Область изобретения
Настоящее изобретение относится к новому производному ацилтиомочевины или его соли, и к его применению.
Предпосылки изобретения
Фермент c-Met представляет собой рецептор тирозин киназы, идентифицированный как прото-онкоген и проявляет свою физиологическую функцию, когда присоединен к HGF, служащему в качестве лиганда. В нормальных тканях c-Met играет роль в регенерации, лечении ран и формировании органов. Однако, во многих раковых клетках (рак клеток почки, рак желудка, рак легкого, колоректальный рак, рак поджелудочной железы, рак яичников, рак клеток печени, рак головы и шеи, меланома и так далее) промотируется проявление сверхэкспрессии, мутации или транслокации c-Met, что приводит к чрезмерно активированному состоянию (непатентный документ 1). В таких условиях c-Met играет некоторую роль в клеточной пролиферации, инфильтрации/метастаз, онкогенезе, образовании новых сосудов и анти-апоптозе (смотри непатентные документы 2, 3 и 4). Кроме того, во многих исследованиях было выявлено, что сверхэкспрессия и повышение уровня активации c-Met в раковых клетках негативно соотносится с предсказанием дальнейшего протекания болезни, и известно, что c-Met является фактором, сопутствующим неблагоприятному прогнозу рака (смотри непатентные документы 5 и 6).
Таким образом, если вводится лекарственное средство, которое специфически ингибирует c-Met в клетках рак/опухоль, в которых c-Met активирован путем сверхэкспрессии, то пролиферация, инфильтрация и метастаз раковых клеток могли бы быть ингибированы более специфически и интенсивно, и, таким образом, как полагают, лекарственное средство будет вносить свой вклад в лечение рака, продление жизни пациентов и улучшение QOL. Между тем, в современной терапии, поскольку выражение уровень и уровень активации c-Met служат в качестве индикаторов для стратификации пациентов, пациенты могли бы получать подходящее лечение, которое является в высшей степени предпочтительным с этической точки зрения.
К настоящему времени широко было изучено применение производных ацилтиомочевины в качестве фармацевтических агентов или других агентов (смотри, например, патентные документы 1 - 7). Однако никогда не сообщалось о производном ацилтиомочевины по настоящему изобретению, представленном формулой (I), где указанное соединение имеет аминокарбонильную группу в качестве заместителя в 6-положении хинолинового кольца и алкокси группу в качестве заместителя в 7-положении хинолинового кольца.
Документы, относящиеся к уровню техники
Непатентные документы
Непатентный документ 1: Cancer Letters, 225, p. 1-26 (2005).
Непатентный документ 2: J. Cell Biol. 111, p. 2097-2108 (1990).
Непатентный документ 3: Semin Cancer Biol, 11, p. 153-165 (2001).
Непатентный документ 4: Am. J. Pathol., 158, p. 1111-1120 (2001).
Непатентный документ 5: Jpn. J. Cancer Res., 87, p. 1063-1069 (1996).
Непатентный документ 6: Cancer, 85(9), p.1894-1902 (1999).
Патентные документы
Патентный документ 1: WO 2001/047890.
Патентный документ 2: WO 2002/032872.
Патентный документ 3: WO 2003/000660.
Патентный документ 4: WO 2005/030140.
Патентный документ 5: WO 2005/121125.
Патентный документ 6: WO 2006/104161.
Патентный документ 7: WO 2006/108059.
Сущность изобретения
Задачи, решаемые изобретением
Объектом настоящего изобретения является противоопухолевый агент, который показывает превосходное ингибирующее c-Met действие и который уменьшает побочные эффекты путем избирательного поражения опухолевых клеток, в которых специфически экспрессируется c-Met.
Средства для решения поставленных задач
Создатели настоящего изобретения провели интенсивные исследования с целью решения вышеуказанной задачи и обнаружили, что по сравнению с вышеуказанными соединениями, которые обладают ингибирующим c-Met действием, производное ацилтиомочевины по настоящему изобретению, представленное формулой (I), соединение, имеющее аминокарбонильную группу в качестве заместителя в 6-положении хинолинового кольца и алкокси группу в качестве заместителя в 7-положении хинолинового кольца, (1) обладает ингибирующим c-Met действием, эквивалентным или более высоким, чем то, которым обладают общеизвестные соединения в исследованиях in vitro, (2) проявляет более высокое избирательное поражающее действие в отношении опухолевых клеток, в которых c-Met сверх экспрессирован или высоко активирован, по сравнению с опухолевыми клетками, в которых c-Met экспрессируется в более низкой степени, и нормальными клетками, и (3) снижает побочные эффекты и проявляет сильную регрессию опухоли в исследованиях in vivo, в которых используются ксенотрансплантатные модели. Другими словами, создатели настоящего изобретения обнаружили, что производное ацилтиомочевины, представленное формулой (I), которое селективно действует на опухолевые клетки, в которых специфически экспрессируется c-Met, снижает побочные эффекты и может быть использовано в качестве превосходного противоопухолевого агента. Настоящее изобретение было создано на базе данного обнаружения.
В соответствии с этим, настоящее изобретение относится к производному ацилтиомочевины, представленному формулой (I):
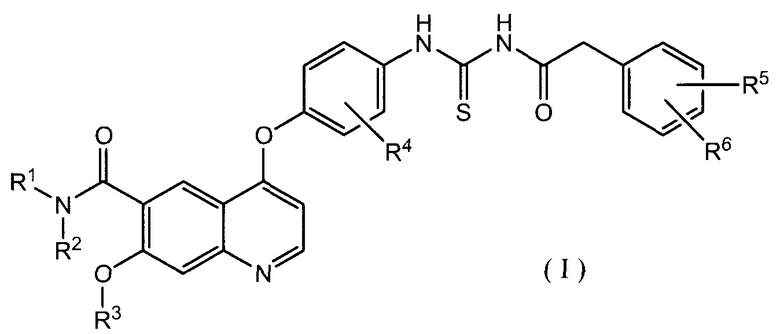
где каждый из R1 и R2, которые могут быть одинаковыми или различными, представляет собой атом водорода, необязательно замещенную C1-6 алкильную группу, необязательно замещенную C3-10 циклоалкильную группу, необязательно замещенную C6-14 ароматическую углеводородную группу или необязательно замещенную насыщенную или ненасыщенную гетероциклическую группу, или R1 и R2 могут образовывать, вместе с атомом азота, к которому они присоединены, необязательно замещенное азотсодержащее гетероциклическое кольцо;
R3 представляет собой C1-6 алкильную группу; и
каждый из R4, R5 и R6, которые могут быть одинаковыми или отличающимися друг от друга, представляет собой атом водорода, атом галогена, необязательно замещенную C1-6 алкильную группу, C1-6 алкокси группу, C1-6 алкиламино группу, необязательно замещенную ароматическую углеводородную группу или необязательно замещенную насыщенную или ненасыщенную гетероциклическую группу, или R5 и R6 могут образовывать кольцо вместе с фенильным кольцом, к которому они присоединены,
или к его соли.
Настоящее изобретение относится также к фармацевтическому агенту, содержащему, в качестве активного ингредиента, производное ацилтиомочевины, представленное формулой (I) или его соль.
Настоящее изобретение относится также к фармацевтической композиции, содержащей производное ацилтиомочевины, представленное формулой (I), или его соль, и фармацевтически приемлемый носитель.
Настоящее изобретение относится также к применению производного ацилтиомочевины, представленного формулой (I), или его соли при получении противоопухолевого агента.
Настоящее изобретение относится также к способу лечения рака, заключающемуся во введении субъекту, при необходимости этого, эффективного количества производного ацилтиомочевины, представленного формулой (I), или его соли.
Осуществление изобретения
В патентном документе 6 описано соединение, подобное соединению по настоящему изобретению, где указанное описанное соединение имеет хинолиновое кольцо и структуру ацилтиомочевины. Однако в патентном документе 6 не описано такое соединение, которое имеет аминокарбонильную группу в качестве заместителя в 6-положении хинолинового кольца, а указанный заместитель является характеристическим признаком настоящего изобретения. Как показано в примерах исследований, описанных здесь далее, соединение по настоящему изобретению, отличающееся тем, что имеет аминокарбонильную группу в качестве заместителя в 6-положении хинолинового кольца, проявляет в исследованиях in vitro ингибирующую активность по отношению к киназе c-Met, эквивалентную или выше, чем активность у подобного соединения, описанного в патентном документе 6 (сравнительное соединение 1). Однако, совершенно неожиданно, при введении в дозе, при которой сравнительное соединение 1 было бы токсичным, соединение по настоящему изобретению не проявляет токсичности (то есть, потери массы тела). Таким образом, доза соединения могла бы быть увеличена, и в in vivo исследованиях на голых мышах наблюдалось эффективное действие по уменьшению опухоли.
Как указано выше, соединение (I) по настоящему изобретению или его соль обладает превосходным ингибирующим c-Met действием в исследованиях in vitro, указанное ингибирующее c-Met действие имеет высокую селективность по отношению к опухолевым клеткам, в которых специфически экспрессируется c-Met, и проявляет сильное уменьшающее опухоль действие в исследованиях in vivo. Поэтому соединение по изобретению является полезным противоопухолевым агентом, уменьшающим побочные эффекты.
Заболевания, которые можно лечить путем введения лекарственного средства, содержащего соединение по настоящему изобретению включают, например, злокачественные опухоли, такие как рак головы и шеи, рак пищевода, рак желудка, рак толстой кишки, рак прямой кишки, рак печени, рак желчного пузыря/желчного протока, рак желчных протоков, рак поджелудочной железы, рак легкого, рак груди, рак яичников, рак шейки матки, рак матки, рак почки, рак мочевого пузыря, рак простаты, тестикулярную опухоль, саркому костей и мягких тканей, лейкемию, злокачественную лимфому, множественную миелому, рак кожи, опухоль мозга и мезотелиому. Кроме того, соединение по изобретению особенно эффективно при лечении пролиферативных заболеваний, включая стимулирование дифференцировки и пролиферацию клеток (например, пролиферативные и иммунологические злокачественные заболевания кожи, включая кератинизацию или воспаление, такие как псориаз); является полезным в качестве иммуносупрессора при лечении иммунологических заболеваний, таких как ревматизм; и при трансплантации органов.
Краткое описание рисунков
Фиг. 1 - график, показывающий действие на массу тела мышей соединения по настоящему изобретению и сравнительного соединения.
Фиг. 2 - график, показывающий противоопухолевые эффекты в исследованиях in vivo соединения по настоящему изобретению и сравнительного соединения.
Подробное описание изобретения
В описании настоящего изобретения, когда выражение "необязательно замещенный" добавляют к описанию структур, это относится к тому факту, что релевантная структура может иметь один или несколько заместителей в положении(ях), которое(ые) может(могут) быть химически замещены.
Вид, число и положение заместителя(ей), представленного(ых) в структуре, специально не ограничиваются. Когда присутствуют два или более заместителей, то они могут быть идентичными или отличаться один от другого. Примеры "заместителя" включают атом галогена, гидроксильную группу, циано группу, нитро группу, C1-6 алканоильную группу, C1-6 алкильную группу, C3-10 циклоалкильную группу, C2-6 алкенильную группу, C1-6 алкокси группу, амино группу, C1-6 алкиламино группу, C1-6 алканоиламино группу, C1-6 алкиламинокарбонильную группу, C1-6 алкилсульфонильную группу, C6-14 ароматическую углеводородную группу, насыщенную или ненасыщенную гетероциклическую группу, насыщенную или ненасыщенную гетероциклил-карбонильную группу и оксо группу. Когда имеется или имеются заместитель(и), их число обычно составляет от 1 до 3.
В формуле (I) "C1-6 алкильная группа" в определении "необязательно замещенная C1-6 алкильная группа", обозначенная как R1 или R2, представляет собой C1-C6 линейную или разветвленную алкильную группу. Примеры алкильной группы включают метильную, этильную, н-пропильную, изопропильную, н-бутильную, втор-бутильную, трет-бутильную, н-пентильную, изопентильную, н-гексильную и изогексильную группы.
В формуле (I) "C3-10 циклоалкильная группа" в определении "необязательно замещенная C3-10 циклоалкильная группа", обозначенная как R1 или R2, представляет собой C3-C10 циклоалкильную группу. Примеры включают циклопропильную, циклобутильную, циклопентильную и циклогексильную группы.
В формуле (I) "C6-14 ароматическая углеводородная группа" в определении "необязательно замещенная C6-14 ароматическая углеводородная группа", обозначенная как R1 или R2, представляет C6-C14 ароматическую углеводородную группу. Примеры включают фенильную и нафтильную группы.
В формуле (I) "насыщенная или ненасыщенная гетероциклическая группа" в определении "необязательно замещенная насыщенная или ненасыщенная гетероциклическая группа", обозначенная как R1 или R2, представляет собой моноциклическую или бициклическую насыщенную или ненасыщенную гетероциклическую группу, содержащую один или два атома, выбранных из атома кислорода, атома азота и атома серы. Примеры включают пирролидинильную, пиперидинильную, пиперазинильную, морфолино, тиоморфолино, гомопиперидинильную, тетрагидротиенильную, имидазолильную, тиенильную, фурильную, пирролильную, оксазолильную, изоксазолильную, тиазолильную, изотиазолильную, пиразолинильную, триазолильную, тетразолильную, пиридильную, пиразильную, пиримидинильную, пиридазильную, индолильную, изоиндолильную, индазолильную, метилендиоксифенильную, этилендиоксифенильную, бензофуранильную, дигидробензофуранильную, бензоимидазолильную, бензоксазольную, бензотиазолильную, пуринильную, хинолильную, изохинолильную, хиназолинильную и хиноксалильную группы. Из указанных, предпочтительными являются 5-7-членные насыщенные гетероциклы, где каждый имеет один или два атома азота; например, пирролидинильная, пиперидинильная, пиперазинильная, гомопиперидинильная и тетрагидротиенильная группы.
В формуле (I) примеры "азот-содержащего гетероциклического кольца" в определении "необязательно замещенное азотсодержащее гетероциклическое кольцо", образованное R1 и R2 вместе с атомом азота, к которому они присоединены, включают азот-содержащие насыщенные гетероциклические группы, такие как пирролидинильная, пиперидинильная, пиперазинильная и морфолино группы. Из указанных, пирролидинильная и пиперидинильная группы являются предпочтительными.
В формуле (I) примеры "C1-6 алкильной группы", обозначенной как R3, включают вышеуказанные алкильные группы. Из указанных, C1-3 алкильные группы являются предпочтительными, при этом метильная группа является более предпочтительной.
В формуле (I) примеры атома галогена, обозначенные как R4, R5 или R6, включают атом фтора, атом брома, атом хлора и атом йода. Из указанных, атом фтора и атом хлора являются предпочтительными.
В формуле (I) "C1-6 алкильная группа" в определении "необязательно замещенная C1-6 алкильная группа", обозначенная как R4, R5 или R6, включает вышеуказанные алкильные группы. Из указанных, метильная группа является предпочтительной.
В формуле (I) "C1-6 алкокси группа" в определении "необязательно замещенная C1-6 алкокси группа", обозначенная как R4, R5 или R6, представляет собой C1-C6 линейную или разветвленную алкокси группу. Примеры включают метокси, этокси, н-пропилокси, изопропилокси, н-бутилокси, втор-бутилокси, трет-бутилокси, н-пентилокси и н-гексилокси группы. Из указанных, C1-3 алкокси группы являются предпочтительными, при этом метокси группа является более предпочтительной.
В формуле (I) "C1-6 алкиламино группа" в определении "необязательно замещенная C1-6 алкиламино группа", обозначенная как R4, R5 или R6, представляет собой амино группу, моно- или ди-замещенную вышеуказанными C1-6 алкильными группами. Примеры включают метиламино, этиламино, диметиламино, метилэтиламино, н-пропиламино, изопропиламино, н-бутиламино, втор-бутиламино, трет-бутиламино, н-пентиламино и н-гексиламино группы.
В формуле (I) "ароматическая углеводородная группа" в определении "необязательно замещенная ароматическая углеводородная группа", обозначенная как R4, R5 или R6, представляет собой вышеуказанную C6-C14 ароматическую углеводородную группу. Примеры предпочтительных членов включают фенильную и нафтильную группы.
В формуле (I) "насыщенный или ненасыщенный гетероцикл" в определении "необязательно замещенный насыщенный или ненасыщенный гетероцикл", обозначенный как R4, R5 или R6, включает вышеуказанные насыщенные или ненасыщенные гетероциклические группы. Примеры предпочтительных членов включают 5-7-членные насыщенные гетероциклы, каждый из которых имеет один или два атома азота, такие как пирролидинильная, пиперидинильная и пиперазинильная группы.
Примеры кольца, образованного вместе с фенильным кольцом, к которому присоединены R5 и R6, включают нафталиновое кольцо, хинолиновое кольцо, хиназолиновое кольцо, индольное кольцо, бензимидазольное кольцо, метилендиоксифенильное кольцо и этилендиоксифенильное кольцо.
Более подробно вышеуказанные заместители описаны ниже. Примеры атома галогена включают вышеуказанные атомы галогена. Примеры C1-6 алканоильной группы включают формильную, ацетильную, пропионильную и бутилильную группы. Примеры C1-6 алкильной группы включают вышеуказанные C1-6 алкильные группы. Примеры C3-10 циклоалкильной группы включают вышеуказанные C3-10 циклоалкильные группы. Примеры C2-6 алкенильной группы включают винильную и 2-пропенильную группу. Примеры C1-6 алкокси группы включают вышеуказанные C1-6 алкокси группы. Примеры C1-6 алкиламино группы включают вышеуказанные C1-6 алкиламино группы. Примеры C1-6 алканоиламино группы включают аминогруппы, каждая из которых замещена вышеуказанными C1-6 алканоильными группами. Примеры C1-6 алкиламинокарбонильных групп включают аминокарбонильные группы, каждая из которых моно- или ди-замещена вышеуказанными C1-6 алкильными группами. Примеры C1-6 алкилсульфонильных групп включают сульфонильные группы, каждая из которых замещена вышеуказанными C1-6 алкильными группами. Примеры C6-14 ароматических углеводородных групп включают вышеуказанные C6-14 ароматические углеводородные группы. Примеры насыщенной или ненасыщенной гетероциклической группы включают вышеуказанные насыщенные или ненасыщенные гетероциклические группы.
R1, предпочтительно, представляет собой атом водорода или C1-3 алкильную группу, при этом атом водорода и метил являются более предпочтительными. Из указанных, атом водорода является особенно предпочтительным.
R2, предпочтительно, представляет собой необязательно замещенную C1-6 алкильную группу, необязательно замещенную C6-14 ароматическую углеводородную группу или необязательно замещенную насыщенную или ненасыщенную гетероциклическую группу.
C1-6 алкильная группа, обозначенная как R2, является, более предпочтительно, C1-4 алкильной группой, при этом метильная, этильная, н-пропильная, изопропильная, н-бутильная и втор-бутильная группы являются особенно предпочтительными. Далее заместитель «C1-6 алкильная группа», обозначенный как R2, будет описан подробно. Заместитель предпочтительно выбран из гидроксильной группы, C3-10 циклоалкильной группы, C1-6 алкокси группы, C1-6 алкиламино группы, C1-6 алканоиламино группы, C1-6 алкилсульфонильной группы, ароматической углеводородной группы, насыщенной или ненасыщенной гетероциклической группы, C1-6 алкиламинокарбонильной группы и насыщенной или ненасыщенной гетероциклил-карбонильной группы. C3-10 циклоалкильной группой более предпочтительно является циклогексильная группа. C1-6 алкокси группа, более предпочтительно, является C1-3 алкокси группой, при этом метокси, этокси и изопропилокси группы являются особенно предпочтительными. C1-6 алкокси группа может дополнительно иметь заместитель. Таким заместителем предпочтительно является гидроксильная группа. C1-6 алкиламино группой, более предпочтительно, является диэтиламино группа. C1-6 алканоиламино группой более предпочтительно является ацетиламино группа. C1-6 алкилсульфонильной группой, более предпочтительно, является метилсульфонильная группа. Ароматической углеводородной группой, более предпочтительно, является фенильная группа. Насыщенной или ненасыщенной гетероциклической группой, более предпочтительно, является 5-7-членная гетероциклическая группа, содержащая 1-4 атома азота и/или атома кислорода, при этом пирролидинильная, морфолино, диоксоланильная, тетрагидропиранильная, пиридильная и тетразолильная группы являются особенно предпочтительными. Насыщенные или ненасыщенные гетероциклические группы могут дополнительно иметь заместитель. Таким заместителем, предпочтительно, является C1-6 алкильная группа (особенно метильная группа) или оксо группа. C1-6 алкиламинокарбонильной группой, более предпочтительно, является этиламинокарбонильная, диметиламино или метилбутиламино группа. C1-6 алкиламинокарбонильная группа может дополнительно иметь заместитель. Таким заместителем, предпочтительно, является гидроксильная группа или C1-6 алкокси группа (особенно метокси группа). Насыщенной или ненасыщенной гетероциклил-карбонильной группой особенно предпочтительно является 5-7-членная насыщенная гетероциклил-карбонильная группа, содержащая 1 или 2 атома азота и/или атома кислорода, при этом пирролидинилкарбонильная и морфолинокарбонильная группы являются особенно предпочтительными. Насыщенная или ненасыщенная гетероциклил-карбонильная группа может дополнительно иметь заместитель. Таким заместителем, предпочтительно, является атом галогена (особенно атом фтора) или C1-6 алкильная группа (особенно метильная группа), которая может иметь гидроксильную группу.
C6-14 ароматической углеводородной группой, обозначенной как R2, более предпочтительно, является фенильная группа. Конкретным примером заместителя C6-14 ароматической углеводородной группы, обозначенной как R2, предпочтительно является C1-6 алкильная группа, при этом метильная группа является более предпочтительной.
Насыщенной или ненасыщенной гетероциклической группой, обозначенной как R2, является 5-7-членный насыщенный гетероцикл, содержащий 1 или 2 атома азота или атома серы, при этом пиперидинильная, гомопиперидинильная и тетрагидротиенильная группы являются более предпочтительными. Конкретным примером заместителя насыщенной или ненасыщенной гетероциклической группы, обозначенной как R2, предпочтительно, является гидроксильная группа, C1-6 алканоильная группа, C1-6 алкоксикарбонильная группа, C1-6 алкиламинокарбонильная группа или оксо группа, при этом гидроксильная группа, ацетильная группа, этиламинокарбонильная группа, трет-бутилоксикарбонильная группа и оксо группа являются более предпочтительными.
R2 представляет собой, особенно предпочтительно, метильную, метоксиэтильную, морфолиноэтильную, морфолинокарбонилметильную, 2-гидрокси-н-бутильную, 2-гидрокси-2-метил-н-пропильную или 1-гидрокси-н-бутан-2-ильную группу. В случае 1-гидрокси-н-бутан-2-ильной группы (S)-форма является особенно предпочтительной.
R4 представляет собой, предпочтительно, атом галогена, при этом атом фтора и атом хлора являются особенно предпочтительными. Положением R4, предпочтительно, является 2-положение или 3-положение, при этом 2-положение является особенно предпочтительным.
Каждый из R5 и R6 представляет собой, предпочтительно, атом водорода, атом галогена, необязательно замещенную C1-6 алкильную группу или C1-3 алкокси группу. Заместителем C1-6 алкильной группы, определенным как R5 или R6, предпочтительно, является атом галогена, при этом атом фтора является более предпочтительным.
В одном предпочтительном случае, один из R5 и R6 представляет собой атом водорода, и другой представляет собой атом водорода, атом галогена, трифторметильную группу или метокси группу. В одном более предпочтительном осуществлении один из R5 и R6 представляет собой атом водорода, и другой представляет собой атом водорода или атом галогена. Когда один из R5 и R6 представляет собой атом водорода и другой представляет собой атом галогена, положением R6 является, предпочтительно, 2-положение или 4-положение.
В настоящем изобретении особенно предпочтительными являются следующие производные ацилтиомочевины и их соли.
4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид,
4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(метоксиэтил)хинолин-6-карбоксамид,
4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(2-морфолиноэтил)хинолин-6-карбоксамид,
4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(2-морфолино-2-оксоэтил)хинолин-6-карбоксамид,
4-(2-фтор-4-(3-(2-(4-фторфенил)ацетил)тиоуреидо)фенокси)-N-(2-гидроксибутил)-7-метоксихинолин-6-карбоксамид,
4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-N-(2-гидрокси-2-метилпропил)-7-метоксихинолин-6-карбоксамид,
(S)-4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-N-(1-гидроксибутан-2-ил)-7-метоксихинолин-6-карбоксамид,
4-(2-фтор-4-(3-(2-(4-фторфенил)ацетил)тиоуреидо)фенокси)-7-метокси-N-(2-морфолиноэтил)хинолин-6-карбоксамид,
(S)-4-(2-фтор-4-(3-(2-(4-фторфенил)ацетил)тиоуреидо)фенокси)-N-(1-гидроксибутан-2-ил)-7-метоксихинолин-6-карбоксамид,
(S)-4-(2-фтор-4-(3-(2-(2-фторфенил)ацетил)тиоуреидо)фенокси)-N-(1-гидроксибутан-2-ил)-7-метоксихинолин-6-карбоксамид,
(S)-4-(4-(3-(2-(4-хлорфенил)ацетил)тиоуреидо)-2-фторфенокси)-N-(1-гидроксибутан-2-ил)-7-метоксихинолин-6-карбоксамид.
Производное ацилтиомочевины по настоящему изобретению, представленное в виде формулы (I), также охватывает его стереоизомер, его оптический изомер и его сольват, такой как гидрат.
Производное ацилтиомочевины по настоящему изобретению, представленное в виде формулы (I), может быть в виде соли. Соль, предпочтительно, представляет собой фармацевтически приемлемую соль. Примеры соли включают соли неорганического основания, соли органического основания, соли с неорганической кислотой, соли с органической кислотой, соли с кислотной аминокислотой и соли с основной аминокислотой.
Конкретные примеры солей неорганических оснований включают соли щелочного металла (например, натрия или калия) и соли щелочноземельного металла (например, магния или кальция).
Примеры органических оснований, образующих соли, включают триметиламин, триэтиламин, пиридин, N-метилпиридин, N-метилпирролидон, этаноламин, диэтаноламин, триэтаноламин и дициклогексиламин.
Примеры неорганических кислот включают хлористоводородную кислоту, серную кислоту, бромистоводородную кислоту, йодистоводородную кислоту, азотную кислоту и фосфорную кислоту.
Примеры органических кислот включают муравьиную кислоту, уксусную кислоту, пропионовую кислоту, малоновую кислоту, янтарную кислоту, глутаровую кислоту, фумаровую кислоту, малеиновую кислоту, молочную кислоту, яблочную кислоту, лимонную кислоту, виноградную кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту и метансульфоновую кислоту.
Примеры кислотных аминокислот включают глютаминовую кислоту и аспарагиновую кислоту, а примеры основных аминокислот включают лизин, аспарагин и орнитин.
Производное ацилтиомочевины по настоящему изобретению, представленное в виде формулы (I), может быть фармацевтически приемлемым пролекарством. Нет конкретных ограничений относительно фармацевтически приемлемого пролекарства, и может быть использовано любое пролекарство в той степени, пока пролекарство может быть преобразовано в соединение, представленное в виде формулы (I), в физиологических условиях in vivo (желудочные кислоты или фермент) посредством гидролиза, окисления или восстановления. Примеры пролекарства включают производные сложных эфиров, которые модифицируют карбоксильную группу, такие как метиловый эфир, этиловый эфир, пропиловый эфир, фениловый эфир, карбоксиоксиметиловый эфир и этоксикарбониловый эфир. Примерами типичных пролекарств являются соединения, которые в физиологических условиях преобразуются в соединения (I), описанные в работе "Development of Drugs, vol. 7, p. 163-198)", опубликованной в Hirokawa Shoten (1990).
Производное ацилтиомочевины по настоящему изобретению, представленное в виде формулы (I), или его соль также охватывает его гидрат, его сольват и его кристаллический полиморф.
Соединение по настоящему изобретению может быть получено в соответствии с нижеописанной схемой. Исходные продукты, требуемые для синтеза соединения по настоящему изобретению, могут быть коммерческими продуктами или могут быть получены способом, описанным в литературе. На схеме заместители имеют такие же значения, какие определены для формулы (I).
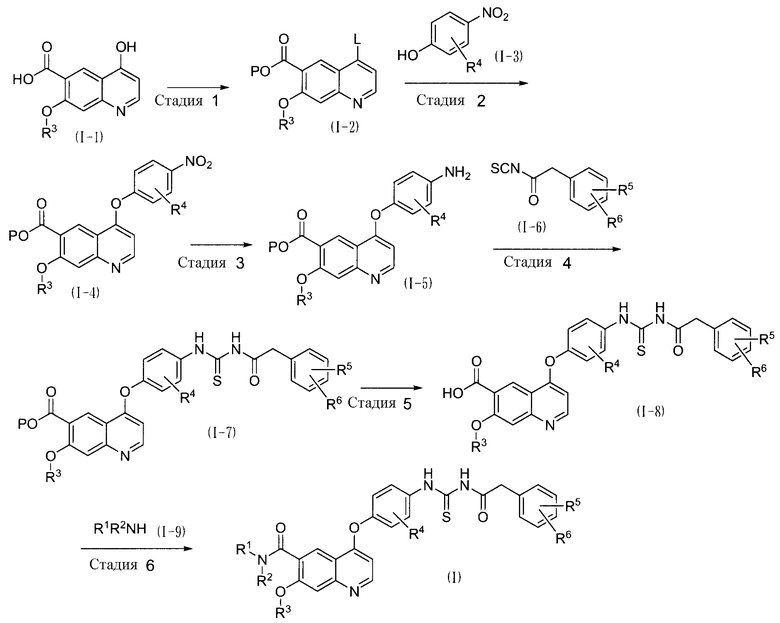
На схеме L представляет собой удаляемую группу, P представляет собой низшую алкильную группу или бензильную группу, имеющую заместитель, особенно метильную, этильную, метоксиметильную, трет-бутильную, бензильную, 4-нитробензильную, 4-метоксибензильную группы и так далее. Другие группы имеют такие же значения, какие определены для формулы (I).
Стадия 1
На стадии 1 соединение (I-2) получают из соединения (I-1). В частности, соединение (I-1), которое может быть получено в соответствии со способом, описанным в документе WO 2002-032872, обрабатывают тионилхлоридом, оксихлоридом фосфора и так далее, служащим в качестве растворителя, вводя, таким образом, галоген в виде удаляемой группы L. Температура реакции составляет от 0°C до температуры кипения, предпочтительно от 80°C до температуры кипения. Время реакции составляет от 0,1 до 100 часов, предпочтительно от 1 до 24 часов. Если требуется, может быть добавлен N,N-диметилформамид, от 0,001 до 1 объема, предпочтительно от 0,002 до 0,1, относительно соединения (I-1).
После завершения вышеуказанной реакции, помимо удаления группы L, карбоксильную группу в 6-положении также преобразовывали в галогенангидрид кислоты. Таким образом, галогенангидрид кислоты подвергали взаимодействию со спиртом P-OH, необязательно, в присутствии основания, вводя таким образом защитную группу P, вследствие чего может быть получено соединение (I-2). Нет никаких конкретных ограничений в отношении растворителя, до той степени, пока растворитель не является реакционно-способным в отношении галогенангидрида кислоты, и в качестве растворителя может быть использовано основание. Примеры спирта P-OH включают метанол, этанол, трет-бутанол, бензиловый спирт, 4-нитробензиловый спирт и 4-метоксибензиловый спирт. Спирт может быть использован в количестве 1 эквивалента по отношению к эквивалентному количеству растворителя, предпочтительно 10 эквивалентов по отношению к эквивалентному количеству растворителя. Примеры основания включают органические амины, такие как триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, лутидин и коллидин; гидрокарбонат натрия, карбонат натрия, метоксид натрия, метоксид калия, этоксид натрия, этоксид калия и трет-бутоксид калия. Основание может быть использовано в относительном количестве от 1 до 200, предпочтительно от 1,5 до 100, по отношению к соединению (I-1). Температура реакции составляет от -30°C до температуры кипения, предпочтительно от 0 до 50°C. Время реакции составляет от 0,1 до 100 часов, предпочтительно от 1 до 24 часов.
Стадия 2
Стадия 2 включает реакцию конденсации между соединением (I-2) и соединением (I-3), что дает таким образом соединение (I-4). Соединение (I-3) может быть использовано в относительном количестве от 1 до 100 эквивалентов, предпочтительно от 1,1 до 10 эквивалентов, по отношению к соединению (I-2). Реакцию конденсации, предпочтительно, осуществляют в присутствии основания. Примеры основания включают органические амины, такие как триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, лутидин и коллидин; и неорганические основания, такие как гидрокарбонат натрия, карбонат натрия, карбонат калия и карбонат цезия. Основание может быть использовано в относительном количестве от 1 до 100 эквивалентов, предпочтительно от 2 до 10 эквивалентов. Нет никаких конкретных ограничений в отношении растворителя, используемого в реакции, до той степени, пока растворитель не вступает легко в реакцию с соединениями (I-2), (I-3) и (I-4) и так далее. Примеры растворителя включают N,N-диметилацетамид, дифениловый эфир, хлорбензол, 1,2-дихлорбензол, N-метилпирролидин-2-он и диметил сульфоксид. Указанные растворители могут быть использованы сами по себе или в сочетании. Температура реакции составляет от -30 до 300°C, предпочтительно от 30 до 200°C. Время реакции составляет от 0,1 до 100 часов, предпочтительно от 0,5 до 24 часов.
Стадия 3
На стадии 3 нитро группу соединения (I-4) восстанавливают, получая таким образом соединение (I-5). Восстановление нитро группы может быть осуществлено с помощью восстанавливающего агента, такого как железо-хлорид аммония или железо-уксусная кислота. Если соединение (I-4) не содержит в качестве группы P Cl, Br или I, или функциональную группу, такую как бензил, 4-нитробензил или 4-метоксибензил, то может быть выбрано каталитическое восстановление. Когда используют железо-хлорид аммония, в качестве растворителя могут быть использованы вода, метанол, этанол, 2-пропанол, тетрагидрофуран, 1,4-диоксан, толуол, метилен хлорид, хлороформ, ацетонитрил, N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидин-2-он, диметил сульфоксид и так далее. Указанные растворители могут быть использованы сами по себе или в сочетании. Температура реакции составляет от 0 до 200°C, предпочтительно от 30 до 100°C. Время реакции составляет от 0,1 до 100 часов, предпочтительно от 0,5 до 24 часов.
Когда используют каталитическое восстановление, примеры катализатора, используемого в реакции, включают 5-10% Pd-C и гидроксид палладия. Катализатор может быть использован в относительном количестве от 0,01 до 10, предпочтительно от 0,02 до 5, по отношению к соединению (I-4). Источник водорода, например, муравьиная кислота, формиат аммония, циклогексен или дициклогексен, могут быть использованы в количестве от 1 до 200 эквивалентов, предпочтительно от 1,1 до 100 эквивалентов. Когда используют водород, давление водорода может быть от 0,01 до 3,0 MPa, и предпочтительно составляет от 0,1 до 1,0 MPa. Примеры растворителя включают метанол, этанол, тетрагидрофуран, этил ацетат, N,N-диметилформамид и диметилформамид, и указанные растворители могут быть использованы сами по себе или в сочетании.
Стадия 4
На стадии 4 соединение (I-7) получают из соединения (I-5) с использованием тиоизоцианата (I-6). Тиоизоцианат (I-6) может быть получен отдельно посредством способа, описанного в документе WO 2005-082855, из галогенангидрида кислоты или карбоновой кислоты. Соединение (I-6) может быть использовано в количестве от 1 до 100 эквивалентов по отношению к соединению (I-5), предпочтительно от 1,1 до 30 эквивалентов. Нет никаких конкретных ограничений в отношении растворителя, используемого в реакции, и могут быть использованы гексан, толуол, тетрагидрофуран, ацетонитрил, N,N-диметилформамид, N-метилпирролидин-2-он, метанол, этанол, изопропанол и так далее. Указанные растворители могут быть использованы сами по себе или в сочетании. Температура реакции составляет от -30 до 200°C, предпочтительно от 0 до 100°C. Время реакции составляет от 0,1 до 100 часов, предпочтительно от 0,5 до 24 часов.
Стадия 5
На стадии 5 карбоновую кислоту (I-8) получают из сложного эфира (I-7). Реакция может быть осуществлена в основных или кислотных условиях, или может быть осуществлено каталитическое восстановление, таким образом, чтобы сложный эфир преобразовывался в карбоновую кислоту.
Когда группа P представляет собой метил или этил, удаление защитных групп, предпочтительно, осуществляют в основных условиях. Примеры основания включают гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия, гидроксид калия и гидроксид лития. Основание может быть использовано в количестве от 1 до 100 эквивалентов, предпочтительно от 1,1 до 30 эквивалентов. Примеры растворителя включают воду, метанол, этанол, изопропанол, тетрагидрофуран, 1,4-диоксан и N,N-диметилформамид. Указанные растворители могут быть использованы сами по себе или в сочетании.
Когда группа P представляет собой, например, трет-бутил, удаление защитных групп предпочтительно осуществляют в кислотных условиях. Примеры кислот включают хлористоводородную кислоту, уксусную кислоту, трифторуксусную кислоту, серную кислоту и толуолсульфоновую кислоту. Кислота может быть использована в концентрации 1н по отношению к растворитель-эквивалентное количество, предпочтительно 2н по отношению к растворитель-эквивалентное количество. Примеры растворителя включают воду, метанол, этанол, изопропанол, этил ацетат, тетрагидрофуран, 1,4-диоксан, метиленхлорид и хлороформ. Указанные растворители могут быть использованы сами по себе или в сочетании.
Когда группа P представляет собой, например, бензил, 4-нитробензил или 4-метоксибензил, удаление защитных групп, предпочтительно, осуществляют посредством каталитического восстановления в присутствии катализатора. Катализатором восстановления может быть 5-10% Pd-C или гидроксид палладия. Катализатор может быть использован в относительном количестве от 0,01 до 10 по отношению к соединению (I-7), предпочтительно от 0,02 до 5. Источник водорода, например, водород, муравьиная кислота, формиат аммония, циклогексен или 1,4-дициклогексен, могут быть использованы в количестве от 1 до 200 эквивалентов, предпочтительно от 1,1 до 100 эквивалентов. Примеры растворителя включают метанол, этанол, изопропанол, тетрагидрофуран, этил ацетат и N,N-диметилформамид, и указанные растворители могут быть использованы сами по себе или в сочетании.
В любом сочетании, температура реакции составляет от -30 до 200°C, предпочтительно от 0 до 100°C, и время реакции составляет от 0,1 до 100 часов, предпочтительно от 0,5 до 24 часов.
Стадия 6
Стадия 6 включает реакцию конденсации между карбоновой кислотой (I-8) и амином (I-9). Стадия 6 для получения соединения (I) может быть осуществлена с участием галогенангидрида кислоты, полученного из карбоновой кислоты (I-8), или с использованием обычно используемого конденсирующего агента.
В способе с участием галогенангидрида кислоты, сначала карбоновую кислоту (I-8) преобразуют в ее хлорангидрид с использованием тионилхлорида, оксихлорида фосфора и так далее, в растворитель-эквивалентном количестве. Температура реакции составляет от -30 до 200°C, предпочтительно от 0 до 100°C. Время реакции составляет от 0,1 до 100 часов, предпочтительно от 1 до 24 часов.
Затем, посредством введения амина (I-9) в полученный таким образом галогенангидрид кислоты, может быть получено соединение (I). Если требуется, может быть использовано основание. Примеры основания включают органические амины, такие как триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, лутидин и коллидин; и неорганические основания, такие как гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия, метоксид натрия, метоксид калия, этоксид натрия, этоксид калия и трет-бутоксид калия. Амин (I-9) может быть использован в количестве от 1 до 100 эквивалентов, предпочтительно от 1,1 до 50 эквивалентов. Примеры растворителя, используемого в реакции, включают тетрагидрофуран, 1,4-диоксан, толуол, метиленхлорид, хлороформ, ацетонитрил, N,N-диметилформамид, N,N-диметилацетамид и диметил сульфоксид.
Альтернативно, при получении соединения (I) может быть использован конденсирующий агент. Примеры конденсирующего агента включают N,N'-дициклогексилкарбодиимид (DCC), N,N'-диизопропилкарбодиимид (DIC), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (WSC), дифенилфосфорил азид (DPPA), гексафторфосфат бензотриазол-1-ил-окситрис(диметиламино)фосфония (BOP), гексафторфосфат бензотриазол-1-ил-окситрипирролидинофосфония (PyBOP), фосфат 7-азабензотриазол-1-ил-окситрис(пирролидино)фосфония (PyAOP), гексафторфосфат бромтрис(пирролидино)фосфония (BroP), гексафторфосфат хлортрис(пирролидин-1-ил)фосфония (PyCroP), 3-(диэтоксифосфорилокси)-1,2,3-бензотриазин-4(3H)-он (DEPBT), гексафторфосфат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU) и гидрохлорид 4-(5,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолин (DMTMM). В сочетании с конденсирующим агентом могут быть использованы дополнительные соединения, такие как 1-гидроксибензотриазол (HOBt), 1-гидрокси-7-азабензотриазол (HOAt) и N-гидроксисукцинимид (HOSu). Указанные дополнительные соединения могут быть использованы в количествах от 0,1 до 100 эквивалентов, предпочтительно от 1 до 10 эквивалентов. Если требуется, может быть использовано основание, такое как триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, лутидин или коллидин в количестве от 0,1 до 100 эквивалентов, предпочтительно от 1 до 10 эквивалентов. Может быть использован амин (I-9) в том же количестве как указано выше. Нет никаких конкретных ограничений в отношении растворителя, и могут быть использованы вода, метанол, этанол, 2-пропанол, тетрагидрофуран, 1,4-диоксан, толуол, метиленхлорид, хлороформ, ацетонитрил, N,N-диметилформамид, N,N,-диметилацетамид, диметил сульфоксид и так далее. Температура реакции составляет от -30 до 200°C, предпочтительно от 0 до 100°C. Время реакции составляет от 0,1 до 100 часов, предпочтительно от 0,5 до 24 часов.
Соединение (I-5) может быть преобразовано в его амид другими, чем указанными выше способами, в соответствии со стадиями 5 и 6, и затем соединение (I) может быть получено в соответствии со стадией 4. Когда группа P представляет собой метил, соединение (I-5) может быть преобразовано в его амид общеизвестным методом, таким как аминолиз, и затем соединение (I) может быть получено в соответствии со стадией 4.
Полученное таким образом соединение по настоящему изобретению и его синтетические промежуточные продукты обычно могут быть выделены и очищены посредством известных средств выделения/очистки (например, перекристаллизация, кристаллизация, перегонка или колоночная хроматография). Обычно соединение по настоящему изобретению и его промежуточные продукты синтеза могут быть преобразованы в их фармацевтически приемлемые соли известным методом, и соли могут быть преобразованы в свободные формы.
Когда соединение (I) по настоящему изобретению используют в виде лекарственного средства, его смешивают с дополнительным фармацевтическим носителем, и смесь может быть сформована в различные лекарственные формы, в соответствии с профилактическими или терапевтическими целями. Могут быть использованы любые формы, и примеры включают пероральные средства, жидкости для инъекций, суппозитории, мази и припарки. Из указанных, предпочтительно используются пероральные средства. Указанные лекарственные формы могут быть получены методами, обычно известными и используемыми в данной области.
В качестве фармацевтических носителей могут быть использованы различные органические и неорганические носители, которые обычно используют для приготовления препаратов. Примеры носителя для твердого препарата включают дополнительное вещество, связующее, разрыхлитель, смазывающее вещество и окрашивающее вещество, а примеры носителя для жидкого препарата включают растворитель, солюбилизирующий агент, суспендирующий агент, агент, придающий тоничность, буфер и успокоительный агент. Если требуется, могут быть также использованы дополнительные соединения для препарата, такие как консервант, антиоксидант, окрашивающее вещество, подсластитель и стабилизатор.
Пероральная твердая форма может быть получена путем смешивания соединения по настоящему изобретению с дополнительным веществом, и с необязательным дополнительным веществом, связующим, разрыхлителем, смазывающим агентом, окрашивающим веществом, ароматизирующим/дезодорирующим агентом и так далее, и далее формования смеси в таблетки, таблетки с покрытием, гранулы, порошок, капсулы и так далее способом, известным в данной области.
Примеры дополнительного вещества включают лактозу, сахарозу, D-маннитол, глюкозу, крахмал, карбонат кальция, каолин, микрокристаллическую целлюлозу и безводную кремниевую кислоту.
Примеры связующего агента включают воду, этанол, 1-пропанол, 2-пропанол, простой сироп, жидкую глюкозу, жидкий α-крахмал, жидкий желатин, D-маннитол, карбоксиметил целлюлозу, гидроксипропил целлюлозу, гидроксипропил крахмал, метил целлюлозу, этил целлюлозу, шеллак, фосфат кальция и поливинилпирролидон.
Примеры разрыхлителя включают сухой крахмал, альгинат натрия, порошкообразный агар, гидрокарбонат натрия, карбонат кальция, лаурилсульфат натрия, моноглицерил стеарат и лактозу.
Примеры смазывающего агента включают очищенный тальк, стеарат натрия (соль), стеарат магния, боракс и полиэтиленгликоль.
Примеры окрашивающего вещества включают оксид титана и оксид железа.
Примеры ароматизирующего/дезодорирующего агента включают сахарозу, апельсиновую корку, лимонную кислоту и виноградную кислоту.
Жидкий пероральный препарат может быть получен путем смешивания соединения по настоящему изобретению с ароматизирующим агентом, буфером, стабилизатором, дезодорантом и так далее, и далее формования смеси в жидкий агент для внутреннего приема, сироп, эликсир и так далее способом, известным в данной области. Ароматизирующим/дезодорирующим агентом, используемым при получении, может быть любой из вышеуказанных агентов. Примеры буфера включают цитрат натрия. Примеры стабилизатора включают трагакант, гуммиарабик и желатин. Если требуется, пероральный препарат может быть покрыт известным способом материалом, устойчивым по отношению к желудочному соку, или материалом для покрытия для сохранения его действия. Примеры такого материала для покрытия включают гидроксипропилметил целлюлозу, этил целлюлозу, гидроксиметил целлюлозу, гидроксипропил целлюлозу, полиоксиэтиленгликоль и Твин 80 (зарегистрированная товарная марка).
Инъекционные растворы могут быть получены путем смешивания соединения по настоящему изобретению с дополнительными соединениями, такими как регулятор pH, буфер, стабилизатор, агент, придающий тоничность, и местный анестезирующий агент, и далее формования смеси способом, известным в данной области, с получением таким образом подкожных, внутримышечных и внутривенных инъекционных жидкостей. Примеры регулятора pH и буфера включают цитрат натрия, ацетат натрия и фосфат натрия. Примеры стабилизатора включают пиросульфит натрия, EDTA, тиогликолевую кислоту и тиомолочную кислоту. Примеры местного анестезирующего агента включают прокаин гидрохлорид и лидокаин гидрохлорид. Примеры агента, придающего тоничность, включают хлорид натрия, глюкозу, D-маннитол и глицерин.
Суппозитории могут быть получены путем смешивания соединения по настоящему изобретению с носителем для препаратов, известным в данной области, таким как полиэтиленгликоль, ланолин, масло какао и триглицерид жирной кислоты, и с необязательным поверхностно-активным веществом, таким как Твин 80 (зарегистрированная товарная марка), и далее формования смеси в суппозитории способом, известным в данной области.
Мази могут быть получены путем смешивания соединения по настоящему изобретению с необязательным дополнительным соединением, обычно используемым в данной области, таким как основа, стабилизатор, увлажнитель и консервант, и далее формования смеси в мази способом, известным в данной области. Примеры основы включают жидкий парафин, белый вазелин, белый воск, октилдодециловый спирт и парафин. Примеры консерванта включают метил п-гидроксибензоат, этил п-гидроксибензоат и пропил п-гидроксибензоат.
Припарки могут быть получены нанесением вышеуказанных мазей, крема, геля, пасты и так далее на обычно используемую подложку обычным способом. Примеры подходящих подложек включают плетеный и неплетеный фабричный хлопок, штапельное волокно или химическое волокно и пленку и пенообразный листок, сделанный из безопасного винилхлорида, полиэтилена, полиуретана и так далее.
Количество соединения по настоящему изобретению, входящее в любое из вышеуказанных стандартных лекарственных форм, изменяется в соответствии с формой лекарственного средства и признаками болезни у пациента, которому вводят указанное соединение. Однако, обычно, в стандартной лекарственной форме, количество предпочтительно составляет от около 0,05 до 1,000 мг (пероральный агент), от около 0,01 до 500 мг (инъекционный раствор) или от около 1 до 1,000 мг (суппозиторий).
Дневная доза лекарственного средства, имеющего любую из вышеуказанных дозированных форм, изменяется в соответствии с признаком болезни, массой тела, возрастом, полом и так далее у пациента, и не может быть определена однозначно. Однако, дневная доза для взрослого (масса тела: 50 кг) составляет обычно от около 0,05 до 5,000 мг, предпочтительно от 0,1 до 1,000 мг. Предпочтительно, лекарственное средство вводят в виде одной дневной дозы, или она может быть разделена (например, на 2 или 3).
Примеры
Подробные варианты осуществления по настоящему изобретению описаны в виде примеров и фармакологических примеров исследования, которые не следует рассматривать как ограничивающие таким образом изобретение.
Пример 1
трет-Бутил 4-хлор-7-метоксихинолин-6-карбоксилат (1a)
4-Гидрокси-7-метоксихинолин-6-карбоновую кислоту, (описанную в WO 2002/032872) (25 г) растворяли в тионил хлориде (100 мл), и к раствору добавляли N,N-диметилформамид (5 мл) с последующим кипячением при нагревании в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении и подвергали азеотропной перегонке с толуолом. Продукт добавляли к раствору трет-бутоксикалия (150 г, 6-кратное количество) в трет-бутаноле (300 мл) на ледяной бане, затем перемешивали в течение 17 часов. Реакционную смесь концентрировали при пониженном давлении, и добавляли (300 мл) воду на ледяной бане, затем экстрагировали н-гексаном (300 мл). Органическую фазу промывали насыщенным солевым раствором (300 мл) и сушили над сульфатом натрия, затем концентрировали при пониженном давлении, получая таким образом соединение 1a (10,5 г, выход: 31%).
1H-ЯМР (CDCl3) δ: 8,73 (1H, д, J=4,2 Гц), 8,50 (1H, с), 7,49 (1H, с), 7,38 (1H, д, J=4,8 Гц), 4,03 (3H, с), 1,64 (9H, с); ESI-MS m/z 294 (МН+).
трет-Бутил 4-(2-фтор-4-нитрофенокси)-7-метоксихинолин-6-карбоксилат (1b)
Соединение 1a (3,60 г) растворяли в N-метилпирролидин-2-оне (14 мл), и к раствору добавляли диизопропилэтиламин (6,55 мл) и 2-фтор-4-нитрофенол (2,89 г). Смесь нагревали до температуры 140°C и перемешивали в течение 4 часов. К реакционной смеси добавляли дистиллированную воду на ледяной бане, и образовавшийся осадок фильтровали, получая таким образом соединение 1b (4,71 г, выход: 93%).
1H-ЯМР (ДМСО-d6) δ: 8,75 (1H, д, J=4,8 Гц), 8,47 (1H, дд, J=10,4 Гц, 2,8 Гц), 8,38 (1H, с), 8,23 (1H, ддд, J=8,8 Гц, 1,2 Гц, 1,2 Гц), 7,74 (1H, т, J=8,0 Гц), 7,55 (1H, с), 6,78 (1H, д, J=5,2 Гц), 3,99 (3H, с), 1,54 (9H, с); ESI-MS m/z 415 (МН+).
трет-Бутил 4-(4-амино-2-фторфенокси)-7-метоксихинолин-6-карбоксилат (1c)
Соединение 1b (400 мг) растворяли в смеси вода-этанол (1:1) (10 мл), и добавляли порошок железа (1,0 г) и хлорид аммония (1,0 г), затем перемешивали при температуре 80°C в течение 2 часов. Реакционную смесь фильтровали через целит для удаления таким образом порошка железа, и к фильтрату добавляли воду (100 мл), затем экстрагировали этилацетатом (50 мл). Органическую фазу промывали насыщенным солевым раствором (100 мл) и сушили над сульфатом натрия, затем концентрировали при пониженном давлении, получая таким образом соединение 1c (335 мг, выход: 93%).
1H-ЯМР (ДМСО-d6) δ: 8,65 (1H, д, J=5,2 Гц), 8,40 (1H, с), 7,48 (1H, с), 7,10 (1H, т, J=9,2 Гц), 6,55 (1H, дд, J=13,2 Гц, 2,8 Гц), 6,48-6,44 (2H, м), 5,51 (2H, с), 3,96 (3H, с), 1,55 (9H, с); ESI-MS m/z 385 (МН+).
трет-Бутил 4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксилат (1d)
Фенилацетил хлорид (1,10 мл) и тиоцианат калия (1,21 г) растворяли в ацетонитриле (15 мл), затем перемешивали при температуре 70°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Затем продукт разделяли с помощью водного насыщенного раствора гидрокарбоната натрия (100 мл) и этилацетата (50 мл). Органический слой промывали насыщенным солевым раствором (100 мл) и сушили над сульфатом натрия, затем концентрировали при пониженном давлении, получая таким образом фенилацетил тиоизоцианат. Данный продукт (фенилацетил тиоизоцианат) не подвергали дополнительной очистке и растворяли в толуоле (8 мл). Раствор (12 мл) соединения 1c в смеси толуол-этанол (5:1) добавляли к толуольному раствору, затем перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении и очищали посредством колоночной хроматографии на силикагеле (элюент: 100% этил ацетат), получая таким образом соединение 1d (620 мг, выход: 53%).
1H-ЯМР (CDCl3) δ: 12,62 (1H, с), 8,70 (1H, с), 8,58 (1H, с), 8,09 (1H, дд, J=11,8 Гц, 2,0 Гц), 7,81 (1H, с), 7,51-7,30 (7H, м), 6,71 (1H, с), 4,18 (3H, с), 3,78 (2H, с), 1,64 (6H, с); ESI-MS m/z 562 (МН+).
Гидрохлорид 4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоновой кислоты (1e)
Соединение 1d (88,0 мг) растворяли в растворе 4н HCl-диоксан, затем перемешивали при температуре 70°C в течение 1 часа. Образовавшийся в реакционной смеси осадок фильтровали, получая таким образом соединение 1e (67,1 мг, выход: 79%).
1H-ЯМР (ДМСО-d6) δ: 12,54 (1H, с), 11,86 (1H, с), 8,98 (1H, д, J=6,4 Гц), 8,70 (1H, с), 8,11 (1H, д, J=12,4 Гц), 7,74-7,73 (1H, м), 7,65-7,60 (2H, м), 7,37-7,32 (4H, м), 7,30-7,25 (1H, м), 6,91 (1H, д, J=6,0 Гц), 4,04 (3H, с), 3,83 (2H, с); ESI-MS m/z 506 (МН+).
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-N-(3-изопропилоксипропил)-7-метоксихинолин-6-карбоксамид (1)
Соединение 1e (13,2 мг), 3-изопропоксипропиламин (9,11 мкл), и N-гидрат гидрохлорида 4-(5,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолина (здесь и далее указываемый как DMTMM·N-гидрат) (8,67 мг) растворяли в тетрагидрофуране (1 мл), затем перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь перегоняли при пониженном давлении, и к остатку добавляли воду. Образовавшийся осадок фильтровали, получая таким образом указанное в заголовке соединение 1 (11,6 мг, выход: 79%).
1H-ЯМР (ДМСО-d6) δ: 12,51 (1H, с), 11,83 (1H, с), 8,69 (1H, д, J=5,6 Гц), 8,54 (1H, с), 8,39 (1H, т, J=4,8 Гц), 8,04 (1H, дд, J=12,4 Гц, J=2,0 Гц), 7,58-7,49 (3H, м), 7,39-7,34 (4H, м), 7,32-7,27 (1H, м), 6,53 (1H, д, J=5,2 Гц), 4,02 (3H, с), 3,84 (2H, с), 3,58-3,50 (1H, м), 3,45 (2H, т, J=6,0 Гц), 3,40-3,36 (2H, м), 1,79-1,68 (2H, м), 1,09(6H, д, J=6,0 Гц); ESI-MS m/z 605 (МН+).
Пример 2
N-((2,2-Диметил-1,3-диоксолан-4-ил)метил)-4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксамид (2)
Способом, подобным описанному в примере 1, из соединения 1e (19,4 мг), (2,2-диметил-1,3-диоксолан-4-ил)метанамина (13,5 мкл) и N-гидрата DMTMM (11,9 мг) получали указанное в заголовке соединение 2 (9,3 мг, выход: 42%).
1H-ЯМР (CDCl3) δ: 12,50 (1H, с), 9,26 (1H, с), 8,66 (1H, дд, J=5,4 Гц, 0,8 Гц), 8,52 (1H, с), 8,23 (1H, т, J=5,6 Гц), 7,96 (1H, дд, J=11,2 Гц, J=2,8 Гц), 7,53 (1H, с), 7,46-7,37 (4H, м), 7,32-7,28 (3H, м), 6,44 (1H, дд, J=7,2 Гц), 4,43-4,38 (1H, м), 4,13-4,09 (1H, м), 4,12 (3H, с), 3,79-3,71 (3H, м), 2,42 (2H, т, J=8,0 Гц), 3,76 (2H, с), 1,49 (3H, с), 1,43 (1H, с), 1,39 (2H, с); ESI-MS m/z 619 (МН+).
Пример 3
N-(2, 3-Дигидроксипропил)-4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксамид (3)
Способом, подобным описанному в примере 1, из соединения 1e (20,1 мг), 3-амино-1,2-пропандиола (8,45 мг) и N-гидрата DMTMM (12,3 мг) получали указанное в заголовке соединение 3 (5,1 мг, выход: 24%).
1H-ЯМР (ДМСО-d6) δ: 12,50 (1H, с), 11,82 (1H, с), 8,69 (1H, с), 8,69 (1H, д, J=5,2 Гц), 8,48 (1H, т, J=5,6 Гц), 8,39 (1H, т, J=4,8 Гц), 8,04 (1H, дд, J=12,0 Гц, J=2,4 Гц), 7,58-7,50 (3H, м), 7,37-7,33 (4H, м), 7,31-7,26 (1H, м), 6,52 (1H, д, J=5,4 Гц), 4,92 (1H, ушир.), 4,65 (1H, ушир.), 4,03 (3H, с), 3,82 (2H, с), 3,65 (1H, т, J=5,6 Гц), 3,52-3,46 (1H, м), 3,43-3,37 (3H, м, J=6,0 Гц); ESI-MS m/z 579 (МН+).
Пример 4
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(пиридин-3-илметил)хинолин-6-карбоксамид (4)
Способом, подобным описанному в примере 1, из соединения 1e (16,7 мг), 3-пиколиламин (7,79 мкл) и N-гидрата DMTMM (10,2 мг) получали указанное в заголовке соединение 4 (8,1 мг, выход: 44%).
1H-ЯМР (ДМСО-d6) δ: 12,50 (1H, с), 11,81 (1H, с), 9,01 (1H, т, J=7,6 Гц), 8,69 (1H, д, J=5,2 Гц), 8,58 (1H, д, J=1,6 Гц), 8,57 (1H, с), 8,45 (1H, дд, J=4,8 Гц, 1,0 Гц), 8,02 (1H, дд, J=12,8 Гц, 1,6 Гц), 7,77 (1H, д, J=8,0 Гц), 7,56-7,48 (4H, м), 7,39-7,33 (6H, м), 7,31-7,26 (1H, м), 6,52 (1H, д, J=5,6 Гц), 4,55 (2H, д, J=6,0 Гц), 4,03 (3H, с), 3,82 (2H, с); ESI-MS m/z 596 (МН+).
Пример 5
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(3-(2-оксопирролидин-1-ил)пропил)хинолин-6-карбоксамид (5)
Способом, подобным описанному в примере 1, из соединения 1e (18,8 мг), N-(3'-аминопропил)-2-пирролидинона (12,2 мкл) и N-гидрата DMTMM (11,5 мг), получали указанное в заголовке соединение 5 (5,5 мг, выход: 25%).
1H-ЯМР (CDCl3) δ: 12,53 (1H, с), 9,24 (1H, с), 8,76 (1H, с), 8,65 (1H, д, J=5,6 Гц), 8,53 (1H, т, J=6,0 Гц), 7,95 (1H, дд, J=12,0 Гц, J=2,4 Гц), 7,52 (1H, с), 7,45-7,37 (4H, м), 7,32-7,30 (2H, м), 7,23 (1H, д, J=8,4 Гц), 6,42 (1H, дд, J=5,2 Гц, 1,2 Гц), 4,17 (3H, с), 3,76 (2H, с), 3,52-3,42 (6H, м), 2,42 (2H, т, J=8,0 Гц), 2,06 (2H, тт, J=7,6 Гц), 1,86 (2H, тт, J=6,0 Гц); ESI-LRMS m/z 630 (МН+).
Пример 6
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид (6)
Способом, подобным описанному в примере 1, соединение 1e (20 мг), 40% водный раствор метиламина (5 мкл) и N-гидрат DMTMM (22 мг) растворяли в тетрагидрофуране (1 мл), затем перемешивали при температуре 30°C в течение 1 часа, получая таким образом указанное в заголовке соединение 6 (18,4 мг, выход: 96%).
1H-ЯМР (ДМСО-d6) δ: 12,51 (1H, с), 11,83 (1H, с), 8,69 (1H, д, J=4,8 Гц), 8,60 (1H, с), 8,38 (1H, д, J=4,8 Гц), 8,03 (1H, дд, J=12,4 Гц, J=2,0 Гц), 7,58-7,50 (4H, м), 7,39-7,34 (4H, м), 6,53 (1H, д, J=5,2 Гц), 4,03 (3H, с), 3,84 (2H, с), 2,84 (3H, д, J=4,8 Гц); ESI-MS m/z 518 (МН+).
Пример 7
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(метоксиэтил)хинолин-6-карбоксамид (7)
Способом, подобным описанному в примере 1, соединение 1e (20 мг), 2-метоксиэтиленамин (6 мг), N-гидрат DMTMM (22 мг) растворяли в этаноле (1 мл), затем перемешивали при температуре 30°C в течение 1 часа, получая таким образом указанное в заголовке соединение 7 (17,3 мг, выход: 83%).
1H-ЯМР (ДМСО-d6) δ: 12,51 (1H, с), 11,83 (1H, с), 8,71-8,69 (1H, м), 8,62 (1H, с), 8,54-8,44 (1H, м), 8,04 (1H, дд, J=12,4 Гц, 1,6 Гц), 7,58-7,50 (3H, м), 7,36-7,34 (4H, м), 7,32-7,27 (1H, м), 6,53 (1H, д, J=4,8 Гц), 4,04 (3H, с), 3,84 (2H, с), 3,50-3,48 (4H, м), 3,30 (3H, с); ESI-MS m/z 562 (МН+).
Пример 8
N-(2-(Диэтиламино)этил)-4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксамид (8)
Способом, подобным описанному в примере 1, из соединения 1e (22,6 мг), 2-(диэтиламино)этиламина (14,8 мкл) и N-гидрата DMTMM (13,8 мг) получали указанное в заголовке соединение 8 (12,3 мг, выход: 49%).
1H-ЯМР (ДМСО-d6) δ: 12,50 (1H, с), 11,83 (1H, с), 8,73-8,70 (1H, м), 8,72 (1H, с), 8,51 (1H, т, J=5,2 Гц), 8,04 (1H, дд, J=12,8 Гц, 1,6 Гц), 7,58-7,50 (3H, м), 7,38-7,33 (4H, м), 7,31-7,27 (1H, м), 6,53 (1H, д, J=6,0 Гц), 4,05 (3H, с), 3,84 (2H, с), 3,42-3,37 (2H, м), 2,67-2,53 (6H, м), 1,01 (6H, т, J=7,2 Гц); ESI-MS m/z 604 (МН+).
Пример 9
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(2-морфолиноэтил)хинолин-6-карбоксамид (9)
Способом, подобным описанному в примере 1, из соединения 1e (9,6 мг), 2-морфолиноэтиламина (5,77 мкл) и N-гидрата DMTMM (5,88 мг) получали указанное в заголовке соединение 9 (3,3 мг, выход: 30%).
1H-ЯМР (CDCl3) δ: 12,53 (1H, с), 9,26 (1H, с), 9,08 (1H, т, J=3,6 Гц), 8,72 (1H, с), 8,66 (1H, д, J=5,0 Гц), 7,95 (1H, дд, J=11,6 Гц, 2,4 Гц), 7,55 (1H, с), 7,45-7,36 (4H, м), 7,32-7,24 (3H, м), 6,44 (1H, дд, J=5,2 Гц, 0,8 Гц), 4,37 (2H, д, J=4,0 Гц), 4,18 (3H, с), 3,79-3,72 (7H, м), 3,77 (2H, с), 3,52 (2H, т, J=4,8 Гц); ESI-MS m/z 618 (МН+).
Пример 10
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-N-(2-(2-гидроксиэтокси)этил)-7-метоксихинолин-6-карбоксамид (10)
Способом, подобным описанному в примере 1, из соединения 1e (9,7 мг), 2-(2-аминоэтокси)этанола (4,44 мкл) и N-гидрата DMTMM (5,94 мг) получали указанное в заголовке соединение 10 (3,0 мг, выход: 28%).
1H-ЯМР (ДМСО-d6) δ: 12,51 (1H, с), 11,83 (1H, с), 8,70 (1H, д, J=5,2 Гц), 8,64 (1H, с), 8,49 (1H, т, J=5,2 Гц), 8,04 (1H, д, J=12,2 Гц), 7,58-7,50 (3H, м), 7,38-7,34 (4H, м), 7,31-7,27 (1H, м), 6,52 (1H, д, J=5,2 Гц), 4,62 (1H, т, J=5,2 Гц), 4,04 (3H, с), 3,84 (2H, с), 3,58 (2H, т, J=5,6 Гц), 3,54-3,47 (6H, м); ESI-MS m/z 592 (МН+).
Пример 11
N-(2-Ацетамидоэтил)-4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксамид (11)
Способом, подобным описанному в примере 1, из соединения 1e (20,7 мг), N-ацетилэтилендиамина (9,75 мг) и N-гидрата DMTMM (12,7 мг) получали указанное в заголовке соединение 11 (5,4 мг, выход: 20%).
1H-ЯМР (ДМСО-d6) δ: 12,49 (1H, с), 11,82 (1H, с), 8,68 (1H, дд, J=5,2 Гц, 2,8 Гц), 8,63 (1H, д, J=2,4 Гц), 8,48 (1H, т, J=5,6 Гц), 8,02 (1H, д, J=12,4 Гц), 7,98 (1H, с), 7,56-7,49 (3H, м), 7,36-7,32 (4H, м), 7,30-7,26 (1H, м), 6,51 (1H, д, J=5,2 Гц), 4,02 (3H, с), 3,82 (2H, с), 3,38-3,35 (2H, м), 3,28-3,22 (2H, с), 1,82 (3H, с); ESI-MS m/z 590 (МН+).
Пример 12
N-(1,3-Дигидроксипропан-2-ил)-4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксамид (12)
Способом, подобным описанному в примере 1, из соединения 1e (37,1 мг), 2-амино-1,3-пропандиола (15,6 мг) и N-гидрата DMTMM (22,7 мг) получали указанное в заголовке соединение 12 (11,5 мг, выход: 29%).
1H-ЯМР (ДМСО-d6) δ: 12,50 (1H, с), 11,82 (1H, с), 8,74 (1H, с), 8,69 (1H, д, J=5,2 Гц), 8,29 (1H, д, J=8,0 Гц), 8,03 (1H, дд, J=12,0 Гц, 2,0 Гц), 7,57-7,50 (3H, м), 7,35-7,33 (4H, м), 7,31-7,26 (1H, м), 6,52 (1H, д, J=5,2 Гц), 4,80 (2H, т, J=5,2 Гц), 4,04 (3H, с), 3,99-3,94 (1H, м), 3,83 (2H, с), 3,61-3,56 (2H, м), 3,54-3,47 (2H, м); ESI-MS m/z 579 (МН+).
Пример 13
трет-Бутил 4-(4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксамидо)-6-карбоксамидо)пиперидин-1-карбоксилат (13)
Способом, подобным описанному в примере 1, из соединения 1e (750 мг), 4-амино-1-Boc-пиперидина (332 мг), триэтиламина (230 мкл) и N-гидрата DMTMM (459 мг) получали указанное в заголовке соединение 13 (446 мг, выход: 52%).
1H-ЯМР (CDCl3) δ: 12,50 (1H, с), 9,24 (1H, с), 8,66 (1H, д, J=5,4 Гц), 8,49 (1H, с), 7,96 (1H, дд, J=11,6 Гц, 2,4 Гц), 7,83 (1H, д, J=7,6 Гц), 7,53 (1H, с), 7,47-7,37 (4H, м), 7,33-7,29 (3H, м), 6,44 (1H, д, J=5,0 Гц, 1,2 Гц), 4,23 (1H, ушир.), 4,11 (3H, с), 4,03-4,01 (1H, м), 3,76 (2H, с), 3,04 (3H, т, J=12,0 Гц), 2,92 (1H, т, J=10,8 Гц), 2,09-1,98 (3H, м), 1,48 (9H, с); ESI-MS m/z 688 (МН+).
Пример 14
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(пиперидин-4-ил)хинолин-6-карбоксамид дигидрохлорид (14)
Соединение 13 (446 мг) растворяли в 4н растворе HCl-диоксана, затем перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении, и остаток подвергали азеотропной перегонке с толуолом, получая таким образом указанное в заголовке соединение 14 (406 мг, выход: 95%).
1H-ЯМР (ДМСО-d6) δ: 12,50 (1H, с), 11,82 (1H, с), 8,67 (1H, д, J=5,2 Гц), 8,44 (1H, с), 8,30 (1H, д, J=7,6 Гц), 8,02 (1H, дд, J=12,8 Гц, 1,6 Гц), 7,57-7,44 (3H, м), 7,37-7,33 (4H, м), 7,31-7,26 (1H, м), 6,51 (1H, д, J=5,2 Гц), 4,04-3,96 (1H, ушир.), 3,99 (3H, с), 3,93 (2H, с), 3,96-3,79 (4H, м), 3,83 (2H, с), 2,92 (2H, ушир.), 1,83 (1H, м); ESI-MS m/z 588 (МН+).
Пример 15
N-(1-(Этилкарбамоил)пиперидин-4-ил)-4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксамид (15)
Соединение 14 (335 мг) суспендировали в тетрагидрофуране (5 мл) и к суспензии добавляли триэтиламин (212 мкл) и этил изоцианат (71,2 мкл), затем перемешивали при комнатной температуре в течение 15 часов. Реакционную смесь концентрировали при пониженном давлении, и образовавшийся твердый продукт фильтровали, получая таким образом указанное в заголовке соединение 15 (271 мг, выход: 81%).
1H-ЯМР (ДМСО-d6) δ: 12,50 (1H, с), 11,82 (1H, с), 8,67 (1H, д, J=5,6 Гц), 8,44 (1H, с), 8,28 (1H, д, J=8,0 Гц), 8,02 (1H, д, J=12,8), 7,56-7,48 (3H, м), 7,35-7,28 (5H, м), 6,51 (1H, д, J=4,8 Гц), 6,45 (1H, т, J=4,8 Гц), 3,99 (4H, с), 3,88 (2H, д, J=12,8 Гц), 3,82 (2H, с), 3,06-2,99 (2H, м), 2,82 (3H, т, J=12,0 Гц), 1,43-1,34 (3H, м), 0,99 (3H, т, J=7,2 Гц); ESI-MS m/z 659 (МН+).
Пример 16
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(2-оксоазепан-3-ил)хинолин-6-карбоксамид (16)
Способом, подобным описанному в примере 1, из соединения 1e (260 мг), DL-α-амино-ε-капролактама (73,7 мг), триэтиламина (134 мкл) и N-гидрата DMTMM (159 мг) получали указанное в заголовке соединение 16 (221 мг, выход: 75%).
1H-ЯМР (ДМСО-d6) δ: 12,50 (1H, с), 11,82 (1H, с), 9,19 (1H, д, J=6,0 Гц), 8,90 (1H, с), 8,71 (1H, д, J=5,2 Гц), 8,05-7,96 (2H, м), 7,60-7,51 (3H, м), 7,37-7,33 (4H, м), 7,31-7,26 (1H, м), 6,53 (1H, д, J=4,4 Гц), 4,65-4,61 (1H, м), 4,10 (3H, с), 3,96 (2H, с), 3,83 (2H, с), 2,07 (1H, д, J=12,8 Гц), 1,94-1,90 (1H, м), 1,79-1,69 (2H, м), 1,47-1,38 (1H, м), 1,29-1,20 (1H, м); ESI-MS m/z 616 (МН+).
Пример 17
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(2-оксо-2-(пирролидин-1-ил)этил)хинолин-6-карбоксамид (17)
Способом, подобным описанному в примере 1, из соединения 1e (304 мг), гидрохлорида 2-амино-1-(пирролидин-1-ил)этанона (120 мг), триэтиламина (235 мкл) и N-гидрата DMTMM (186 мг) получали указанное в заголовке соединение 17 (220 мг, выход: 64%).
1H-ЯМР (ДМСО-d6) δ: 12,50 (1H, с), 11,82 (1H, с), 8,83 (2H, д, J=1,2 Гц), 8,71 (1H, дд, J=5,2 Гц, 1,6 Гц), 8,05-7,99 (1H, м), 7,60-7,53 (3H, м), 7,35-7,33 (4H, м), 7,30-7,28 (1H, м), 6,53 (1H, д, J=5,2 Гц), 4,14 (2H, д, J=4,0 Гц), 4,08 (3H, д, J=1,2 Гц), 3,83 (2H, с), 3,48-3,44 (2H, м), 3,39-3,24 (2H, м), 1,94-1,88 (2H, м), 1,83-1,76 (2H, м); ESI-MS m/z 616 (МН+).
Пример 18
N-(1-Ацетилпиперидин-4-ил)-4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксамид (18)
Способом, подобным описанному в примере 1, из соединения 1e (14,3 мг), гидрохлорида 1-(4-аминопиперидин-1-ил)этанона (5,66 мг), триэтиламина (9,21 мкл) и N-гидрата DMTMM (8,76 мг) получали указанное в заголовке соединение 18 (6,0 мг, выход: 36%).
1H-ЯМР (ДМСО-d6) δ: 12,50 (1H, с), 11,82 (1H, с), 8,67 (2H, д, J=5,2 Гц), 8,45 (1H, с), 8,02 (1H, дд, J=12,4 Гц, 2,4 Гц), 7,56-7,49 (3H, м), 7,37-7,33 (4H, м), 7,31-7,26 (1H, м), 6,52 (1H, д, J=5,2 Гц), 4,23 (1H, д, J=13,2 Гц), 4,10-3,98 (1H, ушир.), 4,00 (3H, с), 3,83 (2H, с), 3,78 (1H, д, J=14,4 Гц), 3,21-3,15 (2H, м), 2,78 (1H, т, J=10,8 Гц), 2,68-2,65 (1H, м), 2,00 (3H, с), 1,93-1,88 (1H, м), 1,86-1,81 (1H, м); ESI-MS m/z 630 (МН+).
Пример 19
трет-Бутил 4-(2-фтор-4-(3-(2-(4-фторфенил)ацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксилат (19a)
4-Фторфенилуксусную кислоту (900 мг) растворяли в тионилхлориде (5 мл), и раствор кипятили при нагревании в течение 2 часов. Реакционную систему концентрировали при пониженном давлении и подвергали азеотропной перегонке с толуолом, получая таким образом 4-фторфенилацетил хлорид в виде сырого продукта. Данный хлорангирид кислоты растворяли в ацетонитриле (20 мл) и к раствору добавляли тиоизоцианат калия (851 мг), затем перемешивали при температуре 70°C в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Затем продукт разделяли с помощью насыщенного водного раствора гидрокарбоната натрия (100 мл) и этилацетата (50 мл). Органический слой промывали насыщенным солевым раствором (100 мл) и сушили над сульфатом натрия, затем концентрировали при пониженном давлении, получая таким образом 4-фторфенилацетил тиоизоцианат. Данный тиоизоцианат не подвергали дополнительной очистке и растворяли в тетрагидрофуране (20 мл). Раствор (20 мл) соединения 1c (374 мг) в тетрагидрофуране добавляли к тиоизоцианатному раствору, и смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении, и образовавшийся твердый продукт фильтровали, получая таким образом соединение 19a (452 мг, выход: 79%).
1H-ЯМР (CDCl3) δ: 12,47 (1H, с), 11,82 (1H, с), 8,73 (1H, с), 8,65 (1H, д, J=4,4 Гц), 7,95 (1H, дд, J=11,2 Гц, 2,8 Гц), 7,49 (1H, с), 7,43-7,40 (1H, м), 7,31-7,25 (3H, м), 7,15 (2H, м), 6,42 (1H, дд, J=5,2 Гц, 1,2 Гц), 4,03 (3H, с), 3,74 (2H, с), 1,64 (9H, с); ESI-MS m/z 580 (МН+).
Гидрохлорид 4-(2-Фтор-4-(3-(2-(4-фторфенил)ацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоновой кислоты (19b)
Соединение 19a (385 мг) растворяли в 4н растворе смеси HCl-диоксан (10 мл) и раствор перемешивали при температуре 80°C в течение 4 часов. Образовавшийся в реакционной смеси осадок фильтровали, получая таким образом соединение 19b (245 мг, выход: 66%).
1H-ЯМР (ДМСО-d6) δ: 12,52 (1H, с), 11,85 (1H, с), 8,94 (1H, д, J=6,0 Гц), 8,68 (1H, с), 8,11 (1H, д, J=12,4 Гц), 7,66 (1H, с), 7,62 (1H, д, J=3,4 Гц), 7,39 (2H, дд, J=8,4 Гц, 5,6 Гц), 7,19 (2H, т, J=8,8 Гц), 6,85 (1H, д, J=6,0 Гц), 4,04 (3H, с), 3,84 (2H, с); ESI-MS m/z 524 (МН+).
4-(2-Фтор-4-(3-(2-(4-фторфенил)ацетил)тиоуреидо)фенокси)-7-метокси-N-(2-оксоазепан-3-ил)хинолин-6-карбоксамид (19)
Способом, подобным описанному в примере 1, из соединения 19b (53,0 мг), DL-α-амино-ε-капролактама (14,3 мг), триэтиламина (38,9 мкл) и N-гидрата DMTMM (30,9 мг) получали указанное в заголовке соединение 19 (16,9 мг, выход: 29%).
1H-ЯМР (ДМСО-d6) δ: 12,47 (1H, с), 11,81 (1H, с), 9,20 (1H, д, J=5,6 Гц), 8,90 (1H, с), 8,71 (1H, д, J=5,2 Гц), 8,04-7,96 (2H, м), 7,60 (1H, с), 7,57-7,51 (2H, м), 7,39-7,36 (2H, м), 7,20-7,15 (2H, м), 6,53 (1H, д, J=5,2 Гц), 4,65-4,61 (1H, м), 4,10 (3H, с), 3,83 (2H, с), 3,48-3,44 (2H, м), 2,08-2,05 (1H, м), 1,94-1,90 (1H, м), 1,79-1,72 (2H, м), 1,44-1,40 (1H, м), 1,29-1,19 (1H, м).
Пример 20
(S)-трет-Бутил 2-(3-фторпирролидин-1-ил)-2-оксоэтилкарбамат (20a)
(S)-3-Фторпирролидин гидрохлорид (535 мг), N-Boc глицин (746 мг), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (1,41 г), 1-гидроксибензотриазол (993 мг) и триэтиламин (1,19 мл) растворяли в тетрагидрофуране (5 мл), и раствор перемешивали при температуре 70°C в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении, и добавляли к остатку водный насыщенный раствор бикарбоната натрия (20 мл), затем экстрагировали этилацетатом(10 мл). Органический слой промывали последовательно 0,1н HCl (10 мл) и насыщенным солевым раствором (10 мл) и сушили над сульфатом натрия, затем концентрировали при пониженном давлении, получая таким образом соединение 20a (98,1 мг, выход: 9,3%).
1H-ЯМР (CDCl3) δ: 5,45 (1H, ушир.), 5,40-5,19 (1H, м), 4,00-3,82 (3H, м), 3,72-3,49 (3H, м), 4,10 (3H, с), 3,83 (2H, с), 3,48-3,44 (2H, м), 2,41-2,24 (1H, м), 2,19-1,91 (1H, м); FAB-MS m/z 247 (МН+).
(S)-2-Амино-1-(3-фторпирролидин-1-ил)этанон гидрохлорид (20b)
Соединение 20a (98,1 мг) растворяли в 4н растворе смеси HCl-1,4-диоксан, и раствор перемешивали при комнатной температуре в течение 4 часов, получая таким образом соединение 20b (33,5 мг, выход: 46%).
1H-ЯМР (ДМСО-d6) δ: 8,28 (3H, ушир.), 5,48-5,22 (1H, м), 3,88-3,27 (6H, м), 2,29-1,89 (2H, м).
(S)-4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-N-(2-(3-фторпирролидин-1-ил)-2-оксоэтил)-7-метоксихинолин-6-карбоксамид (20)
Способом, подобным описанному в примере 1, из соединения 1e (35,3 мг), соединения 20b (14,3 мг), триэтиламина (22,7 мкл) и N-гидрата DMTMM (21,6 мг) получали указанное в заголовке соединение 20 (15,1 мг, выход: 37%).
1H-ЯМР (ДМСО-d6) δ: 12,50 (1H, с), 11,82 (1H, с), 8,83 (1H, т, J=4,4 Гц), 8,81 (1H, с), 8,71 (1H, д, J=5,2 Гц), 7,59 (1H, с), 7,57-7,51 (2H, м), 7,37-7,33 (5H, м), 7,31-7,25 (1H, м), 6,53 (1H, д, J=5,6 Гц), 5,49-27 (1H, м), 4,27-4,13 (2H, м), 4,08 (3H, с), 3,82 (2H, с), 3,86-3,63 (3H, м), 2,32-2,05 (3H, м); ESI-MS m/z 634 (МН+).
Пример 21
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(2-морфолино-2-оксоэтил)хинолин-6-карбоксамид (21)
Способом, подобным описанному в примере 1, из соединения 1e (24,2 мг), гидрохлорида 2-амино-1-морфолиноэтанона (синтезированного в соответствии с описанным в документе J. Med. Chem., 1988, 31(11), 2145-2152) (9,67 мг), триэтиламина (18,7 мкл) и N-гидрата DMTMM (14,8 мг) получали указанное в заголовке соединение 21 (18,4 мг, выход: 65%).
1H-ЯМР (ДМСО-d6) δ: 12,50 (1H, с), 11,82 (1H, с), 8,85-8,83 (1H, м), 8,82 (1H, с), 8,71 (1H, дд, J=5,2 Гц), 8,03 (1H, д, J=12,0 Гц), 7,60 (1H, с), 7,55 (1H, с), 7,55-7,50 (1H, м), 7,35-7,28 (5H, м), 6,52 (1H, д, J=5,2 Гц), 4,24 (2H, д, J=4,8 Гц), 4,08 (2H, с), 3,96 (3H, с), 3,82 (2H, с), 3,59 (2H, д, J=13,2 Гц), 3,53-3,48 (2H, м), 3,15-3,00 (2H, м); ESI-MS m/z 632 (МН+).
Пример 22
N-(2-(Диметиламино)-2-оксоэтил)-4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксамид (22)
Способом, подобным описанному в примере 1, из соединения 1e (37,0 мг), гидрохлорида 2-амино-N,N-диметилацетамида (11,4 мг), N-гидрата DMTMM (22,7 мг) и триэтиламина (23,8 мкл) получали указанное в заголовке соединение 22 (6,8 мг, выход: 17%).
1H-ЯМР (ДМСО-d6) δ: 12,51 (1H, с), 11,82 (1H, с), 8,87-8,84 (2H, м), 8,72 (1H, дд, J=5,4 Гц, 0,6 Гц), 7,60-7,52 (3H, м), 7,38-7,33 (4H, м), 7,31-7,26 (1H, м), 6,54 (1H, д, J=4,8 Гц), 4,21 (2H, д, J=4,8 Гц), 4,09 (3H, с), 3,83 (2H, с), 3,00 (3H, с), 2,89 (3H, м); ESI-MS m/z 590 (МН+).
Пример 23
4-(2-Фтор-4-(3-(2-(4-фторфенил)ацетил)тиоуреидо)фенокси)-N-(2-гидроксибутил)-7-метоксихинолин-6-карбоксамид (23)
Способом, подобным описанному в примере 1, из соединения 19b (25,0 мг), 1-амино-2-бутанола (10,6 мкл) и N-гидрата DMTMM (14,8 мг) получали указанное в заголовке соединение 23 (14,2 мг, выход: 53%).
1H-ЯМР (ДМСО-d6) δ: 12,47 (1H, с), 11,81 (1H, с), 8,69 (1H, д, J=4,0 Гц), 8,65 (1H, с), 8,39 (1H, т, J=8,8 Гц), 8,02 (1H, д, J=11,2 Гц), 7,55-7,49 (3H, м), 7,37 (2H, дд, J=7,0 Гц, 6,0 Гц), 7,17 (2H, т, J=8,8 Гц), 6,52 (1H, д, J=5,4 Гц), 4,80 (1H, д, J=4,8 Гц), 4,03 (3H, с), 3,86 (2H, с), 3,82 (2H, с), 1,51-1,45 (2H, м), 1,39-1,31 (2H, м), 0,90 (3H, т, J=7,2 Гц); ESI-MS m/z 595 (МН+).
Пример 24
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-N-(2-гидрокси-2-метилпропил)-7-метоксихинолин-6-карбоксамид (24)
Способом, подобным описанному в примере 1, из соединения 1e (49,1 мг), 1-амино-2-метилпропан-2-ола (синтезированного в соответствии с описанным в документе Angew. Chem. Int. Ed., 2007, 46(25), 4751-4753) (20,2 мг) и N-гидрата DMTMM (30,1 мг) получали указанное в заголовке соединение 24 (36,1 мг, выход: 69%).
1H-ЯМР (ДМСО-d6) δ: 12,50 (1H, с), 11,80 (1H, с), 8,69 (1H, д, J=5,6 Гц), 8,66 (1H, с), 8,34 (1H, т, J=6,0 Гц), 8,02 (1H, д, J=11,2 Гц), 7,57-7,52 (3H, м), 7,37-7,33 (4H, м), 7,30-7,27 (1H, м), 6,52 (1H, д, J=5,6 Гц), 4,63 (1H, с), 4,04 (3H, с), 3,83 (2H, с), 3,81 (1H, д, J=2,4 Гц), 1,55 (1H, с), 1,14 (6H, с); ESI-MS m/z 577 (МН+).
Пример 25
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-N-((1-гидроксициклогексил)метил)-7-метоксихинолин-6-карбоксамид (25)
Способом, подобным описанному в примере 1, из соединения 1e (17,9 мг), гидрохлорида 1-(аминометил)циклогексанола (синтезированного в соответствии с описанным в документе J. Org. Chem., 1989. 54(24), 5651-5654) (6,57 мг) и N-гидрата DMTMM (11,0 мг) получали указанное в заголовке соединение 25 (8,2 мг, выход: 40%).
1H-ЯМР (CDCl3) δ: 12,51 (1H, с), 9,25 (1H, с), 8,66 (1H, д, J=5,2 Гц), 8,55 (1H, с), 8,23 (1H, т, J=5,4 Гц), 7,96 (1H, дд, J=11,6 Гц, 2,4 Гц), 7,53 (1H, с), 7,46-7,37 (4H, м), 7,33-7,23 (3H, м), 6,44 (1H, дд, J=5,2 Гц, 0,8 Гц), 4,12 (3H, с), 3,76 (2H, с), 3,58 (1H, д, J=5,8 Гц), 1,65-1,52 (10H, м), 1,37 (1H, ушир.); ESI-MS m/z 617 (МН+).
Пример 26
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-N-((4-гидрокситетрагидро-2H-пиран-4-ил)метил)-7-метоксихинолин-6-карбоксамид (26)
Способом, подобным описанному в примере 1, из соединения 1e (241 мг), гидрохлорида 4-(аминоэтил)тетрагидро-2H-пиран-4-ола (синтезированного в соответствии с описанным в документе US 2005/0696358 A1) (89,4 мг), триэтиламина (155 мкл) и N-гидрата DMTMM (147 мг) получали указанное в заголовке соединение 26 (236 мг, выход: 86%).
1H-ЯМР (CDCl3) δ: 12,51 (1H, с), 9,26 (1H, с), 8,67 (1H, д, J=5,4 Гц), 8,45 (1H, с), 8,26 (1H, т, J=6,0 Гц), 7,96 (1H, д, J=11,6 Гц, 2,4 Гц), 7,55 (1H, с), 7,47-7,38 (4H, м), 7,33-7,24 (3H, м), 6,52 (1H, дд, J=5,2 Гц, 0,8 Гц), 4,13 (3H, с), 3,83-3,78 (4H, м), 3,76 (2H, с), 3,61 (2H, д, J=6,4 Гц), 3,30 (1H, ушир.), 1,78 (2H, м), 1,64 (2H, д, J=12,8 Гц); ESI-MS m/z 619 (МН+).
Пример 27
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(2-(метилсульфонил)этил)хинолин-6-карбоксамид (27)
Способом, подобным описанному в примере 1, соединение 1e (25 мг), 2-(метилсульфонил)этанамин (6 мг), триэтиламин (19 мкл) и N-гидрата DMTMM (20 мг) растворяли в этаноле (1 мл) и раствор перемешивали при комнатной температуре в течение 1 часа, получая таким образом указанное в заголовке соединение 27 (20,6 мг, выход: 73%).
1H-ЯМР (ДМСО-d6) δ: 12,50 (1H, с), 11,83 (1H, с), 8,77 (1H, т, J=5,6 Гц), 8,70-8,69 (2H, м), 8,03 (1H, дд, J=12,2 Гц, 1,8 Гц), 7,57-7,50 (4H, м), 7,38-7,34 (4H, м), 7,31-7,26 (1H, м), 6,53 (1H, д, J=5,6 Гц), 4,03 (3H, с), 3,83 (2H, с), 3,76 (2H, дт, J=6,2 Гц), 3,42 (2H, д, J=6,8 Гц), 3,07 (3H, с); ESI-MS m/z 611 (МН+).
Пример 28
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-о-толилхинолин-6-карбоксамид (28)
Способом, подобным описанному для получения соединения 1, из соединения 1e (32 мг), о-толуидина (7,59 мкл) и N-гидрата DMTMM (19,6 мг) получали указанное в заголовке соединение 28 (19,8 мг, выход: 56%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,51 (1H, с), 11,82 (1H, с), 9,96 (1H, с), 8,76 (1H, с), 8,72 (2H, д, J=5,2 Гц), 7,64 (1H, с), 7,81 (1H, д, J=7,2 Гц), 7,58-7,52 (2H, м), 7,36-7,33 (4H, м), 7,30-7,21 (3H, м), 7,12 (1H, т, J=7,6 Гц), 6,55 (1H, д, J=5,6 Гц), 4,12 (3H, с), 3,83 (2H, с), 2,34 (3H, с); ESI-MS m/z 595 (МН+).
Пример 29
(S)-4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-N-(2-гидрокси-1-фенилэтил)-7-метоксихинолин-6-карбоксамид (29)
Способом, подобным описанному для получения соединения 1, из соединения 1e (44 мг), (S)-2-амино-2-фенилэтанола (15,5 мг) и N-гидрата DMTMM (19,1 мг) получали указанное в заголовке соединение 29 (47,0 мг, выход: 86%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,50 (1H, с), 11,82 (1H, с), 8,83 (1H, д, J=8,0 Гц), 8,69 (1H, д, J=5,2 Гц), 8,58 (1H, с), 8,02 (1H, дд, J=11,6 Гц, 1,6 Гц), 7,57 (1H, с), 7,57-7,49 (2H, м), 7,41-7,31 (7H, м), 7,29-7,22 (3H, м), 6,52 (1H, д, J=5,2 Гц), 5,09 (1H, кв, J=7,6 Гц), 5,02 (1H, т, J=5,6 Гц), 4,06 (3H, с), 3,82 (2H, с), 3,70-3,67 (2H, м); ESI-MS m/z 625 (МН+).
Пример 30
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-N-(2-гидрокси-2-(2-метил-2H-тетразол-5-ил)этил)-7-метоксихинолин-6-карбоксамид (30)
Способом, подобным описанному для получения соединения 1, из соединения 1e (30,0 мг), 2-амино-1-(2-метил-2H-тетразол-5-ил)этанола (12,7 мг) и N-гидрата DMTMM (19,7 мг) получали указанное в заголовке соединение 30 (31,2 мг, выход: 83%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,50 (1H, с), 11,82 (1H, с), 8,68 (1H, д, J=5,2 Гц), 8,64 (1H, с), 8,58-8,53 (1H, м), 8,03 (1H, д, J=11,6 Гц), 7,57-7,52 (3H, м), 7,36-7,33 (4H, м), 7,30-7,26 (1H, м), 6,52 (1H, д, J=5,6 Гц), 6,07 (1H, д, J=6,4 Гц), 4,34 (3H, с), 4,00 (3H, с), 3,83―3,82 (3H, м), 3,78-3,70 (2H, м); ESI-MS m/z 649 (МН+).
Пример 31
(S)-4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-N-(1-гидроксибутан-2-ил)-7-метоксихинолин-6-карбоксамид (31)
Способом, подобным описанному для получения соединения 1, из соединения 1e (81,7 мг), (S)-2-аминобутан-1-ола (22,8 мкл) и N-гидрата DMTMM (53,7 мг) получали указанное в заголовке соединение 31 (89,6 мг, выход: 96%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,50 (1H, с), 11,82 (1H, с), 8,68 (1H, д, J=5,1 Гц), 8,55 (1H, с), 8,12 (1H, д, J=8,6 Гц), 8,02 (1H, дд, J=12,0 Гц, 1,4 Гц), 7,58-7,48 (3H, м), 7,36-7,25 (4H, м), 7,31-7,25 (1H, м), 6,51 (1H, д, J=5,4 Гц), 4,75 (1H, т, J=5,6 Гц), 4,01 (3H, с), 3,92-3,85 (1H, м), 3,82 (2H, с), 3,52-3,47 (1H, м), 3,45-3,38 (1H, м), 1,78-1,70 (1H, м), 1,50-1,40 (1H, м), 0,92 (3H, т, J=7,6 Гц); ESI-MS m/z 577 (МН+).
Пример 32
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-N-(1-гидрокситетрагидротиофен-3-ил)-7-метоксихинолин-6-карбоксамид (32)
Способом, подобным описанному для получения соединения 1, из соединения 1e (25,7 мг), гидрохлорида 4-аминотетрагидротиофен-3-ола (16,9 мг), N-гидрата DMTMM (11,5 мг) и триэтиламина (14,2 мкл) получали указанное в заголовке соединение 32 (30,2 мг, выход: 93%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,50 (1H, с), 11,82 (1H, с), 8,81 (1H, с), 8,71 (1H, д, J=5,2 Гц), 8,57 (1H, д, J=7,2 Гц), 8,03 (1H, дд, J=13,2 Гц, 2,4 Гц), 7,58 (1H, с), 7,57-7,50 (2H, м), 7,36-7,25 (5H, м), 6,54 (1H, дд, J=5,2 Гц, 0,8 Гц), 4,37 (2H, д, J=4,8 Гц), 4,06 (3H, с), 3,83 (2H, с), 3,10 (1H, дд, J=12,0 Гц, 4,4 Гц), 3,02 (1H, дд, J=9,6 Гц, 7,2 Гц), 2,80-2,62 (3H, м); ESI-MS m/z 625 (МН+).
Пример 33
трет-Бутил 2-(4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксамидо)ацетат (33a)
Способом, подобным описанному для получения соединения 1, из соединения 1e (65,2 мг), гидрохлорида трет-бутилового эфира глицина (18,9 мг), N-гидрата DMTMM (39,9 мг) и триэтиламина (42,1 мкл) получали соединение 33a (67,2 мг, выход: 90%).
1H-ЯМР (400 Гц, CDCl3) δ: 12,51 (1H, с), 9,28 (1H, д, J=4,8 Гц), 8,66 (1H, д, J=5,6 Гц), 8,59 (1H, ушир.), 8,51 (1H, дд, J=4,8 Гц), 7,95 (1H, дд, J=12,0 Гц, 2,4 Гц), 7,46-7,36 (5H, м), 7,32-7,23 (3H, м), 6,44 (1H, д, J=4,8 Гц, 1,2 Гц), 4,24 (2H, д, J=4,8 Гц), 4,16 (3H, с), 3,76 (2H, с), 1,53 (9H, с); ESI-MS m/z 619 (МН+).
Гидрохлорид 2-(4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксамидо)уксусной кислоты (33b)
Способом, подобным описанному для получения соединения 1e, из соединения 33a (55,7 мг) получали соединение 33b (37,2 мг, выход: 63%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,55 (1H, с), 11,86 (1H, с), 8,98 (1H, д, J=6,0 Гц), 8,87 (1H, дд, J=5,6 Гц), 8,76 (1H, с), 8,12 (1H, дд, J=12,4 Гц, 1,2 Гц), 7,74 (1H, с), 7,64-7,62 (2H, м), 7,36―7,34 (5H, м), 7,32-7,27 (1H, м), 6,91 (1H, д, J=6,4 Гц), 4,09 (3H, с), 4,01 (2H, д, J=5,6 Гц), 3,83 (2H, с); ESI-MS m/z 563 (МН+).
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(2-(2-метоксиэтиламино)-2-оксоэтил)хинолин-6-карбоксамид (33)
Способом, подобным описанному для получения соединения 1, из соединения 33b (50 мг), 2-метоксиэтанамина (11 мкл), N-гидрата DMTMM (34,6 мг) и N-метилморфолина (18,4 мкл) получали указанное в заголовке соединение 33 (12,0 мг, выход: 23%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,51 (1H, с), 11,83 (1H, с), 8,76 (1H, с), 8,75 (1H, т, J=5,4 Гц), 8,71 (1H, д, J=5,4 Гц), 8,06-8,00 (2H, м), 7,59 (1H, с), 7,57-7,50 (2H, м), 7,38-7,26 (5H, м), 6,53 (1H, д, J=5,4 Гц), 4,07 (3H, с), 3,97 (2H, д, J=5,4 Гц), 3,84 (2H, с), 3,39-3,35 (2H, м), 3,30-3,27 (2H, м), 3,25 (3H, с); ESI-MS m/z 620 (МН+).
Пример 34
(S)-4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-N-(2-(2-(гидроксиметил)пирролидин-1-ил)-2-оксоэтил)-7-метоксихинолин-6-карбоксамид (34)
Способом, подобным описанному для получения соединения 1, из соединения 33b (50 мг), (S)-пирролидин-2-илметанола (11 мкл), N-гидрата DMTMM (30 мг) и N-метилморфолина (24 мкл) получали указанное в заголовке соединение 34 (32 мг, выход: 60%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,51 (1H, ушир.с), 11,83 (1H, ушир.с), 8,88-8,83 (2H, м), 8,72 (1H, д, J=5,1 Гц), 8,04 (1H, д, J=12,2 Гц), 7,63-7,51 (3H, м), 7,39-7,26 (5H, м), 6,54 (1H, д, J=5,1 Гц), 4,74 (1H, т, J=5,5 Гц), 4,39-4,22 (1H, м), 4,18-4,13 (1H, м), 4,10 (3H, с), 4,05-3,95 (1H, м), 3,84 (2H, с), 3,56-3,41 (3H, м), 2,02-1,76 (5H, м); ESI-MS m/z 646 (МН+).
Пример 35
N-(2-(Этил(2-гидрокси-2-метилпропил)амино)-2-оксоэтил)-4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксамид (35)
Способом, подобным описанному для получения соединения 1, из соединения 33b (35,0 мг), 1-(этиламино)-2-метилпропан-2-ола (17,1 мг) и N-гидрата DMTMM (19,4 мг) получали указанное в заголовке соединение 35 (12,3 мг, выход: 32%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,50 (1H, с), 11,82 (1H, с), 8,86 (1H, с ушир.), 8,84 (1H, д, J=2,7 Гц), 8,71 (1H, д, J=5,4 Гц), 8,03 (1H, дд, J=11,7 Гц, 2,0 Гц), 7,59 (1H, с), 7,57-7,52 (2H, м), 7,37-7,32 (4H, м), 7,21-7,25 (1H, м), 6,53 (1H, д, J=5,6 Гц), 4,32-4,27 (2H, м), 4,09, 4,07 (3H, с), 3,82 (2H, с), 3,50-3,20 (4H, м), 1,18-1,13 (5H, м), 1,09-1,01 (5H, м); ESI-MS m/z 662 (МН+).
Пример 36
трет-Бутил 2-(4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксамидо)пропаноат (36a)
Способом, подобным описанному для получения соединения 1, из соединения 1e (100 мг), гидрохлорида трет-бутилового эфира аланина (47 мг) и N-гидрата DMTMM (71 мг) получали соединение 36a (109 мг, выход: 87%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,57 (1H, с), 11,89 (1H, с), 8,76 (1H, д, J=5,2 Гц), 8,65 (1H, с), 8,61 (1H, т, J=5,2 Гц), 8,10 (1H, д, J=12,4 Гц), 7,60-7,30 (8H, м), 6,60 (1H, д, J=5,2 Гц), 4,08 (3H, с), 3,90 (2H, с), 3,57 (2H, тд, J=6,5 Гц, J=6,5 Гц), 2,57 (2H, т, J=6,5 Гц), 1,47 (9H, с); ESI-MS m/z 633 (МН+).
Гидрохлорид 2-(4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксамидо)пропановой кислоты (36b)
Способом, подобным описанному для получения соединения 1e, из соединения 36a (95 мг) получали соединение 36b (92 мг, выход: 100%).
1H-ЯМР (400 Гц, ДМСО-d6) δ; 12,55 (1H, с), 11,86 (1H, с), 8,94 (1H, д, J=5,9 Гц), 8,68 (1H, с), 8,65 (1H, т, J=5,9 Гц), 8,11 (1H, д, J=12,4 Гц), 7,69-7,58 (3H, м), 7,38-7,25 (5H, м), 6,87 (1H, д, J=5,9 Гц), 4,05 (3H, с), 3,84 (2H, с), 3,54 (2H, тд, J=6,7 Гц, J=6,7 Гц), 2,55 (2H, т, J=6,7 Гц); ESI-MS m/z 577 (МН+).
N-(3-(Диметиламино)-3-оксопропил)-4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метоксихинолин-6-карбоксамид (36)
Способом, подобным описанному в примере 1, из соединения 36b (30 мг), диметиламин гидрохлорида (6,0 мг), N-гидрата DMTMM (20 мг) и N-метилморфолина (16 мкл) получали указанное в заголовке соединение 36 (21,2 мг, выход: 68%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,51 (1H, с), 11,83 (1H, с), 8,72 (1H, с) 8,70 (1H, д, J=5,2 Гц), 8,66 (1H, т, J=5,9 Гц), 8,04 (1H, д, J=12,2 Гц), 7,58-7,25 (8H, м), 6,53 (1H, д, J=5,2 Гц), 4,04 (3H, с), 3,84 (2H, с), 3,54 (2H, тд, J=6,4 Гц, J=6,4 Гц), 2,97 (3H, с), 2,85 (3H, с), 2,61 (2H, т, J=6,4 Гц); ESI-MS m/z 604 (МН+).
Пример 37
4-(2-Фтор-4-(3-(2-(4-фторфенил)ацетил)тиоуреидо)фенокси)-7-метокси-N-(2-морфолиноэтил)хинолин-6-карбоксамид (37)
Способом, подобным описанному в примере 1, из соединения 19b (523 мг), 2-морфолиноэтанамина (171 мг) и N-гидрата DMTMM (360 мг) получали указанное в заголовке соединение 37 (462 мг, выход: 73%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,47 (1H, с), 11,82 (1H, с), 8,70 (1H, д, J=5,2 Гц), 8,70 (1H, с) 8,53-8,48 (1H, м), 8,02 (1H, д, J=13,0 Гц), 7,59-7,48 (3H, м), 7,43-7,33 (2H, м), 7,24-7,13 (2H, м), 6,53 (1H, д, J=5,2 Гц), 4,07 (3H, с), 3,84 (2H, с), 3,63-3,59 (4H, м), 3,49-3,32 (6H, м), 2,50-2,40 (2H, м); ESI-MS m/z 636 (МН+).
Пример 38
(S)-4-(2-Фтор-4-(3-(2-(4-фторфенил)ацетил)тиоуреидо)фенокси)-N-(1-гидроксибутан-2-ил)-7-метоксихинолин-6-карбоксамид (38)
Способом, подобным описанному в примере 1, из соединения 19b (50 мг), (S)-2-аминобутан-1-ола (12 мкл) и N-гидрата DMTMM (34 мг) получали указанное в заголовке соединение 38 (25 мг, выход: 45%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,48 (1H, с ушир.), 11,82 (1H, с ушир.), 8,69 (1H, д, J=5,2 Гц), 8,57 (1H, с), 8,13 (1H, д, J=8,3 Гц), 8,03 (1H, д, J=13,2 Гц), 7,58-7,50 (3H, м), 7,39 (2H, дд, J=8,5 Гц, J=5,6 Гц), 7,22-7,15 (2H, м), 6,52 (1H, д, J=5,2 Гц), 4,77 (1H, т, J=5,6 Гц), 4,03 (3H, с), 3,93-3,86 (1H, м), 3,84 (2H, с), 3,55-3,48 (1H, м), 3,45-3,40 (1H, м), 1,72-1,63 (1H, м), 1,53-1,42 (1H, м), 0,93 (3H, т, J=7,4 Гц); ESI-MS m/z 595 (МН+).
Пример 39
Метил 4-(2-фтор-4-нитрофенокси)-7-метоксихинолин-6-карбоксилат (39a)
Из метил 4-хлор-7-метоксихинолин-6-карбоксилата (синтезированного в соответствии с описанным в документе WO 2005/080377) (1,00 г), 2-фтор-4-нитрофенола (936 мг) и N,N-диизопропилэтиламина (1,35 мл) получали соединение 39a (1,38 г, выход: 93%).
1H-ЯМР (CDCl3)δ: 8,74 (1H, с), 8,73 (1H, д, J=5,2 Гц), 7,54 (1H, с), 7,45-7,40 (3H, м), 6,49 (1H, дд, J=5,0 Гц, 1,4 Гц), 4,06 (3H, с), 3,98 (3H, с); ESI-MS m/z 373 (МН+).
Метил 4-(4-амино-2-фторфенокси)-7-метоксихинолин-6-карбоксилат (39b)
Способом, подобным описанному для получения соединения 1b, из соединения 39a (275 мг), порошка железа (206 мг) и хлорида аммония (275 мг) получали соединение 39b (188 мг, выход: 74%).
1H-ЯМР (400 Гц, CDCl3) δ: 8,83 (1H, с), 8,63 (1H, д, J=5,2 Гц), 7,48 (1H, с), 7,03 (1H, т, J=8,4 Гц), 6,56 (1H, дд, J=11,6 Гц, 2,8 Гц), 6,50 (1H, ддд, J=8,8 Гц, 2,6 Гц, 1,0 Гц), 6,41 (1H, дд, J=5,0 Гц, 1,2 Гц), 4,04 (3H, с), 3,97 (3H, с), 3,84 (2H, с ушир.); ESI-MS m/z 343 (МН+).
4-(4-Амино-2-фторфенокси)-7-метоксихинолин-6-карбоновая кислота (39c)
Соединение 39b (1,0 г) добавляли к метанолу (10 мл) и затем добавляли 4M водный гидроксид натрия (650 мкл) и воду (400 мкл), далее перемешивали при комнатной температуре в течение 2 часов. После завершения реакции к реакционной смеси добавляли 6н водную соляную кислоту, таким образом устанавливая pH 3, и образовавшийся осадок фильтровали, в результате чего получали соединение 39c (862 мг, выход: 90%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 8,66 (1H, д, J=5,4 Гц), 8,54 (1H, с), 7,48 (1H, с), 7,09 (1H, дд, J=8,8 Гц), 6,55 (1H, дд, J=13,0 Гц, 2,7 Гц), 6,48-6,43 (2H, м), 5,55 (1H, с ушир.), 3,96 (3H, с); ESI-MS m/z 329 (МН+).
4-(4-(3-(2-(2-Фторфенил)ацетилтиоуреидо)-2-фторфенокси)-7-метоксихинолин-6-карбоновая кислота (39d)
Способом, подобным описанному для получения соединения 1d, из соединения 39c (1,79 г), 2-фторфенилацетил тиоизоцианата (1,97 г) и смеси растворителей N,N-диметилацетамида (30 мл), толуола (30 мл) и этанола (6 мл), получали карбоновую кислоту 39d в виде сырого продукта (1,89 г, выход: 89%). Сырой продукт использовали в последующей реакции без дополнительной очистки.
4-(2-Фтор-4-(3-(2-(2-фторфенил)ацетил)тиоуреидо)фенокси)-N-(2-гидрокси-2-метилпропил)-7-метоксихинолин-6-карбоксамид (39)
Способом, подобным описанному для получения соединения 1, из соединения 39d (126 мг), N-гидрата DMTMM (87 мг) и 1-амино-2-метилпропан-2-ола (37 мг) получали указанное в заголовке соединение 39 (89 мг, выход: 62%).
1H-ЯМР (400 Гц, CDCl3) δ: 12,43 (1H, с), 9,26 (1H, с), 8,67 (1H, д, J=5,1 Гц), 8,59 (1H, м), 8,26 (1H, м), 7,97 (1H, дд, J=11,5 Гц, 2,4 Гц), 7,54 (1H, с), 7,44-7,15 (6H, м), 6,44 (1H, дд, J=5,4 Гц, 1,2 Гц), 4,13 (3H, с), 3,79 (2H, с), 3,57 (2H, д, J=5,8 Гц), 2,57 (1H, с), 1,33 (6H, с); ESI-MS m/z 595 (МН+).
Пример 40
(S)-4-(2-Фтор-4-(3-(2-(2-фторфенил)ацетил)тиоуреидо)фенокси)-N-(1-гидроксибутан-2-ил)-7-метоксихинолин-6-карбоксамид (40)
Способом, подобным описанному для получения соединения 1, из соединения 39d (121 мг), N-гидрата DMTMM (83 мг) и (S)-2-аминобутан-1-ола (28 мг) получали указанное в заголовке соединение 40 (84 мг, выход: 61%).
1H-ЯМР (400 Гц, CDCl3) δ: 12,44 (1H, с), 9,25 (1H, с), 8,67 (1H, д, J=5,1 Гц), 8,64 (1H, с), 8,03 (1H, д, J=7,6 Гц), 7,97 (1H, дд, J=11,6 Гц, 2,6 Гц), 7,54 (1H, с), 7,44-7,14 (6H, м), 6,45 (1H, дд, J=5,1 Гц, 1,2 Гц), 4,12 (3H, с), 3,86 (1H, м), 3,79 (2H, с), 3,75 (1H, м), 3,07 (1H, т, J=5,5 Гц), 1,82-1,60 (2H, м), 1,07 (3H, т, J=7,5 Гц); ESI-MS m/z 595 (МН+).
Пример 41
(S)-4-(4-Амино-2-фторфенокси)-N-(1-гидроксибутан-2-ил)-7-метоксихинолин-6-карбоксамид (41a)
Способом, подобным описанному для получения соединения 1, из соединения 39c (300 мг), N-гидрата DMTMM (329 мг) и (S)-2-аминобутан-1-ола (113 мкл) получали соединение 41a (297 мг, выход: 81%).
1H-ЯМР (ДМСО-d6) δ: 8,64 (1H, д, J=5,1 Гц), 8,56 (1H, с), 8,12 (1H, д, J=8,3 Гц), 7,51 (1H, с), 7,09 (1H, т, J=9,0 Гц), 6,56 (1H, дд, J=13,3 Гц, J=2,3 Гц), 6,50-6,43 (2H, м), 5,52 (2H, с), 4,78 (1H, т, J=5,5 Гц), 4,01 (3H, с), 3,95-3,85 (1H, м), 3,56-3,48 (1H, м), 3,46-3,38 (1H, м), 1,74-1,62 (1H, м), 1,54-1,41 (1H, м), 0,93 (3H, т, J=7,4 Гц); ESI-MS m/z 400 (МН+).
(S)-4-(2-Фтор-4-(3-(2-(3-фторфенил)ацетил)тиоуреидо)фенокси)-N-(1-гидроксибутан-2-ил)-7-метоксихинолин-6-карбоксамид (41)
Способом, подобным описанному для получения соединения 1d, из соединения 41a (100 мг) и 3-фторфенилацетил тиоизоцианата (73 мг) получали указанное в заголовке соединение 41 (115 мг, выход: 78%).
1H-ЯМР (ДМСО-d6) δ: 12,44 (1H, с), 11,83 (1H, с), 8,69 (1H, д, J=5,3 Гц), 8,57 (1H, с), 8,13 (1H, д, J=8,3 Гц), 8,03 (1H, д, J=12,2 Гц), 7,59-7,49 (3H, м), 7,44-7,36 (1H, м), 7,23-7,09 (3H, м), 6,52 (1H, д, J=5,3 Гц), 4,77 (1H, т, J=5,5), 4,03 (3H, с), 3,93-3,84 (1H, м), 3,88 (2H, с), 3,54-3,48 (1H, м), 3,45-3,38 (1H, м), 1,72-1,62 (1H, м), 1,54-1,43 (1H, м), 0,93 (3H, т, 7,4 Гц); ESI-MS m/z 595 (МН+).
Пример 42
4-(4-Амино-2-фторфенокси)-N-(2-гидрокси-2-метилпропил)-7-метоксихинолин-6-карбоксамид (42a)
Способом, подобным описанному для получения соединения 1, из соединения 39c (103 мг), N-гидрата DMTMM (104 мг) и 1-амино-2-метилпропан-2-ола (42 мг) получали соединение 42a (66,3 мг, выход: 53%).
1H-ЯМР (400 Гц, CDCl3) δ: 9,27 (1H, с), 8,64 (1H, д, J=5,2 Гц), 8,26 (1H, с ушир.), 7,52 (1H, с), 7,02 (1H, дд, J=8,4 Гц), 6,56 (1H, дд, J=12,0 Гц, 2,8 Гц), 6,50 (1H, ддд, J=8,8 Гц, 2,8 Гц, 0,8 Гц), 6,42 (1H, дд, J=5,4 Гц, 1,2 Гц), 4,12 (3H, с), 3,82 (1H, ушир.), 3,57 (2H, д, J=6,0 Гц), 2,70 (1H, с ушир.), 1,33 (6H, с); ESI-MS m/z 400 (МН+).
4-(4-(3-(2-(4-Хлорфенил)ацетил)тиоуреидо)-2-фторфенокси)-N-(2-гидрокси-2-метилпропил)-7-метоксихинолин-6-карбоксамид (42)
Способом, подобным описанному для получения соединения 1d, из соединения 42a (55 мг) и 4-хлорфенилацетил тиоизоцианата (43,7 мг) получали указанное в заголовке соединение 42 (41,3 мг, выход: 49%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,45 (1H, с), 11,82 (1H, с), 8,71 (1H, д, J=5,4 Гц), 8,67 (1H, с), 8,35 (1H, т, J=6,1 Гц), 8,02 (1H, д, J=11,0 Гц), 7,58-7,49 (3H, м), 7,43-7,32 (4H, м), 6,55 (1H, д, J=5,4 Гц), 4,04 (3H, с), 3,84 (2H, с), 3,36-3,30 (2H, м), 1,98 (1H, ушир.), 1,15 (6H, с); ESI-MS m/z 611, 613 (МН+).
Пример 43
(S)-4-(4-(3-(2-(4-Хлорфенил)ацетил)тиоуреидо)-2-фторфенокси)-N-(1-гидроксибутан-2-ил)-7-метоксихинолин-6-карбоксамид (43)
Способом, подобным описанному для получения соединения 1d, из соединения 41a (63,0 мг) и 4-хлорфенилацетил тиоизоцианата (50,1 мг) получали указанное в заголовке соединение 43 (29,9 мг, выход: 31%).
1H-ЯМР (400 Гц, CD3OD) δ: 8,85 (1H, с), 8,63 (1H, д, J=5,6 Гц), 8,07 (1H, дд, J=12,0 Гц, 2,4 Гц), 7,52 (1H, с), 7,50-7,30 (7H, м), 6,60 (1H, дд, J=5,4 Гц, 1,0 Гц), 4,11 (3H, с), 4,08-4,02 (1H, м ушир.), 3,76 (2H, с), 3,67 (2H, дд, 4,6 Гц), 3,27-3,22 (1H, м), 1,80-1,73 (1H, м), 1,65-1,57 (1H, м), 1,18 (2H, с), 1,04 (3H, т, J=7,6 Гц); ESI-MS m/z 611, 613 (МН+).
Пример 44
4-(4-(3-(2-(2,6-Дифторфенил)ацетил)тиоуреидо)-2-фторфенокси)-7-метоксихинолин-6-карбоновая кислота (44a)
Способом, подобным описанному для получения соединения 1d, из соединения 39c (98 мг), 2,6-дифторфенилацетил тиоизоцианата (128 мг) и смеси растворителей N,N-диметилацетамида (1,5 мл), толуола (1,5 мл) и этанола (300 мкл) получали соединение 44a в виде сырого продукта (143 мг, выход: 89%).
4-(4-(3-(2-(2,6-Дифторфенил)ацетилтиоуреидо)-2-фторфенокси)-7-метокси-N-(2-морфолиноэтил)хинолин-6-карбоксамид (44)
Способом, подобным описанному для получения соединения 1, из соединения 44a (143 мг), N-гидрата DMTMM (95 мг), 2-морфолиноэтанамина (51 мг) и N,N-диметилацетамида (1 мл) получали указанное в заголовке соединение 44 (103 мг, выход: 60%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,35 (1H, с ушир.), 11,98 (1H, с ушир.), 8,70 (1H, д, J=5,3 Гц), 8,70 (1H, с), 8,50 (1H, т, J=5,4 Гц), 8,03 (1H, д ушир., J=13,5 Гц), 7,57 (1H, с), 7,61-7,39 (3H, м), 7,18-7,10 (2H, м), 6,53 (1H, д, J=5,3 Гц), 4,07 (3H, с), 3,98 (2H, с), 3,62-3,58 (4H, м), 3,50-3,47 (2H, м), 3,47-3,20 (4H, м), 2,50-2,47 (2H, м); ESI-MS m/z 654 (МН+).
Пример 45
4-(4-(3-(2-(2,6-Дифторфенил)ацетилтиоуреидо)-2-фторфенокси)-N-(2-гидрокси-2-метилпропил)-7-метоксихинолин-6-карбоксамид (45)
Способом, подобным описанному для получения соединения 1, из соединения 44a (101 мг), N,N-диметилацетамида (600 мкл), N-гидрата DMTMM (68 мг) и 1-амино-2-метилпропан-2-ола (31 мг) получали указанное в заголовке соединение 45 (74 мг, выход: 65%).
1H-ЯМР (400 Гц, CDCl3) δ: 12,38 (1H, с), 9,26 (1H, с), 8,69 (1H, с ушир.), 8,67 (1H, д, J=5,4 Гц), 8,26 (1H, м), 7,97 (1H, дд, J =11,5 Гц, 2,7 Гц), 7,54 (1H, с), 7,43-7,32 (2H, м), 7,04-6,96 (3H, м), 6,44 (1H, дд, J=5,2 Гц, 1,1 Гц), 4,13 (3H, с), 3,84 (2H, с), 3,57 (2H, д, J=5,9 Гц), 2,58 (1H, с), 1,33 (6H, с); ESI-MS m/z 613 (МН+).
Пример 46
4-(4-Амино-2-фторфенокси)-7-метокси-N-метилхинолин-6-карбоксамид (46a)
Соединение 39b (100 мг) растворяли в N-метилпиперидин-2-оне (250 мкл) и добавляли 40%-ный раствор метиламинметанола (250 мкл), затем перемешивали при температуре 40°C в течение 16 часов. Затем к реакционной смеси добавляли воду, и образовавшийся осадок фильтровали, получая таким образом соединение 46a (63,7 мг, выход: 64%).
1H-ЯМР (400 Гц, CDCl3) δ: 9,28 (1H, с), 8,63 (1H, д, J=5,4 Гц), 7,84 (1H, ушир.), 7,50 (1H, с), 7,02 (1H, т, J=8,6 Гц), 6,56 (1H, дд, J=12,0 Гц, 2,4 Гц), 6,50 (1H, ддд, J=8,4 Гц, 2,8 Гц, 0,8 Гц), 6,43 (1H, дд, J=5,2 Гц, 1,2 Гц), 4,11 (3H, с), 3,83, 3,80 (2H, ушир.), 3,08 (3H, д, J=5,0 Гц); ESI-MS m/z 342 (МН+).
4-(2-Фтор-4-(3-(2-(3-метоксифенил)ацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид (46)
Способом, подобным описанному для получения соединения 1d, из соединения 46a (50,0 мг) и 3-метоксифенилацетил изотиоцианата (45,5 мг) получали указанное в заголовке соединение 46 (40,1 мг, выход: 50%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,49 (1H, с), 11,79 (1H, с), 8,70 (1H, д, J=5,4 Гц), 8,59 (1H, с), 8,37 (1H, д, J=4,2 Гц), 8,03 (1H, дд, J=12,0 Гц, 2,4 Гц), 7,58-7,49 (3H, м), 7,42-7,33 (2H, м), 7,22-7,17 (2H, м), 6,54 (1H, д, J=4,4 Гц), 4,02 (3H, с), 3,79 (2H, с), 3,75 (3H, с), 2,83 (3H, д, J=4,8 Гц); ESI-MS m/z 549 (МН+).
Пример 47
4-(2-Фтор-4-(3-(2-(4-трифторметилфенил)ацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид (47)
Способом, подобным описанному для получения соединения 1d, из соединения 46a (50,0 мг) и 4-трифторметилфенилацетил изотиоцианата (53,9 мг) получали указанное в заголовке соединение 47 (41,2 мг, выход: 48%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,42 (1H, с), 11,87 (1H, с), 8,69 (1H, д, J=5,2 Гц), 8,59 (1H, с), 8,36 (1H, д, J=4,8 Гц), 8,02 (1H, дд, J=12,0 Гц, 2,0 Гц), 7,72 (2H, д, J=8,4 Гц), 7,60-7,47 (5H, м), 6,52 (1H, д, J=5,2 Гц), 4,02 (3H, с), 3,96 (2H, с), 2,83 (3H, д, J=4,8 Гц); ESI-MS m/z 587 (МН+).
Пример 48
Метил 4-(2-хлор-4-нитрофенокси)-7-метоксихинолин-6-карбоксилат (48a)
Способом, подобным описанному для получения соединения 1a, из метил 4-хлор-7-метоксихинолин-6-карбоксилата (350 мг), 2-хлор-4-нитрофенола (240 мг), N,N-диизопропилэтиламина (484 мкл) и N-метилпирролидин-2-она (1,5 мл) получали соединение 48a (130 мг, выход: 24%).
1H-ЯМР (400 Гц, CDCl3) δ: 8,73 (1H, с), 8,73 (1H, д, J=5,2 Гц), 8,48 (1H, д, J=2,8 Гц), 8,25 (1H, дд, J=8,8 Гц, 2,4 Гц), 7,55 (1H, с), 7,35 (1H, д, J=8,8 Гц), 6,42 (1H, д, J=4,8 Гц), 4,07 (3H, с), 3,98 (3H, с); ESI-MS m/z 389, 391 (МН+).
Метил 4-(4-амино-2-хлорфенокси)-7-метоксихинолин-6-карбоксилат (48b)
Способом, подобным описанному для получения соединения 1c, из соединения 48a (111 мг), смеси вода-метанол-тетрагидрофуран (1:1:1) (5 мл), порошка железа (49,7 мг) и хлорида аммония (111 мг) получали соединение 48b в виде сырого продукта (31,2 мг, выход: 31%).
ESI-MS m/z 359, 361 (МН+).
4-(4-Амино-2-хлорфенокси)-7-метокси-N-метилхинолин-6-карбоксамид (48c)
Способом, подобным описанному для получения соединения 46a, из соединения 48b (29,0 мг), 40% водного раствора метиламина (200 мкл) и N-метилпирролидин-2-она (200 мкл) получали соединение 48c (27,1 мг, выход: 94%).
1H-ЯМР (400 Гц, CDCl3) δ: 9,30 (1H, с), 8,61 (1H, д, J=5,6 Гц), 7,84 (1H, ушир.), 7,51 (1H, с), 7,02 (1H, д, J=8,4 Гц), 6,83 (1H, д, J=2,8 Гц), 6,64 (1H, дд, J=8,4 Гц, 2,8 Гц), 6,32 (1H, дд, J=5,4 Гц), 4,11 (3H, с), 3,78 (2H, ушир.), 3,08 (3H, д, J=6,0 Гц); ESI-MS m/z 358, 360 (МН+).
4-(2-Хлор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид (48)
Способом, подобным описанному для получения соединения 1d, из соединения 48c (24,0 мг) и фенилацетил тиоизоцианата (17,8 мг) получали указанное в заголовке соединение 48 (28,1 мг, выход: 79%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,49 (1H, с), 11,85 (1H, с), 8,94 (1H, д, J=6,0 Гц), 8,69 (1H, с), 8,47 (1H, д, J=4,8 Гц), 8,21 (1H, д, J=1,6 Гц), 7,78 (1H, дд, J=8,6 Гц, 2,0 Гц), 7,61 (1H, д, J=8,8 Гц), 7,59 (1H, с), 7,35-7,13 (5H, м), 6,69 (1H, д, J=5,8 Гц), 4,07 (3H, с), 3,83 (2H, с), 2,84 (3H, д, J=4,4 Гц); ESI-MS m/z 535, 537 (МН+).
Пример 49
Метил 4-(3-фтор-4-нитрофенокси)-7-метоксихинолин-6-карбоксилат (49a)
Способом, подобным описанному для получения соединения 1a, из метил 4-хлор-7-метоксихинолин-6-карбоксилата (300 мг), 3-фтор-4-нитрофенола (225 мг), N,N-диизопропилэтиламина (415 мкл) и N-метилпирролидин-2-она (1,5 мл) получали соединение 49a (112 мг, выход: 25%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 8,82 (1H, д, J=5,1 Гц), 8,45 (1H, с), 7,67 (1H, дд, J=12,2 Гц, 2,7 Гц), 7,59 (1H, с), 7,32 (1H, дд, J=8,8 Гц, 2,7 Гц), 6,83-6,74 (1H, м), 3,98 (3H, с), 3,84 (3H, с); ESI-MS m/z 373 (МН+).
Метил 4-(4-амино-3-фторфенокси)-7-метоксихинолин-6-карбоксилат (49b)
Способом, подобным описанному для получения соединения 1b, из соединения 49a (102 мг), порошка железа (76,5 мг) и хлорида аммония (100 мг) получали соединение 49b (59,7 мг, выход: 64%).
1H-ЯМР (400 Гц, CDCl3) δ: 8,79 (1H, с), 8,63 (1H, д, J=5,2 Гц), 7,49 (1H, с), 6,91-6,80 (3H, м), 6,44 (1H, д, J=5,2 Гц), 4,05 (3H, с), 3,97 (3H, с), 3,78, 3,75 (2H, ушир.); ESI-MS m/z 343 (МН+).
4-(4-Амино-3-фторфенокси)-7-метокси-N-метилхинолин-6-карбоксамид (49c)
Способом, подобным описанному для получения соединения 46a, из соединения 49b (50,5 мг), 40% водного раствора метиламина (500 мкл) и N-метилпирролидин-2-она (500 мкл) получали соединение 49c (31,2 мг, выход: 62%).
1H-ЯМР (400 Гц, CDCl3) δ: 9,24 (1H, с), 8,62 (1H, д, J=5,6 Гц), 7,86 (1H, с ушир.), 7,50 (1H, с), 6,90-6,79 (3H, м), 6,46 (1H, д, J=5,2 Гц), 4,11 (3H, с), 3,76, 3,74 (2H, ушир.), 3,08 (3H, д, J=5,0 Гц); ESI-MS m/z 342 (МН+).
4-(3-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид (49)
Способом, подобным описанному для получения соединения 1d, из соединения 49c (25,0 мг) и фенилацетил тиоизоцианата (19,5 мг) получали указанное в заголовке соединение 49 (13,5 мг, выход: 36%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,51 (1H, с), 11,89 (1H, с), 8,71 (1H, д, J=4,8 Гц), 8,55 (1H, с), 8,34 (1H, д, J=4,8 Гц), 8,06 (1H, дд, J=8,8 Гц), 7,53 (1H, с), 7,42 (1H, дд, J=10,8 Гц, 2,8 Гц), 7,37―7,25 (5H, м), 7,19-7,15 (1H, м), 6,62 (1H, д, J=5,6 Гц), 4,01 (3H, с), 3,83 (2H, с), 2,82 (3H, д, J=4,8 Гц); ESI-MS m/z 519 (МН+).
Пример 50
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N,N-диметилхинолин-6-карбоксамид (50)
Способом, подобным описанному для получения соединения 1, из соединения 1e (285 мг), 50% водного раствора диметиламина (147 мкл) и N-гидрата DMTMM (174 мг) получали указанное в заголовке соединение 50 (256 мг, выход: 91%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,49 (1H, с), 11,81 (1H, с), 8,66 (1H, дд, J=5,4 Гц, 1,2 Гц), 8,06 (1H, д, J=1,0 Гц), 8,01 (1H, д, J=12,4 Гц), 7,56-7,47 (2H, м), 7,52 (1H, с), 7,38-7,32 (4H, м), 7,31―7,25 (1H, м), 6,52 (1H, д, J=5,1 Гц), 3,97 (3H, д, J=0,8 Гц), 3,83 (2H, с), 3,01 (3H, д, J=1,0 Гц), 2,79 (3H, д, J=1,2 Гц); ESI-MS m/z 533 (МН+).
Пример 51
N-(3-Фтор-4-(7-метокси-6-(4-(пирролидин-1-ил)пиперидин-1-карбонил)хинолин-4-илокси)фенилкарбамотиоил)-2-фенилацетамид (51)
Способом, подобным описанному для получения соединения 1, из соединения 1e (27,6 мг), 4-(пирролидин-1-ил)пиперидина (13,8 мг) и N-гидрата DMTMM (18,1 мг) получали указанное в заголовке соединение 51 (14,1 мг, выход: 39%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,50 (1H, с), 11,83 (1H, с), 8,68 (1H, д, J=5,1 Гц), 8,08 (1H, д, J=14,1), 8,02 (1H, д, J=12,4 Гц), 7,58-7,45(2H, м), 7,53 (1H, с), 7,40-7,33 (4H, м), 7,33-7,26 (1H, м), 6,53 (1H, дд, J=4,6 Гц), 4,40 (1H, д, J=11,7 Гц), 3,98 (3H, д, J=9,3 Гц), 3,84 (2H, с), 3,10-2,90 (2H, м), 2,75-2,60 (4H, м), 2,05-1,92 (1H, м), 1,85-1,67 (6H, м), 1,55-1,20 (3H, м); ESI-MS m/z 656 (МН+).
Пример 52
N-(3-Фтор-4-(6-(3-гидроксипирролидин-1-карбонил)-7-метоксихинолин-4-илокси)фенилкарбамотиоил)-2-фенилацетамид (52)
Способом, подобным описанному для получения соединения 1, из соединения 1e (20,0 мг), пирролидин-3-ола (9,3 мг) и N-гидрата DMTMM (11,8 мг) получали указанное в заголовке соединение 52 (15,0 мг, выход: 71%).
1H-ЯМР (400 Гц, ДМСО-d6) δ: 12,51 (1H, с), 11,85 (1H, с), 8,93 (1H, д, J=6,4 Гц), 8,31 (1H, с), 8,10 (1H, д, J=12,4 Гц), 7,71 (1H, с), 7,61-7,47 (2H, м), 7,40―7,18 (5H, м), 6,87 (1H, д, J=6,1 Гц), 4,33 (1H, ушир.), 4,23 (1H, ушир.), 4,03 (3H, с), 3,83 (2H, с), 2,00-1,80 (4H, м), 1,77-1,72 (2H, м); ESI-MS m/z 575 (МН+).
Сравнительный пример 1
4-(2-Фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-6, 7-диметокси-хинолин-6-карбоксамид (сравнительное соединение 1)
Указанное в заголовке соединение было синтезировано в соответствии с описанным в документе WO 2006/104161.
Пример исследования 1
Тест по определению c-Met ингибирующей активности (in vitro)
Ингибирующую активность соединения в отношении c-Met киназы определяли следующими способами.
Способ A) анализ ингибирования c-Met с использованием AlphaScreenTM
В качестве субстрата был использован биотинилированный пептид, включающий фосфорилированный сайт Pyk2 (Tyr402), который был указан как био-субстрат в документе Clin. Cancer Res. vol. 8, (2), pp. 620-7 (2002). В присутствии соединения по настоящему изобретению, субстрат, c-Met (08-051, Carna bio Co., Ltd) и ATP (конечная концентрация: 20 мкМ) добавляли к реакционному буферу (60 мМ HEPES (pH: 7,5), 5 мМ MgCl2, 5 мМ MnCl2, 3 мкМ Na3VO4 и 1,25 мМ DTT). Смесь оставляли взаимодействовать при комнатной температуре в течение 20 минут. К реакционной смеси добавляли EDTA до конечной концентрация в 50 мМ, таким образом останавливая реакцию. К реакционной смеси добавляли определяющую жидкость, которую получали в соответствии с протоколом AlphaScreenTM Phosphotyrosine (P-Tyr-100) Assay Kit (phosphotyrosine-recognizing antibody-bound, 6760620C, Perkin Elmer). Реакцию проводили в течение одного часа при комнатной температуре. Затем измеряли интенсивность флуоресценции реакционной смеси, используя мульти-меченый счетчик (EnVisionTM, Perkin Elmer). Концентрацию соединения, которая дает 50% ингибирования образования фосфорилированного продукта, обозначали как IC50 (мкМ), и результаты показаны в следующих таблицах.
Способ B) анализ ингибирования c-Met с использованием DeskTop Profiler
Смесь ингибиторов фермента дефосфорилирования (PhosSTOP, #4906837, продукт от фирмы Roche) и смесь ингибиторов протеазы (Complete, Mini, без EDTA, #1836170; продукт от фирмы Roche) добавляли к реакционному буферу (100 мМ HEPES (pH: 7,5), 10 мМ MgCl2, 0,003% Brij-35, 0,04% Твин и 1 мМ DTT). В присутствии соединения по настоящему изобретению добавляли рекомбинантный c-Met (очищенный продукт от фирмы Taiho Pharmaceutical Co., Ltd.), флуоресцентно меченный c-Met субстратный пептид (FL-Пептид 2, #760346, Caliper Life Sciences) (конечная концентрация: 1,5 мкМ), и ATP (конечная концентрация: 43 мкМ), и смесь оставляли взаимодействовать при температуре 28°C в течение 90 минут. К реакционной смеси добавляли EDTA до конечной концентрации 10 мМ, таким образом останавливая реакцию. С помощью DeskTop Profiler (#119900, Caliper Life Sciences) определяли интенсивность каждой флуоресценции субстрата и фосфорилированного продукта, таким образом определяли количество образовавшегося фосфорилированного продукта. Концентрацию соединения, которая давала 50% ингибирования образования фосфорилированного продукта обозначали как IC50 (мкМ), и результаты представлены в следующих таблицах.
Таблица 1
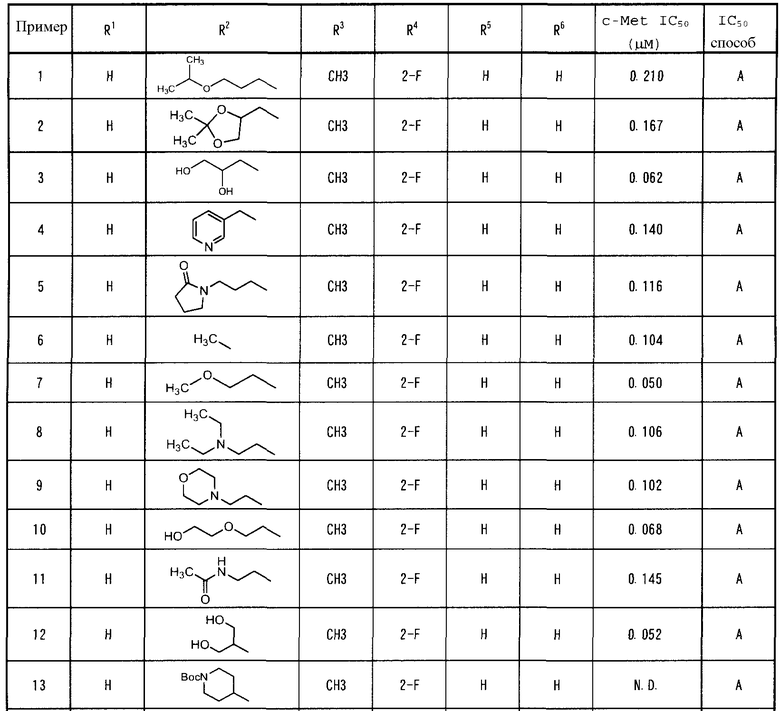
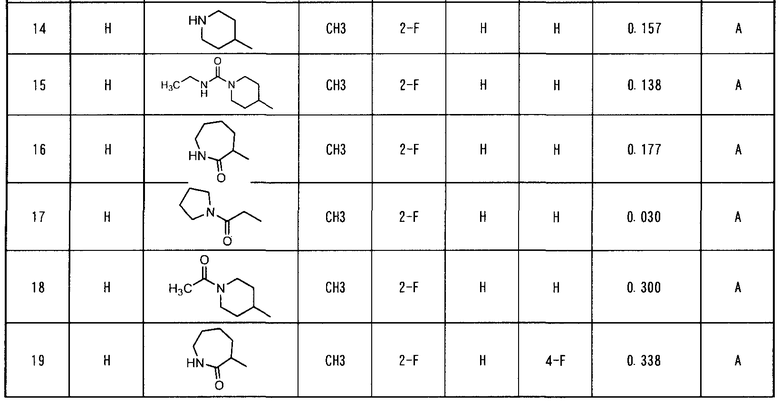
Таблица 2
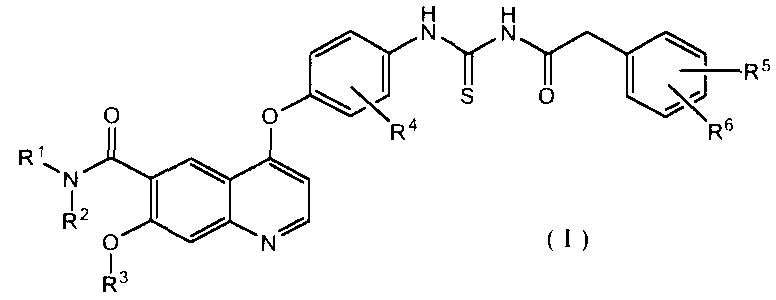
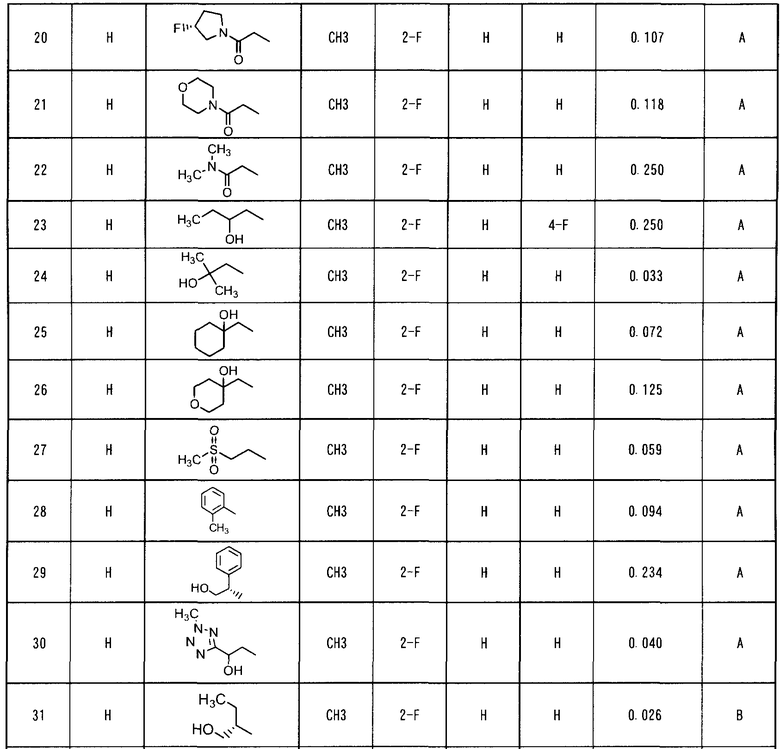
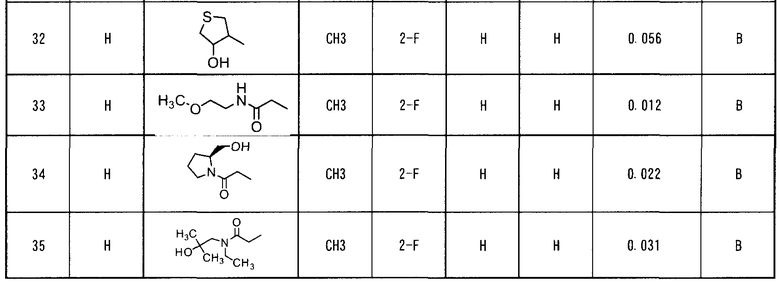
Таблица 3
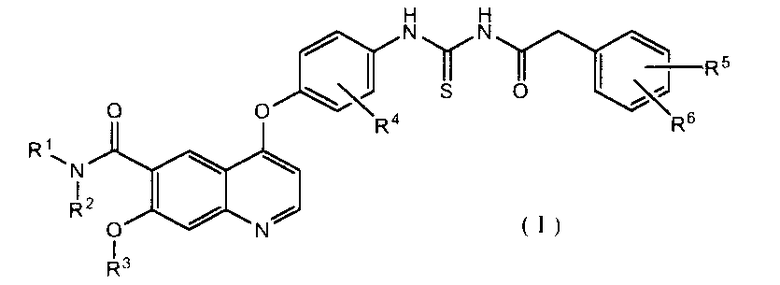
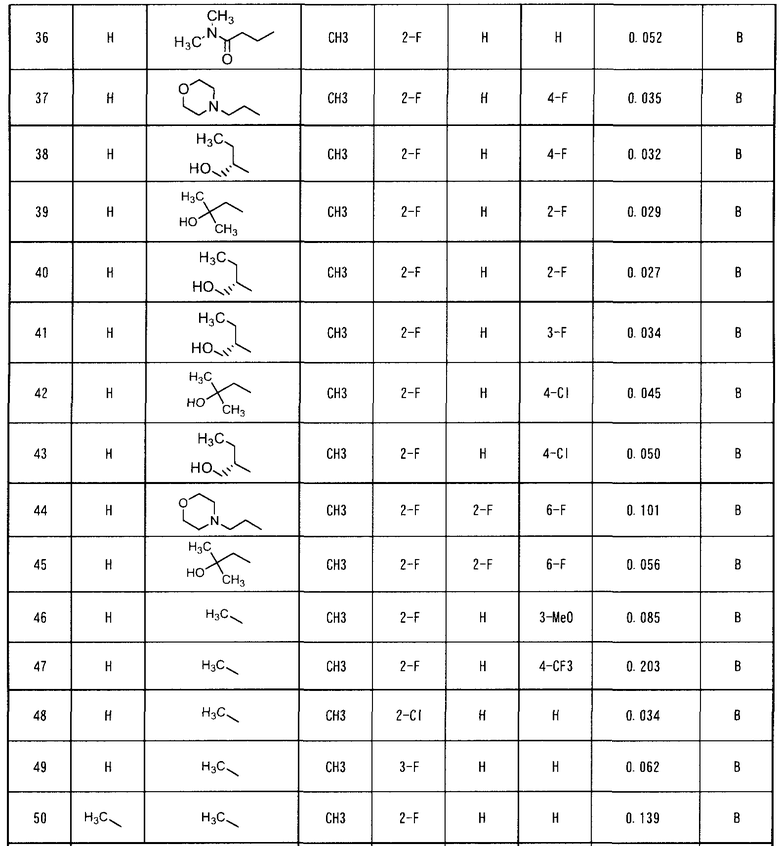
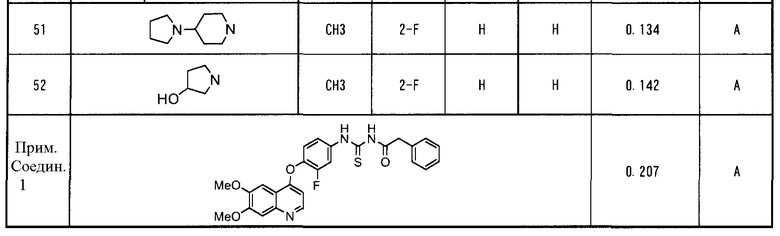
Также было исследовано сравнительное соединение (сравнительное соединение 1). Сравнительное соединение 1 имеет структуру, подобную структуре соединения по настоящему изобретению (описана в примерах патентного документа 6), и известно его применение в качестве лекарственного средства (опублик. в журнале Bioorg. Med. Chem. Lett., 18 (2008), 2793-2798). Исследование показало, что соединение по настоящему изобретению проявляет ингибирующую C-Met активность, которая равна или выше чем у сравнительного соединения 1.
Пример исследования 2
Анализ ингибирования клеточной пролиферации в отношении клеток NUGC4 (штамм раковых клеток желудка человека, в которых c-Met сверх экспрессируется и высоко активирован), исследование in vitro
Суспензию клеток NUGC4 (в 10% FBS-содержащей RPMI 1640 среде (продукт от фирмы Wako Pure Chemical Industries Ltd.) или FBS-содержащую DMEM среду (продукт от фирмы Nacalai Tesque, Inc.)) инокулировали в каждой лунке 96-луночного (плоскодонный) микропланшета в количестве 2×103 клетки (0,1 мл), и планшет инкубировали в инкубаторе в атмосфере 5% CO2 при температуре 37°C в течение одного дня. Каждое соединение по настоящему изобретению и сравнительное соединение 1 растворяли в диметил сульфоксиде до концентрации 30 мМ. Раствор разбавляли 10% FBS-содержащей RPMI 1640 или средой DMEM, до конечной концентрации исследуемого соединения 60, 20, 6, 2, 0,6 или 0,2 мкМ. Полученный таким образом раствор исследуемого соединения добавляли к каждой лунке планшета с культурой клеток NUGC4 при 0,1 мл/лунку, и планшет инкубировали в инкубаторе в атмосфере 5% CO2 при температуре 37°C в течение 3 дней. После культивирования к каждой лунке добавляли по 20 мкл 25% водного раствора глутаральдегида (продукт от фирмы Nacalai Tesque, Inc.), и планшет оставляли при комнатной температуре в течение 20 минут, посредством чего клетки фиксировались. После этого планшет промывали водопроводной водой и сушили. Затем к каждой лунке добавляли по 100 мкл/лунку водный раствор смеси 0,05% кристаллический фиолетовый/20% метанол (продукт от фирмы Wako Pure Chemical Industries Ltd.), и планшет оставляли стоять при комнатной температуре в течение 20 минут, посредством чего клетки окрашивались. После этого планшет промывали водопроводной водой и сушили. К каждой лунке добавляли 0,05 M NaH2PO4/этанол (1/1=об./об.) (100 мкл), таким образом экстрагируя кристаллическим фиолетовым. Абсорбцию экстрагированного кристаллического фиолетового измеряли при 540 нм, используя считывающее устройство для микропланшетов, и абсорбцию использовали в качестве показателя подсчета живых клеток. Процент ингибирования рассчитывали с помощью следующего уравнения, и рассчитывали концентрацию исследуемого соединения, ингибирующую на 50% (IC50 (мкМ)).
Процент ингибирования (%)=(C-T)/C×100
T: Абсорбция в лунке, в которую добавляли исследуемое соединение.
C: Абсорбция в лунке, в которую исследуемое соединение не добавляли.
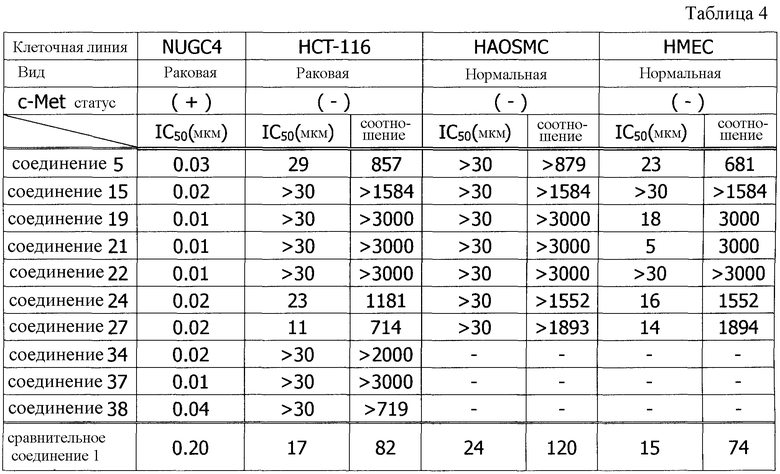
Как видно из таблицы 4, соединение по настоящему изобретению проявляет ингибирующую клеточную пролиферацию активность выше, чем у сравнительного соединения 1, в отношении NUGC4 (штамм раковых клеток желудка человека, в которых c-Met сверх экспрессируется и высоко активирован). Таким образом, подтверждается, что соединение по настоящему изобретению проявляет превосходную противоопухолевую активность.
Такое же исследование in vitro по ингибированию клеточной пролиферации осуществляли в отношении низко экспрессирующих c-Met опухолевых клеток (HCT-116), нормальных клеток (HAOSMC (клетки гладких мышц аорты человека)) и нормальных клеток (клетки HMEC эндотелия капилляров кожи человека). Сравнительное соединение 1 показало IC50 от 15 до 24 мкМ на штаммах указанных клеток, тогда как большинство соединений по настоящему изобретению показало IC50≥30 мкМ. Таким образом, подтверждалось, что соединения по настоящему изобретению проявляют ингибирующую пролиферацию клеток активность, которая равна или ниже, чем у сравнительного соединения 1, в отношении клеточных штаммов с низкой экспрессией c-Met. Другими словами, при сопоставлении со сравнительным соединением 1, отличие (соотношение) между IC50 соединения по настоящему изобретению в отношении клеток с низкой экспрессией c-Met или нормальных клеток и в отношении раковых клеток со сверхэкспрессией c-Met является значительно больше. На основе данного открытия, было подтверждено, что соединение по настоящему изобретению проявляет ингибирующую клеточную пролиферацию активность с высокой клеточной избирательностью.
Пример исследования 3
Исследование по определению дозы для оценки противоопухолевого действия (in vivo)
С целью определения дозы для оценки противоопухолевого действия, каждое из соединения по настоящему изобретению и сравнительное соединение 1 вводили перорально голым мышам (n=3-5/группа) в течение 14 дней (один раз в день). Максимально переносимую дозу рассчитывали, основываясь на изменении массы тела мышей.
В процессе периода введения соединения, рассчитывали процент изменения массы тела мышей (BWC%). Когда наблюдалось уменьшение значения BWC ≥10% в группе, которой вводилось соединение, дозу в этом случае определяли как токсичную дозу лекарства. Таким образом, половинное значение токсичной дозы рассматривали как максимально переносимую дозу.
BWC для мышей рассчитывали с помощью следующего уравнения, и изменение в значение BWC каждой группы в процессе введения показаны на фиг. 1.
BWC (%)=([(масса тела мыши в день измерения массы)―(масса тела мыши в группе)]/( масса тела мыши в группе))×100
Как видно из фиг. 1, не наблюдалось снижения массы тела при введении группе сравнительного соединения 1 (100 мг/кг) в процессе введения, но наблюдалось >10% снижения BWC при введении группе сравнительного соединения 1 (200 мг/кг). Таким образом, токсичную дозу сравнительного соединения 1 определяли как 200 мг/кг, а максимально переносимую дозу определяли как 100 мг/кг. В примере исследования 4 (оценка противоопухолевого действия), доза сравнительного соединения 1 была 100 мг/кг.
Между тем, не наблюдалось потери массы тела в группе, которой вводилось соединение по настоящему изобретению (200 мг/кг). Более того, как показано на фиг. 1, не наблюдалось потери массы тела в группе при введении (400 мг/кг). Таким образом, дозу соединения 1 определяли как 400 мг/кг в примере исследования 4 (оценка противоопухолевого действия).
Пример исследования 4
Оценка противоопухолевого действия на подкожно ксенотрансплантированных моделях, используя штамм рака желудка человека (NUGC4) (in vivo)
Клетки рака желудка человека (NUGC4) (полученного от фирмы ATCC) подкожно трансплантировали голым мышам. Когда объем образовавшейся опухоли у голых мышей достигал от около 100 до около 300 мм3, мышей разбивали на группы (5 или 6/групп) разделением случайным образом таким образом, чтобы объемы опухоли в каждой группе были равными (день 1). Каждое из соединений по настоящему изобретению и сравнительное соединение 1 вводили перорально один раз в день в течение 14 дней.
Основываясь на результатах примера исследования 3, дозу сравнительного соединения 1 устанавливали как 100 мг/кг/день, что является максимально переносимой дозой в течение 14-дневного периода введения по примеру исследования 4 (то есть, максимальная доза, которая дает <10% снижение массы тела в течение периода введения). Дозу соединения по настоящему изобретению устанавливали как 400 мг/кг/день.
С целью сравнения зависимых от времени изменений в профиле пролиферации опухоли для вводимых исследуемых соединений, относительный объем опухоли (RTV) в сравнении с объемом опухоли во время распределения на группы рассчитывали с помощью следующего уравнения. Изменения в значении RTV для каждой группы показаны на фиг. 2.
RTV=(объем опухоли в день измерения объема опухоли)/(объем опухоли при распределении на группы)
В случае, когда значение RTV в группе, которой вводят соединение по изобретению, в последний день оценки было меньше, чем в группе, которой вводят сравнительное соединение 1, и оно являлось статистически значимым (t-критерий Стьюдента), соединение по настоящему изобретению рассматривали как существенно более эффективное, чем сравнительное соединение 1. На фиг. 2, статистически значимый уровень отмечен *.
Как видно из фиг. 2, соединение по настоящему изобретению вызывает эффективное уменьшение опухоли в течение одной недели от начала введения, проявляя более значительное противоопухолевое действие, чем сравнительное соединение 1.
Как описано здесь и выше, соединение по настоящему изобретению проявило C-Met ингибирующее действие, которое равно или выше чем у сравнительного соединения 1 (пример исследования 1), и проявило превосходную специфичность в ингибирующем клеточную пролиферацию действии (пример исследования 2), и это указывает на то, что токсичность в отношении нецелевых клеток, включая нормальные клетки, является небольшой. В исследовании по определению дозы с использованием голых мышей, соединение по настоящему изобретению не вызывало снижения массы тела, даже когда его вводили в дозе 400 мг/кг, что выше токсичной дозы (200 мг/кг) сравнительного соединения 1, и это указывает на то, что соединение по изобретению имело низкую токсичность (пример исследования 3). Кроме того, соединение по настоящему изобретению может быть введено в большой дозе (400 мг/кг), которая значительно выше, чем максимально переносимая доза (100 мг/кг) сравнительного соединения 1. Таким образом, соединение по изобретению показывает превосходную регрессию опухоли (противоопухолевое действие) (пример исследования 4).
| название | год | авторы | номер документа |
|---|---|---|---|
| ХИНОЛИНИЛ-СОДЕРЖАЩЕЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2803116C2 |
| НОВЫЕ ПИРИДИНОВОЕ ПРОИЗВОДНОЕ И ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ (1) | 2005 |
|
RU2330021C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
| НОВОЕ 2-ГЕТЕРОАРИЛ-ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ БЕНЗИМИДАЗОЛА | 2004 |
|
RU2329261C2 |
| НОВЫЕ СОЕДИНЕНИЯ, ИХ ИЗОМЕР ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ В КАЧЕСТВЕ АНТАГОНИСТА ВАНИЛОИДНОГО РЕЦЕПТОРА И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2007 |
|
RU2448108C2 |
| НОВОЕ ИМИДАЗООКСАЗИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2012 |
|
RU2578608C2 |
| АНТАГОНИСТ CASR | 2004 |
|
RU2315036C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2711502C2 |
| АРОМАТИЧЕСКИЕ ПРОИЗВОДНЫЕ 6,7-ДИЗАМЕЩЕННЫХ 3-ХИНОЛИНКАРБОКСАМИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЕ ДЛЯ ИЗГОТОВЛЕНИЯ ЛЕКАРСТВА | 2002 |
|
RU2281940C2 |
| БИАРИЛЬНОЕ ПРОИЗВОДНОЕ И СОДЕРЖАЩЕЕ ЕГО ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2016 |
|
RU2760373C2 |
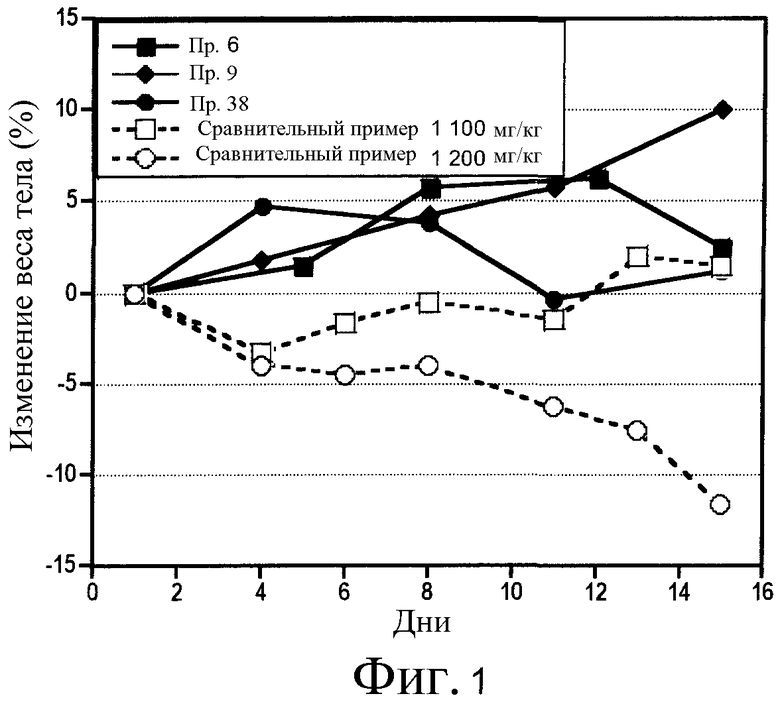
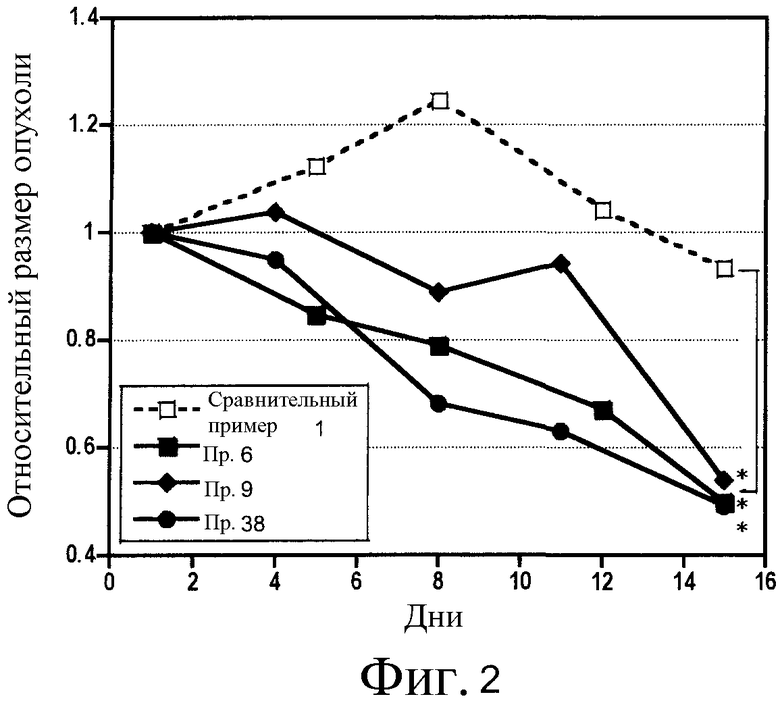
Настоящее изобретение относится к новым производным ацилтиомочевины формулы (I) или к его фармацевтически приемлемой соли, где R1 представляет собой атом водорода или C1-3 алкильную группу; R2 представляет собой атом водорода, необязательно замещенную C1-6 алкильную группу, необязательно замещенную С6-14 ароматическую углеводородную группу или необязательно замещенную насыщенную или ненасыщенную 5-7-членную гетероциклическую группу, содержащую 1 или 2 атома(ов) азота или атома(ов) серы, или R1 и R2 могут образовывать, вместе с атомом азота, к которому они присоединены, необязательно замещенную азотсодержащую насыщенную гетероциклическую группу, выбранную из группы, включающей, пирролидинильную, пиперидинильную, пиперазинильную или морфолино группу; где заместитель выбирают из группы, включающей атом галогена, гидроксильную группу, цианогруппу, нитрогруппу, C1-6 алканоильную группу, C1-6 алкильную группу, С3-10 циклоалкильную группу, С2-6 алкенильную группу, C1-6 алкоксигруппу, аминогруппу, C1-6 алкиламино группу, C1-6 алканоиламиногруппу, C1-6 алкиламинокарбонильную группу, C1-6 алкилсульфонильную группу, С6-14 ароматическую углеводородную группу, насыщенную или ненасыщенную 5-7-членную гетероциклическую группу, содержащую 1-4 атома(ов) азота и/или атома(ов) кислорода, насыщенную или ненасыщенную 5-7-членную гетероциклил-карбонильную группу, содержащую 1 или 2 атома(ов) азота и/или атома(ов) кислорода, и оксо группу; R3 представляет собой C1-6 алкильную группу; и R4 представляет собой атом галогена; R5 и R6, которые могут быть одинаковыми или отличающимися друг от друга, представляют собой атом водорода, атом галогена, C1-3 алкильную группу, которая может быть замещена атомом галогена, или C1-6 алкоксигруппу. Также изобретение относится к фармацевтическому и противоопухолевому агенту на основе соединения формулы (I), применению соединения формулы (I). Технический результат: получены новые производные ацетилтиомочевины, обладающие ингибирующим c-Met действием. 5 н. и 6 з.п. ф-лы, 2 ил., 4 табл., 56 пр.
1. Производное ацилтиомочевины, представленное формулой (I):
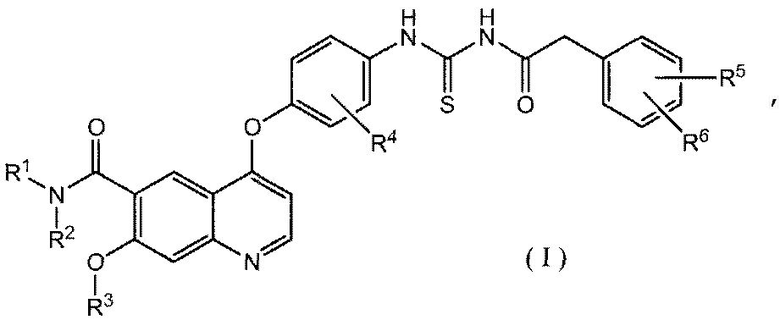
где R1 представляет собой атом водорода или С1-3 алкильную группу;
R2 представляет собой атом водорода, необязательно замещенную C1-6 алкильную группу, необязательно замещенную С6-14 ароматическую углеводородную группу или необязательно замещенную насыщенную или ненасыщенную 5-7-членную гетероциклическую группу, содержащую 1 или 2 атома(ов) азота или атома(ов) серы,
или R1 и R2 могут образовывать вместе с атомом азота, к которому они присоединены, необязательно замещенную азотсодержащую насыщенную гетероциклическую группу, выбранную из группы, включающей пирролидинильную, пиперидинильную, пиперазинильную или морфолиногруппу;
где заместитель выбирают из группы, включающей атом галогена, гидроксильную группу, цианогруппу, нитрогруппу, С1-6 алканоильную группу, С1-6 алкильную группу, С3-10 циклоалкильную группу, С2-6 алкенильную группу, C1-6 алкокси группу, аминогруппу, С1-6 алкиламиногруппу, С1-6 алканоиламиногруппу, C1-6 алкиламинокарбонильную группу, С1-6 алкилсульфонильную группу, С6-14 ароматическую углеводородную группу, насыщенную или ненасыщенную 5-7-членную гетероциклическую группу, содержащую 1-4 атома(ов) азота и/или атома(ов) кислорода, насыщенную или ненасыщенную 5-7-членную гетероциклил-карбонильную группу, содержащую 1 или 2 атома(ов) азота и/или атома(ов) кислорода, и оксогруппу;
R3 представляет собой C1-6 алкильную группу; и
R4 представляет собой атом галогена; R5 и R6, которые могут быть одинаковыми или отличающимися друг от друга, представляют собой атом водорода, атом галогена, C1-3 алкильную группу, которая может быть замещена атомом галогена, или C1-6 алкоксигруппу;
или его фармацевтически приемлемая соль.
2. Производное ацилтиомочевины по п.1 или его фармацевтически приемлемая соль,
где R2 представляет собой атом водорода, необязательно замещенную C1-6 алкильную группу, необязательно замещенную С6-14 ароматическую углеводородную группу или необязательно замещенную насыщенную или ненасыщенную 5-7-членную гетероциклическую группу, содержащую 1 или 2 атома(ов) азота или атома(ов) серы,
или R1 и R2 могут образовывать вместе с атомом азота, к которому они присоединены, необязательно замещенную насыщенную азотсодержащую гетероциклическую группу, выбранную из группы, включающей пирролидинильную, пиперидинильную, пиперазинильную и морфолиногруппу;
R3 представляет собой С1-3 алкильную группу.
3. Производное ацилтиомочевины по п.2 или его фармацевтически приемлемая соль,
где R1 представляет собой атом водорода или метильную группу;
R2 представляет собой необязательно замещенную C1-6 алкильную группу, необязательно замещенную фенильную группу или необязательно замещенную 5-7-членную гетероциклическую группу, которая содержит 1 или 2 атома азота или атома серы, или
R1 и R2 образуют вместе с атомом азота, к которому они присоединены, необязательно замещенную пирролидинильную группу или необязательно замещенную пиперидинильную группу;
R3 представляет собой метильную группу;
R4 представляет собой атом фтора или атом хлора;
R5 представляет собой атом водорода или атом галогена; и
R6 представляет собой атом водорода, атом галогена, трифторметильную группу или метоксигруппу.
4. Производное ацилтиомочевины по п.3 или его фармацевтически приемлемая соль,
где R1 представляет собой атом водорода;
R2 представляет собой C1-6 алкильную группу, которая может иметь заместитель, и указанный заместитель представляет собой гидроксильную группу, С3-10 циклоалкильную группу, C1-6 алкоксигруппу, C1-6 алкиламиногруппу, C1-6 алканоиламиногруппу, C1-6 алкилсульфонильную группу, С6-14 ароматическую углеводородную группу, насыщенную или ненасыщенную 5-7-членную гетероциклическую группу, содержащую 1-4 атома(ов) азота и/или атома(ов) кислорода, C1-6 алкиламинокарбонильную группу или насыщенную или ненасыщенную 5-7-членную гетероциклил-карбонильную группу, содержащую 1 или 2 атома(ов) азота и/или атома(ов) кислорода;
R3 представляет собой метильную группу;
R4 представляет собой атом фтора или атом хлора;
R5 представляет собой атом водорода; и R6 представляет собой атом водорода, атом фтора или атом хлора.
5. Производное ацилтиомочевины по п.4 или его фармацевтически приемлемая соль,
где R2 представляет собой С1-4 алкильную группу, которая может иметь заместитель, и указанный заместитель представляет собой гидроксильную группу, циклогексильную группу, C1-3 алкоксигруппу, C1-6 алкиламиногруппу, ацетиламиногруппу, метилсульфонильную группу, фенильную группу, насыщенную или ненасыщенную 5-7-членную гетероциклическую группу, содержащую от 1 до 4 атома азота и/или атома кислорода, C1-6 алкиламинокарбонильную группу или насыщенную или ненасыщенную 5-7-членную гетероциклил-карбонильную группу, содержащую 1 или 2 атома азота и/или атома кислорода.
6. Производное ацилтиомочевины по п.5 или его фармацевтически приемлемая соль,
где R2 представляет собой метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу или втор-бутильную группу,
где заместитель любой из указанных алкильных групп представляет собой гидроксильную группу, циклогексильную группу, метоксигруппу, этоксигруппу, изопропилоксигруппу, диэтиламиногруппу, ацетиламиногруппу, метилсульфонильную группу, фенильную группу, пирролидинильную группу, морфолиногруппу, диоксоланильную группу, тетрагидропиранильную группу, пиридильную группу, триазолильную группу, этиламинокарбонильную группу, диметиламинокарбонильную группу, метилбутиламинокарбонильную группу, пирролидинилкарбонильную группу или морфолинокарбонильную группу.
7. Производное ацилтиомочевины по п.1 или его фармацевтически приемлемая соль, которое выбрано из группы, содержащей:
4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-метилхинолин-6-карбоксамид;
4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(метоксиэтил)хинолин-6-карбоксамид;
4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(2-морфолиноэтил)хинолин-6-карбоксамид;
4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-7-метокси-N-(2-морфолино-2-оксоэтил)хинолин-6-карбоксамид;
4-(2-фтор-4-(3-(2-(4-фторфенил)ацетил)тиоуреидо)фенокси)-N-(2-гидроксибутил)-7-метоксихинолин-6-карбоксамид;
4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-N-(2-гидрокси-2-метилпропил)-7-метоксихинолин-6-карбоксамид;
(S)-4-(2-фтор-4-(3-(2-фенилацетил)тиоуреидо)фенокси)-N-(1-гидроксибутан-2-ил)-7-метоксихинолин-6-карбоксамид;
4-(2-фтор-4-(3-(2-(4-фторфенил)ацетил)тиоуреидо)фенокси)-7-метокси-N-(2-морфолиноэтил)хинолин-6-карбоксамид;
(S)-4-(2-фтор-4-(3-(2-(4-фторфенил)ацетил)тиоуреидо)фенокси)-N-(1-гидроксибутан-2-ил)-7-метоксихинолин-6-карбоксамид;
(S)-4-(2-фтор-4-(3-(2-(2-фторфенил)ацетил)тиоуреидо)фенокси)-N-(1-гидроксибутан-2-ил)-7-метоксихинолин-6-карбоксамид; и
(S)-4-(4-(3-(2-(4-хлорфенил)ацетил)тиоуреидо)-2-фторфенокси)-N-(1-гидроксибутан-2-ил)-7-метоксихинолин-6-карбоксамид.
8. Фармацевтический агент, обладающий ингибирующим c-Met действием, содержащий в качестве активного ингредиента производное ацилтиомочевины по любому из пп.1-7 или его фармацевтически приемлемую соль.
9. Противоопухолевый агент, содержащий в качестве активного ингредиента производное ацилтиомочевины по любому из пп.1-7 или его фармацевтически приемлемую соль.
10. Фармацевтическая композиция, обладающая ингибирующим c-Met действием, содержащая производное ацилтиомочевины по любому из пп.1-7 или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.
11. Применение производного ацилтиомочевины по любому из пп.1-7 или его фармацевтически приемлемой соли для получения противоопухолевого агента.
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| EP 1870414 A1, 26.12.2007 | |||
| АЗОТСОДЕРЖАЩИЕ АРОМАТИЧЕСКИЕ ПРОИЗВОДНЫЕ, ИХ ПРИМЕНЕНИЕ, ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 2001 |
|
RU2264389C2 |
| ПРОИЗВОДНЫЕ ТИОМОЧЕВИНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ ОБРАТНОЙ ТРАНСКРИПТАЗЫ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА | 1992 |
|
RU2106341C1 |

