Для настоящего изобретения испрашивается приоритет на основании следующей заявки:
[1] CN 201811157825.9, дата подачи заявки 30 сентября 2018 г.
Область техники
[2] Настоящее изобретение относится к ряду хинолинпирролидин-2-оновых соединений и их применению для получения лекарственных средств для лечения связанных с ингибированием ATM заболеваний. Настоящее изобретение, в частности, относится к производному, представленному формулой (I), его таутомерам или его фармацевтически приемлемым композициям.
Уровень техники
[3] Мутантный при атаксии-телеангиэктазии (ataxia telangiectasia mutated, ATM) ген является геном с аутосомно-рецессивным типом наследования. У гомозиготных организмов наблюдается прогрессирующее нейродегенеративное заболевание. Заболевание начинается у пациента в возрасте примерно 1 года и проявляется в виде мозжечковой атаксии. В возрасте примерно 6 лет возникает опухолеподобное расширение мелких кровеносных сосудов в правом и левом глазу, на лице и шее. Часто в результате инфекции наступает смерть пациента. ATM ген является важным геном, связанным с репарацией повреждений ДНК, поэтому пациенты обычно проявляют особую чувствительность к рентгеновскому излучению и значительно сниженную способность к репарации ДНК. Примерно 1% людей являются гетерозиготными по мутантным ATM генам. Несмотря на то, что признаков заболевания у них не наблюдается, они также подвержены повышенному риску рака. ATM ген расположен на хромосоме 11q22-q23 и имеет общую длину 150 т.п.н., кодирующую последовательность длиной 12 т.п.н., и в сумме 66 экзонов. ATM ген является одним из генов человека с наибольшим количеством экзонов, обнаруженных к настоящему времени, он также является одним из наиболее важных генов и относится к генам «домашнего хозяйства».
[4] Продуктом, кодируемым ATM геном, является ATM белок, который представляет собой серин/треониновую протеинкиназу, содержащую 3056 аминокислот, имеет относительную молекулярную массу 370000, в основном расположен в ядре и микросомах и участвует в прохождении клеточного цикла и ответах на контрольные точки клеточного цикла при повреждении ДНК. ATM протеинкиназа, которая является членом семейства киназ, родственных фосфатидилинозитол-3-киназе (PIKK), представляет собой аутофосфорилированный белок и обычно существует в форме неактивного димера. Когда в ДНК происходит двухцепочечный разрыв, ATM протеинкиназа фосфорилируется и деполимеризуется в течение первых нескольких минут, и содержание фосфорилированной протеинкиназы достигает максимального значения через 2-3 часа.
[5] Сигнальный путь ATM белка при репарации повреждений ДНК в основном включает: сигнальный путь ATM-CHK2-Cdc25A/B/C; сигнальный путь ATM-CHK2-p53; сигнальный путь ATM-Nbs1-Smc1/3; и сигнальный путь ATM-p38MAPK-MK2. Процесс, с помощью которого ATM белок распознает двухцепочечные разрывы ДНК и фосфорилируется, включает участие комплексов MRN. M, то есть MRE11 (белок мейотической рекомбинации), обладает нуклеазной активностью и способностью связываться с ДНК; R означает Rad50, обладающий АТФазной активностью; N означает, что NBS1 участвует в локализации комплекса в ядре и способствует нормальной сборке комплекса в точке разрыва ДНК. Различные белки в комплексе MRN должны действовать согласованно друг с другом с целью регуляции связывания ATM белка с точкой разрыва ДНК для обеспечения завершения репарации поврежденной ДНК.
[6] ATM играет ключевую роль в репарации двухцепочечных разрывов ДНК. Поскольку вероятность двухцепочечных разрывов в нормальных клетках мала, селективные ингибиторы АТМ малоэффективны при применении отдельно. Однако, поскольку ATM является ключевым звеном всего пути репарации повреждений ДНК, многие возможные комбинации включают ингибиторы ATM. В настоящее время в доклинических и клинических исследованиях ингибиторы ATM комбинируют с лучевой терапией, химиотерапией и другими нацеленными ингибиторами репарации повреждений ДНК, такими как ингибиторы PARP. AZD0156 от AstraZeneca является первым соединением, которое поступило в клиническое исследование фазы I. В настоящее время AZD1390 и M-3541 от Merck, Германия, также вслед за ним поступают в клиническое исследование фазы I.
[7] Соответствующие заболевания, которые лечат с применением ингибиторов ATM киназы, представляют собой солидные опухоли, при этом указанные солидные опухоли включают, но не ограничиваются ими: рак легкого, рак молочной железы, рак головы и шеи, рак предстательной железы, лимфому, рак яичников, почечно-клеточную карциному, рак пищевода, лейкоз, рак мочевого пузыря, рак желудка, меланому, уротелиальную карциному, опухоль головного мозга, колоректальный рак, рак печени, мезотелиому, карциному внутрипеченочного желчного протока и так далее.
AZD0156
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
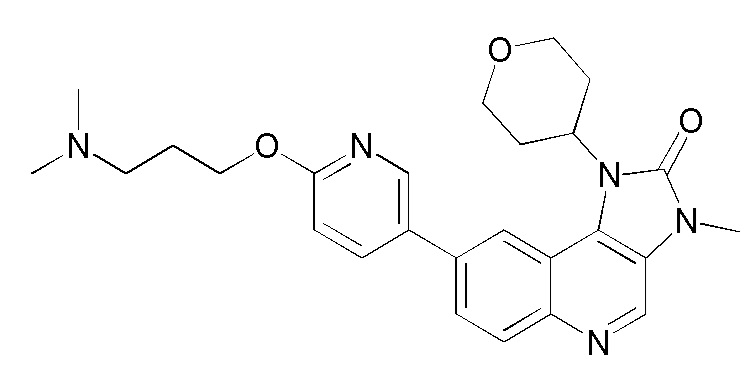
[8] В настоящем изобретении предложено соединение, представленное формулой (I), его изомер или его фармацевтически приемлемая соль,
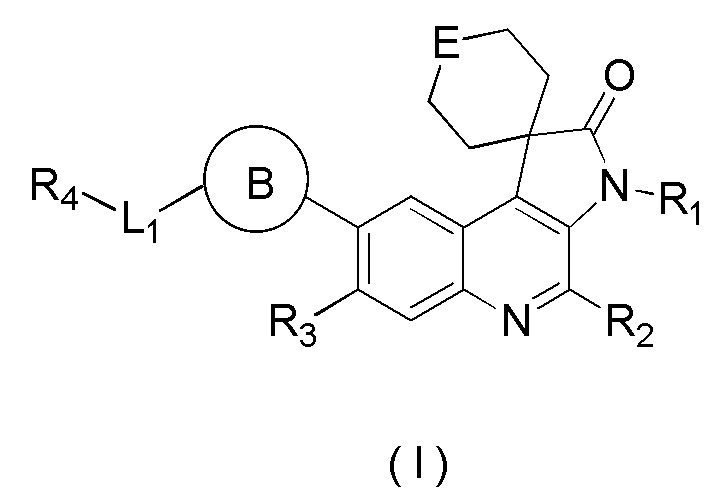
[9] где
[10] E выбран из -N(R5)-, - - и -C(R6)(R7)-;
[11] R1 выбран из C1-3 алкила, C1-3 алкокси и C3-6 циклоалкила, где указанные C1-3 алкил, C1-3 алкокси и C3-6 циклоалкил необязательно замещены 1, 2 или 3 Ra;
[12] R2 выбран из H, F, Cl, Br, I, H и NH2;
[13] R3 выбран из H, F, Cl, Br, I, H, NH2, CN, C1-3 алкила и C1-3 алкокси, где указанные C1-3 алкил и C1-3 алкокси необязательно замещены 1, 2 или 3 Rb;
[14] R4 выбран из C1-6 алкила и N(Rc)(Rd);
[15] R5 выбран из H, C1-6 алкила, C3-6 циклоалкила, C1-6 алкил-C=-, C1-6 алкил--C=- и C3-6 циклоалкил-C=-, где указанные C1-6 алкил, C3-6 циклоалкил, C1-6 алкил-C=-, C1-6 алкил- -C= - и C3-6 циклоалкил-C= - необязательно замещены 1, 2 или 3 Re;
[16] каждый из R6 и R7 независимо выбран из H, F, Cl, Br, I, H, NH2, CN, C1-6 алкила и C1-6 алкокси, где указанные C1-6 алкил или C1-6 алкокси необязательно замещены 1, 2 или 3 Rf;
[17] L1 выбран из одинарной связи, -(CH2)m- и -(CH2)m- -;
[18] m выбран из 1, 2, 3 и 4;
[19] кольцо B выбрано из фенила и 5-6-членного гетероарила, где указанные фенил и 5-6-членный гетероарил необязательно замещены 1, 2 или 3 Rg;
[20] каждый из Ra и Rb независимо выбран из F, Cl, Br, I, H и NH2;
[21] каждый из Rc и Rd независимо выбран из H, C1-3 алкила и C3-6 циклоалкила, где каждый из указанных C1-3 алкила и C3-6 циклоалкила независимо выбран из 1, 2 или 3 R;
[22] или Rc и Rd совместно с атомом N, к которому они присоединены, образуют 4-6-членный гетероциклоалкил, необязательно замещенный 1, 2 или 3 R;
[23] каждый из Re, Rf и Rg независимо выбран из F, Cl, Br, I, H и NH2;
[24] каждый R независимо выбран из F, Cl, Br, I, H и NH2;
[25] и каждый из указанных 5-6-членного гетероарила и 4-6-членного гетероциклоалкила содержит 1, 2, 3 или 4 гетероатома или группы гетероатомов, независимо выбранных из -NH-, - -, -S- и N.
[26] В некоторых вариантах реализации настоящего раскрытия указанный выше R1 выбран из CH3, CH2CH3 и циклопропила, где указанные CH3, CH2CH3 и циклопропил необязательно замещены 1, 2 или 3 Ra, и другие переменные определены в настоящем изобретении.
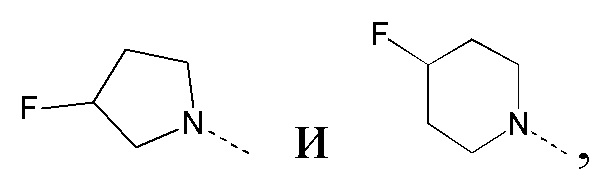
[27] В некоторых вариантах реализации настоящего раскрытия указанный выше R1 выбран из CH3, CH2F, CHF2, CF3, CH2CH3 и циклопропила, и другие переменные определены в настоящем изобретении.
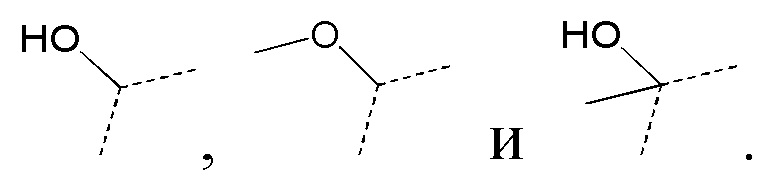
[28] В некоторых вариантах реализации настоящего раскрытия указанный выше R3 выбран из H, F, Cl, Br, I, H, NH2, CN, CH3, CH2CH3 и  , где указанные CH3, CH2CH3 и необязательно замещены 1, 2 или 3 Rb, и другие переменные определены в настоящем изобретении.
, где указанные CH3, CH2CH3 и необязательно замещены 1, 2 или 3 Rb, и другие переменные определены в настоящем изобретении.
[29] В некоторых вариантах реализации настоящего раскрытия указанный выше R3 выбран из H, F, Cl, Br, I, H, NH2, CN, CH3, CH2F, CHF2, CF3, CH2CH3 и , и другие переменные определены в настоящем изобретении.
[30] В некоторых вариантах реализации настоящего раскрытия каждый из указанных выше Rc и Rd независимо выбран из CH3, CH2CH3 и циклопропила, и другие переменные определены в настоящем изобретении.
[31] В некоторых вариантах реализации настоящего раскрытия указанные выше Rc и Rd совместно с атомом N, к которому они присоединены, образуют пирролидил и пиперидинил, где указанные пирролидил и пиперидинил необязательно замещены 1, 2 или 3 R, и другие переменные определены в настоящем изобретении.
[32] В некоторых вариантах реализации настоящего раскрытия указанные выше Rc и Rd совместно с атомом N, к которому они присоединены, образуют  и
и 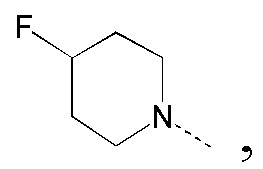 и другие переменные определены в настоящем изобретении.
и другие переменные определены в настоящем изобретении.
[33] В некоторых вариантах реализации настоящего раскрытия указанный выше R4 выбран из CH3, CH2CH3, 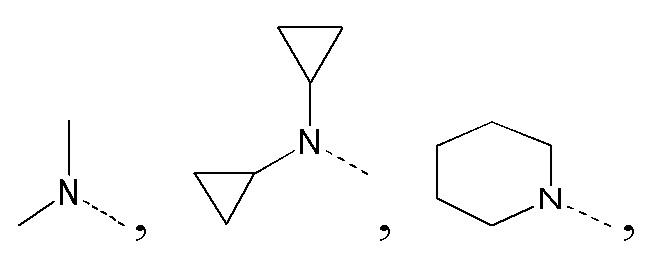
 и другие переменные определены в настоящем изобретении.
и другие переменные определены в настоящем изобретении.
[34] В некоторых вариантах реализации настоящего раскрытия указанный выше R5 выбран из H, CH3, CH3CH2, CH(CH3)2, циклопропила, CH3C= -, CH(CH3)2C= -, CH3C= - и циклопропил-C= -, где указанные CH3, CH3CH2, CH(CH3)2, циклопропил, CH3C= -, CH(CH3)2C= -, CH3C= - и циклопропил-C= - необязательно замещены 1, 2 или 3 Re, и другие переменные определены в настоящем изобретении.
[35] В некоторых вариантах реализации настоящего раскрытия указанный выше R5 выбран из H, CH3, CH2F, CHF2, CF3, CH3CH2, CH2FCH2, CHF2CH2, CF3CH2, CH(CH3)2, циклопропила, CH3C= -, CH(CH3)2C= -, CH3C= - и циклопропил-C= -, и другие переменные определены в настоящем изобретении.
[36] В некоторых вариантах реализации настоящего раскрытия каждый из указанных выше R6 и R7 независимо выбран из H, F, Cl, Br, I, H, NH2, CN, CH3, CH3CH2, CH(CH3)2 и , где указанные CH3, CH3CH2, CH(CH3)2 и необязательно замещены 1, 2 или 3 Rf, и другие переменные определены в настоящем изобретении.
[37] В некоторых вариантах реализации настоящего раскрытия каждый из указанных выше R6 и R7 независимо выбран из H, F, Cl, Br, I, H, NH2, CN, CH3, CH2F, CHF2, CF3, CH3CH2, CH(CH3)2 и , и другие переменные определены в настоящем изобретении.
[38] В некоторых вариантах реализации настоящего изобретения указанный выше E выбран из -, -CF2-, -N(CH3)-, -NH-, 
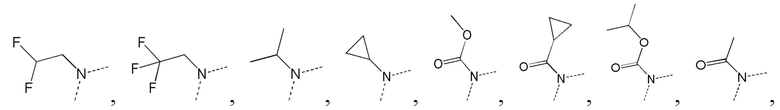
[39] В некоторых вариантах реализации настоящего изобретения указанный выше L1 выбран из одинарной связи, -(CH2)-- и -(CH2)3-, и другие переменные определены в настоящем изобретении.
[40] В некоторых вариантах реализации настоящего изобретения указанное выше кольцо B выбрано из фенила, пиридила, пиразолила, индазолила и имидазолила, где указанные фенил, пиридил, пиразолил, индазолил и имидазолил необязательно замещены 1, 2 или 3 Rg, и другие переменные определены в настоящем изобретении.
[41] В некоторых вариантах реализации настоящего изобретения указанное выше кольцо B выбрано из 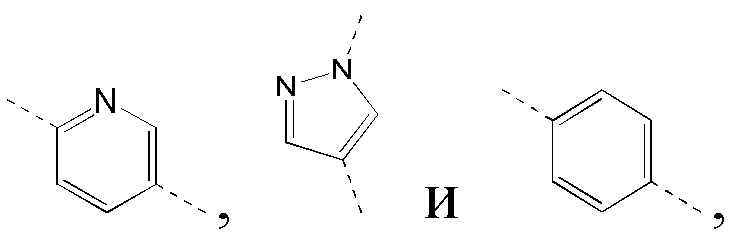 где указанные
где указанные 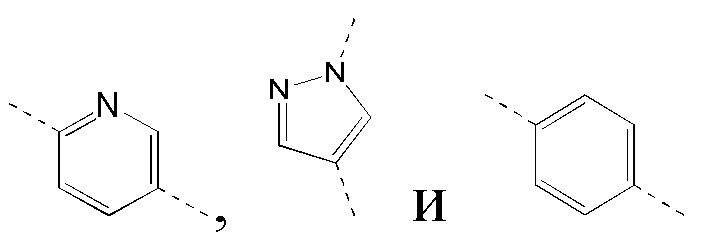 необязательно замещены 1, 2 или 3 Rg, и другие переменные определены в настоящем изобретении.
необязательно замещены 1, 2 или 3 Rg, и другие переменные определены в настоящем изобретении.
[42] В некоторых вариантах реализации настоящего изобретения указанное выше кольцо B выбрано из 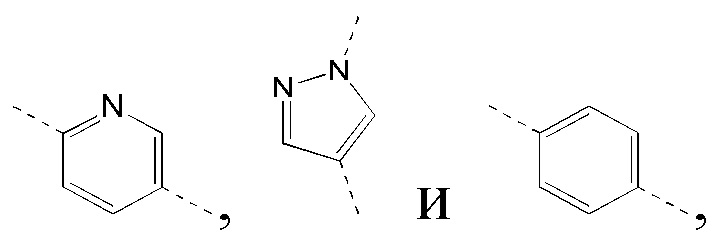 и другие переменные определены в настоящем изобретении.
и другие переменные определены в настоящем изобретении.
[43] В некоторых вариантах реализации настоящего изобретения указанный выше R4-L1- выбран из CH3, CH3CH2-, 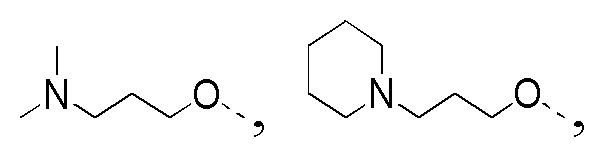
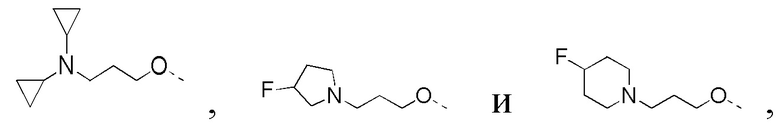 и другие переменные определены в настоящем изобретении.
и другие переменные определены в настоящем изобретении.
[44] Другие решения согласно настоящему изобретению получают с помощью любой комбинации указанных выше переменных.
[45] В некоторых вариантах реализации настоящего изобретения указанное выше соединение, его изомер или его фармацевтически приемлемая соль выбраны из
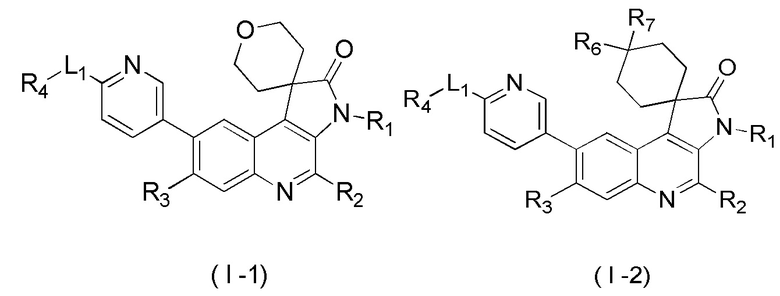
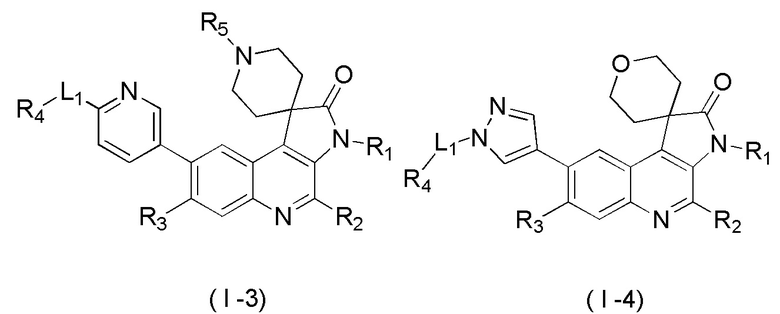
[46] где
[47] R1, R2, R3, R4, R5, R6, R7 и L1 определены в настоящем изобретении.
[48] В настоящем изобретении также предложено следующее соединение, его изомер или его фармацевтически приемлемая соль, где указанное соединение выбрано из
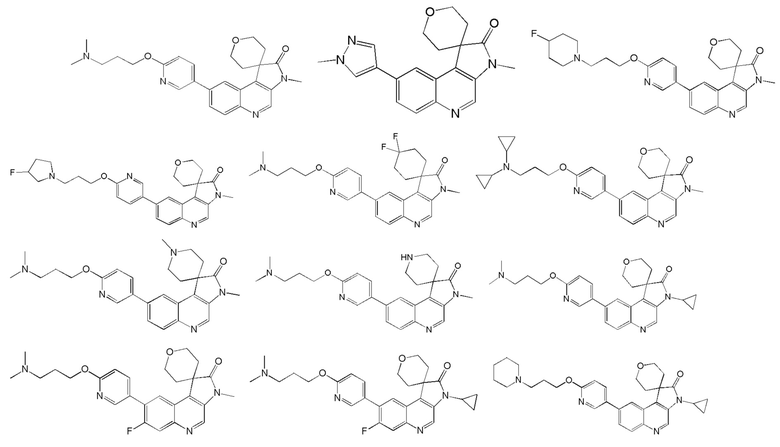
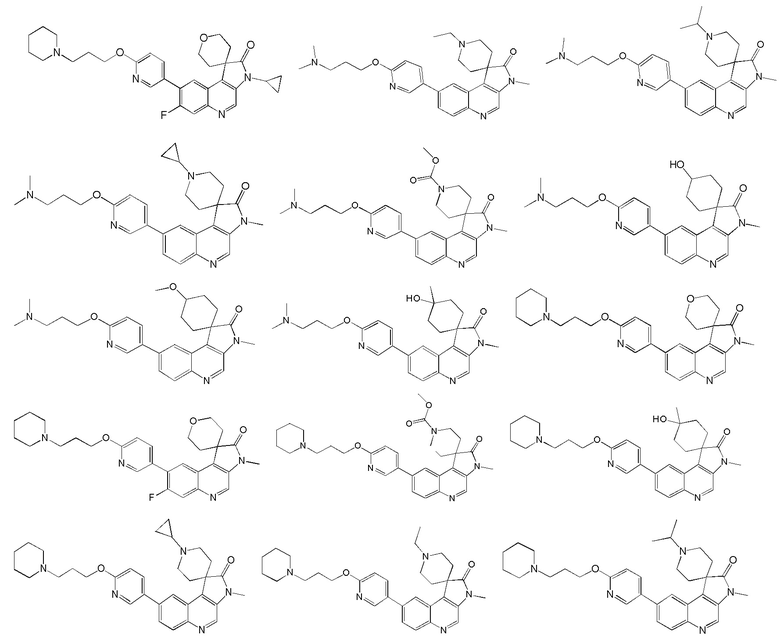
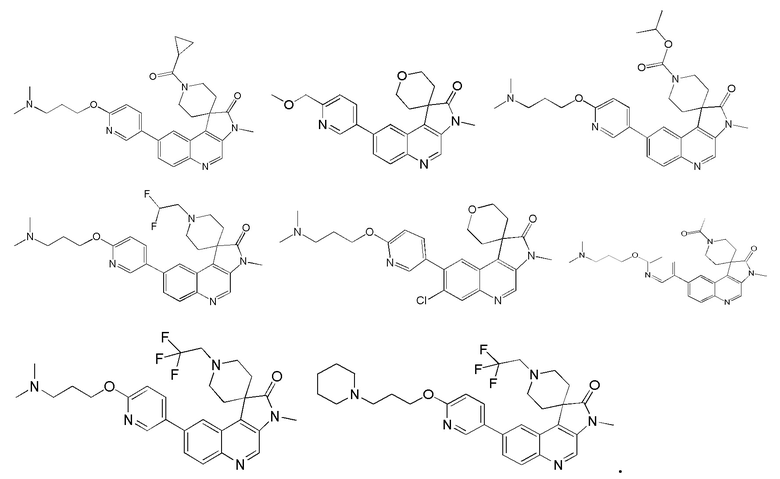
[49] В некоторых вариантах реализации настоящего изобретения указанное выше соединение, его изомер или его фармацевтически приемлемая соль выбраны из
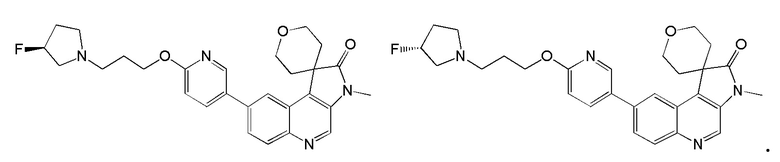
[50] В некоторых вариантах реализации настоящего изобретения предложены способы применения указанного выше соединения, его изомера или его фармацевтически приемлемой соли для получения лекарственных средств, подобных ингибиторам ATM киназы.
[51] В некоторых вариантах реализации настоящего изобретения предложено указанное выше применение, при этом лекарственные средства, подобные ингибиторам ATM киназы, представляют собой лекарственные средства для лечения солидных опухолей.
Определения и описание
[52] Если не указано иное, следующие термины и фразы, используемые в настоящем описании, имеют следующие значения. Конкретный термин или фразу, если они не определены специально, не следует рассматривать как неопределенные или неоднозначные, а следует понимать в их обычном значении. В случае, когда в настоящем описании используется торговое наименование, оно предназначено для обозначения соответствующего продукта или его активного ингредиента.
[53] Термин «фармацевтически приемлемый» в настоящем описании относится к тем соединениям, материалам, композициям и/или лекарственным формам, которые в рамках обоснованного медицинского заключения подходят для применения в контакте с тканями людей и животных без чрезмерной токсичности, раздражения, аллергических реакций или других проблем или осложнений, с учетом разумного отношения пользы и рисков.
[54] Термин «фармацевтически приемлемая соль» относится к соли соединения согласно настоящему изобретению, которая получена из соединения, имеющего конкретные заместители, указанного в настоящем изобретении, и относительно нетоксичных кислот или оснований. В случае, когда соединения согласно настоящему изобретению содержат относительно кислые функциональные группы, соли присоединения основания могут быть получены путем приведения нейтральной формы таких соединений в контакт с достаточным количеством основания либо в чистом растворе, либо в подходящем инертном растворителе. Фармацевтически приемлемые соли присоединения оснований включают соли натрия, калия, кальция, аммония, органических аминов или магния, или схожие соли. В случае, когда соединения согласно настоящему изобретению содержат относительно основные функциональные группы, соли присоединения кислоты могут быть получены путем приведения нейтральной формы таких соединений в контакт с достаточным количеством кислоты либо в чистом растворе, либо в подходящем инертном растворителе. Примеры фармацевтически приемлемых солей присоединения кислоты включают соли неорганических кислот, которые включают, например, соляную кислоту, бромоводородную кислоту, азотную кислоту, угольную кислоту, гидрокарбонат, фосфорную кислоту, моногидрофосфат, дигидрофосфат, серную кислоту, гидросульфат, иодоводородную кислоту и фосфористую кислоту; и соли органических кислот, которые включают, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфокислоту, п-толуолсульфокислоту, лимонную кислоту, винную кислоту и метансульфокислоту; а также включают соли аминокислот (таких как аргинин) и соли органических кислот, таких как глюкуроновая кислота. Некоторые конкретные соединения согласно настоящему изобретению содержат основные и кислотные функциональные группы, и, таким образом, могут быть превращены в любую соль присоединения основания или кислоты.
[55] Фармацевтически приемлемые соли согласно настоящему изобретению могут быть синтезированы из исходного соединения, содержащего кислотные радикалы или основные радикалы, с помощью обычных химических способов. Обычно способ получения таких солей включает: осуществление реакции этих соединений в форме свободной кислоты или свободного основания со стехиометрическим количеством подходящего основания или кислоты в воде или органическом растворителе или их смеси с получением солей.
[56] Структура соединения согласно настоящему изобретению может быть подтверждена обычными способами, хорошо известными специалисту в данной области техники. В случае, когда настоящее изобретение относится к абсолютной конфигурации соединения, указанная абсолютная конфигурация может быть подтверждена с помощью технических средств, общепринятых в данной области техники. Например, при рентгеновской дифракции монокристаллов (SXRD) используют дифрактометр Bruker D8 venture для сбора данных об интенсивности дифракции выращенного монокристалла, источником света является излучение CuKα, и режимом сканирования - сканирование ϕ/ω. После сбора соответствующих данных далее используют прямой метод (Shelxs97) для решения кристаллической структуры, чтобы можно было подтвердить абсолютную конфигурацию.
[57] Соединения согласно настоящему изобретению могут существовать в конкретных геометрических или стереоизомерных формах. Настоящее изобретение предполагает все такие соединения, включая цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры, а также рацемические смеси и другие их смеси, такие как энантиомерно или диастереомерно обогащенные смеси, все из которых включены в объем настоящего изобретения. Дополнительные асимметрические атомы углерода могут присутствовать в заместителе, таком как алкильная группа. Все эти изомеры и их смеси включены в объем настоящего изобретения.
[58] Если не указано иное, термин «энантиомер» или «оптические изомеры» относится к стереоизомерам, которые являются зеркальным отображением друг друга.
[59] Если не указано иное, термин «цис-транс-изомер» или «геометрический изомер» является следствием того факта, что относительно двойных или одинарных связей образующих кольцо атомов углерода свободное вращение невозможно.
[60] Если не указано иное, термин «диастереомеры» относится к стереоизомерам, молекулы которых имеют два или более хиральных центра и не являются зеркальным отображением друг друга.
[61] Если не указано иное, «(D)» или «(+)» означает правовращающий, «(L)» или «(-)» означает левовращающий, и «(DL)» или «(±)» означает рацемический.
[62] Если не указано иное, клиновидная сплошная связь ( ) и клиновидная пунктирная связь (
) и клиновидная пунктирная связь ( ) указывают на абсолютную конфигурацию стереоскопического центра; прямая сплошная связь (
) указывают на абсолютную конфигурацию стереоскопического центра; прямая сплошная связь ( ) и прямая пунктирная связь (
) и прямая пунктирная связь ( ) указывают на относительную конфигурацию стереоскопического центра; волнистой линией (
) указывают на относительную конфигурацию стереоскопического центра; волнистой линией ( ) представлена клиновидная сплошная связь () или клиновидная пунктирная связь (); или волнистой линией () представлена прямая сплошная связь () и прямая пунктирная связь ().
) представлена клиновидная сплошная связь () или клиновидная пунктирная связь (); или волнистой линией () представлена прямая сплошная связь () и прямая пунктирная связь ().
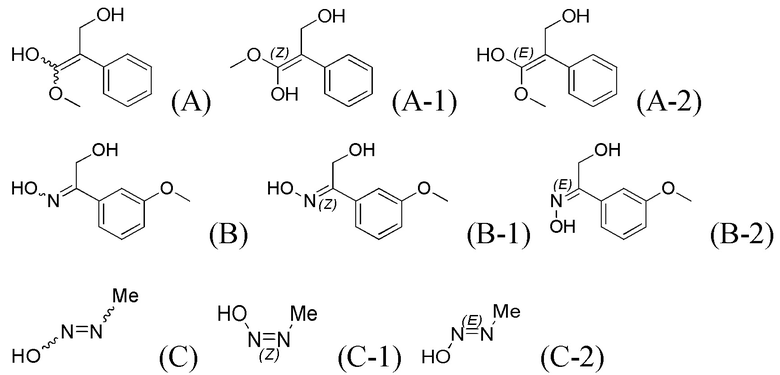
[63] Если не указано иное, когда в соединении имеется структура двойной связи, такая как двойная связь углерод-углерод, двойная связь углерод-азот и двойная связь азот-азот, и каждый из соединенных двойной связью атомов соединен с двумя различными заместителями (в случае двойной связи при атоме азота неподеленная пара электронов атома азота рассматривается как заместитель, связанный с ним), если атом при двойной связи в указанном соединении и его заместитель соединены волнистой линией (), это указывает на (Z) изомер, (E) изомер или смесь двух изомеров соединения. Например, следующая формула (A) указывает, что соединение находится в форме одного изомера формулы (A-1) или формулы (A-2) или смеси двух изомеров формулы (A-1) и формулы (А-2); следующая формула (B) указывает, что соединение находится в форме одного изомера формулы (B-1) или формулы (B-2) или смеси двух изомеров формулы (B-1) и формулы (B-2). Следующая формула (C) указывает, что соединение находится в форме одного изомера формулы (C-1) или формулы (C-2) или смеси двух изомеров формулы (C-1) и формулы (C-2).
[64] Соединения согласно настоящему изобретению могут существовать в конкретных таутомерных формах. Если не указано иное, термин «таутомер» или «таутомерная форма» означает, что при комнатной температуре изомеры с различными функциональными группами находятся в динамическом равновесии и могут быстро превращаться друг в друга. Там, где возможна таутомеризация (например, в растворе), может быть достигнуто химическое равновесие таутомеров. Например, в случае протонных таутомеров (также известных как прототропные таутомеры) возможны взаимные превращения путем миграции протона, такие как кето-енольная изомеризация и имино-енаминная изомеризация. В случае валентных таутомеров возможны некоторые взаимные превращения путем рекомбинации некоторых электронов, образующих связь. Конкретным примером кето-енольной таутомеризации является взаимное превращение двух таутомеров, пентан-2,4-диона и 4-гидроксипент-3-ен-2-она.
[65] Если не указано иное, термины «обогащенный одним изомером», «изомерно обогащенный», «обогащенный одним энантиомером» или «энантиомерно обогащенный» относятся к содержанию одного из изомеров или энантиомеров, составляющему менее 100%, и содержание указанного изомера или энантиомера составляет 60% или более, или 70% или более, или 80% или более, или 90% или более, или 95% или более, или 96% или более, или 97% или более, или 98% или более, или 99% или более, или 99,5% или более, или 99,6% или более, или 99,7% или более, или 99,8% или более, или 99,9% или более.
[66] Если не указано иное, термин «избыток изомера» или «избыток энантиомера» относится к разнице между относительными процентными содержаниями двух изомеров или энантиомеров. Например, если содержание одного изомера или энантиомера составляет 90%, и содержание другого изомера или энантиомера составляет 10%, избыток изомера или энантиомера (значение ee) составляет 80%.
[67] Оптически активные (R)- и (S)-изомеры, а также D- и L-изомеры могут быть получены путем хирального синтеза, или с применением хиральных реагентов, или другими обычными способами. Если желательным является конкретный энантиомер соединения согласно настоящему изобретению, он может быть получен путем асимметрического синтеза или дериватизации с применением хирального вспомогательного вещества, при этом полученную смесь диастереомеров разделяют и вспомогательные группы отщепляют с получением чистого желаемого энантиомера. В качестве альтернативы, когда молекула содержит основную функциональную группу (такую как аминогруппа) или кислотную функциональную группу (такую как карбоксильная группа), могут быть получены диастереомерные соли с подходящими оптически активными кислотой или основанием с последующим разделением диастереомеров обычными способами, известными в данной области техники, и дальнейшим выделением чистых энантиомеров. Помимо этого, разделение энантиомеров и диастереомеров часто выполняют посредством хроматографии с применением хиральных неподвижных фаз, необязательно в сочетании со способами химической дериватизации (например, получение карбаматов из аминов). Соединения согласно настоящему изобретению могут содержать не встречающиеся в природе отношения атомных изотопов одного или более атомов, составляющих соединение. Например, соединения могут быть помечены с помощью радиоактивных изотопов, таких как тритий (3H), иод-125 (125I) или C-14 (14C). В качестве другого примера, атом водорода может быть заменен на атом дейтерия с получением дейтерированных лекарственных средств. Связь, образованная дейтерием и углеродом, прочнее, чем связь, образованная обычным водородом и углеродом. По сравнению с недейтерированными лекарственными средствами, дейтерированные лекарственные средства обладают сниженными токсическими побочными эффектами, повышенной стабильностью лекарственного средства, повышенной эффективностью, более длительным биологическим временем полужизни лекарственных средств и другими преимуществами. Предполагается, что все изотопные варианты соединений согласно настоящему изобретению, являющиеся или не являющиеся радиоактивными, включены в объем настоящего изобретения. «Необязательный» или «необязательно» означает, что описанное далее событие или обстоятельство может, но не обязательно, произойти, и что настоящее описание включает случаи, когда упомянутое событие или обстоятельство происходит, и случаи, когда упомянутое событие или обстоятельство не происходит.
[68] Термин «замещенный» означает, что любые один или более атомов водорода при конкретном атоме заменены на заместители, которые могут включать дейтерий и варианты водорода, при условии, что валентное состояние указанного атома является нормальным и замещенное соединение является стабильным. В случае, когда заместитель представляет собой кислород (то есть =°), это означает, что заменены два атома водорода. Замещение кислородом не встречается в ароматических группах. Термин «необязательно замещенный» означает, что атом может быть или не быть замещенным. Если не указано иное, тип и количество заместителей могут быть выбраны произвольно исходя из того, что они могут быть получены в химии.
[69] В случае, когда какая-либо переменная (такая как R) встречается в составе или структуре соединения более одного раза, ее определение в каждом случае является независимым. Таким образом, например, если группа замещена 0-2 R, указанная группа может быть необязательно замещена не более чем двумя R, и R в каждом случае имеет независимые варианты. Помимо этого, комбинации заместителей и/или их вариантов допустимы, только если такие комбинации приводят к стабильным соединениям.
[70] В случае, когда индекс при соединяющей группе равен 0, например, -(CRR)0-, это означает, что соединяющая группа представляет собой одинарную связь.
[71] В случае, когда одна из переменных выбрана из одинарной связи, это означает, что две группы, с которыми она соединена, связаны непосредственно. Например, когда L представляет собой одинарную связь в A-L-Z, это означает, что указанная структура фактически представляет собой A-Z.
[72] В случае, когда заместитель является вакантным, это означает, что заместитель не существует. Например, когда X является вакантным в A-X, это означает, что структура фактически представляет собой A. Когда для перечисленных заместителей не указывают, через какой атом они связаны с замещенной группой, такие заместители могут быть связаны через любой из их атомов, например, пиридил в качестве заместителя может быть присоединен к замещенной группе через любой атом углерода пиридинового кольца.
[73] Когда для включенной в перечень соединяющей группы не указывают направление ее присоединения, направление присоединения является произвольным, например, соединяющая группа L в 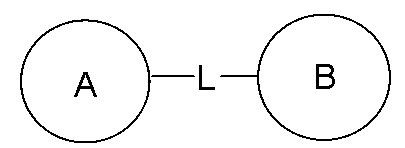 представляет собой -M-W-, в этой ситуации -M-W- может соединять кольцо A и кольцо B в направлении, соответствующем порядку чтения слева направо, с получением
представляет собой -M-W-, в этой ситуации -M-W- может соединять кольцо A и кольцо B в направлении, соответствующем порядку чтения слева направо, с получением 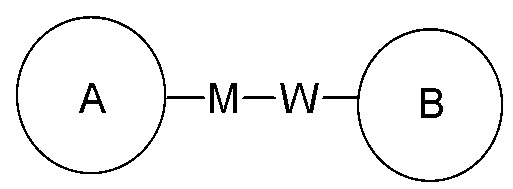 , а также может соединить кольцо A и кольцо B в направлении, противоположном порядку чтения слева направо, с получением
, а также может соединить кольцо A и кольцо B в направлении, противоположном порядку чтения слева направо, с получением 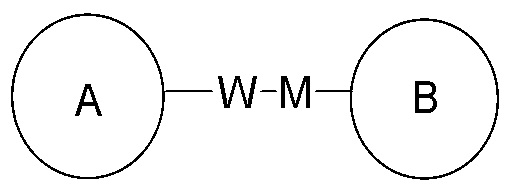 . Комбинации соединяющих групп, заместителей и/или их вариантов допустимы, только если такие комбинации приводят к стабильным соединениям.
. Комбинации соединяющих групп, заместителей и/или их вариантов допустимы, только если такие комбинации приводят к стабильным соединениям.
[74] Если не указано иное, количество атомов в кольце обычно определяется как количество членов кольца. Например, «5-7-членное кольцо» означает «кольцо», содержащее 5-7 атомов, расположенных по кругу.
[75] Если не указано иное, термин «C1-6 алкил» используют для обозначения линейной или разветвленной насыщенной углеводородной группы, состоящей из 1-6 атомов углерода. C1-6 алкил включает C1-5, C1-4, C1-3, C1-2, C2-6, C2-4, C6 и C5 алкил; он может быть одновалентным (таким как метил), двухвалентным (таким как метилен) или многовалентным (таким как метин). Примеры C1-6 алкила включают, но не ограничиваются ими, метил (Ме), этил (Et), пропил (включая н-пропил и изопропил), бутил (включая н-бутил, изобутил, втор-бутил и трет-бутил), пентил (включая н-пентил, изопентил и неопентил) и гексил.
[76] Если не указано иное, термин «C1-3 алкил» используют для обозначения линейной или разветвленной насыщенной углеводородной группы, состоящей из 1-3 атомов углерода. C1-3 алкил включает C1-2 и C2-3 алкил; он может быть одновалентным (таким как метил), двухвалентным (таким как метилен) или многовалентным (таким как метин). Примеры C1-3 алкила включают, но не ограничиваются ими, метил (Ме), этил (Et) и пропил (включая н-пропил и изопропил).
[77] Если не указано иное, термин «C1-6 алкокси» означает алкильные группы, содержащие от 1 до 6 атомов углерода, которые соединены с остальной частью молекулы через один атом кислорода. C1-6 алкокси включает C1-4, C1-3, C1-2, C2-6, C2-4, C6, C5, C4, C3 алкокси и так далее. Примеры C1-6 алкокси включают, но не ограничиваются ими, метокси, этокси, пропокси (включая н-пропокси и изопропокси), бутокси (включая н-бутокси, изобутокси, втор-бутокси и трет-бутокси), пентилокси (включая н-пентилокси, изопентилокси и неопентилокси), гексилокси и так далее.
[78] Если не указано иное, термин «C1-3 алкокси» означает алкильные группы, содержащие от 1 до 3 атомов углерода, которые соединены с остальной частью молекулы через один атом кислорода. Группа C1-3 алкокси включает C1-2, C2-3, C3, C2 алкокси и так далее. Примеры C1-3 алкокси включают, но не ограничиваются ими, метокси, этокси, пропокси (включая н-пропокси и изопропокси) и так далее.
[79] Если не указано иное, «C3-6 циклоалкил» означает насыщенную циклическую углеводородную группу, состоящую из 3-6 атомов углерода, что включает моноциклическую и бициклическую кольцевую систему, и C3-6 циклоалкил включает C3-5, C4-5 и C5-6 циклоалкил; он может быть одновалентным, двухвалентным или многовалентным. Примеры C3-6 циклоалкила включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил и циклогексил.
[80] Если не указано иное, термин «4-6-членный гетероциклоалкил» отдельно или в комбинации с другими терминами, соответственно, означает насыщенную циклическую группу, состоящую из 4-6 атомов - членов кольца, 1, 2, 3 или 4 атома - члена кольца из которых являются гетероатомами, независимо выбранными из °, S и N, а остальные атомы представляют собой атомы углерода, при этом атом азота необязательно кватернизован, и гетероатомы азота и серы могут быть необязательно окислены (то есть представлять собой N° и S(°)p, где p равно 1 или 2). Он включает моноциклическую и бициклическую кольцевую систему, где бициклическая система включает спирокольцевую систему, конденсированную кольцевую систему и мостиковую кольцевую систему. Помимо этого, применительно к «4-6-членным гетероциклоалкилам» гетероатом может занимать положение атома, соединяющего гетероциклический алкил с остальной частью молекулы. 4-6-членный гетероциклоалкил включает 5-6-членный, 4-членный, 5-членный и 6-членный гетероциклоалкил, и так далее. Примеры 4-6-членного гетероциклоалкила включают, но не ограничиваются ими, азетидинил, оксетанил, тиатанил, пирролидинил, пиразолидинил, имидазолидинил, тетрагидротиенил (включая тетрагидротиофен-2-ил, тетрагидротиофен-3-ил и так далее), тетрагидрофуранил (включая тетрагидрофуран-2-ил и так далее), тетрагидропиранил, пиперидинил (включая 1-пиперидинил, 2-пиперидинил и 3-пиперидинил), пиперазинил (включая 1-пиперазинил, 2-пиперазинил и так далее), морфолинил (включая 3-морфолинил, 4-морфолинил и так далее), диоксанил, дитианил, изоксазолидинил, изотиазолидинил, 1,2-оксазинил, 1,2-тиазинил, гексагидропиридазинил, гомопиперазинил или гомопиперидинил и так далее.
[81] Если не указано иное, термины «5-6-членное гетероарильное кольцо» и «5-6-членный гетероарил» в настоящем изобретении могут использоваться взаимозаменяемо, и термин «5-6-членный гетероарил» означает моноциклическую группу, имеющую сопряженную систему π-электронов и состоящую из 5-6 атомов - членов кольца, 1, 2, 3, или 4 атома - члена кольца из которых являются гетероатомами, независимо выбранными из °, S, и N, а остальные атомы представляют собой атомы углерода, при этом атомы азота необязательно кватернизованы, и гетероатомы азота и серы могут быть необязательно окислены (то есть представлять собой N° и S(°)p, где p равно 1 или 2). 5-6-членный гетероарил может быть соединен с остальной частью молекулы через гетероатом или атом углерода. 5-6-членный гетероарил включает 5-членный и 6-членный гетероарил. Примеры 5-6-членного гетероарила включают, но не ограничиваются ими, пирролил (включая N-пирролил, 2-пирролил и 3-пирролил), пиразолил (включая 2-пиразолил и 3-пиразолил), имидазолил (включая N-имидазолил, 2-имидазолил, 4-имидазолил и 5-имидазолил), оксазолил (включая 2-оксазолил, 4-оксазолил и 5-оксазолил), триазолил (1H-1,2,3-триазолил, 2H-1,2,3-триазолил, 1H-1,2,4-триазолил и 4H-1,2,4-триазолил), тетразолил, изоксазолил (3-изоксазолил, 4-изоксазолил и 5-изоксазолил), тиазолил (включая 2-тиазолил, 4-тиазолил и 5-тиазолил), фурил (включая 2-фуранил и 3-фуранил), тиенил (включая 2-тиенил и 3-тиенил), пиридил (включая 2-пиридил, 3-пиридил и 4-пиридил), пиразинил или пиримидинил (включая 2-пиримидил и 4-пиримидил).
[82] Если не указано иное, Cn-n+m или Cn-Cn+m включает любой конкретный случай от n до n + m атомов углерода, например, C1-12 включает C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12, а также включает любой диапазон от n до n + m, например, C1-12 включает C1-3, C1-6, C1-9, C3-6, C3-9, C3-12, C6-9, C6-12 и C9-12; схожим образом, от n-членного до n + m-членного означает, что количество атомов в кольце составляет от n до n + m, например, 3-12-членное кольцо включает 3-членное кольцо, 4-членное кольцо, 5-членное кольцо, 6-членное кольцо, 7-членное кольцо, 8-членное кольцо, 9-членное кольцо, 10-членное кольцо, 11-членное кольцо и 12-членное кольцо, а также включает любой диапазон от n до n + m, например, 3-12-членное кольцо включает 3-6-членное кольцо, 3-9-членное кольцо, 5-6-членное кольцо, 5-7-членное кольцо, 6-7-членное кольцо, 6-8-членное кольцо и 6-10-членное кольцо.
[83] Термин «уходящая группа» относится к функциональной группе или атому, которые могут быть заменены на другую функциональную группу или атом в результате реакции замещения (например, реакции замещения по сродству). Например, типичные уходящие группы включают трифторметансульфонат; хлор, бром и иод; сульфонаты, такие как метансульфонат, тозилат, п-бромбензолсульфонат и п-толуолсульфонат; и ацилокси, такие как ацетокси и трифторацетокси.
[84] Термин «защитная группа» включает, но не ограничивается ими, «защитную группу для аминогруппы», «защитную группу для гидроксильной группы» или «защитную группу для меркаптогруппы». Термин «защитная группа для аминогруппы» относится к защитной группе, которая подходит для предотвращения побочных реакций по атому азота аминогруппы. Типичные защитные группы для аминогруппы включают, но не ограничиваются ими, формил; ацил, такой как алканоил (например, ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такой как трет-бутоксикарбонил (B c); арилметоксикарбонил, такой как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fm c); арилметил, такой как бензил (Bn), трифенилметил (Tr), 1,1-бис-(4'-метоксифенил)метил; силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS). Термин «защитная группа для гидроксильной группы» относится к защитной группе, которая подходит для предотвращения побочных реакций гидроксильной группы. Типичные защитные группы для гидроксильной группы включают, но не ограничиваются ими, алкил, такой как метил, этил и трет-бутил; ацил, такой как алканоил (например, ацетил); арилметил, такой как бензил (Bn), п-метоксибензил (PMB), 9-флуоренилметил (Fm) и дифенилметил (DPM); силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS).
[85] Соединения согласно настоящему изобретению могут быть получены различными синтетическими способами, хорошо известными специалисту в данной области техники, включая перечисленные ниже конкретные варианты реализации, варианты реализации, полученные путем комбинации с другими способами химического синтеза, и эквивалентные альтернативные варианты реализации, хорошо известные специалисту в данной области техники, при этом предпочтительные варианты реализации включают, но не ограничиваются ими, примеры, приведенные в настоящем изобретении.
[86] Растворители, применяемые в настоящем изобретении, являются коммерчески доступными. В настоящем изобретении использованы следующие сокращения: aq означает воду; HATU означает -(7-азабензотриазол-1-ил)-N,N,N',N''-тетраметилмочевины гексафторфосфат; EDC означает N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид; m-CPBA означает 3-хлорпероксибензойную кислоту; eq означает эквивалент; CDI означает карбонилдиимидазол; ДХМ означает дихлорметан; ПЭ означает петролейный эфир; DIAD означает диизопропилазодикарбоксилат; ДМФА означает N,N-диметилформамид; ДМСО означает диметилсульфоксид; EtAc означает этилацетат; EtH означает этанол; MeH означает метанол; CBz означает бензилоксикарбонил, который является защитной группой для аминогруппы; BC означает трет-бутоксикарбонил, который является защитной группой для аминогруппы; HAc означает уксусную кислоту; NaCNBH3 означает цианоборогидрид натрия; к.т. означает комнатную температуру; /N означает в течение ночи; ТГФ означает тетрагидрофуран; Bc2 означает ди-трет-бутилдикарбонат; ТФУ означает трифторуксусную кислоту; DIPEA означает диизопропилэтиламин; SCl2 означает тионилхлорид; CS2 означает дисульфид углерода; Ts°H означает п-толуолсульфокислоту; NFSI означает N-фтор-N-(бензолсульфонил)бензолсульфонамид; NCS означает 1-хлорпирролидин-2,5-дион; n-Bu4NF означает фторид тетрабутиламмония; iPr°H означает 2-пропанол; Тпл. означает температуру плавления; LDA означает диизопропиламид лития; LiHMDS означает гексаметилдисилазид лития; Ксантфос означает 4,5-бис(дифенилфосфино)-9,9-диметилксантен; LiAlH4 означает тетрагидрид лития-алюминия; Pd(dba)2 означает трис(дибензилиденацетон)дипалладий; mCPBA означает м-хлорпероксибензойную кислоту; pd(dppf)Cl2 означает [1,1'-бис(дифенилфосфино)ферроцен]палладия дихлорид; DBU означает 1,8-диазабицикло[5.4.0]ундецен-7-ен; DIPA означает диизопропиламин; n-BuLi означает н-бутиллитий; NBS означает N-бромсукцинимид; MeI означает иодметан; TBAB означает бромид тетрабутиламмония; Pd(PPh3)4 означает тетракис(трифенилфосфин)палладий; MeCN означает ацетонитрил; NaH означает гидрид натрия; TBS°Tf означает (трет-бутилдиметилсилил)трифторметансульфонат.
[87] Соединения названы в ручном режиме или с помощью программного обеспечения ChemDraw®, и коммерчески доступные соединения названы согласно названиям в каталоге поставщика.
Технические результаты
[88] Соединение согласно настоящему изобретению обладает значительным ингибирующим действием в отношении ATM киназы и очень хорошей селективностью действия на киназу, хорошей растворимостью и проникающей способностью, может проникать в головной мозг, и обладает потенциалом для превращения в лекарственное средство для лечения опухолей головного мозга. Комбинация соединения согласно настоящему изобретению и этопозида демонстрирует хороший синергетический эффект, который превосходит эффективность AZD0156 в комбинации с этопозидом.
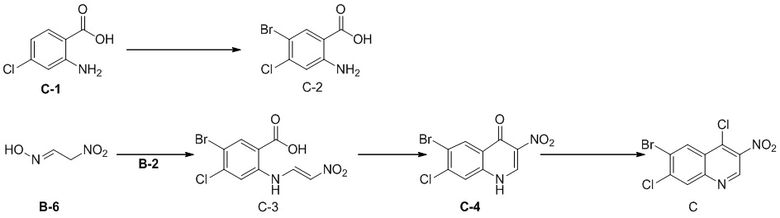
Краткое описание чертежей
[89] Фигура 1: относительное изменение массы.
[90] Фигура 2: кривая роста опухоли.
Подробное описание предпочтительных вариантов реализации
[91] Настоящее изобретение будет подробно описано с помощью следующих примеров, которые, однако, не влекут каких-либо неблагоприятных ограничений настоящего изобретения. Настоящее изобретение было подробно описано в настоящем описании, и в нем также раскрыты его конкретные варианты реализации. Для специалиста в данной области техники, без отступления от сущности и объема настоящего изобретения будут очевидны все изменения и улучшения, которые можно внести в конкретные варианты реализации настоящего изобретения.
Промежуточное соединение A
Способ синтеза:
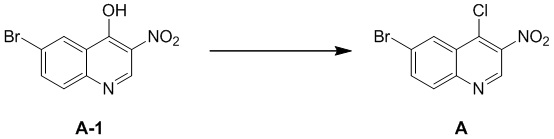
[92] Стадия 1: Синтез промежуточного соединения A
[93] К раствору соединения A-1 (34 г, 126,37 ммоль) в SCl2 (200 мл) добавляли ДМФА (95,00 мг, 1,30 ммоль, 0,1 мл) и реакционный раствор перемешивали при 80 °C в течение 16 часов. После завершения реакции SCl2 удаляли при пониженном давлении, в результате чего получали неочищенный продукт A, который непосредственно применяли на следующей стадии.
[94] МС m/z: 286,7[M+H] +
[95] 1H ЯМР (400 МГц, ДМСО-d6) δ 9,20 (s, 1H), 8,29 (d, J = 2,3 Гц, 1H), 7,94 (dd, J = 2,3, 8,8 Гц, 1H), 7,72 (d, J = 8,8 Гц, 1H)
Промежуточное соединение B
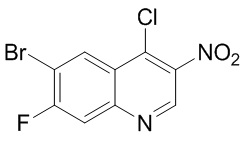
Способ синтеза:
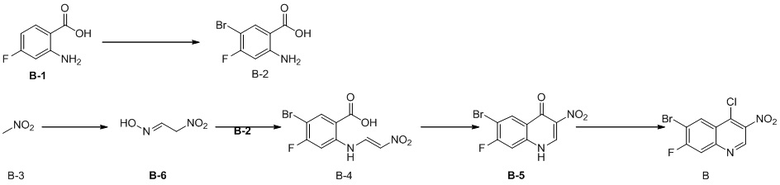
[96] Стадия 1: Синтез соединения B-2
[97] При 0 °C и в защитной атмосфере азота раствор NBS (30,12 г, 169,22 ммоль) в ДМФА (100 мл) добавляли к раствору B-1 (25 г, 161,16 ммоль) в ДМФА (100 мл) и реакционную систему перемешивали при 30 °C в течение 2 ч. После завершения реакции смесь концентрировали при пониженном давлении для удаления реакционного растворителя, затем суспендировали в течение 30 минут с использованием воды (100 мл) и затем промывали ацетонитрилом (10 мл), в результате чего получали B-2.
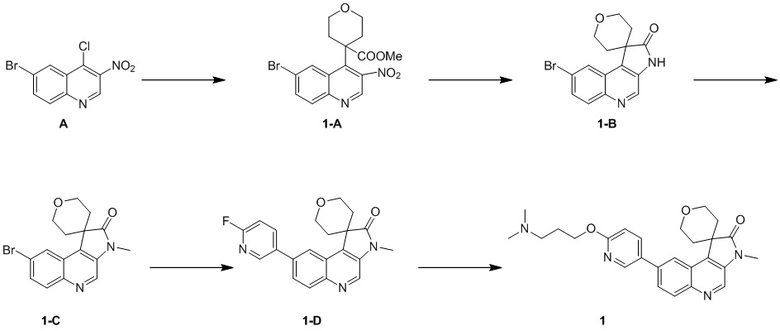
[98] МС m/z: 233,8[M+H] +
[99] 1H ЯМР (400 МГц, ДМСО-d6) δ 7,88 (ушир. d, J = 7,88 Гц, 1 H), 6,69 (ушир. d, J = 11,38 Гц, 1 H)
[100] Стадия 2: Синтез соединения B-6
[101] В круглодонной колбе нитрометан (18 г, 294,89 ммоль, 15,93 мл) (B-3) медленно добавляли к раствору NaH (17,69 г, 442,33 ммоль) в H2 (100 мл) и внутреннюю температуру поддерживали на уровне 30 °C. Затем смесь нагревали до 40 °C и перемешивали в течение 30 минут и охлаждали. Затем медленно добавляли другую часть нитрометана (18,00 г, 294,89 ммоль, 15,93 мл), реакционную систему нагревали до 45 °C и перемешивали в течение 30 минут и затем повышали температуру до 50 °C-55 °C и перемешивали в течение 5 минут, в результате чего получали смешанный раствор B-6, который непосредственно применяли на следующей стадии.
[102] Стадия 3: Синтез соединения B-4
[103] Смешанный раствор B-6 охлаждали до 30 °C и добавляли лед (80 г) и концентрированную соляную кислоту (15 мл). Указанный вышеупомянутый смешанный раствор добавляли к раствору B-2 (34,3 г, 146,57 ммоль) в HCl (12 M, 90 мл) и H2 (200 мл) и перемешивали при 30 °C в течение 12 ч. Твёрдое вещество осаждалось и фильтровали. Остаток на фильтре собирали и затем промывали ацетонитрилом (50 мл), в результате чего получали соединение B-4.
[104] МС m/z: 304,7[M+H] +
[105] 1H ЯМР (400 МГц, ДМСО-d6) δ 12,99 (ушир. d, J = 12,5 Гц, 1H), 8,23 - 8,13 (m, 1H), 8,08 - 7,96 (m, 1H), 7,88 (ушир. d, J = 10,5 Гц, 1H), 6,80 (ушир. s, 1H)
[106] Стадия 4: Синтез соединения B-5
[107] В защитной атмосфере азота раствор B-4 (44 г, 111,06 ммоль) в уксусном ангидриде (397,79 г, 3,90 моль, 364,94 мл) нагревали при 100 °C в течение 1 ч и затем прекращали нагревание. Добавляли ацетат натрия (9,38 г, 114,39 ммоль) и нагревали с обратным холодильником при 150 °C в течение 15 минут. Наконец, добавляли ещё одну порцию ацетата натрия (9,38 г, 114,39 ммоль) и реакционную систему нагревали с обратным холодильником при 150 °C в течение 1 ч. После завершения реакции растворитель удаляли путем концентрирования. Оставшиеся твердые вещества суспендировали с использованием воды (200 мл) в течение 1 ч, затем суспендировали с использованием смешанного раствора EtAc и метанола (55 мл, EtAc: MeH = 10 : 1) в течение 1 ч и фильтровали , в результате чего получали соединение B-5.
[108] МС m/z: 287,0[M+H] +
[109] 1H ЯМР (400 МГц, ДМСО-d6) δ 9,25 (s, 1H), 8,44 (d, J = 7,5 Гц, 1H), 7,63 (ушир. d, J = 9,3 Гц, 1H)
[110] Стадия 5: Синтез промежуточного соединения B
[111] За исключением использования соответствующих исходных веществ, промежуточное соединение B получали с использованием того же способа, как и при получении соединения A в промежуточном соединении A.
[112] МС m/z: 304,7[M+H] +
Промежуточное соединение C
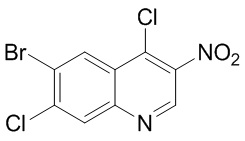
Способ синтеза:
[113] Стадия 1: Синтез соединения C-2
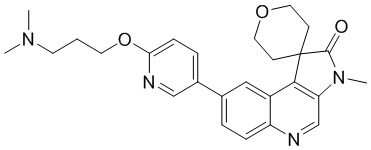
[114] За исключением использования соответствующих исходных веществ, соединение C-2 получали с использованием того же способа, как и при получении соединения B-2 в промежуточном соединении B.
[115] МС m/z: 249,8[M+H] +
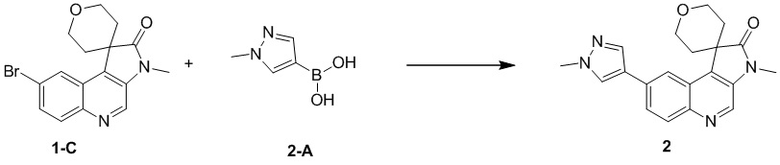
[116] 1H ЯМР (400 МГц, ДМСО-d6) δ 7,90 (ушир. s, 1H), 7,01 (ушир. s, 1H)
[117] Стадия 2: Синтез соединения C-3
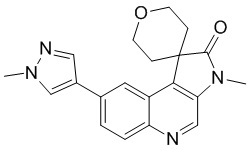
[118] За исключением использования соответствующих исходных веществ, соединение C-3 получали с использованием того же способа, как и при получении соединения B-4 в промежуточном соединении B.
[119] МС m/z: 320,8[M+H] +
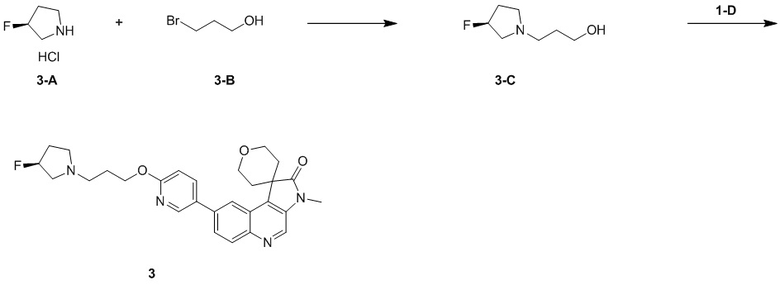
[120] 1H ЯМР (400 МГц, ДМСО-d6) δ 14,71 - 13,60 (m, 1H), 12,91 (ушир. s, 1H), 8,26 - 8,01 (m, 3H), 6,80 (ушир. s, 1H)
[121] Стадия 3: Синтез соединения C-4
[122] За исключением использования соответствующих исходных веществ, соединение C-4 получали с использованием того же способа, как и при получении соединения B-5 в промежуточном соединении B.
[123] МС m/z: 302,7[M+H] +
[124] 1H ЯМР (400 МГц, ДМСО-d6) δ 9,29 (ушир. s, 1H), 8,46 (s, 1H), 7,94 (s, 1H)
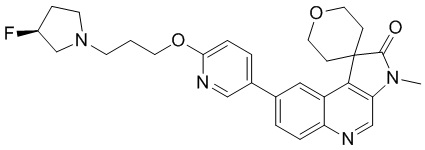
[125] Стадия 4: Синтез промежуточного соединения C
[126] За исключением использования соответствующих исходных веществ, промежуточное соединение C получали с использованием того же способа, как и при получении соединения A в промежуточном соединении A.
[127] МС m/z: 320,7[M+H] +
Пример 1: Соединение 1
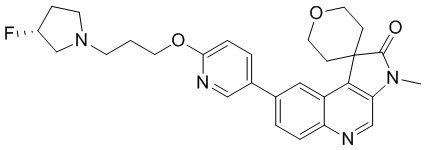
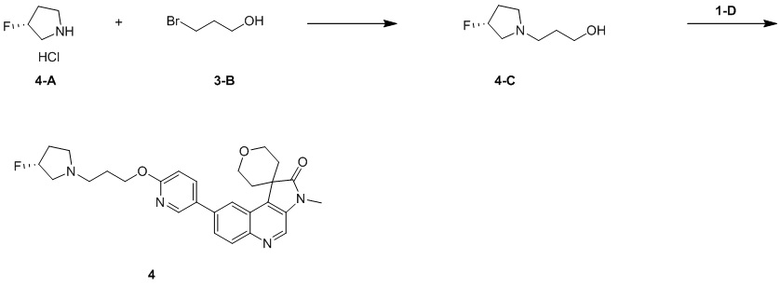
Способ синтеза:
[128] Стадия 1: Синтез соединения 1-A
[129] n-BuLi (2,5 M, 765,23 мкл) медленно по каплям добавляли к раствору DIPA (193,58 мг, 1,91 ммоль, 270,37 мкл) в ТГФ (1 мл) при -60 °C и перемешивали в течение 0,5 ч. Затем по каплям добавляли метилтетрагидропиран-4-карбоксилат (300,88 мг, 2,09 ммоль, 278,59 мкл) при -60 °C и перемешивали в течение 1 ч и раствор промежуточного соединения A (500 мг, 1,74 ммоль) в ТГФ (4 мл) добавляли и перемешивали при -60 °C в течение 2 ч. После завершения реакции реакцию гасили путем добавления 20 мл насыщенного раствора NH4Cl при 0 °C и проводили экстракцию EtAc (30 мл, 10 мл * 3). Органическую фазу собирали, промывали насыщенным солевым раствором (50 мл) и сушили над безводным сульфатом натрия. После удаления осушителя путем фильтрации, растворитель удаляли при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали с помощью колоночной хроматографии (0 до 10% ТГФ/ПЭ), в результате чего получали соединение 1-A.
[130] МС m/z: 394,9[M+H] +
[131] 1H ЯМР (400 МГц, CDCl3) δ 8,75 (s, 1H), 8,52 (d, J = 1,8 Гц, 1H), 8,05 (d, J = 9,0 Гц, 1H), 7,88 (dd, J = 2,0, 9,0 Гц, 1H), 4,00 - 3,89 (m, 4H), 3,69 (s, 3H), 2,52 - 2,46 (m, 2H), 2,43 - 2,31 (m, 2H)
[132] Стадия 2: Синтез соединения 1-B
[133] Zn (330,92 мг, 5,06 ммоль) и NH4Cl (270,70 мг, 5,06 ммоль) добавляли к раствору соединения 1-A (200 мг, 506,07 мкмоль) в H2 (10 мл) и ТГФ (10 мл). Реакционный раствор перемешивали при 70°C в течение 0,5 ч. После завершения реакции реакционный раствор фильтровали через целит. Фильтрат подвергали экстракции EtAc (90 мл, 30 мл * 3). Органическую фазу собирали, промывали насыщенным солевым раствором (50 мл) и сушили над безводным сульфатом натрия. После удаления осушителя путем фильтрации, растворитель удаляли при пониженном давлении, в результате чего получали неочищенный продукт и неочищенный продукт очищали с помощью колоночной хроматографии (0 до 10% ТГФ/ДХМ), в результате чего получали соединение 1-B.
[134] МС m/z: 332,8[M+H] +
[135] 1H ЯМР (400 МГц, CDCl3) δ 8,74 (s, 1H), 8,23 (d, J = 1,8 Гц, 1H), 8,01 (d, J = 9,0 Гц, 1H), 7,69 (dd, J = 2,0, 9,0 Гц, 1H), 4,48 - 4,40 (m, 2H), 4,01 (dd, J = 5,1, 11,7 Гц, 2H), 2,68 (dt, J = 5,1, 13,5 Гц, 2H), 1,75 (ушир. d, J = 14,3 Гц, 2H)
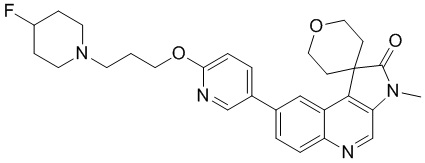
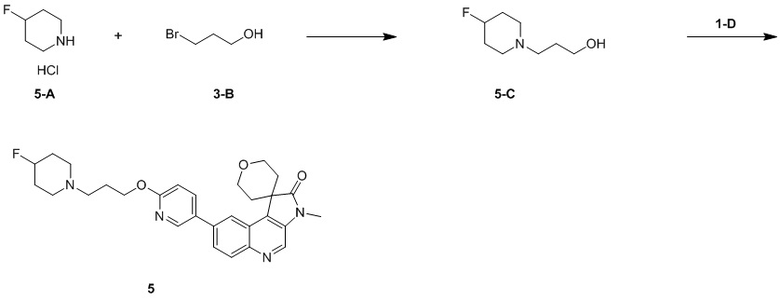
[136] Стадия 3: Синтез соединения 1-C
[137] MeI (1,350 г, 9,51 ммоль, 592,11 мкл), TBAB (9,68 мг, 30,01 мкмоль) и NaH (24,01 мг, 600,28 мкмоль) добавляли к смешанному раствору соединения 1-B (100 мг, 300,14 мкмоль) в ДХМ (5 мл) и H2 (5 мл) и перемешивали при 25 °C в течение 21 ч. После завершения реакции к реакционной системе добавляли 10 мл воды при комнатной температуре для гашения реакции и проводили экстракцию ДХМ (30 мл, 10 мл * 3). Органическую фазу собирали, промывали насыщенным солевым раствором (50 мл) и сушили над безводным сульфатом натрия. После удаления осушителя путем фильтрации, растворитель удаляли при пониженном давлении, в результате чего получали неочищенный продукт и неочищенный продукт очищали с помощью колоночной хроматографии (0 до 10% ТГФ/ДХМ), в результате чего получали соединение 1-C.
[138] МС m/z: 346,9[M+H] +
[139] 1H ЯМР (400 МГц, CDCl3) δ 8,71 (s, 1H), 8,24 (d, J = 2,0 Гц, 1H), 8,01 (d, J = 9,1 Гц, 1H), 7,68 (dd, J = 2,1, 9,1 Гц, 1H), 4,48 (dt, J = 2,1, 12,2 Гц, 2H), 4,00 (dd, J = 5,1, 11,6 Гц, 2H), 3,38 (s, 3H), 2,67 (dt, J = 5,3, 13,4 Гц, 2H), 1,66 (ушир. d, J = 14,3 Гц, 2H)
[140] Стадия 4: Синтез соединения 1-D
[141] В защитной атмосфере азота 1,4-диоксан (3 мл) и H2 (3 мл) добавляли к реакционной системе соединения 1-C (70 мг, 201,61 мкмоль), 2-фторпиридин-5-борной кислоты (42,61 мг, 302,41 мкмоль), Pd(PPh3)4 (23,30 мг, 20,16 мкмоль) и Na2C3 (64,11 мг, 604,83 мкмоль) и перемешивали при 80 °C в течение 3 ч. После завершения реакции к реакционной системе добавляли H2 (20 мл) и EtAc (30 мл) и фильтровали через целит. Органическую фазу собирали, промывали насыщенным солевым раствором (50 мл) и сушили над безводным сульфатом натрия. После удаления осушителя путем фильтрации, растворитель удаляли при пониженном давлении, в результате чего получали неочищенный продукт и неочищенный продукт очищали с помощью колоночной хроматографии (0 до 30% ТГФ/ДХМ), в результате чего получали соединение 1-D.
[142] МС m/z: 364,0[M+H] +
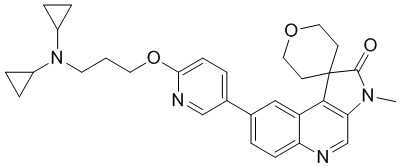
[143] 1H ЯМР (400 МГц, CDCl3) δ 8,75 (s, 1H), 8,56 (d, J = 2,1 Гц, 1H), 8,26 (d, J = 8,9 Гц, 1H), 8,18 (d, J = 1,5 Гц, 1H), 8,11 (dt, J = 2,5, 8,0 Гц, 1H), 7,79 (dd, J = 1,8, 8,8 Гц, 1H), 7,12 (dd, J = 3,0, 8,4 Гц, 1H), 4,50 (ушир. t, J = 11,3 Гц, 2H), 4,00 (dd, J = 4,9, 11,7 Гц, 2H), 3,41 (s, 3H), 2,74 (dt, J = 5,3, 13,4 Гц, 2H), 1,72 (ушир. d, J = 14,1 Гц, 2H)
[144] Стадия 5: Синтез соединения 1
[145] При 0 °C и в защитной атмосфере азота раствор соединения 3-диметиламино-1-пропанола (34,39 мг, 333,34 мкмоль, 38,99 мкл) в ДМФА (2 мл) добавляли NaH (26,67 мг, 666,68 мкмоль, 60% чистота) и затем раствор соединения 1-D (61,45 мг, 166,67 мкмоль) в ДМФА (2 мл) добавляли и перемешивали при 27 °C в течение 2,5 ч. При 0 °C к реакционной системе добавляли 50 мл воды для гашения реакции и проводили экстракцию посредством ДХМ/i-pr H (5/1) (10 мл * 3). Органическую фазу собирали, промывали насыщенным солевым раствором (100 мл) и сушили над безводным сульфатом натрия. После удаления осушителя путем фильтрации, растворитель удаляли при пониженном давлении, в результате чего получали неочищенный продукт и неочищенный продукт подвергали колоночной хроматографии (0 до 10% MeH/ДХМ (добавление водного аммиака)), в результате чего получали соединение 1.
[146] МС m/z: 447,2[M+H] +
[147] 1H ЯМР (400 МГц, CDCl3) δ 8,66 (s, 1H), 8,47 (d, J = 2,5 Гц, 1H), 8,16 (d, J = 8,9 Гц, 1H), 8,12 (d, J = 1,5 Гц, 1H), 7,88 (dd, J = 2,5, 8,6 Гц, 1H), 7,75 (dd, J = 1,7, 8,8 Гц, 1H), 6,85 (d, J = 8,5 Гц, 1H), 4,49 - 4,34 (m, 4H), 3,94 (dd, J = 4,9, 11,6 Гц, 2H), 3,35 (s, 3H), 2,71 (dt, J = 5,1, 13,4 Гц, 2H), 2,50 - 2,43 (m, 2H), 2,25 (s, 6H), 2,02 - 1,94 (m, 2H), 1,65 (ушир. d, J = 14,0 Гц, 2H)
Пример 2: Соединение 2
Способ синтеза:
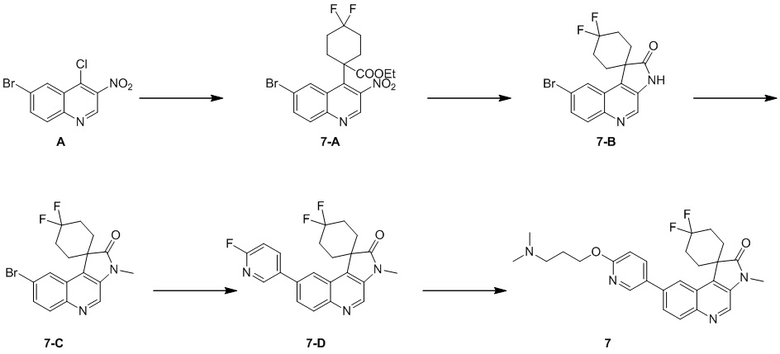
[148] Стадия 1: Синтез соединения 2
[149] В защитной атмосфере азота 1,4-диоксан (3 мл) добавляли к реакционной системе соединения 1-C (200 мг, 576,03 мкмоль), (1-метил-1H-пиразол)-4-борной кислоты (179,78 мг, 864,04 мкмоль), комплекса [1,1'-бис(дифенилфосфино)ферроцен]палладия дихлорида дихлорметана (47,04 мг, 57,60 мкмоль) и ацетата калия (169,59 мг, 1,73 ммоль) и перемешивали при 80 °C в течение 18 ч. После завершения реакции растворитель удаляли при пониженном давлении, в результате чего получали неочищенный продукт и неочищенный продукт очищали с помощью колоночной хроматографии (0 до 10% ТГФ/ДХМ), в результате чего получали соединение 2.
[150] МС m/z: 349,1[M+H] +
[151] 1H ЯМР (400 МГц, CDCl3) δ 8,64 (s, 1H), 8,13 (d, J = 8,9 Гц, 1H), 8,08 (d, J = 1,8 Гц, 1H), 7,91 (s, 1H), 7,83 (s, 1H), 7,75 (dd, J = 1,8, 8,8 Гц, 1H), 4,51 (dt, J = 2,1, 12,2 Гц, 2H), 4,04 - 3,98 (m, 5H), 3,38 (s, 3H), 2,81 - 2,70 (m, 2H), 1,69 (ушир. d, J = 14,3 Гц, 2H)
Пример 3: Соединение 3
Способ синтеза:
[152] Стадия 1: Синтез соединения 3-C
[153] Ацетонитрил (10 мл) добавляли в колбу с 3-A (200 мг, 1,59 ммоль), 3-B (225,80 мг, 1,62 ммоль, 146,62 мкл) и K2C3 (660,37 мг, 4,78 ммоль) и перемешивали при 80 °C в течение 12 ч. Систему фильтровали путем добавления метанола (50 мл) и затем растворитель удаляли из фильтрата при пониженном давлении. Остаточное твердое вещество добавляли к дихлорметану (50 мл), фильтровали и концентрировали при пониженном давлении, в результате чего получали 3-C.
[154] МС m/z: 147,9[M+H] +
[155] 1H ЯМР (400 МГц, CD3D) δ 5,28- 5,07(m, 1 H), 3,72 -3,57(m, 2 H), 3,04-2,87 (m, 2 H), 2,73 - 2,56 (m, 3 H), 2,47-2,39 (m, 1 H), 2,28-1,90(m, 2 H), 1,82-1,69 (m, 2 H)
[156] Стадия 2: Синтез соединения 3
[157] За исключением использования соответствующих исходных веществ, соединение 3 получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[158] МС m/z: 491,2[M+H] +
[159] ee: 100%, RT = 1,955 мин (тип колонки: Chiralpak AD-3 50×3 мм внутр. диаметр, 3 мкм; подвижная фаза: A: C2, B: этанол (0,05% диэтиламина); градиент: доля B в подвижной фазе увеличивается от 5% до 40% в течение 2,5 минут и сохраняется на уровне 40% в течение 0,35 минут и затем доля подвижной фазы снижается от 40% до 5% в течение 0,15 минут; скорость потока: 2,5 мл/мин, температура колонки: 40 °C).
[160] 1H ЯМР (400 МГц, CDCl3) δ 8,71 (s, 1H), 8,51 (d, J = 2,5 Гц, 1H), 8,21 (d, J = 8,8 Гц, 1H), 8,16 (d, J = 1,8 Гц, 1H), 7,93 (dd, J = 2,5, 8,5 Гц, 1H), 7,80 (dd, J = 1,9, 8,9 Гц, 1H), 6,89 (d, J = 8,5 Гц, 1H), 5,28 - 5,09 (m, 1H), 4,53 - 4,42 (m, 4H), 3,99 (dd, J = 4,8, 11,5 Гц, 2H), 3,40 (s, 3H), 2,98 - 2,85 (m, 2H), 2,84 - 2,77 (m, 1H), 2,75 (ушир. d, J = 5,0 Гц, 1H), 2,74 - 2,66 (m, 3H), 2,52 - 2,44 (m, 1H), 2,25 - 1,98 (m, 4H), 1,70 (ушир. d, J = 14,3 Гц, 2H)
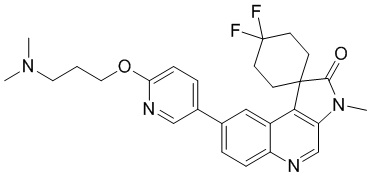
Пример 4: Соединение 4
Способ синтеза:
[161] Стадия 1: Синтез соединения 4-C
[162] За исключением использования соответствующих исходных веществ, соединение 4-C получали с использованием того же способа, как и при получении соединения 3-C в примере 3.
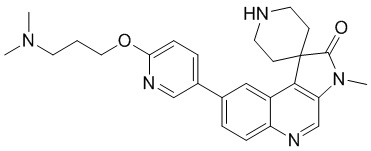
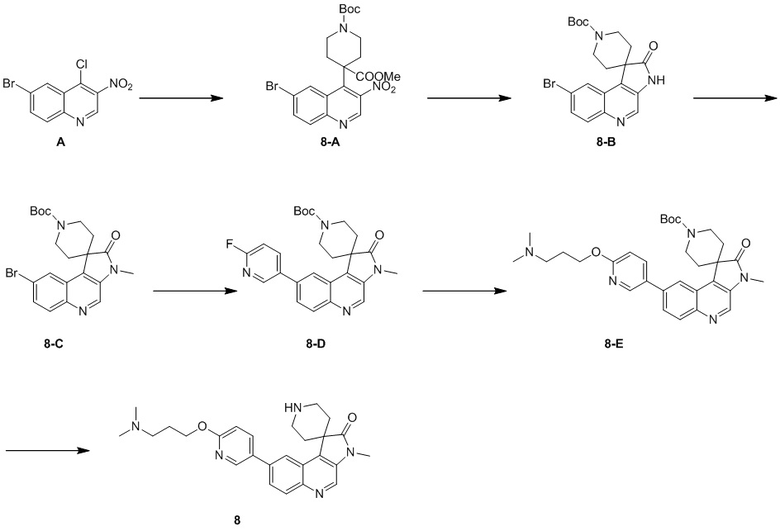
[163] МС m/z: 147,9[M+H] +
[164] 1H ЯМР (400 МГц, CD3D) δ 5,30 - 5,08 (m, 1 H), 3,64 (t, J = 6,28 Гц, 2 H), 3,04 - 2,89 (m, 2 H), 2,62 (ddd, J = 9,66, 5,52, 2,38 Гц, 3 H), 2,42 (td, J = 8,16, 7,03 Гц, 1 H), 2,29 -1,94(m, 2 H), 1,83 - 1,70 (m, 2 H)
[165] Стадия 2: Синтез соединения 4
[166] За исключением использования соответствующих исходных веществ, соединение 4 получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[167] МС m/z: 491,3[M+H] +
[168] ee: 100%, время удерживания = 1,952 мин (тип колонки: Chiralpak AD-3 50 × 3 мм внутр. диаметр, 3 мкм; подвижная фаза: A: C2, B: этанол (0,05% диэтиламина); градиент: 5%-40% B, 2,5 минут 40%-40% B, 0,35 минут; 40%-5% B, 0,15 минут; скорость потока: 2,5 мл/мин; температура колонки: 40 °C).
[169] 1H ЯМР (400 МГц, CDCl3) δ 8,71 (s, 1H), 8,51 (d, J = 2,3 Гц, 1H), 8,21 (d, J = 8,8 Гц, 1H), 8,16 (s, 1H), 7,93 (dd, J = 2,4, 8,7 Гц, 1H), 7,83 - 7,77 (m, 1H), 6,89 (d, J = 8,8 Гц, 1H), 5,28 - 5,08 (m, 1H), 4,54 - 4,42 (m, 4H), 3,99 (ушир. dd, J = 4,6, 11,7 Гц, 2H), 3,40 (s, 3H), 2,98 - 2,85 (m, 2H), 2,84 - 2,66 (m, 5H), 2,53 - 2,43 (m, 1H), 2,26 - 1,97 (m, 4H), 1,71 (ушир. s, 2H)
Пример 5: Соединение 5
Способ синтеза:
[170] Стадия 1: Синтез соединения 5-C
[171] За исключением использования соответствующих исходных веществ, соединение 5-C получали с использованием того же способа, как и при получении соединения 3-C в примере 3.
[172] 1H ЯМР (400 МГц, CDCl3) δ 4,84 - 4,56 (m, 1H), 3,83 - 3,70 (m, 3H), 3,52 (t, J = 6,5 Гц, 1H), 2,64 - 2,57 (m, 3H), 2,10 - 04 (m, 1H), 1,93 - 1,87 (m, 2H), 1,87 - 1,81 (m, 2H), 1,74 - 1,68 (m, 2H)
[173] Стадия 2: Синтез соединения 5
[174] За исключением использования соответствующих исходных веществ, соединение 5 получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[175] МС m/z: 505,3[M+H] +
[176] 1H ЯМР (400 МГц, CDCl3) δ 8,71 (s, 1H), 8,51 (d, J = 2,3 Гц, 1H), 8,22 (d, J = 9,0 Гц, 1H), 8,16 (d, J = 1,5 Гц, 1H), 7,93 (dd, J = 2,5, 8,8 Гц, 1H), 7,80 (dd, J = 1,8, 8,8 Гц, 1H), 6,89 (d, J = 8,5 Гц, 1H), 4,80 - 4,59 (m, 1H), 4,49 (ушир. t, J = 11,4 Гц, 2H), 4,43 (t, J = 6,5 Гц, 2H), 3,99 (dd, J = 4,9, 11,7 Гц, 2H), 3,40 (s, 3H), 2,76 (dt, J = 5,1, 13,5 Гц, 2H), 2,63 (ушир. s, 2H), 2,56 (ушир. t, J = 7,4 Гц, 2H), 2,42 (ушир. s, 2H), 2,08 - 1,99 (m, 2H), 1,88 (ушир. d, J = 4,5 Гц, 4H), 1,70 (ушир. d, J = 14,1 Гц, 2H)
Пример 6: Соединение 6
Способ синтеза:
[177] Стадия 1: Синтез соединения 6-C
[178] За исключением использования соответствующих исходных веществ, соединение 6-C получали с использованием того же способа, как и при получении соединения 3-C в примере 3.
[179] МС m/z: 155,9 [M+H] +
[180] Стадия 2: Синтез соединения 6
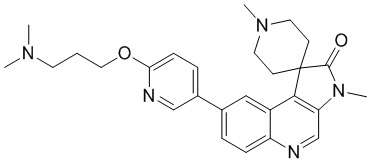
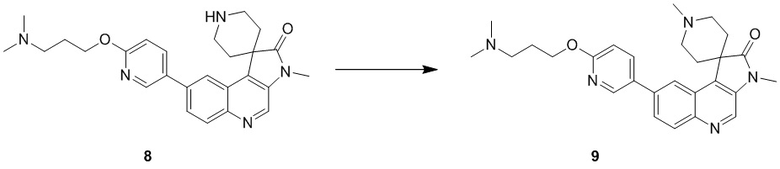
[181] За исключением использования соответствующих исходных веществ, соединение 6 получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[182] МС m/z: 499,3[M+H] +
[183] 1H ЯМР (400 МГц, CDCl3) δ 8,71 (s, 1H), 8,52 (d, J = 2,0 Гц, 1H), 8,22 (d, J = 8,8 Гц, 1H), 8,17 (d, J = 1,8 Гц, 1H), 7,93 (dd, J = 2,5, 8,5 Гц, 1H), 7,80 (dd, J = 1,9, 8,9 Гц, 1H), 6,90 (d, J = 8,5 Гц, 1H), 4,49 (ушир. t, J = 11,2 Гц, 2H), 4,39 (t, J = 6,5 Гц, 2H), 3,99 (dd, J = 4,8, 11,5 Гц, 2H), 3,40 (s, 3H), 2,94 - 2,88 (m, 2H), 2,76 (dt, J = 5,0, 13,4 Гц, 2H), 2,18 - 2,10 (m, 2H), 1,94 - 1,87 (m, 2H), 1,70 (ушир. d, J = 14,3 Гц, 2H), 0,50 - 0,39 (m, 8H)
Пример 7: Соединение 7
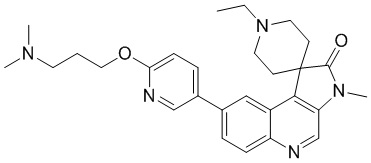
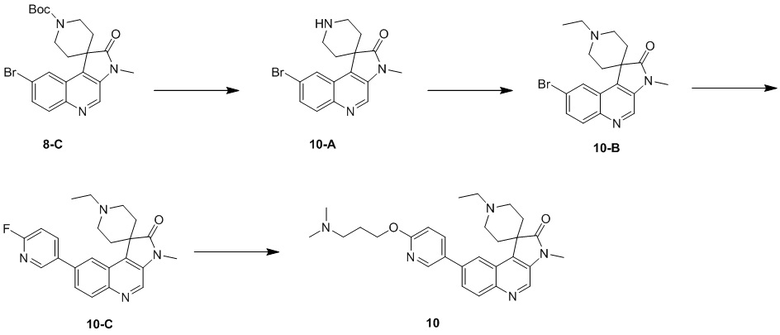
Способ синтеза:
[184] Стадия 1: Синтез соединения 7-A
[185] За исключением использования соответствующих исходных веществ, соединение 7-A получали с использованием того же способа, как и при получении соединения 1-A в примере 1.
[186] МС m/z: 443,0[M+H] +
[187] 1H ЯМР (400 МГц, CDCl3) δ 8,79 (s, 1H), 8,52 (d, J = 1,8 Гц, 1H), 8,05 (d, J = 9,0 Гц, 1H), 7,88 (dd, J = 1,9, 8,9 Гц, 1H), 4,14 (q, J = 7,2 Гц, 2H), 2,66 - 2,57 (m, 2H), 2,53 - 2,39 (m, 3H), 2,38 - 2,27 (m, 1H), 2,21 - 2,09 (m, 2H), 1,08 (t, J = 7,2 Гц, 3H)
[188] Стадия 2: Синтез соединения 7-B
[189] За исключением использования соответствующих исходных веществ, соединение 7-B получали с использованием того же способа, как и при получении соединения 1-B в примере 1.
[190] МС m/z: 366,8[M+H] +
[191] 1H ЯМР (400 МГц, CDCl3) δ 8,78 (s, 1H), 8,05 - 8,02 (m, 2H), 7,70 (dd, J = 2,0, 9,0 Гц, 1H), 2,92 - 2,80 (m, 1H), 2,78 - 2,65 (m, 3H), 2,17 (ушир. s, 2H), 1,94 (ушир. d, J = 12,3 Гц, 2H)
[192] Стадия 3: Синтез соединения 7-C
[193] За исключением использования соответствующих исходных веществ, соединение 7-C получали с использованием того же способа, как и при получении соединения 1-C в примере 1.
[194] 1H ЯМР (400 МГц, CDCl3) δ 8,71 (s, 1H), 8,04 (d, J = 1,8 Гц, 1H), 8,01 (d, J = 9,0 Гц, 1H), 7,68 (dd, J = 1,8, 9,0 Гц, 1H), 3,38 (s, 3H), 2,94 - 2,75 (m, 2H), 2,74 - 2,63 (m, 2H), 2,15 (ушир. s, 2H), 1,89 - 1,79 (m, 2H)
[195] Стадия 4: Синтез соединения 7-D
[196] За исключением использования соответствующих исходных веществ, соединение 7-D получали с использованием того же способа, как и при получении соединения 1-D в примере 1.
[197] МС m/z: 398,0[M+H] +
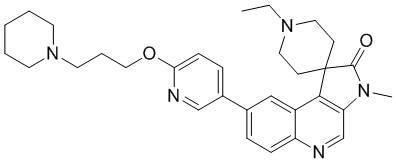
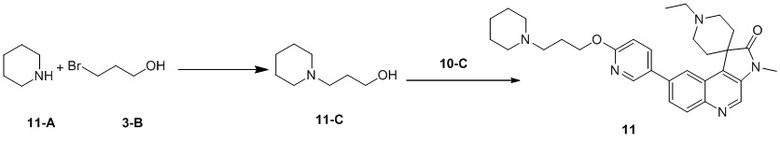
[198] 1H ЯМР (400 МГц, CDCl3) δ 8,75 (s, 1H), 8,56 (d, J = 2,5 Гц, 1H), 8,26 (d, J = 8,8 Гц, 1H), 8,10 (dt, J = 2,5, 8,0 Гц, 1H), 8,06 (d, J = 1,8 Гц, 1H), 7,82 (dd, J = 2,0, 8,8 Гц, 1H), 7,11 (dd, J = 2,9, 8,4 Гц, 1H), 3,41 (s, 3H), 2,95 - 2,73 (m, 4H), 2,15 (ушир. d, J = 5,3 Гц, 2H), 1,90 (ушир. d, J = 11,5 Гц, 2H)
[199] Стадия 5: Синтез соединения 7
[200] За исключением использования соответствующих исходных веществ, соединение 7 получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[201] МС m/z: 481,2[M+H] +
[202] 1H ЯМР (400 МГц, CDCl3) δ 8,70 (s, 1H), 8,50 (d, J = 2,3 Гц, 1H), 8,21 (d, J = 9,0 Гц, 1H), 8,02 (d, J = 1,8 Гц, 1H), 7,91 (dd, J = 2,6, 8,7 Гц, 1H), 7,81 (dd, J = 1,9, 8,9 Гц, 1H), 6,88 (d, J = 8,5 Гц, 1H), 4,43 (t, J = 6,4 Гц, 2H), 3,40 (s, 3H), 2,96 - 2,74 (m, 4H), 2,58 (ушир. t, J = 7,5 Гц, 2H), 2,35 (s, 6H), 2,20 - 2,11 (m, 2H), 2,10 - 2,01 (m, 2H), 1,88 (ушир. d, J = 9,8 Гц, 2H)
Пример 8: Соединение 8
Способ синтеза:
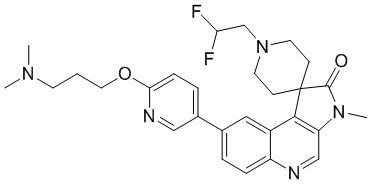
[203] Стадия 1: Синтез соединения 8-A
[204] За исключением использования соответствующих исходных веществ, соединение 8-A получали с использованием того же способа, как и при получении соединения 1-A в примере 1.
[205] 1H ЯМР (400 МГц, CDCl3) δ 8,74 (s, 1H), 8,46 (d, J = 1,8 Гц, 1H), 8,05 (d, J = 8,8 Гц, 1H), 7,88 (dd, J = 2,0, 9,0 Гц, 1H), 4,02 (ушир. s, 2H), 3,66 (s, 3H), 3,36 - 3,22 (m, 2H), 2,53 (ушир. s, 2H), 2,28 - 2,18 (m, 2H), 1,48 (s, 9H)
[206] Стадия 2: Синтез соединения 8-B
[207] За исключением использования соответствующих исходных веществ, соединение 8-B получали с использованием того же способа, как и при получении соединения 1-B в примере 1.
[208] МС: m/z: 432,0[M+H] +
[209] 1H ЯМР (400 МГц, CDCl3) δ 8,73 (s, 1H), 8,07 (s, 1H), 8,02 - 7,98 (m, 2H), 7,68 (dd, J = 2,0, 9,0 Гц, 1H), 4,30 - 4,01 (m, 2H), 3,78 - 3,72 (m, 2H), 2,49 (ушир. s, 2H), 1,78 (ушир. d, J = 13,8 Гц, 2H), 1,62 (s, 9H)
[210] Стадия 3: Синтез соединения 8-C
[211] За исключением использования соответствующих исходных веществ, соединение 8-C получали с использованием того же способа, как и при получении соединения 1-C в примере 1.
[212] МС m/z: 446,0[M+H] +
[213] 1H ЯМР (400 МГц, CDCl3) δ 8,71 (s, 1H), 8,03 - 7,99 (m, 2H), 7,67 (dd, J = 2,1, 9,2 Гц, 1H), 4,30 - 4,02 (m, 2H), 3,75 (ушир. d, J = 6,8 Гц, 2H), 3,38 (s, 3H), 2,48 (ушир. s, 2H), 1,69 (ушир. d, J = 13,1 Гц, 2H), 1,57 (s, 9H)
[214] Стадия 4: Синтез соединения 8-D
[215] За исключением использования соответствующих исходных веществ, соединение 8-D получали с использованием того же способа, как и при получении соединения 1-D в примере 1.
[216] МС m/z: 463,2[M+H] +
[217] 1H ЯМР (400 МГц, CDCl3) δ 8,75 (s, 1H), 8,51 (d, J = 2,5 Гц, 1H), 8,25 (d, J = 8,8 Гц, 1H), 8,06 (dt, J = 2,6, 8,0 Гц, 1H), 7,97 (d, J = 1,8 Гц, 1H), 7,78 (dd, J = 1,8, 8,8 Гц, 1H), 7,06 (dd, J = 3,0, 8,5 Гц, 1H), 4,29 - 4,02 (m, 2H), 3,90 - 3,72 (m, 2H), 3,41 (s, 3H), 2,55 (ушир. s, 2H), 1,76 (ушир. d, J = 14,1 Гц, 2H), 1,50 (s, 9H)
[218] Стадия 5: Синтез соединения 8-E
[219] За исключением использования соответствующих исходных веществ, соединение 8-E получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[220] 1H ЯМР (400 МГц, CDCl3) δ 8,70 (s, 1H), 8,45 (d, J = 2,3 Гц, 1H), 8,20 (d, J = 8,8 Гц, 1H), 7,96 (d, J = 1,5 Гц, 1H), 7,86 (dd, J = 2,5, 8,5 Гц, 1H), 7,78 (dd, J = 1,9, 8,9 Гц, 1H), 6,84 (d, J = 8,5 Гц, 1H), 4,41 (t, J = 6,5 Гц, 2H), 3,81 - 3,77 (m, 2H), 3,39 (s, 3H), 2,57 - 2,52 (m, 2H), 2,50 - 2,45 (m, 2H), 2,28 (s, 6H), 2,03 - 1,96 (m, 2H), 1,74 (ушир. d, J = 13,6 Гц, 2H), 1,71 - 1,65 (m, 2H), 1,51 (s, 9H)
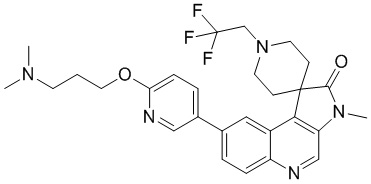
[221] Стадия 6: Синтез соединения 8
[222] Трифторуксусную кислоту (1 мл) добавляли к 8-E (100 мг, 183,26 мкмоль). Реакционный раствор перемешивали при 20 °C в течение 1 ч. Трифторуксусную кислоту удаляли при пониженном давлении, в результате чего получали неочищенный продукт, который отделяли с помощью препаративной хроматографии (кислотная, подвижная фаза: ацетонитрил -вода). Затем добавляли аммиачную воду для доведения pH до 8. Неочищенный продукт получали путем концентрирования при пониженном давлении и очищали с помощью колоночной хроматографии (0 до 10% Me H/ДХМ), в результате чего получали соединение 8.
[223] МС m/z: 446,2[M+H] +
[224] 1H ЯМР (400 МГц, CDCl3) δ 8,69 (s, 1H), 8,51 (d, J = 2,5 Гц, 1H), 8,22 (d, J = 1,5 Гц, 1H), 8,20 (d, J = 8,9 Гц, 1H), 7,93 (dd, J = 2,6, 8,6 Гц, 1H), 7,77 (dd, J = 1,7, 8,8 Гц, 1H), 6,88 (d, J = 8,6 Гц, 1H), 4,42 (t, J = 6,5 Гц, 2H), 3,74 - 3,65 (m, 2H), 3,38 (s, 3H), 3,04 (ушир. dd, J = 3,4, 12,3 Гц, 2H), 2,59 (dt, J = 4,6, 13,3 Гц, 2H), 2,50 - 2,45 (m, 2H), 2,27 (s, 6H), 2,04 - 1,96 (m, 2H), 1,75 (ушир. d, J = 14,0 Гц, 2H)
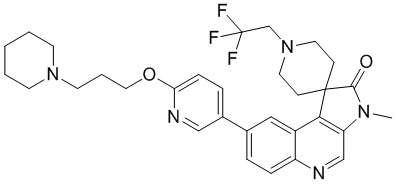
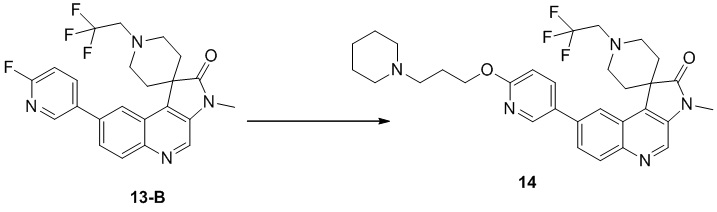
Пример 9: Соединение 9
Способ синтеза:
[225] Стадия 1: Синтез соединения 9
[226] Водный раствор формальдегида (127,51 мг, 1,57 ммоль, 116,98 мкл, 37% чистота) добавляли к раствору соединения 8 (70 мг, 157,11 мкмоль) в муравьиной кислоте (3 мл) и реакционный раствор перемешивали при 60 °C в течение 23 ч. После завершения реакции неочищенный продукт получали путем концентрирования при пониженном давлении. Добавляли 20 мл аммиачной воды и неочищенный продукт получали путем концентрирования при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии (0 до 6% Me H/ДХМ) и затем очищали с помощью препаративной хроматографии (нейтральная, подвижная фаза: ацетонитрил-вода), в результате чего получали соединение 9.
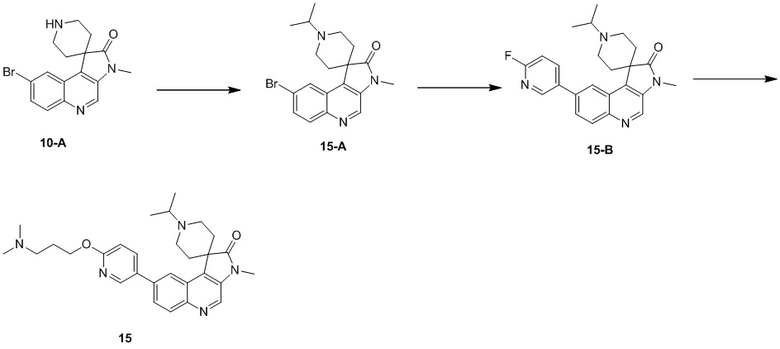
[227] МС m/z: 460,3[M+H] +
[228] 1H ЯМР (400 МГц, CDCl3) δ 8,67 (s, 1H), 8,52 (ушир. s, 1H), 8,26 (ушир. s, 1H), 8,19 (ушир. d, J = 8,8 Гц, 1H), 7,96 (ушир. d, J = 7,8 Гц, 1H), 7,76 (ушир. d, J = 8,5 Гц, 1H), 6,87 (ушир. d, J = 8,5 Гц, 1H), 4,41 (ушир. t, J = 6,1 Гц, 2H), 3,37 (s, 3H), 3,10 - 2,98 (m, 2H), 2,79 (ушир. d, J = 8,8 Гц, 4H), 2,51 - 2,45 (m, 2H), 2,27 (s, 9H), 2,05 - 1,94 (m, 2H), 1,76 (ушир. d, J = 12,8 Гц, 2H)
Пример 10: Соединение 10
Способ синтеза:
[229] Стадия 1: Синтез соединения 10-A
[230] Трифторуксусную кислоту (3 мл) добавляли к соединению 8-C (900 мг, 2,02 ммоль) и реакционный раствор перемешивали при 20 °C в течение 0,5 ч. После завершения реакции неочищенный продукт получали путем концентрирования при пониженном давлении. Добавляли аммиачную воду для доведения pH до 9. Выполняли экстракцию дихлорметаном (90 мл, 30 мл * 3) и органическую фазу концентрировали при пониженном давлении, в результате чего получали неочищенный продукт 10-A, который непосредственно применяли на следующей стадии.
[231] МС m/z: 346,1[M+H] +
[232] 1H ЯМР (400 МГц, CDCl3) δ 8,72 (s, 1H), 8,41 (d, J = 2,0 Гц, 1H), 7,99 (d, J = 9,0 Гц, 1H), 7,69 (dd, J = 2,0, 9,0 Гц, 1H), 3,97 - 3,88 (m, 2H), 3,39 (s, 3H), 3,35 (ушир. d, J = 3,0 Гц, 2H), 2,94 - 2,86 (m, 2H), 1,84 (ушир. d, J = 14,6 Гц, 2H)
[233] Стадия 2: Синтез соединения 10-B
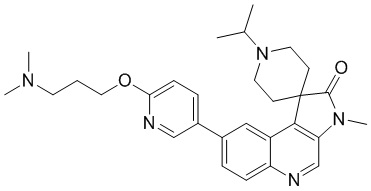
[234] При 20 °C и в защитной атмосфере азота K2C°3 (1,20 г, 8,67 ммоль) добавляли к раствору соединение 10-A (1 г, 2,89 ммоль) и этилбромид (629,82 мг, 5,78 ммоль, 431,38 мкл) в ацетонитриле (20 мл). Реакционный раствор перемешивали при 60 °C в течение 1 ч. Реакцию гасили путем добавления воды (5 мл) при 20°C, затем разбавляли водой (20 мл), проводили экстракцию Et°Ac (90 мл, 30 мл * 3). Органическую фазу собирали, промывали насыщенным раствором хлорида натрия (90 мл, 30 мл * 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт подвергали колоночной хроматографии (0 до 5% Me°H/ДХМ), в результате чего получали соединение 10-B.
[235] МС m/z: 373,9[M+H] +
[236] 1H ЯМР (400 МГц, ДМСО-d6) δ 8,94 (s, 1H), 8,15 (d, J = 1,8 Гц, 1H), 8,03 (d, J = 9,0 Гц, 1H), 7,76 (dd, J = 2,3, 9,0 Гц, 1H), 3,30 (s, 3H), 2,92 - 2,75 (m, 4H), 2,56 - 2,53 (m, 1H), 2,48 - 2,39 (m, 3H), 1,80 - 1,64 (m, 2H), 1,14 - 1,08 (m, 3H)
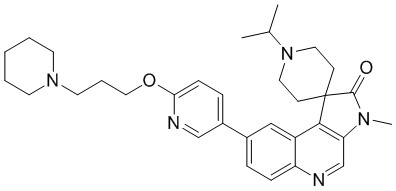
[237] Стадия 3: Синтез соединения 10-C
[238] В защитной атмосфере азота водный раствор соединения 10-B (400 мг, 1,07 ммоль), 2-фторпиридин-5-борную кислоту (301,19 мг, 2,14 ммоль), Na2C°3 (226,55 мг, 2,14 ммоль) и Pd(PPh3)4 (123,50 мг, 106,87 мкмоль) в диоксане (18 мл) и H2° (2 мл) перемешивали при 100 °C в течение 4 ч. Реакционный растворитель удаляли путем концентрирования и неочищенный продукт подвергали колоночной хроматографии (0 до 5% Me°H/ДХМ), в результате чего получали соединение 10-C.
[239] МС m/z: 391,1[M+H] +
[240] Стадия 8: Синтез соединения 10
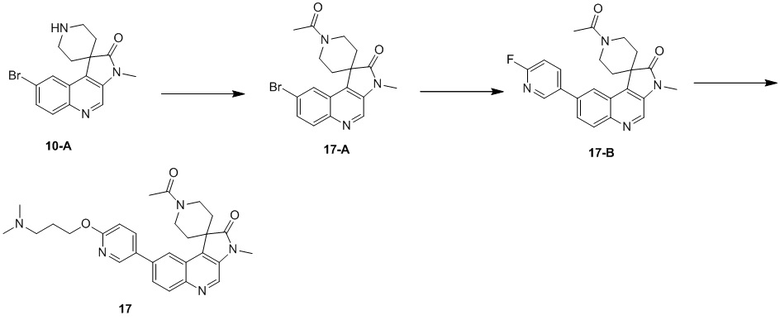
[241] При 20 °C и в защитной атмосфере азота соединение 10-C (200 мг, 512,23 мкмоль) добавляли к раствору 3-диметиламино-1-пропанола (105,69 мг, 1,02 ммоль, 119,82 мкл) и NaH (81,96 мг, 2,05 ммоль, 60% чистота) в ДМФА (10 мл) и реакционный раствор перемешивали в течение 2 ч при 70 °C. Реакционный раствор гасили водой (2 мл) при 20 °C, разбавляли путем добавления воды (10 мл) и подвергали экстракции Et°Ac (10 мл * 3). Органическую фазу собирали, промывали насыщенным раствором хлорида натрия (10 мл * 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали и неочищенный продукт подвергали колоночной хроматографии (0 до 5% MeH/ДХМ), в результате чего получали соединение 10.
[242] МС m/z: 474,2[M+H] +
[243] 1H ЯМР (400 МГц, CDCl3) δ 8,67 (s, 1H), 8,51 (d, J = 2,3 Гц, 1H), 8,26 - 8,23 (m, 1H), 8,19 (d, J = 9,0 Гц, 1H), 7,98 - 7,89 (m, 1H), 7,80 - 7,71 (m, 1H), 6,88 (d, J = 8,8 Гц, 1H), 4,42 (t, J = 6,5 Гц, 2H), 3,37 (s, 3H), 3,03 (ушир. s, 2H), 2,87 (ушир. d, J = 11,5 Гц, 2H), 2,76 (ушир. s, 2H), 2,62 (q, J = 7,1 Гц, 2H), 2,47 (s, 2H), 2,27 (s, 6H), 2,04 - 1,96 (m, 2H), 1,78 (ушир. d, J = 13,8 Гц, 2H), 1,17 (t, J = 7,3 Гц, 3H)
Пример 11: Соединение 11
Способ синтеза:
[244] Стадия 1: Синтез соединения 11-C
[245] За исключением использования соответствующих исходных веществ, соединение 11-C получали с использованием того же способа, как и при получении соединения 3-C в примере 3.
[246] 1H ЯМР (400 МГц, CDCl3) δ 5,96 - 5,45 (m, 1H), 3,78 - 3,70 (m, 2H), 2,51 (t, J = 5,6 Гц, 2H), 2,47 - 2,05 (m, 4H), 1,65 (quin, J = 5,5 Гц, 2H), 1,52 (quin, J = 5,5 Гц, 4H), 1,39 (ушир. s, 2H)
[247] Стадия 2: Синтез соединения 11
[248] За исключением использования соответствующих исходных веществ, соединение 11 получали с использованием того же способа, как и при получении соединения 10 в примере 10.
[249] МС m/z: 514,3[M+H] +
[250] 1H ЯМР (400 МГц, CDCl3) δ 8,67 (s, 1H), 8,53 (d, J = 2,5 Гц, 1H), 8,30 (d, J = 1,3 Гц, 1H), 8,19 (d, J = 8,8 Гц, 1H), 7,97 (dd, J = 2,5, 8,8 Гц, 1H), 7,77 (dd, J = 1,8, 8,8 Гц, 1H), 6,87 (d, J = 8,5 Гц, 1H), 4,40 (t, J = 6,5 Гц, 2H), 3,37 (s, 3H), 3,11 - 2,98 (m, 2H), 2,92 - 2,84 (m, 2H), 2,78 (ушир. s, 2H), 2,62 (q, J = 7,1 Гц, 2H), 2,54 - 2,33 (m, 8H), 2,08 - 1,95 (m, 2H), 1,77 (ушир. d, J = 13,8 Гц, 2H), 1,64 - 1,60 (m, 2H), 1,44 (ушир. d, J = 4,8 Гц, 2H), 1,17 (t, J = 7,2 Гц, 3H)
Пример 12: Соединение 12
Способ синтеза:
[251] Стадия 1: Синтез соединения 12-A
[252] За исключением использования соответствующих исходных веществ, соединение 12-A получали с использованием того же способа, как и при получении соединения 10-B в примере 10.
[253] МС m/z: 410,4 [M+H] +
[254] 1H ЯМР (400 МГц, CDCl3) δ 8,69 (s, 1H), 8,25 (d, J = 1,8 Гц, 1H), 8,00 (d, J = 9,0 Гц, 1H), 7,67 (dd, J = 2,0, 9,0 Гц, 1H), 6,21 - 5,85 (m, 1H), 3,36 (s, 3H), 3,33 - 3,25 (m, 2H), 3,00 - 2,87 (m, 4H), 2,65 (dt, J = 4,6, 13,4 Гц, 2H), 1,73 (ушир. d, J = 14,6 Гц, 2H)
[255] Стадия 2: Синтез соединения 12-B
[256] За исключением использования соответствующих исходных веществ, соединение 12-B получали с использованием того же способа, как и при получении соединения 10-C в примере 10.
[257] МС m/z: 427,2[M+H] +
[258] Стадия 3: Синтез соединения 12
[259] За исключением использования соответствующих исходных веществ, соединение 12 получали с использованием того же способа, как и при получении соединения 10 в примере 10.
[260] МС m/z: 510,4 [M+H] +
[261] 1H ЯМР (400 МГц, CDCl3) δ 8,69 (s, 1H), 8,53 (d, J = 2,3 Гц, 1H), 8,24 - 8,16 (m, 2H), 7,93 (dd, J = 2,5, 8,5 Гц, 1H), 7,78 (dd, J = 2,0, 8,8 Гц, 1H), 6,90 (d, J = 8,5 Гц, 1H), 6,20 - 5,81 (m, 1H), 4,43 (t, J = 6,5 Гц, 2H), 3,38 (s, 3H), 3,31 (ушир. t, J = 11,0 Гц, 2H), 3,00 - 2,93 (m, 1H), 2,92 - 2,81 (m, 3H), 2,75 (dt, J = 4,8, 13,3 Гц, 2H), 2,52 - 2,46 (m, 2H), 2,29 (s, 6H), 2,07 - 1,96 (m, 2H), 1,77 (ушир. d, J = 14,3 Гц, 2H).
Пример 13: Соединение 13
Способ синтеза:
[262] Стадия 1: Синтез соединения 13-A
[263] За исключением использования соответствующих исходных веществ, соединение 13-A получали с использованием того же способа, как и при получении соединения 10-B в примере 10.
[264] МС m/z: 428,1[M+H] +
[265] Стадия 2: Синтез соединения 13-B
[266] За исключением использования соответствующих исходных веществ, соединение 13-B получали с использованием того же способа, как и при получении соединения 10-C в примере 10.
[267] МС m/z: 445,2[M+H] +
[268] 1H ЯМР (400 МГц, CDCl3) δ 8,73 (s, 1H), 8,58 (d, J = 2,3 Гц, 1H), 8,27 - 8,22 (m, 2H), 8,13 (dt, J = 2,6, 8,0 Гц, 1H), 7,79 (dd, J = 1,8, 9,0 Гц, 1H), 7,11 (dd, J = 2,9, 8,4 Гц, 1H), 3,50 - 3,41 (m, 2H), 3,39 (s, 3H), 3,17 (q, J = 9,5 Гц, 2H), 2,94 (ушир. dd, J = 3,9, 10,9 Гц, 2H), 2,76 (dt, J = 4,8, 13,3 Гц, 2H), 1,80 (ушир. s, 2H).
[269] Стадия 3: Синтез соединения 13
[270] За исключением использования соответствующих исходных веществ, соединение 13 получали с использованием того же способа, как и при получении соединения 10 в примере 10.
[271] МС m/z: 528,3[M+H] +
[272] 1H ЯМР (400 МГц, CDCl3) δ 8,68 (s, 1H), 8,53 (d, J = 2,3 Гц, 1H), 8,23 - 8,18 (m, 2H), 7,94 (dd, J = 2,5, 8,8 Гц, 1H), 7,79 (dd, J = 1,6, 8,9 Гц, 1H), 6,90 (d, J = 8,5 Гц, 1H), 4,43 (t, J = 6,4 Гц, 2H), 3,46 (ушир. t, J = 11,2 Гц, 2H), 3,38 (s, 3H), 3,17 (q, J = 9,7 Гц, 2H), 2,92 (ушир. d, J = 11,0 Гц, 2H), 2,78 (dt, J = 4,5, 13,3 Гц, 2H), 2,53 - 2,46 (m, 2H), 2,28 (s, 6H), 2,06 - 1,96 (m, 2H), 1,77 (ушир. s, 2H)
Пример 14: Соединение 14
Способ синтеза:
[273] Стадия 1: Синтез соединения 14
[274] За исключением использования соответствующих исходных веществ, соединение 14 получали с использованием того же способа, как и при получении соединения 10 в примере 10.
[275] МС m/z: 568,3[M+H] +
[276] 1H ЯМР (400 МГц, CDCl3) δ 8,69 (s, 1H), 8,53 (d, J = 2,5 Гц, 1H), 8,24 - 8,18 (m, 2H), 7,94 (dd, J = 2,5, 8,5 Гц, 1H), 7,79 (dd, J = 1,6, 8,9 Гц, 1H), 6,89 (d, J = 8,5 Гц, 1H), 4,42 (t, J = 6,4 Гц, 2H), 3,46 (ушир. t, J = 11,2 Гц, 2H), 3,38 (s, 3H), 3,17 (q, J = 9,5 Гц, 2H), 2,92 (ушир. d, J = 11,3 Гц, 2H), 2,77 (dt, J = 4,6, 13,4 Гц, 2H), 2,58 - 2,51 (m, 2H), 2,46 (ушир. s, 2H), 2,10 - 2,00 (m, 2H), 1,77 (ушир. d, J = 14,1 Гц, 2H), 1,63 (ушир. s, 6H), 1,46 (ушир. s, 2H).
Пример 15: Соединение 15
Способ синтеза:
[277] Стадия 1: Синтез соединения 15-A
[278] За исключением использования соответствующих исходных веществ, соединение 15-A получали с использованием того же способа, как и при получении соединения 10-B в примере 10.
[279] МС m/z: 388,1[M+H] +
[280] 1H ЯМР (400 МГц, ДМСО-d6) δ 8,94 (s, 1H), 8,22 - 8,14 (m, 1H), 8,03 (d, J = 9,0 Гц, 1H), 7,76 (dd, J = 2,0, 9,0 Гц, 1H), 3,30 (s, 3H), 3,19 - 3,04 (m, 2H), 2,92 (ушир. d, J = 8,3 Гц, 1H), 2,83 - 2,66 (m, 2H), 2,43 (ушир. s, 2H), 1,82 - 1,68 (m, 2H), 1,11 (ушир. d, J = 4,8 Гц, 6H)
[281] Стадия 2: Синтез соединения 15-B
[282] За исключением использования соответствующих исходных веществ, соединение 15-B получали с использованием того же способа, как и при получении соединения 10-C в примере 10.
[283] МС m/z: 405,0[M+H] +
[284] 1H ЯМР (400 МГц, CDCl3) δ 8,64 (s, 1H), 8,53 (d, J = 2,0 Гц, 1H), 8,39 (s, 1H), 8,15 (d, J = 8,8 Гц, 2H), 7,70 (dd, J = 1,8, 8,8 Гц, 1H), 6,98 (dd, J = 2,8, 8,5 Гц, 1H), 3,31 (s, 3H), 3,28 - 3,17 (m, 2H), 2,87 (ушир. s, 1H), 2,83 - 2,67 (m, 4H), 1,81 - 1,67 (m, 2H), 1,09 (ушир. d, J = 6,0 Гц, 6H)
[285] Стадия 3: Синтез соединения 15
[286] За исключением использования соответствующих исходных веществ, соединение 15 получали с использованием того же способа, как и при получении соединения 10 в примере 10.
[287] МС m/z: 488,4[M+H] +
[288] 1H ЯМР (400 МГц, CDCl3) δ 8,70 (s, 1H), 8,56 (d, J = 2,3 Гц, 1H), 8,37 (s, 1H), 8,21 (d, J = 8,8 Гц, 1H), 8,03 (ушир. d, J = 6,8 Гц, 1H), 7,85 - 7,76 (m, 1H), 6,91 (d, J = 8,5 Гц, 1H), 4,44 (s, 2H), 3,39 (s, 3H), 3,37 - 3,26 (m, 2H), 2,97 (ушир. s, 1H), 2,81 (ушир. s, 4H), 2,49 (s, 2H), 2,29 (s, 6H), 2,07 - 1,98 (m, 2H), 1,87 - 1,75 (m, 2H), 1,19 (ушир. d, J = 6,5 Гц, 6H)
Пример 16: Соединение 16
Способ синтеза:
[289] Стадия 1: Синтез соединения 16
[290] За исключением использования соответствующих исходных веществ, соединение 16 получали с использованием того же способа, как и при получении соединения 10 в примере 10.
[291] МС m/z: 528,2[M+H] +
[292] 1H ЯМР (400 МГц, CDCl3) δ 8,61 (s, 1H), 8,48 (d, J = 2,4 Гц, 1H), 8,29 (ушир. s, 1H), 8,13 (d, J = 8,9 Гц, 1H), 7,94 (ушир. s, 1H), 7,71 (ушир. d, J = 8,8 Гц, 1H), 6,81 (d, J = 8,6 Гц, 1H), 4,33 (s, 2H), 3,31 (s, 3H), 3,28 - 3,16 (m, 2H), 2,89 (ушир. s, 1H), 2,73 (ушир. s, 4H), 2,43 (ушир. d, J = 7,9 Гц, 2H), 2,36 (ушир. s, 4H), 2,05 (ушир. s, 2H), 1,99 - 1,93 (m, 2H), 1,72 (ушир. d, J = 12,8 Гц, 2H), 1,58 - 1,54 (m, 2H), 1,38 (ушир. d, J = 3,8 Гц, 2H), 1,11 (ушир. d, J = 6,1 Гц, 6H)
Пример 17: Соединение 17
Способ синтеза:
[293] Стадия 1: Синтез соединения 17-A
[294] Соединение 10-A (150 мг, 433,25 мкмоль) растворяли в дихлорметане (10 мл), добавляли N,N-диизопропилэтиламин (111,99 мг, 866,50 мкмоль, 150,93 мкл) и добавляли ацетилхлорид (51,01 мг, 649,87 мкмоль, 46,38 мкл) при 0 °C в атмосфере азота. Смешанную систему нагревали до 30 °C, перемешивали в течение 2 ч и затем концентрировали, в результате чего получали неочищенный продукт, который отделяли и очищали с помощью колоночной хроматографии (ДХМ/ТГФ = 1/0 до 4/1), в результате чего получали соединение 17-A.
[295] МС m/z: 388,1[M+H] +
[296] 1H ЯМР (400 МГц, CDCl3) δ 8,72 (s, 1H), 8,01 (d, J = 9,3 Гц, 1H), 7,98 (d, J = 2,0 Гц, 1H), 7,68 (dd, J = 2,0, 9,0 Гц, 1H), 4,74 (ушир. dd, J = 4,9, 13,7 Гц, 1H), 4,18 (dt, J = 2,8, 13,3 Гц, 1H), 3,84 (ушир. dd, J = 4,6, 13,7 Гц, 1H), 3,60 (dt, J = 2,9, 13,2 Гц, 1H), 3,39 (s, 3H), 2,56 - 2,40 (m, 2H), 2,26 (s, 3H), 1,87 - 1,74 (m, 2H)
[297] Стадия 2: Синтез соединения 17-B
[298] Соединение 17-A (85 мг, 218,93 мкмоль) растворяли в безводном диоксане (10 мл) и воде (2 мл), добавляли 2-фторпиридин-5-борную кислоту (46,27 мг, 328,39 мкмоль) и карбонат натрия (69,61 мг, 656,78 мкмоль) и добавляли тетракистрифенилфосфинпалладий (37,95 мг, 32,84 мкмоль) в атмосфере азота. Смешанную систему перемешивали при 80 °C в течение 4 ч и затем подвергали экстракции дихлорметаном (150 мл, 50 мл * 3). Органическую фазу собирали, промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия и концентрировали, в результате чего получали неочищенный продукт, который отделяли и очищали с помощью колоночной хроматографии (ДХМ : ТГФ = 1/0 до 4/1), в результате чего получали соединение 17-B.
[299] МС m/z: 405,6[M+H] +
[300] 1H ЯМР (400 МГц, CDCl3) δ 8,76 (s, 1H), 8,49 (d, J = 2,5 Гц, 1H), 8,26 (d, J = 8,8 Гц, 1H), 8,09 - 8,01 (m, 1H), 7,92 (d, J = 1,8 Гц, 1H), 7,78 (dd, J = 2,0, 8,8 Гц, 1H), 7,12 (dd, J = 2,9, 8,4 Гц, 1H), 4,71 (ушир. d, J = 8,5 Гц, 1H), 4,20 (dt, J = 2,9, 13,2 Гц, 1H), 3,84 (ушир. d, J = 9,3 Гц, 1H), 3,68 - 3,54 (m, 1H), 3,42 (s, 3H), 2,65 - 2,49 (m, 2H), 2,22 (s, 3H), 1,86 (ушир. dd, J = 14,9, 18,9 Гц, 2H)
[301] Стадия 3: Синтез соединения 17
[302] Гидрид натрия (24,07 мг, 601,71 мкмоль, чистота: 60%) растворяли в N,N-диметилформамиде (10 мл) и добавляли 3-диметиламино-1-пропанол (23,28 мг, 225,64 мкмоль, 26,39 мкл) при 0 °C в атмосфере азота и перемешивали в течение 0,5 ч и затем добавляли раствор 17-B (70 мг, 150,43 мкмоль) в N,N-диметилформамиде (5 мл). Смешанную систему нагревали до комнатной температуры (25 °C) и перемешивали в течение 4 ч и затем гасили путем добавления воды (20 мл). Смешанную систему диспергировали в 100 мл воды и подвергали экстракции дихлорметаном (150 мл, 50 мл * 3). Органическую фазу собирали, промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия и концентрировали, в результате чего получали неочищенный продукт, который отделяли и очищали с помощью препаративной высокоэффективной жидкостной хроматографии (нейтральная, подвижная фаза: ацетонитрил-вода), в результате чего получали соединение 17.
[303] МС m/z: 488,4[M+H] +
[304] 1H ЯМР (400 МГц, CDCl3) δ 8,72 (s, 1H), 8,44 (d, J = 2,0 Гц, 1H), 8,22 (d, J = 8,8 Гц, 1H), 7,90 (d, J = 1,5 Гц, 1H), 7,85 (dd, J = 2,5, 8,5 Гц, 1H), 7,79 (dd, J = 1,9, 8,9 Гц, 1H), 6,90 (d, J = 8,8 Гц, 1H), 4,71 (ушир. d, J = 10,0 Гц, 1H), 4,43 (t, J = 6,4 Гц, 2H), 4,24 - 4,13 (m, 1H), 3,83 (ушир. d, J = 13,8 Гц, 1H), 3,68 - 3,57 (m, 1H), 3,41 (s, 3H), 2,65 - 2,52 (m, 2H), 2,52 - 2,43 (m, 2H), 2,28 (s, 6H), 2,23 (s, 3H), 2,05 - 1,96 (m, 2H), 1,85 (ушир. t, J = 14,2 Гц, 2H)
Пример 18: Соединение 18
Способ синтеза:
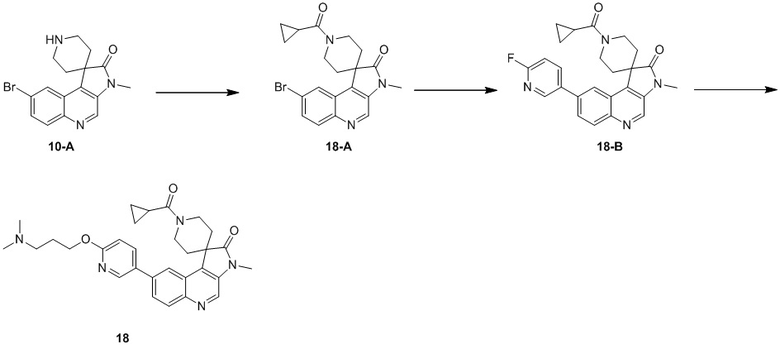
[305] Стадия 1: Синтез соединения 18-A
[306] За исключением использования соответствующих исходных веществ, соединение 18-A получали с использованием того же способа, как и при получении соединения 17-A в примере 17.
[307] МС m/z: 414,1[M+H] +
[308] Стадия 2: Синтез соединения 18-B
[309] За исключением использования соответствующих исходных веществ, соединение 18-B получали с использованием того же способа, как и при получении соединения 17-B в примере 17.
[310] МС m/z: 431,2[M+H] +
[311] 1H ЯМР (400 МГц, CDCl3) δ 8,76 (s, 1H), 8,50 (d, J = 2,5 Гц, 1H), 8,26 (d, J = 9,0 Гц, 1H), 8,05 (dt, J = 2,6, 8,0 Гц, 1H), 7,93 (d, J = 1,5 Гц, 1H), 7,79 (dd, J = 1,9, 8,9 Гц, 1H), 7,11 (dd, J = 2,8, 8,5 Гц, 1H), 4,67 (ушир. d, J = 12,5 Гц, 1H), 4,26 (ушир. d, J = 7,5 Гц, 2H), 3,67 (ушир. t, J = 12,8 Гц, 1H), 3,42 (s, 3H), 2,59 (ушир. d, J = 12,3 Гц, 2H), 1,95 - 1,85 (m, 2H), 1,79 (ушир. d, J = 13,1 Гц, 1H), 1,12 (ушир. s, 1H), 1,00 (ушир. s, 1H), 0,83 (dd, J = 3,6, 7,9 Гц, 2H)
[312] Стадия 3: Синтез соединения 18
[313] За исключением использования соответствующих исходных веществ, соединение 18 получали с использованием того же способа, как и при получении соединения 17 в примере 17.
[314] МС m/z: 514,4[M+H] +
[315] 1H ЯМР (400 МГц, CDCl3) δ 8,72 (s, 1H), 8,45 (d, J = 2,3 Гц, 1H), 8,21 (d, J = 8,8 Гц, 1H), 7,91 (d, J = 1,8 Гц, 1H), 7,86 (dd, J = 2,5, 8,5 Гц, 1H), 7,80 (dd, J = 1,8, 8,8 Гц, 1H), 6,88 (d, J = 8,3 Гц, 1H), 4,67 (ушир. d, J = 11,0 Гц, 1H), 4,43 (t, J = 6,4 Гц, 2H), 4,25 (ушир. d, J = 8,3 Гц, 2H), 3,67 (ушир. t, J = 12,3 Гц, 1H), 3,41 (s, 3H), 2,71 - 2,52 (m, 4H), 2,34 (s, 6H), 2,09 - 2,05 (m, 1H), 2,04 - 2,00 (m, 1H), 1,95 - 1,83 (m, 3H), 1,16 - 1,01 (m, 2H), 0,89 - 0,79 (m, 2H)
Пример 19: Соединение 19
Способ синтеза:
[316] Стадия 1: Синтез соединения 19-A
[317] За исключением использования соответствующих исходных веществ, соединение 19-A получали с использованием того же способа, как и при получении соединения 17-A в примере 17.
[318] 1H ЯМР (400 МГц, CDCl3) δ 8,71 (s, 1H), 8,03 - 7,99 (m, 2H), 7,67 (dd, J = 2,0, 9,0 Гц, 1H), 4,35 - 4,09 (m, 2H), 3,82 (s, 3H), 3,38 (s, 3H), 2,57 - 2,44 (m, 2H), 1,74 (ушир. d, J = 14,1 Гц, 2H), 1,04 (ушир. s, 2H)
[319] Стадия 2: Синтез соединения 19-B
[320] За исключением использования соответствующих исходных веществ, соединение 19-B получали с использованием того же способа, как и при получении соединения 17-B в примере 17.
[321] МС m/z: 421,1[M+H] +
[322] 1H ЯМР (400 МГц, CDCl3) δ 8,75 (s, 1H), 8,51 (d, J = 2,0 Гц, 1H), 8,25 (d, J = 9,0 Гц, 1H), 8,07 (dt, J = 2,5, 7,9 Гц, 1H), 7,97 (s, 1H), 7,78 (dd, J = 1,8, 8,8 Гц, 1H), 7,11 (dd, J = 2,9, 8,4 Гц, 1H), 4,24 (ушир. s, 2H), 3,83 (ушир. s, 2H), 3,79 (s, 3H), 3,41 (s, 3H), 2,64 - 2,51 (m, 2H), 1,79 (ушир. d, J = 14,1 Гц, 2H)
[323] Стадия 3: Синтез соединения 19
[324] За исключением использования соответствующих исходных веществ, соединение 19 получали с использованием того же способа, как и при получении соединения 17 в примере 17.
[325] МС m/z: 504,2[M+H] +
[326] 1H ЯМР (400 МГц, CDCl3) δ 8,71 (s, 1H), 8,46 (d, J = 2,3 Гц, 1H), 8,21 (d, J = 8,8 Гц, 1H), 7,94 (d, J = 1,5 Гц, 1H), 7,88 (dd, J = 2,5, 8,5 Гц, 1H), 7,78 (dd, J = 1,9, 8,9 Гц, 1H), 6,89 (d, J = 8,8 Гц, 1H), 4,42 (t, J = 6,4 Гц, 2H), 4,32 - 4,08 (m, 2H), 3,92 - 3,81 (m, 2H), 3,79 (s, 3H), 3,40 (s, 3H), 2,65 - 2,55 (m, 2H), 2,53 - 2,47 (m, 2H), 2,29 (s, 6H), 2,06 - 1,97 (m, 2H), 1,76 (ушир. s, 2H)
Пример 20: Соединение 20
Способ синтеза:
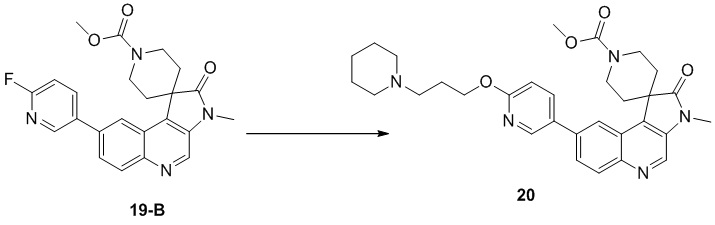
[327] Стадия 1: Синтез соединения 20
[328] За исключением использования соответствующих исходных веществ, соединение 20 получали с использованием того же способа, как и при получении соединения 17 в примере 17.
[329] МС m/z: 544,3[M+H] +
[330] 1H ЯМР (400 МГц, CDCl3) δ 8,71 (s, 1H), 8,46 (s, 1H), 8,21 (d, J = 8,8 Гц, 1H), 7,94 (s, 1H), 7,87 (dd, J = 2,1, 8,7 Гц, 1H), 7,78 (ушир. d, J = 9,0 Гц, 1H), 6,88 (d, J = 8,5 Гц, 1H), 4,41 (ушир. t, J = 6,4 Гц, 2H), 4,33 - 4,07 (m, 2H), 3,94 - 3,81 (m, 2H), 3,79 (s, 3H), 3,40 (s, 3H), 2,66 - 2,37 (m, 8H), 2,10 - 1,98 (m, 2H), 1,78 (ушир. d, J = 12,8 Гц, 6H), 1,45 (ушир. s, 2H)
Пример 21: Соединение 21
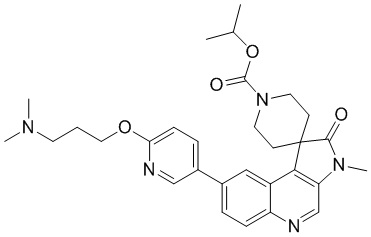
Способ синтеза:
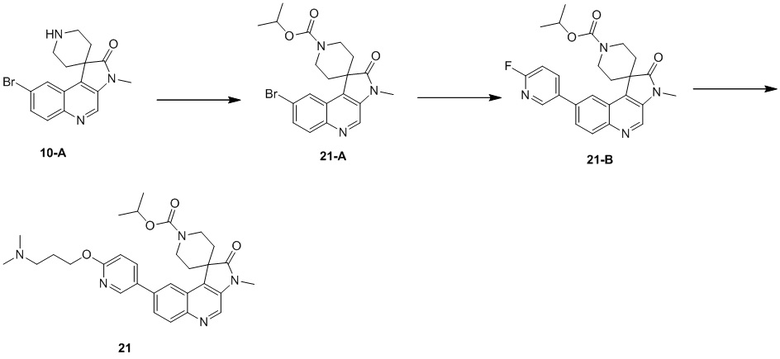
[331] Стадия 1: Синтез соединения 21-A
[332] За исключением использования соответствующих исходных веществ, соединение 21-A получали с использованием того же способа, как и при получении соединения 17-A в примере 17.
[333] МС m/z: 432,2[M+H] +
[334] Стадия 2: Синтез соединения 21-B
[335] За исключением использования соответствующих исходных веществ, соединение 21-B получали с использованием того же способа, как и при получении соединения 17-B в примере 17.
[336] МС m/z: 449,2[M+H] +
[337] 1H ЯМР (400 МГц, CDCl3) δ 8,75 (s, 1H), 8,51 (d, J = 2,5 Гц, 1H), 8,25 (d, J = 8,8 Гц, 1H), 8,09 - 8,02 (m, 1H), 7,96 (d, J = 1,8 Гц, 1H), 7,78 (dd, J = 2,0, 8,8 Гц, 1H), 7,08 (dd, J = 2,9, 8,4 Гц, 1H), 5,02 (td, J = 6,3, 12,4 Гц, 1H), 4,15 (ушир. s, 2H), 3,83 (ушир. s, 2H), 3,41 (s, 3H), 2,57 (ушир. s, 2H), 1,77 (ушир. d, J = 14,1 Гц, 2H), 1,28 (d, J = 6,3 Гц, 6H)
[338] Стадия 3: Синтез соединения 21
[339] За исключением использования соответствующих исходных веществ, соединение 21 получали с использованием того же способа, как и при получении соединения 17 в примере 17.
[340] МС m/z: 532,3[M+H] +
[341] 1H ЯМР (400 МГц, CD3 D) δ 8,77 (s, 1H), 8,45 (d, J = 2,3 Гц, 1H), 8,12 (d, J = 8,8 Гц, 1H), 8,02 (dd, J = 2,5, 8,8 Гц, 1H), 7,96 (d, J = 1,5 Гц, 1H), 7,85 (dd, J = 1,8, 9,0 Гц, 1H), 6,92 (d, J = 8,8 Гц, 1H), 4,97 (td, J = 6,2, 12,5 Гц, 1H), 4,44 (t, J = 6,0 Гц, 2H), 4,12 (ушир. dd, J = 3,5, 13,3 Гц, 2H), 3,84 - 3,71 (m, 2H), 3,38 (s, 3H), 3,13 - 3,06 (m, 2H), 2,74 (s, 6H), 2,60 - 2,49 (m, 2H), 2,23 - 2,13 (m, 2H), 1,73 (ушир. d, J = 14,1 Гц, 2H), 1,27 (d, J = 6,3 Гц, 6H)
Пример 22: Соединение 22
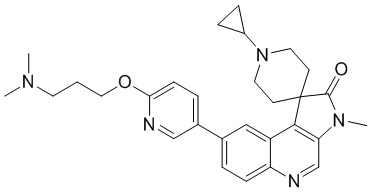
Способ синтеза:
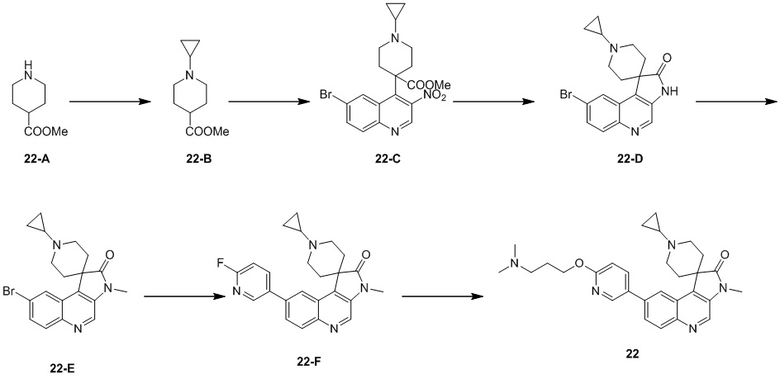
[342] Стадия 1: Синтез соединения 22-B
[343] 1,2-дихлорэтана (100 мл) добавляли к метил-4-пиперидинкарбоксилату (10 г, 69,84 ммоль), циклопропилбороновой кислоте (12,00 г, 139,68 ммоль), пиридину (5,52 г, 69,84 ммоль, 5,64 мл) и карбонату натрия (14,80 г, 139,68 ммоль, 2 экв.), трижды заменяли кислородом и перемешивали в течение 16 ч при 70 °C в атмосфере кислорода. После завершения реакции реакционную смесь охлаждали до 0 °C и гасили путем добавления 200 мл воды. Затем добавляли 100 мл аммиачной воды и проводили экстракцию дихлорметаном (150 мл, 50 мл * 3). Органическую фазу собирали, промывали насыщенным солевым раствором (200 мл) и сушили над безводным сульфатом натрия. После удаления осушителя путем фильтрации, растворитель удаляли при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали с помощью колоночной хроматографии (0 до 10% Et Ac/ПЭ), в результате чего получали соединение 22-B.
[344] 1H ЯМР (400 МГц, CDCl3) δ 3,66 (s, 3H), 3,03 - 2,95 (m, 2H), 2,29 (tt, J = 4,0, 11,2 Гц, 1H), 2,20 (dt, J = 2,5, 11,5 Гц, 2H), 1,91 - 1,83 (m, 2H), 1,73 - 1,62 (m, 2H), 1,59 - 1,52 (m, 1H), 0,46 - 0,40 (m, 2H), 0,40 - 0,36 (m, 2H)
[345] Стадия 2: Синтез соединения 22-C
[346] За исключением использования соответствующих исходных веществ, соединение 22-C получали с использованием того же способа, как и при получении соединения 1-A в примере 1.
[347] МС m/z: 434,0[M+H] +
[348] 1H ЯМР (400 МГц, CDCl3) δ 8,75 (s, 1H), 8,68 (d, J = 1,8 Гц, 1H), 8,03 (d, J = 8,8 Гц, 1H), 7,86 (dd, J = 1,9, 8,9 Гц, 1H), 3,67 (s, 3H), 2,95 (ушир. d, J = 12,3 Гц, 2H), 2,81 - 2,70 (m, 2H), 2,55 (ушир. d, J = 13,1 Гц, 2H), 2,32 - 2,22 (m, 2H), 1,77 - 1,66 (m, 1H), 0,45 (ушир. d, J = 7,0 Гц, 2H), 0,42 (ушир. d, J = 4,3 Гц, 2H)
[349] Стадия 3: Синтез соединения 22-D
[350] Соединение 22-C (1,7 г, 3,91 ммоль) растворяли в ТГФ (70 мл), добавляли раствор NH4Cl (2,09 г, 39,14 ммоль) в воде (70 мл) и затем добавляли порошок цинка (2,56 г, 39,14 ммоль). Реакционный раствор перемешивали при 70 °C в течение 24 ч. После завершения реакции порошок цинка удаляли, и смешанный раствор концентрировали, в результате чего получали неочищенный продукт который непосредственно применяли на следующей стадии.
[351] МС m/z: 371,9[M+H] +
[352] Стадия 4: Синтез соединения 22-E
[353] Раствор гидроксида натрия (322,36 мг, 8,06 ммоль) и тетрабутиламмонийбромида (64,95 мг, 201,47 мкмоль) в воде (20 мл) добавляли к раствору 22-D (1,5 г, 4,03 ммоль) в дихлорметане (40 мл). Затем по каплям добавляли йодметан (3,660 г, 25,79 ммоль, 1,61 мл) и реакционный раствор перемешивали при 20 °C в течение 48 ч. После завершения реакции к реакционной системе добавляли 50 мл воды при комнатной температуре для гашения реакции и проводили экстракцию дихлорметаном (90 мл, 30 мл * 3). Органическую фазу собирали, промывали насыщенным солевым раствором (100 мл) и сушили над безводным сульфатом натрия. После удаления осушителя путем фильтрации, растворитель удаляли при пониженном давлении, в результате чего получали неочищенный продукт соединения 22-E, который непосредственно применяли на следующей стадии.
[354] МС m/z: 386,1[M+H] +
[355] Стадия 5: Синтез соединения 22-F
[356] В защитной атмосфере азота 1,4-диоксан (20 мл) и H2° (20 мл) добавляли к реакционной системе соединения 22-E (1 г, 2,53 ммоль), 2-фторпиридин-5-борной кислоте (534,71 мг, 3,79 ммоль), тетракистрифенилфосфинпалладию (292,34 мг, 252,98 мкмоль) и карбонату натрия (804,41 мг, 7,59 ммоль) и перемешивали при 80 °C в течение 16 ч. После завершения реакции растворитель удаляли при пониженном давлении, в результате чего получали неочищенный продукт в виде соединения 22-F, который непосредственно применяли на следующей стадии.
[357] МС m/z: 403,1[M+H] +
[358] Стадия 6: Синтез соединения 22
[359] За исключением использования соответствующих исходных веществ, соединение 22 получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[360] МС m/z: 486,3[M+H] +
[361] 1H ЯМР (400 МГц, CDCl3) δ 8,68 (s, 1H), 8,51 (d, J = 2,0 Гц, 1H), 8,24 (s, 1H), 8,19 (d, J = 8,8 Гц, 1H), 7,94 (dd, J = 2,0, 8,5 Гц, 1H), 7,76 (ушир. d, J = 8,8 Гц, 1H), 6,87 (d, J = 8,5 Гц, 1H), 4,41 (t, J = 6,4 Гц, 2H), 3,38 (s, 3H), 3,27 (ушир. t, J = 11,4 Гц, 2H), 2,97 (ушир. d, J = 9,5 Гц, 2H), 2,68 (dt, J = 4,4, 13,2 Гц, 2H), 2,47 (ушир. t, J = 7,4 Гц, 2H), 2,27 (s, 6H), 1,98 (ушир. s, 2H), 1,88 (ушир. d, J = 4,3 Гц, 1H), 1,75 (ушир. d, J = 13,8 Гц, 2H), 0,52 (ушир. s, 4H)
Пример 23: Соединение 23
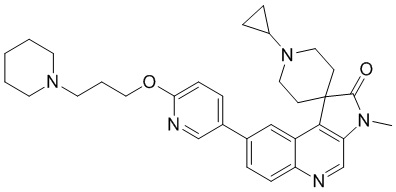
Способ синтеза:
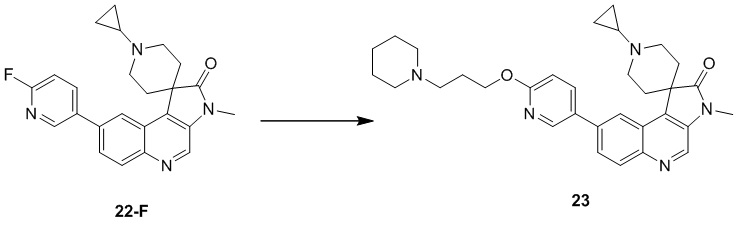
[362] Стадия 1: Синтез соединения 23
[363] За исключением использования соответствующих исходных веществ, соединение 23 получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[364] МС m/z: 526,3[M+H] +
[365] 1H ЯМР (400 МГц, CDCl3) δ 8,68 (s, 1H), 8,51 (d, J = 2,3 Гц, 1H), 8,24 (d, J = 1,5 Гц, 1H), 8,19 (d, J = 9,0 Гц, 1H), 7,94 (dd, J = 2,5, 8,5 Гц, 1H), 7,76 (dd, J = 1,8, 8,8 Гц, 1H), 6,87 (d, J = 8,5 Гц, 1H), 4,40 (t, J = 6,5 Гц, 2H), 3,38 (s, 3H), 3,32 - 3,22 (m, 2H), 3,01 - 2,93 (m, 2H), 2,68 (dt, J = 4,5, 13,3 Гц, 2H), 2,55 - 2,49 (m, 2H), 2,44 (ушир. s, 2H), 2,08 - 1,97 (m, 6H), 1,91 - 1,86 (m, 1H), 1,75 (ушир. d, J = 13,8 Гц, 2H), 1,65 - 1,62 (m, 1H), 1,60 - 1,58 (m, 1H), 1,45 (ушир. s, 2H), 0,55 - 0,49 (m, 4H)
Пример 24: Соединение 24
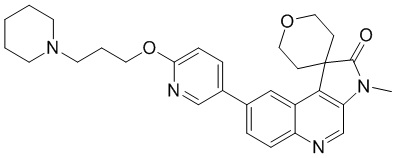
Способ синтеза:
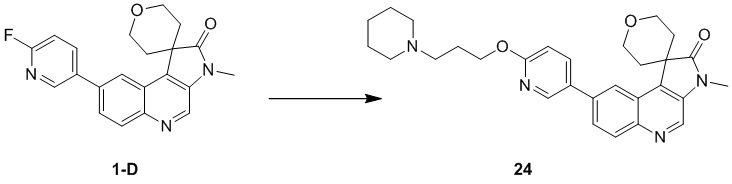
[366] Стадия 1: Синтез соединения 24
[367] Гидрид натрия (101,26 мг, 2,53 ммоль, чистота: 60%) растворяли в N,N -диметилформамиде (10 мл) и добавляли раствор соединения 11-C (181,31 мг, 1,27 ммоль) в N,N -диметилформамиде (5 мл) при 0 °C в атмосфере азота и перемешивали в течение 0,5 ч и затем добавляли раствор 1-D (230 мг, 632,94 мкмоль) в N,N-диметилформамиде (5 мл). Смешанную систему нагревали до комнатной температуры (25 °C), непрерывно перемешивали 2 ч в атмосфере азота, затем гасили путем добавления воды (10 мл) и подвергали экстракции дихлорметаном (150 мл, 50 мл * 3). Органическую фазу собирали и органическую фазу концентрировали, в результате чего получали неочищенный продукт, который отделяли и очищали с помощью препаративной высокоэффективной жидкостной хроматографии ([вода (10 мМ бикарбонат аммония)-ацетонитрил]; ацетонитрил B%: 28%-58%, 7 мин), в результате чего получали соединение 24.
[368] МС m/z: 487,3[M+H] +
[369] 1H ЯМР (400 МГц, CDCl3) δ 8,69 (s, 1H), 8,50 (d, J = 2,0 Гц, 1H), 8,20 (d, J = 8,8 Гц, 1H), 8,15 (s, 1H), 7,91 (dd, J = 2,3, 8,5 Гц, 1H), 7,78 (ушир. d, J = 8,0 Гц, 1H), 6,87 (d, J = 8,5 Гц, 1H), 4,54 - 4,34 (m, 4H), 3,97 (ушир. dd, J = 4,8, 11,5 Гц, 2H), 3,38 (s, 3H), 2,74 (dt, J = 4,9, 13,2 Гц, 2H), 2,54 - 2,46 (m, 2H), 2,42 (ушир. s, 2H), 2,12 (ушир. s, 2H), 2,02 (td, J = 6,9, 14,3 Гц, 2H), 1,68 (ушир. d, J = 13,8 Гц, 2H), 1,62 - 1,53 (m, 4H), 1,43 (ушир. s, 2H)
Пример 25: Соединение 25
Способ синтеза:
[370] Стадия 1: Синтез соединения 25-A
[371] За исключением использования соответствующих исходных веществ, соединение 25-A получали с использованием того же способа, как и при получении соединения 1-A в примере 1.
[372] МС m/z: 465,0[M+H] +
[373] Стадия 2: Синтез соединения 25-B
[374] За исключением использования соответствующих исходных веществ, соединение 25-B получали с использованием того же способа, как и при получении соединения 1-B в примере 1.
[375] МС m/z: 389,4[M+H] +
[376] Стадия 3: Синтез соединения 25-C
[377] Соединение 25-B (3,2 г, 8,22 ммоль) растворяли в тетрагидрофуране (10 мл) и трифторуксусной кислоте (15,40 г, 135,06 ммоль, 10,00 мл) и добавляли воду (10,00 г, 555,08 ммоль, 10 мл). Смешанную систему перемешивали при комнатной температуре (25 °C) в течение 12 ч, затем регулировали до достижения pH = 7 до 8 с использованием гидроксида натрий (1 M) и подвергали экстракции Et°Ac (200 мл, 100 мл * 2). Органическую фазу собирали и концентрировали, в результате чего получали неочищенный продукт, который отделяли и очищали с помощью колоночной хроматографии (дихлорметан/тетрагидрофуран = 1/0 до 4/1), в результате чего получали соединение 25-C.
[378] МС m/z: 345,1[M+H] +
[379] Стадия 4: Синтез соединения 25-D
[380] За исключением использования соответствующих исходных веществ, соединение 25-D получали с использованием того же способа, как и при получении соединения 1-C в примере 1.
[381] МС m/z: 359,1[M+H] +
[382] Стадия 5: Синтез соединения 25-E
[383] Соединение 25-D (0,367 г, 1,02 ммоль) растворяли в метаноле (20 мл) и добавляли борогидрид натрия (77,30 мг, 2,04 ммоль) при 0 °C в атмосфере азота. Смешанную систему нагревали до комнатной температуры (20 °C), непрерывно перемешивали в течение 3 ч, затем гасили путем добавления воды (20 мл) и подвергали экстракции смесью дихлорметан/метанол = 10/1 (50 мл). Органическую фазу собирали и концентрировали, в результате чего получали неочищенный продукт, который отделяли и очищали с помощью колоночной хроматографии (ДХМ/ТГФ = 1/0 до 4/1), в результате чего получали соединение 25-E.
[384] МС m/z: 360,9[M+H] +
[385] Стадия 6: Синтез соединения 25-F
[386] Гидрид натрия (106,29 мг, 2,66 ммоль, чистота: 60%) растворяли в тетрагидрофуране (10 мл) и добавляли раствор 25-E (320 мг, 885,86 мкмоль) в тетрагидрофуране (10 мл) при 0 °C в атмосфере азота и перемешивали при 0 °C в течение 0,5 ч. Затем добавляли иодметан (502,95 мг, 3,54 ммоль, 220,59 мкл) в атмосфере азота. Смешанную систему нагревали до комнатной температуры (25 °C) и перемешивали в течение 2 ч в атмосфере азота. Смешанный раствор гасили путем диспергирования в 50 мл воды, подвергали экстракции дихлорметаном (150 мл, 50 мл * 3) и промывали насыщенным солевым раствором (50 мл). Органическую фазу собирали, сушили над безводным сульфатом натрия и отделяли и очищали с помощью колоночной хроматографии (дихлорметан/тетрагидрофуран = 1/0 до 10/1), в результате чего получали соединение 25-F.
[387] МС m/z 375,1[M+H] +
[388] Стадия 6: Синтез соединения 25-G
[389] За исключением использования соответствующих исходных веществ, соединение 25-G получали с использованием того же способа, как и при получении соединения 1-D в примере 1.
[390] МС m/z: 392,6[M+H] +
[391] Стадия 7: Синтез соединения 25
[392] За исключением использования соответствующих исходных веществ, соединение 25 получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[393] МС m/z: 475,2[M+H] +
[394] 1H ЯМР (400 МГц, CDCl3) δ 8,68 (s, 1H), 8,50 (d, J = 2,5 Гц, 1H), 8,20 (d, J = 8,8 Гц, 1H), 8,01 (s, 1H), 7,90 (dd, J = 2,5, 8,5 Гц, 1H), 7,76 (dd, J = 1,5, 8,8 Гц, 1H), 6,91 (d, J = 8,5 Гц, 1H), 4,43 (t, J = 6,4 Гц, 2H), 3,48 (s, 3H), 3,38 (s, 3H), 2,53 - 2,44 (m, 4H), 2,41 - 2,35 (m, 2H), 2,28 (s, 6H), 2,10 - 1,97 (m, 4H), 1,91 (ушир. d, J = 13,8 Гц, 2H), 1,78 (ушир. s, 2H)
Пример 26: Соединение 26
Способ синтеза:
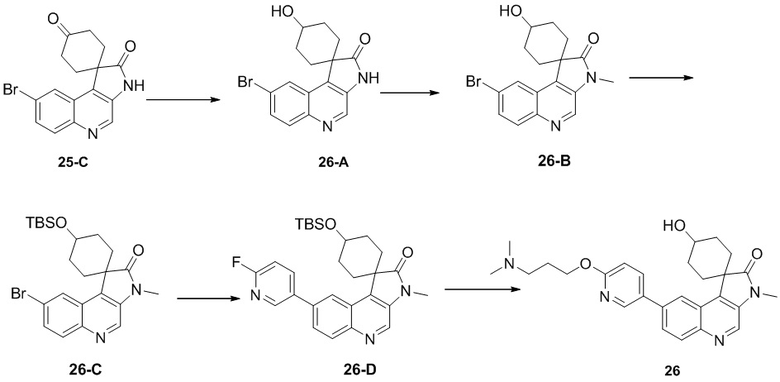
[395] Стадия 1: Синтез соединения 26-A
[396] Соединение 25-C (1,05 г, 3,04 ммоль) растворяли в метаноле (20 мл) и добавляли боргидрид натрия (172,62 мг, 4,56 ммоль) при 0 °C в атмосфере азота. Смешанную систему нагревали до комнатной температуры (20 °C), непрерывно перемешивали в течение 3 ч, затем гасили путем добавления воды (20 мл) и подвергали экстракции дихлорметаном (100 мл, 50 мл * 2). Органическую фазу собирали и концентрировали, в результате чего получали неочищенный продукт, который отделяли и очищали с помощью колоночной хроматографии (дихлорметан/тетрагидрофуран = 1/0 до 4/1), в результате чего получали соединение 26-A.
[397] МС m/z: 347,0[M+H] +
[398] Стадия 2: Синтез соединения 26-B
[399] Соединение 26-A (840 мг, 2,42 ммоль) растворяли в дихлорметане (5 мл), добавляли раствор гидроксида натрия (387,09 мг, 9,68 ммоль) в воде (5 мл) и тетрабутиламмонийбромид (39,00 мг, 120,97 мкмоль) и добавляли иодметан (1,37 г, 9,68 ммоль, 602,44 мкл) при 0 °C в атмосфере азота. Смешанную систему нагревали до комнатной температуры (25 °C) и перемешивали в течение 3 ч в атмосфере азота, и затем смешанный раствор диспергировали в 50 мл воды и подвергали экстракции дихлорметаном (100 мл, 50 мл * 2). Органическую фазу собирали и концентрировали, в результате чего получали неочищенный продукт, который отделяли и очищали с помощью колоночной хроматографии (дихлорметан/тетрагидрофуран = 1/0 до 5/1), в результате чего получали 26-B.
[400] МС m/z: 361,1[M+H] +
[401] Стадия 3: Синтез соединения 26-C
[402] Соединение 26-B (260 мг, 719,76 мкмоль) растворяли в N,N-диметилформамиде (10 мл) и пиридине (56,93 мг, 719,76 мкмоль, 58,09 мкл) и добавляли TBS°Tf (190,26 мг, 719,76 мкмоль, 165,44 мкл) и перемешивали в течение 3 ч при комнатной температуре (25 °C), затем промывали путем добавления 300 мл (100 мл * 3) воды и проводили экстракцию 150 мл (50 мл * 3) дихлорметана. Органическую фазу собирали и концентрировали, в результате чего получали неочищенный продукт, который отделяли и очищали с помощью колоночной хроматографии (дихлорметан/тетрагидрофуран = 1/0 до 10/1), в результате чего получали 26-C.
[403] МС m/z: 474,6[M+H] +
[404] Стадия 4: Синтез соединения 26-D
[405] За исключением использования соответствующих исходных веществ, соединение 26-D получали с использованием того же способа, как и при получении соединения 1-D в примере 1.
[406] МС m/z: 492,3[M+H] +
[407] 1H ЯМР (400 МГц, CDCl3) δ 8,72 (s, 1H), 8,55 (d, J = 2,3 Гц, 1H), 8,24 (d, J = 9,0 Гц, 1H), 8,12 - 8,05 (m, 1H), 8,03 (d, J = 1,8 Гц, 1H), 7,75 (dd, J = 2,0, 8,8 Гц, 1H), 7,12 (dd, J = 2,8, 8,5 Гц, 1H), 3,39 (s, 3H), 2,55 - 2,40 (m, 4H), 1,94 - 1,78 (m, 4H), 0,97 - 0,88 (m, 9H), 0,13 (s, 6H)
[408] Стадия 5: Синтез соединения 26
[409] За исключением использования соответствующих исходных веществ, соединение 26 получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[410] МС m/z: 461,3[M+H] +
[411] 1H ЯМР (400 МГц, CDCl3) δ 8,69 (s, 1H), 8,49 (d, J = 2,3 Гц, 1H), 8,21 (d, J = 8,8 Гц, 1H), 8,00 (d, J = 1,5 Гц, 1H), 7,89 (dd, J = 2,6, 8,7 Гц, 1H), 7,77 (dd, J = 1,8, 8,8 Гц, 1H), 6,90 (d, J = 8,5 Гц, 1H), 4,43 (t, J = 6,5 Гц, 2H), 3,99 - 3,89 (m, 1H), 3,39 (s, 3H), 2,58 - 2,36 (m, 6H), 2,28 (s, 6H), 2,06 - 1,96 (m, 4H), 1,89 (ушир. d, J = 13,8 Гц, 2H)
Пример 27: Соединение 27
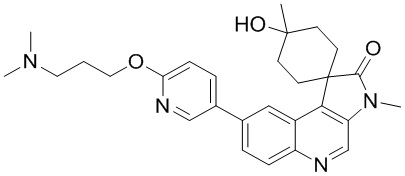
Способ синтеза:
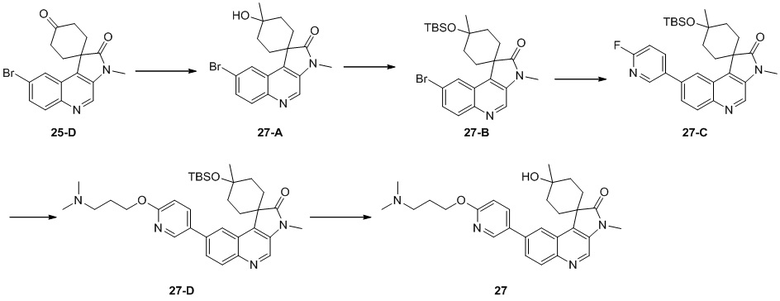
[412] Стадия 1: Синтез соединения 27-A
[413] Соединение 25-D (900 мг, 2,51 ммоль) растворяли в тетрагидрофуране (20 мл) при 0 °C и по каплям добавляли метилмагнийбромид (4 M, 1,88 мл) в атмосфере азота. Повышали температуру смешанной системы до 25 °C и перемешивали в течение 2 ч в атмосфере азота, и затем реакционную систему гасили водой (50 мл) и подвергали экстракции смесью дихлорметан/изопропанол = 10/1 (50 мл * 3). Органическую фазу собирали, отделяли и очищали с помощью колоночной хроматографии (ДХМ/тетрагидрофуран = 1/0 до 3/1), в результате чего получали соединение 27-A.
[414] МС m/z: 375,0[M+H] +
[415] Стадия 2: Синтез соединения 27-B
[416] Соединение 27-A (380 мг, 1,01 ммоль) растворяли в дихлорметане (10 мл) и добавляли TBS°Tf (401,52 мг, 1,52 ммоль, 349,14 мкл) и триэтиламин (204,94 мг, 2,03 ммоль, 281,89 мкл). Смешанную систему перемешивали в течение 12 ч при комнатной температуре (25 °C), затем промывали путем добавления 300 мл (100 мл * 3) воды и подвергали экстракции дихлорметаном (150 мл, 50 мл * 3). Органическую фазу собирали и концентрировали, в результате чего получали неочищенный продукт, который отделяли и очищали с помощью колоночной хроматографии (дихлорметан/тетрагидрофуран = 1/0 до 10/1), в результате чего получали 27-B.
[417] МС m/z: 489,3[M+H] +
[418] 1H ЯМР (400 МГц, CDCl3) δ 8,68 (s, 1H), 8,10 (d, J = 2,0 Гц, 1H), 8,00 (d, J = 9,0 Гц, 1H), 7,66 (dd, J = 2,3, 9,0 Гц, 1H), 3,36 (s, 3H), 2,71 - 2,54 (m, 2H), 2,41 (dt, J = 3,8, 14,2 Гц, 2H), 1,80 - 1,65 (m, 4H), 1,56 (s, 3H), 0,91 (s, 9H), 0,22 - 0,13 (m, 6H)
[419] Стадия 3: Синтез соединения 27-C
[420] За исключением использования соответствующих исходных веществ, соединение 27-C получали с использованием того же способа, как и при получении соединения 1-D в примере 1.
[421] МС m/z: 506,8[M+H] +
[422] Стадия 4: Синтез соединения 27-D
[423] За исключением использования соответствующих исходных веществ, соединение 27-D получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[424] МС m/z: 589,6[M+H] +
[425] Стадия 5: Синтез соединения 27
[426] Соединение 27-D (110 мг, 186,80 мкмоль) растворяли в трифторуксусной кислоты (21,30 мг, 186,80 мкмоль, 13,83 мкл), перемешивали при 60 °C в течение 1 ч, затем регулировали с помощью 1 M раствора гидроксида натрия до pH = 7 до 8, подвергали экстракции дихлорметаном (150 мл, 50 мл * 3) и промывали насыщенным солевым раствором (50 мл). Органическую фазу собирали, сушили над безводным сульфатом натрия и концентрировали, в результате чего получали неочищенный продукт, который отделяли и очищали с помощью препаративной высокоэффективной жидкостной хроматографии (нейтральная, подвижная фаза: ацетонитрил-вода), в результате чего получали соединение 27.
[427] МС m/z: 475,3[M+H] +
[428] 1H ЯМР (400 МГц, CDCl3) δ 8,69 (s, 1H), 8,49 (d, J = 2,5 Гц, 1H), 8,22 (d, J = 8,8 Гц, 1H), 8,06 (d, J = 1,8 Гц, 1H), 7,89 (dd, J = 2,5, 8,5 Гц, 1H), 7,79 (dd, J = 2,0, 8,8 Гц, 1H), 6,89 (d, J = 8,5 Гц, 1H), 4,42 (t, J = 6,5 Гц, 2H), 3,39 (s, 3H), 2,57 (d, J = 9,0 Гц, 4H), 2,51 - 2,45 (m, 2H), 2,28 (s, 6H), 2,04 - 1,96 (m, 2H), 1,87 - 1,75 (m, 4H), 1,58 (s, 3H)
Пример 28: Соединение 28
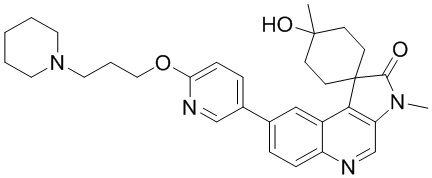
Способ синтеза:
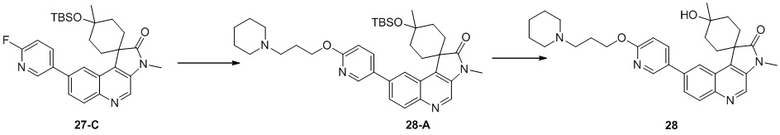
[429] Стадия 1: Синтез соединения 28-A
[430] За исключением использования соответствующих исходных веществ, соединение 28-A получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[431] МС m/z: 629,6[M+H] +
[432] Стадия 2: Синтез соединения 28
[433] За исключением использования соответствующих исходных веществ, соединение 28 получали с использованием того же способа, как и при получении соединения 27 в примере 27.
[434] МС m/z: 515,4[M+H] +
[435] 1H ЯМР (400 МГц, CDCl3) δ 8,69 (s, 1H), 8,49 (d, J = 2,8 Гц, 1H), 8,22 (d, J = 8,8 Гц, 1H), 8,05 (d, J = 1,5 Гц, 1H), 7,89 (dd, J = 2,5, 8,5 Гц, 1H), 7,79 (dd, J = 2,0, 8,8 Гц, 1H), 6,89 (d, J = 8,5 Гц, 1H), 4,41 (t, J = 6,5 Гц, 2H), 3,39 (s, 3H), 2,57 (d, J = 9,3 Гц, 2H), 2,50 (ушир. d, J = 8,0 Гц, 2H), 2,43 (ушир. s, 2H), 2,09 - 1,97 (m, 2H), 1,84 (ушир. d, J = 8,5 Гц, 2H), 1,78 (ушир. d, J = 7,8 Гц, 2H), 1,59 (ушир. s, 8H), 1,57 (ушир. s, 3H), 1,45 (ушир. s, 2H)
Пример 29: Соединение 29
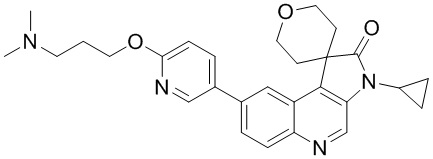
Способ синтеза:
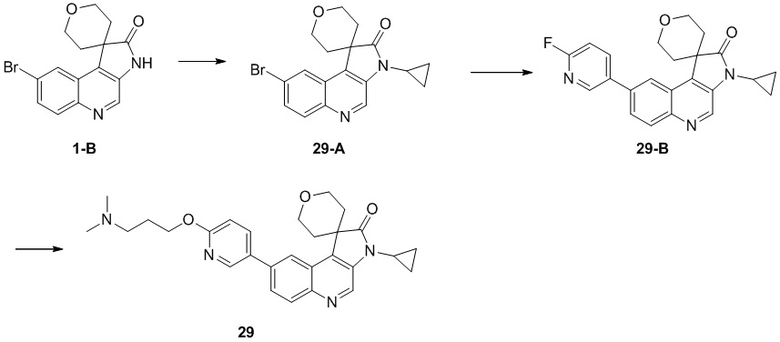
[436] Стадия 1: Синтез соединения 29-A
[437] Соединение 1-B (200 мг, 600,28 мкмоль) растворяли в безводном толуоле (15 мл) и добавляли циклопропилбороновую кислоту (103,12 мг, 1,20 ммоль), карбонат натрия (127,25 мг, 1,20 ммоль), ацетат меди (109,03 мг, 600,28 мкмоль) и пиридин (94,96 мг, 1,20 ммоль, 96,90 мкл). Смешанную систему перемешивали в течение 12 ч при 70 °C и затем фильтровали. Фильтрат собирали и концентрировали, в результате чего получали неочищенный продукт, который отделяли и очищали с помощью колоночной хроматографии (дихлорметан/тетрагидрофуран = 1/0 до 10/1), в результате чего получали соединение 29-A.
[438] МС m/z: 373,0[M+H] +
[439] 1H ЯМР (400 МГц, CDCl3) δ 8,95 (s, 1H), 8,22 (d, J = 1,5 Гц, 1H), 8,01 (d, J = 9,0 Гц, 1H), 7,67 (dd, J = 2,0, 9,0 Гц, 1H), 4,50 - 4,35 (m, 2H), 4,03 - 3,91 (m, 2H), 2,80 (tt, J = 3,6, 7,0 Гц, 1H), 2,64 (dt, J = 5,4, 13,5 Гц, 2H), 1,64 (s, 2H), 1,23 - 1,15 (m, 2H), 1,01 - 0,93 (m, 2H)
[440] Стадия 2: Синтез соединения 29-B
[441] За исключением использования соответствующих исходных веществ, соединение 29-B получали с использованием того же способа, как и при получении соединения 1-D в примере 1.
[442] МС m/z: 390,1[M+H] +
[443] 1H ЯМР (400 МГц, CDCl3) δ 8,99 (s, 1H), 8,55 (d, J = 1,8 Гц, 1H), 8,26 (d, J = 8,8 Гц, 1H), 8,17 (d, J = 1,0 Гц, 1H), 8,11 (dt, J = 2,5, 7,9 Гц, 1H), 7,85 - 7,75 (m, 1H), 7,11 (dd, J = 2,9, 8,4 Гц, 1H), 4,47 (ушир. t, J = 11,3 Гц, 2H), 3,98 (dd, J = 5,0, 11,8 Гц, 2H), 2,83 (tt, J = 3,7, 6,9 Гц, 1H), 2,71 (dt, J = 5,3, 13,4 Гц, 2H), 1,70 (ушир. s, 2H), 1,28 - 1,16 (m, 2H), 1,05 - 0,90 (m, 2H)
[444] Стадия 3: Синтез соединения 29
[445] За исключением использования соответствующих исходных веществ, соединение 29 получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[446] МС m/z: 473,3[M+H] +
[447] 1H ЯМР (400 МГц, CDCl3) δ 8,95 (s, 1H), 8,50 (d, J = 2,5 Гц, 1H), 8,21 (d, J = 8,8 Гц, 1H), 8,15 (d, J = 1,8 Гц, 1H), 7,92 (dd, J = 2,5, 8,5 Гц, 1H), 7,79 (dd, J = 1,8, 8,8 Гц, 1H), 6,89 (d, J = 8,5 Гц, 1H), 4,51 - 4,42 (m, 4H), 3,97 (dd, J = 4,9, 11,7 Гц, 2H), 2,82 (td, J = 3,3, 6,9 Гц, 1H), 2,73 (dt, J = 5,3, 13,7 Гц, 2H), 2,64 (ушир. s, 2H), 2,40 (s, 6H), 2,14 - 2,05 (m, 2H), 1,67 (ушир. d, J = 14,1 Гц, 2H), 1,21 (q, J = 6,8 Гц, 2H), 1,02 - 0,96 (m, 2H)
Пример 30: Соединение 30
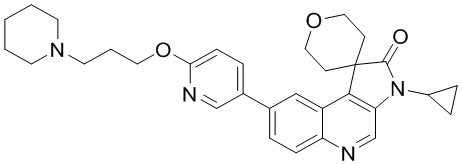
Способ синтеза:
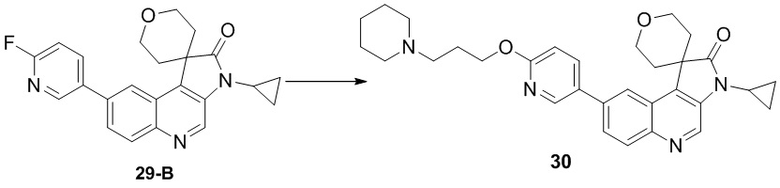
[448] Стадия 1: Синтез соединения 30
[449] За исключением использования соответствующих исходных веществ, соединение 30 получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[450] МС m/z: 513,2[M+H] +
[451] 1H ЯМР (400 МГц, CDCl3) δ 8,95 (s, 1H), 8,50 (d, J = 2,5 Гц, 1H), 8,21 (d, J = 9,0 Гц, 1H), 8,15 (d, J = 1,5 Гц, 1H), 7,92 (dd, J = 2,6, 8,7 Гц, 1H), 7,79 (dd, J = 1,9, 8,9 Гц, 1H), 6,88 (d, J = 8,5 Гц, 1H), 4,51 - 4,39 (m, 4H), 3,97 (dd, J = 4,9, 11,7 Гц, 2H), 2,82 (tt, J = 3,5, 7,0 Гц, 1H), 2,73 (dt, J = 5,3, 13,4 Гц, 2H), 2,55 - 2,48 (m, 2H), 2,43 (ушир. s, 2H), 2,12 - 1,98 (m, 2H), 1,68 (ушир. s, 4H), 1,63 - 1,56 (m, 4H), 1,45 (ушир. d, J = 5,3 Гц, 2H), 1,26 - 1,14 (m, 2H), 1,08 - 0,90 (m, 2H)
Пример 31: Соединение 31
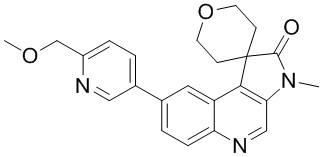
Способ синтеза:
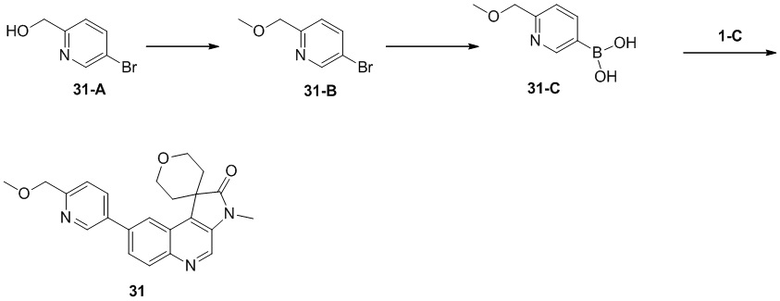
[452] Стадия 1: Синтез соединения 31-B
[453] При 0 °C и в защитной атмосфере азота соединение 31-A (5 г, 26,59 ммоль) медленно добавляли к раствору гидрида натрия (1,60 г, 39,89 ммоль, 60% чистота) в ТГФ (20 мл). Реакционный раствор перемешивали при 25 °C в течение 0,5 ч и затем раствор медленно добавляли иодметан (5,66 г, 39,89 ммоль, 2,48 мл) в ТГФ (10 мл) при 0 °C. Реакционный раствор перемешивали при 20°C в течение 12 ч. При 20 °C, реакционный раствор гасили водой (20 мл), разбавляли путем добавления воды (10 мл) и подвергали экстракции Et°Ac (150 мл, 50 мл * 3). Органическую фазу собирали, промывали насыщенным раствором хлорида натрия (150 мл, 50 мл * 3), сушили над безводным сульфатом натрия, фильтровали и концентрировали, в результате чего получали неочищенный продукт, Остаток очищали с помощью колоночной хроматографии (0 до 10% Et°Ac/ПЭ), в результате чего получали 31-B.
[454] МС m/z: 201,8[M+H] +
[455] 1H ЯМР (400 МГц, CDCl3) δ 8,61 (d, J = 2,0 Гц, 1H), 7,82 (dd, J = 2,3, 8,5 Гц, 1H), 7,33 (d, J = 8,3 Гц, 1H), 4,53 (s, 2H), 3,47 (s, 3H)
[456] Стадия 2: Синтез соединения 31-C
[457] В защитной атмосфере азота смешанный раствор 31-B (4,8 г, 23,76 ммоль), бис(пинаколято)диборона (6,64 г, 26,13 ммоль), ацетата калия (6,99 г, 71,27 ммоль) и Pd(dppf)Cl2 (1,74 г, 2,38 ммоль) в диоксане (40 мл) и воде (8 мл) перемешивали при 100°C в течение 12 ч. Реакционный раствор фильтровали, и фильтрат концентрировали, в результате чего получали остаток, который отделяли и очищали с помощью препаративной тонкослойной хроматографии на силикагелевой пластине, в результате чего получали соединение 31-C.
[458] МС m/z: 167,9[M+H] +
[459] 1H ЯМР (400 МГц, ДМСО-d6) δ 8,88 (s, 1H), 8,49 (d, J = 7,8 Гц, 1H), 7,73 (d, J = 7,8 Гц, 1H), 4,69 (s, 2H), 3,42 (s, 3H).
[460] Стадия 3: Синтез соединения 31
[461] За исключением использования соответствующих исходных веществ, соединение 31 получали с использованием того же способа, как и при получении соединения 1-D в примере 1.
[462] МС m/z: 390,3[M+H] +
[463] 1H ЯМР (400 МГц, CDCl3) δ 8,92 (d, J = 2,0 Гц, 1H), 8,74 (s, 1H), 8,28 - 8,19 (m, 2H), 8,03 (dd, J = 2,3, 8,0 Гц, 1H), 7,84 (dd, J = 2,0, 8,8 Гц, 1H), 7,59 (d, J = 8,0 Гц, 1H), 4,69 (s, 2H), 4,56 - 4,41 (m, 2H), 3,99 (dd, J = 4,9, 11,7 Гц, 2H), 3,54 (s, 3H), 3,40 (s, 3H), 2,75 (dt, J = 5,3, 13,4 Гц, 2H), 1,71 (ушир. d, J = 14,1 Гц, 2H)
Пример 32: Соединение 32
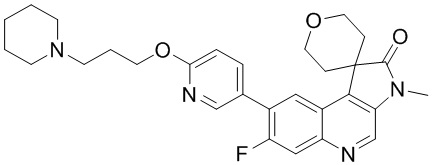
Способ синтеза:
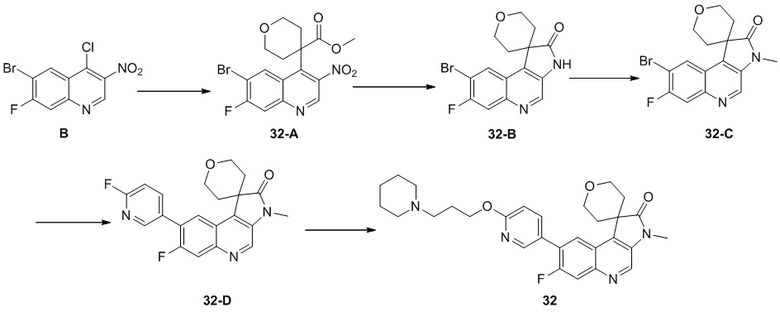
[464] Стадия 1: Синтез соединения 32-A
[465] При -60 °C и в защитной атмосфере азота n-BuLi (2,5 M, 3,93 мл) медленно добавляли к раствору DIPA (993,72 мг, 9,82 ммоль, 1,39 мл) в ТГФ (10 мл), и реакционную систему перемешивали в течение 30 минут при -30 °C, а затем медленно добавляли раствор метилтетрагидропиран-4-карбоксилата (1,49 г, 10,31 ммоль, 1,38 мл) в ТГФ (10 мл). Реакционную систему перемешивали в течение 1 ч при -65 °C и, наконец, медленно добавляли раствор соединения B (1,5 г, 4,91 ммоль) в ТГФ (10 мл). Реакционную систему перемешивали при -65 °C в течение 2 ч. После завершения реакции реакцию гасили путем добавления воды (5 мл) и затем разбавляли путем добавления насыщенного солевого раствора (10 мл) и подвергали экстракции EtAc (30 мл, 10 мл * 3). Органическую фазу собирали, промывали насыщенным солевым раствором (30 мл, 10 мл * 3), сушили над безводным сульфатом натрия и концентрировали, в результате чего получали остаток твердые вещества, который подвергали колоночной хроматографии (0 до 5% ТГФ/ПЭ), в результате чего получали 32-A.
[466] МС m/z: 412,8[M+H] +
[467] Стадия 2: Синтез соединения 32-B
[468] В защитной атмосфере азота порошок цинка (1,14 г, 17,43 ммоль) добавляли к раствору 32-A (720 мг, 1,74 ммоль) и NH4Cl (932,10 мг, 17,43 ммоль, 609,22 мкл) в ТГФ (10 мл) и H2° (10 мл) и реакционную систему перемешивали при 70°C в течение 3 ч. После завершения реакции реакционную систему фильтровали и фильтрат концентрировали, в результате чего получали в остатке твердые вещества. Твердые вещества суспендировали с использованием воды (20мл) в течение 30 минут, в результате чего получали 32-B.
[469] МС m/z: 350,9[M+H] +
[470] Стадия 3: Синтез соединения 32-C
[471] В защитной атмосфере азота раствор йодметана (347,68 мг, 2,45 ммоль, 152,49 мкл) в дихлорметане (10 мл) добавляли к раствору 32-B (500 мг, 1,07 ммоль), TBAB (34,33 мг, 106,50 мкмоль) и NaH (63,90 мг, 1,60 ммоль) в ДХМ (10 мл) и H2° (10 мл), и реакционную систему перемешивали при 30 °C в течение 1 ч. После завершения реакции реакционную систему фильтровали и фильтрат концентрировали, в результате чего получали в остатке твердые вещества. Указанные твердые вещества суспендировали с использованием воды (20 мл) в течение 30 минут, в результате чего получали 32-C.
[472] МС m/z: 364,9[M+H] +
[473] Стадия 4: Синтез соединения 32-D
[474] В защитной атмосфере азота раствор 32-C (200 мг, 547,65 мкмоль), 2-фторпиридин-5-борной кислоты (154,34 мг, 1,10 ммоль), Na2C3 (116,09 мг, 1,10 ммоль), Pd2(dba)3 (50,15 мг, 54,77 мкмоль) и Xphs (50,15 мг, 54,77 мкмоль) в диоксане (18 мл) и воде (2 мл) перемешивали при 100°C в течение 2 ч. Реакционный смешанный раствор концентрировали, в результате чего получали остаточные твердые вещества и твердые вещества подвергали колоночной хроматографии (0 до 50% EtAc/ПЭ), в результате чего получали соединение 32-D.
[475] МС m/z: 382,0[M+H] +
[476] Стадия 5: Синтез соединения 32
[477] При 20 °C и в защитной атмосфере азота 32-D (100 мг, 262,21 мкмоль) добавляли к раствору 1-пиперидинпропанола (75,11 мг, 524,42 мкмоль, 28,71 мкл) и NaH (41,95 мг, 1,05 ммоль, 60% чистота) в ДМФА (10 мл) и реакционную систему перемешивали при 70 °C в течение 2 ч. После завершения реакции реакцию гасили путем добавления воды (2 мл) и концентрировали, в результате чего получали остаточные твердые вещества. Твердые вещества отделяли с помощью колоночной хроматографии (0 до 10% Me°H/ДХМ) и препаративной ВЭЖХ (колонка: B°st°n Prime C18 150 × 30 мм 5 мкм; подвижная фаза: [вода (0,05% аммоний гидроксид об./об.)-ацетонитрил]; ацетонитрил B%: 50%-80%, 8 мин), в результате чего получали соединение 32.
[1] МС m/z: 505,3[M+H] +
[479] 1H ЯМР (400 МГц, CDCl3) δ 8,71 (s, 1H), 8,42 (s, 1H), 8,07 (d, J = 8,0 Гц, 1H), 7,93 - 7,84 (m, 2H), 6,89 (d, J = 8,8 Гц, 1H), 4,56 - 4,34 (m, 4H), 3,97 (dd, J = 4,8, 11,5 Гц, 2H), 3,39 (s, 3H), 2,78 - 2,60 (m, 2H), 2,52 (ушир. s, 2H), 2,50 - 2,33 (m, 4H), 2,10 - 1,99 (m, 2H), 1,69 (ушир. d, J = 14,3 Гц, 2H), 1,60 (ушир. s, 4H), 1,50 - 1,40 (m, 2H)
Пример 33: Соединение 33
Способ синтеза:
[480] Стадия 1: Синтез соединения 33
[481] За исключением использования соответствующих исходных веществ, соединение 33 получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[482] МС m/z: 465,2[M+H] +
[483] 1H ЯМР (400 МГц, CDCl3) δ 8,64 (s, 1H), 8,37 - 8,32 (m, 1H), 7,99 (d, J = 8,0 Гц, 1H), 7,85 - 7,77 (m, 2H), 6,82 (d, J = 8,5 Гц, 1H), 4,45 - 4,35 (m, 4H), 3,90 (dd, J = 4,9, 11,7 Гц, 2H), 3,32 (s, 3H), 2,69 - 2,54 (m, 4H), 2,34 (s, 6H), 2,11 - 1,97 (m, 2H), 1,63 (ушир. d, J = 14,1 Гц, 2H)
Пример 34: Соединение 34
Способ синтеза:
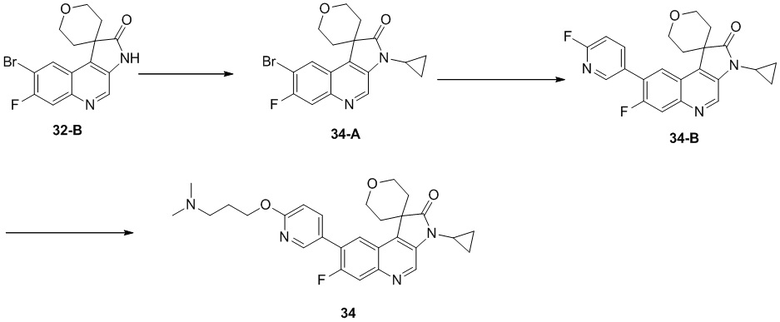
[484] Стадия 1: Синтез соединения 34-A
[485] За исключением использования соответствующих исходных веществ, соединение 34-A получали с использованием того же способа, как и при получении соединения 29-A в примере 29.
[486] 1H ЯМР (400 МГц, CDCl3) δ 8,95 (s, 1H), 8,31 (d, J = 7,3 Гц, 1H), 7,84 (d, J = 9,5 Гц, 1H), 4,50 - 4,37 (m, 2H), 3,98 (dd, J = 5,0, 11,8 Гц, 2H), 2,84 - 2,72 (m, 1H), 2,66 - 2,52 (m, 2H), 1,62 (s, 2H), 0,98 - 0,94 (m, 2H), 0,90 - 0,86 (m, 2H)
[487] Стадия 2: Синтез соединения 34-B
[488] За исключением использования соответствующих исходных веществ, соединение 34-B получали с использованием того же способа, как и при получении соединения 1-D в примере 1.
[489] МС m/z: 408,1[M+H] +
[490] 1H ЯМР (400 МГц, CDCl3) δ 8,92 (s, 1H), 8,42 (s, 1H), 8,01 (d, J = 8,0 Гц, 2H), 7,86 (d, J = 11,4 Гц, 1H), 7,05 (dd, J = 2,9, 8,4 Гц, 1H), 4,38 (dt, J = 1,9, 12,1 Гц, 2H), 3,89 (dd, J = 5,1, 11,7 Гц, 2H), 2,75 (s, 1H), 2,56 (ушир. d, J = 4,6 Гц, 2H), 1,63 (s, 1H), 1,61 - 1,59 (m, 1H), 1,17 - 1,11 (m, 2H), 0,96 - 0,89 (m, 2H)
[491] Стадия 3: Синтез соединения 34
[492] За исключением использования соответствующих исходных веществ, соединение 34 получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[493] МС m/z: 491,3[M+H] +
[494] 1H ЯМР (400 МГц, CDCl3) δ 8,97 (s, 1H), 8,43 (s, 1H), 8,07 (d, J = 8,0 Гц, 1H), 7,96 - 7,85 (m, 2H), 6,91 (d, J = 8,5 Гц, 1H), 4,46 (ушир. t, J = 6,4 Гц, 4H), 4,03 - 3,92 (m, 2H), 2,90 - 2,79 (m, 1H), 2,72 - 2,62 (m, 2H), 2,56 - 2,49 (m, 2H), 2,31 (s, 6H), 2,07 - 2,01 (m, 2H), 1,71 - 1,66 (m, 2H), 1,26 - 1,20 (m, 2H), 1,05 - 0,97 (m, 2H)
Пример 35: Соединение 35
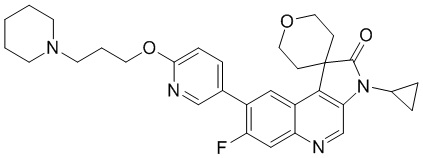
Способ синтеза:
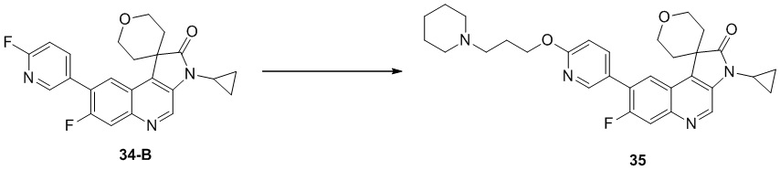
[495] Стадия 1: Синтез соединения 35
[496] За исключением использования соответствующих исходных веществ, соединение 35 получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[497] МС m/z: 531,4[M+H] +
[498] 1H ЯМР (400 МГц, CDCl3) δ 8,97 (s, 1H), 8,43 (s, 1H), 8,07 (d, J = 8,1 Гц, 1H), 7,95 - 7,85 (m, 2H), 6,91 (d, J = 8,6 Гц, 1H), 4,45 (ушир. d, J = 6,8 Гц, 4H), 4,04 - 3,94 (m, 2H), 2,88 - 2,80 (m, 1H), 2,72 - 2,61 (m, 2H), 2,57 - 2,51 (m, 2H), 2,50 - 2,33 (m, 4H), 2,11 - 2,03 (m, 2H), 1,72 - 1,66 (m, 6H), 1,47 (ушир. s, 2H), 1,26 - 1,20 (m, 2H), 1,05 - 0,98 (m, 2H)
Пример 36: Соединение 36
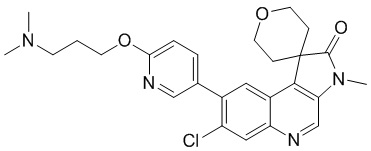
Способ синтеза:
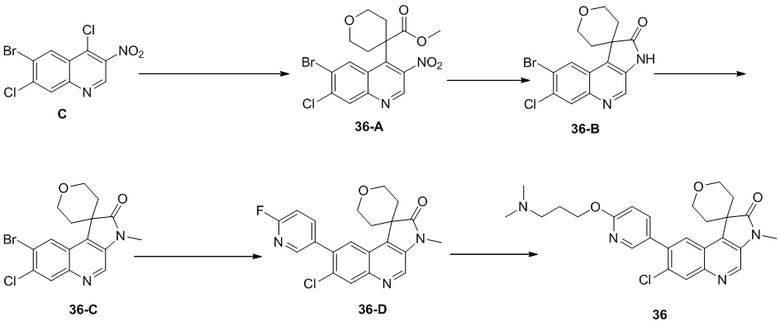
[499] Стадия 1: Синтез соединения 36-A
[500] За исключением использования соответствующих исходных веществ, соединение 36-A получали с использованием того же способа, как и при получении соединения 1-A в примере 1.
[501] МС m/z: 429,0[M+H] +
[502] Стадия 2: Синтез соединения 36-B
[503] За исключением использования соответствующих исходных веществ, соединение 36-B получали с использованием того же способа, как и при получении соединения 1-B в примере 1.
[504] МС m/z: 366,9[M+H] +
[505] Стадия 3: Синтез соединения 36-C
[506] За исключением использования соответствующих исходных веществ, соединение 36-C получали с использованием того же способа, как и при получении соединения 1-C в примере 1.
[507] МС m/z: 381,0[M+H] +
[508] Стадия 4: Синтез соединения 36-D
[509] За исключением использования соответствующих исходных веществ, соединение 36-D получали с использованием того же способа, как и при получении соединения 1-D в примере 1.
[510] МС m/z: 398,0[M+H] +
[511] 1H ЯМР (400 МГц, CDCl3) δ 8,76 (s, 1H), 8,36 (d, J = 2,3 Гц, 1H), 8,32 (s, 1H), 8,01 (s, 1H), 8,00 - 7,94 (m, 1H), 7,10 (dd, J = 2,8, 8,3 Гц, 1H), 4,50 - 4,39 (m, 2H), 3,95 (dd, J = 5,0, 11,8 Гц, 2H), 3,40 (s, 3H), 2,61 (dt, J = 5,3, 13,4 Гц, 2H), 1,68 (ушир. d, J = 14,6 Гц, 2H)
[512] Стадия 5: Синтез соединения 36
[513] За исключением использования соответствующих исходных веществ, соединение 36 получали с использованием того же способа, как и при получении соединения 1 в примере 1.
[514] МС m/z: 481,3[M+H] +
[515] 1H ЯМР (400 МГц, CDCl3) δ 8,66 (s, 1H), 8,27 - 8,17 (m, 2H), 7,93 (s, 1H), 7,73 (dd, J = 2,5, 8,5 Гц, 1H), 6,80 (d, J = 8,5 Гц, 1H), 4,45 - 4,33 (m, 4H), 3,88 (dd, J = 4,9, 11,7 Гц, 2H), 3,33 (s, 3H), 2,75 - 2,65 (m, 2H), 2,57 (dt, J = 5,3, 13,3 Гц, 2H), 2,43 (s, 6H), 2,16 - 2,06 (m, 2H), 1,61 (ушир. d, J = 13,8 Гц, 2H)
Оценка биологического действия
Экспериментальный пример 1: Оценка in vitr°
[516] Все соединения согласно настоящему изобретению, применяемые в экспериментах, получены компанией, проводящей исследование, и их химические названия и структурные формулы приведены в примерах получения каждого соединения. Экспериментальные исследования были проведены британской компанией Eur°fins, и результаты экспериментов были предоставлены указанной компанией. Следующие экспериментальные способы также были выполнены указанной компанией.
Способ проведения эксперимента по исследованию ферментативной активности ATM
[517] Полученную от человека ATM киназу инкубировали в буферном растворе, содержащем 30 нМ GST-cMyc-p53 и Mg/ATP. Концентрация Mg/ATP определялась в соответствии с различными потребностями. Реакцию инициировали путем добавления комплекса Mg/ATP. После инкубирования в течение примерно 30 минут при комнатной температуре добавляли содержащий ЭДТА останавливающий раствор для остановки реакции. В конце добавляли буфер для детекции фосфорилированного р53, содержащий меченное d2 моноклональное антитело к GST и меченное европием антитело к фосфорилированному Ser15. Затем использовали режим флуоресценции с временным разрешением для считывания диска детекции и получали сигнал гомогенной флуоресценции с временным разрешением (HTRF) путем вычисления по формуле HTRF = 10000 × (Em665 нм/Em620 нм).
Способ проведения эксперимента по исследованию ферментативной активности ДНК-ПК
[518] Полученную от человека киназу ДНК-ПК инкубировали в буферном растворе, содержащем 50 нМ GST-cMyc-p53 и Mg/ATP. Концентрация Mg/ATP определялась в соответствии с различными потребностями. Реакцию инициировали путем добавления комплекса Mg/ATP. После инкубирования в течение примерно 30 минут при комнатной температуре добавляли содержащий ЭДТА останавливающий раствор для остановки реакции. В конце добавляли буфер для детекции фосфорилированного р53, содержащий меченное d2 моноклональное антитело к GST и меченное европием антитело к фосфорилированному Ser15. Затем использовали режим флуоресценции с временным разрешением для считывания диска детекции и получали сигнал гомогенной флуоресценции с временным разрешением (HTRF) путем вычисления по формуле HTRF = 10000 x (Em665 нм/Em620 нм).
Таблица 1: Результаты определения активности клеток in vitr° (IC50) для соединений согласно настоящему изобретению
[519] Заключение: Соединение согласно настоящему изобретению обладает значительным ингибирующим действием в отношении ATM киназы и обладает хорошей селективностью действия в отношении киназы ДНК-ПК.
[520] Экспериментальный пример 2: Фармакодинамические исследования in viv° ингибиторов ATM и этопозида, которые действуют синергетически в модели у самок мышей BALB/c nude с подкожным ксенотрансплантатом опухоли из клеток H446 рака легкого человека
Объект эксперимента:
[521] Оценка эффективности in viv исследуемого препарата - ингибитора ATM и этопозида в модели у мышей BALB/c nude с подкожным ксенотрансплантатом опухоли из клеток H446 рака легкого человека при внутрибрюшинном или пероральном введении.
План эксперимента:
Таблица 2. Разделение экспериментальных животных на группы и схема введения для оценки эффективности in viv ингибиторов ATM и этопозида
[522] Примечание: В/Б: внутрибрюшинное введение; П/О: пероральное введение; QD: один раз в день; BIW: дважды в неделю; QD (PG-D0, 3D °n, 4D °ff fr°m PG-D1) × 4W: введение со вторника по четверг, один раз в день, еженедельный цикл, в течение четырех недель; BIW + QD (PG-D0, 3D °n, 4D °ff fr°m PG-D1) × 4 W: этопозид вводили в понедельник, ингибитор ATM вводили со вторника по четверг, один раз в день, еженедельный цикл, в течение четырех недель.
Методы и стадии проведения эксперимента:
[523] 1. Культура клеток
[524] Клетки рака легкого человека H446 (ATCC, Manassas, VA, HTB-171) культивировали в монослое in vitr°. Условия культивирования: среда RPMI-1640 с добавлением 10% фетальной бычьей сыворотки, 100 Ед./мл пенициллина и 100 мкг/мл стрептомицина и культивирование при 37°C и 5% C°2. Два раза в неделю проводили обычную обработку для расщепления панкреатином-ЭДТА с целью пассажа. Когда насыщение клеток достигало 80-90%, клетки собирали, подсчитывали и осуществляли инокуляцию.
[525] 2. Инокуляция клеток опухоли
[526] 0,2 мл 5 × 106 клеток H446 (1 : 1 плюс матригель) подкожно инокулировали в правую часть спины каждой мыши nude. Разделение на группы и введение начинали, когда средний объем опухоли достигал 125 мм3 (см. таблицу).
[527] 3. Получение исследуемых образцов
Таблица 3. Способ получения исследуемых образцов
[528] Примечание: Перед введением животным препараты необходимо осторожно и тщательно перемешать.
Измерение опухоли и показатель при проведении эксперимента:
[529] Показатель при проведении эксперимента предназначен для исследования того, был ли рост опухоли ингибирован, отсрочен или излечен. Диаметр опухоли измеряли дважды в неделю с помощью штангенциркуля. Формула расчета объема опухоли: V = 0,5a × b2, где a и b представляют собой длинный и короткий диаметры опухоли, соответственно.
[530] Для оценки подавляющего рост опухоли действия соединений используют TGI (%). TGI (%) отражает степень подавления роста опухоли. Расчет TGI (%): TGI (%) = [(1-(средний объем опухоли на конец введения в группе лечения - средний объем опухоли на начало введения в этой группе лечения)/(средний объем опухоли на конец введения в контрольной группе с введением растворителя - средний объем опухоли на начало введения в контрольной группе с введением растворителя)] × 100 %.
[531] Скорость пролиферации опухоли T/C (%): где T означает средний объем опухоли, полученный при последнем определении (PG-D26) в группе лечения, и C означает средний объем опухоли, полученный при последнем определении (PG-D26) в контрольной группе.
Статистический анализ:
[532] Были включены среднее значение и стандартная ошибка среднего (С.О.С.) объема опухоли в каждой группе в каждый момент времени (конкретные данные представлены в таблицах). Группа лечения продемонстрировала лучший эффект лечения на 26 день после введения, в конце исследования, поэтому на основе этих данных проводили статистический анализ для оценки различий между группами. Анализ сравнения двух групп проводили с помощью T-критерия, а анализ сравнения трех или более групп проводили с помощью однофакторного дисперсионного анализа (AN°VA). Если наблюдалась неоднородность дисперсии, применяли критерий Геймса-Хоуэлла. Если наблюдалась однородность дисперсии, для анализа применяли критерий Даннета (двусторонний). Все данные анализировали с помощью программного обеспечения SPSS 17.0. p < 0,05 считали значимым различием.
Результаты эксперимента:
Смертность, заболеваемость и изменения массы тела
[533] Массу тела экспериментальных животных используют в качестве контрольного показателя для косвенного определения токсичности лекарственного средства. В этой модели ни одна из групп введения не продемонстрировала значительное снижение массы тела (Фигура 1). Мыши с № 42161 были обнаружены мертвыми в день 15 после введения препаратов в группе с применением комбинации 15 мг/кг этопозида и 5 мг/кг AZD0156. В группе лечения, где этопозид применяли в комбинации с примером 32 и AZD0156, соответственно, у части животных наблюдали снижение массы тела на более чем 10%, но не менее чем 15%. Действие исследуемого препарата - ингибитора ATM и этопозида на массу тела самок мышей BALB/c nude в модели с подкожным ксенотрансплантатом опухоли из клеток H446 представлено на Фигуре 1. Относительное изменение массы тела рассчитывали на основе массы тела животного в начале введения. Точка на графике представляет собой изменение средней массы тела в группе в процентах, и планка погрешностей представляет собой стандартную ошибку среднего (С.О.С.).
Объем опухоли
[534] Изменения объема опухоли в каждой группе после лечения с применением исследуемого препарата - ингибитора ATM и этопозида в модели у самок мышей BALB/c nude с подкожным ксенотрансплантатом опухоли из клеток H446 представлены в таблице 4.
Таблица 4. Объем опухоли в разные временные точки в каждой группе
после введения
[535] Примечание: a. Среднее ± С.О.С.
Кривая роста опухоли
[536] Кривая роста опухоли у мышей с опухолью в модели с опухолевым ксенотрансплантатом H446 после введения исследуемого препарата - ингибитора ATM и этопозида. Кривая роста опухоли представлена на Фигуре 2. Точка на графике представляет собой средний объем опухоли в группе, и столбик погрешностей представляет собой стандартную ошибку среднего (С.О.С.).
Показатель оценки противоопухолевой эффективности (рассчитанный на основе объема опухоли на 26 день после введения)
Таблица 5. Оценка противоопухолевой эффективности исследуемого препарата - ингибитора ATM и этопозида в модели опухолевого ксенотрансплантата H446
[537] Примечание: a. Среднее ± С.О.С. Животное #42161 из группы 5 было обнаружено мертвым в день PG-D15, и данные о нем не учитываются в статистике.
[538] b. Ингибирование роста опухоли рассчитывали с помощью T/C и TGI (TGI (%) = [1-(T26-T0)/(V26-V0)] × 100).
[539] c. p-Значение рассчитывали на основе объема опухоли.
Обсуждение эксперимента:
[540] В этом эксперименте оценивали эффективность исследуемого препарата - ингибитора ATM и этопозида в модели опухолевого ксенотрансплантата рака легкого человека H446. Объем опухоли в каждой группе в разные моменты времени был представлен в таблице 4, таблице 5 и на Фигуре 2. Через 26 дней после введения объем опухоли у мышей с опухолью в контрольной группе с введением растворителя достиг 2782 мм3, а для групп с введением этопозида + примера 32 (15 мг/кг + 5 мг/кг) и этопозида + AZD0156 (15 мг/кг + 5 мг/кг) средний объем опухоли составлял 930 мм3 и 1344 мм3, соответственно, T/C составлял 33,42% и 48,30, соответственно, TGI составлял 69,70% и 54,11%, и p-значение составляло 0,006 и 0,028 по сравнению с контролем с введением растворителя, соответственно.
Заключение по результатам эксперимента:
[541] В эксперименте по оценке эффективности ингибитора ATM и этопозида in viv° в модели опухолевого ксенотрансплантата рака легкого человека H446 комбинация примера 32 и этопозида показала хороший синергетический эффект, который превышал эффективность AZD0156 в комбинации с этопозидом.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДУЛЯТОРЫ КАЛЬПАИНА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2773288C2 |
| ПРОТИВОФИБРОЗНЫЕ ПИРИДИНОНЫ | 2015 |
|
RU2692485C2 |
| СЛИТЫЕ ТРИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ИНТЕГРАЗЫ ВИЧ | 2016 |
|
RU2740187C2 |
| ЗАМЕЩЕННЫЕ 4-(АРИЛАМИНО) СЕЛЕНОФЕНОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2011 |
|
RU2566293C2 |
| ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ, ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, СПОСОБ ПОЛУЧЕНИЯ, КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2732576C2 |
| ПРОИЗВОДНОЕ ПИРИМИДИНА И ПЯТИЧЛЕННОГО АЗОТСОДЕРЖАЩЕГО ГЕТЕРОЦИКЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКИЕ ПРИМЕНЕНИЯ | 2019 |
|
RU2775229C1 |
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ МИМЕТИКОВ ТРО | 2006 |
|
RU2385865C2 |
| КОНДЕНСИРОВАННЫЕ ГЕТЕРОАРИЛЫ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2552114C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
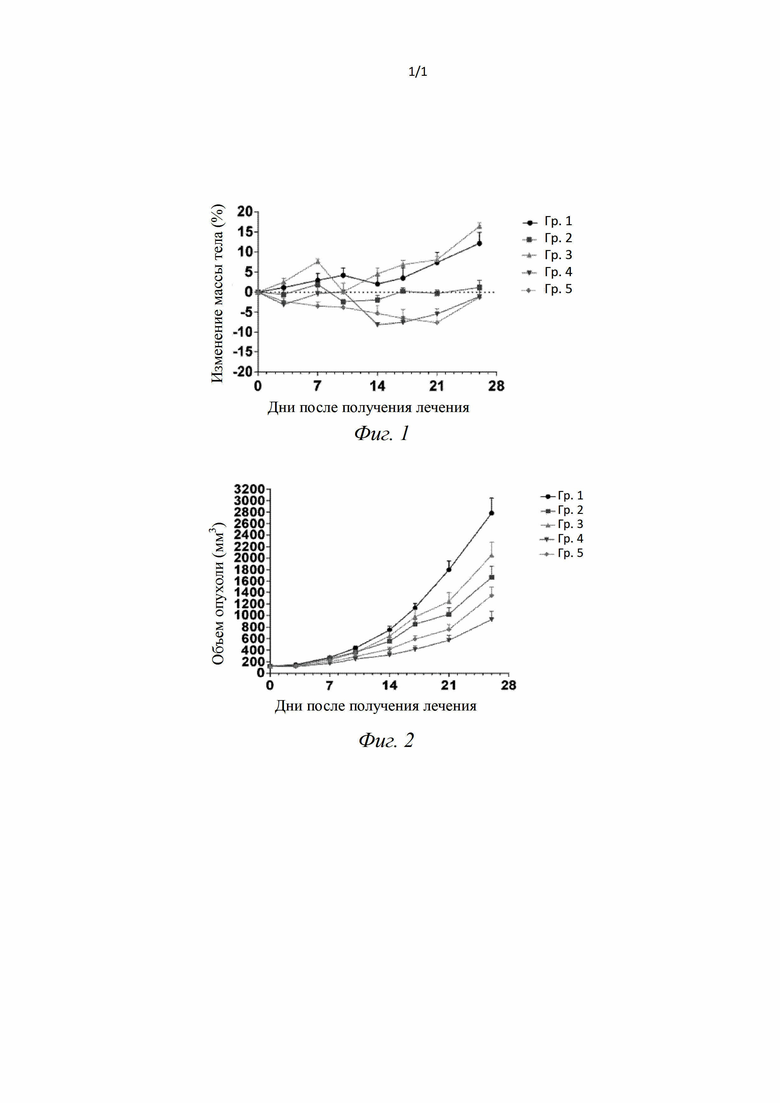
Изобретение относится к области органической химии, а именно к производному хинолинпирролидин-2-она формулы (I), его изомеру или фармацевтически приемлемой соли, где E выбран из -N(R5)-, -O- и -C(R6)(R7)-; R1 выбран из C1-3 алкила и C3-6 циклоалкила; R2 представляет собой H; R3 выбран из H, F, Cl; R4 выбран из C1-6 алкила и N(Rc)(Rd); R5 выбран из H, C1-6 алкила, C3-6 циклоалкила, C1-6 алкил-C=O-, C1-6 алкил-O-C=O- и C3-6 циклоалкил-C=O-, где указанный C1-6 алкил необязательно замещен 1, 2 или 3 Re; каждый из R6 и R7 независимо выбран из H, F, OH, C1-6 алкила и C1-6 алкокси; L1 выбран из одинарной связи и -(CH2)m-O-; m выбран из 1, 2 и 3; кольцо B представляет собой 5-6-членный гетероарил; каждый из Rc и Rd независимо выбран из C1-3 алкила и C3-6 циклоалкила; или Rc и Rd совместно с атомом N, к которому они присоединены, образуют 5-6-членный гетероциклоалкил, необязательно замещенный 1 R; Re представляет собой F; R представляет собой F; и каждый из указанных 5-6-членного гетероарила и 5-6-членного гетероциклоалкила независимо содержит 1 или 2 гетероатома, независимо выбранных из -NH- и N. Также изобретение относится к конкретному производному хинолинпирролидин-2-она и их применению для получения лекарственного средства, способного ингибировать АТМ киназу. Технический результат: получены новые гетероциклические соединения, полезные при лечении заболеваний, связанных с ингибированием АТМ киназы. 3 н. и 16 з.п. ф-лы, 2 ил., 5 табл., 38 пр.
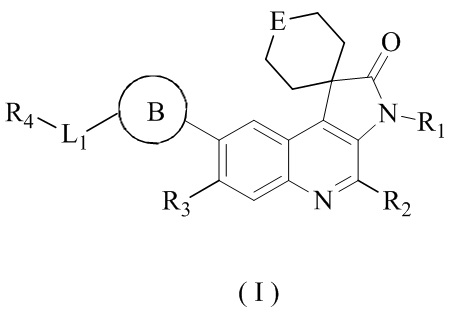
1. Соединение, представленное формулой (I), его изомер или фармацевтически приемлемая соль
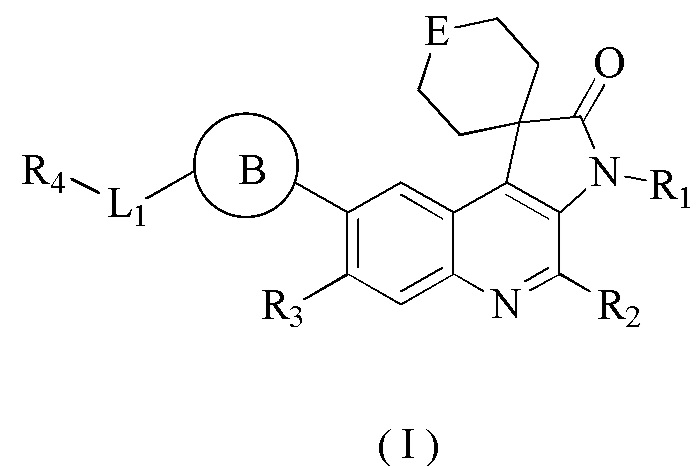 ,
,
где
E выбран из -N(R5)-, -O- и -C(R6)(R7)-;
R1 выбран из C1-3 алкила и C3-6 циклоалкила;
R2 представляет собой H;
R3 выбран из H, F, Cl;
R4 выбран из C1-6 алкила и N(Rc)(Rd);
R5 выбран из H, C1-6 алкила, C3-6 циклоалкила, C1-6 алкил-C=O-, C1-6 алкил-O-C=O- и C3-6 циклоалкил-C=O-, где указанный C1-6 алкил необязательно замещен 1, 2 или 3 Re;
каждый из R6 и R7 независимо выбран из H, F, OH, C1-6 алкила и C1-6 алкокси;
L1 выбран из одинарной связи и -(CH2)m-O-;
m выбран из 1, 2 и 3;
кольцо B представляет собой 5-6-членный гетероарил;
каждый из Rc и Rd независимо выбран из C1-3 алкила и C3-6 циклоалкила;
или Rc и Rd совместно с атомом N, к которому они присоединены, образуют 5-6-членный гетероциклоалкил, необязательно замещенный 1 R;
Re представляет собой F;
R представляет собой F;
и каждый из указанных 5-6-членного гетероарила и 5-6-членного гетероциклоалкила независимо содержит 1 или 2 гетероатома, независимо выбранных из -NH- и N.
2. Соединение, его изомер или фармацевтически приемлемая соль по п. 1, где R1 выбран из CH3, CH2CH3 и циклопропила.
3. Соединение, его изомер или фармацевтически приемлемая соль по п. 1, где каждый из Rc и Rd независимо выбран из CH3, CH2CH3 и циклопропила.
4. Соединение, его изомер или фармацевтически приемлемая соль по п. 1 или 2, где Rc и Rd совместно с атомом N, к которому они присоединены, образуют пирролидил и пиперидинил, причем указанные пирролидил и пиперидинил необязательно замещены 1 R.
5. Соединение, его изомер или фармацевтически приемлемая соль по п. 4, где Rc и Rd совместно с атомом N, к которому они присоединены, образуют  .
.
6. Соединение, его изомер или фармацевтически приемлемая соль по п. 1, где R4 выбран из CH3, CH2CH3,  и
и  .
.
7. Соединение, его изомер или фармацевтически приемлемая соль по любому из пп. 1 или 2, где R5 выбран из H, CH3, CH3CH2, CH(CH3)2, циклопропила, CH3OC=O-, CH(CH3)2OC=O-, CH3C=O- и циклопропил-C=O-, и указанные CH3, CH3CH2, CH(CH3)2 необязательно замещены 1, 2 или 3 Re.
8. Соединение, его изомер или фармацевтически приемлемая соль по п. 7, где R5 выбран из H, CH3, CH2F, CHF2, CF3, CH3CH2, CH2FCH2, CHF2CH2, CF3CH2, CH(CH3)2, циклопропила, CH3OC=O-, CH(CH3)2OC=O-, CH3C=O- и циклопропил-C=O-.
9. Соединение, его изомер или фармацевтически приемлемая соль по любому из пп. 1 или 2, где каждый из R6 и R7 независимо выбран из H, F, OH, CH3, CH3CH2, CH(CH3)2 и  .
.
10. Соединение, его изомер или фармацевтически приемлемая соль по п. 7 или 9, где E выбран из -O-, -CF2-, -N(CH3)-, -NH-, 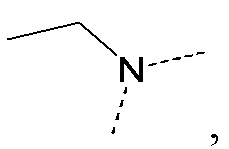
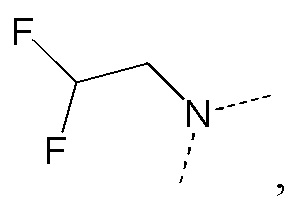
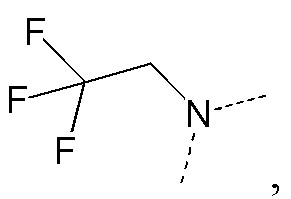
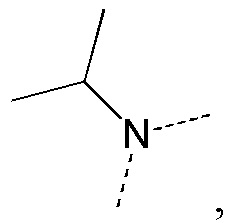
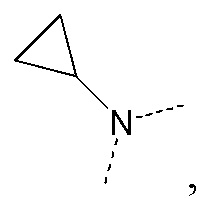
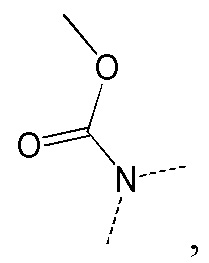
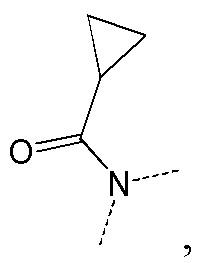
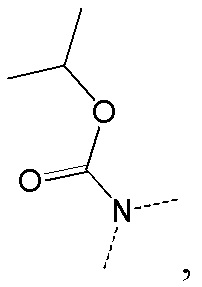
 и
и  .
.
11. Соединение, его изомер или фармацевтически приемлемая соль по п. 1, где L1 выбран из одинарной связи, -(CH2)-O- и -(CH2)3-O-.
12. Соединение, его изомер или фармацевтически приемлемая соль по любому из пп. 1 или 2, где кольцо B выбрано из пиридила, пиразолила и имидазолила.
13. Соединение, его изомер или фармацевтически приемлемая соль по п. 12, где кольцо B выбрано из 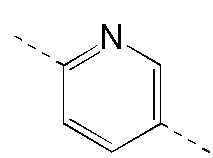 и
и 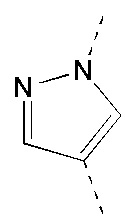 .
.
14. Соединение, его изомер или фармацевтически приемлемая соль по п. 11, где R4-L1- выбран из CH3, CH3OCH2-, 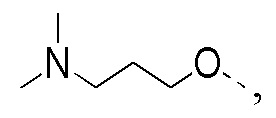
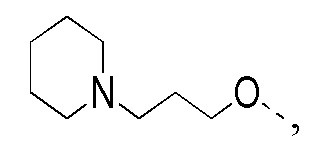
 и
и  .
.
15. Соединение, его изомер или фармацевтически приемлемая соль по любому из пп. 1-11, выбранные из
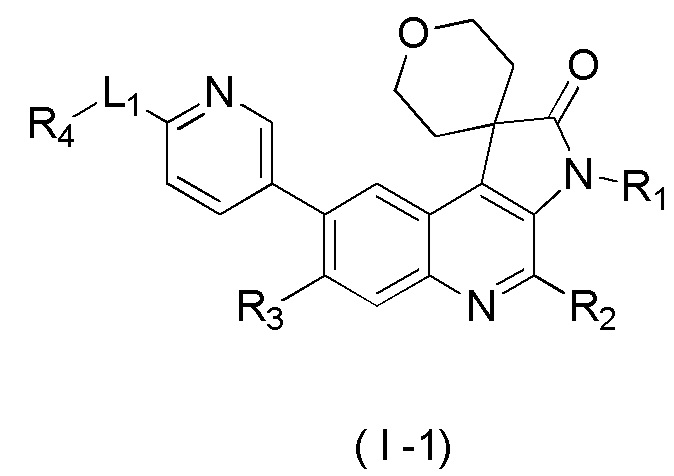
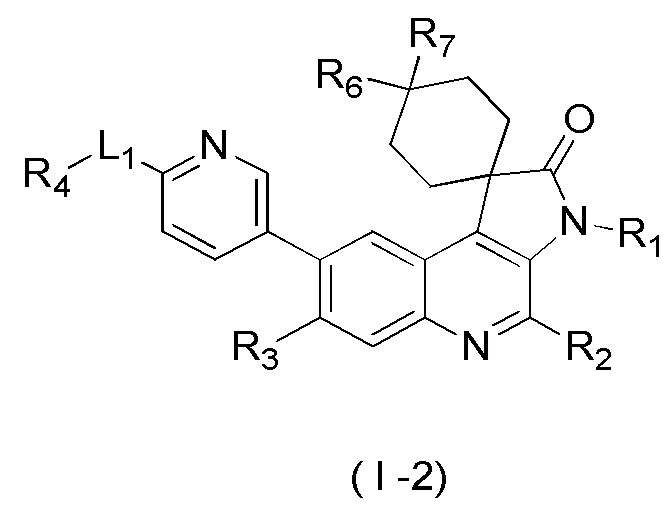
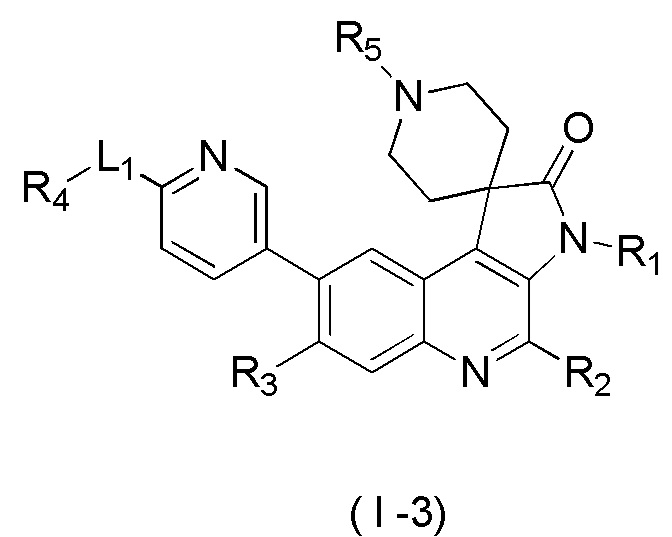
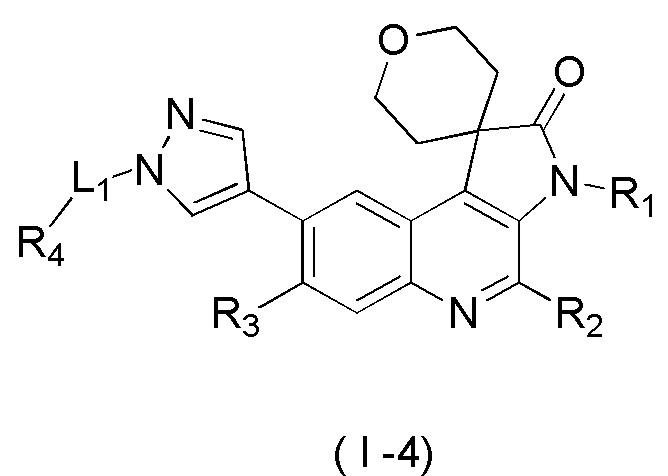 ,
,
где
R1, R2, R3, R4, R5, R6, R7 и L1 такие, как определено в любом из пп. 1-11.
16. Соединение, представленное следующими формулами, его изомер или фармацевтически приемлемая соль, где указанное соединение выбрано из
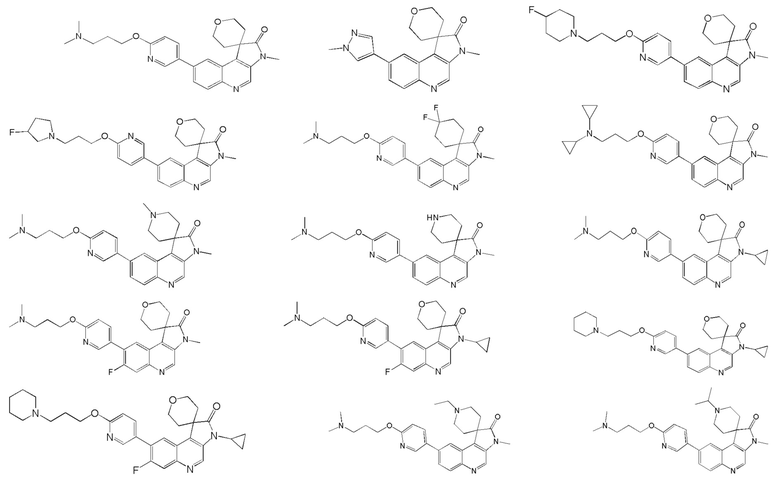
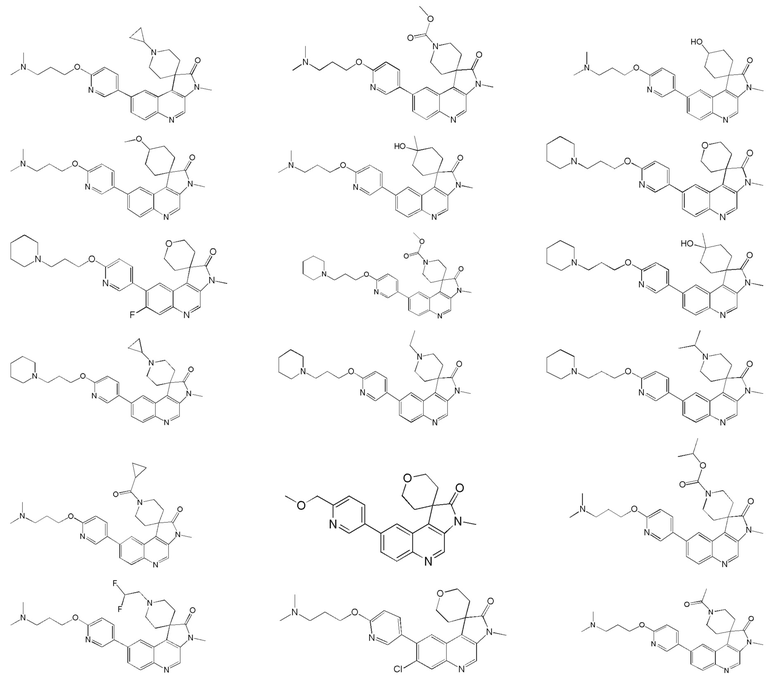
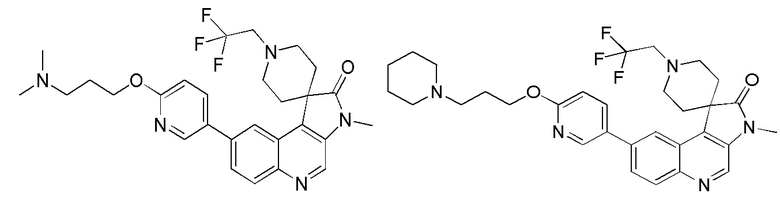 .
.
17. Соединение, его изомер или фармацевтически приемлемая соль по п. 16, выбранные из
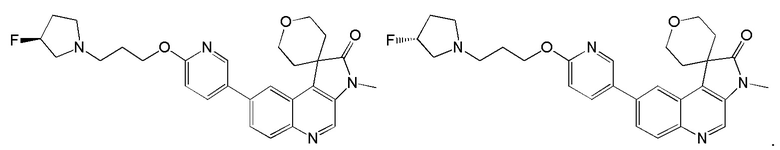
18. Применение соединения, его изомера или его фармацевтически приемлемой соли по любому из пп. 1-17 для получения лекарственного средства, способного ингибировать ATM киназу.
19. Применение по п. 18, где указанное лекарственное средство способно ингибировать солидные опухоли.
| WO 2017076898 A1, 11.05.2017 | |||
| WO 2017076895 A1, 11.05.2017 | |||
| WO 2016155884 A1, 06.10.2016 | |||
| WO 2017194632 A1, 16.11.2017 | |||
| CN 102399218 A, 04.04.2012 | |||
| АНТАГОНИСТЫ ВАНИЛОИДНОГО РЕЦЕПТОРА ПОДТИПА 1(VR1) И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2450006C2 |

