[0001] Настоящая заявка испрашивает приоритет по следующей заявке:
[0002] CN 201811549551.8, поданной 18 декабря 2018 г.
ОБЛАСТЬ ТЕХНИКИ
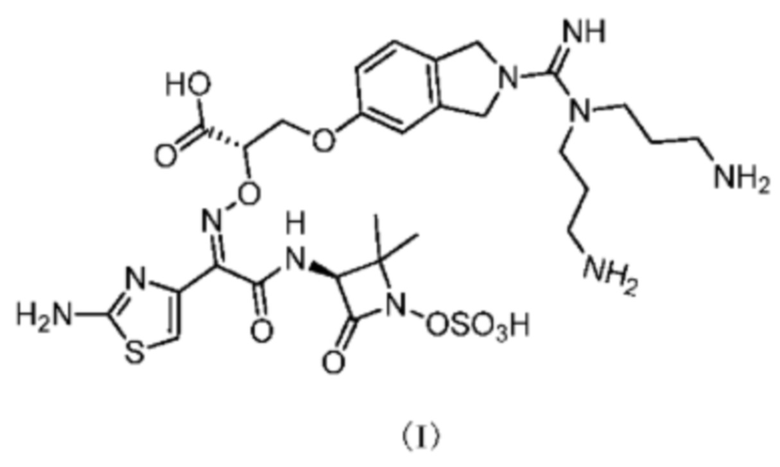
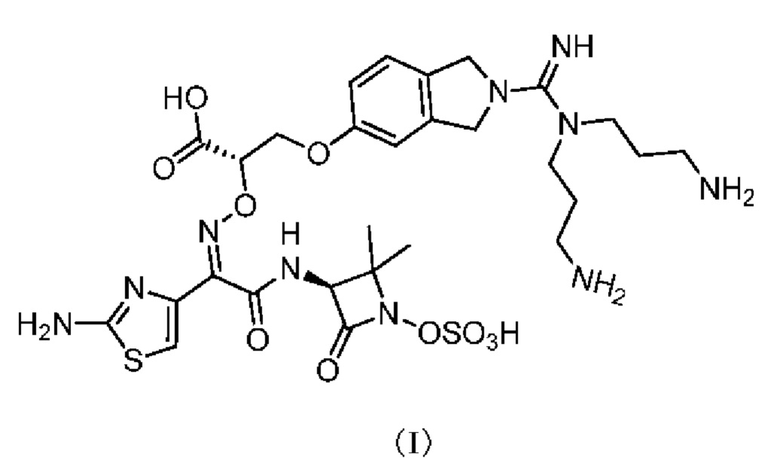
[0003] Настоящее изобретение относится к применению соединения, представленного формулой (I), и его фармацевтически приемлемых солей в области фармации.
УРОВЕНЬ ТЕХНИКИ
[0004] Эксперты и должностные лица системы общественного здравоохранения в целом считают, что появление и распространение лекарственно-устойчивых бактерий является одной из основных проблем общественного здравоохранения в XXI веке. Частота антибиотикорезистентности и ее связь с тяжелыми инфекционными заболеваниями возрастает тревожными темпами. Особую тревогу вызывает растущая распространенность внутрибольничной резистентности к патогенным микроорганизмам. Из более чем двух миллионов внутрибольничных инфекций, которые происходят ежегодно в Соединенных Штатах, от 50% до 60% вызваны устойчивыми к антибиотикам бактериями. Высокий уровень резистентности к широко используемым противомикробным препаратам повышает заболеваемость, смертность и затраты, связанные с внутрибольничными инфекциями. Число больных, умерших от неизлечимых внутрибольничных инфекций, продолжает расти. По оценкам, антибиотикорезистентность ежегодно убивает 700000 человек во всем мире, и некоторые эксперты предсказывают, что это число достигнет 10 миллионов к 2050 году, если не будут предприняты усилия по разработке новых терапевтических средств или терапевтических графиков (Nature, 2017, 543, 15). Варианты лечения инфекций, вызванных мультирезистентными грамотрицательными бактериями, включая Enterobacteriaceae и неферментирующие бактерии, являются особенно ограниченными; еще более серьезным является то, что конвейер развития фармацевтической промышленности включает мало соединений, которые могут пробить резистентность бактерий (Clin. Inf. Dis., 2009, 48, 1-12).
[0005] В течение последних нескольких десятилетий очень успешный и хорошо переносимый класс β-лактамных антибиотиков был основной основой для лечения инфекций, вызванных грамотрицательными патогенами. Из них для лечения инфекций, вызванных грамотрицательными бактериями, особенно широко используются цефалоспорины третьего поколения, карбапенемы и моноциклические лактамы. Однако появление все большего количества β-лактамаз и других механизмов резистентности серьезно угрожает среднесрочной доступности текущих соединений в этих подклассах, особенно лактамазы расширенного спектра действия (БЛРС) и карбапенемазы являются важной движущей силой резистентности к лекарственным препаратам; поэтому существует срочная потребность в новых β-лактамных антибиотиках, которые могут пробить резистентность к лекарственным препаратам, чтобы заполнить указанный пробел.
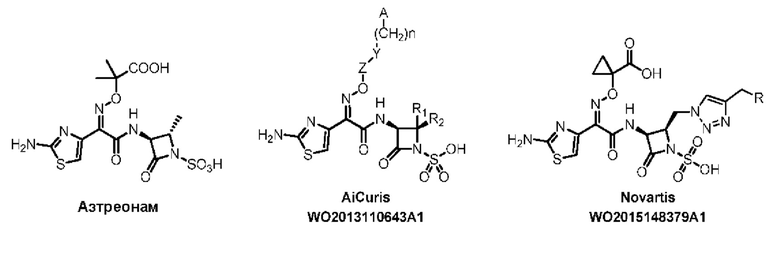
[0006] Поскольку азтреонам является единственным одобренным FDA моноциклическим β-лактамом, используемым во всем мире, а второй аналог (тигемонам) продается только на японском рынке, ценность азтреонама как моноциклического β-лактамного антибиотика далеко не полностью раскрыта (Rev. Infect. Dis., 1985, 7, 579-593). С другой стороны, бактериальная резистентность ухудшает проницаемость азтреонама, усиливает экзоцитоз и уменьшает антибактериальный спектр. Для повышения проницаемости моноциклических β-лактамов для бактерий была введена серия систем захвата носителей железа для моноциклических молекул β-лактама в Basilea (WO 2007065288), Naeja Pharmaceuticals (WO 2002022613) и Squibb&Sons (AS 5290929, EP 531976, EP 484881). Недавно компания Pfizer повторно изучила моноциклический β-лактам, несущий сульфонамидокарбонильную активирующую группу в положении N1 (WO 2010070523). Кроме того, в WO 2008116813 Basilea описывает комбинированную терапию с использованием моноциклических β-лактамов и карбапенемов. AiCuris (WO 2013110643) и Novartis (WO 2015148379) сообщили об исследованиях по усилению активности путем модификации заместителей на молекулах азтреонама, соответственно, соединениями формулы, представленной ниже, где группа А представляет собой ароматическую кольцевую структуру, к которой присоединены амидино и гуанидиновые группы. Novartis (WO 2017050218) также сообщает о солевых формах одного из соединений, которые в настоящее время находятся в стадии доклинической или клинической разработки.
КРАТКОЕ ОПИСАНИЕ
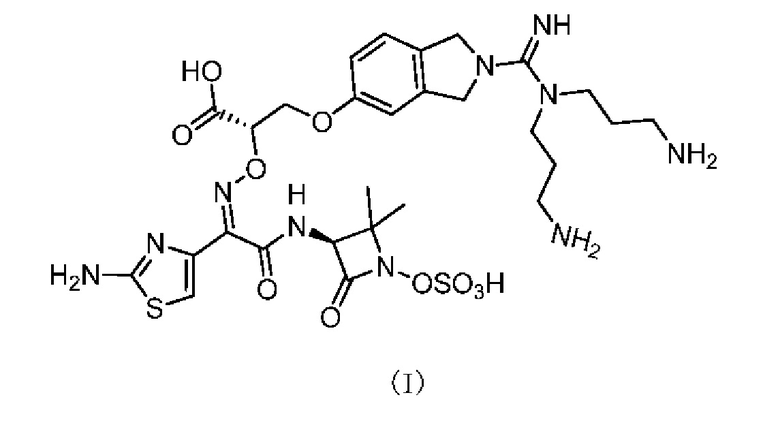
[0007] Согласно настоящему изобретению предложено применение соединения, представленного формулой (I), или его фармацевтически приемлемой соли, для получения лекарственного средства для лечения пневмонии.
[0009] В некоторых вариантах реализации настоящего изобретения, где пневмония в применении, описанном выше, вызвано инфекцией Pseudomonas aeruginosa.
[0010] Согласно настоящему изобретению также предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения, представленного формулой (I), или его фармацевтически приемлемой соли в качестве активного ингредиента, и одно или более фармацевтически приемлемых вспомогательных веществ и/или фармацевтически приемлемых носителей.
[0011] В некоторых вариантах реализации настоящего изобретения, вспомогательное вещество в композиции, описанной выше, представляет собой поверхностный стабилизатор, солюбилизатор, буфер, помутнение, связующее вещество, разрыхлитель или смазывающее вещество.
[0012] В некоторых вариантах реализации настоящего изобретения, поверхностный стабилизатор в композиции, описанной выше, содержит амфотерное поверхностно-активное вещество, неионогенное поверхностно-активное вещество, катионное поверхностно-активное вещество или анионное поверхностно-активное вещество, или их комбинацию.
[0013] В некоторых вариантах реализации настоящего изобретения, фармацевтическая композиция в случае композиции, описанной выше, предназначена для перорального применения.
[0014] В некоторых вариантах реализации настоящего изобретения, фармацевтическая композиция в случае композиции, описанной выше, представляет собой таблетку или капсулу.
[0015] В некоторых вариантах реализации настоящего изобретения, фармацевтическая комбинация в случае композиции, описанной выше, находится в форме инъекционного состава или ингаляционного состава.
[0016] Согласно настоящему изобретению предложено применение композиции, описанной выше, для получения лекарственного средства для лечения пневмонии.
[0017] В некоторых вариантах реализации настоящего изобретения, пневмония, при которой применяют композицию, как описано выше, вызвана инфицированием грамотрицательными бактериями.
[0018] В некоторых вариантах реализации настоящего изобретения, пневмония, при которой применяют композицию, как описано выше, вызвана инфекцией Pseudomonas aeruginosa.
ТЕХНИЧЕСКИЙ РЕЗУЛЬТАТ
[0019] Соединение согласно настоящему изобретению обладает хорошей антибактериальной активностью в отношении грамотрицательных бактерий и, в частности, обладает превосходной антибактериальной активностью в отношении Pseudomonas aeruginosa.
[0020] Определения и описание
[0021] В настоящем описании следующие термины и фразы имеют следующие значения, если не указано иное. Конкретный термин или фразу, если они не определены специально, не следует толковать как неопределенные или неясные, а следует толковать в общем смысле. В случае, когда в настоящем описании используются торговые наименования, они предназначены для обозначения соответствующих товаров или их активных ингредиентов. Термин «фармацевтически приемлемый» в настоящем описании используется для обозначения тех соединений, материалов, композиций и/или лекарственных форм следующим образом: в рамках обоснованного медицинского заключения, подходит для применения в контакте с тканями людей и животных без чрезмерной токсичности, раздражения, аллергической реакции и/или других проблем или осложнений, соответствуя разумному соотношению польза/риск.
[0022] Термин «фармацевтически приемлемые соли» относится к солям соединений согласно настоящему изобретению, которые получают из соединений, содержащих указанные заместители, предложенные в настоящем изобретении, и относительно нетоксичных кислот или оснований. В случае, когда соединения согласно настоящему изобретению содержат относительно кислые функциональные группы, соли присоединения основания могут быть получены путем введения в контакт нейтральной формы таких соединений с достаточным количеством основания в чистом растворе или в подходящем инертном растворителе. Фармацевтически приемлемые соли присоединения оснований включают соли натрия, калия, кальция, аммония, органического аммиака или соли магния, или схожие соли. В случае, когда соединения согласно настоящему изобретению содержат относительно основные функциональные группы, соли присоединения кислоты могут быть получены путем приведения нейтральной формы таких соединений в контакт с достаточным количеством кислоты в чистом растворе или в подходящем инертном растворителе. Примеры фармацевтически приемлемых солей присоединения кислоты включают соли неорганических кислот, включая, например, хлористоводородную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, бикарбонат, фосфорную кислоту, моногидрофосфат, дигидрофосфат, серную кислоту, сульфат водорода, йодистоводородную кислоту, фосфористую кислоту и т.п.; и соли органических кислот, включая кислоты, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфокислоту, п-толуолсульфокислоту лимонную кислоту, винную кислоту, метансульфокислоту и т.п.; также включают соли аминокислот, таких как аргинин, а также соли органических кислот, таких как глюкуроновая кислота (см. Berge et al., «Pharmaceutical Salts», Journal of Pharmaceutical Science 66:1-19 (1977)). Некоторые конкретные соединения согласно настоящему изобретению содержат основные и кислотные функциональные группы, таким образом, их можно превращать в любую соль присоединения основания или кислоты.
[0023] Предпочтительно, нейтральную форму соединения регенерируют путем приведения соли в контакт с основанием или кислотой обычным способом и выделения исходного соединения. Исходная форма соединения отличается от его различных солевых форм по определенным физическим свойствам, таким как растворимость в полярных растворителях.
[0024] В настоящем описании «фармацевтически приемлемая соль» представляет собой производное соединения согласно настоящему изобретению, в котором исходное соединение модифицировано путем солирования с кислотой или основанием. Примеры фармацевтически приемлемых солей включают, но не ограничиваются перечисленными: основную группу, такую как соли аминов и неорганических или органических кислот, кислотные радикалы, такие как щелочные или органические соли карбоновых кислот. Фармацевтически приемлемые соли включают обычные нетоксичные соли или четвертичные аммониевые соли исходного соединения, например, соли, образованные из нетоксичных неорганических или органических кислот. Обычные нетоксичные соли включают, но не ограничиваются ими, соли, полученные из неорганических и органических кислот, выбранных из 2-ацетоксибензойной кислоты, 2-гидроксиэтансульфокислоты, уксусной кислоты, аскорбиновой кислоты, бензосульфокислоты, бензойной кислоты, бикарбонатного радикала, угольной кислоты, лимонной кислоты, этилендиаминтетрауксусной кислоты, этандисульфокислоты, этансульфокислоты, фумаровой кислоты, глюкогептозы, глюконовой кислоты, глутаминовой кислоты, гликолевой кислоты, бромистоводородной кислоты, хлористоводородной кислоты, гидройодида, гидрокси, гидроксинафталина, 2-гидроксиэтансульфокислоты, молочной кислоты, лактозы, додецилсульфокислоты, малеиновой кислоты, яблочной кислоты, манделиновой кислоты, метансульфокислоты, азотной кислоты, оксальной кислоты, памокислоты, пантотеновой кислоты, фенуксусной кислоты, фосфорной кислоты, полигалактопропионовой кислоты, солевой кислоты, стеариновой кислоты, уксусной кислоты, янтарной кислоты, сульфаминовой кислоты, п-аминобензойной кислоты, серной кислоты, таннина, винной кислоты и п-толуолсульфоновой кислоты.
[0025] Фармацевтически приемлемые соли согласно настоящему изобретению могут быть синтезированы из исходного соединения, содержащего кислотный радикал или основную группу, с помощью обычных химических способов. Обычно такие соли получают путем введения этих соединений в форме свободной кислоты или свободного основания в реакцию со стехиометрическим количеством подходящего основания или кислоты в воде или органическом растворителе или их смеси. Как правило, неводные среды, такие как эфир, этилацетат, этанол, изопропиловый спирт или ацетонитрил, являются предпочтительными.
[0026] Помимо солевых форм, соединения, предложенные в настоящем изобретении, существуют в форме пролекарств. Пролекарства соединений согласно настоящему описанию легко превращаются в соединения согласно настоящему изобретению путем химических изменений в физиологических условиях. Кроме того, пролекарства могут быть превращены в соединения согласно настоящему изобретению с помощью химических или биохимических способов в среде in vivo.
[0027] Некоторые соединения согласно настоящему изобретению могут существовать в несольватированных или сольватированных формах, включая гидратированные формы. В общем, сольватированные формы эквивалентны несольватированным формам и предназначены для включения в объем настоящего изобретения.
[0028] Некоторые соединения согласно настоящему изобретению могут содержать асимметрические атомы углерода (оптические центры) или двойные связи. Рацетаты, диастереомеры, геометрические изомеры и отдельные изомеры включены в объем настоящего изобретения.
[0029] Если не указано иное, абсолютная конфигурация стереоцентра обозначена клиновидными сплошными связями  и клиновидными пунктирными связями
и клиновидными пунктирными связями  при этом клиновидные сплошные связи или клиновидные пунктирные связи
при этом клиновидные сплошные связи или клиновидные пунктирные связи  обозначены волнистыми линиями
обозначены волнистыми линиями 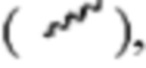 а относительная конфигурация стереоцентра обозначена связями в виде сплошных линий
а относительная конфигурация стереоцентра обозначена связями в виде сплошных линий 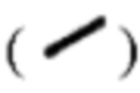 и связями в виде пунктирных линий
и связями в виде пунктирных линий  В случае, когда соединения согласно настоящему описанию содержат олефиновые двойные связи или другие центры геометрической асимметрии, они включают геометрические изомеры Е, Z, если не указано иное. Аналогично, все таутомерные формы включены в объем настоящего изобретения.
В случае, когда соединения согласно настоящему описанию содержат олефиновые двойные связи или другие центры геометрической асимметрии, они включают геометрические изомеры Е, Z, если не указано иное. Аналогично, все таутомерные формы включены в объем настоящего изобретения.
[0030] Соединения согласно настоящему изобретению могут существовать в конкретных геометрических или стереоизомерных формах. Настоящее изобретение включает все такие соединения, включая цис и транс изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереомеры, (D)-изомеры, (b)-изомеры, а также их рацемические и другие смеси, такие как энантиомерно или диастереомерно обогащенные смеси, и все такие смеси входят в объем настоящего изобретения. Дополнительные асимметрические атомы углерода могут присутствовать в заместителях, таких как алкильные группы. Все такие изомеры и их смеси включены в объем настоящего изобретения.
[0031] Оптически активные (R)- и (S)-изомеры, а также D- и L-изомеры могут быть получены путем хирального синтеза или с применением хиральных реагентов или другими обычными способами. Требуемый энантиомер соединения согласно настоящему изобретению может быть получен путем асимметрического синтеза или дериватизации с применением хиральных вспомогательных агентов, где полученную смесь диастереомеров разделяют и вспомогательную группу отщепляют с получением чистого желаемого энантиомера. В качестве альтернативы, когда молекула содержит основную функциональную группу, такую как аминогруппа, или кислотную функциональную группу, такую как карбоксильная группа, образуются диастереомерные соли с подходящими оптически активными кислотой или основанием с последующим разделением диастереомеров обычными способами, известными в данной области техники, и последующим выделением с получением чистых энантиомеров. Кроме того, энантиомеры и диастереомеры, как правило, разделяют с помощью хроматографии с применением хиральной неподвижной фазы, необязательно в комбинации с химической дериватизацией (например, с образованием карбамата из амина).
[0032] Соединение согласно настоящему изобретению может содержать не встречающиеся в природе отношения атомных изотопов одного или более атомов, составляющих соединение. Например, соединения могут быть помечены с помощью радиоизотопов, таких как тритий (3Н), иод -125 (125I) или С-14 (14С). Все изотопные варианты соединений согласно настоящему изобретению, являющихся или не являющихся радиоактивными, включены в объем настоящего изобретения.
[0033] Термин «фармацевтически приемлемый носитель» относится к любому составу или переносящей среде, способным осуществлять доставку эффективного количества активного агента согласно настоящему изобретению без нарушения биологической активности активного агента и без проявления токсических или побочных эффектов в отношении организма-хозяина или пациента. Типичные носители включают воду, масла, вещества растительного и минерального происхождения, кремы, основы для лосьонов, основы для мазей и т.п. Они содержат суспендирующие агенты, агенты, придающие клейкость, усилители проникновения через кожу и т.п. Дополнительную информацию по перевозчикам можно найти в источнике Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005), содержание указанного источника включено в настоящее описание посредством ссылки.
[0034] Термин «вспомогательное вещество» обычно относится к носителю, разбавителю и/или среде, необходимым для приготовления эффективной фармацевтической композиции.
[0035] Для лекарственного средства или фармакологически активного агента термин «эффективное количество» или «терапевтически эффективное количество» относится к количеству лекарственного средства или агента, которое не является токсичным, но достаточным для достижения желаемого эффекта. В случае лекарственных форм для перорального введения согласно настоящему изобретению «эффективное количество» одного активного вещества в композиции относится к количеству, необходимому для достижения желаемого эффекта в комбинации с другим активным веществом в композиции. Определение эффективного количества варьирует от человека к человеку в зависимости от возраста и общего состояния реципиента и также от конкретного активного вещества, и подходящие эффективные количества в каждом случае могут быть определены специалистом в данной области техники согласно установленным экспериментам.
[0036] Термины «активный ингредиент», «терапевтический агент», «активное вещество» или «активный агент» относятся к химическому соединению, которое является эффективным при лечении расстройства, заболевания или состояния, представляющего интерес.
[0037] Растворители, применяемые согласно настоящему изобретению, являются коммерчески доступными.
[0038] В настоящем изобретении используются следующие сокращения:
[0039] вод. представляет собой воду; мин представляет собой минуты; FA представляет собой муравьиную кислоту; m-СРВА представляет собой 3-хлорпероксибензойную кислоту; экв. представляет собой эквивалентное количество; DCC представляет собой N,N'-дициклогексилкарбодиимид; ДХМ представляет собой дихлорметан; РЕ представляет собой петролейный эфир; DIAD представляет собой диизопропилазодикарбоксилат; ДМФА представляет собой N,N-диметилформамид; ВН3⋅SMe2 представляет собой боран-диметилсульфид; ДМСО представляет собой диметилсульфоксид; EtOAc представляет собой этилацетат; EtOH представляет собой этанол; МеОН представляет собой метанол; Cbz представляет собой бензилоксикарбонил, аминозащитную группу; Вое представляет собой трет-бутоксикарбонил, аминозащитную группу; НОАс представляет собой уксусную кислоту; ACN представляет собой ацетонтрил; ВН3 представляет собой цианоборгидрид натрия; к.т. представляет собой комнатную температуру; ТГФ представляет собой тетрагидрофуран; Boc2O представляет собой ди-трет-бутилдикарбонат; ТФУ представляет собой трифторуксусную кислоту; DIPEA представляет собой диизопропилэтиламин; SOCl2 представляет собой тионилхлорид; iPrOH представляет собой 2-пропанол; mp представляет собой температуру плавления; LDA представляет собой диизопропиламид лития; TEMPO представляет собой 2,2,6,6-тетраметилпиперидин-1-оксильный радикал или 2,2,6,6-тетраметилпиперидин оксид; NaClO представляет собой гипохлорит натрия; NaClO2 представляет собой хлорит натрия; HOBt представляет собой 1-гидроксибензотриазол; psi представляет собой фунты на квадратный дюйм; DMF⋅SO3 представляет собой N,N-диметилформамид-сульфотриоксид; KH2PO4 представляет собой дигидрофосфат калия; Bu4HSO4 представляет собой тетрабутиламмония гидросульфат; PPh3 представляет собой трифенилфосфин; NH2NH2⋅H2O представляет собой гидразингидрат; dppf представляет собой 1,1'-бис(дифенилфосфино)ферроцин; Pd2(dba3) представляет собой трис(дибензилиденацетон)дипалладий (0); МИК представляет собой минимальную ингибирующую концентрацию; DMAP представляет собой 4-диметиламинопиридин; BnBr представляет собой бензилбромид; Н2О2 представляет собой пероксид водорода.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0040] На фиг. 1 показаны результаты бактериальной нагрузки на легкие при лечении легочной инфекции Pseudomonas aeruginosa у иммуносупрессивных мышей.
ПОДРОБНОЕ ОПИСАНИЕ
[0041] Следующие примеры подробно описывают настоящее изобретение, но они не предназначены для наложения каких-либо неблагоприятных ограничений на настоящее изобретение. Настоящее изобретение было подробно описано в настоящем описании, и также раскрыты его конкретные варианты реализации. Специалистам в данной области техники будет очевидно, что в конкретных вариантах реализации без отступления от сущности и объема настоящего изобретения могут быть сделаны различные изменения и модификации.
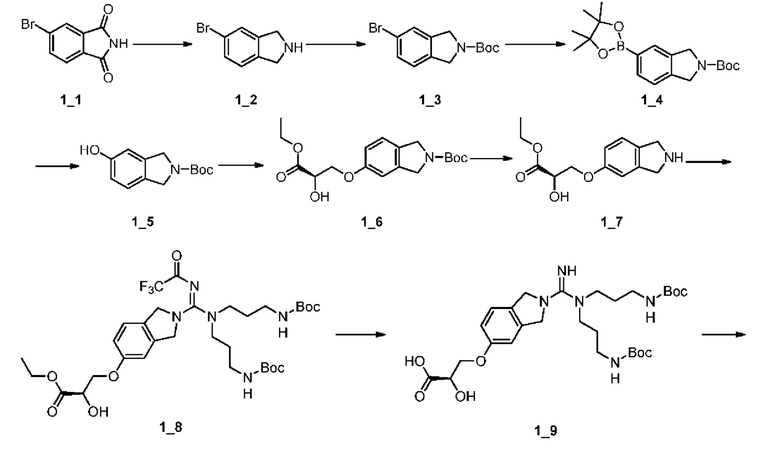
[0042] Синтез промежуточного соединения А1:
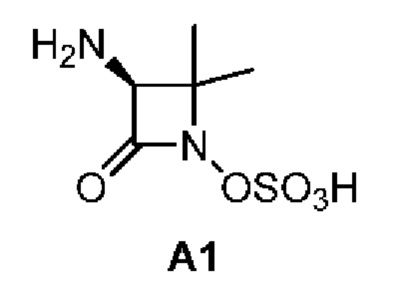
[0043]
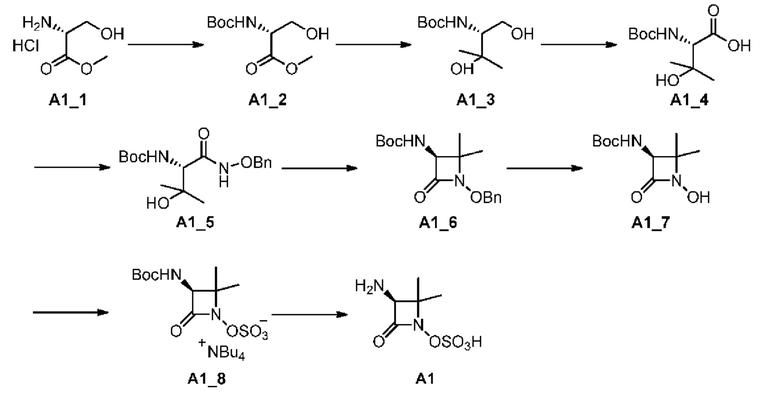
[0044] Стадия 1: КТГФ (1,50 л) добавляли соединение А1_1 (100,00 г, 642,76 ммоль, 1,00 экв.) и триэтиламин (136,59 г, 1,35 моль, 187,10 мл, 2,10 экв.), и полученную смесь охлаждали до 0°С; по каплям добавляли Boc2O (154,31 г, 707,03 ммоль, 162,43 мл, 1,10 экв.) в ТГФ (500,00 мл) при этой температуре, реакционную смесь нагревали до 10°С и перемешивали при этой температуре в течение 10 часов, а затем фильтровали; фильтрат концентрировали при пониженном давлении с получением неочищенного продукта, к которому добавляли насыщенный раствор бикарбоната натрия (300 мл) и экстрагировали смесь этилацетатом (500 мл*2); объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения А1_2.
[0045] 1H ЯМР (400 МГц, CDCl3) δ (ppm): 5,51 (br s, 1H), 4,46-4,31 (m, 1H), 4,03-3,86 (m, 2H), 3,83-3,72 (m, 3H), 2,64 (br s, 1H), 1,46 (s, 9H).
[0046] Стадия 2: A1_2 растворяли в ТГФ (2000 мл), смесь перемешивали при -50°С в течение 10 минут, затем по каплям добавляли MeMgBr (ЗМ, 638,59 мл, 6,00 экв.) в течение 20 минут при -50°С.Полученную смесь перемешивали при 25°С в течение 60 минут, после чего реакционную смесь гасили при 0°С путем добавления разбавленной соляной кислоты (2000 мл, 0,5 М), затем полученную смесь экстрагировали этилацетатом (500 мл*2); объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали, фильтрат концентрировали при пониженном давлении, в результате чего получали неочищенный продукт, который промывали петролейным эфиром/этилацетатом (70 мл, 10/1) путем перемешивания и очищали с помощью колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 10/1 до 1/1 (об./об.)) с получением Соединения А1_3.
[0047] 1H ЯМР (400 МГц, CDCl3) δ (ppm): 5,41-5,23 (m, 1H), 3,96 (br d, J=11,2 Гц, 1H), 3,79-3,70 (m, 1H), 3,40 (br d, J=8,3 Гц, 1H), 2,53-2,39 (m, 2H), 1,39 (s, 9H), 1,28 (s, 3H), 1,18 (s, 3H).
[0048] Стадия 3: A1_3 (30 r, 136,81 ммоль, 1,00 экв.) растворяли в смешанном растворе фосфатно-натриевого буфера (540,00 мл, 0,7 М, 2,76 экв.) и ацетонитрила (300 мл), затем по каплям добавляли TEMPO (2,15 г, 13,68 ммоль, 0,10 экв.), NaClO (81,47 г, 5,47 ммоль, 67,33 мл, чистота 0,5%, 0,04 экв.) и NaClO2 (98,99 г, 1,09 моль, 8,00 экв.) в воде (300 мл) путем перемешивания при 35°С; полученную смесь перемешивали при 35°С в течение 12 часов, затем охлаждали до комнатной температуры и добавляли лимонную кислоту (10 г); полученную смесь экстрагировали этилацетатом (500 мл*4), объединенные органические слои промывали насыщенным водным раствором хлорида натрия (100 мл), сушили над безводным сульфатом натрия и фильтровали, а фильтрат концентрировали при пониженном давлении. К полученному неочищенному продукту добавляли водный карбонат натрия (300 мл, 2 М) и промывали этилацетатом (200 мл*2); водный слой охлаждали до 0°С и доводили рН до 3,0 с помощью разбавленной соляной кислоты (1 М); водный раствор насыщали хлоридом натрия и полученную смесь экстрагировали этилацетатом (500 мл*4); объединенные органические слои промывали насыщенным водным раствором хлорида натрия (50 мл), сушили над безводным сульфатом натрия и фильтровали, а фильтрат концентрировали при пониженном давлении с получением соединения А1_4.
[0049] d6 ЯМР (400 МГц, CDCl3) δ (ppm):5,42 (br d, J=7,8 Гц, 1H), 4,18 (br d, J=8,4 Гц, 1H), 1,39 (s, 9H), 1,30 (s, 3H), 1,22 (s, 3H).
[0050] Стадия 4: Al_4 (48 r, 205,78 ммоль, 1,00 экв.) растворяли в DMF (700 мл), затем добавляли DCC (84,92 г, 411,56 ммоль, 83,25 мл, 2,00 экв.) и HOBt (55,61 г, 411,56 ммоль, 2 экв.), смесь перемешивали при 10°С в течение 0,5 ч, затем О-бензилгидроксиламина гидрохлорид (39,41 г, 246,93 ммоль, 1,20 экв.) и бикарбонат натрия (69,15 г, 823,11 ммоль, 32,01 мл, 4 экв.) в воде; полученную смесь перемешивали при 10°С в течение 1,5 ч, затем реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении; неочищенный продукт разбавляли водой (400 мл) и подвергали экстракции этилацетатом (500 мл*2). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали; фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 6/1 до 3/1 (об./об.)) с получением Соединения А1_5.
[0051] 1H ЯМР (400 МГц, ДМСО-d6) δ (ppm): 11,06 (s, 1H), 7,45-7,32 (m, 5Н), 6,45 (br d, J=9,2 Гц, 1H), 4,80 (d, J=2,6 Гц, 2H), 4,65 (s, 1H), 4,04 (d, J=7,0 Гц, 1H), 3,77 (br d, J=9,2 Гц, 1H), 1,40 (s, 9H), 1,11 (s, 3H), 1,08 (s, 3H);
[0052] ЖХ-МС (ИЭР) m/z: 283 (М-56+1).
[0053] Стадия 5: А1_5 (57 г, 168,44 ммоль, 1 экв.) растворяли в пиридине (600 мл) путем перемешивания при 55°С в течение 12 ч и добавляли по каплям триоксид серы пиридин (187,67 г, 1,18 моль, 7 экв.); затем реакционную смесь концентрировали при пониженном давлении и полученное твердое вещество растворяли в этилацетате (800 мл); к твердому веществу добавляли водный карбонат калия (816,94 мл, 2 М, 9,7 экв.) при 0°С и полученную смесь перемешивали при 100°С в течение 2 ч; затем реакционную смесь охлаждали до комнатной температуры и экстрагировали этилацетатом (400 мл*3); объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении; полученный неочищенный продукт очищали с помощью колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 12/1 до 9/1 (об./об.)) с получением Соединения А1_6.
[0054] 1H ЯМР (400 МГц, CDCl3) δ (ppm): 7,41 (br d, J=1,0 Гц, 5H), 5,02-4,97 (m, 2H), 4,32 (d,.7=6,7 Гц, 1H), 1,50-1,43 (m, 9H), 1,34 (s, 3H), 1,11 (s, 3H);
[0055] ЖХ-МС (ИЭР) m/z: 321,1 (M+1).
[0056] Стадия 6: A1_6 (31 r, 96,76 ммоль, 1,00 экв.) растворяли в метаноле (620 мл) и добавляли Pd/C (3 г, 10%) в атмосфере азота, затем реакционную пробирку заменяли азотом три раза, затем реакционную пробирку насыщали газообразным водородом при 20°С и проводили реакцию при давлении 50 psi (примерно 345 кПа) в течение 1 ч, затем реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения А1_7.
[0057] Стадия 7: к раствору А1_7 (22 г, 95,54 ммоль, 1,00 экв.) в ДМФА (220 мл) добавляли ДМФА⋅SO3 (17,56 г, 114,65 ммоль, 1,2 экв.); смесь перемешивали при 0°С в течение 1 ч и затем разбавляли насыщенным KH2PO4 (200 мл); полученную смесь экстрагировали этилацетатом (100 мл) и Bu4HSO4 (38,93 г, 114,65 ммоль, 1,20 экв.) добавляли к объединенным водным слоям в течение 20 мин при 10°С и полученную водную фазу экстрагировали EtOAc (350 мл*4); объединенные органические фазы концентрировали при пониженном давлении с получением Соединения А1_8.
[0058] Стадия 8: А1_8 (68 г, 123,24 ммоль, 1,00 экв.) добавляли к трифторуксусной кислоте (300 мл) и смесь перемешивали при 15°С в течение 4 ч в атмосфере азота; реакционную смесь разбавляли дихлорметаном (350 мл) и фильтровали и фильтрат концентрировали при пониженном давлении для получения соединения А1.
[0059] 1Н ЯМР (400 МГц, ДМСО-d6) δ (ppm):8,79 (br s, 3Н), 4,18 (br s, 1H), 1,46-1,38 (m, 6Н).
[0060] Синтез промежуточного соединения А2:
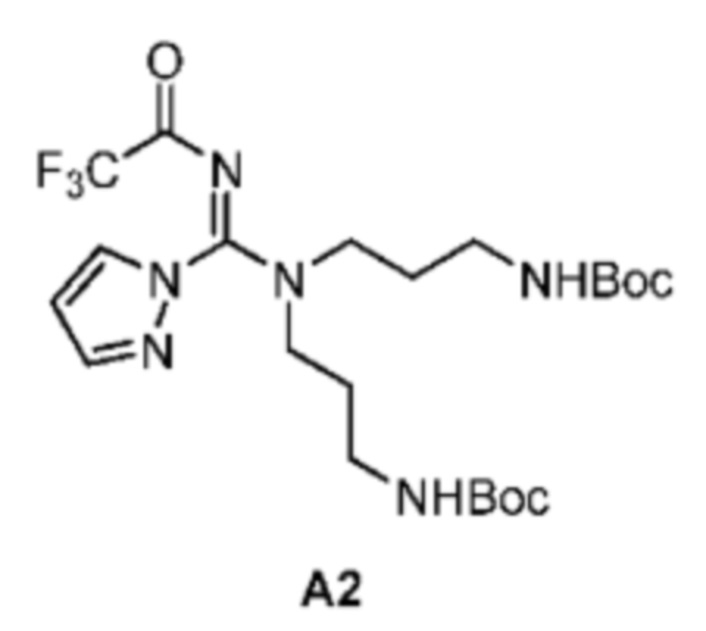
[0061]
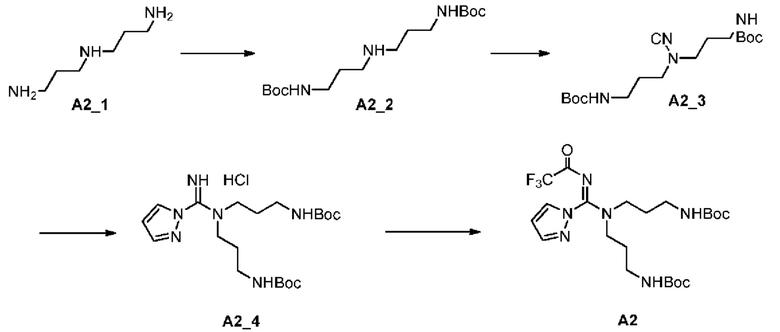
[0062] Стадия 1: к раствору А2_1 (7 г, 53,35 ммоль, 7,54 мл, 1 экв.) в ТГФ (70 мл) медленно по каплям добавляли BOC-ONB (29,80 г, 106,69 ммоль, 2 экв.) и Et3N (11,34 г, 112,03 ммоль, 15,59 мл, 2,1 экв.) в ТГФ (330 мл) при 20°С, и полученную смесь перемешивали при 20°С в течение 11 ч, фильтровали и фильтрат концентрировали при пониженном давлении, полученный остаток разбавляли раствором карбоната калия (100 мл, 2 М) и подвергали экстракции этилацетатом (100 мл*2); органические фазы объединяли и концентрировали при пониженном давлении с получением Соединения А2_2.
[0063] Стадия 2: к раствору А2_2 (15 г, 45,26 ммоль, 1 экв.) в МеОН (150 мл) добавляли BrCN (7,86 г, 74,21 ммоль, 5,46 мл, 1,64 экв.) и ацетат натрия (7,43 г, 90,51 ммоль, 2 экв.) при 0°С, смесь перемешивали при комнатной температуре в течение 2 ч, затем разбавляли насыщенным раствором карбоната натрия (300 мл) и подвергали экстракции этилацетатом (100 мл); органические фазы концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 5/1 до 1/1) с получением соединения А2_3.
[0064] Стадия 3: Соединение А2_3 (4,2 г, 11,78 ммоль, 1 экв.) и пиразола гидрохлорид (1,23 г, 11,78 ммоль, 1 экв.) добавляли к ТГФ (40 мл) и заменяли азотом три раза, затем смесь перемешивали при 75°С в течение 12 ч; реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (100 мл), фильтровали и осадок на фильтре собирали с получением соединения А2_4 после высушивания.
[0065] ЖХМС (ИЭР) m/z: 425,4 (М+1).
[0066] Стадия 4: к раствору Соединения А2_4 (2,1 г, 4,56 ммоль, 1 экв.) в ДХМ (20 мл) при 0°С добавляли TFAA (765,41 мг, 3,64 ммоль, 506,89 мкл, 0,8 экв.) и триэтиламин (1,01 г, 10,02 ммоль, 1,39 мл, 2,2 экв.), смесь перемешивали при 0°С в течение 20 мин, затем разбавляли водой (20 мл), полученную смесь подвергали экстракции ДХМ (50 мл*2), органические слои объединяли и концентрировали при пониженном давлении с получением Соединения А2.
[0067] Синтез промежуточного соединения A3:
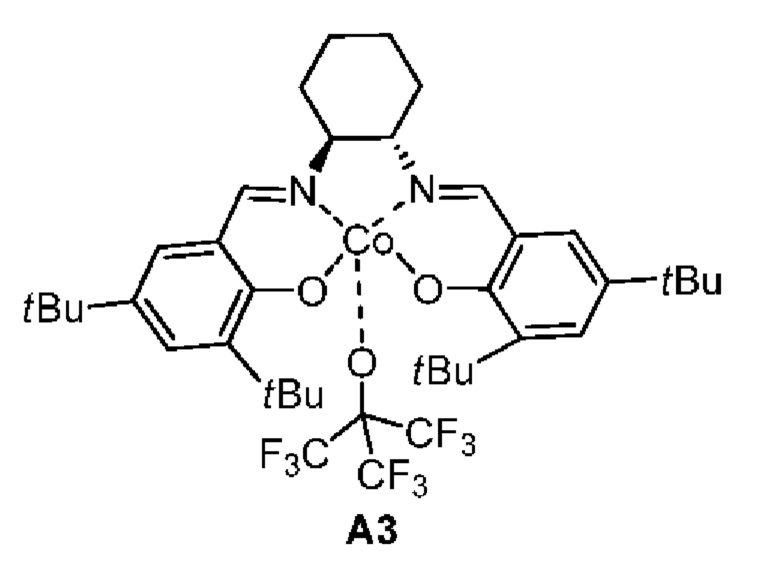
[0068]
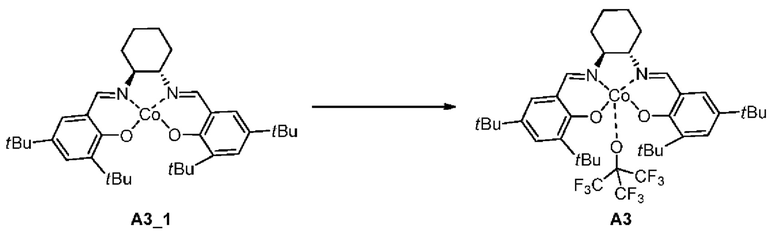
[0069] Стадия 1: Соединение А3_1 добавляли к смешанному раствору 1,1,1,3,3,3-гексафтор-2-(трифторметил)пропан-2-ола (10,16 г, 43,06 ммоль, 10 экв.) и ДХМ (20 мл), реагент перемешивали при комнатной температуре в течение 45 мин (20-25°С) и концентрировали при пониженном давлении с получением соединения A3.
[0070] Пример 1: Получение соединения, представленного формулой (I)
[0071]
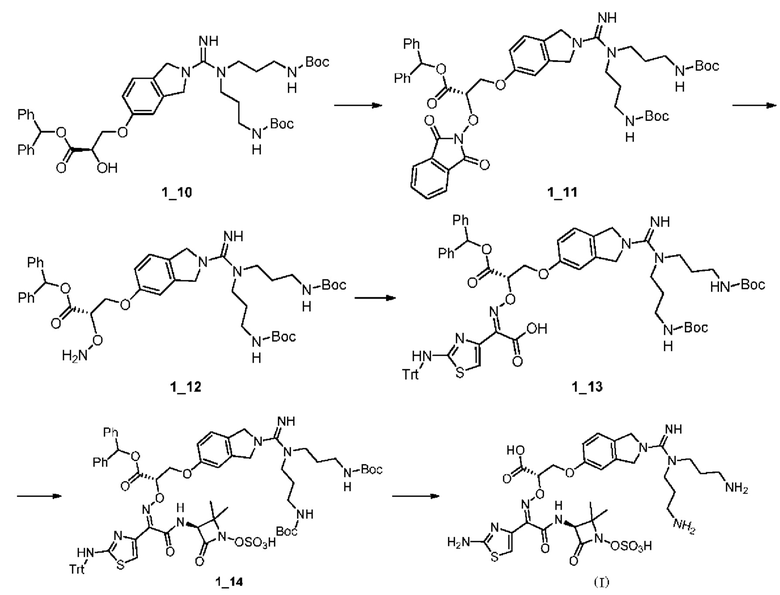
[0072] Стадия 1: к раствору соединения 11 (29 г, 128,30 ммоль, 1 экв.) в ТГФ (300 мл) добавляли ВН3⋅SMe2 (10 М, 38,49 мл, 3 экв.). Смесь подвергали реакции при 80°С в течение 12 часов, затем охлаждали до 0°С и гасили метанолом (100 мл); затем добавляли разбавленную соляную кислоту (90 мл, 1 М) путем перемешивания при 80°С в течение 1 часа, и смесь концентрировали при пониженном давлении для удаления растворителя; остаток разбавляли водой (100 мл) и экстрагировали этилацетатом (150 мл*2); водный слой затем доводили до рН=10-11 водным раствором гидроксида натрия (1 М) и полученную водную фазу экстрагировали этилацетатом (150 мл*2); объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении для получения Соединения 12.
[0073] Стадия 2: к раствору соединения 12 (6 г, 30,29 ммоль, 1 экв.) в дихлорметане (50 мл) добавляли Boc2O (6,61 г, 30,29 ммоль, 6,96 мл, 1 экв.) и триэтиламин (6,13 г, 60,59 ммоль, 8,43 мл, 2 экв.); смесь перемешивали при 20°С в течение 12 ч и концентрировали при пониженном давлении для удаления растворителя; остаток разбавляли водой (100 мл) и подвергали экстракции этилацетатом (50 мл*3); объединенные органические слои концентрировали при пониженном давлении для получения остатка, который очищали с помощью колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 1/0 до 10/1 (об./об.)) с получением соединения 1_3.
[0074] Стадия 3: к Соединению 1_3 (9 г, 30,18 ммоль, 1 экв.) и 4,4,4',4',5,5,5',5' -октаметил-2,2'-бис(1,3,2-диоксаборану) (15,33 г, 60,37 ммоль, 2 экв.) в ДМСО (150 мл) добавляли Pd(dppf)Cl2⋅CH2Cl2 (2,46 г, 3,02 ммоль, 0,1 экв.) и ацетат калия (11,85 г, 120,73 ммоль, 4 экв.); смесь заменяли азотом три раза и перемешивали при 90°С в течение 12 ч; реакционную смесь разбавляли водой (200 мл) и подвергали экстракции этилацетатом (150 мл*3); объединенные органические слои фильтровали и концентрировали фильтрат при пониженном давлении, и остаток очищали с помощью колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 100/1 до 20/1 (об.)) с получением Соединения 1_4.
[0075] Стадия 4: к раствору Соединения 1_4 (11 г, 31,86 ммоль, 1 экв.) в ТГФ (100 мл) добавляли Н2О2 (86,69 г, 764,69 ммоль, 73,47 мл, чистота 30%, 24 экв.) и уксусную кислоту (9,95 г, 165,68 ммоль, 9,48 мл, 5,2 экв.); смесь перемешивали при 20°С в течение 12 ч, затем гасили насыщенным карбонатом натрия (30 мл) и полученную смесь разбавляли водой (10 мл) и подвергали экстракции этилацетатом (20 мл*2); объединенные органические слои концентрировали при пониженном давлении с получением остатка, который очищали с помощью колоночной хроматографии (SiO2, петролейный эфир/этилацетат = 15/1-7/1 (об./об.)) с получением Соединения 1_5. ЖХ-МС (ИЭР) m/z: 180 (М-56+1). 1Н ЯМР (400 МГц, ДМСО-d6) δ (ррт): 7,09 (t, J=6,3 Гц, 1H), 6,72-6,65 (m, 2Н), 4,47 (brt, J=12,7 Гц, 4Н), 1,45 (s, 9Н).
[0076] Стадия 5: к раствору промежуточного соединения 1_5 (6,8 г, 26,69 ммоль, 1 экв.), этилоксиран-2-карбоксилата (7,75 г, 66,72 ммоль, 2,5 экв.) и  молекулярных сит (8 г) в МТВЕ (10 мл) добавляли катализатор A3 (673,34 мг, 800,64 мкмоль, 0,03 экв.), смесь трижды заменяли азотом и перемешивали при 20°С в течение 12 ч; реакционную смесь разбавляли этилацетатом (30 мл), фильтровали и фильтрат концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 6/1 до 3/1 (об./об.)) с получением Соединения 16.
молекулярных сит (8 г) в МТВЕ (10 мл) добавляли катализатор A3 (673,34 мг, 800,64 мкмоль, 0,03 экв.), смесь трижды заменяли азотом и перемешивали при 20°С в течение 12 ч; реакционную смесь разбавляли этилацетатом (30 мл), фильтровали и фильтрат концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 6/1 до 3/1 (об./об.)) с получением Соединения 16.
[0077] Стадию 6: к раствору соединения 16 (6,3 г, 17,32 ммоль, 1 экв.) в ДХМ (20 мл) добавляли ТФУ (14,88 г, 130,51 ммоль, 9,66 мл, 7,53 экв.) при 0°С, смесь перемешивали при 20°С в течение 1 ч и концентрировали при пониженном давлении с получением соли трифторуксусной кислоты Соединения 1_7.
[0078] Стадия 7: к раствору промежуточного соединения А2 (3,8 г, 7,30 ммоль, 1 экв.) в ДМ ФА (30 мл) добавляли триэтиламин (2,95 г, 29,20 ммоль, 4,06 мл, 4 экв.) и соль трифторуксусной кислоты Соединения 1_7 (5,33 г, 14,60 ммоль, 2 экв.); смесь перемешивали при 45°С в течение 2 часов, затем концентрировали при пониженном давлении для удаления ДМФА, остаток разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл); объединенные органические слои промывали насыщенным водным раствором хлорида натрия (25 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка, который очищали с помощью колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 1/1 до 0/1) с получением Соединения 1_8. ЖХМС (ИЭР) m/z: 704,4 (М+1).
[0079] Стадия 8: к раствору Соединения 1_8 (3,3 г, 4,50 ммоль, 1 экв.) в МеОН (20 мл) добавляли NaOH (378,39 мг, 9,46 ммоль, 2,1 экв.); после перемешивания смеси при 20°С в течение 17 ч реакционную смесь доводили до рН=3-4 с помощью разбавленной соляной кислоты (2 М) и концентрировали при пониженном давлении, остаток разбавляли метанолом (20 мл) для растворения, затем фильтровали и концентрировали при пониженном давлении для получения Соединения 19. ЖХМС (ИЭР) m/z: 580,5 (М+1).
[0080] Стадия 9: к раствору Соединения 19 (2 г, 3,45 ммоль, 1 экв.) в МеОН (20 мл) добавляли дифенилдиазометан (1,34 г, 6,90 ммоль, 2 экв.); смесь перемешивали при 20°С в течение 12 часов, затем концентрировали при пониженном давлении и остаток разбавляли водой (20 мл) и экстрагировали ДХМ (40 мл); объединенные органические слои промывали насыщенным водным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия, затем фильтровали и концентрировали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии (SiO2, ДХМ/МеОН = от 20/1 до 10/1 (об./об.)) с получением Соединения 1_10. ЖХМС (ИЭР) m/z: 746,5 (М+1).
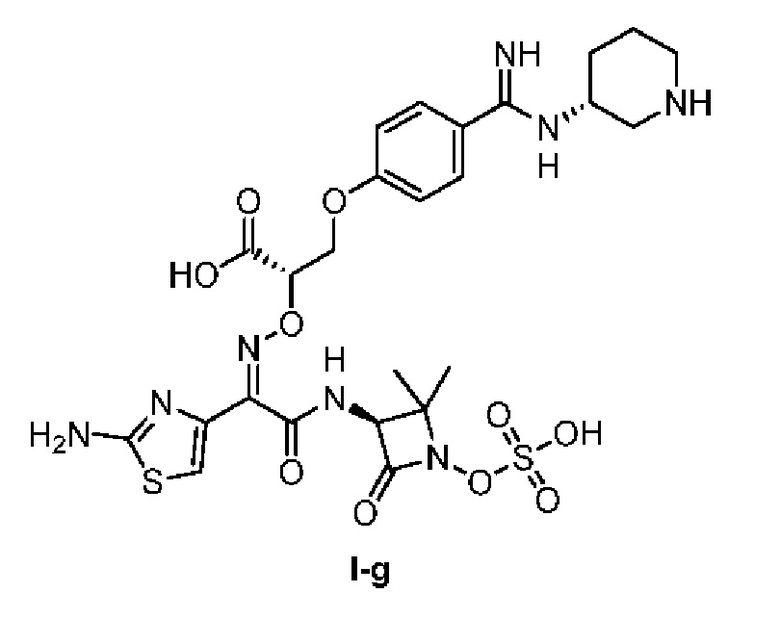
[0081] Стадия 10: к раствору Соединения 1_10 (1,2 г, 1,42 ммоль, 1 экв.) и 2-гидроксиизоиндолин-1,3-диона (278,65 мг, 1,71 ммоль, 1,2 экв.) в ТГФ (12 мл) добавляли PPI13 (560,04 мг, 2,14 ммоль, 1,5 экв.) и DIAD (431,75 мг, 2,14 ммоль, 415,15 мкл, 1,5 экв.) при 0°С; смесь перемешивали при 20°С в течение 1 ч, затем концентрировали при пониженном давлении для удаления ТГФ, и остаток очищали с помощью колоночной хроматографии (SiO2, ДХМ/EtOH=20/1 до 10/1 (об./об.)) с получением Соединения 111. ЖХМС (ИЭР) m/z: 891,5 (М+1).
[0082] Стадия 11: к раствору Соединения 1_11 (1 г, 1,10 ммоль, 1 экв.) в EtOH (10 мл) добавляли NH2NH2⋅Н2О (77,95 мг, 1,32 ммоль, 75,68 мкл, чистота 85%, 1,2 экв.); смесь перемешивали при 20°С в течение 30 мин, фильтровали и фильтрат концентрировали при пониженном давлении, остаток разбавляли водой (10 мл) и экстрагировали ДХМ (20 мл), объединенные органические слои сушили над безводным сульфатом аммония, фильтровали и концентрировали при пониженном давлении с получением Соединения 1_12. ЖХМС (ИЭР) m/z: 761,5 (М+1).
[0083] Стадия 12: к раствору соединения 1 12 (900 мг, 1,00 ммоль, 1 экв.) в ДХМ (5 мл) и ЕЮН (5 мл) добавляли промежуточное соединение А 2 (416,01 мг, 1,00 ммоль, 1 экв.), смесь перемешивали в атмосфере азота при 20°С в течение 1 ч, затем реакционную смесь концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии (SiO2, ДХМ/МеОН = от 20/1 до 10/1 (об./об.)) с получением соединения 1_13. ЖХМС (ИЭР) m/z: 1157,7 (М+1).
[0084] Стадия 13: к раствору Соединения 1_13 (200 мг, 163,39 мкмоль, 1 экв.) в ДМФА (2 мл) добавляли N,N'-диизопропилкарбодиимид (41,24 мг, 326,77 мкмоль, 2 экв.) и HOBt (44,15 мг, 326,77 мкмоль, 2 экв.); после того, как смесь перемешивали при 20°С в течение 1 ч, добавляли промежуточное соединение А1 (48,08 мг, 228,74 мкмоль, 1,4 экв.) и NaHCO3 (54,90 мг, 653,55 мкмоль, 25,42 мкл, 4 экв.) и перемешивали при 20°С в течение 11 ч; реакционную смесь разбавляли водой (8 мл) и собирали твердое вещество фильтрованием с получением Соединения 1_14. ЖХМС (ИЭР) m/z: 1350,2 (М+1).
[0085] Стадия 14: к раствору Соединения 1_14 (220 мг, 163,01 мкмоль, 1 экв.) в ДХМ (1 мл) добавляли ТФУ (1,54 г, 13,51 ммоль, 1 мл, 82,85 экв.) при 0°С и перемешивали в течение 1 ч; реакционную смесь разбавляли петролейным эфиром/этилацетатом (10 мл, 4/1) и собирали твердое вещество путем фильтрования и очищали с помощью препаративной ВЭЖХ (ТФУ, колонка: Phenomenex Synergi С18 150 мм×25 мм×10 мкм; подвижная фаза: [вода (0,1% ТФУ)-ацетонитрил]; ацетонитрил %: 1%-30%, 9 мин) с получением Соединения (I).
[0086] 1H ЯМР (400 МГц, D2O) δ=7,23 (d, J=8,4 Гц, 1H), 7,10 (s, 1H), 6,93-6,85 (m, 2H), 5,19 (dd, J=2,0, 5,7 Гц, 1H), 4,87-4,76 (m, 4H), 4,64 (s, 1H), 4,54-4,48 (m, 1H), 4,44-4,37 (m, 1H), 3,43 (br t, J=7,3 Гц, 4H), 3,04-2,91 (m, 4H), 1,98 (quin, J=7,6 Гц, 4H), 1,41 (s, 3H), 0,97 (s, 3Н) м.д.; ЖХ/МС (ИЭР) m/z: 741,3 (M+1).
[0087] Экспериментальный пример 1: Исследование с соединением, представленным формулой (I), на легочной инфекции Pseudomonas aeruginosa у мышей
[0088] 1. Экспериментальные штаммы
[0089] Pseudomonas aeruginosa РАИ.
[0090] 2. Тестируемые лекарственные средства
[0091] 1) Тестируемое соединение: Соединение, представленное формулой (I),
[0092] (2) Эталонное соединение: Вещество согласно заявке на патент AiCuris WO 2018065636: эталонное соединение I-g, азтреонам (продукт Dalian Meilunbio Biotechnology Co., Ltd.).
[0093]
[0094] 3. Среда
[0095] Агар Мюллера-Хинтона (МНА) и среду TSA приобретали у BD.
[0096] 4. Экспериментальные животные
[0097] 46 самок мышей CD-I (ICR), предоставленных Beijing Vital River Laboratory Animal Technology Co., Ltd., масса тела 23-27 г, возраст 7 недель.
[0098] 5. Методика проведения эксперимента:
[0099] (1) Иммуносупрессивные мыши, индуцированные внутрибрюшинной инъекцией циклофосфамида
[0100] 46 иммуносупрессивных мышей получали путем внутрибрюшинной инъекции циклофосфамида 150 мг/кг в день 1 и день 4.
[0101] (2) Группировка в экспериментах
[0102] Были созданы семь групп, включая группы с высокими, средними и низкими дозами соединения, представленного формулой (I), группы с высокими и средними дозами соединения I-g, группу азтреонама и модельную группу; в каждой группе было 6 животных, остальные 4 животных подвергались легочной инфекции в течение 2 часов, и ткани легких удаляли для подсчета бактериальной нагрузки. Конкретные группы см. в таблице ниже.
[0103]


[0104] (3) Легочная инфекция Pseudomonas aeruginosa
[0105] Мышам интратрахеально вводили 50 мкл бактериальной жидкости (2×103 КОЕ). Четыре модели мышей умерщвляли через 2 часа после инфицирования.
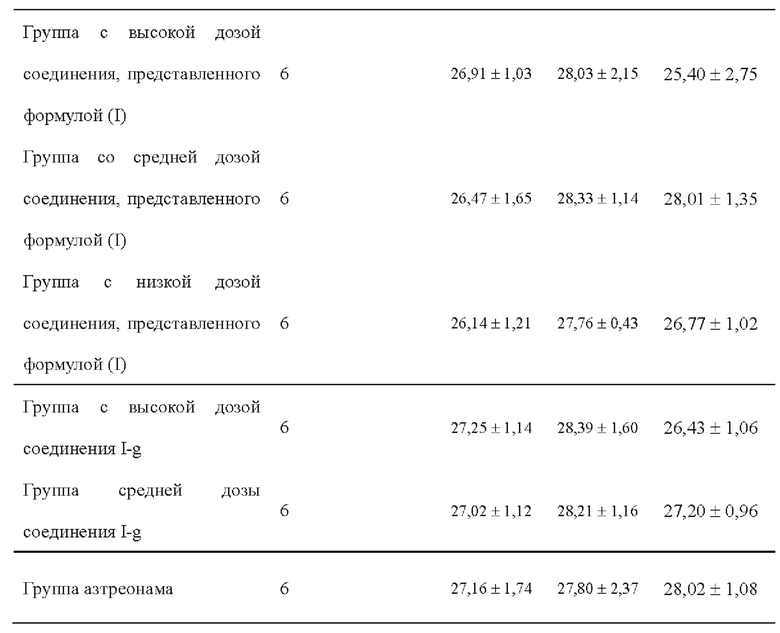
[0106] (4) Введение
[0107] Через 2 часа после инфицирования, мышам внутрибрюшинно вводили инъекцию один раз через 2 часа, 4 часа, 6 часов и 8 часов в соответствии с группами, в общей сложности 4 раза.
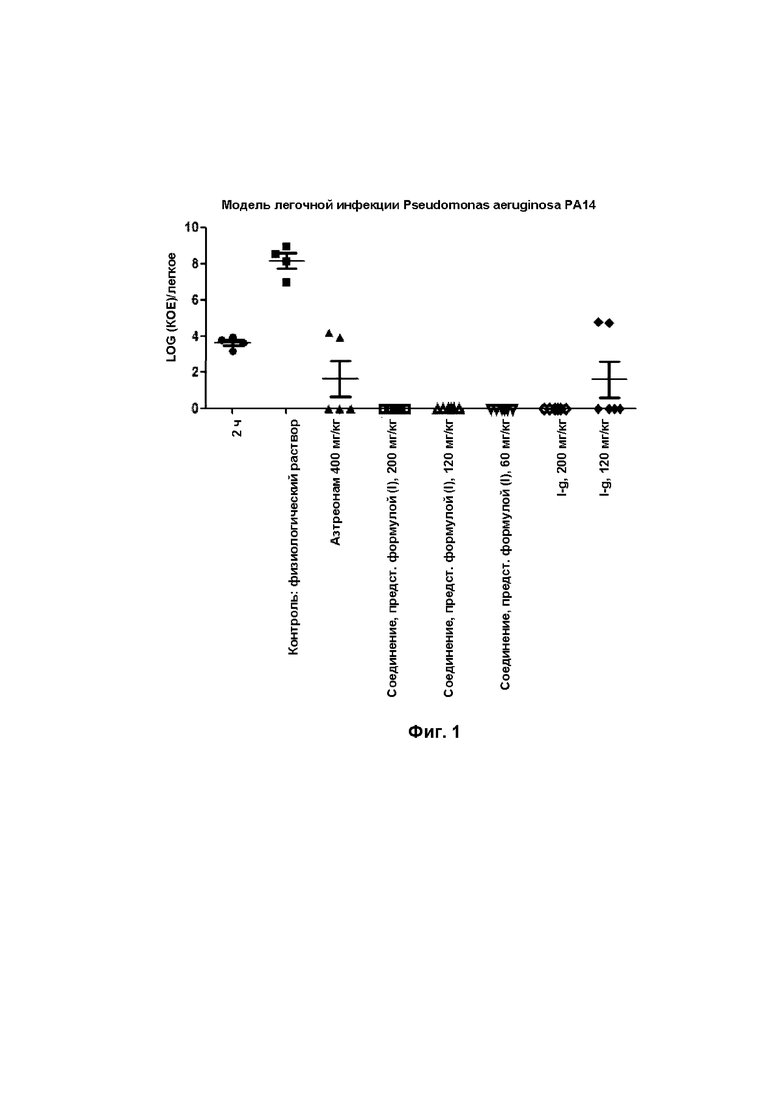
[0108] (5) Подсчет бактерий
[0109] Через 24 ч после инфицирования мышей в каждой группе умерщвляли путем вывиха шейки матки, а ткани легких и почек удаляли в асептических условиях, затем помещали в стерилизованную гомогенатную пробирку, взвешивали, добавляли надлежащее количество физиологического раствора (NS) и гомогенизировали гомогенизатором в течение 1 мин; ткани легких в модельной группе разбавляли в 104, 105 и 10б раз, ткани легких в каждой группе введения разбавляли в 10 и 100 раз, ткани почек в модельной группе разбавляли в 102, 103 и 104 раз, а ткани легких в каждой группе введения разбавляли в 10 раз; серийный разбавитель высевали на планшеты TSA со спиральным покрытием и инкубировали в течение ночи при 37°С для определения количества КОЕ с помощью счетчика колоний.
[0110] (6) Масса тела
[0111] Мышей взвешивали каждый день после начала испытания и регистрировали изменения массы тела.
[0112] (7) Обработка данных
[0113] Графики распределения КОЕ в легочной ткани были сделаны с использованием программного обеспечения для картирования Graphpad Prism. Средние значения КОЕ и массы тела рассчитывали с помощью программного обеспечения SPSS 19.0, а различия между группами анализировали с помощью дисперсионного анализа.
[0114] 6. Результаты эксперимента:
[0115] (1) Бактериальная нагрузка у мышей с ослабленным иммунитетом после легочной инфекции Pseudomonas aeruginosa
[0116] Четыре иммуносупрессивные мыши, которым дважды внутрибрюшинно вводили циклофосфамид, инфицировали Pseudomonas aeruginosa РА14 в количестве 1,06×104 КОЕ; через 2 часа легкие ткани удаляли и гомогенизировали для определения количества бактерий; рассчитывали бактериальную нагрузку у мышей со средней нагрузкой в диапазоне от 5,10×103 КОЕ.
[0117] (2) Изменения массы тела: массы тела каждой группы животных показаны в таблице 2.
[0118]

[0119] (3) Бактериальная нагрузка в легочной ткани мышей после лечения препаратом
[0120] Мышам внутрибрюшинно вводили соединения формулы (I), соединение I-g и азтреонам через 2 ч, 4 ч, 6 чи8 ч после инфицирования; животных умерщвляли через 24 ч, ткани легких удаляли в асептических условиях, погружали в физиологический раствор (NS), гомогенизировали, разбавляли соответствующим образом, а затем 50 мкл разбавителей равномерно высевали на планшеты TSA; планшеты инкубировали в течение ночи в инкубаторе при 37°С для подсчета количества колоний, которые преобразовывали в КОЕ на миллилитр с помощью коэффициента разбавления, и рассчитывали логарифмическое значение бактериальной нагрузки основания 10, а среднее и стандартное отклонение сравнивали в каждой группе. Результаты показаны в таблице 3 и на фиг. 1. Бактериальная нагрузка через 24 часа в модельной группе увеличилась с 1,06×104 КОЕ до 3,34×108 КОЕ (LOG10 бактериальной нагрузки составлял 8,14). Бактериальная нагрузка каждой группы введения была значительно ниже, чем у группы моделирования, и была в основном устранена. Соединение, представленное формулой (I), полностью элиминировали в группах с высокой, средней и низкой дозами.
[0121] Выводы:
[0122] Соединение, представленное формулой (I), оказывает in vivo лечебное действие на легочную инфекцию Pseudomonas aeruginosa у мышей с подавленным иммунитетом, вызванную циклофосфамидом, и может значительно снижать бактериальную нагрузку на ткани легких и устранять Pseudomonas aeruginosa, инфицированные легкими. При этом соединение, представленное формулой (I), полностью элиминировало бактерии, инфицированные в легком, в самой низкой дозе. Масса тела животных в группе введения существенно не изменялась, что указывает на то, что соединение, представленное формулой (I), является безопасным.

| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ FASN | 2014 |
|
RU2686323C2 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2017 |
|
RU2773346C2 |
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ НЕЙРОПРОТЕКТОРНЫМ ДЕЙСТВИЕМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2802457C1 |
| МАКРОЦИКЛИЧЕСКИЕ АНТИБИОТИКИ ШИРОКОГО СПЕКТРА ДЕЙСТВИЯ | 2020 |
|
RU2834484C2 |
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ FASN | 2014 |
|
RU2737434C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ EGFR | 2021 |
|
RU2815022C1 |
| ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ, ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, СПОСОБ ПОЛУЧЕНИЯ, КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2732576C2 |
| ИНГИБИТОРЫ МАТРИКСНОЙ МЕТАЛЛОПРОТЕИНАЗЫ (MMP) И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2797558C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРОВ ERBB | 2019 |
|
RU2810215C2 |
| МОДУЛЯТОРЫ КАЛЬПАИНА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2773288C2 |
Изобретение относится к применению терапевтически эффективного количества соединения, представленного формулой (I), или его фармацевтически приемлемой соли для получения лекарственного средства для лечения пневмонии, при этом пневмония вызвана инфицированием грамотрицательными бактериями, в частности Pseudomonas aeruginosa, фармацевтической композиции и способу лечения такой пневмонии. Технический результат – лечение пневмонии, вызванной инфицированием грамотрицательными бактериями, в частности Pseudomonas aeruginosa. 5 н. и 9 з.п. ф-лы, 1 ил., 3 табл., 2 пр.
1. Применение терапевтически эффективного количества соединения, представленного формулой (I), или его фармацевтически приемлемой соли для получения лекарственного средства для лечения пневмонии, при этом пневмония вызвана инфицированием грамотрицательными бактериями;
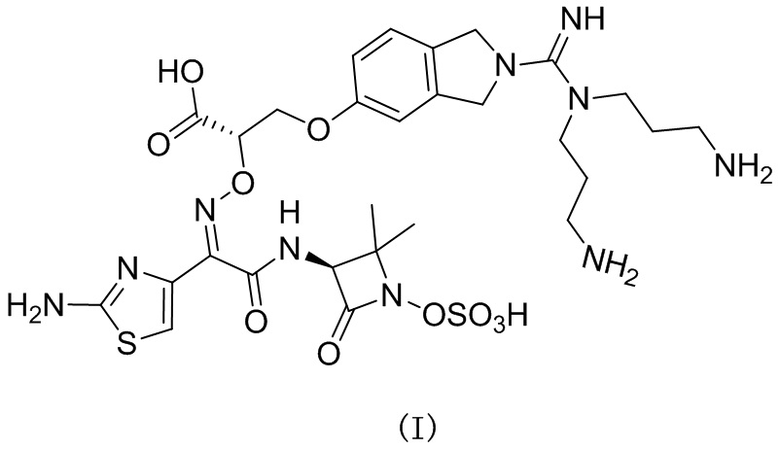 .
.
2. Применение по п. 1, где указанная пневмония вызвана инфекцией Pseudomonas aeruginosa.
3. Фармацевтическая композиция для лечения пневмонии, содержащая терапевтически эффективное количество соединения, представленного формулой (I), или его фармацевтически приемлемой соли в качестве активного ингредиента и одного или более фармацевтически приемлемых вспомогательных веществ и/или фармацевтически приемлемых носителей, при этом пневмония вызвана инфицированием грамотрицательными бактериями;
.
4. Композиция по п. 3, отличающаяся тем, что вспомогательное вещество представляет собой поверхностный стабилизатор, солюбилизатор, буфер, замутнитель, связующее вещество, разрыхлитель или смазывающее вещество.
5. Композиция по п. 4, отличающаяся тем, что поверхностный стабилизатор содержит амфотерное поверхностно-активное вещество, неионогенное поверхностно-активное вещество, катионное поверхностно-активное вещество, анионное поверхностно-активное вещество, или их комбинацию.
6. Композиция по любому из пп. 3-5, отличающаяся тем, что фармацевтическая композиция предназначена для перорального применения.
7. Композиция по п. 6, отличающаяся тем, что фармацевтическая композиция представляет собой таблетку или капсулу.
8. Композиция по любому из пп. 3-5, отличающаяся тем, что фармацевтическая комбинация находится в форме инъекционного препарата или ингаляционного препарата.
9. Применение композиции по любому из пп. 3-8 для получения лекарственного средства для лечения пневмонии, вызванной инфицированием грамотрицательными бактериями.
10. Применение по п. 9, отличающееся тем, что пневмония вызвана инфекцией Pseudomonas aeruginosa.
11. Способ лечения пневмонии, вызванной инфицированием грамотрицательными бактериями, у субъекта, нуждающегося в этом, включающий введение субъекту композиции по любому из пп. 3-8.
12. Способ лечения пневмонии по п. 11, в котором пневмония вызвана инфекцией Pseudomonas aeruginosa.
13. Способ лечения пневмонии у субъекта, нуждающегося в этом, включающий введение субъекту терапевтически эффективного количества соединения, представленного формулой (I), или его фармацевтически приемлемой соли, при этом пневмония вызвана инфицированием грамотрицательными бактериями;
.
14. Способ лечения пневмонии по п. 13, в котором пневмония вызвана инфекцией Pseudomonas aeruginosa.
| WO 2018065636 A1, 12.04.2018 | |||
| CN 108137573 B, 11.06.2021 | |||
| CN 106164072 B, 01.10.2019 | |||
| ПРОИЗВОДНЫЕ 2-ГИДРОКСИЭТИЛ-1Н-ХИНОЛИН-2-ОНА И ИХ АЗАИЗОСТЕРИЧЕСКИЕ АНАЛОГИ С АНТИБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ | 2010 |
|
RU2540862C2 |

