Настоящее изобретение относится к стереоизомерным формам интраконазола и саперконазола, способам получения вышеуказанных стереоизомерных форм, их комплексам с производными циклодекстрина, фармацевтическим композициям, содержащим вышеуказанные комплексы, и способам получения вышеуказанных комплексов и фармацевтических композиций.
Интраконазол или (±)-цис-4-[4-[4-[-4-[[2-(2,4-дихлорфенил)- 2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил] фенил-2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол-3-он представляет собой противогрибковое соединение с широким спектром действия, разработанное для орального, парентерального и локального применения и описанное в патенте США N 4267179.
Егоаналог,содержащийвструктуредвафтора,саперконазолили(±)-цис-4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4- триазол-1-илметил)-1.3-диоксолан-4-ил] -метокси] фенил]-1-пиперазинил] фенил-2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол-3-он обладает улучшенной активностью в отношении Aspergillus spp. и он описан в патенте США N 4916134.
Оба соединения существуют в виде смеси четырех стереоизомеров.
Разработка эффективных фармацевтических композиций интраконазола и саперконазола затруднена из-за того, что названные соединения только весьма незначительно растворяются в воде. Растворимость обоих соединений может быть повышена за счет комплексообразования с циклодекстринами или их производными, как это описано в заявке WO 85/02767 и в патенте США N 4764604.
Неожиданно было установлено, что каждый из индивидуальных стереоизомеров интраконазола и саперконазола обладают более высокой растворимостью в воде, чем диастереомерные смеси вышеуказанных соединений, особенно, когда они образуют комплексы с циклодекстрином или его производными.
В результате могут быть получены фармацевтические композиции, обладающие хорошей биоактивностью, содержащие меньше циклодекстрина в качестве комплексообразующего агента.
Настоящее изобретение относится к стереоизомерным формам интраконазола (X=Cl) и саперконазола (X=F), которые могут быть представлены формулой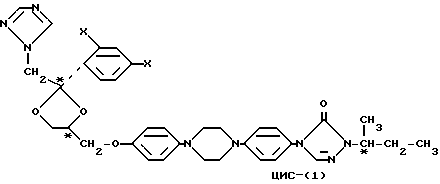
и их фармацевтически приемлемым кислотно-аддитивным солям.
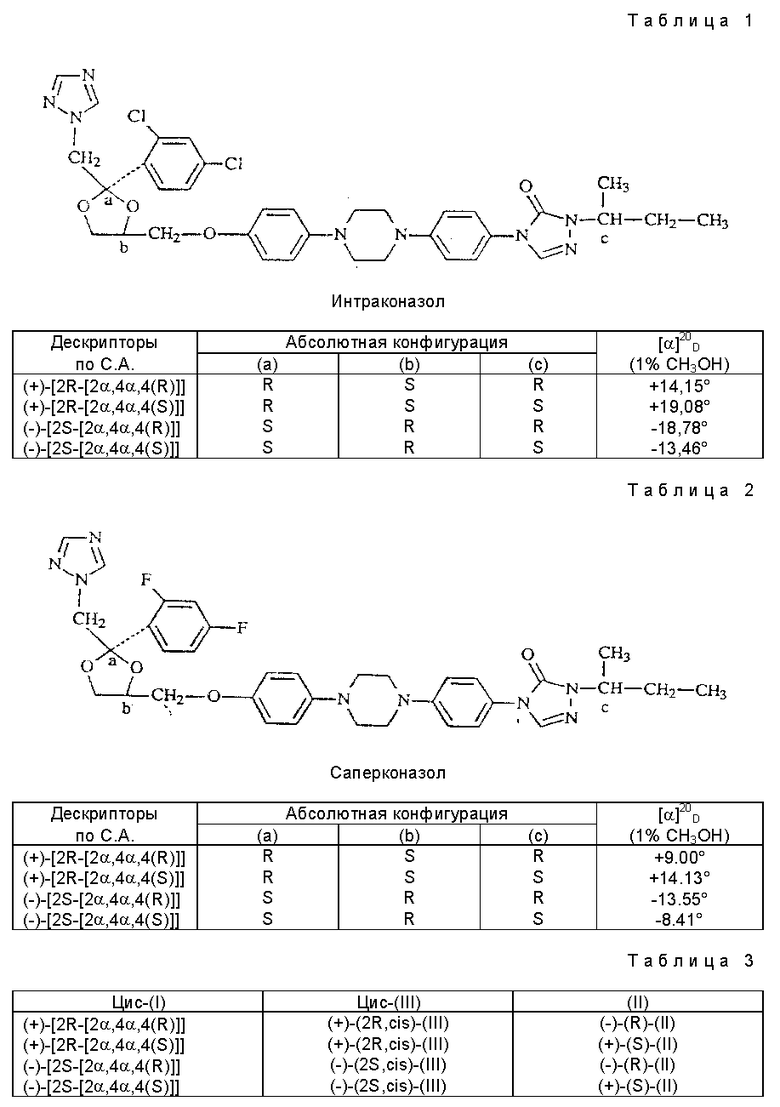
Три звездочки в формуле показывают три хиральных центра, а приставка "цис" означает, что фрагмент (1H-1,2,4-триазол-1-илметил) и замещенный фенокси-фрагмент расположены с одной стороны плоскости 1,3-диоксоланового кольца.
Четыре возможных стереоизомера цис-форм могут быть описаны по различным правилам номенклатуры. Таблицы 1-3 показывают корреляцию среди стереохимических дескрипторов по С.А., абсолютную конфигурацию при каждом хиральном центре и удельное оптическое вращение [α]
Что касается промежуточных соединений, то понятие "энантиомерно чистый" означает, что промежуточные соединения имеют энантиомерную чистоту по меньшей мере от 96 до 100%, более конкретно имеют энантиомерную чистоту от 98 до 100%.
Стереоизомерные формы соединений формулы I имеют основные свойства. Фармацевтически приемлемые кислотно-аддитивные соли, как упоминалось выше, включают терапевтически активные нетоксичные кислотно-аддитивные солевые формы, которые соединения формулы I способны образовывать.
Вышеуказанные солевые формы могут быть получены при обработке основной формы соединений формулы I подходящими кислотами, такими, как неорганические кислоты, например галогенводородные кислоты из числа соляной, бромистоводородной и т.д., серная кислота, азотная кислота, фосфорная кислота и аналогичные кислоты; как органические кислоты, такие как, например, уксусная кислота, пропионовая, гидроксиуксусная, 2-гидроксипропионовая, 2-оксопропионовая кислота, этандикислота, пропандикислота, бутандикислота, (Z)-2-бутендикислота, (E)-2-бутендикислота, 2-гидроксибутандикислота, 2,3-дигидроксибутандикислота, 2-гидрокси-1,2,3-пропантрикарбоновая кислота, метансульфоновая, этансульфоновая, бензолсульфоновая, 4-метилбензолсульфоновая, циклогексилсульфоновая, 2-гидроксибензойная, 4-амино-2-гидроксибензойная кислоты и другие подобные кислоты. При обработке щелочью солевая форма может быть переведена обратно в основную форму.
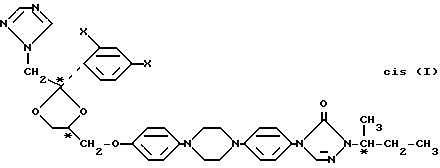
Четыре индивидуальные стериоизомерные формы соединений формулы I могут быть приготовлены O-алкилированием энантиомерного чистого фенола формулы (-)-(R)-(II) или (+)-(S)-(II) с энантиомерно чистым производным 1,3-диоксолана формулы (-)-(2S,цис)-(III) или (+)-(2R,цис)-(II), где заместитель -OR представляет собой удаляемую сульфонилоксигруппу, такую как, например, 4-метилбензолсульфонилокси-группу (тозилат) или метансульфонилокси-группу (мезилат)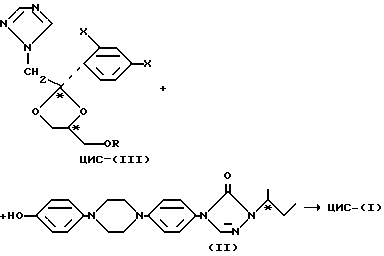
Рассмотренная выше реакция О-алкилирования может быть проведена согласно известным методикам, например при перемешивании и нагревании реагентов в подходящем растворителе, таком как диполярный апротонный растворитель, например N, N-диметилформамид, N,N-диметилацетамид и другие подобные растворители, в присутствии основания, такого как гидроксид щелочного металла, например гидроксид натрия или калия.
Полученные таким образом стереоизомерные формы соединения формулы цис-(I) могут быть дополнительно очищены хорошо известными способами, например жидкостной хроматографией или перекристаллизацией.
Соотношение между стереохимией стереоизомерных форм соединения формулы I, полученного по рассмотренной выше реакции O-алкилирования, и стереохимией исходных продуктов (II) и (III) показано в таблице 3.
Энантиомерно чистый фенол формулы (-)-(R)-(II) может быть получен из (S)-2-бутанола (IV). Энантиомерно чистый (S)-бутанол (IV) может быть превращен в соответствующий (S)- сульфонат, (V) реакцией с 4-метилбензол-сульфонилхлоридом (R-Me) или 1-бром-4-бензолсульфонилхлоридом (R=Br) в пиридине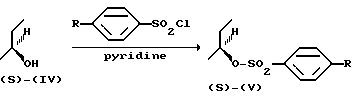
Энантиоселективное сочетание сульфоната (S)-(V) с триазолом (VI) (полученным в соответствии с методикой Примера XVII патента США 426717) протекает с инверсией конфигурации при хиральном центре и дает (-)-(R)-(VII)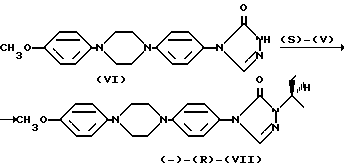
Рассмотренная выше реакция сочетания может быть проведена в инертном растворителе в присутствии основания, такого как гидрид натрия.
Энантиомерная чистота полученного таким образом продукта (-)-(R)-(VII) составляет от приблизительно 65% до приблизительно 75% и может быть увеличена до энантиомерной чистоты более 98% путем превращения (-)-(R)-(VII) в соль R-камфарсульфокислоты а ацетоне с последующей повторной перекристаллизацией соли из смеси этанол/ацетон (2:7, об./об.).
Деалкилирование полученного указанным способом анизола (-)-(R)-(VII) при кипячении в концентрированной бромистоводородной кислоте приводит к энантиомерно чистому фенолу (-)-(R)-(II). Для того, чтобы исключить бромирование на последней стадии, в реакционную смесь добавляют сульфит натрия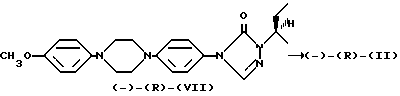
Другая энантиомерная форма (+)-(S)-(II) может быть получена по аналогичной методике из (R)-2-бутанола. Оптическую очистку полученного таким образом промежуточного соединения (+)-(S)-(VII) проводят с помощью соли (S)-камфарсульфонокислоты.
(-)-(R)-(VII) и (+)-(S)-(VII) можно получать также расщеплением соответствующего рацемата (±)-(VII) дробной кристаллизацией с энантиомерно чистой камфарсульфокислоты из смеси этанол/ацетон (1:4, об./об.).
Дополнительная оптическая очистка до энантиомерной чистоты более 98% может быть достигнута при поворотной перекристаллизации соли камфарсульфокислоты, как это описано выше.
Энантиомер (-)-(R)-(VII) получают при перекристаллизации рацемата (+)-(VII) с (R)-камфарсульфокислотой; энантиомер (+)-(S)-(VII) может быть получен аналогично с помощью (S)-камфарсульфокислоты из рацемата (+)-(VII), или предпочтительно из маточной жидкости предыдущей стадии расщепления с использованием (R)-камфарсульфокислоты, которая насыщена энантиомером (+)-(S)-(VII).
Однако рацемат (+)-(VII) может быть расщеплен с помощью жидкостной хроматографии с использованием хиральной неподвижной фазы, такой как производное амилозы, в частности, амилоза-трис-(3,5-диметилфенил)карбамат, нанесенный на микропористую матрицу γ-аминопропил-двуокись кремния (Chiralpak ADТМ, Daicel), или неподвижной фазы Пиркла (Pirkle).
Разделение предпочтительно проводят с использованием в качестве подвижной фазы спирта, такого как метанол или этанол (необязательно денатурированный 1% метанола). Для того, чтобы ускорить разделение, процесс хроматографического элюирования может быть проведен при температурах выше комнатной (приблизительно при 30oC). При использовании технологии с рециклом следовых хроматографических пиков можно еще сократить время разделения.
Для крупномасштабного процесса с успехом может быть использована полностью непрерывная технология с перемещающимся абсорбционным слоем.
По методике, аналогичной методике, описанной выше, на хиральной неподвижной фазе - амилоза-трис-(3,5-диметилфенил)карбамат, нанесенный на микропористую матрицу γ-аминопропил-двуокись кремния (Chiralpak ADТМ, Dalcel), на два энантиомера может быть разделен и рацемат (+)-(II).
Энантиомерно чистый ингредиент (+)-(2R,цис) (III), где заместитель OR представляет собой тозилат, может быть получен из коммерчески доступного (S)-2,2-диметил-1,3-диоксолан-4-ил-метанола (S)-(VIII).
Реакция (S)-(VIII) с 4-метилбензолсульфонилхлоридом в пиридине и гидролиз полученного таким образом продукта в водном растворе кислоты, например 6 н. соляной кислоты, необязательно в смеси с растворителем, таким как спирт или кетон, например ацетон, приводит к соответствующему (R)-1,2-дигидроксипропилтозилату (IX)
Ацетилирование (R)-(IX) 1-(2,4-дигалогенфенил)-2-(1H-1,2,4-триазолил)этанолом (X) в мягких условиях приводит к смеси цис/транс-диоксоланов, из которой после хроматографирования выделяют цис-изомер (+)-(2R, цис)-(III)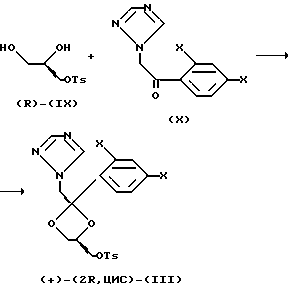
Рассмотренная выше реакция ацетилирования может быть проведена при перемешивании и нагревании реагентов в инертном растворителе, таком как галогенированные углеводороды, например метиленхлорид или хлороформ, в присутствии кислоты, предпочтительно сульфоновой кислоты, такой как метансульфоновая кислота.
По аналогичной методике из (R)-2,2-диметил-1,3-диоксолан-4-илметанола (R)-(VIII) может быть получен (-)-(2S,цис)-(III).
Рацемические соединения формулы (+)-цис-(III)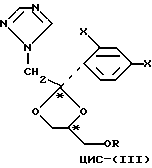
где заместитель X представляет собой атом хлора или фтора, а заместитель R представляет собой 4-метилбензолсульфонил, метансульфонил, а также C1-5-алкилкарбонил, могут быть разделены на индивидуальные энантиомеры жидкостной хроматографией с использованием хиральной неподвижной фазы, такой как неподвижная фаза на основе целлюлозы или амилозы.
Предпочтительным примером такой фазы является целлюлоза-трис-(4-метилбензоат) или в чисто полимерной форме или нанесенной на микропористую матрицу γ- аминопропил - двуокись кремния (Chiralcel ODТМ, Daicel).
В качестве подвижной фазы предпочтительно используют спирт, например метанол или этанол (необязательно денатурированный 1% метанола). Метанол является предпочтительным для производных целлюлозы в чисто полимерной форме.
Для того, чтобы ускорить разделение, процесс хроматографического элюирования может быть проведен при температурах выше комнатной (приблизительно при 30oC). При использовании технологии с рециклом следовых хроматографических пиков можно еще сократить время разделения.
Для крупномасштабного процесса с успехом может быть использована полностью непрерывная технология с перемещающимся абсорбционным слоем.
В случае, когда заместитель R представляет собой C1-5-алкилкарбонил, разделенные энантиомеры превращают в мезилатное или тозилатное производное согласно известным методикам, таким как омыление с последующим сульфонированием.
Индивидуальные стереоизомерные формы итраконазола и саперконазола обладают противогрибковой активностью. Каждый из четырех изомеров вносит вклад в общую активность исходного соединения и ни один не является более активным, чем вышеуказанное исходное соединение. Изомеры (-)-[2S-[ 2α,4α, 4(R)]] и (-)-[2S-[ 2α,4α, 4(S)]] более активны, чем их соответствующие антиподы.
Индивидуальные стереоизомеры итраконазола и саперконазола также могут быть получены при разделении диастереоизомерной смеси с использованием жидкостной хроматографии на хиральной неподвижной фазе, такой как производное амилозы, в частности амилоза-трис-(3,5-диметилфенил)карбамат, нанесенный на микропористую матрицу γ-аминопропил-двуокись кремния (Chiralpak ADTM, Daicel).
Приемлемой подвижной фазой являются спирты, в частности метанол или этанол (необязательно денатурированный 1% метанола). Для того, чтобы ускорить разделение и удержать продукт в растворе, процесс хроматографического элюирования может быть проведен при температурах выше комнатной (приблизительно при 50oC).
Недостатком данного способа является то, что эти соединения имеют плохую растворимость и только небольшие количества могут быть разделены за единицу времени. Более того, расположение 4 пиков препятствует использованию технологии рециркулирования следовых пиков. Следовательно, разделение промежуточных соединений при получении стереоизомерно чистых итраконазола и саперконазола должно быть более предпочтительным, чем разделение конечных продуктов.
Индивидуальные стереоизомерные формы итраконазола и саперконазола отличаются более высокой растворимостью в воде, чем диастереоизомерные смеси вышеуказанных соединений, особенно, если индивидуальные стереоизомеры имеют форму комплекса с производными циклодекстрина.
Приемлемыми производными циклодекстрина являются α-, β- и γ- циклодекстрины или простые эфиры и смешанные простые эфиры циклодекстрина, где одна или несколько гидроксильных групп фрагментов ангидроглюкозы циклодекстрина замещены C1-6-алкилом, в частности метилом, этилом или изопропилом; гидрокси-C1-6-алкилом, в частности гидроксиэтилом, гидроксипропилом или гидроксибутилом; карбокси-C1-6-алкилом, в частности карбоксиметилом или карбоксиэтилом; C1-6-алкилкарбонилом, в частности ацетилом; C1-6-алкилоксикарбонил-C1-6-алкилом или карбокси-C1-6-алкилокси-C1-6-алкилом, в частности карбоксиметоксипропилом или карбоэтоксипропилом; C1-6-алкилкарбонилокси-C1-6-алкилом, в частности 2-ацетилоксипропилом.
В качестве комплексообразующих соединений и/или солюбилизирующих соединений особенно предпочтительны β-ЦД, 2,6-диметил-β-ЦД, 2-гидроксиэтил-β-ЦД, 2-гидроксиэтил-γ-ЦД, 2-гидроксипропил-γ-ЦД и (2-карбоксиметокси)пропил-β-ЦД и особенно 2-гидрокси-β-ЦД.
Понятие смешанные простые эфиры относится к производным циклодекстрина, где по меньшей мере две гидроксильные группы циклодекстрина этерифицированы различными группами, такими как, например, гидроксипропильная и гидроксиэтильная.
Для измерения среднего числа молей алкокси-фрагментов на моль ангидроцеллюлозы используется среднее молярное замещение (М.З.). В производных циклодекстрина, которые используются в композициях настоящего изобретения, М.З. находится в интервале от 0.125 до 10, в частности от 0.3 до 3 или от 0.3 до 1.5.
Предпочтительно М.З. лежит в интервале от приблизительно 0.3 до приблизительно 0.8, в частности приблизительно от 0.35 до 0.5 и наиболее предпочтительно составляет приблизительно 0.4.
Средняя степень замещения (С.З.) относится к числу замещенных гидроксильных групп на один фрагмент ангидроглюкозы. В производных циклодекстрина, используемых в настоящем изобретении, С.З. находится в интервале от 0.125 до 3, предпочтительно от 0.2 до 2, или от 0.2 до 1.5. Предпочтительно С.З. составляет приблизительно от 0.2 до 0.7, в частности от приблизительно 0.35 до 0.5, и наиболее предпочтительно С.З. составляет приблизительно 0.4.
Более предпочтительные гидроксильные производные циклодекстрина, которые могут быть использованы в настоящем изобретении, представляют собой частично замещенные производные циклодекстрина, где средняя степень алкилирования по гидроксильным группам различных положений фрагментов ангидроглюкозы составляет приблизительно от 0 до 20% для 3 положения, от 2 до 70% для 2 положения и приблизительно от 5 до 90% для 6 положения.
Предпочтительно количество незамещенного β- и γ- циклодекстрина составляет менее 5% из расчета на все содержание циклодекстрина и предпочтительно это количество составляет менее 1.5%. Другим производным циклодекстрина, представляющим интерес для настоящего изобретения, является метилированный β-циклодекстрин.
Наиболее предпочтительными для использования в настоящем изобретении производными циклодекстрина являются частично замещенные β- циклодекстриновые эфиры или замещенные эфиры, содержащие гидроксипропильные, гидроксиэтильные заместители и особенно 2-гидроксипропильные и/или 2-(1-гидроксипропильные) заместители.
Наиболее предпочтительным для использования в настоящем изобретении производным циклодекстрина является гидроксипропил -β- циклодекстрин, имеющий М. З. в интервале приблизительно от 0.35 до 0.50 и содержащий менее 1.5% незамещенного β- циклодекстрина.
Замещенные циклодекстрины могут быть приготовлены по методикам, описанным в патенте США N 34959731, EP N 0149197, 017571, патенте США N 4535152, PCT 90/12035 и патенте Великобритании N 2189254.
Другими литературными источниками, в которых описываются циклодекстрины для использования в композициях в соответствии с настоящим изобретением и которые дают указания по получению, очистке и анализу циклодекстринов, являются: "Cyclodextrin technology" - Jozef Szejtli; Kluwer Academic Publishes. - 1988. глава - "Циклодекстрины в фармацевтике"; "Cyclodextrin Chemistry". - M. L.Bender et al., Springer-Verlage, Berlin. - 1978; "Advances in Carbohydrat Chemistry". - Vol.12. Ed. M.L.Wolfrom, Academic Press, New York (1957). - в главе "The Schardiner Dextrin by Dexter French" - стр.189-260; "Cyclodextrins and their Inclusion Complexes". -J.Szejtli, Akademiai Kiado, Budapest, Венгрия (1982); I.Tabushi - Acc. Chem. Research, 1982. 15, p.66-72; W. Sanger, Angewandte Chemie, 92, p.343-362 (1981); A.P.Croft and R.A. Bartsch. - Tetrahedron, 39, p.1417-1474 (1983); Irie et al. - Pharmaceutical Research, 5, p. 713-716 (1988); Pitha et al. - Int. Pharm. J. - 29, 73 (1986); DE 3118218; DE 3317K64; EP-A-94157; US-4659696 и US-4383992.
Особенное внимание должно быть уделено тем источникам, в которых описывается получение и способы очистки, которые дают возможность получать смеси циклодекстрина, в которых количество незамещенного циклодекстрина составляет менее 5% из расчета на общее содержание циклодекстрина.
Комплексы индивидуальных стереоизомерных соединений формулы I с производными циклодекстрина, описанными выше, могут быть легко получены при растворении циклодекстрина или его эфирных производных в воде и добавлении к раствору индивидуального стереоизомерного соединения формулы I при перемешивании или при встряхивании полученной смеси до полного растворения.
С точки зрения хранения может быть полезным дегидратировать полученные таким образом растворы, например, путем вымораживания или при распылительной сушке. Дегидратированные комплексы легко восстанавливаются при добавлении воды или водного раствора циклодекстрина.
Комплексы индивидуальных стереоизомерных соединений формулы I с производными циклодекстрина особенно полезны при получении фармацевтических композиций для орального или локального применения. Такие фармацевтические композиции содержат в качестве активного ингредиента комплекс, описанный выше, и фармацевтически приемлемый носитель.
Вышеуказанный носитель может иметь различную форму в зависимости от желаемого препарата. Такие фармацевтические композиции представляют собой преимущественно дозированные единичные формы, приемлемые предпочтительно для орального, ректального, парентерального введения или введения через кожу.
Например, при приготовлении композиций в виде оральных дозирующих форм может быть использована любая фармацевтическая среда, такая как, например, вода, гликоли, масла, спирты и подобные среды, если готовятся оральные жидкие препараты, такие как суспензии, сиропы, эликсиры или растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие вещества, диспергирующие добавки и другие подобные соединения, если готовятся порошки, пеллеты, капсулы или таблетки.
Из-за простоты применения таблетки и капсулы представляют собой наиболее предпочтительные оральные дозированные формы и в этом случае используются твердые фармацевтические носители. В парентеральных композициях носитель обычно включает воду и составляет по меньшей мере большую его часть, хотя могут быть введены и другие ингредиенты, например добавки, способствующие растворению.
Растворы для инъекций, например, могут быть приготовлены с использованием солевого раствора или раствора смеси соли и глюкозы. Растворы для инъекций также могут быть приготовлены с использованием подходящих жидких носителей, суспендирующих агентов и т.д.
В случае композиций, предназначенных для введения через кожу, носитель необязательно включает агент для ускорения проникновения и/или приемлемый смачивающий агент, необязательно смешанные с другими приемлемыми добавками любой природы в небольшом количестве, и эти вспомогательные добавки не вызывают значительного отрицательного эффекта на коже.
Указанные вспомогательные добавки могут облегчать применение на коже и/или могут способствовать получению желаемых композиций. Эти композиции могут быть введены различными путями, например в виде трасдермальных пятен, мазей. Кислотно-аддитивные соли из-за их повышенной растворимости в воде по сравнению с соответствующими основными формами являются более предпочтительным для получения водных композиций.
Особое преимущество состоит в рецептурировании вышеуказанных фармацевтических композиций в дозирующей единичной форме для простого применения и однородности дозировки.
Используемая в настоящем описании и в формуле изобретения дозированная единичная форма относится к физически дискретным единицам, которые приемлемы в качестве унитарных дозировок, при этом каждая единица содержит предопределенное количество активного ингредиента, рассчитанное для получения желаемого терапевтического эффекта, в сочетании с фармацевтическим носителем.
Примерами таких дозированных единичных форм являются таблетки (в том числе таблетки с покрытием), капсулы, пеллеты, пакеты с порошками, облатки, растворы для инъекций или суспензии, мерная чайная или столовая ложка (teaspoonfuls, tablespoonfuls) и другие, а также их отдельные сочетания.
В конечной композиции содержание циклодекстрина составляет приблизительно от 2.5 до 20% мас., предпочтительно приблизительно от 5 до 20%, более предпочтительно от 5 до 15%, например приблизительно 10%, а остальное количество составляет вода, активный ингредиент и любые другие наполнители.
Например, стабильные фармацевтические композиции могут состоять из воды, циклодекстрина и активного ингредиента без необходимости введения дополнительных стабилизаторов, таких как сыворотка человеческого альбумина, сыворотка бычьего альбумина, лецитин, метилцеллюлоза, полиэтиленгликоль, серосодержащие восстанавливающие агенты, мочевина, аминокислоты и поверхностно-активные вещества.
Может также быть добавлен агент, регулирующий pH, например соляная кислота, уксусная кислота, лимонная кислота, гидроксид натрия, гидроксид калия, а также их любые соли, в частности цитрат натрия. Приемлемое значение pH для рецептурированных стереоизомеров итраконазола и саперконазола лежит в интервале от 6.5 до 7.4, предпочтительно от 6.8 до 7.0.
Подходящими консервантами для рассмотренных выше фармацевтических препаратов являются спирты, например этанол, 1,3-пропандиол, бензиловый спирт или его производные, фенилэтиловый спирт, фенол или производные фенола, такие как бутилпарабен, метилпарабен, м-крезол или хлоркрезол; кислоты, например бензойная кислота, сорбиновая кислота; лимонная кислота, пропионат натрия, динатрий-ЭДТУ, хлоргексидин, диизэтионат гексамидина, гексетидин, необязательно в смеси с бисульфитом натрия, или с пропиленгликолем, или, что менее предпочтительно, четвертичные аммоннийные соли, соединения металлов, такие как оксид цинка, тиомерсал- (thiomersal) или фенил-ртутные соли, например ацетат фенилртути.
Для получения препаратов для инъекций хорошо добавлять изотонирующий агент, например хлористый натрий, хлористый калий, сорбитол.
Также может быть необходимым добавление приемлемого комплексообразователя, такого как хлорид кальция, цитрат, ЭДТУ и другие комплексообразующие агенты с фармацевтически приемлемыми солями металлов. Например, может быть добавлен хлорид кальция в концентрации приблизительно 0,02-2 г/л.
Вышеуказанные композиции могут быть легко получены при растворении циклодекстрина или эфирного производного циклодекстрина в воде с последующим добавлением к полученному раствору соединения формулы I и других адъювантов и компонентов, таких как, например, хлористый натрий, нитрат калия, глюкоза, маннитол, сорбитол, ксилитол и буферы, такие как, например, фосфатный, ацетатный или цитратный буфер; и необязательно концентрированием или сушкой раствора при упаривании в вакууме или лиофилизацией; и необязательно дополнительной перекристаллизацией лиофилизированного остатка с водой.
Предпочтительные препараты в соответствии с настоящим изобретением имеют низкую токсичность и не обладают раздражающим действием, следовательно, позволяют получать медикаменты для инъекций, которые могут быть использованы по схеме повторяющегося применения без опасения возникновения иммуногенных реакций.
Водные препараты настоящего изобретения и наполнители, если это необходимо, могут быть подвергнуты сушке вымораживанием по известным методикам с получением дегидратированной композиции, которая может храниться в течение длительного промежутка времени и перед использованием растворяется.
В вышеуказанных высушенных вымораживанием и при распылительной сушке рецептурах мольное отношение и весовое отношение циклодекстрина к активному ингредиенту могут быть такими же, что и в упомянутых выше водных растворах.
В некоторых случаях удобно восстанавливать вымороженные или высушенные при распылительной сушке рецептуры в водном растворе циклодекстрина, при этом мольное отношение и весовое отношение циклодекстрина и активного ингредиента может быть ниже, чем в вышеупомянутых водных растворах. Следует отметить, что введение ЦД к комплексу никоим образом не влияет на антигрибковые свойства интраконазола или саперконазола, а только улучшает растворимость, облегчая таким образом их введение, т.е. эффективность увеличивается за счет повышения растворимости.
Экспериментальная часть
А. Получение промежуточных соединений
Пример 1
К раствору 40,5 г 1-(2,4-дихлорфенил)-2-(1H-1,2,4-триазол-1- ил)этанона в 300 мл метансульфокислоты и 300 мл метиленхлорида порциями добавляют 41.4 г (S)-1,2,3-пропантриол-4-метилбензолсульфоната (эфир).
Реакционную массу перемешивают в течение 4 дней при температуре кипения с отделением воды. После охлаждения всю массу при перемешивании порциями добавляют (в течение 1 ч) к смеси K2CO3 (3.6 M), 500 г льда и 500 мл хлороформа. Перемешивают в течение 1 ч и затем органический слой отделяют.
Водный слой экстрагируют хлороформом (3 раза) и объединенные экстракты промывают водой (2 раза) и упаривают. Остаток очищают колоночной хроматографией (силикагель, хлороформ). Элюент необходимой фракции упаривают и остаток превращают в 4-метилбензолсульфонатную соль (1:1) в 4-метил-2-пентаноне.
Соль отфильтровывают и сушат, получают 27.0 г (26.9%) 4-метилбензолсульфонатной соли (1: 1) 4-метилбензилсульфоната (сложный эфир)(-)-(2S-цис)-(2,4-дихлорфенил)-2-(1H-1,2,4-триазол-1-илметил) -1,3-диоксолан-4-метанола; т.пл. 194,5oC, [α]
Пример 2
К раствору 12.8 г 1-(2,4-дихлорфенил)-2-(1H-1,2,4-триазол-1-ил)-этанона в 72.2 мл метиленхлорида при перемешивании и при комнатной температуре добавляют по каплям в течение 15 мин 15 мл метансульфоновой кислоты. По окончании добавления к реакционной массе добавляют 14.8 г (2R)-1,2,3-пропантриол-4-метилбензолсульфоната (эфир), и полученную смесь кипятят при перемешивании в течение 48 ч с азеотропной отгонкой воды.
Полученную смесь по каплям добавляют к смеси 195.6 мл метиленхлорида и раствора 50 г карбоната калия в 200 мл воды. После перемешивания органический слой отделяют и сушат, фильтруют и упаривают. Остаток очищают колоночной хроматографией на силикагеле с использованием в качестве элюента хлороформа. Чистые фракции собирают и упаривают.
Остаток превращают в 4-метилбензолсульфонатную соль в 4-метил-2-пентаноне. Продукт отфильтровывают и сушат, получают 6.1 г (18.5%) 4-метилбензолсульфонатной соли (1: 1) 4-метилбензолсульфоната (сложный эфир) (+)-(2R, цис)-2-(2,4-дихлорфенил)-2-(1H-1,2,4-триазол- 1-ил-метил)-1,3-диоксолан-4-метанола; т.пл. 194.0oC; [α]
Пример 3
а) Смесь 8,2 г (R)-(-)-2-бутанола, 31.5 г 1-бром-4-бензолсульфонилхлорида, 1.0 г N,N-диметил-4-пиридинамина, 55.5 мл пиридина и 293 мл метиленхлорида перемешивают в течение 3 дней при комнатной температуре.
Реакционную смесь промывают дважды разбавленной соляной кислотой и один раз водой, сушат, фильтруют и упаривают в вакууме. Остаток очищают колоночной хроматографией на силикагеле с использованием в качестве элюента хлороформа. Чистые фракции собирают и упаривают, получают 11.5 г (35.6%) 4-бромбензолсульфоната (сложный эфир) (R)-2-бутанола (промежуточное соединение 3).
По аналогичной методике из (S)-2-бутанола и 4-метилбензолсульфонилхлорида получают 4-метилбензолсульфонат (S)-2-бутанола (промежуточное соединение 4).
б) Смесь 3.5 г 2,4-дигидро-4-[4-(4-метоксифенил)-1-пиперазинил]фенил]-3H-1,2,4-триазол-3-она, 0.6 г 50%-ной дисперсии гидрида натрия и 100 мл N, N-диметилформамида перемешивают в течение 3 ч при 80oC.
После добавления 3.5 г 4-бромбензолсульфоната (сложный эфир) (R)-2-бетанола (промежуточное соединение 3) перемешивание продолжают в течение 6 ч при той же температуре. После охлаждения к реакционной массе добавляют воду. Отфильтровывают кристаллический продукт и растворяют его в хлороформе и полученный раствор фильтруют, отбрасывая нерастворенную часть.
Полученный раствор сушат и очищают колоночной хроматографией на силикагеле, элюент-хлороформ. Чистые фракции собирают и упаривают. Остаток перекристаллизовывают из 4-метил-2-пентанона. Продукт отфильтровывают и в 2-пропаноне превращают в (S)-7,7-диметил-2-оксобицикло[2.2.1] гептан-1-метансульфонатную соль. Полученную соль отфильтровывают и перекристаллизовывают из 1-бутанола. Продукт отфильтровывают и сушат, получают 1.2 г (13.7%) (+)-(S)-2,4-дигидро-4-/-4-/- 4-(4-метоксифенил)-2-пиперазинил /фенил/-3H-1,2,4-триазол-3-он (S)-7,7-диметил-2-оксобицикло/2.2.1/гептан-1-метансульфонат (1: 2), т. пл. 192.0oC [α]
По аналогичной методике промежуточное соединение 4 превращают в (-)-(R)-2,4-дигидро-4-/4-/ 4-(4-метоксифенил)-1-пиперазинил /фенил/-3H-1,2,4-триазол-2-он (R)-7,7-диметил-2-оксобицикло/2.2.1/гептан-1-метансульфонат (1: 2), т. пл. 193,0oC [α]
в) Смесь 29.4 г (+)-(S)-2,4-дигидро-4-/4-/ 4-(4-метоксифенил)-1-пиперазинил/фенил/-3H-1,2,4-триазол-3-он (S)-7,7- диметил-2-оксобицикло[2.2.1] гептан-1-метансульфоната (1:2) промежуточное соединение (5), 2.0 г сульфита натрия и 151 мл 48%-ного раствора бромистоводородной кислоты в воде кипятят при перемешивании в течение 5 ч.
Реакционную массу охлаждают до комнатной температуры и добавляют воду. Реакционную массу нейтрализуют карбонатом калия до pH 7 при перемешивании в смеси метиленхлорида и 1-бутанола (90:10 по объему). Органический слой отделяют и сушат, фильтруют и упаривают в вакууме. Полученный остаток растирают в метаноле. Осажденный продукт отфильтровывают и очищают колоночной хроматографией на силикагеле, элюент - смесь хлороформа и метанола, 98:2. Чистые фракции собирают и упаривают, остаток кристаллизуют из 4-метил-2-пентанона. Продукт отфильтровывают и сушат, получают 10.4 г (77.7%) (+)-(S)-2,4-дигидро-2-[4-[4-(4-метоксифенил)-1-пиперазинил] фенил] - 2-(1-метилпропил)-3H-1,2,4-триазол-3-она, т. пл. 180.6oC [α]
По аналогичной методике промежуточное соединение 6 превращают в (-)-(R)-2,4-дигидро-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] фенил] -2 -(1-метилпропил)-3H-1,2,4-триазол-3-он; т.пл. 180.4oC; [α]
Пример 4
Смесь 44.6 г 1-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-ил)-этанола, 56.0 г 4-метилбензолсульфоната (сложный эфир) (2S)-1,2,3-пропантриола, 200 мл метансульфоновой кислоты и 150 мл метиленхлорида кипятят при перемешивании с отделением воды.
После охлаждения реакционную массу по каплям добавляют к смеси лед-вода, K2CO3 (экв. ) и метиленхлорида. Органический слой отделяют и водную фазу повторно экстрагируют метиленхлоридом. Объединенные экстракты сушат, фильтруют и упаривают. Остаток очищают колоночной хроматографией (силикагель, элюент - хлороформ).
Желаемую фракцию упаривают и остаток превращают в 2-метил-2-пентаноне в 4-метилбензолсульфонатную соль. Полученную соль перекристаллизовывают из 4-метил-2-пентанона, получают 20.5 г (16.4%) 4-метил-бензолсульфоната (эфир) 4-метилбензолсульфоната (соль 1:1) (-)-(2S,цис)-2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1- илметил)-1,3-диоксолан-4-метанола, т.пл. 182.5oC; [α]
Пример 5
Смесь 40.0 г 1-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-ил)-этанола, 56.0 г 4-метилбензолсульфоната (сложный эфир) (2R)-1,2,3-пропантриола, 250 мл метансульфоновой кислоты и 100 мл метиленхлорида кипятят при перемешивании с отделением воды в течение 24 ч.
После охлаждения реакционную массу по каплям добавляют к смеси лед-вода, K2CO3 и метиленхлорида. Органический слой отделяют, промывают водой, сушат, фильтруют и упаривают. Остаток очищают колоночной хроматографией (силикагель, элюент - хлороформ). Желаемую фракцию упаривают и остаток превращают в 4-метил-2-пентаноне в 4-метилбензолсульфонатную соль.
Полученную соль перекристаллизовывают из ацетонитрила, получают 23.1 г (20.6%) (+)-(2R, цис)-2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол- 1-илметил)-1,3-диоксолан-4-метанол 4-метилбензолсульфоната (эфир) 4-метилбензолсульфоната (соль 1: 1), т. пл. 183.5oC; [α]
Пример 6
Раствор 4 г 2,4-дигидро-4-[4-[4-(4-гидроксифенил)-1- пиперазинил]фенил] -2-(1-метилпропил)-3H-1,2,4-триазол-3-она в 600 мг метанола расщепляют с помощью колоночной жидкостной хроматографии.
Параметры процесса:
колонка: 400 мм x 100 мм вн.д., наполненная 1 кг 20 мкм амилоза-трис-(3,5-диметилфенил)карбаматом (Chiralpak ADTM: Daicel);
подвижная фаза: 150 мл/мин этанола;
температура: 30oC.
Образец в 4 г разделяют в три цикла и получают две фракции, содержащие 95 - 97% от теоретического выхода (т.пл. 158 - 162oC). Оптическая чистота, определенная с помощью высокоэффективной жидкостной хроматографии, составляет 97% (промежуточные соединения 11 и 12).
Пример 7
Раствор 5 г 4-метилбензолсульфоната (эфир) (+)-цис-2- (2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-метанола в 500 мг метанола расщепляют с помощью колоночной жидкостной хроматографии.
Параметры процесса:
колонка: 500 мм x 100 мм вн.д., наполненная 2 кг 20 мкм целлюлоза-трис-(4-метилбензоатом) (Chiralcel ADTM: Daicel);
подвижная фаза: 150 мл/мин этанола;
температура: 30oC.
Образец в 5 г разделяют в три цикла и получают (-) соединение (выход 93 - 95%, чистота по ВЭЖХ - 100%; α (365 нм) = -34.69o (1% MeOH); т.пл. 92 - 94oC) (промежуточное соединение 13) и (+) соединение (выход 95 - 96%, чистота по ВЭЖХ - 98,8%; α (365 нм) = +34.02 (1% MeOH); т.пл. 92 - 94oC) (промежуточное соединение 14).
B. Получение конечных соединений
Пример 8
Смесь 5,2 г (+)-(2R,цис)-2-(2,4-дихлорфенил)-2-(1H-1,2,4-триазол- 1-илметил)-1,3-диоксолан-4-метанола 4-метилбензолсульфоната (сложный эфир) (промежуточное соединение 2), 2.9 г (+)-(S)-2,4-дигидро-4- [4-[4-(4-гидроксифенил)-1-пиперазинил] фенил] -2-(1-метилпропил)-3H- 1,2,4-триазол-3-она (промежуточное соединение 7), 1,0 г чешуек гидроксида натрия и 100 мл N,N-диметилформамида перемешивают в течение 7 ч при 50oC в атмосфере азота.
После охлаждения к реакционной смеси добавляют воду. Образующийся осадок отфильтровывают и растворяют в хлороформе. Органический слой промывают водой, фильтруют и упаривают. Остаток очищают колоночной хроматографией (силикагель, элюент - хлороформ/метанол, 98 : 1,5).
Желаемую фракцию элюента упаривают и остаток растирают в метаноле, получают 3,9 г (79,0%) (+)-[2R-[ 2α,4α, -4(S)]]-4-[4-[4-[4-[[2-(2,4-дихлорфенил)-2- (1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]- 1-пиперазинил] фенил]-2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол- 3-она; т.пл. 175.3oC; [α]
Пример 9
Смесь 6.5 г 4-метилбензолсульфоната (сложный эфир) 4-метилбензолсульфоната (соль, 1 : 1) (-)-(2S,цис)-2-(2,4-дихлорфенил)- 2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-метанола (промежуточное соединение 1), 3,6 г (+)-(S)-2,4-дигидро-4- [4-[4-(4-гидроксифенил)-1-пиперазинил] фенил]-2-(1-метилпропил)-3H- 1,2,4-триазол-3-она (промежуточное соединение 7), 1.0 г чешуек гидроксида натрия и 100 мл N,N-диметилформамида перемешивают в течение ночи при 50oC в атмосфере азота.
После охлаждения к реакционной смеси добавляют воду. Образующийся осадок отфильтровывают и растворяют в метиленхлориде. Органический слой промывают водой, фильтруют и упаривают. Остаток растирают в метаноле, получают 4.6 г (72.5%) (-)-[2S-[ 2α,4α, 4(S)] ]-4-[4-[4-[4-[[2-(2,4-дихлорфенил)-2-(1H-1,2,4-триазол-1-илметил)- 1,3-диоксолан-4-ил] метокси]фенил]-1-пиперазинил] фенил]-2,4-дигидро-2- (1-метилпропил)-3H-1,2,4-триазол-3-она; т.пл. 146.5oC; [α]
Пример 10
Смесь 6.5 г 4-метилбензолсульфоната (сложный эфир) 4-метилбензолсульфоната (соль, 1 : 1) (-)-(2S,цис)-2- (2,4-дихлорфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан- 4-метанола (промежуточное соединение 1), 3,6 г (-)-(R)-2,4- дигидро-4-[4-[4-(4-гидроксифенил)-1-пиперазинил]фенил]-2- (1-метилпропил)-3H-1,2,4-триазол-3-она (промежуточное соединение 8), 1.0 г чешуек гидроксида натрия и 100 мл N,N-диметилформамида перемешивают в течение ночи при 50oC в атмосфере азота.
После охлаждения к реакционной смеси добавляют воду. Образующийся осадок отфильтровывают и растворяют в метиленхлориде. Органический слой промывают водой, фильтруют и упаривают. Остаток растирают в метаноле, получают 4.4 г (69.3%) (-)-[2S-[ 2α,4α, 4(R)] ]-4-[4-[4-[4-[[2-(2,4-дихлорфенил)-2-(1H-1,2,4-триазол-1-илметил)- 1,3-диоксолан-4-ил] метокси]фенил]-1-пиперазинил] фенил]-2,4-дигидро-2- (1-метилпропил)-3H-1,2,4-триазол-3-она; т.пл. 156.6oC; [α]
Пример 11
Смесь 6.5 г 4-метилбензолсульфоната (сложный эфир) 4-метилбензолсульфоната (соль, 1 : 1) (+)-(2R,цис)-2- (2,4-дихлорфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан- 4-метанола (промежуточное соединение 1), 3.6 г (-)-(R)-2,4- дигидро-4-[4-[4-(4-гидроксифенил)-1-пиперазинил]фенил]-2- (1-метилпропил)-3H-1,2,4-триазол-3-она (промежуточное соединение 8), 1.0 г чешуек гидроксида натрия и 100 мл N,N-диметилформамида перемешивают в течение ночи при 50oC в атмосфере азота.
После охлаждения к реакционной смеси добавляют воду. Образующийся осадок отфильтровывают и растворяют в хлороформе. Органический слой промывают водой, фильтруют и упаривают. Остаток растирают в метаноле, сушат в вакууме при 100oC, получают 4.7 г (74%) (+)-[2R-[ 2α,4α, 4(R)]]-4-[4-[4-[4-[[2-(2,4-дихлорфенил)- 2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан и 4-ил]метокси] фенил] 1-пиперазинил]фенил]-2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол- 3-она; т. пл. 148.0oC; [α]
Пример 12
Смесь 11.7 г 4-метилбензолсульфоната (сложный эфир) 4-метилбензолсульфоната (соль, 1 : 1) (-)-(2S,цис)-2- (2,4-дихлорфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан- 4-метанола (промежуточное соединение 9), 6.6 г (+)-(S)-2,4- дигидро-4-[4-[4-(4-гидроксифенил)-1-пиперазинил]фенил]-2- (1-метилпропил)-3H-1,2,4-триазол-3-она (промежуточное соединение 7), 1.5 г чешуек гидроксида натрия и 150 мл N,N-диметилформамида перемешивают в течение ночи при 50oC в атмосфере азота.
После охлаждения к реакционной смеси добавляют воду. Образующийся осадок отфильтровывают и растворяют в хлороформе. Органический слой промывают водой, фильтруют и упаривают. Остаток растирают в метаноле, получают 10.1 г (88.3%) (-)-[2S-[ 2α,4α, 4(S)]]-4-[4-[4-[4-[[2-(2,4-дифторфенил)-2- (1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси] фенил]- 1-пиперазинил] фенил]-2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол- 3-она; т.пл. 179.7oC; [α]
Пример 13
Смесь 9.3 г 4-метилбензолсульфоната (сложный эфир) 4-метилбензолсульфоната (соль, 1 : 1) (-)-(2S,цис)-2-(2,4-дифторфенил)- 2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-метанола (промежуточное соединение 9), 5.5 г (-)-(R)-2,4-дигидро-4- [4-[4-(4-гидроксифенил)-1-пиперазинил] фенил]-2-(1-метилпропил)-3H- 1,2,4-триазол-3-она (промежуточное соединение 8), 1.0 г чешуек гидроксида натрия и 150 мл и N,N-диметилформамида перемешивают в течение ночи при 50oC в атмосфере азота.
После охлаждения к реакционной смеси добавляют 800 мл воды. Образующийся осадок отфильтровывают и растворяют в метиленхлориде. Органический слой промывают водой, фильтруют и упаривают. Остаток очищают с помощью колоночной хроматографии (силикагель, элюент - хлороформ/метанол, 98.5 : 1.5).
Желаемые фракции упаривают и остаток растирают в метаноле, получают 7.7 г (81.8%) (-)-[2S-[ 2α,4α, 4(R)]]-4-[4-[4-[4-[[2- (2,4-дихлорфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан- 4-ил]-метокси]фенил]-1-пиперазинил] фенил]-2,4-дигидро-2- (1-метилпропил)-3H-1,2,4-триазол-3-она; т.пл. 183,3oC; [α]
Пример 14
Смесь 9.9 г 4-метилбензолсульфоната (сложный эфир) 4-метилбензолсульфоната (соль, 1:1) (+)-(2R,цис)-2-(2,4-дифторфенил)- 2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-метанола (промежуточное соединение 10), 5.5 г (-)-(R)-2,4-дигидро-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] фенил]-2-(1-метилпропил)-3H-1,2,4, -триазол-3-она (промежуточное соединение 8), 2.0 г чешуек гидроксида натрия и 150 мл N,N-диметилформамида перемешивают в течение ночи при 50oC в атмосфере азота.
После охлаждения к реакционной смеси добавляют воду. Образующий осадок отфильтровывают и растворяют в метиленхлориде. Органический слой промывают водой, фильтруют и упаривают. Остаток растирают в метаноле, получают 8.3 г (88.1%) (+)-[2R[ 2α,4α, 4(R)] ] -4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил) -1,3-диоксолан-4-ил] метокси]фенил]-1-пиперазинил] фенил]-2,4-дигидро- 2-(1-метилпропил)-3H-1,2,4-триазол-3-она; т.пл. 180.6oC; [α]
Пример 15
Смесь 6.7 г 4-метилбензолсульфоната (сложный эфир) 4-метил-бензолсульфоната (соль, 1:1) (+)-(2R,цис)-2-(2,4-фторфенил)- 2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-метанола (промежуточное соединение 10), 3.9 г (+)-(S)-2,4-дигидро-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] фенил] - 2-(1-метилпропил)-3H-1,2,4-триазол-3-она (промежуточное соединение 7), 1,5 г чешуек гидроксида натрия и 100 мл N,N-диметилформамида перемешивают в течение ночи при 50oC в атмосфере азота.
После охлаждения к реакционной смеси добавляют воду. Образующийся осадок отфильтровывают и растворяют в хлороформе. Органический слой промывают водой, фильтруют и упаривают. Остаток растирают в метаноле, получают 5.6 г (83.2%) (+)-[2R-[ 2α,4α, 4(S)] ]-4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1- илметил)-1,3-диоксолан-4-ил] метокси]фенил]-1-пиперазинил] фенил] -2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол-3-она; т.пл. 182.4oC; [α]
C. Физико-химические примеры
Пример 16
Растворимость индивидуальных стереоизомерных форм итраконазола и саперконазола в воде или в отсутствие или в присутствии различных концентраций 2-гидроксипропил-β-циклодекстрина при комнатной температуре определяют путем приготовления насыщенных растворов с измерение количества растворенного активного ингредиента. Образец исходного раствора, содержащего 0, 2.5, 5, 7.5, 10, 20, 30 или 40% (масс на объем) 2-гидроксипропил -β- циклодекстрина (М.З = 0.4) помещают в 20 мл темную ампулу.
В каждую ампулу добавляют такое количество интраконазола и саперконазола, которого достаточно для получения насыщенного раствора. Ампулы обрабатывают ультразвуком в течение 10 мин и все ампулы проверяют с целью удостовериться, что не осталось нерастворившегося вещества. После растворения всего материала добавляют следующую порцию.
Ампулы закрывают и защищают от света, заворачивая их в алюминиевую фольгу. Затем ампулы встряхивают в подходящей аппаратуре. Через 24 ч ампулы снова проверяют на наличие нерастворившегося остатка и есть ли необходимость в добавлении новой порции. Ампулы встряхивают по меньшей мере 72 ч и затем выдерживают до полного осаждения нерастворившегося материала. Ампулы открывают и измеряют pH насыщенных растворов.
Аликвоту находящейся над осадком жидкости фильтруют с помощью УФ-спектрометрии (255 нм), определяют количество растворенного активного ингредиента, если необходимо, то перед проведением УФ-анализа вышеуказанную аликвоту разбавляют до концентрации, приемлемой для УФ-спектрометрии.
В приведенных таблицах 4, 5 объединены данные по концентрациям (мг/100 мл) интраконазола и его индивидуальных стереоизомерных форм, а также саперканазола и его индивидуальных форм в воде или в отсутствие, или в присутствии различных концентраций 2-гидроксипропил-β-циклодекстрина.
Пример 17
Растворимость соединения 3 в искусственном желудочном соке сравнивают с растворимостью интраконазола. Приблизительно 10 мг любого продукта добавляют при комнатной температуре к 100 мл искусственного желудочного сока (0.2 г NaCl+0.7 мл концентрированной соляной кислоты, разбавленной до 10 мл).
Смесь перемешивают и из каждого раствора отбирают образец раствора через равномерные промежутки времени. Количество растворенного активного ингредиента определяют по методике, описанной в предыдущем примере.
В таблице 6 приведены данные по количеству растворенного соединения (в мл/100 мл).
Пример получения комплекса стериоизомерной формы интраказола с бета-циклодекстрином.
Раствор для орального введения.
В сосуде перемешивают 10 мл пропиленгликоля и 0,95 мл 10 н.HCl Этот раствор нагревают при температуре 40 - 50oC. К смеси добавляют 2,5 г (+)-[2R-[ 2α,4α, 4(S)] ]-4-[4-[4-[4-[[2-(2,4-дихлорфенил)-2-(1H-1,2,4-триазол-1-илметил) -1,3-диоксалан-4-ил] -метокси] фенил]-1-пиперазинил]фенил]-2,4- дигидро-2-(1-метилпропил)-3H-1,2,4-триазол-3-она и перемешивают до гомогенного состояния смеси. Затем раствор охлаждали до комнатной температуры. 2-Гидроксипропил -β- циклодекстрин (2-HP-β-CD), содержащий менее 1,5% незамещенного бета-циклодекстрина (60 г) растворяют в 40 мл очищенной воды в отдельном сосуде и затем добавляют к раствору лекарства. Последний перемешивают до гомогенного состояния. Доводят pH смеси до 1,9-2,1 10 н. раствором NaOH.
Затем добавляют очищенную воду до общего объема 100 мл и перемешивают до гомогенного состояния смеси. Таким образом получен раствор циклодекстринового комплекса интраконазола для орального применения. Этот раствор содержит 25 мг/мл (+)-[2R-[ 2α,4α, 4(S)]]-4-[4-[4-[4-[[2-(2,4-дихлорфенил)-2-(1H-1,2,4-триазол-1- илметил)-1,3-диоксолан-4-ил] метокси]фенил]-1-пиперазинил] фенил]-2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол-3-она.
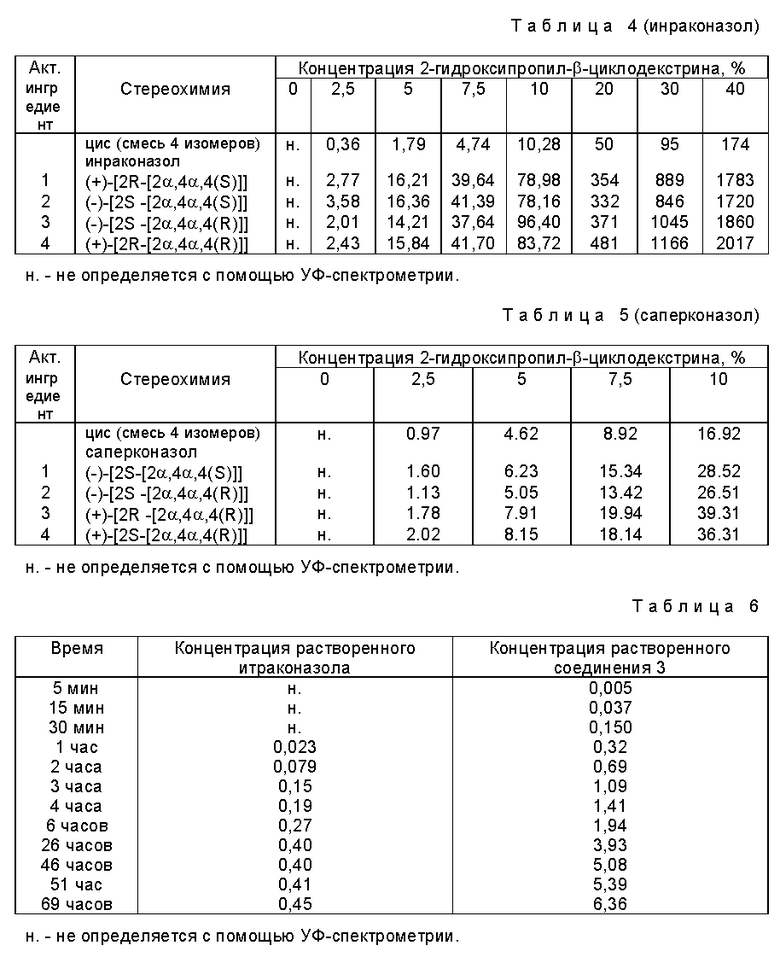
Изобретение относится к стериозомерным формам итраконазола (Х=Сl) и саперконазола (Х= F), которые могут быть представлены формулой цис-(I), приведенной в тексте описания, их фармацевтически приемлемым кислотно-аддитивным солевым формам, способам получения указанных стереоизомерных форм, их комплексам с производными циклодекстрина и фармацевтической композиции, содержащим вышеуказанные комплексы, обладающей антигрибковой активностью. 4 с. и 4 з.п.ф-лы.
где (1H-1,2,4-триазол-1-илметильный) фрагмент и замещенный феноксифрагмент расположены с одной стороны плоскости 1,3-диоксоланового кольца,
или его фармацевтически приемлемая кислотно-аддитивная соль.
(+)-[2R-[2α,4α, 4(S)] ] -4-[4-[4-[4-[[2-(2,4-дихлорфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси]фенил]-1-пиперазинил]фенил]-2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол-3-он;
(-)-[2S-[2α,4α, 4(S)] ]-4-[-4-[4-[4-[[2-(2,4-дихлорфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси]фенил]-1-пиперазинил]фенил]-2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол-3-он;
(-)-[2S-[2α,4α, 4(R)] ]-4-[-4-[4-[4-[[2-(2,4-дихлорфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси]фенил]-1-пиперазинил]фенил]-2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол-3-он;
(+)-[2R-[2α,4α, 4(R)] ] -4-[4-[4-[4-[[2-(2,4-дихлорфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси]фенил]-1-пиперазинил]фенил]-2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол-3-он;
(-)-[2S-[2α,4α, 4(S)] ] -4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси]фенил]-1-пиперазинил]фенил]-2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол-3-он;
(-)-[2S-[2α,4α, 4(R)] ]-4-[-4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси]фенил]-1-пиперазинил]фенил]-2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол-3-он;
(+)-[2R-[2α,4α, 4(R)] ] -4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси]фенил]-1-пиперазинил]фенил]-2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол-3-он;
(+)-[2R-[2α,4α, 4(S)] ] -4-[4-[4-[4-[[2-(2,4-дифторфенил)-2-(1H-1,2,4-триазол-1-илметил)-1,3-диоксолан-4-ил] метокси]фенил]-1-пиперазинил]фенил]-2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол-3-он;
3. Форма по п.2, имеющая стереоизомерную чистоту по меньшей мере от 96 до 100%.
(-)-(R)-(II)
или
(+)-(S)-(II)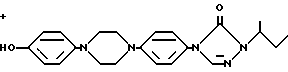
с энантиомерно чистым производным 1,3-диоксолана формулы
(-)-(2S, цис)-(III)
или
(+)-(2R, цис)-(III)
где OR является отщепляемой сульфонильной группой,
и при необходимости проводят дополнительную очистку полученного соединения общей формулы I, а также, если это необходимо, превращают его в фармацевтически приемлемую кислотно-аддитивную солевую форму путем обработки подходящей кислотой или превращают солевую форму в свободную основную форму обработкой щелочью.
| СПОСОБ ОДНОРАСТВОРНОГО ТРАВЛЕНИЯ ФОРМ | 0 |
|
SU283992A1 |
| US 4267179, 1981 | |||
| US 4916134, 1990 | |||
| US 4287195, 1981 | |||
| Станок для растяжения чулок бумагоделательных машин | 1929 |
|
SU23762A1 |
| ИСКРОВОЙ ПРОМЕЖУТОК ВЕНТИ.ЛЬНОГО РАЗРЯДНИКА | 0 |
|
SU237963A1 |
| US 4764604, 1988 | |||
| WO 8502767 A1, 1985 | |||
| WO 9012035 A1, 1990 | |||
| US 4535152, 1983. | |||

