[Область техники]
Настоящее изобретение относится к фармацевтической композиции для предупреждения или лечения острого миелоидного лейкоза или метастатического рака молочной железы, содержащей производное индирубина в качестве активного ингредиента.
[Уровень техники]
FMS-подобная тирозинкиназа-3 (FLT-3), рецепторная тирозинкиназа (RTK), принадлежащая к семейству RTK III типа, играет важную роль в выживании и пролиферации гематопоэтических клеток [1]. Активация FLT3 инициируется связыванием лиганда FLT3, который экспрессируется стромальными клетками, с рецептором. В результате димеризация и аутофосфорилирование рецептора FLT3 запускает последующие пути передачи сигналов, которые классифицируют на сигнальные пути PI3K/AKT, RAS/MAPK и STAT5 [2]. Рецептор FLT3 в высоких концентрациях экспрессируется в 70-100% клеток AML. Важность FLT3 при лейкозе тщательно изучена, и определенная группа мутаций FLT3 отмечена у примерно 1/3 всех пациентов с острым миелоидным лейкозом (AML) [3]. В настоящее время идентифицированы два основных типа мутаций FLT3: мутации внутренней тандемной дупликации (ITD) в околомембранной области и точечные мутации в киназном домене [4]. Указанные мутации имеют неблагоприятное прогностическое значение при безуспешности химиотерапии и рецидивах [5-8]. Кроме того, в недавних исследованиях показано, что мутации FLT3-ITD являются драйверной мутацией для прогрессирования AML и подходящей терапевтической мишенью при AML [9-10]. Таким образом, многими исследователями и фармацевтическими компаниями предприняты попытки поиска ингибиторов FLT3 как потенциальных терапевтических агентов для борьбы с AML.
Описаны несколько потенциальных клинических агентов, направленно воздействующих на FLT3, включая лестауртиниб [11], мидостаурин [12], тандутиниб [13], сорафениб [14], KW-2449 [15] и квизартиниб [16]. Среди них лестауртиниб и мидостаурин являются производными индолокарбазола и хорошо известными многонаправленными ингибиторами тирозинкиназы. Тандутиниб, пиперазинил-хиназолиновое соединение, ингибирует FLT3, а также c-Kit и PDGFR. Большинство указанных ингибиторов, которые изначально были предназначены для воздействия на другие киназы, были перенаправлены на лечение AML благодаря ингибированию мутации FLT3-ITD. Кроме того, обнаружено, что палбоциклиб, который использовали в качестве терапевтического агента в качестве ингибитора FLT3 для лечения рака молочной железы, эффективен при лечении пациентов с AML, и, следовательно, можно сделать вывод, что ингибиторы FLT3 можно использовать для лечения и рака молочной железы, и острого миелоидного лейкоза.
По-видимому, большинство современных ингибиторов FLT3 являются посредственными, главным образом вследствие их низкой эффективности и селективности в отношении мишени, за исключением квизартиниба [17-20]. Таким образом, в настоящее время остро необходима разработка эффективных ингибиторов киназы FLT3.
В настоящем описании цитированы многие научные статьи и патентные документы, и указаны ссылки на них. Содержание цитированных статей и патентных документов включено в настоящий документ посредством ссылки в полном объеме, и уровень развития той области техники, к которой относится настоящее изобретение, а также содержание настоящего изобретения описаны более наглядно.
[Документальные источники известного уровня техники]
[Непатентные документальные источники]
[1] S.D. Lyman, L. James, J. Zappone, P.R. Sleath, M.P. Beckmann, T. Bird, Characterization of the protein encoded by the flt3 (flk2) receptor-like tyrosine kinase gene, Oncogene. 8 (1993) 815-822. https://www.scopus.com/inward/record.uri?eid=2-s2.0-0027409749&partnerID=40&md5=ca39f6e31e1f6fea56547e6d4398e79d.
[2] D.L. Stirewalt, J.P. Radich, The role of FLT3 in haematopoietic malignancies, Nat. Rev. Cancer. 3 (2003) 650-665. https://www.scopus.com/inward/record.uri?eid=2-s2.0-0141465061&partnerID=40&md5=74c5831b6ffd9c07c920cdb2268e0abf.
[3] M. Nakao, S. Yokota, T. Iwai, H. Kaneko, S. Horiike, K. Kashima, Y. Sonoda, T. Fujimoto, S. Misawa, Internal tandem duplication of the flt3 gene found in acute myeloid leukemia, Leukemia. 10 (1996) 1911-1918. https://www.scopus.com/inward/record.uri?eid=2-s2.0-0030451722&partnerID=40&md5=55bd8ef1a79110698496bfafe5d74459.
[4] Y. Yamamoto, H. Kiyoi, Y. Nakano, R. Suzuki, Y. Kodera, S. Miyawaki, N. Asou, K. Kuriyama, F. Yagasaki, C. Shimazaki, H. Akiyama, K. Saito, M. Nishimura, T. Motoji, K. Shinagawa, A. Takeshita, H. Saito, R. Ueda, R. Ohno, T. Naoe, Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies, Blood. 97 (2001) 2434-2439. doi:10.1182/blood.V97.8.2434.
[5] C. Thiede, C. Steudel, B. Mohr, M. Schaich, U. Schäkel, U. Platzbecker, M. Wermke, M. Bornhäuser, M. Ritter, A. Neubauer, G. Ehninger, T. Illmer, Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis, Blood. 99 (2002).
[6] S. Schnittger, C. Schoch, M. Dugas, W. Kern, P. Staib, C. Wuchter, H. Löffler, C.M. Sauerland, H. Serve, T. Büchner, T. Haferlach, W. Hiddemann, Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease, Blood. 100 (2002).
[7] F.M. Abu-Duhier, A.C. Goodeve, G.A. Wilson, M.A. Gari, I.R. Peake, D.C. Rees, E.A. Vandenberghe, P.R. Winship, J.T. Reilly, FLT3 internal tandem duplication mutations in adult acute myeloid leukaemia define a high-risk group, Br. J. Haematol. 111 (2000) 190-195. doi:10.1046/j.1365-2141.2000.02317.x.
[8] L.-Y. Shih, C.-F. Huang, J.-H. Wu, T.-L. Lin, P. Dunn, P.-N. Wang, M.-C. Kuo, C.-L. Lai, H.-C. Hsu, Internal tandem duplication of FLT3 in relapsed acute myeloid leukemia: a comparative analysis of bone marrow samples from 108 adult patients at diagnosis and relapse, Blood. 100 (2002).
[9] C.C. Smith, Q. Wang, C.-S. Chin, S. Salerno, L.E. Damon, M.J. Levis, A.E. Perl, K.J. Travers, S. Wang, J.P. Hunt, P.P. Zarrinkar, E.E. Schadt, A. Kasarskis, J. Kuriyan, N.P. Shah, Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia, Nature. 485 (2012) 260-263. doi:10.1038/nature11016.
[10] J.S. Welch, T.J. Ley, D.C. Link, C.A. Miller, D.E. Larson, D.C. Koboldt, L.D. Wartman, T.L. Lamprecht, F. Liu, J. Xia, C. Kandoth, R.S. Fulton, M.D. McLellan, D.J. Dooling, J.W. Wallis, K. Chen, C.C. Harris, H.K. Schmidt, J.M. Kalicki-Veizer, C. Lu, Q. Zhang, L. Lin, M.D. O’Laughlin, J.F. McMichael, K.D. Delehaunty, L.A. Fulton, V.J. Magrini, S.D. McGrath, R.T. Demeter, T.L. Vickery, J. Hundal, L.L. Cook, G.W. Swift, J.P. Reed, P.A. Alldredge, T.N. Wylie, J.R. Walker, M.A. Watson, S.E. Heath, W.D. Shannon, N. Varghese, R. Nagarajan, J.E. Payton, J.D. Baty, S. Kulkarni, J.M. Klco, M.H. Tomasson, P. Westervelt, M.J. Walter, T.A. Graubert, J.F. DiPersio, L. Ding, E.R. Mardis, R.K. Wilson, The Origin and Evolution of Mutations in Acute Myeloid Leukemia, Cell. 150 (2012) 264-278. doi:10.1016/j.cell.2012.06.023.
[11] B.D. Smith, M. Levis, M. Beran, F. Giles, H. Kantarjian, K. Berg, K.M. Murphy, T. Dauses, J. Allebach, D. Small, Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia, Blood. 103 (2004).
[12] R.M. Stone, D.J. DeAngelo, V. Klimek, I. Galinsky, E. Estey, S.D. Nimer, W. Grandin, D. Lebwohl, Y. Wang, P. Cohen, E.A. Fox, D. Neuberg, J. Clark, D.G. Gilliland, J.D. Griffin, Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412, Blood. 105 (2004).
[13] D.J. DeAngelo, R.M. Stone, M.L. Heaney, S.D. Nimer, R.L. Paquette, R.B. Klisovic, M.A. Caligiuri, M.R. Cooper, J.-M. Lecerf, M.D. Karol, S. Sheng, N. Holford, P.T. Curtin, B.J. Druker, M.C. Heinrich, Phase 1 clinical results with tandutinib (MLN518), a novel FLT3 antagonist, in patients with acute myelogenous leukemia or high-risk myelodysplastic syndrome: safety, pharmacokinetics, and pharmacodynamics, Blood. 108 (2006).
[14] W. Zhang, M. Konopleva, Y. Shi, T. McQueen, D. Harris, X. Ling, Z. Estrov, A. Quintás-Cardama, D. Small, J. Cortes, M. Andreeff, Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia., J. Natl. Cancer Inst. 100 (2008) 184-98. doi:10.1093/jnci/djm328.
[15] Y. Shiotsu, H. Kiyoi, Y. Ishikawa, R. Tanizaki, M. Shimizu, H. Umehara, K. Ishii, Y. Mori, K. Ozeki, Y. Minami, A. Abe, H. Maeda, T. Akiyama, Y. Kanda, Y. Sato, S. Akinaga, T. Naoe, KW-2449, a novel multikinase inhibitor, suppresses the growth of leukemia cells with FLT3 mutations or T315I-mutated BCR/ABL translocation, Blood. 114 (2009).
[16] P.P. Zarrinkar, R.N. Gunawardane, M.D. Cramer, M.F. Gardner, D. Brigham, B. Belli, M.W. Karaman, K.W. Pratz, G. Pallares, Q. Chao, K.G. Sprankle, H.K. Patel, M. Levis, R.C. Armstrong, J. James, S.S. Bhagwat, AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML), Blood. 114 (2009).
[17] S. Knapper, A.K. Burnett, T. Littlewood, W.J. Kell, S. Agrawal, R. Chopra, R. Clark, M.J. Levis, D. Small, A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy, Blood. 108 (2006).
[18] D.J. De Angelo, R.M. Stone, M.L. Heaney, S.D. Nimer, R. Paquette, R. Bruner-Klisovic, M.A. Caligiuri, M.R. Cooper, J.-M. LeCerf, G. Iyer, M.C. Heinrich, B.J. Druker, Phase II Evaluation of the Tyrosine Kinase Inhibitor MLN518 in Patients with Acute Myeloid Leukemia (AML) Bearing a FLT3 Internal Tandem Duplication (ITD) Mutation., Blood. 104 (2015).
[19] F. Ravandi, J.E. Cortes, D. Jones, S. Faderl, G. Garcia-Manero, M.Y. Konopleva, S. O’Brien, Z. Estrov, G. Borthakur, D. Thomas, S.R. Pierce, M. Brandt, A. Byrd, B.N. Bekele, K. Pratz, R. Luthra, M. Levis, M. Andreeff, H.M. Kantarjian, Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia., J. Clin. Oncol. 28 (2010) 1856-62. doi:10.1200/JCO.2009.25.4888.
[20] K.W. Pratz, J. Cortes, G.J. Roboz, N. Rao, O. Arowojolu, A. Stine, Y. Shiotsu, A. Shudo, S. Akinaga, D. Small, J.E. Karp, M. Levis, A pharmacodynamic study of the FLT3 inhibitor KW-2449 yields insight into the basis for clinical response, Blood. 113 (2009).
[21] S.J. Choi, M.J. Moon, S.D. Lee, S.-U. Choi, S.-Y. Han, Y.-C. Kim, Indirubin derivatives as potent FLT3 inhibitors with anti-proliferative activity of acute myeloid leukemic cells, 2010. doi:10.1016/j.bmcl.2010.01.039.
[22] T.-C. Chou, Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies, Pharmacol. Rev. 58 (2006).
[23] K. Pratz, M. Levis, Incorporating FLT3 inhibitors into acute myeloid leukemia treatment regimens, Leuk. Lymphoma. 49 (2008) 852-863. doi:10.1080/10428190801895352.
[24] J.T. DiPiro, R.L. Talbert, G.C. Yee, B.G. Wells, L.M. Posey, Pharmacotherapy A Pathophysiologic Approach 9/E, McGraw-Hill Education2014.
[25] D. Small, FLT3 mutations: biology and treatment., Hematol. Am. Soc. Hematol. Educ. Progr. 2006 (2006) 178-84. doi:10.1182/asheducation-2006.1.178.
[26] S.K. Tasian, J.A. Pollard, R. Aplenc, Molecular Therapeutic Approaches for Pediatric Acute Myeloid Leukemia, Front. Oncol. 4 (2014) 55. doi:10.3389/fonc.2014.00055.
[27] K.W. Pratz, S.M. Luger, Will FLT3 inhibitors fulfill their promise in acute meyloid leukemia, Curr. Opin. Hematol. 21 (2014) 72-78. doi:10.1097/MOH.0000000000000022.
[Описание]
[Техническая задача]
Авторами настоящего изобретения предприняты активные попытки обнаружения новых соединений, обладающих ингибирующей способностью в отношении киназы FLT3. В результате установлено, что определенные соединения-производные индирубина могут эффективно ингибировать киназу FLT3, дополняя настоящее изобретение.
Соответственно, объект настоящего изобретения заключается в обеспечении нового соединения, обладающего ингибирующей способностью в отношении FLT3, его фармацевтически приемлемой соли, сольвата или гидрата.
Другой объект настоящего изобретения заключается в обеспечении фармацевтической композиции для предупреждения или лечения острого миелоидного лейкоза.
Другой объект настоящего изобретения заключается в обеспечении фармацевтической композиции для предупреждения или лечения метастатического рака молочной железы.
Другие объекты и преимущества настоящего изобретения станут понятны из подробного описания в сочетании с прилагаемой формулой изобретения и графическими материалами.
[Техническое решение]
В соответствии с одним аспектом настоящего изобретения, в данном изобретении предложено соединение, представленной химической формулой 1, изображенной ниже, его фармацевтически приемлемая соль, сольват или гидрат:
[Химическая формула 1]
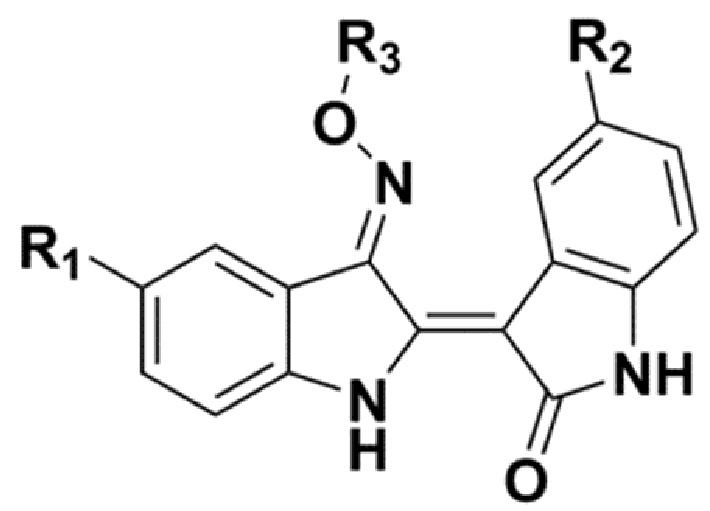
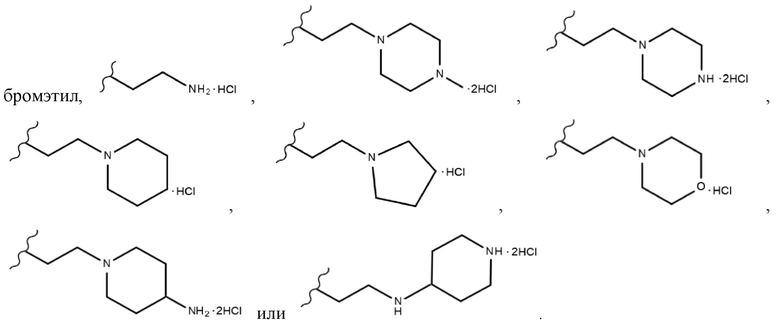
В химической формуле 1 R1 представляет собой водород, фтор или гидрокси, R2 представляет собой водород, галоген, нитро, карбоксил, C1-C4 алкиловый сложный эфир или C1-C4 алкокси, замещенный или незамещенный галогеном, и R3 представляет собой водород, 2-бромэтил, 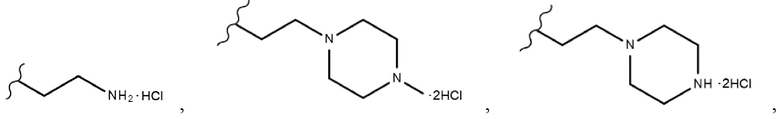
Авторами настоящего изобретения предприняты активные попытки разработки новых ингибиторов киназы FLT3 и в результате установлено, что определенные соединения-производные индирубина могут эффективно ингибировать киназу FLT3.
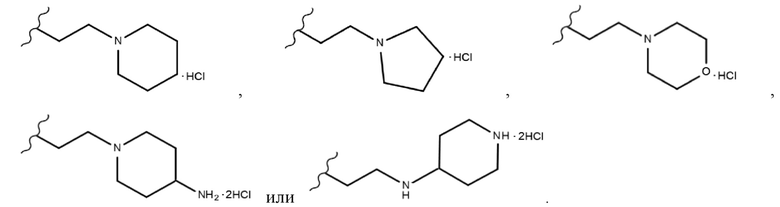
В одном конкретном варианте реализации настоящего изобретения R3 согласно настоящему изобретению представляет собой любой из заместителей, выбранных из группы, состоящей из 2-бромэтила, 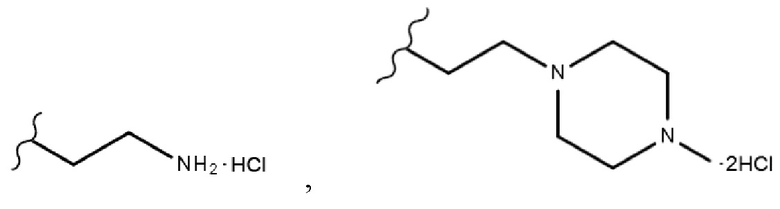 или
или 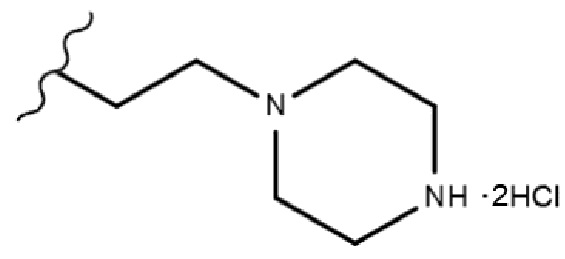 . Более предпочтительно, R3 согласно настоящему изобретению представляет собой любой из заместителей, выбранных из группы, состоящей из , еще более предпочтительно
. Более предпочтительно, R3 согласно настоящему изобретению представляет собой любой из заместителей, выбранных из группы, состоящей из , еще более предпочтительно 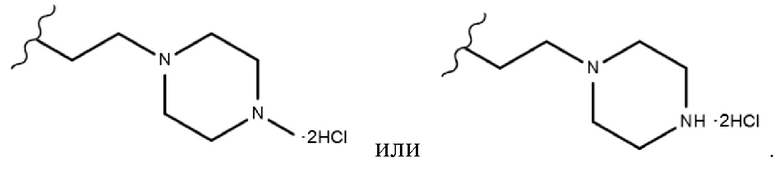 В одном конкретном варианте реализации настоящего изобретения R2 согласно настоящему изобретению представляет собой C1-C4 алкиловый сложный эфир.C1-C4 алкильный заместитель, входящий в состав C1-C4 алкилового сложного эфира, в одном конкретном варианте реализации настоящего изобретения включает неразветвленные или разветвленные C1-C4 алкильные группы и, в частности, он может представлять собой, например, метил, этил, н-пропил, изо-пропил, н-бутил, втор-бутил, изо-бутил или трет-бутил. Если R2 согласно настоящему изобретению представляет собой алкиловый сложноэфирный заместитель соединения, а не карбоксильный заместитель соединения, то он имеет меньшее значение IC50 для киназы FLT3 и, следовательно, может быть более эффективным для ингибирования киназы FLT3.
В одном конкретном варианте реализации настоящего изобретения R2 согласно настоящему изобретению представляет собой C1-C4 алкиловый сложный эфир.C1-C4 алкильный заместитель, входящий в состав C1-C4 алкилового сложного эфира, в одном конкретном варианте реализации настоящего изобретения включает неразветвленные или разветвленные C1-C4 алкильные группы и, в частности, он может представлять собой, например, метил, этил, н-пропил, изо-пропил, н-бутил, втор-бутил, изо-бутил или трет-бутил. Если R2 согласно настоящему изобретению представляет собой алкиловый сложноэфирный заместитель соединения, а не карбоксильный заместитель соединения, то он имеет меньшее значение IC50 для киназы FLT3 и, следовательно, может быть более эффективным для ингибирования киназы FLT3.
В одном конкретном варианте реализации настоящего изобретения R2 согласно настоящему изобретению представляет собой галоген. В частности, галоген может представлять собой фтор, хлор, бром или йод.
В одном конкретном варианте реализации настоящего изобретения R2 согласно настоящему изобретению представляет собой C1-C4 алкокси, замещенный или не замещенный галогеном. C1-C4 алкильный заместитель, входящий в состав C1-C4 алкокси, в одном конкретном варианте реализации настоящего изобретения включает неразветвленные или разветвленные C1-C4 алкильные группы, в частности, он может представлять собой, например, метил, этил, н-пропил, изо-пропил, н-бутил, втор-бутил, изо-бутил или трет-бутил. В частности, C1-C4 алкокси, замещенный галогеном, может представлять собой C1-C4 алкоксифторид и, более конкретно, трифторметокси.
В одном конкретном варианте реализации настоящего изобретения C1-C4 алкиловый сложный эфир согласно настоящему изобретению представляет собой метиловый сложный эфир.
В одном конкретном варианте реализации настоящего изобретения R2 согласно настоящему изобретению представляет собой C1-C4 алкиловый сложный эфир, и R3 представляет собой любой из заместителей, выбранных из группы, состоящей из 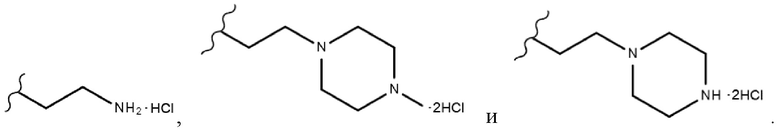
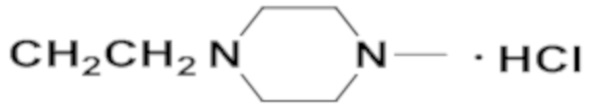
В одном конкретном варианте реализации настоящего изобретения соединение, представленное химической формулой согласно настоящему изобретению, может представлять собой любое соединение, выбранное из группы, состоящей из следующих соединений: метил-(2Z,3E)-2’-оксо-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-5’-карбоксилата дигидрохлорид (соединение 4), метил-(2Z,3E)-3-(гидроксиимино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилат (соединение 1), метил-(2Z,3E)-3-((2-бромэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилат (соединение 2), метил-(2Z,3E)-3-((2-аминоэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилата гидрохлорид (соединение 3), метил-(2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилата дигидрохлорид (соединение 5), метил-(2Z,3E)-3-((2-морфолиноэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилата гидрохлорид (соединение 6), (2Z,3E)-3-((2-бромэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновая кислота (соединение 8), (2Z,3E)-3-((2-аминоэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновой кислоты гидрохлорид (соединение 9), (2Z,3E)-2’-оксо-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-5’-карбоновой кислоты дигидрохлорид (соединение 10), (2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновой кислоты дигидрохлорид (соединение 11), (2Z,3E)-3-((2-морфолиноэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилата гидрохлорид (соединение 12), (2Z,3E)-5-гидрокси-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-5’-нитро-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 14), (2Z,3E)-3-((2-аминоэтокси)имино)-5’-нитро-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 15), (2Z,3E)-5’-нитро-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 16), (2Z,3E)-5-фтор-5’-нитро-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 17), (2Z,3E)-5-фтор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-5’-нитро-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 18), (2Z,3E)-5-фтор-5’-нитро-3-((2-(пиперидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 19), (2Z,3E)-5-фтор-5’-нитро-3-((2-(пирролидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 20), (2Z,3E)-5,5’-дифтор-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 21), (2Z,3E)-5,5’-дифтор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 22), (2Z,3E)-5,5’-дифтор-3-((2-(пиперидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 23), (2Z,3E)-5,5’-дифтор-3-((2-(пирролидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 24), (2Z,3E)-3-((2-(4-аминопиперидин-1-ил)этокси)имино)-5,5’-дифтор-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 25), (2Z,3E)-5,5’-дифтор-3-((2-(пиперидин-4-иламино)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 26), (2Z,3E)-5,5’-дифтор-3-((2-морфолиноэтокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 27), (2Z,3E)-5’-фтор-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 33), (2Z,3E)-5’-фтор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 34), (2Z,3E)-5’-фтор-3-((2-(пиперидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 35), (2Z,3E)-5’-фтор-3-((2-(пирролидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 36), (2Z,3E)-5’-фтор-3-((2-(пиперидин-4-иламино)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 37), (2Z,3E)-5’-хлор-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 38), (2Z,3E)-5’-хлор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 39), (2Z,3E)-5’-бром-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 40), (2Z,3E)-5’-бром-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 41), (2Z,3E)-5’-иод-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 42), (2Z,3E)-5’-иод-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 43), (2Z,3E)-3-((2-(пиперазин-1-ил)этокси)имино)-5’-(трифторметокси)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 44), (2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-5’-(трифторметокси)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 45), (2Z,3E)-5’-метокси-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 46) и (2Z,3E)-5’-метокси-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 47).
В одном конкретном варианте реализации настоящего изобретения соединение, представленное химической формулой 1 согласно настоящему изобретению, может иметь значение IC50 для ингибирования FLT3, составляющее 100 нМ или менее. Более конкретно, соединение, представленное химической формулой 1, может иметь значение IC50 для ингибирования FLT3, составляющее 50 нМ или менее, еще более конкретно 40 нМ или менее, еще более конкретно 35 нМ или менее, еще более конкретно 30 нМ или менее, еще более конкретно 25 нМ или менее, еще более конкретно 20 нМ или менее, еще более конкретно 15 нМ или менее, и еще более конкретно 10 нМ или менее.
В соответствии с другим аспектом настоящего изобретения, в настоящем изобретении предложена фармацевтическая композиция для предупреждения или лечения острого миелоидного лейкоза или метастатического рака молочной железы, содержащая: (a) соединение, представленное химической формулой 1, изображенной ниже, его фармацевтически приемлемую соль, сольват или гидрат; и (b) фармацевтически приемлемый носитель:
[Химическая формула 1]
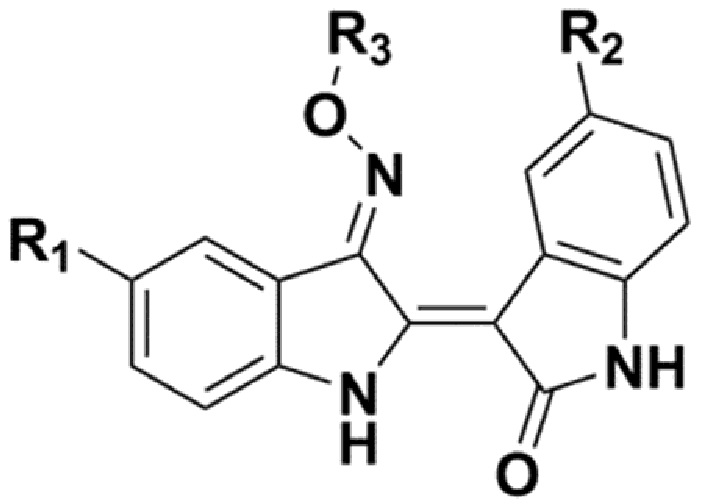
В химической формуле 1 R1 представляет собой водород, фтор или гидрокси, R2 представляет собой водород, галоген, нитро, карбоксил, C1-C4 алкиловый сложный эфир или C1-C4 алкокси, замещенный или незамещенный галогеном, и R3 представляет собой водород, 2-бромэтил, 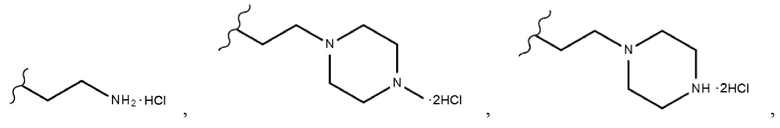
 . В соответствии с одним вариантом реализации настоящего изобретения, соединение, представленное химической формулой 1, может представлять собой любое соединение, выбранное из группы, состоящей из следующих соединений: метил-(2Z,3E)-2’-оксо-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-5’-карбоксилата дигидрохлорид (соединение 4), метил-(2Z,3E)-3-(гидроксиимино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилат (соединение 1), метил-(2Z,3E)-3-((2-бромэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилат (соединение 2), метил-(2Z,3E)-3-((2-аминоэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилата гидрохлорид (соединение 3), метил-(2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилата дигидрохлорид (соединение 5), метил-(2Z,3E)-3-((2-морфолиноэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилата гидрохлорид (соединение 6), (2Z,3E)-3-(гидроксиимино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновая кислота (соединение 7), (2Z,3E)-3-((2-бромэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновая кислота (соединение 8), (2Z,3E)-3-((2-аминоэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновой кислоты гидрохлорид (соединение 9), (2Z,3E)-2’-оксо-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-5’-карбоновой кислоты дигидрохлорид (соединение 10), (2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновой кислоты дигидрохлорид (соединение 11), (2Z,3E)-3-((2-морфолиноэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилата гидрохлорид (соединение 12), (2Z,3E)-5-гидрокси-3-(гидроксиимино)-5’-нитро-[2,3’-бииндолинилиден]-2’-он (соединение 13), (2Z,3E)-5-гидрокси-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-5’-нитро-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 14), (2Z,3E)-3-((2-аминоэтокси)имино)-5’-нитро-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 15), (2Z,3E)-5’-нитро-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 16), (2Z,3E)-5-фтор-5’-нитро-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 17), (2Z,3E)-5-фтор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-5’-нитро-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 18), (2Z,3E)-5-фтор-5’-нитро-3-((2-(пиперидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 19), (2Z,3E)-5-фтор-5’-нитро-3-((2-(пирролидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 20), (2Z,3E)-5,5’-дифтор-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 21), (2Z,3E)-5,5’-дифтор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 22), (2Z,3E)-5,5’-дифтор-3-((2-(пиперидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 23), (2Z,3E)-5,5’-дифтор-3-((2-(пирролидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 24), (2Z,3E)-3-((2-(4-аминопиперидин-1-ил)этокси)имино)-5,5’-дифтор-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 25), (2Z,3E)-5,5’-дифтор-3-((2-(пиперидин-4-иламино)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 26), (2Z,3E)-5,5’-дифтор-3-((2-морфолиноэтокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 27), (2Z,3E)-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 28), (2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 29), (2Z,3E)-3-((2-(пиперидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 30), (2Z,3E)-3-((2-(пирролидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 31), (2Z,3E)-3-((2-(пиперидин-4-иламино)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 32), (2Z,3E)-5’-фтор-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 33), (2Z,3E)-5’-фтор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 34), (2Z,3E)-5’-фтор-3-((2-(пиперидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 35), (2Z,3E)-5’-фтор-3-((2-(пирролидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 36), (2Z,3E)-5’-фтор-3-((2-(пиперидин-4-иламино)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 37), (2Z,3E)-5’-хлор-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 38), (2Z,3E)-5’-хлор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 39), (2Z,3E)-5’-бром-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 40), (2Z,3E)-5’-бром-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 41), (2Z,3E)-5’-иод-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 42), (2Z,3E)-5’-иод-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 43), (2Z,3E)-3-((2-(пиперазин-1-ил)этокси)имино)-5’-(трифторметокси)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 44), (2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-5’-(трифторметокси)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 45), (2Z,3E)-5’-метокси-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 46)и (2Z,3E)-5’-метокси-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 47).
. В соответствии с одним вариантом реализации настоящего изобретения, соединение, представленное химической формулой 1, может представлять собой любое соединение, выбранное из группы, состоящей из следующих соединений: метил-(2Z,3E)-2’-оксо-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-5’-карбоксилата дигидрохлорид (соединение 4), метил-(2Z,3E)-3-(гидроксиимино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилат (соединение 1), метил-(2Z,3E)-3-((2-бромэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилат (соединение 2), метил-(2Z,3E)-3-((2-аминоэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилата гидрохлорид (соединение 3), метил-(2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилата дигидрохлорид (соединение 5), метил-(2Z,3E)-3-((2-морфолиноэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилата гидрохлорид (соединение 6), (2Z,3E)-3-(гидроксиимино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновая кислота (соединение 7), (2Z,3E)-3-((2-бромэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновая кислота (соединение 8), (2Z,3E)-3-((2-аминоэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновой кислоты гидрохлорид (соединение 9), (2Z,3E)-2’-оксо-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-5’-карбоновой кислоты дигидрохлорид (соединение 10), (2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновой кислоты дигидрохлорид (соединение 11), (2Z,3E)-3-((2-морфолиноэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилата гидрохлорид (соединение 12), (2Z,3E)-5-гидрокси-3-(гидроксиимино)-5’-нитро-[2,3’-бииндолинилиден]-2’-он (соединение 13), (2Z,3E)-5-гидрокси-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-5’-нитро-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 14), (2Z,3E)-3-((2-аминоэтокси)имино)-5’-нитро-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 15), (2Z,3E)-5’-нитро-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 16), (2Z,3E)-5-фтор-5’-нитро-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 17), (2Z,3E)-5-фтор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-5’-нитро-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 18), (2Z,3E)-5-фтор-5’-нитро-3-((2-(пиперидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 19), (2Z,3E)-5-фтор-5’-нитро-3-((2-(пирролидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 20), (2Z,3E)-5,5’-дифтор-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 21), (2Z,3E)-5,5’-дифтор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 22), (2Z,3E)-5,5’-дифтор-3-((2-(пиперидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 23), (2Z,3E)-5,5’-дифтор-3-((2-(пирролидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 24), (2Z,3E)-3-((2-(4-аминопиперидин-1-ил)этокси)имино)-5,5’-дифтор-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 25), (2Z,3E)-5,5’-дифтор-3-((2-(пиперидин-4-иламино)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 26), (2Z,3E)-5,5’-дифтор-3-((2-морфолиноэтокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 27), (2Z,3E)-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 28), (2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 29), (2Z,3E)-3-((2-(пиперидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 30), (2Z,3E)-3-((2-(пирролидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 31), (2Z,3E)-3-((2-(пиперидин-4-иламино)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 32), (2Z,3E)-5’-фтор-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 33), (2Z,3E)-5’-фтор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 34), (2Z,3E)-5’-фтор-3-((2-(пиперидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 35), (2Z,3E)-5’-фтор-3-((2-(пирролидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 36), (2Z,3E)-5’-фтор-3-((2-(пиперидин-4-иламино)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 37), (2Z,3E)-5’-хлор-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 38), (2Z,3E)-5’-хлор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 39), (2Z,3E)-5’-бром-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 40), (2Z,3E)-5’-бром-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 41), (2Z,3E)-5’-иод-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 42), (2Z,3E)-5’-иод-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 43), (2Z,3E)-3-((2-(пиперазин-1-ил)этокси)имино)-5’-(трифторметокси)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 44), (2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-5’-(трифторметокси)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 45), (2Z,3E)-5’-метокси-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 46)и (2Z,3E)-5’-метокси-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 47).
Фармацевтическая композиция согласно настоящему изобретению содержит фармацевтически приемлемый носитель, помимо активных ингредиентов. Фармацевтически приемлемый носитель, включенный в фармацевтическую композицию согласно настоящему изобретению, обычно используют в лекарственных формах, и его примеры включают лактозу, декстрозу, сахарозу, сорбит, маннит, крахмал, гуммиарабик, фосфат кальция, альгинат, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, сироп, метилцеллюлозу, метилгидроксибензоат, пропилгидроксибензоат, тальк, стеарат магния и минеральное масло, и т.д., но не ограничиваются ими. Фармацевтическая композиция согласно настоящему изобретению может дополнительно содержать смазывающие вещества, увлажняющие агенты, подсластители, ароматизаторы, эмульгаторы, суспендирующие агенты и консерванты и т.д., помимо вышеуказанных компонентов. Подходящие фармацевтически приемлемые носители и лекарственные формы подробно описаны в публикации Remington, Pharmaceutical Sciences (19е изд., 1995).
Подходящая доза фармацевтической композиции согласно настоящему изобретению может быть прописана различным образом, в соответствии с такими факторами, как способ составления лекарственной формы, прием, используемый для введения, возраст, масса, пол и патологическое состояние пациента, рацион, время введения, способ введения, скорость выведения и аллергические реакции. В то же время доза фармацевтической композиции согласно настоящему изобретению может предпочтительно составлять от 1 до 1000 мг/кг (массы) в сутки.
Фармацевтическую композицию согласно настоящему изобретению можно вводить перорально или парентерально, причем в случае парентерального введения ее можно наносить местно на кожу или вводить внутривенной инъекцией, подкожной инъекцией, внутримышечной инъекцией, внутрибрюшинной инъекцией, трансдермальным введением и т.д. С учетом того что фармацевтическую композицию согласно настоящему изобретению используют для лечения острого миелоидного лейкоза, введение предпочтительно осуществляют посредством внутривенной инъекции или перорального введения.
Фармацевтическая композиция согласно настоящему изобретению может быть составлена с применением фармацевтически приемлемого носителя и/или вспомогательного вещества в соответствии со способом, который может быть без труда осуществлен специалистом в той области техники, к которой относится настоящее изобретение, и она может быть получена в единичной лекарственной форме или может быть получена в упакованном виде в многодозовом контейнере. В частности, лекарственная форма может быть представлена в форме раствора, суспензии или эмульсии в масляной или водорастворимой среде, или может быть в форме экстракта, порошка, гранулы, таблетки или капсулы, и может дополнительно содержать диспергатор или стабилизатор.
Фармацевтическую композицию согласно настоящему изобретению можно вводить перорально. Примеры твердого препарата для перорального введения включают таблетку, пилюлю, порошок, гранулу, капсулу, пастилку и т.д., и такие твердые препараты получают смешиванием соединения согласно настоящему изобретению с по меньшей мере одним вспомогательным веществом, таким как крахмал, карбонат кальция, сахароза или лактоза, или желатин и т.д. Помимо простых вспомогательных веществ, используют также смазывающие вещества, такие как стеарат магния и тальк. Примеры жидкого препарата для перорального введения могут включать суспензию, жидкость для употребления внутрь, эмульсию или сироп и т.д. Помимо традиционных простых разбавителей, таких как вода и жидкий парафин, в жидкий препарат могут быть включены различные вспомогательные вещества, такие как смачивающий агент, подсластитель, ароматизатор и консервант и т.д.
Примеры препарата для парентерального введения включают стерилизованный водный раствор, неводный растворитель, растворитель для получения суспензии, эмульсию, агент для сушки вымораживанием и суппозитории, и т.д. В качестве неводного растворителя и растворителя для получения суспензии можно использовать пропиленгликоль, полиэтиленгликоль, растительное масло, такое как оливковое масло, сложный эфир для инъекций, такой как этилолеат и т.д. В качестве основы для суппозитория можно использовать витепсол, макрогол, твин 61, масло какао, лауриновое масло, глицерин, желатин и т.д.
С учетом того, что фармацевтическая композиция согласно настоящему изобретению представляет собой композицию для предупреждения или лечения острого миелоидного лейкоза или метастатического рака молочной железы, предпочтительно можно вводить ее парентерально, например, посредством внутривенного введения, внутрибрюшинного введения, внутримышечного введения, подкожного введения, местного введения и т.д.
Производное индирубина согласно настоящему изобретению можно использовать в форме фармацевтически приемлемой соли, и в качестве такой соли можно использовать соль присоединения кислоты, образованную фармацевтически приемлемой свободной кислотой. Соль присоединения кислоты получают из: неорганических кислот, таких как хлористоводородная кислота, азотная кислота, фосфорная кислота, серная кислота, бромистоводородная кислота, иодистоводородная кислота, азотистая кислота или фосфористая кислота; нетоксичных органических кислот, таких как алифатический моно- и дикарбоксилат, фенил-замещенный алканоат, гидрокси-алканоат и алкандиоат, ароматические кислоты и алифатические и ароматические сульфоновые кислоты; или органических кислот, таких как уксусная кислота, бензойная кислота, лимонная кислота, молочная кислота, малеиновая кислота, глюконовая кислота, метансульфоновая кислота, 4-толуолсульфоновая кислота, винная кислота и фумаровая кислота. Примеры фармацевтически нетоксичных солей включают сульфат, пиросульфат, бисульфат, сульфит, бисульфит, нитрат, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, хлорид, бромид, иодид, фторид, ацетат, пропионат, деканоат, каприлат, акрилат, формиат, изобутилат, капрат, гептаноат, пропиолат, оксалат, малонат, сукцинат, суберат, себацинат, фумарат, малеат, бутин-1,4-диоат, гексан-1,6-диоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фталат, терефталат, бензолсульфонат, толуолсульфонат, хлорбензолсульфонат, ксилолсульфонат, фенилацетат, фенилпропионат, фенилбутират, цитрат, лактат, β-гидроксибутилат, гликолят, малат, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат или манделат, но не ограничиваясь ими.
Соль присоединения кислоты согласно настоящему изобретению может быть получена традиционным способом. Например, соль присоединения кислоты может быть получена посредством растворения производного индирубина химической формулы 1 в органическом растворителе, таком как метанол, этанол, ацетон, метиленхлорид, ацетонитрил и т.д., добавления к нему органической кислоты или неорганической кислоты с получением осадка, а затем фильтрования и высушивания полученного осадка, или может быть получена отгонкой растворителя и избытка кислоты при пониженном давлении, с последующей сушкой или кристаллизацией в органическом растворителе.
Кроме того, фармацевтически приемлемая соль металла может быть получена с использованием основания. Например, соль щелочного металла или щелочноземельного металла получают посредством растворения соединения в растворе избытка гидроксида щелочного металла или гидроксида щелочноземельного металла, фильтрования нерастворимой соли соединения, а затем выпаривания и сушки фильтрата. В частности, в качестве соли металла, с фармацевтической точки зрения получают соль натрия, калия или кальция. Кроме того, получают соответствующую соль серебра посредством взаимодействия соли щелочного металла или соли щелочноземельного металла с подходящей солью серебра (например, с нитратом серебра). Кроме того, настоящее изобретение включает не только производное индирубина, представленное химической формулой 1, и его фармацевтически приемлемую соль, но и сольват, гидрат, стереоизомер и т.д., которые могут быть получены из них.
В соответствии с другим аспектом настоящего изобретения, в настоящем изобретении предложен способ предупреждения или лечения острого миелоидного лейкоза или метастатического рака молочной железы, включающий: (a) введение индивидууму композиции, содержащей соединение, представленное химической формулой 1, изображенной ниже, его фармацевтически приемлемую соль, сольват или гидрат:
[Химическая формула 1]
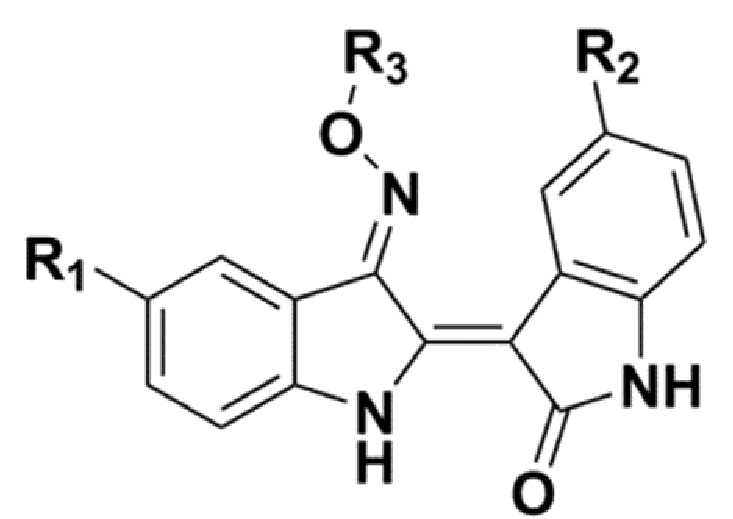
В химической формуле 1 R1 представляет собой водород, фтор или гидрокси, R2 представляет собой водород, галоген, нитро, карбоксил, C1-C4 алкиловый сложный эфир или C1-C4 алкокси, замещенный или незамещенный галогеном, и R3 представляет собой водород, 2-бромэтил,
Термины, использованные в данном контексте, являются такими, как описано выше. Индивидуум, которому вводят лекарственную форму согласно настоящему изобретению, может относиться к любым животным, включая людей. Животные могут включать людей, а также других млекопитающих, таких как коровы, лошади, овцы, свиньи, козы, верблюды, антилопы, собаки, кошки и т.д., нуждающиеся в лечении подобных симптомов, но не ограничиваясь ими.
В данном контексте термин «введение» относится к введению пациенту фармацевтической композиции согласно настоящему изобретению любым подходящим способом, и способ введения согласно настоящему изобретению может включать различные способы, такие как пероральное или парентеральное введение, при условии, что оно может обеспечивать достижение ткани-мишени. Лекарственная форма согласно настоящему изобретению может быть получена в различных формах, в зависимости от требуемого способа введения.
В соответствии с другим аспектом настоящего изобретения, в настоящем изобретении предложено применение соединения, представленного химической формулой 1, изображенной ниже, его фармацевтически приемлемой соли, сольвата или гидрата для получения фармацевтической композиции для предупреждения или лечения острого миелоидного лейкоза или метастатического рака молочной железы:
[Химическая формула 1]

В химической формуле 1 R1 представляет собой водород, фтор или гидрокси, R2 представляет собой водород, галоген, нитро, карбоксил, C1-C4 алкиловый сложный эфир или C1-C4 алкокси, замещенный или незамещенный галогеном, и R3 представляет собой водород, 2-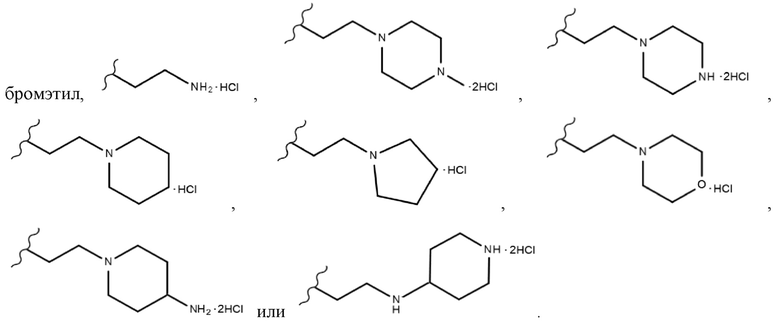
[Полезные эффекты изобретения]
Ниже обобщенно представлены признаки и преимущества настоящего изобретения:
(a) В настоящем изобретении предложена новая композиция для ингибирования FLT3.
(b) В настоящем изобретении предложена фармацевтическая композиция для предупреждения или лечения острого миелоидного лейкоза или метастатического рака молочной железы.
(c) При использовании композиции согласно настоящему изобретению, она может обеспечивать эффективное ингибирование активности киназы FLT3, и ее можно эффективно использовать для предупреждения или лечения острого миелоидного лейкоза или метастатического рака молочной железы.
[Краткое описание графических материалов]
На фиг. 1 показана структура и ингибирующий эффект LDD1937 (соединение 4) в отношении активности киназы FLT3. На изображении (A) показана химическая структура LDD1937, а на (B) показано действие LDD1937 на активность киназы FLT3 in vitro. Ингибирование киназной активности рекомбинантной FLT3 измеряли с помощью анализа методом гомогенной флуоресценции с временным разрешением (HTRF). Ингибирование киназы рассчитывали с использованием 1% ДМСО в качестве отрицательного контроля. Данные представлены как среднее значение ± СОС для трех независимых экспериментов.
На фиг. 2 показана in vivo противоопухолевая эффективность LDD1937 (соединение 4). Клетки MV-4-11 подкожно инокулировали мышам BALB/c nu/nu. По достижении среднего объема опухоли 100 мм3, мышам вводили инъекцию 5 мг/кг или 10 мг/кг LDD1937 или PBS (контроль) в хвостовую вену, ежедневно на протяжении 21 дня. На изображении (A) показаны результаты измерения объема опухоли и расчет объема опухоли. На изображении (B) показаны результаты измерения массы опухоли, проведенного после усыпления мышей на 21 день после введения лекарственного средства. Снимали на фотоаппарат приведенные в качестве примера изображения массы опухоли, вырезанной у животных контрольной группы и у животных с дозой 5 мг/кг, (вклейка). Данные представлены как среднее значение ±СОС. **P<0,01, ***P<0,001 по сравнению с контрольной группой, соответственно.
На фиг. 3 показано изменение массы тела в течение периода введения LDD1937. Измеряли массу мышей, которым вводили 5 мг/кг или 10 мг/кг LDD1937 или PBS (контроль) в течение 21 дня от начала эксперимента.
На фиг. 4 показано изменение массы печени, почек и раковой ткани после введения четырех соединений на протяжении 4 недель. LDD-2614 представляет собой соединение 21, LDD-2633 представляет собой соединение 28, LDD-2634 представляет собой соединение 34, и LDD-2635 представляет собой соединение 33.
На фиг. 5 показано изменение объема раковой ткани после введения четырех соединений на протяжении 4 недель. LDD-2614 представляет собой соединение 21, LDD-2633 представляет собой соединение 28, LDD-2634 представляет собой соединение 34, и LDD-2635 представляет собой соединение 33.
На фиг. 6 показаны изображения размера раковой ткани после введения четырех соединений на протяжении 4 недель. LDD-2614 представляет собой соединение 21, LDD-2633 представляет собой соединение 28, LDD-2634 представляет собой соединение 34, и LDD-2635 представляет собой соединение 33.
[Подробное описание вариантов реализации изобретения]
Далее настоящее изобретение более подробно описано с помощью примеров. Однако указанные примеры приведены лишь для специального пояснения настоящего изобретения, и специалистам в данной области техники понятно, что настоящее изобретение не ограничено приведенными примерами, в соответствии с сущностью настоящего изобретения.
Пример 1: Клеточная культура
Клетки MV4;11 острого миелоидного лейкоза человека приобретали в Американской коллекции типовых культур (ATCC, Роквилл, штат Мэриленд, США, CRL-9591) и выращивали клетки в среде IMDM (Sigma Co., Сент-Луис, штат Миссури, США) с добавлением 10% эмбриональной бычьей сыворотки, 1% пенициллина/стрептомицина и 4 мМ L-глутамина (Life Technology, Гранд Айленд, штат Нью-Йорк). Клетки MDA-MB-231 (метастатический рак молочной железы, ATCC HTB-26), Jurkat (острый T-лимфоцитарный лейкоз человека, ATCC TIB-152) и K-562 (хронический миелогенный лейкоз человека, ATCC CCL-243) выращивали в среде RPMI-1640 (Sigma Co.). Клетки MCF7 (аденокарцинома молочной железы человека, ATCC HTB-22) и PC-3 (аденокарцинома предстательной железы человека, ATCC CRL-1435) выращивали в среде DMEM (Sigma Co.) с добавлением 10% эмбриональной бычьей сыворотки и 1% пенициллина/стрептомицина. Выращенные клетки инкубировали при 3°С с 5% CO2.
Жизнеспособность клеток оценивали с помощью анализа с тетразолием, используя набор для анализа жизнеспособности клеток EZ-Cytox (DaeilLab, Корея). Вкратце, 2000-15000 клеток помещали на 96-луночные планшеты в 100 мкл среды. На следующий день клетки обрабатывали соединениями, а также диметилсульфоксидом (ДМСО) в качестве отрицательного контроля. Через три дня (72 часа) после добавления лекарственного соединения в каждую лунку 96-луночного планшета добавляли 15 мкл реагента из набора EZ-Cytox и затем инкубировали при 37°С в инкубаторе с увлажненным CO2 в течение 4 часов. После инкубации измеряли оптическую плотность (ОП) при длине волны 450 нм, используя многоканальный ридер Victor (Perkin Elmer, Уолтем, штат Массачусетс, США). Значения IC50 рассчитывали по нелинейной регрессии, используя программу Prism версии 5.01 (GraphPad, Ла-Холья, штат Калифония, США).
Пример 2: Синтез и хранение соединений
Разрабатывали и синтезировали соединения 1 - 47.
2-1. Схема синтеза 1
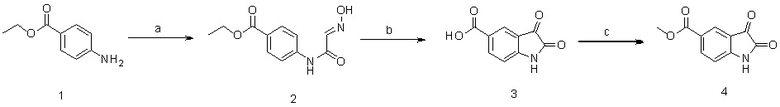
Реагенты и условия реакций: (a) хлоральгидрат, H2NOH·HCl, Na2SO4, вода, H2SO4, 100°С, в течение ночи; (b) конц. H2SO4, 90°С, 25 ми.; (c)H2SO4, 60°С, 1 ч.
Этил-4-аминобензоат нагревали в хлоральгидрате и водном растворе Na2SO4 в течение 1 часа. Затем полученный продукт оставляли взаимодействовать с гидрохлоридом гидроксиламина и затем охлаждали с получением твердого продукта, который отфильтровывали с использованием диэтилового эфира с получением продукта 2. Полученный таким образом продукт нагревали до 90°С в присутствии высокой концентрации серной кислоты с получением продукта, который нейтрализовали 1 н. водным раствором NaOH. Затем полученный продукт экстрагировали этилацетатом (EA) и концентрировали на ротационном испарителе. Из полученной таким образом смеси выделяли осадок, используя смесь диэтилового эфира и ДХМ, и затем фильтровали с получением продукта 3. Полученный продукт оставляли взаимодействовать с серной кислотой и метанолом с получением осадка (продуктов 4a и 4b).
2-2. Схема синтеза 2
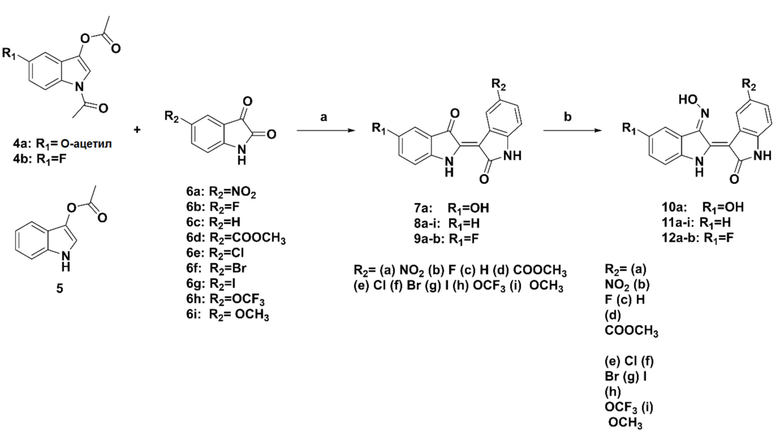
Реагенты и условия реакций: (a) Na2CO3, метанол:вода (2:1), комн. т-ра, 3 ч, (b) гидрохлорид гидроксиламина, пиридин, кипячение с обратным холодильником, 6 ч.
Аналоги изатина 6a-i, растворенные в MeOH, добавляли к индоксил-N,O-диацетату 4a-b или к индоксилацетату 5 и перемешивали смесь в течение 5 минут. Затем к смеси добавляли безводный Na2CO3 (2,5 экв.) и перемешивали при комнатной температуре в течение 3 часов. Затем полученный продукт отфильтровывали, добавляя воду, и несколько раз промывали холодной водой с получением продуктов 7a, 8a-i и 9a-b, в которых положение 5,5’ замещено с целью образования осадков. Затем полученные продукты растворяли в пиридине (0,1 М), добавляли гидрохлорид гидроксиламина (5 экв.) и кипятили с обратным холодильником в течение 6 часов. Затем смесь охлаждали и подкисляли 1 н. HCl с получением осадка, который отфильтровывали и несколько раз промывали водой. Наконец, полученные таким образом осадки очищали посредством осаждения с помощью ДХМ и гексана с получением конечных продуктов 10a, 11a-i и 12a-b.
2-3. Схема синтеза 3
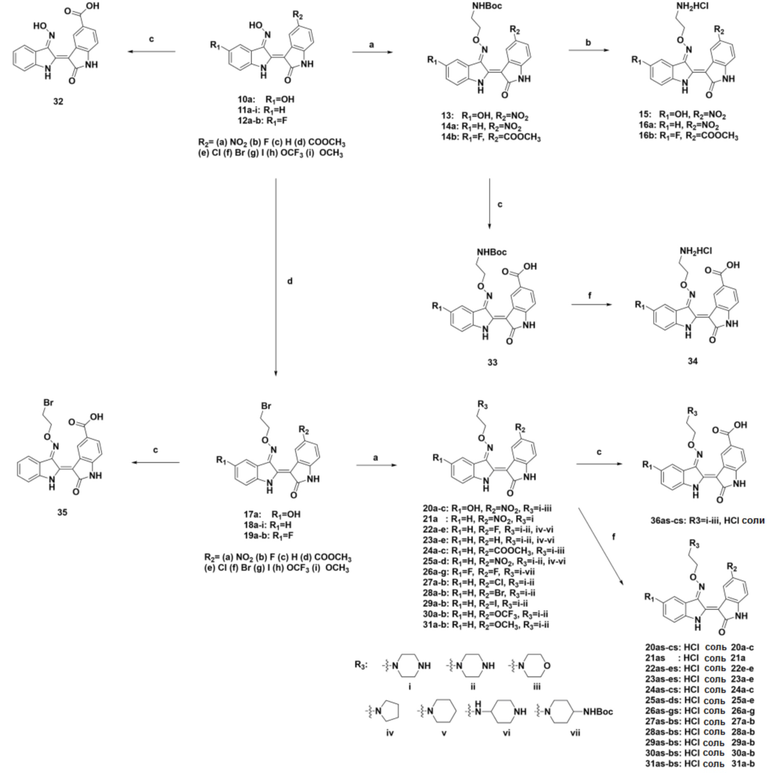
Реагенты и условия реакций: (a) 2-(Boc-амино)этилбромид, K2CO3, ДМФА, комн. т-ра, в течение ночи; (b) 4 г HCl в 1,4-диоксане, ДХМ, комн. т-ра, 3 ч; (c) 1 н. NaOH, 1,4-диоксан, 40°С, в течение ночи, (d) 1,2-дибромэтан, Et3N, ДМФА, комн. т-ра, в течение ночи; (e) амины i-vi, ДМФА, 50°С, в течение ночи; (f) 4 н. HCl в 1,4-диоксане, ДХМ или ТГФ, 0°С, 30 мин или ТФК, 0°С, 30 мин.
Ниже приведены иллюстративные способы синтеза производных индирубина, полученных по схеме синтеза 3:
3-1) Алкиламинирование производные индирубин-3’-оксима
Иллюстративный способ синтеза посредством алкиламинирования производных индирубин-3’-оксима, полученных по схеме синтеза 3, представлен ниже:
Производные индирубин-3’-оксима растворяли в ДМФА (от 1 мл до 4 мл), добавляли 2-(Boc-амино)этилбромид (1,2 экв.) и K2CO3 (от 2 экв. до 3 экв.) и затем перемешивали в течение ночи при комнатной температуре. После подтверждения того, что реакция завершена, ДМФА упаривали на ротационном испарителе и затем добавляли воду с получением осадков, которые выделяли фильтрованием. Осадки очищали колоночной хроматографией с получением производных индирубина, продуктов 13, 14a и 14b. Затем полученные таким образом производные индирубина растворяли в ДХМ (1 мл), добавляли 4 н. раствор HCl в диоксане (от 6 экв. до 18 экв.) при 0°С и перемешивали при комнатной температуре в течение 3 часов. После подтверждения того, что реакция завершена, полученные осадки несколько раз промывали ДХМ посредством фильтрования и сушили с получением чистого твердого вещества. Производные индирубина получали в форме солей (продукты 15, 16a и 16b).
3-2) Алкилирование производных индирубин-3’-оксима
Иллюстративный способ синтеза посредством алкилирования производных индирубин-3’-оксима, полученных по схеме синтеза 3, представлен ниже:
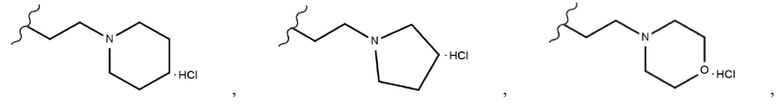
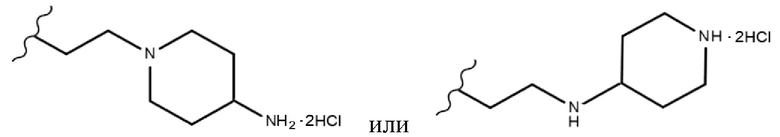
Производные индирубин-3’-оксима растворяли в ДМФА (15), добавляли 1,2-дибромэтан (10 экв.) и ТЭА (от 3 экв. до 10 экв.) и затем перемешивали в течение ночи при комнатной температуре. После подтверждения того, что реакция завершена, добавляли воду с получением осадков, которые несколько раз промывали водой посредством фильтрования и сушили с получением чистого твердого вещества (продукты 17a, 18a-i и 19a-b). Затем полученные таким образом производные индирубина растворяли в ДМФА, добавляли пиперазин, морфолин, пирролидин и пиперидин (от 10 экв. до 30 экв.) и затем перемешивали при 50°С в течение ночи. После подтверждения того, что реакция завершена, к перемешанному раствору добавляли воду с получением твердого вещества, которое выделяли фильтрованием и несколько раз промывали водой. Затем вымывали оставшиеся примеси ацетоном и MeOH с получением требуемых продуктов (продукты 20a-c, 21a, 22a-e, 23a-e, 24a-c, 25a-d, 26a-g, 27a-b, 28a-b, 29a-b, 30a-b и 31a-b). Затем полученные таким образом продукты растворяли в ДХМ или ТГФ, добавляли 4 н. раствор HCl в диоксане при 0°С и затем перемешивали 30 минут. После завершения реакции полученное твердое вещество промывали ДХМ посредством фильтрования с получением требуемых продуктов в форме солей (продукты 20as-cs, 21as, 22as-es, 23as-es, 24as-cs, 25as-ds, 26as-fs, 27as-bs, 28as-bs, 29as-bs, 30as-bs и 31as-bs). Кроме того, таким же получали соединение 26gs в форме соли, используя ТФК.
3-3) Карбоксильные производные индирубин-3’-оксима
Иллюстративный способ синтеза карбоксильных производных индирубин-3’-оксима, полученных по схеме синтеза 3, представлен ниже:
Производные индирубин-3’-оксима (продукты 11d, 14b, 18d и 24a-24c) растворяли в 1,4-диоксане, добавляли 1 н. NaOH и затем перемешивали в течение ночи при 40°С. После подтверждения того, что реакция завершена, полученный продукт охлаждали при комнатной температуре и подкисляли добавлением 1 н. HCl. Затем воду упаривали, и после концентрирования удаляли оставшийся NaCl фильтрованием, используя MeOH, с получением требуемых продуктов в виде твердого вещества (продукты 32, 33, 35 и 31as-cs (соль)). Затем полученный таким образом продукт 33 растворяли в ДХМ, добавляли 4 н. раствор HCl в диоксане при 0°С и затем перемешивали 10 минут. После завершения реакции полученное твердое вещество промывали ДХМ посредством фильтрования с получением требуемого продукта 34 в форме соли.
Подтверждение синтеза соединений
Продукт 11d: Метил-(2Z,3E)-3-(гидроксиимино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилат (соединение 1)
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 11,76 - 11,80 (м, 1 H), 11,05 - 11,10 (м, 1 H), 9,17 - 9,21 (м, 1 H), 8,24 (д, J=8,01 Гц, 1H), 7,73 - 7,78 (м, 1H), 7,36 - 7,42 (м, 2H), 7,00 - 7,06 (м, 1H), 6,92 - 6,98 (м, 1H), 3,79 - 3,86 (м, 3H).
Продукт 18d: Метил-(2Z,3E)-3-((2-бромэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилат (соединение 2)
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 11,68 (с, 1H), 11,18 (с, 1H), 9,36 (д, J=1,6 Гц, 1H), 8,23 (д, J=7,6 Гц, 1H), 7,83 (дд, J=8,0, 1,6 Гц, 1H), 7,48 (м, 2H), 7,10 (м, 1H), 7,01 (д, J=8,0 Гц, 1H), 4,98 (т, J=5,6 Гц, 2H), 4,06 (т, J=5,6 Гц, 2H), 3,86 (с, 3H).
Продукт 16b: Метил-(2Z,3E)-3-((2-аминоэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилата гидрохлорид (соединение 3)
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 11,66 (с, 1H), 11,18 (с, 1H), 9,30 (д, J=1,60 Гц, 1H), 8,24 (д, J=7,78 Гц, 1H), 8,17 (шс, 2H), 7,80 (дд, J=8,13, 1,72 Гц, 1H), 7,41 - 7,48 (м, 2H), 7,04 (ддд, J=7,90, 5,72,2,63 Гц, 1H), 6,98 (д, J=8,01 Гц, 1H), 4,78 - 4,85 (м, 2H), 3,79 - 3,86 (м, 3H), 3,47 (д, J=4,81 Гц, 2H).
Продукт 24as: Метил-(2Z,3E)-2’-оксо-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-5’-карбоксилата дигидрохлорид (соединение 4, LDD1937)
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 11,70 (с, 1H), 11,23 (с, 1H), 9,36 (с, 1H), 8,27 (д, J=7,6 Гц, 1H), 7,84 (д, J=8,4 Гц, 1H), 7,48 (м, 2H), 7,07 (м, 1H), 7,02 (д, J=8,8 Гц, 1H), 5,03 (м, 2H), 3,88 (с, 3H), 3,30 (м, 10H, перекрывается с ДМСО).
Продукт 24bs: Метил-(2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилата дигидрохлорид (соединение 5)
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 11,71 (с, 1H), 11,22 (с, 1H), 9,38 (д, J=1,6 Гц, 1H), 8,24 (д, J=7,6 Гц, 1H), 7,84 (дд, J=8,4, 1,6 Гц, 1H), 7,48 (м, 2H), 7,08 (м, 1H), 7,02 (д, J=8,4 Гц, 1H), 4,96 (м, 2H), 3,87 (с, 3H), 3,30 (м, 10H, перекрывается с водой).
Продукт 24cs: Метил-(2Z,3E)-3-((2-морфолиноэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоксилата гидрохлорид (соединение 6)
1H ЯМР (400 МГц, ДМСО-d6) δ 11,70 (с, 1H), 11,22 (с, 1H), 10,94 (шс, 1H, морфолин N+-H), 9,36 (с, 1H), 8,26 (д, J=7,6 Гц, 1H), 7,84 (дд, J=8,0, 2,0 Гц, 1H), 7,49 (м, 2H), 7,09 (м, 1H), 7,02 (д, J=8,0 Гц, 1H), 5,08 (шс, 2H), 3,97 (м, 2H), 3,80 (с, 3H), 3,81 (м, 4H), 3,56 (м, 2H), 3,18 (м, 2H, частично перекрывается с водой).
Продукт 32: (2Z,3E)-3-(гидроксиимино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновая кислота (соединение 7)
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 11,68 (с, 1H), 11,23 (с, 1H), 8,73 - 8,78 (м, 1H), 8,18 (с, 1H), 8,01 (д, J=5,04 Гц, 1H), 7,49 (м, 2H), 6,95 (д, J=8,0 Гц, 1H).
Продукт 35: (2Z,3E)-3-((2-бромэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновая кислота (соединение 8)
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 11,66 (с, 1H), 11,11 (с, 1H), 9,34 (с, 1H), 8,22 (д, J=7,6 Гц, 1H), 7,80 (д, J=8,0 Гц, 1H), 7,63 (м, 2H), 7,08 (м, 1H), 6,97 (д, J=8,4 Гц, 1H), 4,96 (т, J=5,6 Гц, 2H), 4,03 (т, J=5,6 Гц, 2H).
Продукт 34: (2Z,3E)-3-((2-аминоэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновой кислоты гидрохлорид (соединение 9)
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 11,68 (с, 1H), 11,18 (с, 1H), 9,35 (д, J=1,2 Гц, 1H), 8,26 (д, J=7,6 Гц, 1H), 8,17 (с, 3H, H+), 7,82 (дд,J=8,0, 1,6 Гц, 1H), 7,48 (м, 2H), 7,08 (м, 1H), 7,00 (д, J=8,4 Гц, 1H), 4,83 (т, J=5,2 Гц, 2H), 3,45 (т, J=5,2 Гц, 2H).
Продукт 36as: (2Z,3E)-2’-оксо-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-5’-карбоновой кислоты дигидрохлорид (соединение 10, LDD1940)
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 11,69 (с, 1H), 11,17 (с, 1H), 9,37 (с, 1H), 8,91(шс, 2H, пиперазин N+-H), 8,23 (д, J=7,6 Гц, 1H), 7,82 (д, J=8,4 Гц, 1H), 7,47 (м, 2H), 7,07 (м, 1H), 7,00 (д, J=8,4 Гц, 1H), 4,92 (шс, 2H), 3,05 (м, 10H, перекрывается с водой).
Продукт 36bs: (2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновой кислоты дигидрохлорид (соединение 11)
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 11,68 (с, 1H), 11,16 (с, 1H), 9,36 (с, 1H), 8,23 (д, J=8,0 Гц, 1H), 7,82 (д, J=8,0 Гц, 1H), 7,47 (м, 2H), 7,07 (м, 1H), 6,99 (д, J=8,0 Гц, 1H), 4,93 (шс, 2H), 3,17 (м, 10H, перекрывается с водой), 2,77 (с, 3H).
Продукт 36cs: (2Z,3E)-3-((2-морфолиноэтокси)имино)-2’-оксо-[2,3’-бииндолинилиден]-5’-карбоновой кислоты гидрохлорид (соединение 12)
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 11,67 (с, 1H), 11,27(шс, 1H, морфолин N+-H), 11,18 (с, 1H), 9,34 (с, 1H), 8,25 (д, J=7,6 Гц, 1H), 7,82 (д, J=8,4 Гц, 1H), 7,48 (м, 2H), 7,08 (м, 1H), 6,99 (д, J=8,0 Гц, 1H), 5,06 (шс, 2H), 3,96 (м, 2H), 3,80 (м, 4H), 3,53 (м, 2H), 3,24 (м, 2H, частично перекрывается с водой).
Продукт 10a: (2Z,3E)-5-гидрокси-3-(гидроксиимино)-5’-нитро-[2,3’-бииндолинилиден]-2’-он (соединение 13)
1H ЯМР (300 МГц, ДМСО-d6) δ 13,87 (1H, с, NOH), 11,78 (1H, с, NH), 11,35 (1H, с, N-H), 9,41 (1H, д, J=2,8 Гц), 9,32 (1H, с, O-H), 8,05 (1H, дд, J=11,6, 2,8 Гц), 7,76 (1H, д, J=3,2 Гц), 7,29 (1H, д, J=11,6 Гц), 7,04 (1H, д, J=11,2 Гц) 6,86 (1H, дд, J=11,2, 3,2 Гц)
Продукт 20bs: (2Z,3E)-5-гидрокси-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-5’-нитро-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 14)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 11,66 (с, 1H), 11,46 (с, 1H), 9,56 (д, J=2,4 Гц, 1H), 9,51 (шс, 1H), 8,10 (дд, J=8,8, 2,4 Гц, 1H), 7,75 (с, 1H), 7,32 (д, J=8,8 Гц, 1H), 7,08 (д, J=8,8 Гц, 1H) 6,93 (дд, J= 8,8, 2,4 Гц, 1H), 5,01 (шс, 2H), 3,27 (м, 10H, частично перекрывается с водой), 2,8 (с, 3H).
Продукт 16a: (2Z,3E)-3-((2-аминоэтокси)имино)-5’-нитро-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 15)
1H ЯМР (400 МГц, ДМСО-d6) δ 11,66 (с, 1 H) 11,18 (с, 1 H) 9,30 (д, J=1,60 Гц, 1 H) 8,24 (д, J=7,78 Гц, 1 H) 8,17 (шс, 2 H) 7,80 (дд, J=8,13, 1,72 Гц, 1 H) 7,41 - 7,48 (м, 2 H) 7,04 (ддд, J=7,90, 5,72, 2,63 Гц, 1 H) 6,98 (д, J=8,01 Гц, 1 H) 4,78 - 4,85 (м, 2 H) 3,79 - 3,86 (м, 3 H) 3,47 (д, J=4,81 Гц, 2 H)
Продукт 21as: (2Z,3E)-5’-нитро-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 16)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 11,70 (с, 1H), 11,25 (с, 1H), 9,45 (д, J=2,4 Гц, 1H), 8,23 (д, J=8,4 Гц, 2H), 8,03 (м, 1H), 7,50 (д, J=2,4 Гц, 1H), 7,43 (м, 1H), 7,04 (м, 2H), 4,82 (м, 2 H) 3,81 (м, 3 H) 3,42 (м, 2 H)
Продукт 15as: (2Z,3E)-5-фтор-5’-нитро-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 17)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 11,66 (с, 1H), 11,47 (с, 1H), 9,51 (д, J=2,4 Гц, 1H), 9,48 (с, 1H), 8,24 (шс, 3H), 8,09 (дд, J=8,4, 2,4 Гц, 1H), 7,78 (д, J=2,4 Гц, 1H), 7,30 (д, J=8,4 Гц, 1H), 7,07 (д, J=8,8 Гц, 1H), 6,95 (дд, J=8,4, 2,4 Гц, 1H), 4,87 (т, J=4,4 Гц, 2H), 3,49 (т, J=4,4 Гц, 2H).
Продукт 25bs: (2Z,3E)-5-фтор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-5’-нитро-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 18)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 9,58 (1H, д, J=2,1 Гц), 8,14 (1H, м), 7,97 (1H, дд, J=8,9, 2,4 Гц), 7,57 (1H, м), 7,41 (1H, м), 7,08 (1H, м), 4,87 (2H, м), 2,96 (2H, м), 2,38 (4H, м), 2,15 (3H, м).
Продукт 25ds: (2Z,3E)-5-фтор-5’-нитро-3-((2-(пиперидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 19)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 11,80 (м, 1H), 10,79 (с, 1H), 8,46 (дд, J=11,3, 2,4 Гц, 1H), 8,19 (м, 1H), 7,48 (м, 2H), 7,08 (м, 1H), 7,01 (м, 1H), 6,90 (м, 1H), 4,69 (т, J=6,1 Гц, 2H), 2,90 (м, 2 H), 2,24 (с, 2 H), 1,94 (м, 1 H), 1,58 (м, 3 H), 1,40 (м, 2 H)
Продукт 25cs: (2Z,3E)-5-фтор-5’-нитро-3-((2-(пирролидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 20)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 9,56 (с, 1H), 8,07 д, J= 8,8 Гц, 1H), 7,88 (д, J=8,8 Гц, 1H), 7,48 (с, 1H), 7,34 (с, 1H), 7,03 (д, J=9,6 Гц, 1H), 4,84 (м, 2H), 4,12 (м, 2H).
Продукт 26as: (2Z,3E)-5,5’-дифтор-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 21)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 11,80 (м, 1H), 10,83 (м, 1H), 8,44 (м, 1H), 8,01 (м, 1H), 7,49 (м, 1H), 7,38 (м, 1H), 7,01 (м, 1H), 6,87 (дд, J=4,9 Гц, 1H), 4,75 (м, 2H), 2,90 (м, 2H), 2,74 (м, 3H), 2,47 (м, 3H).
Продукт 26bs: 2Z,3E)-5,5’-дифтор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 22)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 11,77 (м, 1H), 10,81 (м, 1H), 8,44 (м, 1H), 7,99 (дд, J=8,9, 2,8 Гц, 1H), 7,47 (дд, J=8,9, 4,6 Гц, 1H), 7,34 (тд, J=9,0, 2,8 Гц, 1H), 6,98 (тд, J=8,9, 2,8 Гц, 1H), 6,89 (м, 1H), 4,74 (м, 2H), 2,92 (м, 2H), 2,38 (м, 3H), 2,15 (м, 3H).
Продукт 26es: (2Z,3E)-5,5’-дифтор-3-((2-(пиперидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 23)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 11,78 (м, 1H), 10,79 (шс, 1H), 8,41 (дд, J=11,3, 2,8 Гц, 1H), 7,97 (дд, J=8,7, 2,6 Гц, 1H), 7,47 (дд, J=8,7, 4,4 Гц, 1H), 7,38 (м, 1H), 7,01 (м, 1H), 6,89 (м, 1H), 4,69 (т, J=6,0 Гц, 2H), 2,85 (т, J=6,1 Гц, 2H), 1,51 (к, J=5,5 Гц, 4H), 1,42 (м, 2H).
Продукт 26ds: (2Z,3E)-5,5’-дифтор-3-((2-(пирролидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 24)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 10,86 (м, 1H), 8,41 (дд, J=11,3, 2,8 Гц, 1H), 7,92 (дд, J=8,6, 2,8 Гц, 1H), 7,47 (дд, J=8,9, 4,6 Гц, 1H), 7,34 (тд, J=9,1, 2,6 Гц, 1H), 6,97 (тд, J=8,8, 2,6 Гц, 1H), 6,89 (м, 1H), 4,70 (т, J=6,0 Гц, 1H), 3,00 (т, J=6,0 Гц, 2H), 2,60 (м, 4H), 1,69 (дт, J=6,6, 3,1 Гц, 4H).
Продукт 26gs: (2Z,3E)-3-((2-(4-аминопиперидин-1-ил)этокси)имино)-5,5’-дифтор-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 25)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 8,32 (дд, J=11,1, 2,6 Гц, 1H), 8,01 (дд, J=8,7, 2,6 Гц, 1H), 7,48 (д, J=8,9, 4,6 Гц, 1H), 7,40 (м, 1H), 7,04 (м, 1H), 6,88 (дд, J=8,6, 4,9 Гц, 1H), 5,01 (м, 2H), 3,37 (м, 4H), 2,96 (дд, J=11,6 Гц, 2H), 2,15 (м, 4H), 1,73 (м, 1H).
Продукт 26fs: (2Z,3E)-5,5’-дифтор-3-((2-(пиперидин-4-иламино)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 26)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 8,41 (дд, J=11,3, 2,8 Гц, 1H), 7,96 (дд, J=8,7, 2,6 Гц, 1H), 7,47 (дд, J=8,9, 4,3 Гц, 1H), 7,38 (1H, и), 6,97 (дд, J=9,2, 2,8 Гц, 1H), 6,86 (дд, J=8,6, 4,9 Гц, 1H), 4,69 (т, J=6,0 Гц, 2H), 2,93 (м, 4H), 2,15 (м, 2H), 1,72 (м, 2H), 1,33 (м, 2H).
Продукт 26cs: (2Z,3E)-5,5’-дифтор-3-((2-морфолиноэтокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 27)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 9,59 (д, J=2,4 Гц, 1H), 8,13 (м, 1H), 7,97 (д, J=6,1 Гц, 1H), 7,51 (с, 1H), 7,37 (с, 1H), 7,07 (д, J=8,6 Гц, 1H), 4,83 (т, J=5,8 Гц, 1H), 3,17 (с, 2H), 2,94 (м, 4H), 1,53 (м, 5H), 1,38 (дд, J=5,3, 0,8 Гц, 3H).
Продукт 23as: (2Z,3E)-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 28)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 11,84 (1H, м, N-H), 10,78 (1H, с, N-H), 8,62 (1H, д, J=7,9 Гц), 8,17 (1H, д, J=7,6 Гц), 7,46 (2H, м), 7,15 (1H, тд, J=7,6, 1,2 Гц), 7,03 (1H, м), 6,99 (1H, м), 6,90 (1H, д, J=7,6 Гц), 4,69 (2H, т, J=6,0 Гц), 3,46 (4H, м), 2,84 (2H, т, J=6,1 Гц), 2,69 (2H, т, J=4,7 Гц), 2,45 (2H, м).
Продукт 23bs: (2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 29)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 11,70 (1H, м, N-H), 10,78 (1H, с), 8,64 (1H, м), 8,17 (1H, д, J=7,9 Гц), 7,42 (1H, д, J=1,2 Гц), 7,16 (1H, тд, J=7,6, 1,2 Гц), 7,06 (1H, м), 7,00 (1H, м), 6,93 (1H, м), 4,69 (2H, т, J=6,1 Гц), 2,96 (2H, м), 2,54 (4H, м), 2,39 (4H,м), 2,14 (3H, с).
Продукт 23ds: (2Z,3E)-3-((2-(пиперидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 30)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 8,61 (1H, д, J=6,0 Гц), 8,16 (1H, д, J=6,0 Гц), 7,43 (1H, м), 7,15 (1H, м), 7,03 (1H, с), 6,97 (1H, м), 6,89 (1H, д, J=6,0 Гц), 4,69 (2H, м), 2,86 (2H, м), 2,08 (4H, с), 1,52 (3H, м), 1,43 (2H, м).
Продукт 23cs: (2Z,3E)-3-((2-(пирролидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 31)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 11,69 (1H, шс), 10,76 (1H, с), 8,66 (1H, м), 8,18 (1H, м), 7,46 (2H, м), 7,18 (1H, м), 7,06 (1H, м), 6,98 (1H, тд, J=7,7, 1,1 Гц), 6,91 (1H, дд, J=7,7, 0,6 Гц), 4,69 (2H, т, J=6,0 Гц), 2,99 (2H, т, J=6,0 Гц), 2,59 (4H, м), 1,69 (4H, дт, J=6,9, 3,2 Гц).
Продукт 23es: (2Z,3E)-3-((2-(пиперидин-4-иламино)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 32)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 8,62 (1H, д, J=7,6 Гц), 8,17 (1H, д, J=7,7 Гц), 7,46 (2H,м), 7,15 (1H, тд, J=7,56, 1,2 Гц), 7,03 (1H, тд, J=7,2, 1,6 Гц), 7,01 (1H, м), 6,91 (1H, д, J=7,7 Гц), 4,68 (2H, т, J=6,1 Гц), 2,92 (4H, м), 2,17 (2H, м).
Продукт 22as: (2Z,3E)-5’-фтор-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 33)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 11,83 (1H, м, N-H), 10,84 (1H, м, N-H), 8,45 (1H, дд, J=11,3, 2,8 Гц), 8,21 (1H, м), 7,47 (2H, м), 7,09 (1H, м), 6,99 (1H, м), 6,87 (1H, дд, J=8,4, 5,0 Гц), 4,76 (3H, м), 2,91 (2H, м), 2,70 (4H, м), 2,47 (3H, м).
Продукт 22bs: (2Z,3E)-5’-фтор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 34)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 11,84 (1H, м, N-H), 10,81 (1H, м, N-H), 8,47 (1H, дд, J=11,3, 2,7 Гц), 8,20 (1H, м), 7,46 (2H, м), 7,13 (1H, м), 6,99 (1H, м), 6,85 (1H, дд, J=8,4, 5,0 Гц), 4,74 (3H, м), 2,90 (2H, м), 2,72 (4H, м), 2,45 (3H, м), 2,14 (3H, м).
Продукт 22ds: (2Z,3E)-5’-фтор-3-((2-(пиперидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 35)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 11,80 (м, 1H), 10,79 (с, 1H), 8,46 (дд, J=11,3, 2,4 Гц, 1H), 8,19 (м, 1H), 7,48 (м, 2H), 7,08 (м, 1H), 7,01 (м, 1H), 6,90 (м, 1H), 4,69 (т, J=6,1 Гц, 2H), 2,90 (м, 2H), 2,24 (с, 2H), 1,94 (м, 1H), 1,58 (м, 3H), 1,40 (м, 2H).
Продукт 22cs: (2Z,3E)-5’-фтор-3-((2-(пирролидин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она гидрохлорид (соединение 36)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 10,78 (1H, шс), 8,46 (1H, дд, J=11,1, 2,6 Гц), 8,18 (1H, м), 7,47 (2H, м), 7,09 (1H, м), 6,96 (1H, дд, J=9,0, 2,6 Гц), 6,87 (1H, дд, J=8,4, 5,0 Гц), 4,69 (2H, т, J=6,1 Гц), 3,01 (2H, т, J=6,0 Гц), 2,61 (4H, м), 1,68 (4H, дт, J=7,0, 3,2 Гц).
Продукт 22es: (2Z,3E)-5’-фтор-3-((2-(пиперидин-4-иламино)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 37)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.); 8,48 (1H, м), 8,17 (1H, дд, J=7,6, 0,9 Гц), 7,46 (2H, м), 6,98 (1H, д, J=2,8 Гц), 6,89 (1H, м), 4,68 (2H, т, J=6,1 Гц), 2,93 (4H, м), 2,15 (2H, м).
Продукт 27a: (2Z,3E)-5’-хлор-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 38)
1H ЯМР (400 МГц, ДМСО-d6) δ 11,83 (с, 1H), 10,84 (с, 1H), 8,60 (с, 1H), 8,24 (д, J= 7,6 Гц, 1H), 7,45 (м, 2H), 7,14 (м, 2H), 6,88 (д, J= 7,9 Гц, 1H), 4,69 (т, J=6,0 Гц, 2H), 3,46 (м, 4H), 2,84 (т, J=6,1 Гц, 2H), 2,69 (т, J=4,7 Гц, 2H), 2,45 (м, 2H).
Продукт 27b: (2Z,3E)-5’-хлор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 39)
1H ЯМР (400 МГц, ДМСО-d6) δ 11,85 (с, 1H), 10,82 (с, 1H), 8,59 (с, 1H), 8,19 (д, J= 7,6 Гц, 1H), 7,65 (м, 2H), 7,14 (м, 2H), 6,90 (д, J= 7,9 Гц, 1H), 4,74 (м, 2H), 2,91 (м, 2H), 2,40 (м, 3H), 2,15 (м, 3H).
Продукт 28a: (2Z,3E)-5’-бром-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 40)
1H ЯМР (400 МГц, ДМСО-d6) δ 11,82 (с, 1H) , 10,76 (с, 1H), 8,65 (с, 1H), 8,19 (д, J= 8,0 Гц, 1H), 7,45 (м, 2H), 7,28 (д, J=8,2 Гц, 1H), 7,08 (м, 1H), 6,88 (д, J= 8,0 Гц, 1H) 4,72 (м, 3H), 2,87 (м, 2H), 2,70 (м, 4H), 2,47 (м, 3H).
Продукт 28b: (2Z,3E)-5’-бром-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 41)
1H ЯМР (400 МГц, ДМСО-d6) δ 11,82 (с, 1H), 10,77 (с, 1H), 8,65 (с, 1H), 8,19 (д, J= 8,0 Гц, 1H), 7,45 (м, 2H), 7,28 (д, J=8,2 Гц, 1H), 7,08 (м, 1H), 6,88 (д, J= 8,0 Гц, 1H), 4,74 (м, 3H), 2,93 (м, 2H), 2,75 (м, 4H), 2,45 (м, 3H), 2,17 (м, 3H).
Продукт 29a: (2Z,3E)-5’-иод-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 42)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.) 11,83 (с, 1H), 10,80 (с, 1H), 8,85 (с, 1H), 8,20 (д, J= 8,0 Гц, 1H), 7,45 (м, 3H), 7,06 (м, 1H), 6,75 (д, J= 8,0 Гц, 1H) 4,75 (м, 3H), 2,85 (м, 2H), 2,71 (м, 4H), 2,45 (м, 3H).
Продукт 29b: (2Z,3E)-5’-иод-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 43)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.) 11,82 (с, 1H) , 10,77 (с, 1H), 8,66 (с, 1H), 8,21 (д, J= 8,0 Гц, 1H), 7,46 (м, 3H), 7,08 (м, 1H), 6,74 (д, J= 8,0 Гц, 1H), 4,75 (м, 3H), 2,90 (м, 2H), 2,72 (м, 4H), 2,42 (м, 3H), 2,17 (м, 3H).
Продукт 30a: (2Z,3E)-3-((2-(пиперазин-1-ил)этокси)имино)-5’-(трифторметокси)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 44)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.) 11, 73 (с, 1H), 10,79 (с, 1H), 8,57 (с, 1H), 8,27 (д, J= 7,5 Гц, 1H), 7,41 (м, 2H), 6,99 (м, 2H), 6,94 (д, J= 8,4 Гц, 1H) 4,73 (м, 2H), 2,87 (м, 2H), 2,76 (м, 3H), 2,45 (м, 3H).
Продукт 30b: (2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-5’-(трифторметокси)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 45)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.) 11,68 (с, 1H) 10,88 (с, 1H), 8,60 (с, 1H), 8,24 (1H, д, J= 7,5 Гц), 7,40 (2H, м), 7,01 (2H, м), 6,94 (1H, д, J= 8,4 Гц) 4,82 (2H, м), 2,98 (2H, м), 2,41 (4H, м), 2,18 (3H, м).
Продукт 31a: (2Z,3E)-5’-метокси-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 46)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.) 11,74 (с, 1H), 10,49 (с, 1H), 8,35 (д, J= 2,2 Гц, 1H), 8,22 (д, J= 7,4 Гц, 1H), 7,40 (м, 2H), 7,02 (м, 1H), 6,75 (д, J= 8,3 Гц, 1H), 6,70 (дд, J= 7,6 Гц, 2,4 Гц, , 1H), 4,75 (м, 2H), 3,72 (с, 3H), 2,90 (м, 2H), 2,74 (м, 3H), 2,47 (м, 3H).
Продукт 31b: (2Z,3E)-5’-метокси-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-2’-она дигидрохлорид (соединение 47)
1H ЯМР (400 МГц, ДМСО-d6) δ (м.д.) 11,76 (с, 1H), 10,51 (с, 1H), 8,38 (д, J= 2,1 Гц, 1H), 8,23 (д, J= 7,4 Гц, 1H), 7,42 (м, 2H), 7,02 (м, 1H), 6,75 (д, J= 8,3 Гц, 1H), 6,72 (м, 1H), 4,75 (м, 2H), 3,72 (с, 3H), 2,94 (м, 2H), 2,36 (м, 3H), 2,17 (м, 3H).
Полученные таким образом соединения растворяли в ДМСО (Fisher, Уолтем, штат Массачусетс, США) в концентрации 10 ммоль/л и хранили при -20°С.
Пример 3: In vitro анализ киназы
Ингибирование активности киназы FLT3 измеряли с помощью анализа методом гомогенной флуоресценции с временным разрешением (HTRF). Рекомбинантные белки, содержащие домен киназы FLT3, приобретали у компании Carna biosciences (Япония). Оптимальный фермент, АТФ и концентрации субстрата определяли с помощью набора для HTRF KinEASE (Cisbio, Франция) по инструкциям производителя. Ферменты FLT3 последовательно смешивали с разбавленными соединениями и пептидными субстратами в буферном растворе киназы (50 мМ HEPES (рН 7,0), 500 мкМ АТФ, 0,1 мМ ортованадата натрия, 5 мМ MgCl2, 1 мМ DTT, 0,01% альбумина бычьей сыворотки (BSA) и 0,02% NaN3). После добавления реагентов для обнаружения проводили измерение сигнала TR-FRET с помощью многоканального ридера Victor (Perkin Elmer, Уолтем, штат Массачусетс, США). Значения IC50 рассчитывали по нелинейной регрессии с помощью программы Prism версии 5.01 (GraphPad). Анализы киназ JAK2, JAK3, cMET и RET in vitro также осуществляли методом HTRF.
In vitro анализ киназы IRAK4 осуществляли, используя анализ активности киназы LANCE Ultra (Perkin Elmer), который включает пептид ULight-p70S6K (Thr389) (мотив фосфорилирования FLGFTYVAP). Указанный анализ состоит из последовательного смешивания ферментов с разбавленными соединениями, 50 нМ пептидом ULight-p70S6K (Thr389) и 500 мкМ АТФ, которые предварительно разбавлены в буферном растворе киназы (50 мМ HEPES (pH 7,5), 10 мМ MgCl2, 1 мМ ЭГТК, 2 мМ DTT и 0,01% Tween 20). Реакционную смесь киназы инкубировали при 25 °C в течение 90 минут и останавливали добавлением 10 мМ ЭДТК. Для обнаружения фосфо-субстрата добавляли антитело Eu-анти-фосфо-p70S6K (Thr389), разбавленное буферным раствором для обнаружения, до конечной концентрации 2 нМ, и инкубировали продукты реакции при 25°С в течение 1 часа. Сигнал измеряли с помощью многоканального ридера EnVision.
Пример 4: Ксенотрансплантат опухоли у мышей (ксенотрансплантат MV4;11)
Клетки MV4;11 подкожно инкулировали в подвздошную область самок мышей BALB/c nu/nu (бестимусных «голых» мышей) (5×106 клеток/мышь). По достижении среднего объема опухоли 100 мм3 (примерно через 14 дней после инокуляции) мышей случайным образом разделяли на 3 группы (n=10 для контрольной группы, n=6 для групп с дозой 5 мг/кг или 10 мг/кг) и вводили в хвостовую вену инъекцию 5 мг/кг или 10 мг/кг LDD1937 при 20 мл/кг в PBS, или чистый PBS (контроль). Инъекции лекарственного соединения или контрольного PBS вводили ежедневно в течение 21 дня. Размер опухоли измеряли два раза в неделю в течение 21 дня, и рассчитывали объем опухоли по следующему уравнению: V (объем) = X (длина) × D (ширина)2/2. Через 21 день мышей усыпляли и измеряли массу опухоли.
Пример 5: Ксенотрансплантат опухоли у мышей (ксенотрансплантат MDA-MB-231)
6-недельных мышей линии Balb/c nu/nu (самок) приобретали у компании Central Lab. Animal Inc. Клеточную линию MDA-MB-231 приобретали у компании ATCC. Каждой мыши инокулировали 1×107 клеток для получения модели мышей с опухолью, и в качестве растворителя использовали 100 мкл смешанного раствора Corning Matrigel/PBS. Введение лекарства начинали по достижении объема опухоли от 200 мм3 до 300 мм3 (в принципе, объем опухоли измеряли ежедневно, и рассчитывали объем опухоли по следующему уравнению: V (объем) = X (длина) × D (ширина)2/2).
Вещества-кандидаты LDD-2614 (соединение 21), 2633 (соединение 28), 2634 (соединение 34), 2635 (соединение 33) вводили в дозе 20 мг/кг/перорально/сутки, а доксорубицин, который использовали в качестве контрольного препарата, вводили в дозе 5 мг/кг/внутрибрюшинно (один раз в неделю) в течение 4 недель.
Экспериментальные результаты
1. LDD1937 как ингибитор активности киназы FLT3
В таблице 1 представлена ингибирующая активность против FLT3 и MV4;11.
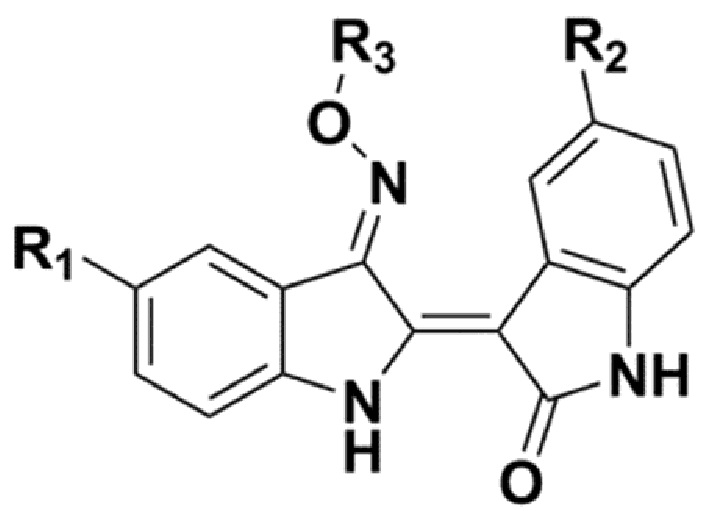
[Таблица 1]
В таблице 2 представлена ингибирующая активность соединений 13 - 37 против FLT3, MV4;11 и MDA-MB-231.
[Таблица 2]
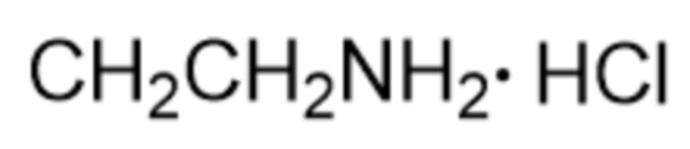
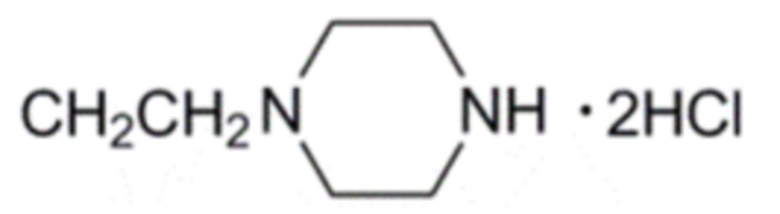
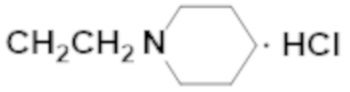
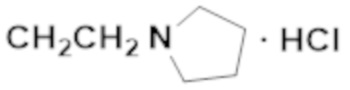
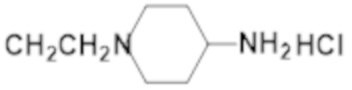
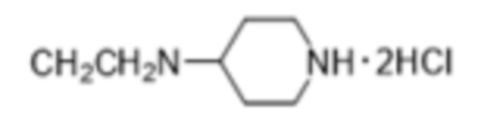
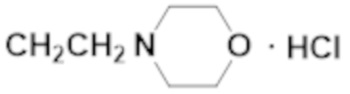
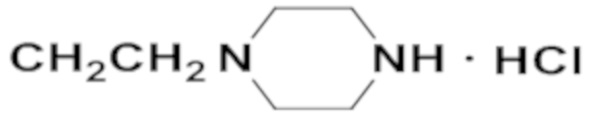
Активность каждого соединения измеряли 3 раза.
Авторами настоящего изобретения ранее описано, что некоторые 5-замещенные производные индирубина являются эффективными ингибиторами FLT3, которые эффективно ингибируют рост клеток острого миелоидного лейкоза [21]. Несмотря на то, что индирубин обладает мощным ингибирующим эффектом в отношении киназ, его слабая растворимость в воде вызывает некоторые физиологические трудности. Для решения проблем с растворимостью указанных производных индирубина, авторами настоящего изобретения разработаны и синтезированы новые аналоги, в молекулах которых содержатся гидрофильные функциональные группы.
Недавно авторами настоящего изобретения обнаружено, что производное индирубина с 5-карбоксильной группой 7, которое эффективно ингибирует киназу FLT3 (IC50=8 нМ), не обладает антипролиферативным действием против клеточной линии лейкоза человека MV4;11, которая экспрессирует FLT3-ITD (см. Таблицу 1). Интересно, что соответствующий аналог 1 с 5-карбоксильной группой, превращенной в сложный эфир, демонстрирует эффективное антипролиферативное действие против клеток MV4;11 (IC50 = 41 нМ), несмотря на очень слабую активность в отношении ингибирования FLT3. Таким образом, авторами настоящего изобретения предпринята попытка оптимизации для повышения активности в отношении ингибирования роста киназы FLT3 и MV4;11 посредством осуществления дополнительной дериватизации в положении 3’ индирубинового скелета.
Описано, что замещение производных индирубиноксима 3’-алкилом для других киназ обеспечивает такие преимущества, как более высокая эффективность и растворимость в воде. Несмотря на то, что в литературе отсутствует описание 3’-алкилзамещенных производных индирубиноксима, содержащих 5-карбоксильную или сложноэфирную группу, в качестве ингибиторов FLT3, авторы настоящего изобретения синтезировали такие аналоги для получения преимущества алкильного замещения в 3’-положении оксима. Ингибирующее действие 3’-замещенных аналогов индирубина описано в таблице 1. В целом, алкил-замещенные соединения демонстрируют усиленную ингибирующую активность в отношении FLT3 и MV4;11 по сравнению с соединениями 1 и 7. Аналоги соединения 1 с 5-сложноэфирным замещением демонстрируют эффективное ингибирующее действие против киназы FLT3, а также клеток MV4;11. Однако 5-карбоксильные производные (9-12) демонстрируют немного более слабую ингибирующую активность, чем 5-сложноэфирные аналоги (3-6), несмотря на сильную ингибирующая активность против киназы FLT3. Ингибирующий эффект против FLT3 и MV4;11 зависит от заместителя в положении R. Ингибирующая активность против FLT3 увеличивается в следующем порядке: морфолин, этилбромид < N-метилпиперазин < амин, пиперазин, а ингибирующая активность против MV4;11 увеличивается в следующем порядке: морфолин, этилбромид < амин, N-метилпиперазин < пиперазин. Этилбромид и морфолин в качестве заместителей в положении R (соединения 2, 6, 8 и 12) не оказывают решающего влияния на ингибирующую способность против FLT3 и MV4;11. Этилпиперазиновые соединения 4 и 10 демонстрируют наиболее эффективное ингибирующее действие против FLT3 (IC50=3 нМ) и MV4;11 (IC50=1 нМ и 40 нМ) в каждой группе. Кроме того, соединение 4 более эффективно ингибирует рост клеток MV4;11, чем CEP-701, который является известным ингибитором FLT3. Неожиданно, все алкил-замещенные серии соединения 1 с 5-метиловым сложным эфиром демонстрируют существенно усиленную ингибирующую активность против киназы FLT3. Этилпиперазин-замещенное соединение 4 демонстрирует наиболее эффективное действие против FLT3 (IC50=3 нМ) и MV4;11 (IC50=1 нМ), активность которого увеличена в 1000 раз и в 40 раз, соответственно, по сравнению с исходным соединением 1. Кроме того, внедрение алкильного заместителя в 3’-положении оксима индирубина с 5-карбоксильной группой 7 существенно увеличивает ингибирующий эффект против MV4;11. Подобно семейству с 5-метиловым сложным эфиром, пиперазин-замещенное соединение 10 демонстрирует наиболее эффективное ингибирующее действие против FLT3 и MV4;11. Однако соединение 10 демонстрирует умеренное ингибирующее действие лишь против клеток MV4;11, несмотря на его высокую ингибирующую активность против киназы FLT3. Авторы настоящего изобретения прогнозируют, что производные с 5-карбоксильной группой будут слишком полярными для прохождения через клеточную мембрану.
В процессе определения ингибиторов рецепторных тирозинкиназ были синтезированы несколько аналогов индирубина и изучена взаимосвязь их структуры с активностью (таблица 1). Из 12 соединений выбрали и дополнительно описали соединение 4 (LDD1937, фиг. 1A), метил-(2Z,3E)-2’-оксо-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3’-бииндолинилиден]-5’-карбоксилата дигидрохлорид. Как показано на фиг. 1B, значение IC50 соединения 4 (LDD1937) против активности киназы FLT3 составило 3 нМ. Измеряли также значения IC50 соединения 4 против активности других киназ (таблица 3). Наблюдали по меньшей мере 170-кратное различие значений IC50 между FLT3 и другими киназами.
[Таблица 3]
Данные означают среднее значение ± ст.откл.
2. Ингибирование пролиферации клеток MV4;11 и инициация селективной апоптотической гибели клеток под действием LDD1937
Клетки MV4;11 представляют собой лейкозные клетки с мутацией рецепторной тирозинкиназы FLT3. Клетки MV4;11 содержат мутацию FLT3 (FLT3-ITD) с внутренней тандемной дупликацией (ITD) в околомембранном домене, которая вызывает конститутивную активацию активности FLT3 [3]. Рост и выживание клеток MV4;11, как известно, зависят от активности FLT3 [23]. Измеряли цитотоксичность LDD1937, и результаты представлены в таблице 4. Для анализа цитотоксичности использовали также иммортализованные T-лимфоциты Jurkat, клетки рака предстательной железы PC-3, клетки рака молочной железы MCF-7 и клетки эритропоэза K562. Клетки MV4;11 демонстрировали наибольшую селективность к действию LDD1937 (GI50 = 1 нМ) по сравнению с другими клеточными линиями. С точки зрения значений GI50, соединение LDD1937 было в 1000 - 2000 раз более эффективным в клетках MV4;11, чем в других клеточных линиях.
[Таблица 4]
Данные означают среднее значение ± ст.откл.
3. In Vivo ингибирование роста опухоли под действием LDD1937
Для оценки эффективности LDD1937 in vivo проводили исследование ксенотрансплантата MV4;11. Клетки MV4;11 вводили подкожной инъекцией мышам BALB/c nu/nu для обеспечения роста опухоли до размера примерно 100 мм3. Кроме того, в течение 3 недель внутривенно вводили LDD1937 или контрольный PBS. Как показано на фиг. 2A, размер опухоли в группе с LDD1937 был существенно меньше, чем в контрольной группе. В частности, в группе с дозой 10 мг/кг опухоль исчезала с 3 дня, по результатам измеренного объема опухоли (фиг. 2A). Иссечение очага инъекции опухоли подтвердило полное исчезновение опухоли в группе с дозой 10 мг/кг. Таким образом, массу опухоли можно было измерить лишь в контрольной группе и в группе с дозой 5 мг/кг, и наблюдали существенное уменьшение массы опухоли в группе с дозой 5 мг/кг LDD1937 (фиг. 2B). Кроме того, не наблюдали существенного различия массы тела между указанными группами в течение всего периода введения (фиг. 3).
4. In Vivo противораковое действие 4 соединений
Для определения in vivo эффективности четырех производных индирубина проводили исследование ксенотрансплантата MDA-MB-231. Выращенные в культуре клетки инокулировали мышам BALB/c nu/nu (самки) до достижения размера опухоли примерно 200 мм3 – 300 мм3. Кроме того, вводили четыре производных индирубина, LDD-2614 (соединение 21), 2633 (соединение 28), 2634 (соединение 34) и 2635 (соединение 33), каждое в дозе 20 мг/кг. В качестве сравнительной группы вводили доксорубицин в дозе 5 мг/кг в течение 4 недель. Как показано на фиг. 4, не наблюдали существенного различия массы печени и почек после введения указанных четырех соединений, но наблюдали некоторые различия массы раковой ткани. В течение того же периода было подтверждено, что объем раковой ткани уменьшился до такого же уровня, как в контрольной группе (фиг. 5). Иссечение очага инъекции опухоли подтвердило, что три соединения 2633 (соединение 28), 2634 (соединение 34) и 2635 (соединение 33) демонстрируют такое же противораковое действие, как в сравнительной группе (фиг. 6).
FMS-подобная тирозинкиназа-3 (FLT-3) принадлежит к семейству рецепторных тирозинкиназ (RTK), и мутации FLT3 наблюдают у 1/3 пациентов с острым миелоидным лейкозом (AML). Как описано выше, авторами настоящего изобретения идентифицирован эффективный ингибитор FLT3 LDD1937, который содержит скелет индирубина. Высокая ингибирующая активность LDD1937 против FLT3 продемонстрирована в in vitro анализе киназы (IC50=3 нМ). Соединение LDD1937 селективно ингибирует рост клеток MV4;11 (GI50=1 нМ) и вызывает апоптотическую гибель клеток. LDD1937 вызывает остановку клеточного цикла на фазе G2/M и увеличивает популяцию клеток на суб-G1 фазе. Фосфорилирование STAT5, следующего элемента передачи сигналов FLT3, существенно уменьшается под действием LDD1937 дозозависимым образом. Фармакокинетические свойства LDD1937 изучали на мышах. Кроме того, изучен in vivo противоопухолевый эффект с использованием ксенотрансплантатов MV4;11. Объем и масса опухоли у мышей nu/nu были существенно снижены по сравнению с контрольной группой, при внутривенном введении 5 мг/кг и 10 мг/кг.
Несмотря на то, что представлено подробное описание определенных частей настоящего изобретения, специалистам в данной области техники понятно, что указанные конкретные технологии являются лишь предпочтительным вариантом реализации, и что объем настоящего изобретения не ограничен ими. Следовательно, существенный объем настоящего изобретения следует определять по сопроводительной формуле изобретения и ее эквивалентам.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 2-ИМИНОПИРРОЛИДИНА | 2002 |
|
RU2270192C2 |
| ИНГИБИТОРЫ СИГМА-РЕЦЕПТОРА | 2005 |
|
RU2417987C2 |
| НОВОЕ ТРИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2009 |
|
RU2470934C1 |
| ПРОИЗВОДНОЕ ПИРРОЛО-ПИРИДИНОВОГО СОЕДИНЕНИЯ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА, ДЛЯ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С ПРОТЕИНКИНАЗОЙ | 2018 |
|
RU2730849C1 |
| 2-АМИНОБЕНЗОКСАЗОЛКАРБОКСАМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-НТ3 | 2007 |
|
RU2448105C2 |
| ПРОИЗВОДНЫЕ 2-ЗАМЕЩЕННЫХ ФЕНИЛ-5, 7-ДИГИДРОКАРБИЛ-3, 7-ДИГИДРОПИРРОЛО [2, 3-d] ПИРИМИДИН-4-ОНОВ, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2004 |
|
RU2323220C2 |
| ДИАМИНОГЕТЕРОЦИКЛИЧЕСКОЕ КАРБОКСАМИДНОЕ СОЕДИНЕНИЕ | 2010 |
|
RU2526253C2 |
| ПРОИЗВОДНЫЕ ГИДРОКСИБЕНЗАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ Hsp90 | 2006 |
|
RU2458919C2 |
| ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2013 |
|
RU2623221C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2011 |
|
RU2652638C2 |
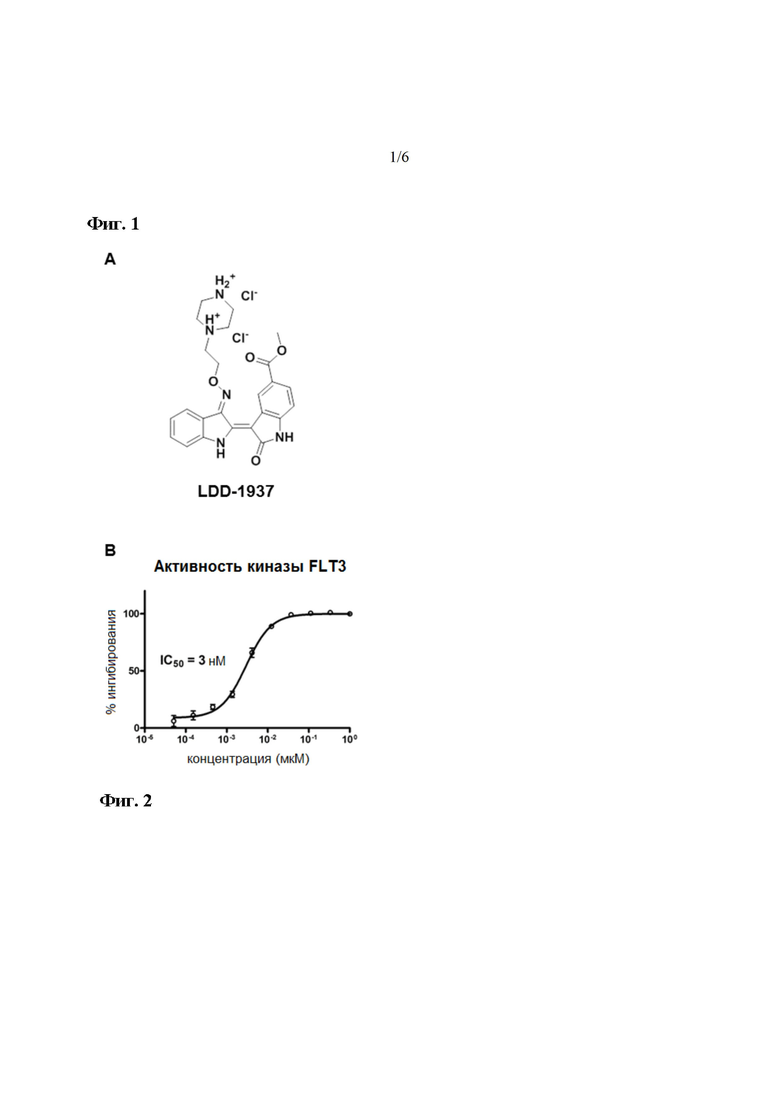
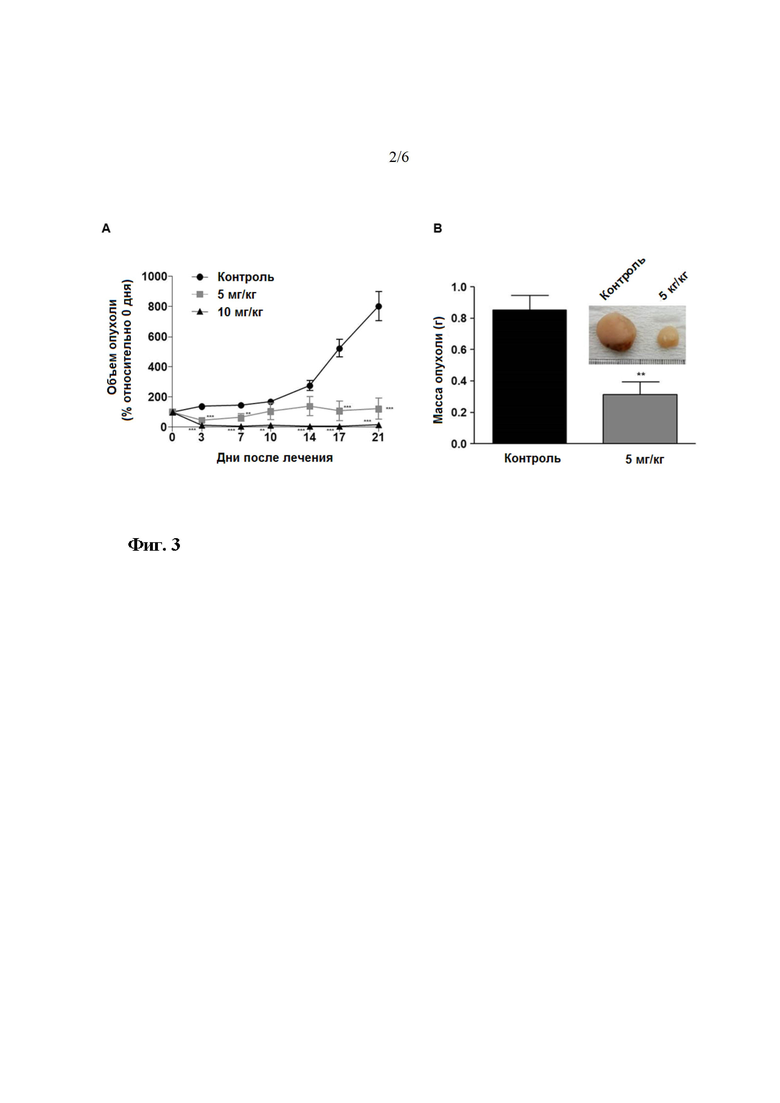
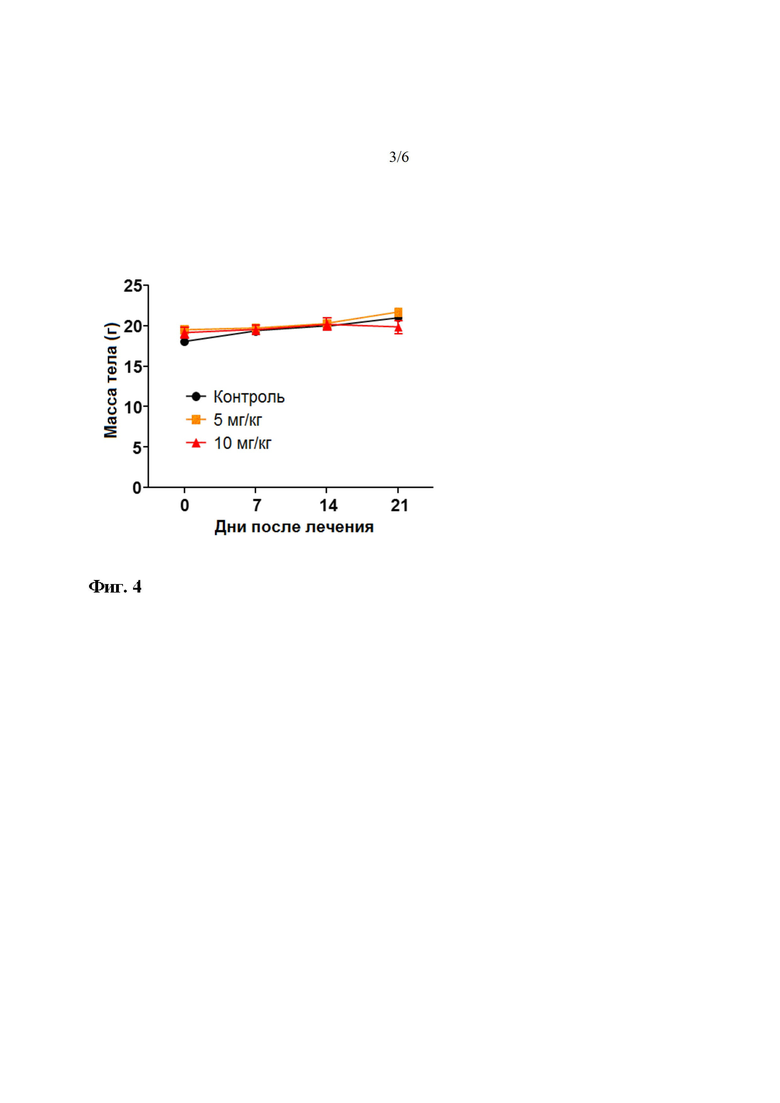
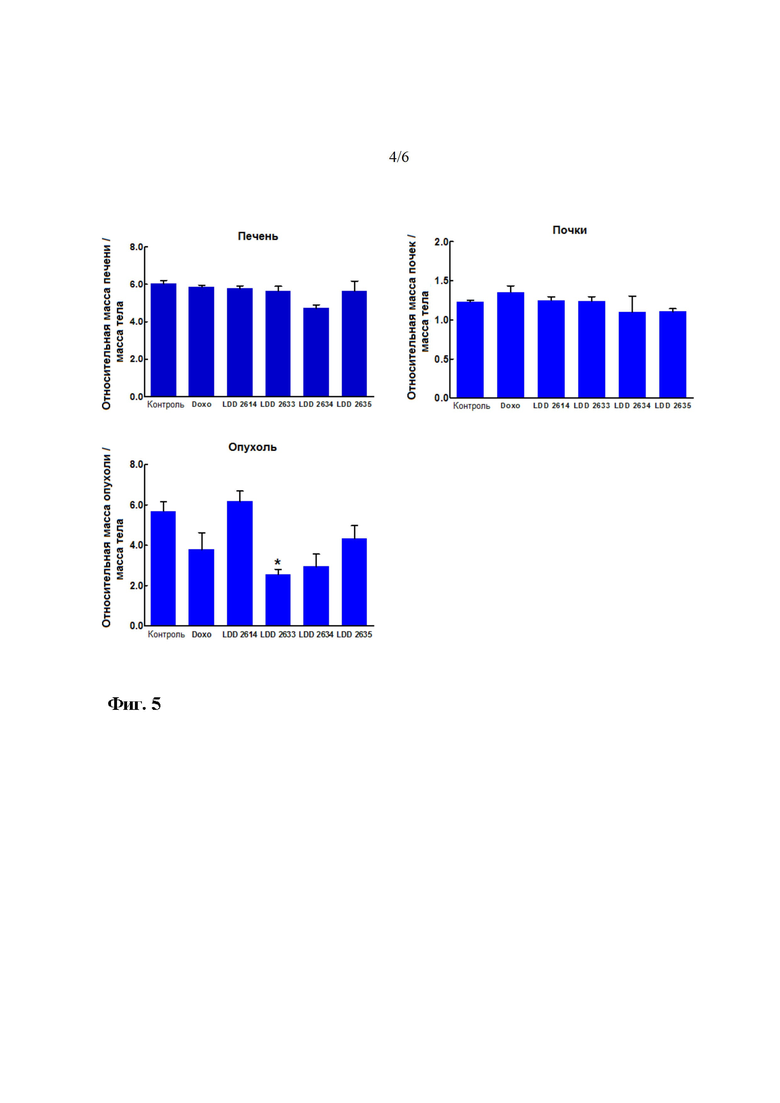
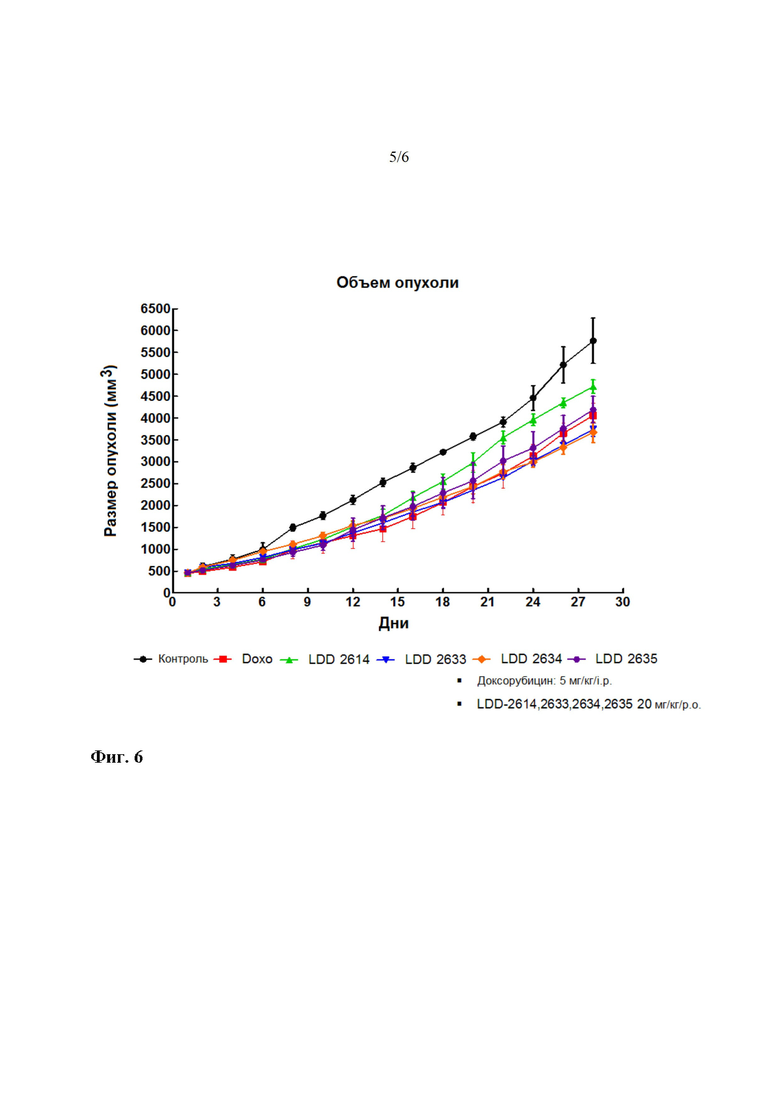
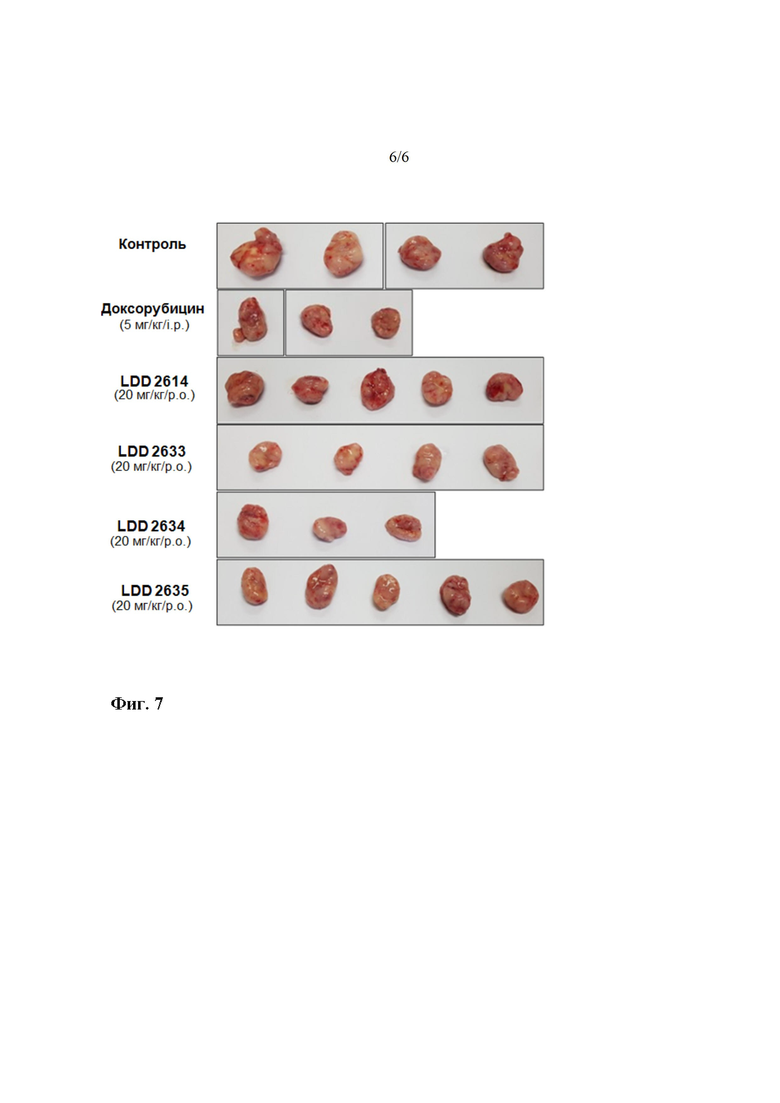
Изобретение относится к соединению, представленному химической формулой 1, или его фармацевтически приемлемой соли, где R1 представляет собой водород, R2 представляет собой фтор или C1-C4 алкиловый сложный эфир, и R3 представляет собой структуры (а), (b), (c) или (d). Также изобретение относится к фармацевтической композиции для предупреждения или лечения заболевания, связанного с ингибированием киназы FLT3, содержащей (a) эффективное количество соединения, представленного химической формулой 1, или его фармацевтически приемлемой соли; и (b) фармацевтически приемлемый носитель. Технический результат - соединения, обладающие ингибирующей способностью в отношении FLT3. 2 н. и 3 з.п. ф-лы, 4 табл., 6 ил., 5 пр.
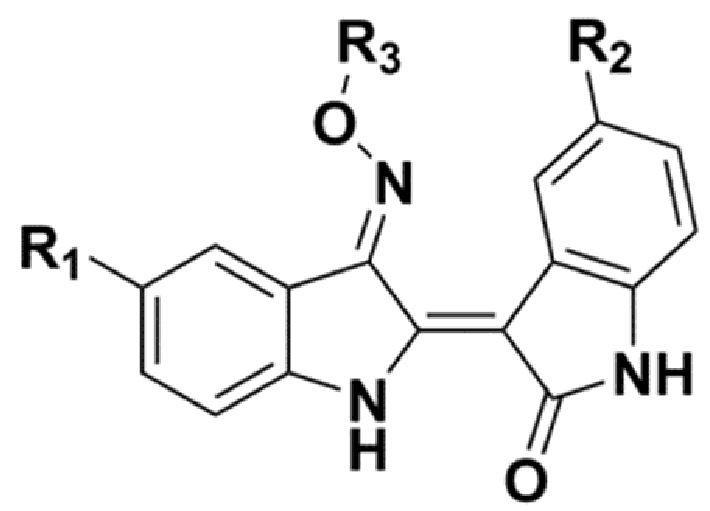
формула 1,
 (а),
(а), (b),
(b),  (c),
(c),  (d)
(d)
1. Соединение, представленное химической формулой 1, изображенной ниже, или его фармацевтически приемлемая соль:
[Химическая формула 1]
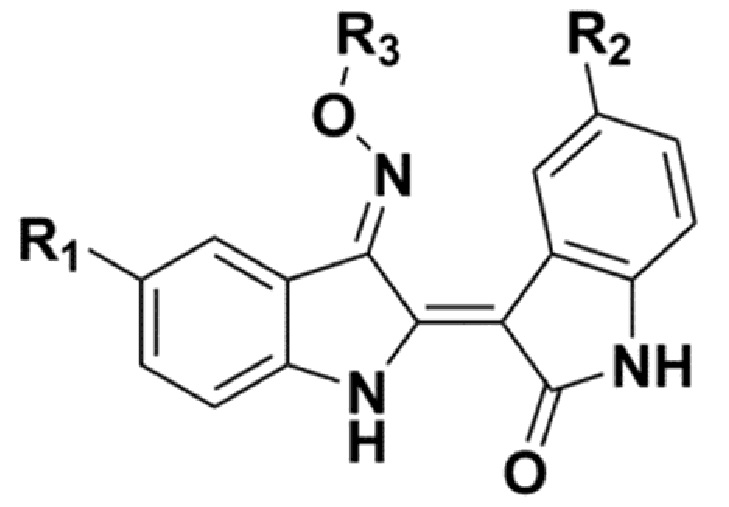
в указанной химической формуле 1
R1 представляет собой водород,
R2 представляет собой фтор или C1-C4 алкиловый сложный эфир, и
R3 представляет собой
 ,
,  ,
,  , или
, или  .
.
2. Соединение или его фармацевтически приемлемая соль по п.1, в котором C1-C4 алкиловый сложный эфир представляет собой метиловый сложный эфир.
3. Соединение или его фармацевтически приемлемая соль по п.1, в котором соединение, представленное химической формулой 1, представляет собой любое соединение, выбранное из группы, состоящей из следующих соединений:
метил-(2Z,3E)-2'-оксо-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3'-бииндолинилиден]-5'-карбоксилат,
метил-(2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-2'-оксо-[2,3'-бииндолинилиден]-5'-карбоксилат,
(2Z,3E)-5'-фтор-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3'-бииндолинилиден]-2'-он,
(2Z,3E)-5'-фтор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3'-бииндолинилиден]-2'-он, и
(2Z,3E)-5'-фтор-3-((2-(пиперидин-4-иламино)этокси)имино)-[2,3'-бииндолинилиден]-2'-он.
4. Фармацевтическая композиция для предупреждения или лечения заболевания, связанного с ингибированием киназы FLT3, содержащая:
(a) эффективное количество соединения, представленного химической формулой 1, изображенной ниже, или его фармацевтически приемлемой соли; и
(b) фармацевтически приемлемый носитель:
[Химическая формула 1]
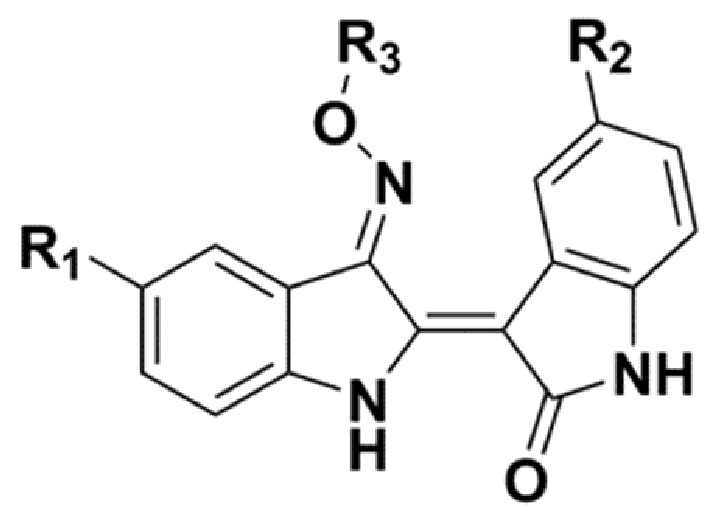
в химической формуле 1
R1 представляет собой водород,
R2 представляет собой фтор или C1-C4 алкиловый сложный эфир, и
R3 представляет собой
, , , или .
5. Фармацевтическая композиция по п.4, в которой соединение, представленное химической формулой 1, представляет собой любое соединение, выбранное из группы, состоящей из следующих соединений:
метил-(2Z,3E)-2'-оксо-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3'-бииндолинилиден]-5'-карбоксилат,
метил-(2Z,3E)-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-2'-оксо-[2,3'-бииндолинилиден]-5'-карбоксилат,
(2Z,3E)-5'-фтор-3-((2-(пиперазин-1-ил)этокси)имино)-[2,3'-бииндолинилиден]-2'-он,
(2Z,3E)-5'-фтор-3-((2-(4-метилпиперазин-1-ил)этокси)имино)-[2,3'-бииндолинилиден]-2'-он,
и
(2Z,3E)-5'-фтор-3-((2-(пиперидин-4-иламино)этокси)имино)-[2,3'-бииндолинилиден]-2'-он.
| US 20150259288 A1, 17.09.2015 | |||
| WO 2005070416 A1, 04.08.2005 | |||
| US 20140275168 A1, 18.09.2014 | |||
| НОВЫЕ ПРОИЗВОДНЫЕ ЦИКЛОМОЧЕВИНЫ, ИХ ПОЛУЧЕНИЕ И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2004 |
|
RU2341523C2 |
| Cheng, X., Merz, K.-H., Vatter, S., Zeller, J., et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

