Перекрестная ссылка на родственные заявки
[0001] В настоящей заявке испрашивается преимущество предварительной заявки на патент США рег. №62/680259, поданной 4 июня 2018, и предварительной заявки на патент США рег. №62/821678, поданной 21 марта 2019, содержание которых включено в настоящее описание в полном объеме.
Заявление о спонсировании правительством на средства Федерального фонда
[0002] Настоящее изобретение было разработано при поддержке Правительства на грант No. DP1 HG007811, выданный Национальным Институтом Здравоохранения. Правительство имеет определенные права на это изобретение.
Область изобретения
[0003] Варианты раскрытия настоящего изобретения относятся к секвенированию нуклеиновых кислот. В частности, варианты описанных здесь способов и композиций относятся к получению индексированных моноклеточных библиотек транскритомов и к получению данных об их последовательностях.
Предпосылки создания изобретения
[0004] Клетки проходят через различные функциональные и молекулярные состояния на различных этапах своего развития, таких как развитие многоклеточного организма и ответ на различные условия, такие как воздействие терапевтического агента. Характеризация пути перехода состояний клеток или судьбы клеток является полезной для понимания клеточных путей, включая развитие и молекулярный ответ клеток на изменение окружающей среды. Так, например, могут быть идентифицированы регуляторы дефектов развития, что позволяет лучше понять механизмы влияния терапевтических агентов на клетки.
[0005] Комбинаторное индексирование отдельных клеток («sci») представляет собой методику, в основе которой используется штриховое кодирование с разделением пулов для уникального мечения нуклеиновых кислот, содержащихся в большом количестве в отдельных клетках или ядрах. Однако, современные геномные методы, проводимые с использованием отдельных клеток, не обладают достаточной пропускной способностью и разрешением для получения общего представления о молекулярных состояниях и траекториях быстро диверсифицирующегося и расширяющегося числа типов клеток, которые обычно присутствуют во время развития многоклеточного организма. Современные геномные методы, проводимые с использованием отдельных клеток, позволяют лишь быстро зафиксировать состояние клетки, а поэтому не могут предоставить информацию о динамике перехода клетки из одного состояния в другое, регулируемой внутренними факторами (например, программой внутреннего клеточного цикла) и внешними факторами (например, ответом клетки на внешний стимул, такой как терапевтический агент).
Сущность
[0006] Настоящее изобретение относится к способам идентификации динамики перехода клетки из одного состояния в другое путем мечения вновь синтезированной РНК. Целые и вновь синтезированные РНК-транскриптомы фиксируют для характеризации динамики транскритомов между временными точками на уровне отдельных клеток. Настоящее изобретение также относится к способам, которые направлены на секвенирование представляющих интерес мРНК отдельных клеток, что позволяет устранить ограничения по силе тока при детектировании изменений количества любого данного транскрипта. Кроме того, настоящее изобретение относится к способам, которые снижают уровень потери клеток и устраняют ограничения по эффективности реакций, что позволяет определить профиль большего количества отдельных клеток, чем это было возможно ранее.
[0007] В одном варианте осуществления изобретения, способ включает получение множества ядер или клеток в первом множестве компартментов, где каждый компартмент содержит субпопуляцию ядер или клеток, и мечение вновь синтезированной РНК в субпопуляциях клеток или ядер, полученных из клеток. Молекулы РНК в каждой субпопуляции ядер или клеток обрабатывают для получения индексированных ядер или клеток, где такая обработка включает добавление к нуклеиновым кислотам РНК, присутствующим в каждой субпопуляции ядер или клеток, первой компартмент-специфической индексной последовательности с получением индексированных нуклеиновых кислот ДНК, присутствующих в индексированных ядрах или клетках, а затем объединение индексированных ядер или клеток для создания объединенных индексированных ядер или клеток.
[0008] В другом варианте осуществления изобретения, способ включает получение множества ядер или клеток в первом множестве компартментов, где каждый компартмент содержит субпопуляцию ядер или клеток. Каждую субпопуляцию подвергают контактированию с обратной транскриптазой и праймером, которые гибридизуются с предварительно определенной нуклеиновой кислотой РНК, что приводит к образованию двухцепочечных нуклеиновых кислот ДНК с праймером и соответствующей нуклеотидной последовательности ДНК из нуклеиновых кислот матричной РНК. Молекулы ДНК в каждой субпопуляции ядер или клеток обрабатывают для получения индексированных ядер или клеток, где такая обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в каждой субпопуляции ядер или клеток, первой компартмент-специфической индексной последовательности с получением индексированных нуклеиновых кислот, присутствующих в индексированных ядрах или клетках, а затем объединение индексированных ядер или клеток для создания объединенных индексированных ядер или клеток.
[0009] В другом варианте осуществления изобретения, способ включает получение множества ядер или клеток в первом множестве компартментов, где каждый компартмент содержит субпопуляцию ядер или клеток. Каждую субпопуляцию подвергают контактированию с обратной транскриптазой и праймером, которые гибридизуются с предварительно определенной нуклеиновой кислотой РНК, что приводит к образованию двухцепочечных нуклеиновых кислот ДНК с праймером и соответствующей нуклеотидной последовательности ДНК из нуклеиновых кислот матричной РНК. Молекулы ДНК в каждой субпопуляции ядер или клеток обрабатывают для получения индексированных ядер или клеток, где такая обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в каждой субпопуляции ядер или клеток, первой компартмент-специфической индексной последовательности с получением индексированных нуклеиновых кислот, присутствующих в индексированных ядрах или клетках, а затем объединение индексированных ядер или клеток для создания объединенных индексированных ядер или клеток. Объединенные в пул индексированные ядра или клетки разделяют, а затем дополнительно обрабатывают для добавления второго компартмент-специфического индекса к молекулам ДНК, объединяют, разделяют и снова обрабатывают для добавления третьего компартмент-специфического индекса к молекулам ДНК.
[0010] Определения
[0011] Используемые здесь термины имеют общепринятое значение, известное специалистам в данной области, если это не оговорено особо. Некоторые используемые здесь термины и их значения приводятся ниже.
[0012] Используемые здесь термины «организм» и «индивидуум» являются синонимами и относятся к микроорганизмам (например, к прокариотическим или эукариотическим), к животным и растениям. Примером животного является млекопитающее, такое как человек.
[0013] Используемый здесь термин «тип клеток» относится к идентификации клеток на основе морфологии, фенотипа, эволюционного развития или других известных или распознаваемых отличительных свойств клеток. Различные типы клеток могут быть получены из одного организма (или из организма одного и того же вида). Типичными типами клеток являются, но не ограничиваются ими, гаметы (включая женские гаметы, например, зрелые яйцеклетки или яйцеклетки, и мужские гаметы, например сперматозоиды), эпителиальные клетки яичника, фибробласты яичника, клетки яичек, клетки мочевого пузыря, иммунные клетки, В-клетки, Т-клетки, природные клетки-киллеры, дендритные клетки, раковые клетки, эукариотические клетки, стволовые клетки, клетки крови, мышечные клетки, жировые клетки, клетки кожи, нервные клетки, костные клетки, клетки поджелудочной железы, эндотелиальные клетки, эпителиальные клетки поджелудочной железы, альфа-клетки поджелудочной железы, бета-клетки поджелудочной железы, эндотелиальные клетки поджелудочной железы, лимфобласты костного мозга, В-лимфобласты костного мозга, макрофаги костного мозга, эритробласты костного мозга, дендритные клетки костного мозга, адипоциты костного мозга, остеоциты костного мозга, хондроциты костного мозга, промиелобласты, мегакариобласты костного мозга, клетки мочевого пузыря, В-лимфоциты головного мозга, глиальные клетки головного мозга, нейроны, астроциты головного мозга, клетки нейроэктодермы, макрофаги головного мозга, микроглиальные клетки головного мозга, эпителиальные клетки головного мозга, нейроны коркового слоя, фибробласты головного мозга, эпителиальные клетки молочной железы, эпителиальные клетки толстой кишки, В-лимфоциты толстой кишки, эпителиальные клетки молочной железы, миоэпителиальные клетки молочной железы, фибробласты молочной железы, энтероциты толстой кишки, эпителиальные клетки шейки матки, эпителиальные клетки протоков молочной железы, эпителиальные клетки языка, дендритные клетки миндалин, В-лимфоциты миндалин, лимфобласты периферической крови, Т-лимфобласты периферической крови, кожные Т-лимфоциты периферической крови, природные клетки-киллеры периферической крови, В-лимфобласты периферической крови, моноциты периферической крови, миелобласты периферической крови, монобласты периферической крови, промиелобласты периферической крови, макрофаги периферической крови, базофилы периферической крови, эпителиальные клетки печени, тучные клетки печени, эпителиальные клетки печени, В-лимфоциты печени, эндотелиальные клетки селезенки, эпителиальные клетки селезенки, В-лимфоциты селезенки, гепатоциты печени, фибробласты печени, эпителиальные клетки легких, эндотелиальные клетки бронхов, фибробласты легких, В-лимфоциты легких, шванновские клетки легких, плоские клетки легких, макрофаги легких, остеобласты легких, нейроэндокринные клетки, клетки альвеолы легких, эпителиальные клетки желудка и фибробласты желудка.
[0014] Используемый здесь термин «ткань» означает скопление или агрегацию клеток, которые вместе выполняют одну или более специфических функций в организме. Клетки могут быть, но необязательно, морфологически сходными. Типичными тканями являются, но не ограничиваются ими, ткани эмбрионов, эпидидимиса, глаз, мышц, кожи, сухожилий, вены, артерий, крови, сердца, селезенки, лимфоузлов, кости, костного мозга, легких, бронхов, трахеи, кишечника, тонкого кишечника, толстой кишки, ободочной кишки, прямой кишки, слюнных желез, языка, желчного пузыря, аппендикса, печени, поджелудочной железы, головного мозга, желудка, кожи, почек, мочеточника, мочевого пузыря, мочеиспускательного канала, гонады, яичек, яичника, матки, фаллопиевых труб, тимуса, гипофиза, щитовидной железы, надпочечников или паращитовидной железы. Ткань может быть получена из любых различных органов человека или другого организма. Ткань может быть здоровой или пораженной заболеванием. Примерами пораженных заболеванием тканей являются, но не ограничиваются ими, злокачественные новообразования репродуктивной ткани, легких, молочной железы, толстой и прямой кишки, предстательной железы, носоглотки, желудка, яичек, кожи, нервной системы, кости, яичника, печени, кроветворных тканей, поджелудочной железы, матки, почек, лимфоидных тканей и т.п. Злокачественные новообразованиями могут быть новообразования различных гистологических подтипов, например, карцинома, аденокарцинома, саркома, фиброаденокарцинома, нейроэндокринная опухоль или недифференцированная опухоль.
[0015] Используемый здесь термин «компартмент» означает область или объем, которые отделяют или изолируют одни элементы от других элементов. Типичными компартментами являются, но не ограничиваются ими, флаконы, пробирки, лунки, капли, болюсы, сферы, сосуды, поверхностные элементы или области или объемы, разделенные физическими силами, такими как поток жидкости, магнетизм, электрический ток или т.п. В одном варианте осуществления изобретения, компартмент представляет собой лунку многолуночного планшета, такого как 96- или 384-луночный планшет. Используемый здесь термин «капли» может включать гидрогелевую сферу, которая представляет собой гранулу для инкапсуляции одного или более ядер или клеток, и включает гидрогелевую композицию. В некоторых вариантах осуществления изобретения, капля представляет собой гомогенную каплю из гидрогелевого вещества или представляет собой полую каплю, имеющую оболочку из полимерного гидрогеля. Гомогенная капля, независимо от того, является ли она гомогенной или полой, может быть подходящей для инкапсуляции в нее одного или более ядер или одной или более клеток. В некоторых вариантах осуществления изобретения, капля представляет собой каплю, стабилизированную поверхностно-активным веществом.
[0016] Используемый здесь термин «транспосомный комплекс» означает фермент интеграции и нуклеиновую кислоту, включающую сайт распознавания интеграции. «Транспосомный комплекс» представляет собой функциональный комплекс, образованный транспозазой и сайтом распознавания транспозазы, который способен катализировать реакцию транспозиции (см., например, Gunderson et al., WO 2016/130704). Примерами ферментов интеграции являются, но не ограничиваются ими, интеграза или транспозаза. Примерами сайтов распознавания интеграции являются, но не ограничиваются ими, сайт распознавания транспозазы.
[0017] Используемый здесь термин «нуклеиновая кислота» употребляется в соответствии с ее применением в данной области и включает природные нуклеиновые кислоты или их функциональные аналоги. Особенно подходящие функциональные аналоги способны гибридизоваться с нуклеиновой кислотой по последовательность-специфическому механизму или могут быть использованы в качестве матрицы для репликации конкретной нуклеотидной последовательности. Природные нуклеиновые кислоты обычно имеют остов, содержащий фосфодиэфирные связи. Структура аналога может иметь чередующуюся связь в остове, включая любую из множества связей, известных специалистам в данной области. Природные нуклеиновые кислоты обычно имеют сахар дезоксирибозу (например, присутствующую в дезоксирибонуклеиновой кислоте (ДНК)) или сахар рибозу (например, присутствующую в рибонуклеиновой кислоте (РНК)). Нуклеиновая кислота может содержать любой из ряда аналогов этих сахарных групп, известных специалистам в данной области. Нуклеиновая кислота может включать природные или неприродные основания. В соответствии с этим, природная дезоксирибонуклеиновая кислота может иметь одно или более оснований, выбранных из группы, состоящей из аденина, тимина, цитозина или гуанина, а рибонуклеиновая кислота может иметь одно или более оснований, выбранных из группы, состоящей из аденина, урацила, цитозина или гуанина. Подходящие неприродные основания, которые могут быть включены в нуклеиновую кислоту, известны специалистам в данной области. Примерами неприродных оснований являются блокированная нуклеиновая кислота (LNA), мостиковая нуклеиновая кислота (BNA) и псевдокомплементарные основания (Trilink Biotechnologies, San Diego, CA). В ДНК-олигонуклеотид могут быть включены основания LNA и BNA, которые повышают силу и специфичность гибридизации олигонуклеотидов. Основания LNA и BNA известны специалистам в данной области и используются в рутинных способах. Если это не оговорено особо, то термин «нуклеиновая кислота» включает природную и неприродную мРНК, некодирующую РНК, например РНК без poly-A на 3'-конце, нуклеиновые кислоты, происходящие от РНК, например, кДНК и ДНК.
[0018] Используемый здесь термин «мишень», если он употребляется по отношению к нуклеиновой кислоте, означает семантический идентификатор нуклеиновой кислоты в описанных здесь способах или композициях и необязательно ограничивает структуру или функцию нуклеиновой кислоты помимо тех, которые были конкретно указаны. Нуклеиновая кислота-мишень может представлять собой, по существу, любую нуклеиновую кислоту с известной или неизвестной последовательностью. Нуклеиновая кислота может представлять собой, например, фрагмент геномной ДНК (например, хромосомной ДНК), внехромосомной ДНК, такой как плазмида, неклеточной ДНК, РНК (например, РНК или некодирующей РНК), белков (например, клеточных белков или белков клеточной поверхности) или кДНК. Секвенирование позволяет определить последовательность всей молекулы или части молекулы-мишени. Мишень может происходить от первичного образца нуклеиновой кислоты, такого как ядро. В одном варианте осуществления изобретения, мишени могут быть обработаны с получением матриц, подходящих для амплификации, путем присоединения универсальных последовательностей к одному или обоим концам каждого фрагмента-мишени. Мишени могут быть также получены из образца первичной РНК посредством обратной транскрипции в кДНК. В одном варианте осуществления изобретения, мишень используется в отношении подгруппы ДНК, РНК или белков, присутствующих в клетке. Целевое секвенирование включает отбор и выделение представляющих интерес генов или областей или белков, обычно, с помощью ПЦР-амплификации (например, область-специфических праймеров) или методом захвата на основе гибридизации или с использованием антител. Нацеленное обогащение может быть осуществлено на различных стадиях этого способа. Так, например, представление РНК-мишени может быть достигнуто с использованием мишень-специфических праймеров на стадии обратной транскрипции или на стадии обогащения на основе гибридизации подгруппы из более сложных библиотек. Примером является секвенирование экзома или анализ L1000 (Subramanian et al., 2017, Cell, 171:1437-1452). Нацеленное секвенирование может включать любой из способов обогащения, известных специалисту в данной области.
[0019] Термин «универсальный», если он используется здесь для описания нуклеотидной последовательности, относится к области последовательности, которая является общей для двух или более молекул нуклеиновой кислоты, где эти молекулы также имеют области последовательности, которые отличаются друг от друга, Универсальная последовательность, которая присутствует в различных членах набора молекул, позволяет захватывать множество различных нуклеиновых кислот благодаря группе универсальных нуклеиновых кислот для захвата, например, для захвата олигонуклеотидов, которые являются комплементарными части универсальной последовательности, например универсальной последовательности для захвата. Неограничивающие примеры универсальных последовательностей для захвата включают последовательности, которые являются идентичными или комплементарными праймерам Р5 и Р7. Аналогичным образом, универсальная последовательность, которая присутствует в различных членах набора молекул, позволяет осуществлять репликацию (например, секвенирование) или амплификацию множества различных нуклеиновых кислот благодаря группе универсальных праймеров, которые являются комплементарными части универсальной последовательности, например универсальной последовательности для заякоривания. В одном варианте осуществления изобретения, универсальные последовательности для заякоривания используются в качестве сайта, с которым гибридизуется универсальный праймер для секвенирования (например, секвенирующий праймер для считывания на дорожке 1 или 2). Следовательно, олигонуклеотид для захвата или универсальный праймер включает последовательность, которая может специфически гибридизоваться с универсальной последовательностью.
[0020] Термины «Р5» и «Р7» могут быть использованы при описании универсальной последовательности для захвата или олигонуклеотида для захвата. Термины «Р5» (праймер Р5) и «Р7» (праймер Р7) относятся к комплементу к Р5 и Р7, соответственно. Следует отметить, что в описанных здесь способах могут быть использованы любая подходящая универсальная последовательность для захвата или олигонуклеотид для захвата, и что использование Р5 и Р7 приводится лишь для примера. Использование олигонуклеотидов для захвата, таких как Р5 и Р7, или их комплементов на проточных ячейках известно специалистам в данной области, и их примеры раскрываются в WO 2007/010251, WO 2006/064199, WO 2005/065814, WO 2015/106941, WO 1998/044151 и WO 2000/018957. Так, например, любой подходящий прямой праймер для амплификации, независимо от того, является ли он иммобилизованным или присутствует в растворе, может быть использован в описанных здесь способах гибридизации с комплементарной последовательностью и амплификации последовательности. Аналогичным образом, любой подходящий обратный праймер для амплификации, независимо от того является ли он иммобилизованным или присутствует в растворе, может быть использован в описанных здесь способах гибридизации с комплементарной последовательностью и амплификации последовательности. Специалисту в данной области известны способы конструирования и использования последовательностей праймеров, которые являются подходящими для захвата и/или амплификации представленных здесь нуклеиновых кислот.
[0021] Используемый здесь термин «праймер» и его производные обычно относятся к любой нуклеиновой кислоте, которая может гибридизоваться с представляющей интерес последовательностью-мишенью. Обычно, праймер функционирует как субстрат, на котором нуклеотиды могут полимеризоваться посредством полимеразы, или с которыми может быть лигирована нуклеотидная последовательность, такая как индекс; однако, в некоторых вариантах осуществления, праймер может встраиваться в синтезированную цепь нуклеиновой кислоты и образовывать сайт, с которым может гибридизоваться другой праймер для инициации синтеза новой цепи, которая является комплементарной синтезированной молекуле нуклеиновой кислоты. Праймер может включать любую комбинацию нуклеотидов или их аналогов. В некоторых вариантах осуществления изобретения, праймер представляет собой одноцепочечный олигонуклеотид или полинуклеотид. Используемые здесь термины «полинуклеотид» и «олигонуклеотид» являются синонимами и означают полимерную форму нуклеотидов любой длины и могут включать рибонуклеотиды, дезоксирибонуклеотиды их аналоги или их смеси. Эти термины следует понимать как эквиваленты, аналоги или любые ДНК, РНК, кДНК или конъюгаты «антитело-олигонуклеотид», полученные из нуклеотидных аналогов, и эти термины могут быть применены к одноцепочечным (таким как смысловые или антисмысловые) полинуклеотидам и двухцепочечным полинуклеотидам. Используемый здесь термин также охватывает кДНК, которая является комплементарной ДНК или ее копией, полученной из матричной РНК, например, под действием обратной транскриптазы. Этот термин относится только к первичной структуре молекулы. Таким образом, этот термин включает трех-, двух- и одноцепочечную дезоксирибонуклеиновую кислоту («ДНК»), а также трех-, двух- и одноцепочечную рибонуклеиновую кислоту («РНК»).
[0022] Используемый здесь термин «адаптер» и его производные, например универсальный адаптер, в общих чертах относится к любому линейному олигонуклеотиду, который может быть присоединен к молекуле нуклеиновой кислоты согласно изобретению. В некоторых вариантах осуществления изобретения, адаптер, по существу, не является комплементарным 3'-концу или 5'-концу любой последовательности-мишени, присутствующей в образце. В некоторых вариантах осуществления изобретения, подходящий адаптер имеет длину в пределах приблизительно 10-100 нуклеотидов, приблизительно 12-60 нуклеотидов или приблизительно 15-50 нуклеотидов. Вообще говоря, адаптер может включать любую комбинацию нуклеотидов и/или нуклеиновых кислот. В некоторых аспектах изобретения, адаптер может включать одну или более расщепляемых групп в одном или более положениях. В другом аспекте изобретения, адаптер может включать последовательность, которая, по существу, идентична или, по существу, комплементарна по меньшей мере части праймера, например, универсального праймера. В некоторых вариантах осуществления изобретения, адаптер может включать штрих-код (также называемый здесь «меткой» или «индексом») для облегчения последующего исправления ошибок, идентификации или секвенирования. Термины «адаптор» и «адаптер» используются как синонимы.
[0023] Используемый здесь термин «каждый», если он употребляется для описания группы объектов, относится к идентификации отдельного объекта в этой группе, но он необязательно относится к каждому предмету в данной группе, если это не оговорено особо.
[0024] Используемый здесь термин «транспорт» относится к перемещению молекулы через жидкость. Этот термин может включать пассивный транспорт, такой как движение молекул вдоль градиента их концентрации (например, пассивная диффузия). Этот термин также может включать активный транспорт, благодаря которому молекулы могут двигаться вдоль градиента концентрации или против градиента концентрации. Таким образом, транспорт может включать приложение энергии для перемещения одной или более молекул в желаемом направлении или в нужное положение, такое как сайт амплификации.
[0025] Используемый здесь термин «амплифицировать», «амплификация» или «реакция амплификации» и их производные, по существу, относятся к любому действию или процессу, в результате которого по меньшей мере часть молекулы нуклеиновой кислоты реплицируется или копируется по меньшей мере в одну дополнительную молекулу нуклеиновой кислоты. Дополнительная молекула нуклеиновой кислоты включает, но необязательно, последовательность, которая, по существу, идентична или, по существу, комплементарна по меньшей мере некоторой части матричной молекулы нуклеиновой кислоты. Матричная молекула нуклеиновой кислоты может быть одноцепочечной или двухцепочечной, а дополнительная молекула нуклеиновой кислоты может независимо быть одноцепочечной или двухцепочечной. Амплификация включает, но необязательно, линейную или экспоненциальную репликацию молекулы нуклеиновой кислоты. В некоторых вариантах осуществления изобретения, такая амплификация может быть осуществлена в изотермических условиях; а в других вариантах осуществления изобретения, такая амплификация может включать реакцию в термоячейке. В некоторых вариантах осуществления изобретения, амплификация представляет собой мультиплексную амплификацию, которая включает одновременную амплификацию множества последовательностей-мишеней в одной реакции амплификации. В некоторых вариантах осуществления изобретения, термин «амплификация» включает амплификацию по меньшей мере некоторой части нуклеиновых кислот на основе ДНК и РНК отдельно или в комбинации. Реакция амплификации может включать любые процессы амплификации, известные специалисту в данной области. В некоторых вариантах осуществления изобретения, реакция амплификации включает полимеразную цепную реакцию (ПЦР).
[0026] Используемый здесь термин «условия амплификации» и его производные обычно относится к условиям, подходящим для амплификации одной или более последовательностей нуклеиновых кислот. Такая амплификация может быть линейной или экспоненциальной. В некоторых вариантах осуществления изобретения, условия амплификации могут включать изотермические условия или, альтернативно, условия реакции в термоячейке или комбинацию изотермических условий и условий реакции в термоячейке. В некоторых вариантах осуществления изобретения, условия, подходящие для амплификации одной или более последовательностей нуклеиновых кислот, включают условия полимеразной цепной реакции (ПЦР). Обычно, условия амплификации относятся к реакционной смеси, которая является достаточной для амплификации нуклеиновых кислот, таких как одна или более последовательностей-мишеней, фланкированных универсальной последовательностью, или для амплификации амплифицированной последовательности-мишени, лигированной с одним или более адаптерами. Вообще говоря, условия амплификации включают катализатор для амплификации или для синтеза нуклеиновой кислоты, например, полимеразу; праймер, который обладает некоторой степенью комплементарности с амплифицируемой нуклеиновой кислотой; и нуклеотиды, такие как дезоксирибонуклеотид-трифосфаты (dNTP), для ускорения удлинения праймера после гибридизации с нуклеиновой кислотой. Условия амплификации могут потребовать гибридизации или отжига праймера с нуклеиновой кислотой, удлинения праймера и стадии денатурации, при которой удлиненный праймер отделяется от последовательности нуклеиновой кислоты в процессе амплификации. Обычно, но необязательно, условия амплификации могут включать реакцию в термоячейке; а в некоторых вариантах осуществления изобретения, условия амплификации включают множество циклов, где повторяются стадии отжига, удлинения и разделения. Обычно, условия амплификации включают катионы, такие как Mg2+ или Mn2+, и могут также включать различные модификаторы ионной силы.
[0027] Используемые здесь термины «повторная амплификация» и их производные обычно относятся к любому процессу, посредством которого по меньшей мере часть амплифицированной молекулы нуклеиновой кислоты дополнительно амплифицируется посредством любой подходящей реакции амплификации (называемой в некоторых вариантах осуществления изобретения «вторичной» амплификацией), что приводит к образованию повторно амплифицированной молекулы нуклеиновой кислоты. Вторичная амплификация необязательно должна быть идентична первоначальному процессу амплификации, в результате которого была получена амплифицированная молекула нуклеиновой кислоты, и при этом не требуется, чтобы повторно амплифицированная молекула нуклеиновой кислоты была полностью идентичной или полностью комплементарной амплифицированной молекуле нуклеиновой кислоты; и единственное, что необходимо, - это то, чтобы повторно амплифицированная молекула нуклеиновой кислоты включала по меньшей мере часть молекулы амплифицированной нуклеиновой кислоты или ее комплемент. Так, например, повторная амплификация, в отличие от первичной амплификации, может включать использование различных условий амплификации и/или различных праймеров, включая различные мишень-специфичные праймеры.
[0028] Используемый здесь термин «полимеразная цепная реакция» («ПЦР») относится к методу Муллиса, см. патенты США №.4683195 и 4683202, где описан способ повышения концентрации сегмента представляющего интерес полинуклеотида в смеси геномной ДНК без клонирования или очистки. Этот способ амплификации представляющего интерес полинуклеотида состоит из введения большого избытка двух олигонуклеотидных праймеров в смесь ДНК, содержащую представляющий интерес полинуклеотид, с последующим проведением серий термоциклов в присутствии ДНК-полимеразы. Два праймера являются комплементарными соответствующим цепям представляющего интерес двухцепочечного полинуклеотида. Сначала смесь денатурируют при более высокой температуре, а затем праймеры гибридизуют с комплементарными последовательностями в представляющей интерес молекуле полинуклеотида. После отжига, праймеры удлиняют посредством полимеразы с образованием новой пары комплементарных цепей. Стадии денатурации, отжига и удлинения праймеров под действием полимеразы могут повторяться много раз (и эти стадии называются термоциклами) с получением высокой концентрации амплифицированного сегмента нужного представляющего интерес полинуклеотида. Длину амплифицированного сегмента нужного представляющего интерес полинуклеотида (ампликона) определяют по относительным положениям праймеров по отношению друг к другу, и, следовательно, эта длина является регулируемым параметром. Благодаря повторению такого процесса, этот метод называется ПЦР. Поскольку нужные амплифицированные сегменты представляющего интерес полинуклеотида становятся преобладающими последовательностями нуклеиновых кислот (с точки зрения концентрации) в смеси, то считается, что они являются «ПЦР-амплифицированными». В модификации обсуждаемого выше способа, молекулы нуклеиновой кислоты-мишени могут быть амплифицированы с помощью ПЦР с использованием множества различных пар праймеров, а в некоторых случаях, одной или более пар праймеров на представляющую интерес молекулу нуклеиновой кислоты-мишени, что позволяет проводить мультиплексную ПЦР-реакцию.
[0029] Как определено в настоящей заявке, «мультиплексная амплификация» относится к селективной и нерандомизированной амплификации двух или более последовательностей-мишеней в образце, проводимой с использованием по меньшей мере одного мишень-специфического праймера. В некоторых вариантах осуществления изобретения, мультиплексную амплификацию осуществляют так, чтобы некоторые или все последовательности-мишени амплифицировались в одном реакционном сосуде. «Плексность» или «множество» для данной мультиплексной амплификации, по существу, относится к числу различных мишень-специфических последовательностей, которые амплифицируются в процессе одной мультиплексной амплификации. В некоторых вариантах осуществления изобретения, плексность может быть приблизительно 12-кратной, 24-кратной, 48-кратной, 96-кратной, 1692-кратной, 384-кратной, 768-кратной, 1536-кратной, 3072-кратной, 6144-кратной или более. Амплифицированные последовательности-мишени также могут быть детектированы несколькими различными методами (например, с помощью гель-электрофореза с последующей денситометрией, путем количественной оценки с использованием биоанализатора или количественной ПЦР; путем гибридизации с меченым зондом; путем включения биотинилированных праймеров с последующим детектированием конъюгата «авидин-фермент»; путем включения 32Р-меченных дезоксинуклеотид-трифосфатов в амплифицированную последовательность-мишень).
[0030] Используемый здесь термин «амплифицированные последовательности-мишени» и их производные обычно относится к последовательности нуклеиновой кислоты, полученной путем амплификации последовательностей-мишеней с использованием мишень-специфических праймеров и с применением описанных здесь способов. Амплифицированные последовательности-мишени могут быть смысловыми (то есть, с положительной цепью) или антисмысловыми (то есть, с отрицательной цепью) по отношению к последовательностям-мишеням.
[0031] Используемые здесь термины «лигирующий», «лигирование» и их производные обычно относятся к способу ковалентного связывания двух или более молекул вместе, например, ковалентному связыванию двух или более молекул нуклеиновой кислоты друг с другом. В некоторых вариантах осуществления изобретения, лигирование включает введение «ников» между соседними нуклеотидами нуклеиновых кислот. В некоторых вариантах осуществления изобретения, лигирование включает образование ковалентной связи между концом первой и концом второй молекулы нуклеиновой кислоты. В некоторых вариантах осуществления изобретения, лигирование может включать образование ковалентной связи между 5'-фосфатной группой одной нуклеиновой кислоты и 3'-гидроксильной группой второй нуклеиновой кислоты, и тем самым образование лигированной молекулы нуклеиновой кислоты. Обычно, в целях раскрытия настоящего изобретения, амплифицированная последовательность-мишень может быть лигирована с адаптером с получением амплифицированной последовательности-мишени, лигированной с адаптером.
[0032] Используемый здесь термин «лигаза» и его производные, по существу, означает любой агент, способный катализировать лигирование двух молекул субстрата. В некоторых вариантах осуществления изобретения, лигаза представляет собой фермент, способный катализировать включение ников между соседними нуклеотидами нуклеиновой кислоты. В некоторых вариантах осуществления изобретения, лигаза представляет собой фермент, способный катализировать образование ковалентной связи между 5'-фосфатом одной молекулы нуклеиновой кислоты и 3'-гидроксилом другой молекулы нуклеиновой кислоты, что будет приводить к образованию лигированной молекулы нуклеиновой кислоты. Подходящими лигазами могут быть, но не ограничиваются ими, ДНК-лигаза Т4, РНК-лигаза Т4 и ДНК-лигаза E. coli.
[0033] Используемый здесь термин «условия лигирования» и его производные обычно относится к условиям, подходящим для лигирования двух молекул друг с другом. В некоторых вариантах осуществления изобретения, условия лигирования являются подходящими для закрытия «ников» или «разрывов» между нуклеиновыми кислотами. Используемые здесь термины «ник» или «разрыв» соответствуют терминам, используемым в литературе. Обычно, ник или разрыв может быть лигирован в присутствии фермента, такого как лигаза, при подходящей температуре и рН. В некоторых вариантах осуществления изобретения, ДНК-лигаза Т4 может соединять «ник» между нуклеиновыми кислотами при температуре приблизительно 70-72°С.
[0034] Используемый здесь термин «проточная ячейка» относится к камере, имеющей твердую поверхность, через которую могут проходить один или более жидких реагентов. Примеры проточных ячеек и родственных жидкостных систем и платформ для детектирования, которые могут быть легко применены в способах согласно изобретению, описаны, например, Bentley et al., Nature 456: 53-59 (2008), в WO 04/018497; в патенте США 7057026; в WO 91/06678; WO 07/123744; в патентах США 7292992; 7211414; 7315019; 7405281 и в заявке на патент США 2008/0108082.
[0035] Используемый здесь термин «ампликон», если он относится к нуклеиновой кислоте, означает продукт копирования нуклеиновой кислоты, где этот продукт имеет нуклеотидную последовательность, которая является идентичной или комплементарной по меньшей мере части нуклеотидной последовательности нуклеиновой кислоты. Ампликон может быть получен любыми различными методами амплификации, в которых используются нуклеиновая кислота или ее ампликон в качестве матрицы, включая, например, удлинение под действием полимеразы, полимеразную цепную реакцию (ПЦР), амплификацию по типу «катящегося кольца» (RCA), удлинение посредством лигирования или лигазную цепную реакцию. Ампликон может представлять собой молекулу нуклеиновой кислоты, имеющую одну копию конкретной нуклеотидной последовательности (например, продукт ПЦР) или множество копий нуклеотидной последовательности (например, конкатемерный продукт RCA). Первый ампликон нуклеиновой кислоты-мишени обычно представляет собой комплементарную копию. Последующие ампликоны являются копиями, которые создаются после продуцирования первого ампликона из нуклеиновой кислоты-мишени или из первого ампликона. Последующий ампликон может иметь последовательность, которая, по существу, комплементарна нуклеиновой кислоте-мишени или, по существу, идентична нуклеиновой кислоте-мишени.
[0036] Используемый здесь термин «сайт амплификации» означает сайт или массив, где могут быть созданы один или более ампликонов. Сайт амплификации может также иметь конфигурацию, при которой он содержит, сохраняет или присоединяет по меньшей мере один ампликон, который генерируется в этом сайте.
[0037] Используемый здесь термин «массив» означает совокупность сайтов, которые могут отличаться друг от друга по их относительному местоположению. Различью молекулы, которые находятся в различных положениях массива, могут отличаться друг от друга в зависимости от местоположения сайтов в массиве. Отдельный сайт массива может включать одну или более молекул определенного типа. Так, например, сайт может включать одну молекулу нуклеиновой кислоты-мишени, имеющую конкретную последовательность, или этот сайт может включать несколько молекул нуклеиновой кислоты, имеющих одну и ту же последовательность (и/или комплементарную последовательность). Сайты массива могут иметь различные признаки на одной и той же подложке. Типичными признаками являются, но не ограничиваются ими, углубления в подложке, сферы (или другие частицы), расположенные в подложке или на подложке, выступы из подложки, складки на подложке или каналы в подложке. Сайты массива могут представлять собой отдельные подложки, каждая из которых содержит различные молекулы. Различные молекулы, прикрепленные к отдельным подложкам, могут быть идентифицированы по положениям подложки на поверхности, с которой они связаны, или по положениям подложки в жидкости или геле. Репрезентативные массивы, в которых отдельные подложки расположены на поверхности, включают, но не ограничиваются ими, массивы, имеющие сферы в лунках.
[0038] Используемый здесь термин «емкость», если он относится к сайту и молекуле нуклеиновой кислоты, означает максимальное количество молекул нуклеиновой кислоты, которое может занимать этот сайт. Так, например, этот термин может относиться к общему количеству молекул нуклеиновой кислоты, которые могут занимать сайт в конкретных условиях. Также могут быть использованы и другие параметры, включая, например, общую массу молекул нуклеиновой кислоты или общее число копий конкретной нуклеотидной последовательности, которая может занимать сайт в конкретных условиях. Обычно, емкость сайта для нуклеиновой кислоты-мишени будет, по существу, эквивалентна емкости сайта для ампликонов нуклеиновой кислоты-мишени.
[0039] Используемый здесь термин «агент для захвата» означает материал, химическое вещество, молекулу или их части, которые способны присоединяться к молекуле-мишени, удерживаться на этой молекуле или связываться с ней (например, с нуклеиновой кислотой-мишенью). Типичными агентами для захвата являются, но не ограничиваются ими, нуклеиновая кислота для захвата (также называемая здесь олигонуклеотидом для захвата), которая комплементарна по меньшей мере части нуклеиновой кислоты-мишени; член пары связывания «рецептор-лиганд» (например, авидин, стрептавидин), биотин, лектин, углевод, белок, связывающийся с нуклеиновой кислотой, эпитоп, антитело и т.п.), способные связываться с нуклеиновой кислотой-мишенью (или связываться с линкерной молекулой, присоединенной к ней), или химический реагент, способный образовывать ковалентную связь с нуклеиновой кислотой-мишенью (или с линкерной молекулой, присоединенной к ней).
[0040] Используемый здесь термин «репортерная часть» может означать любую идентифицируемую метку, маркер, индексы, штрих-коды или группу, которые позволяют определять состав, идентичность и/или источник исследуемого аналита. В некоторых вариантах осуществления изобретения, репортерная часть может включать антитело, которое специфически связывается с белком. В некоторых вариантах осуществления изобретения, антитело может включать детектируемую метку. В некоторых вариантах осуществления изобретения, репортер может включать антитело или аффинный реагент, меченный нуклеиновой кислотой-меткой. Нуклеиновая кислота-метка может быть детектирована, например, с помощью анализа на проксимальное лигирование (PLA) или анализа на проксимальное удлинение (PEA) или считывание на основе секвенирования (Shahi et al. Scientific Reports volume 7, Article number: 44447, 2017) или CITE-seq (Stoeckius et al. Nature Methods 14: 865-868, 2017).
[0041] Используемый здесь термин «клональная популяция» относится к популяции нуклеиновых кислот, которая является гомогенной по отношению к конкретной нуклеотидной последовательности. Гомогенная последовательность обычно имеет длину по меньшей мере 10 нуклеотидов, но может быть даже более длинной, например, ее длина может составлять по меньшей мере 50, 100, 250, 500 или 1000 нуклеотидов. Клональная популяция может быть получена из одной нуклеиновой кислоты-мишени или матричной нуклеиновой кислоты. Обычно, все нуклеиновые кислоты в клональной популяции будут иметь одинаковую нуклеотидную последовательность. Следует отметить, что небольшое количество мутаций (например, из-за артефактов амплификации) может происходить в клональной популяции без отклонения от клональности.
[0042] Используемый здесь термин «уникальный молекулярный идентификатор» или «UMI» относится к молекулярной метке, либо рандомизированной, либо нерандомизированной, либо полурандомизированной, которая может быть присоединена к нуклеиновой кислоте. UMI, при его включении в нуклеиновую кислоту, может быть использован для последующей коррекции смещения амплификации путем прямого подсчета уникальных молекулярных идентификаторов (UMI), которые секвенируют после амплификации.
[0043] Используемый здесь термин «экзогенное» соединение, например, экзогенный фермент, относится к соединению, которое обычно или по своей природе не присутствует в конкретной композиции. Так, например, если конкретная композиция включает клеточный лизат, то экзогенный фермент представляет собой фермент, который обычно или по своей природе не присутствует в клеточном лизате.
[0044] Используемый здесь термин «предоставление» в отношении композиции, изделия, нуклеиновой кислоты или ядра означает получение композиции, изделия, нуклеиновой кислоты или ядра, закупку композиции, изделия, нуклеиновой кислоты или ядра, или получение соединения, композиции, изделия или ядра каким-либо другим образом.
[0045] Термин «и/или» означает один или все из перечисленных элементов или комбинацию любых двух или более из перечисленных элементов.
[0046] Слова «предпочтительный» и «предпочтительно» относятся к вариантам раскрытия изобретения, которые могут давать определенные преимущества при определенных обстоятельствах. Однако, предпочтительными также могут быть и другие варианты осуществления изобретения при тех же самых или других обстоятельствах. Кроме того, при описании одного или более предпочтительных вариантов осуществления не подразумевается, что не могут быть использованы и другие варианты, а поэтому из объема раскрытия изобретения не могут быть исключены и другие варианты.
[0047] Термины «содержит» и их варианты, если эти термины имеются в описании и в формуле изобретения, не имеют ограничивающего значения.
[0048] Следует отметить, что везде, где описанные здесь варианты осуществления изобретения употребляются вместе со словом «включать», «включает» или «включающий» и т.п., также могут быть использованы и другие аналогичные варианты, описанные с употреблением терминов «состоящий из» и/или «состоящий, по существу, из».
[0049] Если это не оговорено особо и не указано иное, то слова «а», «an», «the» и «по меньшей мере один» являются синонимами и означают один или более, чем один.
[0050] Кроме того, в настоящем описании, указание численных диапазонов вплоть до граничных значений включает все числа, включенные в этот диапазон (например, интервал 1-5 включает значения 1; 1,5; 2; 2,75; 3; 3,80; 4; 5 и т.п.).
[0051] Для любого раскрытого здесь способа, который включает отдельные стадии, эти стадии могут быть проведены в любом возможном порядке. И при необходимости, могут быть одновременно проведены две или более стадий в любой комбинации.
[0052] Во всем описании изобретения, термины «один вариант осуществления изобретения», «вариант осуществления изобретения», «определенные варианты осуществления изобретения» или «некоторые варианты осуществления изобретения» и т.п. означают, что конкретные признаки, конфигурации, композиции или характеристики, описанные согласно данному варианту, включены по меньшей мере в один вариант осуществления раскрытия изобретения. Таким образом, употребление таких терминов в различных частях описания изобретения необязательно относится к тому же варианту раскрытия изобретения. Кроме того, конкретные признаки, конфигурации, композиции или характеристики могут быть объединены любым подходящим способом в одном или более вариантах осуществления изобретения.
Краткое описание чертежей
[0053] Нижеследующее подробное описание иллюстративных вариантов раскрытия настоящего изобретения приводится для лучшего понимания изобретения при его чтении со ссылкой на нижеследующие чертежи.
[0054] На фиг. 1 представлена общая блок-схема общего иллюстративного способа комбинаторного индексирования в одной клетке в соответствии с раскрытием изобретения.
[0055] На фиг. 2 представлена общая блок-схема общего иллюстративного способа комбинаторного индексирования в одной клетке в соответствии с раскрытием изобретения.
[0056] На фиг. 3 представлена общая блок-схема общего иллюстративного способа комбинаторного индексирования в одной клетке в соответствии с раскрытием изобретения.
[0057] На фиг. 4 показано, что sci-РНК-seq3 позволяет определить профиль ~2 миллионов клеток от 61 эмбриона мыши на 5 стадиях развития в одном эксперименте. (А) рабочая диаграмма sci-РНК-seq3 и экспериментальная схема. (В) Сравнение пропускной способности эксперимента с недавно полученными отчетами. (С) График рассеяния для числа UMI для мышей и человека, построенный для клеток HEK293T и NIH/3T3. (D) Гистограмма, иллюстрирующая число клеток, профилированных для каждого из 61 эмбриона мыши. (Е) Прямоугольная диаграмма, иллюстрирующая количество генов и обнаруженных UMI на клетку. (F) График рассеяния уникальных ридов, выровненных по генам Xist (chr X) и chrY на мышиный эмбрион. (G) Псевдовременная траектория псевдообъемов профилей РНК-seq мышиных эмбрионов. (Н) Тепловая карта изменений в экспрессии маркерного гена Е9.5-Е13.5 из псевдообъемов профилей РНК-seq мышиных эмбрионов.
[0058] На фиг. 5 показаны результаты анализов на признаки и анализов QC для sci-РНК-seq3. (А) Гистограмма, иллюстрирующая число ОТ-лунок, используемых для каждого из 61 мышиного эмбриона. (В) Гистограмма, иллюстрирующая распределение исходных секвенирующих ридов для каждой ПЦР-лунки в sci-РНК-seq3. (С) Прямоугольная диаграмма, иллюстрирующая число UMI на клетку для клеток HEK293T и NIH/3T3. (D) Прямоугольная диаграмма, иллюстрирующая соотношение ридов, картируемых для ожидаемых видов клеток HEK293T (человека) и NIH/3T3 (мыши). (Е) Прямоугольная диаграмма для сравнения числа UMI на клетку (снижение выборки до 20000 исходных ридов на клетку) для sci-РНК-seq3 и sci-РНК-seq. (F) Корреляция между данными измерений уровней экспрессии генов в профилях объединенных клеток HEK293T для sci-РНК-seq3 и sci-РНК-seq.
[0059] На фиг. 6 показаны результаты анализов на дополнительные признаки и анализов QC для sci-РНК-seq3. (А) График рассеяния, иллюстрирующий корреляцию между количеством используемых ОТ-лунок и количеством выделенных клеток на эмбрион. (B-D) Гистограмма для сравнения числа исходных веквенирующих ридов (В), детектированных генов (С) и UMI (D) на клетку в методах sci-РНК-seq3 и других методах. (Е) Прямоугольная диаграмма, иллюстрирующая число UMI, детектированных на клетку эмбрионов на всех пяти стадиях развития. (F) Гистограмма, иллюстрирующая число мужских и женских эмбрионов, профиль которых был определен на каждой стадии развития.
[0060] На фиг. 7 показаны профили псевдообъема РНК-seq мышиных эмбрионов, которые были легко разделены по стадиям развития. (A) t-SNE объединенных транскритомов отдельных клеток, полученных от каждого из 61 мышиного эмбриона, позволило идентифицировать пять тесно кластеризованных групп, идеально совпадающих по стадиям развития. (В) Псевдовременная траектория профилей псевдообъема РНК-seq мышиных эмбрионов идентична траектории, показанной на фиг. 4G, но отличается тем, что псевдовремя показано цветом. (С) Профили 61 эмбриона были упорядочены по псевдовремени. Три самых ранних и три самых поздних (по псевдовремени) эмбриона Е10.5 показаны на фотографиях и отличаются по морфологии.
[0061] На фиг. 8 проиллюстрирована идентификация клеток основных типов, участвующих в органогенезе мыши. (А) визуализация t-SNE 2026641 клеток мышиных эмбрионов, окрашенных по идентификатору кластера при кластеризации Лювена и аннотированных на основе маркерных генов. То же самое t-SNE представлено ниже на графике, где проиллюстрированы только клетки на каждой стадии развития. Примитивные эритроидные (транзиентные) и окончательные эритроидные (размножающиеся) кластеры показаны в рамке для иллюстрации динамики их пролиферации. (В) Точечный график, иллюстрирующий экспрессию одного выбранного маркерного гена на тип клетки. Размер пятна соответствует проценту клеток каждого типа, а его цвет соответствует среднему уровню экспрессии.
[0062] На фиг. 9 проиллюстрирована идентификация клеток основных типов, участвующих в органогенезе мыши, и соответствующие наборы маркерных генов, специфичных для клеток конкретных типов. (А) Визуализация t-SNE при переходе клетки из Е9.5 в Е13.5, где стадии развития показаны цветом. Аналогичное t-SNE показано на фиг. 8А, но в данном случае, на каждом графике показаны только клетки, полученные в один момент времени. (В) Тепловая карта, иллюстрирующая относительную экспрессию генов по основным идентифицированным типам клеток. (С) Гистограмма, иллюстрирующая число маркерных генов в клетках каждого типа с экспрессией, более, чем в два раза превышающей экспрессию маркерных генов в клетках второго типа с наиболее высоким уровнем экспрессии (FDR 5%).
[0063] На фиг. 10 показано, что клетки, происходящие от реплицированных эмбрионов для одного и того же момента времени, не обнаруживают явных периодических эффектов. (А-Е) визуализация t-SNE клеток мышиных эмбрионов на различных стадиях развития: Е9,5 (А), Е10,5 (В), Е1,5 (С), Е12,5 (D), Е13,5 (Е), окрашенных по идентификатору эмбриона на каждой стадии.
[0064] На фиг. 11 показана динамика изменения чисел клеток конкретных типов в процессе органогенеза у мышей. (А) Гистограмма, иллюстрирующая число клеток, профилированных для клеток каждого типа с разбивкой по стадиям развития. (В) Тепловая карта, иллюстрирующая относительное число клеток каждого типа (строки) в 61 мышином эмбрионе (столбцы). Абсолютное число клеток конкретного типа на эмбрион вычисляли путем умножения доли, которую внесят клетки конкретного типа в данный эмбрион, на общее вычисленное число клеток на этой стадии развития. Для представления данных, эти оценки нормализуют в каждом ряду по максимальному оцененному числу клеток данного типа для всего 61 эмбриона. Эмбрионы отсортировывали слева направо по псевдовремени развития. (С) Линейный график, иллюстрирующий относительное изменение числа клеток для примитивных эритроидных и окончательных эритроидных линий дифференцировки, рассчитанное как на панели В. Пунктирными линиями показана относительная экспрессия маркерных генов для примитивных эритроидов (Hbb-bhl) и окончательных эритроидов (Hbb-bs). Экспериментальные данные для отдельных эмбрионов были упорядочены по псевдовремени развития и сглажены методом Лесса.
[0065] На фиг. 12 проиллюстрирована кластеризация Лювена и визуализация t-SNE подкластеров каждой из клеток 38 основных типов. Поскольку гетерогенность типов клеток была совершенно очевидна для множества из 38 кластеров, представленных на фиг. 8А, то авторами была разработана итеративная стратегия повторяющейся кластеризации Лювена для клеток каждого основного типа в целях идентификации подкластеров. После удаления подкластеров, в которых доминируют один или два эмбриона, и после слияния очень похожих подкластеров было получено всего 655 подкластеров (также называемых «подтипами», чтобы отличить их от клеток 38 основных типов, идентифицированных путем начальной кластеризации).
[0066] На фиг. 13 проиллюстрирована кластеризация Лювена и визуализация t-SNE подкластеров каждой из клеток 38 основных типов на всех стадиях развития. Эта фигура идентична фиг. 12, за исключением того, что клетки были окрашены по стадии развития, а не по ID подкластера.
[0067] На фиг. 14 проиллюстрирована чувствительность детектирования типов клеток в зависимости от клеточного охвата. (А) визуализация t-SNE всех клеток (левый график, n=2026641) и подмножества с более низкой выборкой (правый график, n=50000), окрашенные по идентификаторами кластеров Лювена, как показано на фиг. 8А. (В) визуализация t-SNE всех эндотелиальных клеток, (левый график, n=35878) и клеток подмножества с более низкой выборкой (правый график, n=1173), окрашенных по идентификатору кластеров Лювена, и вычисленных на основе 35878 эндотелиальных клеток. (С) визуализация t-SNE 1173 эндотелиальных клеток, окрашенных по идентификатору кластеров Лювена, и вычисленных на основе 1173 эндотелиальных клеток.
[0068] На фиг. 15 проиллюстрированы клетки 655 подтипов, происходящих от множества эмбрионов и определенных по сериям маркеров. (А) Гистограмма, иллюстрирующая распределение подкластеров по числу клеток (медиана 1869; диапазон 51-65894). (В) Гистограмма, иллюстрирующая распределение подкластеров по числу эмбрионов, от которых происходят эти клетки (>5 клеток, квалифицированных как благоприятствующий фактор). (С) Гистограмма, иллюстрирующая распределение подкластеров по отношению клеток, происходящих от эмбриона, рассматриваемого как наиболее благоприятствующий фактор. (D) Гистограмма, иллюстрирующая распределение подкластеров по числу маркерных генов (с уровнем экспрессии, который по меньшей мере в 1,5 раза превышает уровень экспрессии по сравнению с уровнем экспрессии в клетках других подтипов с наиболее высоким уровнем экспрессии в том же самом основном кластере; 5% FDR).
[0069] На фиг. 16 показана динамика изменения числа клеток конкретых типов в процессе органогенеза у мышей. (А) Тепловая карта, иллюстрирующая относительную экспрессию генов 655 идентифицированных подкластеров. (В) Тепловая карта, иллюстрирующая относительное число клеток каждого подтипа (ряды) в 61 мышином эмбрионе (столбцы). Абсолютное число клеток конкретного подтипа на эмбрион вычисляли как показано на фиг. 11В. (С) Визуализация t-SNE всех 61 эмбрионов мыши только на основе доли клеток 655 подтипов в каждом эмбрионе.
[0070] На фиг. 17 проиллюстрирована идентификация и характеризация подтипов эпителиальных клеток и апикального эктодермального гребня конечности (AER). (А) Визуализация t-SNE и аннотация эпителиальных клеток определенных подтипов на основе маркеров. (В) Визуализация t-SNE всех эпителиальных клеток, окрашенных по уровню экспрессии Fgf8. (С) Изображения дорожек гибридизации in situ Fgf8 в эмбрионах Е10.5 (слева) и Е11.5 (справа). (D) Визуализация t-SNE всех эпителиальных клеток, окрашенных по уровню экспрессии Fndc3a. (Е) Изображения дорожек гибридизации in situ Fndc3a в эмбрионе E10.5. Стрелка: сайт экспрессии гена. (F) Прямоугольная диаграмма, иллюстрирующая долю клеток AER на эмбрион на различных стадиях развития. (G) Псевдовременная траектория моноклеточных транскриптомов AER, окрашенных по стадиям развития. (Н) Линейный график, иллюстрирующий относительную экспрессию маркерных генов AER в течение всего псевдовремени развития.
[0071] На фиг. 18 проиллюстрирована идентификация подтипов мышиного эпителия. Точечный график, иллюстрирующий экспрессию одного выбранного маркерного гена на подтип эпителия. Размер пятна соответствует проценту клеток определенного типа, а его цвет соответствует среднему уровню экспрессии.
[0072] На фиг. 19 проиллюстрирована динамика экспрессии генов в клетках апикального эктодермального гребня конечностей (AER) в течение псевдовремени. (А) Тепловая карта, иллюстрирующая сглаженную дифференциальную экспрессию генов в зависимости от псевдовремени (FDR 1%) в клетках AER, и построенная по отрицательной биномиальной регрессии, а также масштабируемая по проценту от максимальной экспрессии гена. В каждом ряду указаны различные гены, и эти гены распределены по подмножествам, которые имеют активированную (вверху), подавляемую (в середине) или транзиентную динамику (внизу) между Е9.5 и Е13.5. (В-С) Графики, иллюстрирующие значение q, преобразованное в log10, и объединенные баллы для обогащенных членов реактом, вычисленных с помощью enrichR (В) и факторов транскрипции (С) для генов, экспрессия которых значительно снижается при развитии AER. Наилучшие члены пути обогащения (Reactome2016) со значительным уменьшением числа генов включают прохождение клеточного цикла (митотического клеточного цикла, qva1=0,0002) и метаболизма глюкозы (метаболизма углеводов, qva1=0,0002). Наиболее обогащенные TF с мишенями для снижения числа генов включают факторы плюрипотентности, такие как Is11 (qva1 <10-5), Pou5f1 (qva1=0,002) и Nanog (qva1=0,003).
[0073] На фиг. 20 проиллюстрирована характеризация клеточных траекторий во время дифференцировки мезенхимы конечностей. (А) 3D-визуализация UMAP мезенхимальных клеток конечностей, окрашенных по стадиям развития (слева и справа представлены изображения в двух направлениях). (В) График рассеяния, иллюстрирующий нормализованную экспрессию Pitxl и Tbx5 в мезенхимальных клетках конечностей. Показаны только клетки, в которых обнаружены Pitxl и/или Tbx5. (С) График Volkano, иллюстрирующий дифференциально экспрессируемые гены (FDR 5%, показаны красным) между передней и задней конечностями. Гены с наибольшим уровнем дифференциальной экспрессии были помечены. Ось X: log2-преобразованное кратное изменение между передними и задними конечностями для каждого гена. Ось Y: - log10-преобразованное qval исходя из теста на дифференцировку. (D) Та же самая визуализация, как и на панели А, но с окрашиванием по нормализованной экспрессии генов проксимальных маркеров/хондроцитов (Sox6, Sox9), а также маркеров дистальных органов (Hoxdl3, Tfap2b), передних (Рах9, Alx4) или задних (Hand2, Shh) конечностей. (F) Изображения дорожек гибридизации in situ для Hoxdl3 у эмбрионов Е10,5-Е13,5. (G) Та же самая визуализация, как и на панели А и D, но с окрашиванием по нормализованной экспрессии генов Сра2. Их паттерн экспрессии на этой траектории позволил авторам предсказать, что Сра2 является дистальным маркером развития мезенхимы конечностей, таким как Hoxdl3. (Н) Изображения дорожек гибридизации in situ для Сра2 у эмбрионов Е10.5-Е11.5. (I) Объединенные систематизированные результаты траекторий для AER и мезенхимы конечностей.
[0074] На фиг. 21 проиллюстрирована характеризация траекторий изменения клеток во время развития мезенхимы конечности. (А) Тепловая карта, иллюстрирующая гены с наиболее высокими уровнями дифференциальной экспрессии между различными стадиями развития клеток мезенхимальных клеток конечностей. (В) Гистограмма, иллюстрирующая log10-преобразованное скорректированное значение р для обогащенных факторов транскрипции генов с повышенной степенью активации во время развития мезенхимы конечности. (С) Визуализация t-SNE мезенхимальных клеток конечностей, окрашенных для передних конечностей (Tbx5+) и задних конечностей (Pitx1+). Клетки с отсутствием экспрессии или с экспрессией обоих Tbx5 и Pitxl не показаны.
[0075] На фиг. 22 показана экспрессия маркеров, пространственно ограниченных в конечностях. Каждая панель иллюстрирует другой маркерный ген. Цветом показаны количества UMI, которые были масштабированы по размеру библиотеки, логарифмически преобразованы, а затем картированы по Z-показателям для сравнения между генами. Клетки с отсутствием экспрессии данного маркера были исключены для предотвращения превышения точек на графике. (А) Маркер задней конечности Pitxl и маркер передней конечности Tbx5. (В) Первый ряд: маркеры проксимальных конечностей Sox6 (которые также помечают хондроциты) и Sox9. Второй ряд: маркеры дистальных конечностей Hoxdl3 и Tfap2b. Третий ряд: маркеры передних конечностей 68 Рах9 и Alx4. Четвертый ряд: маркеры задних конечностей Shh и Hand2.
[0076] На фиг. 23 показаны модули пространственно ограниченных генов в конечностях. Всего 1191 ген был кластеризован с помощью иерархической кластеризации. Дендрограмма была распределена на 8 модулей с использованием функции границы дерева в R, и была оценена совокупная экспрессия генов в каждом модуле. Цветом показаны совокупные значения UMI для каждого модуля, которые были масштабированы по размеру библиотеки, логарифмически преобразованы, а затем картированы по Z-показателям для сравнения между модулями. Клетки с отсутствием экспрессии данного модуля были исключены для предотвращения превышения точек на графике.
[0077] На фиг. 24 проиллюстрирована характеризация восьми основных траекторий развития, наблюдаемых во время органогенеза у мышей. (А) 3D-визуализация UMAP общего набора данных авторов; вверху: вид в двух направлениях; внизу: увеличенный вид траекторий мезенхимы (слева) и нервной трубки/хорды (справа), окрашенных по стадиям развития. (В) Тепловая карта, иллюстрирующая долю клеток от каждого из 38 основных типов клеток, присвоенных каждой из 8 основных траекторий. В столбцах представлены восемь основных линий дифференцировки, показанных цветом на верхней гистограмме (см. ключ на панели А). (С) 3D-визуализация UMAP эпителиальных субтраекторий, окрашенных по стадиям развития.
[0078] На фиг. 25 проиллюстрирована характеризация восьми основных траекторий развития, наблюдаемых во время органогенеза у мышей. (А) Эта фигура аналогична фиг. 24А, за исключением того, что цвет соответствует 38 основным кластерам клеток. (В-С) Площадь участка, показывающая оцененную долю (В) и оцененное абсолютное число клеток (С) на эмбрион, полученное для каждой из восьми основных траекторий клеток от Е9.5 до Е13.5.
[0079] На фиг. 26 проиллюстрирована визуализация UMAP для восьми основных траекторий клеток, окрашенных по основному идентификатору кластера клеток.
[0080] На фиг. 27 проиллюстрирована визуализация UMAP для восьми основных траекторий клеток, окрашенных по стадиям развития.
[0081] На фиг. 28 проиллюстрирована визуализация UMAP подтипов эпителиальных клеток. Окраска соответствует 29 эпителиальным подтипам, показанным на фиг. 17А.
[0082] На фиг. 29 показано разрешение траекторий клеток в миогенезе. Края на основных графиках, которые определяют траектории, сообщаемые в Monocle 3, показаны в виде голубых отрезков фрагментов. (А) Клетки, предположительно участвующие в миогенезе, были выделены из траектории мезенхимных клеток in silico, а затем использованы для создания субтраектории миоцитов (Методы). (В) Клетки в субтраектории миоцитов, окрашенные по стадиям развития. (С) Клетки в траектории миоцитов, окрашенные по экспрессии выбранных регуляторов транскрипции при миогенезе. Клетки без детектируемой экспрессии для данного гена были исключены из графика. (D) Клетки, классифицированные по стадиям развития в соответствии с маркерами, показаны на панели С (Методы).
[0083] На фиг. 30 проиллюстрировано совместное профилирование общего и вновь синтезированного транскриптома по судьбе sci-клеток. (А) Рабочая диаграмма sci-клеток с ключевыми стадиями, изложенными в описании. (Б) Схема эксперимента. Клетки А549 обрабатывали дексаметазоном в зависимости от времени. Клетки во всех условиях обработки метили S4U за два часа до сбора для оценки судьбы sci-клеток. (С) График Violin, где показано отношение S4U-меченых ридов на клетку за шесть обработок. (D) График Violin, где показано отношение S4U-меченых ридов в экзонных и интронных ридах. Для всех прямоугольных диаграмм: толстые горизонтальные линии; медианы; верхний и нижний края прямоугольника; первый и третий квартили, соответственно; усы, в 1,5 раза превышающие межквартильный интервал; круги, выбросы. (Е) Визуализация UMAP в клетках А549 с использованием целого транскриптома (слева), вновь синтезированного транскриптома (в середине) и их обоих (справа). (F) Визуализация, аналогичная (Е), но с окрашиванием по идентификатору кластера, обнаруженному с использованием целого транскриптома. (G) Визуализация UMAP в клетках А549 по общей информации с окрашиванием по нормализованной экспрессии маркерных генов G2/M на уровне РНК (слева) и на уровне вновь синтезированной РНК (справа). Число UMI для этих генов масштабируют по размеру библиотеки, логарифмически преобразуют, объединяют, а затем картируют по Z-показателям.
[0084] На фиг. 31 показаны результаты анализов на дополнительные признаки и анализов QC для судьбы sci-клеток. (А) График рассеяния для числа UMI у мышей (NIH/3T3) и человека (HEK293T) на клетку в условиях оценки судьбы sci-клеток. (B-D) Прямоугольная диаграмма, где показано отношение S4U-меченных ридов, число UMI и чистота (доля ридов, картируемых по ожидаемым видам) на клетку для клеток HEK293T (число клеток n=932) и клеток NIH/3T3 (число клеток n=438). Для всех прямоугольных диаграмм: толстые горизонтальные линии; медианы; верхний и нижний края прямоугольника; первый и третий квартили, соответственно; усы, в 1,5 раза превышающие межквартильный интервал; круги, выбросы. (E-F) Корреляция (корреляция Спирмена) между оценками измерений экспрессии генов в объединенных профилях клеток HEK293T (Е) и NIH/3T3 (F) по sci-судьбе (ось у) по сравнению с sci-РНК-seq-клетками (ось х).
[0085] На фиг. 32 показана оценка sci-судьбы клеток А549, обработанных дексаметазоном. (А, В) График Violin, где показано число UMI (А) и генов (В) на клетку в шести условиях обработки. Для всех прямоугольных диаграмм: толстые горизонтальные линии; медианы; верхний и нижний края прямоугольника; первый и третий квартили, соответственно; усы, в 1,5 раза превышающие межквартильный интервал; круги, выбросы. (С) График корреляции, где показан коэффициент корреляции Пирсона между различными условиями обработки для объединенного целого транскриптома (вверху справа) и вновь синтезированного транскриптома (внизу слева). (D) Визуализация UMAP в клетках А549 с использованием вновь синтезированного транскриптома с окрашиванием по идентификатору кластера, идентифицированному с использованием вновь синтезированной транскриптомы. (Е) Тепловая карта, показывающая долю клеток от каждого кластера, определенного по всему транскриптому, которые входят в каждый клеточный кластер вновь синтезированного транскриптома. (F-G) Визуализация UMAP в клетках А549 с использованием общего и вновь синтезированного транскриптома с окрашиванием по нормализованной экспрессии маркерных генов S-фазы по экспрессии общей РНК (F) и вновь синтезированной РНК (G). Число UMI для этих генов масштабируют по размеру библиотеки, логарифмически преобразуют, объединяют, а затем картируют по Z-показателям.
[0086] На фиг. 33 проиллюстрирована характеризация модулей TF, инициирующих переход клеток из одного состояния в другое. (А) Идентифицированная связь (синий) между факторами транскрипции (оранжевый) и регулируемыми генами (серый). Модули TF, ассоциированные с прохождением клеточного цикла или GR-ответом, помечены. (В) Визуализация UMAP клеток А549, упорядоченных по модулям TF клеточного цикла с окрашиванием вновь синтезированной мРНК маркеров S-фазы и G2/М-фазы (вверху), трех фаз клеточного цикла (внизу слева) и девяти стадий клеточного цикла с помощью анализа на неконтролируемую кластеризацию (внизу справа). (С) Визуализация UMAP клеток А549, упорядоченных по модулям TF GR-ответа с окрашиванием по времени обработки DEX (слева), активности СЕВРВ и FOXOl (в середине) и идентификатору кластера в анализе на неконтролируемую кластеризацию (справа). Для вычисления активности TF, число вновь синтезированных UMI для этих генов масштабируют по размеру библиотеки, логарифмически преобразуют, объединяют, а затем картируют по Z-показателям. (D) Таблица, где показано наблюдаемое отношение (черный) состояния клетки к комбинаторному состоянию модулей клеточного цикла (ось х) и модулей GR-ответа (ось у). Красное число означает ожидаемое отношение, предполагающее независимый набор. (Е) Тепловая карта, иллюстрирующая долю состояний клеток, определяемых по комбинаторным состояниям модулей TF в каждом из основных кластеров, идентифицированных в анализе на кластеризацию на основе объединенного целого и вновь синтезированного транскриптома.
[0087] На фиг. 34 проиллюстрированы модули TF, инициирующие переход клеток из одного состояния в другое для DEX-обработанных клеток А549. (А) Идентифицированные гены-мишени (серые) от СЕВРВ (оранжевый). Показаны только связи с регулируемым коэффициентом корреляции исходя из LASSO >0,6. (В) Визуализация UMAP клеток А549 по целому и вновь синтезированному транскриптому с окрашиванием по экспрессии СЕВРВ (слева) и активности (справа). (С) похож на (В), но с окрашиванием по экспрессии YOD1 (слева) и по активности YOD1 (справа). (D) аналогичен (В), но с окрашиванием по экспрессии GTF2IRD1 (слева) и по активности GTF2IRD1 (справа). (Е) аналогичен (В), но с окрашиванием по экспрессии E2F1 (слева), по активности E2F1 (в середине) и по совокупной экспрессии целого транскриптома для генов, сцепленных с E2F1 (справа). (F) Тепловая карта, где показано абсолютное значение коэффициента корреляции Пирсона между модулями TF. 29 модулей TF были распределены на пять групп с помощью анализа на иерархическую кластеризацию.
[0088] На фиг. 35 показаны клеточные состояния, характеризуемые комбинаторными состояниями функциональных модулей TF. (А) Схема, иллюстрирующая стратегию характеризации состояний клеток по комбинаторным состояниям функциональных модулей TF. (В) Визуализация Umap для всех клеток с использованием как целого, так и вновь синтезированного транскриптома с окрашиванием по основному идентификатору кластера, идентифицированного с помощью алгоритма кластеризации пиков плотности в области UMAP.
[008 9] На фиг. 36 проиллюстрирована характеризация траекторий перехода клеток из одного состояния а другое для >6000 отдельных клеток. (А) Схема, иллюстрирующая коррекцию памяти, и анализ на слияние клеток для построения траектории перехода клеток из одного состояния в другое, подробно рассматриваемые в описании и Методе. (В) 3D-график для клеток, окрашенных по времени обработки DEX (также в виде z-координат). Координаты х и у соответствуют области UMAP по целому и вновь синтезированному транскриптому на фиг. 30Е (слева). Слитые родительские и дочерние клетки показаны серыми линиями. (С) Аналогично (В), за исключением того, что координаты х и у соответствуют области UMAP по динамике моноклеточного транскриптома в одной клетке в шести временных точках. (D) Линейные графики, иллюстрирующие динамику состояния клеток с точки зрения различных GR-ответов (вверху) и фазы клеточного цикла (внизу) в кластерах для каждой клеточной траектории (слева) или во всех клетках (справа) независимо от анализа на слияние клеток. (Е) Сеть перехода клеток из одного состояния в другое. Узлами являются 27 состояний клеток, охарактеризованных на фиг. 33D, а звенья представляют собой идентифицированные пути перехода клеток из одного состояния в другое. Звенья с низкой вероятностью перехода (<0,1) отфильтровывают. Квадратами с пунктирными линиями показаны примеры состояний с обратимой динамикой перехода. (F) Корреляционный график, иллюстрирующий корреляцию доли состояния клеток между условиями обработки. Положительные корреляции показаны синим цветом, а отрицательные - красным. Форма эллипса коррелирует с коэффициентами корреляции (на эллипсе). (G) График рассеяния, иллюстрирующий корреляцию доли состояния клеток между наблюдаемыми 10-часовыми группами DEX-обработки и прогнозируемыми долями состояний клеток. Прогноз основан на вероятностях перехода клеток из одного состояния в другое и на оценке доли состояния клеток в группе без обработки DEX. Синяя линия соответствует линии линейной регрессии. (Н) График рассеяния, иллюстрирующий корреляцию вероятности перехода клеток из одного состояния в другое, рассчитанной по полным данным (0-10 часов) или частичным данным (0-6 часов) вместе с линией линейной регрессии.
[0090] На фиг. 37 показана оценка уровня детектирования новой РНК и уровня разложения РНК. (А) График рассеяния, иллюстрирующий корреляцию между осью х: различия нормализованного целого транскриптома между клетками без DEX и клетками через 2 часа после обработки DEX, и осью у: различия нормализованного вновь синтезированного транскриптома между клетками без DEX и клетками через 2 часа после обработки DEX. Синей линией обозначена линия линейной регрессии. Полный транскриптом и вновь синтезированный транскриптом в каждый момент времени нормализуют по размеру библиотеки всего транскриптома в данный момент времени. (В) График корреляции, иллюстрирующий корреляцию оценки степени деградации генов между условиями обработки. Положительные корреляции представлены синим цветом, а отрицательные - красным. Форма эллипса коррелирует с коэффициентами корреляции (на эллипсе).
[0091] На фиг. 38 показана сеть перехода клеток из одного состояния в другое для предсказания состояния клеток. (А) График корреляции, иллюстрирующий корреляцию между наблюдаемыми состояниями клеток в каждое время обработки и прогнозируемым состоянием клеток по вероятности перехода клеток из одного состояния в другое и по доле состояний клеток у группы без обработки DEX. Синей линией показана линия линейной регрессии. (В) График рассеяния, иллюстрирующий корреляцию доли состояний клеток между наблюдаемыми 10-часовыми группами обработки DEX и предсказанными значениями. Предсказанные значения основаны на вероятностях перехода клеток из одного состояния в другое, оцененных по частичным данным (0-6 часов), и по доле состояния клеток у группе без обработки DEX. Синей линией показана линия линейной регрессии.
[0092] На фиг. 39 показано, что вероятности перехода клеток из одного состояния в другое регулируются паттерном стабильности перехода в соседнее состояние. (А) График корреляции, иллюстрирующий корреляцию между расстоянием перехода (расстоянием Пирсона) и вероятностью перехода клеток из одного состояния в другое, вместе с красной линией сглаживания Лесса, помеченной ggplot2. (В) 3D-график, иллюстрирующий паттерн нестабильности состояния клеток. Ось X представляет состояния GR-ответов (от состояния «нет» до состояния «низкий» - «высокий»). Ось Y представляет фазы клеточных циклов от G0/G1 до G2/M. Ось Z представляет паттерн нестабильности состояния клеток, определяемый по вероятности скачка каждого состояния клетки в другие состояния через 2 часа. (С) График рассеяния, иллюстрирующий взаимосвязь между нестабильностью состояния клеток и изменением доли клеток до и после 10-часовой обработки DEX, вместе с красной линией сглаживания сглаживания Лесса, помеченной ggplot2. (D) График рассеяния, иллюстрирующий корреляцию между нестабильностью состояния и энтропией перехода из одного состояния в другое вместе с линией линейной регрессии (синего цвета). (Е) Прямоугольная диаграмма, иллюстрирующая анализ на перекрестное подтверждение методом R-квадрат для предсказания вероятности перехода между состояниями только по расстоянию перехода или по комбинации расстояния перехода и паттерна нестабильности состояний с использованием плотно соединенной нейронной сети.
[0093] На фиг. 40 представлен сканирующий снимок экзона-мишени в гене LMO2. Экзон-мишень указан на дорожке «Экзоны-мишени». Следует обратить внимание, что 12 ОТ-праймеров, прошедших через фильтры, охватывают экзон, по возможности, на достаточном уровне. Две нижние дорожки, «Primers_plus/minus» и «Captured_plus/minus», иллюстрируют риды, картированные по ОТ-праймерам и захваченным транскриптам, соответственно. Разница в количестве считываний указывает на число инициации нежелательных событий.
[0094] На фиг. 41 представлена таблица для сравнительного ранжирования генов в базе данных по секвенированию общей ядерной РНК ENCODE и генов в библиотеке для захвата мультиплексной ОТ in situ. Гены-мишени показаны жирным шрифтом. Из 12 наиболее распространенных генов в библиотеке-мишени было выбрано 8 генов-мишеней. В последних двух столбцах - «RANK» означает ранжирование в базе данных авторов изобретения, a «ENCODE», означает ранжирование в базе данных ENCODE. Избыточные РНК, не являющиеся мишенями, включают митохондриальные рибосомные РНК RNR2 и RNR1, а часто встречающиеся ядерные РНК включают IncPHК MALAT1. Следует отметить, что ген LMO2 простирается от 4627-го наиболее детектируемого гена в базе данных ENCODE (из 26281 генов) до 3-го наиболее детектируемого гена в библиотеке авторов.
[0095] На фиг. 42 показано обогащение захваченного транскрипта по сравнению с общей РНК. График рассеяния для ридов в библиотеке мишеней и для ридов в ядерной РНК ENCODE. Гены, на которые нацелены ОТ, обозначены красным; при этом, следует отметить, что обычно они находятся за пределами диагонали, что указывает на то, что они были обогащены по сравнению с ожидаемым уровнем экспрессии в эталонной базе данных. Избыточные ядерные IncPHК MALAT1 и XIST показаны синим цветом: и они находятся выше диагонали, что указывает на успешное обогащение по сравнению с этими РНК. еРНК-мишень не обогащена (оранжевый). Среднее обогащение было в 45,3 раза выше ожидаемого уровня для 9 генов-мишеней в этом эксперименте. В целом, риды генов-мишеней составляют 31% от общего числа ридов, картированных по генам в этой базе данных.
[0096] Схематически представленные чертежи необязательно соответствуют масштабу. Одинаковые номера, используемые на фигурах, относятся к одинаковым компонентам, стадиям и т.п. Однако, следует отметить, что использование числа для ссылки на компонент на данном чертеже не рассматривается как ограничение компонента на другом чертеже, обозначенного тем же номером. Кроме того, использование различных номеров для обозначения компонентов не означает, что различные пронумерованные компоненты не могут быть идентичны другим пронумерованным компонентам или похожи на них.
Подробное описание иллюстративных вариантов осуществления изобретения
[0097] В одном варианте осуществления изобретения, описанный здесь способ может быть применен для создания библиотек секвенирования (sci) и комбинаторного индексирования отдельных клеток, которые включают транскриптомы множества отдельных клеток. Так, например, этот способ может быть применен для получения информации о последовательностях транскриптомов целых клеток, транскритомов вновь синтезированной РНК или их комбинации. В другом варианте осуществления изобретения, описанный здесь способ может быть применен для получения sci-секвенирующих библиотек, которые включают информацию о последовательности субпопуляции нуклеиновых кислот РНК. Так, например, если некодирующая регуляторная область является мишенью для перестановки, то кодирующая область, находящаяся в цис-положении по отношению к регуляторной области, может быть протестирована на измененную экспрессию. В другом примере, эксперименты по клеточной карте могут проводиться с использованием считанных данных, ограниченных числом мРНК, которые являются высокоинформативными.
[0098] Способ может включать одну или более стадий получения выделенных ядер или клеток, распределения субпопуляций выделенных ядер или клеток на компартменты, обработки выделенных ядер или клеток так, чтобы они включали фрагменты нуклеиновой кислоты, и добавление компартмент-специфического индекса к фрагментам нуклеиновой кислоты. Этот способ может включать, но необязательно, обработку клеток в заранее определенных условиях и/или мечение вновь синтезированной РНК в клетках. Этот способ может быть направлен на получение информации, которая включает информацию о клеточном транскриптоме или субпопуляции нуклеиновых кислот РНК. Эти стадии могут выполняться практически в любом порядке и могут быть объединены различными способами. Необязательно, ядра могут быть выделены из клеток после обработки клеток в предварительно определенных условиях и мечения вновь синтезированной РНК.
Получение выделенных ядер или клеток
[0099] Описанный здесь способ может включать получение клеток или ядер, выделенных из множества клеток (фиг. 1, блок 10; фиг. 2, блок 22). Клетки могут происходить от любого(ых) организма(ов) и от клеток любого типа или любой ткани организма(ов). В одном варианте осуществления изобретения, клетки могут представлять собой эмбриональные клетки, например клетки, полученные из эмбриона. В одном варианте осуществления изобретения, клетки или ядра могут быть выделены из раковой или пораженной ткани. Способ может также включать диссоциацию клеток и/или выделение ядер. Количество ядер или клеток может быть равно по меньшей мере двум. Верхний предел зависит от практических ограничений оборудования (например, многолуночных планшетов, числа индексов), используемого в других стадиях описанного здесь способа. Количество ядер или клеток, которые могут быть использованы, не имеет конкретных ограничений и может исчисляться миллиардами. Так, например, в одном варианте осуществления изобретения, число ядер или клеток может составлять не более 100000000, не более 10000000, не более 1000000000, не более 100000000, не более 10000000, не более 1000000, не более 100000, нет более 10000, не более 1000, не более 500 или не более 50. Для специалиста очевидно, что в некоторых вариантах осуществления изобретения, молекулы нуклеиновой кислоты в каждом ядре представляют весь транскриптом этого ядра, например весь транскриптом, недавно синтезированный транскриптом или тот и другой.
[00100] В тех вариантах осуществления изобретения, где используются выделенные ядра, ядра могут быть получены путем экстракции и фиксации. Необязательно и предпочтительно, способ получения выделенных ядер не включает ферментативную обработку. В тех вариантах осуществления, где получают вновь синтезированный транскриптом, ядра не выделяют до тех пор, пока клетка не будет обработана в условиях, подходящих для мечения вновь синтезированных транскриптов.
[00101] В одном варианте осуществления изобретения, ядра выделяют из отдельных клеток, которые являются адгезивными или присутствуют в суспензии. Способы выделения ядер из отдельных клеток известны специалисту в данной области. Ядра обычно выделяют из клеток, присутствующих в ткани. Способ получения выделенных ядер обычно включает получение ткани, выделение ядер из полученной ткани, а затем фиксацию ядер. В одном варианте осуществления изобретения, все стадии осуществляют на льду.
[00102] Получение ткани включает мгновенное замораживание ткани в жидком азоте, а затем уменьшение размера ткани путем разрезания на кусочки диаметром 1 мм или менее. Ткань может быть уменьшена в размерах путем ее измельчения или физического воздействия. Измельчение может быть осуществлено с помощью лезвия бритвы для разрезания ткани на мелкие кусочки. Применение физического воздействия может быть осуществлено путем разбивания ткани молотком или подобным предметом, а полученная композиция из измельченной ткани называется порошком.
[00103] Выделение ядер может быть осуществлено путем инкубирования кусочков ткани или порошка в буфере для лизиса клеток в течение по меньшей мере 1-20 минут, например, 5, 10 или 15 минут. Подходящими буферами являются буферы, которые стимулируют лизис клеток, но сохраняют целостность ядер. Пример буфера для лизиса клеток включает 10 мМ Трис-HCl, рН 7,4, 10 мМ NaCl, 3 мМ MgCl2, 0,1% IGEPAL СА-630, 1% SUPERазы в ингибиторе РНКазы (20 ед/мкл, Ambion) и 1% BSA (20 мг/мл, NEB). В стандартных методах выделения ядер часто используют одно или более экзогенных соединений, таких как экзогенные ферменты, для облегчения выделения. Примерами полезных ферментов, которые могут присутствовать в буфере для лизиса клеток, являются, но не ограничиваются ими, ингибиторы протеазы, ДНКаза, лизоцим, протеиназа К, поверхностно-активные вещества, лизостафин, зимолаза, целлюлаза, протеаза или гликаназа и т.п. (Islam et al., Micromachines (Basel), 2017, 8(3):83; www.sigmaaldrich.com/life-science/biochemicals/biochemical-products.html?TablePage=14573107). В одном варианте осуществления изобретения, один или более экзогенных ферментов не присутствуют в буфере для лизиса клеток, используемом в описанном здесь способе. Так, например, экзогенный фермент, (i) не добавляется в клетки до смешивания клеток и буфера для лизиса, (ii) не присутствует в буфере для лизиса клеток до смешивания с клетками, (iii) не добавляется в смесь клеток и буфера для лизиса клеток, или применяются их комбинации. Для специалиста в данной области очевидно, что эти уровни компонентов могут быть несколько изменены без снижения ценности буфера для лизиса клеток для выделения ядер. Затем экстрагированные ядра очищают путем проведения одного или более раундов промывки буфером для ядер. Пример буфера для ядер включает 10 мМ Трис-HCl, рН 7,4, 10 мМ NaCl, 3 мМ MgCl2, 1% SUPERазы в ингибиторе РНКазы (20 ед/мкл, Ambion) и 1% BSA (20 мг/мл, NEB). Подобно буферу для лизиса клеток, экзогенные ферменты могут также отсутствовать в буфере для ядер, используемом в способе согласно изобретению. Специалисту очевидно, что эти уровни компонентов могут быть несколько изменены без снижения ценности буфера для выделения ядер. Специалисту также очевидно, что BSA и/или поверхностно-активные вещества могут быть использованы в буферах, применяемых для выделения ядер.
[00104] Выделенные ядра фиксируют путем воздействия перекрестно-сшивающего агента. Подходящий пример перекрестно-сшивающего агента включает, но не ограничивается им, параформальдегид. Параформальдегид может присутствовать в концентрации от 1% до 8%, например, 4%. Обработка ядер параформальдегидом может включать добавление параформальдегида к суспензии ядер и инкубирование при 0°С. Необязательно, но предпочтительно, после фиксации проводят промывку в буфере для ядер.
[00105] Выделенные фиксированные ядра могут быть использованы непосредственно или разделены на аликвоты, а затем сразу заморожены в жидком азоте для последующего использования. При подготовке к использованию после замораживания, оттаянные ядра могут быть сделаны проницаемыми, например, с использованием 0,2% тритона-100 в течение 3 минут на льду, а затем быстро обработаны ультразвуком для уменьшения скопления ядер.
[00106] Стандартные методы экстракции ядер из тканей обычно включает инкубирование ткани с тканеспецифическим ферментом (например, трипсином) при высокой температуре (например, при 37°С) в течение периода времени от 30 минут до нескольких часов, а затем клетки подвергают лизису буфером для лизиса клеток в целях экстракции ядер. Описанный здесь метод выделения ядер имеет несколько преимуществ: (1) искусственные ферменты не вводят, и все стадии осуществляют на льду. Это приводит к возможному снижению пертурбации состояний клеток (например, состояния транскриптома). (2) Новый метод был апробирован для большинства типов тканей, включая такни головного мозга, легких, почек, селезенки, сердца, мозжечка и образцы тканей с патологиями, такие как опухолевые ткани. По сравнению с традиционными методами экстракции ядер из тканей, в которых используются различные ферменты для различных типов тканей, новый метод может потенциально уменьшать отклонения при сравнении состояний клеток из различных тканей. (3) Новый метод также снижает стоимость и повышает эффективность за счет исключения стадии обработки ферментом. (4) По сравнению с другими методами экстракции ядер (например, методом измельчения тканей Даунса), новый метод является более надежным для различных типов тканей (например, метод Даунса требует оптимизации циклов Даунса для различных тканей) и позволяет обрабатывать большие фрагменты образцов с высокой производительностью (например, метод Даунса ограничен размером измельчителя).
[00107] Выделенные ядра могут, но необязательно, не содержать нуклеосому или могут быть помещены в условия, способствующие истощению ядер, состоящих из нуклеосом, с получением ядер, обедненных нуклеосомами.
Распределение субпопуляций
[00108] Описанный здесь способ включает распределение субпопуляций выделенных ядер или клеток по множеству компартментов (фиг. 1, блок 11; фиг. 2, блок 23; фиг. 3, блок 32). Способ может включать множество стадий распределения, где популяция выделенных ядер или клеток (также называемая здесь пулом) разделяется на субпопуляции. Обычно, субпопуляции выделенных ядер или клеток, например субпопуляции, присутствующие во множестве компартментов, индексируют с помощью компартмент-специфических индексов, а затем объединяют в пул. В соответствии с этим, способ обычно включает по меньшей мере одну стадию «разделения и объединения», состоящую из сбора выделенных ядер или клеток, их распределения и добавления компартмент-специфического индекса, где число стадий «разделения и объединения» может зависеть от числа различных индексов, которые добавляют к фрагментам нуклеиновых кислот. Каждая начальная субпопуляция ядер или клеток перед индексацией может быть уникальной среди других субпопуляций. Так, например, каждая первая субпопуляция может быть взята из уникального образца или обработана в уникальных условиях. После индексирования, субпопуляции могут быть объединены, разделены на субпопуляции, проиндексированы и снова объединены, по мере необходимости, до тех пор, пока к фрагментам нуклеиновой кислоты не будет добавлено достаточное количество индексов. Этот процесс позволяет присваивать уникальный индекс или комбинацию индексов каждой отдельной клетке или ядру. После завершения индексирования, например, после добавления одного, двух, трех или более индексов, выделенные ядра или клетки могут быть подвергнуты лизису. В некоторых вариантах осуществления изобретения, добавление индекса и лизис могут происходить одновременно.
[00109] Количество ядер или клеток, присутствующих в субпопуляции и, следовательно, в каждом компартменте, может составлять по меньшей мере 1. В одном варианте осуществления изобретения, количество ядер или клеток, присутствующих в субпопуляции, составляет не более, чем 100000000, не более, чем 10000000, не более, чем 1000000, не более, чем 100000, не более, чем 10000, не более, чем 4000, не более, чем 3000, не более, чем 2000 или не более, чем 1000, не более, чем 500 или не более, чем 50. В одном варианте осуществления изобретения, число ядер или клеток, присутствующих в субпопуляции, может составлять от 1 до 1000, от 1000 до 10000, от 10000 до 100000, от 100000 до 1000000, от 1000000 до 10000000 или от 10000000 до 100000000. В одном варианте осуществления изобретения, количества ядер или клеток, присутствующих в каждой субпопуляции, являются приблизительно одинаковыми. Количество ядер, присутствующих в субпопуляции и, следовательно, в каждом компартменте, было частично основано на желании авторов уменьшить наложение индексов, которое представляют собой присутствие двух ядер или клеток, имеющих одинаковую комбинацию индексов, заканчивающихся в одном и том же компартменте на этой стадии метода. Способы распределения ядер или клеток по субпопуляциям известны специалисту в данной области и являются рутинными. Хотя может быть применена цитометрия с использованием клеточного сортинга с активацией флуоресценции (FACS), однако, в некоторых вариантах осуществления изобретения, предпочтительным является использование простого разведения. В одном варианте осуществления изобретения, FACS-цитометрия не используется. Ядра, имеющие различные плоидности, могут быть стробированы и обогащены окрашиванием, например окрашиванием DAPI (4',6-диамидино-2-фенилиндолом). Окрашивание может быть также использовано для отделения отдельных клеток от дублетов во время сортинга.
[00110] Количество компартментов на стадиях распределения (и последующее добавление индекса) может зависеть от используемого формата. Так, например, количество компартментов может составлять от 2 до 96 компартментов (при использовании 96-луночного планшета), от 2 до 384 компартментов (при использовании 384-луночного планшета) или от 2 до 1536 компартментов (при использовании 1536-луночного планшета). В одном варианте осуществления изобретения, может быть использовано множество планшетов. В одном варианте осуществления изобретения, каждый компартмент может представлять собой каплю. Если типом используемого компартмента является капля, которая содержит два или более ядер или клеток, то может быть использовано любое количество капель, например, по меньшей мере 10000, по меньшей мере 100000, по меньшей мере 1000000 или по меньшей мере 10000000 капель. Субпопуляции выделенных ядер или клеток обычно индексируют в компартментах перед объединением.
[00111] В некоторых вариантах осуществления изобретения, компартмент представляет собой каплю или лунку. Транскриптом, вновь синтезированный транскриптом или его субпопуляции в клетке или в ядре могут быть помечены уникальным индексом или комбинацией индексов в капле или в лунке. Индексированные библиотеки, выделенные из отдельных частей капель или лунок, могут быть объединены для дальнейшей обработки и секвенирования. Примерами таких методов являются, но не ограничиваются ими, системы анализа отдельных клеток от 10Х Genomics (Pleasanton, СА), Biorad (Hercules, СА) и CellSee (Ann Arbor, MI).
Обработка в предварительно определенных условиях
[00112] В необязательном варианте осуществления изобретения, каждую субпопуляцию клеток подвергают воздействию агента или пертурбации (Фиг. 1, блок 12). Таким агентом может быть, по существу, любой агент, вызывающий изменение в клетке. Так, например, агент может изменить транскриптом клетки, изменять структуру хроматина клетки, изменить активность белка в клетке, изменять ДНК клетки, изменять состояние метилирования, изменять редактирование ДНК клетки или вызывать другие изменения. Примерами агентов являются, но не ограничиваются ими, соединение, такое как белок (включая антитело), не-рибосомный белок, поликетид, органическая молекула (включая органическую молекулу размером в 900 Дальтон или менее), неорганическая молекула, молекула РНК или РНКи, углевод, гликопротеин, нуклеиновая кислота или их комбинация. В одном варианте осуществления изобретения, агент вызывает генетическую пертурбацию, например, белок, редактирующий ДНК, такой как CRISPR или Talen. В одном варианте осуществления изобретения, агент представляет собой терапевтическое лекарственное средство. В одном варианте осуществления изобретения, клетка может представлять собой клетку дикого типа, а в другом варианте осуществления изобретения, клетка может быть генетически модифицирована так, чтобы она включала генетическую пертурбацию, например, нокин гена или нокаут гена (Szlachta et al., Nat Commun., 2018, 9:4275). Субпопуляции клеток могут подвергаться воздействию одного и того же агента, но различные параметры могут изменяться в различных компартментах, что позволяет тестировать множество парметров в одном эксперименте. Так, например, различные дозы, различные продолжительности обработки и различные типы клеток могут быть протестированы в одном многолуночном планшете. В одном варианте осуществления изобретения, клетки могут экспрессировать белок, обладающий известной активностью, и влияние агента на активность оценивают в различных условиях. Использование индексных последовательностей для мечения фрагментов нуклеиновых кислот позволяет затем идентифицировать нуклеиновые кислоты, происходящие от конкретных субпопуляций ядер или клеток, например, из одной лунки многолуночного планшета.
[00113] Мечение нуклеиновых кислот
[00114] В необязательном варианте осуществления изобретения, нуклеиновые кислоты, такие как РНК, кДНК или ДНК, продуцируемые клеткой, были помечены (фиг. 1, блок 13). Современные методы геномных технологий для отдельных клеток позволяют получить мгновенное изображение состояния клетки, но не дают информацию о динамике клеточного перехода. Авторами настоящего изобретения было обнаружено, что мечение вновь синтезированной РНК позволяет захватывать как весь транскриптом, так и вновь синтезированный транскриптом на уровне отдельных клеток методом индексирования с разделением и объединением, методом комбинаторного индексирования или любым методом индексирования отдельных клеток. Весь транскриптом и вновь синтезированная РНК получают один и тот же уникальный индекс или комбинацию индексов, позволяющую определить имеющиеся (например, ранее существовавшее), и вновь синтезированные нуклеиновые кислоты в одной и той же клетке. Это позволяет охарактеризовать динамику перехода клеток из одного состояния в другое, регулируемую внутренними факторами (например, программой внутреннего клеточного цикла) и внешними факторами (например, реакцией клетки на внешний стимул, такой как терапевтическое лекарственное средство). Кроме того, в некоторых вариантах осуществления изобретения обеспечивается захват как целого транскриптома, так и вновь синтезированного транскриптома на уровне одной клетки, вместе с информацией о разложении транскриптома по сравнению с его прежним состоянием (память прежнего состояния). Память о прежних состояниях каждой клетки может быть скорректирована по степени деградации мРНК (коррекция памяти), так, чтобы каждая клетка могла быть охарактеризована по динамике транскриптома между двумя или более моментами времени.
[00115] Существуют различные методы мечения вновь синтезированной нуклеиновой кислоты для того, чтобы ее можно было отличить от уже существующей нуклеиновой кислоты, и, в основном, может быть применен любой метод. Обычно, метку включают в нуклеиновые кислоты по мере их синтеза. Один из типов методов включает введение нуклеозидного аналога, который добавляет идентифицируемую мутацию. Так, например, добавление нуклеозидного аналога 4-тиоуридина (S4U) в молекулу РНК приводит к точечной мутации во время стадии обратной транскрипции, и тем самым, к образованию мутированной кДНК первой цепи, имеющей замену тимина на цитозин (Sun and Chen, 2018, Metabolic Labeling of Newly Synthesized RNA with 4sU to in Parallel Assess RNA Transcription and Decay. In: Lamande S. (eds) mRNA Decay. Methods in Molecular Biology, vol. 1720. Humana Press, New York, NY). Эта точковая мутация может быть идентифицирована на стадиях секвенирования и анализа путем сравнения последовательности с эталоном. Другой тип метода включает введение меченого гаптеном нуклеотида, который можно использовать для очистки РНК, содержащих гаптен. Примерами являются биотинилированные нуклеотиды (Luo et al., 2011, Nucl. Acids Res., 39 (19): 8559-8571) и нуклеотиды, модифицированные дигоксигенином (поставляемые от Jena Bioscience GmbH). Третий тип метода включает введение нуклеотида, который может быть модифицирован посредством химической реакции, например, нуклеотида, функционализированного путем нажатия кнопки на установке, и добавление гаптена (Bharmal et al., 2010, J. Biomol Tech., 21 (3 Suppl): S43, поставляемые от Jena Bioscience GmbH и Thermo Fisher Scientific). Другой тип метода включает введение мутагенного нуклеотида, такого как, но не ограничивающегося ими, 8-оксо-dGTP и dPTP (поставляемых от Jena Bioscience GmbH).
[00116] Предварительно определенные условия обычно используются для клетки, но не для выделенных ядер; однако, мечение нуклеиновых кислот по мере их синтеза может быть осуществлено с использованием клеток или ядер, выделенных из клеток.
[00117] В некоторых вариантах осуществления изобретения, мечение может быть применено к вновь синтезированной кДНК или ДНК. Мечение может быть использовано для идентификации конкретного состояния или субпопуляции клеток или ядер. Так, например, различные количества меток, например нуклеозидного аналога, нуклеотида, меченного гаптеном, нуклеотида, функционализированного нажатием кнопки и/или мутагенного нуклеотида и/или различные соотношения меток могут быть использованы для специфического мечения РНК, кДНК или ДНК компартмента. В другом варианте осуществления изобретения, метка может быть добавлена в различные моменты времени для фиксации момента времени. Различные метки или различные соотношения меток могут быть добавлены для дифференциального мечения РНК в различные периоды времени. В некоторых вариантах осуществления изобретения, мечение может быть частью схемы индексации для выделения отдельных клеток. Так, например, в стадии удлинения может быть использован уникальный набор нуклеотидов для каждого компартмента. Мечение может происходить на стадии обратной транскрипции, стадии удлинения, стадии гибридизации или стадии амплификации, такой как ПЦР. В некоторых вариантах осуществления изобретения, это позволяет обнаруживать дублеты или множество клеток или столкновения клеток.
Обработка с получением фрагментов нуклеиновых кислот
[00118] В одном варианте осуществления изобретения, обработка выделенных ядер или клеток может быть применена для фрагментации нуклеиновых кислот ДНК в выделенных ядрах или клетках с получением фрагментов нуклеиновых кислот (фиг. 1, блок 14). Фрагментация нуклеиновых кислот может оказаться полезной для получения молекул, длина которых является подходящей для секвенирования описанными здесь способами. Обработка может быть необходима в случае, когда секвенируемые нуклеиновые кислоты-мишени происходят от ДНК, присутствующей в ядрах или клетках; однако, в некоторых вариантах осуществления изобретения, обработка является необязательной, если секвенируемые нуклеиновые кислоты-мишени происходят от РНК (например, от мРНК и/или некодирующей РНК), присутствующей в ядрах или клетках, поскольку в некоторых вариантах осуществления изобретения, молекулы РНК необязательно должны быть фрагментированы. В других вариантах осуществления изобретения, нуклеиновые кислоты, происходящие от молекул РНК, являются фрагментированными. Фрагментация может происходить на любой стадии этого способа. Так, например, иллюстративный способ, показанный на фиг. 2, включает фрагментацию после добавления двух индексов к молекулам нуклеиновой кислоты.
[00119] При обработке нуклеиновых кислот в ядрах или клетках обычно присоединяют нуклеотидную последовательность к одному или обоим концам фрагментов нуклеиновой кислоты, полученных в результате обработки, и нуклеотидная последовательность может включать и обычно включает одну или более универсальных последовательностей. Универсальную последовательность можно использовать, например, в качестве «посадочной полосы» на последующем этапе для отжига нуклеотидной последовательности, которую можно использовать в качестве праймера для добавления другой нуклеотидной последовательности, такой как индекс, к фрагменту нуклеиновой кислоты. Нуклеотидная последовательность такого праймера может, но необязательно, включать индексную последовательность. При обработке нуклеиновых кислот в ядрах или клетках обычно добавляют один или более уникальных молекулярных идентификаторов к одному или обоим концам фрагментов нуклеиновых кислот, полученных в результате обработки.
[00120] Известны различные способы обработки нуклеиновых кислот в ядрах или клетках с получением фрагментов нуклеиновых кислот. Примерами являются ферменты CRISPR и Talen-подобные ферменты, а также ферменты, раскручивающие ДНК (например, геликазы), которые могут образовывать одноцепочечные области, с которыми могут гибридизоваться фрагменты ДНК и инициировать удлинение или амплификацию. Так, например, может быть применена амплификация на основе геликазы (Vincent et al., 2004, EMBO Rep., 5 (8):795-800). В одном варианте осуществления изобретения, удлинение или амплификацию инициируют рандомизированным праймером. В одном варианте осуществления изобретения используется транспосомный комплекс.
[00121] Транспосомный комплекс представляет собой транспозазу, связанную с сайтом распознавания транспозазы, и может встраивать сайт распознавания транспозазы в нуклеиновую кислоту-мишень внутри ядра в процессе, иногда называемом «мечением». При некоторых таких событиях встраивания, одна цепь сайта распознавания транспозазы может быть перенесена в нуклеиновую кислоту-мишень. Такая цепь называется «перенесенной цепью». В одном варианте осуществления изобретения, транспосомный комплекс включает димерную транспозазу, имеющую две субъединицы и две несмежные последовательности транспозона. В другом варианте осуществления изобретения, транспозаза включает димерную транспозазу, имеющую две субъединицы, и непрерывную последовательность транспозона. В одном варианте осуществления изобретения, 5'-конец одной или обеих цепей сайта распознавания транспозазы может быть фосфорилирован.
[00122] Некоторые варианты осуществления изобретения могут включать использование гиперактивной Tn5-транспозазы и сайта распознавания транспозазы типа Tn5 (Goryshin and Reznikoff, J. Biol. Chem., 273: 7367 (1998)) или MuA-транспозазы и сайта распознавания Mu-транспозазы, содержащих концевые последовательности R1 и R2 (Mizuuchi, K., Cell, 35: 785, 1983; Savilahti, H. et al., EMBO J., 14: 4893, 1995). Концевые последовательности мозаичного Tn5 (ME) могут быть также использованы как последовательности, оптимизированные специалистом в данной области.
[00123] Дополнительные примеры систем транспозиции, которые могут быть использованы с определенными вариантами композиций и способов согласно изобретению включают Tn552 Staphylococcus aureus (Colegio et al., J. Bacteriol, 183: 2384-8, 2001; Kirby С et al, Mol. Microbiol., 43: 173-86, 2002), Tyl (Devine & Boeke, Nucleic Acids Res., 22: 3765-72, 1994 и публикация Международной заявки WO 95/23875), транспозон Tn7 (Craig, NL, Science. 271: 1512, 1996; Craig, NL, Review in: Curr Top Microbiol Immunol., 204: 27-48, 1996), Tn/O и IS10 (Kleckner N, et al., Curr Top Microbiol Immunol., 204: 49-82, 1996), транспозазу Mariner (Lampe DJ, et al., EMBO J., 15: 5470-9, 1996); Tel (Plasterk RH, Curr. Topics Microbiol. Immunol., 204: 125-43, 1996); элемент P (Gloor, GB, Methods Mol. Biol., 260: 97-114, 2004); Tn3 (Ichikawa & Ohtsubo, J. Biol. Chem. 265: 18829-32, 1990); бактериальные инсерционные последовательности (Ohtsubo & Sekine, Curr. Top. Microbiol. Immunol. 204: 1-26, 1996); ретровирусы (Brown et al., Proc Natl Acad Sci USA, 86:2525-9, 1989) и ретротранспозон дрожжей (Boeke & Corces, Annu Rev Microbiol. 43: 403-34, 1989). Дополнительные примеры включают IS5, Tn10, Tn903, IS911 и сконструированные варианты ферментов семейства транспозаз (Zhang et al., (2009) PIoS Genet. 5: e1000689. Epub 2009, Oct. 16; Wilson C. et al (2007) J. Microbiol. Methods 71: 332-5).
[00124] Другими примерами интеграз, которые могут быть использованы в описанных здесь способах и композициях, являются ретровирусные интегразы и последовательности распознавания интеграз для таких ретровирусных интеграз, такие как интегразы, происходящие от ВИЧ-1, ВИЧ-2, SIV, PFV-1, RSV.
[00125] Последовательности транспозонов, используемые в описанных здесь способах и композициях, представлены в публикации заявки на патент США №2012/0208705, в публикации заявки на патент США №2012/0208724 и в публикации Международной патентной заявки № WO 2012/061832. В некоторых вариантах осуществления изобретения, последовательность транспозона включает первый сайт распознавания транспозазы и второй сайт распознавания транспозазы. В тех вариантах осуществления изобретения, где комплекс транспосомный комплекс используется для введения индексной последовательности, эта индексная последовательность может присутствовать между сайтами распознавания транспозазы или в транспозоне.
[00126] Некоторые используемые здесь транспосомные комплексы включают транспозазу, имеющую две последовательности транспозона. В некоторых таких вариантах осуществления изобретения, две последовательности транспозона не связаны друг с другом, то есть, другими словами, последовательности транспозонов не являются смежными. Примеры таких транспосом известны специалистам в данной области (см., например, публикацию заявки на патент США №2010/0120098).
[00127] Обычно, мечение применяют для получения фрагментов нуклеиновой кислоты, которые включают различные нуклеотидные последовательности на каждом конце (например, последовательность праймера N5 на одном конце и праймера N7 на другом конце). Это может быть достигнуто путем использования двух типов транспосомных комплексов, где каждый транспосомный комплекс включает различные нуклеотидные последовательности, которые являются частью перенесенной цепи. В некоторых вариантах осуществления изобретения, применяемое здесь мечение позволяет встраивать одну нуклеотидную последовательность во фрагменты нуклеиновой кислоты. Встраивание нуклеотидной последовательности приводит к получению фрагментов нуклеиновой кислоты, имеющих шпилечный лигирующий дуплекс на одном конце и нуклеотидную последовательность, в которую встроен транспосомный комплекс, на другом конце. Нуклеотидная последовательность, включающая транспосомный комплекс, имеет универсальную последовательность. Универсальная последовательность служит в качестве комплементарной последовательности для гибридизации в описанной здесь стадии амплификации для введения другого индекса.
[00128] В некоторых вариантах осуществления изобретения, транспосомный комплекс включает последовательность нуклеиновой кислоты транспозона, которая связывает две субъединицы транспозазы с образованием «петлевого комплекса» или «петлевой транспосомы». В одном примере, транспосома включает димерную транспозазу и последовательность транспозона. Петлевые комплексы могут гарантировать встраивание транспозонов в ДНК-мишень при сохранении информации об упорядочении исходной ДНК-мишени и без фрагментации ДНК-мишени. Очевидно, что петлевые структуры могут встраивать нужные последовательности нуклеиновой кислоты, такие как индексы, в нуклеиновую кислоту-мишень с сохранением физической связи с нуклеиновой кислотой-мишенью. В некоторых вариантах осуществления изобретения, последовательность транспозона петлевого транспосомного комплекса может включать сайт фрагментации, так, чтобы последовательность транспозона могла фрагментироваться с образованием транспосомного комплекса, включающего две последовательности транспозона. Такие транспосомные комплексы могут быть использованы для гарантии того, что соседние фрагменты ДНК-мишени, в которые встраиваются транспозоны, будут иметь комбинации штрих-кодов, которые могут быть однозначно собраны на более поздней стадии анализа.
[00129] В одном варианте осуществления изобретения, фрагментацию нуклеиновых кислот осуществляют с использованием сайта фрагментации, присутствующего в нуклеиновых кислотах. Обычно, сайты фрагментации вводят в нуклеиновые кислоты-мишени с использованием транспосомного комплекса. В одном варианте осуществления изобретения, после фрагментации нуклеиновых кислот, транспозаза остается связанной с фрагментами нуклеиновой кислоты, а поэтому, фрагменты нуклеиновой кислоты, полученные из одной и той же молекулы геномной ДНК, остаются физически связанными (Adey et al., 2014, Genome Res., 24: 2041-2049). Так, например, петлевой транспосомный комплекс может включать сайт фрагментации. Сайт фрагментации может использоваться для физического расщепления, но не информационной ассоциации между индексными последовательностями, которые были встроены в нуклеиновую кислоту-мишень. Расщепление может быть осуществлено биохимическим, химическим или другим способом. В некоторых вариантах осуществления изобретения, сайт фрагментации может включать нуклеотид или нуклеотидную последовательность, которые могут быть фрагментированы различными способами. Примерами сайтов фрагментации являются, но не ограничиваются ими, сайт рестриктирующей эндонуклеазы, по меньшей мере один рибонуклеотид, расщепляемый РНКазой; нуклеотидные аналоги, расщепляемые в присутствии определенного химического агента; диоловая связь, расщепляемая путем обработкой периодатом; дисульфидная группа, расщепляемая химическим восстановителем; расщепляемый фрагмент, который может быть подвергнут фотохимическому расщеплению; и пептид, расщепляемый ферментом пептидазой или другими подходящими способами (см., например, публикацию заявки на патент США №2012/0208705, публикацию заявки на патент США №2012/0208724 и WO 2012/061832).
[00130] Транспосомный комплекс может необязательно включать индексную последовательность, также называемую индексом транспозазы. Индексная последовательность присутствует как часть последовательности транспозона. В одном варианте осуществления изобретения, индексная последовательность может присутствовать на перенесенной цепи, то есть, цепи сайта распознавания транспозазы, которая переносится в нуклеиновую кислоту-мишень.
[00131] После мечения ядер и обработки фрагментов нуклеиновой кислоты может быть проведена стадия очистки для повышения чистоты молекул. При этом может быть проведен любой подходящий метод очистки, такой как электрофорез, эксклюзионная хроматография или т.п. В некоторых вариантах осуществления изобретения, для отделения нужных молекул ДНК, например, от невключенных праймеров и для отбора нуклеиновых кислот по размеру могут быть использованы парамагнитные сферы для твердофазной обратимой иммобилизации. Парамагнитные сферы для твердофазной обратимой иммобилизации являются коммерчески доступными и поставляются Beckman Coulter (Agencourt AMPure XP), Thermofisher (MagJet), Omega Biotek (Mag-Bind), Promega Beads (Promega) и Кара Biosystems (Кара Pure Beads).
Добавление компартмент-специфического индекса
[00132] Индексная последовательность, также называемая меткой или штрих-кодом, являются подходящей в качестве маркера для характеризации компартмента, в котором присутствует конкретная нуклеиновая кислота. В соответствии с этим, индекс представляет собой последовательность нуклеиновой кислоты-метки, которая присоединена к каждой нуклеиновой кислоте-мишени, присутствующей в конкретном компартменте, и присутствие этой метки указывает на компартмент или позволяет идентифицировать компартмент, в котором присутствует популяция выделенных ядер или клеток на определенной стадии этого метода. Добавление индекса к фрагментам нуклеиновой кислоты осуществляют с использованием субпопуляций выделенных ядер или клеток, распределенных по различным компартментам (фиг. 1, блок 15; фиг. 2, блоки 24, 26 и 30; фиг. 3, блоки 33 и 37).
[00133] Индексная последовательность может иметь длину в любое подходящее количество нуклеотидов, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 или более нуклеотидов. Метка из четырех нуклеотидов дает возможность определить мультиплексность 256 образцов в одном массиве, а метка из шести оснований позволяет обрабатывать 4096 образцов в одном массиве.
[00134] В одном варианте осуществления изобретения, добавление индекса достигается в процессе обработки нуклеиновых кислот с получением фрагментов нуклеиновых кислот. Так, например, можно использовать транспосомный комплекс, который включает индекс. В некоторых вариантах осуществления изобретения, индекс добавляют после получения фрагментов нуклеиновой кислоты, содержащих нуклеотидную последовательность на одном или обоих концах, путем обработки. В других вариантах осуществления изобретения, для добавления индекса обработка не требуется. Так, например, индекс может быть добавлен непосредственно к нуклеиновым кислотам РНК без фрагментации нуклеиновых кислот РНК. В соответствии с этим, термин «фрагмент нуклеиновой кислоты» включает нуклеиновые кислоты, которые образуются в результате обработки, и нуклеиновые кислоты РНК, а также нуклеиновые кислоты, происходящие от этих нуклеиновых кислот.
[00135] Способы добавления индекса включают, но не ограничиваются ими, лигирование, удлинение (включая удлинение с использованием обратной транскриптазы), гибридизацию, адсорбцию, специфические или неспецифические взаимодействия праймеров, амплификацию или транспозицию. Нуклеотидная последовательность, которую добавляют к одному или обоим концам фрагментов нуклеиновой кислоты, также может включать одну или более универсальных последовательностей и/или уникальных молекулярных идентификаторов. Универсальная последовательность может быть использована, например, в качестве «посадочной полосы» на последующей стадии для отжига нуклеотидной последовательности, которая может быть использована в качестве праймера для добавления другой нуклеотидной последовательности, такой как другой индекс и/или другая универсальная последовательность, к фрагменту нуклеиновой кислоты. Таким образом, для включения индексной последовательности может быть применен способ, который включает одну, две или более стадий, с использованием, по существу, любой комбинации лигирования, удлинения, гибридизации, адсорбции, специфических или неспецифических взаимодействий праймера, амплификации или транспозиции.
[00136] Так, например, в вариантах осуществления изобретения, в которых используются фрагменты нуклеиновой кислоты, происходящие от мРНК, для добавления индекса к мРНК в одну или две стадии могут быть применены различные методы. Так, например, индекс может быть добавлен с применением методов продуцирования кДНК. Праймер с poly-T-последовательностью на 3'-конце может быть гибридизован с молекулами мРНК и удлинен с использованием обратной транскриптазы. Обработка выделенных ядер или клеток этими компонентами в условиях, подходящих для обратной транскрипции, позволяет осуществлять одностадийное добавление индекса, что будет приводить к образованию популяции индексированных ядер или клеток, где каждое ядро или каждая клетка содержит индексированные фрагменты нуклеиновой кислоты. Альтернативно, праймер с poly-T-последовательностью включает универсальную последовательность вместо индекса, а индекс добавляют в последующей стадии лигирования, удлинения праймера, амплификации, гибридизации или их комбинации. В некоторых вариантах осуществления изобретения, штрих-код добавляют без использования универсальной последовательности. Индексированные фрагменты нуклеиновой кислоты в синтезированной цепи могут включать и обычно включают индексную последовательность, указывающую на конкретный компартмент.
[00137] В вариантах осуществления изобретения, которые включают использование фрагментов нуклеиновой кислоты, происходящих от некодирующей РНК, могут быть применены различные способы добавления индекса к некодирующей РНК в одну или две стадии. Так, например, индекс может быть добавлен с использованием первого праймера, который включает рандомизированную последовательность и праймер для переключения матрицы, где любой праймер может включать индекс. Может быть использована обратная транскриптаза, обладающая терминальной трансферазной активностью, которая приводит к добавлению нематричных нуклеотидов к 3'-концу синтезированной цепи, а праймер для переключения матрицы включает нуклеотиды, которые гибридизуются с нематричными нуклеотидами, добавленными под действием обратной транскриптазы. Примером подходящего фермента обратной транскриптазы является обратная транскриптаза вируса мышиного лейкоза Молони. В конкретном варианте осуществления изобретения, реагент SMARTer™, поставляемый от Takara Bio USA, Inc. (кат. №634926), используют для переключения матрицы в целях добавления индекса к некодирующей РНК и мРНК, если это желательно. Альтернативно, первый праймер и/или праймер для переключения матрицы могут включать универсальную последовательность вместо индекса, и индекс добавляют в последующей стадии лигирования, удлинения праймера, амплификации, гибридизации или их комбинации. Индексированные фрагменты нуклеиновой кислоты на синтезированной цепи могут включать и обычно включают индексную последовательность, указывающую на конкретный компартмент. Другие варианты осуществления изобретения включают 5'- или 3'-профилирование РНК или полноразмерное профилирование РНК.
[00138] В другом варианте осуществления изобретения, специфическая мРНК и/или некодирующая РНК могут быть мишенями для амплификации. Таргетинг позволяет создавать секвенирующие библиотеки, обогащенные последовательностями, которые с большей вероятностью будут давать полезную информацию, и тем самым значительно снижать глубину секвенирования и связанные с этим затраты, а также повышають способность обнаруживать тонкие различия между клетками. Молекулы РНК, включающие одну или более мРНК и/или одну или более некодирующих РНК, могут быть отобраны на вероятность получения полезной информации, а праймеры могут быть использованы для селективного отжига предварительно определенных нуклеиновых кислот РНК и амплификации субпопуляции общих молекул РНК, присутствущих в клетке или в ядре. Специалисту в данной области очевидно, что соответствующие молекулы РНК для отбора зависят от эксперимента. Так, например, при оценке некодирующих пертурбаций, на изменение уровня экспрессии могут быть протестированы только кодирующие области, находящиеся в цис-положении по отношению к разрушаемому регуляторному элементу. Такой подход позволяет уменьшить фон рибосомных ридов в большей степени, чем использование рандомизированного гексамера или poly-T-праймеров. Этот подход также позволяет осуществлять нацеливание на стыки сплайсинга и экзоны, образованные в результате альтернативных событий в сайте инициации транскрипции, и таким образом получить информацию об изоформах, которую трудно получить стандартными sci-методами.
[00139] Нацеленная амплификация молекул РНК может происходить в несколько стадий во время получения библиотеки. В одном варианте осуществления изобретения, нацеленная амплификация множества мишеней происходит во время обратной транскрипции молекул РНК. В эксперименте может быть использовано множество различных праймеров, нацеленных на различные молекулы РНК. В одном варианте осуществления изобретения может быть использовано множество различных праймеров, нацеленных на различные области одной и той же молекулы РНК. Использование множества праймеров, нацеленных на различные области одной и той же молекулы РНК, дает множество возможностей осуществления обратной транскрипции молекулы РНК в кДНК, что будет повышать вероятность обнаружения молекулы РНК.
[00140] В одном варианте осуществления изобретения, праймеры, используемые для нацеленной амплификации, не включают индекс. Если индекс не добавляют во время реакции амплификации, то распределение клеток или ядер по различным компартментам является необязательным, и амплификация может происходить за одну реакцию со всеми молекулами РНК и всеми присутствующими праймерами. В тех вариантах осуществления изобретения, где индекс добавляют во время реакции амплификации, распределение клеток или ядер является полезным, и амплификация может быть осуществлена за одну реакцию в каждом компартменте в присутствии всех молекул РНК и всех праймеров, но, при этом, каждый праймер в компартменте имеет один и тот же компартмент-специфический индекс.
[00141] В одном варианте осуществления изобретения, конструирование праймеров для мультиплексного захвата мишени может быть осуществлено исходя из одного или более из следующих соображений. После отбора РНК для нацеленной амплификации может быть собрана последовательность РНК и могут быть определены все возможные праймеры обратной транскриптазы, то есть, праймеры-кандидаты. Длина любого праймера должна быть достаточной для их функционирования в реакции обратной транскрипции, и может составлять, например, от 20 до 30 нуклеотидов.
[00142] Праймеры-кандидаты могут быть отфильтрованы по различным критериям, включая, но не ограничиваясь ими, содержание GC, локализацию оснований GC в праймере, вероятность нацеливания на нежелательный сайт и картируемость. Подходящее содержание GC составляет 40-60%, что соответствует температурам плавления, которые составляют приблизительно 55-70°С. Предпочтительно, чтобы эти два гуаниновых или цитозиновых основания присутствовали в последних 5 нуклеотидах у 3'-конца праймера, что увеличит вероятность того, что гибридизованный праймер будет хорошим субстратом для удлинения под действием фермента обратной транскриптазы.
[00143] Что касается вероятности нежелательного праймирования, то авторами настоящего изобретения было обнаружено, что хотя РНК-мишени были в высокой степени обогащены, однако, большая часть ридов все еще происходила от других РНК, которые присутствовали в клетках в большом количестве. Большинство этих событий нежелательного праймирования является результатом того, что приблизительно 5-8 пар оснований были комплементарны между 3'-концом праймера и РНК, не являющейся мишенью. Авторами настоящего изобретения было обнаружено, что следует учитывать избыточность конечного гексамера праймера-кандидата в общей клеточной РНК. Было определено, что подходящие праймеры включают последний гексамер, который либо (i) не присутствует в рибосомной РНК, либо (ii) присутствует на низком уровне в общей клеточной РНК.
[00144] Описаны примеры гексамеров, не присутствующих в рибосомной РНК (гексамеры, которые «не были рандомизированы соответствующим образом» или NSR-гексамеры, описаны Armor et al., 2009, Nature Methods, 6(9): 647-49). Было обнаружено, что праймеры, обладающие этим свойством, с гораздо меньшей вероятностью имеют нецелевое праймирование в рибосомной РНК. Одним из методов определения того факта, присутствует ли гексамер на низком уровне в общей клеточной РНК, является метод, который может включать идентификацию избытка каждого гексамера в молекулах РНК в клетке, например, все возрастающую транскрипцию, включая рибосомную транскрипцию, в клетках анализируемого типа в соответствии с описанными здесь способами. Использование праймеров-кандидатов, которые имеют небольшой избыточный уровень, например, в пределах самого низкого квартиля, может снижать уровень таргетинга в нежелательный сайт.
[00145] Праймеры-кандидаты также могут быть оценены по картируемости. Так, например, каждый кандидат может быть выровнен по мишеням с использованием алгоритма типа «галстука-бабочки» и допускает 3 несоответствия. Эта стадия будет гарантировать, что каждый праймер будет иметь только один сайт-мишень в геноме.
[00146] В некоторых вариантах осуществления изобретения, амплификация множества мишеней в одной и той же реакции, также называемой мультиплексным захватом мишеней, с регуляцией температур отжига праймеров обратной транскриптазы, может быть использована для поддержания специфической обратной транскрипции и амплификации нужных РНК-мишеней. Так, например, типичные протоколы обратной транскрипции включают денатурацию смеси РНК и праймера обратной транскрипции и охлаждение до 4°С для обеспечения отжига. Низкая температура отжига создает условия слишком низкой жесткости и приводит к нежелательным событиям отжига. Для повышения вероятности того, что будут наблюдаться только события отжига, когда все праймеры-мишени обратной транскрипции гибридизуются с правильными мишенями, высокая температура должна поддерживаться в течение всего процесса обратной транскрипции. В одном варианте осуществления изобретения, компоненты, например, смесь фиксированных клеток, пула праймеров обратной транскрипции и dNTP при 65°С, гибридизуют при 53°С, добавляют в смесь фермента/буфера для обратной транскрипции, которая была предварительно уравновешена при 53°С до реакции отжига, а затем проводят реакцию удлинения при 53°С в течение 20 минут. Таким образом, уменьшается вероятность отжига праймеров обратной транскрипции при низкой температуре между стадиями денатурации и удлинения. Специалисту в данной области очевидно, что могут быть внесены некоторые изменения, например, изменение температуры или времени без снижения специфичности обратной транскрипции.
[00147] Для добавления индекса к фрагменту нуклеиновой кислоты могут быть применены и другие способы, и способ добавления индекса не имеет конкретных ограничений. Так, например, в одном варианте осуществления изобретения, введение индексной последовательности включает лигирование праймера с одним или обоими концами фрагментов нуклеиновой кислоты. Лигирование лигирующего праймера может облегчаться благодаря присутствию универсальной последовательности на концах фрагментов нуклеиновой кислоты. Примером праймера является шпилечный лигирующий дуплекс. Лигирующий дуплекс может быть лигирован с одним концом или предпочтительно с обоими концами фрагментов нуклеиновой кислоты.
[00148] В другом варианте осуществления изобретения, введение индексной последовательности включает использование одноцепочечных фрагментов нуклеиновой кислоты и синтез второй цепи ДНК. В одном варианте осуществления изобретения, вторую цепь ДНК получают с использованием праймера, который включает последовательности, комплементарные нуклеотидам, присутствующим на концах одноцепочечных фрагментов нуклеиновой кислоты.
[00149] В другом варианте осуществления изобретения, включение индекса происходит в один, два, три или более раундов штрихового кодирования с разделением и объединением, что приводит к созданию библиотек отдельных клеток с одним, двумя, тремя или множеством (например, четырьмя или более) индексами.
[00150] В другом варианте осуществления изобретения, включение индексов и медиатора амплификации (например, универсальной последовательности) является полезным, и позволяет получать секвенирующие библиотеки-мишени для отдельных клеток и/или секвенирующие библиотеки-мишени для отдельных клеток.
Добавление универсальных последовательностей для иммобилизации
В одном варианте осуществления изобретения, добавление нуклеотидов во время стадий обработки и/или индексации приводит к добавлению универсальных последовательностей, подходящих для иммобилизации и секвенирования фрагментов. В другом варианте осуществления изобретения, индексированные фрагменты нуклеиновой кислоты могут быть также обработаны для добавления универсальных последовательностей, подходящих для иммобилизации и секвенирования фрагментов нуклеиновой кислоты. Специалисту в данной области очевидно, что в тех вариантах осуществления изобретения, где компартмент представляет собой каплю, последовательности для иммобилизации фрагментов нуклеиновой кислоты являются необязательными. В одном варианте осуществления изобретения, введение универсальных последовательностей, подходящих для иммобилизации и секвенирования фрагментов, включает лигирование идентичных универсальных адаптеров (также называемых «несоответствующими адапторами», общие признаки которых описаны Gormley et al., в патенте США 7741463 и Bignell et al. в патенте США 8053192) с 5'- т 3'-концами индексированных фрагментов нуклеиновой кислоты. В одном варианте осуществления изобретения, универсальный адаптер включает все последовательности, необходимые для секвенирования, включая последовательности для иммобилизации индексированных фрагментов нуклеиновой кислоты на массиве.
[00152] В одном варианте осуществления изобретения может быть использовано лигирование тупых концов. В другом варианте осуществления изобретения, фрагменты нуклеиновой кислоты получают так, чтобы они имели одиночные выступающие нуклеотиды, например, под действием активности ДНК-полимеразы определенных типов, такой как Taq-полимераза или экзо-(-)-полимераза Кленова, которая обладает не зависимой от матрицы концевой трансферазной активностью, добавляющей один или больше дезоксинуклеотидов, например дезоксиаденозин (А) к 3'-концам индексируемых фрагментов нуклеиновой кислоты. В некоторых случаях, выступающий нуклеотид представляет собой более, чем одно основание. Такие ферменты могут быть использованы для добавления одного нуклеотида «А» к тупому концу 3'-конца каждой цепи фрагментов нуклеиновой кислоты. Таким образом, «А» может быть добавлен к 3'-концу каждой цепи двухцепочечных фрагментов-мишеней посредством реакции с Taq-полимеразой или экзо-(-)-полимеразой Кленова, а дополнительные последовательности, добавляемые к каждому концу фрагмента нуклеиновой кислоты, могут включать совместимый выступающий «Т», присутствующий на 3'-конце каждой области добавляемой двухцепочечной нуклеиновой кислоты. Эта концевая модификация также предотвращает аутолигирование нуклеиновых кислот, а поэтому наблюдается отклонение в сторону образования индексируемых фрагментов нуклеиновых кислот, фланкированных последовательностями, которые добавляются в этом варианте осуществления изобретения.
[00153] В другом варианте осуществления изобретения, если универсальный адаптер, лигированный с индексированными фрагментами нуклеиновой кислоты, не включает все последовательности, необходимые для секвенирования, то может быть проведена стадия амплификации, такая как ПЦР, для дополнительной модификации универсальных адаптеров, присутствующих в каждом индексированном фрагменте нуклеиновой кислоты до иммобилизации и секвенирования. Так, например, исходная реакция удлинения праймера может быть проведена с использованием универсальной якорной последовательности, комплементарной универсальной последовательности, присутствующей в индексируемом фрагменте нуклеиновой кислоты, в котором образуются продукты удлинения, комплементарные обеим цепям каждого отдельного индексируемого фрагмента нуклеиновой кислоты. Обычно, ПЦР добавляет дополнительные универсальные последовательности, такие как универсальная последовательность для захвата.
[00154] После добавления универсальных адаптеров, либо одностадийным способом лигирования, либо путем гибридизации универсального адаптера, включающего все последовательности, необходимые для секвенирования, или двухстадийным способом лигирования универсального адаптера, а затем амплификации для дальнейшей модификации универсального адаптера, конечные индексные фрагменты будут включать универсальную последовательность для захвата и якорную последовательность. Результатом добавления универсальных адаптеров к каждому концу является множество или библиотека проиндексированных фрагментов нуклеиновой кислоты.
[00155] Полученные индексированные фрагменты, взятые вместе, представляют собой библиотеку нуклеиновых кислот, которые могут быть иммобилизованы, а затем секвенированы. Термин «библиотека», также называемый здесь «библиотекой секвенирования», означает набор фрагментов нуклеиновых кислот из отдельных ядер или клеток, содержащих известные универсальные последовательности на их 3'- и 5'-концах. Эта библиотека включает нуклеиновые кислоты из всего транскриптома, нуклеиновые кислоты из вновь синтезированных молекул РНК или их комбинацию и может быть использована для секвенирования всего транскриптома, транскриптома вновь синтезированной РНК или их комбинации.
[00156] Индексированные фрагменты нуклеиновой кислоты могут быть обработаны в условиях, которые позволяют отбирать молекулы предварительно определенного размера, например, длиной от 150 до 400 нуклеотидов, например, от 150 до 300 нуклеотидов. Полученные индексированные фрагменты нуклеиновой кислоты объединяют, а затем, они могут быть, но необязательно, подвергнуты очистке для повышения чистоты молекул ДНК путем удаления по меньшей мере части невключенных универсальных адаптеров или праймеров. При этом может быть проведен любой подходящий способ очистки, такой как электрофорез, эксклюзионная хроматография или т.п. В некоторых вариантах осуществления изобретения, для отделения нужных молекул ДНК, например, от невключенных праймеров, и для отбора нуклеиновых кислот по размеру могут быть использованы парамагнитные сферы для твердофазной обратимой иммобилизации. Парамагнитные сферы для твердофазной обратимой иммобилизации являются коммерчески доступными и поставляются Beckman Coulter (Agencourt AMPure XP), Thermofisher (MagJet), Omega Biotek (Mag-Bind), Promega Beads (Promega) и Kapa Biosystems (Kapa Pure Beads).
[00157] Неограничивающий иллюстративный вариант раскрытия изобретения показан на фиг. 1. В этом варианте осуществления изобретения, способ включает получение множества клеток (фиг. 1, блок 10). Этот способ также включает распределение субпопуляций клеток по множеству компартментов (фиг. 1, блок 11) и обработку клеток в предварительно определенных условиях (фиг. 1, блок 12). Предварительно определенные условия могут отличаться и обычно отличаются между различными компартментами. Так, например, различные компартменты могут включать различные дозы агента, различные пертурбации, различные продолжительности обработки, различные типы клеток и т.п. Вновь синтезированную РНК затем метят (фиг. 1, блок 13). Может быть осуществлено мечение клеток, либо ядра могут быть выделены из клеток, а затем помечены. В некоторых вариантах осуществления изобретения, клеточную РНК обрабатывают с образованием фрагментов (фиг. 1, блок 14). РНК, присутствующую в клетках или ядрах, затем индексируют (фиг. 1, блок 15). При этом возможны различные варианты индексирования РНК. Так, например, в одном варианте осуществления изобретения, все мРНК, присутствующие в клетке, индексируются с использованием праймера, который включает poly-T-область. В другом варианте осуществления изобретения, специфические нуклеиновые кислоты РНК могут быть индексированы.
[00158] Другой неограничивающий иллюстративный вариант раскрытия изобретения показан на фиг. 2 и описан в Примере 1. В этом варианте осуществления изобретения, способ включает получение выделенных ядер из множества клеток (фиг. 2, блок 22). Этот способ также включает распределение субпопуляций выделенных ядер по первому множеству компартментов (фиг. 2, блок 23). Количество компартментов в первой стадии распределения (фиг. 2, блок 23) может зависеть от используемого формата. Так, например, количество компартментов может составлять от 2 до 96 компартментов (при использовании 96-луночного планшета), от 2 до 384 компартментов (при использовании 384-луночного планшета) или от 2 до 1536 компартментов (при использовании 1536-луночного планшета). В качестве альтернативы можно использовать и другие компартменты, такие как капли.
[00159] Способ также включает получение индексируемых ядер (фиг. 2, блок 24). В одном из вариантов осуществления изобретения, получение индексированных ядер включает использование обратной транскриптазы с олиго-dT-праймером для добавления индекса, рандомизированной нуклеотидной последовательности и универсальной последовательности. Индекс в каждом компартменте является уникальным, например, каждый индекс является компартмент-специфическим. Рандомизированная последовательность используется в качестве уникального молекулярного идентификатора (UMI) для мечения уникальных фрагментов нуклеиновых кислот. Рандомизированная последовательность может быть также использована для облегчения удаления дубликатов при последующей обработке. Универсальная последовательность служит в качестве комплементарной последовательности для гибридизации в описанной здесь стадии лигирования. В другом варианте осуществления изобретения, получение индексированных ядер включает использование обратной транскриптазы со специфическими праймерами для нацеливания на заранее определенные молекулы РНК. Обратная транскрипция может приводить к добавлению индекса, рандомизированной нуклеотидной последовательности и универсальной последовательности к молекулам РНК-мишени. Обработка ядер этими компонентами в условиях, подходящих для обратной транскрипции, приводит к образованию популяции индексируемых ядер, где каждое ядро содержит индексированные фрагменты нуклеиновой кислоты. Индексированные фрагменты нуклеиновой кислоты могут включать и обычно включают на синтезированной цепи индексную последовательность, указывающую на конкретный компартмент. Пример индексированного фрагмента нуклеиновой кислоты показан на фиг. 1А Примера 1 (см. раздел «Индексированная обратная транскрипция»).
[00160] Индексированные ядра от множества компартментов могут быть объединены (фиг. 2, блок 25). Субпопуляции этих объединенных индексированных ядер, называемых здесь объединенными индексированными ядрами, затем распределяют по второму множеству компартментов (фиг. 2, блок 25). После распределения ядер по субпопуляциям, в каждый компартмент индексированных фрагментов нуклеиновой кислоты включают вторую индексную последовательность с получением фрагментов с двумя индексами. В результате этого происходит дополнительное индексирование индексированных фрагментов нуклеиновой кислоты (фиг. 2, блок 26).
[00161] В этом иллюстративном варианте осуществления изобретения, введение второй индексной последовательности включает лигирование дуплекса, лигирование шпилечного лигирующего дуплекса с индексированными фрагментами нуклеиновой кислоты в каждом компартменте. Использование шпилечного лигирующего дуплекса для введения универсальной последовательности, индекса или их комбинации в конец фрагмента нуклеиновой кислоты-мишени обычно включает использование одного конца дуплекса в качестве праймера для последующей амплификации. В противоположность этому, шпилечный лигирующий дуплекс, используемый в этом варианте осуществления изобретения, не действует как праймер. Преимущество использования описанного здесь шпилечного лигирующего дуплекса заключается в уменьшении аутолигирования, наблюдаемого для многих шпилечных лигирующих дуплексов, описанных в литературе. В одном варианте осуществления изобретения, лигирующий дуплекс включает пять элементов: 1) универсальную последовательность, которая является комплементарной универсальной последовательности, присутствующей в праймере олиго-dT, 2) второй индекс, 3) i-дезокси-U, 4) нуклеотидную последовательность, которая может образовывать шпильку и 5) обратный комплемент второго индекса. Последовательности второго индекса являются уникальными для каждого компартмента, в который были помещены распределенные индексированные ядра (фиг. 2, блок 25) после добавления первого индекса посредством обратной транскрипции. Пример фрагмента нуклеиновой кислоты с двумя индексами показан на фиг. 1А Примера 1 (см. «Индексированное шпилечное лигирование»).
[00162] Удаление i-дезокси-U, присутствующего в области шпильки шпилечного лигирующего дуплекса, включенного во фрагменты нуклеиновой кислоты, может происходить до, во время или после очистки. Удаление остатка урацила может быть осуществлено любым доступным способом, а в одном из вариантом, используется реагент для урацил-специфического вырезания (USER), поставляемый NEB.
[00163] Субпопуляции этих объединенных ядер с двумя индексами, называемые здесь объединенными ядрами с двумя индексами, затем распределяют по третьему множеству компартментов (фиг. 2, блок 27). В одном варианте осуществления изобретения, в каждую лунку распределяют от 100 до 30000 ядер. В одном варианте осуществления изобретения, число ядер в лунке составляет по меньшей мере 100, по меньшей мере 500, по меньшей мере 1000 или по меньшей мере 5000. В одном варианте осуществления изобретения, число ядер в лунке составляет не более 30000, не более 25000, не более 20000 или не более 15000. В одном варианте осуществления изобретения, число ядер, присутствующих в субпопуляции, может составлять 100-1000, 1000-10000, 10000-20000 или 20000-30000. В одном варианте осуществления изобретения, в каждую лунку помещают 2500 ядер. В одном варианте осуществления изобретения, число ядер, присутствующих в каждой субпопуляции является, приблизительно одинаковым.
[00164] После распределения ядер с двумя индексами по субпопуляциям осуществляют синтез второй цепи ДНК (фиг. 2, блок 28). Нуклеиновые кислоты в ядрах обрабатывают путем мечения (фиг. 2, блок 29). Каждый компартмент, содержащий ядра с двумя индексами, включает транспосомный комплекс. В этом варианте осуществления изобретения, мечение проводят для получения фрагментов нуклеиновой кислоты, которые включают различные нуклеотидные последовательности на каждом конце (например, последовательность праймера N5 на одном конце и праймера N7 на другом конце).
[00165] После мечения ядер, во фрагменты нуклеиновой кислоты с двумя индексами в каждом компартменте вводят третью индексную последовательность для получения фрагментов с тремя индексами, где третья индексная последовательность в каждом компартменте отличается от первой и второй индексных последовательностей в компартментах. Это приводит к дополнительной индексации индексированных фрагментов нуклеиновой кислоты (фиг. 2, блок 30; см. также фиг. 1А примера («обработка USER, индексированная ПЦР»)) до иммобилизации и секвенирования. В одном варианте осуществления изобретения, универсальные последовательности, присутствующие на концах фрагментов нуклеиновой кислоты с двумя индексами (например, нуклеотидная последовательность, встроенная в шпилечный лигирующий дуплекс на одном конце, и нуклеотидная последовательность, встроенная в транспосомный комплекс на другом конце), могут быть использованы для связывания праймеров и для удлинения в реакции амплификации. Обычно, используют два различных праймера. Один праймер гибридизуется с универсальными последовательностями у 3'-конца одной цепи фрагментов нуклеиновой кислоты с двумя индексами, а второй праймер гибридизуется с универсальными последовательностями у 3'-конца другой цепи фрагментов нуклеиновой кислоты с двумя индексами. Таким образом, якорные последовательности (например, сайт, с которым гибридизуется универсальный праймер, такой как секвенирующий праймер для секвенирования рида 1 или рида 2), присутствующие на каждом праймере, могут отличаться. Каждый из подходящих праймеров может включать дополнительные универсальные последовательности, такие как универсальная последовательность для захвата (например, сайт, с которым гибридизуется олигонуклеотид для захвата, где олигонуклеотид для захвата может быть иммобилизован на поверхности твердой подложке). Поскольку каждый праймер включает индекс, то эта стадия приводит к добавлению еще одной последовательности индекса, по одной на каждом конце фрагментов нуклеиновой кислоты с получением фрагментов с тремя индексами. В одном варианте осуществления изобретения, индексированные праймеры, такие как индексированный праймер Р5 и индексированный праймер Р7, могут быть использованы для добавления третьего индекса. Фрагменты с тремя индексами объединяют, а затем они могут быть подвергнуты описанной здесь стадии очистки.
[00166] Для секвенирования может быть получено множество фрагментов с тремя индексами. После объединения фрагментов с тремя индексами и после их очистки, их обогащают, обычно путем иммобилизации и/или амплификации перед секвенированием (фиг. 2, блок 31).
[00167] Другой неограничивающий иллюстративный вариант раскрытия изобретения показан на фиг. 3 и описан в Примере 4. В этом варианте осуществления изобретения, способ включает получение выделенных ядер или клеток (фиг. 3, блок 30). Ядра или клетки могут быть обработаны обратной транскриптазой и специфическими праймерами для нацеливания и обогащения предварительно определенных молекул РНК (фиг. 3, блок 31). Обработка ядер или клеток этими компонентами в условиях, подходящих для обратной транскрипции, позволяет получить популяцию ядер или клеток, где каждое ядро содержит фрагменты нуклеиновой кислоты, обогащенные последовательностями, присутствующими в предварительно определенных молекулах РНК. Этот способ также включает распределение субпопуляций ядер или клеток по первому множеству компартментов (фиг. 3, блок 32). Число компартментов в первой стадии распределения (фиг. 3, блок 32) может зависеть от используемого формата. Так, например, количество компартментов может составлять от 2 до 96 компартментов (при использовании 96-луночного планшета), от 2 до 384 компартментов (при использовании 384-луночного планшета) или от 2 до 1536 компартментов (при использовании 1536-луночного планшета). В качестве альтернативы можно использовать и другие компартменты, такие как капли.
[00168] Этот способ также включает получение индексированных ядер или клеток (фиг. 3, блок 33) путем включения во фрагменты нуклеиновой кислоты в каждом компартменте индексной последовательности для получения индексированных фрагментов.
[00169] В одном варианте осуществления изобретения, введение индексной последовательности включает лигирование шпилечного лигирующего дуплекса с индексированными фрагментами нуклеиновой кислоты в каждом компартменте. Ядра или клетки, содержащие индексированные фрагменты, объединяют, а затем субпопуляции этих объединенных индексированных ядер или клеток распределяют по второму множеству компартментов (фиг. 3, блок 34).
[00170] Распределение индексированных ядер или клеток по субпопуляциям может сопровождаться синтезом второй цепи ДНК (фиг. 3, блок 35). Нуклеиновые кислоты в ядрах или клетках обрабатывают путем мечения (фиг. 3, блок 36). Каждый компартмент, содержащий проиндексированные ядра, включает транспосомный комплекс. В этом варианте осуществления изобретения, мечение проводят для получения фрагментов нуклеиновой кислоты, которые включают различные нуклеотидные последовательности на каждом конце (например, последовательность праймера N5 на одном конце и праймера N7 на другом конце).
[00171] После мечения ядер, во фрагменты нуклеиновой кислоты с двумя индексами в каждом компартменте вводят вторую индексную последовательность для получения фрагментов с двумя индексами, где вторая индексная последовательность в каждом компартменте отличается от первых индексных последовательностей в компартментах. Это приводит к дополнительной индексации индексированных фрагментов нуклеиновой кислоты (фиг. 3, блок 37) до иммобилизации и секвенирования.
[00172] Для секвенирования может быть получено множество фрагментов с двумя индексами, где данные секвенирования обогащают последовательностями, присутствующими в предварительно определенных молекулах РНК. После объединения и очистки фрагментов с двумя индексами, их обогащают обычно путем иммобилизации и/или амплификации перед секвенированием (фиг. 3, блок 38).
Получение иммобилизованных образцов для секвенирования
[00173] Методы присоединения индексированных фрагментов от одного или более источников к субстрату известны специалистам. В одном варианте осуществления изобретения, индексированные фрагменты обогащают с использованием множества олигонуклеотидов для захвата, специфичных к индексированным фрагментам, и олигонуклеотиды для захвата могут быть иммобилизованы на поверхности твердой подложки. Так, например, олигонуклеотиды для захвата могут включать первый член универсальной связывающей пары, а второй член связывающей пары может быть иммобилизован на поверхности твердой подложки. Аналогичным образом, методы амплификации иммобилизованных фрагментов с двумя индексами включают, но не ограничиваются ими, мостиковую амплификацию и кинетическое исключение. Методы иммобилизации и амплификации до секвенирования описаны, например, Bignell et al. (патент США 8053192), Gunderson et al. (WO 2016/130704), Shen et al. (патент США 8895249) и Pipenburg et al. (патент США 9309502).
[00174] Объединенный образец может быть иммобилизован при подготовке для секвенирования. Секвенирование может быть осуществлено с использованием массива отдельных молекул, либо эти молекулы могут быть амплифицированы до секвенирования. Амплификация может быть проведена с использованием одного или более иммобилизованных праймеров. Иммобилизованный(е) праймер(ы) может (могут) представлять собой, например, дорожку на плоской поверхности или на пуле сфер. Пул сфер можно быть выделен с получением эмульсии с одной сферой в каждом «компартменте» эмульсии. При концентрации только одной матрицы на «компартменте» на каждой сфере амплифицируется только одна матрица.
[00175] Используемый здесь термин «твердофазная амплификация» означает любую реакцию амплификации нуклеиновой кислоты, осуществляемую на твердом носителе или в комбинации с твердым носителем, так, чтобы все амплифицированные продукты или их часть иммобилизовались на твердом носителе по мере их образования. В частности, этот термин охватывает твердофазную полимеразную цепную реакцию (твердофазную ПЦР) и твердофазную изотермическую амплификацию, которые являются реакциями, аналогичными стандартной амплификации в жидкой фазе, за исключением того, что один или оба прямых и обратных праймеров амплификации иммобилизованы на твердом носителе. Твердофазная ПЦР охватывает системы, такие как эмульсии, в которых один праймер заякорен на сфере, а другой присутствует в свободном растворе, и образование колоний в твердофазных гелевых матрицах, где один праймер заякорен на поверхности, а другой находится в свободном растворе.
[00176] В некоторых вариантах осуществления изобретения, твердый носитель содержит профилированную поверхность. Термин «профилированная поверхность» относится к расположению различных областей внутри или на открытом слое твердого носителя. Так, например, одна или более областей могут представлять собой элементы, в которых присутствуют один или более праймеров амплификации. Эти элементы могут быть разделены промежуточными областями, где отсутствуют праймеры амплификации. В некоторых вариантах осуществления изобретения, структура может представлять собой элементы в формате х-у, которые находятся в рядах и в столбцах. В некоторых вариантах осуществления изобретения, структура может представлять собой повторяющееся расположение элементов и/или промежуточных областей. В некоторых вариантах осуществления изобретения, структура может представлять собой случайное расположение элементов и/или промежуточных областей. Репрезентативными профилированными поверхностями, которые могут быть использованы в описанных здесь способах и композициях, являются поверхности, описанные в патенте США No. 8778848, 8778849 и 9079148 и в публикации заявки на патент США No. 2014/0243224.
[00177] В некоторых вариантах осуществления изобретения, твердый носитель включает массив лунок или впадин в поверхности. Это может быть получено, в основном, известными методами с применением ряда технологий, включая, но не ограничиваясь ими, фотолитографию, методы штамповки, методы формования и методы микротравления. Как будет понятно специалистам в данной области, применяемая технология будет зависеть от состава и формы подложки-массива.
[00178] Элементы в профилированной поверхности могут представлять собой лунки в массиве лунок (например, микролунки или нанолунки) на стекле, кремнии, пластике или других подходящих твердых носителях со структурированным, ковалентно связанным гелем, таким как сополимер (N-(5-азидоацетамидилпентил)акриламида и акриламида) (PAZAM, см., например, публикацию заявок на патент США №2013/184796, WO 2016/066586 и WO 2015/002813). Этот способ позволяет создать гелевые прокладки, используемые для секвенирования, которые могут быть стабильными в течение раундов секвенирования с большим количеством циклов. Ковалентное связывание полимера с лунками позволяет сохранять гель со структурированными свойствами в течение всего времени полужизни структурированного носителя при различных применениях. Однако, во многих вариантах осуществления изобретения, гель не обязательно должен быть ковалентно связан с лунками. Так, например, в некоторых условиях, не содержащий силана акриламид (SFA, см., например, патент США №8563677), который ковалентно не связан с любой частью структурированного носителя, может быть использован в качестве геля.
[00179] В конкретных вариантах осуществления изобретения, структурированный субстрат может быть изготовлен путем формирования рисунка на твердом носителе с лунками (например, микролунками или нанолунками) путем покрытия структурированного носителя гелевым материалом (например, PAZAM, SFA или его химически модифицированными вариантами, такими как азидолизированный вариант SFA (азидо-SFA) и полировки покрытого гелем носителя, например, посредством химической или механической полировки с сохранением геля в лунках, но удалением или инактивацией почти всего геля из промежуточных областей на поверхности структурированного носителя между лунками. Праймерные нуклеиновые кислоты могут быть присоединены к гелевому материалу. Затем раствор индексированных фрагментов может быть подвергнут контактированию с полированным субстратом, так, чтобы отдельные индексированные фрагменты были «засеяны» в отдельные лунки посредством взаимодействий с праймерами, связанными с гелевым материалом, однако, нуклеиновые кислоты-мишени не будут занимать промежуточные области из-за отсутствия гелевого материала или отсутствия его активности. Амплификация индексированных фрагментов будет ограничена лунками, поскольку отсутствие геля или отсутствие активности геля в промежуточных областях препятствует внешней миграции растущей колонии нуклеиновых кислот. Этот способ может быть легко осуществлен в промышленных масштабах, является масштабируемым и включает применение стандартных методов микро- или нанообработки.
[00180] Хотя раскрытие настоящего изобретения охватывает способы «твердофазной» амплификации, где был иммобилизован только один праймер для амплификации (другой праймер обычно присутствует в свободном растворе), однако, в одном варианте осуществления изобретения, предпочтительно, чтобы твердый носитель был снабжен прямыми и обратными иммобилизованными праймерами. На практике, на твердом носителе будет присутствовать «множество» идентичных прямых праймеров и/или «множество» идентичных обратных праймеров, иммобилизованных на твердом носителе, поскольку процесс амплификации требует избытка праймеров для поддержания амплификации. Ссылки на прямые и обратные праймеры в описании настоящей заявки, соответственно интерпретируются как охватывающие «множество» таких праймеров, если это не противоречит контексту изобретения.
[00181] Как будет понятно специалисту в данной области, любая данная реакция амплификации требует присутствия по меньшей мере одного типа прямого праймера и по меньшей мере одного типа обратного праймера, специфичного для амплифицируемой матрицы. Однако, в определенных вариантах осуществления изобретения, прямой и обратный праймеры могут включать специфичные к матрице части идентичной последовательности и могут иметь полностью идентичные нуклеотидные последовательности и структуры (включая любые не-нуклеотидные модификации). Другими словами, можно осуществить твердофазную амплификацию с использованием только одного типа праймера, и такие способы с одним праймером входят в объем настоящего изобретения. В других вариантах осуществления изобретения могут быть использованы прямые и обратные праймеры, которые содержат идентичные последовательности, специфичные к матрице, но которые отличаются некоторыми другими структурными признаками. Так, например, праймер одного типа может содержать не-нуклеотидную модификацию, которая отсутствует в другом праймере.
[00182] Во всех вариантах раскрытия изобретения, праймеры для твердофазной амплификации предпочтительно иммобилизуют путем ковалентного связывания в одном положении с твердым носителем у 5'-конца или возле 5'-конца праймера, что позволяет части праймера, специфичной к матрице, свободно гибридизоваться с его когнатной матрицей и с 3'-гидроксильной группой, свободной для удлинения праймера. Для этой цели может быть применен любой подходящий метод ковалентного связывания, известный специалистам. Выбранный метод химического связывания будет зависеть от природы твердого носителя и любой его дериватизации или функционализации. Сам праймер может включать группу, которая может представлять собой не-нуклеотидную химическую модификацию, для облегчения связывания. В конкретном варианте осуществления изобретения, праймер может включать серусодержащий нуклеофил, такой как фосфортиоат или тиофосфат, у 5'-конца. В случае полиакриламидных гидрогелей на твердом носителе, этот нуклеофил будет связываться с бромацетамидной группой, присутствующей в гидрогеле. Более конкретный способ связывания праймеров и матриц с твердым носителем представляет собой способ 5'-фосфортиоатного связывания с гидрогелем, состоящим из полимеризованного акриламида и N-(5-бромацетамидилпентил)акриламида (BRAPA), как описано в WO 05/065814.
[00183] В некоторых вариантах раскрытия изобретения могут быть использованы твердые носители, которые включают инертную основу или матрицу (например, предметные стекла, полимерные сферы и т.п.), которые были «функционализированы», например, путем нанесения слоя или покрытия промежуточного вещества, включающего реакционноспособные группы, которые обеспечивают ковалентное связывание с биомолекулами, такими как полинуклеотиды. Примерами таких носителей являются, но не ограничиваются ими, полиакриламидные гидрогели, нанесенные на инертную основу, такую как стекло. В таких вариантах осуществления изобретения, биомолекулы (например, полинуклеотиды) могут быть непосредственно ковалентно связаны с промежуточным веществом (например, гидрогелем), но само промежуточное вещество может быть ковалентно не связано с основой или матрицей (например, со стеклянной основой). Термин «ковалентное связывание с твердым носителем» следует интерпретировать, соответственно, как термин, охватывающий структуру такого типа.
[00184] Объединенные образцы могут быть амплифицированы на сферах, где каждая сфера содержит прямой и обратной праймер амплификации. В конкретном варианте осуществления изобретения, библиотеку индексированных фрагментов используют для получения кластеризованных массивов групп нуклеиновых кислот, аналогичных тем, которые описаны в публикации заявки на патент США №2005/0100900, в патенте США №7115400, в заявке на патент США WO 00/18957 и WO 98/44151, путем твердофазной амплификации, а более конкретно, твердофазной изотермической амплификации. Используемые здесь термины «кластер» и «группа» являются синонимами и означают дискретный сайт на твердом носителе, включающий множество идентичных цепей иммобилизованной нуклеиновой кислоты и множество идентичных цепей иммобилизованной комплементарной нуклеиновой кислоты. Термин «кластеризованный массив» означает массив, образованный такими кластерами или группами. В этом контексте, термин «массив» не следует понимать как упорядоченное расположение кластеров.
[00185] Используемый здесь термин «твердая фаза» или «поверхность» означает любой плоский массив, где праймеры присоединены к плоской поверхности, например, к стеклу, к двуокиси кремния или пластиковым предметным стеклам микроскопа или аналогичных устройств с проточной кюветой; сферы, где один или два праймера присоединены к сферам, а сферы являются амплифицированными; или массив сфер на поверхности после амплификации сфер.
[00186] Кластеризованные массивы могут быть получены с использованием любого способа проведения реакции в термоячейке как описано в WO 98/44151, или способа, в котором поддерживают постоянную температуру, а циклы удлинения и денатурации осуществляют путем замены реагентов. Такие способы изотермической амплификации описаны в патентной заявке WO No. 02/46456 и в публикации заявки на патент США 2008/0009420. Из-за более низких температур, используемых в изотермическом процессе, этот способ является особенно предпочтительным в некоторых вариантах осуществления изобретения.
[00187] Следует отметить, что любые методы амплификации, описанные в настоящей заявке или, по существу, известные специалистам в данной области, могут быть проведены с использованием универсальных или мишень-специфических праймеров для амплификации иммобилизованных фрагментов ДНК. Подходящие способы амплификации включают, но не ограничиваются ими, полимеразную цепную реакцию (ПЦР), амплификацию с заменой цепи (SDA), амплификацию, опосредованную транскрипцией (ТМА) и амплификацию на основе последовательности нуклеиновой кислоты (NASBA), как описано в патенте США No. №8003354. Указанные выше способы амплификации могут быть использованы для амплификации одной или более представляющих интерес нуклеиновых кислот. Так, например, ПЦР, включая мультиплексную ПЦР, SDA, ТМА, NASBA и т.п. могут быть использованы для амплификации фрагментов иммобилизованной ДНК. В некоторых вариантах осуществления изобретения, праймеры, которые являются специфичными к представляющему интерес полинуклеотиду, включены в реакцию амплификации.
[00188] Другие подходящие способы амплификации полинуклеотидов могут включать методы удлинения и лигирования олигонуклеотидов, амплификации по типу «катящегося кольца» (RCA) (Lizardi et al., Nat. Genet. 19: 225-232 (1998)) и анализ на лигирование олигонуклеотидов (OLA) (см., в общих чертах патенты США №№7582420, 5185243, 5679524 и 5573907; ЕР 0320308 В1; ЕР 0336731 В1; ЕР 0439182 Bl; WO 90/01069; WO 89/12696 и WO 89/09835). Очевидно, что эти методики амплификации могут быть разработаны для амплификации иммобилизованных фрагментов ДНК. Так, например, в некоторых вариантах осуществления изобретения, метод амплификации может включать реакции амплификации для лигирования зонда или анализ на лигирование олигонуклеотидов (OLA), которые включают праймеры, специфически нацеленные на представляющую интерес нуклеиновую кислоту. В некоторых вариантах осуществления изобретения, способ амплификации может включать реакцию лигирования-удлинения праймеров, которая включает праймеры, специфически нацеленные на представляющую интерес нуклеиновую кислоту. В качестве неограничивающего примера праймеров для удлинения и лигирования служат праймеры, которые могут быть специально сконструированы для амплификации представляющей интерес нуклеиновой кислоты, и такая амплификация может включать праймеры, используемые для анализа GoldenGate (Illumina, Inc., San Diego, CA), описанного в патентах США No. 7582420 и 7611869.
[00189] Наногранулы ДНК могут быть также использованы в комбинации с описанными здесь способами и композициями. Способы получения и использования наногранул ДНК для геномного секвенирования можно найти, например, в патентах США и публикациях патента США 7910354, в 2009/0264299, 2009/0011943, 2009/0005252, 2009/0155781, 2009/0118488, и, например, в публикации Drmanac et al., 2010, Science 327 (5961): 78-81. Вкратце, после лигирования адапторов для фрагментации ДНК геномной библиотеки с фрагментами, лигированные фрагменты адаптера подвергают циркуляризации путем лигирования с циклической лигазой и проводят амплификацию по типу «катящегося кольца» (как описано Lizardi et al., 1998. Nat. Genet. 19: 225-232 и в US 2007/0099208 Al). Удлиненная конкатемерная структура ампликонов способствует спирализации, и тем самым создает компактные наносферы ДНК. Наносферы ДНК могут быть захвачены на носителях, предпочтительно для создания упорядоченного или структурированного массива так, чтобы поддерживалось расстояние между наносферами, что позволяло бы осуществлять секвенирование отдельных наносфер ДНК. В некоторых вариантах осуществления изобретения, таких как варианты, в которых используются метод Complete Genomics (Mountain View, CA), перед циркуляризацией проводят последовательные раунды лигирования адаптера, амплификации и расщепления для получения конструкций «голова к хвосту», имеющих несколько фрагментов геномной ДНК, разделенных последовательностями адаптера.
[00190] Репрезентативными способами изотермической амплификации, которые могут быть применены в способе раскрытия изобретения, являются, но не ограничиваются ими, амплификация с множеством замен (MDA), описанная, например, Dean et al., Proc. Natl. Акад. Sci. USA 99: 5261-66 (2002), или изотермическая амплификация нуклеиновых кислот с вытеснением цепи, например, описанная в патенте США No. 6214587. Другие методы, которые не основаны на ПЦР и могут быть применены в способе раскрытия изобретения, являются, например, амплификация с вытеснением цепи (SDA), описанная, например, Walker et al., Molecular Methods for Detection Detection, Academic Press, Inc., 1995; в патентах США №№5455166 и 5130238 и Walker et al., Nucl. Acids Res. 20: 1691-96 (1992), или амплификация с вытеснением гиперразветвленной цепи, описанная, например, Lage et al., Genome Res. 13: 294-307 (2003). Методы изотермической амплификации могут быть проведены, например, с использованием полимеразы Phi 29 для вытеснения цепи, или с использованием крупного фрагмента ДНК-полимеразы Bst, 5'→3'-экзо для рандомизированной амплификации геномной ДНК с использованием праймеров. Преимущество использования этих полимераз заключается в их высокой эффективности и активности вытеснения цепи. Высокая эффективность позволяет полимеразам образовывать фрагменты длиной 10-20 т.п.о. Как указывалось выше, более мелкие фрагменты могут быть получены в изотермических условиях с использованием полимераз, имеющих низкую эффективность и активность вытеснения цепи, таких как полимераза Кленова. Дополнительное более подробное описание реакций амплификации и их условий и компонентов приводится в патенте США №7670810.
[00191] Другим способом амплификации полинуклеотидов, который применяется в настоящем изобретении, является ПЦР-мечение, где используется популяция двухдоменных праймеров, имеющих константную 5'-область, за которой следует рандомизированная 3'-область, как описано, например, Grothues et al. Nucleic Acids Res. 21 (5):1321-2 (1993). Первые раунды амплификации проводят для создания множества сайтов инициации на термоденатурированной ДНК на основе отдельной гибридизации исходя из случайно синтезированной 3'-области. Исходя из природы 3'-области, было сделано предположение, что сайты инициации имеют случайное распределение по всему геному. После этого, несвязанные праймеры могут быть удалены, и дальнейшая репликация может быть проведена с использованием праймеров, комплементарных константной 5'-области.
[00192] В некоторых вариантах осуществления изобретения, изотермическая амплификация может быть осуществлена посредством амплификации с кинетическим исключением (KEA), также называемой амплификацией с исключением (ExAmp). Библиотека нуклеиновых кислот согласно изобретению может быть получена методом, включающим стадию взаимодействия реагента для амплификации с получением множества сайтов амплификации, каждый из которых включает по существу клональную популяцию ампликонов из отдельной нуклеиновой кислоты-мишени, которая занимает этот сайт. В некоторых вариантах осуществления изобретения, реакция амплификации продолжается до тех пор, пока не будет образовано достаточное количество ампликонов для заполнения соответствующего сайта амплификации. Заполнение уже занятого сайта до такого уровня препятствует закреплению и амплификации нуклеиновых кислот-мишеней в этом сайте, что приводит к образованию клональной популяции ампликонов в этом сайте. В некоторых вариантах осуществления изобретения, кажущаяся клональность может быть достигнута, даже если сайт амплификации не заполнен до его полной емкости еще до закрепления второй нуклеиновой кислоты в этом сайте. При некоторых условиях, амплификация первой нуклеиновой кислоты-мишени может продолжаться до того момента, когда будет образовано достаточное число копий, которое могло бы в высокой степени превзойти или подавить образование копий второй нуклеиновой кислоты-мишени, которая транспортируется в этот сайт. Так, например, в варианте осуществления изобретения, в котором применяется способ мостиковой амплификации на кольцевом элементе диаметром менее 500 нм, было определено, что после 14 циклов экспоненциальной амплификации первой целевой нуклеиновой кислоты-мишени, контаминация второй нуклеиновой кислотой-мишенью на том же самом сайте будет давать количество контаминирующих ампликонов, которое будет недостаточным для неблагоприятного влияния на секвенирующий анализ посредством синтеза на платформе Illumina для секвенирования.
[00193] В некоторых вариантах осуществления изобретения, сайты амплификации в массиве могут быть, но не обязательно, полностью клональными. Скорее всего, для некоторых применений, отдельный сайт амплификации может быть преимущественно заполнен ампликонами из первого проиндексированного фрагмента и может также иметь низкий уровень контаминирующих ампликонов из второй нуклеиновой кислоты-мишени. Массив может включать один или более сайтов амплификации, которые имеют низкий уровень контаминирующих ампликонов, при условии, что уровень контаминации не будет оказывать негативное влияние на последующее использование массива. Так, например, если массив используется для детектирования, то приемлемым уровнем контаминации будет уровень, который не будет негативно влиять на отношение сигнал/шум или на разрешение метода детектирования. В соответствии с этим, кажущаяся клональность будет, как обычно, иметь отношение к конкретному использованию или применению массива, полученного описанными здесь способами. Типичные уровни контаминации, которые могут быть приемлемыми в отдельном сайте амплификации для конкретных применений, включают, но не ограничиваются ими, максимум 0,1%, 0,5%, 1%, 5%, 10% или 25% контаминирующих ампликонов. Массив может включать один или более сайтов амплификации, имеющих эти репрезентативные уровни контаминирующих ампликонов. Так, например, до 5%, 10%, 25%, 50%, 75% или даже 100% сайтов амплификации в массиве могут иметь несколько контаминирующих ампликонов. При этом очевидно, что в массиве или в другой совокупности сайтов, по меньшей мере 50%, 75%, 80%, 85%, 90%, 95% или 99% или более сайтов могут быть клональными или могут иметь кажущуюся клональность.
[00194] В некоторых вариантах осуществления изобретения, кинетическое исключение может происходить в том случае, когда процесс происходит с достаточно высокой скоростью и позволяет эффективно исключать другое событие или другой процесс. Так, например, можно создать массив нуклеиновых кислот, в котором сайты массива случайным образом заполняются индексированными фрагментами из раствора, а копии индексированных фрагментов образуются в процессе амплификации так, чтобы каждый из фрагментов полностью заполнял эти сайты. В соответствии со способами кинетического исключения согласно изобретению, процессы заполнения и амплификации могут происходить одновременно в условиях, при которых скорость амплификации превышает скорость заполнения. Так, например, относительно высокая скорость, с которой происходит образование копий в сайте, заполненном первой нуклеиновой кислотой-мишенью, будет эффективно исключать заполнение сайта амплификации второй нуклеиновой кислотой. Способы амплификации с кинетическим исключением могут быть осуществлены способом, подробно описанным в публикации заявки на патент США №2013/0338042.
[00195] При кинетическом исключении может использоваться относительно низкая скорость инициации амплификации (например, низкая скорость создания первой копии индексированного фрагмента) по сравнению с относительно высокой скоростью создания последующих копий индексированного фрагмента (или первой копии индексированного фрагмента). В примере, описанном в предыдущем абзаце, кинетическое исключение происходит из-за относительно низкой скорости заполнения индексированного фрагмента (например, относительно медленной диффузии или транспорта) по сравнению с относительно высокой скоростью, с которой происходит амплификация с заполнением сайта копиями индексированного фрагмента. В другом иллюстративном варианте осуществления изобретения, кинетическое исключение может происходить из-за замедления образования первой копии индексированного фрагмента, который заполняет сайт (например, отложенная или медленная активация) по сравнению с относительно высокой скоростью заполнения этого сайта последующими копиями. В этом примере, отдельный сайт может быть заполнен несколькими различными индексированными фрагментами (например, несколько индексированных фрагментов может присутствовать в каждом сайте до амплификации). Однако, образование первой копии любого данного индексированного фрагмента может быть активировано случайным образом, в результате чего средняя скорость формирования первой копии является относительно низкой по сравнению со скоростью, с которой генерируются последующие копии. В этом случае, хотя отдельный сайт может быть заполнен несколькими различными индексированными фрагментами, однако, кинетическое исключение позволяет амплифицировать только один из этих индексированных фрагментов. Более конкретно, после активации первого индексированного фрагмента для амплификации, сайт будет быстро заполняться копиями до полной емкости, что будет предотвращать создание копий второго индексированного фрагмента в этом сайте.
[00196] В одном варианте осуществления изобретения осуществляют способ одновременного (i) транспорта индексированных фрагментов в сайты амплификации со средней скоростью транспорта и (ii) амплификации индексированных фрагментов, которые присутствуют в сайтах амплификации, со средней скоростью амплификации, где средняя скорость амплификации превышает среднюю скорость транспорта (патент США №9166913). Соответственно, в таких вариантах осуществления изобретения, кинетическое исключение может быть достигнуто с использованием относительно низкой скорости транспорта. Так, например, для достижения желаемой средней скорости транспорта может быть выбрана достаточно низкая концентрация индексируемых фрагментов, причем, чем ниже концентрации, тем ниже средняя скорость трансфорта. Альтернативно или дополнительно, для снижения скорости транспорта может быть использован раствор с высокой вязкостью, и/или в растворе могут присутствовать реагенты для молекулярного загущения. Примеры подходящих реагентов для молекулярного загущения включают, но не ограничиваются ими, полиэтиленгликоль (ПЭГ), фиколл, декстран или поливиниловый спирт. Репрезентативные реагенты и препараты для молекулярного загущения описаны в патенте США №7399590, который вводится в настоящее описание посредством ссылки. Другим фактором, который может быть скорректирован для достижения желаемой скорости транспорта, является средний размер нуклеиновых кислот-мишеней.
[00197] Реагент для амплификации может включать дополнительные компоненты, которые облегчают образование ампликона, а в некоторых случаях, увеличивают скорость образования ампликона. Примером является рекомбиназа. Рекомбиназа может облегчать образование ампликона посредством повторяющегося проникновения/удлинения. Более конкретно, рекомбиназа может облегчать проникновение индексируемого фрагмента под действием полимеразы, и удлинение праймера под действием полимеразы с помощью индексированного фрагмента, используемого в качестве матрицы для образования ампликона. Этот процесс может быть повторен как цепная реакция, где ампликоны, образующиеся после каждого раунда проникновения/удлинения, служат в качестве матриц в последующем раунде. Этот процесс может происходить быстрее, чем стандартная ПЦР, поскольку в данном случае не требуется проведения цикла денатурации (например, тепловой или химической денатурации). Таким образом, амплификация, инициируемая рекомбиназой, может быть осуществлена в изотермических условиях. Обычно, для облегчения амплификации, в реагент для амплификации, стимулируемый рекомбиназой, желательно включать АТФ или другие нуклеотиды (или в некоторых случаях, их негидролизуемые аналоги). Смесь рекомбиназы и одноцепочечного связывающегося белка (SSB) являются особенно подходящей, поскольку SSB может также стимулировать амплификацию. Типичными препаратами для амплификации, облегчаемой рекомбиназой, являются препараты, которые являются коммерчески доступными и поставляются в виде наборов TwistAmp от TwistDx (Cambridge, UK). Подходящие компоненты реагента для амплификации, облегчаемой рекомбиназой, и условия реакции описаны в патентах США №№5223414 и 7399590.
[00198] Другим примером компонента, который может быть включен в состав реагента для амплификации в целях облегчения образования ампликона, а в некоторых случаях, увеличения скорости образования ампликона, является геликаза. Геликаза может облегчать образование ампликона под действием цепной реакции образования ампликона. Этот процесс может происходить быстрее, чем стандартная ПЦР, поскольку в данном случае не требуется проведения цикла денатурации (например, тепловой или химической денатурации). Таким образом, амплификация, инициируемая геликазой, может быть осуществлена в изотермических условиях. Смесь геликазы и одноцепочечного связывающегося белка (SSB) являются особенно подходящей, поскольку SSB может также стимулировать амплификацию. Типичными препаратами для амплификации, облегчаемой геликазой, являются коммерчески доступные препараты, которые поставляются в виде наборов IsoAmp от Biohelix (Beverly, MA). Кроме того, примеры подходящих препаратов, которые включают белок геликазу, описаны в патентах США №№7399590 и 7829284.
[00199] Еще один пример компонента, который может быть включен в амплифицирующий реагент для облегчения образования ампликона, а в некоторых случаях, для увеличения скорости образования ампликона, представляет собой ориджин-связывающий белок.
Использование в секвенировании/Методы секвенирования
[00200] После прикрепления индексированных фрагментов к поверхности определяют последовательность иммобилизованных и амплифицированных индексированных фрагментов. Секвенирование может быть осуществлено любым подходящим методом секвенирования, и способы определения последовательности иммобилизованных и амплифицированных индексированных фрагментов, включая повторный синтез цепи, известны специалистам и описаны, например, Bignell et al. (патент США 8, 053, 192), Gunderson et al. (WO 2016/130704), Shen et al. (патент США 8895249) и Pipenburg et al. (патент США 9309502).
[00201] Описанные здесь способы могут быть применены в комбинации с различными методами секвенирования нуклеиновых кислот. Особенно подходящими методами являются методы, где нуклеиновые кислоты присоединяются в фиксированных положениях в массиве, таким образом, что их относительные положения не изменяются, и методы, где получают несколько изображений массивов. Особенно подходящими являются варианты, в которых изображения получают в различных цветовых каналах, например, совпадающих с различными метками, используемыми для того, чтобы отличить нуклеотидное основание одного типа от другого. В некоторых вариантах осуществления изобретения, способ определения нуклеотидной последовательности индексированного фрагмента может быть автоматизированным. Предпочтительные варианты осуществления изобретения включают методы секвенирования посредством синтеза («SBS»).
[00202] Методы SBS обычно включают ферментативное удлинение растущей цепи нуклеиновой кислоты посредством итеративного добавления нуклеотидов к матричной цепи. В традиционных методах SBS, единственный нуклеотидный мономер может быть представлен нуклеотиду-мишени в присутствии полимеразы при каждой доставке. Однако, в описанных здесь способах, нуклеотидный мономер более, чем одного типа, может быть представлен нуклеиновой кислоте-мишени в присутствии полимеразы во время доставки.
[00203] В одном варианте осуществления изобретения, нуклеотидный мономер включает блокированные нуклеиновые кислоты (LNA) или мостиковые нуклеиновые кислоты (BNA). Использование LNA или BNA в нуклеотидном мономере повышает эффективность гибридизации между нуклеотидным мономером и секвенирующей последовательностью праймера, присутствующей на иммобилизованном индексированном фрагменте.
[00204] В SBS могут быть использованы нуклеотидные мономеры, которые имеют терминирующую группу, или нуклеотидные мономеры, которые не содержат терминирующих групп. Способы с использованием нуклеотидных мономеров, в которых отсутствуют терминаторы, включают, например, пиросеквенирование и секвенирование с использованием нуклеотидов, меченных γ-фосфатом, как подробно описано далее в настоящей заявке. В способах с использованием нуклеотидных мономеров, не содержащих терминаторов, число нуклеотидов, добавляемых в каждом цикле, обычно является вариабельным и зависит от последовательности матрицы и способа доставки нуклеотидов. Для методов SBS, в которых используются нуклеотидные мономеры, имеющие терминирующую группу, терминатор может быть фактически необратимым в условиях секвенирования как в случае традиционного секвенирования Сэнгера, в котором используются дидезоксинуклеотиды, либо терминатор может быть обратимым, как это имеет место в методах секвенирования, разработанных фирмой Solexa (в настоящее время называемой Illumina, Inc.).
[00205] В методах SBS могут использоваться нуклеотидные мономеры, которые имеют метку, или нуклеотидные мономеры, которые не имеют метки. Соответственно, события включения могут быть детектированы исходя из свойств метки, таких как флуоресценция метки; характерные признаки нуклеотидного мономера, такие как молекулярная масса или заряд; побочный продукт включения нуклеотида, такой как высвобождаемый пирофосфат или т.п. В тех вариантах осуществления изобретения, где два или более различных нуклеотида присутствуют в реагенте для секвенирования, различные нуклеотиды могут отличаться друг от друга, или, альтернативно, две или более различных меток могут быть неразличимыми в применяемых методах обнаружения. Так, например, различные нуклеотиды, присутствующие в реагенте для секвенирования, могут иметь разные метки, и их можно различить с использованием подходящих оптических приборов, описанных в методах секвенирования, разработанных Solexa (в настоящее время называемой Illumina, Inc.).
[00206] Предпочтительные варианты осуществления изобретения включают методы пиросеквенирования. Пиросеквенирование позволяет детектировать высвобождение неорганического пирофосфата (PPi), по мере включения конкретных нуклеотидов в растущую цепь (Ronaghi, М., Karamohamed, S., Pettersson, В., Uhlen, М. and Nyren, P. (1996) «Real-time DNA sequencing using detection of pyrophosphate release». Analytical Biochemistry 242(1), 84-9; Ronaghi, M. (2001) «Pyrosequencing sheds light on DNA sequencing». Genome Res. 11(1), 3-11; Ronaghi, M., Uhlen, M. and Nyren, P. (1998) «A sequencing method based on real-time pyrophosphate». Science 281(5375), 363; патенты США №№6210891; 6258568 и 6274320). При пиросеквенировании, высвобожденный PPi может быть детектирован путем непосредственного превращения в аденозинтрифосфат (АТФ) под действием АТФ-сульфуразы, а уровень генерируемого АТФ определяют с помощью фотонов, продуцируемых люциферазой. Секвенируемые нуклеиновые кислоты могут быть присоединены к элементам в массиве, и этот массив может быть визуализирован для захвата хемилюминесцентных сигналов, которые вырабатываются в результате включения нуклеотидов в элементы массива. Изображение может быть получено после обработки массива нуклеотидом определенного типа (например, А, Т, С или G). Изображения, полученные после добавления нуклеотидов каждого типа, будут отличаться в зависимости от детектируемых элементов в массиве. Эти различия в изображениях отражают различное содержание последовательностей элементов на массиве. Однако, относительные положения каждого элемента остаются неизменными на изображениях. Изображения могут быть сохранены, обработаны и проанализированы описанными здесь методами. Так, например, изображения, полученные после обработки массива каждым из нуклеотидов различных типов, могут обрабатываться таким же образом, как описано здесь для изображений, полученных из различных каналов детектирования в методах секвенирования на основе обратимых терминаторов.
[00207] В другом репрезентативном SBS, циклическое секвенирование осуществляют путем постадийного добавления обратимых нуклеотидов-терминаторов, содержащих, например, отщепляемую метку или фотообесцвечиваемую метку-краситель, как описано, например, в WO 04/018497 и в патенте США No. №7057026. Этот метод коммерциализирован фирмой Solexa (в настоящее время называемой Illumina Inc.), а также описан в WO 91/06678 и WO 07/123,744. Доступность флуоресцентно-меченных терминаторов, в которых оба конца могут быть обратимыми, а флуоресцентная метка отщеплена, способствует эффективному секвенированию посредством циклической обратимой терминации (CRT). Полимеразы могут быть также сконструированы для эффективного включения модифицированных нуклеотидов и удлинения цепи от этих модифицированных нуклеотидов.
[00208] В некоторых вариантах секвенирования на основе обратимого терминатора, метки, по существу, не ингибируют удлинение в условиях реакции SBS. Однако, детектирующие метки могут быть удалены, например, путем отщепления или разложения. Изображения могут быть сделаны после включения меток в матричные элементы нуклеиновых кислот. В конкретных вариантах осуществления изобретения, каждый цикл включает одновременную доставку четырех различных нуклеотидов в массив, и нуклеотид каждого типа имеет спектрально отличающиеся метки. Затем могут быть получены четыре изображения, каждое из которых получают с использованием канала детектирования, специфичного для одной из четырех различных меток. Альтернативно, нуклеотиды различных типов могут быть добавлены последовательно, и изображение массива может быть получено между каждыми стадиями добавления. В таких вариантах осуществления изобретения, каждое изображение будет демонстрировать элементы нуклеиновой кислоты, которые включают нуклеотиды определенного типа. На различных изображениях будут присутствовать или отсутствовать различные элементы, что обусловлено различными составами последовательности каждого элемента. Однако, относительное положение элементов на изображениях остается неизменным. Изображения, полученные с помощью таких методов SBS с использованием обратимого терминатора, могут быть сохранены, обработаны и проанализированы, как описано в настоящей заявке. После стадии получения изображения, метки и молекулы обратимого терминатора могут быть удалены для последующих циклов добавления и детектирования нуклеотидов. Удаление меток после того, как они были детектированы в конкретном цикле и перед последующим циклом, может оказаться предпочтительным с точки зрения снижения фонового сигнала и перекрестных помех между циклами. Примеры подходящих меток и способов удаления приводятся в настоящей заявке.
[00209] В конкретных вариантах осуществления изобретения, некоторые или все нуклеотидные мономеры могут включать обратимые терминаторы. В таких вариантах осуществления изобретения, обратимые терминаторы/отщепляемые флуорофоры могут включать флуорофоры, связанные с рибозной молекулой посредством 3'-сложноэфирной связи (Metzker, Genome Res. 15: 1767-1776 (2005)). В других методах, химический метод с использованием терминатора отличается от метода расщепления флуоресцентной метки (Ruparel et al., Proc Natl Acad Sci USA 102: 5932-7 (2005)). Ruparel и др. описали получение обратимых терминаторов, в которых используется небольшая 3'-аллильная группа для блокирования удлинения, но, при этом, может быть легко осуществлено деблокирование путем короткой обработки палладиевым катализатором. Флуорофор был присоединен к основанию посредством фоторасщепляемого линкера, который легко расщепляется путем 30-секундного облучения длинноволновым УФ-светом. Таким образом, в качестве расщепляемого линкера может быть использован агент для восстановления дисульфида, либо агент для фоторасщепления. Другим методом обратимой терминации является использование природной терминации, которая будет происходить после помещения объемного красителя на dNTP. Присутствие заряженного объемного красителя на dNTP может действовать как эффективный терминатор благодаря стерическому и/или электростатическому затруднению. Наличие одного события включения предотвращает дальнейшие включения, если краситель не удален. Расщепление красителя удаляет флуорофор и эффективно отменяет терминацию. Примеры модифицированных нуклеотидов также описаны в патентах США No. №№7427673 и 7057026.
[00210] Дополнительные репрезентативные системы и методы SBS, которые могут быть применены вместе с описанными здесь способами и системами, описаны в публикациях заявок на патент США №№. 2007/0166705, 2006/0188901, 2006/0240439, 2006/0281109, 2012/0270305 и 2013/0260372, в патенте США 7057026, в публикации РСТ WO 05/065814, в публикация заявки на патент США №2005/0100900 и в публикациях РСТ WO 06/064199 и WO 07/010251.
[00211] В некоторых вариантах осуществления изобретения может применяться детектирование четырех различных нуклеотидов с использованием менее, чем четырех различных меток. Так, например, SBS может быть осуществлен с применением методов и систем, описанных во включенных материалах публикации заявки на патент США 2013/0079232. В первом примере, пара нуклеотидов может быть детектирована на одной и той же длине волны, но с одной лишь разницей, заключающейся в отличии интенсивности для одного члена пары от интенсивности для другого члена пары или в замене одного члена пары (например, посредством химической модификации, фотохимической модификации или физической модификации), что вызывает появление или исчезновение кажущегося сигнала по сравнению с сигналом, обнаруженным для другого члена пары. В другом примере, три из четырех различных нуклеотидов могут быть детектированы при определенных условиях, в то время как нуклеотид четвертого типа не содержит метку, которая может быть детектирована в этих условиях, или детектируется на минимальном уровне в этих условиях (например, детектируется на минимальном уровне из-за фоновой флуоресценции и т.п.). Включение первых трех нуклеотидов в нуклеиновую кислоту может быть определено по присутствию соответствующих сигналов, а включение нуклеотида четвертого типа в нуклеиновую кислоту может быть определено по отсутствию детектирования или детектированию любого сигнала на минимальном уровне. В третьем примере, нуклеотид одного типа может включать метку(и), которая(ые) детектируется(ются) в двух различных каналах, тогда как нуклеотиды других типов детектируются не более, чем в одном из каналов. Вышеупомянутые три репрезентативные конфигурации не считаются взаимоисключающими и могут использоваться в различных комбинациях. Репрезентативный вариант, который объединяет все три примера, представляет собой метод SBS на основе флуоресценции, в котором используется нуклеотид первого типа, детектируемый в первом канале (например, dATP, имеющий метку, которая детектируется в первом канале при возбуждении на первой длине волны возбуждения), нуклеотид второго типа, который детектируется во втором канале (например, dCTP, имеющий метку, которая детектируется во втором канале при возбуждении на второй длине волны возбуждения), нуклеотид третьего типа, который детектируется в первом и во втором канале (например, dTTP, имеющий по меньшей мере одну метку, которая детектируется в обоих каналах при возбуждении на первой и/или второй длине волны возбуждения), и нуклеотид четвертого типа, который не содержит метки, то есть не детектируется или детектируется на минимальном уровне в любом канале (например, dGTP, не имеющий метки).
[00212] Далее, как описано во включенных материалах публикации заявки на патент США №2013/0079232, данные секвенирования могут быть получены с использованием одного канала. В таких так называемых методах секвенирования с использованием одного красителя, нуклеотид первого типа метят, но метку удаляют после получения первого изображения, а нуклеотид второго типа метят только после получения первого изображения. Нуклеотид третьего типа сохраняет свою метку как на первом, так и на втором изображениях, а нуклеотид четвертого типа остается немеченным на обоих изображениях.
[00213] В некоторых вариантах осуществления изобретения могут быть проведено секвенирование методами лигирования. В таких методах используют ДНК-лигазу для включения олигонуклеотидов и идентификации включения таких олигонуклеотидов. Олигонуклеотиды обычно имеют различные метки, которые коррелируют с идентичностью конкретного нуклеотида в последовательности, с которой гибридизуются эти олигонуклеотиды. Как и в случае других методов SBS, изображения могут быть получены после обработки массива элементов нуклеиновой кислоты мечеными реагентами для секвенирования. Каждое изображение будет показывать элементы нуклеиновой кислоты, имеющие включенные метки конкретного типа. Различные элементы будут присутствовать или отсутствовать на различных изображениях из-за разного содержания последовательности каждого элемента, но относительное положение элементов на этих изображениях останется неизменным. Изображения, полученные методами секвенирования на основе лигирования, могут быть сохранены, обработаны и проанализированы, как описано в настоящей заявке. Репрезентативные системы и методы SBS, которые могут быть применены вместе с описанными здесь методами и системами, описаны в патентах США №№6969488, 6172218 и 6306597.
[00214] В некоторых вариантах осуществления изобретения может быть проведено секвенирование нанопор (Deamer, D. W. & Akeson, М. "Nanopores and nucleic acids: prospects for ultrarapid sequencing." Trends Biotechnol. 18, 147-151 (2000); Deamer, D. and D. Branton, "Characterization of nucleic acids by nanopore analysis", Acc. Chem. Res. 35:817-825 (2002); Li, T, M. Gershow, D. Stein, E. Brandin, and J. A. Golovchenko, "DNA molecules and configurations in a solid-state nanopore microscope" Nat. Mater. 2:611-615 (2003)). В таких вариантах осуществления изобретения, индексированный фрагмент проходит через нанопору. Нанопора может представлять собой синтетическую пору или биологический мембранный белок, такой как α-гемолизин. Если индексируемый фрагмент проходит через нанопору, то каждая пара оснований может быть идентифицирована путем измерения колебаний электропроводности поры (патент США No. 7001792; Soni, G. V. & Meller, «A. Progress toward ultrafast DNA sequencing using solid-state nanopores». Clin. Chem. 53, 1996-2001 (2007); Healy, K. «Nanopore-based single-molecule DNA analysis». Nanomed. 2, 459-481 (2007); Cockroft, S. L., Chu, J., Amorin, M. & Ghadiri, M. R. «A single-molecule nanopore device detects DNA polymerase activity with single-nucleotide resolution». J. Am. Chem. Soc. 130, 818-820 (2008)). Данные, полученные после секвенирования нанопор, могут быть сохранены, обработаны и проанализированы, как описано в настоящей заявке. В частности, данные могут быть обработаны как изображение в соответствии с репрезентативной обработкой оптических изображений и других изображений, описанных в настоящей заявке.
[00215] В некоторых вариантах осуществления изобретения могут применяться способы, включающие мониторинг активности ДНК-полимеразы в реальном времени. Включения нуклеотидов могут быть детектированы по взаимодействиям методами переноса флуоресцентной резонансной энергии (FRET) между флуорофор-содержащей полимеразой и γ-фосфат-меченными нуклеотидами, как описано, например, в патентах США No. 7329492 и 7211414, или включения нуклеотидов могут быть детектированы с помощью волноводов нулевой моды, как описано, например, в патенте США No. 7315019, и с использованием флуоресцентных нуклеотидных аналогов и сконструированных полимераз, как описано, например, в патенте США No. 7405281 и в публикации заявки на патент США №2008/0108082. Облучение может быть ограничено объемом в масштабе гептолитров в окружении поверхностно-связанной полимеразы, так, чтобы включение флуоресцентно меченных нуклеотидов могло наблюдаться на низком фоновом уровне (Levene, М. J. et al. «Zero-mode waveguides for single molecule analysis at high concentrations», Science 299, 682-686 (2003); Lundquist, P. M. et al. «Parallel confocal detection of single molecules in real time», Opt. Lett. 33, 1026-1028 (2008); Korlach, J. et al., «Selective aluminum passivation for targeted immobilization of single DNA polymerase molecules in zero-mode waveguide nano structures)), Proc. Natl. Acad. Sci. USA 105, 1176-1181 (2008)). Изображения, полученные такими способами, могут быть сохранены, обработаны и проанализированы, как описано в настоящей заявке.
[00216] Некоторые варианты SBS включают детектирование протона, высвобождаемого после включения нуклеотида в продукт удлинения. Так, например, при секвенировании на основе детектирования высвобождаемых протонов могут быть использованы электрический детектор и методы с его применением, которые являются коммерчески доступными и разработаны Ion Torrent (Guilford, СТ, a Life Technologies subsidiary) или методы и системы секвенирования, описанные в публикациях заявок на патент США. №2009/0026082; 2009/0127589; 2 010/0137143; и 2010/0282617. Описанные здесь способы амплификации нуклеиновых кислот-мишеней посредством кинетического исключения, могут быть легко применены к субстратам, используемым для обнаружения протонов. Более конкретно, описанные здесь способы могут быть применены для получения клональных популяций ампликонов, которые используются для обнаружения протонов.
[00217] Вышеуказанные способы SBS могут быть преимущественно осуществлены в мультиплексном формате, так, чтобы множество различных индексированных фрагментов было модифицировано одновременно. В конкретных вариантах осуществления изобретения, различные индексированные фрагменты могут быть обработаны в общем реакционном сосуде или на поверхности конкретной основы. Это облегчает доставку реагентов для секвенирования, удаление непрореагировавших реагентов и детектирование событий включения мультиплексным способом. В вариантах осуществления изобретения, в которых используются связанные с поверхностью нуклеиновые кислоты-мишени, индексированные фрагменты могут иметь формат массива. В формате массива, индексированные фрагменты обычно могут быть связаны с поверхностью различимыми способами широкого ряда. Индексированные фрагменты могут быть связаны посредством прямой ковалентной связи, путем прикрепления к сфере или к другой частице или путем связывания с полимеразой или с другой молекулой, которая связана с поверхностью. Массив может включать одну копию индексированного фрагмента на каждом сайте (также называемого элементом), либо несколько копий, имеющих одну и ту же последовательность, могут присутствовать на каждом сайте или в каждом элементе. Множество копий может быть получено методами амплификации, такими как мостиковая амплификация или эмульсионная ПЦР, как описано более подробно в настоящей заявке.
[00218] В описанных здесь способах могут быть использованы массивы, имеющие элементы с различными плотностями, включая, например, по меньшей мере приблизительно 10 элементов/см2, 100 элементов/см2, 500 элементов/см2, 1000 элементов/см2, 5000 элементов/см2, 10000 элементов/см2, 50000 элементов/см2, 100000 элементов/см2, 1000000 элементов/см2, 5000000 элементов/см2 или более.
[00219] Преимущество описанных здесь способов заключается в том, что они одновременно обеспечивают быстрое и эффективное детектирование множества элементов на см2. Соответственно, настоящее изобретение относится к интегрированным системам, позволяющим получать и детектировать нуклеиновые кислоты методами, известными специалистам, такими как методы, описанные в настоящей заявке. Таким образом, интегрированная система согласно изобретению может включать жидкие компоненты, способные доставлять реагенты для амплификации и/или секвенирующие реагенты к одному или более иммобилизованным индексированным фрагментам, где указанная система включает такие компоненты, как насосы, клапаны, резервуары, поточные линии и т.п. Проточная кювета может быть сконфигурирована и/или использована в интегрированной системе для обнаружения нуклеиновых кислот-мишеней. Репрезентативные проточные кюветы описаны, например, в публикации заявок на патент США №2010/0111768 и №13/273666. Как проиллюстрировано для проточных кювет, один или более жидких компонентов интегрированной системы могут быть использованы в способе амплификации и детектирования. В качестве примера может служить вариант секвенирования нуклеиновых кислот, где один или более жидких компонентов интегрированной системы могут быть использованы в описанном здесь способе амплификации и для доставки секвенирующих реагентов в способе секвенирования, таком как способ, проиллюстрированный выше. Альтернативно, интегрированная система может включать отдельные жидкостные системы для осуществления методов амплификации и методов детектирования. Примерами интегрированных систем секвенирования, подходящих для получения амплифицированных нуклеиновых кислот, а также для определения последовательности нуклеиновых кислот являются, но не ограничиваются ими, платформа MiSeqTM (Illumina, Inc., San Diego, CA) и устройства, описанные в патенте США рег. No. 13/273666.
[00220] Настоящее изобретение также относится к композициям. При практическом применении описанных здесь способов могут быть получены различные композиции. Так, например, может быть получена композиция, включающая индексированные фрагменты нуклеиновой кислоты, где индексированные фрагменты нуклеиновой кислоты происходят от вновь синтезированной РНК. В одном варианте осуществления изобретения, вновь синтезированная РНК является меченной. Настоящее изобретение также относится к многолуночному планшету, где лунка многолуночного планшета содержит индексированные фрагменты нуклеиновой кислоты.
[00221] Настоящее изобретение также относится к наборам. В одном варианте осуществления изобретения, набор предназначен для получения секвенирующей библиотеки, где вновь синтезированная РНК является меченной. В другом варианте осуществления изобретения, набор включает описанную здесь нуклеотидную метку. В другом варианте осуществления изобретения, набор включает один или более праймеров для отжига РНК, где по меньшей мере один праймер предназначен для направленной амплификации одной или более предварительно определенных нуклеиновых кислот. В дополнительном варианте осуществления изобретения, набор включает компоненты для добавления по меньшей мере трех индексов к нуклеиновым кислотам. Набор также может включать другие компоненты, подходящие для получения секвенирующей библиотеки. Так, например, набор может включать по меньшей мере один фермент, который опосредует лигирование, удлинение праймера или амплификацию для обработки молекул РНК в целях включения индекса. Набор может включать нуклеиновые кислоты с индексными последовательностями. Набор также может включать другие компоненты, подходящие для добавления индекса к нуклеиновой кислоте, такие как транспосомный комплекс. Набор также может включать один или более праймеров для отжига РНК. Праймеры могут быть предназначены для получения всего транскриптома (например, праймер, который включает poly-T-последовательность) или для нацеленной амплификации одной или более предварительно определенных нуклеиновых кислот.
[00222] Компоненты набора в подходящем упаковочном материале обычно присутствуют в количестве, достаточном для проведения по меньшей мере одного анализа или его применения. При этом, могут быть включены, но необязательно, и другие компоненты, такие как буферы и растворы. Обычно также включены инструкции по применению упакованных компонентов. Используемый здесь термин «упаковочный материал» означает одну или более физических структур, используемых для размещения содержимого набора. Упаковочный материал изготавливают рутинными методами, обычно для обеспечения стерильной среды без примесей. Упаковочный материал может иметь этикетку, на которой указано, что компоненты могут быть использованы для получения секвенирующей библиотеки. Кроме того, упаковочный материал содержит инструкции по применению материалов, содержащихся в наборе. Используемый здесь термин «упаковка» означает контейнер из таких материалов, как стекло, пластик, бумага, фольга и т.п., способных сохранять компоненты набора в фиксированных пределах. «Инструкции по применению» обычно включают общеизвестное выражение, описывающее концентрацию реагента или по меньшей мере один из параметров аналитического метода, таких как относительные количества реагента и образца, подлежащих смешиванию, время хранения смесей реагента/образца, температура, буферные условия и т.п.
Репрезентативны варианты осуществления изобретения
[00223] Вариант 1. Способ получения библиотеки для секвенирования, содержащей нуклеиновые кислоты, происходящие из множества отдельных ядер или клеток, где указанный способ включает:
(a) получение множества ядер или клеток в первом множестве компартментов,
где каждый компартмент содержит субпопуляцию ядер или клеток;
(b) мечение вновь синтезированной РНК в субпопуляциях клеток или ядер, полученных из клеток;
(c) обработку молекул РНК в каждой субпопуляции ядер или клеток с получением индексированных ядер или клеток,
где обработка включает добавление к нуклеиновым кислотам РНК, присутствующим в каждой субпопуляции ядер или клеток, первой компартмент-специфической индексной последовательности с получением индексированных нуклеиновых кислот ДНК, присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию или амплификацию; и
(d) объединение индексированных ядер или клеток с получением объединенных индексированных ядер или клеток.
[00224] Вариант 2. Способ Варианта 1, где обработка включает:
контактирование субпопуляций с обратной транскриптазой и праймером, которые гибридизуются с нуклеиновыми кислотами РНК, с образованием двухцепочечных нуклеиновых кислот ДНК, содержащих праймер, и соответствующей нуклеотидной последовательности ДНК из матричных молекул РНК.
[00225] Вариант 3. Способ вариантов 1 или 2, где праймер содержит поли-Т-нуклеотидную последовательность, которая гибридизуется с поли(А)-хвостом мРНК.
[00226] Вариант 4. Способ любого из вариантов 1-3, где обработка также включает контактирование субпопуляций со вторым праймером, где второй праймер содержит последовательность, которая гибридизуется с предварительно определенной нуклеиновой кислотой ДНК.
[00227] Вариант 5. Способ любого из вариантов 1-4, где второй праймер содержит компартмент-специфический индекс.
[00228] Вариант 6. Способ любого из вариантов 1-5, где праймер содержит последовательность, которая гибридизуется с предварительно определенной нуклеиновой кислотой РНК.
[00229] Вариант 7. Способ любого из вариантов 1-6, где способ включает праймеры в различных компартментах, которые гибридизуются с различными нуклеотидами одной и той же предварительно определенной нуклеиновой кислоты РНК.
[00230] Вариант 8. Способ любого из вариантов 1-7, где праймер включает праймер переключения матрицы.
[00231] Вариант 9. Способ любого из вариантов 1-7, где обработка для добавления первой компартмент-специфической индексной последовательности включает двухстадийный способ добавления нуклеотидной последовательности, содержащей
универсальную последовательность, к нуклеиновым кислотам РНК с получением нуклеиновых кислот ДНК, а затем добавление первой компартмент-специфической индексной последовательности к нуклеиновым кислотам ДНК.
[00232] Вариант 10. Способ получения библиотеки для секвенирования, содержащей нуклеиновые кислоты, происходящие из множества отдельных ядер или клеток, где указанный способ включает:
(a) получение множества ядер или клеток в первом множестве компартментов,
где каждый компартмент содержит субпопуляцию ядер или клеток;
(b) контактирование каждой субпопуляции с обратной транскриптазой и праймером, которые гибридизуются с предварительно определенной нуклеиновой кислотой РНК, с образованием двухцепочечных нуклеиновых кислот ДНК, содержащих праймер, и соответствующей нуклеотидной последовательности ДНК из матричных молекул РНК;
(c) обработку молекул ДНК в каждой субпопуляции ядер или клеток с получением индексированных ядер или клеток,
где обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в каждой субпопуляции ядер или клеток, первой компартмент-специфической индексной последовательности с получением индексированных нуклеиновых кислот ДНК,
присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию или амплификацию; и
(d) объединение индексированных ядер или клеток с получением объединенных индексированных ядер или клеток.
[00233] Вариант 11. Способ варианта 10, где праймер содержит первую компартмент-специфическую индексную последовательность.
[00234] Вариант 12. Способ вариантов 10 или 11, дополнительно включающий, перед контактированием, мечение вновь синтезированной РНК в субпопуляциях клеток или ядер, выделенных из клеток.
[00235] Вариант 13. Способ любого из вариантов 10-12, где обработка для добавления первой компартмент-специфической индексной последовательности включает двухстадийный способ добавления нуклеотидной последовательности, содержащей универсальную последовательность, к нуклеиновым кислотам, а затем добавление первой компартмент-специфической индексной последовательности к нуклеиновым кислотам.
[00236] Вариант 14. Способ любого из вариантов 1-13, где предварительно определенная нуклеиновая кислота РНК представляет собой мРНК.
[00237] Вариант 15. Способ любого из вариантов 1-14, где уже существующие нуклеиновые кислоты РНК и вновь синтезированные нуклеиновые кислоты РНК помечены одинаковыми индексами в одном и том же компартменте.
[00238] Вариант 16. Способ любого из вариантов 1-15, где мечение включает инкубирование множества ядер или клеток в композиции, содержащей нуклеотидную метку, где нуклеотидная метка включена во вновь синтезированную РНК.
[00239] Вариант 17. Способ любого из вариантов 1-16, где нуклеотидная метка содержит нуклеотидный аналог, меченный гаптеном нуклеотид, мутагенный нуклеотид или нуклеотид, который может быть модифицирован посредством химической реакции.
[00240] Вариант 18. Способ любого из вариантов 1-17, где более чем одна нуклеотидная метка включена во вновь синтезированную РНК.
[00241] Вариант 19. Способ любого из вариантов 1-18, где отношения нуклеотидной метки или меток отличаются для различных компартментов или моментов времени.
[00242] Вариант 20. Способ любого из вариантов 1-19, также включающий обработку субпопуляций ядер или клеток в предварительно определенных условиях перед мечением.
[00243] Вариант 21. Способ любого из вариантов 1-20, где предварительно определенное условие включает обработку агентом.
[00244] Вариант 22. Способ любого из вариантов 1-21, где агент включает белок, не-рибосомный белок, поликетид, органическую молекулу, неорганическую молекулу, молекулу РНК или РНКи, углевод, гликопротеин, нуклеиновую кислоту или их комбинацию.
[00245] Вариант 23. Способ любого из вариантов 1-22, где агент включает терапевтическое лекарственное средство.
[00246] Вариант 24. Способ любого из вариантов 1-23, где предварительно определенные условия двух или более компартментов являются различными.
[00247] Вариант 25. Способ любого из вариантов 1-24, где обработку и мечение проводят одновременно или обработку проводят до мечения.
[00248] Вариант 26. Способ любого из вариантов 1-25, дополнительно включающий:
распределение субпопуляций объединенных индексированных ядер или клеток по второму множеству компартментов и добавление к индексированным нуклеиновым кислотам, присутствующим в субпопуляциях ядер или клеток, второй индексной последовательности с получением ядер или клеток с двумя индексами, содержащих фрагменты нуклеиновой кислоты с двумя индексами, где добавление включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию; и
объединение ядер или клеток с двумя индексами с получением объединенных ядер или клеток с двумя индексами.
[00249] Вариант 27. Способ любого из вариантов 1-26, дополнительно включающий:
распределение субпопуляций объединенных ядер или клеток с двумя индексами по третьему множеству компартментов и добавление к индексированным нуклеиновым кислотам, присутствующим в субпопуляциях ядер или клеток, третьей индексной последовательности с получением ядер или клеток с тремя индексами, содержащих фрагменты нуклеиновой кислоты с тремя индексами, где добавление включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию; и
объединение ядер или клеток с тремя индексами с получением объединенных ядер или клеток с тремя индексами.
[00250] Вариант 28. Способ любого из вариантов 1-27, где распределение включает разведение.
[00251] Вариант 29. Способ любого из вариантов 1-27, где распределение включает сортинг.
[00252] Вариант 30. Способ любого из вариантов 1-29, где добавление включает контактирование субпопуляций со шпилечным лигирующим дуплексом в условиях, подходящих для лигирования шпилечного лигирующего дуплекса с концом фрагментов нуклеиновой кислоты, содержащих одну или две индексных последовательности.
[00253] Вариант 31. Способ любого из вариантов 1-30, где добавление включает контактирование фрагментов нуклеиновой кислоты, содержащих одну или более индексных последовательностей, с транспосомным комплексом, где транспосомный комплекс в компартментах содержит транспозазу и универсальную последовательность, где контактирование также включает условия, подходящие для фрагментации фрагментов нуклеиновой кислоты и включения универсальной последовательности во фрагменты нуклеиновой кислоты.
[00254] Вариант 32. Способ любого из вариантов 1-31, где добавление включает лигирование первой компартмент-специфической индексной последовательности, а также добавление второй индексной последовательности с получением ядер или клеток с двумя индексами, содержащих фрагменты нуклеиновой кислоты с двумя индексами, где добавление включает транспозицию.
[00255] Вариант 33. Способ любого из вариантов 1-32, где добавление включает лигирование второй компартмент-специфической индексной последовательности, а также добавление третьей индексной последовательности с получением ядер или клеток с двумя индексами, содержащих фрагменты нуклеиновой кислоты с тремя индексами, где добавление включает транспозицию.
[00256] Вариант 34. Способ любого из вариантов 1-33, где компартмент включает лунку или каплю.
[00257] Вариант 35. Способ любого из вариантов 1-34, где компартменты первого множества компартментов содержат от 50 до 100000000 ядер или клеток.
[00258] Вариант 36. Способ любого из вариантов 1-35, где компартменты второго множества компартментов содержат от 50 до 100000000 ядер или клеток.
[00259] Вариант 37. Способ любого из вариантов 1-36, где компартменты третьего множества компартментов содержат от 50 до 100000000 ядер или клеток.
[00260] Вариант 38. Способ любого из вариантов 1-37, дополнительно включающий получение индексированных нуклеиновых кислот из объединенных индексированных ядер или клеток, с получением библиотеки для секвенирования из множества ядер или клеток.
[00261] Вариант 39. Способ любого из вариантов 1-38, дополнительно включающий получение нуклеиновых кислот с двумя индексами из объединенных ядер или клеток с двумя индексами, с получением библиотеки для секвенирования из множества ядер или клеток.
[00262] Вариант 40. Способ любого из вариантов 1-39, дополнительно включающий получение нуклеиновых кислот с тремя индексами из объединенных ядер или клеток с тремя индексами, с получением библиотеки для секвенирования из множества ядер или клеток.
[00263] Вариант 41. Способ любого из вариантов 1-40, дополнительно включающий:
получение поверхности, содержащей множество сайтов амплификации,
где сайты амплификации включают по меньшей мере две популяции связанных одноцепочечных олигонуклеотидов для захвата, имеющих свободный 3'-конец, и
контактирование поверхности, содержащей сайты амплификации, с фрагментами нуклеиновой кислоты, содержащими одну, две или три индексных последовательности, в условиях, подходящих для получения множества сайтов амплификации, каждый из которых содержит клональную популяцию ампликонов, из отдельного фрагмента, содержащего множество индексов.
[00264] Вариант 42. Способ любого из вариантов 1-41, где добавление компартмент-специфической индексной последовательности включает двухстадийный способ добавления нуклеотидной последовательности, содержащей универсальную последовательность, к нуклеиновым кислотам, а затем добавление первой компартмент-специфической индексной последовательности к нуклеиновым кислотам.
[00265] Вариант 43. Способ получения библиотеки для секвенирования, содержащей нуклеиновые кислоты, происходящие из множества отдельных ядер или клеток, где указанный способ включает:
(a) получение множества ядер или клеток в первом множестве компартментов,
где каждый компартмент содержит субпопуляцию ядер или клеток;
(b) контактирование каждой субпопуляции с обратной транскриптазой и праймером с получением двухцепочечных нуклеиновых кислот ДНК, содержащих праймер, и соответствующей нуклеотидной последовательности ДНК из матричных молекул РНК;
(c) обработку молекул ДНК в каждой субпопуляции ядер или клеток с получением индексированных ядер или клеток,
где обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в каждой субпопуляции ядер или клеток, первой компартмент-специфической индексной последовательности с получением индексированных нуклеиновых кислот, присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию;
(d) объединение индексированных ядер или клеток с получением объединенных индексированных ядер или клеток;
(e) распределение объединенных индексированных ядер или клеток по второму множеству компартментов, где каждый компартмент содержит субпопуляцию ядер или клеток;
(f) обработку молекул ДНК в каждой субпопуляции ядер или клеток с получением ядер или клеток с двумя индексами,
где обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в каждой субпопуляции ядер или клеток, второй компартмент-специфической индексной последовательности с получением нуклеиновых кислот с двумя индексами, присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию;
(g) объединение ядер или клеток с двумя индексами с получением объединенных ядер или клеток с двумя индексами;
(h) распределение объединенных ядер или клеток с двумя индексами по третьему множеству компартментов,
где каждый компартмент содержит субпопуляцию ядер или клеток;
(i) обработку молекул ДНК в каждой субпопуляции ядер или клеток с получением ядер или клеток с тремя индексами,
где обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в каждой субпопуляции ядер или клеток, третьей компартмент-специфической индексной последовательности с получением нуклеиновых кислот с тремя индексами, присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию; и
(j) объединение ядер или клеток с тремя индексами с получением объединенных ядер или клеток с тремя индексами.
[00266] Вариант 44. Способ получения библиотеки для секвенирования, содержащей нуклеиновые кислоты, происходящие из множества отдельных ядер или клеток, где указанный способ включает:
(a) получение множества ядер или клеток;
(b) контактирование множества ядер или клеток с обратной транскриптазой и праймером, с получением двухцепочечных нуклеиновых кислот ДНК, содержащих праймер, и соответствующей нуклеотидной последовательности ДНК из матричных нуклеиновых кислот РНК;
(c) распределение ядер или клеток по первому множеству компартментов,
где каждый компартмент содержит субпопуляцию ядер или клеток;
(d) обработку молекул ДНК в каждой субпопуляции ядер или клеток с получением индексированных ядер или клеток,
где обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в каждой субпопуляции ядер или клеток, первой компартмент-специфической индексной последовательности с получением индексированных нуклеиновых кислот, присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию;
(e) объединение индексированных ядер или клеток с получением объединенных индексированных ядер или клеток;
(f) распределение объединенных индексированных ядер или клеток по второму множеству компартментов, где каждый компартмент содержит субпопуляцию ядер или клеток;
(g) обработку молекул ДНК в каждой субпопуляции ядер или клеток с получением ядер или клеток с двумя индексами,
где обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в каждой субпопуляции ядер или клеток, второй компартмент-специфической индексной последовательности с получением нуклеиновых кислот с двумя индексами, присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию;
(h) объединение ядер или клеток с двумя индексами с получением объединенных ядер или клеток с двумя индексами;
(i) распределение объединенных ядер или клеток с двумя индексами по третьему множеству компартментов, где каждый компартмент содержит субпопуляцию ядер или клеток;
(j) обработку молекул ДНК в каждой субпопуляции ядер или клеток с получением ядер или клеток с тремя индексами,
где обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в субпопуляциях ядер или клеток, третьей компартмент-специфической индексной последовательности с получением нуклеиновых кислот с тремя индексами, присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию; и
(k) объединение ядер или клеток с тремя индексами с получением объединенных ядер или клеток с тремя индексами.
[00267] Вариант 45. Способ любого из вариантов 43 или 44, где праймер гибридизуется с нуклеиновыми кислотами РНК с образованием двухцепочечных нуклеиновых кислот ДНК, содержащих праймер, и соответствующей нуклеотидной последовательности ДНК из матричных молекул РНК.
[00268] Вариант 46. Способ любого из вариантов 43-45, где праймер содержит поли-Т-нуклеотидную последовательность, которая гибридизуется с поли(А)-хвостом мРНК.
[00269] Вариант 47. Способ любого из вариантов 43-46, где контактирование также включает контактирование субпопуляций со вторым праймером, где второй праймер содержит
последовательность, которая гибридизуется с предварительно определенной нуклеиновой кислотой ДНК.
[00270] Вариант 48. Способ любого из вариантов 43-47, где второй праймер содержит компартмент-специфический индекс.
[00271] Вариант 49. Способ любого из вариантов 43-45, где праймер содержит последовательность, которая гибридизуется с предварительно определенной нуклеиновой кислотой РНК.
[00272] Вариант 50. Способ любого из вариантов 43-49, где предварительно определенной нуклеиновой кислотой РНК является мРНК.
[00273] Вариант 51. Способ любого из вариантов 43-50, где праймер включает праймер переключения матрицы.
[00274] Вариант 52. Способ любого из вариантов 43-51, где обработка для добавления одной или более первой, второй или третьей компартмент-специфической индексной последовательности включает двухстадийный способ добавления нуклеотидной последовательности, содержащей универсальную последовательность, к нуклеиновым кислотам, а затем добавление первой компартмент-специфической индексной последовательности к нуклеиновым кислотам ДНК.
[00275] Вариант 53. Способ любого из вариантов 43-52, где праймер содержит первую компартмент-специфическую индексную последовательность.
[00276] Вариант 54. Способ любого из вариантов 43-53, дополнительно включающий, перед контактированием, мечение вновь синтезированной РНК в субпопуляциях клеток или ядер, выделенных из клеток.
[00277] Вариант 55. Способ любого из вариантов 43-54, где уже существующие нуклеиновые кислоты РНК и вновь синтезированные нуклеиновые кислоты РНК помечены одинаковыми индексами в одном и том же компартменте.
[00278] Вариант 56. Способ любого из вариантов 43-55, где мечение включает инкубирование множества ядер или клеток в композиции, содержащей нуклеотидную метку, где нуклеотидная метка включена во вновь синтезированную РНК.
[00279] Вариант 57. Способ любого из вариантов 43-56, где нуклеотидная метка содержит нуклеотидный аналог, меченный гаптеном нуклеотид, мутагенный нуклеотид или нуклеотид, который может быть модифицирован посредством химической реакции.
[00280] Вариант 58. Способ любого из вариантов 43-57, где более чем одна нуклеотидная метка включена во вновь синтезированную РНК.
[00281] Вариант 59. Способ любого из вариантов 43-58, где отношения нуклеотидной метки или меток отличаются для различных компартментов или моментов времени.
[00282] Вариант 60. Способ любого из вариантов 43-59, также включающий обработку субпопуляций ядер или клеток компартментов в предварительно определенных условиях перед мечением.
[00283] Вариант 61. Способ любого из вариантов 43-60, где предварительно определенное условие включает обработку агентом.
[00284] Вариант 62. Способ любого из вариантов 43-61, где агент включает белок, не-рибосомный белок, поликетид, органическую молекулу, неорганическую молекулу, молекулу РНК или РНКи, углевод, гликопротеин, нуклеиновую кислоту или их комбинацию.
[00285] Вариант 63. Способ любого из вариантов 43-62, где агент включает терапевтическое лекарственное средство.
[00286] Вариант 64. Способ любого из вариантов 43-63, где предварительно определенные условия двух или более компартментов являются различными.
[00287] Вариант 65. Способ любого из вариантов 43-64, где обработку и мечение проводят одновременно или обработку проводят до мечения.
[00288] Вариант 66. Способ любого из вариантов 43-65, где одно или более распределений включают разведение.
[00289] Вариант 67. Способ любого из вариантов 43-65, где одно или более распределений включают сортинг.
[00290] Вариант 68. Способ любого из вариантов 43-67, где добавление одной или более из первой, второй или третьей компартмент-специфических индексных последовательностей включает контактирование субпопуляций со шпилечным лигирующим дуплексом в условиях, подходящих для лигирования шпилечного лигирующего дуплекса с концом фрагментов нуклеиновой кислоты.
[00291] Вариант 69. Способ любого из вариантов 43-68, где добавление одной или более из первой, второй или третьей компартмент-специфических индексных последовательностей включает контактирование фрагментов нуклеиновой кислоты с транспосомным комплексом, где транспосомный комплекс в компартментах содержит транспозазу и универсальную последовательность, где контактирование также включает условия, подходящие для фрагментации фрагментов нуклеиновой кислоты и включения нуклеотидной последовательности во фрагменты нуклеиновой кислоты.
[00292] Вариант 70. Способ любого из вариантов 43-69, где добавление первого или второго компартмент-специфического индекса включает лигирование, а добавление последующей компартмент-специфической индексной последовательности включает транспозицию.
[00293] Вариант 71. Способ любого из вариантов 43-70, где компартмент включает лунку или каплю.
[00294] Вариант 72. Способ любого из вариантов 43-71, где компартменты первого множества компартментов содержат от 50 до 100000000 ядер или клеток.
[00295] Вариант 73. Способ любого из вариантов 43-72, где компартменты второго множества компартментов содержат от 50 до 100000000 ядер или клеток.
[00296] Вариант 74. Способ любого из вариантов 43-73, где компартменты третьего множества компартментов содержат от 50 до 100000000 ядер или клеток.
[00297] Вариант 75. Способ любого из вариантов 43-74, дополнительно включающий получение нуклеиновых кислот с тремя индексами из объединенных ядер или клеток с тремя индексами, и тем самым получение библиотеки для секвенирования из множества ядер или клеток.
[00298] Вариант 76. Способ любого из вариантов 43-76, дополнительно включающий:
получение поверхности, содержащей множество сайтов амплификации,
где сайты амплификации включают по меньшей мере две популяции связанных одноцепочечных олигонуклеотидов для захвата, имеющих свободный 3'-конец, и
контактирование поверхности, содержащей сайты амплификации, с фрагментами нуклеиновой кислоты с тремя индексами в условиях, подходящих для получения множества сайтов амплификации, каждый из которых содержит клональную популяцию ампликонов, из отдельного фрагмента, содержащего множество индексов.
[00299] Вариант 77. Способ получения библиотеки для секвенирования, содержащей нуклеиновые кислоты, из множества отдельных клеток, где указанный способ включает:
(a) получение ядер из множества клеток;
(b) распределение субпопуляций ядер по первому множеству компартментов и контактирование каждой субпопуляции с обратной транскриптазой и праймером, где праймер в каждом компартменте содержит первую индексную последовательность, которая отличается от первых индексных последовательностей в других компартментах, с получением индексированных ядер содержащих индексированные фрагменты нуклеиновых кислот;
(c) объединение индексированных ядер с получением объединенных индексированных ядер;
(d) распределение субпопуляций объединенных индексированных ядер по второму множеству компартментов и контактирование каждой субпопуляции со шпилечным лигирующим дуплексом в условиях, подходящих для лигирования шпилечного лигирующего дуплекса с концом индексированных фрагментов нуклеиновой кислоты, содержащих первую индексную последовательность, с получением ядер с двумя индексами, содержащих фрагменты нуклеиновой кислоты с двумя индексами, где шпилечный лигирующий дуплекс содержит вторую индексную последовательность, отличающуюся от вторых индексных последовательностей в других компартментах;
(e) объединение ядер с двумя индексами с получением объединенных ядер с двумя индексами;
(f) распределение субпопуляций объединенных ядер с двумя индексами по третьему множеству компартментов и обработку фрагментов нуклеиновой кислоты с двумя индексами в условиях, подходящих для синтеза второй цепи;
(g) контактирование фрагментов нуклеиновой кислоты с двумя индексами с транспосомным комплексом, где транспосомный комплекс в каждом компартменте содержит транспозазу и универсальную последовательность, где контактирование включает условия, подходящие для фрагментации фрагментов нуклеиновой кислоты с двумя индексами и включения универсальной последовательности во фрагменты нуклеиновой кислоты с двумя индексами с получением фрагментов нуклеиновой кислоты с двумя индексами, содержащих первый и второй индексы на одном конце и универсальную последовательность на другом конце;
(h) включение во фрагменты нуклеиновой кислоты с двумя индексами в каждом компартменте третьей индексной последовательности с получением фрагментов с тремя индексами;
(i) объединение фрагментов с тремя индексами с получением библиотеки для секвенирования, содержащей нуклеиновые кислоты транскриптома, из множества отдельных клеток.
[00300] Вариант 78. Способ варианта 77, где праймер содержит поли-Т-нуклеотидную последовательность, которая гибридизуется с поли(А)-хвостом мРНК.
[00301] Вариант 79. Способ вариантов 77-78, где праймер каждого компартмента содержит последовательность, которая гибридизуется с предварительно определенной мРНК.
[00302] Вариант 80. Способ любого из вариантов 77-79, где способ включает праймеры в различных компартментах, которые гибридизуются с различными нуклеотидами одной и той же предварительно определенной мРНК.
[00303] Вариант 81. Способ получения библиотеки для секвенирования транскриптома, содержащей нуклеиновые кислоты, из множества отдельных клеток, где указанный способ включает:
(a) получение объединенных ядер из множества клеток;
(b) контактирование объединенных ядер с обратной транскриптазой и праймером, содержащим последовательность олиго-dT, которая гибридизуется с поли(А)-хвостом мРНК с образованием объединенных ядер, содержащих фрагменты нуклеиновой кислоты;
(c) распределение субпопуляций объединенных ядер по множеству компартментов и контактирование каждой субпопуляции со шпилечным лигирующим дуплексом в условиях, подходящих для лигирования шпилечного лигирующего дуплекса с концом фрагментов нуклеиновой кислоты с получением индексированных ядер, содержащих индексированные фрагменты нуклеиновой кислоты, где шпилечный лигирующий дуплекс содержит индексную последовательность, отличающуюся от индексных последовательностей в других компартментах;
(d) объединение индексированных ядер с получением объединенных индексированных ядер;
(e) распределение субпопуляций объединенных индексированных ядер по второму множеству компартментов и обработку индексированных фрагментов нуклеиновой кислоты в условиях, подходящих для синтеза второй цепи;
(f) контактирование индексированных фрагментов нуклеиновой кислоты с транспосомным комплексом, где транспосомный комплекс в каждом компартменте содержит транспозазу и универсальную последовательность, где контактирование включает условия, подходящие для фрагментации индексированных фрагментов нуклеиновой кислоты и включения универсальной последовательности в индексированные фрагменты нуклеиновой кислоты с получением индексированных фрагментов нуклеиновой кислоты, содержащих индекс на одном конце и универсальную последовательность на другом конце;
(g) включение в индексированные фрагменты нуклеиновой кислоты в каждом компартменте второй индексной последовательности с получением фрагментов с двумя индексами;
(j) объединение фрагментов с двумя индексами с получением библиотеки для секвенирования, содержащей нуклеиновые кислоты транскриптома, из множества отдельных клеток.
[00304] Вариант 82. Способ выделения ядер, включающий:
(a) мгновенное замораживание ткани в жидком азоте;
(b) уменьшение размера ткани для получения обработанной ткани; и
(c) извлечение ядер из обработанной ткани путем инкубирования в буфере, который стимулирует лизис клеток и сохраняет целостность ядер в отсутствие одного или более экзогенных ферментов.
[00305] Вариант 83. Способ варианта 82, где указанное уменьшение размера включает измельчение ткани, воздействие на ткань физической силы или их комбинацию.
[00306] Вариант 84. Способ варианта 82 или 83, дополнительно содержащий:
(d) обработку извлеченных ядер перекрестносшивающим агентом с получением фиксированных ядер; и
(е) промывку фиксированных ядер.
[00307] Вариант 85. Набор для его применения в целях получения библиотеки для секвенирования, где указанный набор включает нуклеотидную метку и по меньшей мере один фермент, который опосредует лигирование, удлинение праймера или амплификацию.
[00308] Вариант 86. Набор для его применения в целях получения библиотеки для секвенирования, где указанный набор включает праймер, который гибридизуется с предварительно определенной нуклеиновой кислотой, и по меньшей мере один фермент, который опосредует лигирование, удлинение праймера или амплификацию.
Примеры
[00309] Настоящее изобретение проиллюстрировано на следующих примерах. Следует отметить, что конкретные примеры, материалы, количества и процедуры должны интерпретироваться в широком смысле в соответствии с объемом и сущностью раскрытого здесь изобретения.
Пример 1
[00310] Динамический паттерн транскрипции при органогенезе млекопитающих с разрешением отдельных клеток.
[00311] В процессе органогенеза млекопитающих, клетки трех зародышевых слоев трансформируются в эмбрион, который включает большинство основных внутренних и внешних органов. Ключевые регуляторы дефектов развития могут быть исследованы на этой важной стадии, но современные технологии не обладают пропускной способностью и разрешением, достаточными для получения общей информации о молекулярных состояниях и траекториях быстро диверсифицирующегося и расширяющегося числа клеток различных типов. Авторами была предпринята попытка исследовать динамику транскрипции при развитии мышей во время органогенеза при разрешении отдельных клеток. После усовершенствования протокола на основе комбинаторной индексации отдельных клеток («sci-РНК-seq3»), авторами был определен профиль свыше 2 миллионов клеток, полученных из 61 эмбриона мыши, на стадии беременности на дни 9,5-13,5 (Е9.5-Е13.5; 10-15 повторов в каждый момент времени). Авторами были идентифицированы сотни клеток расширяющихся, сжимающихся и переходных типов, многие из которых детектируются только по определенной здесь глубине сканирующего охвата клеток, и были определены соответствующие наборы генов-маркеров, специфичных к клеткам конкретного типа, Некоторые из которых были подтверждены авторами по общему уровню их гибридизация in situ. Авторами была исследована динамика пролиферации и экспрессии генов в клетках конкретных типов в течение определенного периода времени, включая направленные анализы только апикального эктодермального гребня, мезенхимы конечностей и скелетных мышц. С использованием нового алгоритма, авторами были идентифицированы основные пути развития отдельных клеток в процессе мышиного органогенеза, и в них были обнаружены примеры различных путей достижения одной и той же конечной точки, то есть разветвления и конвергенции. Эти данные составляют основной ресурс в биологии развития млекопитающих и представлены так, чтобы облегчить понимание информации, постоянно комментируемой учеными.
[00312] Введение
[00313] Органогенез млекопитающих является удивительным процессом. В течение короткого промежутка времени, клетки трех зародышевых слоев превращаются в сформировавшийся эмбрион, который включает большинство основных внутренних и внешних органов. Хотя человеческие эмбрионы на очень ранней стадии могут быть культивированы и исследованы in vitro1, однако, доступ к материалам, соответствующим более поздним стадиям эмбрионального развития человека, ограничен. А поэтому, большинство исследований органогенеза млекопитающих проводится на организмах-моделях, а, в частности, на мышах.
[00314] По сравнению с людьми, мыши развиваются быстро, всего лишь 21 день между оплодотворением и рождением детенышей. Имплантация мышиных бластоцистов (32-64 клетки) происходит на 4-й день эмбриогенеза (Е4.0). За этой стадией следует гаструляция и образование первичных зародышевых слоев (Е6.5-Е7.5; 660-15К клеток)2,3. В течение этого периода времени происходит образование примитивных полос и распределение отдельных линий дифференцировки эмбриона от передней до задней области4. На ранних стадиях сомита (Е8.0-Е8.5), эмбрион переходит от гаструляции к раннему органогенезу, ассоциированному с формированием нервной пластинки и сердечной трубки (клетки 60К-90К). Классический органогенез начинается на стадии Е9.5. В последующие четыре дня (Е9.5-Е13.5), эмбрион мыши развивается от нескольких сотен тысяч клеток до более чем десяти миллионов клеток, и одновременно развиваются сенсорные органы, желудочно-кишечный тракт и органы дыхания, спинной мозг, скелетная система и кроветворная система. Неудивительно, что этот важный период развития мышей был интенсивно изучен. Действительно, большинство ключевых регуляторов дефектов развития могут быть изучены в течение этого периода времени5,6.
[00315] Общепринятая парадигма исследования органогенеза у мышей включает акцентирование внимания на отдельной системе органов на ограниченной стадии развития и объединение исследований по нокауту генов с фенотипированием по анатомической морфологии, гибридизацией in situ, иммуногистохимией7,8 или, совсем недавно, с профилированием транскриптома или эпигенома9. Хотя такие целенаправленные исследования позволили получить фундаментальное представление о развитии млекопитающих, однако, современные технологии не обладают пропускной способностью и разрешением, достаточными для получения общей информации о динамических молекулярных процессах, происходящих в разнообразных и быстро растущих популяциях и субпопуляциях клеток во время органогенеза.
[00316] «Профилирование методом дробовика», относящееся к молекулярному содержимому отдельных клеток, представляет собой перспективный путь для решения этих проблем и дальнейшего углубления понимания пути развития млекопитающих. Так, например, применение методов секвенирования PHK-seq отдельных клеток недавно выявило высокую гетерогенность в нейронах и миокардиоцитах во время развития мышей10,11. Хотя недавно было издано два транскрипционных атласа отдельных клеток мышей и были представлены важные ресурсы для области12,13, однако, они, в основном, ограничены органами взрослых особей, и пока еще не были предприняты попытки охарактеризовать появление и временную динамику типов клеток млекопитающих во время их развития.
[00317] Комбинаторная индексация отдельных клеток («sci-») представляет собой методологическую основу, в которой используется штриховое кодирование с разделением-объединением для уникального мечения состава нуклеиновых кислот в большом количестве отдельных клеток или ядер14,21. Недавно авторами был разработан «sci-протокол» для транскриптомов («sci-PHK-seq»), и этот протокол был применен для создания 50-кратного «клеточного охвата» методом «дробовика» для нематод Caenorhabditis elegans на стадии L219. Хотя пропускная способность sci-»-методов возрастает экспоненциально с увеличением числа раундов индексации, однако, этот потенциал еще предстоит полностью реализовать в отношении других факторов, таких как скорость потери клеток и ограниченная эффективность реакции на некоторых этапах19,21. Чтобы решить эту проблему, авторами был разработан и тщательно оптимизирован 3-уровневый метод sci-PHK-seq (sci-PHK-seq3), в результате чего был разработан рабочий протокол, который позволяет определить профиль более одного миллиона клеток на эксперимент. Как было показано ранее19, множественные выборки (например, реплики, моменты времени и т.п.) могут быть подвергнуты штриховому кодированию во время первого раунда индексации и одновременно обработаны.
[00318] В данном случае, авторами была предпринята попытка исследовать транскрипционную динамику развития мышей во время органогенеза при разрешении отдельных клеток с использованием sci-PHK-seq3. В одном эксперименте, авторами был определен профиль более 2 миллионов отдельных клеток, полученных из 61 эмбриона мыши на стадии между Е9.5-Е13.5 (10-15 повторов в каждый момент времени). Исходя из этих данных, авторами было идентифицировано 38 основных типов клеток, а также более 600 клеток более гранулярных типов (называемых здесь «подтипами», чтобы отличить их от 38 основных типов клеток). В целом, авторами были обнаружены тысячи новых генов-кандидатов для клеток определенных типов и подтипов, которые были подтверждены авторами как репрезентативные примеры по общему уровню их гибридизации in situ. Авторами была количественно оценена динамика пролиферации и экспрессии генов в клетках размножающихся и транзиентных типов во втором триместре беременности, включая целевые анализы только апикального эктодермального гребня, мезенхимы конечностей и скелетных мышц. С использованием нового алгоритма, авторами были
идентифицированы основные пути развития отдельных клеток в процессе мышиного органогенеза, и в них были обнаружены примеры различных путей достижения одной и той же конечной точки, то есть, разветвления и конвергенции. Эти данные имеются в свободном доступе и представлены так, чтобы облегчить понимание информации, постоянно комментируемой учеными.
[00319] Результаты
[00320] Профилирование 2 миллионов клеток из 61 мышиного эмбриона на 5 стадиях развития с помощью sci-PHK-seq3
[00321] Для увеличения пропускной способности sci-PHK-seq, авторами было исследовано свыше 1000 экспериментальных условий. Что касается первоначального описания авторами данного метода19, то основные усовершенствования, введенные с помощью sci-PHK-seq3 (фиг. 4А, Методы), заключаются в том, что: (i) Авторами была разработана новая стратегия выделения и фиксации ядер, где ядра выделяют непосредственно из свежих тканей без какой-либо ферментативной обработки. Ядра, после их выделения, фиксируют в 4% параформальдегиде, и эти ядра могут храниться в жидком азоте до следующей обработки (ii). По сравнению с предыдущим описанием 3-уровневого индексирования19, авторы перешли от индексированного мечения Tn5 к индексированному шпилечному лигированию. (iii) Несколько отдельных реакций, например, реакция обратной транскрипции были дополнительно оптимизированы для повышения эффективности. (iv) Стадию FACS-сортинга не проводили, а для минимизации агрегации ядер проводили стадии обработки ультразвуком и фильтрации. Стадии получения библиотеки sci-PHK-seq3 могут быть завершены одним человеком за одну неделю, и значительно превышают «экспериментальную» пропускную способность альтернативных протоколов sc-PHK-seq (фиг. 4В).
[00322] Авторами были собраны эмбрионы мышей C57BL/6 между Е9.5-Е13.5, и мгновенно заморожены в жидком азоте, включая 10-15 эмбрионов по меньшей мере из трех независимых пометов на стадию. Впоследствии авторы выделяли ядра из 61 отдельного цельного эмбриона и осуществляли sci-PHK-seq3 (фиг. 4А). Ядра, полученные от каждого эмбриона, помещали в различные лунки в течение первого раунда индексирования так, чтобы профили PHK-seq отдельных ядер могли соответствовать эмбрионам, из которых они были получены (фиг. 5А). В качестве внутреннего контроля, авторами также была добавлена смесь клеток HEK293T и NIH/3T3 в две лунки во время первого раунда индексирования. После завершения протокола sci-PHK-seq3, полученная библиотека была секвенирована за один раунд NovaSeq с получением 11 миллиардов ридов (фиг. 5В).
[00323] Из этого одного эксперимента, авторами было получено 2072011 транскриптомов отдельных клеток (количество уникального молекулярного идентификатора или количество UMI≥200), включая 2058652 клетки из 61 мышиного эмбриона и 13359 клеток, происходящих от клеток HEK293T или NIH/3T3. Можно с уверенностью предсказать, что транскриптомы клеток HEK293T и NIH/3T3 в подавляющем большинстве случаев картированы по геному одного вида или другого вида с 420 (3%) соударениями (фиг. 4С). При глубине секвенирования 23207 ридов на клетку наблюдалась медиана 3676 UMI на клетки HEK293T и 5163 UMI на клетки NIH/3T3, при этом, 3,9% и 2,9% ридов на клетку были картированы у несоответствующих видов, соответственно (фиг. 5C-D). Авторами было проведено сравнение исходного протокола sci-PHK-seq с sci-PHK-seq3 путем снижения объема выборки ранее полученного набора данных19 до эквивалентной глубины секвенирования на клетку HEK293T или NIH/3T3. Протокол sci-PHK-seq3, давал 40-кратное увеличение пропускной способности и продемонстрировал сравнимую эффективность с точки зрения количества UMI, обнаруженных на клетку (фиг. 5Е). Кроме того, профили объединенных транскриптомов отдельных клеток HEK293T, полученных в соответствии с протоколом sci-PHK-seq3 и sci-PHK-seq, были в высокой степени скоррелированными (Пирсон: 0,98, фиг. 5F).
[00324] 2058652 клеток, полученных из эмбрионов, были картированы по 61 отдельному эмбриону на основе их штрихового кодирования в первом раунде (медиана 35272 клеток на эмбрион; фиг. 4D). Количество клеток, выделенных из каждого эмбриона, хорошо коррелировало с количеством лунок в первом раунде, соответствующих этим клеткам (Спирман: 0,75, фиг. 6А). На относительно небольшой глубине секвенирования (~5000 ридов на клетку) была определена медиана для 519 генов (671 UMI) на клетку (фиг .4Е). Этот результат был сравним с результатом или превышал результат других исследований scRNA-seq, в которых различные типы клеток были выделены и аннотированы19,21,22, несмотря на то, что число исходных секвенирующих ридов на клетку составлял менее, чем одну треть (фиг. 6B-D). У эмбрионов на более поздней стадии (Е12.5 и Е13.5) наблюдалось несколько меньшее количество UMI на клетку, что свидетельствует о снижении содержания мРНК на ядро во время развития (фиг. 6Е).
[00325] Исходя из грубых оценок количества клеток на эмбрион в каждый момент времени (Методы) и суммирования всех 10-15 репликатов в каждый момент времени, авторы провели оценку клеточного охвата методом «дробовика» для мышиных эмбрионов, и такая оценка составляла 0,8× на Е9,5 (клетки 200К на эмбрион; в данном случае было профилировано 152К), 0,3× на 10,5 (клетки 1,1М; профилировано 378К), 0,2× на Е11,5 (клетки 2М; профилировано 616К), 0,08 на Е12,5 (клетки 6М; профилировано 475К) и 0,03× на Е13,5 (клетки 13М; профилировано 437К). Таким образом, еще до проведения авторами снижения объема выборки, было обнаружено, что количество клеток, которые были профилированы на каждой стадии, эквивалентны значительному проценту количества клеток отдельных мышиных эмбрионов (3-80%).
[00326] Для проверки качества данных, авторами были объединены транскриптомы отдельных клеток каждого индивидуума, в результате чего был получен 61 «псевдообъемный профиль» мышиных эмбрионов. После подсчета количества UMI, картированных по транскрипту Xist (экспрессируемому только у самок) или по транскриптам на Y-хромосоме, мышиные эмбрионы могли быть легко разделены на мужские (х=31) и женские (n=30) группы (фиг. 4F) с сбалансированным представлением с точки зрения количества мужских и женских репликатов на каждой стадии (фиг. 6F).
[00327] Для дополнительной проверки качества, авторами было проведено t-стохастическое встраивание соседних областей (t-SNE) в «псевдообъемные» транскриптомы 61 эмбриона, в результате чего было получено пять тесно кластеризованных групп, которые идеально совпадали по их стадиям развития (фиг. 7А). Авторы также упорядочили мышиные эмбрионы по «псевдовременной траектории» с использованием Monocle23 на основе наилучших 1000 лучших генов, дифференциально экспрессирующихся в различные моменты времени, и результирующее упорядочение также соответствовало ожидаемому (фиг. 4G). При этом наблюдалось два заметных пробела в «псевдовременной траектории» на уровне эмбрионов, то есть, один пробел между Е9.5 и Е10.5, а другой между Е1 1.5 и Е12.5, что позволяет предположить о наличиии резких изменений в глобальном транскриптоме в этих «окнах». Каждому эмбриону авторы присваивали псевдовремя, которое потенциально отражало более детальную оценку стадии развития (фиг. 7В). Так, например, эмбрионы Е10.5, находящиеся на более ранней стадии развития, морфологически отличались от эмбрионов на более поздней псевдовременной стадии развития (фиг. 7С).
[00328] Авторами также были исследованы изменения во всем транскриптоме во время его развития. 12236 генов дифференциально экспрессировались на различных стадиях развития (данные не приводятся), и авторами был построен график по данным для некоторых наиболее динамичных генов как показано на фиг. 4Н. Как и ожидалось, наблюдалось повышение уровня экспрессии генов гемоглобина взрослого индивидуума, таких как Hbb-bt и Hbb-bs, и снижение экспрессии генов гемоглобина эмбрионов, таких как Hbb-bhl и Hbb-x. Гены с известной ролью в дифференцировке нейронов, включая Cntn424, Neurod225 и Neurod626, имели повышенный уровень экспрессии на более поздних стадиях. Однако, большинство наиболее высокодинамичных генов, например, Slc35f4, Prtg и Trim30a, ранее не были охарактеризованы. Независимо от этого и действительно, по предположениям авторов, мотивация для сбора данных об отдельных клетках заключается в том, что динамика экспрессии генов «всего эмбриона», в основном, обусловлена резкими изменениями относительного числа клеток различных типов, но не изменениями внутри клеток какого-либо одного типа.
[00329] Идентификация и аннотация основных типов и подтипов клеток, присутствующих во время органогенеза у мышей
[00330] Для идентификации клеток основных типов, авторы провели кластеризацию Лювена для 2058652 транскриптомов отдельных клеток (то есть, всех эмбрионов из все моменты времени), в результате чего было идентифицировано 40 различных групп с последующей визуализацией t-SNE (фиг.8А). При этом обнадеживает тот факт, что, хотя и наблюдались явные различия между клетками, полученными в различные моменты времени (фиг. 9А), однако, клетки, полученные из реплицированных эмбрионов в один и тот же момент времени, были распределены одинаково (фиг. 10). На основе наборов генов, специфичных для каждого из этих 40 кластеров, авторы вручную проводили индексирование клеток по их типам путем сравнения с опубликованными маркерными генами (данные не приводятся). Для 37 кластеров, авторы могли достоверно и точно присвоить каждой клетке по одной букве, в то время как два кластера соответствовали определенной эритроидной линии дифференцировки. Один кластер имел аномально высокие уровни UMI, но невысокое число кластер-специфичных генов, что позволяет предположить, что это может быть техническим артефактом клеточных дублетов. Объединение определенных кластеров эритроидной линии дифференцировки и отбрасывание этого предполагаемого кластера-дублета позволило выявить 38 основных типов клеток (фиг. 8А). Для многих кластеров, высокоспецифические маркерные гены позволяют точно дифференцировать клетки по их типам (фиг. 8В, фиг. 9В-С, данные не показаны). Так, например, кластер 6 (эпителиальные клетки) специфически экспрессировал хорошо охарактеризованные маркерные гены Epcam и Trp6321,28, в то время как кластер 29 (гепатоциты) был специфически маркирован по экспрессии Afp и Alb12. Более мелкие кластеры, включая некоторые кластеры, соответствующие в высокой степени специализированным типам клеток, могут быть также легко аннотированы. Так, например, кластер 36 был обогащен транскриптами с высоким уровнем экспрессии во время развития сетчатки, такими как Tyr и Trpm1, что с большой уверенностью позволяет предположить, что эти клетки представляют собой меланоциты29, 30. Кластер 37 был обогащен транскриптами, экспрессирующимися исключительно в развивающемся храсталике. Для кластеров, соответствующих эмбриональной ткани мезенхимы и соединительной ткани, идентификация клеток конкретных типов оказалась более сложной, в основном, потому, что в современной литературе указано меньшее число высокоспецифичных маркерных генов.
[00331] Из 26183 генов, 17789 генов (68%) были дифференциально экспрессированы (FDR 5%) в клетках 38 основных типов (фиг. 9В, данные не приводятся). Из этих клеток, авторами было идентифицировано 2863 клетко-специфических маркеров, подавляющее большинство которых ранее, насколько это известно авторам, не были ассоциированы с соответствующим типом клеток (в среднем 75 маркеров на кластер; фиг. 8В, фиг. 9С). В качестве примера того, насколько эти данные будут полезны для определения новых маркеров экспрессии генов, специфичных к стадиям развития и типам клеток, был рассмотрен ген «звуковой еж» (Shh), который, как было показано, играет важную роль в развитии многих систем органов, включая конечности, срединные структуры головного мозга, таламус, спинной мозг и легкие31. Авторами была детектирована наивысшая экспрессия Shh в кластере 30 (хорда; данные не приводятся), вместе с Ntnl, Slit1 и Spon1, о которых известно, что все они экспрессируются в клетках хорды и в вентральной пластинке нервной трубки в процессе развития32-34. Однако, гены Тох2, Stxbp6, Schip1, Frmd4b, ранее не описанные как маркеры хорды, также были в высокой степени специфичными к кластеру 30.
[00332] Как и ожидалось, авторы наблюдали заметные изменения в соотношениях типов клеток во время органогенеза. В то время как большинство из 38 основных типов клеток пролиферировались экспоненциально, Некоторые из них были транзиентными, и окончательно исчезали на Е13.5 (фиг. 11А-В). Так, например, примитивная эритроидная линия дифференцировки, происходящая из желточного мешка, представленного кластером 26, была охарактеризована по экспрессии Hbb-bhl, а окончательная эритроидная линия дифференцировки, происходящая из печени плода, была помечена по экспрессии Hbb-bs в кластере 22 (данные не приводятся). На стадии Е9.5, авторами были обнаружены, в основном, клетки, соответствующие примитивной эритроидной линии дифференцировки (фиг. 8А). В течение следующих 5 дней, окончательная эритроидная линия дифференцировки стала преобладающим типом клеток в кровообращении плода и, в конечном счете, превратилась исключительно в линию дифференцировки эритроцитов на Е13.5 (фиг. 8А). Соответствующие генные маркеры продемонстрировали сходную динамику (фиг. 11С).
[00333] Для идентифицированных здесь клеток 38 основных типов, медиана составляет 47073 клеток, причем самый большой кластер содержит 144648 клеток (предшественников соединительной ткани; 7,0% от общего набора данных), а самый маленький кластер включает только 1000 клеток (моноцитов/гранулоцитов; 0,05% от общего набора данных). Поскольку гетерогенность клеток определенных типов была совершенно очевидна во многих из этих 38 кластерах, авторы приняли итеративную стратегию, включающую повторную кластеризацию Лювена для каждого основного типа клеток в целях идентификации подкластеров (фиг. 12-13). После удаления подкластеров, в которых доминируют один или два эмбриона, и слияния очень похожих подкластеров (Методы), было идентифицировано всего 655 подкластеров (называемых здесь «подтипами», чтобы отличить их от 38 основных типов клеток; фиг. 12-13). Следует отметить, что чувствительность детектирования клеток определенных типов и подтипов, оцененная авторами в этом исследовании, непосредственно зависит от большого числа профилированных клеток. Так, например, повторение кластеризации Лювена на 2,5% данных, полученных авторами (50000 клеток), выявило только субпопуляцию клеток определенных типов и подтипов (фиг. 14).
[00334] 655 подтипов состоят из медианы 1869 клеток и варьируются от 51 клетки (подтип клеток хорды) до 65894 клеток (подтипа клеток-предшественников соединительной ткани) (фиг. 15А).
[00335] Почти все подтипы (99%) состоят из клеток множества эмбрионов, причем, доминирование какого-либо одного эмбриона не наблюдалось (фиг. 15В-С). Для того, чтобы подтвердить, что эти подтипы входят в совместимые транскрипционные программы, которые отличаются от родственных подтипов, авторами была определена медиана 55 специфических маркеров на подтип (фиг. 15D; следует отметить, что подтип-специфические маркеры определяются по специфичности в пределах соответствующих основных типов клеток, а не в общем наборе данных). Таким образом, для более, чем 38 основных типов клеток, отдельные подтипы продемонстрировали изменяющуюся динамику между Е9.5 и Е13.5. Большинство подтипов (64%) обнаруживали увеличение оцениваемого числа клеток, в то время как 12% клеток имели пониженное число, а 24% показали более сложные паттерны (фиг. 16А-В). Интересно отметить, что авторы могли легко отделить эмбрионы на различных стадиях развития, основываясь исключительно на соотношении клеток, соответствующих каждому подтипу (фиг. 16С).
[00336] Характеризация траекторий экспрессии генов во время развития апикального эктодермального гребня (AER) конечностей
[0 0337] В качестве примера органа, который может быть представлен с подробными аннотациями и исследованиями подтипа, авторы выбрали эпителий (кластер 6), а в частности, апикальный эктодермальный гребень (подкластер 6.25). На основе подтип-специфичных маркерных генов, авторы аннотировали 29 подтипов эпителия (кластер 6; фиг. 17А; фиг. 18А, данные не приводятся). Так, например, эпителиальные клетки подтипа 6.10 были помечены Ос90, то есть, геном, экспрессируемым исключительно в эпителии слухового пузырька35, тогда как эпителиальные клетки подтипа 6.25 показали повышенную экспрессию хорошо охарактеризованных маркерных генов Fgf8, Msx2 и Rspo2, специфичных к апикальному эктодермальному гребню (AER), то есть, в высокой степени специализированному эпителию, участвующему в развитии пальцев36. Для всех эпителиальных подтипов, авторы идентифицировали гены, ранее не известные как маркеры. Так, например, AER также отличался экспрессией Fndc3a, Adamts3, Slc16a10, Snap91 и Pou6f2. Общая гибридизация in situ (WISH) Fgf8 (известного маркера) и Fndc3a (нового маркера) подтвердила, что оба эти гена экспрессируются на самом дистальном конце почечных узлов конечности, представляющем AER при Е10.5 (фиг. 17В-Е).
[00338] Далее авторами была исследована динамика пролиферации клеток и экспрессия генов во время развития AER. Авторами было идентифицировано всего 1237 клеток AER, что составляло лишь 0,0 6% от полученного авторами всего набора данных, но они участвовали в развитии почти каждого эмбриона (45 из 61 с более чем 5 профилированными клетками AER). Хотя клетки AER детектировались во все моменты времени, однако, по наблюдению авторов, они находились на своем пике с точки зрения числа клеток на эмбрион на стадии Е9.5, а затем уменьшались (фиг. 17F), что соответствовало предыдущим отчетам37 и проведенным авторами исследованиям по валидации in situ (фиг. 17С). Для характеризации динамики экспрессии генов в AER во время развития, авторами было осуществлено псевдотемпоральное упорядочение клеток AER на основе 500 наилучших дифференциально экспрессированных генов на стадиях развития, что позволило создать простую траекторию раннего или позднего развития (фиг. 17G). 710 белок-кодирующих генов были дифференциально экспрессированы по псевдовремени развития (FDR 5%) (данные не приводятся). Так, например, Fgf9, о котором известно, что он обнаруживает AER-специфическую экспрессию в почечных узлах конечностей38, продемонстрировал замедленную динамику активации по сравнению с Fgf8 и Fndc3a (фиг. 17Н). В значительной степени активированные гены могут играть важную роль в дифференцировке клеток AER. Так, например, активированные гены включают Rspo2, который, как известно, играет важную роль в сохранении AER и в поддержании роста и формировании паттернов в развитии конечностей39 (фиг. 17Н).
[00339] Авторами также были идентифицированы гены, экспрессия которых значительно снижалась в клетках AER в период времени между стадиями Е9.5 и Е13.5 (169 генов при FDR 1%; фиг. 19А). Такими генами являются Ki67 (ММ67) и инсулиноподобный фактор роста 2 (Igf2), которые оба играют определенную роль в стимуляции пролиферации клеток40,41 (фиг. 17Н). Действительно, в соответствии с прекращением пролиферации AER в этом временном окне развития, анализы уровня пути значительного снижения экспрессии генов выявил сроки, ассоциированные с прохождением клеточного цикла и метаболизмом глюкозы, а также факторы транскрипции, связанные с плюрипотентностью (Is11, Pou5f1, Nanog) (фиг. 19В-С).
[00340] Характеристика траекторий изменения судьбы клеток при развитии мезенхимы конечности
[00341] Затем авторами была предпринята попытка исследовать пути развития, которые проходят клетки различных типов в течение этого важного периода развития млекопитающих, включая переходы клеток из одних типов и подтипов в другие. Большинство современных алгоритмов реконструирования псевдовременной траектории имеют два основных ограничения. Во-первых, при их создании предполагается, что клетки находятся в одном непрерывном многообразии, то есть, без разрывов между субпопуляциями клеток. Однако, поскольку самые ранние эмбрионы согласно изобретению образуются на стадии Е9.5, то полученный авторами набор данных не содержит клетки, соответствующие по меньшей мере некоторым наследственным состояниям. Во-вторых, в этих алгоритмах предполагается, что лежащая в их основе траектория представляет собой дерево, в котором точки ветвления соответствуют решениям судьбы клеток. Однако, известно, что некоторые ткани содержат транскрипционно неразличимые клетки, вносимые транскрипционно различными линиями дифференцировки, то есть сходимостью траекторий, разделенных одним или более событиями ветвления.
[00342] Для устранения этих ограничений, авторами был разработан новый алгоритм, включенный в пакет Monocle42, в целях разрешения множественных непересекающихся траекторий, а также для обеспечения как ветвления, так и сходимости внутри траекторий. Алгоритм Monocle 3 начинается с проецирования клеток на малоразмерное пространство, соответствующее состоянию транскрипции, посредством программы однородной множественной аппроксимации и проецирования (UMAP)43. Затем, с помощью Monocle 3 обнаруживают сообщества сходных клеток с использованием кластеризации Лювена и объединяют соседние сообщества с помощью статистического анализа, включенного в алгоритм аппроксимированного сжатия графов44 (AGA). Важно отметить, что эти процедуры позволяют поддерживать множество непересекающихся сообществ клеток. Последняя стадия в Monocle 3 направлена на разрешение путей развития отдельных клеток и выявление местоположений не только ветвей, но и сходящихся элементов в наборе клеток, которые составляют каждое сообщество, то есть, траекторий. Ранее авторами была описана процедура, называемая построение «L1-графа» для встраивания «основного графа» в проекцию профилей PHK-seq отдельных клеток, так, чтобы каждая клетка находилась возле определенной точки на графе45. Хотя с помощью L1-графа можно изучать траектории с замкнутыми петлями и ветвями, однако, он мог работать только на наборах данных с несколькими сотнями клеток. Чтобы алгоритм мог обрабатывать тысячи или даже миллионы клеток, авторами были внесены два усовершенствования. Во-первых, авторы запустили этот алгоритм на нескольких сотнях центроидов данных, а не на самих клетках. Во-вторых, они ограничили процедуру линейного программирования алгоритма для соблюдения границ между непересекающимися траекториями, определенными тестом AGA.
[00343] Сначала авторами была сделана попытка применить этот новый алгоритм к одному главному типу клеток, кластеру 25, 26559 клеток которого были аннотированы как мезенхима клеток почечных узлов конечностей по экспрессии Hoxd13, Fgf10 и Lmx1b (данные не приводятся). Визуализация траектории клеток этого кластера с помощью Monocle 3 иллюстрирует резкое размножение мезенхимальных клеток конечностей в процессе развития, причем, основной рост наблюдался между стадиями Е10.5 и Е12.5 (фиг. 20A). Экспрессия генов может быть любой, но постоянной во время такого размножения, причем, уровни 4763 белок-кодирующих генов значительно изменяются (FDR 1%; данные не приводятся). Ранние стадии развития мезенхимы конечностей характеризуются некоторыми предполагаемыми генами, такими как Tbx1546 и Gpc347, а более поздние стадии характеризуются Msx148, Epha449 и Dach150 (фиг. 21А), но подавляющее большинство динамически экспрессируемых генов являются новыми. Факторы транскрипции, значительно повышенные во время развития мезенхимы конечностей, включали факторы, участвующие в дифференцировке хондроцитов (например, Sox951 и Yap152), дифференцировке мышц (например, Tead453) и в заживлении ран и регенерации конечностей (например, Smarcd154) (фиг. 21В).
[00344] Интересно отметить, что клетки передних и задних конечностей было нелегко разделить путем неконтролируемой кластеризации (фиг. 21С) или анализа траектории (фиг. 22А), но их можно было различить по взаимоисключающей экспрессии ТЬх5 в передних конечностях (2085 клеток, 7,9% от всех мезенхимальных клеток конечностей) и Pitx1 в задних конечностях (1885 клеток, 7,1% от всех мезенхимальных клеток конечностей), причем, только 22 клетки экспрессировали оба маркера (0,08% от всех мезенхимальных клеток конечностей по сравнению с ~0,6% ожидаемых клеток, если они были независимыми; фиг. 20В)55. Таким образом, 285 генов дифференциально экспрессировались в клетках, относящихся к передней и задней конечностям (фиг. 20С, данные не приводятся). Известные маркерные гены, такие как Tbx4 и гены кластера Нохс (Нохс4-10)56, были активированы в клетках задних конечностей, как и ожидалось, но также были идентифицированы и новые маркеры. Так, например, авторами было обнаружено, что Epha3 и Hs3st3bl имели 5-кратный избыток в передних конечностях, a Pcdh17 и Igf1 имели 3-кратный избыток в задних конечностях.
[00345] Хотя время развития является главной осью изменения траектории мезенхимы конечности в алгоритме Monocle 3 (фиг. 20A), однако, очевидно, существует дополнительная структура. По меньшей мере некоторые из них, по-видимому, соответствуют двум основным пространственным осям развития конечностей: проксимально-дистальной оси (основное направление разрастания) и передне-задней оси (соответствует пяти пальцам)55. Так, например, Sox6 и Sox9 (проксимальный) 57, 58, Hoxd13 и Tfap2b (дистальный) 36, Рах9 и Alx4 (передний) и Shh и Hand2 (задний) были дифференциально распределены по траектории согласно Monocle 3 (фиг. 20D; фиг. 22В). Гибридизация in situ всей линии Hoxd13 (известного дистального маркера) и Сра2 (нового маркера, распределение которого по траектории согласно Monocle 3 было сходным с распределением известных дистальных маркеров) подтвердила, что оба гена экспрессируются в мезенхиме дистальных конечностей на стадиях между Е10.5 и Е13.5 (фиг. 20F-H). Применение критерия Морана для определения пространственной автокорреляции к траектории мезенхимы конечности выявило 1191 значительно различающихся генов (FDR 1%; I>10 по критерию Морана). Эти гены были сгруппированы в восемь паттернов экспрессии, Некоторые из которых совпадают с распределением маркеров на проксимально-дистальной и передне-задней осях (фиг. 23, данные не приводятся).
[00346] Объединенные суммарные результаты, полученные авторами для траекторий AER и мезенхимы конечности, представлены на фиг. 20I. Хотя развитие конечности определяется относительно простыми траекториями, однако, анализы авторов показали, как этот атлас отдельных клеток, участвующих в органогенезе мышей, может использоваться для характеризации пространственно-временной динамики экспрессии генов в конкретных системах.
[00347] Определение и характеристика дифференцировки основных клеточных линий мышиного органогенеза
[00348] Затем, авторами была предпринята попытка идентифицировать основные линии дифференцировки развития и клеточные траектории для всего набора данных. С помощью алгоритма Monocle 3 была сделана выборка 100000 высококачественных клеток (UMI>400) по восьми четко разделенным линиям дифференцировки (фиг. 24А, фиг. 25А). Почти все из 38 основных типов клеток почти исключительно входят в одну из этих восьми групп (фиг. 24В). Исключение составляют три из четырех наименьших кластеров: моноциты/гранулоциты (36 клеток), хрусталик (125 клеток) и мегакариоциты (287 клеток), вероятно, вследствие их малого количества. Двумя наиболее сложными структурами, очевидно, являются мезенхимальная траектория, которая включает все типы мезенхимальных и мышечных клеток (слева, фиг. 24А и фиг. 25А), и траектория нервной трубки/хорды, которая включает хорду, нервную трубку, клетки-предшественники и развивающиеся нейроны и глиальные клетки (справа, фиг. 24А и фиг. 25А). Первая траектория нервного гребня {«нервного гребня 1») включает меланоциты и предшественники шванновских клеток, а вторая траектория нервного гребня {«нервного гребня 2») состоит из сенсорных нейронов. Гемопоэтическая траектория включает мегакариоциты, эритроциты и лимфоциты, а каждая из трех остальных траекторий {печеночная, эндотелиальная, эпителиальная) соответствуют одному главному типу клеток. Хотя предполагаемое количество клеток на эмбрион в каждой из этих линий дифференцировки увеличивается экспоненциально начиная со стадии Е9.5 и до Е13.5, однако, их соотношения остаются относительно стабильными, за исключением гепатоцитов, которые увеличиваются почти в десять раз в этом временном окне развития (0,3% на стадии Е9.5→2,8% на стадии Е13.5) (фиг. 25В-С).
[00349] UMAP позволяет проецировать клетки одного и того же типа в определенные области, но в отличие от t-SNE, также позволяет размещать клетки родственных типов рядом друг с другом. Так, например, ранние мезенхимальные клетки, по-видимому, расходятся из определенной области в миоциты, мезенхиму конечности, хондроциты/остеобласты и соединительные ткани (фиг. 24А, слева). Аналогичным образом, типы клеток, обнаруженные в более поздние моменты развития, такие как глутаматергические нейроны, отделены от ранних предшественников ЦНС (например, радиальных глиальных клеток) «мостиком» из нервных клеток-предшественников (фиг. 24А, справа). С другой стороны, разрывы (например, между восемью основными линиями дифференцировки), вероятно, указывают на отсутствие промежуточных или наследственных состояний между этими группами из-за ограничений в исследованиях авторами на стадиях Е9.5-Е13.5.
[00350] После того, как авторы отдельно проанализировали каждую из восьми основных линий дифференцировки с помощью анализа для оценки траекторий, как описано выше, то есть, по аналогии с итеративной субкластеризацией, то траектории мезенхимы и нервной трубки/хорды были построены снова, как описано выше (фиг. 26-27, верхний ряд), а другие основные линии дифференцировки (эпителиальные, эндотелиальные и т.п.) продемонстрировали множество прерывистых сублиний, что позволило детально раскрыть траектории для подтипов (фиг. 26-27, остальные ряды). Так, например, если авторы аннотировали эпителиальную траекторию с помощью 29 подтипов (фиг. 17А), то они наблюдали несколько отдельных субтраекторий, каждая из которых происходит от основной концентрации клеток, полученных на стадии Е9.5, лежащих в основе субпопуляций эпителиальных клеток определенных подтипов (фиг. 24С, фиг. 28). Так, например, эпителиальные клетки апикального эктодермального гребня (фиг. 17G) образуют линейную субтраекторию от клеток на стадиях Е9.5-Е13.5, которые четко отделены от других эпителиальных субтраекторий (фиг. 24С, нижний центр).
[00351] Реконструирование клеточных траекторий во время миогенеза скелета
[00352] Необходимо провести дополнительные исследования для того, чтобы полностью выяснить взаимосвязь между клеточными типами и подтипами, которые составляют траектории, представленные на фиг. 24, а в частности, являются более сложными. В качестве репрезентативного примера может служить проведенная авторами более детальная оценка развития ткани мышц, которая состоит из различных мезодермальных линий дифференцировки, которые формируются до начала органогенеза. Так, например, внеглазные мышцы образуются из прехордиальной мезодермы, тогда как другие мышцы лица и челюсти образуются из носоглоточной мезодермы. Миогенез скелета формируется благодаря основному набору миогенных регуляторных факторов (MRF), которые активируются различными наборами вышерасположенных генов59. Так, например, Рах3 активирует Myod1 в мышцах туловища, тогда как в области головы, Рах3 является необязательным, a MRF активируются Pitx2 и Tbx160-62. Myod1 или Myf5, в свою очередь, активируют миогенин, который запускает экспрессию множества генов, необходимых для обеспечения сократительной способности скелетной мышцы. Авторами была выдвинута гипотеза, что миогенная траектория, если рассматривать ее в масштабе всего эмбриона, будет иметь несколько точек входа, которые подают клетки на общий путь, соответствующий активации программы экспрессии генов ядра, общей для миотрубок.
[00353] Для проверки этой гипотезы, авторами были выделены миоциты и их предполагаемые клетки-«предки» из мезенхимального пути, сначала путем количественной оценки фракции клеток в каждом узле главного графа, которые были классифицированы как миоциты (кластер 13). Затем, авторами были собраны все узлы «большинства миоцитов», а ребра главного графа были использованы для расширения этого набора узлов в более широкую «окрестность» клеток (фиг. 29А). Затем, авторы повторно запустили Monocle 3 на этой субпопуляции клеток для построения траектории, специфичной для миогенеза. Эта траектория отличалась множественными очаговыми концентрациями клеток Е9.5, причем клетки на более поздних стадиях были распределены по нескольким путям, отходящим наружу (фиг. 29В). Рах3 и Pax7, которые характерны для предшественников скелетных мышц, экспрессировались в клетках, распределенных по широкой полосе главного графа (фиг. 29С). Из этой области графа исходят два параллельных линейных сегмента, на которых клетки экспрессируют либо Myf5, либо Myod. Оба пути входят в общую область, занятую клетками, экспрессирующими Myog или Myh3, маркерами миоцитов и миотрубок, соответственно. Дополнительный путь, пройденный клетками от стадии Е9.5, которые экспрессируют Lhx2, Tbx1 и Pitx2, но очень низкие уровни Рах3, входит в траекторию непосредственно перед сегментами Myf5 и Myod1, возможно, соответствующими мезодерме глотки. Таким образом, динамика MRF и их вышерасположенных активаторов по всей траектории согласуется с мнением о том, что различные мезодермальные линии дифференцировки используют разные факторы для совмещения с базовой программой мышечных генов (фиг. 29D).
[00354] Обсуждение
[00355] В этом исследовании, авторы попытались охарактеризовать развитие млекопитающих путем профилирования транскриптомов отдельных клеток в масштабе всего мышиного эмбриона, ориентируясь на окно, соответствующее классическому органогенезу. В результате профилирования более 2000000 клеток из 61 отдельного эмбриона в одном эксперименте с sci-PHK-seq3, авторами также была разработана техническая основа для небольших лабораторий в целях создания наборов данных PHK-seq для отдельных клеток с беспрецедентной пропускной способностью. Для разрешения ветвления, сходимости и разрывов в траекториях развития, авторы представили Monocle 3, новый алгоритм для определения траектории, которая будет масштабироваться до миллионов клеток.
[00356] Для эмбрионов мышей со средним сроком беременности, авторы идентифицировали 38 основных типов клеток и более 600 подтипов. Каждый из этих типов и подтипов характеризуется экспрессией ряда маркерных генов, подавляющее большинство которых являются новыми, и репрезентативные примеры которых авторы подтвердили путем общей гибридизации in situ. В качестве иллюстрации эффективности глубокого клеточного охвата методом «дробовика» для характеризации редких типов клеток, авторами были выделены маркеры и динамически экспрессируемые гены в апикальном эктодермальном гребне (AER), специализированном эпителии, играющем важную роль в развитии пальцев, но здесь представлено только 0,06% профилированных клеток. 38 основных типов клеток в целом подразделяются на 8 траекторий, включая мезенхимальную траекторию, нервную трубку/хорду, кроветворную, печеночную, эндотелиальную, эпителиальную траекторию и две траектории нервного гребня. Разрыв между этими восьмью траекториями, вероятно, является следствием отсутствия представления наследственных или промежуточных состояний в наборе данных авторов, начиная с Е9.5. Анализ траектории мезенхимы конечности выявил корреляты гетерогенности развития, соответствующие как временной, так и множественной пространственным осям. Сосредоточив свое внимание на траектории субпопуляции мезенхимальных клеток, соответствующей миоцитам и их предшественникам, авторами было идентифицировано множество субтраекторий, которые сходятся в одну общую конечную точку, соответствующую миотрубкам. Этот пример «конвергенции» программ экспрессии отличается от структуры ветвления, присваеваемой большинством алгоритмов для определения траектории развития.
[00357] Исследование авторов имеет несколько ограничений, которые необходимо учитывать. Во-первых, как и в случае других атласов отдельных клеток, данные о транскриптоме отдельных клеток являются недостаточными. Тем не менее, предыдущие исследования показали, что программы транскрипции могут легко различаться в наборах данных транскриптома отдельных клеток на удивительно малой глубине секвенирования63. Возможность определить 655 транскрипционно различных подтипов с медианой 671 UMI на клетку, согласуется с этим представлением, и объединение транскриптомов клеток каждого типа или подтипа позволяет авторам создать репрезентативные профили экспрессии. Во-вторых, хотя авторы достаточно уверены в большинстве сделанных здесь присвоений клеткам определенных типов, но тем не менее, их следует рассматривать как предварительные. Ключевой проблемой является то, что развитие мышей в середине срока беременности (Е9.5-Е13.5) ранее не изучалось ни с точки зрения разрешения отдельных клеток, ни в масштабе всего организма. Существующие атласы транскрипции для отдельных клеток включают представленные отдельные органы взрослых мышей или мышей на поздних эмбриональных стадиях12,13. Хотя на сегодняшний день авторы добились значительных успехов, однако, полная аннотация этих 655 подтипов клеток является текущим проектом, и авторы ожидают, что они получат поддержку при участии исследователей и экспертов в данной области для достижения стабильного консенсуса. С этой целью авторы создали общедоступную базу данных для облегчения аннотации для авторов и исследователей (доступна в Интернете на сайте atlas.gs.washington.edu/mouse-rna/). Уникальная страница для каждого подтипа включает загрузочную матрицу клеток, которые ее составляют, список генов-маркеров, специфичных для этого подтипа, и описание динамики клеток этого подтипа в изученном здесь окне развития.
[00358] Возможно, что, в конечном счете, отдаленная цель ученых в этой области с точки зрения технической перспективы заключается в создании всеобъемлющего, пространственно-временного молекулярного атласа развития млекопитающих при разрешении отдельных клеток. Для достижения этой цели был остановлен выбор на мышах, который имеет несколько преимуществ, включая небольшой размер мыши, доступность ранних временных точек ее развития, врожденный генетический фон и генетическую манипуляцию. Эти данные, представляющие количество клеток, соответствующее значительному проценту клеточного содержимого отдельного эмбриона мыши (от 3 до 80% «клеточного охвата методом дробовика» на стадию), являются мощным ресурсом в области биологии развития, а также могут способствовать дальнейшему продвижению разработки компьютерных методов для разрешения и интерпретации типов клеток или траекторий их развития. Заглядывая в будущее, авторы ожидают, что суммарная оценка транскриптома, дополнительных молекулярных фенотипов64, истории происхождения линии дифференцировки65 и широкая информация помогут сформировать общее представление о развитии млекопитающих.
[00359] В заключение следует отметить, что атласы развития отдельных клеток мышей дикого типа также являются первым шагом к пониманию плейотропных нарушений развития в масштабе организма, а также к детальному изучению тонкой роли генов и регуляторных последовательностей в развитии. Так, например, хотя ~35% нокаутов генов у мышей являются летальными5, однако, многие нокауты, а в частности, нокауты консервативных регуляторных последовательностей, не обнаруживают каких-либо аномалий при обычном фенотипировании66. Авторы предполагают, что sc-PHK-seq в масштабе организма позволяет осуществлять методы обратной генетики, например, потенциально обнаружить ранее пропущенные фенотипы с тонкими дефектами в молекулярных программах или относительные соотношения определенных типов клеток67.
[00360] Методы
[00361] Выделение эмбрионов
[00362] Мышей C57BL/6 получали из Лаборатории Джексона (Bar-Harbor, ME) и подбирали пары для спаривания. День спаривания считался эмбриональным днем (Е) 0.5. Иссечения выполняли как описано ранее69, и все эмбрионы немедленно замораживали в жидком азоте. Все процедуры на животных осуществляли с разрешения руководителей Института, Регуляторных Органов штата и Правительства (в соответствии с протоколом IACUC 4378-01).
[00363] Общая гибридизация in situ
[00364] Экспрессию мРНК в эмбрионах мышей на стадии Е9.5-Е11.5 оценивали путем общей гибридизации in situ (WISH) с использованием меченого дигоксигенином антисмыслового рибозонда, транскрибированного из клонированных геноспецифических зондов (набор для ПЦР-синтеза зонда DIG, Roche). Целые эмбрионы фиксировали в течение ночи в 4% PFA/PBS. Эмбрионы промывали в PBST (0,1% Твина) и постадийно дегидратировали в 25%, 50% и 75% метаноле/PBST и, наконец, хранили при -20°С в 100% метаноле. Протокол WISH осуществляли следующим образом: День 1) Эмбрионы регидратировали на льду на обратимых стадиях обработки метанолом/PBST, промывали в PBST, отбеливали в 6% H202/PBST в течение 1 часа и промывали в PBST. Затем эмбрионы обрабатывали в 10 мкг/мл протеиназы K/PBST в течение 3 минут, инкубировали в глицине/PBST, промывали в PBST и, наконец, повторно фиксировали в течение 2 0 минут 4% PFA/PBS, 0,2% глутаральдегидом и 0,1% Твином 20. После дополнительных стадий промывки PBST, эмбрионы инкубировали при 68°С в буфере L1 (50% деионизованный формамид, 5× SSC, 1% ДСН, 0,1% Твин 20 в DEPC; рН 4,5) в течение 10 минут. Затем эмбрионы инкубировали в течение 2 часов при 68°С в буфере для гибридизации 1 (L1 с 0,1% тРНК и 0,05% гепарином). После этого, эмбрионы инкубировали в течение ночи при 68°С в буфере для гибридизации 2 (в буфере для гибридизации 1 с 0,1% тРНК и 0,05% гепарином и зондом 1:500 DIG). День 2) Удаление несвязанного зонда осуществляли с помощью серии стадий промывки 3 × 30 минут каждая, при 68°С: L1, L2 (50% деионизованный формамид, 2 × SSC, рН 4,5, 0,1% Твина 20 в DEPC; рН 4,5) и L3 (2× SSC, рН 4,5, 0,1% Твин 20 в DEPC; рН 4,5). Затем эмбрионы обрабатывали в течение 1 часа раствором РНКазы (0,1 М NaCl, 0,01 М Триса, рН 7,5, 0,2% Твина 20, 100 мкг/мл РНКазы А в H2O) с последующей промывкой в TBST 1 (140 мМ NaCl, 2,7 мМ KCl, 2 5 мМ Триса-HCl, 1% Твина 20; рН 7,5). Затем эмбрионы блокировали в течение 2 часов при комнатной температуре в блокирующем растворе (TBST 1 с 2% телячьей сывороткой и 0,2% BSA) с последующим инкубированием при 4°С в течение ночи в блокирующем растворе, содержащем 1:5000 антитело против дигоксигенина-АР, 1:5000. День 3) Удаление несвязанного антитела осуществляли с помощью серии стадий промывок TBST 2 (TBST с 0,1% Твина 20 и 0,05% левамизола/тетрамизола) 8×30 минут при комнатной температуре и оставляли на ночь при 4°С. День 4) Окрашивание эмбрионов начинали путем промывки при комнатной температуре буфером со щелочной фосфатазой (0,02 М NaCl, 0,05 М MgCl2, 0,1% Твина 20, 0,1 М Триса-HCl и 0,05% левамизола/тетрамизола в H2O) 3×20 минут с последующим окрашиванием пурпурным субстратом для АР ВМ (Roche). Окрашенные эмбрионы визуализировали под микроскопом Zeiss Discovery V.12 и с помощью цифровой камеры Leica DFC420.
[00365] Культивирование клеток млекопитающих
[00366] Все клетки млекопитающих культивировали при 37°С с 5% CO2 и хранили в DMEM с высоким содержанием глюкозы (Gibco, кат. №11965) для клеток HEK293T и NIH/3T3, а затем добавляли 10% FBS и IX Pen/Strep (Gibco, кат.№15140122; 100 ед./мл пенициллина, 100 мкг/мл стрептомицина). Клетки трипсинизировали 0,25% трипсином-EDTA (Gibco, кат. №25200-056) и распределяли в отношении 1:10 три раза в неделю.
[00367] Выделение и фиксация ядер мышиных эмбрионов
[00368] Мышиные эмбрионы на различных стадиях развития обрабатывали вместе для уменьшения кластер-эффекта. Каждый мышиный эмбрион измельчали на мелкие кусочки лезвием бритвы в 1 мл охлажденного льдом буфера для лизиса клеток (10 мМ Трис-HCl, рН 7,4, 10 мМ NaCl, 3 мМ MgCl2 и 0,1% IGEPAL СА-63070, модифицированного так, чтобы он включал 1% ингибитора Super-РНКазы и 1% BSA) и переносили в верхнюю часть 4 0 мкм-фильтра для клеток (Falcon). Ткани гомогенизировали с помощью резинового наконечника поршня шприца (5 мл, BD) в 4 мл буфера для лизиса клеток. Затем, отфильтрованные ядра переносили в новую пробирку объемом 15 мл (Falcon) и осаждали путем центрифугирования при 500×g в течение 5 минут и один раз промывали 1 мл буфера для лизиса клеток. Ядра фиксировали в 4 мл охлажденного льдом 4% параформальдегида (EMS) в течение 15 минут на льду. После фиксации, ядра два раза промывали в 1 мл буфера для промывки ядер (буфер для лизиса клеток без IGEPAL) и ресуспендировали в 500 мкл буфера для промывки ядер. Образцы распределяли по двум пробиркам по 2 50 мкл в каждой пробирке и мгновенно замораживали в жидком азоте.
[00369] Для оценки контроля качества, клетки HEK293T и NIH/3T3 трипсинизировали, центрифугировали при 300×g в течение 5 минут (4°С) и один раз промывали в 1×PBS. Равное количество клеток и NIH/3T3 объединяли и подвергали лизису с использованием 1 мл охлажденного льдом буфера для лизиса клеток с последующей фиксацией и хранением в таких же условиях, как и для мышиных эмбрионов.
[00370] Получение и секвенирование библиотеки sci-РНК-seq3
[00371] Оттаянные ядра делали проницаемыми с использованием 0,2% тритона Х-100 (в буфере для промывки ядер) в течение 3 минут на льду, а затем быстро обрабатывали ультразвуком (Diagenode, 12 секунд в режиме малой мощности) для уменьшения агломерации ядер. Затем, ядра один раз промывали буфером для промывки ядер и фильтровали через 1 мл-фильтр для клеток Flowmi (Flowmi). Отфильтрованные ядра центрифугировали при 500×g в течение 5 минут и ресуспендировали в буфере для промывки ядер.
[00372] Затем ядра от каждого мышиного эмбриона распределяли по нескольким отдельным лункам в четырех 96-луночных планшетах. Затем регистрировали взаимосвязь между идентификатором лунок и мышиным эмбрионом для последующей обработки данных. Для каждой лунки, 80000 ядер (16 мкл) смешивали с 8 мл 25 мкМ заякоренного праймера олиго-dT (5'-/5Phos/CAGAGCNNNNNNNNN [10 п.о. штрих-код] ТТТТТТТТТТТТТТТТТТТТТТТТТТТТТТ-3', (SEQ ID NO: 1), где «N» представляет собой любое основание; IDT) и 2 мкл 10 мМ dNTP-смеси (Thermo) денатурировали при 55°С в течение 5 минут и сразу помещали на лед. После этого, в каждую лунку добавляли 14 мкл реакционной смеси для реакции первой цепи, содержащей 8 мкл 5× буфера для первой цепи Superscript IV (Invitrogen), 2 мкл 100 мМ DTT (Invitrogen), 2 мкл обратной транскриптазы Superscript IV (200 ед./мкл, Invitrogen), 2 мкл рекомбинантного ингибитора рибонуклеазы RNaseOUT (Invitrogen). Обратную транскрипцию осуществляли путем инкубирования планшетов в градиенте температур (4°С, 2 минуты, 10°С, 2 минуты, 20°С, 2 минуты, 30°С, 2 минуты, 40°С, 2 минуты, 50°С, 2 минуты и 55°С, 10 минут).
[00373] После реакции ОТ, в каждую лунку добавляли 60 мкл буфера для разведения ядер (10 мМ Трис-HCl, рН 7,4, 10 мМ NaCl, 3 мМ MgCl2 и 1% BSA). Ядра из всех лунок объединяли и центрифугировали при 500×g в течение 10 минут. Затем ядра ресуспендировали в буфере для промывки ядер и перераспределяли в четыре других 96-луночных планшета, где каждая лунка включала 4 мкл буфера для лигирования Т4 (NEB), 2 мкл ДНК-лигазы Т4 (NEB), 4 мкл раствора бетаина (5М, Sigma-Aldrich), 6 мкл буфера для промывки ядер, 8 мкл адаптера для лигирования со штрих-кодом (100 мкМ, 5'-GCTCTG [штрих-код 9 п.о. или 10 п.о. А]/i-дезокси-U/ACGACGCTCTTCCGATCT [обратный комплемент штрих-кода А]-3') (SEQ ID NO: 2) и 16 мкл 40% ПЭГ 8000 (Sigma-Aldrich). Реакцию лигирования проводили при 16°С в течение 3 часов.
[00374] После реакции ОТ, в каждую лунку добавляли 60 мкл буфера для разведения ядер (10 мМ Трис-HCl, рН 7,4, 10 мМ NaCl, 3 мМ и 1% BSA). Ядра из всех лунок объединяли и центрифугировали при 600×g в течение 10 минут. Ядра один раз промывали буфером для промывки ядер и два раза фильтровали через 1 мл-фильтр для клеток Flowmi (Flowmi), подсчитывали и перераспределяли по восьми 96-луночным планшетам, где каждая лунка, включала 2500 ядер в 5 мкл буфера для промывки ядер и 5 мкл буфера для элюирования (Qiagen). Затем, в каждую лунку добавляли 1,33 мкл буфера для синтеза второй цепи мРНК (NEB) и 0,66 мкл фермента для синтеза второй цепи мРНК (NEB), и синтез второй цепи проводили при 16°С в течение 180 мин.
[00375] Для мечения, содержимое каждой лунки смешивали с 11 мкл буфера Nextera TD (Illumina) и 1 мкл только фермента [i7] TDE1 (62,5 нМ, Illumina), а затем инкубировали при 55°С в течение 5 минут для проведения мечения. Затем реакцию прекращали путем добавления 24 мкл ДНК-связывающего буфера (Zymo) на лунку и инкубирования при комнатной температуре в течение 5 минут. Затем каждую лунку очищали с использованием 1,5× сфер AMPure ХР (Beckman Coulter). На стадии элюирования, в каждую лунку добавляли 8 мкл воды, не содержащей нуклеазы, 1 мкл 10× буфера USER (NEB), 1 мкл фермента USER (NEB) и инкубировали при 37°С в течение 15 минут. В каждую лунку добавляли еще 6,5 мкл буфера для элюирования. Сферы AMPure ХР удаляли магнитным стержнем, и продукт элюирования переносили в новый 96-луночный планшет.
[00376] Для ПЦР-амплификации, содержимое каждой лунки (16 мкл продукта) смешивали с 2 мкл 10 мкМ индексированного праймера Р5 (5'-AATGATACGGCGACCACCGAGATCTACAC[i5]ACACTCTTTCCCTACACGACGCTCTTCCGATCT-3'; IDT) (SEQ ID NO: 3), 2 мкл 10 мкМ праймера Р7 (5'-CAAGCAGAAGACGGCATACGAGAT[i7]GTCTCGTGGGCTCGG-3', IDT) (SEQ ID NO: 4) и 20 мкл 2× ПЦР-смеси NEBNext High-Fidelity Master Mix (NEB). Амплификацию осуществляли по следующей программе: 72°С в течение 5 минут, 98°С в течение 30 секунд, 12-14 циклов (98°С в течение 10 секунд, 66°С в течение 30 секунд, 72°С в течение 1 минуты) и наконец при 72°С в течение 5 минут.
[00377] После ПЦР, образцы объединяли и очищали с использованием 0,8 объема сфер AMPure ХР. Концентрации библиотек определяли с помощью Qubit (Invitrogen), и библиотеки визуализировали с помощью электрофореза на 6% ТВЕ-ПААГ-геле. Все библиотеки были секвенированы на одной платформе NovaSeq (Illumina) (считывание 1: 34 цикла, считывание 2: 52 цикла, индекс 1: 10 циклов, индекс 2: 10 циклов).
[00378] Обработка секвенирующих ридов
[00379] Базовые запрашиваемые последовательности преобразовывали в формат fastq с использованием bcl2fastq Illumina и подвергали демультиплексированию на основе штрих-кодов ПЦР i5 и i7 с использованием пакета программ для демультиплексирования с максимальным правдоподобием deML71 с параметрами по умолчанию. Дальнейшая обработка последовательностей и создание матрицы для оцифровывания отдельных клеток были аналогичны sci-PHK-seq19, за исключением того, что ОТ-индекс был объединен с индексом шпилечного адаптера, и, таким образом, картированные риды были подразделены на составные клеточные индексы путем демультиплексирования ридов с использованием ОТ-индекса и индекса лигирования (ED<2, включая инсерции и делеции). Вкратце, демультиплексированные риды были отфильтрованы по ОТ-индексу и индексу лигирования (ED<2, включая инсерции и делеции), а адаптер был удален с использованием программы trim_galore/0.4.1 с параметрами по умолчанию. Усеченные риды картировали по эталонному мышиному геному (mm10) для ядер мышиных эмбрионов или химерному эталонному геному человеческого hgl9 и мышиного mm10 для смешанных ядер НЕК293Т и NIH/3T3 с использованием программы STAR/v 2.5.2b72 с параметрами по умолчанию и с аннотациями генов (GENCODE V19 для человека; GENCODE VM11 для мыши). Уникально картированные риды выделяли, и дубликаты удаляли с использованием последовательности уникального молекулярного идентификатора (UMI), индекса обратной транскрипции (ОТ), индекса адаптера шпилечного лигирования и рида 2 с конечными координатами (то есть, риды с идентичными UMI, индексом ОТ, индексом адаптера лигирования и сайтом мечения рассматривались как дубликаты). И наконец, картированные риды распределяли по отдельным клеточным индексам путем последующего демультиплексирования ридов с использованием ОТ-индекса и индекса шпилечного лигирования (ED<2, включая инсерции и делеции). Для эксперимента со смешанными видами вычисляли процент уникально картированных ридов для геномов каждого вида. Клетки с более чем 85% UMI, отнесенных к одному виду, рассматривались как видоспецифичные клетки, а остальные клетки классифицировались как смешанные клетки или «столкновения». Для создания цифровых матриц, авторы вычислили число цепь-специфических UMI для каждой клетки, картированной по экзонным и интронным областям каждого гена с помощью пакета программ Python HTseq73. Для мультикартировнных ридов, эти риды были отнесены к самым близким генам, за исключением случаев, когда другой перемежающийся ген попадал в пределы 100 п.о. до конца ближайшего гена, и в этом случае, рид отбрасывали. Для большинства анализов, авторами были включены ожидаемые UMI для интронных и экзонных цепей на матрицах для экспрессии генов отдельных клеток.
[00380] Анализ всего мышиного эмбриона
[00381] После создания матрицы для подсчета генов отдельных клеток, каждая клетка была приписана к ее исходному мышиному эмбриону исходя из штрих-кода ОТ. Карты ридов для каждого эмбриона объединяли для создания «общей РНК-seq» для каждого эмбриона. Для разделения эмбрионов по половому признаку, авторами были подсчитаны риды, картированные по генам некодирующих РНК, специфичных для самок (Xist), или по генам chr Y (за исключением гена Erdr1, который присутствует в chr X и chr Y). Эмбрионы были легко разделены на женскую группу (с большим числом ридов, картированных по генам Xist, чем число ридов для генов chr Y) и мужскую группу (с большим числом ридов, картированных по генам chr Y, чем по Xist).
[00382] Псевдовременное упорядочение целых эмбрионов мыши осуществляли с помощью Monocle 274. Вкратце, объединенную матрицу для экспрессии генов конструировали как описано выше. Дифференциально экспрессируемые гены в различных условиях развития были идентифицированы с помощью дифференциальной функции GeneTest программы Monocle 274. Лучшие 2000 генов с наименьшим значением q были использованы для построения псевдовременной траектории с использованием Monocle 274. Каждому эмбриону было присвоено псевдовременное значение исходя из его положения на дереве траекторий.
[00383] Кластеризация клеток, визуализация t-SNE и идентификация маркерных генов
[00384] Цифровую матрицу для экспрессии генов создавали на основе исходных данных секвенирования как описано выше. Клетки с менее, чем 200 UMI отбрасывали. Последующий анализ проводили с помощью Monocle274 и пакета программ python scanpy75. Вкратце, определенное число генов, картированных по половым хромосомам, удаляли перед кластеризацией и уменьшением размерности. Стадию предварительной обработки проводили методом, аналогичным подходу, используемому Zheng et al.22, по функции «zheng17 recipe» (n_top-genes=2000) в scanpy75. Данные по размерности были сначала уменьшены с помощью РСА (30 компонентов), а затем с помощью t-SNE, после чего была проведена кластеризация Лювена на 30 основных компонентах (разрешение=1,5). Было идентифицировано 40 кластеров. Затем, авторы отобрали 1000 клеток из каждого кластера, и дифференциально экспрессированные гены в разных кластерах были идентифицированы с помощью дифференциальной функции GeneTest в программе Monocle 274. Гены, специфичные для каждого кластера, идентифицировали так же, как описано в литературе76. Кластеры были отнесены к известным типам клеток на основе кластер-специфических маркеров (Таблица 1). Один кластер имел аномально высокое число UMI, но не имел достаточного количества кластер-специфических генов, что позволяет предположить, что он может быть техническим артефактом клеточных дублетов, а следовательно, может быть удален. Два других кластера, как оказалось, соответствовали окончательной линии дифференцировки эритроидов и были объединены. Профили консенсусной экспрессии для клеток каждого типа были сконструированы как описано в литературе76. Для идентификации маркера гена, специфичного для клеток конкретного типа, авторами был выбран ген, который был дифференциально экспрессирован в клетках различных типов (FDR 5%, анализ на отношение вероятностей), а также максимально экспрессировался в клетке каждого типа по меньшей мере с 2-кратным увеличением по сравнению с клетками других типов, занимающими второе место по максимальной экспрессии.
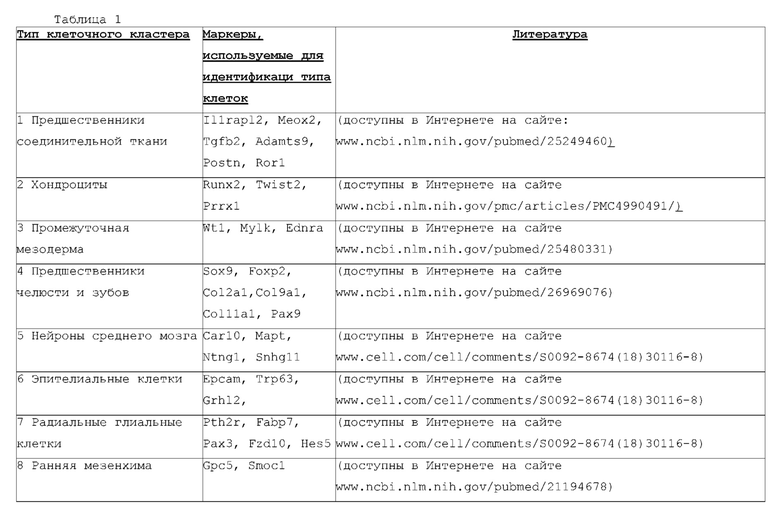
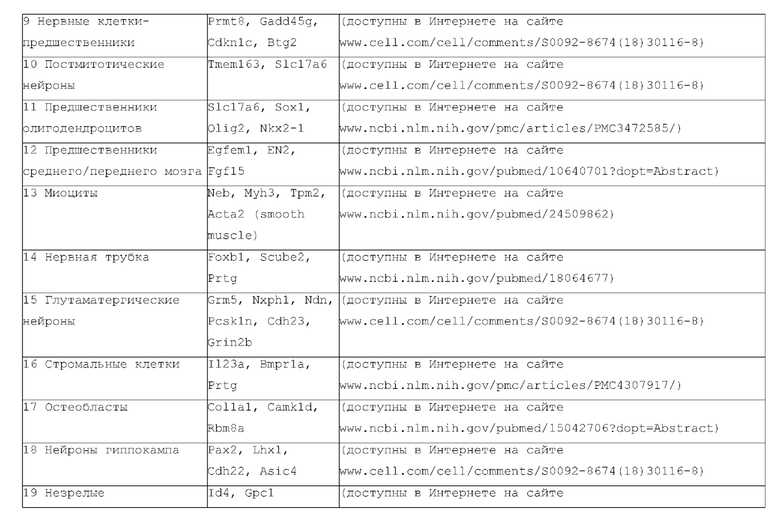
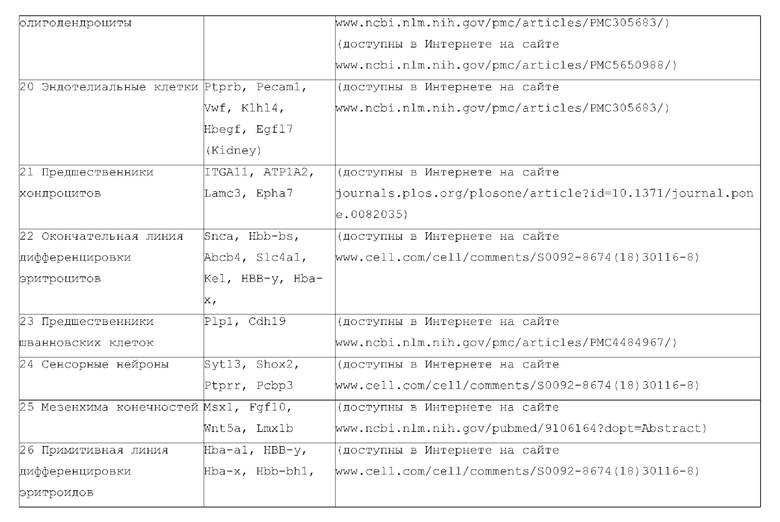
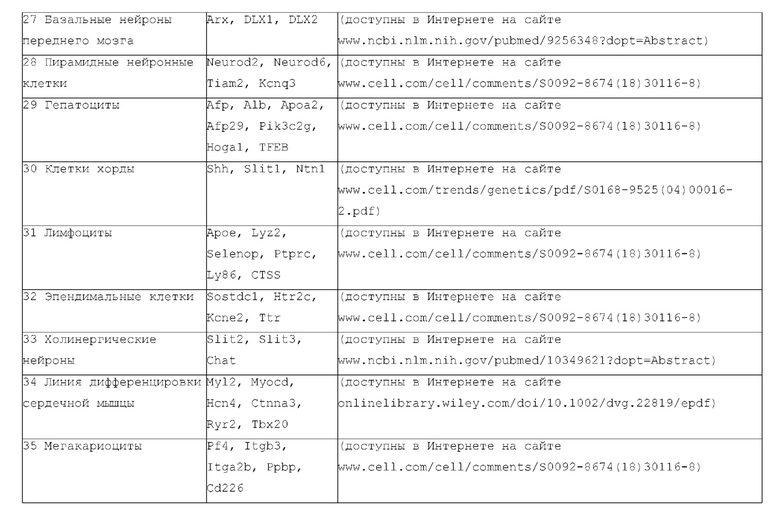

[00385] Для идентификации подкластера, авторы выбирали клетки высокого качества (UMI>400) для каждого основного типа и проводили РСА, t-SNE, кластеризацию Лювена аналогично анализу общего кластера. В высокой степени смещенные подкластеры отфильтровывали, если большинство клеток (>50%) кластера происходили от одного эмбриона. Очень похожие подкластеры были объединены, если их объединенные транскриптомы были в высокой степени скоррелированными (коэффициент корреляции Пирсона>0,95), и если два кластера были близки друг к другу в пространстве t-SNE. Дифференциально экспрессируемые гены во всех подкластерах были идентифицированы для каждого основного типа клеток как описано выше.
[00386] Для оценки числа клеток каждого типа (или подтипа), авторы сначала вычислили долю клеток каждого типа в отдельном эмбрионе, а затем результат умножали на общее число клеток, вычисленное для каждого эмбриона (Е9.5: 200000: Е10.5: 1100000; Е11.5: 2600000; Е12.5: 6100000; Е13.5: 13000000).
[00387] Для идентификации пол-специфических типов клеток (или подтипов), авторы сначала вычислили число клеток каждого типа (или подтипа) для самцов и самок на пяти стадиях развития. Отношение, специфичное для клеток каждого типа у самцов и самок, сравнивали с общим отношением числа клеток у самцов и самок на каждой стадии развития. Затем, авторы применили биномиальный критерий в R, для идентификации клеток определенных типов или подтипов со значительными различиями у самцов и самок для каждого типа клеток (х и n представляют собой число женских клеток и общее число клеток каждого типа на каждой стадии развития, р представляет собой число женских клеток на каждой стадии развития). Значение р преобразовывают в скорректированное значение q по методу Бенджамини и Хохберга с помощью функции коррекции р в R.
[00388]
[00389] Псевдовременной анализ AER и мезенхимы конечностей
[00390] Псевдовременное упорядочение клеток AER, передних или задних конечностей было осуществлено с помощью Monocle 274. Вкратце, дифференциально экспрессируемые гены на пяти стадиях развития были идентифицированы с помощью дифференциальной функции GeneTest в Monocle 274. Лучшие 500 генов с наименьшим значением q были использованы для построения псевдовременной траектории с использованием Monocle 274, где число UMI на клетку было использовано в качестве ковариаты при построении дерева. Каждой клетке присваивали псевдовременное значение на основе его положения вдоль дерева траекторий. Сглаженное изменение экспрессии маркерного гена по псевдовремени было определено путем введения функции plot_genes_in_pseudotim в Monocle 274. Клетки на траектории были сгруппированы по методу, описанному в литературе77. Вкратце, клетки были сначала сгруппированы в аналогичных положениях по псевдовремени с помощью кластеризации k-средних вдоль псевдовременной оси (k=10). Эти кластеры были подразделены на группы, содержащие по меньшей мере 50 и не более, чем 100 клеток. Затем, авторы объединили профили транскриптома клеток в каждой группе. Экспрессия гена по псевдовремени была вычислена методом, описанным в литературе77. Вкратце, гены, удовлетворяющие критерию значимости (FDR 5%) в различных условиях обработки, были отобраны, и был использован натуральный сплайн для построения графика зависимости генов от псевдовремени, где среднее число генов было включено в качестве ковариаты. Экспрессию каждого гена вычитали из наименьшей экспрессии, а затем делили на наибольшую экспрессию. Гены с максимальной экспрессией в течение первых 20% псевдовремени были помечены как активированные гены. Гены с максимальной экспрессией в последние 20% псевдовремени были помечены как репрессированные гены. Другие гены были помечены как временные гены. Обогащенные реактомные элементы (Reactome_2016) и факторы транскрипции (СпЕА_2016) были идентифицированы с использованием пакета программ EnrichR78.
[00391] Заключение о траекториях, определенных с помощью Monocle 3
[00392] Рабочая схема Monocle 3 состоит из 3 основных стадий организации клеток по потенциально прерывистым траекториям, с последующими необязательными статистическими анализами для поиска генов, которые различаются по экспрессии на всех этих траекториях. Monocle 3 также включает пакеты программ по визуализации для облегчения исследования траекторий в трех измерениях.
[00393] Уменьшение размерности с помощью однородной множественной аппроксимации и проецирования (UMAP)
[00394] Сначала программа Monocle 3 была использована для проецирования данных в маломерное пространство, что облегчает изучение основного графа, который описывает клеточные переходы между транскриптомными состояниями. Программа Monocle 3 работает вместе с UMAP, то есть, с недавно предложенным алгоритмом, основанным на римановой геометрии и алгебраической топологии, для уменьшения размерности и визуализации данных79. Качество визуализации, достигаемое с помощью этого алгоритма, может конкурировать с качеством, достигаемым с помощью популярного метода t-SNE (t-стохастического встраивания соседних областей), широко применяемого в транскриптомике для отдельных клеток. Однако, если t-SNE в основном направлен на размещение в высокой степени сходных клеток в одних и тех же областях маломерного пространства, то UMAP также сохраняет отношения расстояний с более широким размахом. Сам алгоритм UMAP также является более эффективным (сложность алгоритма UMAP оценивается по О (N) по сравнению с О(Nlog(N)) для t-SNE). Вкратце, UMAP сначала дает топологическое представление о многомерных данных с локальными множественными аппроксимациями и объединяет их локальные размытые представления упрощенных множеств. Затем UMAP оптимизирует введение нижнего измерения, минимизируя перекрестную энтропию между представлением низкой размерности и представлением высокой размерности.
[00395] Вычислительная эффективность UMAP резко ускоряет анализ данных мышиных эмбрионов. Авторами было обнаружено, что UMAP завершает анализ серии данных для двух миллионов клеток за 3 часа, в то время как анализ t-SNE занимает более 10 часов с 10 центрами (в данном случае используется мультицентровой bh-t-SNE). Несколько деталей реализации этого метода делает UMAP более эффективным. Алгоритмы UMAP и t-SNE имеют две основных стадии: сначала создается промежуточная структура из пространства с высокой размерностью (обычно это пространство с уменьшенным верхним РСА), а затем осуществляется поиск включений с низкой размерностью для представления промежуточной структуры. Во второй стадии, в обоих методах используется способ стохастического понижения решетки с различными функциями потерь для включения данных в маломерное пространство. Для t-SNE требуется функция потери для глобальной нормализации, а для UMAP не требуется функция потери, а используется другая целевая функция. Эта стадия позволяет, по существу, линейно масштабировать UMAP по ряду выборки данных. В Monocle 3, авторы работают с программой имплементации Python UMAP (доступной в Интернете на сайте www.atgithub.com/lmcinnes/umap) от Leland Mclnnes и John Healy с помощью пакета программ reticulate (доступного в Интернете на сайте atcran.r-project.org/web/packages/reticulate/ index.html).
[00396] Распределение клеток по прерывающимся траекториям
[00397] Недавно Вольф и его коллеги предложили идею организовать сбор данных о транскриптоме для отдельных клеток в «абстрактный разбиваемый граф» (AGA), связывающий кластеры клеток, которые могут быть связаны друг с другом с точки зрения времени развития. Вкратце, их алгоритм позволяет построить граф k-ближайших соседей на клетках, а затем идентифицирует «сообщества» клеток методом Лювена, аналогичным предыдущим методам анализа данных CyTOF или сбора данных PHK-seq для отдельных клеток80. Затем с помощью AGA можно построить граф, в котором вершинами являются лювеновские сообщества. Две вершины связаны с ребром в графе AGA, если клетки в соответствующих сообществах являются соседями в графе kNN чаще, чем это можно было бы ожидать в простой биномиальной модели81. Подобные методы были также недавно разработаны и применены для анализа наборов данных в атласе клеток для рыбы-зебры и лягушек82, 83.
[00398] Монокль 3 опирается на следующие идеи: сначала строят граф kNN для клеток в пространстве UMAP, а затем их группируют в лювеновские сообщества, и каждую пару сообществ тестируют на значимое число связей между их соответствующими клетками. Сообщества, которые имеют больше связей, чем это ожидалось при нулевой гипотезе ложной связи (FDR<10%), остаются связанными на графике AGA, а те связи, которые не прошли этот тест, разрываются. Результирующий граф AGA будет иметь один или более компонентов, каждый из которых передается на следующий шаг (L1-граф) в виде отдельной группы клеток, которые будут организованы в траекторию. Алгоритм AGA, по существу останавливается на этом этапе, представляя граф AGA как своего рода крупную траекторию в каждом сообществе, отражающую клетки в различных состояниях, которые они могут принимать по мере их развития. В отличие от этого, как описано в следующем разделе, Monocle 3 использует граф AGA для ограничения пространства главных графов, которые могут формировать конечную траекторию. То есть, Monocle 3 использует крупный граф AGA для изучения мелкой траектории.
[00399] Реализация Monocle 3 вышеупомянутых процедур масштабируется до миллионов клеток. Вкратце, он использует функцию лювеновской кластеризации из пакета программ igraph для обнаружения сообщества. Затем, основные расчеты AGA, сделанные Wolf и др., обрабатываются на компьютере с помощью ряда операций на разреженных матрицах. X можно принять за (разреженную) матрицу, представляющую член сообщества клеток. Каждый столбец X представляет лювенское сообщество, а каждый ряд X соответствует конкретной клетке. Xij=1, если клеткам принадлежит лювенскому сообществу j, а в противном случае Xij=0. Далее, можно получить матрицу смежности А графа kNN, используемую для осуществления кластеризации Лювена, где Aij=1, если клетка i соединяется с j на графе kNN. Затем матрицу М соединения между каждым кластером вычисляют по формуле:
[00400] М=ХТАХ
[00401] После построения М, авторы могут следовать Дополнительному примечанию 3.1 из ссылки81 для вычисления значимости связи между каждой кластеризацией Лювена, и рассмотреть любые кластеры с величиной р более, чем 0,05 по умолчанию, как неразрывные.
[00402] Изучение основного графа
[00403] Монокль 3 позволяет исследовать основной граф, который находится в таком же малоразмерном пространстве, как и данные для представления возможных путей развития клеток. В Monocle 3 используется расширенная реализация алгоритма84 Ll-графа для изучения основного графа. Мао и др. описали два варианта алгоритма Ll-графа84. В первом алгоритме («Алгоритм 1»), эти данные были оптимизированы относительно всех отдельных исходных данных в базе данных. Ранее авторами было показано, что, хотя L1-граф может применяться к данным RNA-seq для отдельных клеток, однако, он позволяет изучать наиболее «шумные» графы, которые не устойчивы к снижению выборки, и этот алгоритм эффективно не масштабируется по наборам данных за пределами нескольких сотен клеток85. Авторы не исследовали «Алгоритм 2», описанный в работе Qiu et al., где сначала осуществляют отбор «ориентировочных» исходных данных с использованием алгоритма кластеризации K-средних. Затем алгоритм оптимизируют по сравнению с этой гораздо меньшей выборкой данных. В Monocle 3 используется алгоритм, который, при его применении к клеткам в пространстве UMAP, является надежным, и с некоторыми ключевыми изменениями, может масштабироваться до миллионов клеток.
[00404] Реализация авторами алгоритма L1-графа имеет несколько ключевых особенностей, которые позволяют проанализировать большие наборы данных и достоверно выделить основной граф. Сначала авторы изучали L1-граф в пространстве UMAP (по умолчанию, в 3-мерном). Авторами была использована кластеризация K-медиоидов для отбора «ориентировочных» клеток в целях ускорения оптимизации. Количество выбранных «ориентировочных» клеток влияет на время работы алгоритма и качество решения: слишком много ориентиров приведет к неразрешимой проблеме линейного программирования. Поэтому, авторами было определено количество ориентиров в зависимости от данных путем присваивания К сообществам, которые были обнаружены среди клеток, и которые были в три раза больше лювенских сообществ, что на практике приводило к быстрым и стабильным решениям.
[00405] Вторая важная оптимизация L1-графа заключается в том, что авторы накладывают ограничения на «допустимое» пространство всех возможных графов W, рассматриваемых при оптимизации. Мао et al. рассмотрели все возможные границы между ориентировочными исходными данными. Однако, даже при наличии всего лишь тысячи «ориентировочных» клеток, задача линейного программирования быстро может стать неосуществимой, поскольку число переменных зависит от числа ребер в графе. В Monocle 3, авторы вводят ребра только в допустимое пространство, которое находится либо в дереве минимального охвата (MST), построенного на «ориентировочных» точках, либо в графе kNN (по умолчанию k=3), построенном на вершинах с нечетной степенью в МСТ. И наконец, авторами были исключены ребра, которые связывали бы клетки в различных соединенных компонентах графа AGA, построенного как описано в предыдущем разделе.
[00406] Идентификация генов, экспрессия которых зависит от траектории
[00407] Для идентификации генов, различающихся по экспрессии в зависимости от траектории развития, авторы позаимствовали статистический критерий, обычно используемый при анализе пространственных данных. Статистический I-критерий Морана представляет собой критерий разнонаправленной и многомерной пространственной автокорреляции. Эта статистический метод позволяет определить пространственные взаимосвязи между исходными данными через граф ближайших соседей, что делает его особенно подходящим для анализа больших наборов данных RNA-seq для отдельных клеток.
[004 08] Т-критерий Морана86 определяют по формуле:
[00409] 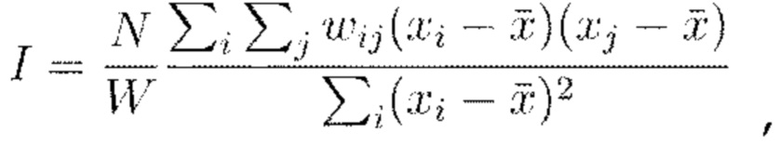
[00410] где N означает число клеток, проиндексированных как i и j, а х означает величину экспрессии представляющего интерес гена;  представляет собой среднее значение экспрессии генов для ближайших соседей клеток i' (или j'); ωij представляет собой весовую матрицу, определенную графом ближайших соседей с нулем по диагонали (то есть, ωii=0), и ωij=1/ki, где ki означает число ближайших соседей, a W равно сумме всех ωij.
представляет собой среднее значение экспрессии генов для ближайших соседей клеток i' (или j'); ωij представляет собой весовую матрицу, определенную графом ближайших соседей с нулем по диагонали (то есть, ωii=0), и ωij=1/ki, где ki означает число ближайших соседей, a W равно сумме всех ωij.
[00411] Для идентификации ближайших соседей, используемых для создания весовой матрицы W, авторами был сначала построен граф ближайших соседей к (по умолчанию 25) (kNN) для всех клеток в пространстве UMAP. Авторами также была проецирована каждая клетка на ближайший узел в основном графе. Затем авторы удалили все ребра из графа kNN, которые соединяют клетки, проецируемые на узлы основного графа, который не является частью ребра.
[00412] В Monocle 3 авторами была реализована функция manifoldTest для идентификации множества скоррелированных генов, которая зависит от модифицированных версий рутинных программ, взятых из пакета программ spdep для реализации I-критерия Морана.
[00413] Библиография
[00414] 1. Fogarty, N. М. Е. et al. Genome editing reveals a role for OCT4 in human embryogenesis. Nature 550, 67-73 (2017).
[00415] 2. Kojima, Y., Tam, О. H. & Tam, P. P. L. Timing of developmental events in the early mouse embryo. Semin. Cell Dev. Biol. 34, 65-75 (2014).
[00416] 3. Tam, P. P. L. & Loebel, D. A. F. Gene function in mouse embryogenesis: get set for gastrulation. Nat. Rev. Genet. 8, 368-381 (2007).
[00417] 4. Rivera-Péres J. A. & Hadjantonakis, A.-K. The Dynamics of Morphogenesis in the Early Mouse Embryo. Cold Spring Harb. Perspect. Biol. 7, a015867 (2014).
[00418] 5. Dickinson, M. E. et al. High-throughput discovery of novel developmental phenotypes. Nature 537, 508-514 (2016).
[00419] 6. Meehan, Т. F. et al. Disease model discovery from 3,328 gene knockouts by The International Mouse Phenotyping Consortium. Nat. Genet. 49, 1231-1238 (2017).
[00420] 7. Shyer, A. E., Huycke, T. R., Lee, C, Mahadevan, L. & Tabin, C. J. Bending gradients: how the intestinal stem cell gets its home. Cell 161, 569-580 (2015).
[00421] 8. Uygur, A. et al. Scaling Pattern to Variations in Size during Development of the Vertebrate Neural Tube. Dev. Cell 37, 127-135 (2016).
[00422] 9. Gorkin, D. et al. Systematic mapping of chromatin state landscapes during mouse development. (2017). doi:10.1101/166652
[00423] 10. Mayer, C. et al. Developmental diversification of cortical inhibitory interneurons. Nature 555, 457-462 (2018).
[00424] 11. Lescroart, F. et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science (2018). doi:10.1126/science.aao4174
[00425] 12. Han, X. et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 172, 1091-1107.e17 (2018).
[00426] 13. The Tabula Muris Consortium, Quake, S. R., Wyss-Coray, T. & Darmanis, S. Transcriptomic characterization of 20 organs and tissues from mouse at single cell resolution creates a Tabula Muris. (2017). doi:10.1101/237446
[00427] 14. Amini, S. et al. Haplotype-resolved whole-genome sequencing by contiguity-preserving transposition and combinatorial indexing. Nat. Genet. 46, 1343-1349 (2014).
[00428] 15. Adey, A. et al. In vitro, long-range sequence information for de novo genome assembly via transposase contiguity. Genome Res. 24, 2041-2049 (2014).
[00429] 16. Cusanovich, D. A. et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910-914 (2015).
[00430] 17. Vitak, S. A. et al. Sequencing thousands of single-cell genomes with combinatorial indexing. Nat. Methods 14, 302-308 (2017).
[00431] 18. Ramani, V. et al. Massively multiplex single-cell Hi-C. Nat. Methods 14, 263-266 (2017).
[00432] 19. Cao, J. et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661-667 (2017).
[00433] 20. Mulqueen, R. M. et al. Scalable and efficient single-cell DNA methylation sequencing by combinatorial indexing. (2017). doi:10.1101/157230
[00434] 21. Rosenberg, A. B. et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science (2018). doi:10.112б/science.aam8999
[00435] 22. Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
[00436] 23. Qiu, X. et al. Reversed graph embedding resolves complex single-cell developmental trajectories. (2017). doi:10.1101/110668
[00437] 24. Fernandez, T. et al. Disruption of contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am. J. Hum. Genet. 74, 1286-1293 (2004).
[00438] 25. Olson, J. M. et al. NeuroD2 is necessary for development and survival of central nervous system neurons. Dev. Biol. 234, 174-187 (2001).
[00439] 26. Uittenbogaard, M., Baxter, К. K. & Chiaramello, A. NeuroD6 Genomic Signature Bridging Neuronal Differentiation to Survival via the Molecular Chaperone Network. J. Neurosci. Res. 88, 33 (2010).
[00440] 27. Yang, A. et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398, 714-718 (1999).
[00441] 28. McQualter, J. L., Yuen, K., Williams, B. & Bertoncello, I. Evidence of an epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proc. Natl. Acad. Sci. U. S. A. 107, 1414-1419 (2010).
[00442] 29. Cichorek, M., Wachulska, M., Stasiewicz, A. & Tymińska A. Skin melanocytes: biology and development. Advances in Dermatology and Allergology 1, 30-41 (2013).
[00443] 30. Tomihari, M., Hwang, S.-H., Chung, J.-S., Cruz, P. D., Jr. & Ariizumi, K. Gpnmb is a melanosome-associated glycoprotein that contributes to melanocyte/keratinocyte adhesion in a RGD-dependent fashion. Exp.Dermatol. 18, 586-595 (2009).
[00444] 31. Varjosalo, M. & Taipale, J. Hedgehog: functions and mechanisms. Genes Dev. 22, 2454-2472 (2008).
[00445] 32. Strahle, U., Lam, C. S., Ertzer, R. & Rastegar, S. Vertebrate floor-plate specification: variations on common themes. Trends Genet. 20, 155-162 (2004).
[00446] 33. Holmes, G. P. et al. Distinct but overlapping expression patterns of two vertebrate slit homologs implies functional roles in CNS development and organogenesis. Mech. Dev. 79, 57-72 (1998).
[00447] 34. Akle, V. et al. F-spondin/sponlb expression patterns in developing and adult zebrafish. PLoS One 7, e37593 (2012).
[00448] 35. Hartman, В. H., Durruthy-Durruthy, R., Laske, R. D., Losorelli, S. & Heller, S. Identification and characterization of mouse otic sensory lineage genes. Front. Cell. Neurosci. 9, 79 (2015).
[00449] 36. Petit, F., Sears, К. E. & Ahituv, N. Limb development: a paradigm of gene regulation. Nat. Rev. Genet. 18, 245-258 (2017).
[00450] 37. Guo, Q., Loomis, C. & Joyner, A. L. Fate map of mouse ventral limb ectoderm and the apical ectodermal ridge. Dev. Biol. 264, 166-178 (2003).
[00451] 38. Lewandoski M, E. al. Fgf8 signalling from the AER is essential for normal limb development. - PubMed - NCBI. Available at: https://www.ncbi.nlm.nih.gov/pubmed/11101846. (Accessed: 22nd April 2018)
[00452] 39. Aoki M, E. al. R-spondin2 expression in the apical ectodermal ridge is essential for outgrowth and patterning in mouse limb development. - PubMed - NCBI. Available at: https://www.ncbi.nlm.nih.gov/pubmed/18067586. (Accessed: 22nd April 2018)
[00453] 40. Gerdes, J., Schwab, U., Lemke, H. & Stein, H. Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int. J. Cancer 31, 13-20 (1983).
[00454] 41. Bergman, D., Halje, M., Nordin, M. & Engström W. Insulin-like growth factor 2 in development and disease: a mini-review. Gerontology 59, 240-249 (2013).
[00455] 42. Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381-386 (2014).
[00456] 43. Mclnnes, L. & Healy, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. (2018).
[00457] 44. Alexander Wolf, F. et al. Graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. bioRxiv 208819 (2017). doi:10.1101/208819
[00458] 45. Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979-982 (2017).
[00459] 46. Singh, M. K. et al. The T-box transcription factor Tbxl5 is required for skeletal development. Mech. Dev. 122, 131-144 (2005).
[00460] 47. Paine-Saunders, S., Viviano, B. L., Zupicich, J., Skarnes, W. C. & Saunders, S. glypican-3 controls cellular responses to Bmp4 in limb patterning and skeletal development. Dev. Biol. 225, 179-187 (2000).
[00461] 48. Hara, K. & Ide, H. Msx1 expressing mesoderm is important for the apical ectodermal ridge (AER)-signal transfer in chick limb development. Dev. Growth Differ. 39, 705-714 (1997).
[00462] 49.  D. G. et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 161, 1012-1025 (2015).
D. G. et al. Disruptions of Topological Chromatin Domains Cause Pathogenic Rewiring of Gene-Enhancer Interactions. Cell 161, 1012-1025 (2015).
[00463] 50. Davis, R. J. et al. Dachl mutant mice bear no gross abnormalities in eye, limb, and brain development and exhibit postnatal lethality. Mol. Cell. Biol. 21, 1484-1490 (2001).
[00464] 51. Akiyama, H., Chaboissier, M.-C, Martin, J. F., Schedl, A. & de Crombrugghe, B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813-2828 (2002).
[00465] 52. Deng, Y. et al. Yap1 Regulates Multiple Steps of Chondrocyte Differentiation during Skeletal Development and Bone Repair. Cell Rep.14, 2224-2237 (2016).
[00466] 53. Joshi, S. et al. TEAD transcription factors are required for normal primary myoblast differentiation in vitro and muscle regeneration in vivo. PLoS Genet. 13, е1006600 (2017).
[00467] 54. Knapp, D. et al. Comparative transcriptional profiling of the axolotl limb identifies a tripartite regeneration-specific gene program. PLoS One 8, e61352 (2013).
[00468] 55. Zeller, R., López-Rios J. & Zuniga, A. Vertebrate limb bud development: moving towards integrative analysis of organogenesis. Nat. Rev. Genet. 10, 845-858 (2009).
[00469] 56. Nishimoto, S., Minguillon, C, Wood, S. & Logan, M. P. 0. A combination of activation and repression by a colinear Hox code controls forelimb-restricted expression of Tbx5 and reveals Hox protein specificity. PLoS Genet. 10, el004245 (2014).
[00470] 57. Vargesson, N., Luria, V., Messina, I., Erskine, L. & Laufer, E. Expression patterns of Slit and Robo family members during vertebrate limb development. Mech. Dev. 106, 175-180 (2001).
[00471] 58. Chimal-Monroy, J. et al. Analysis of the molecular cascade responsible for mesodermal limb chondrogenesis: Sox genes and BMP signaling. Dev. Biol. 257, 292-301 (2003).
[00472] 59. Braun, T. & Gautel, M. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat. Rev. Mol. Cell Biol. 12, 349-361 (2011).
[00473] 60. Tajbakhsh, S., Rocancourt, D., Cossu, G. & Buckingham, M. Redefining the genetic hierarchies controlling skeletal myogenesis: Pax-3 and Myf-5 act upstream of MyoD. Cell 89, 127-138 (1997).
[00474] 61. Harel, I. et al. Distinct origins and genetic programs of head muscle satellite cells. Dev. Cell 16, 822-832 (2009).
[00475] 62. Sambasivan, R. et al. Distinct regulatory cascades govern extraocular and pharyngeal arch muscle progenitor cell fates. Dev. Cell 16, 810-821 (2009).
[00476] 63. Heimberg, G., Bhatnagar, R., El-Samad, H. & Thomson, M. Low Dimensionality in Gene Expression Data Enables the Accurate Extraction of Transcriptional Programs from Shallow Sequencing. Cell Syst 2, 239-250 (2016).
[00477] 64. Cusanovich, D. A. et al. The cis-regulatory dynamics of embryonic development at single cell resolution. (2017). doi:10.1101/166066
[00478] 65. McKenna, A. et al. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353, aaf7907 (2016).
[00479] 66. Osterwalder, M. et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554, 239-243 (2018).
[00480] 67. Dickel, D. E. et al. Ultraconserved Enhancers Are Required for Normal Development. Cell 172, 491-499.el5 (2018).
[00481] 68. Li, D. et al. Formation of proximal and anterior limb skeleton requires early function of Irx3 and Irx5 and is negatively regulated by Shh signaling. Dev. Cell 29, 233-240 (2014).
[00482] 69. Kraft, K. et al. Deletions, Inversions, Duplications: Engineering of Structural Variants using CRISPR/Cas in Mice. Cell Rep.(2015). doi:10.1016/j.celrep.2 015.01.016
[00483] 70. Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213-1218 (2013).
[00484] 71. Renaud, G., Stenzel, U., Maricic, Т., Wiebe, V. & Kelso, J. deML: robust demultiplexing of Illumina sequences using a likelihood-based approach. Bioinformatics 31, 770-772 (2015).
[00485] 72. Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15-21 (2013).
[00486] 73. Anders, S., Pyl, P. T. & Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics btu638 (2014).
[00487] 74. Qiu, X. et al. Reversed graph embedding resolves complex single-cell developmental trajectories. (2017). doi:10.1101/110668
[00488] 75. Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
[00489] 76. Cao, J. et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661-667 (2017).
[00490] 77. Pliner, H. et al. Chromatin accessibility dynamics of myogenesis at single cell resolution. (2017). doi:10.1101/155473
[00491] 78. Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90-7 (2016).
[00492] 79. McInnes, L. & Healy, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. (2018).
[00493] 80. Levine, J. H. et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162, 184-197 (2015).
[00494] 81. Wolf, F. A. et al. Graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. (2017). doi:10.1101/208819
[00495] 82. Wagner, D. E. et al. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science eaar4362 (2018).
[00496] 83. Briggs, J. A. et al. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science eaar5780 (2018).
[00497] 84. Mao, Q., Wang, L., Tsang, I. & Sun, Y. Principal Graph and Structure Learning Based on Reversed Graph Embedding. IEEE Trans. Pattern Anal. Mach. Intell. (2016). doi:10.1109/TPAMI.2 016.2 635657
[00498] 85. Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979-982 (2017).
[00499] 86. Moran, P. A. P. Notes on continuous stochastic phenomena. Biometrika 37, 17-23 (1950).
[00500] Пример 2
[00501] Новый метод выделения ядер из ткани и их фиксации (sc-RNA-seq)
[00502] Реагенты: BSA (с чистотой, используемой в молекулярной биологии, NEB, # B9000S); ингибитор Super-РНКазы (Thermo, №АМ2696); EMS 157-4-100, 4% водный раствор параформальдегида (формальдегида), класса ЕМ, 100 мл (Amazon).
[00503] Буферы: Буфер для ядер (хранится при 4°С): 10 мМ Трис-HCl, рН 7,4, 10 мМ NaCl, 3 мМ MgCl2, 10% IGEPAL СА-630 (хранится при 4°С). Буфер для промывки ядер (каждый раз заменяли свежим): 98 0 мкл буфера для ядер с 10 мкл BSA и 10 мкл ингибитора Super-РНКазы тщательно перемешивали и хранили на льду. Буфер для лизиса ядер (каждый раз заменяли свежим): буфер для промывки ядер с 0,1% IGEPAL СА-630.
[00504] Выделение ядер непосредственно из ткани
[00505] Ткани измельчали на мелкие кусочки лезвием бритвы в 1 мл охлажденного льдом буфера для лизиса клеток (10 мМ Трис-, рН 7,4, 10 мМ NaCl, 3 мМ и 0,1% IGEPAL СА-630, 1% ингибитора Super-РНКазы и 1% BSA) и переносили в верхнюю часть 4 0 мкм-фильтра для клеток (Falcon).
[00506] Ткани гомогенизировали с помощью резинового наконечника поршня шприца (5 мл, BD) в 4 мл буфера для лизиса клеток.
[00507] Затем, отфильтрованные ядра переносили в новую пробирку объемом 15 мл (Falcon) и осаждали путем центрифугирования при 500× g в течение 5 минут и один раз промывали 1 мл буфера для лизиса клеток.
[00508] Фиксация ядер
[00509] Ядра фиксировали в 4 мл охлажденного льдом 4% параформальдегида (EMS) в течение 15 минут на льду.
[00510] После фиксации, ядра два раза промывали в 1 мл буфера для промывки ядер (буфера для лизиса клеток без IGEPAL) и ресуспендировали в 500 мкл буфера для промывки ядер.
[00511] Образцы распределяли на несколько партий и мгновенно замораживали в жидком азоте. Замороженные образцы могут быть перенесены на сухой лед.
[00512] Пример 3
[00513] Характеризация динамики перехода клеток из одного состояния в другое по sci-судьбе
[00514] Развитие живого организма заключается в прохождении клетками различных состояний в строго организованном временном порядке. Несмотря на все возрастающее применение геномных методов для отдельных клеток, количественная оценка динамики перехода клетки из одного состояния в другое остается сложной задачей. В данном случае, авторами вводится понятие «sci-судьба» как высокопроизводительный анализ на основе комбинаторного индексирования, проводимый для профилирования как целого, так и вновь синтезированного транскриптома в каждой из тысяч отдельных клеток. В качестве доказательства этой концепции, авторы применили понятие sci-судьба к модельной системе ответа на кортизол и охарактеризовали более 6000 событий перехода клетки из одного состояния в другое в соответствии с известной динамикой клеточных циклов после активации глюкокортикоидного рецептора. Исходя из этого анализа, авторами было показано, что направление перехода клетки из одного состояния в другое и его вероятность регулируются расстояниями между данными состояниями и природой нестабильности таких состояний. Эта техника и вычислительные методы могут быть легко применимы и к другим биологическим системам для количественной характеризации динамики состояний клеток и расшифровки внутреннего механизма определения судьбы клеток.
[00515] Клетка проходит различные функциональные и молекулярные состояния во время развития многоклеточного организма. Характеризация пути перехода клетки из одного состояния в другое или судьбы клетки является основой для понимания процессов развития и их применения в таких областях, как клеточная инженерия. Несмотря на все возрастающее применение геномных методов для отдельных клеток, эти методы позволяют получить изображения только состояния клетки, а поэтому, не могут предоставить информацию о динамике клеточных переходов (1). Хотя для характеристики клеточных переходов из одного состояния в другое может быть применен метод отслеживания отдельных клеток с помощью времяпролетной микроскопии (2, 3), однако, эти методы имеют ограниченную пропускную способность и могут отслеживать изменения только в нескольких генах, а следовательно, имеют низкую способность к расшифровке сложных систем.
[00516] В настоящей заявке авторами была описана новая стратегия количественной оценки динамики перехода клетки из одного состояния в другое на уровне всего транскриптома. Эта стратегия зависит от новой методики PHK-seq для отдельных клеток на основе комбинаторного индексирования, sci-судьбы. Путем мечения вновь синтезированной мРНК 4-тиоуридином (4, 5), который будет давать точковые мутации C>T во время обратной транскрипции, sci-судьба охватывает как целый транскриптом, так и вновь синтезированный транскриптом на уровне отдельных клеток, вместе с информацией о деградированном транскриптоме в прошлом (память о прежнем состоянии). Память о прежних состояниях каждой клетки затем корректируют по скорости разложения мРНК (методом коррекции памяти), так, чтобы каждая клетка могла быть охарактеризована по динамике транскриптома между двумя временными точками.
[00517] Для характеризации динамики перехода клетки из одного состояния в другое, регулируемой внутренними и внешними факторами, авторами была применено понятие sci-судьба к модельной системе ответа на кортизол, где судьба клетки запускается двумя основными факторами: программой внутреннего клеточного цикла и внешней активацией глюкокортикоидного рецептора (GR), индуцируемой лекарственным средством. Активация GR влияет на активность почти каждой клетки в организме и регулирует гены, контролирующие развитие, обмен веществ и иммунный ответ (6). С помощью sci-судьбы авторы определили динамику всего транскриптома для более чем 6000 отдельных клеток. Исходя из сходства между прошлым и текущим состояниями транскриптома, авторами были построены тысячи траекторий перехода клеток из одного состояния в другое, охватывающих пять временных точек, которые могут быть сгруппированы в три типа судьбы клеток в соответствии с известными паттернами прохождения клеточного цикла при активации GR. Кроме того, авторами были охарактеризованы скрытые клеточные состояния по функциональной активности модулей TF, и был сделан вывод о сети клеточных переходов для прогнозирования состояния клетки. И наконец, авторами было показано, что направление и вероятность перехода клеток из одного состояния в другое регулируются сходством транскриптомов и природой нестабильности соседних состояний. Разработанные здесь теоретические, вычислительные и экспериментальные подходы должны быть легко применимы к другим биологическим системам, в которых динамика клеточных переходов до сих пор неизвестна.
[00518] Общий обзор sci-судьбы
[00519] Анализ sci-судьбы осуществляют путем проведения следующих стадий (фиг.30А): (i) клетки сначала инкубируют с 4-тиоуридином (S4U), широко используемым аналогом тимидина, для мечения вновь синтезированной РНК (7-13). (ii) Клетки собирают, фиксируют 4% параформальдегидом, после чего проводят реакцию алкилирования посредством связывания с тиолом (SH), в результпте которой происходит ковалентное присоединение карбоксиамидетильной группы к S4U посредством нуклеофильного замещения (4). (iii) Клетки распределяют по массе в каждую лунку 4×96-луночных планшетов. Первый молекулярный индекс PHK-seq вводят в мРНК клеток в каждую лунку посредством обратной транскрипции (ОТ) in situ с помощью поли(Т)-праймера, имеющего специфичный к лунке штрих-код и вырожденный уникальный молекулярный идентификатор (UMI). Во время синтеза кДНК, мРНК, меченная модифицированным S4U, имитирует превращение тимина в цитозин (Т>С) и приводит к образованию мутированной кДНК первой цепи. (iv) Клетки из всех лунок объединяют, а затем перераспределяют путем клеточного сортинга с активацией флуоресценции (FACS) во множестве 96-луночных планшетов. Клетки стробируют окрашиванием DAPI (4',6-диамидино-2-фенилиндолом), чтобы отличить одну клетку от дублетов во время сортинга. Двухцепочечную кДНК получают путем разложения РНК и синтеза второй цепи и подвергают транспозиции с Tn5. Затем, кДНК амплифицируют с помощью полимеразной цепной реакции (ПЦР) с комбинацией праймеров, распознающих -адаптер на 5'-конце и ОТ-праймер на 3'-конце. Эти праймеры также имеют специфичный для лунки штрих-код, который вводит второй молекулярный индекс RNA-seq. (v) ПЦР-ампликоны объединяют и подвергают массивному параллельному секвенированию. Как и в случае с другими «sci»-протоколами (14-21), большинство ядер проходит через уникальную комбинацию лунок, а поэтому содержимое каждой лунки помечают уникальной комбинацией штрих-кодов, которые могут быть использованы для группировки ридов, происходящих из одной и той же клетки. Вновь синтезированная мРНК из всего транскриптома идентифицируется по превращениям «Т>С» с поправкой на фоновую ошибку (Метод).
[00520] Для оценки контроля качества, авторы сначала протестировали метод с использованием смеси клеток (человеческих) и NIH/3T3 (мышиных) в четырех условиях: с мечением S4U или без мечения (200 нМ, 6 часов) и с обработкой IAA или без нее (фиг. 31A-D). При мечении S4U и обработке IAA (состояние sci-судьбы), транскриптомы из человеческих/мышиных клеток были в значительной степени видо-специфическими (>99% чистота для человеческих и мышиных клеток, 2,6% соударений) с высоким отношением обнаруженных мутированных ридов Т>С (46% для человеческих клеток и 31% для мышиных клеток в состоянии sci-судьбы по сравнению с 0,8% для человеческих клеток и 0,8% для мышиных клеток в условиях без обработки). Авторами была достигнута почти эквивалентная чистота клеток в четырех условиях, хотя в группах обработки IAA были обнаружены несколько более низкие UMI. Объединенные транскриптомы после оценки sci-судьбы по сравнению с нормальным sci-RNA-seq были в высокой степени скоррелированными (корреляция Спирмена r=0,99; фиг. 31Е-F), что позволяет предположить, что кратковременный процесс мечения и превращения оказывает минимальное влияние на состояние клетки.
[00521] Совместное профилирование общего и вновь синтезированного транскриптома в клетках А549, обработанных дексаметазоном.
[00522] Затем авторы применили анализ sci-судьбы к модели ответа на кортизол, где дексаметазон (DEX), синтетический имитатор кортизола активирует глюкокортикоидный рецептор (GR), который связывается с тысячами положений по всему геному и значительно изменяет состояние клетки за короткий промежуток времени (22-25). Авторы обрабатывали клетки А549, полученные из аденокарциномы легкого, в течение 0, 2, 4, 6, 8 или 10 часов 100 мМ DEX. В каждом состоянии, клетки инкубировали с S4ET (200 нМ) в течение последних двух часов перед сбором клеток в лунках 384 × 192 для оценки sci-судьбы (фиг. 30В). Каждое из этих шести условий представлено в 64 лунках во время первого цикла индексирования так, чтобы условия обработки можно было восстановить на основе первого индекса каждой клетки.
[00523] После фильтрации низкокачественных клеток, потенциальные дублеты и небольшую подгруппу дифференцированных клеток (Метод), авторами были определены профили отдельных клеток для 6680 клеток (медиана для 26176 детектированых мРНК, на клетку) с медианой 20% меченных UMI на клетку (фиг. 30С, фиг. 32А-В). Интронные риды обнаруживали значительно более высокий уровень вновь синтезированной мРНК, чем экзонные риды (65% в интронных ридах и 13% в экзонных ридах, р-величина <2,2е-16, ранговый критерий знаков Уилкоксона; фиг. 30D), что соответствует ожиданиям, что интронные риды будут обогащенными во вновь синтезированном транскриптоме.
[00524] Сначала авторы задались вопросом, передают ли весь транскриптом и вновь синтезированный транскриптом различную информацию по характеризации состояния клетки. Авторами были объединены весь транскриптом и вновь синтезированный транскриптом для каждого условия обработки и была оценена их корреляция. В отличие от всего транскриптома, вновь синтезированный транскриптом обнаруживал резкое различие для групп, не обработанных DEX (О ч.), и DEX-обработанных групп (фиг. 32С). В соответствии с этим, уменьшение размерности с помощью однородной множественной аппроксимации и проецирования (UMAP) (26) для всего или вновь синтезированного транскриптома дает разные результаты (фиг. 30Е): весь транскриптом не может разделять клетки, которые не были обработаны DEX (0 ч.), и клетки с ранней обработкой DEX (2 часа), в то время как вновь синтезированный транскриптом объединяет все DEX-обработанные клетки в одну группу. Клеточные кластеры, идентифицированные по целому или вновь синтезированному транскриптому, не полностью совпадают друг с другом (фиг.30F, фиг. 32D-E). Это и ожидалось, поскольку вновь синтезированный транскриптом непосредственно отражает активность промотора гена или эпигенетический ответ на внешнюю среду, тогда как весь транскриптом, в основном, определяется по остатку мРНК из его прежнего состояния.
[00525] Для характеризации состояния клеток с помощью общей информации, авторы объединили главные компоненты (PC) из целого и вновь синтезированного транскриптома для анализа UMAP. Общая информация разделяет клетки на клетки без DEX-обработки (0 ч.), клетки с ранней обработкой (2 часа) и с поздней обработкой (>2 часов) (фиг. 30Е). Интересно отметить, что два кластера (кластер 1 и 4), характеризующиеся целым транскриптомом, были разделены на четыре отдельные группы по общей информации (фиг. 30F). Авторы оценили уровень экспрессии и скорость нового синтеза генных маркеров, ассоциированных с клеточным циклом (27) (фиг. 30G, фиг. 32F-G): вновь разделенные кластеры по общей информации соответствуют клеткам в фазе G2/M (высокий уровень экспрессии и высокая скорость синтеза маркеров G2/M) и клеткам в ранней фазе G0/G1 (высокий уровень экспрессии и низкая скорость синтеза маркеров G2/M). Это позволяет предположить, что вновь синтезированный транскриптом передает другую информацию о состоянии клетки по сравнению со всем транскриптомом, а общая информация может обеспечивать более высокое разрешение при характеризации состояния клетки.
[00526] Характеризация функциональных модулей TF, позволяющих определить судьбу клеток.
[00527] Далее авторами была сделана попытка
охарактеризовать модули TF, запускающие процесс перехода клеток из одного состояния в другое. Связи между факторами транскрипции (TF) и их регулируемыми генами были идентифицированы в два этапа: для каждого гена, авторы вычислили корреляции между скоростью синтеза мРНК в течение последних двух часов и уровнем экспрессии TF для более, чем 6000 клеток с использованием LASSO (наименьшей абсолютной усадкой и отбором оператора). Эти идентифицированные связи были дополнительно отфильтрованы либо с помощью анализа опубликованных данных CHIP-seq (28), либо анализа на обогащения мотива (29) (Метод). Было идентифицировано всего 986 связей между 29 TF и 532 генами (фиг. 33А, Таблица S1) на основе ковариации TF-гена и подтверждения данных по связыванию ДНК. Для оценки возможности того, что связи являются артефактами регулируемой регрессии, авторы сделали перестановку идентификаторов выборки матрицы экспрессии TF и провели тот же самый анализ. После этой перестановки, связи не были обнаружены.
[00528] Были идентифицированы модули TF, запускающие GR-ответ, включая известные эффекторы GR-ответа, такие как СЕВРВ (30) (фиг. 34А-В), FOXOl (37) и JUNB (32) (фиг. 33А). Авторы также обнаружили несколько новых модулей TF, ассоциированных с GR-ответом, включая YOD1 и GTF2IRD1, с повышенной экспрессией и активностью в DEX-обработанных клетках (фиг. 34C-D). Были идентифицированы основные модули TF, запускающие процесс прохождения клеточного цикла, и эти модули включают E2F1, E2F2, E2F7, BRCA1 и MYBL2 (33). По сравнению с общим уровнем экспрессии, скорость синтеза генов новой РНК, регулируемых модулями TF клеточного цикла, в большей степени коррелирует с экспрессией TF-мишени (фиг. 34Е). Кроме того, авторами были также обнаружены модули TF, ассоциированные с дифференцировкой клеток, такие как GATA3, в основном, экспрессирующиеся в группе популяции покоящихся клеток (34), и модули TF, ассоциированные с реакцией на окислительный стресс, такие как NRF1 (35) и NFE2L2 (NRF2) (36).
[00529] Затем авторы охарактеризовали активность TF путем объединения скоростей синтеза генов новой РНК в каждом модуле TF и вычислили абсолютный коэффициент корреляции между каждой парой TF (фиг. 34F). В высокой степени скоррелированная активность TF указывает на то, что эти пары могут функционировать в комбинации друг с другом. Иерархическая кластеризация разделяет эти 29 модулей TF на пять основных модулей (фиг. 34F): первый модуль представляет собой все модули TF, ассоциированные с клеточным циклом, такие как E2F1 и FOXM1 (33), и представляет собой фактор, запускающий процесс прохождения клеточного цикла. Третий модуль представляет собой все модули TF, ассоциированные с GR-ответом, такие как FOXOl, СЕВРВ, JUNB и RARB (30) (31) (32). Другие группы модулей TF включают три TF (KLF6, TEAD1 и YOD1), совместно регулируемые как клеточным циклом, так и GR-ответом (модуль 2); внутренний путь дифференцировки, включая GATA3 и AR (модуль 3), и TF, ассоциированные с ответом на стресс, такие как NRF1 и NFE2L2 (модуль 5).
[00530] Для идентификации различных состояний клеточного цикла, авторы сначала упорядочили клетки по активности модуля TF, ассоциированного с клеточным циклом. Клетки были упорядочены по сглаженной траектории клеточного цикла, что подтверждалось скоростью синтеза известных маркеров клеточного цикла (27) (фиг. 33 В). Авторы наблюдали разрыв между фазой G2/M и фазой G0/G1, что соответствует резкому изменению состояния клетки во время ее деления. Посредством неконтролируемой кластеризации авторами было идентифицировано девять состояний клеточного цикла, охватывающих фазы клеточного цикла G0/G1, S и G2/M, исходя из уровня экспрессии маркера клеточного цикла (фиг.33 В). Клетки могут быть упорядочены по другой сглаженной траектории с использованием модулей TF ассоциированных с GR-ответом. Эта траектория четко коррелирует со временем обработки DEX и динамикой известной активности TF, регулируемой активацией GR (фиг. 33С). Посредством неконтролируемого анализа на кластеризацию, авторы идентифицировали три клеточных кластера по GR-ответам, что соответствует отсутствию GR-ответа/низкому уровню/высокому уровню GR-ответа (фиг. 33С).
[00531] Затем авторы попытались количественно охарактеризовать скрытые клеточные состояния в системе (фиг. 35А). Девять состояний клеточного цикла и три состояния GR-ответа были показаны на фиг. 33В-С. Были идентифицированы все возможные комбинаторные состояния, причем, наименьшая группа включала 1,1% (74) от всех клеток (фиг. 33D). Наблюдаемая доля клеточных состояний была близка к ожидаемой при условии независимого отбора. Это соответствует низкому коэффициенту корреляции (корреляции Пирсона r=0,004) между активностью этих двух функциональных модулей TF для более, чем 6000 клеток. Для сравнения, путем анализа на снижение размерности и анализа на кластеризацию для всего и вновь синтезированного транскриптома, авторами было идентифицировано 6 основных кластеров (фиг. 35В). Эти основные кластеры могут быть легко определены путем объединения групп этих 27 состояний клеток (фиг. 33Е).
[00532] Характеризация траектории перехода клеток из одного состояния в другое и сети перехода таких состояний
[00533] С использованием всего транскриптома и вновь синтезированного транскриптома, охарактеризованных для каждой клетки, авторы могли сделать вывод о состоянии транскриптома одной клетки до мечения S4U (фиг. 36А). Восстановление предшествующего клеточного транскриптома зависит от двух параметров: скорости детектирования вновь синтезированных ридов с точки зрения sci-судьбы и скорости разложения (или полуразложения) каждой мРНК (Метод). Оба эти параметра можно оценить в одном и том же эксперименте по оценке sci-судьбы.
[00534] Сначала авторы оценивали уровень детектирования sci-судьбы. Авторы предполагают, что время полужизни мРНК является постоянным в различных условиях DEX-обработки. Это предположение было также подтверждено последующей проверкой стабильности. Согласно этому предположению, общий транскриптом, частично разрушенный за 2 часа до S4U-мечения, должен быть таким же в клетках, не обработанных DEX, и в клетках, обработанных DEX через 2 часа. Таким образом, различия в общем транскриптоме (объем) должны совпадать с различиями во вновь синтезированном транскриптоме (объем), скорректированными по степени обнаружения в зависимости от техники. Поскольку весь транскриптом и вновь синтезированный транскриптом были охарактеризованы в данном эксперименте, то авторы могли непосредственно определить уровень детектирования sci-судьбы. Различия во вновь синтезированной мРНК хорошо коррелируют с различиями в уровне экспрессии мРНК (критерий Пирсона, r=0,93, фиг. 37А), что позволяет предположить, что уровень детектирования новой РНК является довольно стабильным по всем генам. Таким образом, авторы использовали медиану скорости захвата новой РНК (82%) для последующего анализа.
[00535] Затем была вычислена степень разложения мРНК за 2 часа. Поскольку популяция клеток А549 может рассматриваться как стабильная без внешней пертурбации, то для клеток после 2-часовой обработки DEX, их прежнее состояние (за 2 часа до мечения S4U) должно быть таким же, как и у клеток, не обработанных DEX. Аналогично, прежнее состояние (перед мечением S4U) для клеток, обработанных DEX на время Т=0/2/4/6/8/10 часов, должно быть аналогично состоянию профилированных клеток на время Т=0/0/2/4/6/8 часов. Что касается всего транскриптома и вновь синтезированного транскриптома, охарактеризованных для всех условий обработки, можно оценить степень разложения мРНК для нескольких тысяч генов в каждом 2-часовом интервале времени. После вышеупомянутой проверки на стабильность было обнаружено, что степень разрушения генов значительно коррелируют со временем обработки DEX (фиг. 37В). Затем авторы использовали усредненную степень разрушения генов для последующего анализа. После определения усредненной степени детектирования новой мРНК и степени разрушения генов, авторы оценивали прежнее состояние транскриптома одной клетки так, чтобы каждая клетка могла быть охарактеризована по динамике транскриптома в двухчасовом интервале.
[00536] Для оценки динамики состояний клеток в течение более длительного интервала (то есть, в течение 10 часов), авторами был разработан потоковый алгоритм для оценки взаимосвязи родительских и дочерних клеток в одной и той же траектории перехода клеток из одного состояния в другое (фиг. 36А): для каждой клетки А (например, для клеток, обработанных DEX через 2 часа) был идентифицирован профиль клетки В в более ранний момент времени (например, для клеток, не обработанных DEX), и текущее состояние клетки В было аналогичным ее прежнему состоянию А, как было определено исходя из недавно разработанной стратегии выравнивания для идентификации общих состояний клеток между двумя наборами данных (27). Состояние В можно рассматривать как родительское состояние А. Аналогично, авторами был также определен профиль другой клетки С в более поздний момент времени (например, для клеток, обработанных DEX через 4 часа), и клетка С имела свое прежнее состояние, аналогичное текущему состоянию клетки А. Клетка С может рассматриваться как будущее состояние клетки А. Применяя ту же самую стратегию на все прежние и последующие состояния, определенные для каждой клетки, авторы построили 6680 траекторий перехода клеток из одного состояния в другое в течение 10 часов и в пяти временных точках (фиг. 36А-В). Следует отметить, что этот анализ основан на предположении, что прежнее и текущее состояния каждой клетки (кроме клеток в начальный и конечный моменты времени) были детально детектированы, что справедливо и для полученных авторами наборов данных, так как было профилировано более 6000 клеток (более 1000 клеток на одно условие), или одна клетка в течение менее одной минуты во время клеточного цикла. Был определен профиль состояния каждой из множества клеток (>50), и таким образом, может быть также зафиксирован стохастический процесс перехода клеток из одного состояния в другое.
[00537] Для подтверждения результата, авторами был проведен анализ на уменьшение размерности и неконтролируемую кластеризацию для траекторий 6680 отдельных клеток, которые были сгруппированы в три кластера траекторий. Авторами была оценена динамика клеточных состояний, охарактеризованных на фиг. 36С. Как и ожидалось, все три траектории показали переход клеток из одного состояния в другое в отсутствии GR-ответа при низком/высоком GR-ответе в течение определенного периода времени (фиг. 36D). Авторы наблюдали различную динамику клеточного цикла во всех этих трех траекториях (фиг. 36D): траектория 1 указывала на снижение фазы G2/M и соответственно, на увеличение фазы G0/G1, и на переход клетки из промежуточных состояний G2/M и G1 в фазу G1. Траектория 2 указывала на переход клетки из промежуточных состояний S и G2/M в фазу G2/M. На траектории 3 авторы наблюдали переход клетки из промежуточной фазы G1 и S в раннюю фазу S во время ранней обработки DEX (0-2 часа), но этот переход ингибировался в условиях более поздней обработки DEX (>2 часа после обработки DEX), что свидетельствует о том, что длительная обработка DEX приводит к остановке фазы G1. Это соответствует изменениям соотношений состояний клеток в течение определенного времени обработки и с данными предыдущих исследований (37,38) (фиг. 36D). Это позволяет предположить, что пути перехода отдельных клеток, характеризуемые sci-судьбой, могут восстанавливать общие направления перехода клеток из одного состояния в другое.
[00538] После определения профиля каждого состояния множества клеток (>70), авторами была определена вероятность перехода клеток из одного состояния в другое во всех 27 скрытых состояниях. Переходы клеток из одного состояния в другое с низкой вероятностью перехода (<0,1) могут быть вызваны редкими событиями или шумом и, таким образом, отфильтрованы. Сеть переходов клеток из одного состояния в другое может быть определена 2 7 состояниями клеток как узлы, а связи указывают на возможные пути перехода (фиг. 36Е). Направление прохождения клеточного цикла может быть легко охарактеризовано по меньшей мере тремя стадиями перехода с необратимыми направлениями перехода во время клеточного цикла (фиг. 36Е). В поздней фазе G1 и в поздней фазе G2/M, авторами было также обнаружено несколько состояний, указывающих на обратимую динамику переходов, которая может указывать на две контрольных точки клеточного цикла в фазах G1/S и G2/M (33). Как и ожидалось, клетки с одинаковым клеточным циклом, но с разными GR-ответами продемонстрировали резко отличающуюся динамику перехода, а клетки с высоким GR-ответом обычно имеют тенденцию к остановке фазы G1 или G2/M.
[00539] В качестве проверки на непротиворечивость, для того, чтобы подтвердить, может ли сеть перехода клеток из одного состояния в другое охватывать динамику перехода клеток из одного состояния в другое, авторы провели анализ для того, чтобы определить, могут ли вероятности перехода указывать на возможность восстановления реальных распределений состояний клетки в разные моменты времени. Действительно, хотя соотношения состояний клетки динамически изменяются в течение 10 часов (фиг. 36F), однако, сеть переходов состояний позволяет точно предсказать соотношения 27 состояний клеток во всех пяти более поздних временных точках, начиная от соотношения состояний DEX-обработанных клеток на 0 часов (фиг. 36G, фиг. 38А). Авторы также вычислили сеть переходов состояний клеток только для части данных (от 0 до 6 часов), которая давала в высокой степени скоррелированные вероятности перехода с полными данными, и точно предсказали состояния клеток за 10 часов (фиг. 36Н, фиг. 38 В).
[00540] Характеризация факторов, регулирующих направления перехода клеток из одного состояния в другое
[00541] Для характеризации факторов, регулирующих вероятность перехода состояний клеток, авторы сначала вычислили расстояние между состояниями клеток по расстоянию Пирсона для объединенного транскриптома (целого и вновь синтезированного) между каждой парой состояний. Как и ожидалось, вероятность перехода клетки из одного состояния в другое отрицательно коррелирует с расстоянием перехода (коэффициент корреляции Спирмена = -0,38, фиг. 39А). Авторы также вычислили нестабильность состояния, определяемую долей клеток, выходящих из данного состояния в течение двух часов (фиг. 39В). Природа нестабильности состояний хорошо коррелирует с направлениями перехода клетки (фиг. 39В): состояния в отсутствии GR-ответа являются более нестабильными по сравнению с состояниями с высоким GR-ответом. В состояниях с высоким GR-ответом, клетки в ранней фазе G1 имеют самую низкую нестабильность, в то время как клетки в промежуточных состояниях G1/S демонстрируют высокий пик нестабильности, что соответствует остановке фазы G1 при более поздней обработке DEX.
[00542] Изменения соотношения состояний клетки через 10 часов хорошо коррелирует с нестабильностью состояния клетки (коэффициент корреляции Спирмена = -0,88, фиг. 39С), что позволяет предположить, что динамика состояния клетки зависит от природы нестабильности состояния клетки. Нестабильность состояния также хорошо коррелирует с энтропией вероятности перехода состояний, что указывает на разнообразие конечных точек перехода состояний (корреляция Пирсона r=0,73, фиг. 39D). Для того, чтобы проверить, может ли вероятность перехода между состояниями быть следствием нестабильности соседних состояний, авторами был построен график зависимости нестабильности от расстояния до ближайших состояний в модели нейронной сети, для предсказания вероятности перехода из каждого состояния в другие состояния. Сочетание нестабильности соседних состояний и расстояний позволило повысить эффективность предсказания вероятности перехода между одним состоянием в другое более, чем в десять раз по сравнению с использованием только расстояний между состояниями (после перекрестной валидации, медианный критерий r2 составляет 0,58 при использовании информации о двух параметрах, и 0,046 при использовании только расстояний между состояниями, р-величина=4,5е-10, двусторонний критерий суммы рангов Уилкоксона, фиг. 39Е), что позволяет предположить, что направления и вероятности перехода состояния клеток зависят от природы стабильности соседнего состояния. При этом, клетки предпочитают переходить в более стабильное непосредственно следующее соседнее состояние.
[00543] Обсуждение
[00544] Авторами настоящей заявки была разработана первая стратегия характеризации динамики перехода состояния клеток на уровне всего транскриптома. Эта стратегия зависит от sci-судьбы, то есть, представляет собой новый высокопроизводительный метод секвенирования РНК-последовательности отдельных клеток на основе комбинаторного индексирования, позволяющий охарактеризовать как весь, так и вновь синтезированный транскриптом в тысячах клеток. Подобно другим «sci»-методам, sci-судьба может быть легко масштабирована до миллионов клеток (39) и потенциально совместима с характеризацией как транскриптома, так и эпигенома (40). По этой sci-судьбе можно охарактеризовать динамику состояния клеток в гораздо более сложной системе (то есть, на уровне развития всего эмбриона), где реальный путь перехода клеток в клетки нескольких сотен типов до сих пор неизвестен. Кроме того, авторами был разработан вычислительный алгоритм потокового типа для оценки степени захвата вновь синтезированной РНК и степени разрушения генов по данным о sci-судьбе (коррекция памяти), и были построены тысячи дифференциальных траекторий для каждой из отдельных клеток, связанных общим прежним и текущим состоянием транскриптома в каждый момент времени.
[00545] Для проверки методов и для того, чтобы определить, как влияют внутренние и внешние факторы на динамику состояний клетки, авторы применили эту стратегию к модельной системе ответа на кортизол, где судьба клеток динамически регулируется активацией GR внутреннего клеточного цикла и внешней активацией GR, вызываемой лекарственным средством. Авторы показали, что вновь синтезированный транскриптом непосредственно связан с ответом эпигенома на внешние стимулы, а совместный анализ целого и вновь синтезированного транскриптома выявил более высокое разрешение при разделении клеточных состояний. Посредством ковариации между экспрессией TF и скоростью синтеза новой РНК в тысячах клеток, авторы идентифицировали до тысячи связей между TF и регулируемыми генами, на что указывают данные связывания ДНК. Авторы также определили 27 «скрытых клеточных состояний», характеризуемых комбинаторным состоянием функциональных модулей TF в прохождении клеточного цикла и GR-ответом, по сравнению только с 6 состояниями, оцениваемыми с помощью стандартного анализа на кластеризацию.
[00546] Путем анализа на коррекцию памяти и клеточных связей, авторы построили более 6000 траекторий перехода отдельных клеток, охватывающих 10 часов, при этом, основные траектории соответствуют известной динамике состояния клетки в клеточном цикле и GR-ответу. Сеть перехода клеток из одного состояния в другое характеризуется вероятностью перехода во всех состояниях клетки, что подтверждается восстановлением динамики 27 состояний клеток во всех пяти временных точках. И наконец, авторами было обнаружено, что вероятности перехода клетки из одного состояния в другое зависят от двух ключевых факторов сети перехода состояния клетки: расстояния между состояниями и природы нестабильности состояний, и оба этих фактора могут быть оценены обычными методами секвенирования -seq для отдельных клеток.
[00547] Эта стратегия, несмотря на ее мощность, имеет несколько ограничений. Во-первых, для точного построения траектории отдельной клетки необходима полная характеризация состояния клетки в каждый момент времени. Для точной оценки вероятности перехода также необходимо провести множество наблюдений для каждого состояния. Эти ограничения могут быть легко устранены с помощью комбинаторной стратегии sci-судьбы, которая позволяет охарактеризовать миллионы клеток в одном эксперименте. Другое ограничение заключается в том, что для систем in vitro требуется проведение множества экспериментов по мечению S4U. Тем не менее, недавние исследования показали, что S4U позволяет осуществлять стабильное мечение клетки конкретного типа посредством специфической транскрипции РНК во множестве тканей мыши (то есть, в ткани головного мозга, кишечника и жировой ткани) (41, 42), что позволяет предположить, что оценка sci-судьбы вместе с дальнейшими оптимизациями для повышения уровня включения S4U и степени детектирования, могут быть применены для определения профиля динамики транскриптома отдельных клеток in vivo.
[00548] Оценка sci-судьбы открывает новые возможности для применения «статических» геномных методов характеризации динамических систем отдельных клеток. По сравнению с традиционными технологиями, основанными на визуализации, оценка sci-судьбы позволяет охарактеризовать динамику состояния клетки на уровне всего транскриптома и обеспечивать всестороннюю характеризацию состояния клетки без отбора маркеров и обнаружения ключевой движущей силы в дифференцировке клеток. И наконец, авторы предполагают, что sci-судьба может быть легко объединена с альтернативными методами отслеживания линии дифференцировки (43-45) для расшифровки подробной динамики перехода состояния клетки в каждое конечное состояние в пределах нескольких сотен линий дифференцировки.
[00549] Материалы и методы.
[00550] Культивирование клеток млекопитающих.
[00551] Все клетки млекопитающих культивировали при 37°С с 5% CO2 и хранили в DMEM с высоким содержанием глюкозы (Gibco, кат. №11965) для клеток НЕК293Т и NIH/3T3, или в среде DMEM/F12 для клеток А549, в которую добавляли 10% FBS и 1× Pen/Strep (Gibco, cat. №15140122; 100 ед./мл пенициллина, 100 мкг/мл стрептомицина). Клетки трипсинизировали 0,25% трипсином-EDTA (Gibco, кат. №25200-056) и распределяли в отношении 1:10 три раза в неделю.
[00552] Обработка образцов для sci-судьбы
[00553] Клетки А549 обрабатывали 100 нМ DEX в течение 0 часов, 2 часов, 4 часов, 6 часов, 8 часов и 10 часов. Клетки во всех условиях обработки инкубировали с 200 мкМ S4U в течение последних двух часов перед сбором клеток. Для клеток НЕК2 93Т и NIH/3T3, клетки инкубировали с 200 мкМ S4U в течение 6 часов перед сбором клеток.
[00554] Все клеточные линии (клетки А549, НЕК293Т и NIH/3T3) трипсинизировали, центрифугировали при 300х g в течение 5 минут (4°С) и один раз промывали в охлажденном льдом PBS. Все клетки фиксировали 4 мл охлажденным льдом 4% параформальдегидом (EMS) в течение 15 минут на льду. После фиксации, клетки осаждали при 500× g в течение 3 минут (4°С) и один раз промывали 1 мл PBSR (1× PBS, рН 7,4, 1% BSA, 1% SuperRnaseln, 1% 10 мМ DTT). После промывки, клетки ресуспендировали в PBSR в количестве 10 миллионов клеток на мл, быстро замораживали и хранили в жидком азоте. Клетки, фиксированные параформальдегидом, оттаивали на водяной бане при 37°С, центрифугировали при 500× g в течение 5 минут и инкубировали с 500 мкл PBSR, включающего 0,2% тритона Х-100, в течение 3 минут на льду. Клетки осаждали и ресуспендировали в 500 мкл воды, не содержащей нуклеазы и включающей 1% SuperRnaseln. Затем, к клеткам добавляли 3 мл 0,1 н и инкубировали в течение 5 минут на льду (21). В клетки добавляли 3,5 мл Трис-HCl (рН=8,0) и 35 мкл 10% Тритона Х-100 для нейтрализации. Клетки осаждали и промывали 1 мл PBSR. Клетки ресуспендировали в 100 мкл PBSR. 100 мкл PBSR с фиксированными клетками инкубировали со смесью, включающей 40 мкл иодацетамида (IAA, 100 мМ), 40 мкл натрий-фосфатного буфера (500 мМ, рН=8,0), 200 мкл ДМСО и 20 мкл H2O, при 50°С в течение 15 минут. Реакцию гасили 8 мкл DTT (1 М) и 8,5 мл PBS (47). Клетки осаждали и ресуспендировали в 100 мкл PBSI (1×PBS, рН 7,4, 1% BSA, 1% SuperRnaseln). Для всех последующих промывок, ядра осаждали центрифугированием при 500× g в течение 5 минут (4°С).
[00555] Следующие стадии осуществляли в соответствии с протоколом sci-RNA-seq для ядер, фиксированных параформальдегидом (15, 16). Вкратце, клетки распределяли по четырем 9б-луночным планшетам. Для каждой лунки, 5000 ядер (2 мкл) смешивали с 1 мкл 25 мМ заякоренного олиго-dT-праймера (5'-ACGACGCTCTTCCGATCTNNNNNNNN[10 п.о.-индекс]
TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTVN-3') (SEQ ID NO: 5), где «N» означает любое основание, а «V» означает «А», «С» или «G»; IDT), и 0,25 мкл смеси 10 мМ dNTP (Thermo) денатурировали при 55°С в течение 5 минут и сразу помещали на лед. После этого, в каждую лунку добавляли 1,75 мкл реакционной смеси для первой цепи, содержащей 1 мкл 5× буфера для первой цепи Superscript IV (Invitrogen), 0,25 мкл 100 мМ DTT (Invitrogen), 0,25 мкл обратной транскриптазы Superscript IV (200 ед./мкл, Invitrogen), 0,25 мкл рекомбинантного ингибитора рибонуклеазы RNaseOUT (Invitrogen). Обратную транскрипцию осуществляли путем инкубирования планшетов в градиенте температур: 4°С, 2 минуты, 10°С, 2 минуты, 2 0°С, 2 минуты, 30°С, 2 минуты, 40°С, 2 минуты, 50°С, 2 минуты, и 55°С, 10 минут. Затем, все клетки (или ядра) объединяли, окрашивали 4',6-диамидино-2-фенилиндолом (DAPI, Invitrogen) в конечной концентрации 3 мкМ и сортировали по 25 ядер на лунку в 5 мкл буфера ЕВ. Клетки стробировали по окрашиванию DAPI так, чтобы синглеты были отделены от дублетов и отсортированы в каждую лунку. Затем, в каждую лунку добавляли 0,66 мкл буфера для синтеза второй цепи мРНК (NEB) и 0,34 мкл фермента для синтеза второй цепи мРНК (NEB), и синтез второй цепи проводили при 16°С в течение 180 минут. Затем содержимое каждой лунки смешивали с 5 мкл буфера Nextera TD (Illumina) и 1 мкл фермента TDE1 только для i7 (25 нМ, Illumina, разведенного в буфере Nextera TD), и инкубировали при 55°С в течение 5 минут для мечения. Реакцию прекращали добавлением 10 мкл ДНК-связывающего буфера (Zymo) и инкубирования при комнатной температуре в течение 5 минут. Затем каждую лунку очищали с использованием 30 мкл сфер AMPure ХР (Beckman Coulter), элюировали в 16 мкл буфера ЕВ (Qiagen), а затем переносили в свежий многолуночный планшет.
[00556] Для ПЦР-реакции, содержимое каждой лунки смешивали с 2 мкл 10 мкМ праймера Р5 (5'-AATGATACGGCGACCACCGAGATCTACAC[i5]ACACTCTTTCCC TACACGACGCTCTTCCGAT СТ-3'; IDT) (SEQ ID NO: 6), 2 мкл 10 мкМ праймера Р7 (5'-CAAGCAGAAGACGGCATACGAGAT[i7]GTCTCGTGGGCTCGG-3', IDT) (SEQ ID NO: 7) и 20 мкл 2× ПЦР-смеси NEBNext High-Fidelity Master Mix (NEB). Амплификацию осуществляли по следующей программе: 72°С в течение 5 минут, 98°С в течение 30 секунд, 18-22 цикла (98°С в течение 10 секунд, 66°С в течение 30 секунд, 72°С в течение 1 минуты) и наконец, при 72°С в течение 5 минут. После ПЦР, образцы объединяли и очищали с использованием 0,8 объема сфер AMPure ХР. Концентрации библиотек определяли с помощью Qubit (Invitrogen), и библиотеки визуализировали с помощью электрофореза на 6% ТВЕ-ПААГ-геле. Все библиотеки были секвенированы на одной платформе NovaSeq 500 (Illumina) с использованием набора для 150 циклов V2 (рид 1: 18 циклов, рид 2: 130 циклов, индекс 1: 10 циклов, индекс 2: 10 циклов).
[00557] Выравнивание ридов и последующая обработка
[00558] Выравнивание ридов и создание матрицы для подсчета генов в целях секвенирования РНК-seq для отдельных клеток осуществляли с использованием алгоритма потокового типа, который был разработан для sci-РНК-seq (48), с небольшими модификациями. Сначала, риды картировали по эталонному геному с помощью STAR/v2.5.2b {49) с аннотациями генов из GENCODE V19 для человека и GENCODE VM11 для мыши. Для экспериментов с клетками НЕК2 93Т и NIH/3T3 был использован индекс, включающий комбинацию хромосом человека (hgl9) и мыши (mm10). Для эксперимента А549 авторы использовали конструкцию человеческого генома hgl9.
[00559] Файлы sam для отдельных клеток были сначала преобразованы в файл tsv выравнивания с использованием функции sam2tsv в jvarkit (50). Затем для создания файла выравнивания для каждой отдельной клетки, мутации, соответствующие фоновым SNP, отфильтровывали. Для оценки эталонного фонового SNP для клеток А549, авторы загрузили данные для спаренных по концам общих PHK-seq для клеток А549 из ENCODE (28) (выбранное имя: ENCFF542FVG, ENCFF538ZTA, ENCFF214JEZ, ENCFF629LOL, ENCFF149CJD, ENCFF006WNO, ENCFF828WTU, ENCFF380VGD). Каждый файл fastq спаренного конца сначала обрезали с использованием программы-адаптера trim_galore/0.4.1 (51) с параметрами по умолчанию, выровненными по конструкции человеческого генома hgl9 с помощью STAR/v2.5.2b {49). Некартированные риды и множество картированных ридов удаляли с помощью samtools/vl.3 (52). Дуплицированные риды отфильтровывали с использованием функции MarkDuplicates в picard/1.105 (53). Недуплицированные риды от всех образцов объединяли и отсортировывали с использованием samtools/vl.3 (52). Фоновые SNP запрашивали по функции mpileup в samtools/vl.3 (52) и функции mpileup2snp в VarScan/2.3.9 {54). Для эксперимента по тестированию НЕК2 93Т и NIH/3T3, фоновый эталонный SNP создавали в аналогичном алгоритме потокового типа вместе с объединенными данными sam для отдельных клеток в контрольных условиях (в условиях без мечения S4U и без обработки IAA).
[00560] Для создания файла выравнивания для отдельных клеток, все мутации с показателем качества ≤13 удаляли. Мутации на обоих концах каждого рида были обусловлены, главным образом, ошибками секвенирования, а поэтому были также отфильтрованы. Для каждого рида авторы оценивали наличие мутаций Т>С (для смысловой цепи) или мутаций A>G (для антисмысловой цепи), и эти мутированные риды помечали как вновь синтезированные риды.
[00561] Каждую клетку охарактеризовали двумя цифровыми матрицами экспрессии генов исходя из данных полного секвенирования и данных о вновь синтезированной РНК, как описано выше. Гены с экспрессией в 5 или менее, чем в 5 клетках, отфильтровывали. Клетки с менее, чем 2000 UMI или более, чем 80000 UMI, отбрасывали. Клетки с оценкой дублетов >0,2 в соответствии с алгоритмом потокового типа для анализа дублетов Scrublet/0,2 (55) удаляли.
[00562] Данные размерности были сначала сокращены с помощью РСА (после отбора наилучших 2000 генов с наибольшей дисперсией) на цифровых матрицах для экспрессии генов исходя из данных полной экспрессии генов или данных экспрессии вновь синтезированных генов с помощью Monocle 3 (55,57). Наилучшие 10 PC были отобраны для анализа на уменьшение размерности с использованием программы однородной множественной аппроксимации и проецирования (UMAP/0.3.2), то есть, недавно предложенного алгоритма, основанного на римановой геометрии и алгебраической топологии, для уменьшения размерности и визуализации данных (26). Для проведения совместного анализа, авторы объединили 10 наилучших PC, вычисленных для всего транскриптома, и 10 наилучших PC, вычисленных для вновь синтезированного транскриптома для каждой отдельной клетки перед уменьшением размерности с помощью UMAP. Клеточные кластеры создавали с помощью алгоритма densityPeak, реализованного в Monocle 3 (56, 57). Сначала, авторы провели анализ UMAP на основе общей информации о всех обработанных клетках, и этот кластер идентифицировали как выброс (724 из 7404 клеток). Эти клетки были помечены GATA3, то есть, маркером дифференцированных клеток (34), отличающихся высоким уровнем экспрессии, и были отфильтрованы перед проведением последующего анализа.
[00563] Анализ на связывание фактора транскрипции (TF) с регуляторными генами
[00564] Авторы поставили своей целью идентифицировать связи между TF и регуляторными генами на основе их ковариации. Были детектированы клетки с более, чем 10000 UMI, и были отобраны гены с вновь синтезированными ридами, детектированными в более, чем 10% всех клеток. Экспрессию всего гена и число вновь синтезированных генов на клетку нормализовали по клетко-специфическим факторам размера библиотеки, вычисленным с помощью матрицы экспрессии всего гена с использованием алгоритма estimateSizeFactors в Monocle 3 (56, 57), а затем подвергали логарифмическому преобразованию, центрировали, и масштабировали с помощью функции scale() в R. Для каждого детектированного гена была построена регрессионная модель LASSO с помощью пакета программ glmnet (58) для предсказания нормализованных уровней экспрессии исходя из нормализованной экспрессии 853 TF, аннотированных в данных «motifAnnotations_hgnc» из пакета программ RcisTarget {29), путем построения следующей модели:
[00565] Gi=β0+βtTi
[00566] где Gi представляет собой скорректированную величину экспрессии гена i. Эту величину вычисляли путем подсчета количества вновь синтезированных мРНК для каждой клетки, нормализованного путем оценки клетко-специфического фактора размера клетки (SGi) с помощью estimateSizeFactors в Monocle 3 (56, 57) на матрице для общей экспрессии в каждой клетке, и подвергали логарифмическому преобразованию:
[00567] 
[00568] Для упрощения последующего сравнения генов, авторы стандартизировали ответ Gi перед построением модели для каждого гена i с функцией scale () в R.
[00569] Аналогично Gi, Ti представляет собой скорректированную величину экспрессии TF для каждой клетки. Эту величину вычисляли путем подсчета общего уровня экспрессии TF для каждой клетки, нормализованного путем оценки клетко-специфического фактора размера клетки (SGi) с помощью estimateSizeFactors в Monocle 3 [56, 57) на матрице для общей экспрессии в каждой клетке, и подвергали логарифмическому преобразованию:
[00570]
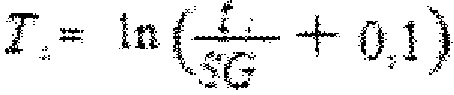
[00571] Перед построением, Ti стандартизируют с помощью функции scale () в R.
[00572] Способ согласно изобретению направлен на TF, которые могут регулировать каждый ген, путем поиска субпопуляции, которая может быть использована для предсказания его экспрессии в регрессионной модели. Однако, TF с экспрессией, коррелирующей с экспрессией гена, не гарантирует, что он будет регулировать этот ген: если ген А специфически экспрессируется в клетке в состоянии 1, а ген TF В специфически экспрессируется в клетке типа 2. Хотя отрицательные корреляции между экспрессией TF и скоростью синтеза нового гена могут укзывать на активность репрессора транскрипции, однако, авторы почувствовали, что более вероятным объяснением негативных связей, о которых сообщает glmnet, является наличие взаимоисключающих паттернов экспрессии, специфичной к состоянию клетки и активности TF. Таким образом, во время предсказания, авторы исключили TF, экспрессия которого негативно коррелировала с уровнем синтеза гена, а также с низким коэффициентом корреляции (≤0,03). Авторами было идентифицировано всего 6103 связи между TF и регуляторными генами.
[00573] В целях идентификации предполагаемых мишеней для прямого связывания, авторы выбрали точку пересечения связей с TF, охарактеризованными в эксперименте ENCODE Chip-seq (28). Из 1086 связей с TF, охарактеризованных в ENCODE, 807 связей были подтверждены в TF-сайтах связывания рядом с промоторами генов {59), что в 4,3 раза увеличивало отношение вероятностей (количество подтвержденных связей по сравнению с неподтвержденными связями) по сравнению с фоном (отношение вероятностей = 2,89 для связей, идентифицированных методом регрессии LASSO и 0,67 для фона, величина р<2.2е-16, точный критерий Фишера). Были сохранены только наборы генов со значимым обогащением сайтов связывания только с TF Chip-seq (точный критерий Фишера, уровень ложного обнаружения 5%), и эти наборы были сокращены для удаления генов непрямых мишеней без сохранения данных связывания с TF. В этом методе была сохранена 591 связь.
[00574] Для дополнительного подтверждения связей генов TF, авторами был также использован пакет программ SCENIC (29), то есть, алгоритм потокового типа для построения сетей регуляторных генов на основе обогащения мотивов TF-мишеней вокруг промоторов генов (10 т.п.о.). Каждый модуль совместной экспрессии, идентифицированный посредством регрессии LASSO, анализировали с помощью анализа на цис-регуляторный мотив с использованием RcisTarget(29). Только модули со значимым обогащением мотивов правильного регулятора TF были сохранены и сокращены для удаления генов непрямых мишеней без сохранения мотива. Авторами были отфильтрованы связи гена TF по трем пороговым значениям коэффициента корреляции (0,3, 0,4 и 0,5), и были объединены все связи, подтвержденные с помощью RcisTarget(29). Методом анализа мотивов было подтверждено всего 509 связей. После объединения обоих методов, авторами было идентифицировано всего 986 регуляторных генов TF по ковариации между уровнем экспрессии TF и синтеза генов, на что указывали данные по связыванию ДНК или анализ мотивов. Для оценки вероятности того, что эти связи будут представлять собой артефакты регулированной регрессии, авторами была проведена перестановка идентификаторов выборки матрицы для экспрессии TF и был проведен тот же анализ. После такой перестановки связи не обнаруживались.
[00575] Упорядочение клеток по функциональным модулям TF
[00576] Для вычисления активности TF в каждой клетке, количества вновь синтезированных UMI для генов в модуле TF-мишени были масштабированы по размеру библиотеки, подвергнуты логарифмиеческому преобразованию и объединены, а затем картированы по Z-показателям. Поскольку было высказано предположение, что TF, обладающие в высокой степени скоррелированной или нескоррелированной активностью, могут функционировать в биологическом процессе в связанной форме, то авторы вычислили абсолютный коэффициент корреляции Пирсона между каждой парой активности TF, и на основании этого кластеризовали TF методом кластеризации ward.d2 в пакете программ pheatmap/1.0.12(60). Пять функциональных модулей TF были идентифицированы и аннотированы на основе их функций.
[00577] Для характеризации состояний клеток по размеру каждого функционального модуля TF, клетки были упорядочены по активности TF, ассоциированных с клеточным циклом (модуль 1 TF) или TF, ассоциированных с GR-ответом (модуль 3 TF), методом UMAP (метрика = «косинус», n_neighbords = 30, min_dist = 0,01). Траекторию прохождения клеточного цикла подтверждали по маркерам генов клеточного цикла в Seurat/2.3.4(27). Три фазы клеточного цикла были идентифицированы с помощью алгоритма densityPeak, реализованного в Monocle 3 (56,57) по координатам UMAP, упорядоченным по модулям TF клеточного цикла. Поскольку каждая главная фаза клеточного цикла по-прежнему сохраняла вариабельную активность TF и экспрессию маркера клеточного цикла, то авторы сегментировали каждую фазу на ранние/средние/поздние состояния с помощью кластеризации посредством k-средних (k=3) и получили всего девять состояний клеточного цикла. Три состояния GR-ответа были идентифицированы с помощью алгоритма densityPeak, реализованного в Monocle 3 (56,51).
[00578] Восстановление прежнего состояния транскриптома по sci-судьбе
[00579] Для идентификации прежнего состояния транскриптома (состояния клетки перед S4U-мечением), авторы выдвинули гипотезу, что время полужизни мРНК является постоянным при различных условиях обработки DEX. Это предположение дополнительно подтверждали последующей проверкой стабильности. Согласно этому предположению, общий транскриптом, частично разрушенный за 2 часа до S4U-мечения, должен быть таким же в клетках, не обработанных DEX, и в клетках, обработанных DEX через 2 часа. Таким образом, различия в общем транскриптоме (по объему) должны совпадать с различиями во вновь синтезированном транскриптоме (по объему), скорректированном по степени обнаружения в зависимости от техники:
[00580] A0h/S0h - (N0h/S0h)/α=A2h/S2h ~ (N2h/S2h)/α
[00581] A0h означает число объединенных UMI для всех клеток в группе без DEX-обработки; S0h означает размер библиотеки (общее количество клеток для UMI) при отсутствии обработки DEX; N0h означает число объединенных вновь синтезированных UMI для всех клеток в группе без DEX-обработки; A2h означает число объединенных UMI для всех клеток в группе DEX-обработки через 2 часа; S2h означает размер библиотеки (общее количество UMI для клеток) в группе DEX-обработки через 2 часа; N2h означает число объединенных вновь синтезированных UMI для всех клеток в группе DEX-обработки через 2 часа; а означает степень детектирования sci-судьбы. Теоретически, для каждого гена может быть определена одна степень детектирования. Однако, для генов с незначительными различиями скорости нового синтеза между двумя состояниями, оцененный параметр а находится в пределах уровня шума. Таким образом, авторами были отобраны гены, обнаруживающие более высокие различия в нормализованной скорости нового синтеза между двумя состояниями: сначала была протестирована серия пороговых величин для фильтрации генов и рассчитан α для каждого гена. Затем авторами был построен график зависимости между пороговым значением и соотношением генов с выходящими за пределы величинами α, (<0 или>1). Авторами был выбран порог, который в точке изгиба на графике соответствовал 186 выбранным генам. Различия во вновь синтезированной мРНК этих генов сильно коррелируют с различиями в уровне экспрессии мРНК (критерий Пирсона, r=0,93, фиг. 37А), что позволяет предположить, что уровень детектирования новой РНК является довольно стабильным по всем генам. Таким образом, медиана скорости захвата вновь синтезированной РНК по sci-судьбе составляет 82%.
[00582] Затем была вычислена степень разложения мРНК за каждые 2 часа. Поскольку популяция клеток А549 может рассматриваться как стабильная без внешней пертурбации, то для клеток после 2-часовой обработки DEX, их прежнее состояние (за 2 часа до мечения S4U) должно быть таким же, как и у клеток, не обработанных DEX. Аналогично, прежнее состояние (перед мечением S4U) для клеток, обработанных DEX на время Т=0/2/4/6/8/10 часов, должно быть аналогично состоянию профилированных клеток на время Т=0/0/2/4/6/8 часов.
[00583] At1/St1 - (Nt1/St1)/α=At0/St0⋅β
[00584] At1 означает число объединенных UMI для всех клеток на t1; St1 означает размер библиотеки (общее количество клеток для UMI) на t1; Nt1 означает число объединенных вновь синтезированных UMI для всех клеток на t1; α означает оцененную степень детектирования sci-судьбы; At0 означает число объединенных UMI для всех клеток на t0; St0 означает размер библиотеки (общее количество UMI для клеток) на t0; β=1-степень геноспецифического разложения на время t0-t1, и соответствует времени полужизни мРНК γ, где β вычисляется по формуле:
[00585] β=1-(1/2) (t1-t0)/γ
[00586] Степень деградации гена β может быть вычислена после каждой DEX-обработки с 2-часовым интервалом. После вышеупомянутый проверки стабильности было обнаружено, что степень деградации генов сильно коррелируют в различные периоды времени DEX-обработки (фиг. 35В). Затем, авторы использовали усредненную степень деградации генов для последующего анализа.
[00587] Исходя из оцененной степени детектирования и деградации генов, прежнее состояние транскриптома каждой клетки может быть вычислено по формуле:
[00588] at1-nt1/α=at0⋅β
[00589] at1 означает число UMI для отдельных клеток на t1; nt1 означает число вновь синтезированных UMI для отдельных клеток на t1; а означает оцененную степень детектирования sci-судьбы; β=1 - степень геноспецифического разложения на время t0-t1. at0 означает оцененное число UMI для отдельных клеток за прошедший момент времени t0, где все отрицательные величины были приведены к 0.
[00590] Анализ на связывание для построения траектории состояния отдельной клетки.
[00591] Посредством анализа на связывание, авторами была поставлена цель идентифицировать связанные родительские и дочерние клетки в одной и той же траектории. Технически, для клеток на t1, авторы объединили прежнее состояние транскриптома (перед мечением S4U, за 2 часа до t1 в проведенном авторами эксперименте) в одну группу 1, а состояние всего транскриптома на t0 (за 2 часа до t1) во вторую группу 2. Если предположить, что какой-либо явный апоптоз клеток отсутствовал, то эти две группы должны иметь сходное распределение клеточных состояний. Авторами была применена стратегия множественного выравнивания для идентификации общих клеточных состояний между двумя наборами данных, исходя из общих источников вариабельности (27). Этот анализ основан на другом предположении, что прежнее и текущее состояние каждой клетки (кроме клеток в начальный и конечный моменты времени) детально детектируются, что подтверждается полученными авторами наборами данных по мере характеризации более 6000 клеток (более 1000 клеток на одно условие), или одной клетки в течение менее одной минуты во время прохождения клеточного цикла. После введения алгоритма потокового типа, клеточные состояния, начиная с t0, и прежние состояния, начиная с t1, выравнивают в одном и том же пространстве UMAP. Необоснованность вышеприведенных допущений может быть обнаружена по выбросам во время выравнивания двух наборов данных. Для каждой клетки А на t1, авторами были выбраны ее ближайшие соседи на t0 как родительское состояние при выравнивании в пространстве UMAP. Аналогично, для каждой клетки на t0, авторами были выбраны ее ближайшие соседи на t1 как состояния дочерних клеток. Следует отметить, что связь необязательно должна быть двунаправленной: родительское состояние одной клетки может быть связано с состоянием другой дочерней клетки. Поскольку родительское состояние и дочернее состояние были идентифицированы для каждой клетки (кроме клеток на 0 часов и 10 часов), то авторы затем идентифицировали связанную родительскую клетку родителя каждой клетки и аналогично связанную дочернюю клетку потомка каждой клетки. Таким образом, каждая отдельная клетка может быть охарактеризована по пути перехода клетки из одного состояния в другое во всех пяти временных точках, охватывающих период 10 часов. Поскольку множество клеток (>50) было охарактеризовано в каждом состоянии, то может быть также зафиксирован стохастический процесс перехода клетки из одного состояния в другое.
[00592] Уменьшение размерности и анализ на кластеризацию для оценки динамики транскриптома отдельных клеток.
[00593] Для уменьшения размерности в динамике транскриптома отдельных клеток, для каждого состояния было отобрано 5 наилучших PC для всего транскриптома и 5 наилучших PC для вновь синтезированного транскриптома, и эти PC были объединены во временном порядке для каждой траектории состояния отдельной клетки для анализа UMAP. Основные типы траекторий клеток были идентифицированы с помощью алгоритма кластеризации пиков плотности {61).
[00594] Принимая во внимание соотношение состояния клетки в начальный момент времени (обработка на 0 часов) и вероятности перехода клетки из одного состояния в другое, оцененные по полученным данным, авторами сначала было предсказано распределение состояний клетки через 2 часа, если допустить, что процесс перехода клетки из одного состояния в другое при DEX-обработке представляет собой динамику Маркова, которая является автономной для клеток и не зависит от времени. Аналогично, распределение состояний клеток в более поздний момент времени может быть вычислено на основе прогнозируемого распределения состояний клеток за 2 часа до этого.
[00595] Предсказание вероятности перехода клетки из одного состояния в другое по нестабильности состояния
[00596] Нестабильность состояния клетки определяется как вероятность перехода каждого состояния в другие состояния через 2 часа. Для вычисления расстояния переходов между клетками, авторы сначала отобрали равное количество (n=50) клеток в каждом состоянии и объединили полный транскриптом и вновь синтезированный транскриптом для всех клеток в этом состоянии. Каждое состояние клетки может быть определено по общей информации, объединяющей весь и вновь синтезированный транскриптом. Расстояние между состояниями клеток вычисляют как коэффициент корреляции Пирсона для общей информации между двумя состояниями.
[00597] Для предсказания вероятности перехода между состояниями, авторами была построена 3-слойная нейронная сеть (число элементов: 128, 128, 26 с активацией сопротивления на каждом слое; функция потери: cosine_proximity, размер партии: 128, периоды: 80) с помощью программы Keras/2.2.4(62). Для ввода авторы использовали нестабильность состояния в текущий момент, нормализованную нестабильность состояния для других 26 состояний (масштабируемых по нестабильности текущего состояния) и расстояние перехода (квадрат) от текущего состояния до других 26 состояний (в том же порядке следования состояний как и в векторе нестабильности состояний). Во избежание избыточной аппроксимации модели, авторы сделали перестановку порядка состояний нестабильности по 200 раз для каждого ввода, причем, порядок расстояний перехода между состояниями поддерживался таким же, как и при нестабильности состояний. Для оценки производительности модели, авторы применили программу проверки достоверности данных «с сохранением/без сохранения» функций путем обучения 26 состояниям, и проверки модели в прежнем состоянии с предсказанием вероятностей перехода состояний во все остальные 26 состояний. Для предсказания вероятности перехода только по расстоянию перехода используется одна и та же модель обучения и проверки с заменой всех нестабильностей исходного состояния на 1.
[00598] Библиография
[00599] 1. N. Moris, С. Pina, А. М. Arias, Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev. Genet. 17, 693-703 (2016).
[00600] 2. A. Filipczyk et al., Network plasticity of pluripotency transcription factors in embryonic stem cells. Nat. Cell Biol. 17, 1235-1246 (2015).
[00601] 3. S. Hormoz et al., Inferring Cell-State Transition Dynamics from Lineage Trees and Endpoint Single-Cell Measurements. Cell Syst. 3, 419-433.e8 (2016).
[00602] 4. V. A. Herzog et al., Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods. 14, 1198-1204 (2017).
[00603] 5. J. A. Schofield, E. E. Duffy, L. Kiefer, M. C. Sullivan, M. D. Simon, TimeLapse-seq: adding a temporal dimension to RNA sequencing through nucleoside recoding. Nat. Methods. 15, 221-225 (2018).
[00604] 6. J. C. Buckingham, Glucocorticoids: exemplars of multi-tasking. Br. J. Pharmacol. 147, S258 (2006).
[00605] 7. M. D. Cleary, C. D. Meiering, E. Jan, R. Guymon, J. C. Boothroyd, Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nat. Biotechnol. 23, 232-237 (2005).
[00606] 8. L. Dolken et al., High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14, 1959-1972 (2008).
[00607] 9. C. Miller et al., Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 7, 458-458 (2014).
[00608] 10. E. E. Duffy et al., Tracking Distinct RNA Populations Using Efficient and Reversible Covalent Chemistry. Mol. Cell. 59, 858-866 (2015).
[00609] 11. B. Schwalb et al., TT-seq maps the human transient transcriptome. Science. 352, 1225-1228 (2016).
[00610] 12. M. Rabani et al., Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. 29, 436-442 (2011).
[00611] 13. M. R. Miller, K. J. Robinson, M. D. Cleary, C. Q. Doe, TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat. Methods. 6, 439-441 (2009).
[00612] 14. D. A. Cusanovich et al., Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 348, 910-914 (2015).
[00613] 15. J. Cao et al., Comprehensive single-cell transcriptional profiling of a multicellular organism. Science. 357, 661-667 (2017).
[00614] 16. J. Cao et al., Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 361, 1380-1385 (2018).
[00615] 17. V. Ramani et al., Massively multiplex single-cell Hi-C (2016), doi:10.1101/065052.
[00616] 18. R. M. Mulqueen et al., Highly scalable generation of DNA methylation profiles in single cells. Nat. Blotechnol. 36, 428-431 (2018).
[00617] 19. S. A. Vitak et al., Sequencing thousands of single-cell genomes with combinatorial indexing. Nat. Methods. 14, 302-308 (2017).
[00618] 20. Y. Yin et al., High-throughput mapping of meiotic crossover and chromosome mis-segregation events in interspecific hybrid mice (2018),, doi:10.1101/338053.
[00619] 21. A. B. Rosenberg et al., Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science. 360, 176-182 (2018).
[00620] 22. Т. E. Reddy et al., Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 19, 2163-2171 (2009).
[00621] 23. S. John et al., Chromatin accessibility predetermines glucocorticoid receptor binding patterns. Nat. Genet. 43, 264-268 (2011).
[00622] 24. Т. E. Reddy, J. Gertz, G. E. Crawford, M. J. Garabedian, R. M. Myers, The Hypersensitive Glucocorticoid Response Specifically Regulates Period 1 and Expression of Circadian Genes. Mol. Cell. Biol. 32, 3756-3767 (2012).
[00623] 25. С.M. Vockley et al., Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome. Cell. 166, 1269-1281.el9 (2016).
[00624] 26. L. Mclnnes, J. Healy, N. Saul, L. GroBberger, UMAP: Uniform Manifold Approximation and Projection. Journal of Open Source Software. 3, 861 (2018).
[00625] 27. A. Butler, P. Hoffman, P. Smibert, E. Papalexi, R. Satija, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411-420 (2018).
[00626] 28. The ENCODE Project Consortium, The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 306, 636-640 (2004).
[00627] 29. S. Aibar et al., SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 14, 1083-1086 (2017).
[00628] 30. M. Boruk, J. G. A. Savory, R. J. G. Hache, AF-2-Dependent Potentiation of CCAAT Enhancer Binding Proteinβ - Mediated Transcriptional Activation by Glucocorticoid Receptor. Mol. Endocrinol. 12, 1749-1763 (1998).
[00629] 31. W. Qin et al., Identification of functional glucocorticoid response elements in the mouse FoxO1 promoter. Biochem. Biophys. Res. Commun. 450, 979-983 (2014).
[00630] 32. C. S. Sheela Rani, N. Elango, S.-S. Wang, K. Kobayashi, R. Strong, Identification of an Activator Protein-1-Like Sequence as the Glucocorticoid Response Element in the Rat Tyrosine Hydroxylase Gene. Mol. Pharmacol. 75, 589 (2009).
[00631] 33. M. Fischer, G. A. Muller, Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit. Rev. Biochem. Mol. Biol. 52, 638-662 (2017).
[00632] 34. J. Chou, S. Provot, Z. Werb, GATA3 in development and cancer differentiation: cells GATA have it! J. Cell. Physiol. 222, 42-49 (2010).
[00633] 35. J. Y. C. Madhurima Biswas, Role of Nrfl in antioxidant response element-mediated gene expression and beyond. Toxicol. Appl. Pharmacol. 244, 16 (2010).
[00634] 36. I.-G. Ryoo, M.-K. Kwak, Regulatory crosstalk between the oxidative stress-related transcription factor Nfe212/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 359, 24-33 (2018).
[00635] 37. J. M. Harmon, M. R. Norman, B. J. Fowlkes, E. B. Thompson, Dexamethasone induces irreversible G1 arrest and death of a human lymphoid cell line. J. Cell. Physiol. 98, 2 67-278 (1979).
[00636] 38. A. K. Greenberg et al., Glucocorticoids inhibit lung cancer cell growth through both the extracellular signal-related kinase pathway and cell cycle regulators. Am. J. Respir. Cell Mol. Biol. 27, 320-328 (2002).
[00637] 39. J. Cao et al., Comprehensive single-cell transcriptional profiling of a multicellular organism. Science. 357, 661-667 (2017).
[00638] 40. J. Cao et al., Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 361, 1380-1385 (2018).
[00639] 41. W. Matsushima et al., SLAM-ITseq: sequencing cell type-specific transcriptomes without cell sorting. Development. 145 (2018), doi:10.1242/dev.164640.
[00640] 42. U. Sharma et al., Small RNAs are trafficked from the epididymis to developing mammalian sperm (2017), doi:10.1101/194522.
[00641] 43. A. McKenna et al., Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science. 353, aaf7907 (2016).
[00642] 44. B. Raj et al., Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol. 36, 442-450 (2018).
[00643] 45. K. L. Frieda et al., Synthetic recording and in situ readout of lineage information in single cells. Nature. 541, 107-111 (2017).
[00 644] 46. H. Wickham, ggplot2: Elegant Graphics for Data Analysis (Springer, 2016).
[00645] 47. M. Muhar et al., SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science. 360, 800-805 (2018).
[00646] 48. J. Cao et al., Comprehensive single-cell transcriptional profiling of a multicellular organism. Science. 357, 661-667 (2017).
[00647] 49. A. Dobin et al., STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15-21 (2013).
[00648] 50. P. Lindenbaum, JVarkit: java-based utilities for Bioinformatics. figshare (2015).
[00649] 51. FelixKrueger, FelixKrueger/TrimGalore. GitHub, (доступна на https://github.com/FelixKrueger/TrimGalore).
[00650] 52. H. Li et al., The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25, 2078-2079 (2009).
[00651] 53. Picard Tools - By Broad Institute, (доступна на http://broadinstitute.github.io/picard/).
[00652] 54. D. C. Koboldt et al., VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568-576 (2012).
[00653] 55. S. L. Wolock, R. Lopez, A. M. Klein, Scrublet: computational identification of cell doublets in single-cell transcriptomic data (2018),, doi:10.1101/357368.
[00654] 56. X. Qiu et al., Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods. 14, 979-982 (2017).
[00655] 57. cole-trapnell-lab, cole-trapnell-lab/monocle-release. GitHub, (доступна на https://github.com/cole-trapnell-lab/monocle-release).\
[00656] 58. J. Friedman, T. Hastie, R. Tibshirani, Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 33 (2010),
doi:10.18637/jss.v033.i01.
[00657] 59. Dataset - ENCODE Transcription Factor Binding Site Profiles, (доступна на http://amp.pharm.mssm.edu/Harmonizome/dataset/ENCODE+Transcripti on+Factor+Binding+Site+Profiles).
[00658] 60. raivokolde, raivokolde/pheatmap.GitHub, (доступна на https://github.com/raivokolde/pheatmap).
[00659] 61. A. Rodriguez, A. Laio, Clustering by fast search and find of density peaks. Science. 344, 1492-1496 (2014).
[00660] 62. keras-team, keras-team/keras. GitHub, (доступна на https://github.com/keras-team/keras
[00661] Пример 4
[00662] Захват мультиплексного транскрипта
[00663] Большинство методов секвенирования РНК отдельных клеток выполняется при охвате от 15000 до 50000 уникальных ридов на клетку (Ziegenhain et al. 2017), тогда как общее содержание мРНК в отдельных клетках может варьироваться от 50000 до 300000 молекул (Marinov et al. 2014). Кроме того, в большинстве этих методов используются олиго(dT)-праймеры для обратной транскрипции (ОТ), и эти методы направлены на секвенирование у 3'-конца РНК. Это означает, что такие методы имеют ограниченные возможности для обнаружения изменений в численности любого данного транскрипта. Недавние исследования, в которых было охарактеризовано большое количество клеток (Gasperini et al. 2019; Cao et al. 2019), потребовали очень высокой глубины секвенирования, причем, запуск программ Illumina NovaSeq, использованных в этих исследованиях, стоит 30000 долларов каждая, что делает такие эксперименты недоступными для большинства ученых.
[00664] Однако, в обоих случаях, количество операций считывания, необходимых для получения биологической информации из имеющихся данных, является относительно небольшим. При получении информации о некодирующих пертурбациях в отдельных клетках, анализ на изменение уровня экспрессии проводили только для генов, расположенных в цис-положении по отношению к регуляторному элементу, подвергнутому дизрупции (Xie et al. 2017; Gasperini et al. 2018). Хотя в экспериментах по созданию клеточного атласа, для кластеризации сходных клеток использовали общие паттерны экспрессии, однако, присвоение клеткам конкретных типов проводили с использованием небольшого числа ключевых генов факторов транскрипции. Таким образом, возможность получить данные считывания для транскриптов генов, которые являются наиболее информативными в этих экспериментах, позволяет значительно снизить необходимую глубину секвенирования и увеличить мощность анализа для обнаружения незначительных различий между клетками.
[00665] Авторы сосредоточили свое внимание на секвенирование отдельных клеток для идентификации представляющих интерес мРНК с использованием специфических ОТ-праймеров, а не олиго(dT)-праймеров. Подобный метод был недавно использован, в основном, для специфической последовательности всех известных стыков сплайсинга в дрожжах, что привело к 100-кратному обогащению областями-мишенями по сравнению с областями, не являющимися мишенями (Xu et al., 2018). Пул ОТ-праймеров, перекрывающих представляющие интерес транскрипты, позволяет сократить число считываний транскриптомной библиотеки (sci-PHK-seq) до сотен захваченных транскриптов на эксперимент.
[00666] Эти манипуляции с sciPHK-seq имеют множество преимуществ по сравнению с олиго-(dT)-праймированием. Во-первых, эти манипуляции позволяют осуществлять прямое секвенирование областей генома, которые были определены авторами как наиболее информативные для каждого эксперимента. Во-вторых, они дают возможность осуществлять обратную транскрипцию каждой молекулы РНК в кДНК, что увеличивает вероятность детектирования на молекулу РНК. В-третьих, этот подход позволяет осуществлять нацеливание только на те ампликоны, которые были уникально картированы, и могут уменьшить фон рибосомных ридов в большей степени, чем альтернативые методы с использованием рандомизированного гексамера или олиго(dT)-праймирования. В-четвертых, это позволяет осуществлять нацеливание на информативные области мРНК, такие как области стыка сплайсинга и экзоны, образующиеся в результате альтернативных событий в сайтах инициации транскрипции, и таким образом получить информацию об изоформе, которую нелегко обнаружить с помощью обычного sciPHK-seq.
[00667] sciPHK-seq идеально подходит для модификации несколькими ОТ-праймерами. В большинстве методов PHK-seq для отдельных клеток используются сферы, связанные с уникальными олиго-идентификаторами, для введения штрих-кодов, идентифицирующих клетки, в транскриптом каждой клетки, обычно путем захвата мРНК посредством гибридизации с их поли(А)-хвостом. Хотя такие сферы были модифицированы для добавления небольшого количества специфических ОТ-праймеров в целях увеличения охвата нескольких транскриптов (Saikia et al. 2018), однако, эту стратегию будет трудно масштабировать до сотен транскриптов-мишеней или сделать быстрые замены между экспериментами. Таким образом, адаптивность комбинаторной индексации отдельных клеток будет полезной при разработке мультиплексного ОТ-секвенирования PHK-seq в отдельных клетках.
[00668] Рабочая схема для этого аспекта аналогична трехуровневому протоколу sciPHK-seq, описанному в Примерах 1 и 3, но в некоторых вариантах, она не включает стадию ОТ.
[00669] 1. Конструирование пула От-праймеров. В одном аспекте, эти праймеры будут синтезированы отдельно, а затем объединены. Для нацеливания на >384 ампликонов может быть синтезирована библиотека праймеров, амплифицированных в виде двухцепочечной ДНК с последующей обработкой для получения одноцепочечных праймеров, как описано в литературе (Xu et al. 2018). Эта вторая стратегия позволяет добавлять множество уникальных индексов к ОТ-праймерам (что позволяет индексировать sciPHK-seq при ОТ и конечной ПЦР).
[00670] 2. Проведение мультиплексной ОТ с использованием пула праймеров. Эта реакция может представлять собой либо отдельную реакцию с тысячами клеток (если на этой стадии не проводили индексирование), либо множество параллельных реакций, которые будут добавлять в высокой степени специфический индекс при обратной транскрипции.
[00671] 3. Лигирование шпилечного адаптера для добавления индекса, специфичного для лунки.
[00672] 4. Объединение всех клеток и проведение синтеза второй цепи.
[00673] 5. Распределение клеток по множеству лунок и проведение мечения для добавления второго постоянного ПЦР-фрагмента.
[00674] 6. ПЦР-амплификация, добавление конечного индекса, специфичного для лунки.
[00675] 7. Секвенирование.
[00676] Рабочая схема конструирования праймера:
[00677] 1. Сбор последовательностей для всех экзонов из генов-мишеней.
[00678] 2. Компьютерный анализ всех возможных ОТ-праймеров размером в 25 п.о.
[00679] 3. Фильтрация ОТ-праймеров-кандидатов по:
[00680] а. содержанию GC 40-60%, соответствующему температурам плавления приблизительно от 55 до 70 градусов.
[00681] b. по меньшей мере по 2 G или С в последних 5 нуклеотидах праймера для повышения вероятности того, что гибридизованный ОТ-праймер будет хорошим субстратом для удлинения под действием фермента обратной транскриптазы.
[00682] с. вероятности нецелевого праймирования. В первом эксперименте согласно изобретению, авторы обнаружили, что хотя гены-мишени согласно изобретению были в высокой степени обогащены, однако, большая часть ридов все еще происходила от других РНК, которые присутствуют в клетках в избыточном количестве. Большинство из этих событий нецелевого праймирования были результатом ~ 5-8 п.о. - комплементарности между 3'-концом праймера и РНК, не являющейся мишенью. Таким образом, разработанный авторами алгоритм конструирования праймера был сделан с учетом избытка конечного гексамера ОТ-праймера в общей клеточной РНК. Авторами были включены только те ОТ-праймеры, в которых этот последний гексамер:
[00683] i. Вообще не присутствует в рибосомной РНК. Этот гексамер происходит от серии описанных ранее гексамеров, которые «не были рандомизированы соответствующий образом, или гексамеров NSR (Armour et al. 2009). Праймеры, которые проходят через этот фильтр, с гораздо меньшей вероятностью будут нецелевыми праймерами в рибосомной РНК.
[00684] ii. Присутствует в общей клеточной РНК в низком количестве. Авторы подсчитали количество всех 4096 возможных гексамеров в ридах PRO-seq, картированных по человеческому геному (Core et al., 2014). PRO-seq позволяет измерить все возрастающую транскрипцию в клетках, включая рибосомную транскрипцию. Авторами были использованы только ОТ-праймеры, которые заканчиваются гексамерами, присутствующими в нижнем квартиле избытка в этом наборе данных. Это позволяет сохранить несколько гексамеров, которые, хотя и присутствуют в рибосомной РНК, но встречаются в клетках не так часто, как РНК.
[00685] Этот фильтр избыточного содержания резко изменяет выбор праймера. Наблюдается только ~ 17% перекрывание между праймерами, выбранными с помощью алгоритма потокового типа согласно изобретению в присутствии или в отсутствии этого фильтра. В будущем, варианты разработанного авторами алгоритма конструирования потокового типа позволят улучшить фильтрацию объектов, не являющихся мишенью. По мере сбора данных для большего числа праймеров, авторы должны иметь возможность оценивать большее число нецелевых событий праймирования.
[00686] 4. Фильтрация кандидатов по картируемости. Авторы выравнивали каждого кандидата по hgl9 с использованием программы «галстук-бабочка», что позволило определить 3 несоответствия. Эта стадия гарантирует, что каждый праймер будет иметь только один сайт-мишень в геноме.
[00687] 5. Выбор из возможных праймеров, которые прошли через эти фильтры, набора, который наиболее равномерно располагается по всему гену.
[00688] Для каждого гена, на который осуществляется нацеливание, авторы решают, сколько необходимо сконструировать праймеров на один экзон. Авторами был включен первый и последний праймер, которые проходят фильтры для каждого экзона, а затем были собраны внутренние праймеры, которые наиболее равномерно покрывают экзон, путем минимизации расстояния от положений праймеров, которые бы точно разделяли экзон на n кусков.
[00689] Так, например, для экзона с 300 п. о., где авторы провели поиск 3 праймеров, были взяты праймеры, наиболее близкие к положениям 1, 150 и 300, которые прошли все фильтры до этой точки.
[00690] 6. Для данного пилотного эксперимента, ОТ-праймеры были упорядочены в 384-луночных планшетах и объединены для создания эквимолярной смеси всех праймеров. Затем эту смесь фосфорилировали полинуклеотид-киназой Т4 для лигирования индексированного шпилечного олигонуклеотида во время создания библиотеки sciPHK-seq (Cao et al., 2019). Это гораздо более выгодно, чем заказывать фосфорилированные олигонуклеотиды. OT-праймеры в 2 5 п. о. также добавляют уникальный молекулярный идентификатор (UMI) в 8 п. о. и фрагменты в 6 п. о. для отжига шпилечного олигонуклеотида, который добавляет специфический для лунки индекс (для комбинаторной индексации) и ПЦР-фрагмент.
[00691] Этот процесс может быть итеративным, когда каждый ОТ-праймер заказывается отдельно: более низкое нецелевое отношение было достигнуто в более поздних экспериментах путем селективного повторного объединения праймеров, которые, как было обнаружено, имеют благоприятные скорости захвата в первом эксперименте. Каждый рид для секвенирования Illumina охватывает ОТ-праймер 25 п. о. и захваченную молекулу РНК, что позволяет картировать ОТ-праймеры и захваченные молекулы отдельно для вычисления скорости нацеливания для каждого праймера.
[00692] Более поздние раунды могут включать больше ОТ-праймеров путем их синтеза в массиве. Библиотека праймеров может быть размножена с помощью ПЦР и сделана одноцепочечной путем селективной экзонуклеолитической деградации цепи, которая не включает блокирующую группу в ПЦР-праймере (Xu et al., 2018). Большой массив можно использовать для синтеза нескольких пулов праймеров: если каждый пул имеет специфический ПЦР-фрагмент, то один массив может быть использован для получения десятков пулов из тысяч праймеров, каждый из которых может быть селективно амплифицирован.
[00693] Мультиплексная обратная транскрипция:
[00694] Мультиплексный захват мишени может быть осуществлен в несколько стадий в соответствии с протоколом создания библиотеки PHK-seq. Однако авторы считают, что обратную транскрипцию легче всего проводить параллельно. В высокой степени мультиплексные ПЦР-реакции редко бывают успешными. Реакции ПЦР включают множество (10-20) циклов. Это означает, что проблемы с нецелевым отжигом усугубляются после экспоненциального роста во время этих циклов, которые часто опережают циклы целевого отжига. В мультиплексной ПЦР, для каждой мишени получают два специфических ПЦР-праймера. Цель состоит в том, чтобы эти два праймера специфически амплифицировали только свою мишень. Однако, в большом пуле праймеров будет несколько комбинаций, которые гибридизуются с другими праймерами, присутствующими в пуле. Поскольку концентрация праймеров намного выше, чем концентрация молекул матрицы, то эти димеры праймеров будут доминировать в пуле к концу ПЦР. Невозможность осуществления в высокой степени мультиплексной ПЦР является причиной того, что многие целевые протоколы амплификации, такие как секвенирование экзома, часто используют молекулярные инверсионные зонды для захвата мишеней (Hiatt et al. 2013). В таких протоколах, специфичность к мишени достигается с помощью одной стадии отжига между зондом и мишенью. Целевые специфические зонды добавляют ПЦР-фрагменты, которые затем используются в общей целевой ПЦР-амплификации. Методы комбинаторной индексации в одной клетке основаны на индексации в несколько стадий во время создания библиотеки: метод инверсионного зонда для захвата мишеней из кДНК не позволяет выполнить достаточное количество стадий индексации.
[00695] Для захвата мультиплексной мишени авторы использовали специфический праймер обратной транскрипции, после чего проводили ПЦР-реакцию, которая амплифицирует все молекулы, подвергаемые обратной транскрипции. Таким образом, стратегия авторов аналогична использованию зондов молекулярной инверсии для целевой амплификации ДНК: одна стадия (обратная транскрипция) избирательно нацеливается на представляющие интерес транскрипты и добавляет общий ПЦР-фрагмент, который может быть использован для амплификации всех молекул-мишеней во время ПЦР. Таким образом, высокая специфичность при обратной транскрипции играет очень важную роль. Поддержание высокой температуры после отжига ОТ-праймеров облегчает мультиплексное специфическое праймирование. Обычные протоколы обратной транскрипции включают денатурацию смеси РНК и праймера обратной транскрипции и охлаждение до 4 градусов для обеспечения отжига. Эта низкая температура отжига является слишком мягкой для событий нецелевого отжига. Авторы должны убедиться, что единственными событиями отжига, которые могут быть расширены, являются события, для которых все в высокой степени оспецифичные ОТ-праймеры, разработанные авторами, нашли свои мишени. Таким образом, авторы поддерживали высокую температуру в течение осуществления всего протокола, как и в других мультиплексных методах специфической обратной транскрипции (Xu et al. 2018). Авторами была проведена денатурация смеси фиксированных клеток, пула ОТ-праймеров и dNTP при 65°С, отжиг при 53°С, а затем добавление смеси фермента/буфера для обратной транскрипции, которую предварительно уравновешивали при 53°С, для проведения реакции отжига, а затем проводили цикл удлинения при 53°С в течение 20 минут. Таким образом, ОТ-праймеры не имели возможности осуществлять отжиг при низкой температуре между стадиями денатурации и удлинения.
[00696] Остальные стадии этого метода соответствуют методам, описанным в Примерах 1 и 3. Шпилечный адаптер лигируют in situ с добавлением клеточного индекса. Клетки объединяют, промывают и распределяют по новым лункам для проведения последней стадии индексации. В этих лунках проводят синтез второй цепи. Затем, двухцепочечную кДНК метят с добавлением второго общего ПЦР-фрагмента (первый фрагмент получают путем лигирования, а второй путем мечения). ДНК выделяют из клеток путем связывания со сферами Ampure, а затем проводят ПЦР с добавлением второго индекса.
[00697] Предварительные результаты:
[00698] Все результаты, представленные на фиг. 40-42, получены из общей библиотеки (без комбинаторного индексирования отдельных клеток) in situ (все стадии осуществляют в ядрах, фиксированных параформальдегидом), полученной с использованием пула ОТ-праймеров, нацеленных на гены в локусе LMO2 в клетках К562.
[00699] Библиография:
[00700] Armour, Christopher D., John С.Castle, Ronghua Chen, Tomas Babak, Patrick Loerch, Stuart Jackson, Jyoti K. Shah, et al. 2009. "Digital Transcriptome Profiling Using Selective Hexamer Priming for cDNA Synthesis." Nature Methods 6 (9): 647-49.
[00701] Cao, Junyue, Malte Spielmann, Xiaojie Qiu, Xingfan Huang, Daniel M. Ibrahim, Andrew J. Hill, Fan Zhang, et al. 2019. "The Single-Cell Transcriptional Landscape of Mammalian Organogenesis." Nature 566 (7745): 496-502.
[00702] Core, Leighton J., Andre L. Martins, Charles G. Danko, Colin T. Waters, Adam Siepel, and John T. Lis. 2014. "Analysis of Nascent RNA Identifies a Unified Architecture of Initiation Regions at Mammalian Promoters and Enhancers." Nature Genetics 46 (12): 1311-20.
[00703] Gasperini, Molly, Andrew J. Hill, Jose L. McFaline-Figueroa, Beth Martin, Seungsoo Kim, Melissa D. Zhang, Dana Jackson, et al. 2019. "A Genome-Wide Framework for Mapping Gene Regulation via Cellular Genetic Screens." Cell 176 (6): 1516.
[00704] Gasperini, Molly, Andrew Hill, Jose L. McFaline-Figueroa, Beth Martin, Cole Trapnell, Nadav Ahituv, and Jay Shendure. 2018. "crisprQTL Mapping as a Genome-Wide Association Framework for Cellular Genetic Screens." bioRxiv. https://doi.org/10.1101/314344.
[00705] Hiatt, Joseph В., Colin C. Pritchard, Stephen J. Salipante, Brian J. O'Roak, and Jay Shendure. 2013. "Single Molecule Molecular Inversion Probes for Targeted, High-Accuracy Detection of Low-Frequency Variation." Genome Research 23 (5): 843-54.
[00706] Marinov, Georgi K., Brian A. Williams, Ken McCue, Gary P. Schroth, Jason Gertz, Richard M. Myers, and Barbara J. Wold. 2014. "From Single-Cell to Cell-Pool Transcriptomes: Stochasticity in Gene Expression and RNA Splicing." Genome Research 24 (3): 496-510.
[00707] Saikia, Mridusmita, Philip Burnham, Sara H. Keshavjee, Michael F. Z. Wang, Pablo Moral-Lopez, Meleana M. Hinchman, Charles G. Danko, John S. L. Parker, and Iwijn De Vlaminck. 2018. "Simultaneous Multiplexed Amplicon Sequencing and Transcriptome Profiling in Single Cells." bioRxiv. https://doi.org/10.1101/328328.
[00708] Xie, Shiqi, Jialei Duan, Boxun Li, Pei Zhou, and Gary C. Hon. 2017. "Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells." Molecular Cell 66 (2): 285-99.e5.
[00709] Xu, Hansen, Benjamin J. Fair, Zach Dwyer, Michael Gildea, and Jeffrey A. Pleiss. 2018. "Multiplexed Primer Extension Sequencing Enables High Precision Detection of Rare Splice Isoforms." bioRxiv. https://doi.org/10.1101/331629.
[00710] Ziegenhain, Christoph, Beate Vieth, Swati Parekh, Bjorn Reinius, Amy Guillaumet-Adkins, Martha Smets, Heinrich Leonhardt, Holger Heyn, Ines Hellmann, and Wolfgang Enard. 2017. "Comparative Analysis of Single-Cell RNA Sequencing Methods." Molecular Cell 65 (4): 631-43.e4.
[00711] Полное раскрытие всех цитируемых здесь патентов, патентных заявок и публикаций, а также материалов, доступных в электронной форме (включая, например, представление нуклеотидных последовательностей, например, в GenBank и RefSeq, и представление аминокислотных последовательностей, например, в SwissProt, PIR, PRF, PDB, и трансляции из аннотированных кодирующих областей в GenBank и RefSeq), включено в настоящее описание посредством ссылки в полном объеме. Дополнительные материалы, цитируемые в публикациях (такие как дополнительные таблицы, дополнительные чертежи, дополнительные материалы и методы и/или дополнительные экспериментальные данные), также включены в настоящее описание посредством ссылки в полном объеме. В случае, если существует какое-либо несоответствие между раскрытием настоящей заявки и раскрытием(ями) любого документа, включенного в настоящую заявку посредством ссылки, следует отдать предпочтение описанию настоящей заявки. Вышеизложенное подробное описание и примеры приведены лишь для лучшего понимания изобретения. Однако, они не должны рассматриваться как ограничение объема изобретения. Раскрытие настоящего изобретения не ограничивается конкретными представленными и описанными здесь деталями, поскольку для специалиста в данной области очевидно, что в настоящее изобретение могут быть включены изменения, определенные формулой изобретения.
[00712] Если это не оговорено особо, то все числа, выражающие количества компонентов, молекулярные массы и т.п., используемые в описании и формуле изобретения, во всех случаях следует понимать как «приблизительные». В соответствии с этим, если это не оговорено особо, то числовые параметры, представленные в описании и в формуле изобретения, являются приблизительными значениями, которые могут варьироваться в зависимости от желаемых свойств, которые должны быть достигнуты в настоящем изобретении. Каждый числовой параметр должен рассматриваться по меньшей мере как число, обозначаемое значащими цифрами с применением обычных методов округления, но, тем не менее, он не должен интерпретироваться как попытка ограничить доктрину эквивалентов объемом формулы изобретения.
[00713] Несмотря на то, что числовые интервалы и параметры, определяющие широкий объем раскрытия изобретения, являются приблизительными, однако, числовые значения, указанные в конкретных примерах, сообщаются с максимально возможной точностью. При этом, все числовые значения, по своей сути содержат интервал, который обязательно является результатом стандартного отклонения, обнаруживаемого в их соответствующих экспериментальных измерениях.
[00714] Все заголовки представлены для удобства читателя и не должны рассматриваться как ограничение смысла текста, следующего за заголовком, если это не оговорено особо.
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> ILLUMINA, INC.
UNIVERSITY OF WASHINGTON
<120> КРУПНОМАСШТАБНЫЕ МОНОКЛЕТОЧНЫЕ БИБЛИОТЕКИ ТРАНСКРИПТОМОВ
И СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ
<130> IP-1722-PCT
<140> PCT/US2019/035422
<141> 2019-06-04
<150> 62/821,678
<151> 2019-03-21
<150> 62/680,259
<151> 2018-06-04
<160> 11
<170> PatentIn version 3.5
<210> 1
<211> 54
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
праймер
<220>
<221> modified_base
<222> (7)..(24)
<223> a, c, t, g, неизвестные или другие
<400> 1
cagagcnnnn nnnnnnnnnn nnnntttttt tttttttttt tttttttttt tttt 54
<210> 2
<211> 45
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательност: синтетический
олигонуклеотид
<220>
<223> Описание Combined ДНК/RNA Molecule: синтетический
олигонуклеотид
<220>
<221> modified_base
<222> (7)..(16)
<223> a, c, t, g, неизвестные или другие
<220>
<221> misc_feature
<222> (7)..(16)
<223> Эта область может включать 9-10 оснований
<220>
<221> modified_base
<222> (36)..(45)
<223> a, c, t, g, неизвестные или другие
<220>
<221> misc_feature
<222> (36)..(45)
<223> Эта область может включать 9-10 оснований
<400> 2
gctctgnnnn nnnnnnuacg acgctcttcc gatctnnnnn nnnnn 45
<210> 3
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
праймер
<400> 3
aatgatacgg cgaccaccga gatctacac 29
<210> 4
<211> 24
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
праймер
<400> 4
caagcagaag acggcatacg agat 24
<210> 5
<211> 68
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
праймер
<220>
<221> modified_base
<222> (19)..(36)
<223> a, c, t, g, неизвестные или другие
<220>
<221> modified_base
<222> (68)..(68)
<223> a, c, t, g, неизвестные или другие
<400> 5
acgacgctct tccgatctnn nnnnnnnnnn nnnnnntttt tttttttttt tttttttttt 60
ttttttvn 68
<210> 6
<211> 29
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
праймер
<400> 6
aatgatacgg cgaccaccga gatctacac 29
<210> 7
<211> 24
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
праймер
<400> 7
caagcagaag acggcatacg agat 24
<210> 8
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
праймер
<400> 8
acactctttc cctacacgac gctcttccga tct 33
<210> 9
<211> 15
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
праймер
<400> 9
gtctcgtggg ctcgg 15
<210> 10
<211> 33
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
праймер
<400> 10
acactctttc cctacacgac gctcttccga tct 33
<210> 11
<211> 15
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Описание искусственной последовательности: синтетический
праймер
<400> 11
gtctcgtggg ctcgg 15
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ВЫСОКОПРОИЗВОДИТЕЛЬНЫЕ БИБЛИОТЕКИ ОДИНОЧНЫХ КЛЕТОК И СПОСОБЫ ПОЛУЧЕНИЯ И ИСПОЛЬЗОВАНИЯ | 2020 |
|
RU2838946C2 |
| ВЫСОКОПРОИЗВОДИТЕЛЬНЫЕ БИБЛИОТЕКИ ОДИНОЧНЫХ ЯДЕР И ОДИНОЧНЫХ КЛЕТОК И СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИСПОЛЬЗОВАНИЯ | 2020 |
|
RU2838545C2 |
| ВЫСОКОПРОИЗВОДИТЕЛЬНОЕ СЕКВЕНИРОВАНИЕ ОДИНОЧНОЙ КЛЕТКИ СО СНИЖЕННОЙ ОШИБКОЙ АМПЛИФИКАЦИИ | 2019 |
|
RU2744175C1 |
| ВЫСОКОПРОИЗВОДИТЕЛЬНОЕ СЕКВЕНИРОВАНИЕ ОДИНОЧНОЙ КЛЕТКИ СО СНИЖЕННОЙ ОШИБКОЙ АМПЛИФИКАЦИИ | 2019 |
|
RU2833615C2 |
| ПОЛНОГЕНОМНЫЕ БИБЛИОТЕКИ ОТДЕЛЬНЫХ КЛЕТОК ДЛЯ БИСУЛЬФИТНОГО СЕКВЕНИРОВАНИЯ | 2018 |
|
RU2770879C2 |
| СИСТЕМА АНАЛИЗА ДЛЯ ОРТОГОНАЛЬНОГО ДОСТУПА К БИОМОЛЕКУЛАМ И ИХ МЕЧЕНИЯ В КЛЕТОЧНЫХ КОМПАРТМЕНТАХ | 2017 |
|
RU2771892C2 |
| АНАЛИЗ МНОЖЕСТВА АНАЛИТОВ С ИСПОЛЬЗОВАНИЕМ ОДНОГО АНАЛИЗА | 2019 |
|
RU2824049C2 |
| СПОСОБ И КОМПОЗИЦИЯ ДЛЯ АНАЛИЗА КЛЕТОЧНЫХ КОМПОНЕНТОВ | 2016 |
|
RU2761432C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ОПРЕДЕЛЕНИЯ ЛИГАНДОВ НА МАТРИЦАХ С ИСПОЛЬЗОВАНИЕМ ИНДЕКСОВ И ШТРИХКОДОВ | 2020 |
|
RU2825578C1 |
| СПОСОБ И ПРОДУКТ ДЛЯ ЛОКАЛИЗОВАННОЙ ИЛИ ПРОСТРАНСТВЕННОЙ ДЕТЕКЦИИ НУКЛЕИНОВОЙ КИСЛОТЫ В ОБРАЗЦЕ ТКАНИ | 2012 |
|
RU2603074C2 |
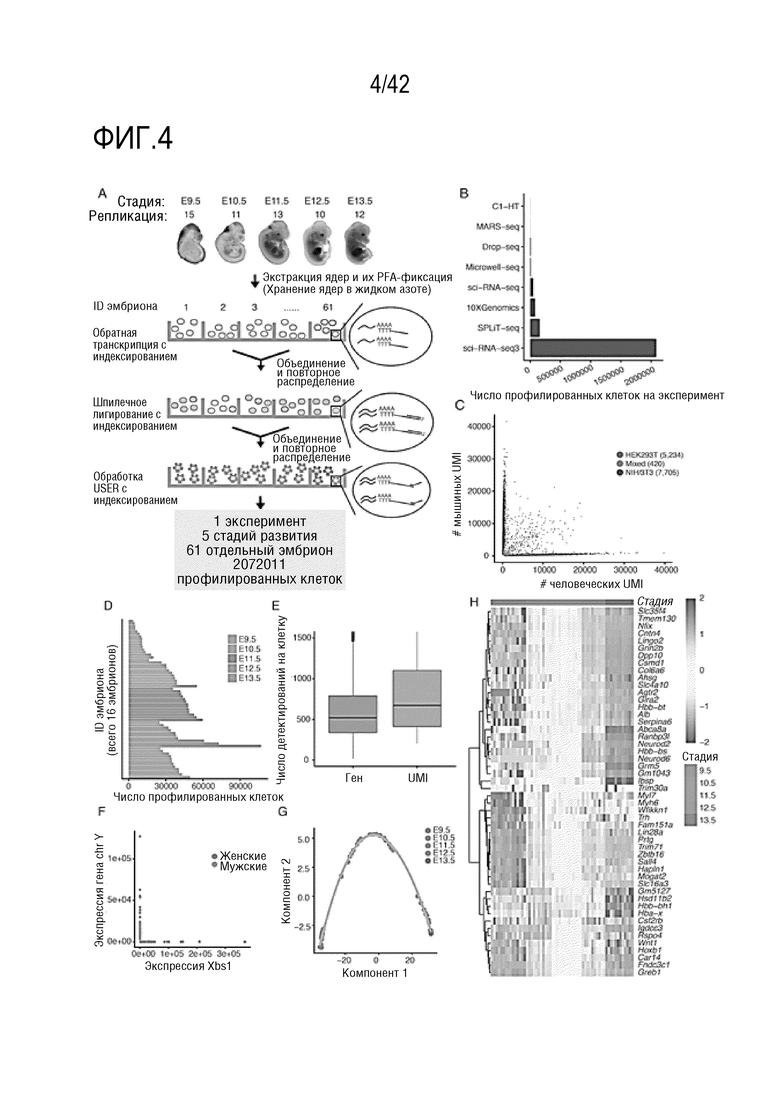
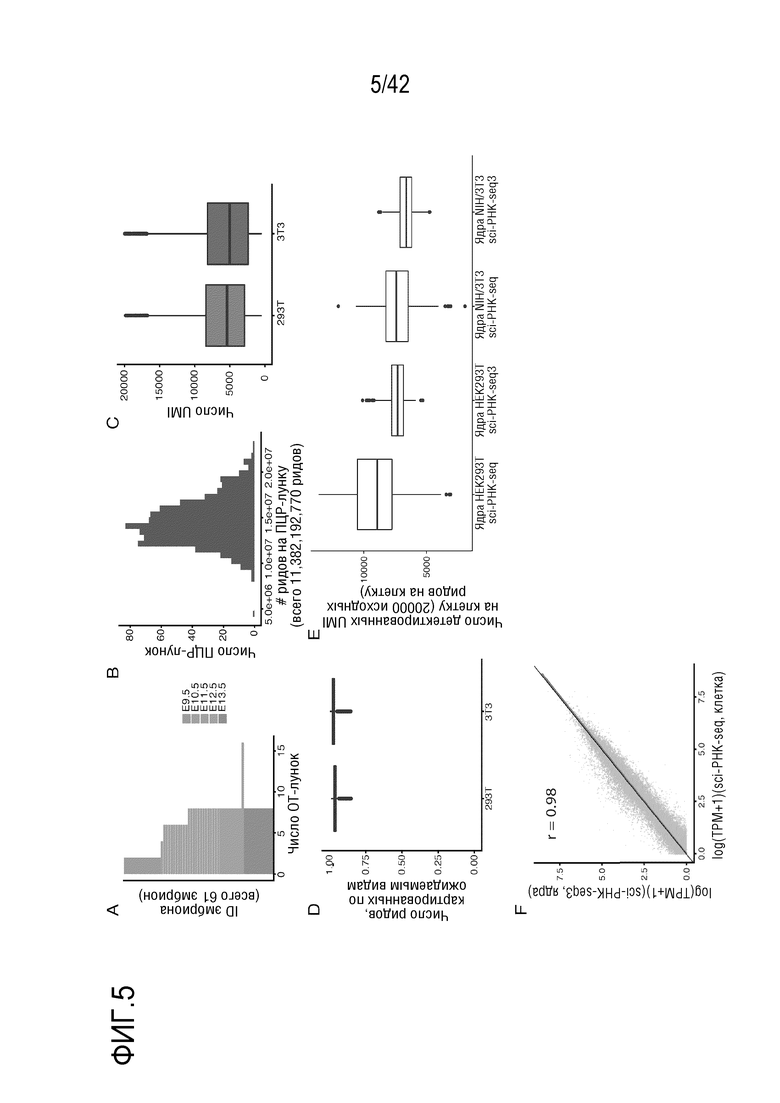
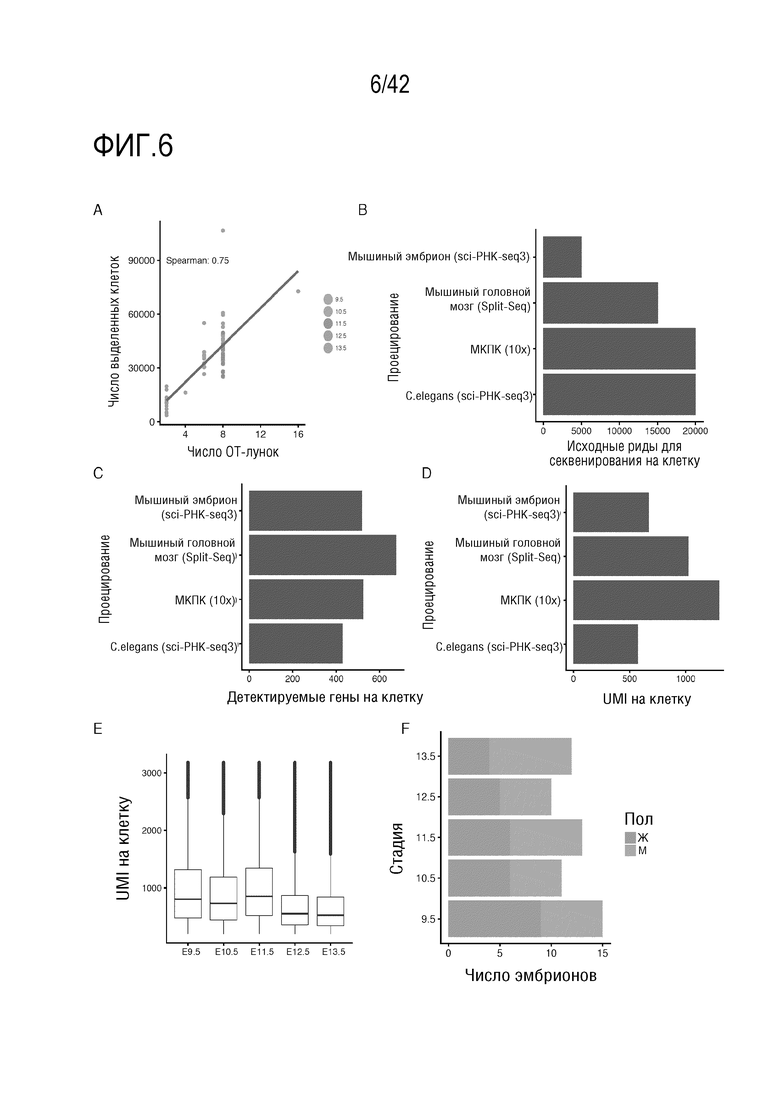
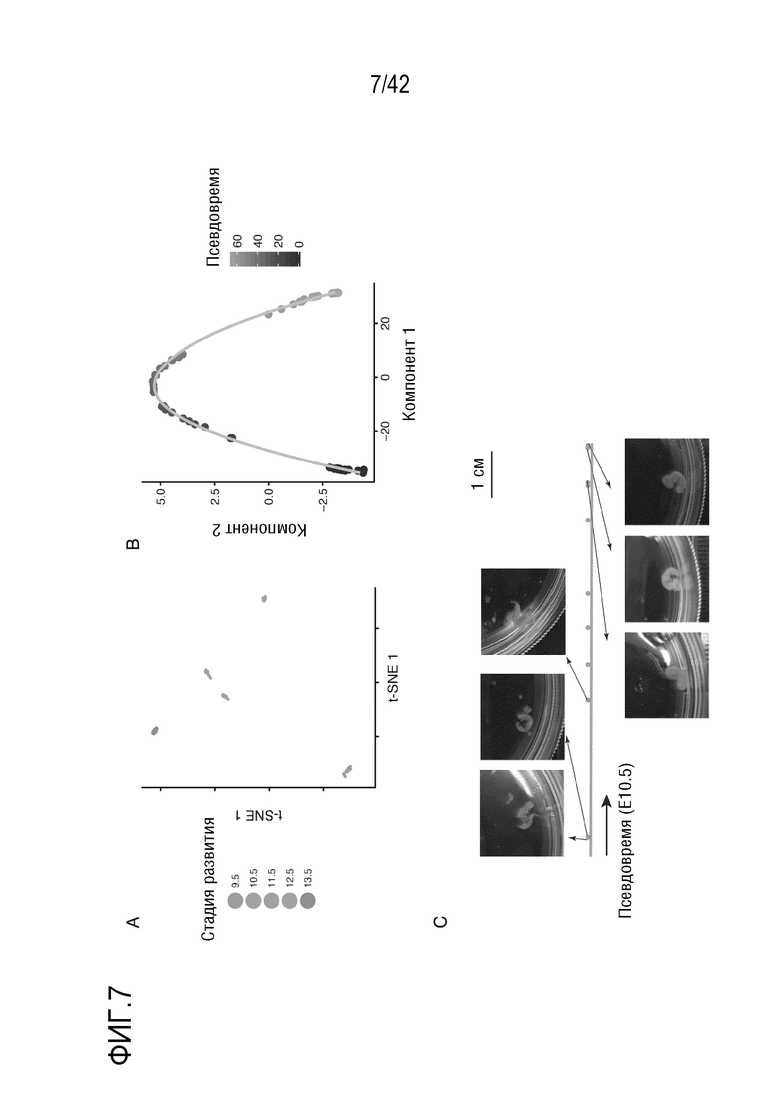
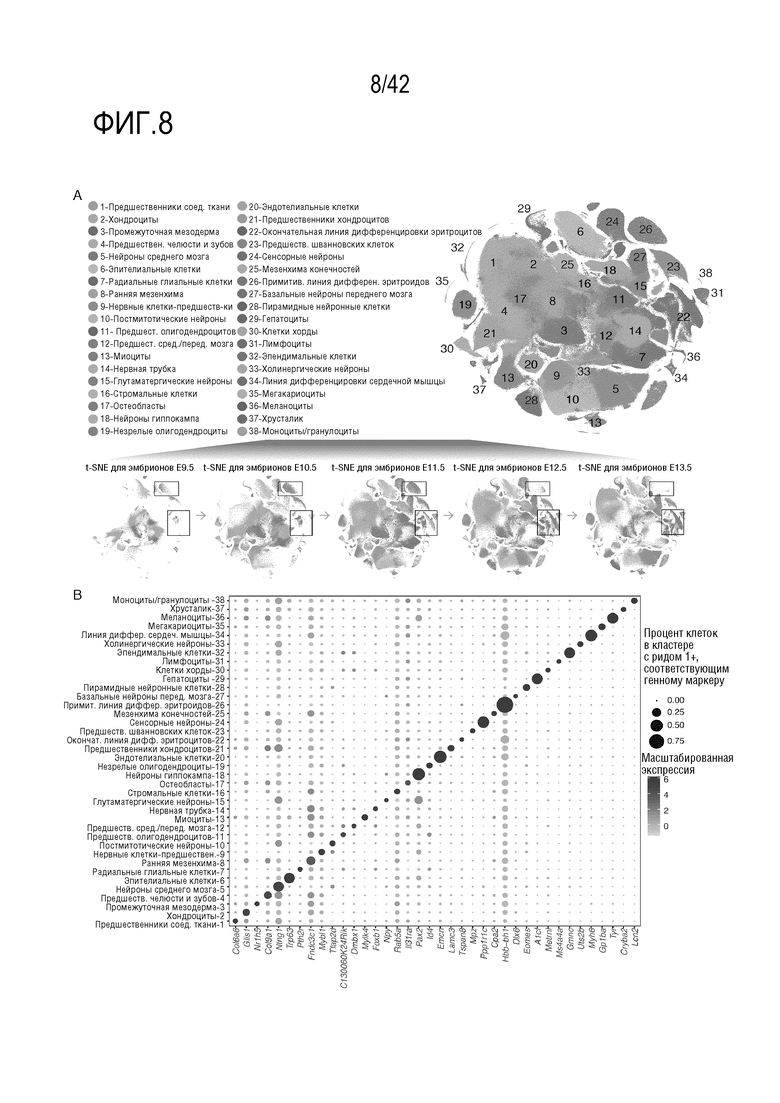
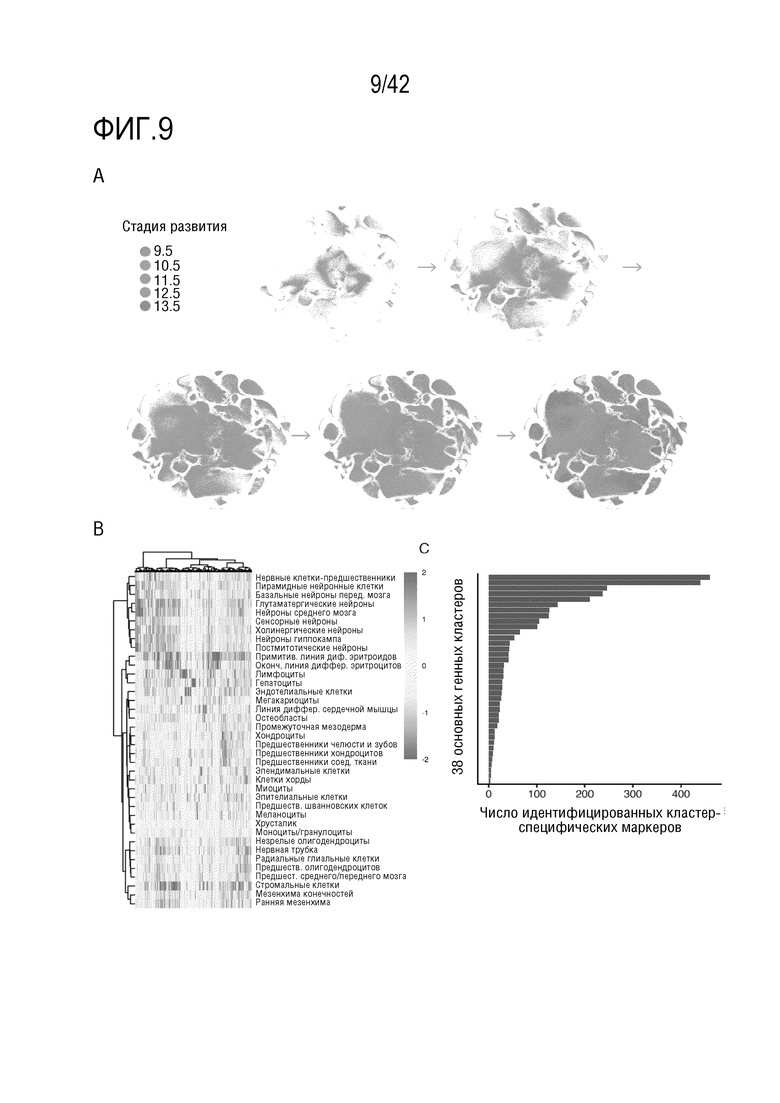
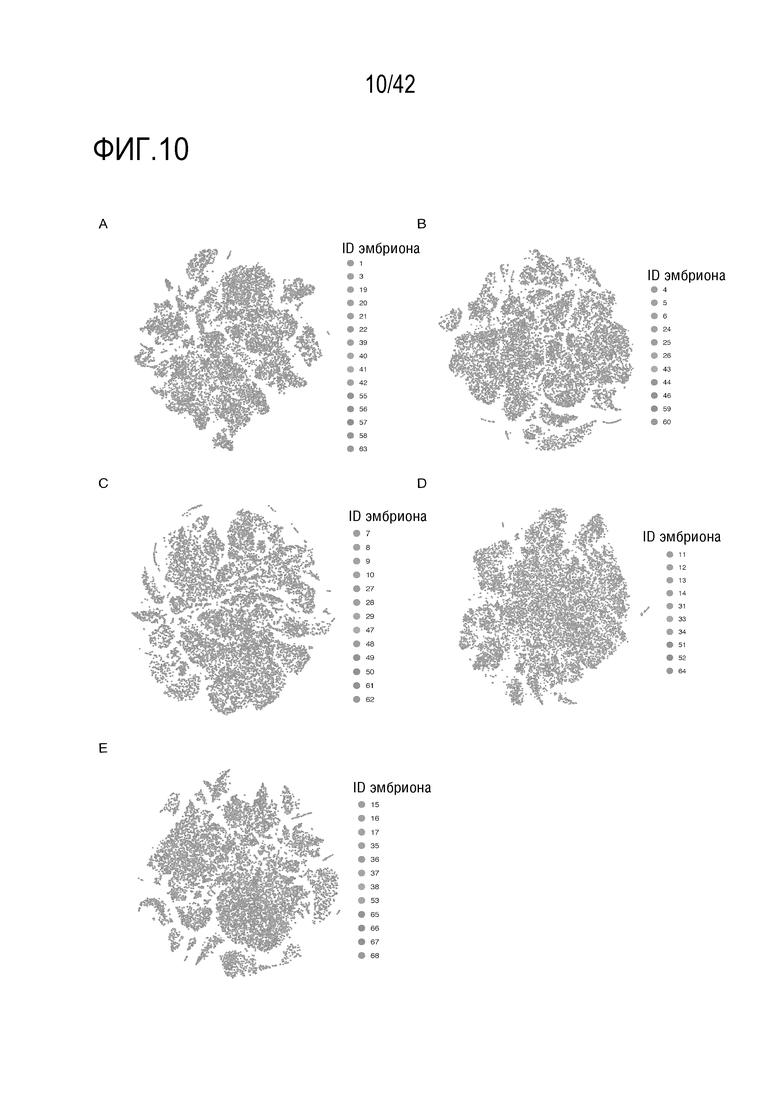
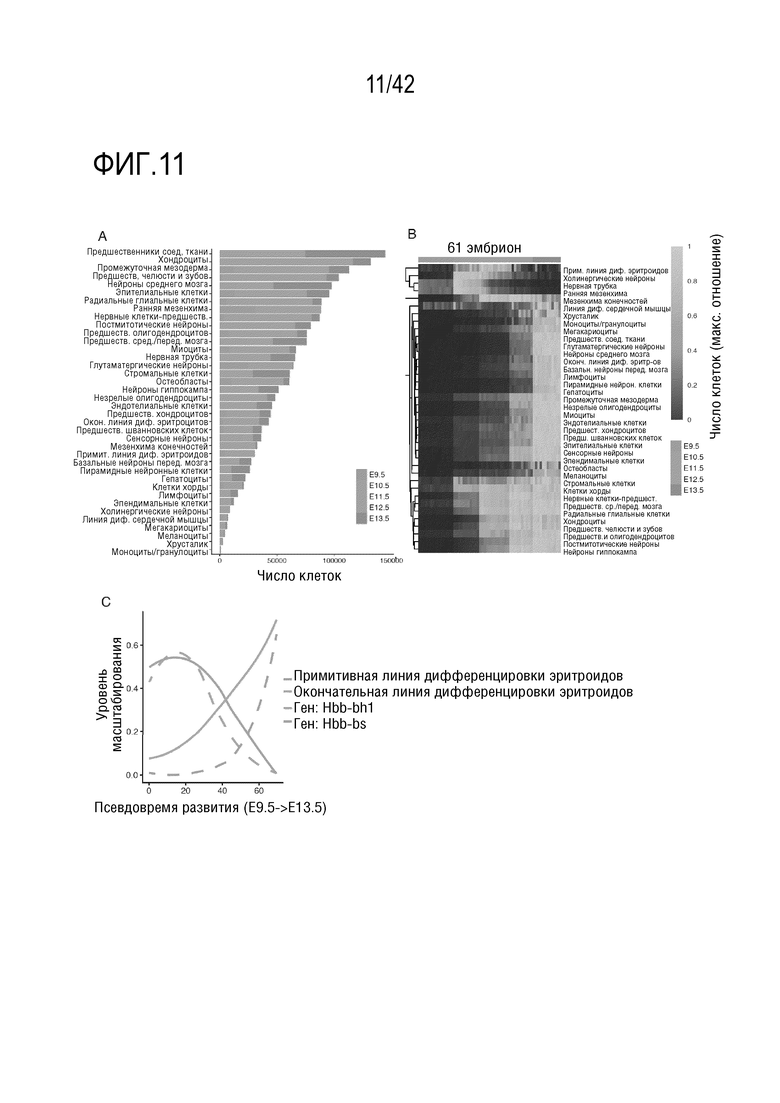


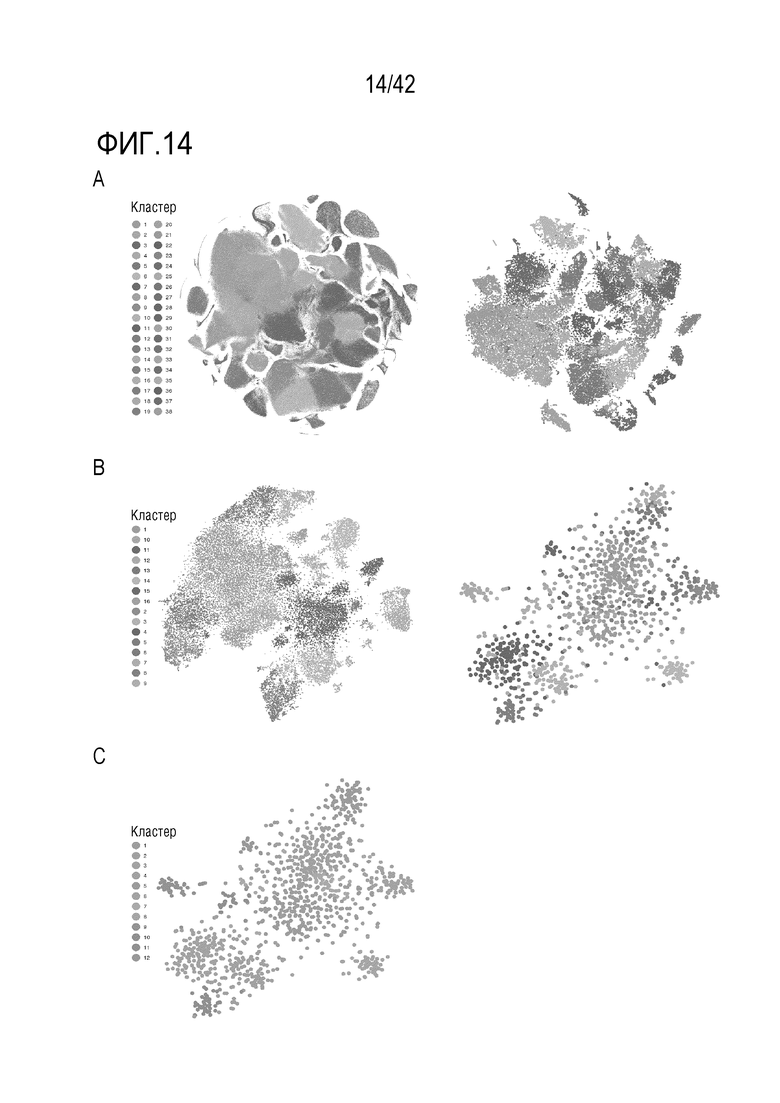
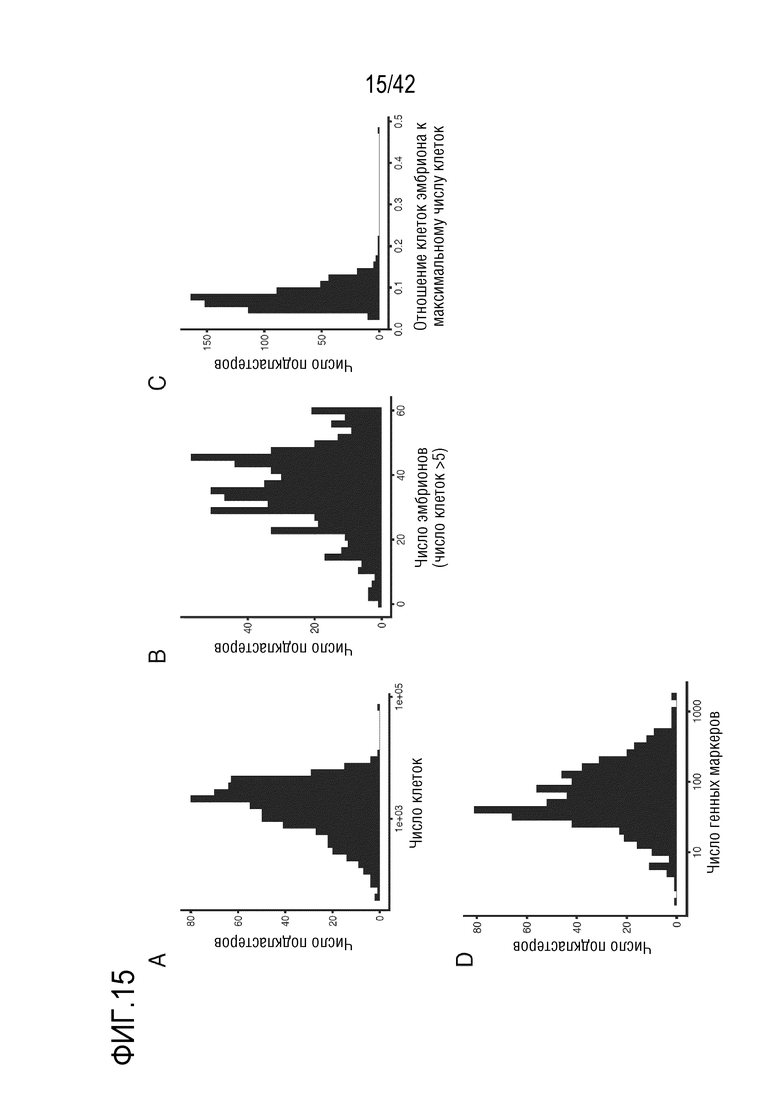
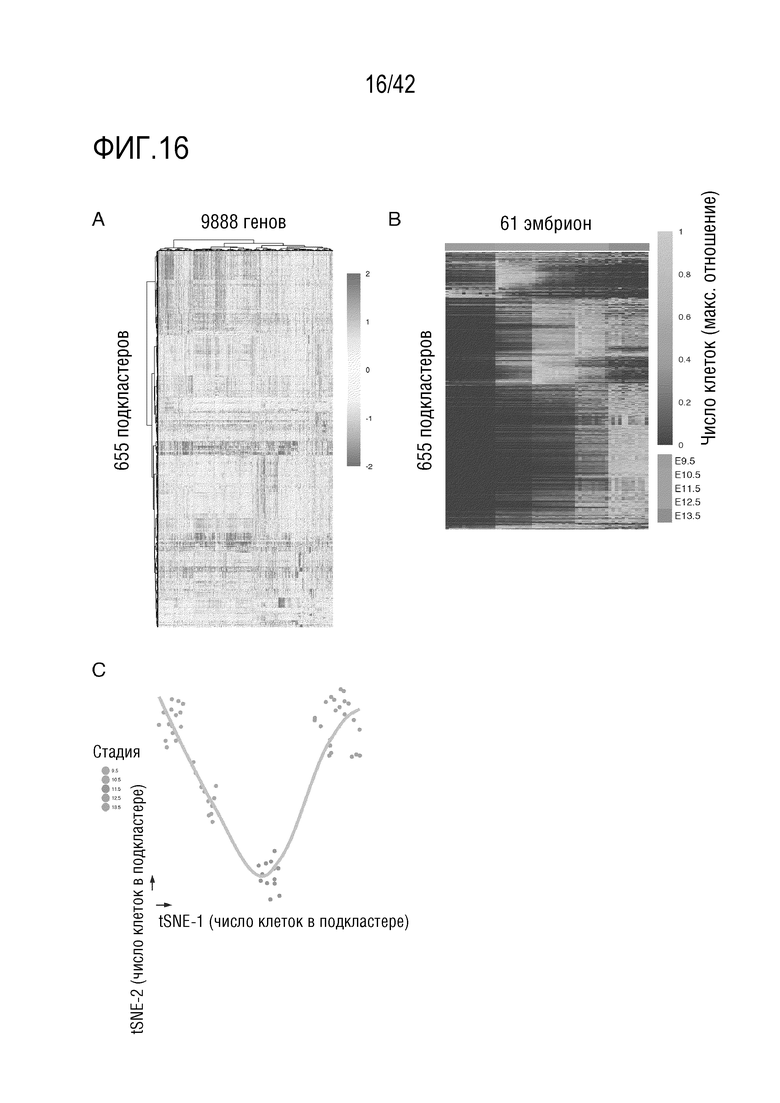
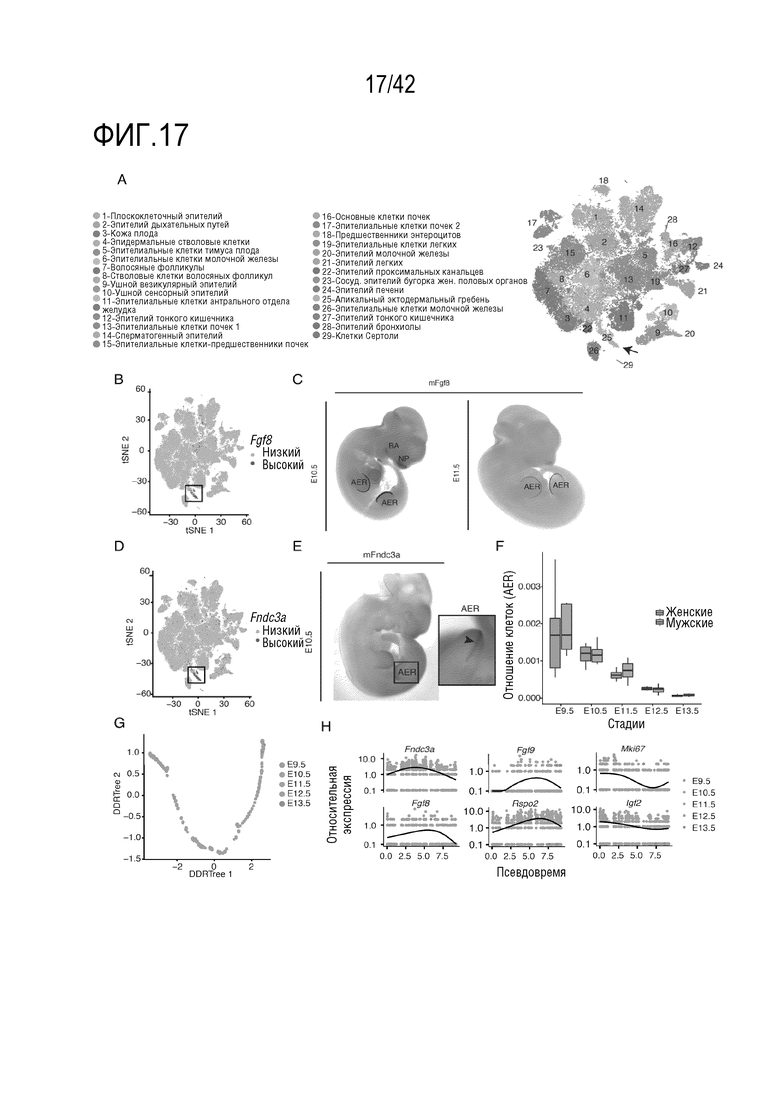
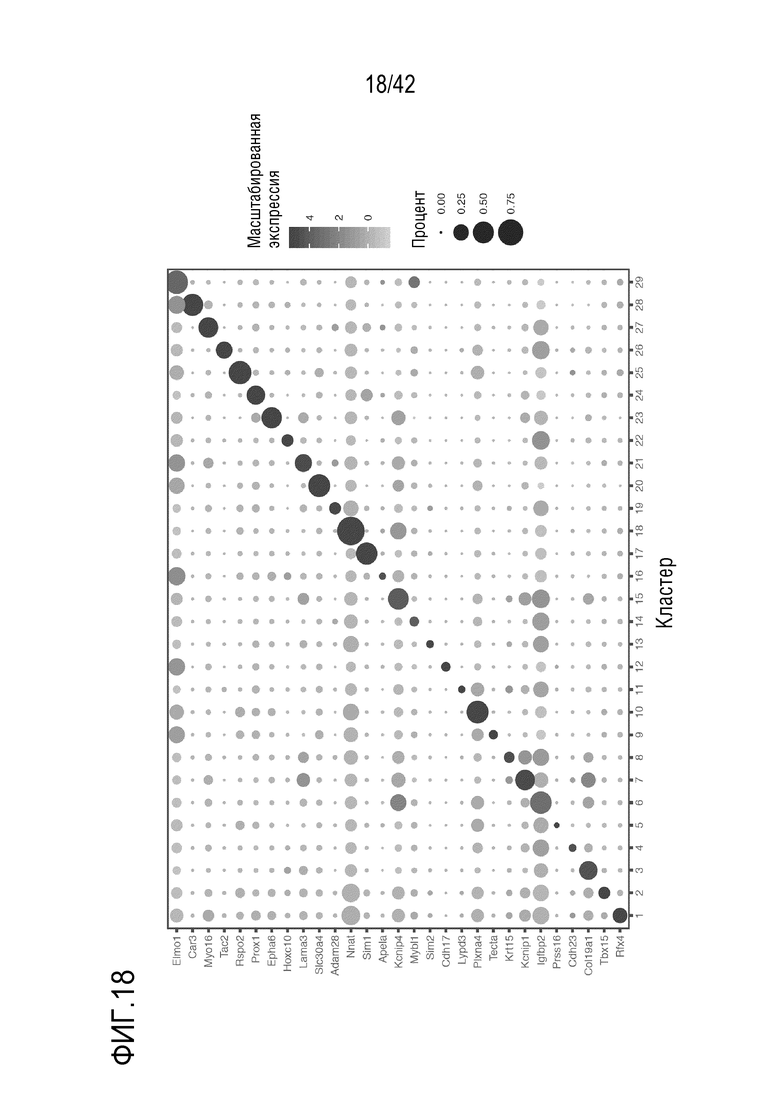
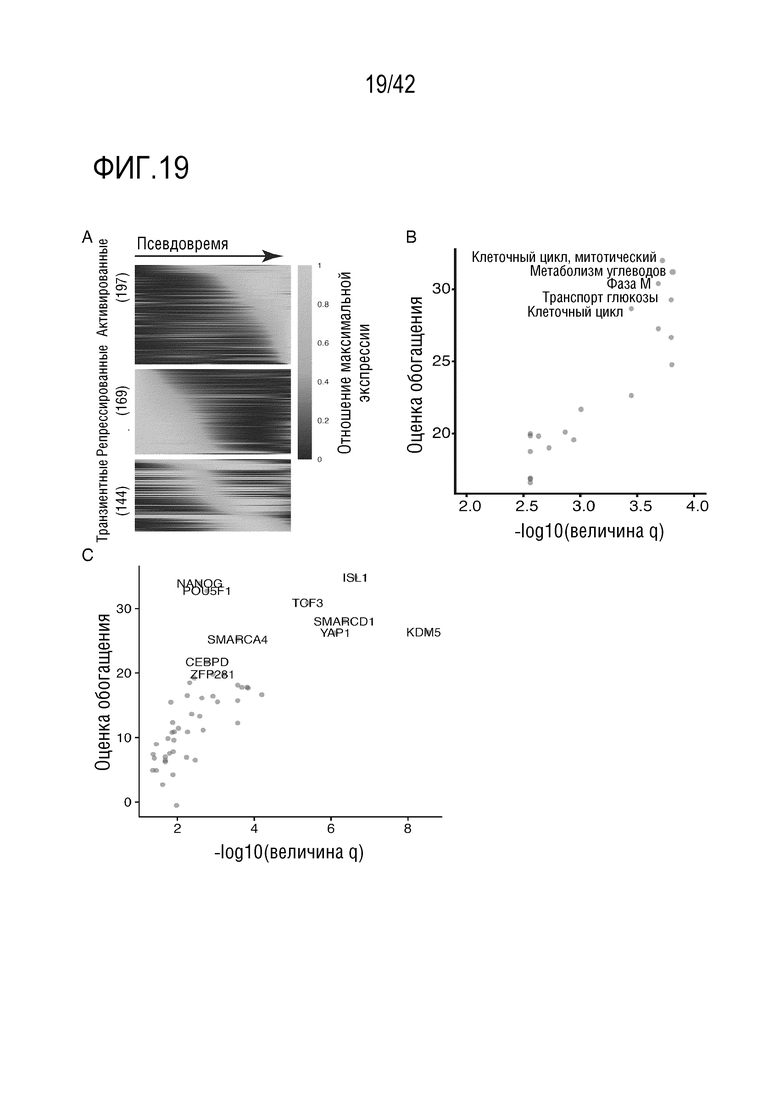
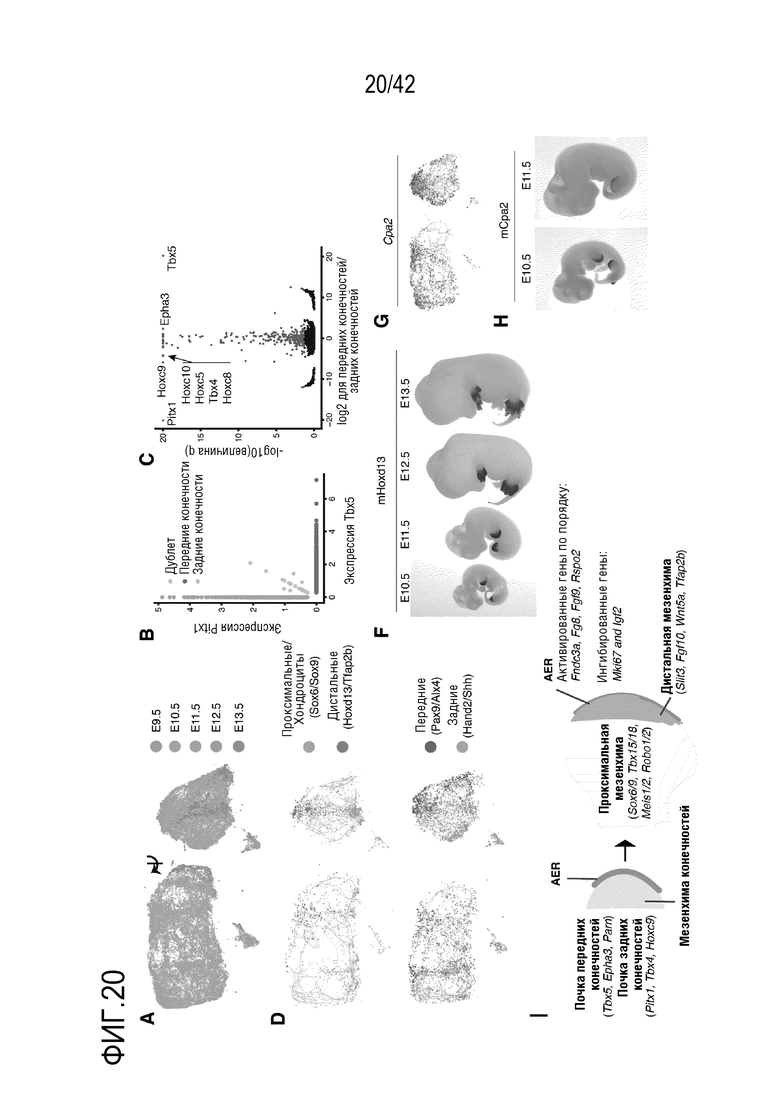
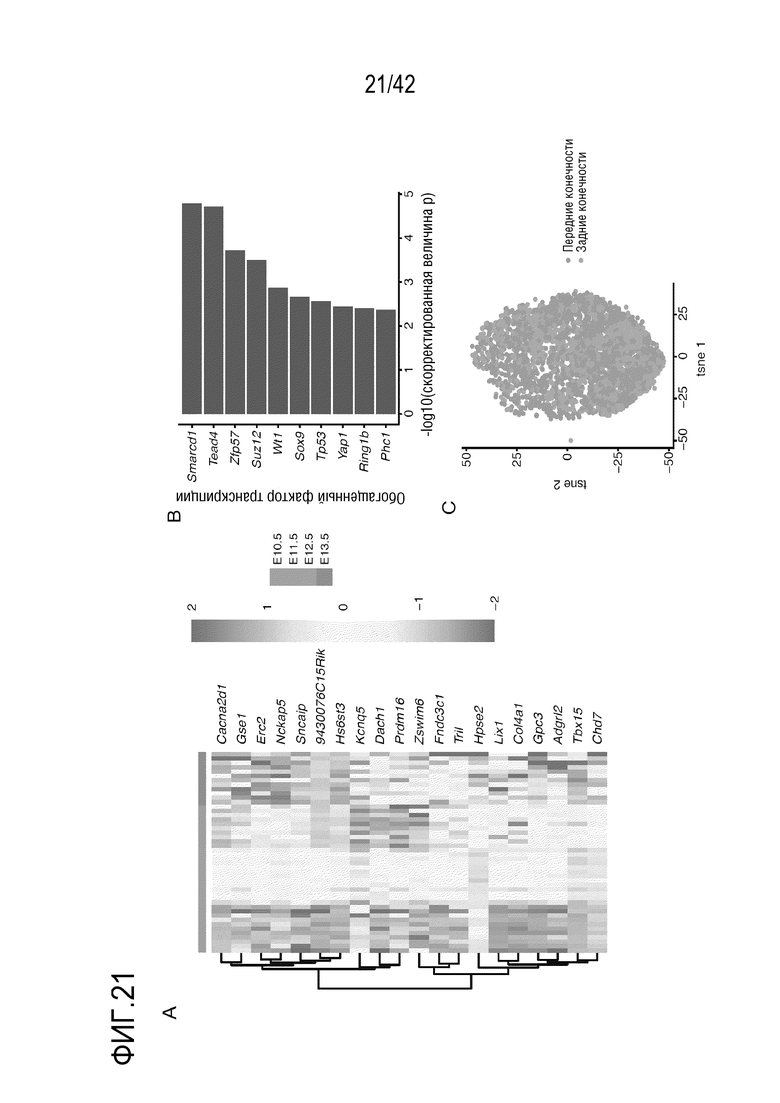
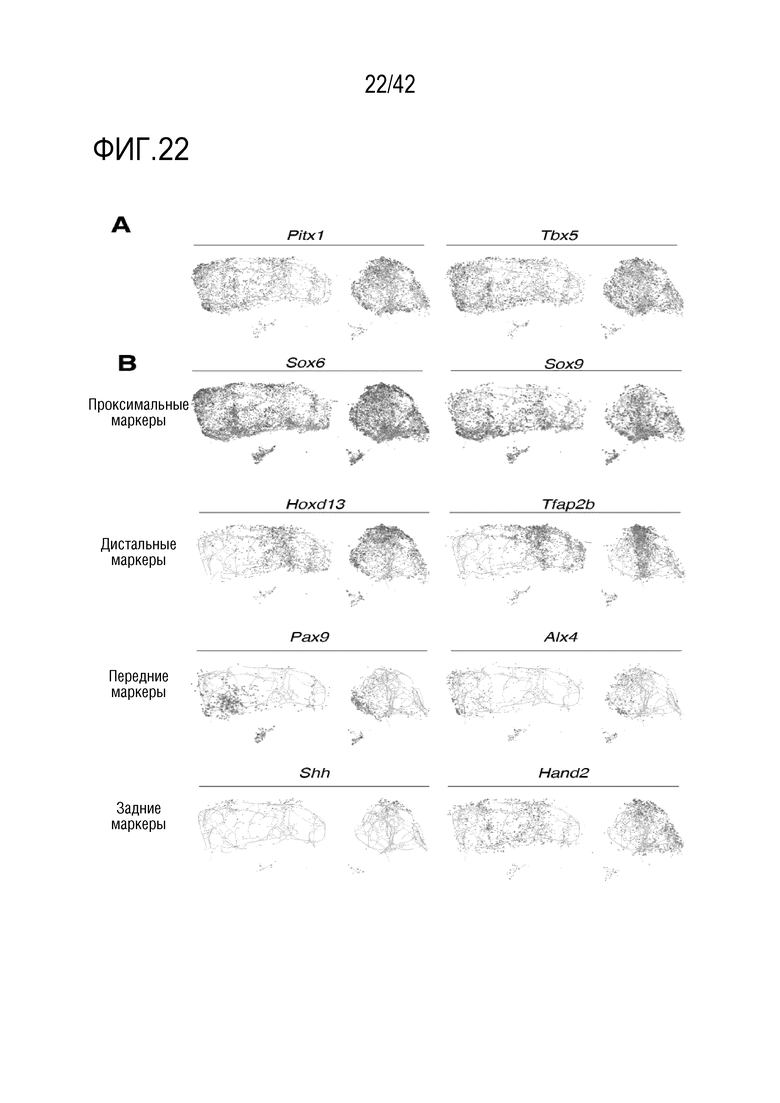


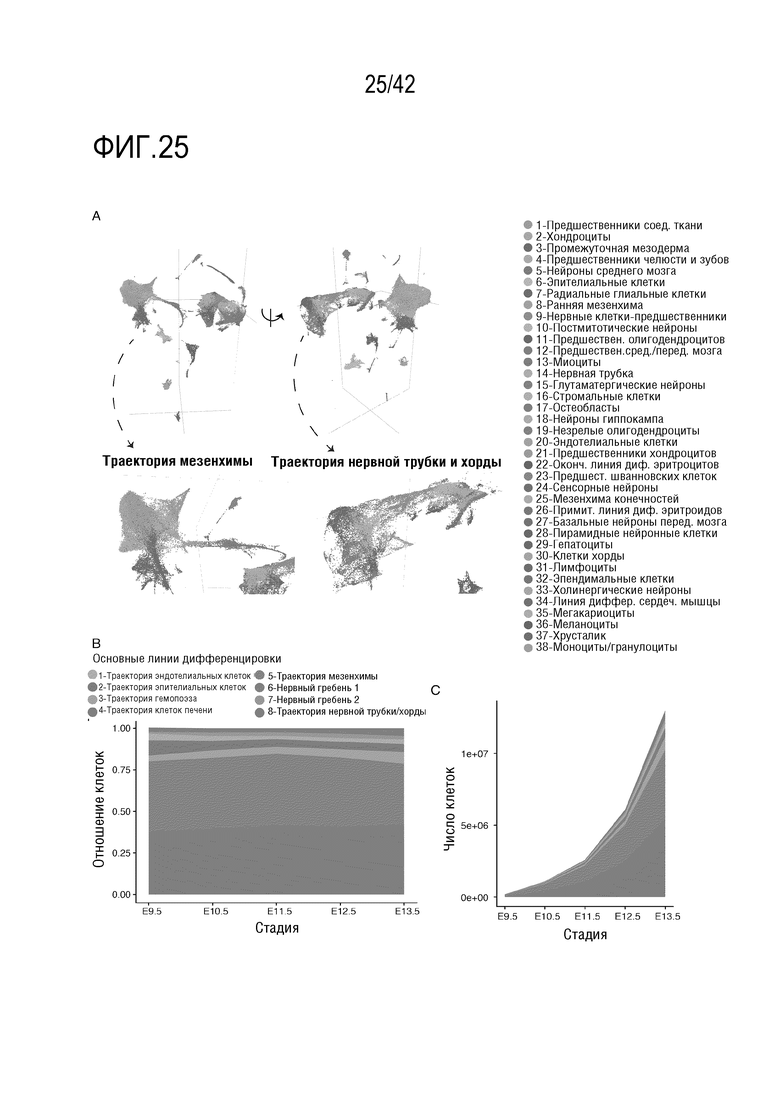

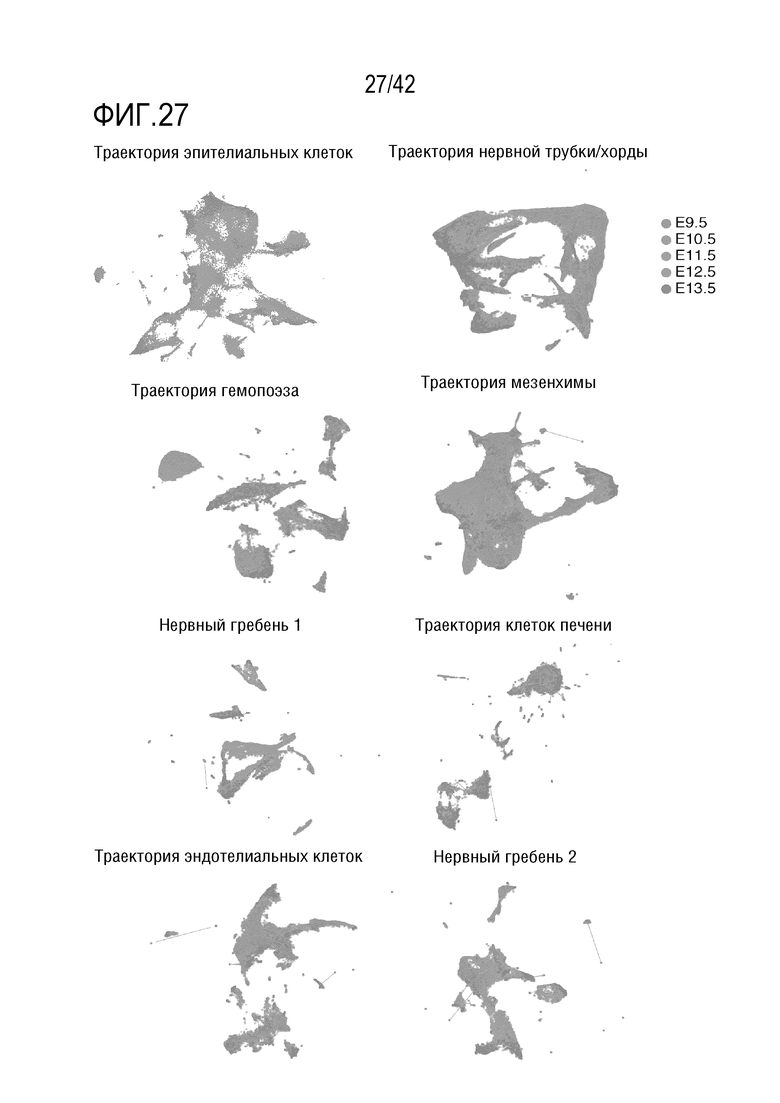

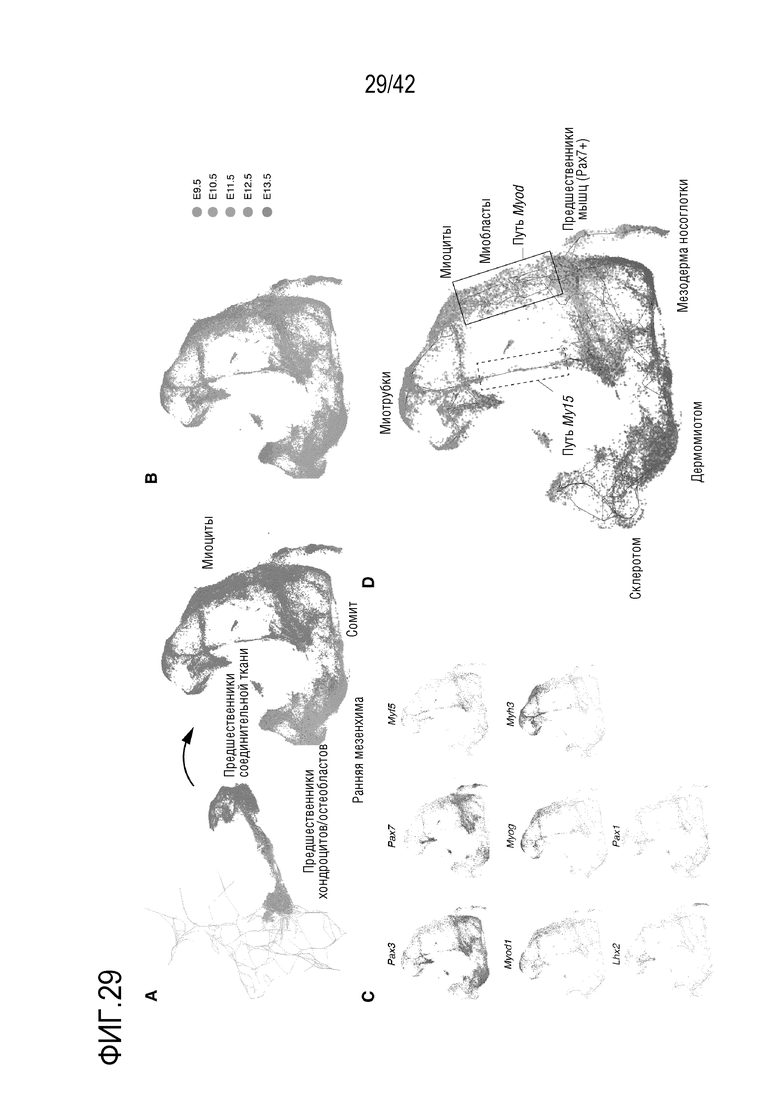
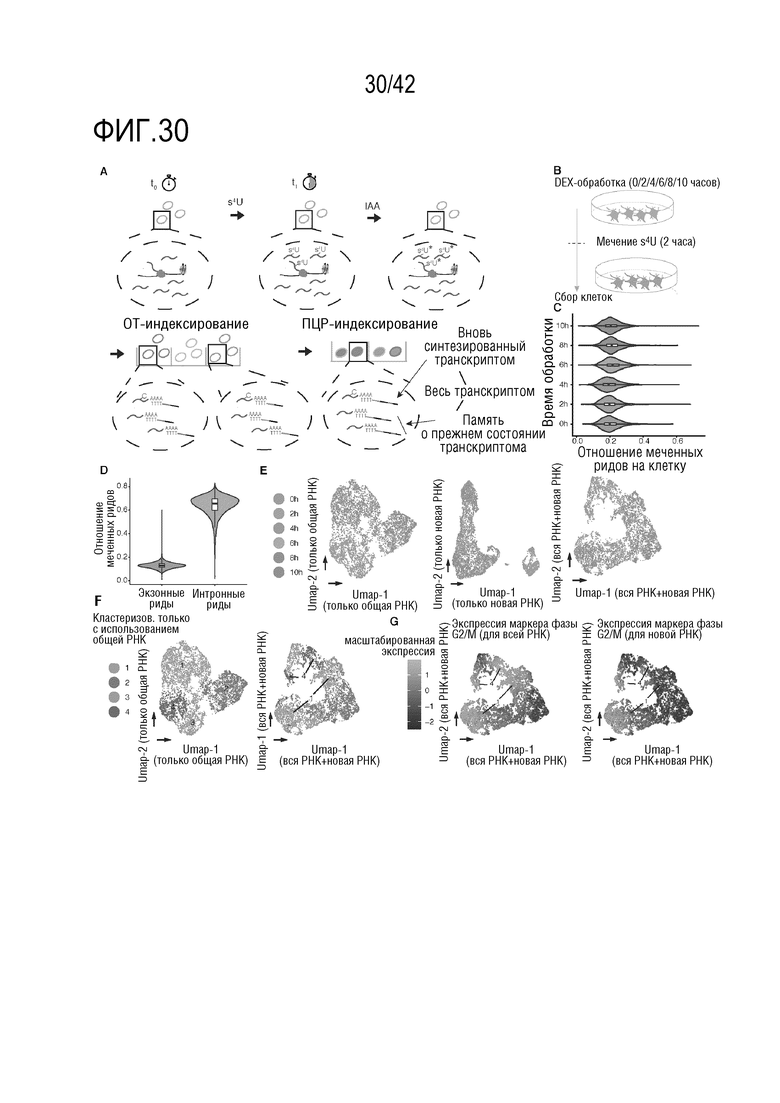
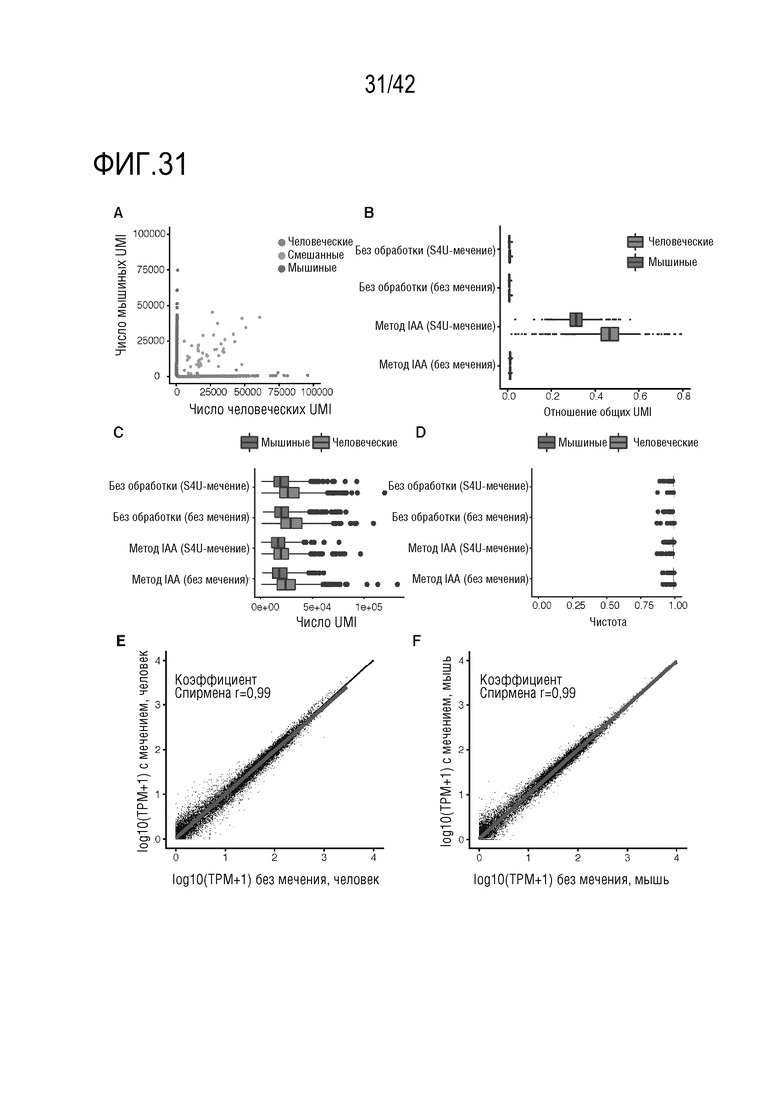
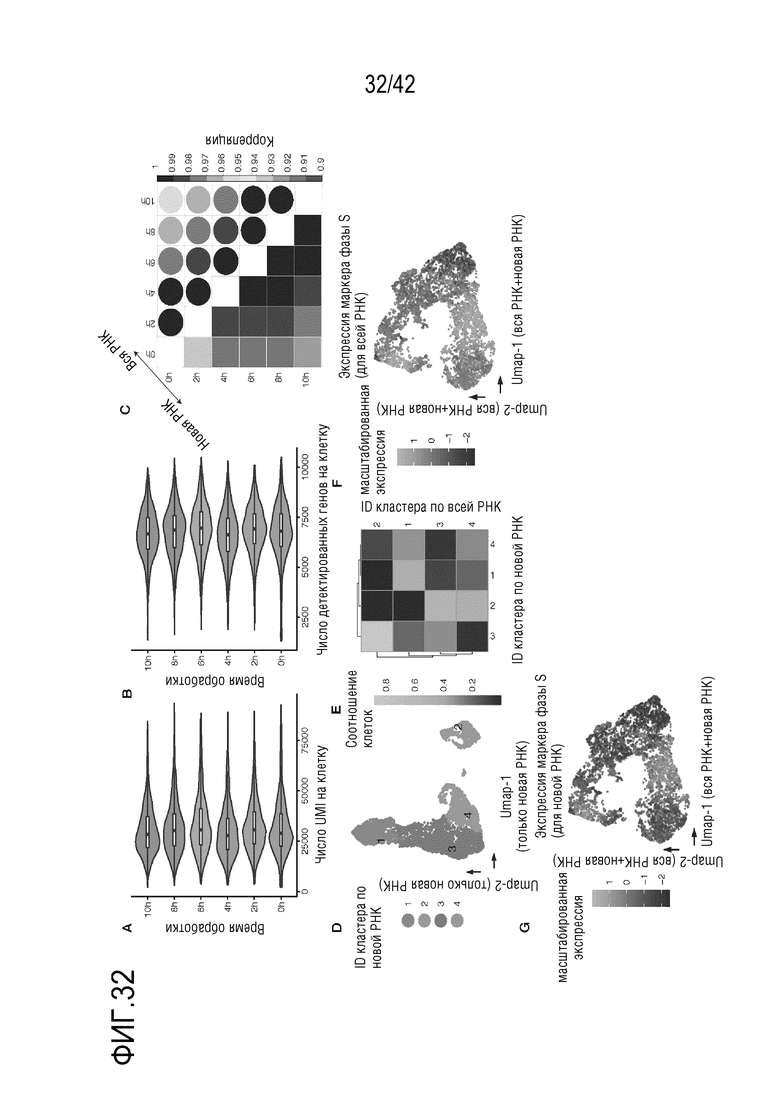
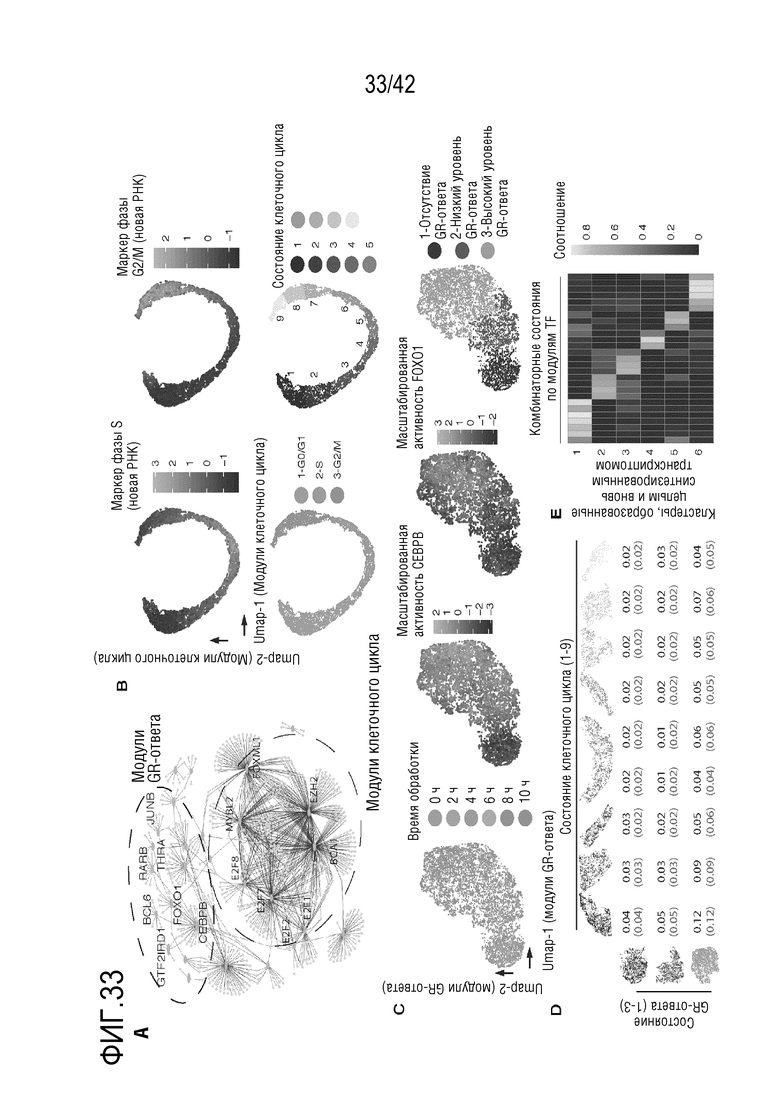
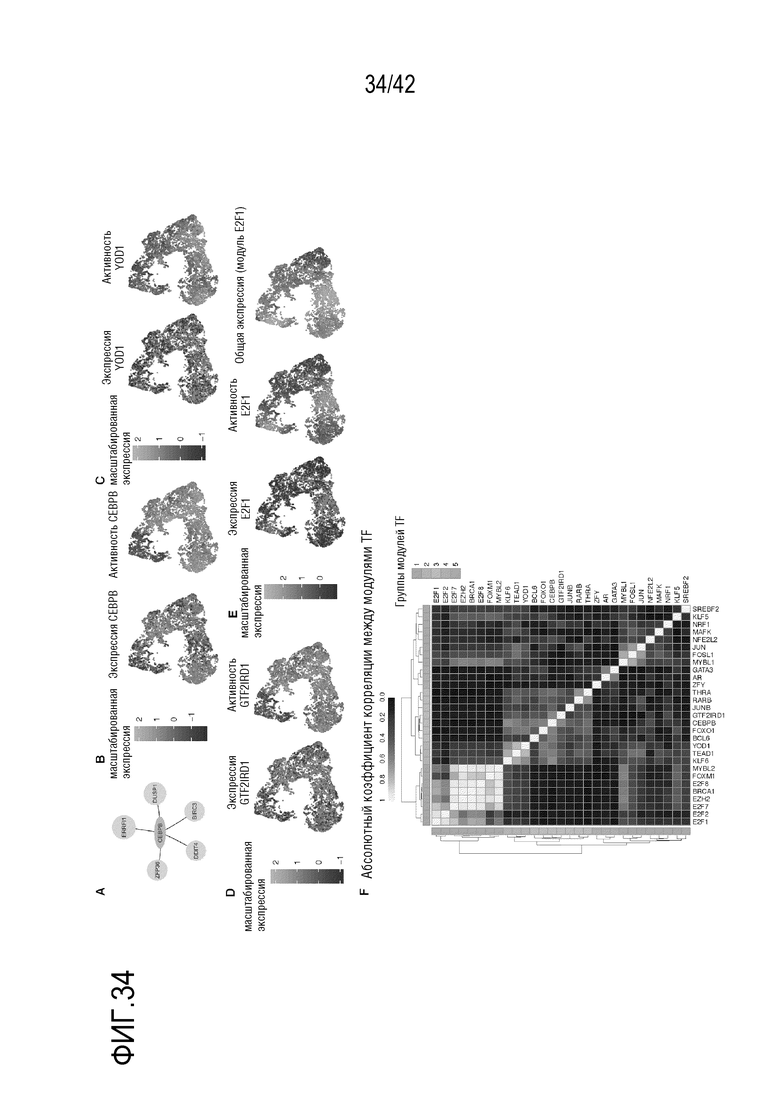
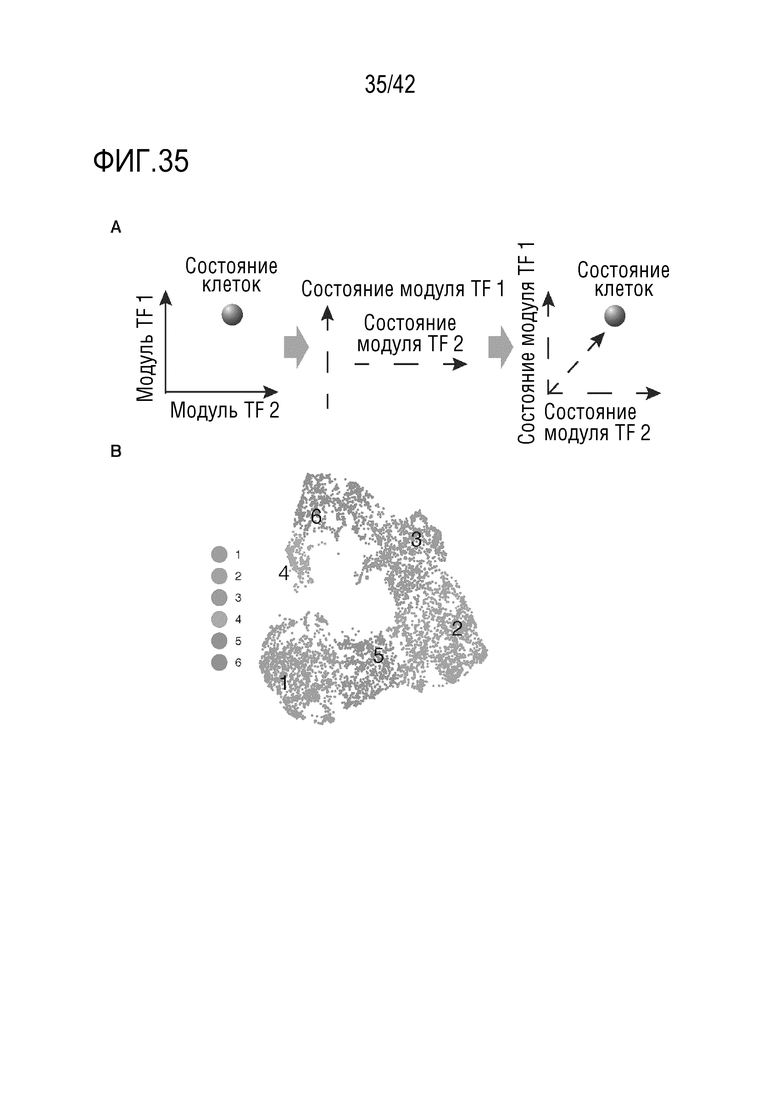

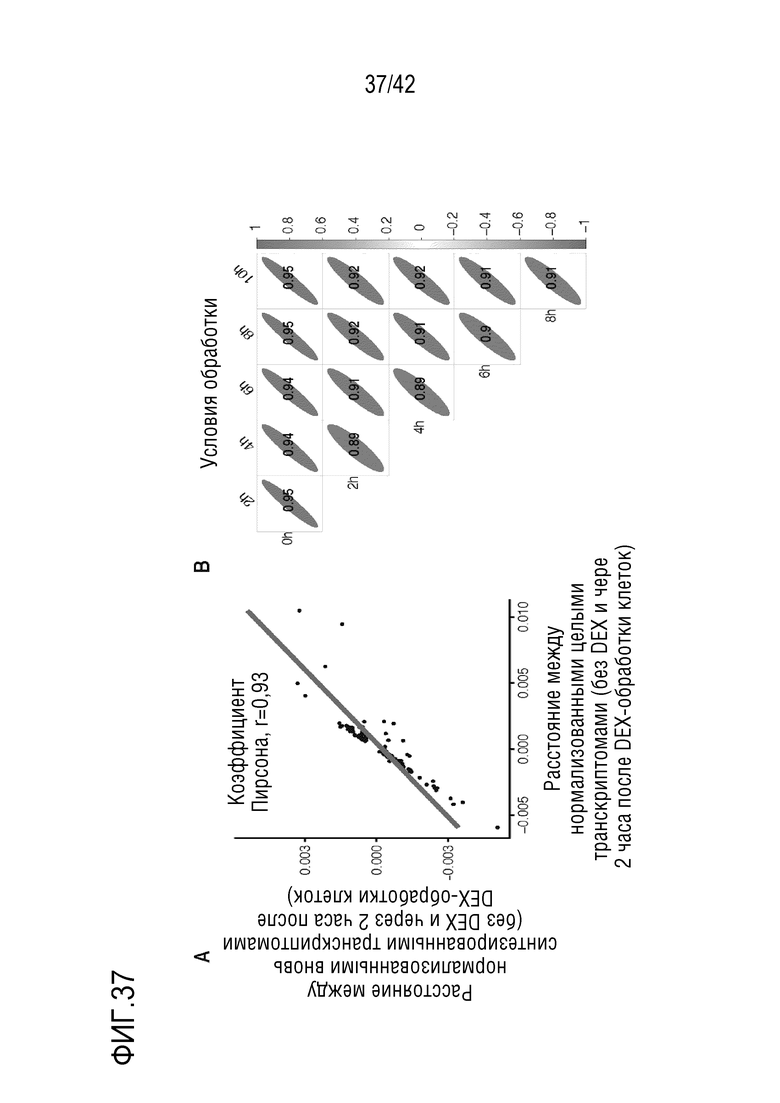
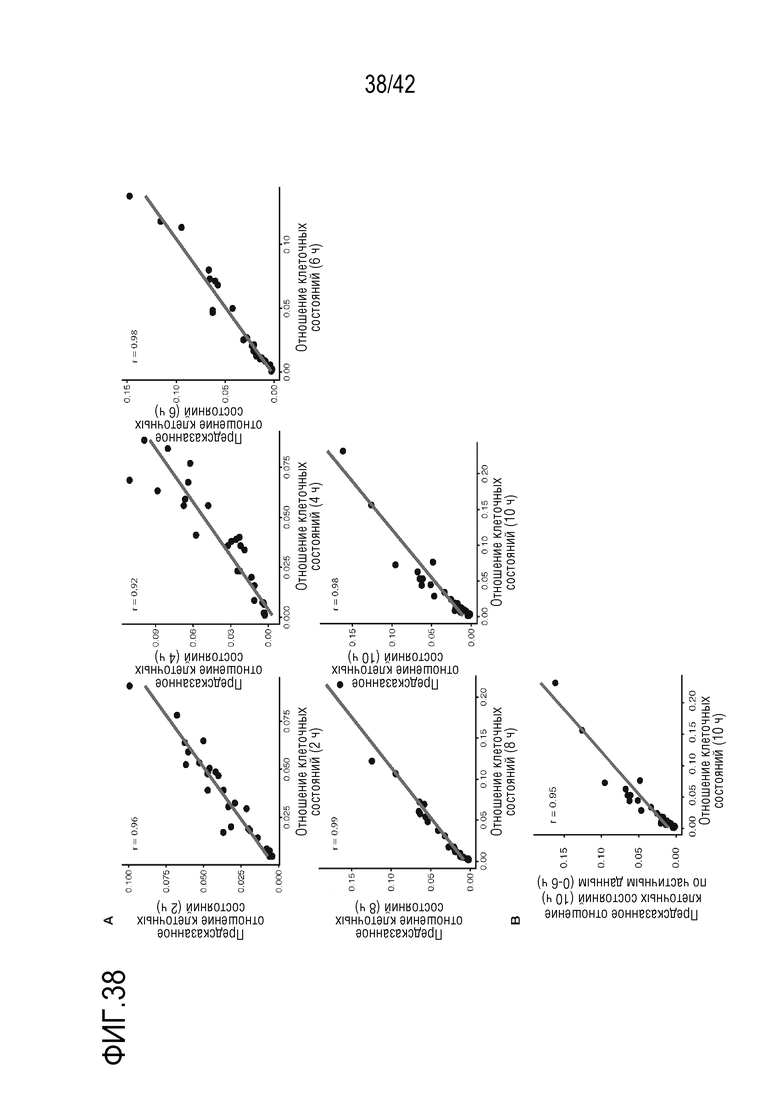
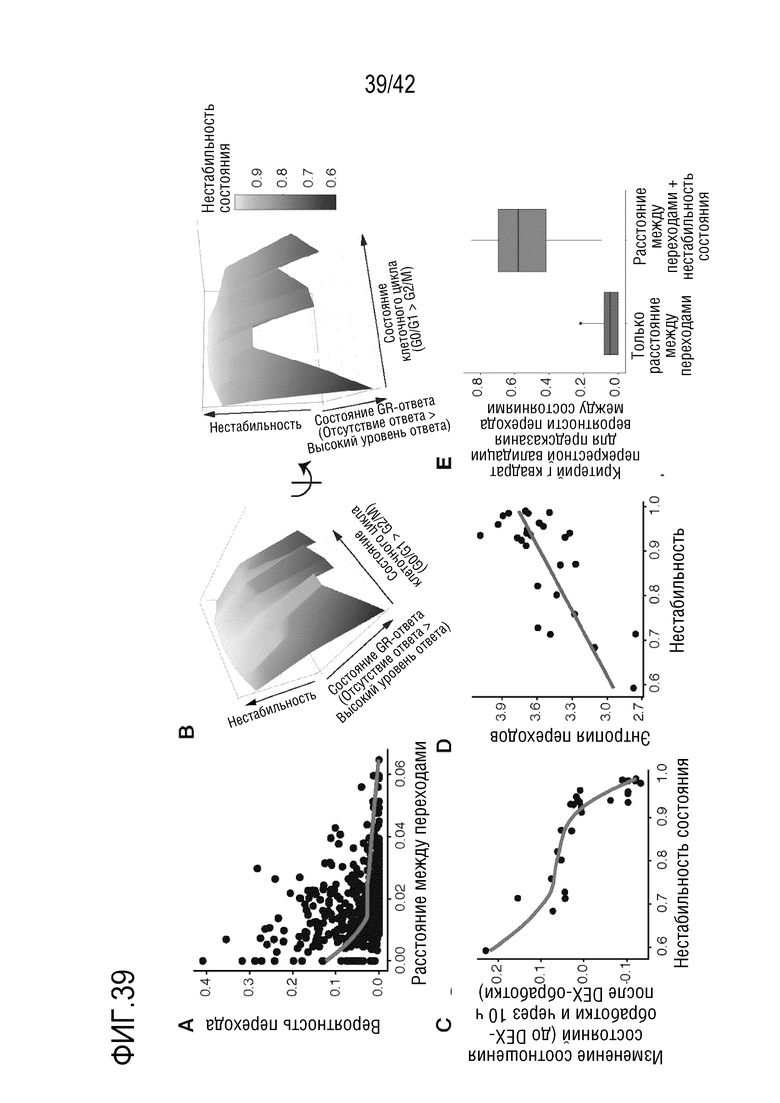

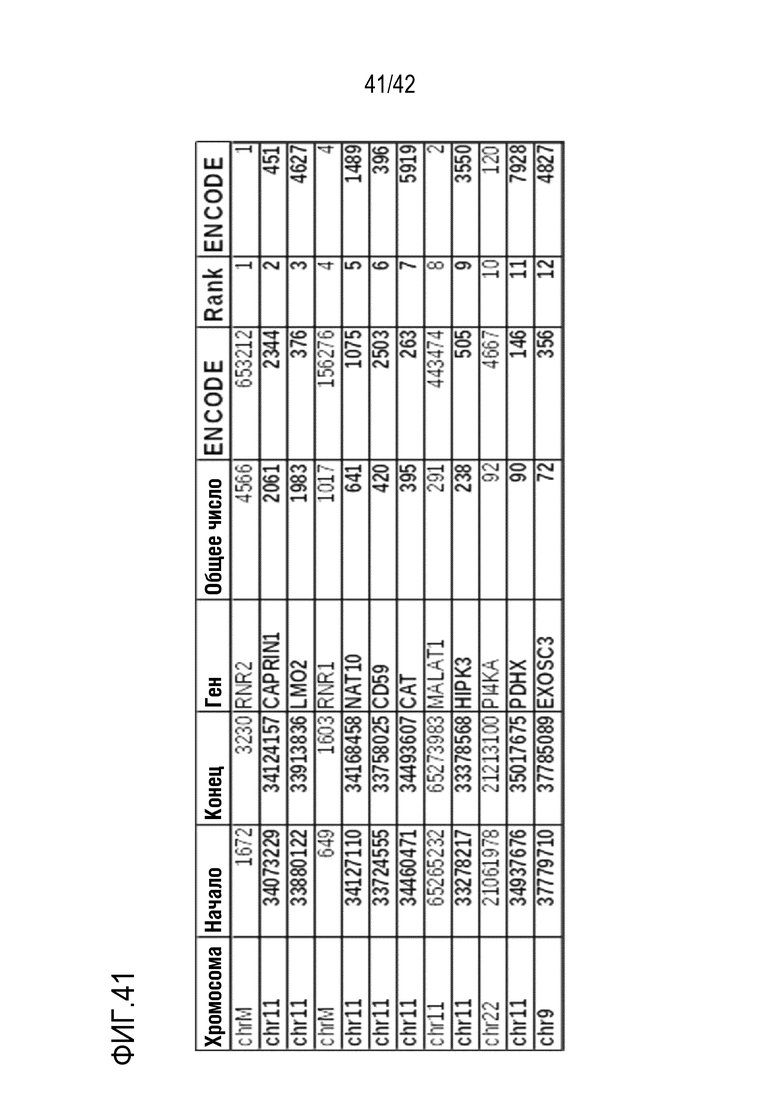
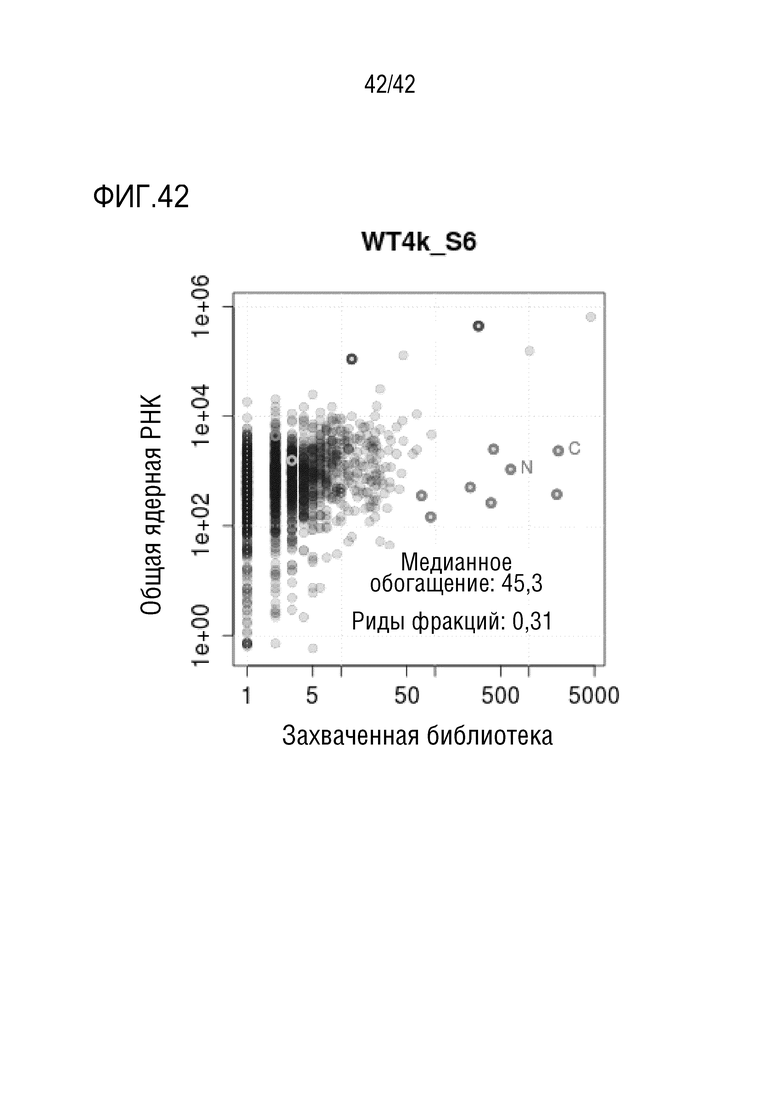
Изобретение относится к области биотехнологии. Предложены варианты способа получения библиотеки для секвенирования, содержащей нуклеиновые кислоты, происходящие из множества отдельных ядер или клеток. Изобретение обеспечивает возможность оценки транскрипционной динамики сложных биологических систем. 6 н. и 15 з.п. ф-лы, 42 ил., 1 табл., 4 пр.
1. Способ получения библиотеки для секвенирования, содержащей нуклеиновые кислоты, происходящие из множества отдельных ядер или клеток, где указанный способ включает:
(а) получение множества ядер или клеток в первом множестве компартментов,
где каждый компартмент содержит субпопуляцию ядер или клеток;
(b) мечение вновь синтезированной РНК в субпопуляциях клеток или ядер, полученных из клеток;
(c) обработку молекул РНК в каждой субпопуляции ядер или клеток с получением индексированных ядер или клеток,
где обработка включает добавление к нуклеиновым кислотам РНК, присутствующим в каждой субпопуляции ядер или клеток, первой компартмент-специфической индексной последовательности с получением индексированных нуклеиновых кислот ДНК, присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию или амплификацию; и
(d) объединение индексированных ядер или клеток с получением объединенных индексированных ядер или клеток.
2. Способ по п. 1, где обработка включает:
контактирование субпопуляций с обратной транскриптазой и праймером, которые гибридизуются с нуклеиновыми кислотами РНК, с образованием двухцепочечных нуклеиновых кислот ДНК, содержащих праймер, и соответствующей нуклеотидной последовательности ДНК из матричных молекул РНК, и необязательно
а) где праймер содержит поли-Т-нуклеотидную последовательность, которая гибридизуется с поли(А)-хвостом мРНК, где необязательно обработка также включает контактирование субпопуляций со вторым праймером, где второй праймер содержит последовательность, которая гибридизуется с предварительно определенной нуклеиновой кислотой ДНК, где необязательно второй праймер содержит компартмент-специфический индекс;
b) где праймер содержит последовательность, которая гибридизуется с предварительно определенной нуклеиновой кислотой РНК, где необязательно способ включает праймеры в различных компартментах, которые гибридизуются с различными нуклеотидами одной и той же предварительно определенной нуклеиновой кислоты РНК, или
с) где праймер включает праймер переключения матрицы.
3. Способ по п. 1, где обработка для добавления первой компартмент-специфической индексной последовательности включает двухстадийный способ добавления нуклеотидной последовательности, содержащей универсальную последовательность, к нуклеиновым кислотам РНК с получением нуклеиновых кислот ДНК, а затем добавление первой компартмент-специфической индексной последовательности к нуклеиновым кислотам ДНК.
4. Способ получения библиотеки для секвенирования, содержащей нуклеиновые кислоты, происходящие из множества отдельных ядер или клеток, где указанный способ включает:
(а) получение множества ядер или клеток в первом множестве компартментов,
где каждый компартмент содержит субпопуляцию ядер или клеток;
(b) мечение вновь синтезированной РНК в субпопуляциях клеток или ядер, полученных из клеток;
(c) контактирование каждой субпопуляции с обратной транскриптазой и праймером, которые гибридизуются с предварительно определенной нуклеиновой кислотой РНК, с образованием двухцепочечных нуклеиновых кислот ДНК, содержащих праймер, и соответствующей нуклеотидной последовательности ДНК из матричных молекул РНК;
(d) обработку молекул ДНК в каждой субпопуляции ядер или клеток с получением индексированных ядер или клеток,
где обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в каждой субпопуляции ядер или клеток, первой компартмент-специфической индексной последовательности с получением индексированных нуклеиновых кислот, присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию или амплификацию; и
(e) объединение индексированных ядер или клеток с получением объединенных индексированных ядер или клеток.
5. Способ по п. 4, где
а) праймер содержит первую компартмент-специфическую индексную последовательность, или
b) где обработка для добавления первой компартмент-специфической индексной последовательности включает двухстадийный способ добавления нуклеотидной последовательности, содержащей универсальную последовательность, к нуклеиновым кислотам, а затем добавление первой компартмент-специфической индексной последовательности к нуклеиновым кислотам.
6. Способ по п. 2 или 5, где предварительно определенная нуклеиновая кислота РНК представляет собой мРНК.
7. Способ по п. 1 или 5, где
а) уже существующие нуклеиновые кислоты РНК и вновь синтезированные нуклеиновые кислоты РНК помечены одинаковыми индексами в одном и том же компартменте,
b) где мечение включает инкубирование множества ядер или клеток в композиции, содержащей нуклеотидную метку, где нуклеотидная метка включена во вновь синтезированную РНК, необязательно
i) где нуклеотидная метка содержит нуклеотидный аналог, меченный гаптеном нуклеотид, мутагенный нуклеотид или нуклеотид, который может быть модифицирован посредством химической реакции, или
ii) где более чем одна нуклеотидная метка включена во вновь синтезированную РНК, где необязательно отношения нуклеотидной метки или меток отличаются для различных компартментов или моментов времени,
c) также включает обработку субпопуляций ядер или клеток в предварительно определенных условиях перед мечением, необязательно
- где предварительно определенное условие включает обработку агентом, необязательно
i) где агент включает белок, не-рибосомный белок, поликетид, органическую молекулу, неорганическую молекулу, молекулу РНК или РНКи, углевод, гликопротеин, нуклеиновую кислоту или их комбинацию, или
ii) где агент включает терапевтическое лекарственное средство,
- где предварительно определенные условия двух или более компартментов являются различными,
- где обработку и мечение проводят одновременно или обработку проводят до мечения.
8. Способ по п. 1 или 4, дополнительно включающий:
распределение субпопуляций объединенных индексированных ядер или клеток по второму множеству компартментов и добавление к индексированным нуклеиновым кислотам, присутствующим в субпопуляциях ядер или клеток, второй индексной последовательности с получением ядер или клеток с двумя индексами, содержащих фрагменты нуклеиновой кислоты с двумя индексами, где добавление включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию;
объединение ядер или клеток с двумя индексами с получением объединенных ядер или клеток с двумя индексами.
9. Способ по п. 8, дополнительно включающий:
распределение субпопуляций объединенных ядер или клеток с двумя индексами по третьему множеству компартментов и добавление к индексированным нуклеиновым кислотам, присутствующим в субпопуляциях ядер или клеток, третьей индексной последовательности с получением ядер или клеток с тремя индексами, содержащих фрагменты нуклеиновой кислоты с тремя индексами, где добавление включает лигирование, гибридизацию, удлинение праймера, амплификацию или транспозицию;
объединение ядер или клеток с тремя индексами с получением объединенных ядер или клеток с тремя индексами, необязательно
а) где распределение включает разведение,
b) где распределение включает сортинг,
с) где добавление включает контактирование субпопуляций со шпилечным лигирующим дуплексом в условиях, подходящих для лигирования шпилечного лигирующего дуплекса с концом фрагментов нуклеиновой кислоты, содержащих одну или две индексные последовательности,
d) где добавление включает контактирование фрагментов нуклеиновой кислоты, содержащих одну или более индексных последовательностей, с транспосомным комплексом, где транспосомный комплекс в компартментах содержит транспозазу и универсальную последовательность, где контактирование также включает условия, подходящие для фрагментации фрагментов нуклеиновой кислоты и включения универсальной последовательности во фрагменты нуклеиновой кислоты.
10. Способ по п. 1 или 4, где добавление включает лигирование первой компартмент-специфической индексной последовательности, а также включает добавление второй индексной последовательности с получением ядер или клеток с двумя индексами, содержащих фрагменты нуклеиновой кислоты с двумя индексами, где добавление включает транспозицию.
11. Способ по п. 8, где добавление включает лигирование второй компартмент-специфической индексной последовательности, а также включает добавление третьей индексной последовательности с получением ядер или клеток с двумя индексами, содержащих фрагменты нуклеиновой кислоты с тремя индексами, где добавление включает транспозицию.
12. Способ по любому из пп. 1, 8 или 9, а) где компартмент включает лунку или каплю, b) где компартменты первого множества компартментов содержат от 50 до 100000000 ядер или клеток,
необязательно по п.8, i) где компартменты второго множества компартментов содержат от 50 до 100000000 ядер или клеток, ii) также включает получение нуклеиновых кислот с двумя индексами из объединенных ядер или клеток с двумя индексами и тем самым получение библиотеки для секвенирования из множества ядер или клеток,
необязательно по п. 9, i) где компартменты третьего множества компартментов содержат от 50 до 100000000 ядер или клеток, ii) также включает получение нуклеиновых кислот с тремя индексами из объединенных ядер или клеток с тремя индексами и тем самым получение библиотеки для секвенирования из множества ядер или клеток.
13. Способ по п. 1 или 4, дополнительно включающий получение индексированных нуклеиновых кислот из объединенных индексированных ядер или клеток и тем самым получение библиотеки для секвенирования из множества ядер или клеток.
14. Способ по любому из пп. 1-11, дополнительно включающий:
получение поверхности, содержащей множество сайтов амплификации,
где сайты амплификации включают по меньшей мере две популяции связанных одноцепочечных олигонуклеотидов для захвата, имеющих свободный 3’-конец, и
контактирование поверхности, содержащей сайты амплификации, с фрагментами нуклеиновой кислоты, содержащими одну, две или три индексные последовательности, в условиях, подходящих для получения множества сайтов амплификации, каждый из которых содержит клональную популяцию ампликонов, из отдельного фрагмента, содержащего множество индексов.
15. Способ по любому из пп. 8 или 9, где добавление компартмент-специфической индексной последовательности включает двухстадийный способ добавления нуклеотидной последовательности, содержащей универсальную последовательность, к нуклеиновым кислотам, а затем добавление компартмент-специфической индексной последовательности к нуклеиновым кислотам.
16. Способ получения библиотеки для секвенирования, содержащей нуклеиновые кислоты, происходящие из множества отдельных ядер или клеток, где указанный способ включает:
(а) получение множества ядер или клеток в первом множестве компартментов,
где каждый компартмент содержит субпопуляцию ядер или клеток;
(b) контактирование каждой субпопуляции с обратной транскриптазой и праймером с получением двухцепочечных нуклеиновых кислот ДНК, содержащих праймер, и соответствующей нуклеотидной последовательности ДНК из матричных молекул РНК;
(c) обработку молекул ДНК в каждой субпопуляции ядер или клеток с получением индексированных ядер или клеток,
где обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в каждой субпопуляции ядер или клеток, первой компартмент-специфической индексной последовательности с получением индексированных нуклеиновых кислот, присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию; и
(d) объединение индексированных ядер или клеток с получением объединенных индексированных ядер или клеток;
(е) распределение объединенных индексированных ядер или клеток по второму множеству компартментов,
где каждый компартмент содержит субпопуляцию ядер или клеток;
(f) обработку молекул ДНК в каждой субпопуляции ядер или клеток с получением ядер или клеток с двумя индексами,
где обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в каждой субпопуляции ядер или клеток, второй компартмент-специфической индексной последовательности с получением нуклеиновых кислот с двумя индексами, присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию; и
(g) объединение ядер или клеток с двумя индексами с получением объединенных ядер или клеток с двумя индексами;
(h) распределение объединенных ядер или клеток с двумя индексами по третьему множеству компартментов,
где каждый компартмент содержит субпопуляцию ядер или клеток;
(i) обработку молекул ДНК в каждой субпопуляции ядер или клеток с получением ядер или клеток с тремя индексами,
где обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в каждой субпопуляции ядер или клеток, третьей компартмент-специфической индексной последовательности с получением нуклеиновых кислот с тремя индексами, присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию; и
(j) объединение ядер или клеток с тремя индексами с получением объединенных ядер или клеток с тремя индексами.
17. Способ получения библиотеки для секвенирования, содержащей нуклеиновые кислоты, происходящие из множества отдельных ядер или клеток, где указанный способ включает:
(а) получение множества ядер или клеток;
(b) контактирование множества ядер или клеток с обратной транскриптазой и праймером, с получением двухцепочечных нуклеиновых кислот ДНК, содержащих праймер, и соответствующей нуклеотидной последовательности ДНК из матричных нуклеиновых кислот РНК;
(c) распределение ядер или клеток по первому множеству компартментов,
где каждый компартмент содержит субпопуляцию ядер или клеток;
(d) обработку молекул ДНК в каждой субпопуляции ядер или клеток с получением индексированных ядер или клеток,
где обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в каждой субпопуляции ядер или клеток, первой компартмент-специфической индексной последовательности с получением индексированных нуклеиновых кислот, присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию; и
(e) объединение индексированных ядер или клеток с получением объединенных индексированных ядер или клеток;
(f) распределение объединенных индексированных ядер или клеток по второму множеству компартментов,
где каждый компартмент содержит субпопуляцию ядер или клеток;
(g) обработку молекул ДНК в каждой субпопуляции ядер или клеток с получением ядер или клеток с двумя индексами,
где обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в каждой субпопуляции ядер или клеток, второй компартмент-специфической индексной последовательности с получением нуклеиновых кислот с двумя индексами, присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию; и
(h) объединение ядер или клеток с двумя индексами с получением объединенных ядер или клеток с двумя индексами;
(i) распределение объединенных ядер или клеток с двумя индексами по третьему множеству компартментов,
где каждый компартмент содержит субпопуляцию ядер или клеток;
(j) обработку молекул ДНК в каждой субпопуляции ядер или клеток с получением ядер или клеток с тремя индексами,
где обработка включает добавление к нуклеиновым кислотам ДНК, присутствующим в субпопуляциях ядер или клеток, третьей компартмент-специфической индексной последовательности с получением нуклеиновых кислот с тремя индексами, присутствующих в индексированных ядрах или клетках,
где обработка включает лигирование, удлинение праймера, гибридизацию, амплификацию или транспозицию; и
(k) объединение ядер или клеток с тремя индексами с получением объединенных ядер или клеток с тремя индексами.
18. Способ по п. 16 или 17,
а) где праймер гибридизуется с нуклеиновыми кислотами РНК с образованием двухцепочечных нуклеиновых кислот ДНК, содержащих праймер, и соответствующей нуклеотидной последовательности ДНК из матричных молекул РНК, необязательно
i) где праймер содержит поли-Т-нуклеотидную последовательность, которая гибридизуется с поли(А)-хвостом мРНК, необязательно, где контактирование также включает контактирование субпопуляций со вторым праймером, где второй праймер содержит последовательность, которая гибридизуется с предварительно определенной нуклеиновой кислотой ДНК, необязательно где второй праймер содержит компартмент-специфический индекс, или
ii) где праймер содержит последовательность, которая гибридизуется с предварительно определенной нуклеиновой кислотой РНК, необязательно где предварительно определенной нуклеиновой кислотой РНК является мРНК, или
b) где праймер включает праймер переключения матрицы, или
с) где обработка для добавления одной или более первой, второй или третьей компартмент-специфической индексной последовательности включает двухстадийный способ добавления нуклеотидной последовательности, содержащей универсальную последовательность, к нуклеиновым кислотам, а затем добавление первой компартмент-специфической индексной последовательности к нуклеиновым кислотам ДНК, или
d) где праймер содержит первую компартмент-специфическую индексную последовательность, или
е) дополнительно включающий, перед контактированием, мечение вновь синтезированной РНК в субпопуляциях клеток или ядер, выделенных из клеток, необязательно,
i) где уже существующие нуклеиновые кислоты РНК и вновь синтезированные нуклеиновые кислоты РНК помечены одинаковыми индексами в одном и том же компартменте, или
ii) где мечение включает инкубирование множества ядер или клеток в композиции, содержащей нуклеотидную метку, где нуклеотидная метка включена во вновь синтезированную РНК, или
iii) где нуклеотидная метка содержит нуклеотидный аналог, меченный гаптеном нуклеотид, мутагенный нуклеотид или нуклеотид, который может быть модифицирован посредством химической реакции, или
iv) где более чем одна нуклеотидная метка включена во вновь синтезированную РНК, необязательно, где отношения нуклеотидной метки или меток отличаются для различных компартментов или моментов времени, или
v) также включающий обработку субпопуляции ядер или клеток компартментов в предварительно определенных условиях перед мечением, необязательно а) где предварительно определенное условие включает обработку агентом, b) где предварительно определенные условия двух или более компартментов являются различными, или с) где обработку и мечение проводят одновременно или обработку проводят до мечения, необязательно, где агент включает белок, не-рибосомный белок, поликетид, органическую молекулу, неорганическую молекулу, молекулу РНК или РНКи, углевод, гликопротеин, нуклеиновую кислоту или их комбинацию,
- где агент включает терапевтическое лекарственное средство,
f) где одно или более распределений включают разведение,
g) где одно или более распределений включают сортинг,
h) где добавление одной или более из первой, второй или третьей компартмент-специфических индексных последовательностей включает контактирование субпопуляций со шпилечным лигирующим дуплексом в условиях, подходящих для лигирования шпилечного лигирующего дуплекса с концом фрагментов нуклеиновой кислоты,
i) где добавление одной или более из первой, второй или третьей компартмент-специфических индексных последовательностей включает контактирование фрагментов нуклеиновой кислоты с транспосомным комплексом, где транспосомный комплекс в компартментах содержит транспозазу и универсальную последовательность, где контактирование также включает условия, подходящие для фрагментации фрагментов нуклеиновой кислоты и включения нуклеотидной последовательности во фрагменты нуклеиновой кислоты,
j) где добавление первого или второго компартмент-специфического индекса включает лигирование, а добавление последующей компартмент-специфической индексной последовательности включает транспозицию,
k) где компартмент включает лунку или каплю,
l) где компартменты первого множества компартментов содержат от 50 до 100000000 ядер или клеток,
m) где компартменты второго множества компартментов содержат от 50 до 100000000 ядер или клеток,
n) где компартменты третьего множества компартментов содержат от 50 до 100000000 ядер или клеток,
o) дополнительно включающий получение нуклеиновых кислот с тремя индексами из объединенных ядер или клеток с тремя индексами и тем самым получение библиотеки для секвенирования из множества ядер или клеток,
p) дополнительно включающий:
получение поверхности, содержащей множество сайтов амплификации,
где сайты амплификации включают по меньшей мере две популяции связанных одноцепочечных олигонуклеотидов для захвата, имеющих свободный 3’-конец, и
контактирование поверхности, содержащей сайты амплификации, с фрагментами нуклеиновой кислоты с тремя индексами в условиях, подходящих для получения множества сайтов амплификации, каждый из которых содержит клональную популяцию ампликонов, из отдельного фрагмента, содержащего множество индексов.
19. Способ получения библиотеки для секвенирования, содержащей нуклеиновые кислоты, из множества отдельных клеток, где указанный способ включает:
(а) получение ядер из множества клеток;
(b) распределение субпопуляций ядер по первому множеству компартментов и контактирование каждой субпопуляции с обратной транскриптазой и праймером, где праймер в каждом компартменте содержит первую индексную последовательность, которая отличается от первых индексных последовательностей в других компартментах, с получением индексированных ядер, содержащих индексированные фрагменты нуклеиновых кислот;
(c) объединение индексированных ядер с получением объединенных индексированных ядер;
(d) распределение субпопуляций объединенных индексированных ядер по второму множеству компартментов и контактирование каждой субпопуляции со шпилечным лигирующим дуплексом в условиях, подходящих для лигирования шпилечного лигирующего дуплекса с концом индексированных фрагментов нуклеиновой кислоты, содержащих первую индексную последовательность, с получением ядер с двумя индексами, содержащих фрагменты нуклеиновой кислоты с двумя индексами, где шпилечный лигирующий дуплекс содержит вторую индексную последовательность, отличающуюся от вторых индексных последовательностей в других компартментах;
(e) объединение ядер с двумя индексами с получением объединенных ядер с двумя индексами;
(f) распределение субпопуляций объединенных ядер с двумя индексами по третьему множеству компартментов и обработку фрагментов нуклеиновой кислоты с двумя индексами в условиях, подходящих для синтеза второй цепи;
(g) контактирование фрагментов нуклеиновой кислоты с двумя индексами с транспосомным комплексом, где транспосомный комплекс в каждом компартменте содержит транспозазу и универсальную последовательность, где контактирование включает условия, подходящие для фрагментации фрагментов нуклеиновой кислоты с двумя индексами и включения универсальной последовательности во фрагменты нуклеиновой кислоты с двумя индексами с получением фрагментов нуклеиновой кислоты с двумя индексами, содержащих первый и второй индексы на одном конце и универсальную последовательность на другом конце;
(i) включение во фрагменты нуклеиновой кислоты с двумя индексами в каждом компартменте третьей индексной последовательности с получением фрагментов с тремя индексами;
(j) объединение фрагментов с тремя индексами с получением библиотеки для секвенирования, содержащей нуклеиновые кислоты транскриптома, из множества отдельных клеток.
20. Способ по п. 19,
- где праймер содержит поли-Т-последовательность, которая гибридизуется с поли(А)-хвостом мРНК, или
- где праймер каждого компартмента содержит последовательность, которая гибридизуется с предварительно определенной мРНК, необязательно, где способ включает праймеры в различных компартментах, которые гибридизуются с различными нуклеотидами одной и той же предварительно определенной мРНК.
21. Способ получения библиотеки для секвенирования транскриптома, содержащей нуклеиновые кислоты, из множества отдельных клеток, где указанный способ включает:
(а) получение объединенных ядер из множества клеток;
(b) контактирование объединенных ядер с обратной транскриптазой и праймером, содержащим последовательность олиго-dT, которая гибридизуется с поли(А)-хвостом мРНК с образованием объединенных ядер, содержащих фрагменты нуклеиновой кислоты;
(c) распределение субпопуляций объединенных ядер по множеству компартментов и контактирование каждой субпопуляции со шпилечным лигирующим дуплексом в условиях, подходящих для лигирования шпилечного лигирующего дуплекса с концом фрагментов нуклеиновой кислоты с получением индексированных ядер, содержащих индексированные фрагменты нуклеиновой кислоты, где шпилечный лигирующий дуплекс содержит индексную последовательность, отличающуюся от индексных последовательностей в других компартментах;
(d) объединение индексированных ядер с получением объединенных индексированных ядер;
(e) распределение субпопуляций объединенных индексированных ядер по второму множеству компартментов и обработку индексированных фрагментов нуклеиновой кислоты в условиях, подходящих для синтеза второй цепи;
(f) контактирование индексированных фрагментов нуклеиновой кислоты с транспосомным комплексом, где транспосомный комплекс в каждом компартменте содержит транспозазу и универсальную последовательность, где контактирование включает условия, подходящие для фрагментации индексированных фрагментов нуклеиновой кислоты и включения универсальной последовательности в индексированные фрагменты нуклеиновой кислоты с получением индексированных фрагментов нуклеиновой кислоты, содержащих индекс на одном конце и универсальную последовательность на другом конце;
(g) включение в индексированные фрагменты нуклеиновой кислоты в каждом компартменте второй индексной последовательности с получением фрагментов с двумя индексами;
(h) объединение фрагментов с двумя индексами с получением библиотеки для секвенирования, содержащей нуклеиновые кислоты транскриптома, из множества отдельных клеток.
| JUNYUE CAO et al., Comprehensive single-cell transcriptional profiling of a multicellular organism, Science, 18 Aug 2017, Vol.357, Issue 6352, pp.661-667 | |||
| WO 2015200609 A1, 30.12.2015 | |||
| WO 2016130704 A2, 18.08.0216 | |||
| ЗАДЕСЕНЕЦ К.С | |||
| и др., Полногеномное секвенирование геномов эукариот: от секвенирования фрагментов ДНК к сборке генома, ГЕНЕТИКА, |

