Настоящее изобретение относится к области аналитических и биоорганических исследований, предназначено для диагностики агрессивности глиомы и первичного обнаружения и рецидивирования опухоли мочевого пузыря, а также при развитии области трансляционных исследований в будущем для других приложений путем обнаружения мутаций в промоторе гена теломеразной обратной транскриптазы человека. Представленное изобретение может быть использовано в гематологии и онкологической диагностике, в биохимических исследованиях для молекулярного узнавания, качественного и количественного обнаружения мутаций в промоторе гена каталитической субъединицы теломеразы человека с высокими чувствительностью, селективностью и экспрессностью.
Уровень техники
Теломеразная обратная транскриптаза человека (hTERT) - фермент, основной функцией которого является добавление особых повторяющихся последовательностей нуклеотидов на концы хромосом, которые укорачиваются из-за недорепликации ДНК. В норме теломераза активна в стволовых, половых и некоторых других клетках организма, которым необходимо постоянно делиться, однако в дифференцированных соматических клетках уровень экспрессии и активность теломеразы ограничены. Активация теломеразы в соматических клетках является критическим событием для их перерождения в опухолевые клетки и наблюдается в 85% случаев онкологических заболеваний [Shay J. W., Wright W. E. Senescence and immortalization: role of telomeres and telomerase // Carcinogenesis. 2005. V.26. P. 867-874]. Рак мочевого пузыря является наиболее частым онкологическим заболеванием мочевыделительной системы, и последние исследования показали, что мутации в промоторе hTERT являются самыми частыми мутациями на всех стадиях заболевания. [Allory Y. et al. Telomerase reverse transcriptase promoter mutations in bladder cancer: high frequency across stages, detection in urine, and lack of association with outcome // Eur. Urol. 2014. V. 65. P. 360-366]. Из уровня техники известны решения, позволяющие осуществлять молекулярную диагностику онкологических заболеваний. Одной из причин возникновения рака является появление соматических мутаций в клетке, которые нарушают ее нормальное функционирование. Некоторые генетические изменения являются характеристическими для определенного вида опухолевого поражения и могут служить маркерами заболевания. Обнаружение и определение поврежденных участков ДНК позволяет проводить прогнозирование и достоверную диагностику широкого класса заболеваний на ранних стадиях. Например, известно, что присутствие определенных соматических мутаций в онкогенах определяет скорость развития некоторых онкологических поражений. Например, при наличии соматических мутаций в онкогенах KRAS и BRAF колоректальный рак развивается в кратчайшие сроки, быстро метастазирует и трудно поддается химиотерапии. Наличие тех или иных маркеров позволяет определить точную область поражения и стадию заболевания, позволяет предсказать ответ на определенный вид терапий, а также обеспечивает возможность мониторинга течения болезни и рецидивирования. В настоящее время биопсия ткани является стандартной процедурой отбора биологического материала для анализа опухоли на наличие специфических маркеров, однако данный подход обладает рядом ограничений. Во-первых, для получения материала необходимо хирургическое вмешательство, что ограничивает частоту проведения данной процедуры. Во-вторых, пространственная гетерогенность опухоли может приводить к ненадежным результатам детектирования онкомаркеров, особенно при однократном тестировании. Наконец, возникает необходимость мониторинга течения болезни во времени и для этих целей частое проведение инвазивного вмешательства не представляется возможным. Также известно, что позднее диагностирование онкологического заболевания - одна из причин, которая уменьшает шансы пациента выжить. Острая необходимость в использовании более доступных биоматериалов для анализа, а также необходимость развития неинвазивных методов диагностики, привели к разработке диагностических подходов с использованием свободно циркулирующей ДНК (сцДНК), а точнее - циркулирующей опухолевой ДНК (цоДНК), присутствующей в физиологических жидкостях. Таким образом, исследование легко доступной сцДНК с помощью высокочувствительных методов открывает широкие возможности для диагностики пациентов, больных раком. Для ранней диагностики заболеваний и своевременного лечения требуется определение ДНК с поврежденными фрагментами с высокой чувствительностью и точностью, поскольку даже небольшое изменение качественного и количественного состава биологических жидкостей могут быть вызваны началом широкого набора серьезных заболеваний.
В настоящее время используемые методы основаны на идентификации последовательностей ДНК путем гибридизации комплементарных последовательностей с флуоресцентным методом обнаружения. В большинстве методов определения малых количеств нуклеиновых кислот, как правило, используется предварительная амплификация фрагментов ДНК в полимеразной цепной реакции (ПЦР). Известное из уровня техники решение, описанное в работе [S. Garczyk, N. Ortiz- , U. Schneider, I. Lurje, K. Guricova, N.T.Gaisa, E. Lorsy, K. Lindemann-Docter, A. Heidenreich, R
, U. Schneider, I. Lurje, K. Guricova, N.T.Gaisa, E. Lorsy, K. Lindemann-Docter, A. Heidenreich, R  . Next-generation sequencing reveals potential predictive biomarkers and targets of therapy for urothelial carcinoma in situ of the urinary bladder. Pathology. 2020. V. 190. P. 323-332], основано на секвенировании нового поколения (СНП) и позволяет анализировать одновременно миллионы копий цоДНК. Промотор гена TERT был проверен на наличие двух известных повторяющихся мутаций: С124 и С146C. Частота вариативности аллеля была установлена в 5%. В качестве эталонного генома для дизайна панели использовали hg19; hg38 служил эталонным геномом для аннотации мутаций.
. Next-generation sequencing reveals potential predictive biomarkers and targets of therapy for urothelial carcinoma in situ of the urinary bladder. Pathology. 2020. V. 190. P. 323-332], основано на секвенировании нового поколения (СНП) и позволяет анализировать одновременно миллионы копий цоДНК. Промотор гена TERT был проверен на наличие двух известных повторяющихся мутаций: С124 и С146C. Частота вариативности аллеля была установлена в 5%. В качестве эталонного генома для дизайна панели использовали hg19; hg38 служил эталонным геномом для аннотации мутаций.
Однако предложенная стратегия, как полноэкзомное и полногеномное секвенирование, обычно позволяет сгенерировать от 30 до 100 фрагментов ДНК, что приводит к слишком низкой чувствительности для анализа редких мутации в цоДНК во всем объеме сцДНК. Процесс СНП сложен в плане постановки эксперимента, затратен по времени, обладает не очень высокой чувствительностью и требует сложного биоинформатического анализа.
Известное из уровня техники решение, описанное в работе [J. J. G. Pritchard, G. Hamilton, C. D.Hurst, S. Fraser, C. Orange, M. A.Knowles, R. J. Jones, H. Y. Leung, T. Iwata. Monitoring of urothelial cancer disease status after treatment by digital droplet PCR liquid biopsy assays. Urologic Oncology: Seminars and Original Investigations. 2020. doi: 10.1016/j.urolonc.2020.05.012], основано на Сэнгер-секвенировании и цкПЦР. Сэнгер-секвенирование позволило выявить точечные мутации у 70% пациентов, чьи биообразцы были проанализированы методом цкПЦР. Случаи ремиссии и рецидивов, отслеживаемые посредством анализа мутаций PIK3CA E542K и TP53 Y163C в плазме и моче, совпадали с клиническими наблюдениями в течение 48 месяцев от начала химиотерапии. Разработанная методика, основанная на цкПЦР, позволила детектировать мутацию промотора обратной транскриптазы (TERT) теломеразы (-124). Предложенный метод выявления этой мутации способен отличить мутант от аллелей дикого типа.
Однако для постановки цкПЦР необходимо знать заранее маркерные генетические и эпигенетические изменения, кроме того, цкПЦР обладает ограниченным потенциалом для мультиплексного анализа. Предел обнаружения ДНК, выделенных из опухолевых тканей, для таких подходов, как секвенирование по методу Сэнгера, не превышает 5% (пятипроцентное содержание мутантной аллели на фоне последовательностей дикого типа).
Известное из уровня техники решение, описанное в патенте РФ №2463354 от 10.10.2012 «Способ диагностики рака мочевого пузыря с помощью онкомаркера tfdp1 (варианты) и набор для его осуществления», состоит в получении биоматериала и выделении рибонуклеиновых кислот (РНК), синтезе комплементарных дезоксирибонуклеиновых кислот (кДНК) на матрице РНК, нормировании концентрации кДНК TFDP1 по контрольному гену, проведении количественной ПЦР-амплификации фрагмента гена TFDP1. Далее определяют количество амплифицированного фрагмента ДНК TFDP1 для образца биоматериала. При концентрации кДНК гена TFDP1 в физиологических жидкостях, превышающей 1.5% концентрации кДНК гена бета-актина АСТВ, диагностируют наличие переходноклеточного рака мочевого пузыря. В предложенное решение входит набор для осуществления описанного способа методом ПЦР, включающий два праймера с определенной последовательностью при молярном соотношении 1:1. Способ-вариант предусматривает проведение диагностики методом иммуноферментного анализа (ИФА). Получают образцы мочи и крови пациента, выделяют смесь белковых компонентов мочи и крови, проводят ИФА с моноклональными антителами, и/или поликлональными антителами, и/или их фрагментами против рекомбинантного белка TFDP1 и/или его уникальных фрагментов длиной свыше 8 аминокислот. Диагностируют рак мочевого пузыря при концентрации белка TFDP1 в исследуемых образцах, превышающей 5-кратную концентрацию белка TFDP1 в контроле. Способ (вариант) позволяет с достоверностью диагностировать рак мочевого пузыря, в том числе на ранней стадии прогрессии опухолевой трансформации.
Однако в предложенном решении диагностика рака мочевого пузыря проводится по содержанию в крови и моче матричных рибонуклеиновых кислот (мРНК) гена TFDP1 и/или белка TFDP1. Кроме того, для описанного решения характерны все ограничения, связанные с использованием ПЦР: (1) чувствительность напрямую зависит от эффективности амплификации ПЦР ДНК-фрагмента образца, резко снижающейся при наличии повреждений ДНК или ее фрагментации; (2) при необходимости определения нескольких молекул-мишеней мультиплексно и на фоне большого количества ДНК со схожей последовательностью, точность ПЦР становится неудовлетворительной для постановки диагноза при существенном снижении чувствительности анализа; (3) возрастающий предел обнаружения (ПО) мутаций приводит к повышению вероятности получения ложно-положительных результатов; (4) существенное фрагментирование ДНК будет зависеть от процедур пробоотбора и пробоподготовки; (5) для детектирования фрагментов длиной менее 50 нуклеотидов, выделение и специфическая гибридизация праймеров для амплификации практически невозможны; (6) идентификации последовательности ДНК ПЦР-методами препятствует образование неканонических структур ДНК-матрицей, например G(гуанин)-квадруплексов.
На данный момент спектроскопия гигантского комбинационного рассеяния (спектроскопия ГКР) является возможным эффективным аналитическим инструментом и обладает большим потенциалом в области исследования биомолекул, в частности нуклеиновых кислот. Основные преимущества метода спектроскопии ГКР применительно к анализу биомолекул, по сравнению с уже существующими методами, состоят в (1) высокой чувствительность метода, вплоть до детектирования единичных молекул; (2) сигналы ГКР содержат информацию о молекулярных отпечатках пальцев всех компонентов анализируемой биологической системы; (3) спектроскопия ГКР устойчива к фотодеградации и тушению по сравнению с флуоресцентными методами и подходит для длительного мониторинга; (4) ширина пиков в 10-100 раз меньше по сравнению с пиками спектров флуоресценции органических меток и квантовых точек, что обеспечивает возможность мультиплексного анализа при одной длине волны возбуждения; (5) ГКР-активные наносубстраты могут быть подобраны и сконструированы с различным покрытием, различной формы и размера в зависимости от аналитических целей. Во многих работах показана принципиальная возможность использования прямой ГКР-спектроскопии для обнаружения однонуклеотидных замен [Xu L.J., Lei Z.C., Li J., Zong C., Yang C. J., Ren, B. Label-Free Surface-Enhanced Raman Spectroscopy Detection of DNA with Single-Base Sensitivity // Journal of the American Chemical Society. 2015. V. 137. P. 5149-5154]. Однако ГКР-спектры ДНК без использования молекул репортеров были получены преимущественно с использованием синтетических олигонуклеотидов. Расшифровка спектров ГКР, полученных в результате анализа клинических образцов, все еще сопряжена с рядом трудностей, так как сигналы, соответствующие большому количеству нецелевого генетического материала, могут преобладать над слабыми сигналами молекул мишеней, содержащихся в низких концентрациях в исследуемом образце. Другой подход к детектированию анализируемого вещества - непрямая спектроскопия ГКР - заключается в использовании ГКР-активных молекул-репортеров и регистрации изменения сигнала, которое происходит в результате взаимодействия ГКР-активной молекулы с анализируемым веществом. Данный способ позволяет преодолеть трудности, которые часто возникают при регистрации собственных колебательных спектров нуклеиновых кислот, а именно сложность интерпретации результатов в присутствии большого количества нецелевых молекул или генетического материала, если говорить об анализе клинических образцов. С другой стороны, утрачивается информация о структурных перестройках и химических особенностях анализируемых молекул, которая содержится в собственных характеристических спектрах ГКР.
Наиболее близким к заявляемому является способ измерения связанных с раком веществ, включая свободную ДНК, происходящую от раковых клеток, посредством спектроскопии КР (RU2723160, 09.06.2020). Предложенный способ определения ДНК в биологический жидкостях заключается в подготовке наночастиц благородных металлов, выступающих в роли ГКР-усилителя; гибридизации мишени с направленно выбранным ковалентно ГКР-меченным комплементарным олигонуклеотидом; нанесении пробы с исследуемым соединением на поверхность наночастиц жидкой пробы с исследуемым соединением с образованием направленно упорядоченной за счет нековалентных взаимодействий структуры, включающей определяемую ДНК, ГКР-метку и олигонуклеотид; детектировании полученного гибридизированного ГКР-меченного дуплекса методом спектроскопии гигантского комбинационного рассеяния, и о качественном и количественном содержании ДНК судят по положению и интенсивности полос на регистрируемых спектрах. Способ применим для определения малых количеств ДНК на фоне большого количества посторонних мешающих компонентов анализируемых реальных биологических жидкостей: крови, мочи, церебральной жидкости и др. Данный результат достигается за счет дополнительного селективного связывания фрагментов ДНК с направленно выбранными ковалентно ГКР-меченными олигонуклеотидами на поверхности наночастиц. Кроме того, за счет направленного структурирования и упорядочивания, а также контроля расстояния от ГКР-метки до поверхности наночастиц регистрируется воспроизводимый на всей сенсорной поверхности усиленный сигнал ГКР. Усиление сигнала комбинационного рассеяния в 103 - 1014 раз достигается за счет дополнительного оборачивания гибридизованного дуплекса ДНК вокруг металлических наночастиц, выступающих в качестве ГКР-усилителя, благодаря нековалентным взаимодействиям. Предлагаемый способ позволяет надежно количественно определять необходимые ДНК на уровне 10-14 М.
Однако в известном способе предложено определение мутантной последовательности ДНК, не позволяющей диагностировать рак мочевого пузыря. Для достоверного детектирования и определения мутаций C228T и C250T на уровне ультранизких концентраций, что позволит диагностирование заболевания на ранних стадиях (до ок. 6,000 поврежденных фрагментов в анализируемом образце), необходимо использование определенного рабочего расстояния от ГКР-метки до металлической наноструктуры. В отсутствии мутантной последовательности ДНК свободный зонд образует шпильку. В случае присутствия мутантного фрагмента ДНК определяемая последовательность взаимодействует с комплементарной последовательностью зонда, шпилька раскрывается, вследствие чего наблюдается резкий рост интенсивности сигнала ГКР по сравнению с ситуацией, когда метка находилась в составе шпильки. При этом использование комплементарной последовательностью зонда с любой длиной из диапазона 1-5 нм не приводит к возможности высокочувствительного определения мутаций C228T и C250T на уровне ультранизких концентраций. Более того, в изобретении описан способ анализа и его принцип без оптимизированной ГКР-платформы, позволяющего достоверно определять биомаркеры - мутантные последовательности ДНК C228T и C250T.
Технической проблемой является отсутствие наборов и простых способов диагностики рака мочевого пузыря на ранних стадиях развития опухоли.
Раскрытие изобретения
Техническим результатом заявляемой группы изобретений является разработка набора, позволяющего с высокой точностью проводить диагностику рака мочевого пузыря на ранних стадиях развития опухоли.
Заявляемая группа изобретений позволяет проводить идентификацию двух мутантных последовательностей ДНК C228T и C250T и их селективное определение в биологической пробе для выявления у пациентов рака мочевого пузыря (1) на ранних стадиях: начиная с I стадии (при начальных стадиях опухолевого процесса: стадия Т1 или более поверхностные опухоли), когда опухоль является неинвазивной; (2) с высокими чувствительностью (низким пределом обнаружения от концентрации 1⋅10-14 М, что составляет ок. 6,000 поврежденных фрагментов ДНК); (3) селективностью по отношению к иным фрагментам ДНК; и (4) экспрессностью (время пробоподготовки нескольких образцов одновременно - до 60 мин и самого анализа - 10 с).
Диагностика рака мочевого пузыря осуществляется за счет селективного узнавания и определения мутаций C228T и C250T в последовательности ДНК, что включает в себя использование набора из планарного наноструктурированного ГКР-сенсора и водных растворов, меченных красителем с пределом обнаружения не более 1 мкМ, например родамином 6Ж, олигонуклетидов: GnuS-C228T-R6G и GnuS-C250T-R6G (где Gnus - последовательность, состоящая из 15 - 20 аминокислот, из которых не менее 20% составляют цитозин (C) и гуанин (G), например последовательности SEQ ID №1 - №4). В качестве красителя с пределом обнаружения не более 1 мкМ используют родаминовые, цианиновые, ксантеновые и диазо-красители, метиленовый синий и кристаллический фиолетовый.
Способ заключается в заборе мочи пациента, последующем отборе 20 мкл жидкой пробы для гибридизации предположительно содержащихся там мутантных последовательностей ДНК C228T и C250T с комплементарными им олигонуклеотидами GnuS-C228T-метка или GnuS-C250T-метка, ковалентно модифицированным коммерчески доступной ГКР-меткой - например, родамином 6Ж (R6G). Далее 10 мкл полученной смеси наносят на планарную сенсорную поверхность. Иммобилизация зонда в настоящем изобретении основывается на специфическом электростатическом взаимодействии между якорной олигонуклеотидной последовательностью Gnus, входящей в состав зондов GnuS-C228T-метка и GnuS-C250T-метка и металлическими наночастицами. Благодаря высокой энергии связывания серебряных кластеров с цитозином (C) и гуанином (G), входящих в состав якорной последовательности, которая в несколько раз превышает энергию образования Уотсон-Криковских пар, серебряные частицы оказываются внутри якорной последовательности. Такое взаимодействие, в отличие от ковалентных сшивок, обеспечивает дополнительную структуризацию поверхности, что обеспечивает образование «горячих точек» (англ. “hot spots”) и усиление сигнала. Усиление сигнала комбинационного рассеяния в 102 - 108 раз достигается за счет создания дополнительных «горячих точек» на поверхности благодаря тому, что серебряные частицы оказываются внутри якорной последовательности. В составе индикаторной системы для детектирования мутаций C228Т и C250Т в промоторе гена hTERT использовали конструкцию, состоящую из дезоксиолигонуклеотида, обеспечивающего иммобилизацию зонда на последовательности наночастиц серебра (Gnus), с которым связана последовательность с ГКР-меткой (R6G), комплементарная последовательности аналита (Фиг. 11). Предложенный подход позволяет использовать метод спектроскопии ГКР с высокими коэффициентами чувствительности (до 105 -106 у.е.), низкими пределами обнаружения (ПО до 3 фМ), высоким соотношением сигнал/шум (отсутствие влияние матрицы реальных образцов на полезные сигналы), высокой селективностью (по отношению к неповрежденным фрагментам ДНК или мутациям иного происхождения), широким диапазоном определяемых концентраций (ДОК от 1⋅10-14 до 1·10-7 М), высокими воспроизводимостью и прецизионностью (коэффициенты корреляции градуировочных зависимостей составляет не менее 0.97, относительное стандартное отклонение при нижней определяемой концентрации не более 0.10) для качественного и количественного определения мутаций C228Т и C250Т в целях диагностики онкологических заболеваний. За счет направленного структурирования и упорядочивания, а также контроля расстояния от ГКР-метки до поверхности наночастиц регистрируется воспроизводимый на всей сенсорной поверхности усиленный сигнал ГКР. Предлагаемый способ определения ДНК демонстрирует отсутствие сигналов на спектрах гигантского комбинационного рассеяния для нормальных последовательностей ДНК и мутаций иного типа, что позволяет надежно селективно количественно определять характерные для рака мочевого пузыря мутации C228Т и C250Т с ПО до 10-15 М.
Технический результат достигается заявляемым набором, состоящим из (1) твердофазного планарного сенсора: диэлектрического химически инертного материала и наноструктурированного покрытия на основе наночастиц благородных металлов толщиной 0.1-10 мкм, и (2) 1 мкМ водных растворов, меченных красителем с пределом обнаружения не более 1 мкМ олигонуклеотидов: GnuS-C228T-метка и GnuS-C250T-метка (где Gnus - последовательность, состоящая из 15 - 20 аминокислот, из которых не менее 20% составляют цитозин (C) и гуанин (G), например последовательности SEQ ID №1 - №4) для детектирования и определения характерных для рака мочевого пузыря мутаций ДНК C228T и C250T в биологических пробах. Технический результат достигается способом детектирования и определения мутаций ДНК C228T и C250T, заключающимся в (1) нанесении на планарную сенсорную поверхность предварительно подготовленной жидкой пробы, полученной при гибридизации раствора анализируемой биологической жидкости пациента с комплементарным олигонуклеотидом GnuS-C228T-метка или GnuS-C250T-метка, ковалентно модифицированным коммерчески доступной ГКР-меткой; (2) облучении монохроматическим пучком лазера с длиной волны 485 - 785 нм и регистрации сигнала комбинационного рассеяния, при этом о качественном и количественном содержании мутаций ДНК C228T и C250T в изучаемом образце судят по положению и интенсивности полос на регистрируемых спектрах, характерных для метки-красителя.
При этом в качестве наночастиц благородных металлов используют серебро, золото, платину.
При этом наночастицы имеют размеры 20-90 нм и характеризуются полосой плазмонного резонанса 420-650 нм.
При этом в качестве материала-носителя для наночастиц используют силикатное стекло.
При этом используют 10-500 мкмоль благородного металла в расчете на 1 мм2 поверхности носителя с последующей сушкой.
При этом для нанесения пробы и однократного анализа используют поверхность площадью 4×4 мм2.
При этом количество ГКР-меченного олигонуклеотида используют в мольном соотношении от 1:1 к мутациям ДНК C228T и C250T.
При этом гибридизацию проводят путем нагрева и выдерживания в течение 5-10 мин при температуре 95°С, с последующим охлаждением до комнатной температуры.
При этом подготовленные жидкие пробы наносят путем накапывания на поверхность наночастиц.
При этом используют жидкие пробы объемом 10 мкл с концентрацией мутаций ДНК C228T и C250T 10-15 - 10-2 М.
При этом при проведении анализа используют монохроматическое лазерное излучение с длиной волны 514 нм и мощностью, не превышающей 25% от номинальной величины излучения, в течение до 10 - 60 с.
При этом о качественном и количественном содержании мутаций ДНК C228T и C250T в образце судят по положению и интенсивности полос при 1385, 1470 и 1590 см-1 с допустимой величиной погрешности не более 10 см-1.
При использовании родамина 6Ж в качестве метки-красителя о наличии и содержании мутаций ДНК C228T и C250T в образце судят по положению и интенсивности полос при 1385, 1470 и 1590 см-1 с допустимой величиной погрешности не более 10 см-1.
Для определения количественного содержания мутаций предварительно строят калибровочные кривые.
Представленный способ пригоден для работы с портативным, коммерчески доступным, серийным оборудованием вне лабораторных условий. Таким образом, с использованием заявляемого оптического планарного сенсора возможно высокочувствительное и селективное качественное и количественное определение присутствующих в образце мутаций ДНК C228T и C250T «у постели больного».
Таким образом, использование предлагаемого способа анализа позволяет узнавать о присутствии мутаций C228T и C250T и судить об их количественном содержании с высокой селективностью, широким диапазоном определяемых концентраций, высокой воспроизводимостью и прецизионностью.
Краткое описание чертежей
Изобретение поясняется чертежами.
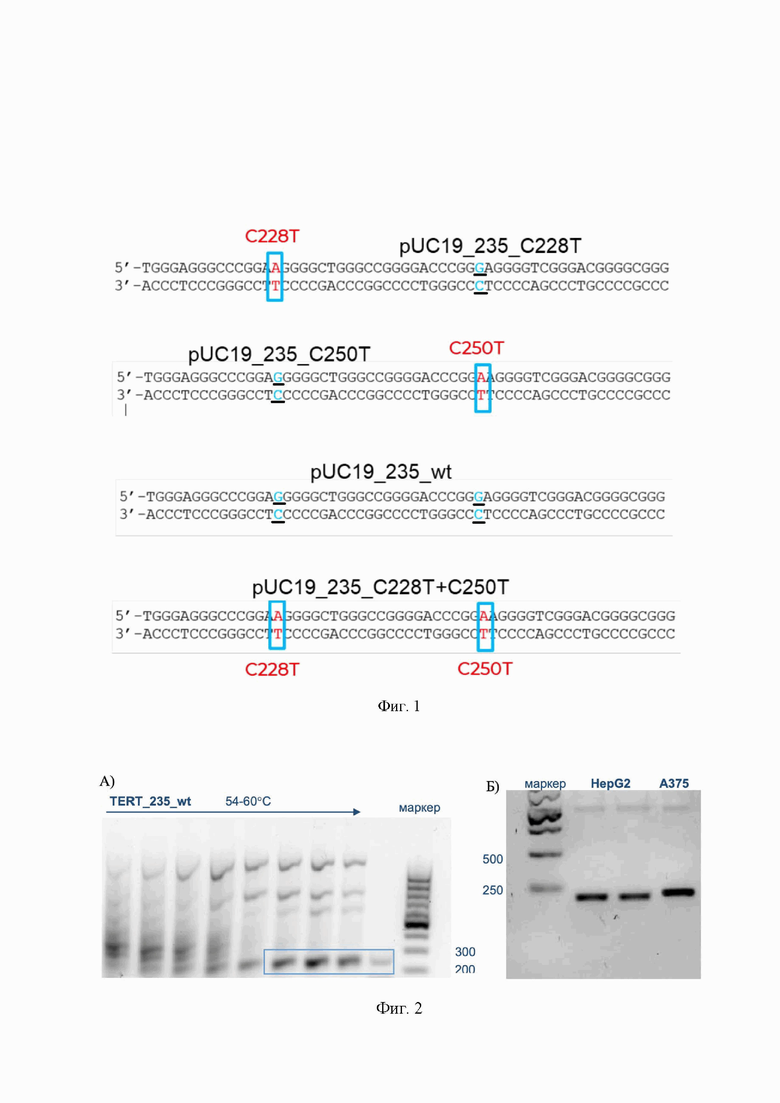
На фиг. 1 представлены фрагменты клонируемых вставок. Подчеркнутые буквы - нуклеотиды, соответствующие дикому типу.
На фиг. 2 представлены А) подбор условий ПЦР-амплификации промотора гена hTERT с геномной ДНК человека (целевой продукт выделен рамкой); Б) продукты ПЦР-амплификации вставки с геномов клеточных линий HepG2 и A375.
На фиг. 3 представлена карта используемого вектора pUC19 с отмеченными сайтами клонирования HindIII и KpnI.
На фиг. 4 представлены хроматограммы, полученные с помощью секвенирования по Сэнгеру, для фрагментов вставок. Мутантные нуклеотиды выделены рамкой - 4б; нуклеотиды, соответствующие дикому типу, выделены рамкой - 4а.
На фиг. 5 представлена схема детектирования однонуклеотидных замен с помощью флуоресцентных TaqMan зондов. Зонды с меткой FAM комплементарны последовательностям мутантного типа, зонды с меткой R6G комплементарны последовательностям дикого типа.
На фиг. 6 представлены химические структуры используемых флуоресцентных меток и длины волн, соответствующие максимуму испускания флуоресценции.
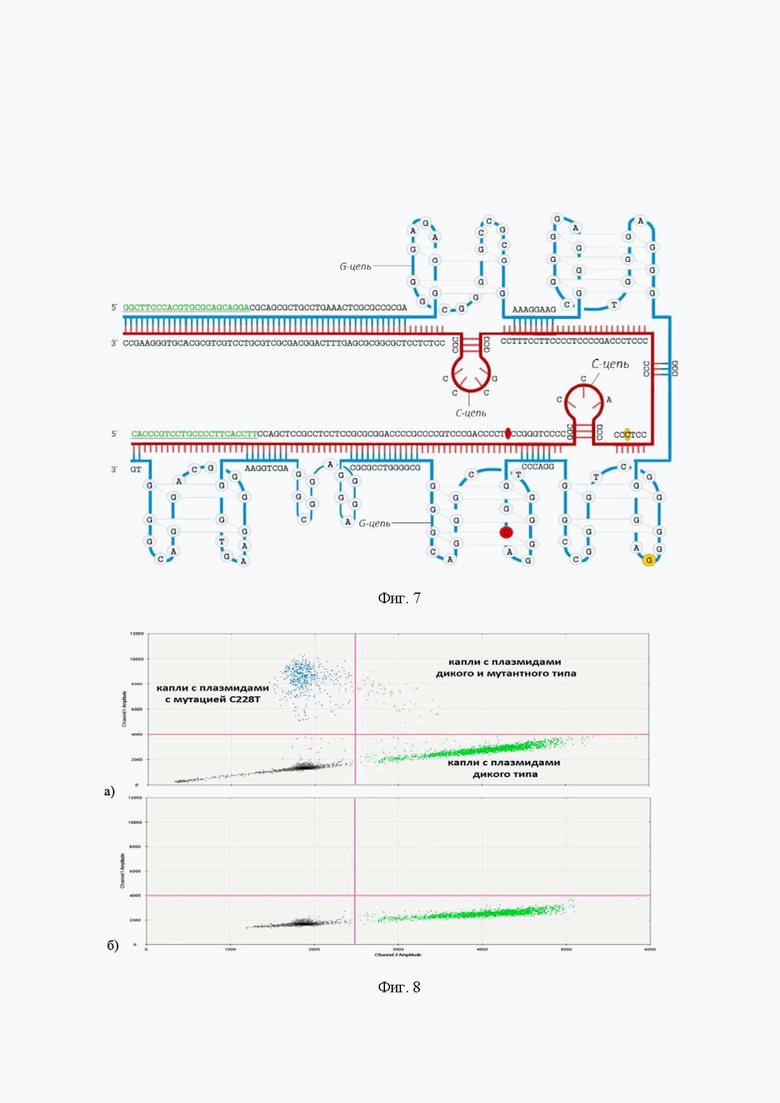
На фиг. 7 представлена схема структуры фрагмента промоторной области гена hTERT, содержащей сайты -124 и -146 от ATG (отмечены кружками), в которых возможно возникновение мутаций C228T и С250Т [Zvereva M., Pisarev E., Hosen I., Kisil O., Matskeplishvili S., Kubareva E., Kamalov D., Tivtikyan A., Manel A., Vian E., Kamalov A., Ecke T., Calvez-Kelm F.L. Activating telomerase TERT promoter mutations and their application for the detection of bladder cancer. Int. J. Mol. Sci. 2020, 21(17), 6034]. Синим цветом обозначена G-цепь, красным С-цепь. Подчеркнуты участки, соответствующие праймерам, с помощью которых данный фрагмент был амплифицирован. Структура была получена с использованием программы “QGRS Mapper” при следующих параметрах: максимальная длина G - квадруплекса составила 30 нуклеотидов, минимальный размер нуклеотидной группы - 2 нуклеотида, размер петли - от 0 до 36 нуклеотидов.
На фиг. 8 представлены а) результат цкПЦР, приведенный в виде двумерного картирования сигнала по двум независимым каналам. Соотношение плазмид мутантного типа к плазмидам дикого типа в данном эксперименте pUC19_235_C228Т : pUC19_235_wt = 1:4; б) результат цкПЦР для системы со 100% содержанием плазмиды pUC19_235_wt. Выше пороговой линии капли, соответствующие мутантной плазмиде pUC19_235_C228Т, отсутствуют.
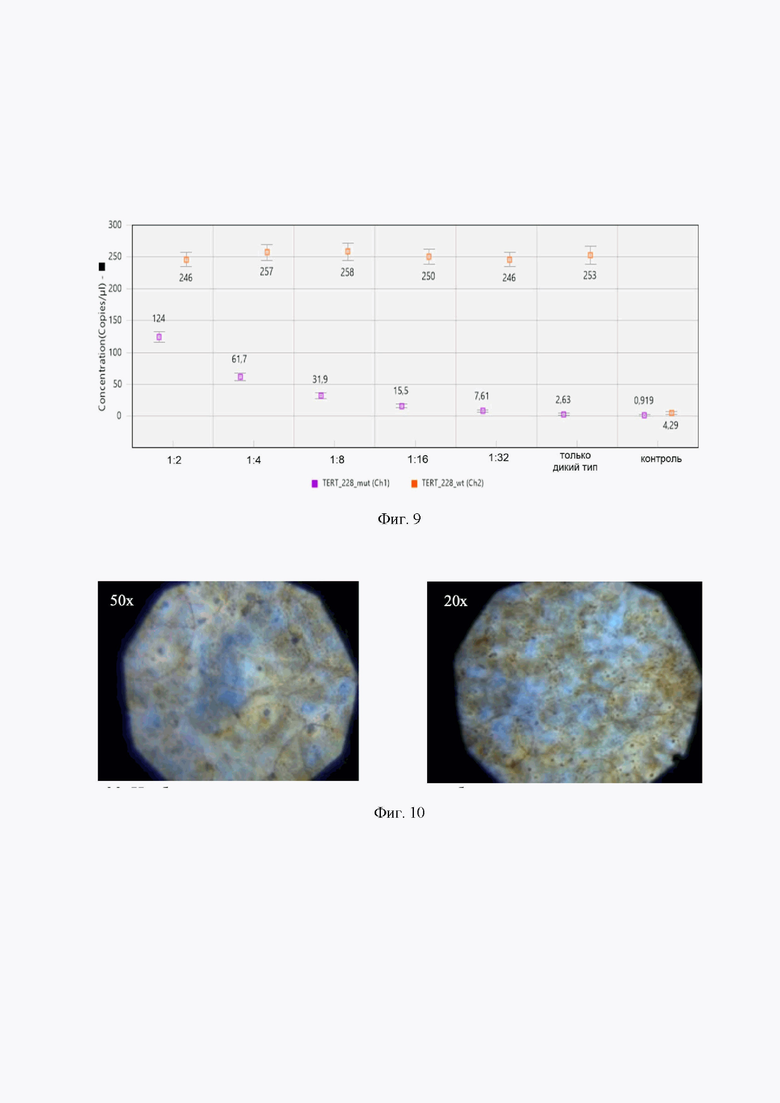
На фиг. 9 представлены уровни аналитической чувствительности метода на примере обнаружения мутантной плазмиды pUC19_235_C228Т на фоне большого содержания плазмиды дикого типа pUC19_235_wt. По оси абсцисс соотношение количества плазмид мутантного типа к плазмидам дикого типа. Концентрация плазмиды дикого типа во всех случаях составляет ~5000 копий в реакционной смеси. Раститровка по концентрациям плазмиды, содержащей мутацию С228Т, с исходной концентрацией ~2500 копий в реакционной смеси с шагом в 2 раза. Чувствительность обнаружения составила 2.03%.
На фиг. 10 представлены оптические микрофотографии поверхности наночастиц серебра при различных увеличениях объектива.
На фиг. 11 представлены а) схематичное изображение системы для детектирования фрагментов последовательности промотора гена hTERT, состоящей из иммобилизованного на поверхности наночастиц зонда с меткой R6G за счет якорной последовательности Gnus; б) сиквенсы последовательностей Gnus, GnuS-C250Т-R6G - зонда для детекции последовательности с мутацией С250Т и GnuS-C228Т-R6G - зонда для детекции последовательности с мутацией С228Т. Синим выделена последовательность, обеспечивающая иммобилизацию. Зеленым выделены участки, образующие дуплекс с аналитами.
На фиг. 12 представлены спектры ГКР, полученные с использованием λex = 514 нм для зонда GnuS-C228T-R6G, нанесенного в объеме 10 мкл и в концентрациях 10-8 - 10-14 М на планарную поверхность наночастиц серебра. Номера на подписи соответствуют отрицательному десятичному логарифму концентрации образца: 8 - 10-8 М, 10 - 10-10 М, 12 - 10-12 М, 14 - 10-14 М. Рамками выделены положения характеристических пиков.
На фиг. 13 представлены спектры ГКР, полученные с использованием λex = 514 нм для системы GnuS-C228T-R6G с A228, нанесенной в объеме 10 мкл и в концентрациях 10-8 - 10-14 М на планарную поверхность наночастиц серебра. Номера на подписи соответствуют отрицательному десятичному логарифму концентрации образца: 8 - 10-8 М, 10 - 10-10 М, 12 - 10-12 М, 14 - 10-14 М. Рамками выделены положения характеристических пиков.
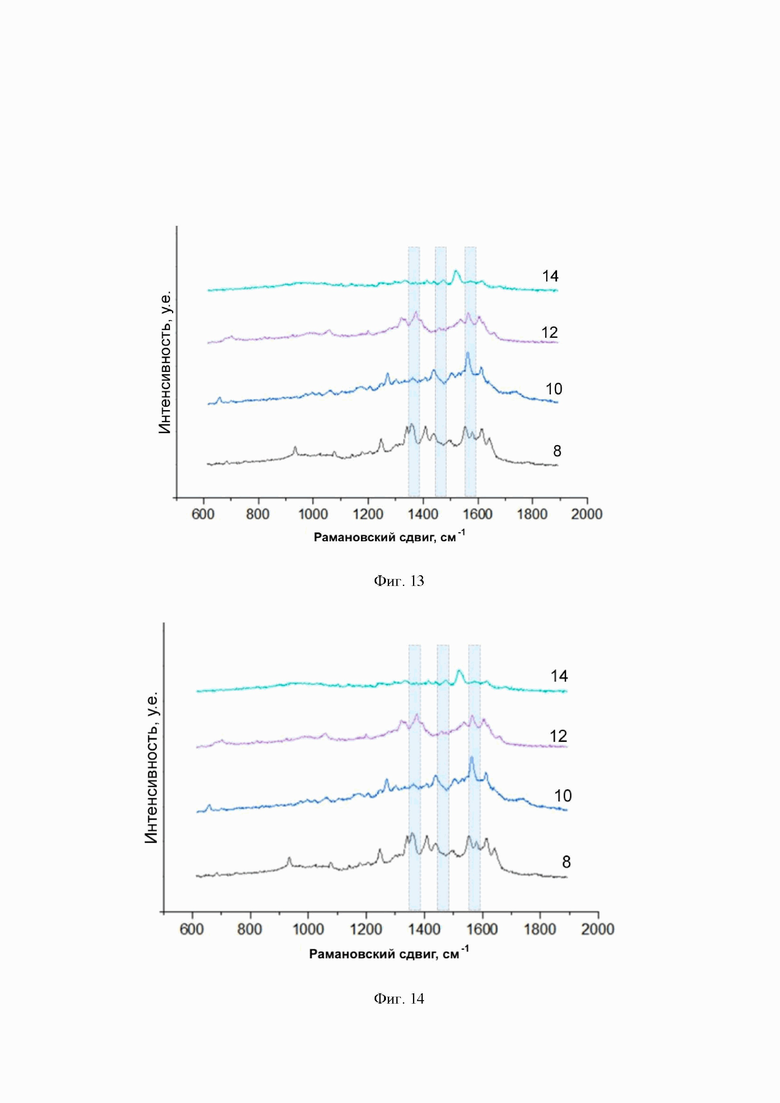
На фиг. 14 представлены спектры ГКР, полученные с использованием λex = 514 нм для зонда GnuS-C250T-R6G, нанесенного в объеме 10 мкл и в концентрациях 10-8 - 10-14 М на планарную поверхность наночастиц серебра. Номера на подписи соответствуют отрицательному десятичному логарифму концентрации образца: 8 - 10-8 М, 9 - 10-9 М, 10 - 10-10 М, 11 - 10-11 М, 12 - 10-12 М, 13 - 10-13 М, 14 - 10-14 М. Голубыми рамками выделены положения характеристических пиков.
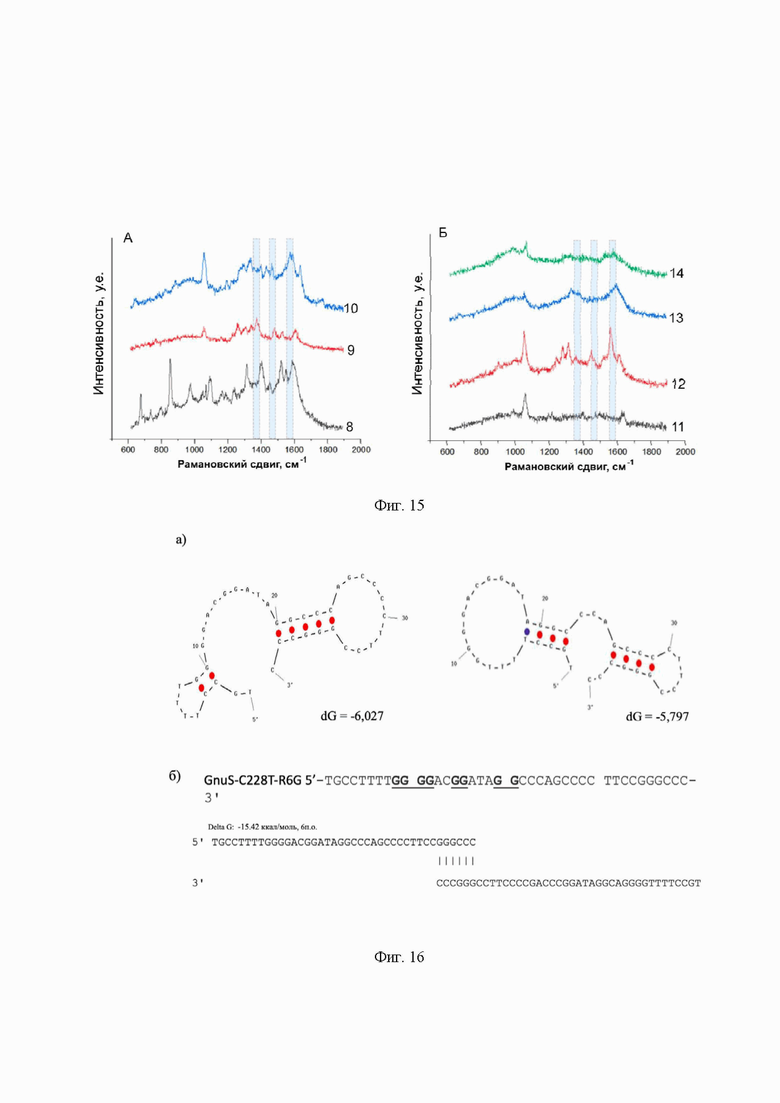
На фиг. 15 представлены спектры ГКР, полученные с использованием λex = 514 нм для системы GnuS-C250T-R6G с A250, нанесенной в объеме 10 мкл и в концентрациях 10-8 - 10-14 М на планарную поверхность наночастиц серебра. Номера на подписи соответствуют отрицательному десятичному логарифму концентрации образца: 8 - 10-8 М, 9 - 10-9 М, 10 - 10-10 М, 11 - 10-11 М, 12 - 10-12 М, 13 - 10-13 М, 14 - 10-14 М. Голубыми рамками выделены положения характеристических пиков.
На фиг. 16 представлены а) вторичные структуры, образованные последовательностью зонда GnuS-C228Т-R6G. Метка R6G находится на 3' - конце цепи; б) место наиболее вероятного образования G-квадруплекса.
Осуществление изобретения
В настоящем изобретении приняты следующие обозначения и термины:
ВЭЖХ - высокоэффективная жидкостная хроматография,
ГКР - гигантское комбинационное рассеяние,
Спектр ГКР - спектр комбинационного рассеяния с сигналами, усиленными за счет эффекта поверхностного плазмонного резонанса на наноструктурах благородных металлов,
ДМАА - диметиламиламин,
ДМСО - диметилсульфоксид,
ДМФА - диметилформамид,
ДНК - дезоксирибонуклеиновая кислота,
ДОК - диапазон определяемых концентраций,
ИФА - иммуноферментный анализ,
кДНК - комплементарная дезоксирибонуклеиновая кислота,
мРНК - матричная рибонуклеиновая кислота,
НЧС - наночастицы серебра,
ОФ-ВЭЖХ - обращено-фазовая ВЭЖХ,
ПО - предел обнаружения,
Промотор - участок олигонуклеотидной последовательности, узнаваемый РНК-полимеразой как стартовая площадка для начала транскрипции,
ПЦР - полимеразная цепная реакция,
РНК - рибонуклеиновая кислота,
СНП - секвенирование нового поколения,
Спектр ГКР - спектр комбинационного рассеяния с сигналами, усиленными за счет эффекта поверхностного плазмонного резонанса на наноструктурах благородных металлов,
сцДНК - свободно циркулирующая ДНК дезоксирибонуклеиновая кислота,
Трис-HCl - раствор трисаминометана с соляной кислотой,
цкПЦР - цифровая капельная полимеразная цепная реакция,
цоДНК - циркулирующая опухолевая дезоксирибонуклеиновая кислота,
ЭДТА - этилендиаминтетрацетат натрия,
A - аденин,
ACTB - англ. actin, cytoplasmic a - высококонсервативный белок, который полимеризуется с образованием нитей, которые образуют сшитые сети в цитоплазме клеток,
BRAF - Серин/треониновая протеинкиназа B-raf; англ. Serine/threonine-protein kinase B-raf; КФ:2.7.11.25,
C - цитозин,
ESI-MS - масс-спектрометрия с ионизацией электрораспылением,
FAM - флуоресцеин (3’,6’-дигидрокси-3H-спиро[2-бензофуран-1,9’-ксантен]-3-он),
G - гуанин,
hTERT - теломераза обратной транскриптазы,
KRAS - протоонкоген, представитель семейства белков Ras (это семейство генов, а также белки, которые они кодируют - малые G-белки),
PAGE - денатурирующий полиакриловый гель,
R6G - родамин 6Ж (этил-2-[3-(этиламино)-6-этилимино-2,7-диметилксантен-9-ил]бензоат гидрохлорид),
SERS - англ. surface enhanced Raman spectroscopy,
T - тимин,
TAMRA - раствор трет-бутиламин/вода, 1:3 об./об.,
TBTA - трис(бензилтриазолил)амин,
TFDP1 - англ. Transcription Factor Dp-1 - ген, кодирующим белок. Заболевания, связанные с TFDP1, включают колоректальный рак и гепатоцеллюлярную карциному.
Химические символы / сокращения имеют свои обычные значения: °С (градус (градусы) Цельсия), нм (нанометр (нанометры)), мкм (микрометр (микрометры)), см (сантиметр (сантиметр)), мин (минута (минуты)), с (секунда (секунды)), мкл (микролитр (микролитры)), мкг (микрограмм (микрограммы)), М (моль (моли) в литре), л (литр (литры)), мл (миллилитр (миллилитры)), мкл (микролитр (микролитры)), г (грамм (граммы)), мг (миллиграмм (миллиграммы)), моль (моли), ммоль (миллимоль (миллимоли)), масс. % (массовый процент (массовые проценты)), м.м. (молекулярная масса).
Представленные ниже примеры конкретного осуществления изобретения приведены для предоставления специалистам в данной области техники полного описания проведения и применения анализа по изобретению, но не ограничивают предполагаемый авторами изобретения объем изобретения.
Все приведенные ниже реагенты являются коммерчески доступными. Все процедуры, если не оговорено особо, осуществляли при комнатной температуре или температуре окружающей среды, то есть в диапазоне от 18 до 25°C. В ходе всех экспериментов для приготовления водных растворов и промывки суспензий использовали деионизированную воду высокой чистоты с удельным сопротивлением не менее 18.2 МОм⋅см, очищенную с использованием установки «Milli-Q», «Millipore». Спектры ГКР зарегистрированы при помощи микроскопа «InVia Reflex» («Renishaw», Великобритания) в конфокальном режиме с использованием Ar+ лазера (длина волны - 514.5 нм, 20 мВт). Пропускание широкополосного фильтра нейтральной плотности составляло 10% для спектров ГКР. Время накопления сигнала - 10 с. Юстировку прибора проводили с использованием монокристаллических пластин кремния в качестве стандарта. Фокусное расстояние используемого объектива 250 мм, максимальный размер пятна лазера на объекте - ок. 300 мкм. Все спектры были сняты с использованием 50×-го объектива «Leica».
Заявляемый тест-набор состоит из двух ключевых компонентов: планарного наноструктурированного ГКР-сенсора и растворов меченных красителем с пределом обнаружения не более 1 мкМ, например родамином 6Ж, олигонуклетидов: GnuS-C228T-метка и GnuS-C250T-метка (где Gnus - последовательность, состоящая из 15 - 20 аминокислот, из которых не менее 20% составляют цитозин (C) и гуанин (G), например последовательности SEQ ID №1 - №4). Использование предложенного набора заключается в нанесении на планарную сенсорную поверхность предварительно подготовленной жидкой пробы, полученной при гибридизации раствора анализируемой биологической жидкости пациента с комплементарным олигонуклеотидом GnuS-C228T-метка или GnuS-C250T-метка, ковалентно модифицированным коммерчески доступной ГКР-меткой; при этом о качественном и количественном содержании мутаций ДНК C228T и C250T в изучаемом образце судят по положению и интенсивности полос на регистрируемых спектрах ГКР.
Пример 1. Способ селективного детектирования мутаций C228T и C250T методом спектроскопии ГКР
Синтез наноструктурированной поверхности серебра. В качестве ГКР-усилителя использовали планарный оптический ГКР-сенсор, представляющий собой нанесенные на поверхность тонкой стеклянной пластинки наночастицы серебра толщиной 0.4 - 1.5 мкм. Получение наноструктурированной поверхности серебра осуществлялось пиролизом аэрозоля аммиачного комплекса. К 100 мл свежеприготовленного 0.17 М водного раствора нитрата серебра (2.93 г) по каплям со стеклянной палочки при постоянном перемешивании добавляли ~30 мл 0.1 М водного раствора гидроксида натрия до полного выпадения черно-коричневого осадка оксида серебра (I). Затем осадок Ag2O тщательно промывали деионизованной водой (трижды порциями по 100 мл воды), далее добавляли 100 мл воды и по каплям 7 мл концентрированного водного раствора аммиака при постоянном перемешивании для образования водного раствора аммиачного комплекса серебра. После полного растворения осадка дополнительно добавляли 25 мл концентрированного водного раствора аммиака для предотвращения выпадения оксида серебра (I) в процессе аэрозольного распыления аммиачного комплекса. Бóльшие количества аммиака существенно ухудшали микроструктуру образующихся в процессе пиролиза серебряных колец. Раствор разбавляли до 170 мл водой. Полученный прозрачный 0.1 М раствор комплекса серебра фильтровали через мембранный фильтр, затем разбавляли в 8 раз водой до концентрации [Ag(NH3)2]OH 12.5 мМ. В контейнер установки ультразвукового аэрозольного ингалятора переносили 50 мл конечного раствора. Два покровных стекла предварительно отмывали травильным раствором пиранья (H2SO4: H2O2 = 3 : 1) и нагревали на дне химического стакана до 290 - 320°С. На небулайзер устанавливали патрубок-переходник с диаметром ~1 см. Использовали параметры распыления: 7 на «УЗ» и 1 на «Воздух». Таким образом, серебро наносили на поверхность покровного стекла ультразвуковым серебряным дождем аммиачного комплекса серебра (I). Аэрозольные капли диаметром 1-5 мм служили микрореакторами и обеспечивали формирование кольцевых кластеров наночастиц серебра диаметром 90±5 нм (Фиг. 10). Морфология получаемых сенсорных наноструктурированных поверхностей контролировалась методом сканирующей электронной микроскопии с полевой эмиссией. Важное свойство предложенных серебряных сенсоров заключалось в реализации широкой полосы возбуждения плазмонного резонанса, которая покрывала весь видимый диапазон длин волн, начиная с 420 нм. Это позволило получить резонансно усиленный сигнал ГКР от широкого набора меток, включая родамин 6Ж (R6G), флуоресцеин (FAM) и цианин (Cy3, Cy5 и др.) (Фиг. 6). Для получения резонансно-усиленного сигнала ГКР необходимо, чтобы область частот возбуждения плазмонного резонанса перекрывалась с полосой поглощения ГКР-метки. Поскольку коммерчески доступные аргоновый и гелий-неоновый лазеры излучают в области видимого света с наиболее сильной интенсивностью при длинах волн 514.5 и 633 нм соответственно, в качестве наиболее подходящей ГКР-активной молекулы-репортера выбрали родамин 6G (λmax ≈ 524 нм).
В качестве планарного сенсорного элемента для усиления сигнала комбинационного рассеяния может быть использована любая шероховатая металлическая поверхность, характеризующаяся шероховатостью не менее 2 нм и обладающая эффектом поверхностного плазмонного резонанса, что было подтверждено на примере серебра и золота.
Наиболее перспективная стратегия создания систем детектирования последовательностей нуклеиновых кислот с помощью спектроскопии ГКР заключается в использовании разработанной аналитической системы, в которой регистрация целевой ДНК происходит за счет комплементарных взаимодействий мишени с ГКР-меченым зондом, иммобилизованном на поверхности наночастиц металла.
Синтез меченых последовательностей олигонуклеотидов. Для получения меченых последовательностей олигонуклеотидов использовали автоматизированную методику синтеза, ранее описанную и приведенную в работах авторов «Способ обнаружения и определения ДНК с заданной последовательностью методом спектроскопии гигантского комбинационного рассеяния» Патент РФ № 2723160 от 09.06.2020 г.; V. M. Farzan, E. A. Ulashchik, Y. V. Martynenko-Makaev, M. V. Kvach, I. O. Aparin, V. A. Brylev, T. A. Prikazchikova, S. Y. Maklakova, A. G. Majouga, A. V. Ustinov, G. A. Shipulin, V. V. Shmanai, V. A. Korshun and T. S. Zatsepin. Automated solid-phase click synthesis of oligonucleotide conjugates: From small molecules to diverse n‐acetylgalactosamine clusters. Bioconjug. Chem. 2017. V. 28. P. 2599-2607]. 5’-Алкин-модифицированные олигонуклеотиды получали в синтезаторах олигонуклеотидов ASM-2000 («Biosset») или MM-12 («Bioautomation») с использованием амидофосфитного метода. Защищенные 2’-дезоксирибонуклеозид-3'-фосфорамидиты, Unylinker-CPG (500 Å) и S-этилтио-1H-тетразол были приобретены у «ChemGenes». Раствор азида 6-R6G (100 мМ) и медного катализатора CuI⋅P(OEt)3 (100 мМ) в апротонном биполярном растворителе (диметилсулфоксиде (ДМСО), диметилфорамиде (ДМФА) или диметиламиламине (ДМАА), 100 мкл) добавляли к 5’-алкин-модифицированным олигонуклеотидам, связанным с твердой фазой в колонке (1 мкмоль) 5 раз каждые 30 мин. Через 3 ч твердый носитель тщательно промывали (10 × 150 мкл ДМСО, затем 3 × 150 мкл ацетонитрила) с последующим снятием защиты у цианоэтильных групп (2 × 10% диэтиламина в ацетонитриле (10 мин) и полное снятие защиты с использованием смеси TAMRA (трет-бутиламин/вода, 1:3 об./об.) при 55°С в течение ночи. Раствор выпаривали и аликвоту анализировали с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) и масс-спектрометрии с электрораспылительной ионизацией (ESI-MS).
Полученные олигонуклеотиды дважды очищали денатурирующим полиакриловым гелем (PAGE), а затем методом обращенно-фазовой ВЭЖХ и (ОФ-ВЭЖХ). Гель-электрофорез денатурированных олигонуклеотидов проводили в 15% PAGE, содержащем 7 М мочевины в трис-боратном буфере (50 мМ Трис-HCl, 50 мМ борная кислота, 1 мМ этилендиаминтетрацетат натрия (ЭДТА), рН 8.3). Олигонуклеотиды извлекали из геля путем электроэлюции с помощью Elutrap (Whatman) в трис-боратном буфере (5 мМ Трис-HCl, 5 мМ борная кислота, 0.1 мМ ЭДТА, рН 8.3). Анализ методом ВЭЖХ и очистку олигонуклеотидов проводили с использованием хроматографической колонны 2.1×50 мм Jupiter С18 (5 мкм, «Phenomenex»), буфера А: 10 мМ диизопропиламин и 15 мМ 1,1,1,3,3,3-гексафторизопропанол и буфера Б: 10 мМ диизопропиламин, 15 мМ 1,1,1,3,3,3-гексафторизопропанол и 80% ацетонитрил. Соли промывали буфером А (4 цикла), а затем 100% буфера Б (2 цикла) при скорости потока 0.3 мл/мин и температуре 45°C. Структуру полученных олигонуклеотидов подтверждали методом ESI-MS с использованием системы Agilent 1260-Bruker Maxis Impact. Масс-спектрометрический анализ олигонуклеотидов проводился в отрицательном режиме (капиллярное напряжение 3500 В), и регистрируемые спектры пересчитывали методом максимальной энтропии.
Полученный 5’-алкин-модифицированных олигонуклеотид модифицировали малыми молекулами ГКР-индикаторов. Полученный олигонуклеотид готовили в твердой фазе и связывали с 5'-алкинфосфорамидитом. Клик-модификацию проводили на твердой фазе путем добавления 10 мМ раствора азида, 100 мМ CuI⋅P(OEt)3 4×100 мкл (азида 2 экв.) в течение 4 ч с последующим промыванием. В большинстве случаев олигонуклеотиды отщепляли от носителя и снимали защиту в синтезаторе, используя 1,2-этилендиамин/толуол, 1:1 об./об., в течение 2 ч при комнатной температуре с последующим промыванием (4×150 мкл ацетонитрила) и элюирование 50% ацетонитрилом в воде или снятие защиты с использованием раствора АМА (концентрированный водный аммиак и 40% водный метиламин, 1:1 об./об.) в течение 30 мин при 65°С. Для R6G-меченных олигонуклеотидов снятие защиты проводили с использованием раствора TAMRA при 55°C в течение ночи. Для FAM-меченных олигонуклеотидов использовали две схемы снятия защиты (концентрированный водный раствор аммиака при комнатной температуре в течение 2 и 18 ч). Для переключения фазы раствора 5'-алкин-модифицированный олигонуклеотид (2 нмоль) растворяли в 1 М ацетате триэтиламмония (30 мкл). Затем добавляли 20 мкл раствора медного катализатора в 60% ДМСО (15 мМ трис(бензилтриазолил)амин (TBTA) и 13 мМ сульфата меди (II)), а затем 4 мкл 10 мМ азида FAM в ДМСО и 140 мкл 80% ДМСО. Раствор дегазировали и дважды заполняли аргоном в вакуумном центрифужном концентраторе, а затем добавляли дегазированный раствор аскорбиновой кислоты (0.25 М, 6 мкл). Реакционную смесь оставляли на ночь при комнатной температуре, и олигонуклеотидный продукт осаждали 4% LiClO4 в ацетоне (1 мл). Очистку и анализ методом ВЭЖХ проводили, как описано выше.
Обычно для иммобилизации зонда на металлической поверхности используют ковалентные взаимодействия между модифицированным олигонуклеотидом и наночастицами металла. Иммобилизация зонда в настоящем изобретении основывалась на специфическом электростатическом взаимодействии между якорной олигонуклеотидной последовательностью Gnus, входящей в состав зонда и содержащей не менее 20% цитозиновых и гуаниновых нуклеотидов, и металлическими наночастицами. Благодаря высокой энергии связывания серебряных кластеров с цитозином (C) и гуанином (G), входящих в состав якорной последовательности, которая в несколько раз превышает энергию образования Уотсон-Криковских пар, серебряные частицы оказываются внутри якорной последовательности. Такое взаимодействие, в отличие от ковалентных сшивок, обеспечивает дополнительную структуризацию поверхности, что обеспечивает образование горячих точек и усиление сигнала [Bossert N., de Bruin D.,  et al. Fluorescence-tunable Ag-DNA biosensor with tailored cytotoxicity for live-cell applications // Sci. Rep. 2016. V. 6. P. 37897]. В приведенных примерах в качестве последовательности GnuS рассматривали 5’-TGCCTTTTGGGGACGGATA-3’. При этом при использовании иных последовательностей GnuS (5’-AATGATCTTAACCAAGTATA-3’, 5’-TCGGTTGGGGAGCCTAT-3’ и 5’-TGGCTAATCCCCTCGGATA-3’), т.е. при сохранении длины зонда от 15 до 20 нуклеиновых кислот и содержании цитозина и гуанина не менее 20% обеспечивались аналогичные чувствительность, селективность, экспрессность и достоверность анализа.
et al. Fluorescence-tunable Ag-DNA biosensor with tailored cytotoxicity for live-cell applications // Sci. Rep. 2016. V. 6. P. 37897]. В приведенных примерах в качестве последовательности GnuS рассматривали 5’-TGCCTTTTGGGGACGGATA-3’. При этом при использовании иных последовательностей GnuS (5’-AATGATCTTAACCAAGTATA-3’, 5’-TCGGTTGGGGAGCCTAT-3’ и 5’-TGGCTAATCCCCTCGGATA-3’), т.е. при сохранении длины зонда от 15 до 20 нуклеиновых кислот и содержании цитозина и гуанина не менее 20% обеспечивались аналогичные чувствительность, селективность, экспрессность и достоверность анализа.
В качестве системы детектирования мутаций C228Т и C250Т в промоторе гена hTERT использовали конструкцию, состоящую из дезоксиолигонуклеотида, обеспечивающего иммобилизацию зонда на последовательности наночастиц серебра (Gnus), с которым связана последовательность с ГКР-меткой (R6G), комплементарная последовательности аналита (Фиг. 11). Аналитами являлись последовательности A-228 и A-250, содержащие фрагмент промотора гена hTERT, с однонуклеотидными заменами С228Т и С250Т соответственно.
Из стоковых водных растворов олигонуклеотидов GnuS-C228T-R6G, Gnus-С250Т-R6G, А-C250T и А-C228T с концентрациями 1 мM готовили рабочие водные растворы для каждого образца с концентрациями 10-8 - 10-14 М. В целях регистрации индивидуальных спектров ГКР для олигонуклеотидов GnuS-C228T-R6G и Gnus-С250Т-R6G рабочие растворы наносили на поверхность наночастиц серебра в объеме 10 мкл и регистрировали спектры ГКР после полного высыхания подложки при комнатной температуре. Для регистрации спектров систем GnuS-C228T-R6G + А-C228T и Gnus-С250Т-R6G + А-C250T (Таблица 1) предварительно смешивали компоненты до достижения итоговых концентраций растворов 10-8 - 10-14 М. После смешения выдерживали при T = 22°C ок. 10 мин, затем нагревали смесь при T = 95°C в течение 5 мин, после чего оставляли смесь охладиться до T = 22°C в течение ок. 60 мин. Затем полученный раствор наносили на поверхность наночастиц серебра в объеме 10 мкл и регистрировали спектры ГКР после полного высыхания подложки при комнатной температуре.
Полученные спектры ГКР для индивидуальных растворов зондов GnuS-C250Т-R6G и GnuS-C228Т-R6G, нанесенных в объеме 10 мкл с помощью дозатора на планарную поверхность наночастиц серебра при четырех различных концентрациях, а также спектры систем зонд + аналит: GnuS-C250Т-R6G + A250 и GnuS-C228Т-R6G + A228 продемонстрировали возможность селективного детектирования мутаций C250Т и C228Т (Фиг. 12 - 15). Перед нанесением системы зонд + аналит проводилось предварительное нагревание смеси при 94°С в течение 5 мин, а после - постепенное охлаждение до комнатной температуры.
В случае спектров ГКР, полученных отдельно для зондов GnuS-C250Т-R6G и GnuS-C228Т-R6G, характеристические пики либо отсутствуют, либо их интенсивность находится на уровне фона. В случае использования полных разработанных индикаторных систем GnuS-C250Т-R6G + A250 и GnuS-C228Т-R6G + A250 наблюдали интенсивные пики, характерные для ГКР-метки R6G при 1380, 1470 и 1590 см-1. Спектры ГКР для предложенных детектирующих систем GnuS-C250Т-R6G + A250 и GnuS-C228Т-R6G + A250 имели неизменное положение характеристических пиков: 1383, 1470 и 1590 см-1, вне зависимости от концентрации мутаций в анализируемом образце.
В отсутствии анализируемой молекулы свободный зонд образовывал шпильку (якорь GnuS не был вовлечен в образование шпильки), а метка находилась на близком расстоянии от поверхности. В зависимости от того, насколько близкое это расстояние и находится ли метка в горячей точке, сигнал мог как возникать, так и затухать [«Способ обнаружения и определения ДНК с заданной последовательностью методом спектроскопии гигантского комбинационного рассеяния» Патент РФ № 2723160 от 09.06.2020 г.]. При внесении аналита в систему он взаимодействовал с комплементарной последовательностью зонда, шпилька раскрывалась, вследствие чего и наблюдалось изменение интенсивности сигнала ГКР по сравнению с ситуацией, когда метка находилась в составе шпильки. Таким образом, при раскрытии шпильки ковалентная связь с меткой сохранялась, изменялась структура ДНК, что приводило к изменению взаиморасположения метки и поверхности. В отсутствии анализируемой молекулы зонд образовывал сложную вторничную структуру (шпильки, внутримолекулярные димеры, а также внутренние G-квадруплексы) (Фиг. 16). В образование этой вторичной структуры был вовлечен 5’-конец зонда, который отвечает за иммобилизацию на поверхности наночастицы. При попытке гибридизации определяемых поврежденных последовательностей ДНК с неподходящей по последовательности меченым олигонуклеотидом: GnuS-C250Т-R6G с A228 и GnuS-C228Т-R6G с A250 аналитический сигнал методом спектроскопии ГКР зарегистрировать не удалось. Этот факт дополнительно продемонстрировал, что без эффективной иммобилизации и образования структурированных агрегатов получить существенного усиления сигнала было невозможно, полное отсутствие или низкая интенсивность сигналов для прочих фрагментов ДНК свидетельствовала о достаточной для анализа селективности методики.
Исключительно в присутствии С228Т и С250Т мутантных фрагментов ДНК происходило взаимодействие зонда с аналитом с образованием дуплекса; и 5’-конец зонда, вовлеченный в образование вторичной структуры, высвобождался, и происходила эффективная иммобилизация системы {зонд + аналит} на поверхности наночастиц, что приводит к наблюдаемому увеличению интенсивности сигнала. Созданная система позволила детектировать мутации С228Т и С250Т в промоторе гена hTERT с помощью спектроскопии ГКР, выводы о количественном содержании определяемых фрагментов обеспечило аналит-зависимое усиление сигнала ГКР. В случае нанесения на поверхность зонда GnuS-C228Т-R6G в присутствии мутации С250 взаимодействия не происходило и аналитического сигнала на спектрах ГКР не наблюдали, как и в случае с GnuS-C250Т-R6G в присутствии мутации С228, что подтвердило кросс-селективность разработанной методики. Важно, что при использовании олигонуклеотидов, меченных ГКР-меткой, отличающейся от предложенной длины, достижение необходимой чувствительности определения мутантных фрагментов ДНК не было возможным, что было показано на олигонуклетидах с зондами RnuS и PnuS (Таблица 2). Согласно рассчитанным в соответствии с химическими структурами длинами, расстояние от наноструктурированной поверхности до ГКР-метки составил 4 нм для GnuS, 5 нм для RnuS и 3 нм для PnuS. Использование красителей-меток c последовательностью Gnus из ряда родаминовых, цианиновых, ксантеновых и диазо-красителей, метиленового синего и кристаллического фиолетового, обеспечивало достижение необходимого уровня чувствительности (Таблица 2). Пробоподготовка всех образцов (до количества, которое необходимо проанализировать), заключающаяся в гибридизации определяемого фрагмента ДНК с мечеными олигонуклеотидами, занимала около 60 мин. При этом никаких дополнительных манипуляций, кроме включения/выключения термостата, от лаборанта в этот момент не требуется, поэтому целесообразно проводить эту процедуру для нескольких проб одновременно. Общее время анализа занимало не более 1 мин с достаточным временем накопления спектра - 10 с.
Воспроизводимость аналитических сигналов составляла менее 5%. Полученный ПО для мутантных фрагментов ДНК - 10-14 М, что составляет ок. 6,000 поврежденных фрагментов в 1 мкл анализируемого образца. Достигнутый ПО позволяет диагностировать рак мочевого пузыря, начиная с I стадии (при начальных стадиях опухолевого процесса: стадия Т1 или более поверхностные опухоли), когда опухоль является неинвазивной. В случае использования метки-красителя R6G, вывод о качественном составе и количественном содержании осуществляли по наличию характеристических полос: 1383, 1470 и 1590 см-1, на спектрах ГКР и интенсивности полученных сигналов по градуировочным зависимостям соответственно.
Таблица 1. Определяемые последовательности нуклеиновых кислот, последовательности ГКР-меченных олигонуклеотидов и их краткие обозначения

Таблица 2. Метрологические характеристики (с количеством параллельных измерений n = 5 и доверительной вероятностью P = 0.95) определения мутантных последовательностей ДНК с ковалентно ГКР-меченными олигонуклеотидами комплементарных последовательностей различной длины (Параметры съемки спектров ГКР: длина волны возбуждения - 514 нм, мощность лазера - 10%, время экстинкции - 10 с)
I = (a±Δa)lgc + (b±Δb)*
* - вид зависимости интенсивности аналитического сигнала при характеристическом рамановском сдвиге от концентрации определяемой ДНК (моль/л);
** - стандартное относительное отклонение для измерений при минимальной концентрации (cн) из диапазона определяемых содержаний.
Пример 2. Валидация результатов определения мутаций C228T и C250T методом спектроскопии ГКР с помощью метода цкПЦР
Поскольку процедуры клинической диагностики заболеваний должны выполняться согласно определенным стандартам, необходимо проводить предварительную оценку диагностических методов: определять их аналитическую чувствительность и другие характеристики. Определение возможностей самого метода на клинических образцах или на клеточных линиях не дает в полной мере достоверных результатов, так как опухолевые образцы по своей природе неоднородны, а клеточные линии содержат не 100% мутантной аллельной фракции. Поэтому для внедрения цкПЦР или предложенной методики на основе ГКР-спектроскопии в клиническую практику необходимо создание единой стандартной аналитической системы для сравнения и валидации результатов, полученных различными лабораторными методами в различных лабораторных условиях.
В качестве доклинических испытаний, наиболее приближенным к клиническим, получили модельные референсные конструкции путем клонирования фрагментов генома человека и внесения определяемых мутаций: С228Т и С250Т.
Клонирование. Были получены четыре плазмиды, содержащие все необходимые вставки:
1. дикого типа (pUC19_235_wt),
2. с мутацией С228Т (pUC19_235_C228Т),
3. с мутацией С250Т(pUC19_235_С250Т),
4. одновременно с обеими мутациями С228Т и С250Т (pUC19_235_С228Т+С250Т).
Для получения вставок дикого типа в качестве матрицы для ПЦР использовали Human Genomic DNA 11691112001 Roche, а для вставок с мутацией С228T в положении -146 использовали ДНК клеточной линии HepG2 (содержание мутантной ДНК 70%), для вставок с мутацией С250Т в положении -126 использовали ДНК клеточной линии А375. (Фиг. 1). Клонируемые вставки представляли собой GC-богатые фрагменты (77% GC для фрагмента 235 п.о.), поэтому их амплификация была затруднена из-за сложной вторичной структуры (предположительно образование G-квадруплексов). Для амплификации фрагмента дикого типа с геномной ДНК человека в реакционную смесь добавляли 7-деаза-2’-дезоксигуанозин-5’-трифосфат (7-deaza-dGTP) до финальной концентрации 200 мкМ (Фиг. 2, А). Продукт ПЦР нужной длины очищали с помощью разделения методом гель-электрофореза и выделения из агарозного геля.
Полимеразная цепная реакция (ПЦР). Для амплификации фрагментов дикого типа и фрагмента с мутацией С228Т составляли следующую ПЦР-смесь:
Тщательно перемешивали и помещали в прибор для проведения ПЦР T100 PCR Thermal Cycler фирмы Bio Rad. Параметры амплификации: 95°C в течение 4 мин - 1 цикл; 95°C - 30 с, 58°C - 20 c, 72°C - 30 c - 39 циклов; 72°C в тчение 5 мин - 1 цикл.
Амплификация фрагмента c генома клеточных линий HepG2 и A375 проходила с использованием Q5 ДНК-полимеразы (New England Biolabs). Состав реакционных смесей:
Параметры амплификации: 98°C в течение 30 с - 1 цикл; 98°C - 7 с, 68°C - 15 c, 72°C - 10 c - 35 циклов; 72°C в течение 2 мин - 1 цикл.
В случае использования в качестве матриц для амплификации клонируемого фрагмента ДНК клеточных линий HepG2 и A375 подход с добавлением 7-deaza-dGTP не был успешен и не удавалось получить количественно ПЦР-продукт даже в случае проведения двухэтапного ПЦР. Для решения этой задачи была использована Q5 полимераза, точность которой более чем в 100 раз выше, чем у с Taq полимеразы, в буфере для амплификации GC-богатой матрицы с добавлением энхансера. Обычно в состав GC-энхансеров входят такие соединения, как ДМСО и глицерин, которые способствуют нарушению сложной вторичной структуры и увеличивают эффективность амплификации (Фиг. 2, Б).
Полученные ПЦР-продукты обработали эндонуклеазами рестрикции HindIII и KpnI, и легировали по липким концам с плазмидой pUC19 (Фиг. 3), линеаризованной с помощью тех же эндонуклеаз рестрикции. Полученную лигазную смесь трансформировали в компетентные клетки JM109 E.coli и высевали на чашку Петри с 5-бром-4-хлор-3-индолил-бета-D-галактопиранозидом (X-gal) и изопропил-бета-галактопиранозидом (ИПТГ) для бело-голубой селекции. На следующий день высевали ночные культуры из белых колоний. Из ночных культур выделяли плазмиду и после проведения рестриктного анализа с помощью эндонуклеазы рестрикции PvuII, подтверждавшего наличие вставки, секвенировали вставку методом Сэнгера (Фиг. 4).
Разделение фрагментов ДНК в агарозном геле, выделение с геля. После проведения ПЦР проводили очистку полученных ПЦР-продуктов в 1.2% агарозном геле. Для приготовления агарозного геля добавляли 1.0 г легкоплавкой агарозы к 100 мл раствора 1xTBE. Визуализацию полос обеспечивали за счет добавления в агарозу 2.5 мкл флуоресцентного красителя SybrGreen I (10000x). Напряжение составляло ~100 - 150 В. Разделение фрагментов ДНК контролировали с помощью камеры с ультрафиолетовой лампой.
Выделение ДНК из агарозного геля проводили с помощью набора реагентов «Cleanup mini» компании «Евроген». Из геля вырезали нужную полосу, визуализированную SybrGreen I, и помещали ее в пробирку. К гелю добавляли 3-хкратный объем «Связывающего буфера» (из расчета, что 100 мкг геля имеет объем 100 мкл), инкубировали при 55°C до полного растворения геля. Полученный раствор переносили в колонку для связывания ДНК, колонку ставили в собирательную пробирку. Собирательную пробирку с колонкой центрифугировали при максимальных оборотах в настольной центрифуге в течение 30 с (все дальнейшие центрифугирования проводили при максимальных оборотах). Проскок выливали, добавляли в колонку 700 мкл промывочного раствора и центрифугировали 30 с. Проскок выливали и центрифугировали колонку в течение 1 мин. После этого помещали колонку в новую пробирку, добавляли 30 мкл элюирующего буфера и центрифугировали 30 с. Полученную ДНК хранили при температуре -20°C.
Приготовление векторов и вставок. Перед легированием вектор и вставки обрабатывались эндонуклеазами рестрикции FastDigest KpnI и FastDigest HindIII, «Thermo Scientific».
Смешивали все необходимые компоненты реакции и инкубировали в течение 1 ч при 37°C. После этого ферменты инактивировали нагреванием при 80°C в течение 10 мин. После этого рестриктную смесь с плазмидной ДНК наносили на агарозный гель и выделяли нужный фрагмент с геля, описанным выше способом.
Лигирование. Лигирование проводили с помощью T4 DNA ligase («Thermo Scientific»). К 10-икратному T4 DNA ligase buffer добавляли 50 - 100 нг линеаризованной плазмиды и вставку в молярном соотношении с плазмидой 5:1, T4 ДНК лигазу до концентрации 0.05 ед./мкл, нужное количество воды. Лигазную смесь оставляли на ночь при температуре +4°C.
Получение компетентных клеток E.coli и их трансформация. 250 мл среды LB инокулировали свежей колонией клеток и растили культуру при 18°C и умеренном перемешивании до A600 ~0.4. Клетки охлаждали во льду и собирали центрифугированием при 5000 об/мин 10 мин при 4°C в роторе JA-14 (Beckman). После этого клетки дважды промывали буфером TB. Для этого осадок клеток ресуспендировали в 30 мл охлажденного TB буфера, инкубировали при слабом перемешивании во льду в течение 10 мин и центрифугировали при 5000 об/мин 10 мин при 4°C. После последнего центрифугирования клетки снова ресуспендировали в буфере TB, добавляли ДМСО до 3.5% и затем - до конечной концентрации 7% с 10-иминутным интервалом при 0°C при слабом перемешивании. Аликвоты суспензии клеток по 100 мкл замораживали в жидком азоте и хранили при температуре -80°C.
Для трансформации суспензию компетентных клеток размораживали на льду, добавляли 20 - 50 нг плазмиды или 5 - 10 мкл лигазной смеси, выдерживали на льду 20 мин. Смесь помещали в термостат, нагретый до 42°C, выдерживали в течение 40 - 50 с, после этого добавляли 300 мкл среды LB и инкубировали в течение 1 ч при 37°C. Половину клеток высевали на чашку Петри с твердой средой LB, содержавшей ампициллин. Инкубировали при 37°C в течение 16 ч. Выбор колоний со вставкой проводили с помощью бело-голубой селекции.
Выделение плазмидной ДНК. Плазмиды из ночной культуры выделяли с помощью набора реагентов Plasmid Miniprep фирмы Евроген из E. coli. Для этого растили 2 - 5 мл ночной культуры с одной колонии, клетки осаждали в течение 2 мин при 8000g в настольной микроцентрифуге. Отбирали среду, клетки ресуспендировали в 250 мкл «Ресуспендирующего раствора», добавляли 250 мкл «Лизирующего раствора» и осторожно перемешивали, пока лизат не становился прозрачным. Добавляли 350 мкл «Нейтрализующего раствора», перемешивали, инкубировали в течение 1 мин. От дебриса избавлялись центрифугированием при максимальных оборотах 10 мин в настольной микроцентрифуге (дальнейшие центрифугирования выполняли при максимальных оборотах). Супернатант перемещали в колонку в собирательной пробирке и центрифугировали в течение 30 с. Удаляли фильтрат из собирательной пробирки. Делали две промывки по 300 мкл «Промывочного раствора» с центрифугированием колонки по 30 с. После последней промывки центрифугировали пустую колонку в течение 1 мин. После этого колонку переносили в новую пробирку, добавляли 50 мкл «Элюирующего раствора», инкубировали при комнатной температуре в течение 1 мин и центрифугировали в течение 1 мин. Полученные плазмиды хранили при температуре -20°C.
Рестриктный анализ. Для подтверждения наличия вставки проводился рестриктный анализ плазмид с использованием рестриктазы FastDigest PvuII, «Thermo Scientific». Для рестрикции брали 200 нг плазмиды, 2 мкл 10× буфера FastDigest Green и 5 ед. активности фермента на 20 мкл реакции. Инкубировали при 37°C в течение 1 ч. Продукты анализировали в 1.2 масс.% агарозном геле.
Сайт-направленный мутагенез. Для направленного мутагенеза проводили ПЦР-амплификацию полученной плазмиды pUC19_235_C228Т с использованием полимераз Pfu DNA Polymerase и DreamTaq DNA Polymerase, «Thermo Scientific».
В пробирке емкостью 0.2 мл составляли следующую ПЦР-смесь:
Параметры амплификации: 95°C в течение 30 с - 1 цикл; 95°C - 50 с, 65°C - 50 c, 68°C - 4мин - 18 циклов; 68°C в течение 7 мин - 1 цикл.
После ПЦР в реакционную смесь вносили 0.5 мкл DpnI (10 U/μl, Thermo Scientific) и инкубировали при 37°C в течение 16 ч. Инактивировали фермент при 80°C 20 мин. Далее трансформировали компетентные клетки E.coli 10 мкл реакционной смеси и высевали на чашку Петри. Белые колонии пересевали в жидкую среду, выделяли из клеток плазмиды и проверяли наличие вставки с помощью секвенирования методом Сэнгера.
Определение первичной структуры плазмидной ДНК. Нуклеотидные последовательности при создании и использовании генно-инженерных конструкций определяли с помощью автоматического секвенатора ABI prism 3100-Avant genetic Analyzer (4-х капиллярный).
ПЦР в реальном времени. ПЦР в реальном времени проводилось с использованием Maxima hot start PCR Master mix, «Thermo Scientific». 2xPCR Master mix добавляли праймеры до концентрации 0.9 мкМ, 5 - 50 нг матрицы, 7-deaza-dGTP, флуоресцентные зонды до концентрации 250 нМ и доводили до нужного объема. Температура отжига и параметры амплификации варьировались в зависимости от эксперимента, время элонгации рассчитывали при условии, что Taq полимераза синтезирует ДНК со скоростью 1000 нуклеотидов в минуту.
Цифровая капельная ПЦР. Для постановки цкПЦР готовили следующую реакционную смесь, объемом 22 мкл:
ПЦР-амплификация для обнаружения мутаций С228Т и С250Т происходила отдельно, параметры амплификации: 95°C в течение 10 мин; 40 циклов 94°C - 30 с, скорость изменения температуры 2.5°C/сек., 54°C для C228T (64°C для C250T) в течение 30 с; 98°C - 10 мин, затем хранение при температуре 12°C после завершения амплификации. До регистрации флуоресцентного сигнала плашку с ПЦР-продуктом хранили не более недели при температуре +4°C.
Генерация капель осуществлялась с помощью автоматического генератора QX200, Biorad. Интенсивность флуоресценции в каждой капле измеряли с помощью QX200 Droplet reader, Biorad. Анализ результатов проводили на программном обеспечении QuantaSoftTM Analysis Pro 1.0.596 software, Biorad.
Согласно принципиальной схеме обнаружения мутаций (Фиг. 5) для детектирования каждой из мутаций использовали 2 пары флуоресцентных зондов: одна пара зондов с меткой родамин 6Ж (R6G) (Фиг. 6) к смысловой и матричной цепи дикого типа (TERT_250_wt_prb1, TERT_250_wt_prb2 для дикого типа в положении С250Т, TERT_228_wt_prb1, TERT_228_wt_prb2 для дикого типа в положении С228Т), вторая пара зондов с меткой флуоресцеин (FAM) к смысловой и матричной цепи мутантного типа (TERT_250_mut_prb1 и TERT_250_mut_prb2 на мутацию С250Т и TERT_228_mut_prb1 и TERT_228_mut_prb2 на мутацию С228Т). Вначале ПЦР температуру увеличивали для того, чтобы двойная спираль ДНК денатурировала. В этот момент сигнал от флуоресцентного красителя на 5’-конце зонда гасился нефлуоресцентным гасителем, расположенным на 3’-конце пробы. На следующем этапе температура снижалась, позволяя праймерам и зондам образовывать дуплексы с целевой последовательностью. Taq-полимераза синтезировала комплементарную ДНК на денатурировавшей цепи. Когда полимераза достигала зонда, она расщепляла зонд за счет своей 5’ → 3’ экзонуклеазной активности, отделяя флуорофор от гасителя. С каждым циклом ПЦР высвобождалось все больше молекул флуорофора, что приводило к возрастанию интенсивности флуоресценции, пропорциональной количеству молекул синтезируемого ампликона. Использовали рекомендованный размер ампликона 60 - 200 п.о. (65 п.о. для С228Т и 92 п.о. для C250Т), выбирали участок для амплификации с 40-60% содержанием GC-пар и избегали регионов со сложной вторичной структурой. Поскольку содержание GC-пар в получаемых ампликонах 77-80% (Фиг. 7), для эффективной работы полимераз в реакционную смесь добавляли GC-энхансеров, нарушающие вторичную структуру и способствующие нормальному разделению цепей ДНК. Для этих целей использовали 7-deaza-dGTP, который может включаться в состав синтезируемой цепи ДНК-полимеразами. Поскольку в седьмом положении цикла в 7-deaza-dGTP отсутствует атом азота, включение 7-deaza-dGTP в состав ДНК препятствовало образованию G-квадруплексов в GC-богатых участках. На Фигуре 8 представлен результат анализа системы, состоящей из плазмиды pUC19_235_wt дикого типа в количестве 5000 копий на 20 мкл реакционной смеси, и плазмиды pUC19_235_C228Т в количестве 1250 копий на 20 мкл реакционной смеси. Анализ результатов эксперимента проводился с помощью программного обеспечения QuantaSoftTM analysis pro. Пороговое значение для канала 1 (мутантная проба С228Т), составило 4000. Для канала 2 (проба дикого типа С228Т) пороговое значение находилось на уровне 2500. Область с синими точками представляет собой капли с плазмидами мутантного типа, область с зелеными точками - капли с плазмидами дикого типа, область с оранжевыми точками - капли, содержащие плазмиды обоих типов (Фиг. 8).
При оптимизации системы обнаружения мутации С228Т, состоящей из плазмид pUC19_235_C228Т и pUC19_235_wt, максимальное разрешение между кластерами достигалось при температуре 54°С. При этой температуре при 100% содержании в системе лишь плазмиды дикого типа в количестве 2500 положительных капель не наблюдается ложноположительных результатов (капель) в FAM-канале. Таким образом, подобранные условия обеспечивают 100% селективность обнаружения при условии отсутствия контаминации анализируемой смеси последовательностями мутантного типа. Далее был проведен анализ систем, содержащих плазмиду с мутацией С228Т на фоне избытка плазмиды дикого типа, концентрация которой постоянна и составляет 5000 копий на 20 мкл реакционной смеси. (Фиг. 9). Делая поправку на специфичность и контрольный образец, чувствительность составила 2%.
Подобранные условия (температурный режим, концентрация 7-deaza-dGTP, флуоресцентные TaqMan зонды и праймеры) проведения цифровой капельной ПЦР, позволили достичь селективного обнаружения последовательности промотора гена hTERT с мутациями С228Т и C250T, что было необходимо для валидации результатов, полученных методом спектроскопии ГКР. Однако ГКР-спектроскопия позволяет достичь более низких пределов обнаружения и наиболее важно, что использование этого метода не требует предварительной энзиматической амплификации мишени.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Федеральное государственное бюджетное образовательное учреждение
высшего профессионального образования "Московский государственный
университет имени М.В.Ломоносова" (МГУ)
<120> Набор для диагностики рака мочевого пузыря с помощью мутаций С228Т
и C250T в промоторе гена HTERT и способ ее осуществления
<160> SEQ ID NО:1
<210> 1
<211> 19
<212> GnuS
<213> Artificial sequence
<400> 1
1 TGCCTTTTGG GGACGGATA
<160> SEQ ID NО:2
<210> 2
<211> 20
<212> GnuS
<213> Artificial sequence
<400> 2
1 AATGATCTTA ACCAAGTATA
<160> SEQ ID NО:3
<210> 3
<211> 17
<212> GnuS
<213> Artificial sequence
<400> 3
1 TCGGTTGGGG AGCCTAT
<160> SEQ ID NО:4
<210> 3
<211> 19
<212> GnuS
<213> Artificial sequence
<400> 4
1 TGGCTAATCC CCTCGGATA
<---



Изобретение относится к области биотехнологии. Предложены набор и способ для диагностики рака мочевого пузыря с помощью мутаций C228T и C250T в промоторе гена hTERT. Набор включает сенсор и растворы меченных красителем с пределом обнаружения не более 1 мкМ олигонуклетидов GnuS-C228T-метка и GnuS-C250T-метка. Способ включает забор биологического образца, где одну часть образца смешивают с раствором олигонуклеотида GnuS-C228T-метка, а другую часть с раствором GnuS-C250T-метка, проведение их гибридизации с последующим определением мутаций ДНК C228T и C250T. Изобретения обеспечивают достижение высокой точности проведения диагностики рака мочевого пузыря на ранних стадиях развития опухоли. 2 н. и 16 з.п. ф-лы, 16 ил., 2 табл., 2 пр.
1. Набор для диагностики рака мочевого пузыря с помощью мутаций C228T и C250T в промоторе гена hTERT с использованием спектроскопии гигантского комбинационного рассеяния (спектроскопия ГКР), включающий твердофазный планарный сенсор, выполненный из диэлектрического химически инертного материала и покрытия на основе наночастиц благородных металлов, толщиной 1-10 мкм; и растворы меченных красителем с пределом обнаружения не более 1 мкМ олигонуклетидов GnuS-C228T-метка и GnuS-C250T-метка, где Gnus – последовательность, состоящая из 15-20 аминокислот, из которых не менее 20% составляют цитозин (C) и гуанин (G).
2. Набор по п.1, характеризующийся тем, что Gnus представляет собой нуклеотидную последовательность SEQ ID №1 - №4.
3. Набор по п.1, характеризующийся тем, что покрытие имеет наноструктуру с шероховатостью не менее 2 нм.
4. Набор по п.1, характеризующийся тем, что краситель с пределом обнаружения не более 1 мкМ представляет собой родаминовый, цианиновый, ксантеновый и диазо-краситель, метиленовый синий, кристаллический фиолетовый.
5. Набор по п.1, характеризующийся тем, что наночастицы благородных металлов выполенены из серебра, платины, золота.
6. Набор по п.2, характеризующийся тем, что наночастицы имеют размеры 20-90 нм.
7. Набор по п.1, характеризующийся тем, что диэлектрический химически инертный материал для покрытия наночастицами представляет собой силикатное стекло.
8. Набор по п.1, характеризующийся тем, что концентрация GnuS-C228T-метка или GnuS-C250T-метка олигонуклеотида в растворе составляет от 10-6 до 10-10 М.
9. Способ диагностики рака мочевого пузыря с использованием набора по п.1, включающий забор биологического образца, одну часть которого смешивают с раствором олигонуклеотида GnuS-C228T-метка, а другую часть с раствором GnuS-C250T-метка, проводят их гибридизацию с последующим определением мутаций ДНК C228T и C250T, посредством нанесения полученных смесей на поверхность сенсора, облучением монохроматическим пучком лазера с длиной волны 485-785 нм, с регистрацией сигнала комбинационного рассеяния, при этом о качественном и количественном содержании мутаций ДНК C228T и C250T судят по положению и интенсивности полос, характерных для метки-красителя.
10. Способ по п.9, характеризующийся тем, что в качестве биологического образца используют мочу пациента.
11. Способ по п.9, характеризующийся тем, что количество ГКР-меченного олигонуклеотида используют в объемном соотношении от 1:5 до 5:1 к анализируемому образцу, содержащему мутации ДНК C228T и C250T.
12. Способ по п.9, характеризующийся тем, что проводят предварительную гибридизацию последовательностей нуклеиновых кислот с раствором, меченных красителем.
13. Способ по п.12, характеризующийся тем, что гибридизацию проводят путем смешения анализируемого и раствора, меченного красителем, их нагрева и выдерживания в течение 5-10 мин при температуре 95°С, с последующим охлаждением до комнатной температуры.
14. Способ по п.9, характеризующийся тем, что используют анализируемые биологические образцы, смешанные с раствором олигонуклеотида GnuS-C228T-метка или GnuS-C250T-метка путем их накапывания на поверхность наночастиц.
15. Способ по п.9, характеризующийся тем, что используют образцы объемом 10 мкл с концентрацией мутаций ДНК C228T и C250T 10-15 – 10-2 М.
16. Способ по п.9, характеризующийся тем, что используют монохроматическое лазерное излучение мощностью, не превышающей 200 мBт, в течение до 10-60 с.
17. Способ по п.10, характеризующийся тем, что о наличии и содержании мутаций ДНК C228T и C250T в образце судят по положению и интенсивности полос при 1385, 1470 и 1590 см-1 с допустимой величиной погрешности не более 10 см-1.
18. Способ по п.9, характеризующийся тем, что для определения количественного содержания мутаций предварительно строят калибровочные кривые.
| СПОСОБ ОБНАРУЖЕНИЯ И ОПРЕДЕЛЕНИЯ ДНК С ЗАДАННОЙ ПОСЛЕДОВАТЕЛЬНОСТЬЮ МЕТОДОМ СПЕКТРОСКОПИИ ГИГАНТСКОГО КОМБИНАЦИОННОГО РАССЕЯНИЯ | 2019 |
|
RU2723160C1 |
| Токарный резец | 1924 |
|
SU2016A1 |
| Способ восстановления спиралей из вольфрамовой проволоки для электрических ламп накаливания, наполненных газом | 1924 |
|
SU2020A1 |

