Область техники
Настоящее изобретение относится к твердым формам и кристаллическим формам соединения формулы (I), как низкомолекулярного ингибитора BRD4, и к способу его получения, а также относится к его применению для получения лекарственного препарата для лечения заболеваний, связанных с BRD4.
Уровень техники
Ацетилирование гистонов регулирует транскрипцию генов и структуру хромосом, а также играет важную роль в эпигенетике. Белки семейства BET (бромодомен и экстратерминальный домен), как «считыватели» гена распознавания ацетилирования гистонов, могут специфически связываться с остатками ацетилированного лизина и привлекать другие факторы транскрипции. Участвуя в белок-белковых взаимодействиях, образуется медиаторный комплекс, фосфорилирующий РНК-полимеразу, которая активирует транскрипцию генов и регулирует с-Мус и другие гены, регулирующие последующие звенья сигнальных каскадов. Важную роль в пролиферации раковых клеток играет усиление экспрессии специфических генов (например, с-Мус), от которых она сильно зависит. В ходе исследований было установлено, что опухолевые клетки находятся в сильной зависимости от работы специфических генов, что делает их очень чувствительными к ингибиторам BET. В присутствии ингибиторов BET белки BET не связываются с ацетилированным лизином гистона, и тем самым блокируют экспрессию Мус факторами транскрипции, что приводит к подавлению роста опухоли.
Семейство белков BET включает в себя 4 представителя: BRD2, BRD3, BRD4 и BRDT, каждый из которых состоит из двух N-концевых тандемных областей (BD1 и BD2), экстратерминального домена, нескольких консервативных участков (А, В, затравочная область) и С-концевого мотива (СКМ). Наиболее изученным представителем среди них является BRD4. Было установлено, что возникновение гематологических опухолей, включая лимфомы (например, острая миелолимфома и др.), лейкозы (например, острый лимфобластный лейкоз и др.), миеломы (например, множественные миеломы и др.) и солидные опухоли, такие как нейроцитома, глиомы, рак молочной железы (например, трижды негативный рак молочной железы и др.), опухоли желудочно-кишечного тракта (например, рак толстого кишечника и прямой кишки и др.), рак предстательной железы и др., связано со сверхэкспрессией BRD4, но до сих пор на рынке не было одобрено ни одного препарата, направленно действующего на BRD4.
Раскрытие сущности изобретения
Согласно одному аспекту настоящего изобретения настоящее изобретение обеспечивает соединение формулы (I) в твердой форме,

В некоторых вариантах реализации настоящего изобретения соединение в твердой форме находится в кристаллической форме.
В некоторых вариантах реализации настоящего изобретения кристаллическая форма представляет собой кристаллическую форму А соединения формулы (I), имеющую порошковую рентгеновскую дифрактограмму, содержащую характерные дифракционные пики при следующих углах 2θ: 7,03±0,2°, 11,28±0,2°, 14,00±0,2°.
В некоторых вариантах реализации настоящего изобретения кристаллическая форма А имеет порошковую рентгеновскую дифрактограмму, содержащую характерные дифракционные пики при следующих углах 2θ: 7,03±0,2°, 11,28±0,2°, 20,07±0,2°.
В некоторых вариантах реализации настоящего изобретения кристаллическая форма А имеет порошковую рентгеновскую дифрактограмму, содержащую характерные дифракционные пики при следующих углах 2θ: 7,03±0,2°, 19,48±0,2°, 20,07±0,2°, 26,05±0,2°.
В некоторых вариантах реализации настоящего изобретения кристаллическая форма А имеет порошковую рентгеновскую дифрактограмму, содержащую характерные дифракционные пики при следующих углах 2θ: 7,03±0,2°, 11,28±0,2°, 17,31±0,2°, 19,48±0,2°, 20,07±0,2°, 26,05±0,2°.
В некоторых вариантах реализации настоящего изобретения кристаллическая форма А имеет порошковую рентгеновскую дифрактограмму, содержащую характерные дифракционные пики при следующих углах 2θ: 7,03±0,2°, 11,28±0,2°, 14,00±0,2°, 15,11±0,2°, 17,31±0,2°, 19,48±0,2°, 20,07±0,2°, 22,86±0,2°.
В некоторых вариантах реализации настоящего изобретения кристаллическая форма А имеет порошковую рентгеновскую дифрактограмму, содержащую характерные дифракционные пики при следующих углах 2θ: 7,03±0,2°, 11,28±0,2°, 14,00±0,2°, 15,11±0,2°, 17,31±0,2°, 19,48±0,2°, 20,07±0,2°, 26,05±0,2°.
В некоторых вариантах реализации настоящего изобретения кристаллическая форма А имеет порошковую рентгеновскую дифрактограмму, содержащую характерные дифракционные пики при следующих углах 2θ: 7,03±0,2°, 11,28±0,2°, 12,39±0,2°, 14,00±0,2°, 15,11±0,2°, 17,31±0,2°, 19,48±0,2°, 20,07±0,2°, 22,86±0,2°, 26,05±0,2°.
В некоторых вариантах реализации настоящего изобретения кристаллическая форма А имеет порошковую рентгеновскую дифрактограмму, содержащую характерные дифракционные пики при следующих углах 2θ: 7,03±0,2°, 11,28±0,2°, 12,39±0,2°, 14,00±0,2°, 14,87±0,2°, 15,11±0,2°, 17,31±0,2°, 19,48±0,2°, 20,07±0,2°, 22,86±0,2°.
В некоторых вариантах реализации настоящего изобретения кристаллическая форма А имеет порошковую рентгеновскую дифрактограмму, содержащую характерные дифракционные пики при следующих углах 2θ: 7,03±0,2°, 11,28±0,2°, 12,39±0,2°, 14,00±0,2°, 14,87±0,2°, 15,11±0,2°, 17,31±0,2°, 19,48±0,2°, 20,07±0,2°, 22,86±0,2°, 26,05±0,2°.
В некоторых вариантах реализации настоящего изобретения кристаллическая форма А имеет порошковую рентгеновскую дифрактограмму, содержащую характерные дифракционные пики при следующих углах 2θ: 7,03±0,2°, 11,28±0,2°, 12,39±0,2°, 14,00±0,2°, 14,87±0,2°, 15,11±0,2°, 17,31±0,2°, 17,68±0,2°, 19,48±0,2°, 20,07±0,2°, 22,86±0,2°.
В некоторых вариантах реализации настоящего изобретения кристаллическая форма А имеет порошковую рентгеновскую дифрактограмму, содержащую характерные дифракционные пики при следующих углах 2θ: 7,03±0,2°, 11,28±0,2°, 12,39±0,2°, 14,00±0,2°, 14,87±0,2°, 15,11±0,2°, 17,31±0,2°, 17,68±0,2°, 19,48±0,2°, 20,07±0,2°, 22,86±0,2°, 26,05±0,2°.
В некоторых вариантах реализации настоящего изобретения, кристаллическая форма А имеет дифрактограмму XRPD, по существу, как показано на фиг. 1.
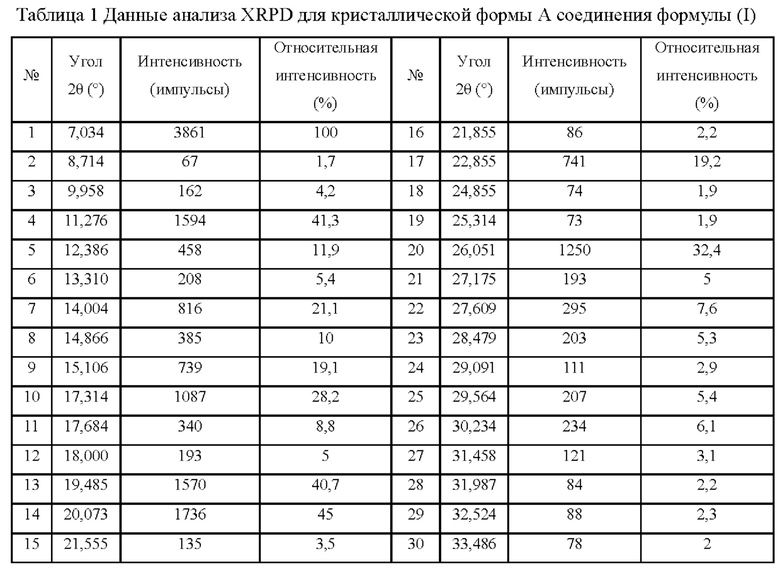
В некоторых вариантах реализации настоящего изобретения данные анализа дифрактограммы XRPD для кристаллической формы А приведены в таблице 1:
В некоторых вариантах реализации настоящего изобретения кристаллическая форма А имеет кривую дифференциальной сканирующей калориметрии (ДСК) с возникновением эндотермического пика при 289,22±3°С.
В некоторых вариантах реализации настоящего изобретения кривая ДСК кристаллической формы А, по существу, как показано на фиг. 2.
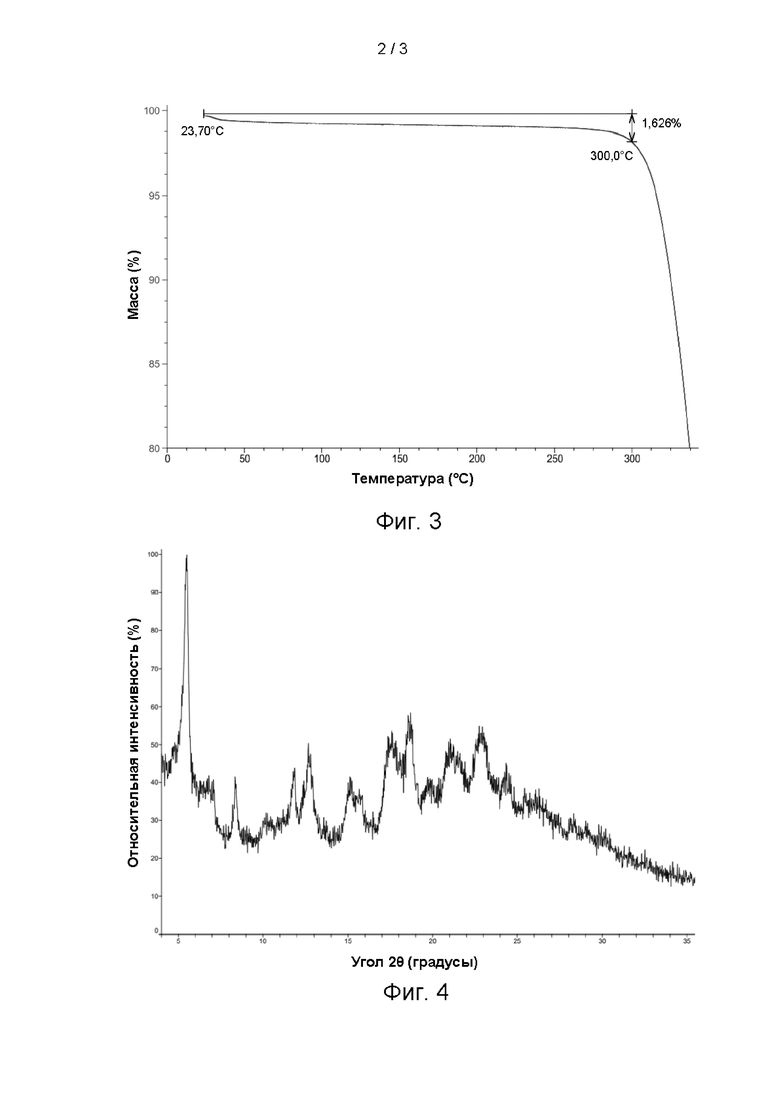
В некоторых вариантах реализации настоящего изобретения потеря массы кристаллической формы А по данным термограммы термогравиметрического анализа (ТГА) составляет 1,626% при 300,00±3°С.
В некоторых вариантах реализации настоящего изобретения термограмма ТГА кристаллической формы А, по существу, как показано на фиг. 3.
В некоторых вариантах реализации настоящего изобретения, кристаллическая форма представляет собой кристаллическую форму В соединения формулы (I), имеющую порошковую рентгеновскую дифрактограмму, содержащую характерные дифракционные пики при следующих углах 2θ: 5,50±0,2°, 8,36±0,2°, 11,87±0,2°.
В некоторых вариантах реализации настоящего изобретения, кристаллическая форма В имеет порошковую рентгеновскую дифрактограмму, содержащую характерные дифракционные пики при следующих углах 2θ: 5,50±0,2°, 8,36±0,2°, 12,66±0,2°.
В некоторых вариантах реализации настоящего изобретения кристаллическая форма В имеет порошковую рентгеновскую дифрактограмму, содержащую характерные дифракционные пики при следующих углах 2θ: 5,50±0,2°, 8,36±0,2°, 11,87±0,2°, 12,39±0,2°, 12,66±0,2°, 15,11±0,2°, 17,35±0,2°, 18,70±0,2°.
В некоторых вариантах реализации настоящего изобретения кристаллическая форма В имеет дифрактограмму XRPD, по существу, как показано на фиг. 4.
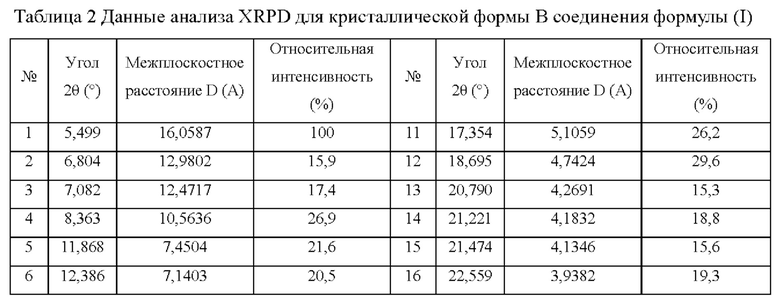
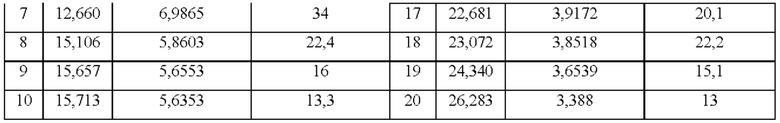
В некоторых вариантах реализации настоящего изобретения данные анализа дифрактограммы XRPD для кристаллической формы В приведены в таблице 2.
В другом аспекте настоящего изобретения настоящее изобретение также обеспечивает способ получения соединения формулы (I) в твердой форме, при этом твердая форма представляет собой кристаллическую форму А, включающий:
(1) добавление соединения формулы (I) в растворитель с образованием суспензии или раствора;
(2) перемешивание суспензии или раствора в термомиксере с поддержанием постоянной температуры, затем разделение и сушку для получения кристаллической формы А соединения формулы (I).
В некоторых вариантах реализации настоящего изобретения разделение на этапе (2) в способе получения представляет собой центрифугирование или фильтрацию.
В некоторых вариантах реализации настоящего изобретения разделение на этапе (2) в способе получения представляет собой центрифугирование.
В некоторых вариантах реализации настоящего изобретения растворитель в способе получения представляет собой простой растворитель, выбранный из группы, состоящей из С1-4алкил-O-С1-4алкила, С1-4алкилС(=O)ОС1-4алкила, С1-4алкил-CN, С1-4алкил-ОН или C1-4алкилС(=O)С1-4алкила.
В некоторых вариантах реализации настоящего изобретения простой растворитель в способе получения представляет собой метил-трет-бутиловый эфир, этилацетат, ацетонитрил, этанол, ацетон, метанол или метилэтилкетон.
В некоторых вариантах реализации настоящего изобретения растворитель в способе получения представляет собой смешанный растворитель из С1-4алкилС(=O)С1-4алкила и воды или смешанный растворитель из C1-4алкил-ОН и воды.
В некоторых вариантах реализации настоящего изобретения для смешанного растворителя, состоящего из С1-4алкилС(=O)С1-4алкила и воды, в способе получения, отношение объема С1-4алкилС(=O)С1-4алкила к объему воды составляет 1-5:1, предпочтительно 2:1; или для смешанного растворителя, состоящего из C1-4алкил-ОН и воды, отношение объема C1-4алкил-ОН к объему воды составляет 1-5:1, предпочтительно 3:1.
В некоторых вариантах реализации настоящего изобретения смешанный растворитель в способе получения представляет собой смешанный растворитель из ацетона и воды или смешанный растворитель из этанола и воды.
В некоторых вариантах реализации настоящего изобретения смешанный растворитель в способе получения представляет собой смешанный растворитель ацетон-вода отношением объемов 2:1, или смешанный растворитель этанол-вода отношением объемов 3:1.
Термин «С1-4алкил» относится к любой группе с прямой или разветвленной цепью, содержащей от 1 до 4 атомов углерода, такой как метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил и т.п. И каждый «С1-4алкил» может быть одинаковым или разным.
В некоторых вариантах реализации настоящего изобретения отношение массы соединения к объему растворителя в способе получения составляет 1 г: 5-15 мл или 1 г: 5-12 мл, или 1 г: 5-10 мл.
В некоторых вариантах реализации настоящего изобретения температура перемешивания в способе получения составляет 25°С-45°С.
В некоторых вариантах реализации настоящего изобретения время перемешивания в способе получения составляет от 12 часов до 50 часов или от 12 часов до 48 часов, или от 12 часов до 24 часов.
В другом аспекте настоящего изобретения настоящее изобретение также обеспечивает способ получения соединения формулы (I) в твердой форме, при этом твердая форма представляет собой кристаллическую форму В, включающий:
(1) добавление соединения формулы (I) в растворитель с образованием суспензии или раствора;
(2) перемешивание суспензии или раствора в термомиксере с поддержанием постоянной температуры, затем разделение и сушку для получения кристаллической формы В соединения формулы (I).
В некоторых вариантах реализации настоящего изобретения разделение на стадии (2) в способе получения представляет собой центрифугирование или фильтрование.
В некоторых вариантах реализации настоящего изобретения разделение на стадии (2) в способе получения представляет собой центрифугирование.
В некоторых вариантах реализации настоящего изобретения растворитель в способе получения представляет собой тетрагидрофуран.
В некоторых вариантах реализации настоящего изобретения отношение массы соединения к объему растворителя в способе получения составляет 1 г: 5-10 мл.
В некоторых вариантах реализации настоящего изобретения температура перемешивания в способе получения составляет 25°С-45°С.
В некоторых вариантах реализации настоящего изобретения время перемешивания в способе получения составляет от 12 часов до 50 часов или от 12 часов до 48 часов, или от 12 часов до 24 часов.
В другом аспекте настоящего изобретения настоящее изобретение обеспечивает фармацевтическую композицию, содержащую, как описано выше, соединение формулы (I) в твердой форме или кристаллическую смесь любых двух или более кристаллических форм.
В другом аспекте настоящего изобретения настоящее изобретение также обеспечивает применение соединения формулы (I) в твердой форме или фармацевтической композиции, описанной выше, для получения лекарственного препарата для лечения заболеваний, связанных с BRD4.
В некоторых аспектах настоящего изобретения применение характеризуется тем, что заболевания, связанные с BRD4, включают опухоли.
В некоторых вариантах реализации настоящего изобретения применение характеризуется тем, что опухоли включают гематологические опухоли и солидные опухоли на поздних стадиях, при этом гематологические опухоли включают лейкоз, лимфому и миелому, и солидные опухоли на поздних стадиях включают нейроцитому, глиому, рак молочной железы, опухоль желудочно-кишечного тракта и рак предстательной железы; предпочтительно, лейкоз представляет собой острый лимфобластный лейкоз или лимфома представляет собой острую миелоидную лимфому, или рак молочной железы представляет собой трижды негативный рак молочной железы, или опухоль желудочно-кишечного тракта представляет собой рак толстого кишечника и прямой кишки.
В другом аспекте настоящего изобретения настоящее изобретение также относится к соединению формулы (I) в твердой форме или фармацевтической композиции, описанной выше, для применения в лечении заболеваний, связанных с BRD4.
В некоторых вариантах реализации настоящего изобретения твердая форма соединения формулы (I) или фармацевтическая композиция, описанная выше, где заболевания, связанные с BRD4, включают опухоли.
В некоторых вариантах реализации настоящего изобретения твердая форма соединения формулы (I) или фармацевтическая композиция, описанная выше, где опухоли включают гематологические опухоли и солидные опухоли на поздних стадиях, при этом гематологические опухоли включают лейкоз, лимфому и миелому, и солидные опухоли на поздних стадиях включают нейроцитому, глиому, рак молочной железы, опухоль желудочно-кишечного тракта и рак предстательной железы; предпочтительно лейкоз представляет собой острый лимфобластный лейкоз или лимфома представляет собой острую миелоидную лимфому, или рак молочной железы представляет собой трижды негативный рак молочной железы, или опухоль желудочно-кишечного тракта представляет собой рак толстого кишечника и прямой кишки.
В другом аспекте настоящего изобретения настоящее изобретение также относится к способу лечения заболевания, связанного с BRD4, у нуждающегося в этом субъекта, включающему введение субъекту соединения формулы (I) в твердой форме или фармацевтической композиции, описанной выше.
В некоторых вариантах реализации настоящего изобретения, способ лечения заболевания у субъекта, где заболевание включает опухоли.
В некоторых вариантах реализации настоящего изобретения, способ лечения заболевания у субъекта, где опухоли включают гематологические опухоли и солидные опухоли на поздних стадиях, при этом гематологические опухоли включают лейкоз, лимфому и миелому, и солидные опухоли на поздних стадиях включают нейроцитому, глиому, рак молочной железы, опухоль желудочно-кишечного тракта и рак предстательной железы; предпочтительно, лейкоз представляет собой острый лимфобластный лейкоз или лимфома представляет собой острую миелоидную лимфому, или рак молочной железы представляет собой трижды негативный рак молочной железы, или опухоль желудочно-кишечного тракта представляет собой рак толстого кишечника и прямой кишки.
Термин «субъект» включает всех представителей животных, включая, но не ограничиваясь, млекопитающих (например, мышей, крыс, представителей семейства кошачьих, обезьян, представителей семейства псовых, лошадей, свиней и т.д.) и человека.
Термин «по существу, как показано на фиг.» означает, что по меньшей мере 50% или по меньшей мере 60%, или по меньшей мере 70%, или по меньшей мере 80%, или по меньшей мере 90%, или по меньшей мере 95%, или по меньшей мере 99% пиков в порошковой рентгеновской дифрактограмме или кривой ДСК, или термограмме ТГА показаны на соответствующем чертеже.
Определения и описания
Следующие термины и фразы, используемые в настоящей заявке, имеют следующие значения, если не указано иное. Конкретная фраза или термин не должны рассматриваться как неопределенные или неясные в отсутствие четкого определения и должны трактоваться в соответствии с общепринятым значением. Если в настоящей заявке используется название торговой марки, подразумевается, что оно относится к соответствующему коммерческому продукту или его действующему веществу.
Промежуточные соединения настоящего изобретения могут быть получены с помощью различных методов синтеза, хорошо известных специалистам в этой области техники, включая частные варианты реализации настоящего изобретения, приведенные в качестве примеров ниже, варианты реализации настоящего изобретения, созданные сочетаниями методов синтеза с другими методами химического синтеза, и их эквиваленты, хорошо известные специалистам в этой области техники, и предпочтительные варианты реализации, включающие примеры настоящего изобретения, но не ограничивающиеся ими.
Химические реакции частных вариантов реализации настоящего изобретения проводятся с использованием подходящих растворителей, которые совместимы с химическими реакциями, представленными в настоящей заявке, а также реагентами и материалами, необходимыми для этого. Для получения соединений настоящего изобретения специалистам в этой области техники иногда необходимо варьировать или выбирать стадии синтеза или схемы реакций на основе существующих вариантов реализации настоящего изобретения.
Настоящее изобретение будет подробно описано ниже на примерах, которые являются исключительно иллюстративными и никак не ограничивают настоящее изобретение каким-либо образом.
Все растворители, применяемые в настоящем изобретении, являются коммерчески доступными и могут применяться без дополнительной очистки.
Растворитель, применяемый в настоящем изобретении, можно приобрести в продаже. В настоящем изобретении используются следующие сокращения: DCM - дихлорметан; ДМФА - N,N-диметилформамид; ДМСО - диметилсульфоксид; EtOH - этанол; МеОН - метанол; TFA - трифторуксусная кислота; TsOH - п-толуолсульфоновая кислота; mp - температура плавления; EtSO3H - этансульфоновая кислота; MeSO3H - метансульфоновая кислота; АТФ - аденозинтрифосфат; HEPES - 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота; ЭДТА - этилендиаминтетрауксусная кислота; MgCl2 - дихлорид магния; MnCl2 - дихлорид марганца; ДТТ - дитиотреитол.
Технические результаты изобретения
Соединение формулы (I), описанное в настоящей заявке, обладает хорошей стабильностью кристаллической формы и является простым в применении для получения лекарственных препаратов; кристаллические формы настоящего изобретения демонстрируют превосходную эффективность в отношении BRD4, имеют лучшие фармакокинетические свойства и высокую скорость перорального всасывания, обладают такими характеристиками, как высокая активность, хорошая метаболическая стабильность, хорошая растворимость, пригодность для перорального приема и т.п. и могут обеспечить более эффективное лечение заболеваний, вызванных аномальной экспрессией BRD4.
1.1 Порошковая рентгеновская дифракция (порошковый рентгеновский дифрактометр, XRPD)
Модель прибора: рентгеновский дифрактометр Bruker D8 ADVANCE
Способ проведения испытания: приблизительно 10-20 мг образца брали для определения дифракции XRPD.
Подробное описание параметров дифракции XRPD приведено ниже:
Рентгеновский генератор: Cu, kα, 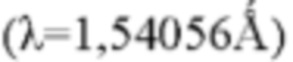
Напряжение в трубке: 40 кВ, ток в трубке: 40 мА
Эмиссионная щель: 1 град
Щель, ограничивающая высоту: 10 мм
Щель рассеивания: 1 град
Приемная щель: 0,15 мм
Монохроматор: стационарный монохроматор
Диапазон сканирования: для кристаллической формы А: 4-33 град; и для кристаллической формы В: 4-35 град
Скорость сканирования: 10 град/мин
1.2 Дифференциальная сканирующая калориметрия (дифференциальный сканирующий калориметр, ДСК)
Модель прибора: дифференциальный сканирующий калориметр ТА Q2000
Способ проведения испытания: образец (0,5-1 мг) помещали в алюминиевую ячейку ДСК для испытания и образец нагревали от 30°С до 300°С в условиях подачи 50 мл/мин N2 при скорости нагревания 10°С/мин.
1.3 Термогравиметрический анализ (термогравиметрический анализатор, ТГА)
Модель прибора: термогравиметрический анализатор ТА Q5000IR
Способ проведения испытания: образец (2-5 мг) помещали в платиновый тигель ТГА для испытания и образец нагревали от комнатной температуры до температуры, при которой потери массы составляли 20% в условиях подачи 25 мл/мин N2 при скорости нагревания 10°С/мин.
1.4 Анализ динамической сорбции паров (ДСП) в настоящей заявке
Модель прибора: Анализатор динамической сорбции паров SMS DVS Advantage
Условия проведения испытания: образец (10-20 мг) помещали в лотки для образцов ДСП для испытания.
Подробное описание параметров ДСП:
Температура: 25°С
Балансировка: dm/dt=0,01%/мин (быстрая: 10 мин, продолжительная: 180 мин)
Сушка: сушка в течение 120 мин при относительной влажности (RH) 0%.
Шаг RH (%) в испытании: 10%
Диапазон шага RH (%) в испытании: 0% - 90% - 0%
Оценка гигроскопичности образцов была проведена следующим образом:

Описание чертежей
Фиг. 1 представляет собой дифрактограмму XRPD излучения Cu-Kα кристаллической формы А соединения формулы (I).
Фиг. 2 представляет собой кривую ДСК кристаллической формы А соединения формулы (I).
Фиг. 3 представляет собой термограмму ТГА кристаллической формы А соединения формулы (I).
Фиг. 4 представляет собой дифрактограмму XRPD излучения Cu-Kα кристаллической формы В соединения формулы (I).
Фиг. 5 представляет собой изотерму ДСП кристаллической формы А соединения формулы (I).
Частные варианты реализации изобретения
Для более полного понимания настоящего изобретения ниже приведены конкретные примеры, которые являются исключительно иллюстративными и никак не ограничивают настоящее изобретение каким-либо образом.
Пример 1: Получение соединения формулы (I)
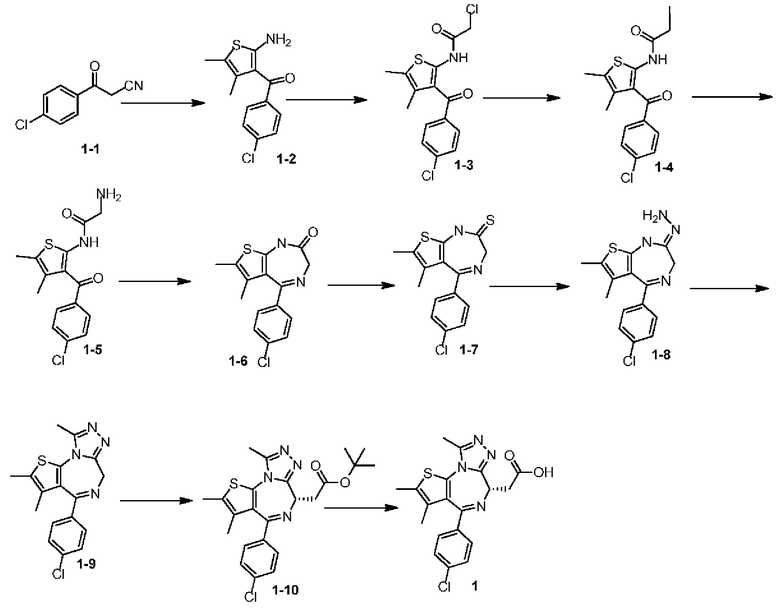
Этап 1:
Соединение 1-1 (25,00 г, 139,20 ммоль, 1,00 экв.), 2-бутанон (11,04 г, 153,12 ммоль, 13,63 мл, 1,10 экв.) и морфолин (12,13 г, 139,20 ммоль, 12,25 мл, 1,00 экв.) растворяли в этаноле (200,00 мл), затем добавляли сублимированную серу (4,46 г, 139,20 ммоль, 1,00 экв.). Суспензию нагревали до 70°С и перемешивали в течение 12 часов в защитной атмосфере азота. Реакционную смесь сначала выпаривали при пониженном давлении с получением желтого масла, к которому добавляли воду (500 мл) и экстрагировали этилацетатом (200 мл×4). Объединенные органические фазы собирали, промывали насыщенным солевым раствором (200 мл), сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении. Полученное неочищенное вещество очищали на колонке с силикагелем (петролейный эфир/этилацетат=10/1) с получением соединения 1-2. 1Н ЯМР (400 МГц, CDCl3) δ ppm 7,47 (d, J=8,0 Гц, 2H), 7,38 (d, J=8,0 Гц, 2H), 6,43 (br s, 2H), 2,13 (s, 3Н), 1,56 (s, 3Н).
Этап 2:
Соединение 1-2 (10,00 г, 37,63 ммоль, 1,00 экв.) растворяли в хлороформе (100,00 мл) и по каплям добавляли 2-хлорацетилхлорид (6,37 г, 56,45 ммоль, 4,49 мл, 1,50 экв.), затем реакционную смесь перемешивали при 70°С в течение 1 ч. Реакционную смесь промывали насыщенным раствором бикарбоната натрия (100 мл) и насыщенным солевым раствором (50 мл), затем сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении. Полученное неочищенное вещество перекристаллизовывали из метанола (40 мл) с получением соединения 1-3. 1H ЯМР (400 МГц, CDCl3) δ ppm 11,81 (br s, 1H), 7,58 (dd, J=2,0, 6,4 Гц, 2H), 7,45 (dd, J=2,2, 8,6 Гц, 2H), 4,25 (s, 2H), 2,29 (s, 3H), 1,72 (s, 3H).
Этап 3:
Соединение 1-3 (11,00 г, 32,14 ммоль, 1,00 экв.) и йодид натрия (9,63 г, 64,28 ммоль, 2,00 экв.) добавляли в тетрагидрофуран (50,00 мл) и смесь перемешивали при 60°С в течение 2 ч. Реакционную смесь выпаривали при пониженном давлении с получением соединения 1-4, которое использовали на следующем этапе непосредственно без очистки. Жидкостная хроматография-масс-спектрометрия (ЖХМС) с ионизацией распылением в электрическом поле (ESI) m/z: 433,9 (М+1).
Этап 4:
Соединение 1-4 (14,00 г, 32,28 ммоль, 1,00 экв.) растворяли в тетрагидрофуране (100,00 мл), охлаждали до -60°С и насыщали газообразным аммиаком в течение 30 минут. Реакционную смесь медленно нагревали до 20°С и перемешивали в течение 3 часов. Реакционную смесь выпаривали при пониженном давлении. Полученное твердое вещество растворяли в этилацетате (150 мл), промывали водой (50 мл×3) и насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и затем выпаривали при пониженном давлении с получением соединения 1-5, которое использовали на следующем этапе непосредственно без очистки. ЖХМС (ESI) m/z: 322,9 (М+1), 344,9 (M+Na).
Этап 5:
Соединение 1-5 (10,00 г, 30,98 ммоль, 1,00 экв.) растворяли в изопропаноле (150,00 мл) и ледяной уксусной кислоте (50,00 мл) и перемешивали при 90°С в течение 3 ч. Растворитель удаляли из реакционного раствора при пониженном давлении. Оставшуюся смесь растворяли в хлороформе (20 мл), промывали насыщенным раствором бикарбоната натрия (20 мл) и насыщенным солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и затем выпаривали при пониженном давлении. Полученный неочищенный продукт перекристаллизовывали из этилацетата (50 мл) с получением соединения 1-6. 1H ЯМР (400 МГц, CDCl3) δ ppm 8,98 (br s, 1H), 7,46 (d, J=8,4 Гц, 2H), 7,35 (d, J=8,4 Гц, 2H), 4,80 (d, J=8,8 Гц, 1H), 3,93 (d, J=8,6 Гц, 1H), 2,28 (s, 3Н), 1,59 (s, 3Н).
Этап 6:
Пентасульфид фосфора (17,07 г, 76,79 ммоль, 8,17 мл, 3,60 экв.) добавляли к непрерывно перемешиваемой суспензии карбоната натрия (4,07 г, 38,39 ммоль, 1,80 экв.) в 1,2-дихлорэтане (200,00 мл), перемешивали при 20°С в течение 1 часа, затем добавляли соединение 1-6 (6,50 г, 21,33 ммоль, 1,00 экв.). Полученную суспензию выдерживали при 65°С в течение 5 часов. Реакционную смесь охлаждали до 20°С и фильтровали, и осадок после фильтрации растворяли в этилацетате (2 л), промывали насыщенным солевым раствором (500 мл), сушили над сульфатом натрия, фильтровали и затем выпаривали при пониженном давлении. Полученное неочищенное вещество очищали на колонке с силикагелем (петролейный эфир/этилацетат=5/1) с получением соединения 1-7.
Этап 7:
К суспензии соединения 1-7 (3,50 г, 10,91 ммоль, 1,00 экв.) в метаноле (5,00 мл) добавляли гидразин гидрат (1,67 г, 32,72 ммоль, 1,62 мл, 98% чистоты, 3,00 экв.) при 0°С и реакционную смесь перемешивали при 0°С в течение 1 ч. Реакционную смесь фильтровали и осадок после фильтрации сушили в печи с получением соединения 1-8, которое использовали на следующем этапе напрямую. ЖХМС (ESI) m/z: 318,9 (М+1).
Этап 8:
К смеси соединения 1-8 (2,50 г, 7,84 ммоль, 1,00 экв.) в толуоле (100,00 мл) добавляли триэтил ортоацетат (3,82 г, 23,52 ммоль, 4,29 мл, 3,00 экв.). Реакционную смесь перемешивали при 80°С в течение 1 ч. Реакционную смесь выпаривали при пониженном давлении и полученное неочищенное вещество перекристаллизовывали из этилацетата (10 мл) с получением соединения 1-9. ЖХМС (ESI) m/z: 344,9 (М+1).
Этап 9:
К раствору соединения 1-9 (1,50 г, 4,38 ммоль, 1,00 экв.) в тетрагидрофуране (180 мл) добавляли LiHMDS (1М, 8,76 мл, 2,00 экв.) по каплям при 70°С. Реакционную смесь перемешивали при этой температуре в течение 1 часа, затем по каплям добавляли раствор трет-бутил 2-бромацетата (1,28 г, 6,57 ммоль, 970,82 мкл, 1,50 экв.) в тетрагидрофуране (20 мл). После добавления реакционную смесь медленно нагревали до 20°С и перемешивали в течение 5 часов. Реакционную смесь гасили насыщенным раствором NH4Cl (50 мл), экстрагировали этилацетатом (100 мл), промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и затем выпаривали при пониженном давлении. Полученное неочищенное вещество очищали с помощью колоночной флэш-хроматографии и полученное соединение разделяли с помощью сверхкритической флюидной хроматографии (СФК) с получением соединения 1-10 (основный-EtOH, колонка: AS (250 мм×30 мм, 5 мкм), подвижная фаза В: 30%, скорость потока (мл/мин): 55) ([α]25D +54 (С 0,6, CHCl3)). ЖХМС (ESI) m/z: 457,0 (М+1).
Этап 10:
Соединение 1-10 (150,00 мг, 328,23 мкмоль, 1,00 экв.) растворяли в дихлорметане (5,00 мл) и трифторуксусной кислоте (1,00 мл) и реакционную смесь перемешивали в течение 4 ч при 20°С. Реакционную смесь выпаривали при пониженном давлении с получением соединения 1, которое использовали на следующем этапе непосредственно. ЖХМС (ESI) m/z: 401,0 (М+1).

Этап 11:
Соединение 2 (0,78 г, 3,66 ммоль, 1 экв.), трет-бутилкарбамат (643,39 мг, 5,49 ммоль, 1,5 экв.), трис(дибензилиденацетон)дипалладий (335,29 мг, 366,15 мкмоль, 0,1 экв.), карбонат цезия (2,39 г, 7,32 ммоль, 2 экв.) и 4,5-бис(дифенилфосфино)-9,9-диметилксантен (211,86 мг, 366,15 мкмоль, 0,1 экв.) добавляли в 1,4-диоксан (10 мл) и выдерживали при 100°С в течение 12 часов в защитной атмосфере азота. К реакционной смеси добавляли воду (20 мл), добавляли этилацетат (20 мл), нерастворимое вещество отделяли фильтрованием, водную фазу экстрагировали этилацетатом (10 мл), объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении. Очистку проводили с использованием колонки для флэш-хроматографии с получением соединения 3. ЖХМС (ESI) m/z: 250,1 (М+1).
Этап 12:
Трифторуксусную кислоту (3,85 г, 33,77 ммоль, 2,5 мл, 18,30 экв.) добавляли к соединению 3 (0,46 г, 1,85 ммоль, 1 экв.) в безводном дихлорметане (20 мл) и выдерживали при 20°С в течение 12 часов после добавления. Реакционную смесь промывали водой (20 мл), рН водной фазы доводили до 7 насыщенным раствором бикарбоната натрия, водную фазу экстрагировали дихлорметаном (10 мл×2), объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении. Получали соединение 4, которое использовали на следующем этапе без дополнительной очистки. ЖХМС (ESI) m/z: 149,8 (М+1).
Этап 13:
POCl3 (76,50 мг, 498,90 мкмоль, 46,36 мкл, 2 экв.) добавляли к раствору соединения 1 (100 мг, 249,45 мкмоль, 1 экв.) и соединения 4 (44,65 мг, 299,34 мкмоль, 1,2 экв.) в пиридине (2 мл) при 0°С, после добавления температуру повышали до 20°С в течение 1,5 часов. Реакционную смесь гасили добавлением воды (3 мл) и рН водной фазы доводили до 7 с помощью 2н соляной кислоты. Водную фазу экстрагировали дихлорметаном (5 мл×3), органическую фазу сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении. Очистку проводили методом тонкослойной хроматографии (дихлорметан/метанол=10/1) с получением соединения формулы (I) в виде стеклянной пасты или пены, прилипшей к стенке флакона. ЖХМС (ESI) m/z: 532,1 (М+1). 1H ЯМР (400 МГц, CDCl3) δ ppm 9,98 (br s, 1H), 7,94 (d, J=6,8 Гц, 1H), 7,67 (d, J=8,0 Гц, 1H), 7,32-7,36 (m, 3H), 7,24-7,27 (m, 2H), 5,08-5,16 (m, 2H), 4,57-4,61 (m, 1H), 3,78-3,84 (m, 1H), 3,45-3,50 (m, 1H), 2,63 (s, 3H), 2,36 (s, 3H), 1,63 (s, 3H).
Пример 2: Получение кристаллической формы А соединения формулы (I)
Около 50 мг соединения формулы (I) взвешивали и добавляли в стеклянные флаконы объемом 1,5 мл, соответственно, и добавляли соответствующее количество растворителей (см. таблицу 3) до образования суспензии, которую запечатывали герметизирующей пленкой и перемешивали в термомиксере с поддержанием постоянной температуры при 40°С в течение 48 часов. Затем образцы центрифугировали в центрифуге и центрифугированное твердое вещество помещали в вакуумную печь на ночь для высушивания при 30°С с получением кристаллической формы А соединения формулы (I).

Пример 3: Получение кристаллической формы В соединения формулы (I)
Около 50 мг соединения формулы (I) взвешивали и добавляли в стеклянный флакон объемом 1,5 мл и добавляли тетрагидрофуран (0,2 мл) до образования суспензии, которую запечатывали герметизирующей пленкой. Смесь перемешивали в термомиксере с поддержанием постоянной температуры при 40°С в течение 48 часов. Затем образцы центрифугировали в центрифуге и центрифугированное твердое вещество помещали в вакуумную печь на ночь для высушивания при 30°С с получением кристаллической формы В соединения формулы (I).
Экспериментальный пример 1: Испытание на стабильность в твердом состоянии кристаллической формы А соединения формулы (I)
В соответствии с руководством по испытаниям на стабильность активных фармацевтических ингредиентов (АФИ) и составов (The general guidelines of the Chinese Pharmacopoeia 2015 edition volume IV 9001), стабильность кристаллической формы A соединения формулы (I) исследовали в режиме ускоренных (40°С/75% RH, герметично) и длительных (25°С/60% RH, герметично) испытаний.
1,5 г кристаллической формы А соединения формулы (I) взвешивали, помещали на дно стеклянной бутыли для образцов и распределяли тонким слоем, соответственно, при этом образцы для испытаний в условиях 25°C/60%RH и 40°C/75%RH упаковывали в двухслойные пакеты из полиэтилена низкой плотности (ПЭНП). Каждый слой пакетов из ПЭНП запечатывали отдельно и затем пакет из ПЭНП помещали в пакет из алюминиевой фольги, и запечатывали. Образцы, находившиеся в разных условиях, отбирали и исследовали на 90-й день, результат испытания сравнивали с первоначальным результатом испытания на 0-й день, и результаты испытаний представлены в следующей таблице 4:
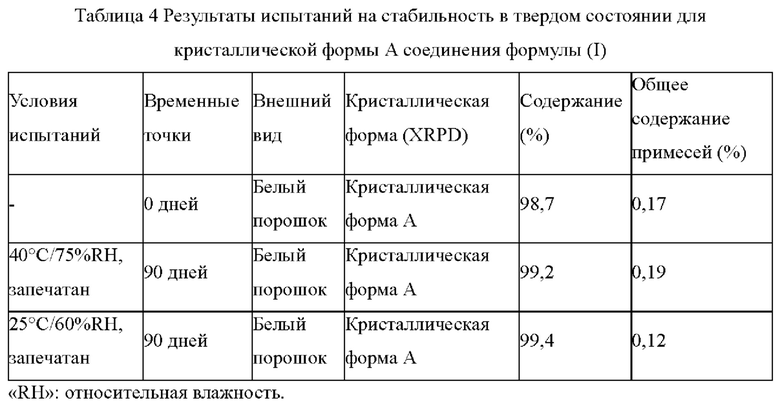
Заключение: кристаллическая форма А соединения формулы (I) обладает хорошей стабильностью.
Экспериментальный пример 2: Исследование гигроскопичности кристаллической формы А соединения формулы (I)
Материалы для эксперимента:
Динамический анализатор сорбции паров SMS DVS Advantage
Способ проведения эксперимента:
10-15 мг кристаллической формы А соединения формулы (I) помещали на лоток для образцов ДСП для проведения испытания.
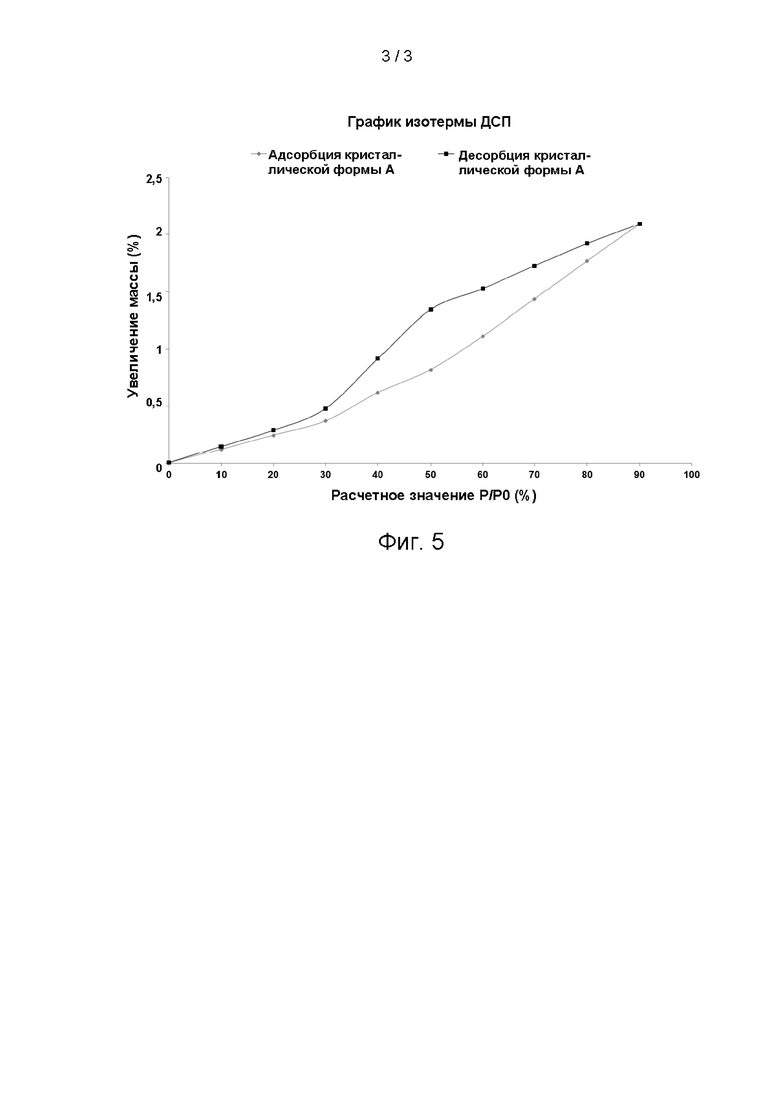
Результаты эксперимента:
Изотерма ДСП кристаллической формы А соединения формулы (I) показана на фиг. 5, ΔW=1,789%.
Заключение эксперимента:
Кристаллическая форма А соединения формулы (I) характеризуется увеличением массы за счет сорбции влаги на 1,789% при 25°С и 80% RH и, таким образом, является мало гигроскопичной.
Пример 3: Анализ биохимической активности BRD4
Подготовка эксперимента:
1) для экспериментов брали белки BRD4-BD1 и BRD4-BD2 компании BPS; а также полипептиды компании ANASPEC; проявляющие реагенты компании Perkinelmer;
2) скрининг соединений проводили с применением экспериментального механизма TR-FRET;
3) проводили испытания соединений.
Этапы эксперимента представлены ниже:
1) Подготовка планшетов для соединений:
Подготовка планшетов для соединений в эксперименте осуществляли с помощью Эхо-метода:
Соединение разбавляли с помощью Эхо-метода до 10 концентраций при 3-кратном уменьшении каждой последующей концентрации: 20000, 6666,67, 2222,22, 740,74, 246,91, 82,305, 27,435, 9,145, 3,048, 1,016 нМ.
2) Подготовка реагентов для реакции:
Необходимые реагенты готовили в день проведения эксперимента:
a) подготовка 1× аналитического буфера (буферный раствор для испытания);
b) подготовка 3× раствора компонентов для эксперимента:
1. Реагенты помещали на лед для самопроизвольного расплавления с целью последующего использования;
2. 1× аналитического буфера (буферный раствор для испытания) брали для приготовления «раствора А» (раствор белка), «раствора В» (раствор полипептида) и «раствора С» (раствор реагента для анализа) для экспериментов, чтобы компоненты в реакционной системе образовали 3× растворы, и количество растворов А, В, С было достаточным для проведения необходимого количества экспериментов.
3) Этапы проведения эксперимента представлены ниже:
Аналитические планшеты - это планшеты, содержащие градиентные концентрации соединений и соответствующие растворы ДМСО, подготовленные перед экспериментом с помощью Эхо-метода:
a) вынимали аналитический планшет, добавляли 5 мкл/лунка «раствора А» (раствор белка) в колонки 2-23 аналитического планшета, а затем добавляли 5 мкл/лунка 1× буфера для анализа в колонки 1 и 24 аналитического планшета в качестве Min контроля в экспериментальной системе;
b) центрифугировали при 1000 об/мин в течение 30 секунд;
c) инкубировали планшет при 23°С в течение 20 минут;
d) добавляли 5 мкл/лунка «раствора В» (раствор полипептида) в колонки 1-24 аналитического планшета после 20 минут инкубации;
e) центрифугировали при 1000 об/мин в течение 30 секунд;
f) инкубировали планшет при 23°С в течение 20 минут;
g) добавляли 5 мкл/лунка «раствора С» (раствор реагента для анализа) в колонки 1-24 аналитического планшета после 20 минут инкубации;
h) центрифугировали при 1000 об/мин в течение 30 секунд;
i) инкубировали планшет при 23°С в течение 40 минут;
j) считывали планшеты на планшетном анализаторе EnVision.
4) Анализ данных:
a) рассчитывали значение Z' для каждого аналитического планшета с использованием соответствующих Мах контроля (максимальный контроль) и Min контроля (минимальный контроль) для каждого аналитического планшета и следили за тем, чтобы значение Z' для каждого аналитического планшета было больше 0,5;
b) рассчитывали значение IC50 по сигналу исследуемых соединений с помощью XLFIT5 и следили за тем, чтобы оно оставалось в пределах 3-кратного среднего значения согласно литературным данным, результаты представлены в таблице 5.

5) Заключение:
Соединение формулы (I) оказывает значительное ингибирующее действие на BRD4-BD1 и BRD4-BD2.
Пример 4: Исследование фармакодинамики соединения формулы (I) in vivo в раковых клетках MDA-MB-231_luc подкожной ксенотрансплантационной опухолевой модели рака молочной железы человека
1. Планирование эксперимента

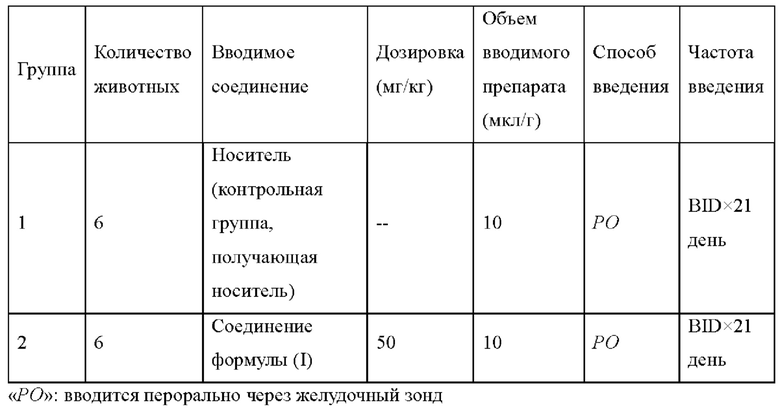
2. Материалы для эксперимента
2.1 Животные для эксперимента
Вид: мышь
Порода: голая мышь BALB/c
Возраст в неделях и масса: возраст 6-8 недель, масса тела 18-22 грамма
Пол: самка
Поставщик: Shanghai SIPPR-Bk Laboratory Animal Co., Ltd.
3. Порядок и этапы эксперимента
3.1 Культура клеток
Раковые клетки молочной железы человека MDA-MB-231_luc культивировали в монослое in vitro. Условия культивирования: культуральная среда RPMI-1640 (поставщик: Gibco; артикул: 22400-089; номер производственной партии: 4868546) с 10% фетальной бычьей сывороткой, 100 ед/мл пенициллина и 100 мкг/мл стрептомицина. Культуру выращивали при 37°С в 5% СО2. Традиционную обработку желудочно-кишечного тракта смесью панреатин-ЭДТА проводили два раза в неделю для лучшего усвоения препарата. Когда клетки достигали фазы экспоненциального роста, клетки собирали, считали и инокулировали.
3.2 Инокуляция опухолевых клеток
0,2 мл 10×106 клеток MDA-MB-231_luc подкожно инокулировали в правую часть спины каждой голой мыши (PBS:Матригель=1:1). Разделение на группы и введение препарата начинали, когда средний объем опухоли достигал 100-150 мм3.
3.3 Измерение опухоли и показатели эксперимента
Показатели эксперимента позволяли выяснить, был ли рост опухоли подавлен, замедлен или опухоль была вылечена. Диаметр опухоли измеряли дважды в неделю с помощью штангенциркуля. Для расчета объема опухоли использовали следующее уравнение: V=0,5а×b2, где а и b представляли собой большой и малый диаметры опухоли, соответственно.
Эффект ингибирования роста опухоли от применения соединения оценивали по TGI (%) или относительной скорости пролиферации опухоли Т/С (%). TGI (%) отражает скорость ингибирования роста опухоли. Расчет TGI (%) производили следующим образом: TGI (%)=[(1-(средний объем опухоли в конце цикла введения препарата в группе, получавшей лечение-средний объем опухоли в начале цикла введения препарата в этой группе, получавшей лечение))/(средний объем опухоли в конце лечения в контрольной группе, получавшей носитель-средний объем опухоли в начале лечения в контрольной группе, получавшей носитель)]×100%.
Относительную скорость пролиферации опухоли Т/С (%) рассчитывали в соответствии с приведенным далее уравнением: Т/С %=Trtv/Crtv×100% (Trtv: RTV группы, получавшей лечение; Crtv: RTV группы отрицательного контроля). Относительный объем опухоли (RTV) рассчитывали в соответствии с результатами измерения опухоли. Уравнение расчета выглядело следующим образом RTV=Vt/V0, где V0 - средний объем опухоли, измеренный при распределении по группам и введении препарата (т.е. d0) и Vt - средний объем опухоли на момент измерения. Trtv и Crtv получали на основе данных, собранных в тот же день.
В конце эксперимента измеряли массу опухоли и рассчитывали процент Т/Смассы. Тмассы и Смассы представляли собой массу опухоли в группе, получавшей препарат, и контрольной группе, получавшей носитель, соответственно.
3.4 Статистический анализ
Статистический анализ включал определение среднего значения и стандартной ошибки среднего (SEM) объема опухоли каждой группы в каждой временной точке. Группа, получавшая препарат, показала наилучший результат от лечения на 21-й день после введения препарата в конце эксперимента, поэтому статистический анализ проводили на основе этих данных для оценки различий между группами. Для сравнения двух групп проводили Т-тест, а сравнение трех и более групп проводили с помощью одностороннего ANOVA. Если значение F значительно отличалось, применяли тест Геймса-Хоуэлла. Если значение F не имело существенных отличий, для проведения анализа применяли тест Даннета (2-сторонний). Полный анализ данных проводили с помощью программного обеспечения SPSS 17,0. р<0,05 считали значительным расхождением.
4. Заключение эксперимента
На 21-й день после введения препарата, для соединения формулы (I), скорость ингибирования роста опухоли составила TGI=54,85%, Т/С=52,99%, р<0,05; не было значительного изменения массы тела животных, и они хорошо переносили препарат.
Пример 5: Исследование фармакодинамики соединения формулы (I) in vivo в раковых клетках РС-3 подкожной ксенотрансплантационной опухолевой модели рака предстательной железы человека
1. Планирование эксперимента
Способ получения исследуемого вещества был таким же, как представлено в таблице 6, и распределение животных по группам и режим дозирования были такими же, как представлено в таблице 7.
2. Материалы для эксперимента
2.1 Животные для эксперимента
Вид: мышь
Порода: голая мышь BALB/c
Возраст в неделях и масса: возраст 6-8 недель, масса тела 18-22 грамма
Пол: самец
Поставщик: Shanghai SIPPR-Bk Laboratory Animal Co., Ltd.
3. Порядок и этапы эксперимента
3.1 Культура клеток
Раковые клетки РС-3 предстательной железы человека культивировали в монослое in vitro. Условия культивирования: культуральная среда F-12K (поставщик: Gibco; артикул: 21127-022; номер производственной партии: 1868870) с 10% фетальной бычьей сывороткой, 100 ед/мл пенициллина и 100 мкг/мл стрептомицина. Культуру выращивали при 37°С в 5% СО2. Традиционную обработку желудочно-кишечного тракта смесью панреатин-ЭДТА проводили два раза в неделю для лучшего усвоения препарата. Когда клетки достигали фазы экспоненциального роста, клетки собирали, считали и инокулировали.
3.2 Инокуляция опухолевых клеток
0,1 мл 10×106 клеток РС-3 подкожно инокулировали в правую часть спины каждой лысой мыши. Разделение на группы и введение препарата начинали, когда средний объем опухоли достигал 100-150 мм3.
3.3 Измерение опухоли, показатели эксперимента и статистический анализ были такими же, как представлено в модели MDA-MB-231.
4. Заключение эксперимента
На 21-й день после введения препарата, по сравнению с контрольной группой, получавшей носитель, исследуемое соединение формулы (I) обладало значительным ингибирующим действием на опухоль (Т/С=44,63%, TGI=58,4%, p=0,033); животные хорошо переносили препарат.
Пример 6: Противоопухолевое действие соединения формулы (I) in vivo в раковых клетках МС38 толстой кишки мыши в модели трансплантации опухоли животным.
1. Планирование эксперимента


2. Материал для эксперимента
2.1 Животные для эксперимента
Вид: мышь
Порода: мышь C57BL6
Возраст в неделях и масса: возраст 6-7 недель, масса тела 16-20 грамм
Пол: самка
Поставщик: Shanghai SLAC Laboratory Animal Co., Ltd.
3. Порядок и этапы эксперимента
3.1 Культура клеток
Раковые клетки толстой кишки мыши МС38 (OBiO Technology (Shanghai) Corp., Ltd.) культивировали в монослое in vitro. Условия культивирования: культуральная среда DMEM (Gibco; артикул: 12100) с 10% фетальной бычьей сывороткой. Культуру выращивали при 37°С в 5% CO2 в инкубаторе. Традиционную обработку желудочно-кишечного тракта 0,25% смесью панреатин-ЭДТА проводили два раза в неделю для лучшего усвоения препарата. Когда клетки достигали фазы экспоненциального роста, и плотность составляла 80%-90%, клетки собирали, считали и инокулировали.
3.2 Инокуляция опухолевых клеток
0,1 мл 2×105 клеток МС38 подкожно инокулировали в правую часть спины каждой мыши. Случайное разделение на группы и введение препарата проводили в соответствии с объемом опухоли, когда средний объем опухоли достигал около 70 мм3.
3.3 Измерение опухоли
Диаметр опухоли измеряли два раза в неделю с помощью штангенциркуля. Для расчета объема опухоли использовали следующее уравнение: V=0,5×а×b2, где а и b представляли собой большой и малый диаметры опухоли, соответственно.
Эффект ингибирования роста опухоли от применения соединения оценивали по TGI (%) или относительной скорости пролиферации опухоли Т/С (%). Относительная скорость пролиферации опухоли Т/С (%)=Trtv/Crtv×100% (Trtv: RTV группы, получавшей лечение; Crtv: RTV группы отрицательного контроля). Относительный объем опухоли (RTV) рассчитывали в соответствии с результатами измерения опухоли. Уравнение для расчета: RTV=Vt/V0, где V0 - средний объем опухоли, измеренный при разделении на группы и введении препарата (т.е. D0), и Vt - средний объем опухоли на момент измерения. Trtv и Crtv получали на основе данных, собранных в тот же день.
TGI (%) отражает скорость ингибирования роста опухоли. TGI (%)=[(1-(средний объем опухоли в конце цикла введения препарата в группе, получавшей лечение-средний объем опухоли в начале цикла введения препарата в этой группе, получавшей лечение))/(средний объем опухоли в конце лечения в контрольной группе, получавшей носитель-средний объем опухоли в начале лечения в контрольной группе, получавшей носитель)]×100%.
В конце эксперимента измеряли массу опухоли и рассчитывали процент Тмассы/Смассы. Тмассы и Смассы представляли собой массу опухоли в группе, получавшей препарат, и контрольной группе, получавшей носитель, соответственно.
3.4 Статистический анализ
Статистический анализ проводили с помощью программного обеспечения SPSS на основании объема опухоли и массы опухоли в конце эксперимента. Для сравнения двух групп проводили Т-тест, а сравнение трех и более групп проводили с помощью одностороннего ANOVA. Если дисперсия была однородной (значение F существенно не отличалось), для анализа использовали метод LSD. Если дисперсия не была однородной (значение F значительно отличалось), для анализа использовали метод Геймса-Хоуэлла. p<0,05 считали значительным расхождением.
4. Заключение эксперимента
На 20-й день после введения исследуемого соединения формулы (I), для группы введения препарата в дозировке 15 мг/кг: относительная скорость пролиферации опухоли составила Т/С=33,68%, скорость ингибирования роста опухоли составила TGI=68,81%, р<0,0001; для группы введения препарата в дозировке 25 мг/кг: относительная скорость пролиферации опухоли составила Т/С=27,59%, TGI=75,21%, р<0,0001; и для группы введения препарата в дозировке 50 мг/кг: Т/С=10,04%, TGI=93,46%, р<0,0001. Значительный эффект ингибирования опухоли наблюдали в каждой группе животных с хорошей переносимостью, которым вводили препарат.
Пример 7 Фармакокинетическое исследование in vivo соединения формулы (I) на мышах
В качестве подопытных животных брали самок мышей Balb/c. Соединение формулы (I) вводили мышам внутривенно и внутрижелудочно, затем методом жидкостной хроматографии с тандемной масс-спектрометрией (ЖХ-МС/МС) определяли концентрацию лекарственного вещества в плазме крови в разные временные точки. Изучали фармакокинетическое действие соединения формулы (I) in vivo на мышах и оценивали его фармакокинетические характеристики.
1. Описание эксперимента
1.1 Экспериментальное лекарственное вещество: Соединение формулы (I)
1.2 Животные для эксперимента: Шестнадцать здоровых взрослых самок мышей Balb/c разделяли на четыре группы по массе тела по четыре мыши в каждой группе. Животных приобретали у Shanghai Lingchang BioTech Co., Ltd. из Shanghai SLAC Laboratory Animal Co., Ltd., лицензия на выращивание животных № SCXK (Shanghai) 2013-0018.
1.3 Приготовление лекарственного вещества
Брали соответствующее количество образца, добавляли 5% конечного объема ДМСО и затем 95% конечного объема 20% HP-β-CD. Смесь перемешивали с помощью ультразвука до получения прозрачного раствора концентрацией 0,5 мг/мл. После фильтрации ее применяли для внутривенного введения.
Брали соответствующее количество образца и растворяли в 0,5% растворе натрий карбоксиметилцеллюлозы. Смесь перемешивали с помощью ультразвука до получения гомогенной суспензии концентрацией 0,5 мг/мл, которую использовали для внутрижелудочного введения.
1.4 Введение препарата
Восемь самок мышей Balb/c разделили на две группы. После ночного голодания первой группе вводили препарат внутривенно в объеме 2,5 мл/кг и дозировке 1 мг/кг. Второй группе вводили препарат внутрижелудочно в объеме 5 мл/кг и дозировке 3 мг/кг.
2. Эксперимент
После внутривенного введения препарата у самок мышей Balb/c брали 30 мкл крови в каждой временной точке: 0,0833, 0,25, 0,5, 1, 2, 4, 8 и 24 часа, и помещали в пробирки, содержащие 2 мкл ЭДТА К2; после внутрижелудочного введения препарата у самок мышей Balb/c брали 30 мкл крови в каждой временной точке: 0,0833, 0,25, 0,5, 1, 2, 4, 8 и 24 часа, и помещали в пробирки, содержащие 2 мкл ЭДТА К2. Пробирку центрифугировали при 3000 g в течение 15 минут для отделения плазмы и отделенную плазму хранили при -60°С. Животных разрешали кормить через 2 часа после введения препарата.
Метод ЖХ-МС/МС применяли для измерения содержания исследуемого соединения в плазме крови после внутривенного и внутрижелудочного введения препарата мышам. Линейный диапазон метода составлял 2,00-6000 нмоль/л; образцы плазмы анализировали после осаждения белка обработкой ацетонитрилом. Результаты исследования фармакокинетических параметров представлены в таблице 8.


Заключение эксперимента: соединение формулы (I) обладает сильным действием при пероральном введении, низкой скоростью выведения лекарственного вещества и высокой биодоступностью при пероральном введении.
Пример 8 Фармакокинетическое исследование соединения формулы (I) in vivo на крысах
В качестве подопытных животных брали самцов крыс SD. Соединение формулы (I) вводили крысам внутривенно и внутрижелудочно и затем методом ЖХ-МС/МС определяли концентрацию лекарственного вещества в плазме крови в разные временные точки. Изучали фармакокинетическое действие соединения формулы (I) in vivo на крысах и оценивали его фармакокинетические характеристики.
1. Описание эксперимента
1.1 Экспериментальное лекарственное вещество: Соединение формулы (I) (кристаллическая форма А)
1.2 Животные для эксперимента: 4 здоровых взрослых самцов крыс SD разделяли на 2 группы по массе по 2 крысы в каждой группе. Животных приобретали у Beijing Weitonglihua Laboratory Animals Ltd., лицензия на выращивание животных № SCXK (Beijing) 2016-0006.
1.3 Приготовление лекарственного вещества
Брали соответствующее количество образца, добавляли 5% конечного объема ДМСО и затем 95% конечного объема 20% HP-β-CD. Смесь перемешивали с помощью ультразвука до получения прозрачного раствора концентрацией 0,5 мг/мл. После фильтрации его использовали для внутривенного введения.
Брали соответствующее количество образца и растворяли в 0,5% растворе натрий карбоксиметилцеллюлозы. Смесь перемешивали с помощью ультразвука до получения гомогенной суспензии концентрацией 1 мг/мл, которую использовали для внутрижелудочного введения.
1.4 Введение препарата
Четырех самцов крыс SD разделяли на две группы. После ночного голодания первой группе вводили препарат внутривенно в объеме 4 мл/кг и дозировке 2 мг/кг. Второй группе вводили препарат внутрижелудочно в объеме 10 мл/кг и дозировке 10 мг/кг.
2. Эксперимент
После внутривенного введения препарата у самцов крыс SD брали 100 мкл крови в каждой временной точке: 0,0833, 0,25, 0,5, 1, 2, 4, 6, 8 и 24 часа, и помещали в пробирки, содержащие 2 мкл ЭДТА К2; после внутрижелудочного введения препарата у крыс SD брали 100 мкл крови в каждой временной точке: 0,0833, 0,25, 0,5, 1, 2, 4, 6, 8 и 24 часа, и помещали в пробирки, содержащие 2 мкл ЭДТА К2. Пробирку центрифугировали при 3000 g в течение 15 минут для отделения плазмы и отделенную плазму хранили при -60°С. Животных разрешали кормить через 2 часа после введения препарата.
Метод ЖХ-МС/МС применяли для измерения содержания исследуемого соединения в плазме крови после внутривенного и внутрижелудочного введения препарата крысам. Линейный диапазон метода составил 2,00-6000 нмоль/л; образцы плазмы анализировали после осаждения белка обработкой ацетонитрилом. Результаты исследования фармакокинетических параметров представлены в таблице 9.


Заключение эксперимента: соединение формулы (I) обладает сильным действием при пероральном введении, низкой скоростью выведения лекарственного вещества и высокой биодоступностью при пероральном введении.
Пример 9: Анализ влияния соединения формулы (I) на калиевый канал hERG
Влияние соединения формулы (I) на ток калиевого канала hERG исследовали на клетках СНО (клетки яичника китайского хомячка), стабильно экспрессирующих калиевый канал hERG, с применением полностью автоматической технологии пэтч-клемп QPatch.
1. Описание эксперимента
1.1 Экспериментальное лекарственное вещество: соединение формулы (I)
1.2 Экспериментальная система: клеточная линия CHO-hERG
1.3 Подготовка клеток
Клетки CHO-hERG культивировали в колбе для культивирования объемом 175 см2. После увеличения клеточной плотности до 60-80% культуральную среду удаляли, клетки промывали один раз 7 мл фосфатного буферного раствора (PBS) и затем добавляли 3 мл Detachin для расщепления.
После завершения расщепления добавляли 7 мл культуральной среды для нейтрализации и затем смесь центрифугировали. Супернатант удаляли и затем добавляли 5 мл культуральной среды для ресуспендирования, чтобы довести концентрацию клеток до 2~5×106/мл.
1.4 Приготовление раствора
Способ приготовления раствора представлен в таблице 10 ниже.
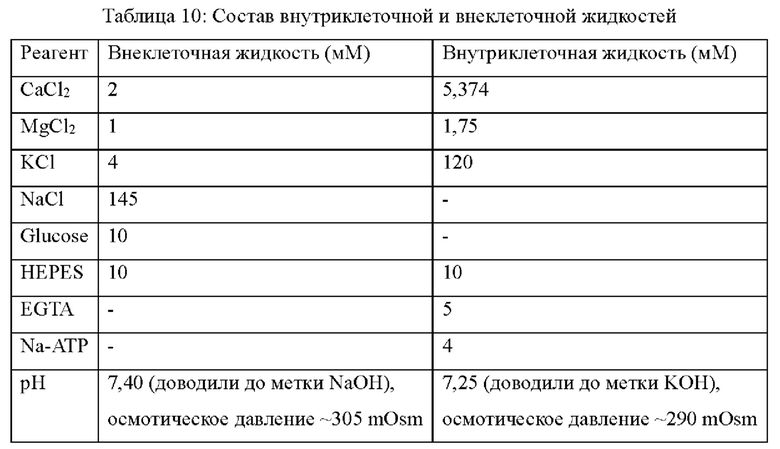
2. Эксперимент
20 мМ базового раствора соединения разбавляли внеклеточной жидкостью и 5 мкл 20 мМ базового раствора соединения добавляли к 2495 мкл внеклеточной жидкости, разбавляли до 40 мкМ при 500-кратном разбавлении и затем подвергали последовательному 3-кратному серийному разбавлению внеклеточной жидкостью, содержащей 0,2% ДМСО, с получением необходимой конечной концентрации для проведения испытания. Максимальная концентрация в испытании составляла 40 мкМ, все концентрации, которые исследовали, в свою очередь составляли: 40, 13,33, 4,44, 1,48, 0,49, 0,16 мкМ, соответственно, всего 6 концентраций. Конечная концентрация ДМСО в испытании не превышала 0,2%, что никак не влияло на калиевый канал hERG.
Формирование одноклеточного высокоимпедансного уплотнения и целой клетки автоматически завершали с помощью прибора QPatch. После получения записи в режиме целой клетки, клетки зажимали напряжением в -80 милливольт. Сначала клетки подвергали предварительному воздействию напряжения в -50 милливольт в течение 50 миллисекунд, затем их подвергали деполяризации при +40 милливольт в течение 5 секунд, затем их подвергали реполяризации при -50 милливольт в течение 5 секунд, после чего напряжение восстанавливали до -80 милливольт. Такую стимуляцию напряжением проводили каждые 15 секунд. Данные регистрировали в течение 2 минут, затем вводили внеклеточную жидкость, после чего данные регистрировали в течение 5 минут. Затем начинали вводить лекарственное вещество. Введение исследуемого соединения начинали с самой низкой концентрации, каждую концентрацию исследовали в течение 2,5 минут. После непрерывного введения всех концентраций в качестве положительного контроля вводили 3 мкМ цизаприда. При введении каждой концентрации исследовали по меньшей мере три клетки (n≥3). Экспериментальные данные анализировали с помощью программного обеспечения XLFit.
3. Заключение
Результаты показывают, что соединение формулы (I) ингибирует калиевый ток hERG при IC50>40 мкМ.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛЬ ИНГИБИТОРА LSD1 И ЕЁ ПОЛИМОРФНАЯ ФОРМА | 2019 |
|
RU2794977C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА МИМЕТИКА SMAC, ПРИМЕНЯЕМОГО В КАЧЕСТВЕ ИНГИБИТОРА IAP, И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2020 |
|
RU2819398C2 |
| ИНГИБИРОВАНИЕ ЦИКЛИЧЕСКОГО AMP-ЧУВСТВИТЕЛЬНОГО ЭЛЕМЕНТ-СВЯЗЫВАЮЩЕГО БЕЛКА (CREB) | 2020 |
|
RU2831142C2 |
| СОЛИ И КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ДИАЗАБЕНЗОФЛУОРАНТРЕНОВЫХ СОЕДИНЕНИЙ | 2017 |
|
RU2762189C2 |
| СОЛЬ АГОНИСТА ОПИОИДНОГО РЕЦЕПТОРА (MOR), КРИСТАЛЛИЧЕСКАЯ ФОРМА I ЕГО ФУМАРАТНОЙ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2018 |
|
RU2779119C2 |
| СОЛИ ПРОИЗВОДНОГО ИНДАЗОЛА И ИХ КРИСТАЛЛЫ | 2017 |
|
RU2747399C2 |
| ПОЛИМОРФНЫЕ ФОРМЫ ИКОТИНИБА И ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2710013C2 |
| ТВЕРДЫЕ ФОРМЫ ПЛАДИЕНОЛИДПИРИДИНОВЫХ СОЕДИНЕНИЙ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2743349C2 |
| ФУМАРАТ ПИРИДИЛАМИНА И ЕГО КРИСТАЛЛЫ | 2015 |
|
RU2684278C1 |
| СПОСОБЫ ЛЕЧЕНИЯ ДЕТСКИХ РАКОВЫХ ЗАБОЛЕВАНИЙ | 2017 |
|
RU2751636C2 |

Изобретение относится к соединению формулы (I), где соединение находится в кристаллической форме, где кристаллическая форма соединения формулы (I) представляет собой кристаллическую форму A или кристаллическую форму B. Кристаллическая форма A имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики рентгеновской дифракции при следующих углах 2θ: 7,03±0,2°, 11,28±0,2°, 14,00±0,2°, 15,11±0,2°, 17,31±0,2°, 19,48±0,2°, 20,07±0,2°, 22,86±0,2°. Кристаллическая форма B имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики рентгеновской дифракции при следующих углах 2θ: 5,50±0,2°, 8,36±0,2°, 11,87±0,2°, 12,39±0,2°, 12,66±0,2°, 15,11±0,2°, 17,35±0,2°, 18,70±0,2°. Способ получения соединения формулы (I) в кристаллической форме A включает (1) добавление соединения формулы (I) в растворитель с образованием суспензии или раствора; где растворитель выбран из группы, состоящей из метил-трет-бутилового эфира, этилацетата, ацетонитрила, этанола, ацетона, метанола, метилэтилкетона, смешанного растворителя ацетон-вода с отношением объемов 2:1 и смешанного растворителя этанол-вода с отношением объемов 3:1; (2) перемешивание суспензии или раствора в термомиксере с поддержанием постоянной температуры при 25-45°C, затем центрифугирование и сушку с получением кристаллической формы A соединения формулы (I). Способ получения соединения формулы (I) в кристаллической форме B включает (1) добавление соединения формулы (I) в тетрагидрофуран с образованием суспензии или раствора; (2) перемешивание суспензии или раствора в термомиксере с поддержанием постоянной температуры при 25-45°C, затем центрифугирование и сушку с получением кристаллической формы B соединения формулы (I). Технический результат – соединение формулы (I) в кристаллической форме для лечения заболеваний, связанных с BRD4. 5 н. и 8 з.п. ф-лы, 5 ил., 10 табл., 9 пр.
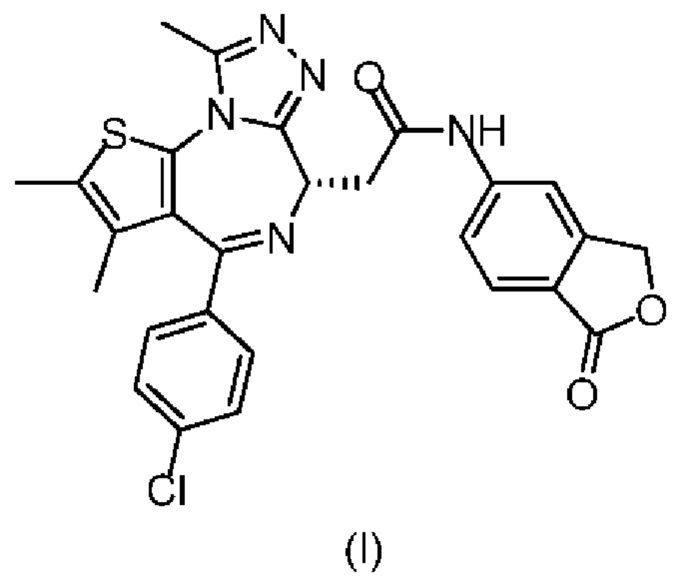
1. Соединение формулы (I)
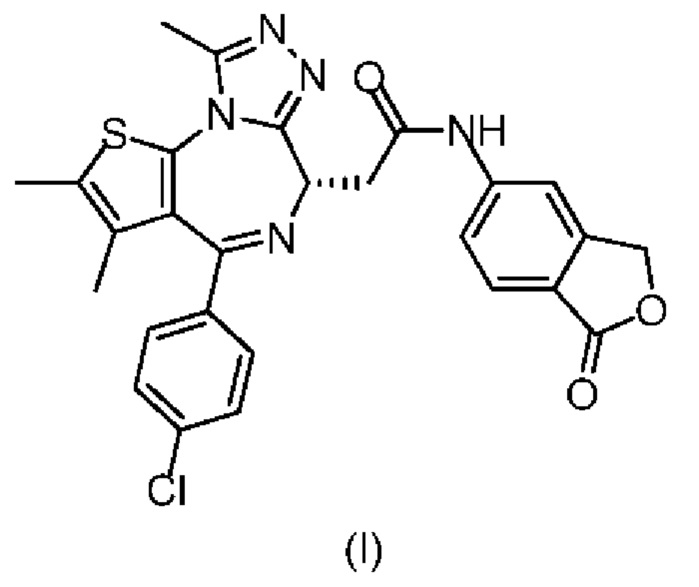 ,
,
где соединение находится в кристаллической форме, где кристаллическая форма соединения формулы (I) представляет собой кристаллическую форму A или кристаллическую форму B,
где кристаллическая форма A имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики рентгеновской дифракции при следующих углах 2θ: 7,03±0,2°, 11,28±0,2°, 14,00±0,2°, 15,11±0,2°, 17,31±0,2°, 19,48±0,2°, 20,07±0,2°, 22,86±0,2°;
где кристаллическая форма B имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики рентгеновской дифракции при следующих углах 2θ: 5,50±0,2°, 8,36±0,2°, 11,87±0,2°, 12,39±0,2°, 12,66±0,2°, 15,11±0,2°, 17,35±0,2°, 18,70±0,2°.
2. Соединение формулы (I) по п. 1, где кристаллическая форма A имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики рентгеновской дифракции при следующих углах 2θ: 7,03±0,2°, 11,28±0,2°, 12,39±0,2°, 14,00±0,2°, 15,11±0,2°, 17,31±0,2°, 19,48±0,2°, 20,07±0,2°, 22,86±0,2°, 26,05±0,2°.
3. Соединение формулы (I) по п. 1, где кристаллическая форма A имеет кривую дифференциальной сканирующей калориметрии (ДСК) с возникновением эндотермического пика при 289,22±3°C.
4. Соединение формулы (I) по п. 1, где потеря массы кристаллической формы A составляет 1,626% при 300,00±3°C по данным термограммы термогравиметрического анализа (ТГА).
5. Способ получения соединения формулы (I) в кристаллической форме A по любому из пп. 1-4, включающий:
(1) добавление соединения формулы (I) в растворитель с образованием суспензии или раствора;
где растворитель выбран из группы, состоящей из метил-трет-бутилового эфира, этилацетата, ацетонитрила, этанола, ацетона, метанола, метилэтилкетона, смешанного растворителя ацетон-вода с отношением объемов 2:1 и смешанного растворителя этанол-вода с отношением объемов 3:1;
(2) перемешивание суспензии или раствора в термомиксере с поддержанием постоянной температуры при 25-45°C, затем центрифугирование и сушку с получением кристаллической формы A соединения формулы (I).
6. Способ по п. 5, где отношение массы соединения к объему растворителя составляет 1 г : 5-10 мл.
7. Способ получения соединения формулы (I) в кристаллической форме B по п. 1, включающий:
(1) добавление соединения формулы (I) в тетрагидрофуран с образованием суспензии или раствора;
(2) перемешивание суспензии или раствора в термомиксере с поддержанием постоянной температуры при 25-45°C, затем центрифугирование и сушку с получением кристаллической формы B соединения формулы (I).
8. Способ по п. 7, где отношение массы соединения к объему тетрагидрофурана составляет 1 г : 5-10 мл.
9. Фармацевтическая композиция для лечения заболеваний, связанных с BRD4, где композиция содержит терапевтически эффективное количество соединения формулы (I) по любому из пп. 1-4.
10. Применение соединения формулы (I) по любому из пп. 1-4 или фармацевтической композиции по п. 9 для получения лекарственного препарата для лечения заболеваний, связанных с BRD4.
11. Применение по п. 10, где заболевания, связанные с BRD4, включают опухоли.
12. Применение по п. 11, где опухоли включают гематологические опухоли и солидные опухоли на поздних стадиях, при этом гематологические опухоли включают острый лимфобластный лейкоз, лимфому и миелому, и солидные опухоли на поздних стадиях включают нейроцитому, рак молочной железы и рак предстательной железы.
13. Применение по п. 12, где лимфома представляет собой острую миелоидную лимфому или рак молочной железы представляет собой трижды негативный рак молочной железы.
| US 5712274 A, 27.01.1998 | |||
| CN 101910182 A, 08.12.2010 | |||
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| КОНЪЮГАТЫ И МАЛЫЕ МОЛЕКУЛЫ, ВЗАИМОДЕЙСТВУЮЩИЕ С РЕЦЕПТОРОМ CD16а | 2013 |
|
RU2519546C1 |
| MINO R | |||
| CAIRA: "Crystalline Polymorphism of Organic Compounds", TOPICS IN CURRENT CHEMISTRY, 1998, vol.198, pp.163-208. | |||

