ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[001] Настоящая заявка испрашивает приоритет на основании предварительной заявки на патент США № 62/188846, поданной 6 июля 2015 года и предварительной заявки на патент США № 62/218672, поданной 15 сентября 2015, содержание каждой из которых в полном объеме включено в настоящий документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
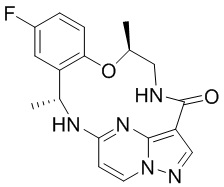
[002] Настоящее изобретение относится к полиморфным формам (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5Н)-она, подходящим для лечения заболеваний, таких как рак, у млекопитающих. Также настоящее изобретение относится к композициям, содержащим указанные полиморфные формы, к способам применения указанных композиций для лечения заболеваний, таких как рак, у млекопитающих, в частности у человека.
УРОВЕНЬ ТЕХНИКИ
[003] Соединение (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5Н)-он (также именуемое в настоящем описании как «соединение I»), представленное формулой I,
I
[004] представляет собой высокоактивный низкомолекулярный мультитаргетный ингибитор киназ, демонстрирующий активность в отношении киназы ALK (киназы анапластической лимфомы) дикого типа и мутантной киназы ALK, киназы ROS1 (рецепторной тирозинкиназы протоонкогена ROS1) дикого типа и мутантной киназы ROS1, киназ семейства TRK (тропомиозин-родственные рецепторные тирозинкиназы), киназы JAK2 из семейства Янус-киназ, SRC (семейства Src белковых тирозинкиназ (SFK)) и FAK (фокальной адгезионной киназы). Соединение I обладает свойствами, включая противоопухолевые свойства, которые фармакологически опосредуются через ингибирование рецепторов тирозинкиназы. Соединение I описано в международной заявке № PCT/US2015/012597, которая включена в настоящее описание посредством ссылки во всей полноте.
[005] Протеинкиназы являются ключевыми регуляторами роста, пролиферации и выживания клеток. Было показано, что различные заболевания, такие как рак, боль, неврологические заболевания, аутоиммунные заболевания и воспаление, опосредуются рецепторными тирозинкиназами, такими как ALK, ROS1, TRK, JAK2, SRC и FAK. Например, генетические и эпигенетические изменения могут накапливаться в раковых клетках и приводить к аномальной активации путей передачи сигналов, что приводит к злокачественным процессам. Manning, G. et al., Science 2002, 298, 1912-1934. Фармакологическое ингибирование этих сигнальных путей открывает перспективные возможности для направленной терапии рака. Sawyers, C., Nature 2004, 432, 294-297.
[006] Хотя соединение I применяют при лечении заболеваний, связанных с рецепторными тирозинкиназами, такими как ALK, ROS1, TRK, JAK2, SRC и FAK, было бы предпочтительно иметь полиморфные формы, обладающие улучшенными свойствами, такими как улучшенная кристалличность, растворимость и/или сниженная гигроскопичность, с сохранением при этом химической и энантиомерной стабильности.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[007] В одном аспекте настоящее изобретение относится к кристаллической форме (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5H)-она.
[008] В другом варианте реализации кристаллическая полиморфная форма (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5Н)-она является безводной.
[009] В другом варианте реализации настоящего изобретения кристаллическая полиморфная форма 1 соединения I может быть представлена формулой
I.
[010] В еще одном варианте реализации настоящего изобретения кристаллическая полиморфная форма имеет порошковую рентгенограмму, содержащую пик при угле дифракции (2θ) 27,4±0,1. В еще одном варианте реализации настоящего изобретения кристаллическая полиморфная форма имеет порошковую рентгенограмму, содержащую пики при углах дифракции (2θ) 9,4±0,1 и 27,4±0,1. В еще одном варианте реализации настоящего изобретения кристаллическая полиморфная форма имеет порошковую рентгенограмму, содержащую пики при углах дифракции (2θ) 9,4±0,1, 18,8±0,1 и 27,4±0,1. В еще одном варианте реализации настоящего изобретения кристаллическая полиморфная форма имеет порошковую рентгенограмму, содержащую пики при углах дифракции (2θ) 9,4±0,1, 16,5±0,1, 18,8±0,1 и 27,4±0,1. В еще одном варианте реализации настоящего изобретения кристаллическая полиморфная форма имеет порошковую рентгенограмму, содержащую пики при углах дифракции (2θ) 9,4±0,1, 16,5±0,1, 18,8±0,1, 22,8±0,1 и 27,4±0,1. В еще одном варианте реализации настоящего изобретения кристаллическая полиморфная форма имеет порошковую рентгенограмму, содержащую пики при углах дифракции (2θ) 9,4±0,1, 16,1±0,1, 16,5±0,1, 18,8±0,1, 22,8±0,1 и 27,4±0,1. В еще одном варианте реализации настоящего изобретения кристаллическая полиморфная форма имеет порошковую рентгенограмму, содержащую пики при углах дифракции (2θ) 9,4±0,1, 16,1±0,1, 16,5±0,1, 18,8±0,1, 21,2±0,1, 22,8±0,1 и 27,4±0,1.
[011] В еще одном аспекте кристаллическая форма имеет порошковую рентгенограмму, содержащую пики с углами дифракции (2θ) по существу такими же, как показано на фиг. 1.
[012] Настоящее изобретение также относится к фармацевтической композиции, содержащей полиморфную форму 1 соединения I формулы
I.
[013] Настоящее изобретение также относится к капсуле, содержащей описанные в настоящем документе фармацевтические композиции.
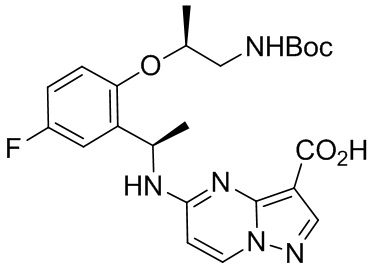
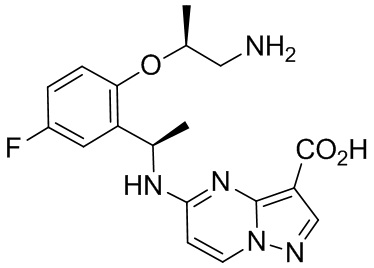
[014] В другом аспекте настоящее изобретение относится к способу лечения заболевания, особенно рака, у млекопитающего, включая человека, причем указанный способ включает введение указанному млекопитающему терапевтически эффективного количества полиморфной формы 1 соединения (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10] бензоксатриазациклотридецин-4(5H)-она, описанного в настоящем документе, или фармацевтической композиции, содержащей полиморфную форму 1 соединения (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5H)-она, описанного в настоящем документе.
[015] В одном варианте реализации настоящее изобретение относится к способу лечения аномального роста клеток у млекопитающего, включая человека, нуждающегося в таком лечении, включающему введение указанному млекопитающему терапевтически эффективного количества полиморфной формы 1 свободного основания соединения I. В другом варианте реализации настоящего изобретения аномальный рост клеток опосредуется по меньшей мере одной генетически измененной тирозинкиназой.
[016] В другом варианте реализации настоящего изобретения указанный аномальный рост клеток опосредуется ALK, ROS1, TRK, JAK2, SRC, FAK или их комбинацией. В другом варианте реализации аномальный указанный рост клеток опосредуется мутантной ALK дикого типа. В другом варианте реализации указанный аномальный рост клеток опосредуется мутантной ROS1 дикого типа. В другом варианте реализации указанный аномальный рост клеток опосредуется мутантной TRK дикого типа. В другом варианте реализации указанный аномальный рост клеток опосредуется мутантной JAK2 дикого типа. В другом варианте реализации указанный аномальный рост клеток опосредуется мутантной SRC дикого типа. В другом варианте реализации указанный аномальный рост клеток опосредуется мутантной FAK дикого типа.
[017] В другом варианте реализации указанный аномальный рост клеток представляет собой рак. В другом варианте реализации указанный рак выбран из группы, состоящей из рака легкого, немелкоклеточного рака легкого, мелкоклеточного рака легкого, рака кости, рака поджелудочной железы, рака кожи, рака головы или шеи, гепатоцеллюлярной карциномы, кожной или внутриглазной меланомы, рака матки, рака яичников, рака прямой кишки, рака анальной области, рака желудка, рака толстой кишки, рака молочной железы, карциномы фаллопиевых труб, карциномы эндометрия, карциномы шейки матки, карциномы влагалища, карциномы вульвы, болезни Ходжкина, рака желудка и рака пищевода и желудка, рака эндокринной системы, рака щитовидной железы, рака паращитовидной железы, рака надпочечников, саркомы мягких тканей, рака уретры, рака полового члена, рака предстательной железы, хронического или острого лейкоза, лимфоцитарных лимфом, таких как анапластическая крупноклеточная лимфома, рака мочевого пузыря, рака почки или мочеточника, почечно-клеточной карциномы, карциномы почечной лоханки, новообразований центральной нервной системы (ЦНС), глиобластомы, первичной лимфомы ЦНС, опухолей оси позвоночника, глиомы ствола головного мозга, аденомы гипофиза, воспалительных миофибробластических опухолей и их комбинаций.
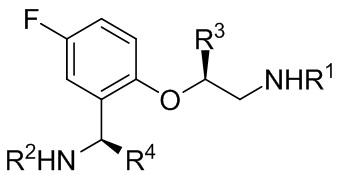
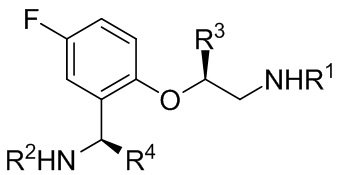
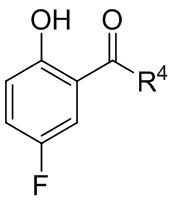
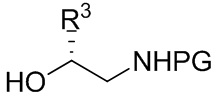
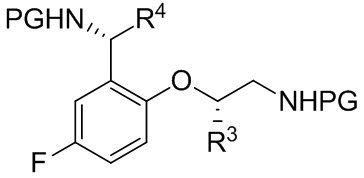
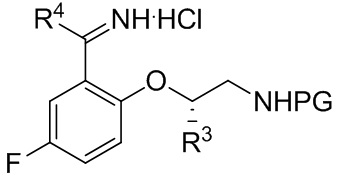
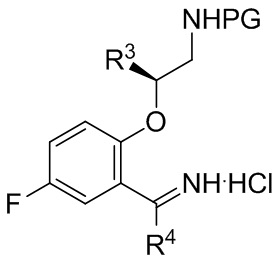
[018] В другом аспекте настоящее изобретение относится к соединению формулы II
II
[019] где каждый из R1 и R2 независимо представляет собой H или PG, и каждый из R3 и R4 независимо представляет собой C1-C4 алкил.
[020] В другом аспекте настоящее изобретение относится к способам получения соединения формулы B
 .
.
B
[021] Дополнительные варианты реализации, признаки и преимущества настоящего изобретения будут очевидны из последующего подробного описания и описания вариантов реализации настоящего изобретения. Соединения согласно настоящему изобретению могут быть описаны как варианты реализации в любом из нижеследующих пронумерованных пунктов. Следует понимать, что любой из описанных здесь вариантов реализации можно использовать совместно с любыми другими вариантами реализации, описанными в настоящем документе, в той степени, в которой варианты реализации не противоречат друг другу.
[022] 1. Кристаллическая полиморфная форма (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5Н)-она.
[023] 2. Кристаллическая полиморфная форма по пункту 1, отличающаяся тем, что указанная кристаллическая форма представляет собой полиморфную форму свободного основания (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5Н)-она.
[024] 3. Кристаллическая полиморфная форма по пунктам 1 или 2, имеющая порошковую рентгенограмму, содержащую пик при угле дифракции (2θ) 27,4±0,1.
[025] 4. Кристаллическая полиморфная форма по любому из пунктов 1-3, имеющая порошковую рентгенограмму, содержащую пики при углах дифракции (2θ) 9,4±0,1 и 27,4±0,1.
[026] 5. Кристаллическая полиморфная форма по любому из пунктов 1-4, имеющая порошковую рентгенограмму, содержащую пики при углах дифракции (2θ) 9,4±0,1, 18,8±0,1 и 27,4±0,1.
[027] 6. Кристаллическая полиморфная форма по любому из пунктов 1-5, имеющая порошковую рентгенограмму, содержащую пики при углах дифракции (2θ) 9,4±0,1, 16,5±0,1, 18,8±0,1 и 27,4±0,1.
[028] 7. Кристаллическая полиморфная форма по любому из пунктов 1-6, имеющая порошковую рентгенограмму, содержащую пики при углах дифракции (2θ) 9,4±0,1, 16,5±0,1, 18,8±0,1, 22,8±0,1 и 27,4±0,1.
[029] 8. Кристаллическая полиморфная форма по любому из пунктов 1-7, имеющая порошковую рентгенограмму, содержащую пики при углах дифракции (2θ) 9,4±0,1, 16,1±0,1, 16,5±0,1, 18,8±0,1, 22,8±0,1 и 27,4±0,1.
[030] 9. Кристаллическая полиморфная форма по любому из пунктов 1-8, имеющая порошковую рентгенограмму, содержащую пики при углах дифракции (2θ) 9,4±0,1, 16,1±0,1, 16,5±0,1, 18,8±0,1, 21,2±0,1, 22,8±0,1 и 27,4±0,1.
[031] 10. Кристаллическая полиморфная форма (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5H)-она, имеющий порошковую рентгенограмму, по существу такую же, как показано на фиг. 1.
[032] 11. Фармацевтическая композиция, содержащая кристаллическую полиморфную форму по любому из предшествующих пунктов.
[033] 12. Способ лечения заболевания у млекопитающего, включающий введение указанному млекопитающему терапевтически эффективного количества кристаллической полиморфной формы по любому из пунктов 1-10.
[034] 13. Способ по пункту 12, отличающийся тем, что указанное млекопитающее представляет собой человека.
[035] 14. Способ по пункту 12 или 13, отличающийся тем, что указанное заболевание выбрано из группы, состоящей из рака, боли, неврологических заболеваний, аутоиммунных заболеваний и воспаления.
[036] 15. Способ по любому из пунктов 12-14, отличающийся тем, что указанное заболевание представляет собой рак.
[037] 16. Способ по пункту 15, отличающийся тем, что указанный рак выбран из групп, состоящих из рака легкого, немелкоклеточного рака легкого, мелкоклеточного рака легкого, рака кости, рака поджелудочной железы, рака кожи, рака головы или шеи, гепатоцеллюлярной карциномы, кожной или внутриглазной меланомы, рака матки, рака яичников, рака прямой кишки, рака анальной области, рака желудка, рака толстой кишки, рака молочной железы, карциномы фаллопиевых труб, карциномы эндометрия, карциномы шейки матки, карциномы влагалища, карциномы вульвы, болезни Ходжкина, рака желудка и рака пищевода и желудка, рака эндокринной системы, рака щитовидной железы, рака паращитовидной железы, рака надпочечников, саркомы мягких тканей, рака уретры, рака полового члена, рака предстательной железы, хронического или острого лейкоза, лимфоцитарных лимфом, таких как анапластическая крупноклеточная лимфома, рака мочевого пузыря, рака почки или мочеточника, почечно-клеточной карциномы, карциномы почечной лоханки, новообразований центральной нервной системы (ЦНС), глиобластомы, первичной лимфомы ЦНС, опухолей оси позвоночника, глиомы головного мозга, аденомы гипофиза, воспалительных миофибробластических опухолей и их комбинаций.
[038] 17. Способ по пункту 16, отличающийся тем, что указанный рак представляет собой немелкоклеточный рак легкого.
[039] 18. Соединение формулы II
II
[040] где каждый из R1 и R2 независимо представляет собой H или PG, и каждый из R3 и R4 независимо представляет собой C1-C4 алкил.
[041] 19. Соединение по пункту 18, отличающееся тем, что R1 и R2 представляют собой PG.
[042] 20. Соединение по пункту 18, отличающееся тем, что R2 представляет собой Н.
[043] 21. Соединение по пункту 18 или 19, отличающееся тем, что R1 представляет собой Н.
[044] 22. Соединение по пункту 18 или 20, отличающееся тем, что R1 представляет PG.
[045] 23. Соединение по пункту 18 или 21, отличающееся тем, что R2 представляет PG.
[046] 24. Соединение по любому из пунктов 18-23, отличающееся тем, что R3 и R4 представляют собой метил.
[047] 25. Соединение по любому из пунктов 18-24, отличающееся тем, что PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts.
[048] 26. Соединение по любому из пунктов 18-25, отличающееся тем, что PG представляет собой Boc.
[049] 27. Соединение формулы B-14
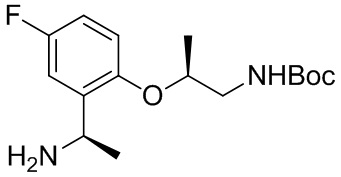
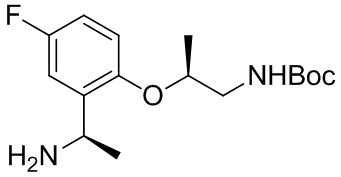
B-14.
[050] 28. Способ получения соединения формулы I
I
[051] включающий
[052] а. приведение соединения формулы А
A
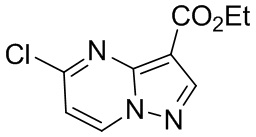
[053] в контакт с соединением формулы B-14
B-14
[054] в присутствии основания с получением соединения формулы С
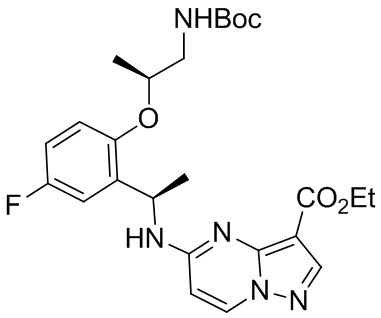 ; или
; или
C
[055] b. приведение соединения формулы С в контакт с неорганическим основанием с получением соединения формулы D
 ; или
; или
D
[056] c. приведение соединения формулы D в контакт с кислотой с получением соединения формулы E
 ; или
; или
E
[057] d. приведение соединения формулы E в контакт с основанием в присутствии фосфинатного реагента с получением соединения формулы I.
[058] 29. Способ получения соединения формулы В
B
[059] где
[060] PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и
[061] каждый из R3 и R4 независимо представляет собой C1-C4 алкил;
[062] включающий
[063] а. приведение соединения формулы B-1
B-1
[064] где R4 представляет собой C1-C4 алкил; в контакт с соединением формулы B-2R
B-2R
[065] где R3 представляет собой C1-C4 алкил, и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; в присутствии азодикарбоксилатного реагента и фосфинового реагента с получением соединения формулы B-3
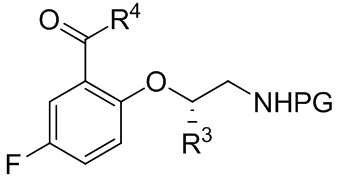
B-3
[066] где PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; или
[067] b. приведение соединения формулы B-3
B-3
[068] где PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; в контакт с (R)-2-метил-2-пропансульфинамидом с получением соединения формулы B-5
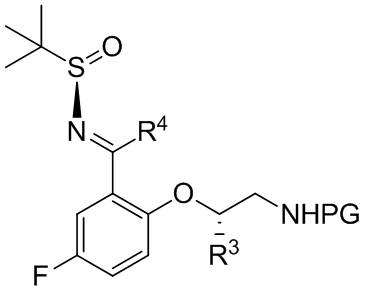
B-5
[069] где PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; или
[070] с. приведение соединения формулы B-5
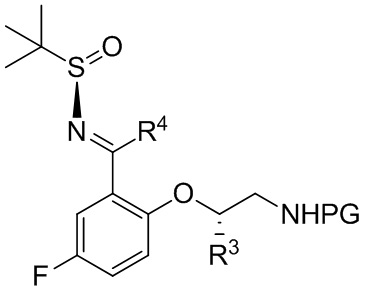
B-5
[071] где PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; в контакт с восстанавливающим агентом с получением соединения формулы B-6
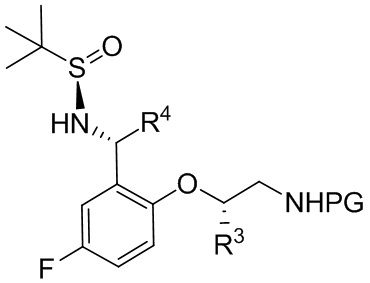
B-6
[072] где PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; или
[073] d. приведение соединения формулы B-6
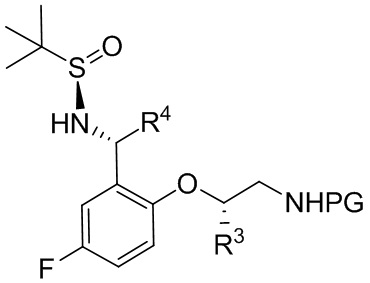
B-6
[074] где PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; в контакт с йодным реагентом с получением соединения формулы B.
[075] 30. Способ получения соединения формулы В
B
[076] где
[077] PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и
[078] каждый из R3 и R4 независимо представляет собой C1-C4 алкил;
[079] включающий
[080] a. взаимодействие соединения формулы B-7
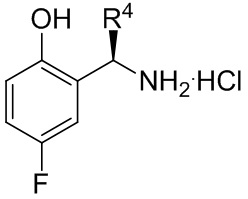
B-7
[081] где R4 представляет собой C1-C4 алкил; в условиях, подходящих для получения соединения формулы B-8
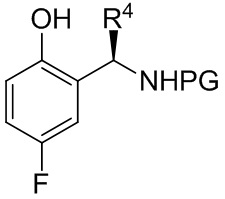
B-8
[082] где R4 представляет собой C1-C4 алкил; и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; или
[083] b. приведение соединения формулы B-8
B-8
[084] где R4 представляет собой C1-C4 алкил; и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; в контакт с соединением формулы B-2R
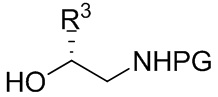
B-2R
[085] где R3 представляет собой C1-C4 алкил, и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; в присутствии азодикарбоксилатного реагента и фосфинового реагента с получением соединения формулы B-9
B-9
[086] где каждая PG независимо выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts, при условии, что все PG разные; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; или
[087] с. приведение соединения формулы B-9
B-9
[088] где каждая PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; при условии, что все PG разные; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; в контакт с неорганическим основанием с получением соединения формулы B.
[089] 31. Способ получения соединения формулы В
B
[090] где
[091] PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и
[092] каждый из R3 и R4 независимо представляет собой C1-C4 алкил;
[093] включающий
[094] a. взаимодействие соединения формулы B-10
B-10
[095] с соединением формулы B-2S
B-2S
[096] где R3 представляет собой C1-C4 алкил, и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; в основания с получением соединения формулы B-11
B-11
[097] где R3 представляет собой C1-C4 алкил, и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; или
[098] b. приведение соединения формулы B-11
B-11
[099] где R3 представляет собой C1-C4 алкил, и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; в контакт с нуклеофилом с получением соединения формулы B-12
B-12
[0100] где каждый из R3 и R4 независимо представляет собой C1-C4 алкил, и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; или
[0101] с. приведение соединения формулы B-12
B-12
[0102] где каждый из R3 и R4 независимо представляет собой C1-C4 алкил, и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; в контакт с восстанавливающим агентом с получением соединения формулы B.
[0103] 30. Способ получения соединения формулы В
B
[0104] где
[0105] PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и
[0106] каждый из R3 и R4 независимо представляет собой C1-C4 алкил;
[0107] включающий
[0108] a. взаимодействие соединения формулы B-12
B-12
[0109] где PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; в условиях, подходящих для получения соединения формулы B-13
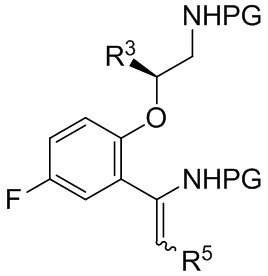
B-13
[0110] где каждая PG независимо выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts, при условии, что все PG разные; R3 представляет собой C1-C4 алкил; и R5 представляет собой C1-C3 алкил; или
[0111] b. приведение соединения формулы B-13
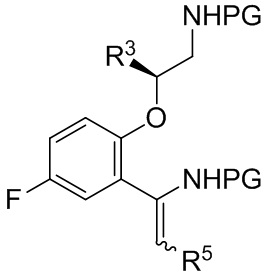
B-13
[0112] где каждая PG независимо выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts, при условии, что все PG разные; R3 представляет собой C1-C4 алкил; и R5 представляет собой C1-C3 алкил; в контакт с восстанавливающим агентом с получением соединения формулы B-9
B-9
[0113] где каждая PG независимо выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts, при условии, что все PG разные; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; или
[0114] с. приведение соединения формулы B-9
B-9
[0115] где каждая PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; при условии, что все PG разные; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; в контакт с неорганическим основанием с получением соединения формулы B.
ОПРЕДЕЛЕНИЯ
[0116] Используемый в настоящем документе термин «алкил» включает необязательно разветвленную цепь атомов углерода, содержащую от 1 до 4 атомов углерода, и подобные, которые могут упоминаться как «низший алкил». Иллюстративные алкильные группы включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил.
[0117] При использовании в настоящем документе, если не указано иное, термин «аномальный рост клеток» относится к росту клеток, который не зависит от нормальных регуляторных механизмов (например, отсутствие контактного торможения).
[0118] При использовании в настоящем документе, если не указано иное, термин «лечить» означает обращение вспять, облегчение, ингибирование прогресса (то есть, терапию, направленную на излечение) или предотвращение расстройства или состояния, к которому применяется такой термин, или одного или более симптомов указанного расстройства или состояния. При использовании в настоящем документе, если не указано иное, термин «лечение» относится к акту лечения, в том смысле, в каком термин «лечить» определен выше. «Профилактическое» лечение предназначено отсрочить развитие заболевания, симптома заболевания или медицинского состояния, подавить симптомы, которые могут появиться, или снизить риск развития или рецидива заболевания или симптома. «Терапия (лечение), направленная на лечение» включает снижение тяжести или подавление ухудшения существующего заболевания, симптома или состояния. Таким образом, лечение включает улучшение или предотвращение ухудшения существующих симптомов заболевания, предотвращение появления дополнительных симптомов, улучшение или предотвращение основополагающих системных причин симптомов, подавление расстройства или заболевания, например, остановку развития указанного расстройства или заболевания, облегчение указанного расстройства или заболевания, регресс указанного расстройства или заболевания, облегчение состояния, вызванного указанным заболеванием или расстройством, или прекращение симптомов указанного заболевания или расстройства.
[0119] Термин «субъект» относится к нуждающемуся в таком лечении пациенту, представляющему собой млекопитающее, такое как человек.
[0120] Используемый в настоящем документе термин «по существу один и тот же» применительно к положениям пика дифракции рентгеновских лучей означает, что учитывается типичное положение пика и вариабельность интенсивности. Например, специалист в данной области техники поймет, что положения пика (2θ) будут демонстрировать некоторую изменчивость от аппарата к аппарату, обычно до 0,1°. Кроме того, специалист в данной области техники поймет, что относительная интенсивность пиков будет обладать изменчивостью между различными аппаратами, а также изменчивостью из-за степени кристалличности, предпочтительной ориентации, поверхности подготовленного образца и других факторов, известных специалистам в данной области, и должна использоваться только в качестве качественной меры.
[0121] Используемый в настоящем документе термин «защитная группа» или «PG» относится к любой группе, про которую специалисту в данной области известно, что она может быть введена в молекулу посредством химической модификации функциональной группы, такой как амин или гидроксил, с получением хемоселективности в последующей химической реакции. Очевидно, что такие защитные группы могут быть впоследствии удалены из функциональной группы в более поздней точке синтеза, чтобы обеспечить дополнительную возможность прохождения реакции в указанных функциональных группах или, в случае конечного продукта, открыть указанную функциональную группу. Защитные группы описаны, например, в Wuts, P. G. M., Greene, T. W., Greene, T. W., & John Wiley & Sons. (2006). Greene's protective groups in organic synthesis. Hoboken, N.J: Wiley-Interscience. Специалист в данной области техники легко поймет условия химического процесса, при которых указанные защитные группы могут быть добавлены на функциональную группу. Соответствующие защитные аминогруппы, подходящие согласно настоящему изобретению, включают, но не ограничиваются ими, 9-флуоренилметилкарбонил (FMOC), трет-бутилкарбонил (Boc), бензилоксикарбонил (Cbz), ацетил (Ac), трифторацетил, фталимид, бензил (Bn), трифенилметил (тритил, Tr), бензилиден и п-толуолсульфонил (тозиламид, Ts).
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0122] На фиг. 1 приведена порошковая рентгенограмма кристаллической формы свободного основания (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5H)-она, полиморфная форма 1.
[0123] На фиг. 2 приведена термограмма дифференциальной сканирующей калориметрии (ДСК) кристаллической формы свободного основания (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5H)-она, полиморфная форма 1. (a) кривая ТГ; (b) кривая ТГ%.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0124] Прежде чем настоящее изобретение будет описано далее, следует понимать, что настоящее изобретение не ограничено конкретными описанными вариантами реализации, поскольку они могут, разумеется, меняться. Следует также понимать, что используемая здесь терминология предназначена только для описания конкретных вариантов реализации и не предназначена для ограничения настоящего изобретения, поскольку его объем ограничен только прилагаемой формулой изобретения.
[0125] Если не определено иначе, все технические и научные термины, используемые в настоящем документе, имеют значение, которое обычно понимается специалистом в данной области техники, к которой относится настоящее изобретение. Все патенты, заявки на патент, опубликованные заявки на патент и другие публикации, упоминаемые в настоящем документе, включены в настоящий документ в качестве ссылки во всей их полноте. Если определение, приведенное в настоящем разделе, противоречит или иным образом не согласуется с определением, изложенном в патенте, заявке на патент или другой публикации, включенной в настоящее описание посредством ссылки, то определение, изложенное в этом разделе, превалирует над определением, включенным в настоящее описание посредством ссылки.
[0126] Используемое в настоящем описании и в прилагаемой формуле изобретения существительное в единственном числе также относится и к существительному во множественном числе, если в контексте ясно не указано иначе. Кроме того, следует отметить, что формула изобретения может быть составлена так, чтобы исключить любой необязательный элемент. Таким образом, настоящее заявление предназначено для использования в качестве предварительного основания для использования такой исключительной терминологии, как «исключительно», «только» и т. п. относительно элементов формулы изобретения, или для использования «отрицательного» ограничения.
[0127] Уникальная физическая форма свободного основания (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5Н)-она была получена в соответствии с описанными в настоящем документе способами. Рентгенограмма полиморфной формы 1 свободного основания, полученная порошковой рентгеновской дифракцией (PXRD), приведена на фиг. 1, при этом соответствующие данные приведены в таблице 1.
Таблица 1
[0128] Термограмма ДКС для кристаллической полиморфной формы 1 приведена на фиг. 2.
[0129] В одном аспекте соединения и фармацевтические композиции согласно настоящему изобретению направлены на киназы тирозиновых рецепторов, в частности, ALK, ROS1, TRK, JAK2, SRC или FAK. Таким образом, указанные соединения и фармацевтические композиции можно применять для предотвращения, обращения вспять, замедления или ингибирования активности одной или нескольких указанных киназ. В некоторых вариантах реализации настоящего изобретения описаны способы лечения заболеваний, опосредованных одной или несколькими рецепторными тирозинкиназами.
[0130] Примерами указанных заболеваний являются рак, боль, неврологические заболевания, аутоиммунные заболевания и воспаление.
[0131] В некоторых вариантах реализации настоящего изобретения описаны способы лечения рака, включающие введение терапевтически эффективного количества кристаллической полиморфной формы 1 соединения I. Указанный рак выбран, но не ограничивается этим, из рака легкого и подобных, немелкоклеточного рака легкого, мелкоклеточного рака легкого, рака кости, рака поджелудочной железы, рака кожи, рака головы или шеи, гепатоцеллюлярной карциномы, кожной или внутриглазной меланомы, рака матки, рака яичников, рака прямой кишки, рака анальной области, рака желудка, рака толстой кишки, рака молочной железы, карциномы фаллопиевых труб, карциномы эндометрия, карциномы шейки матки, карциномы влагалища, карциномы вульвы, болезни Ходжкина, рака желудка и рака пищевода и желудка, рака эндокринной системы, рака щитовидной железы, рака паращитовидной железы, рака надпочечников, саркомы мягких тканей, рака уретры, рака полового члена, рака предстательной железы, хронического или острого лейкоза, лимфоцитарных лимфом, таких как анапластическая крупноклеточная лимфома, рака мочевого пузыря, рака почки или мочеточника, почечно-клеточной карциномы, карциномы почечной лоханки, новообразований центральной нервной системы (ЦНС), таких как глиобластома, первичная лимфома ЦНС, опухоли оси позвоночника, глиомы ствола головного мозга и подобных, аденомы гипофиза, воспалительных миофибробластических опухолей и их комбинаций. В других вариантах реализации настоящего изобретения указанные способы предназначены для лечения рака легкого или немелкоклеточного рака легкого.
[0132] В некоторых вариантах реализации настоящего изобретения описаны способы лечения или профилактики боли, включающие введение терапевтически эффективного количества кристаллической полиморфной формы 1 соединения I. Боль включает, например, боль по любой причине или любой этиологии, включая боль при раке, боль от химиотерапевтического лечения, невралгическую боль, боль от травмы или по другим причинам.
[0133] В некоторых вариантах реализации настоящего изобретения описаны способы лечения аутоиммунного заболевания, включающие введение терапевтически эффективного количества кристаллической полиморфной формы 1 соединения I. Аутоиммунные заболевания включают, например, ревматоидный артрит, синдром Шегрена, диабет типа I и волчанку. Примерами неврологических заболеваний являются болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склероз и болезнь Хантингтона.
[0134] В некоторых вариантах реализации настоящего изобретения описаны способы лечения воспалительных заболеваний, включающие введение терапевтически эффективного количества кристаллической полиморфной формы 1 соединения I. Примерами воспалительных заболеваний являются атеросклероз, аллергия и воспаление от инфекции или травмы.
[0135] В способах лечения согласно настоящему изобретению термин «эффективное количество» означает количество или дозу, достаточные для того, чтобы в целом приводить к желаемому терапевтическому эффекту у пациентов, нуждающихся в таком лечении. Эффективные количества или дозы соединений согласно настоящему изобретению могут быть установлены обычными методами, такими как моделирование, эскалация дозы или клинические испытания с учетом обычных факторов, например, способа или пути введения или доставки лекарственного средства, фармакокинетики агента, тяжести и течения инфекции, состояния здоровья пациента, расстройства, веса пациента, а также мнения лечащего врача. Примерная доза находится в диапазоне от примерно 0,1 мг до 1 г ежедневно или от примерно 1 мг до 50 мг ежедневно или от примерно 50 до 250 мг ежедневно или от примерно 250 мг до 1 г ежедневно. Общую дозу можно назначать в виде единичной дозы или в разделенных единицах дозировки (например, дважды в сутки (BID), три раза в сутки (TID), четыре раза в сутки (QID)).
[0136] После того, как произошло улучшение в течении заболевания у пациента, дозу можно корректировать для профилактического или поддерживающего лечения. Например, дозировку, частоту введения, или и то, и другое, можно уменьшать в зависимости от симптомов до уровня, при котором сохраняется желаемый терапевтический или профилактический эффект. Конечно, если симптомы уменьшены до соответствующего уровня, лечение можно прекратить. Однако, если симптомы повторяются, пациентам может потребоваться периодическое лечение на длительной основе. Пациентам также может потребоваться хроническое лечение на длительной основе.
Фармацевтические композиции
[0137] Настоящее изобретение также относится к фармацевтическим композициям, содержащим полиморфную форму свободного основания 1 соединения I, описанного в настоящем документе. Фармацевтические композиции согласно настоящему изобретению могут, например, быть в форме, пригодной для перорального введения, в виде таблетки, капсулы, пилюли, порошка, композиций с замедленным высвобождением, раствора, суспензии, для парентеральной инъекции в виде стерильного раствора, суспензии или эмульсии, для местного введения в виде мази или крема или для ректального введения в виде суппозитория. Фармацевтическая композиция может быть в стандартных лекарственных формах, подходящих для однократного введения точных доз. Фармацевтическая композиция может содержать обычные фармацевтически приемлемые вспомогательные вещества. Кроме того, фармацевтические композиции, описанные в настоящем документе, могут содержать другие лекарственные или фармацевтические агенты, носители, разбавители и т. д.
[0138] Фармацевтически приемлемое вспомогательное вещество представляет собой вещество, которое нетоксично и в остальном биологически пригодно для введения субъекту. Такие вспомогательные вещества облегчают введение описанных в настоящем документе соединений и совместимы с активным ингредиентом. Примеры фармацевтически приемлемых вспомогательных веществ включают стабилизаторы, смазывающие вещества, поверхностно-активные вещества, разбавители, антиоксиданты, связующие вещества, красители, наполнители, эмульгаторы или модификаторы вкуса. В предпочтительных вариантах реализации фармацевтические композиции согласно настоящему изобретению представляют собой стерильные композиции. Фармацевтические композиции можно получать с применением известных способов приготовления, которые становятся доступными специалистам в данной области.
[0139] Стерильные композиции также относятся к настоящему изобретению, включая композиции, которые согласуются с национальными и местными нормами, относящимся к указанным композициям.
[0140] Фармацевтические композиции и соединения, описанные в настоящем документе, могут быть составлены в виде растворов, эмульсий, суспензий или дисперсий в подходящих фармацевтических растворителях или носителях, или в виде пилюль, таблеток, пастилок, суппозиториев, саше, драже, гранул, порошков, порошков для восстановления или капсул с твердыми носителями в соответствии с общепринятыми способами, известными в данной области техники для получения различных лекарственных форм. Фармацевтические композиции согласно настоящему изобретению можно вводить подходящим путем введения, таким как пероральный, парентеральный, ректальный, назальный, местный или глазной, или посредством ингаляции. В некоторых вариантах реализации настоящего изобретения указанные композиции составлены для внутривенного или перорального введения.
[0141] Для перорального введения соединения согласно настоящему изобретению могут быть представлены в твердой форме, такой как таблетка или капсула, или в виде раствора, эмульсии или суспензии. Для приготовления пероральных композиций соединения согласно настоящему изобретению можно составлять таким образом, чтобы получать дозу, например, от примерно 0,1 мг до 1 г в сутки или от примерно 1 мг до 50 мг в сутки или от примерно 50 до 250 мг в сутки или примерно 250 мг до 1 г в сутки. Пероральные таблетки могут включать активный(ые) ингредиент(ы), смешанный(ые) с совместимыми фармацевтически приемлемыми вспомогательными веществами, такими как разбавители, дезинтегрирующие агенты, связывающие агенты, смазывающие агенты, подсластители, ароматизаторы, красители и консерванты. Подходящие инертные наполнители включают карбонат натрия и кальция, фосфат натрия и кальция, лактозу, крахмал, сахар, глюкозу, метилцеллюлозу, стеарат магния, маннит, сорбит и тому подобное. Примеры жидких пероральных вспомогательных веществ включают этанол, глицерин, воду и тому подобное. Типичными дезинтегрирующими агентами являются крахмал, поливинилпирролидон (ПВП), крахмалгликолят натрия, микрокристаллическая целлюлоза и альгиновая кислота. Связывающие агенты могут включать крахмал и желатин. Смазывающий агент, если он присутствует, может представлять собой стеарат магния, стеариновую кислоту или тальк. Если необходимо, указанные таблетки могут быть покрыты материалом, таким как глицерилмоностеарат или глицерилдистеарат, для задержки абсорбции в желудочно-кишечном тракте или могут быть покрыты энтеросолюбильным покрытием.
[0142] Капсулы для перорального введения включают твердые и мягкие желатиновые капсулы. Для приготовления твердых желатиновых капсул активный(ые) ингредиент(ы) можно смешивать с твердым, полутвердым или жидким разбавителем. Мягкие желатиновые капсулы можно получать путем смешивания активного ингредиента с водой, маслом, таким как арахисовое масло или оливковое масло, жидким парафином, смесью моно- и диглицеридов короткоцепочечных жирных кислот, полиэтиленгликолем 400 или пропиленгликолем.
[0143] Жидкости для перорального введения могут быть в форме суспензий, растворов, эмульсий или сиропов или могут быть лиофилизированы или представлены в виде сухого продукта для восстановления водой или другим подходящим носителем перед применением. Такие жидкие композиции могут необязательно содержать: фармацевтически приемлемые вспомогательные вещества, такие как суспендирующие агенты (например, сорбит, метилцеллюлоза, альгинат натрия, желатин, гидроксиэтилцеллюлоза, карбоксиметилцеллюлоза, гель стеарата алюминия и тому подобное); неводные носители, например, масло (например, миндальное масло или фракционированное кокосовое масло), пропиленгликоль, этиловый спирт или воду; консерванты (например, метил- или пропил-п-гидроксибензоат или сорбиновую кислоту); смачивающие агенты, такие как лецитин; и, при желании, ароматизаторы или красители.
[0144] Для парентерального применения, включая внутривенный, внутримышечный, внутрибрюшинный, интраназальный или подкожный пути, агенты согласно настоящему изобретению могут быть предоставлены в стерильных водных растворах или суспензиях, забуференных до подходящего рН и изотоничности, или в парентерально приемлемом масле. Подходящие водные носители включают раствор Рингера и изотонический хлорид натрия. Такие формы могут быть представлены в виде единичной дозы, такой как ампулы или одноразовые инъекционные устройства, в многодозовых формах, таких как флаконы, из которых можно набирать соответствующую дозу, или в твердой форме или предварительно концентрированной, которую можно применять для приготовления композиции для инъекций. Примерные дозы для инфузии составляют от 1 до 1000 мкг/кг/мин агента, смешанного с фармацевтическим носителем, в течение периода от нескольких минут до нескольких дней.
[0145] Для назального, ингаляционного или перорального введения предлагаемые фармацевтические композиции можно вводить, используя, например, состав для распыления, также содержащий подходящий носитель. Композиции согласно настоящему изобретению могут быть составлены для ректального введения в виде суппозитория.
[0146] Для местных применений соединения согласно настоящему изобретению предпочтительно составлены в виде кремов или мазей или аналогичного носителя, подходящего для местного применения. Для местного введения предлагаемые соединения можно смешивать с фармацевтическим носителем в концентрации от примерно 0,1% до примерно 10% лекарственного средства по отношению к носителю. Для другого способа введения агентов согласно настоящему изобретению можно применять композицию патча для осуществления трансдермальной доставки.
[0147] Способы получения различных фармацевтических композиций с определенным количеством активного соединения известны или будут очевидны для специалистов в данной области. Примеры см. в Remington's Pharmaceutical Sciences, Mack Publishing Company, Easter, Pa., 15th Edition (1975).
Комбинации лекарственных средств
[0148] Соединения согласно настоящему изобретению, описанные в настоящем документе, можно применять в фармацевтических композициях или способах в комбинации с одним или более дополнительных активных ингредиентов при лечении описанных в настоящем документе заболеваний и расстройств. Дополнительные активные ингредиенты включают другие терапевтические средства или агенты, которые смягчают неблагоприятные эффекты терапии, направленной на заболевания. Такие комбинации могут служить для повышения эффективности, облегчения других симптомов заболевания, уменьшения одного или нескольких побочных эффектов или уменьшения требуемой дозы соединения согласно настоящему изобретению. Дополнительные активные ингредиенты можно вводить в отдельной фармацевтической композиции или их можно включать в одну фармацевтическую композицию с соединением согласно настоящему изобретению. Дополнительные активные ингредиенты можно вводить одновременно с введением соединения согласно настоящему изобретению, или до введения, или после введения.
[0149] Агенты для комбинаций включают дополнительные активные ингредиенты, про которые известно, что они эффективны при лечении описанных здесь заболеваний и расстройств, в том числе агенты, активные против другой мишени, связанной с указанным заболеванием, или признанные таковыми. Например, композиции и составы согласно настоящему изобретению, а также способы лечения могут дополнительно содержать другие лекарственные средства или фармацевтические препараты, например, другие активные агенты, подходящие для лечения или паллиативного лечения целевых заболеваний или связанных с ними симптомов или состояний. Для показаний рака дополнительные агенты включают, но не ограничиваются ими, ингибиторы киназы, такие как ингибиторы EGFR (например, эрлотиниб, гефитиниб), ингибиторы Raf (например, вемурафениб), ингибиторы VEGFR (например, сунитиниб), ингибиторы ALK (например, кризотиниб), стандартные химиотерапевтические агенты, такие как алкилирующие агенты, антиметаболиты, противоопухолевые антибиотики, ингибиторы топоизомеразы, препараты платины, ингибиторы митоза, антитела, гормональная терапия или кортикостероиды. Для показаний боли подходящие дополнительные агенты включают противовоспалительные средства, такие как НПВС. Фармацевтические композиции согласно настоящему изобретению могут дополнительно содержать один или более таких активных агентов, и способы лечения могут дополнительно включать введение эффективного количества одного или нескольких таких активных агентов.
Способы синтеза
[0150] В некоторых вариантах реализации настоящее изобретение относится к способу получения соединения формулы I
I
[0151] включающему
[0152] (а) приведение в контакт соединения формулы А
A
[0153] с соединением формулы B
B-14
[0154] в присутствии основания с получением соединения формулы С
; или
C
[0155] (b) приведение соединения формулы С в контакт с неорганическим основанием с получением соединения формулы D
; или
D
[0156] (c) приведение в контакт соединения формулы D с кислотой с получением соединения формулы E
; или
E
[0157] (d) приведение в контакт соединения формулы E с основанием в присутствии фосфинатного реагента с получением соединения формулы I.
[0158] Будет понятно, что настоящее изобретение включает способы получения соединения формулы I, описанного в параграфах выше, включающие более одного из тех этапов, которые перечислены в качестве альтернативы. Соответственно, настоящее изобретение относится к способу получения соединения формулы I, включающему стадии (a) и (b). Альтернативно настоящее изобретение относится к способу получения соединения формулы I, включающему стадии (b) и (c). Альтернативно настоящее изобретение относится к способу получения соединения формулы I, включающему стадии (c) и (d). Альтернативно настоящее изобретение относится к способу получения соединения формулы I, включающему стадии (a), (b) и (c). Альтернативно настоящее изобретение относится к способу получения соединения формулы I, включающему стадии (b), (c) и (d). Альтернативно настоящее изобретение относится к способу получения соединения формулы I, включающему стадии (a), (b), (c) и (d).
[0159] На первом этапе (а) указанное основание может представлять собой любое органическое основание, такое как аминовое основание. Подходящие аминовые основания включают, но не ограничиваются ими, DIEA, TEA, трибутиламин, 2,6-лутидин, 2,2,6,6-тетраметилгуанидин и тому подобное. В некоторых вариантах реализации настоящего изобретения стадия (а) может быть проведена в присутствии полярного протонного растворителя, такого как спиртовой растворитель. Подходящие полярные протонные растворители включают, но не ограничиваются ими, MeOH, EtOH, iPrOH, n-BuOH, втор-BuOH и тому подобное. В некоторых вариантах реализации настоящего изобретения указанный полярный протонный растворитель представляет собой n-BuOH. В некоторых вариантах реализации настоящего изобретения стадию (а) можно осуществлять при температуре от примерно 50°C до примерно 150°С. В некоторых вариантах реализации настоящего изобретения температура составляет примерно 120°C.
[0160] На стадии (b) указанное неорганическое основание может представлять собой любое неорганическое основание, такое как гидроксидное основание. Подходящие гидроксидные основания включают, но не ограничиваются ими, гидроксид натрия, гидроксид лития и тому подобное. В некоторых вариантах реализации настоящего изобретения стадию (b) можно проводить в присутствии полярного протонного растворителя, полярного апротонного растворителя или их смеси. Подходящие полярные протонные растворители включают, но не ограничиваются ими, MeOH, EtOH, iPrOH, n-BuOH, втор-BuOH, H2O и тому подобное. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. В некоторых вариантах реализации настоящего изобретения стадию (b) можно проводить в смеси растворителей, такой как ТГФ/МеОН/Н2О. В некоторых вариантах реализации настоящего изобретения стадию (а) можно осуществлять при температуре от примерно 30°C до примерно 100°C. В некоторых вариантах реализации настоящего изобретения температура составляет примерно 70°C.
[0161] На стадии (с) указанная кислота может представлять собой сильную неорганическую кислоту, такую как HCl, такую как 2М HCl. В некоторых вариантах реализации настоящего изобретения указанная кислота может представлять собой раствор сильной кислоты в полярном апротонном растворителе, таком как Et2O. Например, кислота, подходящая для использования на стадии (с), может включать 2М HCl в Et2O. В некоторых вариантах реализации настоящего изобретения стадию (с) можно проводить в присутствии дополнительного полярного апротонного растворителя. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. В некоторых вариантах реализации настоящего изобретения указанный полярный протонный растворитель представляет собой ДХМ. В некоторых вариантах реализации настоящего изобретения стадию (а) можно осуществлять при температуре от примерно 0°C до примерно 50°C. В некоторых вариантах реализации настоящего изобретения температура составляет примерно 25°C.
[0162] На первом этапе (d) указанное основание может представлять собой любое органическое основание, такое как аминовое основание. Подходящие аминовые основания включают, но не ограничиваются ими, DIEA, TEA, трибутиламин, 2,6-лутидин, 2,2,6,6-тетраметилгуанидин и тому подобное. Подходящие фосфитатные реагенты включают фосфинатные реагенты, про которые специалистам в данной области техники известно, что они пригодны для получения активированного сложного эфира карбоновой кислоты, такого как пентафторфенилдифенилфосфинат (FDPP). В некоторых вариантах реализации настоящего изобретения стадию (d) можно осуществлять в присутствии дополнительного полярного апротонного растворителя или смеси полярных апротонных растворителей. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. В некоторых вариантах реализации настоящего изобретения указанный полярный апротонный растворитель представляет собой смесь ДМФА и ДХМ. В некоторых вариантах реализации настоящего изобретения стадию (а) можно осуществлять при температуре от примерно -20°C до примерно 50°C. В некоторых вариантах реализации настоящего изобретения температура составляет от примерно 0°C до примерно 25°C.
[0163] В некоторых вариантах реализации настоящее изобретение относится к способу (способ А) получения соединения формулы B
B
[0164] где
[0165] PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и
[0166] каждый из R3 и R4 независимо представляет собой C1-C4 алкил;
[0167] включающему
[0168] (а) приведение соединения формулы B-1
B-1
[0169] где R4 представляет собой C1-C4 алкил; в контакт с соединением формулы B-2R
B-2R
[0170] где R3 представляет собой C1-C4 алкил, и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; в присутствии азодикарбоксилатного реагента и фосфинового реагента с получением соединения формулы B-3
B-3
[0171] где PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; или
[0172] (b) приведение соединения формулы B-3
B-3
[0173] где PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; в контакт с (R)-2-метил-2-пропансульфинамидом с получением соединения формулы B-5
B-5
[0174] где PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; или
[0175] (c) приведение соединения формулы B-5
B-5
[0176] где PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; в контакт с восстанавливающим агентом с получением соединения формулы B-6
B-6
[0177] где PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; или
[0178] (d) приведение соединения формулы B-6
B-6
[0179] где PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; в контакт с йодным реагентом с получением соединения формулы B.
[0180] Будет понятно, что настоящее изобретение включает способы получения соединения формулы B по способу A, как описано в параграфах выше, включающие более одного из тех этапов, которые перечислены в качестве альтернативы. Соответственно, настоящее изобретение относится к способу получения соединения B, включающему стадии (a) и (b). Альтернативно настоящее изобретение относится к способу получения соединения B, включающему стадии (b) и (c). Альтернативно настоящее изобретение относится к способу получения соединения B, включающему стадии (c) и (d). Альтернативно настоящее изобретение относится к способу получения соединения B, включающему стадии (a), (b) и (c). Альтернативно настоящее изобретение относится к способу получения соединения B, включающему стадии (b), (c) и (d). Альтернативно настоящее изобретение относится к способу получения соединения B, включающему стадии (a), (b), (c) и (d).
[0181] На стадии (а) способа А азодикарбоксилатный реагент может представлять собой любой такой реагент, известный в данной области техники. Подходящие азодикарбоксилатные реагенты включают, но не ограничиваются ими, DEAD, диизопропилазодикарбоксилат (DIAD), ди-(4-хлорбензил)азодикарбоксилат (DCAD) и тому подобное. На стадии (а) способа А фосфиновый реагент может представлять собой любой органофосфиновый реагент, известный в данной области техники, включая, но не ограничиваясь ими, трифенилфосфин, трибутилфосфин и тому подобное. В некоторых вариантах реализации настоящего изобретения стадию (а) способа А можно проводить в присутствии дополнительного полярного апротонного растворителя. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. В некоторых вариантах реализации настоящего изобретения указанный полярный протонный растворитель представляет собой ДХМ. В некоторых вариантах реализации настоящего изобретения стадию (а) способа А можно осуществлять при температуре от примерно -20°C до примерно 50°C. В некоторых вариантах реализации настоящего изобретения температура составляет от примерно 0°C до примерно 25°C. В некоторых вариантах реализации каждый из R3 и R4 представляет собой метил. В некоторых вариантах осуществления PG представляет собой Boc.
[0182] В некоторых вариантах осуществления соединение формулы B-2 может быть получено путем приведения (R)-1-аминопропан-2-ола в контакт с (Boc)2O в присутствии аминового основания.
[0183] В некоторых вариантах реализации настоящего изобретения стадию (b) способа А можно проводить в присутствии кислоты Льюиса. В некоторых вариантах реализации настоящего изобретения стадию (b) способа А можно проводить в присутствии акцептора воды. В некоторых вариантах реализации настоящего изобретения указанные кислота Льюиса и поглотитель воды могут быть одним и тем же реагентом. В некоторых вариантах реализации настоящего изобретения указанные кислота Льюиса и поглотитель воды могут быть разными реагентами. Подходящие кислоты Льюиса включают, но не ограничиваются ими, сульфат меди (II), сульфат магния, тетраэтоксититан, тетраизопропоксититан и тому подобное. Подходящие поглотители воды включают, но не ограничиваются ими, п-толуолсульфонат пиридиния, сульфат магния, сульфат натрия, тетраэтоксититан, тетраизопропоксититан и тому подобное. В некоторых вариантах реализации настоящего изобретения указанные кислота Льюиса и акцептор воды представляют собой тетраэтоксититан. В некоторых вариантах реализации настоящего изобретения стадию (b) способа А можно проводить в присутствии дополнительного полярного апротонного растворителя. В некоторых вариантах реализации настоящего изобретения стадию (b) способа А можно проводить в присутствии смеси полярных апротонных растворителей. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. В некоторых вариантах реализации настоящего изобретения указанный полярный апротонный растворитель представляет собой смесь ТГФ и 2-метил-ТГФ. В некоторых вариантах реализации настоящего изобретения стадию (b) способа А можно осуществлять при температуре от примерно 0°C до примерно 80°C. В некоторых вариантах реализации настоящего изобретения температура составляет от примерно 15°C до примерно 65°C. В некоторых вариантах реализации каждый из R3 и R4 представляет собой метил. В некоторых вариантах осуществления PG представляет собой Boc.
[0184] На стадии (с) способа А указанный восстанавливающий агент может представлять собой любой элемент или соединение, общеизвестные в данной области техники, которые теряют (или «отдают») электрон другим химическим веществом в окислительно-восстановительной химической реакции, включая, но не ограничиваясь ими, гидридные реагенты, элементный водород, силановые реагенты, эфирные реагенты Ганча и тому подобное. Подходящие восстанавливающие агенты включают, но не ограничиваются ими, NaBH4, LiAlH4 и H2. В некоторых вариантах реализации настоящего изобретения указанный восстанавливающий агент на стадии (с) способа А представляет собой NaBH4. В некоторых вариантах реализации настоящего изобретения стадию (с) способа А можно проводить в присутствии полярного органического растворителя. В некоторых вариантах реализации настоящего изобретения стадию (с) способа А можно проводить в присутствии смеси полярных растворителей. В некоторых вариантах реализации указанный полярный органический растворитель или смесь полярных органических растворителей могут быть полярными апротонными или полярными протонными. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. Подходящие полярные протонные растворители включают, но не ограничиваются ими, MeOH, EtOH, iPrOH, n-BuOH, втор-BuOH, H2O и тому подобное. В некоторых вариантах реализации настоящего изобретения указанный полярный апротонный растворитель представляет собой смесь ТГФ и H2O. В некоторых вариантах реализации настоящего изобретения стадию (с) способа А можно осуществлять при температуре от примерно -78°C до примерно 30°C. В некоторых вариантах реализации настоящего изобретения температура составляет от примерно -50°C до примерно 25°C. В некоторых вариантах реализации каждый из R3 и R4 представляет собой метил. В некоторых вариантах осуществления PG представляет собой Boc.
[0185] На стадии (d) способа А йодный реагент может представлять собой любой йодный реагент, известный в данной области техники, подходящий для снятия защиты с сульфинамида. В некоторых вариантах реализации указанный йодный реагент представляет собой I2. В некоторых вариантах реализации настоящего изобретения стадию (d) способа А можно проводить в присутствии полярного органического растворителя. В некоторых вариантах реализации настоящего изобретения стадию (d) способа А можно проводить в присутствии смеси полярных растворителей. В некоторых вариантах реализации указанный полярный органический растворитель или смесь полярных органических растворителей могут быть полярными апротонными или полярными протонными. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. Подходящие полярные протонные растворители включают, но не ограничиваются ими, MeOH, EtOH, iPrOH, n-BuOH, втор-BuOH, H2O и тому подобное. В некоторых вариантах реализации настоящего изобретения указанный полярный апротонный растворитель представляет собой смесь ТГФ и H2O. В некоторых вариантах реализации настоящего изобретения стадию (d) способа А можно осуществлять при температуре от примерно 0°C до примерно 80°C. В некоторых вариантах реализации настоящего изобретения температура составляет от примерно 25°C до примерно 60°C. В некоторых вариантах реализации настоящего изобретения температура составляет примерно 0°C. В некоторых вариантах реализации каждый из R3 и R4 представляет собой метил. В некоторых вариантах осуществления PG представляет собой Boc.
[0186] Альтернативно в некоторых вариантах реализации настоящее изобретение относится к способу (способ B) получения соединения формулы B
B
[0187] где
[0188] PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и
[0189] каждый из R3 и R4 независимо представляет собой C1-C4 алкил;
[0190] включающему
[0191] (а) взаимодействие соединения формулы B-7
B-7
[0192] где R4 представляет собой C1-C4 алкил; в условиях, подходящих для получения соединения формулы B-8
B-8
[0193] где R4 представляет собой C1-C4 алкил; и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; или
[0194] (b) приведение соединения формулы B-8
B-8
[001] где R4 представляет собой C1-C4 алкил; и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; в контакт с соединением формулы B-2R
B-2R
[0195] где R3 представляет собой C1-C4 алкил, и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; в присутствии азодикарбоксилатного реагента и фосфинового реагента с получением соединения формулы B-9
B-9
[0196] где каждая PG независимо выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts, при условии, что все PG разные; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; или
[0197] (c) приведение соединения формулы B-9
B-9
[0198] где каждая PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; при условии, что все PG разные; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; в контакт с неорганическим основанием с получением соединения формулы B.
[0199] Будет понятно, что настоящее изобретение включает способы получения соединения формулы B по способу В, как описано в параграфах выше, включающие более одного из тех этапов, которые перечислены в качестве альтернативы. Соответственно, настоящее изобретение относится к способу получения соединения B, включающему стадии (a) и (b). Альтернативно настоящее изобретение относится к способу получения соединения B, включающему стадии (b) и (c). Альтернативно настоящее изобретение относится к способу получения соединения B, включающему стадии (a), (b) и (c).
[0200] На стадии (а) способа В соединение формулы В-7 можно подвергнуть взаимодействию в условиях, подходящих для введения аминовой защитной группы (или PG). Понятно, что такие условия общеизвестны специалистам в данной области техники и могут быть использованы любые такие условия, совместимые с функциональностью соединения формулы B-7 и остальной частью процесса, описанного в способе B. Подходящие защитные группы (или PG) включают, но не ограничиваются ими, FMOC, Boc, Cbz, Ac, трифторацетил, фталимид, Bn, тритил, бензилиден и Ts. В некоторых вариантах реализации настоящего изобретения PG представляет собой трифторацетил. В некоторых вариантах реализации настоящего изобретения соединение формулы B-7 можно подвергнуть взаимодействию в условиях, подходящих для введения трифторацетила.
[0201] В некоторых вариантах реализации настоящего изобретения стадия (а) способа В включает приведение соединения формулы В-7 в контакт с трифторуксусным ангидридом в присутствии органического основания, такого как аминовое основание. Подходящие аминовые основания включают, но не ограничиваются ими, DIEA, TEA, трибутиламин, 2,6-лутидин, 2,2,6,6-тетраметилгуанидин и тому подобное. В некоторых вариантах реализации настоящего изобретения стадию (а) способа В можно проводить в присутствии полярного апротонного растворителя. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. В некоторых вариантах реализации настоящего изобретения указанный полярный протонный растворитель представляет собой ДХМ. В некоторых вариантах реализации настоящего изобретения стадию (а) способа В можно осуществлять при температуре от примерно -20°C до примерно 25°C. В некоторых вариантах реализации настоящего изобретения температура составляет примерно 0°C. В некоторых вариантах реализации настоящего изобретения R4 представляет собой метил. В некоторых вариантах реализации настоящего изобретения PG представляет собой трифторацетил.
[0202] На стадии (b) способа B азодикарбоксилатный реагент может представлять собой любой такой реагент, известный в данной области техники. Подходящие азодикарбоксилатные реагенты включают, но не ограничиваются ими, DEAD, диизопропилазодикарбоксилат (DIAD), ди-(4-хлорбензил)азодикарбоксилат (DCAD) и тому подобное. На стадии (b) способа B указанный фосфиновый реагент может представлять собой любой органофосфиновый реагент, известный в данной области техники, включая, но не ограничиваясь ими, трифенилфосфин, трибутилфосфин и тому подобное. В некоторых вариантах реализации настоящего изобретения стадию (b) способа В можно проводить в присутствии полярного апротонного растворителя. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. В некоторых вариантах реализации настоящего изобретения указанный полярный протонный растворитель представляет собой ДХМ. В некоторых вариантах реализации настоящего изобретения стадию (b) способа В можно осуществлять при температуре от примерно -20°C до примерно 50°C. В некоторых вариантах реализации настоящего изобретения температура составляет от примерно 0°C до примерно 25°C. В некоторых вариантах реализации каждый из R3 и R4 представляет собой метил. В некоторых вариантах реализации одна PG представляет собой Boc и одна PG представляет собой трифторацетил.
[0203] В некоторых вариантах реализации настоящего изобретения стадию (с) способа В можно проводить в условиях, подходящих для удаления одной из групп PG в соединении формулы В-9, тогда как другая группа PG остается в неизмененном виде. На стадии (c) способа В указанное неорганическое основание может представлять собой любое неорганическое основание, такое как гидроксидное основание. Подходящие гидроксидные основания включают, но не ограничиваются ими, гидроксид натрия, гидроксид лития и тому подобное. В вариантах реализации настоящего изобретения стадию (с) способа В можно проводить в присутствии полярного протонного растворителя, полярного апротонного растворителя или их смеси. Подходящие полярные протонные растворители включают, но не ограничиваются ими, MeOH, EtOH, iPrOH, n-BuOH, втор-BuOH, H2O и тому подобное. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. В некоторых вариантах реализации настоящего изобретения стадию (с) способа В можно проводить в смеси растворителей, такой как ТГФ/МеОН. В некоторых вариантах реализации настоящего изобретения стадию (с) способа В можно осуществлять при температуре от примерно 30°C до примерно 100°C. В некоторых вариантах реализации настоящего изобретения температура составляет примерно 50°C. В некоторых вариантах реализации каждый из R3 и R4 представляет собой метил. В некоторых вариантах реализации одна PG представляет собой Boc и одна PG представляет собой трифторацетил.
[0204] Альтернативно в некоторых вариантах реализации настоящее изобретение относится к способу (способ C) получения соединения формулы B
B
[0205] где
[0206] PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и
[0207] каждый из R3 и R4 независимо представляет собой C1-C4 алкил;
[0208] включающему
[0209] (а) взаимодействие соединения формулы B-10
B-10
[0210] с соединением формулы B-2S
B-2S
[0211] где R3 представляет собой C1-C4 алкил, и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; в основания с получением соединения формулы B-11
B-11
[0212] где R3 представляет собой C1-C4 алкил, и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; или
[0213] (b) приведение соединения формулы B-11
B-11
[0214] где R3 представляет собой C1-C4 алкил, и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; в контакт с нуклеофилом с получением соединения формулы B-12
B-12
[0215] где каждый из R3 и R4 независимо представляет собой C1-C4 алкил, и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; или
[0216] (c) приведение соединения формулы B-12
B-12
[0217] где каждый из R3 и R4 независимо представляет собой C1-C4 алкил, и PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; в контакт с восстанавливающим агентом с получением соединения формулы B.
[0218] Будет понятно, что настоящее изобретение включает способы получения соединения формулы B по способу С, как описано в параграфах выше, включающие более одного из тех этапов, которые перечислены в качестве альтернативы. Соответственно, настоящее изобретение относится к способу получения соединения B, включающему стадии (a) и (b). Альтернативно настоящее изобретение относится к способу получения соединения B, включающему стадии (b) и (c). Альтернативно настоящее изобретение относится к способу получения соединения B, включающему стадии (a), (b) и (c).
[0219] На стадии (а) способа С указанное основание может представлять собой любое сильное ненуклеофильное основание, известное специалисту в данной области техники. Подходящие сильные ненуклеофильные основания включают, но не ограничиваются ими, KHMDS, трет-бутоксид калия, диизопропиламид лития, 1,5,7-триазабицикло(4,4,0)дец-5-ен (TBD), 7-метил-1,5,7-триазабицикло(4.4.0)дец-5-ен (MTBD), 1,8-диазабицикло[5.4.0]-ундец-7-ен (DBU), 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,1,3,3-тетраметилгуанидин (TMG) и тому подобное. В некоторых вариантах реализации настоящего изобретения сильное ненуклеофильное основание представляет собой KHMDS. В некоторых вариантах реализации настоящего изобретения стадию (а) способа С можно проводить в присутствии дополнительного полярного апротонного растворителя. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. В некоторых вариантах реализации настоящего изобретения указанный полярный апротонный растворитель представляет собой ТГФ. В некоторых вариантах реализации настоящего изобретения стадию (а) способа С можно осуществлять при температуре от примерно -20°C до примерно 40°C. В некоторых вариантах реализации настоящего изобретения температура составляет от примерно 0°C до примерно 25°C. В некоторых вариантах реализации настоящего изобретения R3 представляет собой метил. В некоторых вариантах осуществления одна PG представляет собой Boc.
[0220] На стадии (b) способа С указанный нуклеофил может представлять собой любой нуклеофил, способный доставлять нуклеофильный атом углерода в функциональную группу нитрила. Подходящие нуклеофилы включают, но не ограничиваются ими, алкилметаллгалогенидные реагенты, такие как реактивы Гриньяра, и литийорганические реагенты. В некоторых вариантах реализации настоящего изобретения указанный нуклеофил на стадии (b) способа C представляет собой C1-C4 алкил MgBr. В некоторых вариантах реализации настоящего изобретения указанный нуклеофил на стадии (b) способа C представляет собой MeMgBr. В некоторых вариантах реализации настоящего изобретения стадию (b) способа С можно проводить в присутствии дополнительного полярного апротонного растворителя. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. В некоторых вариантах реализации настоящего изобретения указанный полярный апротонный растворитель представляет собой ТГФ. В некоторых вариантах реализации настоящего изобретения стадия (b) способа C дополнительно включает добавление спиртового растворителя, такого как метанол или этанол, для гашения реакции. В некоторых вариантах реализации настоящего изобретения стадия (b) способа C дополнительно включает добавление сильной неорганической кислоты с образованием иминиевой соли. В некоторых вариантах реализации настоящего изобретения указанная сильная кислота представляет собой эфирный раствор HCl. В некоторых вариантах реализации настоящего изобретения стадию (b) способа С можно осуществлять при температуре от примерно -80°C до примерно 40°C. В некоторых вариантах реализации настоящего изобретения температура составляет от примерно -78°C до примерно 25°C. В некоторых вариантах реализации R3 и R4 представляет собой метил. В некоторых вариантах осуществления одна PG представляет собой Boc.
[0221] На стадии (с) способа C указанный восстанавливающий агент может представлять собой любой элемент или соединение, общеизвестные в данной области техники, которые теряют (или «отдают») электрон другим химическим веществом в окислительно-восстановительной химической реакции, включая, но не ограничиваясь ими, гидридные реагенты, элементный водород, силановые реагенты, эфирные реагенты Ганча и тому подобное. Подходящие восстанавливающие агенты включают H2 и эфир Ганча. В некоторых вариантах реализации настоящего изобретения удобно приводить восстановителем в контакт в присутствии катализатора, такого как иридиевый катализатор, рутениевый катализатор, родиевый катализатор, палладиевый катализатор и тому подобное. Специалисту в данной области техники будет понятно, что катализатор может представлять собой любую известную в данной области техники систему катализаторов, пригодную для того, чтобы способствовать восстановлению иминиевой соли до амина. В некоторых вариантах реализации настоящего изобретения указанный катализатор представляет собой [Ir(COD)Cl]2/(S, S)-f-бинафан. В некоторых вариантах реализации настоящего изобретения указанный восстановитель на стадии (с) способа С представляет собой Н2 в присутствии [Ir(COD)Cl]2 и (S, S)-f-бинафана. В некоторых вариантах реализации настоящего изобретения Н2 применяют при давлении от примерно 2 атмосфер до примерно 15 атмосфер. В некоторых вариантах реализации настоящего изобретения Н2 применяют при давлении примерно 10 атмосфер.
[0222] В некоторых вариантах реализации настоящего изобретения стадия (c) способа C может быть проведена в присутствии полярного протонного растворителя, такого как спиртовой растворитель. Подходящие полярные протонные растворители включают, но не ограничиваются ими, MeOH, EtOH, iPrOH, n-BuOH, втор-BuOH и тому подобное. В некоторых вариантах реализации настоящего изобретения указанный полярный протонный растворитель представляет собой MeOH. C вариантах реализации настоящего изобретения стадию (с) способа С можно проводить в присутствии дополнительного полярного апротонного растворителя. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. В некоторых вариантах реализации настоящего изобретения указанный полярный протонный растворитель представляет собой ДХМ. В некоторых вариантах реализации настоящего изобретения стадия (c) способа C может быть проведена в смеси полярного протонного растворителя и полярного апротонного растворителя. C вариантах реализации настоящего изобретения стадию (с) способа С можно проводить в смеси ДХМ и MeOH.
[0223] Альтернативно в некоторых вариантах реализации настоящее изобретение относится к способу (способ D) получения соединения формулы B
B
[0224] где
[0225] PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и
[0226] каждый из R3 и R4 независимо представляет собой C1-C4 алкил;
[0227] включающему
[0228] (а) взаимодействие соединения формулы B-12
B-12
[0229] где PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; в условиях, подходящих для получения соединения формулы B-13
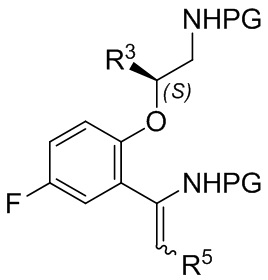
B-13
[0230] где каждая PG независимо выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts, при условии, что все PG разные; R3 представляет собой C1-C4 алкил; и R5 представляет собой C1-C3 алкил; или
[0231] (b) приведение соединения формулы B-13
B-13
[0232] где каждая PG независимо выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts, при условии, что все PG разные; R3 представляет собой C1-C4 алкил; и R5 представляет собой C1-C3 алкил; в контакт с восстанавливающим агентом с получением соединения формулы B-9
B-9
[0233] где каждая PG независимо выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts, при условии, что все PG разные; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; или
[0234] (c) приведение соединения формулы B-9
B-9
[0235] где каждая PG выбрана из группы, состоящей из FMOC, Boc, Cbz, Ac, трифторацетила, фталимида, Bn, тритила, бензилидена и Ts; при условии, что все PG разные; и каждый из R3 и R4 независимо представляет собой C1-C4 алкил; в контакт с неорганическим основанием с получением соединения формулы B.
[0236] Будет понятно, что настоящее изобретение включает способы получения соединения формулы B по способу D, как описано в параграфах выше, включающие более одного из тех этапов, которые перечислены в качестве альтернативы. Соответственно, настоящее изобретение относится к способу получения соединения B, включающему стадии (a) и (b). Альтернативно настоящее изобретение относится к способу получения соединения B, включающему стадии (b) и (c). Альтернативно настоящее изобретение относится к способу получения соединения B, включающему стадии (a), (b) и (c).
[0237] В некоторых вариантах реализации настоящего изобретения стадия (а) способа D включает приведение соединения формулы В-12 в контакт с трифторуксусным ангидридом в присутствии органического основания, такого как аминовое основание. Подходящие аминовые основания включают, но не ограничиваются ими, DIEA, TEA, трибутиламин, 2,6-лутидин, 2,2,6,6-тетраметилгуанидин и тому подобное. В некоторых вариантах реализации настоящего изобретения стадию (а) способа D можно проводить в присутствии полярного апротонного растворителя. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. В некоторых вариантах реализации настоящего изобретения указанный полярный протонный растворитель представляет собой ДХМ. В некоторых вариантах реализации настоящего изобретения стадию (а) способа D можно осуществлять при температуре от примерно -20°C до примерно 25°C. В некоторых вариантах реализации настоящего изобретения температура составляет примерно 0°C. В некоторых вариантах реализации настоящего изобретения R4 представляет собой метил. В некоторых вариантах реализации настоящего изобретения PG представляет собой трифторацетил.
[0238] На стадии (с) способа D указанный восстанавливающий агент может представлять собой любой элемент или соединение, общеизвестные в данной области техники, которые теряют (или «отдают») электрон другим химическим веществом в окислительно-восстановительной химической реакции, включая, но не ограничиваясь ими, гидридные реагенты, элементный водород, силановые реагенты, эфирные реагенты Ганча и тому подобное. Подходящие восстанавливающие агенты включают H2 и эфир Ганча. В некоторых вариантах реализации настоящего изобретения удобно приводить восстановителем в контакт в присутствии катализатора, такого как иридиевый катализатор, рутениевый катализатор, родиевый катализатор, палладиевый катализатор и тому подобное. Специалисту в данной области техники будет понятно, что катализатор может представлять собой любую известную в данной области техники систему катализаторов, пригодную для того, чтобы способствовать восстановлению иминиевой соли до амина. В некоторых вариантах реализации настоящего изобретения указанный катализатор представляет собой [((R,R)-Me-DuPHOS)-Rh-(COD)]BF4. В некоторых вариантах реализации настоящего изобретения указанный восстановитель на стадии (b) способа D представляет собой H2 в присутствии [((R,R)-Me-DuPHOS)-Rh-(COD)]BF4. В некоторых вариантах реализации настоящего изобретения Н2 применяют при давлении от примерно 2 фунтов на квадратный дюйм (~13,8 кПа) до примерно 100 фунтов на квадратный дюйм (~689,5 кПа). В некоторых вариантах реализации настоящего изобретения Н2 применяют при давлении примерно 90 фунтов на квадратный дюйм (~620,5 кПа).
[0239] В некоторых вариантах реализации настоящего изобретения стадия (b) способа D быть проведена в присутствии полярного протонного растворителя, такого как спиртовой растворитель. Подходящие полярные протонные растворители включают, но не ограничиваются ими, MeOH, EtOH, iPrOH, n-BuOH, втор-BuOH и тому подобное. В некоторых вариантах реализации настоящего изобретения указанный полярный протонный растворитель представляет собой MeOH. В некоторых вариантах реализации настоящего изобретения стадию (b) способа D можно проводить в присутствии дополнительного полярного апротонного растворителя. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. В некоторых вариантах реализации настоящего изобретения указанный полярный протонный растворитель представляет собой ДХМ. В некоторых вариантах реализации настоящего изобретения стадия (b) способа D быть проведена в смеси полярного протонного растворителя и полярного апротонного растворителя. B реализации настоящего изобретения стадию (b) способа D можно проводить в смеси ДХМ и MeOH.
[0240] В некоторых вариантах реализации настоящего изобретения стадию (с) способа D можно проводить в условиях, подходящих для удаления одной из групп PG в соединении формулы В-9, тогда как другая группа PG остается в неизмененном виде. На стадии (c) способа D неорганическое основание может представлять собой любое неорганическое основание, такое как карбонатное основание или гидроксид. Подходящие гидроксидные основания включают, но не ограничиваются ими, гидроксид натрия, гидроксид лития и тому подобное. Подходящие карбонатные основания включают, но не ограничиваются ими, карбонат калия, бикарбонат натрия, карбонат натрия и тому подобное. В некоторых вариантах осуществления неорганическое основание представляет собой карбонат калия. В вариантах реализации настоящего изобретения стадию (с) способа D можно проводить в присутствии полярного протонного растворителя, полярного апротонного растворителя или их смеси. Подходящие полярные протонные растворители включают, но не ограничиваются ими, MeOH, EtOH, iPrOH, n-BuOH, втор-BuOH, H2O и тому подобное. Подходящие полярные апротонные растворители включают, но не ограничиваются ими, ТГФ, 2-метил-ТГФ, Et2O, ДХМ, EtOAc, ДМФА, CH3CN, ацетон, HMPT, ДМСО и т.п. В некоторых вариантах реализации настоящего изобретения стадию (c) способа D можно проводить в MeOH. В некоторых вариантах реализации настоящего изобретения стадию (с) способа D можно осуществлять при температуре от примерно 30°C до примерно 100°C. В некоторых вариантах реализации настоящего изобретения температура составляет примерно 50°C. В некоторых вариантах реализации каждый из R3 и R4 представляет собой метил. В некоторых вариантах реализации одна PG представляет собой Boc и одна PG представляет собой трифторацетил.
ПРИМЕРЫ
[0241] Приведенные ниже примеры и составы дополнительно иллюстрируют конкретные аспекты вариантов осуществления раскрытия. Следует понимать, что объем настоящего изобретения никоим образом не ограничен рамками следующих примеров.
[0242] Аббревиатуры
[0243] В приведенных в настоящем документе примерах использованы материалы, включая, но не ограничиваясь ими, те, которые описаны следующими сокращениями, известными специалистам в данной области:
[0244] Синтез кристаллической полиморфной формы 1 соединения I
[0245] Соединение I получали в соответствии со следующей схемой синтеза:
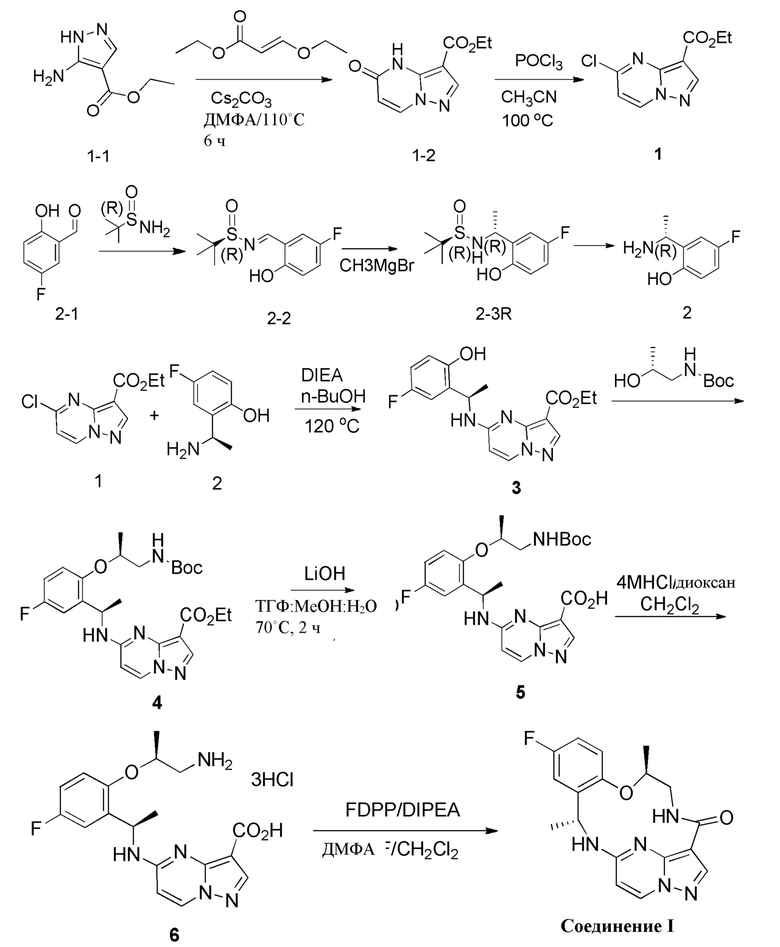
[0246] Пример 1: Получение 5-хлорпиразоло[1,5-a]пиримидин-3-карбоксилата (1).
[0247] Стадия 1: Получение этил 5-оксо-4Н-пиразоло[1,5-a]пиримидин-3-карбоксилата (1-2)
[0248] К смеси этил 5-амино-1Н-пиразол-4-карбоксилата (Sigma-Aldrich, 150,00 г, 1,08 ммоль) и этил (E)-3-этоксипроп-2-еноата (Sigma-Aldrich, 292,16 г, 2,03 моль) в ДМФА (3,2 л) добавляли Cs2СО3 (656,77 г, 2,02 моль) одной порцией при температуре 20°С в атмосфере N2. Смесь перемешивали при температуре 110°C в течение 6 часов. ТСХ (PE:EtOAc=1:1) показала завершение реакции. Указанную смесь охлаждали до температуры 20 °С и фильтровали через слой целита. Остаток на фильтре промывали этилацетатом (3×30 мл). Фильтрат добавляли к H2O (2 л) и подкисляли HOAc до рН=4. Полученный осадок отфильтровывали с получением 1-2 (173,00 г, 834,98 ммоль, выход 86,36%) в виде белого твердого вещества: 1H ЯМР (400 MГц, ДМСО-d6) δ 8,54 (d, J=7,91 Гц, 1H), 8,12 (s, 1H), 6,13 (d, J=7,91 Гц, 1H), 4,27 (q, J=7,11 Гц, 2H), 1,28 (t, J=7,09 Гц, 3H).
[0249] Стадия 2: Получение 5-хлорпиразоло[1,5-a]пиримидин-3-карбоксилата (1).
[0250] К смеси 1-2 (158,00 г, 762,59 ммоль) в MeCN (1,6 л) добавляли POCl3 (584,64 г, 3,81 моль) при температуре 20°С в атмосфере N2. Указанную смесь перемешивали при температуре 100°С в течение 2 часов. ТСХ (PE:EA=1:1) показала завершение реакции. Смесь охлаждали до температуры 20°C и выливали в ледяную воду (5000 мл) порциями при температуре 0°C и перемешивали в течение 20 минут. Осадок фильтровали и сушили с получением 1 (110,00 г, 487,52 ммоль, выход 63,93%) в виде белого твердого вещества: 1H ЯМР (400 MГц, ДМСО-d6) δ 9,33 (d, J=7,28 Гц, 1H), 8,66 (s, 1H), 7,41 (d, J=7,15 Гц, 1H), 4,31 (q, J=7,15 Гц, 2H), 1,32 (t, J=7,09 Гц, 3H).
[0251] Пример 2: Получение (R)-2-(1-аминоэтил) -4-фторфенола (2).
[0252] Стадия 1: Получение (R)-N-(5-фтор-2-гидроксибензилиден)-2-метилпропан-2-сульфинамида (2-2)
[0253] К раствору (R)-2-метилпропан-2-сульфинамида (Sigma-Aldrich, 150,00 г, 1,24 моль, 1,00 экв.) и 5-фтор-2-гидроксибензальдегида (2-1) (Sigma-Aldrich, 173,74 г, 1,24 моль, 1,00 экв.) в ДХМ (2,00 л) добавляли Cs2CO3 (646,43 г, 1,98 моль, 1,60 экв.). Смесь перемешивали при температуре 16°C в течение 16 часов. ТСХ (PE:EtOAc=5:1) показала завершение реакции. Реакционную смесь гасили добавлением H2O (1000 мл) при температуре 0°C и затем экстрагировали EtOAc (500 мл × 4). Объединенные органические слои промывали солевым раствором (1000 мл) и сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 2-2 (230,00 г, 945,33 ммоль, выход 76,24%). 1H ЯМР (CDCl3, 400 MГц) δ 8,64 (s, 1H), 7,22-7,11 (m, 2H), 7,03-6,95 (m, 1H), 1,28 (s, 9H).
[0254] Стадия 2: Получение (R)-N-((R)-1-(5-фтор-2-гидроксифенил)этил)-2-метилпропан-2-сульфинамида (2-3R)
[0255] К раствору (R)-N-(5-фтор-2-гидроксибензилиден)-2-метилпропан-2-сульфинамида (2-2) (200,00 г, 822,03 ммоль, 1,00 экв.) в ТГФ (2,5 л) добавляли MeMgBr (490,09 г, 4,11 моль, 5,00 экв.) по каплям при температуре -65°С в атмосфере N2 в течение 30 мин. Затем смесь нагревали до комнатной температуры и перемешивали в течение 18 часов. ТСХ (PE: EtOAc=1: 1) показала завершение реакции с образованием двух диастереомеров. Реакционную смесь гасили добавлением H2O (2 л) при температуре 0°C, смесь экстрагировали EtOAc (500 мл ×3). Объединенные органические слои промывали солевым раствором (500 мл) и сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=от 50/1 до 1:1) с получением (R)-N-((R)-1-(5-фтор-2-гидроксифенил)этил)-2-метилпропан-2-сульфинамида (2-3R) (125 г, верхнее, менее полярное пятно с Rf: 0,5, PE: EA=1:1). 1H ЯМР (CDCl3, 400 MГц) δ: 9,17 (s, 1H), 6,68 (dd, J=3,0, 8,8 Гц, 1H), 6,47 (dt, J=3,0, 8,4 Гц, 1H), 6,31 (dd, J=4,8, 8,8 Гц, 1H), 5,11 (d, J=8,0 Гц, 1H), 4,28 (квинт., J=7,2 Гц, 1H), 1,43 (d, J=6,8 Гц, 3H), 1,20 (s, 9H).
[0256] Стадия 3: Получение (R)-2-(1-аминоэтил)-4-фторфенола (2)
[0257] Раствор (R)-N-((R)-1-(5-фтор-2-гидроксифенил)этил)-2-метилпропан-2-сульфинамида (2-3R) (125 г, 481.99 ммоль, 1,00 экв.) в HCl/диоксане (1,5 л, 4 н.) перемешивали при температуре окружающей среды в течение 2 часов. ТСХ (PE:EtOAc=2:1) показала завершение реакции. Смесь фильтровали с получением HCl-соли (R)-2-(1-аминоэтил)-4-фторфенола (2) (85 г, 443,56 ммоль, выход 90,03%) в виде белого твердого вещества. 1H ЯМР (d-ДМСО, 400 MГц) δ 10,24 (s, 1H), 8,48 (шир. s., 3H), 7,31 (dd, J=2,9, 9,7 Гц, 1H), 7,05-6,99 (m, 1H), 6,98-6,93 (m, 1H), 4,59-4,45 (m, 1H), 1,46 (d, J=6,8 Гц, 3H).
[0258] Пример 3: Получение (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5Н)-она (соединение I).
[0259] Стадия 1: Получение этил (R)-5-((1-(5-фтор-2-гидроксифенил)этил)амино)пиразоло[1,5-a]пиримидин-3-карбоксилата (3)
[0260] К раствору (R)-2-(1-аминоэтил)-4-фторфенола (2) (85 г, 443,56 ммоль, 1,00 экв.) и этил 5-хлорпиразоло[1,5-a]пиримидин-3-карбоксилата (1) (100,08 г, 443,56 ммоль, 1,00 экв.) в н-BuOH (2 л) добавляли DIEA (343,96 г, 2,66 моль, 6,00 экв.). Указанную смесь перемешивали при температуре 120°С в течение 2 часов. ТСХ (PE: EtOAc=1:1) показала завершение реакции. Реакционную смесь разбавляли H2O (500 мл) при температуре 16°C и экстрагировали EtOAc (500 мл ×3). Объединенные органические слои промывали солевым раствором (500 мл) и сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=10/1 до 1:3) с получением этил (R)-5-((1-(5-фтор-2-гидроксифенил)этил)амино)пиразоло[1,5-a]пиримидин-3-карбоксилата (3) (122 г, 349,34 ммоль, выход 78,76%, чистота > 99%) в виде белого твердого вещества. 1H ЯМР (CDCl3, 400 MГц) δ 9,28 (шир. s., 1H), 8,26 (s, 1H), 8,14 (d, J=7,5 Гц, 1H), 6,95-6,89 (m, 2H), 6,87-6,80 (m, 1H), 6,18 (d, J=7,5 Гц, 1H), 5,98 (d, J=8,3 Гц, 1H), 5,71-5,54 (m, 1H), 4,50-4,35 (m, 2H), 1,60 (d, J=6,8 Гц, 3H), 1,42 (t, J=7,2 Гц, 3H).
[0261] Стадия 2: Получение этил 5-(((R)-1-(2-(((S)-1-((трет-бутоксикарбонил)амино)пропан-2-ил)окси)-5-фторфенил)этил)амино)пиразоло [1,5-a]пиримидин-3-карбоксилата (4)
[0262] Смесь этил (R)-5-((1-(5-фтор-2-гидроксифенил)этил)амино)пиразоло[1,5-a]пиримидин-3-карбоксилата (3) (10,00 г, 29,04 ммоль) и трет-бутил (R)-(2-гидроксипропил)карбамата (Combi-Blocks, 7,63 г, 43,56 ммоль) азеотропно сушили из ДХМ/толуола и затем повторно растворяли в ДХМ (11,62 мл). К указанному раствору добавляли PPh3 (11,43 г, 43,56 ммоль) и смесь перемешивали до полного растворения исходных материалов. К раствору добавляли DEAD (8,81 г, 43,56 ммоль) в течение 5 мин при перемешивании. Реакционную перемешивали в течение 3 часов. Реакционную смесь разбавляли ДХМ (125 мл) с последующим добавлением водного раствора NaOH (2М, 100 мл). Смесь интенсивно перемешивали в течение 12 часов и слои разделяли. Водный слой экстрагировали ДХМ (3×50 мл). Объединенные экстракты промывали солевым раствором (50 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией (система Teledyne ISCO, диоксид кремния (330 г), 0-40% этилацетат в гексане с получением этил 5-(((R)-1-(2-(((S)-1-((трет-бутоксикарбонил)амино)пропан-2-ил)окси)-5-фторфенил)этил)амино)-пиразоло[1,5-a]пиримидин-3-карбоксилата (4) (8,88 г, выход 60,9%). ЖХ-МС m/z 502,2 (М+Н)+. 1H ЯМР (400 MГц, хлороформ-d) δ 8,24 (s, 1H), 8,21 (d, J=7,6 Гц, 1H), 7,04 (d, J=8,4 Гц, 1H), 6,87 (d, J=6,0 Гц, 2H), 6,13 (d, J=7,2 Гц, 1H), 5,91 (шир. s., 1H), 4,58 (d, J=3,6 Гц, 1H), 4,43-4,28 (m, 2H), 3,52-3,34 (m, 2H), 1,54 (d, J=6,8 Гц, 3H), 1,47-1,36 (m, 12H), 1,30 (d, J=6,4 Гц, 3H).
[0263] Стадия 3: Получение 5-(((R)-1-(2-(((S)-1-((трет-бутоксикарбонил)амино)пропан-2-ил)окси)-5-фторфенил)этил)амино)пиразоло [1,5-a]пиримидин-3-карбоновой кислоты (5)
[0264] К раствору этил 5-(((R)-1-(2-(((S)-1-((трет-бутоксикарбонил)амино)пропан-2-ил)окси)-5-фторфенил)этил)амино)пиразоло[1,5-a]пиримидин-3-карбоксилата (4) (6,98 г, 13,92 ммоль, 1 экв.) в метаноле (65 мл) и ТГФ (20 мл) добавляли LiOH (2М, 47,9 мл, 95,8 ммоль). Смесь нагревали при температуре 70°С в течение 3 часов, охлаждали до температуры окружающей среды и затем гасили водным раствором HCl (2М, 95,8 мл) с получение значения рН <5. Реакционную смесь экстрагировали CH2Cl2 (3×50 мл) и сушили над Na2SO4. После фильтрования, выпаривания и сушки в высоком вакууме получали белое твердое вещество, 5-(((R)-1-(2-(((S)-1-((трет-бутоксикарбонил)амино)пропан-2-ил)окси)-5-фторфенил)этил)амино)пиразоло[1,5-a]пиримидин-3-карбоновую кислоту (5), которое использовали на следующей стадии без дополнительной очистки. ЖХ-МС m/z 474,2 (М+Н)+.
[0265] Стадия 4: Получение 5-(((R)-1-(2-(((S)-1-аминопропан-2-ил)окси)-5-фторфенил)этил)амино)пиразоло [1,5-a]пиримидин-3-карбоновой кислоты (6)
[0266] К раствору 5-(((R)-1-(2-(((S)-1-((трет-бутоксикарбонил)амино)пропан-2-ил)окси)-5-фторфенил)этил)амино)пиразоло[1,5-a]пиримидин-3-карбоновой кислоты (5) (6,59 г, 13,92 ммоль) в CH2Cl2 (130 мл) добавляли HCl в диоксане (4М, 30,4 мл). Перемешивание продолжали при комнатной температуре в течение 2 часов, пока ЖХМС не показала завершение реакции. Реакционную смесь концентрировали и сушили в высоком вакууме с получением соединения 6 в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. ЖХ-МС m/z 374,2 (М+Н)+.
[0267] Стадия 5: Получение (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5Н)-она (соединение I).
[0268] 5-(((R)-1-(2-(((S)-1-аминопропан-2-ил)окси)-5-фторфенил)этил)амино)пиразоло[1,5-a]пиримидин-3- карбоновую кислоту (6) (5,20 г, 13,93 ммоль) растворяли в ДМФА (75 мл) с получением раствора А. К раствору основания Хьюнига (DIPEA) (14,40 г, 111,4 ммоль) в ДМФА (150 мл) и ДХМ (350 мл) добавляли раствор А (25 мл) и одну треть от общего количества FDPP (5,62 г, 14,63 ммоль) последовательно. Реакционную смесь перемешивали в течение 1 часа, и ЖХ-МС показала завершение реакции сочетания. Тот же процесс повторяли еще 2 раза. Конечный раствор перемешивали при комнатной температуре в течение 63 часов (или пока ЖХМС не показала завершение реакции). Реакцию гасили добавлением водного раствора Na2CO3 (2М, 150 мл) и смесь перемешивали в течение 15 мин и экстрагировали ДХМ (3 × 150 мл). Объединенные экстракты сушили над Na2SO4, концентрировали при пониженном давлении и очищали флэш-хроматографией (система Teledyne ISCO, двуокись кремния (220 г), 0-7,5% метанола в дихлорметане) с получением (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10] бензоксатриазациклотридецин-4(5Н)-1 (соединение I) (4,38 г, 12,33 ммоль, выход 88,5%) в виде белого твердого вещества. ЖХ-МС: m/z [M+H]+ 356,2. 1H ЯМР (500 MГц, ДМСО-d6) δ ppm 9,82 (dd, J=8,02, 2,29 Гц, 1 H), 8,81 (d, J=6,87 Гц, 1 H), 8,58 (d, J=7,45 Гц, 1 H), 8,04 (s, 1 H), 7,12 (dd, J=9,45, 3,15 Гц, 1 H), 6,99-7,05 (m, 1 H), 6,94-6,99 (m, 1 H), 6,36 (d, J=7,45 Гц, 1 H), 5,53 (m, 1 H), 4,45-4,52 (m, 1 H), 3,90 (ddd, J=13,46, 8,31, 4,01 Гц, 1 H), 3,10-3,17 (m, 1 H), 1,46 (d, J=6,30 Гц, 3 H), 1,44 (d, J=7,45 Гц, 3 H).
[0269] Пример 4: Получение кристаллической полиморфной формы 1 соединения I.
[0270] Твердое соединение I (5,55 г), полученное непосредственно из фракций очистки из примера 3, стадия 5, повторно растворяли в EA:ДХМ:MeOH (200:150:40) и указанный раствор концентрировали до объема примерно 70 мл с удалением большей части ДХМ и метанола. Образовалось белое кристаллическое твердое вещество. Указанное белое кристаллическое твердое вещество фильтровали с получением кристаллической полиморфной формы 1 соединения I. Аналит. вычислено для C18H18FN5O2: C, 60,84; H, 5,11; N, 19,71. Обнаружено: С 60,54; Н, 5,48; N 19,88.
[0271] Крупномасштабный синтез соединения I
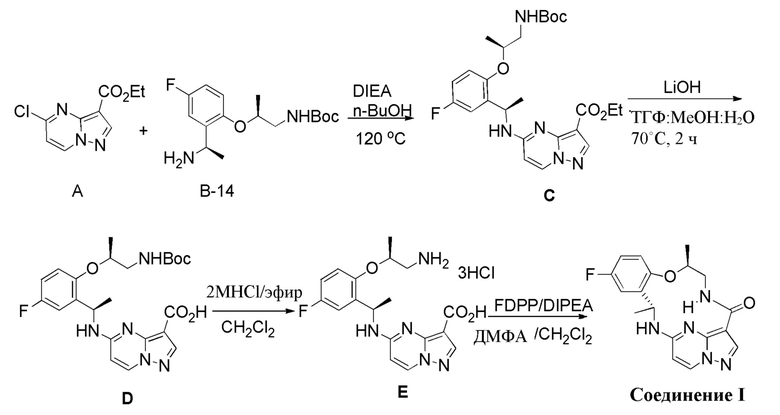
[0272] Пример 5: Синтез соединения С
[0273] К раствору А (126,40 г, 560,22 ммоль, 1,00 экв.) и B-14 (175,00 г, 560,22 ммоль, 1,00 экв.) в н-BuOH (1,70 л) добавляли DIEA (485,09 г, 3,75 моль, 655,53 мл, 6,70 экв.) при температуре 25°С. Указанную смесь перемешивали при температуре 120°С в течение 5 часов. ЖХМС показала, что исходный материал полностью израсходован. Растворитель удаляли и к остатку добавляли воду (1 л) и затем разбавляли EtOAc (1 л), экстрагировали EtOAc (2 л x 3). Объединенные органические слои промывали солевым раствором (1 л), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=10/1 до 1:1) с получением соединения С (174,00 г, 346,92 ммоль, выход 61,93%) в виде белого твердого вещества. ЖХ-МС: m/z 502,2 (M+H+). 1H ЯМР (400 MГц, хлороформ-d) δ 8,22 (s, 1H), 8,19 (d, J=7,6 Гц, 1H), 7,04 (d, J=8,4 Гц, 1H), 6,86 (d, J=5,2 Гц, 2H), 6,14 (d, J=6,4 Гц, 1H), 6,06 (шир. s., 1H), 5,52 (шир. s., 2H), 4,57 (d, J=3,2 Гц, 1H), 4,40-4,28 (m, 2H), 3,51-3,31 (m, 2H), 1,53 (d, J=6,8 Гц, 3H), 1,47-1,32 (m, 12H), 1,29 (d, J=6,0 Гц, 3H).
[0274] Пример 6: Синтез соединения D
[0275] К раствору 5-(((R)-1-(2-(((S)-1-((трет-бутоксикарбонил)амино)пропан-2-ил)окси)-5-фторфенил)этил)амино)пиразоло[1,5-a]пиримидин-3-карбоксилата (C) (140,00 г, 279,13 ммоль, 1 экв.) в метаноле (644 мл) и ТГФ (252 мл) добавляли водный раствор LiOH (3,3М, 504 мл, 1,6632 ммоль, 5,95 экв.). Прозрачный раствор нагревали при температуре 70°С в течение 2,5 часов. Реакционную смесь охлаждали на ледяной бане и затем гасили с помощью водн. HCl (3,3 М, 504 мл) для получением значения рН<5. Реакционную смесь экстрагировали CH2Cl2 (1 л и 2×500 мл). Объединенные экстракты промывали солевым раствором и сушили над Na2SO4. После фильтрования, выпаривания и сушки в высоком вакууме получали белое твердое вещество, 5-(((R)-1-(2-(((S)-1-((трет-бутоксикарбонил)амино)пропан-2-ил)окси)-5-фторфенил)этил)амино)пиразоло[1,5-a]пиримидин-3-карбоновую кислоту (D) (140,78 г). Указанный продукт использовали на следующем этапе без дополнительной очистки.
[0276] Пример 7: Синтез соединения E
[0277] К раствору 5-(((R)-1-(2-(((S)-1-((трет-бутоксикарбонил)амино)пропан-2-ил)окси)-5-фторфенил)этил)амино)пиразоло[1,5-a]пиримидин-3-карбоновой кислоты (D) (132,17 г, 279,13 ммоль) в CH2Cl2 (659 мл) добавляли HCl в диэтиловом эфире (2М, 497 мл) при температуре окружающей среды. Реакционную смесь перемешивали при температуре окружающей среды в течение 22 часов и добавляли дополнительный раствор HCl в диэтиловом эфире (2М, 100 мл) и перемешивали в течение 5 часов. Твердый продукт фильтровали, промывали диэтиловым эфиром и сушили в высоком вакууме с получением соединения Е в виде 3 HCl-соли, которую непосредственно использовали на следующей стадии.
[0278] Пример 8: Синтез соединения I из соединения E
[0279] 5-(((R)-1-(2 -(((S)-1-аминопропан-2-ил)окси)-5-фторфенил)этил)амино)пиразоло[1,5-а]пиримидин-3- карбоновую кислоту (E) (208 г, 557,07 ммоль) (полученную из 280 г C) растворяли в ДМФА (1,1 л) и основании Хьюнига (300 мл) с получением раствора А (~ 1700 мл). К двум 4-литровым реакционным колбам добавляли ДМФА (608 мл), ДХМ (3,19 л) и основание Хьюнига (500 мл), соответственно. В каждую реакционную колбу добавляли раствор А (160 мл) с последующим добавлением FDPP (20 г, 36,61 ммоль). Реакционную смесь перемешивали в течение одного часа, и ЖХМС показала, что соединение Е было полностью израсходовано. Тот же процесс повторяли до тех пор, пока не добавили в две указанные реакционные колбы весь раствор А. После последнего добавления к каждой колбе добавляли дополнительный FDPP (10 г) и конечные растворы перемешивали при температуре окружающей среды в течение ночи. Реакционный раствор из одной реакционной колбы концентрировали до 1,5 л и затем разбавляли ДХМ (4 л) и промывали водным раствором Na2CO3 (2М, 3 л). Водный слой экстрагировали ДХМ (3×700 мл). Объединенные органические слои промывали водным раствором Na2CO3 (1М, 2 л), водой (2 л) и сушили над Na2SO4. Ту же самую процедуру обработки применяли ко второй реакционной колбе. Объединенные растворы фильтровали и концентрировали при пониженном давлении до объема ~ 600 мл. Во время концентрации наблюдали большое количество осадка, который перемешивали при температуре 0°C в течение 0,5 часа и затем фильтровали с получением твердого продукта (145 г). Фильтрат конденсировали досуха и остаток повторно растворяли в ДХМ (1 л) и промывали водным раствором HCl (0,4 М, 500 мл), Na2CO3 (2М, 1 л), водой (1 л), и сушили над Na2SO4. После фильтрования раствор концентрировали до примерно 100 мл и отделяли осадок, который фильтровали и промывали диэтиловым эфиром (50 мл) с получением дополнительного продукта (10,7 г). Объединенное твердое вещество повторно растворяли в 10% метаноле в ДХМ (2 л) и фильтровали с получением прозрачного раствора, который дополнительно разбавляли метанолом (500 мл). Раствор концентрировали при пониженном давлении до ~ 400 мл и охлаждали при температуре 0 o C в течение 1 часа. Твердое вещество отфильтровывали и промывали холодным метанолом (2×60 мл) и диэтиловым эфиром (2×75 мл) и сушили в высоком вакууме с получением (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]-бензоксатриазациклотридецин-4(5Н)-она (соединение I) (145,302 г). Фильтрат концентрировали до ~ 120 мл и затем охлаждали при температуре 0°C в течение 30 минут с получением дополнительного вещества (4,815 грамма). В общей сложности получали 150,12 г (выход 75,8% для трех стадий: гидролиз, де-boc и циклизация) (соединение I) с чистотой >98%. ЖХ-МС: m/z [M+H]+ 356,2. 1H ЯМР (500 MГц, ДМСО-d6) δ ppm 9,82 (dd, J=8,02, 2,29 Гц, 1 H), 8,81 (d, J=6,87 Гц, 1 H), 8,58 (d, J=7,45 Гц, 1 H), 8,04 (s, 1 H), 7,12 (dd, J=9,45, 3,15 Гц, 1 H), 6,99-7,05 (m, 1 H), 6,94-6,99 (m, 1 H), 6,36 (d, J=7,45 Гц, 1 H), 5,53 (m, 1 H), 4,45-4,52 (m, 1 H), 3,90 (ddd, J=13,46, 8,31, 4,01 Гц, 1 H), 3,10-3,17 (m, 1 H), 1,46 (d, J=6,30 Гц, 3 H), 1,44 (d, J=7,45 Гц, 3 H).
[0280] Синтез соединения A
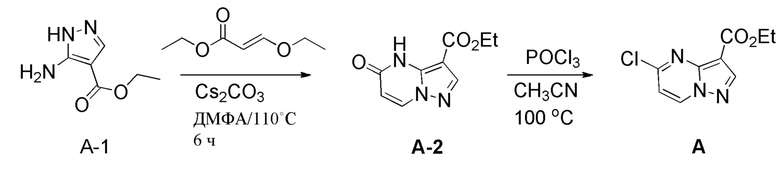
[0281] Пример 9: Получение этил 5-оксо-4Н-пиразоло[1,5-a]пиримидин-3-карбоксилата (А-2)
[0282] Этиловый эфир 3-амино-1Н-пиразол-4-карбоновой кислоты (A-1) (Langchem Inc, 2,0 кг, 12,9 моль) загружали в 50-литровый реактор с рубашкой. Затем добавляли ДМФА (для промышленного применения, 20 л) с последующим добавлением этил-3-этоксиакрилата (LightChem, 3,5 кг, 24,5 моль, 1,9 экв.) и Cs2CO3 (NuoTai Chem, 8,0 кг, 24,5 моль, 1,9 экв.). Реакционную смесь нагревали до температуры 110~115°С в течение 40 мин и перемешивали при этой температуре в течение ночи. Пар-ионная хроматография на ЖХМС показала, что почти все исходные материалы были израсходованы. Затем указанный раствор охлаждали до температуры окружающей среды в течение 1 часа с получением смеси. Полученное твердое вещество собирали фильтрованием и промывали EtOAc (6 л). Твердое вещество собирали и растворяли в воде (20 л). Раствор затем подкисляли ледяной уксусной кислотой (6,5 л) до рН 4. Во время подкисления экзотермической реакции не наблюдалось. Полученное твердое вещество собирали фильтрованием и промывали водой (10 л). Твердое вещество сушили при температуре 50°С в вакууме в течение 15 часов с получением А-2 (2,3 кг, >99,9%, выход 87%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,54 (d, J=7,91 Гц, 1H), 8,12 (s, 1H), 6,13 (d, J=7,91 Гц, 1H), 4,27 (q, J=7,11 Гц, 2H), 1,28 (t, J=7,09 Гц, 3H).
[0283] Пример 10: Получение 5-хлорпиразоло[1,5-a]пиримидин-3-карбоксилата (А).
[0284] А-2 (2,3 кг, 11,1 моль) загружали в 50-литровый реактор с рубашкой. Добавляли MeCN (для промышленного применения, 23 л) и охлаждали до температуры 15-20°С при перемешивании. В смесь в течение 10 мин добавляли чистый POCl3 (8,5 кг, 55,5 моль, 5,0 экв.) без изменений в температуре реактора. Реакционную смесь нагревали до температуры 100~105°С в течение 1 часа и затем перемешивали при этой температуре в течение 2 часов. Пар-ионная хроматография на ЖХМС показала, что исходный материал был израсходован. Затем реакционный раствор переносили в реактор объемом 100 л с рубашкой, содержащий ледяную воду (50 л, 5°C), в течение 1 часа. Скорость добавления контролировали так, чтобы экзотермическая реакция не превышала внутреннюю температуру 30°C. Полученную суспензию перемешивали при температуре 15-20°С в течение 30 мин. Образовавшееся твердое вещество отфильтровывали и сушили при температуре 45°С в вакууме в течение 36 часов с получением A (1,8 кг, чистота 99,9% по ЖХМС, выход 72%) в виде белого твердого вещества: 1H ЯМР (400 MГц, ДМСО-d6) δ 9,33 (d, J=7,28 Гц, 1H), 8,66 (s, 1H), 7,41 (d, J=7,15 Гц, 1H), 4,31 (q, J=7,15 Гц, 2H), 1,32 (t, J=7,09 Гц, 3H).
[0285] Синтез соединения B - способ A
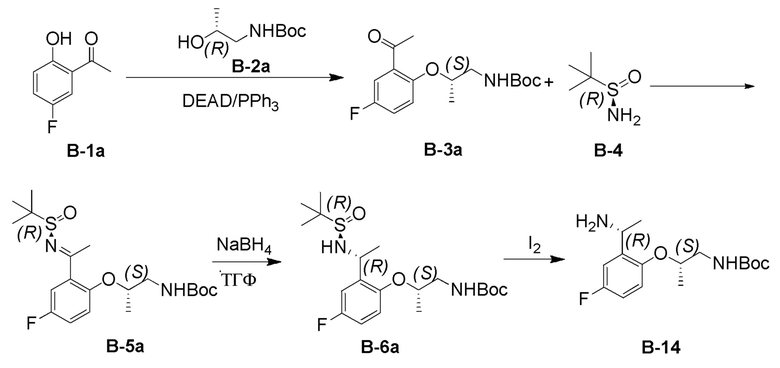
[0286] Пример 11: Получение трет-бутил (R)-(2-гидроксипропил)карбамата (B-2a)
[0287] К раствору (R)-1-аминопропан-2-ола (600,00 г, 7,99 моль, 631,58 мл, 1,00 экв.) и TEA (808,33 г, 7,99 моль, 1,11 л, 1,00 экв.) в ДХМ (3,00 л) добавляли (Boc)2O (1,74 кг, 7,99 моль, 1,84 л, 1,00 экв.). Реакционную смесь перемешивали при температуре 25°C в N2 течение 5 часов. ТСХ показала завершение реакции. Реакционную смесь распределяли между насыщенным NaHCO3 (500 мл) и ДХМ (1 л) и затем промывали солевым раствором (1 л). Органический слой сушили над безводным Na2SO4 и выпаривали с получением соединения B-2a (1,37 кг, 7,82 моль, выход 97,86%) в виде масла. 1H ЯМР (400 MГц, хлороформ-d) δ 5,00 (шир. s., 1H), 3,89 (s, 1H), 3,25 (dd, J=2,8, 10,4 Гц, 1H), 3,00 (td, J=6,8, 13,6 Гц, 1H), 2,71-2,50 (m, 1H), 1,44 (s, 9H), 1,17 (d, J=6,4 Гц, 3H).
[0288] Пример 12: Получение трет-бутил (S)-(2-(2-ацетил-4-фторфенокси)пропил)карбамата (B-3a)
[0289] К раствору B-1a (500,00 г, 3,24 моль, 1,00 экв.), B-2a (851,57 г, 4,86 моль, 1,50 экв.) и PPh3 (1,27 кг, 4,86 моль, 1,50 экв.) в дихлорметане (1,5 л) добавляли DEAD (902,79 г, 5,18 моль, 940,41 мл, 1,60 экв.) по каплям при температуре 0 °С. Раствор перемешивали при температуре 25 °С в течение 4 часов. ТСХ показала, что было обнаружено одно большое новое пятно с большей полярностью, и что исходный материал был полностью израсходован. К смеси добавляли петролейный эфир (1,5 л), затем фильтровали твердое вещество, растворитель фильтрата удаляли и остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=от 20/1 до 10:1) с получением B-3a (680,00 г, 2,18 моль, выход 67,28%) в виде красного масла. 1H ЯМР (400 MГц, хлороформ-d) δ 7,38 (dd, J=3,2, 8,8 Гц, 1H), 7,13 (ddd, J=3,2, 7,2, 8,8 Гц, 1H), 6,97 (dd, J=4,0, 8,8 Гц, 1H), 5,06 (шир. s., 1H), 4,63-4,52 (m, 1H), 3,52-3,39 (m, 1H), 3,38-3,27 (m, 1H), 2,59 (s, 3H), 1,42 (s, 9H), 1,32 (d, J=6,4 Гц, 3H).
[0290] Пример 13: Получение трет-бутил ((S)-2-(2-((E)-1-(((R)-трет-бутилсульфинил)имино)-этил)-4-фторфенокси)пропил)карбамата (B-5a)
[0291] К смеси B-4 (219,98 г, 1,82 моль, 1,50 экв.), диглиму (162,35 г, 1,21 моль, 172,71 мл, 1,00 экв.) и B-3a (376,00 г, 1,21 моль, 1,00 экв.) в ТГФ (1,88 л) и 2-метилтетрагидрофуране (1,88 л) добавляли тетраэтоксититан (552,03 г, 2,42 моль, 501,85 мл, 2,00 экв.) одной порцией при температуре 20°С в атмосфере N2. Смесь перемешивали при температуре 60°C в течение 12 часов. ТСХ показала, что оставалось примерно 15% исходного материала. Указанную смесь охлаждали до температуры 20°С. Добавляли воду (2 л). Водную фазу экстрагировали этилацетатом (2000 мл ×3). Объединенную органическую фазу промывали насыщенным солевым раствором (1 л), сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением B-5a (520,00 г, неочищенный) в виде красного масла, которое использовали на следующей стадии без дополнительной очистки.
[0292] Пример 14: Получение трет-бутил ((S)-2-(2-((R)-1-(((R)-трет-бутилсульфинил)амино)-этил)-4-фторфенокси)пропил)карбамата (B-6a)
[0293] К раствору B-5a (520,00 г, 1,25 моль, 1,00 экв.) в ТГФ/H2O (3,82 л/78 мл) добавляли NaBH4 (142,37 г, 3,76 моль, 3,00 экв.) при температуре -50°С, затем указанную реакционную смесь нагревали до температуры 25°С и перемешивали при температуре 25°С в течение 12 часов. ТСХ показала, что исходный материал полностью израсходован. К смеси добавляли воду (1 л) и экстрагировали EtOAc (2 л ×2). Органический слой промывали насыщенной NaCl (1 л) и сушили над Na2SO4. Удаляли растворитель и остаток очищали колоночной хроматографией (SiO2, петролейный эфир/этилацетат=от 20/1 до 10:1) с получением B-6a (270,00 г, 570,40 ммоль, выход 45,47%). 1H ЯМР (400 MГц, хлороформ-d) δ 7,06 (dd, J=3,2, 9,2 Гц, 1H), 6,95 (dt, J=3,2, 8,4 Гц, 1H), 6,80 (dd, J=4,4, 9,2 Гц, 1H), 6,70 (шир. s., 1H), 4,93 (d, J=6,0 Гц, 1H), 4,57-4,46 (m, 1H), 3,68-3,65 (m, 1H), 3,59-3,57 (m, 1H), 3,22-3,10 (m, 1H), 1,47 (d, J=6,8 Гц, 3H), 1,40 (s, 9H), 1,27-1,25 (m, 3H), 1,22 (s, 9H).
[0294] Пример 15: Получение трет-бутил ((S)-2-(2-((R)-1-аминоэтил)-4-фторфенокси)пропил)карбамата (B-14)
К раствору B-6a (270,00 г, 570,40 ммоль, 1,00 экв.) и молекулярного йода (28,95 г, 114,08 ммоль, 22,98 мл, 0,20 экв.) в ТГФ (2,16 л) добавляли H2O (540,00 мл) при температуре 25°С под N2. Смесь перемешивали при температуре 50°C в течение 3 часов. ТСХ показала, что исходный материал полностью израсходован. Смесь концентрировали с получением B-14 (330,00 г, неочищенный) в виде белого твердого вещества. ЖХ-МС: m/z 313,2 (M+H+). 1H ЯМР (400 MГц, хлороформ-d) δ 7,08 (dd, J=2,8, 9,4 Гц, 1H), 6,91-6,79 (m, 2H), 5,72 (шир. s., 1H), 4,55-4,32 (m, 2H), 3,52-3,41 (m, 1H), 3,31-3,19 (m, 1H), 1,42 (s, 9H), 1,38 (d, J=6,8 Гц, 3H), 1,29 (d, J=6,0 Гц, 3H).
[0295] Синтез соединения B-14 - способ B
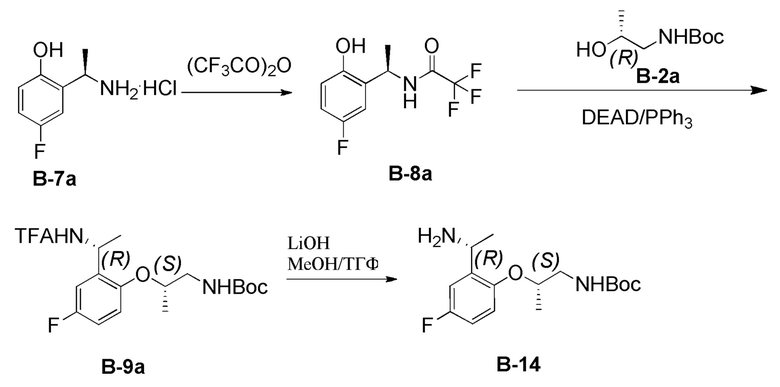
[0296] Пример 16: Получение (R)-2,2,2-трифтор-N-(1-(5-фтор-2-гидроксифенил)-этил)ацетамида (B-8a)
[0297] К раствору (R)-2-(1-аминоэтил)-4-фторфенола гидрохлорида (NetChem, 1,00 г, 5,22 ммоль) и триэтиламина (1,58 г, 15,66 ммоль) в ДХМ (26,10 мл) при температуре 0 °С добавляли трифторуксусный ангидрид (1,26 г, 6,00 ммоль). Реакционный раствор перемешивали в течение 2 часов при температуре 0 °C, затем гасили добавлением 0,5 М водного раствора HCl (100 мл). Указанную смесь экстрагировали ДХМ (3×50 мл). Объединенные экстракты промывали 0,5 М раствором HCl (2×50 мл), водой (100 мл) и солевым раствором (50 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением B-8a (1,203 г, выход 91,5%). ЖХ-МС: m/z 252 (M+H+).
[0298] Пример 17: Получение трет-бутил ((S)-2-(4-фтор-2-((R)-1-(2,2,2-трифторацетамидо)этил)фенокси)пропил)карбамата (B-9a)
[0299] Смесь (R)-2,2,2-трифтор-N-(1-(5-фтор-2-гидроксифенил)этил)ацетамид (1,20 г, 4,78 ммоль) и трет-бутила (R)-(2-гидроксипропил)карбамата (1,68 г, 9,56 ммоль) азеотропно сушили из ДХМ:толуола. Затем остаток повторно растворяли в ДХМ (2,00 мл) и к раствору добавляли PPh3 (2,57 г, 9,80 ммоль). Смесь перемешивали до полного растворения всех реагентов. Раствор охлаждали до температуры 0°С и при перемешивании очень медленно добавляли DIAD (1,98 г, 9,80 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 часов, а затем нагревали до температуры 35°С и перемешивали в течение 16 часов. Смесь концентрировали при пониженном давлении и использовали на следующей стадии без очистки. ЖХ-МС: m/z 431 (M+Na+).
[0300] Пример 18: Получение трет-бутил ((S)-2-(2-((R)-1-аминоэтил)-4-фторфенокси)пропил)карбамата (B-14)
[0301] К раствору неочищенного трет-бутил ((S)-2-(4-фтор-2-((R)-1-(2,2,2-трифторацетамидо)этил)фенокси)пропил)карбамата (1,95 г, 4,77 ммоль) в MeOH (15 мл) и ТГФ (5 мл) добавляли 2 М водный раствор LiOH (7,03 мл). Смесь нагревали при температуре 50°С в течение 6 часов, охлаждали до комнатной температуры и разбавляли водой (100 мл) и 2 М раствором NaOH (25 мл) и экстрагировали ДХМ (3×75 мл). Объединенные экстракты промывали 2 М раствором NaOH (75 мл), сушили над Na2SO4, концентрировали при пониженном давлении и сушили в высоком вакууме. Остаток растворяли в 1:1 ДХМ:гексан (100 мл) и экстрагировали 0,5 М HCl в вода:MeOH 9:1 (3×60 мл). Объединенные водные экстракты промывали 1:3 ДХМ:гексан (100 мл), нейтрализовывали 2 М раствором NaOH (100 мл) и экстрагировали ДХМ (3×100 мл). Объединенные экстракты ДХМ сушили над Na2SO4, концентрировали при пониженном давлении и сушили в высоком вакууме с получением белого твердого вещества B-14 (797,6 мг, выход 53% для комбинированных трех стадий). ЖХ-МС: m/z 313,2 (M+H+). 1H ЯМР (400 MГц, хлороформ-d) δ 7,08 (dd, J=2,8, 9,4 Гц, 1H), 6,91-6,79 (m, 2H), 5,72 (шир. s., 1H), 4,55-4,32 (m, 2H), 3,52-3,41 (m, 1H), 3,31-3,19 (m, 1H), 1,42 (s, 9H), 1,38 (d, J=6,8 Гц, 3H), 1,29 (d, J=6,0 Гц, 3H).
[0302] Синтез соединения B-14 - способ C
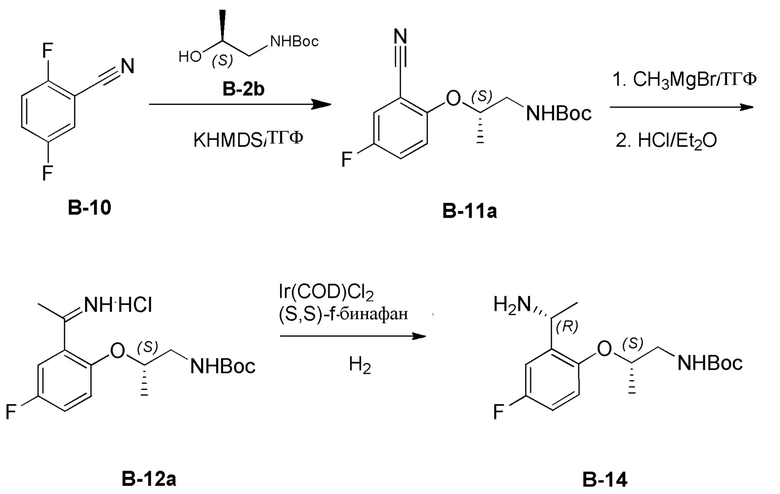
[0303] Пример 19: Получение трет-бутил (S)-(2-(2-циано-4-фторфенокси)пропил)карбамата (B-11a)
[0304] К раствору трет-бутил (S)-(2-гидроксипропил)карбамата (1,32 г, 7,55 ммоль) и 2,5-дифторбензонитрила (Aldrich, 1,00 г, 7,19 ммоль) в ТГФ (48 мл) при температуре 0°С добавляли KHMDS (1 М, 7,55 мл). Указанную реакционную смесь нагревали до комнатной температуры и перемешивали в течение 18 часов под азотом. Раствор концентрировали, разбавляли ДХМ (150 мл), промывали 0,1 М HCl (3×150 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Флэш-хроматография (система ISCO, диоксид кремния (24 г), 0-25% этилацетат в гексане) давала B-11a (1,43 г, 4,86 ммоль, выход 67,58%). ЖХ-МС: m/z 317 (M+Na+). 1H ЯМР (500 MГц, ДМСО-d6) δ ppm 7,72 (dd, J=8,31, 3,15 Гц, 1 H), 7,51-7,59 (m, 1 H), 7,34 (dd, J=9,45, 4,30 Гц, 1 H), 7,08 (t, J=5,73 Гц, 1 H), 4,58 (секст., J=5,96 Гц, 1 H), 3,15-3,25 (m, 1 H), 3,06-3,13 (m, 1 H), 1,36 (s, 9 H), 1,24 (d, J=5,73 Гц, 3 H).
[0305] Пример 20: Получение трет-бутил (S)- (2-(4-фтор-2-(1-иминоэтил)фенокси)пропил)-карбамат гидрохлорида (B-12a)
[0306] К раствору трет-бутил (S)-(2-(2-циано-4-фторфенокси)пропил)карбамата (100,00 мг, 0,34 ммоль) в ТГФ (1,70 мл) добавляли MeMgBr (3 М, 0,34 мл) при температуре -78°C. Раствор нагревали до комнатной температуры и перемешивали в течение 4 часов. Реакцию гасили МеОН (475,20 мг, 14,83 ммоль) при температуре -78°С, затем нагревали до комнатной температуры и перемешивали в течение 2 часов. Реакционный раствор фильтровали через слой целита, концентрировали досуха при пониженном давлении и сушили в высоком вакууме. Затем указанный остаток повторно растворяли в МТБЭ:ДХМ (1:3, 5 мл) и охлаждали до температуры 0°С с последующим добавлением эфирного раствора HCl (2 М, 0,17 мл). Указанный реакционный раствор нагревали до комнатной температуры и перемешивали в течение 1 часа. После концентрирования остаток суспендировали в МТВЕ (4 мл) и фильтровали. Твердое вещество промывали МТБЭ и сушили в высоком вакууме с получением B-12а (62,7 мг, выход 52,7%). ЖХ-МС: m/z 334 (M+Na+). 1H ЯМР (500 MГц, ДМСО-d6) δ ppm 12,61 (bs, 1H), 11,70 (bs, 1H), 7,77 (dd, J=9,45, 3,15 Гц, 1 H), 7,61-7,68 (m, 1 H), 7,43 (dd, J=9,45, 4,30 Гц, 1 H), 7,26 (t, J=5,73 Гц, 1 H), 4,68-4,76 (m, 1 H), 3,14-3,24 (m, 1 H), 3,13-3,25 (m, 1 H), 2,81 (s, 3 H), 1,36 (s, 9 H), 1,25 (d, J=6,30 Гц, 3 H).
[0307] Пример 21: Получение трет-бутил ((S)-2-(2-((R)-1-аминоэтил)-4-фторфенокси)-пропил)карбамата гидрохлорида (B-14)
[0308] К хорошо перемешанному раствору [Ir(COD)Cl]2 (Strem Chemicals, 2,1 мг, 0,003 ммоль) и (S,S)-f-бинафана (Strem Chemicals, 5,1 мг, 0,006 ммоль) в CH2Cl2 (1 мл) добавляли трет-бутил (S)-(2-(4-фтор-2-(1-иминоэтил)фенокси)пропил)карбамата гидрохлорид (0,3 ммоль) в МеОН (2 мл). Затем реакционный сосуд помещали в стальной автоклав. Инертную атмосферу заменяли на H2 и реакционную смесь перемешивали при 10 атмосферах H2 (150 фунтов на квадратный дюйм) при температуре окружающей среды в течение 12 часов. Полученную смесь концентрировали в вакууме и растворяли в насыщенном водном растворе NaHCO3 (5 мл). После перемешивания в течение 10 минут указанную смесь экстрагировали CH2Cl2 (3×2 мл), сушили над Na2SO4, концентрировали и сушили в высоком вакууме с получением соединения B-14. 1H ЯМР (400 MГц, хлороформ-d) δ 7,08 (dd, J=2,8, 9,4 Гц, 1H), 6,91-6,79 (m, 2H), 5,72 (шир. s., 1H), 4,55-4,32 (m, 2H), 3,52-3,41 (m, 1H), 3,31-3,19 (m, 1H), 1,42 (s, 9H), 1,38 (d, J=6,8 Гц, 3H), 1,29 (d, J=6,0 Гц, 3H).
[0309] Синтез соединения B-14 - способ D
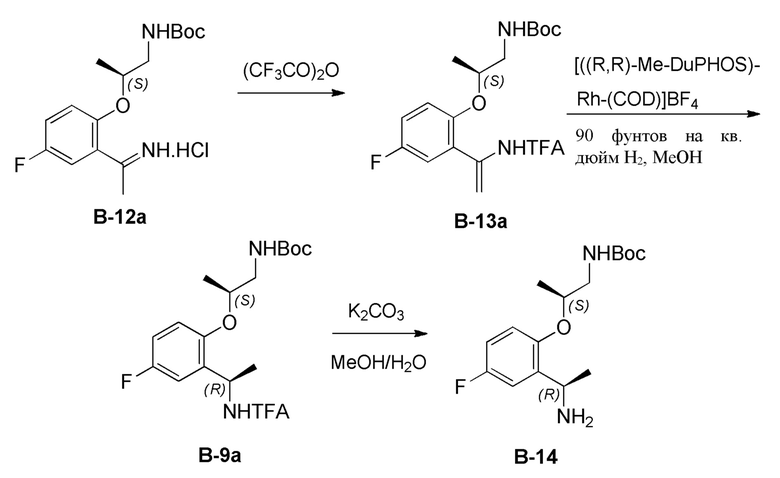
[0310] Пример 22: Получение трет-бутил (S)-2-(4-фтор-2-(1-(2,2,2-трифторацетамидо)-винил)фенокси)пропил)карбамата (B-13a)
[0311] К раствору трет-бутил (S)-(2-(4-фтор-2-(1-иминоэтил)фенокси)пропил)карбамата гидрохлорида (25,00 мг, 0,072 ммоль) и трифторуксусного ангидрида (17,41 мг, 0,083 ммоль) в ДХМ (0,36 мл) при температуре 0°С добавляли триэтиламин (43,76 мг, 0,432 ммоль). Реакционный раствор перемешивали в течение 3 часов при 0°С, гасили добавлением 0,5 М водного раствора HCl (25 мл), экстрагировали ДХМ (150 мл). Экстракт промывали 0,5 М водным раствором HCl (25 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Флэш-хроматография (система ISCO, диоксид кремния (24 г), 0-50% этилацетат в гексане) давала B-13a (15,70 мг, 0,038 ммоль, выход 53,60%). ЖХ-МС: m/z 429 (M+Na+). 1H ЯМР (500 MГц, ДМСО-d6) δ ppm 10,70 (s, 1 H) 7,13-7,22 (m, 1 H) 7,06-7,13 (m, 2 H) 6,92 (t, J=6,01 Гц, 1 H) 5,66 (s, 1 H) 5,18 (s, 1 H) 4,42 (q, J=6,30 Гц, 1 H) 3,17 (dt, J=13,75, 6,01 Гц, 1 H) 2,91-3,01 (m, 1 H) 1,35 (s, 9 H) 1,13 (d, J=6,30 Гц, 3 H).
[0312] Пример 23: Получение трет-бутил ((S)-2-(4-фтор-2-((R)-1-(2,2,2-трифторацетамидо)этил)фенокси)пропил)карбамата (B-9a)
[0313] Трет-бутил (S)-(2-(4-фтор-2-(1-(2,2,2-трифторацетамидо)винил)фенокси)пропил)карбамат (3,00 ммоль) и [((R,R)-Me-DuPHOS)-Rh-(COD)]BF4 (Strem Chemicals, 0,2 мол.%) помещали в стеклянный сосуд высокого давления, который затем продували водородом три раза. Затем добавляли дегазованный метанол (10 мл) и сосуд далее продували водородом и загружали водородом до достижения давления 90 фунтов на квадратный дюйм (~620,5 кПа). После перемешивания в течение 20 часов реакционную смесь выпаривали с получением остатка. Указанный остаток растворяли в EtOAc (5 мл) и раствор фильтровали через короткую силикатную пробку для удаления остатков катализатора. Затем растворитель выпаривали с получением соединения В-9а.
[0314] Пример 24: Получение трет-бутил ((S)-2-(2-((R)-1-аминоэтил)-4-фторфенокси)пропил)карбамата гидрохлорида (B-14)
[0315] К раствору трет-бутил ((S)-2-(4-фтор-2-((R)-1-(2,2,2-трифторацетамидо)этил)фенокси)пропил)карбамата (1,00 ммоль) в метаноле (30 мл) и воде (10 мл) добавляли K2CO3 (3,00 ммоль). Указанную смесь нагревали при температуре 60°C до полного завершения гидролиза. Реакционную смесь трижды экстрагировали дихлорметаном (30 мл). Объединенные экстракты сушили над Na2SO4, фильтровали, концентрировали и сушили в высоком вакууме с получением соединения B-14. 1H ЯМР (400 MГц, хлороформ-d) δ 7,08 (dd, J=2,8, 9,4 Гц, 1H), 6,91-6,79 (m, 2H), 5,72 (шир. s., 1H), 4,55-4,32 (m, 2H), 3,52-3,41 (m, 1H), 3,31-3,19 (m, 1H), 1,42 (s, 9H), 1,38 (d, J=6,8 Гц, 3H), 1,29 (d, J=6,0 Гц, 3H).
[0316] Исследование кристаллической полиморфной формы 1 соединения I
[0317] Пример 25: Порошковая рентгеновская дифракция (PXRD) кристаллической полиморфной формы 1 соединения I.
[0318] Образец соединения I, кристаллическая полиморфная форма 1, измеряли на приборе Bruker D8 Advance, снабженном линейным детектором 1-D Lynxeye с использованием Cu-излучения (1,54178 Å). Образец вращали во время сбора данных для того, чтобы ограничить пики предпочтительной ориентации. Данные собирали на углах 2°-50° 2θ с шагом 0,02° и скоростью сканирования 0,25 сек на шаг. Результаты показаны на фиг. 1.
[0319] Пример 26: Дифференциальная сканирующая калориметрия (ДСК) кристаллической полиморфной формы 1 соединения I.
[0320] Измерения ДСК проводили с использованием дифференциального сканирующего калориметра Seiko, модель SSC/5200. Образец 7,16 мг соединения I, кристаллическая полиморфная форма 1, уравновешивали при температуре 30°C, а затем нагревали до температуры 380°C со скоростью 10°C/мин. Образец соединения I, кристаллическая полиморфная форма 1, показал температуру плавления 345,5°C. Результаты показаны на фиг. 2.
Биологические примеры
Пример 27: Анализы связывания киназы.
[001] Анализы связывания киназы проводили на DiscoveRx с использованием общего протокола KINOMEscan Kd (Fabian, M. A. et al., ʺA small molecule-kinase interaction map for clinical kinase inhibitors,ʺ Nat. Biotechnol. 2005, 23(3):329-36). Для большинства анализов помеченные киназой фаговые штаммы T7 были получены на хозяине E. coli, полученном из штамма BL21. E. coli выращивали до логарифмической фазы, заражали фагом T7 и инкубировали при встряхивании при температуре 32°C до лизиса. Лизаты центрифугировали и фильтровали для удаления клеточного дебриса. Оставшиеся киназы были получены в клетках HEK-293 и затем помечены ДНК для обнаружения qPCR. Магнитные гранулы, покрытые стрептавидином, обрабатывали биотинилированными маломолекулярными лигандами в течение 30 минут при комнатной температуре с получением аффинных смол для киназных анализов. Лигандированные гранулы блокировали избытком биотина и промывали блокирующим буфером (SeaBlock (Pierce), 1% BSA, 0,05% Tween 20, 1 мМ DTT) для удаления несвязанного лиганда и для уменьшения неспецифического связывания. Реакции связывания собирали путем комбинирования киназ, лигандированных аффинных гранул и тестируемых соединений в 1x связывающем буфере (20% SeaBlock, 0,17x PBS, 0,05% Tween 20, 6 мМ DTT). Все реакции проводили в 96-луночных планшетах из полистирола в конечном объеме 0,135 мл. Планшеты для анализа инкубировали при комнатной температуре с встряхиванием в течение 1 часа и аффинные гранулы промывали промывочным буфером (1×PBS, 0,05% Tween 20). Затем гранулы повторно суспендировали в буфере для элюирования (1×PBS, 0,05% Tween 20, 0,5 мкМ небиотинилированного аффинного лиганда) и инкубировали при комнатной температуре с встряхиванием в течение 30 минут. Концентрацию киназы в элюатах измеряли с помощью qPCR. По данным указанного метода, соединение I имело сродство связывания с JAK2 Kd=0,082 нМ и ALK Kd=5,7 нМ.
Пример 28: Создание стабильной клеточной линии EML4-ALK Ba/F3 и анализ пролиферации клеток.
[002] Ген EML4-ALK дикого типа (вариант 1) синтезировали в GenScript и клонировали в плазмиду pCDH-CMV-MCS-EF1-Puro (System Biosciences, Inc). Клеточную линию Ba/F3-EML4-ALK дикого типа получали путем инфицирования клеток Ba/F3 лентивирусом, содержащим EML4-ALK дикого типа. Стабильные клеточные линии выбирали путем обработки пуромицином, после чего удаляли IL-3. 5000 клеток высевали в 384-луночный белый планшет за ночь до обработки соединениями. Пролиферацию клеток измеряли при помощи системы CellTiter-Glo на основе люциферазы для определения АТФ (Promega) по протоколу производителя после 48 часов инкубации с различными концентрациями соединения. Определение IC50 выполнялось с использованием программного обеспечения GraphPad Prism (GraphPad, Inc., Сан Диего, Калифорния, США). Данные для соединения I представлены в таблице 2.
Пример 29: Анализы пролиферации клеток
[003] Клетки колоректальной клеточной линии KM 12 (несущие эндогенный гибридный ген TPM3-TRKA) культивировали в среде DMEM, дополненной 10% фетальной бычьей сыворотки и 100 ед/мл пенициллина/стрептомицина. 5000 клеток высевали в 384-луночный белый планшет за 24 часа до обработки соединениями. Пролиферацию клеток измеряли при помощи системы CellTiter-Glo на основе люциферазы для определения АТФ (Promega) по протоколу производителя после 72 часов инкубации. Определение IC50 выполнялось с использованием программного обеспечения GraphPad Prism (GraphPad, Inc., Сан Диего, Калифорния, США).
[004] Клетки колоректальной клеточной линии KM12 (несущие эндогенный гибридный ген TPM3-TRKA) культивировали в среде DMEM, дополненной 10% фетальной бычьей сывороткой и 100 ед/мл пенициллина/стрептомицина. Клетки клеточной линии SET-2 с эссенциальной тромбоцитемией (несущие эндогенную точечную мутацию JAK2 V618F) или клеточной линии лимфомы Т-клеток Karpas-299 (несущие эндогенный гибридный ген NPM-ALK) культивировали в среде RPMI, дополненной 10% фетальной бычьей сыворотки и 100 U/мл пенициллина/стрептомицина. 5000 клеток высевали в 384-луночный белый планшет за 24 часа до обработки соединениями. Пролиферацию клеток измеряли при помощи системы CellTiter-Glo на основе люциферазы для определения АТФ (Promega) по протоколу производителя после 72 часов инкубации. Определение IC50 выполнялось с использованием программного обеспечения GraphPad Prism (GraphPad, Inc., Сан Диего, Калифорния, США).
[005] Данные для соединения I представлены в таблице 2.
Таблица 2
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СИНТЕЗА ПРОТИВООПУХОЛЕВОГО СОЕДИНЕНИЯ И ЕГО ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2021 |
|
RU2837452C1 |
| СПОСОБ ПОЛУЧЕНИЯ АМИНОКИСЛОТНЫХ СОЕДИНЕНИЙ | 2013 |
|
RU2643146C2 |
| СПОСОБ ПРОИЗВОДСТВА ДЕГАРЕЛИКСА И ЕГО ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2011 |
|
RU2602042C2 |
| ЖИДКОФАЗНЫЕ ПУТИ ДЛЯ ГЕКСАПЕПТИДОВ WNT | 2019 |
|
RU2801268C2 |
| ИНГИБИТОРЫ ГИСТОНДЕАЦЕТИЛАЗЫ | 2016 |
|
RU2720678C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНПИРРОЛИДИН-2-ОНА И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2771314C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ АЗОТИСТОГО ИПРИТА | 2016 |
|
RU2715233C2 |
| ПРОИЗВОДНЫЕ ИЛИДА СУЛЬФОКСОНИЯ В КАЧЕСТВЕ ЗОНДОВ ДЛЯ ЦИСТЕИНОВОЙ ПРОТЕАЗЫ | 2019 |
|
RU2815018C2 |
| СИСТЕМЫ ДОСТАВКИ ЛЕКАРСТВ НА ОСНОВЕ МАЙТАНЗИНОИДА | 2018 |
|
RU2783076C2 |
| Синтез соединений хиназолина | 2021 |
|
RU2836663C1 |
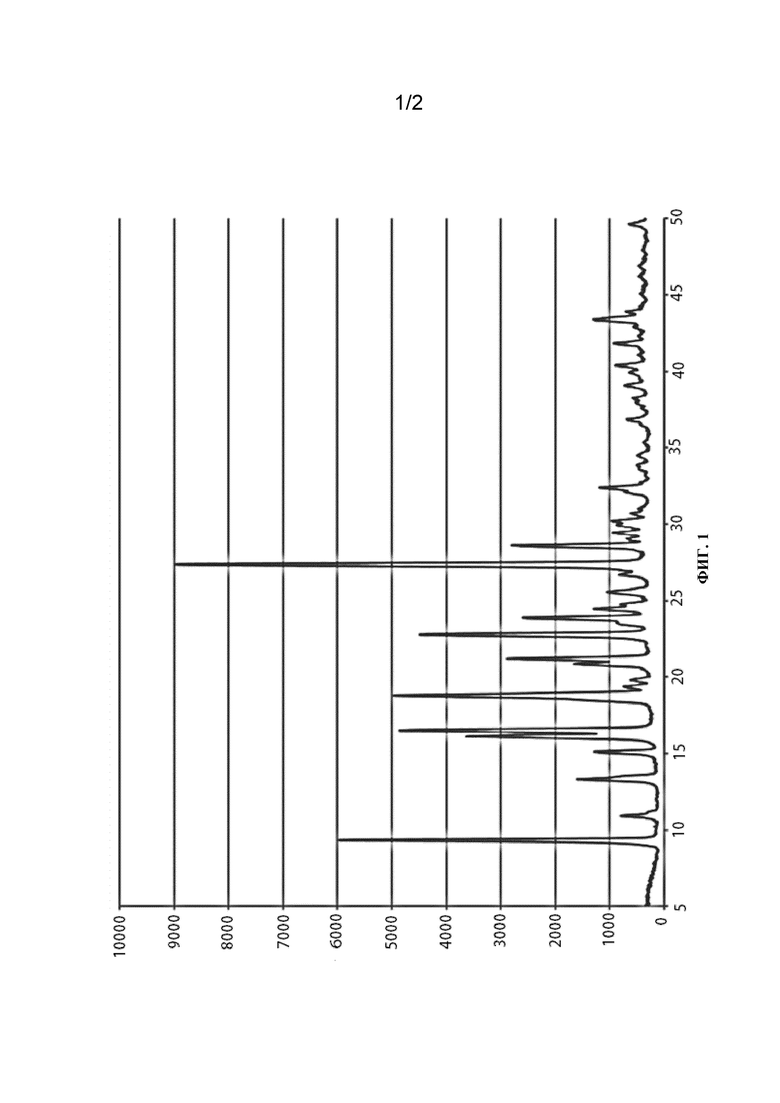
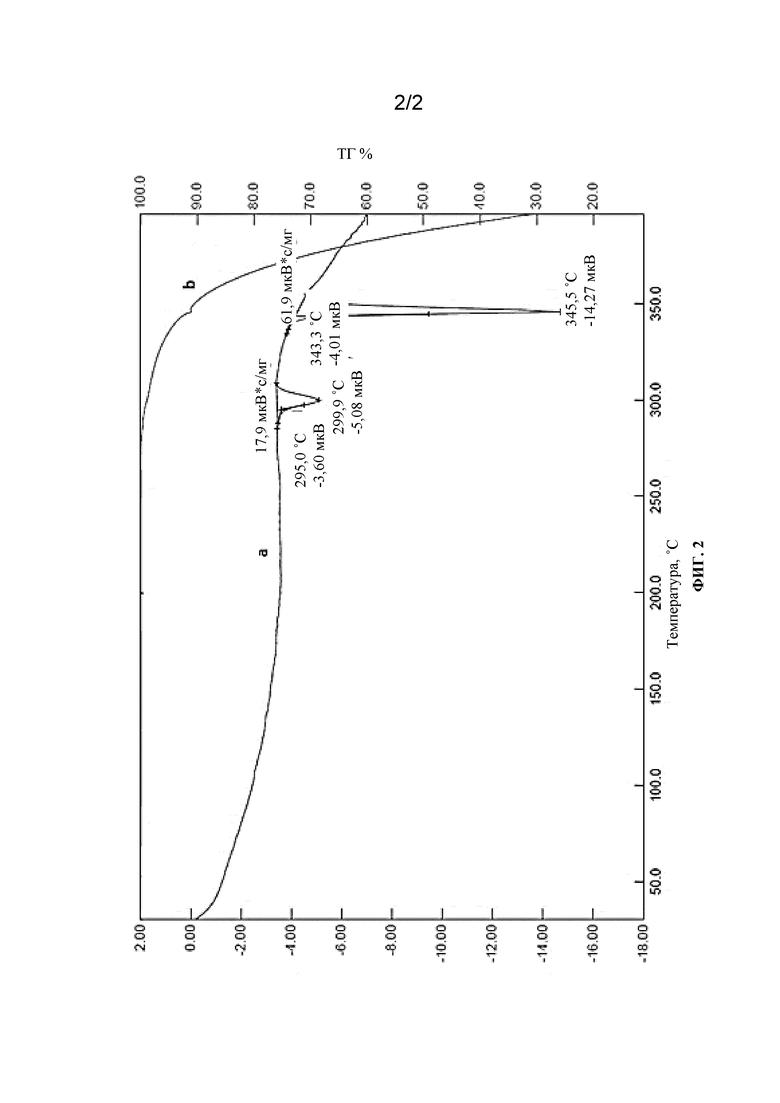
Изобретение относится к кристаллической полиморфной форме (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5Н)-она в форме свободного основания, имеющей порошковую рентгенограмму, содержащую пики при углах дифракции (2θ) 9,4±0,1, 16,1±0,1, 16,5±0,1, 18,8±0,1, 21,2±0,1, 22,8±0,1 и 27,4±0,1. Изобретение также относится к фармацевтической композиции, обладающей свойствами ингибитора тирозинкиназных рецепторов ALK, ROS1, TRK, JAK2, SRC и/или FAK, на основе указанного соединения. Технический результат – получена новая кристаллическая форма (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5Н)-она и фармацевтическая композиция на его основе, которые могут найти применение в медицине для лечения заболеваний, таких как рак. 4 н. и 5 з.п. ф-лы, 2 ил., 2 табл., 29 пр.
1. Кристаллическая полиморфная форма (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5Н)-она в форме свободного основания, имеющая порошковую рентгенограмму, содержащую пики при углах дифракции (2θ) 9,4±0,1, 16,1±0,1, 16,5±0,1, 18,8±0,1, 21,2±0,1, 22,8±0,1 и 27,4±0,1.
2. Кристаллическая полиморфная форма (7S,13R)-11-фтор-7,13-диметил-6,7,13,14-тетрагидро-1,15-этенпиразоло[4,3-f][1,4,8,10]бензоксатриазациклотридецин-4(5H)-она, имеющая порошковую рентгенограмму, по существу такую, как показано на фиг. 1.
3. Фармацевтическая композиция, обладающая свойствами ингибитора тирозинкиназных рецепторов ALK, ROS1, TRK, JAK2, SRC и/или FAK, содержащая терапевтически эффективное количество кристаллической полиморфной формы по п.1 или 2 и одно или несколько фармацевтически приемлемых вспомогательных веществ.
4. Фармацевтическая композиция по п.3, где одно или несколько фармацевтически приемлемых вспомогательных веществ выбраны из группы, состоящей из стабилизаторов, смазывающих веществ, поверхностно-активных веществ, разбавителей, антиоксидантов, связующих веществ, красителей, наполнителей, эмульгаторов или модификаторов вкуса.
5. Фармацевтическая композиция по п.3, где одно или несколько фармацевтически приемлемых вспомогательных веществ выбраны из группы, состоящей из разбавителей, дезинтегрирующих агентов, связывающих агентов, смазывающих агентов, подсластителей, ароматизаторов, красителей и консервантов.
6. Фармацевтическая композиция по любому из пп.3-5, где одно или несколько фармацевтически приемлемых вспомогательных веществ выбраны из группы, состоящей из лактозы, стеарата магния, маннита, глицерина, крахмалгликолята натрия, микрокристаллической целлюлозы, сорбита, альгината натрия, гидроксиметилцеллюлозы и карбоксиметилцеллюлозы.
7. Способ лечения рака, опосредованного активностью тирозинкиназных рецепторов ALK, ROS1, TRK, JAK2, SRC и/или FAK, у субъекта, включающий введение указанному млекопитающему терапевтически эффективного количества кристаллической полиморфной формы по п. 1.
8. Способ по п. 7, отличающийся тем, что указанный рак выбран из групп, состоящих из рака легкого, немелкоклеточного рака легкого, мелкоклеточного рака легкого, рака кости, рака поджелудочной железы, рака кожи, рака головы или шеи, гепатоцеллюлярной карциномы, кожной или внутриглазной меланомы, рака матки, рака яичников, рака прямой кишки, рака анальной области, рака желудка, рака толстой кишки, рака молочной железы, карциномы фаллопиевых труб, карциномы эндометрия, карциномы шейки матки, карциномы влагалища, карциномы вульвы, болезни Ходжкина, рака желудка и рака пищевода и желудка, рака эндокринной системы, рака щитовидной железы, рака паращитовидной железы, рака надпочечников, саркомы мягких тканей, рака уретры, рака полового члена, рака предстательной железы, хронического или острого лейкоза, лимфоцитарных лимфом, таких как анапластическая крупноклеточная лимфома, рака мочевого пузыря, рака почки или мочеточника, почечно-клеточной карциномы, карциномы почечной лоханки, новообразований центральной нервной системы (ЦНС), глиобластомы, первичной лимфомы ЦНС, опухолей оси позвоночника, глиомы ствола головного мозга, аденомы гипофиза, воспалительных миофибробластических опухолей и их комбинаций.
9. Способ по п. 8, отличающийся тем, что указанный рак представляет собой немелкоклеточный рак легкого.
| US 2013203776 A1, 08.08.2013 | |||
| US 2014206605 A1, 24.07.2014 | |||
| US 2013245021 A1, 19.09.2013 | |||
| ЕА 201100450 А1, 31.10.2011. |

