Область техники, к которой относится изобретение
Настоящее изобретение относится к области лекарственных средств, в частности, к производному иминомочевины и к способу его получения и применению.
Предпосылки создания настоящего изобретения
Индоламин-2,3-диоксигеназа, мономерный гем-содержащий фермент, способен катализировать окислительное раскрытие индольного кольца L-триптофана с формированием кинуренина. Высокая экспрессия индоламин-2,3-диоксигеназы приводит к истощению локального запаса триптофана в клетках, что индуцирует арест цикла Т-клеток в G1 фазе, ингибируя тем самым пролиферацию Т-клеток. С другой стороны, распад индоламин-2,3-диоксигеназа-зависимого триптофана приводит к увеличению уровня кинуренина, а также индуцирует апоптоз Т-клеток, опосредованный свободными радикалами кислорода. В-третьих, усиление экспрессии индоламин-2,3-диоксигеназы в дендритных клетках усиливает локальную иммуносупрессию, опосредованную регуляторными Т-клетками (Treg), посредством распада локального запаса триптофана, усиливая тем самым периферическую иммунную толерантность организма к опухольспецифическому антигену. Индоламин-2,3-диоксигеназа стала наиболее важной низкомолекулярной регуляторной мишенью для противоопухолевой иммунотерапии.
В исследованиях было обнаружено, что индоламин-2,3-диоксигеназа связана со многими физиологическими процессами в организме человека. В 1998 году Munn et al. обнаружили, что плод способен выживать в организме матери с отличающимся генотипом в период беременности без отторжения за счет того, что плацентарные плазмодитрофобластные клетки синтезируют индоламин-2,3-диоксигеназу, которая ингибирует через кровоток реакцию отторжения материнских Т-клеток в отношении плода. После дополнительной подкожной имплантации беременным мышам капсулы с замедленным высвобождением, содержащей ингибитор индоламин-2,3-диоксигеназы 1-метилтриптофан, они обнаружили, что эмбрион был отторгнут и абортирован (Munn DH, Zhou M, Attwood JT, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science, 1998, 281 (5380): 1191-3). Кроме того, некоторые заболевания, обусловленные аномальными иммунными ответами, такие как реакция отторжения трансплантата и аутоиммунные заболевания, также тесно связаны с индоламин-2,3-диоксигеназой.
Несмотря на то, что в последние годы был достигнут значительный прогресс в лечении опухолей, клиническая эффективность до сих пор неудовлетворительна. Ускользание от иммунного ответа представляет собой один из основных биологических механизмов онкогенеза и метастазирования опухолей, и стал важным фактором, оказывающим влияние на терапевтический эффект в отношении опухолей. Индоламин-2,3-диоксигеназа, как иммунный регуляторный фермент, может эффективно ингибировать функцию Т-клеток, усиливать функцию Treg клеток и индуцировать дисфункцию NK-клеток, в то время как опухолевые клетки могут использовать указанные врожденные механизмы иммунорегуляции в организме для ускользания от идентификации и уничтожения иммунной системой (Jia Yunlong, Wang Yu, Chinese Journal of Cancer Biotherapy, 2004, 21 (6): 693-7). С целью обеспечения пациентов с опухолями оптимальной пользы от лечения, необходимо рационально корректировать стратегию лечения в отношении ускользания опухолей от иммунного ответа. Ингибитор индоламин-2,3-диоксигеназы согласно настоящему изобретению может эффективно регулировать иммунную систему пациента, блокировать ускользание опухолевых клеток от иммунного ответа, и характеризуется хорошим терапевтическим эффектом в отношении большинства спонтанных опухолей. Основываясь на регулировании иммунной системы, в дополнение к лечению опухолей, ингибитор индоламин-2,3-диоксигеназы согласно настоящему изобретению также может лечить другие заболевания, связанные с иммунитетом, такие как хроническая инфекция и СПИД.
Индоламин-2,3-диоксигеназа также тесно связана с неврологическими заболеваниями. Она может снижать уровень 5-гидрокситриптамина и быть причиной психических заболеваний, таких как депрессия и тревожность. Она также может обуславливать накопление в головном мозге нейротоксичных метаболитов, таких как хинолиновая кислота, что тесно связано с возникновением нейродегенеративных заболеваний, таких как болезнь Альцгеймера. Индоламин-2,3-диоксигеназа может влиять на функцию головного мозга посредством по меньшей мере двух механизмов: 1) при воспалительном ответе, катаболизм триптофана может приводить к снижению концентраций циркулирующего триптофана, приводя тем самым к снижению уровня 5-гидрокситриптамина, что приводит к депрессии; 2) индоламин-2,3-диоксигеназа катаболизирует триптофан до продуктов, которые участвуют в пути метаболизма кинуренина, приводя к накоплению кинуренина и нейротоксичной хинолиновой кислоты (Kong Linglei, Kuang Chunxiang, Yang Qing, Chinese Journal of Medicinal Chemistry, 2009, 19(2): 147-154).
Содержание настоящего изобретения
Настоящее изобретение относится к соединению или его фармацевтически приемлемой соли, к композиции, содержащей соединение или его фармацевтически приемлемую соль, и к способу ингибирования активности индоламин-2,3-диоксигеназы (IDO) путем применения настоящего соединения или его фармацевтически приемлемой соли или к способу лечения заболевания, характеризующегося патологическим путем метаболизма триптофана, опосредованным индоламин-2,3-диоксигеназой, путем использования соединения или его фармацевтически приемлемой соли, и к применению соединения или фармацевтически приемлемой соли для производства лекарственного средства для ингибирования активности индоламин-2,3-диоксигеназы или для лечения заболевания, характеризующегося патологическим путем метаболизма триптофана, опосредованным индоламин-2,3-диоксигеназой.
Соединение или его фармацевтически приемлемая соль обладает превосходной ингибирующей активностью в отношении индоламин-2,3-диоксигеназы, и эта активность существенно выше в сравнении с другими ингибиторами IDO. Кроме того, путем измерения массы тела мышей до и после введения соединения или его фармацевтически приемлемой соли, было обнаружено, что в сравнении с другими ингибиторами IDO, соединение согласно настоящему изобретению или его фармацевтически приемлемая соль могут существенно улучшать качество жизни мышей в процессе лечения опухоли и существенно снижать побочные эффекты. В клинической практике, соединение согласно настоящему изобретению или его фармацевтически приемлемая соль будут улучшать не только качество жизни пациента, но также существенно улучшат комплаентность пациента к лекарственным средствам и эффективность лекарств. Соединение согласно настоящему изобретению или его фармацевтически приемлемая соль могут существенно улучшить нарушения обучаемости и памяти у животных и усиливать способность приобретать знания и способность к пространственной памяти, обладают положительной терапевтической ценностью для нейродегенеративных заболеваний, таких как синдром Альцгеймера, и превосходят другие ингибиторы IDO. Соединение согласно настоящему изобретению или его фармацевтически приемлемая соль могут усиливать функцию DC по стимулированию пролиферации Т-клеток, вследствие чего их можно использовать для лечения опухолевых заболеваний, аутоиммунных заболеваний, реакции отторжения трансплантата и инфекционных заболеваний, и оно превосходит другие ингибиторы IDO.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
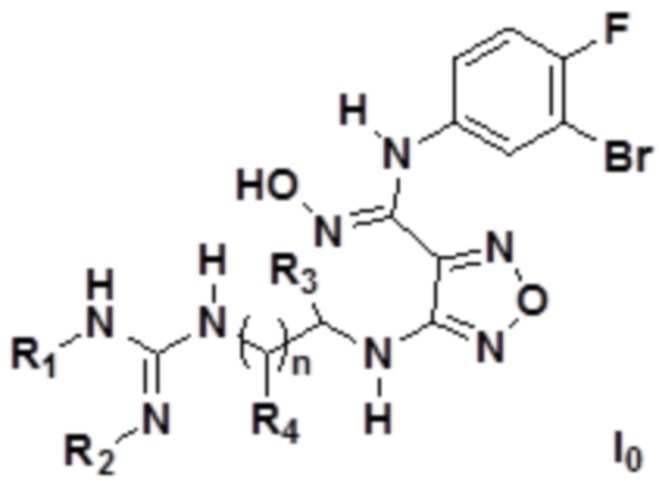
Согласно некоторым вариантам осуществления, изобретение относится к соединению, представленному формулой I0, или его фармацевтически приемлемой соли:
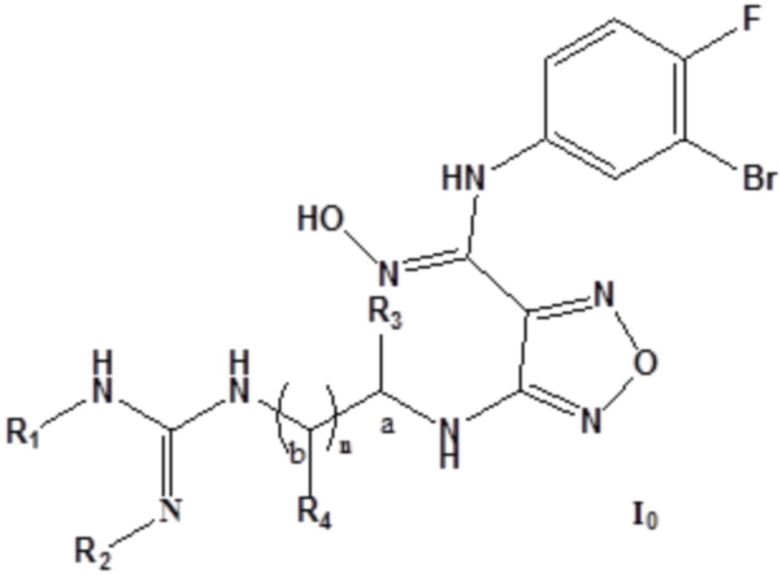
где каждый из R1 и R2 независимо выбирают из группы, состоящей из: H, замещенного или незамещенного C1-10алкила, альдегидной группы, замещенного или незамещенного карбонила, циано, CF3, замещенного или незамещенного C1-10алкокси, замещенного или незамещенного сульфонила, замещенного или незамещенного C3-10циклоалкила, замещенного или незамещенного C2-10алкенила, замещенного или незамещенного C6-20арила, замещенного или незамещенного C3-14гетероарила;
каждый из R3 и R4 независимо представляет собой монозаместитель, выбранный из группы, состоящей из: H, замещенного или незамещенного C1-10алкила, замещенного или незамещенного C3-10циклоалкила, циано, замещенного или незамещенного C1-10алкокси, замещенного или незамещенного сульфонила, замещенного или незамещенного C6-20арила, замещенного или незамещенного C3-14гетероарила; или
каждый из R3 и R4 независимо выбирают из дизаместителей, формирующих посредством этого вместе с атомом C в a- или b-положении следующие группы:  ,
,  или
или 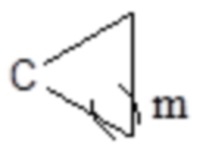 , где C представляет собой атом C в a- или b-положении, m равен целому числу, выбранному из 0-6, такому как 0 или 1, или 2, или 3, или 4, или 5, или 6; кроме того, группа, сформированная R3 и R4 вместе с атомом C в a- или b-положении, представляет собой C=CH2,
, где C представляет собой атом C в a- или b-положении, m равен целому числу, выбранному из 0-6, такому как 0 или 1, или 2, или 3, или 4, или 5, или 6; кроме того, группа, сформированная R3 и R4 вместе с атомом C в a- или b-положении, представляет собой C=CH2,  ,
,  ,
, 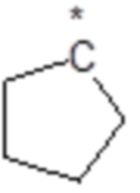 ,
, 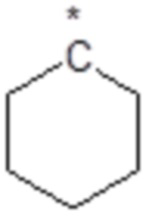 или
или 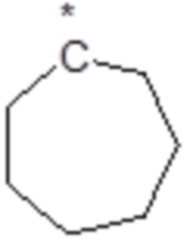 ;
;
n равен целому числу, выбранному из 0-6, такое как 0, 1, 2, 3, 4, 5 или 6; предпочтительно, n равен 0, 1, 2 или 3.
Согласно некоторым вариантам осуществления, настоящее изобретение относится к описанному выше соединению, представленному формулой I0, или его фармацевтически приемлемой соли, где каждый из R1 и R2 независимо выбирают из группы, состоящей из: C1-6алкила, карбонила, C1-6алкокси, сульфонила, амидино, сульфинила, который необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: галогена, гидрокси, карбокси, карбонила, альдегидной группы, циано, амино, арила, гетероарила, C1-6алкила, C3-12циклоалкила, C2-6алкенила и C3-12циклоалкенила, где карбокси, карбонил, альдегидная группа, циано, амино, арил, гетероарил, C3-12циклоалкил, C2-6алкенил, C3-12циклоалкенил в качестве заместителя C1-6алкила, карбонила, C1-6алкокси, сульфонила, амидино или сульфинила необязательно замещен одним или более заместителями, выбранными из группы, состоящей из: H, галогена, C1-6алкила, карбонила, C1-6алкокси, сульфинила и сульфонила, причем галоген выбирают из группы, состоящей из F, Cl, Br и I.
Согласно некоторым вариантам осуществления, настоящее изобретение относится к описанному выше соединению, представленному формулой I0, или его фармацевтически приемлемой соли, где каждый из R1 и R2 независимо выбирают из группы, состоящей из: H, метила, этила, пропила, изопропила, R5C(O)-, R5S(O)x-; R5 выбирают из группы, состоящей из C1-10алкила, C3-12циклоалкила, замещенного C1-10алкил и замещенного C3-12циклоалкила, где замещенный C1-10алкил или замещенный C3-12циклоалкил замещен гидрокси, циано, C1-6алкилом, C3-12циклоалкилом, C1-6алкокси, арилом или гетероарилом, где x равен 1 или 2;
каждый из R3 и R4 независимо представляет собой монозаместитель, выбранный из группы, состоящей из: H, замещенного или незамещенного C1-10алкила, замещенного или незамещенного C3-10циклоалкила, циано, замещенного или незамещенного C1-10алкокси, замещенного или незамещенного сульфонила, замещенного или незамещенного C6-20арила, замещенного или незамещенного C3-14гетероарила; или
каждый из R3 и R4 независимо выбирают из дизаместителей, формирующих посредством этого вместе с атомом C в a- или b-положении следующие группы: , , C=CH2, , , или .
Согласно некоторым вариантам осуществления, настоящее изобретение относится к описанному выше соединению, представленному формулой I0, или его фармацевтически приемлемой соли, где каждый из R1 и R2 независимо выбирают из группы, состоящей из: H, метила, этила, пропила, изопропила, R5C(O)-, R5S(O)x-; R5 выбирают из группы, состоящей из C1-10алкила, C3-12циклоалкила, замещенного C1-6алкила и замещенного C3-8циклоалкила, причем замещенный C1-6алкил или замещенный C3-8циклоалкил замещен гидрокси, циано, C1-6алкилом, C3-12циклоалкилом, C1-6алкокси, арилом или гетероарилом; x равен 2; R3 и R4 оба представляют собой H.
Согласно некоторым вариантам осуществления, настоящее изобретение относится к описанному выше соединению, представленному формулой I0, или его фармацевтически приемлемой соли, где если R2, R3 и R4 в формуле I0 соответственно представляют собой H, то соединение представлено формулой I:
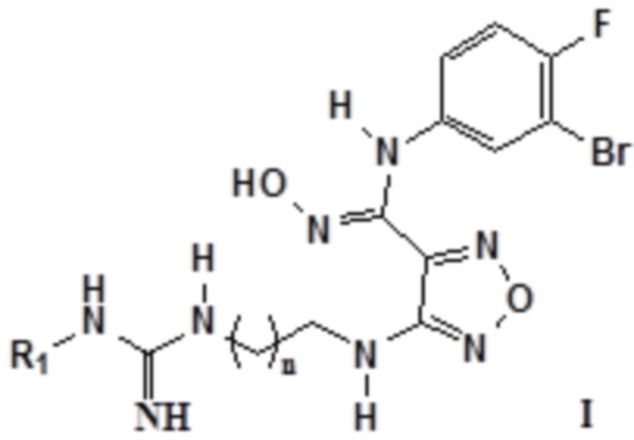 ,
,
где R1 выбирают из группы, состоящей из: H, амино, сульфонила, нитро, карбонила, амидино, C1-6алкила, замещенного амидино, замещенного C1-6алкила, замещенного C1-6алкокси, замещенного карбонила, замещенного сульфонила и замещенного сульфинила, где замещенный амидино, замещенный C1-6алкил, замещенный C1-6алкокси, замещенный карбонил, замещенный сульфонил или замещенный сульфинил замещен галогеном, гидрокси, карбокси, карбонилом, альдегидной группой, циано, амино, арилом, гетероарилом, C3-12циклоалкилом, C2-6алкенилом, C3-12циклоалкенилом; n равен целому числу, выбранному из 0-6, такому как 0, 1, 2, 3, 4, 5 или 6; предпочтительно, n равен 1 или 2.
Согласно некоторым вариантам осуществления, настоящее изобретение относится к описанному выше соединению, представленному формулой I0, или его фармацевтически приемлемой соли, где если R1 в формуле I представляет собой H, то соединение представлено формулой II:
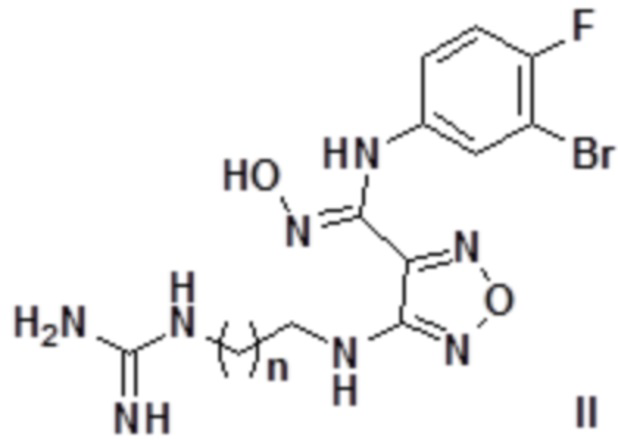 ,
,
где n равен целому числу, выбранному из 0-6, такому как 0, 1, 2, 3, 4, 5 или 6.
Согласно некоторым вариантам осуществления, настоящее изобретение относится к описанному выше соединению, представленному формулой I0, или его фармацевтически приемлемой соли, где если R1 в формуле I представляет собой R0-замещенный карбонил, то соединение представлено формулой III:
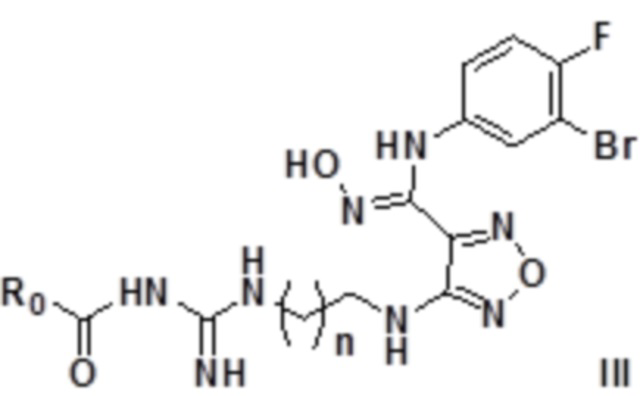 ,
,
где R0 выбирают из группы, состоящей из H, C1-6алкила, замещенного C1-6алкила и замещенного C1-6алкокси, где замещенный C1-6алкил или замещенный C1-6алкокси замещен галогеном, гидрокси, карбокси, карбонилом, альдегидной группой, циано, амино, арилом, гетероарилом, C3-12циклоалкилом, C2-6алкенилом, C3-12циклоалкенилом, n равен целому числу, выбранному из 0-6, такому как 0, 1, 2, 3, 4, 5 или 6.
Кроме того, R0 выбирают из группы, состоящей из C1-6алкила, замещенного C1-6алкила и замещенного C1-6алкокси, где замещенный C1-6алкил или замещенный C1-6алкокси замещен галогеном, гидрокси, карбокси, карбонилом, альдегидной группой, циано, амино, арилом, гетероарилом, C3-12циклоалкилом, C2-6алкенилом, C3-12циклоалкенилом.
Согласно некоторым вариантам осуществления, настоящее изобретение относится к описанному выше соединению, представленному формулой I0, или его фармацевтически приемлемой соли, где если R1 в формуле I0 представляет собой H, то соединение представлено формулой IV:
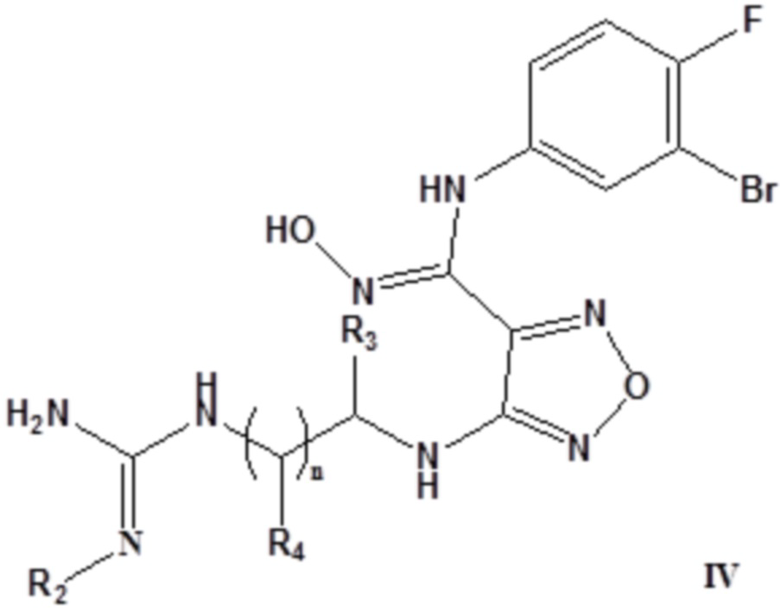 ,
,
где R2 выбирают из группы, состоящей из H, метила, этила, пропила, изопропила, R5C(O)-, R5S(O)m-; R5 выбирают из группы, состоящей из H, C1-10алкила, C3-12циклоалкила, замещенного C1-10алкил и замещенного C3-12циклоалкила, где замещенный C1-10алкил и замещенный C3-12циклоалкил замещен гидрокси, циано, CF3, C1-6алкилом, C3-10циклоалкилом, алкокси, арилом или гетероарилом; m выбирают из 1 или 2;
каждый из R3 и R4 независимо выбирают из группы, состоящей из H, C1-10алкила, C3-12циклоалкила, замещенного C1-10алкила, замещенного C3-12циклоалкила, замещенного C1-10алкокси, замещенного сульфонила и замещенного C3-14гетероарила, где замещенный C1-10алкил, замещенный C3-12циклоалкил, замещенный C1-10алкокси, замещенный сульфонил или замещенный C3-14гетероарил замещен гидрокси, циано, галогеном, C1-6алкилом, C3-10циклоалкилом, алкокси, арилом или гетероарилом;
где n равен целому числу, выбранному из 0-6, такому как 0, 1, 2, 3, 4, 5 или 6;
кроме того, R2 выбирают из группы, состоящей из H, R5C(O)-, R5S(O)m-; R5 выбирают из группы, состоящей из H, C1-10алкила, C3-12циклоалкила, замещенного C1-10алкила и замещенного C3-12циклоалкила, где замещенный C1-10алкил или замещенный C3-12циклоалкил замещен гидрокси, циано, CF3, C1-6алкилом, C3-10циклоалкилом, C1-6алкокси, арилом или гетероарилом; m выбирают из 1 или 2;
кроме того, R2 выбирают из группы, состоящей из R5C(O)- и R5S(O)x-; R5 выбирают из группы, состоящей из C1-6алкила, C3-8циклоалкила, замещенного C1-6алкила и замещенного C3-8циклоалкила, где замещенный C1-6алкил или замещенный C3-8циклоалкил замещен гидрокси, циано, C1-6алкилом, C3-8циклоалкилом, C1-6алкокси, арилом или гетероарилом; x равен 2; R3 и R4 оба представляют собой H.
Согласно некоторым вариантам осуществления, изобретение относится к указанному ниже соединению или его фармацевтически приемлемой соли:
 ,
,  ,
,  ,
,  ,
,  ,
, 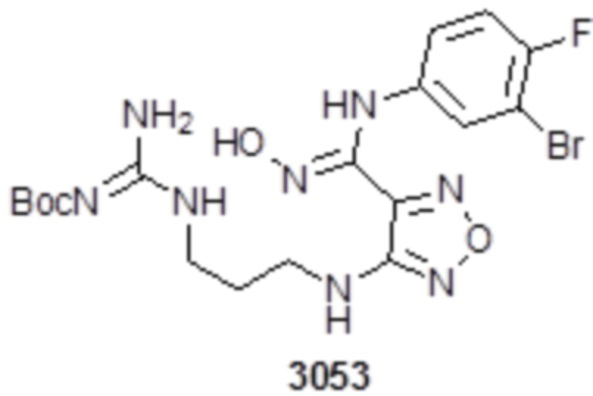 ,
, 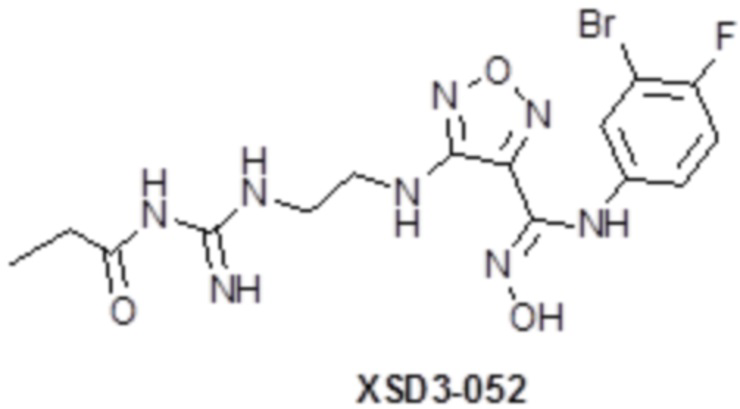 ,
, 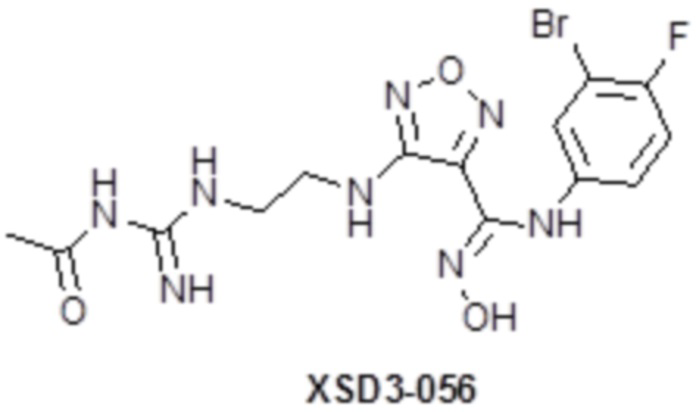 ,
, 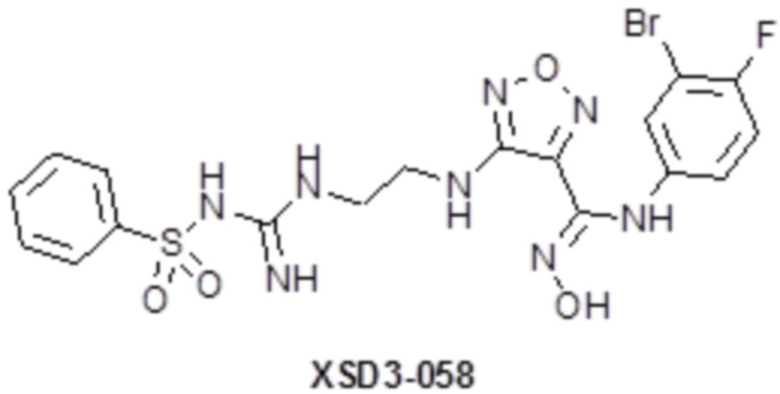 ,
, 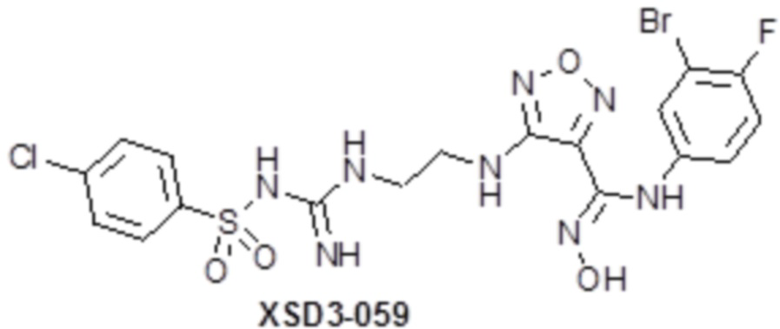 ,
, 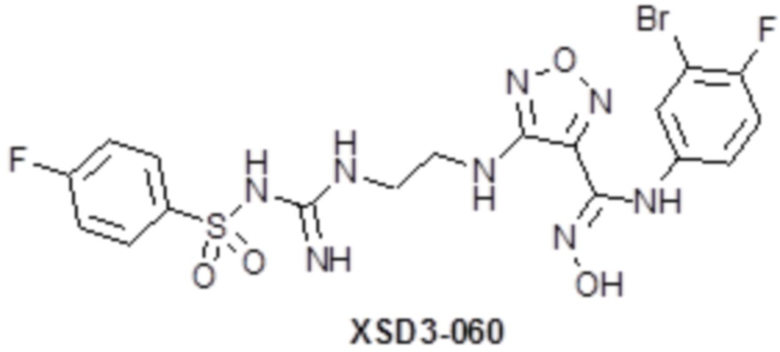 ,
, 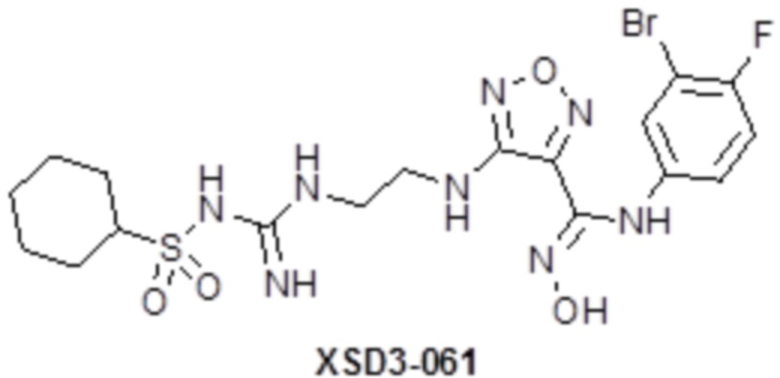 ,
, 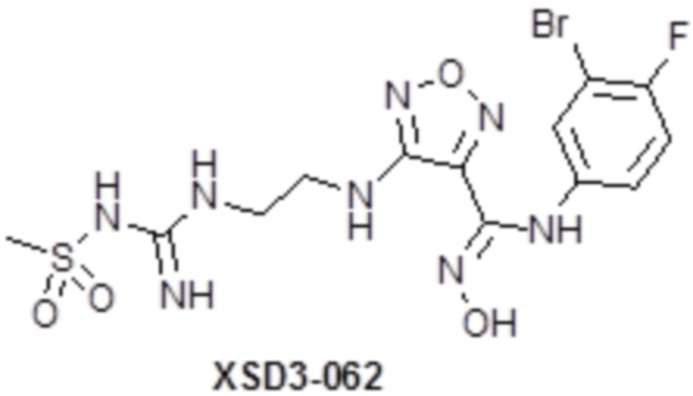 ,
, 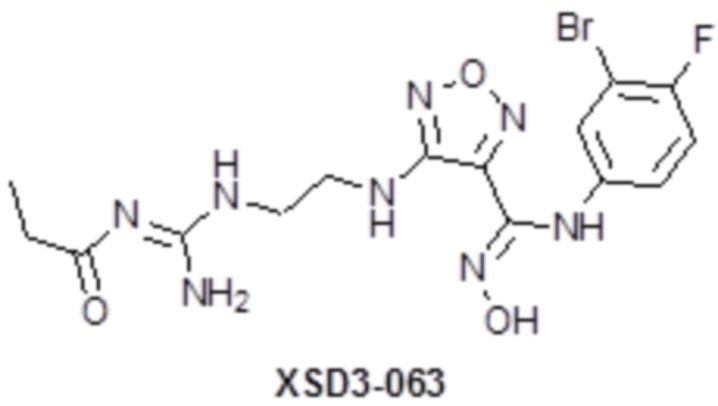 ,
, 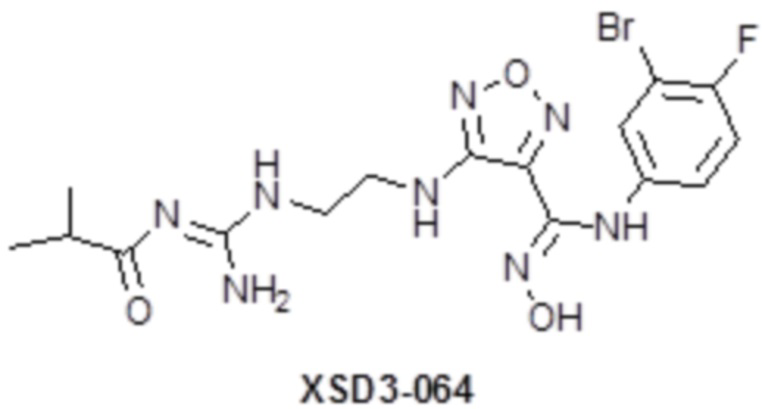 ,
, 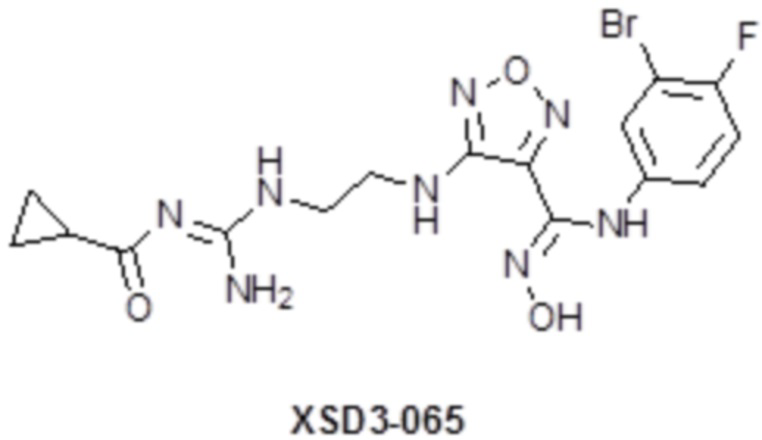 ,
, 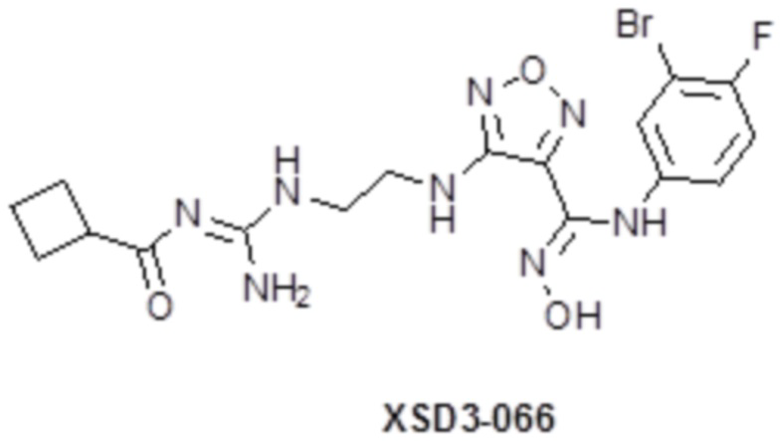 ,
, 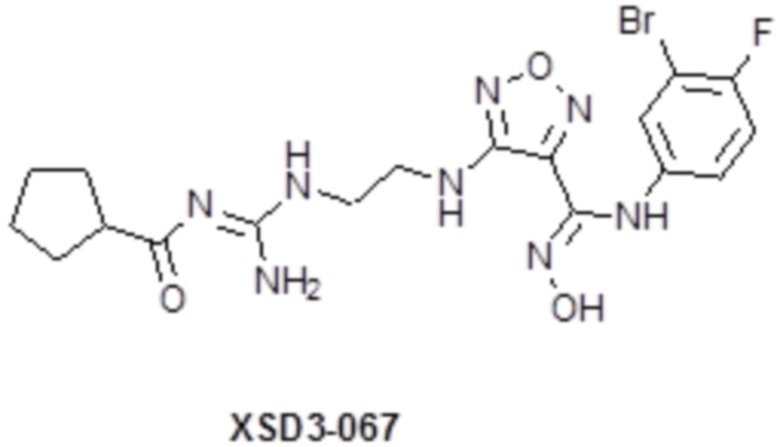 ,
, 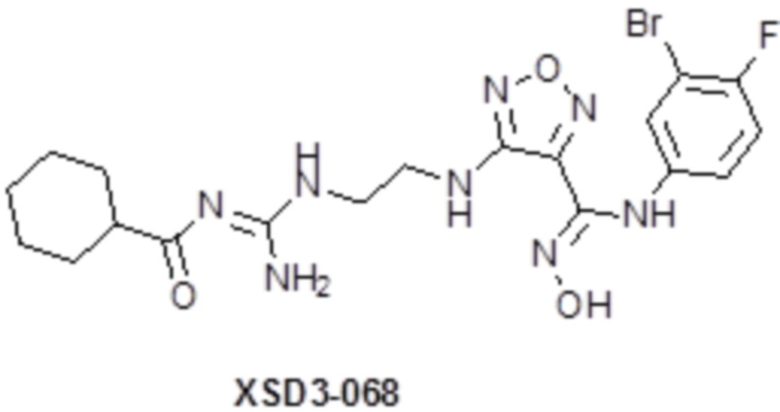 ,
, 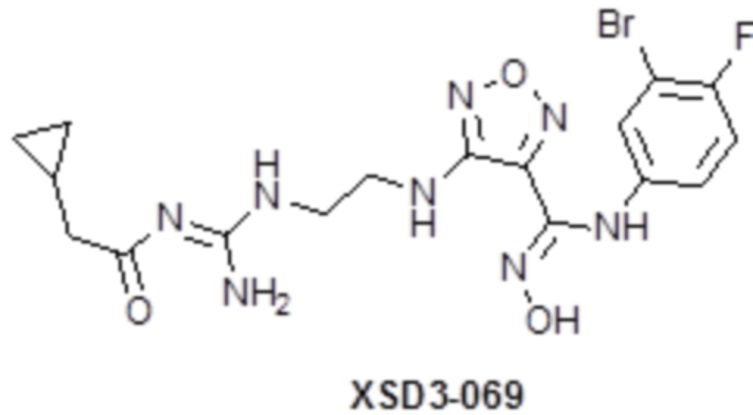 ,
, 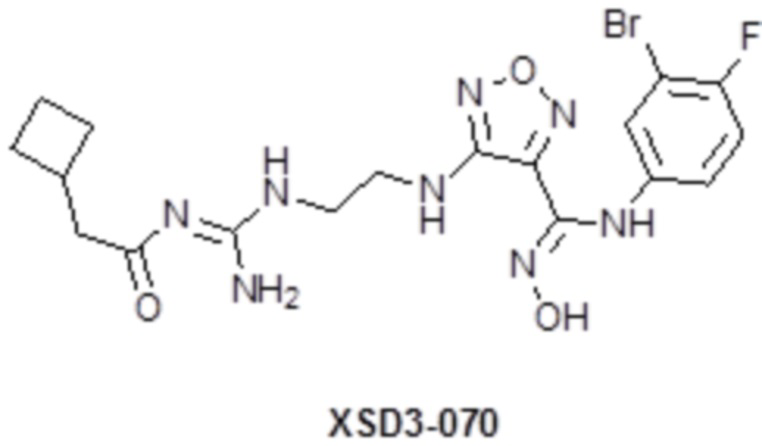 ,
, 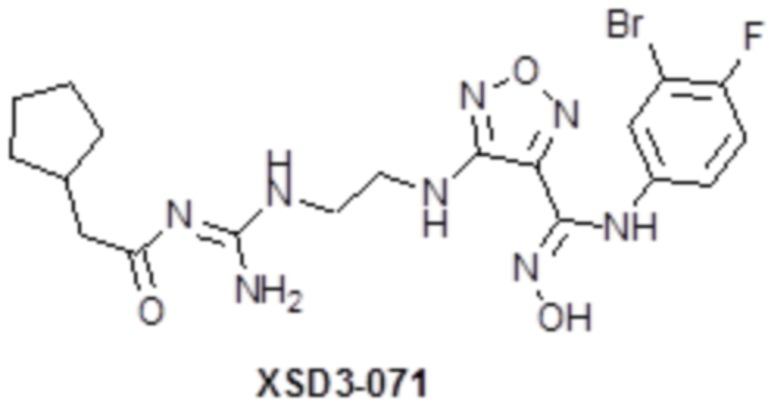 ,
, 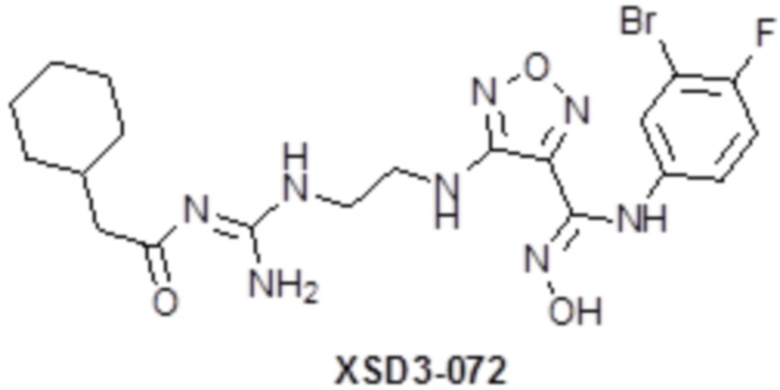 ,
, 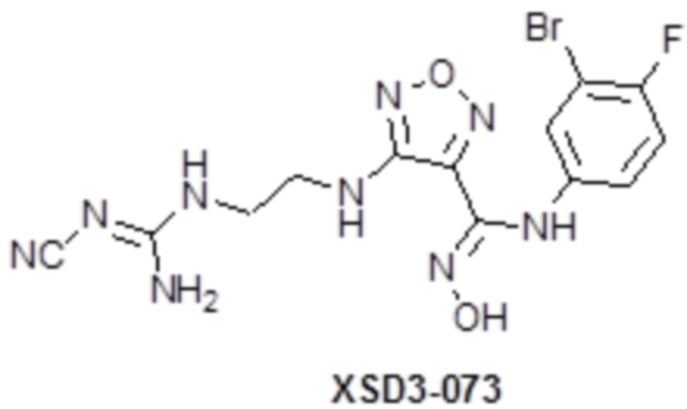 ,
, 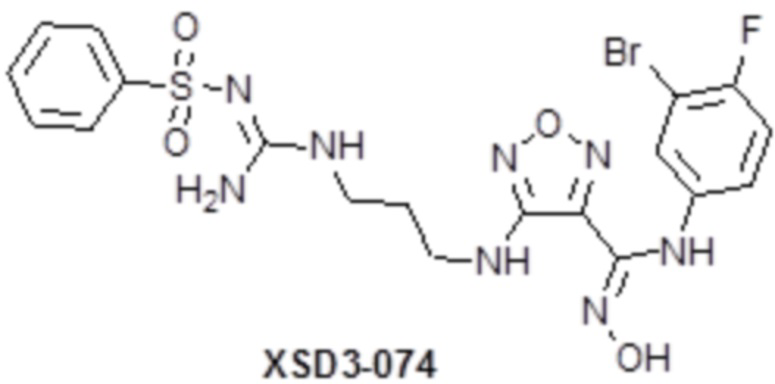 ,
, 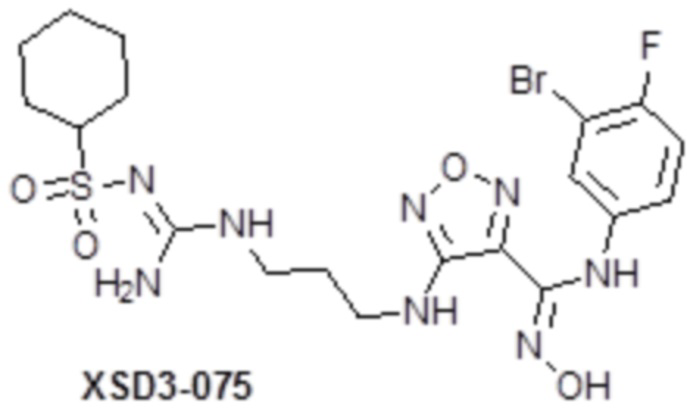 ,
, 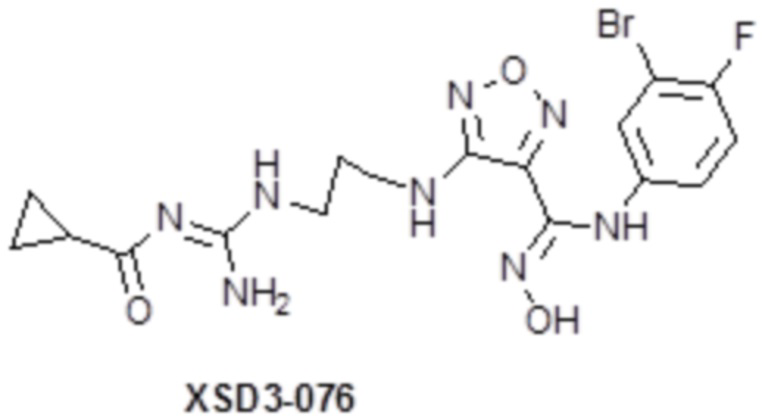 ,
, 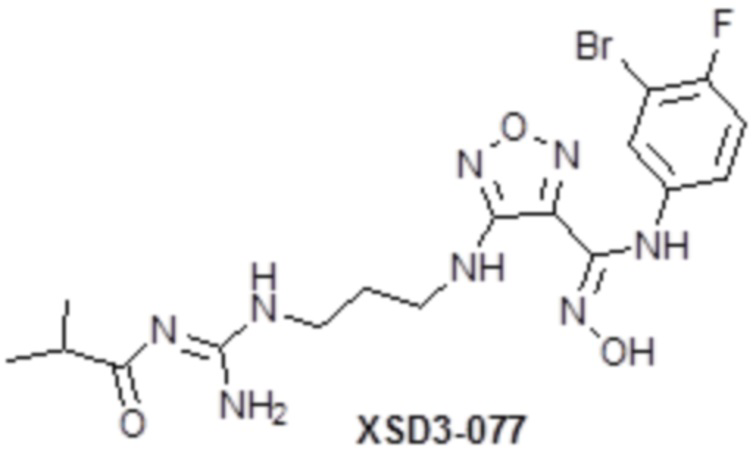 ,
, 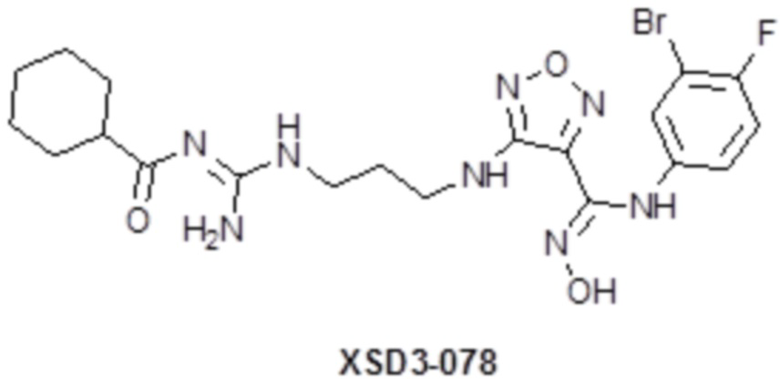 ,
, 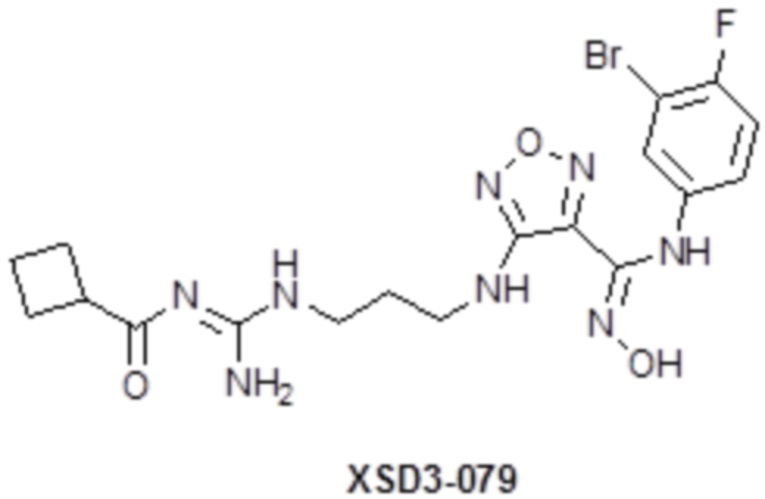 ,
, 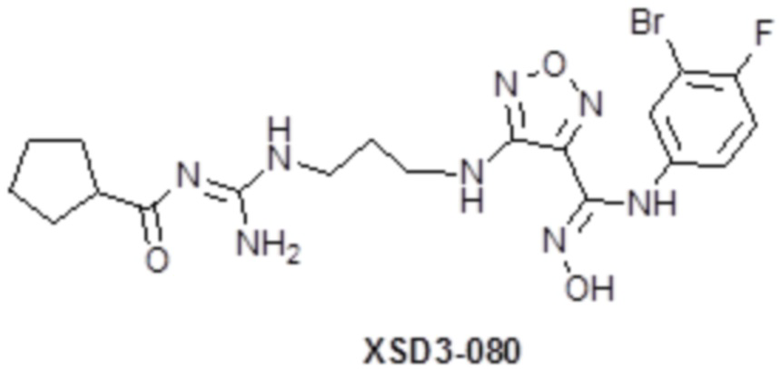 ,
, 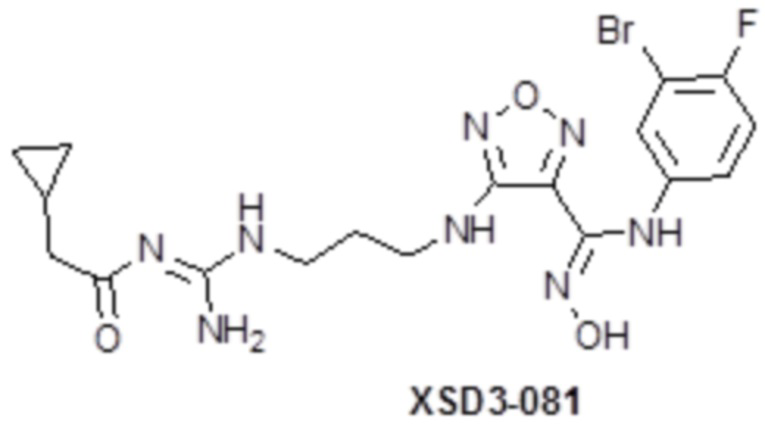 ,
, 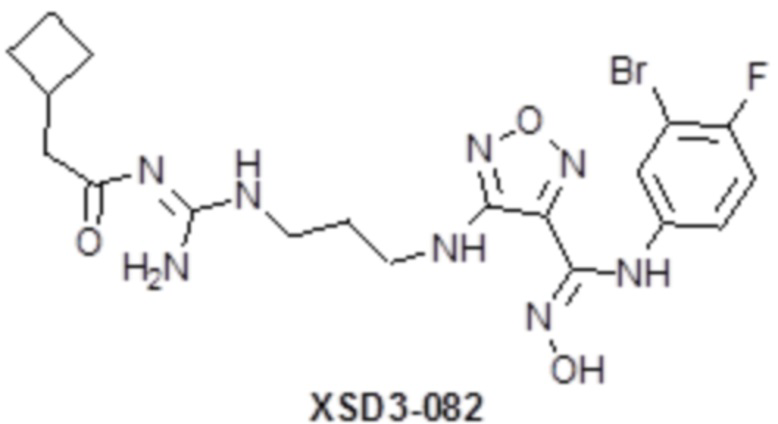 ,
, 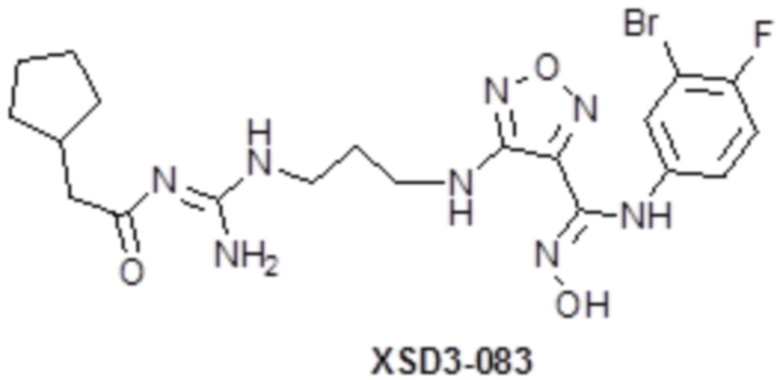 ,
, 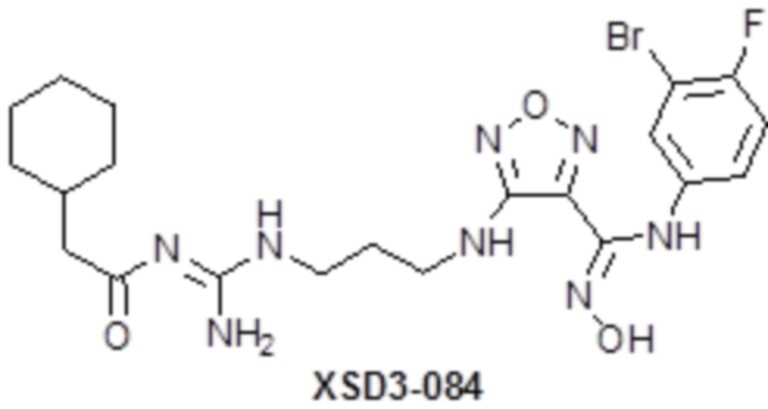 ,
, 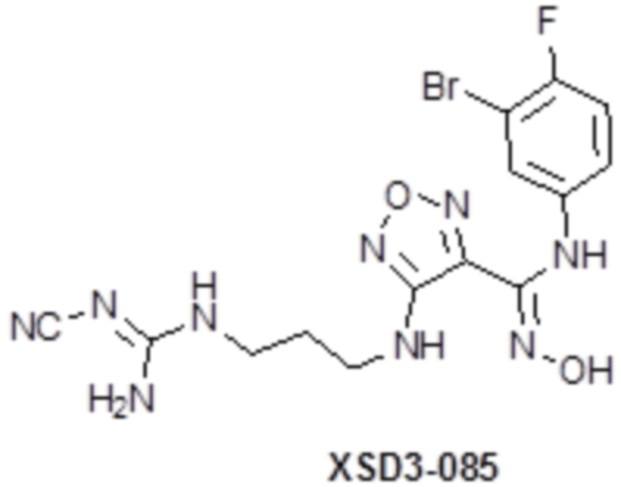 ,
, 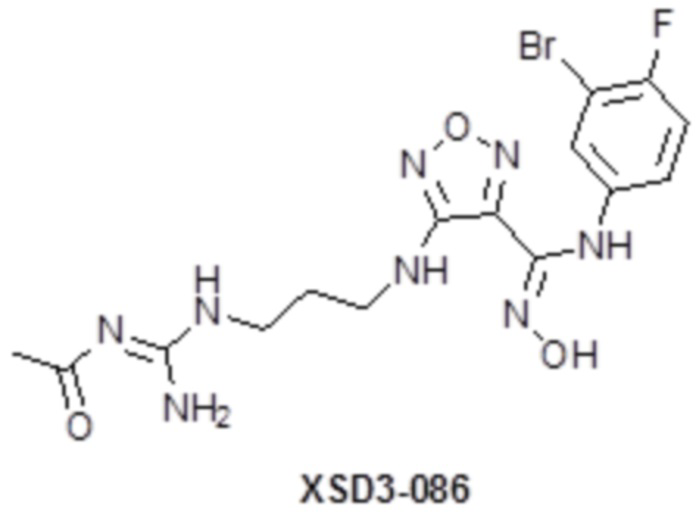 ,
,  ,
,  ,
,  ,
,  ,
, 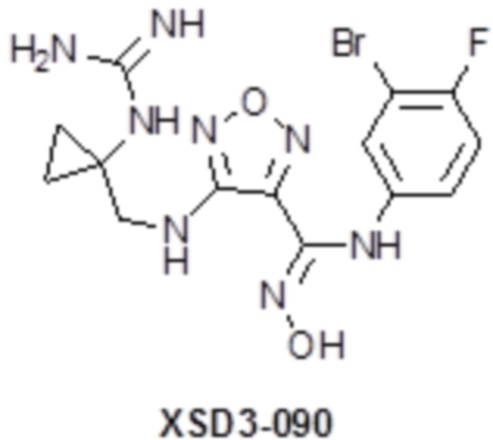 ,
, 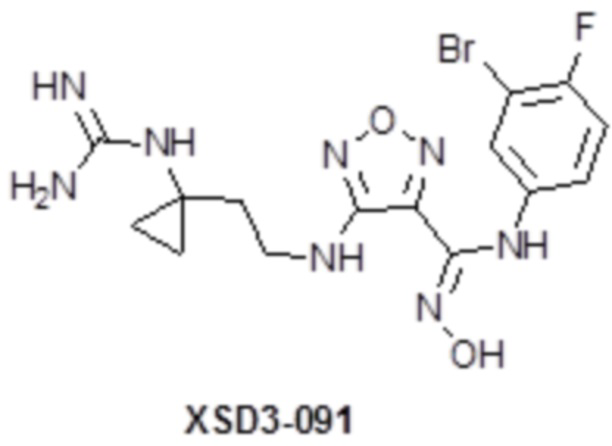 ,
, 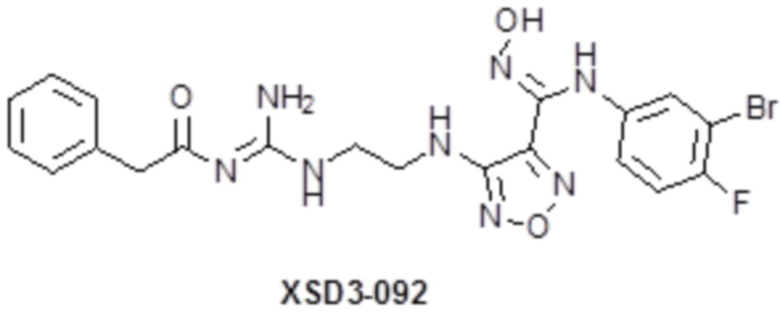 ,
, 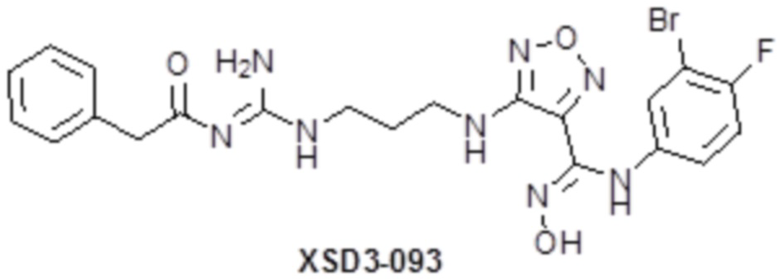 ,
,  ,
,  ,
,  ,
,  ,
, 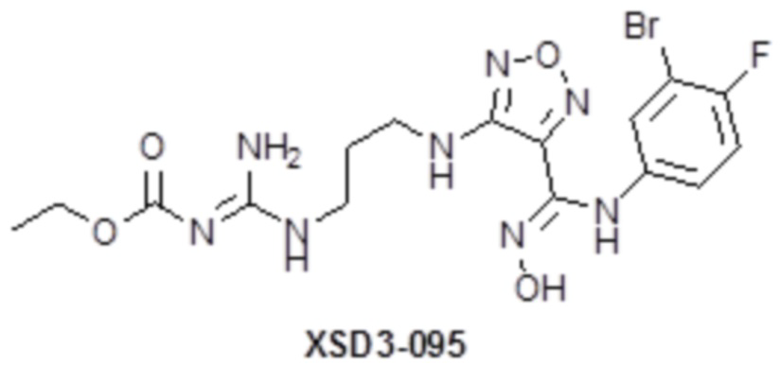 ,
, 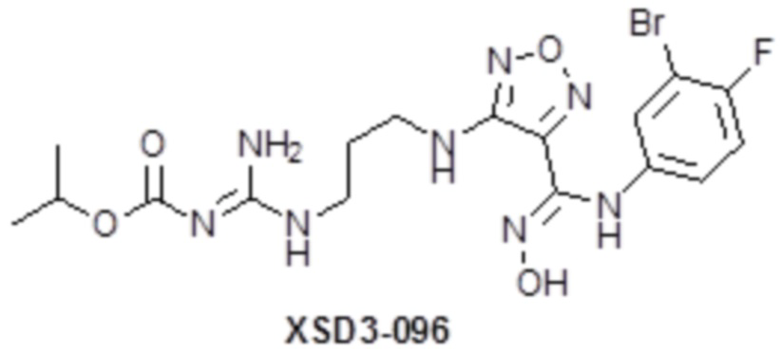 ,
, 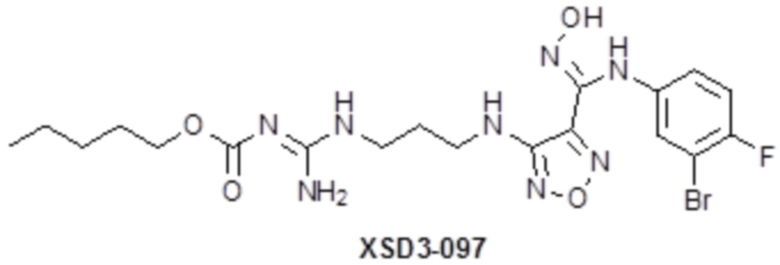 ,
, 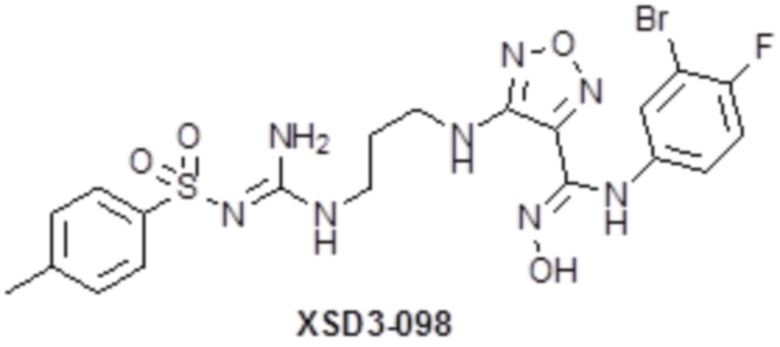 ,
,  .
.
Исходя из общей формулы или способа синтеза общей формулы согласно настоящему изобретению, могут быть получены соединения, не ограниченные указанными конкретными соединениями, и, с учетом методологических принципов получения общей формулы или способа синтеза общей формулы согласно настоящему изобретению, все конкретные соединения, которые могут быть получены специалистами в данной области техники без необходимости в творческих трудозатратах, подпадают под объем настоящего изобретения.
Согласно некоторым вариантам осуществления, настоящее изобретение относится к способу синтеза описанного выше соединения, представленного формулой I0:
 ,
,
т.е. к общему способу синтеза I, который характеризуется следующими стадиями:
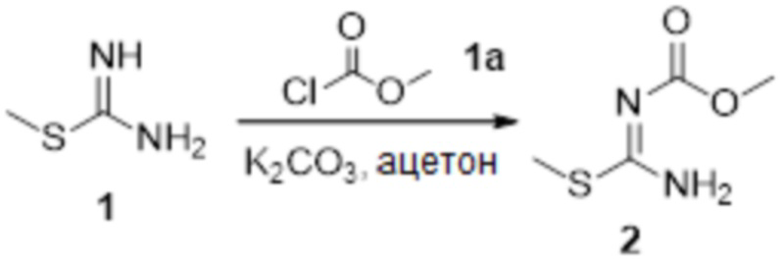
1) Окисление соединения 1a с получением соединения 2a:
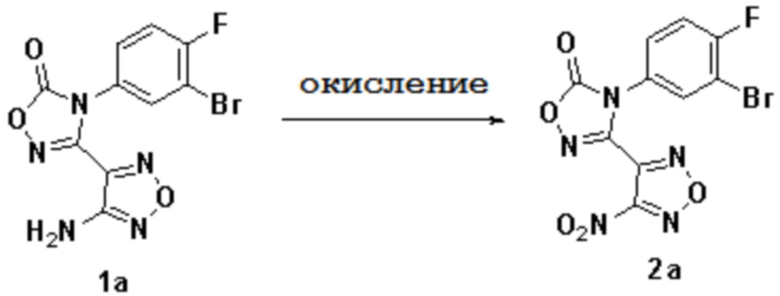 ,
,
причем используемый выше окислитель включает без ограничения по меньшей мере один из пероксида водорода, озона, пероксиуксусной кислоты;
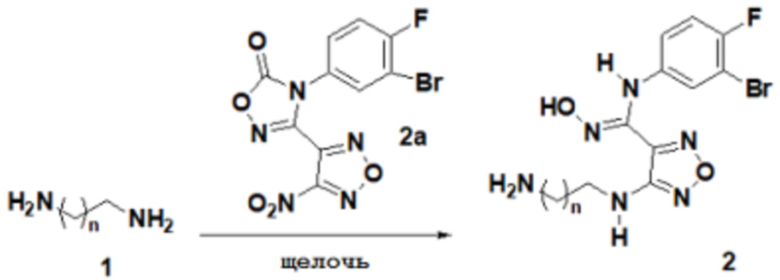
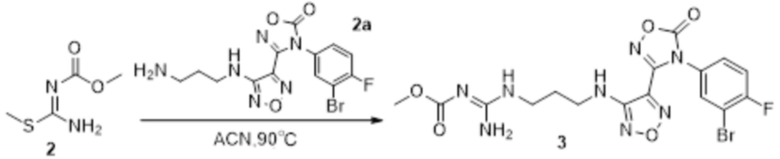
2) Взаимодействие соединения, представленного формулой 1, с соединением, представленным формулой 2a, в щелочных условиях с получением соединения, представленного формулой 2:
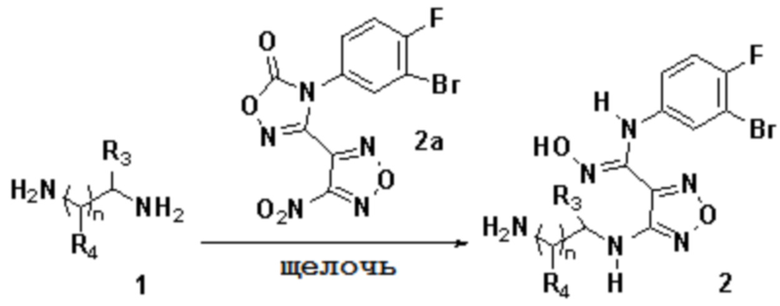 ,
,
причем используемая выше щелочь включает без ограничения гидроксиды щелочных металлов, предпочтительно гидроксид натрия, гидроксид калия, гидроксид бария;
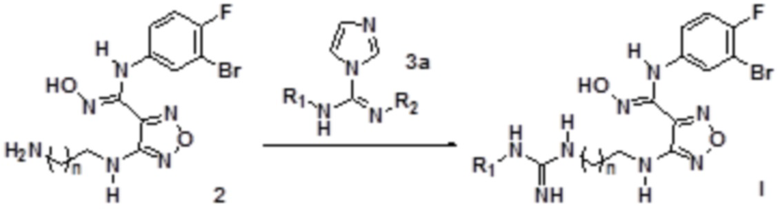
3) Взаимодействие соединения, представленного формулой 2, с соединением, представленным формулой 3a с получением соединения, представленного формулой I0:
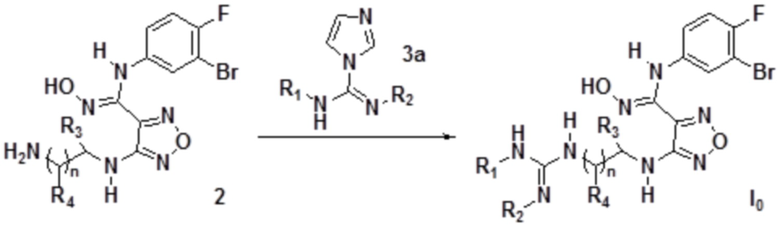
где каждый из R1 и R2 независимо выбирают из группы, состоящей из: H, замещенного или незамещенного C1-10алкила, альдегидной группы, замещенного или незамещенного карбонила, циано, CF3, замещенного или незамещенного C1-10алкокси, замещенного или незамещенного сульфонила, замещенного или незамещенного C3-10циклоалкил, замещенного или незамещенного C2-10алкенила, замещенного или незамещенного C6-20арила, замещенного или незамещенного C3-14гетероарила;
каждый из R3 и R4 независимо представляет собой монозаместитель, выбранный из группы, состоящей из: H, замещенного или незамещенного C1-10алкила, замещенного или незамещенного C3-10циклоалкила, циано, замещенного или незамещенного C1-10алкокси, замещенного или незамещенного сульфонила, замещенного или незамещенного C6-20арила и замещенного или незамещенного C3-14гетероарила;
или каждый из R3 и R4 независимо выбирают из дизаместителей, формирующих посредством этого вместе с атомом C в a- или b-положении следующие группы: , или , где C представляет собой атом C в a- или b-положении, m равен целому числу, выбранному из 0-6;
n равен целому числу, выбранному из 0-6.
Согласно некоторым вариантам осуществления, настоящее изобретение относится к способу синтеза описанного выше соединения, представленного формулой I:
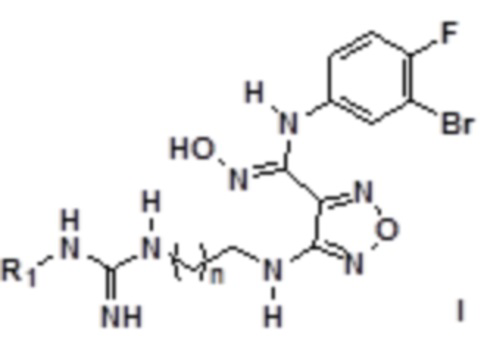 ,
,
т.е. к общему способу синтеза IA, который характеризуется следующими стадиями:
1) Окисление соединения 1a с получением соединения 2a:
;
2) Взаимодействие соединения, представленного формулой 1, с соединением 2a в щелочных условиях с получением соединения, представленного формулой 2:
 ;
;
3) Взаимодействие соединения, представленного формулой 2 с соединением, представленным формулой 3a с получением соединения, представленного формулой I:
 ,
,
где R1 выбирают из группы, состоящей из: H, амино, сульфонила, нитро, карбонила, амидино, C1-6алкила, замещенного амидино, замещенного C1-6алкила, замещенного C1-6алкокси, замещенного карбонила, замещенного сульфонила и замещенного сульфинила, где замещенный амидино или замещенный C1-6алкил, замещенный C1-6алкокси, замещенный карбонил, замещенный сульфонил или замещенный сульфинил замещен галогеном, гидрокси, карбокси, карбонилом, альдегидной группой, циано, амино, арилом, гетероарилом, C3-12циклоалкилом, C2-6алкенилом, C3-12циклоалкенилом; n равен целому числу, выбранному из 0-6;
использованный выше окислитель предпочтительно представляет собой без ограничения по меньшей мере один из пероксида водорода, озона или пероксиуксусной кислоты;
щелочь предпочтительно выбирают без ограничения из гидроксидов щелочных металлов, предпочтительно гидроксида натрия, гидроксида калия или гидроксида бария.
Согласно некоторым вариантам осуществления, настоящее изобретение относится к способу синтеза описанного выше соединения, представленного формулой II:
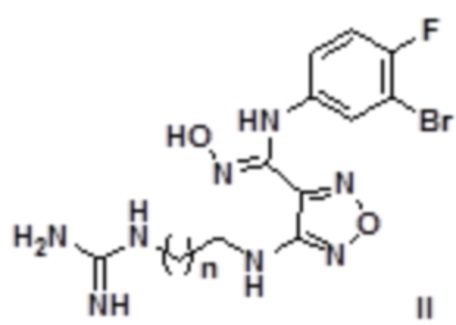 ,
,
т.е. к общему способу синтеза II, который характеризуется следующими стадиями:
1) Окисление соединения, представленного формулой 2, соединением 3a' с получением соединения, представленного формулой IIa:
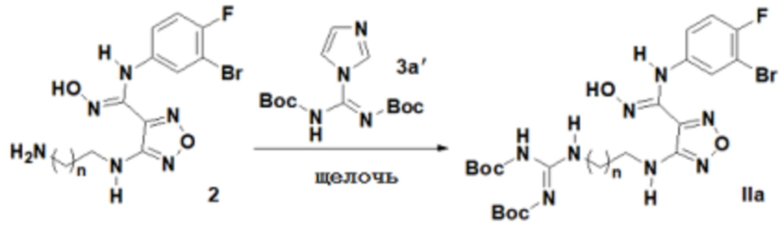 ;
;
2) Снятие защиты с соединения, представленного формулой IIa, в кислых условиях с последующим проведением реакции в щелочных условиях с получением соединения, представленного формулой II:
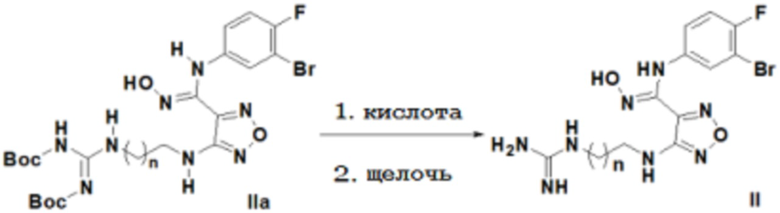 ,
,
где n равен 0, 1, 2, 3 или 4,
щелочь выбирают без ограничения из гидроксидов щелочных металлов и щелочноземельных металлов, предпочтительно гидроксида натрия, гидроксида калия, гидроксида бария.
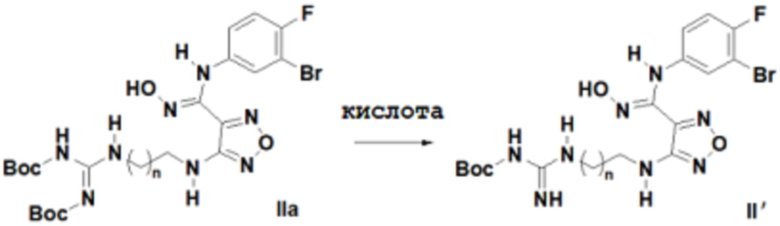
Согласно некоторым вариантам осуществления, настоящее изобретение относится к способу синтеза описанного выше соединения, представленного формулой II':
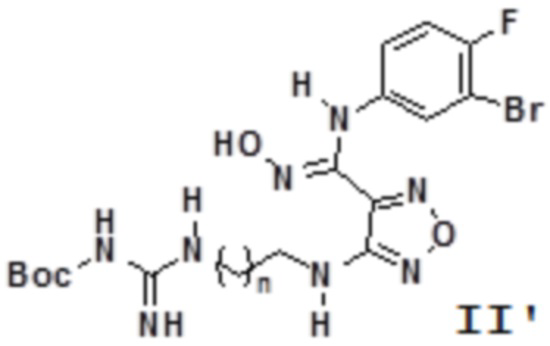 ,
,
т.е. к общему способу синтеза III, который характеризуется следующими стадиями:
 ,
,
n равен 0, 1, 2, 3 или 4,
соединение, представленное формулой IIa, вступает в реакцию в кислых условиях с получением соединения, представленного формулой II'.
Согласно некоторым вариантам осуществления, настоящее изобретение относится к способу синтеза описанного выше соединения, представленного формулой III:
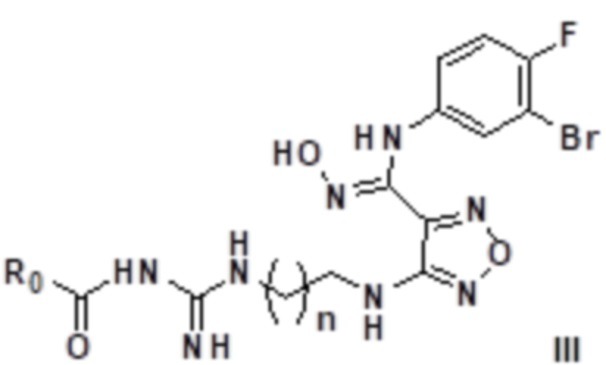 ,
,
т.е. к общему способу синтеза IV, который характеризуется следующими стадиями:
1) Осуществление взаимодействия соединения, представленного формулой II, с соединением, представленным формулой 4a, с получением соединения, представленного формулой IIIa:
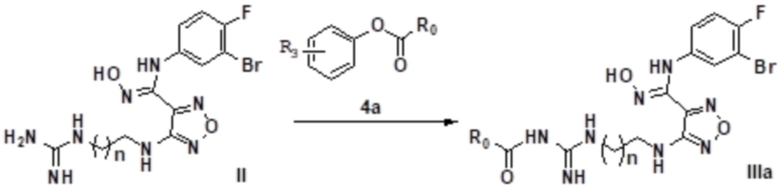 ;
;
где R3 выбирают из группы, состоящей из H, OH, CN, CH3-mXm, нитро, C1-9алкила, C1-9алкокси, C3-9циклоалкокси, C3-12циклоалкила, C1-6гетероалкила, 3-12-членного гетероциклоалкила, арила, гетероарила, замещенного C1-6алкила, замещенного C1-9алкокси, замещенного арила, замещенного гетероарила и замещенного карбонила, где замещенный C1-6алкил, замещенный C1-9алкокси, замещенный арил, замещенный гетероарил или замещенный карбонил замещен галогеном, гидрокси, карбокси, карбонилом, альдегидной группой, циано, амино, сульфонилом, арилом, гетероарилом, C3-12циклоалкилом, C3-12циклоалкенилом, m равен 1, 2 или 3; предпочтительно, m равен 2 или 3;
2) Взаимодействие соединения, представленного формулой IIIa в щелочных условиях с получением соединения, представленного формулой III:
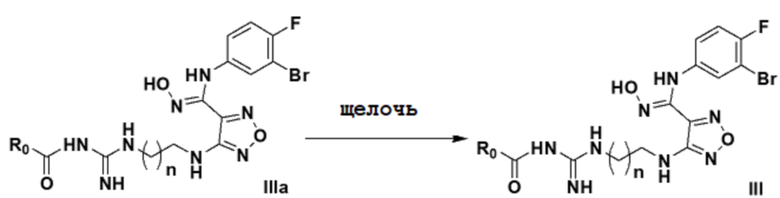 .
.
Более определенно, R3 выбирают из группы, состоящей из H, OH, CN, CH3-mXm, нитро, C1-9алкила, C3-9циклоалкокси, C3-12циклоалкила, C1-6гетероалкила, 3-12-членного гетероциклоалкила, арила, гетероарила, замещенного C1-6алкила, замещенного C1-9алкокси, замещенного арила и замещенного гетероарила, где замещенный C1-6алкил, замещенный C1-9алкокси, замещенный арил или замещенный гетероарил замещен галогеном, гидрокси, карбокси, карбонилом, альдегидной группой, циано, амино, сульфонилом, арилом, гетероарилом, C3-12циклоалкилом, C3-12циклоалкенилом. Кроме того, R3 выбирают из группы, состоящей из H, OH, CN, CF3, CHCl2, CH2Cl, нитро, метила, этила, пропила, изопропила, бутила, изобутила, втор-бутила, трет-бутила, пентила, 1-метилбутила, 2-метилбутила, 3-метилбутила, 1,1-диметилпропила, 2,2-диметилпропила, 1,2-диметилпропила, 1-этилпропила, гексила, пентилметила, пентилэтила, пентилпропила, пентилбутила, гексилметила, гексилэтила, гексилпропила, циклопропила, циклобутила, циклопентила, циклогексила, циклопропилметила, циклопропилэтила, циклопропилпропила, циклобутилметила, циклобутилэтила, циклобутилпропила, циклопентилметила, циклопентилэтила, циклопентилпропила, циклогексилметила, циклогексилэтила, циклогексилпропила, пара-метоксибензила (PMB), бензила (Bn); или
R3 является любым, выбранным из группы, состоящей из замещенного фурана, пиррола, тиофена, пиразола, имидазола, оксазола, тиофена, изоксазола, изотиазола, пиридина, пирана, тиопирана, пиридазина, пиридина, пиразина и пиперазина, или представляет собой дизаместитель на бензольном кольце, т.е. формирует вместе с бензольным кольцом бензофуран, бензопиррол, бензопиперазин; или
группу 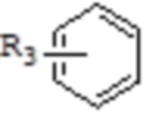 в формуле 4a выбирают из группы, состоящей из карбазола, акридина, феназина или фенотиазина.
в формуле 4a выбирают из группы, состоящей из карбазола, акридина, феназина или фенотиазина.
Способ синтеза согласно настоящему изобретению является лишь одним путем получения каждого синтезируемого целевого соединения или его промежуточного продукта, при котором каждая стадия и номер, такой как 1a, 2a, 3a, 4a, 1, 2, 3, и т.д., являются независимыми, и их получение не ограничивается способом согласно настоящему изобретению.
Если не указано иное, то растворитель, используемый на каждой стадии реакций согласно настоящему изобретению, описанных выше и далее в настоящем документе, представляет собой растворитель, общепринятый в данной области техники, и принцип отбора заключается в том, чтобы растворитель был способен растворять химические реагенты, но не участвовал во взаимодействии, не экстрагировал продукт или не позволял соответствующему продукту кристаллизоваться в растворе, с тем чтобы быть разделенным с примесями. Примеры растворителя включают воду, галогенированные алканы, алкиламины, алифатические углеводороды, сложные эфира, спирты, ароматические углеводороды, простые эфиры, гетероциклические растворители. Более определенно, растворитель выбирают без ограничения из следующих: метанол, этанол, пропанол, изопропил, диэтиловый эфир, этилацетат, уксусная кислота, циклогексан, дихлорметан, хлороформ, тетрагидрофуран, пиридин, диэтиламин, триэтиламин, диметилформамид, толуол и смеси по меньшей мере из двух из них.
Если не указано иное, то в каждой из реакций согласно настоящему изобретению, описанной выше и далее в настоящем документе, если реагент присутствует в избыточном количестве, то завершение реакции может проводиться путем добавления вещества, которое взаимодействует с избыточным реагентом.
Если не указано иное, то в каждой из реакций согласно настоящему изобретению, описанной выше и далее в настоящем документе, способ очистки продукта на каждой стадии реакций выбирают из группы, состоящей из экстрагирования, кристаллизации, удаления растворителя, колоночной хроматографии, причем все манипуляции при их проведении являются общепринятыми в данной области техники методиками, и специалист в данной области техники может манипулировать ими в соответствии с определенными условиями.
Цифры, используемые в общей формуле согласно настоящему изобретению, используются для удобного описания общей формулы, и в конкретных вариантах осуществления они могут быть модифицированы в другие цифры, такие как 1, 2, 3, и т.д., для удобства описания, и представляют собой выражения общей формулы и общих уравнений реакций, которые не влияют на суть структурной формулы и уравнения реакций с ее участием.
Для соединений, охватываемых формулами I-III, и их конкретных примеров соединений, хиральные или цис-транс изомеры и смеси указанных изомеров соединений в любом соотношении также подпадают под объем соединений, охватываемых формулами I-III, и их конкретных примеров соединений.
Согласно некоторым вариантам осуществления, настоящее изобретение относится к фармацевтической композиции, содержащей описанные выше соединения, т.е. соединения, охватываемые формулами I-III, и описанные выше конкретные соединения, или их фармацевтически приемлемую соль, и один или более фармацевтически приемлемых адъювантов.
Используемый в настоящем описании термин «фармацевтически приемлемая соль» относится к аддитивным солям, сформированным с фармацевтически приемлемой кислотой или щелочью, или к их сольвату. Такие фармацевтически приемлемые соли включают соли кислот, где кислоты включают соляную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту, бромистоводородную кислоту, йодистоводородную кислоту, муравьиную кислоту, уксусную кислоту, пара-толуолсульфоновую кислоту, сульфиновую кислоту, метансульфоновую кислоту, бензойную кислоту, фумаровую кислоту, лимонную кислоту, винную кислоту, малеиновую кислоту, жирную кислоту. Соли добавления нетоксичной фармацевтически приемлемой щелочи включают соли щелочей, и такие щелочи включают щелочи натрия, калия, кальция, магния, алюминия, аммония.
Согласно некоторым вариантам осуществления, настоящее изобретение относится к описанному выше соединению или его фармацевтически приемлемой соли, которое способно ингибировать активность индоламин-2,3-диоксигеназы, и может быть использовано для производства лекарственного средства для лечения заболевания, характеризующегося патологическим путем метаболизма триптофана, опосредованным индоламин-2,3-диоксигеназой; лекарственное средство используют для лечения злокачественной опухоли, инфекционного заболевания, нейродегенеративного заболевания, депрессии, тревожности или возрастной катаракты;
где злокачественную опухоль выбирают из группы, состоящей из злокачественной опухоли легкого, злокачественной опухоли печени, злокачественной опухоли толстого кишечника, злокачественной опухоли поджелудочной железы, злокачественной опухоли молочной железы, злокачественной опухоли предстательной железы, злокачественной опухоли головного мозга, злокачественной опухоли яичников, злокачественной опухоли шейки матки, злокачественной опухоли яичек, злокачественной опухоли почки, злокачественной опухоли головы и шеи, лимфомы, меланомы или лейкоза;
нейродегенеративное заболевание относится к болезни Альцгеймера;
инфекционное заболевание относится к инфекции, вызванной бактерией, грибком, вирусом или паразитом.
Согласно некоторым вариантам осуществления, настоящее изобретение относится к способу ингибирования активности индоламин-2,3-диоксигеназы путем использования описанного выше соединения или его фармацевтически приемлемой соли или для лечения заболевания, характеризующегося патологическим путем метаболизма триптофана, опосредованным индоламин-2,3-диоксигеназой, причем способ включает введение терапевтически эффективного количества соединения или его фармацевтически приемлемой соли. Заболевание выбирают из группы, состоящей из злокачественной опухоли, инфекционного заболевания, нейродегенеративного заболевания, депрессии, тревожности или возрастной катаракты; где злокачественную опухоль выбирают из группы, состоящей из злокачественной опухоли легкого, злокачественной опухоли печени, злокачественной опухоли толстого кишечника, злокачественной опухоли поджелудочной железы, злокачественной опухоли молочной железы, злокачественной опухоли предстательной железы, злокачественной опухоли головного мозга, злокачественной опухоли яичников, злокачественной опухоли шейки матки, злокачественной опухоли яичек, злокачественной опухоли почки, злокачественной опухоли головы и шеи, лимфомы, меланомы или лейкоза; нейродегенеративное заболевание относится к болезни Альцгеймера; инфекционное заболевание относится к инфекции, вызванной бактерией, грибком, вирусом или паразитом.
В соответствии с примерами согласно настоящему изобретению, результаты теста на активность демонстрируют, что соединения, полученные посредством настоящего изобретения, обладают превосходной ингибирующей активностью в отношении индоламин-2,3-диоксигеназы, и эта активность существенно выше, чем у соединения INCB024360. Результаты тестов in vivo демонстрируют, что соединения согласно настоящему изобретению обладают высокой степенью ингибирования в отношении опухолей, и терапевтический эффект в отношении опухолей значительно лучше, чем у соединения INCB024360 и других ингибиторов IDO. Кроме того, путем измерения массы тела мышей до и после введения, было обнаружено, что в сравнении с другими ингибиторами IDO, ингибиторы индоламин-2,3-диоксигеназы согласно настоящему изобретению могут существенно снижать побочные эффекты в процессе лечения опухоли, существенно улучшать качество жизни мышей, а в клинической практике не только улучшать качество жизни пациента, но также существенно улучшать комплаентность пациента к лекарственным средствам и эффективность лекарств.
Соединения согласно настоящему изобретению могут существенно улучшать нарушения обучаемости и памяти, усиливать способность приобретать знания и способность к пространственной памяти, и обладают положительной терапевтической ценностью при нейродегенеративных заболеваниях, таких как синдром Альцгеймера, и их эффект превосходит другие ингибиторы IDO.
В эксперименте на реакцию пролиферации Т-клеток было обнаружено, что соединение согласно настоящему изобретению может усиливать функцию DC по стимулированию пролиферации Т-клеток, вследствие чего оно может быть использовано для лечения опухолевых заболеваний, аутоиммунных заболеваний, реакции отторжения трансплантата и инфекционных заболеваний, и несомненно превосходит другие ингибиторы IDO.
Если ингибитор индоламин-2,3-диоксигеназы согласно настоящему изобретению используют для производства лекарственного средства для лечения заболевания, характеризующегося патологическим путем метаболизма триптофана, опосредованным индоламин-2,3-диоксигеназой, то он демонстрирует следующие технические преимущества:
(1) Противоопухолевый эффект является заметным. Соединение согласно настоящему изобретению обладает существенной ингибирующей активностью в отношении индоламин-2,3-диоксигеназы, и в тесте in vivo показано, что степень ингибирования опухоли соединением согласно настоящему изобретению значительно выше, чем таковая для циклофосфамида в качестве лекарства положительного контроля и соединения INCB024360.
(2) Побочный эффект снижен. Соединение согласно настоящему изобретению представляет собой ингибитор индоламин-2,3-диоксигеназы, который обращает вспять ингибирование пролиферации Т-клеток и регулирует иммунную функцию организма посредством ингибирования активности индоламин-2,3-диоксигеназы, выполняя тем самым функции мониторинга и устранения опухолевых клеток иммунной системой человека. Основываясь на таком особенном механизме действия, это соединение не влияет негативно на рост нормальных клеток человеческого организма, при этом ингибируя рост опухолевых клеток, существенно снижая тем самым побочные эффекты. Кроме того, соединение обладает значительным терапевтическим эффектом в отношении аутоиммунных заболеваний, реакции отторжения трансплантата и инфекционных заболеваний, ассоциированных с пролиферацией Т-клеток.
(3) Лечение болезни Альцгеймера и других нейродегенеративных заболеваний значимо эффективно, нарушения обучаемости и памяти у животных могут быть существенно улучшены, и могут быть существенно усилены способность к приобретению знаний и способность к пространственной памяти.
Конкретные модели для выполнения изобретения
Настоящее изобретение более детально описано ниже в отношении конкретных вариантов осуществления, но настоящее изобретение ими не ограничивается. Кроме того, с учетом методологических принципов получения общей формулы или способа синтеза общей формулы (общий способ синтеза I, общий способ синтеза IA, общий способ синтеза II, общий способ синтеза III, общий способ синтеза IV) и конкретных вариантов осуществления настоящего изобретения, все конкретные соединения, полученные специалистами в данной области техники без необходимости в творческих трудозатратах, подпадают под объем настоящего изобретения.
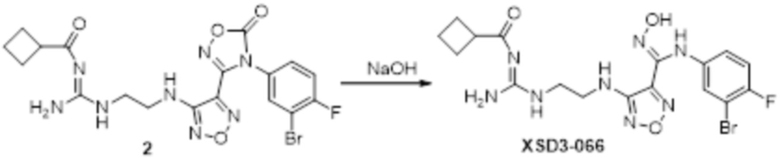
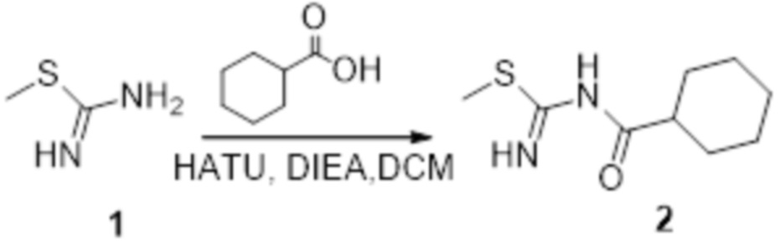
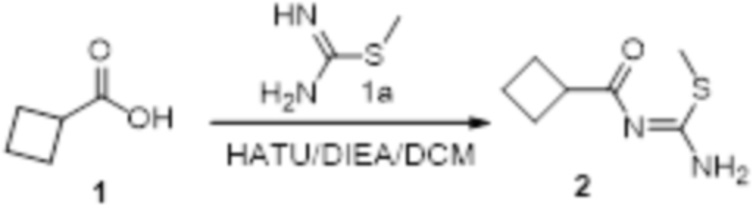
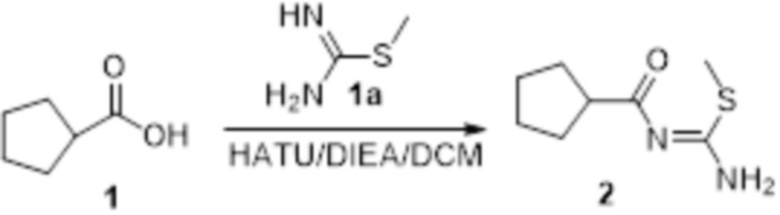
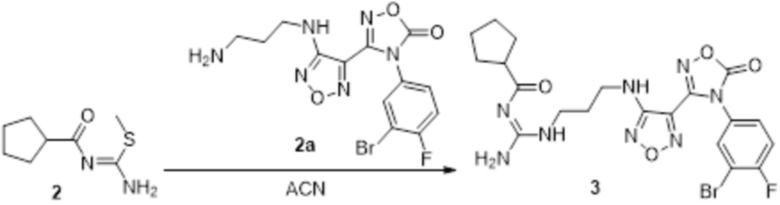
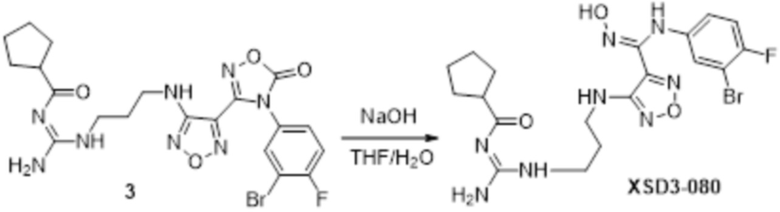
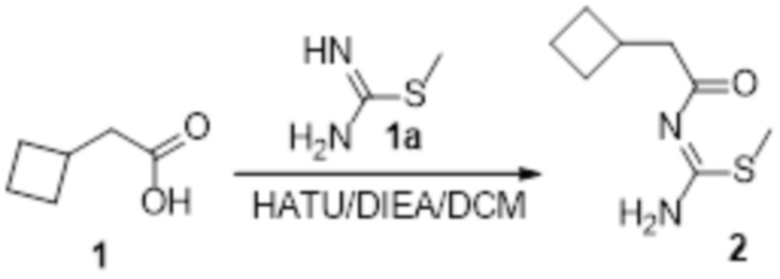
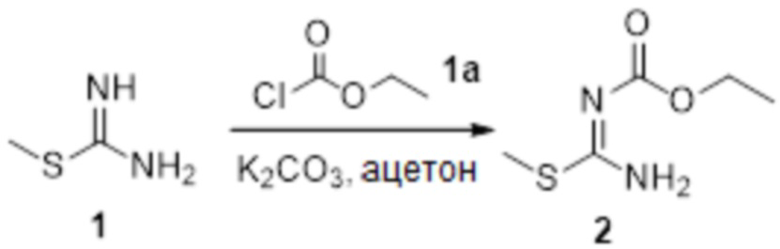
Пример 1
Реакция 1
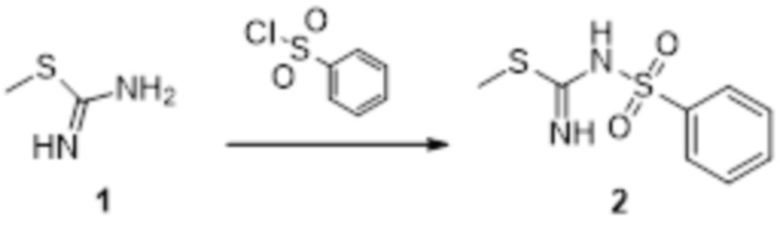
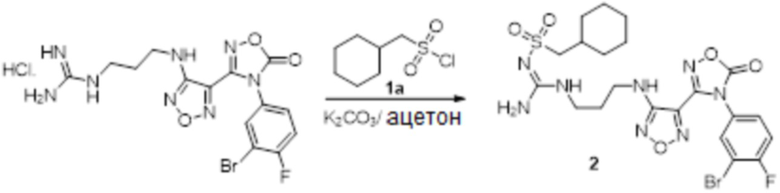
Соединение 1 (903 мг, 10 ммоль) растворяли в ацетоне (10 мл), затем при комнатной температуре добавляли карбонат калия (2,76 г, 20 ммоль), перемешивали при комнатной температуре в течение 0,5 ч, по каплям добавляли бензолсульфонилхлорид, перемешивали при комнатной температуре в течение ночи, гасили добавлением воды, полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 900 мг белого порошка.
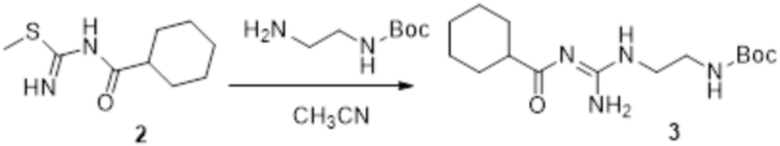
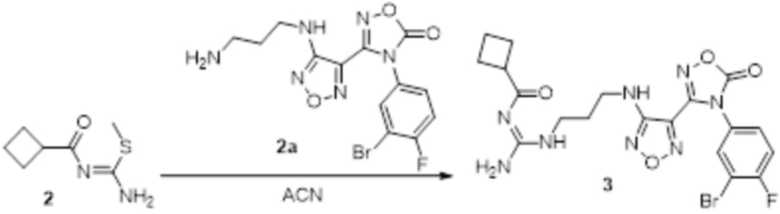
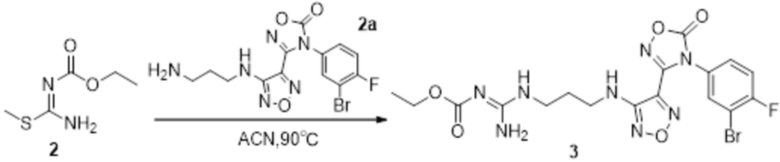
Реакция 2
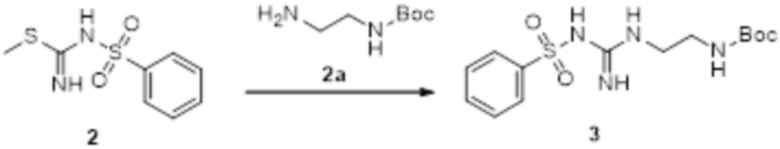
Соединение 2 (230 мг, 1 ммоль) растворяли в ацетонитриле (5 мл), перемешивали при комнатной температуре в течение 0,5 ч, затем добавляли соединение 2a (320 мг, 2 ммоль), нагревали при 60°C в течение 24 ч, полученное вещество сразу упаривали досуха в условиях пониженного давления и подвергали очистке методом препаративной HPLC с получением 210 мг целевого продукта.
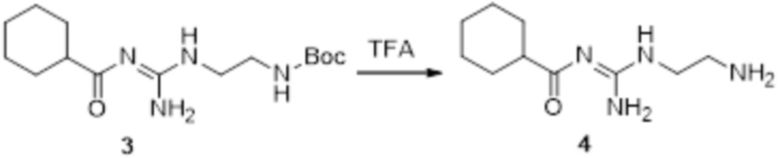
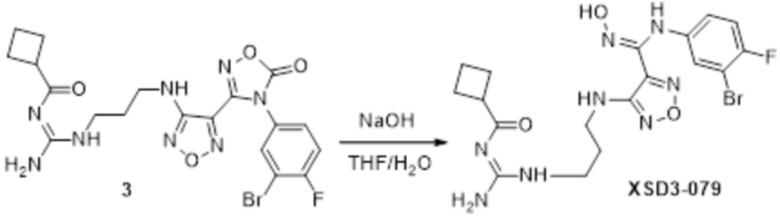
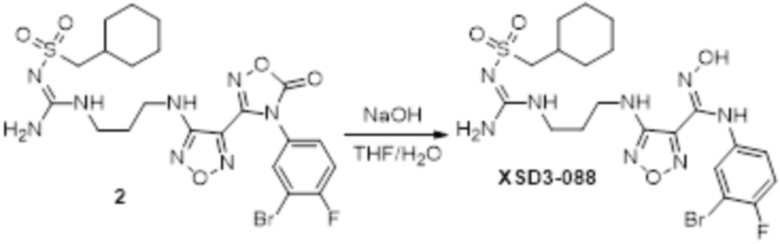
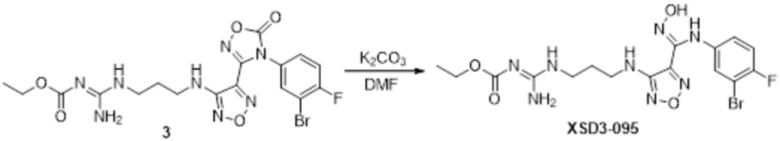
Реакция 3
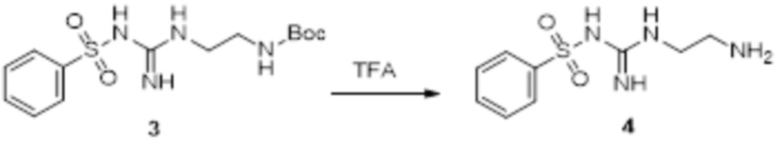
Соединение 3 (171 мг, 0,5 ммоль) растворяли в дихлорметане (10 мл), перемешивали при комнатной температуре в течение 5 мин, добавляли трифторуксусную кислоту (5 мл), осуществляли взаимодействие при комнатной температуре в течение 2 ч, а затем полученное вещество сразу упаривали досуха в условиях пониженного давления с получением целевого продукта 4, которое сразу использовали в виде неочищенного продукта на следующей стадии.
Реакция 4
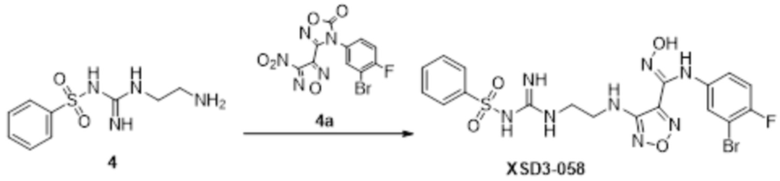
Соединение 4 (120 мг, 0,5 ммоль) растворяли в тетрагидрофуране, добавляли соединение 4a (120 мг, 0,5 ммоль), перемешивали смесь при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), и перемешивали смесь при комнатной температуре в течение 10 мин. Реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением 5 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,45 (с, 1H), 8,89 (с, 1H), 7,76-7,77 (м, 2H), 7,48-7,52 (м, 3H), 7,10-7,20 (м, 3H), 6,75-6,77 (м, 2H), 6,27 (с, 1H), 3,73-3,77 (м, 2H), 2,52-2,69 (м, 2H).
Чистота согласно HPLC: @214 нм 99,5%, @254 нм 100%.
LC-MS: m/z 541 [M+1].
Опираясь на общий способ синтеза I и Пример 1, синтезировали следующие соединения.
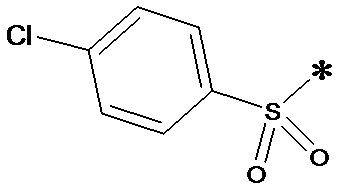 , n равен 1, R2, R3, R4 представляют собой H.
, n равен 1, R2, R3, R4 представляют собой H.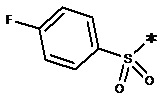 , n равен 1, R2, R3, R4 представляют собой H.
, n равен 1, R2, R3, R4 представляют собой H.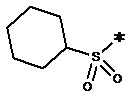 , n равен 1, R2, R3, R4 представляют собой H.
, n равен 1, R2, R3, R4 представляют собой H.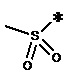 , n равен 1, R2, R3, R4 представляют собой H.
, n равен 1, R2, R3, R4 представляют собой H.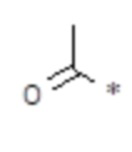 (метилкарбонил), n равен 2, R2, R3, R4 представляют собой H.
(метилкарбонил), n равен 2, R2, R3, R4 представляют собой H.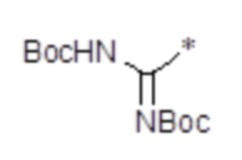 , n равен 1, R2, R3, R4 представляют собой H.
, n равен 1, R2, R3, R4 представляют собой H.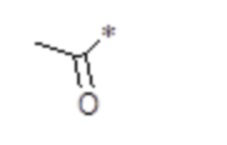 (метилкарбонил), n равен 1, R2, R3, R4 представляют собой H.
(метилкарбонил), n равен 1, R2, R3, R4 представляют собой H.
Пример 10
Реакция 1
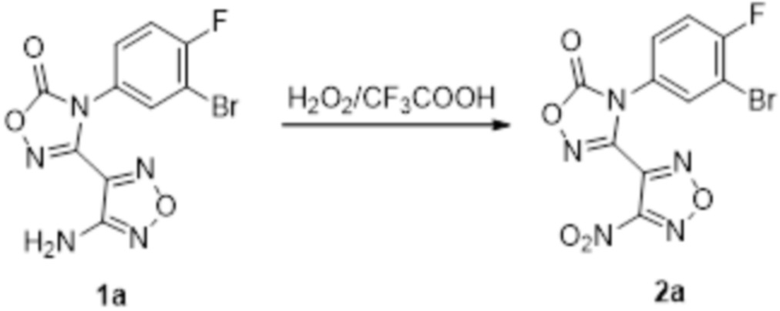
Соединение 1a (682 мг, 2 ммоль) растворяли в трифторуксусной кислоте (13 мл), затем при комнатной температуре по каплям добавляли 30% H2O2, смесь подвергали реакции в течение ночи при 50°C, и реакционный раствор изменялся из мутного до прозрачного желтого. После завершения реакции, реакционную смесь гасили добавлением насыщенного раствора сульфата натрия. После того, как KI-крахмальная индикаторная бумага становилась бесцветной, полученное вещество экстрагировали этилацетатом (50 мл × 2), и органическую фазу сушили над безводным сульфатом натрия, концентрировали, а затем подвергали очистке методом колоночной хроматографии (петролейный эфир:этилацетат = 1:1) с получением бледно-желтого твердого соединения 2a (500 мг, выход 67%).
Реакция 2
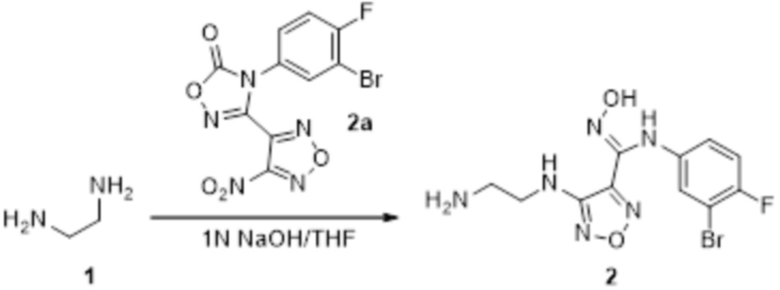
К раствору соединения 2a (170 мг, 0,5 ммоль) в тетрагидрофуране (10 мл) добавляли этилендиамин 1 (30 мг, 0,5 ммоль), затем добавляли 1н NaOH (0,4 мл), реакционный раствор перемешивали при комнатной температуре в течение 0,5 ч, и сразу реакционный раствор использовали для получения соединения 2 (120 мг).
Реакция 3
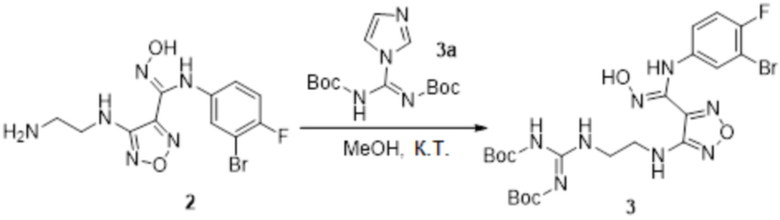
Смешанный раствор соединения 2 (120 мг, 0,33 ммоль) и соединения 3a (100 мг, 0,33 ммоль) в метаноле (10 мл) перемешивали при комнатной температуре в течение ночи, реакционный раствор концентрировали и подвергали очистке методом колоночной хроматографии (петролейный эфир:этилацетат = 1:1) с получением соединения 3 (42 мг, 23%).
Реакция 4
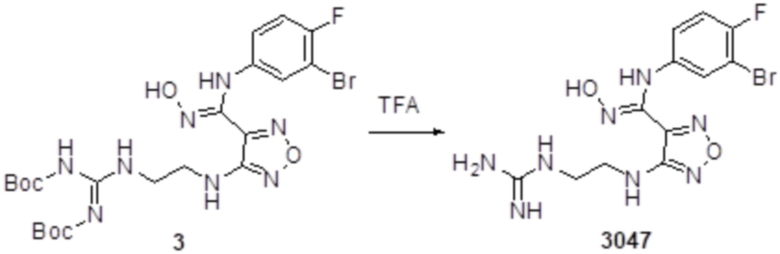
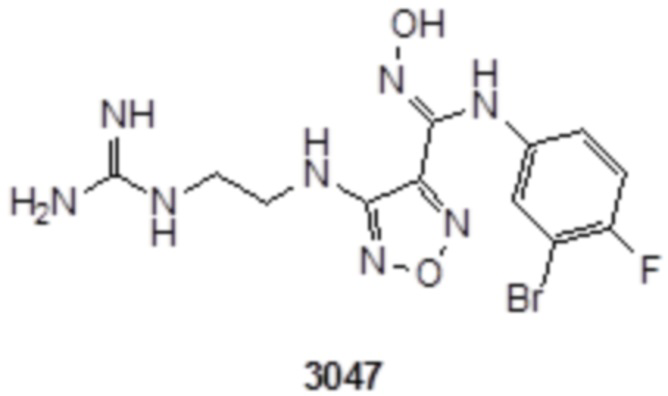
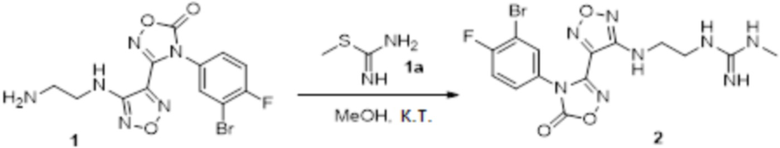
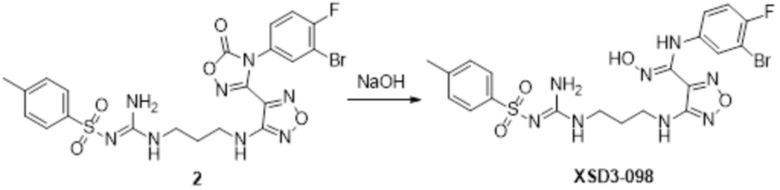
К раствору соединения 3 (31 мг, 0,05 ммоль) в дихлорметане (3 мл) добавляли трифторуксусную кислоту (0,6 мл), и перемешивали при комнатной температуре в течение ночи. Реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением соединения XSD3-047 (9 мг, выход 68%). Для целевого соединения: 1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,4 (с, 1H), 8,88 (с, 1H), 7,22-7,11 (м, 2H), 6,71-6,5 (м, 3H), 6,38-6,21 (м, 2H), 3,37 (м, 3H), 3,01 (м, 2H). MS: m/z 401,2 [M+1].
Пример 11
Реакция 1
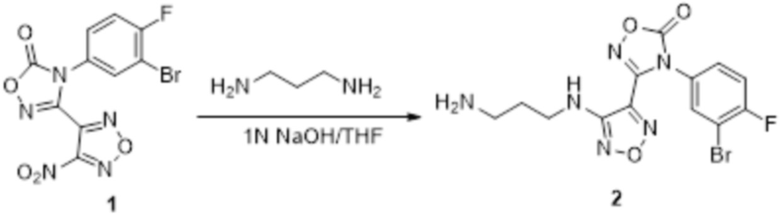
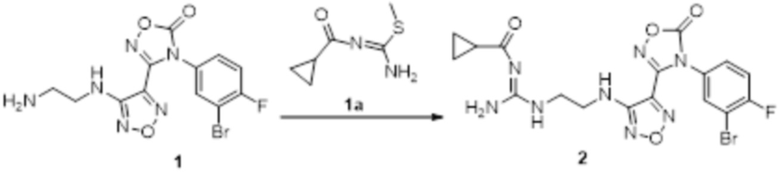
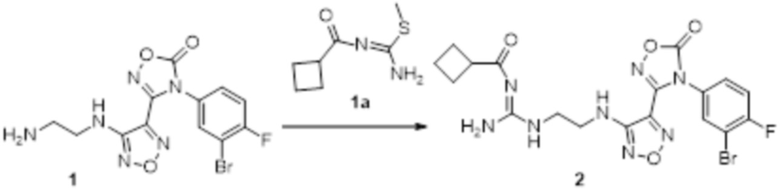
К раствору соединения 1 (170 мг, 0,5 ммоль, способ синтеза которого описан для синтеза соединения 2a в итоговом отчете для XSD3-047) в тетрагидрофуране (10 мл) добавляли 1,3-диаминопропан (35 мг, 0,5 ммоль), реакционный раствор перемешивали при комнатной температуре в течение 0,5 ч, и реакционный раствор сразу использовали для получения 130 мг целевого продукта.
Реакция 2
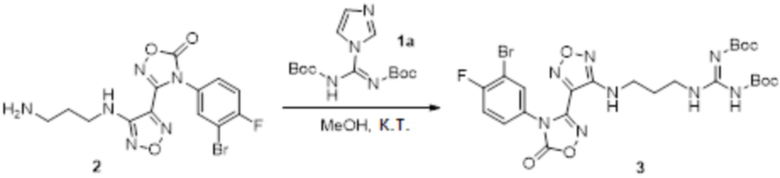
Смешанный раствор соединения 2 (130 мг, 0,33 ммоль) и соединения 1a (100 мг, 0,33 ммоль) в метаноле (10 мл) перемешивали при комнатной температуре в течение ночи, реакционный раствор концентрировали и подвергали очистке методом колоночной хроматографии (петролейный эфир:этилацетат = 1:1) с получением соединения 3 (78 мг, 45%).
Реакция 3
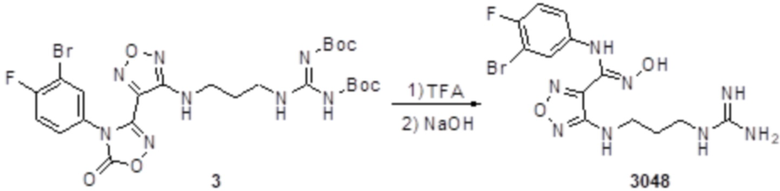
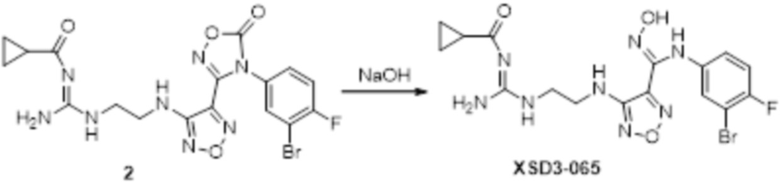
К раствору соединения 3 (31 мг, 0,05 ммоль) в дихлорметане (3 мл) добавляли трифторуксусную кислоту (0,6 мл), перемешивали при комнатной температуре в течение ночи, затем корректировали до pH=12 добавлением 1н раствора NaOH, непрерывно перемешивали в течение 20 мин, и отслеживали развитие реакции методом LCMS. После завершения реакции, реакционный раствор корректировали до нейтральных значений добавлением 0,5н HCl, реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением целевого продукта (9 мг, выход 68%). Для целевого продукта: 1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,4 (с, 0,6 H), 8,88 (с, 1H), 7,6 (с, 1H), 7,4-6,7 (м, 6H), 6,7 (с, 1H), 6,24 (с, 1H), 3,19-3,14 (м, 4H), 1,75 (м, 2H). MS: m/z 415,2 [M+1].
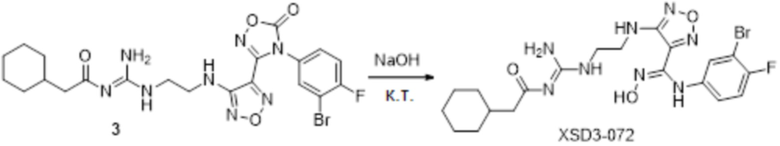
Пример 12
Реакция 1
Смешанный раствор соединения 1 (384 мг, 1 ммоль, способ получения которого описан в примере получения соединения 3047) и соединения 1a (100 мг, 1,1 ммоль) в тетрагидрофуране (10 мл) перемешивали при комнатной температуре в течение 48 ч, и реакционный раствор сразу использовали для получения целевого продукта 2 (89 мг, выход 20%).
Реакция 2
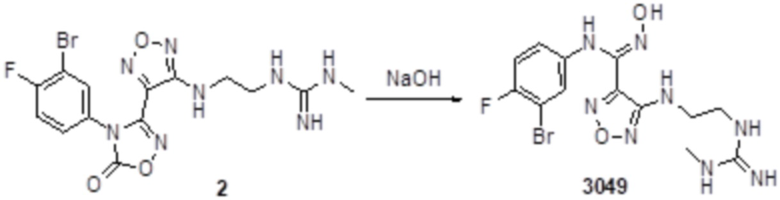
Соединение 2 (89 мг, 0,2 ммоль) растворяли в THF (10 мл), затем корректировали до pH=12 добавлением 1н раствора NaOH, и непрерывно перемешивали в течение 20 мин. Развитие реакции отслеживали методом LCMS. После завершения реакции, реакционный раствор корректировали до нейтральных значений добавлением 0,5н HCl, реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением целевого продукта (13 мг, выход 15%). Для целевого соединения: 1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,47 (с, 0,6 H), 8,92 (с, 1H), 7,50-7,09 (м, 5H), 6,70 (с, 1H), 6,28 (с, 1H), 2,72-2,50 (м, 4H), 2,50 (м, 3H). MS: m/z 416,2 [M+1].
Пример 13
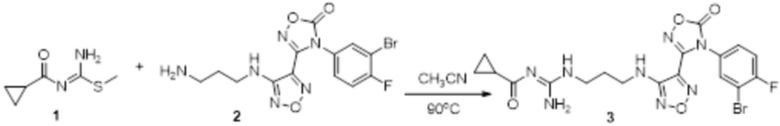
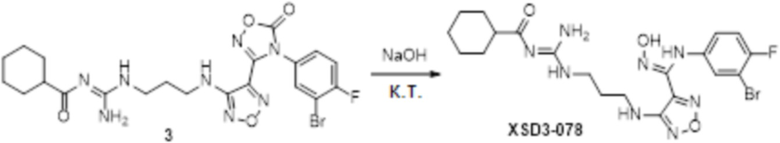
Реакция 1
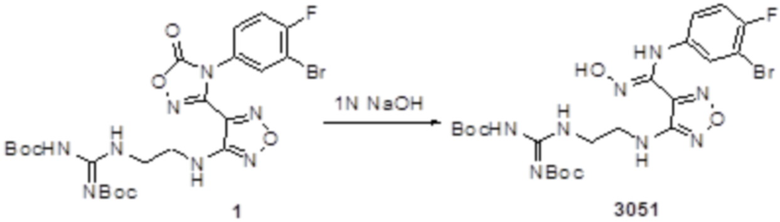
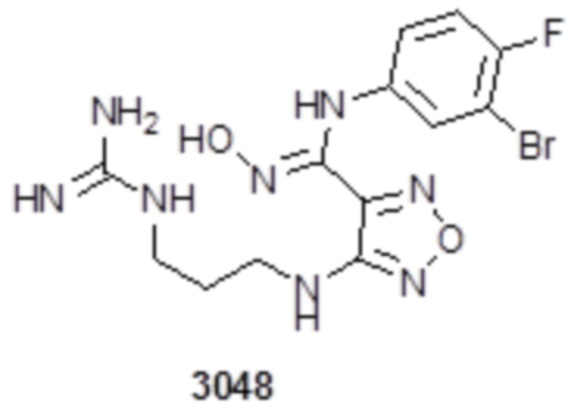
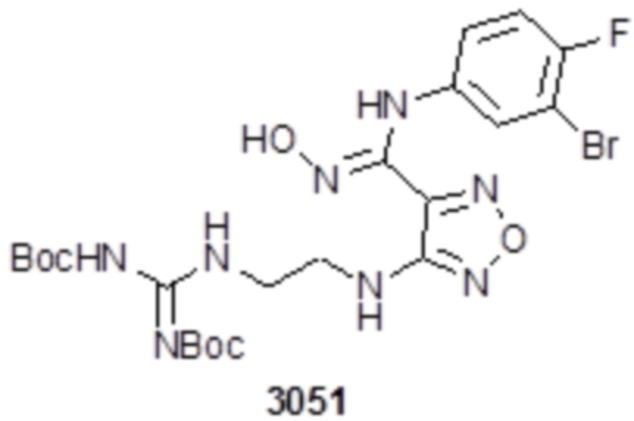
Соединение 1 (102 мг, 0,016 ммоль, способ синтеза которого описан в итоговом отчете для XSD3-047) растворяли в THF (10 мл), затем при комнатной температуре добавляли 1н раствор NaOH (0,1 мл), перемешивали при комнатной температуре в течение 0,2 ч, и отслеживали развитие реакции методом LCMS. После завершения реакции, pH корректировали до нейтральных значений добавлением 0,5н раствора HCl, с получением 52 мг целевого соединения. Для целевого соединения: 1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,45 (с, 1H), 8,89 (с, 1H), 8,24 (м, 1H), 7,76-7,77 (м, 2H), 7,14-7,20 (м, 2H), 7,18 (с, 1H), 6,31 (с, 1H), 3,33-3,67 (м, 4H), 1,44-1,48 (м, 18H). MS: m/z 601,2 [M+1].
Пример 14
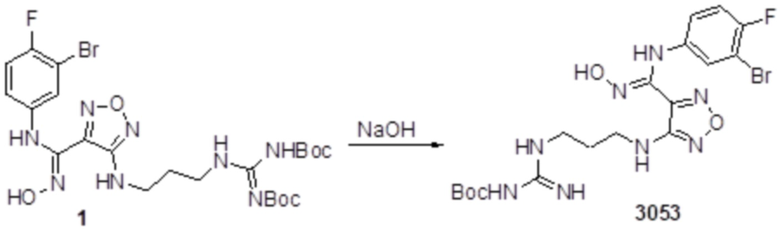
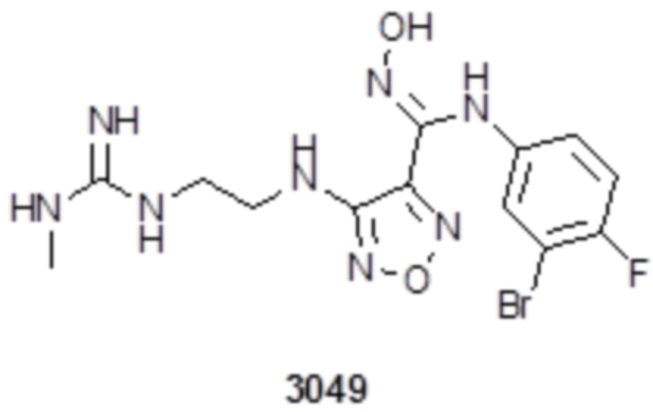
Соединение 1 (52 мг, 0,1 ммоль, способ синтеза которого описан в итоговом отчете для XSD3-048) растворяли в THF (10 мл), затем при комнатной температуре добавляли TFA (0,5 мл), перемешивали при комнатной температуре в течение 12 ч, и отслеживали развитие реакции методом LCMS. После очистки методом препаративной HPLC, получали 15 мг целевого соединения. Для целевого соединения: 1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,44 (с, 1H), 10,85 (с, 1H), 8,91 (с, 1H), 8,49 (с, 1H), 7,18-7,09 (м, 3H), 6,78-6,75 (м, 1H), 6,28-6,25 (м, 1H), 3,33-3,19 (м, 4H), 1,85-1,78 (м, 2H), 1,5 (с, 3H). Результат охарактеризования методом LC-MS: m/z 515 [M+1].
Пример 15
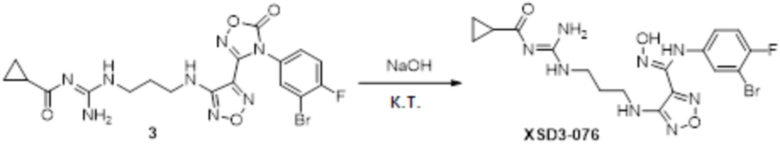
Реакция 1
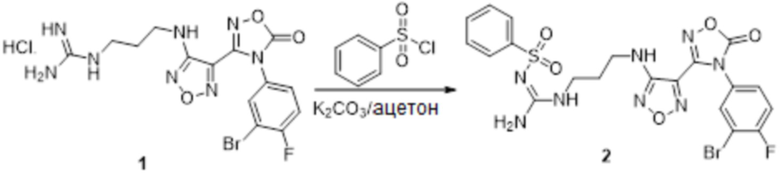
Соединение 1 (100 мг, 0,23 ммоль) растворяли в ацетоне (10 мл), затем добавляли карбонат калия (0,276 г, 2,0 ммоль), перемешивали при комнатной температуре в течение 0,5 ч, по каплям добавляли бензолсульфонилхлорид, перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили добавлением воды, полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, который сразу использовали на следующей стадии.
Реакция 2
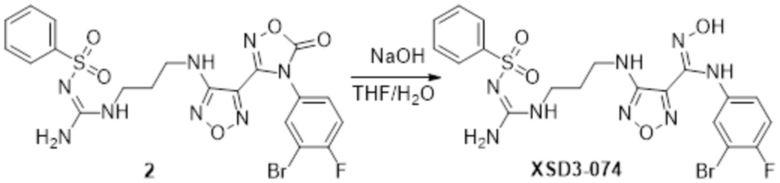
Соединение 2 (120 мг, 0,5 ммоль) растворяли в смеси тетрагидрофуран/вода, по каплям добавляли 1н гидроксид натрия (1 мл), и перемешивали при комнатной температуре в течение 10 мин. Реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением 20 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,45 (с, 1H), 8,90 (с, 1H), 7,75-7,77 (м, 2H), 7,46-7,52 (м, 3H), 7,12-7,20 (м, 1H), 7,10-7,11 (м, 1H), 6,75-6,78 (м, 2H), 6,19 (с, 1H), 4,25 (м, 2H), 3,11-3,19 (м, 4H),1,67-1,70 (м, 2H).
Чистота согласно HPLC: @214 нм 99,2%, @254 нм 99,3%.
LC-MS: m/z 555 [M+1].
Пример 16
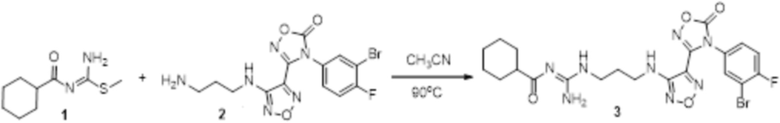
Реакция 1
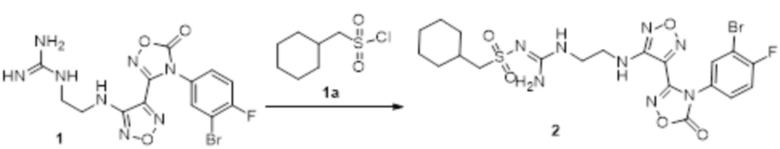
Соединение 1 (106 мг, 0,25 ммоль) растворяли в ацетоне (10 мл), добавляли карбонат калия (138 мг, 1 ммоль), по каплям добавляли соединение 1a (49 мг, 0,25 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды, полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 100 мг неочищенного продукта.
Реакция 2
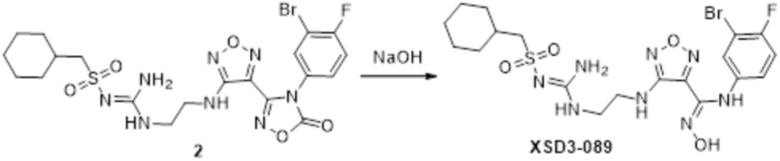
Соединение 2 (100 мг) растворяли в метаноле, перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), перемешивали при комнатной температуре в течение 10 мин. Реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением 12 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,45 (с, 1H), 8,90 (с, 1H), 7,10-7,21 (м, 2H), 6,76-7,17 (м, 4H), 6,28 (м, 1H), 3,33-3,40 (м, 4H), 2,76 (м, 2H), 0,98-1,86 (м, 11H).
Чистота согласно HPLC: @214 нм 93,7%, @254 нм 97,6%.
LC-MS: m/z 563 [M+1].
Пример 17
Реакция 1
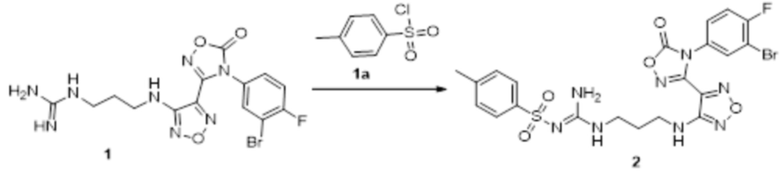
Соединение 1 (110 мг, 0,25 ммоль) растворяли в ацетоне (10 мл), добавляли карбонат калия (138 мг, 1 ммоль), по каплям добавляли соединение 1a (47,5 мг, 0,25 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия, концентрировали с получением 100 мг неочищенного продукта.
Реакция 2
Соединение 2 (100 мг) растворяли в метаноле, перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), перемешивали при комнатной температуре в течение 10 мин, реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением 15 мг целевого продукта.
1H-ЯМР (400 МГц, CDCl3): δ (м.д.): 9,96 (с, 1H),7,77-7,79 (м, 2H), 6,18-7,32 (м, 9H), 3,31-3,42 (м, 4H),2,40 (с, 3H), 1,76 (м, 2H).
Чистота согласно HPLC: @214 нм 98,6%, @254 нм 98,9%.
LC-MS: m/z 571 [M+1].
Пример 18
Реакция 1
Соединение 1 (96 мг, 10 ммоль) и соединение 1a (48 мг, 0,25 ммоль) растворяли в ацетонитриле (10 мл), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 100 мг неочищенного продукта.
Реакция 2
Соединение 2 (100 мг) растворяли в метаноле, перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), перемешивали при комнатной температуре в течение 10 мин. Реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением 15 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,76 (с, 1H), 11,45 (с, 1H), 8,68-9,04 (м, 3H), 7,09-7,20 (м, 2H), 6,73-6,77 (м, 1H), 6,33-6,36 (м, 1H), 3,53-3,54 (м, 2H), 3,44-3,46 (м, 2H), 1,18(м, 1H), 1,00-1,02 (м, 2H), 0,90-0,92 (м, 2H).
Чистота согласно HPLC: @214 нм 98,8%, @254 нм 99,8%.
LC-MS: m/z 471 [M+1].
Пример 19
Реакция 1
Соединение 1 (96 мг, 0,25 ммоль) и соединение 1a (48 мг, 0,25 ммоль) растворяли в ацетонитриле (10 мл), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия, и концентрировали с получением 100 мг неочищенного продукта.
Реакция 2
Соединение 2 (100 мг) растворяли в метаноле, перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), перемешивали при комнатной температуре в течение 10 мин. Реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением 12 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,48 (с, 1H), 11,28 (с, 1H), 8,75-8,92 (м, 3H), 7,09-7,20 (м, 2H), 6,73-6,77 (м, 1H), 6,35-6,38 (м, 1H), 3,53-3,54 (м, 2H), 3,44-3,46 (м, 2H), 3,24-3,28 (м, 2H), 2,13-2,20 (м, 3H),1,77-1,98 (м, 2H).
Чистота согласно HPLC: @214 нм 97,9%, @254 нм 98,6%.
LC-MS: m/z 485 [M+1].
Пример 20
Реакция 1
Соединение 1 (500 мг, 5,5 ммоль) растворяли в 3 мл дихлорметана, добавляли гексафторфосфат O-(7-азабензотриазол)-N,N,N',N'-тетраметилурония (128 мг, 0,336 ммоль), циклогексанкарбоновую кислоту (800 мг, 6,25 ммоль) и диизопропилэтиламин (1,16 г, 896 ммоль) в указанной последовательности, осуществляли взаимодействие при комнатной температуре в течение ночи под защитным слоем азота, гасили добавлением воды, полученное вещество экстрагировали этилацетатом (50 мл × 5), жидкие экстракты объединяли, промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и упаривали досуха в условиях пониженного давления с получением 40 мг продукта, который сразу использовали на следующей стадии.
Реакция 2
Соединение 2 (530 мг, 2,65 ммоль) растворяли в ацетонитриле (5 мл), добавляли соединение 2a (850 мг, 5,3 ммоль), перемешивали при комнатной температуре в течение 24 ч и сразу упаривали досуха в условиях пониженного давления с получением 300 мг продукта, который сразу использовали на следующей стадии.
Реакция 3
Соединение 3 (300 мг, 0,99 ммоль) растворяли в дихлорметане (10 мл), перемешивали при комнатной температуре в течение 5 мин, добавляли трифторуксусную кислоту (5 мл), осуществляли взаимодействие при комнатной температуре в течение 2 ч, и сразу упаривали полученное вещество досуха в условиях пониженного давления с получением 200 мг целевого продукта 4, который представлял собой неочищенный продукт и который сразу использовали на следующей стадии.
Реакция 4
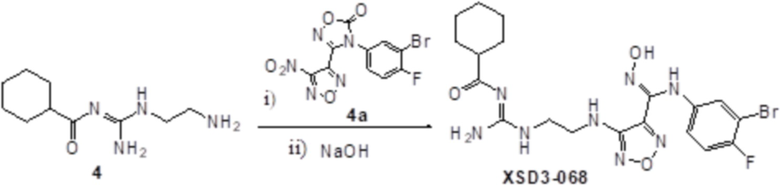
Соединение 4 (200 мг, 0,99 ммоль) растворяли в тетрагидрофуране, добавляли соединение 4a (250 мг, 1,0 ммоль), перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), перемешивали при комнатной температуре в течение 10 мин. Реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением 3 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,90 (с, 1H), 11,55 (с, 1H), 8,67-9,12 (м, 3H), 7,10-7,20 (м, 2H),6,73-6,77 (м, 1H), 6,37 (с, 1H), 3,33-3,54 (м, 4H),2,16-2,21 (м, 1H), 1,56-1,81 (м, 6H), 1,15-1,36 (м, 1H).
Чистота согласно HPLC: @214 нм 97,7%, @254 нм 98,0%.
LC-MS: m/z 511 [M+1].
Пример 21
Реакция 1:
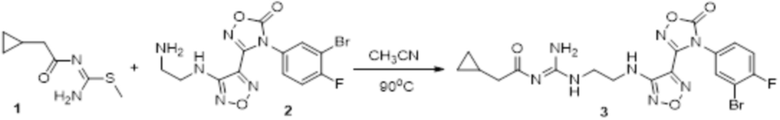
Соединение 1 (35 мг, 0,2 ммоль) и соединение 2 (90 мг, 0,22 ммоль) растворяли в 10 мл ацетонитрила и перемешивали при 90°C в течение 16 ч. Реакционный раствор сразу упаривали досуха в условиях пониженного давления с получением соединения 3, которое представляло собой неочищенный продукт и которое сразу использовали на следующей стадии (100 мг).
Реакция 2:
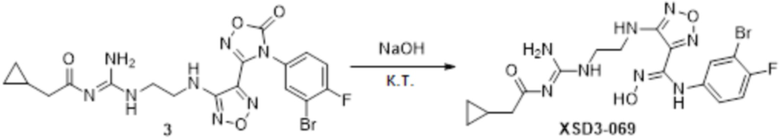
Соединение 3 (100 мг, 0,1 ммоль) растворяли в 1,5н растворе NaOH (2 мл) в THF (5 мл), перемешивали при комнатной температуре в течение 0,5 часа, и подвергали полученное вещество очистке методом препаративной HPLC с получением целевого соединения (30 мг, выход: 30%).
1H-ЯМР (DMSO, 400M): 11,40 (с, 1H), 11,33 (с, 1H), 9,09 (с, 1H), 8,93 (с, 1H), 8.,7 (с, 1H), 7,09-7,20 (м, 2H), 6,73-6,77 (м, 1H), 6,36 (с, 1H), 3,45-3,55 (м, 4H), 2,31-2,33 (м, 2H), 0,95-0,99 (м, 1H), 0,47-0,51 (м, 2H), 0,16-0,20(м, 2H).
Чистота согласно HPLC: @214 нм 98,8%, @254 нм 98,9%.
LC-MS: m/z 483,1 [M+1].
Пример 22
Реакция 1:
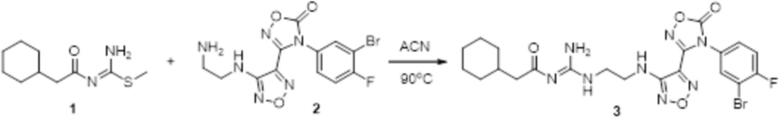
Соединение 1 (44 мг, 0,2 ммоль) и соединение 2 (90 мг, 0,22 ммоль) растворяли в 10 мл ацетонитрила и перемешивали при 90°C в течение 16 ч. Реакционный раствор сразу упаривали досуха в условиях пониженного давления с получением соединения 3, которое представляло собой неочищенный продукт и которое сразу использовали на следующей стадии (100 мг).
Реакция 2:
Соединение 3 (100 мг, 0,1 ммоль) растворяли в 1н растворе NaOH (4 мл) в тетрагидрофуране (5 мл), перемешивали при комнатной температуре в течение 1 часа и подвергали очистке методом препаративной HPLC с получением целевого соединения (35 мг, выход: 35%).
1H-ЯМР (DMSO, 400M): 11,40 (с, 1H), 11,33 (с, 1H), 9,18 (с, 1H), 9,02 (с, 1H), 8,78 (с, 1H, 7,10-7,17 (м, 2H), 6,73-6,76 (м, 1H), 6,35-6,38 (м, 1H), 3,46-3,65 (м, 4H), 2,26-2,38 (м, 2H), 1,47-1,69 (м, 5H), 0,80-1,22 (м, 6H),
Чистота согласно HPLC: @214 нм 96,5%, @254 нм 96,5%.
LC-MS: m/z 527,2 [M+1].
Пример 23
Реакция 1:
Соединение 1 (35 мг, 0,26 ммоль) и соединение 2 (90 мг, 0,22 ммоль) растворяли в 10 мл ацетонитрила и перемешивали при 90°C в течение 20 ч. Реакционный раствор сразу упаривали досуха в условиях пониженного давления с получением соединения 4, которое представляло собой неочищенный продукт и которое сразу использовали на следующей стадии (110 мг).
Реакция 2:
Соединение 3 (100 мг, 0,2 ммоль) растворяли в 1н раствор NaOH (1,5 мл) в тетрагидрофуране (5 мл), перемешивали при комнатной температуре в течение 0,5 часа, и подвергали полученное вещество очистке методом препаративной HPLC с получением целевого соединения (15 мг, выход: 15%).
1H-ЯМР (DMSO, 400M): 12,50 (с, 1H), 11,50 (ушир., 1H), 9,19 (с, 1H), 8,65-8,91 (м, 2H), 7,12-7,21 (м, 2H), 6,73-6,77 (м, 1H), 6,36 (с, 1H), 3,24-3,36 (м, 4H), 1,77-1,84 (м, 3H), 0,90-1,02 (м, 4H).
Чистота согласно HPLC: @214 нм 98,6%, @254 нм 98,3%.
LC-MS: m/z 481,1 [M+1].
Пример 24
Реакция 1:
Соединение 1 (42 мг, 0,2 ммоль) и соединение 2 (90 мг, 0,22 ммоль) растворяли в 10 мл ацетонитрила и перемешивали при 90°C в течение 20 ч. Реакционный раствор сразу упаривали досуха в условиях пониженного давления с получением соединения 4, которое представляло собой неочищенный продукт и которое сразу использовали на следующей стадии (120 мг).
Реакция 2:
Соединение 3 (110 мг, 0,2 ммоль) растворяли в 1н растворе NaOH (5 мл) в тетрагидрофуране (5 мл), перемешивали при комнатной температуре в течение 1 часа, и подвергали полученное вещество очистке методом препаративной HPLC с получением целевого соединения (35 мг, выход: 35%).
1H-ЯМР (DMSO, 400M): 11,44 (с, 1H), 11,35 (с, 1H), 9,06 (с, 1H), 8,92 (с, 1H), 8,64 (с, 3H), 7,16-7,21 (т, 1H),7,09-7,11 (кв, 1H), 6,74-6,78 (м, 1H), 6,26-6,29 (м, 1H), 3,23-3,34 (м, 4H), 2,49-2,50 (м, 1H), 1,61-1,81 (м, 7H), 1,17-1,32 (м, 5H).
Чистота согласно HPLC: @214 нм 99,4%, @254 нм 99,8%.
LC-MS: m/z 527,2 [M+1].
Пример 25
Реакция 1
При одновременном выполнении, в двух сосудах соединение 1 (500 мг, 8,77 ммоль) растворяли в DCM, добавляли соединение 1a (3,3 г, 21,93 ммоль), HATU (4,01 г, 5,52 ммоль) и DIEA (3,71 г, 56,5 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды, полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и подвергали очистке методом колоночной хроматографии с получением 128 мг продукта.
Реакция 2
Соединение 2 (128 мг, 0,743 ммоль) растворяли в ACN, добавляли соединение 2a (300 мг, 0,752 ммоль), перемешивали при 90°C в течение ночи. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия, и концентрировали с получением 300 мг неочищенного продукта.
Реакция 3
Соединение 3 (300 мг) растворяли в THF/H2O, перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), и перемешивали при комнатной температуре в течение 10 мин. После завершения гидролиза согласно данным LCMS, по каплям добавляли 1M HCl до нейтральных значений среды, реакционный раствор экстрагировали этилацетатом, экстракт концентрировали и подвергали очистке методом препаративной HPLC (способ в кислых условиях) с получением 25 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,341 (с, 2H), 8,649-8,923 (м, 4H), 6,274-7,188 (м, 4H), 3,255-3,324 (м, 4H), 1,694-2,067 (м, 9H).
Чистота согласно HPLC: @214 нм 99,05%, @254 нм 98,1%.
LC-MS: m/z 497[M+1].
Пример 26
Реакция 1:
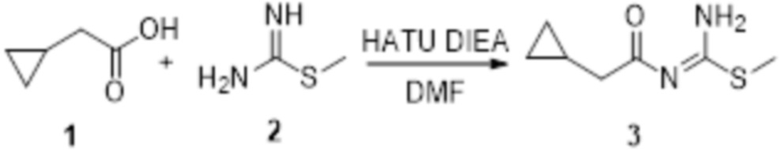
Соединение 1 (1,0 г, 10 ммоль) и соединение 2 (2,7 мг, 30 ммоль) растворяли в 20 мл DMF и перемешивали при комнатной температуре, затем добавляли HATU (3,8 г, 10 ммоль), DIEA (6,5 г, 50 ммоль), и перемешивали в течение 10 ч. Реакционный раствор добавляли к 60 мл воды, экстрагировали этилацетатом, экстракт упаривали досуха в условиях пониженного давления и подвергали очистке методом колоночной хроматографии с получением соединения 3 (0,4 г, выход: 24%).
Реакция 2:
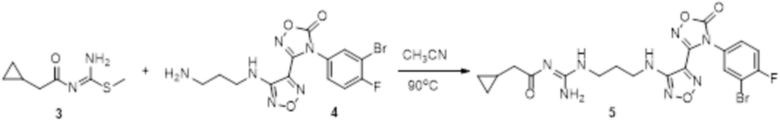
Соединение 3 (35 мг, 0,2 ммоль) и соединение 4 (90 мг, 0,22 ммоль) растворяли в 10 мл ацетонитрила и перемешивали при 90°C в течение 20 ч, реакционный раствор сразу упаривали досуха в условиях пониженного давления с получением соединения 5, которое представляло собой неочищенный продукт и которое сразу использовали на следующей стадии (120 мг).
Реакция 3:
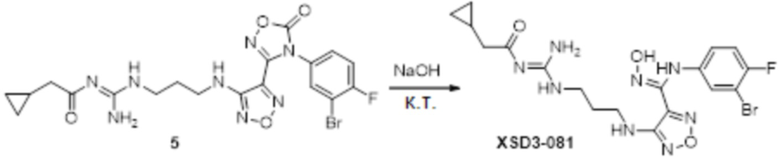
Соединение 3 (110 мг, 0,2 ммоль) растворяли в 1н растворе NaOH (4 мл) в тетрагидрофуране (5 мл), перемешивали при комнатной температуре в течение 1 часа, и подвергали очистке полученное вещество методом препаративной HPLC с получением целевого соединения (15 мг, выход: 15%).
1H-ЯМР (DMSO, 400M): 11,43 (с, 1H), 11,27 (с, 1H), 9,06 (с, 1H), 8,92 (с, 1H), 8,64 (с, 3H), 7,16-7,21 (т, 1H), 7,09-7,11 (кв, 1H), 6,74-6,78 (м, 1H), 6,26-6,29 (м, 1H), 3,23-3,35 (м, 4H), 2,32-2,34 (м, 2H), 1,80-1,84 (м, 2H), 0,95-1,01 (м, 1H), 0,49-0,51 (м, 5H),0,18-0,19 (м, 2H).
Чистота согласно HPLC: @214 нм 96,5%, @254 нм 97,9%.
LC-MS: m/z 497,2 [M+1].
Пример 27
Реакция 1:
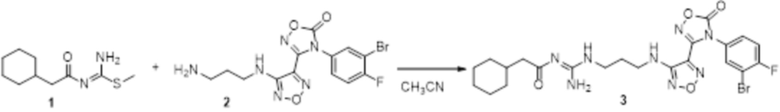
Соединение 1 (45 мг, 0,2 ммоль) и соединение 2 (90 мг, 0,22 ммоль) растворяли в 10 мл ацетонитрила и перемешивали при 90°C в течение 20 ч, реакционный раствор сразу упаривали досуха в условиях пониженного давления с получением соединения 4, которое представляло собой неочищенный продукт и которое сразу использовали на следующей стадии (120 мг, неочищ., выход >99%).
Реакция 2:
Соединение 3 (110 мг, 0,2 ммоль) растворяли в 1н растворе NaOH (4 мл) в тетрагидрофуране (5 мл) и перемешивали при комнатной температуре в течение 1 часа, полученное вещество подвергали очистке методом препаративной HPLC с получением целевого соединения (55 мг, выход = 50%).
1H-ЯМР (DMSO, 400M): 11,53 (с, 1H), 11,44 (с, 1H), 9,13 (с, 1H), 8,92 (с, 1H), 8,67 (с, 1H), 7,16-7,21 (т, 1H), 7,09-7,11 (кв, 1H), 6,74-6,78 (м, 1H), 6,26-6,29 (м, 1H), 3,23-3,34 (м, 4H), 2,27-2,28 (м, 2H), 1,66-1,84 (м, 8H), 0,91-1,17 (м, 5H).
LC-MS: m/z 583,2 [M+1].
Пример 28
Реакция 1
Соединение 1 (106 мг, 0,25 ммоль) растворяли в ацетоне (10 мл), добавляли карбонат калия (138 мг, 1 ммоль), по каплям добавляли соединение 1a (43,5 мг, 0,25 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 100 мг неочищенного продукта.
Реакция 2
Соединение 2 (100 мг) растворяли в метаноле, перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), перемешивали при комнатной температуре в течение 10 мин, реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением 9 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,48 (с, 1H), 11,44 (с, 1H), 8,92-8,95 (м, 3H), 7,27-7,34 (м, 5H), 7,09-7,17 (м, 2H), 6,74-6,76 (м, 1H), 6,32-6,35 (м, 1H), 3,75-3,77 (м, 2H), 3,50-3,56 (м, 2H), 3,44-3,46 (м, 2H).
Чистота согласно HPLC: @214 нм 97,9%, @254 нм 99,2%.
LC-MS: m/z 521 [M+1].
Пример 29
Реакция 1
Соединение 1 (110 мг, 0,25 ммоль) растворяли в ацетоне (10 мл), добавляли карбонат калия (138 мг, 1 ммоль), по каплям добавляли соединение 1a (43,5 мг, 0,25 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 100 мг неочищенного продукта.
Реакция 2
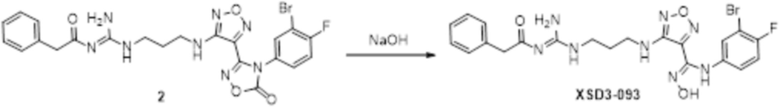
Соединение 2 (100 мг) растворяли в метаноле, перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), перемешивали при комнатной температуре в течение 10 мин, реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением 9 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,64 (с, 1H), 11,42 (с, 1H), 9,03-9,07 (м, 3H), 7,27-7,34 (м, 5H), 7,09-7,17 (м, 2H), 6,74-6,76 (м, 1H), 6,32-6,35 (м, 1H), 3,75-3,77 (м, 2H), 3,14-3,34 (м, 4H), 1,80-1,84 (м, 2H).
Чистота согласно HPLC: @214 нм 96,0%, @254 нм 96,0%.
LC-MS: m/z 535 [M+1].
Пример 30
Реакция 1
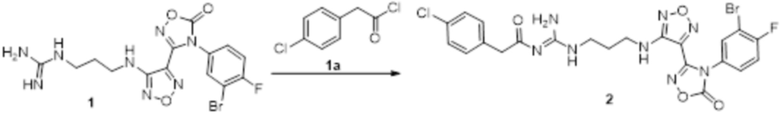
Соединение 1 (330 мг, 0,75 ммоль) растворяли в ацетоне (30 мл), добавляли карбонат калия (414 мг, 3 ммоль), по каплям добавляли соединение 1a (130,5 мг, 0,75 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 300 мг неочищенного продукта.
Реакция 2
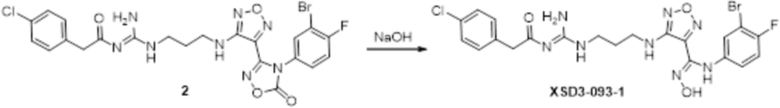
Соединение 2 (300 мг) растворяли в метаноле, перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), перемешивали при комнатной температуре в течение 10 мин, реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением 59 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,64 (с, 1H), 11,42 (с, 1H), 9,03-9,07 (м, 4H), 7,32-7,35 (м, 2H), 7,15-7,32 (м, 3H), 7,08-7,11 (м, 1H) 6,74-6,77 (м, 1H), 6,25-6,28 (м, 1H), 3,75-3,77 (м, 2H), 3,14-3,34 (м, 4H), 1,80-1,84 (м, 2H).
Чистота согласно HPLC: @214 нм 99,90%, @254 нм 99,70%.
LC-MS: m/z 569[M+1].
Пример 31
Реакция 1
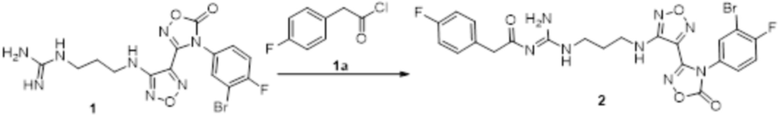
Соединение 1 (330 мг, 0,75 ммоль) растворяли в ацетоне (30 мл), добавляли карбонат калия (414 мг, 3 ммоль), по каплям добавляли соединение 1a (130,5 мг, 0,75 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 300 мг неочищенного продукта.
Реакция 2
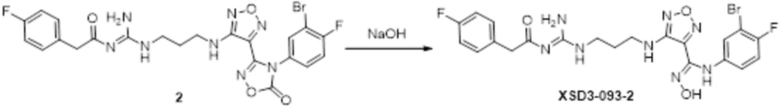
Соединение 2 (300 мг) растворяли в метаноле, перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), перемешивали при комнатной температуре в течение 10 мин, реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением 35 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,64 (с, 1H), 11,42 (с, 1H), 9,03-9,07 (м, 4H), 7,32-7,35 (м, 2H),7,15-7,32 (м, 3H), 7,08-7,11 (м, 1H), 6,74-6,77 (м, 1H), 6,25-6,28 (м, 1H), 3,75-3,77 (м, 2H),3,14-3,34 (м, 4H), 1,80-1,84 (м, 2H).
Чистота согласно HPLC: @214 нм 99,40%, @254 нм 99,25%.
LC-MS: m/z 553[M+1].
Пример 32
Реакция 1
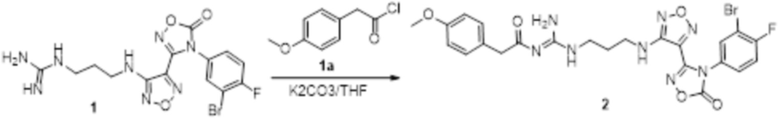
Соединение 1 (330 мг, 0,75 ммоль) растворяли в тетрагидрофуране (10 мл), добавляли карбонат калия (414 мг, 3 ммоль), перемешивали при комнатной температуре в течение 20 мин, по каплям добавляли раствор соединения 1a (184 мг, 1 ммоль) в тетрагидрофуране (1 мл), перемешивали при комнатной температуре в течение 2 ч, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 500 мг неочищенного продукта.
Реакция 2

Соединение 2 (500 мг) растворяли в тетрагидрофуране (20 мл), перемешивали при комнатной температуре в течение 5 мин, по каплям добавляли 1н гидроксид натрия (5 мл), перемешивали при комнатной температуре в течение 30 мин. Реакционный раствор экстрагировали водой и этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт растворяли в ацетонитриле и подвергали очистке методом препаративной HPLC с получением 29 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,91(с, 1H), 11,43 (с, 1H), 9,04 (с, 1H), 8,90 (с, 1H), 8,70 (с, 2H), 7,25-7,23 (м, 2H), 7,20-7,16 (м, 1H), 7,11-7,09 (м, 1H), 6,91-6,88 (м, 2H), 6,78-6,75 (м, 1H), 6,25 (м, 1H), 3,73 (с, 3H), 3,68 (с, 2H), 3,28-3,23 (м, 4H), 1,83-1,80 (м, 2H).
Чистота согласно HPLC: @214 нм 99,2%, @254 нм 99,7%.
LC-MS: m/z 563,2[M+1].
Пример 33
Реакция 1
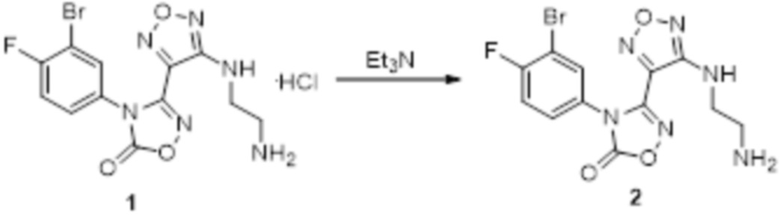
Соединение 1 (150 мг, 0,47 ммоль) растворяли в DCM, добавляли Et3N, перемешивали при комнатной температуре в течение 1 ч, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 100 мг неочищенного продукта.
Реакция 2
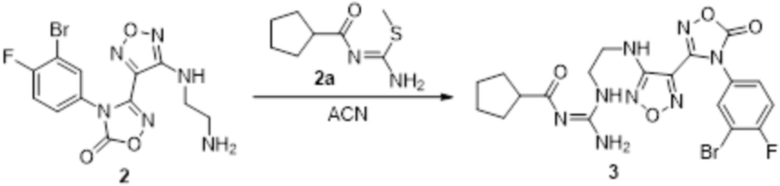
Соединение 2 (100 мг, 1,08 ммоль) растворяли в ACN, добавляли соединение 2a (120 мг, 1,06 ммоль), перемешивали при 90°C в течение ночи. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 100 мг неочищенного продукта.
Реакция 3
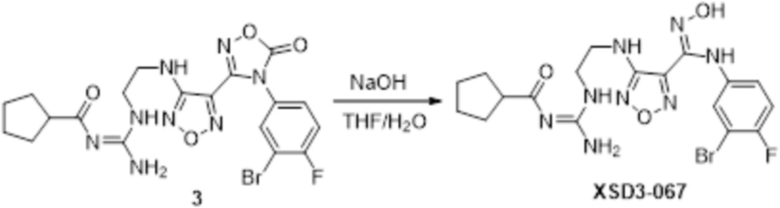
Соединение 3 (100 мг) растворяли в THF/H2O, перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), перемешивали при комнатной температуре в течение 10 мин, реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением 8 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,579 (с, 1H), 11,480 (с, 1H), 8,718-8,917 (м, 3H), 6,286-7,187 (м, 4H), 3,255-3,324 (м, 4H), 2,499-2,507 (м, 1H), 1,570-1,843 (м,8H).
Чистота согласно HPLC: @214 нм 97,3%, @254 нм 98,1%.
LC-MS: m/z 497[M+1].
Пример 34
Реакция 1
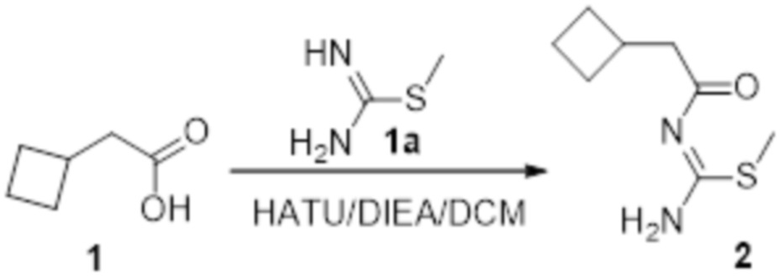
Соединение 1 (500 мг, 8,77 ммоль) растворяли в DCM, добавляли соединение 1a (3,3 г, 21,93 ммоль), HATU (4,01 г, 5,52 ммоль) и DIEA (3,71 г, 56,5 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и подвергали очистке методом колоночной хроматографии с получением 120 мг целевого продукта.
Реакция 2
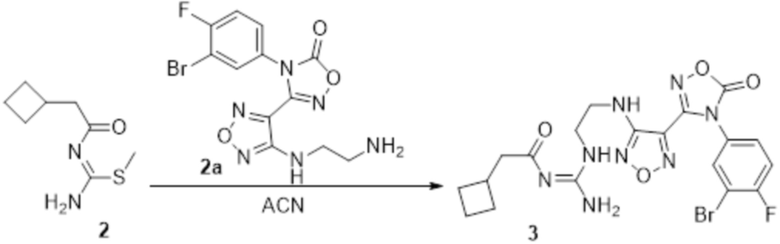
Соединение 2 (120 мг, 1,08 ммоль) растворяли в ACN, добавляли соединение 2a (100 мг, 1,06 ммоль), перемешивали при 90°C в течение ночи. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 100 мг неочищенного продукта.
Реакция 3
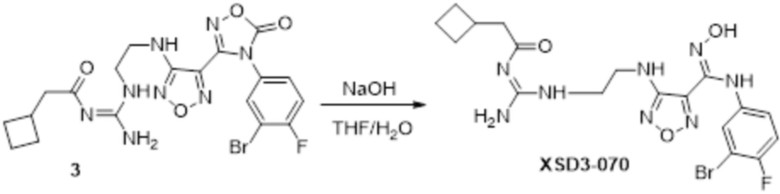
Соединение 3 (100 мг) растворяли в THF/H2O, перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), перемешивали при комнатной температуре в течение 10 мин, реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением 12 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 12,041 (с, 1H), 8,649-8,923 (м, 3H), 6,274-7,188 (м, 4H), 3,255-3,324 (м, 4H), 2,509-2,683 (м, 2H), 1,594-2,067 (м,6H).
Чистота согласно HPLC: @214 нм 95,9%, @254 нм 95,4%.
LC-MS: m/z 497[M+1].
Пример 35
Реакция 1
Соединение 1 (1 г, 8,77 ммоль) растворяли в DCM, добавляли соединение 1a (6,3 г, 21,93 ммоль), HATU (4,01 г, 10,52 ммоль) и DIEA (5,71 г, 56,5 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и подвергали очистке методом колоночной хроматографии с получением 200 мг продукта.
Реакция 2
Соединение 2 (200 мг, 1,08 ммоль) растворяли в ACN, добавляли соединение 2a (420 мг, 1,06 ммоль), перемешивали при 90°C в течение ночи. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 100 мг неочищенного продукта.
Реакция 3
Соединение 3 (100 мг) растворяли в THF/H2O, перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), перемешивали при комнатной температуре в течение 10 мин, реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением 12 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,579 (с, 1H), 11,480 (с, 1H), 8,718-8,917 (м, 3H), 6,286-7,187 (м, 4H), 3,255-3,324 (м, 4H), 2,499-2,507 (м, 1H), 1,570-1,843 (м,10H).
Чистота согласно HPLC: @214 нм 91,2%, @254 нм 96,8%.
LC-MS: m/z 511[M+1].
Пример 36
Реакция 1
Соединение 1 (500 мг, 8,77 ммоль) растворяли в DCM, добавляли соединение 1a (3,3 г, 21,93 ммоль), HATU (4,01 г, 10,52 ммоль) и DIEA (5,71 г, 56,5 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и подвергали очистке методом колоночной хроматографии с получением 120 мг целевого продукта.
Реакция 2
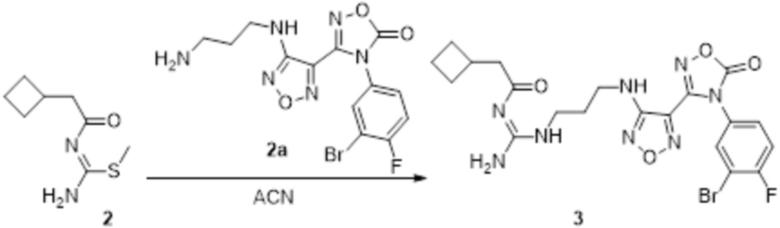
Соединение 2 (120 мг, 1,08 ммоль) растворяли в ACN, добавляли соединение 2a (100 мг, 1,06 ммоль), перемешивали при 90°C в течение ночи. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 100 мг неочищенного продукта.
Реакция 3
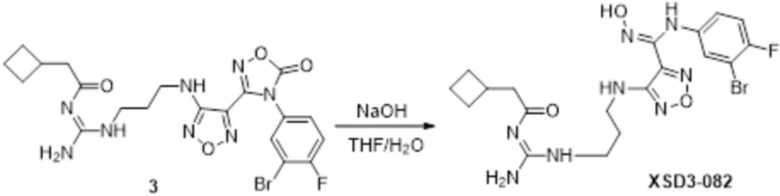
Соединение 3 (100 мг) растворяли в THF/H2O, перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), перемешивали при комнатной температуре в течение 10 мин, реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением 12 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,441 (с, 2H), 8,649-8,923 (м, 3H), 6,274-7,188 (м, 4H), 3,255-3,324 (м, 4H), 1,694-2,067 (м, 11H).
Чистота согласно HPLC: @214 нм 99,1%, @254 нм 99,3%.
LC-MS: m/z 511[M+1].
Пример 37
Реакция 1
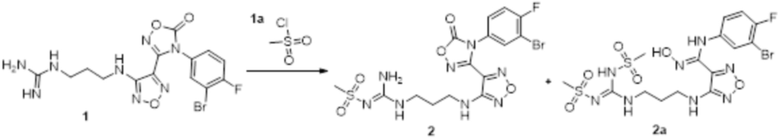
Соединение 1 (110 мг, 0,25 ммоль) растворяли в ацетоне (10 мл), добавляли карбонат калия (138 мг, 1 ммоль), по каплям добавляли соединение 1a (113 мг, 1 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 100 мг неочищенного продукта.
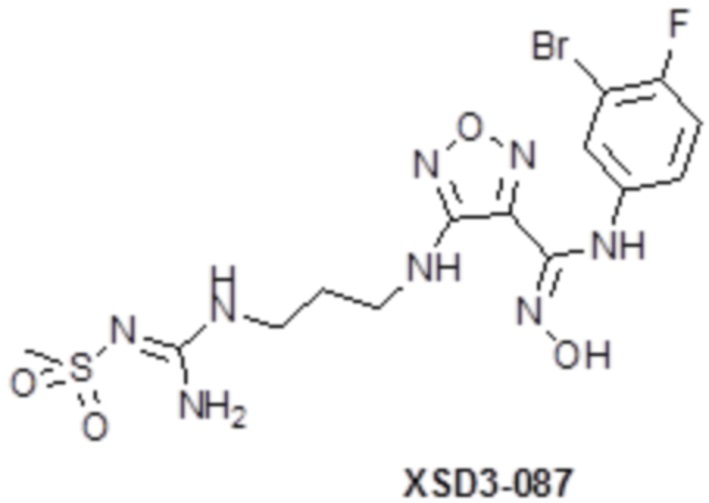
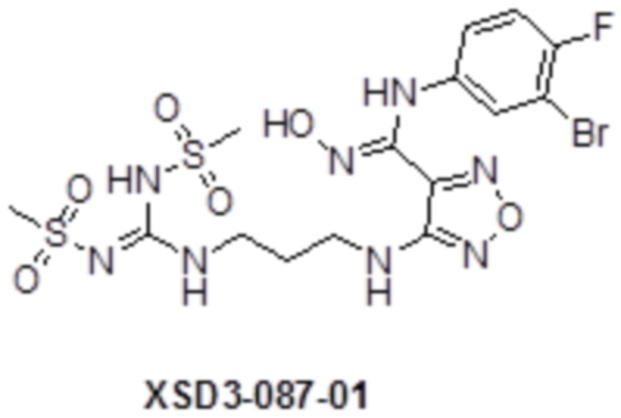
Реакция 2
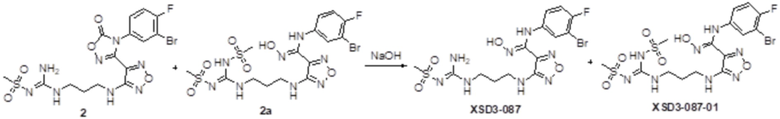
Соединения 2 и 2a (100 мг) растворяли в метаноле, перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), перемешивали при комнатной температуре в течение 10 мин. Реакционный раствор концентрировали и подвергали очистке методом препаративной HPLC с получением целевых продуктов XSD3-087 (15 мг) и XSD3-087-01 (17 мг).
XSD3-087 1H-ЯМР (400 МГц, DMSO): δ (м.д.): 11,41 (с, 1H), 8,90 (м, 1H), 6,18-7,32 (с, 1H), 7,10-7,21 (м, 2H),7,10-7,12 (м, 2H), 6,76-6,79 (м, 1H), 6,23 (м, 1H), 3,12-3,23 (м, 4H), 6,76-6,79 (м, 1H), 2,67 (с, 3H), 1,72-1,75 (м, 2H).
Чистота согласно HPLC: @214 нм 93,5%, @254 нм 94,6%.
LC-MS: m/z 492 [M+1].
XSD3-087-01 1H-ЯМР (400 МГц,DMSO): δ (м.д.): 11,43 (с, 1H), 8,90(м, 1H), 8,01 (м, 2H), 7,10-7,20 (с, 2H), 6,85 (м, 1H), 6,23 (м, 1H), 3,72-3,76 (м, 2H), 3,40 (с, 3H), 3,24-3,26 (м, 2H), 2,93 (с, 3H), 1,87-1,91 (м, 2H).
Чистота согласно HPLC: @214 нм 98%, @254 нм 98,6%.
LC-MS: m/z 570 [M+3].
Пример 38
Реакция 1
Соединение 1 (100 мг, 0,21 ммоль) растворяли в ацетоне (10 мл), добавляли карбонат калия (78 мг, 0,55 ммоль), по каплям добавляли соединение 1a (41 мг, 0,23 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 100 мг неочищенного продукта.
Реакция 2
Соединение 2 (100 мг) растворяли в THF/H2O, перемешивали при комнатной температуре в течение 10 мин, по каплям добавляли 1н гидроксид натрия (1 мл), перемешивали при комнатной температуре в течение 10 мин. Реакционный раствор концентрировали и подвергали препаративной HPLC с получением 5 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,421 (с, 1H), 8,897 (с, 1H), 7,153-7,197 (м, 2H), 7,105-7,120 (м, 1H), 6,746-7,098 (м, 1H), 6,220 (с, 1H), 4,078 (с, 2H), 3,104-3,129 (м, 4H), 2,735-2,761 (м, 2H), 1,535-1,853 (м, 7H), 0,976-1,202 (м, 6H).
Чистота согласно HPLC: @214 нм 99,8%, @254 нм 99,3%.
LC-MS: m/z 575[M+1].
Пример 39
Реакция 1
Соединение 1 (1,0 г, 3,6 ммоль) растворяли в ацетоне (16 мл), добавляли карбонат калия (993 мг, 7,2 ммоль), по каплям добавляли соединение 1a (254 мг, 2,7 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 300 мг неочищенного продукта.
Реакция 2
Соединение 2a (100 мг) растворяли в ацетонитриле, добавляли соединение 2 (40 мг, 0,27 ммоль), перемешивали при 90°C в течение 12 ч, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 80 мг неочищенного продукта.
Реакция 3
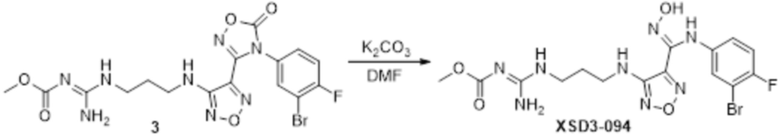
Соединение 3 (80 мг) растворяли в DMF, перемешивали при комнатной температуре в течение 10 мин, добавляли карбонат калия (132 мг, 0,96 ммоль), перемешивали при комнатной температуре в течение 12 ч, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия, концентрировали с получением 60 мг неочищенного продукта, который подвергали очистке методом препаративной HPLC с получением 14,2 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,53 (с, 1H), 8,91 (с, 1H), 8,58-8,27 (м, 3H), 7,38-7,12 (м, 2H), 6,78 (с, 1H), 6,28 (с, 1H), 3,65 (с, 3H), 3,28-3,24 (м, 4H), 1,79 (с, 2H).
Чистота согласно HPLC: @214 нм 94,5%, @254 нм 95,4%.
LC-MS: m/z 473 [M+1].
Пример 40
Реакция 1
Соединение 1 (1,0 г, 3,6 ммоль) растворяли в ацетоне (16 мл), добавляли карбонат калия (993 мг, 7,2 ммоль), по каплям добавляли соединение 1a (294 мг, 2,7 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 400 мг неочищенного продукта.
Реакция 2
Соединение 2a (100 мг) растворяли в ацетонитриле, добавляли соединение 2 (42 мг, 0,27 ммоль), перемешивали при 90°C в течение 12 ч, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 66 мг неочищенного продукта.
Реакция 3
Соединение 3 (66 мг) растворяли в DMF, перемешивали при комнатной температуре в течение 10 мин, добавляли карбонат калия (108 мг, 0,78 ммоль), перемешивали при комнатной температуре в течение 12 ч, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 50 мг неочищенного продукта, который подвергали очистке методом препаративной HPLC с получением 26,5 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,47 (с, 1H), 11,24 (с, 1H), 8,92 (с, 1H), 8,62 (с, 2H), 7,21-7,09 (м, 2H), 6,79-6,75 (м, 1H), 6,29-6,25 (м, 1H), 4,25-4,20 (м, 2H), 3,32-3,24 (м, 4H), 1,85-1,80 (м, 2H), 1,28-1,24 (м, 3H).
Чистота согласно HPLC: @214 нм 97,8%, @254 нм 98,6%.
LC-MS: m/z 487 [M+1].
Пример 41
Реакция 1
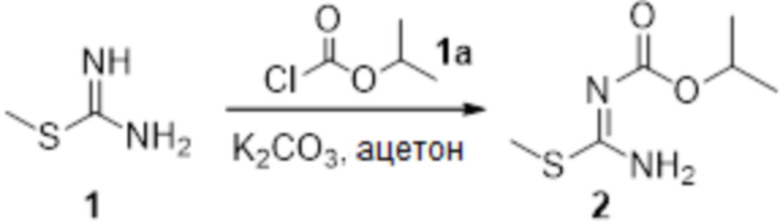
Соединение 1 (1,0 г, 3,6 ммоль) растворяли в ацетоне (16 мл), добавляли карбонат калия (993 мг, 7,2 ммоль), по каплям добавляли соединение 1a (732 мг, 6,0 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 360 мг неочищенного продукта.
Реакция 2
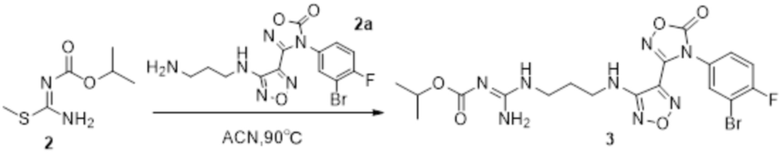
Соединение 2a (100 мг) растворяли в ацетонитриле, добавляли соединение 2 (45 мг, 0,27 ммоль), перемешивали при 90°C в течение 12 ч, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 51 мг неочищенного продукта.
Реакция 3
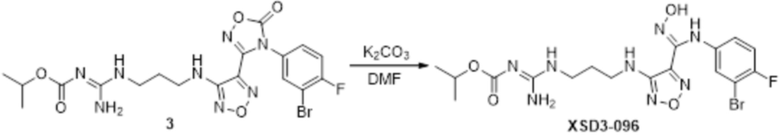
Соединение 3 (51 мг) растворяли в DMF, перемешивали при комнатной температуре в течение 10 мин, добавляли карбонат калия (108 мг, 0,78 ммоль), перемешивали при комнатной температуре в течение 12 ч, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 50 мг неочищенного продукта, который подвергали очистке методом препаративной HPLC с получением 10,6 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,48 (с, 1H), 11,18 (с, 1H), 8,92 (с, 1H), 8,60 (с, 2H), 7,21-7,09 (м, 2H), 6,79-6,75 (м, 1H), 6,30-6,25 (м, 1H), 4,98-4,91 (м, 1H), 3,32-3,24 (м, 4H), 1,83-1,78 (м, 2H), 1,28-1,27 (м, 6H).
Чистота согласно HPLC: @214 нм 95,2%, @254 нм 98,0%.
LC-MS: m/z 501 [M+1].
Пример 42
Реакция 1
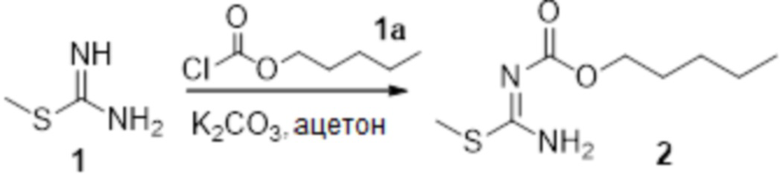
Соединение 1 (1,0 г, 3,6 ммоль) растворяли в ацетоне (16 мл), добавляли карбонат калия (993 мг, 7,2 ммоль), по каплям добавляли соединение 1a (540 мг, 3,6 ммоль), перемешивали при комнатной температуре в течение ночи, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 330 мг неочищенного продукта.
Реакция 2

Соединение 2a (100 мг) растворяли в ацетонитриле, добавляли соединение 2 (50 мг, 0,27 ммоль), перемешивали при 90°C в течение 12 ч, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали с получением 90 мг неочищенного продукта.
Реакция 3
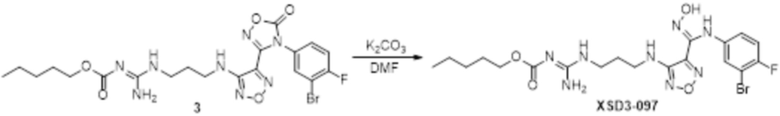
Соединение 3 (90 мг) растворяли в DMF, перемешивали при комнатной температуре в течение 10 мин, добавляли карбонат калия (132 мг, 0,96 ммоль), перемешивали при комнатной температуре в течение 12 ч, гасили добавлением воды. Полученное вещество экстрагировали этилацетатом, сушили над безводным сульфатом натрия, и концентрировали с получением 70 мг неочищенного продукта, который подвергали очистке методом препаративной HPLC с получением 32,5 мг целевого продукта.
1H-ЯМР (400 МГц, DMSO-d6): δ (м.д.): 11,51 (с, 1H), 11,24 (с, 1H), 8,91 (с, 1H), 8,55-8,27 (м, 3H), 7,21-7,09 (м, 2H), 6,79-6,75 (м, 1H), 6,29-6,25 (м, 1H), 4,25-4,20 (м, 2H), 3,32-3,24 (м, 4H), 1,80 (с, 2H), 1,57 (с, 2H), 1,28 (с, 4H), 0,88-0,85 (м, 3H).
1H-ЯМР (400 МГц, CDCl3): δ (м.д.): 7,23-7,20 (м, 1H), 7,10 (с, 1H), 7,04-7,00 (м, 1H), 6,92-6,88 (м, 1H), 5,85 (с, 1H), 4,09-4,06 (м, 2H), 3,58-3,53 (м, 2H), 3,38-3,35 (м, 2H), 1,98-1,97 (м, 2H), 1,70-1,63 (м, 2H), 1,38-1,35 (м, 4H), 0,91-0,88 (м, 3H).
Чистота согласно HPLC: @214 нм 98,1%, @254 нм 97,8%.
LC-MS: m/z 529[M+1]
Опираясь на способ синтеза I и описанные выше Примеры, синтезировали следующие соединения.
 , n равен 2, R1, R3, R4 представляют собой H.
, n равен 2, R1, R3, R4 представляют собой H. , n равен 1, R1, R3, R4 представляют собой H.
, n равен 1, R1, R3, R4 представляют собой H. , n равен 2, R1, R3, R4 представляют собой H., n равен 2, R1, R3, R4 представляют собой H.
, n равен 2, R1, R3, R4 представляют собой H., n равен 2, R1, R3, R4 представляют собой H. , n равен 1, R1, R3, R4 представляют собой H.
, n равен 1, R1, R3, R4 представляют собой H.
Пример 55: Определение ингибирующей активности в отношении индоламин-2,3-диоксигеназы и IC50
Конструирование плазмиды, содержащей ген индоламин-2,3-диоксигеназы человека, ее экспрессирование в E. coli, экстрагирование и очистку проводили в соответствии со способом, описанным в Littlejohn et al. (Takikawa O, Kuroiwa T, Yamazaki F, et al. J. Biol. Chem. 1988, 263, 2041-2048). В 96-луночном планшете перемешивали 50 мМ буферный раствор фосфата калия (рН 6,5), 20 мМ аскорбат, 20 мкМ метиленовый синий и очищенный белок индоламин-2,3-диоксигеназы человека, и добавляли к смеси 200 мкМ L-триптофан и ингибитор. Реакцию проводили при 37°C в течение 60 минут, затем реакцию останавливали посредством добавления 30% трихлоруксусной кислоты, смесь инкубировали при 65°C в течение 15 минут для гидролиза N-формилкинуренина до кинуренина, и центрифугировали при 3400 об/мин в течение 5 минут для удаления выпавшего в осадок белка. Супернатант помещали в новый 96-луночный планшет, добавляли раствор 2% (мас./об) пара-диметиламинобензальдегида в уксусной кислоте, осуществляли взаимодействие путем инкубирования при 25°C в течение 10 минут, и проводили считывание на спектрофотометре при 480 нм. Контрольные лунки не содержали ингибитор индоламин-2,3-диоксигеназы или саму индоламин-2,3-диоксигеназу, и их использовали в качестве необходимых нелинейных параметров регрессии для определения IC50 для каждого соединения. Нелинейную регрессию и определение значений IC50 проводили с использованием программного обеспечения GraphPad Prism 4. Те соединения, значения IC50 для которых были менее чем 10 мкМ, рассматривались как эффективные ингибиторы в данном методе анализе. Соединения Примеров согласно настоящему изобретению обладали превосходной ингибирующей активностью в отношении индоламин-2,3-диоксигеназы.
Таблица 1: Значения IC50 соединений
Примечание: INCB024360 представлял собой ингибитор индоламин-2,3-диоксигеназы следующего строения:
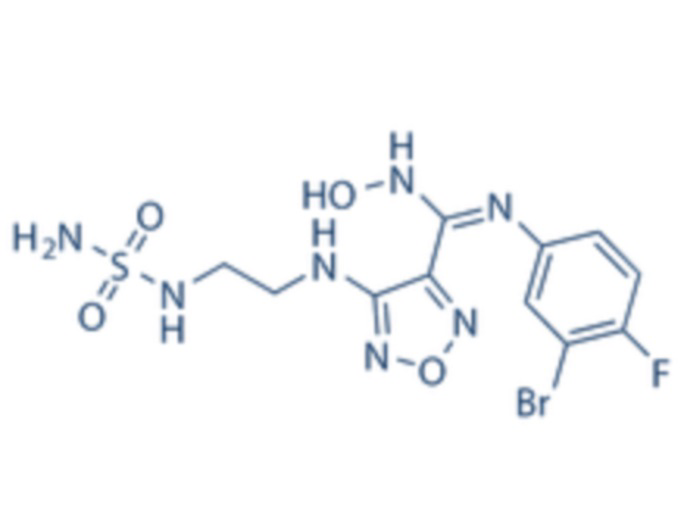 .
.
Значения IC50 для следующих соединений определяли способом, описанным в Примере 55, и конкретные результаты представлены в Таблице 2:
Таблица 2: Значения IC50 соединений
Пример 56: Тест на противоопухолевую активность ингибиторов индоламин-2,3-диоксигеназы in vivo
1. Деление животных на группы и способ тестирования
Были взяты клетки LLC в фазе логарифмического роста, жизнеспособность клеток определяли способом окрашивания трипановым синим, концентрацию жизнеспособных клеток доводили до 1×107 клеток/мл, и вводили гомологичным мышам C57BL6 путем подкожной инъекции в дозе 0,2 мл/мышь. После формирования опухолей мышей случайным образом разделяли на контрольную группу, группу с введением циклофосфамида (CTX), группу с введением соединения INCB024360, группу с введением соединения 3047, в соответствии с массой их опухолей и массой тела; в каждой группе было 10 мышей, причем в группе с введением CTX препарат вводили путем интраперитонеальной инъекции в дозе 150 мг/кг, группу с введением соединения INCB024360 и группу с введением соединения 3047 подвергали интрагастральному введению, контрольная группа получала такой же объем нормального солевого раствора в то же самое время, и частота введения для каждой группы составляла однократно в сутки. Тест завершали после 21 суток введения.
Спустя 24 часа после последнего введения, животных взвешивали и умерщвляли, извлекали опухоли и взвешивали их, и рассчитывали среднюю степень ингибирования опухоли (I) в соответствии со следующей формулой: I=(1 - средняя масса опухоли в группе с введением лекарства/средняя масса опухоли в контрольной группе) × 100%
2. Статистические данные и способ обработки
Экспериментальные данные анализировали посредством spss16.0 и однофакторного дисперсионного анализа, и различие с p<0,05 считали статистически значимым.
3. Результаты теста и обсуждение
Таблица 3: Результаты ингибирования соединениями опухолей LLC у мышей
В сравнении с контрольной группой, ##P<0,01;
В сравнении с группой с введением CTX и группой с введением INCB024360, #P<0,05;
Как видно из Таблицы 3, масса опухоли в каждой группе с введением лекарства существенно отличалась от таковой в контрольной группе (P<0,01); группа с введением соединения 3047 существенно отличалась от группы с введением циклофосфамида и группы с введением INCB024360 (P<0,05). Этот результат указывает на то, что соединение согласно настоящему изобретению превосходит существующие химиотерапевтические лекарства циклофосфамид и соединение INCB024360 по терапевтическому эффекту в отношении опухолей.
Таблица 4: Эффекты соединений на массу тела мышей
В сравнении с группой с введением циклофосфамида, ##P<0,01
Как видно из Таблицы 4, группа с введением соединения 3047 не продемонстрировала существенной разницы по массе тела в сравнении с контрольной группой, но показала значительное отличие в сравнении с группой с введением CTX. Этот результат указывает на то, что соединение согласно настоящему изобретению увеличивает массу тела у мышей при контроле роста опухоли, то есть побочный эффект был снижен, и качество жизни мышей было существенно улучшено. Клинически это означает, что качество жизни пациентов может быть улучшено, и комплаентность пациента к лекарственным средствам и эффективность лекарств могут быть существенно усилены.
Кроме того, мы также тестировали клетки Colon26 злокачественной опухоли толстого кишечника мышей, клетки Hepa1-6 злокачественной опухоли печени мышей, клетки 4T1 злокачественной опухоли молочной железы мышей и другие линии клеток, и результаты продемонстрировали, что соединения согласно настоящему изобретению обладают значительными ингибирующими эффектами в отношении указанных опухолей.
Противоопухолевую активность следующих соединений in vivo определяли способом Примера 56, и конкретные результаты представлены в следующей таблице:
Таблица 5: Результаты ингибирования соединением опухолей LCC у мышей
В сравнении с контрольной группой, ##P<0,01.
Как видно из Таблицы 5, масса опухоли в каждой группе с введением лекарства существенно отличалась от таковой для контрольной группы (P<0,01). Такой результат указывает на то, что соединение согласно настоящему изобретению обладает значимым терапевтическим эффектом в отношении опухоли.
Был протестирован эффект каждого соединения на массу тела мыши. Существенной разницы по массе тела между группой с введением соединения XSD3-058 и группой с введением соединения XSD3-079 в сравнении с контрольной группой обнаружено не было. Такой результат указывает на то, что соединения согласно настоящему изобретению могут увеличивать массу тела у мышей, регулируя рост опухоли, снижать побочные эффекты лекарства и значительно улучшать качество жизни мыши. Клинически, соединения согласно настоящему изобретению могут улучшать качество жизни пациентов и значительно усиливать комплаентность пациента к лекарственным средствам и эффективность лекарств.
Пример 57: Определение поведенческих изменений в тесте водного лабиринта Морриса у мышей с болезнью Альцгеймера
1. Деление животных на группы и способы тестирования
Согласно настоящему изобретению, для формирования моделей AD в соответствии со способом Richardson et al. с однократной инъекцией агрегированного Aβ1-42 в билатеральную область CA3 гиппокампа отбирали мышей в возрасте 9 месяцев, затем разделяли их на контрольную группу, группу с введением соединения INCB024360, группу с введением соединения 3047; всего 10 мышей на группу, пять самцов и пять самок. Анализ поведения мыши проводили с использованием водного лабиринта Морриса (программное обеспечение для мониторинга и анализа Ethovision XT, система водного лабиринта Морриса производства Noldus, Netherlands). Процедуру тестирования в водном лабиринте делили на две части, тест на обнаружение скрытой платформы в течение 5 последующих суток, и пространственный тест на 6-е сутки, причем перед каждым тестом мышам осуществляли введения в соответствии с тестируемыми группами и разработанными дозами. Мышей тренировали 4 раза в сутки, каждый раз погружая мышей в разные области, водный лабиринт был разделен на области 1, 2, 3 и 4 в соответствии с югом, востоком, севером и западом, и платформа представляла собой область 5, расположенную в области 4. Каждое время плавания составляло 60 с, интервал для каждой тренировки составлял приблизительно 1 час, и если мышь не находила платформу, то латентный период считали равным 60 с. Тест на обнаружение скрытой платформы использовали для определения способности мышей к обучению, тогда как пространственный тест использовали для определения у мышей способности к пространственной памяти.
2. Статистические данные и способ обработки
Латентный период спасения в тесте на обнаружение скрытой платформы анализировали посредством дисперсионного анализа множества измерений с использованием программного обеспечения для статистического анализа SPSS16.0; и анализировали время плавания в каждом квадранте и количество пересечений целью в пространственном тесте посредством однофакторного дисперсионного анализа. Данные выражали в виде среднего значения ± стандартное отклонение, и значимый уровень различия в двустороннем тесте задавали как P=0,05.
3. Результаты теста и обсуждение
Таблица 6: Латентный(ые) период(ы) каждой группы животных по поиску платформы в тесте со скрытой платформой
В сравнении с контрольной группой, #P<0,05, ##P<0,01,
В сравнении с группой с введением INCB024360, #P<0,05, ##P<0,01.
Таблица 7: Время и количество остановок в области платформы для каждой группы животных
В сравнении с контрольной группой, #P<0,05, ##P<0,01,
В сравнении с группой с введением INCB024360, #P<0,05.
Как может быть видно из Таблиц 6 и 7, соединение 3047 может значительно улучшать нарушение обучаемости и памяти у животных, значительно улучшать способность к приобретению знаний и способность к пространственной памяти, и превосходит соединение INCB024360, указывая на то, что разработка соединения согласно настоящему изобретению имеет огромное значение при лечении синдрома Альцгеймера.
Эффект следующих соединений на поведение мышей с болезнью Альцгеймера тестировали посредством способа Примера 57. Конкретные результаты представлены в следующей таблице:
Таблица 8: Латентный(ые) период(ы) по поиску платформы в тесте со скрытой платформой для каждой группы животных
В сравнении с контрольной группой, #P<0,05, ##P<0,01.
Таблица 9: Время и количество остановок в области платформы для каждой группы животных
В сравнении с контрольной группой, ##P<0,01.
Как может быть видно из Таблиц 8 и 9, соединения согласно настоящему изобретению могут значительно улучшать нарушение обучаемости и памяти, и значительно улучшать способность к приобретению знаний и способность к пространственной памяти, и результаты указывают на то, что разработка соединения согласно настоящему изобретению имеет огромное значение при лечении синдрома Альцгеймера.
Пример 58: Стимулированный DC Т-клеточный пролиферативный ответ после лечения лекарством
Дендритная клетка (DC) представляет собой наиболее мощную антигенпрезентирующую клетку (APC), которая может эффективно активировать интактные Т-клетки для пролиферации, что является наиболее важным различием между DC и другими APC. DC является инициатором иммунного ответа. DC стала одной из самых актуальных точек приложения современных иммунологических исследований по причине ее ключевой роли в CD4+, CD8+ T-клеточном иммунном ответе. На сегодняшний день, исследование DC в основном сфокусировано на профилактическом и лечебном эффекте при опухолевых заболеваниях, аутоиммунных заболеваниях, реакции отторжения трансплантата и борьбе с инфекцией.
1. Выделение и культивирование дендритных клеток периферической крови человека
Лейкоцитарный слой периферической крови человека извлекали и разбавляли тем же объемом 0,01 моль/л PBS. PBMC выделяли стандартным образом с помощью лимфоцитарной сепарационной среды, и корректировали концентрацию клеток до 3×106 мл-1 в полной среде RPMI1640; клетки вносили в 6-луночный планшет по 3 мл на лунку культивировали в инкубаторе при 5% CO2, 37°C в течение 2 часов, 3 раза промывали PBS для удаления неадгезирующих клеток, затем добавляли культуральную среду, содержащую IL-4 (100 ед/мл), GM-CSF (150 нг/мл) и TNF-α (500 ед/мл), а затем культивировали стандартным образом, обновляя среду наполовину через каждые сутки. После 8 суток культивирования, клетки использовали для идентификации и эксперимента.
2. Приготовление Т-клеток
Слой PBMC человека отделяли посредством способа, описанного на этапе 1. Макрофаги удаляли адгезивным способом, В-клетки удаляли при помощи шприца с нейлоновой ватой, и корректировали полученные Т-клетки до концентрации клеток 1×106 клеток/мл.
3. Приготовление DC
Зрелые DC с чистотой в 99% центрифугировали, добавляли RPMI1640 с получением концентраций клеток 1×105, 4×104, 2×104/мл, и вносили в 96-луночный планшет в количестве 100 мкл на лунку, по две лунки на каждую концентрацию. По отдельности добавляли соединение INCB024360 и соединение 3047, и культивировали в течение 2 суток.
4. Метод анализа пролиферации Т-клеток (MLR)
В каждой из описанных выше групп с введением лекарства к DC добавляли Т-клетки в количестве 100 мкл на лунку, культивировали в инкубаторе при 5% CO2, 37°C в течение 72 часов; за 6 часов до окончания культивирования в каждой лунке аккуратно удаляли 100 мкл культурального раствора, добавляли 10 мкл MTT (5 мг/мл), затем культивировали в инкубаторе в течение 6 часов, далее добавляли 100 мкл 0,01 моль/л HCl-10% SDS, оставляли на ночь при 37°C, и определяли оптическую плотность (A570 нм) в микропланшете, чтобы продемонстрировать уровень пролиферации Т-клеток.
5. Результаты эксперимента и анализ
Таблица 10: Эффект соединения 3047 на стимулированную DC пролиферацию Т-клеток
В сравнении с контрольной группой, #P<0,05, ##P<0,01,
В сравнении с INCB024360 группой, #P<0,05.
Как может быть видно из Таблицы 10, по сравнению с контрольной группой, количество Т-клеток в группе с введением соединения 3047 и группе с введением INCB024360 было существенно увеличено, демонстрируя значимые различия (#P<0,05, ##P<0,01); и по сравнению с группой с введением соединения INCB024360, группа с введением соединения 3047 имела более значительный эффект на пролиферацию Т-клеток, демонстрируя значимое различие (#P<0,05). Это указывает на то, что соединение согласно настоящему изобретению обладает значительным эффектом усиления стимулированной DC пролиферации Т-клеток, и этот эффект существенно лучше, чем таковой для соединения INCB024360, а потому может быть использовано для лечения опухолевых заболеваний, аутоиммунных заболеваний, реакции отторжения трансплантата и инфекционных заболеваний.
Стимулированные DC Т-клеточные пролиферативные ответы после лечения следующими соединениями определяли способом Примера 58, и конкретные результаты представлены в следующей таблице:
Таблица 11: Эффект каждого соединения на стимулированную DC пролиферацию Т-клеток
В сравнении с контрольной группой, ##P<0,01.
Как может быть видно из Таблицы 11, по сравнению с контрольной группой, количество Т-клеток в каждой группе с введением соединения было существенно увеличено, демонстрируя значимое различие (##P<0,01), указывая на то, что соединение согласно настоящему изобретению может значительно усиливать стимулированную DC пролиферацию Т-клеток, а потому может быть использовано для лечения заболеваний, связанных с IDO, таких как опухолевые заболевания, аутоиммунные заболевания, реакция отторжения трансплантата и инфекционные заболевания.
| название | год | авторы | номер документа |
|---|---|---|---|
| Регулятор на основе азотсодержащего гетероциклического производного, способ его получения и его применение | 2020 |
|
RU2829553C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ДЕЙСТВУЮЩИЕ НА БЕЛКИ CRBN | 2019 |
|
RU2801068C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ИММУНОСУПРЕССИИ, ОПОСРЕДУЕМОЙ МЕТАБОЛИЗИРОВАНИЕМ ТРИПТОФАНА | 2014 |
|
RU2667509C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ СУЛЬФОНАМИДА, ОБЛАДАЮЩИЕ СЕЛЕКТИВНОЙ NOX-ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ | 2019 |
|
RU2809030C2 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ДИГИДРОНАФТИРИДИНОНА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ, И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2809869C1 |
| ЗАМЕЩЕННЫЕ ДИГИДРОПИРАЗОЛОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ HIF-ПРОЛИЛ-4-ГИДРОКСИЛАЗЫ | 2009 |
|
RU2509080C9 |
| ХИНОЛИНИЛ-СОДЕРЖАЩЕЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2803116C2 |
| АРИЛ-ИЛИ ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2632193C2 |
| ФОСФОНООКСИМЕТИЛОВЫЕ ЭФИРЫ ТАКСАНОВЫХ ПРОИЗВОДНЫХ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ПРОТИВООПУХОЛЕВАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ РОСТА ОПУХОЛИ У МЛЕКОПИТАЮЩИХ | 1993 |
|
RU2136673C1 |
| ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ И 7-ДЕАЗАПУРИНОВЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2606514C2 |
Изобретение относится к соединению, представленному формулой I, или его фармацевтически приемлемой соли, которые могут найти применение для лечения заболеваний, характеризующихся патологическим путем метаболизма триптофана, опосредованным индоламин–2,3–диоксигеназой. В формуле I R1 выбирают из группы, состоящей из H, C1–6алкила и C1-6алкил-C(O)-; n равен целому числу, выбранному из 0–6. Изобретение относится также к конкретным соединениям, способам синтеза соединений, представленных формулами II и II’, фармацевтической композиции для ингибирования индоламин–2,3–диоксигеназы, содержащей предлагаемые соединения, и способу лечения заболевания, характеризующегося патологическим путем метаболизма триптофана, опосредованным индоламин–2,3–диоксигеназой. 6 н. и 7 з.п. ф-лы, 11 табл., 58 пр.
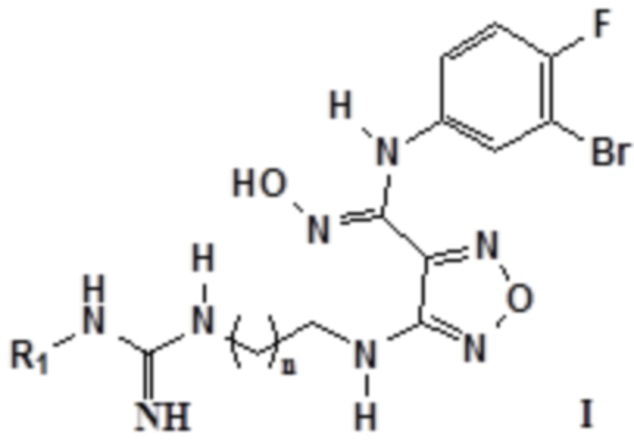
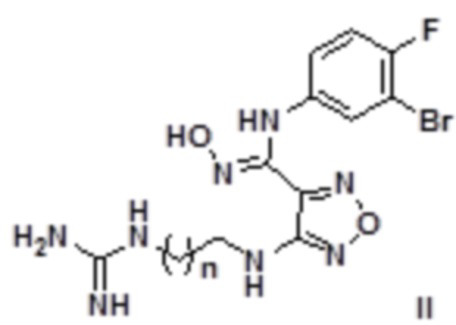
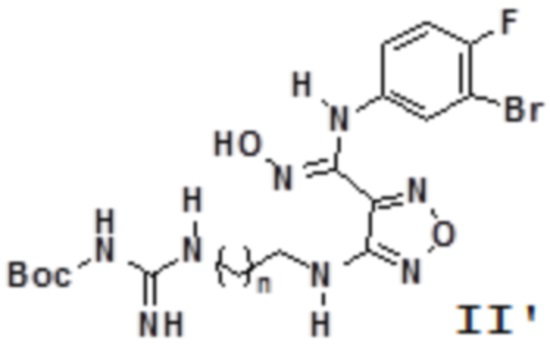
1. Соединение, представленное формулой I, или его фармацевтически приемлемая соль
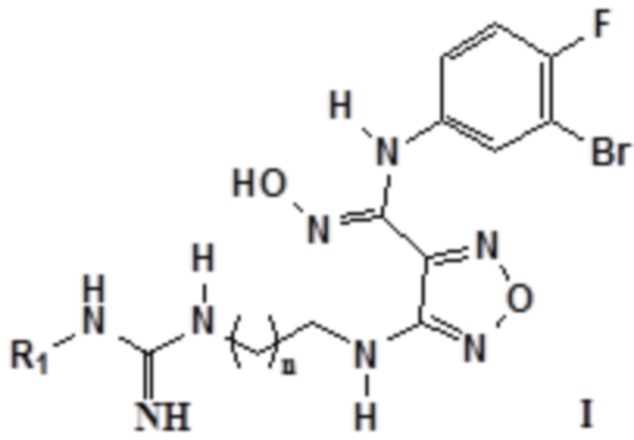 ,
,
где R1 выбирают из группы, состоящей из H, C1–6алкила и C1-6алкил-C(O)-; n равно целому числу, выбранному из 0–6.
2. Соединение или его фармацевтически приемлемая соль по п. 1, отличающееся тем, что R1 выбирают из группы, состоящей из H и C1-6алкила.
3. Соединение или его фармацевтически приемлемая соль по п. 1, отличающееся тем, что R1 выбирают из группы, состоящей из H, метила, этила, пропила, изопропила, бутила.
4. Соединение или его фармацевтически приемлемая соль по п. 1, отличающееся тем, что n выбирают из 0, 1, 2, 3 и 4.
5. Соединение или его фармацевтически приемлемая соль по п. 1, отличающееся тем, что если R1 в формуле I представляет собой H, то соединение представлено формулой II
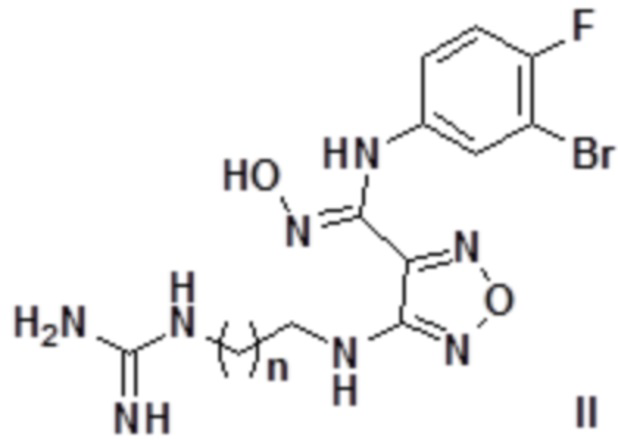 .
.
6. Соединение или его фармацевтически приемлемая соль по п. 5, отличающееся тем, что n равно 0, 1, 2 или 3.
7. Соединение или его фармацевтически приемлемая соль по п. 1, отличающееся тем, что если R1 в формуле I представляет собой R0–замещенный карбонил, то соединение представлено формулой III
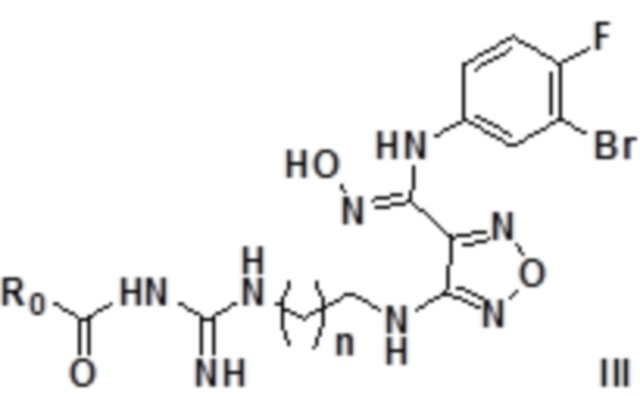 ,
,
где R0 представляет собой C1–6алкил.
8. Соединение или его фармацевтически приемлемая соль, где соединение выбирают из группы, состоящей из:
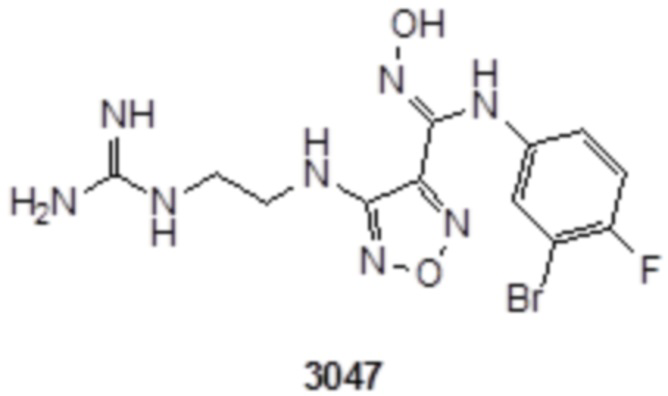 ,
, 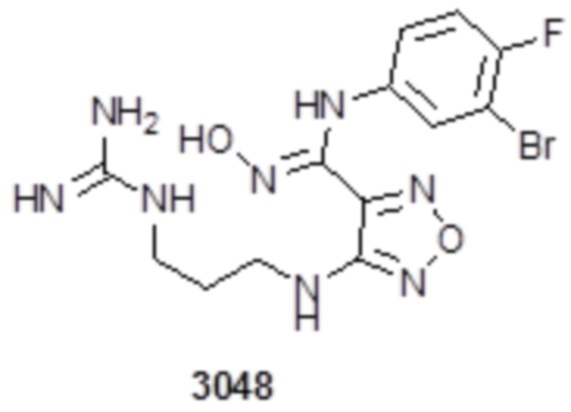 ,
, 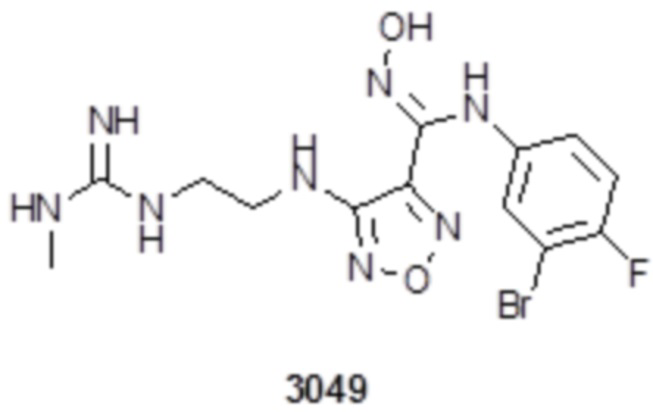 ,
, 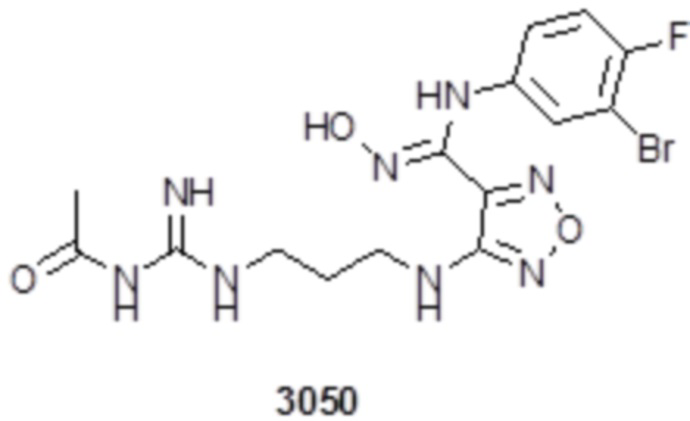 ,
, 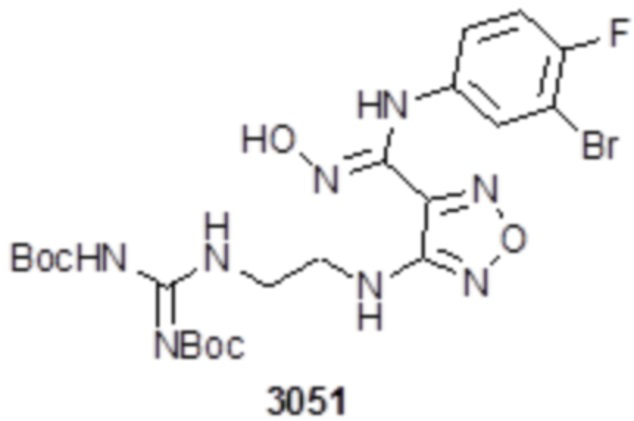 ,
, 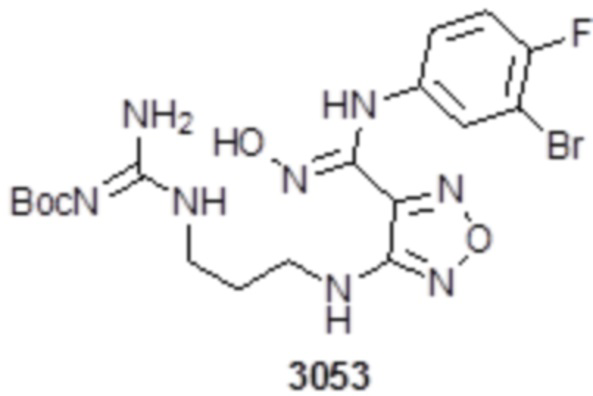 .
.
9. Способ синтеза для получения соединения, представленного следующей формулой
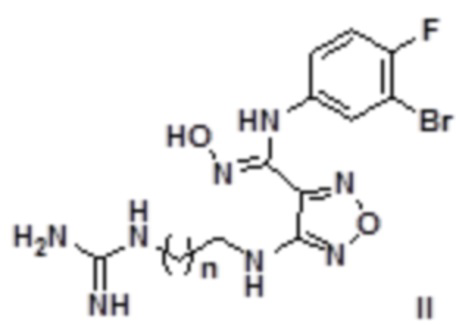 (формула II),
(формула II),
где n выбирают из 0, 1, 2, 3 и 4;
отличающийся тем, что он включает следующие стадии:
1) взаимодействие соединения, представленного формулой 2, с соединением 3a' с получением соединения, представленного формулой IIa
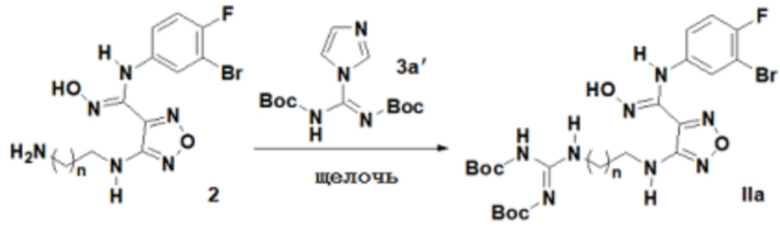 ;
;
2) проведение снятия защиты с соединения, представленного формулой IIa, в кислых условиях с последующим проведением реакции в щелочных условиях с получением соединения, представленного формулой II
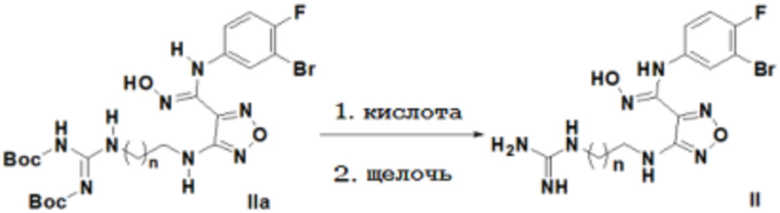 .
.
10. Способ синтеза для получения соединения, представленного следующей формулой
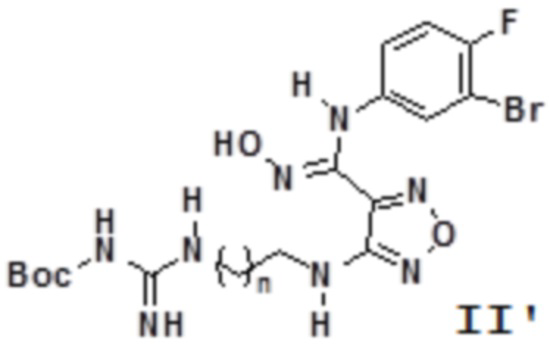 (формула II’),
(формула II’),
отличающийся тем, что он включает следующую стадию:
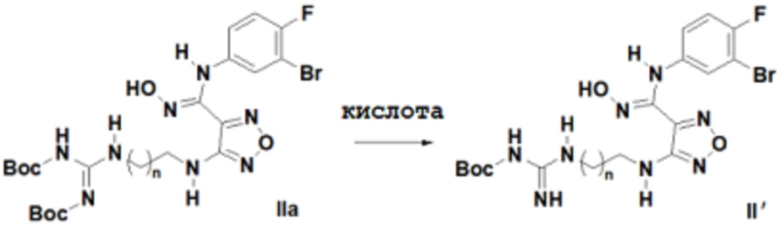 ,
,
где n выбирают из 0, 1, 2, 3 или 4;
реакцию соединения, представленного формулой IIa, проводят в кислых условиях с получением соединения, представленного формулой II’.
11. Фармацевтическая композиция для ингибирования индоламин–2,3–диоксигеназы, содержащая соединение или его фармацевтически приемлемую соль по любому из пп. 1–8 и один или более фармацевтически приемлемых адъювантов.
12. Способ лечения заболевания, где способ включает введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества по меньшей мере одного из соединения или его фармацевтически приемлемой соли по любому из пп. 1–8, или его фармацевтической композиции по п. 11, где заболевание представляет собой заболевание, характеризующееся патологическим путем метаболизма триптофана, опосредованным индоламин–2,3–диоксигеназой, и заболевание представляет собой злокачественную опухоль или болезнь Альцгеймера.
13. Способ по п. 12, где злокачественную опухоль выбирают из группы, состоящей из злокачественной опухоли легкого, злокачественной опухоли печени, злокачественной опухоли толстого кишечника, злокачественной опухоли поджелудочной железы, злокачественной опухоли молочной железы, злокачественной опухоли предстательной железы, злокачественной опухоли головного мозга, злокачественной опухоли яичников, злокачественной опухоли шейки матки, злокачественной опухоли яичек, злокачественной опухоли почки, злокачественной опухоли головы и шеи, лимфомы, меланомы и лейкоза.
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Токарный резец | 1924 |
|
SU2016A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| RU 2015124002 A, 10.01.2017. | |||

