Область техники, к которой относится настоящее изобретение
[0001]
Настоящее изобретение относится к фенилдифторметилзамещенному соединению пролинамида, которое оказывает ингибирующее действие на катепсин S и, как ожидается, будет применяться в качестве активного ингредиента фармацевтической композиции, например, фармацевтической композиции для профилактики и/или лечения аутоиммунного заболевания, включая в себя системную красную волчанку (SLE) и волчаночный нефрит, аллергии или отторжения трансплантата органа, костного мозга или ткани.
Предшествующий уровень техники настоящего изобретения
[0002]
Катепсин S представляет собой лизосомальную цистеиновую протеазу, экспрессируемую главным образом в антигенпрезентирующих клетках, таких как дендритные клетки, макрофаги и В-клетки, и обуславливает деградацию инвариантной цепи, связанной с молекулами главного комплекса гистосовместимости класса II (МНС класса II) во время производства. Молекулы МНС класса II связываются с аутологичным или неаутологичным пептидом, включенным внеклеточно, и индуцируют секрецию различных цитокинов, презентируя аутологичный пептид или неаутологичный пептид CD4-позитивным Т-клеткам. Было подтверждено, что ингибирование или делеция катепсина S ингибирует загрузку антигенного пептида в молекулы МНС класса II, и, кроме того, подавление презентации антигена CD4-позитивным Т-клеткам снижает иммунный ответ против чужеродных антигенов ("Immunity", 1999, vol. 10, No. 2, p. 207-217). Считается, что в случае такого аутоиммунного заболевания, как SLE, описанная выше презентация антигена происходит в отношении патогенного аутологичного пептида, и поэтому считается, что существует высокая вероятность того, что ингибитор катепсина S будет применим для лечения аутоиммунного заболевания. ("Journal of Clinical Investigation", 1998, Vol. 101, No. 11, p. 2351-2363).
[0003]
Соответственно, ожидается, что ингибитор катепсина S является перспективным в качестве средства для профилактики и/или лечения аутоиммунного заболевания, включая в себя SLE и волчаночный нефрит, или средства для профилактики и/или лечения аллергии или отторжения трансплантата органа, костного мозга или ткани.
[0004]
В патентном документе 1 раскрывается, что соединение формулы (А) проявляет ингибирующее действие на катепсин S и применимо для лечения различных метаболических заболеваний или иммунных заболеваний, таких как SLE.
[Хим. 1]
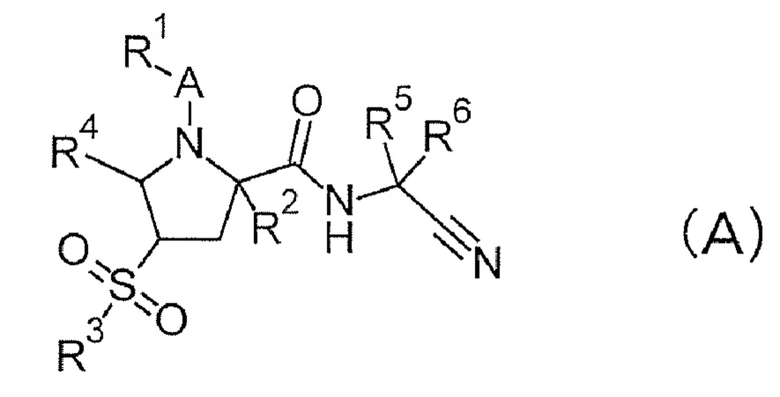
(Обратитесь к настоящей публикации в отношении символов в формуле.)
В патентном документе 2 раскрывается, что соединение формулы (В) проявляет ингибирующее действие на катепсин S и применимо для лечения различных метаболических заболеваний или иммунных заболеваний, таких как SLE.
[Хим. 2]
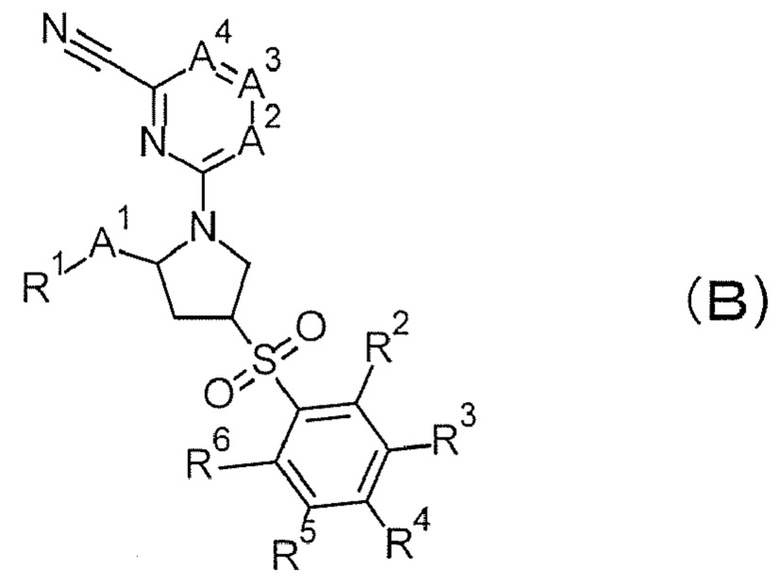
(Обратитесь к настоящей публикации в отношении символов в формуле.)
В непатентном документе 1 раскрывается, что соединение формулы (С) препятствует развитию SLE и волчаночного нефрита.
[Хим. 3]
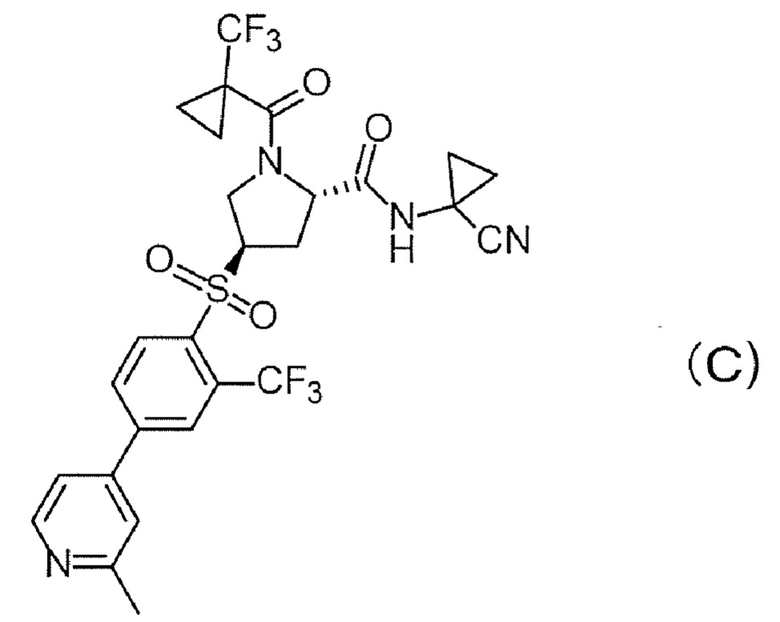
В патентном документе 3 раскрывается, что соединение формулы (D) проявляет ингибирующее действие на катепсин S и применимо для лечения сахарного диабета и т.п.
[Хим. 4]
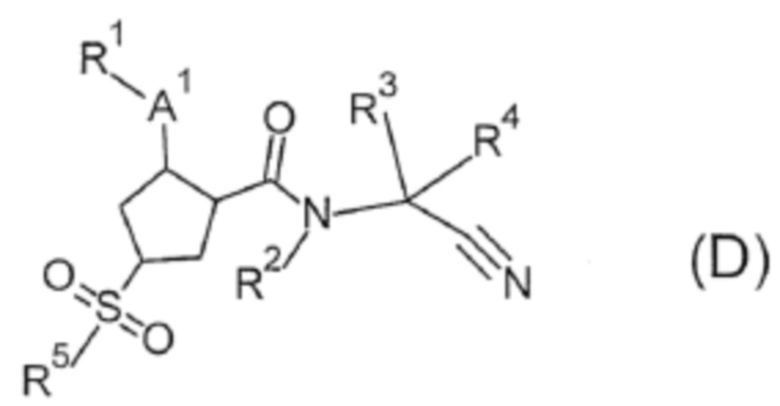
((Обратитесь к настоящей публикации в отношении символов в формуле.) В патентном документе 4 раскрывается что соединение формулы (Е) проявляет ингибирующее действие на катепсин S и применимо для лечения сахарного диабета и т.п.
[Хим. 5]
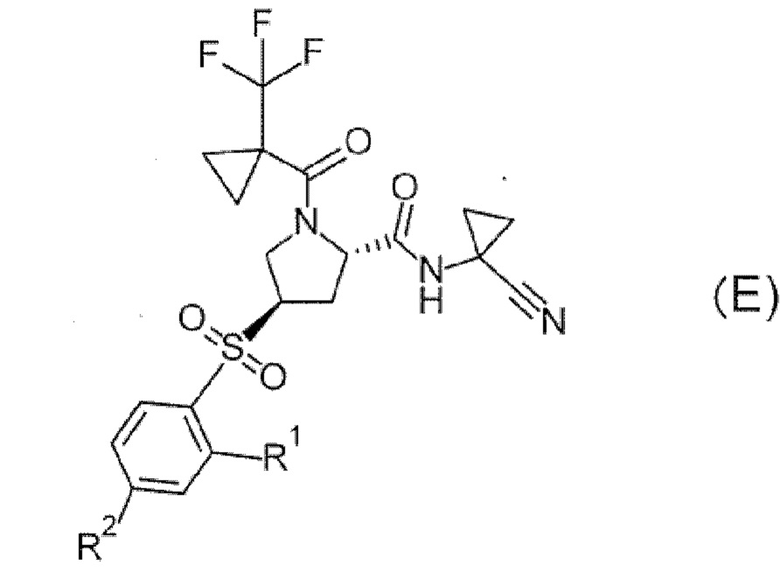
(Обратитесь к настоящей публикации в отношении символов в формуле.)
Материалы, использованные при экспертизе заявки
Патентный документ
[0005]
[Патентный документ 1] WO 2010/121918
[Патентный документ 2] WO 2012/059507
[Патентный документ 3] WO 2010/142650
[Патентный документ 4] WO 2017/144483
Не патентный документ
[0006]
[Не патентный документ 1] «Annals of the Rheumatic Diseases)), 2015, Vol. 74, p.452-463
Раскрытие настоящего изобретения
Решаемые настоящим изобретением проблемы
[0007]
Предусмотрено соединение, которое обладает ингибирующим эффектом на катепсин S и, как ожидается, будет применимо в качестве активного ингредиента фармацевтической композиции, например, фармацевтической композиции для профилактики и/или лечения аутоиммунного заболевания, включая в себя SLE и волчаночный нефрит, аллергии или отторжения трансплантата органа, костного мозга или ткани.
Средства для решения проблем
[0008]
В результате интенсивных исследований соединения, оказывающего ингибирующее действие на катепсин S, авторы настоящего изобретения обнаружили, что фенилдифторметилзамещенное соединение пролинамида оказывает ингибирующее действие на катепсин S, таким образом, настоящее изобретение является полным.
То есть настоящее изобретение относится к соединению формулы (I) или его соли и относится к фармацевтической композиции, содержащей соединение формулы (I) или его соль и один или несколько вспомогательных веществ.
[Хим. 6]
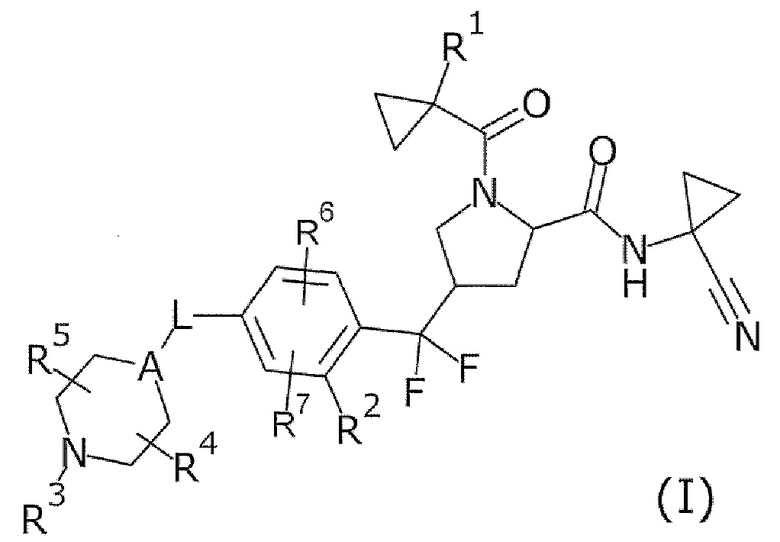
(В формуле
R1 представляет собой низший алкил или галогеновый низший алкил,
R2 представляет собой галоген или галогеновый низший алкил,
L представляет собой связь или -CH2-,
А представляет собой СН или N,
R3 представляет собой Н или низший алкил,
R4 и R5 одинаковые или отличаются друг от друга и представляют собой Н или низший алкил, и
R6 и R7 одинаковые или отличаются друг от друга и представляют собой Н, низший алкил или галоген.)
Если не описано иное, в случае, если символы в химических формулах в настоящем описании также используются в других химических формулах, эти же символы обладают тем же значением.
[0009]
Настоящее изобретение дополнительно относится к фармацевтической композиции для предотвращения и/или лечения аутоиммунного заболевания, включая в себя SLE и волчаночный нефрит, аллергии или отторжение трансплантата органа, костного мозга или ткани, содержащей соединение формулы (I) или его соль. Фармацевтическая композиция включает в себя средство для предотвращения и/или лечения аутоиммунного заболевания, включая в себя SLE и волчаночный нефрит, аллергии или отторжение трансплантата органа, костного мозга или ткани, причем она содержит соединение формулы (I) или его соль.
Настоящее изобретение еще дополнительно относится к:
(1) соединению формулы (I) или его соли, которое представляет собой ингибитор катепсина S;
(2) соединению формулы (I) или его соли для применения в качестве ингибитора катепсина S;
(3) ингибитору катепсина S, содержащему соединение формулы (I) или его соль;
(4) применению соединения формулы (I) или его соли для изготовления фармацевтической композиции для предотвращения и/или лечения аутоиммунного заболевания, включая в себя SLE и волчаночный нефрит, аллергии или отторжение трансплантата органа, костного мозга или ткани;
(5) применению соединения формулы (I) или его соли для предотвращения и/или лечения аутоиммунного заболевания, включая в себя SLE и волчаночный нефрит, аллергии или отторжение трансплантата органа, костного мозга или ткани;
(6) соединению формулы (I) или его соли для применения при предотвращении и/или лечении аутоиммунного заболевания, включая в себя SLE и волчаночный нефрит, аллергии или отторжение трансплантата органа, костного мозга или ткани; и
(7) способу предотвращения и/или лечения аутоиммунного заболевания, включая в себя SLE и волчаночный нефрит, аллергии или отторжение трансплантата органа, костного мозга или ткани, причем способ включает в себя введение субъекту эффективного количества соединения формулы (I) или его соли.
«Субъект» относится к людям или другим животным, нуждающимся в предотвращении или лечении заболевания, и согласно одному варианту осуществления субъект относится к людям, нуждающимся в предотвращении или лечении заболевания.
Осуществление настоящего изобретения
[0010]
Соединение формулы (I) или его соль обладает ингибирующим действием на катепсин S и может быть использовано в качестве средства для предотвращения и/или лечения аутоиммунного заболевания, включая в себя SLE и волчаночный нефрит, аллергии или отторжение трансплантата органа, костного мозга или ткани.
Варианты осуществления настоящего изобретения
[0011]
Далее настоящее изобретение будет описано подробно.
[0012]
Термин «низший алкил» относится к неразветвленному или разветвленному алкилу, содержащему атомы углерода в количестве от 1 до 6 (далее также называется как C1-6), такому как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил и н-гексил, и относится к C1-4 алкилу согласно одному варианту осуществления, метилу или этилу согласно одному варианту осуществления и метилу согласно одному варианту осуществления.
[0013]
Термин «галоген» означает F, Cl, Br и I.
[0014]
Термин «галогеновый низший алкил» представляет собой C1-6 алкил, замещенный одним или несколькими атомами галогена, и относится к C1-6 алкилу, замещенному атомами галогена в количестве от 1 до 5 согласно одному варианту осуществления и относится к CF3 согласно одному варианту осуществления.
[0015]
Некоторые аспекты настоящего изобретения показаны ниже.
(1) Соединение или его соль, в котором формула (I) представлена следующей формулой (Ia).
[Хим. 7]
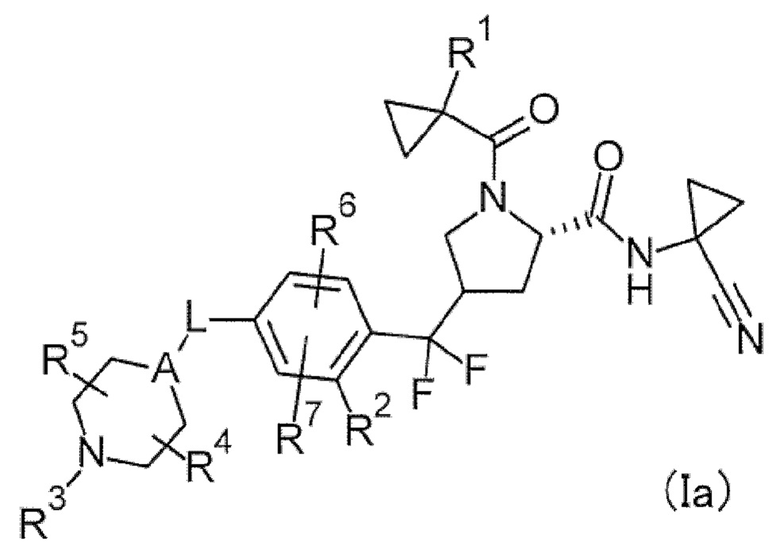
(2) Соединение или его соль, в котором формула (I) представлена следующей формулой (Ib).
[Хим. 8]
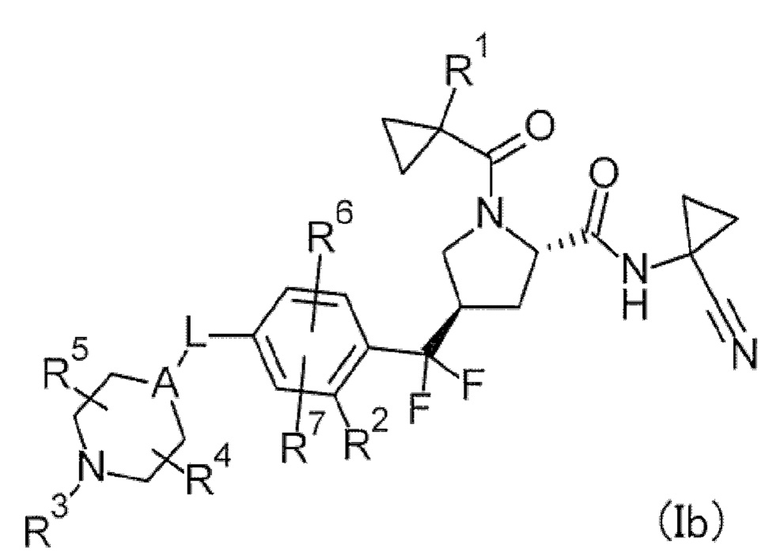
(3) Соединение или его соль, в котором R1 представляет собой низший алкил или галогеновый низший алкил; соединение или его соль, в котором R1 представляет собой галогеновый низший алкил; соединение или его соль, в котором R1 представляет собой низший алкил; соединение или его соль, в котором R1 представляет собой метил или CF3; или соединение или его соль, в котором R1 представляет собой метил.
(4) Соединение или его соль, в котором R2 представляет собой галоген или галогеновый низший алкил; соединение или его соль, в котором R2 представляет собой галоген; соединение или его соль, в котором R2 представляет собой галогеновый низший алкил; или соединение или его соль, в котором R2 представляет собой CF3.
(5) Соединение или его соль, в котором А представляет собой СН или N; соединение или его соль, в котором А представляет собой СН; или соединение или его соль, в котором А представляет собой N.
(6) Соединение или его соль, в котором L представляет собой связь или -СН2-; соединение или его соль, в котором L представляет собой связь; или соединение или его соль, в котором L представляет собой -СН2-.
(7) Соединение или его соль, в котором R3 представляет собой Н или низший алкил; соединение или его соль, в котором R3 представляет собой Н; соединение или его соль, в котором R3 представляет собой низший алкил; соединение или его соль, в котором R3 представляет собой метил или этил; или соединение или его соль, в котором R3 представляет собой метил.
(8) Соединение или его соль, в котором R4 представляет собой Н или низший алкил; соединение или его соль, в котором R4 представляет собой низший алкил; или соединение или его соль, в котором R4 представляет собой Н.
(9) Соединение или его соль, в котором R5 представляет собой Н или низший алкил; соединение или его соль, в котором R5 представляет собой низший алкил; или соединение или его соль, в котором R5 представляет собой Н.
(10) Соединение или его соль, в котором R6 представляет собой Н, низший алкил или галоген; соединение или его соль, в котором R6 представляет собой низший алкил; соединение или его соль, в котором R6 представляет собой галоген; или соединение или его соль, в котором R6 представляет собой Н.
(11) Соединение или его соль, в котором R7 представляет собой Н, низший алкил или галоген; соединение или его соль, в котором R7 представляет собой низший алкил; соединение или его соль, в котором R7 представляет собой галоген; или соединение или его соль, в котором R7 представляет собой Н.
(12) Соединение или его соль, которое представлено двумя или несколькими не противоречивыми комбинациями среди вариантов осуществления, описанных в (1)-(11).
[0016]
Примеры соединения или его соли по настоящему изобретению, представленные комбинациями в вышеуказанном варианте осуществления (12), включают в себя следующие варианты осуществления.
(13) Соединение формулы (I) или его соль, в котором R1 представляет собой низший алкил или галогеновый низший алкил, R2 представляет собой галоген или галогеновый низший алкил, L представляет собой связь или -CH2-, А представляет собой СН или N, R3 представляет собой Н или низший алкил, R4 представляет собой Н или низший алкил, R5 представляет собой Н или низший алкил, R6 представляет собой Н и R7 представляет собой Н.
(14) Соединение формулы (Ia) или его соль, в котором R1 представляет собой низший алкил или галогеновый низший алкил, R2 представляет собой галоген или галогеновый низший алкил, L представляет собой связь или -CH2-, А представляет собой СН или N, R3 представляет собой Н или низший алкил, R4 представляет собой Н или низший алкил, R5 представляет собой Н или низший алкил, R6 представляет собой Н, низший алкил или галоген и R7 представляет собой Н, низший алкил или галоген.
(15) Соединение формулы (I) или его соль, в котором R1 представляет собой низший алкил или галогеновый низший алкил, R2 представляет собой галоген или галогеновый низший алкил, L представляет собой -CH2-, А представляет собой СН или N, R3 представляет собой Н или низший алкил, R4 представляет собой Н или низший алкил, R5 представляет собой Н или низший алкил, R6 представляет собой Н, низший алкил или галоген и R7 представляет собой Н, низший алкил или галоген.
(16) Соединение формулы (I) или его соль, в котором R1 представляет собой низший алкил или галогеновый низший алкил, R2 представляет собой галогеновый низший алкил, L представляет собой -CH2-, А представляет собой N, R3 представляет собой низший алкил, R4 представляет собой Н, R5 представляет собой Н, R6 представляет собой Н и R7 представляет собой Н.
(17) Соединение формулы (Ia) или его соль, в котором R1 представляет собой низший алкил или галогеновый низший алкил, R2 представляет собой галогеновый низший алкил, L представляет собой -CH2-, А представляет собой N, R3 представляет собой низший алкил, R4 представляет собой Н, R5 представляет собой Н, R6 представляет собой Н и R7 представляет собой Н.
(18) Соединение формулы (Ib) или его соль, в котором R1 представляет собой низший алкил или галогеновый низший алкил, R2 представляет собой галогеновый низший алкил, L представляет собой -CH2-, А представляет собой N, R3 представляет собой низший алкил, R4 представляет собой Н, R5 представляет собой Н, R6 представляет собой Н и R7 представляет собой Н.
[0017]
Конкретные примеры соединений, включенных в настоящее изобретение, включают в себя соединения или его соли, выбранные из следующей группы:
(4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-[(1-метилциклопропил)карбонил]-L-пролинамид,
(4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамид,
(4R)-N-(1-цианоциклопропил)-4-[{4-[(4-этилпиперазин-1-ил)метил]-2-(трифторметил)фенил}(дифтор)метил]-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамид и
(4R)-N-(1-цианоциклопропил)-4-[{4-[(4-этилпиперазин-1-ил)метил]-2-(трифторметил)фенил}(дифтор)метил]-1-[(1-метилциклопропил)карбонил]-L-пролинамид.
[0018]
Конкретные примеры соединений, включенных в настоящее изобретение, включают в себя соединение или его соль, которое является кристаллом, содержащим (4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-[(1-метилциклопропил)карбонил]-L-пролинамид (далее в некоторых случаях называется как «соединение А») и янтарную кислоту в молярном соотношении 1:2 и которое характеризуется любым из следующих аспектов.
(1) Соединение характеризуется пиками приблизительно 2θ(°) 2,7, 5,3, 9,8, 10,4, 13,5, 14,0, 15,1, 16,6, 17,4 и 24,4 порошковой рентгеновской дифракцией.
(2) Соединение характерным образом обладает пиками приблизительно 2θ(°) 5,3, 9,8, 15,1, 16,6 и 24,4 порошковой рентгеновской дифракцией.
(3) Соединение характеризуется эндотермическим пиком приблизительно 134,3°С дифференциальной сканирующей калориметрией (анализ DSC).
Кристалл, содержащий соединение А и янтарную кислоту в молярном соотношении 1:2, также включает в себя кристалл дисукцината соединения А и сокристалл янтарной кислоты моносукцинат соединения А.
[0019]
Таутомеры и геометрические изомеры могут существовать в соединении формулы (I) в зависимости от типов заместителя. Согласно настоящему описанию соединение формулы (I) может быть описано только в одной форме изомера, но настоящее изобретение также включает в себя изомеры, отличные от нее, и включает в себя форму, полученную разделением изомеров, или их смесь.
Кроме того, соединение формулы (I) может содержать центр асимметрии или ось хиральности в некоторых случаях, и основанные на нем энантиомеры (оптические изомеры) могут существовать. Соединение формулы (I) или его соль включает в себя любой выделенный отдельный энантиомер, такой как (R) форма или (S) форма, и их смесь (включая рацемическую смесь или не рацемическую смесь). Согласно одному варианту осуществления энантиомер является «стереохимически чистым». Термин «стереохимически чистый» относится к степени чистоты, в которой специалисты настоящей области техники смогут распознать, что энантиомер является в основном стереохимически чистым. Согласно другому варианту осуществления энантиомер представляет собой, например, соединение со стереохимической чистотой 90% э. и. (энантиомерный избыток) или более, 95% э. и. или более, 98% э. и. или более или 99% э. и. или более.
[0020]
Соль соединения формулы (I) представляет собой фармацевтически приемлемую соль соединения формулы (I) и в зависимости от типов заместителей в некоторых случаях может быть образована кислотно-аддитивная соль. Ее конкретные примеры включают в себя кислотно-аддитивную соль неорганической кислоты, такой как хлористоводородная кислота, бромистоводородная кислота, йодоводородная кислота, серная кислота, азотная кислота и фосфорная кислота; и органической кислоты, такой как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, миндальная кислота, виннокаменная кислота, дибензоилвиннокаменная кислота, дитолуоилвиннокаменная кислота, лимонная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, пара-толуолсульфоновая кислота, аспарагиновая кислота и глутаминовая кислота, и т.п. Кроме того, соль соединения формулы (I) также включает в себя сокристалл соединения формулы (I) и кислоту.
[0021]
Настоящее изобретение дополнительно включает в себя вещества с кристаллическим полиморфизмом, различные гидраты и сольваты соединения формулы (I) и его соль. Настоящее изобретение дополнительно включает в себя соединение формулы (I) или его соль, которое является фармацевтически приемлемым и помечено одним или несколькими радиоактивными или не радиоактивными изотопами. Примеры подходящих изотопов, используемых для изотопной маркировки соединения по настоящему изобретению, включают в себя изотопы, такие как водород (2Н, 3Н и т.п.), углерод (11С, 13С, 14С и т.п.), азот (13N, 15N и т.п.), кислород (15O, 17O, 18O и т.п.), фтор (18F и т.п.), хлор (36Cl и т.п.), йод (123I, 125I, и т.п.), фосфор (32Р и т.п.) и серу (35S и т.п.).
Меченое изотопом соединение по настоящему изобретению может быть использовано для исследования и т.п. распределения в ткани лекарственных средств и/или субстратов. Например, радиоактивные изотопы, такие как тритий (3Н) и углерод 14 (14С), могут быть использованы для этой цели с точки зрения легкости маркировки и удобства определения.
Замещение более тяжелыми изотопами, например, замещение водорода дейтерием (2Н), является преимущественными в отношении обработки с улучшением метаболической стабильности в некоторых случаях (например, увеличение периода полураспада in vivo, снижение необходимой дозы, снижение взаимодействия между лекарственными средствами).
Замещение изотопами с позитронным изучением (11C, 18F, 15O, 13N и т.п.) может быть использовано в испытании позитронно-эмиссионной томографией (PET) для исследования загрузки субстратного рецептора.
Меченое изотопом соединение по настоящему изобретению обычно может быть получено способами из известного уровня техники, известного специалистам настоящей области техники, или таким же способом получения как в примерах или примерах получения с использованием подходящих реагентов, которые являются изотопно мечеными вместо не меченых изотопом реагентов.
[0022]
(Способ получения)
Соединение формулы (I) и его соль могут быть получены применением различных известных способов синтеза с применением их основной структуры или характеристик на основе типов заместителей. В зависимости от типов функциональных групп для технологии производства в некоторых случаях является эффективным заменить функциональную группу соответствующей защитной группой (группой, которая может быть легко превращена в функциональную группу) заранее на стадии из исходного вещества в промежуточное соединение. Примеры такой защитной группы включают в себя защитную группу и т.п. и описаны в « Protective Groups in Organic Synthesis (4th edition, 2006)» by Wuts (PGM. Wuts) and Greene (T.W. Greene). Защитная группа может быть соответствующим образом выбрана и использована согласно таким реакционным условиям. В таком способе требуемое соединение может быть получено введением защитной группы для проведения реакции, а затем удалением защитной группы при необходимости.
Protective Groups in Organic Synthesis (4th edition, 2006)» by Wuts (PGM. Wuts) and Greene (T.W. Greene). Защитная группа может быть соответствующим образом выбрана и использована согласно таким реакционным условиям. В таком способе требуемое соединение может быть получено введением защитной группы для проведения реакции, а затем удалением защитной группы при необходимости.
Фармацевтически приемлемое пролекарство представляет собой соединение, содержащее группу, которая может быть превращена в аминогруппу, гидроксильную группу, карбоксильную группу или т.п.путем сольволиза или при физиологических условиях. Примеры группы, образующей пролекарство, включают в себя группу, описанную в Prog. Med., 5, 2157-2161 (1985) и «Pharmaceutical Research and Development, Drug Design, Hirokawa Publishing Company» (Hirokawa-Shoten Ltd., 1990), Vol. 7, Molecular Design 163-198.
Кроме того, подобно защитной группе, пролекарство соединения формулы (I) может быть получено путем введения конкретной группы на стадии из исходного вещества в промежуточное соединение или дополнительным проведением реакции с применением полученного соединения формулы (I). Реакция может быть проведена применением способа, известного специалисту настоящей области техники, такого как общая эстерификация, амидирование и дегидратация.
В дальнейшем описан характерный способ получения соединения формулы (I). Каждый способ получения также может быть проведен при ссылке на справочный документ, присоединенные к объяснению. Способ получения по настоящему изобретению не ограничен показанными ниже примерами.
[0023]
Согласно настоящему описанию могут быть использованы следующие аббревиатуры.
DMF = N,N-диметилформамид, DMSO = диметилсульфоксид, EtOAc = этилацетат, МеОН = метанол, MeCN = ацетонитрил, THF = тетрагидрофуран, TEA = триэтиламин, DIPEA = N,N-диизопропилэтиламин, NMM = N-метилморфолин, XPhos = 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил, RuPhos = 2-дициклогексилфосфино-2',6'-диизопропоксибифенил, XantPhos = 4,5-бис(дифенилфосфино)-9,9-диметилксантен, HATU = O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат, солевой раствор = насыщенный водный раствор хлорида натрия, MgSO4 = безводный сульфат магния.
[0024]
В структурных формулах и группах согласно настоящему описанию могут быть использованы следующие аббревиатуры.
Ас = ацетил, ВОС = трет-бутоксикарбонил, t-Bu = трет-бутил, Me = метил, Et = этил, CF3 = трифторметил, Ms = метансульфонил, Ts = пара-толуолсульфонил, Tf = трифторметансульфонил, Ph = фенил.
[0025]
(Способ получения 1)
[Хим. 9]
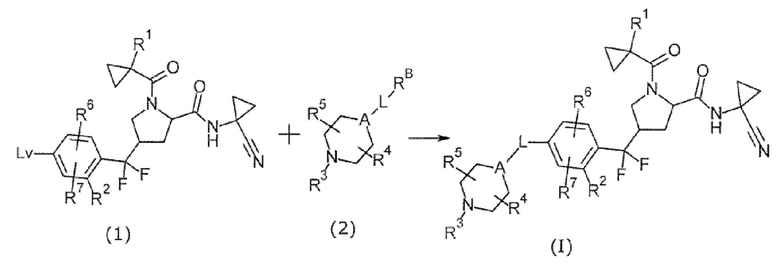
(В формуле RB представляет собой -BF3-Y+, -В(OR)3, и т.п. Lv представляет собой уходящую группу. Y представляет собой щелочной металл, такой как Na или K. R может быть Н или низший алкил или два R могут образовывать низший алкилен вместе.)
Соединение формулы (I) может быть получено реакцией сочетания между соединением (1) и соединением (2). Примеры уходящей группы включают в себя галоген, TfO и т.п.
В этой реакции соединение (1) и соединение (2) использовали в эквивалентных количествах или любое из них в избыточном количестве. Их смесь смешивали в растворителе, инертном в течение реакции, в присутствии основания и палладиевого катализатора при комнатной температуре до нагревания до температуры образования флегмы, согласно одному варианту осуществления от комнатной температуры до 150°С, обычно в течение от 0,1 часа до 5 суток.
Примеры растворителя включают в себя, но без конкретного ограничения, ароматические углеводороды, такие как толуол, эфиры, такие как THF и 1,4-диоксан, галогенированные углеводороды, такие как дихлорметан, спирты, DMF, DMSO, EtOAc, MeCN, Н2О и смеси растворителей. В качестве примера основания предпочтительным является неорганическое основание, такое как CS2CO3, K3PO4, K2CO3, Na2CO3 и KOH. Примеры палладиевого катализатора включают в себя палладиевый катализатор, введенный в систему при помощи ацетата палладия и фосфинового лиганда, такого как XPhos и RuPhos, тетракис(трифенилфосфина)палладия, дихлорбис(трифенилфосфин)палладия, 1,1'-бис(дифенилфосфино)ферроценпалладия (II) дихлорида и т.п.
[Документ]
Edited by A. d. Meijere and F. Diederich, «Metal-Catalyzed Cross-Coupling Reactions», 1st edition, VCH Publishers Inc., 1997
Edited by The Chemical Society of Japan, «5th Edition, Courses in Experimental Science (Vol. 14)», Maruzen, 2005
[0026]
(Способ получения 2)
[Хим. 10]
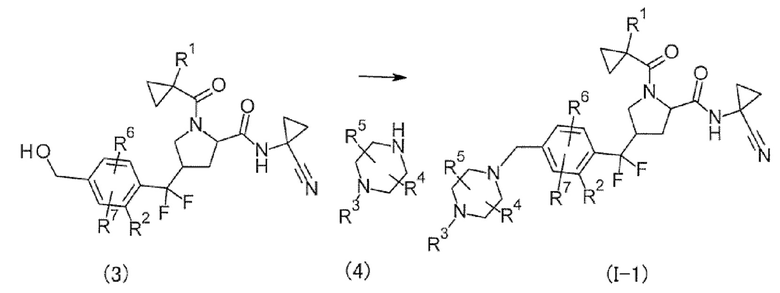
(В формуле соединение формулы (I-1) представляет собой соединение формулы (I), в котором L представляет собой CH2 и А представляет собой N.)
Соединение формулы (I-1) может быть получено введением уходящей группы в соединение (3), а затем путем осуществления взаимодействия с соединением (4).
В этой реакции соединение, полученное осуществлением взаимодействия соединения (3) с галогенированным сульфонильным соединением, таким как MsCl или TsCl, в растворителе, инертном в течение реакции, в присутствии основания, и соединение (4) использовали в эквивалентных количествах или любое из них в избыточном количестве. Их смесь смешивали в растворителе, инертном в течение реакции, в присутствии основания при охлаждении льдом до нагревания с обратным холодильником, предпочтительно при от 0°С до 120°С, обычно в течение от 0,1 часа до 5 суток.
Примеры растворителя включают в себя, но без конкретного ограничения, ароматические углеводороды, такие как толуол, эфиры, такие как 1,4-диоксан, галогенированные углеводороды, такие как дихлорметан, DMF, DMSO, EtOAc, MeCN, и смеси растворителей. Примеры основания включают в себя органическое основание, такое как TEA, DIPEA и NMM, неорганическое основание, такое как K2CO3, Na2CO3 и KOH и т.п.
[0027]
(Способ получения 3)
[Хим. 11]
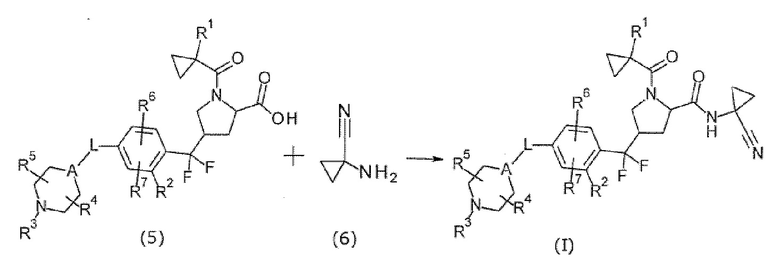
Соединение формулы (I) может быть получено реакцией амидирования между соединением (5) и соединением (6). В этой реакции соединение (5) и соединение (6) использовали в эквивалентных количествах или любое из них в избыточном количестве. Их смесь смешивали в растворителе, инертном в течение реакции, в присутствии конденсатора при охлаждении до нагревания, предпочтительно при от -20°С до 60°С, обычно в течение от 0,1 часа до 5 суток. Примеры растворителя включают в себя, но без конкретного ограничения, ароматические углеводороды, такие как толуол, эфиры, такие как THF и 1,4-диоксан, галогенированные углеводороды, такие как дихлорметан, спирты, DMF, DMSO, EtOAc, MeCN, и смеси растворителей. Примеры конденсатора включают в себя HATU, 1-(3-диметиламинопропил)-3-этилкарбодиимид или его гидрохлорид, дициклогексилкарбодиимид, 1,1'-карбонилдиимидазол, дифенилфосфорил азид и т.п. В некоторых случаях является предпочтительным использование добавки (например, 1-гидроксибензотриазола) для реакции. В некоторых случаях является предпочтительным проведение реакции в присутствии органического основания, такого как TEA, DIPEA и NMM, или неорганического основания, такого как K2CO3, Na2CO3 и KOH и т.п., чтобы позволить реакции проходить плавно.
Более того, может быть использован способ, при котором соединение (5) превращали в реакционноспособное производное, а затем проводили реакцию с соединением (6). Примеры реакционноспособного производного карбоновой кислоты включают в себя галогенангидрид, полученный путем осуществления взаимодействия с галогенирующим средством, таким как оксихлорид фосфора и тионилхлорид, смешанный кислотный ангидрид, полученный путем осуществления взаимодействия с изобутилхлорформиатом или т.п., активный сложный эфир, полученный путем конденсации с 1-гидроксибензотриазолом или т.п., и т.п. Реакцию между такими реакционноспособными производными и соединением (6) проводили в растворителе, инертном в течение реакции, таком как галогенированные углеводороды, ароматические углеводороды, эфиры или т.п. при условиях от охлаждения до нагревания, предпочтительно при от -20°С до 60°С.
[Документ]
S.R. Sandler and W. Karo, «Organic Functional Group Preparations», 2nd Edition, Vol. 1, Academic Press, Inc., 1991
Edited by The Chemical Society of Japan, «Courses in Experimental Chemistry (5th Edition)», Vol. 16 (2005) (Maruzen)
[0028]
(Синтез исходного вещества 1)
[Хим. 12]
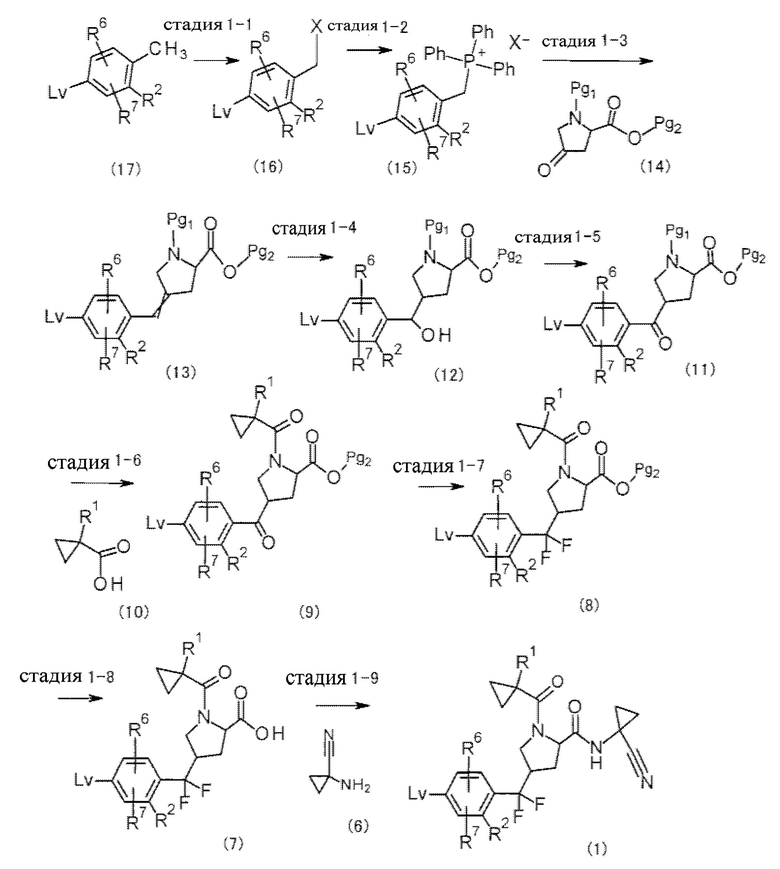
(В формуле Lv представляет собой уходящую группу, Pg1 и Pg2 представляют собой защитные группы и X представляет собой галоген. Перекрестные двойные связи в соединении (13) представляют собой смесь геометрических изомеров.)
Примеры Pg1 включают в себя группу ВОС и т.п., и примеры Pg2 включают в себя группу трет-Bu, группу Et, группу Me и т.п.
Стадии, представленные стадией 1-1 - стадией 1-3, представляют собой реакцию получения соединения (15), используемого в качестве реагента Виттига (фосфорный илид), из соединения (17), и реакцию получения соединения (13) реакцией Виттига соединения (15) и соединения (14), соответственно. На стадии 1-1 и стадии 1-2 соединение (17) оставляли взаимодействовать с галогенирующим средством, таким как N-бромсукцинимид или бром, с образованием галогенида (16), а затем смесь с трифенилфосфином перемешивали в растворителе, инертном в течение реакции, при от охлаждения до нагревания с обратным холодильником, согласно одному варианту осуществления при от 0°С до 120°С, обычно в течение от приблизительно 0,1 часа до 5 суток. В случае дигалогенирования в течение реакции галогенирования моногалогенид (16) может быть получен путем осуществления взаимодействия с диэтилфосфонатом. На стадии 1-3 смесь соединения (14) и соединения (15) перемешивали в растворителе, инертном в течение реакции, в присутствии основания при от охлаждения до нагревания с обратным холодильником, предпочтительно при от 0°С до 100°С, обычно в течение от приблизительно 0,1 часа до 10 суток. Примеры растворителя включают в себя ароматические углеводороды, эфиры, галогенированные углеводороды, такие как дихлорметан, спирты, DMF, DMSO, EtOAc, MeCN и их смесь. Примеры основания включают в себя органические основания, такие как метилат натрия, трет-бутилат калия, н-бутиллитий, гексаметилдисилазид лития, неорганическое основание, такое как K2CO3, Na2CO3 и KОН и т.п.
[Документ]
Wittig, G et al., U. Ber., 1954, Vol. 87, p. 1318
Стадия, представленная стадией 1-4, представляет собой реакцию получения соединения (12) реакцией окисления, которая возникает после гидроборирования алкена соединения (13). В этой реакции, реагент, который получали смешиванием смеси соединения (13) и комплекса боран-THF, 9-борабицикло[3.3.1]нонана, дисиамилборана, тексилборана или т.п., в растворителе, инертном в течение реакции, предпочтительно при от 10°С до 80°С обычно в течение от 0,1 часа до 3 суток, обрабатывали эквивалентным количеством или избыточным количеством окислителя в растворителе, инертном в течение реакции, в присутствии основания при от охлаждения до нагревания с обратным холодильником, предпочтительно при от -20°С до 80°С, обычно в течение от 0,1 часа до 3 суток, и тем самым может быть получено соединение (12).
Примеры растворителя включают в себя ароматические углеводороды, эфиры, такие как THF, галогенированные углеводороды, DMF, DMSO, EtOAc, MeCN, и их смесь. Примеры основания включают в себя NaOH, K2CO3, Na2CO3 и KOH и т.п. Примеры окислителя включают в себя пероксид водорода, гидропероксид кумена, перуксусную кислоту, пербензойную кислоту, мета-хлорпербензойную кислоту, оксон, активированный диоксид марганца, хромовую кислоту, перманганат калия, перйодат натрия и т.п.
[Документ]
J. Am. Chem., Soc, 1956, Vol. 78, p. 5694-5695
J. Org. Chem., 1986, Vol. 51, p. 439-445
Стадия, представленная стадией 1-5, представляет собой реакцию получения соединения (11) реакцией окисления соединения (12). В этой реакции соединение (12) обрабатывали эквивалентным количеством или избыточным количеством окислителя в растворителе, инертном в течение реакции, при от охлаждения до нагревания с обратным холодильником, предпочтительно при от -20°С до 80°С, обычно в течение от 0,1 часа до 3 суток. В этой реакции подходящим образом использовали окисление DMSO, такое как окисление при помощи тетрапропиламмония перрутената (ТРАР), или окисление Сверна, или окисление с применением реагента Десса-Мартина. При окислении ТРАР соединение (12) обрабатывали в присутствии тетрапропиламмония перрутената, который являлся катализатором окисления, молекулярного сита 4А, что являлось дегидратирующим средством, и N-метилморфолин-М-оксида (NMMO), который являлся реокислителем. Примеры растворителя включают в себя ароматические углеводороды, эфиры, галогенированные углеводороды, такие как дихлорметан, DMF, DMSO, EtOAc, MeCN, и их смесь. Примеры других окислителей включают в себя пероксид водорода, гидропероксид кумена, перуксусную кислоту, пербензойную кислоту, мета-хлорпербензойную кислоту, оксон, активированный диоксид марганца, хромовую кислоту, перманганат калия, перйодат натрия и т.п.
[Документ]
J. Chem. Soc, Chem. Commun., 1987, p. 1625-1627
Edited by The Chemical Society of Japan, «5th Edition, Courses in Experimental Science (Vol. 14)», Maruzen, 2005
Стадия, представленная стадией 1-6, представляет собой реакцию снятия защитной группы Pg1 соединения (11), а затем конденсацию с соединением (10), и тем самым получение соединения (9). Стадия может быть проведена таким же способом, что в способе получения 3 после проведения реакции снятия защитных групп при ссылке на способ, описанный в « Protective Groups in Organic Synthesis)), 4th edition, 2006, и т.п.
Стадия, представленная стадией 1-7, представляет собой реакцию получения соединения (8) фторированием соединения (9). В этой реакции соединение (9) перемешивали в растворителе, инертном в течение реакции, в присутствии фторирующего средства при от охлаждения до нагревания, предпочтительно при от -20°С до 120°С, обычно в течение от 0,1 часа до 10 суток. Примеры растворителя включают в себя ароматические углеводороды, эфиры, галогенированные углеводороды, такие как дихлорметан, DMF, DMSO, EtOAc, MeCN и их смесь. Примеры фторирующего средства включают в себя 4-трет-бутил-2,6-диметилфенилсеры трифторид, фторид водорода, диэтиламиносеры трифторид (DAST), тетрафторид серы (SF4), бис(2-метоксиэтил)аминосеры трифторид и т.п.
[Документ]
J. Am. Chem. Soc, 2010, Vol. 132, p. 18199-18205
Стадия, представленная стадией 1-8, представляет собой реакцию получения соединения (7) снятием защитных групп с соединения (8). Эта реакция может быть упомянута, например, в способе, описанном в « Protective Groups in Organic Synthesis)), 4th edition, 2006, и т.п.
Стадия, представленная стадией 1-9, представляет собой реакцию получения соединения (1) конденсацией соединения (7) и соединения (6). Стадия может быть проведена таким же способом, что и способ получения 3.
[0029]
(Синтез исходного вещества 2)
[Хим. 13]
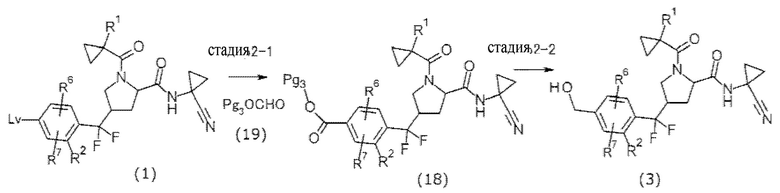
(В формуле Pg3 представляет собой защитную группу.)
Примеры Pg3 включают в себя 2,4,6-трихлорфенил.
Стадия, представленная стадией 2-1, представляет собой реакцию получения соединения (18) путем осуществления взаимодействия соединения (1) и соединения (19). Стадия может быть проведена таким же способом, что и способ получения 1, за исключением того, что соединение (19) использовали вместо соединения (2).
В качестве растворителя предпочтительным является толуол или бензотрифторид. В качестве основания предпочтительным является TEA или трибутиламин. В качестве палладиевого катализатора предпочтительным является палладиевый катализатор, доведенный in situ ацетатом палладия и фосфиновым лигандом, таким как XantPhos.
[Документ]
Organic Letters, 2012, Vol. 14, No. 20, pp. 5370-5373.
Стадия, представленная стадией 2-2, представляет собой реакцию получения соединения (3) реакцией восстановления соединения (18). В этой реакции соединение (18) обрабатывали эквивалентным количеством или избыточным количеством восстановителя в растворителе, инертном в течение реакции, при от охлаждения до нагревания, предпочтительно при от -20°С до 80°С, обычно в течение от 0,1 часа до 3 часов. Примеры растворителя включают в себя ароматические углеводороды, эфиры, галогенированные углеводороды, спирты, такие как МеОН, DMF, DMSO, EtOAc, MeCN и их смеси. Примеры восстановителя включают в себя гидридные восстановители, такие как боргидрид натрия и боргидрид лития.
[Документ]
М. Hudlicky, «Reductions in Organic Chemistry, 2nd Edition (ACS Monograph: 188)», ACS, 1996
R.C. Larock, «Comprehensive Organic Transformations)), 2nd Edition, VCH Publishers, Inc., 1999
T.J. Donohoe «Oxidation and Reduction in Organic Synthesis (Oxford Chemistry Primers 6)», Oxford Science Publications, 2000
Edited by The Chemical Society of Japan, «Courses in Experimental Chemistry (5th Edition))), Vol. 14 (2005) (Maruzen)
[0030]
(Синтез исходного вещества 3)
[Хим. 14]
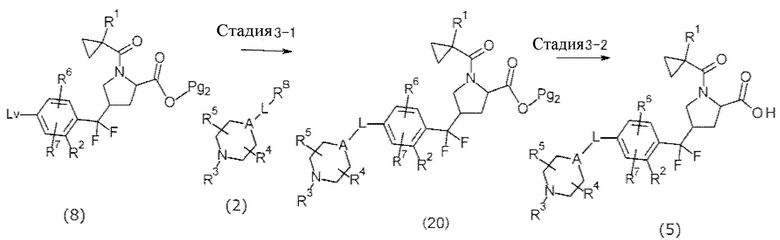
Стадия, показанная на стадии 3-1, представляет собой реакцию получения соединения (20) реакцией сочетания соединения (8) и соединения (2), и может быть проведена таким же способом, что и в способе получения 1.
Стадия, представленная стадией 3-2, представляет собой реакцию получения соединения (5) снятием защитных групп с соединения (20), и может быть проведена таким же способом, что и стадия 1-8 в синтезе исходного вещества 1.
[0031]
Соединение формулы (I) выделяли в виде соединения в свободной форме, его соли, гидрата, сольвата или вещества, обладающего кристаллическим полиморфизмом, и очищали. Соль соединения формулы (I) также может быть получена подверганием соединения реакции образования соли общего способа.
Выделение и очистку проводили с применением обычных химических операций, таких как экстракция, фракционная кристаллизация и различная фракционная хроматография.
Различные изомеры могут быть получены выбором соответствующего исходного соединения или могут быть разделены с применением различия в физико-химических свойствах между изомерами. Например, оптический изомер может быть получен общим способом оптического расщепления рацемической формы (например, фракционной кристаллизацией, приводящей к диастереомерной соли с оптически активным основанием или кислотой, хроматографией с применением хиральной колонки или т.п., и т.п.) или может быть получен из соответствующего оптически активного исходного соединения.
[0032]
Фармакологическая активность соединения формулы (I) может быть подтверждена следующими исследованиями или хорошо известными улучшенными исследованиями.
[0033]
Пример 1 исследования: Измерение in vitro ингибирующей человеческий катепсин S активности
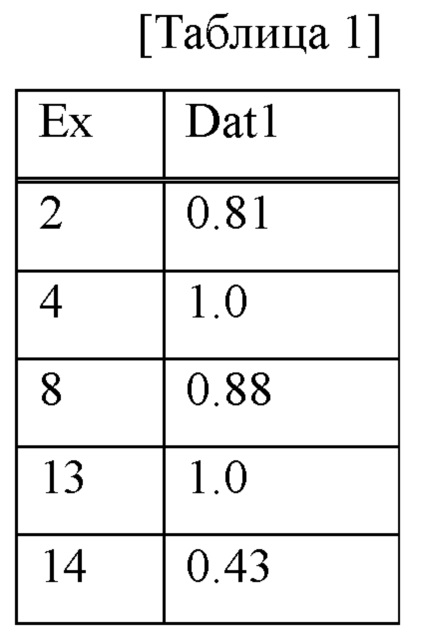
В 96-луночный планшет добавляли 5 мкл фермента человеческого катепсина S (R&D 1183-CY-010) до 20 нг/лунку. Затем с помощью буфера для анализа (ацетат натрия в концентрации 50 мМ, хлорид натрия в концентрации 250 мМ и дитиотреитол в концентрации 5 мМ (DTT), рН=4,5) исследуемое соединение (раствор ДМСО в концентрации 10 мМ) разбавляли посредством серий 10-кратных разведений с 5 стадиями, чтобы конечная концентрация составляла от 0,1 нМ до 1 мкМ, или посредством серий 3-кратных разведений с 7 стадиями, чтобы конечная концентрация составляла от 0,01 нМ до 10 нМ, и 10 мкл добавляли в лунку (конечная концентрация ДМСО составляла 0,1%) с последующим добавлением 25 мкл синтетического субстрата VVR-AMC (Peptide Institute 3211-V), так что конечная концентрация составляет 40 мкМ, и, таким образом, инициировали ферментативную реакцию. Интенсивность флуоресценции (длина волны возбуждения: 380 нм, длина волны флуоресценции: 460 нм) измеряли при температуре 37°С в течение 5-10 минут от начала реакции с использованием спектрофлуорофотометра (SPECTRAMAX GEMINI, Molecular Devices), и скорости реакции при линейности (5 минут) получали для каждой концентрации исследуемого соединения. Степень ингибирования при каждой концентрации определяли путем подавления скорости реакции во время отсутствия добавления фермента без добавления исследуемого соединения и скорости реакции во время добавления фермента без добавления исследуемого соединения посредством 100% ингибирования и 0% ингибирования, соответственно, и, следовательно, значение IC50 рассчитывали с помощью способа нелинейной регрессии сигмовидной Emax. Результаты показаны в таблице 1. В таблице Ех представляет собой № соединения примера, a Dat1 представляет собой значение IC50 (нМ) ингибирующей активности человеческого катепсина S.
[0034]
[0035]
Пример 2 исследования: Оценка ингибирующего действия на экспрессию МНС класса II in vitro с использованием мышиных спленоцитов (система оценки клеток)
В антигенпрезентирующих клетках ингибирование катепсина S подавляет экспрессию молекул МНС класса II. В результате подавление презентации антигена CD4-позитивным Т-клеткам вызывает ухудшение иммунного ответа на чужеродные антигены. Что касается увеличения экспрессии МНС класса II в В-клетках, было изучено ингибирующее действие соединения формулы (I).
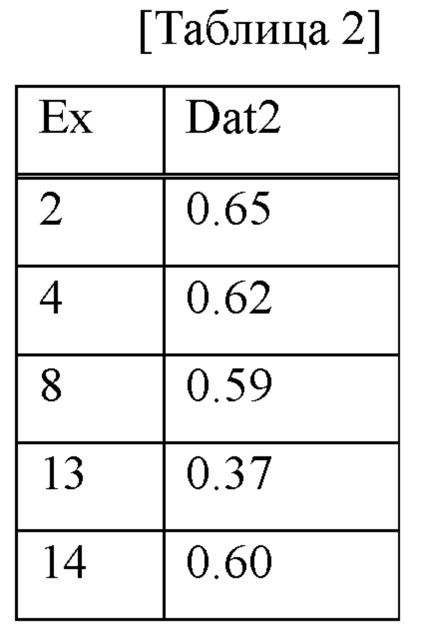
Спленоциты, собранные от самцов мышей C57BL/6J (Charles River Laboratories Japan, Inc.), высевали в 96-луночный планшет по 1×105 клеток/лунку. Средой RPMI 1640 (содержащей 10% эмбриональной бычьей сыворотки (FCS), 5×10-5 М 2-меркаптоэтанола, 50 МЕ/мл пенициллина и 50 мкг/мл стрептомицина), раствор с ДМСО в концентрации 10 мМ исследуемого соединения разбавляли посредством серий 5-кратных разведений с 9 стадиями, чтобы конечная концентрация составляла от 0,026 нМ до 10 мкМ, или разбавляли посредством серий 5-кратных разведений с 12 стадиями, чтобы конечная концентрация составляла от 0,205 пМ до 10 мкМ (конечная концентрация ДМСО составляет 0,1%), и добавляли. В то же время в лунку добавляли LPS (Sigma L4005), чтобы конечная концентрация составляла 2 мкг/мл, и клетки культивировали при температуре 37°С при 5% СО2 в течение 48 часов. После культивирования клетки окрашивали меченным биотином антителом YAe (EBIOSCIENCE 13-5741-85) при температуре 4°С в течение 20 минут и промывали. Затем клетки дополнительно окрашивали меченым флуорохромом FITC антителом к В220 мыши (BD BIOSCIENCES 553088) и меченым флуорохромом РЕ стрептавидином (BD BIOSCIENCES 554061) в течение 20 минут при температуре 4°С. Поэтому уровень экспрессии (интенсивность флуоресценции YAe-биотин/стрептавидин-РЕ) МНС класса II, связанного с пептидом Еа, в В220-положительных В-клетках измеряли с использованием системы проточной цитометрии (Guava EasyCyte Plus System, Millipore). Степень ингибирования при каждой концентрации определяли путем подавления значения во время отсутствия стимуляции LPS без добавления исследуемого соединения и значения во время стимуляции LPS без добавления исследуемого соединения посредством 100% ингибирования и 0% ингибирования, соответственно, и, следовательно, значение IC50 рассчитывали с помощью способа нелинейной регрессии сигмоидной Emax. Результаты показаны в таблице 2. В таблице Ех представляет собой № соединения примера, a Dat2 представляет собой значение IC50 (нМ).
[0036]
[0037]
Пример 3 исследования: Оценка эффекта ингибирования экспрессии МНС класса II ex vivo с использованием периферической крови мыши
Эффект ингибирования экспрессии молекул МНС класса II оценивали в системе ех vivo.
Исследуемое соединение вводили перорально самцам мышей C57BL/6J (Charles River Laboratories Japan, Inc.) и исследовали действие ингибирования в отношении увеличения экспрессии МНС класса II в В-клетках периферической крови после перорального введения. То есть 10 мл/кг исследуемого соединения, растворенного в наполнителе [30% пропиленгликолевый растворитель {пропиленгликоль: гидрогенизированное касторовое масло (НСО40): Твин 80=4:2:1}/HCl (2 эквивалента по отношению к исследуемому соединению)/вода], вводили перорально самцам мышей C57BL/6J (доза: 0,3 мг/кг, 4 субъекта на группу), и периферическую кровь получали через 6 часов. К 90 мкл периферической крови добавляли 10 мкл PBS или 10 мкл (конечная концентрация 100 мкг/мл) LPS (Sigma L4005), и культивирование проводили при температуре 37°С при 5% СО2 в течение 15 часов. После культивирования клетки окрашивали меченым флуорохромом FITC антителом к IA/IE мыши (BD BIOSCIENCES 553623) и меченым флуорохромом РЕ антителом к В220 мыши (BD BIOSCIENCES 553090) в течение 30 минут при охлаждении, а затем проводили гемолиз и фиксацию в течение 11-12 минут при температуре 37°С с использованием буфера (BD BIOSCIENCES Phosflow Lyse/Fix Buffer 558049). После промывания с использованием системы проточной цитометрии (FACSCanto II, BD BIOSCIENCES) уровень экспрессии МНС класса II на поверхности В220-позитивных В-клеток измеряли со средней интенсивностью флуоресценции FITC (в дальнейшем именуемой MFI) в качестве показателя. Разницу между MFI стимуляции LPS и MFI без стимуляции определяли как ΔMFI и, следовательно, степень ингибирования ΔMFI в соответствии с введением исследуемого соединения рассчитывали, устанавливая ΔMFI мышей, которым вводили 10 мл/кг наполнителя, как 1.
[0038]
В этом исследовании соединение примера 8 показало 41% ингибирования (0,3 мг/кг), что ингибировало увеличение экспрессии МНС класса II.
[0039]
Пример 4 исследования: Оценка ингибирующего действия на возникновение SLE-подобного заболевания в модели самопроизвольного возникновение с использованием мышей F1 NZB/W
Мышей NZB/W F1 (Japan SLC, Inc.) использовали в качестве модели SLE, самопроизвольно развивающегося заболевания, близкого к человеку "JAMA", 1966, Vol. 195, p. 145; "Advances in Immunology", 1985, Vol. 37, p. 269-390; "Journal of Biomedicine and Biotechnology", 2011, Vol. 2011, 271694).
Самок мышей NZB/W F1 делили на группы в соответствии со значениями белка в моче (значение, скорректированное на креатинин), значением антител к двухцепочечной ДНК (дцДНК) (IgG) в плазме, уровнем экспрессии МНС класса II в В-клетках периферической крови и массе тела в возрасте 19 недель. В это время были исключены особи со значениями белка в моче (скорректированное значение креатинина) 3 или более. В возрасте 20 недель исследуемое соединение вводили перорально два раза в день, а затем с течением времени проводили сбор мочи и сбор крови. Изменения в значении белка в моче и значении антитела к дцДНК оценивали с течением времени.
В вышеуказанном исследовании значение антитела IgG к дцДНК в плазме, производимого в связи с SLE-подобным заболеванием, измеряли способом ИФА (ALPHA DIAGNOSTIC 5120). В средних геометрических значениях значение антитела в возрасте 28 недель и в возрасте 32 недель для каждой особи, среднее геометрическое значение для 10 субъектов, которым вводили наполнитель, составляло 216662 Ед/мл, тогда как среднее геометрическое значение для 10 субъектов, которым вводили 1 мг/кг два раза в день соединения примера 2 составляло 26827 Ед/мл, что представляет собой низкое значение, характеризующееся значительной разницей (значение Р=0,0009, множественное сравнение Даннетта). Кроме того, измеряли значения белка в моче (скорректированное на креатинин значение) в возрасте 40 недель. Среднее геометрическое значение для 10 субъектов, которым вводили наполнитель, составляло 13,0, тогда как среднее геометрическое значение для 10 субъектов, которым вводили 1 мг/кг два раза в день соединения примера 2 составляло 0,7, что представляет собой низкое значение, характеризующееся значительной разницей (значение Р=0,0005, множественное сравнение Даннетта). На основании этого было подтверждено, что соединение примера 2 обладает действием подавления возникновения SLE-подобного заболевания у самок мышей NZB/W F1.
[0040]
Пример 5 исследования: Оценка терапевтического действия на SLE-подобное заболевание в модели индуцированного poly (I:С) возникновения с использованием мышей NZB/WF1
poly (I:С), который является лигандом для Toll-подобного рецептора 3, вводят мышам NZB/W F1 (Japan SLC, Inc.), и, следовательно, можно ускорить увеличение протеинурии, связанной с SLE-подобным заболеванием. Введение исследуемого соединения начинается с состояния, когда протеинурия индуцируется введением poly (I:С), и затем оценивают терапевтический эффект на SLE-подобное заболевание.
200 мкг poly (I:C) (InvivoGen tlrl-picw-250) вводят мышам NZB/W F1 в возрасте 22 недель три раза в неделю в течение 4 недель, что в сумме составляет 12 раз. В последующие 2 недели особей, у которых значение белка в моче (скорректированное на креатинин значение) стало по существу от 2 до 50, включают в исследование и распределяют на основе значения белка в моче. После группировки исследуемое соединение вводят перорально два раза в день в течение 5 недель, мочу собирают с течением времени, и, следовательно, оценивают изменение значения белка в моче с течением времени.
[0041]
На основании приведенных выше результатов ожидается, что соединение формулы (I) или его соль будут применять в качестве средства для профилактики и/или лечения аутоиммунного заболевания, включая в себя SLE и волчаночный нефрит, аллергии или отторжение трансплантата органа, кости костного мозга или ткани.
[0042]
Фармацевтическая композиция, содержащая одно или несколько соединений формулы (I) или их соли в качестве активного ингредиента, может быть получена с использованием вспомогательного вещества, как правило, используемого в настоящей области, т.е. фармацевтического средства, фармацевтического носителя и т.п., в соответствии с обычно используемыми способами.
Введение может быть в любой форме перорального введения с таблетками, пилюлями, капсулами, гранулами, порошками, растворами и т.п., такими инъекциями, как внутрисуставные, внутривенные и внутримышечные инъекции, и парентеральным введением через суппозиторий, глазные капли, глазную мазь, трансдермальный раствор, мазь, трансдермальный пластырь, трансмукозальный раствор, трансмукозальный пластырь, ингалятор и т.п.
[0043]
В качестве твердой композиции для перорального применения применяют таблетки, порошки, гранулы и т.п. В таких твердых композициях один или несколько активных ингредиентов смешивают по меньшей мере с одним инертным вспомогательным веществом. Композиция может содержать неактивные добавки, такие как смазывающие вещества и разрыхлители, стабилизаторы и солюбилизирующие средства в соответствии с общими способами. Таблетки или пилюли могут быть покрыты сахарной оболочкой или пленкой растворимого в желудке или растворимого в кишечнике вещества, если необходимо.
Жидкие композиции для перорального введения включают в себя фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры и т.п.и включают в себя, как правило, используемые инертные разбавители, такие как очищенная вода или этанол. Жидкая композиция может содержать солюбилизирующее средство, смачивающее средство, такой адъювант, как суспендирующее средство, подсластитель, вкусоароматическую добавку, ароматизатор и консервант в дополнение к инертному разбавителю.
[0044]
Инъекции для парентерального введения содержат стерильные водные или неводные растворы, суспензии или эмульсии. Примеры водного растворителя включают в себя дистиллированную воду для инъекций или физиологический раствор. Примеры неводных растворителей включают в себя такие спирты, как этанол. Такая композиция может дополнительно содержать тонизирующее средство, консервант, смачивающее средство, эмульгирующее средство, диспергирующее средство, стабилизирующее средство или солюбилизирующее средство. Их стерилизуют, например, путем фильтрации через задерживающий бактерии фильтр и смешивания стерилизующего средства или облучения. Они также могут быть использованы посредством приготовления стерильной твердой композиции и растворения или суспендирования композиции в стерильной воде или стерильном инъецируемом растворителе перед применением.
[0045]
Примеры наружных препаратов включают в себя мази, пластыри, кремы, желе, горячие компрессы, спреи, лосьоны, глазные капли, глазные мази и т.п.Включена обычно используемая мазевая основа, лосьонная основа, водные или неводные растворы, суспензии, эмульсии и т.п.
[0046]
Трансмукозальные средства, такие как ингаляторы и трансназальные препараты, являются твердыми, жидкими или полутвердыми и могут быть изготовлены в соответствии с известными способами предшествующего уровня техники. Например, могут быть надлежащим образом добавлены хорошо известные вспомогательные вещества и, кроме того, регуляторы рН, консерванты, поверхностно-активные вещества, смазывающие вещества, стабилизаторы, загустители и т.п. Для введения можно использовать устройство для правильной ингаляции или инсуффляции. Например, с использованием известного устройства, такого как дозирующее ингаляторное устройство или небулайзер, соединение можно вводить отдельно или в виде порошка составленной смеси, или в виде раствора или суспензии в комбинации с фармацевтически приемлемым носителем. Ингалятор сухого порошка и т.п.может представлять собой устройство для однократного или многократного введения, и может применяться сухой порошок или порошкообразная капсула. Альтернативно, форма введения может представлять собой подходящее средство выброса, такое как аэрозольный баллон под давлением, с использованием подходящего газа, такого как хлорфторалкан или диоксид углерода.
[0047]
В случае перорального введения ежедневная доза составляет приблизительно от 0,001 до 100 мг/кг, предпочтительно от 0,1 до 30 мг/кг и более предпочтительно от 0,1 до 10 мг/кг массы тела, что является подходящим, и эту дозу вводят один раз или делят на 2-4 дозы. В случае внутривенного введения подходящая суточная доза составляет приблизительно от 0,0001 до 10 мг/кг массы тела, и эту дозу вводят от одной до нескольких доз в день. Что касается трансмукозального средства, приблизительно от 0,001 до 100 мг/кг массы тела вводят от одной до нескольких доз в день. Дозу определяют в соответствии с индивидуальными случаями с учетом симптомов, возраста, пола и т.п.
[0048]
Фармацевтическая композиция по настоящему изобретению содержит от 0,01% до 100 мас. % и от 0,01 до 50 мас. % согласно одному варианту осуществления одного или нескольких соединений формулы (I) или их соли, которая является активным ингредиентом, хотя ее масса может варьировать в зависимости от пути введения, лекарственной формы, места введения, типов вспомогательных веществ и добавок.
[0049]
Соединение формулы (I) можно применять в комбинации с различными средствами для лечения или средствами для профилактики заболеваний, при которых соединение формулы (I), как считается, проявляет эффективность. Комбинацию можно вводить одновременно или раздельно по очереди или через желаемый интервал времени. Средство для одновременного введения может представлять собой смешивающее средство или может быть составлено отдельно.
[Примеры]
[0050]
Далее способ получения соединения формулы (I) будет объяснен более подробно на основе примеров. Настоящее изобретение не ограничено соединениями, описанными в следующих примерах. Кроме того, каждый способ получения исходного соединения показан в примерах получения. Более того, способ получения соединения формулы (I) не ограничен только способами получения конкретных примеров, показанных ниже. Соединение формулы (I) может быть получено комбинацией таких способов получения или способами, которые очевидны специалистам настоящей области техники.
[0051]
Начальная температура кривой DSC, полученная измерением при следующих условиях, показана в следующих таблицах как точка плавления.
DSC измерение проводили с применением алюминиевой кюветы для образцов в состоянии не покрытия кюветы для образцов в условиях измерения диапазона температуры: от комнатной температуры до 300°С, увеличение скорости температуры: 10°С/мин и скорость потока азота: 50 мл/мин с применением DSC Q2000 (измерено ТА Instruments.).
[0052]
Порошковую рентгеновскую дифракцию проводили с применением RINT-TTR II (измерено RIGAKU Corporation) при условиях лампы: Си, ток лампы: 300 мА, напряжение на лампе: 50 кВ, ширина образца: 0,020°, скорость сканирования: 4°/мин, длина волны: 1,5418 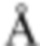 , измеренный диапазон дифракционного угла: (2θ): 2,5-40°.
, измеренный диапазон дифракционного угла: (2θ): 2,5-40°.
Что касается образца порошковой рентгеновской дифракции, поскольку природа его данных, пространство кристаллической решетки и все образцы являются важными в идентификации кристаллической идентичности, интервал погрешности дифракционного угла (2θ(°)) при порошковой рентгеновской дифракции обычно составляет ±0,2°, но дифракционный угол и дифракционная интенсивность могут меняться в зависимости от направления роста кристалла, размера зерен и условий измерения, и таким образом образцы не должны быть точно интерпретированы.
[0053]
Следующие аббревиатуры иногда использовали в примерах, примерах получения и таблицах, описанных позже.
Прим. получ. = № примера получения., прим. = № примера, син. = способ получения (обозначающий, что получение проводили тем же способом, что и в описанных №примера или №примера получения), стр. = структурная формула, данн. = физико-химические данные величина, ESI+ = m/z значение ESI-MS (представляющее [М+Н]+, если конкретно не указано иное), ESI- = m/z значение ESI-MS (представляющее [М-Н]-, если конкретно не указано иное), ЯМР1: δ (ppm) пика в 1Н ЯМР в DMSO-d6 при комнатной температуре, ЯМР2: δ (ppm) пика в 1Н ЯМР в DMSO-d6 при 80°С, ЯМР3: δ (ppm) пика в 1Н ЯМР в DMSO-d6 при 60°С, [α]D23,5: D линия, удельное вращение при 23,5°С, т.пл.: точка плавления, 2θ: дифракционный угол пика при порошковой рентгеновской дифракции.
HCl в структурной формуле представляет собой гидрохлорид, а число перед HCl представляет собой молярное отношение. Например, 2HCl означает дигидрохлорид. Подобным образом, SA представляет собой сукцинат и 2SA означает дисукцинат (включая сокристалл, содержащий соединение и янтарную кислоту в молярном соотношении 1:2).
Символ gm, отмеченный в № примера получения и № примера, представляет собой смесь геометрических изомеров. Подобным образом, символ em представляет собой смесь эпимеров при 4-положении пирролидинового кольца или смесь эпимеров при 3-положении пирролидинового кольца, а символ dm представляет собой смесь диастереомеров с разными стерическими конфигурациями при α-положении бензильной группы и 4-положении пирролидинового кольца.
[0054]
В целях удобства концентрация моль/л представлена как М. Например, 1 М водный раствор гидроксида натрия означает 1 моль/л водного раствора гидроксида натрия.
[0055]
Пример получения 1
N-бромсукцинимид (148,9 г) и 47% бромистоводородную кислоту (5 мл) добавляли к MeCN (700 мл) раствору 4-бром-1-метил-2-(трифторметил)бензола (100 г) при комнатной температуре в атмосфере газообразного аргона. К смеси добавляли 2,2'-азобис(изобутиронитрил) (3,43 г), перемешивали при 70°С в течение 10 минут, а затем перемешивали при 100°С всю ночь. Смесь охлаждали до комнатной температуры. Смесь охлаждали на льду, добавляли насыщенный водный раствор тиосульфата натрия и смесь перемешивали в течение 10 минут. Воду и EtOAc добавляли к смеси. Органический слой отделяли и промывали насыщенным водным раствором бикарбоната натрия и солевым раствором. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. К остатку добавляли н-гексан и осажденное твердое вещество отделяли фильтрацией. Фильтрат концентрировали при пониженном давлении. DIPEA (79 мл) и диэтилфосфонат (54 мл) добавляли к раствору THF (500 мл) остатка при охлаждении льдом. Смесь перемешивали при комнатной температуре в течение 2 часов. Смесь охлаждали на льду и к ней добавляли воду. К смеси добавляли EtOAc и органический слой отделяли и промывали хлористоводородной кислотой (1 М), водой, насыщенным водным раствором бикарбоната натрия и солевым раствором. Органический слой сушили над MgSO4 и концентрировали при пониженном давлении. Трифенилфосфин (115 г) добавляли к толуольному (700 мл) раствору остатка и смесь перемешивали всю ночь при 100°С. Осадок собирали фильтрацией и промывали толуолом с получением [4-бром-2-(трифторметил)бензил](трифенил)фосфоний бромида (198,51 г) в виде твердого вещества.
[0056]
Пример получения 2
Трет-бутилат калия (27,5 г) добавляли к дихлорметановому (600 мл) раствору [4-бром-2-(трифторметил)бензил](трифенил)фосфоний бромида (150 г) в атмосфере газообразного аргона при охлаждении льдом и смесь перемешивали при комнатной температуре в течение 6 часов. Смесь охлаждали на льду и к ней добавляли дихлорметановый (150 мл) раствор 1-трет-бутилового сложного эфира 2-метилового сложного эфира (2S)-4-оксопирролидин-1,2-дикарбоновой кислоты (50 г), и смесь перемешивали при комнатной температуре в течение 4 суток. Насыщенный водный раствор хлорида аммония добавляли к смеси и перемешивали в течение 15 минут.
Органический слой отделяли и водный слой экстрагировали хлороформом. Объединенные органические слои промывали солевым раствором. Органический слой сушили над MgSO4 и концентрировали при пониженном давлении. EtOAc (40 мл) и н-гексан (200 мл) добавляли к остатку и смесь перемешивали при комнатной температуре в течение 1 часа. Осадок отделяли фильтрацией и фильтрат концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на силикагеле (элюент; н-гексан/EtOAc) с получением 1-трет-бутилового сложного эфира 2-метилового сложного эфира (2S)-4-[4-бром-2-(трифторметил)бензилиден]пирролидин-1,2-дикарбоновой кислоты (смесь геометрических изомеров) (51,74 г) в виде масляного вещества.
[0057]
Пример получения 3
Комплекс борана-THF (1 М THF раствор, 167 мл) добавляли к THF (100 мл) раствору 1-трет-бутилового сложного эфира 2-метилового сложного эфира (2S)-4-[4-бром-2-(трифторметил)бензилиден]пирролидин-1,2-дикарбоновой кислоты (смесь геометрических изомеров) (25,9 г) в атмосфере газообразного аргона при охлаждении льдом. Смесь перемешивали при 30°С в течение 30 минут. Смесь охлаждали с применением бани с ледяной водой, к которой добавляли хлорид натрия, и к ней добавляли МеОН (27 мл). К смеси добавляли смесь водного раствора NaOH (1 М, 112 мл) и 30% водного раствора пероксида водорода (18 мл) и смесь перемешивали при комнатной температуре в течение 1 часа. Смесь охлаждали на льду и EtOAc и добавляли 14% водный раствор тиосульфата натрия. Смесь перемешивали при комнатной температуре в течение 1 часа. Органический слой отделяли и промывали солевым раствором. Органический слой сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на силикагеле (элюент; н-гексан/EtOAc) с получением 1-трет-бутилового сложного эфира 2-метилового сложного эфира (2S)-4-{[4-бром-2-(трифторметил)фенил](гидрокси)метил}пирролидин-1,2-дикарбоновой кислоты (смесь диастереомеров с различными стерическими конфигурациями в α-положении бензильной группы и 4-положении пирролидинового кольца) (20,96 г) в виде твердого вещества.
[0058]
Пример получения 4
4-метилморфолин N-оксид (6,1 г) и молекулярное сито 4А (8,7 г) добавляли к дихлорметановому (105 мл) раствору 1-трет-бутилового сложного эфира 2-метилового сложного эфира (2S)-4-{[4-бром-2-(трифторметил)фенил](гидрокси)метил}пирролидин-1,2-дикарбоновой кислоты (смесь диастереомеров, содержащих различные стерические конфигурации при α-положении бензильной группы и 4-положении пирролидинового кольца) (20,96 г) в атмосфере газообразного азота при охлаждении льдом, и смесь перемешивали в течение 10 минут. Тетрапропиламмония перрутенат (1,5 г) добавляли к смеси при охлаждении льдом и смесь перемешивали при комнатной температуре в течение 1 часа. EtOAc добавляли к смеси и концентрировали при пониженном давлении. Осадок фильтровали с применением силикагеля и промывали EtOAc. Насыщенный водный раствор тиосульфата натрия добавляли к фильтрату и смесь перемешивали. Органический слой отделяли и промывали солевым раствором. Органический слой сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на силикагеле (элюент; н-гексан/EtOAc) с получением 1-трет-бутилового сложного эфира 2-метилового сложного эфира (2S)-4-[4-бром-2-(трифторметил)бензоил]пирролидин-1,2-дикарбоновой кислоты (смесь эпимеров при 4-положении пирролидинового кольца) (19,6 г) в виде масляного вещества.
[0059]
Пример получения 5
Хлорид водорода (4 М раствор 1,4-диоксана, 395 мл) добавляли к МеОН (395 мл) суспензии 1-трет-бутилового сложного эфира 2-метилового сложного эфира (2S)-4-[4-бром-2-(трифторметил)бензоил]пирролидин-1,2-дикарбоновой кислоты (смесь эпимеров при 4-положении пирролидинового кольца) (78,9 г) при охлаждении льдом и смесь перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали при пониженном давлении. Толуол добавляли к смеси и смесь концентрировали при пониженном давлении. 1-Метилциклопропанкарбоновую кислоту (20 г), HATU (76 г) и DIPEA (86 мл) добавляли к дихлорметановому (630 мл) раствору осадка в атмосфере газообразного азота при охлаждении льдом и смесь перемешивали в течение 5 часов при комнатной температуре. Смесь охлаждали на льду и к ней добавляли насыщенный водный раствор бикарбоната натрия. Хлороформ добавляли к смеси и органический слой отделяли и промывали хлористоводородной кислотой (1 М), насыщенным водным раствором бикарбоната натрия и солевым раствором. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на аминосиликагеле (элюент; н-гексан/EtOAc) и методом колоночной хроматографии на силикагеле (элюент; н-гексан/EtOAc) с получением метилового сложного эфира 4-[4-бром-2-(трифторметил)бензоил]-1-[(1-метилциклопропил)карбонил]-L-пролина (смесь эпимеров при 4-положении пирролидинового кольца) (57,65 г) в виде масляного вещества.
[0060]
Пример получения 6
С применением реакционного сосуда, полученного из Teflon (зарегистрированный товарный знак), 4-трет-бутил-2,6-диметилфенилсеры трифторид (138,6 г) и комплекс фторида водорода и пиридина (101,5 мл) добавляли к дихлорметановому (320 мл) раствору метилового сложного эфира 4-[4-бром-2-(трифторметил)бензоил]-1-[(1-метилциклопропил)карбонил]-L-пролина (смесь эпимеров при 4-положении пирролидинового кольца) (64 г) в атмосфере газообразного аргона и смесь перемешивали в течение 15 часов при комнатной температуре. Перемешивание в реакционном сосуде останавливали и смесь оставляли отстаиваться при комнатной температуре в течение 75 часов. Смесь добавляли к смеси льда и 28% водного аммиачного раствора (1400 мл). Хлороформ добавляли к смеси и смесь перемешивали в течение 8 часов. Органический слой отделяли и водный слой экстрагировали хлороформом. Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия, хлористоводородной кислотой (1 М), насыщенным водным раствором бикарбоната натрия и солевым раствором. Органический слой сушили над MgSO4 и концентрировали при пониженном давлении. К остатку добавляли н-гексан и смесь перемешивали при комнатной температуре в течение 1 часа. Нерастворимое вещество отделяли фильтрацией и фильтрат концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на силикагеле (элюент; н-гексан/EtOAc). Полученное масляное вещество смешивали с н-гексаном (253 мл) и EtOAc (2,5 мл). Смесь нагревали и при осаждении твердого вещества смесь оставляли охлаждаться и перемешивали в течение 15 часов при комнатной температуре. Осадок собирали фильтрацией и промывали смесью н-гексана и EtOAc (99:1) с получением метилового сложного эфира (4R)-4-{[4-бром-2-(трифторметил)фенил](дифтор)метил}-1-[(1-метилциклопропил)карбонил]-L-пролина (26,6 г) в виде твердого вещества.
[0061]
Пример получения 7
Водный раствор гидроксида лития гидрата (1 М, 82 мл) добавляли к смеси метилового сложного эфира (4R)-4-{[4-бром-2-(трифторметил)фенил](дифтор)метил}-1-[(1-метилциклопропил)карбонил]-L-пролина (26,57 г) и THF (265 мл) при охлаждении льдом и смесь перемешивали при комнатной температуре в течение 2 часов. Смесь охлаждали на льду и к ней добавляли хлористоводородную кислоту (1 М, приблизительно 85 мл). EtOAc добавляли к смеси и органический слой отделяли и промывали солевым раствором. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением (4R)-4-{[4-бром-2-(трифторметил)фенил](дифтор)метил}-1-[(1-метилциклопропил)карбонил]-L-пролина (26,03 г) в виде твердого вещества.
[0062]
Пример получения 8
1-Аминоциклопропанкарбонитрила гидрохлорид (7,19 г), HATU (21,9 г) и DIPEA (23,5 мл) добавляли к дихлорметановому (258 мл) раствору (4R)-4-{[4-бром-2-(трифторметил)фенил](дифтор)метил}-1-[(1-метилциклопропил)карбонил]-L-пролина (25,80 г) при охлаждении льдом. Смесь перемешивали при комнатной температуре в течение 15 часов. Хлороформ и насыщенный водный раствор хлорида аммония добавляли к смеси. Органический слой отделяли и промывали насыщенным водным раствором бикарбоната натрия и солевым раствором. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на силикагеле (элюент; н-гексан/EtOAc) с получением (4R)-4-{[4-бром-2-(трифторметил)фенил](дифтор)метил}-N-(1-цианоциклопропил)-1-[(1-метилциклопропил)карбонил]-L-пролинамида (28,68 г) в виде твердого вещества.
[0063]
Пример получения 9
С применением реакционного сосуда, полученного из Teflon (зарегистрированный товарный знак), 4-трет-бутил-2,6-диметилфенилсеры трифторид (25 г) и комплекс фторида водорода и пиридина (18 мл) добавляли к дихлорметановому (64 мл) раствору метилового сложного эфира 4-[4-бром-2-(трифторметил)бензоил]-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролина (смесь эпимеров при 4-положении пирролидинового кольца) (12,9 г) в атмосфере газообразного аргона и смесь перемешивали в течение 9 часов при комнатной температуре. Перемешивание в реакционном сосуде останавливали и смесь оставляли отстаиваться при комнатной температуре в течение 14 часов. Смесь перемешивали при комнатной температуре в течение 11 часов. Перемешивание в реакционном сосуде останавливали и смесь оставляли отстаиваться при комнатной температуре в течение 14 часов. Смесь добавляли к смеси льда, 28% водного аммиачного раствора (220 мл) и хлороформа и перемешивали в течение 3 часов. Органический слой отделяли и водный слой экстрагировали хлороформом. Объединенные органические слои сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. К остатку добавляли н-гексан и смесь перемешивали при комнатной температуре в течение 1 часа. Нерастворимое вещество отделяли фильтрацией и фильтрат концентрировали при пониженном давлении. Толуол добавляли к осадку и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на силикагеле (элюент; н-гексан/EtOAc) с получением метилового сложного эфира 4-{[4-бром-2-(трифторметил)фенил](дифтор)метил}-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролина (смесь эпимеров при 4-положении пирролидинового кольца) (11,67 г) в виде масляного вещества.
[0064]
Пример получения 10
Метиловый сложный эфир 4-{[4-бром-2-(трифторметил)фенил](дифтор)метил}-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролина (смесь эпимеров при 4-положении пирролидинового кольца) (3,69 г) очищали методом колоночной хроматографии (колонка: CHIRALPAK IA, элюент: н-гексан/EtOAc) с получением метилового сложного эфира (4R)-4-{[4-бром-2-(трифторметил)фенил](дифтор)метил}-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролина (1,91 г) в виде масляного вещества.
[0065]
Пример получения 11
Смесь 1,4-диоксанового (10 мл) метилового сложного эфира 4-{[4-бром-2-(трифторметил)фенил](дифтор)метил}-1-[(1-метилциклопропил)карбонил]-L-пролина (смесь эпимеров при 4-положении пирролидинового кольца) (478 мг), трифтор[(4-метилпиперазин-1-ил)метил]борат(1-) калия (435 мг), ацетата палладия (II) (23 мг), RuPhos (97 мг) и K3PO4 (839 мг) и воды (0,072 мл) перемешивали всю ночь при 80°С в атмосфере газообразного аргона. Насыщенный водный раствор бикарбоната натрия добавляли к смеси. Хлороформ добавляли к смеси и органический слой отделяли. Органический слой промывали солевым раствором и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на аминосиликагеле (элюент; хлороформ/МеОН) с получением метилового сложного эфира 4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-[(1-метилциклопропил)карбонил]-L-пролина (смесь эпимеров при 4-положении пирролидинового кольца) (340 мг) в виде масляного вещества.
[0066]
Пример получения 12
Трифенилфосфин (9,75 г) добавляли к толуоловому (70 мл) раствору 4-бром-1-(бромметил)-2-хлорбензола (10 г) при комнатной температуре и смесь перемешивали при 80°С в течение 6 часов. Смесь охлаждали на льду и перемешивали в течение 30 минут. Осадок собирали фильтрацией и промывали толуолом с получением (4-бром-2-хлорбензил)(трифенил)фосфоний бромида (18,9 г) в виде твердого вещества.
[0067]
Пример получения 13
2,4,6-Трихлорфенилформиат (1 г), ацетат палладия (II) (120 мг), XantPhos (300 мг) и DIPEA (1,5 мл) добавляли к толуоловому (30 мл) раствору 4-{[4-бром-2-(трифторметил)фенил](дифтор)метил}-N-(1-цианоциклопропил)-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида (смесь эпимеров при 4-положении пирролидинового кольца) (2,1 г) при 100°С в атмосфере газообразного аргона и смесь перемешивали в течение 1 часа. Смесь охлаждали на льду и к ней добавляли хлороформ и солевой раствор. Нерастворимое вещество отделяли фильтрацией при помощи целита и органический слой фильтрата отделяли. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на силикагеле (элюент: н-гексан/EtOAc) с получением 2,4,6-трихлорфенилового сложного эфира 4-{[(5S)-5-[(1-цианоциклопропил)карбамоил]-1-{[1-(трифторметил)циклопропил]карбонил}пирролидин-3-ил](дифтор)метил}-3-(трифторметил)бензойной кислоты (смесь эпимеров при 3-положении пирролидинового кольца) (2,4 г) в виде твердого вещества.
[0068]
Пример получения 14
Боргидрид натрия (103 мг) добавляли к смеси 2,4,6-трихлорфенилового сложного эфира 4-{[(5S)-5-[(1-цианоциклопропил)карбамоил]-1-{[1-(трифторметил)циклопропил]карбонил}пирролидин-3-ил](дифтор)метил}-3-(трифторметил)бензойной кислоты (смесь эпимеров при 3-положении пирролидинового кольца) (1 г) и THF (20 мл) при охлаждении льдом. МеОН (2 мл) добавляли к смеси при охлаждении льдом и смесь перемешивали при комнатной температуре в течение 30 минут. Воду добавляли к смеси. EtOAc добавляли к смеси и органический слой отделяли и промывали солевым раствором. Органический слой сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на силикагеле (элюент; н-гексан/EtOAc) с получением N-(1-цианоциклопропил)-4-{дифтор[4-(гидроксиметил)-2-(трифторметил)фенил]метил}-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида (смесь эпимеров при 4-положении пирролидинового кольца) (290 мг) в виде твердого вещества.
[0069]
Пример получения 15
Смесь 1,2-дибромэтана (0,032 мл) и хлор(триметил)силана (0,047 мл) добавляли к суспензии цинка (365 мг) в N,N-диметилацетамиде (2 мл) при комнатной температуре в атмосфере газообразного аргона и смесь перемешивали при 50°С в течение 15 минут. К смеси добавляли N,N-диметилацетамидный (2 мл) раствор трет-бутилового сложного эфира 4-йодпиперидин-1-карбоновой кислоты (1,73 г) при комнатной температуре и смесь перемешивали при 50°С в течение 15 минут. Вышеуказанную смесь добавляли к N,N-диметилацетамидной (5 мл) суспензии метилового сложного эфира 4-{[4-бром-2-(трифторметил)фенил](дифтор)метил}-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролина (смесь эпимеров при 4-положении пирролидинового кольца) (1 г), комплекса 1,1'-бис(дифенилфосфино)ферроцен-палладия(II) дихлорида-дихлорметана (151 мг) и йодида меди (I) (71 мг) при комнатной температуре в атмосфере газообразного аргона и смесь перемешивали при 80°С всю ночь. Смесь охлаждали до комнатной температуры, EtOAc добавляли и нерастворимое вещество отделяли фильтрацией. Фильтрат промывали водой и солевым раствором и органический слой сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на силикагеле (элюент: н-гексан/EtOAc) с получением трет-бутилового сложного эфира 4-[4-{дифтор[(5S)-5-(метоксикарбонил)-1-{[1-(трифторметил)циклопропил]карбонил}пирролидин-3-ил]метил}-3-(трифторметил)фенил]пиперидин-1-карбоновой кислоты (смесь эпимеров при 3-положении пирролидинового кольца) (1,15 г) в виде масляного вещества.
[0070]
Пример получения 16
Трифторуксусную кислоту (1,5 мл) добавляли к дихлорметановому (15 мл) и MeCN (7,5 мл) раствору трет-бутилового сложного эфира 4-[4-{[(5S)-5-[(1-цианоциклопропил)карбамоил]-1-{[1-(трифторметил)циклопропил]карбонил}пирролидин-3-ил](дифтор)метил}-3-(трифторметил)фенил]пиперидин-1-карбоновой кислоты (смесь эпимеров при 3-положении пирролидинового кольца) (675 мг) при охлаждении льдом и смесь перемешивали при комнатной температуре в течение 3 часов. Трифторуксусную кислоту (2,25 мл) добавляли к смеси при комнатной температуре и смесь перемешивали в течение 4 часов. Смесь добавляли к насыщенному водному раствору бикарбоната натрия. Хлороформ добавляли к смеси и органический слой отделяли и промывали солевым раствором. Органический слой сушили над MgSO4 и концентрировали при пониженном давлении с получением N-(1-цианоциклопропил)-4-{дифтор[4-(пиперидин-4-ил)-2-(трифторметил)фенил]метил}-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида (смесь эпимеров при 4-положении пирролидинового кольца) (509 мг) в виде твердого вещества.
[0071]
Пример получения 17
Карбонат калия (5,5 г) добавляли к дихлорметановому (80 мл) раствору (4-бром-2-хлорбензил)(трифенил)фосфоний бромида (18,9 г) при комнатной температуре в атмосфере газообразного азота и смесь перемешивали в течение 20 минут. К смеси добавляли 1-трет-бутиловый сложный эфир 2-метиловый сложный эфир (2S)-4-оксопирролидин-1,2-дикарбоновой кислоты (6,5 г) и 18-crown-6 (110 мг) при комнатной температуре и смесь перемешивали при нагревании с обратным холодильником в течение 3 суток. Смесь охлаждали на льду и к ней добавляли насыщенный водный раствор бикарбоната натрия. Органический слой отделяли и промывали солевым раствором. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на силикагеле (элюент; н-гексан/EtOAc) с получением 1-трет-бутилового сложного эфира 2-метилового сложного эфира (2S)-4-(4-бром-2-хлорбензилиден)пирролидин-1,2-дикарбоновой кислоты (смесь геометрических изомеров) (5,17 г) в виде масляного вещества.
[0072]
Пример получения 18
Бис(2-метоксиэтил)аминосеры трифторид (15 мл) добавляли к метиловому сложному эфиру 4-(4-бром-2-хлорбензоил)-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролина (смесь эпимеров при 4-положении пирролидинового кольца) (2,8 г) в атмосфере газообразного аргона и смесь перемешивали при 90°С в течение 3 часов. Смесь охлаждали на льду и добавляли к смеси льда и насыщенного водного раствора бикарбоната натрия. EtOAc добавляли к смеси и органический слой отделяли и промывали солевым раствором. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на силикагеле (элюент; н-гексан/EtOAc) с получением метилового сложного эфира 4-[(4-бром-2-хлорфенил)(дифтор)метил]-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролина (смесь эпимеров при 4-положении пирролидинового кольца) (2,37 г) в виде масляного вещества.
[0073]
Пример получения 19
Гидроксид лития гидрат (20 мг) добавляли к МеОН (0,7 мл) и THF (0,7 мл) раствору трет-бутилового эфира 4-(4-{дифтор[(5S)-5-(метоксикарбонил)-1-{[1-(трифторметил)циклопропил]карбонил}пирролидин-3-ил]метил}-3-фторфенил)пиперидин-1-карбоновой кислоты (смесь эпимеров при 3-положении пирролидинового кольца) (105 мг) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали при пониженном давлении с получением (2S)-4-[{4-[1-(трет-бутоксикарбонил)пиперидин-4-ил]-2-фторфенил}(дифтор)метил]-1-{[1-(трифторметил)циклопропил]карбонил}пирролидин-2-карбоксилата лития (смесь эпимеров при 4-положении пирролидинового кольца) (102 мг) в виде твердого вещества.
[0074]
Пример 1
Трифтор[(4-метилпиперазин-1-ил)метил]борат (1-) калия (4,1 г), XPhos (892 мг), карбонат цезия (12 г) и ацетат палладия (II) (214 мг) добавляли к 1,4-диоксановому (50 мл) и водному (10 мл) раствору (4R)-4-{[4-бром-2-(трифторметил)фенил](дифтор)метил}-N-(1-цианоциклопропил)-1-[(1-метилциклопропил)карбонил]-L-пролинамида (5 г) при комнатной температуре в атмосфере газообразного аргона и смесь перемешивали в течение 3 часов при нагревании с обратным холодильником. Смесь охлаждали до комнатной температуры и к ней добавляли воду. Нерастворимое вещество отделяли фильтрацией при помощи целита и промывали хлороформом. Воду и хлороформ добавляли к фильтрату. Органический слой отделяли и промывали солевым раствором. Органический слой сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на силикагеле (элюент; н-гексан/EtOAc и хлороформ/МеОН) с получением (4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-[(1-метилциклопропил)карбонил]-L-пролинамида (4,7 г) в виде твердого вещества.
[0075]
Пример 2
Хлороводород (4 М 1,4-диоксановый раствор, 1,39 мл) добавляли к EtOAc (7 мл) и этанольному (7 мл) раствору (4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-[(1-метилциклопропил)карбонил]-L-пролинамида (1,43 г) и смесь перемешивали при комнатной температуре в течение 30 минут. Смесь концентрировали при пониженном давлении. Этанол добавляли к остатку и смесь концентрировали при пониженном давлении. EtOAc добавляли к остатку и смесь перемешивали приблизительно 1 час. Смесь концентрировали при пониженном давлении. Этанол (10 мл) добавляли к осадку и нагревали при 95°С с получением раствора. Смесь перемешивали всю ночь при комнатной температуре. Осадок собирали фильтрацией и промывали этанолом с получением (4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-[(1-метилциклопропил)карбонил]-L-пролинамида дигидрохлорида (516 мг) в виде твердого вещества.
[0076]
Пример 4
Смесь (4R)-4-{[4-бром-2-(трифторметил)фенил](дифтор)метил}-N-(1-цианоциклопропил)-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида (627,4 мг), трифтор[(4-метилпиперазин-1-ил)метил]бората (1-) калия (705 мг), ацетата палладия (II) (25 мг), RuPhos (105 мг), K3PO4 (905 мг), 1,4-диоксана (13 мл) и воды (1,3 мл) перемешивали при 80°С в течение 6 часов в атмосфере газообразного аргона. Насыщенный водный раствор бикарбоната натрия и хлороформ добавляли к смеси. Органический слой отделяли и промывали солевым раствором. Органический слой сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на аминосиликагеле (элюент; хлороформ/МеОН) с получением (4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида (491,4 мг) в виде масляного вещества.
[0077]
Пример 7
Хлористоводородную кислоту (6 М, 0,678 мл) добавляли к этанольному (6 мл) раствору трет-бутилового сложного эфира 4-{4-[{(3R,5S)-5-[(1-цианоциклопропил)карбамоил]-1-[(1-метилциклопропил)карбонил]пирролидин-3-ил}(дифтор)метил]-3-(трифторметил)бензил}пиперазин-1-карбоновой кислоты (339 мг) и перемешивали при 50°С в течение 1 часа. Смесь перемешивали при 60°С в течение 5 часов. Смесь охлаждали на льду и к ней добавляли водный раствор NaOH (1 М, 5 мл). Хлороформ добавляли к смеси и органический слой отделяли и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на аминосиликагеле (элюент; хлороформ/МеОН). Хлороводород (4 М 1,4-диоксановый раствор, 0,285 мл) добавляли к раствору 1,4-диоксана (3 мл) и этанола (5 мл) полученного масляного вещества и смесь перемешивали при комнатной температуре в течение 5 минут. Смесь концентрировали при пониженном давлении. EtOAc добавляли к остатку и осадок собирали фильтрацией и промывали EtOAc с получением (4R)-N-(1-цианоциклопропил)-4-{дифтор[4-(пиперазин-1-илметил)-2-(трифторметил)фенил]метил}-1-[(1-метилциклопропил)карбонил]-L-пролинамида дигидрохлорида (190 мг) в виде твердого вещества.
[0078]
Пример 8
Смесь 4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-[(1-метилциклопропил)карбонил]-L-пролина (смесь эпимеров при 4-положении пирролидинового кольца) (415 мг), 1-аминоциклопропанкарбонитрила гидрохлорида (117 мг), HATU (375 мг), DIPEA (0,338 мл) и DMF (8 мл) перемешивали всю ночь при комнатной температуре. Воду добавляли к смеси и смесь экстрагировали хлороформом. Органический слой концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на аминосиликагеле (элюент; хлороформ/МеОН). Полученное масляное вещество очищали методом высокоэффективной жидкостной хроматографии с обращенной фазой (элюент; 0,1% водный раствор муравьиной кислоты/MeCN). Хлороводород (4 М 1,4-диоксановый раствор, 0,115 мл) добавляли к дихлорметановому (1 мл) раствору полученного остатка и смесь перемешивали при комнатной температуре в течение 5 минут. Смесь концентрировали при пониженном давлении с получением N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-[(1-метилциклопропил)карбонил]-L-пролинамида дигидрохлорида (смесь эпимеров при 4-положении пирролидинового кольца) (62 мг) в виде твердого вещества.
[0079]
Пример 9
Трифторуксусную кислоту (0,11 мл) добавляли к дихлорметановому (0,42 мл) и MeCN (0,11 мл) раствору трет-бутилового сложного эфира 4-[4-{[(3R,5S)-5-[(l-цианоциклопропил)карбамоил]-1-{[1-(трифторметил)циклопропил]карбонил}пирролидин-3-ил](дифтор)метил}-3-(трифторметил)бензил]пиперазин-1-карбоновой кислоты (35 мг) при охлаждении льдом и смесь перемешивали всю ночь при комнатной температуре. Смесь концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на аминосиликагеле (элюент; хлороформ/МеОН). Хлороводород (4 М 1,4-диоксановый раствор, 0,015 мл) добавляли к 1,4-диоксановому (0,35 мл) раствору полученного остатка и смесь перемешивали при комнатной температуре в течение 5 минут. Смесь концентрировали при пониженном давлении и к осадку добавляли диизопропиловый эфир. Осадок собирали фильтрацией с получением (4R)-N-(1-цианоциклопропил)-4-{дифтор[4-(пиперазин-1-илметил)-2-(трифторметил)фенил]метил}-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида дигидрохлорида (22 мг) в виде твердого вещества.
[0080]
Пример 10
Метансульфохлорид (0,011 мл) и TEA (0,039 мл) добавляли к дихлорметановому (2 мл) раствору N-(1-цианоциклопропил)-4-{дифтор[4-(гидроксиметил)-2-(трифторметил)фенил]метил}-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида (смесь эпимеров при 4-положении пирролидинового кольца) (50 мг) и смесь перемешивали при комнатной температуре в течение 30 минут. Смесь концентрировали при пониженном давлении. DMF (2 мл), 1-этилпиперазин (0,024 мл) и карбонат калия (86 мг) добавляли к остатку и смесь перемешивали при комнатной температуре в течение 20 часов. Воду и EtOAc добавляли к смеси и органический слой отделяли и промывали водой и солевым раствором. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на аминосиликагеле (элюент: н-гексан/хлороформ и хлороформ/МеОН) с получением N-(1-цианоциклопропил)-4-[{4-[(4-этилпиперазин-1-ил)метил]-2-(трифторметил)фенил}(дифтор)метил]-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида (смесь эпимеров при 4-положении пирролидинового кольца) (52 мг) в виде твердого вещества.
[0081]
Пример 12
Хлороводород (4 М EtOAc раствор, 0,455 мл) добавляли к EtOAc (12 мл) и этаноловому (1,2 мл) раствору (4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида (565,9 мг) и смесь перемешивали при комнатной температуре всю ночь. Осадок собирали фильтрацией и промывали EtOAc с получением (4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида дигидрохлорида (508 мг) в виде твердого вещества.
[0082]
Пример 15
Уксусную кислоту (0,010 мл) и триацетоксиборогидрид натрия (50 мг) добавляли к дихлорэтановому (1 мл) раствору N-(1-цианоциклопропил)-4-{дифтор[2-фтор-4-(пиперидин-4-ил)фенил]метил}-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида (смесь эпимеров при 4-положении пирролидинового кольца) (32 мг) в атмосфере газообразного аргона при охлаждении льдом и смесь перемешивали несколько минут. Ацетальдегид (0,030 мл) добавляли к смеси при охлаждении льдом и смесь перемешивали в течение 30 минут. Насыщенный водный раствор бикарбоната натрия добавляли к смеси при охлаждении льдом и органический слой отделяли и промывали солевым раствором. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на аминосиликагеле (элюент; н-гексан/EtOAc) с получением N-(1-цианоциклопропил)-4-{[4-(1-этилпиперидин-4-ил)-2-фторфенил](дифтор)метил}-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида (смесь эпимеров при 4-положении пирролидинового кольца) (27 мг) в виде твердого вещества.
[0083]
Пример 17
Уксусную кислоту (0,019 мл) и ацетальдегид (0,045 мл) добавляли к дихлорэтановому (1,9 мл) раствору N-(1-цианоциклопропил)-4-{дифтор[4-(пиперидин-4-ил)-2-(трифторметил)фенил]метил}-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида (смесь эпимеров при 4-положении пирролидинового кольца) (95 мг) при комнатной температуре и смесь перемешивали в течение 5 минут. Триацетоксиборгидрид натрия (136 мг) добавляли к смеси при комнатной температуре и смесь перемешивали в течение 2 часов. Воду и хлороформ добавляли к смеси и органический слой отделяли и промывали солевым раствором. Органический слой сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали методом колоночной хроматографии на силикагеле (элюент; хлороформ/МеОН). Хлороводород (4 М 1,4-диоксановый раствор, 0,025 мл) добавляли к дихлорметановому (2 мл) раствору полученного масляного вещества и смесь перемешивали в течение 30 минут. Смесь концентрировали при пониженном давлении с получением N-(1-цианоциклопропил)-4-{[4-(1-этилпиперидин-4-ил)-2-(трифторметил)фенил](дифтор)метил}-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида гидрохлорида (смесь эпимеров при 4-положении пирролидинового кольца) (60,6 мг) в виде твердого вещества.
[0084]
Пример 18
Янтарную кислоту (104 мг) добавляли к 2-пропаноловому (2,5 мл) раствору (4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-[(1-метилциклопропил)карбонил]-L-пролинамида (250 мг) при комнатной температуре. Смесь перемешивали при 60°С в течение 5 минут, а затем перемешивали при комнатной температуре всю ночь. 2-Пропанол (2,5 мл) добавляли к смеси и перемешивали в течение 10 минут. Осадок собирали фильтрацией и промывали небольшим количеством 2-пропанола с получением кристалла (281 мг), содержащего 1:2 (4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-[(1-метилциклопропил)карбонил]-L-пролинамид и янтарную кислоту (называемый в настоящей заявке в некоторых случаях «дисукцинат»).
[0085]
Соединения примеров получения и примеров, показанные в следующих таблицах, получали таким же способом, что и в описанных выше примерах получения или примерах.
[0086]
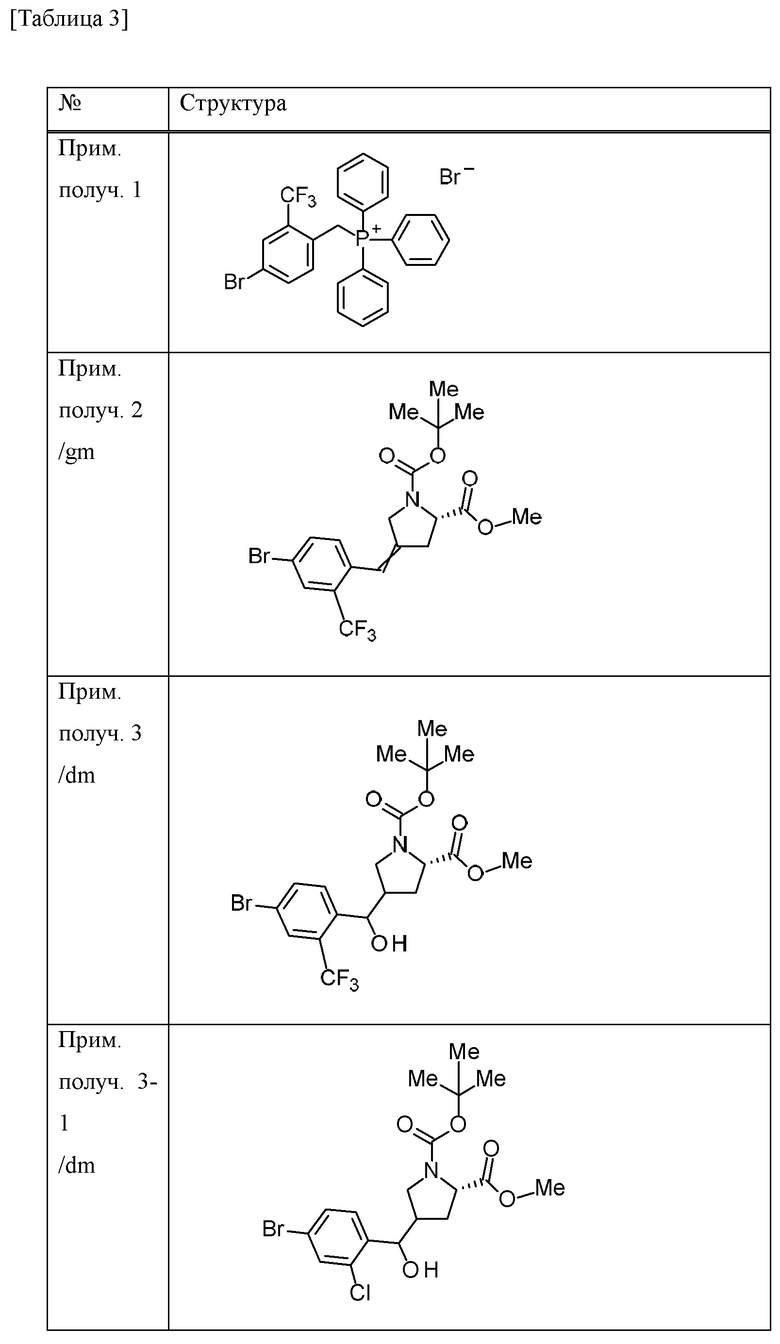
[0087]
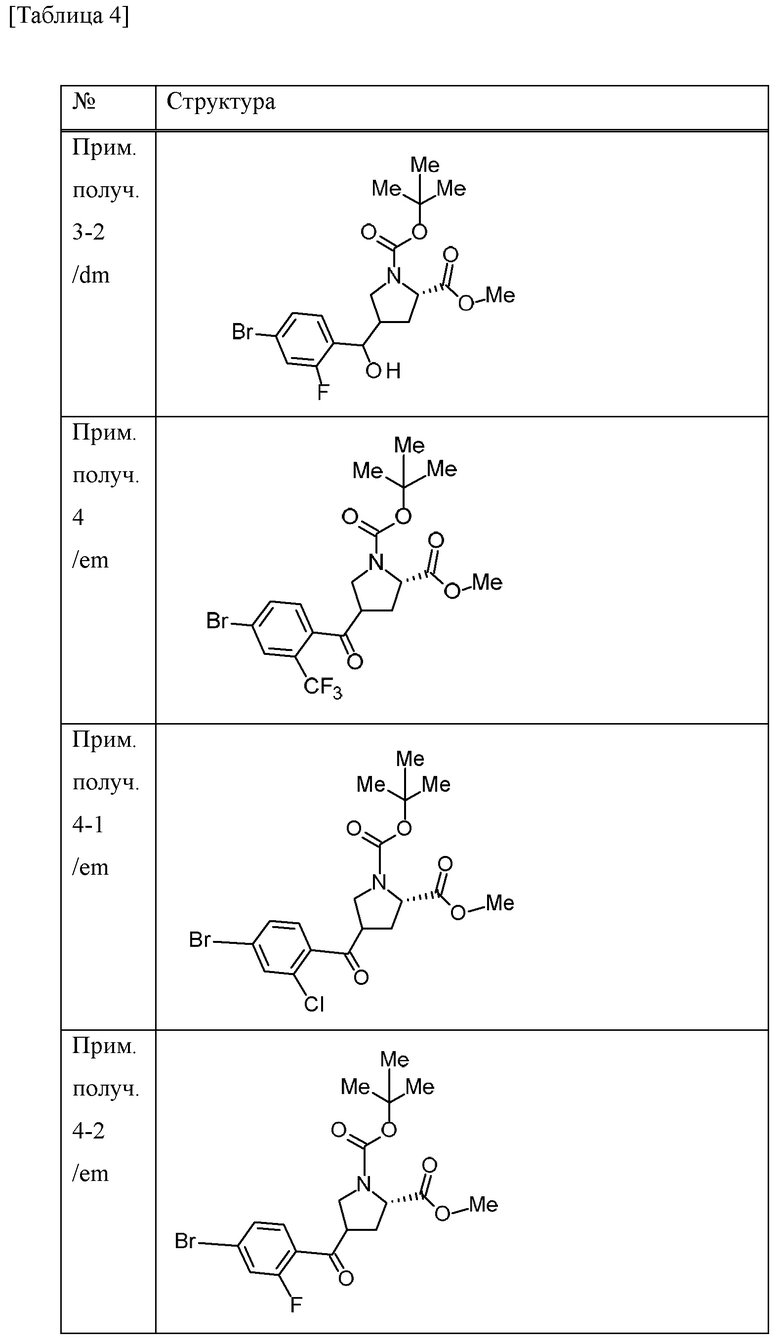
[0088]
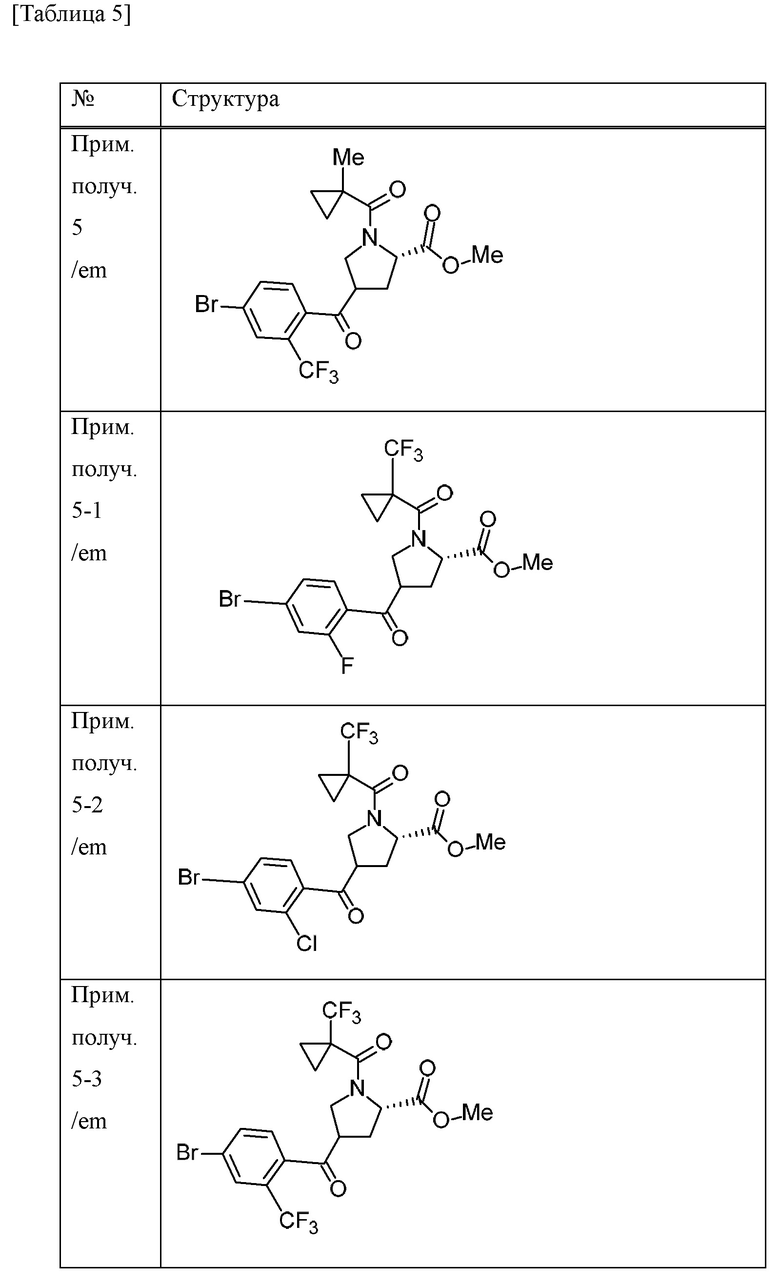
[0089]
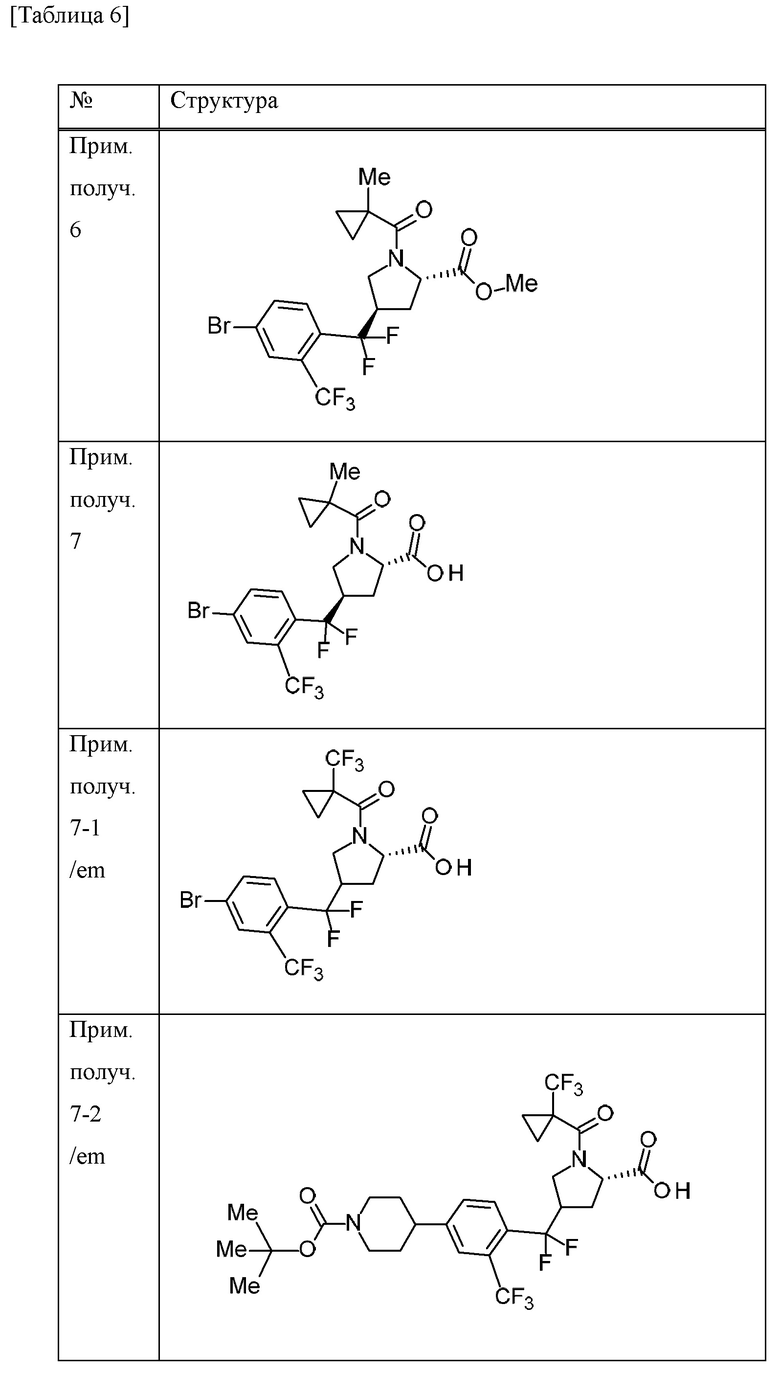
[0090]
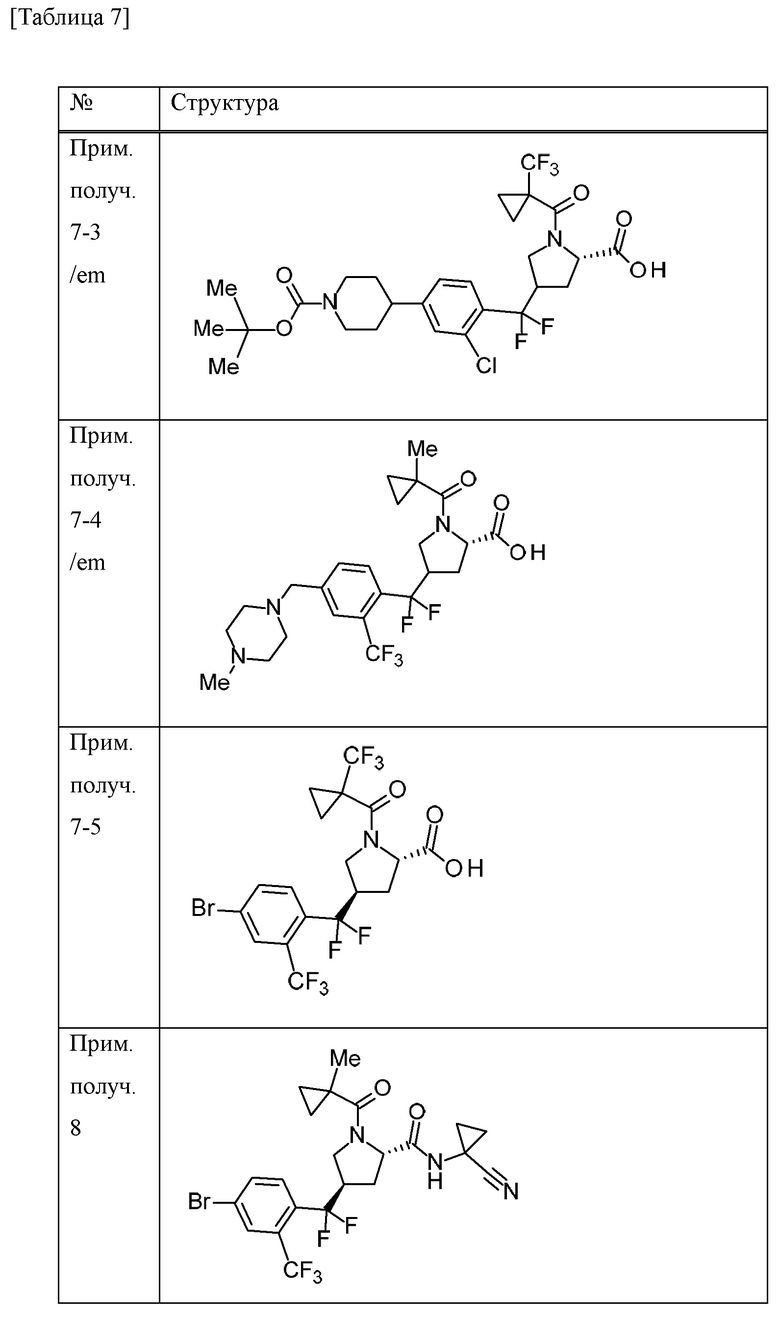
[0091]
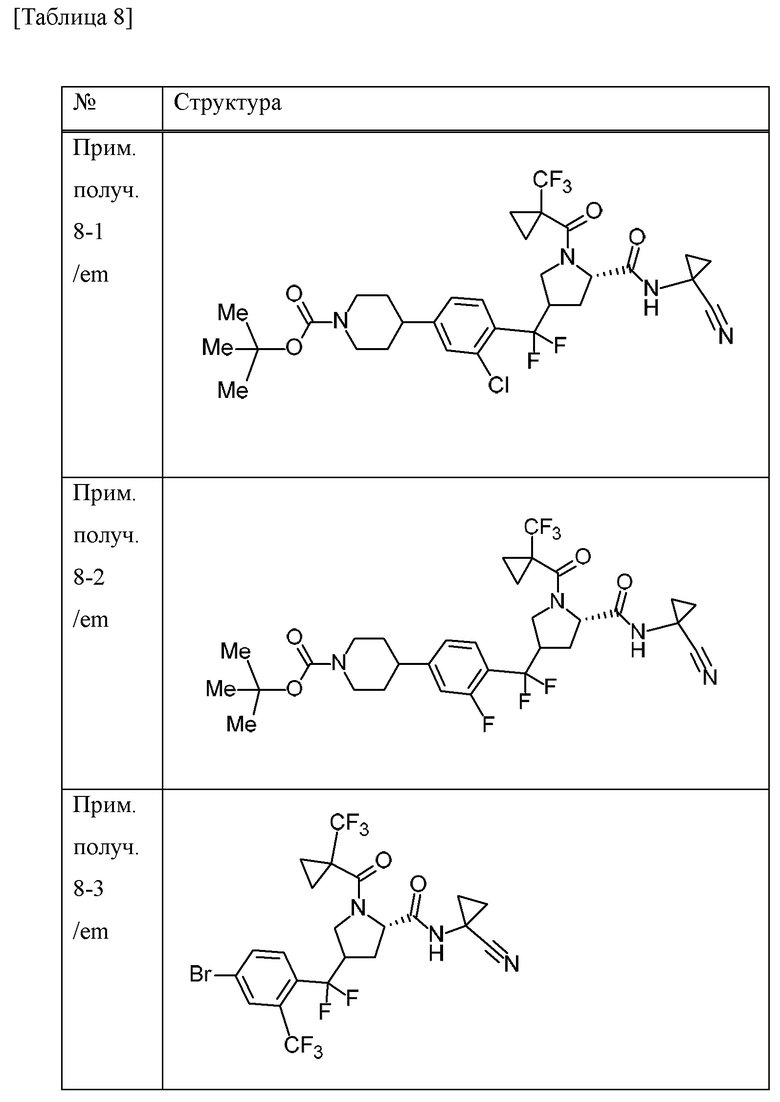
[0092]
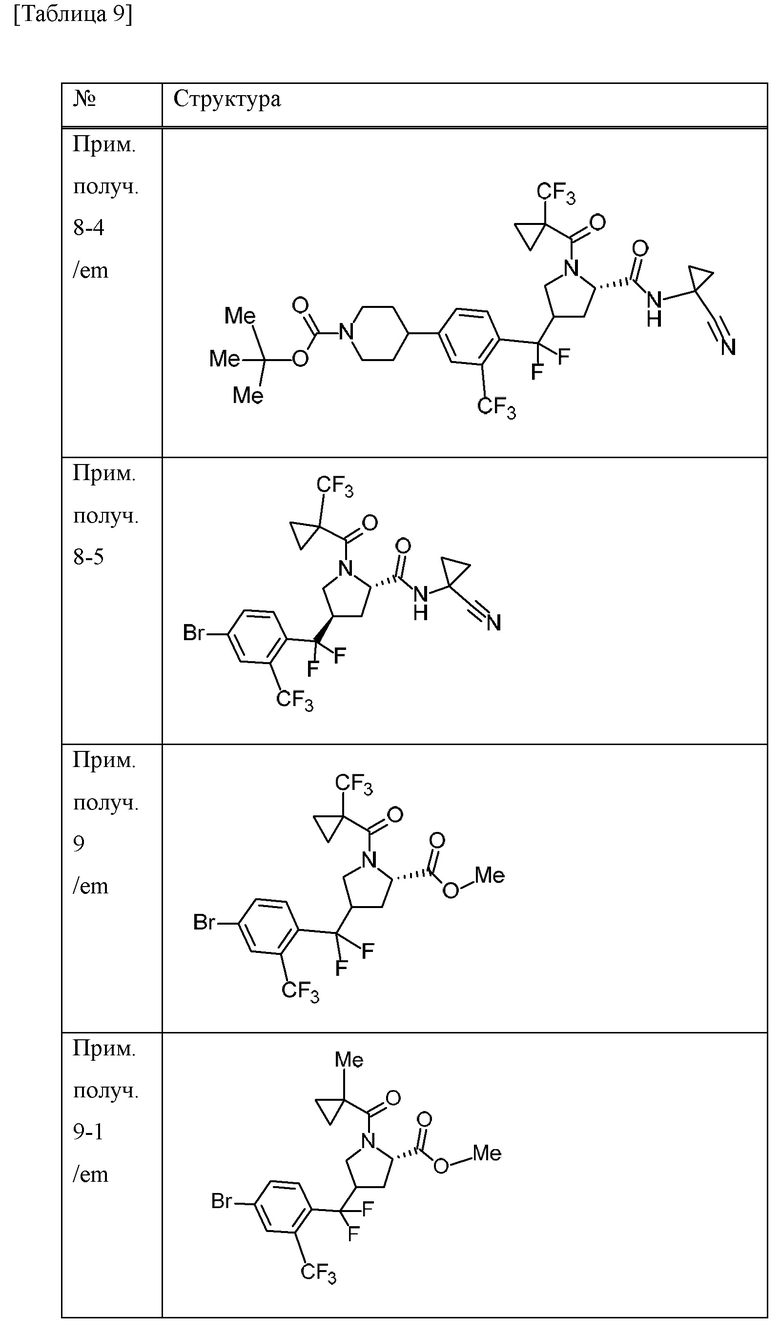
[0093]
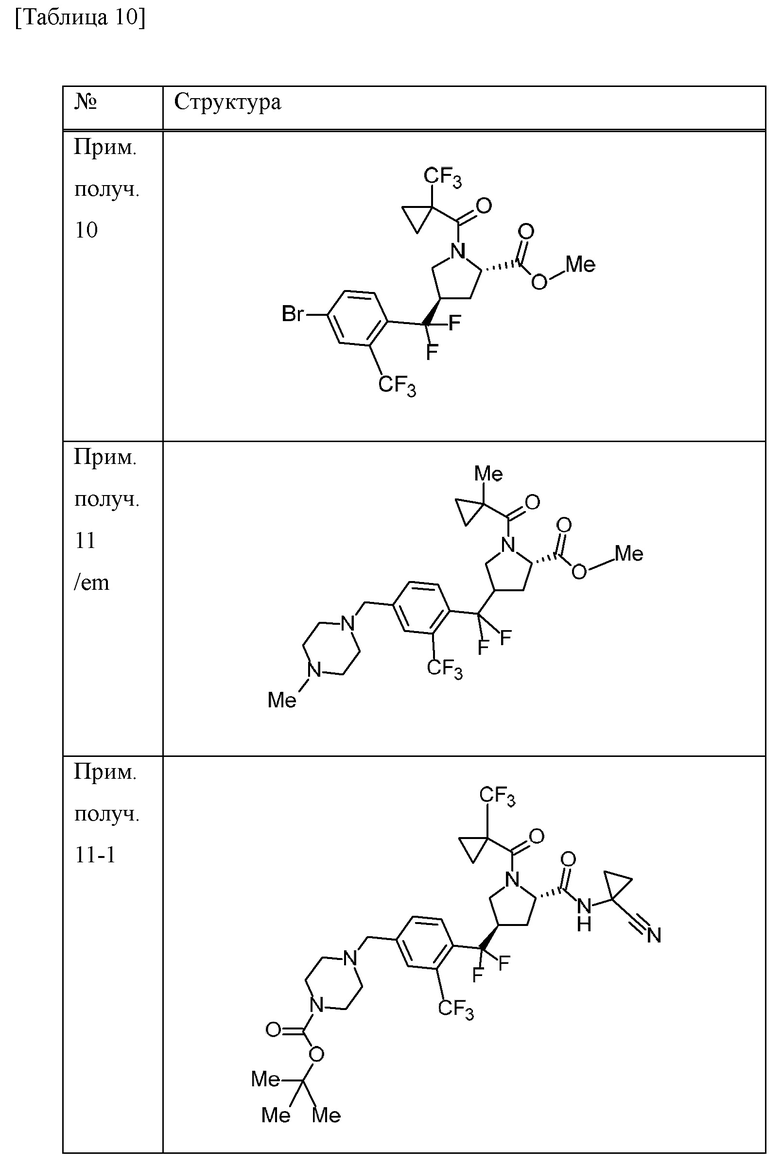
[0094]
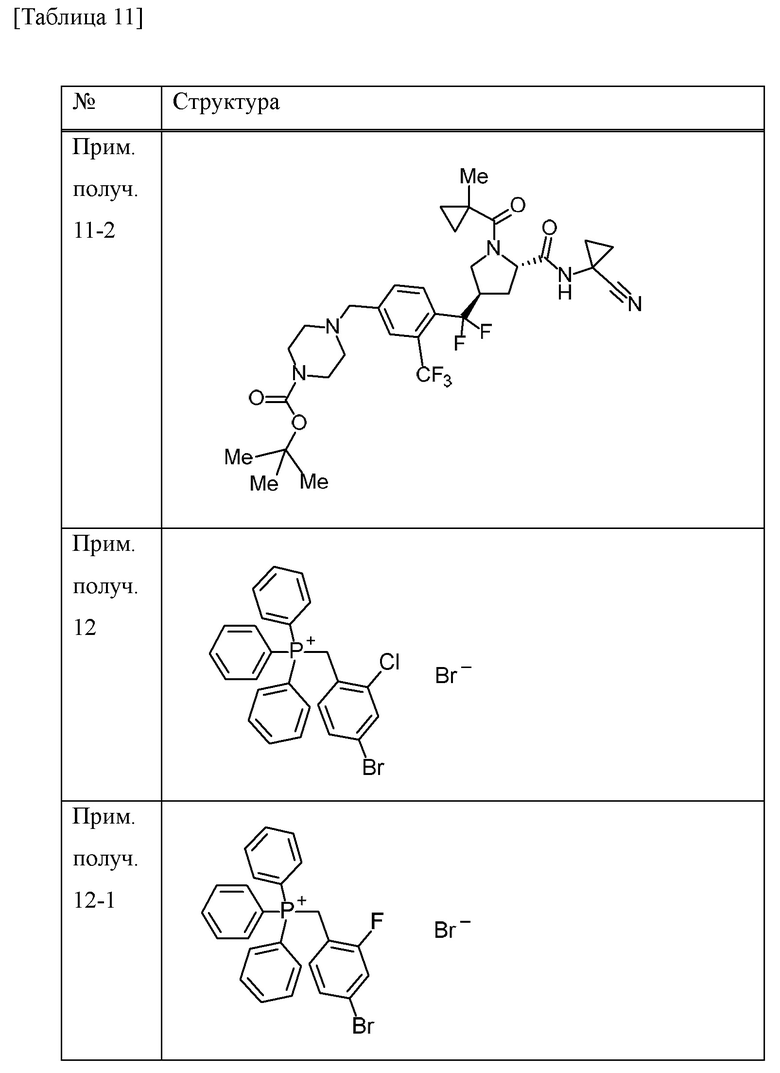
[0095]
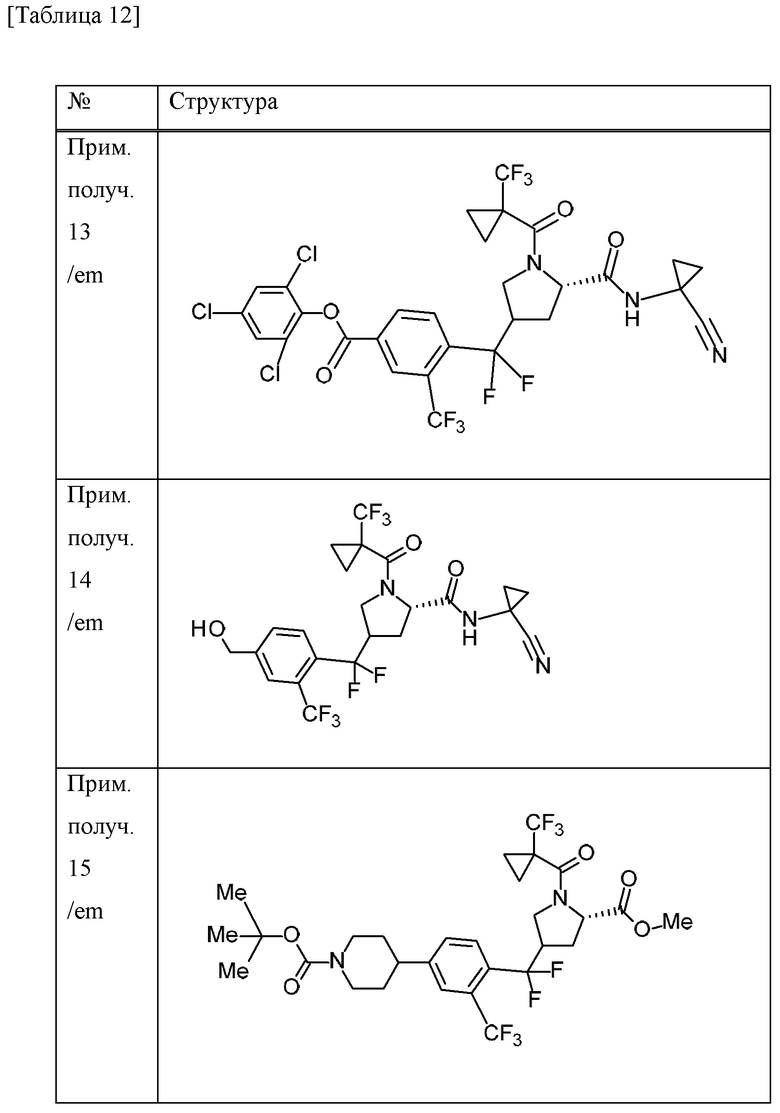
[0096]
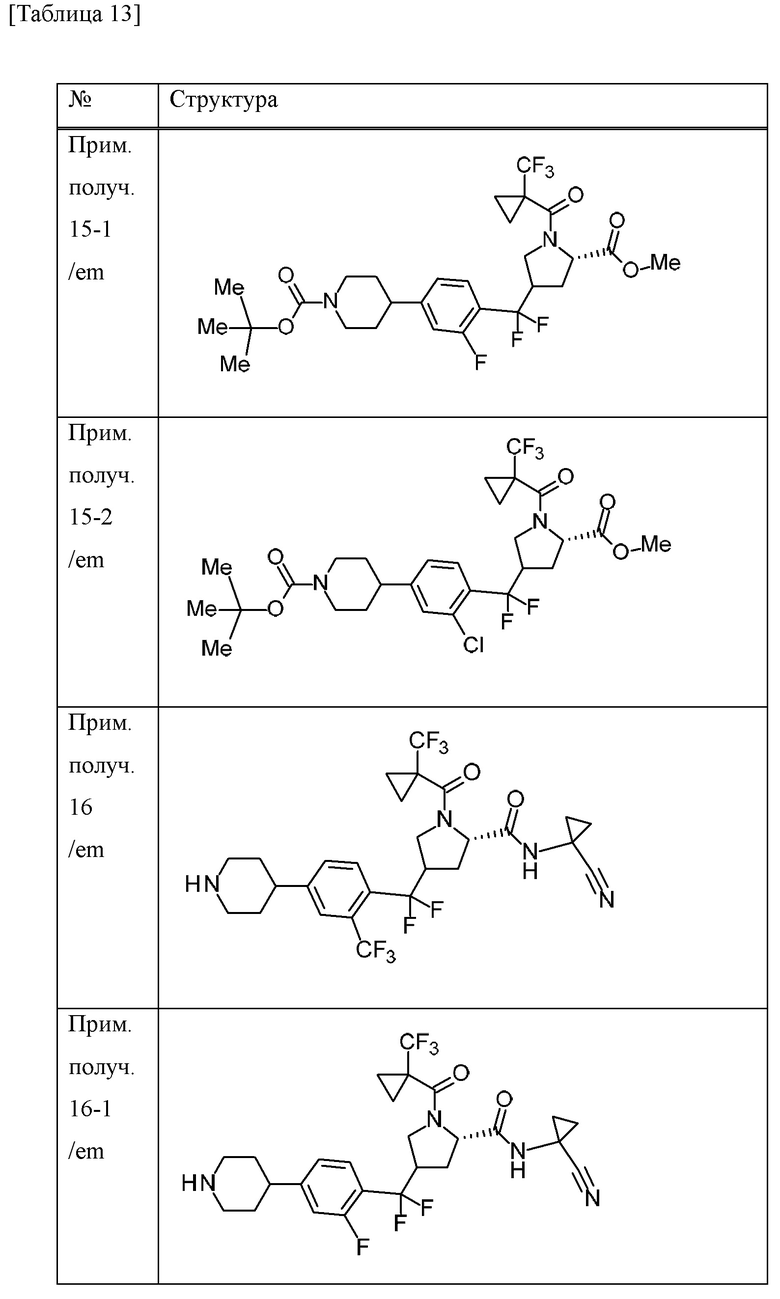
[0097]
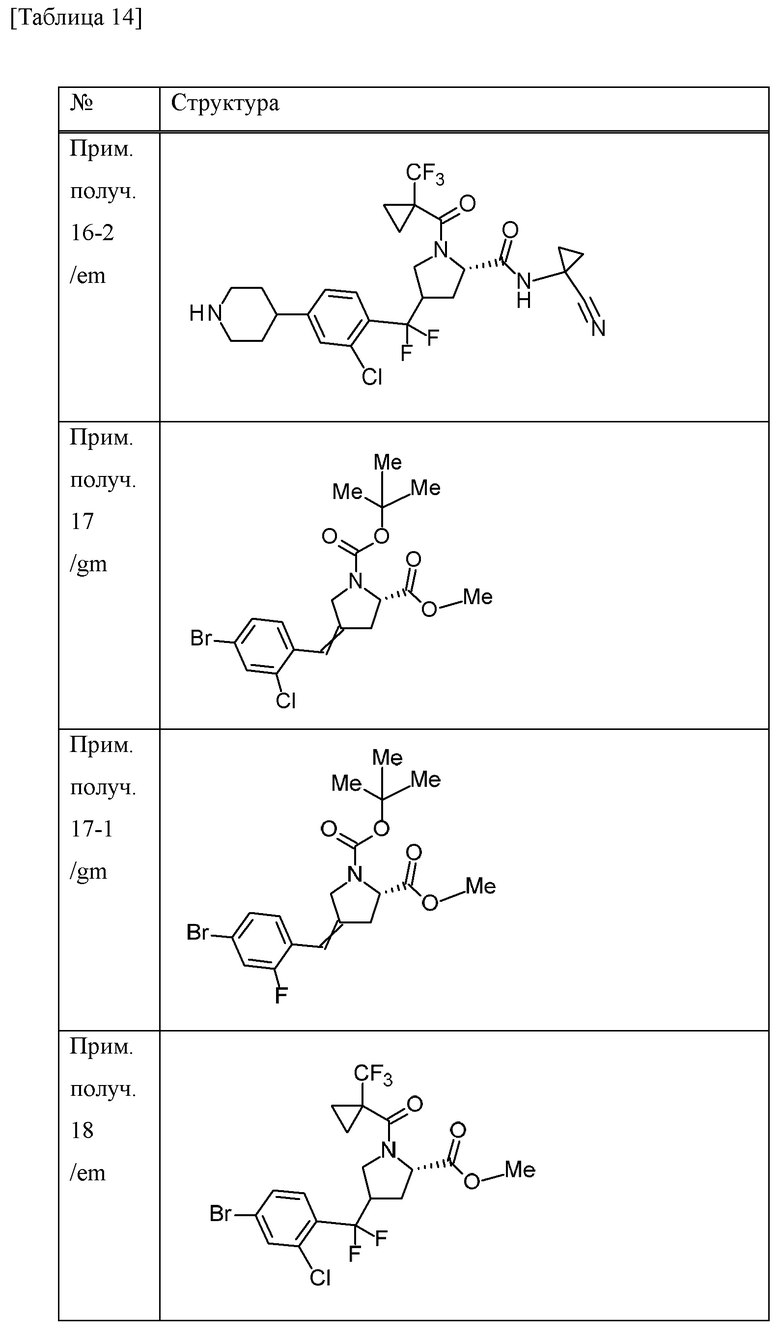
[0098]
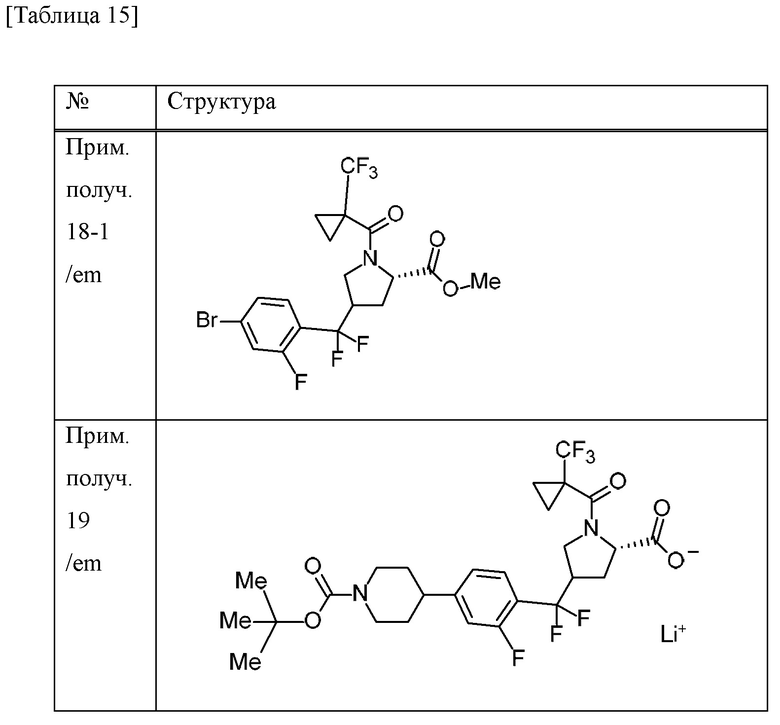
[0099]
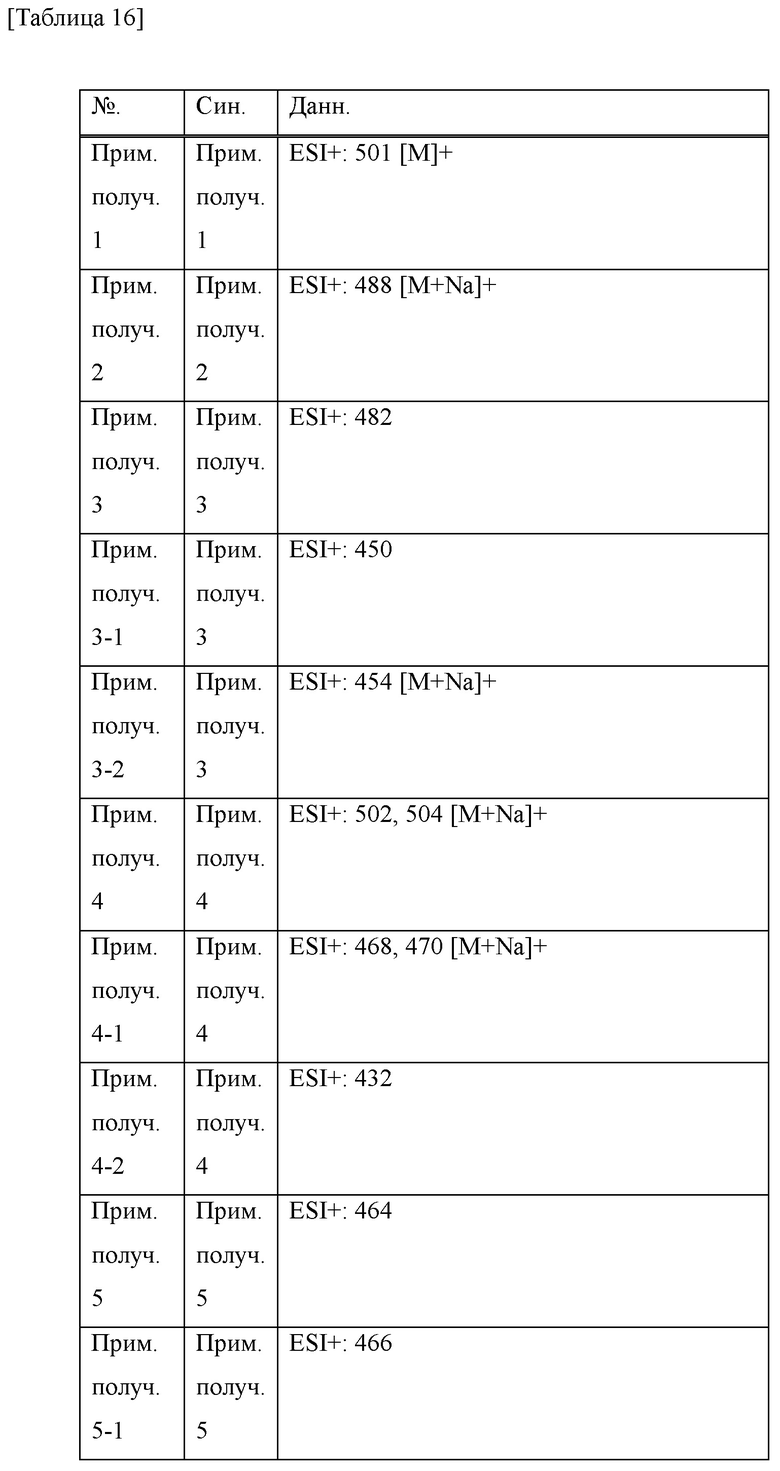
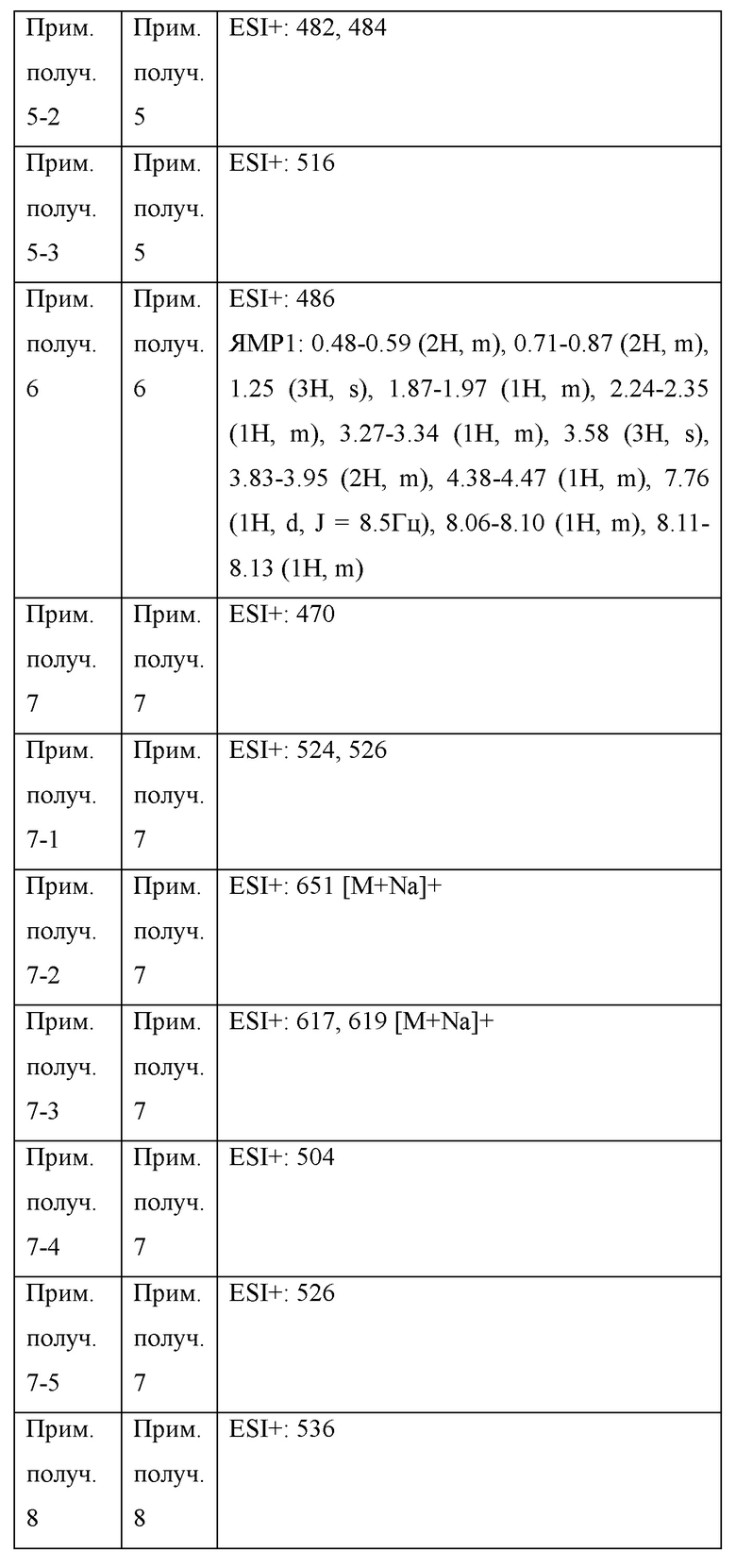
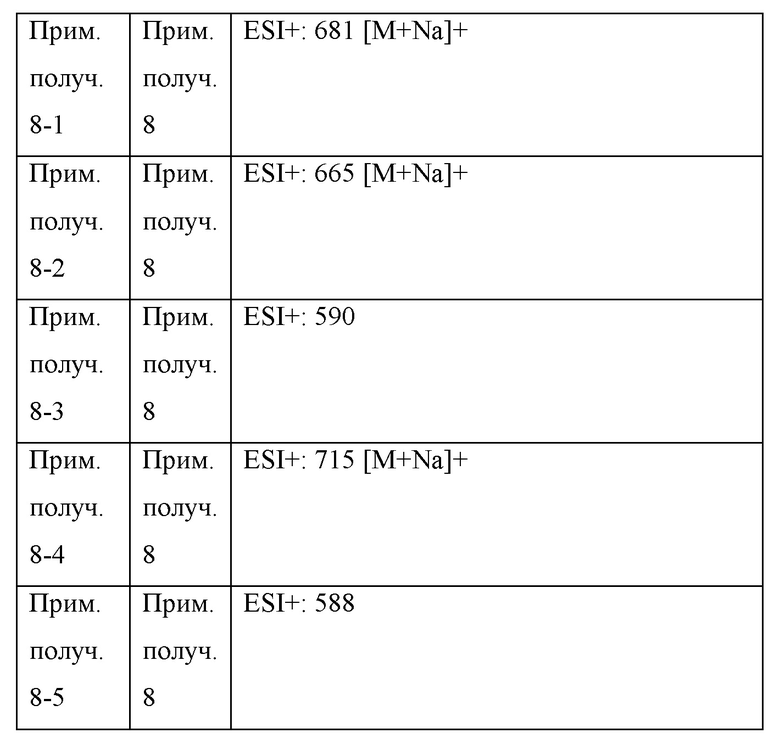
[0100]
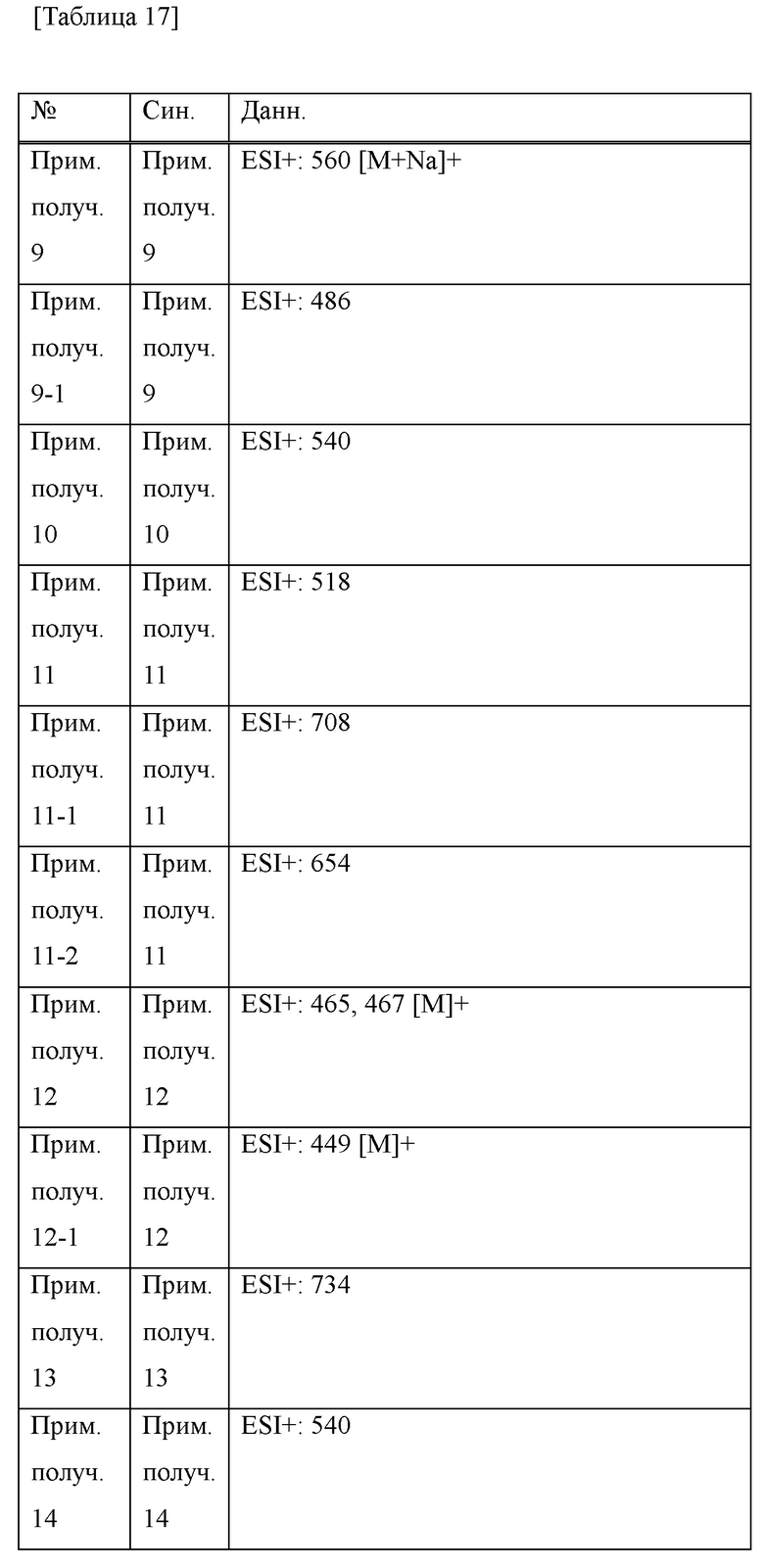

[0101]
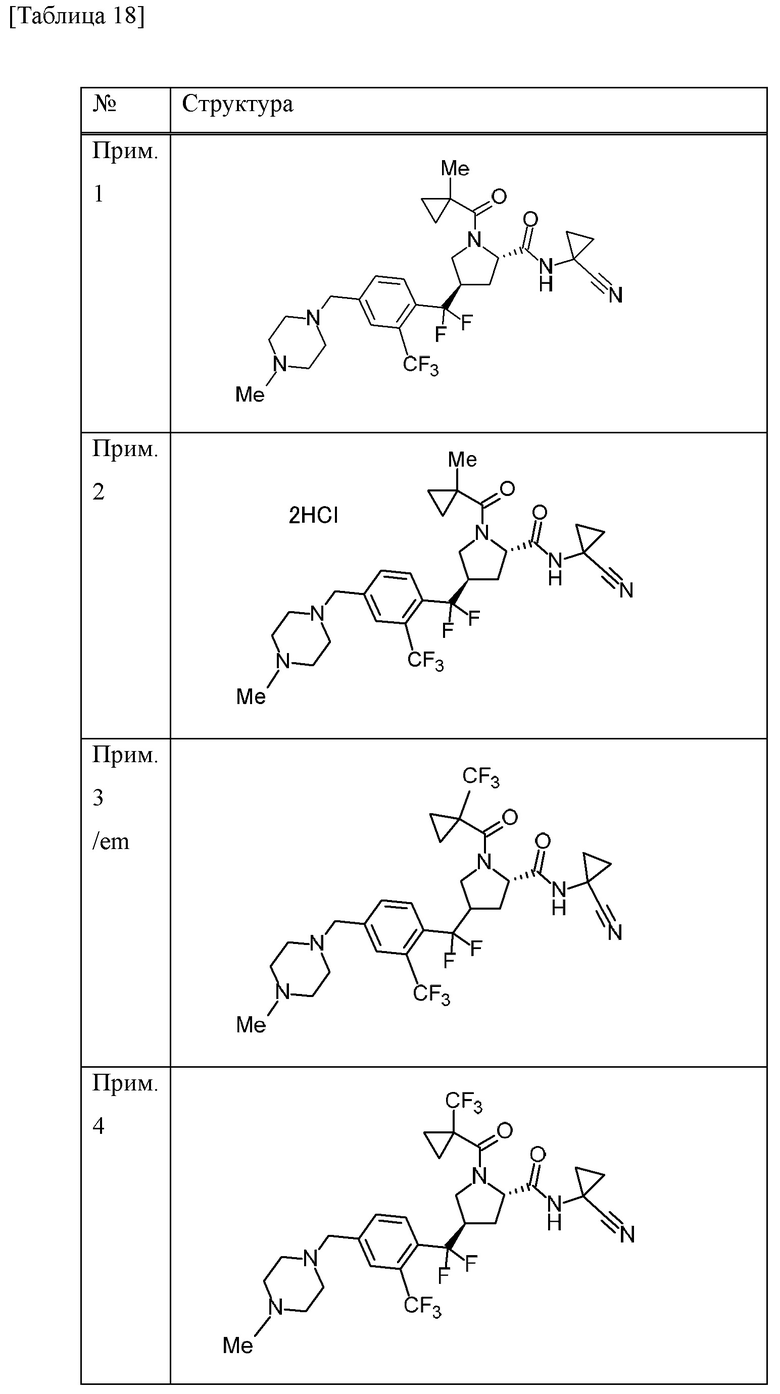
[0102]
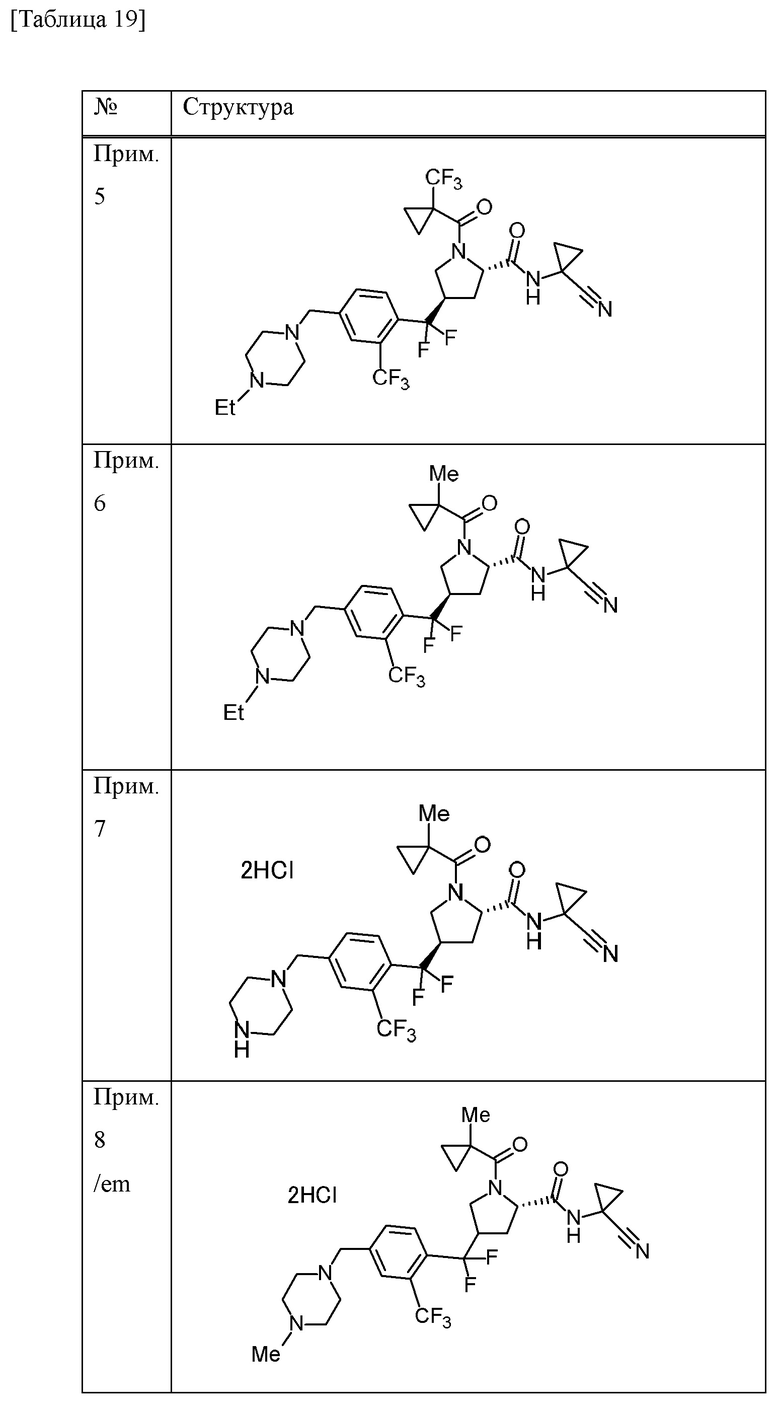
[0103]
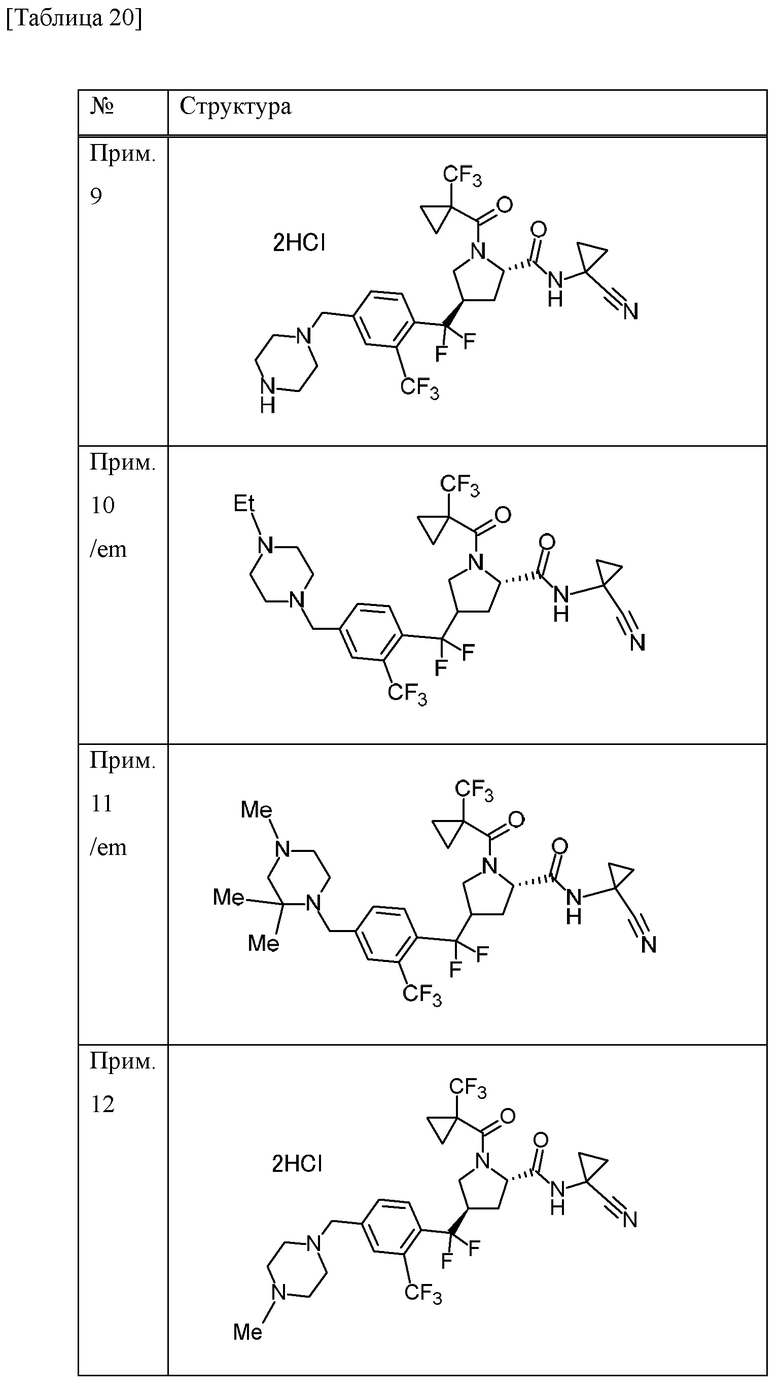
[0104]
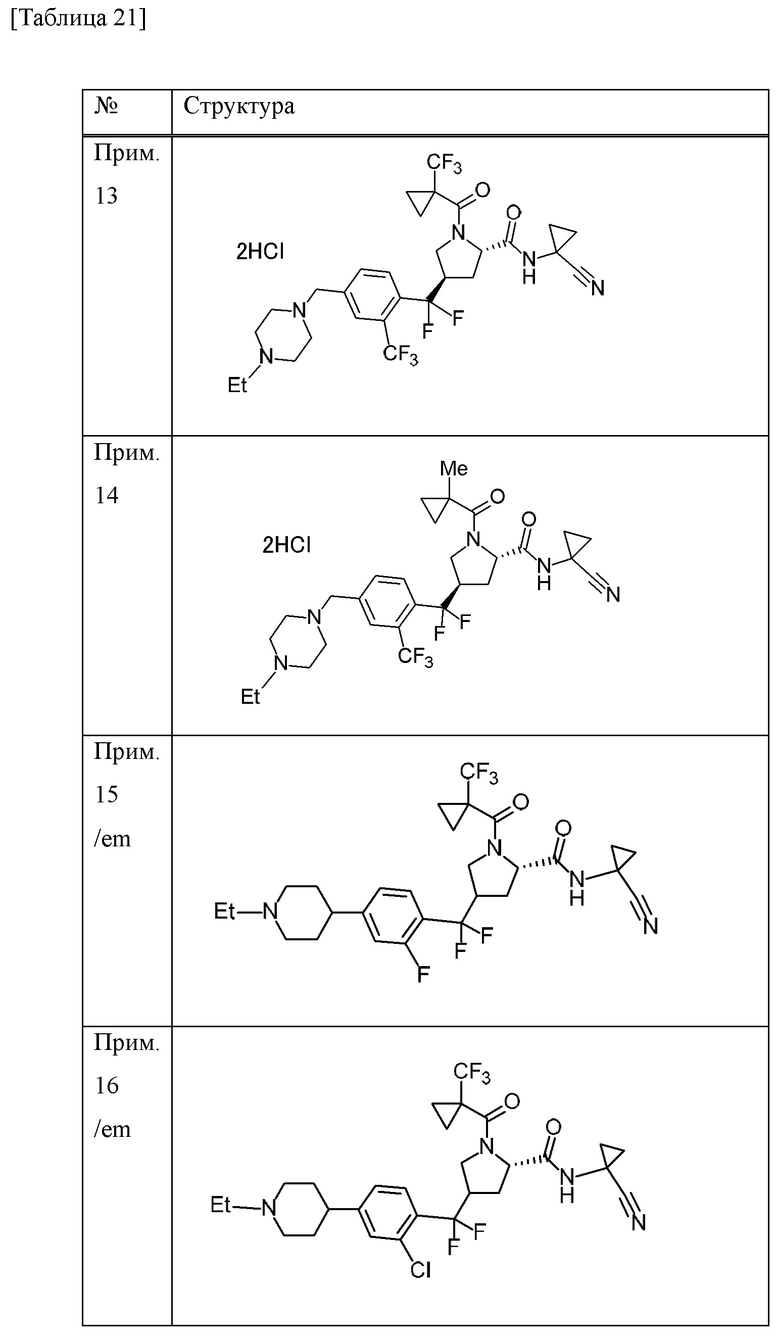
[0105]
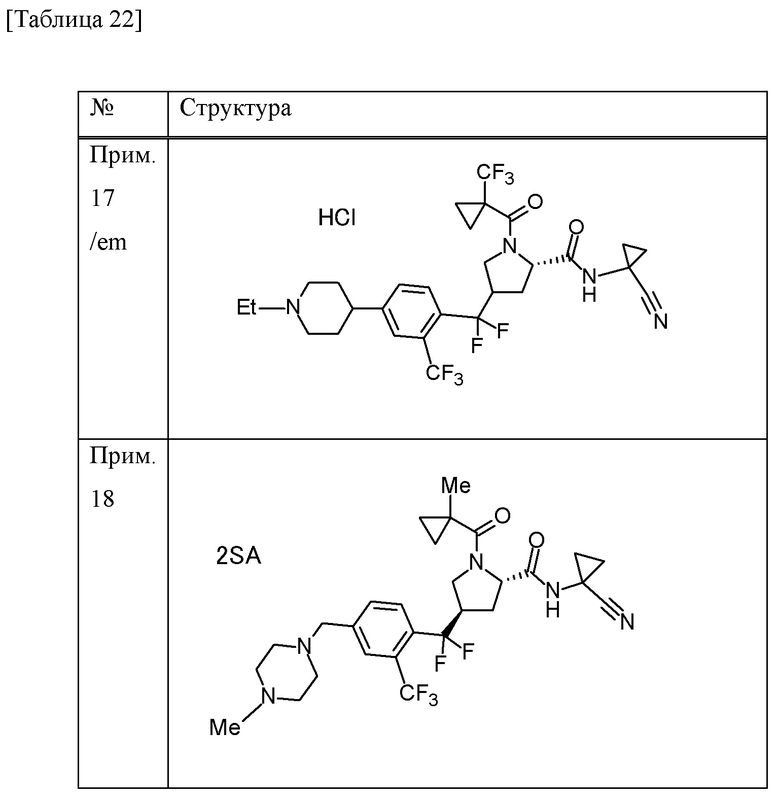
[0106]
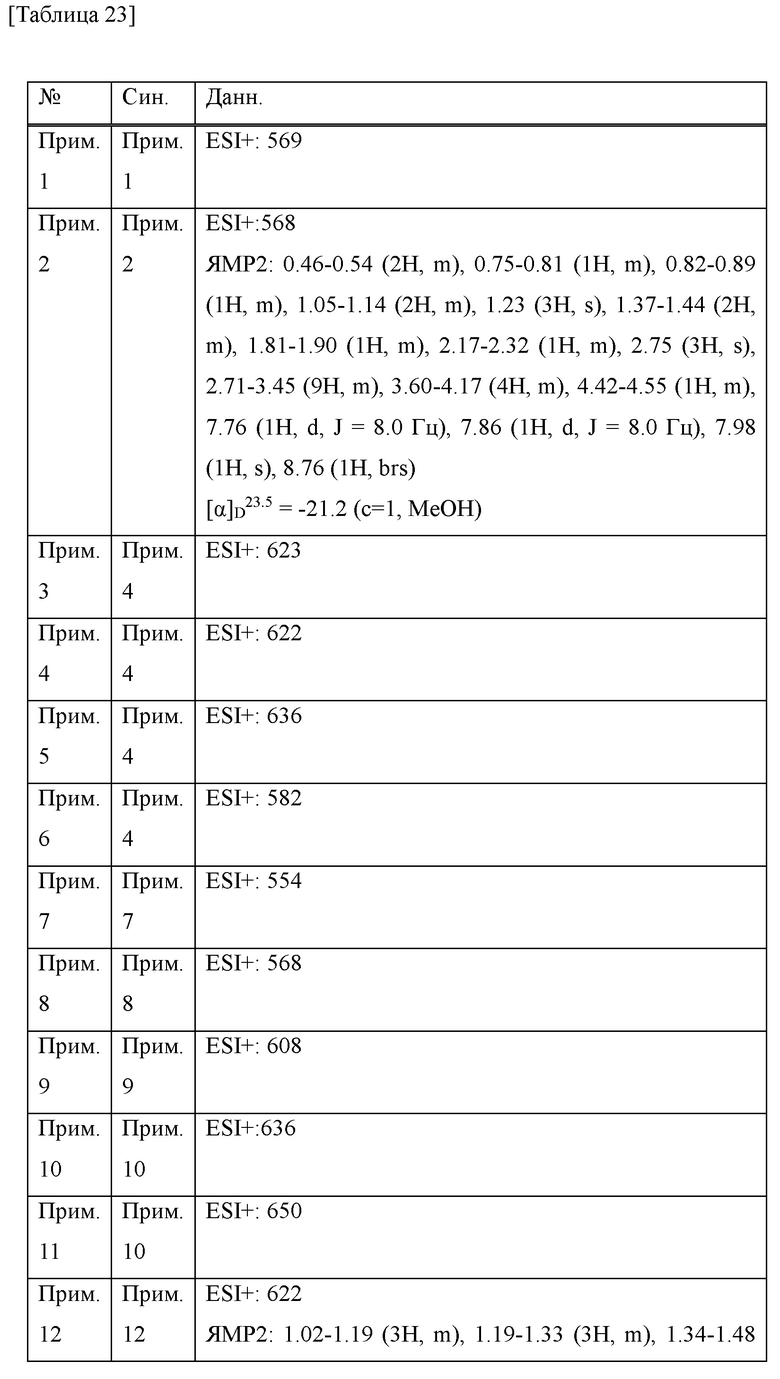

[0107]
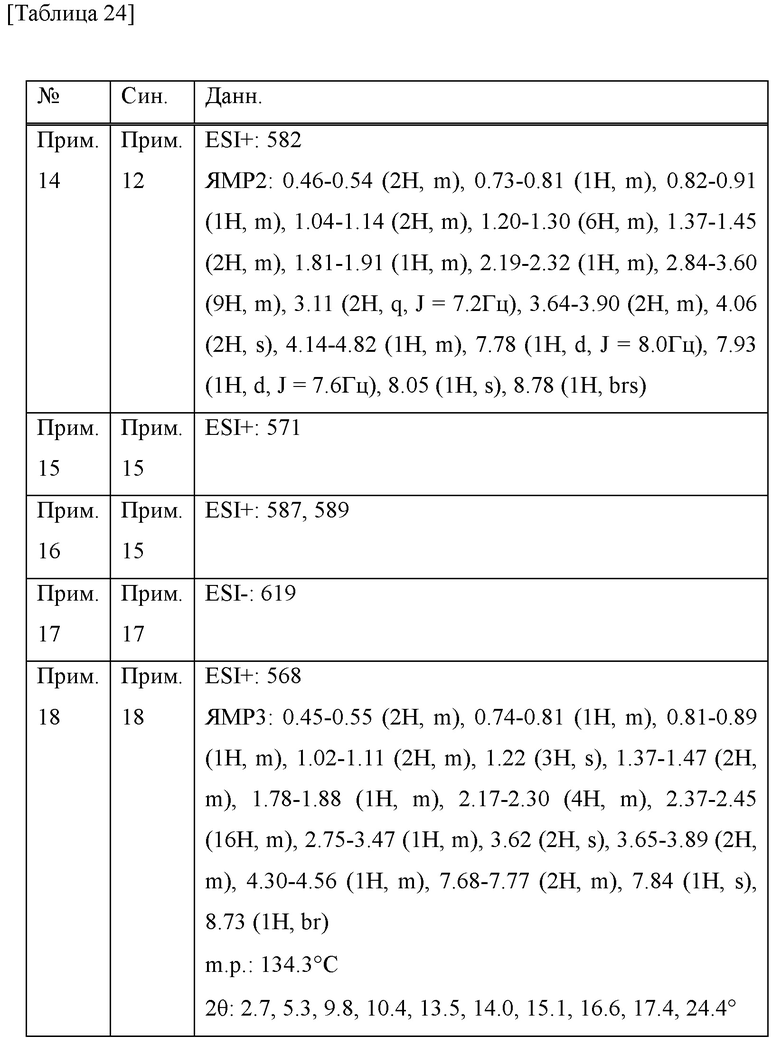
Промышленная применимость
[0108]
Фенилдифторметилзамещенное соединение пролинамида по настоящему изобретению оказывает ингибирующее действие на катепсин S и ожидается в качестве средства для профилактики и/или лечения аутоиммунного заболевания, включая в себя SLE и волчаночный нефрит, аллергии или отторжения трансплантата органа, костного мозга или ткани.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ КАТЕПСИН-ЦИСТЕИНПРОТЕАЗЫ | 2003 |
|
RU2312861C2 |
| ИНГИБИТОРЫ ЦИСТЕИНПРОТЕАЗ КАТЕПСИНОВ | 2014 |
|
RU2692799C2 |
| АГОНИСТЫ РЕЦЕПТОРА МЕЛАНОКОРТИНА | 2007 |
|
RU2411240C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ АЗЕТИДИНА | 2013 |
|
RU2629114C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ МОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ | 2019 |
|
RU2769507C1 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2014 |
|
RU2653504C2 |
| АРИЛ-N-АРИЛЬНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РНК-ВИРУСНОЙ ИНФЕКЦИИ | 2020 |
|
RU2821414C2 |
| З-АМИНОПИРИДИНЫ В КАЧЕСТВЕ АГОНИСТОВ GPBAR1 | 2012 |
|
RU2594886C2 |
| ИНГИБИТОРЫ ОКСАЗИНМОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ (MAGL) | 2019 |
|
RU2794334C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА | 2011 |
|
RU2561276C2 |
Изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, где в формуле R1 представляет собой низший алкил или галогеновый низший алкил, R2 представляет собой галоген или галогеновый низший алкил, L представляет собой связь или -CH2-, А представляет собой СН или N, R3 представляет собой Н или низший алкил, R4 и R5 одинаковые или отличаются друг от друга и представляют собой Н или низший алкил, R6 и R7 одинаковые или отличаются друг от друга и представляют собой Н, низший алкил или галоген. Также изобретение относится к фармацевтической композиции для предотвращения или лечения аутоиммунного заболевания посредством ингибирования катепсина S, содержащей эффективное количество соединения или его фармацевтически приемлемой соли соединения формулы I и одно или несколько вспомогательных веществ. Соединения формулы I предназначены для предотвращения или лечения аутоиммунного заболевания, посредством ингибирования катепсина S, причем аутоиммунное заболевание, выбрано из SLE и волчаночного нефрита, аллергии или отторжений трансплантата органа, костного мозга или ткани. Технический результат – фенилдифторметилзамещенное соединение пролинамида, оказывающее ингибирующее действие на катепсин S. 5 н. и 19 з.п. ф-лы, 24 табл., 18 пр.
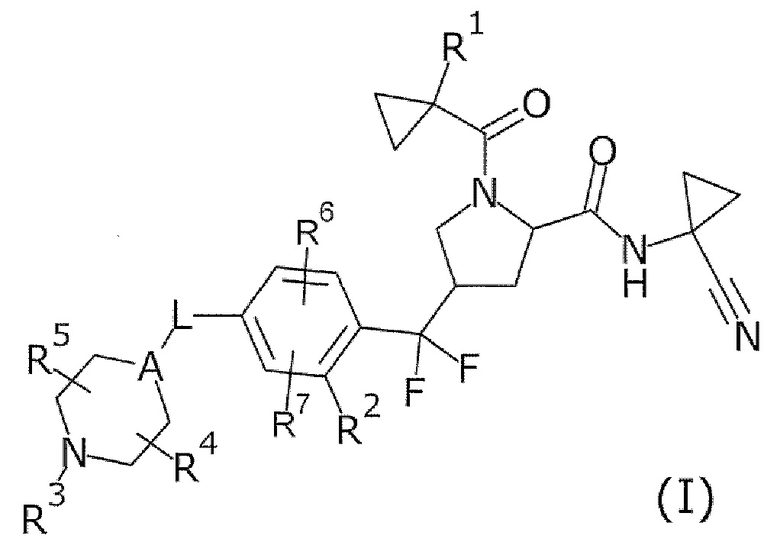
1. Соединение формулы (I) или его фармацевтически приемлемая соль:
[Хим. 15]
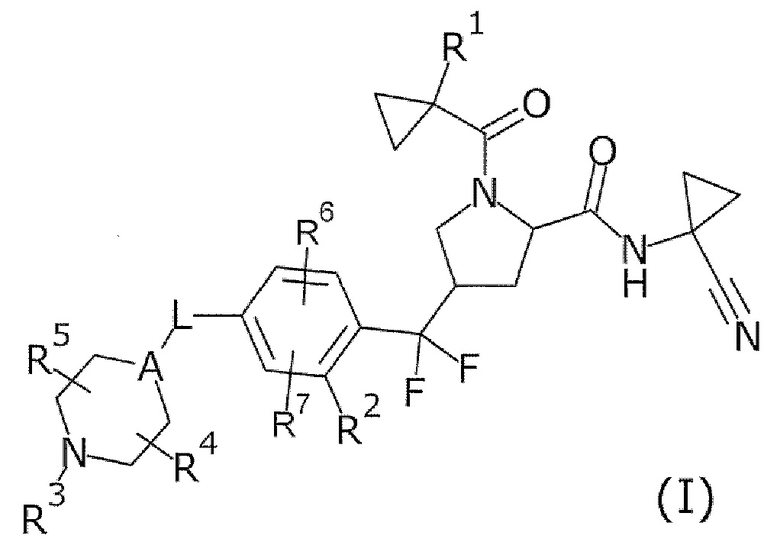
в формуле
R1 представляет собой низший алкил или галогеновый низший алкил,
R2 представляет собой галоген или галогеновый низший алкил,
L представляет собой связь или -CH2-,
А представляет собой СН или N,
R3 представляет собой Н или низший алкил,
R4 и R5 одинаковые или отличаются друг от друга и представляют собой Н или низший алкил,
R6 и R7 одинаковые или отличаются друг от друга и представляют собой Н, низший алкил или галоген.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где L представляет собой -CH2-.
3. Соединение или его фармацевтически приемлемая соль по п. 2, где R2 представляет собой галогеновый низший алкил, А представляет собой N, R3 представляет собой низший алкил, R4 представляет собой Н, R5 представляет собой Н, R6 представляет собой Н и R7 представляет собой Н.
4. Соединение или его фармацевтически приемлемая соль по п. 1, где формула (I) представляет собой формулу (Ia)
[Хим. 16]
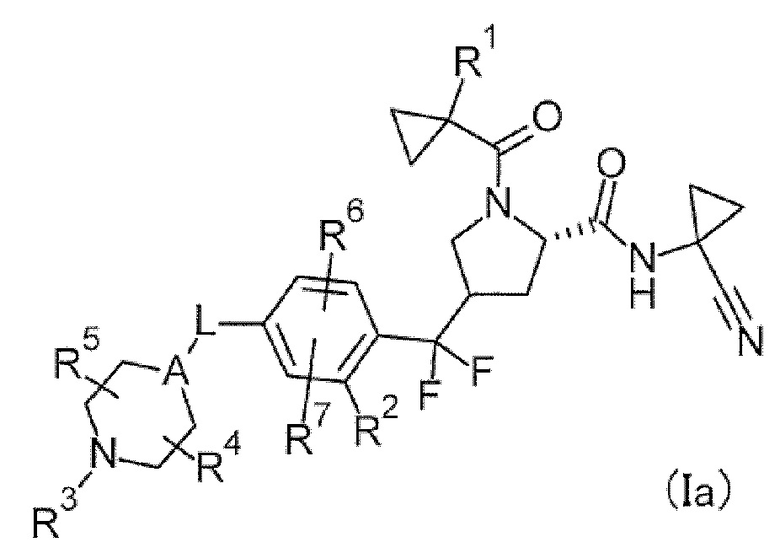
5. Соединение или его фармацевтически приемлемая соль по п. 1, где формула (I) представляет собой формулу (Ib)
[Хим. 17]
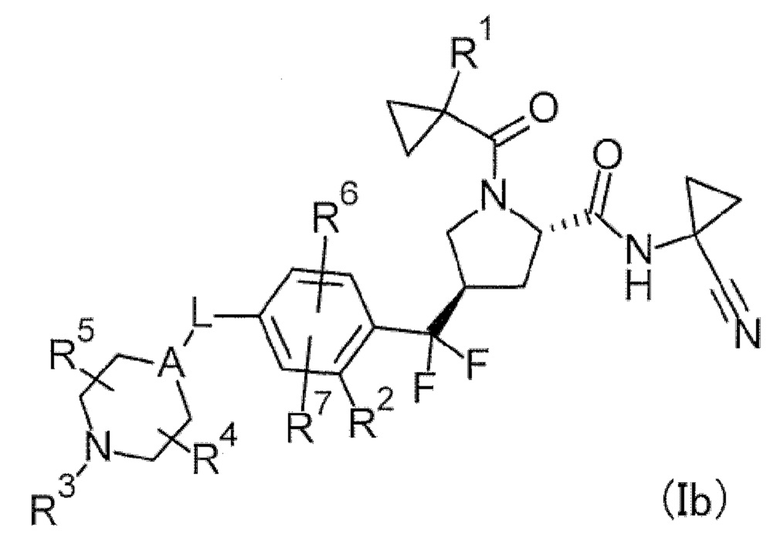
6. Соединение или его фармацевтически приемлемая соль по п. 3,
где формула (I) представляет собой формулу (Ib).
7. Соединение или его фармацевтически приемлемая соль по п. 1, которое выбрано из группы, состоящей из:
(4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-[(1-метилциклопропил)карбонил]-L-пролинамида,
(4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида,
(4R)-N-(1-цианоциклопропил)-4-[{4-[(4-этилпиперазин-1-ил)метил]-2-(трифторметил)фенил}(дифтор)метил]-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамида,
(4R)-N-(1-цианоциклопропил)-4-[{4-[(4-этилпиперазин-1-ил)метил]-2-(трифторметил)фенил}(дифтор)метил]-1-[(1-метилциклопропил)карбонил]-L-пролинамида, и
его фармацевтически приемлемая соль.
8. Соединение или его фармацевтически приемлемая соль по п. 7, которое представляет собой (4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамид или его фармацевтически приемлемую соль.
9. Соединение или его фармацевтически приемлемая соль по п. 7, которое представляет собой (4R)-N-(1-цианоциклопропил)-4-[{4-[(4-этилпиперазин-1-ил)метил]-2-(трифторметил)фенил}(дифтор)метил]-1-{[1-(трифторметил)циклопропил]карбонил}-L-пролинамид или его фармацевтически приемлемую соль.
10. Соединение или его фармацевтически приемлемая соль по п. 7, которое представляет собой (4R)-N-(1-цианоциклопропил)-4-[{4-[(4-этилпиперазин-1-ил)метил]-2-(трифторметил)фенил}(дифтор)метил]-1-[(1-метилциклопропил)карбонил]-L-пролинамид или его фармацевтически приемлемую соль.
11. Соединение или его фармацевтически приемлемая соль по п. 7, которое представляет собой (4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-[(1-метилциклопропил)карбонил]-L-пролинамид или его фармацевтически приемлемую соль.
12. Соединение или его фармацевтически приемлемая соль по п. 11, которое представляет собой (4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-[(1-метилциклопропил)карбонил]-L-пролинамида дисукцинат.
13. Соединение или его фармацевтически приемлемая соль по п. 12, которое представляет собой кристалл (4R)-N-(1-цианоциклопропил)-4-(дифтор{4-[(4-метилпиперазин-1-ил)метил]-2-(трифторметил)фенил}метил)-1-[(1-метилциклопропил)карбонил]-L-пролинамида дисукцината.
14. Соединение или его фармацевтически приемлемая соль по п. 13, которое представляет собой кристалл, характеризующийся пиками приблизительно 2θ(°) 2,7, 5,3, 9,8, 10,4, 13,5, 14,0, 15,1, 16,6, 17,4 и 24,4 порошковой рентгеновской дифракцией при использовании Cu лампы.
15. Соединение или его фармацевтически приемлемая соль по п. 14, которое представляет собой кристалл, характеризующийся эндотермическим пиком 134,3°С дифференциальной сканирующей калориметрии.
16. Фармацевтическая композиция для предотвращения или лечения аутоиммунного заболевания посредством ингибирования катепсина S, содержащая эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-15 и одно или несколько вспомогательных веществ.
17. Фармацевтическая композиция по п. 16, причем аутоиммунное заболевание, выбрано из SLE и волчаночного нефрита, аллергии или отторжений трансплантата органа, костного мозга или ткани.
18. Фармацевтическая композиция по п. 16, причем соединение или его фармацевтически приемлемая соль представляет собой соединение или его фармацевтически приемлемую соль по п. 7.
19. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-15 для изготовления фармацевтической композиции для предотвращения или лечения аутоиммунного заболевания, посредством ингибирования катепсина S, причем аутоиммунное заболевание выбрано из SLE и волчаночного нефрита, аллергии или отторжений трансплантата органа, костного мозга или ткани.
20. Применение по п. 19, причем соединение или его фармацевтически приемлемая соль представляет собой соединение или его фармацевтически приемлемую соль по п. 7.
21. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-15 для предотвращения или лечения аутоиммунного заболевания, посредством ингибирования катепсина S, причем аутоиммунное заболевание выбрано из SLE и волчаночного нефрита, аллергии или отторжений трансплантата органа, костного мозга или ткани.
22. Применение по п. 21, причем соединение или его фармацевтически приемлемая соль представляет собой соединение или его фармацевтически приемлемую соль по п. 7.
23. Способ предотвращения или лечения аутоиммунного заболевания, посредством ингибирования катепсина S, причем аутоиммунное заболевание, выбрано из SLE и волчаночного нефрита, аллергии или отторжений трансплантата органа, костного мозга или ткани, причем способ включает в себя введение субъекту эффективного количества соединения или его соли по любому из пп. 1-15.
24. Способ по п. 23, причем соединение или его фармацевтически приемлемая соль представляет собой соединение или его фармацевтически приемлемую соль по п. 7.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| US 20160244417 A1, 25.08.2016 | |||
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА | 2011 |
|
RU2548684C2 |
| Lee-Dutra, A., Wiener, D | |||
| K., & Sun, S.: "Cathepsin S inhibitors: 2004 - 2010", Expert Opinion on Therapeutic Patents, 2011, v.21(3), p.311-337 | |||
| Hardegger, L | |||
| A., | |||

