Данное изобретение относится к области органического синтеза и касается, в частности, усовершенствованного промышленного способа получения N-пропил-N-(2-(2,4,6-трихлорфенокси)этил)-1H-имидазол-1-карбоксамида. Прохлораз применяется в сельском хозяйстве в качестве фунгицида против широкого диапазона заболеваний, повреждающих зерновые культуры, фрукты и овощи.
Среди применяемых в настоящее время фунгицидных препаратов наибольшее распространение получили системные азольные фунгициды, а именно производные 1,2,4-триазола и имидазола, по механизму действия являющиеся ингибиторами биосинтеза стероидов (ИБС), которые имеют низкие нормы расхода, малотоксичны и обладают широким спектром действия. Наиболее широко применяемыми азольными фунгицидами являются тебуконазол, пропиконазол, эпоксиконазол, триадимефон, прохлораз.
Первым патентом, который описывал как методы получения, так и применение в качестве фунгицидных препаратов N-арилоксиэтил-N-алкилкарбамоилимидазолы, был патент компании Boots Company Limited (GB 1469772 А, кл. A01N 47/18, опубл. 06.04.1977). В качестве примеров приведены методики получения N-(2-феноксиэтил)-N-пропилкарбамоилимидазола. Первую стадию N-алкилирования пропиламина бромфенетолом проводят при соотношении реагентов 4:1 при комнатной температуре в течение недели. На следующей стадии в насыщенный фосгеном этилацетат прибавляют по каплям полученный раствор вторичного амина в этилацетате в течение 45 мин, далее кипятят реакционную массу при постоянном пропускании фосгена в течение 3 ч. На заключительной стадии к раствору имидазола в тетрагидрофуране добавляют полученный карбамоилхлорид, далее кипятят в течение 24 ч, выделяют конечный продукт. Недостатками известной схемы синтеза являются использование высокотоксичного фосгена, длительность проведения процесса.
Четырехстадийная схема синтеза прохлораза описана в ирландском патенте (IE 64136, кл. C07D 233/56, опубл. 12.07.1995). На первой стадии получают 2'-бром-2,4,6-трихлорфенетол конденсацией 2,4,6-трихлорфенола с дибромэтаном в присутствии карбоната калия, в ацетоне при 45°С в течение 40 ч. Стадию N-алкилирования пропиламина с полученным бромфенетолом проводят при соотношении реагентов 5,1:1 при температуре кипения в течение 6 ч. Последующую стадию фосгенирования полученного вторичного амина проводят в толуоле, в токе фосгена при температуре кипения в течение 1 ч. Завершающую стадию конденсации полученного карбамоилхлорида с избытком имидазола проводят также в толуоле при температуре кипения в течение 12 ч. Недостатками известного способа являются длительность проведения синтеза, использование высокотоксичного газообразного реагента - фосгена, а также органических растворителей ацетона и толуола, являющихся прекурсорами. Использование прекурсоров подлежит контролю в Российской Федерации (Постановление Правительства РФ от 30 июня 1998 г. №681 «Об утверждении перечня наркотических средств, психотропных веществ и их прекурсоров, подлежащих контролю в Российской Федерации»).
Наиболее близким аналогом изобретения является способ получения прохлораза, описанный в китайской заявке CN 1224716 А (кл. A01N 47/38, опубл. 04.08.1999, пример 1). Данный способ также является четырехстадийным и включает реакции алкилирования (1), аминирования (2), ацилирования (3) и амидирования (4). На первой стадии смешивают 2,4,6-трихлорфенол, воду и дибромэтан, доводят реакционную смесь до кипения, прикапывают гидроксид натрия в течение 1 ч, охлаждают смесь после 8 ч кипячения. На второй стадии смешивают н-пропиламин и дихлорэтан, смесь нагревают до 55°С, прикапывают в течение 40 минут выделенный полупродукт первой стадии, предварительно растворив его в дихлорэтане, далее реакционную смесь выдерживают 8 часов. На третьей стадии смешивают выделенный полупродукт второй стадии и дихлорэтан, нагревают до 30°С, прикапывают раствор фосгена в дихлорэтане, после окончания прикапывания перемешивают реакционную массу в течение 30 минут при температуре не более 40°С, затем постепенно повышают температуру до 50°С в течение 30 минут, далее в течение 1 часа пропускают сухой воздух, удаляют остатки фосгена. На четвертой стадии к полупродукту третьей стадии добавляют имидазол, нагревают реакционную смесь до 80°С, выдерживают 6 ч, охлаждают и выделяют конечный продукт. Выход прохлораза (в расчете на исходный трихлорфенол) составляет 55,6%, чистота - 87,3%. Недостатком указанного способа является использование высокотоксичного газообразного реагента - фосгена, недостаточная чистота конечного продукта.
Целью изобретения является создание наиболее технологичного и безопасного процесса получения прохлораза.
Техническим результатом является повышение общего выхода прохлораза до 62% и его чистоты до 99% при одновременной оптимизации технологического процесса (сокращение времени проведения стадий синтеза, упрощение схемы производства), а также повышении его безопасности.
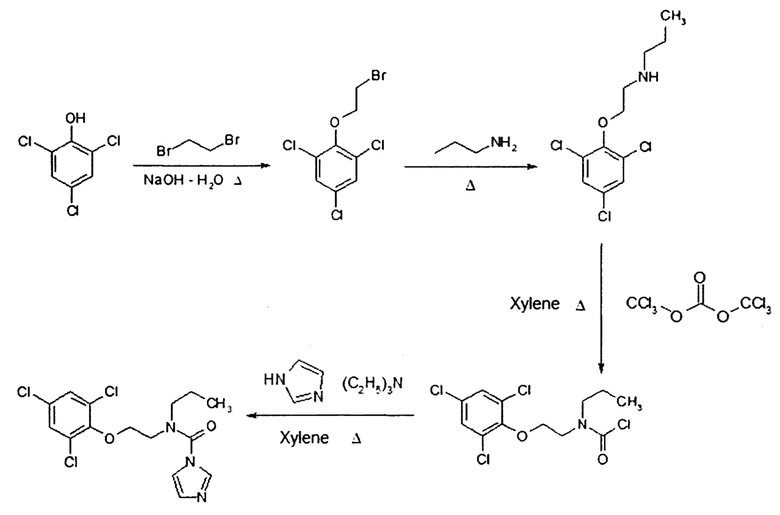
Объектом настоящего изобретения является промышленный способ получения прохлораза, включающий следующие последовательные стадии:
(а) интенсивное перемешивание 2,4,6-трихлорфенола, воды и избытка дибромэтана, доведение реакционной массы до 100°С, равномерное добавление раствора гидроксида натрия по каплям в течение 6 часов, кипячение полученной смеси в течение 2 часов, ее охлаждение до температуры 20°С, выделение полупродукта 2-(2-бромэтокси)-1,3,5-трихлорбензола известным способом;
(б) интенсивное перемешивание избытка н-пропиламина и полученного на стадии (а) раствора полупродукта при температуре 20°С, кипячение реакционной смеси в течение 6 часов при температуре 48°С, упаривание избыточного н-пропиламина, растворение остатка в орто-ксилоле, очистка реакционной массы газообразным хлороводородом, фильтрование полученной суспензии, выделение о-ксилольного раствора полупродукта N-(2-(2,4,6-трихлорфенокси)этил)пропан-1-амина известным способом;
(в) нагрев трифосгена в орто-ксилоле до 100°С, прикапывание при интенсивном перемешивании полученного на стадии (б) о-ксилольного раствора полупродукта, по окончании прикапывания перемешивание реакционной смеси при нагревании в течение 8 часов, охлаждение реакционной смеси до 80°С;
(г) равномерное добавление при 80°С к образовавшемуся на стадии (в) полупродукту нагретого не ниже 60°С о-ксилольного раствора имидазола и триэтиламина, перемешивание при температуре 80°С в течение 3 часов, охлаждение реакционной смеси до комнатной температуры, выделение продукта N-пропил-N-(2-(2,4,6-трихлорфенокси)этил)-1Н-имидазол-1-карбоксамида известным способом.
Технический результат достигается предлагаемым последовательным четырехстадийным превращением 2,4,6-трихлорфенола в 2-(2-бромэтокси)-1,3,5-трихлорбензол, далее - в N-(2-(2,4,6-трихлорфенокси)этил)пропан-1-амин, далее - в N-(2,4,6-трихлорфеноксиэтил)-N-пропилкарбамоил хлорид, далее - в N-пропил-N-(2-(2,4,6-трихлорфенокси)этил)-1Н-имидазол-1-карбоксамид в соответствии со схемой:
Использование предлагаемого способа имеет следующие преимущества перед аналогами:
- Использование на первой стадии высокореакционного дибромэтана и замена ацетона на воду позволяет повысить температуру реакционной смеси до 100°С и, соответственно, существенно сократить время проведения реакции алкилирования.
- Прикапывание раствора гидроксида натрия на первой стадии в течение 6 часов позволяет повысить выход конечного продукта, уменьшить примеси.
- Использование ортоксилола (вместо толуола) на стадии очистки и выделения полупродукта второй стадии позволяет увеличить выход и чистоту конечного продукта. Гидрохлорид полупродукта второй стадии лучше выпадает из орто-ксилола, чем из толуола.
- Очистку и выделение полупродукта второй стадии, а также стадии фосгенирования и конденсации осуществляют без использования прекурсора (толуол заменили на ортоксилол). Таким образом, не требуется предоставлять отчет перед контролирующими органами по покупке, расходам и остаткам толуола.
- На третьей стадии фосген заменили на трифосген. Применение трифосгена позволяет избежать потерь низкокипящего фосгена как на стадии непосредственного ацилирования вторичного амина в ксилольном растворе при 100°С, так и при последующем выдерживании реакционной массы. Кроме того, с трифосгеном удобнее работать: можно точно взвесить необходимое количество вещества для проведения реакции.
- Объединение третьей и четвертой стадий (реакции фосгенирования и конденсации проводят в одном реакторе) позволило упростить схему производства, поскольку не нужно выделять образующийся на 3-й стадии промежуточный продукт (N-(2,4,6-трихлорфеноксиэтил)-N-пропил карбамоил хлорид).
Изобретение иллюстрируется следующими примерами.
Пример 1. Синтез полупродукта 2-(2-бромэтокси)-1,3,5-трихлорбензола
В реактор, снабженный рубашкой обогрева, верхнеприводной мешалкой, обратным холодильником и капельной воронкой (или дозирующим насосом), загружают 914 мл воды, 1200 г трихлорфенола, 2237,9 г дибромэтана (свежий и/или рецикловый) и интенсивно перемешивают. Далее включают нагрев, доводят реакционную смесь до кипения (t=95°С - 100°С). И при этой температуре начинают добавлять раствор гидроксида натрия, полученный из 269,2 г гидроксида натрия и 860 мл технической подготовленной воды, через капельную воронку в течение 6 часов, поддерживая равномерное кипение реакционной массы. По окончании добавления раствора гидроксида натрия кипятят полученную смесь в течение 2 часов. Далее охлаждают реакционную массу до 20°С. Осадок отфильтровывают. Фильтрат отстаивают до полного расслоения. Обрабатывают нижний органический и верхний неорганический слои. Выделяют полупродукт (2-(2-бромэтокси)-1,3,5-трихлорбензол) известным способом и анализируют. Выход полупродукта составил 80,8%.
1Н ЯМР-спектр, (CDCl3, 5, м.д., J/Гц): 3.71 т (2Н, OCH2CH2Br, J=6.6), 4.33 т (2Н, OCH2CH2Br, J=6.6), 7.34 с (2Н, 2CHAr).
Технический продукт используют без дополнительной очистки на следующей стадии.
Пример 2. Синтез полупродукта N-(2-(2,4,6-трихлорфенокси)этил)пропан-1-амина В реактор, снабженный рубашкой обогрева, верхнеприводной мешалкой и обратным холодильником, загружают 70% н-пропиламина (1191 мл). При интенсивном перемешивании и температуре 20°С загружают 1300 г 2-(2-бромэтокси)-1,3,5-трихлорбензола в жидком виде. Остатки 2-(2-бромэтокси)-1,3,5-трихлорбензола смывают из сборника остатками (оставшимися 30% пропиламина) пропиламина. Полученную реакционную смесь кипятят в течение 6 часов при температуре 48-50°С. Избыток пропиламина удаляют из реакционной смеси. Далее переносят реакционную массу после упаривания пропиламина обратно в реактор, растворяют в орто-ксилоле и полученный раствор насыщают газообразным хлористым водородом. В результате барботирования реакционной массы хлористым водородом выпадает осадок N-(2-(2,4,6-трихлорфенокси)этил)-1-пропиламин гидрохлорида. Полученную суспензию фильтруют. Осадок промывают. Получают о-ксилольный раствор полупродукта N-(2-(2,4,6-трихлорфенокси)этил)пропан-1-амин и анализируют. Выход полупродукта составил 84,4%.
1Н ЯМР-спектр, (CDC13, δ, м.д., J/Гц): 0.95 т (3Н, СН3, J=7.3), 1.56 кв (2Н, СН2СН2СН3, J=7.3), 1.93 уш. с.(1H, NH), 2.67 т (2Н, СН2СН2СН3, J=7.3), 3.03 т (2Н, OCH2CH2N, J=5.1), 4.15 т (2Н, OCH2CH2N r, J=5.1), 7.30 м (2Н, 2CHAr).
Технический продукт используют на следующей стадии.
Пример 3. Синтез конечного продукта N-пропил-N-(2-(2,4,6-трихлорфенокси)этил)-1Н-имидазол-1-карбоксамида (прохлораза)
В реактор, снабженный рубашкой обогрева, верхнеприводной мешалкой, обратным холодильником и системой дегазации, загружают 183,57 г трифосгена, остатки трифосгена смывают 912 мл орто-ксилола. Включают перемешивание, нагревают реакционную массу до кипения (t=100°С). При интенсивном перемешивании прикапывают продукт второй стадии (о-ксилольный раствор N-(2-(2,4,6-трихлорфенокси)этил)пропан-1-амина). По окончании прикапывания перемешивают реакционную смесь при нагревании в течение 8 часов. В результате реакции образуется полупродукт N-(2,4,6-трихлорфеноксиэтил)-N-пропилкарбамоил хлорид. Охлаждают реактор до 80°С
Отдельно готовят раствор имидазола в ксилоле. Для этого в колбу, снабженную магнитной мешалкой, вносят 119,4 г имидазола, 3649 мл орто-ксилола и 421,5 г триэтиламина. Нагревают полученный раствор до 80°С и перемешивают до полного растворения имидазола.
Нагретый орто-ксилольный раствор имидазола равномерно вносят в реактор, содержащий продукт реакции фосгенирования, при этом поддерживают раствор подогретым (не ниже 60°С) во избежание кристаллизации имидазола. Перемешивают реакционную смесь при 80°С 3 часа. Охлаждают до комнатной температуры. Реакционную массу обрабатывают, выделяют продукт N-пропил-N-(2-(2,4,6-трихлорфенокси)этил)-1Н-имидазол-1-карбоксамид (прохлораз) известным способом. Полученный конечный продукт анализируют.
Выход продукта по данной стадии - 90,3%.
1Н ЯМР-спектр, (CDCl3, δ, м.д., J/Гц): 0.94 т (3Н, СН3, J=7.3), 1.55-1.85 м (2Н, СН2СН2СН3, А,Б), 3.27 т (2Н, СН2СН2СН3,Б), 3.56 т (2Н, СН2СН2СН3, J=7.3, А), 3.65 т (2Н, OCH2CH2N, Б), 3.88 т (2Н, OCH2CH2N, J=7.2, А), 4.11 т (2Н, OCH2CH2N,Б), 4.23 т (2Н, OCH2CH2N, J=5.1, А), 7.12 с (1Н, CHIm), 7.31 с (1Н, CHIm), 7.34 с (2Н, 2CHAr), 7.96 с (1Н, CHIm).
Общий выход прохлораза составил 61,6%, а чистота - 99%.
Таким образом, при использовании предлагаемого способа увеличивается выход конечного продукта, повышается его чистота, оптимизируется технологический процесс с точки зрения безопасности и технологичности.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения моноэфиров гексадииндиола-1,6 | 1973 |
|
SU447398A1 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 1992 |
|
RU2045525C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 1989 |
|
RU2024510C1 |
| Способ получения N-(2-гидроксиэтил)-О-изопропилкарбамата | 2016 |
|
RU2651792C1 |
| Способ получения 3-(4-аминоэтоксибензоил) бензо( @ )тиофенов или их солей | 1982 |
|
SU1155157A3 |
| СПОСОБ БОРЬБЫ С ГРИБКОВЫМИ ИНФЕКЦИЯМИ РАСТЕНИЙ | 1989 |
|
RU2067832C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2-(ХЛОРФЕНОКСИ)-ПРОПИОНОВОЙ КИСЛОТЫ | 2017 |
|
RU2662441C1 |
| РАЗВЕТВЛЕННЫЕ АЛКИЛАМИНОПРОИЗВОДНЫЕ ТИАЗОЛА ИЛИ ИХ ВОЗМОЖНЫЕ СТЕРЕОИЗОМЕРЫ ИЛИ ИХ СОЛИ ПРИСОЕДИНЕНИЯ С МИНЕРАЛЬНОЙ ИЛИ ОРГАНИЧЕСКОЙ КИСЛОТОЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АКТИВНОСТЬЮ В ОТНОШЕНИИ ФАКТОРА ВЫСВОБОЖДЕНИЯ КОРТИКОТРОПНОГО ГОРМОНА, И АЛКИЛАМИНО-ПРОИЗВОДНЫЕ ТИАЗОЛА В КАЧЕСТВЕ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 1993 |
|
RU2102389C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИКАРБОНАТОВ | 2008 |
|
RU2378296C2 |
| Способ получения галоидангидридов сульфаминовой кислоты | 1976 |
|
SU619102A3 |
Изобретение относится к области органического синтеза и касается, в частности, усовершенствованного промышленного способа получения прохлораза. Данное соединение применяется в сельском хозяйстве в качестве фунгицида против широкого диапазона заболеваний, повреждающих зерновые культуры, фрукты и овощи. Предлагаемый способ включает последовательные стадии (а)-(г). На стадии (а) осуществляют интенсивное перемешивание 2,4,6-трихлорфенола, воды и избытка дибромэтана, доведение реакционной массы до 100°С, равномерное добавление раствора гидроксида натрия по каплям в течение 6 ч, кипячение полученной смеси в течение 2 ч, ее охлаждение до температуры 20°С и выделение полупродукта 2-(2-бромэтокси)-1,3,5-трихлорбензола. На стадии (б) происходит интенсивное перемешивание избытка н-пропиламина и полученного на стадии (а) раствора полупродукта при температуре 20°С, кипячение реакционной смеси в течение 6 ч при температуре 48°С, упаривание избыточного н-пропиламина, растворение остатка в орто-ксилоле, очистка реакционной массы газообразным хлороводородом, фильтрование полученной суспензии и выделение о-ксилольного раствора полупродукта N-(2-(2,4,6-трихлорфенокси)этил)пропан-1-амина. На стадии (в) осуществляют нагрев трифосгена в орто-ксилоле до 100°С, прикапывание при интенсивном перемешивании полученного на стадии (б) о-ксилольного раствора полупродукта, по окончании прикапывания перемешивание реакционной смеси при нагревании в течение 8 ч и охлаждение реакционной смеси до 80°С. На стадии (г) осуществляют равномерное добавление при 80°С к образовавшемуся на стадии (в) полупродукту нагретого не ниже 60°С о-ксилольного раствора имидазола и триэтиламина, перемешивание при температуре 80°С в течение 3 ч, охлаждение реакционной смеси до комнатной температуры и выделение продукта прохлораза. Изобретение позволяет повысить общий выход прохлораза до 62% и его чистоту до 99% при одновременной оптимизации технологического процесса и повышении его безопасности. 3 пр.
Промышленный способ получения прохлораза, включающий следующие последовательные стадии:
(а) интенсивное перемешивание 2,4,6-трихлорфенола, воды и избытка дибромэтана, доведение реакционной массы до 100°С, равномерное добавление раствора гидроксида натрия по каплям в течение 6 ч, кипячение полученной смеси в течение 2 ч, ее охлаждение до температуры 20°С, выделение полупродукта 2-(2-бромэтокси)-1,3,5-трихлорбензола;
(б) интенсивное перемешивание избытка н-пропиламина и полученного на стадии (а) раствора полупродукта при температуре 20°С, кипячение реакционной смеси в течение 6 ч при температуре 48°С, упаривание избыточного н-пропиламина, растворение остатка в орто-ксилоле, очистка реакционной массы газообразным хлороводородом, фильтрование полученной суспензии, выделение о-ксилольного раствора полупродукта N-(2-(2,4,6-трихлорфенокси)этил)пропан-1-амина;
(в) нагрев трифосгена в орто-ксилоле до 100°С, прикапывание при интенсивном перемешивании полученного на стадии (б) о-ксилольного раствора полупродукта, по окончании прикапывания перемешивание реакционной смеси при нагревании в течение 8 ч, охлаждение реакционной смеси до 80°С;
(г) равномерное добавление при 80°С к образовавшемуся на стадии (в) полупродукту нагретого не ниже 60°С о-ксилольного раствора имидазола и триэтиламина, перемешивание при температуре 80°С в течение 3 ч, охлаждение реакционной смеси до комнатной температуры, выделение продукта прохлораза.
| Образец для ультразвуковой дефектоскопии слоистых материалов | 1983 |
|
SU1224716A1 |
| CN 110092757 A, 06.08.2019 | |||
| Способ получения производных имидазола | 1974 |
|
SU585810A3 |

