Область техники
Изобретение относится к биомедицинским технологиям, а именно к анализу данных РНК-секвенирования. Изобретение может быть использовано при диагностике заболеваний, связанных с нарушением альтернативного сплайсинга.
Уровень техники
Созревание мРНК у эукариот включает в себя стадию сплайсинга - вырезания участков пре-мРНК называемых интронами и сшивание оставшихся участков называющихся экзонами. Интроны с обоих сторон ограничены сайтами сплайсинга, с 5’-конца интрона находится донорный сайт, с 3’-конца интрона - акцепторный. Все экзоны (кроме первых и последних) также ограничены с обоих сторон сайтами сплайсинга. В случае, если один и тот же фрагмент пре-мРНК в некоторых случаях вставляется в зрелую. мРНК или исключаются из нее говорят об альтернативный сплайсинге (АС). Альтернативный сплайсинг отдельного фрагмента РНК в данном биологическом образце характеризуется его частотой включения - отношением концентраций транскриптов данного гена содержащих данные фрагмент к суммарной концентрации всех транскриптов гена. Известно, что играет ключевую роль в развитии и функционировании нервной, мышечной, иммунной и других систем. Нарушения альтернативного сплайсинга связаны с такими заболеваниями как аутизм, болезнь Альцгеймера или миотоническая дистрофия. Таким образом, изучение альтернативного сплайсинга имеет и прикладное и фундаментальное значение. Подобные исследования обычно включают в себя определение частот включения участков мРНК в каждом отдельном биологическом образце и сравнение этих частот включения между двумя группами образцов, например между образцами полученными от больных и здоровых доноров или между образцами выделенными из различных органов. Развитие методов секвенирования нового поколения позволило анализировать АС в масштабе всего генома. Массовое секвенирование РНК (РНК-Сек) позволяет получить десятки миллионов коротких прочтений РНК. Выравнивание этих фрагментов на геном позволяет определить какие участки генома транскрибируются и после процессинга пре-мРНК входят в зрелые мРНК. Сравнение выравнивай прочтений РНК-Сек с геномной аннотацией (набором геномных координат генов, транскриптов и составляющих их экзонов) может позволить вычислить частоты включения экзонов всех экспрессирующихся генов. На данный момент существует несколько методов, позволяющих исследовать альтернативный сплайсинг (АС) при помощи данных РНК-Сек, однако каждый из них обладает некоторыми недостатками. Методы Cuffdiff2 [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3869392/], MISO [https://www.ncbi.nlm.nih.gov/pubmed/21057496], MATS [https://www.ncbi.nlm.nih.gov/pubmed/25480548] работают на уровне транскриптов или генов и не позволяют найти конкретный экзон, что необходимо для поиска причин паталогических изменений АС. Метод DEXseq [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3460195/] направлен на выявление дифференциального использования экзонов не только в следствии АС, но также в следствии использования альтернативных стартов или концов транскрипции. Поэтому использование DEXseq для поиска патологических изменений АС может приводить к ложно-положительным результатам.
Сущность изобретения
Задачей настоящего изобретения является создание способа обнаружения отличий в результатах альтернативного сплайсинга (АС) у различных групп субъектов при помощи анализа данных РНК-Сек.
Указанная задача решается путем создания способа подсчета частоты включения экзона в данных РНК-секвенирования, включающего следующие стадии:
а) получают данные РНК-секвенирования, состоящие из набора прочтений, из по меньшей мере одного образца, при этом образец получают из субъекта, принадлежащего к определенному биологическому виду;
б) получают информацию о референсной геномной ДНК организма, принадлежащего к указанному биологическому виду, которая содержит последовательности генов;
в) осуществляют разбиение указанных последовательностей генов на сегменты, при этом каждый сегмент содержит фрагмент гена между двумя ближайшими сайтами сплайсинга;
г) картируют полученные наборы прочтений на указанные последовательности генов, и для каждого сегмента определяют прочтения, содержащие последовательность, которая соответствует последовательности этого сегмента;
д) для каждого сегмента определяют количество «включающих» прочтений, подтверждающих наличие данного сегмента в данных РНК-секвенирования, также определяют количество «исключающих» прочтений, опровергающих наличие данного сегмента в данных РНК-секвенирования, и на основании этих двух чисел вычисляют частоту включения экзона, соответствующего данному сегменту.
В предпочтительном варианте, данный способ характеризуется тем, что частоту включения экзона вычисляют по формуле:
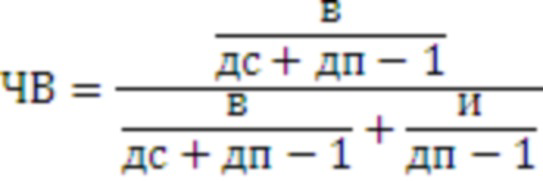 ,
,
где в и и обозначают количество включающих и исключающих прочтений, а дс и дп обозначают длину сегмента и прочтения в нуклеотидах, соответственно.
Указанная задача также решается путем создания способа определения статистически значимых отличий в частоте включения экзона в данных РНК-секвенирования, полученных из по меньшей мере двух различных образцов, включающего следующие стадии:
а) получают данные РНК-секвенирования, состоящие из набора прочтений, из по меньшей мере двух различных образцов, при этом образцы получают из субъектов, принадлежащих к одному биологическому виду;
б) получают информацию о референсной геномной ДНК организма, принадлежащего к указанному биологическому виду, которая содержит последовательности генов;
в) осуществляют разбиение указанных последовательностей генов на сегменты, при этом каждый сегмент содержит фрагмент гена между двумя ближайшими сайтами сплайсинга;
г) для каждого образца картируют полученные наборы прочтений на указанные последовательности генов, и для каждого сегмента определяют прочтения, содержащие последовательность, которая соответствует последовательности этого сегмента;
д) в каждом образце для каждого сегмента определяют количество «включающих» прочтений, подтверждающих наличие данного сегмента в данных РНК-секвенирования, также определяют количество «исключающих» прочтений, опровергающих наличие данного сегмента в данных РНК-секвенирования, и на основании этих двух чисел вычисляют частоту включения экзона, соответствующего данному сегменту;
е) определяют статистически значимые отличия в частоте включения экзона в по меньшей мере одном образце по сравнению с другими образцами.
В предпочтительном варианте, данный способ характеризуется тем, что частоту включения экзона в каждом образце вычисляют по формуле:
,
где в и и обозначают количество включающих и исключающих прочтений, а дс и дп обозначают длину сегмента и прочтения в нуклеотидах, соответственно.
Техническим результатом настоящего изобретения является увеличение скорости анализа данных РНК-секвенирования и уменьшение времени, которое затрачивает специалист для интерпретации получаемых результатов. Получаемые результаты могут быть использованы в медицинской диагностике при изучении нарушений АС и их ассоциаций с определенными заболеваниями или состояниями.
Краткое описание чертежей
Фиг. 1. Схема предложенного метода анализа АС. Аннотация генома, выравнивание прочтений и информация о разбиении образцов на сравниваемые группы является входными данными алгоритма. Алгоритм осуществляет разбиение генов на сегменты, подсчет прочтений и статистический анализ для поиска сегментов со статистически значимыми отличиями АС между группами.
Подробное раскрытие изобретения
В описании данного изобретения термины «включает» и «включающий» интерпретируются как означающие «включает, помимо всего прочего». Указанные термины не предназначены для того, чтобы их истолковывали как «состоит только из». Если не определено отдельно, технические и научные термины в данной заявке имеют стандартные значения, общепринятые в научной и технической литературе.
Прочтение (рид) - это короткая (от 30 до 500 нт) нуклеотидная последовательность, полученная в результате применения методов массового секвенирования к ДНК или РНК, выделенной из биологического образца. Наиболее часто применяемые технологии массового секвенирования позволяют получить несколько десятков миллионов прочтений на один биологический образец, обычно длина прочтения составляет около 100 нуклеотидов.
Картировать (или выравнивать) прочтение РНК-Сек на последовательности генов из геномной ДНК организма означает определить место в геномной ДНК, с которого был транскрибирован фрагмент, в результате секвенирования которого получилось данное прочтение.
Входными данными для анализа АС предлагаемым алгоритмом являются выравнивание данных РНК-сек на геном анализируемого организма и аннотация генома. Одновременно может анализироваться произвольное количество образцов РНК-сек полученных от одной или нескольких особей одного вида. Мощность метода зависит от количества прочтений в каждом образце, рекомендуется иметь хотя бы 30 млн прочтений на образец, однако метод может работать и с меньшим числом прочтений. Предлагаемый алгоритм для анализа АС состоит из трех стадий:
1. Разбиения всех генов, присутствующих в аннотации, на сегменты - фрагменты генов между двумя ближайшими сайтами сплайсинга.
В рамках этой процедуры рассматриваются сайты сплайсинга данного гена. Несколько сайтов с идентичными координатами и типами (донорный/акцепторный) схлопываются в один. Участок между двумя соседними сайтами сплайсинга является сегментом. Далее, все сегменты классифицируются на константные экзоны или интроны (сегменты, являющиеся экзонами или интронами соответственно во всех мРНК гена проходящих через данный участок) и альтернативные (сегменты, являющиеся экзонами в одних мРНК и интронами в других). Альтернативные сегменты разбиваются на четыре основных типа: кассетные экзоны (начинается с акцепторного сайта и кончается донорным), альтернативный донорный/акцепторный сегменты (начинается и кончается донорным/акцепторным сайтом) и удержанные интроны - начинаются с донорного сайта и кончаются акцепторным.
2. Подсчет числа прочтений
Для каждого сегмента в каждом образце подсчитывается два числа: количество прочтений, подтверждающих включение данного сегмента в мРНК («включающие» прочтения, то есть прочтения выравнивание которых пересекает сегмент хотя бы на один нуклеотид) и количество прочтений, подтверждающих исключение данного сегмента из мРНК («исключающие» прочтения, то есть прочтения выравнивающиеся на границу пары экзонов, один из которых находится до, а другой после данного сегмента). В ходе данной процедуры исключаются прочтения, которые выравниваются в несколько мест генома. Для устранения эффекта непроцессированных мРНК из подсчета исключающих прочтений и включающих прочтений для всех сегментов кроме удержанных интронов не учитываются прочтения пересекающиеся с интронами.
Для вычисления ЧВ (частота включения сегмента) число включающих и исключающих прочтений нормируется на количество различных позиций на которые могли бы потенциально выровняться прочтения:
где в и и обозначают количество включающих и исключающих прочтений, а дс и дп обозначают длину сегмента и прочтения соответственно. Предпочтительным является использование данных РНК-Сек с постоянной длинной прочтения, в ином случае используется средняя (на данный образец) длина прочтения.
3. Поиск сегментов со статистически значимыми отличиями ЧВ между тестовой и контрольной группами
Поиск сегментов с статистически значимыми отличиями в частотах включения осуществляется при помощи обобщенных линейных моделей с биномиальным распределением. Для учета биологической вариабельности используется тест на квази-отношение правдоподобий. Для коррекции на множественное тестирование применяется поправка Бенджамини-Хохберга. Сегменты с корректированным p-значением меньше 0.05 считаются статистически значимо отличными между сравниваемыми группами. Потенциально, благодаря использованию линейных моделей метод позволяет производить сравнение как двух выборок, так и анализировать более сложные экспериментальные дизайны с большим числом ковариатов.
Результатом работы алгоритма является список всех аннотированных сегментов, частоты их включения во всех сравниваемых образцах, разность средних частот включения между сравниваемыми группами образцов и p-значения для этого сравнения.
Примеры применения
Метод был применен для анализа изменений АС в ходе развития мозга человека и других приматов. Было показано что частоты включения сотен экзонов меняются в ходе постнатального развития мозга. При этом, некоторые изменения продолжаются и в старении, в частности, сплайсинг двух кассетных экзонов в транскриптах генов APP и MAPT, вовлеченных в болезнь Альцгеймера.
Несмотря на то, что изобретение описано со ссылкой на раскрываемые варианты воплощения, для специалистов в данной области должно быть очевидно, что конкретные подробно описанные случаи приведены лишь в целях иллюстрирования настоящего изобретения, и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения. Должно быть, понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КЛАССИФИКАЦИЯ САЙТОВ СПЛАЙСИНГА НА ОСНОВЕ ГЛУБОКОГО ОБУЧЕНИЯ | 2018 |
|
RU2780442C2 |
| МОДИФИЦИРОВАНИЕ ЭКСПРЕССИИ ГЕНА FSHβ С ПОМОЩЬЮ ГОМОЛОГИЧНОЙ РЕКОМБИНАЦИИ | 1999 |
|
RU2229309C2 |
| СПОСОБ ОЦЕНКИ СТАТУСА 14-ГО ЭКЗОНА ГЕНА MET ПО ДАННЫМ РНК СЕКВЕНИРОВАНИЯ | 2023 |
|
RU2817869C1 |
| ГЕНОМНАЯ ИНФРАСТРУКТУРА ДЛЯ ЛОКАЛЬНОЙ И ОБЛАЧНОЙ ОБРАБОТКИ И АНАЛИЗА ДНК И РНК | 2017 |
|
RU2804029C2 |
| ГЕНОМНАЯ ИНФРАСТРУКТУРА ДЛЯ ЛОКАЛЬНОЙ И ОБЛАЧНОЙ ОБРАБОТКИ И АНАЛИЗА ДНК И РНК | 2017 |
|
RU2761066C2 |
| МОДУЛЯТОРЫ И МОДУЛЯЦИЯ РНК РЕЦЕПТОРА КОНЕЧНЫХ ПРОДУКТОВ ГЛУБОКОГО ГЛИКИРОВАНИЯ | 2020 |
|
RU2820247C2 |
| Способ конструирования минигенов млекопитающих и рекомбинантная плазмида pgC1HDR, кодирующая миниген ингибитора С1 эстеразы человека, предназначенная для получения гуманизированных по гену Serping1 мышей | 2022 |
|
RU2805177C1 |
| ПРОПУСК ЭКЗОНОВ С ПОМОЩЬЮ ПРОИЗВОДНЫХ ПЕПТИДО-НУКЛЕИНОВЫХ КИСЛОТ | 2017 |
|
RU2786637C2 |
| СИНТЕТИЧЕСКИЕ 5 UTR (НЕТРАНСЛИРУЕМЫЕ ОБЛАСТИ), ЭКСПРЕССИОННЫЕ ВЕКТОРЫ И СПОСОБ ПОВЫШЕНИЯ ТРАНСГЕННОЙ ЭКСПРЕССИИ | 2008 |
|
RU2524431C2 |
| Система направленного изменения сплайсинга в гене MARK2 | 2023 |
|
RU2810907C1 |
Изобретение относится к биотехнологии. Описан способ обнаружения отличий в частоте включения экзона при помощи анализа данных РНК-секвенирования. Согласно способу: получают данные РНК-секвенирования, состоящие из набора прочтений, из по меньшей мере одного образца, при этом образец получают из субъекта, принадлежащего к определенному биологическому виду; б) получают информацию о референсной геномной ДНК организма, принадлежащего к указанному биологическому виду, которая содержит последовательности генов; в) осуществляют разбиение указанных последовательностей генов на сегменты, при этом каждый сегмент содержит фрагмент гена между двумя ближайшими сайтами сплайсинга; г) картируют полученные наборы прочтений на указанные последовательности генов, и для каждого сегмента определяют прочтения, содержащие последовательность, которая соответствует последовательности этого сегмента; д) для каждого сегмента определяют количество «включающих» прочтений, подтверждающих наличие данного сегмента в данных РНК-секвенирования, также определяют количество «исключающих» прочтений, опровергающих наличие данного сегмента в данных РНК-секвенирования, и на основании этих двух чисел вычисляют частоту включения экзона, соответствующего данному сегменту. Техническим результатом изобретения является увеличение скорости анализа данных РНК-секвенирования и уменьшение времени, которое затрачивает специалист для интерпретации получаемых результатов. Получаемые результаты могут быть использованы в медицинской диагностике при изучении нарушений альтернативного сплайсинга и их ассоциаций с определенными заболеваниями или состояниями. 2 н.п. ф-лы, 1 ил.
1. Способ подсчета частоты включения экзона в данных РНК-секвенирования, включающий следующие стадии:
а) получают данные РНК-секвенирования, состоящие из набора прочтений, из по меньшей мере одного образца, при этом образец получают из субъекта, принадлежащего к определенному биологическому виду;
б) получают информацию о референсной геномной ДНК организма, принадлежащего к указанному биологическому виду, которая содержит последовательности генов;
в) осуществляют разбиение указанных последовательностей генов на сегменты, при этом каждый сегмент содержит фрагмент гена между двумя ближайшими сайтами сплайсинга;
г) картируют полученные наборы прочтений на указанные последовательности генов, и для каждого сегмента определяют прочтения, содержащие последовательность, которая соответствует последовательности этого сегмента;
д) для каждого сегмента определяют количество «включающих» прочтений, подтверждающих наличие данного сегмента в данных РНК-секвенирования, также определяют количество «исключающих» прочтений, опровергающих наличие данного сегмента в данных РНК-секвенирования, и на основании этих двух чисел вычисляют частоту включения экзона, соответствующего данному сегменту, по формуле:
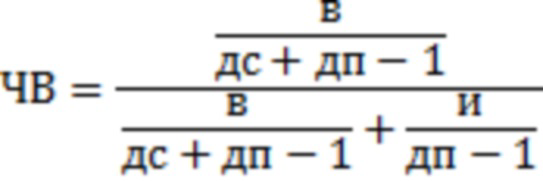 ,
,
где в и и обозначают количество включающих и исключающих прочтений, а дс и дп обозначают длину сегмента и прочтения в нуклеотидах, соответственно.
2. Способ определения статистически значимых отличий в частоте включения экзона в данных РНК-секвенирования, полученных из по меньшей мере двух различных образцов, включающий следующие стадии:
а) получают данные РНК-секвенирования, состоящие из набора прочтений, из по меньшей мере двух различных образцов, при этом образцы получают из субъектов, принадлежащих к одному биологическому виду;
б) получают информацию о референсной геномной ДНК организма, принадлежащего к указанному биологическому виду, которая содержит последовательности генов;
в) осуществляют разбиение указанных последовательностей генов на сегменты, при этом каждый сегмент содержит фрагмент гена между двумя ближайшими сайтами сплайсинга;
г) для каждого образца картируют полученные наборы прочтений на указанные последовательности генов, и для каждого сегмента определяют прочтения, содержащие последовательность, которая соответствует последовательности этого сегмента;
д) в каждом образце для каждого сегмента определяют количество «включающих» прочтений, подтверждающих наличие данного сегмента в данных РНК-секвенирования, также определяют количество «исключающих» прочтений, опровергающих наличие данного сегмента в данных РНК-секвенирования, и на основании этих двух чисел вычисляют частоту включения экзона, соответствующего данному сегменту, по формуле:,
где в и и обозначают количество включающих и исключающих прочтений, а дс и дп обозначают длину сегмента и прочтения в нуклеотидах, соответственно;
е) определяют статистически значимые отличия в частоте включения экзона в по меньшей мере одном образце по сравнению с другими образцами.
| Wang, Weichen, et al | |||
| "Identifying differentially spliced genes from two groups of RNA-seq samples." Gene, 2013, 518(1): 164-170 (см | |||
| Устройство для отыскания металлических предметов | 1920 |
|
SU165A1 |
| Liu, Ruolin, Ann E | |||
| Loraine, and Julie A | |||
| Dickerson | |||
| "Comparisons of computational methods for differential alternative splicing detection using RNA-seq in | |||

