Область техники, к которой относится изобретение
Изобретение относится к новым соединениям, композициям, содержащим эти соединения, и их применению в терапии, например при лечении микобактериальных инфекций или при лечении вызванных микобактериями заболеваний, таких как туберкулез (также известный как TB).
Предпосылки создания изобретения
В соответствии с отчетом, опубликованным Всемирной Организации Здравоохранения в 2014 г., каждый год почти десять миллионов людей инфицируются туберкулезом (ТВ), вызывая 1,5 миллиона смертей в год. Несмотря на доступные методы лечения туберкулеза, заболеваемость по-прежнему начинает расти вследствие инфекции Mycobacterium tuberculosis из-за того, что бактериальный агент, вызывающий TB, становятся устойчивыми ко многим препаратам первой линии, таким как изониазид и рифампицин.
Этионамид, структурный аналог изониазида, который является таким же эффективным, как изониазид, часто назначают для лечения резистентного ко многим лекарственным средствам TB (MDR TB). Тем не менее недостатком, связанным с использованием этионамида, является то, что для получения приемлемой концентрации лекарственного средства в крови, требуется доза до 1 г/день, что связанно с серьезными побочными эффектами, включая нейротоксичность и фатальную гепатотоксичность. Таким образом, существует потребность в снижении клинической дозы и уменьшении воздействия этионамида.
Следовательно, одна цель настоящего изобретения состоит в создании новых соединений, которые могут усиливать активность препаратов, используемых при лечении туберкулеза, в частности, лекарственных средств, которые активируются через сигнальный путь EthA, как и этионамид. Еще одной целью настоящего изобретения является предоставление новых соединений для лечения туберкулеза.
В публикации РСТ WO 2014/096369 описаны спироизоксазолиновые соединения, несущие в качестве заместителей группы арила, циклоалкила или гетероарила. Считается, что такие соединения полезны при лечении TB.
Сущность изобретения
В первом аспекте настоящего изобретения предлагается соединение формулы (I):
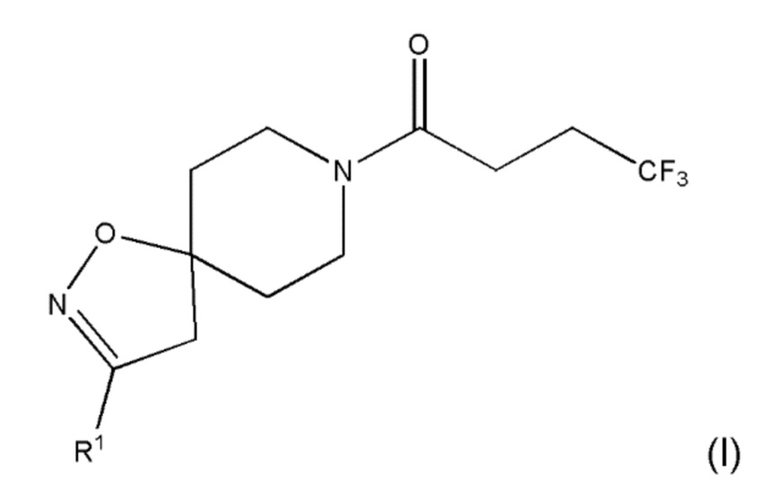 ,
,
где R1 представляет собой галоген; циано; неразветвленный С1-6 алкил; разветвленный С3-4 алкил; неразветвленный С1-6 алкокси; разветвленный С3-4 алкокси; метил, замещенный одним или несколькими атомами фтора; этил, замещенный одним или несколькими атомами фтора; метокси, замещенный одним или несколькими атомами фтора; или этокси, замещенный одним или несколькими атомами фтора;
или его фармацевтически приемлемая соль.
Во втором аспекте настоящего изобретения предлагается соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии, в частности, для применения при лечении туберкулеза.
В соответствии с третьим аспектом настоящего изобретения предлагается способ лечения микобактериальной инфекции у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В соответствии с четвертым аспектом настоящего изобретения, предлагается способ лечения заболевания, вызванного микобактериальной инфекцией, у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В пятом аспекте настоящего изобретения предлагается применение соединения формулы (I) или его фармацевтически приемлемой соли при изготовлении лекарственного средства, применяемого при лечении микобактериальной инфекции или заболевания, вызванного микобактериальной инфекцией.
В соответствии с шестым аспектом настоящего изобретения предлагается соединение формулы (I) или его фармацевтически приемлемая соль для применения при лечении микобактериальной инфекции или для применения при лечении заболевания, вызванного микобактериальной инфекцией.
В седьмом аспекте настоящего изобретения предлагается фармацевтическая композиция, содержащая (а) соединение формулы (I) или его фармацевтически приемлемую соль; и (b) фармацевтически приемлемый эксципиенты.
В восьмом аспекте настоящего изобретения предлагается комбинация (а) соединения формулы (I) или его фармацевтически приемлемой соли; и (b) по меньшей мере одного другого агента против микобактерий.
Подробное описание изобретения
Как указано выше, один из аспектов настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли:
,
где R1 представляет собой галоген; циано; неразветвленный С1-6 алкил; разветвленный С3-4 алкил; неразветвленный С1-6 алкокси; разветвленный С3-4 алкокси; метил, замещенный одним или несколькими атомами фтора; этил, замещенный одним или несколькими атомами фтора; метокси, замещенный одним или несколькими атомами фтора; или этокси, замещенный одним или несколькими атомами фтора.
В одном варианте осуществления настоящее изобретение относится к соединению формулы (I).
В одном варианте осуществления изобретения R1 представляет собой фтор, хлор или бром; циано; неразветвленный С1-6 алкил; разветвленный С3-4 алкил; неразветвленный С1-6 алкокси; разветвленный С3-4 алкокси; метил, замещенный одним или несколькими атомами фтора; этил, замещенный одним или несколькими атомами фтора; метокси, замещенный одним или несколькими атомами фтора; или этокси, замещенный одним или несколькими атомами фтора.
В одном варианте осуществления изобретения R1 представляет собой фтор, хлор или бром; циано; неразветвленный С1-6 алкил; разветвленный С3-4 алкил; неразветвленный С1-6 алкокси; разветвленный С3-4 алкокси; моно-, ди- или трифторметил; моно-, ди- или трифторметокси; 2-фторэтил; 2,2-дифторэтил; 2,2,2-трифторэтил; 2-фторэтокси; 2,2-дифторэтокси; или 2,2,2-трифторэтокси.
В одном варианте осуществления изобретения R1 представляет собой хлор или бром; циано; неразветвленный С1-6 алкил; разветвленный С3-4 алкил; неразветвленный С1-6 алкокси; разветвленный С3-4 алкокси; моно-, ди- или трифторметил; моно-, ди- или трифторметокси; 2-фторэтил; 2,2-дифторэтил; 2,2,2-трифторэтил; 2-фторэтокси; 2,2-дифторэтокси; или 2,2,2-трифторэтокси.
В одном варианте осуществления изобретения R1 представляет собой фтор, хлор, бром, циано, неразветвленный С1-6 алкил, разветвленный С3-4 алкил, неразветвленный С1-6 алкокси, разветвленной С3-4 алкокси, трифторметил, трифторметокси, 2,2,2-трифторэтил или 2,2,2-трифторэтокси.
В одном варианте осуществления изобретения R1 представляет собой хлор, бром, циано, неразветвленный С1-6 алкил, разветвленный С3-4 алкил, неразветвленный С1-6 алкокси, разветвленной С3-4 алкокси, трифторметил, трифторметокси, 2,2,2-трифторэтил или 2,2,2-трифторэтокси.
В одном варианте осуществления изобретения R1 представляет собой фтор, хлор, бром, циано, неразветвленный С1-6 алкил, разветвленный С3-4 алкил, неразветвленный С1-6 алкокси, разветвленной С3-4 алкокси, трифторметил, 2,2,2-трифторэтил или 2,2,2-трифторэтокси.
В одном варианте осуществления изобретения R1 представляет собой хлор, бром, циано, неразветвленный С1-6 алкил, разветвленный С3-4 алкил, неразветвленный С1-6 алкокси, разветвленной С3-4 алкокси, трифторметил, 2,2,2-трифторэтил или 2,2,2-трифторэтокси.
В одном варианте осуществления изобретения R1 представляет собой фтор, бром, циано, неразветвленный C1-4 алкил, неразветвленный С1-6 алкокси, разветвленный С4 алкокси, трифторметил, 2,2,2-трифторэтил или 2,2,2-трифторэтокси.
В одном варианте осуществления изобретения R1 представляет собой бром, циано, неразветвленный C1-4 алкил, неразветвленный С1-6 алкокси, разветвленный С4 алкокси, трифторметил, 2,2,2-трифторэтил или 2,2,2-трифторэтокси.
В одном варианте осуществления изобретения R1 представляет собой фтор, бром, циано, метил, этил, трифторметил, 2,2,2-трифторэтокси, метокси, этокси, н-пропокси, изо-бутокси или гексилокси.
В одном варианте осуществления изобретения R1 представляет собой бром, циано, метил, этил, трифторметил, 2,2,2-трифторэтокси, метокси, этокси, н-пропокси, изо-бутокси или гексилокси.
В одном варианте осуществления изобретения R1 представляет собой трифторметил.
Конкретные соединения, которые являются полезными в рамках настоящего изобретения, включают следующие соединения:
4,4,4-трифтор-1-[3-(2,2,2-трифторэтокси)-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил]бутан-1-он;
4,4,4-трифтор-1-(3-пропокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он;
4,4,4-трифтор-1-(3-изобутокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он;
4,4,4-трифтор-1-(3-гексилокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он;
1-(3-бром-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-он;
1-(3-этокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-он;
4,4,4-трифтор-1-(3-метокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он;
4,4,4-трифтор-1-(3-(трифторметил)-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он;
4,4,4-трифтор-1-(3-метил-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он;
1-(3-этил-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-он;
8-(4,4,4-трифторбутаноил)-1-окса-2,8-диазаспиро[4,5]дец-2-ene-3-карбонитрил; и
4,4,4-трифтор-1-(3-фтор-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он.
Термины и определения
Как используется в данном описании, термин «галоген» означает фтор, хлор, бром или йод. Более конкретно, этот термин предназначен для обозначения фтора, хлора или брома.
Как используется в данном описании, термин «циано» относится к группе -CN.
Как используется в данном описании, термин «неразветвленный С1-6 алкил» относится к алкильной группе с линенйной цепью, содержащей от одного до шести атомов углерода. Таким образом, термин «неразветвленный С1-6 алкил» охватывает метил, этил, н-пропил, н-бутил, н-пентил и н-гексил.
Как используется в данном описании, термин «разветвленный С3-4 алкил» относится к алкильной группе с разветвленной цепью, содержащей три или четыре атома углерода. Таким образом, термин «разветвленный С3-4 алкил» охватывает изопропил, втор-бутил и изобутил.
Как используется в данном описании, термин «неразветвленный С1-6 алкокси» относится к алкоксигруппам с линейной цепью, содержащим от одного до трех атомов углерода. Таким образом, термин «неразветвленный С1-6 алкокси» охватывает метокси, этокси, н-пропокси, н-бутокси, н-пентилокси и н-гексилокси.
Как используется в данном описании, термин «разветвленный С3-4 алкокси» относится к алкоксигруппам с разветвленной цепью, содержащим три или четыре атома углерода. Таким образом, термин «разветвленный С3-4 алкокси» охватывает изо-пропокси, втор-бутокси и изо-бутокси.
Как используется в данном описании, термин «метил, замещенный одним или несколькими атомами фтора» относится к метильной группе, которая замещена одним, двумя или тремя атомами фтора. Таким образом, термин «метил, замещенный одним или несколькими атомами фтора» охватывает моно-фторметил (-CH2F), ди-фторметил (-CHF2) и трифторметил (-CF3).
Как используется в данном описании, термин «этил, замещенный одним или несколькими атомами фтора» относится к этильной группе, которая замещена одним, двумя, тремя, четырьмя или пятью атомами фтора. Таким образом, термин "этил, замещенный одним или несколькими атомами фтора" охватывает 2-фторэтил (-CH2CH2F), 2,2-дифторэтил (-CH2CHF2) и 2,2,2-трифторэтил (-CH2CF3).
Как используется в данном описании, термин «метокси, замещенный одним или несколькими атомами фтора» относится к группе, в которой метокси углерод метильной группы замещен одним, двумя или тремя атомами фтора. Таким образом, термин «метил, замещенный одним или несколькими атомами фтора» охватывает монофторметокси (-OCH2F), дифторметокси (-OCHF2) и трифторметокси (-OCF3).
Как используется в данном описании, термин «этокси, замещенный одним или несколькими атомами фтора» относится к группе, в которой этокси углерод этильной группы замещен одним, двумя, тремя, четырьмя или пятью атомами фтора.
Таким образом, термин "этил, замещенный одним или несколькими атомами фтора" охватывает 2-фторэтокси (-OCH2CH2F), 2,2-дифторэтокси (-OCH2CHF2) и 2,2,2-трифторэтокси (-OCH2CF3).
Следует иметь в виду, что соединение формулы (I) могут существовать в различных таутомерных формах. Все возможные таутомеры находлятся в пределах объема настоящего изобретения.
Термин «соединения по изобретению», используемый в данном описании, означает соединение формулы (I) или его фармацевтически приемлемую соль. Термин «соединение по изобретению» означает любое одно из соединений по изобретению, определенное выше.
Кроме того, следует понимать, что указания, такие как «соединение формулы (I) или его фармацевтически приемлемая соль» или «соединения по изобретению», подразумевают соединение формулы (I) его фармацевтически приемлемую соль или сольват соединения формулы (I) или их любую фармацевтически приемлемую комбинацию. Таким образом, в качестве неограничивающего примера, используемого в данном описании с целью иллюстрации, указание «соединение формулы (I) или его фармацевтически приемлемая соль» охватывает фармацевтически приемлемую соль соединения формулы (I), которая присутствует в виде сольвата, и это указание также охватывает смесь соединения формулы (I) и фармацевтически приемлемой соли соединения формулы (I).
Следует понимать, что в данном описании ссылки на соединение формулы (I) или его фармацевтически приемлемая соль охватывают соединение формулы (I) в виде свободного основания или в виде его фармацевтически приемлемой соли. Таким образом, в одном варианте осуществления настоящее изобретение относится к соединению формулы (I). В другом варианте осуществления настоящее изобретение относится к фармацевтически приемлемой соли соединения формулы (I).
Термин «фармацевтический приемлемый» относится к соединениям (включая соль), материалам, композициям и лекарственным формам, которые в пределах объема знаний в области медицины подходят для использования при контакте с тканями человека и животных без чрезмерной токсичности, раздражения или других проблем или осложнений, соразмерных с разумным соотношением польза/риск.
Фармацевтически приемлемые соли включают, среди прочего, такие, которые описаны в Berge, J. Pharm. Sci, 1977, 66, 1-19, или такие, которые перечислены в справочнике P.H. Stahl & C.G. Wermuth: Handbook of Pharmaceutical Salts; Properties, Selection and Use, Second Edition Stahl/Wermuth: Wiley-VCH/VHCA, 2011 (см. http://www.wilev.com/WilevCDA/WilevTitle/productCd-3906390519 html).
Подходящие фармацевтически приемлемые соли могут включать кислотно-аддитивные соли. Такие соли могут быть образованы путем взаимодействия с соответствующей кислотой, возможно в подходящем растворителе, таком как органический растворитель, с получением соли, которая может быть выделена путем кристаллизации и фильтрации.
Типовые фармацевтически приемлемые кислотно-аддитивные соли включают, но без ограничения, 4-ацетамидобензоат, ацетат, адипат, альгинат, аскорбат, аспартат, бензолсульфонат (безилат), бензоат, бисульфат, битартрат, бутират, кальций-эдетат, камфорат, камфоросульфонат (камсилат), капрат (деканоат), капроат (гексаноат), каприлат (октаноат), циннамат, цитрат, цикламат, диглюконат, 2,5-дигидроксибензоат, дисукцинат, додецилсульфат (эстолат), эдетат (этилендиаминтетраацетат), эстолат (лаурилсульфат), этан-1,2-дисульфонат (эдисилат), этансульфонат (эзилат), формиат, фумарат, галактарат (мукат), гентизат (2,5-дигидроксибензоат), глюкогептонат (глюцептат), глюконат, глюкуронат, глутамат, глутарат, глицерофосфонат, гликолят, гексилрезорцинат, гиппурат, гидрабамин (N, N'- ди(дегидроабиэтил)этилендиамин), гидробромид, гидрохлорид, гидроиодид, гидроксинафтоат, изобутират, лактат, лактобионат, лаурат, малат, малеат, малонат, манделат, метансульфонат (мезилат), метилсульфат, мукат, нафталин-1,5-дисульфонат (нападизилат), нафталин-2-сульфонат (напсилилат), никотинат, нитрат, олеат, пальмитат, п-аминобензолсульфонат, п-аминосалицилат, памоат (эмбонат), пантотенат, пектинат, персульфат, фенилацетат, фенилэтилбарбитурат, фосфат, полигалактуронат, пропионат, п-толуолсульфонат (тозилат), пироглутамат, пируват, салицилат, себацинат, стеарат, субацетат, сукцинат, сульфамат, сульфат, таннат, тартрат, теоклат (8-хлортеофиллинат), тиоцианат, треэтиодид, ундеканоат, ундециленат и валерат.
Используемый в данном описании термин «терапевтически эффективное количество» означает любое количество, которое, при сравнении с соответствующим субъектом, который не получил такое количество, приводит к улучшению лечения, выздоровлению, профилактике или облегчению тяжести заболевания, расстройства или побочного эффекта, или к снижению развития болезни или расстройства.
Соответствующее «терапевтически эффективное количество» будет зависеть от целого ряда факторов, в том числе, например, от возраста и массы субъекта, определенного состояния, требующего лечения и его тяжесть, природы препарата, а также от способа введения, и оно будет, в конечном счете, определяться в соответствии с мнением лечащего врача.
Соединения формулы (I), где m представляет собой 0, может содержать один или несколько асимметрических центров (также называемых хиральными центрами), и, следовательно, они могут существовать в виде отдельных энантиомеров, диастереоизомеров или других стереоизомерных форм или в виде их смесей. Хиральные центры, такие как хиральные атомы углерода, также могут присутствовать в заместителе, таком как алкильная группа. В случае, когда не указана стереохимия хирального центра, присутствующего в соединении формулы (I), или в какой-либо химической структуре, представленной в данном документе, то такая структура охватывает любые стереоизомеры и все их смеси. Таким образом, соединения формулы (I), содержащие один или более хиральных центров, могут быть использованы в виде рацемических модификаций, включая рацемические смеси и рацематы, энантиомерно-обогащенные смеси или в виде энантиомерно-чистых индивидуальных стереоизомеров.
Получение соединений
Соединения по изобретению могут быть получены различными методами, в том числе стандартными химическими методами. Любые указанные выше определения имеют приведенные выше значения, если не указано иное. Иллюстративные методы общего синтеза показаны ниже на следующих схемах, которые могут быть легко адаптированы для получения других соединений по изобретению. Конкретные соединения по изобретению могут быть получены в соответствии с экспериментальными процедурами, описанными в разделе Примеры.
Конкретные соединения настоящего изобретения могут быть получены в соответствии с экспериментальными процедурами, описанными в разделе Примеры.
Общие процедуры, используемые для синтеза соединений формулы (I), показаны ниже на схемах реакций 1-7, и они проиллюстрированы в Примерах.
Получение соединений формулы (Iа)
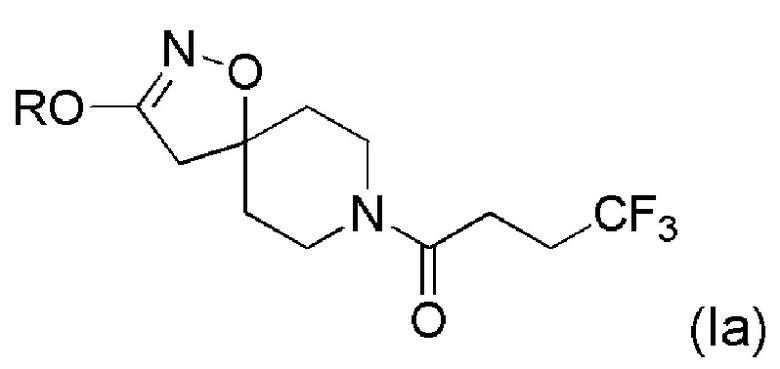
Соединения формулы (Iа), которые являются алкоксиспиро-соединениями формулы (I), могут быть получены по Схеме 1 (ниже) путем удаления защитной группы Вос из защитной группы алкоксиспиро-соединения формулы (II), например, с помощью TFA, и дальнейшей связи солевой аминогруппы TFA с 4,4,4-трифторбутановой кислотой. Промежуточное соединение формулы (II) может быть получено взаимодействием соответствующих коммерчески доступных спиртов (ROH) с N-Вос-защищенного галогенированного спиро-соединения формулы (IV), схема синтеза которого показана ниже.
Схема 1
Получение соединений формулы (I), где R1 представляет собой неразветвленный С1-6 алкокси; С3-4 разветвленный алкокси; метокси, замещенный одним или несколькими атомами фтора; или этокси, замещенный одним или несколькими атомами фтора
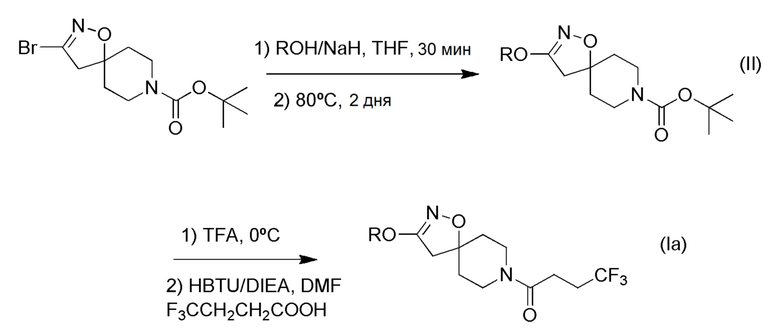
Альтернативно, соединения формулы (Iа), которые являются алкоксиспиро-соединениями, могут быть получены по Схеме 2 (ниже) путем взаимодействия соответствующих коммерчески доступных спиртов (ROH) с галогенированным трифторбутанамидным спиро-соединением формулы (IIIa) в присутствии подходящего основания, такого как карбонат калия.
Схема 2
Альтернативное получение соединений формулы (I), где R1 представляет собой неразветвленный C1-6 алкокси; разветвленный C3-4 алкокси; метокси, замещенный одним или несколькими атомами фтора; или этокси, замещенный одним или несколькими атомами фтора
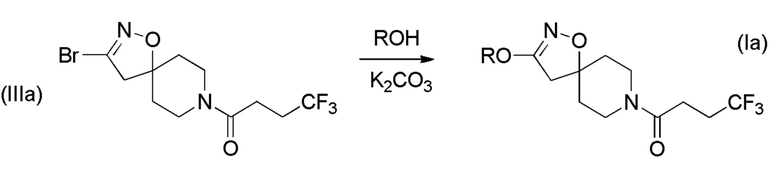
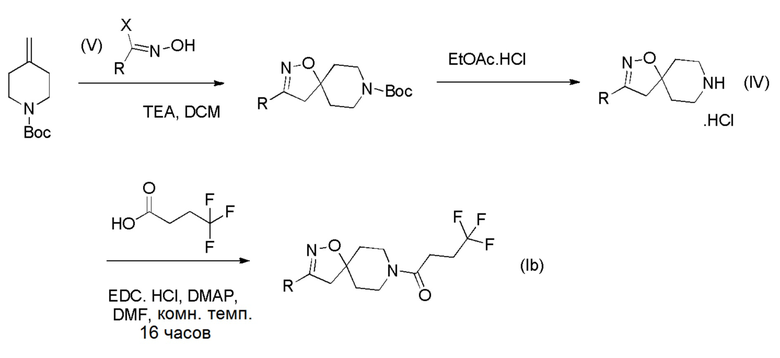
Получение соединений формулы (Ib)
Соединения формулы (b), которые являются алкилспиро-соединениями, могут быть получены в соответствии со Схемой 3 (ниже) путем связывания соединения формулы (IV) с 4,4,4-трифторбутановой кислотой. Соединения формулы (IV) могут быть получены путем циклизации коммерчески доступного трет-бутил-4-метиленпиперидин-1-карбоксилата с оксимами соединений формулы (V), с последующим отщеплением N-Вос-защитной группы кислотой, такой как хлористоводородная кислота.
Схема 3
Получение соединений формулы (I), где R1 представляет собой неразветвленный C1-6 алкил или разветвленный C3-4 алкил
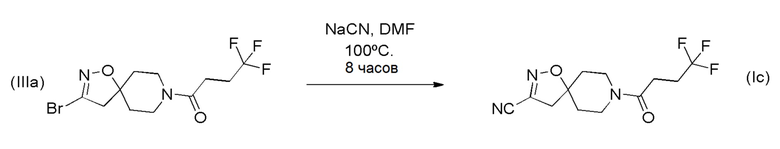
Получение соединения формулы (Ic)
Циано-соединение формулы (Ic) может быть получено путем нуклеофильного замещения соединения (IIIa), например, цианидом натрия, в подходящем растворителе, таком как DMF, в соответствии со Схемой 4 (ниже).
Схема 4
Получение соединений формулы (I), где R1 представляет собой CN
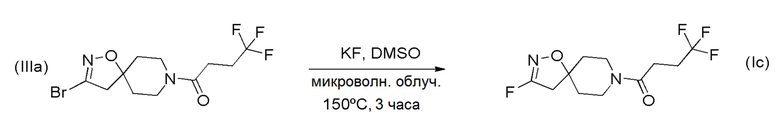
Получение соединения формулы (Id)
Фтор-соединение формулы (Id) может быть получено путем замещения галогена в соединении (IIIa) с использованием, например, фторида калия в подходящем растворителе, таком как DMSO, в соответствии со Схемой 5 (ниже).
Схема 5
Получение соединений формулы (I), где R1 представляет F
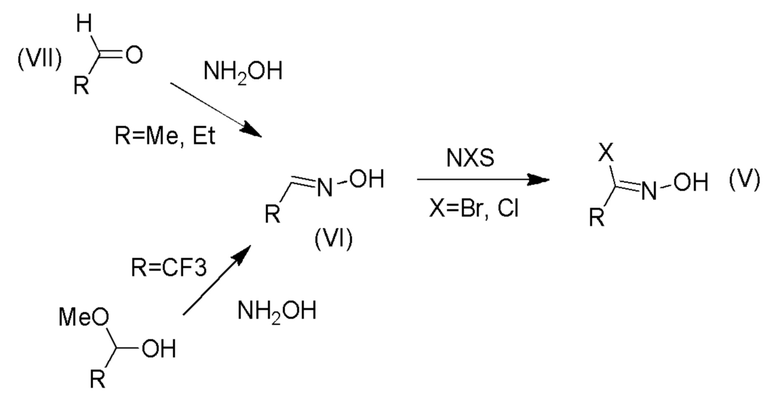
Получение промежуточных соединений (IIIa) и (V)
Промежуточные оксимы формулы (V) могут быть получены путем реакции галогенирования промежуточных соединений формулы (VI) с использованием N-галогенсукцинимида, например, таких как NBS или NCS, как показано на Схеме 6 (ниже). Промежуточные оксимы формулы (VI), где R представляет собой метил или этил, могут быть получены взаимодействием соответствующего альдегида формулы (VII с, например, гидроксиламином. Альтернативно, промежуточное соединение, представляющее собой трифторметилоксим, где R представляет собой трифторметил, может быть получено путем взаимодействия 2,2,2-трифтор-1-метоксиэтанола с гидроксиламином.
Схема 6
Получение промежуточного соединения V
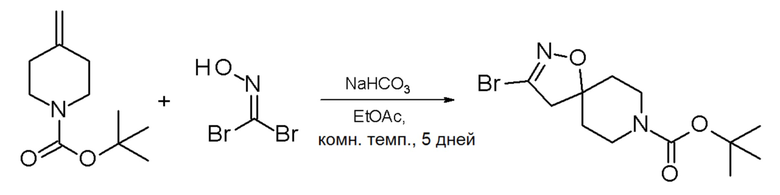
Галогенированное N-Вос-защищенное промежуточное спиро- соединение формулы (III) может быть получено взаимодействием коммерчески доступного трет-бутил-4-метиленпиперидин-1-карбоксилата и дибромида гидроксикарбонимида в присутствии, например, бикарбонат натрия (Схема 7).
Схема 7
Получение промежуточного соединения III
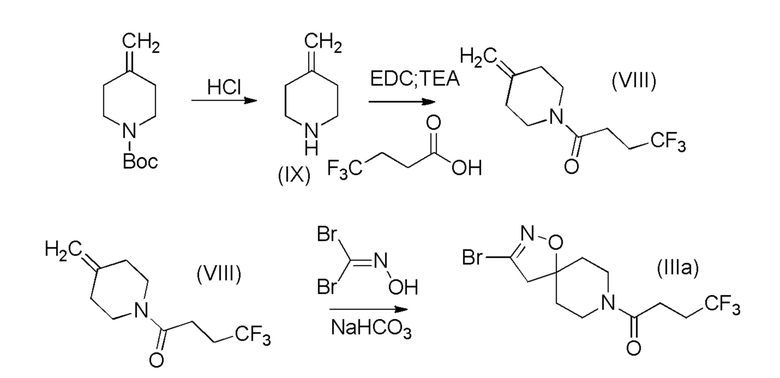
Аналогичным образом может быть получено промежуточное соединение (IIIa) путем взаимодействия алкена (VIII) с дибромидом гидроксикарбонимида в тех же условиях, как описано выше. Промежуточное соединение (VIII) может быть получено путем реакции сочетания гидрохлорида промежуточного амино-соединения (IX) с 4,4,4-трифторбутановой кислоты в стандартных условиях. И, наконец, промежуточное амино-соединение (IX) может быть получено из коммерчески доступного трет-бутил-4-метиленпиперидин-1-карбоксилата обработкой хлористым водородом. Это показано на Схеме 8 (ниже).
Схема 8
Получение промежуточного соединения IIIa
Способы применения
В одном аспекте настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения его в терапии.
В другом аспекте настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения его при лечении микобактериальной инфекции. Микобактериальная инфекция является инфекцией, которая вызвана микобактериями.
Микобактерия может быть членом одной из следующих групп микобактерий: комплекса Mycobacterium tuberculosis (MTC), комплекса Mycobacterium avium (MAC), клада Mycobacterium gordonae, клада Mycobacterium kansasii, клада Mycobacterium chelonae, клада Mycobacterium fortuitum, клада Mycobacterium parafortuitum или клада Mycobacterium vaccae. Микобактерии могут также относиться к Mycobacterium ulcerans или Mycobacterium leprae.
В одном варианте осуществления микобактерии являются членом комплекса Mycobacterium tuberculosis (MTC).
Члены комплекса Mycobacterium tuberculosis (MTC) включают Mycobacterium tuberculosis, Mycobacterium africanum, Mycobacterium bovis, Mycobacterium bovis BCG, Mycobacterium canetti, Mycobacterium caprae, Mycobacterium microti и Mycobacterium pinnipedii. Эти микобактерии являются возбудителями туберкулеза у человека и животных. Микобактерии туберкулеза являются основной причиной туберкулеза у людей.
В одном варианте осуществления изобретения, инфекция является инфекцией, вызываемой Mycobacterium tuberculosis. Другими словами, микобактериальная инфекция вызвана инфекцией Mycobacterium tuberculosis.
В одном варианте осуществления изобретения, Mycobacterium tuberculosis имеют множественную лекарственную устойчивость. В другом варианте Mycobacterium tuberculosis устойчивы к этионамиду.
Члены комплекса Mycobacterium avium (MAC) включают Mycobacterium avium, Mycobacterium avium paratuberculosis, Mycobacterium avium silaticum, Mycobacterium avium hominissuis, Mycobacterium columbiense и Mycobacterium indicus pranii.
Члены клада Mycobacterium gordonae включают Mycobacterium asiaticum and Mycobacterium gordonae.
Члены клада Mycobacterium kansasii включают Mycobacterium gastri и Mycobacterium kansasii.
Члены клада Mycobacterium chelonae включают Mycobacterium abscessus, Mycobacterium bolletii и Mycobacterium chelonae.
Члены клада Mycobacterium fortuitum включают Mycobacterium boenickei, Mycobacterium brisbanense, Mycobacterium cosmeticum, Mycobacterium fortuitum, Mycobacterium fortuitum subspecies acetamidolyticum, Mycobacterium houstonense, Mycobacterium mageritense, Mycobacterium neworleansense, Mycobacterium peregrinum, Mycobacterium porcinum, Mycobacterium senegalense и Mycobacterium septicum.
Члены клада Mycobacterium parafortuitum включают Mycobacterium austroafricanum, Mycobacterium diernhoferi, Mycobacterium frederiksbergense, Mycobacterium hodleri, Mycobacterium neoaurum и Mycobacterium parafortuitum.
Таким образом, микобактериальные инфекции могут быть вызваны инфекцией микобактериями, выбранными из следующих: Mycobacterium tuberculosis, Mycobacterium africanum, Mycobacterium bovis, Mycobacterium bovis BCG, Mycobacterium canetti, Mycobacterium caprae, Mycobacterium microti, Mycobacterium pinnipedii, Mycobacterium avium, Mycobacterium avium paratuberculosis, Mycobacterium avium silaticum, Mycobacterium avium hominissuis, Mycobacterium columbiense, Mycobacterium indicus pranii, Mycobacterium asiaticum, Mycobacterium gordonae, Mycobacterium gastri, Mycobacterium kansasii, Mycobacterium abscessus, Mycobacterium bolletii, Mycobacterium chelonae, include Mycobacterium boenickei, Mycobacterium brisbanense, Mycobacterium cosmeticum, Mycobacterium fortuitum, Mycobacterium fortuitum subspecies acetamidolyticum, Mycobacterium houstonense, Mycobacterium mageritense, Mycobacterium neworleansense, Mycobacterium peregrinum, Mycobacterium porcinum, Mycobacterium senegalense, Mycobacterium septicum, Mycobacterium austroafricanum, Mycobacterium diernhoferi, Mycobacterium frederiksbergense, Mycobacterium hodleri, Mycobacterium neoaurum, Mycobacterium parafortuitum, Mycobacterium ulcerans и Mycobacterium leprae.
В другом аспекте настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения при лечении заболевания, вызванного микобактериальной инфекцией, где микобактерия выбрана из микобактерий, которые описаны выше. Заболевания, вызванные микобактериальной инфекцией, включают, но без ограничения, туберкулез (например, вызванный Mycobacterium tuberculosis), лепру (например, вызванную Mycobacterium leprae), болезнь Джона (например, вызванную Mycobacterium avium subspecies paratuberculosis), язву Бурули или Бернсдейла (например, вызванную Mycobacterium ulceran), болезнь Крона (например, вызванную Mycobacterium avium subspecies paratuberculosis), заболевание легких или инфекцию легких, пневмонию, инфекции бурсы, синовия, сухожильных влагалищ, локализованный абсцесс, лимфаденит, инфекции кожи и мягких тканей, синдром леди Уиндермир (например, вызванный комплексом Mycobacterium avium (MAC), заболевание легких, вызванное комплексом Mycobacterium avium (MAC), диссеминированная инфекция, вызванная комплексом Mycobacterium avium intracellulare (DMAIC), легочная болезнь джакузи (например, вызванную комплексом Mycobacterium avium), мастит, вызванный MAC, пиомиозит, вызванный MAC, или гранулему.
В одном варианте осуществления изобретения, заболевание представляет собой туберкулез. Таким образом, один аспект настоящего изобретения относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения при лечении туберкулеза.
В другом варианте осуществления настоящее изобретение относится к способу лечения микобактериальной инфекции у млекопитающего, нуждающегося в этом, где указанное лечение включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Как описано здесь, микобактериальные инфекции является инфекциями, вызванными микобактериями. Микобактерии являются такими, как описано выше.
В одном варианте осуществления изобретения, микобактериальная инфекция является инфекцией, вызванная Mycobacterium tuberculosis.
В другом варианте осуществления, настоящее изобретение относится к способу лечения заболевания, вызванного микобактериальной инфекцией у млекопитающего, нуждающегося в этом, где указанное лечение включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В одном варианте осуществления изобретения, заболевание представляет собой туберкулез. Таким образом, изобретение предоставляет способ лечения туберкулеза у млекопитающего, нуждающегося в этом, где указанное лечение включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В одном варианте осуществления настоящего изобретения, млекопитающим является человек.
Специалистам в данной области техники понятно, что указания в отношении лечение относятся к лечению установленных патологических состояний. Тем не менее, соединения по изобретению могут, в зависимости от состояния, также быть полезны при профилактике некоторых заболеваний. Таким образом, в одном варианте осуществления, настоящее изобретение обеспечивает лечение или предотвращение заболевания, такого как туберкулез (TB). В другом варианте осуществления, настоящее изобретение обеспечивает лечение заболевания, такого как туберкулез. В другом варианте осуществления, настоящее изобретение обеспечивает предотвращение заболевания, такого как туберкулез.
В другом варианте осуществления, настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли при изготовлении лекарственного средства, применяемого при лечении микобактериальной инфекции или при лечении заболевания, вызванного микобактериальной инфекцией.
Кроме того, здесь также описано применение соединения формулы (I) или его фармацевтически приемлемой соли при изготовлении лекарственного средства, применяемого при лечении туберкулеза.
В одном варианте осуществления изобретения, соединение формулы (I) или его фармацевтически приемлемую соль, применяемые при лечении туберкулеза, вводят совместно с тиоамидом. В другом варианте осуществления изобретения, тиоамид представляет собой этионамид. В альтернативном варианте осуществления изобретения, тиоамид представляет собой протионамид.
Таким образом, в одном варианте осуществления изобретения предлагается фармацевтическая композиция для применения при лечении туберкулеза, где указанная композиция содержит (а) соединение формулы (I); (b) тиоамид, например, этионамид или протионамид; и, необязательно, (c) фармацевтически приемлемый эксципиент.
В другом варианте осуществления, настоящее изобретение относится к способу лечения микобактериальной инфекции у млекопитающего, нуждающегося в этом, где указанное лечение включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли в комбинация с тиоамидом, где указанный тиоамид может представлять собой этионамид. В альтернативном варианте осуществления изобретения, тиоамид представляет собой протионамид. Как описано здесь, микобактериальные инфекции является инфекциями, вызванными микобактериями. Микобактерии являются такими, как описано выше.
В одном варианте осуществления изобретения, микобактериальной инфекцией является инфекция, вызванная Mycobacterium tuberculosis.
В другом варианте осуществления, настоящее изобретение относится к способу лечения заболевания, вызванного микобактериальной инфекцией, у млекопитающего, нуждающегося в этом, где указанное лечение включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтический приемлемой соли в сочетании с тиоамидом, где указанный тиоамид может представлять собой этионамид. В альтернативном варианте осуществления изобретения, тиоамид представляет собой протионамид.
В одном варианте осуществления изобретения, заболевание представляет собой туберкулез. Таким образом, здесь также предоставляется способ лечения туберкулеза у млекопитающего, нуждающегося в этом, где указанное лечение включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли в сочетании с тиоамидом, где указанный тиоамид может представлять собой этионамид. В альтернативном варианте осуществления изобретения, тиоамид представляет собой протионамид.
В другом варианте осуществления настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли в сочетании с тиоамидом (например, этионамидом) при изготовлении лекарственного средства, применяемого при лечении микобактериальной инфекции или при лечении заболевания, вызванного микобактериальной инфекцией. В альтернативном варианте осуществления изобретения, тиоамид представляет собой протионамид.
Кроме того, здесь описано применение соединения формулы (I) или его фармацевтически приемлемой соли в сочетании с тиоамидом (например, этионамидом) при изготовлении лекарственного средства, применяемого при лечении туберкулеза. В альтернативном варианте осуществления изобретения, тиоамид представляет собой протионамид.
В одном варианте осуществления изобретения, соединение формулы (I), используемое в описанных выше способах и при лечении, представляет собой 4,4,4-трифтор-1-(3-(трифторметил)-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он, имеющий следующую структуру:
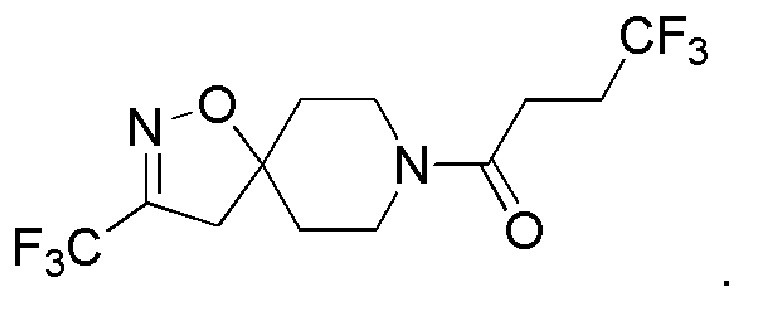
Фармацевтические композиции
Соединения формулы (I) и их фармацевтически приемлемые соли, перед их введением пациенту, как правило, но не обязательно, могут быть представлены в виде фармацевтических композиций. Соответственно, в другом аспекте настоящего изобретения предлагается фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент.
Фармацевтические композиции могут быть введены любым подходящим способом, например пероральным (в том числе буккальным или сублингвальным), ректальным, ингаляционным, интраназальным, местным (включая трансбуккальным, сублингвальным или трансдермальным) или парентеральным (включая подкожным, внутримышечным, внутривенным или внутрикожным) путем. В частности, фармацевтические композиции по изобретению могут быть введены перорально или внутривенно.
Подходящие фармацевтически приемлемые эксципиенты включают следующие виды эксципиентов: носители, разбавители, наполнители, связующие, дезинтегранты, лубриканты, глиданты, гранулирующие агенты, покрывающие агенты, смачивающие агенты, растворители, сорастворители, суспендирующие агенты, эмульгаторы, подсластители, флаворанты, вещества, маскирующие вкус, красители, вещества, предотвращающие слипание, увлажнители, хелатирующие агенты, пластификаторы, агенты, повышающие вязкость, антиоксиданты, консерванты, стабилизаторы, поверхностно-активные вещества и буферирующие агенты.
Подходящие способы получения препаратов и композиций из соединений по изобретению известны специалистам в данной области техники, и они описаны в справочнике Remington: The Science and Practice of Pharmacy, 21st Edition, 2006.
Фармацевтические композиции могут быть представлены в виде единичных дозированных форм, содержащих заранее определенное количество активного ингредиента в единичной дозированной форме. Предпочтительные единичные дозированные формы композиций содержат суточную дозу активного ингредиента или его суб-дозу, или подходящую часть дозы. Следовательно, такие единичные дозированные формы можно вводить более одного раза в день. Предпочтительными единичными дозированными формами композиции являются такими, которые содержат суточную дозу активного ингредиента или его суб-дозу (для введения чаще, чем один раз в день), как описано здесь выше, или подходящую часть дозы.
Когда соединения по изобретению или их фармацевтически приемлемые соли используют при лечении туберкулеза, то они могут применяться отдельно или в сочетании с дополнительным терапевтическим агентом, таким как дополнительным агентом против микобактерий, например, агентом для борьбы с туберкулезом и/или противовирусным агентом, включая антиретровирусные агенты.
Например, настоящее изобретение относится к соединениям формулы (I) или их фармацевтически приемлемым солям в комбинации с дополнительным агентом против туберкулеза. В одном варианте осуществления изобретения, такая комбинация содержит два, три, четыре, пять, шесть или семь дополнительных противотуберкулезных агентов. Например, при лечении туберкулеза с множественной лекарственной устойчивостью, пациентам обычно вводят комбинацию из четырех или более лекарственных препаратов. Например, при лечении туберкулеза, чувствительного к лекарственным средствам, пациентам обычно вводят комбинацию из трех или четырех лекарственных препаратов.
Дополнительный противотуберкулезный агент представляет собой средство или агент, находящийся на стадии разработке, или средство, одобренное или рекомендованное для лечения туберкулеза, и оно может быть выбрано из изониазида, рифампицина, пиразинамида, этамбутола, моксифлоксацина, рифапентина, клофазимина, этионамида, протионамида, изоксила, триацетазона, рифабутина, диарилхинолина, такого как бедаквилин (TMC207) или TBAJ-587, нитроимидазо-оксазина PA-824, деламанида (OPC-67683), оксазолидинона, такого как линезолид, тедизолид, радезолид, сутезолид (PNU-100480), позизолид (AZD-5847) или TBI-223, аналога EMB SQ109, OPC-167832, GSK3036656 (также известного как GSK070), GSK2556286, GSK3211830, бензотиазинона, такого как BTZ043 или PBTZ169, азаиндола, такого как TBA-7371, динитробензамида или бета-лактама, такого как меропенем, фаропенем, эртапенем, тебипенем, или бета-лактамные комбинации, такие как аугментин (амоксициллин-клавуланат).
В одном варианте осуществления изобретения, противотуберкулезный агент/средство может быть выбрано из изониазида, рифампина, пиразинамида, этамбутола, моксифлоксацина, рифапентина, клофазимина, этионамида, протионамида, изоксазолила, тиазетазозона, бедаквилина (TMC207), нитроимидазо-оксазина РА-824, деламанида (OPC-67683), оксазолидинона, такого как линезолид, тедизолид, радезолид, сутезолид (PNU-100480) или позизолид (AZD-5847), аналога EMB SQ109, OPC-167832, GSK3036656 (также известный как GSK070), GSK2556286, GSK3211830 и бензотиазинона или динитробензамида.
В соответствии с настоящим изобретением комбинация может дополнительно содержать противовирусный агент, включая антиретровирусный агент.
Такие антиретровирусные агенты/средства могут быть выбраны из зидовудина, диданозина, ламивудина, залцитабина, абакавира, ставудина, адефовира, дипивоксил адефовира, фозивудина, тодоксила, эмтрицитабина, аловудина, амдоксовира, элвуцитабина, невирапина, делавирдина, эфавиренца, ловирида, иммунокала, олтипраза, каправирина, лерсивирина, GSK2248761, ТМС-278, ТМС-125, этравирина, саквинавира, ритонавира, индинавира, нелфинавира, ампренавира, фозампренавира, бреканавира, дарунавира, атазанавира, типранавира, палинавира, лазинавира, энфувиртида, Т-20, Т-1249, PRO-542, PRO-140, TNX-355, BMS-806, BMS-663068 и BMS-626529, 5-геликса, ралтегравира, элвитегравира, GSK1349572, GSK1265744, викривирока (Sch-C), Sch-D, TAK779, маравирока, TAK449, диданозина, тенофовира, лопинавира или дарунавира.
Соединение по изобретению (т.е. соединение формулы (I) или его фармацевтически приемлемая соль) может использоваться в комбинации с противотуберкулезным агентом, который активируется через сигнальный путь EthA. Специалист в данной области техники может определить, является ли конкретное соединение активируемым через сигнальный путь EthA, используя для этого, например, метод, описанный в публикации: "Activation of the prodrug ethionamide is regulated by mycobacteria" A. R. Baulard et al., Journal of Biological Chemistry, 2000, pp. 28326-28331.
Более конкретно, противотуберкулезный агент/средство может быть выбрано из семейства тиоамидов, таких как этионамид, протионамид, изоксил и тиазетазон.
В одном варианте осуществления изобретения, соединение по изобретению (т.е. соединение формулы (I) или его фармацевтический приемлемая соль) используется в комбинации с этионамидом. В этом варианте осуществления изобретения, соединения по изобретению (т.е. соединение формулы (I) или его фармацевтически приемлемая соль) показали способность потенцировать активность этионамида.
Комбинации, для удобства их использования, могут быть представлены в форме фармацевтической композиции или препарата. Таким образом, здесь также представляется фармацевтическая композиция, содержащая (а) соединение по изобретению (т.е. соединение формулы (I) или его фармацевтически приемлемая соль), как описано здесь, вместе с (b) одним или несколькими фармацевтически приемлемыми носителями, как описано здесь, и (c) по меньшей мере одним другим противотуберкулезным лекарственным агентом/средством, и (d) необязательно, противовирусным агентом/средством, включая антиретровирусные агенты.
Соединение по изобретению (т.е. соединение формулы (I) или его фармацевтически приемлемая соль) и дополнительный терапевтический агент можно вводить вместе или по-отдельности, и при введении по-отдельности введение можно выполнять раздельно или последовательно в любом порядке (этим же или другим путем введения). Количество соединения по изобретению (т.е. соединение формулы (I) или его фармацевтически приемлемая соль) и дополнительный терапевтически активный агент (или агенты) и соответствующие временные показатели введения выбирают таким образом, чтобы достичь желаемого комбинированного терапевтического эффекта.
Примеры
Далее изобретение будет проиллюстрировано с помощью следующих неограничивающих примеров. Несмотря на то, что ниже описаны конкретные варианты осуществления изобретения, специалисту в данной области техники понятно, что могут быть сделаны различные изменения и модификации. Указания в отношении способов получения препаратов, полученных способом, аналогичным или подобным способом, или общим способом получения других препаратов, могут включать изменения обычных параметров, таких как время, температура, условия обработки, незначительные изменения количеств реагентов и т.п.
Сокращения
Ниже приведены определения некоторых сокращений и символов, используемые здесь. Следует иметь в виду, что этот список не является исчерпывающим, и смысл используемых сокращений и символов, не вошедших в перечень определений, приведенный ниже, является очевидным для специалистов в данной области техники. В описании изобретения химические элементы идентифицированы в соответствии с периодической таблицей элементов.
ACN/MeCN ацетонитрил
anh., безводн. безводный
CDCl3 дейтерированный хлороформ
CD2Cl2 дейтерированный дихлорметан
CyHex циклогексан
DCM дихлорметан
DIPEA диизопропилэтиламин
DMAP 4-диметиламинопиридин
DMF диметилформамид
DMSO-d6 дейтерированный диметилсульфоксид
EDC N-(3-диметиламинопропил)-N'-этилкарбодиимид
EtOAc этилацетат
Ex., Прим. Пример
HBTU гексафторфосфат N, N,N′,N′-тетраметил-O-(1H-бензотриазол-1-ил)урония
HPLC, ВЭЖХ высокоэффективная жидкостная хроматографии
Int. промежуточное соединение
M молярность
MS масс-спектроскопия
min, мин минуты
N нормальность
NaH гидрид натрия
NMR, ЯМР спектр ядерного магнитного резонанса
TFA трифторуксусная кислота
TEA триэтиламин
THF тетрагидрофуран
TLC, ТСХ тонкослойная жидкостная хроматография
rt, комн. темп. комнатная температура
UPLC сверхпроизводительная жидкостная хроматография
При записи результатов протонного ядерного магнитного резонанса (1Н-ЯМР) химические сдвиги (δ) представлены в миллионных долях по отношению к тетраметилсилану (TMS), используемому в качестве внутреннего стандарта. Для данных ЯМР использованы следующие сокращения: с=синглет, д=дублет, т=триплет, кв=квартет, м=мультиплет, дд=двойной дублет, дт=двойной триплет, набл. = наблюдаемый, ш=широкий, уш. - уширенный. Масс-спектры получали с использованием методов ионизации с электрораспылением (ES). Все температуры приведены в градусах Цельсия.
В некоторых из нижеприведенных примеров получения промежуточных соединений и соединений по изобретению, исходные материалы идентифицированы путем ссылки на другие номера примеров промежуточных соединений или примеров соединений по изобретению. Это не означает, что материал какого-либо конкретного промежуточного соединения или соединения по примеру обязательно используется на последующей стадии описываемого примера, поскольку такое указание используется для обозначения названия соответствующего соединения.
Промежуточные соединения
Промежуточное соединение 1: трет-бутил-3-бром-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-карбоксилат
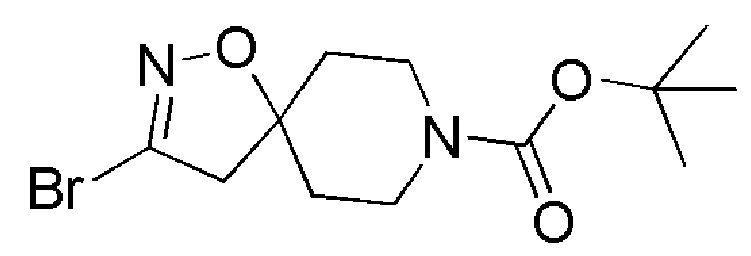
К суспензии трет-бутил-4-метиленпиперидин-1-карбоксилата (FLUOROCHEM, 2 г, 10,14 ммоль) и бикарбонат натрия (8,52 г, 101,38 ммоль) в EtOAc (50 мл) добавляли дибромформальдоксим (FLUOROCHEM, 5 г, 24,74 ммоль). Реакционную смесь перемешивали в течение 5 дней при комнатной температуре. Добавляли целит, полученную суспензию фильтровали под вакуумом и промывали EtOAc. Растворитель выпаривали и остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента градиент циклогексан/EtOAc (циклогексан/EtOAc от 95/5 до 90/10), с получением указанного в заголовке соединения (7,29 г, 92%) в виде белого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ м.д.: 3,72-3,64 (м, 2H), 3,40-3,31 (м, 2H), 2,95 (с, 2H), 1,92-1,84 (м, 2H), 1,72-1,65 (м, 2H), 1,45 (с, 9H); [ES+MS] m/z 319, 321 (MH+).
Промежуточное соединение 2: трет-бутил-3-(2,2,2-трифторэтокси)-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-карбоксилат
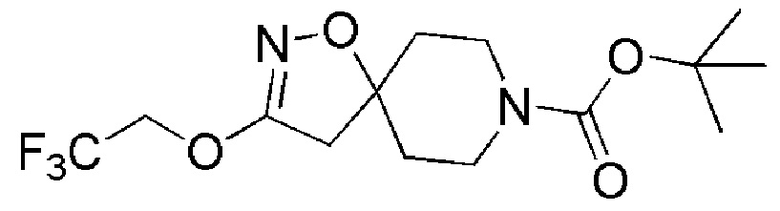
К раствору 2,2,2-трифторэтанола (SIGMA-ALDRICH, 282 мг, 2,82 ммоль) в 2 мл THF при 0°С добавляли NaH (SIGMA-ALDRICH, 1 мг 12,8, 2,82 ммоль) и смесь перемешивали в течение 30 минут. Затем при 0°C добавляли промежуточное соединение 1, трет-бутил-3-бром-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-карбоксилат (300 мг, 0,94 ммоль), и смесь перемешивали при 80°С в течение 2 дней. Раствор промывали насыщенным раствором хлорида аммония и экстрагировали с помощью EtOAc (x2). Органические слои сушили над (безводн.) MgSO4 и фильтровали. Растворитель выпаривали под вакуумом. Остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента градиент циклогексан/EtOAc (0-20% EtOAc в течение 30 минут и 20% EtOAc в течение 10 минут), с получением указанного в заголовке соединения (200 мг, 63%).
[ES+ MS] m/z 339 (MH+).
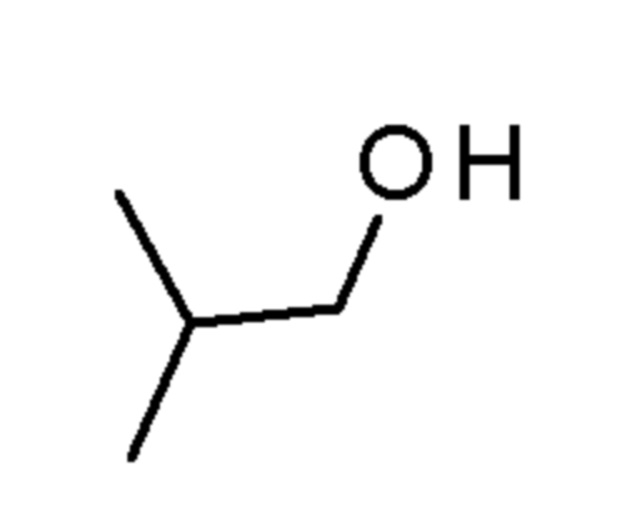
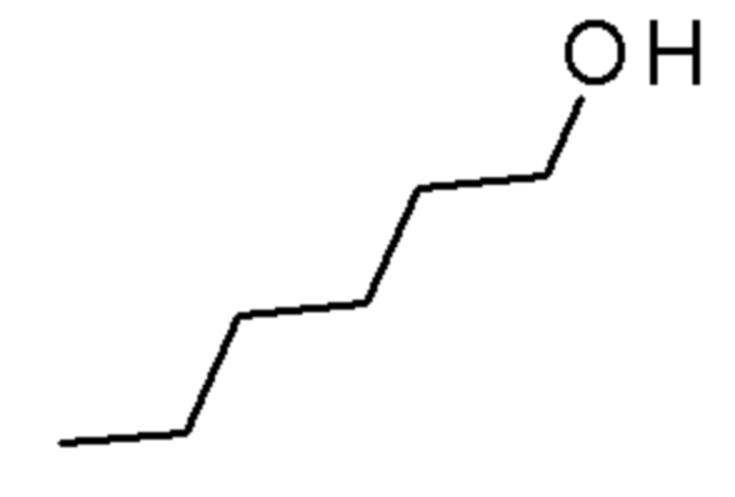
Промежуточные соединения 3-5 получали способами, аналогично способу, описанному для промежуточного соединения 2, но с заменой спирта (2,2,2-трифторэтанола) на соединения, указанные в Таблице 1. Продукты очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента градиент циклогексан/EtOAc (0-10% EtOAc в течение 30 минут и 10% EtOAc в течение 10 минут).
Таблица 1
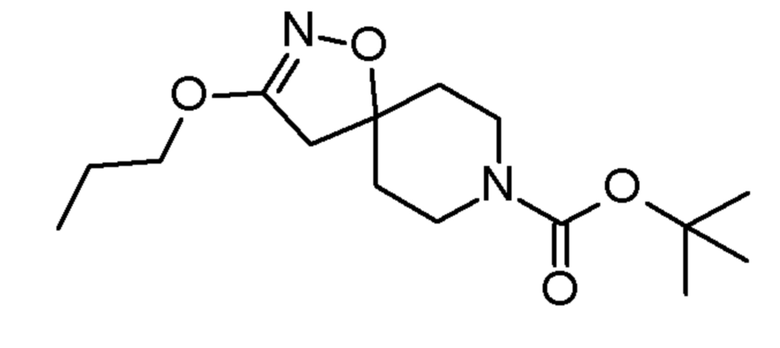
SIGMA-ALDRICH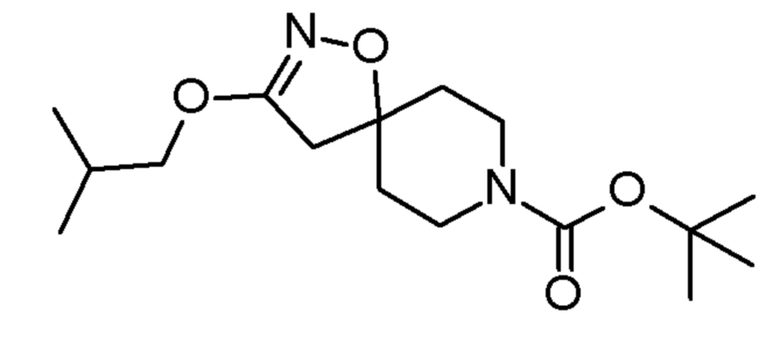
SIGMA-ALDRICH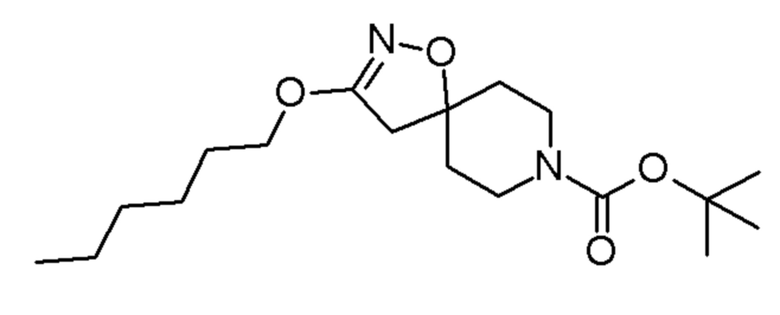
ACROS
Промежуточное соединение 6: гидрохлорид 4-метиленпиперидина
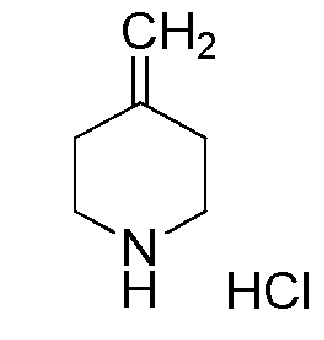
К раствору трет-бутил-4-метиленпиперидин-1-карбоксилата (APOLLO, 10 г, 50,7 ммоль) в диоксане (130 мл) при 0°С, добавляли 4М раствор HCl в диоксане (ALFA-AESAR, 130 мл, 507 ммоль, 10 экв.), и смесь перемешивали при комнатной температуре в течение ночи. Контроль с помощью UPLC и TLC показал, что реакция завершена. Растворитель удаляли в вакууме, получая целевое соединение, гидрохлорид 4-метиленпиперидина, которое использовали на следующей стадии без дополнительной очистки (7,6 г, 100%).
1H ЯМР (400МГц, DMSO-d6) δ м.д.: 9,19 (уш. с, 2H), 4,86 (с, 2H), 3,06 (т, J=6,0 Гц, 4H), 2,41 (т, J=6,0 Гц, 4H).
Промежуточное соединение 7: 4,4,4-трифтор-1-(4- метиленпиперидин-1-ил)бутан-1-он
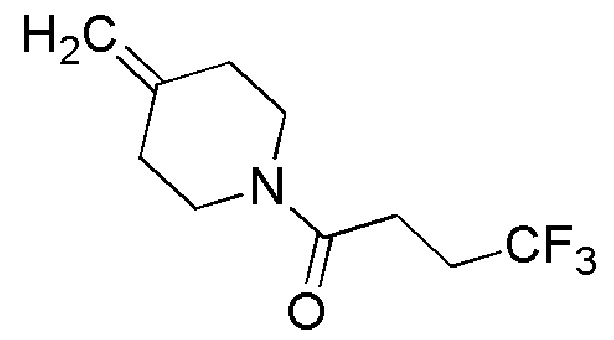
Раствор EDC.HCI (ALFA-AESAR, 19,4 г, 101,3 ммоль), ТЭА (ALFA-AESAR, 28 мл, 202,5 ммоль) и 4,4,4-trifluorobutyric кислоты (ALFA-AESAR, 14,4 г, 101,3 ммоль) в DCM (200 мл) перемешивали при комнатной температуре в течение 10 мин, затем добавляли промежуточное соединение 6 (6,77 г, 50,6 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Смесь промывали водой, разделяли на 2 фазы, и затем водную фазу экстрагировали с помощью DCM. Собранный органический слой сушили над Na2SO4 (безводн.), фильтровали и упаривали. Полученный неочищенный продукт очищали с помощью флэш-хроматографии (Si SNAP 340, циклогексан/EtOAc, от 9/1 до 6/4), получая целевой продукт (10,34 г, 92%) в виде бледно-желтого масла.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 4,83 (с, 2H), 3,70-3,59 (м, 2H), 3,51-3,44 (м, 2H), 2,65-2,47 (м, 4H), 2,30-2,21 (м, 4H); [ES+MS] m/z 222 (MH+).
Промежуточное соединение 8: оксим 2,2,2-трифторацетальдегида
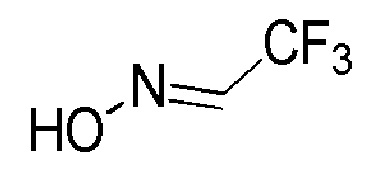
К перемешиваемому раствору 2,2,2-трифтор-1-метоксиэтанола (ALFA-AESAR, 20 г, 153,8 ммоль) и гидрохлорида гидроксиламина (AVRA, 12,8 г, 184,5 ммоль) в метаноле (30 мл) при 0°С добавляли воду (70 мл), а затем при 0°C медленно добавляли 50%-й раствор гидроксида натрия (FINAR, 36 мл). Температуру реакционной смеси повышали до 26°C и перемешивали в течение 16 часов при той же температуре. Реакцию контролировали с помощью TLC. По завершении реакции к реакционной смеси добавляли н-гексан (250 мл), и слои разделяли. Водный слой подкисляли (до рН=3) 6М HCl, а затем экстрагировали диэтиловым эфиром (3 × 500 мл). Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4 (безводн.), фильтровали и концентрировали при пониженном давлении, с получением указанного в заголовке соединения в виде бесцветной жидкости (18 г, неочищенный продукт).
1H ЯМР (400 МГц, CDCl3) δ м.д.: 9,11 (уш. с, 1H), 7,54-7,46 (м, 1H).
Промежуточное соединение 9: 2,2,2-трифтор-N-гидроксиацетимидоилбромид
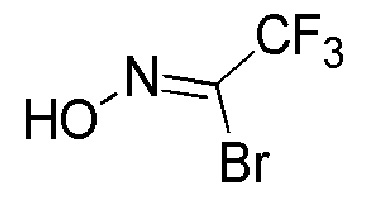
К перемешиваемому раствору промежуточного соединения 8 (18 г, 159,2 ммоль) в DMF (90 мл) при 0°C добавляли раствор N-бромсукцинимида (AVRA, 31,1 г, 175,2 ммоль) в DMF (90 мл). Реакционную смесь перемешивали при 26°С в течение 3 часов. Реакцию контролировали с помощью TLC. По завершении реакции реакционную смесь выливали в воду со льдом (300 мл) и экстрагировали диэтиловым эфиром (3 × 400 мл). Органический слой промывали насыщенным солевым раствором (300 мл), сушили над Na2SO4 (безводн.), фильтровали, и концентрировали при пониженном давлении, с получением указанного в заголовке соединения (25 г, неочищенный продукт) в виде масла коричневого цвета.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 11,55 (уш. с, 1H).
Промежуточное соединение 10: оксим пропиональдегида
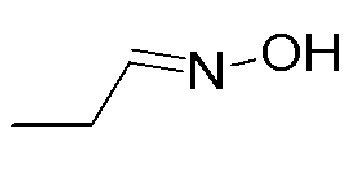
К перемешиваемому раствору пропиональдегида (ALFA-AESAR, 2 г, 34,43 ммоль) и K2CO3 (CHEMLABS, 9,36 г, 68,87 ммоль) в DCM (30 мл), добавляли гидрохлорид гидроксиламина (AVRA, 2,86 г, 41,32 ммоль). Полученную реакционную смесь перемешивали в течение 16 часов при температуре 26°C. Реакцию контролировали с помощью TLC. По окончании реакции реакционную смесь разбавляли водой (200 мл) и экстрагировали с помощью DCM (2 × 50 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 (безводн.), фильтровали, и концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,0 г) в виде твердого вещества белого цвета. Полученное неочищенное соединение использовали на следующей стадии без какой-либо дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ: 8,50 (уш. с, 1H), 6,70 (т, J=5,4 Гц, 1H), 2,26-2,22 (м, 2H), 1,14-1,09 (м, 3H).
Промежуточное соединение 11: N-гидроксипропионимидоилхлорид
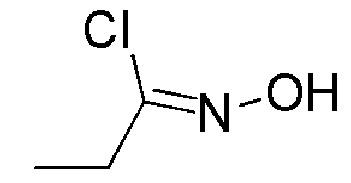
К перемешиваемому раствору промежуточного соединения 10 (1 г, 13,681 ммоль) в DMF (10 мл) при 26°С добавляли по порциям N-хлорсукцинимид (AVRA, 2,19 г, 16,41 ммоль), реакционную смесь перемешивали в течение 30 минут при температуре 26°C. Реакцию контролировали с помощью TLC. По окончании реакции реакционную смесь разбавляли водой (100 мл) и экстрагировали с помощью EtOAc (2 × 50 мл), объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 (безводн.), фильтровали и концентрировали при пониженном давлении, с получением указанного в заголовке соединения в виде бесцветной жидкости (1,0 г, 68,4%).
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,32 (уш. с, 1H), 2,53 (кв, J=7,4 Гц, 2H), 1,21 (т, J=7,4 Гц, 3H).
Промежуточное соединение 12: N-гидроксиацетимидоилхлорид
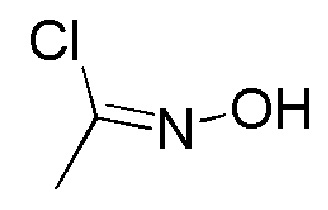
К раствору оксима (Е)-ацетальдегида (TCI, 1 г, 16,949 ммоль) в DMF (10 мл) при 0°С добавляли порциями N-хлорсукцинимид (AVRA, 2,9 г, 22,03 ммоль). Температуру реакционной смеси повышали до 26°C, и перемешивали в течение 3 часов при этой же температуре. Реакцию контролировали с помощью TLC. По окончании реакции реакционную смесь разбавляли водой со льдом (100 мл) и экстрагировали диэтиловым эфиром (2 × 300 мл). Органический слой промывали насыщенным солевым раствором (200 мл), сушили над Na2SO4 (безводн.), фильтровали, и фильтрат концентрировали при пониженном давлении, с получением указанного в заголовке соединения (1,2 г, неочищенный продукт) в виде бесцветной жидкости.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,48 (уш. с, 1H), 2,27 (с, 3H).
Промежуточное соединение 13: трет-бутил-3-(трифторметил)-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-карбоксилат
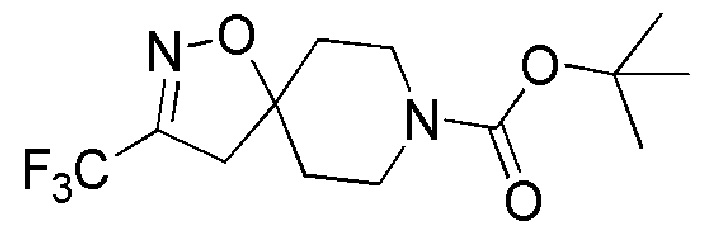
К перемешиваемому раствору промежуточного соединения 9 (6 г, 30,41 ммоль) в DCM (60 мл) при 0°С добавляли порциями бикарбонат калия (ALFA-AESAR, 6,09 г, 60,83 ммоль). Затем при 0°C добавляли по каплям трет-бутил-4-метиленциклогексанкарбоксилат (OAKWOOD, 5,86 г, 30,415 ммоль). Реакционную смесь перемешивали при 26°C в течение 16 часов. Реакцию контролировали с помощью TLC. По окончании реакции реакционную смесь выливали в воду (200 мл), и экстрагировали EtOAc (3 × 250 мл). Органический слой промывали насыщенным солевым раствором (200 мл) и сушили над Na2SO4 (безводн.), затем фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель, 100-200 меш), элюируя градиентом 0-10% EtOAc в петролейном эфире. Собирали чистые фракции и концентрировали их при пониженном давлении, с получением указанного в заголовке соединения (6,5 г, 56%) в виде светло-желтой жидкости.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 3,80-3,63 (м, 2H), 3,42-3,35 (м, 2H), 2,91 (кв, J=1,5 Гц, 2H), 1,94-1,89 (м, 2H), 1,77-1,67 (м, 2H), 1,47 (с, 9H); [ES- MS] m/z 307 (M-H).
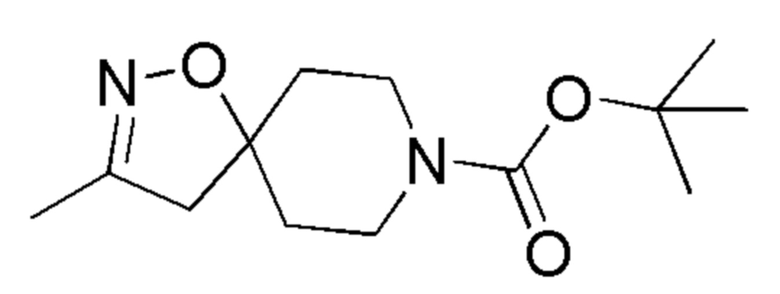
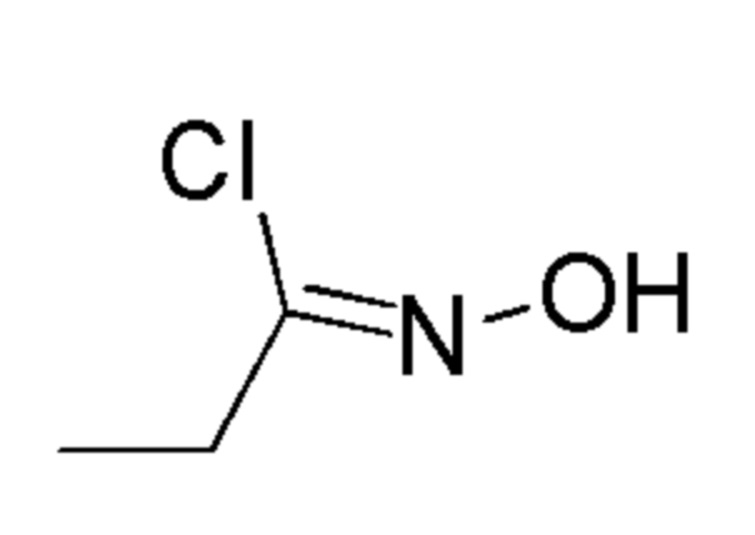
Промежуточные соединения 14 и 15 получали способами, аналогично способу, описанному для промежуточного соединения 13, но вместо промежуточного соединения 9 использовали соединения, указанные в Таблице 2, а также с заменой бикарбоната калия на TEA.
Таблица 2
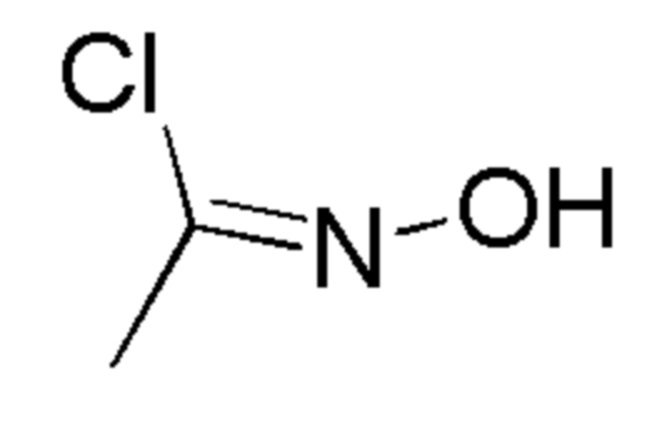
Промежут. соед.
12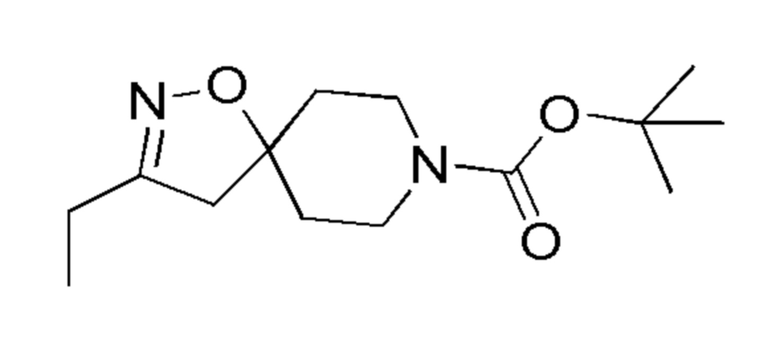
Промежут. соед.
11
Промежуточное соединение 16: гидрохлорид 3-(трифторметил)-1-окса-2,8-диазаспиро[4,5]дец-2-ена
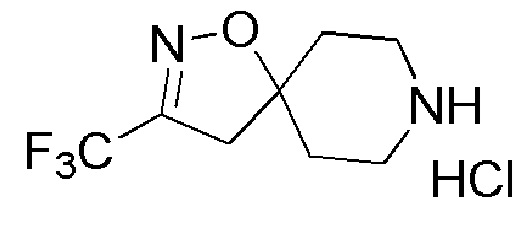
К перемешиваемому раствору промежуточного соединения 13 (3 г, 9,73 ммоль) в EtOAc (30 мл) при 0°C добавляли по каплям 4М раствор HCl в EtOAc (SYMAX, 30 мл). Реакционную смесь перемешивали при 26°С в течение 2 часов. Реакцию контролировали с помощью TLC. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Остаток промывали диэтиловым эфиром (150 мл) и сушили, с получением указанного в заголовке соединения (2,21 г, неочищенный продукт) в виде не совсем белого твердого вещества.
1H ЯМР (400 МГц, DMSO-d6) δ м.д.: 9,42 (уш. с, 2H), 3,28 (с, 2H), 3,20-3,04 (м, 4H), 2,14-2,00 (м, 4H); [ES+MS] m/z 209 (MH+).
Промежуточные соединения 17 и 18 получали способами, аналогично способу, описанному для промежуточного соединения 16, но вместо промежуточного соединения 13 использовали соединения, указанные в Таблице 3.
Таблица 3
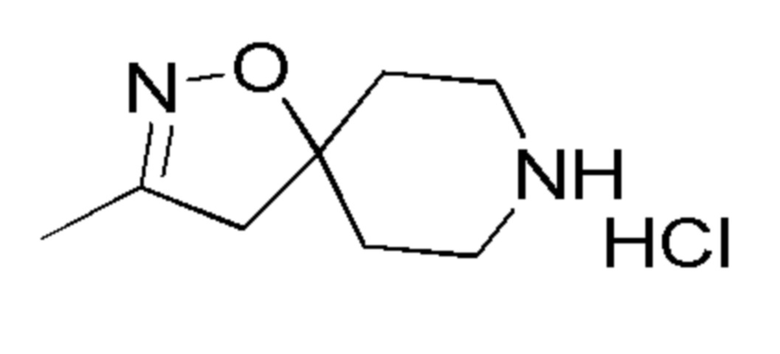
14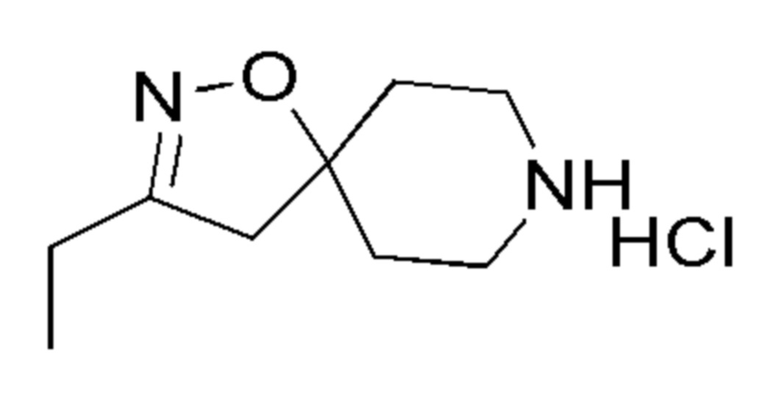
15
Примеры
Пример 1: 4,4,4-трифтор-1-[3-(2,2,2-трифторэтокси)-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил]бутан-1-он
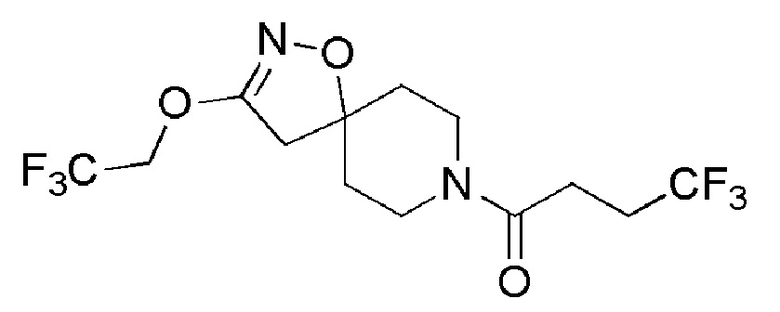
Промежуточное соединение 2 (200 мг, 0,59 ммоль) растворяли при 0°C в 2 мл DCM и добавляли по каплям TFA (2 мл). Смесь перемешивали при 0°C в течение 10 минут. К реакционной смеси добавляли насыщенный раствор бикарбоната натрия, и продукт экстрагировали с помощью EtOAc. Органический слой сушили над MgSO4 (безводн.), фильтровали, и затем концентрировали досуха с получением остатка желтого цвета.
К раствору DIPEA (ALFA-AESAR, 0,3 мл, 1,76 ммоль) в DMF (5 мл) в атмосфере аргона при 0°С добавляли HBTU (SIGMA-ALDRICH, 669 мг, 1 ммоль 0,76) и 4,4,4-трифторбутановую кислоту (ALFA-AESAR, 125 мг, 0,88 ммоль). Добавляли указанный выше остаток, и смесь перемешивали в атмосфере аргона при комнатной температуре в течение ночи. Затем под вакуумом удаляли DMF. Остаток очищали с помощью HPLC (изократический градиент, ACN/Н2О формиат аммония, рН 3,8: 36/64), с получением указанного в заголовке соединения (137 мг, 64%).
1H ЯМР (300 МГц, CD2Cl2) δ м.д.: 4,54 (кв, J=8,3 Гц, 2H), 4,13-4,05 (м, 1H), 3,66-3,45 (м, 2H), 3,35-3,26 (м, 1H), 2,87 (с, 2H), 2,68-2,37 (м, 4H), 2,10-1,84 (м, 2H), 1,78-1,66 (м, 2H); [ES+MS] m/z 363 (MH+).
Соединения по Примерам 2-4 получали способами, аналогично способу, описанному в Примере 1, заменяя промежуточное соединение 2 на соединения, указанные в Таблице 4. Также указаны изменения на стадии очистки.
Таблица 4
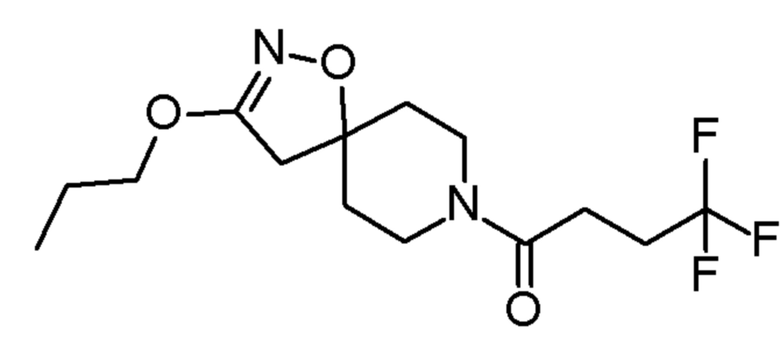 4,4,4-трифтор-1-(3-пропокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он
4,4,4-трифтор-1-(3-пропокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он
см. примечание a
3
0,51 ммоль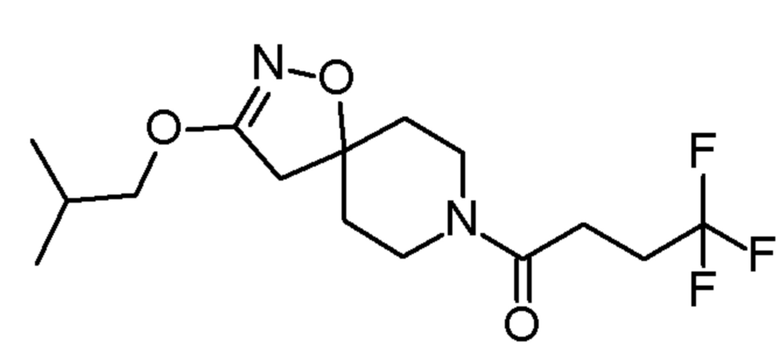 4,4,4-трифтор-1-(3-изобутокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он
4,4,4-трифтор-1-(3-изобутокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он
см. примечание b
4
0,67 ммоль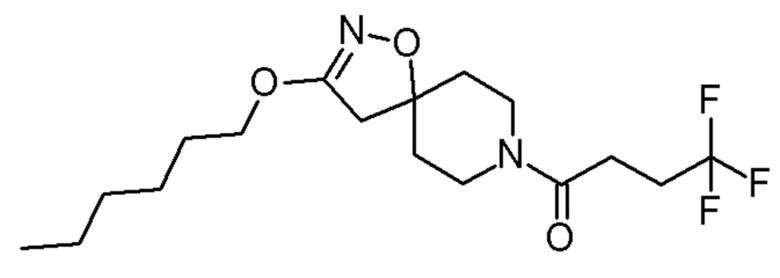 4,4,4-трифтор-1-(3-гексилокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он
4,4,4-трифтор-1-(3-гексилокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он
см. примечание c
5
0,77 ммоль
a очистка с помощью HPLC (pH 3,8, изократический градиент, ACN/H2O формиат аммония: 33/67)
b очистка с помощью HPLC (pH 3,8, изократический градиент, ACN/H2O формиат аммония: 39/61)
c очистка с помощью HPLC (pH 3,8, изократический градиент, ACN/H2O формиат аммония: 54/46)
Пример 5 и промежуточное соединение 19: 1-(3-бром-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-он
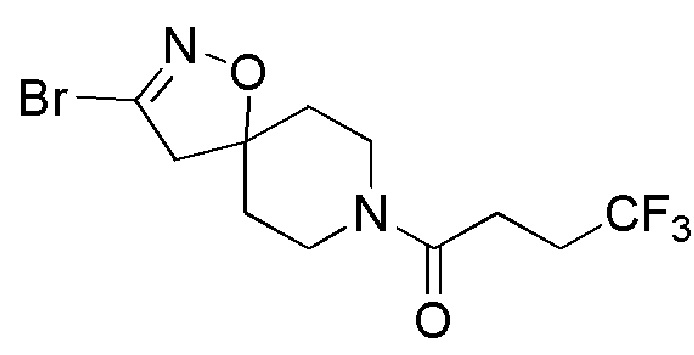
К суспензии промежуточного соединения 7 (9,3 г, 42 ммоль) и бикарбоната натрия (ALFA-AESAR, 35,3 г, 420 ммоль) в EtOAc (300 мл) добавляли дибромформальдоксим (COMBI-BLOCKS, 10,2 г, 50 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 2-х дней. Затем добавляли целит, полученную суспензию фильтровали под вакуумом и промывали EtOAc, растворитель выпаривали и остаток (около 20 г) очищали с помощью флэш-хроматографии (Si SNAP 340, CyHex/EtOAc, от 8/2 до 1/1), с получением указанного в заголовке соединения (12,23 г, 85%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 4,32-4,20 (м, 1H), 3,69-3,60 (м, 1H), 3,59-3,47 (м, 1H), 3,31-3,20 (м, 1H), 3,00 (с, 2H), 2,67-2,43 (м, 4H), 2,07-1,96 (м, 2H), 1,78-1,68 (м, 2H); [ES+MS] m/z 343, 345 (MH+).
Пример 6: 1-(3-этокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-он
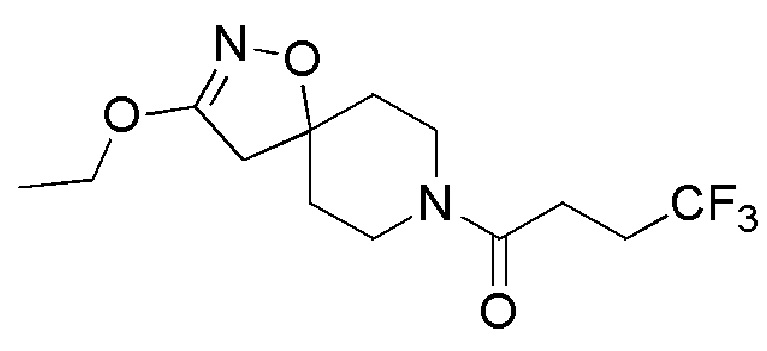
К перемешиваемому раствору промежуточного соединения 19 (0,100 г, 0,29 ммоль) в этаноле (4 мл) добавляли твердый карбонат калия (ALFA-AESAR, 0,101 г, 0,73 ммоль), и полученную суспензию энергично перемешивали при 70°С в запаянном флаконе в течение 20 часов. Контроль с помощью UPLC показал наличие исходного материала. Затем к реакционной смеси добавляли K3PO4 (ALFA-AESAR, 0,168 г, 0,73 ммоль) и нагревали при 70°С в течение дополнительных 4 часов до полного расхода исходного материала. Суспензию фильтровали, фильтрат выпаривали, получая неочищенный материал, который далее очищали с помощью полупрепаративной LCMS, с получением указанного в заголовке соединения (0,0427 г, 47%) в виде бледно-желтого твердого вещества.
1H ЯМР (400 МГц, DMSO-d6) δ м.д.: 4,08 (кв, J=7,03 Гц, 2H), 3,65-3,55 (м, 1H), 3,50-3,37 (м, 3H), 2,84 (с, 2H), 2,65-2,56 (м, 2H), 2,49-2,40 (м, 2H), 1,78-1,56 (м, 4H), 1,27 (т, J=7,03 Гц, 3H); [ES+MS] m/z 309 (MH+).
Пример 7: 4,4,4-трифтор-1-(3-метокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он
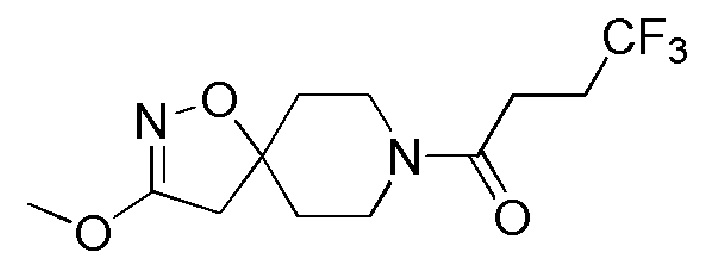
К перемешиваемому раствору промежуточного соединения 19 (1 г, 2,914 ммоль) в метаноле (10 мл) при 0°C добавляли порциями метоксид натрия (AVRA, 786 мг, 14,5 ммоль). Реакционную смесь нагревали до 70°C в течение 16 часов. Реакцию контролировали с помощью TLC. По завершении реакции реакционную смесь гасили водой (100 мл) и экстрагировали EtOAc (3 × 200 мл). Органический слой промывали насыщенным солевым раствором (200 мл), сушили над Na2SO4 (безводн.), фильтровали и упаривали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель, 100-200 меш), элюируя смесью 0-30% EtOAc в петролейном эфире. Собирали чистые фракции и концентрировали их при пониженном давлении, с получением указанного в заголовке соединения (800 мг, 93%) в виде не совсем белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 4,24-4,17 (м, 1H), 3,86 (с, 3H), 3,63-3,49 (м, 2H), 3,30-3,21 (м, 1H), 2,75 (с, 2H), 2,62-2,44 (м, 4H), 2,08-1,95 (м, 2H), 1,71-1,61 (м, 2H); [ES+MS] m/z 295 (MH+).
Пример 8: 4,4,4-трифтор-1-(3-(трифторметил)-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он
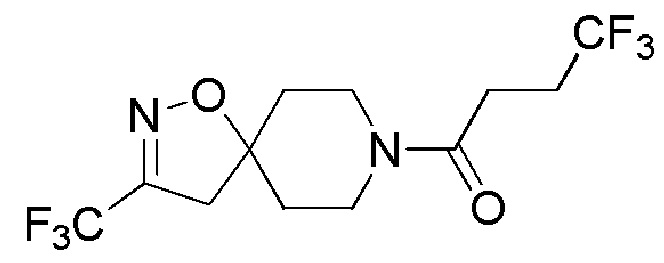
К перемешиваемому раствору промежуточного соединения 16 (2,2 г, 9,016 ммоль) и 4,4,4-трифторбутановой кислоты (Oakwood, 1,53 г, 10,819 ммоль) в DMF (25 мл) при 0°C добавляли EDC.HCI (SILVERY, 4,3 г, 22,54 ммоль) и DMAP (AVRA, 3,2 г, 27,04 ммоль). Реакционную смесь перемешивали при 26°C в течение 16 часов. Реакцию контролировали с помощью TLC. По окончании реакции реакционную смесь выливали в ледяную воду (200 мл) и экстрагировали EtOAc (3 × 250 мл). Органический слой промывали насыщенным солевым раствором (200 мл), сушили над Na2SO4 (безводн.), фильтровали и фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт (3,2 г) очищали с помощью колоночной хроматографии (силикагель, 100-200 меш), элюируя смесью 0-20% EtOAc в петролейном эфире. Собирали чистые фракции и концентрировали при пониженном давлении, с получением указанного в заголовке соединения (1,7 г, 56%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 4,33-4,25 (м, 1H), 3,74-3,62 (м, 1H), 3,60-3,49 (м, 1H), 3,33-3,21 (м, 1H), 2,94 (с, 2H), 2,64-2,45 (м, 4H), 2,07-1,94 (м, 2H), 1,80-1,68 (м, 2H); [ES+MS] m/z 333 (MH+).
Соединения по Примерам 9 и 10 получали способами, аналогично способу, описанному в Примере 8, заменяя промежуточное соединение 16 на соединения, указанные в Таблице 5.
Таблица 5
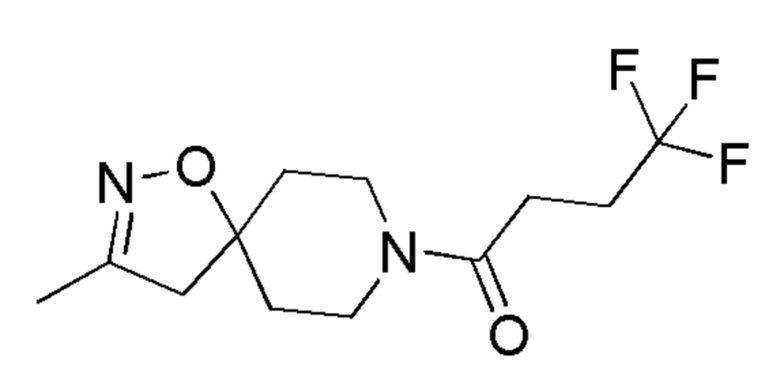
4,4,4-трифтор-1-(3-метил-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он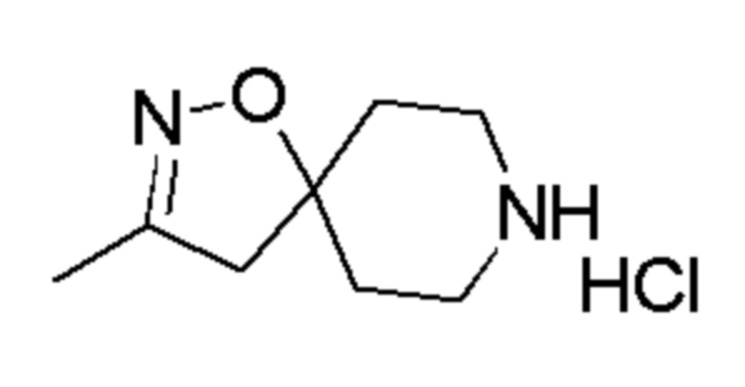
1,83 ммоль
Промежут. соед.
17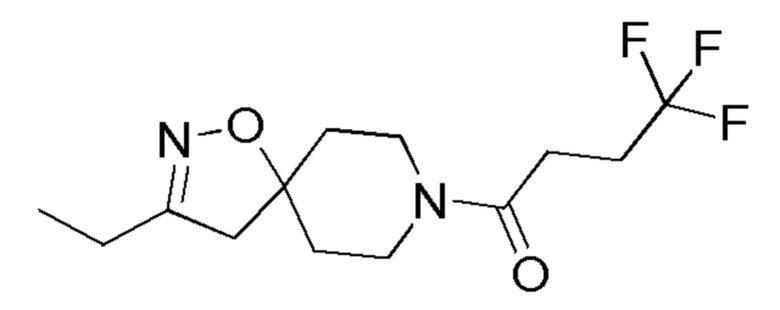
1-(3-этил-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-он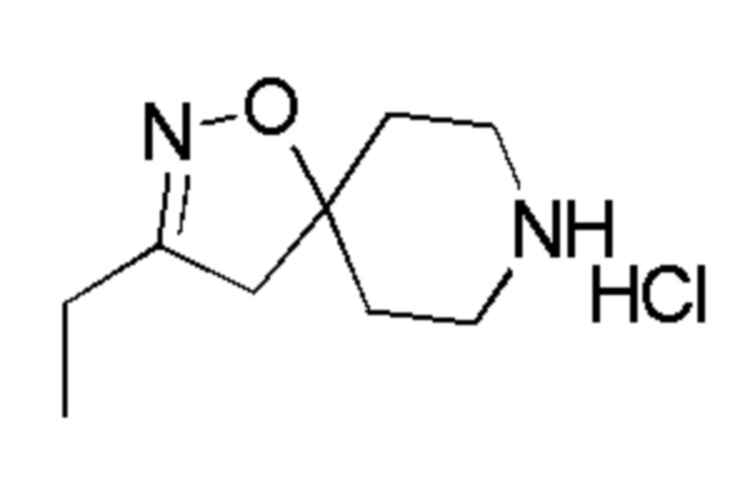
2,1 ммоль
Промежут. соед.
18
Пример 11: 8-(4,4,4-трифторбутаноил)-1-окса-2,8-диазаспиро[4,5]дец-2-ен-3-карбонитрил
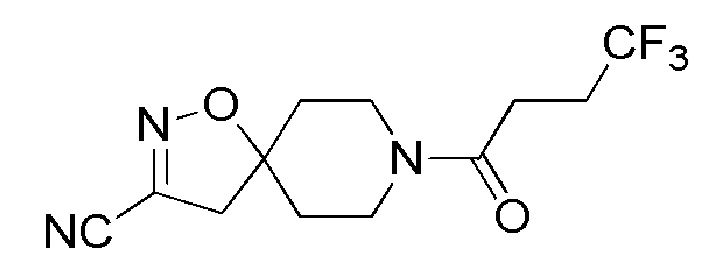
К перемешиваемому раствору промежуточного соединения 1 (5,5 г, 17,231 ммоль) в EtOAc (50 мл) при 0°C добавляли 4М раствор HCl в EtOAc (SYMAX, 50 мл). Затем реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакцию контролировали с помощью TLC. По окончании реакции реакционную смесь концентрировали при пониженном давлении. Остаток растирали с диэтиловым эфиром (3 × 20 мл), и твердое вещество сушили с получением смеси (4,0 г, неочищенный продукт) гидрохлорида 3-хлор-1-окса-2,8-диазаспиро[4,5]дец-2-ена (основной компонент: [ES+MS] m/z 175 (MH+)) и 3-бром-1-окса-2,8-диазаспиро[4,5]дец-2-ена (минорный компонент: [ES+ MS] m/z 219, 221 (MH+)) в виде бледно-желтого твердого вещества.
К перемешиваемому раствору этой смеси (500 мг) и 4,4,4-трифторбутановой кислоты (MATRIX, 500 мг, 3,554 ммоль) в DMF (10 мл) при 0°C добавляли DMAP (AVRA, 860 мг, 4,109 ммоль) и EDC.HCl (ASHVARSHA, 1,13 г, 5,92 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакцию контролировали с помощью TLC. По окончании реакции реакционную смесь выливали в ледяную воду (50 мл) и экстрагировали EtOAc (2 × 50 мл). Органический слой промывали 1N HCl (50 мл) и насыщенным солевым раствором (100 мл), сушили над Na2SO4 (безводн.), фильтровали, и фильтрат выпаривали при пониженном давлении, с получением 600 мг смеси соединений 1-(3-хлор-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-она и 1-(3-бром-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-она в виде вязкой жидкости коричневого цвета.
К раствору указанной выше смеси соединений (200 мг) в DMFА (5 мл) при 26°С добавляли цианид натрия (ASHVARSHA, 65 мг, 1,342 ммоль). Реакционную смесь нагревали до 100°C и перемешивали в течение 8 часов при той же температуре. Ход реакции контролировали с помощью TLC. По окончании реакции реакционную смесь гасили холодной водой со льдом (5 мл), и экстрагировали EtOAc (2 × 50 мл). Органический слой промывали насыщенным солевым раствором (2 × 50 мл), сушили над Na2SO4 (безводн.), фильтровали, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии (силикагель, 100-200 меш), элюируя 80% EtOAc в петролейном эфире. Собирали чистые фракции, и их концентрировали при пониженном давлении, с получением указанного в заголовке соединения (150 мг, 78%) в виде не совсем белого твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 4,35-4,25 (м, 1H), 3,75-3,63 (м, 1H), 3,59-3,49 (м, 1H), 3,32-3,21 (м, 1H), 2,96 (с, 2H), 2,64-2,43 (м, 4H), 2,05-1,92 (м, 2 H), 1,80-1,68 (м, 2H); [ES+MS] m/z 290 (MH+).
Пример 12: 4,4,4-трифтор-1-(3-фтор-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он
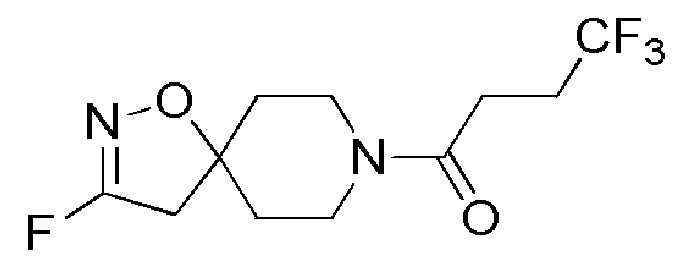
К смеси промежуточных продуктов, как и в Примере 11 (1-(3-хлор-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-он и 1-(3-бром-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-он) (3 г) в DMSO (20 мл) при 26°C добавляли порциями фторид калия (COMBIBLOCKS, 1,7 г, 30,131 ммоль). Реакционную смесь нагревали до 150°C и перемешивали в течение 3 часов в условиях воздействия микроволнового излучения. Ход реакции контролировали с помощью TLC. По завершении реакции реакционную смесь разбавляли водой (200 мл) и экстрагировали EtOAc (2 × 150 мл). Органический слой промывали насыщенным солевым раствором (150 мл), сушили над Na2SO4 (безводн.), фильтровали, и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью препаративной HPLC (колонка Kromosil) градиент от 0% до 100% ACN/H2O (0,2% муравьиной кислоты), 16 мин. Собирали чистые фракции, и удаляли растворитель путем лиофилизации, с получением указанного в заголовке соединения (11 мг, 0,4%) в виде бледно-желтой смолы.
1H ЯМР (400 МГц, DMSO-d6) δ м.д.: 3,61-3,41 (м, 4H), 3,14 (d, J=4,9 Гц, 2H), 2,68-2,58 (м, 2H), 2,55-2,44 (м, 2H), 1,84 (т, J=5,8 Гц, 2H), 1,74 (т, J=5,9 Гц, 2H); [ES+MS] m/z 283 (MH+).
Биологическая активность
Измерение ингибирования роста штаммов M. tuberculosis GFP при использовании комбинации этионамида (ETH) и соединений по Примерам 1-11
1. Конструирование микобактерий рекомбинантных штаммов.
Штамм M. tuberculosis H37Rv-GFP. Рекомбинантный штамм M. tuberculosis H37Rv, экспрессирующий зеленый флуоресцентный белок (GFP-H37Rv), получали путем трансформации интегративной плазмиды pNIP48 (Abadie et al., 2005; Cremer et al., 2002). В этой плазмиде, полученной из микобактериофага MS6, ген GFP был клонирован под контролем сильного микобактериального промотора pBlaF, и она конститутивно экспрессирует GFP. Эта плазмида также содержит ген устойчивости к гигромицину.
Штамм M. tuberculosis W4-E1-GFP (мутанатный). Штамм M. tuberculosis Е1 получен из штамма Beijing W4, который подвергали селекции на чашках с агаром, содержащим этионамид (20 мкг/мл). Этот штамм имеет мутацию Gly343Ala в EthA. Штамм W4-Е1 трансформировали с использованием pNIP48, как описано выше, с получением флуоресцентного штамма W4-E1-GFP.
2. Рост и подготовка флуоресцентных микобактерий
Бактериальные культуры хранили при -80°C, и их инокулировали в 5 мл среды Middlebrook 7H9, дополненной коктейлем олеиновая кислота-альбумин-декстроза-каталаза (OADC, Difco, Sparks MD, США) и 50 мкг/мл гигромицина (Invitrogen, Carlsbad, CA, США) в колбы для культуры тканей объемом 25 см2. Колбы инкубировали при 37°С без встряхивания в течение 7 дней. Затем культуры разбавляли свежей культуральной средой до достижения оптической плотности OD600, равной 0,1. Эти разбавленные культуры в объеме 50 мл переносили в культуральные флаконы (75 см2), и их инкубировали в течение 7 дней при 37°C без встряхивания.
3. Подготовка микропланшетов
Этионамид (Sigma, E6005) разводили в DMSO до концентраций 0,1 мг/мл и 0,8 мг/мл; аликвоты хранили в замороженном состоянии при -20°С. Испытуемые соединения ресуспендировали в DMSO до конечной концентрации 10 мкМ. Этионамид и испытуемые соединения переносили в полипропиленовый 384-луночный планшет малого объема (Corning, кат. No. 3672), и их использовали для подготовки аналитических планшетов. Десять 3-кратны серийных разведений соединений (как правило, в диапазоне от 30 до 4,5 e-3 мкМ) приготавливали в черных 384-луночных полистироловых планшетах Greiner с прозрачным дном (Greiner, кат. No. 781091), используя ручные жидкостные дозаторы Echo 550 (Labcyte). Компенсировали объем DMSO, чтобы концентрация во всех лунках составляла 0,3%.
Затем в 384-луночные планшеты переносили этионамид, используя дозаторы Echo. Конечная концентрация ETH для анализов, включающих H37Rv-GFP, составляла 0,1 мкг/мл, и 0,8 мкг/мл для анализов, включающих W4-E1-GFP. Конечное количество DMSO в планшете для анализа составляло <1% об./об. для каждой лунки.
Контрольные образцы в планшетах для анализа включали DMSO в концентрации 0,3% (отрицательный контроль) и INH в количестве 1 мкг/мл (положительный контроль). Референсный планшет включал рифампицин, INH (изониазид) и ETH (этионамид) в диапазоне концентраций от 30 до 1,8 e-3 мкг/мл (15 точек, разведения 2x).
Культуры H37Rv-GFP или W4-E1-GFP, предназначенные для добавления в планшеты для анализа, промывали два раза в PBS (Gibco, 14190), ресуспендировали в свежей культуральной среде (без гигромицина), и выращивали в течение 5 дней при температуре 37°C.
Наконец, культуры разводили до оптической плотности при 600 нм (OD600), равной 0,02 с использованием свежей культуральной среды без добавления гигромицина, и в каждый планшет для анализа переносили 50 мкл полученной культуры. Планшеты инкубировали при 37°С в течение 5 дней. Флуоресцентный сигнал регистрировали на планшет-ридере Victor 3 MultiLabel (Perkin Elmer), при длине волдны возбуждения 485 нм и длине волны излучения 535 нм.
Результаты
Показатель EC50_H37Rv отражает способность соединений по изобретению усиливать активность этионамида в отношении штаммов H37Rv, а показатель EC50_мутант отражает способность соединений по изобретению потенцировать активность этионамида против штаммов микобактерий туберкулеза, устойчивых к этионамиду.
Все соединения по Примерам 1-12 испытывали в основном в соответствии с процедурой, описанной выше, и установлено, что все они имеют среднее значение показателя EC50_H37Rv <0,75 мкМ и среднее значение показателя EC50_мутант <3,0 мкМ.
Соединения по Примерам 1, 2, 5-8, 10 и 11 имеют среднее значение показателя EC50_H37Rv <0,20 мкМ, и среднее значение показателя EC50_мутант <1,0 мкМ.
Соединения по Примерам 6-8 имеют среднее значение показателя EC50_H37Rv <0,04 мкМ, и 11 имеют среднее значение показателя EC50_H37Rv <0,20 мкМ, и среднее значение показателя EC50_мутант <0,45 мкМ. Соединение по Примеру 8 имеет среднее значение показателя EC50_H37Rv, равное 0,038 мкМ, и среднее значение показателя EC50_мутант, равное 0,14 мкМ.
Для сравнения, в том же самом анализе, как описано выше, было испытано соединение по Примеру 10 документа WO 2014/096369, и установлено, что это соединение имеет среднее значение показателя EC50_H37Rv, равное 0,89 мкМ, и среднее значение показателя EC50_мутант, равное 3,5 мкМ.
Анализ ингибирования Mycobacterium tuberculosis H37Rv в человеческих макрофагах ТНР-1 (внутриклеточный анализ) в условиях in vitro
Внутриклеточный скрининг является ценным инструментом для идентификации новых соединений против туберкулеза, которые активны в макрофагах человека. Это ex vivo анализ может отражать физиологические условия, имитирующие заболевание, и позволять оценивать благоприятный вклад для клеток-хозяев (Sorrentino, F. et al. (2016) Antimicrob. Agents Chemother. 60 (1), 640-645).
Процедура осуществляли в соответствии с описанием, представленным в публикации Sorrentino, F. et al. (2016) Antimicrob. Agents Chemother. 60 (1), 640-645, за исключением того, что перед высеванием инфицированных клеток ТНР-1 в 384-луночные планшеты, инфицированные макрофаги на последней стадии промывки отфильтровывали с использованием фильтра с размером пор 40 мкм для удаления клеточных комков и получения суспензии из одиночных клеток.
Соединения по Примерам испытывали в целом в соответствии с протоколом указанного выше анализа (но при отсутствии этионамида), и установлено, что у соединений по Примерам 1, 2 и 4-10 значение IC50 составляло менее 0,5 мкМ. Для соединений по Примерам 5, 6, 7, 8 и 10 установлено, что значение IC50 составляет менее 0,1 мкМ. Для соединений по Примерам 6, 7 и 8 установлено, что значение IC50 меньше или равно 0,05 мкМ.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ | 2018 |
|
RU2767652C2 |
| АГОНИСТЫ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2015 |
|
RU2678835C2 |
| ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ СПИРОПИПЕРИДИНИЛА И ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2021 |
|
RU2839891C1 |
| СПИРО-КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ПИПЕРИДИНА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КАЛИЕВОГО КАНАЛА НАРУЖНОГО МЕДУЛЛЯРНОГО СЛОЯ | 2013 |
|
RU2642066C2 |
| ОКСАЗОЛИДИНОНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ПРОТИВОБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2016 |
|
RU2794494C2 |
| ИНГИБИТОРЫ МУТАЦИИ HER2 | 2022 |
|
RU2834124C2 |
| БЕНЗАМИДНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2801647C2 |
| ТЕТРАЗАМЕЩЕННЫЕ АЛКЕНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2733741C2 |
| Новые замещенные соединения имидазопиридина в качестве ингибиторов индоламин-2,3-диоксигеназы и/или триптофан-2,3-диоксигеназы | 2017 |
|
RU2741911C2 |
| N-(2-ЦИАНОГЕТЕРОЦИКЛИЛ)ПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2669922C2 |
Изобретение относится к соединениям или к их фармацевтически приемлемой соли, выбранным из 4,4,4-трифтор-1-[3-(2,2,2-трифторэтокси)-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил]бутан-1-она; 4,4,4-трифтор-1-(3-пропокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-она; 4,4,4-трифтор-1-(3-изобутокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-она; 4,4,4-трифтор-1-(3-гексилокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-она; 1-(3-бром-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-она; 1-(3-этокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-она; 4,4,4-трифтор-1-(3-метокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-она; 4,4,4-трифтор-1-(3-(трифторметил)-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-она; 4,4,4-трифтор-1-(3-метил-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-она; 1-(3-этил-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-она; 8-(4,4,4-трифторбутаноил)-1-окса-2,8-диазаспиро[4,5]дец-2-ен-3-карбонитрила; и 4,4,4-трифтор-1-(3-фтор-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-она. Изобретение также относится к фармацевтической композиции и комбинации для применения в лечении инфекции Mycobacterium tuberculosis на основе указанных соединений. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине при лечении туберкулеза. 5 н. и 3 з.п. ф-лы, 5 табл., 12 пр.
1. Соединение формулы (I) или его фармацевтически приемлемая соль
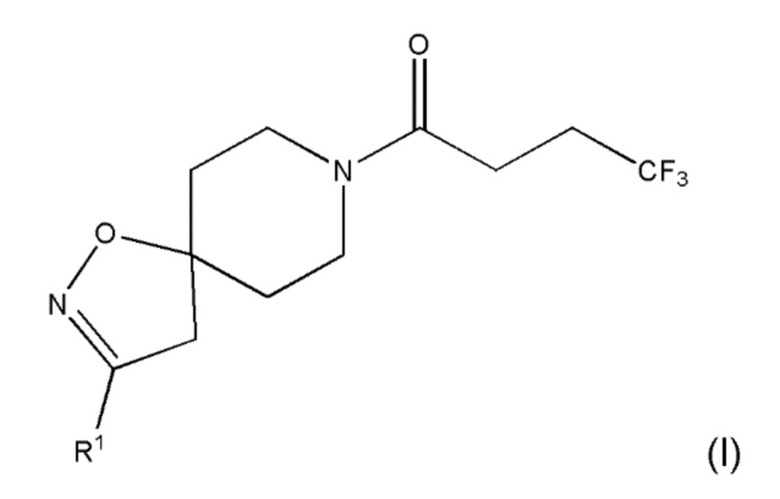 ,
,
где соединение выбрано из следующих соединений:
4,4,4-трифтор-1-[3-(2,2,2-трифторэтокси)-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил]бутан-1-она;
4,4,4-трифтор-1-(3-пропокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-она;
4,4,4-трифтор-1-(3-изобутокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-она;
4,4,4-трифтор-1-(3-гексилокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-она;
1-(3-бром-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-она;
1-(3-этокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-она;
4,4,4-трифтор-1-(3-метокси-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-она;
4,4,4-трифтор-1-(3-(трифторметил)-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-она;
4,4,4-трифтор-1-(3-метил-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-она;
1-(3-этил-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)-4,4,4-трифторбутан-1-она;
8-(4,4,4-трифторбутаноил)-1-окса-2,8-диазаспиро[4,5]дец-2-ен-3-карбонитрила; и
4,4,4-трифтор-1-(3-фтор-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-она.
2. Соединение или его фармацевтически приемлемая соль по п.1, где соединение представляет собой 4,4,4-трифтор-1-(3-(трифторметил)-1-окса-2,8-диазаспиро[4,5]дец-2-ен-8-ил)бутан-1-он, имеющий следующую структуру
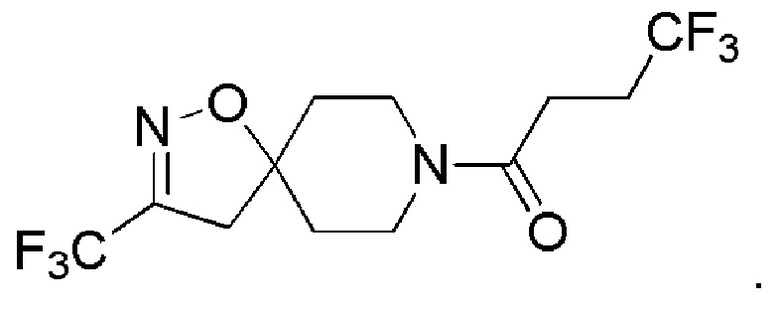
3. Соединение или его фармацевтически приемлемая соль по любому из пп. 1, 2 для применения при лечении микобактериальной инфекции или для применения при лечении заболевания, вызванного микобактериальной инфекцией, где микобактериальная инфекция является инфекцией, вызванной Mycobacterium tuberculosis.
4. Соединение или его фармацевтически приемлемая соль по любому из пп. 1, 2 для применения при лечении туберкулеза.
5. Способ лечения микобактериальной инфекции, вызванной Mycobacterium tuberculosis, у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по любому из пп. 1, 2.
6. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1, 2 для изготовления лекарственного средства, применяемого при лечении микобактериальной инфекции Mycobacterium tuberculosis, или заболевания, вызванного микобактериальной инфекцией Mycobacterium tuberculosis.
7. Фармацевтическая композиция для применения в лечении инфекции Mycobacterium tuberculosis, содержащая: (а) эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли, определенное в любом из пп. 1, 2; и (b) фармацевтически приемлемый эксципиент.
8. Комбинация для применения в лечении инфекции Mycobacterium tuberculosis, (а) соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп. 1, 2 и (b) другого агента против микобактерий, который представляет собой этионамид.
| WO 2014096369 A1, 26.06.2014 | |||
| NICOLAS BLONDIAUX ET AL., Science, 355, 2017, pp.1206-1211 | |||
| KAI LV ET AL, ACS Med | |||
| Chem | |||
| Lett., vol.2005, no | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Кровля из глиняных обожженных плит с арматурой из проволочной сетки | 1921 |
|
SU120A1 |
| ПРОИЗВОДНЫЕ 2,3-ДИГИДРО-6-НИТРОИМИДАЗО [2, 1-b] ОКСАЗОЛА | 2003 |
|
RU2326121C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СПИРОСОЕДИНЕНИЯ | 2010 |
|
RU2506266C2 |

