Приоритет
CN 201910722102.7, с датой подачи 6 августа 2019 г.
Техническая область
Обеспечены кристаллические формы ингибитора ATR и способы их получения, а также применение для изготовления лекарственного средства для лечения, связанного с ATR заболевания.
Предпосылки создания изобретения
ATR (Ataxia Telangiectasia-mutated и Rad3-родственная протеинкиназа) принадлежит к семейству PIKK (киназа, родственная по отношению к фосфатидилинозитол-3-киназе) и участвует в репарации повреждений ДНК для поддержания стабильности генов. Протеинкиназа ATR имеет синергетический ответ на повреждение ДНК, стресс репликации и нарушения клеточного цикла. ATR и ATM принадлежат к семейству PIKK серин/треониновых протеинкиназ, и они являются общим компонентом клеточного цикла и репарации повреждений ДНК, а другие члены включают Chk1, BRCA1, р53. ATR в основном отвечает за стресс репликации ДНК (остановку репликационной вилки) и репарацию одноцепочечного разрыва.
В случае разрыва двухцепочечной ДНК или остановки репликационной вилки, ATR активируется структурой одноцепочечной ДНК. ДНК-полимераза остается в процессе репликации ДНК, и репликационная геликаза продолжает раскручиваться на переднем конце репликационной вилки ДНК, что приводит к образованию длинной одноцепочечной ДНК (ssDNA), которая затем связывается одноцепочечной ДНК и RPA (репликационный белок A). ATR/ATR действующий белковый комплекс рекрутируется RPA во время стресса репликации или повреждения ДНК к участку повреждения, комплекс RPA-одноцепочечная ДНК активирует комплекс RAD17/rfc2-5 для связывания с участком повреждения, соединение ДНК-одноцепочечная ДНК активирует гетеротример Rad9-HUS-RAD1 (9-1-1), 9-1-1, в свою очередь, рекрутирует TopBP1 для активации ATR. Как только ATR активируется, ATR способствует репарации ДНК через нижележащие мишени, стабилизируя и перезапуская остановленные репликационные вилки и временную остановку клеточного цикла. Эти функции достигаются с помощью ATR через опосредование нижележащей целевой Chk1. ATR действует в качестве контрольной точки для повреждения ДНК в клеточном цикле во время S-фазы. Она может опосредовать деградацию CDC25A через Chk1, тем самым задерживая процесс репликации ДНК и предоставляя время для репарации репликационной вилки. ATR также является основным регулятором контрольной точки клеточного цикла G2/M, предотвращая преждевременное вступление клеток в митоз до завершения репликации ДНК или повреждения ДНК. Эта ATR-зависимая остановка клеточного цикла G2/M в основном опосредована двумя механизмами: 1. Деградация CDC25A; 2. Фосфорилирование Cdc25C с помощью Chk1 для связывания с белком 14-3. Связывание Cdc25C с белком 14-3-3 способствует его экспорту из ядра и цитоплазматической изоляции, тем самым ингибируя его способность дефосфорилировать и активировать ядерный Cdc2, что, в свою очередь, препятствует вступлению в митоз.
Мутации гена ATR встречаются очень редко, и лишь небольшое число пациентов с синдромом Секеля имеют мутации гена ATR, которые характеризуются отставанием в росте и микроцефалией. Нарушение путей, связанных с ATR, может привести к нестабильности генома, и белок ATR активируется большинством видов химиотерапии рака. Кроме того, дупликация гена ATR описывается как фактор риска развития рабдомиосаркомы.
ATR необходима для саморепликации клеток и активируется в S-фазе, чтобы регулировать начало репликации и восстанавливать поврежденные репликационные вилки. Повреждение репликационных вилок может повысить чувствительность раковых клеток к противораковым агентам на основе платины и гидроксимочевины, и снизить резистентность раковых клеток. Следовательно, ингибирование ATR может стать эффективным методом лечения рака в будущем.
Краткое описание изобретения
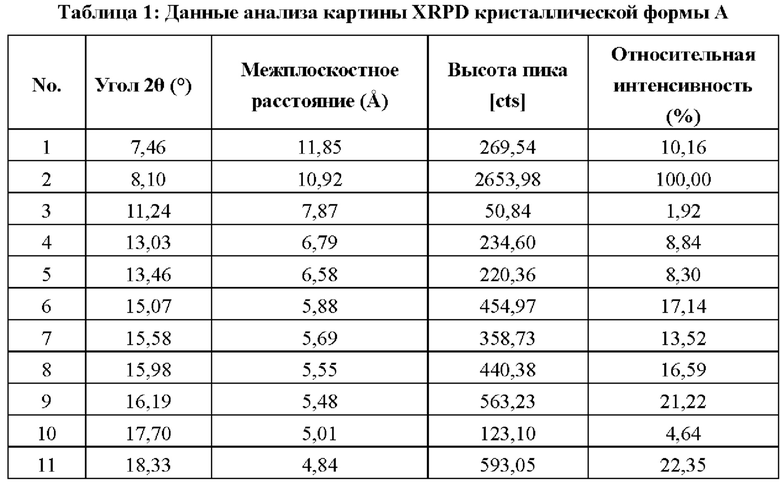
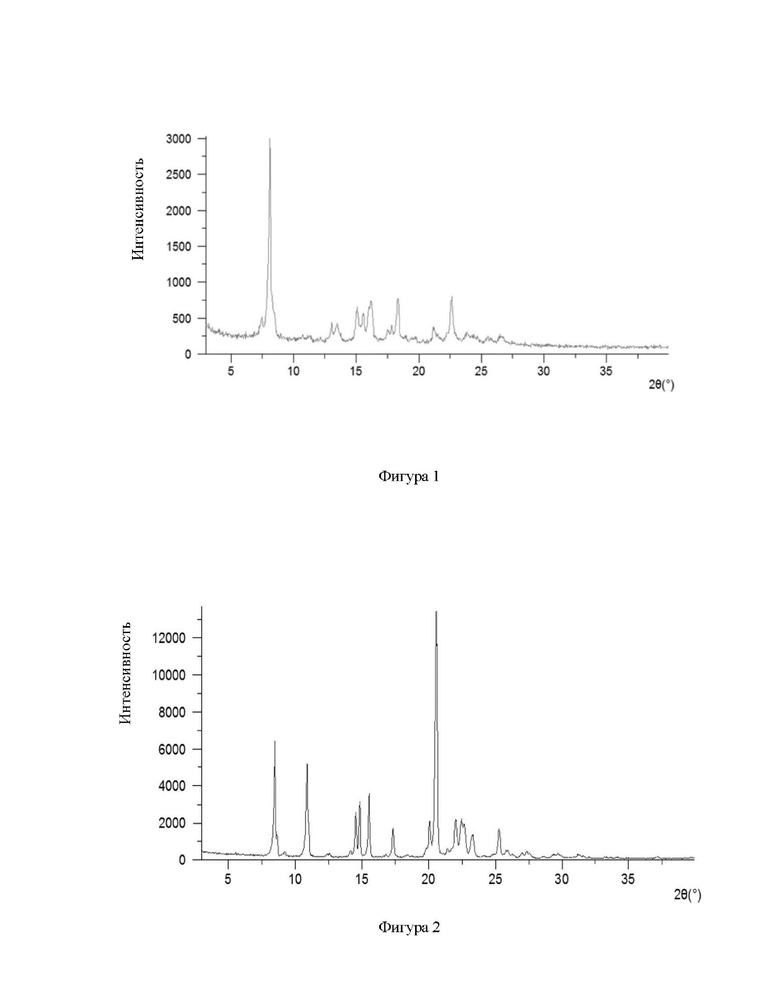
Обеспечена кристаллическая форма А соединения формулы (I), при этом кристаллическая форма А имеет картину порошковой рентгеновской дифракции (XRPD), имеющую характерные дифракционные пики при следующих углах 2θ: 8,10±0,20°, 18,33±0,20° и 22,63±0,20°.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая форма А имеет картину порошковой рентгеновской дифракции (XRPD), имеющую характерные дифракционные пики при следующих углах 2θ: 7,46±0,20°, 8,10±0,20°, 13,03±0,20°, 15,07±0,20°, 15,58±0,20°, 16,19±0,20°, 18,33±0,20° и 22,63±0,20°.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая форма А имеет картину порошковой рентгеновской дифракции (XRPD), имеющую характерные дифракционные пики при следующих углах 2θ: 7,46±0,20°, 8,10±0,20°, 13,03±0,20°, 13,46±0,20°, 15,07±0,20°, 15,58±0,20°, 16,19±0,20°, 18,33±0,20°, 21,17±0,20° и 22,63±0,20°.
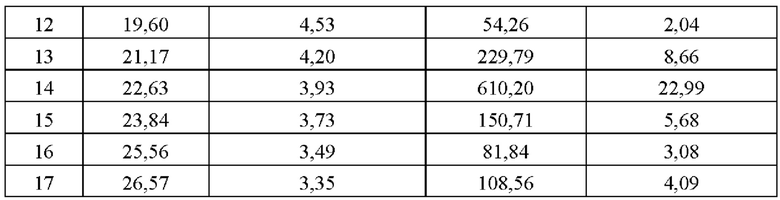
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая форма А имеет картину порошковой рентгеновской дифракции (XRPD), имеющую характерные дифракционные пики при следующих углах 2θ: 7,46°, 8,10°, 11,24°, 13,93°, 13,46°, 15,97°, 15,58°, 15,98°, 16,19°, 17,79°, 18,33°, 19,69°, 21,17°, 22,63°, 23,84°, 25,56° и 26,57°.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая форма А имеет картину XRPD, показанную на фигуре 1.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая форма А имеет картину XRPD с данными анализа, показанными в таблице 1:
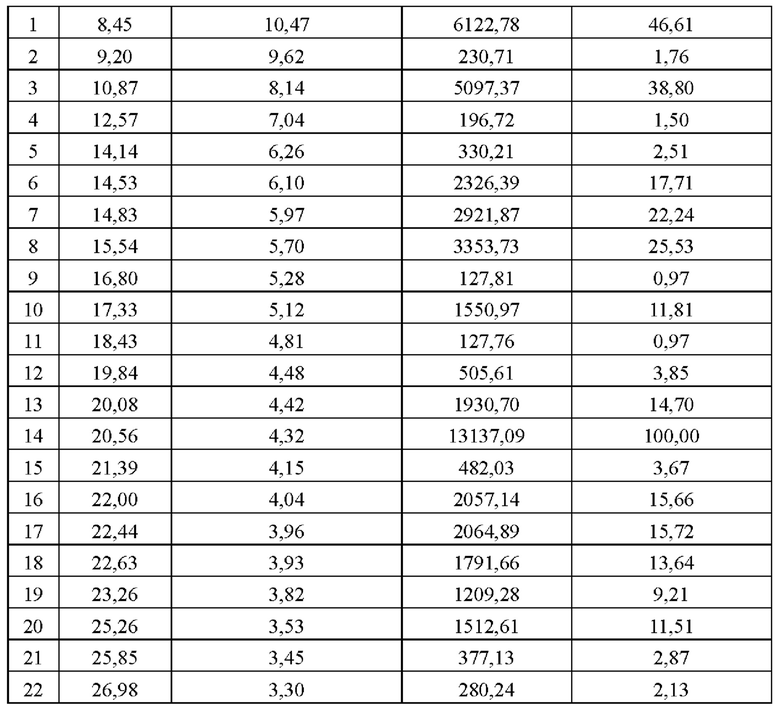
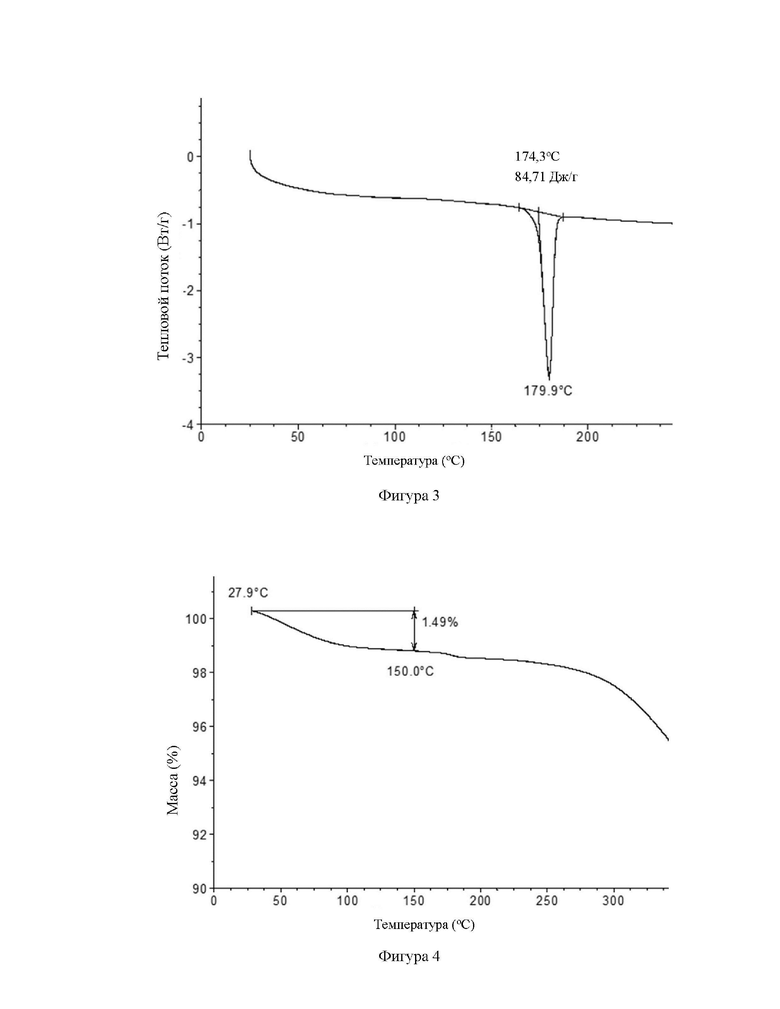
Обеспечена кристаллическая форма В соединения формулы (I), где кристаллическая форма В имеет картину порошковой рентгеновской дифракции (XRPD), имеющую характерные дифракционные пики при следующих углах 2θ: 8,45±9,20°, 10,87±9,20° и 20,56±0,20°.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая форма В имеет картину порошковой рентгеновской дифракции (XRPD), имеющую характерные дифракционные пики при следующих углах 2θ: 8,45±9,29°, 19,87±0,29°, 14,83±0,20°, 15,54±0,20°, 17,33±9,29°, 29,56±0,29°, 22,99±9,20° и 22,63±0,29°.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая форма В имеет картину порошковой рентгеновской дифракции (XRPD), имеющую характерные дифракционные пики при следующих углах 2θ: 8,45±9,29°, 10,87±0,20°, 14,83±0,20°, 15,54±0,20°, 17,33±0,20°, 29,08±0,29°, 29,56±9,29°, 22,00±9,29°, 22,63±9,29° и 25,26±9,29°.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая форма В имеет картину порошковой рентгеновской дифракции (XRPD), имеющую характерные дифракционные пики при следующих углах 8,45°, 9,20°, 19,87°, 12,57°, 14,14°, 14,53°, 14,83°, 15,54°, 16,80°, 17,33°, 18,43°, 19,84°, 29,08°, 20,56°, 21,39°, 22,00°, 22,44°, 22,63°, 23,26°, 25,26°, 25,85° и 26,98°.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая форма В имеет картину XRPD, показанную на фигуре 2.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая форма В имеет картину XRPD с данными анализа, показанными в таблице 2:

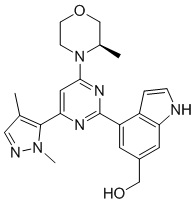
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая форма В имеет кривую дифференциальной сканирующей калориметрии (DSC), имеющую одну точку начала эндотермического пика при 174,3±3°С.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая форма В имеет картину DSC, показанную на фигуре 3.
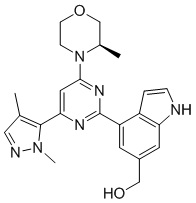
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая форма В имеет кривую термогравиметрического анализа (TGA), где потеря массы при 150°С±3°С составляет 1,49%.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием кристаллическая форма В имеет картину TGA, показанную на фигуре 4.
Обеспечен способ получения кристаллической формы А соединения формулы (I), включающий:
1) добавление соединения формулы (I) в этанольный растворитель;
2) добавление воды;
3) перемешивание в течение 100-120 ч;
4) проведение перекристаллизации при комнатной температуре с получением кристаллической формы А.
Обеспечен способ получения кристаллической формы В соединения формулы (I), включающий:
1) добавление соединения формулы (I) в растворитель;
2) нагревание до температуры при перемешивании в течение 2,5-120 ч;
3) проведение перекристаллизации при комнатной температуре с получением кристаллической формы В.
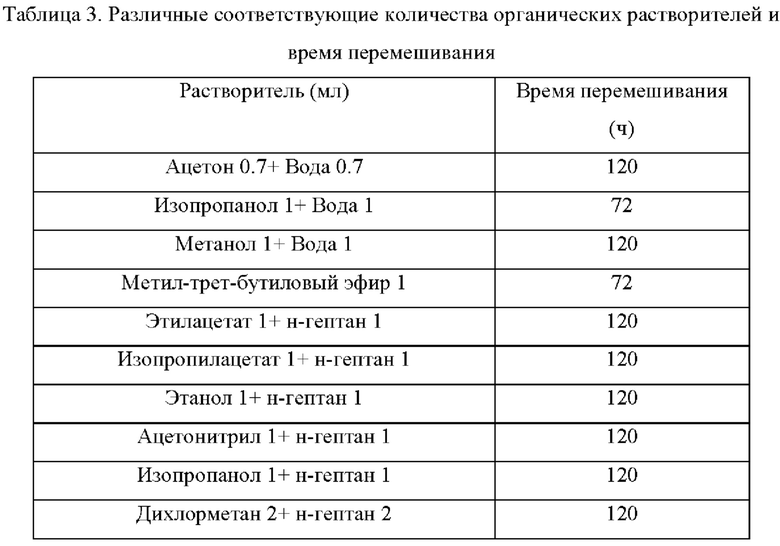
В некоторых вариантах осуществления в соответствии с настоящим раскрытием растворитель представляет собой: метанол, метил-трет-бутиловый эфир, смесь метанол/вода (об./об., 1:0,3-1), смесь ацетон/вода (об./об., 1:1), смесь изопропанол/вода (об./об., 1:1), смесь этилацетат/н-гептан (об./об., 1:1), смесь изопропилацетат/н-гептан (об./об., 1:1), смесь этанол/н-гептан (об./об, 1:1), смесь ацетонитрил/н-гептан (об/об, 1:1), смесь изопропанол/н-гептан (об/об, 1:1) или смесь дихлорметан/н-гептан (об/об, 1:1).
В некоторых вариантах осуществления в соответствии с настоящим раскрытием температура составляет 25-70°С.
Обеспечен способ получения кристаллической формы В соединения формулы (I), включающий:
1) добавление соединения формулы (I) в спиртовой растворитель;
2) добавление воды;
3) перемешивание в течение 15-20 ч;
4) проведение перекристаллизации при комнатной температуре с получением кристаллической формы В.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием объемное соотношение спиртового растворителя и воды составляет 1:1-1:4.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием спиртовой растворитель выбирают из группы, состоящей из метанола.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием диапазон концентрации соединения формулы (I) выбирают из группы, состоящей из 25 мг/мл-50 мг/мл.
Также обеспечено применение соединения формулы (I), кристаллической формы А или кристаллической формы В для изготовления лекарственного средства для лечения заболевания, связанного с ATR.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием лекарственное средство предназначено для лечения солидной опухоли или опухоли кроветворной системы.
В некоторых вариантах осуществления в соответствии с настоящим раскрытием лекарственное средство предназначено для применения в лечении колоректального рака, рака желудка, рака пищевода, первичной перитонеальной карциномы, адренокортикальной карциномы, почечной светлоклеточной карциномы, рака предстательной железы, уротелиальной карциномы мочевого пузыря, рака яичников, рака молочной железы, карциномы эндометрия, карциномы фаллопиевой трубы, немелкоклеточного рака легкого или мелкоклеточного рака легкого.
Технический эффект
Кристаллическая форма А и кристаллическая форма В соединения формулы (I) в соответствии с настоящим раскрытием являются стабильными, менее подвержены воздействию света, тепла и влаги, обладают хорошей эффективностью лекарственного средства in vivo и являются многообещающими для применения в качестве лекарственного средства.
Определение и описание
Если не указано иное, следующие термины и фразы имеют следующие определения. Конкретный термин или фраза не должны рассматриваться как неопределенные или неясные без конкретного определения и их следует понимать в обычном смысле. Используемое в настоящем документе торговое наименование относится к соответствующему изделию или активному ингредиенту.
Промежуточные соединения по настоящему изобретению могут быть получены различными способами синтеза, хорошо известными специалисту в данной области, включая конкретные варианты осуществления, перечисленные ниже, варианты осуществления, образованные комбинацией с другими способами химического синтеза, и эквивалентные альтернативные варианты осуществления, хорошо известные специалисту в данной области. Предпочтительные варианты осуществления включают, но без ограничения, приведенные ниже примеры.
Химическая реакция, описанная в конкретных вариантах осуществления, проводится в подходящем растворителе, и этот растворитель должен быть подходящим для химических изменений по настоящему раскрытию и для требуемых реагентов и материалов. Чтобы получить соединение по настоящему раскрытию, специалист в данной области может модифицировать или выбрать стадию синтеза или схему реакции на основе существующих вариантов осуществления.
Настоящее раскрытие будет подробно описано и примеры не предназначены для ограничения настоящего раскрытия.
Используемые в настоящем документе растворители являются коммерчески доступными и могут использоваться без дополнительной очистки.
Используемые в настоящем документе растворители могут быть коммерчески доступными. В настоящем документе используются следующие сокращения: EtOH обозначает этанол; МеОН обозначает метанол; TFA обозначает трифторуксусную кислоту; TsOH обозначает п-толуолсульфоновую кислоту; т.пл. обозначает температуру плавления; EtSO3H обозначает этансульфоновую кислоту; MeSO3H обозначает метансульфоновую кислоту; THF обозначает тетрагидрофуран; EtOAc обозначает этилацетат.
Рентгеновский порошковый дифрактометр (XRPD)
Прибор:
Способ тестирования: приблизительно 10-20 мг образца используют для детекции XRPD.
Подробные параметры XRPD следующие:
Источник излучения: Cu, kα (Kα1=1,540598 Å, Kα2=1,544426 Å, отношение интенсивностей Kα2/Kα1: 0,5)
Напряжение на рентгеновской трубке: 45 кВ, ток на рентгеновской трубке: 40 мА
Щель расходимости: фиксированная 1/8 град
1-я щель Соллера: 0,04 рад
2-я щель Соллера: 0,04 рад
Приемная щель: нет
Антирассеивающая щель: 7,5 мм
Время измерения: 5 мин
Диапазон углов сканирования: 3-40 градусов
Угол ширины шага: 0,0263 градусов
Время/шаг: 46,665 сек
Скорость вращения диска образцов: 15 об/мин
Дифференциальный сканирующий калориметр (DSC)
Прибор: Дифференциальный сканирующий калориметр ТА Q200/Q2000/2500
Способ тестирования: Образец (приблизительно 1-5 мг) помещают в алюминиевую кювету для тестирования DSC, в потоке азота 50 мл/мин и нагревают от 25°С (комнатная температура) до разложения со скоростью нагревания 10°С/мин.
Термогравиметрический анализатор (TGA)
Прибор: Термогравиметрический анализатор ТА Q5000/5500
Способ тестирования: Образец (приблизительно 1-5 мг) помещают в алюминиевые кюветы для тестирования TGA, в потоке азота 10 мл/мин и нагревают от комнатной температуры до 350°С со скоростью нагревания 10°С/мин.
Краткое описание чертежей
На фигуре 1 показана картина XRPD при излучении Cu-Kα кристаллической формы А соединения формулы (I);
на фигуре 2 показана картина XRPD при излучении Cu-Kα кристаллической формы В соединения формулы (I);
на фигуре 3 показана картина DSC кристаллической формы В соединения формулы (I);
на фигуре 4 показана картина TGA кристаллической формы В соединения формулы (I).
Подробное описание
Чтобы лучше понять настоящее раскрытие, ниже будет представлено дополнительное описание со ссылкой на конкретные примеры, которые не являются каким-либо его ограничением.
Пример 1: Получение соединения формулы (I)
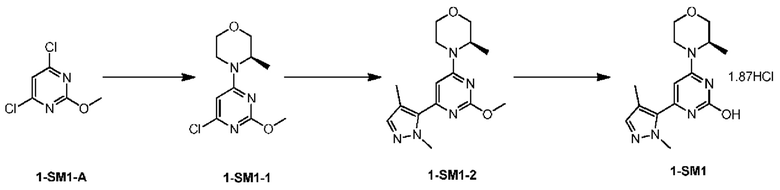
Стадия 1: Получение соединения 1-SM1-2
В 50 л реактор при комнатной температуре вносили 4,0 л диметилсульфоксида, к которому последовательно добавляли 1-SM1-A (1500,69 г, 8,39 моль), (R)-3- метилморфолин (854,97 г, 8,45 моль), карбонат калия (2891 г, 20,92 моль), а затем снова добавляли 6,0 л диметилсульфоксида для разбавления. После добавления реакционную систему перемешивали при 95°С в течение 3 ч. После обнаружения полного превращения в 1-SM1-1 температуру понижали до 45°С, и реактор продували азотом в течение 5 мин, а затем добавляли сложный пинаколовый эфир 1,4-диметилпиразол-5-бороновой кислоты (1952,44 г, 8,79 моль), тетракис(трифенилфосфин)палладий (192,98 г, 0,167 моль). После добавления вносили 2,0 л диметилсульфоксида для промывания внутренней стенки, а затем перемешивали в течение 12 ч при 104°С в атмосфере азота. После завершения реакции температуру понижали до 40°С, и реакционную систему фильтровали. Осадок на фильтре промывали 20,0 л этилацетата и фильтрат выливали в реактор. В реактор добавляли 15,0 л воды и реакционную систему перемешивали в течение 2 мин, и оставляли отстаиваться для разделения жидкости. Водную фазу снова экстрагировали 10,0 л этилацетата и органические фазы объединяли, промывали водой (10,0 л), насыщенным солевым раствором (8,0 л * 2), соответственно. Органические фазы концентрировали с получением неочищенного соединения 1-SM1-2, которое использовали непосредственно для следующей реакции. MS m/z: 304.0[М+Н]
Стадия 2: Получение соединения 1-SM1
Хлорид водорода (1344,0 г, 36,82 моль) пропускали через 6,0 л 1,4-диоксана при -40°С для дальнейшего использования. В 50 л реактор вносили 5,0 л 1,4-диоксана. Неочищенный продукт 1-SM1-2, полученный путем концентрирования, растворяли в 5,0 л 1,4-диоксана и вносили в реактор, в который при перемешивании добавляли 15,0 л 1,4-диоксана для разбавления. Температуру повышали до 70°С и к реакционной жидкости медленно добавляли вышеупомянутую смесь хлористоводородной кислоты/1,4-диоксана (1344 г, 6,0 л) и реакцию проводили при 98°С в течение 15 ч. Температуру снижали до 40°С и реакционную систему фильтровали. Осадок на фильтре промывали 15,0 л этилацетата, и твердое вещество выливали в реактор и суспендировали в 15,0 л этилацетата в течение 30 мин. Реакционную систему фильтровали и осадок на фильтре промывали 5,0 л этилацетата. Твердое вещество сушили в вакуумном сушильном шкафу с получением соединения 1-SM1.
MS m/z: 290.1[М+Н]+
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1.34 (br d, J=6.52 Гц, 3H) 2.08 (s, 3H) 3.47-3.58 (m, 2H) 3.64-3.70 (m, 1H) 3.72-3.78 (m, 1H) 3.87 (s, 3H) 3.97 (br s, 1 H) 4.07-4.32 (m, 1H) 4.47 (br s, 1H) 6.61 (s, 1H) 7.44 (s, 1H)
Стадия 3: Получение соединения 1-SM2
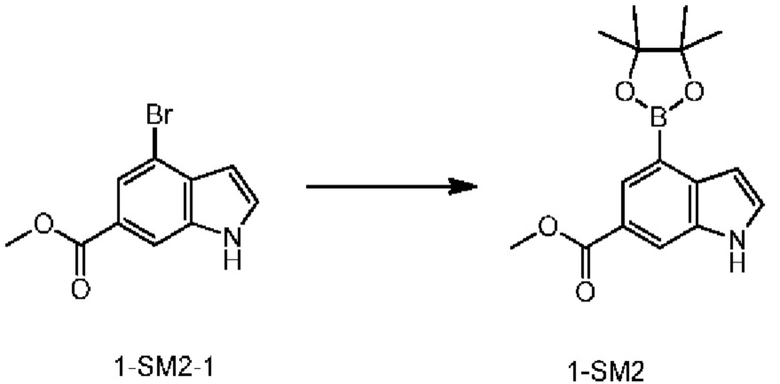
К раствору соединения 1-SM2-1 (2 г, 7,87 ммоль), бис(пинаколато)дибора (4,00 г, 15,74 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (0,3 г, 410,00 мкмоль) в 1,4-диоксане (25,0 мл) добавляли ацетат калия (2,32 г, 23,61 ммоль) и реакционную систему трижды продували азотом. Реакционную систему нагревали при 100°С при перемешивании в течение 8 ч и фильтровали. Раствор концентрировали с получением неочищенного продукта, который разделяли колоночной хроматографией с получением соединения 1-SM2.
MS-ESI m/z: 302.1 [М+Н]+.
Стадия 4: Получение соединения 1-1
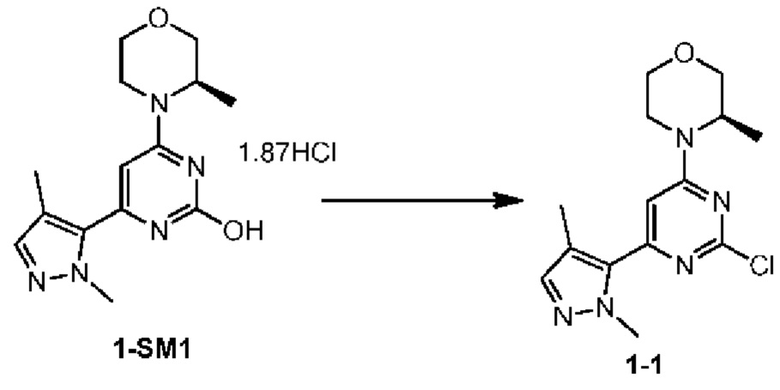
При комнатной температуре в 50 л реактор вносили 15,0 л толуола, и последовательно добавляли соединение 1-SM1 (1500,0 г, 4,23 моль), триэтиламин (1175,0 мл, 8,45 моль), и при перемешивании порциями добавляли оксихлорид фосфора (1178,0 мл, 12,68 моль). После добавления реакционный раствор перемешивали при 103-108°С в течение 1 ч 40 мин. После обнаружения завершения реакции температуру снижали до 45°С. Реакционный раствор переносили в резервуар для временного хранения и в реактор добавляли 15,0 л очищенной воды. При перемешивании реакционный раствор порциями добавляли в очищенную воду, поддерживая температуру при 20-40°С. После добавления рН доводили до 6-7 с помощью 12,0 л водного раствора гидроксида натрия (4М) и поддерживали температуру при 20-40°С. После регулирования рН в реактор при равномерном перемешивании добавляли 7,5 л этилацетата для расслоения. Водную фазу экстрагировали 15,0 л этилацетата. Органические фазы объединяли, промывали 12,0 л насыщенного солевого раствора, концентрировали при пониженном давлении до полного отсутствия фракций с получением неочищенного продукта. Неочищенный продукт растворяли в 1,5 л метил-трет-бутилового эфира, к которому порциями при перемешивании добавляли 12,0 л н-гептана, смесь перемешивали в течение 5 мин и фильтровали. Осадок на фильтре промывали 5,0 л н-гептана, твердое вещество помещали в лоток и оставляли для высыхания с получением соединения 1-1.
MS m/z: 308.0[М+Н]+
1Н ЯМР (400 МГц, DMSO-d6) δ ppm 1.29 (br d, J=6.78 Гц, 3H) 2.17 (s, 3H) 3.24-3.32 (m, 1H) 3.52 (br s, 1H) 3.63-3.69 (m, 1H) 3.78 (br d, J=11.54 Гц, 1H) 3.96 (s, 3H) 4.01 (br s, 1H) 4.14 (br s, 1H) 4.47 (br s, 1H) 6.89 (s, 1H) 7.42 (s, 1H)
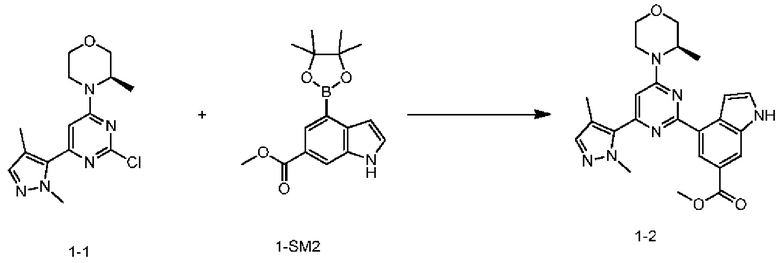
Стадия 5: Получение соединения 1-2
При 20-30°С в атмосфере азота 2,1 л диметилсульфоксида при перемешивании вносили в 10 л стеклянный реактор, в который последовательно добавляли соединение 1-1 (0,21 кг), соединение 1-SM2 (0,306 кг), водный раствор карбоната натрия (1,3 М, 1,05 л), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (0,00749 кг) с помощью капельной воронки. Реакционную систему нагревали до внутренней температуры 60-70°С и поддерживали эту температуру в течение 4-16 ч. Систему охлаждали до 40-45°С, добавляли по каплям 5,25 л воды в течение 30 мин, а затем перемешивали в течение 30 мин. Систему фильтровали с помощью вакуумного фильтра и осадок на фильтре промывали 2,1 л воды. Сушка в вакууме при 45°С обеспечила получение неочищенного продукта. К неочищенному продукту, полученному на предыдущей стадии, добавляли 2,625 л этилацетата. После равномерного перемешивания до растворения снова добавляли 10,5 л метил-трет-бутилового эфира при дополнительном перемешивании в течение 30 мин. После фильтрования с использованием воронки Бюхнера со слоем целита, слой целита снова промывали смешанным раствором 2,1 л этилацетата и метил-трет-бутилового эфира (объемное соотношение 1:4). Фильтраты объединяли и органическую фазу концентрировали с получением концентрата. Собирали верхний черный осадок на целите, к которому добавляли 1,5 л этилацетата. После перемешивания при комнатной температуре в течение 1 ч фильтрацию выполняли с использованием воронки Бюхнера со слоем целита, осадок на фильтре промывали 0,5 л этилацетата и фильтрат концентрировали. Концентраты, полученные на двух предыдущих стадиях, объединяли.
К полученному ранее концентрату добавляли 1,5 л этилацетата для растворения, а затем медленно по каплям добавляли в 4,5 л раствора н-гептана при перемешивании (1,5 ч), после чего перемешивали еще в течение 2 ч. Систему фильтровали, осадок на фильтре промывали смешанным раствором 0,4 л этилацетата и н-гептана (объемное соотношение 1:3). После сушки в вакууме осадок на фильтре помещали в одногорлую колбу объемом 2 л, в которую добавляли 0,8 л изопропилацетата. После кипячения с обратным холодильником в течение 4 ч реакционную систему медленно охлаждали до комнатной температуры и перемешивали в течение ночи. Реакционную систему фильтровали с использованием воронки Бюхнера и осадок на фильтре промывали 0,3 л изопропилацетата. Твердое вещество собирали и сушили в вакууме с получением продукта.
Продукт растворяли в 4,2 л этилацетата и при перемешивании добавляли 42 г активированного угля. Реакционную систему перемешивали в течение ночи при кипячении с обратным холодильником, фильтровали в горячем состоянии и фильтровали с использованием воронки Бюхнера со слоем целита. Слой целита снова промывали 2,0 л этилацетата и фильтраты объединяли. Органическую фазу концентрировали до 3,0 л. К полученной выше органической фазе добавляли 1,2 л этилацетата и 43 г активированного угля, перемешивали при кипячении с обратным холодильником в течение 8 часов и затем фильтровали в горячем состоянии. Реакционную систему фильтровали с использованием воронки Бюхнера со слоем целита. Слой целита промывали 2,0 л этилацетата и фильтраты объединяли. Органическую фазу концентрировали и сушили в вакууме с получением соединения 1-2.
MS m/z: 447.0 [М+Н]+
1Н ЯМР (CHCl3-d, 400 МГц): δ=8.98 (d, J=1.3 Гц, 1H), 8.57 (br s, 1H), 8.27 (s, 1H), 7.58 (d, J=2.0 Гц, 1H), 7.49 (t, J=2.8 Гц, 1H), 7.42 (s, 1H), 6.50 (s, 1H), 4.48 (br s, 1H), 4.29 (br d, J=12.5 Гц, 1H), 4.17 (s, 3H), 4.13 (dd, J=11.9, 2.9 Гц, 1H), 3.98 (s, 3H), 3.87-3.93 (m, 1H), 3.80-3.87 (m, 1H), 3.69 (td, J=11.9, 3.0 Гц, 1H), 3.44 (td, J=12.8, 3.8 Гц, 1H), 2.25 (s, 3H), 1.44 ppm (d, J=7.0 Гц, 3H)
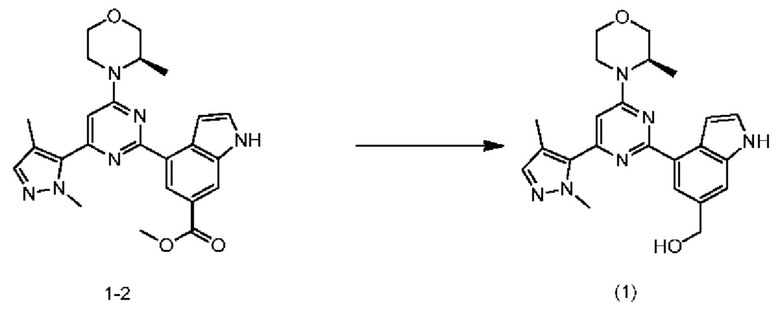
Стадия 6: Получение соединения формулы (1)
Алюмогидрид лития (63,0 мл, 2,5 М) при 20°С добавляли к соединению 1-2 (35,0 г, 78,39 ммоль) в тетрагидрофуране (50,0 мл) и реакционную систему перемешивали при 20°С в течение 1 ч. При 0-5°С 6,9 мл воды, 6,9 мл 15% гидроксида натрия и 20,7 мл воды последовательно медленно добавляли к реакционному раствору, который затем фильтровали. Фильтрат концентрировали с получением неочищенного продукта, который разделяли колоночной хроматографией (этилацетат/петролейный эфир: 50-100%) с получением продукта. Вышеупомянутый продукт растворяли в 20,0 мл диметилсульфоксида при комнатной температуре, медленно добавляли по каплям в 400 мл перемешиваемой воды, фильтровали и сушили с получением соединения формулы (I).
MS m/z: 419.1 [М+Н]+
1Н ЯМР (400 МГц, CHCl3-d) δ ppm 8.39 (br s, 1H), 8.28 (s, 1H), 7.58 (s, 1H), 7.51 (br s, 1H), 7.42 (s, 1H), 7.35 (t, J=2.76 Гц, 1H), 6.49 (s, 1H), 4.89 (s, 2H), 4.51 (br s, 1H), 4.30 (br d, J=14.05 Гц, 1H), 4.11-4.18 (m, 4H), 3.81-3.94 (m, 2H), 3.70 (td, J=11.86, 3.14 Гц, 1H), 3.44 (td, J=12.86, 3.89 Гц, 1H), 2.26 (s, 3H), 1.45 (d, J=6.78 Гц, 3H)
Пример 2: Получение кристаллической формы А соединения формулы (1)
Взвешивали около 500,0 мг соединения формулы (I) и растворяли в 5 мл этанола, в который по каплям добавляли 15 мл очищенной воды. После добавления реакционную систему помещали на магнитную мешалку (20°С) и перемешивали в течение 120 ч. Суспензию фильтровали с получением твердого вещества, которое сушили в течение ночи в вакуумном сушильном шкафу с получением кристаллической формы А соединения формулы (I).
1H ЯМР (400 МГц, CHCl3-d) δ=8.39 (br s, 1H), 8.25 (d, J=1.3 Гц, 1H), 7.53 (s, 1H), 7.49 (t, J=2.3 Гц, 1H), 7.40 (s, 1H), 7.31 (t, J=2.8 Гц, 1H), 6.46 (s, 1H), 4.86 (s, 2H), 4.48 (br d, J=4.8 Гц, 1H), 4.28 (br d, J=12.5 Гц, 1H), 4.15-4.07 (m, 4H), 3.91-3.86 (m, 1H), 3.84-3.79 (m, 1H), 3.67 (dt, J=3.0, 11.9 Гц, 1H), 3.42 (dt, J=3.9, 12.9 Гц, 1H), 2.23 (s, 3H), 1.42 (d, J=7.0 Гц, 3H)
Пример 3: Получение кристаллической формы В соединения формулы (1)
Около 100 мг соединения формулы (1) добавляли в разные стеклянные бутыли, в которые добавляли соответствующие количества органических растворителей или смесей растворителей (таблица 3). Указанные выше образцы перемешивали в смесителе при постоянной температуре (40°С) (время перемешивания см. в таблице 3) (защита от света). Затем твердое вещество отфильтровывали и помещали в вакуумный сушильный шкаф (40°С) для сушки в течение ночи. Все обработки обеспечили получение кристаллической формы В.
Пример 4: Получение кристаллической формы В соединения формулы (1)
Соединение формулы (1) (см. массу в таблице 4) медленно добавляли в растворитель метанол при (60-70°С) (см. объем в таблице 4), а затем медленно добавляли воду (см. объем в таблице 4). После перемешивания в течение 0,5 ч при 60°С температуру снижали до 55°С и перемешивали в течение 0,5 ч. Затем температуру снижали до 50°С и перемешивали в течение 0,5 ч. Затем температуру снижали до 45°С и перемешивали в течение 0,5 ч. Затем температуру снижали до 40°С и перемешивали в течение 0,5 ч. Затем температуру снижали до 35°С и перемешивали в течение 0,5 ч. Затем температуру снижали до 30°С и перемешивали в течение 0,5 ч. Затем температуру снижали до 20-25°С и перемешивали в течение 10 ч. Твердое вещество отфильтровывали с получением кристаллической формы В.

Пример 5: Получение кристаллической формы В соединения формулы (1)
Методика эксперимента: Около 5,5 г соединения формулы (1) медленно добавляли в 50 мл метанольного растворителя при (60-70°С). После перемешивания при 60°С в течение 0,5 ч температуру снижали до 25°С и перемешивали в течение 2 ч, и твердое вещество отфильтровывали с получением кристаллической формы В.
Пример 6: Получение кристаллической формы В соединения формулы (1)
900,0 г соединения формулы (I) растворяли в 9,0 л метанола, к которому медленно по каплям добавляли 9,0 л очищенной воды при комнатной температуре (25°С). Реакционную систему дополнительно перемешивали в течение 20 ч и фильтровали при пониженном давлении. Осадок на фильтре промывали 6,0 л очищенной воды и твердое вещество сушили в вакууме с получением кристаллической формы В соединения формулы (I).
MS m/z: 419.0 [М+Н]+
1Н ЯМР (CHCl3-d, 400 МГц): δ=8.60 (br s, 1H), 8.21 (s, 1H), 7.45 (br s, 1H), 7.42 (br s, 1H), 7.40 (s, 1H), 7.25 (br d, J=2.5 Гц, 1H), 6.45 (s, 1H), 4.81 (br s, 2H), 4.47 (br d, J=5.8 Гц, 1H), 4.27 (br d, J=13.8 Гц, 1H), 4.07-4.13 (m, 4H), 3.85-3.91 (m, 1H), 3.78-3.84 (m, 1H), 3.66 (td, J=11.9, 3.0 Гц, 1H), 3.41 (td, J=12.8, 3.8 Гц, 1H), 2.22 (s, 3H), 1.41 ppm (d, J=6.8 Гц, 3H)
Пример 7: Тесты на стабильность в твердом состоянии кристаллической формы А в условиях высокой температуры и высокой влажности
Два образца кристаллической формы А параллельно взвешивали, каждый около 100 мг, помещали на дно стеклянной бутыли для образцов и распределяли тонким слоем. Образцы запечатывали алюминиевой фольгой, и в алюминиевой фольге делали несколько небольших отверстий, чтобы обеспечить полный контакт образца с окружающим воздухом. Образцы помещали в контейнер с постоянной температурой и влажностью в условиях 40°С/75% влажности. Образцы в указанных выше условиях отбирали и тестировали на 30-й день, и результаты тестирования сравнивали с первоначальным результатом теста в день 0. В таблице 5 ниже показаны результаты теста:
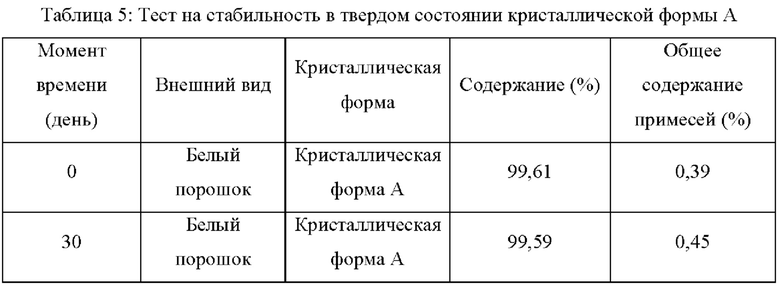
Вывод: Кристаллическая форма А соединения формулы (I) обладает хорошей стабильностью и является простой для изготовления лекарственных средств
Пример 8: Тесты на физическую стабильность в твердом состоянии кристаллической формы А в различных условиях температуры, влажности и освещенности
Параллельно взвешивали четыре образца кристаллической формы А, каждый около 100 мг, помещали на дно стеклянной бутыли для образцов и распределяли тонким слоем. Образцы запечатывали алюминиевой фольгой, и в алюминиевой фольге делали несколько небольших отверстий, чтобы обеспечить полный контакт образца с окружающим воздухом. Четыре подготовленных образца помещали в условия 25°С/92,5% относительной влажности, 60°С, 40°С/75% и освещенности, соответственно, и исследовали физическую стабильность образцов на 10-й день. В то же время отдельно взвешивали образец кристаллической формы А массой около 100 мг, помещали на дно стеклянной бутыли для образцов, закрывали завинчивающейся крышкой и хранили при -20°С для использования в качестве контроля. На 10-й день все образцы вынимали, температуру возвращали к комнатной температуре и наблюдали за появлением изменений во внешнем виде образцов. XRPD использовали для детекции кристаллических форм образцов. Путем сравнения образцов ускоренного режима тестирования с контрольным образцом, определяли физическую стабильность в твердом состоянии кристаллической формы А соединения формулы (I). В следующей таблице 6 показаны результаты эксперимента на физическую стабильность в твердом состоянии кристаллической формы А.
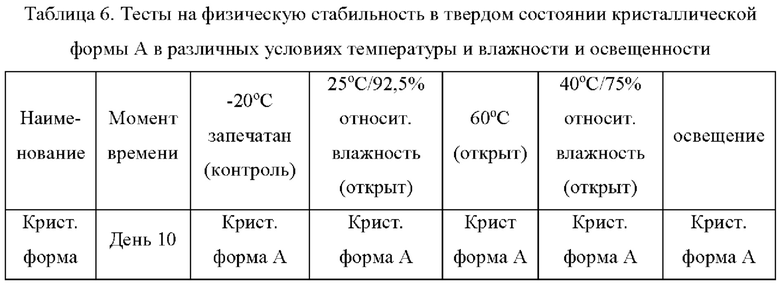
Вывод: Кристаллическая форма А соединения формулы (I) обладает хорошей стабильностью и является простой для изготовления лекарственных средств
Пример 9: Тесты на физическую стабильность в твердом состоянии кристаллической формы В в условиях высокой температуры, высокой влажности и освещенности
Два образца кристаллической формы В взвешивали параллельно для каждой группы, помещали на дно стеклянной бутыли для образцов и распределяли тонким слоем. Образцы запечатывали алюминиевой фольгой, и в алюминиевой фольге делали несколько небольших отверстий, чтобы обеспечить полный контакт образца с окружающим воздухом. Образцы помещали в камеру с постоянной температурой и влажностью или в световой бокс с различной влажностью. Образцы, помещенные в вышеуказанные условия, отбирали и тестировали на день 5, день 10, день 30, через 1 месяц, 3 месяца или 6 месяцев. Результаты теста сравнивали с результатами первоначального теста на день 0. Результаты теста показаны в таблице 7-11 ниже: 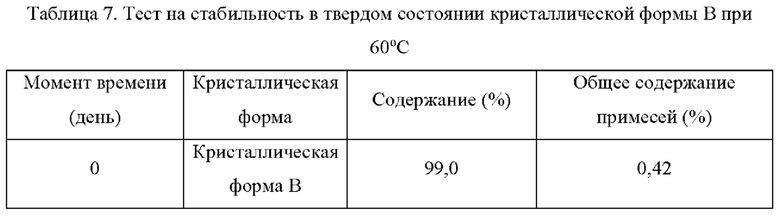
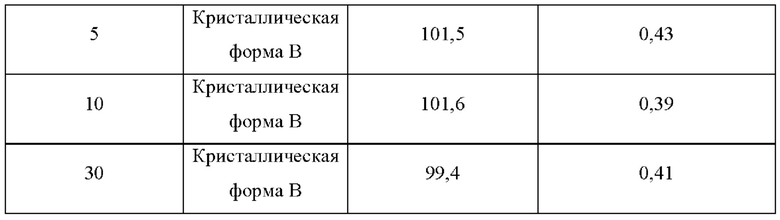
Вывод: Кристаллическая форма В соединения формулы (I) обладает хорошей стабильностью в условиях высокой температуры и является простой для изготовления лекарственных средств
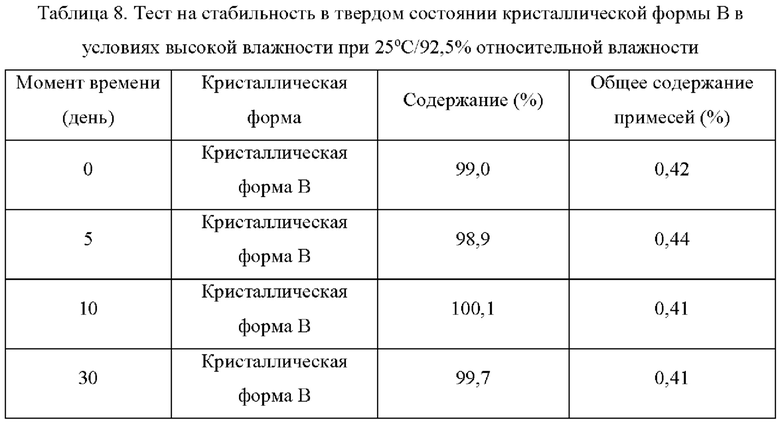
Вывод: Кристаллическая форма В соединения формулы (I) обладает хорошей стабильностью в условиях высокой влажности и является простой для изготовления лекарственных средств
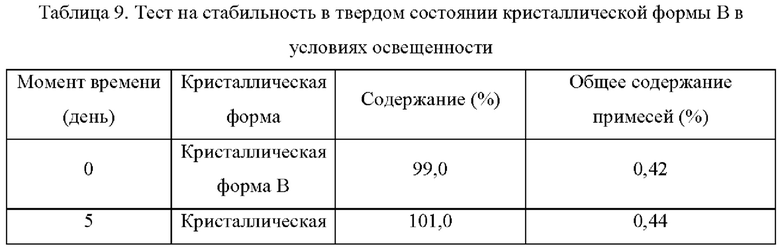

Вывод: Кристаллическая форма В соединения формулы (I) обладает хорошей стабильностью в условиях освещенности
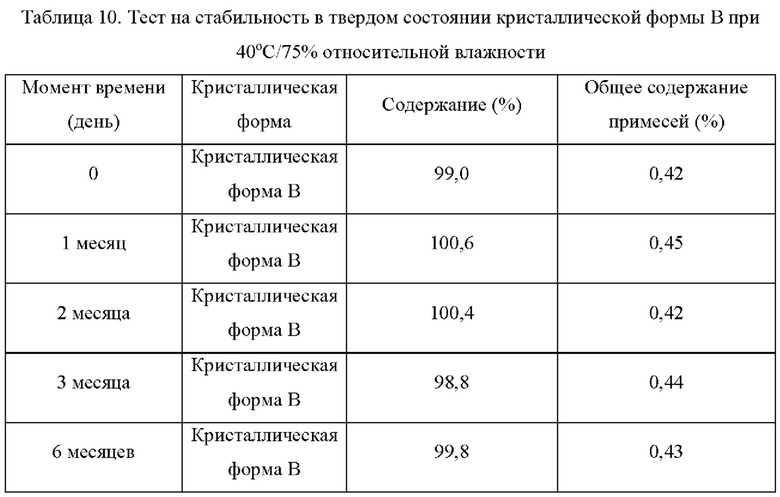
Вывод: Кристаллическая форма В соединения формулы (I) обладает хорошей стабильностью и является простой для изготовления лекарственных средств 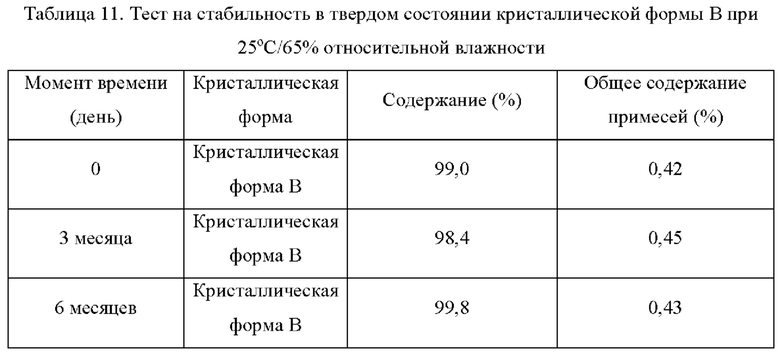
Вывод: Кристаллическая форма В соединения формулы (I) обладает хорошей стабильностью и является простой для производства лекарственных средств
Экспериментальный пример 1: Оценка in vitro
Значения IC50 определяли для оценки ингибирующей активности тестируемого соединения в отношении киназы ATR человека.
ATR/ATRIP(h) инкубировали в аналитическом буфере, содержащем 50 нМ GST-сМус-р53 и Mg/ATP (в требуемой концентрации). Реакцию инициировали добавлением смеси Mg/ATP. После инкубации в течение 30 мин при комнатной температуре для прекращения реакции добавляли стоп-раствор, содержащий EDTA. Наконец, добавляли детектирующий буфер, содержащий d2-меченное анти-GST моноклональное антитело и меченное европием анти-фосфо Ser15 антитело против фосфорилированного р53. Затем считывали планшет в режиме флуоресценции с временным разрешением и выполняли гомогенную флуоресценцию с временным разрешением.
Сигнал флуоресценции (HTRF) определяли по формуле: HTRF=HTRF=10000×(Ет665 нм / Ет620 нм).
Программу XLFit версии 5.3 (ID Business Solutions) использовали для анализа данных IC50. Нелинейный регрессионный анализ использовали для подбора S-образной кривой доза-эффект (переменный наклон). Результат теста показан в таблице 12:

Вывод: Рассматриваемое соединение формулы (I) обладает хорошим ингибирующим эффектом в отношении киназы ATR.
Экспериментальный пример 2: Тест на жизнеспособность клеток in vitro
Эффект соединения на ингибирование клеточной пролиферации исследовали в этом тесте путем обнаружения эффекта соединения на клеточную активность in vitro в опухолевых клеточных линиях LoVo.
Детекция жизнеспособности клеток CellTiter-Glo с использованием люминесцентного анализа
Следующие стадии осуществляли в соответствии с инструкциями к набору Promega CellTiter-Glo Luminescence Cell Viability Detection Kit (Promega-G7573).
(1). Буфер CellTiter-Glo расплавляли и оставляли для доведения температуры до комнатной температуры.
(2). Субстрат CellTiter-Glo оставляли для доведения температуры до комнатной температуры.
(3). Буфер CellTiter-Glo добавляли во флакон с субстратом CellTiter-Glo для растворения субстрата и приготовления рабочего раствора CellTiter-Glo.
(4). Перемешивание на вортексе выполняли медленно для полного растворения.
(5). Планшет с клеточной культурой вынимали и оставляли на 30 мин для уравновешивания до комнатной температуры.
(6). В каждую лунку добавляли по 50 мкл (половина объема культуральной среды в каждой лунке) рабочего раствора CellTiter-Glo. Планшет оборачивали алюминиевой фольгой для защиты от света.
(7). Культуральный планшет встряхивали на орбитальном шейкере в течение 2 мин для индукции лизиса клеток.
(8). Культуральный планшет выдерживали при комнатной температуре в течение 10 мин для стабилизации люминесцентного сигнала.
(9). Люминесцентный сигнал детектировали на планшетном ридере SpectraMax i3x компании Molecular Devices.
Анализ данных
Для расчета скорости ингибирования (IR) тестируемого соединения использовали следующую формулу: IR (%)=(1-(RLU соединения-RLU бланкового контроля)/(RLU контрольного носителя-RLU бланкового контроля))*100%.
Скорости ингибирования различных концентраций соединений рассчитывали в Excel, а затем использовали программное обеспечение GraphPad Prism для построения кривой ингибирования и расчета соответствующих параметров, включая минимальную скорость ингибирования, максимальную скорость ингибирования и IC50.
Результаты теста показаны в таблице 13:
Таблица 13. Результаты теста in vitro на ингибирование пролиферации клеток LoVo

Вывод: Рассматриваемое соединение формулы (1) TR обладает хорошим ингибирующим эффектом на опухолевые клетки LoVo с мутацией в сигнальном пути ATM.
Экспериментальный пример 3: Исследование фармакокинетических свойств in vivo
Тестируемые образцы: На основании вышеуказанных тестов некоторые из этих соединений, обладающие высокой активностью и репрезентативными структурами, отбирали для дополнительных тестов.
Методика эксперимента: Целью данного исследования является определение фармакокинетических параметров соединения и расчет его биодоступности при кормлении через зонд у самок мышей Balb/c Nude.
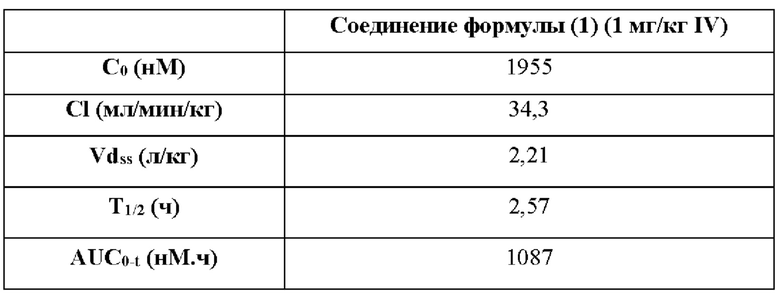
В этом проекте использовали шесть самок мышей Balb/c Nude, трем мышам инъецировали внутривенно дозу 1 мг/кг, при этом образцы плазмы собирали в 0 ч (до введения) и через 0,0833, 0,25, 0,5, 1, 2, 4, 6, 8 и 24 ч после введения, а остальным трем мышам вводили внутрижелудочно дозу 10 мг/кг или 25 мг/кг, при этом образцы плазмы собирали в 0 ч (до введения) и через 0,5, 1, 2, 3, 4, 6, 8, 24 ч после введения. На собранных образцах проводили анализ LC-MS/MS, и получали данные. На основании собранных данных анализа вычисляли соответствующие фармакокинетические параметры с помощью программного обеспечения Phoenix WinNonlin 6.2.1. Результаты теста представлены в таблицах 14.1 и 14.2.
14.1 Результаты введения путем внутривенной инъекции
14.2 Результаты внутрижелудочного введения
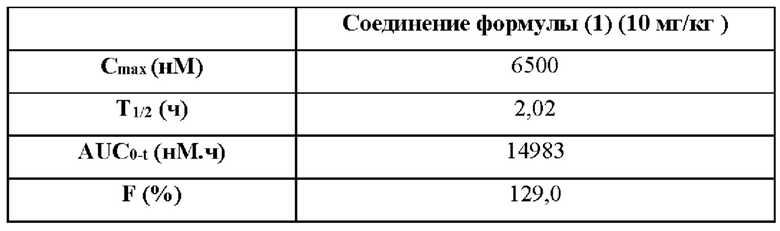
Примечание: С0 (нМ) обозначает концентрацию лекарственного средства в организме в момент времени 0 мин; С1 (мл/мин/кг) обозначает скорость выведения лекарственного средства из организма; Vdss (л/кг) обозначает объем распределения лекарственного средства в организме; Т1/2 (ч) обозначает период полувыведения; AUC0-t (нМ⋅ч) обозначает объем воздействия лекарственного средства в организме; Cmax (нМ) обозначает самую высокую концентрацию лекарственного средства в организме; F обозначает биодоступность.
Вывод: рассматриваемое соединение формулы (1) обладает хорошей абсорбцией и воздействием при внутрижелудочном введении и является подходящим для перорального введения.
Экспериментальный пример 4: Исследование эффективности in vivo при колоректальном раке LoVo CDX
Цель: LoVo представляют собой опухолевые клетки колоректальной аденокарциномы с мутацией MRE11A (MRE11A является ключевым компонентом сигнального пути ATM для репарации двухцепочечных разрывов ДНК), которые чувствительны к ингибитору ATR. В этом тесте используют модель рака прямой кишки LoVo CDX для проверки ингибирующего эффекта ингибитора ATR на опухоль с дефектным сигнальным путем ATM.
Экспериментальная методика:
1. Экспериментальные животные
Виды: мышь
Штамм: мыши BALB/c nude
Поставщик: Beijing Weitong Lihua Laboratory Animal Technology Co., Ltd.
Возраст в неделях и вес: возраст 6-8 недель, 18-22 г
Пол: женский
2. Культура клеток
Клетки LoVo рака толстой кишки человека (ЕСАСС, каталог: 87060101), монослойная культура in vitro, условия культивирования: среда Хэма F-12, содержащая 10% эмбриональной телячьей сыворотки, 100 ЕД/мл пенициллина, 100 мкг/мл стрептомицина и 2 мМ глутамина, при 37°С в атмосфере 5% СО2.
Панкреатин-EDTA использовали для нормального переваривания и отхождения кала два раза в неделю. Когда насыщение клеток составляло 80%-90%, клетки собирали, подсчитывали и высевали. Клетки LoVo инокулировали подкожно в правую часть спины каждой голой мыши в количестве 0,1 мл (10×106), и групповое введение начинали, когда средний объем опухоли достигал 173 мм3.
3. Приготовление тестируемого вещества и дозировка
Взвешивали 25,51 мг соединения формулы (1) и растворяли в 0,500 мл DMSO. Добавляли 2,000 мл пропиленгликоля и 2,500 мл деионизированной воды, перемешивали на вортексе, хорошо смешивали и доводили до рН=6,0 с получением прозрачного раствора.
Дозировка: 25 мг/кг всех тестируемых соединений вводили через зонд дважды в сутки с интервалом 8 ч в течение суток.
4. Измерение опухоли и экспериментальные индикаторы
Диаметр опухоли измеряли штангенциркулем два раза в неделю. Формула для расчета объема опухоли: V=0,5а×b2, где а и b представляют собой большой диаметр и короткий диаметр опухоли, соответственно.
Противоопухолевую эффективность соединения оценивали по скорости ингибирования роста опухоли TGI (%) или относительной скорости пролиферации опухоли Т/С (%).
Относительная скорость пролиферации опухоли Т/С (%)=TRTV/CRTV×100% (TRTV: среднее значение RTV в лечебной группе; CRTV: среднее значение RTV в группе отрицательного контроля). В соответствии с результатами измерения опухоли рассчитывали относительный объем опухоли (RTV), и формула для расчета представляла собой RTV=Vt/V0, где V0 представляет собой объем опухоли, измеренный на момент группового введения (т.е. DO), Vt представляет собой объем опухоли при измерении в определенный момент времени, и TRTV и CRTV используют данные в один и тот же день.
TGI (%) представляет скорость ингибирования роста опухоли. TGI (%)=[1-(средний объем опухоли в конце лечения в определенной лечебной группе - средний объем опухоли в начале лечения в лечебной группе) / (средний объем опухоли в конце лечения в контрольной группе, получавшей растворитель - средний объем опухоли в начале лечения в контрольной группе, получавшей растворитель)]×100%.
После завершения эксперимента определяли массу опухоли и рассчитывали процент массы Т/С, где масса Т и масса С представляют массу опухоли в группе введения и контрольной группе, получавшей носитель, соответственно.
5. Результаты теста
В этом тесте оценивали эффективность соединения в модели ксенотрансплантата опухоли колоректального рака человека, при этом в качестве эталона использовали контрольную группу, получавшую растворитель. На 17-й день введения группа, получавшая соединение формулы (1) (25 мг/кг), имела Т/С и TGI 27,8% и 90,7%, соответственно, по сравнению с контрольной группой, получавшей носитель.
6. Заключение
В этом эксперименте рассматриваемое соединение формулы (1) демонстрирует ингибирующий эффект на рост клеток LoVo колоректального рака человека у мышей, несущих подкожный ксенотрансплант опухоли.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКАЯ ФОРМА МИМЕТИКА SMAC, ПРИМЕНЯЕМОГО В КАЧЕСТВЕ ИНГИБИТОРА IAP, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2020 |
|
RU2819398C2 |
| СПОСОБ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ ИНГИБИТОРА ПОВЕРХНОСТНОГО АНТИГЕНА ВИРУСА ГЕПАТИТА В | 2020 |
|
RU2823673C1 |
| ИНГИБИТОР ATR И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2799047C2 |
| АНАЛОГ ПИРИДО[1,2-A]ПИРИМИДОНА, ЕГО КРИСТАЛЛИЧЕСКАЯ ФОРМА, ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2016 |
|
RU2753696C2 |
| БОРАТНОЕ ПРОИЗВОДНОЕ АЗЕТИДИНА | 2019 |
|
RU2802287C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА И СОЛЕВАЯ ФОРМА ИНГИБИТОРА TGF-βRI И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2018 |
|
RU2750702C1 |
| СОЕДИНЕНИЕ ИНГИБИТОРА BRD4 В ТВЕРДОЙ ФОРМЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2793346C1 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ATR КИНАЗЫ | 2014 |
|
RU2687276C2 |
| ИНДАЗОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ FGFR, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2719428C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРОВ ATR КИНАЗЫ (ВАРИАНТЫ) | 2014 |
|
RU2720408C2 |
Изобретение относится к кристаллической форме B соединения формулы (I), где кристаллическая форма В имеет картину порошковой рентгеновской дифракции, имеющую характерные дифракционные пики при следующих углах 2θ: 8.45±0.20°, 10.87±0.20°, 14.83±0.20°, 15.54±0.20° и 20.56±0.20°. Способ получения кристаллической формы В соединения формулы (I) по изобретению осуществляют путем (1) добавления соединения формулы (I) в спиртовой растворитель, (2) добавления воды, (3) перемешивания в течение 15-20 часов, (4) проведения перекристаллизации при комнатной температуре с получением кристаллической формы В, где спиртовой растворитель представляет собой метанол; объемное соотношение спиртового растворителя и воды составляет 1:1-1:4, диапазон концентрации соединения формулы (I) выбирают из группы, состоящей из 25-50 мг/мл. Также изобретение относится к применению кристаллической формы В соединения формулы (I) по изобретению для изготовления лекарственного средства для лечения заболевания, связанного с ATR. Технический результат - кристаллическая форма В соединения формулы I, обладающая стабильностью, меньшей подверженностью к воздействию света, тепла и влаги, хорошей эффективностью лекарственного средства in vivo и подходящая для применения в качестве лекарственного средства. 3 н. и 10 з.п. ф-лы, 4 ил., 14 табл., 13 пр.
 (I)
(I)
1. Кристаллическая форма B соединения формулы (I), где
кристаллическая форма В имеет картину порошковой рентгеновской дифракции, имеющую характерные дифракционные пики при следующих углах 2θ: 8.45±0.20°, 10.87±0.20°, 14.83±0.20°, 15.54±0.20° и 20.56±0.20°
 (I).
(I).
2. Кристаллическая форма В по п. 1, где
кристаллическая форма В имеет картину порошковой рентгеновской дифракции, имеющую характерные дифракционные пики при следующих углах 2θ: 8.45±0.20°, 10.87±0.20°, 14.83±0.20°, 15.54±0.20°, 17.33±0.20°, 20.56±0.20°, 22.00±0.20° и 22.63±0.20°.
3. Кристаллическая форма В по п. 2, где
кристаллическая форма B имеет картину порошковой рентгеновской дифракции, имеющую характерные дифракционные пики при следующих углах 2θ: 8.45±0.20°, 10.87±0.20°, 14.83±0.20°, 15.54±0.20°, 17.33±0.20°, 20.08±0.20°, 20.56±0.20°, 22.00±0.20°, 22.63±0.20° и 25.26±0.20°.
4. Кристаллическая форма В по п. 3, где
кристаллическая форма B имеет картину порошковой рентгеновской дифракции, имеющую характерные дифракционные пики при следующих углах 2θ: 8.45°, 9.20°, 10.87°, 12.57°, 14.14°, 14.53°, 14.83°, 15.54°, 16.80°, 17.33°, 18.43°, 19.84°, 20.08°, 20.56°, 21.39°, 22.00°, 22.44°, 22.63°, 23.26°, 25.26°, 25.85° и 26.98°.
5. Кристаллическая форма В по п. 4, где
кристаллическая форма B имеет картину XRPD, показанную на фиг. 2.
6. Кристаллическая форма В по любому из пп. 1-5, где
кристаллическая форма В имеет кривую дифференциальной сканирующей калориметрии (DSC), имеющую одну точку начала эндотермического пика при 174,3±3°С.
7. Кристаллическая форма В по п. 6, где
кристаллическая форма B имеет картину DSC, показанную на фиг. 3.
8. Кристаллическая форма В по любому из пп. 1-5, где
кристаллическая форма В имеет кривую термогравиметрического анализа (TGA), где потеря массы при 150°С±3°С составляет 1,49%.
9. Кристаллическая форма В по п. 8, где
кристаллическая форма B имеет картину TGA, показанную на фиг. 4.
10. Способ получения кристаллической формы В соединения формулы (I) по любому из пп. 1-9, включающий:
1) добавление соединения формулы (I) в спиртовой растворитель;
2) добавление воды;
3) перемешивание в течение 15-20 часов;
4) проведение перекристаллизации при комнатной температуре с получением кристаллической формы В;
где спиртовой растворитель представляет собой метанол; объемное соотношение спиртового растворителя и воды составляет 1:1-1:4; диапазон концентрации соединения формулы (I) выбирают из группы, состоящей из 25-50 мг/мл.
11. Применение кристаллической формы В по любому из пп. 1-9 для изготовления лекарственного средства для лечения заболевания, связанного с ATR.
12. Применение по п. 11, где
лекарственное средство предназначено для лечения солидной опухоли или опухоли кроветворной системы.
13. Применение по п. 11, где
лекарственное средство предназначено для лечения колоректального рака, рака желудка, рака пищевода, первичной перитонеальной карциномы, адренокортикальной карциномы, почечной светлоклеточной карциномы, рака предстательной железы, уротелиальной карциномы мочевого пузыря, рака яичников, рака молочной железы, карциномы эндометрия, карциномы фаллопиевой трубы, немелкоклеточного рака легкого или мелкоклеточного рака легкого.
| WO 2019050889 A1, 14.03.2019 | |||
| CN 103068391 A, 24.04.2013 | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| WO 2006005915 A1, 19.01.2006 | |||
| WO 2019036641 A1, 21.02.2019 | |||
| Приспособление для стягивания тормозных тяг | 1930 |
|
SU22087A1 |
| MINO R.CAIRA, Crystalline polymorphism of organic compounds, TOPICS IN CURRENT CHEMISTRY, Springer Verlag Berlin Heidelberg, 1998, V.198, p.163-208 | |||
| Вакуумный генератор с ртутным катодом | 1928 |
|
SU14706A1 |

