединения Int-12a
К смеси соединения 1 (25 г, 69,4 ммоль), гидрохлорида этанамина (11,31 г, 139 ммоль) и 6-хлор-бензотриазол-1-ил-окси-трис-пирролидинoфосфоний гексафторфосфата (61,6 г, 111 ммоль) в N,N-диметилформамиде (150 мл) добавляли N,N-диизопропилэтиламин (36,3 мл, 208 ммоль) при 0°C. Смесь оставляли для перемешивания при 25°C в течение 16 часов. Смесь разбавляли водой и экстрагировали этилацетатом. Объединенные органические части промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием колоночной хроматографии на силикагеле (петролейный эфир:этилацетат=1,5:1) с получением Int-12a. 1H ЯМР (400 МГц, CDCl3): δ 7,66 (шир.с, 1H), 7,30-7,50 (м, 5H), 6,60 (с, 1H), 5,38 (с, 2H), 4,73 (с, 1H), 4,41-4,63 (м, 2H), 3,75-3,85 (м, 1H), 3,50-3,60 (м, 1H), 3,20-3,30 (м, 2H), 1,50-1,75 (м, 6H), 0,95 (т, J=7,6 Гц, 3H). Масса, рассчитанная для C21H25NO6: 387,2, найдено 388,2 (M+H)+.
Стадия B - Синтез промежуточного соединения Int-12b
К раствору Int-12a (20 г, 46,5 ммоль) в этаноле (70 мл) добавляли гидроксид аммония (28% водный раствор, 17,89 мл, 465 ммоль). Полученную смесь оставляли для перемешивания при 25°C в течение 16 часов. Смесь концентрировали в вакууме с получением Int-12b, которое использовали без дополнительной очистки. 1H ЯМР (400 МГц, CD3OD): δ 7,25-7,50 (м, 5H), 6,58 (с, 1H), 5,41 (с, 2H), 4,50-5,00 (м, 3H), 3,80-4,00 (м, 1H), 3,50-3,60 (м, 1H), 3,26 (q, J=7,6 Гц, 2H), 1,50-1,70 (м, 6H), 1,02 (т, J=7,0 Гц, 3H). Масса, рассчитанная для C21H26N2O5: 386,2, найдено 387,2 (M+H)+.
Стадия C - Синтез промежуточного соединения Int-12c
К раствору Int-12b (19 г, 41,8 ммоль) в N,N-диметилформамиде (80 мл) добавляли K2CO3 (11,55 г, 84 ммоль) и O-(2,4-динитрофенил)гидроксиламин (12,48 г, 62,7 ммоль) при 0°C. Полученную смесь оставляли для перемешивания при 25°C в течение 48 часов. Смесь фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием колоночной хроматографии на силикагеле (этилацетат в петролейном эфире 33% до 100%, затем MeOH в этилацетате: 0 до 15%) с получением Int-12c. 1H ЯМР (400 МГц, CD3OD): δ 7,25-7,55 (м, 5H), 6,65 (с, 1H), 5,12 (с, 2H), 4,50-4,80 (м, 3H), 3,83-3,86 (м, 1H), 3,55-3,58 (м, 1H), 3,20-3,30 (м, 2H), 1,55-2,02 (м, 6H), 1,12 (т, J=7,2 Гц, 3H). Масса, рассчитанная для C21H27N3O5: 401,2, найдено 402,1 (M+H)+.
Стадия D - Синтез промежуточного соединения Int-12d
К раствору Int-12c (11 г, 20,90 ммоль) в THF (40 мл) и AcOH (4,00 мл) добавляли параформальдегид (1,556 мл, 20,90 ммоль). Полученную смесь нагревали при кипячении с обратным холодильником в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме и полученный остаток обрабатывали насыщенным водным раствором NaHCO3 (2×50 мл). Водный слой экстрагировали этилацетатом и объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали. Полученный остаток очищали с использованием колоночной хроматографии на силикагеле (петролейный эфир в этилацетате: 25% до 80%) с получением Int-12d. 1H ЯМР (400 МГц, CD3OD): δ 7,47-7,49 (м, 2H), 7,30-7,34 (м, 3H), 6,68 (с, 1H), 5,20 (с, 2H), 4,75-4,78 (м, 2H), 4,60 (д, J=14,8 Гц, 1H), 4,47 (с, 2H), 3,77- 3,90 (т, J=5,2 Гц, 1H), 3,49-3,56 (м, 3H), 1,60-1,78 (м, 6H), 1,19-1,26 (м, 3H). Масса, рассчитанная для C22H27N3O5: 413,2, найдено 414,2 (M+H)+.
Стадия E - Синтез промежуточного соединения Int-12e
К раствору Int-12d (8,14 г, 17,72 ммоль) в дихлорметане (40 мл) добавляли ди-трет-бутилдикарбонат (8,14 мл, 35,4 ммоль), триэтиламин (5,38 г, 53,2 ммоль) и 4-диметиламинопиридин (0,216 г, 1,772 ммоль). Смесь оставляли для перемешивания при 20°C в течение 16 часов и затем концентрировали в вакууме. Полученный остаток очищали с использованием колоночной хроматографии на силикагеле (этилацетат в петролейном эфире: 25% до 50%) с получением Int-12e. 1H ЯМР (400 МГц, CDCl3): δ 7,61 (д, J=7,2 Гц, 2H), 7,27-7,35 (м, 3H), 6,60 (д, J=10,8 Гц, 1H), 5,44 (д, J=10,8 Гц, 1H), 5,32 (д, J=10,8 Гц, 1H), 5,15 (д, J=12,8 Гц, 1H), 4,32-4,50 (м, 2H), 3,61-3,65 (м, 2H), 3,40-3,54 (м, 2H), 1,52-2,05 (м, 6H), 1,43 (с, 9H), 1,22 (т, J=7,0 Гц, 3H). Масса, рассчитанная для C27H35N3O7: 513,2, найдено 514,3 (M+H)+.
Стадия F - Синтез промежуточного соединения Int-12f
К раствору Int-12e (6 г, 11,68 ммоль) в этилацетате (50 мл) добавляли раствор HCl в этилацетате (4 M, 3 мл) и смесь оставляли для перемешивания при 20°C в течение 10 мин. Смесь концентрировали в вакууме и полученный остаток очищали с использованием хроматографии на силикагеле (этилацетат: MeOH=100: 2) с получением Int-12f. 1H ЯМР (400 МГц, CD3OD): δ 7,45-7,50 (м, 2H), 7,29-7,40 (м, 3H), 6,68 (с, 1H), 5,10-5,30 (м, 2H), 4,37-4,60 (м, 2H), 3,50-3,60 (м, 1H), 3,30-3,40 (м, 1H), 1,46 (с, 9H), 1,20 (т, J=6,8 Гц, 3H).
Стадия G - Синтез промежуточного соединения Int-12g
К раствору Int-12f (4,4 г, 10,25 ммоль) в дихлорметане (100 мл) добавляли N,N-диизопропилэтиламин (23,26 мл, 133 ммоль), DMSO (14,54 мл, 205 ммоль) и комплекс пиридин-триоксид серы (19,55 г, 123 ммоль). Смесь оставляли для перемешивания при 20°C в течение 16 часов. Смесь промывали водным раствором HCl (0,5 M, 50 мл), сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением Int-12g. 1H ЯМР (400 МГц, CD3OD): δ 9,80 (с, 1H), 7,50-7,60 (м, 2H), 7,26-7,34 (м, 3H), 6,86 (с, 1H), 4,80-5,50 (м, 4H), 3,50-3,70 (м, 1H), 3,25-3,35 (м, 1H), 1,40 (с, 9H), 1,21 (т, J=7,4 Гц, 3H). Масса, рассчитанная для C22H25N3O6: 427,2, найдено 428,1 (M+H)+.
Стадия H - Синтез промежуточного соединения Int-12h
К раствору триметилсульфоний иодида (344 мг, 1,684 ммоль) в N,N-диметилформамиде (8 мл) добавляли NaH (135 мг, 3,37 ммоль) и смесь оставляли для перемешивания при 20°C в течение 1,5 часов. Смесь обрабатывали раствором Int-12g (200 мг, 0,421 ммоль) в N,N-диметилформамиде (12,00 мл) и перемешивали при 20°C в течение 30 мин. Смесь обрабатывали водой и фильтровали. Фильтрат очищали с использованием препаративной ОФ-ВЭЖХ с получением Int-12h. 1H ЯМР (400 МГц, CD3OD): δ 7,51-7,52 (м, 2H), 7,34-7,36 (м, 3H), 7,20 (с, 1H), 5,55-5,58 (д, J=7,2 Гц, 1H), 5,29 (с, 2H), 4,63 (с, 2H), 3,83-3,85 (м, 1H), 3,62-3,68 (т, J=7,2 Гц, 2H), 3,41-3,43 (м, 1H), 1,24-1,28 (д, J=7,0 Гц, 3H). Масса, рассчитанная для C18H19N3O4: 341,1, найдено 341,9 (M+H)+.
Стадия I - Синтез промежуточного соединения Int-12i
К раствору Int-12h (60 мг, 0,176 ммоль) в ацетонитриле (8 мл) добавляли N-бромсукцинимид (37,5 мг, 0,211 ммоль). Смесь оставляли для перемешивания при 25°C в течение 30 мин, концентрировали в вакууме и полученный остаток очищали с использованием препаративной ТСХ на силикагеле (100% этилацетат) с получением Int-12i. 1H ЯМР (400 МГц, CD3OD) δ 7,52-7,53 (м, 2H), 7,31-7,32 (м, 3H), 5,49-7,52 (м, 1H), 5,26 (дд, J=6,4, 6,4 Гц, 2H), 4,63 (д, J=6,4 Гц, 1H), 4,36 (д, J=6,4 Гц, 1H), 3,58-3,61 (м, 3H), 3,35-3,39 (м, 1H), 1,23 (т, J=14 Гц, 3H); Масса, рассчитанная для C18H18BrN3O4: 419,0, 421,0, найдено 420,0, 422,0 (M+H)+.
Стадия J - Синтез промежуточного соединения Int-12j
К раствору Int-12i (50 мг, 0,119 ммоль) в N,N-диметилформамиде (3 мл) добавляли гидрид натрия (2,86 мг, 0,119 ммоль) при 0°C. Смесь оставляли для перемешивания при 0°C в течение 30 мин, обрабатывали иодметаном (16,89 мг, 0,119 ммоль) и перемешивали при 0°C в течение 20 мин. Добавляли воду и смесь экстрагировали этилацетатом. Объединенные органические слои сушили над Na2SO4, концентрировали в вакууме и полученный остаток очищали с использованием препаративной ТСХ (100% этилацетат) с получением Int-12j. Масса, рассчитанная для C19H20BrN3O4: 433,1, 435,1, найдено 434,1, 436,1 (M+H)+.
Стадия K - Синтез промежуточного соединения Int-12k-1 (энантиомер A) и Int-12k-2 (энантиомер B)
К раствору Int-12j (40 мг, 0,092 ммоль) в метаноле (3 мл) и диметилсульфоксиде (1 мл) добавляли Pd(PPh3)4 (11,60 мг, 0,010 ммоль), (2,4-дифторфенил)метанамин (26,4 мг, 0,184 ммоль) и N,N-диизопропилэтиламин (11,90 мг, 0,092 ммоль). Смесь оставляли для перемешивания при 90°C в течение 40 минут в атмосфере оксида углерода (1 атм). Смесь охлаждали до комнатной температуры, разбавляли водным раствором HCl (1N, 4 мл) и экстрагировали этилацетатом. Объединенные органические части промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Неочищенное вещество очищали с использованием препаративной ТСХ на силикагеле (100% этилацетат) с получением Int-12k в виде рацемата. 1H ЯМР (400 МГц, CDCl3) δ 7,54-7,56 (м, 2H), 7,23-7,32 (м, 4H), 6,71-6,78 (м, 2H), 6,03 (с, 1H), 5,26 (дд, J=8, 8 Гц, 2H), 4,54-4,64 (м, 2H), 4,30 (д, J=6,4 Гц, 1H), 4,16 (д, J=6,4 Гц, 1H), 3,70-3,72 (м, 1H), 3,58-3,59 (м, 2H), 3,45 (с, 3H), 3,39-3,41 (м, 1H), 2,88-2,90 (м, 1H), 1,23 (т, J=14 Гц, 3H); Масса, рассчитанная для C27H26F2N4O5: 524,2, найдено 525,2 (M+H)+.
Разделение энантиомеров осуществляли методом СФХ (AD, 250 мм × 30 мм, 10 мкм, SC-CO2/метанол 55/45 при 80 мл/мин) с получением Int-12k-1 (энантиомер A) и Int-12k-2 (энантиомер B).
Стадия L - Синтез соединения 22
К раствору Int-12k-1 (энантиомер A) (15 мг, 0,028 ммоль) в N,N-диметилформамиде (3 мл) добавляли хлорид лития (12,12 мг, 0,286 ммоль). Смесь оставляли для перемешивания при 110°C в течение 30 мин, охлаждали до комнатной температуры и непосредственно очищали с использованием препаративной ОФ-ВЭЖХ с получением соединения 22. 1H ЯМР 0346110-0182-1: (400 МГц, CD3OD) δ 7,40-7,46 (м, 1H), 6,91-6,98 (м, 2H), 5,93-5,94 (м, 1H), 4,82-4,83 (м, 1H), 4,50 (дд, J=8, 8 Гц, 2H), 4,45 (д, J=9,8 Гц, 1H), 3,91 (д, J=9,8 Гц, 1H), 3,61-3,92 (м, 2H), 3,46 (с, 3H), 3,14-3,17 (м, 1H), 1,25 (т, J=14,0 Гц, 3H); Масса, рассчитанная для C20H20F2N4O5: 434,1, найдено 435,2 (M+H)+.
Пример 13
Получение соединения 23
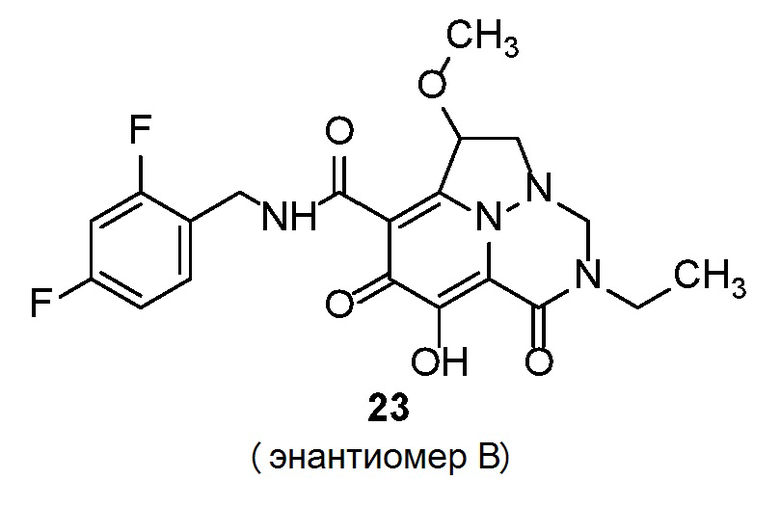
Соединение 23 получали из Int-12k-2 (энантиомер B) с использованием способов, описанных в Примере 12. 1H ЯМР (400 МГц, CD3OD) δ 7,40-7,46 (м, 1H), 6,91-6,98 (м, 2H), 5,93-5,95 (м, 1H), 4,82-4,85 (м, 1H), 4,50 (дд, J=8,0, 8,0 Гц, 2H), 4,45 (д, J=9,8 Гц, 1H), 3,91 (д, J=9,8 Гц, 1H), 3,61-3,92 (м, 2H), 3,46 (с, 3H), 3,14-3,19 (м, 1H), 1,25 (т, J=14,0 Гц, 3H); Масса, рассчитанная для C20H20F2N4O5: 434,1, найдено 435,2 (M+H)+.
Следующие соединения по настоящему изобретению получали с использованием способов, описанных в Примерах 12 и 13, с соответствующими заменами взаимодействующих веществ и/или реагентов.
[M+H]+
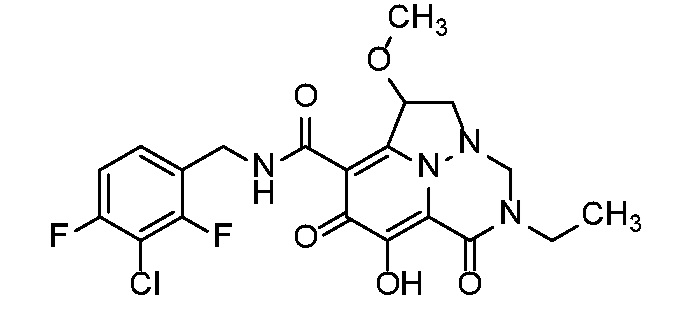
найдено 469,1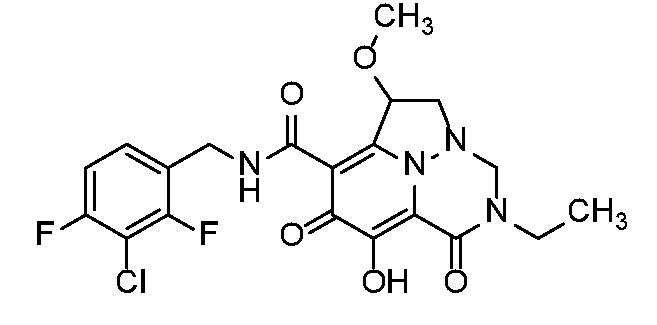
найдено 469,1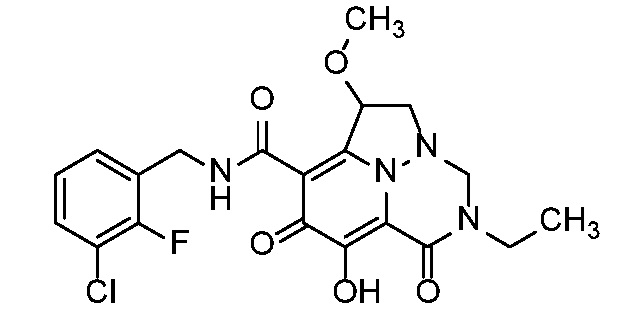
найдено 451,1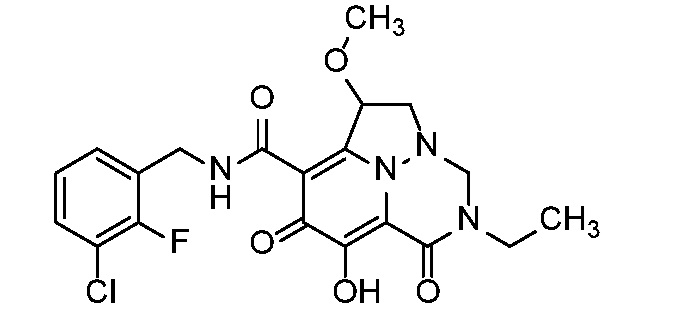
найдено 451,1
b СФХ (OJ, 250 мм × 30 мм, 10 мкм, SC-CO2/этанол 65:35 при 80 мл/мин)
Пример 14
Получение соединения 28
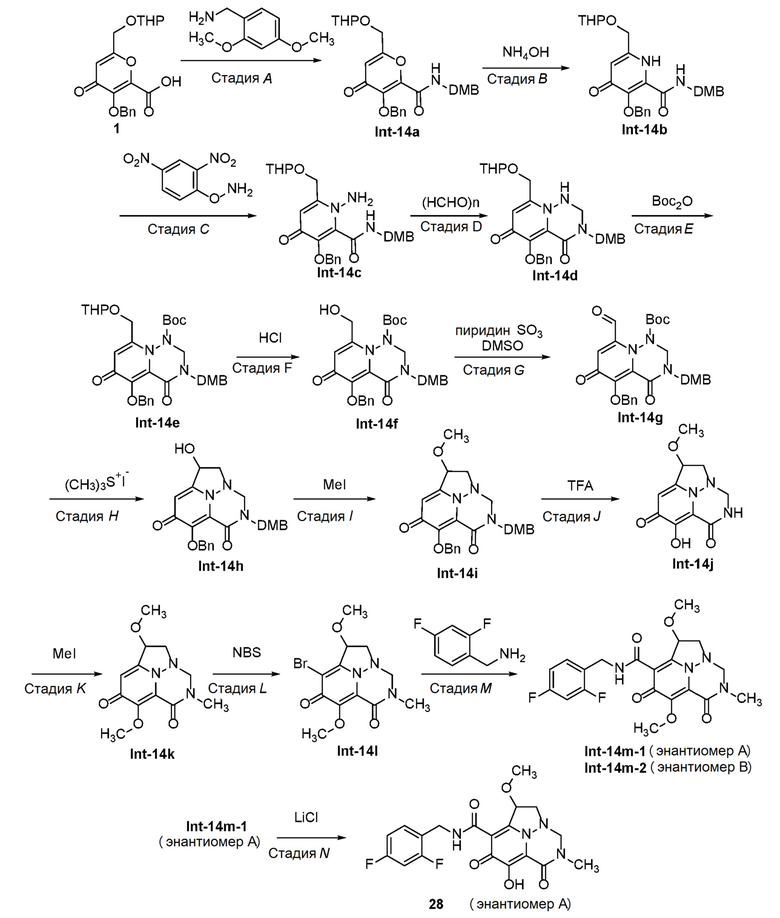
Стадия A - Синтез промежуточного соединения Int-14a
К раствору соединения 1 (53,75 г, 149,21 ммоль), (2,4-диметоксифенил)метанамина (49,88 г, 298,42 ммоль), HOAT(26,40 г, 193,9 ммоль) и HATU (73,75 г, 193,93 ммоль) в N,N-диметилформамиде (150 мл) добавляли N,N-диизопропилэтиламин (57,83 г, 447,2 ммоль). Смесь оставляли для перемешивания при 20°C в течение 16 часов. Смесь разбавляли водой (150 мл), экстрагировали этилацетатом. Органические фазы промывали насыщенным солевым раствором (200 мл), сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием колоночной хроматографии на силикагеле (100% этилацетат:петролейный эфир=1:1,5) с получением Int-14a. 1H ЯМР (400 МГц, CD3OD) δ 7,11-7,33 (м, 5H), 6,42-6,62 (м, 3H), 5,24 (с, 2H), 4,73 (с, 1H), 4,33-4,56 (м, 4H), 3,64-3,87 (м, 7H), 3,50 (дд, J=5,2, 10,4 Гц, 2H), 1,61 (дд, J=5,1, 10,4 Гц, 6H). Масса, рассчитанная для C28H31NO8: 509,2, найдено 510,2. (M+H)+.
Стадия B - Синтез промежуточного соединения Int-14b
Раствор Int-14a (30,1 г, 59,08 ммоль) и гидроксида аммония (28% водный раствор, 40 мл) в этаноле (30 мл) оставляли для перемешивания при 20°C в течение 16 часов. Смесь концентрировали в вакууме с получением Int-14b, которое использовали без дополнительной очистки. 1H ЯМР (400 МГц, CD3OD) δ 7,07-7,29 (м, 5H), 6,39-6,60 (м, 3H), 5,30 (с, 2H), 4,57-4,72 (м, 3H), 4,35-4,46 (м, 2H), 3,70-3,94 (м, 7H), 3,38-3,59 (м, 2H), 1,57 (с, 6H). Масса, рассчитанная для C28H32N2O7: 508,2, найдено 509,2. (M+H)+.
Стадия C - Синтез промежуточного соединения Int-14c
К раствору Int-14b (30,1 г, 47,34 ммоль) и K2CO3 (13,09 г, 94,8 ммоль) в N,N-диметилформамиде (100 мл) добавляли O-(2,4-динитрофенил)гидроксиламин (14,15 г, 70,95 ммоль) по порциям при перемешивании при 20°C. Реакционную смесь оставляли для перемешивания при 20°C в течение 48 часов. Смесь фильтровали и фильтрат очищали с использованием препаративной ОФ-ВЭЖХ (вода с 0,05% NH4OH/ацетонитрил) с получением Int-14c. 1H ЯМР (400 МГц, CD3OD) δ 7,17-7,35 (м, 5H), 6,58-6,66 (м, 1H), 6,45 (д, J=1,6 Гц, 1H), 6,26 (дд, J=2,0, 8,2 Гц, 1H), 5,00-5,14 (м, 2H), 4,55-4,81 (м, 3H), 4,41 (с, 2H), 3,63-3,94 (м, 7H), 3,54 (д, J=11,3 Гц, 2H), 1,50-1,85 (м, 6H). Масса, рассчитанная для C28H33N3O7: 523,2, найдено 524,2. (M+H)+.
Стадия D - Синтез промежуточного соединения Int-14d
Параформальдегид (0,614 г, 20,53 ммоль) добавляли к раствору Int-14c (10,75 г, 20,53 ммоль) в тетрагидрофуране (50 мл) и уксусной кислоте (10,75 мл). Смесь оставляли для перемешивания при 80°C в течение 18 часов. Смесь концентрировали в вакууме с получением Int-14d, которое использовали без дополнительной очистки. 1H ЯМР (400 МГц, CD3OD) δ 7,18-7,34 (м, 5H), 6,62 (с, 1H), 6,56 (д, J=2,0 Гц, 1H), 6,50 (дд, J=2,0, 8,2 Гц, 1H), 5,12-5,21 (м, 2H), 4,53-4,73 (м, 5H), 4,41 (с, 2H), 3,73-3,84 (м, 7H), 3,41-3,58 (м, 2H), 1,52-1,74 (м, 6H). Масса, рассчитанная для C29H33N3O7: 535,2, найдено 536,2. (M+H)+.
Стадия E - Синтез промежуточного соединения Int-14e
К раствору Int-14d (6,02 г, 11,2 ммоль), триэтиламина (4,69 мл, 33,71 ммоль) и ди-трет-бутилдикарбоната (5,220 мл, 22,49 ммоль) в дихлорметане (50 мл) добавляли 4-диметиламинопиридин (0,137 г, 1,12 ммоль). Смесь оставляли для перемешивания при 25°C в течение 16 часов. Смесь концентрировали в вакууме и полученный остаток очищали с использованием колоночной хроматографии на силикагеле (петролейный эфир:этилацетат=1:1 до 1:2) с получением Int-14e. 1H ЯМР (400 МГц, CD3OD) δ 7,07-7,48 (м, 6H), 6,42-6,69 (м, 3H), 5,11-5,35 (м, 3H), 4,60-4,75 (м, 3H), 4,35-4,53 (м, 2H), 3,72-3,92 (м, 6H), 3,37-3,64 (м, 3H), 1,54-1,92 (м, 6H), 1,07-1,49 (м, 9H). Масса, рассчитанная для C34H41N3O9: 635,3, найдено 636,2. (M+H)+.
Стадия F - Синтез промежуточного соединения Int-14f
К раствору Int-14e (4,95 г, 9,10 ммоль) в этилацетате (10 мл) добавляли HCl в этилацетате (4 M, 4,14 мл) при 0°C. Смесь оставляли для перемешивания при 25°C в течение 10 мин. Смесь концентрировали в вакууме и очищали с использованием колоночной хроматографии на силикагеле (этилацетат:метанол=100:2) с получением Int-14f. 1H ЯМР (400 МГц, CD3OD) δ 6,99-7,26 (м, 6H), 6,45-6,62 (м, 3H), 6,15-6,31 (м, 2H), 4,66-4,79 (м, 4H), 4,25-4,41 (м, 2H), 3,74 (д, J=2,0 Гц, 6H), 1,06-1,56 (м, 9H). Масса, рассчитанная для C29H33N3O8: 551,2, найдено 552,1. (M+H)+.
Стадия G - Синтез промежуточного соединения Int-14g
К раствору Int-14f (2,75 г, 4,99 ммоль), диметилсульфоксида (7,09 мл, 99,7 ммоль) и N,N-диизопропилэтиламина (11,33 мл, 64,8 ммоль) в дихлорметане (50 мл) добавляли комплекс триоксид серы-пиридин (9,53 г, 59,9 ммоль). Смесь оставляли для перемешивания при 25°C в течение 16 часов. Смесь разбавляли водным раствором HCl (1 N, 15 мл), промывали водным раствором HCl (1 N, 3 × 20 мл) и насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением Int-14g, которое использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 9,75 (с, 1H), 7,45 (д, J=3,5 Гц, 2H), 7,19-7,32 (м, 3H), 6,81 (с, 1H), 6,30-6,51 (м, 3H), 5,48 (д, J=10,5 Гц, 2H), 5,30 (д, J=5,5 Гц, 1H), 4,83-4,90 (м, 1H), 4,55 (д, J=13,7 Гц, 1H), 4,38 (д, J=14,1 Гц, 1H), 3,76 (д, J=9,8 Гц, 6H), 1,29 (с, 9H). Масса, рассчитанная для C29H31N3O8: 549,2, найдено 550,1. (M+H)+.
Стадия H - Синтез промежуточного соединения Int-14h
К раствору триметилсульфоний иодида (1093 мг, 5,33 ммоль) в N,N-диметилформамиде (26 мл) добавляли NaH (215 мг, 5,33 ммоль) при перемешивании в атмосфере азота и смесь оставляли для перемешивания при 25°C в течение 2 часов. К смеси добавляли по каплям раствор Int-14g (2330 мг, 5,33 ммоль) в N,N-диметилформамиде (3 мл) при 0°C и смесь оставляли для перемешивания при 0°C в течение 10 минут в атмосфере азота. Смесь разбавляли водой (2 мл) при 0°C и фильтровали. Фильтрат непосредственно очищали с использованием препаративной ОФ-ВЭЖХ с получением Int-14h. 1H ЯМР (400 МГц, CD3OD) δ 7,48 (д, J=5,1 Гц, 2H), 7,29 (д, J=5,9 Гц, 3H), 6,65 (с, 1H), 5,37 (т, J=6,3 Гц, 1H), 5,23 (с, 2H), 4,48 (с, 4H), 3,73 (с, 4H), 3,62 (с, 1H), 3,55 (т, J=4,9 Гц, 2H), 3,34 (с, 3H), 3,23 (с, 1H). Масса, рассчитанная для C25H25N3O6: 463,2, найдено 464,1 (M+H)+.
Стадия I - Синтез промежуточного соединения Int-14i
К раствору Int-14h (600 мг, 1,294 ммоль) и гидрида натрия (156 мг, 6,48 ммоль) в N,N-диметилформамиде (10 мл) добавляли иодметан (1102 мг, 7,76 ммоль). Смесь оставляли для перемешивания при 25°C в течение 16 часов. Смесь разбавляли водой (10 мл) и экстрагировали этилацетатом. Органическую фазу сушили над безводным Na2SO4 и фильтровали. Фильтрат концентрировали в вакууме и полученный остаток очищали с использованием препаративной ТСХ на силикагеле (дихлорметан:метанол=10:1) с получением Int-14i. 1H ЯМР (400 МГц, CD3OD) δ 7,25 (д, J=8,6 Гц, 1H), 6,71 (с, 1H), 6,41-6,58 (м, 2H), 5,03-5,13 (м, 1H), 4,88-4,91 (м, 2H), 4,55-4,69 (м, 3H), 4,40 (д, J=10,6 Гц, 1H), 3,54-4,00 (м, 9H), 3,43 (с, 3H). Масса, рассчитанная для C26H27N3O6: 477,2, найдено 478,2 (M+H)+.
Стадия J - Синтез промежуточного соединения Int-14j
Смесь Int-14i (200 мг, 0,498 ммоль) в трифторуксусной кислоте (5 мл) перемешивали в условиях микроволнового облучения при 110°C в течение 1 часа. Смесь концентрировали в вакууме и полученный остаток очищали с использованием препаративной ТСХ на силикагеле (дихлорметан:метанол=10:1) с получением Int-14j. 1H ЯМР (400 МГц, CD3OD) δ 5,28 (с, 1H), 4,37-4,63 (м, 3H), 4,00 (с, 3H), 3,66 (с, 2H), 3,50 (с, 3H). Масса, рассчитанная для C11H13N3O4: 251,1, найдено 252,2 (M+H)+.
Стадия K - Синтез промежуточного соединения Int-14k
К раствору Int-14j (60 мг, 0,238 ммоль) и K2CO3 (66,0 мг, 00,478 ммоль) в N,N-диметилформамиде (2 мл) добавляли MeI (0,044 мл, 0,716 ммоль). Смесь оставляли для перемешивания при 25°C в течение 16 часов. Смесь очищали с использованием препаративной ОФ-ВЭЖХ с получением Int-14k. 1H ЯМР (400 МГц, CD3OD) δ 5,40-5,55 (м, 1H), 4,73-4,90 (м, 3H), 4,12-4,30 (м, 3H), 3,80-3,92 (м, 2H), 3,55-3,64 (м, 3H), 3,19-3,27 (м, 3H). Масса, рассчитанная для C12H15N3O4: 265,1, найдено 266,2 (M+H)+.
Стадия L - Синтез промежуточного соединения Int-14l
К раствору Int-14k (40 мг, 0,159 ммоль) в ацетонитриле (3 мл) добавляли N-бромсукцинимид (42,5 мг, 0,239 ммоль). Смесь оставляли для перемешивания при 20°C в течение 3 часов. Смесь непосредственно очищали с использованием препаративной ТСХ на силикагеле (дихлорметан:метанол=10: 1) с получением Int-14l. 1H ЯМР (400 МГц, CD3OD) δ 5,30 (с, 1H), 4,67 (с, 1H), 4,64 (с, 1H), 3,92 (с, 3H), 3,52 (с, 3H), 3,31-3,34 (м, 2H), 3,15 (с, 3H). Масса, рассчитанная для C12H14BrN3O4: 343,0, 345,0, найдено 344,1, 346,1 (M+H)+.
Стадия M - Синтез промежуточного соединения Int-14m-1 (энантиомер A) и Int-14m-2 (энантиомер B)
К раствору Int-14l (30 мг, 0,087 ммоль), (2,4-дифторфенил)метанамина (37,42 мг, 0,261 ммоль) и N,N-диизопропилэтиламина (0,076 мл, 0,436 ммоль) в диметилсульфоксиде (1 мл) и метаноле (3 мл) добавляли Pd(Ph3P)4 (50,4 мг, 0,0435 ммоль) одной порцией. Смесь оставляли для перемешивания при 90°C в атмосфере оксида углерода (1 атм) в течение 5 часов. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат непосредственно очищали с использованием препаративной ОФ-ВЭЖХ с получением Int-14m в виде рацемата. 1H ЯМР (400 МГц, CD3OD) δ: 7,40-7,50 (м, 1H), 6,85-7,00 (м, 2H), 6,94 (д, J=5,6 Гц, 1H), 4,58-4,70 (м, 2H), 4,40 (д, J=10,4 Гц, 1H), 3,94 (с, 3H), 3,85 (д, J=10,4 Гц, 1H), 3,46 (с, 3H), 3,10-3,15 (м, 4H), 2,70 (д, J=12,0 Гц, 1H). Масса, рассчитанная для C20H20F2N4O5: 434,1, найдено 435,2 (M+H)+.
Разделение энантиомеров осуществляли методом СФХ (AD, 250 мм × 30 мм, 10 мкм, SC-CO2/i-PrOH=60/40 при 80 мл/мин) с получением Int-14m-1 (энантиомер A) и Int-14m-2 (энантиомер B)
Стадия N - Синтез соединения 28
Раствор Int-14m-1 (3,0 мг, 6,9 мкмоль) и хлорида лития (2,9 мг, 0,069 ммоль) в N,N-диметилформамиде (2 мл) оставляли для перемешивания при 80°C в течение 5 часов. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрат непосредственно очищали с использованием препаративной ОФ-ВЭЖХ с получением соединения 28. 1H ЯМР (400 МГц, CD3OD) δ 7,42 (д, J=7,2 Гц, 1H), 6,93 (д, J=10,8 Гц, 2H), 5,94 (с, 1H), 4,61 (д, J=14,0 Гц, 2H), 4,44 (с, 2H), 3,81-4,00 (м, 2H), 3,37-3,56 (м, 3H), 3,04-3,24 (м, 3H). Масса, рассчитанная для C19H18F2N4O5: 420,1, найдено 421,2 (M+H)+.
Пример 15
Получение соединения 29
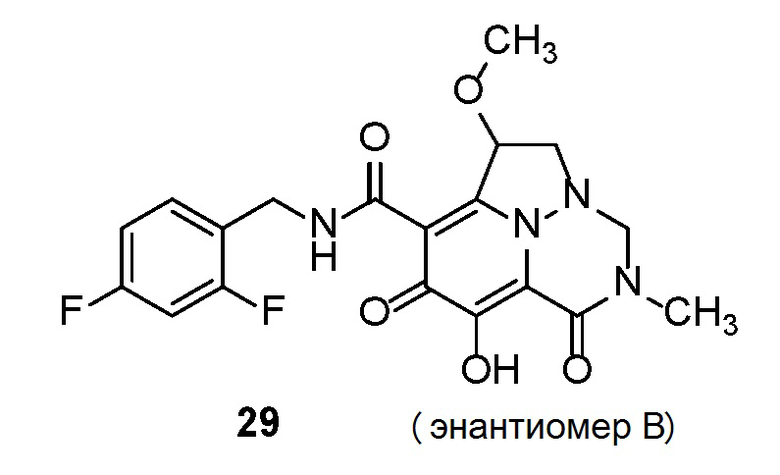
Соединение 29 получали из Int-14m-2 (энантиомер B) с использованием способов, описанных в Примере 14. 1H ЯМР (400 МГц, CD3OD) δ 7,42 (дд, J=7,6, 14,7 Гц, 1H), 6,85-7,01 (м, 2H), 5,93 (д, J=5,2 Гц, 1H), 4,53-4,67 (м, 2H), 4,44 (д, J=8,4 Гц, 2H), 3,89 (д, J=10,4 Гц, 2H), 3,39-3,57 (м, 3H), 3,03-3,21 (м, 3H). Масса, рассчитанная для C19H18F2N4O5: 420,1, найдено 421,2 (M+H)+.
Следующие соединения по настоящему изобретению получали с использованием способов, описанных в Примерах 14 и 15, с соответствующими заменами взаимодействующих веществ и/или реагентов.
[M+H]+
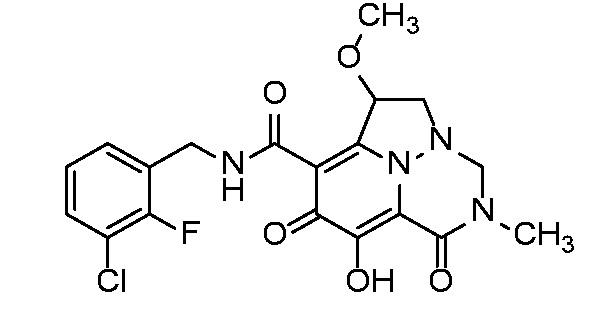
найдено 437,2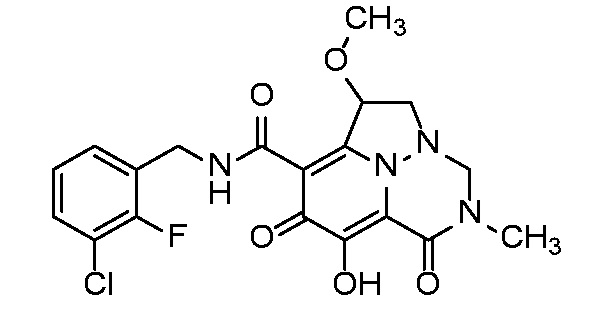
найдено 437,2
найдено 437,2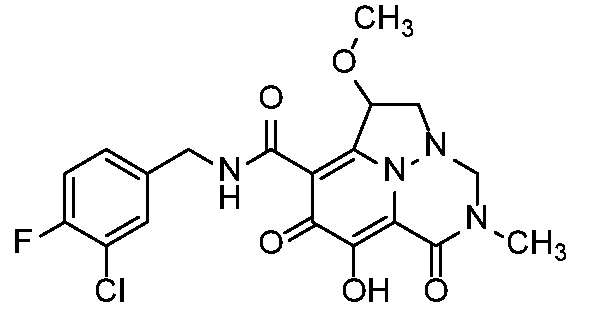
найдено 437,2
b СФХ (OJ, 250 мм × 30 мм, 10 мкм, SC-CO2/этанол 65:35 при 80 мл/мин)
Пример 16
Получение соединения 34
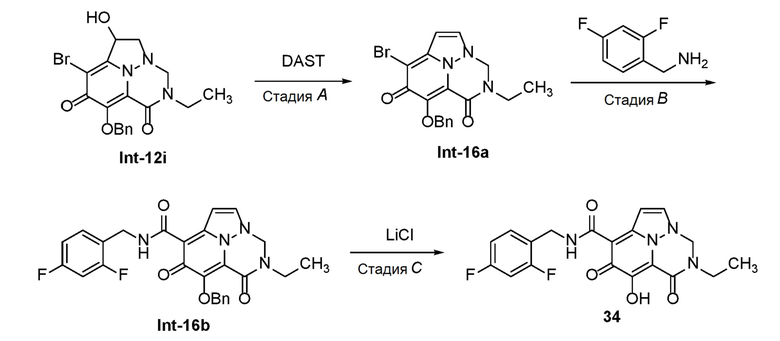
Стадия A - Синтез промежуточного соединения Int-16a
Раствор Int-12i (50 мг, 0,119 ммоль) и трифторида диэтиламиносеры (DAST) (19,2 мг, 0,12 ммоль) в дихлорметане (5 мл) оставляли для перемешивания при 20°C в течение 16 часов. Смесь разбавляли водой (5 мл) и экстрагировали дихлорметаном. Объединенные органические части промывали насыщенным солевым раствором (15 мл), сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ (этилацетат:метанол=10:1) с получением Int-16a. 1H ЯМР (400 МГц, CD3OD) δ 7,98 (шир.с, 1H), 7,60 (д, J=6,2 Гц, 2H), 7,31 (д, J=7,8 Гц, 3H), 6,59-6,68 (м, 1H), 5,51 (шир.с, 2H), 5,29 (с, 2H), 3,68 (д, J=7,0 Гц, 2H), 1,26 (т, J=6,8 Гц, 3H). Масса, рассчитанная для C18H16BrN3O3: 401,0, 403,0, найдено 402,1, 404,1 (M+H)+.
Стадия B - Синтез промежуточного соединения Int-16b
К смеси Int-16a (40 мг, 0,28 ммоль), N,N-диизопропилэтиламина (0,081 мл, 0,49 ммоль), (оксиди-2,1-фенилен)бис(дифенилфосфин) (DPEphos) (15,1 мг, 0,03 ммоль) добавляли диметилсульфоксид (2 мл). Смесь оставляли для перемешивания при 20°C в течение 5 минут и затем обрабатывали добавлением Pd(OAc)2 (4,2 мг, 0,02 ммоль). Смесь перемешивали в атмосфере оксида углерода (1 атм) при 90°C в течение 3 часов. Смесь разбавляли водным раствором HCl (1 N, 3 мл) и экстрагировали этилацетатом. Объединенные органические части промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Неочищенное вещество очищали с использованием препаративной ТСХ на силикагеле (этилацетат:метанол=9: 1) с получением Int-16b. 1H ЯМР (400 МГц, CD3OD) δ 8,00 (шир.с, 1H), 7,54-7,65 (м, 1H), 7,18-7,53 (м, 5H), 6,92 (шир.с, 2H), 6,41-6,61 (м, 1H), 5,40-5,62 (м, 2H), 5,15-5,40 (м, 2H), 4,61 (шир.с, 2H), 3,65 (шир.с, 1H), 3,44 (шир.с, 1H), 1,26 (шир.с, 3H). Масса, рассчитанная для C26H22F2N4O4: 492,2 найдено, 493,3 (M+H)+.
Стадия C - Синтез соединения 34
Раствор хлорида лития (8,1 мг, 0,19 ммоль) и Int-16b (18,7 мг, 0,04 ммоль) в N,N-диметилформамиде (2 мл) оставляли для перемешивания при 80°C в течение 3 часов. Смесь фильтровали и фильтрат непосредственно очищали с использованием препаративной ОФ-ВЭЖХ с получением соединения 34. 1H ЯМР (400 МГц, CD3OD) δ 7,84-8,05 (м, 1H), 7,43 (шир.с, 2H), 6,91-6,98 (м, 1H), 5,62-5,74 (м, 1H), 4,85-4,89 (м, 2H), 4,65 (шир.с, 2H), 3,69-3,85 (м, 2H), 1,30 (д, J=11,7 Гц, 3H). Масса, рассчитанная для C19H16F2N4O4: 402,1, найдено 403,2 (M+H)+.
Пример 17
Получение соединения 35
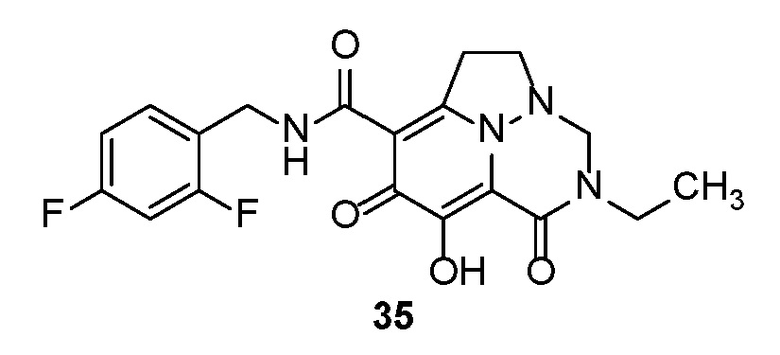
Смесь соединения 34 (30 мг, 0,061 ммоль) и Pd/C (5 мг) в метаноле (10 мл) оставляли для перемешивания при 20°C в атмосфере водорода (1 атм) в течение 13 часов. Смесь фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ОФ-ВЭЖХ с получением соединения 35. 1H ЯМР (400 МГц, диметилсульфоксид-d6) δ 10,77-10,80 (м, 1H), 7,33-7,41 (м, 1H), 7,19-7,23 (м, 1H), 7,00-7,05 (м, 1H), 4,46-4,48 (м, 4H), 3,45-3,90 (м, 2H), 2,63 (с, 2H), 2,29 (м, 2H), 1,08-1,14 (м, 3H). Масса, рассчитанная для C19H18F2N4O4: 404,1, найдено 405,2 (M+H)+.
Пример 18
Получение соединения 36
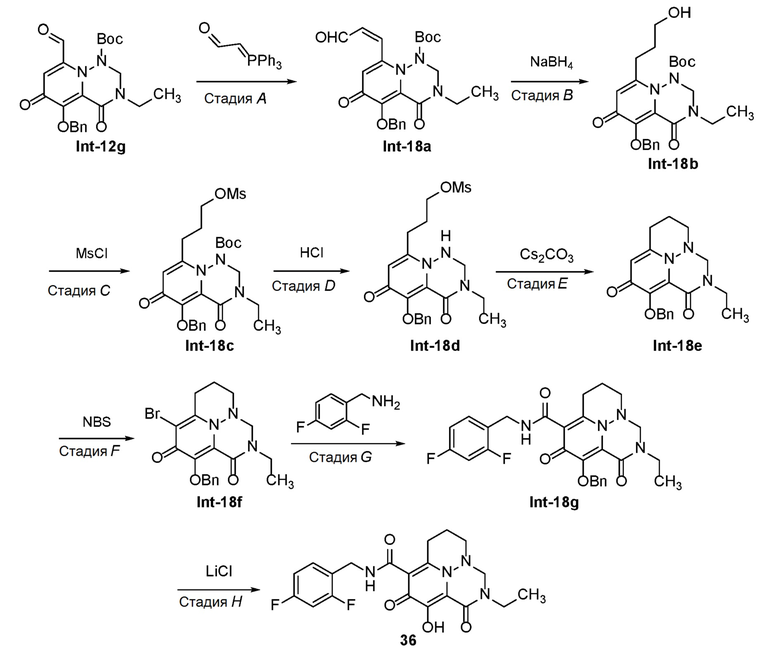
Стадия A - Синтез промежуточного соединения Int-18a
К раствору 2-(трифенилфосфоранилиден)ацетальдегида (256 мг, 0,842 ммоль) в тетрагидрофуране (15 мл) добавляли Int-12g (300 мг, 0,702 ммоль) при 10°C в атмосфере азота. Смесь оставляли для перемешивания при 10°C в течение 18 часов. Смесь гасили водой (30 мл) и экстрагировали этилацетатом. Объединенные органические части сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме с получением Int-18a. 1H ЯМР (400 МГц, CD3OD) δ 9,95-9,97 (м, 1H), 7,51-7,60 (м, 2H), 7,21-7,24 (м, 3H), 6,52 (с, 1H), 5,31-5,33 (м, 1H), 5,14-5,17 (м, 1H), 5,02-5,05 (м, 1H), 4,81-4,84 (м, 1H), 3,53-3,60 (м, 2H), 3,18-3,26 (м, 2H), 1,28 (с, 9H), 1,08-1,14 (м, 3H). Масса, рассчитанная для C24H27N3O6: 453,2, найдено 454,1 (M+H)+.
Стадия B - Синтез промежуточного соединения Int-18b
К раствору Int-18a (200 мг, 0,442 ммоль) в тетрагидрофуране (6 мл) добавляли NaBH4 (50,06 мг, 1,324 ммоль). Смесь оставляли для перемешивания при 10°C в течение 3 часов. Смесь гасили водой (15 мл) и экстрагировали этилацетатом. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ на силикагеле (этилацетат:метанол=10:1) с получением Int-18b. 1H ЯМР (400 МГц, CD3OD) δ 7,46-7,47 (м, 2H), 7,32-7,34 (м, 3H), 6,45 (с, 1H), 5,17-5,34 (м, 3H), 4,79-4,82 (м, 1H), 3,62-3,63 (м, 3H), 3,55-3,59 (м, 1H), 3,16-3,17 (м, 1H), 2,74-2,77 (м, 1H), 2,65-2,68 (м, 1H), 1,81-1,87 (м, 2H), 1,47 (с, 9H), 1,20-1,24 (м, 3H). Масса, рассчитанная для C24H31N3O6: 457,2, найдено 458,1 (M+H)+.
Стадия C - Синтез промежуточного соединения Int-18c
К раствору Int-18b (100 мг, 0,218 ммоль) в дихлорметане (4 мл) добавляли триэтиламин (66,4 мг, 0,656 ммоль) с последующим добавлением метансульфонилхлорида (50,08 мг, 0,438 ммоль). Смесь оставляли для перемешивания при 10°C в течение 18 часов. Реакционную смесь гасили водой (10 мл), экстрагировали этилацетатом. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением Int-18c. 1H ЯМР (400 МГц, CDCl3) δ 7,53-7,55 (м, 2H), 7,20-7,26 (м, 3H), 6,19 (с, 1H), 5,29-5,31 (м, 1H), 5,14-5,15 (м, 1H), 5,00-5,03 (м, 1H), 4,66-4,69 (м, 1H), 4,15-4,19 (м, 2H), 3,52-3,56 (м, 1H), 3,29-3,30 (м, 1H), 3,06-3,07 (м, 1H), 2,92 (с, 3H), 2,63-2,67 (м, 1H), 2,46-2,50 (м, 1H), 1,40 (с, 9H), 1,12-1,37 (м, 3H). Масса, рассчитанная для C25H33N3O8S: 535,2, найдено 536,2 (M+H)+.
Стадия D - Синтез промежуточного соединения Int-18d
Раствор Int-18c (100 мг, 0,185 ммоль) в 1% растворе HCl в метаноле (5 мл) оставляли для перемешивания при 55°C в течение 16 часов. Смесь концентрировали в вакууме с получением Int-18d, которое использовали без дополнительной очистки. Масса, рассчитанная для C20H25N3O6S: 435,1, найдено 436,1 (M+H)+.
Стадия E - Синтез промежуточного соединения Int-18e
К раствору Int-18d (80 мг, 0,185 ммоль) в N,N-диметилформамиде (1 мл) добавляли карбонат цезия (297,5 мг, 0,920 ммоль). Смесь оставляли для перемешивания при 55°C в течение 4 часов и затем фильтровали и фильтрат концентрировали в вакууме с получением Int-18e, которое использовали без дополнительной очистки. 1H ЯМР 0356873-0093-1a: (400 МГц, CD3OD) δ 7,39-7,40 (м, 2H), 7,23-7,24 (м, 3H), 6,33 (с, 1H), 5,10 (с, 2H), 4,36-4,50 (м, 3H), 3,46-3,56 (м, 3H), 3,15-3,19 (м, 2H), 1,97-2,00 (м, 2H), 1,10-1,13 (м, 3H). Масса, рассчитанная для C19H21N3O3: 339,2, найдено 339,9 (M+H)+.
Стадия F - Синтез промежуточного соединения Int-18f
К раствору Int-18e (40 мг, 0,117 ммоль) в ацетонитриле (3 мл) добавляли N-бромсукцинимид (105 мг, 0,589 ммоль). Смесь оставляли для перемешивания при 20°C в течение 10 мин. Неочищенный продукт очищали с использованием препаративной ТСХ на силикагеле (100% этилацетат) с получением Int-18f. 1H ЯМР (400 МГц, CD3OD) δ 7,49-7,50 (м, 2H), 7,31-7,33 (м, 3H), 5,20 (с, 2H), 4,59 (с, 2H), 3,52-3,54 (м, 2H), 3,26-3,28 (м, 2H), 3,06-3,10 (м, 2H), 2,09-2,12 (м, 2H), 1,19-1,22(м, 3H). Масса, рассчитанная для C19H20BrN3O3: 417,1, 419,1, найдено 418,1, 420,1 (M+H)+.
Стадия G - Синтез промежуточного соединения Int-18g
К раствору Int-18f (33 мг, 0,079 ммоль) в диметилсульфоксиде (1 мл) и метаноле (4 мл) добавляли (2,4-дифторфенил)метанамин (22,4 мг, 0,16 ммоль), DIPEA (0,3 мл) и Pd(PPh3)4 (44 мг). Смесь оставляли для перемешивания при 80°C в течение 1 часа, охлаждали до комнатной температуры, разбавляли водой (3 мл) и экстрагировали этилацетатом. Объединенные органические части сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ на силикагеле (этилацетат:метанол=14:1) с получением Int-18g. 1H ЯМР (400 МГц, CD3OD) δ 7,39-7,49 (м, 3H), 7,21-7,23 (м, 3H), 6,84-6,89 (м, 2H), 5,11 (с, 2H), 4,60 (с, 2H), 4,49 (с, 2H), 3,46-3,48 (м, 2H), 3,42-3,44 (м, 2H), 3,15-3,17 (м, 2H), 1,93-1,98 (м, 2H), 1,10-1,14 (м, 3H). Масса, рассчитанная для C27H26F2N4O4: 508,2, найдено 509,3 (M+H)+.
Стадия H - Синтез соединения 36
К раствору Int-18g (13,2 мг, 0,026 ммоль) в N,N-диметилформамиде (5 мл) добавляли LiCl (11,04 мг, 0,259 ммоль). Смесь нагревали до 100°C в течение 30 мин, охлаждали до комнатной температуры и фильтровали. Фильтрат непосредственно очищали с использованием препаративной ОФ-ВЭЖХ с получением соединения 36. 1H ЯМР (400 МГц, CD3OD) δ 7,50-7,56 (м, 1H), 6,92-6,95 (м, 2H), 4,80 (с, 2H), 4,55 (с, 2H), 3,57-3,62 (м, 2H), 3,42-3,44 (м, 2H), 3,20-3,23 (м, 2H), 2,01-2,04 (м, 2H), 1,22-1,25 (м, 3H). Масса, рассчитанная для C20H20F2N4O4: 418,1, найдено 419,0 (M+H)+.
Пример 19
Получение соединения 37
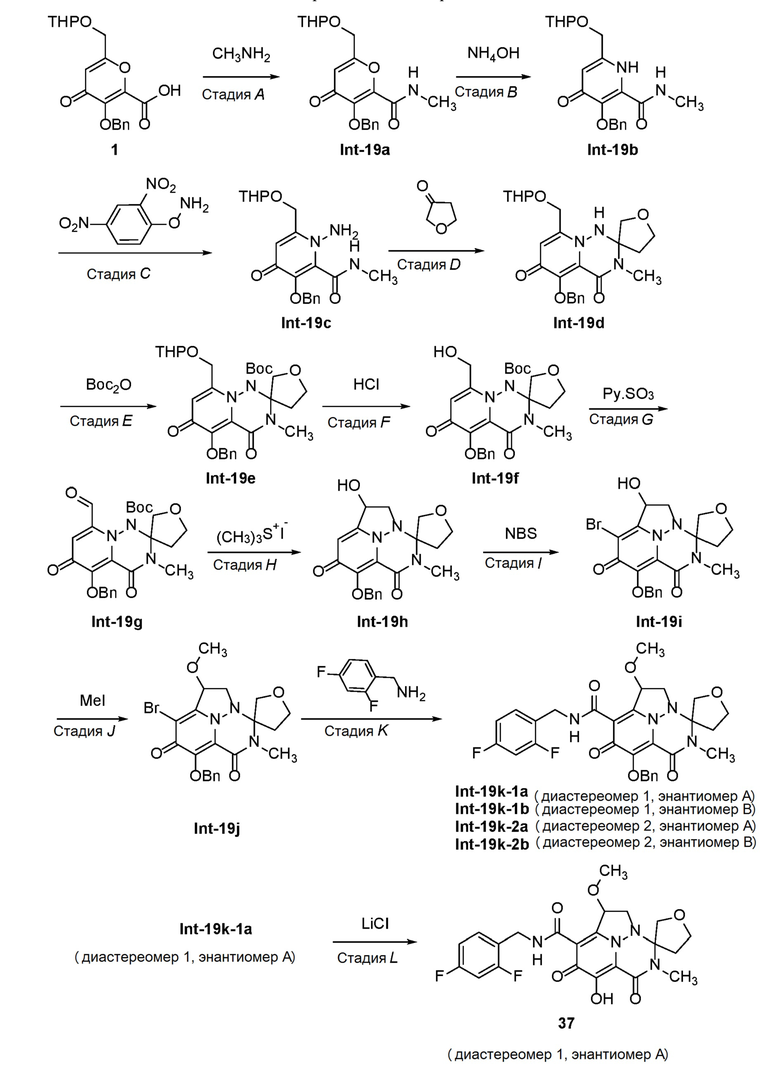
Стадия A - Синтез промежуточного соединения Int-19a
К перемешиваемому раствору соединения 1 (10 г, 27,8 ммоль), 6-хлор-бензотриазол-1-ил-окси-трис-пирролидинoфосфоний гексафторфосфата (24,63 г, 44,4 ммоль) и гидрохлорида метанамина (3,75 г, 55,5 ммоль) в N,N-диметилформамиде (100 мл) добавляли N,N-диизопропилэтиламин (14,54 мл, 83 ммоль) при 0°C. Полученную смесь оставляли для перемешивания при 20°C в течение 16 часов. Смесь выливали в воду (500 мл) и экстрагировали этилацетатом. Объединенные органические части промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием колоночной хроматографии на силикагеле (этилацетат в петролейном эфире: 0 до 50%) с получением Int-19a. 1H ЯМР (400 МГц, CDCl3) δ 8,02 (шир.с, 1H), 7,39-7,41 (м, 5H), 6,61(с, 1H), 5,38 (с, 2H), 4,74-4,75 (м, 1H), 4,62 (д, J=15,2 Гц, 1H), 4,44 (д, J=15,2 Гц, 1H), 3,79-3,84 (м, 1H), 3,53-3,56 (м, 1H), 3,89 (с, 3H), 1,56-3,04 (м, 6H). Масса, рассчитанная для C20H23NO6: 373,2, найдено 374,2 (M+H)+.
Стадия B - Синтез промежуточного соединения Int-19b
Раствор Int-19a (9,85 г, 20,06 ммоль) и гидроксида аммония (28% водный раствор, 170 мл) в этаноле (100 мл) оставляли для перемешивания при 20°C в течение 16 часов. Смесь концентрировали в вакууме с получением Int-19b, которое использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 8,20 (шир.с, 1H), 7,27-7,41 (м, 5H), 6,40(с, 1H), 5,46 (с, 2H), 4,68-4,69(м, 1H), 4,65 (д, J=15,2 Гц, 1H), 4,55 (д, J=15,2 Гц, 1H), 3,99-4,00 (м, 1H), 3,57-3,60 (м, 1H), 3,88 (с, 3H), 1,59-1,88 (м, 6H). Масса, рассчитанная для C20H24N2O5: 372,2, найдено 373,1 (M+H)+.
Стадия C - Синтез промежуточного соединения Int-19c
К перемешиваемому раствору Int-19b (9,35 г, 20,09 ммоль) и K2CO3 (3,78 г, 20,09 ммоль) в N,N-диметилформамиде (50 мл) добавляли O-(2,4-динитрофенил)гидроксиламин (8,00 г, 40,2 ммоль) при 0°C. Полученную смесь оставляли для перемешивания при 20°C в течение 24 часов. Смесь фильтровали и фильтрат очищали с использованием препаративной ОФ-ВЭЖХ (вода с 0,05% NH4OH/ацетонитрил) с получением Int-19c. 1H ЯМР (400 МГц, CD3OD) δ 7,28-7,41 (м, 5H), 6,63 (с, 1H), 5,11 (с, 2H), 4,85-4,86 (м, 1H), 4,76-4,84 (м, 1H), 3,82-3,88 (м, 1H), 3,54-3,57 (м, 1H), 3,82 (с, 3H), 1,57-1,58 (м, 6H). Масса, рассчитанная для C20H25N3O5: 387,2, найдено 388,1 (M+H)+.
Стадия D - Синтез промежуточного соединения Int-19d
К раствору Int-19c (168 мг, 0,412 ммоль) и уксусной кислоты (0,1 мл) в тетрагидрофуране (2 мл) добавляли дигидрофуран-3(2H)-он (643 мг, 7,47 ммоль). Полученную смесь нагревали при 75°C в течение 20 часов. Смесь фильтровали и фильтрат очищали с использованием препаративной ОФ-ВЭЖХ (вода с 0,05% NH4OH/ацетонитрил) с получением Int-19d. 1H ЯМР (400 МГц, CD3OD) δ 7,36 (д, J=3,2 Гц, 2H), 7,29 (т, J=5,6 Гц, 3H), 6,66 (с, 1H), 5,25 (с, 2H), 4,48-4,87 (м, 2H), 4,50-4,56 (м,1H), 3,98-3,99 (м, 2H), 3,81-3,86 (м, 2H), 3,53-3,55 (м, 2H), 3,02 (с, 3H), 1,56-1,89 (м, 8H). Масса, рассчитанная для C24H29N3O6: 455,2, найдено 456,0 (M+H)+.
Стадия E - Синтез промежуточного соединения Int-19e
К раствору Int-19d (2 г, 4,53 ммоль) в дихлорметане (50 мл) добавляли 4-диметиламинопиридин (0,055 г, 0,453 ммоль), триэтиламин (1,263 мл, 9,06 ммоль) и ди-трет-бутилдикарбонат (3,104 мл, 9,06 ммоль). Смесь оставляли для перемешивания при 20°C в течение 16 часов. Смесь концентрировали в вакууме и полученный остаток очищали с использованием колоночной хроматографии на силикагеле (петролейный эфир/этилацетат=1:1) с получением Int-19e. 1H ЯМР (400 МГц, CD3OD) δ 7,63 (д, J=13,8 Гц, 2H), 7,27-7,35 (м, 3H), 6,60-6,65 (м, 1H), 5,45 (д, J=10,8 Гц, 1H), 5,21 (д, J=10,8 Гц, 1H), 4,61-4,74 (м, 2H), 4,51-4,54 (м, 1H), 3,81-3,98 (м, 2H), 3,71-3,79 (м, 2H), 3,53-3,55 (м, 2H), 3,10 (с, 3H), 1,40-3,08 (м, 8H), 1,39 (с, 9H). Масса, рассчитанная для C29H37N3O8: 555,3, найдено 556,1 (M+H)+.
Стадия F - Синтез промежуточного соединения Int-19f
К раствору Int-19e (1,8 г, 3,24 ммоль) в этилацетате (20 мл) добавляли раствор HCl в этилацетате (4 M, 1 мл), смесь оставляли для перемешивания при 20°C в течение 10 минут. Смесь концентрировали в вакууме и полученный остаток очищали с использованием колоночной хроматографии на силикагеле (этилацетат:метанол=100:2) с получением Int-19f. 1H ЯМР (400 МГц, CD3OD) δ 7,26-7,32 (м, 5H), 6,68 (с, 1H), 5,42-5,44 (м, 1H), 5,19-5,28 (м, 1H), 4,52-4,56 (м, 1H), 4,36-4,40 (м, 1H), 3,96-4,04 (м, 2H), 3,62-3,75 (м, 2H), 3,08 (с, 3H), 3,17-3,20 (м, 2H), 1,38 (с, 9H). Масса, рассчитанная для C24H29N3O7: 471,2, найдено 473,1 (M+H)+.
Стадия G - Синтез промежуточного соединения Int-19g
К раствору Int-19f (1,1 г, 3,333 ммоль) в дихлорметане (20 мл) добавляли N,N-диизопропилэтиламин (5,30 мл, 30,3 ммоль), диметилсульфоксид (3,31 мл, 46,7 ммоль) и комплекс триоксид серы-пиридин (4,46 г, 28,0 ммоль), смесь оставляли для перемешивания при 20°C в течение 16 часов. Смесь промывали водным раствором HCl (0,5 M, 10 мл), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали в вакууме с получением Int-19g, которое использовали без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 9,79 (с, 1H), 7,41-7,43 (м, 1H), 7,19-7,23 (м, 1H), 6,79 (с, 1H), 5,55-5,61 (м, 1H), 5,27-5,31 (м, 1H), 3,56-3,59 (м, 2H), 3,00-3,05 (м, 2H), 3,55 (с, 3H), 3,17-3,20 (м, 2H), 1,36 (с, 9H). Масса, рассчитанная для C24H27N3O7: 469,2, найдено 488,3 (M+H3O)+.
Стадия H - Синтез промежуточного соединения Int-19h
Раствор триметилсульфоний иодида (348 мг, 1,704 ммоль) в N,N-диметилформамиде (8 мл) обрабатывали гидридом натрия (147 мг, 3,66 ммоль) при 20°C. Смесь перемешивали в атмосфере азота при 20°C в течение 1,5 часов. К смеси добавляли по каплям раствор Int-19g (200 мг, 0,426 ммоль) в N,N-диметилформамиде (12 мл) при 20°C. Смесь перемешивали при 20°C в течение 10 минут в атмосфере азота и затем разбавляли водой (3 мл) при 0°C. Смесь непосредственно очищали с использованием препаративной ОФ-ВЭЖХ с получением Int-19h. 1H ЯМР (400 МГц, CD3OD) δ 7,30-7,45 (м, 5H), 7,06 (с, 1H), 5,43-5,47 (м, 1H), 5,32 (м, 2H), 3,63-3,99 (м, 6H), 3,10-3,15 (м, 3H), 3,09 (с, 3H). Масса, рассчитанная для C20H21N3O5: 383,1, найдено 384,1 (M+H)+.
Стадия I - Синтез промежуточного соединения Int-19i
Раствор Int-19h (84 мг, 0,219 ммоль) в ацетонитриле (3 мл) обрабатывали N-бромсукцинимидом (58,5 мг, 0,329 ммоль). Смесь оставляли для перемешивания при 20°C в течение 10 минут и затем непосредственно очищали с использованием препаративной ТСХ на силикагеле (этилацетат:метанол=14:1) с получением Int-19i. 1H ЯМР (400 МГц, CD3OD) δ 7,42-7,43 (м, 2H), 7,29-7,30 (м, 3H), 5,45-5,49 (м, 1H), 5,35-5,37 (м, 1H), 5,23-5,33 (м, 1H), 3,59-4,08 (м, 6H), 3,63-3,99 (м, 4H), 3,13 (с, 3H), 3,10-3,15 (м, 3H). Масса, рассчитанная для C20H20BrN3O5: 461,1, 463,1, найдено 462,1, 464,2 (M+H)+.
Стадия J - Синтез промежуточного соединения Int-19j
К раствору Int-19i (100 мг, 0,216 ммоль) в N,N-диметилформамиде (5 мл) добавляли гидрид натрия (26,0 мг, 0,649 ммоль) и иодметан (307 мг, 3,163 ммоль) при 18°C, смесь оставляли для перемешивания при 18°C в течение 5 минут. Добавляли воду (5 мл) и смесь экстрагировали этилацетатом. Объединенные органические части промывали насыщенным солевым раствором и концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ на силикагеле (этилацетат:метанол=14:1) с получением Int-19j. 1H ЯМР (400 МГц, CD3OD) δ 7,39-7,42 (м, 2H), 7,26-7,28 (м, 3H), 5,33-5,38 (м, 1H), 5,18-5,23 (м, 2H), 3,52-3,41 (м, 2H), 3,50 (с, 3H), 3,11 (с, 3H), 3,10-3,15 (м, 3H). Масса, рассчитанная для C21H22BrN3O5: 475,1, 477,1, найдено 476,1, 478,1 (M+H)+.
Стадия K - Синтез промежуточного соединения Int-19k-1a, Int-19k-1b, Int-19k-2a и Int-19k-2b
Раствор Int-19j (90 мг, 0,189 ммоль) в диметилсульфоксиде (2 мл) и метаноле (2 мл) обрабатывали (2,4-дифторфенил)метанамином (54,1 мг, 0,378 ммоль), N,N-диизопропилэтиламином (0,066 мл, 0,378 ммоль) и Pd(Ph3P)4 (21,83 мг, 0,019 ммоль). Смесь нагревали при 90°C в атмосфере оксида углерода (1 атм) в течение 3 часов. Добавляли этилацетат (10 мл) и реакционную смесь фильтровали. Фильтрат промывали водным раствором HCl (0,5 M, 4 мл), сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали с использованием препаративной ТСХ на силикагеле (100% этилацетат) с получением Int-19k-1 (диастереомер 1, более высокий Rf на силикагеле) и Int-19k-2 (диастереомер 2, более низкий Rf на силикагеле).
Int-19k-1 (диастереомер 1) 1H ЯМР (400 МГц, CD3OD) δ 7,19-7,62 (м, 7H), 6,83-6,88 (м, 1H), 5,81-5,82 (м, 1H), 5,27-5,31 (м, 1H), 5,13-5,16 (м, 1H), 4,49-4,55 (м, 2H), 3,73-4,05 (м, 6H), 3,55 (с, 3H), 3,04 (с,3H), 3,09-3,12 (м, 2H). Масса, рассчитанная для C29H28F2N4O6: 566,2, найдено 567,3 (M+H)+.
Int-19k-2 (диастереомер 2) 1H ЯМР (400 МГц, CD3OD) δ 7,19-7,66 (м, 7H), 6,86-6,88 (м, 1H), 5,76-5,77 (м, 1H), 5,24-5,27 (м, 1H), 5,13-5,16 (м, 1H), 4,53-4,59 (м, 2H), 3,46-4,02 (м, 6H), 3,35 (с, 3H), 3,04 (с,3H), 3,05-3,23 (м, 2H). Масса, рассчитанная для C29H28F2N4O6: 566,2, найдено 567,3 (M+H)+.
Разделение Int-19k-1 (диастереомер 1) осуществляли методом СФХ (OJ, 250мм × 30мм, 5мкм, 30% метанола с 0,05% NH4OH в SC-CO2, 80 мл/мин) с получением Int-19k-1a (диастереомер 1, энантиомер A) и Int-19k-1b (диастереомер 1, энантиомер B)
Разделение Int-19k-2 (диастереомер 2) осуществляли методом СФХ (OJ, 250мм × 30мм, 5мкм, 30% метанола с 0,05% NH4OH в SC-CO2, 80 мл/мин) с получением Int-19k-2a (диастереомер 2, энантиомер A) и Int-19k-2b (диастереомер 2, энантиомер B)
Стадия L - Синтез соединения 37 (диастереомер 1, энантиомер A)
К раствору Int-19k-1a (диастереомер 1, энантиомер A) (5 мг, 8,83 мкмоль) в N,N-диметилформамиде (1 мл) добавляли хлорид лития (0,374 мг, 8,83 мкмоль). Смесь нагревали при 80°C в течение 3 часов, охлаждали до комнатной температуры и непосредственно очищали с использованием препаративной ОФ-ВЭЖХ с получением соединения 37. 1H ЯМР (400 МГц, CD3OD) δ 7,40-7,46 (м, 1H), 6,90-6,98 (м, 2H), 5,93 (д, J=4,4 Гц, 1H), 4,60-4,68 (м, 3H), 4,32 (д, J=11,6 Гц, 1H), 3,84-4,12 (м, 4H), 3,45 (с, 3H), 3,21 (с, 3H), 3,47-3,52 (м, 1H), 3,06-3,14 (м, 1H). Масса, рассчитанная для C22H22F2N4O6: 476,2, найдено 477,2 (M+H)+.
Следующие соединения по настоящему изобретению получали с использованием способов, описанных в Примере 19, с соответствующими заменами взаимодействующих веществ и/или реагентов.
[M+H]+
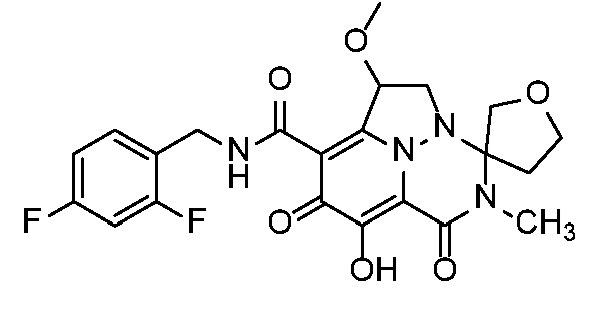
энант. B
найдено 477,2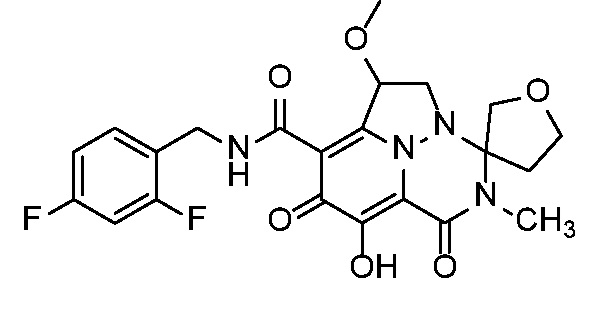
энант. A
найдено 477,2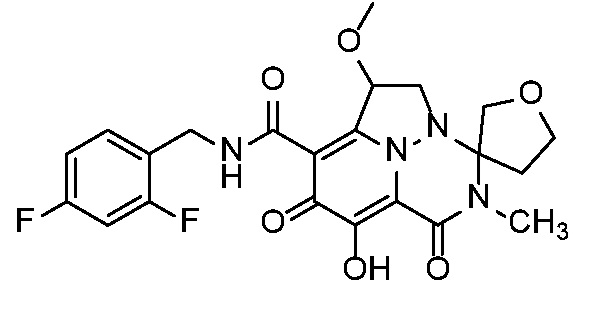
энант. B
найдено 477,2
Пример 20
Получение соединения Int-20f
Стадия А - Синтез соединения Int-20a
К раствору 3-гидроксипиколиновой кислоты (340 г, 2,44 моль) в 2,8 л MeOH, перемешиваемому при 15°С, добавляли H2SО4 (720 г, 7,33 моль). Реакционную смесь нагревали до 65°C на масляной бане и перемешивали в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь нейтрализовали до рН=7 медленным добавлением насыщенного водного раствора Na2CО3. Полученную смесь экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SО4. После фильтрования фильтрат концентрировали под вакуумом с получением Int-20а. Неочищенное вещество использовали в следующей реакции без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3 ) δ 10,62 (с, 1H), 6,28 (д, J= 4.4 Гц, 2H), 4,05 (с, 3H).
Стадия В - Синтез соединения Int-20b
К смеси соединения Int-20а (50 г, 327 ммоль) в Н2О (5,0 л), перемешиваемой при 15°С, добавляли бром (157 г, 979 ммоль). Смесь перемешивали при температуре 15°С в течение 5 часов. Полученную смесь фильтровали, фильтровальную лепешку промывали водой и сушили под вакуумом с получением соединения Int-20b. Неочищенное вещество использовали в следующей реакции без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3 ) δ 11,37 (с, 1H), 7,87 (с, 1H), 4,07 (с, 3H).
Стадия С - Синтез соединения Int-20с
К раствору соединения Int-20b (200 г, 643 ммоль) в ацетоне (4,0 л) при перемешивании при 15°C добавляли Cs2CО3 (377 г, 1,160 моль) с последующим добавлением по каплям иодометана (274 г, 1930 ммоль). Реакционную смесь нагревали при 60°С в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь фильтровали. Фильтровальную лепешку промывали ацетоном и очищали хроматографией на силикагеле, элюируя петролейным эфиром:EtOAc=25:1~10:1, с получением соединения Int-20с. 1H ЯМР (400 МГц, CDCl3) δ 7,85 (с, 1Н), 3,99 (с, 3Н), 3,98 (с, 3Н).
Стадия D - Синтез соединения Int-20d
К раствору соединения Int-2°C (350 г, 1080 ммоль, 1,0 экв) в THF (1,8 л), перемешиваемому при 15°C, добавляли Н2О (350 мл) с последующим добавлением моногидрата гидроксида лития (54 г, 1300 ммоль). Реакционную смесь перемешивали при 25°С в течение 2 часов. Растворитель удаляли под вакуумом с получением соединения Int-20d в виде желтого твердого вещества. Неочищенное вещество использовали в следующей реакции без дополнительной очистки. 1H ЯМР (400 МГц, DMSO-d6) δ 7,73 (с, 1H), 3,83 (с, 3H).
Стадия Е - Синтез соединения Int-20е
К раствору соединения Int-20d (240 г, 757 ммоль) и DMF (1,50 л), перемешиваемого при 0~5°С, медленно добавляли NaН (115 г, 2,88 моль, 60% масс.). Смесь перемешивали при 0~5°С в течение 30 мин, затем добавляли раствор (4-метоксифенил)метанола (157 г, 1,14 моль) в DMF (1,50 л). Реакционную смесь перемешивали при 0~5°С в течение 30 мин, затем нагревали до 15°С и перемешивали в течение 2 часов. Реакцию гасили путем добавления 1 л насыщенного водного раствора NН4Cl и подкисляли 4 N водным раствором НCl до рН=4~5. Полученную смесь экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным Na2SО4, затем концентрировали под вакуумом с получением соединения Int-20e. Масса, рассчитанная для C15H14NBrO5: 367,0, найдено 389,8 (M+Na)+.
Стадия F - Синтез соединения Int-20f
К смеси соединения 6 (290 г, 788 ммоль) и К2CО3 (272 г, 1970 ммоль) в DMF (2,5 л), перемешиваемой при 15°с, медленно добавляли иодометан (355 г, 2360 ммоль). Реакционную смесь перемешивали при 15°С в течение 12 часов. Реакционную смесь разбавляли 1,5 л воды и экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным Na2SО4, затем концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле, элюируя смесью петролейный эфир:этилацетат:дихлорметан=10:1~2:1. Фракции, содержащие продукт, объединяли и концентрировали под вакуумом. Остаток перекристаллизовали из этилацетата/петролейного эфира. Твердое вещество собирали фильтрованием, промывали петролейным эфиром и сушили под вакуумом с получением соединения Int-20f. 1Н ЯМР (400 МГц, CDCl3): δ 7,35 (д, J=8,8 Гц, 2Н), 7,16 (с, 1Н), 6,95 (д, J=8,8 Гц, 2Н), 5,10 (с, 2Н), 3,95 (с, 3Н), 3,91 (с, 3Н), 3,84 (с, 3Н).
Пример 21
Получение соединения 41
Стадия А - Синтез соединения Int-21a
К раствору соединения Int-20f (10 г, 26,2 ммоль) в толуоле (100 мл) добавляли аллилтрибутилстаннан (17,33 г, 52,3 ммоль) и тетракис(трифенилфосфин)палладий(0) (1,512 г, 1,308 ммоль) при 25°С. Раствор дегазировали и трижды продували азотом и полученную смесь перемешивали при 110°С в течение 16 часов под баллоном азота. Реакционную смесь гасили водой (40 мл) и экстрагировали при помощи EtOAc (50 мл ×3). Объединенную органическую фазу сушили над безводным Na2SО4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле, элюируя смесью петролейный эфир/EtOAc=3/1, с получением соединения Int-21а. 1Н ЯМР (400 МГц, CDCl3) δ: 7,33-7,35 (м, 2Н), 6,88-6,94 (м, 3Н), 5,94-6,04 (м, 1Н), 5,13-5,17 (м, 2Н), 5,08 (с, 2Н), 3,94 (с, 3Н), 3,88 (с, 3Н), 3,82 (с, 3Н), 3,54 (д, J=6,8 Гц, 2Н). Масса, рассчитанная для C19H21NO5: 343.1, найдено 344,1 (M+H)+.
Стадия B - Синтез соединения Int-21b
Раствор соединения Int-21а (6,2 г, 18,06 ммоль) в дихлорметане (60 мл) барботировали газообразным О3 при -78°С в течение 15 мин. Затем добавляли борогидрид натрия (1,025 г, 27,1 ммоль) и перемешивали при 0°С в течение 1 часа. Реакционную смесь гасили водой (40 мл) и экстрагировали этилацетатом (40 мл ×3). Объединенную органическую фазу сушили над безводным Na2SО4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле, элюируя смесью дихлорметан/MeOH=10/1, с получением соединения Int-21b. Масса, рассчитанная для C18H21NO6: 347,1, найдено 348,1 (M+H)+.
Стадия C - Синтез соединения Int-21c
К раствору соединения Int-21b (2,4 г, 6,91 ммоль) в THF (35 мл), перемешиваемому при 25°С, добавляли 2N раствор метанамина в THF (34,5 мл, 69,1 ммоль). Реакционную смесь перемешивали при 25°C в течение 16 часов. Полученную смесь концентрировали в вакууме и остаток очищали хроматографией на силикагеле, элюируя смесью дихлорметан/MeOH=10/1, с получением соединения Int-21с. 1H ЯМР (400 МГц, CDCl3) δ: 7,41 (с, 1Н), 7,34 (д, J=8,4 Гц, 2Н), 6,92 (м, 3Н), 5,09 (с, 2Н), 3,99 (т, J=5,6 Гц, 2Н), 3,89 (с, 3Н), 3,82 (с, 3Н) 2,98 (т, J=5,2 Гц, 2Н), 2,93 (т, J=5,6 Гц, 3Н). Масса, рассчитанная для C18H22N2O5: 346,1, найдено 347,0 (M+H)+.
Стадия D - Синтез соединения Int-21d
К перемешиваемому раствору соединения Int-21с (2,3 г, 6,64 ммоль) в дихлорметане (30 мл), перемешиваемому при 0°С, добавляли триэтиламин (2,78 мл, 19,92 ммоль) и метансульфоновый ангидрид (1,735 г, 9,96 ммоль). Смесь перемешивали при 25°С в течение 2 часов. Смесь гасили водой (20 мл) и экстрагировали при помощи EtOAc (30 мл ×3). Объединенную органическую фазу сушили над безводным Na2SО4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле, элюируя смесью дихлорметан/MeOH=10/1, с получением соединения Int-21d. 1H ЯМР (400 МГц, CDCl3) δ: 7,64 (с, 1Н), 7,34 (д, J=8,4 Гц, 2Н), 6,92 (м, 3Н), 5,10 (с, 2Н), 4,65 (т, J=6,0 Гц, 2Н), 3,92 (с, 3Н), 3,82 (с, 3Н) 3,12 (т, J=6,0 Гц, 2Н), 2,97 (д, J=5,2 Гц, 3Н), 2,89 (с, 3Н). Масса, рассчитанная для C19H24N2O7S: 424,1, найдено 424,9 (M+H) +.
Стадия E - Синтез соединения Int-21e
К раствору соединения Int-21d (2,1 г, 4,95 ммоль) в МеОН (25 мл) добавляли Pd/C (0,526 г, 0,495 ммоль) при 25°С. Реакционную смесь перемешивали при 25°С в течение 3 часов под баллоном с водородом. Полученную смесь фильтровали и фильтрат концентрировали в вакууме. Остаток очищали хроматографией на силикагеле, элюируя смесью дихлорметан/MeOH=10/1, с получением соединения Int-21е. 1H ЯМР (400 МГц, CDCl3) δ: 8,31 (с, 1Н), 6,40 (с, 1Н), 4,49 (т, J=6,0 Гц, 2Н), 4,14 (с, 3Н), 3,10 (с, 3Н), 3,04 (м, 5Н). Масса, рассчитанная для C11H16N2O6S: 304,1, найдено 304,9 (M+H)+.
Стадия F - Синтез соединения Int-21f
К раствору соединения Int-21e (300 мг, 0,986 ммоль) в MeCN (6 мл) добавляли карбонат цезия (1285 мг, 3,94 ммоль) и О-(2,4-динитрофенил)гидроксиламин (393 мг, 1,972 ммоль) при 25°С. Реакционную смесь перемешивали при 25°С в течение 3 часов. Полученную смесь фильтровали и фильтрат очищали препаративной ВЭЖХ (колонка: Waters XSELECT C18 150 мм × 30 мм × 5 мкм, Условия: вода (0,1% TFA)-ACN Начало В 0% Завершение В 20% Время градиента (мин) 10, 100% В время удерживания мин) 2, Скорость потока (мл/мин) 25) с получением соединения Int-21f. Масса, рассчитанная для C10H13N3O3: 223,1, найдено 224,0 (М+Н)+.
Стадия G - Синтез соединения Int-21g
К раствору соединения Int-21f (60 мг, 0,269 ммоль) в 1,1-дихлорэтане (3 мл) добавляли диметоксиметан (614 мг, 8,06 ммоль) и метансульфокислоту (155 мг, 1,613 ммоль) при 25°С. Раствор перемешивали при 120°С в течение 4 часов под баллоном азота. Смесь концентрировали в вакууме и остаток очищали препаративной ВЭЖХ (колонка: Waters XSELECT C18 150 мм × 30 мм × 5 мкм, Условия: вода (0,1% TFA)-ACN Начало B 0% Завершение B 20% Время градиента(мин) 10, 100% В время удержания (мин) 2, Скорость потока(мл/мин) 25) с получением соединения Int-21g. Масса, рассчитанная для C11H13N3O3: 235,1, найдено 236,1 (M+H)+.
Стадия H - Синтез соединения Int-21h
К раствору соединения Int-21g (37 мг, 0,157 ммоль) в МеОН (4 мл) добавляли NIS (70,8 мг, 0,315 ммоль) и m-CPBA (27,1 мг, 0,157 ммоль) при 25°С. Реакционную смесь перемешивали при 70°С в течение 1 часа под баллоном азота, а затем охлаждали до комнатной температуры. Смесь фильтровали и фильтрат очищали препаративной ВЭЖХ (колонка: Waters XSELECT C18 150 мм × 30 мм × 5 мкм, Условия: вода (0,1% TFA)-ACN Начало B 0% Завершение B 40% Время градиента(мин) 10 100% время удержания B (мин) 2 Скорость потока (мл/мин) 25) с получением соединения Int-21h в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ: 4,59 (с, 2H), 3,92 (с, 3H), 3,54 (м, 2H), 3,46 (м, 2H), 3,15 (с, 3H). Масса, рассчитанная для C11H12IN3O3: 361,0, найдено 362,0 (M+H)+.
Стадия I - Синтез соединения Int-21i
К раствору соединения Int-21h (28 мг, 0,078 ммоль) в DMSO (3 мл) добавляли (2,4,6-трифторфенил)метанамин (125 мг, 0,775 ммоль), DIPEA (0,135 мл, 0,775 ммоль) и Pd(PPh3)4 (17,92 мг, 0,016 ммоль). Смесь дегазировали и продували при помощи СО три раза. Полученную смесь перемешивали при 90°С под баллоном с оксидом углерода в течение 2 часов. Смесь гасили водой (10 мл) и экстрагировали при помощи EtOAc (20 мл × 3). Объединенный органический слой сушили над безводным Na2SО4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле, элюируя смесью дихлорметан/MeOH=10/1, с получением соединения Int-21i. 1H ЯМР (400 МГц, CDCl3) δ: 10,82 (с, 1Н), 6,59 (м, 2Н), 4,54 (м, 2Н), 4,34 (с, 2Н), 4,00 (с, 3Н), 3,97 (м, 2Н), 3,32 (м, 2Н), 3,12 (с, 3Н). Масса, рассчитанная для C19H17F3N4O4: 422,1, найдено 422,9 (M+H)+.
Стадия J - Синтез соединения 41
К раствору соединения Int-21i (18 мг, 0,043 ммоль) в АСN (2 мл), перемешиваемому при 25°С, добавляли бромид магния (23,54 мг, 0,128 ммоль). Смесь перемешивали при 25°С в течение 1 часа под баллоном азота. Реакционную смесь разбавляли при помощи МеОН (1 мл) и полученный раствор очищали препаративной ВЭЖХ (колонка: Boston Green ODS 150 мм × 30 мм, 5 мкм, Условия: вода (0,1% TFA)-ACN Начало B 30%, Завершение B 60%, Время градиента(мин) 10, 100% B время удерживания (мин) 2, Скорость потока (мл/мин) 25) с получением соединения 41 в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ: 10,75 (с, 1H), 6,65 (т, J=8,0 Гц, 2H), 4,62 (д, J=5,6 Гц, 2H), 4,46 (с, 2H), 3,95 (м, 2H), 3,41 (м, 2H), 3,18 (с, 3H). Масса, рассчитанная для C18H15F3N4O4: 408,1, найдено 408,9 (M+H) +.
Пример 22
Получение соединения 42 и соединения 43
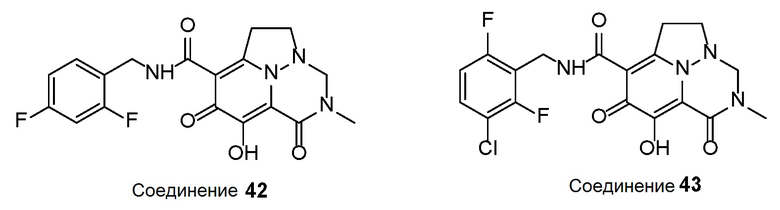
Соединения 42 и 43 были получены с использованием способов, описанных в Примере 21, заменяя (2,4,6-трифторфенил)метанамин соответствующими бензиламинами на стадии I.
Соединение 42: 1Н ЯМР (400 МГц, CDCl3) δ: 10,80 (с, 1Н), 7,35 (м, 1Н), 6,82 (м, 2Н), 4,60 (д, J=4,8 Гц, 2Н), 4,48 (с, 2Н), 3,94 (м, 2Н), 3,42 (м, 2Н), 3,19 (с, 3Н). Масса, рассчитанная для C18H16F2N4O4: 390,1, найдено 391,0 (M+H)+.
Соединение 43: 1Н ЯМР (400 МГц, CDCl3) δ: 10,80 (с, 1Н), 7,29 (м, 1Н), 6,85 (м, 1Н), 4,70 (д, J=5,6 Гц, 2Н), 4,47 (с, 2Н), 3,95 (м, 2Н), 3,42 (м, 2Н), 3,19 (с, 3Н). Рассчитано для C18H15ClF2N4O4: 424,1, найдено 425,0 (M+H)+.
Анализ на ингибирование репликации ВИЧ
Этот анализ может быть полезным для оценки способности соединения по настоящему изобретению ингибировать репликацию ВИЧ. Анализ представляет собой кинетический анализ, в котором используют репортерную клеточную линию (MT4-gag-GFP) для определения количества новых клеток, инфицированных за каждый цикл репликации.
MT4-GFP клетки (250000 клеток/мл) массово инфицировали ВИЧ-1 (NL4-3 штамм) при низкой множественности заражения (MOI) в RPMI+10% FBS в течение 24 часов. Клетки затем промывали один раз в RPMI+10% FBS и ресуспендировали в RPMI+0% нормальная человеческая сыворотка (NHS). Испытываемые соединения подвергали серийному разведению в DMSO на ECHO. Инфицированные MT4-GFP клетки добавляли в 384-луночный покрытый поли-D-лизином черный планшет с прозрачным дном, в который добавляли разведения испытываемых соединений. Клетки высевали при плотности 8000 клеток на лунку и конечную концентрацию DMSO доводили до 0,4%. Инфицированные клетки (содержащие зеленый GFP клетки) затем подсчитывали через 24 и через 48 часов после инкубации с использованием Acumen eX3. Коэффициент репродукции вируса (R0) определяли с использованием количества инфицированных клеток в точке времени 48 часов, деленного на количество инфицированных клеток в точке времени 24 часа. Процент ингибирования вирусного роста рассчитывали по формуле[1-(R-Rтройная терапия)/(RDMSO-Rтройная терапия)]×100. Активность соединений IP или IC50 можно определить с использованием анализа кривой доза-ответ с 4 параметрами.
Иллюстративные соединения по настоящему изобретению испытывали с использованием этого протокола анализа, и результаты представлены в таблице ниже.
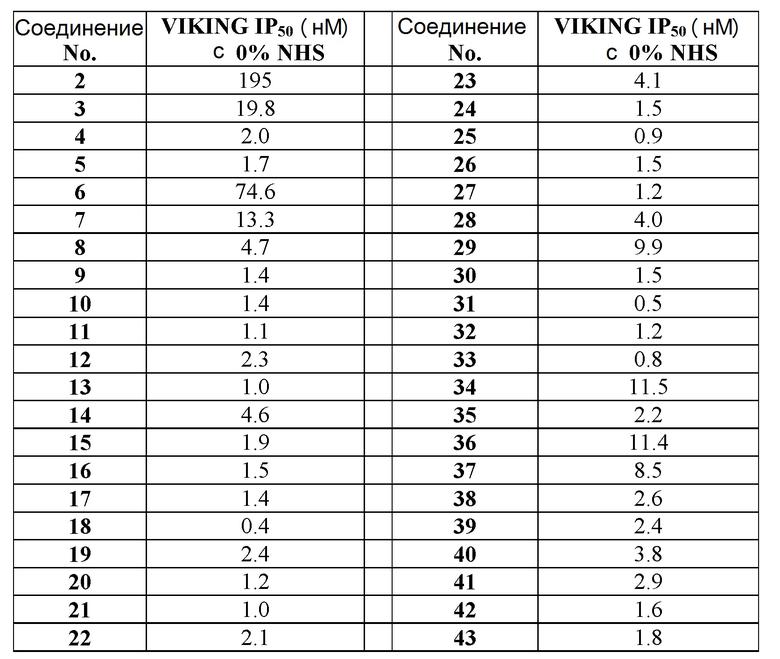
Лечение или профилактика ВИЧ-инфекции
Трициклические гетероциклические соединения могут быть полезны для ингибирования ВИЧ, ингибирования интегразы ВИЧ, лечения ВИЧ-инфекции и/или уменьшения вероятности или тяжести симптомов ВИЧ-инфекции и ингибирования вирусной репликации ВИЧ и/или продуцирования вируса ВИЧ в клеточной системе. Например, трициклические гетероциклические соединения могут быть полезны для лечения ВИЧ-инфекции после того, как возникло подозрение на заражение после контакта с ВИЧ таким путем, как переливание крови, обмен жидкостей организма, укусы, случайный укол иглой или контакт с кровью субъекта во время операции или других медицинских процедур.
Соответственно, в одном варианте осуществления изобретение обеспечивает способы лечения ВИЧ-инфекции у субъекта, включающие введение субъекту эффективного количества по меньшей мере одного трициклического гетероциклического соединения или его фармацевтически приемлемой соли или пролекарства. В конкретном варианте осуществления вводимое количество является эффективным для лечения или профилактики ВИЧ-инфекции у субъекта. В другом конкретном варианте осуществления вводимое количество является эффективным для ингибирования репликации вируса ВИЧ и/или продуцирования вируса у субъекта. В одном варианте осуществления произошло прогрессирование ВИЧ-инфекции до СПИДа.
Трициклические гетероциклические соединения также полезны для подготовки и осуществления скрининговых анализов на противовирусные соединения. Например, трициклические гетероциклические соединения могут быть полезны для идентификации резистентных клеточных линий ВИЧ, содержащих мутации, которые являются отличными инструментами для скрининга на более сильные противовирусные соединения. Кроме того, трициклические гетероциклические соединения могут быть полезны для установления или определения сайта связывания других противовирусных средств с интегразой ВИЧ.
Композиции и комбинации по настоящему изобретению могут быть полезны для лечения субъекта, страдающего инфекцией, связанной с любым генотипом ВИЧ.
Комбинированная терапия
В другом варианте осуществления представленные способы для лечения или профилактики ВИЧ-инфекции также могут включать введение одного или нескольких дополнительных терапевтических средств, которые не являются трициклическими гетероциклическими соединениями.
В одном варианте осуществления дополнительное терапевтическое средство представляет собой противовирусное средство.
В другом варианте осуществления дополнительное терапевтическое средство представляет собой иммуномодулятор, такой как иммуносупрессивное средство.
Соответственно, в одном варианте осуществления настоящее изобретение обеспечивает способы для лечения вирусной инфекции у субъекта, при этом такой способ включает введение субъекту: (i) по меньшей мере одного трициклического гетероциклического соединения (может включать два или больше разных трициклических гетероциклических соединений) или его фармацевтически приемлемой соли или пролекарства и (ii) по меньшей мере одного дополнительного терапевтического средства, которое является отличным от трициклического гетероциклического соединения, где вводимые количества вместе являются эффективными для лечения или профилактики вирусной инфекции.
При введении комбинированной терапии по изобретению субъекту терапевтические средства в комбинации или фармацевтическую композицию или композиции, включающие терапевтические средства, можно вводить в любом порядке, например, последовательно, параллельно, вместе, одновременно и т.п. Количества различных активных веществ в такой комбинированной терапии могут быть разными (разная величина доз) или одинаковыми количествами (одинаковая величина доз). Таким образом, в целях неограничивающей иллюстрации, трициклическое гетероциклическое соединение и дополнительное терапевтическое средство могут присутствовать в фиксированных количествах (с фиксированной величиной доз) в одной единице дозирования (например, капсула, таблетка и т.п.).
В одном варианте осуществления по меньшей мере одно трициклическое гетероциклическое соединение вводят в период времени, когда дополнительное терапевтическое средство(средства) проявляют их профилактический или терапевтический эффект, или наоборот.
В другом варианте осуществления по меньшей мере одно трициклическое гетероциклическое соединение и дополнительное терапевтическое средство(средства) вводят в дозах, обычно используемых, когда такие средства используют в виде монотерапии для лечения вирусной инфекции.
В другом варианте осуществления по меньшей мере одно трициклическое гетероциклическое соединение и дополнительное терапевтическое средство(средства) вводят в более низких дозах, чем дозы, обычно используемые, когда такие средства используют в виде монотерапии для лечения вирусной инфекции.
Еще в одном варианте осуществления по меньшей мере одно трициклическое гетероциклическое соединение и дополнительное терапевтическое средство(средства) действуют синергически, и их вводят в более низких дозах, чем дозы, обычно используемые, когда такие средства используют в виде монотерапии для лечения вирусной инфекции.
В одном варианте осуществления по меньшей мере одно трициклическое гетероциклическое соединение и дополнительное терапевтическое средство(средства) присутствуют в одной и той же композиции. В одном варианте осуществления эта композиция является подходящей для перорального введения. В другом варианте осуществления эта композиция является подходящей для внутривенного введения. В другом варианте осуществления эта композиция является подходящей для подкожного введения. Еще в одном варианте осуществления эта композиция является подходящей для парентерального введения.
Вирусные инфекции и связанные с вирусом расстройства, которые можно лечить или предотвратить с использованием комбинированных терапевтических способов по настоящему изобретению, включают, но не ограничиваются этим, инфекции и расстройства, перечисленные выше.
В одном варианте осуществления вирусная инфекция представляет собой ВИЧ-инфекцию.
В другом варианте осуществления вирусная инфекция представляет собой СПИД.
По меньшей мере одно трициклическое гетероциклическое соединение и дополнительное терапевтическое средство(средства) могут действовать аддитивно или синергически. Синергическая комбинация может позволить использовать более низкие дозы одного или нескольких средств и/или менее частое введение одного или нескольких средств комбинированной терапии. Более низкая дозировка или менее частое введение одного или нескольких средств может снизить токсичность терапии без снижения эффективности терапии.
В одном варианте осуществления введение по меньшей мере одного трициклического гетероциклического соединения и дополнительного терапевтического средства(средств) может ингибировать резистентность вирусной инфекции к этим средствам.
Как отмечено выше, настоящее изобретение также направлено на применение соединения формулы I с одним или несколькими анти-ВИЧ средствами. "Анти-ВИЧ средство" представляет собой любое средство, которое является, непосредственно или опосредованно, эффективным для ингибирования обратной транскриптазы ВИЧ или другого фермента, необходимого для репликации или инфекции ВИЧ, лечения или профилактики ВИЧ-инфекции и/или лечения, профилактики или задержки начала или прогрессирования СПИДа. Должно быть понятно, что анти-ВИЧ средство является эффективным для лечения, профилактики или отсрочки начала развития или прогрессирования ВИЧ-инфекции или СПИДа и/или заболеваний или состояний, возникающих в результате этого или связанных с этим. Например, соединения по настоящему изобретению можно эффективно вводить, как в период до контакта, так и/или после контакта с вирусом, в сочетании с эффективными количествами одного или нескольких анти-ВИЧ средств, выбранных из противовирусных средств против ВИЧ, иммуномодуляторов, противоинфекционных средств или вакцин, полезных для лечения ВИЧ-инфекции или СПИДа. Подходящие противовирусные средства против ВИЧ для использования в комбинации с соединениями по настоящему изобретению включают, например, следующие средства, перечисленные в таблице A:
EI=ингибитор входа; FI=ингибитор слияния; PI=ингибитор протеазы; nRTI=нуклеозидный ингибитор обратной транскриптазы; II=ингибитор интегразы; nnRTI=ненуклеозидный ингибитор обратной транскриптазы. Некоторые из лекарственных средств, перечисленных в таблице, используют в солевой форме; например, абакавир сульфат, индинавир сульфат, атазанавир сульфат, нелфинавир мезилат.
В одном варианте осуществления одно или несколько анти-ВИЧ лекарственных средств выбраны из, ламивудина, абакавира, ритонавира, дарунавира, атазанавира, эмтрицитабина, тенофовира, рилпивирина и лопинавира.
В другом варианте осуществления соединение формулы (I) используют в комбинации с ламивудином.
Еще в одном варианте осуществления соединение формулы (I) используют в комбинации с атазанавиром.
В другом варианте осуществления соединение формулы (I) используют в комбинации с дарунавиром.
В другом варианте осуществления соединение формулы (I) используют в комбинации с рилпивирином.
В одном варианте осуществления соединение формулы (I) используют в комбинации с ламивудином и абакавиром.
В другом варианте осуществления соединение формулы (I) используют в комбинации с дарунавиром.
В другом варианте осуществления соединение формулы (I) используют в комбинации с эмтрицитабином и тенофовиром.
Еще в одном варианте осуществления, соединение формулы (I) используют в комбинации с атазанавиром.
В другом варианте осуществления соединение формулы (I) используют в комбинации с ритонавиром и лопинавиром.
В одном варианте осуществления соединение формулы (I) используют в комбинации с абакавиром и ламивудином.
В другом варианте осуществления соединение формулы (I) используют в комбинации с лопинавиром и ритонавиром.
В одном варианте осуществления настоящее изобретение обеспечивает фармацевтические композиции, включающие (i) соединение формулы (I) или его фармацевтически приемлемую соль или пролекарство; (ii) фармацевтически приемлемый носитель; и (iii) одно или несколько дополнительных анти-ВИЧ средств, выбранных из ламивудина, абакавира, ритонавира и лопинавира или фармацевтически приемлемых солей или пролекарств, где присутствующие количества компонентов (i) и (iii) вместе эффективны для лечения или профилактики ВИЧ-инфекциии или для лечения, профилактики или отсрочки начала или прогрессирования СПИДа у субъекта, нуждающегося в этом.
В другом варианте осуществления настоящее изобретение обеспечивает способ для лечения или профилактики ВИЧ-инфекциии или для лечения, профилактики или отсрочки начала или прогрессирования СПИДа у субъекта, нуждающегося в этом, который включает введение субъекту (i) соединения формулы (I) или его фармацевтически приемлемой соли или пролекарства и (ii) одного или нескольких дополнительных анти-ВИЧ средств, выбранных из ламивудина, абакавира, ритонавира и лопинавира или их фармацевтически приемлемых солей или пролекарств, где вводимые количества компонентов (i) и (ii) вместе эффективны для лечения или профилактики ВИЧ-инфекции или для лечения, профилактики или отсрочки начала или прогрессирования СПИДа у субъекта, нуждающегося в этом.
Должно быть понятно, что объем комбинаций соединений по настоящему изобретению с анти-ВИЧ средствами не ограничивается противовирусными средствами против ВИЧ, перечисленными в таблице А, но включает в принципе любую комбинацию с любой фармацевтической композицией, полезной для лечения или профилактики СПИДа. Антивирусные средства против ВИЧ и другие средства обычно используют в этих комбинациях при их обычных диапазонах доз и схемах введения, как известно в данной области техники, включая, например, дозы, описанные в Physician's Desk Reference, Thomson PDR, Thomson PDR, 57-е издание (2003), 58-е издание (2004), 59-е издание (2005) и т.п. Диапазоны доз для соединения по изобретению в этих комбинациях такие же, как описано выше.
Дозы и схемы введения других средств, используемых в комбинированной терапии по настоящему изобретению для лечения или профилактики ВИЧ-инфекции, может определить лечащий врач с учетом разрешенных к применению доз и схемы введения, указанных во вкладыше в упаковку; возраста, пола и общего состояния здоровья субъекта; и типа и тяжести вирусной инфекции или связанного с этим заболевания или расстройства. При введении в комбинации трициклическое гетероциклическое соединение (соединения) и другое средство (средства) можно вводить одновременно (т.е. в одной и той же композиции или в отдельных композициях одно за другим) или последовательно. Это особенно полезно, когда компоненты комбинации вводят в соответствии с разными схемами введения, например, один компонент вводят один раз в день, а другой компонент вводят каждые шесть часов, или когда фармацевтические композиции являются разными, например, одна представляет собой таблетку, а другая капсулу. Поэтому набор, включающий отдельные лекарственные формы, является предпочтительным.
Композиции и введение
При введении субъекту трициклические гетероциклические соединения можно вводить в качестве компонента композиции, которая включает фармацевтически приемлемый носитель или наполнитель. Настоящее изобретение обеспечивает фармацевтические композиции, включающие эффективное количество по меньшей мере одного трициклического гетероциклического соединения и фармацевтически приемлемый носитель. В фармацевтических композициях и способах по настоящему изобретению активные ингредиенты типично вводят в смеси с подходящими веществами-носителями, подходящим образом выбранными для предполагаемой формы введения, т.е. для пероральных таблеток, капсул (заполненных либо твердым, либо полутвердым, либо жидким веществом), порошков для восстановления, гелей для перорального введения, эликсиров, диспергируемых гранул, сиропов, суспензий и т.п., и в соответствии с обычной фармацевтической практикой. Например, для перорального введения в форме таблеток или капсул активный лекарственный компонент может быть объединен с любым пероральным нетоксичным фармацевтически приемлемым инертным носителем, таким как лактоза, крахмал, сахароза, целлюлоза, стеарат магния, дикальцийфосфат, сульфат кальция, тальк, маннит, этиловый спирт (жидкие формы) и т.п. Препараты в твердой форме включают порошки, таблетки, диспергируемые гранулы, капсулы, облатки и суппозитории. Порошки и таблетки могут состоять на около 0,5-95 процентов из композиции по настоящему изобретению. Таблетки, порошки, облатки и капсулы можно использовать в виде твердых лекарственных форм, подходящих для перорального введения.
Кроме того, когда это желательно или необходимо, подходящие связующие, смазывающие вещества, разрыхлители и красители также могут быть включены в смесь. Подходящие связующие включают крахмал, желатин, природные сахара, сахаристые вещества из кукурузы, природные и синтетические смолы, такие как аравийская камедь, альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль и воски. Из смазывающих веществ для использования в этих лекарственных формах можно указать борную кислоту, бензоат натрия, ацетат натрия, хлорид натрия и т.п. Разрыхлители включают крахмал, метилцеллюлозу, гуаровую камедь и т.п. Подсластители и отдушки и консерванты также могут быть включены в случае необходимости.
Жидкие формы препаратов включают растворы, суспензии и эмульсии и могут включать воду или водно-пропиленгликолевые растворы для парентерального введения.
Жидкие формы препаратов также могут включать растворы для интраназального введения.
Также включены твердые формы препаратов, которые предназначены для преобразования непосредственно перед применением в жидкие препараты для перорального или парентерального введения. Такие жидкие формы включают растворы, суспензии и эмульсии.
Для получения суппозиториев низкоплавкий воск, такой как смесь глицеридов жирных кислот, или масло какао сначала расплавляют и в нем гомогенно диспергируют активный ингредиент при перемешивании. Расплавленную гомогенную смесь затем разливают в формы удобного размера, оставляют для охлаждения и, таким образом, отверждают.
Кроме того, композиции по настоящему изобретению можно сформулировать в виде формы с замедленным высвобождением для обеспечения контролируемой скорости высвобождения любого одного или нескольких компонентов или активных ингредиентов для оптимизации терапевтических эффектов, т.е. противовирусной активности и т.п. Подходящие лекарственные формы для замедленного высвобождения включают многослойные таблетки, содержащие слои с различными скоростями разложения, или полимерные матрицы с контролируемым высвобождением, пропитанные активными компонентами и сформированные в виде таблеток или капсул, содержащих такие пропитанные или инкапсулированные пористые полимерные матрицы.
В одном варианте осуществления одно или несколько трициклических гетероциклических соединений вводят перорально.
В другом варианте осуществления одно или несколько трициклических гетероциклических соединений вводят внутривенно.
В одном варианте осуществления фармацевтический препарат, включающий по меньшей мере одно трициклическое гетероциклическое соединение, находится в стандартной лекарственной форме. В такой форме препарат подразделен на единичные дозы, содержащие эффективные количества активных компонентов.
Композиции можно получить в соответствии с традиционными способами смешивания, гранулирования или нанесения покрытия, соответственно, и композиции по настоящему изобретению могут содержать, в одном варианте осуществления, от около 0,1% до около 99% трициклического гетероциклического соединения (соединений) по массе или объему. В различных вариантах осуществления композиции по настоящему изобретению могут содержать, в одном варианте осуществления, от около 1% до около 70% или от около 5% до около 60% трициклического гетероциклического соединения (соединений) по массе или объему.
Соединения формулы I можно вводить перорально при дозе в диапазоне от 0,001 до 1000 мг/кг массы тела млекопитающего (например, человека) в день в виде одной дозы или дробных доз. Один диапазон доз составляет от 0,01 до 500 мг/кг массы тела в день перорально в виде одной дозы или дробных доз. Другой диапазон доз составляет от 0,1 до 100 мг/кг массы тела в день перорально в виде одной дозы или дробных доз. Для перорального введения композиции могут быть представлены в форме таблеток или капсул, содержащих от 1,0 до 500 мг активного ингредиента, в частности, 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400 и 500 мг активного ингредиента для симптоматической корректировки дозировки для субъекта, подлежащего лечению. Конкретный уровень доз и частота введения для любого конкретного субъекта могут варьироваться и будут зависеть от различных факторов, включая активность конкретного используемого соединения, метаболическую стабильность и продолжительность действия этого соединения, возраст, массу тела, общее состояние здоровья, пол, режим питания, способ и время введения, скорость экскреции, комбинацию лекарственных средств, тяжесть конкретного состояния и хозяина, принимающего лечение.
Для удобства общую суточную дозу можно разделить и вводить в несколько приемов в течение дня, если это желательно. В одном варианте осуществления суточную дозу вводят в один прием. В другом варианте осуществления общую суточную дозу вводят в виде двух дробных доз в течение 24-часового периода. В другом варианте осуществления общую суточную дозу вводят в виде трех дробных доз в течение 24-часового периода. И еще в одном варианте осуществления общую суточную дозу вводят в виде четырех дробных доз в течение 24-часового периода.
Стандартные дозы трициклических гетероциклических соединений можно вводить с различной частотой. В одном варианте осуществления стандартную дозу трициклического гетероциклического соединения можно вводить один раз в день. В другом варианте осуществления стандартную дозу трициклического гетероциклического соединения можно вводить два раза в неделю. В другом варианте осуществления стандартную дозу трициклического гетероциклического соединения можно вводить один раз в неделю. В еще одном варианте осуществления стандартную дозу трициклического гетероциклического соединения можно вводить один раз в две недели. В другом варианте осуществления стандартную дозу трициклического гетероциклического соединения можно вводить один раз в месяц. В еще одном варианте осуществления стандартную дозу трициклического гетероциклического соединения можно вводить один раз в два месяца. В другом варианте осуществления стандартную дозу трициклического гетероциклического соединения можно вводить один раз каждые 3 месяца. В следующем варианте осуществления стандартную дозу трициклического гетероциклического соединения можно вводить один раз каждые 6 месяцев. В другом варианте осуществления стандартную дозу трициклического гетероциклического соединения можно вводить один раз в год.
Количество и частоту введения трициклических гетероциклических соединений будут регулировать в соответствии с решением лечащего врача с учетом таких факторов, как возраст, состояние и размер субъекта, а также тяжесть симптомов, подлежащих лечению. Композиции по изобретению могут также включать одно или несколько дополнительных терапевтических средств, выбранных из перечисленных в настоящей заявке выше.
Наборы
В одном аспекте настоящее изобретение обеспечивает набор, включающий терапевтически эффективное количество по меньшей мере одного трициклического гетероциклического соединения или фармацевтически приемлемой соли или пролекарства указанного соединения и фармацевтически приемлемый носитель, наполнитель или разбавитель.
В другом аспекте настоящее изобретение обеспечивает набор, включающий определенное количество по меньшей мере одного трициклического гетероциклического соединения или фармацевтически приемлемой соли или пролекарства указанного соединения и определенное количество по меньшей мере одного дополнительного терапевтического средства, указанного выше, где количества двух или более активных ингредиентов дают желаемый терапевтический эффект. В одном варианте осуществления одно или несколько трициклических гетероциклических соединений и одно или несколько дополнительных терапевтических средств представлены в одном и том же контейнере. В одном варианте осуществления одно или несколько трициклических гетероциклических соединений и одно или несколько дополнительных терапевтических средств представлены в отдельных контейнерах.
Настоящее изобретение не должно ограничиваться конкретными вариантами осуществления, раскрытыми в примерах, которые предназначены для иллюстрации нескольких аспектов изобретения, и любые варианты осуществления, которые являются функционально эквивалентными, охватываются объемом настоящего изобретения. Действительно, различные модификации изобретения в дополнение к тем, которые показаны и описаны в настоящей заявке, станут очевидными для специалистов в данной области техники и предполагаются как входящие в объем прилагаемой формулы изобретения.
В настоящей заявке процитирован ряд ссылочных документов, полное раскрытие которых включено в настоящую заявку посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| СЛИТЫЕ ТРИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ИНТЕГРАЗЫ ВИЧ | 2016 |
|
RU2740187C2 |
| МОДУЛЯТОРЫ 5HT РЕЦЕПТОРА | 2003 |
|
RU2317982C2 |
| СОЕДИНЕНИЯ | 2016 |
|
RU2725616C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ СОСТОЯНИЙ, ОПОСРЕДОВАННЫХ СХСR4 И CCR5 | 2001 |
|
RU2277092C2 |
| ТРИАРИЛЬНЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2840345C2 |
| АНТИПРОЛИФЕРАТИВНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2811969C2 |
| СПИРОПИПЕРИДИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1993 |
|
RU2168512C2 |
| ИНГИБИТОР LSD1, А ТАКЖЕ СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2763898C2 |
| ЛЕЧЕНИЕ ИНФЕКЦИЙ H. PYLORI С ПРИМЕНЕНИЕМ ИНГИБИТОРОВ MTAN | 2015 |
|
RU2663803C2 |
| ПРОИЗВОДНЫЕ ТИОМОЧЕВИНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ ОБРАТНОЙ ТРАНСКРИПТАЗЫ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА | 1992 |
|
RU2106341C1 |
Изобретение относится к соединению или его фармацевтически приемлемой соли, имеющему формулу (I), в которой А представляет собой -CH(R2)-; X представляет собой 5-членный моноциклический гетероарил, включающий 2 кольцевых гетероатома, представляющих собой N, или -N(R5)C(O)-; Y выбран из -О-, -N(R5)- или -CH(R3)- или -A-Y- представляет собой -C(R2)=CH-; Z представляет собой -С(=О), -CH(R4)- или связь таким образом, что: (i) когда Y представляет собой -О- или -N(R5)-, тогда Z представляет собой -С(=O), (ii) когда Y представляет собой -CH(R3)-, тогда Z представляет собой связь или -CH(R4), и (iii) когда -А-Y- представляет собой -C(R2)=CH-, тогда Z представляет собой связь; R1 представляет собой фенильную группу, которая замещена 1-3 группами, каждая из которых независимо выбрана из галогена; R2 выбран из Н, -O-(C1-C6алкил); R3 выбран из Н; в каждом случае R4 независимо выбран из Н; в каждом случае R5 независимо представляет собой Н или C1-C6алкил; R7A представляет собой Н; R7B представляет собой Н или R7A и R7B вместе с общим для них атомом углерода, к которому они присоединены, объединяются с образованием спироциклической 4-7-членной моноциклической гетероциклоалкильной группы, содержащей 1 гетероатом, представляющий собой О; и R8 выбран из C1-C6алкила, -(C1-С6алкилен)-О-(С1-С6алкил) и -(C1-С6алкилен)-С3-С7циклоалкила. Изобретение также относится к индивидуальным соединениям, фармацевтической композиции, способу ингибирования интегразы ВИЧ у субъекта, способу лечения ВИЧ-инфекции или лечения, профилактики или отсрочки начала или прогрессирования СПИДа у субъекта. Технический результат: получены новые соединения формулы (I), ингибирующие интегразу ВИЧ и которые могут найти применение при лечении ВИЧ-инфекции. 5 н. и 11 з.п. ф-лы, 1 табл., 22 пр.
 (I)
(I)
1. Соединение, имеющее формулу:
(I)
или его фармацевтически приемлемая соль,
где:
А представляет собой -CH(R2)-;
X представляет собой 5-членный моноциклический гетероарил, включающий 2 кольцевых гетероатома, представляющих собой N, или
-N(R5)C(O)-;
Y выбран из -О-, -N(R5)- или -CH(R3)- или -A-Y- представляет собой -C(R2)=CH-;
Z представляет собой -С(=О), -CH(R4)- или связь таким образом, что: (i) когда Y представляет собой -О- или -N(R5)-, тогда Z представляет собой -С(=O), (ii) когда Y представляет собой -CH(R3)-, тогда Z представляет собой связь или -CH(R4), и (iii) когда -А-Y- представляет собой -C(R2)=CH-, тогда Z представляет собой связь;
R1 представляет собой фенильную группу, которая замещена 1-3 группами, каждая из которых независимо выбрана из галогена;
R2 выбран из Н, -O-(C1-C6алкил);
R3 выбран из Н;
в каждом случае R4 независимо выбран из Н;
в каждом случае R5 независимо представляет собой Н или C1-C6алкил;
R7A представляет собой Н;
R7B представляет собой Н,
или R7A и R7B вместе с общим для них атомом углерода, к которому они присоединены, объединяются с образованием спироциклической 4-7-членной моноциклической гетероциклоалкильной группы, содержащей 1 гетероатом, представляющий собой О; и
R8 выбран из C1-C6алкила, -(C1-С6алкилен)-О-(С1-С6алкил) и -(C1-С6алкилен)-С3-С7циклоалкила.
2. Соединение по п. 1, имеющее формулу (Ia):
(Ia)
или его фармацевтически приемлемая соль,
где:
X представляет собой диазолил или -N(R5)C(O)-;
R1 представляет собой фенильную группу, которая необязательно замещена 1-3 группами, каждая из которых независимо выбрана из Cl и F;
R2 представляет собой H или -O-(C1-C6 алкил);
в каждом случае R4 независимо выбран из H;
в каждом случае R5 независимо представляет собой H или C1-C6 алкил;
R7A представляет собой H;
R7B представляет собой H или R7A и R7B вместе с общим для них атомом углерода, к которому они присоединены, объединяются с образованием спироциклической 4-7-членной моноциклической гетероциклоалкильной группы, содержащей 1 гетероатом, представляющий собой О; и
R8 выбран из C1-C6 алкила, -(C1-C6 алкилен)-O-(C1-C6 алкил) и -(C1-C6 алкилен)-C3-C7 циклоалкила.
3. Соединение по любому из пп. 1, 2, где X представляет собой –NHC(O)-.
4. Соединение по любому из пп. 1, 2, где X представляет собой 5-членный моноциклический гетероарил, включающий 2 кольцевых гетероатома, представляющих собой N.
5. Соединение по любому из пп. 1-4, где R1 представляет собой фенил, который замещен 1-3 группами, каждая из которых независимо выбрана из F и Cl.
6. Соединение по п. 5, где R1 выбран из:
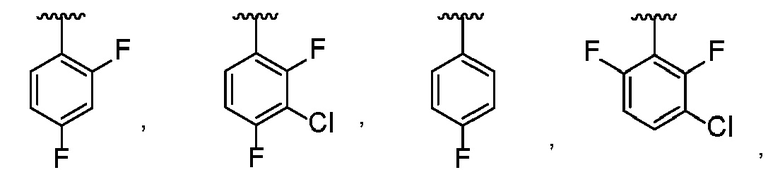
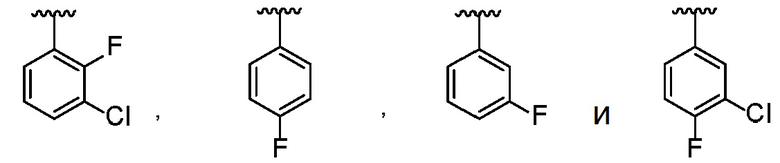 .
.
7. Соединение по любому из пп. 1-6, где R7A и R7B каждый представляет собой H.
8. Соединение по любому из пп. 1-6, где R7A и R7B вместе с общим для них атомом углерода, к которому они присоединены, объединяются с образованием 4-7-членной гетероциклоалкильной группы, содержащей 1 гетероатом, представляющий собой О.
9. Соединение по любому из пп. 1-8, где R8 представляет собой метил, этил, изопропил, -CH2CH2OCH3 и -CH2-циклопропил.
10. Соединение, выбранное из группы, состоящей из
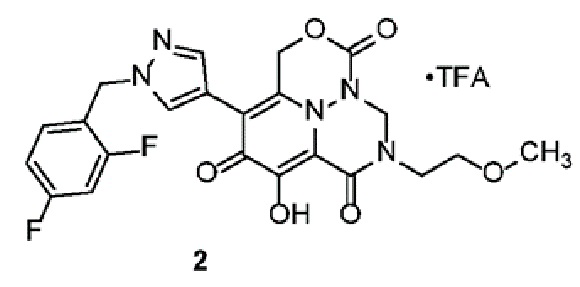
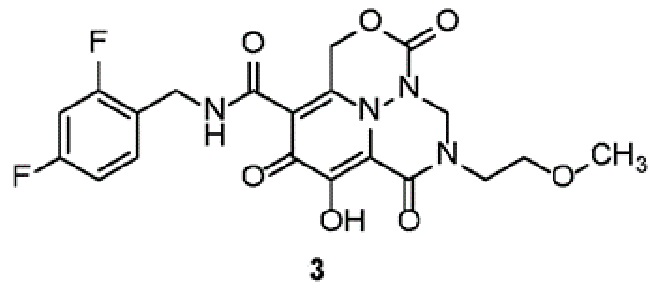
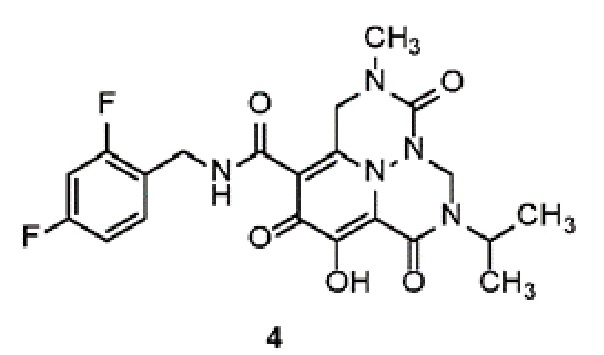
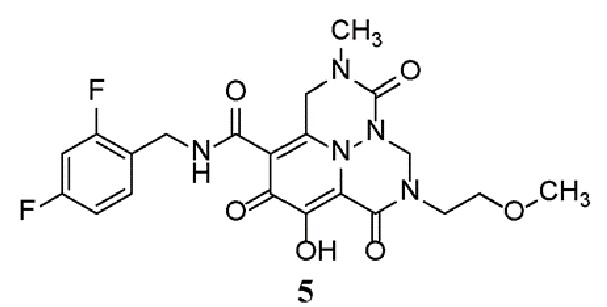
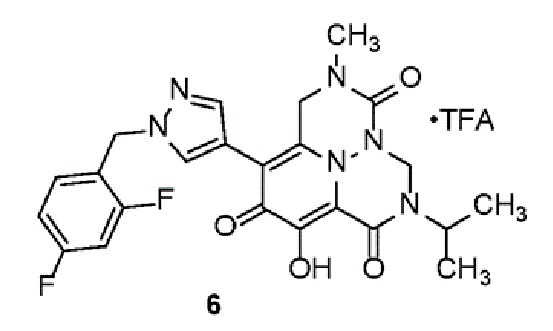
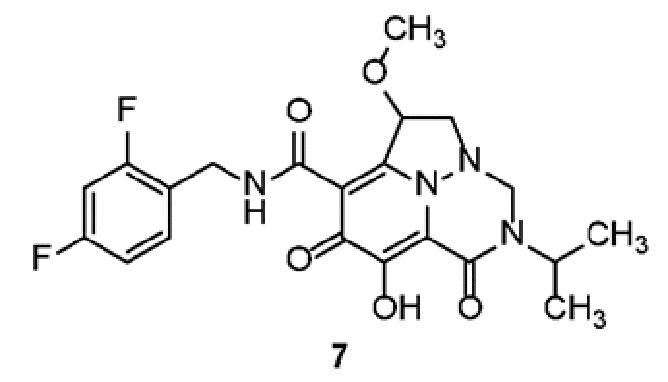
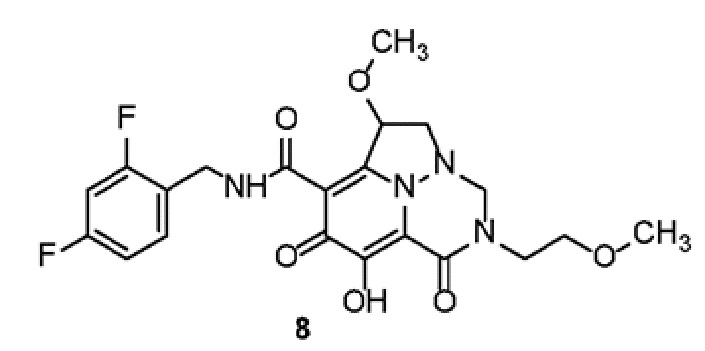
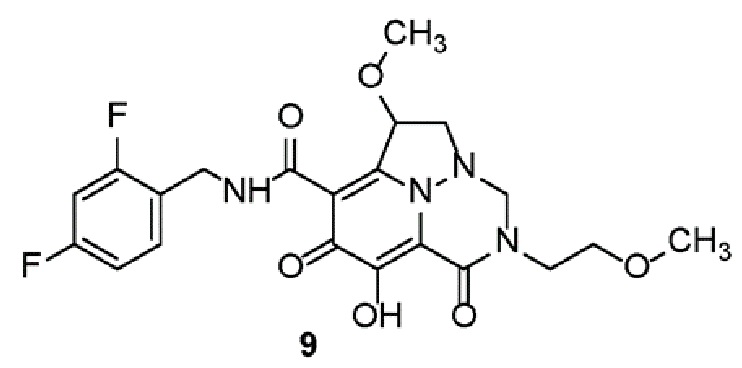
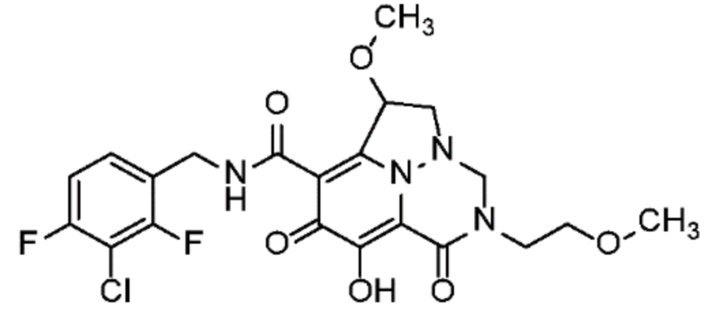
энантиомер A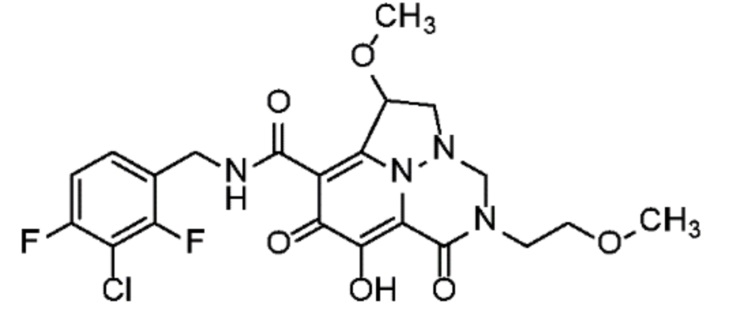
энантиомер B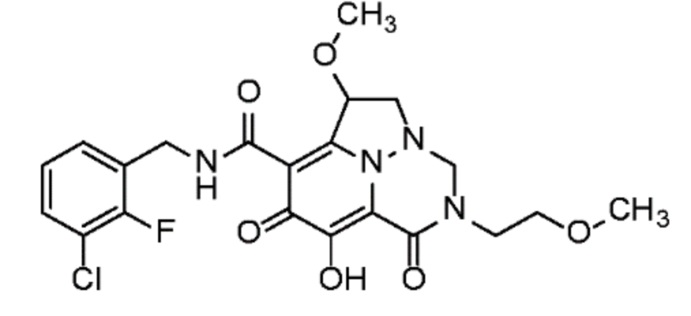
энантиомер A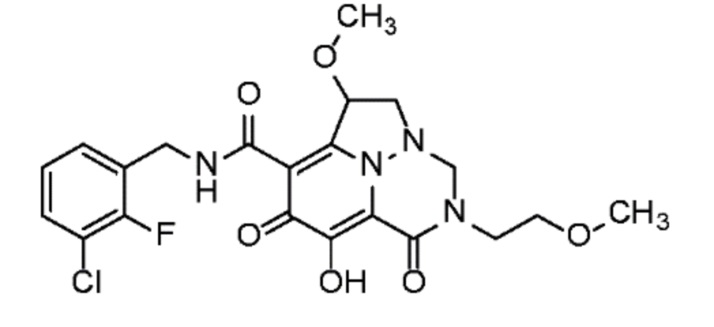
энантиомер B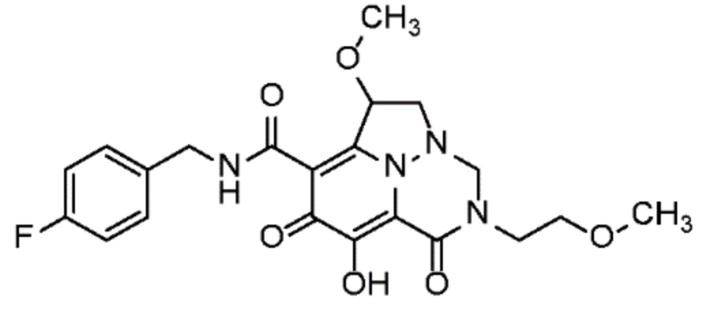
энантиомер A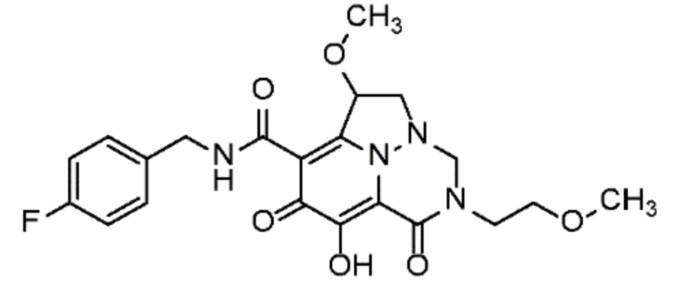
энантиомер B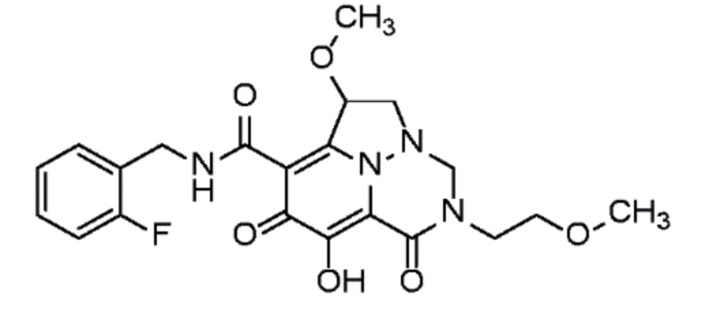
энантиомер B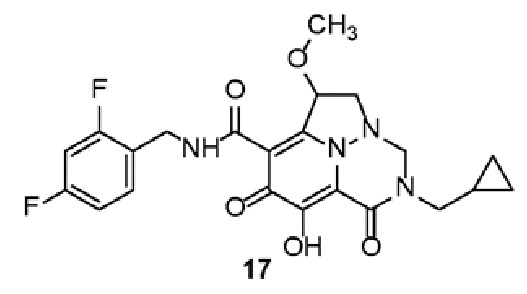
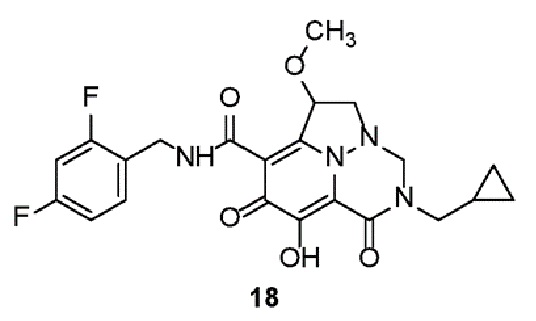
энантиомер B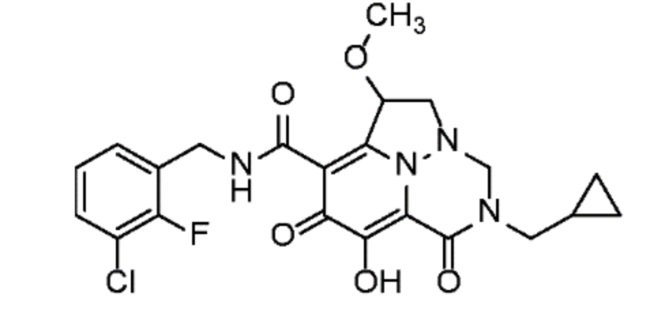
энантиомер А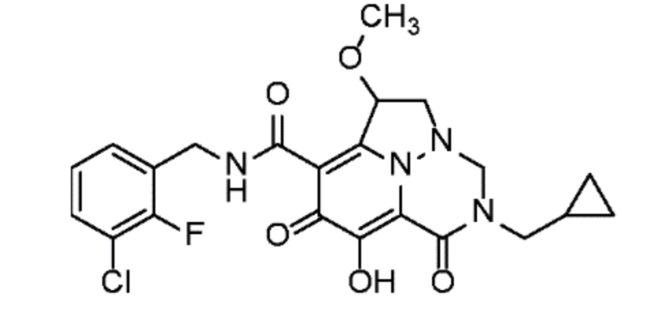
энантиомер B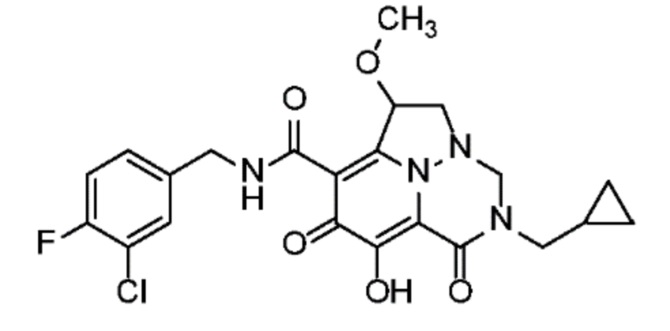
энантиомер B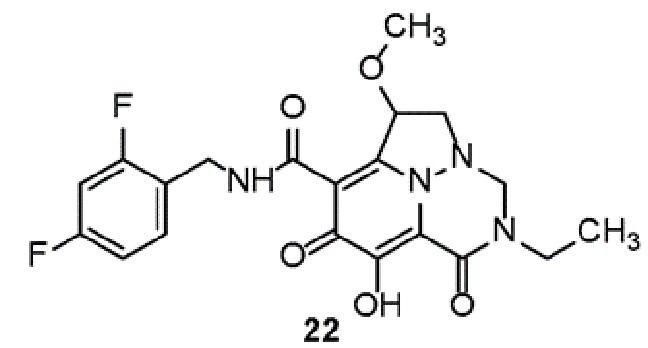
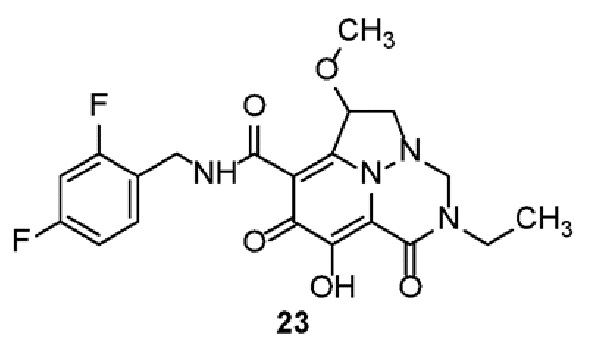
энантиомер B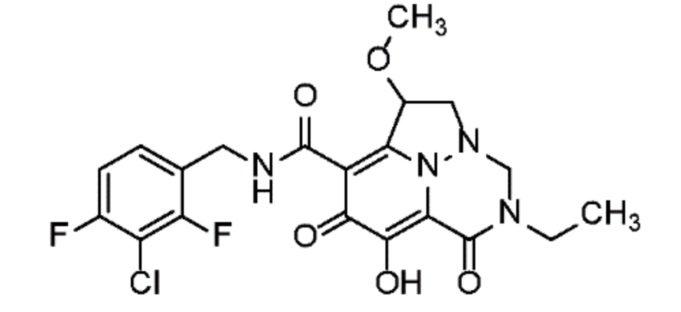
энантиомер Aa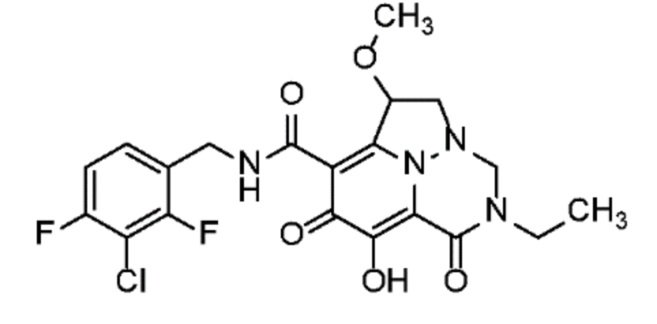
энантиомер Ba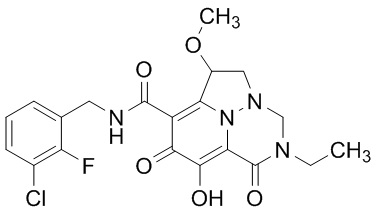
энантиомер Ab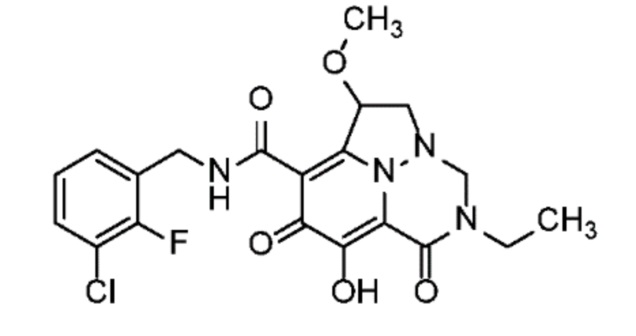
энантиомер Bb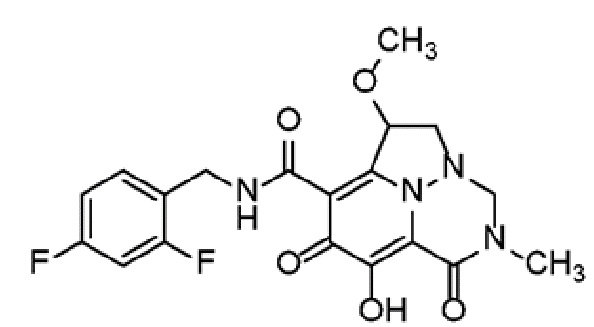
энантиомер A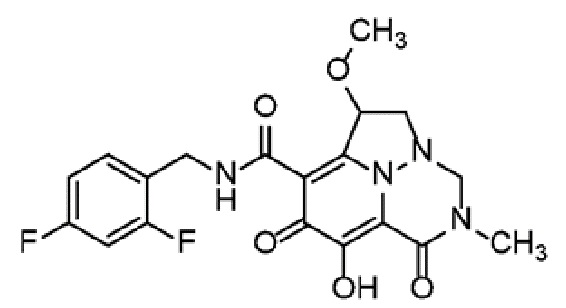
энантиомер B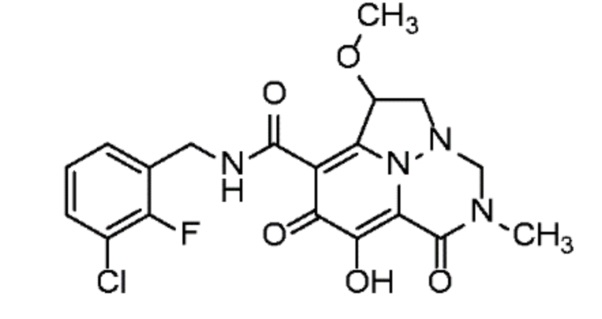
энантиомер Aa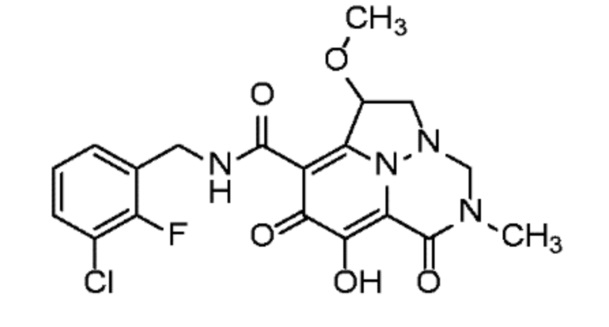
энантиомер Ba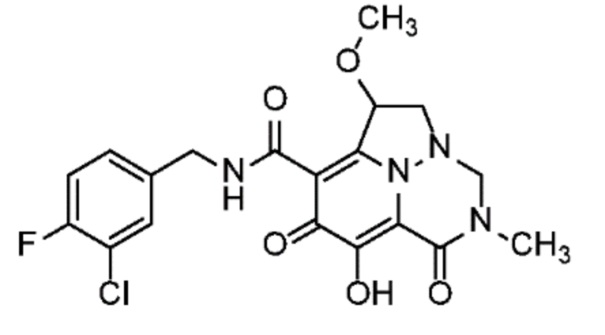
энантиомер Ab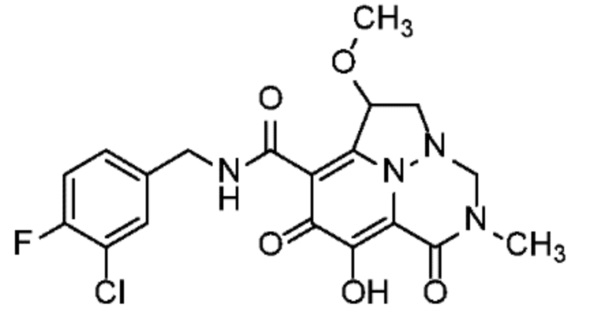
энантиомер Bb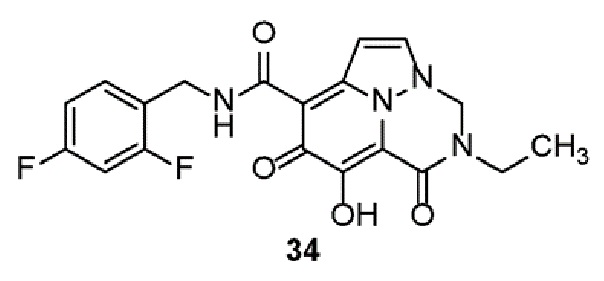
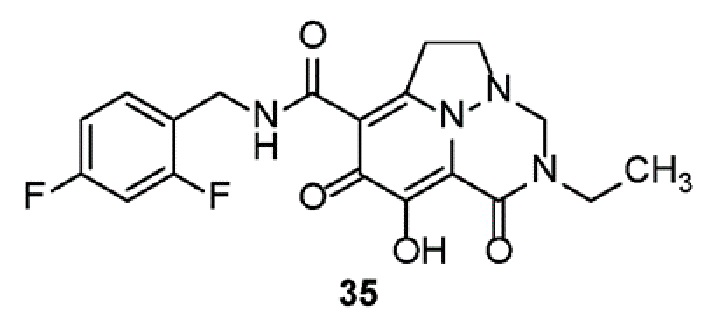
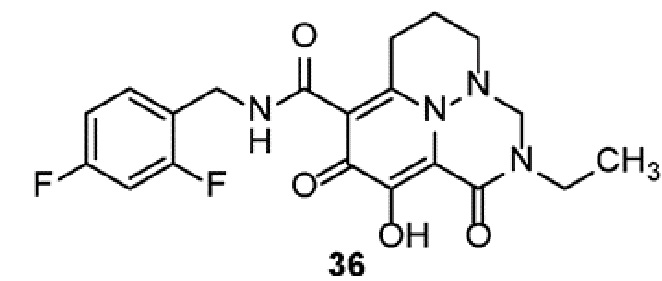
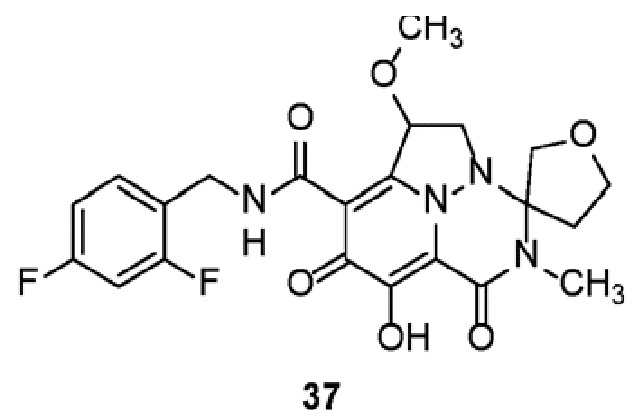
диастереомер 1, энантиомер A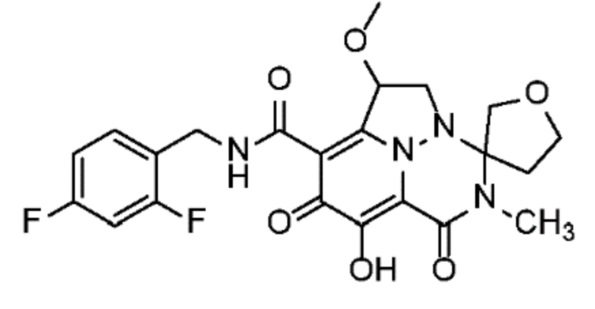
диастереомер 1, энантиомер B
диастереомер 2, энантиомер A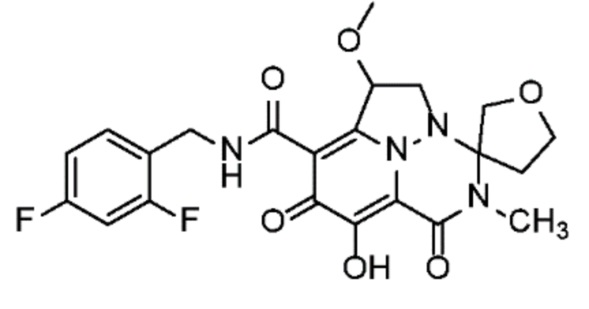
диастереомер 2, энантиомер B
или его фармацевтически приемлемая соль.
11. Фармацевтическая композиция для ингибирования интегразы ВИЧ, включающая эффективное количество соединения по любому из пп. 1-10 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
12. Способ ингибирования интегразы ВИЧ у субъекта, нуждающегося в этом, который включает введение субъекту эффективного количества соединения по любому из пп. 1-10 или его фармацевтически приемлемой соли.
13. Способ лечения ВИЧ-инфекции или лечения, профилактики или отсрочки начала или прогрессирования СПИДа у субъекта, нуждающегося в этом, который включает введение субъекту эффективного количества соединения по любому из пп. 1-9 или его фармацевтически приемлемой соли.
14. Соединение по любому из пп. 1-10 или его фармацевтически приемлемая соль для применения в получении лекарственного средства для ингибирования интегразы ВИЧ, для лечения или профилактики ВИЧ-инфекции или для лечения, профилактики или отсрочки начала или прогрессирования СПИДа у субъекта, нуждающегося в этом.
15. Фармацевтическая композиция по п. 11, дополнительно включающая одно или несколько дополнительных терапевтических средств, выбранных из ламивудина, абакавира, ритонавира, дарунавира, атазанавира, эмтрицитабина, тенофовира, рилпивирина и лопинавира.
16. Способ по п. 13, дополнительно включающий введение субъекту одного или нескольких дополнительных терапевтических средств, выбранных из абакавира, ламивудина, ритонавира и лопинавира, где вводимые количества соединения по любому из пп. 1-10 и одного или нескольких дополнительных терапевтических средств вместе эффективны для лечения ВИЧ-инфекции или для лечения, профилактики или отсрочки начала или прогрессирования СПИДа.
| T | |||
| Kawasuji et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Bi- and Tricyclic Derivatives Result in Superior Antiviralang Pharmacokinetic Profiles" Journal of Medicinal Chemistry, vol | |||
| Приспособление для разматывания лент с семенами при укладке их в почву | 1922 |
|
SU56A1 |
| EA 200801144 A1, 30.10.2008 | |||
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| WO | |||

