Ссылка на родственные заявки
Согласно настоящей заявке испрашивается приоритет и преимущество заявки на выдачу патента Китая № 201910389970.8, поданной в Национальное управление интеллектуальной собственности КНР 10 мая 2019 года, раскрытие которой во всей своей полноте включено в настоящий документ посредством ссылки.
Область техники, к которой относится изобретение
Настоящая заявка относится к области фармацевтической химии, и, в частности, к кристаллической форме миметика SMAC в качестве ингибитора IAP, и к способу ее получения, а также к применению кристаллической формы при получении лекарственного препарата для лечения формы рака, при которой можно получить благоприятный клинический результат от ингибирования cIAP1.
Уровень техники
Запрограммированная гибель клеток играет ключевую роль в регулировании количества клеток и устранении подвергнутых стрессу или поврежденных клеток из нормальных тканей. Действительно, механизмы сигнальной сети апоптоза, присущие большинству клеток, создают главный барьер против развития и прогрессирования рака у человека. Однако у раковых клеток есть общая черта, заключающаяся в том, что они не могут инициировать программу апоптоза и лишены надлежащего процесса апоптоза из-за утраты нормальных механизмов апоптоза. Большинство современных средств противораковой терапии, в том числе химиотерапия, лучевая терапия и иммунотерапия, оказывают свое действие непрямым путем, индуцируя апоптоз у раковых клеток. Поэтому безуспешная инициации программ апоптоза из-за дефектов в нормальных механизмах апоптоза у раковых клеток обычно связана с повышенной устойчивостью к индуцируемому химиотерапией, лучевой терапией или иммунотерапией апоптозу. Следовательно, целенаправленное воздействие на ключевые отрицательные регуляторы, которые играют важную роль в прямой супрессии апоптоза раковых клеток, было бы очень многообещающей стратегией терапии для разработки новых противораковых лекарственных средств.
К данному моменту были выявлены два важных класса отрицательных регуляторов апоптоза. Первым классом регуляторных факторов являются белки семейства Bcl-2, например, два высокоактивных антиапоптозных белка под названием Bcl-2 и Bcl-XL.
Вторым классом отрицательных регуляторов клеточного апоптоза являются ингибиторы белков апоптоза (IAP). IAP были впервые обнаружены у бакуловирусов благодаря их функциональной способности замещать белок P35. К таким белкам относятся следующие белки: XIAP, cIAP1, cIAP2, ML-IAP, ILP-2, NAIP, аполлон и сурвивин. В частности, сцепленный с Х-хромосомой ингибитор апоптоза (XIAP) супрессирует апоптоз путем прямого ингибирования каспазы-3, каспазы-7 и каспазы-9. CIAP преимущественно ингибируют апоптоз путем блокировки каскадов с участием рецепторов смерти. При разрушении молекул cIAP субстрат NIK (NF-κB-индуцирующая киназа) ускользает от разрушения и, таким образом, накапливается и неклассическим путем активирует NF-κB. Активированная NF-κB способствует секреции TNFα, который связывается с TNF-R1 (рецептором 1 TNF), инициируя каскад с участием рецепторов смерти. В результате разрушения молекул cIAP происходит повышенная секреция RIPK1 (взаимодействующей с рецептором протеинкиназы 1) и образуется проапоптотический комплекс RIPK1-FADD-каспаза-8 с FADD (связанный с FAS домен смерти) и каспазой-8, который затем активирует каспазу-3 и инициирует апоптоз.
Сверхэкспрессия cIAP1 и cIAP2 из-за частой амплификации хромосомы на участке 11q21-q23, который охватывает два гена, была замечена при различных злокачественных заболеваниях, в том числе при нейробластоме, почечно-клеточном раке, колоректальном раке и раке желудка.
Краткое описание настоящего изобретения
В одном аспекте настоящая заявка относится к кристаллической форме соединения с формулой (I),
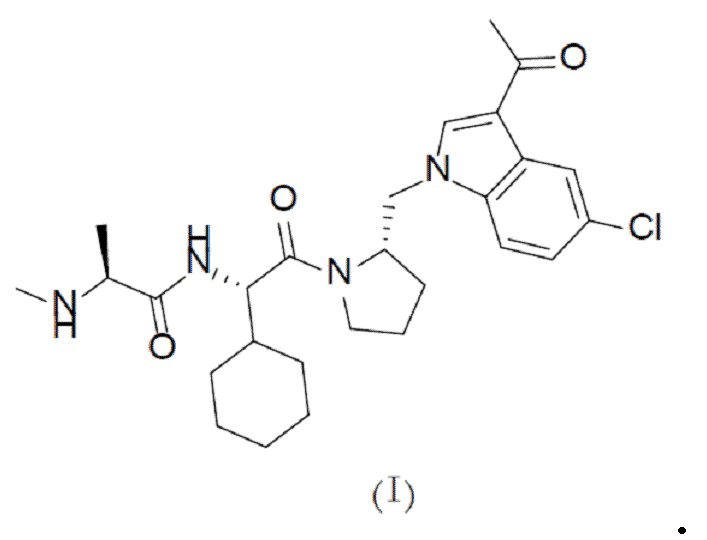
В другом аспекте настоящая заявка относится к кристаллической композиции с соединением с формулой (I), содержащей 50% или более, предпочтительно 75% или более, более предпочтительно 90% или более и наиболее предпочтительно 95% или более по массе кристаллической формы соединения с формулой (I).
В другом аспекте настоящая заявка относится к фармацевтической композиции, содержащей терапевтически эффективное количество кристаллической формы соединения с формулой (I) или кристаллической композиции с соединением с формулой (I); при этом фармацевтическая композиция может дополнительно содержать по меньшей мере один фармацевтически приемлемый носитель или другое вспомогательное вещество.
В другом аспекте настоящая заявка относится к применению кристаллической формы соединения с формулой (I), кристаллической композиции с соединением с формулой (I) или фармацевтической композиции, которые описаны выше, при получении лекарственного препарата для лечения формы рака, при которой можно получить благоприятный клинический результат от ингибирования cIAP1.
В другом аспекте настоящая заявка относится к применению кристаллической формы соединения с формулой (I), кристаллической композиции с соединением с формулой (I) или фармацевтической композиции, которые описаны выше, при лечении формы рака, при которой можно получить благоприятный клинический результат от ингибирования cIAP1 у млекопитающего.
В другом аспекте настоящая заявка относится к способу лечения формы рака, при которой можно получить благоприятный клинический результат от ингибирования cIAP1 у млекопитающего, предусматривающему введение нуждающемуся в том млекопитающему терапевтически эффективного количества кристаллической формы соединения с формулой (I), кристаллической композиции с соединением с формулой (I) или фармацевтической композиции, которые описаны выше.
В другом аспекте настоящая заявка относится к кристаллической форме соединения с формулой (I), кристаллической композиции с соединением с формулой (I) или фармацевтической композиции, которые описаны выше, для применения при лечении формы рака, при которой можно получить благоприятный клинический результат от ингибирования cIAP1 у млекопитающего.
Краткое описание изобретения
Один аспект настоящей заявки заключается в получении кристаллической формы соединения с формулой (I),
Описанная в настоящем документе кристаллическая форма может быть представлена в форме отличного от сольвата соединения или сольвата, например гидрата.
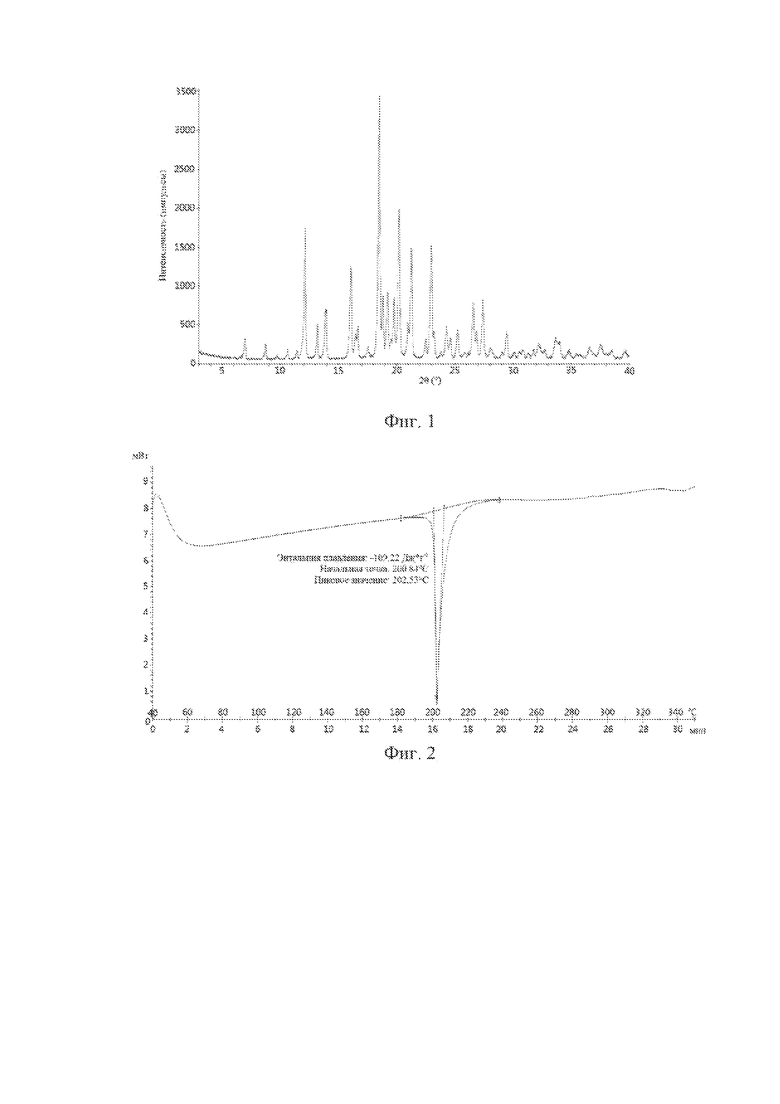
В одном варианте осуществления по настоящей заявке кристаллическая форма соединения с формулой (I) представляет собой кристаллическую форму A, характеризующуюся дифракционными пиками при следующих значениях 2θ на порошковой рентгеновской дифрактограмме, полученной с помощью Cu-Kα-излучения: 12,1°±0,200°, 16,1°±0,200°, 18,5°±0,200°, 20,2°±0,200°, 21,3°±0,200° и 23,0°±0,200°.
В одном варианте осуществления по настоящей заявке кристаллическая форма A характеризуется дифракционными пиками при следующих значениях 2θ на порошковой рентгеновской дифрактограмме, полученной с помощью Cu-Kα-излучения: 12,1°±0,200°, 16,1°±0,200°, 18,5°±0,200°, 18,8°±0,200°, 19,2°±0,200°, 19,8°±0,200°, 20,2°±0,200°, 21,3°±0,200°, 23,0°±0,200°, 26,6°±0,200° и 27,4°±0,200°.
В одном варианте осуществления по настоящей заявке кристаллическая форма A характеризуется дифракционными пиками при следующих значениях 2θ на порошковой рентгеновской дифрактограмме, полученной с помощью Cu-Kα-излучения: 7,0°±0,200°, 8,7°±0,200°, 12,1°±0,200°, 13,2°±0,200°, 13,9°±0,200°, 16,1°±0,200°, 16,7°±0,200°, 18,5°±0,200°, 18,8°±0,200°, 19,2°±0,200°, 19,8°±0,200°, 20,2°±0,200°, 21,0°±0,200°, 21,3°±0,200°, 23,0°±0,200°, 24,3°±0,200°, 25,3°±0,200°, 26,6°±0,200° и 27,4°±0,200°.
В одном варианте осуществления по настоящей заявке кристаллическая форма A характеризуется дифракционными пиками при следующих значениях 2θ на порошковой рентгеновской дифрактограмме, полученной с помощью Cu-Kα-излучения: 7,0°±0,200°, 8,7°±0,200°, 12,1°±0,200°, 13,2°±0,200°, 13,9°±0,200°, 16,1°±0,200°, 16,5°±0,200°, 16,7°±0,200°, 18,5°±0,200°, 18,8°±0,200°, 19,2°±0,200°, 19,8°±0,200°, 20,2°±0,200°, 21,0°±0,200°, 21,3°±0,200°, 23,0°±0,200°, 23,2°±0,200°, 24,3°±0,200°, 25,3°±0,200°, 26,6°±0,200°, 26,9°±0,200°, 27,4°±0,200° и 29,4°±0,200°.
В одном варианте осуществления по настоящей заявке кристаллическая форма A характеризуется дифракционными пиками при следующих значениях 2θ на порошковой рентгеновской дифрактограмме, полученной с помощью Cu-Kα-излучения: 7,0°±0,200°, 8,7°±0,200°, 9,7°±0,200°, 10,6°±0,200°, 11,4°±0,200°, 12,1°±0,200°, 13,2°±0,200°, 13,9°±0,200°, 16,1°±0,200°, 16,5°±0,200°, 16,7°±0,200°, 17,5°±0,200°, 18,5°±0,200°, 18,8°±0,200°, 19,2°±0,200°, 19,5°±0,200°, 19,8°±0,200°, 20,2°±0,200°, 21,0°±0,200°, 21,3°±0,200°, 22,5°±0,200°, 23,0°±0,200°, 23,2°±0,200°, 23,8°±0,200°, 24,3°±0,200°, 24,6°±0,200°, 25,3°±0,200°, 25,8°±0,200°, 26,6°±0,200°, 26,9°±0,200°, 27,4°±0,200°, 28,1°±0,200°, 29,1°±0,200°, 29,4°±0,200°, 30,1°±0,200°, 30,6°±0,200°, 30,8°±0,200°, 31,3°±0,200°, 31,8°±0,200°, 32,2°±0,200°, 32,8°±0,200°, 33,7°±0,200°, 34,0°±0,200°, 34,8°±0,200°, 35,5°±0,200°, 36,6°±0,200°, 37,6°±0,200°, 38,6°±0,200° и 39,6°±0,200°.
В одном варианте осуществления по настоящей заявке в таблице 1 представлены положения и значения интенсивности дифракционных пиков на порошковой рентгеновской дифрактограмме, полученной с помощью Cu-Kα-излучения,, для кристаллической формы А соединения с формулой (I), которая раскрыта в настоящем документе:
Таблица 1. Данные по характеристикам порошковой рентгеновской дифрактограммы у кристаллической формы A
В одном варианте осуществления по настоящей заявке кристаллическая форма А соединения с формулой (I) характеризуется порошковой рентгеновской дифрактограммой, полученной с помощью Cu-Kα-излучения, которая представлена на фиг. 1.
В одном варианте осуществления по настоящей заявке кристаллическая форма A соединения с формулой (I) характеризуется на диаграмме дифференциальной сканирующей калориметрии (DSC) эндотермическим пиком на уровне 202,5°C.
В одном варианте осуществления по настоящей заявке кристаллическая форма A соединения с формулой (I) характеризуется на диаграмме дифференциальной сканирующей калориметрии (DSC) начальной точкой эндотермического пика на уровне 200,8°C.
В одном варианте осуществления по настоящей заявке кристаллическая форма А соединения с формулой (I) характеризуется диаграммой дифференциальной сканирующей калориметрии (DSC), которая представлена на фиг. 2.
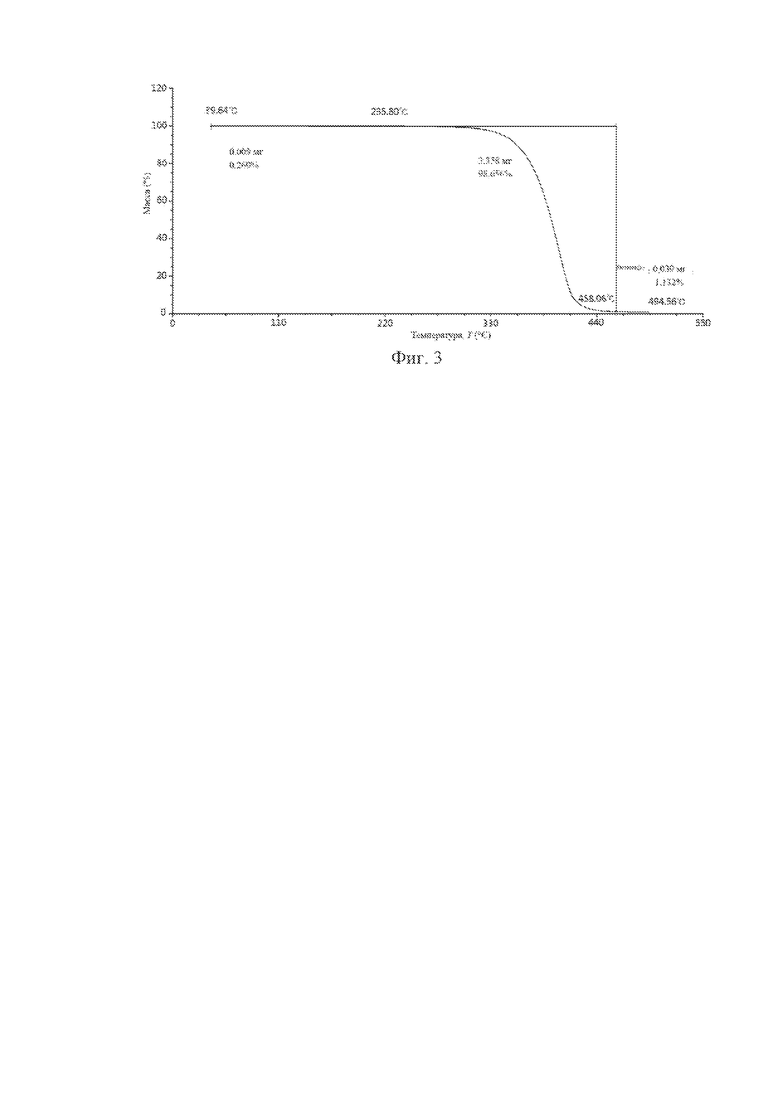
В одном варианте осуществления по настоящей заявке кристаллическая форма А соединения с формулой (I) характеризуется графиком термогравиметрического анализа (TGA), который представлен на фиг. 3.
В другом аспекте настоящая заявка относится к способу получения кристаллической формы A соединения с формулой (I), предусматривающему следующее:
(а) растворение соединения с формулой (I) в растворителе с получением суспензии или раствора;
(b) нагревание суспензии или раствора в колбе с обратным холодильником с получением осветленного раствора; и
(c) фильтрование осветленного раствора перед охлаждением для получения осадка, фильтрование и сушку с получением кристаллической формы A соединения с формулой (I).
В одном варианте осуществления по настоящей заявке растворитель со стадии (а) выбирают из группы, состоящей из метанола, этанола, изопропанола, этилацетата, ацетонитрила, ацетона, тетрагидрофурана, воды и смешанного растворителя, состоящего из воды и любого из перечисленных выше; предпочтительно метанола, этанола и изопропанола и смешанного из них растворителя с водой; более предпочтительно этанола.
В другом аспекте настоящая заявка дополнительно относится к способу получения кристаллической формы A соединения с формулой (I), предусматривающему следующее:
(d) растворение соединения с формулой (I) в растворителе и перемешивание с обработкой ультразвуком с получением суспензии или раствора; и
(e) нагревание и перемешивание суспензии или раствора на термостатическом шейкере, центрифугирование и сушку с получением кристаллической формы А соединения с формулой (I).
В одном варианте осуществления по настоящей заявке растворитель со стадии (d) выбирают из группы, состоящей из метанола, этанола, изопропанола, этилацетата, ацетонитрила, ацетона, тетрагидрофурана, воды и смешанного растворителя, состоящего из воды и любого из перечисленных выше; предпочтительно метанола, этанола, этилацетата, ацетонитрила, ацетона, тетрагидрофурана, воды, смешанного растворителя из воды и метанола, смешанного растворителя из воды и этанола и смешанного растворителя из воды и ацетона. Объемное соотношение воды к метанолу, воды к этанолу или воды к ацетону выбирают из 1:1–1:5, предпочтительно 1:1–1:3.
В одном варианте осуществления по настоящей заявке мольно-объемное соотношение соединения с формулой (I) к растворителю на стадии (а) составляет 1 ммоль : 1–10 мл, предпочтительно 1 ммоль : 2–6 мл, более предпочтительно 1 ммоль : 2–4 мл.
В одном варианте осуществления по настоящей заявке мольно-объемное соотношение соединения с формулой (I) к растворителю на стадии (d) составляет 1 ммоль : 1-15 мл, предпочтительно 1 ммоль : 4-10 мл, более предпочтительно 1 ммоль : 8-10 мл.
В одном варианте осуществления по настоящей заявке температура кипячения в колбе с обратным холодильником на стадии (b) составляет 60–120°C, предпочтительно 80–90°C.
В одном варианте осуществления по настоящей заявке температура перемешивания на стадии (e) составляет 30-50°C, предпочтительно 40-50°C.
В другом аспекте настоящая заявка относится к кристаллической композиции с соединением с формулой (I), содержащей 50% или более, предпочтительно 75% или более, более предпочтительно 90% или более и наиболее предпочтительно 95% или более по массе кристаллической формы соединения с формулой (I). Кристаллическая композиция также может содержать небольшие количества других кристаллических или аморфных форм соединения с формулой (I).
В другом аспекте настоящая заявка относится к фармацевтической композиции, содержащей терапевтически эффективное количество кристаллической формы соединения с формулой (I) или кристаллической композиции с соединением с формулой (I); при этом фармацевтическая композиция может дополнительно содержать по меньшей мере один фармацевтически приемлемый носитель или другое вспомогательное вещество.
В другом аспекте настоящая заявка относится к применению кристаллической формы соединения с формулой (I), кристаллической композиции с соединением с формулой (I) или фармацевтической композиции, которые описаны выше, при получении лекарственного препарата для лечения формы рака, при которой можно получить благоприятный клинический результат от ингибирования cIAP1.
В другом аспекте настоящая заявка относится к применению кристаллической формы соединения с формулой (I), кристаллической композиции с соединением с формулой (I) или фармацевтической композиции, которые описаны выше, при лечении формы рака, при которой можно получить благоприятный клинический результат от ингибирования cIAP1 у млекопитающего.
В другом аспекте настоящая заявка относится к способу лечения формы рака, при которой можно получить благоприятный клинический результат от ингибирования cIAP1 у млекопитающего, предусматривающему введение нуждающемуся в том млекопитающему терапевтически эффективного количества кристаллической формы соединения с формулой (I), кристаллической композиции с соединением с формулой (I) или фармацевтической композиции, которые описаны выше.
В другом аспекте настоящая заявка относится к кристаллической форме соединения с формулой (I), кристаллической композиции с соединением с формулой (I) или фармацевтической композиции, которые описаны выше, для применения при лечении формы рака, при которой можно получить благоприятный клинический результат от ингибирования cIAP1 у млекопитающего.
В одном варианте осуществления по настоящей заявке млекопитающим является человек.
В одном варианте осуществления по настоящей заявке форма рака, при которой можно получить благоприятный клинический результат от ингибирования cIAP1, выбрана из рака молочной железы.
В одном варианте осуществления по настоящей заявке форма рака, при которой можно получить благоприятный клинический результат от ингибирования cIAP1, выбрана из трижды негативного рака молочной железы.
Кристаллическая форма соединения с формулой (I), которая раскрыта в настоящем документе, характеризуется превосходными эффектами по меньшей мере в одном аспекте биологической активности, безопасности, биодоступности и др., и кристаллическая форма А соединения с формулой (I) характеризуется высокой стабильностью, низкой гигроскопичностью и хорошей ингибирующей активностью в отношении cIAP1, таким образом обладая хорошим потенциалом для разработки лекарственных средств. Кристаллическая форма А соединения с формулой (I) также характеризуется хорошей фармакокинетикой, которая может быть подтверждена доклиническими (например, на крысах SD и собаках породы бигль) и клиническими испытаниями, и подходит для применения в качестве лекарственного препарата.
В настоящей заявке фармацевтическая композиция может быть составлена в виде определенной лекарственной формы, а путь введения предпочтительно является пероральным, парентеральным (в том числе подкожным, внутримышечным и внутривенным), ректальным и т. д. Например, к подходящим лекарственным формам для перорального введения относятся таблетки, капсулы, гранулы, порошковые формы, пилюли, порошки, пастилки, сиропы или суспензии; к подходящим лекарственным формам для парентерального введения относятся водные или неводные растворы или эмульсии для инъекций; к подходящим лекарственным формам для ректального введения относятся суппозитории с гидрофильными или гидрофобными носителями.
В настоящей заявке порошковую рентгеновскую дифрактограмму образца снимают в следующих условиях: прибор: рентгеновский дифрактометр Bruker D8 ADVANCE; анод: Cu:Kα; длина волны λ = 1,54056 Ǻ; диапазон 2θ: 3-40°; щель рассеивания: 0,60 мм; щель детектора: 10,50 мм; противорассеивающая щель: 7,10 мм; размер шага: 0,02°; время шага: 0,12 с; скорость вращения образца: 15 об./мин; напряжение и ток в трубке с медным анодом: 40 кВ, 40 мА.
В настоящей заявке DSC-диаграмму снимают в следующих условиях: прибор: дифференциальный сканирующий калориметр TA Q2000; температурный диапазон: 30–300°C; скорость нагрева: 10°C/мин.
В настоящей заявке TGA-термогравиметрический анализ проводят в следующих условиях: прибор: термогравиметрический анализатор TA Q5000; температурный диапазон: 25–300°C; скорость нагрева: 10°C/мин.
Следует отметить, что на порошковой рентгеновской дифрактограмме положение и относительная интенсивность пика могут варьировать в зависимости от измерительных приборов, способов/условий измерения и ряда других факторов. В случае любой конкретной кристаллической формы положение пика может характеризоваться погрешностью, как и результат измерения 2θ может характеризоваться погрешностью ±0,2°. Поэтому данную погрешность необходимо учитывать при изучении каждой кристаллической формы, и кристаллические формы в пределах данной погрешности входят в объем настоящей заявки.
Следует отметить, что для одной и той же кристаллической формы положение эндотермического пика на DSC-диаграмме может варьировать в зависимости от измерительных приборов, способов/условий измерения и ряда других факторов. В случае любой конкретной кристаллической формы начальная точка эндотермического пика может иметь погрешность ±5°C. Поэтому данную погрешность необходимо учитывать при изучении каждой кристаллической формы, и кристаллические формы в пределах данной погрешности входят в объем настоящей заявки.
Следует отметить, что для одной и той же кристаллической формы положение температуры потери массы на TGA-диаграмме может варьировать в зависимости от измерительных приборов, способов/условий измерения и ряда других факторов. В случае любой конкретной кристаллической формы температура потери массы может иметь погрешность ±5°C. Поэтому данную погрешность необходимо учитывать при изучении каждой кристаллической формы, и кристаллические формы в пределах данной погрешности входят в объем настоящей заявки.
Определения и описание
В контексте настоящего описания и формулы изобретения из настоящей заявки представленные далее термины, если не указано иное, имеют указанные далее значения.
Термин «млекопитающее» охватывает человека, домашних животных, таких как лабораторные млекопитающие и домашние питомцы (например, кошек, собак, свиней, коров, овец, коз, лошадей, кроликов), а также неодомашненных млекопитающих, таких как дикие млекопитающие.
Термин «фармацевтическая композиция» относится к составу с раскрываемым в настоящем документе соединением с общепризнанным в настоящей области техники наполнителем для доставки биологически активного соединения млекопитающему, такому как человек. К наполнителю относятся все фармацевтически приемлемые по своему назначению носители. Фармацевтическая композиция облегчает введение соединения в организм.
Термин «терапевтически эффективное количество» относится к количеству лекарственного средства или лекарственного препарата, которое является достаточным для достижения требуемого эффекта, но не является токсичным. Результат определения эффективного количества варьирует от человека к человеку. Он зависит от возраста и общего состояния пациента, а также от конкретного применяемого действующего вещества. В одном случае подходящее эффективное количество может быть определено специалистом в настоящей области техники по результатам стандартных тестов.
Термин «фармацевтически приемлемые носители» относится к веществам, которые вводят вместе с активным ингредиентом, которые не оказывают значительного раздражающего действия на организм и не ухудшают биологическую активность и свойства действующего соединения. Дополнительную информацию о носителях можно получить из публикации Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005), содержание которой включено в настоящий документ посредством ссылки.
В контексте настоящего документа термин «комнатная температура» относится к 20-30°C.
Краткое описание чертежей
На фиг. 1 представлена рентгеновская порошковая дифрактограмма (XRPD) кристаллической формы A соединения с формулой (I) из примера 1.
На фиг. 2 представлена диаграмма результатов дифференциальной сканирующей калориметрии (DSC) кристаллической формы A соединения с формулой (I) из примера 1.
На фиг. 3 представлена диаграмма результатов термогравиметрического анализа (TGA) кристаллической формы A соединения с формулой (I) из примера 1.
Подробное описание изобретения
Последующие конкретные примеры представлены для того, чтобы специалисты в настоящей области техники смогли четче понять и реализовать на практике настоящее изобретение. Данные конкретные примеры не следует рассматривать как ограничение объема настоящего изобретения, а лишь как иллюстративное описание и наглядное представление сути настоящей заявки.
Раскрываемые в настоящем документе промежуточные соединения можно получить рядом способов синтеза, которые хорошо известны специалистам в настоящей области, включая конкретные варианты осуществления, перечисленные ниже, варианты осуществления, образованные их комбинациями с другими способами химического синтеза, и их эквиваленты, известные специалистам в настоящей области техники. Предпочтительные варианты осуществления включают без ограничения примеры, которые раскрыты в настоящем документе.
Химические реакции из раскрываемых в настоящем документе вариантов осуществления проводят в подходящем растворителе, который должен быть пригодным для химических превращений по настоящей заявке и требуемых для этого реагентов и материалов. Для того, чтобы получить раскрываемые в настоящем документе соединения, специалистам в настоящей области техники иногда необходимо будет модифицировать или подобрать процедуру синтеза или схему реакции на основе существующих вариантов осуществления.
Все растворители, применяемые в настоящей заявке, доступны в продаже и могут быть использованы без дополнительной очистки.
Пример 1: получение кристаллической формы A соединения с формулой (I)
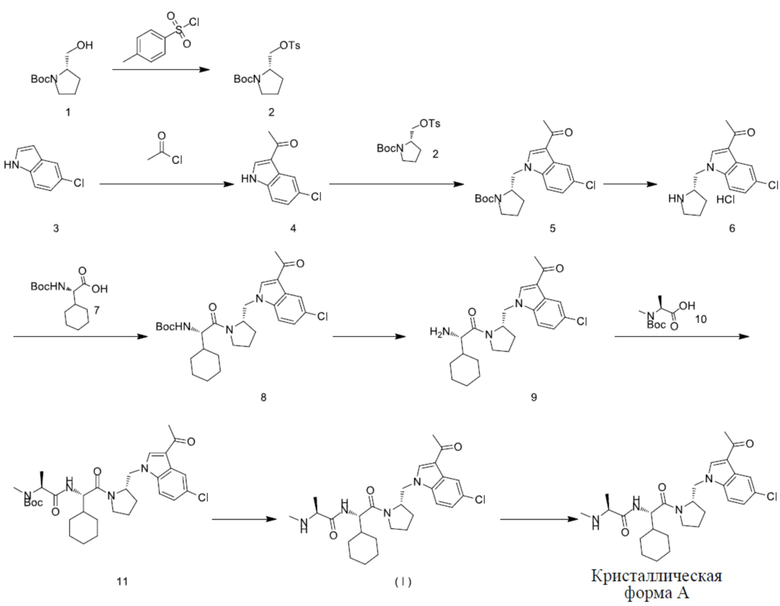
Стадия 1
В реакционный сосуд вносили дихлорметан (16,0 л) и охлаждали его до 0°C перед добавлением Соединения 1 (2,0 кг, 9,94 моля, 1,0 экв.). Перемешивали смесь и вносили триэтиламин (4,42 л, 29,81 моля, 3,0 экв.) и 4-диметиламинопиридин (61,00 г, 496,86 ммоля, 0,05 экв.). Перед внесением в реакционный сосуд дихлорметана (4 л) порциями вносили н-толуолсульфонилхлорид (2,18 кг, 11,43 моля, 1,15 экв.) с внутренней температурой, контролируемой на уровне 0-10°C. Смесь перемешивали 16 ч при 20°C. В реакционный сосуд вносили воду (20 л), смесь перемешивали в течение 5 минут, давали отстояться и разделяли. Водную фазу удаляли, а органическую фазу промывали 10% водным раствором лимонной кислоты (15,0 л × 2) и солевым раствором (15,0 л × 2), сушили над безводным сульфатом натрия (2,0 кг) в течение 1 ч и профильтровывали. Фильтрат концентрировали на роторном испарителе с получением соединения 2. 1H ЯМР (400 МГц, DMSO-d6) δ 7,78 (br d, J = 8,2 Гц, 2H), 7,47 (br d, J = 7,9 Гц, 2H), 4,13-3,93 (m, 2H), 3,92-3,81 (m, 1H), 3,22-3,11 (m, 2H), 1,96-1,80 (m, 2H), 1,74-1,65 (m, 2H), 1,40 (s, 3H), 1,38-1,23 (m, 9H).
Стадия 2
В сухой реакционный сосуд вносили дихлорметан (1,30 л) и соединение 3 (1,28 кг, 8,49 моля, 1,0 экв.) и перемешивали до растворения. Содержимое системы охлаждали до внутренней температуры 0°C и в течение 0,5 часа в атмосфере азота по каплям добавляли ацетилхлорид (606,0 мл, 8,49 моля, 1,0 экв.). Затем в реакционный сосуд в течение 3 часов по каплям добавляли 1 М диэтилалюминийхлорид в н-гексане (8,50 л, 8,49 моля, 1,0 экв.) при внутренней температуре 0–5°C. После завершения добавления систему инкубировали в течение 1 ч. Реакционный раствор медленно порциями выливали в ледяную воду (20,0 л) при перемешивании, при этом в осадок выпадало большое количество красного твердого вещества. Погашенную смесь переносили в роторный испаритель и концентрировали при 30°C до удаления дихлорметана и н-гексана. К остатку добавляли 2-метилтетрагидрофуран (20,0 л) и смесь переносили в сепаратор жидкости, перемешивали в течение 15 минут и разделяли для удаления водной фазы. Органическую фазу промывали солевым раствором (20 л × 2), сушили над безводным сульфатом натрия (3,0 кг) и подвергали фильтрации. Осадок на фильтре промывали 2-метилтетрагидрофураном (2 л) и смыв подвергали фильтрации. Фильтрат переносили в роторный испаритель и концентрировали. Остаток вносили в 50-литровый реакционный сосуд, добавляли смесь этилацетата:н-гептана (об./об.) = 1:1 (10,0 л), перемешивали в течение 10 ч и подвергали фильтрации. Осадок на фильтре промывали смесью этилацетата:н-гептана (об./об.) = 1:1 (300 мл), высушивали и переносили в печь для сушки при пониженном давлении при температуре 35°C с получением соединения 4. 1H ЯМР (400 МГц, DMSO-d6) δ 12,10 (br s, 1H), 8,38 (d, J = 3,2 Гц, 1H), 8,16 (d, J = 2,2 Гц, 1H), 7,50 (d, J = 8,7 Гц, 1H), 7,23 (dd, J = 8,6, 2,1 Гц, 1H), 2,46 (s, 3H).
Стадия 3
В реакционный сосуд в атмосфере азота вносили N,N-диметилацетамид (6,0 л), после чего вносили соединение 2 (3,95 кг, 11,12 моль, 1,30 экв.) и соединение 4 (1,73 кг, 8,56 моль, 1,0 экв.) при температуре 25°C и в течение 10 минут порциями добавляли карбонат цезия (4,18 кг, 12,85 моль, 1,50 экв.). Стенку сосуда промывали N,N-диэтилацетамидом (1,0 л) и инкубировали при 80–85°C в течение 16 часов. Реакционный раствор охлаждали до 25°C, медленно добавляли воду (30 л), вносили этилацетат (20 л), перемешивали и переносили в сепаратор для отделения жидкости. Водную фазу экстрагировали этилацетатом (10,0 мл × 1). Органические фазы объединяли, промывали водой (15,0 л × 2) и солевым раствором (15,0 л × 2), сушили над безводным сульфатом натрия (3,0 кг) в течение 1 часа и подвергали фильтрации. Осадок на фильтре промывали этилацетатом (2,0 л), а фильтрат переносили в роторный испаритель для концентрирования с получением неочищенного продукта. Неочищенный продукт переносили в реакционный сосуд, добавляли смесь этилацетата:н-гептана (об./об.) = 1:2 (9,0 л), перемешивали в течение 2 часов и подвергали фильтрации. Осадок на фильтре очищали смесью этилацетата:н-гептана (об./об.) = 1:2 (500 мл), высушивали, переносили в печь и сушили при пониженном давлении и при температуре 35°C с получением соединения 5. 1H ЯМР (400 МГц, DMSO-d6) δ 8,43-8,28 (m, 1H), 8,17 (s, 1H), 7,74-7,52 (m, 1H), 7,27 (br s, 1H), 4,40-4,05 (m, 3H), 3,31-3,11 (m, 2H), 2,50-2,35 (m, 3H), 1,90-1,69 (m, 4H), 1,39 (br d, J = 6,7 Гц, 5H), 1,08 (br s, 4H).
Стадия 4
В сухой реакционный сосуд вносили этилацетат (16,5 л), после чего вносили соединение 5 (1,65 кг, 4,38 моля, 1,0 экв.). Содержимое системы перемешивали для растворения и охлаждали до внутренней температуры 0°C, по каплям в течение 1 часа добавляли свежеприготовленный хлористый водород в этилацетате (15,0 л, 4,0 моль/л). Реакционный сосуд охлаждали до комнатной температуры и инкубировали при 20–30°C в течение 16 часов. Реакционный раствор подвергали фильтрации, осадок на фильтре промывали этилацетатом (5 л × 2) и высушивали с получением неочищенного продукта. Неочищенный продукт суспендировали в этилацетате и перемешивали в течение 4 часов. Смесь подвергали фильтрации и сушили с получением соединения 6. 1H ЯМР (400 МГц, DMSO-d6) δ 10,00 (br s, 1H), 9,54 (br s, 1H), 8,76 (s, 1H), 8,16 (d, J = 1,8 Гц, 1H), 7,84 (d, J = 8,8 Гц, 1H), 7,30 (dd, J = 8,7, 1,8 Гц, 1H), 4,80-4,72 (m, 1H), 4,62 (br d, J = 5,4 Гц, 1H), 3,92 (br s, 1H), 3,27 (br d, J = 6,8 Гц, 1H), 3,18-3,00 (m, 1H), 2,44 (s, 3H), 2,17-1,63 (m, 4H).
Стадия 5
В реакционный сосуд последовательно вносили соединение 7 (2,13 кг, 8,30 моля, 1,05 экв.) и N,N-диметилформамид (12,50 л) и охлаждали до внутренней температуры 0-10°C, после чего добавляли гексафторфосфат бензотриазол-N,N,N,N-тетраметилурония (3,15 кг, 8,30 моль, 1,05 экв.) с получением белой суспензии. По каплям добавляли N,N-диизопропилэтиламин (3,06 кг, 23,71 моля, 3,0 экв.) с внутренней температурой, контролируемой на уровне 10°C или ниже, до тех пор, пока твердое вещество постепенно не растворялось с получением прозрачного раствора. После добавления внутреннюю температуру контролировали на уровне 5–10°C и содержимое системы перемешивали в течение 0,5 ч реакции. Порциями вносили соединение 6 (2,50 кг, 7,90 моля, 1,0 экв.) с внутренней температурой, контролируемой на уровне 5-10°C, во время чего наблюдали белый дым. После внесения температуру постепенно повышали до 25–30°C, после чего содержимое системы инкубировали в течение 1,5 ч. При перемешивании половину реакционного раствора медленно по каплям добавляли в реакционный сосуд, содержащий воду (30,0 л). После осаждения большого количества твердого вещества смесь перемешивали в течение 30 минут и подвергали фильтрации, а осадок на фильтре высушивали. Другую половину реакционного раствора обрабатывали тем же способом. Осадки на фильтре объединяли и сушили при 50°C под вакуумом до постоянной массы с получением соединения 8. 1H ЯМР (400 МГц, DMSO-d6) δ 8,47-8,41 (m, 1H), 8,15 (d, J = 2,1 Гц, 1H), 7,97-7,91 (m, 1H), 7,32 (dd, J = 8,7, 2,0 Гц, 1H), 6,87 (br d, J = 8,4 Гц, 1H), 4,49-4,30 (m, 2H), 4,15-3,89 (m, 2H), 3,62 (br dd, J = 8,0, 5,0 Гц, 2H), 2,44 (s, 3H), 2,17-1,97 (m, 1H), 1,96-1,85 (m, 1H), 1,78-1,52 (m, 9H), 1,38-1,31 (m, 9H), 1,20-1,06 (m, 4H).
Стадия 6
В 50-литровый реакционный сосуд вносили этилацетат (17,70 л) и охлаждали до 0–5°C, после чего добавляли соединение 8 (1,70 кг, 3,37 моля, 1,0 экв.) с получением белой суспензии. С помощью перистальтического насоса вносили свежеприготовленный хлористый водород в этилацетате (17,70 л, 4 моль/л, 21,0 экв.), во время чего твердое вещество постепенно растворялось с получением красно-бурого раствора. После внесения температуру постепенно повышали до 10–25°C и содержимое системы инкубировали в течение 2,5 ч, при этом в осадок выпадало большое количество красновато-бурого твердого вещества. Реакционный раствор подвергали фильтрации, а осадок на фильтре и твердое вещество на стенках сосуда растворяли в метаноле (7,0 л). Органическую фазу переносили в роторный испаритель и концентрировали при пониженном давлении с получением твердого вещества. Твердое вещество суспендировали в этилацетате (8,85 л) и перемешивали в течение ночи. Смесь подвергали фильтрации, осадок на фильтре сушили при 35–45°C под вакуумом до постоянной массы с получением соединения 9. 1H ЯМР (400 МГц, DMSO-d6) δ 8,50 (s, 1H), 8,30 (br d, J = 3,1 Гц, 3H), 8,15 (d, J = 2,1 Гц, 1H), 7,89 (d, J = 8,8 Гц, 1H), 7,31 (dd, J = 2,1, 8,7 Гц, 1H), 4,56-4,45 (m, 1H), 4,45-4,36 (m, 1H), 4,15 (dd, J = 13,8, 8,3 Гц, 1H), 3,86 (br t, J = 5,1 Гц, 1H), 3,70-3,62 (m, 1H), 3,61-3,56 (m, 1H), 2,44 (s, 3H), 2,19-2,03 (m, 1H), 1,96-1,87 (m, 1H), 1,83-1,66 (m, 5H), 1,60 (br d, J = 10,3 Гц, 3H), 1,14-0,93 (m, 4H).
Стадия 7
В реакционный сосуд вносили N,N-диметилформамид (11,88 л) и охлаждали до 0-5°C, после чего вносили соединение 10 (1,36 кг, 6,71 моля, 1,05 экв.) и гексафторфосфат бензотриазол-N,N,N,N-тетраметилурония (2,54 кг, 6,71 моля, 1,05 экв.). Твердое вещество полностью не растворяли и таким образом получали белую суспензию. Добавляли N,N-диизопропилэтиламин (2,48 кг, 19,16 моля и 3,0 эквивалента) при температуре, контролируемой на уровне 0–5°C, до постепенного растворения твердого вещества с получением прозрачного раствора. После добавления содержимое системы инкубировали при 0–5°C в течение 30 минут и медленно добавляли соединение 9 (2,9 кг, 6,39 моля и 1,0 экв.), во время чего наблюдали выделение белого дыма и явление выделения теплоты. Температуру реакции контролировали на уровне 0–5°C путем контроля скорости подачи, а после добавления содержимое системы инкубировали при 25–30°C в течение 23 часов. Реакционный раствор обрабатывали за 5 порций. Реакционный раствор (приблизительно 5,0 л) по каплям добавляли к 35,0 л воды при помешивании, и в осадок выпадало большое количество белого твердого вещества. Смесь подвергали фильтрации после перемешивания в течение 30 мин, а осадки на фильтре высушивали и объединяли. Осадок на фильтре и твердое вещество на стенках реакционного сосуда растворяли в этилацетате (30,0 л), сушили над безводным сульфатом натрия и подвергали фильтрации. Фильтрат концентрировали при пониженном давлении с получением соединения 11. 1H ЯМР(400 МГц, CDCl3) δ 8,34 (d, J = 2,0 Гц, 1H), 7,73 (s, 1H), 7,67 (br d, J = 8,7 Гц, 1H), 7,29-7,22 (m, 1H), 6,78 (br s, 1H), 4,78-4,45 (m, 4H), 3,88-3,55 (m, 4H), 2,84-2,76 (m, 3H), 2,50 (s, 3H), 2,06-1,94 (m, 2H), 1,73 (br d, J = 10,3 Гц, 6H), 1,51 (s, 9H), 1,39 (d, J = 6,7 Гц, 2H), 1,33 (br d, J = 7,0 Гц, 3H), 1,16 (br s, 4H).
Стадия 8
В реакционный сосуд вносили этилацетат (10,0 л) и охлаждали до 0–5°C. Соединение 11 (3,84 кг, 6,03 моля, 1,0 экв.) растворяли в этилацетате (9,20 л) и вносили в реакционный сосуд. С помощью перистальтического насоса вносили свежеприготовленный хлористый водород в этилацетате (4 моля/л, 19,20 л) со скоростью потока 0,50 л/мин. После добавления содержимое системы постепенно нагревали при комнатной температуре до 25–30°C и инкубировали в течение 19 ч, при этом на внутренней стенке сосуда в осадок выпадало большое количество твердого вещества. В реакционный сосуд вносили простой метил-трет-бутиловый эфир (10,0 л). Реакционный раствор перемешивали в течение 2,0 ч и подвергали фильтрации, а осадок на фильтре высушивали. Осадок на фильтре и твердое вещество на стенке сосуда растворяли в воде (30,0 л) и охлаждали до 5–10°C. Смесь дважды экстрагировали этилацетатом (10,0 л × 2) и собирали водную фазу. Водную фазу обрабатывали за две порции. В реакционный сосуд вносили 15,0 л воды и охлаждали до внутренней температуры 5–10°C. По каплям добавляли 10% водный раствор карбоната калия до pH 9, при этом реакционный раствор из лилового становился бесцветным и выпадал осадок твердого вещества. Смесь перемешивали в течение 30 минут и подвергали фильтрации. Если твердое вещество было слишком мелким для фильтрации, то перед фильтрацией в смесь мог быть добавлен этилацетат (1,50 л) и произведено перемешивание в течение 30 минут. Осадок на фильтре высушивали, переносили в реакционный сосуд, добавляли изопропанол и перемешивали в течение 2,0 ч. Смесь подвергали фильтрации, а осадок на фильтре высушивали под вакуумом при 45°C до постоянной массы с получением соединения с формулой (I). 1H ЯМР (400 МГц, DMSO-d6) δ 8,50-8,39 (m, 1H), 8,20-8,09 (m, 1H), 7,96-7,82 (m, 2H), 7,32 (dd, J = 8,7, 2,1 Гц, 1H), 4,58-4,34 (m, 3H), 4,14-4,02 (m, 1H), 3,79-3,54 (m, 2H), 3,06-2,88 (m, 1H), 2,48-2,41 (m, 3H), 2,24-2,13 (m, 3H), 2,09-1,95 (m, 3H), 1,93-1,85 (m, 1H), 1,79-1,54 (m, 8H), 1,12-1,01 (m, 6H), 0,99-0,86 (m, 1H).
Стадия 9
В реакционный сосуд вносили этанол (14,0 л). Затем в реакционный сосуд при помешивании вносили соединение с формулой (I) (1,74 кг, 3,41 моля, 1,0 экв.). Твердое вещество полностью не растворяли и таким образом получали суспензию. Этанол в сосуде кипятили с обратным холодильником на циркуляционной бане при 90°C, при этом твердое вещество полностью растворялось. Затем смесь инкубировали в течение 15 минут и подвергали быстрой фильтрации. Фильтрат переносили в реакционный сосуд и постепенно охлаждали до комнатной температуры. В осадок выпадало большое количество твердого вещества. Реакционный раствор подвергали фильтрации, а осадок на фильтре высушивали при 35-45°C под вакуумом до постоянной массы с получением кристаллической формы A соединения с формулой (I). 1H ЯМР (400 МГц, DMSO-d6) δ 8,47-8,39 (m, 1H), 8,19-8,08 (m, 1H), 7,97-7,67 (m, 2H), 7,38-7,20 (m, 1H), 4,59-4,29 (m, 3H), 4,17-4,00 (m, 1H), 3,63 (td, J = 7,6, 3,8 Гц, 2H), 2,97 (q, J = 6,8 Гц, 1H), 2,47-2,39 (m, 3H), 2,24-2,14 (m, 3H), 2,11-1,96 (m, 2H), 1,94-1,81 (m, 1H), 1,78-1,49 (m, 8H), 1,25-0,87 (m, 8H).
Пример 2: получение кристаллической формы A соединения с формулой (I)
Примерно 50 мг соединения с формулой (I) переносили в 1,5-мл виалы для жидкостного хроматографа, добавляли соответствующее количество растворителя или смешанного растворителя (см. таблицу 2) и перемешивали или растворяли с обработкой ультразвуком. Образцы суспензии перемешивали (в защищенном от света месте) на термостатическом шейкере (40°C) в течение 60 ч. Затем образцы жидкостей быстро центрифугировали, а твердые вещества сушили в течение ночи в вакуумном сушильном шкафу при 35°C. Высушенные образцы подвергали XRPD-анализу.
Таблица 2: получение кристаллической формы А соединения с формулой (I)
Экспериментальный пример 1: исследование гигроскопичности кристаллической формы А соединения с формулой (I)
Условия
Модель прибора: SMS DVS Advantage
Условия тестирования: кристаллическую форму А соединения с формулой (I) (10-15 мг) помещали в лоток для образцов для DVS для тестирования.
Параметры DVS были следующими:
температура: 25°C;
балансировка: dm/dt = 0,01%/минута (наиболее короткий: 10 минут, наиболее длинный: 180 минут);
сушка: сушка при 0% RH в течение 120 минут;
градиент измерения RH (%): 10 %;
диапазон градиента измерения RH (%): 0%–90%–0%.
В таблице 3 представлены критерии оценки гигроскопичности.
Таблица 3: критерии оценки гигроскопичности
Примечание: гигроскопичное увеличение массы при 25°C/80% RH.
Результаты: кристаллическая форма А соединения с формулой (I) характеризовалась гигроскопичным увеличением массы на уровне 0,72% при 25°C и 80% RH.
Вывод: кристаллическая форма А соединения с формулой (I) является слабогигроскопичной, ее легко хранить, и она не подвержена поглощению влаги из воздуха, деформации, плесневению и т. д., поэтому обладает хорошим потенциалом для разработки лекарственных средств.
Экспериментальный пример 2: тест на стабильность кристаллической формы А соединения с формулой (I)
Процедуры
Фактор влияния в ходе эксперимента: получали 12 частей кристаллической формы А соединения с формулой (I), каждая из которых содержала 1,50 г, причем для каждого условия исследовали по 3 части. Образцы помещали в открытые бутыли для взвешивания. В ходе высокотемпературного теста бутыли помещали в отдельные эксикаторы с разными условиями, а затем для проверки помещали в соответствующие тестеры для испытания стабильности; для теста с высокой влажностью бутыли исследовали в тестерах для испытания стабильности в соответствующих условиях.
Тест на стабильность при облучении: получали 4 части кристаллической формы А соединения с формулой (I), каждая из которых содержала 1,50 г. 2 части были образцами для облучения (одна для 5-суточного облучения, другая для 10-суточного облучения), две другие части были контрольными (одна как 5-суточный контроль, а другая как 10-суточный контроль). Облучаемые образцы равномерно распределяли в чистых бутылях для взвешивания без защитного покрытия, бутыли закрывали прозрачными крышками и помещали в прибор для тестирования с облучением. Контроли упаковывали таким же образом, как и облучаемые образцы, но снаружи закрывали алюминиевой пленкой.
Ускоренный тест на долгосрочное хранение: получали 22 части кристаллической формы А соединения с формулой (I), каждая из которых содержала 1,50 г. Образцы отдельно помещали в двухслойный пакет из LDPE (полиэтилена низкой плотности), причем каждый слой пакета из LDPE запечатывали отдельно. Затем пакет помещали в пакет из алюминиевой фольги для запечатывания термосваркой. В моменты осмотра забирали соответствующие тестируемые образцы и закрывали их крышками для бутылей. Образцы в 0-е сутки оттаивали до комнатной температуры и сразу проводили с ними анализ. Примерно 10 мг тестируемого образца подвергали XRPD. Соединения помещали в описанные далее условия и в разные моменты времени забирали образцы для определения их свойств, XRPD-диаграммы, содержания и родственных веществ. Условия и тестируемые образцы представлены в таблицах 4 и 5.
Таблица 4: результаты определения содержания кристаллической формы А и родственных веществ в тестах на стабильность под воздействием облучения и других факторов влияния
Таблица 5: результаты определения содержания кристаллической формы А и родственных веществ в ускоренных тестах на стабильность
Вывод
По результатам XRPD наблюдали, что кристаллическая форма A соединения с формулой (I) соответствовала форме в 0-е сутки, что позволяло сделать вывод, что кристаллическая форма A соединения с формулой (I) была стабильной в различных условиях тестирования.
Экспериментальный пример 3: анализ связывания cIAP1 BIR3 и XIAP BIR3
Материалы
Буферная система (буфер для BIR3 cIAP1 или BIR3 XIAP): 0,1 моль/л фосфата калия, рН 7,5, 0,1% бычьего сывороточного альбумина; 0,005% тритона Х-100 и 1% диметилсульфоксида.
Зонд: ARPFAQ-K(5-FAM)-NH2.
Цель
cIAP1-BIR3-His: RBC (кат. № APT-11-370), выделенный домен BIR3 человеческого cIAP1 (аминокислоты 258–363; cIAP1 BIR3), экспрессируемый E. coli в виде слитого с GST белка.
XIAP-BIR3-His: RBC (кат. № APT-11-374), выделенный домен BIR3 из XIAP (аминокислоты 255-356; XIAP BIR3), экспрессируемый E. coli в виде слитого с GST белка.
Условие: 5 нM ARPFAQ-K(5-FAM)-NH2, 20 нM cIAP1 BIR3 и 30 нM XIAP BIR3.
Процедуры
Сначала готовили свежий буфер cIAP1 BIR3 или XIAP BIR3 и смешивали с 2-кратным раствором cIAP1 BIR3 или XIAP BIR3. Раствор тестируемого соединения, полностью растворенного в 100% DMSO посредством обработки ультразвуком, вносили в буфер, содержащий cIAP1 BIR3 или XIAP BIR3. В смесь добавляли 2-кратные количества зондов, перемешивали и инкубировали в течение 60 минут в темноте при комнатной температуре. Измеряли флуоресцентную поляризацию и рассчитывали значение mP, в итоге получая значение IC50.
Результаты показаны в таблице 6.
Вывод
Раскрываемые в настоящем документе соединения с формулой (I) характеризовались хорошей связывающей активностью в отношении cIAP1 BIR3 и хорошей избирательностью в отношении cIAP1 и XIAP.
Экспериментальный пример 4: in vitro анализ жизнеспособности клеток
Материалы
Среда RPMI 1640 (Invitrogen-22400089), фетальная бычья сыворотка (Invitrogen-10099141), трипсин 0,05% EDTA·4Na (Invitrogen-25300062), набор для люминесцентного анализа жизнеспособности клеток (Promega-G7573), фосфатно-солевой буферный раствор Дульбекко (HyClone-SH30028.01B) и 384-луночные планшеты (Corning-6007680). Многофункциональный анализатор Envision Multi-label Analyzer.
Процедуры
1. 30 мкл клеточной суспензии MDA-MB-231, содержавшей 250 клеток MDA-MB-231, вносили в лунки 384-луночного планшета.
2. Вносили 20 мкл тестируемого соединения (тестируемое соединение подвергали 5-кратному серийному разведению от 10 мкМ до 10-й концентрации), а затем планшеты инкубировали в термостате с атмосферой диоксида углерода в течение 7 суток.
3. Планшеты с клетками выдерживали при комнатной температуре в течение 30 минут.
4. В каждую лунку вносили 20 мкл реагента Promega CellTiter-Glo.
5. Спустя 10 минут образцы детектировали с помощью многофункционального анализатора Envision.
Результаты: см. таблицу 6.
Вывод
Раскрываемое в настоящем документе соединение с формулой (I) характеризуется хорошей антипролиферативной активностью в отношении клетки MDA-MB-231.
Таблица 6
Экспериментальный пример 5: in vivo исследование эффективности 1
In vivo исследование эффективности проводили на бестимусных мышах BALB/c путем подкожной имплантации ксенотрансплантата, полученного из линии опухолевых клеток человека (CDX) от пациентов с трижды негативным раком молочной железы MDA-MB-231.
Процедуры
Бестимусных мышей BALB/c, самок возрастом 6-8 недель, массой приблизительно 18-22 г, содержали в специальной стерильной среде и в отдельных вентилируемых клетках (по 3 мыши на клетку). Все клетки, подстилка и вода были продезинфицированы перед использованием. Все животные имели свободный доступ к стандартным сертифицированным коммерческим лабораторным кормам. Для исследования у компании Shanghai BK Laboratory Animal Co., LTD были приобретены 48 мышей. Каждой мыши в правый бок подкожно имплантировали раковые клетки (10×106 клеток в 0,2 мл фосфатно-солевого буферного раствора) для роста опухоли. Когда средний объем опухоли достигал приблизительно 147 мм3, мышам вводили тестируемый препарат. Тестируемые соединения вводили перорально с суточной дозой 30 мг/кг. Объем опухоли измеряли каждые 3 суток двумерным штангенциркулем, объем измеряли в мм3 и рассчитывали по следующей формуле: V = 0,5 × a × b2, где a и b соответственно обозначали длинный и короткий диаметр опухоли. Противоопухолевую эффективность определяли путем деления среднего увеличения объема опухоли у животных, обрабатываемых соединением, на среднее увеличение объема опухоли у животных без обработки.
Результаты: см. таблицу 7.
Вывод
В модели CDX трижды негативного рака молочной железы MDA-MB-231 для оценки эффективности in vivo раскрываемое в настоящем документе соединение с формулой (I) характеризовалось лучшей эффективностью.
Таблица 7
(мг/кг)
Экспериментальный пример 6: in vivo исследование эффективности 2
In vivo исследование эффективности проводили на бестимусных мышах BALB/c путем подкожной имплантации ксенотрансплантата, полученного из линии опухолевых клеток человека (CDX) от пациентов с трижды негативным раком молочной железы MDA-MB-231.
Процедуры
Бестимусных мышей BALB/c, самок возрастом 6-8 недель, массой приблизительно 18-22 г, содержали в специальной стерильной среде и в отдельных вентилируемых клетках (по 3 мыши на клетку). Все клетки, подстилка и вода были продезинфицированы перед использованием. Все животные имели свободный доступ к стандартным сертифицированным коммерческим лабораторным кормам. Для исследования у компании Shanghai BK Laboratory Animal Co., LTD были приобретены 48 мышей. Каждой мыши в правый бок подкожно имплантировали опухолевые клетки (10×106 клеток в 0,2 мл фосфатного буфера) для роста опухоли. Когда средний объем опухоли достигал приблизительно 110 мм3, мышам вводили тестируемый препарат. Тестируемые соединения вводили перорально с суточной дозой 30 мг/кг. Объем опухоли измеряли каждые 3 суток двумерным штангенциркулем, объем измеряли в мм3 и рассчитывали по следующей формуле: V = 0,5 × a × b2, где a и b соответственно обозначали длинный и короткий диаметр опухоли. Противоопухолевую эффективность определяли путем деления среднего увеличения объема опухоли у животных, обрабатываемых соединением, на среднее увеличение объема опухоли у животных без обработки.
Результаты: см. таблицу 8.
Вывод
В модели CDX трижды негативного рака молочной железы MDA-MB-231 для оценки эффективности in vivo раскрываемое в настоящем документе соединение с формулой (I) характеризовалось лучшей эффективностью.
Таблица 8
(мг/кг)
| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2014 |
|
RU2696310C1 |
| КРИСТАЛЛИЧЕСКАЯ ИЛИ АМОРФНАЯ ФОРМА АГОНИСТОВ FXR, ПРЕДСТАВЛЯЮЩИХ СОБОЙ ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2800751C2 |
| СОЕДИНЕНИЕ ИНГИБИТОРА BRD4 В ТВЕРДОЙ ФОРМЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2793346C1 |
| СОЛИ ПРОИЗВОДНОГО ИНДАЗОЛА И ИХ КРИСТАЛЛЫ | 2017 |
|
RU2747399C2 |
| СОЛИ И КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ДИАЗАБЕНЗОФЛУОРАНТРЕНОВЫХ СОЕДИНЕНИЙ | 2017 |
|
RU2762189C2 |
| СОЛЬ ИНГИБИТОРА SYK И ЕЕ КРИСТАЛЛИЧЕСКАЯ ФОРМА | 2019 |
|
RU2818103C2 |
| КРИСТАЛЛИЧЕСКИЙ ИНГИБИТОР FGFR4 И ЕГО ПРИМЕНЕНИЕ | 2016 |
|
RU2763328C2 |
| АНАЛОГ ПИРИДО[1,2-A]ПИРИМИДОНА, ЕГО КРИСТАЛЛИЧЕСКАЯ ФОРМА, ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2016 |
|
RU2753696C2 |
| ПОЛИМОРФНАЯ ФОРМА ДИАРИЛЬНОГО МАКРОЦИКЛА | 2016 |
|
RU2765181C2 |
| ТВЕРДЫЕ ФОРМЫ ПЛАДИЕНОЛИДПИРИДИНОВЫХ СОЕДИНЕНИЙ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2743349C2 |
Изобретение относится к кристаллической форме соединения с формулой (I), где кристаллическая форма представляет собой кристаллическую форму А соединения с формулой (I), которая на порошковой рентгеновской дифрактограмме, полученной с помощью Cu-Kα-излучения, характеризуется дифракционными пиками при следующих значениях 2θ 12,1±0,200°, 16,1±0,200°, 18,5±0,200°, 20,2±0,200°, 21,3±0,200° и 23,0±0,200°. Также изобретение относится к фармацевтической композиции для лечения трижды негативного рака молочной железы, содержащей терапевтически эффективное количество кристаллической формы соединения с формулой (I) и фармацевтически приемлемый носитель. Технический результат - кристаллическая форма соединения с формулой (I) для применения в качестве ингибитора в отношении cIAP1, обладающего такими свойствами, как фармакокинетика, биодоступность, низкая гигроскопичность, высокая стабильность. 2 н. и 6 з.п. ф-лы, 3 ил., 8 табл., 8 пр.
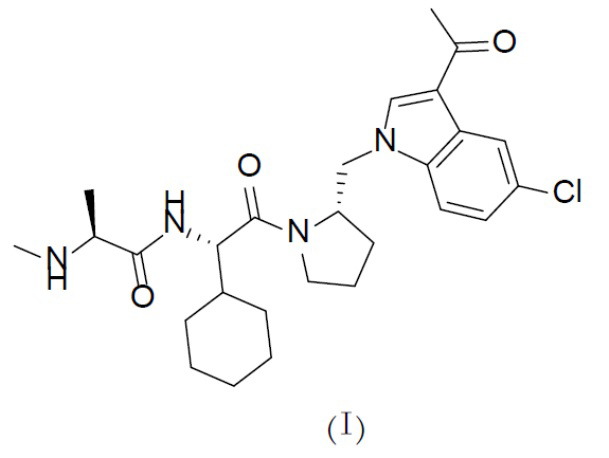
1. Кристаллическая форма соединения с формулой (I)
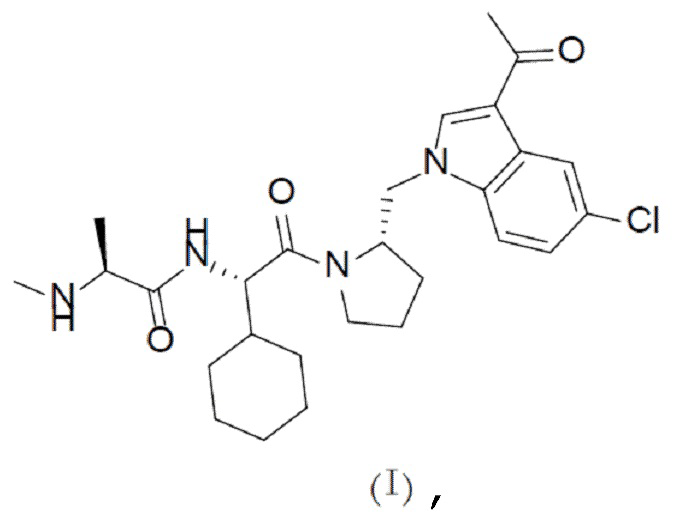
где кристаллическая форма представляет собой кристаллическую форму А соединения с формулой (I), которая на порошковой рентгеновской дифрактограмме, полученной с помощью Cu-Kα-излучения, характеризуется дифракционными пиками при следующих значениях 2θ: 12,1°±0,200°, 16,1°±0,200°, 18,5°±0,200°, 20,2°±0,200°, 21,3°±0,200° и 23,0°±0,200°.
2. Кристаллическая форма А соединения с формулой (I) по п. 1, которая на порошковой рентгеновской дифрактограмме, полученной с помощью Cu-Kα-излучения, характеризуется дифракционными пиками при следующих значениях 2θ: 12,1°±0,200°, 16,1°±0,200°, 18,5°±0,200°, 18,8°±0,200°, 19,2°±0,200°, 19,8°±0,200°, 20,2°±0,200°, 21,3°±0,200°, 23,0°±0,200°, 26,6°±0,200° и 27,4°±0,200°.
3. Кристаллическая форма А соединения с формулой (I) по п. 2, которая на порошковой рентгеновской дифрактограмме, полученной с помощью Cu-Kα-излучения, характеризуется дифракционными пиками при следующих значениях 2θ: 7,0°±0,200°, 8,7°±0,200°, 12,1°±0,200°, 13,2°±0,200°, 13,9°±0,200°, 16,1°±0,200°, 16,7°±0,200°, 18,5°±0,200°, 18,8°±0,200°, 19,2°±0,200°, 19,8°±0,200°, 20,2°±0,200°, 21,0°±0,200°, 21,3°±0,200°, 23,0°±0,200°, 24,3°±0,200°, 25,3°±0,200°, 26,6°±0,200° и 27,4°±0,200°.
4. Кристаллическая форма А соединения с формулой (I) по п. 3, которая на порошковой рентгеновской дифрактограмме, полученной с помощью Cu-Kα-излучения, характеризуется дифракционными пиками при следующих значениях 2θ: 7,0°±0,200°, 8,7°±0,200°, 12,1°±0,200°, 13,2°±0,200°, 13,9°±0,200°, 16,1°±0,200°, 16,5°±0,200°, 16,7°±0,200°, 18,5°±0,200°, 18,8°±0,200°, 19,2°±0,200°, 19,8°±0,200°, 20,2°±0,200°, 21,0°±0,200°, 21,3°±0,200°, 23,0°±0,200°, 23,2°±0,200°, 24,3°±0,200°, 25,3°±0,200°, 26,6°±0,200°, 26,9°±0,200°, 27,4°±0,200° и 29,4°±0,200°.
5. Кристаллическая форма А соединения с формулой (I) по п. 4, которая на порошковой рентгеновской дифрактограмме, полученной с помощью Cu-Kα-излучения, характеризуется дифракционными пиками при следующих значениях 2θ: 7,0°±0,200°, 8,7°±0,200°, 9,7°±0,200°, 10,6°±0,200°, 11,4°±0,200°, 12,1°±0,200°, 13,2°±0,200°, 13,9°±0,200°, 16,1°±0,200°, 16,5°±0,200°, 16,7°±0,200°, 17,5°±0,200°, 18,5°±0,200°, 18,8°±0,200°, 19,2°±0,200°, 19,5°±0,200°, 19,8°±0,200°, 20,2°±0,200°, 21,0°±0,200°, 21,3°±0,200°, 22,5°±0,200°, 23,0°±0,200°, 23,2°±0,200°, 23,8°±0,200°, 24,3°±0,200°, 24,6°±0,200°, 25,3°±0,200°, 25,8°±0,200°, 26,6°±0,200°, 26,9°±0,200°, 27,4°±0,200°, 28,1°±0,200°, 29,1°±0,200°, 29,4°±0,200°, 30,1°±0,200°, 30,6°±0,200°, 30,8°±0,200°, 31,3°±0,200°, 31,8°±0,200°, 32,2°±0,200°, 32,8°±0,200°, 33,7°±0,200°, 34,0°±0,200°, 34,8°±0,200°, 35,5°±0,200°, 36,6°±0,200°, 37,6°±0,200°, 38,6°±0,200° и 39,6°±0,200°.
6. Кристаллическая форма А соединения с формулой (I) по любому из пп. 1-5, которая на диаграмме дифференциальной сканирующей калориметрии характеризуется эндотермическим пиком при 202,5°C.
7. Кристаллическая форма А соединения с формулой (I) по любому из пп.1-5, которая на диаграмме дифференциальной сканирующей калориметрии характеризуется начальной точкой эндотермического пика при 200,8°C.
8. Фармацевтическая композиция для лечения трижды негативного рака молочной железы, содержащая терапевтически эффективное количество кристаллической формы соединения с формулой (I) по п. 1 и фармацевтически приемлемый носитель.
| WO 2009094287 A1, 30.07.2009 | |||
| УСТАНОВКА ДЛЯ ЦЕНТРИФУГИРОВАНИЯ ЖИДКОГО ЗАТВЕРДЕВАЮЩЕГО МАТЕРИАЛА | 1993 |
|
RU2102229C1 |
| ИМИДАЗОПИРИДИНОВЫЕ ИНГИБИТОРЫ IAP | 2007 |
|
RU2466131C2 |
| ИНГИБИТОРЫ IAP | 2005 |
|
RU2401840C2 |
| EA 200701467 A1, 28.12.2007 | |||
| WO 2008014252 A2, 31.01.2008 | |||
| Francisco Sarabia | |||
| Нефтяной конвертер | 1922 |
|
SU64A1 |
| Bioorganic & Medicinal Chemistry, 2005, vol | |||
| Насос | 1917 |
|
SU13A1 |

