Перекрестная ссылка на родственную заявку
По настоящей заявке испрашивается приоритет следующей заявки:
китайская заявка № 201810085704.1, поданная 29 января 2018 года.
Область техники, к которой относится изобретение
Представлены кристаллическая Форма соединения 1H-имидазо[4,5-b]пиридин-2(3H)-она и способ ее получения, а также применение кристаллической формы для получения лекарственного средства для лечения заболевания, связанного с PDE4.
Предпосылки создания изобретения
Фактор некроза опухоли (TNFα) представляет собой цитокин, высвобождаемый преимущественно моноцитами и макрофагами в ответ на иммунную стимуляцию. TNFα может промотировать большинство процессов клеточной дифференциации, рекрутинга, пролиферации и разрушения белка. TNFα имеет защитный эффект против инфекционных агентов, опухолей и повреждения ткани при низком уровне. Однако чрезмерное высвобождение TNFα может также вызывать заболевание. Например, при введении млекопитающим или человеку TNFα может вызывать или обострять воспаление, лихорадку, сердечно-сосудистые проблемы, кровотечение, свертывание крови и острые реакции, подобные острой инфекции и шоку. Продукция избыточного или неконтролируемого TNFα у животных или людей часто указывает на следующие заболевания: эндотоксемия и/или синдром токсического шока, кахексия, респираторный стресс-синдром у взрослых, рак (такой как солидные опухоли и гематологические опухоли), болезнь сердца (такая как застойная сердечная недостаточность), вирусная инфекция, генетическое заболевание, воспалительное заболевание, аллергическое заболевание или аутоиммунное заболевание.
Рак представляет собой заболевание с особой деструктивностью, и повышение уровня TNFα в крови указывает на риск рака или метастазирования. Как правило, раковые клетки не могут выжить в системе кровообращения здорового субъекта, и одна из причин заключается в том, что внутренняя стенка кровеносных сосудов служит барьером для экстравазации раковых клеток. Исследования показали, что ELAM-1 на эндотелиальных клетках может опосредовать адгезию клеток рака толстой кишки к эндотелию, подвергаемому действию цитокинов.
Циклический аденозинмонофосфат (cAMP) играет роль во многих заболеваниях и расстройствах. Увеличение концентрации cAMP в лейкоцитах в процессе воспаления подавляет активацию лейкоцитов и впоследствии высвобождает воспалительные регуляторные факторы, включая TNFα и NF-κB. Повышенный уровень cAMP также приводит к релаксации гладких мышц дыхательных путей.
Основной клеточный механизм инактивации cAMP обусловлен разрушением cAMP семейством изозимов, называемых циклическими нуклеотидфосфодиэстеразами (PDE). Известно, что семейство PDE включает 11 членов. К настоящему времени доказано, что ингибирование фермента PDE4 является особенно эффективным для ингибирования высвобождения медиаторов воспаления и релаксации гладких мышц дыхательных путей, и поэтому фермент PDE4 стал одной из популярных мишеней для лекарственных средств. Согласно другому генетическому кодированию, семейство PDE-4 можно разделить на 4 подтипа (PDE-4A, B, C, D). Среди них экспрессия PDE-4A, PDE-4B и PDE-4D в воспалительных клетках (таких как B-клетки, T-клетки и нейтрофилы) сильнее, чем в PDE-4C. Ингибирование фермента PDE4 приводит к повышению уровней cAMP, регулируя тем самым уровень TNFα так, чтобы лечить заболевания.
Сущность изобретения
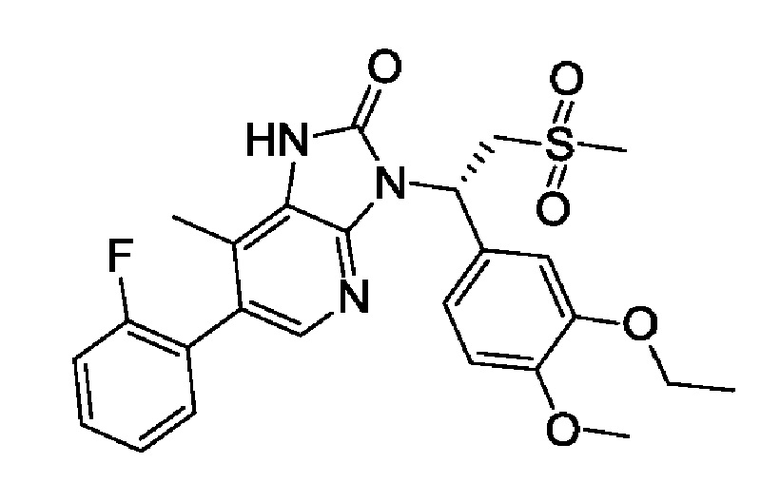
В одном аспекте представлена кристаллическая Форма A соединения 1, где кристаллическая Форма A имеет рентгеновскую порошковую дифрактограмму, имеющую характеристические дифракционные пики при следующих углах 2θ: 14,10±0,2°, 19,07±0,2°, 21,79±0,2°.
Соединение 1
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая Форма A соединения 1 имеет рентгеновскую порошковую дифрактограмму, имеющую характеристические дифракционные пики при следующих углах 2θ: 10,69±0,2°, 12,31±0,2°, 13,45±0,2°, 14,10±0,2°, 14,62±0,2°, 19,07±0,2°, 20,33±0,2°, 21,79±0,2°.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая Форма A соединения 1 имеет рентгеновскую порошковую дифрактограмму, имеющую характеристические дифракционные пики при следующих углах 2θ: 6,25±0,2°, 8,93±0,2°, 10,69±0,2°, 12,31±0,2°, 13,45±0,2°, 14,10±0,2°, 14,62±0,2°, 18,16±0,2°, 19,07±0,2°, 20,33±0,2°, 21,79±0,2°.
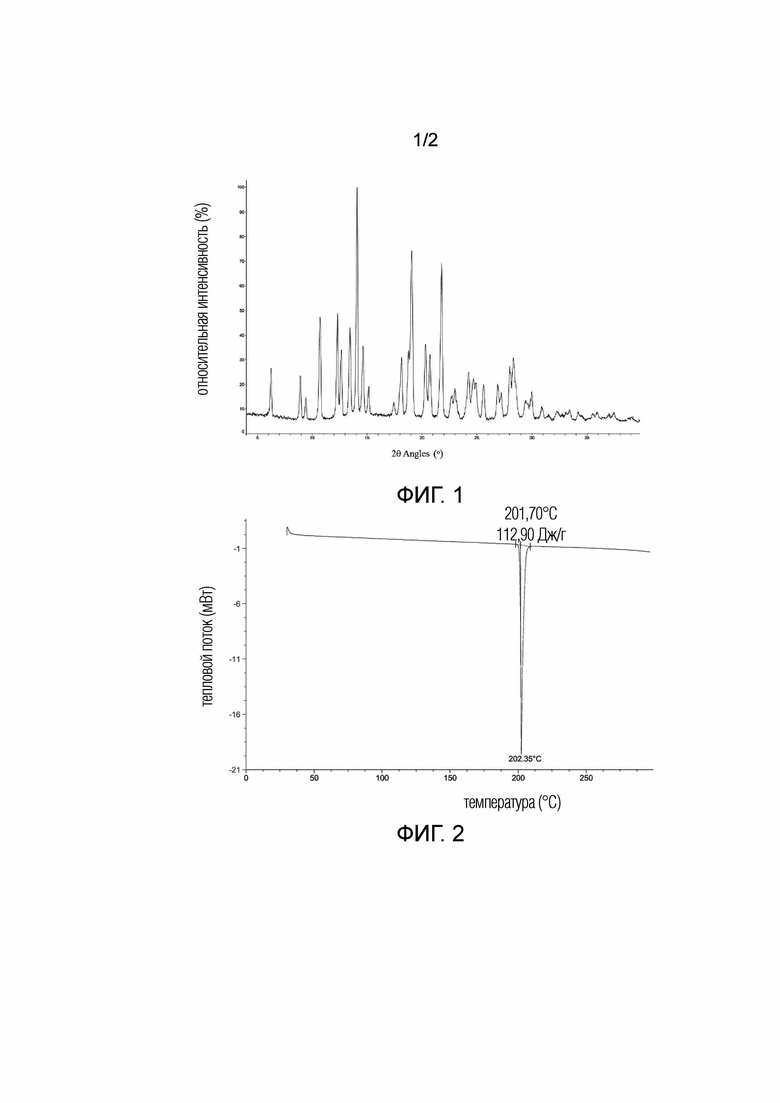
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая Форма A соединения 1 имеет рентгеновскую порошковую дифрактограмму, показанную на Фиг. 1.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая Форма A соединения 1 имеет рентгеновскую порошковую дифрактограмму с данными анализа, показанными в Таблице 1
Данные анализа рентгеновской порошковой дифрактограммы кристаллической Формы A соединения 1
расстояние (Å)
расстояние (Å)
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая Форма A соединения 1 имеет кривую дифференциальной сканирующей калориметрии, имеющую точку начала эндотермического пика при 201,70°C±2°C.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая Форма A соединения 1 имеет кривую DSC, показанную на Фиг. 2.
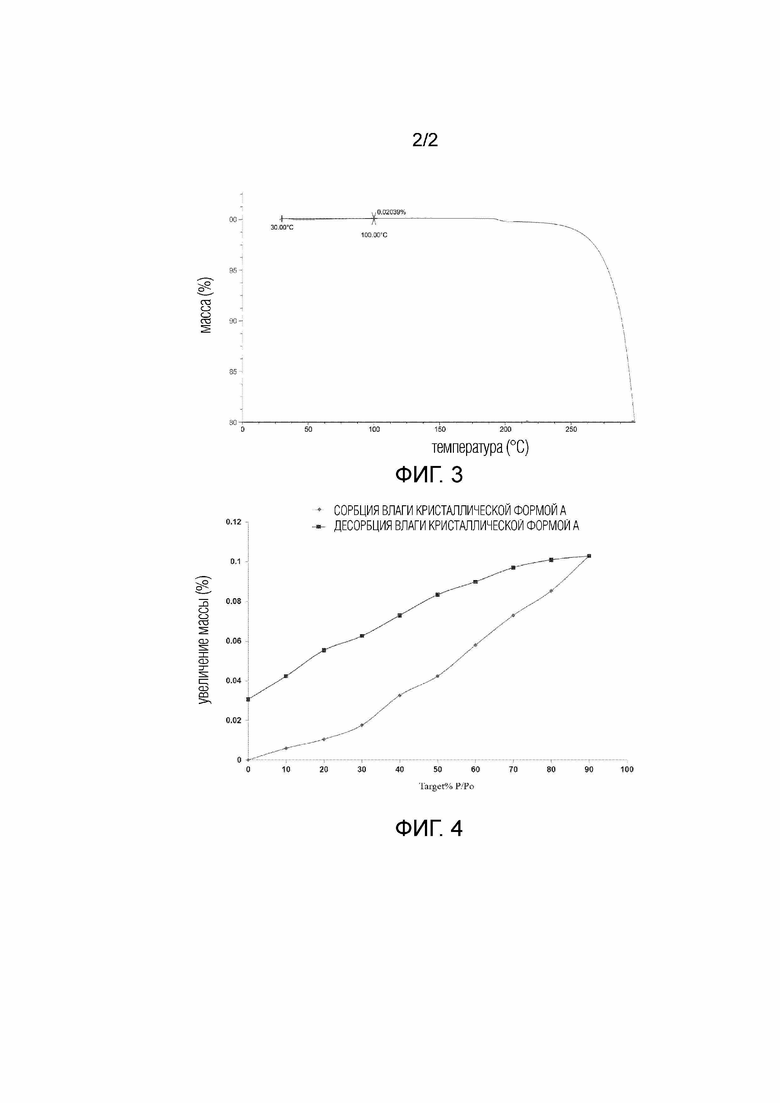
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая Форма A соединения 1 имеет кривую термогравиметрического анализа, где потеря массы при 100,00±2°C составляет 0,02039%.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая Форма A соединения 1 имеет кривую TGA, показанную на Фиг. 3.
В другом аспекте представлен способ получения кристаллической Формы A, включающий добавление соединения 1 в спиртовой растворитель, кетоновый растворитель, эфирный растворитель, смешанный растворитель, состоящий из спиртового растворителя и воды, смешанный растворитель, состоящий из кетонового растворителя и воды, или смешанный растворитель, состоящий из эфирного растворителя и воды; нагревание для растворения и затем охлаждение для кристаллизации с получением кристаллической Формы A.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием спиртовой растворитель выбран из группы, состоящей из метанола, этанола и изопропанола.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кетоновый растворитель выбран из группы, состоящей из ацетона и бутанона.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием эфирный растворитель выбран из группы, состоящей из диметилового эфира гликоля.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием смешанный растворитель, состоящий из спиртового растворителя и воды, выбран из группы, состоящей из смешанного растворителя, состоящего из этанола и воды.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием в смешанном растворителе, состоящем из спиртового растворителя и воды, объемное соотношение спиртового растворителя и воды выбрано из группы, состоящей из 1:0,2-1,5.
Еще в одном аспекте представлено применение кристаллической формы A соединения 1 для получения лекарственного средства для лечения заболевания, связанного с PDE4 рецептором.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием заболевание, связанное с PDE4, включает псориаз, псориатический артрит, хроническую обструктивную пневмонию, анкилозирующий спондилит, воспалительное заболевание кишечника.
Технический эффект
Кристаллическая Форма A соединения 1 обладает хорошей стабильностью, низкой гигроскопичностью и потенциальной возможностью создания лекарства. Кристаллическая Форма A соединения 1 демонстрирует хорошую стабильность в спиртовом растворителе, ацетонитриле, ацетоне, этилацетате, тетрагидрофуране, смешанном растворителе, состоящем из спиртового растворителя и воды, смешанном растворителе, состоящем из ацетонитрила и воды, или смешанном растворителе, состоящем из ацетона и воды. Кристаллическая Форма A соединения 1 демонстрирует хорошую стабильность в условиях ускоренного испытания 40ºC/относительной влажности 75%. кристаллическая Форма A соединения 1 демонстрирует хорошую стабильность в условиях долгосрочного хранения при 25ºC/относительной влажности 60%.
Соединение 1 демонстрирует отличную активность in vitro, направленную на ингибирование фосфодиэстеразы подтипа 4B (PDE4B). Кроме того, соединение 1 демонстрирует отличную активность in vitro, направленную на ингибирование продукции TNFα в hPBMC, которая выше, чем у Апремиласта. Соединение 1 в трех дозовых группах 0,3, 1 и 3 мг/кг существенно улучшает симптомы коллаген-индуцированного артрита. Кроме того, соединение 1 в дозовых группах 1 мг/кг и 3 мг/кг демонстрирует существенное улучшение патологии артрита. Эти три дозовые группы демонстрируют очевидную взаимосвязь доза-эффект при оценке патологии артрита. Терапевтический эффект соединения 1 при 3 мг/кг (клиническая оценка и оценка патологии артрита) лучше, чем у Апремиласта при 5 мг/кг.
Общие определения
Если не указано иное, следующие термины и фразы имеют следующие определения. Конкретный термин или фраза не должны рассматриваться как неопределенные или неясные без конкретного определения и должны пониматься в соответствии с обычными значениями. Используемое торговое наименование должно относиться к соответствующему изделию или активному ингредиенту.
Промежуточные соединения могут быть получены различными способами синтеза, хорошо известными специалистам в данной области, включая конкретные варианты осуществления, перечисленные ниже, варианты осуществления в комбинации с другими способами химического синтеза и эквивалентные альтернативы, хорошо известные специалистам в данной области техники. Предпочтительные варианты осуществления включают, но не ограничиваются этим, Примеры, представленные ниже.
В конкретных вариантах осуществления химическую реакцию осуществляют в подходящем растворителе, и растворитель должен быть подходящим для химических изменений настоящего раскрытия и требуемых реагентов и материалов. Для получения соединения по настоящему изобретению специалист в данной области может модифицировать или выбрать стадию синтеза или схему реакции на основе представленных вариантов осуществления.
Настоящее раскрытие будет описано подробно, и Примеры не должны рассматриваться как его ограничение.
Растворители, используемые в настоящем изобретении, являются коммерчески доступными и могут использоваться без дальнейшей очистки.
Используются следующие аббревиатуры: DMF: диметилформамид; MsOH: метансульфоновая кислота; EtOH: этанол; NaOH: гидроксид натрия.
Соединения названы вручную или с использованием программного обеспечения ChemDraw®. Провайдеры используют названия соединений по каталогу.
Рентгеновский порошковый дифрактометр, XRPD
Устройство: Рентгеновский дифрактометр BRUKER D8 advance
Метод испытания: Для XRPD детекции используют около 10-20 мг образца.
Конкретные XRPD параметры являются следующими:
Излучающая трубка: Cu, kα, (λ=1,54056Ǻ).
Напряжение излучающей трубки: 40 кВ, ток излучающей трубки: 40 мА
Щель расходимости: 0,60 мм
Щель детектора: 10,50 мм
Антирассеивающая щель: 7,10 мм
Диапазон сканирования: 4-40 град.
Размер шага: 0,02 град.
Время/шаг: 0,12 сек
Скорость вращения предметного столика: 15 об/мин
Дифференциальный сканирующий калориметр, DSC
Устройство: Дифференциальный сканирующий калориметр TA Q2000
Метод испытания: Образец (около 1 мг) помещают в DSC алюминиевую чашу для испытания, под 50 мл/мин потоком N2, нагревают от 25°C до 350°C при скорости нагрева 10°C/мин.
Термогравиметрический анализатор, TGA
Устройство: Термогравиметрический анализатор TA Q5000IR
Метод испытания: Образец (2-5 мг) помещают в TGA платиновую чашу для испытания, под 25 мл/мин потоком N2, нагревают от комнатной температуры до 350°C при скорости нагрева 10°C/мин.
Динамическая сорбция паров, DVS
Устройство: Анализатор динамической сорбции паров SEM Advantage-1
Условия испытания: Образец (10-20 мг) помещают в DVS диск образцов для испытания.
Конкретные DVS параметры являются следующими:
Температура: 25°C
Весы: dm/dt=0,01%/мин (мин.: 10 мин, макс.: 180 мин)
Сушка: сушка при 0% RH в течение 120 мин
RH (%) тестируемый градиент: 10%
RH (%) тестируемый диапазон градиента: 0% - 90% - 0%
Гигроскопичность подразделяют на следующие категории:
Метод определения содержания
Устройство: Высокоэффективный жидкостный хроматограф Agilent 1260 с DAD детектором или высокоэффективный жидкостный хроматограф Shimadzu LC-20A с PDA детектором
Конкретные хроматографические параметры являются следующими:
Хроматографическая колонка: Agilent Eclipse plus C18 (4,6мм × 150мм, 3,5мкм)
Температура колонки: 40°C
Скорость потока: 1,0 мл/мин
Длина волны детектора: 230 нм
Объем вводимой пробы: 10 мкл
Время регистрации хроматограммы: 60 мин
Подвижная фаза A: 0,04% водный раствор трифторуксусной кислоты (об/об)
Подвижная фаза B: ацетонитрил
Разбавитель: ацетонитрил:очищенная вода=3:1(об/об)
Раствор для промывки: ацетонитрил:очищенная вода =3:1(об/об)
Процедура градиентного элюирования:
Краткое описание чертежей
Фиг. 1 демонстрирует рентгеновскую порошковую дифрактограмму, полученную с использованием Cu-Kα излучения, кристаллической формы A соединения 1.
Фиг. 2 демонстрирует кривую DSC кристаллической формы A соединения 1.
Фиг. 3 демонстрирует кривую TGA кристаллической формы A соединения 1.
Фиг. 4 демонстрирует кривую DVS кристаллической формы A соединения 1.
Примеры
Следующие Примеры представлены для лучшей иллюстрации для лучшего понимания настоящего изобретения. Конкретные варианты осуществления не следует рассматривать как ограничение настоящего изобретения.
Пример 1: Получение кристаллической Формы A соединения 1
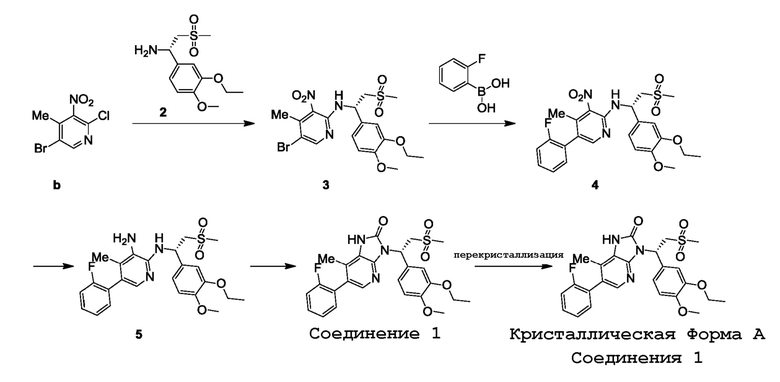
Стадия 1: Синтез соединения 3
При комнатной температуре соединение b (10,00 г, 39,77 ммоль), соединение 2 (9,78 г, 35,79 ммоль) и диизопропиламин (10,28 г, 79,53 ммоль, 13,89 мл) растворяли в N,N-диметилформамиде (200,00 мл), смесь продували азотом три раза и реакционную смесь нагревали до 120°C в защитной атмосфере азота при перемешивании в течение 16 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, добавляли воду (400 мл) и экстрагировали этилацетатом (200 мл ×3). Органические фазы объединяли и промывали насыщенным солевым раствором (100 мл ×3), сушили над безводным сульфатом натрия и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии (элюент: этилацетат/петролейный эфир=1/4-1/2, объемное отношение) с получением целевого соединения 3.
MS-ESI m/z: 509,8 [M+Na]+, 511,8 [M+Na+2]+. 1H ЯМР (400МГц, CDCl3) δ: 8,27 (с, 1H), 7,27 (д, J=6,8 Гц, 1H), 6,90-6,86 (м, 2H), 6,81 (д, J=8,0 Гц, 1H), 5,72 (кв., J=6,4 Гц, 1H), 4,04 (кв., J=6,8 Гц, 2H), 3,80 (с, 3H), 3,68 (дд, J=6,6, 14,6 Гц, 1H), 3,40 (дд, J=6,4, 14,8 Гц, 1H), 2,52 (с, 3H), 2,45 (с, 3H), 1,40 (т, J=7,0 Гц, 3H).
Стадия 2: Синтез соединения 4
При комнатной температуре соединение 3 (12,10 г, 24,78 ммоль) и o-фторфенилбороновую кислоту (5,20 г, 37,17 ммоль) растворяли в диоксане (150,00 мл) и воде (50,00 мл), затем добавляли карбонат калия (10,27 г, 74,34 ммоль) и комплекс дихлорида [1,1'-бис(дифенилфосфино)ферроцен]палладия с дихлорметаном (2,02 г, 2,48 ммоль) в защитной атмосфере азота. Реакционную смесь нагревали в защитной атмосфере азота до 80°C при перемешивании в течение 14 ч. После завершения реакции реакционную смесь охлаждали до комнатной температуры, добавляли воду (500 мл) и экстрагировали этилацетатом (300 мл ×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии (элюент: этилацетат/петролейный эфир=1/10-1/4, объемное отношение) с получением целевого соединения 4.
MS-ESI m/z: 504,1 [M+H]+. 1H ЯМР (400МГц, CDCl3) δ: 8,07 (с, 1H), 7,42 (д, J=6,8 Гц, 1H), 7,38-7,30 (м, 1H), 7,18-7,06 (м, 3H), 6,98-6,89 (м, 2H), 6,83 (д, J=8,4 Гц, 1H), 5,82 (кв., J=6,6 Гц, 1H), 4,13-4,00 (м, 3H), 3,81 (с, 3H), 3,43 (дд, J=6,4, 14,7 Гц, 1H), 2,55 (с, 3H), 2,23 (с, 3H), 1,41 (т, J=7,0 Гц, 3H).
Стадия 3: Синтез соединения 5
При комнатной температуре к соединению 4 (9,20 г, 18,27 ммоль) и хлориду аммония (9,77 г, 182,70 ммоль) добавляли метанол (200,00 мл) и добавляли цинковый порошок (11,95 г, 182,70 ммоль) 20 партиями при 0°C. Реакционную смесь перемешивали при 0°C в течение 16 ч. После завершения реакции реакционную смесь фильтровали для удаления цинкового порошка и фильтрат концентрировали при пониженном давлении с получением остатка. Остаток растворяли в дихлорметане (200 мл). Суспензию фильтровали для удаления нерастворимых веществ и фильтрат концентрировали при пониженном давлении с получением соединения 5.
MS-ESI m/z: 474,0 [M+H]+. 1H ЯМР (400МГц, CDCl3) δ: 7,55 (с, 1H), 7,42-7,32 (м, 1H), 7,20 (д, J=4,8 Гц, 2H), 7,13 (т, J=9,0 Гц, 1H), 7,08-7,03 (м, 2H), 6,88 (д, J=8,8 Гц, 1H), 5,70 (с, 1H), 4,19-4,07 (м, 2H), 4,00-3,90 (м, 1H), 3,87 (с, 3H), 3,58 (дд, J=6,0, 14,4 Гц, 1H), 2,78 (с, 3H), 2,05 (с, 3H), 1,47 (т, J=7,2 Гц, 3H).
Стадия 4: Синтез соединения 1
При комнатной температуре соединение 5 (9,30 г, 19,64 ммоль) и триэтиламин (19,87 г, 196,40 ммоль) растворяли в тетрагидрофуране (200,00 мл). Реакционную смесь охлаждали до 0°C и добавляли по каплям раствор трифосгена (2,33 г, 7,86 ммоль) в тетрагидрофуране (50,00 мл). Тетрагидрофурановый (50,00 мл) раствор добавляли по каплям к описанному выше реакционному раствору. После добавления реакционную смесь перемешивали при 0°C в течение 3 ч в защитной атмосфере азота. После завершения реакции к реакционной смеси добавляли воду (200 мл) и экстрагировали этилацетатом (100 мл ×3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали для удаления осушителя. Фильтрат концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии (элюент: этилацетат/петролейный эфир=1/3-2/1, объемное отношение) с получением соединения 1.
1H ЯМР (400МГц, CDCl3) δ: 10,13-10,01 (м, 1H), 7,87 (с, 1H), 7,39-7,32 (м, 1H), 7,31 (д, J=1,6 Гц, 1H), 7,18-7,15 (м, 3H), 7,11 (т, J=9,2 Гц, 1H), 6,74 (д, J=8,4 Гц, 1H), 6,14 (дд, J=4,8, 9,6 Гц, 1H), 4,86 (дд, J=9,4, 14,6 Гц, 1H), 4,09-3,97 (м, 2H), 3,88 (дд, J=4,4, 14,8 Гц, 1H), 3,76 (с, 3H), 2,70 (с, 3H), 2,16 (с, 3H), 1,35 (т, J=7,0 Гц, 3H).
Стадия 5: Получение кристаллической Формы A соединения 1
При комнатной температуре к соединению 1 (6,45 г, 12,91 ммоль) добавляли воду (160,00 мл) и этанол (170,00 мл) и реакционную смесь перемешивали при 90°C в течение 0,5 ч. Реакционный раствор постепенно становился прозрачным в процессе перемешивания. Реакционную смесь медленно охлаждали до 20°C при перемешивании и перемешивание продолжали при 20°C в течение 16 ч, в течение этого времени осаждалось много белых твердых частиц. Белые твердые частицы собирали фильтрованием и сушили в вакуумной печи при 45°C в течение 18 ч с получением кристаллической Формы A соединения 1.
MS-ESI m/z: 500,2 [M+H]+. 1H ЯМР (400МГц, CDCl3) δ: 9,97 (с, 1H), 7,94 (с, 1H), 7,46-7,39 (м, 1H), 7,38 (д, J=1,6 Гц, 1H), 7,26-7,22 (м, 3H), 7,17 (т, J=9,0 Гц, 1H), 6,81 (д, J=8,0 Гц, 1H), 6,22 (дд, J=4,8, 9,6 Гц, 1H), 4,93 (дд, J=9,6, 14,8 Гц, 1H), 4,13-4,03 (м, 2H), 3,95 (дд, J=4,8, 14,8 Гц, 1H), 3,83 (с, 3H), 2,77 (с, 3H), 2,23 (с, 3H), 1,42 (т, J=7,0 Гц, 3H).
Экспериментальный пример 1: Испытания стабильности кристаллической Формы A в различных растворителях
50 мг кристаллической Формы A несколькими порциями добавляли в один растворитель или смешанные растворители, указанные в следующей таблице, перемешивали при 40°C в течение 2 дней и центрифугировали. Твердые вещества во всех образцах собирали и сушили в вакуумной сушильной печи (40°C) в течение ночи. Кристаллические формы тестировали для XRPD. Результаты показаны в Таблице 2.
Испытания стабильности кристаллической Формы A в различных растворителях
Заключение: Кристаллическая Форма A соединения 1 демонстрирует хорошую стабильность в спиртовом растворителе, ацетонитриле, ацетоне, этилацетате, тетрагидрофуране, смешанном растворителе, состоящем из спиртового растворителя и воды, смешанном растворителе, состоящем из кетонового растворителя и воды, смешанном растворителе, состоящем из ацетонитрила и воды, или смешанном растворителе, состоящем из ацетона и воды.
Экспериментальный пример 2: Испытание на гигроскопичность кристаллической Формы A соединения 1
Материалы:
Анализатор динамической сорбции паров SEM DVS Advantage-1
Процедуры:
10-20 мг кристаллической Формы A соединения 1 помещали в диск для образцов DVS для испытания.
Результаты:
DVS кривая кристаллической Формы A соединения 1 показана на Фиг. 4, △W= 0,08%.
Заключение:
Кристаллическая Форма A соединения 1 имела гигроскопичное увеличение массы 0,08% при 25°C и 80% RH, что было меньше 0,2%, показывая отсутствие гигроскопичности или небольшую гигроскопичность.
Экспериментальный пример 3: Испытания стабильности в твердом состоянии кристаллической Формы A соединения 1 при высокой температуре, высокой влажности и в условиях сильного освещения.
В соответствии с "Guidelines for Stability Test of APIs and Preparations" (Chinese Pharmacopoeia 2015 Edition Four General Principles 9001), кристаллическую Форму A соединения 1 испытывали на стабильность при высокой температуре (60°C, открытый образец), высокой влажности (комнатная температура/относительная влажность 92,5%, открытый образец) и сильного освещения (4500±500люкс, 90мквт/см2, плотно закрыт).
Отвешивали 1,5 г кристаллической Формы A соединения 1 и помещали на открытое предметное стекло и распределяли тонким слоем. Образцы помещали в условия высокой температуры и высокой влажности в эксикатор для контроля и образцы брали на 5, 10 и 30 день для испытания и результаты испытаний сравнивали с исходными результатами испытаний дня 0. Образцы, которые помещали в условия сильного освещения, закрывали прозрачной крышкой с герметизирующей пленкой и образцы брали на 5 и 10 день для испытания и результаты испытаний сравнивали с исходными результатами испытаний в день 0. Результаты испытаний показаны в следующей Таблице 3.
Результаты испытаний стабильности в твердом состоянии кристаллической Формы A соединения 1 в условиях высокой температуры, высокой влажности и сильного освещения
Заключение: Кристаллическая Форма A соединения 1 показала хорошую стабильность в условиях высокой температуры, высокой влажности или сильного освещения
Экспериментальный пример 4: Испытание стабильности в твердом состоянии кристаллической Формы A соединения 1 в условиях ускоренного испытания
В соответствии с "Guidelines for Stability Test of APIs and Preparations" (Chinese Pharmacopoeia 2015, Volume IV, General Principles 9001), кристаллическую Форму A соединения 1 испытывали на стабильность в условиях ускоренных испытаний при высокой температуре и высокой влажности (40°C/относительная влажность 75%, образец плотно закрыт).
Отвешивали 1,4 г кристаллической Формы A соединения 1 и помещали в двухслойный низкой плотности полиэтиленовый пакет. Каждый слой полиэтиленового пакета с низкой плотностью загибали и запечатывали соответственно и затем пакет помещали в пакет из алюминиевой фольги и запечатывали термосваркой. Образцы брали на 1, 2, 3 и 6 месяц для испытания и результаты испытаний сравнивали с исходными результатами испытаний дня 0. Испытание повторяли три раза, каждый раз с другой партией кристаллической Формы A соединения 1. Результаты испытаний показаны в следующей Таблице 4.
Результаты испытаний стабильности в твердом состоянии кристаллической Формы A соединения 1 в условиях ускоренного испытания (40°C/относительная влажность 75%, плотно закрытый образец)
Заключение: Кристаллическая Форма A соединения 1 показала хорошую стабильность в условиях ускоренного испытания 40°C/относительная влажность 75%.
Экспериментальный пример 5: Испытание стабильности в твердом состоянии кристаллической Формы A соединения 1 в условиях долгосрочного хранения
В соответствии с "Guidelines for Stability Test of APIs and Preparations" (Chinese Pharmacopoeia 2015, Volume IV, General Principles 9001), кристаллическую Форму A соединения 1 испытывали на стабильность в условиях долгосрочного хранения (25°C/относительная влажность 60%, плотно закрытый образец).
Отвешивали 1,4 г кристаллической Формы A соединения 1 и помещали в двухслойный низкой плотности полиэтиленовый пакет. Каждый слой полиэтиленового пакета с низкой плотностью загибали и запечатывали соответственно и затем пакет помещали в пакет из алюминиевой фольги и запечатывали термосваркой. Образцы брали на 3, 6, 9, 12 и 18 месяц для испытания и результаты испытаний сравнивали с исходными результатами испытаний дня 0. Испытание повторяли три раза, каждый раз с другой партией кристаллической Формы A соединения 1. Результаты испытаний показаны в следующей Таблице 5.
Результаты испытаний стабильности в твердом состоянии кристаллической Формы A соединения 1 в условиях долгосрочного хранения (25°C/относительная влажность 60%, плотно закрытый образец)
Заключение: Кристаллическая Форма A соединения 1 показала хорошую стабильность в условиях долгосрочного хранения 25°C/относительная влажность 60%.
Пример испытаний 1: Ингибиторная активность соединения 1 в отношении фосфодиэстеразы подтипа 4B (PDE4B фермент)
Этот биологический эксперимент, основанный на поляризации флуоресценции, использовали для определения экспрессии AMP/GMP, т.е. для демонстрации активности фермента путем отслеживания связывания антител AMP/GMP.
Агенты:
Экспериментальный буферный раствор: 10 мМ буферного раствора тригидроксиметиламинометан-хлористоводородная кислота (Tris-HCl) (pH 7,5), 5 мМ MgCl2, 0,01% полиоксиэтиленлаурилового эфира (Brij 35), 1 мМ дитиотреитола (DTT) и 1% DMSO.
Ферменты: Рекомбинантный гуманизированный PDE4B (Genebank Accession Number NM_002600; amino acid 305 terminal) экспрессировали с использованием бакуловируса в клетках насекомого Sf9 с использованием N-концевой GST метки. Мол.масса=78 кДа.
Субстрат фермента: 1 мкМ cAMP
Детекция: Transcreener®AMP2/GMP2 антитело и AMP2/GMP2 AlexaFluor633 мечение.
Процедуры:
1. Растворение рекомбинантного человеческого PDE4B фермента и субстрата фермента (1 мкМ cAMP) в свежеприготовленном экспериментальном буфере;
2. Перенос полученного буферного раствора PDE4B фермента в реакционные лунки;
3. Добавление соединения 1, растворенного при помощи 100% DMSO, в реакционную лунку с буферным раствором PDE4B фермента акустическим методом (эхо 550 нанолитровый диапазон) и осуществление инкубации в течение 10 минут при комнатной температуре;
4. Затем добавление буферного раствора субстрата фермента в указанные реакционные лунки для инициирования реакции;
5. Осуществление инкубации в течение 1 ч при комнатной температуре;
6. Добавление смеси для детекции (Transcreener®AMP2/GMP2 антитело и AMP2/GMP2 AlexaFluor633 метка) для остановки реакции и осуществление инкубации в течение 90 мин при медленном перемешивании. Диапазон определения поляризации флуоресценции возбуждение/эмиссия=620/688.
Анализ данных: Сигнал поляризации флуоресценции преобразовывали в нМ в соответствии с AMP/GMP стандартной кривой и контрольной % активностью фермента относительно DMSO, рассчитанной с использованием программы Excel. Подгонку кривой осуществляли с использованием программы GraphPad Prism (графически представляющей медицинские данные).
Результаты in vitro скринингового теста соединения в соответствии с настоящим раскрытием
Заключение: Соединение 1 показало отличную in vitro активность ингибирования фосфодиэстеразы подтипа 4B (PDE4B).
Пример испытаний 2: Оценка ингибирования продукции TNFα в мононуклеарных клетках периферической крови человека (hPBMC) in vitro
Активность соединения 1, направленная на ингибирование липополисахарид (LPS)-индуцированной продукции TNFα в человеческих мононуклеарных клетках периферической крови.
Процедуры:
1. Сбор нормальной цельной крови человека, которую подвергали антикоагуляционной обработке с использованием пробирки с антикоагулянтом EDTA-K2;
2. Отделение PBMC центрифугированием в градиенте плотности фиколла, подсчет и доведение клеточной концентрации до 2×106/мл;
3. В U-донный 96-луночный планшет добавляли 2×105 клеток/лунка, добавляли LPS 1 нг/мл, различные концентрации соединения при 100 мкМ, 10 мкМ, 1 мкМ, 100 нМ, 10 нМ, 1 нМ, 100 пМ, 10 пМ, 200 мкл системы/лунка, в дублируемые лунки;
4. Осуществление инкубации в течение 24 ч, сбор супернатанта;
5. Детекция уровня TNFα в супернатанте методом ELISA, подгонка кривой ингибирования и расчет IC50 с использованием программы Graphpad Prism.
Ингибиторная активность соединения в соответствии с настоящим раскрытием в отношении продукции TNFα в hPBMC
Заключение: Соединение 1 показало отличную in vitro активность ингибирования продукции TNFα в hPBMC, которая была выше, чем у Апремиласта.
Пример испытаний 3: Модель CIA in vivo
Цель:
Модель коллаген-индуцированного артрита у мышей представляет собой животную модель, используемую для оценки эффективности лекарственного лечения псориатического артрита, и патогенез и симптомы в значительной степени коррелируют с псориатическим артритом. Ряд симптомов, подобных псориатическому артриту человека, таких как покраснение и отек сустава, повреждение суставного хряща и повреждение суставной капсулы, вызывается в модели путем инъекции коллагена типа II для активации реактивности В-клеток, Т-клеток на костный коллаген, и активированные В-клетки и Т-клетки проникают в суставы, вызывая воспаление суставов. При доклинической оценке соединений-кандидатов для лечения псориатического артрита часто используют коллаген-индуцированный артрит у мышей для оценки эффективности.
Целью этого эксперимента было исследование терапевтического эффекта соединения 1 на коллаген-индуцированный артрит у мышей для предоставления соответствующей доклинической фармакодинамической информации для последующих клинических исследований.
Процедуры:
1. Иммунизация с использованием коллагена II типа/полного адъюванта Фрейнда
Получение уксусной кислоты: Разбавление уксусной кислоты до 100 мМ, фильтрование через 0,22 мкм мембранный фильтр и хранение при 4°C.
Раствор бычьего коллагена II типа: Растворение бычьего коллагена II типа (CII) в растворе уксусной кислоты, который хранили при 4°C в течение ночи.
Получение эмульсии: Смешивание CII раствора, который хранили в течение ночи, с равным объемом полного адъюванта Фрейнда и гомогенизация смеси с использованием высокоскоростного гомогенизатора до образования стабильной эмульсии из раствора.
Получение липополисахарида (LPS): Отвешивание LPS, добавление нормального солевого раствора и смешивание до получения стабильного раствора с концентрацией 0,3 мг/кг.
2. Индукция артрита:
Мышей рандомизированно разделяли на разные группы обработки. День первой иммунизации определяли как день 0, а последующие дни обозначали последовательно.
После того, как DBA/1 мышей анестезировали изофлураном, полученную эмульсию коллагена вводили подкожно в хвост.
В день 23 интраперитонеально вводили 100 мкл раствора LPS.
Мышам в нормальной группе иммунизацию не осуществляли.
3. Введение и схема введения доз
В день 27, когда средняя клиническая оценка достигала примерно 1, отбирали 60 мышей с умеренным началом заболевания и их снова рандомизированно разделяли в соответствии с массой тела и оценкой, по 10 мышей в каждой группе.
В CIA модели обычно используют дексаметазон в качестве положительного контрольного лекарственного средства в 0,3 мг/кг дозовой группе. Кроме того, соответствующую схему введения доз соединения 1 и контрольного соединения Апремиласта определяли в соответствии с результатами предварительных экспериментов. Группа 1 включала нормальных мышей без какой-либо обработки; Группа 2 представляла собой контрольную группу введения носителя; Группа 3 представляла собой группу введения дексаметазона при дозе 0,3 мг/кг; Группа 4 представляла собой группу введения Апремиласта при дозе 5 мг/кг; Группа 5, Группа 6 и Группа 7 представляли собой группы введения Соединения 1 при дозах 0,3, 1 и 3 мг/кг, соответственно. Введение осуществляли один раз в день всего в течение 11 дней. Объем внутрижелудочного введения составлял 10 мл/кг.
Группы и схема введения доз
4. Определение показателя инцидентности артрита
Клиническое наблюдение: от 7 дней до иммунизации до 23 дней после иммунизации основное состояние здоровья и изменения массы тела мышей DBA/1 наблюдали ежедневно (регистрировали один раз в неделю). После 23-го дня состояние здоровья, заболеваемость и изменение массы тела мышей наблюдали ежедневно (регистрировали не менее трех раз в неделю) до конца эксперимента.
Клиническая оценка: после инъекции LPS заболеваемость мышей наблюдали каждый день. После начала заболевания у мышей (клинические симптомы артрита) их оценивали в соответствии с тяжестью состояния (покраснение и отек, деформация суставов) в соответствии с 0-4-балльным стандартом, с максимальной оценкой 4 для каждой конечности и максимальной оценкой 16 для каждого животного. Стандарт оценки показан в Таблице 9. Оценку осуществляли по меньшей мере три раза в неделю.
Стандарт клинической оценки артрита
Патология: На 38 день мышь умерщвляли. Брали две задние конечности мыши, пропитывали 10% раствором формалина, декальцинировали раствором муравьиной кислоты, заливали парафином, делали срезы, окрашивали гематоксилин-эозином (HE) и наблюдали микрофотографическим методом. Степень повреждения сустава оценивали по четырем параметрам: инфильтрация воспалительными клетками, образование паннуса, повреждение хряща и резорбция кости, и оценивали по 0-4 балльному стандарту. Стандарт оценки представлен в таблице 10:
Стандарт оценки патологии артрита
5. Статистическая обработка
Экспериментальные данные выражали как среднее значение ± стандартная ошибка (Среднее значение ± SEM), массу тела и клиническую оценку анализировали с использованием двухфакторного дисперсионного анализа, патологическую оценку и AUC анализировали с использованием t-критерия, и значение p <0,05 считалось значимым.
Экспериментальные результаты:
1. Клиническая оценка:
В день 25 после первой иммунизации (День 2 после второй иммунизации) у мышей начиналось развитие клинических симптомов артрита. Введение начинали на 27 день. Средняя клиническая оценка контрольной группы введения носителя постепенно повышалась и достигала 8,3 пунктов в день 36, указывая на успешное установление модели коллаген-индуцированного артрита.
По сравнению с контрольной группой введения носителя, соединение 1 при 0,3, 1 и 3 мг/кг может существенно снижать клиническую оценку артрита у мышей в конечной точке эксперимента (день 37), и клинические средние оценки при трех дозах падали до 3,6 (p<0,0001), 4,3 (p<0,001) и 3,5 (p<0,0001). Следовательно, соединение 1 может эффективно уменьшать коллаген-индуцированный артрит при такой низкой дозе как 0,3 мг/кг. В группе введения 0,3 мг/кг дексаметазона могла существенно подавляться клиническая оценка коллаген-индуцированного артрита. С дня 30 клиническая оценка поддерживалась при 0, что существенно отличалось от контрольной группы введения носителя (p<0,0001), и оставалась на этом уровне до конца эксперимента. В группе введения 5 мг/кг Апремиласта также подавлялось повышение клинической оценки, и было показано существенное отличие от контрольной группы введения носителя с дня 33, и оно продолжалось до конца эксперимента. До дня 37 средняя оценка клинических симптомов составляла 4,2, что было ниже на 3,7 (p<0,001) по сравнению с контрольной группой введения носителя.
Путем анализа кривой клинической оценки каждого животного в каждой группе рассчитывали площадь под кривой (AUC). С использованием среднего значения площади под кривой между группами рассчитывали процент ингибирования в каждой дозовой группе относительно контрольной группы введения носителя. По сравнению с контрольной группой введения носителя, в группе введения дексаметазона и группе введения Апремиласта существенно снижались клинические оценки артрита у животных, и проценты ингибирования составили 96,4% (p<0,0001) и 41,3% (p<0,05), соответственно. Соединение 1 при трех дозах 0,3, 1 и 3 мг/кг может существенно уменьшать площадь под кривой клинической оценки артрита у животных, и проценты ингибирования составили 43,9% (p<0,05), 39,4% (p<0,05) и 51,7% (p<0,01), соответственно. Группа введения соединения 1 при 1 мг/кг имела процент ингибирования, сопоставимый с группой введения Апремиласта при 5 мг/кг (p<0,05 для обеих групп), тогда как группа введения соединения 1 при 3 мг/кг имела лучшие проценты ингибирования, чем группа введения Апремиласта при 5 мг/кг (p значения были < 0,01 и <0,05, соответственно)
2. Гистопатологическая оценка
Две задние конечности из каждой группы мышей брали в виде срезов для H.E. окрашивания, и получали общую оценку обеих задних конечностей. Артрит у мышей в контрольной группе введения носителя имел общую патологическую оценку 20,20±1,15. По сравнению с контрольной группой введения носителя, контрольное соединение Апремиласт при дозе 5 мг/кг также существенно снижало патологическую оценку артрита у мышей, которая могла снижаться до 13,90±1,89 (p<0,05). Соединение 1 при дозе 1 и 3 мг/кг могло существенно снижать патологические оценки артрита у мышей, которые могли снижаться до 14,00±2,43 (p<0,05) и 9,20±1,83 (p<0,0001), соответственно. Группа введения соединения 1 при 1 мг/кг имела патологическую оценку артрита, сопоставимую с группой введения Апремиласта при 5 мг/кг (p<0,05 для обеих групп), тогда как группа введения соединения 1 при 3 мг/кг имела лучшую патологическую оценку артрита, чем в группе введения Апремиласта при 5 мг/кг (p значения <0,0001 и <0,05, соответственно).
3. Заключение
Соединение 1 в трех дозовых группах 0,3, 1 и 3 мг/кг существенно улучшало симптомы коллаген-индуцированного артрита.
Было существенное улучшение патологии артрита в 1 мг/кг и 3 мг/кг дозовых группах, и эти три дозовые группы показали очевидную взаимосвязь доза-эффект в патологической оценке артрита. Терапевтический эффект соединения 1 при 3 мг/кг (клиническая оценка и патологическая оценка артрита) был лучше, чем у Апремиласта при 5 мг/кг.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОР PDE4 | 2017 |
|
RU2743126C2 |
| [4-(5-АМИНОМЕТИЛ-2-ФТОРФЕНИЛ)-ПИПЕРИДИН-1-ИЛ]-[7-ФТОР-1-(2-МЕТОКСИЭТИЛ)-4-ТРИФТОРМЕТОКСИ-1Н-ИНДОЛ-3-ИЛ]-МЕТАНОН КАК ИНГИБИТОР ТРИПТАЗЫ ТУЧНЫХ КЛЕТОК | 2009 |
|
RU2509766C2 |
| КРИСТАЛЛЫ ПРОИЗВОДНОГО ЦИКЛИЧЕСКОГО АМИНА И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2719384C1 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА МИМЕТИКА SMAC, ПРИМЕНЯЕМОГО В КАЧЕСТВЕ ИНГИБИТОРА IAP, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2020 |
|
RU2819398C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ИНГИБИТОРА ATR И ЕЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2832707C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ИНГИБИТОР ГИСТОНДЕАЦЕТИЛАЗЫ И МЕТОТРЕКСАТ | 2019 |
|
RU2772018C1 |
| НОВЫЕ КРИСТАЛЛЫ УРАЦИЛЬНОГО СОЕДИНЕНИЯ | 2016 |
|
RU2686722C1 |
| ТВЕРДАЯ ФОРМА, КРИСТАЛЛИЧЕСКАЯ ФОРМА И КРИСТАЛЛИЧЕСКАЯ ФОРМА А АГОНИСТА FXR, И СПОСОБ ИХ ПОЛУЧЕНИЯ, И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2804320C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2017 |
|
RU2797392C2 |
| СОЕДИНЕНИЕ ИНГИБИТОРА BRD4 В ТВЕРДОЙ ФОРМЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2793346C1 |
Изобретение относится к кристаллической Форме A соединения 1, где кристаллическая Форма A характеризуется рентгеновской порошковой дифрактограммой, имеющей характеристические дифракционные пики при следующих углах 2θ: 10,69±0,2°, 12,31±0,2°, 13,45±0,2°, 14,10±0,2°, 14,62±0,2°, 19,07±0,2°, 20,33±0,2°, 21,79±0,2°. Кристаллическую форму A соединения 1 получают путем добавления соединения 1 в спиртовой растворитель, кетоновый растворитель, смешанный растворитель, состоящий из спиртового растворителя и воды, смешанный растворитель, состоящий из кетонового растворителя и воды; спиртовой растворитель выбран из группы, состоящей из метанола, этанола; кетоновый растворитель выбран из группы, состоящей из ацетона и нагревания для растворения и затем охлаждение для кристаллизации с получением кристаллической Формы A. При этом смешанный растворитель, состоящий из спиртового растворителя и воды, состоит из смешанного растворителя, состоящего из этанола и воды. Технический результат – кристаллическая форма А соединения 1H-имидазо[4,5-b]пиридин-2(3H)-она (1), обладающая хорошей стабильностью, предназначенная для применения при получении лекарственного средства для лечения заболевания, связанного с PDE4 рецептором. 2 н. и 8 з.п. ф-лы, 4 ил., 10 табл., 9 пр.
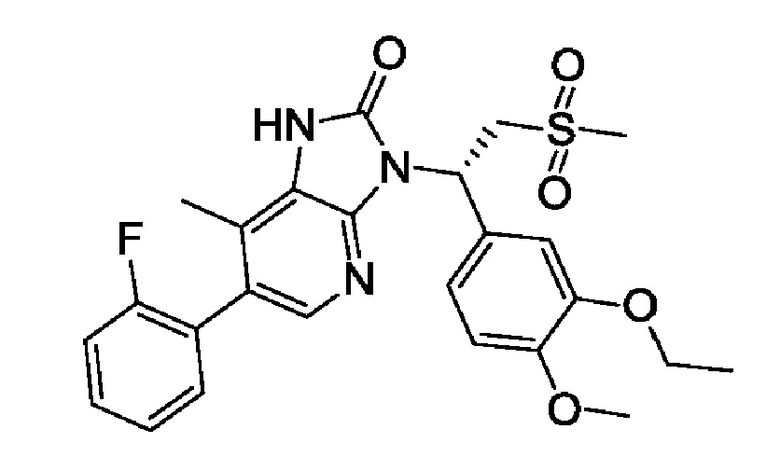
(1)
1. Кристаллическая Форма A соединения 1, где
кристаллическая Форма A характеризуется рентгеновской порошковой дифрактограммой, имеющей характеристические дифракционные пики при следующих углах 2θ: 10,69±0,2°, 12,31±0,2°, 13,45±0,2°, 14,10±0,2°, 14,62±0,2°, 19,07±0,2°, 20,33±0,2° 21,79±0,2°
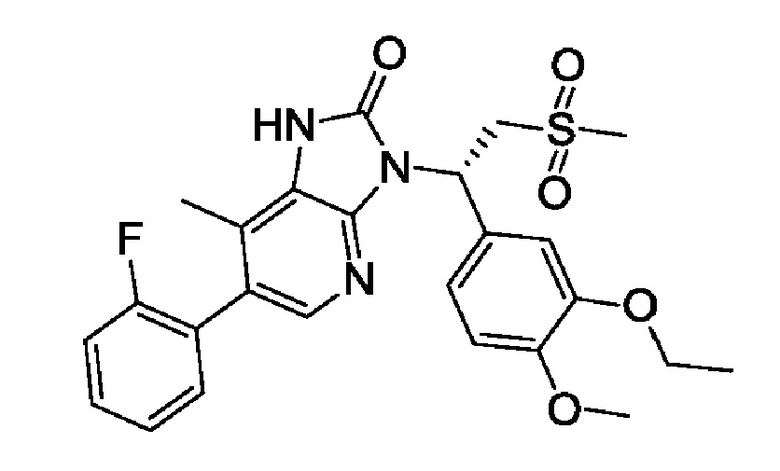
Соединение 1.
2. Кристаллическая Форма A соединения 1 по п. 1, где
кристаллическая Форма A характеризуется рентгеновской порошковой дифрактограммой, имеющей характеристические дифракционные пики при следующих углах 2θ: 6,25±0,2°, 8,93±0,2°, 10,69±0,2°, 12,31±0,2°, 13,45±0,2°, 14,10±0,2°, 14,62±0,2°, 18,16±0,2°, 19,07±0,2°, 20,33±0,2°, 21,79±0,2°.
3. Кристаллическая Форма A соединения 1 по п. 2, где
кристаллическая Форма A характеризуется рентгеновской порошковой дифрактограммой, показанной на Фиг. 1.
4. Кристаллическая Форма A соединения 1 по любому из пп. 1-3, где
кристаллическая Форма A характеризуется кривой дифференциальной сканирующей калориметрии, имеющую точку начала эндотермического пика при 201,70°C±2°C.
5. Кристаллическая Форма A соединения 1 по п. 4, где кристаллическая Форма A характеризуется кривой DSC, показанной на Фиг. 2.
6. Кристаллическая Форма A соединения 1 по любому из пп. 1-3, где кристаллическая Форма A характеризуется кривой термогравиметрического анализа, где потеря массы при 100,00±2°C составляет 0,02039%.
7. Кристаллическая Форма A соединения 1 по п. 6, где кристаллическая Форма A характеризуется кривой TGA, показанную на Фиг. 3.
8. Способ получения кристаллической Формы A соединения 1 по п. 1, включающий
добавление соединения 1 в спиртовой растворитель, кетоновый растворитель, смешанный растворитель, состоящий из спиртового растворителя и воды, смешанный растворитель, состоящий из кетонового растворителя и воды; спиртовой растворитель выбран из группы, состоящей из метанола, этанола; кетоновый растворитель выбран из группы, состоящей из ацетона и
нагревание для растворения и затем охлаждение для кристаллизации с получением кристаллической Формы A.
9. Способ получения кристаллической Формы A соединения 1 по п. 8, где
смешанный растворитель, состоящий из спиртового растворителя и воды, выбран из группы, состоящей из смешанного растворителя, состоящего из этанола и воды.
10. Способ получения кристаллической Формы A соединения 1 по п. 9, где
в смешанном растворителе, состоящем из спиртового растворителя и воды, объемное отношение спиртового растворителя к воде выбрано из группы, состоящей из 1:0,2-1,5.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| CN 105407888 A, 16.03.2016 | |||
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| MINO R | |||
| CAIRA: "Crystalline Polymorphism of Organic Compounds", TOPICS IN CURRENT CHEMISTRY, 1998, vol.198, pp.163-208 | |||
| BARBARA RODRIGUEZ-SPONG et al.: "General | |||

