[Область техники]
Настоящее изобретение относится к композиции для лечения фиброзного заболевания, которая включает соединение бензгидрилтиоацетамида в качестве активного ингредиента, и, более конкретно, к композиции для лечения фиброзного заболевания, которая подавляет экспрессию белков канала KCa2.3 в клеточной мембране, и имеет отличный эффект при лечении, в частности, фиброза печени и фиброза легких.
[Уровень техники]
Фиброз - это явление чрезмерного накопления внеклеточного матрикса, такого как коллаген, в ткани, которое возникает в процессе повреждения и восстановления тканей. Фиброз может возникать во всех органах тела, и он легко возникает особенно при серьезном и обширном повреждении и когда процесс повреждения и восстановления тканей повторяется, как при хронических заболеваниях. Когда возникает фиброз, поврежденная ткань заменяется фиброзной тканью, что ухудшает функции органа. Следовательно, когда фиброз распространяется широко, функция органа значительно ухудшается, что вызывает различные типы заболеваний. В частности, когда фиброз возникает во внутренних органах, которые напрямую влияют на жизнь, таких как печень, легкие, почки и сердце, он может иметь фатальные последствия для здоровья.
Обычно процесс фиброза может включать 1) воздействие вызывающих фиброз заболеваний (обычно хронических заболеваний) или материалов и 2) возникающий в результате фиброзный процесс (воспаление, фиброз и ангиогенез). Когда воспаление и повреждение возникают из-за заболевания или материала, вызывающего фиброз, фиброз и ангиогенез ускоряются факторами роста и цитокинами, которые секретируются клетками, участвующими в этом процессе. Следовательно, фиброзные заболевания можно лечить, устраняя причины фиброза (заболевания или материалы) или подавляя фиброзный процесс.
Однако полностью устранить причины фиброза практически невозможно. Причины многих фиброзных заболеваний, таких как идиопатический фиброз легких, неизвестны. Даже если известны причины фиброзных заболеваний, таких как хронический вирусный гепатит, стеатогепатит, диабет, вызывающий фиброз сердца или почек, и старение, часто вызывающее различные типы фиброзных заболеваний, часто невозможно полностью вылечить эти заболевания. Следовательно, лечение фиброзных заболеваний требует одновременного лечения для подавления фиброзного процесса (воспаления, фиброза, ангиогенеза), а также лечения заболевания, являющегося его причиной. Однако до сих пор не было разработано терапевтического средства для подавления фиброзного процесса.
В фиброзном процессе очень важны образование миофибробластов и активация звездчатых клеток печени (в печени активированные звездчатые клетки печени служат миофибробластами). Формирование миофибробластов, включая активацию звездчатых клеток печени, индуцируется активацией фибробластов или гладкомышечных клеток или эндотелиально-мезенхимальным переходом эндотелиальных клеток. Кроме того, когда образуются миофибробласты, количество миофибробластов значительно увеличивается из-за активной пролиферации клеток, увеличивается выработка внеклеточного матрикса, такого как коллаген, и стимулируется ангиогенез из-за активной пролиферации эндотелиальных клеток сосудов. Такой фиброзный процесс, то есть образование миофибробластов (включая активацию звездчатых клеток печени), пролиферация миофибробластов, образование внеклеточного матрикса, активация эндотелиальных клеток сосудов и ангиогенез, происходят через внутриклеточные Са2+-зависимые сигнальные пути. Следовательно, Са2+ играет очень важную роль в фиброзном процессе.
Для увеличения Са2+ в фибробластах, звездчатых клетках печени и эндотелиальных клетках сосудов существенно важны Са2+ активированные K+ каналы, то есть «каналы KCa». Гиперполяризация, вызванная активацией K+ каналов, способствует притоку Са2+ через входные каналы Са2+ в этих клетках. Каналы KCa, играющие такую роль в этих клетках, - это канал KCa2.3 и канал KCa3.1. Эти два канала K+ похожи по структуре и функциям, но есть разница в клетках, в которых эти каналы распространены.
Поскольку мРНК обнаруживается в большинстве клеток ткани, каналы KCa2.3, возможно, распределены в большинстве тканей организма (Naunyn Schmiedebergs Arch Pharmacol.2004; 369 (6): 602-15) и широко распространены в печени, нервах и эндотелиальных клетках сосудов. С другой стороны, канал KCa3.1 обычно распространен в эндотелиальных клетках сосудов, фибробластах, иммунных клетках и эритроцитах (Curr Med Chem. 2007; 14 (13): 1437-57; Expert Opin Ther Targets. 2013; 17 (10): 1203-1220).
Как описано выше, каналы KCa2.3 или KCa3.1, которые, как считается, вносят значительный вклад в прогрессирование фиброза, способствуя притоку Са2+ через каналы входа Са2+, изучаются в качестве основных мишеней терапевтических агентов при фиброзных заболеваниях. В частности, сообщалось, что селективный ингибитор канала KCa2.3, апамин, оказывает ингибирующее действие на эндотелиально-мезенхимальный переход, который имеет решающее значение для фиброзного процесса, и оказывает терапевтическое действие на фиброз печени и фиброз желчевыводящих путей (Biochem Biophys. Res Commun. 2014; 450 (1): 195-201; Int J. Mol Med. 2017; 39 (5): 1188-1194).
Ингибиторы ионных каналов, которые были разработаны к настоящему времени, подавляют функции клетки посредством ингибирования активности ионного канала (ингибирования потока ионов через белок канала). Поскольку количество канальных белков, экспрессируемых в клеточной мембране, влияет на функцию клетки, функции клеток также можно регулировать путем уменьшения количества канальных белков, экспрессируемых в клеточной мембране (ингибирование экспрессии канального белка в клеточной мембране). До сих пор не было разработано лекарственных средств для регулирования уровня экспрессии канального белка в клеточной мембране, и молекулы, регулирующие уровень экспрессии, могут быть новым терапевтическим средством от различных заболеваний (Chem Med С hem. 2012, 7 (10): 1741-1755). В частности, поскольку экспрессия канала K+Са2.3 увеличивается за счет факторов роста при фиброзных заболеваниях, лекарственные средства для ингибирования экспрессии белков канала KCa2.3 могут быть разработаны в качестве терапевтических агентов для лечения фиброзных заболеваний.
В то же время в патентах США №4066686 и 4177290 производное бензгидрилсульфенилацетамида, включенное в настоящее изобретение, предлагается в качестве лекарственного средства для лечения расстройств центральной нервной системы, и это соединение было разработано в качестве лекарственного средства для лечения нарколепсии компанией Lafon, Франция, и продается под непатентованным наименованием «модафинил». Адрафинил, известный как предшественник модафинила, то есть дифенилметилтиоацетогидроксамовая кислота, также был разработан как лекарство, обладающее такой же эффективностью, как модафинил (CNS Drug Reviews Vol5, №3 193-212, 1999).
Кроме того, в соответствии с патентом США № 4927855 было высказано предположение, что R-изомер модафинила (лафон), то есть (-)-бензгидрилсульфенилацетамид, действует в качестве антидепрессанта оказывая терапевтическое действие на гиперсомнию и болезнь Альцгеймера; согласно патенту США №6180678 было высказано предположение, что R-модафинил (ветохинол, Франция) эффективен при лечении поведенческих проблем у пожилых собак, улучшении обучаемости, контроле мочевого пузыря и улучшении памяти, и согласно патенту США № 9637447 было высказано предположение, что 2-[бис(4-фторфенил)метансульфенил]ацетамид, известный под общим названием «лауфлумид», эффективен против синдрома дефицита внимания с гиперактивностью (ADHD), нарколепсии, эпилепсии и летаргии.
Кроме того, изобретатели сообщили в корейских патентах № 10-1345860 и 10-1414831 и соответствующем патенте США № 9259412, что модафинил и его производные могут быть использованы в качестве лекарственных средств для лечения сосудистых заболеваний и заболеваний, опосредованных каналом KCa3.1, то есть рака и аутоиммунных заболеваний, за счет увеличения уровня цАМФ для расслабления кровеносных сосудов и подавления тока KCa3.1.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[Техническая проблема]
В процессе изучения фармацевтической активности соединений бензгидрилтиоацетамида, включая производные бензгидрилсульфенилацетамида, авторы обнаружили, что такие соединения неожиданно подавляют экспрессию канала KCa2.3 в клеточной мембране и, кроме того, оказывают терапевтическое действие на фиброзные заболевания у мышиных моделей.
Настоящее изобретение направлено на обеспечение новой композиции для лечения фиброзных заболеваний, которая включает соединение бензгидрилтиоацетамида или его фармацевтически приемлемую соль в качестве активного ингредиента. Для справки, термин «соединение бензгидрилтиоацетамида», используемый в данном документе, используется как понятие, включающее «соединение бензгидрилсульфенилацетамида».
Техническое решение
Композиция для лечения фиброзного заболевания согласно настоящему изобретению включает соединение бензгидрилтиоацетамида, представленное приведенной ниже формулой А, или его фармацевтически приемлемую соль в качестве активного ингредиента.
[Формула А]
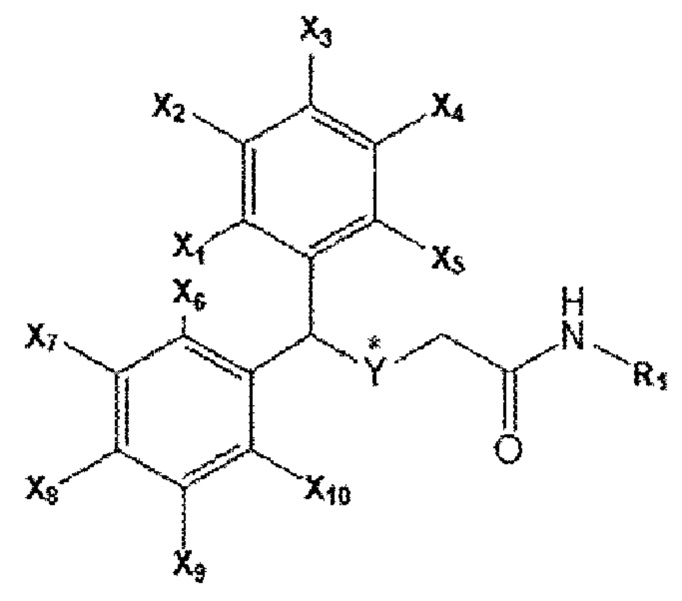
[В формуле А каждый из X1 ~ Х10 может независимо представлять собой водород (Н) или фтор (F), все из которых могут быть одинаковыми или отличаться друг от друга; Y представляет собой серу (S) или сульфоксид (S=O), * указывает на хиральное положение; R1 является любым из водорода, метильной группы, этильной группы, метоксигруппы, этоксигруппы, гидроксильной группы и углеродного соединения, содержащего от 3 до 6 атомов углерода.]
В соединении формулы А каждый из X1 ~ Х10 независимо представляет собой водород (Н) или фтор (F), Y представляет собой серу (S), a R1 представляет собой водород (Н).
В соединении формулы А каждый из X1 ~ Х10 независимо представляет собой водород (Н) или фтор (F), Y представляет собой сульфоксид (S=О), a R1 представляет собой водород (Н).
Соединение формулы А имеет эффект подавления экспрессии белка канала KCa2.3 в клеточной мембране.
Соединение формулы А эффективно при лечении, в частности, фиброза печени и фиброза легких.
Преимущества изобретения
Было подтверждено, что соединение бензгидрилтиоацетамида согласно настоящему изобретению обладает эффектом подавления экспрессии белка канала KCa2.3 в эксперименте in vitro для культивирования клеток, а также обладает эффектом подавления воспаления и фиброза и улучшения функций печени в эксперименте in vivo на мышиных моделях, в которых были индуцированы заболевания печени и легких.
Соответственно, соединение бензгидрилтиоацетамида согласно настоящему изобретению может быть эффективно использовано в качестве фармацевтической композиции для лечения различных типов воспалительных и фиброзных заболеваний, которые возникают в организме человека, и, в частности, воспалительных и фиброзных заболеваний в печени и легких, и ожидается, что при необходимости оно будет реализовано в виде лекарственного средства для животных.
Описание чертежей
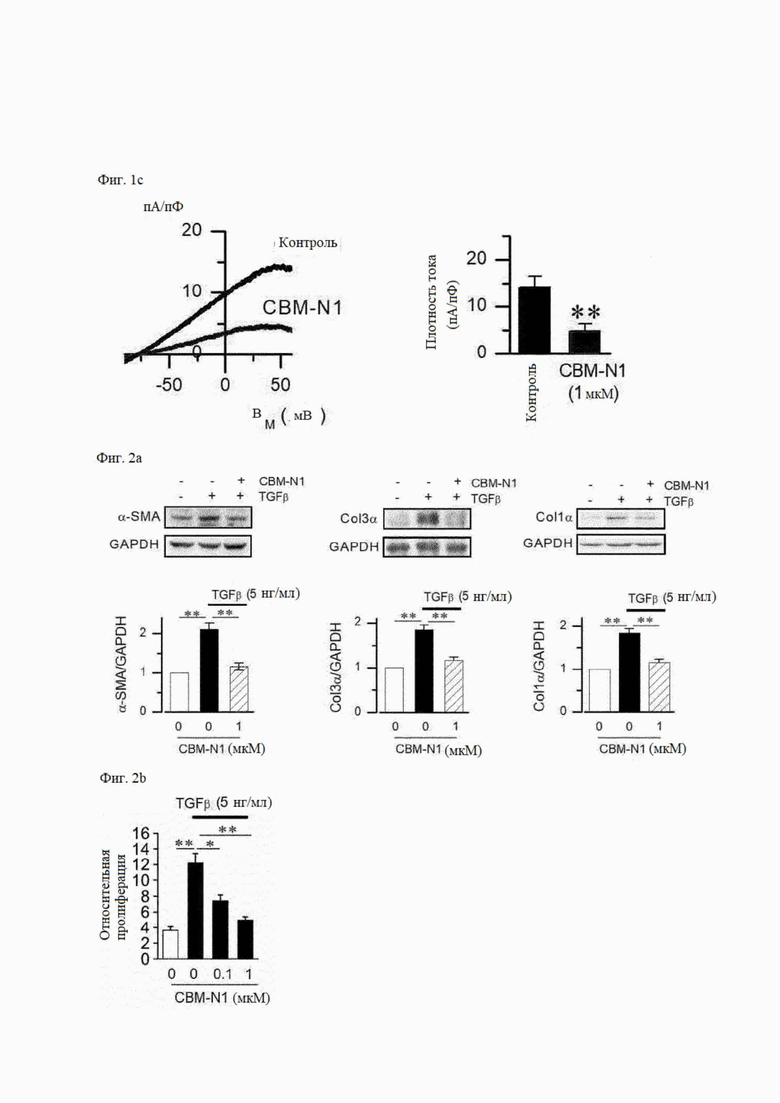
На фиг. 1А-1С показаны эффекты PDGF, TGFβ и соединения формулы А1 согласно настоящему изобретению на экспрессию каналов KCa2.3 и KCa3.1 в эндотелиальных клетках сосудов, фибробластах и звездчатых клетках печени.
На фиг. 2А и 2В показаны эффекты соединения формулы А1 на экспрессию маркера фиброза (фиг. 2А) и пролиферацию клеток (фиг. 2В) в фибробластах, подвергшихся воздействию TGFβ, индуцирующего увеличение экспрессии канала KCa2.3 и фиброз.
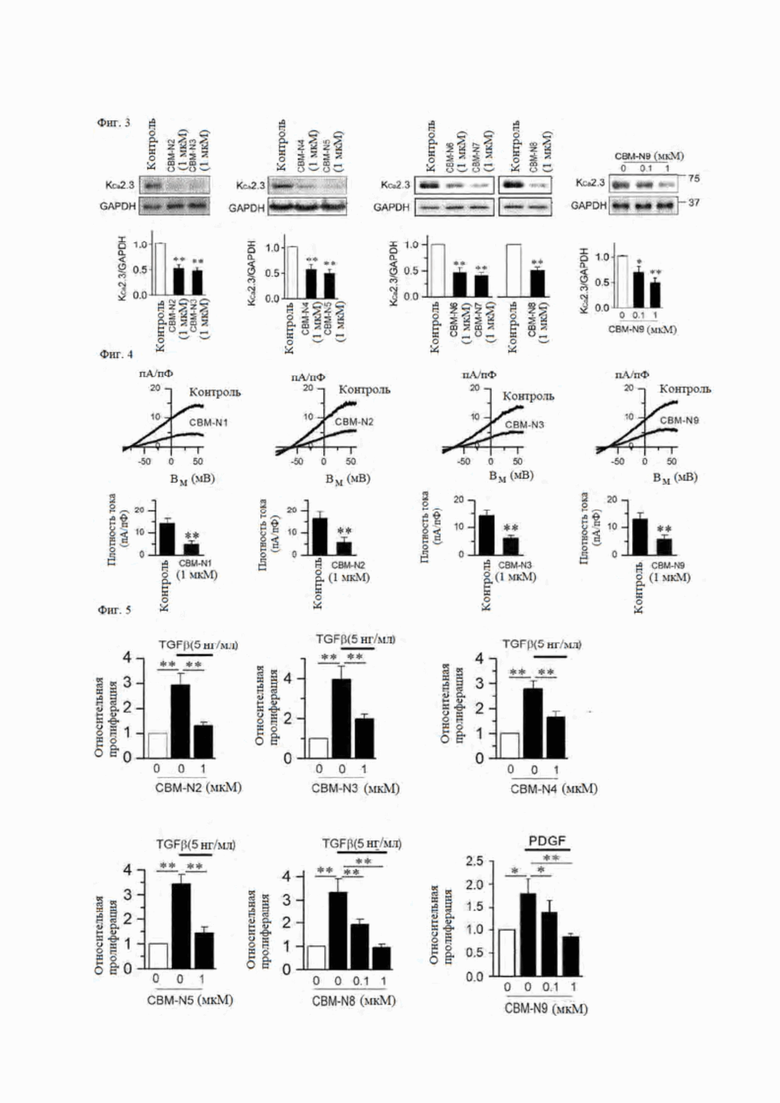
На фиг. 3 показано влияние соединении формул А1-А9 согласно настоящему изобретению на экспрессию канала KCa2.3 в звездчатых клетках печени.
На фиг. 4 показан ток KCa2.3 в звездчатых клетках печени, сниженный в экспрессирующемся канале KCa2.3 из-за воздействия соединений формул А2-А4 и А9 согласно настоящему изобретению в течение 24 часов.
На фиг. 5 показано влияние соединений формул А2-А5, А8 и А9 согласно настоящему изобретению на пролиферацию клеток в фибробластах, подвергнутых воздействию TGFβ или PDGF, индуцирующих фиброз, в течение 24 часов.
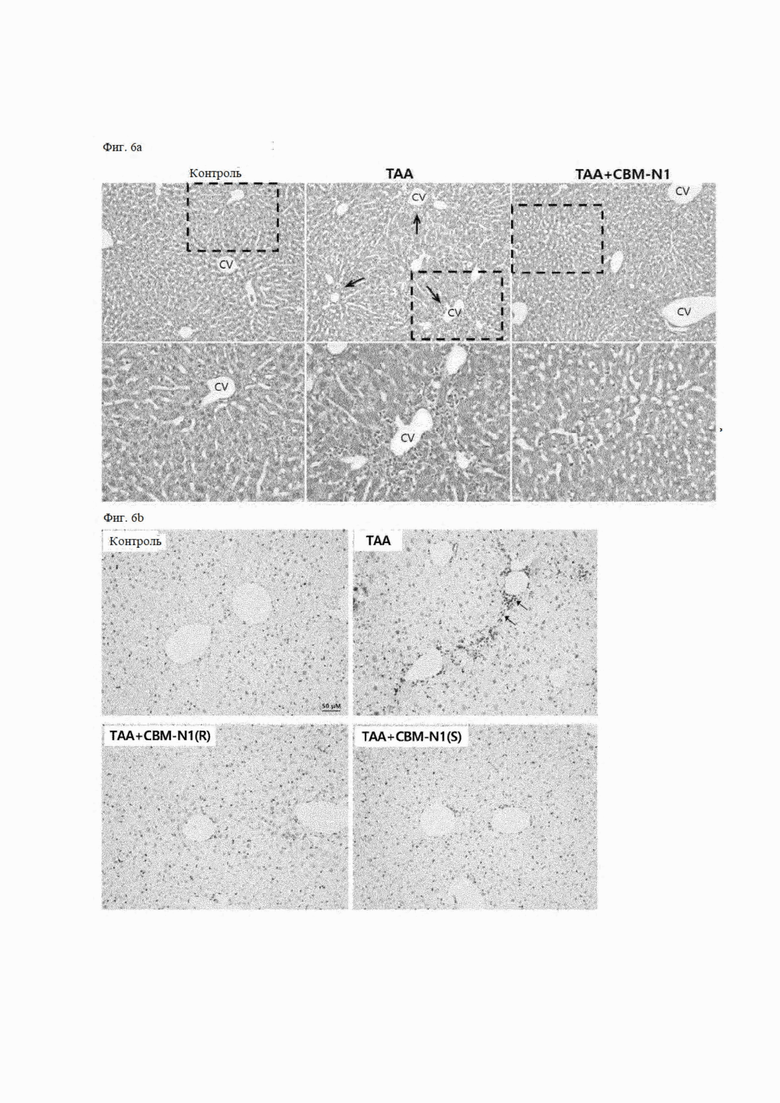
На фиг. 6A-6D показано влияние соединения формулы А1 согласно настоящему изобретению и его изомеров на ингибирование воспаления и ингибирование фиброза на мышиных моделях ТАА-индуцированного заболевания печени с помощью гистологического или иммуногистохимического метода.
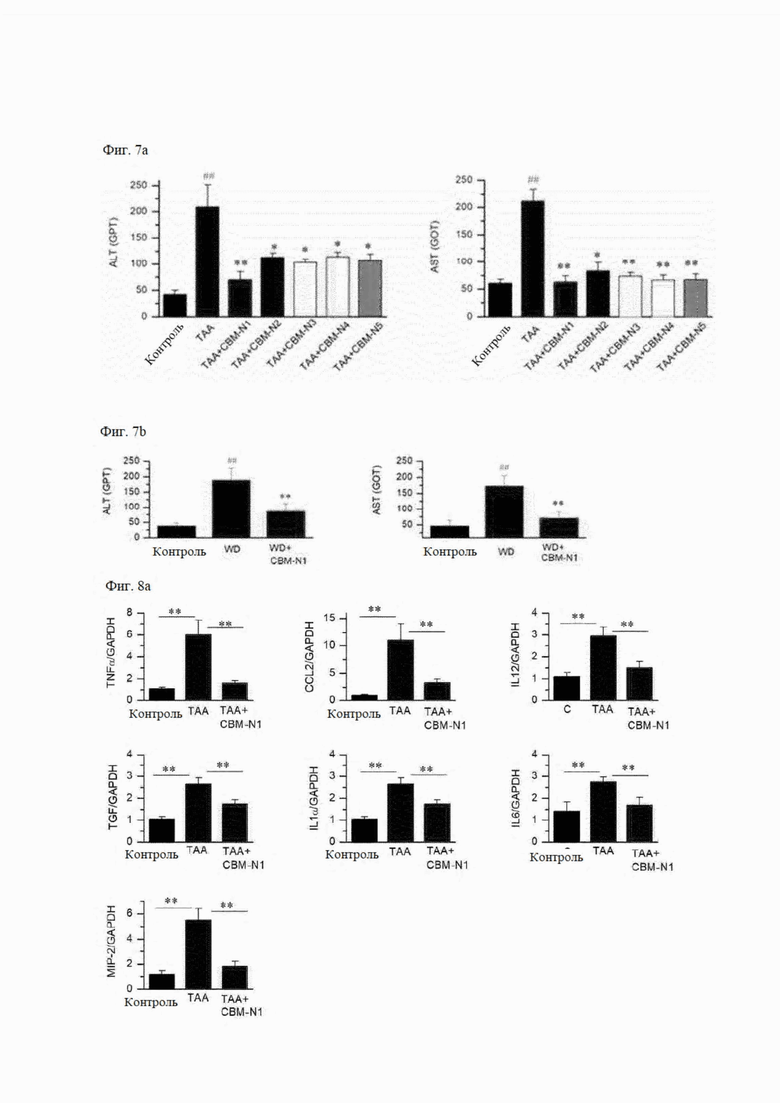
На фиг. 7А и 7В показаны результаты тестирования функций печени в зависимости от наличия или отсутствии введения соединений формул А1-А5 согласно настоящему изобретению на мышиных моделях заболеваний печени, вызванных ТАА или западной диетой (фиг. 7А или 7В).
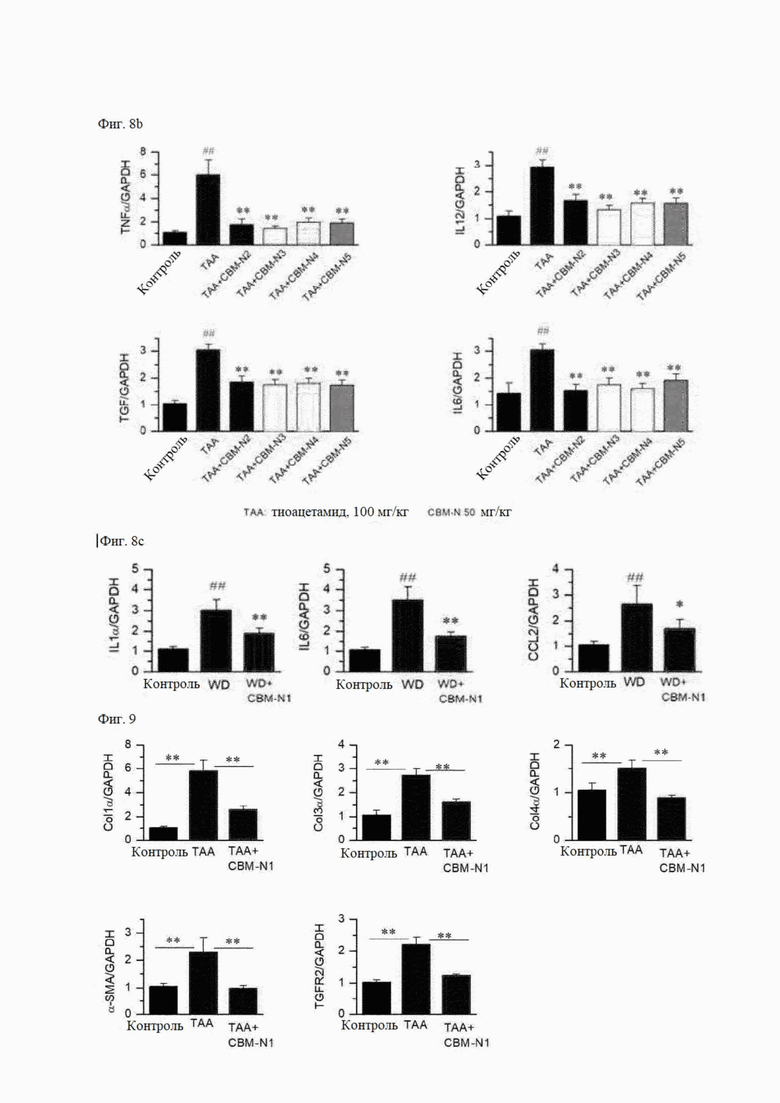
На фиг. 8А и 8В показано изменение экспрессии мРНК воспалительных цитокинов в результате введения соединений формул А1 и А2-А5 согласно настоящему изобретению в мышиных моделях ТАА-индуцированного заболевания печени, и на фиг. 8С показано изменение экспрессии мРНК воспалительных цитокинов в результате введения соединения формулы А1 в мышиных моделях заболевания печени, индуцированного западной диетой (WD).
На фиг. 9 показано изменение экспрессии мРНК маркеров фиброза в зависимости от наличия или отсутствия введения соединения формулы А1 на мышиных моделях с ТАА-индуцированным заболеванием печени.
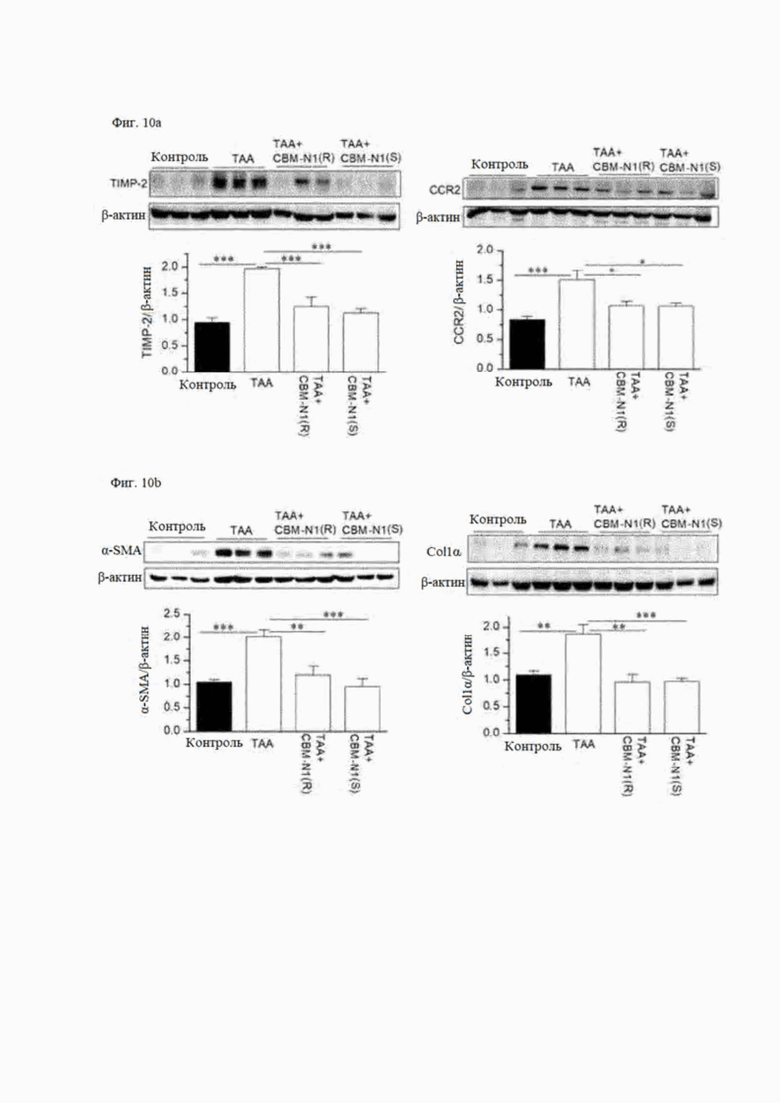
На фиг. 10А и 10В показано действие R-изомера и S-изомера соединения формулы А1 на экспрессию белков маркера воспаления (фиг. 10А) и маркера фиброза (фиг. 10В) на мышиных моделях с ТАА-индуцированным заболеванием печени.
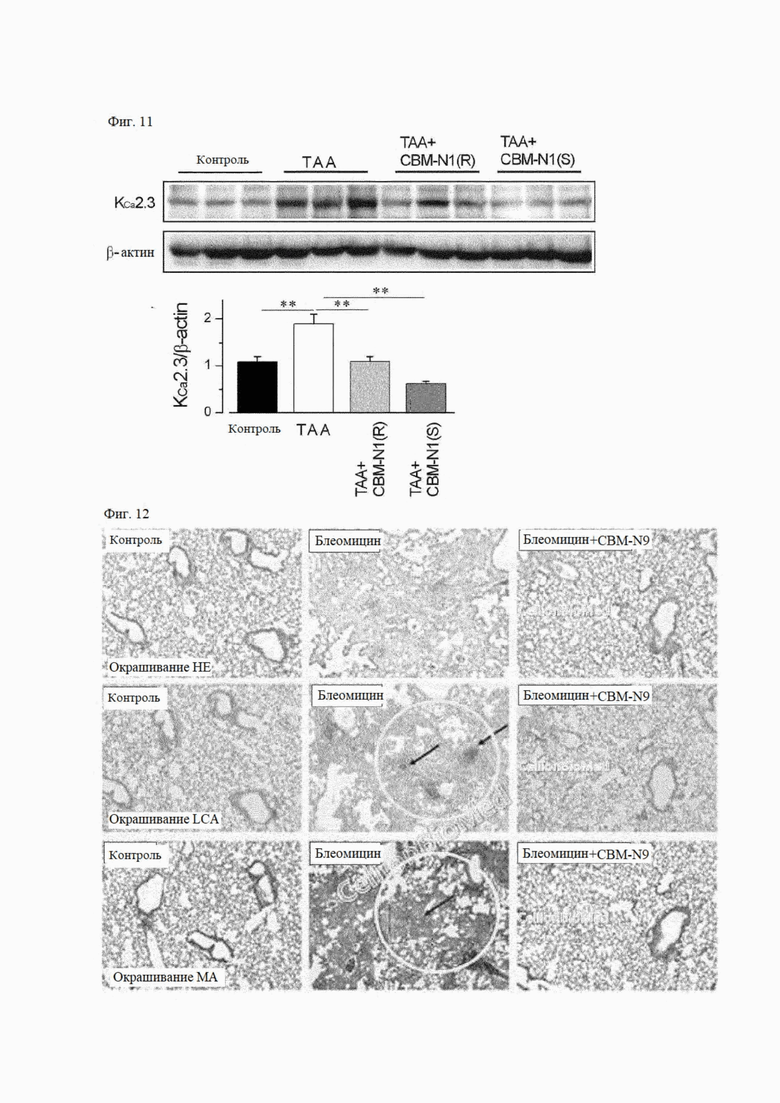
На фиг. 11 показано действие R-изомера и S-изомера соединения формулы А1 на экспрессию белка канала KCa2.3 на мошиных моделях ТАА-индуцированного заболевания печени
На фиг. 12 показано действие соединения формулы А9 на воспаление легких и фиброз на мышиных моделях индуцированного блеомицином фиброза легких.
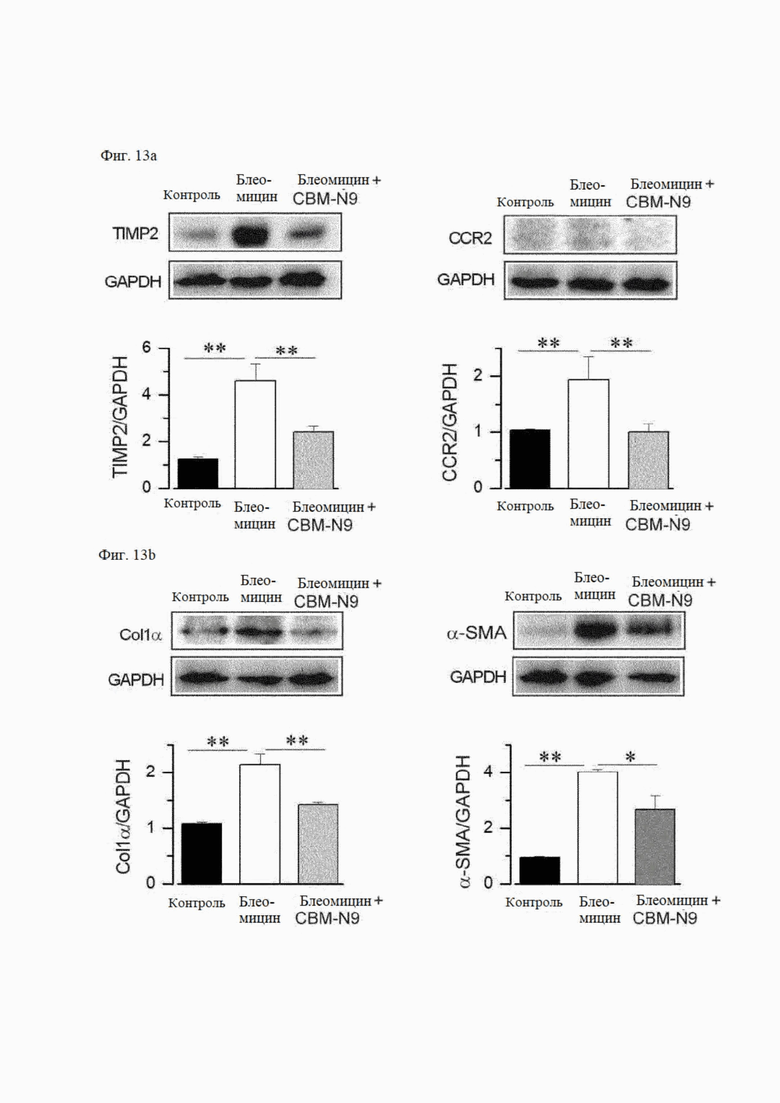
На фиг. 13А и 13В показано действие соединения формулы А9 на экспрессию белков маркера воспаления (фиг. 13А) и маркера фиброза (фиг. 13В) на мышиных моделях индуцированного блеомицином фиброза легких.
Осуществление изобретения
Соединение бензгидрилтиоацетамида согласно настоящему изобретению, представленное формулой А, включает, в частности, соединения формул А1-А9, представленных ниже.
[Формула А1]
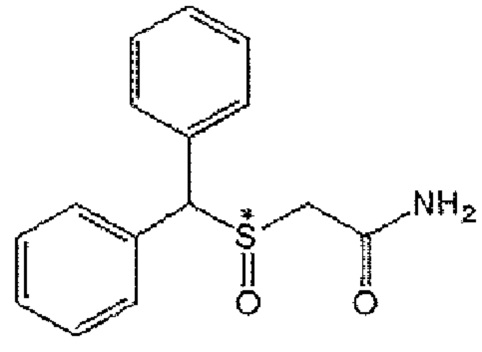
Соединение формулы А1 известно под общим названием «модафинил» и в настоящее время используется в качестве лекарственного средства для лечения гипнолепсии, а клинические испытания для лечения других психических заболеваний продолжаются. Химическое название модафинила - 2-(бензгидрилсульфенил)ацетамид, и он может быть синтезирован известным способом или приобретен.
[Формула А2]
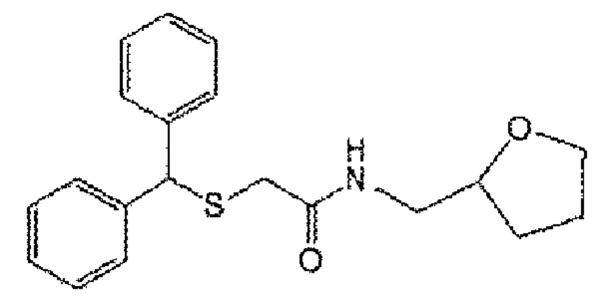
[Формула A3]
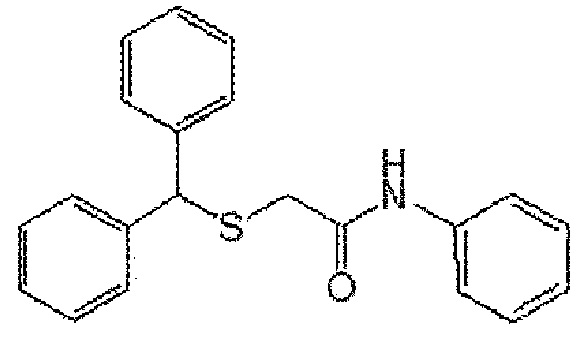
[Формула А4]
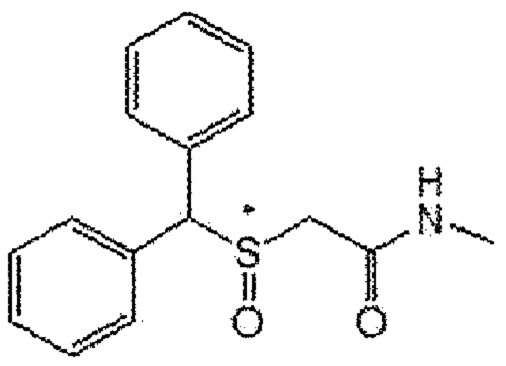
[Формула A5]
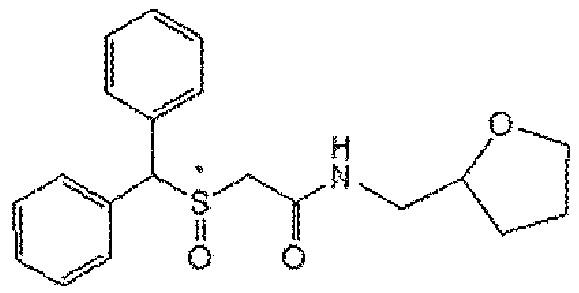
[Формула А6]
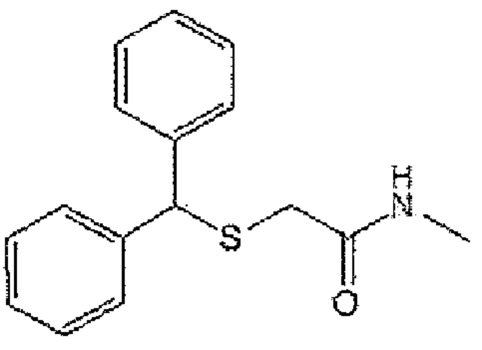
[Формула А7]
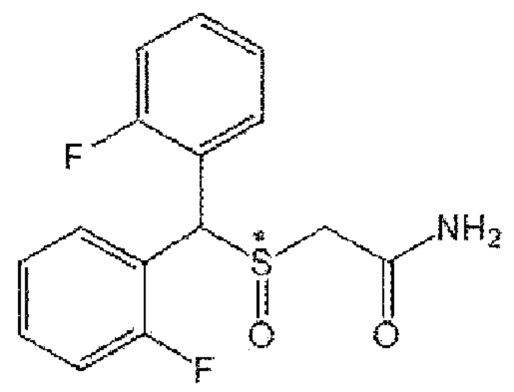
[Формула А8]
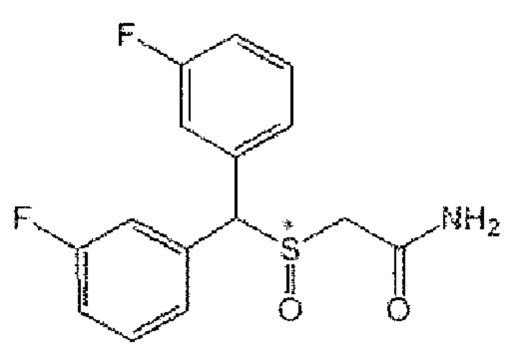
[Формула А9]
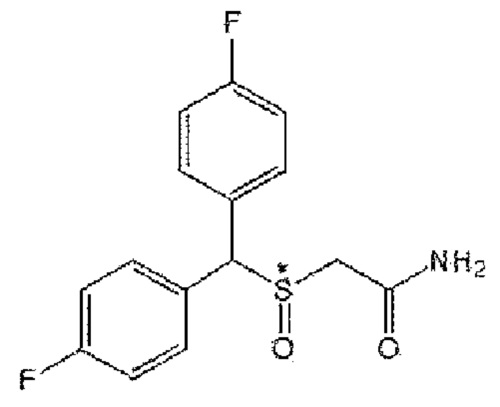
Все соединения формул А2-А9 обладают эффектом подавления экспрессии белка канала KCa2.3 в клеточной мембране в соответствии с тем же механизмом, что и модафинил, и, кроме того, оказывают терапевтическое действие на фиброзные заболевания в организме человека. Среди них соединение формулы А9 известно под непатентованным наименованием «лауфлумид».
Химические названия соединений формул от А1 до А9 следующие. Кодовые названия, перечисленные в скобках в конце каждого химического названия, являются кодовыми названиями, используемыми авторами в следующих примерах.
1) Формула А1, 2-(бензгидрилсульфенил)ацетамид (CBM-N1)
2) Формула А2; 2-(бензгидрилтио)-N-[(тетрагидрофуран-2-ил)метил]ацетамид (CBM-N2)
3) Формула A3; 2-(бензгидрилтио)-N-фенилацетамид (CBM-N3)
4) Формула А4; 2-(бензгидрилсульфенил)-N-метилацетамид (CBM-N4)
5) Формула A3; 2-(бензгидрилсульфенил)-N-[(тетрагидрофуран-2-ил)метил]ацетамид (CBM-N5)
6) Формула А6; 2-(бензгидрилтио)-ен-метилацетамид (CBM-N6)
7) Формула А7; 2-[бис(2-фторфенил)метансульфенил]ацетамид (CBM-N7)
8) Формула А8; 2-[бис(3-фторфенил)метансульфенил]ацетамид (CBM-N8)
9) Формула А9; 2-[бис(4-фторфенил)метансульфенил]ацетамид (CBM-N9).
Соединения формул А2-А6 могут быть синтезированы способами, раскрытыми в патенте Кореи №10-1345860. или коммерчески доступны, но эффективные способы получения соединений формул А7-А9 неизвестны. Таким образом, в настоящем изобретении способы получения соединений формул А7-А9 были описаны в качестве примеров.
Фармацевтическая композиция согласно настоящему изобретению включает фармацевтически приемлемую соль соединения формулы А. Здесь «фармацевтически приемлемая соль» может обычно включать соль металла, соль с органическим основанием, соль с неорганической кислотой, соль с органической кислотой или соль с основной или кислой аминокислотой. Кроме того, фармацевтическая композиция согласно настоящему изобретению может включать как сольват, так и гидрат соединения формулы А, и может включать все доступные стереоизомеры и, кроме того, включать кристаллическую или аморфную форму каждого соединения.
Фармацевтическая композиция согласно настоящему изобретению может быть приготовлена в форме таблетки, пилюли, порошка, гранулы, капсулы, суспензии, жидкости для внутреннего применения, эмульсии, сиропа, аэрозоля или стерильного раствора для инъекций по общепринятой методике. Кроме того, фармацевтическая композиция по настоящему изобретению может быть введена перорально или парентерально в соответствии с целью применения, и парентеральное введение может осуществляться путем кожной инъекции для наружного применения, внутрибрюшинной инъекции, интраректальной инъекции, подкожной инъекции, внутривенной инъекции, внутримышечной инъекции или внутрисердечной инъекции.
Доза фармацевтической композиции согласно настоящему изобретению может варьироваться в зависимости от массы тела пациента, возраста, пола, состояния здоровья, диеты, продолжительности введения, способа введения, скорости выведения и тяжести заболевания. Суточная доза составляет предпочтительно от 0,2 до 20 мг/кг, и более предпочтительно от 0,5 до 10 мг/кг в расчете на активный ингредиент, и ее можно вводить один или два раза в день, но настоящее изобретение этим не ограничивается.
Примеры
1) Синтез соединений
1-1) Синтез соединения формулы А9
Способ синтеза соединения (лауфлумида) формулы А9 будет описан со ссылкой на следующую схему реакции. 24 г 4,4'-бисфторбензгидрола (1) помещали в 500 мл круглодонную колбу, растворяли в 150 мл добавленной трифторуксусной кислоты и перемешивали с 12,05 г добавленной тиголовой кислоты в течение приблизительно 2 часов с последующим подтверждением прекращения реакции методом тонкослойной хроматографии. Продукт реакции подвергали вакуумной перегонке для удаления трифторуксусной кислоты, нейтрализовали и экстрагировали этилацетатным органическим растворителем. Полученный экстракт сушили сульфатом магния, получая при этом 34,8 г соединения (II), которое представляет собой липкое масло желтого цвета, с количественным выходом.
34,8 г соединения (II) растворяли в 250 мл безводного этанола и добавляли 4,2 г концентрированной серной кислоты с последующим кипячением с обратным холодильником в течение 8 часов. Затем полученный продукт охлаждали до комнатной температуры, концентрировали для удаления этанола, растворяли в метиленхлориде и дважды промывали водой. Полученный продукт снова промывали 5% раствором NaHCO3 и сушили безводным сульфатом магния, получая при этом 39,1 г соединения (III), которое представляет собой масло желтого цвета, с количественным выходом.
34,3 г соединения (III) помещали в круглодонную колбу (500 мл), добавляли 210 мл метанола, 21,4 мл кислотного катализатора (кислотный катализатор получали растворением 4 г серной кислоты в 90 мл изопропилового спирта), и медленно добавляли 35% раствор H2O2 с последующим перемешиванием в течение ночи при комнатной температуре. Затем добавляли 70 г хлорида натрия (NaCl), трижды экстрагировали раствором метиленхлорида, сушили безводным сульфатом магния и концентрировали, получая таким образом соединение (IV) с количественным выходом.
В круглодонную колбу (100 мл) добавляли 5,1 г соединения (IV) с 13 мл метанола, добавляли 1,3 г хлорида аммония (NH4Cl) и затем добавляли 98 мл концентрированного раствора гидроксида аммония (NH4OH). После перемешивания в течение ночи белый раствор эмульсионного типа фильтровали, получая при этом 4 г твердого порошка. 4 г твердого порошка растворяли в 28 г изопропилового спирта, кипятили с обратным холодильником и охлаждали до комнатной температуры, получая тем самым 2,1 г 2-[бис(4-фторфенил)метансульфенил]ацетамида, который представляет собой белое кристаллическое соединение, представленное Формулой А9.
1Н ЯМР (DMSO-d6): δ 7,68 (bs, 1Н); 7,56-7,51 (m, 4Н), 7,33 (bs, 1Н), 7,29-7,24 (m, 4Н), 5,4 (s, 1H); 3,4 (d, J=13,6 Hz, 1H); 3,16 (d, J=13,6 Hz, 1H)
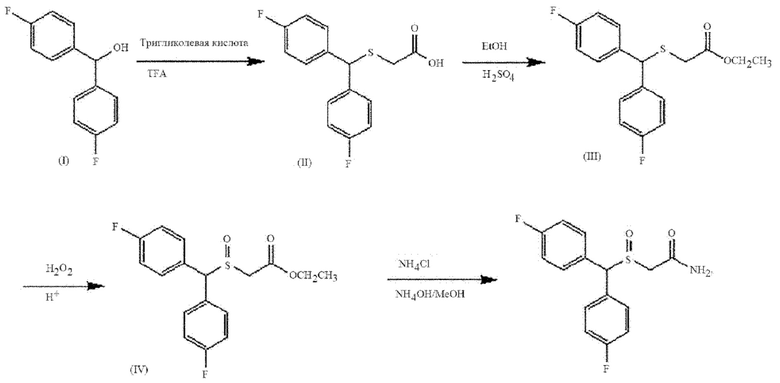
1-2) Синтез соединения формулы A8
3,3'-бисфторбензгидрол был синтезирован обычным методом (Tetrahedron Lett, vol 58, 442, 2017, ЕР 1433744, J. Med. Chem. vol 40, 851, 1997). Это соединение использовали в качестве исходного материала, и соединение формулы А8, то есть 2-[бис(3-фторфенил)метансульфенил]ацетамид, синтезировали способом синтеза соединения формулы А9.
1Н ЯМР (DMSO-d6): δ 7,68 (bs, 1Н); 7,5-7,2 (m, 9H), 5,4 (s, 1H); 3,4 (d, 1H); 3,16 (d, Hz, 1H)
1-3) Синтез соединения формулы A7
2,2'-бисфторбензгидрол был синтезирован обычным методом (ЕР 1,661930, J. Med. Chem. vol 51, # 4, 976, 2008). Это соединение использовали в качестве исходного материала, и соединение формулы А7, то есть 2-[бис(2-фторфенил)метансульфенил]ацетамид, синтезировали способом синтеза соединения формулы А9.
1Н ЯМР (DMSO-d6): δ 7,68 (bs, 1Н); 7,5-7,2 (m, 9H), 7,33 (bs, 1H), 5,4 (s, 1H); 3,4 (d, 1H); 3,16 (d, 1H)
2. Экспериментальный способ
2-1) Культура клеток
Фибробласты (CRL-2795; American Type Culture Collection, VA) культивировали в среде Игла, модифицированной Дульбекко (Hyclone, Logan, UT), эндотелиальные клетки микрососудов матки человека (PromoCell GmbH, Геидельберг, Германия) культивировали в среде MV2 (PromoCell GmbH), а звездчатые клетки печени человека (Innoprot, Bizkia, Испания) культивировали в среде Р60126 (Innoprot).
Все клетки поддерживали в условиях 5% увлажненного углекислого газа при 37°С. Культивированные клетки подвергали воздействию каждого из PDGF, TGFβ и соединений формул А1-А9 (CBM-N1 ~ N9) в течение 24 часов с последующим проведением экспериментов
2-2) Создание мышиных моделей болезни печени.
Чтобы подтвердить эффекты соединений формул А1-А5 (CBM-N1 ~ N5) согласно настоящему изобретению на воспаление и фиброз печени, последующие эксперименты были выполнены на мышах C57BL/6 дикого типа (приобретенных у Orient Bio). Во-первых, чтобы вызвать заболевание печени у мышей, мышам вводили тиоацетамид (ТАА) (эксперимент А) или мышей выращивали на западной диете, вызывающей ожирение печени (эксперимент В). Мышей разделили на здоровую контрольную группу, группу с вызванным заболеванием, и труппу, в которой вводили лекарственное средство, и среди этих трех групп от 15 до 100 мышей использовали в каждой труппе для эксперимента А, и 10 мышей использовали в каждой группе для эксперимента В. Способ медикаментозного лечения мышей в каждой группе следующий.
(1) Здоровый контроль: здоровым контрольным мышам в эксперименте А внутрибрюшинно вводили три раза в неделю такое же количество растворителя ТАА. которое использовалось, когда ТАА вводили группе с вызванным заболеванием, и группу здорового контроля в эксперименте В держали на нормальном рационе питания. В обоих экспериментах А и В растворитель CBM-N1 вводили через пероральную трубку в том же количестве, что и при инъекции CBM-N1, пять раз в неделю. Растворителем ТАА была дистиллированная вода, растворитель CBM-N1 и его производное представляли собой смесь ДМСО и дистиллированной воды в соотношении 1:1. На прилагаемых фигурах С или Контроль относится к здоровому контролю.
(2) Группа с вызванным заболеванием: группе с вызванным заболеванием в эксперименте А внутрибрюшинно вводили ТАА в дозе 100 мг/кг три раза в неделю, группу с вызванным заболеванием в эксперименте В держали на западной диете (WD, 45% насыщенных жиров. 0,2% холестерина и вода, содержащая фруктозу и глюкозу). Все растворители CBM-N1 в экспериментах А и В вводили через пероральную трубку в том же количестве, что и при введении CBM-N1 пять раз в неделю. На прилагаемых фигурах ТАА относится к группе, в которой заболевание вызывается TAA, a WD относится к группе, в которой заболевание вызывается западной диетой.
(3) Группа, в которой вводили лекарственное средство: в эксперименте А соединения формул 1-5 (50 м г/к г/день. 5 раз в неделю) вводили с ТАА (100 мг/кг, 3 раза в неделю) и в эксперименте В CBM-N1-N5 (50 мг/кг/день, 5 раз/неделю) вводили с западной диетой. Мышей, получавших лечение описанным выше способом в течение 16 недель, немедленно умерщвляли чрезмерным введением анестетика, а затем извлекали печень и кровь. На прилагаемых фигурах ТАА + CBM-N1, ТАА + CBM-N2, ТАА + CBM-N3, ТАА + CBM-N4 и ТАА + CBM-N5 относятся к группам с вызванным заболеванием, которым вводили соединения формул А1-А5, соответственно.
2-3) Создание мышиных моделей легочного воспаления и фиброза
Чтобы подтвердить действие соединения формулы А9 (CBM-N9) на воспаление легких и фиброз, вызванные блеомицином, следующие эксперименты были выполнены на мышах C57BL/6 дикого типа. Сначала мышей разделили на здоровую контрольную группу, группу с вызванным заболеванием и группу, получавшую лекарственное средство, и в каждую из трех групп включали по десять мышей. Способ медикаментозного лечения мышей в каждой группе выглядит следующим образом.
(1) Здоровый контроль: такое же количество дистиллированной воды, которое использовалось при применении блеомицина в группе с вызванным заболеванием, вводили интратрахеально. Кроме того, растворитель CBM-N9 вводили внутрибрюшинно в том же количестве, что и при введении CBM-N9 группе с вызванным заболеванием, как описано ниже, пять раз в неделю.
(2) Группа с вызванным заболеванием: 1,5 единицы блеомицина вводили интратрахеально. Кроме того, растворитель CBM-N9 вводили внутрибрюшинно в том же количестве, что и при введении CBM-N9, пять раз в неделю.
(3) Группа, получавшая лекарственное средство: 1,5 единицы блеомицина вводили интратрахеально. Кроме того, CBM-N9 (50 мг/кг) вводили внутрибрюшинно пять раз в неделю.
Мышей, получавших лекарственное средство в течение 4 недель таким же образом, как описано выше, немедленно умерщвляли чрезмерным введением анестетика, а затем извлекали легкие.
2-4) Подготовка парафиновых образцов ткани печени и тканей легких и наблюдение за их морфологическими изменениями
Для гистологического подтверждения терапевтического эффекта соединения формулы А1 (CBM-N1) или соединения формулы А9 (CBM-N9) на каждой мышиной модели получали парафиновый образец ткани. Ткани печени и легких фиксировали раствором параформальдегида и делали срезы толщиной 1-2 мм. Срезы тканей залили парафином, нарезали пластинами толщиной 4 мкм, чтобы удалить парафин с помощью ксилола, и ксилол удалили этанолом с последующей промывкой водопроводной водой. Полученные ткани подвергали окрашиванию гематоксилином и эозином (окрашивание Н&Е) или иммуногистохимии.
(1) Окрашивание Н&Е: сначала ядра окрашивали (синий) раствором для окрашивания гематоксилином Харриса в течение 5 минут и затем окрашивали (розовый) раствором эозина.
(2) Иммуногистохимия маркеров воспаления: маркеры воспаления (CD82 и CD45) окрашивали специфическими антителами, и лимфоидные клетки окрашивали в коричневый цвет.
(3) Иммуногистохимия маркеров фиброза: коллаген окрашивали трихромным окрашиванием по Массону, или волокна ретикулина окрашивали ретикулином.
2-5) Тест функции печени
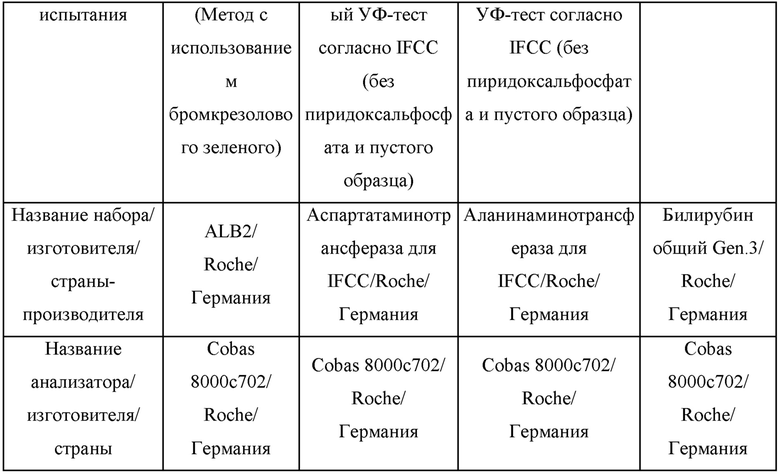
Аминотрансфераза аспарагиновой кислоты (AST, GOT) и аланинаминотрансфераза (ALT, GPT), общий билирубин и концентрации альбумина измеряли с использованием крови, взятой из мышиных моделей заболевания печени. Способ тестирования функции печени показан в таблице 1 ниже.
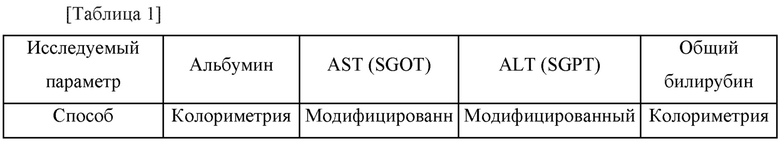
2-6) Анализ полимеразной цепной реакции в реальном времени (ПЦР в реальном времени)
Уровни экспрессии мРНК факторов воспаления или фиброза в извлеченной ткани печени измеряли с помощью ПЦР в реальном времени. РНК ткани печени выделяли с помощью реагента TRIzol (Центр молекулярных исследований, Цинциннати, Огайо), а одноцепочечную к ДНК синтезировали с использованием полимеразы BcaBEST (TakaraShuzo) с последующей полимеразной цепной реакцией.
Последовательности праймеров (SEQ ID NO: 1-30) воспалительных цитокинов и маркеров фиброза, используемых здесь, показаны в таблицах 2-4 ниже.

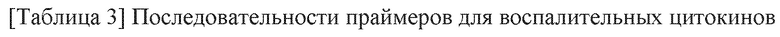


2-7) Вестерн-блоттинг
Клетки лизировали буферным раствором для экстракции белка, концентрацию белка в супернатанте определяли анализом Брэдфорда, и 30 мкг белка загружали в гель SDS-PAGE (полиакриламидный гель в присутствии додецилсульфата натрия) и затем переносили на нитроцеллюлозную мембрану. Нитроцеллюлозную мембрану блокировали 5% содержащим BSA (бычий сывороточный альбумин) TBST (смесь трис-забуференного физиологического раствора и полисорбата 20) (10 мМ трис-HCl, 150 мМ NaCl и 0,1% (об./об.) твин-20, рН 7,6) при комнатной температуре в течение 1 часа. Б лоты инкубировали в течение ночи с первичными антителами, а затем инкубировали со вторичными антителами, конъюгированными с пероксидазой хрена, в течение 1 часа. Полосы визуализировали методом хемилюминесценции. Сбор и обработку данных проводили с помощью анализатора изображений (LAS-3000) и программного обеспечения IMAGE CAUSE (Fuji film, Япония).
2-8) Метод тестирования пролиферации клеток МТТ
Клетки высевали в 96-луночные планшеты из расчета 2×104 клеток/лунку, а затем подвергали воздействию TGFβ, который способствует пролиферации клеток, в течение 24 часов. Кроме того, в каждую лунку добавляли 0,1 мг МТТ с последующей выдержкой в течение 4 часов при 37°С. После этого культуральную среду удаляли, клетки лизировали диметилсульфоксидом, а затем измеряли оптическую плотность при 590 нм с использованием следующих устройств.
2-9) Электрофизиологический анализ
Ток целых клеток через клеточную мембрану в изолированных и культивируемых единичных клетках измеряли с использованием метода фиксации потенциала. Линейной изменение напряжения измеряли от -100 мВ до +100 мВ с использованием стеклянного микроэлектрода в клетках с фиксацией напряжения целой клетки, и полученный ток усиливали с помощью усилителя (ЕРС-10, HEKA, Lambrecht, Германия) с последующей записью с частотой дискретизации от 1 до 4 кГц.
Стандартный внешний раствор содержал 150 мМ NaCl, 6 мМ KCl, 1,5 мМ CaCl2, 1 мМ MgCl2, 10 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазин этансульфоновая кислота) и 10 мМ глюкозы при рН 7,4 (титрованный NaOH), и раствор стеклянного микроэлектрода (пипетки) содержал 40 мМ KCl, 100 мМ K-аспартата, 2 мМ MgCl2, 0,1 мМ EGTA (этиленгликольтетрауксусная кислота), 4 мМ Na2ATP и 10 мМ HEPES при рН 7.2 (титрованный КОН). Концентрацию свободного Са2+ в растворе для пипетки доводили до 1 мкМ, добавляя соответствующее количество Са2+ в присутствии 5 мМ EGTA (рассчитано с помощью CaBuf; G. Droogmans, Leuven, Бельгия).
Ток KCa2.3 выделяли следующим способом. Среди токов, зарегистрированных при введении 1 мкМ Са2+ в клетки с фиксированным напряжением цельной клетки с использованием стеклянного электрода и применением 1-этил-2-бензимидазолинона (1-EBIO, 100 мкМ), активирующего ток KCa2.3, ток, ингибируемый а па ми ном (200 нМ), который является ингибитором каналов KCa2.3, был определен как ток KCa2.3, и зарегистрированный ток был разделен на емкость ячейки и нормализован.
2-10) Статистический анализ
Экспериментальные результаты выражали как среднее ± стандартное отклонение (SEM). Статистический анализ проводился с использованием t-критерия Стьюдента, и Р≤0,05 определяли как значимое различие.
3) Результаты экспериментов с использованием культивированных клеток
Эксперименты проводились для определения влияния соединений формул А1-А9 (CBM-N1 ~ N9) согласно настоящему изобретению на экспрессию канала KCa2.3 in vitro, а также для определения того, могут ли соединения формул А1-А9 (CBM-N1 ~ N9) подавлять фиброз.
3-1) Влияние факторов роста и CBM-N1 на каналы KCa2.1 и KCa3.1
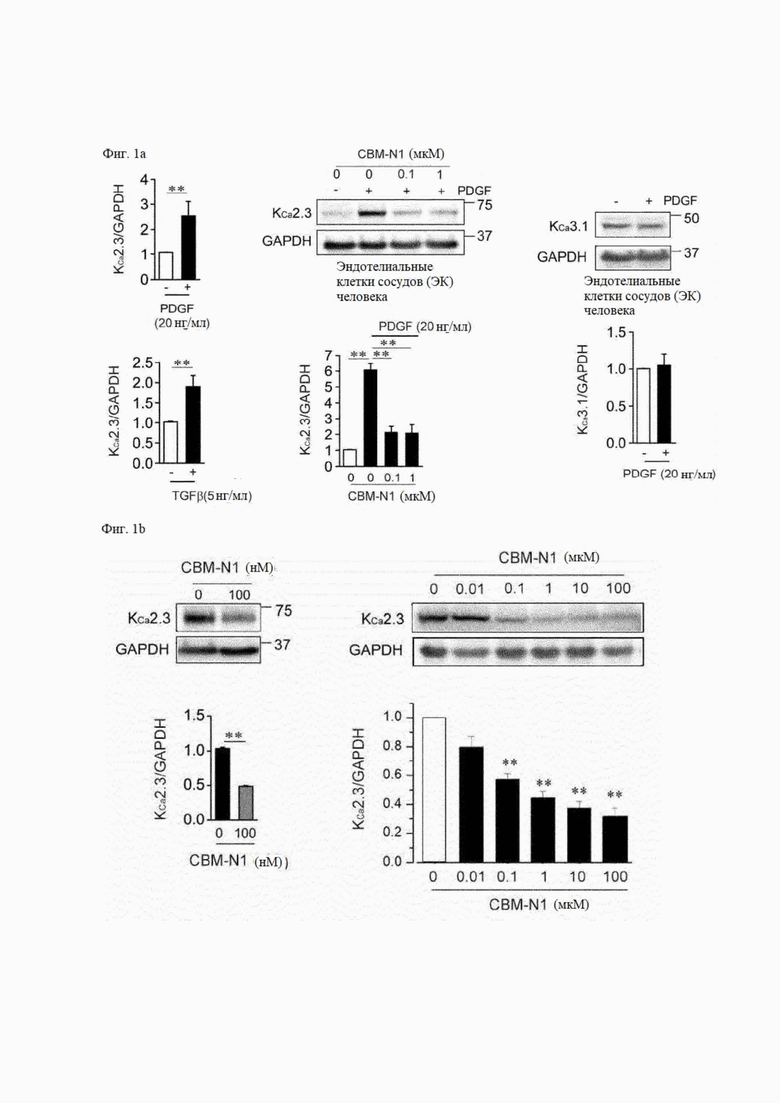
На фиг. 1А показано влияние каждого из PDGF, TGFβ и CBM-N1 на экспрессию канала KCa2.3 или канала KCa3.1 в эндотелиальных клетках сосудов. Когда эндотелиальные клетки сосудов подвергались воздействию PDGF (20 нг/мл) или TGFβ (5 нг/мл) в течение 24 часов, экспрессия мРНК (левая панель) и белка (средняя панель) канала KCa2.3 увеличивались. С другой стороны, экспрессия канала KCa3.1 не увеличивалась из-за обработки PDGF (правая панель). Кроме того, когда клетки, в которых экспрессия белка канала KCa2.3 увеличивалась с помощью PDGF, обрабатывали CBM-N1 в течение 24 часов, экспрессия белка канала KCa2.3 значительно снижалась (средняя панель).
На фиг. 1В показано влияние CBM-N1 на экспрессию белка канала KCa2.3 в фибробластах (левая панель) или звездчатых клетках печени (правая панель). В результате уровень экспрессии стабильного канала KCa2.3 снижался в фибробластах после обработки CBM-N1 и снижался в звездчатых клетках печени в зависимости от концентрации.
На фиг. 1С показано влияние ингибирования экспрессии канала KCa2.3 в звездчатых клетках печени при воздействии CBM-N1 на ток KCa2.3. Плотность тока KCa2.3 при мембранном потенциале +50 мВ сравнивали между клетками, подвергнутыми воздействию CBM-N1 в течение 24 часов, и клетками, не подвергавшимися воздействию CBM-N1 Плотность тока KCa2.3 составляла 18,98±4,17 пА/пФ в клетках, не подвергавшихся воздействию CBM-N1. и 7.77±2,65 мВ/пФ в клетках, подвергнутых воздействию CBM-N1. То есть ток KCa2.3 был значительно снижен за счет ингибирования экспрессии канала KCa2.3 из-за CBM-N1. Как описано выше, снижение тока KCa2.3 в клетках, в которых экспрессия белка канала KCa2.3 была снижена с помощью CBM-N1, означает снижение экспрессии белка канала в клеточной мембране с помощью CBM-N1.
3-2) Ингибирующее действие CBM-N1 на фиброз
На фиг. 2 показано ингибирующее действие CBM-N1 на индуцированный TGFβ фиброз в фибробластах. Эффект индуцирования фиброза с помощью TGFβ определяли по уровням экспрессии маркеров фиброза (фиг. 2А) и эффекту индукции пролиферации клеток (фиг. 2В). Когда фибробласты подвергались действию TGFβ (5 нг/мл) в течение 24 часов, количество маркеров фиброза, таких как α-актин гладких мышц (α-SMA), белки коллагена 1α (Col1α) и коллагена 3α (Col3α), увеличивалось, и стимулировалась пролиферация клеток. Когда фибробласты подвергались действию TGFβ + CBM-N1 в течение 24 часов, уровни экспрессии маркеров фиброза снижались и пролиферация клеток подавлялась. Эти результаты показывают, что CBM-N1 ингибирует фиброз, вызванный фактором, вызывающим фиброз.
3-3) Влияние CBM-N1 и его производных (CBM-N2 - CBM-N9) на экспрессию канала KCa2.3 и пролиферацию клеток
На фиг. 3 показано влияние производных CBM-N1 на экспрессию белка канала KCa2.3 в звездчатых клетках печени. В частности, при воздействии на клетки CBM-N2, CBM-N5, CBM-N8 и CBM-N9 в течение 24 часов экспрессия канала KCa2.3 была значительно снижена.
На фиг. 4 показано влияние ингибирования экспрессии канала KCa2.3 CBM-N2 - CBM-N4 и CBM-N9 в звездчатых клетках печени на ток KCa2.3. В результате сравнения плотностей тока KCa2.3 при мембранном потенциале +50 мВ между клетками, в которых экспрессия канала KCa2.3 ингибируется воздействием соединения в течение 24 часов, и клетками, не подвергавшимися воздействию соединения, из-за воздействия CBM-N2 - CBM-N4 и CBM-N9 плотности тока KCa2.3 были значительно снижены в клетках, в которых экспрессия канала KCa2.3 была снижена.
На фиг. 5 показано влияние CBM-N2 - CBM-N5, CBM-N8 и CBM-N9 на TGFβ или PDGF-индуцированную пролиферацию клеток в фибробластах. Когда клетки подвергались воздействию фактора, вызывающего фиброз, такого как TGFβ (5 нг/мл) или PDGF (20 нг/мл) в течение 24 часов, пролиферация фибробластов значительно увеличивалась, а пролиферация клеток значительно снижалась под воздействием CBM-N2 - CBM-N5, CBM-N8 и CBM-N9.
4) Результаты, полученные на мышиных моделях с заболеванием печени
Эти эксперименты были выполнены для определения терапевтических эффектов соединений формул А1-А5 (CBM-N1 - CBM-N5) на мышиных моделях заболевания печени.
4-1) Гистологический или иммуногистологический анализ
На фиг. 6А показаны результаты окрашивания тканей печени Н&Е (гематоксилин-эозином), и часть, представленная пунктирной линией на верхней панели, была увеличена и показана на нижней панели. В труппе с индуцированным заболеванием (ТАА), ТАА-индуцированное воспаление возникает в области около центральной вены (CV), что может быть подтверждено клетками воспаления, имеющими большие ядра, сконцентрированные около CV (стрелки на фиг. 6А). В частности, по сравнению с группой здорового контроля в группе с вызванным заболеванием, количество воспалительных клеток значительно увеличилось, а в труппе, получавшей лекарственное средство (ТАА + CBM-N1), по сравнению с группой с вызванным заболеванием, количество воспалительных клеток значительно уменьшилось.
На фиг. 6В показаны результаты окрашивания маркера воспаления CD82 в ткани печени, в которых не наблюдали клеток, окрашенных в коричневый цвет, в ткани печени здорового контроля (Контроль), что указывает на отсутствие лимфоидных клеток. С другой стороны, в ткани печени группы, в которой заболевание вызвано ТАА (ТАА), между CV было обнаружено много клеток, окрашенных в коричневый цвет. Однако очень небольшое количество клеток, окрашенных в коричневый цвет, было обнаружено в ткани печени (ТАА + CBM-N1 (R) или ТАА + CBM-N1 (S)) в группе, получавшей лекарственное средство, обработанной ТАА и R-изомером или S-изомером CBM-N1.
На фиг. 6С показаны результаты окрашивания трихромом по Массону коллагеновых волокон в тканях печени, причем коллаген был окрашен в синий цвет. Ткань печени в здоровом контроле (контроль) демонстрирует здоровое состояние, в котором фиброз еще не прогрессировал, тогда как ткань печени в группе, вызванной заболеванием (ТАА), окрашена в синий цвет (указана стрелками) рядом с CV или между CV, демонстрируя прогрессирование фиброза. Однако в ткани печени в группе, получавшей лекарственное средство (ТАА + CBM-N1 (R), ТАА + CBM-N1 (S)), которой вводили ТАА и R-изомер или S-изомер CBM-N1, наблюдали очень незначительный фиброз вокруг CV.
На фиг. 6D показаны результаты окрашивания волокон ретикулина в тканях печени, при этом волокна ретикулина были окрашены в черный цвет. Волокна ретикулина не наблюдались в ткани печени из здорового контроля (контроль), тогда как в ткани печени в группе с вызванным заболеванием (ТАА), волокна ретикулина (обозначены стрелками) наблюдались вокруг CV и между CV. Кроме того, в ткани печени (ТАА + CBM-N1 (R) или (ТАА + CBM-N1 (S)) в группе, получавшей лекарственное средство, ТАА и R-изомер или S-изомер CBM-N1, волокна ретикулина очень слабо наблюдались только около CV.
4-2) Проверка функции печени с помощью анализов ALT и AST в крови
На фиг. 7А показаны результаты тестирования функции печени для здорового контроля, группы с вызванным ТАА заболеванием, и групп, которым вводили лекарственное средство, обработанных CBM-N1 - CBM-N5. В здоровом контроле (Контроль), группе с вызванным заболеванием (ТАА), и группах, которым вводили лекарственное средство (ТАА + CBM-N1, ТАА + CBM-N2, ТАА + CBM-N3, ТАА + СВМ-N4 и ТАА + CBM-N5), уровни ALT составляли 41,6±7,9 ед./л, 209,0±42,4 ед./л, 70,3±14,7 ед./л, 113,4±7,9 ед./л, 103,0±6,9 ед./л, 114,1±8,8 ед./л и 106,4±12,8 ед./л соответственно, а уровни AST в крови составляли 60,4±7,5 ед./л, 211,1±22,4 ед./л, 62,7±11,6 ед./л, 83,6±14,8 ед./л, 73,4±7,4 ед./л, 66,6±9,4 ед./л и 67,7±10,2 ед./л, соответственно.
Кроме того, на фиг. 7В показан результат тестирования группы с заболеванием, вызванным западной диетой, и в группе здорового контроля (контроль), группы, в которой заболевание было вызвано западной диетой (WD), и группы, в которой вводили лекарственное средство (WD + CBM-N1), уровни ALT составляли 38,7±9,7 ед./л, 189,8±37,6 ед./л и 87,2±24,7 ед./л, а уровни AST в крови составляли 47,4±18,2 ед./л, 173,5±31,5 ед./л и 71,4±19,8 ед./л соответственно.
Из результатов испытании можно видеть, что дисфункция печени, вызванная ТАА или западной диетой, значительно восстанавливается соединениями формул А1-А5 (50 мг/кг/день).
4-3) ГИДР в реальном времени на маркеры воспаления
На фиг. 8 показаны результаты сравнения экспрессии мРНК воспалительных цитокинов в здоровом контроле, группе с вызванным заболеванием, и группе, в которой вводили лекарственное средство. В качестве маркеров воспаления используется фактор некроза опухоли альфа (TNFα), хемоаттрактантный белок-1 (CCL2), интерлейкин-12 (IL12), трансформирующий фактор роста (TGF), IL1α, IL6 и макрофагальный белок воспаления-2 (MIP-2), которые усиливаются при воспалении. Уровень мРНК фактора воспаления увеличился в группе с вызванным заболеванием (ТАА) по сравнению со здоровым контролем (контроль), и снизился в группе, получавшей CBM-N1 (ТАА + СВМ-N1), по сравнению с группой с вызванным заболеванием (ТАА) (фиг. 8А).
Уровень мРНК фактора воспаления также снизился в группах, получавших СВМ-N2 - CBM-N5 (ТАА + CBM-N2, ТАА + CBM-N3, ТАА + CBM-N4 и ТАА + CBM-N5) по сравнению с группой с вызванным заболеванием (фиг. 8В). Следовательно, можно видеть, что соединения формул А1-А5 снижают экспрессию воспалительного цитокина, тем самым оказывая терапевтический эффект на воспалительное заболевание печени, вызванное ТАА.
Кроме того, на фиг. 8С показаны результаты сравнения экспрессии мРНК воспалительных цитокинов в здоровом контроле (контроль), труппе, в которой заболевание было вызвано западной диетой (WD), и группой, в которой вводили лекарственное средство (WD + CBM-N1). Здесь в качестве воспалительных цитокинов измеряли CCL2, IL6 и IL1α. Уровни экспрессии мРНК этих факторов воспаления увеличивались в группе с вызванным заболеванием по сравнению со здоровым контролем и снижались в труппе, получавшей лекарственное средство, по сравнению с труппой с вызванным заболеванием.
4-4) ПЦР в реальном времени на маркеры фиброза
На фиг. 9 показаны результаты сравнения уровней экспрессии мРНК маркеров фиброза в здоровом контроле, труппе с вызванным заболеванием, и труппе, в которой вводили лекарственное средство. В качестве маркеров фиброза использовали Col1α, Col3α, Col4α, α-SMA и рецептор 2 трансформирующего фактора роста (TGFR2). Маркеры фиброза увеличились в группе с вызванным заболеванием (ТАА), по сравнению со здоровым контролем (контроль), что указывает на прогрессирование фиброза из-за воспаления. Однако было подтверждено, что в группе, получавшей CBM-N1 (ТАА + CBM-N1), уровни этих факторов фиброза снизились.
4-5) Влияние на экспрессию белка маркера воспаления или белков маркера фиброза
На фиг. 10А показано влияние R-изомера и S-изомера CBM-N1 на экспрессию белков маркеров воспаления в мышиных моделях ТАА-опосредованного заболевания печени. По сравнению с контролем (контроль) уровни экспрессии белков TIMP-2 и CCR2 значительно увеличились в ткани печени группы с вызванным заболеванием (ТАА), что указывает на прогрессирование воспаления. Однако в группе, получавшей R-изомер или S-изомер CBM-N1 [ТАА + CBM-N1 (R), ТАА + CBM-N1 (S)], уровни экспрессии белков TIMP-2 и CCR2 снизились, подтверждая, что воспаление было подавлено.
На фиг. 10В показано влияние R-изомера или S-изомера CBM-N1 на экспрессию белков маркеров фиброза в мышиных моделях ТАА-опосредованного заболевания печени. По сравнению со здоровым контролем (контроль) уровни экспрессии белков α-SMA и Col 1 а значительно увеличились в ткани печени группы с вызванным заболеванием (ТАА), что указывает на прогрессирование фиброза. Однако уровни экспрессии белков a-SMA и Col1α значительно снизились в группе, получавшей R-изомер или S-изомер CBM-N1, что подтверждает ингибирование фиброза.
4-6) Влияние на экспрессию белка канала KCa2.3
На фиг. 11 показано влияние R-изомера или S-изомера CBM-N1 на экспрессию белка канала KCa2.3 в мышиных моделях ТАА-опосредованного заболевания печени. По сравнению со здоровым контролем (контроль) уровень экспрессии белка канала KCa2.3 значительно увеличился в ткани печени в группе с вызванным заболеванием (ТАА). Однако уровень экспрессии белка канала KCa2.3 значительно снизился в группе, получавшей R-изомер или S-изомер CBM-N1 Эти результаты показывают, что вызванное ТАА заболевание печени связано с увеличением экспрессии KCa2.3, а терапевтический эффект CBM-N1 связан со снижением экспрессии KCa2.3.
5) Результат эксперимента с CBM-N9 на мышиных моделях заболевания легких
Этот эксперимент был проведен для определения терапевтического эффекта соединения формулы А9 (CBM-N9) по настоящему изобретению на мышиных моделях индуцированного блеомицином заболеванием легких.
5-1) Гистологический или иммуногистологический анализ
На фиг. 12 показаны результаты окрашивания Н&Е в легочной ткани, иммуногистохимии на CD45 (общий антиген лейкоцитов, окрашивание LCA) и окрашивания трихромом Массона на коллаген. Было подтверждено, что степень воспаления и фиброза увеличивалась в группе с вызванным заболеванием (блеомицин) по сравнению со здоровым контролем, и снижалась в группе, получавшей CBM-N9 (блеомицин + CBM-N9), по сравнению с группой с вызванным заболеванием.
5-2) Анализ экспрессии белков маркеров воспаления или фиброза
На фиг. 13А показано влияние CBM-N9 на экспрессию маркеров воспаления на мышиных моделях заболеваний легких. Уровни экспрессии белков маркеров воспаления, таких как TIMP-2 и CCR2, в легочной ткани значительно увеличились в группе с вызванным заболеванием (блеомицин) по сравнению со здоровым контролем (контроль), что привело к прогрессированию воспаления. Уровни экспрессии белков TIMP-2 и CCR2 значительно снизились в группе, получавшей лекарственное средство CBM-N9 (блеомицин + CBM-N9), что подтверждает ингибирование воспаления.
На фиг. 13В показано влияние CBM-N9 на экспрессию белков-маркеров фиброза на мышиных моделях болезни легких. Уровни экспрессии белков маркеров фиброза, таких как α-SMA и Col1α, значительно увеличились в ткани легких группы с вызванным заболеванием (блеомицин) по сравнению со здоровым контролем (контроль), что указывает на прогрессирование фиброза легких. С другой стороны, уровни экспрессии белков TIMP-2 и CCR2 значительно снизились в группе, получавшей лекарственное средство (блеомицин + CBM-N9), подтверждая ингибирование фиброза.
Оценка и выводы
Как видно выше, когда в культуру клеток вводили соединения формул А1-А9 согласно настоящему изобретению in vitro в течение длительного времени (24 часа или 16 недель), эффект ингибирования фиброза проявлялся в снижении экспрессии белка канала KCa2.3. В частности, было подтверждено, что, когда культивируемые звездчатые клетки печени, фибробласты и эндотелиальные клетки сосудов подвергались воздействию соединений формул А1-А9 в течение 24 часов, экспрессия канала KCa2.3 в клеточной мембране ингибировалась, и экспрессия факторов, связанных с фиброзом (α-SMA, Col1α и т.д.), и пролиферация клеток с помощью факторов роста, вызывающих фиброз, подавлялись.
Кроме того, было подтверждено, что соединения формул А1-А9 согласно настоящему изобретению обладают ингибирующим действием на воспаление и фиброз в группе с вызванным заболеванием печени, даже в эксперименте in vivo на мышиных моделях. В частности, в результате введения соединений формул А1-А9 в мышиных моделях заболевания печени или легких в течение 16 недель значительно подавлялись воспаление и фиброз.
Между тем, как раскрыто в патентах Кореи № 10-1345860 и 10-1414831 и соответствующем ему патенте США № 9259412, соединения формул А1-А5 по настоящему изобретению обладают эффектами ингибирования активности канала KCa3.1 за счет фосфорилирования канала KCa3.1, индуцированного цАМФ Однако считается, что подавление активности канала KCa3.1 за счет увеличения цАМФ будет слабо влиять на лечение фиброза.
Настоящее изобретение относится к эффекту, проявляющемуся при воздействии соединений формул А1-А5 в течение короткого времени (в течение нескольких минут), и повышению из-за этих соединений уровня цАМФ, который достигает наивысшего уровня примерно за 20 минут, а затем его резкому снижению, при этом уровень цАМФ становится аналогичным уровню до введения лекарственного средства в течение трех часов (Endocrinology 144 (4): 1292-1300). Таким образом, это связано с тем, что эффект, вызванный цАМФ, проявляется только в течение короткого времени, например, не больше приблизительно одного часа, и, как в случае настоящего изобретения, вряд ли продлится в течение 24 часов или 16 недель.
Кроме того, как подтверждено на фиг. 1А настоящего изобретения, при воздействии фактора, индуцирующего фиброз, PDGF, в течение 24 часов, экспрессия канала KCa2.3 значительно увеличивалась, тогда как экспрессия канала KCa3.1 не увеличивалась. В соответствии с этим результатом можно сделать вывод, что при фиброзном процессе усиление экспрессии каналов KCa2.3 является очень важным условием, и эффект подавления фиброза соединениями формул А1-А9 согласно настоящему изобретению является результатом снижение экспрессии канала KCa2.3.
Между тем в настоящем изобретении из-за практических ограничений описанные выше эксперименты не проводились для всех соединений, принадлежащих к соединениям формулы А. Однако, принимая во внимание химическую активность соединений формулы А и метаболические механизмы in vivo, логично сделать вывод, что все соединения формулы А обладают фармакологическими эффектами, такими же или подобными соединениям формул А1-А9.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРИМЕНЕНИЕ ИНГИБИТОРОВ VAP-1 ДЛЯ ЛЕЧЕНИЯ ФИБРОЗНЫХ БОЛЕЗНЕЙ | 2010 |
|
RU2580626C2 |
| ПРИМЕНЕНИЕ ИНГИБИТОРОВ VAP-1 ДЛЯ ЛЕЧЕНИЯ ФИБРОЗНЫХ БОЛЕЗНЕЙ | 2010 |
|
RU2667963C1 |
| УСОВЕРШЕНСТВОВАННОЕ СОЕДИНЕНИЕ ДЛЯ ЛЕЧЕНИЯ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ | 2018 |
|
RU2794975C2 |
| КОМПОЗИЦИЯ ДЛЯ ВОССТАНОВЛЕНИЯ НОРМАЛЬНОЙ ТКАНИ ИЗ ФИБРОЗНОЙ ТКАНИ | 2011 |
|
RU2650796C2 |
| МАТЕРИАЛЫ И СПОСОБЫ ЛЕЧЕНИЯ ХРОНИЧЕСКИХ ФИБРОЗНЫХ ЗАБОЛЕВАНИЙ | 2006 |
|
RU2446825C2 |
| ИНГИБИТОРЫ HDAC ДЛЯ ЛЕЧЕНИЯ ИДИОПАТИЧЕСКОГО ЛЕГОЧНОГО ФИБРОЗА И ДРУГИХ ВОСПАЛИТЕЛЬНЫХ НАРУШЕНИЙ ЛЕГКИХ | 2021 |
|
RU2833349C1 |
| ПРИМЕНЕНИЕ ЧИГЛИТАЗАРА И РОДСТВЕННЫХ ЕМУ СОЕДИНЕНИЙ | 2019 |
|
RU2769446C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ НЕАЛКОГОЛЬНОГО СТЕАТОГЕПАТИТА | 2021 |
|
RU2803733C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ НЕАЛКОГОЛЬНОЙ ЖИРОВОЙ ИНФИЛЬТРАЦИИ ПЕЧЕНИ, СОДЕРЖАЩАЯ ЛИГАНД GPR119 В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2019 |
|
RU2768943C1 |
| ЛЕЧЕНИЕ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ У ЧЕЛОВЕКА | 2020 |
|
RU2811365C2 |
Изобретение относится к применению фармацевтической композиции, содержащей эффективное количество соединения формулы А или его фармацевтически приемлемой соли в качестве активного ингредиента и обладающей эффектом подавления экспрессии белка канала KCa2.3 в клеточной мембране для лечения фиброза печени или фиброза легких. В формуле A каждый из X1 ~ X10 независимо представляет собой водород (H) или фтор (F), все из которых могут быть одинаковыми или отличаться друг от друга; Y представляет собой серу (S) или сульфоксид (S=O), * указывает на хиральное положение; R1 является любым из водорода, метильной группы, гидроксильной группы, фенильной группы и (тетрагидрофуран-2-ил)метила. Технический результат – лечение фиброза печени и фиброза легких за счет использования фармацевтической композиции, содержащей эффективное количество соединения формулы А или его фармацевтически приемлемой соли, которое подавляет экспрессию белка канала KCa2.3 в клеточной мембране. 5 з.п. ф-лы, 13 ил., 4 табл., 2 пр.
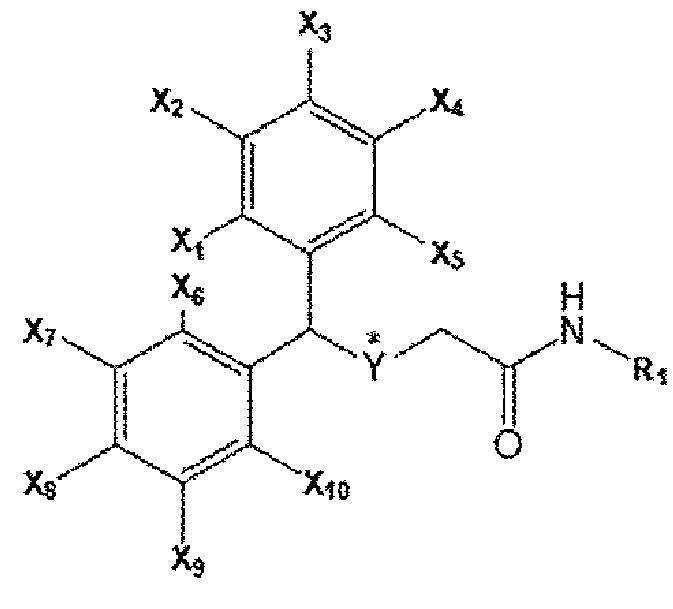
формула А
1. Применение фармацевтической композиции, содержащей эффективное количество соединения бензгидрилтиоацетамида, представленного приведенной ниже формулой А, или его фармацевтически приемлемой соли в качестве активного ингредиента, и обладающей эффектом подавления экспрессии белка канала KCa2.3 в клеточной мембране,
[Формула А]
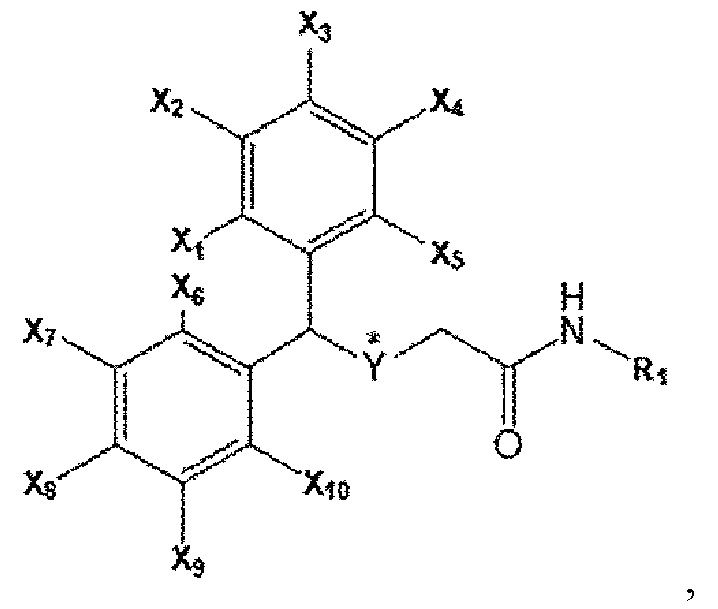
где в формуле A каждый из X1 ~ X10 независимо представляет собой водород (H) или фтор (F), все из которых могут быть одинаковыми или отличаться друг от друга; Y представляет собой серу (S) или сульфоксид (S=O), * указывает на хиральное положение; R1 является любым из водорода, метильной группы, гидроксильной группы, фенильной группы и (тетрагидрофуран-2-ил)метила, для лечения фиброзного заболевания, выбранного из фиброза печени или фиброза легких.
2. Применение по п. 1, где в соединении формулы A каждый из X1 ~ X10 независимо представляет собой водород (H) или фтор (F), Y представляет собой серу (S), и R1 представляет собой водород (H).
3. Применение по п. 1, где в соединении формулы A каждый из X1 ~ X10 независимо представляет собой водород (H) или фтор (F), Y представляет собой сульфоксид (S=O), и R1 представляет собой водород (H).
4. Применение по п. 1, где соединение формулы A представляет собой любое соединение, выбранное из группы, состоящей из 2-(бензгидрилсульфинил)ацетамида, 2-(бензгидрилтио)-N-[(тетрагидрофуран-2-ил)метил]ацетамида, 2-(бензгидрилтио)-N-фенилацетамида, 2-(бензгидрилсульфинил)-N-метилацетамида, 2-(бензгидрилсулфинил)-N-[(тетрагидрофуран-2-ил)метил]ацетамида, 2-(бензгидрилтио)-ен-метилацетамида, 2-[бис(2-фторфенил)метансульфинил]ацетамида, 2-[бис(3-фторфенил)метансульфинил]ацетамида и 2-[бис(4-фторфенил)метансульфинил]ацетамида.
5. Применение по п. 1, где соединение формулы A представляет собой 2-(бензгидрилсульфинил)ацетамид (модафинил).
6. Применение по п. 1, где соединение формулы A представляет собой 2-[бис(4-фторфенил)метансульфинил]ацетамид (лауфлумид).
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| KR 20130080364 A, 12.07.2013 | |||
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| EA 201591775 A1, 31.05.2016 | |||
| Приспособление для ретуши негативов | 1926 |
|
SU9949A1 |

