Область техники, к которой относится изобретение
[0001]
Настоящее изобретение относится к фармацевтической композиции, содержащей 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль и гидроксипропил-β-циклодекстрин, в частности, к фармацевтической композиции для перорального введения.
Предпосылки создания изобретения
[0002]
Циклодекстрины и их производные широко известны как эксципиент для улучшения растворимости гидрофобного соединения в воде. Однако большинство из них представляют собой жидкие составы, в которых гидрофобное соединение, все или его часть, включено в циклодекстрин. Поэтому твердый состав, в который физически добавлен циклодекстрин или его производное для улучшения растворимости, недостаточно хорошо известен.
[0003]
На сегодняшний день, в качестве соединения, обладающего превосходной ингибирующей активностью в отношении c-Met/VEGFR2 и проявляющего противоопухолевую активность, было сообщение о 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамиде (далее также называемом "соединение 1") (PTL 1 и 2 и NPL 1 и 2). Также сообщалось, что соединение 1 полезно в качестве терапевтического средства от остеопороза (PTL 3). Кроме того, также сообщалось о мезилатах соединения 1 и их кристаллах (PTL 4).
[0004]
Однако в этих сообщениях не упоминается фармацевтическая композиция, содержащая соединение 1 или его фармацевтически приемлемую соль и циклодекстрин или его производное.
Перечень цитируемых документов
Патентная литература
[0005]
PTL 1: WO 2009/125597
PTL 2: WO 2013/100014
PTL 3: WO 2015/046484
PTL 4: WO 2016/175305
Непатентная литература
[0006]
NPL 1: Molecular Cancer Therapeutics; 12(12); pp. 2685-96, 2013
NPL 2: European Journal of Cancer; 48(6); p. 94; 2012
Сущность изобретения
Техническая задача
[0007]
Настоящее изобретение обеспечивает фармацевтическую композицию, которая обладает отличной стабильностью, распадаемостью и абсорбируемостью, которую можно легко получить, и которая содержит соединение 1 или его фармацевтически приемлемую соль.
Решение задачи
[0008]
В свете этого, автор настоящего изобретения обнаружил, что путем добавления гидроксипропил-β-циклодекстрина (HP-β-CD) к соединению 1 или его фармацевтически приемлемой соли можно получить фармацевтическую композицию, которая обладает отличной стабильностью, обладает отличной стабильностью, распадаемостью и абсорбируемостью и ее можно легко получить, и таким образом, создал настоящее изобретение.
[0009]
Точнее, настоящее изобретение относится к следующим [1]-[35].
[1] Фармацевтическая композиция, содержащая 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль и гидроксипропил-β-циклодекстрин.
[2] Фармацевтическая композиция в соответствии с пунктом [1], где композиция характеризуется пиками при по меньшей мере 5 или более углах дифракции 2θ (±0,2°), выбранных из 6,5, 7,8, 9,6, 12,4, 18,8, 21,2, 23,0, 24,5 и 26,0 (А°), определенные методом порошковой рентгеновской дифрактометрии.
[3] Фармацевтическая композиция в соответствии с пунктом [1] или [2], где композиция характеризуется пиками при углах дифракции 2θ (±0,2°) 6,5, 7,8, 9,6, 12,4, 18,8, 21,2, 23,0, 24,5 и 26,0 (А°), определенные методом порошковой рентгеновской дифрактометрии.
[4] Фармацевтическая композиция в соответствии с любым из пунктов [1]-[3], где композиция характеризуется пиками при значениях химических сдвигов [δ (м.д.)] 162,6, 130,4, 103,1, 82,7, 73,3, 41,9 и 19,9 в 13С-ЯМР твердого тела.
[5] Фармацевтическая композиция в соответствии с любым из пунктов [1]-[4], где композиция характеризуется пиками при по меньшей мере 5 или более полосах поглощения, выбранных из 1663, 1352, 1225, 1156, 1032, 720 и 553 (см-1) в инфракрасном спектре поглощения.
[6] Фармацевтическая композиция в соответствии с любым из пунктов [1]-[5], где гидроксипропил-β-циклодекстрин содержится в количестве 0,1-5,5 массовых частей в расчете на 1 массовую часть 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамида или его фармацевтически приемлемой соли.
[7] Фармацевтическая композиция в соответствии с любым из пунктов [1]-[6], дополнительно содержащая производное кремниевой кислоты.
[8] Фармацевтическая композиция в соответствии с любым из пунктов [1]-[7], дополнительно содержащая производное целлюлозы.
[9] Фармацевтическая композиция в соответствии с любым из пунктов [1]-[8], где фармацевтическая композиция представляет собой таблетку или гранулу.
[10] Фармацевтическая композиция в соответствии с любым из пунктов [1]-[9], где фармацевтическая композиция предназначена для перорального введения.
[11] Фармацевтическая композиция в соответствии с любым из пунктов [1]-[10], где фармацевтическая композиция представляет собой таблетку.
[12] Фармацевтическая композиция в соответствии с любым из пунктов [1]-[10], где фармацевтическая композиция представляет собой таблетку, имеющую максимальный диаметр 5 мм или меньше.
[13] Фармацевтическая композиция, содержащая 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль и гидроксипропил-β-циклодекстрин, при этом фармацевтическая композиция получена путем физического смешивания.
[14] Фармацевтическая композиция в соответствии с пунктом [13], где физическое смешивание представляет собой способ получения, который не включает стадию, на которой 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль преобразовывают в состояние раствора при получении фармацевтической композиции.
[15] Фармацевтическая композиция в соответствии с пунктом [13] или [14], где физическое смешивание представляет собой смешивание или гранулирование.
[16] Фармацевтическая композиция в соответствии с любым из пунктов [13]-[15], где физическое смешивание представляет собой смешивание, метод сухого гранулирования или метод влажного гранулирования.
[17] Фармацевтическая композиция в соответствии с любым из пунктов [13]-[16], где физическое смешивание представляет собой смешивание, способ гранулирования путем дробления, способ гранулирования в псевдоожиженном слое, способ гранулирования во вращающемся слое, способ экструзионного гранулирования или способ гранулирования с большим усилием сдвига.
[18] Фармацевтическая композиция в соответствии с любым из пунктов [13]-[17], где физическое смешивание представляет собой способ гранулирования в псевдоожиженном слое.
[19] Фармацевтическая композиция в соответствии с любым из пунктов [13]-[18], где композиция характеризуется пиками при по меньшей мере 5 или более углах дифракции 2θ (±0,2°), выбранными из 6,5, 7,8, 9,6, 12,4, 18,8, 21,2, 23,0, 24,5 и 26,0 (А°), определенными методом порошковой рентгеновской дифрактометрии.
[20] Фармацевтическая композиция в соответствии с любым из пунктов [13]-[19], где композиция характеризуется пиками при углах дифракции 2θ (±0,2°) 6,5, 7,8, 9,6, 12,4, 18,8, 21,2, 23,0, 24,5 и 26,0 (А°), определенными методом порошковой рентгеновской дифрактометрии.
[21] Фармацевтическая композиция в соответствии с любым из пунктов [13]-[20], где композиция характеризуется пиками при значениях химических сдвигов [δ (м.д.)] 162,6, 130,4, 103,1, 82,7, 73,3, 41,9 и 19,9 в 13С-ЯМР твердого тела.
[22] Фармацевтическая композиция в соответствии с любым из пунктов [13]-[21], где композиция характеризуется пиками при по меньшей мере 5 или более полосах поглощения, выбранных из 1663, 1352, 1225, 1156, 1032, 720 и 553 (см-1) в инфракрасном спектре поглощения.
[23] Фармацевтическая композиция в соответствии с любым из пунктов [13]-[22], где гидроксипропил-β-циклодекстрин содержится в количестве 0,1-5,5 массовых частей в расчете на 1 массовую часть 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамида или его фармацевтически приемлемой соли.
[24] Фармацевтическая композиция в соответствии с любым из пунктов [13]-[23], дополнительно содержащая производное кремниевой кислоты.
[25] Фармацевтическая композиция в соответствии с любым из пунктов [13]-[24], дополнительно содержащая производное целлюлозы.
[26] Фармацевтическая композиция в соответствии с любым из пунктов [13]-[25], где фармацевтическая композиция представляет собой гранулу.
[27] Фармацевтическая композиция в соответствии с любым из пунктов [13]-[26], где фармацевтическая композиция предназначена для перорального введения.
[28] Фармацевтическая композиция в соответствии с любым из пунктов [13]-[27], где фармацевтическая композиция представляет собой таблетку.
[29] Фармацевтическая композиция в соответствии с любым из пунктов [13]-[28], где фармацевтическая композиция представляет собой таблетку, имеющую максимальный диаметр 5 мм или меньше.
[30] Способ получения фармацевтической композиции, которую можно получить путем осуществления физического смешивания 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамида или его фармацевтически приемлемой соли и гидроксипропил-β-циклодекстрина.
[31] Способ получения в соответствии с пунктом [30], где физическое смешивание представляет собой способ получения, который не включает стадию, на которой 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль преобразовывают в состояние раствора при получении фармацевтической композиции.
[32] Способ получения в соответствии с пунктом [30] или [31], где физическое смешивание представляет собой смешивание или гранулирование.
[33] Способ получения в соответствии с любым из пунктов [30]-[32], где физическое смешивание представляет собой смешивание, метод сухого гранулирования или метод влажного гранулирования.
[34] Способ получения в соответствии с любым из пунктов [30]-[33], где физическое смешивание представляет собой смешивание, метод гранулирования путем дробления, метод гранулирования в псевдоожиженном слое, метод гранулирования во вращающемся слое, метод экструзионного гранулирования или метод гранулирования с большим усилием сдвига.
[35] Способ получения в соответствии с любым из пунктов [30]-[34], где физическое смешивание представляет собой метод гранулирования в псевдоожиженном слое.
[0010]
Настоящее изобретение также относится к следующим аспектам.
- Фармацевтическая композиция для профилактики и/или лечения опухоли, содержащая 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль и гидроксипропил-β-циклодекстрин.
- Противоопухолевое средство, содержащее 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль и гидроксипропил-β-циклодекстрин.
- Применение 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамида или его фармацевтически приемлемой соли и гидроксипропил-β-циклодекстрина для получения противоопухолевого средства.
- Способ профилактики и/или лечения опухоли, включающий стадию введения субъекту фармацевтической композиции, содержащей 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль и гидроксипропил-β-циклодекстрин, в эффективном количестве для лечения и/или профилактики.
- Применение фармацевтической композиции, содержащей 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль и гидроксипропил-β-циклодекстрин, для профилактики и/или лечения опухоли.
- Фармацевтическая композиция для ингибирования с-Met и/или VEGFR2, содержащая 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль и гидроксипропил-β-циклодекстрин.
- Ингибитор для с-Met и/или VEGFR2, содержащий 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль и гидроксипропил-β-циклодекстрин.
- Применение 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамида или его фармацевтически приемлемой соли и гидроксипропил-β-циклодекстрина для получения ингибитора для с-Met и/или VEGFR2.
- Применение фармацевтической композиции, содержащей 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль и гидроксипропил-β-циклодекстрин для ингибирования с-Met и/или VEGFR2.
Этих аспекты могут включать указанные выше характерные признаки изобретения, представленного в настоящей заявке.
Полезные эффекты изобретения
[0011]
В соответствии с настоящим изобретением, предоставляется фармацевтическая композиция, которая обладает отличной стабильностью, распадаемостью и абсорбируемостью, которую можно легко получить и которая содержит соединение 1 или его фармацевтически приемлемую соль и производное циклодекстрина.
Краткое описание чертежей
[0012]
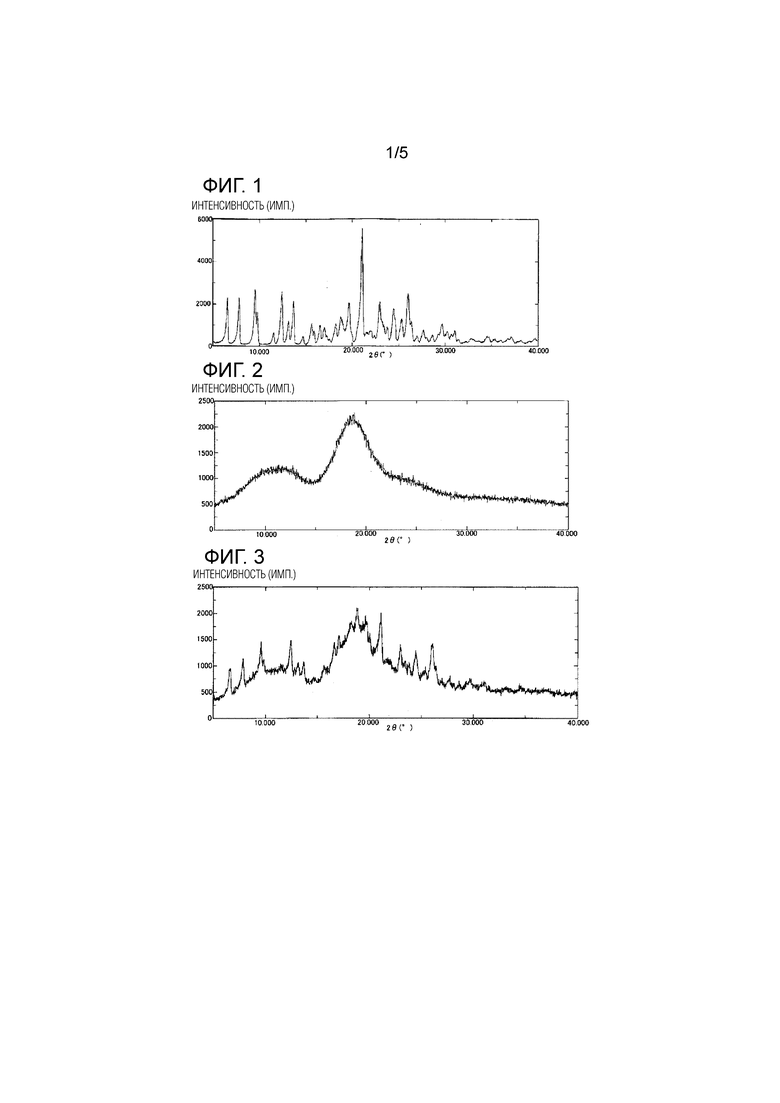
[Фиг. 1] Фиг. 1 представляет результаты измерения методом порошковой рентгеновской дифракционной спектроскопии (XRD) мезилатной соли соединения 1.
[Фиг. 2] Фиг. 2 представляет результаты XRD измерения НР-β-CD.
[Фиг. 3] Фиг. 3 представляет результаты XRD измерения физически смешанного продукта мезилатной соли соединения 1 и НР-β-CD.
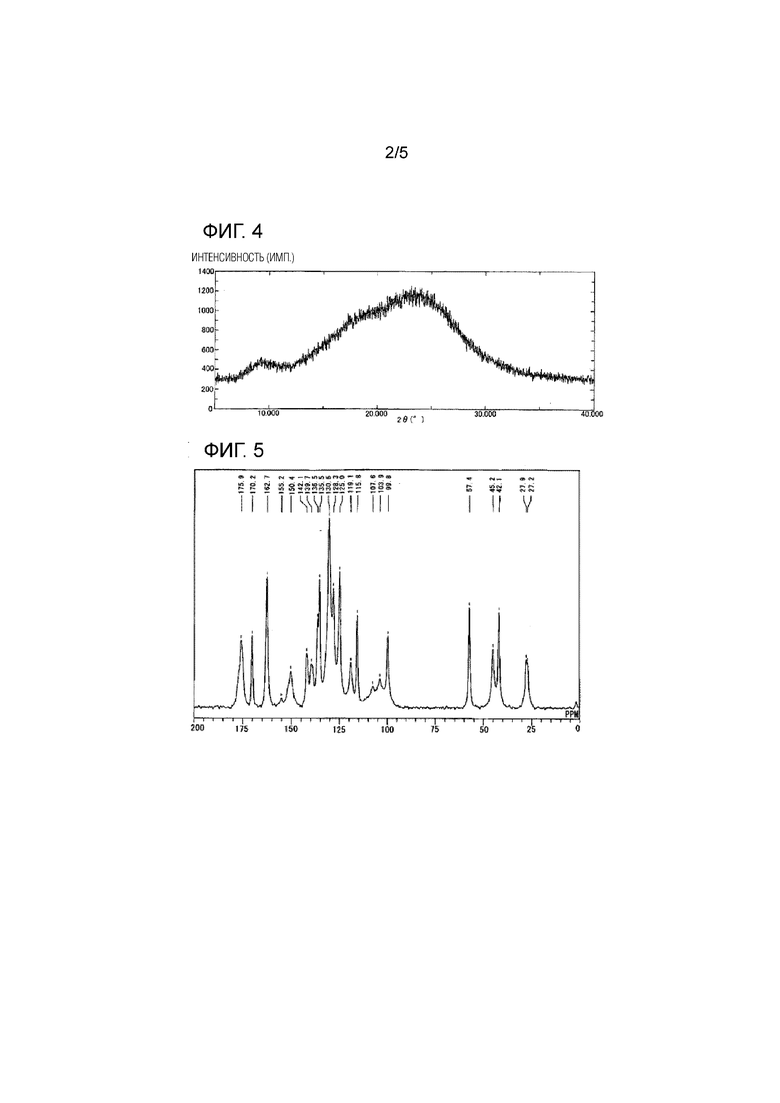
[Фиг. 4] Фиг. 4 представляет результаты XRD измерения высушенного распылением продукта мезилатной соли соединения 1 и HP-β-CD.
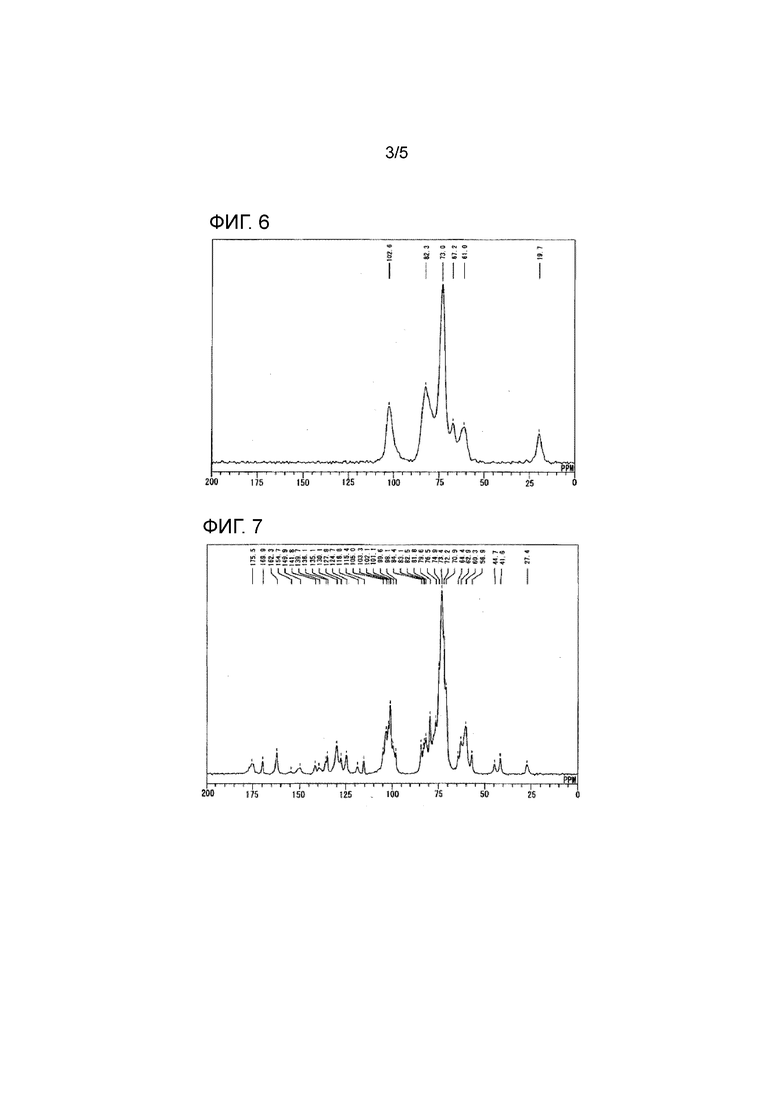
[Фиг. 5] Фиг. 5 представляет результаты измерения методом твердотельного протонного ядерного магнитного резонанса (13С-ЯМР) мезилатной соли соединения 1.
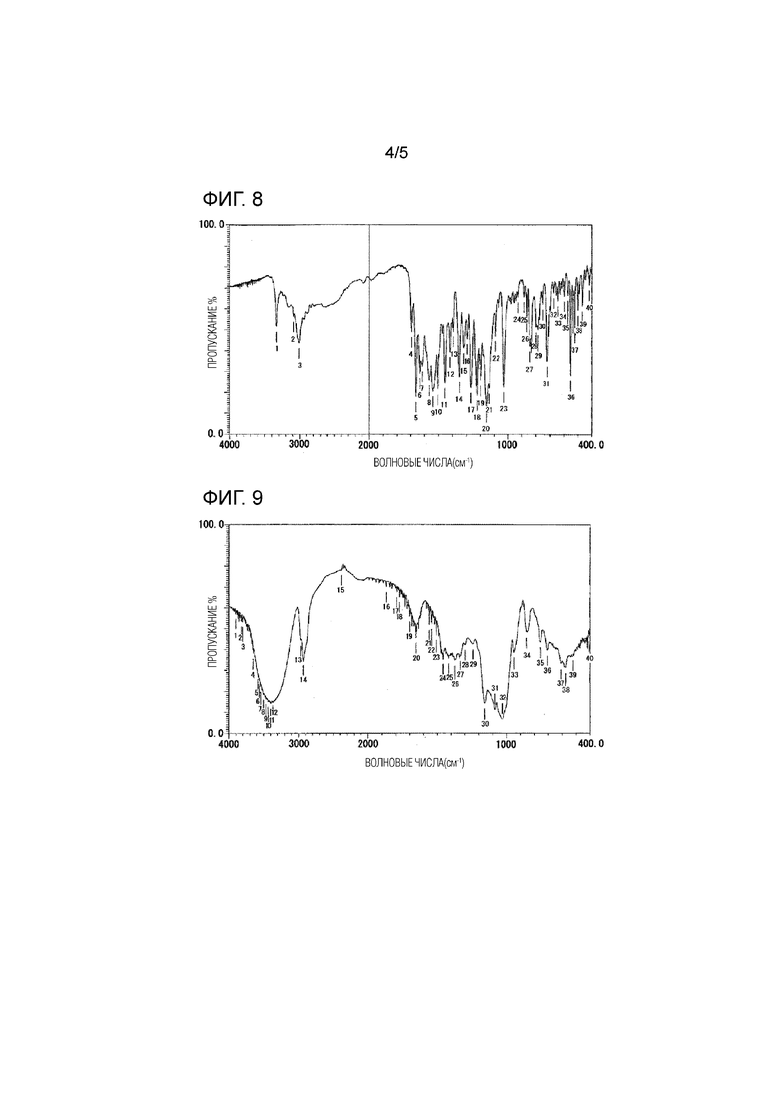
[Фиг. 6] Фиг. 6 представляет результаты 13С-ЯМР измерения HP-β-CD.
[Фиг. 7] Фиг. 7 представляет результаты 13С-ЯМР измерения физически смешанного продукта мезилатной соли соединения 1 и HP-β-CD.
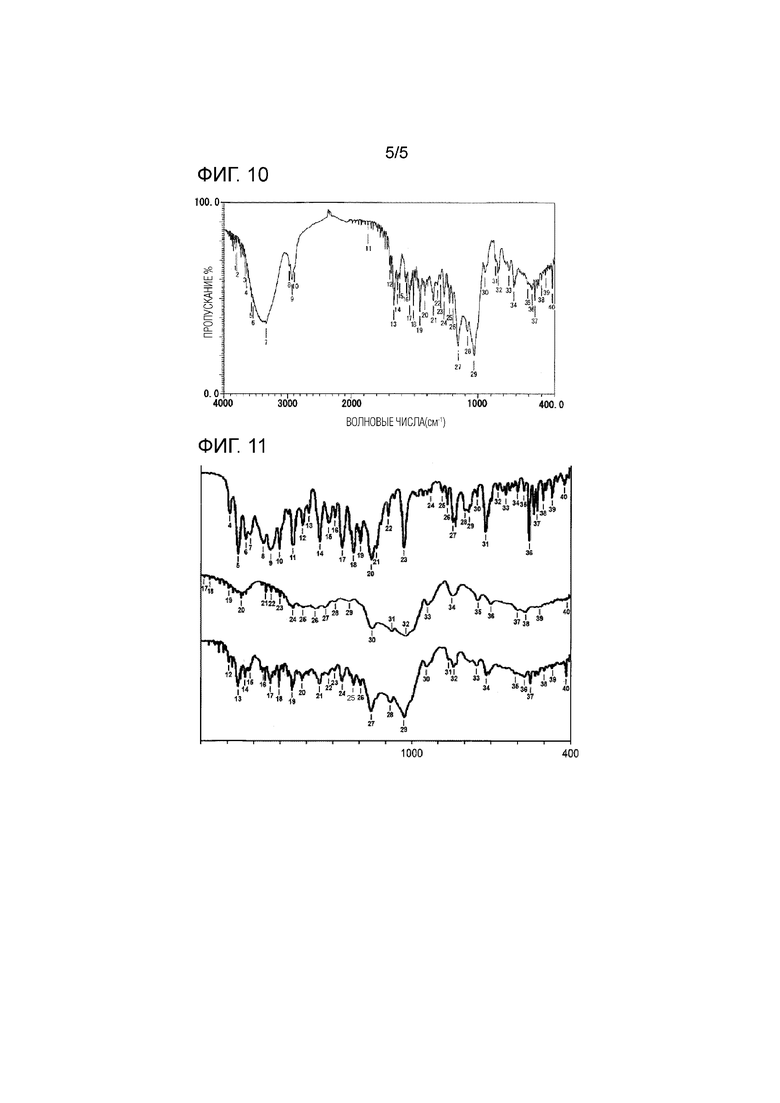
[Фиг. 8] Фиг. 8 представляет результаты измерения методом инфракрасной абсорбционной спектроскопии (ИК) мезилатной соли соединения 1.
[Фиг. 9] Фиг. 9 представляет результаты ИК-измерения HP-β-CD.
[Фиг. 10] Фиг. 10 представляет результаты ИК-измерения физически смешанного продукта мезилатной соли соединения 1 и HP-β-CD.
[Фиг. 11] Фиг. 11 представляет результаты ИК-измерения в характеристических областях мезилатной соли соединения 1, HP-β-CD и физически смешанного продукта мезилатной соли соединения 1 и НР-β-CD в указанном порядке сверху.
Описание вариантов осуществления
[0013]
Активным ингредиентом фармацевтической композиции по настоящему изобретению является соединение 1. Соединение 1 представляет собой 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид, и его структура показана ниже.
[0014]
[Химическая формула 1]
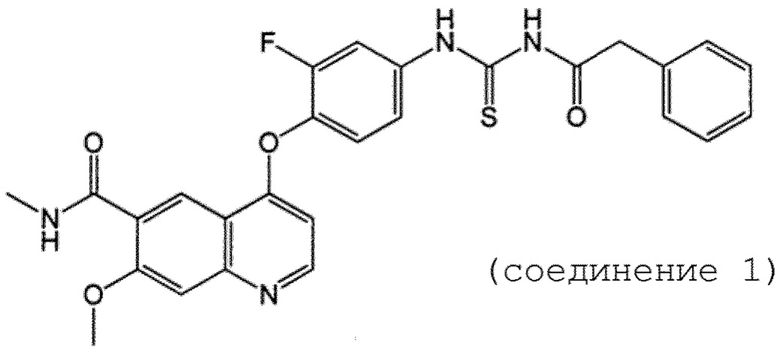
[0015]
В качестве фармацевтически приемлемой соли соединения 1 для использования в настоящем изобретении можно указать такие соли, как кислотно-аддитивные соли, и предпочтительной является мезилатная соль, и более предпочтительным является моно-мезилат.
[0016]
Соединение 1 или его фармацевтически приемлемая соль может представлять собой сольват (например, гидрат или т.п.) или несольватированную форму, и в настоящем изобретении обе формы включены в "соединение 1 или его фармацевтически приемлемую соль". Соединение 1 или его фармацевтически приемлемую соль можно получить способом, описанным, например, в PTL 1 или 4.
[0017]
Соединение 1 или его фармацевтически приемлемая соль для использования в настоящем изобретении содержится в количестве предпочтительно 67% масс. или меньше, более предпочтительно от 5 до 40% масс., еще более предпочтительно от 10 до 20% масс., в расчете на общую массу фармацевтической композиции.
[0018]
В производное циклодекстрина включены не только α-циклодекстрин (α-CD), β-циклодекстрин (β-CD) и γ-циклодекстрин (γ-CD), но также гидроксипропил-β-циклодекстрин (HP-β-CD), сульфобутиловый эфир-β-циклодекстрин (SBE-β-CD) и т.п. Однако, с точки зрения растворимости, стабильности, абсорбирумости, и т.д., производное циклодекстрина по настоящему изобретению представляет собой гидроксипропил-β-циклодекстрин. Количество гидроксипропил-β-циклодекстрина для использования в настоящем изобретении может быть любым при условии, что абсорбирумость соединения 1 или его фармацевтически приемлемой соли, которые можно использовать в настоящем изобретении, улучшается и токсичность гидроксипропил-β-циклодекстрина не появляется.
[0019]
Количество гидроксипропил-β-циклодекстрина для использования в настоящем изобретении предпочтительно составляет 30% масс. или больше, более предпочтительно от 60 до 95% масс., еще более предпочтительно от 76 до 85% масс., в расчете на общую массу фармацевтической композиции.
[0020]
Количество гидроксипропил-β-циклодекстрина для использования в настоящем изобретении предпочтительно составляет от 0,1 до 5,5 массовых частей, более предпочтительно от 4,0 до 5,0 массовых частей, в расчете на 1 массовую часть соединения 1 или его фармацевтически приемлемой соли.
[0021]
В качестве примера фармацевтической композиции по настоящему изобретению можно указать композицию, в которой соединение 1 или его фармацевтически приемлемая соль и гидроксипропил-β-циклодекстрин не образуют клатрат или частично образуют клатрат. Например, методом порошковой рентгеновской дифракционной спектроскопии, ЯМР твердого тела, ИК и т.д. можно подтвердить, что в фармацевтической композиции по настоящему изобретению присутствует композиция, в которой соединение 1 или его фармацевтически приемлемая соль и гидроксипропил-β-циклодекстрин не образуют клатрат.
[0022]
Кроме того, ошибка пика при угле дифракции 2θ в спектре рентгеновской порошковой дифракции в настоящем изобретении составляет около ±0,2°. Это ошибка, вызванная оборудованием, используемым для измерения, получением образца, методом анализа данных или т.п. Поэтому, когда кристалл подвергают XRD измерению в настоящем изобретении, учитывается ошибка ±0,2° полученного дифракционного угла 2θ. Кроме того, по той же причине в настоящем изобретении ошибка пика при химическом сдвиге (м.д.) в спектре твердотельного 13С-ЯМР составляет около ±1,0 м.д., и ошибка пика при полосе поглощения (см-1) в инфракрасном спектре поглощения составляет около ±2 см-1.
[0023]
Фармацевтическая композиция по настоящему изобретению характеризующаяся пиками при по меньшей мере 5 или более углах дифракции (2θ±0,2°), выбранными из 6,5, 7,8, 9,6, 12,4, 18,8, 21,2, 23,0, 24,5 и 26,0 (А°), определенными методом порошковой рентгеновской дифрактометрии, предпочтительно характеризуется пиками при углах дифракции (2θ±0,2°) 6,5, 7,8, 9,6, 12,4, 18,8, 21,2, 23,0, 24,5 и 26,0 (А°).
[0024]
Фармацевтическая композиция по настоящему изобретению предпочтительно характеризуется пиками при значениях химических сдвигов [δ (м.д.)] 162,6, 130,4, 103,1, 82,7, 73,3, 41,9 и 19,9 в спектре твердотельного 13С-ЯМР.
[0025]
Фармацевтическая композиция по настоящему изобретению предпочтительно характеризуется пиками при по меньшей мере 5 или более полосах поглощения, выбранными из 1663, 1352, 1225, 1156, 1032, 720 и 553 (см-1) в инфракрасном спектре поглощения, более предпочтительно характеризуется пиками при полосах поглощения 1663, 1352, 1225, 1156, 1032, 720 и 553 (см-1).
[0026]
Фармацевтическую композицию по настоящему изобретению можно получить путем физического смешивания. Физическое смешивание относится к способу получения, который не включает стадию, на которой соединение 1 или его фармацевтически приемлемую соль преобразовывают в состояние раствора при получении фармацевтической композиции. В качестве примера физического смешивания можно указать смешивание с образованием однородной композиции путем применения соответствующей процедуры для двух или более типов твердых веществ, содержащих соединение 1 или его фармацевтически приемлемую соль, гранулирования, которое осуществляют когда соединение 1 или его фармацевтически приемлемая соль находится в твердом состоянии и т.п.
[0027]
Примеры способа гранулирования, который можно использовать при получении фармацевтической композиции по настоящему изобретению, включают способ сухого гранулирования и способ влажного гранулирования. Конкретные примеры способа сухого гранулирования включают способ гранулирования путем дробления. Кроме того, примеры способа влажного гранулирования включают способ гранулирования в псевдоожиженном слое, способ гранулирования во вращающемся слое, способ экструзионного гранулирования и способ гранулирования с большим усилием сдвига, и предпочтительным является способ гранулирования в псевдоожиженном слое.
[0028]
При физическом смешивании можно добавить растворитель, если это необходимо. В качестве примера растворителя можно указать воду, этанол, смешанный водо-этанольный раствор и т.п., и предпочтительным растворителем является вода. Когда такой растворитель используют при физическом смешивании, фармацевтическую композицию по настоящему изобретению можно использовать как таковую или после ее сушки.
[0029]
При физическом смешивании можно дополнительно добавить флюидизатор или т.п. Примеры флюидизатора включают производные кремниевой кислоты, такие как светлая безводная кремниевая кислота, силикат кальция, алюмометасиликат магния, тальк, силикат алюминия и гидратированный диоксид кремния, и предпочтительной является светлая безводная кремниевая кислота.
При физическом смешивании можно добавить связующее. В качестве примера связующего можно указать производное целлюлозы, крахмал, повидон или поливиниловый спирт. Примеры производного целлюлозы включают гидроксипропилцеллюлозу, гипромеллозу и метилцеллюлозу, и предпочтительной является гидроксипропилцеллюлоза.
[0030]
В данном случае, количество флюидизатора, используемого в настоящем изобретении, обычно составляет 10% масс. или менее, предпочтительно от 0,1 до 2% масс., более предпочтительно от 0,2 до 1% масс., в расчете на общую массу фармацевтической композиции.
[0031]
Примеры фармацевтической композиции по настоящему изобретению включают таблетку, гранулу, порошок и мелкую гранулу, и предпочтительной является таблетка или гранула. В таблетку, гранулу, порошок и мелкую гранулу включен порошкообразный гранулированный материал, который быстро растворяется в полости рта и который можно принимать без воды.
[0032]
Кроме того, в качестве фармацевтической композиции по настоящему изобретению можно указать таблетку. Таблетку также можно получить с использованием общеизвестного эксципиента, и также возможно получить таблетку из вышеуказанной гранулы с использованием общеизвестного способа. В качестве формы таблетки можно использовать обычную форму, такую как цилиндрическая форма, форма диска, линзовидная форма или стержнеобразная форма. Размер таблетки конкретно не ограничивается, при условии, что человек может принимать ее перорально, однако максимальный диаметр (диаметр) предпочтительно составляет 15 мм или менее, более предпочтительно 10 мм или менее. Кроме того, учитывая ее абсорбируемость и то, что ее вводят детям, и т.д., максимальный диаметр еще более предпочтительно составляет 5 мм или менее. Кроме того, с учетом абсорбируемости и простоты получения фармацевтической композиции по настоящему изобретению, предпочтительной является таблетка цилиндрической формы с максимальным диаметром 4 мм или менее. Нижний предел максимального диаметра таблетки конкретно не ограничивается, но обычно составляет 2 мм или более, учитывая удобство обращения с таблеткой.
[0033]
Кроме того, когда фармацевтическая композиция по настоящему изобретению представлена в виде таблетки цилиндрической формы диаметром 5 мм или менее, масса соединения 1 на таблетку составляет 10 мг или менее, с точки зрения размера и абсорбируемости таблетки. После получения таблетки иногда осуществляют процедуру упаковки множества таблеток в одну упаковку. В этом случае, если существует большая разница в диаметре и толщине цилиндрической формы, требуется время для регулирования упаковываемого количества при помощи счетной пластины с отверстиями, и во время этого процесса несколько таблеток может попасть в отверстие счетной пластины, или таблетки не могут попасть в отверстие, и, следовательно, может возникнуть избыток или недостаток упаковываемого количества. Поэтому, в случае таблетки, имеющей цилиндрическую форму диаметром 5 мм или менее, отношение толщины к диаметру цилиндрической формы обычно составляет, например, от 60 до 140%, предпочтительно от 80 до 120%.
[0034]
Кроме того, в фармацевтической композиции по настоящему изобретению, помимо соединения 1 или его фармацевтически приемлемой соли и гидроксипропил-β-циклодекстрина, при необходимости можно использовать другие эксципиенты. Эксципиенты конкретно не ограничиваются, при условии, что их обычно используют в лекарственных продуктах в области фармацевтики, и в качестве примера можно указать, например, флюидизатор, разбавитель, связующее, разрыхлитель, смазывающее вещество, покрывающий агент, краситель, отдушку, агент, маскирующий вкус, и т.п., однако она не ограничивается этим.
[0035]
Фармацевтическая композиция по настоящему изобретению полезна в качестве противоопухолевого средства, поскольку соединение 1 обладает превосходной ингибирующей активностью в отношении с-Met и ингибирующей активностью в отношении VEGFR2. Рак, который является мишенью, конкретно не ограничен, однако его примеры включают рак головы и шеи, рак желудочно-кишечного тракта [например, рак пищевода, рак желудка, желудочно-кишечные стромальные опухоли, рак двенадцатиперстной кишки, рак печени, рак желчных путей (например, рак желчного пузыря и желчного протока и т.д.), рак поджелудочной железы, рак тонкой кишки, рак толстой кишки (например, колоректальный рак, рак толстой кишки, рак прямой кишки и т.д.) и т.д.], рак легких, рак молочной железы, рак яичников, рак матки (например, рак шейки матки, рак эндометрия и т.д.), рак почки, рак мочевого пузыря, рак предстательной железы, уротелиальный рак, саркому кости и мягких тканей, рак крови (например, В-клеточная лимфома, хронический лимфоцитарный лейкоз, периферическая Т-клеточная лимфома, миелодиспластический синдром, острый миелогенный лейкоз, острый лимфоцитарный лейкоз и т.д.), множественную миелому, рак кожи и мезотелиому.
Примеры
[0036]
Далее настоящее изобретение будет дополнительно конкретно описано со ссылкой на примеры, однако изобретение не ограничивается ими. Хотя изобретение в достаточной степени описано с помощью примеров, должно быть понятно, что специалисты в данной области могут осуществлять различные изменения и модификации. Следовательно, такие изменения или модификации включены в изобретение, если они не являются отступлением от объема изобретения.
В качестве различных типов реагентов, используемых в примерах, использовались коммерчески доступные продукты, если не указано иное.
[0037]
<Измерения методом порошковой рентгеновской дифракционной спектроскопии (XRD)>
Метод порошковой рентгеновской дифракции осуществляли путем легкого дробления соответствующего количества испытываемого образца в агатовой ступке, в соответствии с необходимостью, и последующего измерения в соответствии со следующими условиями испытания.
[0038]
Устройство: RINT-2100 Ultima/PC (изготовитель Rigaku Corporation)
Нацеливание: CuKα
Диапазон сканирования: 5,0-40,0°
Ширина выборки: 0,02°
Скорость сканирования: 2°/мин
Работу с устройством, включающим обработку данных, осуществляли в соответствии со способом и процедурой, установленными для каждого устройства.
[0039]
<Измерения методом протонного ядерного магнитного резонанса (13C-ЯМР)>
13С-ЯМР измерения осуществляли на СМХ-300 Infinity (75.188829 МГц, изготовитель Chemagnetic, Inc.) с использованием тетраметилсилана в качестве внутреннего стандарта в случае, когда тетраметилсилан содержался в дейтерированном растворителе, и с использованием ЯМР растворителя в качестве внутреннего стандарта в других случаях. В каждом полученном спектре 13С-ЯМР все δ значения выражены в м.д.
[0040]
<Измерения методом инфракрасной абсорбционной спектрометрии (ИК)>
ИК измерения осуществляли на FT-730 (HORIBA, Ltd.) способом с использованием KBr.
[0041]
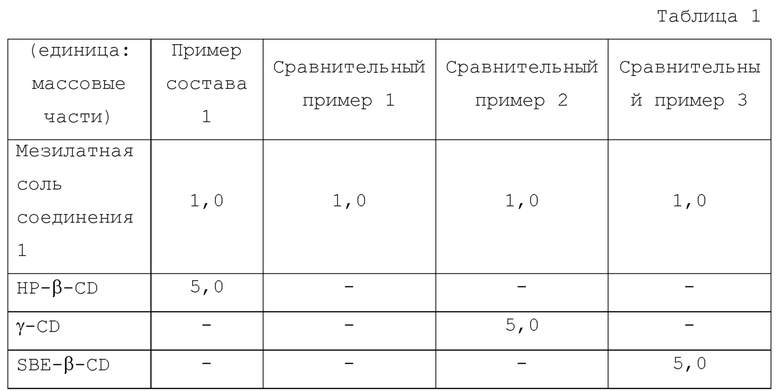
[Пример испытания 1] Испытание на растворимость 1
<Пример состава 1>
HP-β-CD (0,5925 г) растворяли в разбавленном McIlvaine буфере при рН 3,0 (50 мл) с последующим нагреванием до 37°С, таким образом получали раствор для испытания.
<Сравнительный пример 1>
Раствор для испытания получали путем нагревания разбавленного McIlvaine буфера при рН 3,0 (50 мл) до 37°С.
<Сравнительный пример 2>
Раствор для испытания получали таким же способом, как в Примере состава 1, с использованием γ-CD (0,5925 г) вместо HP-β-CD (0,5925 г).
<Сравнительный пример 3>
Раствор для испытания получали таким же способом, как в Примере состава 1, с использованием SBE-β-CD (0,5925 г) вместо HP-β-CD (0,5925 г).
[0042]
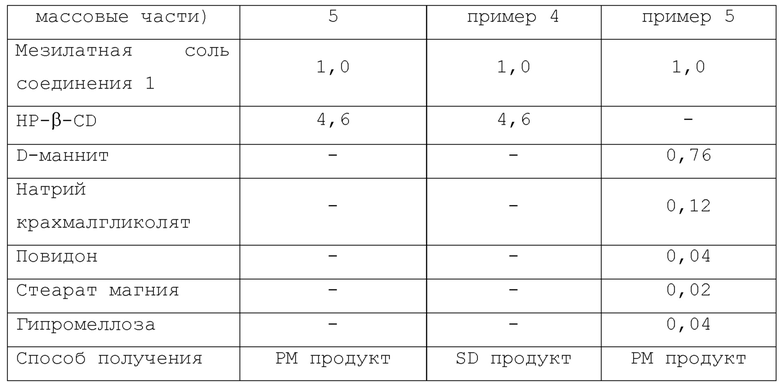
Что касается Примера состава 1, Сравнительного примера 1 и Сравнительного примера 2, измеряли растворимость соединения 1 в зависимости от времени. К каждому из испытываемых растворов добавляли мезилатную соль соединения 1 (0,1185 г) с последующим перемешиванием при 37°С с использованием магнитной мешалки. Композиции Примера состава 1, Сравнительного примера 1, Сравнительного примера 2 и Сравнительного примера 3 показаны в Таблице 1.
[0043]
[0044]
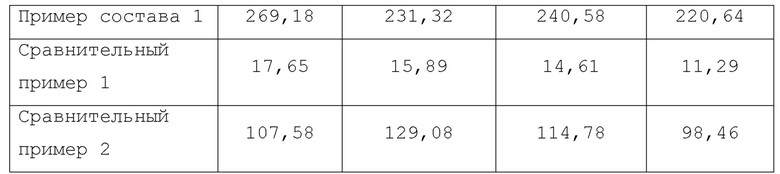
Концентрацию соединения 1 в испытываемом растворе измеряли через 30, 60, 120 и 240 минут после начала испытания с использованием жидкостной хроматографии (ВЭЖХ) в следующих условиях.
[0045]
Устройство: Alliance 2690 (изготовитель Waters, Inc.)
Подвижная фаза А: 10 мМ водного раствора Na2HPO4 (рН 6,5)
Подвижная фаза В: ацетонитрил
Градиент: подвижная фаза А/подвижная фаза В=6/4 (об/об)
Колонка: L-column 2 ODS, 100 мм × 3,0 мм, в.д.: 3 мкм
Длина волны: 240 нм
Работу с устройством, включающим обработку данных, осуществляли в соответствии со способом и процедурой, установленными для каждого устройства. Результаты показаны в Таблице 2.
[0046]

[0047]
Как показано в Таблице 2, было обнаружено, что когда присутствует одинаковое количество производного циклодекстрина, HP-β-CD показывает более высокую растворимость, чем γ-CD. С другой стороны, в случае SBE-β-CD, ошибка измерений концентрации соединения 1 в каждый момент измерения является большой, и трудно предсказать его абсорбирумость, когда его вводят человеку, и поэтому, было обнаружено, что SBE-β-CD не подходит для формулирования композиции с соединением 1.
[0048]
[Пример испытания 2] Испытание на растворимость 2
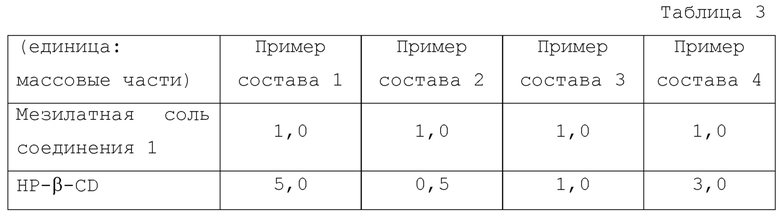
<Пример состава 2>
Раствор для испытания получали таким же способом, как в Примере состава 1, с использованием HP-β-CD (0,0593 г) вместо HP-β-CD (0,5925 г).
<Пример состава 3>
Раствор для испытания получали таким же способом, как в Примере состава 1, с использованием HP-β-CD (0,1185 г) вместо HP-β-CD (0,5925 г).
<Пример состава 4>
Раствор для испытания получали таким же способом, как в Примере состава 1, с использованием HP-β-CD (0,3555 г) вместо HP-β-CD (0,5925 г).
[0049]
Что касается Примеров состава 1-4, растворимость соединения 1 в зависимости от времени измеряли таким же способом, как в Примере испытания 1. К каждому из испытываемых растворов добавляли мезилатную соль соединения 1 (0,1185 г) с последующим перемешиванием при 37°С с использованием магнитной мешалки. Композиции Примеров состава 1-4 показаны в Таблице 3.
[0050]
[0051]
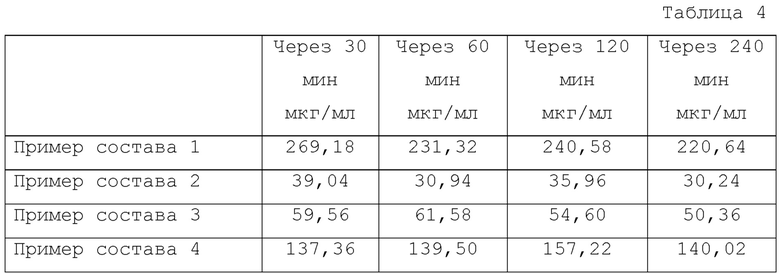
Концентрацию соединения 1 в испытываемом растворе измеряли через 30, 60, 120 и 240 минут после начала испытания с использованием жидкостной хроматографии (ВЭЖХ) в тех же условиях. Результаты показаны в Таблице 4.
[0052]
[0053]
Как показано в Таблице 4, было обнаружено, что растворимость мезилатной соли соединения 1 улучшается в присутствии HP-β-CD, и она показывает более высокую растворимость, когда HP-β-CD присутствует в большем количестве.
[0054]
[Пример испытания 3] XRD измерение
<Пример состава 5>
Физически смешанный продукт (26,6 г, РМ продукт) мезилатной соли соединения 1 получали путем смешивания мезилатной соли соединения 1 (5,0 г) и HP-β-CD (22,8 г) в ступке.
[0055]
Результаты измерений методом рентгеновской порошковой дифрактометрии (XRD) мезилатной соли соединения 1 показаны на Фиг. 1, результаты XRD измерений HP-β-CD показаны на Фиг. 2, результаты XRD измерений физически смешанного продукта мезилатной соли соединения 1 и HP-β-CD, полученного в Примере состава 5, показаны на Фиг. 3, и результаты XRD измерений высушенного распылением продукта мезилатной соли соединения 1 и HP-β-CD, полученного в Сравнительном примере 4, описанном ниже, показаны на Фиг. 4.
[0056]
На основании этих результатов было обнаружено, что при XRD измерении физически смешанного продукта мезилатной соли соединения 1 и HP-β-CD продукт характеризуется пиками при углах дифракции (2θ±0,2°) 6,5, 7,8, 9,6, 12,4, 18,8, 21,2, 23,0, 24,5 и 26,0 (А°), отражающими мезилатную соль соединения 1.
[0057]
[Пример испытания 4] 13С-ЯМР измерения
Результаты измерений методом твердотельного протонного ядерного магнитного резонанса (13С-ЯМР) мезилатной соли соединения 1 показаны на Фиг. 5, результаты 13С-ЯМР измерений HP-β-CD показаны на Фиг. 6, и результаты 13С-ЯМР измерений физически смешанного продукта мезилатной соли соединения 1 и HP-β-CD, полученного в Примере состава 5, показаны на Фиг. 7.
[0058]
На основании этих результатов было обнаружено, что при измерении методом твердотельного протонного ядерного магнитного резонанса (13С-ЯМР) физически смешанного продукта мезилатной соли соединения 1 и HP-β-CD продукт характеризуется пиками при значениях химических сдвигов [δ (м.д.)] 162,6, 130,4, 103,1, 82,7, 73,3, 41,9 и 19,9, отражающи мезилатную соль соединения 1.
[0059]
[Пример испытания 5] ИК-измерения
Результаты измерений методом инфракрасной абсорбционной спектроскопии (IR) мезилатной соли соединения 1 показаны на Фиг. 8, результаты ИК-измерений HP-β-CD показаны на Фиг. 9, и результаты ИК-измерений физически смешанного продукта мезилатной соли соединения 1 и HP-β-CD, полученного в Примере состава 5, показаны на Фиг. 10. Кроме того, результаты ИК-измерений в их характеристических областях показаны на Фиг. 11.
[0060]
На основании этих результатов было обнаружено, что при измерении методом инфракрасной абсорбционной спектроскопией физически смешанного продукта мезилатной соли соединения 1 и НР-β-CD продукт характеризуется пиками при полосах поглощения (см-1) 1663, 1352, 1225, 1156, 1032, 720 и 553, отражающие мезилатную соль соединения 1.
[0061]
[Пример испытания 6] Испытание на стабильность
<Сравнительный пример 4>
Мезилатную соль соединения 1 (5,0 г) и HP-β-CD (22,8 г) растворяли в смешанном растворе воды (100,0 г), этанола (250,0 г) и дихлорметана (150,0 г) с последующей распылительной сушкой с использованием распылительной сушилки (GB22, изготовитель Yamato Scientific Co., Ltd.), таким образом был получен высушенный распылением продукт (19,2 г, SD продукт) мезилатной соли соединения 1.
[0062]
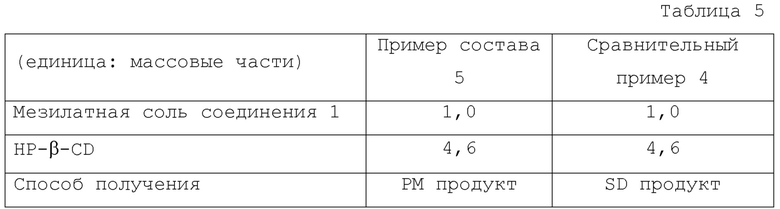
Композиции и способы получения Примера состава 5 и Сравнительного примера 4 показаны в Таблице 5.
[0063]
[0064]
Что касается Примера состава 5 и Сравнительного примера 4, оценивали изменение общего родственных примесей соединения 1 в каждом составе в зависимости от времени. Каждый состав обертывали полиэтиленовой/целлофановой ламинированной пленкой и затем помещали в алюминиевый пакет с осушителем и дезоксигенирующим агентом и концентрацию соединения 1 в препаратах, хранимых при 5, 25 и 40°С в течение 1 месяца, и составах, хранимых при 60°С в течение 1 недели, измеряли с использованием жидкостной хроматографии (ВЭЖХ) в следующих условиях.
[0065]
Устройство: Alliance 2690 (изготовитель Waters, Inc.)
Подвижная фаза А: 10 мМ водного раствора Na2HPO4 (рН 6,5)
Подвижная фаза В: ацетонитрил
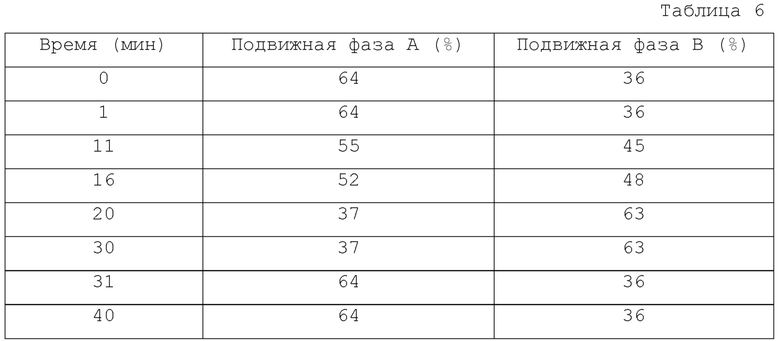
Градиент: показан в Таблице 6
Колонка: L-column 2 ODS, 150 мМ × 4,6 мМ, в.д.: 5 мкм
Длина волны: 220 нм
[0066]
[0067]
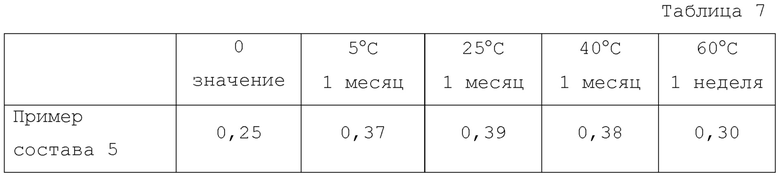
Работу с устройством, включающим обработку данных, осуществляли в соответствии со способом и процедурой, установленными для каждого устройства. Результаты показаны в Таблице 7.
[0068]

[0069]
Как показано в Таблице 7, было обнаружено, что физически смешанный продукт мезилатной соли соединения 1 и HP-β-CD обладает отличной стабильностью по сравнению с высушенным распылением продуктом.
[0070]
[Пример испытания 7] Испытание на абсорбирумость
<Сравнительный пример 5>
Гранулирование осуществляли с использованием гранулятора с большим усилием сдвига (FM-VG-25, изготовитель Powrex Corporation) при добавлении 14% раствора повидона (466 г) к мезилатной соли соединения 1 (1564,2 г), D-манниту (1188 г) и натрий крахмалгликоляту (33 г) с получением, таким образом, влажного порошка. Влажный порошок сушили с использованием гранулятора с псевдоожиженным слоем (NFLO-5, изготовитель Freund Corporation), а затем смешивали с использованием миксера (CV-20, изготовитель Tokuju Corporation) с натрий крахмалгликолятом (158,4 г) и стеаратом магния (26,4 г), получая таким образом гранулы для таблетирования. Гранулы для таблетирования формовали в таблетки, используя таблетировочную машину (VELG 0512SW2MZ, изготовитель Kikusui Seisakusho Ltd.), и затем на них наносили распылением раствор для покрытия, полученный путем добавления гипромеллозы (64,8 г), макрогола 6000 (8,1 г), оксида титана (8,1 г) и желтого оксида железа (0,081 г), с использованием машины для нанесения покрытий (DRC-300, изготовитель Powrex Corporation), таким образом, были получены таблетки с покрытием.
[0071]
Композиции и способы получения Примера состава 5, Сравнительного примера 4 и Сравнительного примера 5 показаны в Таблице 8.
[0072]

[0073]
Что касается Примера состава 5, Сравнительного примера 4 и Сравнительного примера 5, каждый состав вводили животным в следующих условиях и оценивали абсорбирумость.
[0074]
Используемые животные: собаки породы бигль (3 особи мужского пола, Kitayama Labes Co., Ltd.)
Условия кормления: голодали в течение 20 часов с предыдущего дня
Доза: 100 мг/животное (в расчете на соединение 1)
Способ введения: вводили с 50 мл воды
Предварительная обработка: Пентагастрин вводили внутримышечно (10 мкг/0,1 мл/кг) за 30 минут до введения состава и после этого вводили два раза с интервалом 45 минут. Внутривенную инъекцию атропин сульфата вводили внутривенно (20 мкг/0,04 мл/кг) за 30 минут до введения состава.
[0075]
Результаты показаны в Таблице 9.
[0076]

[0077]
Как показано в Таблице 9, было обнаружено, что абсорбция мезилатной соли соединения 1 улучшается при добавлении HP-β-CD, и физически смешанный продукт мезилатной соли соединения 1 и HP-β-CD показывает абсорбирумость, которая сопоставима с высушенным распылением продуктом.
[0078]
[Пример испытания 8] Испытание на абсорбирумость 2
<Пример состава 6>
Гранулирование осуществляли с использованием гранулятора с псевдоожиженным слоем (FL-LABO (special), изготовитель Freund Corporation), распыляя при этом 5% раствор гидроксипропилцеллюлозы (1000 г) на мезилатную соль соединения 1 (296, 3 г), HP-β-CD (1350 г) и светлую безводную кремниевую кислоту (8,8 г), получая таким образом гранулированный материал. Стеарат магния (10 г) добавляли к гранулированному материалу и смешивали в полиэтиленовом мешке, получая таким образом гранулы.
[0079]
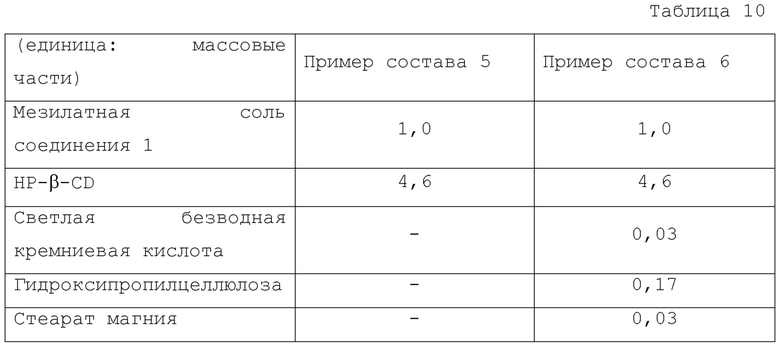
Композиции Примера состава 5 и Примера состава 6 показаны в Таблице 10.
[0080]
[0081]
Что касается Примера состава 5 и Примера состава 6, каждый состав вводили животным в следующих условиях и оценивали абсорбирумость.
[0082]
Используемые животные: собаки породы бигль (3 особи мужского пола, Kitayama Labes Co., Ltd.)
Условия кормления: голодали в течение 20 часов с предыдущего дня
Доза: 400 мг/животное (в расчете на соединение 1)
Способ введения: вводили с 50 мл воды
Предварительная обработка: Пентагастрин вводили внутримышечно (10 мкг/0,1 мл/кг) за 30 минут до введения состава и после этого вводили два раза с интервалом 45 минут. Внутривенную инъекцию атропин сульфата вводили внутривенно (20 мкг/0,04 мл/кг) за 30 минут до введения состава.
[0083]
Результаты показаны в Таблице 11.
[0084]

[0085]
Как показано в Таблице 11, было обнаружено, что гранулы, полученные путем гранулирования мезилатной соли соединения 1 и НР-β-CD, показывают абсорбирумость, сопоставимую с физически смешанным продуктом вышеуказанных.
[0086]
[Пример испытания 9] Испытание на абсорбирумость 3
<Пример состава 7>
Гранулирование осуществляли с использованием гранулятора с псевдоожиженным слоем (NFLO-5, изготовитель Freund Corporation), распыляя при этом 5% раствор гидроксипропилцеллюлозы (3000 г) на мезилатную соль соединения 1 (888,8 г), HP-β-CD (4050 г) и светлую безводную кремниевую кислоту (26,3 г), получая таким образом гранулированный материал. Две партии гранулированного материала смешивали с использованием смесителя (CV-20, изготовитель Tokuju Corporation) со стеаратом магния (60 г), получая таким образом гранулы.
[0087]
<Пример состава 8>
Гранулирование осуществляли с использованием гранулятора с псевдоожиженным слоем (NFLO-5, изготовитель Freund Corporation), распыляя при этом 5% раствор гидроксипропилцеллюлозы (3000 г) на мезилатную соль соединения 1 (888, 8 г), HP-β-CD (4050 г) и светлую безводную кремниевую кислоту (26,3 г), получая таким образом гранулированный материал. Стеарат магния (2,16 г) добавляли к части (396,4 г) гранулированного материала и смешивали в полиэтиленовом мешке, получая таким образом гранулы. Гранулы формовали в таблетки с диаметром 4 мм с использованием таблетировочной машины (VELG 0512SW2MZ, изготовитель Kikusui Seisakusho Ltd.) и затем на них наносили распылением раствор для покрытия, полученный путем добавления воды (480,0 г), гипромеллозы (32,0 г), макрогола 6000 (4,0 г), оксида титана (4,0 г) и желтого оксида железа (0,2 г), с использованием машины для нанесения покрытий (HC-FZ-LABO, изготовитель Freund Corporation), таким образом получали таблетки с покрытием из таблеток с диаметром 4 мм.
[0088]
<Пример состава 9>
Часть (292,1 г) гранул, полученных в Примере состава 7, формовали в таблетки с диаметром 3,5 мм с использованием таблетировочной машины (VELG 0512SW2MZ, изготовитель Kikusui Seisakusho Ltd.) и затем на них наносили распылением раствор для покрытия, полученный путем добавления воды (540,0 г), гипромеллозы (48,0 г), макрогола 6000 (6,0 г), оксида титана (6,0 г) и желтого оксида железа (0,3 г), с использованием машины для нанесения покрытий (HC-FZ-LABO, изготовитель Freund Corporation), таким образом получали таблетки с покрытием из таблеток с диаметром 3,5 мм.
[0089]
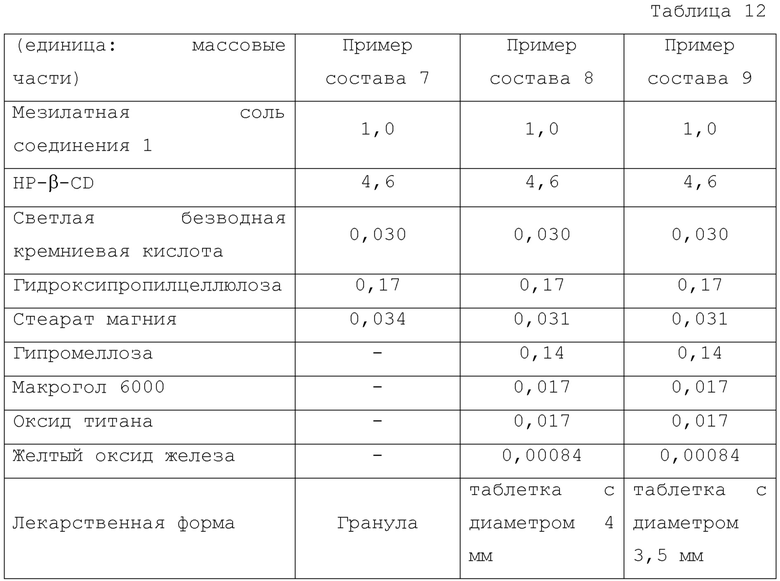
Композиции и лекарственные формы для Примеров состава 7-9 показаны в Таблице 12.
[0090]
[0091]
Что касается указанных выше, каждый состав вводили животным в следующих условиях и оценивали абсорбирумость.
[0092]
Используемые животные: собаки породы бигль (6 особи мужского пола, Kitayama Labes Co., Ltd.)
Условия кормления: голодали в течение 20 часов с предыдущего дня
Доза: 200 мг/животное (в расчете на соединение 1)
Способ введения: вводили с 50 мл воды
Предварительная обработка: Внутривенную инъекцию атропин сульфата вводили внутривенно (20 мкг/0,04 мл/кг) за 30 минут до введения состава. Когда испытание осуществляли при низком внутрижелудочном рН, пентагастрин вводили внутримышечно (10 мкг/0,1 мл/кг) за 30 минут до введения состава и после этого вводили два раза с интервалом 45 минут, а когда испытание осуществляли при высоком внутрижелудочном рН, внутривенно вводили омепразол (1 мг/0,25 мл/кг) за 30 минут до введения состава и через 60 минут после его введения один раз.
В результате было обнаружено, что таблетки, содержащие мезилатную соль соединения 1 и HP-β-CD, показывают абсорбирумость, сопоставимую с гранулами, на которую не влияет внутрижелудочный рН.
[0093]
Хотя настоящее изобретение было подробно описано со ссылкой на конкретные варианты осуществления, специалистам в данной области техники будет очевидно, что возможны различные изменения и модификации без отступления от сути и объема изобретения. Настоящая заявка основана на патентной заявке Японии, поданной 15 февраля 2017 года (патентная заявка №2017-026203), полное содержание которой включено в настоящую заявку посредством ссылки. Кроме того, все ссылочные документы, цитируемые в настоящей заявке, включены посредством ссылки во всей их полноте.
Группа изобретений относится к области медицины и фармацевтики. Первое изобретение группы – стабильная фармацевтическая композиция для перорального введения, включающая 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль и гидроксипропил-β-циклодекстрин, где гидроксипропил-β-циклодекстрин содержится в количестве 3-5,5 массовых частей на 1 массовую часть 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамида или его фармацевтически приемлемой соли. Второе изобретение группы – указанная фармацевтическая композиция, которая при этом получена путем физического смешивания, т.е. способом, не включающим стадию, на которой 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль преобразовывают в состояние раствора. Третье изобретение группы – способ получения указанных композиций, включающий физическое смешивание 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамида или его фармацевтически приемлемой соли и гидроксипропил-β-циклодекстрина, где способ не включает стадию, на которой 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль преобразовывают в состояние раствора. Группа изобретений обеспечивает повышение стабильности, распадаемости и абсорбируемости композиции. 3 н. и 32 з.п. ф-лы, 11 ил., 12 табл., 9 пр.
1. Стабильная фармацевтическая композиция для перорального введения, включающая:
4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль; и
гидроксипропил-β-циклодекстрин, где гидроксипропил-β-циклодекстрин содержится в количестве 3-5,5 массовых частей на 1 массовую часть 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамида или его фармацевтически приемлемой соли.
2. Фармацевтическая композиция по п. 1, где композиция характеризуется пиками при по меньшей мере 5 или более углах дифракции 2θ (±0,2°), выбранными из 6,5, 7,8, 9,6, 12,4, 18,8, 21,2, 23,0, 24,5 и 26,0 (А°), определенными методом порошковой рентгеновской дифрактометрии.
3. Фармацевтическая композиция по п. 1 или 2, где композиция характеризуется пиками при углах дифракции 2θ (±0,2°) 6,5, 7,8, 9,6, 12,4, 18,8, 21,2, 23,0, 24,5 и 26,0 (А°), определенными методом порошковой рентгеновской дифрактометрии.
4. Фармацевтическая композиция по п. 1 или 2, где композиция характеризуется пиками при значениях химических сдвигов [δ, м.д.] 162,6, 130,4, 103,1, 82,7, 73,3, 41,9 и 19,9 в 13C-ЯМР твердого тела.
5. Фармацевтическая композиция по п. 1 или 2, где композиция характеризуется пиками при по меньшей мере 5 или более полосах поглощения, выбранными из 1663, 1352, 1225, 1156, 1032, 720 и 553 (см-1), в инфракрасном спектре поглощения.
6. Фармацевтическая композиция по п. 1 или 2, дополнительно включающая производное кремниевой кислоты.
7. Фармацевтическая композиция по п. 1 или 2, дополнительно включающая производное целлюлозы.
8. Фармацевтическая композиция по п. 1 или 2, где фармацевтическая композиция представляет собой таблетку или гранулу.
9. Фармацевтическая композиция по п. 1 или 2, где фармацевтическая композиция предназначена для перорального введения.
10. Фармацевтическая композиция по п. 1 или 2, где фармацевтическая композиция представляет собой таблетку.
11. Фармацевтическая композиция по п. 1 или 2, где фармацевтическая композиция представляет собой таблетку, имеющую максимальный диаметр 5 мм или менее.
12. Фармацевтическая композиция по п. 1, где количество гидроксипропил-β-циклодекстрина составляеет от 60 до 95 % масс., предпочтительно от 76 до 85 % масс. от общей массы композиции.
13. Фармацевтическая композиция по п. 1 или 12, дополнительно включающая псевдоожижающий агент.
14. Фармацевтическая композиция по п. 13, где количество гидроксипропил-β-циклодекстрина составляет от 60 до 95 % масс. от общей массы композиции и количество псевдоожижающего агента составляет от 0,1 до 2 % масс. от общей массы фармацевтической композиции.
15. Фармацевтическая композиция по п. 13 или 14, где количество гидроксипропил-β-циклодекстрина составляет от 76 до 85 % масс. от общей массы фармацевтической композиции и количество псевдоожижающего агента составляет от 0,2 до 1 % масс. от общей массы фармацевтической композиции.
16. Стабильная фармацевтическая композиция для перорального введения, включающая:
4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль; и
гидроксипропил-β-циклодекстрин, где гидроксипропил-β-циклодекстрин содержится в количестве 3-5,5 массовых частей на 1 массовую часть 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамида или его фармацевтически приемлемой соли,
при этом фармацевтическая композиция получена путем физического смешивания,
где физическое смешивание представляет собой способ получения, который не включает стадию, на которой 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль преобразовывают в состояние раствора при получении фармацевтической композиции.
17. Фармацевтическая композиция по п. 16, где физическое смешивание представляет собой смешивание или гранулирование.
18. Фармацевтическая композиция по п. 16, где физическое смешивание представляет собой смешивание, способ сухого гранулирования.
19. Фармацевтическая композиция по п. 16, где физическое смешивание представляет собой смешивание, способ гранулирования путем дробления, способ гранулирования в псевдоожиженном слое, способ гранулирования во вращающемся слое, способ экструзионного гранулирования или способ гранулирования c высоким усилием сдвига.
20. Фармацевтическая композиция по п. 16, где физическое смешивание представляет собой способ гранулирования в псевдоожиженном слое.
21. Фармацевтическая композиция по п. 16, где композиция характеризуется пиками при по меньшей мере 5 или более углах дифракции 2θ (±0,2°), выбранными из 6,5, 7,8, 9,6, 12,4, 18,8, 21,2, 23,0, 24,5 и 26,0 (А°), определенными методом порошковой рентгеновской дифрактометрии.
22. Фармацевтическая композиция по п. 16, где композиция характеризуется пиками при углах дифракции 2θ (±0,2°) 6,5, 7,8, 9,6, 12,4, 18,8, 21,2, 23,0, 24,5 и 26,0 (А°), определенными методом порошковой рентгеновской дифрактометрии.
23. Фармацевтическая композиция по п. 16, где композиция характеризуется пиками при значениях химических сдвигов [δ, м.д.] 162,6, 130,4, 103,1, 82,7, 73,3, 41,9 и 19,9 в 13C-ЯМР твердого тела.
24. Фармацевтическая композиция по п. 16, где композиция характеризуется пиками при по меньшей мере 5 или более полосах поглощения, выбранными из 1663, 1352, 1225, 1156, 1032, 720 и 553 (см-1), в инфракрасном спектре поглощения.
25. Фармацевтическая композиция по п. 16, где гидроксипропил-β-циклодекстрин содержится в количестве 3-5,5 массовых частей в расчете на 1 массовую часть мезилатной соли 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамида.
26. Фармацевтическая композиция по п. 16, дополнительно включающая производное кремниевой кислоты.
27. Фармацевтическая композиция по п. 16, дополнительно включающая производное целлюлозы.
28. Фармацевтическая композиция по п. 16, где фармацевтическая композиция представляет собой гранулу.
29. Фармацевтическая композиция по п. 16, где фармацевтическая композиция предназначена для перорального введения.
30. Фармацевтическая композиция по п. 16, где фармацевтическая композиция представляет собой таблетку.
31. Фармацевтическая композиция по п. 16, где фармацевтическая композиция представляет собой таблетку, имеющую максимальный диаметр 5 мм или менее.
32. Способ получения фармацевтической композиции по любому из пп. 1-31, включающий физическое смешивание 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамида или его фармацевтически приемлемой соли и гидроксипропил-β-циклодекстрина,
где физическое смешивание представляет собой способ получения, который не включает стадию, на которой 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид или его фармацевтически приемлемую соль преобразовывают в состояние раствора при получении фармацевтической композиции.
33. Способ получения по п. 32, где физическое смешивание представляет собой смешивание или гранулирование.
34. Способ получения по п. 32, где физическое смешивание представляет собой смешивание или способ сухого гранулирования.
35. Способ получения по п. 32, где физическое смешивание представляет собой смешивание, способ гранулирования путем дробления, способ гранулирования во вращающемся слое, способ экструзионного гранулирования или способ гранулирования с высоким усилием сдвига.
| WO 2016175305 A1, 03.11.2016 | |||
| Handbook of Pharmaceutical Excipients | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Eds: ROWE R.C | |||
| et al | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| Приспособление для охлаждения воды | 1921 |
|
SU888A1 |
| ARUN R | |||
| et al | |||
| Cyclodextrins as Drug Carrier Molecule: A Review // Sci Pharm, 2008, Vol | |||
| Аппарат, предназначенный для летания | 0 |
|
SU76A1 |
| ДРОВОПИЛЬНО-ДРОВОКОЛЬНОЕ УСТРОЙСТВО | 1923 |
|
SU567A1 |
| YOKOO M | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

