ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к способу получения высших алканонов, предпочтительно 6-ундеканона. В частности, настоящее изобретение относится к получению высших алканонов, предпочтительно 6-ундеканона, с использованием комбинированного биотехнологического и химического способа.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
6-Ундеканон обычно обнаруживается в травах и специях и входит в состав Osmanthus fragrans (османтус душистый). 6-Ундеканон представляет собой диалкиловый кетон формулы (CH3(CH2)4)2CO. Его, в частности, применяют в пищевой промышленности в качестве усилителя вкуса. В других отраслях промышленности 6-ундеканон применяют в качестве растворителя и в красках и покрытиях.
В промышленности 6-ундеканон обычно получают из карбоновых кислот путем приведения соответствующих карбоновых кислот в контакт с различными катализаторами на основе оксидов металлов при повышенных температурах. Например, в патенте США № 4754074 описан способ образования диалкиловых кетонов с использованием диоксида марганца, нанесенного на оксид алюминия, среди прочих примеров катализаторов, которые можно применять для получения диэтилкетона из пропионовой кислоты. В данной области было хорошо известно, что при получении кетонов из карбоновых кислот можно применять другие катализаторы, такие как оксиды свинца, железа, циркония, марганца, тория и неодима. Данные катализаторы можно применять без значительной потери атомов углерода, несмотря на то, что для эффективности способа необходим газообразный водород под высоким давлением при высоких температурах реакции 250-450°C. Данные условия повышают затраты на получение кетонов.
Кроме того, во всех этих осуществляемых способах полученный диалкиловый кетон всегда образуется из карбоновой кислоты в качестве исходного материала. Например, получение 6-ундеканона начинается с гексановой кислоты. Гексановую кислоту в основном получают исключительно из масел или жиров растительного и животного происхождения. Жиры животного происхождения в качестве сырьевых материалов все еще обладают слабой приемлемостью для клиентов, а масла растительного происхождения, которые содержат карбоновые кислоты с короткой и средней длиной цепи, либо сложно получать, либо их получают только в тропических районах, что также зачастую является результатом уничтожения дождевых лесов. Кроме того, конкретные сырьевые материалы из масел или жиров растительного и животного происхождения обладают специфическими, но определенными профилями жирных кислот, что приводит к сопряженному получению. Таким образом, трудно получить чистую гексановую кислоту в качестве субстрата для производства 6-ундеканона.
Соответственно, в данной области существует потребность в более эффективных средствах производства 6-ундеканона и других высших алканонов из других исходных материалов или в других исходных точках. В частности, в данной области существует потребность в способе получения 6-ундеканона из другого источника сырьевого материала, который обеспечивает эффективное и продуктивное получение.
ГРАФИЧЕСКИЕ МАТЕРИАЛЫ ПО ИЗОБРЕТЕНИЮ
На фиг. 1 показан микробный метаболический путь для удлинения углеродной цепи, например, (a) получение масляной кислоты (C4) с помощью рода Clostridium и Butyrivibrio (Kim BH, et al. Appl Environ Microbiol. 1984;48(4):764-70) и (b) получение гексановой кислоты, предполагаемое у Megasphaera elsdenii и Clostridium kluyveri (Khan MA. Melbourne: Victoria University; 2006).
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении предприняты попытки решить вышеупомянутые проблемы путем обеспечения полного цикла получения высших алканонов, предпочтительно 6-ундеканона, который предусматривает как биотехнологические, так и химические способы. В частности, получение высших алканонов, предпочтительно 6-ундеканона, начинается с простого субстрата, такого как этанол и/или ацетат, с получением гексановой кислоты с использованием биотехнологических способов. Затем образованную гексановую кислоту можно экстрагировать, и экстрагированную гексановую кислоту подвергнуть химической стадии, в ходе которой гексановая кислота преобразуется до высших алканонов, предпочтительно до 6-ундеканона. Преимущество данного способа заключается в том, что в начале используется недорогой и легкодоступный сырьевой материал для получения высших алканонов, предпочтительно 6-ундеканона. Сырьевой материал, представляющий собой ацетат и/или этанол, также получают с использованием способов, в которых не уничтожаются ни животные, ни растения. Кроме того, применение биотехнологической стадии для получения гексановой кислоты с последующей стадией экстракции согласно любому аспекту настоящего изобретения приводит к высокому и чистому выходу гексановой кислоты, которую затем можно легко применять для получения высших алканонов, предпочтительно 6-ундеканона, с использованием химической стадии.
В соответствии с одним аспектом настоящего изобретения представлен способ получения высших алканонов, предпочтительно 6-ундеканона, из этанола и/или ацетата, при этом способ включает
(a) приведение этанола и/или ацетата в контакт с по меньшей мере одним микроорганизмом, способным осуществлять удлинение углеродной цепи с получением гексановой кислоты и/или ее сложного эфира из этанола и/или ацетата;
(b) экстрагирование гексановой кислоты и/или ее сложного эфира из (a) с применением по меньшей мере одного экстрагирующего вещества в водной среде, где экстрагирующее вещество содержит по меньшей мере один алкилфосфиноксид и по меньшей мере один алкан, содержащий по меньшей мере 12 атомов углерода; или по меньшей мере один триалкиламин и по меньшей мере один алкан, содержащий по меньшей мере 12 атомов углерода; и
(c) приведение экстрагированной гексановой кислоты и/или ее сложного эфира из (b) в контакт с по меньшей мере одним катализатором кетонизации и далее с дополнительной алкановой кислотой, содержащей от 1 до 22 атомов углерода, при подходящих условиях реакции для химической кетонизации гексановой кислоты и далее дополнительной алкановой кислоты до высших алканонов, предпочтительно 6-ундеканона.
Применяемый в данном документе термин «высший алканон/алканол/алкановая кислота» относится к алканону/алканолу/алкановой кислоте, содержащим по меньшей мере шесть атомов углерода.
Микроорганизм на стадии (a), способный осуществлять удлинение углеродной цепи с получением гексановой кислоты, может представлять собой любой организм, который способен осуществлять удлинение углеродной цепи в соответствии с фиг. 1 (Jeon et al. Biotechnol Biofuels (2016) 9:129). Путь удлинения углеродной цепи также раскрыт у Seedorf, H., et al., 2008. Микроорганизмы согласно любому аспекту настоящего изобретения также могут предусматривать микроорганизмы, которые в их форме дикого типа не способны к удлинению углеродной цепи, но приобрели этот признак в результате генетической модификации. В частности, микроорганизм на стадии (a) может быть выбран из группы, состоящей из Clostridium carboxidivorans и Clostridium kluyveri. Более конкретно, микроорганизм согласно любому аспекту настоящего изобретения может представлять собой Clostridium kluyveri.
Стадия экстракции на стадии (b) согласно любому аспекту настоящего изобретения обеспечивает повышение выхода относительно количества применяемых экстрагирующих веществ. Например, менее 50% по весу экстрагирующего вещества можно применять для экстракции того же количества гексановой кислоты, как если бы применяли только чистые алканы. Следовательно, при малом объеме экстрагирующего вещества можно экстрагировать больший выход гексановой кислоты. Экстрагирующее вещество также не является пагубным для микроорганизмов. Соответственно, экстрагирующее вещество согласно любому аспекту настоящего изобретения может присутствовать, если гексановую кислоту получают биотехнологическим способом согласно любому аспекту настоящего изобретения. Следовательно, водную среду согласно любому аспекту настоящего изобретения можно рециклировать на стадию (a), в частности, после стадии (b) отделения гексановой кислоты. Данная стадия рециклизации обеспечивает рециклизацию и повторное применение микроорганизмов, поскольку экстрагирующее вещество согласно любому аспекту настоящего изобретения не является токсичным для микроорганизмов. Данная стадия рециклизации водной среды в способе согласно любому аспекту настоящего изобретения обладает дополнительным преимуществом в обеспечении возможности подвергать экстракции остаток гексановой кислоты, который не был экстрагирован на стадии (b) в первом цикле, в течение дополнительного времени или столько раз, сколько водную среду используют повторно. Кроме того, гексановая кислота может быть легко отделена от экстрагирующего вещества согласно любому аспекту настоящего изобретения путем дистилляции. Причина состоит в том, что гексановая кислота минимально подвергается дистилляции при значительно более низкой точке кипения, нежели экстрагирующее вещество, и после отделения путем дистилляции экстрагирующее вещество легко можно использовать повторно.
Способ согласно любому аспекту настоящего изобретения может включать стадию экстрагирования выделенной гексановой кислоты из водной среды. Выделенная гексановая кислота может относится к гексановой кислоте, которая может быть отделена от среды, в которой была получена гексановая кислота. В одном примере гексановую кислоту можно получать в водной среде (например, среде для ферментации, где гексановая кислота продуцируется специфическими клетками из источника углерода). Выделенная гексановая кислота может относится к гексановой кислоте, экстрагированной из водной среды. В частности, стадия экстрагирования обеспечивает отделение избыточной воды от водной среды, что приводит, таким образом, к образованию смеси, содержащей экстрагированную гексановую кислоту.
Экстрагирующее вещество также может называться «средой для экстракции» или «экстрагентом». Экстрагирующее вещество можно применять для экстрагирования/выделения гексановой кислоты, полученной согласно способу по настоящему изобретению, из водной среды, где гексановая кислота была изначально получена. В конце стадии экстрагирования избыточную воду из водной среды можно удалять с получением, таким образом, экстрагирующего вещества, содержащего экстрагированную гексановую кислоту. В частности, в конце стадии экстрагирования с экстракцией и удалением гексановой кислоты остаток может представлять собой среду для ферментации с клетками, применяемыми для получения гексановой кислоты, и затем эти клетки вместе со средой для ферментации можно использовать повторно для стадии (а). Специалист в данной области сможет определить, необходимо ли повторное добавление среды для ферментации и/или клеток после первого цикла. В частности, первый цикл согласно любому аспекту настоящего изобретения предусматривает один круг стадий (a)-(c). Затем среду и/или клетки можно рециклировать из второго цикла далее. Экстрагирующее вещество может содержать комбинацию соединений, которая может приводить к эффективным способам экстрагирования гексановой кислоты из водной среды. В частности, экстрагирующее вещество может содержать
- по меньшей мере один алкилфосфиноксид и по меньшей мере один алкан, содержащий по меньшей мере 12 атомов углерода; или
- по меньшей мере один триалкиламин и по меньшей мере один алкан, содержащий по меньшей мере 12 атомов углерода.
Экстрагирующее вещество согласно любому аспекту настоящего изобретения может эффективно экстрагировать гексановую кислоту в экстрагирующее вещество. Данное экстрагирующее вещество из смеси алкилфосфиноксида или триалкиламина и по меньшей мере одного алкана может считаться подходящим для способа согласно любому аспекту настоящего изобретения, поскольку смесь эффективно работает при экстрагировании необходимой гексановой кислоты в присутствии среды для ферментации. В частности, смесь алкилфосфиноксида или триалкиламина и по меньшей мере одного алкана может рассматриваться как действующая лучше, чем любой известный в настоящее время в уровне техники способ экстракции гексановой кислоты, поскольку она не требует какого-либо специального оборудования для осуществления процесса, и с ней относительно легко обеспечить высокий выход продукта. Кроме того, экстрагирующее вещество согласно любому аспекту настоящего изобретения также не является токсичным для микроорганизмов со стадии (a).
Алкан в экстрагирующем веществе может содержать по меньшей мере 12 атомов углерода. В частности, алкан может содержать порядка 12-18 атомов углерода. В одном примере алкан может быть выбран из группы, состоящей из додекана, тридекана, тетрадекана, пентадекана, гексадекана, гептадекана и октадекана. В дополнительном примере экстрагирующее вещество может содержать смесь алканов.
Алкилфосфиноксиды имеют общую формулу OPX3, где X представляет собой алкил. Подходящие алкилфосфиноксиды согласно любому аспекту настоящего изобретения включают алкильную группу, состоящую из линейного, разветвленного или циклического углеводорода, при этом углеводород состоит из от 1 до приблизительно 100 атомов углерода и от 1 до приблизительно 200 атомов водорода. В частности, «алкил», используемый по отношению к алкилфосфиноксиду согласно любому аспекту настоящего изобретения, может относиться к углеводородной группе, содержащей от 1 до 20 атомов углерода, зачастую от 4 до 15 атомов углерода или от 6 до 12 атомов углерода, и которая может состоять из прямых цепей, циклических звеньев, разветвленных цепей или их смесей. Алкилфосфиноксид может иметь от одной до трех алкильных групп при каждом атоме фосфора. В одном примере алкилфосфиноксид имеет три алкильные группы при атоме P. В некоторых примерах алкильная группа может содержать атом кислорода вместо одного атома углерода алкильной группы C4-C15 или C6-C12, при условии, что атом кислорода не присоединен к атому P алкилфосфиноксида. Как правило, алкилфосфиноксид выбран из группы, состоящей из триоктилфосфиноксида, трибутилфосфиноксида, гексилфосфиноксида, октилфосфиноксида и их смесей. Еще более конкретно алкилфосфиноксид может представлять собой триоктилфосфиноксид (TOPO).
Триалкиламины представляют собой органохимические соединения, полученные из аммиака (NH3), в которых три атома водорода замещены алкильными радикалами. Примерами триалкиламинов являются диметилэтиламин, метилдиэтиламин, триэтиламин, диметил-н-пропиламин, диметил-изо-пропиламин, метилди-н-пропиламин, диметилбутиламин, триоктиламин и т. п. В частности, триалкиламин, применяемый в экстрагирующем веществе согласно любому аспекту настоящего изобретения, может быть нерастворим в воде и может представлять собой триоктиламин.
В одном примере экстрагирующее вещество согласно любому аспекту настоящего изобретения может представлять собой комбинацию алкилфосфиноксида или триалкиламина и по меньшей мере одного алкана. В частности, алкан может содержать по меньшей мере 12 атомов углерода. Более конкретно, алкан может содержать 12-18 атомов углерода. В одном примере алкан может быть выбран из группы, состоящей из додекана, тридекана, тетрадекана, пентадекана, гексадекана, гептадекана и октадекана. В дополнительном примере экстрагирующее вещество может содержать смесь алканов. Еще более конкретно, экстрагирующее вещество согласно любому аспекту настоящего изобретения может представлять собой комбинацию TOPO и тетрадекана или гексадекана.
Триоктилфосфиноксид (TOPO) представляет собой фосфорорганическое соединение формулы OP(C8H17)3. TOPO может быть частью экстрагирующего вещества вместе с по меньшей мере одним алканом согласно любому аспекту настоящего изобретения. В частности, смесь TOPO и алкана, содержащего по меньшей мере 12 атомов углерода, может предусматривать весовое соотношение, составляющее от приблизительно 1:100 до 1:10, TOPO относительно алкана. Более конкретно, весовое соотношение TOPO и алкана в экстрагирующем веществе согласно любому аспекту настоящего изобретения может составлять приблизительно 1:100, 1:90, 1:80, 1:70, 1:60, 1:50, 1:40, 1:30, 1:25, 1:20, 1:15 или 1:10. Еще более конкретно, весовое соотношение TOPO и алкана может быть выбрано в диапазоне от 1:90 до 1:10, от 1:80 до 1:10, от 1:70 до 1:10, от 1:60 до 1:10, от 1:50 до 1:10, от 1:40 до 1:10, от 1:30 до 1:10 или от 1:20 до 1:10. Весовое соотношение TOPO и алкана может составлять от 1:40 до 1:15 или от 1:25 до 1:15. В одном примере весовое соотношение TOPO и алкана может составлять приблизительно 1:15. В примере алкан может представлять собой гексадекан и, следовательно, весовое соотношение TOPO и гексадекана может составлять приблизительно 1:15.
В другом примере, если экстрагирующее вещество содержит алкилфосфиноксид или триалкиламин, который является более растворимым в алкане, применяемом в экстрагирующем веществе, по сравнению с растворимостью TOPO в алкане, содержащем по меньшей мере 12 атомов углерода, то весовое соотношение алкилфосфиноксида (отличного от TOPO) или триалкиламина и алкана может составлять 1:1, 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1 или 10:1. В одном примере экстрагирующее вещество может представлять собой тригексилфосфиноксид, и соотношение тригексилфосфиноксида и алкана может составлять 1:1. В других примерах экстрагирующее вещество может представлять собой алкилфосфиноксид с меньшей длиной цепи, и соотношение алкилфосфиноксида с меньшей длиной цепи и алкана может составлять 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1 или 10:1. В данном случае алкилфосфиноксид с меньшей длиной цепи относится к фосфиноксиду с C1-C4алкильной группой. В другом примере экстрагирующее вещество может представлять собой триалкиламин, при этом известно, что он более растворим в алканах, чем фосфиноксид. Например, триалкиламин может представлять собой триоктиламин (TOA), который может присутствовать в экстрагирующем веществе с алканом согласно любому аспекту настоящего изобретения в соотношении не более 1:1. Амины с меньшей длиной цепи можно применять в еще более высоких соотношениях. В других примерах экстрагирующее вещество может представлять собой триалкиламин с меньшей длиной цепи, и соотношение триалкиламина с меньшей длиной цепи и алкана может составлять 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1 или 10:1. В данном случае алкилфосфиноксид с меньшей длиной цепи относится к фосфиноксиду с C1-C4алкильной группой.
Термин «приблизительно», используемый в данном документе, относится к изменениям в пределах 20 процентов. В частности, термин «приблизительно», используемый в данном документе, относится к +/- 20%, более конкретно +/-10%, еще более конкретно +/- 5% от данного измерения или значения.
На стадии (a) согласно любому аспекту настоящего изобретения этанол и/или ацетат приводят в контакт с по меньшей мере одним видом микроорганизмов, способных к осуществлению удлинения углеродной цепи с получением гексановой кислоты и/или ее сложного эфира из этанола и/или ацетата. В одном примере источник углерода может представлять собой этанол в комбинации с по меньшей мере одним другим источником углерода, выбранным из группы, состоящей из ацетата и бутирата. В частности, источником углерода может быть этанол и ацетат. В другом примере источник углерода может представлять собой комбинацию этанола и масляной кислоты. В одном примере углеродным субстратом может быть этанол сам по себе. В другом примере углеродным субстратом может быть ацетат сам по себе.
Источник ацетата и/или этанола может изменяться в зависимости от доступности. В одном примере этанол и/или ацетат может быть продуктом ферментации синтез-газа или любого углевода, известного из уровня техники. В частности, источник углерода для получения ацетата и/или этанола может быть выбран из группы, состоящей из спиртов, альдегидов, глюкозы, сахарозы, фруктозы, декстрозы, лактозы, ксилозы, пентозы, полиола, гексозы, этанола и синтез-газа.
Смеси источников можно использовать в качестве источника углерода.
Еще более конкретно, источником углерода может быть синтез-газ. Синтез-газ можно превращать в этанол и/или ацетат в присутствии по меньшей мере одного вида ацетогенных бактерий.
Применительно к источнику субстратов, содержащих диоксид углерода и/или монооксид углерода, специалисту в данной области техники будет понятно, что существует множество возможных источников для обеспечения CO и/или CO2 в качестве источника углерода. Можно увидеть, что на практике в качестве источника углерода по настоящему изобретению можно применять любой газ или любую смесь газов, которые способны предоставить микроорганизмам достаточные количества углерода, так чтобы мог быть образован ацетат и/или этанол из источника CO и/или CO2.
Как правило, для ацетогенной клетки по настоящему изобретению источник углерода предусматривает по меньшей мере 50% по весу, по меньшей мере 70% по весу, в частности по меньшей мере 90% по весу CO2 и/или CO, где процентное содержание по весу, %, относится ко всем источникам углерода, которые доступны для клетки согласно любому аспекту настоящего изобретения. Может быть предусмотрен источник, представляющий собой углеродный материал.
Примеры источников углерода в газообразных формах включают отработанные газы, такие как синтез-газ, топочный газ и газы от переработки нефтепродуктов, полученные при дрожжевой ферментации или ферментации с помощью клостридий. Эти отработанные газы образуются при газификации содержащих целлюлозу материалов или газификации угля. В одном примере эти отработанные газы не обязательно получают в виде побочных продуктов других процессов, но могут специально получать для применения со смешанной культурой по настоящему изобретению.
Согласно любому аспекту настоящего изобретения для получения ацетата и/или этанола, применяемых на стадии (а) согласно любому аспекту настоящего изобретения, источником углерода может быть синтез-газ. Синтез-газ можно, например, получать в виде побочного продукта газификации угля. Соответственно, микроорганизм согласно любому аспекту настоящего изобретения может быть способен к превращению вещества, которое представляет собой отходы производства, в ценный ресурс.
В другом примере синтез-газ может быть побочным продуктом газификации широко доступных, недорогих сельскохозяйственных сырьевых материалов для применения со смешанной культурой по настоящему изобретению с получением замещенных и незамещенных органических соединений.
Существуют многочисленные примеры сырьевых материалов, которые можно превращать в синтез-газ, поскольку почти все формы растительности могут быть использованы для этой цели. В частности, сырьевые материалы выбраны из группы, состоящей из многолетних трав, таких как мискантус, остатков кукурузы, отходов переработки, таких как опилки, и т. п.
В целом, синтез-газ можно получать в устройстве для газификации сухой биомассы в основном за счет пиролиза, частичного окисления и парового риформинга, где первичными продуктами синтез-газа являются CO, H2 и CO2. Синтез-газ также может быть продуктом электролиза CO2. Специалист в данной области техники узнает подходящие условия для проведения электролиза CO2 для получения синтез-газа, содержащего СО в требуемом количестве.
Как правило, часть синтез-газа, полученного в процессе газификации, сначала обрабатывают, чтобы оптимизировать значения выхода продукта и предотвратить образование смолы. Крекинг нежелательных смолы и CO в синтез-газе можно выполнять с использованием извести и/или доломита.
Общая эффективность, производительность по этанолу и/или ацетату и/или общий захват углерода в способе по настоящему изобретению могут зависеть от стехиометрии CO2, CO и H2 в непрерывном газовом потоке. Подводимые непрерывные газовые потоки могут состоять из CO2 и H2. В частности, в непрерывном газовом потоке диапазон концентраций CO2 может составлять приблизительно 10-50%, в частности 3% по весу, и H2 будет в пределах от 44% до 84%, в частности от 64 до 66,04% по весу. В другом примере непрерывный газовый поток также может предусматривать инертные газы, подобные N2, вплоть до концентрации N2, составляющей 50% по весу.
Более конкретно, источник углерода, содержащий CO и/или CO2, контактирует с ацетогенными клетками в непрерывном газовом потоке. Еще более конкретно, непрерывный газовый поток содержит синтез-газ. Эти газы можно подавать, например, с применением отдельных сопел, которые открываются в водную среду, фритт, мембран внутри трубы подачи газа в водную среду и т. п.
Специалист в данной области техники поймет, что может быть необходимо контролировать состав и скорости потока струй через определенные промежутки времени. Контроль состава потока может быть достигнут путем изменения пропорций составляющих его струй для достижения целевого или требуемого состава. Состав и скорость потока смешанного потока можно контролировать любыми средствами, известными из уровня техники. В одном примере систему адаптируют для непрерывного контроля скоростей потока и составов по меньшей мере двух струй и объединения их для получения единой смешанной струи субстрата в непрерывном газовом потоке оптимального состава и средств для прохождения оптимизированного потока субстрата в ферментер.
Согласно любому аспекту настоящего изобретения восстановитель, например водород, можно подавать вместе с источником углерода. В частности, этот водород можно подавать, когда подают и/или используют СO и/или CO2. В одном примере газообразный водород является частью синтез-газа, присутствующего согласно любому аспекту настоящего изобретения. В другом примере, где газообразного водорода в синтез-газе недостаточно для способа по настоящему изобретению, можно подавать дополнительный газообразный водород.
Термин «ацетогенные бактерии», используемый в данном документе, относится к микроорганизму, который способен осуществлять путь Вуда-Льюнгдаля и, таким образом, способен превращать CO, CO2 и/или водород в ацетат. Эти микроорганизмы включают микроорганизмы, которые в их форме дикого типа не осуществляют путь Вуда-Льюнгдаля, но приобрели этот признак в результате генетической модификации. Такие микроорганизмы включают без ограничения клетки E. coli. Эти микроорганизмы также могут быть известны как карбоксидотрофные бактерии. В настоящее время, из уровня техники известен 21 род различных ацетогенных бактерий (Drake et al., 2006), и они также могут включать некоторые Clostridia (Drake & Kusel, 2005). Эти бактерии способны использовать диоксид углерода или монооксид углерода в качестве источника углерода с водородом в качестве источника энергии (Wood, 1991). Кроме того, спирты, альдегиды, карбоновые кислоты, а также многочисленные гексозы также могут быть применены в качестве источника углерода (Drake et al., 2004). Восстановительный путь, который ведет к образованию ацетата, называется путем ацетил-CoA или Вуда-Льюнгдаля. В частности, ацетогенные бактерии могут быть выбраны из группы, состоящей из Acetoanaerobium notera (ATCC 35199), Acetonema longum (DSM 6540), Acetobacterium carbinolicum (DSM 2925), Acetobacterium malicum (DSM 4132), вида Acetobacterium № 446 (Morinaga et al., 1990, J. Biotechnol., Vol. 14, p. 187-194), Acetobacterium wieringae (DSM 1911), Acetobacterium woodii (DSM 1030), Alkalibaculum bacchi (DSM 22112), Archaeoglobus fulgidus (DSM 4304), Blautia producta (DSM 2950, ранее Ruminococcus productus, ранее Peptostreptococcus productus), Butyribacterium methylotrophicum (DSM 3468), Clostridium aceticum (DSM 1496), Clostridium autoethanogenum (DSM 10061, DSM 19630 и DSM 23693), Clostridium carboxidivorans (DSM 15243), Clostridium coskatii (ATCC № PTA-10522), Clostridium drakei (ATCC BA-623), Clostridium formicoaceticum (DSM 92), Clostridium glycolicum (DSM 1288), Clostridium ljungdahlii (DSM 13528), Clostridium ljungdahlii C-01 (ATCC 55988), Clostridium ljungdahlii ERI-2 (ATCC 55380), Clostridium ljungdahlii O-52 (ATCC 55989), Clostridium mayombei (DSM 6539), Clostridium methoxybenzovorans (DSM 12182), Clostridium ragsdalei (DSM 15248), Clostridium scatologenes (DSM 757), Clostridium species ATCC 29797 (Schmidt et al., 1986, Chem. Eng. Commun., Vol. 45, p. 61-73), Desulfotomaculum kuznetsovii (DSM 6115), Desulfotomaculum thermobezoicum подвида thermosyntrophicum (DSM 14055), Eubacterium limosum (DSM 20543), Methanosarcina acetivorans C2A (DSM 2834), Moorella sp. HUC22-1 (Sakai et al., 2004, Biotechnol. Let., Vol. 29, p. 1607-1612), Moorella thermoacetica (DSM 521, ранее Clostridium thermoaceticum), Moorella thermoautotrophica (DSM 1974), Oxobacter pfennigii (DSM 322), Sporomusa aerivorans (DSM 13326), Sporomusa ovata (DSM 2662), Sporomusa silvacetica (DSM 10669), Sporomusa sphaeroides (DSM 2875), Sporomusa termitida (DSM 4440) и Thermoanaerobacter kivui (DSM 2030, ранее Acetogenium kivui).
Более конкретно, можно применять штамм ATCC BAA-624 Clostridium carboxidivorans. Еще более конкретно, можно применять бактериальный штамм Clostridium carboxidivorans, помеченный «P7» и «P11», который описан, например, в U.S. 2007/0275447 и U.S. 2008/0057554.
Другим особенно подходящим видом бактерий может являться Clostridium ljungdahlii. В частности, штаммы, выбранные из группы, состоящей из Clostridium ljungdahlii PETC, Clostridium ljungdahlii ERI2, Clostridium ljungdahlii COL и Clostridium ljungdahlii O-52, могут быть использованы при превращении синтез-газа в гексановую кислоту. Эти штаммы, например, описаны в WO 98/00558, WO 00/68407, ATCC 49587, ATCC 55988 и ATCC 55989.
В одном примере получение гексановой кислоты из ацетата и/или этанола, который получен из синтез-газа, может предусматривать применение ацетогенных бактерий совместно с микроорганизмом, способным к удлинению углеродной цепи. Например, Clostridium ljungdahlii можно применять одновременно с Clostridium kluyveri. В другом примере одиночная клетка ацетогенных бактерий может быть способна к активности обоих организмов. Например, ацетогенные бактерии могут представлять собой C. carboxidivorans, которые могут быть способны к осуществлению как пути Вуда-Льюнгдаля, так и пути удлинения углеродной цепи.
Этанол и/или ацетат, применяемые на стадии (а) согласно любому аспекту настоящего изобретения, могут быть продуктом ферментации синтез-газа или могут быть получены с помощью других способов. Этанол и/или ацетат затем можно приводить в контакт с микроорганизмом на стадии (а).
Термин «приведение в контакт», используемый в данном документе, означает осуществление прямого контакта между микроорганизмом и этанолом и/или ацетатом. В одном примере этанол представляет собой источник углерода, и приведение в контакт на стадии (а) предусматривает приведение этанола в контакт с микроорганизмом из стадии (а). Контакт может являться прямым контактом или непрямым, что может предусматривать отделение мембраной или т. п. клеток от этанола, или случаи, когда клетки и этанол могут содержаться в двух различных отделениях и т. д. Например, на стадии (b) гексановая кислота и экстрагент могут быть в различных отделениях.
Микроорганизмы, способные производить гексановую кислоту согласно любому аспекту настоящего изобретения, могут быть культивированы с любыми культуральной средой, субстратами, условиями и способом, широко известными в области культивирования бактерий. Это обеспечивает получение гексановой кислоты с применением биотехнологического способа. В зависимости от микроорганизма, который применяют для получения гексановой кислоты, изменяются подходящие среда для роста, значение pH, температура, скорость перемешивания, уровень инокулята и/или аэробные, микроаэробные или анаэробные условия. Специалисту в данной области техники будут понятны другие условия, необходимые для осуществления способа согласно любому аспекту настоящего изобретения. В частности, условия в контейнере (например, ферментере) можно изменять в зависимости от применяемых микроорганизмов. Изменение условий таким образом, чтобы они подходили для оптимального функционирования микроорганизмов, находится в пределах знаний специалиста в данной области техники.
В одном примере способ, в частности стадию (а) согласно любому аспекту настоящего изобретения, можно осуществлять в водной среде с pH от 5 до 8, от 5,5 до 8 или от 5,5 до 7. Давление может составлять от 1 до 10 бар. Микроорганизмы можно культивировать при температуре, находящейся в диапазоне от приблизительно 20°C до приблизительно 80°C. В одном примере микроорганизм можно культивировать при 37°C.
В некоторых примерах для роста микроорганизма и для продуцирования им гексановой кислоты водная среда может содержать любые питательные вещества, ингредиенты и/или добавки, подходящие для выращивания микроорганизма или для стимулирования продуцирования гексановой кислоты. В частности, водная среда может содержать по меньшей мере одно из следующего: источников углерода, источников азота, таких как аммонийная соль, дрожжевой экстракт или пептон; минералов; солей; кофакторов; буферных средств; витаминов и любых других компонентов и/или экстрактов, которые могут стимулировать рост бактерий. Подлежащая применению культуральная среда должна соответствовать требованиям конкретных штаммов. Описания культуральных сред для различных микроорганизмов приведено в «Manual of Methods for General Bacteriology».
Термин «водный раствор» или «среда» включает любой раствор, содержащий воду, в основном воду, в качестве растворителя, который можно применять для сохранения клетки согласно любому аспекту настоящего изобретения по меньшей мере временно в метаболически активном и/или жизнеспособном состоянии, и содержит, если такое необходимо, любые дополнительные субстраты. Специалисту в данной области техники известны способы получения многочисленных водных растворов, обычно называемых средами, которые можно применять для сохранения культуры и/или клеток, например LB-среда в случае E. coli, можно применять ATCC1754-среду в случае C. ljungdahlii. Преимущественным является применение в качестве водного раствора минимальной среды, т. е. среды с достаточно простым составом, которая содержит только минимальный набор солей и питательных веществ, необходимых для сохранения клетки в метаболически активном и/или жизнеспособном состоянии, в отличие от сложных сред, во избежание необязательных загрязнений продуктов нежелательными побочными продуктами. Например, в качестве минимальной среды можно применять среду M9. Клетки инкубируют с источником углерода достаточно долго для получения необходимого продукта. Например, в течение по меньшей мере 1, 2, 4, 5, 10 или 20 часов. Выбранная температура должна быть такой, чтобы клетки согласно любому аспекту настоящего изобретения оставались каталитически компетентными и/или метаболически активными, например, от 10 до 42°C, предпочтительно от 30 до 40°C, в частности от 32 до 38°C, в случае, если клетка представляет собой клетку C. ljungdahlii. Водная среда согласно любому аспекту настоящего изобретения также предусматривает среду, в которой получают гексановую кислоту. В основном это относится к среде, в которой раствор содержит в основном воду. В одном примере водная среда, в которой клетки используют для получения гексановой кислоты, представляет собой именно ту среду, которая контактирует со средой для экстракции с целью экстракции гексановой кислоты.
В соответствии с любым аспектом настоящего изобретения предпочтительно, чтобы экстракция осуществлялась на стадии (b), при этом одновременно происходила ферментация на стадии (a). Это обозначает экстрагирование гексановой кислоты и/или ее сложного эфира in situ на стадии (b).
На стадии (b) согласно любому аспекту настоящего изобретения гексановую кислоту в водной среде можно приводить в контакт с экстрагирующим веществом в течение времени, необходимого для экстракции гексановой кислоты из водной среды в экстрагирующее вещество. Специалист в данной области техники может быть способен определить количество времени, необходимого для достижения равновесия распределения, и надлежащее скопление пузырьков, которое может понадобиться для оптимизации способа экстракции. В некоторых примерах необходимое время может зависеть от количества гексановой кислоты, которую можно экстрагировать. В частности, время, необходимое для экстракции гексановой кислоты из водной среды в экстрагирующее вещество может занимать всего несколько минут. Согласно любому аспекту настоящего изобретения, где экстракцию проводят на стадии (b) по мере прохождения ферментации на стадии (a), время экстракции может быть эквивалентным времени ферментации.
Соотношение применяемого экстрагирующего вещества и количества гексановой кислоты, подлежащей экстракции, может варьировать в зависимости от того, как быстро осуществляют экстракцию. В одном примере количество экстрагирующего вещества равно количеству водной среды, содержащей гексановую кислоту. После стадии приведения экстрагирующего вещества в контакт с водной средой две фазы (водную и органическую) разделяют с применением любых средств, известных из уровня техники. В одном примере две фазы можно разделять с применением разделительной воронки. Две фазы также можно разделять с применением смесителей-отстойников, пульсационных колонок и т. п. В одном примере отделение экстрагента от гексановой кислоты можно проводить с применением дистилляции с учетом того, что гексановая кислота подвергается перегонке при значительно более низкой точке кипения, нежели экстрагирующее вещество. Специалист в данной области техники сможет выбрать наилучший способ отделения экстрагирующего вещества от необходимой гексановой кислоты на стадии (b) в зависимости от характеристик гексановой кислоты. В частности, стадия (с) согласно любому аспекту настоящего изобретения предусматривает извлечение гексановой кислоты из стадии (b). Приведение гексановой кислоты в контакт с органическим экстрагирующим веществом приводит к образованию двух фаз, две фазы (водную и органическую) разделяют с применением любых средств, известных из уровня техники. В одном примере две фазы можно разделять с применением разделительной воронки. Две фазы также можно разделять с применением смесителей-отстойников, пульсационных колонок, разделения по температуре и т. п. В одном примере, в котором алкановая кислота представляет собой гексановую кислоту, отделение экстрагента от гексановой кислоты можно проводить с применением дистилляции с учетом того, что гексановая кислота подвергается дистилляции при значительно более низкой точке кипения, нежели экстрагент.
Стадия (b) оканчивается органическим абсорбентом, доступным снова для рециклизации или повторного применения.
Соответственно, способ экстракции гексановой кислоты согласно любому аспекту настоящего изобретения можно применять вместе с любым биотехнологическим способом получения гексановой кислоты. Это особенно преимущественно поскольку, как правило, во время процесса ферментации с получением гексановой кислоты с применением биологических способов, гексановую кислоту будут оставлять для сбора в водной среде, и после достижения определенных значений концентрации в среде для ферментации как раз целевой продукт (гексановая кислота) может подавлять активность и продуктивность микроорганизма. Таким образом, это ограничивает общий выход процесса ферментации. При применении данного способа экстракции гексановую кислоту экстрагируют по мере ее продуцирования, таким образом существенно снижая ингибирование конечным продуктом.
Способ согласно любому аспекту настоящего изобретения также является более эффективным и сокращающим затраты, чем традиционные способы удаления гексановой кислоты, в частности, из способа ферментации по мере ее продуцирования, поскольку не существует первичного упора на дистилляцию и/или осаждение с извлечением гексановой кислоты. Способ дистилляции или осаждения может приводить к более высоким производственным затратам, более низкому выходу и более высокой степени образования отходов производства, что снижает общую эффективность способа. С помощью способа согласно любому аспекту настоящего изобретения предпринимают попытки преодолеть эти недостатки.
В частности, смесь микроорганизма и источника углерода согласно любому аспекту настоящего изобретения можно применять в любом известном биореакторе или ферментере для осуществления любого аспекта настоящего изобретения. В одном примере представлен полный способ согласно любому аспекту настоящего изобретения, который начинается с биотехнологического получения гексановой кислоты из ацетата и/или этанола и заканчивается экстракцией гексановой кислоты в одном контейнере. Следовательно может не быть стадии разделения между стадией получения гексановой кислоты и стадией экстрагирования гексановой кислоты. Это сохраняет время и затраты. В частности, во время процесса ферментации микроорганизм можно выращивать в водной среде и в присутствии экстрагирующего вещества. Таким образом, способ согласно любому аспекту настоящего изобретения предусматривает однореакторное устройство для получения гексановой кислоты. Кроме того, поскольку гексановую кислоту экстрагируют по мере продуцирования, никакого ингибирования конечным продуктом не происходит, что обеспечивает поддержание выхода гексановой кислоты. Дополнительную стадию разделения можно проводить с удалением гексановой кислоты. Можно применять любой способ разделения, известный из уровня техники, такой как с использованием воронки, колонки, дистилляции и т. п. Оставшиеся экстрагирующее вещество и/или клетки затем можно использовать повторно.
В другом примере способ экстракции гексановой кислоты может происходить в качестве отдельной стадии и/или в другом реакторе. После осуществления ферментации, при которой уже получили необходимую гексановую кислоту, подлежащую экстрагированию, экстрагирующее вещество согласно любому аспекту настоящего изобретения можно добавлять к среде для ферментации или среду для ферментации можно добавлять в реактор, содержащий экстрагирующее вещество. Затем можно экстрагировать необходимую гексановую кислоту с помощью любого способа разделения, известного из уровня техники, такого как с использованием воронки, колонки, дистилляции и т. п. Оставшееся экстрагирующее вещество затем можно использовать повторно. Среду для ферментации с клетками также можно использовать повторно.
Другое преимущество способа заключается в том, что экстрагирующее вещество можно использовать повторно. Следовательно, как только гексановую кислоту отделяют от экстрагирующего вещества, экстрагирующее вещество можно рециклировать и повторно применять, снижая количество отходов.
Стадия (c) способа согласно любому аспекту настоящего изобретения предусматривает (c) приведение в контакт экстрагированной гексановой кислоты и/или ее сложного эфира из (b) с по меньшей мере одним катализатором кетонизации и далее с другой алкановой кислотой, содержащей от 1 до 22 атомов углерода, при подходящих условиях реакции для химической кетонизации гексановой кислоты до высших алканонов, предпочтительно 6-ундеканона.
Термин «допонительная алкановая кислота, содержащая 1-22 атомов углерода» не охватывает гексановую кислоту. Предпочтительно дополнительная алкановая кислота, содержащая 1-22 атомов углерода, выбрана из прямоцепочечных алкановых кислот, содержащих 4-18, предпочтительно 5-12 атомов углерода.
Катализатор кетонизации согласно любому аспекту настоящего изобретения может представлять собой катализатор на основе оксида металла или смеси на его основе. Кетонизация обеспечивает реакцию гексановой кислоты с дополнительной алкановой кислотой и/или гексановой кислотой, предпочтительно обеспечивает димеризацию двух молекул гексановой кислоты до одной молекулы кетона с удалением одной молекулы воды и одной молекулы диоксида углерода. Механизм, который может быть задействован в кетонизации гексановой кислоты, где может быть образован гексановый ангидрид ((CH3(CH2)4)COOCO(CH2)4CH3), раскрыт по меньшей мере в Woo, Y., Ind. Eng. Chem. Res. 2017, 56: 872-880. Кетонизация гексановой кислоты в присутствии различных катализаторов на основе оксидов металлов также показана в Wang, S. J. Phys. Chem. C 2017, 121, 18030-18046.
Катализатор кетонизации, применяемый согласно любому аспекту настоящего изобретения, может представлять собой гетерогенный катализатор для эффективного производства кетона с более высокой плотностью энергии, предпочтительно C11, из биологически продуцируемой гексановой кислоты согласно стадии (а). В частности, катализатор кетонизации может представлять собой любой катализатор на основе оксида металла или смеси на его основе, выбранные из группы, состоящей из катализатора на основе оксида металла или смесей на его основе, при этом он выбран из группы, состоящей из катализатора на основе гетерополикислоты (H3PW12O40), катализатора на основе оксида ниобия (Nb2O5), катализатора на основе оксида титана (TiO2), катализатора на основе оксида церия (CeO2), смешанного катализатора на основе оксидов цинка и хрома (Zn-Cr), катализатора на основе оксида марганца (MnOx), катализатора на основе оксида лантана (La2O3), катализатора на основе оксида магния (MgO), катализатора на основе оксида железа (FeO, FeO2, Fe2O3, Fe3O4, Fe4O5, Fe5O6, Fe5O7), смешанного катализатора на основе оксидов кремния и алюминия (SiyAlzO), катализатора на основе оксида алюминия (Al2O3) и катализатора на основе диоксида циркония (ZrO2). В MnOx «x» может составлять 1, 2 или 4. В SiyAlzO «y» и «z» могут относиться к любому числу, при этом соотношение z/y представляет собой любое число от 0 до 1. В одном примере кетонизацию осуществляют на гексановой кислоте, как раскрыто в Pham T. N., ACS Catal. 2013, 3: 2456-2473, с использованием подходящего гетерогенного металлического катализатора гидрогенизации и подходящих условий реакции. Раскрытые условия могут варьировать в зависимости от применяемого катализатора для эффективного выхода высших алканонов, предпочтительно 6-ундеканона. В еще одном примере согласно тому, что раскрыто в Gliński, M. et al, Polish J. Chem. 2004, 78: 299-302, для кетонизации гексановой кислоты до 6-ундеканона можно применять катализатор на основе MnO2 и/или Al2O3. В следующем примере при кетонизации гексановой кислоты до 6-ундеканона можно применять катализатор на основе Nb2O5, как раскрыто в US 6265618 B1, в частности, в примере 3. Специалист в данной области техники простым способом проб и ошибок сможет определить, исходя из уровня техники, подходящий катализатор и подходящие условия для получения высших алканонов, предпочтительно 6-ундеканона, из гексановой кислоты и далее другой алкановой кислоты. В Orozco, L.M et al ChemSusChem, 2016, 9(17): 2430-2442 и Orozco, L.M et al Green Chemistry, 2017, 19(6): 1555-1569 также раскрыт другой катализатор, который можно применять в качестве катализатора кетонизации согласно любому аспекту настоящего изобретения.
Специалист в данной области техники сможет определить подходящие условия для применения различных катализаторов кетонизации на стадии (с). В частности, катализатор кетонизации может представлять собой катализатор на основе аэрогеля на основе диоксида циркония, и его можно применять в кетонизации гексановой кислоты, как раскрыто в Woo, Y., Ind. Eng. Chem. Res. 2017, 56: 872-880. Катализатор на основе аэрогеля на основе диоксида циркония не только может обеспечивать эффективное получение высших алканонов, предпочтительно 6-ундеканона, но также предотвращает вымывание катализаторов. У Lee, Y. et al в Applied Catalysis A: General. 2015, 506: 288-293 раскрыты различные катализаторы кетонизации и их эффективность в кетонизации гексановой кислоты. Специалист в данной области техники легко может применять способ, описанный у Lee Y., et al, для определения подходящего катализатора кетонизации и/или условий для применения при кетонизации гексановой кислоты. В частности, подходящие условия реакции на стадии (c) предусматривают температуры реакции 100-500°C, 100-450°C, 100-400°C, 100-350°C, 100-300°C, 100-250°C, 100-200°C, 150-500°C, 150-450°C, 150-400°C, 150-350°C, 150-300°C, 150-250°C, 150-200°C, 200-500°C, 200-450°C, 200-400°C, 200-350°C, 200-300°C, 200-250°C, 250-500°C, 250-450°C, 250-400°C, 250-350°C, 250-300°C и т. п. Более конкретно, высшие алканоны, предпочтительно 6-ундеканон, могут быть получены из гексановой кислоты с использованием катализатора на основе аэрогеля на основе диоксида циркония в качестве катализатора кетонизации при температурах реакции 150-250°C.
В соответствии с другим аспектом настоящего изобретения представлен способ получения высших алканолов, предпочтительно 6-ундеканола, из этанола и/или ацетата, при этом способ включает
(d) приведение высшего алканона, предпочтительно 6-ундеканона, полученного согласно любому аспекту настоящего изобретения, в контакт с по меньшей мере одним металлическим катализатором гидрогенизации для каталитической гидрогенизации высшего алканона, предпочтительно 6-ундеканона, до высших алканолов, предпочтительно 6-ундеканола. Высшие алканолы, предпочтительно 6-ундеканол (C11H24O), вторичный спирт, являются результатом каталитической гидрогенизации высшего алканона, предпочтительно 6-ундеканона, при этом молекулу водорода добавляют между двойной связью углерод-кислород для получения далее высшего алканола, предпочтительно 6-ундеканола, в качестве конечного продукта.
В частности, данный аспект настоящего изобретения включает (а) приведение этанола и/или ацетата в контакт с по меньшей мере одним микроорганизмом, способным осуществлять удлинение углеродной цепи, с образованием гексановой кислоты и/или ее сложного эфира из этанола и/или ацетата; (b) экстрагирование гексановой кислоты и/или ее сложного эфира из (а) с использованием по меньшей мере одного экстрагирующего вещества в водной среде, где экстрагирующее вещество содержит по меньшей мере один алкилфосфиноксид и по меньшей мере один алкан, содержащий по меньшей мере 12 атомов углерода; или по меньшей мере один триалкиламин и по меньшей мере один алкан, содержащий по меньшей мере 12 атомов углерода; (c) приведение экстрагированной гексановой кислоты и/или ее сложного эфира из (b) в контакт с по меньшей мере одним катализатором кетонизации и далее с дополнительной алкановой кислотой, содержащей от 1 до 22 атомов углерода, при подходящих условиях реакции для химической кетонизации гексановой кислоты и далее дополнительной алкановой кислоты до высшего алканона, предпочтительно 6-ундеканона, и (d) приведение высшего алканона, предпочтительно 6-ундеканона, в контакт с по меньшей мере одним металлическим катализатором гидрогенизации для каталитической гидрогенизации высшего алканона, предпочтительно 6-ундеканона, до высших алканолов, предпочтительно 6-ундеканола.
Металлический катализатор гидрогенизации может представлять собой гомогенный или гетерогенный катализатор. Гомогенные металлические катализаторы могут представлять собой комплексы металлов, которые известны в данной области техники. В частности, металлический катализатор гидрогенизации может представлять собой гетерогенный катализатор. Некоторые преимущества применения многофазных каталитических реакций с использованием твердых катализаторов включают легкое разделение катализаторов и продуктов, легкое извлечение, рециклизацию катализатора и относительно мягкие рабочие условия. Существуют также явные экономические и экологические стимулы в использовании гетерогенных катализаторов. В частности, металлический катализатор гидрогенизации может быть выбран из группы, состоящей из катализатора на основе рутения (Ru), катализатора на основе рения (Re), катализатора на основе никеля (Ni), катализатора на основе железа (Fe), катализатора на основе кобальта (Co), катализатора на основе палладия (Pd) и катализатора на основе платины (Pt). Более конкретно, катализатор может быть выбран из группы, состоящей из катализатора на основе Ni, Pd и Pt. В одном примере металлический катализатор гидрогенизации, применяемый согласно любому аспекту настоящего изобретения, может представлять собой наночастицы никеля, как описано в Alonso, F. Tetrahedron, 2008, 64: 1847-52. В другом примере клешневидные комплексы PNP с железом (II) можно применять в качестве металлического катализатора гидрогенизации для гидрогенизации высших алканонов до высших спиртов, предпочтительно 6-ундеканона до 6-ундеканола, как раскрыто в Gorgas, N., Organometallics, 2014, 33 (23): 6905-6914. В еще одном примере наночастицы магнетита катализатора на основе рутения (Ru), катализатора на основе рения (Re), катализатора на основе никеля (Ni), катализатора на основе железа (Fe), кобальта (Co), палладия (Pd) или катализатора на основе платины (Pt), как описано в Tariq Shah M., et al., ACS Applied Materials & Interfaces, 2015: 7(12), 6480-9, можно применять в качестве гетерогенного металлического катализатора гидрогенизации согласно любому аспекту настоящего изобретения. В еще одном примере комплекс медь-фосфин применяют в качестве гомогенного металлического катализатора гидрогенизации согласно любому аспекту настоящего изобретения, как раскрыто в Chen, J-X., Tetrahedron, 2000, 56: 2153-2166. В следующем примере гетерогенный катализатор на основе Pt, в частности, катализатор на основе Pt/Al2O3, как раскрыто в Journal of Molecular Catalysis A: Chemical, 2014, 388-389: 116-122, можно применять для гидрогенизации высшего алканона до высшего спирта, предпочтительно 6-ундеканона до 6-ундеканола. В ChemSusChem, 2017: 10(11), 2527-2533 также раскрыты различные гетерогенные катализаторы, такие как Pt/C, Ru/C и Pd/C, которые можно применять в комбинации с кислотным катализатором или без него для гидрогенизации 6-ундеканона до 6-ундеканола. На основании вышеизложенного специалист в данной области техники сможет определить подходящий катализатор гидрогенизации, который можно применять в любом аспекте настоящего изобретения с получением высшего алканола из высшего алканона, предпочтительно 6-ундеканола из 6-ундеканона.
Специалист в данной области техники легко сможет определить подходящий металлический катализатор гидрогенизации и соответственно изменить условия для эффективного получения высших алканолов, предпочтительно 6-ундеканола, посредством гидрогенизации 6-ундеканона.
В соответствии с еще одним аспектом настоящего изобретения представлен способ получения алкановых кислот, предпочтительно лауриновой кислоты, из этанола и/или ацетата, при этом способ включает
(e) приведение высшего алканола, предпочтительно 6-ундеканола, полученного согласно любому аспекту настоящего изобретения, в контакт с CO2 и гомогенным катализатором карбоксилирования, обеспечивающим карбоксилирование высшего алканола, предпочтительно 6-ундеканола, до алкановой кислоты, предпочтительно лауриновой кислоты.
В частности, данный аспект настоящего изобретения предусматривает (а) приведение этанола и/или ацетата в контакт с по меньшей мере одним микроорганизмом, способным осуществлять удлинение углеродной цепи, с получением гексановой кислоты и/или ее сложного эфира из этанола и/или ацетата; (b) экстрагирование гексановой кислоты и/или ее сложного эфира из (а) с использованием по меньшей мере одного экстрагирующего вещества в водной среде, где экстрагирующее вещество содержит по меньшей мере один алкилфосфиноксид и по меньшей мере один алкан, содержащий по меньшей мере 12 атомов углерода; или по меньшей мере один триалкиламин и по меньшей мере один алкан, содержащий по меньшей мере 12 атомов углерода; (c) приведение экстрагированной гексановой кислоты и/или ее сложного эфира из (b) в контакт с по меньшей мере одним катализатором кетонизации и далее с дополнительной алкановой кислотой, содержащей от 1 до 22 атомов углерода, при подходящих условиях реакции для химической кетонизации гексановой кислоты и далее дополнительной алкановой кислоты до высшего алканона, предпочтительно 6-ундеканона, и (d) приведение высшего алканона, предпочтительно 6-ундеканона, в контакт с по меньшей мере одним металлическим катализатором гидрогенизации для каталитической гидрогенизации высшего алканона, предпочтительно 6-ундеканона, до высших алканолов, предпочтительно 6-ундеканола, и (e) приведение высшего алканола, предпочтительно 6-ундеканола, полученного согласно любому аспекту настоящего изобретения, в контакт с CO2 и гомогенным катализатором карбоксилирования, обеспечивающим карбоксилирование высшего алканола, предпочтительно 6-ундеканола, до алкановой кислоты, предпочтительно лауриновой кислоты.
Алкановые кислоты, предпочтительно лауриновая кислота (C12H24O2), также известная как додекановая кислота, может быть получена из высшего алканола, предпочтительно 6-ундеканола, в присутствии катализатора карбоксилирования. Возможный способ по меньшей мере раскрыт на схеме 1.
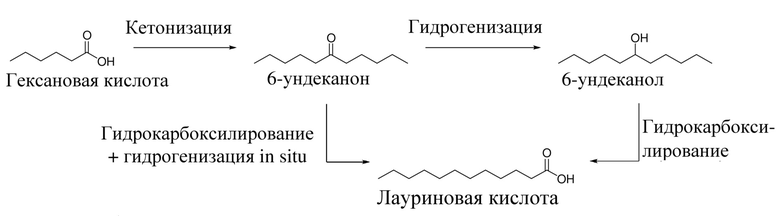
Схема 1. Образование 6-ундеканона, 6-ундеканола и лауриновой кислоты из гексановой кислоты
Катализатор карбоксилирования может быть выбран из группы, состоящей из катализаторов на основе Rh, Ni, Ru, Co, Pt, Pd и Fe. Специалист в данной области техники сможет определить подходящие условия и лиганды, которые могут быть необходимы для применения в сочетании с катализатором карбоксилирования для эффективного и действенного превращения высшего алканола, предпочтительно 6-ундеканола, полученного в соответствии с любым аспектом настоящего изобретения, в алкановую кислоту, предпочтительно лауриновую кислоту. В частности, катализатор карбоксилирования согласно любому аспекту настоящего изобретения может представлять собой гомогенный катализатор карбоксилирования. В одном примере стадия (e) согласно любому аспекту настоящего изобретения предусматривает (ei) приведение высшего алканола, предпочтительно 6-ундеканола, полученного согласно любому аспекту настоящего изобретения, в контакт с по меньшей мере одним катализатором на основе никеля и диоксидом углерода при атмосферном давлении для карбоксилирования высшего алканола до высшей алкановой кислоты, предпочтительно 6-ундеканола до лауриновой кислоты. Стадия (ei) может предусматривать образование промежуточного бромида. Подробности способа на стадии (ei) дополнительно раскрыты в Juliá-Hernández, Nature, 2017, 545: 84-88.
В другом примере стадия (e) согласно любому аспекту настоящего изобретения предусматривает (eii) приведение высшего алканола, предпочтительно 6-ундеканола, полученного согласно любому аспекту настоящего изобретения, в контакт с азотом, CO2 и гомогенным катализатором на основе Rh для карбоксилирования высшего алканола до высшей алкановой кислоты, предпочтительно 6-ундеканола до лауриновой кислоты. Данный способ каталитического гидрокарбоксилирования (схема 2) дополнительно подробно описан в Ostapowicz, T.G et al. Angew. Chem. Int, Ed., 2013, 52:12119-23.
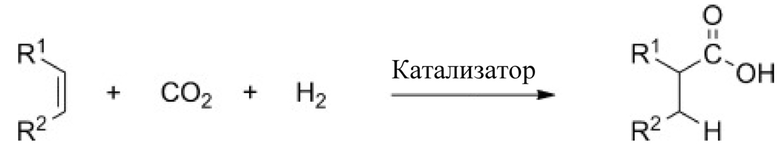
Схема 2. Каталитическое гидрокарбоксилирование олефинов с CO2
В частности, стадию (eii) можно осуществлять при повышенных температурах. Более конкретно, повышенная температура для стадии (eii) может находиться в диапазоне от 100 до 300°C. Например, температура для стадии (eii) может находиться в диапазоне 100-250°C, 100-200°C, 150-300°C или 150-250°C. Кроме того, подходящие условия для стадии (eii) могут включать парциальное давление CO2 в диапазоне от 0,1 до 200 бар, в частности, парциальное давление CO2 может составлять 0,1-150, 0,1-100, 0,1-60 бар и т. п. Специалист в данной области техники сможет определить подходящие условия для карбоксилирования, при которых катализатор не ингибируется и обеспечивается хороший выход высшей алкановой кислоты из высшего алканола, предпочтительно лауриновой кислоты из 6-ундеканола.
Другие примеры катализаторов карбоксилирования, которые можно применять на стадии (e) согласно любому аспекту настоящего изобретения могут быть описаны по меньшей мере в Journal of Molecular Catalysis A: Chemical, 2003, 197: 61-64, J. Am. Chem. Soc. 1985, 107: 3568-3572, J. Am. Chem. Soc. 1985, 107: 3565-3567 и Journal of Molecular Catalysts, 1987, 40: 71-82.
В одном примере металлический катализатор гидрогенизации на стадии (c) также может быть способен осуществлять карбоксилирование на стадии (e) согласно любому аспекту настоящего изобретения. Следовательно, один катализатор может обеспечивать образование алкановой кислоты непосредственно из высшего алканона, предпочтительно лауриновой кислоты непосредственно из 6-ундеканона. В частности, катализатор, который можно применять для преобразования высшего алканона до алкановой кислоты, предпочтительно 6-ундеканона до лауриновой кислоты (т. е. на стадиях (d) и (e)) может представлять собой гомогенный металлический катализатор. Более конкретно, гомогенный металлический катализатор, который можно применять для осуществления стадий (d) и (e) может представлять собой катализатор на основе Rh. Возможные примеры гомогенного катализатора на основе Rh и подходящие условия для применения описаны по меньшей мере в Organic Letters, 2008, 10(20): 4697-4700, Organometallics, 2008, 27(21): 5494-5503, European Journal of Organic Chemistry, 2002, 10: 1685-1689, European Journal of Inorganic Chemistry, 2001, 1: 289-296 и Angewandte Chemie, International Edition, 1998, 37(8): 1100-1103.
ПРИМЕРЫ
Вышеизложенное описывает предпочтительные варианты осуществления, которые, как будет понятно специалистам в данной области техники, могут подвергаться изменениям или модификациям при разработке, конструировании или эксплуатации без отклонения от объема формулы изобретения. Подразумевается, что эти изменения, например, охвачены объемом формулы изобретения.
Пример 1
Получение с помощью Clostridium kluyveri гексановой кислоты из ацетата и этанола
Для биологического превращения этанола и ацетата в гексановую кислоту применяли бактерию Clostridium kluyveri. Все стадии культивирования проводили в анаэробных условиях в устойчивых к давлению стеклянных колбах, которые можно герметично закрывать пробкой из бутилкаучука.
Для получения предварительной культуры 100 мл среды DMSZ52 (pH = 7,0; 10 г/л K-ацетата, 0,31 г/л K2HPO4, 0,23 г/л KH2PO4, 0,25 г/л NH4Cl, 0,20 г/л MgSO4 x 7H2O, 1 г/л дрожжевого экстракта, 0,50 мг/л резазурина, 10 мкл/л HCl (25%, 7,7 M), 1,5 мг/л FeCl2 x 4H2O, 70 мкг/л ZnCl2 x 7H2O, 100 мкг/л MnCl2 x 4H2O, 6 мкг/л H3BO3, 190 мкг/л CoCl2 x 6H2O, 2 мкг/л CuCl2 x 6H2O, 24 мкг/л NiCl2 x 6H2O, 36 мкг/л Na2MO4 x 2H2O, 0,5 мг/л NaOH, 3 мкг/л Na2SeO3 x 5H2O, 4 мкг/л Na2WO4 x 2H2O, 100 мкг/л витамина B12, 80 мкг/л п-аминобензойной кислоты, 20 мкг/л D(+)-биотина, 200 мкг/л никотиновой кислоты, 100 мкг/л D-Ca-пантотената, 300 мкг/л гидрохлорида пиридоксина, 200 мкг/л тиамин-HCl x 2H2O, 20 мл/л этанола, 2,5 г/л NaHCO3, 0,25 г/л цистеин-HCl x H2O, 0,25 г/л Na2S x 9H2O) в колбе объемом 250 мл инокулировали 5 мл замороженной криокультуры Clostridium kluyveri и инкубировали при 37°C в течение 144 ч. до OD600 нм > 0,2.
Для получения основной культуры 200 мл свежей среды DMSZ52 в колбе объемом 500 мл инокулировали подвергнутыми центрифугированию клетками из предварительной культуры до OD600 нм, составляющей 0,1. Данную растущую культуру инкубировали при 37°C в течение 27 ч. до OD600 нм > 0,6. Затем суспензию клеток центрифугировали, промывали с помощью буфера для получения (pH 6,0; 0,832 г/л K-ацетата, 5,0 г/л этанола) и снова центрифугировали.
Для получения культуры для продуцирования 200 мл буфера для получения в колбе объемом 500 мл инокулировали промытыми клетками из основной культуры до OD600 нм, составляющей 0,2. Культуру закрывали пробкой из бутилкаучука и инкубировали в течение 71 ч. при 37°C и 100 об./мин. на открытой водяной бане со встряхиванием. Образцы отбирали в начале и в конце периода культивирования. Их тестировали в отношении оптической плотности, значения pH и различных аналитов (тестировали с помощью ЯМР).
Из результатов видно, что в фазе получения количество ацетата снижалось от 0,54 г/л до 0,03 г/л, и количество этанола снижалось от 5,6 г/л до 4,9 г/л. Кроме того, концентрация масляной кислоты повышалась от 0,05 г/л до 0,28 г/л, и концентрация гексановой кислоты повышалась от 0,03 г/л до 0,79 г/л.
Пример 2
Получение с помощью Clostridium kluyveri гексановой кислоты из масляной кислоты и этанола
Для биологического превращения этанола и масляной кислоты в гексановую кислоту применяли бактерию Clostridium kluyveri. Все стадии культивирования проводили в анаэробных условиях в устойчивых к давлению стеклянных колбах, которые можно герметично закрывать пробкой из бутилкаучука.
Для получения предварительной культуры 100 мл среды DMSZ52 (pH = 7,0; 10 г/л K-ацетата, 0,31 г/л K2HPO4, 0,23 г/л KH2PO4, 0,25 г/л NH4Cl, 0,20 г/л MgSO4 x 7H2O, 1 г/л дрожжевого экстракта, 0,50 мг/л резазурина, 10 мкл/л HCl (25%, 7,7 M), 1,5 мг/л FeCl2 x 4H2O, 70 мкг/л ZnCl2 x 7H2O, 100 мкг/л MnCl2 x 4H2O, 6 мкг/л H3BO3, 190 мкг/л CoCl2 x 6H2O, 2 мкг/л CuCl2 x 6H2O, 24 мкг/л NiCl2 x 6H2O, 36 мкг/л Na2MO4 x 2H2O, 0,5 мг/л NaOH, 3 мкг/л Na2SeO3 x 5H2O, 4 мкг/л Na2WO4 x 2H2O, 100 мкг/л витамина B12, 80 мкг/л п-аминобензойной кислоты, 20 мкг/л D(+)-биотина, 200 мкг/л никотиновой кислоты, 100 мкг/л D-Ca-пантотената, 300 мкг/л гидрохлорида пиридоксина, 200 мкг/л тиамин-HCl x 2H2O, 20 мл/л этанола, 2,5 г/л NaHCO3, 0,25 г/л цистеин-HCl x H2O, 0,25 г/л Na2S x 9H2O) в колбе объемом 250 мл инокулировали 5 мл замороженной криокультуры Clostridium kluyveri и инкубировали при 37°C в течение 144 ч. до OD600 нм > 0,3.
Для получения основной культуры 200 мл свежей среды DMSZ52 в колбе объемом 500 мл инокулировали подвергнутыми центрифугированию клетками из предварительной культуры до OD600 нм, составляющей 0,1. Данную растущую культуру инкубировали при 37°C в течение 25 ч. до OD600 нм > 0,4. Затем суспензию клеток центрифугировали, промывали с помощью буфера для получения (pH 6,16; 4,16 г/л K-ацетата, 10,0 г/л этанола) и снова центрифугировали.
Для получения культур для продуцирования 200 мл буфера для получения в колбе объемом 500 мл инокулировали промытыми клетками из основной культуры до OD600 нм, составляющей 0,2. В первой культуре в начале добавляли 1,0 г/л масляной кислоты в буфер для получения, во второй культуре не добавляли масляную кислоту в буфер для получения. Культуры закрывали пробкой из бутилкаучука и инкубировали в течение 71 ч. при 37°C и 100 об./мин. на открытой водяной бане со встряхиванием. Образцы отбирали в начале и в конце периода культивирования. Их тестировали в отношении оптической плотности, значения pH и различных аналитов (тестировали с помощью ЯМР).
Из результатов видно, что в фазе получения дополненной масляной кислотой культуры количество ацетата снижалось от 3,1 г/л до 1,1 г/л, и количество этанола снижалось от 10,6 г/л до 7,5 г/л. Кроме того, концентрация масляной кислоты повышалась от 1,2 г/л до 2,2 г/л, и концентрация гексановой кислоты повышалась от 0,04 г/л до 2,30 г/л.
В фазе получения недополненной культуры количество ацетата снижалось от 3,0 г/л до 1,3 г/л, и количество этанола снижалось от 10,2 г/л до 8,2 г/л. Кроме того, концентрация масляной кислоты повышалась от 0,1 г/л до 1,7 г/л, и концентрация гексановой кислоты повышалась от 0,01 г/л до 1,40 г/л.
Пример 3
Культивирование Clostridium kluyveri в присутствии декана и TOPO
Бактерию Clostridium kluyveri культивировали с целью биологического превращения этанола и ацетата в гексановую кислоту. Для экстракции in situ полученной гексановой кислоты для культивирования добавляли смесь декана с триоктилфосфиноксидом (TOPO). Все стадии культивирования проводили в анаэробных условиях в устойчивых к давлению стеклянных колбах, которые можно герметично закрывать пробкой из бутилкаучука.
Для получения предварительной культуры 250 мл среды Veri01 (pH 7,0; 10 г/л ацетата калия, 0,31 г/л K2HPO4, 0,23 г/л KH2PO4, 0,25 г/л NH4Cl, 0,20 г/л MgSO4 X 7H2O, 10 мкл/л HCl (7,7 M), 1,5 мг/л FeCl2 X 4H2O, 36 мкг/л ZnCl2, 64 мкг/л MnCl2 X 4H2O, 6 мкг/л H3BO3, 190 мкг/л CoCl2 X 6H2O, 1,2 мкг/л CuCl2 X 6H2O, 24 мкг/л NiCl2 X 6H2O, 36 мкг/л Na2MO4 X 2H2O, 0,5 мг/л NaOH, 3 мкг/л Na2SeO3 X 5H2O, 4 мкг/л Na2WO4 X 2H2O, 100 мкг/л витамина B12, 80 мкг/л п-аминобензойной кислоты, 20 мкг/л D(+)-биотина, 200 мкг/л никотиновой кислоты, 100 мкг/л D-Ca-пантотената, 300 мкг/л гидрохлорида пиридоксина, 200 мкг/л тиамин-HCl x 2H2O, 20 мл/л этанола, 2,5 г/л NaHCO3, 65 мг/л глицина, 24 мг/л гистидина, 64,6 мг/л изолейцина, 93,8 мг/л лейцина, 103 мг/л лизина, 60,4 мг/л аргинина, 21,64 мг/л L-цистеин-HCl, 21 мг/л метионина, 52 мг/л пролина, 56,8 мг/л серина, 59 мг/л треонина, 75,8 мг/л валина) инокулировали 10 мл живой культуры Clostridium kluyveri до стартовой OD600 нм, составляющей 0,1.
Культивирование проводили в устойчивой к давлению стеклянной колбе объемом 1000 мл при 37°C, 150 об./мин. и интенсивности вентиляции, составляющей 1 л/ч. со 100% CO2 во встряхивателе на открытой водяной бане в течение 671 ч. Газ высвобождали в свободное пространство над продуктом в реакторе. Значение pH поддерживали при 6,2 с помощью автоматического добавления 100 г/л раствора NaOH. Свежую среду непрерывно подавали в реактор со степенью разбавления, составляющей 2,0 д-1, и ферментационный бульон непрерывно удаляли из реактора с помощью половолоконной полиэфирсульфоновой мембраны KrosFlo® с размером пор, составляющим 0,2 мкм (Spectrumlabs, Ранчо Домингез, США), с удержанием клеток в реакторе.
Для получения основной культуры 100 мл свежей среды Veri01 в колбе объемом 250 мл инокулировали подвергнутыми центрифугированию клетками из предварительной культуры до OD600 нм, составляющей 0,1. Добавляли дополнительный 1 мл смеси 6% (вес/вес) TOPO в декане. Культуру закрывали пробкой из бутилкаучука и инкубировали при 37°C и 150 об./мин. во встряхивателе на открытой водяной бане в течение 43 ч. в атмосфере 100% CO2.
Во время культивирования отбирали несколько образцов по 5 мл для определения OD600 нм, значения pH и степени образования продукта. Определение концентраций продукта выполняли с помощью полуколичественного анализа с применением 1H-ЯМР-спектроскопии. В качестве внутреннего количественного стандарта использовали триметилсилилпропионат натрия (T(M)SP).
Во время основного культивирования концентрация бутирата повышалась от 0,14 г/л до 2,12 г/л, и концентрация гексанoата повышалась от 0,22 г/л до 0,91 г/л, при этом концентрация этанола снижалась от 15,04 до 11,98 г/л, и концентрация ацетата снижалась от 6,01 до 4,23 г/л.
OD600 нм снижалась за это время от 0,111 до 0,076.
Пример 4
Культивирование Clostridium kluyveri в присутствии тетрадекана и TOPO
Бактерию Clostridium kluyveri культивировали с целью биологического превращения этанола и ацетата в гексановую кислоту. Для экстракции in situ полученной гексановой кислоты для культивирования добавляли смесь тетрадекана с триоктилфосфиноксидом (TOPO). Все стадии культивирования проводили в анаэробных условиях в устойчивых к давлению стеклянных колбах, которые можно герметично закрывать пробкой из бутилкаучука.
Предварительное культивирование Clostridium kluyveri проводили в устойчивой к давлению стеклянной колбе объемом 1000 мл в 250 мл среды EvoDM24 (pH 5,5; 0,429 г/л Mg-ацетата, 0,164 г/л Na-ацетата, 0,016 г/л Ca-ацетата, 2,454 г/л K-ацетата, 0,107 мл/л H3PO4 (8,5%), 0,7 г/л ацетата NH4, 0,35 мг/л Co-ацетата, 1,245 мг/л Ni-ацетата, 20 мкг/л d-биотина, 20 мкг/л фолиевой кислоты, 10 мкг/л пиридоксин-HCl, 50 мкг/л тиамин-HCl, 50 мкг/л рибофлавина, 50 мкг/л никотиновой кислоты, 50 мкг/л Ca-пантотената, 50 мкг/л витамина B12, 50 мкг/л п-аминобензоата, 50 мкг/л липоевой кислоты, 0,702 мг/л (NH4)2Fe(SO4)2 x 4H2O, 1 мл/л KS-ацетата (93,5 мМ), 20 мл/л этанола, 0,37 г/л уксусной кислоты) при 37°C, 150 об./мин. и интенсивности вентиляции, составляющей 1 л/ч., со смесью, содержащей 25% CO2 и 75% N2, во встряхивателе на открытой водяной бане. Газ высвобождали в свободное пространство над продуктом в реакторе. Значение pH поддерживали при 5,5 с помощью автоматического добавления 2,5 M раствора NH3. Свежую среду непрерывно подавали в реактор со степенью разбавления, составляющей 2,0 д-1, и ферментационный бульон непрерывно удаляли из реактора с помощью половолоконной полиэфирсульфоновой мембраны KrosFlo® с размером пор, составляющим 0,2 мкм (Spectrumlabs, Ранчо Домингез, США), с удержанием клеток в реакторе и поддержанием OD600 нм, составляющей ~1,5.
Для получения основной культуры 100 мл среды Veri01 (pH 6,5; 10 г/л ацетата калия, 0,31 г/л K2HPO4, 0,23 г/л KH2PO4, 0,25 г/л NH4Cl, 0,20 г/л MgSO4 X 7H2O, 10 мкл/л HCl (7,7 M), 1,5 мг/л FeCl2 X 4H2O, 36 мкг/л ZnCl2, 64 мкг/л MnCl2 X 4H2O, 6 мкг/л H3BO3, 190 мкг/л CoCl2 X 6H2O, 1,2 мкг/л CuCl2 X 6H2O, 24 мкг/л NiCl2 X 6H2O, 36 мкг/л Na2MO4 X 2H2O, 0,5 мг/л NaOH, 3 мкг/л Na2SeO3 X 5H2O, 4 мкг/л Na2WO4 X 2H2O, 100 мкг/л витамина B12, 80 мкг/л п-аминобензойной кислоты, 20 мкг/л D(+)-биотина, 200 мкг/л никотиновой кислоты, 100 мкг/л D-Ca-пантотената, 300 мкг/л гидрохлорида пиридоксина, 200 мкг/л тиамин-HCl x 2H2O, 20 мл/л этанола, 2,5 г/л NaHCO3, 65 мг/л глицина, 24 мг/л гистидина, 64,6 мг/л изолейцина, 93,8 мг/л лейцина, 103 мг/л лизина, 60,4 мг/л аргинина, 21,64 мг/л L-цистеин-HCl, 21 мг/л метионина, 52 мг/л пролина, 56,8 мг/л серина, 59 мг/л треонина, 75,8 мг/л валина, 2,5 мл/л HCl 25%) в колбе объемом 250 мл инокулировали подвергнутыми центрифугированию клетками из предварительной культуры до OD600 нм, составляющей 0,1. Добавляли дополнительный 1 мл смеси 6% (вес/вес) TOPO в тетрадекане. Культуру закрывали пробкой из бутилкаучука и инкубировали при 37°C и 150 об./мин. во встряхивателе на открытой водяной бане в течение 47 ч. в атмосфере 100% CO2.
Во время культивирования отбирали несколько образцов по 5 мл для определения OD600 нм, значения pH и степени образования продукта. Определение концентраций продукта выполняли с помощью полуколичественного анализа с применением 1H-ЯМР-спектроскопии. В качестве внутреннего количественного стандарта использовали триметилсилилпропионат натрия (T(M)SP).
Во время основного культивирования концентрация бутирата повышалась от 0,05 г/л до 3,78 г/л, и концентрация гексанoата повышалась от 0,09 г/л до 4,93 г/л, при этом концентрация этанола снижалась от 15,52 до 9,36 г/л, и концентрация ацетата снижалась от 6,36 до 2,49 г/л.
OD600 нм повышалась за это время от 0,095 до 0,685.
Пример 5
Культивирование Clostridium kluyveri в присутствии гексадекана и TOPO
Бактерию Clostridium kluyveri культивировали с целью биологического превращения этанола и ацетата в гексановую кислоту. Для экстракции in situ полученной гексановой кислоты для культивирования добавляли смесь гексадекана с триоктилфосфиноксидом (TOPO). Все стадии культивирования проводили в анаэробных условиях в устойчивых к давлению стеклянных колбах, которые можно герметично закрывать пробкой из бутилкаучука.
Для получения предварительной культуры 250 мл среды Veri01 (pH 7,0; 10 г/л ацетата калия, 0,31 г/л K2HPO4, 0,23 г/л KH2PO4, 0,25 г/л NH4Cl, 0,20 г/л MgSO4 X 7H2O, 10 мкл/л HCl (7,7 M), 1,5 мг/л FeCl2 X 4H2O, 36 мкг/л ZnCl2, 64 мкг/л MnCl2 X 4H2O, 6 мкг/л H3BO3, 190 мкг/л CoCl2 X 6H2O, 1,2 мкг/л CuCl2 X 6H2O, 24 мкг/л NiCl2 X 6H2O, 36 мкг/л Na2MO4 X 2H2O, 0,5 мг/л NaOH, 3 мкг/л Na2SeO3 X 5H2O, 4 мкг/л Na2WO4 X 2H2O, 100 мкг/л витамина B12, 80 мкг/л п-аминобензойной кислоты, 20 мкг/л D(+)-биотина, 200 мкг/л никотиновой кислоты, 100 мкг/л D-Ca-пантотената, 300 мкг/л гидрохлорида пиридоксина, 200 мкг/л тиамин-HCl x 2H2O, 20 мл/л этанола, 2,5 г/л NaHCO3, 65 мг/л глицина, 24 мг/л гистидина, 64,6 мг/л изолейцина, 93,8 мг/л лейцина, 103 мг/л лизина, 60,4 мг/л аргинина, 21,64 мг/л L-цистеин-HCl, 21 мг/л метионина, 52 мг/л пролина, 56,8 мг/л серина, 59 мг/л треонина, 75,8 мг/л валина) инокулировали 10 мл живой культуры Clostridium kluyveri до стартовой OD600 нм, составляющей 0,1.
Культивирование проводили в устойчивой к давлению стеклянной колбе объемом 1000 мл при 37°C, 150 об./мин. и интенсивности вентиляции, составляющей 1 л/ч. со 100% CO2 во встряхивателе на открытой водяной бане в течение 671 ч. Газ высвобождали в свободное пространство над продуктом в реакторе. Значение pH поддерживали при 6,2 с помощью автоматического добавления 100 г/л раствора NaOH. Свежую среду непрерывно подавали в реактор со степенью разбавления, составляющей 2,0 д-1, и ферментационный бульон непрерывно удаляли из реактора с помощью половолоконной полиэфирсульфоновой мембраны KrosFlo® с размером пор, составляющим 0,2 мкм (Spectrumlabs, Ранчо Домингез, США), с удержанием клеток в реакторе.
Для получения основной культуры 100 мл свежей среды Veri01 в колбе объемом 250 мл инокулировали подвергнутыми центрифугированию клетками из предварительной культуры до OD600 нм, составляющей 0,1. Добавляли дополнительный 1 мл смеси 6% (вес/вес) TOPO в гексадекане. Культуру закрывали пробкой из бутилкаучука и инкубировали при 37°C и 150 об./мин. во встряхивателе на открытой водяной бане в течение 43 ч. в атмосфере 100% CO2.
Во время культивирования отбирали несколько образцов по 5 мл для определения OD600 нм, значения pH и степени образования продукта. Определение концентраций продукта выполняли с помощью полуколичественного анализа с применением 1H-ЯМР-спектроскопии. В качестве внутреннего количественного стандарта использовали триметилсилилпропионат натрия (T(M)SP).
Во время основного культивирования концентрация бутирата повышалась от 0,14 г/л до 2,86 г/л, и концентрация гексанoата повышалась от 0,20 г/л до 2,37 г/л, при этом концентрация этанола снижалась от 14,59 до 10,24 г/л, и концентрация ацетата снижалась от 5,87 до 3,32 г/л.
OD600 нм повышалась за это время от 0,091 до 0,256.
Пример 6
Культивирование Clostridium kluyveri в присутствии гептадекана и TOPO
Бактерию Clostridium kluyveri культивировали с целью биологического превращения этанола и ацетата в гексановую кислоту. Для экстракции in situ полученной гексановой кислоты для культивирования добавляли смесь гептадекана с триоктилфосфиноксидом (TOPO). Все стадии культивирования проводили в анаэробных условиях в устойчивых к давлению стеклянных колбах, которые можно герметично закрывать пробкой из бутилкаучука.
Для получения предварительной культуры 250 мл среды Veri01 (pH 7,0; 10 г/л ацетата калия, 0,31 г/л K2HPO4, 0,23 г/л KH2PO4, 0,25 г/л NH4Cl, 0,20 г/л MgSO4 X 7H2O, 10 мкл/л HCl (7,7 M), 1,5 мг/л FeCl2 X 4H2O, 36 мкг/л ZnCl2, 64 мкг/л MnCl2 X 4H2O, 6 мкг/л H3BO3, 190 мкг/л CoCl2 X 6H2O, 1,2 мкг/л CuCl2 X 6H2O, 24 мкг/л NiCl2 X 6H2O, 36 мкг/л Na2MO4 X 2H2O, 0,5 мг/л NaOH, 3 мкг/л Na2SeO3 X 5H2O, 4 мкг/л Na2WO4 X 2H2O, 100 мкг/л витамина B12, 80 мкг/л п-аминобензойной кислоты, 20 мкг/л D(+)-биотина, 200 мкг/л никотиновой кислоты, 100 мкг/л D-Ca-пантотената, 300 мкг/л гидрохлорида пиридоксина, 200 мкг/л тиамин-HCl x 2H2O, 20 мл/л этанола, 2,5 г/л NaHCO3, 65 мг/л глицина, 24 мг/л гистидина, 64,6 мг/л изолейцина, 93,8 мг/л лейцина, 103 мг/л лизина, 60,4 мг/л аргинина, 21,64 мг/л L-цистеин-HCl, 21 мг/л метионина, 52 мг/л пролина, 56,8 мг/л серина, 59 мг/л треонина, 75,8 мг/л валина) инокулировали 10 мл живой культуры Clostridium kluyveri до стартовой OD600 нм, составляющей 0,1.
Культивирование проводили в устойчивой к давлению стеклянной колбе объемом 1000 мл при 37°C, 150 об./мин. и интенсивности вентиляции, составляющей 1 л/ч. со 100% CO2 во встряхивателе на открытой водяной бане в течение 671 ч. Газ высвобождали в свободное пространство над продуктом в реакторе. Значение pH поддерживали при 6,2 с помощью автоматического добавления 100 г/л раствора NaOH. Свежую среду непрерывно подавали в реактор со степенью разбавления, составляющей 2,0 д-1, и ферментационный бульон непрерывно удаляли из реактора с помощью половолоконной полиэфирсульфоновой мембраны KrosFlo® с размером пор, составляющим 0,2 мкм (Spectrumlabs, Ранчо Домингез, США), с удержанием клеток в реакторе.
Для получения основной культуры 100 мл свежей среды Veri01 в колбе объемом 250 мл инокулировали подвергнутыми центрифугированию клетками из предварительной культуры до OD600 нм, составляющей 0,1. Добавляли дополнительный 1 мл смеси 6% (вес/вес) TOPO в гептадекане. Культуру закрывали пробкой из бутилкаучука и инкубировали при 37°C и 150 об./мин. во встряхивателе на открытой водяной бане в течение 43 ч. в атмосфере 100% CO2.
Во время культивирования отбирали несколько образцов по 5 мл для определения OD600 нм, значения pH и степени образования продукта. Определение концентраций продукта выполняли с помощью полуколичественного анализа с применением 1H-ЯМР-спектроскопии. В качестве внутреннего количественного стандарта использовали триметилсилилпропионат натрия (T(M)SP).
Во время основного культивирования концентрация бутирата повышалась от 0,15 г/л до 2,82 г/л, и концентрация гексанoата повышалась от 0,19 г/л до 2,85 г/л, при этом концентрация этанола снижалась от 14,34 до 9,58 г/л, и концентрация ацетата снижалась от 5,88 до 3,20 г/л.
OD600 нм повышалась за это время от 0,083 до 0,363.
Пример 7
Культивирование Clostridium kluyveri в присутствии додекана и TOPO
Бактерию Clostridium kluyveri культивировали с целью биологического превращения этанола и ацетата в гексановую кислоту. Для экстракции in situ полученной гексановой кислоты для культивирования добавляли смесь додекана с триоктилфосфиноксидом (TOPO). Все стадии культивирования проводили в анаэробных условиях в устойчивых к давлению стеклянных колбах, которые можно герметично закрывать пробкой из бутилкаучука.
Для получения предварительной культуры 250 мл среды Veri01 (pH 7,0; 10 г/л ацетата калия, 0,31 г/л K2HPO4, 0,23 г/л KH2PO4, 0,25 г/л NH4Cl, 0,20 г/л MgSO4 X 7H2O, 10 мкл/л HCl (7,7 M), 1,5 мг/л FeCl2 X 4H2O, 36 мкг/л ZnCl2, 64 мкг/л MnCl2 X 4H2O, 6 мкг/л H3BO3, 190 мкг/л CoCl2 X 6H2O, 1,2 мкг/л CuCl2 X 6H2O, 24 мкг/л NiCl2 X 6H2O, 36 мкг/л Na2MO4 X 2H2O, 0,5 мг/л NaOH, 3 мкг/л Na2SeO3 X 5H2O, 4 мкг/л Na2WO4 X 2H2O, 100 мкг/л витамина B12, 80 мкг/л п-аминобензойной кислоты, 20 мкг/л D(+)-биотина, 200 мкг/л никотиновой кислоты, 100 мкг/л D-Ca-пантотената, 300 мкг/л гидрохлорида пиридоксина, 200 мкг/л тиамин-HCl x 2H2O, 20 мл/л этанола, 2,5 г/л NaHCO3, 65 мг/л глицина, 24 мг/л гистидина, 64,6 мг/л изолейцина, 93,8 мг/л лейцина, 103 мг/л лизина, 60,4 мг/л аргинина, 21,64 мг/л L-цистеин-HCl, 21 мг/л метионина, 52 мг/л пролина, 56,8 мг/л серина, 59 мг/л треонина, 75,8 мг/л валина) инокулировали 10 мл живой культуры Clostridium kluyveri до стартовой OD600 нм, составляющей 0,1.
Культивирование проводили в устойчивой к давлению стеклянной колбе объемом 1000 мл при 37°C, 150 об./мин. и интенсивности вентиляции, составляющей 1 л/ч. со 100% CO2 во встряхивателе на открытой водяной бане в течение 671 ч. Газ высвобождали в свободное пространство над продуктом в реакторе. Значение pH поддерживали при 6,2 с помощью автоматического добавления 100 г/л раствора NaOH. Свежую среду непрерывно подавали в реактор со степенью разбавления, составляющей 2,0 д-1, и ферментационный бульон непрерывно удаляли из реактора с помощью половолоконной полиэфирсульфоновой мембраны KrosFlo® с размером пор, составляющим 0,2 мкм (Spectrumlabs, Ранчо Домингез, США), с удержанием клеток в реакторе.
Для получения основной культуры 100 мл свежей среды Veri01 в колбе объемом 250 мл инокулировали подвергнутыми центрифугированию клетками из предварительной культуры до OD600 нм, составляющей 0,1. Добавляли дополнительный 1 мл смеси 6% (вес/вес) TOPO в додекане. Культуру закрывали пробкой из бутилкаучука и инкубировали при 37°C и 150 об./мин. во встряхивателе на открытой водяной бане в течение 43 ч. в атмосфере 100% CO2.
Во время культивирования отбирали несколько образцов по 5 мл для определения OD600 нм, значения pH и степени образования продукта. Определение концентраций продукта выполняли с помощью полуколичественного анализа с применением 1H-ЯМР-спектроскопии. В качестве внутреннего количественного стандарта использовали триметилсилилпропионат натрия (T(M)SP).
Во время основного культивирования концентрация бутирата повышалась от 0,14 г/л до 2,62 г/л, и концентрация гексанoата повышалась от 0,22 г/л до 2,05 г/л, при этом концентрация этанола снижалась от 14,62 до 10,64 г/л, и концентрация ацетата снижалась от 5,92 до 3,54 г/л.
OD600 нм повышалась за это время от 0,091 до 0,259.
Пример 8
Определение коэффициента распределения гексановой кислоты между водой и смесью гексадекана и TOPO
Во время всех стадий эксперимента отбирали образцы из обеих фаз для определения значения pH и концентрации гексановой кислоты с помощью высокоэффективной жидкостной хроматографии (HPLC). С помощью 100 г водного раствора 5 г/кг гексановой кислоты и 33 г смеси 6% триоктилфосфиноксида (TOPO) в гексадекане заполняли разделительную воронку и смешивали в течение 1 минуты при 37°C. Затем воронку помещали в кольцо на штативе и эмульсию оставляли отстаиваться с самопроизвольным отделением. Измеряли значение pH водной фазы. Затем добавляли 1 M раствор NaOH в воронку и смешивали. Стадию разделения и отбора образцов повторяли до тех пор, пока не достигали значения pH, составляющего 6,2, в водной фазе. Отбирали образцы из обеих фаз для последующего анализа на данный момент времени. Водную фазу можно анализировать непосредственно с помощью HPLC. Для анализа органической фазы разбавленную гексановую кислоту сперва повторно экстрагировали в воду (pH 12,0 путем добавления 1 M NaOH), а затем анализировали с помощью HPLC. Коэффициент распределения KD гексановой кислоты в системе воды и 6% TOPO в гексадекане рассчитывали из концентраций гексановой кислоты в обеих фазах.
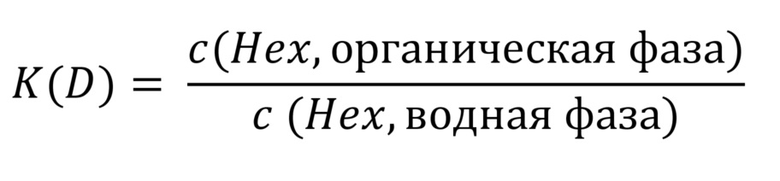
KD для гексановой кислоты в системе воды и 6% TOPO в гексадекане при значении pH 6,2 составлял 4,7.
Пример 9
Определение коэффициента распределения гексановой кислоты между водой и смесью гептадекана и TOPO
Во время всех стадий эксперимента отбирали образцы из обеих фаз для определения значения pH и концентрации гексановой кислоты с помощью высокоэффективной жидкостной хроматографии (HPLC). С помощью 100 г водного раствора 5 г/кг гексановой кислоты и 33 г смеси 6% триоктилфосфиноксида (TOPO) в гептадекане заполняли разделительную воронку и смешивали в течение 1 минуты при 37°C. Затем воронку помещали в кольцо на штативе и эмульсию оставляли отстаиваться с самопроизвольным отделением. Измеряли значение pH водной фазы. Добавляли 1 M раствор NaOH в воронку и смешивали. Стадию разделения и отбора образцов повторяли до тех пор, пока не достигали значения pH, составляющего 6,2, в водной фазе. Отбирали образцы из обеих фаз для последующего анализа на данный момент времени. Водную фазу можно анализировать непосредственно с помощью HPLC. Для анализа органической фазы разбавленную гексановую кислоту сперва повторно экстрагировали в воду (pH 12,0 путем добавления 1 M NaOH), а затем анализировали с помощью HPLC. Коэффициент распределения KD гексановой кислоты в системе воды и 6% TOPO в гептадекане рассчитывали из концентраций гексановой кислоты в обеих фазах.
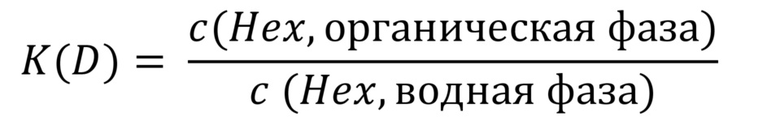
KD для гексановой кислоты в системе воды и 6% TOPO в гептадекане при значении pH 6,2 составлял 5,0.
Пример 10
Определение коэффициента распределения гексановой кислоты между водой и смесью тетрадекана и TOPO
Во время всех стадий эксперимента отбирали образцы из обеих фаз для определения значения pH и концентрации гексановой кислоты с помощью высокоэффективной жидкостной хроматографии (HPLC). С помощью 130 г водного раствора 5 г/кг гексановой кислоты плюс 0,5 г/кг уксусной кислоты и 15 г смеси 6% триоктилфосфиноксида (TOPO) в тетрадекане заполняли разделительную воронку и смешивали в течение 1 минуты при 37°C. Затем воронку помещали в кольцо на штативе и эмульсию оставляли отстаиваться с самопроизвольным отделением. Измеряли значение pH водной фазы. Добавляли 1 M раствор NaOH в воронку и смешивали. Стадию разделения и отбора образцов повторяли до тех пор, пока не достигали значения pH, составляющего 6,2, в водной фазе. Отбирали образцы из обеих фаз для последующего анализа на данный момент времени. Водную фазу можно анализировать непосредственно с помощью HPLC. Для анализа органической фазы разбавленную гексановую кислоту сперва повторно экстрагировали в воду (pH 12,0 путем добавления 1 M NaOH), а затем анализировали с помощью HPLC. Коэффициент распределения KD гексановой кислоты в системе воды и 6% TOPO в тетрадекане рассчитывали из концентраций гексановой кислоты в обеих фазах.
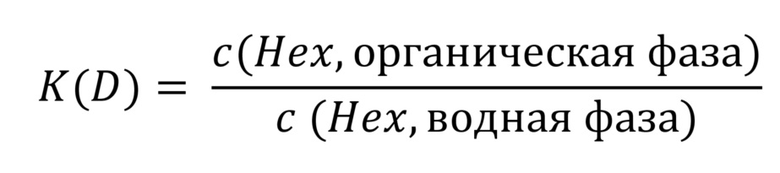
KD для гексановой кислоты в системе воды и 6% TOPO в тетрадекане при значении pH 6,9 составлял 1,3.
Пример 11
Культивирование Clostridium kluyveri с экстракцией in situ гексановой кислоты
Бактерию Clostridium kluyveri культивировали с целью биологического превращения этанола и ацетата в гексановую кислоту. Для экстракции in situ полученной гексановой кислоты смесь тетрадекана с триоктилфосфиноксидом (TOPO) непрерывно пропускали в процессе культивирования. Все стадии культивирования проводили в анаэробных условиях в устойчивых к давлению стеклянных колбах, которые можно герметично закрывать пробкой из бутилкаучука.
Предварительное культивирование Clostridium kluyveri проводили в устойчивой к давлению стеклянной колбе объемом 1000 мл в 250 мл среды EvoDM45 (pH 5,5; 0,004 г/л Mg-ацетата, 0,164 г/л Na-ацетата, 0,016 г/л Ca-ацетата, 0,25 г/л K-ацетата, 0,107 мл/л H3PO4 (8,5%), 2,92 г/л ацетата NH4, 0,35 мг/л Co-ацетата, 1,245 мг/л Ni-ацетата, 20 мкг/л d-биотина, 20 мкг/л фолиевой кислоты, 10 мкг/л пиридоксин-HCl, 50 мкг/л тиамин-HCl, 50 мкг/л рибофлавина, 50 мкг/л никотиновой кислоты, 50 мкг/л Ca-пантотената, 50 мкг/л витамина B12, 50 мкг/л п-аминобензоата, 50 мкг/л липоевой кислоты, 0,702 мг/л (NH4)2Fe(SO4)2 x 4 H2O, 1 мл/л KS-ацетата (93,5 мМ), 20 мл/л этанола, 0,37 г/л уксусной кислоты) при 37°C, 150 об./мин. и интенсивности вентиляции, составляющей 1 л/ч., со смесью, содержащей 25% CO2 и 75% N2, во встряхивателе на открытой водяной бане. Газ высвобождали в свободное пространство над продуктом в реакторе. Значение pH поддерживали при 5,5 с помощью автоматического добавления 2,5 M раствора NH3. Свежую среду непрерывно подавали в реактор со степенью разбавления, составляющей 2,0 д-1, и ферментационный бульон непрерывно удаляли из реактора с помощью половолоконной полиэфирсульфоновой мембраны KrosFlo® с размером пор, составляющим 0,2 мкм (Spectrumlabs, Ранчо Домингез, США), с удержанием клеток в реакторе и поддержанием OD600 нм, составляющей ~1,5.
Для получения основной культуры 150 мл среды EvoDM39 (pH 5,8; 0,429 г/л Mg-ацетата, 0,164 г/л Na-ацетата, 0,016 г/л Ca-ацетата, 2,454 г/л K-ацетата, 0,107 мл/л H3PO4 (8,5%), 1,01 мл/л уксусной кислоты, 0,35 мг/л Co-ацетата, 1,245 мг/л Ni-ацетата, 20 мкг/л d-биотина, 20 мкг/л фолиевой кислоты, 10 мкг/л пиридоксин-HCl, 50 мкг/л тиамин-HCl, 50 мкг/л рибофлавина, 50 мкг/л никотиновой кислоты, 50 мкг/л Ca-пантотената, 50 мкг/л витамина B12, 50 мкг/л п-аминобензоата, 50 мкг/л липоевой кислоты, 0,702 мг/л (NH4)2Fe(SO4)2 x 4H2O, 1 мл/л KS-ацетата (93,5 мМ), 20 мл/л этанола, 8,8 мл раствора NH3 (2,5 моль/л), 27,75 мл/л уксусной кислоты (144 г/л))
в колбе объемом 1000 мл инокулировали 100 мл клеточного бульона из предварительной культуры до OD600 нм, составляющей 0,71.
Культивирование проводили при 37°C, 150 об./мин. и интенсивности вентиляции, составляющей 1 л/ч., со смесью 25% CO2 и 75% N2 во встряхивателе на открытой водяной бане в течение 65 ч. Газ высвобождали в свободное пространство над продуктом в реакторе. Значение pH поддерживали при 5,8 с помощью автоматического добавления 2,5 M раствора NH3. Свежую среду непрерывно подавали в реактор со степенью разбавления, составляющей 0,5 д-1, и ферментационный бульон непрерывно удаляли из реактора путем поддержания OD600 нм, составляющей ~0,5. К ферментационному бульону добавляли дополнительные 120 г смеси 6% (вес/вес) TOPO в тетрадекане. Затем данную органическую смесь непрерывно подавали в реактор и органическую фазу также непрерывно удаляли из реактора со степенью разбавления, составляющей 1 д-1.
Во время культивирования отбирали несколько образцов по 5 мл из обеих, водной и органической, фаз для определения OD600 нм, значения pH и степени образования продукта. Определение концентраций продукта выполняли с помощью полуколичественного анализа с применением 1H-ЯМР-спектроскопии. В качестве внутреннего количественного стандарта использовали триметилсилилпропионат натрия (T(M)SP).
Во время основного культивирования в водной фазе достигали концентрации в равновесном состоянии, составляющей 8,18 г/л этанола, 3,20 г/л ацетата, 1,81 г/л бутирата и 0,81 г/л гексаноата. Значение OD600 нм оставалось постоянным на уровне 0,5. В органической фазе достигали концентрации в равновесном состоянии, составляющей 0,43 г/кг этанола, 0,08 г/кг ацетата, 1,13 г/кг бутирата и 8,09 г/кг гексаноата. После эксперимента клетки оставались жизнеспособными, несмотря на перенос для дополнительных этапов культивирования.
Коэффициент распределения KD субстратов и продуктов в системе водной среды и 6% TOPO в тетрадекане рассчитывали из концентраций в обеих фазах.
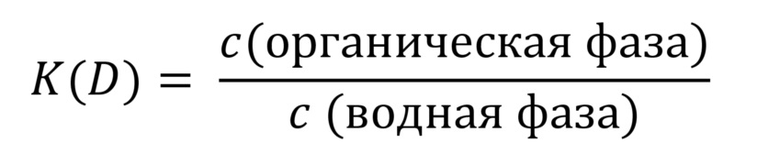
KD в равновесном состоянии составлял 0,05 для этанола, 0,03 для уксусной кислоты, 0,62 для масляной кислоты и 9,99 для гексановой кислоты.
Пример 12
Кетонизация гексановой кислоты
Кетонизацию проводили в нагретом реакторе непрерывного действия с проточным слоем. Сначала в реактор загружали оксид магния на диоксиде кремния (50 вес.%, 14,00 г) и нагревали в потоке аргона (54 мл/мин.) при 330°C в течение одного часа. Температуру повышали до 360°C. Затем смесь гексановой кислоты в тетрадекане (об./об.: 3/1) непрерывно загружали в реактор со скоростью 3,3 мл/ч. Выходящий газообразный поток собирали двумя охлаждающими ловушками, которые охлаждали водой и смесью сухого льда и изопропанола. Собранные фракции взвешивали и анализировали их состав посредством газовой хроматографии (GC). Всего в реактор загружали 370,65 г гексановой кислоты, что соответствует максимальному теоретическому выходу 271,70 г 6-ундеканона и 28,75 г воды и 70,21 г диоксида углерода в качестве побочных продуктов. Полученное количество 6-ундеканона составляло 267,67 г, и количество воды составляло 28,32 г. Это соответствует извлечению 99% массы при полной конверсии. Высокая продуктивность и селективность подтверждены регулярными измерениями посредством GC, поскольку обнаружены только следы гексановой кислоты и никаких побочных продуктов.
Пример 13
Перекрестная кетонизация гексановой кислоты с пальмитиновой кислотой
Техническая процедура перекрестной кетонизации идентична отдельной кетонизации гексановой кислоты (пример 12), за исключением состава исходного субстрата. Исходный материал состоит из гексановой кислоты (116,16 г, 1,00 моль) и пальмитиновой кислоты (256,43 г, 1,00 моль) в качестве субстратов и тетрадекана (124,20 г) в качестве внутреннего стандарта. Исходный субстрат добавляют со скоростью 3,3 мл/ч. и обеспечивают реакцию при температуре 360°C. Наличие двух алкановых кислот обеспечивает получение двух смесей продукта в виде трех кетонов: 6-ундеканона, 6-геникозанона и 16-гентриаконтанона. При полной конверсии полученные количества составляют 42,58 г 6-ундеканона, 155,29 г 6-геникозанона и 112,71 г 16-хентриаконтанона.
Пример 14
Перекрестная кетонизация гексановой кислоты с уксусной кислотой
Техническая процедура перекрестной кетонизации идентична отдельной кетонизации гексановой кислоты (пример 12), за исключением состава исходного субстрата. Исходный материал состоит из гексановой кислоты (232,32 г, 2,00 моль) и пальмитиновой кислоты (120,10 г, 2,00 моль) в качестве субстратов и тетрадекана (117,47 г) в качестве внутреннего стандарта. Исходный субстрат добавляют со скоростью 3,3 мл/ч. и обеспечивают реакцию при температуре 360°C. Наличие двух алкановых кислот обеспечивает получение двух смесей продукта в виде трех кетонов: 2-пропанона, 2-гептанона и 6-ундеканона. При полной конверсии полученные количества составляют 29,04 г 2-пропанона, 114,19 г 2-гептанона и 85,15 г 6-ундеканона.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЭКСТРАКЦИЯ АЛКАНОВЫХ КИСЛОТ | 2019 |
|
RU2785987C2 |
| ЭКСТРАКЦИЯ АЛИФАТИЧЕСКИХ СПИРТОВ | 2020 |
|
RU2807367C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСШИХ СПИРТОВ | 2016 |
|
RU2709952C2 |
| АЭРОБНЫЙ СПОСОБ ПОЛУЧЕНИЯ СПИРТОВ | 2016 |
|
RU2724532C2 |
| СПОСОБ НАКОПЛЕНИЯ ИЗБЫТОЧНОЙ ЭНЕРГИИ | 2014 |
|
RU2663728C2 |
| ПРОИЗВОДНЫЕ 5-ГИДРОКСИМЕТИЛОКСАЗОЛИДИН-2-ОНА ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ КИШЕЧНЫХ ЗАБОЛЕВАНИЙ | 2009 |
|
RU2527769C2 |
| Способ получения производных катехина | 1984 |
|
SU1424729A3 |
| РЕГУЛИРОВАНИЕ ПРОВОДИМОСТИ В ПРОЦЕССЕ АНАЭРОБНОЙ ФЕРМЕНТАЦИИ | 2014 |
|
RU2639503C2 |
| РЕКОМБИНАНТНАЯ КЛЕТКА, ПРОДУЦИРУЮЩАЯ 2-ГИДРОКСИИЗОМАСЛЯНУЮ КИСЛОТУ | 2009 |
|
RU2514672C9 |
| СПОСОБ БИОЛОГИЧЕСКОГО ПРОИЗВОДСТВА н-БУТАНОЛА | 2007 |
|
RU2461627C2 |
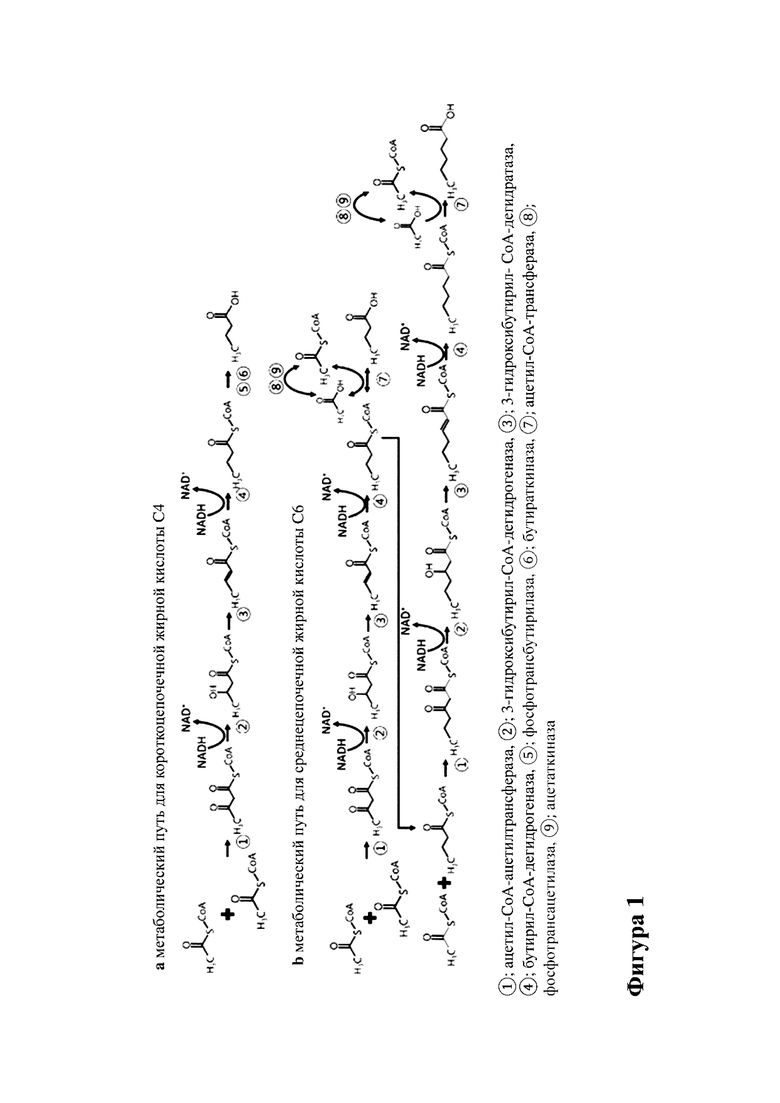
Группа изобретений относится к получению 6-ундеканона, 6-ундеканола и лауриновой кислоты с использованием комбинированного биотехнологического и химического способа. Приводят этанол и/или ацетат в контакт с микроорганизмом, выбранным из Clostridium carboxidivorans и Clostridium kluyveri, способным осуществлять удлинение углеродной цепи с получением гексановой кислоты и/или ее сложного эфира из этанола и/или ацетата. Экстрагируют гексановую кислоту и/или ее сложный эфир с применением экстрагирующего вещества в водной среде. Экстрагирующее вещество содержит алкилфосфиноксид и алкан, содержащий по меньшей мере 12 атомов углерода. Приводят гексановую кислоту и/или ее сложный эфир в контакт с катализатором кетонизации и с дополнительной алкановой кислотой, содержащей от 1 до 22 атомов углерода, при подходящих условиях реакции для химической кетонизации гексановой кислоты и далее дополнительной алкановой кислоты до 6-ундеканона. Далее для получения 6-ундеканола приводят 6-ундеканон в контакт с металлическим катализатором гидрогенизации. Для получения лауриновой кислоты приводят 6-ундеканол в контакт с CO2 и гомогенным катализатором карбоксилирования. Изобретения обеспечивают эффективное получение 6-ундеканола и его производных и сохранение жизнеспособности клеток микроорганизмов. 3 н. и 11 з.п. ф-лы, 1 ил., 14 пр.
1. Способ получения 6-ундеканона из этанола и/или ацетата, при этом способ включает (a) приведение этанола и/или ацетата в контакт с по меньшей мере одним микроорганизмом, способным осуществлять удлинение углеродной цепи с получением гексановой кислоты и/или ее сложного эфира из этанола и/или ацетата;
(b) экстрагирование гексановой кислоты и/или ее сложного эфира из (a) с применением по меньшей мере одного экстрагирующего вещества в водной среде, где экстрагирующее вещество содержит по меньшей мере один алкилфосфиноксид и по меньшей мере один алкан, содержащий по меньшей мере 12 атомов углерода; и
(c) приведение экстрагированной гексановой кислоты и/или ее сложного эфира из (b) в контакт с по меньшей мере одним катализатором кетонизации и далее с дополнительной алкановой кислотой, содержащей от 1 до 22 атомов углерода, при подходящих условиях реакции для химической кетонизации гексановой кислоты и далее дополнительной алкановой кислоты до 6-ундеканона;
где микроорганизм на стадии (a) выбран из группы, состоящей из Clostridium carboxidivorans и Clostridium kluyveri.
2. Способ по п. 1, где катализатор кетонизации из (c) представляет собой катализатор на основе оксида металла или смеси на его основе.
3. Способ по любому из пп. 1 или 2, где катализатор на основе оксида металла или смеси на его основе выбраны из группы, состоящей из катализатора на основе гетерополикислоты (H3PW12O40), катализатора на основе оксида титана (TiO2), катализатора на основе оксида церия (CeO2), смешанного катализатора на основе оксидов цинка и хрома (Zn-Cr), катализатора на основе оксида марганца (MnO2), катализатора на основе оксида лантана (La2O3), катализатора на основе оксида магния (MgO), катализатора на основе оксида железа (FeO, FeO2, Fe2O3, Fe3O4, Fe4O5, Fe5O6, Fe5O7), смешанного катализатора на основе оксидов кремния и алюминия (Si-Al) и катализатора на основе диоксида циркония (ZrO2).
4. Способ по любому из предыдущих пунктов, где катализатор кетонизации представляет собой катализатор на основе аэрогеля на основе диоксида циркония.
5. Способ по любому из предыдущих пунктов, где подходящие условия реакции на стадии (c) предусматривают температуру реакции 150-350°C.
6. Способ по любому из предыдущих пунктов, где алкилфосфиноксид выбран из группы, состоящей из триоктилфосфиноксида, гексилфосфиноксида, октилфосфиноксида и смесей на его основе, и алкан выбран из группы, состоящей из пентадекана, гексадекана, гептадекана, октадекана и тетрадекана.
7. Способ по любому из предыдущих пунктов, где алкилфосфиноксид представляет собой триоктилфосфиноксид (TOPO), и алкан представляет собой тетрадекан.
8. Способ по п. 7, где весовое соотношение TOPO и тетрадекана составляет от 1:100 до 1:10.
9. Способ по любому из предыдущих пунктов, где значение pH водной среды на стадии (b) поддерживают на уровне от 5,5 до 8.
10. Способ получения 6-ундеканола из этанола и/или ацетата, при этом способ включает
(d) приведение 6-ундеканона, полученного в соответствии с любым из пп. 1-8, в контакт с по меньшей мере одним металлическим катализатором гидрогенизации для каталитической гидрогенизации 6-ундеканона до 6-ундеканола.
11. Способ по п. 10, где металлический катализатор гидрогенизации выбран из группы, состоящей из катализатора на основе рутения (Ru), катализатора на основе рения (Re), катализатора на основе никеля (Ni), катализатора на основе железа (Fe), катализатора на основе кобальта (Co) и катализатора на основе платины (Pt).
12. Способ получения лауриновой кислоты из этанола и/или ацетата, при этом способ включает
(e) приведение 6-ундеканола, полученного в соответствии с любым из пп. 10 или 11, в контакт с CO2 и гомогенным катализатором карбоксилирования, способным обеспечивать карбоксилирование 6-ундеканола до лауриновой кислоты.
13. Способ по п. 12, где стадия (e) предусматривает либо
(ei) приведение 6-ундеканола, полученного в соответствии с любым из пп. 10 или 11, в контакт с по меньшей мере одним катализатором на основе никеля и диоксидом углерода при атмосферном давлении для карбоксилирования 6-ундеканола до лауриновой кислоты; либо
(eii) приведение 6-ундеканола, полученного в соответствии с любым из пп. 10 или 11, в контакт с водородом, CO2 и гомогенным катализатором на основе Rh для карбоксилирования 6-ундеканола до лауриновой кислоты.
14. Способ по любому из предыдущих пунктов, где экстрагирующее вещество используют повторно.
| US 6265618 B1, 24.07.2001 | |||
| JEON BYOUNG SEUNG ET AL | |||
| In situ extractive fermentation for the production of hexanoic acid from galactitol by Clostridium sp | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| WEIMER P | |||
| J | |||
| ET AL | |||
| Isolation, characterization, and quantification of Clostridium | |||

