Область техники, к которой относится изобретение
Настоящее изобретение касается производных имидазохинолина и фармацевтических композиций, содержащих производные имидазохинолина. Производные имидазохинолина могут применяться в качестве агонистов толл-подобного рецептора, в частности в качестве агонистов TLR7, и промотируют выработку определенных цитокинов.
Предшествующий уровень техники
Толл-подобные рецепторы (TLR), включающие семейство из 13 рецепторов с разными специализациями, 11 из которых найдены у человека, являются частью клеточной системы распознавания патогенов, которая выработалась для защиты против различных инфекций (бактерии, вирусы, грибы). Активация TLR приводит к цитокиновому ответу, например с высвобождением интерферонов и активацией специализированных иммунных клеток. Функциональное экспрессирование TLR в тканях сильно различается. Часть рецепторов расположена на поверхности клеток, такие как TLR4 (стимулируется E.coli липосахаридом LPS), например на эпителиальных клетках, или TLR3, 7, 8 и 9, расположенные на эндосомальных мембранах в определенных иммунных клетках. Последние активируются нуклеиновыми кислотами, но распознают их различные типы. Например, TLR9 активируется одноцепочечной ДНК, содержащей последовательности CpG, TLR7 и 8 активируются одноцепочечной РНК, и TLR3 активируется двухцепочечной РНК.
Были найдены некоторые низкомолекулярные (SMOL) TLR7 или TLR8 агонисты. Эти агонисты можно сгруппировать на молекулы пуринового типа, такие как 7-тиа-8-оксогуанозин (TOG, изаторибин), или имидазохинолиновые соединения, такие как имиквимод. Имиквимод на настоящий момент является единственным одобренным определенным TLR7 агонистом, который продается в виде 5%-ного крема (компанией Aldara). Он обеспечивает примерно 80% 5-летний клиренс поверхностной базальноклеточной карциномы, которая представляет собой самый распространенный вид рака в мире. Имиквимод активирует TLR7. Функциональное экспрессирование TLR7, судя по всему, ограничивается специфическими иммунными клетками, а именно - известно, что человеческие плазмацитоидные дендритные клетки, B-клетки и, вероятно, эозинофилы активируются TLR7 агонистами.
На протяжении последних нескольких лет во всем мире предпринимаются большие усилия, направленные на использование сильной иммунной активации, вызываемой TLR7, 8 или 9 агонистами, для лечения рака. Иммунотерапия рака, однако, имеет большую историю неудач. Но в последние годы знания об иммунном надзоре над раком и работе определенных иммунных клеток существенно выросли. TLR7, TLR8 или TLR9 агонисты находятся в стадии клинических разработок в качестве средства монотерапии или комбинированной терапии рака, или в качестве адъювантов для вакцин.
Подход к иммунотерапии рака с применением TLR агонистов отличается от более ранних попыток с применением, например, цитокинов, интерферонов или моновалентных вакцин. Активация иммунитета, вызываемая TLR агонистом, является плейотропной и работает с участием определенных иммунных клеток (главным образом дендритных клеток и В-клеток, а также других клеток), которые обеспечивают врожденный и адаптивный иммунный ответ. Кроме того, вырабатывается не только один интерферон, а сразу много разных изоформ, и не только тип I (альфа, бета), но также (косвенно) тип II (гамма, NK-клетки). По меньшей мере для местного нанесения Aldara прекрасно подтверждает предполагаемый принцип работы. Это показывает, что антигены высвобождаются опухолью и что иммуная терапия в принципе может работать при раковых показаниях, и даже в качестве средства монотерапии. Однако для системного введения еще предстоит доказать клинический принцип работы для TLR7-агонистов. В случае поздних стадий рака и системного введения (в особенности подкожного или внутривенного введения) достаточно понятно, что такие TLR агонисты могут обеспечить более высокую, т.е. синергетическую, эффективность в комбинации с другими терапевтическими вмешательствами.
В случае ранних стадий рака ситуация может быть иной. Метастазы опухоли являются тяжелым аспектом в развитии опухоли у пациентов, главным образом потому, что опухоли обнаруживают слишком поздно, когда метастазы уже появились. Принятые способы терапии опухолей включают главным образом цитотоксические лекарственные средства с достаточно узким терапевтическим окном. Поэтому для лечения ранних стадий опухолей, когда подавление распространения метастазов еще возможно, имеется высокая потребность в новых терапевтических средствах с хорошей переносимостью и профилем безопасности.
Активация иммунной системы и, в частности, активация сигнального пути толл-подобного рецептора (TLR) позволяет развивать новые перспективные подходы. TLR9 агонистический CpG-ODN, такой как H2006 или H1826, и TLR7 агонисты, такие как производное гуанозина - изаторибин, или производное имиквимода, были протестированы на мышиной модели метастазов рака почки в легкие. Все протестированные молекулы практически полностью подавляли развитие метастазов в легкие, при хорошей переносимости. Это дает убедительное обоснование для клинических испытаний таких молекул для подавления развития метастазов рака и указывает на возможность системного введения таких лекарственных средств. Однако, TLR7 агонисты SMOL-типа имеют преимущество, заключающееся в разработанном и экономичном синтезе, по сравнению с TLR9 агонистами, относящимися к типу нуклеиновых кислот, и хорошо подходят для наружного применения.
В US-B-6,573,273 описаны имидазохинолиновые и тетрагидроимидазохинолиновые соединения, которые содержат мочевинную, тиомочевинную, ацилмочевинную, сульфонилмочевинную или карбаматную функциональную группу. Утверждается, что эти соединения могут применяться в качестве иммуномодуляторов.
В US-B-6,677,349 описаны имидазохинолиновые и тетрагидроимидазохинолиновые соединения, которые содержат сульфонамидную функциональную группу в положении 1. Утверждается, что эти соединения могут применяться в качестве иммуномодуляторов.
В US-A-2003/0144283 и WO-A-00/76505 описаны имидазохинолиновые и тетрагидроимидазохинолиновые соединения, которые содержат амидную функциональную группу в положении 1. Утверждается, что эти соединения могут применяться в качестве иммуномодуляторов.
В WO-A-2005/051324 описаны имидазохинолиновые, пиридиновые и нафтиридиновые кольцевые системы, замещенные в положении 1 оксимной или особой N-оксидной функциональной группой. Утверждается, что эти соединения могут применяться в качестве иммуномодуляторов.
В WO-A-2009/118296 описаны имидазохинолиновые соединения. Эти соединения описаны как агонисты толл-подобного рецептора/TLR7 активаторы.
Краткое описание изобретения
В настоящем изобретении описаны имидазохинолин-4-аминовые соединения с определенными специфическими заместителями, их физиологически функциональные производные, сольваты и соли, как будет описано далее по тексту. Указанные соединения являются агонистами или активаторами для TLR7 и могут работать как соединения, запускающие выработку цитокинов. Эти соединения имеют общую формулу (I):
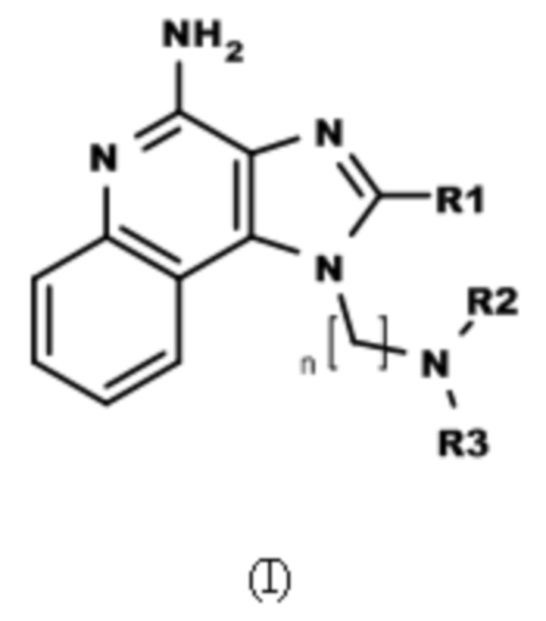
где R1, R2, R3 и n имеют указанные выше значения.
В другом аспекте, в настоящем изобретении описаны способы получения некоторых соединений, имеющих формулу (I), их физиологически функциональных производных, сольватов или солей, как подробно описано ниже.
В другом аспекте, в настоящем изобретении описаны способы лечения или предотвращения некоторых медицинских состояний, включающие введение соединений, имеющих формулу (I), их физиологически функциональных производных, сольватов или солей, субъекту, нуждающемуся в этом, как подробно описано ниже.
В другом аспекте, в настоящем изобретении описано применение соединений, имеющих формулу (I), их физиологически функциональных производных, сольватов или солей, в производстве лекарственного средства для лечения или предотвращения некоторых медицинских состояний, как подробно описано ниже.
В другом аспекте, в настоящем изобретении описаны соединения, имеющие формулу (I), их физиологически функциональные производные, сольваты или соли, для применения в качестве лекарственного средства, в частности для применения в лечении или предотвращении некоторых медицинских состояний, как подробно описано ниже.
В другом аспекте, в настоящем изобретении описаны фармацевтические композиции, содержащие соединения, имеющие формулу (I), их физиологически функциональные производные, сольваты или соли и один или больше фармацевтически приемлемых эксципиентов.
Соединения, имеющие формулу (I), могут применяться, например, в качестве TLR7 активаторов.
Подробное описание изобретения
Было обнаружено, что производные имидазохинолина, имеющие формулу (I), их физиологически функциональные производные, сольваты или соли, более подробно описанные ниже, представляют собой особенно эффективные TLR7 агонисты и обладают неожиданными и весьма благоприятными свойствами.
В настоящем изобретении описаны соединения, имеющие формулу (I):
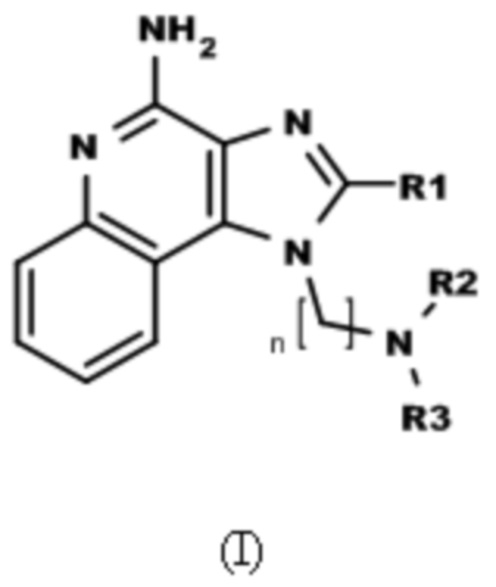
где
R1 выбран из группы, состоящей из -H, C1-6-алкила, C1-6-алкокси-группы, C1-3-алкокси-C1-3-алкила, C1-6-алкилтио-группы, C1-3-алкилтио-C1-3-алкила, C1-3-алкиламино-C1-3-алкила, 4-10-членного гетероциклоалкила, C3-10-циклоалкила, C6-10-арила, C6-10-арил-C1-2-алкила и 5-10-членного гетероарила,
где указанный C1-6-алкил, C1-6-алкокси-группа, C1-3-алкокси-C1-3-алкил, C1-6-алкилтио-группа, C1-3-алкилтио-C1-3-алкил, C1-3-алкиламино-C1-3-алкил, C6-10-арил, 4-10-членный гетероциклоалкил, C3-10-циклоалкил, C6-10-арил-C1-2-алкил и 5-10-членный гетероарил необязательно замещен одной или больше группами, независимо выбранными из группы, состоящей из C1-4-алкила, -OH, атома галогена, -CO-N(R4)2, -N(R4)2, -CO-R4, -COO-R4, -N3, -NO2 и -CN;
R2 выбран из группы, состоящей из -CO-R5, -CONH-R5 и -COO-R5;
R3 представляет собой тетрагидропиран-4-ил, который необязательно замещен одной или больше группами, независимо выбранными из группы, состоящей из C1-4-алкила, -OH и атома галогена;
R4 каждый независимо выбран из группы, состоящей из H и C1-4-алкил;
n представляет собой целое число от 3 до 6; и
R5 выбран из группы, состоящей из -H, C1-6-алкила, C1-6-алкокси-группы, C1-3-алкокси-C1-3-алкила, C1-6-алкилтио-группы, C1-3-алкилтио-C1-3-алкила, C1-3-алкиламино-C1-3-алкила, C6-10-арила, 4-10-членного гетероциклоалкила, C3-10-циклоалкил и 5-10-членного гетероарила,
где указанный C1-6-алкил, C1-6-алкокси-группа, C1-3-алкокси-C1-3-алкил, C1-6-алкилтио-группа, C1-3-алкилтио-C1-3-алкил, C1-3-алкиламино-C1-3-алкил, C6-10-арила, 4-10-членный гетероциклоалкил, C3-10-циклоалкил и 5-10-членный гетероарил необязательно замещен одной или больше группами, независимо выбранными из группы, состоящей из C1-4-алкила, -OH, атом галогена, -CO-N(R4)2, -N(R4)2, -CO-R4, -COO-R4, -N3, -NO2 и -CN;
или их физиологически функциональные производные, сольваты или соли.
В частных вариантах осуществления настоящего изобретения, R1 выбран из группы, состоящей из C1-6-алкила, C1-3-алкокси-C1-3-алкила, C1-3-алкилтио-C1-3-алкила и C1-3-алкиламино-C1-3-алкила, более предпочтительно C1-6-алкила, C1-3-алкокси-C1-3-алкила и C1-3-алкиламино-C1-3-алкила, где указанный C1-6-алкил, C1-3-алкокси-C1-3-алкил, C1-3- алкилтио-C1-3-алкил или C1-3-алкиламино-C1-3-алкил необязательно замещен одной или больше группами, независимо выбранными из группы, состоящей из -OH и галогена.
Еще более предпочтительно, R1 выбран из группы, состоящей из этила, метила, пропила, бутила, метоксиэтила и этиламинометила, каждый из которых необязательно замещен одной или больше группами, независимо выбранными из группы, состоящей из -OH и галогена, и еще более предпочтительно каждый из которых является незамещенным.
И еще более предпочтительно, R1 выбран из группы, состоящей из этила, пропила, бутила, метоксиэтила и этиламинометила, каждый из которых необязательно замещен одной или больше группами, независимо выбранными из группы, состоящей из -OH и галогена, и еще более предпочтительно каждый из которых является незамещенным.
И еще более предпочтительно, R1 выбран из группы, состоящей из этила, метоксиэтила и этиламинометила, каждый из которых необязательно замещен одной или больше группами, независимо выбранными из группы, состоящей из -OH и галогена, и еще более предпочтительно каждый из которых является незамещенным.
И еще более предпочтительно, R1 является незамещенным метоксиэтилом.
Еще более предпочтительно, R1 является незамещенным этиламинометилом.
В частных вариантах осуществления настоящего изобретения, R2 выбран из группы, состоящей из -CO-R5 и -CONH-R5, более предпочтительно -CO-R5.
В частных вариантах осуществления настоящего изобретения, R3 является незамещенным тетрагидропиран-4-илом.
В частных вариантах осуществления настоящего изобретения, каждый R4 независимо выбран из группы, состоящей из H и метила, более предпочтительно H.
В частных вариантах осуществления настоящего изобретения, n равен 4.
В частных вариантах осуществления настоящего изобретения, R5 выбран из группы, состоящей из H, C1-4-алкила, C1-3-алкокси-C1-3-алкила, C1-3-алкиламино-C1-3-алкила, фенила, 5-6-членного гетероциклоалкила, C5-6-циклоалкила и 5-6-членного гетероарила, более предпочтительно H, C1-4-алкила, C1-3-алкокси-C1-3-алкила, фенила и C5-6-циклоалкила, еще более предпочтительно H, C1-4-алкила, где указанный C1-4-алкил, C1-3-алкокси-C1-3-алкил, C1-3-алкиламино-C1-3-алкил, фенил, 5-6-членный гетероциклоалкил, C5-6-циклоалкил и 5-6-членный гетероарил необязательно замещен одной или больше группами, независимо выбранными из группы, состоящей из C1-2-алкила, -OH, атома галогена, -CO-N(R4)2, -N(R4)2, -CO-R4, -COO-R4, -N3, -NO2 и -CN, более предпочтительно - из C1-2-алкила, -OH, атома галогена, NH2, -COMe, -COOH, -COOMe и -CN, еще более предпочтительно - из метила, -OH и галогена.
Более предпочтительно, R5 выбран из группы, состоящей из H, метила, этила, пропила и бутила, еще более предпочтительно - из H, метила или этила, и еще более предпочтительно - из H или метила.
В частных вариантах осуществления настоящего изобретения, R2 представляет собой -CO-R5, где R5 выбран из группы, состоящей из C1-4-алкила, C1-3-алкокси-C1-3-алкила и C1-3-алкиламино-C1-3-алкила, более предпочтительно - из C1-4-алкила и C1-3-алкокси-C1-3-алкила, более предпочтительно R5 представляет собой C1-4-алкил, где указанный C1-4-алкил, C1-3-алкокси-C1-3-алкил и C1-3-алкиламино-C1-3-алкил необязательно замещен одной или больше группами, независимо выбранными из группы, состоящей из C1-2-алкила, -OH, атома галогена, -CO-N(R4)2, -N(R4)2, -CO-R4, -COO-R4, -N3, -NO2 и -CN, более предпочтительно - из C1-2-алкила, -OH, атома галогена, NH2, -COMe, -COOH, -COOMe и -CN, еще более предпочтительно - из метила, -OH и галогена. В одном варианте осуществления, R2 представляет собой CO-R5, где R5 выбран из группы, состоящей из C1-4-алкила, C1-3-алкокси-C1-3-алкила, и C1-3-алкиламино-C1-3-алкила, более предпочтительно - из C1-4-алкила и C1-3-алкокси-C1-3-алкила, более предпочтительно R5 представляет собой C1-4-алкил, где указанный C1-4-алкил, C1-3-алкокси-C1-3-алкил и C1-3-алкиламино-C1-3-алкил является незамещенным.
В частных вариантах осуществления настоящего изобретения, R2 представляет собой -CONH-R5, где R5 выбран из группы, состоящей из H, C1-4-алкила, C1-3-алкокси-C1-3-алкила и C1-3-алкиламино-C1-3-алкила, более предпочтительно - из H и C1-4-алкила, где указанный C1-4-алкил, C1-3-алкокси-C1-3-алкил и C1-3-алкиламино-C1-3-алкил необязательно замещен одной или больше группами, независимо выбранными из группы, состоящей из C1-2-алкила, -OH, атома галогена, -CO-N(R4)2, -N(R4)2, -CO-R4, -COO-R4, -N3, -NO2 и -CN, более предпочтительно - из C1-2-алкила, -OH, атома галогена, NH2, -COMe, -COOH, -COOMe и -CN, еще более предпочтительно -из метила, -OH и галогена. В одном варианте осуществления, R2 представляет собой -CONH-R5, где R5 выбран из группы, состоящей из H, C1-4-алкила, C1-3-алкокси-C1-3-алкила и C1-3-алкиламино-C1-3-алкила, более предпочтительно - из H и C1-4-алкила, где указанный C1-4-алкил, C1-3-алкокси-C1-3-алкил и C1-3-алкиламино-C1-3-алкил является незамещенным.
В одном варианте осуществления, R2 представляет собой -CONH2, -CO-Me, -COOMe или -COOH, предпочтительно -CONH2, -CO-Me или -COOMe, более предпочтительно -CONH2 или -CO-Me.
В частных вариантах осуществления настоящего изобретения, R1 выбран из группы, состоящей из C1-6-алкила, C1-3-алкокси-C1-3-алкила, C1-3-алкилтио-C1-3-алкила и C1-3-алкиламино-C1-3-алкила, более предпочтительно - из C1-6-алкила, C1-3-алкокси-C1-3-алкила и C1-3-алкиламино-C1-3-алкила, где указанный C1-6-алкил, C1-3-алкокси-C1-3-алкил, C1-3- алкилтио-C1-3-алкил или C1-3-алкиламино-C1-3-алкил необязательно замещен одной или больше группами, независимо выбранными из группы, состоящей из -OH и атома галогена; и R2 представляет собой -CO-R5, где R5 выбран из группы, состоящей из C1-4-алкила, C1-3-алкокси-C1-3-алкила и C1-3-алкиламино-C1-3-алкила, более предпочтительно - из C1-4-алкила и C1-3-алкокси-C1-3-алкила, более предпочтительно R5 представляет собой C1-4-алкил, где указанный C1-4-алкил, C1-3-алкокси-C1-3-алкил и C1-3-алкиламино-C1-3-алкил необязательно замещен одной или больше группами, независимо выбранными из группы, состоящей из C1-2-алкила, -OH, атома галогена, -CO-N(R4)2, -N(R4)2, -CO-R4, -COO-R4, -N3, -NO2 и -CN, более предпочтительно - из C1-2-алкила, -OH, атома галогена, NH2, -COMe, -COOH, -COOMe и -CN, еще более предпочтительно - из метила, -OH и галогена. В одном варианте осуществления, R2 представляет собой CO-R5, где R5 выбран из группы, состоящей из C1-4-алкила, C1-3-алкокси-C1-3-алкила и C1-3-алкиламино-C1-3-алкила, более предпочтительно - из C1-4-алкила и C1-3-алкокси-C1-3-алкила, более предпочтительно R5 представляет собой C1-4-алкил, где указанный C1-4-алкил, C1-3-алкокси-C1-3-алкил и C1-3-алкиламино-C1-3-алкил является незамещенным. В этих вариантах осуществления, предпочтительно что R3 представляет собой незамещенный тетрагидропиран-4-ил и/или n равен 4.
В частных вариантах осуществления настоящего изобретения, R1 выбран из группы, состоящей из C1-6-алкила, C1-3-алкокси-C1-3-алкила, C1-3-алкилтио-C1-3-алкила и C1-3-алкиламино-C1-3-алкила, более предпочтительно - из C1-6-алкила, C1-3-алкокси-C1-3-алкила и C1-3-алкиламино-C1-3-алкила, где указанный C1-6-алкил, C1-3-алкокси-C1-3-алкил, C1-3- алкилтио-C1-3-алкил или C1-3-алкиламино-C1-3-алкил необязательно замещен одной или больше группами, независимо выбранными из группы, состоящей из -OH и атома галогена; и R2 представляет собой -CONH-R5, где R5 выбран из группы, состоящей из H, C1-4-алкила, C1-3-алкокси-C1-3-алкила и C1-3-алкиламино-C1-3-алкила, более предпочтительно - из H и C1-4-алкила, где указанный C1-4-алкил, C1-3-алкокси-C1-3-алкил,и C1-3-алкиламино-C1-3-алкил необязательно замещен одной или больше группами, независимо выбранными из группы, состоящей из C1-2-алкила, -OH, атома галогена, -CO-N(R4)2, -N(R4)2, -CO-R4, -COO-R4, -N3, -NO2 и -CN, более предпочтительно - из C1-2-алкила, -OH, атома галогена, NH2, -COMe, -COOH, -COOMe и -CN, еще более предпочтительно - из метила, -OH и галогена. В одном варианте осуществления, R2 представляет собой -CONH-R5, где R5 выбран из группы, состоящей из H, C1-4-алкила, C1-3-алкокси-C1-3-алкила и C1-3-алкиламино-C1-3-алкила, более предпочтительно - из H и C1-4-алкила, где указанный C1-4-алкил, C1-3-алкокси-C1-3-алкил и C1-3-алкиламино-C1-3-алкил является незамещенным. В этих вариантах осуществления, предпочтительно что R3 представляет собой незамещенный тетрагидропиран-4-ил и/или n равен 4.
Данные ниже определения предназначены для дополнительного определения некоторых терминов, применяющихся в контексте настоящего изобретения. Если для какого-либо термина, применяющегося в настоящем тексте, не приведено специального определения, это не значит, что термин не является определенным. В этом случае такие термины следует понимать в соответствии с их стандартным значением, известным квалифицированным специалистам в области, к которой относится настоящее изобретение, в частности в области органической химии, фармацевтики и медицины.
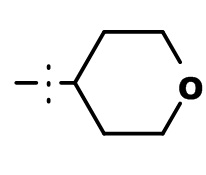
Термин “тетрагидропиран-4-ил” означает группу, имеющую приведенную ниже формулу, где прерванная связь указывает место присоединения к остальной части молекулы.
При использовании в настоящем тексте, термины “алкил” и префикс “алк” охватывают как линейные, так и разветвленные группы и включают соответствующие алкановые, алкеновые и алкиновые группы. Очевидно, что алкеновые и алкиновые группы не могут состоять из одного атома углерода, и такие несуществующие группы не охватываются настоящим изобретением; соответственно и логично, что такие термины как C1-x-алкил (где x представляет собой целое число, указанное в контексте) включают соответствующие C1-x-алканил, C2-x-алкенил и C2-x-алкинил. Предпочтительные алкильные группы содержат в сумме до 5, предпочтительно до 4, более предпочтительно до 3 атомов углерода. В частных вариантах осуществления, алкильная группа выбрана из группы, состоящей из -CH3, -C2H5, -CH=CH2, -C≡CH, -C3H7, -CH(CH3)2, -CH2-CH=CH2, -C(CH3)=CH2, -CH=CH-CH3, -C≡C-CH3, -CH2-C≡CH, -C4H9, -CH2-CH(CH3)2, -CH(CH3)-C2H5, -C(CH3)3, -C5H11, -C6H13, -C2H4-CH=CH2, -CH=CH-C2H5, -CH=C(CH3)2, -CH2-CH=CH-CH3, -CH=CH-CH=CH2, -C2H4-C≡CH, -C≡C-C2H5, -CH2-C≡C-CH3, -C≡C-CH=CH2, -CH=CH-C≡CH, -C≡C-C≡CH, -C2H4-CH(CH3)2, -CH(CH3)-C3H7, -CH2-CH(CH3)-C2H5, -CH(CH3)-CH(CH3)2, -C(CH3)2-C2H5, -CH2-C(CH3)3, -C3H6-CH=CH2, -CH=CH-C3H7, -C2H4-CH=CH-CH3, -CH2-CH=CH-C2H5, -CH2-CH=CH-CH=CH2, -CH=CH-CH=CH-CH3, -CH=CH-CH2-CH=CH2, -C(CH3)=CH-CH=CH2, -CH=C(CH3)-CH=CH2, -CH=CH-C(CH3)=CH2, -CH2-CH=C(CH3)2, C(CH3)=C(CH3)2, -C3H6-C≡CH, -C≡C-C3H7, -C2H4-C≡C-CH3, -CH2-C≡C-C2H5, -CH2-C≡C-CH=CH2, -CH2-CH=CH-C≡CH, -CH2-C≡C-C≡CH, -C≡C-CH=CH-CH3, -CH=CH-C≡C-CH3, -C≡C-C≡C-CH3, -C≡C-CH2-CH=CH2, -CH=CH-CH2-C≡CH, -C≡C-CH2-C≡CH, -C(CH3)=CH-CH=CH2, -CH=C(CH3)-CH=CH2, -CH=CH-C(CH3)=CH2, -C(CH3)=CH-C≡CH, -CH=C(CH3)-C≡CH, -C≡C-C(CH3)=CH2, -C3H6-CH(CH3)2, -C2H4-CH(CH3)-C2H5, -CH(CH3)-C4H9, -CH2-CH(CH3)-C3H7, -CH(CH3)-CH2-CH(CH3)2, -CH(CH3)-CH(CH3)-C2H5, -CH2-CH(CH3)-CH(CH3)2, -CH2-C(CH3)2-C2H5, -C(CH3)2-C3H7, -C(CH3)2-CH(CH3)2, -C2H4-C(CH3)3, -CH(CH3)-C(CH3)3, -C4H8-CH=CH2, -CH=CH-C4H9, -C3H6-CH=CH-CH3, -CH2-CH=CH-C3H7, -C2H4-CH=CH-C2H5, -CH2-C(CH3)=C(CH3)2, -C2H4-CH=C(CH3)2, -C4H8-C≡CH, -C≡C-C4H9, -C3H6-C≡C-CH3, -CH2-C≡C-C3H7 и -C2H4-C≡C-C2H5, еще более предпочтительно - из метила, этила, н-пропила, изопропила, н-бутила, изобутила, втор-бутила и трет-бутила, и еще более предпочтительно - из метила, этила, н-пропила и изопропила, и еще более предпочтительно - из метила и этила. В одном варианте осуществления, термин "алкил" относится только к алканильным группам (т.е., исключая алкенильные и алкинильные группы), в частности к указанным выше алканильным группам, т.е. -CH3, -C2H5, -C3H7 и т.д.). Все перечисленные выше алкильные группы, если не указано иное, необязательно замещены в соответствии с приведенными в вариантах осуществления настоящего изобретения подробностями, т.е. один или больше атомов водорода необязательно заменены на заместитель, указанный в соответствующем варианте осуществления. Особенно предпочтительно, алкильные группы являются незамещенными, если не указано иное.
Частной формой алкильной группы является галогеналкильная группа, которая представляет собой описанную выше алкильную группу, в которой один или больше, предпочтительно по меньшей мере половина, более предпочтительно все атомы водорода в углеводородной цепочке заменены на атомы галогена. Галогеналкильная группа предпочтительно выбрана из группы, состоящей из -C(R7)3, -CH2-C(R7)3, -C(R7)2-CH3, -C(R7)2-C(R7)3, -C(R7)2-CH(R7)2, -CH2-CH(R7)2, -CH(R7)-C(R7)3, -CH(R7)-CH3, и -C2H4-C(R7)3, более предпочтительно из -C(R7)3, где R7 представляет собой галоген, в частности F. Более предпочтительно, галогеналкил выбран из группы, состоящей из -CF3, -CHF2, -CH2CF3 и -CF2Cl, еще более предпочтительно галогеналкил представляет собой -CF3.
Термин “алкинил” означает алкильную группу, содержащую по меньшей мере два атома углерода и углерод-углеродную тройную связь. Замещенный алкинил соответствует данным выше определениям. Термин “алкенил” означает алкильную группу, содержащую по меньшей мере два атома углерода и углерод-углеродную двойную связь.
При использовании в настоящем тексте, «гетероарильная» группа означает ароматическую моно- или бициклическую кольцевую систему, в которой один или больше атомов углерода заменены гетероатомами, независимо выбранными из группы, состоящей из O, N и S, где, в случае моноциклического гетероарила, указанный моноциклический гетероарил может быть опционально сконденсирован с циклоалкильным или гетероциклоалкильным кольцом, и где общее число атомов в цикле гетероарильной группы составляет от пяти до десяти, более предпочтительно пять или шесть. Точка присоединения гетероарильной группы к остальной части молекулы может быть расположена в моно- или бициклической углеводородной кольцевой системе или в опционально приконденсированном циклоалкильном или гетероциклоалкильном кольце. Примерами гетероарильной группы являются тиадиазол, тиазол-2-ил, тиазол-4-ил, тиазол-5-ил, изотиазол-3-ил, изотиазол-4-ил, изотиазол-5-ил, оксазол-2-ил, оксазол-4-ил, оксазол-5-ил, изоксазол-3-ил, изоксазол-4-ил, изоксазол-5-ил, 1,2,4-оксадиазол-3-ил, 1,2,4-оксадиазол-5-ил, 1,2,5-оксадиазол-3-ил, бензоксазол-2-ил, бензоксазол-4-ил, бензоксазол-5-ил, бензизоксазол-3-ил, бензизоксазол-4-ил, бензизоксазол-5-ил, 1,2,5-оксадиазол-4-ил, 1,3,4-оксадиазол-2-ил, 1,2,4-тиадиазол-3-ил, 1,2,4-тиадиазол-5-ил, 1,3,4-тиадиазол-2-ил, изотиазол-3-ил, изотиазол-4-ил, изотиазол-5-ил, бензизотиазол-3-ил, бензизотиазол-4-ил, бензизотиазол-5-ил, 1,2,5-тиадиазол-3-ил, 1-имидазолил, 2-имидазолил, 1,2,5-тиадиазол-4-ил, 4-имидазолил, бензимидазол-4-ил, 1-пирролил, 2-пирролил, 3-пирролил, 2-фуранил, 3-фуранил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиранил, 3-пиранил, 4-пиранил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, пирид-5-ил, пирид-6-ил, 3-пиридазинил, 4-пиридазинил, 2-пиразинил, 1-пиразолил, 3-пиразолил, 4-пиразолил, 1,2,3-триазол-4-ил, 1,2,3-триазол-5-ил, 1,2,4-триазол-3-ил, 1,2,4-триазол-5-ил, 1H-тетразол-2-ил, 1H-тетразол-3-ил, тетразолил, акридил, феназинил, карбазолил, феноксазинил, индолизин, 2-индолил, 3-индолил, 4-индолил, 5-индолил, 6-индолил, 7-индолил, 1-изоиндолил, 3-изоиндолил, 4-изоиндолил, 5-изоиндолил, 6-изоиндолил, 7-изоиндолил, 2-индолинил, 3-индолинил, 4-индолинил, 5-индолинил, 6-индолинил, 7-индолинил, бензо[b]фуранил, бензофуразан, бензотиофуразан, бензотриазол-1-ил, бензотриазол-4-ил, бензотриазол-5-ил, бензотриазол-6-ил, бензотриазол-7-ил, бензотриазин, бензо[b]тиофенил, бензимидазолил, бензотиазолил, хиназолинил, хиноксазолинил, циннолин, хинолинил, тетрагидрохинолинил, изохинолинил, тетрагидроизохинолинил, пурин, фталазин, птеридин, тиатетраазаинден, тиатриазаинден, изотиазолoпиразин, 6-пиримидинил, 2,4-диметокси-6-пиримидинил, бензимидазол-2-ил, 1H-бензимидазолил, бензимидазол-4-ил, бензимидазол-5-ил, бензимидазол-6-ил, бензимидазол-7-ил, тетразол, тетрагидро-тиено[3,4-d]имидазол-2-он, пиразоло[5,1-c][1,2,4]триазин, изотиазолoпиримидин, пиразолотриазин, пиразолопиримидин, имидазопиридазин, имидазопиримидин, имидазопиридин, имидазолoтриазин, триазолoтриазин, триазолoпиридин, триазолoпиразин, триазолoпиримидин или триазолoпиридазин. Все указанные выше гетероарильные группы, если не указано иное, необязательно замещены в соответствии с приведенными в вариантах осуществления настоящего изобретения подробностями, т.е. один или больше атомов водорода необязательно заменены на заместитель, указанный в соответствующем варианте осуществления. Особенно предпочтительно, гетероарильные группы являются незамещенными если не указано иное.
При использовании в настоящем тексте, “циклоалкильная» группа означает неароматическую моно- или бициклическую кольцевую систему, в которой одно из колец представляет собой фенильное кольцо, например 1,2,3,4-тетрагидронафталин. Указанный циклоалкил предпочтительно является моноциклическим. Указанный циклоалкил предпочтительно является полностью насыщенным. Указанный циклоалкил содержит от 3 до 10 атомов углерода, более предпочтительно от 5 до 7 атомов углерода. Еще более предпочтительно, циклоалкил выбран из группы, состоящей из циклопропила, циклобутила, циклопентила, циклогексила, циклогептила, 1-норборнила, 2-норборнила, 7-норборнила, 1-адамантила, 2-адамантила, 1,2-дигидронафтила, 1,2,3,4-тетрагидронафтила, 2,3-дигидроинденила, 1,6-дигидропенталенила, 1,6a-дигидропенталенила, и еще более предпочтительно циклоалкил выбран из группы, состоящей из циклопропила, циклопентила, циклогексила и адамантила. Все указанные выше циклоалкильные группы, если не указано иное, необязательно замещены в соответствии с приведенными в вариантах осуществления настоящего изобретения подробностями, т.е. один или больше атомов водорода необязательно заменены на заместитель, указанный в соответствующем варианте осуществления. Особенно предпочтительно, циклоалкильные группы являются незамещенными, если не указано иное.
При использовании в настоящем тексте, «гетероциклоалкильная» группа означает неароматическую моно- или бициклическую полностью насыщенную или частично ненасыщенную углеводородную кольцевую систему, в которой один или больше атомов углерода заменены гетероатомом, независимо выбранным из группы, состоящей из N, O и S. Гетероциклоалкил предпочтительно не содержит ароматических колец. Гетероциклоалкил предпочтительно является моноциклическим. Гетероциклоалкил предпочтительно является полностью насыщенным. Гетероциклоалкил предпочтительно содержит в сумме от 4 до 10 атомов в цикле, более предпочтительно - в сумме от 5 до 10 атомов в цикле, еще более предпочтительно в сумме от 5 до 7 атомов в цикле, и еще более предпочтительно в сумме 5 или 6 атомов в цикле. Еще более предпочтительно гетероциклоалкил выбран из группы, состоящей из морфолинила, пиперидинила, диоксанила, пиперазинила, тиоморфолинила, пиперидинила, пирролидинила, тетрагидрофуранила, изоксазолидинила, тиоморфолинила, тетрагидротиофуранила и тетрагидропиранила, более предпочтительно - выбран из группы, состоящей из морфолинила, пиперидинила, диоксанила, пиперазинила, тиоморфолинила, пиперидинила и пирролидинила. Все указанные выше гетероциклоалкильные группы, если не указано иное, необязательно замещены в соответствии с приведенными в вариантах осуществления настоящего изобретения подробностями, т.е. один или больше атомов водорода необязательно заменены на заместитель, указанный в соответствующем варианте осуществления. Особенно предпочтительно, гетероциклоалкильные группы являются незамещенными, если не указано иное.
При использовании в настоящем тексте, «галоген» предпочтительно означает фтор, хлор, бром или иод, в особенности хлор
При использовании в настоящем тексте, «алкокси-группа» означает O-алкильную группу, где алкильная группа соответствует данному выше определению. Алкокси-группа в частности выбрана из группы, состоящей из метокси-, этокси- и пропокси-группы, более предпочтительно из метокси-группы. Указанные выше алкокси-группы необязательно замещены одним или больше атомами галогена, в частности одним или больше атомами фтора.
При использовании в настоящем тексте, «алкилтио-группа» означает -S-алкильную группу, где алкильная группа соответствует данному выше определению, в частности метилтио-группу. Указанные выше алкилтио-группы необязательно замещены одним или больше атомами галогена, в частности одним или больше атомами фтора.
При использовании в настоящем тексте, «алкоксиалкильная» группа означает алкильную группу, замещенную O-алкильной группой, где алкильная группа соответствует указанному выше определению, в частности выбрана из группы, состоящей из метоксиэтила, этоксиметила, метоксиметила, пропоксиметила и метоксипропила, более предпочтительно метоксиэтил. Указанные алкоксиалкильные группы необязательно замещены одним или больше атомами галогена, в частности одним или больше атомами фтора.
При использовании в настоящем тексте, «алкилтиоалкильная» группа означает алкильную группу, замещенную S-алкильной группой, где алкильная группа соответствует указанному выше определению, в частности выбрана из группы, состоящей из метилтиоэтила, этилтиометила, метилтиометила, пропилтиометила и метилтиопропила, более предпочтительно метилтиоэтил. Указанные алкилтиоалкильные группы необязательно замещены одним или больше атомами галогена, в частности одним или больше атомами фтора.
При использовании в настоящем тексте, «алкиламиноалкильная» группа означает алкильную группу, связанную с NH-алкильной группой или N-диалкильной группой, где алкильная группа соответствует указанному выше определению, в частности выбрана из группы, состоящей из метиламиноэтила, этиламинометила, метиламинометила, пропиламинометила и метиламинопропила, более предпочтительно этиламинометил. Указанные алкиламиноалкильные группы необязательно замещены одним или больше атомами галогена, в частности одним или больше атомами фтора.
При использовании в настоящем тексте, «арильная» группа означает ароматическую моно- или бициклическую углеводородную кольцевую систему, в которой общее число атомов в цикле составляет от шести до десяти, в частности шесть. Примерами арильной группы являются фенил и нафтил, более предпочтительно фенил. Все указанные выше арильные группы, если не указано иное, необязательно замещены в соответствии с приведенными в вариантах осуществления настоящего изобретения подробностями, т.е. один или больше атомов водорода необязательно заменены на заместитель, указанный в соответствующем варианте осуществления. Особенно предпочтительно, арильные группы являются незамещенными, если не указано иное.
«Арилалкильная» группа, также широко известная как аралкильная группа, означает линейный или разветвленный алкил, определение которому дано в настоящем тексте, замещенный арильной группой, определение которой приведено в настоящем тексте. Примеры арилалкильных групп включают стирил, бензил, фенилэтил, 1-(нафталин-2-ил)этил, в частности арилалкильная группа представляет собой стирил или бензил, более предпочтительно бензил. Указанная арилалкильная группа может быть замещенной, в частности в своей ароматической части, аналогично описанному выше для арильной группы.
Следует понимать, что определения терминов “алкил”, “арил”, “арилалкил”, “гетероциклоалкил”, “циклоалкил”, “гетероарил”, “алкокси”, “алкилтио”, “алкоксиалкил”, “алкилтиоалкил”, “алкиламиноалкил” и т.п., относятся также, в случае применимости, к частным представителям этих групп, конкретизированным в вариантах осуществления настоящего изобретения. Например, определение термина “алкил” относится также, в случае применимости, к таким представителям этих групп, как “C1-6-алкил”, “C1-4-алкил”, “C1-2-алкил”, метил, этил и т.п.. Это означает, например, что указание того, что термин “алкил” охватывает “алканил”, “алкенил” и “алкинил”, относится также к “C1-2-алкилу”, который в свою очередь охватывает метил, этил, этенил и этинил.
Атом азота (N), при упоминании, например, в контексте терминов “гетероарил”, “гетероциклоалкил” и “гетероцикл”, может включать N-оксид, в частности где это химически допустимо с точки зрения стабильности и/или соблюдения валентности.
Атом серы (S), при упоминании, например, в контексте терминов “гетероарил”, “гетероциклоалкил” и “гетероцикл”, может включать S(O) и/или S(O)2-производные, в частности где это химически допустимо с точки зрения стабильности и/или соблюдения валентности.
При использовании в настоящем тексте, термин “замещенный” означает, что один или больше атомов водорода, связанных с атомом углерода или гетероатомом в химической группе или веществе, заменены на группу-заместитель; например, замещенный арил включает 4-гидроксифенил, где H-атом фенильной группы в положении 4 заменен на гидроксильную группу. Замещаемый(-ые) атом(ы) водорода могут быть присоединены к атому углерода или гетероатому и могут быть в явном виде изображены в формуле, например в -NH- группе, или могут быть не изображены в явном виде, но присутствовать, например как в типичной «цепочечной» записи, которая широко применяется для изображения, например, углеводородов. Квалифицированному специалисту будет легко понятно, что исключены такие заместители или точки замещения, которые приводят к соединениям нестабильным и/или недостижимым известными к настоящему моменту способами синтеза. Конкретная группа-заместитель может быть выбрана из группы, состоящей из C1-4-алкила, -OH, атома галогена, -CO-N(Ri)2, -N(Ri)2, -CO-Ri, -COO-Ri, -N3, -NO2 и -CN, где каждый Ri независимо выбран из группы, состоящей из H и C1-4-алкила.
Если не указано иное, упоминание соединений по настоящему изобретению включает их физиологически функциональные производные, сольваты или соли, описанные в настоящем тексте, а также соли указанных физиологически функциональных производных, сольваты солей и физиологически функциональных производных, и опционально сольваты солей физиологически функциональных производных.
При использовании в настоящем тексте, термин “физиологически функциональное производное” соединения по настоящему изобретению означает, например, пролекарство указанного соединения, где по меньшей мере одна из следующих групп дериватизована как указано далее: карбоксильная (-COOH) группа превращена в сложный эфир (имеющий, например, формулу -COOR8, где R8 выбран из группы, состоящей из -H, алкила (такого как C1-6-алкил), алкокси-группы (такой как C1-6-алкокси-группа), алкоксиалкила (такого как C1-3-алкокси-C1-3-алкил), алкилтио-группы (такой как C1-6-алкилтио-группа), алкилтиоалкила (такого как C1-3-алкилтио-C1-3-алкил), алкиламиноалкила (такого как C1-3-алкиламино-C1-3-алкил), арила (такого как C6-10-арил), гетероциклоалкила (такого как 4-10-членный гетероциклоалкил), циклоалкила (такого как C3-10-циклоалкил) и гетероарила (такого как 5-10-членный гетероарил), где указанный алкил, алкокси-группа, алкоксиалкил, алкилтио-группа, алкилтиоалкил, алкиламиноалкил, арил, гетероциклоалкил, циклоалкил и гетероарил необязательно замещены одной или больше группами, независимо выбранными из группы, состоящей из C1-4-алкила, -OH, атома галогена, -CO-N(R9)2, -N(R9)2, -CO-R9, -COO-R9, -N3, -NO2 и -CN, где каждый R9 независимо выбран из группы, состоящей из H и C1-4-алкила); гидроксильная (-OH) группа превращена в сложный эфир (имеющий, например, формулу -COOR8, как описано выше); карбоксильная группа превращена в амид (имеющий, например, формулу -CONH-R8, где R8 соответствует данному выше определению); амин (-NH2) превращен в амид (имеющий, например, формулу -CONH-R8, где R8 соответствует данному выше определению); и гидроксильная группа превращена в фосфатный эфир (имеющий, например, формулу -OP(O)(OR10)2, где R10 каждый независимо выбран из группы, состоящей из H и C1-4-алкила).
Соединения по настоящему изобретению следует понимать как включающие все их таутомерные формы, даже если они не показаны в явном виде в представленных в настоящем тексте формулах, включая формулу (I).
Описанные в настоящем тексте соединения, имеющие формулу (I), следует понимать как охватывающие, где это применимо, все стереоизомеры указанных соединений, если не указано иное. Термин “стереоизомер” при использовании в настоящем тексте означает соединение, содержащее по меньшей мере один стереогенный центр, который может иметь R- или S-конфигурацию согласно правилам ИЮПАК, и охватывает энантиомеры и диастереомеры, как хорошо понятно квалифицированным специалистам в данной области. Следует понимать, что у соединений, содержащих более одного стереогенного центра, каждый из индивидуальных стереогенных центров может независимо от других иметь R- или S-конфигурацию. Термин “стереоизомер” при использовании в настоящем тексте относится также к солям описанных в настоящем тексте соединений с оптически активными кислотами или основаниями. Настоящее изобретение включает также все смеси указанных выше стереоизмеров, независимо от их соотношений, включая рацематы.
В настоящем изобретении, соли соединений по настоящему изобретению предпочтительно представляют собой фармацевтически приемлемые соли соединений по настоящему изобретению. Фармацевтически приемлемые соли - это соли, которые в целом считаются квалифицированными специалистами в данной области подходящими для применения в медицине, например потому, что они не наносят вреда субъектам, которым могут вводить такие соли, или дают побочные эффекты, приемлемые при соответствующем способе введения. Обычно такими фармацевтически приемлемыми солями являются соли, которые считают приемлемыми регулирующие государственные органы, такие как Комиссия США по контролю за лекарствами и продуктами питания (FDA), Европейское агентство лекарственных средств (EMA) или Министерство здравоохранения Японии, Агентство фармацевтики и медицинского оборудования (PMDA). Однако, настоящее изобретение в принципе охватывает также соли соединений по настоящему изобретению, которые не являются фармацевтически приемлемыми, например в качестве интермедиатов при получении соединений по настоящему изобретению или их физиологически функциональных производных, или в качестве интермедиатов при получении фармацевтически приемлемых солей соединений по настоящему изобретению или их физиологически функциональных производных. Такие соли включают нерастворимые в воде соли и, в особенности, водорастворимые соли.
В каждом случае квалифицированный специалист может легко определить, может ли конкретное соединение по настоящему изобретению или его физиологически функциональное производное образовывать соль, т.е. содержит ли соединение по настоящему изобретению или его физиологически функциональное производное группу, которая может нести заряд, такую как, например, амино-группа, карбоксильная группа и т.д.
Примерами солей соединений по настоящему изобретению являются соли с кислотами или соли с основаниями, в особенности с фармацевтически приемлемыми неорганическими и органическими кислотами и основаниями, широко применяемыми в фармацевтике, которые являются либо нерастворимыми в воде, либо, предпочтительно, водорастворимыми солями с кислотами. Соли с основаниями могут, в зависимости от заместителей в соединениях по настоящему изобретению, также быть подходящими. Соли с кислотами можно, например, получить путем смешивания раствора соединения по настоящему изобретению с раствором фармацевтически приемлемой кислоты, такой как соляная кислота, серная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, уксусная кислота, бензойная кислота, лимонная кислота, винная кислота, угольная кислота или фосфорная кислота. Аналогично, фармацевтически приемлемые соли с основаниями могут включать соли щелочных металлов (например, соли натрия или калия); соли щелочно-земельных металлов (например, соли кальция или магния); и соли с подходящими органическими лигандами (например, аммониевые, четвертичные аммониевые и аминные катионы, полученные с применением таких противоионов, как галогенид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, алкилсульфонат и арилсульфонат). Иллюстративные примеры фармацевтически приемлемых солей включают (но не ограничиваются только ими) ацетат, адипат, альгинат, аргинат, аскорбат, аспартат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, бутират, кальция эдетат, камфорат, камфорсульфонат, камсилат, карбонат, хлорид, цитрат, диглюконат, дигидрохлорид, додецилсульфат, эдетат, эдисилат, этансульфонат, формиат, фумарат, галактат, галактуронат, глюконат, глутамат, глицерофосфат, гемисульфат, гептаноат, гексаноат, гексилрезорцинат, гидробромид, гидрохлорид, гидроиодид, 2-гидрокси-этансульфонат, гидроксинафтоат, иодид, изобутират, изотионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, манделат, метансульфонат (мезилат), метилсульфат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, пантотенат, пектинат, персульфат, 3-фенилпропионат, фосфат/дифосфат, фталат, пикрат, пивалат, полигалактуронат, пропионат, салицилат, стеарат, сульфат, суберат, сукцинат, таннат, тартрат, тозилат, ундеканоат, валерат и т.п. (см, например, S. M. Berge et al., "Pharmaceutical Salts", J. Pharm. Sci., 66, pp. 1-19 (1977)).
Соли, которые не являются фармацевтически приемлемыми и которые могут быть получены, например, в качестве промежуточных продуктов при получении соединений по настоящему изобретению в промышленном масштабе, также охватываются настоящим изобретением и, при необходимости, могут быть превращены в фармацевтически приемлемые соли по методикам, известным квалифицированным специалистам в данной области.
Как известно специалистам в данной области, соединения по настоящему изобретению, а также их соли, могут содержать, например после выделения в кристаллическом виде, различные количества растворителей. Поэтому в объем настоящего изобретения включены сольваты и, в особенности, гидраты соединений по настоящему изобретению, а также сольваты, и в особенности гидраты, солей и/или физиологически функциональных производных соединений по настоящему изобретению. Более предпочтительно, настоящее изобретение охватывает гидраты соединений, солей и/или физиологически функциональных производных по настоящему изобретению, содержащие стехиометрически одну, две или половину молекулы воды.
При использовании в настоящем тексте, термин “комнатная температура”, “rt” или “r.t.” означает температуру от 20 до 25°C, в частности около 22°C, если не указано иное.
При использовании в настоящем тексте, термин “стабильное” обозначает соединение, в котором химическая структура не изменяется, когда соединение хранится при температуре примерно от -80°C до +40°C, в частности примерно от -80°C до +25°C, в отсутствие света, влаги или других химически агрессивных факторов, по меньшей мере в течение одной недели, предпочтительно по меньшей мере одного месяца, более предпочтительно по меньшей мере шести месяцев, еще более предпочтительно по меньшей мере одного года, и/или соединение, которое в стандартных условиях по ИЮПАК и в отсутствие света, влаги или других химически агрессивных факторов сохраняет свою структуру достаточно долго для того, чтобы его можно было использовать для терапевтического или профилактического введения пациентам, т.е. по меньшей мере в течение одной недели. Соединения, не обладающие стабильностью в описанных выше терминах, не охватываются настоящим изобретением. В частности, соединения, которые в стандартных условиях по ИЮПАК самопроизвольно разлагаются менее чем за один день, считаются нестабильными соединениями. Квалифицированный специалист в данной области на основе общих представлений легко может понять, какие соединения и какие заместители соответствуют стабильным соединениям.
Соединение, в частности соединение, имеющее формулу (I), является селективным в отношении выбранной мишени (в частности, TLR7), если оно способно связываться и проявлять активность (в частности, агонистическую активность) в отношении выбранной мишени, при этом не связываясь и не проявляя активность (в частности, агонистическую и антагонистическую активность) в отношении других мишеней, т.е. не демонстрирует заметной агонистической активности в отношении других мишеней в стандартных тестах. Согласно настоящему изобретению, соединение, имеющее формулу (I), является селективным в отношении TLR7 , если оно проявляет агонистическую активность в отношении TLR7, но не способно (в значительной степени) проявлять агонистическую активность в отношении других мишеней, в частности TLR8. Предпочтительно, соединение, в частности соединение, имеющее формулу (I), является селективным в отношении TLR7, если его агонистическая активность в отношении других мишеней (в частности TLR8) не превосходит существенно его агонистическую активность в отношении не-TLR белков, таких как LDL рецептор, инсулиновый рецептор или трансферриновый рецептор, или любого другого специфичного полипептида. Предпочтительно, соединение, в частности соединение, имеющее формулу (I), является селективным в отношении выбранной мишени (в частности TLR7), если его агонистическая активность (значение EC50) в отношении указанной мишени по меньшей мере 2-кратно, 3-кратно, 4-кратно, 5-кратно, 10-кратно, 15-кратно, 20-кратно, 25-кратно, 30-кратно, 35-кратно, 40-кратно, 45-кратно, 50-кратно, 60-кратно, 70-кратно, 80-кратно, 90-кратно, 100-кратно или 103-кратно ниже, чем его агонистическая активность в отношении мишени, в отношении которой оно не является селективным (в частности, TLR8). Например, если EC50 для соединения, в частности EC50 для соединения, имеющего формулу (I), в отношении мишени, для которой это соединение является селективным, составляет 1 мкМ, то значение EC50 для мишени, в отношении которой данное соединение не является селективным, должно составлять по меньшей мере 2 мкМ, 3 мкМ, 4 мкМ, 5 мкМ, 10 мкМ, 15 мкМ, 20 мкМ, 25 мкМ, 30 мкМ, 35 мкМ, 40 мкМ, 45 мкМ, 50 мкМ, 60 мкМ, 70 мкМ, 80 мкМ, 90 мкМ, 100 мкМ или 1 мM.
Следует понимать, что настоящее изобретение включает все комбинации указанных выше групп-заместителей. В частности, настоящее изобретение включает все комбинации перечисленных выше групп-заместителей.
Соединения по настоящему изобретению и их соли, содержащие двойную связь, могут существовать в виде Е-изомеров и Z-изомеров. Оба указанные виды изомеров включены в настоящее изобретение. Z-изомер представляет собой геометрический изомер, в котором атомы углерода, соединенные двойной связью, имеют две главные по старшинству группы по одну сторону от двойной связи. Е-изомер представляет собой геометрический изомер, в котором атомы углерода, соединенные двойной связью, имеют две главные по старшинству группы по разные стороны от двойной связи.
Некоторые из соединений и солей по настоящему изобретению могут существовать в разных кристаллических формах (полиморфы), которые все включены в объем настоящего изобретения.
Далее по тексту термин “соединение”, если иное не указано в явном виде, следует понимать как охватывающий описанные в настоящем тексте их физиологически функциональные производные, сольваты и соли.
При использовании в настоящем тексте, термин “лечение” включает полное или частичное излечивание заболевания, профилактику заболевания, облегчение степени тяжести заболевания или остановку развития определенного заболевания.
Термины “медицинское состояние”, “болезнь” и “нарушение” используются в настоящем тексте взаимозаменяемо и означают пролиферативные патологические состояния, включая пролиферативные заболевания, такие как рак, в частности такие патологические состояния (включая разные формы рака), которые описаны в настоящем тексте. Предпочтительно заболевание отличается тем, что его можно лечить посредством агонизации TLR7.
При использовании в настоящем тексте, термин “пролиферативное заболевание” включает заболевание, отличающееся нарушением регуляции роста, пролиферации, дифференцировки, адгезии и/или миграции клеток. Частным примером пролиферативных заболеваний является рак. Термин “раковая клетка” означает аномальную клетку, которая растет в ходе быстрой неконтролируемой пролиферации и продолжает расти после того, как исчезает стимул, инициировавший изначальный рост.
При использовании в настоящем тексте, термин “лекарственное средство” включает описанные в настоящем тексте соединения, имеющие формулу (I), их фармацевтически приемлемые соли или физиологически функциональные производные, которые вводят субъекту в чистом виде, а также композиции, содержащие по меньшей мере одно соединение по настоящему изобретению, его фармацевтически приемлемую соль или физиологически функциональное производное, которые подходят для введения субъекту.
Соединения по настоящему изобретению и их фармацевтически приемлемые соли и физиологически функциональные производные можно вводить животным, в частности млекопитающим, и в особенности человеку, в качестве терапевтических средств в чистом виде, в виде смесей или, в особенности, в форме фармацевтических препаратов или композиций, которые делают возможным энтеральное (например, пероральное) или парентеральное введение, и которые содержат в качестве действующего вещества терапевтически эффективное количество по меньшей мере одного соединения по настоящему изобретению или его соли или физиологически функционального производного, и в дополнение - например, один или больше компонентов, выбранных из группы, состоящей из обычно применяющихся адъювантов, фармацевтически приемлемых эксципиентов, носителей, буферных добавок, разбавителей, растворителей, диспергаторов, эмульгаторов, солюбилизаторов, гелеобразователей, мазевых основ, антиоксидантов, консервантов, стабилизаторов, наполнителей, связующих веществ, загустителей, комплексообразователей, разрыхлителей, усилителей проникновения, полимеров, лубрикантов, покрывающих агентов, пропеллентов, регуляторов тоничности, поверхностно-активных веществ, красителей, отдушек, подсластителей, красителей и/или других общеизвестных фармацевтических вспомогательных веществ.
Фармацевтические композиции, медицинское применение и способы лечения по настоящему изобретению могут включать более одного соединения по настоящему изобретению.
Фармацевтические композиции, содержащие соединение по настоящему изобретению, или его фармацевтически приемлемую соль или физиологически функциональное производное, могут дополнительно содержать одно или больше дополнительных терапевтически активных веществ, которые не являются соединениями, имеющими формулу (I) по настоящему изобретению. При использовании в настоящем тексте, термин “терапевтически активное вещество” означает вещество, которое при введении может вызывать медицинский эффект у пациента. Такой медицинский эффект может включать медицинский эффект, описанный для соединений, имеющих формулу (I) по настоящему изобретению, но может также, в случае терапевтически активных веществ, которые вводятся совместно с соединениями по настоящему изобретению, включать другие медицинские субстанции, такие как, например (но не ограничиваясь только ими) иринотекан, оксалиплатин, гемцитабин, капецитабин, 5-фторурацил, цетуксимаб (Erbitux), панитумумаб (Vectibix), бевацизумаб (Avastin), винкристин, винбластин, винорелбин, виндезин, таксол, амсакрин, этопозид, этопозид фосфат, тенипозид, актиномицин, антрациклины, доксорубицин, валрубицин, идарубицин, эпирубицин, блеомицин, пликамицин, митомицин, мехлорэтамин, циклофосфамид, хлорамбуцил, ифосфамид, бортезомиб, иматиниб, афатиниб, акситиниб, босутиниб, кобиметиниб, дасатиниб, эрлотиниб, лапатиниб, ленватиниб, пазопаниб, сорфениб, сунитиниб, вемурафениб и другие ингибиторы киназ, вориностат, панобиностат, белиностат и другие ингибиторы гистон деацетилазы.
Термин “фармацевтически приемлемая” хорошо известен квалифицированным специалистам и означает, что соответствующее соединение не является вредным для субъекта, которому вводят указанное соединение или композицию, содержащую данное соединение, что указанное соединение является стабильным и что данное соединение химически совместимо (т.е. не реагирует) с другими ингредиентами соответствующей фармацевтической композиции.
Лекарственные средства и фармацевтические композиции по настоящему изобретению, содержащие по меньшей мере одно соединение по настоящему изобретению или его фармацевтически приемлемую соль или физиологически функциональное производное, включают таковые, подходящие для перорального, ректального, бронхиального, назального, наружного, буккального, сублингвального, вагинального или парентерального (включая чрезкожное, внутрикожное, подкожное, внутримышечное, внутрилегочное, внутрисосудистое, внутричерепное, интраперитонеальное, внутривенное, внутриартериальное, интрацеребральное, внутриглазное, внутригрудинное, внутрикоронарное, трансуретральное инъецирование или инфузию) введение, или таковые в форме, подходящей для введения посредством вдыхания или вдувания, включая введение порошков и жидких аэрозолей, или посредством систем с контролируемым высвобождением (например, замедленного высвобождения, рН-контролируемого высвобождения, отложенного высвобождения, повторяющегося высвобождения, пролонгированного высвобождения). Подходящие примеры систем с контролируемым высвобождением включают полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие соединение по настоящему изобретению, при этом указанные матрицы могут иметь вид формованных изделий, например пленок или микрокапсул, или коллоидные носители лекарственных средств, например полимерные наночастицы, или твердые дозированные формы с контролируемым высвобождением, например таблетки с ядром или многослойные таблетки. Частным способом введения по настоящему изобретению является внутривенное введение.
Соединения по настоящему изобретению можно, в частности, вводить в состав препаратов для парентерального введения (например, посредством инъекции, например болюсной инъекции, или непрерывного вливания), и их можно выпускать в дозированной форме для однократного применения в ампулах, предзаполненных шприцах, инфузиях малого объема, или в контейнерах на несколько доз с добавлением консерванта. Композиции в таких формах могут иметь вид суспензий, растворов или эмульсий в масляных или водных носителях и могут содержать вспомогательные вещества, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, действующее вещество может иметь вид порошка, полученного асептическим выделением стерильного твердого продукта или лиофилизацией из раствора, для разбавления подходящим носителем, например стерильной апирогенной водой, непосредственно перед применением.
Можно также применять любые из других общеупотребимых дозированных форм, таких как таблетки, таблетки для рассасывания, парентеральные препараты, сиропы, кремы, мази, аэрозольные препараты, чрезкожные пластыри, чрезслизистые пластыри и т.п.. Фармацевтические композиции по настоящему изобретению можно, например, вводить в состав таблеток, таблеток с покрытием (драже), пилюль, саше, капсул (каплетов), гранул, порошков, суппозиториев, растворов (например, стерильных растворов), эмульсий, суспензий, мазей, кремов, лосьонов, паст, масел, гелей, спреев и пластырей (например, чрезкожных терапевтических систем). Кроме того, фармацевтические композиции можно вводить в состав, например, липосомных систем доставки, систем, в которых действующее вещество скомбинировано с моноклональными антителами, и систем, в которых действующее вещество скомбинировано с полимерами (например, растворимыми или биоразлагаемыми полимерами).
Таблетки, таблетки с покрытием (драже), пилюли, саше, капсулы (каплеты), гранулы, растворы, эмульсии и суспензии являются, например, подходящими для перорального введения. В частности, указанные препараты можно адаптировать таким образом, чтобы они представляли собой форму для приема внутрь, форму с немедленным высвобождением, форму с замедленным высвобождением, форму с повторяющимся высвобождением, форму с пролонгированным высвобождением или форму с длительным высвобождением. Указанные формы можно приготовить, например, посредством нанесения покрытия на таблетки, путем разделения таблеток на несколько отделений, разделенных слоями, разрушающимися в разных условиях (например, при разном уровне рН), или посредством связывания действующего вещества с биоразлагаемым полимером.
Ингаляционное введение в частности осуществляют с применением аэрозоля. Аэрозоль представляет собой жидкостно-газовую дисперсию, твердо-газовую дисперсию или смешанную дисперсию жидкости/твердого вещества в газе.
Размер частиц аэрозоля (твердых, жидких или твердых/жидких частиц) в частности составляет менее 100 мкм, более предпочтительно - в диапазоне от 0.5 до 10 мкм, еще более предпочтительно - в диапазоне от 2 до 6 мкм (значение ID50, измеренное методом лазерной дифракции).
Аэрозоль можно генерировать посредством аэрозоль-вырабатывающих устройств, таких как порошковый ингалятор, дозирующий аэрозольный ингалятор и небулайзер. В зависимости от вида действующего вещества, которое необходимо ввести, аэрозоль-вырабатывающее устройство может содержать действующее вещество в виде порошка, раствора или дисперсии. Раствор может содержать, помимо растворителя, например, одно или больше из следующих вспомогательных веществ: пропелленты, солюбилизаторы (сорастворители), поверхностно-активные вещества, стабилизаторы, буферные добавки, регуляторы тоничности, консерванты и отдушки. Дисперсия может содержать, помимо диспергатора, например, одно или больше из следующих вспомогательных веществ: пропелленты, поверхностно-активные вещества, стабилизаторы, буферные добавки, консерванты и отдушки. Примеры носителей включают (но не ограничиваются только ими) сахариды, например, лактозу и глюкозу. Примеры пропеллентов включают (но не ограничиваются только ими) фторуглероды, например, 1,1,1,2-тетрафторэтан и 1,1,1,2,3,3,3-гептафторпропан.
Частные примеры аэрозоль-вырабатывающих устройств, которые можно применять для ингаляционного ведения, включают (но не ограничиваются только ими) ингаляторы Cyclohaler®, Diskhaler®, Rotadisk®, Turbohaler®, Autohaler®, Turbohaler®, Novolizer®, Easyhaler®, Aerolizer®, Jethaler®, Diskus®, Ultrahaler® и Mystic®. Аэрозоль-вырабатывающие устройства можно комбинировать с разделителями или расширителями, например, Aerochamber®, Nebulator®, Volumatic® и Rondo®, для повышения эффективности ингаляций.
Приготовление лекарственных средств или фармацевтических композиций, содержащих соединения по настоящему изобретению, и их применение можно осуществлять по методикам, хорошо известным квалифицированным специалистам в области медицины.
Фармацевтически приемлемые носители, используемые в приготовлении фармацевтических композиций или лекарственных средств, содержащих соединение по настоящему изобретению, его фармацевтически приемлемую соль или физиологически функциональное производное, могут быть твердыми или жидкими. Твердые фармацевтические композиции, содержащие соединение по настоящему изобретению, его фармацевтически приемлемую соль или физиологически функциональное производное, включают порошки, таблетки, пилюли, капсулы, саше, суппозитории и диспергируемые гранулы. Твердый носитель может включать один или больше компонентов, которые могут также играть роль разбавителей, отдушек, солюбилизаторов, лубрикантов, суспендирующих агентов, связующих веществ, консервантов, разрыхлителей для таблеток или инкапсулирующего материала.
В порошках носитель представляет собой тонкоизмельченное твердое вещество, которое находится в смеси с тонкоизмельченным действующим веществом. В таблетках действующее вещество смешивают с носителем, имеющим необходимую связующую способность, в нужных пропорциях и прессуют в той форме и размере, которые необходимы. Таблеточную смесь можно гранулировать, просеивать и прессовать, или прессовать напрямую. Подходящими носителями являются карбонат магния, стеарат магния, тальк, сахар, лактоза, пектин, декстрин, крахмал, желатин, трагакантовая камедь, метилцеелюлоза, натрия карбоксиметилцелюлоза, низкоплавкий воск, масло какао и т.п.. Термин "препарат" включает готовый состав из действующего вещества и инкапсулирующего вещества как носителя, дающий капсулу, в которой действующее вещество, с носителями или без них, окружено носителем, который в свою очередь ассоциирован с ним. Схожим образом, включены саше и таблетки для рассасывания. Таблетки, порошки, капсулы, пилюли, саше и таблетки для рассасывания могут применяться в качестве твердых форм, подходящих для перорального введения.
Для приготовления суппозиториев сначала плавят низкоплавкий воск, такой как смесь триглицеридов жирных кислот или масло какао, и в полученном расплаве гомогенно диспергируют действующее вещество, например путем перемешивания. Затем расплавленную гомогенную смесь выливают в формы нужного размера, оставляют охлаждаться и затвердевать. Композиции, подходящие для вагинального введения, могут выпускаться в виде пессариев, тампонов, кремов, гелей, пластырей, пен или спреев, содержащих, помимо действующего вещества, подходящие носители, известные в данной области применения. Жидкие препараты включают растворы, суспензии и эмульсии, например водные или водно-пропиленгликолевые растворы. Например, препараты для парентерального инъекционного введения могут представлять собой растворы в водном растворе полиэтиленгликоля.
Водные растворы, подходящие для перорального введения, можно приготовить путем растворения действующего вещества в воде и добавления, например, подходящих красителей, отдушек, стабилизирующих агентов и загустителей, при необходимости. Водные суспензии, подходящие для перорального введения, можно приготовить путем диспергирования тонкоизмельченного действующего вещества в воде с вязким материалом, таким как природные или синтетические смолы, полимеры, метилцеллюлоза, натрия карбоксиметилцеллюлоза или другие хорошо известные суспендирующие агенты.
Также включены в объем настоящего изобретения твердые препараты, которые необходимо непосредственно перед применением превращать в жидкий препарат для перорального введения. Такие жидкие препараты включают растворы, суспензии и эмульсии. Эти препараты могут содержать, помимо действующего вещества, например, красители, отдушки, стабилизаторы, буферные добавки, искусственные или натуральные подсластители, диспергирующие добавки, загустители, солюбилизаторы и т.п.
В одном варианте осуществления настоящего изобретения, лекарственное средство наносят наружно, например в форме чрезкожных терапевтических систем (например, пластырей) или препаратов для наружного применения (например, липосомы, кремы, мази, лосьоны, гели, дисперсии, суспензии, спрей, раствор, пена, порошок). Это может быть полезно для уменьшения нежелательных побочных эффектов и, где это приемлемо, для ограничения лечебного воздействия до тех областей, где это необходимо.
В частности, лекарственное средство может содержать носители или эксципиенты, включая (но не ограничиваясь только ими) липофильную основу (такую как, например, вазелин, парафины, триглицериды, воска, полиалкилсилоксаны), масла (оливковое масло, арахисовое масло, касторовое масло, триглицеридное масло), эмульгатор (такой как, например, лецитин, фосфатидилглицеролы, алкиловые спирты, лаурилсульфат натрия, полисорбаты, холестерин, сорбитановые сложные эфиры с жирными кислотами, сложные эфиры полиоксиэтилена и жирных кислот, полоксамеры), консерванты (например, бензалкония хлорид, хлорбутанол, парабен или тиомерсал), отдушки, буферные добавки (например, соли уксусной кислоты, лимонной кислоты, борной кислоты, фосфорной кислоты, винной кислоты, трометамол или троламин), растворители (например, полиэтиленгликоли, глицерин, этанол, изопропанол или пропиленгликоль) или солюбилизаторы, агенты для создания депо-эффекта, соли для изменения осмотического давления, носители для пластырей (например, полипропилен, этилен-винилацетат сополимер, полиакрилаты, силикон) или антиоксиданты (например, аскорбат, токоферол, бутилгидроксианизол, сложные эфиры галловой кислоты или бутилгидрокситолуол).
Мази и кремы можно, например, создавать на водной или масляной основе с добавлением подходящих загустителей и/или гелеобразователей. Лосьоны можно создавать на водной или масляной основе, и обычно они содержат также один или больше эмульгаторов, стабилизаторов, диспергирующих агентов, суспендирующих агентов, загустителей или красителей.
Композиции, подходящие для наружного применения в ротовой полости, включают таблетки для рассасывания, содержащие действующее вещество в ароматизированной основе, обычно в сахаре или смоле акации или трагакантовой камеди; пастилки, содержащие действующее вещество в инертной основе, такой как желатин и глицерин или сахароза и смола акации; и ополаскиватели для рта, содержащие действующее вещество в подходящем жидком носителе.
Растворы или суспензии можно применять непосредственно в носовой полости известными методами, например с помощью капельной пипетки или в виде спрея. Композиции можно выпускать в форме дозировки для однократного или многократного введения. В последнем случае, используя пипетку, пациент может проводить дозирование надлежащего, заранее отмеренного объема раствора или супензии. В случае спрея это можно обеспечить, например, с помощью дозирующего спреера.
Введение в дыхательные пути можно также осуществить с помощью аэрозольного препарата, в котором действующее вещество находится в упаковке под давлением с подходящим пропеллентом, таким как хлорфторуглерод (CFC), например дихлордифторметан, трихлорфторметан или дихлортетрафторэтан, диоксид углерода или другой подходящий газ. Аэрозоль может также содержать поверхностно-активное вещество, такое как лецитин. Дозировку лекарственного средства можно контролировать с помощью дозирующего клапана.
Альтернативно, лекарственное средство можно выпускать в форме сухого порошка, например порошкообразной смеси соединения с подходящей порошковой основой, такой как лактоза, крахмал, производные крахмала, такие как гидроксипропилцеллюлоза, и поливинилпирролидон (PVP). Удобно, когда порошкообразный носитель образует гель в носовой полости. Порошкообразную композицию можно выпускать в дозированных формах для однократного приема, например, в капсулах или картриджах из, например, желатина, или в блистерах, из которых порошок можно принимать через ингалятор.
В композициях для введения через дыхательные пути, включая интраназальные композиции, соединение обычно имеет малый размер частиц, например порядка 5 микрон или меньше. Такой размер частиц можно обеспечить методами, известными в данной области техники, например, посредством микронизации.
При желании можно применять композиции, адаптированные для достижения замедленного высвобождения действующего вещества.
Фармацевтические препараты предпочтительно находятся в дозированной форме. В таких формах препараты подразделены на однократные дозировки, содержащие необходимое количество действующего вещества. Дозированные формы могут представлять собой упакованные препараты, в которых упаковка содержит дискретные количества препарата, такие как упакованные таблетки, капсулы и порошки в виалах или ампулах. Также дозированной формой могут являться сами капсулы, таблетки, саше или таблетки для рассасывания, либо в упаковке может содержаться их определенное количество. Таблетки или капсулы для перорального приема и жидкости для внутривенного введения или непрерывной инфузии представляют собой предпочтительные композиции.
Дополнительные детали по методикам приготовления препаратов и их введению можно найти в 21-м издании Remington's Pharmaceutical Sciences (Maack Publishing Co. Easton, Pa.).
Соединения по настоящему изобретению можно применять в комбинации с лучевой терапией или в комбинации с лучевой терапией и другими действующими веществами, уже известными в лечении медицинских состояний, описанных в настоящем тексте, в тех случаях, когда отмечается благотворный дополнительный или мультиплицирующий эффект.
Для приготовления фармацевтических препаратов можно применять фармацевтически инертные неорганические или органические эксципиенты. Для приготовления пилюлей, таблеток, таблеток с покрытием и твердых желатиновых капсул, например, можно применять лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли и т.д.. Эксципиенты для мягких желатиновых капсул и суппозиториев представляют собой, например, жиры, воска, полутвердые и жидкие полиолы, природные или гидрогенизированные масла и т.д.. Подходящими эксципиентами для приготовления растворов и сиропов являются, например, вода, сахароза, инвертированный сахар, глюкоза, полиолы и т.д.. Подходящими эксципиентами для приготовления растворов для инъекций являются, например, вода, спирты, глицерин, полиолы или растительные масла.
Термин "терапевтически эффективное количество" означает количество соединения, достаточное для проявления терапевтического эффекта, такого как активация TLR7. Это может служить причиной выделения цитокинов, противоопухолевой активности и/или противовирусной активности. Хотя точное количество действующего вещества, применяемое в фармацевтической композиции по настоящему изобретению, будет зависеть от факторов, известных квалифицированным специалистам в данной области, таких как физическая и химическая природа соединения, а также природа носителя и предполагаемый режим введения, ожидается, что композиции по настоящему изобретению будут содержать достаточно действующего вещества для обеспечения надлежащей дозировки для субъекта. Указанная дозировка может варьироваться в широких пределах и должна подбираться под индивидуальные условия в каждом отдельном случае. Для медицинского применения по настоящему изобретению, надлежащая дозировка будет варьироваться в зависимости от пути введения, конкретного состояния, которое подвергается лечению, и желаемого эффекта. В целом, однако, удовлетворительные результаты достигаются при дозировке соединения по настоящему изобретению примерно от 1 нг/кг до 100 мг/кг веса тела субъекта, предпочтительно от 100 нг/кг до 10 мг/кг, более предпочтительно от 1 мкг/кг до 1 мг/кг. Дозы можно вводить один раз в две недели, один или несколько раз в неделю, или от 2 до 4 раз в сутки в виде разделенных доз или в форме для замедленного высвобождения.
Неожиданно, соединения по настоящему изобретению являются селективными агонистами для TLR7 (особенно в сравнении с TLR8). В частности, авторы настоящего изобретения обнаружили, что двойное замещение в аминогруппе NR2R3 в формуле (I) коррелирует с TLR7 селективностью, в то время как моно-замещение в аминогруппе NR2R3 в формуле (I) (т.е., R2 = H) дает соединения, селективные к TLR8. Таким образом, в одном варианте осуществления, соединения по настоящему изобретению подходят для лечения медицинского состояния, заболевания или нарушения, которое можно лечить путем агонизации TLR7.
Соединения по настоящему изобретению предпочтительно подходят для лечения вирусных заболеваний и пролиферативных заболеваний, в частности гиперпролиферативных заболеваний, таких как доброкачественные и злокачественные формы новообразований, включая рак.
Примерами типов рака в контексте настоящего изобретения являются гепатокарцинома, адренокортикальная карцинома, виды рака, связанные со СПИД, включая вызванную СПИДом лимфому, рак ануса, базальноклеточная карцинома, рак желчных протоков, рак костей, различные виды рака мозга, включая глиому ствола мозга, церебральная астроцитома, злокачественная глиома, эпендимома, медуллобластома, супратенториальная примитивная нейроэктодермальная опухоль, глиома зрительных путей и гипоталамуса, рак груди, аденомы/карциноиды бронхов, лимфома Беркитта, рак желудочно-кишечного тракта, рак с неустановленной первичной локализацией, лимфома центральной нервной системы, рак шейки матки, хронические миелопролиферативные заболевания, рак кишечника, колоректальный рак, рак желудка, кожная Т-клеточная лимфома, рак эндометрия, эпендимома, рак пищевода, экстракраниальная эмбрионально-клеточная опухоль, внегонадная эмбрионально-клеточная опухоль, эмбрионально-клеточная опухоль яичников, рак глаза, включая внутриглазную меланому и ретинобластому, рак желчного пузыря, желудочно-кишечная карциноидная опухоль, гестационная трофобластическая опухоль, глиома, детская глиома ствола мозга, рак головы и шеи, рак крови, печеночноклеточный рак у взрослых и детей (первичный), гипофарингеальный рак, рак островков поджелудочной железы или рак поджелудочной железы, рак почки, рак гортани, острый лимфобластный лейкоз, острый миелоидный лейкоз взрослых и детей, хронический лимфолейкоз, хронический миелолейкоз, лейкоз ворсистых клеток, рак губ и ротовой полости, рак печени, рак легких, включая немелкоклеточный рак легких и мелколеточный рак легких, лимфома Ходжкина, неходжкинская лимфома, первичная лимфома центральной нервной системы, макроглобулинемия Вальденстрёма, карцинома из клеток Меркеля, мезотелиома, метастатический плоскоклеточный рак шеи с неизвестной первичной локализацией, множественная миелома/новообразование плазмацитов, фунгоидный микоз, миелодиспластический синдром, миелодиспластические миелопролиферативные заболевания, множественная миелома, хронические миелопролиферативные заболевания, рак носовой полости и синуса, рак носоглотки, нейробластома, рак ротовой полости, рак ротоглотки, остеосаркома/злокачественная фиброзная гистиоцитома костей, рак яичника, эпителиальный рак яичника, пограничная опухоль яичника, рак поджелудочной железы, рак паращитовидных желез, рак пениса, феохромоцитома, пинеобластома и супратенториальная примитивная нейроэктодермальная опухоль, опухоль гипофиза, новообразование плазмацитов/множественная миелома, плевролегочная бластома, рак простаты, рак почки, рак почечной лоханки и уретры, рак переходных клеток, рабдомиосаркома, рак слюнных желез, саркома Юинга, саркома Капоши, саркома мягких тканей, саркома матки, синдром Сезари, рак кожи, включая меланому и немеланомный рак кожи, рак тонкого кишечника, плоскоклеточный рак, рак желудка, супратенториальная примитивная нейроэктодермальная опухоль, рак яичка, тимома, тимома и рак вилочковой железы, рак щитовидной железы и трофобластическая болезнь, рак матки и эндометрия, саркома матки, рак вагины, рак вульвы и опухоль Вильмса.
В более предпочтительном варианте осуществления настоящего изобретения, соединения по настоящему изобретению можно применять для лечения следующих типов рака: рак простаты, мочевого пузыря, почки (ренальный рак), мышц, яичника, кожи, желудка, поджелудочной железы, груди, шейки матки, кишечника, соединительной ткани, плаценты, костей, мозга, матки, слюнных желез или яичек.
Примеры видов рака включают (но не ограничиваются только ими) рак груди, мочевого пузыря, костей, мозга, центральной и периферической нервной системы, кишечника, желез внутренней секреции, пищевода, эндометрия, половых клеток, головы и шеи, почки, печени, легких, глотки и гортани, мезотелиому, саркому, рак яичника, поджелудочной железы, простаты, прямой кишки, почки, тонкого кишечника, мягких тканей, яичек, желудка, кожи, уретры, вагины и вульвы; наследственные виды рака, ретинобластому и опухоль Вильмса; лейкемию, лимфому, неходжкинскую лимфому, хронический и острый миелоидный лейкоз, острый лимфобластный лейкоз, лимфому Ходжкина, множественную миелому и Т-клеточную лимфому; миелодиспластический синдром, новообразования плазмацитов, паранеопластический синдром, злокачественные опухоли без выявленного первичного очага и СПИД-ассоциированные злокачественные опухоли.
В частности, TLR7 агонисты могут применяться для лечения рака кожи, груди, кишечника, желудка, поджелудочной железы или почки. Чувствительность конкретного вида рака к активации TLR7 можно оценить (не ограничиваясь только этими исследованиями) путем измерения уменьшения массы первичной или метастатической опухоли (минорная, частичная или полная рецессия), по изменениям в гемограмме, по изменениям концентрации гормонов или цитокинов в крови, по ингибированию дальнейшего роста опухолевой массы, стабилизации заболевания у пациента, по оценке биомаркеров или суррогатных маркеров, релевантных для данного заболевания, по увеличению общей выживаемости пациентов, по увеличению времени развития заболевания у пациентов, по увеличенной выживаемости без прогрессирования у пациентов, по увеличенной безрецидивной выживаемости у пациентов, по улучшенному качеству жизни у пациентов или по модулированию сопутствующих заболеваний (например (но не ограничиваясь только ими) боль, кахексия, мобилизация, госпитализация, изменения в гемограмме, потеря веса, заживление ран, лихорадка).
Соединения по настоящему изобретению можно вводить как единственное терапевтическое средство в режиме лечения, либо его можно вводить в комбинации с другим и/или другими действующими веществами, включая дополнительные противораковые средства, модуляторы иммунного ответа, противовирусные средства, антибиотики, противолихорадочные средства и т.п.
Фармацевтические композиции по настоящему изобретению могут содержать одно или больше, предпочтительно одно или два, более предпочтительно одно соединение по настоящему изобретению. Аналогично, медицинское применение по настоящему изобретению может включать одно или больше, предпочтительно одно или два, более предпочтительно одно соединение по настоящему изобретению.
Фармацевтические композиции, содержащие действующее вещество и по меньшей мере одно вспомогательное вещество, можно производить методами, известными квалифицированным специалистам в данной области, например, путем растворения, смешивания, гранулирования, изготовления драже, растирания, эмульгирования, инкапсулирования, заключения в матрикс и лиофилизации.
В случае наружного нанесения, подходящими фармацевтическими препаратами являются, например, мази, кремы, лосьоны, пасты, гели, порошки, растворы, эмульсии, суспензии, масла, спреи и пластыри (например, чрезкожные терапевтические системы).
Для парентеральных способов введения, таких как, например, внутривенное, внутриартериальное, внутримышечное, подкожное, внутрикожное, интраперитонеальное и внутригрудинное введение, предпочтительно применяют растворы (например, стерильные растворы, изотонические растворы). Их, в частности, вводят путем инъекции или инфузии.
В случае интраназального введения, предпочтительными препаратами являются, например, спреи и растворы, применяемые в капельной форме.
Для внутриглазного введения примерами препаратов являются растворы для введения в капельной форме, гели и мази.
В целом, фармацевтические композиции по настоящему изобретению можно вводить таким образом, чтобы дозировка действующего вещества находилась в диапазоне, подходящем для активаторов TLR7. В частности, дозировка находится в диапазоне от 0.001 до 200 мг, предпочтительно от 0.01 мг до 20 мг, более предпочтительно от 0.1 мг до 4 мг, и еще более предпочтительно от 0.2 мг до 2 мг действующего вещества в неделю, из расчета на среднего взрослого пациента, имеющего вес тела 70 кг. Следует отметить, что дозировка зависит, например, от конкретного применяемого соединения, вида животного, подвергающегося лечению, возраста, веса тела, общего состояния здоровья, пола и диеты субъекта, а также от способа и времени введения, скорости экскреции, степени тяжести подлежащего излечению заболевания и лекарственных средств, применяемых в комбинации.
Цитокины, выработка которых может индуцироваться при введении соединений по настоящему изобретению, в целом включают интерферон (IFN) и/или фактор некроза опухоли-α (ФНО-α), а также некоторые интерлейкины (IL). Цитокины, биосинтез которых может индуцироваться под действием соединений по настоящему изобретению, включают IFN-α, ФНО-α, IL-1, IL-6, IL-10 и IL-12, и различные другие цитокины. Наряду с другими эффектами, цитокины ингибируют продуцирование вируса и рост опухолевых клеток, что делает соединения по настоящему изобретению пригодными для лечения опухолей и вирусных заболеваний.
В дополнение к способности индуцировать выработку цитокинов, соединения по настоящему изобретению влияют на другие аспекты естественного иммунного ответа. Например, может стимулироваться активность естественных клеток-киллеров, и этот эффект может быть следствием индуцирования выработки цитокинов. Соединения могут также активировать макрофаги, которые в свою очередь стимулируют секрецию оксида азота и выработку дополнительных цитокинов. Кроме того, соединения могут вызывать пролиферацию и дифференциацию T- и/или B-лимфоцитов.
Эффекты модификации иммунного ответа делают соединения по настоящему изобретению пригодными для использования в лечении широкого ряда патологических состояний. Благодаря своей способности индуцировать выработку цитокинов, таких как IFN-α и/или ФНО-α, и IL-12, данные соединения особенно хорошо подходят для использования в лечении вирусных заболеваний и опухолей. Эта иммуномодулирующая активность говорит о том, что соединения по настоящему изобретению могут применяться в лечении таких заболеваний, как (но не ограничиваясь только ими) вирусные заболевания, включая генитальные бородавки; обычные бородавки; подошвенные бородавки; гепатит В; гепатит С; герпес типа I и типа II; контагиозный моллюск; ВИС; цитомегаловирус; вирус варицелла-зорус; внутриэпителиальные новообразования, такие как внутриэпителиальные новообразования в шейке матки; папилломавирус человека (HPV) и связанные с ним новообразования; грибковые заболевания, например кандидоз, аспергиллез и криптококковый менингит; новообразования, например, базальноклеточная карцинома, лейкоз ворсистых клеток, саркома Капоши, почечноклеточный рак, плоскоклеточный рак, миелогенный лейкоз, множественная миелома, меланома, неходжкинская лимфома, кожная Т-клеточная лимфома и другие виды рака; паразитические заболевания, например pneumocystis carnii, криптоспоридиоз, гистоплазмоз, токсоплазмоз, инфекция, вызванная трипаносомами, и лейшманиоз; и бактериальные инфекции, например, туберкулез и инфекция, вызываемая Mycobacterium avium.
В настоящем изобретении описан также способ лечения вирусной инфекции у животного, включающий введение животному эффективного количества соединения, имеющего формулу (I). Количеством, эффективным для лечения или ингибирования вирусной инфекции, является количество, которое вызывает уменьшение степени выраженности одного или больше проявлений вирусной инфекции, таких как вирусные бляшки, концентрация вируса, скорость продуцирования вируса и смертность в сравнении с контрольными животными, к которым не применялось лечение. Точное количество будет варьироваться в соответствии с факторами, известными в данной области, но оно ожидается равным дозе, указанной выше в отношении активации TLR7, или дозе от примерно 100 нг/кг до примерно 50 мг/кг, в частности от примерно 10 мкг/кг до примерно 5 мг/кг. Количеством, эффективным для лечения нвообразования, является количество, которое вызывает уменьшение размера опухоли или числа очагов опухолей. И снова, точное количество будет варьироваться в зависимости от факторов, известных в данной области, но оно ожидается равным дозе, указанной выше в отношении активации TLR7, или дозе от примерно 100 мг/кг до примерно 50 мг/кг, в частности от примерно 10 мг/кг до примерно 5 мг/кг.
Соединения по настоящему изобретению можно получить, например, как описано ниже и согласно указанным ниже стадиям, или, в частности, методами, описанными в качестве примеров ниже в разделе, посвященном примерам.
Соединения по настоящему изобретению выделяют и очищают известными способами, например путем отгонки растворителя в вакууме и перекристаллизации полученного остатка из подходящего растворителя, или посредством применения стандартных методов очистки, таких как колоночная хроматография на подходящем носителе.
Соли соединений, имеющих формулу (I) по настоящему изобретению, можно получить путем растворения свободного соединения в подходящем растворителе (например, в кетоне, таком как ацетон, метилэтилкетон или метил-изобутилкетон, в простом эфире, таком как диэтиловый эфир, тетрагидрофуран или дикосан, в хлорированном углеводороде, таком как метиленхлорид или хлороформ, или в низкомолекулярном спирте, таком как метанол, этанол или изопропанол), который содержит желаемую кислоту или основание, в который добавляют желаемую кислоту или основание. Кислоты или основание можно применять при получении солей, в зависимости от того - одно- или многооснованя кислота или основание бьерется, и в зависимости от того какую соль собираются получить, в эквимольном количественном соотношении или в другом количестве. Соли получают путем фильтрования, переосаждения, осаждения нерастворителем для соли или путем упаривания растворителя. Полученные соли можно превратить в свободные соединения, которые, в свою очередь, можно превратить в соли. Таким образом, фармацевтически неприемлемые соли, которые можно получить, например, в качестве промежуточных продуктов при производстве в промышленном масштабе, можно превратить в фармацевтически приемлемые соли способами, известными квалифицированным специалистам в данной области.
Соединения, имеющие формулу (I) по настоящему изобретению, можно превратить в их N-оксиды, например, с помощью пероксида водорода в метаноле или с помощью м-хлорпербензойной кислоты в дихлорметане. Квалифицированному специалисту в данной области известны условия реакции N-окисления.
Чистые диастереомеры и чистые энантиомеры соединений и солей по настоящему изобретению, которые имеют формы таких стереоизомеров, можно получить, например, асимметрическим синтезом, с применением хиральных исходных соединений в синтезе и путем расщепления смесей энантиомеров и диастереомеров, получаемых в процессе синтеза.
Смеси энантиомеров и диастереомеров можно расщепить на чистые энантиомеры и чистые диастереомеры способами, известными квалифицированным специалистам в данной области. В частности, смеси диастереомеров разделяют кристаллизацией, в частности дробной кристаллизацией, или хроматографически. Смеси энантиомеров можно разделить, например, путем образования диастереомеров с хиральным вспомогательным агентом, разделения полученных диастереомеров и снятия хирального вспомогательного агента. В качестве хиральных вспомогательных агентов, например, можно использовать хиральные кислоты для разделения энантимерных оснований, и хиральные основания можно применять для разделения энантиомерных кислот через образование диастереомерных солей. Кроме того, можно получать диастереомерные производные, такие как диастереомерные сложные эфиры, из смесей энантиомеров спиртов или смесей энантимеров кислот, соответственно, используя хиральные кислоты или хиральные спирты, соответственно, в качестве хиральных вспомогательных агентов. Кроме того, можно применять диастереомерные комплексы или диастереомерные клатраты для разделения смесей энантиомеров. Альтернативно, смеси энантиомеров можно разделять с применением хиральных колонок в хроматографии. Другим подходящим методом разделения энантиомеров является энзиматическое расщепление.
Как будет понятно квалифицированному специалисту в данной области, настоящее изобретение не ограничивается описанными в настоящем тексте частными вариантами осуществления, но включает все модификации указанных вариантов осуществления, которые охватываются сутью и объемом настоящего изобретения, в соответствии с Формулой изобретения.
Приведенные далее примеры иллюстрируют настоящее изобретение более подробно, не ограничивая его каким-либо образом. Другие соединения по настоящему изобретению, для которых получение не описано в явном виде, могут быть получены аналогично.
Соединения, указанные в приведенных примерах, представляют собой частные варианты осуществления настоящего изобретения.
При использовании в настоящем тексте, термин “включая” следует понимать как “включая, но не ограничиваясь только перечисленным».
Примеры
Получение соединений по настоящему изобретению
Общее
Аббревиатуры: CH2Cl2 = дихлорметан, ДМФА = N,N-диметилформамид, ДМА = N,N-диметилацетамид, ДМСО = диметилсульфоксид, ТГФ = тетрагидрофуран, CH3CN = ацетонитрил, EtOAc = этилацетат, MeOH = метанол, EtOH = этанол, AcOH = уксусная кислота, HCO2H = муравьиная кислота, HCl = соляная кислота, NaOH = гидроксид натрия, LiOH = гидроксид лития, NaHCO3 = бикарбонат натрия, DIPEA = N,N-диизопропилэтиламин или N-этил-N-изопропилпропан-2-амин, RT = комнатная температура, STAB = триацетоксиборгидрид натрия, PPA = пропионовая кислота, TBDMS = трет-бутилдиметилсилил, Boc = трет-бутилоксикарбонил, Cbz = бензилоксикарбонил, Me = метил, Et = этил, ВЭЖХ = высокоэффективная жидкостная хроматография, МС = масс-спектрометрия, ТСХ = тонкослойная хроматография.
Реакции под действием микроволнового излучения проводили с применением системы Biotage® Initiator Robot Sixty. Если не указано иное, все ТСХ проводили на пластинках с SiO2 (силикагель с флуоресцентным индикатором F254), а все очистки методом препаративной обращенно-фазной ВЭЖХ/МС проводили на колонке XTerra RP18 5мкМ 19x150 мм с градиентом 0.1% HCO2H/H2O/15-95% CH3CN. Образцы загружали на обращенно-фазную колонку с применением системы ДМСО/AcOH/H2O = 1/1/1 или CH3CN/MeOH/ДМФА = 80/15/5. Если не указано иное, все растворы HCl, NaOH, KOH или LiOH являются водными.
Соединения характеризовали методом 1Н-ЯМР в d6-диметилсульфоксиде или d-хлороформе на ЯМР-приборе (Bruker) с рабочей частотой 300 МГц или 400 МГц, и методом масс-спектрометрии, которую обычно проводили методом ВЭЖХ/МС в быстром градиенте на C18-носителе с применением режима ESI (электроспрей-ионизация) или APCI (химическая ионизация при атмосферном давлении). Значения [M+H]+ или [M-H]- - это значения, найденные в соответствующей ВЭЖХ/МС хроматограмме для конкретного соединения при протонировании и депротонировании. Все эти значения находятся в допустимых границах +/- 0.2 относительно вычисленных значений точной массы.
ЯМР-характеристики относятся к значениям химического сдвига (δ), выраженным в миллионных долях (м.д.). Относительная площадь пика в 1H ЯМР спектре соответствует числу атомов водорода для данного функционального типа в молекуле. Природа сдвига, а именно мультиплетность, обозначена следующим образом: синглет (с), уширенный синглет (ушир.с), дублет (д), триплет (т), квартет (кв), квинтет (квинт.) и мультиплет (м).
Получение интермедиатов и билдинг-блоков
Схема 1: синтез веществ A-C
Вещество A
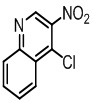
4-хлор-3-нитрохинолин
Стадия 1: 4-гидроксихинолин (250 г, 1.72 моль) растворяли в пропионовой кислоте (200 мл), и полученную смесь перемешивали при 125°C. Затем добавляли по каплям азотную кислоту (158 мл, 3.79 моль, 2.2 экв.), поддерживая температуру реакционной смеси 125°C. После окончания добавления, реакционную смесь перемешивали при 125°C в течение 60 минут и затем охлаждали до комнатной температуры. Выпавший осадок отфильтровывали и промывали последовательно этанолом, водой и снова этанолом. Полученное твердое вещество перекристаллизовывали из горячего этанола, охлаждали, отфильтровывали и сушили при пониженном давлении, получая 252.3 г (77%) 3-нитрохинолин-4-ола в виде бежевого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6) δ 12.96 (ушир.с, 1H), 9.17 (с, 1H), 8.25 (дд, 1H), 7.83-7.68 (м, 2H), 7.51 (м, 1H); MS (ESI+) m/z 191.1 [M+H]+
Стадия 2: POCl3 (661 мл, 7.21 моль, 18.3 экв.) нагревали до 60°C и порциями добавляли 3-нитрохинолин-4-ол (75 г, 0.39 моль). Полученную суспензию перемешивали при 120°C в течение 3 часов, затем охлаждали до комнатной температуры и удаляли растворитель при пониженном давлении. Сырой остаток выливали на смесь льда (1 кг) и CH2Cl2 (500 мл). Водный слой отбрасывали, а из органической фазы при упаривании кристаллизовалось твердое вещество. Полученное твердое вещество отфильтровывали и промывали дихлорметаном, получая 40.4 г (50%) целевого продукта в виде светло-бежевого твердого вещества. Дальнейшей очистки не проводилось.
MS (ESI+) m/z 209.0 211.0 [M+H]+
Альтернативные условия для Стадии 2:
Стадия 2: В круглодонную колбу помещали магнитную мешалку, 3-нитрохинолин-4-ол (41.82 г, 219.90 ммоль), безводный CH2Cl2 (1.05 л) и безводный ДМФА (8.51 мл, 109.95 ммоль, 0.5 экв.), получая суспензию бежевого цвета. После добавления тионилхлорида (34.01 г, 285.87 ммоль, 1.3 экв.) реакционную смесь кипятили в течение 5 часов. Прогресс реакции отслеживали методом ТСХ (петролейный эфир/EtOAc 1:1) и ВЭЖХ/МС. Реакционную смесь затем охлаждали до комнатной температуры и использовали в следующей стадии без дополнительной очистки.
MS (ESI+) m/z 208.9 210.8 [M+H]+
Вещество B
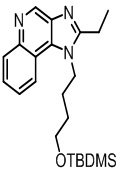
1-(4-((трет-Бутилдиметилсилил)окси)бутил)-2-этил-1H-имидазо[4,5-c]хинолин
Стадия 1: 4-хлор-3-нитрохинолин (вещество A) (15 г, 71.91 ммоль) растворяли в CH2Cl2 (100 мл) и добавляли триэтиламин (19.99 мл, 143.81 ммоль, 2 экв.) порциями при комнатной температуре. 4-Амино-1-бутанол (8.68 мл, 93.48 ммоль, 1.3 экв.) добавляли по каплям в полученный раствор (осторожно! экзотермическая реакция!), реакционную смесь перемешивали при кипячении в течение 2 часов и затем при комнатной температуре в течение ночи. Мониторинг реакции методом ВЭЖХ/МС показал ее полное прохождение. Полученный раствор разделяли между CH2Cl2 и насыщенным водным раствором хлорида аммония, слои разделяли, и водный слой экстрагировали один раз CH2Cl2. Объединенные органические слои сушили над Na2SO4, фильтровали и упаривали при пониженном давлении, получая 12.8 г (68%) целевого продукта в виде темно-желтого твердого вещества. Полученное вещество использовали далее без дополнительной очистки.
1H ЯМР (300 МГц, ДМСО-d6) δ 9.14-9.00 (м, 2H), 8.52 (д, 1H), 7.99-7.74 (м, 2H), 7.58 (м, 1H), 7.28 (ушир.с, 1H), 3.65 (м, 2H), 3.41 (т, 2H), 1.75 (м, 2H), 1.49 (м, 2H); MS (ESI+) m/z 262.1 [M+H]+
Стадия 2: В раствор 4-((3-нитрохинолин-4-ил)амино)бутан-1-ола (42.8 г, 163.80 ммоль) и 4-диметиламинопиридина (800 мг, 6.54 ммоль, 0.04 экв.) в хлороформе (350 мл) добавляли по каплям триэтиламин (34.2 мл, 245.70 ммоль, 1.5 экв.) при комнатной температуре, затем порциями добавляли трет-бутилдиметилсилилхлорид (32.1 г, 212.94 ммоль, 1.3 экв.). Полученную смесь перемешивали в течение ночи при комнатной температуре. Мониторинг реакции методом ВЭЖХ/МС показал ее полное прохождение. Смесь упаривали при пониженном давлении, остаток суспендировали в этилацетате и отфильтровывали. Фильтрат разделяли между этилацетатом и насыщенным водным раствором хлорида аммония, слои разделяли и экстрагировали водный слой один раз этилацетатом. Объединенные органические слои сушили над Na2SO4, фильтровали и упаривали при пониженном давлении, получая 54.7 г (88%) указанного в заголовке соединения в виде желто-зеленого твердого вещества. Полученное вещество использовали далее без дополнительной очистки.
1H ЯМР (300 МГц, ДМСО-d6) δ 9.14-8.95 (м, 2H), 8.51 (д, 1H), 7.94-7.77 (м, 2H), 7.58 (м, 1H), 3.67 (м, 2H), 3.57 (т, 2H), 1.75 (м, 2H), 1.51 (м, 2H), 0.81 (с, 9H), -0.02 (с, 6H); MS (ESI+) m/z 376.1 [M+H]+
Стадия 3: В аппарат Парра емкостью 500 мл (сосуд для проведения реакций под давлением, Parr Instrument GmbH, Germany) загружали N-(4-((трет-бутилдиметилсилил)окси)бутил)-3-нитрохинолин-4-амин (35.6 г, 94.79 ммоль), 5% платину на угле (18.5 г, 4.74 ммоль, 0.05 экв.) и толуол (150 мл). Реактор помещали в шейкер Парра и подавали водород под давлением 50 фунт/кв.дюйм (3.5 кг/см2). Протекание реакции отслеживали методом ТСХ (CH2Cl2/MeOH 100:5), полное прохождение реакции было зарегистрировано через 1 час. Катализатор удаляли фильтрованием через тонкий слой CELITE® Hyflo Supercel. Осадок на фильтре промывали толуолом (3x100 мл) и объединяли фильтраты. Летучие компоненты удаляли при пониженном давлении, получая 31.65 г (96%) целевого продукта в виде темного масла.
1H ЯМР (300 МГц, ДМСО-d6) δ 8.36 (с, 1H), 7.99 (м, 1H), 7.72 (м, 1H), 7.33 (м, 2H), 5.31-4.45 (м, 3H), 3.54 (т, 2H), 3.19 (м, 2H), 1.52 (м, 4H), 0.82 (с, 9H), -0.02 (с, 6H); MS (ESI+) m/z 346.1 [M+H]+
Стадия 4: В круглодонную колбу помещали магнитную мешалку, N4-(4-((трет-бутилдиметилсилил)окси)бутил)хинолин-3,4-диамин (8.99 г, 26.02 ммоль), триэтил ортопропионат (9.17 г, 52.03 ммоль, 2 экв.) и толуол (76 мл). Реакционную смесь кипятили для удаления побочного продукта - этанола - до тех пор, пока ТСХ (бензол/метанол/ацетон 1:1:8) и ВЭЖХ/МС не показали полную конверсию через 20 часов. Реакционную смесь охлаждали, и летучие компоненты удаляли при пониженном давлении, получая 9.97 г (количественный выход) 1-(4-((трет-бутилдиметилсилил)окси)бутил)-2-этил-1H-имидазо[4,5-c]хинолина в виде густого темно-коричневого масла. Полученное вещество использовали в следующей стадии без дополнительной очистки.
1H ЯМР (300 МГц, CDCl3) δ 9.30 (с, 1H), 8.27 (дд, 1H), 8.19 (дд, 1H), 7.64 (м, 2H), 4.57 (т, 2H), 3.71 (т, 2H), 3.01 (кв, 2H), 2.06 (м, 2H), 1.73 (м, 2H), 1.55 (т, 3H), 0.87 (с, 9H), 0.04 (с, 6H); MS (ESI+) m/z 384.2 [M+H]+
Вещество C
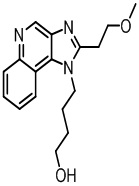
4-(2-(2-Метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)бутан-1-ол
В круглодонную колбу помещали магнитную мешалку, N4-(4-((трет-бутилдиметилсилил)окси)бутил)хинолин-3,4-диамин (5.00 г, 14.47 ммоль), 3-метоксипропановую кислоту (1.81 г, 17.36 ммоль, 1.2 экв.), 2-(3H-[1,2,3]триазолo[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизоурония гексафторфосфат (V) (HATU, 6.60 г, 17.36 ммоль, 1.2 экв.), N-этил-N-изопропилпропан-2-амин (DIPEA, 2.24 г, 17.36 ммоль, 1.2 экв.) и безводный 1-метил-2-пирролидон (NMP, 60 мл). Полученный раствор перемешивали при 120°C до тех пор, пока ТСХ мониторинг (бензол/метанол/ацетон 1:1:8) не подтвердил полную конверсию через 20 часов. Масса целевого продукта детектировалась методом ВЭЖХ/МС. Реакционную смесь разводили десятикратным объемом воды и экстрагировали этилацетатом в жидкость-жидкостном экстракторе. Органический слой упаривали при пониженном давлении и очищали сырой продукт методом флэш-хроматографии на силикагеле (CH2Cl2/MeOH от 95:5 до 90:10), получая 9.48 г (количественный выход, все еще содержит примеси) красновато-коричневого масла. Полученное вещество использовали в следующей стадии без дополнительной очистки.
MS (ESI+) m/z 300.0 [M+H]+
Схема 2: синтез вещества D
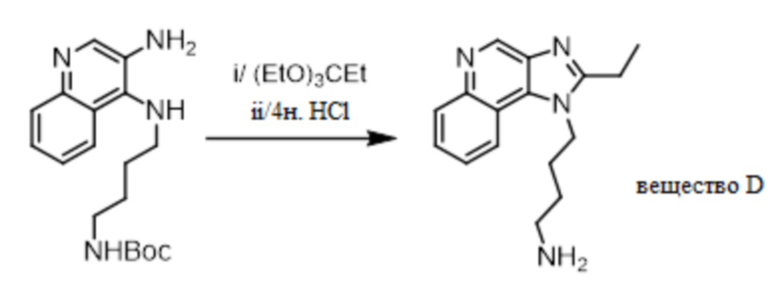
Вещество D
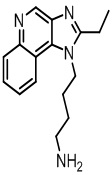
4-(2-Этил-1H-имидазо[4,5-c]хинолин-1-ил)бутан-1-амин
В круглодонную колбу помещали магнитную мешалку, коммерчески доступный трет-бутил (4-((3-аминохинолин-4-ил)амино)бутил)карбамат (3.00 г, 9.08 ммоль), триэтил ортопропионат (3.20 г, 18.16 ммоль, 2 экв.) и толуол (50 мл). Реакционную смесь кипятили для удаления побочного продукта этанола до тех пор, пока ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 90:9:1) и ВЭЖХ/МС показали полную конверсию через 24 часа. Реакционную смесь затем оставляли охлаждаться до комнатной температуры и упаривали при пониженном давлении. Остаток растворяли в безводном 1,4-диоксане (50 мл), добавляли 4н. раствор HCl в 1,4-диоксане (50 мл), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь упаривали в вакууме, и остаток растворяли в насыщенном растворе бикарбоната натрия (100 мл), замораживали и лиофилизовывали. Лиофилизат экстрагировали абсолютным этанолом (3x100 мл). Объединенные экстракты фильтровали и упаривали при пониженном давлении, получая 2.146 г (88%) 4-(2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутан-1-амина в виде красновато-желтого масла.
1H ЯМР (300 МГц, CDCl3) δ 9.30 (с, 1H), 8.28 (м, 1H), 8.18 (м, 1H), 7.64 (м, 2H), 4.55 (т, 2H), 3.01 (кв, 2H), 2.79 (т, 2H), 2.02 (м, 2H), 1.72-1.49 (м, 5H); MS (ESI+) m/z 269.4 [M+H]+
Схема 3: Синтез вещества E
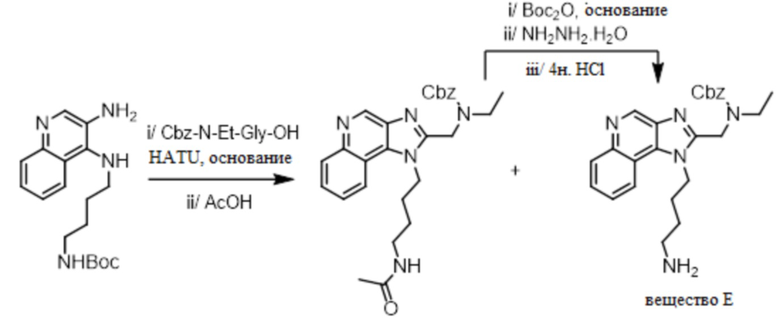
Вещество E
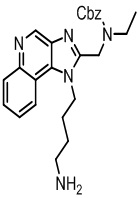
Бензил ((1-(4-аминобутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамат
Стадия 1: В круглодонную колбу помещали магнитную мешалку, коммерчески доступный трет-бутил (4-((3-аминохинолин-4-ил)амино)бутил)карбамат (40.00 г, 121.05 ммоль), 2-{[(бензилокси)карбонил](этил)амино}уксусную кислоту (Cbz-N-Et-Gly-OH, 34.46 г, 145.27 ммоль, 1.2 экв.), 2-(3H-[1,2,3]триазолo[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизоурония гексафторфосфат(V) (HATU, 55.23 г, 145.27 ммоль, 1.2 экв.), триэтиламин (36.75 г, 363.16 ммоль, 3 экв.), 4-диметиламинопиридин (1.48 г, 12.11 ммоль, 0.1 экв.) и безводный ДМФА (800 мл), получая красновато-желтый раствор. Реакционную смесь перемешивали при комнатной температуре до тех пор, пока ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) и ВЭЖХ/МС показали почти полную конверсию примерно через 10-12 часов. Растворитель упаривали при пониженном давлении, остаток растворяли в этилацетате (500 мл) и промывали водой (3x300 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Полученный сырой темно-желтый интермедиат растворяли в ледяной уксусной кислоте (400 мл), и полученную смесь кипятили в течение 36 часов и мониторили методом ВЭЖХ/МС. Реакционную смесь затем упаривали, отгоняя азеотроп толуол-уксусная кислота при пониженном давлении, и остаток суспендировали в насыщенном водном растворе NaHCO3 (200 мл) и фильтровали. Фильтрат экстрагировали хлороформом (3x200 мл), водный слой доводили до pH 10-11 добавлением 3M раствора NaOH и экстрагировали хлороформом (9x100 мл). Осадок на фильтре и водный слой отбрасывали. Объединенные органические слои промывали последовательно насыщенным раствором хлорида натрия (400 мл) и сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии на силикагеле (CHCl3/MeOH/32%-ный раствор аммиака 290:9:1), получая 13.82 г (24%) бензил ((1-(4-ацетамидобутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамата в виде красновато-желтого масла и 8.75 г (~80% чистота, 16%) бензил ((1-(4-аминобутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамата в виде красновато-желтого масла.
1H ЯМР (300 МГц, CDCl3) δ 9.28 (с, 1H), 8.28 (дд, 1H), 8.08 (дд, 1H), 7.67 (м, 2H), 7.43-7.28 (м, 5H), 6.00 (ушир.с, 1H), 5.22 (с, 2H), 4.92 (с, 2H), 4.61 (ушир.с, 2H), 3.43 (кв, 2H), 3.22 (м, 2H), 1.94 (с, 3H), 1.92-1.59 (м, 4H), 1.10 (т, 3H); MS (ESI+) m/z 474.2 [M+H]+
Стадия 2: В круглодонную колбу помещали магнитную мешалку, ставили обратный холодильник, загружали бензил ((1-(4-ацетамидобутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамат (13.20 г, 27.87 ммоль), 4-диметиламинопиридин (0.68 г, 5.57 ммоль, 0.2 экв.) и тетрагидрофуран (420 мл), получая желтый раствор. Добавляли ди-трет-бутилдикарбонат (18.25 г, 83.62 ммоль, 3 экв.), и смесь кипятили в течение 16 часов. ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) и ВЭЖХ/МС показали неполную конверсию. Дополнительное количество ди-трет-бутил дикарбоната (6.08 г, 27.87 ммоль, 1 экв.) и дополнительное кипячение в течение еще 2 часов потребовались для достижения полной конверсии. Добавляли метанол (420 мл) и гидразин моногидрат (11.16 г, 222.98 ммоль, 8 экв.), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Дополнительное количество гидразина моногидрата (2.79 г, 55.75 ммоль, 2 экв.) и дополнительное перемешивание в течение ночи потребовалось для достижения полной конверсии согласно данным ТСХ (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) и ВЭЖХ/МС. Реакционную смесь затем выливали в дихлорметан (800 мл), промывали последовательно 1н. раствором HCl (250 мл), 10%-ным водным раствором сульфата меди(II) (250 мл) и насыщенным водным раствором NaHCO3 (250 мл), сушили над MgSO4, отфильтровывали и упаривали при пониженном давлении. Остаток растворяли в 1,4-диоксане (400 мл) и добавляли 4н. раствор HCl в 1,4-диоксане (200 мл) при комнатной температуре на 60 минут, до тех пор, пока ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) не подтвердил полную конверсию. Реакционную смесь затем разбавляли водой (600 мл), значение pH доводили до 10-11 добавлением 3M раствора NaOH, и смесь экстрагировали дихлорметаном (3x250 мл). Объединенные органические слои упаривали при пониженном давлении. Остаток объединяли с водным слоем и упаривали, отгоняя азеотроп втор-бутанол-вода при пониженном давлении. Сырой остаток экстрагировали дихлорметаном, содержащим 10% абсолютного этанола (2x500 мл). Объединенные органические слои сушили над MgSO4, отфильтровывали и упаривали в вакууме, получая 12.80 г (80-90% чистота, количественный выход) бензил ((1-(4-аминобутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамата в виде темно-желтого масла. Полученный продукт использовали в следующей стадии без дополнительной очистки.
MS (ESI+) m/z 432.2 [M+H]+
Схема 4: синтез Примера 1
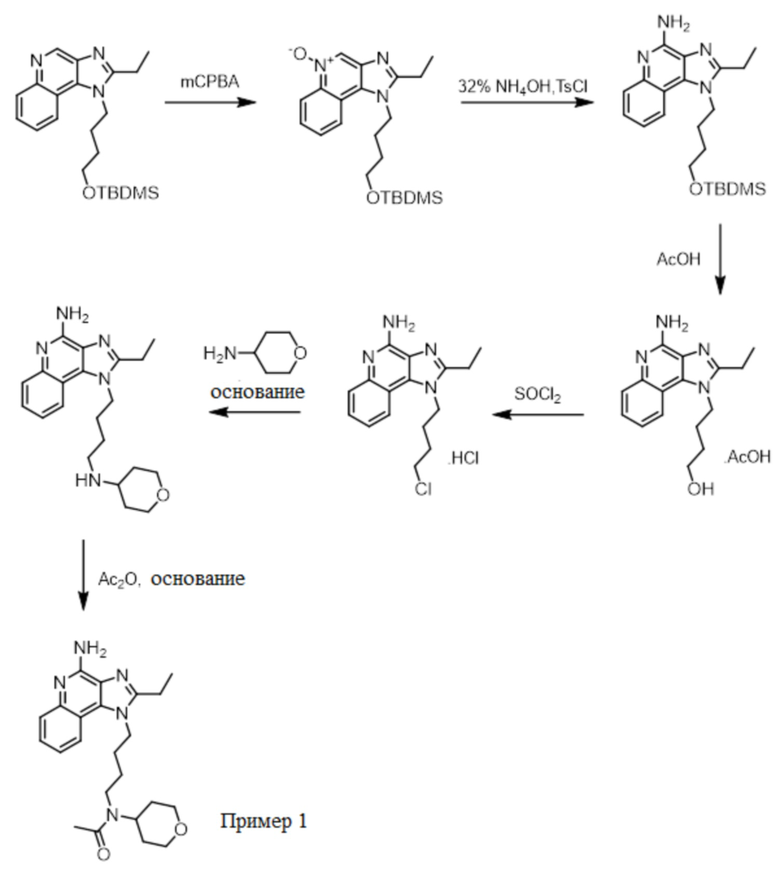
Пример 1
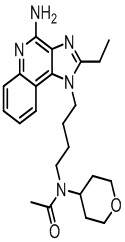
N-(4-(4-Амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-N-(тетрагидро-2H-пиран-4-ил)ацетамид
Стадия 1: В круглодонную колбу помещали магнитную мешалку, 1-(4-((трет-бутилдиметилсилил)окси)бутил)-2-этил-1H-имидазо[4,5-c]хинолин (см. вещество B) (9.97 г, 25.99 ммоль) и хлороформ (130 мл). Добавляли порциями твердую 3-хлорпербензойную кислоту (4.93 г, 28.59 ммоль, 1.1 экв.) в раствор в течение 15 минут, и смесь перемешивали при комнатной температуре в течение 1 часа. ТСХ мониторинг (бензол/метанол/ацетон 1:1:8) и ВЭЖХ/МС показали полное исчезновение исходного вещества после дополнительного добавления 3-хлорпербензойной кислоты (1.35 г, 7.80 ммоль, 0.3 экв.) и дополнительного перемешивания в течение 30 минут. Реакционную смесь разделяли между хлороформом и насыщенным водным раствором бикарбоната натрия, и слои разделяли. Органическую фазу последовательно промывали насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия, сушили над Na2SO4, отфильтровывали и упаривали при пониженном давлении, получая 9.64 г (93%) 1-(4-((трет-бутилдиметилсилил)окси)бутил)-2-этил-1H-имидазо[4,5-c]хинолин 5-оксида в виде бежево-коричневого твердого вещества. Полученное вещество использовали в следующей стадии без дополнительной очистки.
MS (ESI+) m/z 400.1 [M+H]+
Стадия 2: В круглодонную колбу помещали магнитную мешалку, 1-(4-((трет-бутилдиметилсилил)окси)бутил)-2-этил-1H-имидазо[4,5-c]хинолин 5-оксид (9.64 г, 24.12 ммоль), хлороформ (100 мл) и раствор аммиака (32%, 100 мл). 4-Метилбензол-1-сульфонилхлорид (5.52 г, 28.95 ммоль, 1.2 экв.) добавляли в полученную двухфазную смесь в один прием, и реакционную смесь интенсивно перемешивали при комнатной температуре до тех пор, пока ТСХ мониторинг (бензол/метанол/ацетон 1:1:8) и ВЭЖХ/МС показали полную конверсию через 2 часа. Смесь разделяли между хлороформом (150 мл) и насыщенным водным раствором хлорида натрия (250 мл). Органический слой отделяли от водного слоя и промывали насыщенным водным раствором хлорида натрия (2x150 мл), сушили над Na2SO4, фильтровали и упаривали при пониженном давлении, получая 8.57 г (89%) 1-(4-((трет-бутилдиметилсилил)окси)бутил)-2-этил-1H-имидазо[4,5-c]хинолин-4-амина в виде рыже-коричневого твердого вещества. Полученное вещество использовали в следующей стадии без дополнительной очистки.
MS (ESI+) m/z 399.1 [M+H]+
Стадия 3: В круглодонную колбу помещали магнитную мешалку, 1-(4-((трет-бутилдиметилсилил)окси)бутил)-2-этил-1H-имидазо[4,5-c]хинолин-4-амин (8.57 г, 21.50 ммоль), тетрагидрофуран (90 мл), воду (90 мл) и уксусную кислоту (270 мл), и полученный красноватый раствор перемешивали при 60°C до тех пор, пока ТСХ мониторинг (бензол/метанол/ацетон 1:1:8) и ВЭЖХ/МС показали полную конверсию через 16 часов. Реакционную смесь затем оставляли охлаждаться до 0°C и доводили до pH 8, добавляя по каплям 10M раствор NaOH (~ 333 мл). Водный слой экстрагировали смесью этилацетата и этанола (5x200 мл). Органические слои объединяли, упаривали при пониженном давлении, и полученное твердое вещество экстрагировали диметилформамидом, чтобы удалить ацетат натрия. Экстракт упаривали в вакууме, получая 6.78 г (92 %) 4-(4-амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутан-1-ол ацетата в виде бежево-коричневого твердого вещества.
MS (ESI+) m/z 285.1 [M+H]+
Стадия 4: Тионилхлорид (7.04 г, 59.20 ммоль, 3 экв.) добавляли в суспензию 4-(4-амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутан-1-ола ацетата (6.78 г, 19.73 ммоль) в дихлорэтане (300 мл), и полученную желто-бежевую смесь перемешивали при комнатной температуре в течение ночи. ВЭЖХ/МС мониторинг показал полную конверсию. Смесь охлаждали до 0°C и медленно добавляли метанол (25 мл). Летучие компоненты удаляли при пониженном давлении, остаток промывали последовательно ацетоном и диэтиловым эфиром, и сушили в высоком вакууме, получая 5.021 г (75%) 1-(4-хлорбутил)-2-этил-1H-имидазо[4,5-c]хинолин-4-амина гидрохлорида в виде желто-бежевого твердого вещества. Промывные растворы фильтровали, остаток промывали последовательно ацетоном и диэтиловым эфиром и сушили в высоком вакууме, получая 0.401 г (6%) второй порции продукта.
1H ЯМР (300 МГц, ДМСО-d6) δ 13.88 (с, 1H), 8.74 (ушир.с, 2H), 8.26 (д, 1H), 7.83 (д, 1H), 7.72 (т, 1H), 7.58 (т, 1H), 4.62 (м, 2H), 3.71 (м, 2H), 3.02 (кв, 2H), 1.94 (м, 4H), 1.41 (т, 3H); MS (ESI+) m/z 303.1 [M+H]+
Стадия 5: В суспензию 1-(4-хлорбутил)-2-этил-1H-имидазо[4,5-c]хинолин-4-амина гидрохлорида (150 мг, 0.442 ммоль) в безводном ДМА (4 мл) добавляли DIPEA (229 мг, 1.79 ммоль, 4 экв.) и тетрагидро-2H-пиран-4-амин (134 мг, 1.326 ммоль, 3 экв.), и смесь перемешивали при 100°C в течение 4.5 дней. ВЭЖХ/МС мониторинг показал образование целевого продукта. Смесь упаривали в вакууме, и остаток очищали методом препаративной ВЭЖХ, получая 67 мг (42%) 2-этил-1-(4-((тетрагидро-2H-пиран-4-ил)амино)бутил)-1H-имидазо[4,5-c]хинолин-4-амина в виде желто-бежевого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6) δ 8.06 (дд, 1H), 7.62 (дд, 1H), 7.42 (ддд, 1H), 7.26 (ддд, 1H), 6.53 (ушир.с, 2H), 4.54 (т, 2H), 3.84 (м, 2H), 3.25 (дд, 2H), 2.96 (кв, 2H), 2.85 (м, 1H), 2.75 (т, 2H), 1.93-1.73 (м, 4H), 1.63 (м, 2H), 1.38 (т, 3H), 1.33 (м, 2H); MS (ESI+) m/z 368.1 [M+H]+
Стадия 6: В грушевидную колбу помещали магнитную мешалку, 2-этил-1-(4-((тетрагидро-2H-пиран-4-ил)амино)бутил)-1H-имидазо[4,5-c]хинолин-4-амин (43 мг, 0.117 ммоль; получен ионообменной хроматографией на Agilent StratoSpheres PL-HCO3 MP смола для удаления следов муравьиной кислоты), 2M раствор NaOH (88 мкл, 0.176 ммоль, 1.5 экв.) и воду (1 мл), получая светло-желтую суспензию. После добавления уксусного ангидрида (22 мкл, 0.234 ммоль, 2 экв.), полученную смесь перемешивали при комнатной температуре до тех пор, пока ТСХ мониторинг (CH2Cl2/MeOH 9:1) и ВЭЖХ/МС показали полное исчезновение исходного вещества через два дня. Реакционную смесь затем напрямую подвергали очистке методом препаративной ТСХ (SiO2 20 см², CH2Cl2/MeOH 9:1), получая 10.4 мг (22%) не совсем белого твердого вещества.
1H ЯМР (300 МГц, CD3OD) (смесь ротамеров) δ 8.09 (м, 1H), 7.71 (д, 1H), 7.50 (т, 1H), 7.35 (т, 1H), 4.59 (квинт, 2H), 4.28 (м, 0.4H), 4.01-3.77 (м, 2.6H), 3.42 (м, 2H), 3.28 (м, 2H), 3.03 (м, 2H), 2.11 (с, 1.8H), 2.07 (с, 1.2H), 2.02-1.54 (м, 8H), 1.53-1.43 (м, 3H); MS (ESI+) m/z 410.1 [M+H]+
Схема 5: синтез Примера 2 и 4
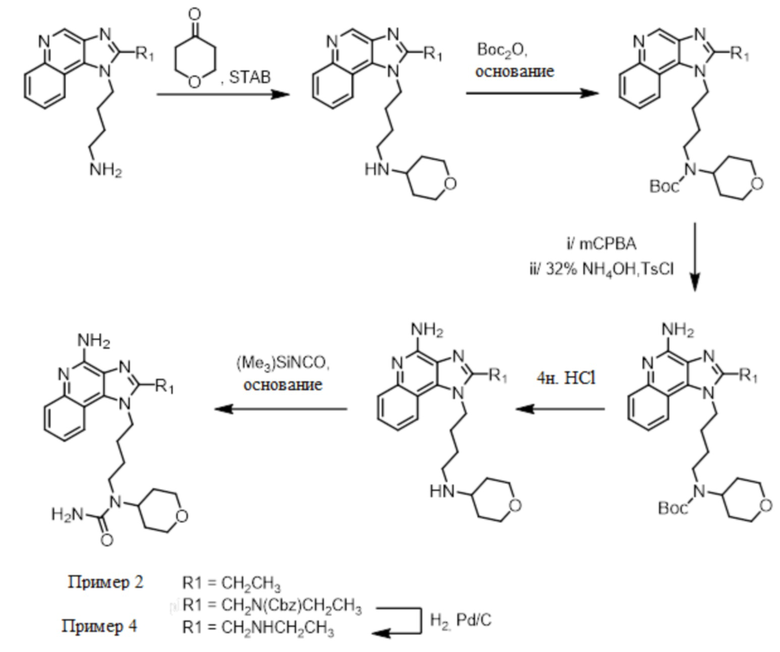
Пример 2
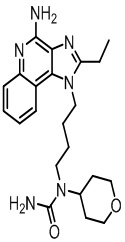
1-(4-(4-Амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-1-(тетрагидро-2H-пиран-4-ил)мочевина
Стадия 1: В круглодонную колбу помещали магнитную мешалку, 4-(2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутан-1-амин (см. вещество D) (1.430 г, 5.329 ммоль), тетрагидро-4H-пиран-4-он (533 мг, 5.329 ммоль, 1 экв.) и 1,2-дихлорэтан (30 мл). После добавления триацетоксиборгидрида натрия (1.581 г, 7.460 ммоль, 1.4 экв.) и уксусной кислоты (320 мг, 5.329 ммоль, 1 экв.), смесь перемешивали при комнатной температуре в течение ночи. Мониторинг реакции методом ВЭЖХ/МС и ТСХ (CHCl3/MeOH/32%-ный раствор аммиака 90:9:1) показал полное исчезновение исходного вещества. Реакционную смесь гасили добавлением 1M раствора NaOH (30 мл), слои разделяли и водную фазу экстрагировали 1,2-дихлорэтаном (3x30 мл). Объединенные органические слои упаривали в вакууме и маслянистый остаток сушили отгоняя азеотроп толуол-вода при пониженном давлении и затем в высоком вакууме. Сырой продукт подвергали колоночной хроматографии на силикагеле (CHCl3/MeOH/32%-ный раствор аммиака от 440:9:1 до 190:9:1), получая 1.652 г (88%) N-(4-(2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)тетрагидро-2H-пиран-4-амина в виде темно-желтого масла.
1H ЯМР (300 МГц, CDCl3) δ 9.30 (с, 1H), 8.27 (м, 1H), 8.19 (м, 1H), 7.64 (м, 2H), 4.55 (т, 2H), 3.96 (м, 2H), 3.37 (м, 2H), 3.00 (кв, 2H), 2.72 (т, 2H), 2.63 (м, 1H), 2.03 (м, 2H), 1.79 (м, 2H), 1.68 (м, 2H), 1.54 (т, 3H), 1.39 (м, 2H); MS (ESI+) m/z 353.4 [M+H]+
Стадия 2: В круглодонную колбу помещали магнитную мешалку, N-(4-(2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)тетрагидро-2H-пиран-4-амин (824 мг, 2.338 ммоль), триэтиламин (473 г, 4.675 ммоль, 2 экв.) и 1,2-дихлорэтан (15 мл), получая желтый раствор. После добавления ди-трет-бутил дикарбоната (510 мг, 2.338 ммоль, 1 экв.) реакционную смесь перемешивали при комнатной температуре в течение ночи. Мониторинг реакции методом ВЭЖХ/МС и ТСХ (CHCl3/MeOH/32%-ный раствор аммиака 90:9:1) показал полное исчезновение исходного вещества. Реакционную смесь промывали 5%-ным раствором лимонной кислоты (15 мл) и насыщенным водным раствором хлорида натрия (15 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали при пониженном давлении, получая 1.006 г (95%) трет-бутил (4-(2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)(тетрагидро-2H-пиран-4-ил)карбамата в виде красно-коричневого масла.
1H ЯМР (300 МГц, CDCl3) δ 9.30 (с, 1H), 8.28 (дд, 1H), 8.13 (дд, 1H), 7.64 (м, 2H), 4.54 (т, 2H), 3.98 (м, 2H), 3.39 (т, 2H), 3.16 (м, 2H), 3.00 (кв, 2H), 1.95 (м, 2H), 1.82-1.49 (м, 9H), 1.40 (с, 9H); MS (ESI+) m/z 453.6 [M+H]+
Стадия 3: В круглодонную колбу помещали магнитную мешалку, трет-бутил (4-(2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)(тетрагидро-2H-пиран-4-ил)карбамат (1.002 г, 2.214 ммоль) и дихлорметан (15 мл), получая темно-желтый раствор. Добавляли порциями твердую 3-хлорпербензойную кислоту (0.955 г, 5.535 ммоль, 2.5 экв.) в раствор в течение 5 минут, и реакционную смесь перемешивали при комнатной температуре до тех пор, пока ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) и ВЭЖХ/МС показали полную конверсию через 3 часа. Добавляли в полученный раствор раствор аммиака (32%, 15 мл), затем п-толуолсульфонилхлорид (1.013 г, 5.313 ммоль, 2.4 экв.) и двухфазную смесь интенсивно перемешивали при комнатной температуре в течение ночи. Мониторинг реакции методом ВЭЖХ/МС и ТСХ (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) показал полное исчезновение исходного вещества. Образовавшиеся два слоя разделяли, и водную фазу экстрагировали дихлорметаном (50 мл). Объединенные органические слои промывали раствором бикарбоната натрия (насыщенный раствор бикарбоната натрия:вода = 1:1, 1x50 мл) и упаривали в вакууме. Остаток подвергали колоночной хроматографии на силикагеле (CHCl3/MeOH/32%-ный раствор аммиака 540:9:1), получая 613 мг (59%) трет-бутил (4-(4-амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)(тетрагидро-2H-пиран-4-ил)карбамата в виде желто-бежевого твердого вещества.
1H ЯМР (300 МГц, CDCl3) (смесь ротамеров) δ 7.90 (дд, 1H), 7.82 (дд, 1H), 7.50 (ддд, 1H), 7.30 (ддд, 1H), 5.42 (ушир.с, 2H), 4.46 (т, 2H), 3.98 (м, 2.5H) 3.39 (т, 2H), 3.18 (2.4H), 2.94 (кв, 2H), 1.93 (м, 2H), 1.82-1.53 (м, 6H), 1.48 (т, 3H), 1.41 (с, 9H); MS (ESI+) m/z 468.3 [M+H]+
Стадия 4: В круглодонную колбу помещали магнитную мешалку, трет-бутил (4-(4-амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)(тетрагидро-2H-пиран-4-ил)карбамат (602 мг, 1.287 ммоль) и безводный 1,4-диоксан (10 мл), получая желто-бежевую суспензию. Добавляли 4н. раствор HCl в 1,4-диоксане (10 мл), и реакционную смесь перемешивали при комнатной температуре 2.5 дня. Мониторинг реакции методом ВЭЖХ/МС и ТСХ (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) показал полное исчезновение исходного вещества. Выпавший осадок отфильтровывали, промывали безводным Et2O (3x20 мл) и деионизировали с помощью ионообменной хроматографии на Agilent StratoSpheres PL-HCO3 MP SPE картридже (содержит нанесенный на полимер четвертичный амин (HCO3- форма) для удаления солей с ТФУК или HCl посредством твердофазной экстракции(SPE) - Agilent, order No. PL3540-G603) для выделения свободного амина. Сырой продукт очищали методом флэш-хроматографии на силикагеле (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1), получая 363 мг 2-этил-1-(4-((тетрагидро-2H-пиран-4-ил)амино)бутил)-1H-имидазо[4,5-c]хинолин-4-амина в виде желто-бежевого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 7.97 (д, 1H), 7.82 (д, 1H), 7.50 (дт, 1H), 7.31 (дт, 1H), 5.39 (ушир.с, 2H), 4.48 (т, 2H), 3.95 (м, 2H), 3.37 (дт, 2H), 2.94 (кв, 2H), 2.70 (т, 2H), 2.63 (м, 1H), 2.01 (м, 2H), 1.79 (м, 2H), 1.65 (м, 2H), 1.48 (т, 3H), 1.37 (м, 2H); MS (ESI+) m/z 368.2 [M+H]+
Стадия 5: В круглодонную колбу помещали магнитную мешалку, 2-этил-1-(4-((тетрагидро-2H-пиран-4-ил)амино)бутил)-1H-имидазо[4,5-c]хинолин-4-амин (367 мг, 0.999 ммоль), триэтиламин (101 мг, 0.999 ммоль, 1 экв.) и безводный хлороформ (5 мл). Полученный желтый раствор охлаждали до 0-4°C в ледяной бане, добавляли триметилсилил изоцианат (127 мг, 1.099 ммоль, 1.1 экв.), и смесь перемешивали при комнатной температуре. Мониторинг реакции методом ВЭЖХ/МС и ТСХ (CHCl3/MeOH/32%-ный раствор аммиака 90:9:1) показал неполное исчезновение исходного вещества через 1 час. Добавляли дополнительное количество триметилсилил изоцианата (127 мг, 1.099 ммоль, 1 экв.) каждые 30 минут в течение следующих 5 часов для достижения полной конверсии. Реакционную смесь упаривали при пониженном давлении, и остаток очищали методом препаративной ТСХ (SiO2 20 cm², CHCl3/MeOH/32%-ный раствор аммиака 90:9:1), получая 273 мг (67%) не совсем белого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6) δ 8.02 (дд, 1H), 7.61 (дд, 1H), 7.41 (ддд, 1H), 7.25 (ддд, 1H), 6.41 (ушир.с, 2H), 5.76 (ушир.с, 2H), 4.49 (т, 2H), 3.96 (м, 1H), 3.85 (дд, 2H), 3.29 (м, 2H), 3.07 (т, 2H), 2.95 (кв, 2H), 1.79 (м, 2H), 1.71-1.53 (м, 4H), 1.44 (м, 2H), 1.38 (т, 3H); MS (ESI+) m/z 411.2 [M+H]+
Пример 4
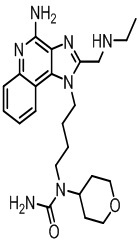
1-(4-(4-Амино-2-((этиламино)метил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-1-(тетрагидро-2H-пиран-4-ил)мочевина
Стадии 1-5: Получали аналогично описанному в Примере 2, используя бензил ((1-(4-аминобутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамат (см. вещество E) (3.5 г, 8.11 ммоль) в качестве исходного вещества, получая 249 мг (5% общий выход за 5 стадий) бензил ((4-амино-1-(4-(1-(тетрагидро-2H-пиран-4-ил)уреидо)бутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамата в виде коричневого масла.
MS (ESI+) m/z 574.4 [M+H]+
Стадия 6: В круглодонную колбу, оснащенную септой для заполнения колбы газом, помещали магнитную мешалку, бензил ((4-амино-1-(4-(1-(тетрагидро-2H-пиран-4-ил)уреидо)бутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамат (245 мг, 0.427 ммоль) и метанол (2 мл), получая бледно-желтый раствор. После добавления 10% Pd/C (45 мг, 0.470 ммоль), колбу подсоединяли к шарику, наполненному водородом, и поочередно вакуумировали и заполняли водородом три раза. Затем систему заполняли водородом, и реакционную смесь перемешивали при атмосферном давлении при комнатной температуре в течение ночи. Добавляли дополнительное количество 10% Pd/C (270 мг, 2.82 ммоль, 6 экв.) в течение следующих 20 часов для достижения полной конверсии по данным ВЭЖХ/МС мониторинга. Затем систему продували аргоном, и катализатор удаляли фильтрованием через тонкий слой CELITE®. Осадок на фильтре промывали метанолом до полного смывания продукта с фильтра, фильтраты объединяли, упаривали при пониженном давлении и сушили в высоком вакууме. Остаток очищали методом препаративной ТСХ (SiO2 20 см², CHCl3/MeOH/32%-ный раствор аммиака 100:10:1), получая 54 мг (28%) белого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6) δ 8.03 (д, 1H), 7.61 (дд, 1H), 7.43 (ддд, 1H), 7.26 (ддд, 1H), 6.46 (ушир.с, 2H), 5.77 (ушир.с, 2H), 4.61 (т, 2H), 4.04 (с, 2H), 4.02-3.79 (м, 3H), 3.34 (м, 2H), 3.09 (т, 2H), 2.63 (кв, 2H), 1.85 (м, 2H), 1.74-1.53 (м, 4H), 1.46 (м, 2H), 1.06 (т, 3H); MS (ESI+) m/z 440.3 [M+H]+
Схема 6: синтез Примера 3
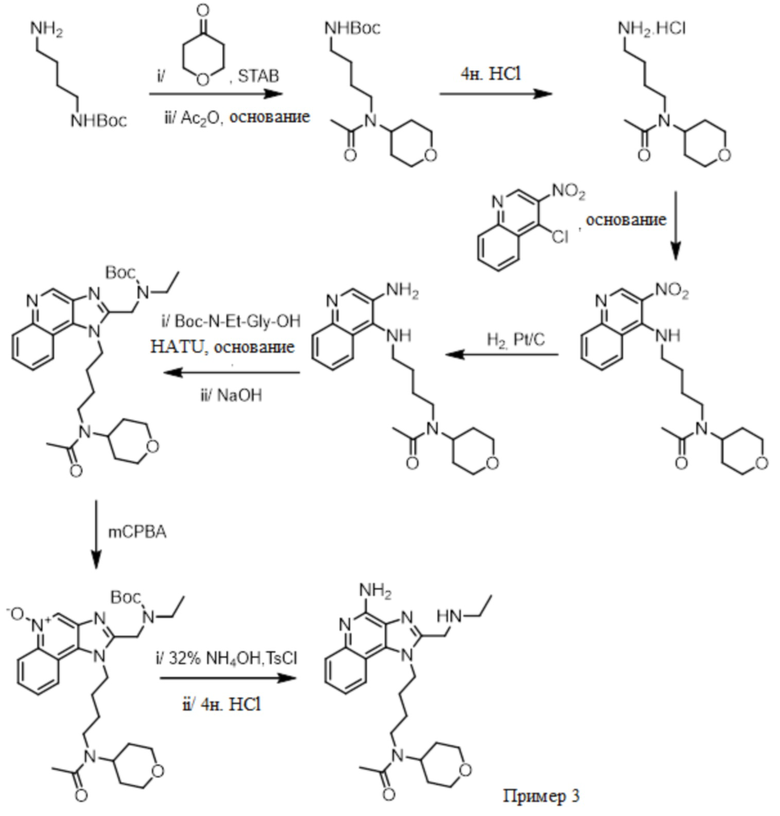
Пример 3
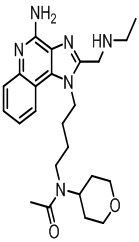
N-(4-(4-Амино-2-((этиламино)метил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-N-(тетрагидро-2H-пиран-4-ил)ацетамид
Стадия 1: Трехгорлую 2-литровую круглодонную колбу, оснащенную октагональной 50 мм x 21 магнитной мешалкой KOMET™, снабжали обратным холодильником, подсоединенным к барботеру с минеральным маслом, и двумя стеклянными пробками. Добавляли N-Boc-путресцин (56.41 г, 299.65 ммоль, 1.5 экв.), CH2Cl2 (500 мл) и молекулярные сита 4 , порошок с размером зерен <5 микрон (Aldrich) (90 г), и смесь перемешивали при комнатной температуре 5 минут. Триацетоксиборгидрид натрия (84.68 г, 399.53 ммоль, 2 экв.) добавляли порциями, и полученную смесь нагревали при 40°C в течение 5 минут при перемешивании. Быстро добавляли раствор оксан-4-она (20.00 г, 199.77 ммоль) в CH2Cl2 (500 мл), и полученную смесь кипятили в течение ночи. Прогресс реакции отслеживали методом ТСХ (CHCl3/MeOH/32%-ный раствор аммиака 90:9:1) и пятна детектировали обработкой нингидриновым реагентом и хлор/о-толуидиновым реагентом. Добавляли дополнительно количество оксан-4-она (4.00 г, 39.96 ммоль, 0.2 экв.) в течение двух следующих дней для достижения полной конверсии. Реакционную смесь затем оставляли охлаждаться до комнатной температуры, добавляли триэтиламин (75.80 г, 749.12 ммоль, 3.75 экв.) и уксусный ангидрид (45.89 г, 449.47 ммоль, 2.25 экв.), и смесь перемешивали при комнатной температуре в течение ночи. Прогресс реакции отслеживали методом ТСХ (CHCl3/MeOH/32%-ный раствор аммиака 90:9:1). Реакционную смесь затем выливали в холодную воду (1.4 л), молекулярные сита удаляли фильтрованием, и дихлорметановый слой отделяли от водного слоя. Водный слой экстрагировали дихлорметаном (3x250 мл), объединенные органические слои промывали насыщенным водным раствором хлорида натрия (2x250 мл), сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Полученный сырой продукт очищали методом флэш-хроматографии на силикагеле (CHCl3/MeOH/32%-ный раствор аммиака 440:9:1), получая 62.8 г (количественный выход) трет-бутил (4-(N-(тетрагидро-2H-пиран-4-ил)ацетамидо)бутил)карбамата в виде оранжевого масла.
, порошок с размером зерен <5 микрон (Aldrich) (90 г), и смесь перемешивали при комнатной температуре 5 минут. Триацетоксиборгидрид натрия (84.68 г, 399.53 ммоль, 2 экв.) добавляли порциями, и полученную смесь нагревали при 40°C в течение 5 минут при перемешивании. Быстро добавляли раствор оксан-4-она (20.00 г, 199.77 ммоль) в CH2Cl2 (500 мл), и полученную смесь кипятили в течение ночи. Прогресс реакции отслеживали методом ТСХ (CHCl3/MeOH/32%-ный раствор аммиака 90:9:1) и пятна детектировали обработкой нингидриновым реагентом и хлор/о-толуидиновым реагентом. Добавляли дополнительно количество оксан-4-она (4.00 г, 39.96 ммоль, 0.2 экв.) в течение двух следующих дней для достижения полной конверсии. Реакционную смесь затем оставляли охлаждаться до комнатной температуры, добавляли триэтиламин (75.80 г, 749.12 ммоль, 3.75 экв.) и уксусный ангидрид (45.89 г, 449.47 ммоль, 2.25 экв.), и смесь перемешивали при комнатной температуре в течение ночи. Прогресс реакции отслеживали методом ТСХ (CHCl3/MeOH/32%-ный раствор аммиака 90:9:1). Реакционную смесь затем выливали в холодную воду (1.4 л), молекулярные сита удаляли фильтрованием, и дихлорметановый слой отделяли от водного слоя. Водный слой экстрагировали дихлорметаном (3x250 мл), объединенные органические слои промывали насыщенным водным раствором хлорида натрия (2x250 мл), сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Полученный сырой продукт очищали методом флэш-хроматографии на силикагеле (CHCl3/MeOH/32%-ный раствор аммиака 440:9:1), получая 62.8 г (количественный выход) трет-бутил (4-(N-(тетрагидро-2H-пиран-4-ил)ацетамидо)бутил)карбамата в виде оранжевого масла.
1H ЯМР (300 МГц, CDCl3) (смесь ротамеров) δ 4.81-4.43 (м, 1.6H), 4.01 (м, 2H), 3.71 (м, 0.4H), 3.56-3.30 (м, 2H), 3.29-3.02 (м, 4H), 2.20-2.00 (м, 3H), 1.93-1.35 (м, 17H). LC-MS анализ не проводили, т.к. соединение не детектируется в УФ.
Стадия 2: В круглодонную колбу помещали магнитную мешалку, раствор трет-бутил (4-(N-(тетрагидро-2H-пиран-4-ил)ацетамидо)бутил)карбамата (62.81 г, 199.76 ммоль) в дихлорметане (1.3 л). Осторожно добавляли 4н. раствор HCl в 1,4-диоксане (1.3 л), и полученную смесь перемешивали при комнатной температуре в течение ночи. Прогресс реакции отслеживали методом ТСХ (CHCl3/MeOH/32%-ный раствор аммиака 240:9:1), и пятна визуализировали с помощью нингидринового реагента. Реакционную смесь упаривали в вакууме, и полученное твердое вещество растирали в диэтиловом эфире, отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме, получая 51.46 г N-(4-аминобутил)-N-(тетрагидро-2H-пиран-4-ил)ацетамида гидрохлорида в виде бежевого твердого вещества. Полученный продукт использовали в следующей стадии без дополнительной очистки.
1H ЯМР (300 МГц, ДМСО-d6) (смесь ротамеров) δ 7.93 (м, 3H, NH3+), 4.25 (м, 0.45H), 3.99-3.71 (м, 2.55H), 3.45-3.24 (м, 2H), 3.23-3.03 (м, 2H), 2.88-2.65 (м, 2H), 2.06 (с, 1.6H), 2.01 (с, 1.4H), 1.89-1.63 (м, 2H), 1.63-1.33 (м, 6H). LC-MS анализ не проводили, т.к. соединение не детектируется в УФ.
Стадия 3: В круглодонную колбу помещали магнитную мешалку, N-(4-аминобутил)-N-(тетрагидро-2H-пиран-4-ил)ацетамид гидрохлорид (неочищенный, 51.46 г, 199.91 ммоль), триэтиламин (121.37 г, 1199.43 ммоль, 6 экв.) и дихлорметан (2.1 л), полученный бледно-желтый раствор охлаждали до 0-4°C в ледяной бане. Осторожно добавляли раствор 4-хлор-3-нитрохинолина (см. вещество A) (45.87 г, 219.90 ммоль) в CH2Cl2 (1.05 л), полученного на предыдущей стадии, смесь перемешивали при 0-4°C в течение 10 минут и затем при комнатной температуре в течение ночи. Прогресс реакции отслеживали методом ТСХ (CHCl3/MeOH/32%-ный раствор аммиака 80:18:2 и 240:9:1) и ВЭЖХ/МС, пятна визуализировали с помощью нингидринового реагента. Реакционную смесь разделяли между водой (6 л) и смесью дихлорметана и метанола (9:1, 1 л). Водный слой отделяли от органического слоя и экстрагировали смесью дихлорметана и метанола (9:1, 3x1 л). Объединенные органические слои сушили над Na2SO4, фильтровали и упаривали в вакууме. Полученный сырой продукт очищали методом флэш-хроматографии на силикагеле (CHCl3/MeOH/32%-ный раствор аммиака 540:9:1), получая 72.89 г (94%) N-(4-((3-нитрохинолин-4-ил)амино)бутил)-N-(тетрагидро-2H-пиран-4-ил)ацетамида в виде красно-желтого масла.
MS (ESI+) m/z 387.0 [M+H]+
Стадия 4: В аппарат Парра объемом 250 мл (сосуд для проведения реакций под давлением, Parr Instrument GmbH, Germany) помещали N-(4-((3-нитрохинолин-4-ил)амино)бутил)-N-(тетрагидро-2H-пиран-4-ил)ацетамид (16.89 г, 43.71 ммоль), 5% платину на угле (8.60 г, 2.19 ммоль, 5 моль.%) и толуол (90 мл). Сосуд помещали в шейкер Парра под давлением 50 фунт/кв.дюйм (3.5 кг/см2). Протекание реакции отслеживали методом ТСХ (CHCl3/MeOH/32%-ный раствор аммиака 240:9:1), реакция завершалась за один час. Катализатор удаляли фильтрованием через тонкий слой CELITE® Hyflo Supercel, осадок на фильтре несколько раз промывали этанолом, и фильтраты объединяли. Летучие компоненты удаляли при пониженном давлении, и сырой продукт очищали методом флэш-хроматографии на силикагеле (CHCl3/MeOH/32%-ный раствор аммиака от 240:9:1 до 140:9:1), получая 14.28 г (92%) N-(4-((3-аминохинолин-4-ил)амино)бутил)-N-(тетрагидро-2H-пиран-4-ил)ацетамида в виде красно-оранжевого масла.
1H ЯМР (300 МГц, CDCl3) (смесь ротамеров) δ 8.49 (с, 0.4H), 8.45 (с, 0.6H), 8.02-7.92 (м, 1H), 7.91-7.73 (м, 1H), 7.53-7.38 (м, 1H), 4.55 (м, 0.6H), 4.16-3.05 (м, 11.4H), 2.14 (с, 1.8H), 2.04 (с, 1.2H), 1-90-1.44 (м, 8H); MS (ESI+) m/z 357.0 [M+H]+
Стадия 5: В круглодонную колбу помещали магнитную мешалку, N-(4-((3-аминохинолин-4-ил)амино)бутил)-N-(тетрагидро-2H-пиран-4-ил)ацетамид (1.00 г, 2.81 ммоль), N-Boc-N-этилглицин (0.68 г, 3.37 ммоль, 1.2 экв.), 2-(3H-[1,2,3]триазолo[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизоурония гексафторфосфат(V) (HATU, 1.28 г, 3.37 ммоль, 1.2 экв.), триэтиламин (0.85 г, 8.42 ммоль, 3 экв.), 4-диметиламинопиридин (0.03 г, 0.28 ммоль, 0.1 экв.) и безводный ДМФА (20 мл), получая красно-желтый раствор. Реакционную смесь перемешивали при комнатной температуре до тех пор, пока ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) и ВЭЖХ/МС показал полную конверсию примерно через 10-12 часов. Растворитель удаляли при пониженном давлении, и остаток, растворенный в этилацетате, промывали водой. Органический слой сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток растворяли в этаноле (20 мл), добавляли 2M раствор NaOH (4.91 мл, 9.82 ммоль, 3.5 экв.), и полученную смесь кипятили до тех пор, пока ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) и ВЭЖХ/МС показал полную конверсию через 18 часов. Реакционную смесь упаривали в вакууме, и остаток разделяли между дихлорметаном (100 мл) и 1M раствором HCl (50 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали в вакууме. Полученный сырой продукт очищали методом флэш-хроматографии на силикагеле (CHCl3/MeOH/32%-ный раствор аммиака 240:9:1), получая 1.24 г (84%) трет-бутил этил((1-(4-(N-(тетрагидро-2H-пиран-4-ил)ацетамидо)бутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)карбамат в виде желто-оранжевого масла.
MS (ESI+) m/z 524.8 [M+H]+
Стадия 6: В круглодонную колбу помещали магнитную мешалку, трет-бутил этил((1-(4-(N-(тетрагидро-2H-пиран-4-ил)ацетамидо)бутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)карбамат (1.24 г, 2.37 ммоль) и хлороформ (100 мл). Добавляли в раствор порциями твердую 3-хлорпербензойную кислоту (1.02 г, 5.92 ммоль, 2.5 экв.) в течение 15 минут, и реакционную смесь перемешивали при комнатной температуре в течение ночи до тех пор, пока ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) и ВЭЖХ/МС показали полную конверсию. Полученный раствор разделяли между хлороформом (100 мл) и насыщенным водным раствором бикарбоната натрия (100 мл), и слои разделяли. Органический слой промывали последовательно насыщенным водным раствором бикарбоната натрия (100 мл) и насыщенным водным раствором хлорида натрия (100 мл), сушили над Na2SO4, фильтровали и упаривали при пониженном давлении, получая 1.33 г (количественный выход) 2-(((трет-бутоксикарбонил)(этил)амино)метил)-1-(4-(N-(тетрагидро-2H-пиран-4-ил)ацетамидо)бутил)-1H-имидазо[4,5-c]хинолин 5-оксида в виде красно-коричневого масла. Полученный продукт использовали в следующей стадии без дополнительной очистки.
MS (ESI+) m/z 540.0 [M+H]+
Стадия 7: В круглодонную колбу помещали магнитную мешалку, 2-(((трет-бутоксикарбонил)(этил)амино)метил)-1-(4-(N-(тетрагидро-2H-пиран-4-ил)ацетамидо) бутил)-1H-имидазо[4,5-c]хинолин 5-оксид (1.33 г, 2.46 ммоль), хлороформ (30 мл) и раствор аммиака (32%, 30 мл). Добавляли в полученную двухфазную смесь п-толуолсульфонилхлорид (0.47 г, 2.46 ммоль, 1 экв.) в один прием, и реакционную смесь интенсивно перемешивали при комнатной температуре в течение ночи до тех пор, пока ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) и ВЭЖХ/МС показали полную конверсию. Реакционную смесь разбавляли хлороформом (70 мл), и фазы разделяли. Водный слой доводили до pH 7 добавлением 2M раствора HCl и экстрагировали хлороформом (2x100 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (150 мл), сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии на силикагеле (CHCl3/MeOH/32%-ный раствор аммиака 240:9:1), получая 430 мг желто-бежевого твердого вещества. Его растирали в горячих растворителях (последовательно этилацетат, ацетон и метанол), охлаждали и отфильтровывали для удаления побочных продуктов. Часть полученного твердого продукта растворяли в хлороформ (25 мл) и добавляли раствор 4н. раствор HCl в 1,4-диоксане (25 мл) при комнатной температуре на ночь для снятия защитной Boc-группы. После упаривания летучих компонентов в вакууме, остаток очищали методом флэш-хроматографии на силикагеле (CHCl3/MeOH/32%-ный раствор аммиака 90:9:1), получая 202 мг не совсем белого твердого вещества.
1H ЯМР (300 МГц, CDCl3) (смесь ротамеров) δ 7.91 (т, 1H), 7.83 (м, 1H), 7.52 (м, 1H), 7.33 (т, 1H), 5.42 (ушир.с, 2H), 4.59 (м, 2.4H), 4.10 (с, 2H), 4.01 (м, 2H), 3.71 (м, 0.6H), 3.42 (м, 2H), 3.25 (дт, 2H), 2.79 (кв, 2H), 2.13 (с, 1.8H), 2.09 (с, 1.2H), 1.98 (м, 2H), 1.86-1.53 (м, 6H), 1.18 (т, 3H); MS (ESI+) m/z 439.3 [M+H]+
Схема 7: синтез Примера 5 и 6
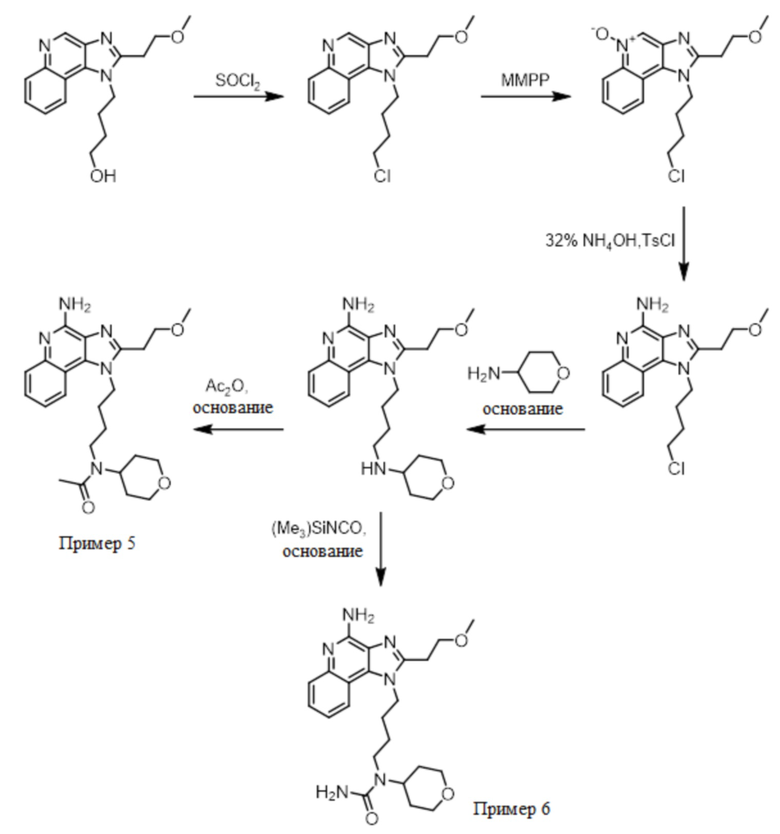
Пример 5
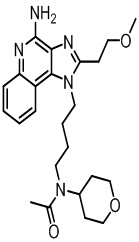
N-(4-(4-Амино-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-N-(тетрагидро-2H-пиран-4-ил)ацетамид
Стадия 1: В круглодонную колбу помещали магнитную мешалку, 4-(2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)бутан-1-ол (см. вещество C) (9.48 г неочищенного продукта, соответствует ~14.47 ммоль исходного соединения ) и дихлорэтан (400 мл), получая желто-коричневый раствор. После добавления тионилхлорида (6.89 мл, 95.00 ммоль, 6.5 экв.) смесь перемешивали при комнатной температуре 12 часов до тех пор, пока ТСХ мониторинг (CH2Cl2/MeOH 9:1) и ВЭЖХ/МС не подтвердили полную конверсию. Летучие компоненты удаляли при пониженном давлении, остаток растворяли в дихлорметане, подщелачивали триэтиламином (7.45 г, 73.67 ммоль, 5 экв.) и очищали методом флэш-хроматографии на силикагеле (CH2Cl2/MeOH 95:5), получая 3.35 г (72%, из расчета на 14.47 ммоль исходного соединения) 1-(4-хлорбутил)-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолина в виде красновато-коричневого масла.
MS (ESI+) m/z 317.9 319.8 [M+H]+
Стадия 2: В реакционную колбу помещали магнитную мешалку, 1-(4-хлорбутил)-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолина гидрохлорид (3.35 г, 9.46 ммоль) и воду (300 мл), получая красновато-коричневую суспензию. После добавления магния монопероксифталата гексагидрата (MMPP) (4.68 г, 9.46 ммоль, 1 экв.) смесь перемешивали при 60°C. Через 2 часа добавляли магния монопероксифталата гексагидрат (4.68 г, 9.46 ммоль, 1 экв.), и смесь дополнительно перемешивали при 60°C в течение 90 минут до тех пор, пока ТСХ мониторинг (CH2Cl2/MeOH 9:1) и ВЭЖХ/МС не подтвердили полную конверсию. Реакционную смесь экстрагировали хлороформом (3x100 мл), и объединенные органические слои промывали насыщенным раствором бикарбоната натрия (1x50 мл). Бикарбонатный слой экстрагировали хлороформом (3x50 мл), объединенные органические слои промывали насыщенным водным раствором хлорида натрия (100 мл), сушили над Na2SO4, фильтровали и упаривали в вакууме получая 1.75 г (55%) 1-(4-хлорбутил)-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин 5-оксида в виде темно-желтого масла. Полученное вещество использовали в следующей стадии без дополнительной очистки.
MS (ESI+) m/z 333.9 335.8 [M+H]+
Стадия 3: В круглодонную колбу помещали магнитную мешалку, 1-(4-хлорбутил)-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин 5-оксид (1.75 г, 5.24 ммоль), хлороформ (50 мл) и раствор аммиака (32%, 50 мл). Добавляли в полученную двухфазную смесь 4-метилбензол-1-сульфонилхлорид (1.20 г, 6.29 ммоль, 1.2 экв.) в один прием, и смесь интенсивно перемешивали при комнатной температуре в течение ночи. ТСХ мониторинг (CH2Cl2/MeOH 9:1) и ВЭЖХ/МС подтвердили полную конверсию и образование целевого продукта. Смесь разбавляли хлороформом (100 мл) и насыщенным водным раствором хлорида натрия (150 мл). Органический слой отделяли от водного слоя и промывали насыщенным водным раствором хлорида натрия (2x100 мл), сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Оставшееся коричнево-желтое масло очищали методом препаративной ВЭЖХ, получая 586 мг (34%) 1-(4-хлорбутил)-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-4-амина в виде желтого твердого вещества.
MS (ESI+) m/z 332.9 334.8 [M+H]+
Стадия 4: В герметично закрытой виале перемешивали смесь1-(4-хлорбутил)-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-4-амина (150 мг, 0.451 ммоль), иодида натрия (68 мг, 0.451 ммоль, 1 экв.), N-этил-N-изопропилпропан-2-амина (116 мг, 0.901 ммоль, 2 экв.), 4-аминотетрагидропирана (137 мг, 1.352 ммоль, 3 экв.) и молекулярных сит 4 в виде порошка с размером зерен <5 микрон (Aldrich) (750 мг) в безводном ДМА (4.5 мл) при 100°C в течение 4 дней, до тех пор пока ВЭЖХ/МС мониторинг не подтвердил полную конверсию и образование целевого продукта. Реакционную смесь фильтровали и упаривали при пониженном давлении, получая 302 мг 2-(2-метоксиэтил)-1-(4-((тетрагидро-2H-пиран-4-ил)амино)бутил)-1H-имидазо[4,5-c]хинолин-4-амина в виде коричневого остатка. Полученное вещество использовали в следующей стадии без дополнительной очистки.
MS (ESI+) m/z 398.0 [M+H]+
Стадия 5: В грушевидную колбу помещали магнитную мешалку, 2-(2-метоксиэтил)-1-(4-((тетрагидро-2H-пиран-4-ил)амино)бутил)-1H-имидазо[4,5-c]хинолин-4-амин (302 мг, 0.760 ммоль), 2н. раствор NaOH (3.799 мл, 7.597 ммоль, 10 экв.) и воду (5 мл), получая желтый раствор. После добавления уксусного ангидрида (0.776 г, 7.597 ммоль, 10 экв.) реакционную смесь перемешивали при комнатной температуре 24 часа, прогресс реакции отслеживали методом ВЭЖХ/МС. Затем добавляли 2н. раствор NaOH (15.194 мл, 30.388 ммоль, 40 экв.) и уксусный ангидрид (3.102 г, 30.388 ммоль, 40 экв.), и смесь дополнительно перемешивали при комнатной температуре еще 24 часа. Реакционную смесь фильтровали, и продукт выделяли методом препаративной ВЭЖХ, получая 89 мг (27%) коричневато-красного твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6) δ 8.02 (дд, 1H), 7.61 (д, 1H), 7.42 (т, 1H), 7.24 (т, 1H), 6.42 (с, 2H), 4.54 (м, 2H), 4.24-3.68 (м, 5H), 3.45-3.09 (м, 9H), 2.01 (д, 3H), 1.89-1.34 (м, 8H); MS (ESI+) m/z 439.9 [M+H]+
Пример 6
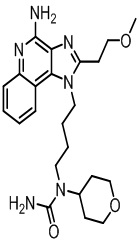
1-(4-(4-Амино-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-1-(тетрагидро-2H-пиран-4-ил)мочевина
В круглодонную колбу помещали магнитную мешалку, 2-(2-метоксиэтил)-1-(4-((тетрагидро-2H-пиран-4-ил)амино)бутил)-1H-имидазо[4,5-c]хинолин-4-амин (получен в Примере 5, Стадии 1-4) (281 мг, 0.706 ммоль), триэтиламин (71 мг, 0.706 ммоль, 1 экв.) и безводный хлороформ (5 мл), получая желтый раствор. Полученный раствор охлаждали до 0-4°C в ледяной бане, и после добавления триметилсилил изоцианата (89 мг, 0.776 ммоль, 1.1 экв.) смесь перемешивали при комнатной температуре. Мониторинг реакции методом ВЭЖХ/МС и ТСХ (CHCl3/MeOH/32%-ный раствор аммиака 90:9:1) показал неполное исчезновение исходного вещества через 1 час. Добавляли дополнительное количество триметилсилил изоцианата (81 мг, 0.706 ммоль, 1 экв.) каждые 30 минут в течение следующих 3.5 часов для достижения полной конверсии. Реакцию гасили добавлением воды (5 мл) и перемешивали дополнительно при комнатной температуре 30 минут. Смесь разбавляли добавлением 100 мл абсолютного этанола и затем упаривали при пониженном давлении примерно до половины объема. Добавляли еще 100 мл абсолютного этанола, и раствор упаривали в вакууме. Полученный сырой продукт очищали методом ТСХ (SiO2 20 см², CHCl3/MeOH/32%-ный раствор аммиака 90:9:1), получая 91 мг (29%) не совсем белого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6) δ 8.02 (дд, 1H), 7.61 (дд, 1H), 7.42 (ддд, 1H), 7.25 (ддд, 1H), 6.42 (с, 2H), 5.77 (ушир.с, 2H), 4.52 (т, 2H), 4.04-3.76 (м, 5H), 3.38-3.24 (м, 5H), 3.19 (т, 2H), 3.08 (т, 2H), 1.88-1.53 (м, 6H), 1.45 (м, 2H); MS (ESI+) m/z 441.5 [M+H]+
Схема 8: синтез Примера 7-11 (для иллюстративных целей)
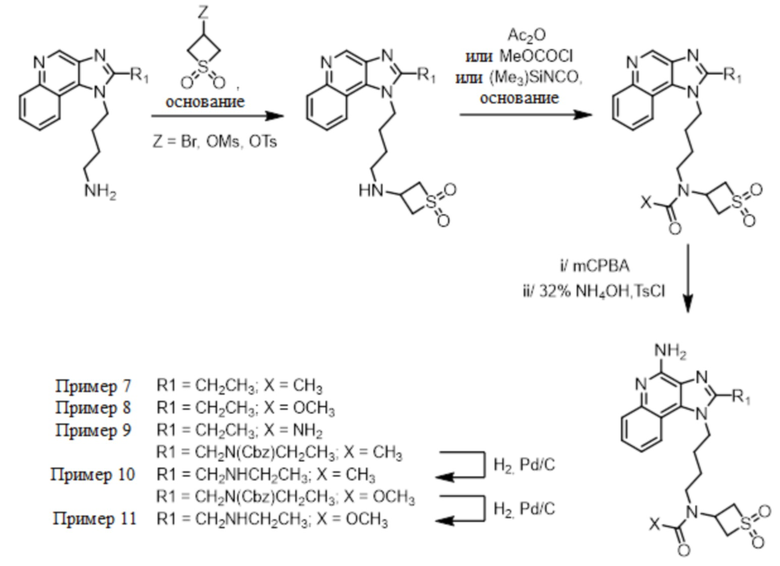
Пример 7 (для иллюстративных целей)
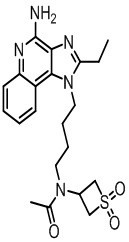
N-(4-(4-Амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-N-(1,1-диоксидотиетан-3-ил)ацетамид
Стадия 1: 4-(2-Этил-1H-имидазо[4,5-c]хинолин-1-ил)бутан-1-амин (см. вещество D) (10.4 г, 38.7 ммоль) суспендировали в смеси MeOH/ТГФ (1:1.5 об/об, 150 мл) и добавляли по каплям раствор триэтиламина (51.9 мл, 373.5 ммоль, 9.65 экв.) в смеси MeOH/ТГФ (1:1 об/об, 20 мл). Затем добавляли порциями 3-бромтиетан 1,1-диоксид (9.3 г, 50.3 ммоль, 1.3 экв.), и полученный раствор перемешивали при 70°C до тех пор, пока ТСХ мониторинг (CH2Cl2/MeOH 9:1) и ВЭЖХ/МС показали почти полную конверсию через 18 часов. Реакционную смесь затем оставляли охлаждаться до комнатной температуры и упаривали при пониженном давлении. Коричневый остаток разделяли между CH2Cl2 и водой, органическую фазу несколько раз промывали водой и, объединенные водные фазы экстрагировали несколько раз дихлорметаном. Объединенные органические фазы сушили над MgSO4, отфильтровывали и упаривали. Полученное твердое вещество затем промывали последовательно этилацетатом и петролейным эфиром и сушили в вакууме, получая 3-((4-(2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)амино)тиетан 1,1-диоксид (5.68 г, 39%) в виде бежевого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6) δ 9.14 (с, 1H), 8.36 (м, 1H), 8.14 (м, 1H), 7.69 (м, 2H), 4.60 (т, 2H), 4.25 (м, 2H), 3.85 (м, 2H), 3.58 (м, 1H), 3.01 (кв, 2H), 2.50 (м, 2H), 1.87 (м, 2H), 1.57 (м, 2H), 1.42 (т, 3H); MS (ESI+) m/z 373.1 [M+H]+
Стадия 2: В раствор 3-((4-(2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)амино)тиетан 1,1-диоксида (3.14 г, 8.42 ммоль) в CH2Cl2 (30 мл) при 0°C добавляли триэтиламин (3.5 мл, 25.3 ммоль, 3 экв.), растворенный в CH2Cl2 (5 мл), и уксусный ангидрид (3.1 мл, 33.7 ммоль, 4 экв.), растворенный в CH2Cl2 (5 мл). Смесь перемешивали 1 час при 0°C и затем при комнатной температуре 18 часов до тех пор, пока ВЭЖХ/МС мониторинг показал полную конверсию. Реакционную смесь отфильтровывали и разделяли фильтрат между CH2Cl2 и водой. Органическую фазу три раза промывали водой, и объединенные водные фазы промывали два раза дихлоремтаном. Объединенные органические слои сушили над MgSO4, отфильтровывали и упаривали. Полученное твердое вещество затем промывали последовательно этилацетатом и петролейным эфиром и сушили в вакууме, получая N-(1,1-диоксидотиетан-3-ил)-N-(4-(2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)ацетамид (2.16 г, 62%) в виде не совсем белого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6) δ 9.14 (с, 1H), 8.35 (м, 1H), 8.15 (м, 1H), 7.69 (м, 2H), 4.72-4.12 (м, 7H), 3.39 (т, 2H), 3.02 (кв, 2H), 2.03 (с, 3H), 1.76 (м, 4H), 1.42 (т, 3H); MS (ESI+) m/z 415.0 [M+H]+
Стадия 3: В перемешиваемый на магнитной мешалке раствор N-(1,1-диоксидотиетан-3-ил)-N-(4-(2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)ацетамида (330 мг, 0.796 ммоль) в CH2Cl2 (20 мл) добавляли пероксиуксусную кислоту (38-40%-ный раствор в уксусной кислоте, 398 мкл, 2.388 ммоль, 3 экв.) при комнатной температуре, и полученную смесь перемешивали при кипячении в течение 3 часов. Полноту конверсии мониторили методом ВЭЖХ/МС. Смесь затем охлаждали до комнатной температуры, разбавляли водой (40 мл) и перемешивали 5 минут. Органический растворитель упаривали, а водную фазу лиофилизовывали, получая сырой 1-(4-(N-(1,1-диоксидотиетан-3-ил)ацетамидо)бутил)-2-этил-1H-имидазо[4,5-c]хинолин 5-оксид (340 мг) в виде белого порошка (MS (ESI+) m/z 430.8 [M+H]+). Этот твердый продукт растворяли в CH2Cl2 (15 мл) и добавляли избыток 32%-ного водного раствора гидроксида аммония (1 мл). Затем добавляли по каплям тозилхлорид (151 мг, 0.790 моль, 1 экв.) в CH2Cl2 (3 мл) в интенсивно перемешиваемую смесь при комнатной температуре и продолжали перемешивание до тех пор. пока ТСХ (CH2Cl2/MeOH 9:1) и ВЭЖХ/МС мониторинг показали полную конверсию через 1.5 часа. Смесь упаривали при пониженном давлении, и остаток диспергировали ультразвуком в MeOH (5 мл). Полученное твердое вещество отфильтровывали и промывали метанолом, получая 160 мг (47%) белого порошка. Альтернативно, эти две последовательные химические реакции проводили в одном реакционном сосуде без выделения N-оксидного интермедиата.
1H ЯМР (300 МГц, ДМСО-d6) δ 8.02 (д, 1H), 7.62 (дд, 1H), 7.42 (ддд, 1H), 7.25 (ддд, 1H), 6.43 (ушир.с, 2H), 4.93-4.13 (м, 7H), 3.39 (м, 2H), 2.95 (кв, 2H), 2.03 (с, 3H), 1.74 (м, 4H), 1.38 (т, 3H); MS (ESI+) m/z 430.2 [M+H]+
Пример 8 (для иллюстративных целей)
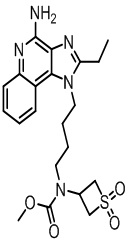
Метил (4-(4-амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)(1,1-диоксидотиетан-3-ил)карбамат
Получали аналогично описанному в Примере 7, исходя из 600 мг (1.61 ммоль) 3-((4-(2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)амино)тиетан 1,1-диоксида (со стадии 1) и используя метил хлорформиат (2 экв.) для образования карбамата, получая 190 мг (23% общий выход за 3 стадии) тозилатной соли в виде не совсем белого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6) δ 8.08 (д, 1H), 7.72 (д, 1H), 7.55 (т, 1H), 7.47 (д, 0.8H, 2xCH тозилата), 7.40 (т, 1H), 7.10 (д, 0.8H, 2xCH тозилата), 4.62-4.24 (м, 7H), 3.60 (с, 3H), 3.33 (м, 2H), 2.98 (кв, 2H), 2.28 (с, 1.2H, CH3 тозилата), 1.84-1.57 (м, 4H), 1.39 (т, 3H); MS (ESI+) m/z 446.0 [M+H]+
Пример 9 (для иллюстративных целей)
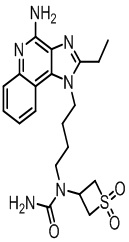
1-(4-(4-Амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-1-(1,1-диоксидотиетан-3-ил)мочевина
Получали аналогично описанному в Примере 7, исходя из 80 мг (0.215 ммоль) 3-((4-(2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)амино)тиетан 1,1-диоксида (со стадии 1) и используя триметилсилил изоцианат (4 экв.) для образования мочевины, получая 8.5 мг (9% общий выход за 3 стадии) бежевого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6) (смесь ротамеров) δ 8.17 (д, 1H), 8.08 (ушир.с, 0.5H), 7.77 (д, 1H), 7.64 (т, 1H), 7.50 (т, 1H), 6.14 (ушир.с, 2H), 5.39 (ушир.с, 1.5H), 4.67-4.14 (м, 7H), 3.50 (м, 2H), 2.99 (м, 2H), 1.78 (м, 2H), 1.66 (м, 2H), 1.40 (м, 3H); MS (ESI+) m/z 431.2 [M+H]+
Пример 10 (для иллюстративных целей)
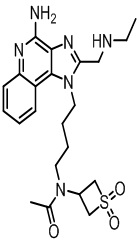
N-(4-(4-Амино-2-((этиламино)метил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-N-(1,1-диоксидотиетан-3-ил)ацетамид
Стадии 1-3: Получали аналогично описанному в Примере 7, используя бензил ((1-(4-аминобутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамат (см. вещество E) (1 г, 2.317 ммоль) в качестве исходного вещества, получая 261 мг (19% за 3 стадии) бензил ((4-амино-1-(4-(N-(1,1-диоксидотиетан-3-ил)ацетамидо)бутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамата в виде не совсем белой пены.
1H ЯМР (300 МГц, CDCl3) δ 7.85 (м, 2H), 7.53 (ддд, 1H), 7.43-7.28 (м, 6H), 5.46 (ушир.с, 2H), 5.23 (с, 2H), 4.84 (с, 2H), 4.66-4.19 (м, 7H), 3.55-3.22 (м, 4H), 2.07 (с, 3H), 1.97-1.56 (м, 4H), 1.10 (т, 3H); MS (ESI+) m/z 573.3 [M+H]+
Стадия 4: В круглодонную колбу, оснащенную септой для заполнения колбы газом, помещали магнитную мешалку, бензил ((4-амино-1-(4-(N-(1,1-диоксидотиетан-3-ил)ацетамидо)бутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамат (261 мг, 0.440 ммоль) и метанол (20 мл), получая бесцветный раствор. После добавления 10% палладия на активированном угле (469 мг, 0.440 ммоль, 1 экв.), колбу подсоединяли к шарику, наполненному водородом, и поочередно вакуумировали и заполняли водородом три раза. Затем систему заполняли водородом, и реакционную смесь перемешивали при атмосферном давлении при комнатной температуре в течение ночи. ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 90:9:1) и ВЭЖХ/МС показали неполную конверсию. Дополнительное количество 10% палладия на активированном угле (0.469 г, 0.440 ммоль) и перемешивание в течение ночи потребовалось для достижения полной конверсии. Затем систему продували аргоном и удаляли катализатор фильтрованием через тонкий слой CELITE® Hyflo Supercel на крупнопористом (пористость = 16-40 мкМ) стеклянном фильтре. Осадок на фильтре промывали метанолом (3x50 мл), объединенные фильтраты фильтровали через 0.45 мкМ PTFE мембрану для удаления остатков катализатора, и упаривали при пониженном давлении. Остаток очищали методом препаративной ТСХ (SiO2 20 см², CHCl3/MeOH/32%-ный раствор аммиака 90:9:1), получая 108 мг (53%) не совсем белого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 7.86 (м, 2H), 7.53 (ддд, 1H), 7.34 (ддд, 1H), 5.70 (ушир.с, 2H), 4.64 (т, 2H), 4.61-4.21 (м, 5H), 4.10 (с, 2H), 3.42 (т, 2H), 2.79 (кв, 2H), 2.10 (с, 3H), 2.03 (м, 2H), 1.78 (м, 2H), 1.17 (т, 3H); MS (ESI+) m/z 459.2 [M+H]+
Пример 11
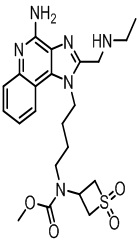
Метил (4-(4-амино-2-((этиламино)метил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)(1,1-диоксидотиетан-3-ил)карбамат
Получали аналогично описанному в Примере 10, исходя из 1 г (0.317 ммоль) бензил ((1-(4-аминобутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамата (см. вещество E) и используя метил хлорформиат (1 экв.) для образования карбамата, получая 131 мг (12% общий выход за 4 стадии) не совсем белого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 7.86 (м, 2H), 7.52 (ддд, 1H), 7.33 (ддд, 1H), 5.62 (ушир.с, 2H), 4.60 (т, 2H), 4.54-4.35 (м, 3H), 4.33-4.18 (м, 2H), 4.09 (с, 2H), 3.73 (с, 3H), 3.39 (т, 2H), 2.78 (кв, 2H), 1.99 (м, 2H), 1.73 (м, 2H), 1.17 (т, 3H); MS (ESI+) m/z 475.2 [M+H]+
Схема 9: синтез Примера 12 (для иллюстративных целей)
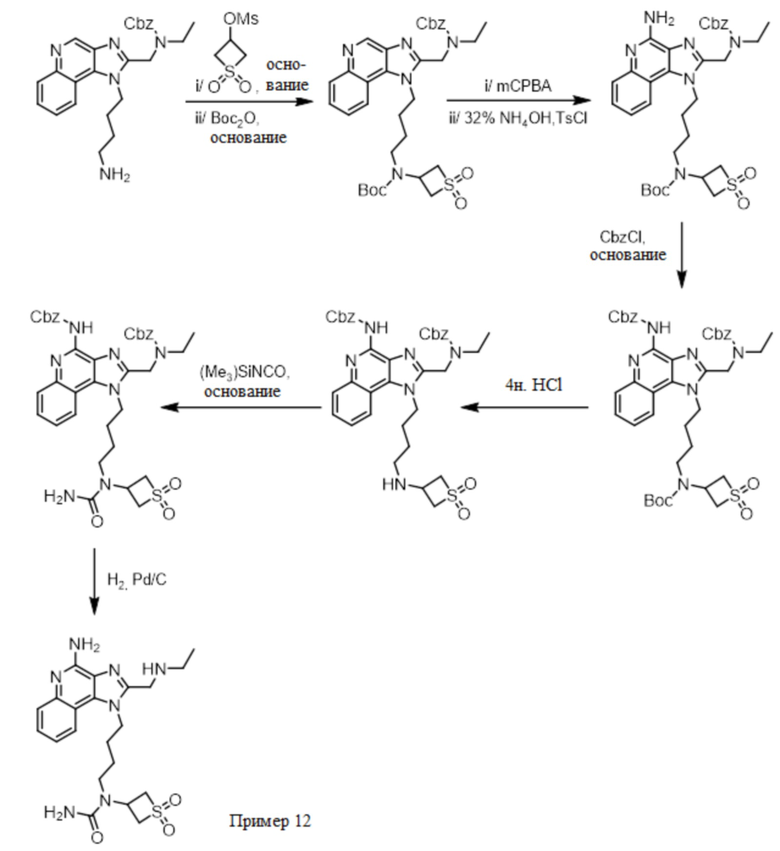
Пример 12
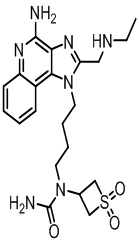
1-(4-(4-Амино-2-((этиламино)метил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-1-(1,1-диоксидотиетан-3-ил)мочевина
Стадия 1: В 50-миллилитровую двугорлую круглодонную колбу, оснащенную обратным холодильником, помещали магнитную мешалку, бензил ((1-(4-аминобутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамат (см. вещество E) (1.000 г, 2.317 ммоль), 1,1-диоксидотиетан-3-ил метансульфонат (557 мг, 2.781 ммоль, 1.2 экв.), DIPEA (1.797 г, 13.904 ммоль, 6 экв.) и смесь ТГФ/вода (4:1 об/об, 20 мл), получая желтую суспензию, которую кипятили до тех пор, пока ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 90:9:1) и ВЭЖХ/МС показали полное исчезновение исходного вещества через 2 часа. Реакционную смесь затем оставляли охлаждаться до комнатной температуры. Затем добавляли ди-трет-бутил дикарбонат (Boc2O) (506 мг, 2.317 ммоль, 1 экв.), N,N-диизопропилэтиламин (599 мг, 4.635 ммоль, 2 экв.) и 4-диметиламинопиридин (28 мг, 0.232 ммоль), и полученную смесь перемешивали при комнатной температуре. Через 18 часов ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) и ВЭЖХ/МС показали неполную конверсию. Добавляли еще Boc2O (506 мг, 2.317 ммоль, 1 экв.), и реакционную смесь дополнительно перемешивали при 60°C в течение ночи. ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) и ВЭЖХ/МС все еще показывали неполную конверсию. Добавляли еще Boc2O (1.011 г, 4.635 ммоль, 2 экв.) и DIPEA (599 мг, 4.635 ммоль, 2 экв.) каждые 80 минут на протяжении следующих 4 часов, и смесь дополнительно перемешивали при 60°C для достижения полной конверсии. Реакционную смесь затем упаривали при пониженном давлении, остаток растворяли в CH2Cl2 (100 мл) и промывали последовательно 10%-ным водным раствором CuSO4 (2x50 мл) и насыщенным водным раствором NaHCO3 (2x50 мл). Органическую фазу сушили над MgSO4, фильтровали и упаривали при пониженном давлении, получая 1.48 г (количественный выход, сырой продукт) трет-бутил (4-(2-((((бензилокси)карбонил)(этил)амино)метил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)(1,1-диоксидотиетан-3-ил)карбамата в виде красновато-желтого масла, которое использовали в следующей стадии без дополнительной очистки.
MS (ESI+) m/z 636.4 [M+H]+
Стадия 2: В круглодонную колбу помещали магнитную мешалку, неочищенный трет-бутил (4-(2-((((бензилокси)карбонил)(этил)амино)метил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)(1,1-диоксидотиетан-3-ил)карбамат (1.480 г, 2.328 ммоль) и CH2Cl2 (20 мл), получая красновато-желтый раствор. Добавляли порциями твердую 3-хлорпербензойную кислоту (1.004 г, 5.820 ммоль, 2.5 экв.) в течение 5 минут, и полученную смесь перемешивали при комнатной температуре 3 часа. ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) и ВЭЖХ/МС показали полную конверсию. Добавляли в раствор аммиака (32%, 20 мл), затем п-толуолсульфонилхлорид (1.065 г, 5.587 ммоль, 2.4 экв.), и двухфазную смесь интенсивно перемешивали при комнатной температуре в течение ночи. ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) и ВЭЖХ/МС показали полную конверсию. Образовавшиеся два слоя разделяли, органический слой промывали водой (20 мл), сушили над MgSO4, отфильтровывали и упаривали в вакууме, получая 1.515 г трет-бутил (4-(4-амино-2-((((бензилокси)карбонил) (этил)амино)метил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)(1,1-диоксидотиетан-3-ил)карбамата в виде красновато-желтого масла, которое использовали в следующей стадии без дополнительной очистки.
MS (ESI+) m/z 651.4 [M+H]+
Стадия 3: В круглодонную колбу с септой и системой подачи аргона помещали неочищенный трет-бутил (4-(4-амино-2-((((бензилокси)карбонил)(этил)амино)метил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)(1,1-диоксидотиетан-3-ил)карбамат (1.515 г, 2.328 ммоль) и CH2Cl2 (20 мл), получая красновато-желтый раствор. Затем добавляли триэтиламин (283 мг, 2.793 ммоль, 1.2 экв.), 4-диметиламинопиридин (28 мг, 0.233 ммоль, 0.1 экв.) и бензил хлорформиат (477 мг, 2.793 ммоль, 1.2 экв.), и реакционную смесь перемешивали при комнатной температуре 2 часа. Дополнительное количество бензил хлорформиата (2.54 г, 14.89 ммоль, 6.4 экв.) и триэтиламина (1.51 г, 14.89 ммоль, 6.4 экв.) добавляли порциями в течение следующих 3 часов, и реакционную смесь дополнительно перемешивали при комнатной температуре в течение ночи до тех пор, пока ТСХ мониторинг (CHCl3/MeOH/32%-ный раствор аммиака 140:9:1) и ВЭЖХ/МС показали полную конверсию. Затем смесь выливали в насыщенный водный раствор NH4Cl (100 мл) и экстрагировали дихлорэтаном (3x50 мл). Объединенные органические слои сушили над MgSO4, отфильтровывали и упаривали в вакууме. Остаток очищали методом флэш-хроматографии на силикагеле (30% EtOAc/EtOH 3:1 в петролейном эфире, содержащем 2% концентрированного раствора аммиака), получая 257 мг (14%) бензил ((4-(((бензилокси)карбонил)амино)-1-(4-((трет-бутоксикарбонил)(1,1-диоксидотиетан-3-ил)амино)бутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамата в виде желтого масла.
MS (ESI+) m/z 785.6 [M+H]+
Стадия 4: В 10-миллилитровую круглодонную колбу, оснащенную магнитной мешалкой, помещали бензил ((4-(((бензилокси)карбонил)амино)-1-(4-((трет-бутоксикарбонил)(1,1-диоксидотиетан-3-ил)амино)бутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамат (257 мг, 0.327 ммоль) и безводный 1,4-диоксан (5 мл), получая желтый раствор. После добавления 4н. раствора HCl в 1,4-диоксане (2.5 мл) реакционную смесь перемешивали при комнатной температуре 60 минут. ТСХ мониторинг (50% EtOAc/EtOH 3:1 в петролейном эфире, содержащем 2% концентрированного раствора аммиака) показали полную конверсию. Реакционную смесь упаривали в вакууме, остаток растворяли в метаноле и деионизировали с помощью ионообменной хроматографии на Agilent StratoSpheres PL-HCO3 MP SPE картридже (см.выше), получая 230 мг (количественный выход, сырой продукт) бензил ((4-(((бензилокси)карбонил)амино)-1-(4-((1,1-диоксидотиетан-3-ил)амино)бутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил) карбамата в виде желтого масла. Полученное вещество использовали в следующей стадии без дополнительной очистки.
MS (ESI+) m/z 685.4 [M+H]+
Стадия 5: В круглодонную колбу помещали магнитную мешалку, бензил ((4-(((бензилокси)карбонил)амино)-1-(4-((1,1-диоксидотиетан-3-ил)амино)бутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил)карбамат (230 мг, 0.336 ммоль), триэтиламин (47 мкл, 0.336 ммоль, 1 экв.) и безводный хлороформ (5 мл), и полученный желтый раствор охлаждали до 0-4°C в ледяной бане. После добавления триметилсилил изоцианата (46 мкл, 0.336 ммоль, 1 экв.) реакционную смесь перемешивали при комнатной температуре. Прогресс реакции отслеживали методом ТСХ (70% EtOAc/EtOH 3:1 в петролейном эфире, содержащем 2% концентрированного раствора аммиака) и ВЭЖХ/МС. Через 45 минут добавляли дополнительное количество триметилсилил изоцианата (одну каплю) и перемешивали при комнатной температуре еще 2 часа для достижения полной конверсии. Реакционную смесь затем гасили добавлением воды (1 мл) и перемешивали при комнатной температуре еще 30 минут. Затем смесь разбавляли абсолютным этанолом (20 мл) и упаривали при пониженном давлении примерно до половины объема (~10 мл). Добавляли абсолютный этанол (20 мл), и раствор упаривали в вакууме. Остаток растворяли в небольшом количестве метанола, наносили на EXtrelut®NT (order No. 1.15092.1000, Merck KgaA) и очищали методом флэш-хроматографии на силикагеле (40% EtOAc/EtOH 3:1 в петролейном эфире, содержащем 2% концентрированного раствора аммиака), получая 110 мг (45%) бензил ((4-(((бензилокси)карбонил)амино)-1-(4-(1-(1,1-диоксидотиетан-3-ил)уреидо)бутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил) карбамата в виде светло-желтого твердого вещества.
MS (ESI+) m/z 728.4 [M+H]+. Не проводили исследований методом ЯМР из-за нерастворимости полученного соединения в обычных ЯМР растворителях.
Стадия 6: В круглодонную колбу, оснащенную септой для заполнения колбы газом, помещали магнитную мешалку, бензил ((4-(((бензилокси)карбонил)амино)-1-(4-(1-(1,1-диоксидотиетан-3-ил)уреидо)бутил)-1H-имидазо[4,5-c]хинолин-2-ил)метил)(этил) карбамат (97 мг, 0.133 ммоль) и метанол (20 мл), получая бесцветную суспензию. После добавления 10% палладия на активированном угле (142 мг, 0.133 ммоль, 1 экв.) колбу подсоединяли к шарику, наполненному водородом, и поочередно вакуумировали и заполняли водородом три раза. Затем систему заполняли водородом, и реакционную смесь перемешивали при атмосферном давлении при комнатной температуре в течение ночи. ТСХ мониторинг (70% EtOAc/EtOH 3:1 в петролейном эфире, содержащем 2% концентрированного раствора аммиака) и ВЭЖХ/МС показал полное исчезновение исходного вещества. Систему продували аргоном и удаляли катализатор фильтрованием через тонкий слой CELITE® Hyflo Supercel на крупнопористом (пористость = 16-40 мкМ) стеклянном фильтре. Осадок на фильтре промывали MeOH (3x50 мл), объединенные фильтраты фильтровали через 0.45 мкМ PTFE мембрану для удаления остатков катализатора и упаривали при пониженном давлении, получая 30 мг (49%) желто-бежевого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6) δ 8.03 (дд, 1H), 7.61 (дд, 1H), 7.43 (ддд, 1H), 7.27 (ддд, 1H), 6.46 (с, 2H), 6.12 (с, 2H), 4.60 (т, 2H), 4.56-4.37 (м, 3H), 4.33-4.19 (м, 2H), 4.03 (с, 2H), 3.25 (м, 2H), 2.63 (кв, 2H), 1.83 (м, 2H), 1.66 (м, 2H), 1.06 (т, 3H); MS (ESI+) m/z 460.1 [M+H]+
Схема 10: синтез Примера 13-15 (для иллюстративных целей)
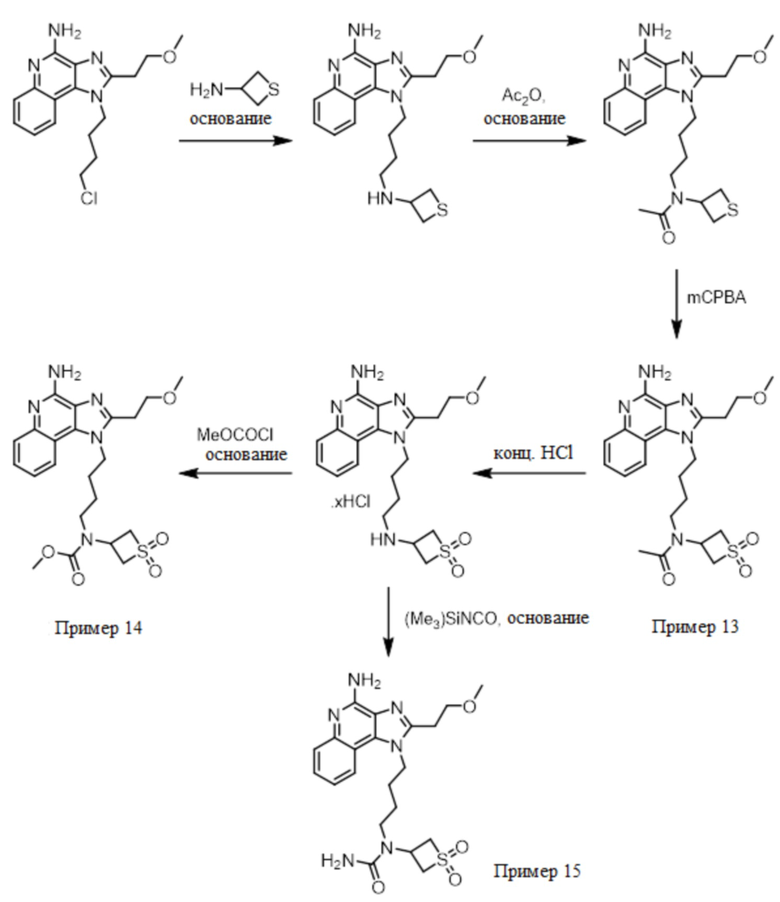
Пример 13 (для иллюстративных целей)
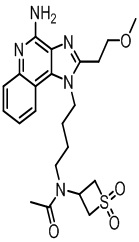
N-(4-(4-Амино-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-N-(1,1-диоксидотиетан-3-ил)ацетамид
Стадия 1: В раствор 1-(4-хлорбутил)-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-4-амин (получен в Примере 5, Стадии 1-3) (286 мг, 0.859 ммоль) в безводном ДМА (10 мл) добавляли иодид натрия (129 мг, 0.859 ммоль, 1 экв.), N-этил-N-изопропилпропан-2-амин (449 мкл, 2.578 ммоль, 3 экв.), тиетан-3-амин гидрохлорид (341 мг, 2.578 ммоль, 3 экв.) и молекулярные сита 4 в виде порошка с размером зерен <5 микрон (Aldrich) (1.5 г). Желтую реакционную смесь перемешивали при 100°C до тех пор, пока ВЭЖХ/МС мониторинг не подтвердил полную конверсию и образование целевого продукта через 4 дня. Реакционную смесь отфильтровывали и упаривали при пониженном давлении, получая 504 мг (неочищенный) 2-(2-метоксиэтил)-1-(4-(тиетан-3-иламино)бутил)-1H-имидазо[4,5-c]хинолин-4-амин в виде коричневого остатка. Полученный сырой продукт использовали в следующей стадии без дополнительной очистки.
MS (ESI+) m/z 385.9 [M+H]+
Стадия 2: В грушевидную колбу помещали магнитную мешалку, неочищенный 2-(2-метоксиэтил)-1-(4-(тиетан-3-иламино)бутил)-1H-имидазо[4,5-c]хинолин-4-амин (504 мг, 1.3 ммоль), 2M раствор NaOH (6.536 мл, 13.07 ммоль, 10 экв.) и воду (5 мл), получая коричневатую суспензию. После добавления уксусного ангидрида (1.22 мл, 13.07 ммоль, 10 экв.) реакционную смесь перемешивали при комнатной температуре в течение ночи. ВЭЖХ/МС мониторинг показал полное исчезновение исходного вещества. Реакционную смесь отфильтровывали и выделяли продукт методом препаративной ВЭЖХ, получая 91 мг (16%) N-(4-(4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-N-(тиетан-3-ил)ацетамида в виде желто-бежевого твердого вещества.
MS (ESI+) m/z 427.9 [M+H]+
Стадия 3: В грушевидную колбу помещали магнитную мешалку, N-(4-(4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-N-(тиетан-3-ил)ацетамид (91 мг, 0.213 ммоль), хлороформ (5 мл) и метанол (5 мл), получая светло-желтый раствор. Добавляли порциями в раствор твердую 3-хлорпербензойную кислоту (110 мг, 0.638 ммоль, 3 экв.), и реакционную смесь перемешивали при комнатной температуре в течение ночи. ВЭЖХ/МС мониторинг показал неполное прохождение реакции. Добавляли еще 3-хлорпербензойную кислоту (73 мг, 0.426 ммоль, 2 экв.) и перемешивали в течение ночи для достижения полной конверсии исходного вещества. Полученный раствор разделяли между хлороформом (50 мл) и насыщенным водным раствором NaHCO3 (50 мл). Слои разделяли, органический слой промывали последовательно насыщенным водным раствором NaHCO3 (50 мл) и насыщенным водным раствором хлорида натрия (50 мл), сушили над Na2SO4, отфильтровывали и упаривали при пониженном давлении. Полученный сырой продукт очищали методом препаративной ВЭЖХ, получая 6.4 мг (7%) не совсем белого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6) δ 8.03 (д, 1H), 7.64 (дд, 1H), 7.46 (ддд, 1H), 7.30 (ддд, 1H), 6.42 (с, 2H), 4.66-4.16 (м, 7H), 3.84 (т, 2H), 3.39 (т, 2H), 3.30 (с, 3H), 3.20 (т, 2H), 2.03 (с, 3H), 1.86-1.62 (м, 4H); MS (ESI+) m/z 459.8 [M+H]+
Пример 14 (для иллюстративных целей)
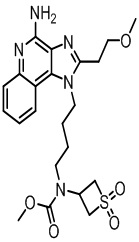
Метил (4-(4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)(1,1-диоксидотиетан-3-ил)карбамат
Стадия 1: N-(4-(4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-N-(1,1-диоксидотиетан-3-ил)ацетамид (Пример 13) (440 мг, 0.95 ммоль) суспендировали в метаноле (12 мл), после чего добавляли концентрированную HCl (2.5 мл). Смесь перемешивали при 100°C в условиях микроволнового облучения до тех пор, пока ВЭЖХ/МС мониторинг показал почти полную конверсию через 2.5 часа. Реакционную смесь разбавляли дихлорметаном (200 мл) и промывали насыщенным водным раствором NaHCO3. Водную фазу экстрагировали дихлорметаном дважды, и объединенные органические фазы сушили над Na2SO4, отфильтровывали и упаривали при пониженном давлении, получая 380 мг (87%) 3-((4-(4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)амино)тиетан 1,1-диоксида гидрохлорида в виде светло-желтого аморфного вещества, которое использовали в следующей стадии без дополнительной очистки.
MS (ESI+) m/z 418.4 [M+H]+
Стадия 2: Метил хлорформиат (21 мкл, 0.27 ммоль, 1 экв.) добавляли при комнатной температуре в суспензию 3-((4-(4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)амино)тиетан 1,1-диоксида гидрохлорида (114 мг, 0.27 ммоль) и K2CO3 (94 мг, 0.81 ммоль, 3 экв.) в H2O (3 мл), и полученную смесь перемешивали в течение ночи при комнатной температуре. ВЭЖХ/МС мониторинг показал неполную конверсию. Потребовалось дополнительно количество метил хлорформиата (21 мкл, 0.27 ммоль, 1 экв.) и перемешивание в течение 1 часа для достижения полной конверсии исходного вещества. Реакционную смесь затем разбавляли дихлорметаном (200 мл) и промывали насыщенным водным раствором NaHCO3. Водную фазу отбрасывали, органическую фазу сушили над Na2SO4, отфильтровывали и упаривали при пониженном давлении. Полученный сырой продукт очищали методом препаративной ТСХ (SiO2 20 см², CH2Cl2/метанол/32%-ный раствор аммиака от 190:9:1 до 230:18:2, затем CH2Cl2/метанол/32%-ный раствор аммиака 150:9:1), получая 50 мг (38%) белого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6) δ 8.0 (д, 1H), 7.62 (дд, 1H), 7.42 (ддд, 1H), 7.26 (ддд, 1H), 6.43 (с, 2H), 4.59-4.24 (м, 7H), 3.83 (т, 2H), 3.59 (с, 3H), 3.33 (т, 2H), 3.30 (с, 3H), 3.18 (т, 2H), 1.69 (м, 4H); MS (ESI+) m/z 476.5 [M+H]+
Пример 15 (для иллюстративных целей)
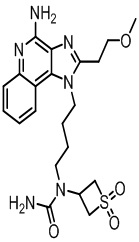
1-(4-(4-Амино-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-1-(1,1-диоксидотиетан-3-ил)мочевина
3-((4-(4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)амино) тиетан 1,1-диоксида гидрохлорид (получен в Примере 14, Стадия 1) (270 мг, 0.64 ммоль) и триэтиламин (404 мкл, 2.91 ммоль, 4.5 экв.) смешивали в хлороформе (10 мл). Добавляли триметилсилил изоцианат (96 мкл, 0.71 ммоль, 1.1 экв.) при 0°C, и смесь перемешивали при комнатной температуре 2 часа. Протекание реакции отслеживали методом ТСХ (CH2Cl2/CHCl3/метанол/32%-ный раствор аммиака 100:80:18:2) и ВЭЖХ/МС. Потребовалось дополнительное количество триметилсилил изоцианата (192 мкл, 1.40 ммоль, 2.2 экв.) и перемешивание при комнатной температуре в течение ночи для достижения полной конверсии исходного вещества. Реакционную смесь отфильтровывали, фильтрат разбавляли дихлорметаном (200 мл) и промывали насыщенным водным раствором NaHCO3. Водную фазу дважды экстрагировали дихлорметаном, объединенные органические слои сушили над Na2SO4, отфильтровывали и упаривали при пониженном давлении. Полученный сырой продукт очищали методом препаративной ТСХ (CH2Cl2/метанол/32%-ный раствор аммиака 150:9:1, затем 180:18:2), получая 34 мг (11%) не совсем белого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6) δ 8.04 (д, 1H), 7.63 (д, 1H), 7.44 (т, 1H), 7.29 (т, 1H), 6.57 (ушир.с, 2H), 6.13 (с, 2H), 4.73-4.17 (м, 7H), 3.84 (т, 2H), 3.30 (м, 5H), 3.19 (т, 2H), 1.78 (м, 2H), 1.65 (м, 2H); MS (ESI+) m/z 461.4 [M+H]+
Пример A - In vitro тестирование:
1) Анализ индуцирования выработки IFN-α - индуцирование выработки IFN-α TLR-агонистами в одноядерных клетках периферической крови человека (PBMC)
Сокращения: BSA = бычий сывороточный альбумин; PBS = фосфатный буфер с pH 7.4; PE = фикоэритрин; EDC = 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид.
Материалы и оборудование: Прибор Bio Plex 200 Luminex, и программа BioPlex Manager; фильтровальные планшеты MultiScreen (Millipore, MABVN1250); реакционные пробирки объемом 1.5 мл со слабым связыванием (Sarstedt, 72.706.600); активационный буфер: 0.1M раствор Na-дигидрофосфата, 0.1M раствор ди-Na гидрофосфата pH 6.2; раствор сульфо-NHS (N-гидроксисульфосукцинимид): 50 мг/мл в воде (свежеприготовленный); раствор EDC: 50 мг/мл в воде (свежеприготовленный); буфер для связывания: PBS; отмывочный буфер: PBS + 0.05% Tween20; блокирующий буфер: PBS + 10 мг/мл BSA + 0.05% азид натрия; аналитический буфер: PBS + 10 мг/мл BSA;
Другие материалы и оборудование (пипетки, реакционные сосуды, шейкер и т.д.) являются стандартным лабораторным оборудованием; вода имела уровень качества MilliQ, буферные растворы фильтровали в стерильных условиях перед использованием.
Преинкубация клеток:
В каждой лунке с U-образным дном 96-луночного планшета преинкубировали 200000 клеток PBMC в среде (RPMI1640 -Gibco #61870-044 + 10% FBS + 1% L-глутамин) в течение 5 часов при 37°C и 5% CO2, после чего добавляли соединения по настоящему изобретению или контроль, соответственно. После добавления соединений по настоящему изобретению или контроля, клетки инкубировали еще 24 часа при 37°C и 5% CO2.
Luminex тест, Стадия 1: Связывание антител с бусинами:
- перенести суспензию (бусины хранят в буферном растворе от производителя) примерно 5 миллионов микросфер MicroPlex Microspheres (= бусины) в реакционную пробирку со слабым связыванием
- центрифугировать суспензию бусин 1 минуту при 10000g, отбросить супернатант и ресуспендировать полученную пеллету в 160 мкл активационного буфера, повторить один раз
- добавить 20 мкл раствора сульфо-NHS и 20 мкл раствора EDC, перемешать на вихревой мешалке
- инкубировать 20 минут без света
- центрифугировать суспензию бусин 1 минуту при 10000g, отбросить супернатант и ресуспендировать полученную пеллету в 500мкл буфера для связывания, повторить один раз
- добавить захватывающее антитело (CaptureAK eBioscience # BMS160, 5 мкг на 1 миллион бусин)
- инкубировать 2 часа при комнатной температуре в отсутствие света при встряхивании
- центрифугировать суспензию бусин 1 минуту при 10000g, отбросить супернатант и ресуспендировать полученную пеллету в 500мкл отмывочного буфера, повторить один раз
- центрифугировать суспензию бусин 1 минуту при 10000g, отбросить супернатант и ресуспендировать полученную пеллету в 100мкл блокирующего буфера
Luminex тест, Стадия 2: Измерение
- добавить 100мкл аналитического буфера в каждую лунку фильтровального планшета (неиспользуемые лунки заклеить липкой пленкой), затем удалить буфер
- добавить примерно 2000 бусин на лунку на аналит (на аналит, применяют разные типы бусин с разным цветом флуоресценции), суспендировать в 50мкл аналитического буфера
- удалить буфер и промыть два раза по 100мкл отмывочного буфера на лунку
- на лунку, добавить 50мкл раствора образца со стадии преинкубирования (надосадочный раствор над клетками), или стандарт (IFN-α standard eBioscience #BMS216MST - градиент концентрации, наивысшая стандартная концентрация 2500 пг/мл) или аналитический буфер (как холостые опыты)
- встряхнуть планшет
- инкубировать планшет 2 часа при комнатной температуре в отсутствие света при встряхивании
- промыть три раза по 100мкл отмывочного буфера на лунку
- добавить 25мкл смеси для детектирования антител (DetectionAK eBioscience # BMS1016BT разбавленный 1:1000) в аналитическом буфере на лунку, затем перемешать планшет на вихревой мешалке
- инкубировать планшет 1 час при комнатной температуре в отсутствие света при встряхивании
- промыть три раза по 100мкл отмывочного буфера на лунку
- добавить 50мкл Стрептавидин/PE (разбавленный 1:200 в аналитическом буфере) на лунку, затем перемешать планшет на вихревой мешалке
- инкубировать планшет 10 минут при комнатной температуре в отсутствие света при встряхивании
- промыть три раза по 100мкл отмывочного буфера на лунку
- добавить 100мкл аналитического буфера на лунку, затем перемешать планшет на вихревой мешалке
- начать Luminex измерение согласно инструкциям производителя, замеряя значение по меньшей мере для 50 бусин на лунку на аналит.
Соединения тестировали в 8 разных концентрациях (1 мкМ, 0.3 мкМ, 0.1 мкМ, 0.03 мкМ, 0.01 мкМ, 0.003 мкМ, 0.001 мкМ, 0.0003 мкМ), применяя полулогарифмическое разведение. Высвобождение интерферона-альфа (IFN-α) дает колоколообразное распределение в выбранном окне разбавлений и измеряется в пг/мл. Наивысшая точка в кривой концентрация-ответ (т.е. максимум колоколообразной кривой) соответствует максимальному высвобождению IFN-α. Для всех протестированных соединений максимальная стимуляция происходит при разных концентрациях.
Соединения из Примеров 1-6 показали следующие результаты:
Таблица 1
(пг/мл)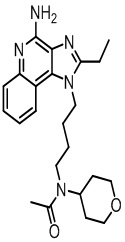
Для сравнения, 3-{ацетил[4-(4-амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил]амино}-2,5-ангидро-1,3,4-тридеоксипентинол, соединение сравнения, описанное в WO-A-2009/118296 как пример III, показало всего около 50% от максимального высвобождения IFN-α для Примера 1 по настоящему изобретению. Кроме того, как можно увидеть из данных в Таблице 2, соответствующие соединения сравнения, имеющие 2-метилтетрагидрофуран-3-ильную группу вместо тетрагидропиран-4-ильной группы в положении R3, показали гораздо более низкое максимальное высвобождение IFN-α.
Таблица 2
(пг/мл)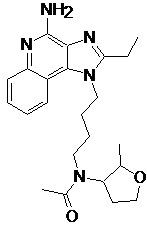
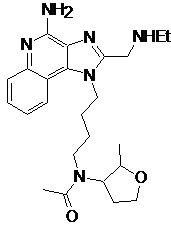
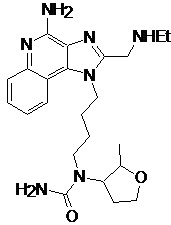
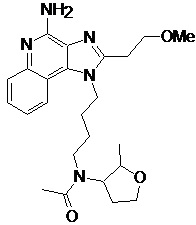
2) Тест стимулирования TLR:
Соединения тестировали на предмет стимулирования человеческих TLR7 и TLR8, соответственно, через экспрессию репортёрного гена, которую исследовали в линиях клеток, устойчиво трансфицированных человеческим TLR7 и TLR8, соответственно, и коэкспрессирующих NFkB/AP-1 индуцируемый репортёрный ген SEAP. Стимулирование TLR лигандом приводит к активации NFkB/AP-1 и усилению секреции SEAP, концентрацию которого можно измерить в надосадочном растворе с помощью системы детектирования QuantiBlue™ (InvivoGen).
Линии клеток представляли собой либо HEK-Blue-hTLR7 (InvivoGen #hkb-htlr7), либо HEK-Blue-hTLR8 (InvivoGen #hkb-htlr8). Клетки выращивали согласно инструкциям производителя.
Анализ:
- приготовить суспензии каждой из линий клеток с Nv = 0.11×106 клеток/мл в среде (DMEM 4,5г/л глюкоза, 2-4 мM L-глутамин + 10% FBS) без антибиотиков (Nv = число живых клеток),
- добавить 180 мкл полученных суспензий на лунку в 96-луночный микропланшет с плоскодонными лунками (coster, # 3506)
- инкубировать планшеты в течение ночи при 37°C и 5% CO2
- приготовить образцы и контрольные образцы в виде растворов в ДМСО: R848, CL075 (агонист TLR 7/8 от InvivoGen) и гардиквимод, каждый в финальной концентрации 10мкM, ODN2006 (InvivoGen) и ODN2216 (InvivoGen) каждый в финальной концентрации 2мкM; соединения по настоящему изобретению тестировали в сериях разбавлений с финальными концентрациями 30, 10, 3, 1 мкM и т.д.,
- добавить в лунки 20 мкл растворов соединений и контрольных образцов (см. выше),
- инкубировать планшеты 20-24 часа при 37°C и 5% CO2
- приготовить раствор QuantiBlue™ (InvivoGen #10H19-MM) согласно инструкциям производителя: солюбилизировать порошок из одного пакетика в 100 мл воды со степенью очистки MilliQ, нагревать при 37°C в водяной бане в течение примерно 30 минут, профильтровать полученный раствор через фильтровальную бумагу,
- добавить 100 мкл приготовленного раствора QuantiBlue™ в каждую лунку 96-луночного микропланшета с плоскодонными лунками,
- добавить на лунку 25 мкл надосадочного раствора культур клеток из индуцированных лунок,
- инкубировать планшеты 1 - 1,5 часа при 37°C (до появления интенсивного синего цвета),
- считать OD при 620 нм на планшет-ридере,
- вычислить значение EC50 на основе полученных результатов.
Соединение из Примера 1 показало значение EC50 = 44 нM в тесте на TLR7 с репортёрным геном. Для сравнения, 3-{ацетил[4-(4-амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил]амино}-2,5-ангидро-1,3,4-тридеоксипентинол, соединение сравнения, описанное в WO-A-2009/118296 как пример III, показало в 10 раз большее значение EC50 = 440 нM в тесте на TLR7 с репортёрным геном.
Кроме того, соединение из Примера 1 не показало активности в отношении TLR8 в описанном выше тесте на TLR8 с репортёрным геном. Однако, соответствующее соединение сравнения без ацетильной группы в положении R2 (т.е., 2-этил-1-(4-((тетрагидро-2H-пиран-4-ил)амино)бутил)-1H-имидазо[4,5-c]хинолин-4-амин, описанное в WO-A-2009/118296 как пример V), показало активность в отношении TLR8 в таком же тесте на TLR8 с репортерным геном, но не продемонстрировало никакой активности в отношении TLR7 вплоть до концентрации 30 мкМ в описанном выше тесте на TLR7 с репортерным геном. Таким образом, полученные данные демонстрируют, что соединения по настоящему изобретению являются селективными агонистами TLR7 по сравнению с TLR8. В частности, авторы настоящего изобретения обнаружили, что двойное замещение в аминогруппе NR2R3 в Формуле (I) связано с TLR7-селективностью, в то время как моно-замещение в аминогруппе NR2R3 в Формуле (I) (т.е., когда R2 представляет собой H) дает соединения, которые селективны к TLR8.
Пример B - In vivo тестирование:
В описанных ниже экспериментах, соединения по настоящему изобретению тестировали на яванских макаках в in vivo животной модели. Определяли - вызывают ли исследуемые соединения (и при какой дозировке) секрецию IFN-α.
В частности, определенную однократную дозу испытуемого соединения вводили внутривенно яванским макакам в ходе 30-минутной инфузии. Через несколько интервалов времени после начала внутривенного введения соединения брали у животных образцы крови (0.5 мл для примерно 0.25 мл плазмы) из vena cephalica antebrachii или vena saphena в K3EDTA пробирки. Образцы крови хранили на измельченном льду до центрифугирования. Получали плазму центрифугированием при 4°C и примерно 1800 g в течение 10 минут, отбирали аликвоты в помеченные микропробирки и хранили замороженными при температуре -70°C или ниже. Образцы плазмы оттаивали, разводили и использовали для определения уровня IFN-α с применением набора IFN-α Elisa Kit (например, VeriKineTM Cynomolgus/Rhesus IFN-α ELISA Kit) согласно инструкциям производителя.
Полученные результаты показывают, что соединения по настоящему изобретению вызывают секрецию IFN-α in vivo у яванских макак, в то время как введение носителя не приводит к возникновению детектируемых концентраций IFN-α. Пиковые концентрации IFN-α в крови обычно достигаются примерно через 150 минут после начала введения соединения. В частности, введение однократной дозы, равной 1, 3 и 10 мг/кг, соединения из Примера 1 по настоящему изобретению дает пиковые концентрации IFN-α в плазме от примерно 80000 до примерно 100000 пг/мл. Самой низкой дозировкой, протестированной для соединения из Примера 1, было 0.1 мг/кг, что приводило к секреции IFN-α до уровня примерно 15000 пг/мл, а дозировка 0.3 мг/кг приводила к пиковой концентрации IFN-α в плазме от примерно 40000 до примерно 80000 пг/мл. Введение однократной дозы, равной 10, 3 или 1 мг/кг, соединения из Примера 5 по настоящему изобретению приводило к пиковой концентрации IFN-α в плазме от примерно 40000 до примерно 120000 пг/мл, от примерно 12000 до примерно 110000 пг/мл, и от примерно 12000 до примерно 120000 пг/мл, соответственно. Самой низкой дозировкой, протестированной для соединения из Примера 5, было 0.03 мг/кг, что приводило к пиковой концентрации IFN-α в плазме от примерно 390 до примерно 10800 пг/мл.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 4-АМИНО-ИМИДАЗОХИНОЛИНА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, ОПОСРЕДОВАННЫХ АГОНИСТАМИ TLR7 И/ИЛИ TLR8 | 2015 |
|
RU2698902C2 |
| ИНГИБИТОРЫ КИНАЗ, ПРИМЕНИМЫЕ ДЛЯ ЛЕЧЕНИЯ МИЕЛОПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ И ДРУГИХ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2007 |
|
RU2482112C2 |
| СОЕДИНЕНИЯ ДЛЯ ИНГИБИРОВАНИЯ КИНАЗЫ EGFR, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2020 |
|
RU2832925C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗО[4,5-С]ПИРИДИНА В КАЧЕСТВЕ АГОНИСТОВ TOLL-ПОДОБНОГО РЕЦЕПТОРА | 2020 |
|
RU2785124C1 |
| ИМИДАЗОХИНОЛИНЗАМЕЩЕННЫЙ СЛОЖНЫЙ ЭФИР ФОСФОРНОЙ КИСЛОТЫ В КАЧЕСТВЕ АГОНИСТА, ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2020 |
|
RU2812182C1 |
| НОВЫЕ СОЕДИНЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2785126C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| ПРОИЗВОДНОЕ РЕЗОРЦИНА В КАЧЕСТВЕ ИНГИБИТОРА HSP90 | 2016 |
|
RU2697703C2 |
| НОВЫЕ ИНГИБИТОРЫ БАКТЕРИАЛЬНЫХ ГЛУТАМИНИЛОВЫХ ЦИКЛАЗ ДЛЯ ИСПОЛЬЗОВАНИЯ В ЛЕЧЕНИИ ПЕРИОДОНТАЛЬНЫХ И СВЯЗАННЫХ С НИМИ ЗАБОЛЕВАНИЙ | 2019 |
|
RU2805298C2 |
| 2-ПИРИДИЛЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ALK5 И/ИЛИ ALK4 | 2012 |
|
RU2612958C2 |
Изобретение относится к соединению, имеющему общую формулу (I), или его фармацевтически приемлемым солям, которые могут найти применение в качестве агонистов толл-подобного рецептора TLR7. В формуле (I) (i) R1 выбран из группы, состоящей из C1-6-алкила, C1-3-алкокси-C1-3-алкила и C1-3-алкиламино-C1-3-алкила; R2 выбран из -CONH2; R3 представляет собой тетрагидропиран-4-ил и n представляет собой целое число от 3 до 6; или (ii) R1 выбран из группы, состоящей из C1-3-алкокси-C1-3-алкила или C1-3-алкиламино-C1-3-алкила; R2 выбран из -CO-R5; R3 представляет собой тетрагидропиран-4-ил; n представляет собой целое число от 3 до 6 и R5 выбран из C1-6-алкила. Изобретение относится также к конкретным соединениям, фармацевтической композиции для индуцирования выработки интерферона-альфа (IFN-α), содержащей предлагаемые соединения, способу лечения рака и их применению для изготовления лекарственного средства для лечения рака. 5 н. и 6 з.п. ф-лы, 2 табл., 15 пр.
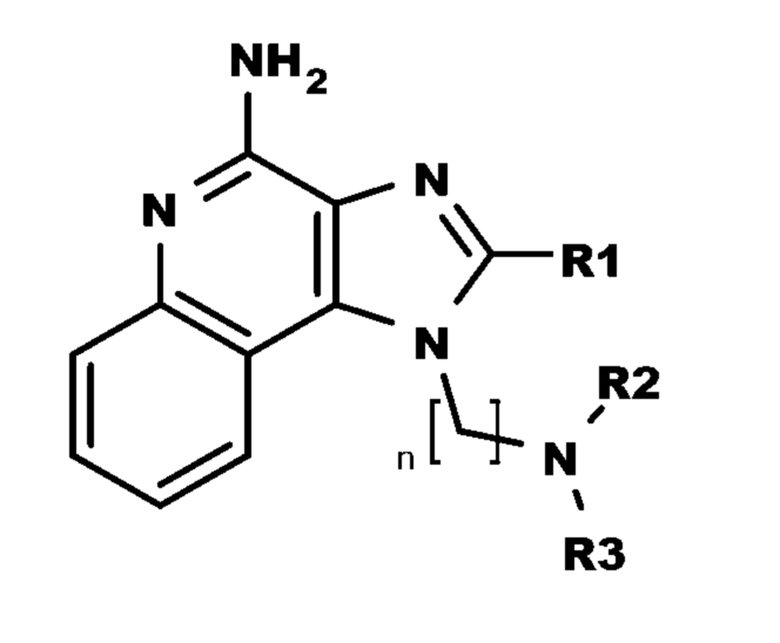
1. Соединение, имеющее общую формулу (I)
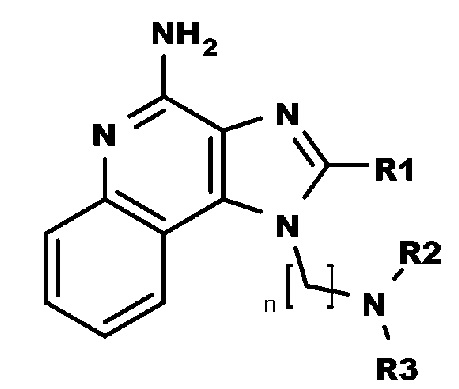 ,
,
где
(i)
R1 выбран из группы, состоящей из C1-6-алкила, C1-3-алкокси-C1-3-алкила и C1-3-алкиламино-C1-3-алкила;
R2 выбран из -CONH2;
R3 представляет собой тетрагидропиран-4-ил и
n представляет собой целое число от 3 до 6;
или его фармацевтически приемлемые соли;
или где
(ii)
R1 выбран из группы, состоящей из C1-3-алкокси-C1-3-алкила или C1-3-алкиламино-C1-3-алкила;
R2 выбран из -CO-R5;
R3 представляет собой тетрагидропиран-4-ил;
n представляет собой целое число от 3 до 6 и
R5 выбран из C1-6-алкила;
или его фармацевтически приемлемые соли.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где R1 выбран из группы, состоящей из этила, метила, пропила, бутила, метоксиэтила и этиламинометила, и R2 выбран из -CONH2.
3. Соединение или его фармацевтически приемлемая соль по п. 1, где R1 выбран из метоксиэтила или этиламинометила и R2 выбран из -CO-R5.
4. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-3, где n представляет собой целое число от 3 до 5.
5. Соединение или его фармацевтически приемлемая соль по п. 4, где n представляет собой 4.
6. Соединение или его фармацевтически приемлемая соль, выбранное из группы, состоящей из следующих:
N-(4-(4-амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-N-(тетрагидро-2H-пиран-4-ил)ацетамид;
1-(4-(4-амино-2-этил-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-1-(тетрагидро-2H-пиран-4-ил)мочевина;
N-(4-(4-амино-2-((этиламино)метил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-N-(тетрагидро-2H-пиран-4-ил)ацетамид;
1-(4-(4-амино-2-((этиламино)метил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-1-(тетрагидро-2H-пиран-4-ил)мочевина;
N-(4-(4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-N-(тетрагидро-2H-пиран-4-ил)ацетамид;
1-(4-(4-амино-2-(2-метоксиэтил)-1H-имидазо[4,5-c]хинолин-1-ил)бутил)-1-(тетрагидро-2H-пиран-4-ил)мочевина.
7. Фармацевтическая композиция для индуцирования выработки интерферона-альфа (IFN-α), содержащая эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 и один или больше фармацевтически приемлемых эксципиентов.
8. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-6 или фармацевтическая композиция по п. 7 для применения в качестве средства для индуцирования выработки интерферона-альфа (IFN-α).
9. Соединение или его фармацевтически приемлемая соль по любому из пп. 1-6 или фармацевтическая композиция по п. 7 для применения в лечении рака, который можно лечить путем агонизации толл-подобного рецептора TLR7, где лечение относится к полному или частичному излечению рака, облегчению рака или прекращению его прогрессирования.
10. Способ лечения рака, который включает введение эффективного количества соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 или фармацевтической композиции по п. 7 субъекту, нуждающемуся в этом, где рак представляет собой рак, который можно лечить путем агонизации толл-подобного рецептора TLR7, где лечение относится к полному или частичному излечению рака, облегчению рака или прекращению его прогрессирования.
11. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 для изготовления лекарственного средства для лечения рака, который можно лечить путем агонизации толл-подобного рецептора TLR7, где лечение относится к полному или частичному излечению рака, облегчению рака или прекращению его прогрессирования.
| Колосоуборка | 1923 |
|
SU2009A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| ИМИДАЗОХИНОЛИНЫ В КАЧЕСТВЕ ДВОЙНЫХ ИНГИБИТОРОВ ЛИПИДКИНАЗЫ И mTOR | 2008 |
|
RU2478387C2 |

