Электроно-донорные соединения являются одним из ключевых компонентов титан-магниевых катализаторов для получения полипропилена. Использование этих соединений в составе катализатора в качестве внутренних доноров позволяет радикально увеличить его стереоспецифичность, и, как следствие, стереорегулярность получаемого полимера. При этом, состав донора влияет на молекулярно-массовое распределение (ММР) полипропилена, которое определяет способ переработки и дальнейшего использования полимера. Так, для получения изделий методом литья под давлением требуется полипропилен с узким молекулярно-массовым распределением, тогда как для получения пленочных и трубных марок используется полипропилен с широким ММР. Одним из первых классов электроно-донорных соединений, введение которых в состав титан-магниевого катализатора позволило получить высокостереорегулярный полипропилен с широким ММР, были 2,3-дизамещенные эфиры янтарной кислоты - сукцинаты (WO 2000063261). Высокая эффективность применения сукцинатов в качестве внутренних доноров позволяет выделить катализаторы, имеющие в составе эти соединения, в отдельное, шестое поколение катализаторов для полимеризации пропилена (Adv Polym Sci, 2013, 257, 81; ACS Catal. 2017, 7, 4509).
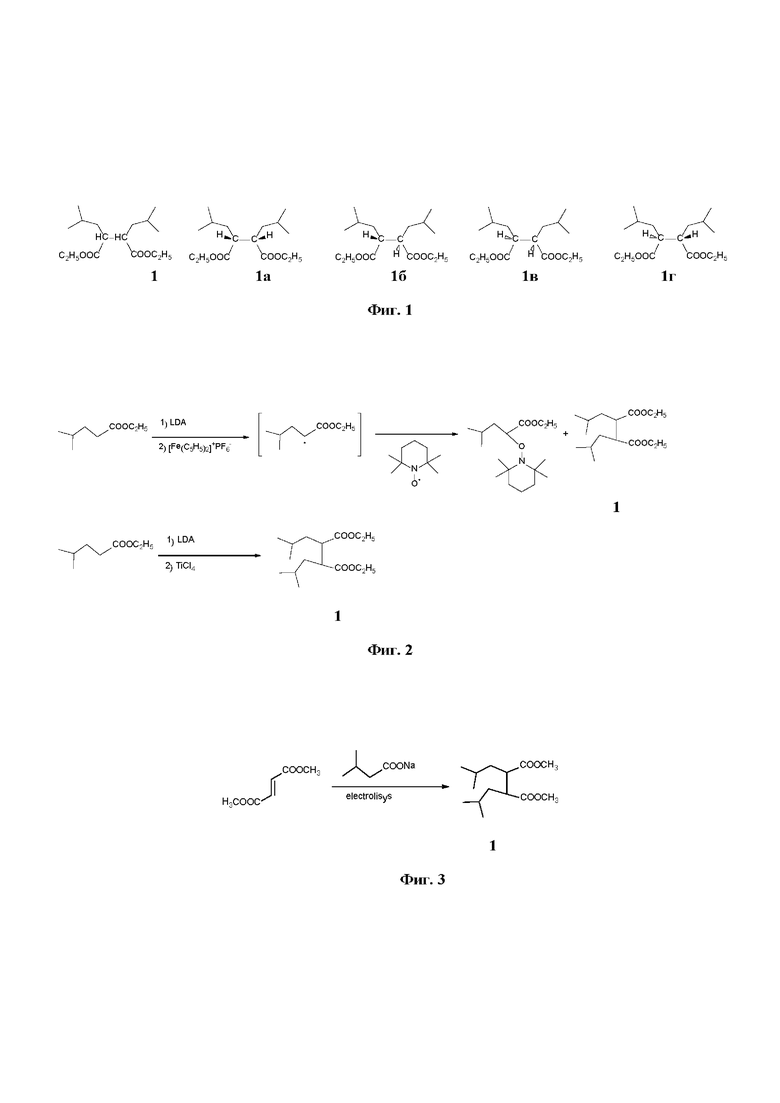
Перспективными внутренними донорами являются диэфиры 2,3-диизобутил-сукцината, в частности, диэтил 2,3-диизобутил-сукцинат (соединение 1) (WO 2000063261; WO 2013029767). Соединение 1 обладает двумя стереоцентрами и способно существовать в виде четырех стереоизомеров 1а-г (Фиг. 1 . Соединения 1а и 1в, а также 1б и 1г попарно являются зеркальными изомерами, поэтому при анализе в ахиральных условиях наблюдаются только две формы соединения 1: анти- (или эритро-), условно отображаемую формулой 1а, и син- (или трео-), условно отображаемую формулой 1б.
В литературе описаны несколько способов получения соединения 1. В работе (J.Org.Chem., 1998, 63, 7130) описано получение сукцината 1 в результате реализации многостадийного процесса, включающего взаимодействие этилового эфира изокапроновой кислоты с диизопропиламидом лития при -78°С, одноэлектронное окисление полученного аниона действием гексафторфосфата ферроцена, и взаимодействие полученного радикала с ТЕМРО (Фиг. 2). Общий выход анти- и син- форм соединения 1 при этом не превышал 23%. В патенте CN 103145553 заявлен похожий способ получения соединения 1, основанный на взаимодействии этилового эфира изокапроновой кислоты с диизопропиламидом лития при -78°С с последующей димеризацией под действием TiCl4 с выходом 84%.
В работе [Can.J.Chem., 1980, vol. 58, p. 1101-1105] приводится еще один вариант получения соединения 1. По этому способу, взаимодействием диметилового эфира фумаровой кислоты с изобутильным радикалом, генерируемым при анодном окислении натриевой соли изовалериановой кислоты, получена смесь изомеров соединения 1 с выходом 37% (Фиг. 3).
Эти способы синтеза соединения 1 обладают значительными недостатками, такими как использование высокоопасных дорогостоящих реагентов (бутиллитий, диизопропиламид лития, гексафторфосфат ферроцена), требуют использования глубокого охлаждения и инертной атмосферы или сложного оборудования, необходимого для проведения электролиза, и, как правило, приводят к соединению 1 с невысоким выходом.
Наиболее близким к предложенному нами способу получения соединения 1, прототипом, является многостадийный синтез соединения 1, включающий алкилирование малонового эфира галогенацетатом с последующими двумя последовательными алкилированиями галогеналкилами, гидролизом, декарбоксилированием и этерификацией (Фиг. 4) (WO 2013029767). В этом варианте используются недорогие доступные реактивы, не требуется применения сложного оборудования.
Однако, воспроизведение методики, предложенной в WO 2013029767, показало, что только две первые стадии процесса воспроизводятся, тогда как третья стадия приводит к образованию смеси исходного изобутилтриэфира и его изо-бутилового эфира (Фиг. 5) в близком соотношении вместо целевого соединения 1 (см. Пример 1), что, очевидно, связано с пониженной С-Н кислотностью реакционного центра в изобутилтриэфире. Независимое описание успешного применения этой последовательности для синтеза соединения 1 в литературе также отсутствует. Следовательно, данная последовательность реакций или не позволяет получить заявленный продукт 1, или отличается низкой воспроизводимостью, что делает её неприменимой для практического использования.
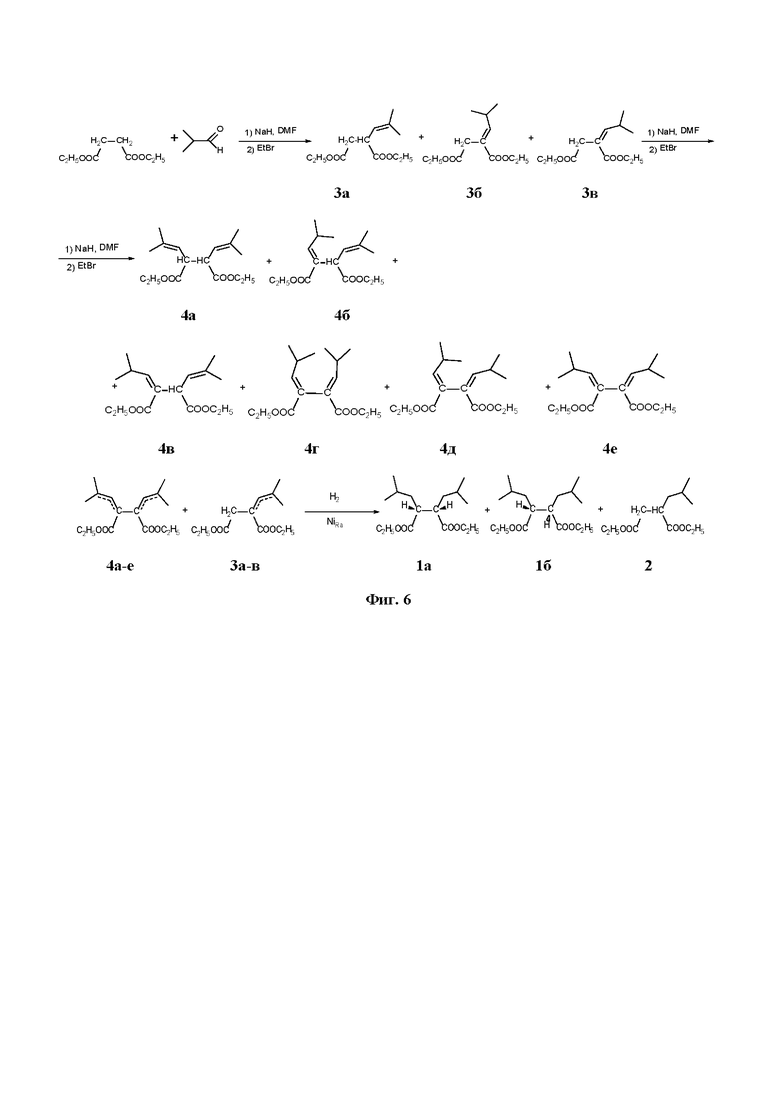
Целью настоящего изобретения является способ получения дизамещенного сукцината 1 со степенью чистоты более 95%, без существенной примеси монозамещенного 2-изобутилсукцината 2, который снижает стереорегулирующую способность катализатора полимеризации олефинов. Общая схема предлагаемого нами способа получения диэфира 2,3-диизобутил-сукцината изображена на Фиг. 6. Процесс получения диэфира 2,3-диизобутил-сукцината заключается в двойной конденсации изобутилового альдегида с соответствующим диэфиром янтарной кислоты с образованием смеси моно- и бис-этилиденовых соединений 3 и 4 и последующее их гидрирование ведущее к 2,3-диизобутил-сукцинату 1.
На первой стадии диэфир янтарной кислоты (например, диэтилсукцинат) вводится в реакцию с изо-бутиловым альдегидом, взятым с избытком 0-30%, в апротонном растворителе, таком как толуол, этилбензол, ксилол, ДМФА, N,N-диметилацетанилид, 1-метил-2-пирролидон, тетрагидрофуран, предпочтительно, ДМФА, или в безводном протонном растворителе, таком как этанол, изопропанол, трет-бутанол. В качестве основания используется 1-1.3 эквивалента гидрида металла формулы MeHn либо алкоксида металла (RO)nMe, где Me - это металл первой или второй группы Периодической системы, n - валентность металла, R - это остаток спиртовой группы С1-С4. Реакция проводится при нагревании в диапазоне температур 40 - 100 °С в течение 3-5 ч. Получающийся в ходе реакции in situ моноэфир 2-изобутилиденсукцината взаимодействием с 1.1 - 1.2 эквивалента алкилирующего агента (алкилгалогенид, диалкилсульфат) в течение 30 мин - 2 ч превращают в диэфир 3а-в. После экстракционной обработки вещество может быть очищено вакуумной перегонкой.
На второй стадии сумма алкилиденсукциновых эфиров 3а-в вводится в реакцию с изо-бутиловым альдегидом, взятым с избытком 0-30%, в апротонном растворителе, таком как толуол, этилбензол, ксилол, ДМФА, N,N-диметилацетанилид, 1-метил-2-пирролидон, тетрагидрофуран, или в безводном протонном растворителе, таком как этанол, изопропанол, трет-бутанол. В качестве основания используется 1-1.3 эквивалента гидрида металлов формулы MeHn либо алкоксида металла (RO)nMe, где Me - это металл первой или второй группы Периодической системы, n - валентность металла, R - это остаток спиртовой группы С1-С4. Реакция проводится при нагревании в диапазоне температур 40 - 100 °С в течение 3-5 ч. Получающийся в ходе реакции in situ моноэфир 2,3-бис-изобутилиденсукцината взаимодействием с 1.1 - 1.2 эквивалента алкилирующего агента (алкилгалогенид, диалкилсульфат) в течение 30 мин - 2 ч превращают в диэфир 4а-е. После экстракционной обработки продукт может быть очищен вакуумной перегонкой.
Первая и вторая стадия процесса могут быть объединены и проведены "в одном сосуде" без выделения промежуточных продуктов 3а-в. Для этого к реакционной массе, полученной на первой стадии, прибавляется 1-1.3 эквивалента используемого основания и 1-1.3 эквивалента изо-бутилового альдегида. Реакция проводится при нагревании в диапазоне температур 40 - 100°С в течение 3-5 ч. Получающийся в ходе реакции in situ моноэфир 2,3-бис-изобутилиденсукцината взаимодействием с 1.1 - 1.2 эквивалента алкилирующего агента (алкилгалогенид, диалкилсульфат) в течение 30 мин - 2 ч превращают в диэфиры 4а-е. После экстракционной обработки продукт может быть очищен вакуумной перегонкой.
На третьей стадии полученная смесь изомерных бис-изобутилидензамещенных сукцинатов 4а-е без предварительного разделения гидрируется водородом при температурах 100-200°С и давлении 60 - 200 бар на катализаторах гидрирования: 5-10 % палладия или платины на угле, или скелетном никеле с образованием анти- и син- изомеров 1а,б. В качестве растворителя используются низшие спирты С1-С3 или линейные или разветвленные углеводороды С5-С8. После очистки продукта вакуумной перегонкой получается 2,3-бис-изобутилсукцинат 1 с чистотой более 95%.
Таким образом, нами предлагается новый способ синтеза диэфира 2,3-диизобутил-сукцината 1, позволяющий получить целевой продукт с чистотой не менее 95% с использованием недорогих коммерчески доступных реагентов и растворителей, обычного химического оборудования.
Полученные диэфиры 2,3-диизобутил-сукцината, в частности, диэтил 2,3-диизобутил-сукцинат 1, могут быть использованы для приготовления титан-магниевых катализаторов полимеризации пропилена в изотактический полипропилен в качестве стереорегулирующего электронодонорного компонента, что подтверждается Примерами 7-9.
Изобретение иллюстрируется следующими примерами.
Пример 1. Подход к синтезу диэтил 2,3-диизобутил-сукцината 1 по методике WO 2013029767
Суспензию NaH в масле (60 %) (64 мг (1.61 ммоль)) промыли 2 раза гексаном по 5 мл, декантировали, остатки растворителя отогнали на роторном испарителе. Добавили 1 мл сухого ДМФА, колбу продули аргоном. При комнатной температуре и перемешивании прибавили по каплям 324 мг (1.07 ммоль) триэтил 1-изобутилэтан-1,1,2-трикарбоксилата, синтезированного по методике (WO 2013029767), перемешивали 30 мин при комнатной температуре. Прибавили по каплям 220 мг (1.61 ммоль) изобутилбромида. Перемешивали реакционную смесь при Т = 85 °С в течение 8 ч. Реакционную смесь охладили, разбавили 5 мл воды и экстрагировали 3 * 5 мл хлористого метилена. Объединенную органическую фазу промыли 2 мл насыщенного раствора NaCl, сушили MgSO4. Осушитель отфильтровали, растворитель отогнали на ротационном испарителе. Вместо ожидаемого триэтил 1,2-бис-изобутилэтан-1,1,2-трикарбоксилата получили 177 мг смеси исходного соединения и его изо-бутилового эфира в соотношении 48 : 52, по данным ЯМР 1Н.
Пример 2. Синтез соединений 3а-в в этаноле.
В инертной атмосфере прибавили 3 мл этанола к 145 мг натрия (6.28 ммоль) и перемешивали до растворения. К полученному раствору прибавили раствор 1.044 г (6 ммоль) диэтилсукцината и 0.411 г (5.71 ммоль) изомасляного альдегида в 1 мл этанола. Реакционную смесь кипятили в течение 7.5 ч. Прибавили 0.654 г (6 ммоль) бромистого этила, кипятили реакционную смесь в течение 2 ч. Реакционную смесь охладили, упарили, разбавили водой и экстрагировали МТБЭ. Объединенную органическую фазу сушили MgSO4. Осушитель отфильтровали, растворитель отогнали на ротационном испарителе. Получили 0.509 г смеси соединений 3а-в, выход 44 %.
Пример 3. Синтез соединений 3а-в в ДМФА.
К 0.18 моль NaH добавили 35 мл ДМФА, охладили до 0 °С, прибавили раствор 30 мл (0.18 моль) диэтилсукцината и 18.5 мл (0.198 моль) изомасляного альдегида в 40 мл ДМФА. Перемешивали реакционную смесь при 60 °С в течение 3 ч. Прибавили 16 мл (0.207 моль) бромистого этила, перемешивали реакционную смесь при 60 °С в течение 2 ч. Реакционную смесь охладили, разбавили 450 мл воды и экстрагировали МТБЭ. Объединенную органическую фазу промыли насыщенным раствором NaCl, сушили MgSO4. Осушитель отфильтровали, растворитель отогнали на ротационном испарителе. Получили 36.19 г смеси соединений 3а-в в соотношении 54 : 28 : 17 по данным ХМС, выход 88 %.
Характеристичные сигналы спектра ЯМР 1Н смеси соединений 3а-в, ЯМР 1Н (400 МГц, CDCl3, δ, м.д., J/Гц): соединение 3а: 2.39 дд (1Н, Н-3, 16.4, 6.0), 2.79 дд (1Н, Н-3, 16.4, 9.0), 3.65 ддд (1Н, Н-2, 9.5, 9.0, 6.0), 5.01 дм (1Н, Н-1', 9.5). Соединение 3б (E-изомер): 3.31 с (2Н, Н-3), 6.73 дм (1Н, Н-2', 10.2). Соединение 3в (Z-изомер): 3.20 с (2Н, Н-3), 5.79 дм (1Н, Н-2', 9.8).
Пример 4. Синтез соединения 1 с чистотой 75%.
А) К 0.25 моль NaH добавили 30 мл ДМФА, прибавили раствор 42 мл (0.25 моль) диэтилсукцината и 26 мл (0.275 моль) изомасляного альдегида в 50 мл ДМФА при комнатной температуре. Перемешивали реакционную смесь при 60 °С в течение 3 ч. Прибавили 25 мл (0.288 моль) бромистого этила, перемешивали реакционную смесь при Т60 °С в течение 2 ч. Добавили 0.288 моль NaH и 27 мл (0.288 моль) изомасляного альдегида в 30 мл ДМФА. Перемешивали реакционную смесь при 60 °С в течение 3 ч. Прибавили 25 мл (0.288 моль) бромистого этила, перемешивали реакционную смесь при 60 °С в течение 2 ч. Реакционную смесь охладили, разбавили водой и экстрагировали МТБЭ. Объединенную органическую фазу промыли насыщенным раствором NaCl, сушили MgSO4. Осушитель отфильтровали, растворитель отогнали на ротационном испарителе. Получили 30.39 г смеси соединений 4а-е, выход 43%. По данным ХМС соотношение соединений 4а-е в смеси составило 16.7 : 15 : 6.8 : 6.5 : 4 : 1.
Б) В стальной автоклав ёмкостью 0.5 л поместили раствор 29.39 г (0.104 моль) соединений 4а-е в 60 мл EtOH (96 %) и суспензию никеля Реннея, приготовленную из 5.00 г сплава Реннея (И.Б. Репинская, М.С. Шварцберг, Избранные методы синтеза органических соединений, Новосибирск: Изд-во Новосиб. Ун-та, 2000, с. 80), в 20 мл EtOH (96 %). Гидрирование проводили при давлении водорода 100 атм. при 180 °С и времени реакции 20 ч. Отогнали растворитель, получили 23.98 г масла, которое перегнали в вакууме, отбирали фракцию с Ткип = 120-140 °С (3 мм рт.ст.), получили 13.35 г соединения 1 с чистотой 60 %. Полученную фракцию перегнали в вакууме повторно, отбирали фракцию с Ткип = 100 – 120 °С (1 мм рт.ст.), получили 6.62 г соединения 1 с чистотой 75 % по ХМС, выход 22 %.
Пример 5. Синтез соединения 1 с чистотой 96%.
А) К 0.159 моль NaH добавили 30 мл ДМФА, охладили до 0 °С, прибавили раствор 36.19 г (0.159 моль) соединения 3 (смесь изомеров) и 19.1 мл (0.206 моль) изомасляного альдегида в 40 мл ДМФА. Перемешивали реакционную смесь при 60 °С в течение 3 ч. Прибавили 14.5 мл (0.190 моль) бромистого этила, перемешивали реакционную смесь при °С в течение 2 ч. Реакционную смесь охладили, разбавили 450 мл воды и экстрагировали МТБЭ. Объединенную органическую фазу промыли насыщенным раствором NaCl, сушили MgSO4. Осушитель отфильтровали, растворитель отогнали на ротационном испарителе. Получили 41.79 г смеси соединений 4а-е в соотношении 45 : 29 : 10 : 7 : 5 : 3 по данным ХМС, выход 94 %. После перегонки в вакууме (5 мм рт.ст.) получили 26.75 г соединения 4 с Ткип = 126-158 °С, выход 60 %.
Б) В стальной автоклав ёмкостью 0.5 л поместили раствор 26.75 г (0.095 моль) соединения 4 в 70 мл EtOH (96 %) и суспензию никеля Реннея, приготовленную из 4.50 г сплава Реннея, в 20 мл EtOH (96 %). Гидрирование проводили при давлении водорода 100 атм и Т = 180 °С и времени реакции t = 24 ч. Растворитель отогнали на ротационном испарителе, получили 23.86 г масла. Вещество перегнали в вакууме, отбирали фракцию с Ткип = 121-148 °С (3 мм рт.ст.), получили 19.97 г соединения 1 с чистотой 93 %. Полученную фракцию перегнали в вакууме повторно, отбирали фракцию с Ткип = 100 - 116 °С (1 мм рт.ст.), получили 16.70 г соединения 1 с чистотой 96 % по ХМС, выход 61 %.
ЯМР 1Н (400 МГц, CDCl3, δ, м.д., J/Гц): 0.84, 0.87 д (6Н+6Н, 4СН3, 6.1), 0.99-1.10 м (2H, H-1'), 1.22, 1.24 т (3Н+3Н, 2ОСН2СН3, 7.0), 1.37-1.69 м (4Н, 2Н-1', 2H-2'), 2.60-2.71 м (2Н, Н-2, Н-3), 4.06-4.20 м (4Н, 2ОСН2).
Пример 6. Синтез соединения 1 с чистотой 99%.
А) К 0.12 моль NaH добавили 20 мл ДМФА, охладили до 0 °С и прибавили раствор 20 мл (0.12 моль) диэтилсукцината и 12.2 мл (0.132 моль) изомасляного альдегида в 25 мл ДМФА. Перемешивали реакционную смесь при 60 °С в течение 3 ч. Прибавили 10.4 мл (0.138 моль) бромистого этила, перемешивали реакционную смесь при 60 °С в течение 2 ч. Добавили 0.138 моль NaH в 25 мл ДМФА. Прибавили раствор 12.8 мл (0.138 моль) изомасляного альдегида в 13 мл ДМФА. Перемешивали реакционную смесь при 60 °С в течение 3 ч. Прибавили 10.4 мл (0.138 моль) бромистого этила, перемешивали реакционную смесь при 60 °С в течение 2 ч. Реакционную смесь охладили, разбавили водой и экстрагировали МТБЭ. Объединенную органическую фазу промыли насыщенным раствором NaCl, сушили MgSO4. Осушитель отфильтровали, растворитель отогнали на ротационном испарителе. Получили 27.97 г смеси соединений 4а-е, выход 83%. После перегонки в вакууме (5 мм рт.ст.) получили 17.98 г вещества 4 с Ткип = 128-158 °С, выход 53 %. По данным ХМС соотношение соединений 4а-е в смеси составило 33 : 29 : 8 : 6 : 2 : 3.
Б) В стальной автоклав ёмкостью 0.5 л поместили раствор 17.98 г (0.064 моль) соединения 4 в 100 мл EtOH (96%) и суспензию никеля Реннея, приготовленную из 3.00 г сплава Реннея, в 20 мл EtOH (96%). Гидрирование проводили при давлении водорода 100 атм и Т = 180 °С и времени реакции t = 24 ч. Растворитель отогнали на ротационном испарителе, получили 17.22 г масла, которое перегнали в вакууме, отбирали фракцию с Ткип = 123-133 °С (3 мм рт.ст.), получили 7.31 г соединения 1 с чистотой 93%. Полученную фракцию перегнали в вакууме повторно, отбирали фракцию с Ткип = 122 - 128 °С (3 мм рт.ст.), получили 5.64 г соединения 1 с чистотой >99% по ХМС, выход 31 %.
Пример 7. Приготовление катализатора полимеризации пропилена с использованием в качестве внутреннего донора соединения 1 с чистотой 75 %.
Приготовление катализатора проводили в три стадии: (1) приготовление магнийорганического соединения (продукт I); (2) приготовление магнийсодержащего носителя (продукт II); (3) приготовление катализатора. Эти стадии проводились согласно примеру 1 в пат. RU 2152404 C1.
Приготовление промежуточного продукта I
В трехгорлую колбу, снабженную обратным холодильником, капельной воронкой и мешалкой, загружают порошок магния (26 г, 1.07 г/атом). Колбу продувают азотом, нагревают порошок магния 1 ч при 80°С и добавляют смесь 173 мл ди-н-бутилового эфира и 80 мл хлорбензола. Затем последовательно в реакционную смесь добавляют 0.03 г йода и 3 мл хлористого н-бутана. После исчезновения йодного окрашивания повышают температуру реакционной смеси до 97°С и в течение 2.5 часов дозируют 250 мл хлорбензола. Полученную в результате этого процесса темную реакционную смесь перемешивают в течение 8 ч при 97°С. Затем перемешивание и нагревание прекращают, твердому продукту дают осесть в течение 48 ч. После декантации раствора над осадком получают раствор продукта I с концентрацией 1.0 моль Mg/л.
Приготовление промежуточного продукта II
Раствор продукта I (100 мл, 1.0 моль/л), полученного, как описано выше, загружают в реактор. Этот раствор охлаждают до 0°С и в течение 2 ч добавляют раствор 11.2 мл тетраэтоксисилана (ТЭС) в 38 мл ди-н-бутилового эфира при перемешивании. После добавления раствора, реакционную смесь выдерживают 0.5 ч при 0°С, затем повышают температуру до 60°С и выдерживают еще 1 ч при 60°С. После этого перемешивание и нагревание прекращают и твердому продукту дают осесть в течение 30 мин. Жидкость над осадком удаляют декантированием. Твердое вещество промывают 5 раз, используя 150 мл гептана. Получают светло-желтое вещество – 13.5 г продукта II, суспендированного в 40 мл гептана.
Приготовление катализатора
Реактор на 500 мл продувают азотом и загружают в него 300 мл четыреххлористого титана при комнатной температуре. Затем реактор нагревают до 115°С на глицериновой бане и добавляют суспензию, содержащую 12 г продукта II в 40 мл гептана. После этого в реактор добавляют 4.2 г стереорегулирующего электронодонорного агента 1, полученного в примере 4 (диэтил 2,3-диизобутил-сукцинат с чистотой 75 %), и реакционную смесь перемешивают при 115°С в течение 1.5 ч. После этого перемешивание прекращают и твердому веществу дают осесть в течение 30 мин. Жидкость над осадком удаляют декантированием, после чего в реактор добавляют 300 мл хлорбензола, реактор нагревают до 100°С и выдерживают в течение 20 мин. Затем перемешивание прекращают, твердому веществу дают осесть в течение 30 мин. Жидкость над осадком удаляют декантированием, после чего в реактор добавляют смесь 150 мл четыреххлористого титана и 150 мл хлорбензола. Реакционную смесь нагревают до 115°С и перемешивают в течение 30 мин, после чего следует вышеописанная процедура осаждения и декантации. Последний цикл повторяют еще раз. Полученное твердое вещество промывают 5 раз, используя 300 мл гептана при 60°С, и после этого получают катализатор, суспендированный в гептане.
Полимеризация пропилена
Полимеризацию пропилена проводили согласно примеру 1 в пат. RU 2152404 C1. Реактор из нержавеющей стали для полимеризации продувают азотом и затем заполняют осушенным гептаном (290 мл), не содержащим кислорода. Затем в реактор вводят раствор 1.2 ммоль триэтилалюминия в 5 мл гептана, 0.06 ммоль циклогексилметилдиметоксисилана (донор Д2), растворенного в 5 мл гептана и 0.01 г катализатора в виде суспензии в 1 мл гептана. Далее добавляют 33 мл (при нормальных условиях) водорода и пропилен до достижения давления 0.05 МПа. Включают перемешивание и реактор быстро нагревают до температуры полимеризации 70°С, подают пропилен до давления 0.6 Мпа и в этих условиях осуществляют процесс полимеризации в течение 1 ч. После этого давление снижают до атмосферного, сливают содержимое реактора и выделяют порошок полимера из гептана. После этого порошок полипропилена высушивают в вакуумном шкафу и взвешивают.
Результаты полимеризации приведены в Таблице 1.
Пример 8.
Катализатор готовят аналогично примеру 7 за исключением того, что в качестве стереорегулирующего электронодонорного агента используется соединение 1, полученное в примере 5 (диэтил 2,3-диизобутил-сукцинат с чистотой 96 %).
Полимеризацию проводят аналогично примеру 7. Результаты полимеризации приведены в Таблице 1.
Пример 9.
Катализатор готовят аналогично примеру 7 за исключением того, что в качестве стереорегулирующего электронодонорного агента используется соединение 1, полученное в примере 6 (диэтил 2,3-диизобутил-сукцинат с чистотой 99 %).
Полимеризацию проводят аналогично примеру 7. Результаты полимеризации приведены в таблице 1.
Таблица 1. Результаты экспериментов по полимеризации пропилена

1) 1 - диэтил 2,3-ди-изобутил-сукцинат.
2) Определение количества фракции ПП, растворимой в ксилоле (XS) проводили согласно методике, описанной в стандарте США ASTM D-5492.
3) Индекс изотактичности (ИИ) порошка полипропилена рассчитывали как 100 - XS.
4) Полидисперсность полипропилена (Mw/Mn) определяли при температуре 160°C методом гельпроникающей хроматографии.
Катализаторы имеют средний размер частиц 17 мкм (по данным лазерной дифракции) и высокую активность (выход ПП). Катализаторы 2 и 3, приготовленные с образцами диэтил 2,3-диизобутил-сукцината, имеющими чистоту ≥ 95%, позволяют получать ПП с требуемой высокой изотактичностью (величины ИИ > 97%). Приготовленные катализаторы позволяют получать также ПП с требуемым широким молекулярно-массовым распределением (величина Mw/Mn находится в области 5-6).
Список использованных источников
G. Morini, G. Balbontin, Y. Gulevich, R. Kelder, H. Duijghuisen, P. Klusener, F. Korndorffer. Components and catalysts for the polymerization of olefins // Patent WO 2000063261 A1.
T. Taniike, M. Terano. The Use of Donors to Increase the Isotacticity of Polypropylene. In: Kaminsky W. (eds) Polyolefins: 50 years after Ziegler and Natta I. // Advances in Polymer Science, vol 257, 81-98. Springer, Berlin, Heidelberg.
A. Vittoria, A. Meppelder, N. Friederichs, V. Busico, R. Cipullo. Demystifying Ziegler–Natta Catalysts: The Origin of Stereoselectivity // ACS Catal. 2017, 7, 7, 4509-4518.
J.B. Sainani, M. Davadra. Process for preparing di-substitued succinates // Patent WO 2013/029767 A1.
U. Jahn. Highly Efficient Generation of Radicals from Ester Enolates by the Ferrocenium Ion. Application to Selective r-Oxygenation and Dimerization Reactions of Esters // J. Org. Chem., 1998, v. 63, 7130-7131.
G. Zhanxian, Ch. Qiuju. Synthesis and application of 2,3-hydrocarbyl substituted diester succinate // Patent CN 103145553 A.
Ph.J. Champagne, R.N. Renaud. Electrochemical oxidation of carboxylic acid anions in the presence of some mono- and di-substituted olefins // Can. J. Chem., 1980, vol. 58, p. 1101-1105.
И.Б. Репинская, М.С. Шварцберг. Избранные методы синтеза органических соединений, Новосибирск: Изд-во Новосиб. Ун-та, 2000, с. 80.
В.А. Захаров, Г.Д. Букатов, С.А. Сергеев. Способ получения катализатора, применяемого для полимеризации олефинов // Patent RU 2152404 C1.
| название | год | авторы | номер документа |
|---|---|---|---|
| Диэтил 2,2-диалкилмалонат и 2,2-диалкил-1,3-диметоксипропан, содержащие 6,6-диметилбицикло[3.1.1]гептановый фрагмент природного происхождения, способ их получения и титан-магниевый катализатор полимеризации пропилена, содержащий эти соединения в своем составе | 2023 |
|
RU2819723C1 |
| 1,3-Диметоксипропаны, содержащие 6,6-диметилбицикло[3.1.1]гептановый фрагмент природного происхождения и н-алкильный заместитель, способ их получения и титан-магниевый катализатор полимеризации пропилена, содержащий эти соединения в своем составе | 2024 |
|
RU2839765C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕРАНИАЛЯ | 2015 |
|
RU2579122C1 |
| Способ получения трет-бутилового спирта | 2019 |
|
RU2715430C1 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА ДЛЯ РЕАКЦИЙ, ИДУЩИХ ПО КИСЛОТНО-ОСНОВНОМУ МЕХАНИЗМУ | 2021 |
|
RU2773703C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,3-ЭПОКСИПИНАНА ИЗ СКИПИДАРА | 2010 |
|
RU2425040C1 |
| Способ безводородного гидрирования сульфатного скипидара в проточном режиме | 2020 |
|
RU2736503C1 |
| Способ очистки трифторида азота от примеси тетрафторида углерода | 2020 |
|
RU2744357C1 |
| Катализатор и способ получения высших 2-кетонов С5-С10 | 2022 |
|
RU2790246C1 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА ДЛЯ РЕАКЦИЙ, ИДУЩИХ ПО КИСЛОТНО-ОСНОВНОМУ МЕХАНИЗМУ | 2021 |
|
RU2773701C1 |

Изобретение относится к способу получения диалкил 2,3-ди-изобутил-сукцинатов формулы 1 из диэфира янтарной кислоты, включающему стадии двойной конденсации изобутилового альдегида с диэфиром янтарной кислоты в апротонном растворителе или в безводном протонном растворителе, предпочтительно ДМФА; в качестве основания используется 1-1.3 эквивалента гидрида металла формулы MeHn либо алкоксида металла (RO)nMe, где Me – это металл первой или второй группы Периодической системы, n – валентность металла, R – это остаток спиртовой группы С1-С4, реакция проводится при нагревании в диапазоне температур 40-100°С в течение 3-5 ч, с образованием смеси моно- и бис-этилиденовых соединений, с промежуточной этерификацией и последующей стадией гидрирования смеси диенов. Технический результат – разработан новый способ получения диалкил 2,3-ди-изобутил-сукцинатов с хорошим выходом и высокой чистотой. Полученные по этому способу соединения могут быть использованы в качестве стереорегулирующих компонентов в составе нанесенных титан-магниевых катализаторов для получения полипропилена. 4 з.п. ф-лы, 6 ил., 1 табл., 9 пр.

1. Способ получения диалкил 2,3-ди-изобутил-сукцинатов формулы 1 из диэфира янтарной кислоты,

включающий стадии двойной конденсации изобутилового альдегида с диэфиром янтарной кислоты в апротонном растворителе или в безводном протонном растворителе, предпочтительно ДМФА; в качестве основания используется 1-1.3 эквивалента гидрида металла формулы MeHn либо алкоксида металла (RO)nMe, где Me – это металл первой или второй группы Периодической системы, n – валентность металла, R – это остаток спиртовой группы С1-С4, реакция проводится при нагревании в диапазоне температур 40–100°С в течение 3-5 ч, с образованием смеси моно- и бис-этилиденовых соединений, с промежуточной этерификацией и последующей стадией гидрирования смеси диенов.
2. Способ по п. 1, отличающийся тем, что на первой стадии проводится взаимодействие диэфира янтарной кислоты с изо-бутиловым альдегидом, получающийся в ходе реакции моноэфир 2-изобутилиденсукцината взаимодействием с алкилирующим агентом превращают в диэфир 2-изобутилиденсукцината.
3. Способ по п. 1, отличающийся тем, что на второй стадии смесь 2-изобутилиденсукциновых диэфиров вводится в реакцию с изо-бутиловым альдегидом, получающийся в ходе реакции моноэфир 2,3-бис-изобутилиденсукцината взаимодействием с алкилирующим агентом превращают в диэфир бис-изобутилидензамещенных сукцината.
4. Способ по п. 1, отличающийся тем, что на третьей стадии смесь изомерных бис-изобутилидензамещенных сукцинатов без предварительного разделения гидрируется водородом при температурах 100-200°С и давлении 60-200 бар на катализаторах гидрирования: палладий, платина на угле, 5-10%, скелетный никель.
5. Способ по п. 1, отличающийся тем, что первая и вторая стадии конденсации изобутилового альдегида с диэфиром янтарной кислоты проводятся «в одном сосуде», без выделения промежуточных 2-изобутилиденсукциновых диэфиров.
| WO 2013029767 A1, 07.03.2013 | |||
| WO 2000063261 A1, 26.10.2000 | |||
| CN 103145553 B, 01.06.2016 | |||
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА, ПРИМЕНЯЕМОГО ДЛЯ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 1996 |
|
RU2152404C1 |

