Настоящая заявка испрашивает приоритет по заявке на патент Китая № CN201710978352.8, поданной 18 октября 2017 г., содержание которой включено в данный документ посредством ссылки во всей своей полноте.
Область техники, к которой относится изобретение
Настоящее изобретение относится к фармацевтической области, при этом оно относится к способу получения ингибитора тирозина и его промежуточного соединения.
Предшествующий уровень техники
В последние годы уровень смертности от опухолей в Китае увеличивается. Уровень смертности от злокачественных опухолей у городских жителей составляет приблизительно 100-200/100000, и рак серьезно угрожает жизни и качеству жизни людей. Химиотерапия или лучевая терапия с использованием традиционных лекарственных средств для химиотерапии является высокотоксичной и слабо специфичной в отношении пролиферации злокачественных опухолей. Следовательно, поиск эффективных и малотоксичных противоопухолевых лекарственных средств в настоящее время является очень сложной и важной задачей в науках о жизни. Рецепторные тирозинкиназы представляют собой класс трансмембранных белков, вовлеченных в передачу сигнала, которые экспрессируются в различных клетках и регулируют рост, дифференцировку и неоваскуляризацию клеток. Исследования показали, что более 50% протоонкогенов и онкогенных продуктов обладают тирозинкиназной активностью, а их аномальная экспрессия вызывает онкогенез. Кроме того, они также тесно связаны с инвазией и метастазированием опухоли, неоваскуляризацией опухоли и резистентностью опухоли к химиотерапии. Ингибиторы тирозинкиназы появились на рынке с 2001 года и стали новым классом противораковых лекарственных средств.
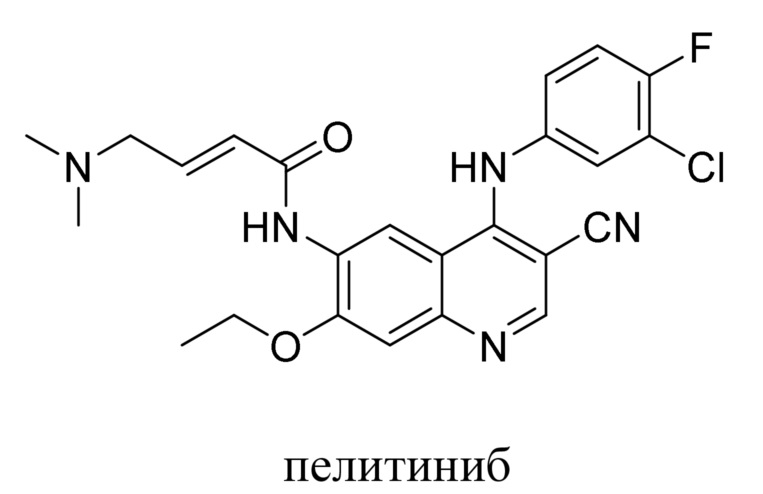
В предшествующем уровне техники было раскрыто множество ингибиторов тирозинкиназы, таких как канертиниб (CI-1033), BIBW-2992, нератиниб (HKI-272) и пелитиниб (EKB-569) и т. д.
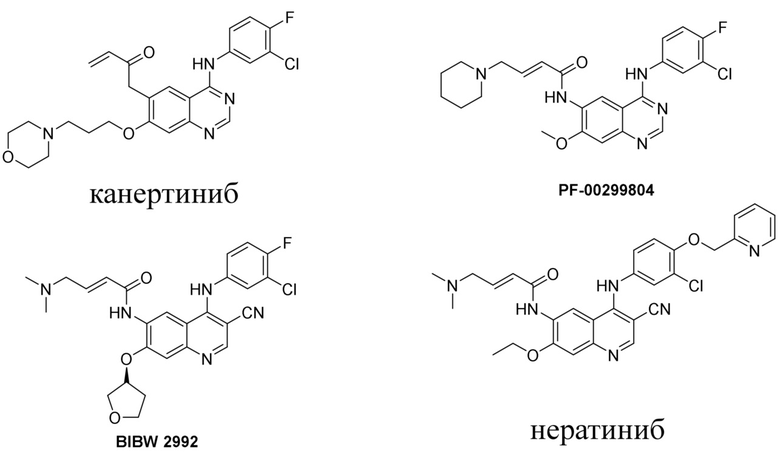
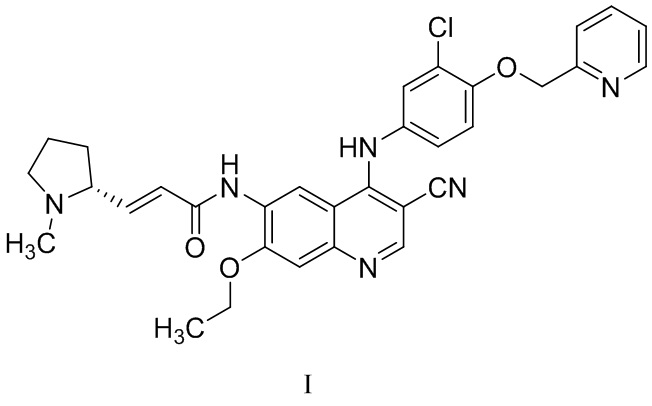
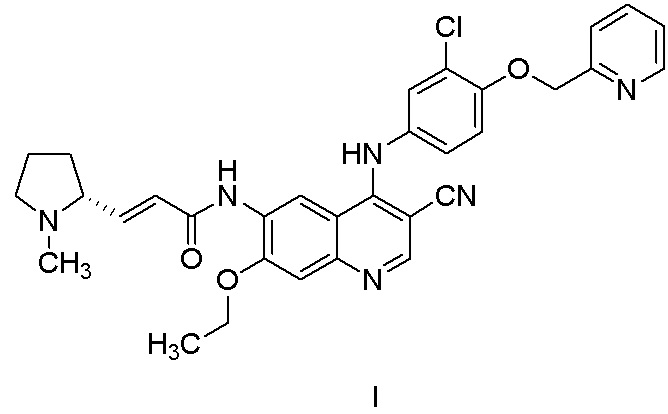
В WO2011029265 раскрыт эффективный ингибитор тирозинкиназы и способ его получения. Его химическим названием является (R,E)-N-(4-((3-хлор-4-(пиридин-2-илметокси)фенил)амино)-3-циано-7-этоксихинолин-6-ил)-3-(1-метилпирролидин-2-ил)акриламид, и его структура представлена формулой I. Соединение обладает очевидными фармакодинамическими преимуществами. В CN102933574A описана дималеатная форма соединения, которая обладает улучшенными физическими и химическими свойствами, фармакокинетическими свойствами и биодоступностью.
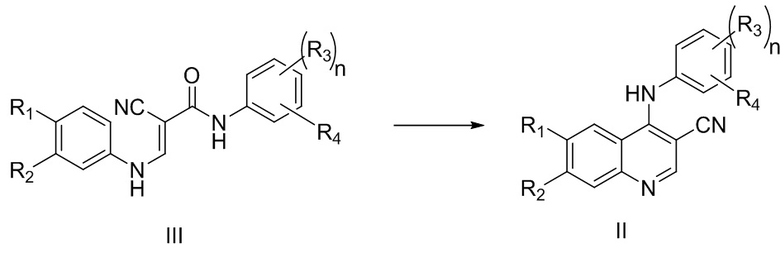
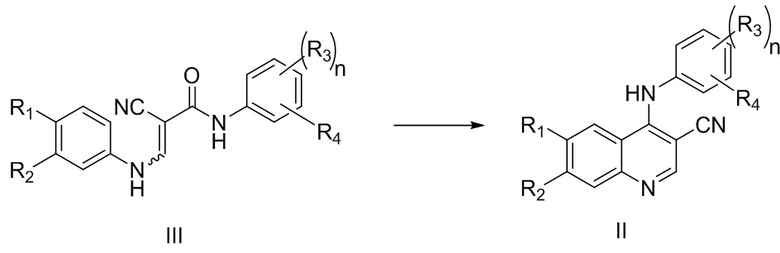
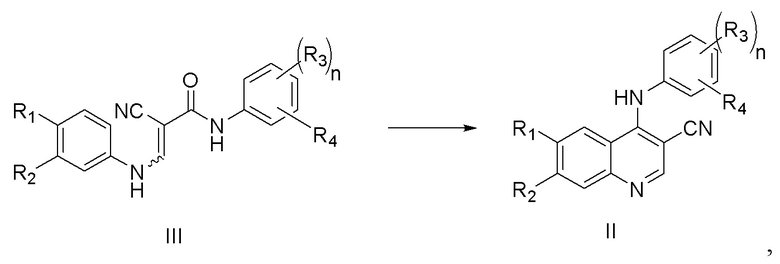
В процессе получения многочисленных существующих ингибиторов тирозинкиназы получение цианохинолинового соединения, представленного формулой II, из соединения, представленного формулой III, является очень важной стадией, которая влияет на эффективность и график всего производственного процесса.
В CN101180269A описан способ получения соединения, представленного формулой II, с использованием 10 эквивалентов оксихлорида фосфора в качестве исходного материала и метанола для катализа реакции. Если способ используется для промышленного производства, то выход реакции является низким, время реакции является длительным, продукт реакции является вязким, последующая обработка является очень сложной, а чистота является невысокой. Вместе с тем из-за сильной коррозионной активности оксихлорида фосфора и его сильной способности вызывать раздражение, при использовании его в больших количествах физическое здоровье производственных рабочих серьезно ухудшается; при этом реакция между метанолом и оксихлоридом фосфора является бурной, а температуру системы трудно контролировать, что при крупномасштабном производстве легко может привести к несчастным случаям на производстве. Следовательно, существует острая необходимость в способе получения соединения, представленного формулой II, с высоким выходом, хорошей чистотой продукта, применением небольшого количества оксихлорида фосфора и мягких условий реакции.
Содержание настоящего изобретения
Для преодоления недостатков предшествующего уровня техники, цель настоящего изобретения заключается в обеспечении нового способа получения ингибитора тирозина и его промежуточного соединения.
Один аспект настоящего изобретения предусматривает способ получения соединения, представленного формулой II,
 ,
,
где
R1 выбран из группы, состоящей из водорода, алкила, галогена, гидроксила, нитро, циано, алкокси, фталимидо,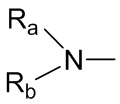 ,
,  и
и  , где каждый из алкила, алкокси необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидроксила, амино, нитро и циано;
, где каждый из алкила, алкокси необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидроксила, амино, нитро и циано;
каждый из Ra и Rb независимо выбран из группы, состоящей из водорода, защитной группы для аминогруппы, алкила, циклоалкила, алканоила, алкенила, алкинила, арила и гетероарила, где каждый из алкила, циклоалкила, алканоила, алкенила, алкинила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидроксила, амино, нитро и циано;
R2 выбран из группы, состоящей из атома водорода, галогена, гидроксила и алкокси;
каждый из R3 независимо выбран из группы, состоящей из алкила, галогена, гидроксила, нитро, циано и алкокси;
R4 характеризуется следующей структурой: 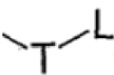 ,
,
T выбран из группы, состоящей из -(CH2)r-, -O(CH2)r-, -NH(CH2)r- и -S(CH2)r;
L выбран из группы, состоящей из арила и гетероарила, предпочтительно фенила, фурила, тиенила, пиридила, пирролила, N-алкилпирролила, пиримидинила, пиразинила, имидазолила и тетразолила, при этом каждый из арила и гетероарила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена и алкила;
n равняется 0, 1, 2, 3 или 4; r равняется 0, 1 или 2;
при этом способ включает стадию осуществления реакции соединения, представленного формулой III, с оксихлоридом фосфора в присутствии катализатора, где катализатор включает катализатор A, при этом катализатор A может представлять собой одно или более, выбранных из группы, состоящей из воды, фосфорной кислоты и фосфата.
Фосфат может представлять собой соль иона металла или соль аммония и может представлять собой, например, фосфат натрия, гидрофосфат динатрия, фосфат калия, фосфат железа(II), дигидрофосфат натрия, дигидрофосфат калия, фосфат цинка, фосфат железа (III), фосфат аммония и т. п.
В некоторых вариантах осуществления катализатор A представляет собой предпочтительно одно или более, выбранных из группы, состоящей из воды, фосфорной кислоты, фосфата натрия, гидрофосфата динатрия, фосфата калия, фосфата железа(II), дигидрофосфата натрия и дигидрофосфата калия.
Растворитель, используемый в реакции, может представлять собой традиционный растворитель, такой как одно или более, выбранных из группы, состоящей из диметилформамида, 1-метил-2-пирролидона, тетрагидрофурана, метилтетрагидрофурана, диоксана, толуола, ксилола, диметилсульфоксида, простого эфира, изопропилового эфира, метил-трет-бутилового эфира, ацетонитрила и пропионитрила, предпочтительно ацетонитрила.
Температура реакции может составлять от 0°C до 200°C, предпочтительно от 20°C до 100°C и более предпочтительно от 40°C до 80°C.
В некоторых вариантах осуществления молярное соотношение соединения, представленного формулой III, и оксихлорида фосфора может составлять от 1:1 до 1:20, предпочтительно от 1:1 до 1:8.
В некоторых вариантах осуществления молярное соотношение соединения, представленного формулой III, и катализатора A может составлять от 1:0,01 до 1:1, предпочтительно от 1:0,1 до 1:0,7.
В некоторых вариантах осуществления катализатор дополнительно предусматривает катализатор B, и при этом катализатор B может представлять собой одно или более, выбранных из группы, состоящей из металлического катализатора и кислоты Льюиса.
Металлический катализатор может представлять собой переходный металл и т. п., такой как железо, кобальт, никель, медь, серебро, платина, золото, палладий, родий, цинк и т. п.
Кислота Льюиса может представлять собой хлорид алюминия, хлорид железа(III), трифторид бора, пентафторид сурьмы, пентахлорид ниобия, хлорид цинка, хлорид меди и т. п.
Предпочтительный катализатор B может представлять собой одно или более, выбранных из группы, состоящей из железа, кобальта, никеля, меди, серебра, платины, золота, палладия, родия, цинка, хлорида алюминия, хлорида железа(III), трифторида бора, пентафторида сурьмы, пентахлорида ниобия, хлорида цинка и хлорида меди, более предпочтительно хлорида алюминия, хлорида железа(III) или хлорида цинка.
В некоторых вариантах осуществления катализатор, используемый в реакции, может включать воду и железо, воду и медь, воду и серебро, воду и платину, воду и золото, воду и хлорид алюминия, воду и хлорид железа(III), воду и трифторид бора, воду и хлорид цинка, воду и хлорид меди, фосфорную кислоту и железо, фосфорную кислоту и медь, фосфорную кислоту и серебро, фосфорную кислоту и платину, фосфорную кислоту и золото, фосфорную кислоту и хлорид алюминия, фосфорную кислоту и хлорид железа(III), фосфорную кислоту и трифторид бора, фосфорную кислоту и хлорид цинка, фосфорную кислоту и хлорид меди, гидрофосфат динатрия и железо, гидрофосфат динатрия и медь, гидрофосфат динатрия и серебро, гидрофосфат динатрия и платину, гидрофосфат динатрия и золото, гидрофосфат динатрия и хлорид алюминия, гидрофосфат динатрия и хлорид железа(III), гидрофосфат динатрия и трифторид бора, гидрофосфат динатрия и хлорид цинка, гидрофосфат динатрия и хлорид меди, фосфат натрия и железо, фосфат натрия и медь, фосфат натрия и серебро, фосфат натрия и платину, фосфат натрия и золото, фосфат натрия и хлорид алюминия, фосфат натрия и хлорид железа(III), фосфат натрия и трифторид бора, фосфат натрия и хлорид цинка, фосфат натрия и хлорид меди, дигидрофосфат калия и железо, дигидрофосфат калия и медь, дигидрофосфат калия и серебро, дигидрофосфат калия и платину, дигидрофосфат калия и золото, дигидрофосфат калия и хлорид алюминия, дигидрофосфат калия и хлорид железа(III), дигидрофосфат калия и трифторид бора, дигидрофосфат калия и хлорид цинка и дигидрофосфат калия и хлорид меди, предпочтительно фосфорную кислоту и железо, фосфорную кислоту и хлорид железа(III), фосфорную кислоту и хлорид цинка, фосфорную кислоту и хлорид алюминия, гидрофосфат динатрия и хлорид алюминия, гидрофосфат динатрия и хлорид железа(III), гидрофосфат динатрия и хлорид цинка или дигидрофосфат калия и хлорид цинка.
В некоторых предпочтительных вариантах осуществления катализатор, используемый в реакции, может включать фосфорную кислоту и железо, фосфорную кислоту и хлорид железа(III), фосфорную кислоту и хлорид алюминия, фосфорную кислоту и хлорид цинка, гидрофосфат динатрия и хлорид цинка, дигидрофосфат натрия и хлорид алюминия, дигидрофосфат калия и хлорид алюминия, дигидрофосфат калия и хлорид железа(III) или дигидрофосфат калия и хлорид цинка.
В некоторых вариантах осуществления молярное соотношение соединения, представленного формулой III, и катализатора B может составлять от 1:0,01 до 1:1, предпочтительно от 1:0,1 до 1:0,7.
В некоторых вариантах осуществления соединение, представленное формулой III, представляет собой
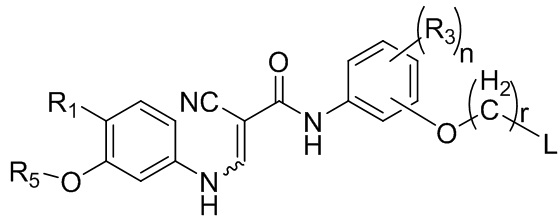 ,
,
соединение, представленное формулой II, представляет собой
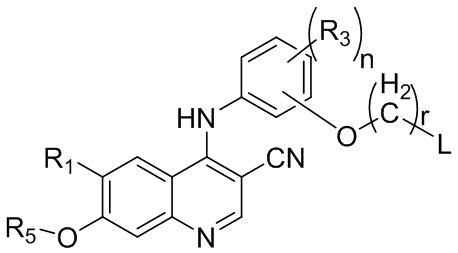 ,
,
где
R1 выбран из группы, состоящей из фталимидо, , и ;
каждый из Ra и Rb независимо выбран из группы, состоящей из водорода, защитной группы для аминогруппы, алкила, циклоалкила, алканоила, алкенила, алкинила, арила и гетероарила, где каждый из алкила, циклоалкила, алканоила, алкенила, алкинила, арила и гетероарила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидроксила, амино, нитро и циано;
каждый из R3 независимо выбран из группы, состоящей из алкила, галогена, гидроксила, нитро, циано и алкокси;
R5 представляет собой C1-C6алкил;
L выбран из группы, состоящей из фенила, фурила, тиенила, пиридила, пирролила, N-алкилпирролила, пиримидинила, пиразинила, имидазолила и тетразолила, при этом каждый из фенила, фурила, тиенила, пиридила, пирролила, N-алкилпирролила, пиримидинила, пиразинила, имидазолила и тетразолила необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена и алкила;
n равняется 0, 1, 2, 3 или 4; r равняется 0, 1 или 2.
В некоторых вариантах осуществления соединение, представленное формулой III, представляет собой
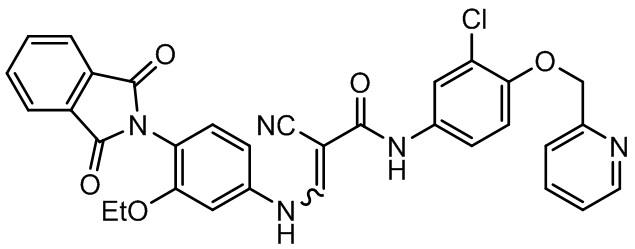 ,
,
соединение, представленное формулой II, представляет собой
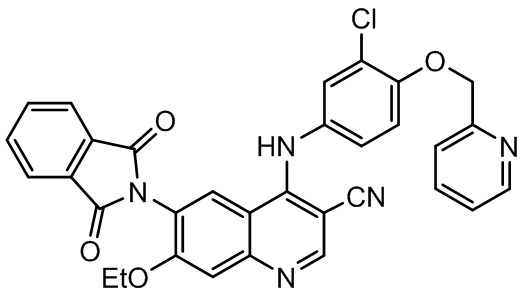 .
.
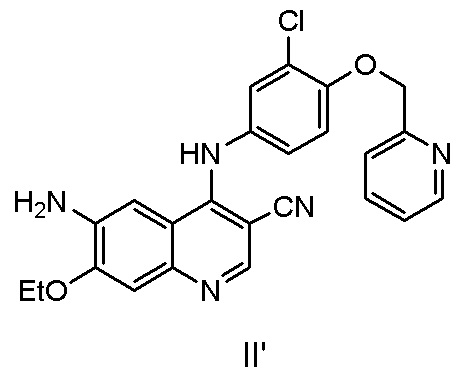
Другой аспект настоящего изобретения дополнительно предусматривает способ получения соединения, представленного формулой II’, включающий стадию осуществления реакции соединения, представленного формулой II, с реагентом для удаления защитной группы.
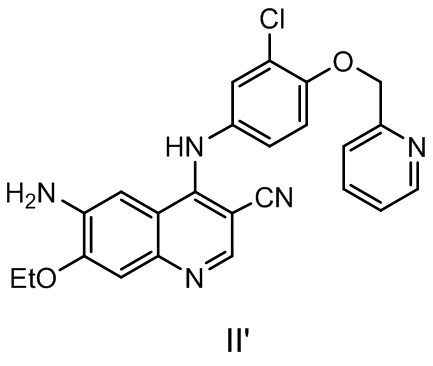
Реагент для удаления защитной группы может представлять собой гидразин, NaBH4, KBH4, алкилкарбоновую кислоту (такую как муравьиная кислота, уксусная кислота, пропионовая кислота и т. п.), неорганическую кислоту (хлористоводородную кислоту, серную кислоту и т. п.), аммиак или аминозамещенный C1-C6алкиловый спирт (аминометанол, аминоэтанол и т. п.), предпочтительно аминозамещенный C1-C6алкиловый спирт и более предпочтительно аминоэтанол. Растворитель, используемый в реакции, может представлять собой традиционный растворитель, такой как одно или более, выбранных из группы, состоящей из диметилформамида, 1-метил-2-пирролидона, диметилсульфоксида, тетрагидрофурана, метилтетрагидрофурана, диоксана, толуола, ксилола, диметилсульфоксида, простого эфира, изопропилового эфира, метил-трет-бутилового эфира, ацетонитрила, пропионитрила, метанола, этанола, изопропилового спирта и воды.
Температура реакции может составлять от 0°C до 200°C, предпочтительно от 20°C до 100°C и более предпочтительно от 40°C до 80°C.
Соединение, представленное формулой II, может быть получено с помощью способа в соответствии с настоящим изобретением. Если реагентом для удаления защитной группы является аминоспирт, то соединение, представленное формулой II', полученное в результате реакции, имеет высокую чистоту, побочные продукты легко удаляются, последующая обработка является простой, и способ является пригодным для промышленного производства.
В некоторых вариантах осуществления молярное соотношение соединения, представленного формулой II, и реагента для удаления защитной группы может составлять от 1:1 до 1:30, предпочтительно от 1:1 до 1:15.
Другой аспект настоящего изобретения дополнительно предусматривает способ получения нератиниба или его фармацевтически приемлемой соли, включающий стадию получения соединения, представленного формулой II, в соответствии с настоящим изобретением.
Кроме того, способ может дополнительно включать стадию получения соединения, представленного формулой II', в соответствии с настоящим изобретением.
Кроме того, способ может дополнительно включать стадию осуществления реакции соединения, представленного формулой II', с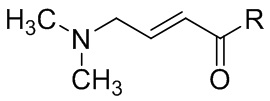 , где R выбран из группы, состоящей из гидроксила, алкокси и галогена. Эту реакцию можно проводить с помощью способов, раскрытых в предшествующем уровне техники (например, CN100537518C).
, где R выбран из группы, состоящей из гидроксила, алкокси и галогена. Эту реакцию можно проводить с помощью способов, раскрытых в предшествующем уровне техники (например, CN100537518C).
Другой аспект настоящего изобретения дополнительно предусматривает способ получения соединения, представленного формулой I, или его фармацевтически приемлемой соли, включающий стадию получения соединения, представленного формулой II, в соответствии с настоящим изобретением.
Кроме того, способ может дополнительно включать стадию получения соединения, представленного формулой II', в соответствии с настоящим изобретением.
Кроме того, способ может дополнительно включать стадию осуществления реакции соединения, представленного формулой II', с 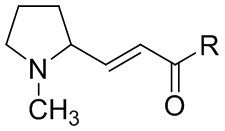 , где R выбран из группы, состоящей из гидроксила, алкокси и галогена. Эту реакцию можно проводить с помощью способов, раскрытых в предшествующем уровне техники.
, где R выбран из группы, состоящей из гидроксила, алкокси и галогена. Эту реакцию можно проводить с помощью способов, раскрытых в предшествующем уровне техники.
В некоторых вариантах осуществления фармацевтически приемлемая соль соединения, представленного формулой I, может представлять собой п-толуолсульфонат, мезилат, малеат, сукцинат или малат, предпочтительно малеат, более предпочтительно дималеат. Соль соединения, представленного формулой I, может быть получена с помощью способов, раскрытых в предшествующем уровне техники (например, CN102933574A).
Другой аспект настоящего изобретения дополнительно предусматривает способ получения соединения, представленного формулой I, включающий:
1) осуществление реакции соединения, представленного формулой IV, с оксихлоридом фосфора в присутствии катализатора с получением соединения, представленного формулой IV’,
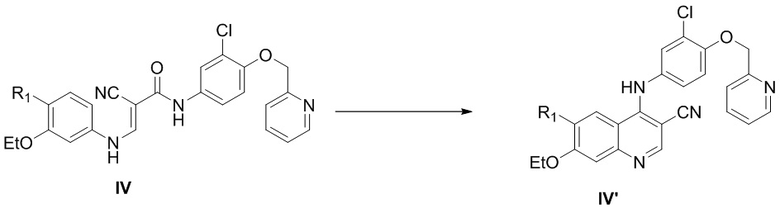 ,
,
где
R1 выбран из группы, состоящей из фталимидо и ;
каждый из Ra и Rb независимо выбран из группы, состоящей из водорода и защитной группы для аминогруппы, и при этом Ra и Rb не являются одновременно водородом;
катализатор выбран из группы, состоящей из воды, фосфорной кислоты, фосфата натрия, гидрофосфата динатрия, фосфата калия, фосфата железа(II), дигидрофосфата натрия, дигидрофосфата калия, фосфорной кислоты и железа, фосфорной кислоты и хлорида железа(III), фосфорной кислоты и хлорида алюминия, фосфорной кислоты и хлорида цинка, гидрофосфата динатрия и хлорида цинка, дигидрофосфата натрия и хлорида алюминия, дигидрофосфата калия и хлорида алюминия, дигидрофосфата калия и хлорида железа(III) и дигидрофосфата калия и хлорида цинка;
2) осуществление реакции соединения, представленного формулой IV’, с реагентом для удаления защитной группы с получением соединения, представленного формулой II’,
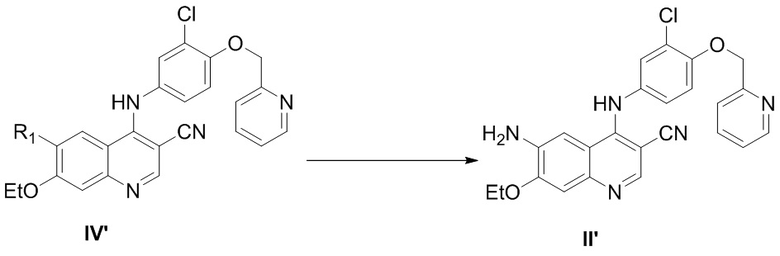 ,
,
при этом средство для удаления защитной группы выбрано из группы, состоящей из гидразина, NaBH4, KBH4, алкилкарбоновой кислоты, неорганической кислоты, аммиака и аминозамещенного C1-C6алкилового спирта, предпочтительно аминозамещенного C1-C6алкилового спирта, более предпочтительно аминоэтанола;
3) осуществление реакции соединения, представленного формулой II’, с с получением соединения, представленного формулой I,
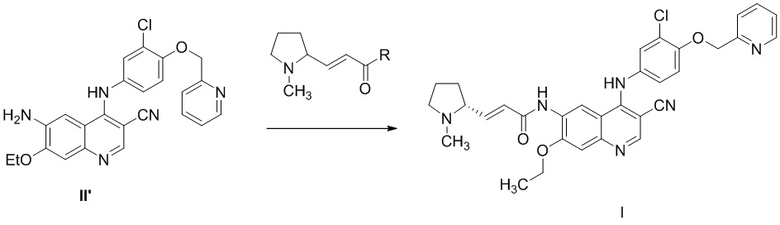 ,
,
где R выбран из группы, состоящей из гидроксила, алкокси и галогена.
Фармацевтически приемлемая соль соединения, представленного формулой I, выбрана из группы, состоящей из п-толуолсульфоната, мезилата, малеата, сукцината и малата, предпочтительно малеата, более предпочтительно дималеата.
В некоторых вариантах осуществления R представляет собой гидроксил, и реакцию в способе проводят в присутствии конденсирующего средства, и при этом конденсирующее средство выбрано из группы, состоящей из DCC, EDC, BOP, HBTU и EEDQ, предпочтительно EEDQ.
В способе получения промежуточного соединения ингибитора тирозинкиназы по настоящему изобретению выход и чистота реакции неожиданно значительно улучшаются, тогда как количество оксихлорида фосфора снижается за счет выбора различных катализаторов или комбинаций катализаторов. Благодаря улучшению выхода и чистоты реакции реакционный раствор может быть непосредственно введен в следующую реакцию после простой последующей обработки, что сокращает время обработки продукта в процессе производства и значительно повышает эффективность производства. Кроме того, способ получения по настоящему изобретению характеризуется мягкими и контролируемыми условиями реакции, что значительно снижает риск безопасности промышленного производства.
Если не указано иное, термины, используемые в описании и формуле изобретения, имеют значения, описанные ниже.
Термин «алкил» относится к насыщенной алифатической углеводородной группе, которая представляет собой группу с прямой или разветвленной цепью, содержащую от 1 до 20 атомов углерода, предпочтительно алкильную группу, содержащую от 1 до 12 атомов углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их различные разветвленные изомеры. Более предпочтительно низший алкил содержит от 1 до 6 атомов углерода, при этом неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т. п. Алкил может быть замещенным или незамещенным, и если алкил замещен, заместитель(заместители) может(могут) быть замещен(замещены) в любой доступной точке присоединения. Заместителем предпочтительно является одна или более группы, независимо выбранные из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидроксила, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклилокси, циклоалкилтио, гетероциклоалкилтио, оксо, карбоксильной и карбоксилатной группы.
Термин «циклоалкил» относится к насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе заместителей, содержащей от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т. п. Полициклический циклоалкил включает циклоалкил, содержащий спирокольцо, конденсированное кольцо или кольцо с мостиковой связью.
Термин «гетероциклил» относится к насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе заместителей, содержащей от 3 до 20 атомов кольца, один или более из которых представляют собой гетероатомы, выбранные из группы, состоящей из N, O и S(O)m (где m представляет собой целое число от 0 до 2), но без -O-O-, -O-S- или -S-S- в кольце, а остальные атомы кольца представляют собой атомы углерода. Предпочтительно гетероциклил содержит от 3 до 12 атомов кольца, где от 1 до 4 атомов представляют собой гетероатомы; более предпочтительно гетероциклил содержит от 3 до 6 атомов кольца. Неограничивающие примеры моноциклического гетероциклила включают пирролидинил, имидазолил, тетрагидрофуранил, тетрагидротиенил, дигидроимидазолил, дигидрофурил, дигидропиразолил, дигидропирролил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и т. п. и предпочтительно пиперидинил или пирролидинил. Полициклический гетероциклил включает гетероциклил, содержащий спирокольцо, конденсированное кольцо или кольцо с мостиковой связью.
Термин «арил» относится к 6-14-членному полностью углеродному моноциклическому кольцу или конденсированному полициклическому кольцу (то есть кольцо имеет смежную пару атомов углерода с другим кольцом), имеющему сопряженную систему π-электронов, предпочтительно к 6-10-членному арилу, например, фенилу и нафтилу. Арильное кольцо может быть конденсировано с гетероарильным кольцом, гетероциклильным кольцом или циклоалкильным кольцом, где кольцо, присоединенное к исходной структуре, представляет собой арильное кольцо, и неограничивающие примеры включают:
 и
и  .
.
Арил может быть замещенным или незамещенным, и если он замещен, то заместителем(заместителями) является(являются) предпочтительно одна или более группы, независимо выбранные из следующей группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидроксила, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, карбоксильной и карбоксилатной группы, предпочтительно фенила.
Термин «гетероарил» относится к 5-14-членной гетероароматической системе, содержащей от 1 до 4 гетероатомов, выбранных из группы, состоящей из O, S и N. Гетероарил предпочтительно представляет собой 5-12-членный гетероарил, например, имидазолил, фурил, тиенил, тиазолил, пиразолил, оксазолил, пирролил, тетразолил, пиридил, пиримидинил, тиадиазол, пиразинил и т. п., предпочтительно имидазолил, пиразолил, пиримидинил или тиазолил; более предпочтительно пиразолил или тиазолил. Гетероарильное кольцо может быть конденсировано с арильным кольцом, гетероциклильным кольцом или циклоалкильным кольцом, где кольцо, присоединенное к исходной структуре, представляет собой гетероарильное кольцо, и неограничивающие примеры включают:
 и
и  .
.
Гетероарил может быть необязательно замещенным или незамещенным, и если он замещен, то заместителем(заместителями) является(являются) предпочтительно одна или более группы, независимо выбранные из следующей группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидроксила, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, карбоксильной и карбоксилатной группы.
Термин «алкокси» относится к -O-(алкил) или -O-(незамещенный циклоалкил), где определение алкила является таким, как указано выше. Неограничивающие примеры алкокси включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси. Алкокси может быть необязательно замещенным или незамещенным, и если он замещен, то заместителем(заместителями) является(являются) предпочтительно одна или более группы, независимо выбранные из следующей группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио, карбоксильной и карбоксилатной группы.
Термин «галоген» относится к фтору, хлору, брому или йоду.
«Защитная группа для аминогруппы» представляет собой подходящую защитную группу для аминогруппы, известную в уровне техники, см. «защитная группа для аминогруппы» в литературе («Protective Groups in Organic Synthesis», 5Th. Ed. T. W Greene & P. G. M Wuts), при этом предпочтительно защитная группа для аминогруппы может представлять собой C1-10-алкилацил или -арилацил, например формил, ацетил, бензоил, фталоил и т. п.; она может представлять собой C1-6алкилсульфонил или C6-10арилсульфонил; она также может представлять собой C1-6алкоксикарбонил или C6-10арилоксикарбонил, Boc или Cbz; она также может представлять собой алкил, например тритил (Tr), 2,4-диметоксибензил (DMB), п-метоксибензил (PMB) или бензил (Bn).
«Необязательный» или «необязательно» означает, что событие или обстоятельство, описанное далее, может произойти, но не обязательно произойдет, и такое описание включает случай, в котором событие или обстоятельство происходит или не происходит. Например, «гетероциклил, необязательно замещенный алкилом» означает, что алкильная группа может присутствовать, но не обязательно присутствует, и такое описание включает случай, когда гетероциклил замещен алкилом, и случай, когда гетероциклил не замещен алкилом.
В химической структуре соединения по настоящему изобретению связь « » не указывает конфигурацию, то есть, если в химической структуре присутствует конфигурационная изомерия, то связь «» может представлять собой «
» не указывает конфигурацию, то есть, если в химической структуре присутствует конфигурационная изомерия, то связь «» может представлять собой « » или «
» или « », или она может содержать обе конфигурации «» и «».
», или она может содержать обе конфигурации «» и «».
Если не указано иное, сокращения, используемые в описании и формуле изобретения, имеют значения, описанные ниже.
Подробное описание предпочтительного варианта осуществления
Настоящее изобретение будет подробно объяснено ниже в сочетании с конкретными примерами, чтобы специалисты в данной области могли более полно понять настоящее изобретение. Следующие примеры дополнительно иллюстрируют настоящее изобретение, однако настоящее изобретение ими не ограничивается.
Условия проведения испытаний для оборудования, применяемого в вариантах осуществления, являлись следующими.
1. Спектрометр ядерного магнитного резонанса (ЯМР)
Модель прибора: Bruker Avance III 400NMR, внутренний стандарт: малеиновая кислота; реагент: d6-DMSO.
Условия проведения испытаний количественного анализа методом ЯМР: аккуратно взвешивали определенное количество малеиновой кислоты и образца и добавляли его в одну пробирку для ЯМР, добавляли d6-DMSO со встряхиванием до полного растворения, а затем проводили количественное определение с помощью спектроскопии на ядрах водорода.
Вариант осуществления 1
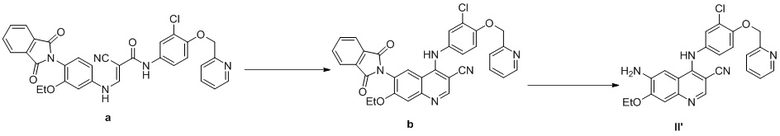
Стадия 1
Соединение a (1,0 экв., 40,0 г, полученное в соответствии со способом, раскрытым в CN101180269A), 390 мл ацетонитрила и 85% фосфорной кислоты (0,6 экв., 3,96 г) последовательно добавляли в реакционную колбу объемом 1 л, перемешивали и нагревали до 60°C; в реакционный раствор добавляли по каплям оксихлорид фосфора (5,0 экв., 51,62 г) и нагревали до температуры образования флегмы в течение 4 ч. после добавления по каплям. Реакционную смесь охлаждали до 10°C и последовательно добавляли по каплям 200 мл воды и 160 мл аммиачной воды с доведением pH до 8. Реакционный раствор непосредственно применяли на следующей стадии без дополнительной очистки.
Стадия 2
Аминоэтанол (10,0 экв., 41,13 г) добавляли в реакционный раствор и смесь нагревали до 50°C в течение 3 ч., а затем охлаждали до комнатной температуры. Осадок на фильтре фильтровали посредством отсасывания и промывали с помощью водного раствора ацетонитрила, осадок на фильтре суспендировали с водным раствором этанола и путем фильтрования с отсасыванием получали 14,05 г соединения, представленного формулой II’, с чистотой 94% согласно HPLC и выходом 46,6% за две стадии.
Вариант осуществления 2
Стадия 1
520 кг ацетонитрила, хлорид цинка (0,3 экв., 4522,5 г) и 85% фосфорную кислоту (0,6 экв., 6,5 кг) добавляли в реактор объемом 3000 л и добавляли 65,70 кг соединения a при перемешивании, реакционный раствор нагревали до 60°C и добавляли по каплям оксихлорид фосфора (5,0 экв., 84,8 кг), после добавления по каплям смесь нагревали до температуры образования флегмы в течение 35 ч. Реакционный раствор охлаждали до температуры ниже 20°C и добавляли по каплям 320 кг воды для гашения реакции. После завершения добавления по каплям, добавляли по каплям аммиак с доведением pH до 8. Реакционный раствор непосредственно применяли на следующей стадии без дополнительной очистки.
Стадия 2
Аминоэтанол (10,0 экв., 70,0 кг) добавляли в реакционный раствор и смесь нагревали до 60°C с осуществлением реакции в течение 4 ч. Реакционный раствор охлаждали до температуры ниже 20°C, фильтровали на центрифуге и осадок на фильтре промывали с помощью 200 кг очищенной воды, после промывания осадок на фильтре суспендировали с 130 кг 95% этанола и 250 кг воды в качестве смешанного растворителя при комнатной температуре в течение 2 ч., а затем фильтровали до сухого состояния с получением 23,08 кг соединения, представленного формулой II' (чистота 93% согласно HPLC, общий выход 46,8% за две стадии).
Вариант осуществления 3
Стадия 1
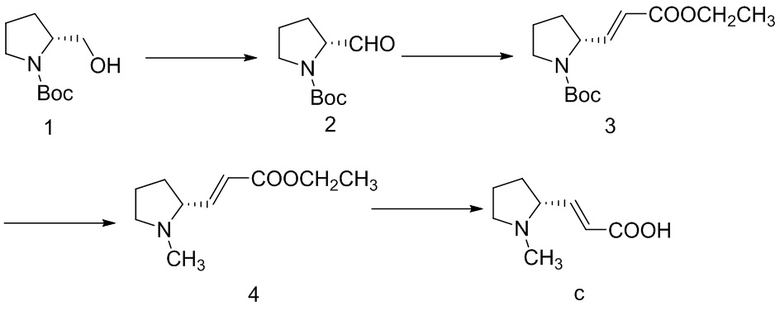
8,0 кг соединения 1, 264 кг дихлорметана, 13,0 кг безводного ацетата натрия добавляли в реактор объемом 300 л и перемешивали, реакционную систему охлаждали до 0°C с помощью замороженного солевого раствора и добавляли партиями 17,14 кг PCC в защитной атмосфере азота. Замороженный солевой раствор удаляли после добавления и обеспечивали нагревание реакционной смеси естественным путем с осуществлением реакции в течение 5 ч. После того, как обнаружение с помощью TLC (этилацетат: петролейный эфир = 1: 3) показало, что реакция завершена,, реакционный раствор фильтровали, фильтрат концентрировали при пониженном давлении с получением черного масла. Продукт элюировали с помощью колоночной хроматографии (элюент: этилацетат:петролейный эфир = 1:3), требуемый компонент собирали и концентрировали при пониженном давлении и добавляли 64 кг этилацетата для обеспечения растворения, смесь промывали с помощью 0,5 н. разбавленного раствора хлористоводородной кислоты и промывали с помощью воды, затем с помощью насыщенного солевого раствора, высушивали над безводным сульфатом натрия и концентрировали с получением 6,42 кг бледно-желтого масла.
114 кг дихлорметана и 3,05 кг 60% гидроксида натрия добавляли в реактор объемом 300 л и равномерно перемешивали, реакционную систему охлаждали с помощью замороженного солевого раствора и добавляли по каплям 7,66 кг триэтилфосфорилацетата, завершали добавление через приблизительно 30 минут и реакционный раствор перемешивали при комнатной температуре до образования пузырьков. Медленно добавляли раствор 6,4 кг соединения 2, полученного на предыдущей стадии, в дихлорметане (85 кг) и завершали добавление через приблизительно 1 ч. и обеспечивали осуществление реакции в смеси при комнатной температуре в течение 1,5-2 ч. После того, как обнаружение TLC показало, что реакция завершена, реакционный раствор охлаждали с помощью замороженного солевого раствора и медленно добавляли водный раствор хлорида аммония (1,26 кг хлорида аммония растворяли в 4,0 кг воды) до прекращения образования пузырьков, смесь перемешивали в течение приблизительно 0,5 ч., а затем по каплям медленно добавляли очищенную воду до тех пор, пока реакционная смесь не становилась прозрачной, затем слои разделяли и водную фазу экстрагировали один раз дихлорметаном, органические фазы объединяли и промывали с помощью насыщенного водного раствора гидрокарбоната натрия и насыщенного солевого раствора и органическую фазу высушивали над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, представляющего собой сложный эфир, который элюировали посредством колоночной хроматографии (элюент: этилацетат:петролейный эфир = 1:8) и требуемый компонент собирали и концентрировали при пониженном давлении с получением 4,82 кг соединения 3 с выходом 45,0%.
Стадия 2
4,8 кг соединения 3 и 58,6 кг муравьиной кислоты добавляли в реактор объемом 100 л и перемешивали при комнатной температуре в течение 15 мин., а затем добавляли 2,63 кг параформальдегида. Реакционную смесь слабо нагревали с обратным холодильником при 90°C в течение 3-4 часов. Обнаружение с помощью TLC показало, что исходный материал исчез, и затем большую часть муравьиной кислоты в реакционном растворе концентрировали (оставалось приблизительно 1/5) и добавляли 1 М хлористоводородную кислоту для доведения pH до 1,0, реакционный раствор промывали этилацетатом, а затем добавляли насыщенный водный раствор карбоната калия для доведения pH до 8,0, экстрагировали с помощью этилацетата, органические фазы объединяли и промывали с помощью насыщенного водного раствора хлорида натрия, высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали с получением всего 2,42 кг соединения 4 с выходом 73,5%.
Стадия 3
2,4 кг соединения 4 добавляли в реакционную колбу объемом 20 л, затем добавляли 5,9 кг метанола и контролировали температуру реакции до значения не выше 30°C, добавляли партиями 1,49 кг гидроксида калия и завершали добавление через приблизительно 1,5 ч. и обеспечивали осуществление реакции в смеси при 30°C в течение 2 ч. После того, как обнаружение TLC показало, что реакция завершена, pH реакционной смеси доводили до 4-5 с помощью 4 н. хлористоводородной кислоты в метаноле на ледяной бане, смесь фильтровали и фильтрат концентрировали до сухого состояния. Добавляли 2,7 кг ацетонитрила с перемешиванием и кристаллизацией и фильтровали с получением 1,06 кг соединения c с выходом 52,1%.
Вариант осуществления 4
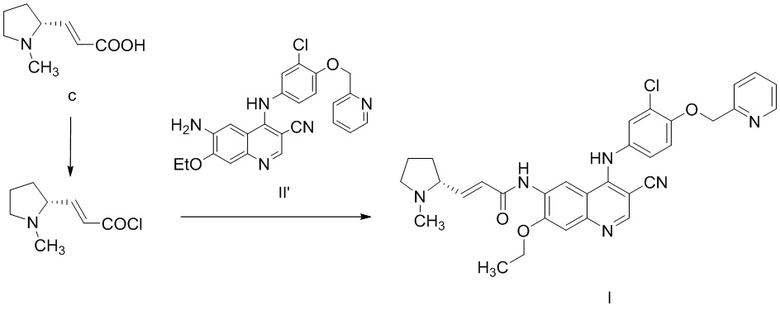
1,0 кг соединения c и 9,4 кг ацетонитрила добавляли в реакционную колбу объемом 20 л и добавляли по каплям 30 г N,N-диметилформамида. Медленно добавляли по каплям 630 г оксалилхлорида на бане с ледяной водой. После добавления по каплям реакционный раствор перемешивали при 20°C в течение 5 ч. Небольшое количество твердого вещества оставалось в нижней части реакционного раствора, и реакционный раствор непосредственно подвергали реакции конденсации на следующей стадии без обработки.
1,15 кг соединения II' растворяли в 7,2 кг N-метилпирролидона и смесь перемешивали в течение 10 мин. Реакционный раствор из предыдущей стадии добавляли по каплям на бане с ледяной водой и перемешивали при комнатной температуре в течение ночи. Обнаружение с помощью TLC показало, что реакция завершена. Реакционный раствор выливали в теплую воду (45,0 кг) при приблизительно 40°C и перемешивали и медленно добавляли 10% раствор гидроксида натрия с доведением pH до 10. Осажденное желтое твердое вещество фильтровали и полученный осадок на фильтре суспендировали с теплой водой (приблизительно 5,0 кг) при 40°C и затем фильтровали, осадок на фильтре растворяли в дихлорметане и отделяли воду, затем органическую фазу высушивали и концентрировали и очищали с помощью колоночной хроматографии. При градиентном элюировании исходный элюент представлял собой дихлорметан:метанол = 25:1 и в конце увеличивался до 15: 1. Требуемый компонент собирали и концентрировали с получением 1,12 кг соединения I с выходом 74,5%.
Сравнительный вариант осуществления 1
Стадия 1
В соответствии со способом варианта осуществления 5 в CN101180269A масса материала, представляющего собой соединение a, составляла 27 кг, масса материала, представляющего собой оксихлорид фосфора, составляла 69,7 кг (10,0 экв.) и количество метанола составляло 13,5 л для обеспечения получения 20 кг соединения b (чистота составляла 35% и была определена путем количественного определения с помощью спектроскопии на ядрах водорода (способ с применением внутреннего стандарта), а фактический выход составлял 26,7%).
Стадия 2
В соответствии со способом варианта осуществления 6 в CN101180269A соединение b, полученное на предыдущей стадии, вводили в реакцию с получением 4,0 кг соединения II' с чистотой HPLC 93% (общий выход составлял 19,7% за две стадии).
Сравнительный вариант осуществления 2
Соединение a (1,0 экв., 40,0 г), 390 мл ацетонитрила и 85% фосфорной кислоты (0,6 экв., 3,96 г) последовательно добавляли в реакционную колбу объемом 1 л, перемешивали и нагревали до 60°C; добавляли оксихлорид фосфора (5,0 экв., 51,62 г) в реакционный раствор и после добавления по каплям нагревали до температуры образования флегмы в течение 4 ч. Реакционную смесь охлаждали до 10°C и последовательно добавляли по каплям 200 мл воды и 160 мл аммиачной воды с доведением pH до 8.
30 мл гидроксида аммония и 50 мл этанола добавляли в реакционный раствор, описанный выше, и реакционную смесь нагревали до температуры образования флегмы и обеспечивали осуществление реакции в течение 4 ч., а затем реакционную смесь охлаждали до комнатной температуры, фильтровали посредством отсасывания и осадок на фильтре промывали водным раствором ацетонитрила, затем осадок на фильтре суспендировали с водным раствором этанола в течение 2 ч. и фильтровали посредством отсасывания с получением 12 г продукта (выход за две стадии 43%, HPLC: 93%), при этом продукт содержал следы (приблизительно 0,3%) побочного продукта, представляющего собой фталимид, который было трудно удалить в последующих реакциях.
Сравнительный вариант осуществления 3
Соединение a (1,0 экв., 80,0 г), 800 мл ацетонитрила и 40 мл метанола последовательно добавляли в реакционную колбу объемом 3 л и перемешивали и нагревали до 60°C; в реакционный раствор добавляли по каплям оксихлорид фосфора (5,0 экв., 103,2 г), после завершения добавления по каплям смесь нагревали до 65°C-70°C и обеспечивали осуществление реакции в течение 54 часов. Обнаружение TLC показало, что все еще оставалось большое количество непрореагировавшего соединения a, при этом последующая обработка была трудной, а выход реакции не мог быть рассчитан.
Поскольку настоящее изобретение было описано в соответствии с его конкретными вариантами осуществления, определенные модификации и эквивалентные изменения будут очевидны для специалистов в данной области техники и включены в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ТРИЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ ДВОЙНОГО ИНГИБИТОРА ФДЭ3/ФДЭ4 | 2021 |
|
RU2838571C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРА PARP И ЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2019 |
|
RU2777597C2 |
| СОЕДИНЕНИЕ 3,4-ДИГИДРОИЗОХИНОЛИНА И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2825312C1 |
| ХИМИЧЕСКИ СВЯЗАННЫЙ КЕРАМИЧЕСКИЙ РАДИАЦИОННО-ЗАЩИТНЫЙ МАТЕРИАЛ И СПОСОБ ЕГО ПОДГОТОВКИ | 2006 |
|
RU2446490C2 |
| Способ извлечения фосфорной кислоты из ферментационного бульона или жидких отходов ферментации и её повторного применения | 2018 |
|
RU2748950C1 |
| ПОЛУЧЕНИЕ ПРОИЗВОДНЫХ ГИДРОКСИ-БЕНЗИЛБЕНЗОЛА | 2014 |
|
RU2671493C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ БИЦИКЛИЧЕСКИХ АМИДИНОГИДРАЗОНОВ (ВАРИАНТЫ) И БИЦИКЛИЧЕСКИЕ ГИДРОКСИАМИДИНЫ | 1993 |
|
RU2126381C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ ТРИЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ SERD, ИМЕЮЩИХ ЗАМЕЩЕННЫЙ ФЕНИЛЬНЫЙ ИЛИ ПИРИДИНИЛЬНЫЙ ФРАГМЕНТ | 2021 |
|
RU2818455C1 |
| ВУЛКАНИЗУЮЩИЕСЯ КОМПОЗИЦИИ НА ОСНОВЕ СОДЕРЖАЩИХ ЭПОКСИДНЫЕ ГРУППЫ НИТРИЛЬНЫХ КАУЧУКОВ | 2012 |
|
RU2622655C2 |
| КАТАЛИЗАТОР ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ, СОДЕРЖАЩИЙ ЦИКЛОТРИВЕРАТРИЛЕН И ЕГО ПРОИЗВОДНЫЕ | 2018 |
|
RU2740916C1 |
Изобретение относится к способу получения соединения, представленного формулой II, которое может найти применение в способе получения эффективного ингибитора тирозинкиназы и его промежуточных соединений. Способ включает стадию осуществления реакции соединения, представленного формулой III, с оксихлоридом фосфора в присутствии катализатора. Катализатор включает катализатор A, при этом катализатор A представляет собой фосфорную кислоту. В указанных формулах R1 представляет собой фталимидо; R2 выбран из группы, состоящей из С1-6алкокси; каждый из R3 независимо выбран из группы, состоящей из галогена; R4 характеризуется следующей структурой: 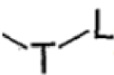 ; T представляет собой -O(CH2)r-; L выбран из группы, состоящей из 6-членного гетероарила, содержащего 1 гетероатом, выбранный из N; n равняется 0, 1, 2, 3 или 4; r равняется 1 или 2. Предлагаемый способ характеризуется высоким выходом, хорошей чистотой продукта и мягкими условиями реакции. Изобретение относится также к способу получения соединения, представленного формулой II', и вариантам способа получения соединения, представленного формулой I. 4 н. и 16 з.п. ф-лы, 4 пр.
; T представляет собой -O(CH2)r-; L выбран из группы, состоящей из 6-членного гетероарила, содержащего 1 гетероатом, выбранный из N; n равняется 0, 1, 2, 3 или 4; r равняется 1 или 2. Предлагаемый способ характеризуется высоким выходом, хорошей чистотой продукта и мягкими условиями реакции. Изобретение относится также к способу получения соединения, представленного формулой II', и вариантам способа получения соединения, представленного формулой I. 4 н. и 16 з.п. ф-лы, 4 пр.
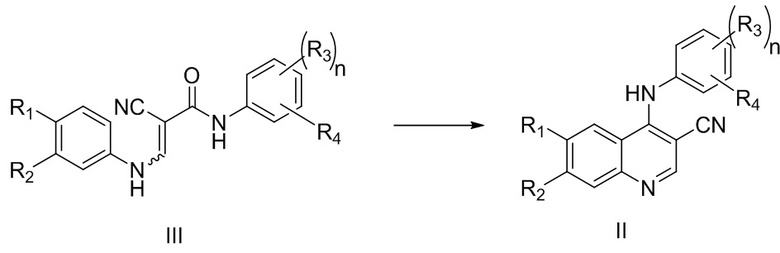
1. Способ получения соединения, представленного формулой II, включающий стадию осуществления реакции соединения, представленного формулой III, с оксихлоридом фосфора в присутствии катализатора
где R1 представляет собой фталимидо;
R2 выбран из группы, состоящей из С1-6алкокси;
каждый из R3 независимо выбран из группы, состоящей из галогена;
R4 характеризуется следующей структурой: 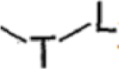 ;
;
T представляет собой -O(CH2)r-;
L выбран из группы, состоящей из 6-членного гетероарила, содержащего 1 гетероатом, выбранный из N;
n равняется 0, 1, 2, 3 или 4; r равняется 1 или 2;
катализатор включает катализатор A, при этом катализатор A представляет собой фосфорную кислоту.
2. Способ по п. 1, где молярное соотношение соединения, представленного формулой III, и катализатора A составляет от 1:0,01 до 1:1
3. Способ по п. 1, где молярное соотношение соединения, представленного формулой III, и катализатора A составляет от 1:0,1 до 1:0,7.
4. Способ по п. 1, где соединение, представленное формулой II, представляет собой
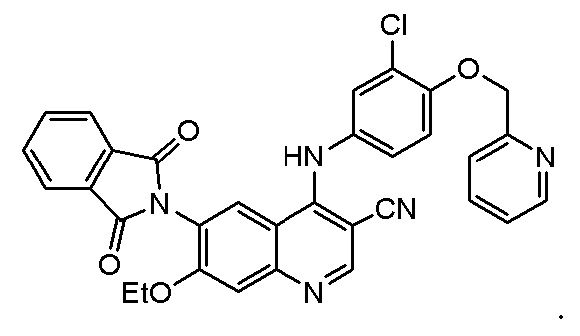
5. Способ по п. 1, где катализатор дополнительно предусматривает катализатор B, при этом катализатор B выбран из одного или более, выбранных из группы, состоящей из хлорида алюминия, хлорида железа(III) или хлорида цинка.
6. Способ по п. 5, где катализатор включает фосфорную кислоту и хлорид железа(III), фосфорную кислоту и хлорид алюминия, фосфорную кислоту и хлорид цинка.
7. Способ по п. 5, где молярное соотношение соединения, представленного формулой III, и катализатора B составляет от 1:0,01 до 1:1.
8. Способ по п. 7, где молярное соотношение соединения, представленного формулой III, и катализатора B составляет от 1:0,1 до 1:0,7.
9. Способ получения соединения, представленного формулой II', включающий стадию осуществления реакции соединения, представленного формулой III, с оксихлоридом фосфора в присутствии катализатора с получением соединения, представленного формулой II, и дополнительно включающий стадию осуществления реакции соединения, представленного формулой II, с реагентом для удаления защитной группы
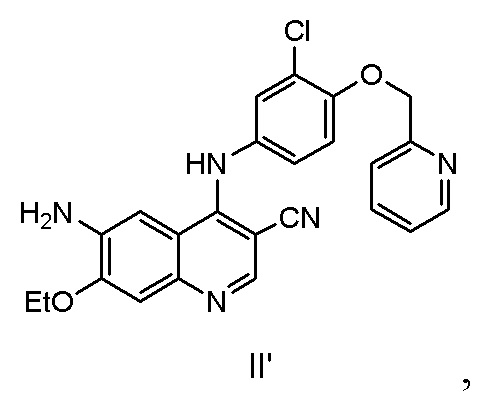
где реагент для удаления защитной группы представляет собой аминозамещенный C1-C6алкиловый спирт;
при этом формула III, формула II и катализатор такие, как определено в п. 1.
10. Способ по п. 9, где реагент для удаления защитной группы представляет собой аминоэтанол.
11. Способ получения соединения, представленного формулой I, или его фармацевтически приемлемой соли, включающий стадию осуществления реакции соединения, представленного формулой III, с оксихлоридом фосфора в присутствии катализатора с получением соединения, представленного формулой II,
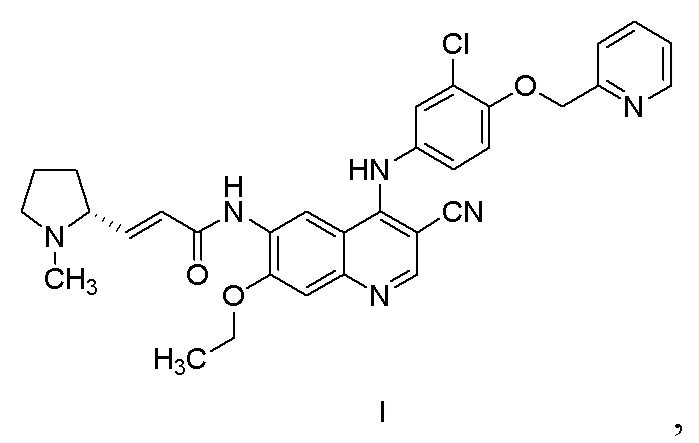
при этом формула III, формула II и катализатор такие, как определено в п. 1.
12. Способ по п. 11, где способ дополнительно включает стадию получения соединения, представленного формулой II', как определено в п. 9.
13. Способ по п. 12, где способ дополнительно включает стадию осуществления реакции соединения, представленного формулой II', с  , где R выбран из группы, состоящей из гидроксила, алкокси и галогена.
, где R выбран из группы, состоящей из гидроксила, алкокси и галогена.
14. Способ получения соединения, представленного формулой I, включающий:
1) осуществление реакции соединения, представленного формулой IV, с оксихлоридом фосфора в присутствии катализатора с получением соединения, представленного формулой IV',
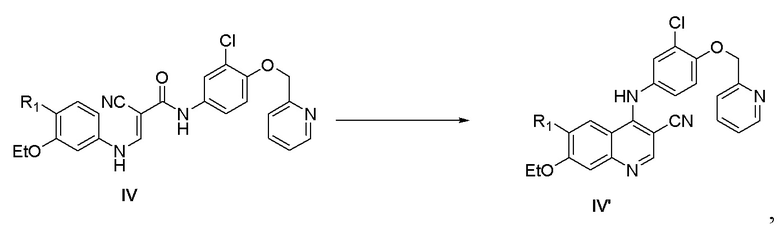
где R1 выбран из группы, состоящей из фталимидо;
катализатор выбран из группы, состоящей из фосфорной кислоты, фосфорной кислоты и хлорида железа(III), фосфорной кислоты и хлорида алюминия, фосфорной кислоты и хлорида цинка;
2) осуществление реакции соединения, представленного формулой IV', с реагентом для удаления защитной группы с получением соединения, представленного формулой II',
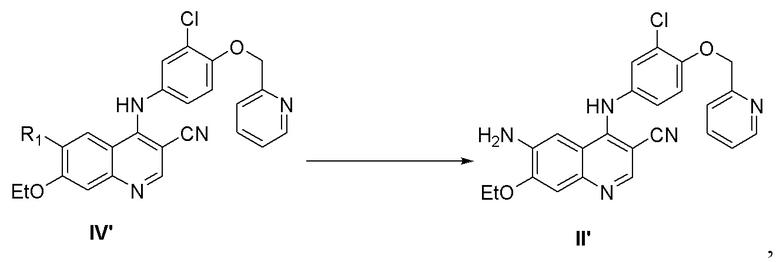
при этом средство для удаления защитной группы представляет собой аминозамещенный C1-C6алкиловый спирт;
3) осуществление реакции соединения, представленного формулой II', с с получением соединения, представленного формулой I,
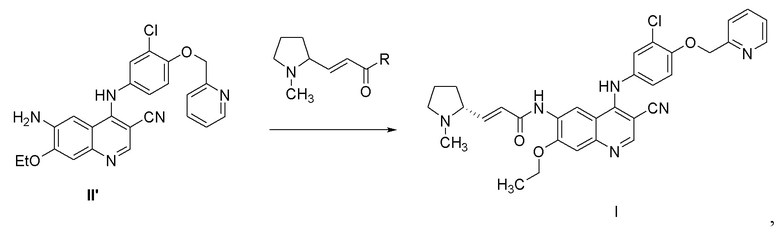
где R выбран из группы, состоящей из гидроксила, алкокси и галогена.
15. Способ по п. 14, где на стадии 2) средство для удаления защитной группы представляет собой аминоэтанол.
16. Способ по п. 11 или 14, где фармацевтически приемлемая соль соединения, представленного формулой I, выбрана из группы, состоящей из п-толуолсульфоната, мезилата, малеата, сукцината и малата.
17. Способ по п. 16, где фармацевтически приемлемая соль соединения, представленного формулой I, представляет собой малеат.
18. Способ по п. 16, где фармацевтически приемлемая соль соединения, представленного формулой I, представляет собой дималеат.
19. Способ по п. 14, где R представляет собой гидроксил и реакцию в способе проводят в присутствии конденсирующего средства, при этом конденсирующее средство выбрано из группы, состоящей из DCC, EDC, BOP, HBTU и EEDQ.
20. Способ по п. 14, где R представляет собой гидроксил и реакцию в способе проводят в присутствии конденсирующего средства, при этом конденсирующее средство представляет собой EEDQ.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| N | |||
| GU ET AL., The Wittig-Horner reaction for the synthesis of neratinib, RES | |||
| CHEM | |||
| INTERMED., 2013, vol | |||
| Машина для изготовления проволочных гвоздей | 1922 |
|
SU39A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| ЭЛЕКТРОЛИТИЧЕСКИЙ АППАРАТ ДЛЯ ПОЛУЧЕНИЯ СВИНЦОВЫХ БЕЛИЛ | 1924 |
|
SU3105A1 |
| ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ (Е)-N-[4-[[3-ХЛОР-4-(2-ПИРИДИЛМЕТОКСИ)ФЕНИЛ]АМИНО]-3-ЦИАНО-7-ЭТОКСИ-6-ХИНОЛИЛ]-3-[(2R)-1-МЕТИЛПИРРОЛИДИН-2-ИЛ]ПРОП-2-ЕНАМИДА, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ РАКА | 2012 |
|
RU2583056C2 |

